User login
Treatment appears feasible for acute PE

Image courtesy of the
Medical College of Georgia
New research suggests that ultrasound-facilitated, catheter-directed, low-dose thrombolysis can produce positive results in patients with acute pulmonary embolism (PE).
In the SEATTLE II study, this treatment prompted significant decreases in thrombus burden, pulmonary hypertension, and the right ventricular-to-left ventricular (RV/LV) diameter ratio.
Major bleeds occurred in 10% of patients, but there were no cases of intracranial hemorrhage.
These results were published in JACC: Cardiovascular Interventions.
The SEATTLE II study was designed to evaluate ultrasound-facilitated, catheter-directed, low-dose thrombolysis using the EKOS EkoSonic® Endovascular System. The research was sponsored by EKOS Corporation, the company developing the system.
The trial included 150 patients diagnosed with acute massive (n=31) or submassive (n=119) PE. Patients received low-dose (24 mg) tissue plasminogen activator for 24 hours with a unilateral catheter or for 12 hours with bilateral catheters.
By 48 hours after treatment initiation, the mean RV/LV diameter ratio had significantly decreased, from 1.55 to 1.13 (P<0.0001).
The mean pulmonary artery systolic pressure decreased significantly as well, from 51.4 mm Hg to 36.9 mm Hg (P<0.0001).
And there was a significant decrease in modified Miller angiographic obstruction index score, from 22.5 to 15.8 (P<0.0001).
There were no intracranial hemorrhages and no fatal bleeding events. Major bleeds occurred in 15 patients (10%) and consisted of 1 severe bleed and 16 moderate bleeds.
Six of the major bleeds occurred in patients with comorbidities known to be associated with an increased risk of bleeding during thrombolytic therapy.
There were 3 serious adverse events that were considered potentially related to the EkoSonic Endovascular System. And 2 serious adverse events were potentially related to tissue plasminogen activator.
At 30 days post-treatment, there were 4 deaths. Three occurred in-hospital, and 1 was directly attributed to PE.
The researchers pointed out that 31 patients presented with massive PE, syncope, and hypotension. And all 31 survived the 30-day follow-up period.
“The SEATTLE II findings establish a new rationale for considering ultrasound-facilitated, catheter-directed, low-dose thrombolysis in both massive and submassive PE,” said study author Gregory Piazza, MD, of Brigham and Woman’s Hospital in Boston, Massachusetts.
“Without any intracranial hemorrhage and using a much-reduced lytic dose, a substantial and clinically meaningful reduction of the RV/LV ratio was achieved.” ![]()
Image courtesy of the
Medical College of Georgia
New research suggests that ultrasound-facilitated, catheter-directed, low-dose thrombolysis can produce positive results in patients with acute pulmonary embolism (PE).
In the SEATTLE II study, this treatment prompted significant decreases in thrombus burden, pulmonary hypertension, and the right ventricular-to-left ventricular (RV/LV) diameter ratio.
Major bleeds occurred in 10% of patients, but there were no cases of intracranial hemorrhage.
These results were published in JACC: Cardiovascular Interventions.
The SEATTLE II study was designed to evaluate ultrasound-facilitated, catheter-directed, low-dose thrombolysis using the EKOS EkoSonic® Endovascular System. The research was sponsored by EKOS Corporation, the company developing the system.
The trial included 150 patients diagnosed with acute massive (n=31) or submassive (n=119) PE. Patients received low-dose (24 mg) tissue plasminogen activator for 24 hours with a unilateral catheter or for 12 hours with bilateral catheters.
By 48 hours after treatment initiation, the mean RV/LV diameter ratio had significantly decreased, from 1.55 to 1.13 (P<0.0001).
The mean pulmonary artery systolic pressure decreased significantly as well, from 51.4 mm Hg to 36.9 mm Hg (P<0.0001).
And there was a significant decrease in modified Miller angiographic obstruction index score, from 22.5 to 15.8 (P<0.0001).
There were no intracranial hemorrhages and no fatal bleeding events. Major bleeds occurred in 15 patients (10%) and consisted of 1 severe bleed and 16 moderate bleeds.
Six of the major bleeds occurred in patients with comorbidities known to be associated with an increased risk of bleeding during thrombolytic therapy.
There were 3 serious adverse events that were considered potentially related to the EkoSonic Endovascular System. And 2 serious adverse events were potentially related to tissue plasminogen activator.
At 30 days post-treatment, there were 4 deaths. Three occurred in-hospital, and 1 was directly attributed to PE.
The researchers pointed out that 31 patients presented with massive PE, syncope, and hypotension. And all 31 survived the 30-day follow-up period.
“The SEATTLE II findings establish a new rationale for considering ultrasound-facilitated, catheter-directed, low-dose thrombolysis in both massive and submassive PE,” said study author Gregory Piazza, MD, of Brigham and Woman’s Hospital in Boston, Massachusetts.
“Without any intracranial hemorrhage and using a much-reduced lytic dose, a substantial and clinically meaningful reduction of the RV/LV ratio was achieved.” ![]()
Image courtesy of the
Medical College of Georgia
New research suggests that ultrasound-facilitated, catheter-directed, low-dose thrombolysis can produce positive results in patients with acute pulmonary embolism (PE).
In the SEATTLE II study, this treatment prompted significant decreases in thrombus burden, pulmonary hypertension, and the right ventricular-to-left ventricular (RV/LV) diameter ratio.
Major bleeds occurred in 10% of patients, but there were no cases of intracranial hemorrhage.
These results were published in JACC: Cardiovascular Interventions.
The SEATTLE II study was designed to evaluate ultrasound-facilitated, catheter-directed, low-dose thrombolysis using the EKOS EkoSonic® Endovascular System. The research was sponsored by EKOS Corporation, the company developing the system.
The trial included 150 patients diagnosed with acute massive (n=31) or submassive (n=119) PE. Patients received low-dose (24 mg) tissue plasminogen activator for 24 hours with a unilateral catheter or for 12 hours with bilateral catheters.
By 48 hours after treatment initiation, the mean RV/LV diameter ratio had significantly decreased, from 1.55 to 1.13 (P<0.0001).
The mean pulmonary artery systolic pressure decreased significantly as well, from 51.4 mm Hg to 36.9 mm Hg (P<0.0001).
And there was a significant decrease in modified Miller angiographic obstruction index score, from 22.5 to 15.8 (P<0.0001).
There were no intracranial hemorrhages and no fatal bleeding events. Major bleeds occurred in 15 patients (10%) and consisted of 1 severe bleed and 16 moderate bleeds.
Six of the major bleeds occurred in patients with comorbidities known to be associated with an increased risk of bleeding during thrombolytic therapy.
There were 3 serious adverse events that were considered potentially related to the EkoSonic Endovascular System. And 2 serious adverse events were potentially related to tissue plasminogen activator.
At 30 days post-treatment, there were 4 deaths. Three occurred in-hospital, and 1 was directly attributed to PE.
The researchers pointed out that 31 patients presented with massive PE, syncope, and hypotension. And all 31 survived the 30-day follow-up period.
“The SEATTLE II findings establish a new rationale for considering ultrasound-facilitated, catheter-directed, low-dose thrombolysis in both massive and submassive PE,” said study author Gregory Piazza, MD, of Brigham and Woman’s Hospital in Boston, Massachusetts.
“Without any intracranial hemorrhage and using a much-reduced lytic dose, a substantial and clinically meaningful reduction of the RV/LV ratio was achieved.” ![]()
Study reveals approaches to aid, prevent apoptosis
apoptosis in cancer cells
Scientists say they have gained new insight into the role Bax plays in apoptosis.
The Bax protein is known to be a key regulator of apoptosis, mediating the release of cytochrome c to the cytosol via oligomerization in the outer mitochondrial membrane before pore formation.
But the exact mechanism of Bax assembly was previously unclear.
Now, research published in Nature Communications has provided some clarity.
Katia Cosentino, PhD, of the Max Planck Institute for Intelligent Systems in Stuttgart, Germany, and her colleagues conducted this research, examining how the mitochondrial membrane becomes permeable.
The team’s experiments on artificial membrane systems showed that Bax is initially inserted into the membrane as a single molecule.
Once inserted, one Bax molecule will join up with a second Bax molecule to form a stable complex, the Bax dimers. From these dimers, larger complexes are formed.
“Surprisingly, Bax complexes have no standard size, but we observed a mixture of different-sized Bax species, and these species are mostly based on dimer units,” Dr Cosentino said.
She and her colleagues noted that these Bax complexes form the pores through which cytochrome c exits the mitochondrial membrane.
But the process of pore formation is finely controlled by other proteins. Some (such as cBid) enable the assembly of Bax elements, while others (such as Bcl-xL) induce their dismantling.
“The differing size of the Bax complexes in the pore formation is likely part of the reason why earlier investigations on pore formation conveyed contradictory results,” Dr Cosentino said.
She and her colleagues believe that, based on these findings, they can make some initial recommendations for medical intervention in the apoptotic process.
They think that, to promote apoptosis, it should be enough to initiate the first step of activating Bax proteins because the subsequent steps of self-organization will then happen automatically.
Conversely, the team’s findings suggest apoptosis can be prevented when drugs force the dismantling of the Bax dimers into their individual elements. ![]()
apoptosis in cancer cells
Scientists say they have gained new insight into the role Bax plays in apoptosis.
The Bax protein is known to be a key regulator of apoptosis, mediating the release of cytochrome c to the cytosol via oligomerization in the outer mitochondrial membrane before pore formation.
But the exact mechanism of Bax assembly was previously unclear.
Now, research published in Nature Communications has provided some clarity.
Katia Cosentino, PhD, of the Max Planck Institute for Intelligent Systems in Stuttgart, Germany, and her colleagues conducted this research, examining how the mitochondrial membrane becomes permeable.
The team’s experiments on artificial membrane systems showed that Bax is initially inserted into the membrane as a single molecule.
Once inserted, one Bax molecule will join up with a second Bax molecule to form a stable complex, the Bax dimers. From these dimers, larger complexes are formed.
“Surprisingly, Bax complexes have no standard size, but we observed a mixture of different-sized Bax species, and these species are mostly based on dimer units,” Dr Cosentino said.
She and her colleagues noted that these Bax complexes form the pores through which cytochrome c exits the mitochondrial membrane.
But the process of pore formation is finely controlled by other proteins. Some (such as cBid) enable the assembly of Bax elements, while others (such as Bcl-xL) induce their dismantling.
“The differing size of the Bax complexes in the pore formation is likely part of the reason why earlier investigations on pore formation conveyed contradictory results,” Dr Cosentino said.
