User login
Registration and Discount Package Information
Attendees may register for the Annual Meeting in three ways, although online registration is strongly encouraged.
1. Internet registration
Go to https://registration.experientevent.com/ShowAAT171/Attendee/Login.aspx
2. Register by phone
Call the AATS/Experient Customer Service Desk
(800) 424-5249 - Toll-free within the USA
(847) 996-5829 - International
3. Mail/fax registration
Send the meeting registration form, along with a check or credit card information, to:
AATS/Experient
5202 Presidents Court, Suite G100
Frederick, MD 21703
Fax: (301) 694-5124 (fax requires credit card information)
You cannot email a copy of your registration form.
Registration Cancellation Policy
Written requests for cancellations and refunds for registration must be received by April 26, 2017. Refunds will be subject to a $50 administrative fee and will be processed after the meeting. Refunds are not available after April 26, 2017. Requests can be sent to [email protected].
Information for International Travelers
Please be sure to check with your local embassy or consulate regarding the required travel documents for visiting Boston. Travel documents may take time to prepare in order to gain access to the United States. Refunds for registration fees will not be issued by the AATS if attendees are unable to travel into the United States due to inadequate travel documents.
For more information, please visit: http://travel.state.gov/content/visas/english/visit/visitor.html.
For information on what to expect if you are applying for a Visa to the United States and to review the Visa Application Guidelines, go to the U.S. Department of State’s website. Additionally, the International Visitors Office (IVO) of the National Academies has resources on all visa-related issues for the scientific community.
The AATS provides several documents that document your participation in the meeting. Official Letters of Invitation are available for meeting attendees. A personalized letter of invitation can be generated after you have been fully registered. Once your registration is fully paid and all verification documents have been received, a personalized letter of invitation can be generated for you. Please contact [email protected] with your request. Please note that you will receive an email confirmation, which you can also bring to your visa interview.
Please contact [email protected] with any further questions pertaining to invitation letters.
Attendees may register for the Annual Meeting in three ways, although online registration is strongly encouraged.
1. Internet registration
Go to https://registration.experientevent.com/ShowAAT171/Attendee/Login.aspx
2. Register by phone
Call the AATS/Experient Customer Service Desk
(800) 424-5249 - Toll-free within the USA
(847) 996-5829 - International
3. Mail/fax registration
Send the meeting registration form, along with a check or credit card information, to:
AATS/Experient
5202 Presidents Court, Suite G100
Frederick, MD 21703
Fax: (301) 694-5124 (fax requires credit card information)
You cannot email a copy of your registration form.
Registration Cancellation Policy
Written requests for cancellations and refunds for registration must be received by April 26, 2017. Refunds will be subject to a $50 administrative fee and will be processed after the meeting. Refunds are not available after April 26, 2017. Requests can be sent to [email protected].
Information for International Travelers
Please be sure to check with your local embassy or consulate regarding the required travel documents for visiting Boston. Travel documents may take time to prepare in order to gain access to the United States. Refunds for registration fees will not be issued by the AATS if attendees are unable to travel into the United States due to inadequate travel documents.
For more information, please visit: http://travel.state.gov/content/visas/english/visit/visitor.html.
For information on what to expect if you are applying for a Visa to the United States and to review the Visa Application Guidelines, go to the U.S. Department of State’s website. Additionally, the International Visitors Office (IVO) of the National Academies has resources on all visa-related issues for the scientific community.
The AATS provides several documents that document your participation in the meeting. Official Letters of Invitation are available for meeting attendees. A personalized letter of invitation can be generated after you have been fully registered. Once your registration is fully paid and all verification documents have been received, a personalized letter of invitation can be generated for you. Please contact [email protected] with your request. Please note that you will receive an email confirmation, which you can also bring to your visa interview.
Please contact [email protected] with any further questions pertaining to invitation letters.
Attendees may register for the Annual Meeting in three ways, although online registration is strongly encouraged.
1. Internet registration
Go to https://registration.experientevent.com/ShowAAT171/Attendee/Login.aspx
2. Register by phone
Call the AATS/Experient Customer Service Desk
(800) 424-5249 - Toll-free within the USA
(847) 996-5829 - International
3. Mail/fax registration
Send the meeting registration form, along with a check or credit card information, to:
AATS/Experient
5202 Presidents Court, Suite G100
Frederick, MD 21703
Fax: (301) 694-5124 (fax requires credit card information)
You cannot email a copy of your registration form.
Registration Cancellation Policy
Written requests for cancellations and refunds for registration must be received by April 26, 2017. Refunds will be subject to a $50 administrative fee and will be processed after the meeting. Refunds are not available after April 26, 2017. Requests can be sent to [email protected].
Information for International Travelers
Please be sure to check with your local embassy or consulate regarding the required travel documents for visiting Boston. Travel documents may take time to prepare in order to gain access to the United States. Refunds for registration fees will not be issued by the AATS if attendees are unable to travel into the United States due to inadequate travel documents.
For more information, please visit: http://travel.state.gov/content/visas/english/visit/visitor.html.
For information on what to expect if you are applying for a Visa to the United States and to review the Visa Application Guidelines, go to the U.S. Department of State’s website. Additionally, the International Visitors Office (IVO) of the National Academies has resources on all visa-related issues for the scientific community.
The AATS provides several documents that document your participation in the meeting. Official Letters of Invitation are available for meeting attendees. A personalized letter of invitation can be generated after you have been fully registered. Once your registration is fully paid and all verification documents have been received, a personalized letter of invitation can be generated for you. Please contact [email protected] with your request. Please note that you will receive an email confirmation, which you can also bring to your visa interview.
Please contact [email protected] with any further questions pertaining to invitation letters.
Epilepsy or Seizure Disorder? The Effect of Cultural and Socioeconomic Factors on Self-Reported Prevalence
Barbara L. Kroner, PhD, MPH
Disclosure Information:
Dr. Kroner has no disclosures.
Dr. Kroner was lead author on the screening survey study discussed in this article (Kroner BL, et al. Epilepsy or seizure disorder? The effect of cultural and socioeconomic factors on self-reported prevalence. Epilepsy Behav. 2016;62:214-217).
Approximately three-quarters of 6420 adult residents of the District of Columbia (DC) screened for epilepsy said they were diagnosed with a seizure disorder rather than epilepsy when given both choices in our population-based survey. The term ‘seizure disorder’ was chosen consistently more often than ‘epilepsy’ in all demographic subgroups, including gender, age, income, education, race, and ethnicity—suggesting there may be a general lack of understanding among patients about what epilepsy means. An implication of these findings is that, although a wealth of information about epilepsy is publicly available for or disseminated by medical providers to people with epilepsy, they may not be getting the message.
Epilepsy can be a complex diagnosis, and people may not associate their seizure condition with the term epilepsy for a variety of reasons. Such reasons include stigma, symptomatic etiology, cultural background, and the terminology used by their medical providers. When there is a known cause for the seizures, such as injury, cancer, stroke, or underlying syndrome, the affected individuals may be more inclined to associate their diagnosis with descriptive phrases that include the term seizure disorder. Medical providers may also tend to use these phrases more often with certain demographic populations, such as the elderly and those with diverse cultural backgrounds.
We conducted the survey in DC, one of the nation’s most culturally, racially, and economically diverse populations, to estimate the prevalence and incidence of epilepsy in various demographic categories associated with the clinical term epilepsy or the more generic term seizure disorder. A single-page, bilingual epilepsy survey was mailed to a representative sample of 20,000 households; it included the standard epilepsy screening question, “Ever diagnosed with epilepsy or a seizure disorder?” Rather than the traditional Yes/No answer choices, we provided answer choices of No, Yes epilepsy, and Yes seizure disorder. A positive response to the screening question, “Currently taking any medicine to control seizures?” was categorized as having active epilepsy or seizure disorder. All response data then were weighted to the DC population size at the time the survey was conducted and to reflect the sampling design. Respondents indicating a diagnosis of epilepsy or seizure disorder were sent a follow-up survey about the etiology of the seizures. The follow-up surveys were reviewed by an epileptologist, who categorized the etiology for each respondent as symptomatic or not symptomatic.
A total of 6420 adults responded to the screening survey, for an overall adjusted response rate of 37%. Among the respondents, there were more females (60.5%) than males (39.5%) and slightly more blacks (45.2%) than whites (44.4%). Half of the adults were at least 50 years of age, and 40% had attended at least some graduate school. There were 107 respondents who reported they had received a diagnosis of epilepsy or a seizure disorder at some time in their life. Subsequent weighted estimates and 95% confidence intervals (CI) for individual lifetime prevalence were 0.54% (95% CI, 0.34-0.74) for epilepsy and 1.30% (95% CI, 0.98-1.62) for seizure disorder, for an overall lifetime prevalence of 1.84% (95% CI, 1.47-2.21) for either condition. Adults with active epilepsy were also significantly more likely to identify their condition as seizure disorder than as epilepsy—0.70% (95% CI, 0.47-0.94) vs 0.21% (95% CI, 0.08-0.33).
For lifetime prevalence, seizure disorder was reported 2 to 3 times more often than epilepsy in almost all demographic subgroups except those 18 to 30 years of age, in whom the lifetime prevalence rates were equal for the 2 conditions. The prevalence of seizure disorder was significantly higher than epilepsy (non-overlapping 95% CI) among non-Hispanic blacks, females, those 50 years of age or older, those with only a high school education, those living in low-income neighborhoods, and those who had resided in DC for at least 5 years.
Similarly, the prevalence of active seizure disorder was higher than the prevalence of active epilepsy in all subgroups, including those 18 to 30 years of age. Active seizure disorder was significantly higher than active epilepsy among the same 6 demographic subgroups that were identified for lifetime prevalence: non-Hispanic blacks, females, those 50 years of age or older, those with only a high school education, those living in low-income neighborhoods, and those who had resided in DC for at least 5years. In 4 subgroups (young adults, Hispanic, race other than white or black, and those residing in DC for less than 5 years), all of the affected respondents identified their condition as seizure disorder, not as epilepsy.
Additional surveys about epilepsy and seizure history were completed by 70 (65.4%) of the 107 respondents. Of these 70 respondents, 22 (31.4%) reported they were diagnosed with epilepsy and 48 (68.6%) reported they were diagnosed with seizure disorder. Symptomatic seizure etiology was noted by 35 (50%) of the respondents, and these were significantly more likely to identify their condition as seizure disorder than as epilepsy (29 [60.4%] vs 6 [27.3%]; P=.02). Specific causes of seizures in both groups included trauma only, stroke only, drugs or alcohol only, trauma plus stroke, alcohol or drug use, and chronic medical condition such as multiple sclerosis, hypertension, congenital arteriovenous malformation, migraines, and brain tumor. Of note, in the overall group of 19 respondents who had seizures due to trauma, 3 had gunshot wounds to the head, which might occur less frequently in the general adult epilepsy population than in DC. Because of the small numbers involved, we were not able to calculate prevalence rates for the symptomatic respondents stratified by demographic characteristics.
The consistently higher number of respondents who self-identified with seizure disorder rather than with epilepsy across all subgroups, including the highly educated, suggests that those who chose seizure disorder are unlikely to be false positive cases of epilepsy. In addition to respondents in the lower socioeconomic groups, females identified significantly more often with seizure disorder than with epilepsy, a finding that may be related to social factors such as stigma. Also, cultural factors and beliefs likely influenced the preference for reporting seizure disorder over epilepsy.
In summary, our findings suggest that both epilepsy and seizure disorder and perhaps other culturally sensitive terms need to be used with patients in clinical practice to ensure they understand their diagnosis and to maximize adherence to treatment and disease management. Patients should understand the synonymy of the 2 terms, particularly because of the overabundance of information available to the public and on the internet that is labeled as epilepsy-specific. In addition, epilepsy education and awareness campaigns ideally should include a more diverse definition of epilepsy in their messages to reach the broadest population of affected individuals, many of whom are in socioeconomic groups at high risk for epilepsy.
To read the full article about this study, please click here.
Barbara L. Kroner, PhD, MPH
Disclosure Information:
Dr. Kroner has no disclosures.
