User login
When Are Patients With Head and Neck Cancer at Risk for Aspiration Pneumonia?
Aspiration pneumonia (AP) is a common late adverse effect of chemoradiotherapy (CRT) and bioradiotherapy for head and neck cancer (HNC). Evaluating the long-term risk factors of AP is difficult because patients’ characteristics vary according to the multimodal therapies they receive, say researchers from Shizuoka Cancer Center in Japan. They conducted a study (the first to their knowledge) to identify specific factors that might help predict which patients have the highest risk of infection.
Related: Lean Six Sigma Applied to Tracking Head/Neck Cancer Patients
The researchers’ retrospective analysis covered nearly 9 years of data. Of the 305 patients in the study, 65 (21%) developed AP after CRT or bioradiotherapy. The median time from end of treatment to AP was 161 days. Nearly all (95%) the patients had Eastern Cooperative Oncology Group performance status of 0 to 1. Most had received standard chemotherapeutic regimens with platinum or cetuximab with supportive care.
The researchers found 5 independent risk factors: habitual alcoholic consumption, poor oral hygiene, coexisting malignancies, hypoalbuminemia before treatment, and use of sleeping pills at the end of treatment. Of those, only hypoalbuminemia was a familiar factor consistent with previous reports.
The finding that oral hygiene predicted AP was unexpected, because at the study hospital, HNC patients who undergo radiotherapy are routinely referred to dentists and receive systematic oral care during the treatment. Of 193 patients with poor oral hygiene before treatment, 135 had been followed up by dentists 3 months after the treatment. Of the 135 patients, 87 whose oral hygiene had improved had a significantly lower frequency of AP than did the 48 patients who had poor oral hygiene (18% vs 54%). The researchers say this suggests that continuous oral management is required in high-risk patients even after treatment.
The researchers also say unnecessary posttreatment administration of sleeping pills might increase the risk of AP. “Notably,” they say, of the 94 patients who used sleeping pills at the end of treatment, 83 continued to use them after the treatment. However, the researchers point to a study that found 31% of patients who had received radiotherapy or CRT had insomnia during the treatment, but about half of them recovered after the treatment.
Seven of 11 patients who had multiple HNCs or coexisting cervical esophageal cancers developed AP. So did 6 of 18 patients who underwent surgical or endoscopic resection for esophageal and gastric cancer. Three of those 6 developed AP within 1 week postresection. The researchers suggest that postsurgical immunosuppression and anesthesia or sedation before endoscopy might impair swallowing function. They also suggest that clinicians may want to consider swallowing exercises for high- or moderate-risk patients to improve swallowing function.
Related: Faster Triage of Veterans With Head and Neck Cancer
The researchers note that AP has been found to be a “significant prognostic factor,” citing a study that found that AP accounted for 19% of noncancer-related deaths of patients with HNC who received CRT. Therefore, they expected AP to be strongly associated with patient survival in their study. However, it wasn’t—perhaps because of the relatively low number of deaths during the follow-up period. Nonetheless, AP “tended to be associated” with increased risk of death.
Source:
Kawai S, Yokota T, Onozawa Y, et al. BMC Cancer. 2017;17:59.
doi: 10.1186/s12885-017-3052-8.
Aspiration pneumonia (AP) is a common late adverse effect of chemoradiotherapy (CRT) and bioradiotherapy for head and neck cancer (HNC). Evaluating the long-term risk factors of AP is difficult because patients’ characteristics vary according to the multimodal therapies they receive, say researchers from Shizuoka Cancer Center in Japan. They conducted a study (the first to their knowledge) to identify specific factors that might help predict which patients have the highest risk of infection.
Related: Lean Six Sigma Applied to Tracking Head/Neck Cancer Patients
The researchers’ retrospective analysis covered nearly 9 years of data. Of the 305 patients in the study, 65 (21%) developed AP after CRT or bioradiotherapy. The median time from end of treatment to AP was 161 days. Nearly all (95%) the patients had Eastern Cooperative Oncology Group performance status of 0 to 1. Most had received standard chemotherapeutic regimens with platinum or cetuximab with supportive care.
The researchers found 5 independent risk factors: habitual alcoholic consumption, poor oral hygiene, coexisting malignancies, hypoalbuminemia before treatment, and use of sleeping pills at the end of treatment. Of those, only hypoalbuminemia was a familiar factor consistent with previous reports.
The finding that oral hygiene predicted AP was unexpected, because at the study hospital, HNC patients who undergo radiotherapy are routinely referred to dentists and receive systematic oral care during the treatment. Of 193 patients with poor oral hygiene before treatment, 135 had been followed up by dentists 3 months after the treatment. Of the 135 patients, 87 whose oral hygiene had improved had a significantly lower frequency of AP than did the 48 patients who had poor oral hygiene (18% vs 54%). The researchers say this suggests that continuous oral management is required in high-risk patients even after treatment.
The researchers also say unnecessary posttreatment administration of sleeping pills might increase the risk of AP. “Notably,” they say, of the 94 patients who used sleeping pills at the end of treatment, 83 continued to use them after the treatment. However, the researchers point to a study that found 31% of patients who had received radiotherapy or CRT had insomnia during the treatment, but about half of them recovered after the treatment.
Seven of 11 patients who had multiple HNCs or coexisting cervical esophageal cancers developed AP. So did 6 of 18 patients who underwent surgical or endoscopic resection for esophageal and gastric cancer. Three of those 6 developed AP within 1 week postresection. The researchers suggest that postsurgical immunosuppression and anesthesia or sedation before endoscopy might impair swallowing function. They also suggest that clinicians may want to consider swallowing exercises for high- or moderate-risk patients to improve swallowing function.
Related: Faster Triage of Veterans With Head and Neck Cancer
The researchers note that AP has been found to be a “significant prognostic factor,” citing a study that found that AP accounted for 19% of noncancer-related deaths of patients with HNC who received CRT. Therefore, they expected AP to be strongly associated with patient survival in their study. However, it wasn’t—perhaps because of the relatively low number of deaths during the follow-up period. Nonetheless, AP “tended to be associated” with increased risk of death.
Source:
Kawai S, Yokota T, Onozawa Y, et al. BMC Cancer. 2017;17:59.
doi: 10.1186/s12885-017-3052-8.
Aspiration pneumonia (AP) is a common late adverse effect of chemoradiotherapy (CRT) and bioradiotherapy for head and neck cancer (HNC). Evaluating the long-term risk factors of AP is difficult because patients’ characteristics vary according to the multimodal therapies they receive, say researchers from Shizuoka Cancer Center in Japan. They conducted a study (the first to their knowledge) to identify specific factors that might help predict which patients have the highest risk of infection.
Related: Lean Six Sigma Applied to Tracking Head/Neck Cancer Patients
The researchers’ retrospective analysis covered nearly 9 years of data. Of the 305 patients in the study, 65 (21%) developed AP after CRT or bioradiotherapy. The median time from end of treatment to AP was 161 days. Nearly all (95%) the patients had Eastern Cooperative Oncology Group performance status of 0 to 1. Most had received standard chemotherapeutic regimens with platinum or cetuximab with supportive care.
The researchers found 5 independent risk factors: habitual alcoholic consumption, poor oral hygiene, coexisting malignancies, hypoalbuminemia before treatment, and use of sleeping pills at the end of treatment. Of those, only hypoalbuminemia was a familiar factor consistent with previous reports.
The finding that oral hygiene predicted AP was unexpected, because at the study hospital, HNC patients who undergo radiotherapy are routinely referred to dentists and receive systematic oral care during the treatment. Of 193 patients with poor oral hygiene before treatment, 135 had been followed up by dentists 3 months after the treatment. Of the 135 patients, 87 whose oral hygiene had improved had a significantly lower frequency of AP than did the 48 patients who had poor oral hygiene (18% vs 54%). The researchers say this suggests that continuous oral management is required in high-risk patients even after treatment.
The researchers also say unnecessary posttreatment administration of sleeping pills might increase the risk of AP. “Notably,” they say, of the 94 patients who used sleeping pills at the end of treatment, 83 continued to use them after the treatment. However, the researchers point to a study that found 31% of patients who had received radiotherapy or CRT had insomnia during the treatment, but about half of them recovered after the treatment.
Seven of 11 patients who had multiple HNCs or coexisting cervical esophageal cancers developed AP. So did 6 of 18 patients who underwent surgical or endoscopic resection for esophageal and gastric cancer. Three of those 6 developed AP within 1 week postresection. The researchers suggest that postsurgical immunosuppression and anesthesia or sedation before endoscopy might impair swallowing function. They also suggest that clinicians may want to consider swallowing exercises for high- or moderate-risk patients to improve swallowing function.
Related: Faster Triage of Veterans With Head and Neck Cancer
The researchers note that AP has been found to be a “significant prognostic factor,” citing a study that found that AP accounted for 19% of noncancer-related deaths of patients with HNC who received CRT. Therefore, they expected AP to be strongly associated with patient survival in their study. However, it wasn’t—perhaps because of the relatively low number of deaths during the follow-up period. Nonetheless, AP “tended to be associated” with increased risk of death.
Source:
Kawai S, Yokota T, Onozawa Y, et al. BMC Cancer. 2017;17:59.
doi: 10.1186/s12885-017-3052-8.
Drug may be new option for difficult-to-treat DLBCL, doc says

WASHINGTON, DC—Selinexor has demonstrated the potential to become a new oral treatment option for patients with difficult-to-treat diffuse large B-cell lymphoma (DLBCL), according to a presenter at the AACR Annual Meeting 2017.
Interim results from the phase 2b SADAL study showed that selinexor produced a 28.6% overall response rate (ORR), with an 11.1% complete response (CR) rate, in a heavily pretreated, older DLBCL population.
Responses were observed in GCB and non-GCB subtypes, and the median duration of response exceeded 7 months.
The most common adverse events (AEs) were fatigue, thrombocytopenia, nausea, anorexia, and vomiting.
Marie Maerevoet, MD, of the Institute Jules Bordet in Brussels, Belgium, presented data from the SADAL study as abstract CT132/13.*
The trial is sponsored by Karyopharm Therapeutics, the company developing selinexor.
Patients and treatment
The study enrolled 72 patients with relapsed or refractory DLBCL. At least 14 weeks had elapsed since their most recent systemic anti-DLBCL therapy.
The patients received selinexor—an oral selective inhibitor of nuclear export (SINE™) compound—at 60 mg or 100 mg twice weekly (days 1 and 3 each week) of each 28-day cycle.
60 mg arm
There were 37 patients in the 60 mg arm. Their median age was 71 (range, 38-87), and most (n=24) were male. Forty-nine percent of these patients (n=18) had GCB DLBCL.
Fourteen percent of patients had high-risk disease (according to the revised international prognostic index). Forty-three percent had high-intermediate-risk, 30% had low-intermediate-risk, and 14% had low-risk disease.
The patients had received a median of 3 prior treatment regimens (range, 2-5). Twenty-seven percent had received a prior transplant.
100 mg arm
There were 35 patients in the 100 mg arm. Their median age was 68 (range, 32-82), and most (n=23) were male. Fifty-one percent of patients (n=18) had GCB DLBCL.
Eleven percent of patients had high-risk, 40% had high-intermediate-risk, 37% had low-intermediate-risk, and 6% had low-risk disease. For 6% of patients, their risk group was unknown.
The patients had received a median of 3 prior treatment regimens (range, 2-5). Forty-six percent had received a prior transplant.
Safety
All 72 patients were evaluable for safety. The most common AEs across both dosing groups were fatigue (65%), thrombocytopenia (54%), nausea (51%), anorexia (49%), vomiting (35%), and anemia (32%).
These events were primarily grades 1 and 2 and were managed with dose modifications and/or standard supportive care.
The 60 mg dose was better tolerated than the 100 mg dose, with fewer dose interruptions and modifications required in the 60 mg arm.
Grade 3/4 AEs that were more common in the 100 mg arm than the 60 mg arm were fatigue (26% vs 11%), thrombocytopenia (46% vs 32%), and anorexia (11% vs 3%).
Efficacy
Sixty-three patients were analyzed for response. The ORR was 28.6% (18/63), with a CR rate of 11.1% (n=7) and a partial response (PR) rate of 17.5% (n=11).
The rate of stable disease (SD) was 14.3% (n=9), and the rate of progressive disease (PD) was 46% (n=29). Seven patients (11.1%) were not evaluable (NE).
The best responses as of March 1, 2017, according to subtype and selinexor dose, were as follows:
| Category | N | ORR | CR | PR | SD | PD | NE |
| 60 mg | 32 | 9 (28.1%) | 4 (12.5%) | 5 (15.6%) | 3 (9.4%) | 17 (53.1%) | 3 (9.4%) |
| 100 mg | 31 | 9 (29.0%) | 3 (9.7%) | 6 (19.4%) | 6 (19.4%) | 12 (38.7%) | 4 (12.9%) |
| GCB subtype | 32 | 8 (25.0%) | 3 (9.4%) | 5 (15.6%) | 6 (18.8%) | 13 (40.6%) | 5 (15.6%) |
| Non-GCB subtype | 31 | 10 (32.3%) | 4 (12.9%) | 6 (19.4%) | 3 (9.7%) | 16 (51.6%) | 2 (6.5%) |
The median duration of response was greater than 7 months. The median time to response was 2 months.