She and her colleagues believe that, based on these findings, they can make some initial recommendations for medical intervention in the apoptotic process.
They think that, to promote apoptosis, it should be enough to initiate the first step of activating Bax proteins because the subsequent steps of self-organization will then happen automatically.
Conversely, the team’s findings suggest apoptosis can be prevented when drugs force the dismantling of the Bax dimers into their individual elements. ![]()
apoptosis in cancer cells
Scientists say they have gained new insight into the role Bax plays in apoptosis.
The Bax protein is known to be a key regulator of apoptosis, mediating the release of cytochrome c to the cytosol via oligomerization in the outer mitochondrial membrane before pore formation.
But the exact mechanism of Bax assembly was previously unclear.
Now, research published in Nature Communications has provided some clarity.
Katia Cosentino, PhD, of the Max Planck Institute for Intelligent Systems in Stuttgart, Germany, and her colleagues conducted this research, examining how the mitochondrial membrane becomes permeable.
The team’s experiments on artificial membrane systems showed that Bax is initially inserted into the membrane as a single molecule.
Once inserted, one Bax molecule will join up with a second Bax molecule to form a stable complex, the Bax dimers. From these dimers, larger complexes are formed.
“Surprisingly, Bax complexes have no standard size, but we observed a mixture of different-sized Bax species, and these species are mostly based on dimer units,” Dr Cosentino said.
She and her colleagues noted that these Bax complexes form the pores through which cytochrome c exits the mitochondrial membrane.
But the process of pore formation is finely controlled by other proteins. Some (such as cBid) enable the assembly of Bax elements, while others (such as Bcl-xL) induce their dismantling.
“The differing size of the Bax complexes in the pore formation is likely part of the reason why earlier investigations on pore formation conveyed contradictory results,” Dr Cosentino said.
She and her colleagues believe that, based on these findings, they can make some initial recommendations for medical intervention in the apoptotic process.
They think that, to promote apoptosis, it should be enough to initiate the first step of activating Bax proteins because the subsequent steps of self-organization will then happen automatically.
Conversely, the team’s findings suggest apoptosis can be prevented when drugs force the dismantling of the Bax dimers into their individual elements. ![]()
New proteasome inhibitor exhibits activity against MM

A novel, cancer-selective proteasome inhibitor has shown early promise for treating multiple myeloma (MM) and breast cancer, according to researchers.
The drug, known as VR23, is a quinoline-sulfonyl hybrid proteasome inhibitor.
In preclinical experiments, VR23 preferentially targeted MM cells and breast cancer cells, demonstrated synergy with bortezomib or paclitaxel, and shrank both MM and breast cancer tumors in mice.
Hoyun Lee, PhD, of the Advanced Medical Research Institute of Canada (AMRIC) in Sudbury, Ontario, and his colleagues described these experiments in Cancer Research.
The team noted that VR23 is structurally distinct from other known proteasome inhibitors, and it “potently inhibits” the activities of trypsin-like proteasomes, chymotrypsin-like proteasomes, and caspase-like proteasomes.
In several experiments, VR23 proved active against breast cancer.
In experiments with MM cell lines, VR23 exhibited activity as a single agent and demonstrated synergy with bortezomib. VR23 proved more effective against bortezomib-resistant cells than bortezomib-naïve cells.
When the researchers introduced treatments to bortezomib-naïve RPMI-8226 cells, they found the cell growth rate was 79.3% with VR23, 12.5% with bortezomib, and 1.6% with both drugs. In bortezomib-resistant RPMI-8226 cells, the cell growth rate was 47% with VR23, 109.7% with bortezomib, and -8.6% with both drugs.
In KAS6/1 cells, the cell growth rate was 65% with VR23, 92% with bortezomib, and 26.5% with both drugs. In bortezomib-resistant ANBL6 cells, the cell growth rate was 94% with VR23, 102.9% with bortezomib, and 48.9% with both drugs.
The researchers also tested VR23 in mice engrafted with RPMI-8226 MM cells. At 24 days after treatment, the tumor volume for VR23-treated mice was 19.1% that of placebo-treated mice.
The team noted that VR23 selectively killed cancer cells via apoptosis. Cancer cells exposed to the drug underwent an abnormal centrosome amplification cycle caused by the accumulation of ubiquitinated cyclin E.
Dr Lee and his colleagues are now planning to work with the US National Cancer Institute to test VR23 in additional cancers. The team is hoping to progress to clinical trials with the drug in the next 3 years.
AMRIC has applied for international intellectual property protection for VR23 and licensed commercial rights to Ramsey Lake Pharmaceutical Corporation (www.ramseylakepharma.com), an operation of AMRIC. ![]()
A novel, cancer-selective proteasome inhibitor has shown early promise for treating multiple myeloma (MM) and breast cancer, according to researchers.
The drug, known as VR23, is a quinoline-sulfonyl hybrid proteasome inhibitor.
In preclinical experiments, VR23 preferentially targeted MM cells and breast cancer cells, demonstrated synergy with bortezomib or paclitaxel, and shrank both MM and breast cancer tumors in mice.
Hoyun Lee, PhD, of the Advanced Medical Research Institute of Canada (AMRIC) in Sudbury, Ontario, and his colleagues described these experiments in Cancer Research.
The team noted that VR23 is structurally distinct from other known proteasome inhibitors, and it “potently inhibits” the activities of trypsin-like proteasomes, chymotrypsin-like proteasomes, and caspase-like proteasomes.
In several experiments, VR23 proved active against breast cancer.
In experiments with MM cell lines, VR23 exhibited activity as a single agent and demonstrated synergy with bortezomib. VR23 proved more effective against bortezomib-resistant cells than bortezomib-naïve cells.
When the researchers introduced treatments to bortezomib-naïve RPMI-8226 cells, they found the cell growth rate was 79.3% with VR23, 12.5% with bortezomib, and 1.6% with both drugs. In bortezomib-resistant RPMI-8226 cells, the cell growth rate was 47% with VR23, 109.7% with bortezomib, and -8.6% with both drugs.
In KAS6/1 cells, the cell growth rate was 65% with VR23, 92% with bortezomib, and 26.5% with both drugs. In bortezomib-resistant ANBL6 cells, the cell growth rate was 94% with VR23, 102.9% with bortezomib, and 48.9% with both drugs.
The researchers also tested VR23 in mice engrafted with RPMI-8226 MM cells. At 24 days after treatment, the tumor volume for VR23-treated mice was 19.1% that of placebo-treated mice.
The team noted that VR23 selectively killed cancer cells via apoptosis. Cancer cells exposed to the drug underwent an abnormal centrosome amplification cycle caused by the accumulation of ubiquitinated cyclin E.
Dr Lee and his colleagues are now planning to work with the US National Cancer Institute to test VR23 in additional cancers. The team is hoping to progress to clinical trials with the drug in the next 3 years.
AMRIC has applied for international intellectual property protection for VR23 and licensed commercial rights to Ramsey Lake Pharmaceutical Corporation (www.ramseylakepharma.com), an operation of AMRIC. ![]()
A novel, cancer-selective proteasome inhibitor has shown early promise for treating multiple myeloma (MM) and breast cancer, according to researchers.
The drug, known as VR23, is a quinoline-sulfonyl hybrid proteasome inhibitor.
In preclinical experiments, VR23 preferentially targeted MM cells and breast cancer cells, demonstrated synergy with bortezomib or paclitaxel, and shrank both MM and breast cancer tumors in mice.
Hoyun Lee, PhD, of the Advanced Medical Research Institute of Canada (AMRIC) in Sudbury, Ontario, and his colleagues described these experiments in Cancer Research.
The team noted that VR23 is structurally distinct from other known proteasome inhibitors, and it “potently inhibits” the activities of trypsin-like proteasomes, chymotrypsin-like proteasomes, and caspase-like proteasomes.
In several experiments, VR23 proved active against breast cancer.
In experiments with MM cell lines, VR23 exhibited activity as a single agent and demonstrated synergy with bortezomib. VR23 proved more effective against bortezomib-resistant cells than bortezomib-naïve cells.
When the researchers introduced treatments to bortezomib-naïve RPMI-8226 cells, they found the cell growth rate was 79.3% with VR23, 12.5% with bortezomib, and 1.6% with both drugs. In bortezomib-resistant RPMI-8226 cells, the cell growth rate was 47% with VR23, 109.7% with bortezomib, and -8.6% with both drugs.
In KAS6/1 cells, the cell growth rate was 65% with VR23, 92% with bortezomib, and 26.5% with both drugs. In bortezomib-resistant ANBL6 cells, the cell growth rate was 94% with VR23, 102.9% with bortezomib, and 48.9% with both drugs.
The researchers also tested VR23 in mice engrafted with RPMI-8226 MM cells. At 24 days after treatment, the tumor volume for VR23-treated mice was 19.1% that of placebo-treated mice.
The team noted that VR23 selectively killed cancer cells via apoptosis. Cancer cells exposed to the drug underwent an abnormal centrosome amplification cycle caused by the accumulation of ubiquitinated cyclin E.
Dr Lee and his colleagues are now planning to work with the US National Cancer Institute to test VR23 in additional cancers. The team is hoping to progress to clinical trials with the drug in the next 3 years.
AMRIC has applied for international intellectual property protection for VR23 and licensed commercial rights to Ramsey Lake Pharmaceutical Corporation (www.ramseylakepharma.com), an operation of AMRIC. ![]()
Corticosteroids Show Benefit in Community-Acquired Pneumonia
Clinical question: Does corticosteroid treatment shorten systemic illness in patients admitted to the hospital for community-acquired pneumonia (CAP)?
Background: Pneumonia is the third-leading cause of death worldwide. Studies have yielded conflicting data about the benefit of adding systemic corticosteroids for treatment of CAP.
Study design: Double-blind, multicenter, randomized, placebo-controlled trial.
Setting: Seven tertiary-care hospitals in Switzerland.
Synopsis: A group of 784 patients hospitalized for CAP were randomized to receive either oral prednisone 50 mg daily for seven days or placebo, with the primary endpoint being time to stable vital signs. The intention-to-treat analysis found that the median time to clinical stability was 1.4 days earlier in the prednisone group (hazard ratio 1.33; 95% CI, 1.15–1.50, P <0.0001) and that length of stay and IV antibiotics were reduced by one day; this effect was valid across all PSI classes and was not dependent on age. Pneumonia-associated complications in the two groups did not differ at 30 days, though the prednisone group had a higher incidence of hyperglycemia requiring insulin. Because all study locations were in a single, fairly homogenous northern European country, care should be taken when hospitalists apply these findings to their patient population, and the risks of hyperglycemia requiring insulin should be taken into consideration.