Dr. Kroner was lead author on the screening survey study discussed in this article (Kroner BL, et al. Epilepsy or seizure disorder? The effect of cultural and socioeconomic factors on self-reported prevalence. Epilepsy Behav. 2016;62:214-217).
Approximately three-quarters of 6420 adult residents of the District of Columbia (DC) screened for epilepsy said they were diagnosed with a seizure disorder rather than epilepsy when given both choices in our population-based survey. The term ‘seizure disorder’ was chosen consistently more often than ‘epilepsy’ in all demographic subgroups, including gender, age, income, education, race, and ethnicity—suggesting there may be a general lack of understanding among patients about what epilepsy means. An implication of these findings is that, although a wealth of information about epilepsy is publicly available for or disseminated by medical providers to people with epilepsy, they may not be getting the message.
Epilepsy can be a complex diagnosis, and people may not associate their seizure condition with the term epilepsy for a variety of reasons. Such reasons include stigma, symptomatic etiology, cultural background, and the terminology used by their medical providers. When there is a known cause for the seizures, such as injury, cancer, stroke, or underlying syndrome, the affected individuals may be more inclined to associate their diagnosis with descriptive phrases that include the term seizure disorder. Medical providers may also tend to use these phrases more often with certain demographic populations, such as the elderly and those with diverse cultural backgrounds.
We conducted the survey in DC, one of the nation’s most culturally, racially, and economically diverse populations, to estimate the prevalence and incidence of epilepsy in various demographic categories associated with the clinical term epilepsy or the more generic term seizure disorder. A single-page, bilingual epilepsy survey was mailed to a representative sample of 20,000 households; it included the standard epilepsy screening question, “Ever diagnosed with epilepsy or a seizure disorder?” Rather than the traditional Yes/No answer choices, we provided answer choices of No, Yes epilepsy, and Yes seizure disorder. A positive response to the screening question, “Currently taking any medicine to control seizures?” was categorized as having active epilepsy or seizure disorder. All response data then were weighted to the DC population size at the time the survey was conducted and to reflect the sampling design. Respondents indicating a diagnosis of epilepsy or seizure disorder were sent a follow-up survey about the etiology of the seizures. The follow-up surveys were reviewed by an epileptologist, who categorized the etiology for each respondent as symptomatic or not symptomatic.
A total of 6420 adults responded to the screening survey, for an overall adjusted response rate of 37%. Among the respondents, there were more females (60.5%) than males (39.5%) and slightly more blacks (45.2%) than whites (44.4%). Half of the adults were at least 50 years of age, and 40% had attended at least some graduate school. There were 107 respondents who reported they had received a diagnosis of epilepsy or a seizure disorder at some time in their life. Subsequent weighted estimates and 95% confidence intervals (CI) for individual lifetime prevalence were 0.54% (95% CI, 0.34-0.74) for epilepsy and 1.30% (95% CI, 0.98-1.62) for seizure disorder, for an overall lifetime prevalence of 1.84% (95% CI, 1.47-2.21) for either condition. Adults with active epilepsy were also significantly more likely to identify their condition as seizure disorder than as epilepsy—0.70% (95% CI, 0.47-0.94) vs 0.21% (95% CI, 0.08-0.33).
For lifetime prevalence, seizure disorder was reported 2 to 3 times more often than epilepsy in almost all demographic subgroups except those 18 to 30 years of age, in whom the lifetime prevalence rates were equal for the 2 conditions. The prevalence of seizure disorder was significantly higher than epilepsy (non-overlapping 95% CI) among non-Hispanic blacks, females, those 50 years of age or older, those with only a high school education, those living in low-income neighborhoods, and those who had resided in DC for at least 5 years.
Similarly, the prevalence of active seizure disorder was higher than the prevalence of active epilepsy in all subgroups, including those 18 to 30 years of age. Active seizure disorder was significantly higher than active epilepsy among the same 6 demographic subgroups that were identified for lifetime prevalence: non-Hispanic blacks, females, those 50 years of age or older, those with only a high school education, those living in low-income neighborhoods, and those who had resided in DC for at least 5years. In 4 subgroups (young adults, Hispanic, race other than white or black, and those residing in DC for less than 5 years), all of the affected respondents identified their condition as seizure disorder, not as epilepsy.
Additional surveys about epilepsy and seizure history were completed by 70 (65.4%) of the 107 respondents. Of these 70 respondents, 22 (31.4%) reported they were diagnosed with epilepsy and 48 (68.6%) reported they were diagnosed with seizure disorder. Symptomatic seizure etiology was noted by 35 (50%) of the respondents, and these were significantly more likely to identify their condition as seizure disorder than as epilepsy (29 [60.4%] vs 6 [27.3%]; P=.02). Specific causes of seizures in both groups included trauma only, stroke only, drugs or alcohol only, trauma plus stroke, alcohol or drug use, and chronic medical condition such as multiple sclerosis, hypertension, congenital arteriovenous malformation, migraines, and brain tumor. Of note, in the overall group of 19 respondents who had seizures due to trauma, 3 had gunshot wounds to the head, which might occur less frequently in the general adult epilepsy population than in DC. Because of the small numbers involved, we were not able to calculate prevalence rates for the symptomatic respondents stratified by demographic characteristics.
The consistently higher number of respondents who self-identified with seizure disorder rather than with epilepsy across all subgroups, including the highly educated, suggests that those who chose seizure disorder are unlikely to be false positive cases of epilepsy. In addition to respondents in the lower socioeconomic groups, females identified significantly more often with seizure disorder than with epilepsy, a finding that may be related to social factors such as stigma. Also, cultural factors and beliefs likely influenced the preference for reporting seizure disorder over epilepsy.
In summary, our findings suggest that both epilepsy and seizure disorder and perhaps other culturally sensitive terms need to be used with patients in clinical practice to ensure they understand their diagnosis and to maximize adherence to treatment and disease management. Patients should understand the synonymy of the 2 terms, particularly because of the overabundance of information available to the public and on the internet that is labeled as epilepsy-specific. In addition, epilepsy education and awareness campaigns ideally should include a more diverse definition of epilepsy in their messages to reach the broadest population of affected individuals, many of whom are in socioeconomic groups at high risk for epilepsy.
To read the full article about this study, please click here.
Barbara L. Kroner, PhD, MPH
Disclosure Information:
Dr. Kroner has no disclosures.
Dr. Kroner was lead author on the screening survey study discussed in this article (Kroner BL, et al. Epilepsy or seizure disorder? The effect of cultural and socioeconomic factors on self-reported prevalence. Epilepsy Behav. 2016;62:214-217).
Approximately three-quarters of 6420 adult residents of the District of Columbia (DC) screened for epilepsy said they were diagnosed with a seizure disorder rather than epilepsy when given both choices in our population-based survey. The term ‘seizure disorder’ was chosen consistently more often than ‘epilepsy’ in all demographic subgroups, including gender, age, income, education, race, and ethnicity—suggesting there may be a general lack of understanding among patients about what epilepsy means. An implication of these findings is that, although a wealth of information about epilepsy is publicly available for or disseminated by medical providers to people with epilepsy, they may not be getting the message.
Epilepsy can be a complex diagnosis, and people may not associate their seizure condition with the term epilepsy for a variety of reasons. Such reasons include stigma, symptomatic etiology, cultural background, and the terminology used by their medical providers. When there is a known cause for the seizures, such as injury, cancer, stroke, or underlying syndrome, the affected individuals may be more inclined to associate their diagnosis with descriptive phrases that include the term seizure disorder. Medical providers may also tend to use these phrases more often with certain demographic populations, such as the elderly and those with diverse cultural backgrounds.
We conducted the survey in DC, one of the nation’s most culturally, racially, and economically diverse populations, to estimate the prevalence and incidence of epilepsy in various demographic categories associated with the clinical term epilepsy or the more generic term seizure disorder. A single-page, bilingual epilepsy survey was mailed to a representative sample of 20,000 households; it included the standard epilepsy screening question, “Ever diagnosed with epilepsy or a seizure disorder?” Rather than the traditional Yes/No answer choices, we provided answer choices of No, Yes epilepsy, and Yes seizure disorder. A positive response to the screening question, “Currently taking any medicine to control seizures?” was categorized as having active epilepsy or seizure disorder. All response data then were weighted to the DC population size at the time the survey was conducted and to reflect the sampling design. Respondents indicating a diagnosis of epilepsy or seizure disorder were sent a follow-up survey about the etiology of the seizures. The follow-up surveys were reviewed by an epileptologist, who categorized the etiology for each respondent as symptomatic or not symptomatic.
A total of 6420 adults responded to the screening survey, for an overall adjusted response rate of 37%. Among the respondents, there were more females (60.5%) than males (39.5%) and slightly more blacks (45.2%) than whites (44.4%). Half of the adults were at least 50 years of age, and 40% had attended at least some graduate school. There were 107 respondents who reported they had received a diagnosis of epilepsy or a seizure disorder at some time in their life. Subsequent weighted estimates and 95% confidence intervals (CI) for individual lifetime prevalence were 0.54% (95% CI, 0.34-0.74) for epilepsy and 1.30% (95% CI, 0.98-1.62) for seizure disorder, for an overall lifetime prevalence of 1.84% (95% CI, 1.47-2.21) for either condition. Adults with active epilepsy were also significantly more likely to identify their condition as seizure disorder than as epilepsy—0.70% (95% CI, 0.47-0.94) vs 0.21% (95% CI, 0.08-0.33).
For lifetime prevalence, seizure disorder was reported 2 to 3 times more often than epilepsy in almost all demographic subgroups except those 18 to 30 years of age, in whom the lifetime prevalence rates were equal for the 2 conditions. The prevalence of seizure disorder was significantly higher than epilepsy (non-overlapping 95% CI) among non-Hispanic blacks, females, those 50 years of age or older, those with only a high school education, those living in low-income neighborhoods, and those who had resided in DC for at least 5 years.
Similarly, the prevalence of active seizure disorder was higher than the prevalence of active epilepsy in all subgroups, including those 18 to 30 years of age. Active seizure disorder was significantly higher than active epilepsy among the same 6 demographic subgroups that were identified for lifetime prevalence: non-Hispanic blacks, females, those 50 years of age or older, those with only a high school education, those living in low-income neighborhoods, and those who had resided in DC for at least 5years. In 4 subgroups (young adults, Hispanic, race other than white or black, and those residing in DC for less than 5 years), all of the affected respondents identified their condition as seizure disorder, not as epilepsy.
Additional surveys about epilepsy and seizure history were completed by 70 (65.4%) of the 107 respondents. Of these 70 respondents, 22 (31.4%) reported they were diagnosed with epilepsy and 48 (68.6%) reported they were diagnosed with seizure disorder. Symptomatic seizure etiology was noted by 35 (50%) of the respondents, and these were significantly more likely to identify their condition as seizure disorder than as epilepsy (29 [60.4%] vs 6 [27.3%]; P=.02). Specific causes of seizures in both groups included trauma only, stroke only, drugs or alcohol only, trauma plus stroke, alcohol or drug use, and chronic medical condition such as multiple sclerosis, hypertension, congenital arteriovenous malformation, migraines, and brain tumor. Of note, in the overall group of 19 respondents who had seizures due to trauma, 3 had gunshot wounds to the head, which might occur less frequently in the general adult epilepsy population than in DC. Because of the small numbers involved, we were not able to calculate prevalence rates for the symptomatic respondents stratified by demographic characteristics.
The consistently higher number of respondents who self-identified with seizure disorder rather than with epilepsy across all subgroups, including the highly educated, suggests that those who chose seizure disorder are unlikely to be false positive cases of epilepsy. In addition to respondents in the lower socioeconomic groups, females identified significantly more often with seizure disorder than with epilepsy, a finding that may be related to social factors such as stigma. Also, cultural factors and beliefs likely influenced the preference for reporting seizure disorder over epilepsy.
In summary, our findings suggest that both epilepsy and seizure disorder and perhaps other culturally sensitive terms need to be used with patients in clinical practice to ensure they understand their diagnosis and to maximize adherence to treatment and disease management. Patients should understand the synonymy of the 2 terms, particularly because of the overabundance of information available to the public and on the internet that is labeled as epilepsy-specific. In addition, epilepsy education and awareness campaigns ideally should include a more diverse definition of epilepsy in their messages to reach the broadest population of affected individuals, many of whom are in socioeconomic groups at high risk for epilepsy.