Among responders, the median time on treatment was 9 months, with a median follow-up of 13 months. As of the data cutoff date, 9 responders remained on treatment, including 6 patients with a CR.
The median overall survival was 8 months for all patients. As of the cutoff date, the median survival for the responders had not been reached.
“With the impressive and durable responses observed to date, including in both the GCB and non-GCB subtypes of DLBCL, single-agent selinexor is demonstrating the potential to become a new oral option for this difficult-to-treat patient population who are not candidates for transplantation and whose disease is unlikely to respond to further chemotherapy or targeted agents,” Dr Maerevoet said.
Trial update
As a result of the interim data from SADAL, and in consultation with the US Food and Drug Administration (FDA), Karyopharm is amending the study protocol.
SADAL will become a single-arm study focusing solely on single-agent selinexor dosed at 60 mg twice weekly.
The study is also being amended to reduce the 14-week treatment-free period to 8 weeks in patients who achieved at least a PR on their most recent therapy. Patients who were refractory to or did not achieve at least a PR on their prior therapy will continue with the 14-week treatment-free period.
Karyopharm plans to enroll up to an additional 90 patients to the new 60 mg single-arm cohort and expects to report top-line results from the SADAL study in mid-2018.
The FDA recently lifted a partial clinical hold placed on the SADAL trial and other trials of selinexor.
The FDA had placed the hold due to a lack of information in the investigator’s brochure, including an incomplete list of serious adverse events associated with selinexor. ![]()
*Data in the abstract differ from the presentation.
WASHINGTON, DC—Selinexor has demonstrated the potential to become a new oral treatment option for patients with difficult-to-treat diffuse large B-cell lymphoma (DLBCL), according to a presenter at the AACR Annual Meeting 2017.
Interim results from the phase 2b SADAL study showed that selinexor produced a 28.6% overall response rate (ORR), with an 11.1% complete response (CR) rate, in a heavily pretreated, older DLBCL population.
Responses were observed in GCB and non-GCB subtypes, and the median duration of response exceeded 7 months.
The most common adverse events (AEs) were fatigue, thrombocytopenia, nausea, anorexia, and vomiting.
Marie Maerevoet, MD, of the Institute Jules Bordet in Brussels, Belgium, presented data from the SADAL study as abstract CT132/13.*
The trial is sponsored by Karyopharm Therapeutics, the company developing selinexor.
Patients and treatment
The study enrolled 72 patients with relapsed or refractory DLBCL. At least 14 weeks had elapsed since their most recent systemic anti-DLBCL therapy.
The patients received selinexor—an oral selective inhibitor of nuclear export (SINE™) compound—at 60 mg or 100 mg twice weekly (days 1 and 3 each week) of each 28-day cycle.
60 mg arm
There were 37 patients in the 60 mg arm. Their median age was 71 (range, 38-87), and most (n=24) were male. Forty-nine percent of these patients (n=18) had GCB DLBCL.
Fourteen percent of patients had high-risk disease (according to the revised international prognostic index). Forty-three percent had high-intermediate-risk, 30% had low-intermediate-risk, and 14% had low-risk disease.
The patients had received a median of 3 prior treatment regimens (range, 2-5). Twenty-seven percent had received a prior transplant.
100 mg arm
There were 35 patients in the 100 mg arm. Their median age was 68 (range, 32-82), and most (n=23) were male. Fifty-one percent of patients (n=18) had GCB DLBCL.
Eleven percent of patients had high-risk, 40% had high-intermediate-risk, 37% had low-intermediate-risk, and 6% had low-risk disease. For 6% of patients, their risk group was unknown.
The patients had received a median of 3 prior treatment regimens (range, 2-5). Forty-six percent had received a prior transplant.
Safety
All 72 patients were evaluable for safety. The most common AEs across both dosing groups were fatigue (65%), thrombocytopenia (54%), nausea (51%), anorexia (49%), vomiting (35%), and anemia (32%).
These events were primarily grades 1 and 2 and were managed with dose modifications and/or standard supportive care.
The 60 mg dose was better tolerated than the 100 mg dose, with fewer dose interruptions and modifications required in the 60 mg arm.
Grade 3/4 AEs that were more common in the 100 mg arm than the 60 mg arm were fatigue (26% vs 11%), thrombocytopenia (46% vs 32%), and anorexia (11% vs 3%).
Efficacy
Sixty-three patients were analyzed for response. The ORR was 28.6% (18/63), with a CR rate of 11.1% (n=7) and a partial response (PR) rate of 17.5% (n=11).
The rate of stable disease (SD) was 14.3% (n=9), and the rate of progressive disease (PD) was 46% (n=29). Seven patients (11.1%) were not evaluable (NE).
The best responses as of March 1, 2017, according to subtype and selinexor dose, were as follows:
| Category | N | ORR | CR | PR | SD | PD | NE |
| 60 mg | 32 | 9 (28.1%) | 4 (12.5%) | 5 (15.6%) | 3 (9.4%) | 17 (53.1%) | 3 (9.4%) |
| 100 mg | 31 | 9 (29.0%) | 3 (9.7%) | 6 (19.4%) | 6 (19.4%) | 12 (38.7%) | 4 (12.9%) |
| GCB subtype | 32 | 8 (25.0%) | 3 (9.4%) | 5 (15.6%) | 6 (18.8%) | 13 (40.6%) | 5 (15.6%) |
| Non-GCB subtype | 31 | 10 (32.3%) | 4 (12.9%) | 6 (19.4%) | 3 (9.7%) | 16 (51.6%) | 2 (6.5%) |
The median duration of response was greater than 7 months. The median time to response was 2 months.
Among responders, the median time on treatment was 9 months, with a median follow-up of 13 months. As of the data cutoff date, 9 responders remained on treatment, including 6 patients with a CR.
The median overall survival was 8 months for all patients. As of the cutoff date, the median survival for the responders had not been reached.
“With the impressive and durable responses observed to date, including in both the GCB and non-GCB subtypes of DLBCL, single-agent selinexor is demonstrating the potential to become a new oral option for this difficult-to-treat patient population who are not candidates for transplantation and whose disease is unlikely to respond to further chemotherapy or targeted agents,” Dr Maerevoet said.
Trial update
As a result of the interim data from SADAL, and in consultation with the US Food and Drug Administration (FDA), Karyopharm is amending the study protocol.
SADAL will become a single-arm study focusing solely on single-agent selinexor dosed at 60 mg twice weekly.
The study is also being amended to reduce the 14-week treatment-free period to 8 weeks in patients who achieved at least a PR on their most recent therapy. Patients who were refractory to or did not achieve at least a PR on their prior therapy will continue with the 14-week treatment-free period.
Karyopharm plans to enroll up to an additional 90 patients to the new 60 mg single-arm cohort and expects to report top-line results from the SADAL study in mid-2018.
The FDA recently lifted a partial clinical hold placed on the SADAL trial and other trials of selinexor.
The FDA had placed the hold due to a lack of information in the investigator’s brochure, including an incomplete list of serious adverse events associated with selinexor. ![]()
*Data in the abstract differ from the presentation.
WASHINGTON, DC—Selinexor has demonstrated the potential to become a new oral treatment option for patients with difficult-to-treat diffuse large B-cell lymphoma (DLBCL), according to a presenter at the AACR Annual Meeting 2017.
Interim results from the phase 2b SADAL study showed that selinexor produced a 28.6% overall response rate (ORR), with an 11.1% complete response (CR) rate, in a heavily pretreated, older DLBCL population.
Responses were observed in GCB and non-GCB subtypes, and the median duration of response exceeded 7 months.
The most common adverse events (AEs) were fatigue, thrombocytopenia, nausea, anorexia, and vomiting.
Marie Maerevoet, MD, of the Institute Jules Bordet in Brussels, Belgium, presented data from the SADAL study as abstract CT132/13.*
The trial is sponsored by Karyopharm Therapeutics, the company developing selinexor.
Patients and treatment
The study enrolled 72 patients with relapsed or refractory DLBCL. At least 14 weeks had elapsed since their most recent systemic anti-DLBCL therapy.
The patients received selinexor—an oral selective inhibitor of nuclear export (SINE™) compound—at 60 mg or 100 mg twice weekly (days 1 and 3 each week) of each 28-day cycle.
60 mg arm
There were 37 patients in the 60 mg arm. Their median age was 71 (range, 38-87), and most (n=24) were male. Forty-nine percent of these patients (n=18) had GCB DLBCL.
Fourteen percent of patients had high-risk disease (according to the revised international prognostic index). Forty-three percent had high-intermediate-risk, 30% had low-intermediate-risk, and 14% had low-risk disease.
The patients had received a median of 3 prior treatment regimens (range, 2-5). Twenty-seven percent had received a prior transplant.
100 mg arm
There were 35 patients in the 100 mg arm. Their median age was 68 (range, 32-82), and most (n=23) were male. Fifty-one percent of patients (n=18) had GCB DLBCL.
Eleven percent of patients had high-risk, 40% had high-intermediate-risk, 37% had low-intermediate-risk, and 6% had low-risk disease. For 6% of patients, their risk group was unknown.
The patients had received a median of 3 prior treatment regimens (range, 2-5). Forty-six percent had received a prior transplant.
Safety
All 72 patients were evaluable for safety. The most common AEs across both dosing groups were fatigue (65%), thrombocytopenia (54%), nausea (51%), anorexia (49%), vomiting (35%), and anemia (32%).
These events were primarily grades 1 and 2 and were managed with dose modifications and/or standard supportive care.
The 60 mg dose was better tolerated than the 100 mg dose, with fewer dose interruptions and modifications required in the 60 mg arm.
Grade 3/4 AEs that were more common in the 100 mg arm than the 60 mg arm were fatigue (26% vs 11%), thrombocytopenia (46% vs 32%), and anorexia (11% vs 3%).
Efficacy
Sixty-three patients were analyzed for response. The ORR was 28.6% (18/63), with a CR rate of 11.1% (n=7) and a partial response (PR) rate of 17.5% (n=11).
The rate of stable disease (SD) was 14.3% (n=9), and the rate of progressive disease (PD) was 46% (n=29). Seven patients (11.1%) were not evaluable (NE).
The best responses as of March 1, 2017, according to subtype and selinexor dose, were as follows:
| Category | N | ORR | CR | PR | SD | PD | NE |
| 60 mg | 32 | 9 (28.1%) | 4 (12.5%) | 5 (15.6%) | 3 (9.4%) | 17 (53.1%) | 3 (9.4%) |
| 100 mg | 31 | 9 (29.0%) | 3 (9.7%) | 6 (19.4%) | 6 (19.4%) | 12 (38.7%) | 4 (12.9%) |
| GCB subtype | 32 | 8 (25.0%) | 3 (9.4%) | 5 (15.6%) | 6 (18.8%) | 13 (40.6%) | 5 (15.6%) |
| Non-GCB subtype | 31 | 10 (32.3%) | 4 (12.9%) | 6 (19.4%) | 3 (9.7%) | 16 (51.6%) | 2 (6.5%) |
The median duration of response was greater than 7 months. The median time to response was 2 months.
Among responders, the median time on treatment was 9 months, with a median follow-up of 13 months. As of the data cutoff date, 9 responders remained on treatment, including 6 patients with a CR.
The median overall survival was 8 months for all patients. As of the cutoff date, the median survival for the responders had not been reached.
“With the impressive and durable responses observed to date, including in both the GCB and non-GCB subtypes of DLBCL, single-agent selinexor is demonstrating the potential to become a new oral option for this difficult-to-treat patient population who are not candidates for transplantation and whose disease is unlikely to respond to further chemotherapy or targeted agents,” Dr Maerevoet said.
Trial update
As a result of the interim data from SADAL, and in consultation with the US Food and Drug Administration (FDA), Karyopharm is amending the study protocol.
SADAL will become a single-arm study focusing solely on single-agent selinexor dosed at 60 mg twice weekly.
The study is also being amended to reduce the 14-week treatment-free period to 8 weeks in patients who achieved at least a PR on their most recent therapy. Patients who were refractory to or did not achieve at least a PR on their prior therapy will continue with the 14-week treatment-free period.
Karyopharm plans to enroll up to an additional 90 patients to the new 60 mg single-arm cohort and expects to report top-line results from the SADAL study in mid-2018.
The FDA recently lifted a partial clinical hold placed on the SADAL trial and other trials of selinexor.
The FDA had placed the hold due to a lack of information in the investigator’s brochure, including an incomplete list of serious adverse events associated with selinexor. ![]()
*Data in the abstract differ from the presentation.
Extremely short and long telomeres linked to risk of leukemia
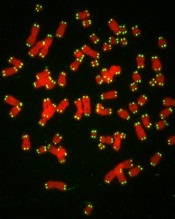
WASHINGTON, DC—Telomere length may predict cancer risk and could be a target for future therapies, according to research presented at the AACR Annual Meeting 2017.
The study suggests that, overall, longer-than-expected telomeres are associated with an increased risk of cancer.
However, investigators also found an increased risk of certain cancers, including leukemia, among patients who had very short or very long telomeres.