Bottom line: Systemic steroids may reduce the time to clinical stability in patients with CAP.
Citation: Blum CA, Nigro N, Briel M, et al. Adjunct prednisone therapy for patients with community-acquired pneumonia: a multicentre, double-blind, randomised, placebo-controlled trial. Lancet. 2015;385(9977):1511–1518.
Clinical question: Does corticosteroid treatment shorten systemic illness in patients admitted to the hospital for community-acquired pneumonia (CAP)?
Background: Pneumonia is the third-leading cause of death worldwide. Studies have yielded conflicting data about the benefit of adding systemic corticosteroids for treatment of CAP.
Study design: Double-blind, multicenter, randomized, placebo-controlled trial.
Setting: Seven tertiary-care hospitals in Switzerland.
Synopsis: A group of 784 patients hospitalized for CAP were randomized to receive either oral prednisone 50 mg daily for seven days or placebo, with the primary endpoint being time to stable vital signs. The intention-to-treat analysis found that the median time to clinical stability was 1.4 days earlier in the prednisone group (hazard ratio 1.33; 95% CI, 1.15–1.50, P <0.0001) and that length of stay and IV antibiotics were reduced by one day; this effect was valid across all PSI classes and was not dependent on age. Pneumonia-associated complications in the two groups did not differ at 30 days, though the prednisone group had a higher incidence of hyperglycemia requiring insulin. Because all study locations were in a single, fairly homogenous northern European country, care should be taken when hospitalists apply these findings to their patient population, and the risks of hyperglycemia requiring insulin should be taken into consideration.
Bottom line: Systemic steroids may reduce the time to clinical stability in patients with CAP.
Citation: Blum CA, Nigro N, Briel M, et al. Adjunct prednisone therapy for patients with community-acquired pneumonia: a multicentre, double-blind, randomised, placebo-controlled trial. Lancet. 2015;385(9977):1511–1518.
Clinical question: Does corticosteroid treatment shorten systemic illness in patients admitted to the hospital for community-acquired pneumonia (CAP)?
Background: Pneumonia is the third-leading cause of death worldwide. Studies have yielded conflicting data about the benefit of adding systemic corticosteroids for treatment of CAP.
Study design: Double-blind, multicenter, randomized, placebo-controlled trial.
Setting: Seven tertiary-care hospitals in Switzerland.
Synopsis: A group of 784 patients hospitalized for CAP were randomized to receive either oral prednisone 50 mg daily for seven days or placebo, with the primary endpoint being time to stable vital signs. The intention-to-treat analysis found that the median time to clinical stability was 1.4 days earlier in the prednisone group (hazard ratio 1.33; 95% CI, 1.15–1.50, P <0.0001) and that length of stay and IV antibiotics were reduced by one day; this effect was valid across all PSI classes and was not dependent on age. Pneumonia-associated complications in the two groups did not differ at 30 days, though the prednisone group had a higher incidence of hyperglycemia requiring insulin. Because all study locations were in a single, fairly homogenous northern European country, care should be taken when hospitalists apply these findings to their patient population, and the risks of hyperglycemia requiring insulin should be taken into consideration.
Bottom line: Systemic steroids may reduce the time to clinical stability in patients with CAP.
Citation: Blum CA, Nigro N, Briel M, et al. Adjunct prednisone therapy for patients with community-acquired pneumonia: a multicentre, double-blind, randomised, placebo-controlled trial. Lancet. 2015;385(9977):1511–1518.
Infection, Acute Kidney Injury Raise 30-Day Readmission Risk for Sepsis Survivors
Nearly one-third of hospitalized patients who survive severe sepsis or septic shock require readmission within 30 days, according to a new report in the Journal of Hospital Medicine. Between 660,000 and 750,000 sepsis hospitalizations occur annually, with the direct costs surpassing $24 billion, the paper notes.
The study, which examined 1697 sepsis survivors from 2008 to 2012, links several clinical factors with increased 30-day readmission risk, including infection with Bacteroides spp and extended-spectrum beta-lactamases organisms and puts sepsis survivors with mild-to-moderate acute kidney injury (AKI) at nearly double the risk of readmission.
Study lead author Marya Zilberberg, MD, MPH, notes that efforts to reduce AKI in sepsis survivors might affect patient outcomes but that more research needs to be done to help decrease readmission rates.
“The hypothesis that reducing the occurrence of AKI would in turn reduce the risk of a 30-day hospitalization needs to be validated,” Dr. Zilberberg writes in an email to The Hospitalist. “Furthermore, it is a difficult goal among the critically ill. So, what this means to me is … this quality metric may be yet another that is putting the cart (the metric) before the horse (evidence to support its use).”
Dr. Zilberberg, founder and president of EviMed Research Group, LLC, an evidence-based medicine and outcomes research firm based in Goshen, Mass., says the study’s results were not completely unexpected as resistant infections are associated with worsening of all outcomes, and “we just showed that 30-day readmission was not immune to that,” she notes.
“We are not sure what effective strategies [there] may be to achieve this goal,” she notes. “In general, delivery of best-known care is the best that can be done. The most that can be said from our study is that antimicrobial resistance is bad even vis-à-vis this outcome, so reducing the burden of antimicrobial resistance, in addition to AKI prevention, is a strategy that might impact this outcome, along with many others.”
Visit our website for more information on sepsis and HM.
Nearly one-third of hospitalized patients who survive severe sepsis or septic shock require readmission within 30 days, according to a new report in the Journal of Hospital Medicine. Between 660,000 and 750,000 sepsis hospitalizations occur annually, with the direct costs surpassing $24 billion, the paper notes.
The study, which examined 1697 sepsis survivors from 2008 to 2012, links several clinical factors with increased 30-day readmission risk, including infection with Bacteroides spp and extended-spectrum beta-lactamases organisms and puts sepsis survivors with mild-to-moderate acute kidney injury (AKI) at nearly double the risk of readmission.
Study lead author Marya Zilberberg, MD, MPH, notes that efforts to reduce AKI in sepsis survivors might affect patient outcomes but that more research needs to be done to help decrease readmission rates.
“The hypothesis that reducing the occurrence of AKI would in turn reduce the risk of a 30-day hospitalization needs to be validated,” Dr. Zilberberg writes in an email to The Hospitalist. “Furthermore, it is a difficult goal among the critically ill. So, what this means to me is … this quality metric may be yet another that is putting the cart (the metric) before the horse (evidence to support its use).”
Dr. Zilberberg, founder and president of EviMed Research Group, LLC, an evidence-based medicine and outcomes research firm based in Goshen, Mass., says the study’s results were not completely unexpected as resistant infections are associated with worsening of all outcomes, and “we just showed that 30-day readmission was not immune to that,” she notes.
“We are not sure what effective strategies [there] may be to achieve this goal,” she notes. “In general, delivery of best-known care is the best that can be done. The most that can be said from our study is that antimicrobial resistance is bad even vis-à-vis this outcome, so reducing the burden of antimicrobial resistance, in addition to AKI prevention, is a strategy that might impact this outcome, along with many others.”
Visit our website for more information on sepsis and HM.
Nearly one-third of hospitalized patients who survive severe sepsis or septic shock require readmission within 30 days, according to a new report in the Journal of Hospital Medicine. Between 660,000 and 750,000 sepsis hospitalizations occur annually, with the direct costs surpassing $24 billion, the paper notes.
The study, which examined 1697 sepsis survivors from 2008 to 2012, links several clinical factors with increased 30-day readmission risk, including infection with Bacteroides spp and extended-spectrum beta-lactamases organisms and puts sepsis survivors with mild-to-moderate acute kidney injury (AKI) at nearly double the risk of readmission.
Study lead author Marya Zilberberg, MD, MPH, notes that efforts to reduce AKI in sepsis survivors might affect patient outcomes but that more research needs to be done to help decrease readmission rates.
“The hypothesis that reducing the occurrence of AKI would in turn reduce the risk of a 30-day hospitalization needs to be validated,” Dr. Zilberberg writes in an email to The Hospitalist. “Furthermore, it is a difficult goal among the critically ill. So, what this means to me is … this quality metric may be yet another that is putting the cart (the metric) before the horse (evidence to support its use).”
Dr. Zilberberg, founder and president of EviMed Research Group, LLC, an evidence-based medicine and outcomes research firm based in Goshen, Mass., says the study’s results were not completely unexpected as resistant infections are associated with worsening of all outcomes, and “we just showed that 30-day readmission was not immune to that,” she notes.
“We are not sure what effective strategies [there] may be to achieve this goal,” she notes. “In general, delivery of best-known care is the best that can be done. The most that can be said from our study is that antimicrobial resistance is bad even vis-à-vis this outcome, so reducing the burden of antimicrobial resistance, in addition to AKI prevention, is a strategy that might impact this outcome, along with many others.”
Visit our website for more information on sepsis and HM.
Is Your Electronic Health Record Putting You at Risk for a Documentation Audit?
A group of 3 busy orthopedists attended coding education each year and did their best to accurately code and document their services. As a risk-reduction strategy, the group engaged our firm to conduct an audit to determine whether they were documenting their services properly and to provide feedback about how they could improve.
What we found was shocking to the surgeons, but all too common, as we review thousands of orthopedic visit notes every year: The same examination had been documented for all visits, with physicians stating in their notes that the examination was medically necessary. In addition, their documentation supported Current Procedural Terminology (CPT) code 99214 at every visit, with visit frequencies of 2 weeks to 4 months.
The culprit of all this sameness? The practice’s electronic health record (EHR).
“Practices with EHRs often have a large volume of visit notes that look almost identical for a patient who is seen for multiple visits,” explains Mary LeGrand, RN, MA, CCS-P, CPC, KarenZupko & Associates consultant and coding educator. “And that is putting physicians at higher risk of being audited or of not passing an audit.”
According to LeGrand, this is because physicians are using the practice’s EHR to “pull forward” the patient’s previous visit note for the current visit, but failing to customize it for the current visit. The unintended consequence of this workflow efficiency is twofold:
1. It creates documentation that looks strikingly similar to, if not exactly like, the patient’s last billed visit note. This is often referred to as note “cloning.”
2. It creates documentation that includes a lot of unnecessary detail that, even if delivered and documented, doesn’t match the medical necessity of the visit, based on the history of present illness statements.
Both of these things can come back to bite you.
Zero in on the Risk
If your practice has an EHR, it is important that you evaluate whether certain workflow efficiency features are putting the practice at risk. You do not necessarily need to dump the EHR, but you may need to take action to reduce the risk of using these features.