To read the full article about this study, please click here.
BRCA2 mutations linked to greater risk for pancreatic cancer
MIAMI BEACH – Although population-wide screening for pancreatic cancer is considered unfeasible and costly, new evidence suggests a benefit to screening a select population: people who test positive for BRCA2 genetic mutations.
Cross-sectional imaging of 117 people with BRCA2 mutations revealed pancreatic abnormalities in 10 patients, including a patient with pancreatic cancer whose only symptom was unexplained weight loss.
Pancreatic cancer is not as common as are some other malignancies, with an incidence estimated between 1% and 3%. However, it is a particularly deadly form of cancer, with only 7.7% of people living to 5 years after diagnosis, according to data from the National Cancer Institute.
A relatively low incidence is a good thing, but it also limits widespread screening. “There is a low predictive value of screening the population at large, and it is not considered cost effective,” said Eugene P. Ceppa, MD, a general surgeon at IU Health University Hospital, Indianapolis. However, patients at high risk for pancreatic adenocarcinoma might be worth targeting for screening, he added.

“This represents a 21% increase in the chance of pancreatic cancer in these patients,” Dr. Ceppa said.
Buoyed by these and other findings, Dr. Ceppa and his colleagues launched a study of their own. “Our hypothesis is that screening all BRCA2s would identify more patients with pancreatic cancer,” he said at the annual meeting of the Americas Hepato-Pancreato-Biliary Association.
Dr. Ceppa and coinvestigators reviewed electronic medical records at their institution from 2005 to 2015. They identified 204 BRCA mutation carriers, and after excluding 87 BRCA1 positive patients, further assessed the 117 with documented BRCA2 mutations. A total 47 people (40%) of this group had undergone cross-sectional imaging. The images were initially reviewed, and then re-reviewed for the study, by radiologists with specific expertise in pancreatology.
The cross-sectional imaging revealed pancreatic abnormalities in 10 people, including 1 patient with a pancreatic ductal adenocarcinoma located in the head of the pancreas. Another nine patients had intraductal papillary mucinous neoplasms (IPMNs). There were no significant demographic or clinical differences between the groups of patients with and without the imaging abnormalities, Dr. Ceppa said.
The investigators also compared the patients with BRCA2 mutations against a historical cohort representing the general population. They found 21% of patients with BRCA2 had a defined pancreatic abnormality, compared with 8% in the general population. The difference was statistically significant (P = .007).
Interestingly, the same comparison also revealed a rate of IPMN of 19%, versus 1%, respectively (P less than .001). “BRCA2 mutation carriers have significantly higher incidence of IPMN than the general population,” Dr. Ceppa said.
The study results support a high-risk screening protocol in asymptomatic BRCA patients regardless of family history, he said. In fact, a high-risk screening protocol implemented at his institution in 2013 led to a 14% detection rate of pancreatic cancer among BRCA2-positive patients, compared with a 3% rate in the general population.
“Your most significant finding might be the more IPMN patients – but how do we follow them, and will it be cost effective?” asked invited discussant Matthew J. Weiss, MD, of Johns Hopkins Medicine in Baltimore.
One of the most notable impacts of instituting the high-risk screening protocol has been an increase in patient referrals from other specialists at Dr. Ceppa’s institution. “I’ve looked at every single breast surgeon in our department, and I know how each of them are referring,” he explained.
Following initial screening of BRCA2 mutation patients, Dr. Ceppa repeats screening at 6 months, 1 year, and then annually. “However, some insurers may balk at our recommendations for frequency of screening,” he noted.
Dr. Ceppa and Dr. Weiss had no relevant financial disclosures.
MIAMI BEACH – Although population-wide screening for pancreatic cancer is considered unfeasible and costly, new evidence suggests a benefit to screening a select population: people who test positive for BRCA2 genetic mutations.
Cross-sectional imaging of 117 people with BRCA2 mutations revealed pancreatic abnormalities in 10 patients, including a patient with pancreatic cancer whose only symptom was unexplained weight loss.
Pancreatic cancer is not as common as are some other malignancies, with an incidence estimated between 1% and 3%. However, it is a particularly deadly form of cancer, with only 7.7% of people living to 5 years after diagnosis, according to data from the National Cancer Institute.
A relatively low incidence is a good thing, but it also limits widespread screening. “There is a low predictive value of screening the population at large, and it is not considered cost effective,” said Eugene P. Ceppa, MD, a general surgeon at IU Health University Hospital, Indianapolis. However, patients at high risk for pancreatic adenocarcinoma might be worth targeting for screening, he added.
“This represents a 21% increase in the chance of pancreatic cancer in these patients,” Dr. Ceppa said.
Buoyed by these and other findings, Dr. Ceppa and his colleagues launched a study of their own. “Our hypothesis is that screening all BRCA2s would identify more patients with pancreatic cancer,” he said at the annual meeting of the Americas Hepato-Pancreato-Biliary Association.
Dr. Ceppa and coinvestigators reviewed electronic medical records at their institution from 2005 to 2015. They identified 204 BRCA mutation carriers, and after excluding 87 BRCA1 positive patients, further assessed the 117 with documented BRCA2 mutations. A total 47 people (40%) of this group had undergone cross-sectional imaging. The images were initially reviewed, and then re-reviewed for the study, by radiologists with specific expertise in pancreatology.
The cross-sectional imaging revealed pancreatic abnormalities in 10 people, including 1 patient with a pancreatic ductal adenocarcinoma located in the head of the pancreas. Another nine patients had intraductal papillary mucinous neoplasms (IPMNs). There were no significant demographic or clinical differences between the groups of patients with and without the imaging abnormalities, Dr. Ceppa said.
The investigators also compared the patients with BRCA2 mutations against a historical cohort representing the general population. They found 21% of patients with BRCA2 had a defined pancreatic abnormality, compared with 8% in the general population. The difference was statistically significant (P = .007).
Interestingly, the same comparison also revealed a rate of IPMN of 19%, versus 1%, respectively (P less than .001). “BRCA2 mutation carriers have significantly higher incidence of IPMN than the general population,” Dr. Ceppa said.
The study results support a high-risk screening protocol in asymptomatic BRCA patients regardless of family history, he said. In fact, a high-risk screening protocol implemented at his institution in 2013 led to a 14% detection rate of pancreatic cancer among BRCA2-positive patients, compared with a 3% rate in the general population.
“Your most significant finding might be the more IPMN patients – but how do we follow them, and will it be cost effective?” asked invited discussant Matthew J. Weiss, MD, of Johns Hopkins Medicine in Baltimore.
One of the most notable impacts of instituting the high-risk screening protocol has been an increase in patient referrals from other specialists at Dr. Ceppa’s institution. “I’ve looked at every single breast surgeon in our department, and I know how each of them are referring,” he explained.
Following initial screening of BRCA2 mutation patients, Dr. Ceppa repeats screening at 6 months, 1 year, and then annually. “However, some insurers may balk at our recommendations for frequency of screening,” he noted.
Dr. Ceppa and Dr. Weiss had no relevant financial disclosures.
MIAMI BEACH – Although population-wide screening for pancreatic cancer is considered unfeasible and costly, new evidence suggests a benefit to screening a select population: people who test positive for BRCA2 genetic mutations.
Cross-sectional imaging of 117 people with BRCA2 mutations revealed pancreatic abnormalities in 10 patients, including a patient with pancreatic cancer whose only symptom was unexplained weight loss.
Pancreatic cancer is not as common as are some other malignancies, with an incidence estimated between 1% and 3%. However, it is a particularly deadly form of cancer, with only 7.7% of people living to 5 years after diagnosis, according to data from the National Cancer Institute.
A relatively low incidence is a good thing, but it also limits widespread screening. “There is a low predictive value of screening the population at large, and it is not considered cost effective,” said Eugene P. Ceppa, MD, a general surgeon at IU Health University Hospital, Indianapolis. However, patients at high risk for pancreatic adenocarcinoma might be worth targeting for screening, he added.
“This represents a 21% increase in the chance of pancreatic cancer in these patients,” Dr. Ceppa said.
Buoyed by these and other findings, Dr. Ceppa and his colleagues launched a study of their own. “Our hypothesis is that screening all BRCA2s would identify more patients with pancreatic cancer,” he said at the annual meeting of the Americas Hepato-Pancreato-Biliary Association.
Dr. Ceppa and coinvestigators reviewed electronic medical records at their institution from 2005 to 2015. They identified 204 BRCA mutation carriers, and after excluding 87 BRCA1 positive patients, further assessed the 117 with documented BRCA2 mutations. A total 47 people (40%) of this group had undergone cross-sectional imaging. The images were initially reviewed, and then re-reviewed for the study, by radiologists with specific expertise in pancreatology.
The cross-sectional imaging revealed pancreatic abnormalities in 10 people, including 1 patient with a pancreatic ductal adenocarcinoma located in the head of the pancreas. Another nine patients had intraductal papillary mucinous neoplasms (IPMNs). There were no significant demographic or clinical differences between the groups of patients with and without the imaging abnormalities, Dr. Ceppa said.
The investigators also compared the patients with BRCA2 mutations against a historical cohort representing the general population. They found 21% of patients with BRCA2 had a defined pancreatic abnormality, compared with 8% in the general population. The difference was statistically significant (P = .007).
Interestingly, the same comparison also revealed a rate of IPMN of 19%, versus 1%, respectively (P less than .001). “BRCA2 mutation carriers have significantly higher incidence of IPMN than the general population,” Dr. Ceppa said.
The study results support a high-risk screening protocol in asymptomatic BRCA patients regardless of family history, he said. In fact, a high-risk screening protocol implemented at his institution in 2013 led to a 14% detection rate of pancreatic cancer among BRCA2-positive patients, compared with a 3% rate in the general population.
“Your most significant finding might be the more IPMN patients – but how do we follow them, and will it be cost effective?” asked invited discussant Matthew J. Weiss, MD, of Johns Hopkins Medicine in Baltimore.
One of the most notable impacts of instituting the high-risk screening protocol has been an increase in patient referrals from other specialists at Dr. Ceppa’s institution. “I’ve looked at every single breast surgeon in our department, and I know how each of them are referring,” he explained.
Following initial screening of BRCA2 mutation patients, Dr. Ceppa repeats screening at 6 months, 1 year, and then annually. “However, some insurers may balk at our recommendations for frequency of screening,” he noted.
Dr. Ceppa and Dr. Weiss had no relevant financial disclosures.
Key clinical point: Although general population screening for pancreatic cancer is considered costly, with a low predictive value, targeting screening to patients with BRCA2 mutations could detect more cases of this deadly disease.
Major finding: People with BRCA2 mutations had a significantly greater incidence of intraductal papillary mucinous neoplasms, 19%, versus 1% in the general population (P less than .001).
Data source: Retrospective study of electronic medical records of 117 patients with BRCA2 mutations at a single academic institution.
Disclosures: Dr. Ceppa and Dr. Weiss had no relevant financial disclosures.
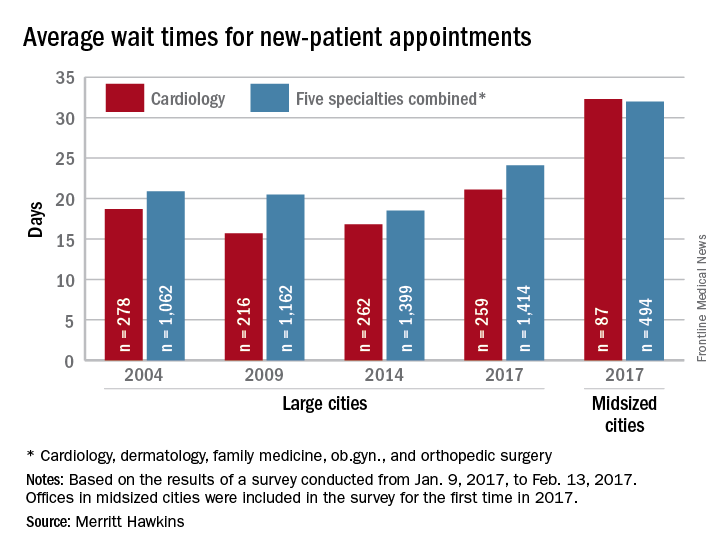
Wait times for cardiologist visits up 4.3 days since 2014
New patients are waiting 4.3 days longer for an appointment with a cardiologist in 2017 than they did in 2014, according to physician recruitment firm Merritt Hawkins.