“Telomeres and cancer clearly have a complex relationship,” said study investigator Jian-Min Yuan, MD, PhD, of the University of Pittsburgh Cancer Institute in Pennsylvania.
“Our hope is that by understanding this relationship, we may be able to predict which people are most likely to develop certain cancers so they can take preventive measures and perhaps be screened more often, as well as develop therapies to help our DNA keep or return its telomeres to a healthy length.”
Dr Yuan and his colleagues presented their research as abstract 2267/21.*
The team analyzed blood samples and health data on 26,540 individuals enrolled in the Singapore Chinese Health Study, which has followed the health outcomes of participants since 1993. As of the end of 2015, 4060 participants had developed cancer (77 with leukemia).
The investigators divided participants into 5 groups, based on the length of their telomeres.
The group with the longest telomeres (quartile 5) had a significantly higher risk of developing any cancer than the group with the shortest telomeres (quartile 1), after taking into account the effects of age, sex, education, body mass index, smoking habits, and alcohol consumption. The hazard ratio (HR) was 1.33 (P<0.0001).
In particular, the group with the longest telomeres had a significantly increased risk of colorectal cancer (HR=1.37, P=0.010), breast cancer (HR=1.39, P=0.007), prostate cancer (HR=1.55, P=0.011), and pancreatic cancer (HR=2.58, P=0.009).
On the other hand, the risk of liver cancer decreased as telomere length increased. The HR was 0.72 for patients in quartile 5 compared to those in quartile 1 (Plinear=0.056).
The risk of 3 cancers was greatest for both the groups with extremely short and extremely long telomeres—quartiles 1 and 5, respectively.
For leukemia, the HR was 2.15 for quartile 1 and 1.68 for quartile 5, when compared to quartile 3 (Pnon-linear=0.015).
For bladder cancer, the HRs were 1.72 and 2.17, respectively (Pnon-linear=0.01). And for stomach cancer, the HRs were 1.63 and 1.55, respectively (Pnon-linear=0.020).
“We had the idea for this study more than 7 years ago, but it took the laboratory 3 months to finish quantifying telomere length for just 100 samples, which was not enough to draw any meaningful conclusions,” Dr Yuan said.
“Not even a decade later, we’ve been able to run nearly 30,000 samples in a year and draw these really robust insights, showing how far technology has come. Even more complicated will be linking telomere length to genome-wide analyses, which is on the horizon. We’re on the cusp of gaining a truly remarkable understanding of the complicated biological phenomena that lead to cancer.” ![]()
*The presentation differed from the abstract.
WASHINGTON, DC—Telomere length may predict cancer risk and could be a target for future therapies, according to research presented at the AACR Annual Meeting 2017.
The study suggests that, overall, longer-than-expected telomeres are associated with an increased risk of cancer.
However, investigators also found an increased risk of certain cancers, including leukemia, among patients who had very short or very long telomeres.
“Telomeres and cancer clearly have a complex relationship,” said study investigator Jian-Min Yuan, MD, PhD, of the University of Pittsburgh Cancer Institute in Pennsylvania.
“Our hope is that by understanding this relationship, we may be able to predict which people are most likely to develop certain cancers so they can take preventive measures and perhaps be screened more often, as well as develop therapies to help our DNA keep or return its telomeres to a healthy length.”
Dr Yuan and his colleagues presented their research as abstract 2267/21.*
The team analyzed blood samples and health data on 26,540 individuals enrolled in the Singapore Chinese Health Study, which has followed the health outcomes of participants since 1993. As of the end of 2015, 4060 participants had developed cancer (77 with leukemia).
The investigators divided participants into 5 groups, based on the length of their telomeres.
The group with the longest telomeres (quartile 5) had a significantly higher risk of developing any cancer than the group with the shortest telomeres (quartile 1), after taking into account the effects of age, sex, education, body mass index, smoking habits, and alcohol consumption. The hazard ratio (HR) was 1.33 (P<0.0001).
In particular, the group with the longest telomeres had a significantly increased risk of colorectal cancer (HR=1.37, P=0.010), breast cancer (HR=1.39, P=0.007), prostate cancer (HR=1.55, P=0.011), and pancreatic cancer (HR=2.58, P=0.009).
On the other hand, the risk of liver cancer decreased as telomere length increased. The HR was 0.72 for patients in quartile 5 compared to those in quartile 1 (Plinear=0.056).
The risk of 3 cancers was greatest for both the groups with extremely short and extremely long telomeres—quartiles 1 and 5, respectively.
For leukemia, the HR was 2.15 for quartile 1 and 1.68 for quartile 5, when compared to quartile 3 (Pnon-linear=0.015).
For bladder cancer, the HRs were 1.72 and 2.17, respectively (Pnon-linear=0.01). And for stomach cancer, the HRs were 1.63 and 1.55, respectively (Pnon-linear=0.020).
“We had the idea for this study more than 7 years ago, but it took the laboratory 3 months to finish quantifying telomere length for just 100 samples, which was not enough to draw any meaningful conclusions,” Dr Yuan said.
“Not even a decade later, we’ve been able to run nearly 30,000 samples in a year and draw these really robust insights, showing how far technology has come. Even more complicated will be linking telomere length to genome-wide analyses, which is on the horizon. We’re on the cusp of gaining a truly remarkable understanding of the complicated biological phenomena that lead to cancer.” ![]()
*The presentation differed from the abstract.
WASHINGTON, DC—Telomere length may predict cancer risk and could be a target for future therapies, according to research presented at the AACR Annual Meeting 2017.
The study suggests that, overall, longer-than-expected telomeres are associated with an increased risk of cancer.
However, investigators also found an increased risk of certain cancers, including leukemia, among patients who had very short or very long telomeres.
“Telomeres and cancer clearly have a complex relationship,” said study investigator Jian-Min Yuan, MD, PhD, of the University of Pittsburgh Cancer Institute in Pennsylvania.
“Our hope is that by understanding this relationship, we may be able to predict which people are most likely to develop certain cancers so they can take preventive measures and perhaps be screened more often, as well as develop therapies to help our DNA keep or return its telomeres to a healthy length.”
Dr Yuan and his colleagues presented their research as abstract 2267/21.*
The team analyzed blood samples and health data on 26,540 individuals enrolled in the Singapore Chinese Health Study, which has followed the health outcomes of participants since 1993. As of the end of 2015, 4060 participants had developed cancer (77 with leukemia).
The investigators divided participants into 5 groups, based on the length of their telomeres.
The group with the longest telomeres (quartile 5) had a significantly higher risk of developing any cancer than the group with the shortest telomeres (quartile 1), after taking into account the effects of age, sex, education, body mass index, smoking habits, and alcohol consumption. The hazard ratio (HR) was 1.33 (P<0.0001).
In particular, the group with the longest telomeres had a significantly increased risk of colorectal cancer (HR=1.37, P=0.010), breast cancer (HR=1.39, P=0.007), prostate cancer (HR=1.55, P=0.011), and pancreatic cancer (HR=2.58, P=0.009).
On the other hand, the risk of liver cancer decreased as telomere length increased. The HR was 0.72 for patients in quartile 5 compared to those in quartile 1 (Plinear=0.056).
The risk of 3 cancers was greatest for both the groups with extremely short and extremely long telomeres—quartiles 1 and 5, respectively.
For leukemia, the HR was 2.15 for quartile 1 and 1.68 for quartile 5, when compared to quartile 3 (Pnon-linear=0.015).
For bladder cancer, the HRs were 1.72 and 2.17, respectively (Pnon-linear=0.01). And for stomach cancer, the HRs were 1.63 and 1.55, respectively (Pnon-linear=0.020).
“We had the idea for this study more than 7 years ago, but it took the laboratory 3 months to finish quantifying telomere length for just 100 samples, which was not enough to draw any meaningful conclusions,” Dr Yuan said.
“Not even a decade later, we’ve been able to run nearly 30,000 samples in a year and draw these really robust insights, showing how far technology has come. Even more complicated will be linking telomere length to genome-wide analyses, which is on the horizon. We’re on the cusp of gaining a truly remarkable understanding of the complicated biological phenomena that lead to cancer.” ![]()
*The presentation differed from the abstract.
Method may help MM patients avoid bone marrow biopsies

Engineers say they have devised a microfluidic technique to capture and count circulating plasma cells from small samples of blood.
They believe the technique, described in Scientific Reports, may allow patients with multiple myeloma (MM) to avoid bone marrow biopsies in favor of conventional blood draws.
“Procedures of the traditional tissue biopsy are painful, associated with complications such as potential infections, and often available only in central hospitals, which require patients to travel long distances,” said Mohammad Qasaimeh, PhD, of New York University Abu Dhabi in United Arab Emirates.
“Capturing plasma cells from blood samples can serve as a liquid biopsy, which can be performed in clinics as often as required, and serve as a diagnostic and prognostic test during and after chemotherapy treatment. Moreover, captured cells can be used for drug testing and, thus, serve as a tool for personalized medicine.”
About the technique
The current microfluidic technique builds on a design that was developed by George Whitesides, PhD, of Harvard University in Cambridge, Massachusetts.
Dr Whitesides and his colleagues fabricated a microchip, the channel of which they etched with repeating, V-shaped grooves, similar to a herringbone pattern.
The grooves cause any fluid flowing through the microchip to swirl about in eddies, rather passing straight through. The cells within the fluid therefore have a higher chance of making contact with the floor of the device.
Researchers have since reproduced this microfluidic design, coating the microchip’s floor with certain molecules to attract cells of interest.
Qasaimeh and colleagues used the microfluidic herringbone design to capture circulating plasma cells. They coated the channels of a microchip with CD138, an antibody expressed on the membranes of plasma cells.
The team then flowed 1 mL samples of blood through the device. The herringbone grooves circulated the blood in the microfluidic channels, where the antibodies grabbed onto any passing plasma cells and let the rest of the blood flow out of the device.
Once the cells were isolated in the microchip, the researchers could count the cells, as well as determine the kinds of antibodies each cell secretes.
Capturing cells
The researchers tested the device using blood samples from healthy donors as well as MM patients.
The team observed very low numbers of circulating plasma cells in healthy samples—2 to 5 cells per mL of blood—and substantially higher counts in MM patients—45 to 184 cells per mL.
The researchers noted that MM patients in remission also exhibited higher counts of circulating plasma cells—20 to 24 cells per mL—than healthy donors.
Rahit Karnik, PhD, of Massachusetts Institute of Technology in Cambridge, said this device may reveal more subtle information about a patient’s state, even in remission.
“When patients go into remission, their antibody levels can look normal,” Dr Karnik said. “But we detect a level of circulating plasma cells that is above the baseline. It’s hard to tell whether these cells are cancerous, but at least this technique is giving us more information. With the ease of a blood draw, this may enable us to track cancer in a much better way.”
Analyzing antibodies
The researchers also looked at antibodies produced by the plasma cells captured in the device.
The team determined the ratio of plasma cells producing kappa- and lambda-type antibodies and compared them to conventional blood tests for the same antibodies, for both healthy subjects and MM patients.
Both sets of results matched, which validates the microfluidic device’s accuracy.
Next steps
Dr Karnik said, in the future, researchers may use this design to perform genetic tests on the captured cells or look for mutations in the cells that may further characterize MM.
“We can capture and stain these cells in the device, which opens the possibility of studying whether there are new mutations in the cells,” Dr Karnik said.
“With cancers like multiple myeloma, even for patients in remission, cancer can recur. Detecting the level or mutation of plasma cells in blood might provide an early detection method for these patients.” ![]()
Engineers say they have devised a microfluidic technique to capture and count circulating plasma cells from small samples of blood.
They believe the technique, described in Scientific Reports, may allow patients with multiple myeloma (MM) to avoid bone marrow biopsies in favor of conventional blood draws.
“Procedures of the traditional tissue biopsy are painful, associated with complications such as potential infections, and often available only in central hospitals, which require patients to travel long distances,” said Mohammad Qasaimeh, PhD, of New York University Abu Dhabi in United Arab Emirates.
“Capturing plasma cells from blood samples can serve as a liquid biopsy, which can be performed in clinics as often as required, and serve as a diagnostic and prognostic test during and after chemotherapy treatment. Moreover, captured cells can be used for drug testing and, thus, serve as a tool for personalized medicine.”
About the technique
The current microfluidic technique builds on a design that was developed by George Whitesides, PhD, of Harvard University in Cambridge, Massachusetts.
Dr Whitesides and his colleagues fabricated a microchip, the channel of which they etched with repeating, V-shaped grooves, similar to a herringbone pattern.
The grooves cause any fluid flowing through the microchip to swirl about in eddies, rather passing straight through. The cells within the fluid therefore have a higher chance of making contact with the floor of the device.
Researchers have since reproduced this microfluidic design, coating the microchip’s floor with certain molecules to attract cells of interest.
Qasaimeh and colleagues used the microfluidic herringbone design to capture circulating plasma cells. They coated the channels of a microchip with CD138, an antibody expressed on the membranes of plasma cells.
The team then flowed 1 mL samples of blood through the device. The herringbone grooves circulated the blood in the microfluidic channels, where the antibodies grabbed onto any passing plasma cells and let the rest of the blood flow out of the device.
Once the cells were isolated in the microchip, the researchers could count the cells, as well as determine the kinds of antibodies each cell secretes.