In a pre-EHR practice, physicians began each visit with a blank piece of paper or dictated the entire visit. Then along came EHR vendors who, in an effort to make things easier and more efficient, created visit templates and the ability to “pull forward” the last visit note and use it as a basis for the current visit. The intention was always that physicians would modify it based on the current visit. But the reality is that physicians are busy, editing is time-consuming, and the unintended consequence is cloning.
“If you pull in unnecessary history or exam information from a previous visit that’s not relevant to the current visit, you can get dinged in an audit for not customizing the note to the patient’s specific presenting complaint,” LeGrand explains, “or, for attempting to bill a higher-level code by unintentionally padding the note with irrelevant information. What is documented for ‘reference’ has to be separated from what can be used to select the level of service.”
Your first documentation risk-reduction strategy is to review notes and look for signs of cloning.
LeGrand explains that a practice may be predisposed to cloning simply because of the way the EHR templates and workflow were set up when the system was implemented. “But,” she says, “‘the EHR made me do it’ defense won’t hold water, because it’s still the physician’s responsibility to customize or remove the information from templates and make the note unique to the visit.”
Yes, physician time is precious. But the reality is that the onus is on the physician to integrate EHR features with clinic workflow and to follow documentation rules.
The second documentation risk-reduction strategy is to make sure the level of evaluation and management (E/M) service billed is supported by medical necessity, not only by documentation artifacts that were relevant to the patient in the past but irrelevant to his or her current presenting complaint or condition.
“Medicare won’t pay for services that aren’t supported by medical necessity,” says LeGrand, “and you can’t achieve medical necessity by simply documenting additional E/M elements.”
This has always been the rule, LeGrand says. “But with the increased use of EHRs, and templates that automatically document visit elements and drive visits to a higher level of service, the Centers for Medicare & Medicaid Services [CMS] and private payers have added scrutiny to medical necessity reviews. They want to validate that higher-level visits billed indeed required a higher level of history and/or exam.”
To do this, the Office of the Inspector General (OIG) has supplemented its audit team with registered nurses. “The nurses assist certified coders by determining whether medical necessity has been met,” explains LeGrand.
Look at a patient who presents with toe pain. You take a detailed family history, conduct a review of systems (ROS), bill a high-level code, and document all the elements to support it. LeGrand explains, “There is no medical necessity to support doing an eye exam for a patient with toe pain in the absence of any other medical history, or performing a ROS to correlate an eye exam with toe pain. So, even if you do it and document it, the higher-level code won’t pass muster in an audit because the information documented is not medically necessary.”
According to LeGrand, the extent of the history and examination should be based on the presenting problem and the patient’s condition. “If an ankle sprain patient returns 2 weeks after the initial evaluation of the injury with a negative medical or surgical history, and the patient has been treated conservatively, it’s probably not necessary to conduct a ROS that includes 10 organ systems,” she says. “If your standard of care is to perform this level of service, no one will fault you for your care delivery; however, if you also choose a level of service based on this system review, without relevance to the presenting problem, and you bill a higher level of service than is supported by the nature of the presenting problem or the plan of care, the documentation probably won’t hold up in an audit where medical necessity is valued into the equation.”
On the other hand, LeGrand adds, if a patient presents to the emergency department after an automobile accident with an open fracture and other injuries, and the surgeon performs a complete ROS, the medical necessity would most likely be supported as the surgeon is preparing the patient for surgery.
Based on LeGrand’s work with practices, this distinction about medical necessity is news to many nonclinical billing staff. “They confuse medical necessity with medical decision-making, an E/M code documentation component, and incorrectly bill for a high-level visit because medical decision-making elements meet the documentation requirements—yet the code is not supported by medical necessity of the presenting problem.”
Talk with your billing team to make sure all staff members understand this critical difference. They must comprehend that the medically necessary level of service is determined by a number of clinical factors, not medical decision-making. Describe some of these clinical factors, which include, but are not limited to, chief complaint, clinical judgment, standards of practice, acute exacerbations/onsets of medical conditions or injuries, and comorbidities.
EHR Dos and Don’ts
LeGrand recommends the following best practices for using EHR documentation features:
1. DON’T simply cut and paste from a previous note. “This is what leads to verbose notes that have little to do with the patient you are documenting,” she says. “If you don’t cut and paste, you’ll avoid the root cause of this risk.”
2. DON’T pull forward information from previous visit notes that have nothing to do with the nature of the patient’s problem. “We understand that this takes extra time because physicians must review the previous note,” LeGrand says. “So if you don’t have time to review the past note, just don’t pull it forward. Start fresh with a new drop-down menu and select elements pertinent to the current visit. Or, dictate or type a note relevant to the current condition and presenting problems.”
How you choose to work this into your process will vary depending on which EHR system you use. “One surgeon I work with dictates everything because the drop-down menus and templates are cumbersome,” LeGrand says. “Some groups find it faster to use the EHR templates that they have customized. Others find their EHR’s point-and-click features most efficient for customizing quickly.”
3. DO customize your EHR visit templates if the use of templates is critical to your efficiency. “This is the most overlooked step in the EHR implementation process because it takes a fair amount of time to do,” LeGrand says. She suggests avoiding the use of multisystem examination templates created for medicine specialties altogether, and insists, “Don’t assume ‘that is how the vendor built it so we have to use it.’ Customize a template for each of your visit types so you can document in the EHR in the same fashion as when you used a paper system. Doing so will save you loads of documentation time.”
4. DO review your E/M code distribution. Generate a CPT frequency report for each physician and for the practice as a whole. Compare the data with state and national usage in orthopedics as a baseline. The American Academy of Orthopaedic Surgeon’s Code-X tool enables easy comparison of your practice’s E/M code usage with state and national data for orthopedics. Simply generate a CPT frequency report from your practice management system and enter the E/M data. Line graphs are automatically generated, making trends and patterns easy to see (Figure).
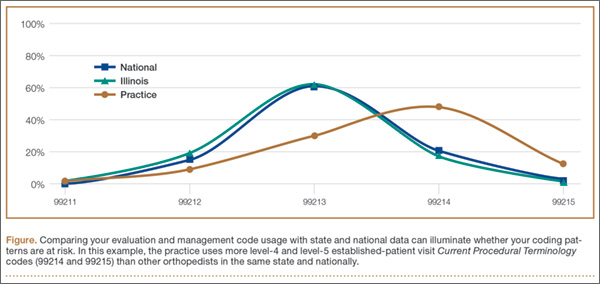
“Identify your outliers, pull charts randomly, and review the notes,” recommends LeGrand. “Make sure there is medical necessity for the level of code that’s been billed and that documentation supports it.”
You may be surprised to find you are an outlier on inpatient hospital codes, or your distribution of level-2 or -3 codes varies from your practice, state, or national data. Orthopedic surgeons don’t typically report high volumes of CPT codes 99204, 99205 or 99215, but if your practice does and you are an outlier, best to pay attention before someone else does.
5. DO select auditors with the right skill sets. Evaluating medical necessity in the note requires a clinical background. “If internal documentation reviews are conducted by the billing team, that’s fine,” LeGrand advises. “Just add a physician assistant or nurse to your internal review team. They can provide clinical oversight and review the note when necessary for medical necessity.”
If you are contracting with external auditors or consultants, verify auditor credentials and skill sets to ensure they can abstract and incorporate medical necessity into the review. “Auditors must be able to do more than count elements,” LeGrand says. “They must have clinical knowledge, and expertise in orthopedics is critical. This knowledge should be used to verify that medical necessity is present in every note.” LeGrand is quick to point out that not every note will be at risk, based on the amount of work performed and documented and the level of service billed. “But medical necessity must always be present.”
The addition of nurses to the OIG’s audit team is a big change and will refine the auditing process by adding more clinical scrutiny. The EHR documentation features are intended to improve efficiency, but only a clinician can determine and document unique visit elements and medical necessity.
Address these intersections of risk by ensuring your documentation meets medical necessity as well as E/M documentation elements. Conduct internal audits bi-annually to verify that E/M usage patterns align with peers and physician documentation is appropriate. And be sure there is clinical expertise on your audit team, whether it is internal or external. CMS now has it, and your practice should too. ◾
A group of 3 busy orthopedists attended coding education each year and did their best to accurately code and document their services. As a risk-reduction strategy, the group engaged our firm to conduct an audit to determine whether they were documenting their services properly and to provide feedback about how they could improve.
What we found was shocking to the surgeons, but all too common, as we review thousands of orthopedic visit notes every year: The same examination had been documented for all visits, with physicians stating in their notes that the examination was medically necessary. In addition, their documentation supported Current Procedural Terminology (CPT) code 99214 at every visit, with visit frequencies of 2 weeks to 4 months.
The culprit of all this sameness? The practice’s electronic health record (EHR).
“Practices with EHRs often have a large volume of visit notes that look almost identical for a patient who is seen for multiple visits,” explains Mary LeGrand, RN, MA, CCS-P, CPC, KarenZupko & Associates consultant and coding educator. “And that is putting physicians at higher risk of being audited or of not passing an audit.”
According to LeGrand, this is because physicians are using the practice’s EHR to “pull forward” the patient’s previous visit note for the current visit, but failing to customize it for the current visit. The unintended consequence of this workflow efficiency is twofold:
1. It creates documentation that looks strikingly similar to, if not exactly like, the patient’s last billed visit note. This is often referred to as note “cloning.”
2. It creates documentation that includes a lot of unnecessary detail that, even if delivered and documented, doesn’t match the medical necessity of the visit, based on the history of present illness statements.
Both of these things can come back to bite you.
Zero in on the Risk
If your practice has an EHR, it is important that you evaluate whether certain workflow efficiency features are putting the practice at risk. You do not necessarily need to dump the EHR, but you may need to take action to reduce the risk of using these features.
In a pre-EHR practice, physicians began each visit with a blank piece of paper or dictated the entire visit. Then along came EHR vendors who, in an effort to make things easier and more efficient, created visit templates and the ability to “pull forward” the last visit note and use it as a basis for the current visit. The intention was always that physicians would modify it based on the current visit. But the reality is that physicians are busy, editing is time-consuming, and the unintended consequence is cloning.
“If you pull in unnecessary history or exam information from a previous visit that’s not relevant to the current visit, you can get dinged in an audit for not customizing the note to the patient’s specific presenting complaint,” LeGrand explains, “or, for attempting to bill a higher-level code by unintentionally padding the note with irrelevant information. What is documented for ‘reference’ has to be separated from what can be used to select the level of service.”
Your first documentation risk-reduction strategy is to review notes and look for signs of cloning.