The average wait time for a new patient to see a cardiologist for a checkup was 21.1 days in 2017, an almost 26% increase from the 16.8 days reported in 2014. Investigators called and made appointments with 259 randomly selected cardiologists in 15 large cities in January and February. It was the fourth such survey the company has conducted since 2004.
This year, the survey also included cardiologists in 15 midsized cities for the first time. The average wait time in these cities was even longer: 32.3 days for the 87 offices contacted. Odessa, Tex., had the longest average wait in a midsized city – 63 days – while Albany, N.Y., and Cedar Rapids, Iowa, had a shortest-for-the-group wait of 10 days, Merritt Hawkins reported. In the large cities, the longest average wait was 45 days (Boston) and the shortest wait was 12 days (Dallas and Houston).
The survey also included four other specialties – dermatology, family medicine, ob.gyn., and orthopedic surgery – and the average wait time for a new-patient appointment for all 1,414 physicians in all five specialties in the 15 large cities was 24.1 days, an increase of 30% from 2014. The average wait time for all specialties in the midsized cities was 32 days for the 494 offices surveyed, the company said.
“Physician appointment wait times are the longest they have been since we began conducting the survey,” Mark Smith, president of Merritt Hawkins, said in a written statement. “Growing physician appointment wait times are a significant indicator that the nation is experiencing a shortage of physicians.”
New patients are waiting 4.3 days longer for an appointment with a cardiologist in 2017 than they did in 2014, according to physician recruitment firm Merritt Hawkins.
The average wait time for a new patient to see a cardiologist for a checkup was 21.1 days in 2017, an almost 26% increase from the 16.8 days reported in 2014. Investigators called and made appointments with 259 randomly selected cardiologists in 15 large cities in January and February. It was the fourth such survey the company has conducted since 2004.
This year, the survey also included cardiologists in 15 midsized cities for the first time. The average wait time in these cities was even longer: 32.3 days for the 87 offices contacted. Odessa, Tex., had the longest average wait in a midsized city – 63 days – while Albany, N.Y., and Cedar Rapids, Iowa, had a shortest-for-the-group wait of 10 days, Merritt Hawkins reported. In the large cities, the longest average wait was 45 days (Boston) and the shortest wait was 12 days (Dallas and Houston).
The survey also included four other specialties – dermatology, family medicine, ob.gyn., and orthopedic surgery – and the average wait time for a new-patient appointment for all 1,414 physicians in all five specialties in the 15 large cities was 24.1 days, an increase of 30% from 2014. The average wait time for all specialties in the midsized cities was 32 days for the 494 offices surveyed, the company said.
“Physician appointment wait times are the longest they have been since we began conducting the survey,” Mark Smith, president of Merritt Hawkins, said in a written statement. “Growing physician appointment wait times are a significant indicator that the nation is experiencing a shortage of physicians.”
New patients are waiting 4.3 days longer for an appointment with a cardiologist in 2017 than they did in 2014, according to physician recruitment firm Merritt Hawkins.
The average wait time for a new patient to see a cardiologist for a checkup was 21.1 days in 2017, an almost 26% increase from the 16.8 days reported in 2014. Investigators called and made appointments with 259 randomly selected cardiologists in 15 large cities in January and February. It was the fourth such survey the company has conducted since 2004.
This year, the survey also included cardiologists in 15 midsized cities for the first time. The average wait time in these cities was even longer: 32.3 days for the 87 offices contacted. Odessa, Tex., had the longest average wait in a midsized city – 63 days – while Albany, N.Y., and Cedar Rapids, Iowa, had a shortest-for-the-group wait of 10 days, Merritt Hawkins reported. In the large cities, the longest average wait was 45 days (Boston) and the shortest wait was 12 days (Dallas and Houston).
The survey also included four other specialties – dermatology, family medicine, ob.gyn., and orthopedic surgery – and the average wait time for a new-patient appointment for all 1,414 physicians in all five specialties in the 15 large cities was 24.1 days, an increase of 30% from 2014. The average wait time for all specialties in the midsized cities was 32 days for the 494 offices surveyed, the company said.
“Physician appointment wait times are the longest they have been since we began conducting the survey,” Mark Smith, president of Merritt Hawkins, said in a written statement. “Growing physician appointment wait times are a significant indicator that the nation is experiencing a shortage of physicians.”
Visit some of Toronto’s best during CHEST 2017
Get ready to visit the metropolitan hub of Canada. Explore new grounds with the chest medicine community for current pulmonary, critical care, and sleep medicine topics presented by world-renowned faculty in a variety of innovative instruction formats.
You will have access to our cutting-edge education, Oct 28 - Nov 1, but don’t forget to take advantage of all that Toronto has to offer.
Food
![]()
Le Petit Dejeune offers an ever-changing menu that ranges from less expensive items, like soup, sandwiches, and salads, to some pricier stuffed crepes, quiche, and eggs florentine. While most Sundays, Saving Grace is packed, but there’s only a 15-minute wait, and the atmosphere is quite pleasant. Looking for the perfect cinnamon bun? Rosen’s Cinnamon Buns is the place to go. But you have to look closely for the bakery’s name, since the sign above the window still advertises the hair salon that used to reside in the same spot!
Nature parks
One of the city’s largest and oldest parks, High Park is Toronto’s version of New York City’s Central Park. There’s plenty to enjoy, such as Grenadier Pond, numerous ravine-based hiking trails, playgrounds, athletic areas, restaurants, a museum, and even a zoo!
If you want a different type of nature excursion, there is always beautiful Niagara Falls, Ontario, which is just a short drive from Toronto. Don’t miss seeing the Tesla monument in Queen Victoria Park, or go 10 minutes north of the Falls to the Botanical Gardens, home to the Butterfly Conservatory with over 2,000 butterflies.
Relaxation
After eventful days of absorbing all the new science CHEST 2017 has to offer, you may want to relax your mind and body. Elmwood spa, located in downtown Toronto, is where “four spacious floors of treatment and renewal options mean that Elmwood Spa can provide the convenience and flexibility to cater to demanding schedules,” according to Elmwood.
Learn more about Toronto opportunities at blogTO.com, and find out more about CHEST 2017 at chestmeeting.chestnet.org.
Get ready to visit the metropolitan hub of Canada. Explore new grounds with the chest medicine community for current pulmonary, critical care, and sleep medicine topics presented by world-renowned faculty in a variety of innovative instruction formats.
You will have access to our cutting-edge education, Oct 28 - Nov 1, but don’t forget to take advantage of all that Toronto has to offer.
Food
![]()
Le Petit Dejeune offers an ever-changing menu that ranges from less expensive items, like soup, sandwiches, and salads, to some pricier stuffed crepes, quiche, and eggs florentine. While most Sundays, Saving Grace is packed, but there’s only a 15-minute wait, and the atmosphere is quite pleasant. Looking for the perfect cinnamon bun? Rosen’s Cinnamon Buns is the place to go. But you have to look closely for the bakery’s name, since the sign above the window still advertises the hair salon that used to reside in the same spot!
Nature parks
One of the city’s largest and oldest parks, High Park is Toronto’s version of New York City’s Central Park. There’s plenty to enjoy, such as Grenadier Pond, numerous ravine-based hiking trails, playgrounds, athletic areas, restaurants, a museum, and even a zoo!
If you want a different type of nature excursion, there is always beautiful Niagara Falls, Ontario, which is just a short drive from Toronto. Don’t miss seeing the Tesla monument in Queen Victoria Park, or go 10 minutes north of the Falls to the Botanical Gardens, home to the Butterfly Conservatory with over 2,000 butterflies.
Relaxation
After eventful days of absorbing all the new science CHEST 2017 has to offer, you may want to relax your mind and body. Elmwood spa, located in downtown Toronto, is where “four spacious floors of treatment and renewal options mean that Elmwood Spa can provide the convenience and flexibility to cater to demanding schedules,” according to Elmwood.
Learn more about Toronto opportunities at blogTO.com, and find out more about CHEST 2017 at chestmeeting.chestnet.org.
Get ready to visit the metropolitan hub of Canada. Explore new grounds with the chest medicine community for current pulmonary, critical care, and sleep medicine topics presented by world-renowned faculty in a variety of innovative instruction formats.
You will have access to our cutting-edge education, Oct 28 - Nov 1, but don’t forget to take advantage of all that Toronto has to offer.
Food
![]()
Le Petit Dejeune offers an ever-changing menu that ranges from less expensive items, like soup, sandwiches, and salads, to some pricier stuffed crepes, quiche, and eggs florentine. While most Sundays, Saving Grace is packed, but there’s only a 15-minute wait, and the atmosphere is quite pleasant. Looking for the perfect cinnamon bun? Rosen’s Cinnamon Buns is the place to go. But you have to look closely for the bakery’s name, since the sign above the window still advertises the hair salon that used to reside in the same spot!
Nature parks
One of the city’s largest and oldest parks, High Park is Toronto’s version of New York City’s Central Park. There’s plenty to enjoy, such as Grenadier Pond, numerous ravine-based hiking trails, playgrounds, athletic areas, restaurants, a museum, and even a zoo!
If you want a different type of nature excursion, there is always beautiful Niagara Falls, Ontario, which is just a short drive from Toronto. Don’t miss seeing the Tesla monument in Queen Victoria Park, or go 10 minutes north of the Falls to the Botanical Gardens, home to the Butterfly Conservatory with over 2,000 butterflies.
Relaxation
After eventful days of absorbing all the new science CHEST 2017 has to offer, you may want to relax your mind and body. Elmwood spa, located in downtown Toronto, is where “four spacious floors of treatment and renewal options mean that Elmwood Spa can provide the convenience and flexibility to cater to demanding schedules,” according to Elmwood.
Learn more about Toronto opportunities at blogTO.com, and find out more about CHEST 2017 at chestmeeting.chestnet.org.
Is there clinical benefit to activity trackers?
SAN DIEGO – In the opinion of Ann R. Garment, MD, wearable activity trackers such as the Fitbit may have a role in improving the physical fitness of patients, but alone they do not promote moderate-to-vigorous physical activity or improve other health outcomes.
An estimated one in five Americans owns some type of wearable technology independent of their smartphones.
“However, these devices come with some challenges,” Dr. Garment, of New York University–Langone Medical Center’s division of general internal medicine, said at the annual meeting of the American College of Physicians. “First, you need to know how to use them. You have to power them up regularly. They can be expensive. And for those that interface with the Internet, there are privacy concerns.”

A 2014 PricewaterhouseCoopers survey of 1,000 U.S. consumers found that 56% believe that their life expectancy will grow an additional 10 years by wearing a device that tracks vital signs, 46% believe that it will help them lose weight, and 42% believe that it will help them dramatically improve their athletic ability.
General guidelines call for 30-60 minutes per day of moderate-to-vigorous physical activity (MVPA) at a pace of 100 steps per minute, in at least 10-minute bouts of activity. However, many studies set a goal of 10,000 steps per day, which does not necessarily need to be done as MVPA.
To assess the impact of wearable technology on helping patients to achieve weight loss, researchers studied 471 patients who were placed on a low-calorie diet, prescribed increases in physical activity, and had group counseling sessions (JAMA 2016 Sep 20;316[11]:1161-71).
At 6 months, the researchers randomized patients into either the standard intervention arm, which had access to materials on a website, telephone counseling sessions, and text message prompts; or to the enhanced intervention arm, which included all of those interventions plus the addition of a wearable device and accompanying web interface to monitor diet and physical activity.

In a separate, larger study, researchers randomly assigned 800 employees from 13 organizations in Singapore to one of four arms: control (no activity tracker or incentives), Fitbit Zip activity tracker without additional incentives, tracker plus charity incentives (money went to charity based on how much they exercised), or tracker plus cash incentives (money went to participants based on how much they exercised) (Lancet Diabetes Endocrinol. 2016 Dec;4[12]:983-95).