Capturing cells
The researchers tested the device using blood samples from healthy donors as well as MM patients.
The team observed very low numbers of circulating plasma cells in healthy samples—2 to 5 cells per mL of blood—and substantially higher counts in MM patients—45 to 184 cells per mL.
The researchers noted that MM patients in remission also exhibited higher counts of circulating plasma cells—20 to 24 cells per mL—than healthy donors.
Rahit Karnik, PhD, of Massachusetts Institute of Technology in Cambridge, said this device may reveal more subtle information about a patient’s state, even in remission.
“When patients go into remission, their antibody levels can look normal,” Dr Karnik said. “But we detect a level of circulating plasma cells that is above the baseline. It’s hard to tell whether these cells are cancerous, but at least this technique is giving us more information. With the ease of a blood draw, this may enable us to track cancer in a much better way.”
Analyzing antibodies
The researchers also looked at antibodies produced by the plasma cells captured in the device.
The team determined the ratio of plasma cells producing kappa- and lambda-type antibodies and compared them to conventional blood tests for the same antibodies, for both healthy subjects and MM patients.
Both sets of results matched, which validates the microfluidic device’s accuracy.
Next steps
Dr Karnik said, in the future, researchers may use this design to perform genetic tests on the captured cells or look for mutations in the cells that may further characterize MM.
“We can capture and stain these cells in the device, which opens the possibility of studying whether there are new mutations in the cells,” Dr Karnik said.
“With cancers like multiple myeloma, even for patients in remission, cancer can recur. Detecting the level or mutation of plasma cells in blood might provide an early detection method for these patients.” ![]()
Engineers say they have devised a microfluidic technique to capture and count circulating plasma cells from small samples of blood.
They believe the technique, described in Scientific Reports, may allow patients with multiple myeloma (MM) to avoid bone marrow biopsies in favor of conventional blood draws.
“Procedures of the traditional tissue biopsy are painful, associated with complications such as potential infections, and often available only in central hospitals, which require patients to travel long distances,” said Mohammad Qasaimeh, PhD, of New York University Abu Dhabi in United Arab Emirates.
“Capturing plasma cells from blood samples can serve as a liquid biopsy, which can be performed in clinics as often as required, and serve as a diagnostic and prognostic test during and after chemotherapy treatment. Moreover, captured cells can be used for drug testing and, thus, serve as a tool for personalized medicine.”
About the technique
The current microfluidic technique builds on a design that was developed by George Whitesides, PhD, of Harvard University in Cambridge, Massachusetts.
Dr Whitesides and his colleagues fabricated a microchip, the channel of which they etched with repeating, V-shaped grooves, similar to a herringbone pattern.
The grooves cause any fluid flowing through the microchip to swirl about in eddies, rather passing straight through. The cells within the fluid therefore have a higher chance of making contact with the floor of the device.
Researchers have since reproduced this microfluidic design, coating the microchip’s floor with certain molecules to attract cells of interest.
Qasaimeh and colleagues used the microfluidic herringbone design to capture circulating plasma cells. They coated the channels of a microchip with CD138, an antibody expressed on the membranes of plasma cells.
The team then flowed 1 mL samples of blood through the device. The herringbone grooves circulated the blood in the microfluidic channels, where the antibodies grabbed onto any passing plasma cells and let the rest of the blood flow out of the device.
Once the cells were isolated in the microchip, the researchers could count the cells, as well as determine the kinds of antibodies each cell secretes.
Capturing cells
The researchers tested the device using blood samples from healthy donors as well as MM patients.
The team observed very low numbers of circulating plasma cells in healthy samples—2 to 5 cells per mL of blood—and substantially higher counts in MM patients—45 to 184 cells per mL.
The researchers noted that MM patients in remission also exhibited higher counts of circulating plasma cells—20 to 24 cells per mL—than healthy donors.
Rahit Karnik, PhD, of Massachusetts Institute of Technology in Cambridge, said this device may reveal more subtle information about a patient’s state, even in remission.
“When patients go into remission, their antibody levels can look normal,” Dr Karnik said. “But we detect a level of circulating plasma cells that is above the baseline. It’s hard to tell whether these cells are cancerous, but at least this technique is giving us more information. With the ease of a blood draw, this may enable us to track cancer in a much better way.”
Analyzing antibodies
The researchers also looked at antibodies produced by the plasma cells captured in the device.
The team determined the ratio of plasma cells producing kappa- and lambda-type antibodies and compared them to conventional blood tests for the same antibodies, for both healthy subjects and MM patients.
Both sets of results matched, which validates the microfluidic device’s accuracy.
Next steps
Dr Karnik said, in the future, researchers may use this design to perform genetic tests on the captured cells or look for mutations in the cells that may further characterize MM.
“We can capture and stain these cells in the device, which opens the possibility of studying whether there are new mutations in the cells,” Dr Karnik said.
“With cancers like multiple myeloma, even for patients in remission, cancer can recur. Detecting the level or mutation of plasma cells in blood might provide an early detection method for these patients.” ![]()
FDA approves drugs more quickly than EMA

The US Food and Drug Administration (FDA) reviews and approves new medicines in a shorter timeframe than the European Medicines Agency (EMA), according to an analysis published in NEJM.
The FDA has faced pressure from the public, politicians, and industry to accelerate review and approval of new medicines.
The FDA’s review process is currently being considered and reexamined as part of negotiations to reauthorize the law that directs funds to the agency—the Prescription Drug User Fee Act—which is due for reauthorization by October 2017.
To inform this debate, a group of researchers compared review times for new drugs approved by the FDA and the EMA between 2011 and 2015.
The results showed that the FDA approved more new drugs than the EMA—170 versus 144—during this time period.
The median review time was 306 days for FDA-approved drugs and 383 days for EMA-approved drugs (P<0.001).
Over the study period, the FDA approved 53 drugs intended to treat cancer/hematologic diseases, and the EMA approved 50. The median review times were 206 days and 379 days, respectively (P<0.001).
The FDA approved 74 orphan drugs, and the EMA approved 36. The median review times were 294 days and 403 days, respectively (P<0.001).
The current analysis is an update to a prior analysis, published in 2012, which showed the FDA approved new medicines more quickly than the EMA and Health Canada.
“The gap we had identified, where the FDA was 2 to 3 months faster, now it’s about 3 to 4 months faster,” said Joseph Ross, MD, of the Yale School of Medicine in New Haven, Connecticut, an author of both studies.
“This is more information that should inform upcoming debates. The FDA is already making decisions quickly, and increasing its regulatory speed shouldn’t be our number-one priority.” ![]()
The US Food and Drug Administration (FDA) reviews and approves new medicines in a shorter timeframe than the European Medicines Agency (EMA), according to an analysis published in NEJM.
The FDA has faced pressure from the public, politicians, and industry to accelerate review and approval of new medicines.
The FDA’s review process is currently being considered and reexamined as part of negotiations to reauthorize the law that directs funds to the agency—the Prescription Drug User Fee Act—which is due for reauthorization by October 2017.
To inform this debate, a group of researchers compared review times for new drugs approved by the FDA and the EMA between 2011 and 2015.
The results showed that the FDA approved more new drugs than the EMA—170 versus 144—during this time period.
The median review time was 306 days for FDA-approved drugs and 383 days for EMA-approved drugs (P<0.001).
Over the study period, the FDA approved 53 drugs intended to treat cancer/hematologic diseases, and the EMA approved 50. The median review times were 206 days and 379 days, respectively (P<0.001).
The FDA approved 74 orphan drugs, and the EMA approved 36. The median review times were 294 days and 403 days, respectively (P<0.001).
The current analysis is an update to a prior analysis, published in 2012, which showed the FDA approved new medicines more quickly than the EMA and Health Canada.
“The gap we had identified, where the FDA was 2 to 3 months faster, now it’s about 3 to 4 months faster,” said Joseph Ross, MD, of the Yale School of Medicine in New Haven, Connecticut, an author of both studies.
“This is more information that should inform upcoming debates. The FDA is already making decisions quickly, and increasing its regulatory speed shouldn’t be our number-one priority.” ![]()
The US Food and Drug Administration (FDA) reviews and approves new medicines in a shorter timeframe than the European Medicines Agency (EMA), according to an analysis published in NEJM.
The FDA has faced pressure from the public, politicians, and industry to accelerate review and approval of new medicines.
The FDA’s review process is currently being considered and reexamined as part of negotiations to reauthorize the law that directs funds to the agency—the Prescription Drug User Fee Act—which is due for reauthorization by October 2017.
To inform this debate, a group of researchers compared review times for new drugs approved by the FDA and the EMA between 2011 and 2015.
The results showed that the FDA approved more new drugs than the EMA—170 versus 144—during this time period.
The median review time was 306 days for FDA-approved drugs and 383 days for EMA-approved drugs (P<0.001).
Over the study period, the FDA approved 53 drugs intended to treat cancer/hematologic diseases, and the EMA approved 50. The median review times were 206 days and 379 days, respectively (P<0.001).
The FDA approved 74 orphan drugs, and the EMA approved 36. The median review times were 294 days and 403 days, respectively (P<0.001).
The current analysis is an update to a prior analysis, published in 2012, which showed the FDA approved new medicines more quickly than the EMA and Health Canada.
“The gap we had identified, where the FDA was 2 to 3 months faster, now it’s about 3 to 4 months faster,” said Joseph Ross, MD, of the Yale School of Medicine in New Haven, Connecticut, an author of both studies.
“This is more information that should inform upcoming debates. The FDA is already making decisions quickly, and increasing its regulatory speed shouldn’t be our number-one priority.” ![]()
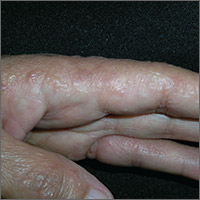
Itchy rash on sides of fingers
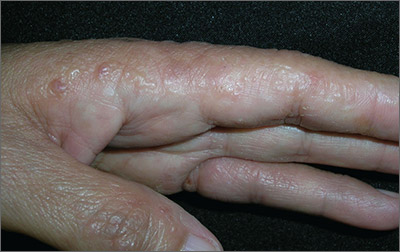
The FP recognized the multiple tapioca-like vesicles on the sides of the patient’s fingers as pompholyx, also known as dyshidrotic eczema. One of the classic findings of dyshidrotic eczema is the vesicles that resemble the small spheres in tapioca pudding. (The FP considered scabies, but ruled it out because no one in the family had a similar rash and there were no signs of any burrows.)
The term “pompholyx” means bubble, while the term “dyshidrotic” means “difficult sweating,” as problems with sweating were once believed to be the cause of this condition. Some dermatologists prefer the name “vesicular hand dermatitis.” Regardless of the terminology, this can be a very mild condition that is a minor nuisance or a very severe chronic condition that impairs the patient’s quality of life.
The FP explained that dyshidrotic eczema is not curable and may be worsened by stress and substances that come into contact with the hands. He discussed the possibility of avoiding washing the dishes and other “wet work,” but the patient said that wasn’t possible, so he told her to use nitrile gloves with cotton liners. (Using gloves without cotton liners often leads to sweating in the gloves that can worsen dyshidrotic eczema.)
The FP prescribed 0.1% triamcinolone cream to be applied twice daily. During a follow-up visit one month later, the patient’s rash had improved considerably. She was still working on lifestyle changes to minimize her contact with water and other substances. If the patient’s condition hadn’t improved with basic treatment, she would have been referred to a dermatologist for patch testing to rule out contact dermatitis.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Hand eczema. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill;2013:597-602.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP recognized the multiple tapioca-like vesicles on the sides of the patient’s fingers as pompholyx, also known as dyshidrotic eczema. One of the classic findings of dyshidrotic eczema is the vesicles that resemble the small spheres in tapioca pudding. (The FP considered scabies, but ruled it out because no one in the family had a similar rash and there were no signs of any burrows.)
The term “pompholyx” means bubble, while the term “dyshidrotic” means “difficult sweating,” as problems with sweating were once believed to be the cause of this condition. Some dermatologists prefer the name “vesicular hand dermatitis.” Regardless of the terminology, this can be a very mild condition that is a minor nuisance or a very severe chronic condition that impairs the patient’s quality of life.
The FP explained that dyshidrotic eczema is not curable and may be worsened by stress and substances that come into contact with the hands. He discussed the possibility of avoiding washing the dishes and other “wet work,” but the patient said that wasn’t possible, so he told her to use nitrile gloves with cotton liners. (Using gloves without cotton liners often leads to sweating in the gloves that can worsen dyshidrotic eczema.)
The FP prescribed 0.1% triamcinolone cream to be applied twice daily. During a follow-up visit one month later, the patient’s rash had improved considerably. She was still working on lifestyle changes to minimize her contact with water and other substances. If the patient’s condition hadn’t improved with basic treatment, she would have been referred to a dermatologist for patch testing to rule out contact dermatitis.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Hand eczema. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill;2013:597-602.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP recognized the multiple tapioca-like vesicles on the sides of the patient’s fingers as pompholyx, also known as dyshidrotic eczema. One of the classic findings of dyshidrotic eczema is the vesicles that resemble the small spheres in tapioca pudding. (The FP considered scabies, but ruled it out because no one in the family had a similar rash and there were no signs of any burrows.)