LeGrand explains that a practice may be predisposed to cloning simply because of the way the EHR templates and workflow were set up when the system was implemented. “But,” she says, “‘the EHR made me do it’ defense won’t hold water, because it’s still the physician’s responsibility to customize or remove the information from templates and make the note unique to the visit.”
Yes, physician time is precious. But the reality is that the onus is on the physician to integrate EHR features with clinic workflow and to follow documentation rules.
The second documentation risk-reduction strategy is to make sure the level of evaluation and management (E/M) service billed is supported by medical necessity, not only by documentation artifacts that were relevant to the patient in the past but irrelevant to his or her current presenting complaint or condition.
“Medicare won’t pay for services that aren’t supported by medical necessity,” says LeGrand, “and you can’t achieve medical necessity by simply documenting additional E/M elements.”
This has always been the rule, LeGrand says. “But with the increased use of EHRs, and templates that automatically document visit elements and drive visits to a higher level of service, the Centers for Medicare & Medicaid Services [CMS] and private payers have added scrutiny to medical necessity reviews. They want to validate that higher-level visits billed indeed required a higher level of history and/or exam.”
To do this, the Office of the Inspector General (OIG) has supplemented its audit team with registered nurses. “The nurses assist certified coders by determining whether medical necessity has been met,” explains LeGrand.
Look at a patient who presents with toe pain. You take a detailed family history, conduct a review of systems (ROS), bill a high-level code, and document all the elements to support it. LeGrand explains, “There is no medical necessity to support doing an eye exam for a patient with toe pain in the absence of any other medical history, or performing a ROS to correlate an eye exam with toe pain. So, even if you do it and document it, the higher-level code won’t pass muster in an audit because the information documented is not medically necessary.”
According to LeGrand, the extent of the history and examination should be based on the presenting problem and the patient’s condition. “If an ankle sprain patient returns 2 weeks after the initial evaluation of the injury with a negative medical or surgical history, and the patient has been treated conservatively, it’s probably not necessary to conduct a ROS that includes 10 organ systems,” she says. “If your standard of care is to perform this level of service, no one will fault you for your care delivery; however, if you also choose a level of service based on this system review, without relevance to the presenting problem, and you bill a higher level of service than is supported by the nature of the presenting problem or the plan of care, the documentation probably won’t hold up in an audit where medical necessity is valued into the equation.”
On the other hand, LeGrand adds, if a patient presents to the emergency department after an automobile accident with an open fracture and other injuries, and the surgeon performs a complete ROS, the medical necessity would most likely be supported as the surgeon is preparing the patient for surgery.
Based on LeGrand’s work with practices, this distinction about medical necessity is news to many nonclinical billing staff. “They confuse medical necessity with medical decision-making, an E/M code documentation component, and incorrectly bill for a high-level visit because medical decision-making elements meet the documentation requirements—yet the code is not supported by medical necessity of the presenting problem.”
Talk with your billing team to make sure all staff members understand this critical difference. They must comprehend that the medically necessary level of service is determined by a number of clinical factors, not medical decision-making. Describe some of these clinical factors, which include, but are not limited to, chief complaint, clinical judgment, standards of practice, acute exacerbations/onsets of medical conditions or injuries, and comorbidities.
EHR Dos and Don’ts
LeGrand recommends the following best practices for using EHR documentation features:
1. DON’T simply cut and paste from a previous note. “This is what leads to verbose notes that have little to do with the patient you are documenting,” she says. “If you don’t cut and paste, you’ll avoid the root cause of this risk.”
2. DON’T pull forward information from previous visit notes that have nothing to do with the nature of the patient’s problem. “We understand that this takes extra time because physicians must review the previous note,” LeGrand says. “So if you don’t have time to review the past note, just don’t pull it forward. Start fresh with a new drop-down menu and select elements pertinent to the current visit. Or, dictate or type a note relevant to the current condition and presenting problems.”
How you choose to work this into your process will vary depending on which EHR system you use. “One surgeon I work with dictates everything because the drop-down menus and templates are cumbersome,” LeGrand says. “Some groups find it faster to use the EHR templates that they have customized. Others find their EHR’s point-and-click features most efficient for customizing quickly.”
3. DO customize your EHR visit templates if the use of templates is critical to your efficiency. “This is the most overlooked step in the EHR implementation process because it takes a fair amount of time to do,” LeGrand says. She suggests avoiding the use of multisystem examination templates created for medicine specialties altogether, and insists, “Don’t assume ‘that is how the vendor built it so we have to use it.’ Customize a template for each of your visit types so you can document in the EHR in the same fashion as when you used a paper system. Doing so will save you loads of documentation time.”
4. DO review your E/M code distribution. Generate a CPT frequency report for each physician and for the practice as a whole. Compare the data with state and national usage in orthopedics as a baseline. The American Academy of Orthopaedic Surgeon’s Code-X tool enables easy comparison of your practice’s E/M code usage with state and national data for orthopedics. Simply generate a CPT frequency report from your practice management system and enter the E/M data. Line graphs are automatically generated, making trends and patterns easy to see (Figure).
“Identify your outliers, pull charts randomly, and review the notes,” recommends LeGrand. “Make sure there is medical necessity for the level of code that’s been billed and that documentation supports it.”
You may be surprised to find you are an outlier on inpatient hospital codes, or your distribution of level-2 or -3 codes varies from your practice, state, or national data. Orthopedic surgeons don’t typically report high volumes of CPT codes 99204, 99205 or 99215, but if your practice does and you are an outlier, best to pay attention before someone else does.
5. DO select auditors with the right skill sets. Evaluating medical necessity in the note requires a clinical background. “If internal documentation reviews are conducted by the billing team, that’s fine,” LeGrand advises. “Just add a physician assistant or nurse to your internal review team. They can provide clinical oversight and review the note when necessary for medical necessity.”
If you are contracting with external auditors or consultants, verify auditor credentials and skill sets to ensure they can abstract and incorporate medical necessity into the review. “Auditors must be able to do more than count elements,” LeGrand says. “They must have clinical knowledge, and expertise in orthopedics is critical. This knowledge should be used to verify that medical necessity is present in every note.” LeGrand is quick to point out that not every note will be at risk, based on the amount of work performed and documented and the level of service billed. “But medical necessity must always be present.”
The addition of nurses to the OIG’s audit team is a big change and will refine the auditing process by adding more clinical scrutiny. The EHR documentation features are intended to improve efficiency, but only a clinician can determine and document unique visit elements and medical necessity.
Address these intersections of risk by ensuring your documentation meets medical necessity as well as E/M documentation elements. Conduct internal audits bi-annually to verify that E/M usage patterns align with peers and physician documentation is appropriate. And be sure there is clinical expertise on your audit team, whether it is internal or external. CMS now has it, and your practice should too. ◾
A group of 3 busy orthopedists attended coding education each year and did their best to accurately code and document their services. As a risk-reduction strategy, the group engaged our firm to conduct an audit to determine whether they were documenting their services properly and to provide feedback about how they could improve.
What we found was shocking to the surgeons, but all too common, as we review thousands of orthopedic visit notes every year: The same examination had been documented for all visits, with physicians stating in their notes that the examination was medically necessary. In addition, their documentation supported Current Procedural Terminology (CPT) code 99214 at every visit, with visit frequencies of 2 weeks to 4 months.
The culprit of all this sameness? The practice’s electronic health record (EHR).
“Practices with EHRs often have a large volume of visit notes that look almost identical for a patient who is seen for multiple visits,” explains Mary LeGrand, RN, MA, CCS-P, CPC, KarenZupko & Associates consultant and coding educator. “And that is putting physicians at higher risk of being audited or of not passing an audit.”
According to LeGrand, this is because physicians are using the practice’s EHR to “pull forward” the patient’s previous visit note for the current visit, but failing to customize it for the current visit. The unintended consequence of this workflow efficiency is twofold:
1. It creates documentation that looks strikingly similar to, if not exactly like, the patient’s last billed visit note. This is often referred to as note “cloning.”
2. It creates documentation that includes a lot of unnecessary detail that, even if delivered and documented, doesn’t match the medical necessity of the visit, based on the history of present illness statements.
Both of these things can come back to bite you.
Zero in on the Risk
If your practice has an EHR, it is important that you evaluate whether certain workflow efficiency features are putting the practice at risk. You do not necessarily need to dump the EHR, but you may need to take action to reduce the risk of using these features.
In a pre-EHR practice, physicians began each visit with a blank piece of paper or dictated the entire visit. Then along came EHR vendors who, in an effort to make things easier and more efficient, created visit templates and the ability to “pull forward” the last visit note and use it as a basis for the current visit. The intention was always that physicians would modify it based on the current visit. But the reality is that physicians are busy, editing is time-consuming, and the unintended consequence is cloning.
“If you pull in unnecessary history or exam information from a previous visit that’s not relevant to the current visit, you can get dinged in an audit for not customizing the note to the patient’s specific presenting complaint,” LeGrand explains, “or, for attempting to bill a higher-level code by unintentionally padding the note with irrelevant information. What is documented for ‘reference’ has to be separated from what can be used to select the level of service.”
Your first documentation risk-reduction strategy is to review notes and look for signs of cloning.
LeGrand explains that a practice may be predisposed to cloning simply because of the way the EHR templates and workflow were set up when the system was implemented. “But,” she says, “‘the EHR made me do it’ defense won’t hold water, because it’s still the physician’s responsibility to customize or remove the information from templates and make the note unique to the visit.”
Yes, physician time is precious. But the reality is that the onus is on the physician to integrate EHR features with clinic workflow and to follow documentation rules.
The second documentation risk-reduction strategy is to make sure the level of evaluation and management (E/M) service billed is supported by medical necessity, not only by documentation artifacts that were relevant to the patient in the past but irrelevant to his or her current presenting complaint or condition.
“Medicare won’t pay for services that aren’t supported by medical necessity,” says LeGrand, “and you can’t achieve medical necessity by simply documenting additional E/M elements.”
This has always been the rule, LeGrand says. “But with the increased use of EHRs, and templates that automatically document visit elements and drive visits to a higher level of service, the Centers for Medicare & Medicaid Services [CMS] and private payers have added scrutiny to medical necessity reviews. They want to validate that higher-level visits billed indeed required a higher level of history and/or exam.”
To do this, the Office of the Inspector General (OIG) has supplemented its audit team with registered nurses. “The nurses assist certified coders by determining whether medical necessity has been met,” explains LeGrand.
Look at a patient who presents with toe pain. You take a detailed family history, conduct a review of systems (ROS), bill a high-level code, and document all the elements to support it. LeGrand explains, “There is no medical necessity to support doing an eye exam for a patient with toe pain in the absence of any other medical history, or performing a ROS to correlate an eye exam with toe pain. So, even if you do it and document it, the higher-level code won’t pass muster in an audit because the information documented is not medically necessary.”