The researchers tied incentives to weekly steps, and the primary outcome was minutes of MVPA per week, measured via a sealed accelerometer and assessed on an intention-to-treat basis at 6 months (end of intervention) and 12 months (after a 6-month postintervention follow-up period).
They found that the cash incentive was most effective at increasing MVPA at 6 months, but this effect was not sustained 6 months after the incentives were discontinued. At 12 months, the researchers “identified no evidence of improvements in health outcomes, either with or without incentives, calling into question the value of these devices for health promotion,” they wrote.
For her part, Dr. Garment concluded that activity trackers alone are not sufficient for increasing MVPA or improving health outcomes.
“Though it’s possible it would do better if it were coupled with other modalities or sustained incentives, we need more research before we can recommend it to our patients,” she said. “Perhaps one of the best things we can be doing for our health and recommending for our patients is to, in the words of food author Michael Pollan, ‘eat food, not too much, mostly plants.’ ”
Dr. Garment reported having no financial disclosures.
SAN DIEGO – In the opinion of Ann R. Garment, MD, wearable activity trackers such as the Fitbit may have a role in improving the physical fitness of patients, but alone they do not promote moderate-to-vigorous physical activity or improve other health outcomes.
An estimated one in five Americans owns some type of wearable technology independent of their smartphones.
“However, these devices come with some challenges,” Dr. Garment, of New York University–Langone Medical Center’s division of general internal medicine, said at the annual meeting of the American College of Physicians. “First, you need to know how to use them. You have to power them up regularly. They can be expensive. And for those that interface with the Internet, there are privacy concerns.”
A 2014 PricewaterhouseCoopers survey of 1,000 U.S. consumers found that 56% believe that their life expectancy will grow an additional 10 years by wearing a device that tracks vital signs, 46% believe that it will help them lose weight, and 42% believe that it will help them dramatically improve their athletic ability.
General guidelines call for 30-60 minutes per day of moderate-to-vigorous physical activity (MVPA) at a pace of 100 steps per minute, in at least 10-minute bouts of activity. However, many studies set a goal of 10,000 steps per day, which does not necessarily need to be done as MVPA.
To assess the impact of wearable technology on helping patients to achieve weight loss, researchers studied 471 patients who were placed on a low-calorie diet, prescribed increases in physical activity, and had group counseling sessions (JAMA 2016 Sep 20;316[11]:1161-71).
At 6 months, the researchers randomized patients into either the standard intervention arm, which had access to materials on a website, telephone counseling sessions, and text message prompts; or to the enhanced intervention arm, which included all of those interventions plus the addition of a wearable device and accompanying web interface to monitor diet and physical activity.
In a separate, larger study, researchers randomly assigned 800 employees from 13 organizations in Singapore to one of four arms: control (no activity tracker or incentives), Fitbit Zip activity tracker without additional incentives, tracker plus charity incentives (money went to charity based on how much they exercised), or tracker plus cash incentives (money went to participants based on how much they exercised) (Lancet Diabetes Endocrinol. 2016 Dec;4[12]:983-95).
The researchers tied incentives to weekly steps, and the primary outcome was minutes of MVPA per week, measured via a sealed accelerometer and assessed on an intention-to-treat basis at 6 months (end of intervention) and 12 months (after a 6-month postintervention follow-up period).
They found that the cash incentive was most effective at increasing MVPA at 6 months, but this effect was not sustained 6 months after the incentives were discontinued. At 12 months, the researchers “identified no evidence of improvements in health outcomes, either with or without incentives, calling into question the value of these devices for health promotion,” they wrote.
For her part, Dr. Garment concluded that activity trackers alone are not sufficient for increasing MVPA or improving health outcomes.
“Though it’s possible it would do better if it were coupled with other modalities or sustained incentives, we need more research before we can recommend it to our patients,” she said. “Perhaps one of the best things we can be doing for our health and recommending for our patients is to, in the words of food author Michael Pollan, ‘eat food, not too much, mostly plants.’ ”
Dr. Garment reported having no financial disclosures.
SAN DIEGO – In the opinion of Ann R. Garment, MD, wearable activity trackers such as the Fitbit may have a role in improving the physical fitness of patients, but alone they do not promote moderate-to-vigorous physical activity or improve other health outcomes.
An estimated one in five Americans owns some type of wearable technology independent of their smartphones.
“However, these devices come with some challenges,” Dr. Garment, of New York University–Langone Medical Center’s division of general internal medicine, said at the annual meeting of the American College of Physicians. “First, you need to know how to use them. You have to power them up regularly. They can be expensive. And for those that interface with the Internet, there are privacy concerns.”
A 2014 PricewaterhouseCoopers survey of 1,000 U.S. consumers found that 56% believe that their life expectancy will grow an additional 10 years by wearing a device that tracks vital signs, 46% believe that it will help them lose weight, and 42% believe that it will help them dramatically improve their athletic ability.
General guidelines call for 30-60 minutes per day of moderate-to-vigorous physical activity (MVPA) at a pace of 100 steps per minute, in at least 10-minute bouts of activity. However, many studies set a goal of 10,000 steps per day, which does not necessarily need to be done as MVPA.
To assess the impact of wearable technology on helping patients to achieve weight loss, researchers studied 471 patients who were placed on a low-calorie diet, prescribed increases in physical activity, and had group counseling sessions (JAMA 2016 Sep 20;316[11]:1161-71).
At 6 months, the researchers randomized patients into either the standard intervention arm, which had access to materials on a website, telephone counseling sessions, and text message prompts; or to the enhanced intervention arm, which included all of those interventions plus the addition of a wearable device and accompanying web interface to monitor diet and physical activity.
In a separate, larger study, researchers randomly assigned 800 employees from 13 organizations in Singapore to one of four arms: control (no activity tracker or incentives), Fitbit Zip activity tracker without additional incentives, tracker plus charity incentives (money went to charity based on how much they exercised), or tracker plus cash incentives (money went to participants based on how much they exercised) (Lancet Diabetes Endocrinol. 2016 Dec;4[12]:983-95).
The researchers tied incentives to weekly steps, and the primary outcome was minutes of MVPA per week, measured via a sealed accelerometer and assessed on an intention-to-treat basis at 6 months (end of intervention) and 12 months (after a 6-month postintervention follow-up period).
They found that the cash incentive was most effective at increasing MVPA at 6 months, but this effect was not sustained 6 months after the incentives were discontinued. At 12 months, the researchers “identified no evidence of improvements in health outcomes, either with or without incentives, calling into question the value of these devices for health promotion,” they wrote.
For her part, Dr. Garment concluded that activity trackers alone are not sufficient for increasing MVPA or improving health outcomes.
“Though it’s possible it would do better if it were coupled with other modalities or sustained incentives, we need more research before we can recommend it to our patients,” she said. “Perhaps one of the best things we can be doing for our health and recommending for our patients is to, in the words of food author Michael Pollan, ‘eat food, not too much, mostly plants.’ ”
Dr. Garment reported having no financial disclosures.
EXPERT ANALYSIS AT ACP INTERNAL MEDICINE
Avelumab produces durable responses in Merkel cell carcinoma
Avelumab (Bavencio) is the first drug to receive approval from the Food and Drug Administration for Merkel cell carcinoma, and new findings show that it elicited durable responses in this hard-to-treat population.
The majority of responses were durable beyond 1 year, with an objective response rate of 33%.
“Merkel cell carcinoma is rare, aggressive skin cancer with a poor prognosis,” said lead author Howard L. Kaufman, MD, a surgical oncologist at the Rutgers Cancer Institute of New Jersey in New Brunswick, who discussed the findings during a presscast held at the annual meeting of the American Association for Cancer Research.
Even though Merkel cell carcinoma is a chemosensitive disease, long-term survival beyond 6 months has not been reported with chemotherapy, explained Dr. Kaufman.
In this phase II trial, Dr. Kaufman and his colleagues assessed the use of avelumab, a fully human anti–PD-L1 monoclonal antibody, in 88 patients with metastatic Merkel cell carcinoma that had progressed after treatment with chemotherapy.
All patients received 10 mg/kg avelumab as an intravenous infusion over one hour every 2 weeks. The primary endpoint was the best objective response and secondary endpoints included progression-free and overall survival.
At a median follow-up of 10.4 months, the response rate was 31.8% (28 responses), and this included 8 complete responses and 20 partial responses.
The estimated proportion of patients with duration of response of 6 months or longer was 92%, and the 6-month progression-free survival rate was 40%.
“The purpose of the presentation now is to report on longer 1-year follow-up data,” said Dr. Kaufman.
In the updated results, the objective response rate was 33.0% (95% confidence interval, 23.3%-43.8%) as two more patients moved into a total response. There were now a total of 10 (11.4%) complete responses and 19 (21.6%) partial responses.
The 6-month durable response rate was 30.6% (95% CI, 20.9%-40.3%), and the median duration of response has not yet been reached (range, 2.8–23.3-plus months; 95% CI, 18.0–not estimable). Responses were ongoing in 21 patients at the time of this analysis.
The estimated proportion of patients with a duration of response lasting 1 year or longer was 74% (95% CI, 53%-87%), and estimated 1-year progression-free survival was 30% (95% CI, 21%-41%). The 1-year overall survival rate was 52% (95% CI, 41%-62%) and median overall survival was 12.9 months (95% CI, 7.5–not estimable).
The maturing survival data suggest that there may be a long-term benefit for a proportion of patients.
“The findings of long-term responses and well-tolerated safety profile suggest that avelumab could be an important new agent for patients with Merkel cell carcinoma who have failed prior chemotherapy,” said Dr. Kaufman. “Given these results, it will be interesting to determine whether response rates could be increased by giving avelumab prior to chemotherapy or in combination with other treatments.”
Avelumab (Bavencio) is the first drug to receive approval from the Food and Drug Administration for Merkel cell carcinoma, and new findings show that it elicited durable responses in this hard-to-treat population.
The majority of responses were durable beyond 1 year, with an objective response rate of 33%.
“Merkel cell carcinoma is rare, aggressive skin cancer with a poor prognosis,” said lead author Howard L. Kaufman, MD, a surgical oncologist at the Rutgers Cancer Institute of New Jersey in New Brunswick, who discussed the findings during a presscast held at the annual meeting of the American Association for Cancer Research.
Even though Merkel cell carcinoma is a chemosensitive disease, long-term survival beyond 6 months has not been reported with chemotherapy, explained Dr. Kaufman.
In this phase II trial, Dr. Kaufman and his colleagues assessed the use of avelumab, a fully human anti–PD-L1 monoclonal antibody, in 88 patients with metastatic Merkel cell carcinoma that had progressed after treatment with chemotherapy.
All patients received 10 mg/kg avelumab as an intravenous infusion over one hour every 2 weeks. The primary endpoint was the best objective response and secondary endpoints included progression-free and overall survival.
At a median follow-up of 10.4 months, the response rate was 31.8% (28 responses), and this included 8 complete responses and 20 partial responses.
The estimated proportion of patients with duration of response of 6 months or longer was 92%, and the 6-month progression-free survival rate was 40%.
“The purpose of the presentation now is to report on longer 1-year follow-up data,” said Dr. Kaufman.
In the updated results, the objective response rate was 33.0% (95% confidence interval, 23.3%-43.8%) as two more patients moved into a total response. There were now a total of 10 (11.4%) complete responses and 19 (21.6%) partial responses.
The 6-month durable response rate was 30.6% (95% CI, 20.9%-40.3%), and the median duration of response has not yet been reached (range, 2.8–23.3-plus months; 95% CI, 18.0–not estimable). Responses were ongoing in 21 patients at the time of this analysis.
The estimated proportion of patients with a duration of response lasting 1 year or longer was 74% (95% CI, 53%-87%), and estimated 1-year progression-free survival was 30% (95% CI, 21%-41%). The 1-year overall survival rate was 52% (95% CI, 41%-62%) and median overall survival was 12.9 months (95% CI, 7.5–not estimable).
The maturing survival data suggest that there may be a long-term benefit for a proportion of patients.