The term “pompholyx” means bubble, while the term “dyshidrotic” means “difficult sweating,” as problems with sweating were once believed to be the cause of this condition. Some dermatologists prefer the name “vesicular hand dermatitis.” Regardless of the terminology, this can be a very mild condition that is a minor nuisance or a very severe chronic condition that impairs the patient’s quality of life.
The FP explained that dyshidrotic eczema is not curable and may be worsened by stress and substances that come into contact with the hands. He discussed the possibility of avoiding washing the dishes and other “wet work,” but the patient said that wasn’t possible, so he told her to use nitrile gloves with cotton liners. (Using gloves without cotton liners often leads to sweating in the gloves that can worsen dyshidrotic eczema.)
The FP prescribed 0.1% triamcinolone cream to be applied twice daily. During a follow-up visit one month later, the patient’s rash had improved considerably. She was still working on lifestyle changes to minimize her contact with water and other substances. If the patient’s condition hadn’t improved with basic treatment, she would have been referred to a dermatologist for patch testing to rule out contact dermatitis.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Hand eczema. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill;2013:597-602.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
“Cold Turkey” Works Best for Smoking Cessation

A 43-year-old man has a 35–pack-year smoking history and currently smokes one pack of cigarettes a day. He is eager to quit smoking since a close friend of his was recently diagnosed with lung cancer. He asks whether he should quit “cold turkey” or gradually. What do you recommend?
Between 2013 and 2014, one in five American adults reported using tobacco products some days or every day, and 66% of smokers in 2013 made at least one attempt to quit.2,3 The risks of tobacco use and the benefits of cessation are well established, and behavioral and pharmacologic interventions (both alone and in combination) increase smoking cessation rates.4 The US Preventive Services Task Force recommends that health care providers address tobacco use and cessation with patients at regular office visits and offer behavioral and pharmacologic interventions.5 Current guidelines, however, make no specific recommendations regarding gradual versus abrupt smoking cessation methods.5
A previous Cochrane review of 10 RCTs demonstrated no significant difference in quit rates between gradual cigarette reduction and abrupt cessation. The meta-analysis was limited, however, by differences in patient populations, outcome definitions, and types of interventions (both pharmacologic and behavioral).6
In a retrospective cohort study, French investigators reviewed an online database of more than 60,000 smokers who presented to nationwide cessation services. The researchers found that older participants (those 45 and older) and heavy smokers (≥ 21 cigarettes/d) were more likely to quit gradually than abruptly.7
STUDY SUMMARY
“Cold turkey” is better than gradual cessation at six months
A noninferiority RCT was conducted in England to assess whether gradual smoking cessation is as successful as abrupt cessation.1 The primary outcome was abstinence from smoking at four weeks, assessed using the Russell Standard. This set of six criteria (including validation by exhaled CO concentrations of < 10 ppm) is used by the National Centre for Smoking Cessation and Training to decrease variability of reported smoking cessation rates in English studies.8
Participants were recruited via letters from their primary care practice inviting them to participate in a smoking cessation study. The 697 subjects were randomized to either the abrupt-cessation group or the gradual-cessation group. Baseline characteristics were similar between groups.
All participants were asked to schedule a quit date for two weeks after their enrollment. Patients assigned to the gradual-cessation group were provided nicotine replacement patches (21 mg/d) and their choice of short-acting nicotine replacement therapy (NRT; gum, lozenges, nasal spray, sublingual tablets, inhalator, or mouth spray) to use in the two weeks leading up to the quit date. They were given instructions to reduce smoking by half of the baseline amount by the end of the first week, and to a quarter of baseline by the end of the second week.
Patients randomly assigned to the abrupt-cessation group were instructed to continue their current smoking habits until the cessation date; during those two weeks they were given nicotine patches (because the other group received them, and some evidence suggests that precessation NRT increases quit rates) but no short-acting NRT.
Following the cessation date, treatment in both groups was identical, including behavioral support, nicotine patches (21 mg/d), and the patient’s choice of short-acting NRT. Behavioral support consisted of visits with a research nurse at the patient’s primary care practice at the following intervals: weekly for two weeks before the quit date; the day before the quit date; weekly for four weeks after the quit date; and eight weeks after the quit date.
The chosen noninferiority margin was equal to a relative risk (RR) of 0.81 (19% reduction in effectiveness) of quitting gradually, compared with abrupt cessation of smoking. Quit rates in the gradual-reduction group did not reach the threshold for noninferiority; in fact, four-week abstinence was significantly more likely in the abrupt-cessation group than in the gradual-cessation group (49% vs 39.2%; RR, 0.80; number needed to treat [NNT], 10). Similarly, secondary outcomes of eight-week and six-month abstinence rates showed superiority of abrupt over gradual cessation. Six months after the quit date, 15.5% of the gradual-cessation group and 22% of the abrupt-cessation group remained abstinent (RR, 0.71; NNT, 15).
Patient preference plays a role
The investigators also found a difference in successful cessation based on the participants’ preferred method of cessation. Participants who preferred abrupt cessation were more likely to be abstinent at four weeks than participants who preferred gradual cessation (52.2% vs 38.3%).
Patients with a baseline preference for gradual cessation were equally as likely to successfully quit when allocated to abrupt cessation against their preference as when they were allocated to gradual cessation. Four-week abstinence was seen in 34.6% of patients who preferred and were allocated to gradual cessation and in 42% of patients who preferred gradual but were allocated to abrupt cessation.
WHAT’S NEW
Higher quality study; added element of preference
This large, well-designed, noninferiority study showed that abrupt cessation is superior to gradual cessation. The size and design of the study, including a standardized method of assessing cessation and a standardized intervention, make this a higher quality study than those in the Cochrane meta-analysis.6 This study also showed that participants who preferred gradual cessation were less likely to be successful—regardless of the method to which they were assigned.

CAVEATS
Generalizability limited by race and number of cigarettes smoked
Patients lost to follow-up at four weeks (35 in the abrupt-cessation group and 48 in the gradual-cessation group) were assumed to have continued smoking, which may have biased the results toward abrupt cessation. That said, the large number of study participants, along with the relatively small number lost to follow-up, minimizes this weakness.
The majority of participants were white, which may limit generalizability to nonwhite populations. In addition, participants smoked an average of 20 cigarettes per day and, as noted previously, an observational study of tobacco users in France found that heavy smokers (≥ 21 cigarettes/d) were more likely to quit gradually than abruptly. Therefore, results may not be generalizable to heavy smokers.7
CHALLENGES TO IMPLEMENTATION
Considerable investment in behavioral support
One significant challenge is the implementation of such a structured tobacco cessation program in primary care. Both abrupt- and gradual-cessation groups were given considerable behavioral support from research nurses. Participants in this study were seen by a nurse seven times in the first six weeks of the study, and the intervention included nurse-created reduction schedules.
Even if patients in the study preferred one method of cessation to another, they were receptive to quitting either gradually or abruptly. In clinical practice, patients are often set in their desired method of cessation. In that setting, our role is then to inform them of the data and support them in whatever method they choose.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Copyright © 2017. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2017;66[3]:174-176).
1. Lindson-Hawley N, Banting M, West R, et al. Gradual versus abrupt smoking cessation: a randomized, controlled noninferiority trial. Ann Intern Med. 2016;164:585-592.
2. Hu SS, Neff L, Agaku IT, et al. Tobacco product use among adults—United States, 2013-2014. MMWR Morb Mortal Wkly Rep. 2016;65:685-691.
3. Lavinghouze SR, Malarcher A, Jama A, et al. Trends in quit attempts among adult cigarette smokers–United States, 2001-2013. MMWR Morb Mortal Wkly Rep. 2015;64:1129-1135.
4. Patnode CD, Henderson JT, Thompson JH, et al. Behavioral counseling and pharmacotherapy interventions for tobacco cessation in adults, including pregnant women: a review of reviews for the US Preventive Services Task Force. Ann Intern Med. 2015;163:608-621.
5. Siu AL; US Preventive Services Task Force. Behavioral and pharmacotherapy interventions for tobacco smoking cessation in adults, including pregnant women: US Preventive Services Task Force Recommendation Statement. Ann Intern Med. 2015;163:622-634.
6. Lindson-Hawley N, Aveyard P, Hughes JR. Reduction versus abrupt cessation in smokers who want to quit. Cochrane Database Syst Rev. 2012;11:CD008033.
7. Baha M, Le Faou AL. Gradual versus abrupt quitting among French treatment-seeking smokers. Prev Med. 2014;63: 96-102.
8. West R, Hajek P, Stead L, et al. Outcome criteria in smoking cessation trials: proposal for a common standard. Addiction. 2005;100:299-303.
A 43-year-old man has a 35–pack-year smoking history and currently smokes one pack of cigarettes a day. He is eager to quit smoking since a close friend of his was recently diagnosed with lung cancer. He asks whether he should quit “cold turkey” or gradually. What do you recommend?
Between 2013 and 2014, one in five American adults reported using tobacco products some days or every day, and 66% of smokers in 2013 made at least one attempt to quit.2,3 The risks of tobacco use and the benefits of cessation are well established, and behavioral and pharmacologic interventions (both alone and in combination) increase smoking cessation rates.4 The US Preventive Services Task Force recommends that health care providers address tobacco use and cessation with patients at regular office visits and offer behavioral and pharmacologic interventions.5 Current guidelines, however, make no specific recommendations regarding gradual versus abrupt smoking cessation methods.5
A previous Cochrane review of 10 RCTs demonstrated no significant difference in quit rates between gradual cigarette reduction and abrupt cessation. The meta-analysis was limited, however, by differences in patient populations, outcome definitions, and types of interventions (both pharmacologic and behavioral).6
In a retrospective cohort study, French investigators reviewed an online database of more than 60,000 smokers who presented to nationwide cessation services. The researchers found that older participants (those 45 and older) and heavy smokers (≥ 21 cigarettes/d) were more likely to quit gradually than abruptly.7
STUDY SUMMARY
“Cold turkey” is better than gradual cessation at six months
A noninferiority RCT was conducted in England to assess whether gradual smoking cessation is as successful as abrupt cessation.1 The primary outcome was abstinence from smoking at four weeks, assessed using the Russell Standard. This set of six criteria (including validation by exhaled CO concentrations of < 10 ppm) is used by the National Centre for Smoking Cessation and Training to decrease variability of reported smoking cessation rates in English studies.8
Participants were recruited via letters from their primary care practice inviting them to participate in a smoking cessation study. The 697 subjects were randomized to either the abrupt-cessation group or the gradual-cessation group. Baseline characteristics were similar between groups.
All participants were asked to schedule a quit date for two weeks after their enrollment. Patients assigned to the gradual-cessation group were provided nicotine replacement patches (21 mg/d) and their choice of short-acting nicotine replacement therapy (NRT; gum, lozenges, nasal spray, sublingual tablets, inhalator, or mouth spray) to use in the two weeks leading up to the quit date. They were given instructions to reduce smoking by half of the baseline amount by the end of the first week, and to a quarter of baseline by the end of the second week.
Patients randomly assigned to the abrupt-cessation group were instructed to continue their current smoking habits until the cessation date; during those two weeks they were given nicotine patches (because the other group received them, and some evidence suggests that precessation NRT increases quit rates) but no short-acting NRT.
Following the cessation date, treatment in both groups was identical, including behavioral support, nicotine patches (21 mg/d), and the patient’s choice of short-acting NRT. Behavioral support consisted of visits with a research nurse at the patient’s primary care practice at the following intervals: weekly for two weeks before the quit date; the day before the quit date; weekly for four weeks after the quit date; and eight weeks after the quit date.
The chosen noninferiority margin was equal to a relative risk (RR) of 0.81 (19% reduction in effectiveness) of quitting gradually, compared with abrupt cessation of smoking. Quit rates in the gradual-reduction group did not reach the threshold for noninferiority; in fact, four-week abstinence was significantly more likely in the abrupt-cessation group than in the gradual-cessation group (49% vs 39.2%; RR, 0.80; number needed to treat [NNT], 10). Similarly, secondary outcomes of eight-week and six-month abstinence rates showed superiority of abrupt over gradual cessation. Six months after the quit date, 15.5% of the gradual-cessation group and 22% of the abrupt-cessation group remained abstinent (RR, 0.71; NNT, 15).
Patient preference plays a role
The investigators also found a difference in successful cessation based on the participants’ preferred method of cessation. Participants who preferred abrupt cessation were more likely to be abstinent at four weeks than participants who preferred gradual cessation (52.2% vs 38.3%).
Patients with a baseline preference for gradual cessation were equally as likely to successfully quit when allocated to abrupt cessation against their preference as when they were allocated to gradual cessation. Four-week abstinence was seen in 34.6% of patients who preferred and were allocated to gradual cessation and in 42% of patients who preferred gradual but were allocated to abrupt cessation.
WHAT’S NEW
Higher quality study; added element of preference
This large, well-designed, noninferiority study showed that abrupt cessation is superior to gradual cessation. The size and design of the study, including a standardized method of assessing cessation and a standardized intervention, make this a higher quality study than those in the Cochrane meta-analysis.6 This study also showed that participants who preferred gradual cessation were less likely to be successful—regardless of the method to which they were assigned.