According to LeGrand, the extent of the history and examination should be based on the presenting problem and the patient’s condition. “If an ankle sprain patient returns 2 weeks after the initial evaluation of the injury with a negative medical or surgical history, and the patient has been treated conservatively, it’s probably not necessary to conduct a ROS that includes 10 organ systems,” she says. “If your standard of care is to perform this level of service, no one will fault you for your care delivery; however, if you also choose a level of service based on this system review, without relevance to the presenting problem, and you bill a higher level of service than is supported by the nature of the presenting problem or the plan of care, the documentation probably won’t hold up in an audit where medical necessity is valued into the equation.”
On the other hand, LeGrand adds, if a patient presents to the emergency department after an automobile accident with an open fracture and other injuries, and the surgeon performs a complete ROS, the medical necessity would most likely be supported as the surgeon is preparing the patient for surgery.
Based on LeGrand’s work with practices, this distinction about medical necessity is news to many nonclinical billing staff. “They confuse medical necessity with medical decision-making, an E/M code documentation component, and incorrectly bill for a high-level visit because medical decision-making elements meet the documentation requirements—yet the code is not supported by medical necessity of the presenting problem.”
Talk with your billing team to make sure all staff members understand this critical difference. They must comprehend that the medically necessary level of service is determined by a number of clinical factors, not medical decision-making. Describe some of these clinical factors, which include, but are not limited to, chief complaint, clinical judgment, standards of practice, acute exacerbations/onsets of medical conditions or injuries, and comorbidities.
EHR Dos and Don’ts
LeGrand recommends the following best practices for using EHR documentation features:
1. DON’T simply cut and paste from a previous note. “This is what leads to verbose notes that have little to do with the patient you are documenting,” she says. “If you don’t cut and paste, you’ll avoid the root cause of this risk.”
2. DON’T pull forward information from previous visit notes that have nothing to do with the nature of the patient’s problem. “We understand that this takes extra time because physicians must review the previous note,” LeGrand says. “So if you don’t have time to review the past note, just don’t pull it forward. Start fresh with a new drop-down menu and select elements pertinent to the current visit. Or, dictate or type a note relevant to the current condition and presenting problems.”
How you choose to work this into your process will vary depending on which EHR system you use. “One surgeon I work with dictates everything because the drop-down menus and templates are cumbersome,” LeGrand says. “Some groups find it faster to use the EHR templates that they have customized. Others find their EHR’s point-and-click features most efficient for customizing quickly.”
3. DO customize your EHR visit templates if the use of templates is critical to your efficiency. “This is the most overlooked step in the EHR implementation process because it takes a fair amount of time to do,” LeGrand says. She suggests avoiding the use of multisystem examination templates created for medicine specialties altogether, and insists, “Don’t assume ‘that is how the vendor built it so we have to use it.’ Customize a template for each of your visit types so you can document in the EHR in the same fashion as when you used a paper system. Doing so will save you loads of documentation time.”
4. DO review your E/M code distribution. Generate a CPT frequency report for each physician and for the practice as a whole. Compare the data with state and national usage in orthopedics as a baseline. The American Academy of Orthopaedic Surgeon’s Code-X tool enables easy comparison of your practice’s E/M code usage with state and national data for orthopedics. Simply generate a CPT frequency report from your practice management system and enter the E/M data. Line graphs are automatically generated, making trends and patterns easy to see (Figure).
“Identify your outliers, pull charts randomly, and review the notes,” recommends LeGrand. “Make sure there is medical necessity for the level of code that’s been billed and that documentation supports it.”
You may be surprised to find you are an outlier on inpatient hospital codes, or your distribution of level-2 or -3 codes varies from your practice, state, or national data. Orthopedic surgeons don’t typically report high volumes of CPT codes 99204, 99205 or 99215, but if your practice does and you are an outlier, best to pay attention before someone else does.
5. DO select auditors with the right skill sets. Evaluating medical necessity in the note requires a clinical background. “If internal documentation reviews are conducted by the billing team, that’s fine,” LeGrand advises. “Just add a physician assistant or nurse to your internal review team. They can provide clinical oversight and review the note when necessary for medical necessity.”
If you are contracting with external auditors or consultants, verify auditor credentials and skill sets to ensure they can abstract and incorporate medical necessity into the review. “Auditors must be able to do more than count elements,” LeGrand says. “They must have clinical knowledge, and expertise in orthopedics is critical. This knowledge should be used to verify that medical necessity is present in every note.” LeGrand is quick to point out that not every note will be at risk, based on the amount of work performed and documented and the level of service billed. “But medical necessity must always be present.”
The addition of nurses to the OIG’s audit team is a big change and will refine the auditing process by adding more clinical scrutiny. The EHR documentation features are intended to improve efficiency, but only a clinician can determine and document unique visit elements and medical necessity.
Address these intersections of risk by ensuring your documentation meets medical necessity as well as E/M documentation elements. Conduct internal audits bi-annually to verify that E/M usage patterns align with peers and physician documentation is appropriate. And be sure there is clinical expertise on your audit team, whether it is internal or external. CMS now has it, and your practice should too. ◾

Cementing Multihole, Metal, Modular Acetabular Shells Into Cages in Revision Total Hip Arthroplasty
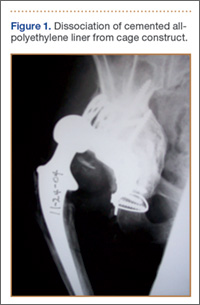
Although the number of total hip arthroplasties (THAs) being performed in the United States is increasing, revision THAs are more common.1 Many acetabular revisions can be successfully performed with standard or jumbo cementless acetabular cups, but major osseous deficiencies typically require reconstruction with a cage or cup/cage that bridges gaps in the pelvis and obtains fixation of the arthroplasty components.2,3 Cages and rings have been combined with all-polyethylene acetabular components (ie, all-polyethylene cups, or APCs) to reconstruct pelvic bone defects, but complications, including APC dissociation (Figure 1) and postoperative instability, can occur despite stable fixation of cage to pelvis.4 The incidence of dislocations with pelvic reconstruction rings using APCs has been reported to be 11%.4 If an APC has to be replaced because of wear, then major surgery may be required to extract the worn cup and cement a new cup in its place.
In this article, we describe a technique in which a metal, multihole acetabular shell is cemented into the cage or ring construct, avoiding some of the complications associated with traditional techniques by permitting use of a variety of liners.
Materials and Methods
We retrospectively reviewed the cases of all of Dr. Bolanos’ patients who underwent acetabular revision THA with cage reconstruction between February 1, 1998 and October 9, 2006. During this period, we were cementing a modular metal shell into the cage instead of an APC or polyethylene liner. All patients who underwent revision THA with cage reconstruction during the study period were included. Bone defects were treated with structural or morselized bone allograft. Every reconstruction involved use of an antiprotrusio cage or ring secured to the pelvis with screws, and a multihole acetabular shell cemented into place with a polyethylene liner applied. Elevated rims, lateralized liners, and constrained liners were used as needed to optimize stability. Femoral components were retained. Cage size was based on matching the osseous deficiencies. Shell size was determined by the inner diameter of the corresponding cage. Liner size was based on matching the shell and femoral head. During this period, none of the patients had other reconstructive techniques, such as trabecular metal augmentation, in combination with a modular acetabular shell, cup/cage reconstruction, or custom triflange components.
Patients engaged in protected weight-bearing ambulation for 3 months after surgery and were then permitted full, unrestricted activity. The primary outcome was mechanical failure of the reconstruction, or reoperation (Table). All reconstructions in this series consisted of acetabular revisions for aseptic loosening.
Surgical Technique
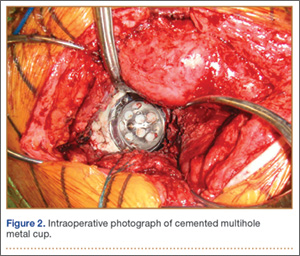
Six consecutive cases of pelvic discontinuity and 7 cases of segmental acetabular bone loss required use of cages or rings. Reconstruction cages were used to secure fixation to the ilium and ischium. With the technique described in this article, we used screws with rounded, prominent heads rather than flat heads between the cup and the cage or ring (Synthes, 6.5 mm) to ensure adequate cement mantle. The rounded screw heads were left prominent to approximate the function of cement pegs found on APCs. Screws were placed into the anterior, superior, medial, and posterior aspects of the cage to ensure adequate cement mantle between cup and cage. This was confirmed with trial placement of the cup into the cage before cementation and observation of the uniformity of the space between cup and cage. Trial placement also confirmed that the screws did not interfere with appropriate positioning of the cup. A multihole, metal acetabular cup was then cemented in the cage or ring such that cement extruded around the shell and into the holes of the cup and the cage, securing the cup to the cage. Use of a multihole, metal shell resulted in excellent cement fixation because the multiple holes created multiple circumferential cement pegs. Various liner options could then be used to optimize stability of the reconstruction. In some cases, excessive cement extruded into the interior aspect of the shell and hardened before curettage. If the excess cement could interfere with complete seating/locking of the liner, then a high-speed burr was used to easily remove cement (Figure 2). Polyethylene liners were then inserted into the shell. Femoral reconstruction was then performed, if needed, and stability of the arthroplasty checked. This technique allows the surgeon to then select from a variety of polyethylene liners as needed to optimize stability. Liners with elevated rims, lateralized liners, and constrained liners could be interchangeable options with this technique.
Results
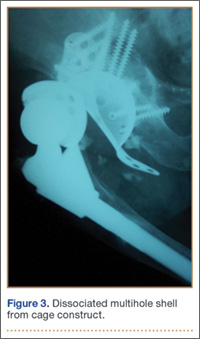
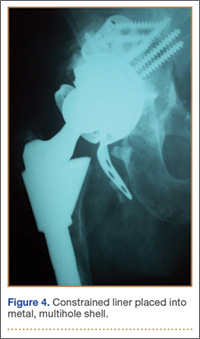
Thirteen patients with major osseous deficiencies of the pelvis were treated using this technique. At mean follow-up of 64.2 months (range, 3-133 months), 10 of the 13 patients had favorable outcomes without further surgery. One patient developed recurrent aseptic loosening that required re-revision, another patient developed recurrent instability that required acetabular liner and femoral head exchange, and a third patient with poor balance fell multiple times. This patient’s ninth fall resulted in dissociation of the acetabular shell from the cage (Figure 3), treated with placement of another cemented multihole metal shell with a standard liner. As dislocations recurred, the liner was changed to a constrained liner (Figure 4). The patient did not have any further dislocations or other hip-related problems. Integrity of cemented shell-cage fixation was maintained in 12 of the 13 patients at final follow-up.