“The findings of long-term responses and well-tolerated safety profile suggest that avelumab could be an important new agent for patients with Merkel cell carcinoma who have failed prior chemotherapy,” said Dr. Kaufman. “Given these results, it will be interesting to determine whether response rates could be increased by giving avelumab prior to chemotherapy or in combination with other treatments.”
Avelumab (Bavencio) is the first drug to receive approval from the Food and Drug Administration for Merkel cell carcinoma, and new findings show that it elicited durable responses in this hard-to-treat population.
The majority of responses were durable beyond 1 year, with an objective response rate of 33%.
“Merkel cell carcinoma is rare, aggressive skin cancer with a poor prognosis,” said lead author Howard L. Kaufman, MD, a surgical oncologist at the Rutgers Cancer Institute of New Jersey in New Brunswick, who discussed the findings during a presscast held at the annual meeting of the American Association for Cancer Research.
Even though Merkel cell carcinoma is a chemosensitive disease, long-term survival beyond 6 months has not been reported with chemotherapy, explained Dr. Kaufman.
In this phase II trial, Dr. Kaufman and his colleagues assessed the use of avelumab, a fully human anti–PD-L1 monoclonal antibody, in 88 patients with metastatic Merkel cell carcinoma that had progressed after treatment with chemotherapy.
All patients received 10 mg/kg avelumab as an intravenous infusion over one hour every 2 weeks. The primary endpoint was the best objective response and secondary endpoints included progression-free and overall survival.
At a median follow-up of 10.4 months, the response rate was 31.8% (28 responses), and this included 8 complete responses and 20 partial responses.
The estimated proportion of patients with duration of response of 6 months or longer was 92%, and the 6-month progression-free survival rate was 40%.
“The purpose of the presentation now is to report on longer 1-year follow-up data,” said Dr. Kaufman.
In the updated results, the objective response rate was 33.0% (95% confidence interval, 23.3%-43.8%) as two more patients moved into a total response. There were now a total of 10 (11.4%) complete responses and 19 (21.6%) partial responses.
The 6-month durable response rate was 30.6% (95% CI, 20.9%-40.3%), and the median duration of response has not yet been reached (range, 2.8–23.3-plus months; 95% CI, 18.0–not estimable). Responses were ongoing in 21 patients at the time of this analysis.
The estimated proportion of patients with a duration of response lasting 1 year or longer was 74% (95% CI, 53%-87%), and estimated 1-year progression-free survival was 30% (95% CI, 21%-41%). The 1-year overall survival rate was 52% (95% CI, 41%-62%) and median overall survival was 12.9 months (95% CI, 7.5–not estimable).
The maturing survival data suggest that there may be a long-term benefit for a proportion of patients.
“The findings of long-term responses and well-tolerated safety profile suggest that avelumab could be an important new agent for patients with Merkel cell carcinoma who have failed prior chemotherapy,” said Dr. Kaufman. “Given these results, it will be interesting to determine whether response rates could be increased by giving avelumab prior to chemotherapy or in combination with other treatments.”
Key clinical point: Treatment with avelumab resulted in an objective response rate of 33% in patients with progressive Merkel cell carcinoma.
Major finding: The estimated proportion of patients with a duration of response lasting 1 year or longer was 74% and estimated 1-year progression-free survival was 30%.
Data source: Updated results from a phase II study that included 88 patients with progressive Merkel cell carcinoma.
Disclosures: This study was funded by EMD Serono. Dr. Kaufman has served on advisory boards for Amgen, Celldex, Compass Therapeutics, EMD Serono, Merck, Prometheus, and Turnstone Biologics.
Nivolumab boosts 5-year survival in advanced NSCLC
Early data show that treatment with the immune checkpoint inhibitor nivolumab (Opdivo) resulted in a 5-year overall survival rate of 16% among patients with advanced non–small-cell lung cancer (NSCLC).
In comparison, the 5-year survival rate for patients with advanced lung and bronchus cancer, according to SEER data, is 4.3%, and for those with advanced NSCLC, 4.9%.
“This is the first report of the long-term survival rate in patients with metastatic NSCLC treated with an immune checkpoint inhibitor,” said Julie Brahmer, MD, of the Bloomberg Kimmel Institute for Cancer Immunotherapy at Johns Hopkins, Baltimore.
For a small subset of patients, immunotherapy can work for a very long time, explained Dr. Brahmer, who discussed her findings during a presscast at the annual meeting of the American Association for Cancer Research.
The 5-year overall survival rate that was reported in this study was much higher than what has been seen for this patient population who receive the standard of care. Statistics show that the majority of patients with advanced disease will die within a year of their diagnosis, Dr. Brahmer pointed out.
The findings presented at the meeting are updated results from the phase Ib CA209-003 dose-escalation cohort expansion trial that comprised 129 patients with heavily pretreated, advanced NSCLC . The cohort was randomized to receive nivolumab once every 2 weeks for up to 2 years at one of three dose levels: 1 mg/kg, 3 mg/kg, or 10 mg/kg.
A previous analysis of the data showed promising activity, and findings from subsequent clinical trials led to the approval of nivolumab for use in the second line setting of advanced NSCLC.
Dr. Brahmer now reported findings based on 5-year results of this phase Ib trial. “This analysis is based on a minimum follow up of 58 months,” she said.
The overall 5-year survival rates for squamous NSCLC were 16%, and the rates for nonsquamous were 15%.
At 1 year, overall survival was 42%. At 2 years, it was 24%, and at 3 years, 18%.
“After 3 years, the survival curve has plateaued out, which is similar to what has been seen in the past in other diseases treated with immunotherapy,” Dr. Brahmer noted.
Within the cohort, there were 16 patients who had survived for at least 5 years. Of this group, 12 achieved a partial response, 2 patients had stable disease, and 2 had progressive disease.
Dr. Brahmer pointed out that there was nothing different or unusual among the 16 patients who survived for 5 years, compared with the rest of the cohort. Their characteristics were similar to others in the study, most of them were former smokers, and they had very similar rates of different histologies.
One interesting note was that within that group, there were two patients with EGFR mutations. “We usually don’t expect them to do well with immunotherapy,” she said.
Dr. Brahmer received research funding from, and is an adviser to, Bristol-Myers Squibb, which funded the study.
Early data show that treatment with the immune checkpoint inhibitor nivolumab (Opdivo) resulted in a 5-year overall survival rate of 16% among patients with advanced non–small-cell lung cancer (NSCLC).
In comparison, the 5-year survival rate for patients with advanced lung and bronchus cancer, according to SEER data, is 4.3%, and for those with advanced NSCLC, 4.9%.
“This is the first report of the long-term survival rate in patients with metastatic NSCLC treated with an immune checkpoint inhibitor,” said Julie Brahmer, MD, of the Bloomberg Kimmel Institute for Cancer Immunotherapy at Johns Hopkins, Baltimore.
For a small subset of patients, immunotherapy can work for a very long time, explained Dr. Brahmer, who discussed her findings during a presscast at the annual meeting of the American Association for Cancer Research.
The 5-year overall survival rate that was reported in this study was much higher than what has been seen for this patient population who receive the standard of care. Statistics show that the majority of patients with advanced disease will die within a year of their diagnosis, Dr. Brahmer pointed out.
The findings presented at the meeting are updated results from the phase Ib CA209-003 dose-escalation cohort expansion trial that comprised 129 patients with heavily pretreated, advanced NSCLC . The cohort was randomized to receive nivolumab once every 2 weeks for up to 2 years at one of three dose levels: 1 mg/kg, 3 mg/kg, or 10 mg/kg.
A previous analysis of the data showed promising activity, and findings from subsequent clinical trials led to the approval of nivolumab for use in the second line setting of advanced NSCLC.
Dr. Brahmer now reported findings based on 5-year results of this phase Ib trial. “This analysis is based on a minimum follow up of 58 months,” she said.
The overall 5-year survival rates for squamous NSCLC were 16%, and the rates for nonsquamous were 15%.
At 1 year, overall survival was 42%. At 2 years, it was 24%, and at 3 years, 18%.
“After 3 years, the survival curve has plateaued out, which is similar to what has been seen in the past in other diseases treated with immunotherapy,” Dr. Brahmer noted.
Within the cohort, there were 16 patients who had survived for at least 5 years. Of this group, 12 achieved a partial response, 2 patients had stable disease, and 2 had progressive disease.
Dr. Brahmer pointed out that there was nothing different or unusual among the 16 patients who survived for 5 years, compared with the rest of the cohort. Their characteristics were similar to others in the study, most of them were former smokers, and they had very similar rates of different histologies.
One interesting note was that within that group, there were two patients with EGFR mutations. “We usually don’t expect them to do well with immunotherapy,” she said.
Dr. Brahmer received research funding from, and is an adviser to, Bristol-Myers Squibb, which funded the study.
Early data show that treatment with the immune checkpoint inhibitor nivolumab (Opdivo) resulted in a 5-year overall survival rate of 16% among patients with advanced non–small-cell lung cancer (NSCLC).
In comparison, the 5-year survival rate for patients with advanced lung and bronchus cancer, according to SEER data, is 4.3%, and for those with advanced NSCLC, 4.9%.
“This is the first report of the long-term survival rate in patients with metastatic NSCLC treated with an immune checkpoint inhibitor,” said Julie Brahmer, MD, of the Bloomberg Kimmel Institute for Cancer Immunotherapy at Johns Hopkins, Baltimore.
For a small subset of patients, immunotherapy can work for a very long time, explained Dr. Brahmer, who discussed her findings during a presscast at the annual meeting of the American Association for Cancer Research.
The 5-year overall survival rate that was reported in this study was much higher than what has been seen for this patient population who receive the standard of care. Statistics show that the majority of patients with advanced disease will die within a year of their diagnosis, Dr. Brahmer pointed out.
The findings presented at the meeting are updated results from the phase Ib CA209-003 dose-escalation cohort expansion trial that comprised 129 patients with heavily pretreated, advanced NSCLC . The cohort was randomized to receive nivolumab once every 2 weeks for up to 2 years at one of three dose levels: 1 mg/kg, 3 mg/kg, or 10 mg/kg.
A previous analysis of the data showed promising activity, and findings from subsequent clinical trials led to the approval of nivolumab for use in the second line setting of advanced NSCLC.
Dr. Brahmer now reported findings based on 5-year results of this phase Ib trial. “This analysis is based on a minimum follow up of 58 months,” she said.
The overall 5-year survival rates for squamous NSCLC were 16%, and the rates for nonsquamous were 15%.
At 1 year, overall survival was 42%. At 2 years, it was 24%, and at 3 years, 18%.
“After 3 years, the survival curve has plateaued out, which is similar to what has been seen in the past in other diseases treated with immunotherapy,” Dr. Brahmer noted.
Within the cohort, there were 16 patients who had survived for at least 5 years. Of this group, 12 achieved a partial response, 2 patients had stable disease, and 2 had progressive disease.
Dr. Brahmer pointed out that there was nothing different or unusual among the 16 patients who survived for 5 years, compared with the rest of the cohort. Their characteristics were similar to others in the study, most of them were former smokers, and they had very similar rates of different histologies.
One interesting note was that within that group, there were two patients with EGFR mutations. “We usually don’t expect them to do well with immunotherapy,” she said.
Dr. Brahmer received research funding from, and is an adviser to, Bristol-Myers Squibb, which funded the study.
Key clinical point: Treatment with nivolumab resulted in a 5-year overall survival rate that is much higher than what is reported for this patient population receiving standard-of-care treatment.
Major finding: Nivolumab yielded a 5-year survival rate of 16% in a cohort of patients with advanced NSCLC.
Data source: Updated results from a phase Ib study that included 129 patients with advanced NSCLC.
Disclosures: Dr. Brahmer received research funding from, and is an adviser to, Bristol-Myers Squibb, which funded the study.
Pulmonary Perspectives: Ensuring quality for EBUS bronchoscopy with varying levels of practitioner experience
Dr. Mahajan and colleagues present a compelling case for requiring minimum standards to perform an EBUS-guided bronchoscopy. Their opinion piece epitomizes the classic tension between physicians with advanced training and those who can only have practice-based training. A middle ground may exist, as perhaps competence could be achieved by simulation, clinical cases performed, and observation by a regional expert? Physicians in practice must have a pathway to adopt new technology whether it is thoracic ultrasound or endobronchial ultrasound, but it must be done in a safe manner. As a referring physician, I would only send my patients who required mediastinal staging to a pulmonologist who I knew performed EBUS regularly.