CAVEATS
Generalizability limited by race and number of cigarettes smoked
Patients lost to follow-up at four weeks (35 in the abrupt-cessation group and 48 in the gradual-cessation group) were assumed to have continued smoking, which may have biased the results toward abrupt cessation. That said, the large number of study participants, along with the relatively small number lost to follow-up, minimizes this weakness.
The majority of participants were white, which may limit generalizability to nonwhite populations. In addition, participants smoked an average of 20 cigarettes per day and, as noted previously, an observational study of tobacco users in France found that heavy smokers (≥ 21 cigarettes/d) were more likely to quit gradually than abruptly. Therefore, results may not be generalizable to heavy smokers.7
CHALLENGES TO IMPLEMENTATION
Considerable investment in behavioral support
One significant challenge is the implementation of such a structured tobacco cessation program in primary care. Both abrupt- and gradual-cessation groups were given considerable behavioral support from research nurses. Participants in this study were seen by a nurse seven times in the first six weeks of the study, and the intervention included nurse-created reduction schedules.
Even if patients in the study preferred one method of cessation to another, they were receptive to quitting either gradually or abruptly. In clinical practice, patients are often set in their desired method of cessation. In that setting, our role is then to inform them of the data and support them in whatever method they choose.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Copyright © 2017. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2017;66[3]:174-176).
A 43-year-old man has a 35–pack-year smoking history and currently smokes one pack of cigarettes a day. He is eager to quit smoking since a close friend of his was recently diagnosed with lung cancer. He asks whether he should quit “cold turkey” or gradually. What do you recommend?
Between 2013 and 2014, one in five American adults reported using tobacco products some days or every day, and 66% of smokers in 2013 made at least one attempt to quit.2,3 The risks of tobacco use and the benefits of cessation are well established, and behavioral and pharmacologic interventions (both alone and in combination) increase smoking cessation rates.4 The US Preventive Services Task Force recommends that health care providers address tobacco use and cessation with patients at regular office visits and offer behavioral and pharmacologic interventions.5 Current guidelines, however, make no specific recommendations regarding gradual versus abrupt smoking cessation methods.5
A previous Cochrane review of 10 RCTs demonstrated no significant difference in quit rates between gradual cigarette reduction and abrupt cessation. The meta-analysis was limited, however, by differences in patient populations, outcome definitions, and types of interventions (both pharmacologic and behavioral).6
In a retrospective cohort study, French investigators reviewed an online database of more than 60,000 smokers who presented to nationwide cessation services. The researchers found that older participants (those 45 and older) and heavy smokers (≥ 21 cigarettes/d) were more likely to quit gradually than abruptly.7
STUDY SUMMARY
“Cold turkey” is better than gradual cessation at six months
A noninferiority RCT was conducted in England to assess whether gradual smoking cessation is as successful as abrupt cessation.1 The primary outcome was abstinence from smoking at four weeks, assessed using the Russell Standard. This set of six criteria (including validation by exhaled CO concentrations of < 10 ppm) is used by the National Centre for Smoking Cessation and Training to decrease variability of reported smoking cessation rates in English studies.8
Participants were recruited via letters from their primary care practice inviting them to participate in a smoking cessation study. The 697 subjects were randomized to either the abrupt-cessation group or the gradual-cessation group. Baseline characteristics were similar between groups.
All participants were asked to schedule a quit date for two weeks after their enrollment. Patients assigned to the gradual-cessation group were provided nicotine replacement patches (21 mg/d) and their choice of short-acting nicotine replacement therapy (NRT; gum, lozenges, nasal spray, sublingual tablets, inhalator, or mouth spray) to use in the two weeks leading up to the quit date. They were given instructions to reduce smoking by half of the baseline amount by the end of the first week, and to a quarter of baseline by the end of the second week.
Patients randomly assigned to the abrupt-cessation group were instructed to continue their current smoking habits until the cessation date; during those two weeks they were given nicotine patches (because the other group received them, and some evidence suggests that precessation NRT increases quit rates) but no short-acting NRT.
Following the cessation date, treatment in both groups was identical, including behavioral support, nicotine patches (21 mg/d), and the patient’s choice of short-acting NRT. Behavioral support consisted of visits with a research nurse at the patient’s primary care practice at the following intervals: weekly for two weeks before the quit date; the day before the quit date; weekly for four weeks after the quit date; and eight weeks after the quit date.
The chosen noninferiority margin was equal to a relative risk (RR) of 0.81 (19% reduction in effectiveness) of quitting gradually, compared with abrupt cessation of smoking. Quit rates in the gradual-reduction group did not reach the threshold for noninferiority; in fact, four-week abstinence was significantly more likely in the abrupt-cessation group than in the gradual-cessation group (49% vs 39.2%; RR, 0.80; number needed to treat [NNT], 10). Similarly, secondary outcomes of eight-week and six-month abstinence rates showed superiority of abrupt over gradual cessation. Six months after the quit date, 15.5% of the gradual-cessation group and 22% of the abrupt-cessation group remained abstinent (RR, 0.71; NNT, 15).
Patient preference plays a role
The investigators also found a difference in successful cessation based on the participants’ preferred method of cessation. Participants who preferred abrupt cessation were more likely to be abstinent at four weeks than participants who preferred gradual cessation (52.2% vs 38.3%).
Patients with a baseline preference for gradual cessation were equally as likely to successfully quit when allocated to abrupt cessation against their preference as when they were allocated to gradual cessation. Four-week abstinence was seen in 34.6% of patients who preferred and were allocated to gradual cessation and in 42% of patients who preferred gradual but were allocated to abrupt cessation.
WHAT’S NEW
Higher quality study; added element of preference
This large, well-designed, noninferiority study showed that abrupt cessation is superior to gradual cessation. The size and design of the study, including a standardized method of assessing cessation and a standardized intervention, make this a higher quality study than those in the Cochrane meta-analysis.6 This study also showed that participants who preferred gradual cessation were less likely to be successful—regardless of the method to which they were assigned.
CAVEATS
Generalizability limited by race and number of cigarettes smoked
Patients lost to follow-up at four weeks (35 in the abrupt-cessation group and 48 in the gradual-cessation group) were assumed to have continued smoking, which may have biased the results toward abrupt cessation. That said, the large number of study participants, along with the relatively small number lost to follow-up, minimizes this weakness.
The majority of participants were white, which may limit generalizability to nonwhite populations. In addition, participants smoked an average of 20 cigarettes per day and, as noted previously, an observational study of tobacco users in France found that heavy smokers (≥ 21 cigarettes/d) were more likely to quit gradually than abruptly. Therefore, results may not be generalizable to heavy smokers.7
CHALLENGES TO IMPLEMENTATION
Considerable investment in behavioral support
One significant challenge is the implementation of such a structured tobacco cessation program in primary care. Both abrupt- and gradual-cessation groups were given considerable behavioral support from research nurses. Participants in this study were seen by a nurse seven times in the first six weeks of the study, and the intervention included nurse-created reduction schedules.
Even if patients in the study preferred one method of cessation to another, they were receptive to quitting either gradually or abruptly. In clinical practice, patients are often set in their desired method of cessation. In that setting, our role is then to inform them of the data and support them in whatever method they choose.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Copyright © 2017. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2017;66[3]:174-176).
1. Lindson-Hawley N, Banting M, West R, et al. Gradual versus abrupt smoking cessation: a randomized, controlled noninferiority trial. Ann Intern Med. 2016;164:585-592.
2. Hu SS, Neff L, Agaku IT, et al. Tobacco product use among adults—United States, 2013-2014. MMWR Morb Mortal Wkly Rep. 2016;65:685-691.
3. Lavinghouze SR, Malarcher A, Jama A, et al. Trends in quit attempts among adult cigarette smokers–United States, 2001-2013. MMWR Morb Mortal Wkly Rep. 2015;64:1129-1135.
4. Patnode CD, Henderson JT, Thompson JH, et al. Behavioral counseling and pharmacotherapy interventions for tobacco cessation in adults, including pregnant women: a review of reviews for the US Preventive Services Task Force. Ann Intern Med. 2015;163:608-621.
5. Siu AL; US Preventive Services Task Force. Behavioral and pharmacotherapy interventions for tobacco smoking cessation in adults, including pregnant women: US Preventive Services Task Force Recommendation Statement. Ann Intern Med. 2015;163:622-634.
6. Lindson-Hawley N, Aveyard P, Hughes JR. Reduction versus abrupt cessation in smokers who want to quit. Cochrane Database Syst Rev. 2012;11:CD008033.
7. Baha M, Le Faou AL. Gradual versus abrupt quitting among French treatment-seeking smokers. Prev Med. 2014;63: 96-102.
8. West R, Hajek P, Stead L, et al. Outcome criteria in smoking cessation trials: proposal for a common standard. Addiction. 2005;100:299-303.
1. Lindson-Hawley N, Banting M, West R, et al. Gradual versus abrupt smoking cessation: a randomized, controlled noninferiority trial. Ann Intern Med. 2016;164:585-592.
2. Hu SS, Neff L, Agaku IT, et al. Tobacco product use among adults—United States, 2013-2014. MMWR Morb Mortal Wkly Rep. 2016;65:685-691.
3. Lavinghouze SR, Malarcher A, Jama A, et al. Trends in quit attempts among adult cigarette smokers–United States, 2001-2013. MMWR Morb Mortal Wkly Rep. 2015;64:1129-1135.
4. Patnode CD, Henderson JT, Thompson JH, et al. Behavioral counseling and pharmacotherapy interventions for tobacco cessation in adults, including pregnant women: a review of reviews for the US Preventive Services Task Force. Ann Intern Med. 2015;163:608-621.
5. Siu AL; US Preventive Services Task Force. Behavioral and pharmacotherapy interventions for tobacco smoking cessation in adults, including pregnant women: US Preventive Services Task Force Recommendation Statement. Ann Intern Med. 2015;163:622-634.
6. Lindson-Hawley N, Aveyard P, Hughes JR. Reduction versus abrupt cessation in smokers who want to quit. Cochrane Database Syst Rev. 2012;11:CD008033.
7. Baha M, Le Faou AL. Gradual versus abrupt quitting among French treatment-seeking smokers. Prev Med. 2014;63: 96-102.
8. West R, Hajek P, Stead L, et al. Outcome criteria in smoking cessation trials: proposal for a common standard. Addiction. 2005;100:299-303.
Dual targeting effective in HER2+ metastatic colorectal cancer
The combination of trastuzumab (Herceptin) and lapatinib (Tykerb) resulted in an overall objective response rate of 30% in heavily pretreated patients with HER2-positive metastatic colorectal cancer, according to findings presented at the annual meeting of the American Association for Cancer Research.
Of 33 patients, 2 patients achieved complete responses and 8 had partial responses, while 13 patients achieved stable disease. This brought the total clinical benefit rate to 70% at the time of data cutoff on Feb. 28.
“There is a large unmet need in colorectal cancer,” said study author, Silvia Marsoni, MD, director of the precision medicine unit at the Istituto di Candiolo, Fondazione del Piemonte per l’Oncologia–IRCCS in Turin, Italy.
“The combination of drugs really had a great impact on the tumor,” said Dr. Marsoni, who discussed the findings at a presscast. “One of these complete responders is still alive without evidence of disease at almost 36 months from the beginning of his treatment. This was a very good duration of response.”
HER2 amplification is found in 5% of RAS wild-type metastatic colorectal cancer cases. Dual HER2 blockade with both trastuzumab and lapatinib had inhibited tumor growth in preliminary studies, but that has not been the case for either drug used alone.
The HERACLES trial was conducted at four centers in Italy and enrolled 33 patients with RAS wild-type, HER2-positive tumors that were refractory to standard of care treatments, including the epidermal growth factor receptor (EGFR) inhibitors cetuximab and panitumumab.
“These patients were heavily pretreated and 75% had four or more prior treatments,” explained Dr. Marsoni.
All participants received trastuzumab intravenously with a 4 mg/kg loading dose followed by 2 mg/kg weekly, and lapatinib was given orally at 1,000 mg daily. Treatment continued until disease progression.
The primary endpoint was the objective response rate, and secondary endpoints included progression-free survival and safety.
The two patients who achieved a complete response continue to be disease free, and both had tumors that were refractory to cetuximab and resistant to all standard treatment.
Historically, the response rate in metastatic colorectal cancer after second-line treatment is very low, at less than 5% with chemotherapy and about 10%-20% with anti-EGFR therapy.
“There was really no toxicity,” explained Dr. Marsoni. “We used a lapatinib dose that was about 20% less than what is used in breast cancer to avoid GI toxicity.”
Only six patients (18%) experienced grade 3 side effects, which included fatigue, skin rash, and elevated bilirubin.
“In conclusion, we can say that, even if HER2-positive patients in colon cancer are only 3% of the population, they are still a little and important piece of the cake that can be actively targeted with a new chemotherapy-free regimen,” said Dr. Marsoni. “We have a new potential treatment for this population.”
The combination of trastuzumab (Herceptin) and lapatinib (Tykerb) resulted in an overall objective response rate of 30% in heavily pretreated patients with HER2-positive metastatic colorectal cancer, according to findings presented at the annual meeting of the American Association for Cancer Research.