Discussion
We have described a novel technique that facilitates reconstruction of major osseous deficiencies of the pelvis. The technique involves cementation of a multihole, metal acetabular shell into a cage or ring, permitting use of modular liners. The modularity in this approach to major hip reconstruction provides stability-optimization options that are not available with APCs. So far, the technique has demonstrated more advantages than disadvantages, so the indications for its use would be whenever a cage is used for pelvic reconstruction. Traditional techniques involve cementing an APC into the cage or ring. Use of multihole, metal shells for this purpose has several theoretical advantages. Multiple holes and the textured surface allow more interdigitation of cement with cup than APCs do; this interdigitation may improve the durability of the cemented interface. Cement also extrudes through the holes of the cage to secure the cup to the pelvis, as is done with cementation of APCs. Introduction of trabecular metal shells may also provide an even more secure bond to the shell, compared with APCs, though durability of a cemented trabecular metal interface has not been established. In addition, mechanical alignment guides cannot fasten as securely onto some APCs.
Nonmodular, cemented, metal-backed acetabular components, which were commonly used in hip arthroplasties at one time, were abandoned because of their relatively high loosening rate and because of advantages noted with modular components.5 The nonmodular components had been developed because of their theoretical advantages of improved distribution of forces into the cement mantle.5,6 However, those models had a relatively smooth metallic surface, which probably did not bond as well to cement as the shells used with the technique described in this article.
Dislocations can occur because of inadequately placed cups. Metallic cups can be improperly positioned, as can APCs. An advantage of the technique we have described over APCs is that liners with raised rims can be inserted with the apex placed wherever needed to best address instability. Dislocations can also occur because of factors such as inadequate offset and cognitive impairments. Our technique allows use of offset liners and constrained liners. Although these options may not prevent further dislocations, they often mitigate instability issues. Constrained liners and lateralized liners can be easily placed, and elevated rims can be swiveled as needed for stability. As use of cementless, metal-backed, modular acetabular components is common in primary THAs, most surgeons are familiar with the modular liner options available with use of the technique described in this article.
In this setting, modular, metal acetabular shells have the advantage of allowing surgeons to use the alignment guides they are accustomed to using. Modularity is another significant advantage over APCs. When an APC wears down, the component must be extracted to permit implantation of a new APC. With metal shells, a worn liner can be exchanged relatively easily. Modularity also gives surgeons many more options for addressing instability. Elevated rims can be moved, head sizes can be changed, and lateralized or constrained liners can be implanted easily. By comparison, with APCs, stability can be addressed only by modifying the femoral component or taking hip precautions which restrict range of motion of the hip. Modification of the femoral component is not possible with nonmodular femoral components in place (Figure 5). A potential disadvantage of this technique is increased cost associated with use of another component.
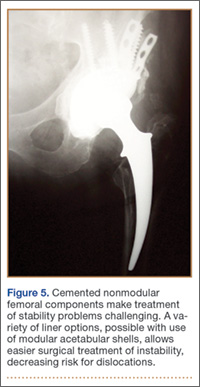
This small series of patients has had an excellent rate of success with cementation of multihole, metal-backed acetabular components into a cage or ring. These components may offer more secure fixation than APCs to cement extruded into the multiple holes, and improved metallurgy, such as trabecular metal. Surgeons who want to use modular components may prefer this technique because it allows them to select from various liner options. Surgeons should consider this technique for patients who need major pelvic reconstruction, though a larger study with longer follow-up is needed to determine its long-term durability.
Although the novel technique we have described has been helpful in our experience, this study had several limitations—small series, retrospective study, relatively short follow-up, lack of control group and functional data—that may have affected its conclusions. Further study and follow-up are needed to better determine the utility of this technique in clinical practice.
1. Kurtz SM, Ong KL, Schmier J, Zhao K, Mowat F, Lau E. Primary and revision arthroplasty surgery caseloads in the United States from 1990 to 2004. J Arthroplasty. 2009;24(2):195-203.
2. Berry DJ, Lewallen DG, Hanssen AD, Cabanela ME. Pelvic discontinuity in revision total hip arthroplasty. J Bone Joint Surg Am. 1999;81(12):1692-1702.
3. Pieringer H, Auersperg V, Böhler N. Reconstruction of severe acetabular bone-deficiency: the Burch-Schneider antiprotrusio cage in primary and revision total hip arthroplasty. J Arthroplasty. 2006;21(4):489-496.
4. Goodman S, Saastamoinen H, Shasha N, Gross A. Complications of ilioischial reconstruction rings in revision total hip arthroplasty. J Arthroplasty. 2004;19(4):436-446.
5. Cates HE, Faris PM, Keating EM, Ritter MA. Polyethylene wear in cemented metal-backed acetabular cups. J Bone Joint Surg Br. 1993;75(2):249-253.
6. Vasu R, Carter DR, Harris WH. Stress distribution in the acetabular region—I. Before and after total joint replacement. J Biomech. 1982;15(3):155-164.
Although the number of total hip arthroplasties (THAs) being performed in the United States is increasing, revision THAs are more common.1 Many acetabular revisions can be successfully performed with standard or jumbo cementless acetabular cups, but major osseous deficiencies typically require reconstruction with a cage or cup/cage that bridges gaps in the pelvis and obtains fixation of the arthroplasty components.2,3 Cages and rings have been combined with all-polyethylene acetabular components (ie, all-polyethylene cups, or APCs) to reconstruct pelvic bone defects, but complications, including APC dissociation (Figure 1) and postoperative instability, can occur despite stable fixation of cage to pelvis.4 The incidence of dislocations with pelvic reconstruction rings using APCs has been reported to be 11%.4 If an APC has to be replaced because of wear, then major surgery may be required to extract the worn cup and cement a new cup in its place.
In this article, we describe a technique in which a metal, multihole acetabular shell is cemented into the cage or ring construct, avoiding some of the complications associated with traditional techniques by permitting use of a variety of liners.
Materials and Methods
We retrospectively reviewed the cases of all of Dr. Bolanos’ patients who underwent acetabular revision THA with cage reconstruction between February 1, 1998 and October 9, 2006. During this period, we were cementing a modular metal shell into the cage instead of an APC or polyethylene liner. All patients who underwent revision THA with cage reconstruction during the study period were included. Bone defects were treated with structural or morselized bone allograft. Every reconstruction involved use of an antiprotrusio cage or ring secured to the pelvis with screws, and a multihole acetabular shell cemented into place with a polyethylene liner applied. Elevated rims, lateralized liners, and constrained liners were used as needed to optimize stability. Femoral components were retained. Cage size was based on matching the osseous deficiencies. Shell size was determined by the inner diameter of the corresponding cage. Liner size was based on matching the shell and femoral head. During this period, none of the patients had other reconstructive techniques, such as trabecular metal augmentation, in combination with a modular acetabular shell, cup/cage reconstruction, or custom triflange components.
Patients engaged in protected weight-bearing ambulation for 3 months after surgery and were then permitted full, unrestricted activity. The primary outcome was mechanical failure of the reconstruction, or reoperation (Table). All reconstructions in this series consisted of acetabular revisions for aseptic loosening.
Surgical Technique
Six consecutive cases of pelvic discontinuity and 7 cases of segmental acetabular bone loss required use of cages or rings. Reconstruction cages were used to secure fixation to the ilium and ischium. With the technique described in this article, we used screws with rounded, prominent heads rather than flat heads between the cup and the cage or ring (Synthes, 6.5 mm) to ensure adequate cement mantle. The rounded screw heads were left prominent to approximate the function of cement pegs found on APCs. Screws were placed into the anterior, superior, medial, and posterior aspects of the cage to ensure adequate cement mantle between cup and cage. This was confirmed with trial placement of the cup into the cage before cementation and observation of the uniformity of the space between cup and cage. Trial placement also confirmed that the screws did not interfere with appropriate positioning of the cup. A multihole, metal acetabular cup was then cemented in the cage or ring such that cement extruded around the shell and into the holes of the cup and the cage, securing the cup to the cage. Use of a multihole, metal shell resulted in excellent cement fixation because the multiple holes created multiple circumferential cement pegs. Various liner options could then be used to optimize stability of the reconstruction. In some cases, excessive cement extruded into the interior aspect of the shell and hardened before curettage. If the excess cement could interfere with complete seating/locking of the liner, then a high-speed burr was used to easily remove cement (Figure 2). Polyethylene liners were then inserted into the shell. Femoral reconstruction was then performed, if needed, and stability of the arthroplasty checked. This technique allows the surgeon to then select from a variety of polyethylene liners as needed to optimize stability. Liners with elevated rims, lateralized liners, and constrained liners could be interchangeable options with this technique.
Results
Thirteen patients with major osseous deficiencies of the pelvis were treated using this technique. At mean follow-up of 64.2 months (range, 3-133 months), 10 of the 13 patients had favorable outcomes without further surgery. One patient developed recurrent aseptic loosening that required re-revision, another patient developed recurrent instability that required acetabular liner and femoral head exchange, and a third patient with poor balance fell multiple times. This patient’s ninth fall resulted in dissociation of the acetabular shell from the cage (Figure 3), treated with placement of another cemented multihole metal shell with a standard liner. As dislocations recurred, the liner was changed to a constrained liner (Figure 4). The patient did not have any further dislocations or other hip-related problems. Integrity of cemented shell-cage fixation was maintained in 12 of the 13 patients at final follow-up.
Discussion
We have described a novel technique that facilitates reconstruction of major osseous deficiencies of the pelvis. The technique involves cementation of a multihole, metal acetabular shell into a cage or ring, permitting use of modular liners. The modularity in this approach to major hip reconstruction provides stability-optimization options that are not available with APCs. So far, the technique has demonstrated more advantages than disadvantages, so the indications for its use would be whenever a cage is used for pelvic reconstruction. Traditional techniques involve cementing an APC into the cage or ring. Use of multihole, metal shells for this purpose has several theoretical advantages. Multiple holes and the textured surface allow more interdigitation of cement with cup than APCs do; this interdigitation may improve the durability of the cemented interface. Cement also extrudes through the holes of the cage to secure the cup to the pelvis, as is done with cementation of APCs. Introduction of trabecular metal shells may also provide an even more secure bond to the shell, compared with APCs, though durability of a cemented trabecular metal interface has not been established. In addition, mechanical alignment guides cannot fasten as securely onto some APCs.