Nitin Puri, MD, FCCP
Endobronchial ultrasound (EBUS) bronchoscopy is a tool that has transformed the diagnosis and staging of lung cancer. Through real-time ultrasound imaging, EBUS provides clear images of lymph nodes and proximal lung masses that can be adequately sampled through transbronchial needle aspiration. EBUS is a minimally invasive, outpatient procedure that can also be used for diagnosing benign disease within the chest. Large studies investigating the use of EBUS for mediastinal staging have shown the procedure to be highly sensitive and specific while harboring an excellent safety profile.1 As a result, EBUS has essentially replaced mediastinoscopy for the staging of lung cancer.
EBUS bronchoscopy was primarily offered at major academic centers when first released and was performed by physicians who were formally trained in the procedure during interventional pulmonology or thoracic surgery fellowships. Over time, the tool has been adopted by established general pulmonologists without formal training in EBUS. Some of these pulmonologists only develop their skills by attending 1- to 2-day courses, which is insufficient supervision to become competent in this important procedure.
An ongoing debate continues as to how many supervised EBUS bronchoscopies should be performed prior to being considered proficient.2 As procedural competence has been associated with the number of EBUS procedures performed, the learning curve required to master EBUS is an important component of proficiency. While most consider learning curves to be variable, evidence produced by Fernandez-Villar and colleagues revealed that EBUS performance continues to improve up to 120 procedures.3 This analysis was performed in unselected consecutive patients based on diagnostic yield, procedure length, number of lymph nodes passes performed in order to obtain adequate samples, and the number of lymph nodes studied per patient. The learning curve was evaluated based on consecutive groups of 20 patients, the number of adequate samples obtained, and the diagnostic accuracy. Their results indicated that the diagnostic effectiveness of EBUS-TBNA improves with increasing number of procedures performed, allowing for access to a greater number of lymph nodes without necessarily increasing the length of the procedure, and by reducing the number of punctures at each nodal station. Based on their results, the first 20 procedures performed yielded a 70% accuracy, 21 to 40 procedures performed resulted in 81.8% accuracy, 41 to 60 procedures performed resulted in 83.3% accuracy, 61 to 80 procedures performed resulted in 89.8% accuracy, 81 to 100 procedures performed resulted in 90.5% accuracy, and 101 to 120 procedures performed resulted in 94.5% accuracy.
While the American Thoracic Society (ATS) and the American College of Chest Physicians (CHEST) both recommend a minimum number of 40 to 50 supervised EBUS bronchoscopies prior to performing the procedure independently, along with 20 procedures per year for maintenance of competency, most institutions do not track the number of EBUS procedures performed and they do not follow the ATS or CHEST recommendations.4,5 As a result, a number of physicians are independently performing EBUS without adequate experience, resulting in possibly poor quality care. Unfortunately, some short courses, intended to generate interest and encourage attendees to pursue further training, are mistakenly assumed to be sufficient by the novice user.
As the number of interventional pulmonary fellowships continues to expand, the growing number of subspecialized pulmonologists with extensive training in EBUS grows. During a dedicated interventional pulmonary fellowship, fellows perform well above the number of EBUS bronchoscopies suggested by the ATS and CHEST in a single year. Recently published accreditation guidelines require a minimum of 100 cases per interventional pulmonary fellow.6 These fellowship-trained interventional pulmonologists are then tested to become board-certified in a wide array of minimally invasive procedures, including EBUS. As a result, a model has developed where both board-certified interventional pulmonologists with extensive training in EBUS and general pulmonologists not meeting ATS or CHEST minimum requirements practice at the same institution. Proponents of a more liberal access to credentialing in EBUS have suggested that adhering to competency requirements constitutes a “barrier to entry” in which incumbent practitioners benefit from limiting competition. However, like any other regulatory metric, the rationale is to prevent asymmetric information. In this example, the physician knows more than the patient. The patient cannot make an informed decision on which provider to choose and what are the minimum requirements that are likely to produce the most useful information (ie, complete staging). For these reasons, it is imperative that regulations protect the patient.
Without question, EBUS bronchoscopy should not be performed only by board-certified interventional pulmonologists. Instead, hospital credentialing committees should adhere to both the ATS and CHEST recommendations for the number of supervised cases necessary prior to performing EBUS independently. As EBUS use continues to grow, fellows in 3- or 4-year pulmonary and critical care fellowships will be likely capable of meeting the minimal number of observed cases, but, if these numbers are not achieved, additional training should be required. Understandably, this could be challenging for physicians who are unable to take time away from their practice to gain this training. However, if these numbers cannot be met, credentialing requirements should be enforced.
Even more challenging than establishing quality measures for EBUS, is to ensure the highest level of care delivery for patients when there exist multiple levels of experience in the same institution. Undoubtedly, patients undergoing EBUS bronchoscopy, or any procedure for that matter, would want the most skilled physician who has attained certification in the procedure. Unfortunately, no formal certification of EBUS exists outside of gaining board certification in interventional pulmonology. To ensure excellence in care, physicians performing EBUS should be involved in quality improvement initiatives and review pathologic yields along with complications on a regular basis in a group setting. Unlike emergency interventions, EBUS bronchoscopy is an entirely elective procedure.
The advent of EBUS bronchoscopy has revolutionized the diagnosis and staging of lung cancer. As use of EBUS continues to become more widespread, the incidence of high volume and low volume proceduralists will become a more commonly encountered scenario. Guidelines have been set by the professional pulmonary societies based on the data and observations available. At the local level, stringent guidelines need to be established by hospitals to ensure a high level of quality with appropriate oversight. Patients undergoing EBUS deserve a physician who is skilled in the procedure and has performed at least the minimum number of procedures to provide the adequate care.
Dr. Mahajan is Medical Director, Interventional Pulmonology, Inova Heart and Vascular Institute - Inova Fairfax Hospital, and Associate Professor, Virginia Commonwealth Medical School; Dr. Khandhar is Medical Director, Thoracic Surgery, Inova Heart and Vascular Institute - Inova Fairfax Hospital, and Assistant Clinical Professor, Virginia Commonwealth Medical School; Falls Church, VA. Dr. Folch is Co-Director, Interventional Pulmonology Chief, Complex Chest Diseases Center, Harvard Medical School, Massachusetts General Hospital, Boston, MA.
References
1. Gomez M, Silvestri GA. Endobronchial ultrasound for the diagnosis and staging of lung cancer. Proc Am Thorac Soc. 2009;6(2):180-186.
2. Folch E, Majid A. Point: Are >50 Supervised Procedures Required to Develop Competency in Performing Endobronchial Ultrasound-Guided Transbronchial Needle Aspiration for Mediastinal Staging? Yes. Chest. 2013;143(4):888-891.
3. Fernandez-Villar A, Leiro-Fernandez V, Botana-Rial M, Represas-Represas C, Nunez-Delgado M. The endobronchial ultrasound-guided transbronchial needle biopsy learning curve for mediastinal and hilar lymph node diagnosis. Chest. 2012; 141(1):278-279.
4. Ernst A, Silvestri GA, Johnstone D. Interventional pulmonary procedures: Guidelines from the American College of Chest Physicians. Chest. 2003;123(5):1693-1717.
5. Bolliger CT, Mathur PN, Beamis JF, et al. ERS/ATS statement on interventional pulmonology. European Respiratory Society/American Thoracic Society. Eur Respir J. 2002;19(2):356-373.
6. Mullon JJ, Burkhart KM, Silvestri G. Interventional Pulmonology Fellowship Accreditation Standards: Executive Summary of the Multi-society Interventional Pulmonology Fellowship Accreditation Committee. Chest. 2017. doi:10.1016/j.chest.2017.01.024.
Dr. Mahajan and colleagues present a compelling case for requiring minimum standards to perform an EBUS-guided bronchoscopy. Their opinion piece epitomizes the classic tension between physicians with advanced training and those who can only have practice-based training. A middle ground may exist, as perhaps competence could be achieved by simulation, clinical cases performed, and observation by a regional expert? Physicians in practice must have a pathway to adopt new technology whether it is thoracic ultrasound or endobronchial ultrasound, but it must be done in a safe manner. As a referring physician, I would only send my patients who required mediastinal staging to a pulmonologist who I knew performed EBUS regularly.
Nitin Puri, MD, FCCP
Endobronchial ultrasound (EBUS) bronchoscopy is a tool that has transformed the diagnosis and staging of lung cancer. Through real-time ultrasound imaging, EBUS provides clear images of lymph nodes and proximal lung masses that can be adequately sampled through transbronchial needle aspiration. EBUS is a minimally invasive, outpatient procedure that can also be used for diagnosing benign disease within the chest. Large studies investigating the use of EBUS for mediastinal staging have shown the procedure to be highly sensitive and specific while harboring an excellent safety profile.1 As a result, EBUS has essentially replaced mediastinoscopy for the staging of lung cancer.
EBUS bronchoscopy was primarily offered at major academic centers when first released and was performed by physicians who were formally trained in the procedure during interventional pulmonology or thoracic surgery fellowships. Over time, the tool has been adopted by established general pulmonologists without formal training in EBUS. Some of these pulmonologists only develop their skills by attending 1- to 2-day courses, which is insufficient supervision to become competent in this important procedure.
An ongoing debate continues as to how many supervised EBUS bronchoscopies should be performed prior to being considered proficient.2 As procedural competence has been associated with the number of EBUS procedures performed, the learning curve required to master EBUS is an important component of proficiency. While most consider learning curves to be variable, evidence produced by Fernandez-Villar and colleagues revealed that EBUS performance continues to improve up to 120 procedures.3 This analysis was performed in unselected consecutive patients based on diagnostic yield, procedure length, number of lymph nodes passes performed in order to obtain adequate samples, and the number of lymph nodes studied per patient. The learning curve was evaluated based on consecutive groups of 20 patients, the number of adequate samples obtained, and the diagnostic accuracy. Their results indicated that the diagnostic effectiveness of EBUS-TBNA improves with increasing number of procedures performed, allowing for access to a greater number of lymph nodes without necessarily increasing the length of the procedure, and by reducing the number of punctures at each nodal station. Based on their results, the first 20 procedures performed yielded a 70% accuracy, 21 to 40 procedures performed resulted in 81.8% accuracy, 41 to 60 procedures performed resulted in 83.3% accuracy, 61 to 80 procedures performed resulted in 89.8% accuracy, 81 to 100 procedures performed resulted in 90.5% accuracy, and 101 to 120 procedures performed resulted in 94.5% accuracy.
While the American Thoracic Society (ATS) and the American College of Chest Physicians (CHEST) both recommend a minimum number of 40 to 50 supervised EBUS bronchoscopies prior to performing the procedure independently, along with 20 procedures per year for maintenance of competency, most institutions do not track the number of EBUS procedures performed and they do not follow the ATS or CHEST recommendations.4,5 As a result, a number of physicians are independently performing EBUS without adequate experience, resulting in possibly poor quality care. Unfortunately, some short courses, intended to generate interest and encourage attendees to pursue further training, are mistakenly assumed to be sufficient by the novice user.
As the number of interventional pulmonary fellowships continues to expand, the growing number of subspecialized pulmonologists with extensive training in EBUS grows. During a dedicated interventional pulmonary fellowship, fellows perform well above the number of EBUS bronchoscopies suggested by the ATS and CHEST in a single year. Recently published accreditation guidelines require a minimum of 100 cases per interventional pulmonary fellow.6 These fellowship-trained interventional pulmonologists are then tested to become board-certified in a wide array of minimally invasive procedures, including EBUS. As a result, a model has developed where both board-certified interventional pulmonologists with extensive training in EBUS and general pulmonologists not meeting ATS or CHEST minimum requirements practice at the same institution. Proponents of a more liberal access to credentialing in EBUS have suggested that adhering to competency requirements constitutes a “barrier to entry” in which incumbent practitioners benefit from limiting competition. However, like any other regulatory metric, the rationale is to prevent asymmetric information. In this example, the physician knows more than the patient. The patient cannot make an informed decision on which provider to choose and what are the minimum requirements that are likely to produce the most useful information (ie, complete staging). For these reasons, it is imperative that regulations protect the patient.