Of 33 patients, 2 patients achieved complete responses and 8 had partial responses, while 13 patients achieved stable disease. This brought the total clinical benefit rate to 70% at the time of data cutoff on Feb. 28.
“There is a large unmet need in colorectal cancer,” said study author, Silvia Marsoni, MD, director of the precision medicine unit at the Istituto di Candiolo, Fondazione del Piemonte per l’Oncologia–IRCCS in Turin, Italy.
“The combination of drugs really had a great impact on the tumor,” said Dr. Marsoni, who discussed the findings at a presscast. “One of these complete responders is still alive without evidence of disease at almost 36 months from the beginning of his treatment. This was a very good duration of response.”
HER2 amplification is found in 5% of RAS wild-type metastatic colorectal cancer cases. Dual HER2 blockade with both trastuzumab and lapatinib had inhibited tumor growth in preliminary studies, but that has not been the case for either drug used alone.
The HERACLES trial was conducted at four centers in Italy and enrolled 33 patients with RAS wild-type, HER2-positive tumors that were refractory to standard of care treatments, including the epidermal growth factor receptor (EGFR) inhibitors cetuximab and panitumumab.
“These patients were heavily pretreated and 75% had four or more prior treatments,” explained Dr. Marsoni.
All participants received trastuzumab intravenously with a 4 mg/kg loading dose followed by 2 mg/kg weekly, and lapatinib was given orally at 1,000 mg daily. Treatment continued until disease progression.
The primary endpoint was the objective response rate, and secondary endpoints included progression-free survival and safety.
The two patients who achieved a complete response continue to be disease free, and both had tumors that were refractory to cetuximab and resistant to all standard treatment.
Historically, the response rate in metastatic colorectal cancer after second-line treatment is very low, at less than 5% with chemotherapy and about 10%-20% with anti-EGFR therapy.
“There was really no toxicity,” explained Dr. Marsoni. “We used a lapatinib dose that was about 20% less than what is used in breast cancer to avoid GI toxicity.”
Only six patients (18%) experienced grade 3 side effects, which included fatigue, skin rash, and elevated bilirubin.
“In conclusion, we can say that, even if HER2-positive patients in colon cancer are only 3% of the population, they are still a little and important piece of the cake that can be actively targeted with a new chemotherapy-free regimen,” said Dr. Marsoni. “We have a new potential treatment for this population.”
The combination of trastuzumab (Herceptin) and lapatinib (Tykerb) resulted in an overall objective response rate of 30% in heavily pretreated patients with HER2-positive metastatic colorectal cancer, according to findings presented at the annual meeting of the American Association for Cancer Research.
Of 33 patients, 2 patients achieved complete responses and 8 had partial responses, while 13 patients achieved stable disease. This brought the total clinical benefit rate to 70% at the time of data cutoff on Feb. 28.
“There is a large unmet need in colorectal cancer,” said study author, Silvia Marsoni, MD, director of the precision medicine unit at the Istituto di Candiolo, Fondazione del Piemonte per l’Oncologia–IRCCS in Turin, Italy.
“The combination of drugs really had a great impact on the tumor,” said Dr. Marsoni, who discussed the findings at a presscast. “One of these complete responders is still alive without evidence of disease at almost 36 months from the beginning of his treatment. This was a very good duration of response.”
HER2 amplification is found in 5% of RAS wild-type metastatic colorectal cancer cases. Dual HER2 blockade with both trastuzumab and lapatinib had inhibited tumor growth in preliminary studies, but that has not been the case for either drug used alone.
The HERACLES trial was conducted at four centers in Italy and enrolled 33 patients with RAS wild-type, HER2-positive tumors that were refractory to standard of care treatments, including the epidermal growth factor receptor (EGFR) inhibitors cetuximab and panitumumab.
“These patients were heavily pretreated and 75% had four or more prior treatments,” explained Dr. Marsoni.
All participants received trastuzumab intravenously with a 4 mg/kg loading dose followed by 2 mg/kg weekly, and lapatinib was given orally at 1,000 mg daily. Treatment continued until disease progression.
The primary endpoint was the objective response rate, and secondary endpoints included progression-free survival and safety.
The two patients who achieved a complete response continue to be disease free, and both had tumors that were refractory to cetuximab and resistant to all standard treatment.
Historically, the response rate in metastatic colorectal cancer after second-line treatment is very low, at less than 5% with chemotherapy and about 10%-20% with anti-EGFR therapy.
“There was really no toxicity,” explained Dr. Marsoni. “We used a lapatinib dose that was about 20% less than what is used in breast cancer to avoid GI toxicity.”
Only six patients (18%) experienced grade 3 side effects, which included fatigue, skin rash, and elevated bilirubin.
“In conclusion, we can say that, even if HER2-positive patients in colon cancer are only 3% of the population, they are still a little and important piece of the cake that can be actively targeted with a new chemotherapy-free regimen,” said Dr. Marsoni. “We have a new potential treatment for this population.”
FROM THE AACR ANNUAL MEETING
Key clinical point: Dual targeting resulted in a 30% objective response rate in heavily pretreated patients with HER2-positive colorectal cancer.
Major finding: Combination trastuzumab and lapatinib resulted in a 70% clinical benefit in heavily pretreated colorectal cancer patients.
Data source: Multicenter prospective study that included 33 patients with HER2-positive metastatic colorectal cancer who had been heavily pretreated.
Disclosures: The study was funded by the Associazione Italiana per la Ricerca sul Cancro, Fondazione Oncologia Niguarda Onlus, and Roche. The study’s lead investigator, Salvatore Siena, MD, has relationships with Amgen, Bayer, Celgene, Eli Lilly, Merck, Merrimack, Novartis, Roche, and Sanofi.
Weight fluctuations linked to higher mortality, CV events
Weight fluctuations – like the typical pattern of weight loss followed by partial or total regain that affects most people attempting to lose weight – are strongly associated with higher mortality, more cardiovascular events, and new-onset diabetes, according to an analysis of the Treating to New Targets trial published online April 6 in the New England Journal of Medicine.
Weight loss is commonly prescribed as a lifestyle intervention to improve health, especially cardiovascular health. However, “the usual pattern for most patients ... is weight loss followed by weight gain” (also termed weight cycling), which has been linked to poor cardiovascular outcomes, especially when the pattern is repeated over time, said Sripal Bangalore, MD, of New York University, and his coinvestigators.

The primary outcome measure – the composite rate of death from coronary heart disease, nonfatal MI, resuscitated cardiac arrest, revascularization, or angina – was significantly associated with weight fluctuations in a dose-dependent fashion so that greater degrees of variability in body weight were linked to higher event rates (N Engl J Med. 2017 April 6. doi: 10.1056/NEJMoa1606148).
“When compared with the lowest quintile, patients with the highest quintile of variability had an increase in the risk of any coronary event of 64%, an increase in the risk of any cardiovascular event of 85%, an increase in the risk of death of 124%, an increase in the risk of MI of 117%, an increase in the risk of stroke of 136%, and an increase in the risk of new-onset diabetes of 78%, independent of traditional risk factors,” Dr. Bangalore and his associates reported.
This association remained strong regardless of the patients’ weight at baseline, consistent among those of normal body weight and those who were overweight or obese. And the association also persisted across multiple sensitivity analyses.
This study was supported by Pfizer. Dr. Bangalore reported serving as a consultant to Pfizer, Daiichi-Sankyo, Boehringer Ingelheim, Merck, Menarini, Gilead, and Abbott Vascular; his associates reported ties to numerous industry sources.
Weight fluctuations – like the typical pattern of weight loss followed by partial or total regain that affects most people attempting to lose weight – are strongly associated with higher mortality, more cardiovascular events, and new-onset diabetes, according to an analysis of the Treating to New Targets trial published online April 6 in the New England Journal of Medicine.
Weight loss is commonly prescribed as a lifestyle intervention to improve health, especially cardiovascular health. However, “the usual pattern for most patients ... is weight loss followed by weight gain” (also termed weight cycling), which has been linked to poor cardiovascular outcomes, especially when the pattern is repeated over time, said Sripal Bangalore, MD, of New York University, and his coinvestigators.
The primary outcome measure – the composite rate of death from coronary heart disease, nonfatal MI, resuscitated cardiac arrest, revascularization, or angina – was significantly associated with weight fluctuations in a dose-dependent fashion so that greater degrees of variability in body weight were linked to higher event rates (N Engl J Med. 2017 April 6. doi: 10.1056/NEJMoa1606148).
“When compared with the lowest quintile, patients with the highest quintile of variability had an increase in the risk of any coronary event of 64%, an increase in the risk of any cardiovascular event of 85%, an increase in the risk of death of 124%, an increase in the risk of MI of 117%, an increase in the risk of stroke of 136%, and an increase in the risk of new-onset diabetes of 78%, independent of traditional risk factors,” Dr. Bangalore and his associates reported.
This association remained strong regardless of the patients’ weight at baseline, consistent among those of normal body weight and those who were overweight or obese. And the association also persisted across multiple sensitivity analyses.
This study was supported by Pfizer. Dr. Bangalore reported serving as a consultant to Pfizer, Daiichi-Sankyo, Boehringer Ingelheim, Merck, Menarini, Gilead, and Abbott Vascular; his associates reported ties to numerous industry sources.
Weight fluctuations – like the typical pattern of weight loss followed by partial or total regain that affects most people attempting to lose weight – are strongly associated with higher mortality, more cardiovascular events, and new-onset diabetes, according to an analysis of the Treating to New Targets trial published online April 6 in the New England Journal of Medicine.
Weight loss is commonly prescribed as a lifestyle intervention to improve health, especially cardiovascular health. However, “the usual pattern for most patients ... is weight loss followed by weight gain” (also termed weight cycling), which has been linked to poor cardiovascular outcomes, especially when the pattern is repeated over time, said Sripal Bangalore, MD, of New York University, and his coinvestigators.
The primary outcome measure – the composite rate of death from coronary heart disease, nonfatal MI, resuscitated cardiac arrest, revascularization, or angina – was significantly associated with weight fluctuations in a dose-dependent fashion so that greater degrees of variability in body weight were linked to higher event rates (N Engl J Med. 2017 April 6. doi: 10.1056/NEJMoa1606148).
“When compared with the lowest quintile, patients with the highest quintile of variability had an increase in the risk of any coronary event of 64%, an increase in the risk of any cardiovascular event of 85%, an increase in the risk of death of 124%, an increase in the risk of MI of 117%, an increase in the risk of stroke of 136%, and an increase in the risk of new-onset diabetes of 78%, independent of traditional risk factors,” Dr. Bangalore and his associates reported.
This association remained strong regardless of the patients’ weight at baseline, consistent among those of normal body weight and those who were overweight or obese. And the association also persisted across multiple sensitivity analyses.
This study was supported by Pfizer. Dr. Bangalore reported serving as a consultant to Pfizer, Daiichi-Sankyo, Boehringer Ingelheim, Merck, Menarini, Gilead, and Abbott Vascular; his associates reported ties to numerous industry sources.
Key clinical point: Weight fluctuations are strongly associated with higher mortality, more cardiovascular events, and new-onset diabetes.
Major finding: Patients with the highest quintile of weight variability had increased risks of coronary events (64%), cardiovascular events (85%), death (124%), MI (117%), stroke (136%), and new-onset diabetes (78%).
Data source: A secondary analysis of data for 9,509 adults with coronary artery disease participating in the Treating to New Targets trial of cholesterol therapy.
Disclosures: This study was supported by Pfizer. Dr. Bangalore reported serving as a consultant to Pfizer, Daiichi-Sankyo, Boehringer Ingelheim, Merck, Menarini, Gilead, and Abbott Vascular; his associates reported ties to numerous industry sources.
Transplantation plus VRD tops VRD alone in myeloma
Adding high-dose chemotherapy and autologous stem cell transplantation to two cycles of bortezomib, lenalidomide, and dexamethasone (VRD) significantly improved progression-free survival in adults with newly diagnosed multiple myeloma, according to new analyses from the multicenter, randomized, phase III IFM 2009 Study.
Median PFS was 50 months in the transplantation group, versus 36 months when patients only received five cycles of VRD for consolidation (adjusted hazard ratio for disease progression or death, 0.65; 95% confidence interval, 0.53 to 0.80; P less than .001), reported Michel Attal, MD, of Institut Universitaire du Cancer de Toulouse-Oncopole in Toulouse, France, and his associates. “Transplantation was also associated with a higher rate of complete response, a lower rate of minimal residual disease detection, and a longer median time to progression,” they wrote (N Engl J Med. 2017 April 6. doi: 10.1056/NEJMoa1611750).
Transplantation did not improve 4-year overall survival (81% with transplantation plus VRD and 82% with VRD only), perhaps because VRD-only patients had the option of salvage transplantation if their disease progressed, the investigators said. In addition, grade 3 or 4 neutropenias were significantly more common with transplantation than otherwise (92% versus 47%), as were grade 3 or 4 gastrointestinal disorders (28% versus 7%) and infections (20% versus 9%). However, the groups did not significantly differ in terms of treatment-related deaths, second primary cancers, thromboembolic events, or peripheral neuropathy.