Without question, EBUS bronchoscopy should not be performed only by board-certified interventional pulmonologists. Instead, hospital credentialing committees should adhere to both the ATS and CHEST recommendations for the number of supervised cases necessary prior to performing EBUS independently. As EBUS use continues to grow, fellows in 3- or 4-year pulmonary and critical care fellowships will be likely capable of meeting the minimal number of observed cases, but, if these numbers are not achieved, additional training should be required. Understandably, this could be challenging for physicians who are unable to take time away from their practice to gain this training. However, if these numbers cannot be met, credentialing requirements should be enforced.
Even more challenging than establishing quality measures for EBUS, is to ensure the highest level of care delivery for patients when there exist multiple levels of experience in the same institution. Undoubtedly, patients undergoing EBUS bronchoscopy, or any procedure for that matter, would want the most skilled physician who has attained certification in the procedure. Unfortunately, no formal certification of EBUS exists outside of gaining board certification in interventional pulmonology. To ensure excellence in care, physicians performing EBUS should be involved in quality improvement initiatives and review pathologic yields along with complications on a regular basis in a group setting. Unlike emergency interventions, EBUS bronchoscopy is an entirely elective procedure.
The advent of EBUS bronchoscopy has revolutionized the diagnosis and staging of lung cancer. As use of EBUS continues to become more widespread, the incidence of high volume and low volume proceduralists will become a more commonly encountered scenario. Guidelines have been set by the professional pulmonary societies based on the data and observations available. At the local level, stringent guidelines need to be established by hospitals to ensure a high level of quality with appropriate oversight. Patients undergoing EBUS deserve a physician who is skilled in the procedure and has performed at least the minimum number of procedures to provide the adequate care.
Dr. Mahajan is Medical Director, Interventional Pulmonology, Inova Heart and Vascular Institute - Inova Fairfax Hospital, and Associate Professor, Virginia Commonwealth Medical School; Dr. Khandhar is Medical Director, Thoracic Surgery, Inova Heart and Vascular Institute - Inova Fairfax Hospital, and Assistant Clinical Professor, Virginia Commonwealth Medical School; Falls Church, VA. Dr. Folch is Co-Director, Interventional Pulmonology Chief, Complex Chest Diseases Center, Harvard Medical School, Massachusetts General Hospital, Boston, MA.
References
1. Gomez M, Silvestri GA. Endobronchial ultrasound for the diagnosis and staging of lung cancer. Proc Am Thorac Soc. 2009;6(2):180-186.
2. Folch E, Majid A. Point: Are >50 Supervised Procedures Required to Develop Competency in Performing Endobronchial Ultrasound-Guided Transbronchial Needle Aspiration for Mediastinal Staging? Yes. Chest. 2013;143(4):888-891.
3. Fernandez-Villar A, Leiro-Fernandez V, Botana-Rial M, Represas-Represas C, Nunez-Delgado M. The endobronchial ultrasound-guided transbronchial needle biopsy learning curve for mediastinal and hilar lymph node diagnosis. Chest. 2012; 141(1):278-279.
4. Ernst A, Silvestri GA, Johnstone D. Interventional pulmonary procedures: Guidelines from the American College of Chest Physicians. Chest. 2003;123(5):1693-1717.
5. Bolliger CT, Mathur PN, Beamis JF, et al. ERS/ATS statement on interventional pulmonology. European Respiratory Society/American Thoracic Society. Eur Respir J. 2002;19(2):356-373.
6. Mullon JJ, Burkhart KM, Silvestri G. Interventional Pulmonology Fellowship Accreditation Standards: Executive Summary of the Multi-society Interventional Pulmonology Fellowship Accreditation Committee. Chest. 2017. doi:10.1016/j.chest.2017.01.024.
Dr. Mahajan and colleagues present a compelling case for requiring minimum standards to perform an EBUS-guided bronchoscopy. Their opinion piece epitomizes the classic tension between physicians with advanced training and those who can only have practice-based training. A middle ground may exist, as perhaps competence could be achieved by simulation, clinical cases performed, and observation by a regional expert? Physicians in practice must have a pathway to adopt new technology whether it is thoracic ultrasound or endobronchial ultrasound, but it must be done in a safe manner. As a referring physician, I would only send my patients who required mediastinal staging to a pulmonologist who I knew performed EBUS regularly.
Nitin Puri, MD, FCCP
Endobronchial ultrasound (EBUS) bronchoscopy is a tool that has transformed the diagnosis and staging of lung cancer. Through real-time ultrasound imaging, EBUS provides clear images of lymph nodes and proximal lung masses that can be adequately sampled through transbronchial needle aspiration. EBUS is a minimally invasive, outpatient procedure that can also be used for diagnosing benign disease within the chest. Large studies investigating the use of EBUS for mediastinal staging have shown the procedure to be highly sensitive and specific while harboring an excellent safety profile.1 As a result, EBUS has essentially replaced mediastinoscopy for the staging of lung cancer.
EBUS bronchoscopy was primarily offered at major academic centers when first released and was performed by physicians who were formally trained in the procedure during interventional pulmonology or thoracic surgery fellowships. Over time, the tool has been adopted by established general pulmonologists without formal training in EBUS. Some of these pulmonologists only develop their skills by attending 1- to 2-day courses, which is insufficient supervision to become competent in this important procedure.
An ongoing debate continues as to how many supervised EBUS bronchoscopies should be performed prior to being considered proficient.2 As procedural competence has been associated with the number of EBUS procedures performed, the learning curve required to master EBUS is an important component of proficiency. While most consider learning curves to be variable, evidence produced by Fernandez-Villar and colleagues revealed that EBUS performance continues to improve up to 120 procedures.3 This analysis was performed in unselected consecutive patients based on diagnostic yield, procedure length, number of lymph nodes passes performed in order to obtain adequate samples, and the number of lymph nodes studied per patient. The learning curve was evaluated based on consecutive groups of 20 patients, the number of adequate samples obtained, and the diagnostic accuracy. Their results indicated that the diagnostic effectiveness of EBUS-TBNA improves with increasing number of procedures performed, allowing for access to a greater number of lymph nodes without necessarily increasing the length of the procedure, and by reducing the number of punctures at each nodal station. Based on their results, the first 20 procedures performed yielded a 70% accuracy, 21 to 40 procedures performed resulted in 81.8% accuracy, 41 to 60 procedures performed resulted in 83.3% accuracy, 61 to 80 procedures performed resulted in 89.8% accuracy, 81 to 100 procedures performed resulted in 90.5% accuracy, and 101 to 120 procedures performed resulted in 94.5% accuracy.
While the American Thoracic Society (ATS) and the American College of Chest Physicians (CHEST) both recommend a minimum number of 40 to 50 supervised EBUS bronchoscopies prior to performing the procedure independently, along with 20 procedures per year for maintenance of competency, most institutions do not track the number of EBUS procedures performed and they do not follow the ATS or CHEST recommendations.4,5 As a result, a number of physicians are independently performing EBUS without adequate experience, resulting in possibly poor quality care. Unfortunately, some short courses, intended to generate interest and encourage attendees to pursue further training, are mistakenly assumed to be sufficient by the novice user.
As the number of interventional pulmonary fellowships continues to expand, the growing number of subspecialized pulmonologists with extensive training in EBUS grows. During a dedicated interventional pulmonary fellowship, fellows perform well above the number of EBUS bronchoscopies suggested by the ATS and CHEST in a single year. Recently published accreditation guidelines require a minimum of 100 cases per interventional pulmonary fellow.6 These fellowship-trained interventional pulmonologists are then tested to become board-certified in a wide array of minimally invasive procedures, including EBUS. As a result, a model has developed where both board-certified interventional pulmonologists with extensive training in EBUS and general pulmonologists not meeting ATS or CHEST minimum requirements practice at the same institution. Proponents of a more liberal access to credentialing in EBUS have suggested that adhering to competency requirements constitutes a “barrier to entry” in which incumbent practitioners benefit from limiting competition. However, like any other regulatory metric, the rationale is to prevent asymmetric information. In this example, the physician knows more than the patient. The patient cannot make an informed decision on which provider to choose and what are the minimum requirements that are likely to produce the most useful information (ie, complete staging). For these reasons, it is imperative that regulations protect the patient.
Without question, EBUS bronchoscopy should not be performed only by board-certified interventional pulmonologists. Instead, hospital credentialing committees should adhere to both the ATS and CHEST recommendations for the number of supervised cases necessary prior to performing EBUS independently. As EBUS use continues to grow, fellows in 3- or 4-year pulmonary and critical care fellowships will be likely capable of meeting the minimal number of observed cases, but, if these numbers are not achieved, additional training should be required. Understandably, this could be challenging for physicians who are unable to take time away from their practice to gain this training. However, if these numbers cannot be met, credentialing requirements should be enforced.
Even more challenging than establishing quality measures for EBUS, is to ensure the highest level of care delivery for patients when there exist multiple levels of experience in the same institution. Undoubtedly, patients undergoing EBUS bronchoscopy, or any procedure for that matter, would want the most skilled physician who has attained certification in the procedure. Unfortunately, no formal certification of EBUS exists outside of gaining board certification in interventional pulmonology. To ensure excellence in care, physicians performing EBUS should be involved in quality improvement initiatives and review pathologic yields along with complications on a regular basis in a group setting. Unlike emergency interventions, EBUS bronchoscopy is an entirely elective procedure.
The advent of EBUS bronchoscopy has revolutionized the diagnosis and staging of lung cancer. As use of EBUS continues to become more widespread, the incidence of high volume and low volume proceduralists will become a more commonly encountered scenario. Guidelines have been set by the professional pulmonary societies based on the data and observations available. At the local level, stringent guidelines need to be established by hospitals to ensure a high level of quality with appropriate oversight. Patients undergoing EBUS deserve a physician who is skilled in the procedure and has performed at least the minimum number of procedures to provide the adequate care.
Dr. Mahajan is Medical Director, Interventional Pulmonology, Inova Heart and Vascular Institute - Inova Fairfax Hospital, and Associate Professor, Virginia Commonwealth Medical School; Dr. Khandhar is Medical Director, Thoracic Surgery, Inova Heart and Vascular Institute - Inova Fairfax Hospital, and Assistant Clinical Professor, Virginia Commonwealth Medical School; Falls Church, VA. Dr. Folch is Co-Director, Interventional Pulmonology Chief, Complex Chest Diseases Center, Harvard Medical School, Massachusetts General Hospital, Boston, MA.
References
1. Gomez M, Silvestri GA. Endobronchial ultrasound for the diagnosis and staging of lung cancer. Proc Am Thorac Soc. 2009;6(2):180-186.
2. Folch E, Majid A. Point: Are >50 Supervised Procedures Required to Develop Competency in Performing Endobronchial Ultrasound-Guided Transbronchial Needle Aspiration for Mediastinal Staging? Yes. Chest. 2013;143(4):888-891.
3. Fernandez-Villar A, Leiro-Fernandez V, Botana-Rial M, Represas-Represas C, Nunez-Delgado M. The endobronchial ultrasound-guided transbronchial needle biopsy learning curve for mediastinal and hilar lymph node diagnosis. Chest. 2012; 141(1):278-279.
4. Ernst A, Silvestri GA, Johnstone D. Interventional pulmonary procedures: Guidelines from the American College of Chest Physicians. Chest. 2003;123(5):1693-1717.
5. Bolliger CT, Mathur PN, Beamis JF, et al. ERS/ATS statement on interventional pulmonology. European Respiratory Society/American Thoracic Society. Eur Respir J. 2002;19(2):356-373.
6. Mullon JJ, Burkhart KM, Silvestri G. Interventional Pulmonology Fellowship Accreditation Standards: Executive Summary of the Multi-society Interventional Pulmonology Fellowship Accreditation Committee. Chest. 2017. doi:10.1016/j.chest.2017.01.024.
Participate in CHEST Foundation’s NetWorks Challenge