Before the advent of immunomodulatory drugs and proteasome inhibitors, several randomized trials showed that the benefits of high-dose chemotherapy with autologous stem cell transplantation outperformed conventional chemotherapy in multiple myeloma. For IFM 2009, 700 newly diagnosed patients aged up to 65 years received three 21-day cycles of VRD induction: bortezomib 1.3 mg/m2 on days 1, 4, 8, and 11; lenalidomide 25 mg on days 1-14; and dexamethasone 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12. Consolidation consisted of either five more VRD cycles with dexamethasone reduced to 10 mg/day, or high-dose (200 mg/m2) melphalan plus transplantation followed by two VRD cycles with the reduced dexamethasone dose. Both arms received 1 year of lenalidomide maintenance (10 mg/day for 3 months, followed by 15/mg day, depending on side effects).
Transplantation was associated with a significantly higher likelihood of complete response (59% versus 48%; P = .03) and undetectable minimal residual disease (79% versus 65%; P less than .001). Undetectable minimal residual disease, in turn, predicted longer PFS (adjusted hazard ratio, 0.30; P = .001). In contrast, age, sex, isotype of the monoclonal component, International Staging System disease stage, and cytogenetic risk profile did not significantly affect PFS among transplant patients.
The PFS benefit of transplantation “must be weighed against the increased risk of toxic effects associated with high-dose chemotherapy plus transplantation, especially since we found that later transplantation might be as effective as early transplantation in securing long- term survival,” the researchers said. “Our results suggest that the use of a combination therapy that incorporates newer proteasome inhibitors, next-generation immunomodulatory drugs, and potent monoclonal antibodies along with transplantation tailored according to minimal residual disease detection could further improve outcomes among adults up to 65 years of age who have multiple myeloma.”
Celgene, Janssen, the French Ministry of Health Programme Hospitalier de Recherche Clinique, and the French National Research Agency funded the study. Dr. Attal had no conflicts. Nine coinvestigators disclosed ties to Celgene, Janssen, and several other pharmaceutical companies. Two coinvestigators disclosed ownership or founder roles in OncoPep, one of whom disclosed patent licensure to OncoPep for a multipeptide vaccine. The other investigators had no conflicts.
In the IFM 2009 trial, the planned treatments were realistic. The combination of immunomodulatory drugs and proteasome inhibitors is associated with high response rates similar to those achieved with transplantation, whereas previous trials had used less effective chemotherapy regimens as comparators.
[Transplantation] resulted in a deeper and longer initial treatment response than did nontransplantation. However, the benefits of transplantation were more modest than some might have hoped, and it did not appear to be curative.

A question remains as to whether all patients with multiple myeloma should undergo immediate autologous stem-cell transplantation, since the use of delayed transplantation resulted in similar overall survival, with some patients not needing transplantation to date. The observed lack of a survival benefit for early transplantation was consistent with previous results that suggested salvage transplantation is an equalizer in this regard.
Charles A. Schiffer, MD, and Jeffrey A. Zonder, MD, are with Karmanos Cancer Institute, Wayne State University, Detroit, Mich. Dr. Zonder is a member of the editorial advisory board of Hematology News. Their comments were taken from an editorial (N Engl J Med. 2017 April 6. doi: 10.1056/NEJMe1700453).
In the IFM 2009 trial, the planned treatments were realistic. The combination of immunomodulatory drugs and proteasome inhibitors is associated with high response rates similar to those achieved with transplantation, whereas previous trials had used less effective chemotherapy regimens as comparators.
[Transplantation] resulted in a deeper and longer initial treatment response than did nontransplantation. However, the benefits of transplantation were more modest than some might have hoped, and it did not appear to be curative.
A question remains as to whether all patients with multiple myeloma should undergo immediate autologous stem-cell transplantation, since the use of delayed transplantation resulted in similar overall survival, with some patients not needing transplantation to date. The observed lack of a survival benefit for early transplantation was consistent with previous results that suggested salvage transplantation is an equalizer in this regard.
Charles A. Schiffer, MD, and Jeffrey A. Zonder, MD, are with Karmanos Cancer Institute, Wayne State University, Detroit, Mich. Dr. Zonder is a member of the editorial advisory board of Hematology News. Their comments were taken from an editorial (N Engl J Med. 2017 April 6. doi: 10.1056/NEJMe1700453).
In the IFM 2009 trial, the planned treatments were realistic. The combination of immunomodulatory drugs and proteasome inhibitors is associated with high response rates similar to those achieved with transplantation, whereas previous trials had used less effective chemotherapy regimens as comparators.
[Transplantation] resulted in a deeper and longer initial treatment response than did nontransplantation. However, the benefits of transplantation were more modest than some might have hoped, and it did not appear to be curative.
A question remains as to whether all patients with multiple myeloma should undergo immediate autologous stem-cell transplantation, since the use of delayed transplantation resulted in similar overall survival, with some patients not needing transplantation to date. The observed lack of a survival benefit for early transplantation was consistent with previous results that suggested salvage transplantation is an equalizer in this regard.
Charles A. Schiffer, MD, and Jeffrey A. Zonder, MD, are with Karmanos Cancer Institute, Wayne State University, Detroit, Mich. Dr. Zonder is a member of the editorial advisory board of Hematology News. Their comments were taken from an editorial (N Engl J Med. 2017 April 6. doi: 10.1056/NEJMe1700453).
Adding high-dose chemotherapy and autologous stem cell transplantation to two cycles of bortezomib, lenalidomide, and dexamethasone (VRD) significantly improved progression-free survival in adults with newly diagnosed multiple myeloma, according to new analyses from the multicenter, randomized, phase III IFM 2009 Study.
Median PFS was 50 months in the transplantation group, versus 36 months when patients only received five cycles of VRD for consolidation (adjusted hazard ratio for disease progression or death, 0.65; 95% confidence interval, 0.53 to 0.80; P less than .001), reported Michel Attal, MD, of Institut Universitaire du Cancer de Toulouse-Oncopole in Toulouse, France, and his associates. “Transplantation was also associated with a higher rate of complete response, a lower rate of minimal residual disease detection, and a longer median time to progression,” they wrote (N Engl J Med. 2017 April 6. doi: 10.1056/NEJMoa1611750).
Transplantation did not improve 4-year overall survival (81% with transplantation plus VRD and 82% with VRD only), perhaps because VRD-only patients had the option of salvage transplantation if their disease progressed, the investigators said. In addition, grade 3 or 4 neutropenias were significantly more common with transplantation than otherwise (92% versus 47%), as were grade 3 or 4 gastrointestinal disorders (28% versus 7%) and infections (20% versus 9%). However, the groups did not significantly differ in terms of treatment-related deaths, second primary cancers, thromboembolic events, or peripheral neuropathy.
Before the advent of immunomodulatory drugs and proteasome inhibitors, several randomized trials showed that the benefits of high-dose chemotherapy with autologous stem cell transplantation outperformed conventional chemotherapy in multiple myeloma. For IFM 2009, 700 newly diagnosed patients aged up to 65 years received three 21-day cycles of VRD induction: bortezomib 1.3 mg/m2 on days 1, 4, 8, and 11; lenalidomide 25 mg on days 1-14; and dexamethasone 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12. Consolidation consisted of either five more VRD cycles with dexamethasone reduced to 10 mg/day, or high-dose (200 mg/m2) melphalan plus transplantation followed by two VRD cycles with the reduced dexamethasone dose. Both arms received 1 year of lenalidomide maintenance (10 mg/day for 3 months, followed by 15/mg day, depending on side effects).
Transplantation was associated with a significantly higher likelihood of complete response (59% versus 48%; P = .03) and undetectable minimal residual disease (79% versus 65%; P less than .001). Undetectable minimal residual disease, in turn, predicted longer PFS (adjusted hazard ratio, 0.30; P = .001). In contrast, age, sex, isotype of the monoclonal component, International Staging System disease stage, and cytogenetic risk profile did not significantly affect PFS among transplant patients.
The PFS benefit of transplantation “must be weighed against the increased risk of toxic effects associated with high-dose chemotherapy plus transplantation, especially since we found that later transplantation might be as effective as early transplantation in securing long- term survival,” the researchers said. “Our results suggest that the use of a combination therapy that incorporates newer proteasome inhibitors, next-generation immunomodulatory drugs, and potent monoclonal antibodies along with transplantation tailored according to minimal residual disease detection could further improve outcomes among adults up to 65 years of age who have multiple myeloma.”
Celgene, Janssen, the French Ministry of Health Programme Hospitalier de Recherche Clinique, and the French National Research Agency funded the study. Dr. Attal had no conflicts. Nine coinvestigators disclosed ties to Celgene, Janssen, and several other pharmaceutical companies. Two coinvestigators disclosed ownership or founder roles in OncoPep, one of whom disclosed patent licensure to OncoPep for a multipeptide vaccine. The other investigators had no conflicts.
Adding high-dose chemotherapy and autologous stem cell transplantation to two cycles of bortezomib, lenalidomide, and dexamethasone (VRD) significantly improved progression-free survival in adults with newly diagnosed multiple myeloma, according to new analyses from the multicenter, randomized, phase III IFM 2009 Study.
Median PFS was 50 months in the transplantation group, versus 36 months when patients only received five cycles of VRD for consolidation (adjusted hazard ratio for disease progression or death, 0.65; 95% confidence interval, 0.53 to 0.80; P less than .001), reported Michel Attal, MD, of Institut Universitaire du Cancer de Toulouse-Oncopole in Toulouse, France, and his associates. “Transplantation was also associated with a higher rate of complete response, a lower rate of minimal residual disease detection, and a longer median time to progression,” they wrote (N Engl J Med. 2017 April 6. doi: 10.1056/NEJMoa1611750).
Transplantation did not improve 4-year overall survival (81% with transplantation plus VRD and 82% with VRD only), perhaps because VRD-only patients had the option of salvage transplantation if their disease progressed, the investigators said. In addition, grade 3 or 4 neutropenias were significantly more common with transplantation than otherwise (92% versus 47%), as were grade 3 or 4 gastrointestinal disorders (28% versus 7%) and infections (20% versus 9%). However, the groups did not significantly differ in terms of treatment-related deaths, second primary cancers, thromboembolic events, or peripheral neuropathy.
Before the advent of immunomodulatory drugs and proteasome inhibitors, several randomized trials showed that the benefits of high-dose chemotherapy with autologous stem cell transplantation outperformed conventional chemotherapy in multiple myeloma. For IFM 2009, 700 newly diagnosed patients aged up to 65 years received three 21-day cycles of VRD induction: bortezomib 1.3 mg/m2 on days 1, 4, 8, and 11; lenalidomide 25 mg on days 1-14; and dexamethasone 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12. Consolidation consisted of either five more VRD cycles with dexamethasone reduced to 10 mg/day, or high-dose (200 mg/m2) melphalan plus transplantation followed by two VRD cycles with the reduced dexamethasone dose. Both arms received 1 year of lenalidomide maintenance (10 mg/day for 3 months, followed by 15/mg day, depending on side effects).
Transplantation was associated with a significantly higher likelihood of complete response (59% versus 48%; P = .03) and undetectable minimal residual disease (79% versus 65%; P less than .001). Undetectable minimal residual disease, in turn, predicted longer PFS (adjusted hazard ratio, 0.30; P = .001). In contrast, age, sex, isotype of the monoclonal component, International Staging System disease stage, and cytogenetic risk profile did not significantly affect PFS among transplant patients.
The PFS benefit of transplantation “must be weighed against the increased risk of toxic effects associated with high-dose chemotherapy plus transplantation, especially since we found that later transplantation might be as effective as early transplantation in securing long- term survival,” the researchers said. “Our results suggest that the use of a combination therapy that incorporates newer proteasome inhibitors, next-generation immunomodulatory drugs, and potent monoclonal antibodies along with transplantation tailored according to minimal residual disease detection could further improve outcomes among adults up to 65 years of age who have multiple myeloma.”
Celgene, Janssen, the French Ministry of Health Programme Hospitalier de Recherche Clinique, and the French National Research Agency funded the study. Dr. Attal had no conflicts. Nine coinvestigators disclosed ties to Celgene, Janssen, and several other pharmaceutical companies. Two coinvestigators disclosed ownership or founder roles in OncoPep, one of whom disclosed patent licensure to OncoPep for a multipeptide vaccine. The other investigators had no conflicts.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: High-dose chemotherapy and autologous stem cell transplantation plus bortezomib, lenalidomide, and dexamethasone (VRD) outperformed consolidation with VRD only in adults with newly diagnosed multiple myeloma.
Major finding: Median progression-free survival was 50 months with transplantation plus VRD and 36 months with VRD only (adjusted hazard ratio for disease progression or death, 0.65; P less than .001).
Data source: A randomized, multicenter, open-label, phase III trial of 700 patients with multiple myeloma.
Disclosures: Celgene, Janssen, the French Ministry of Health Programme Hospitalier de Recherche Clinique, and the French National Research Agency funded the study. Dr. Attal had no conflicts. Nine coinvestigators disclosed ties to Celgene, Janssen, and several other pharmaceutical companies. Two coinvestigators disclosed ownership or founder roles in OncoPep, one of whom disclosed patent licensure to OncoPep for a multipeptide vaccine. The other investigators had no conflicts.