Nonmodular, cemented, metal-backed acetabular components, which were commonly used in hip arthroplasties at one time, were abandoned because of their relatively high loosening rate and because of advantages noted with modular components.5 The nonmodular components had been developed because of their theoretical advantages of improved distribution of forces into the cement mantle.5,6 However, those models had a relatively smooth metallic surface, which probably did not bond as well to cement as the shells used with the technique described in this article.
Dislocations can occur because of inadequately placed cups. Metallic cups can be improperly positioned, as can APCs. An advantage of the technique we have described over APCs is that liners with raised rims can be inserted with the apex placed wherever needed to best address instability. Dislocations can also occur because of factors such as inadequate offset and cognitive impairments. Our technique allows use of offset liners and constrained liners. Although these options may not prevent further dislocations, they often mitigate instability issues. Constrained liners and lateralized liners can be easily placed, and elevated rims can be swiveled as needed for stability. As use of cementless, metal-backed, modular acetabular components is common in primary THAs, most surgeons are familiar with the modular liner options available with use of the technique described in this article.
In this setting, modular, metal acetabular shells have the advantage of allowing surgeons to use the alignment guides they are accustomed to using. Modularity is another significant advantage over APCs. When an APC wears down, the component must be extracted to permit implantation of a new APC. With metal shells, a worn liner can be exchanged relatively easily. Modularity also gives surgeons many more options for addressing instability. Elevated rims can be moved, head sizes can be changed, and lateralized or constrained liners can be implanted easily. By comparison, with APCs, stability can be addressed only by modifying the femoral component or taking hip precautions which restrict range of motion of the hip. Modification of the femoral component is not possible with nonmodular femoral components in place (Figure 5). A potential disadvantage of this technique is increased cost associated with use of another component.
This small series of patients has had an excellent rate of success with cementation of multihole, metal-backed acetabular components into a cage or ring. These components may offer more secure fixation than APCs to cement extruded into the multiple holes, and improved metallurgy, such as trabecular metal. Surgeons who want to use modular components may prefer this technique because it allows them to select from various liner options. Surgeons should consider this technique for patients who need major pelvic reconstruction, though a larger study with longer follow-up is needed to determine its long-term durability.
Although the novel technique we have described has been helpful in our experience, this study had several limitations—small series, retrospective study, relatively short follow-up, lack of control group and functional data—that may have affected its conclusions. Further study and follow-up are needed to better determine the utility of this technique in clinical practice.
Although the number of total hip arthroplasties (THAs) being performed in the United States is increasing, revision THAs are more common.1 Many acetabular revisions can be successfully performed with standard or jumbo cementless acetabular cups, but major osseous deficiencies typically require reconstruction with a cage or cup/cage that bridges gaps in the pelvis and obtains fixation of the arthroplasty components.2,3 Cages and rings have been combined with all-polyethylene acetabular components (ie, all-polyethylene cups, or APCs) to reconstruct pelvic bone defects, but complications, including APC dissociation (Figure 1) and postoperative instability, can occur despite stable fixation of cage to pelvis.4 The incidence of dislocations with pelvic reconstruction rings using APCs has been reported to be 11%.4 If an APC has to be replaced because of wear, then major surgery may be required to extract the worn cup and cement a new cup in its place.
In this article, we describe a technique in which a metal, multihole acetabular shell is cemented into the cage or ring construct, avoiding some of the complications associated with traditional techniques by permitting use of a variety of liners.
Materials and Methods
We retrospectively reviewed the cases of all of Dr. Bolanos’ patients who underwent acetabular revision THA with cage reconstruction between February 1, 1998 and October 9, 2006. During this period, we were cementing a modular metal shell into the cage instead of an APC or polyethylene liner. All patients who underwent revision THA with cage reconstruction during the study period were included. Bone defects were treated with structural or morselized bone allograft. Every reconstruction involved use of an antiprotrusio cage or ring secured to the pelvis with screws, and a multihole acetabular shell cemented into place with a polyethylene liner applied. Elevated rims, lateralized liners, and constrained liners were used as needed to optimize stability. Femoral components were retained. Cage size was based on matching the osseous deficiencies. Shell size was determined by the inner diameter of the corresponding cage. Liner size was based on matching the shell and femoral head. During this period, none of the patients had other reconstructive techniques, such as trabecular metal augmentation, in combination with a modular acetabular shell, cup/cage reconstruction, or custom triflange components.
Patients engaged in protected weight-bearing ambulation for 3 months after surgery and were then permitted full, unrestricted activity. The primary outcome was mechanical failure of the reconstruction, or reoperation (Table). All reconstructions in this series consisted of acetabular revisions for aseptic loosening.
Surgical Technique
Six consecutive cases of pelvic discontinuity and 7 cases of segmental acetabular bone loss required use of cages or rings. Reconstruction cages were used to secure fixation to the ilium and ischium. With the technique described in this article, we used screws with rounded, prominent heads rather than flat heads between the cup and the cage or ring (Synthes, 6.5 mm) to ensure adequate cement mantle. The rounded screw heads were left prominent to approximate the function of cement pegs found on APCs. Screws were placed into the anterior, superior, medial, and posterior aspects of the cage to ensure adequate cement mantle between cup and cage. This was confirmed with trial placement of the cup into the cage before cementation and observation of the uniformity of the space between cup and cage. Trial placement also confirmed that the screws did not interfere with appropriate positioning of the cup. A multihole, metal acetabular cup was then cemented in the cage or ring such that cement extruded around the shell and into the holes of the cup and the cage, securing the cup to the cage. Use of a multihole, metal shell resulted in excellent cement fixation because the multiple holes created multiple circumferential cement pegs. Various liner options could then be used to optimize stability of the reconstruction. In some cases, excessive cement extruded into the interior aspect of the shell and hardened before curettage. If the excess cement could interfere with complete seating/locking of the liner, then a high-speed burr was used to easily remove cement (Figure 2). Polyethylene liners were then inserted into the shell. Femoral reconstruction was then performed, if needed, and stability of the arthroplasty checked. This technique allows the surgeon to then select from a variety of polyethylene liners as needed to optimize stability. Liners with elevated rims, lateralized liners, and constrained liners could be interchangeable options with this technique.
Results
Thirteen patients with major osseous deficiencies of the pelvis were treated using this technique. At mean follow-up of 64.2 months (range, 3-133 months), 10 of the 13 patients had favorable outcomes without further surgery. One patient developed recurrent aseptic loosening that required re-revision, another patient developed recurrent instability that required acetabular liner and femoral head exchange, and a third patient with poor balance fell multiple times. This patient’s ninth fall resulted in dissociation of the acetabular shell from the cage (Figure 3), treated with placement of another cemented multihole metal shell with a standard liner. As dislocations recurred, the liner was changed to a constrained liner (Figure 4). The patient did not have any further dislocations or other hip-related problems. Integrity of cemented shell-cage fixation was maintained in 12 of the 13 patients at final follow-up.
Discussion
We have described a novel technique that facilitates reconstruction of major osseous deficiencies of the pelvis. The technique involves cementation of a multihole, metal acetabular shell into a cage or ring, permitting use of modular liners. The modularity in this approach to major hip reconstruction provides stability-optimization options that are not available with APCs. So far, the technique has demonstrated more advantages than disadvantages, so the indications for its use would be whenever a cage is used for pelvic reconstruction. Traditional techniques involve cementing an APC into the cage or ring. Use of multihole, metal shells for this purpose has several theoretical advantages. Multiple holes and the textured surface allow more interdigitation of cement with cup than APCs do; this interdigitation may improve the durability of the cemented interface. Cement also extrudes through the holes of the cage to secure the cup to the pelvis, as is done with cementation of APCs. Introduction of trabecular metal shells may also provide an even more secure bond to the shell, compared with APCs, though durability of a cemented trabecular metal interface has not been established. In addition, mechanical alignment guides cannot fasten as securely onto some APCs.
Nonmodular, cemented, metal-backed acetabular components, which were commonly used in hip arthroplasties at one time, were abandoned because of their relatively high loosening rate and because of advantages noted with modular components.5 The nonmodular components had been developed because of their theoretical advantages of improved distribution of forces into the cement mantle.5,6 However, those models had a relatively smooth metallic surface, which probably did not bond as well to cement as the shells used with the technique described in this article.
Dislocations can occur because of inadequately placed cups. Metallic cups can be improperly positioned, as can APCs. An advantage of the technique we have described over APCs is that liners with raised rims can be inserted with the apex placed wherever needed to best address instability. Dislocations can also occur because of factors such as inadequate offset and cognitive impairments. Our technique allows use of offset liners and constrained liners. Although these options may not prevent further dislocations, they often mitigate instability issues. Constrained liners and lateralized liners can be easily placed, and elevated rims can be swiveled as needed for stability. As use of cementless, metal-backed, modular acetabular components is common in primary THAs, most surgeons are familiar with the modular liner options available with use of the technique described in this article.
In this setting, modular, metal acetabular shells have the advantage of allowing surgeons to use the alignment guides they are accustomed to using. Modularity is another significant advantage over APCs. When an APC wears down, the component must be extracted to permit implantation of a new APC. With metal shells, a worn liner can be exchanged relatively easily. Modularity also gives surgeons many more options for addressing instability. Elevated rims can be moved, head sizes can be changed, and lateralized or constrained liners can be implanted easily. By comparison, with APCs, stability can be addressed only by modifying the femoral component or taking hip precautions which restrict range of motion of the hip. Modification of the femoral component is not possible with nonmodular femoral components in place (Figure 5). A potential disadvantage of this technique is increased cost associated with use of another component.
This small series of patients has had an excellent rate of success with cementation of multihole, metal-backed acetabular components into a cage or ring. These components may offer more secure fixation than APCs to cement extruded into the multiple holes, and improved metallurgy, such as trabecular metal. Surgeons who want to use modular components may prefer this technique because it allows them to select from various liner options. Surgeons should consider this technique for patients who need major pelvic reconstruction, though a larger study with longer follow-up is needed to determine its long-term durability.
Although the novel technique we have described has been helpful in our experience, this study had several limitations—small series, retrospective study, relatively short follow-up, lack of control group and functional data—that may have affected its conclusions. Further study and follow-up are needed to better determine the utility of this technique in clinical practice.
1. Kurtz SM, Ong KL, Schmier J, Zhao K, Mowat F, Lau E. Primary and revision arthroplasty surgery caseloads in the United States from 1990 to 2004. J Arthroplasty. 2009;24(2):195-203.
2. Berry DJ, Lewallen DG, Hanssen AD, Cabanela ME. Pelvic discontinuity in revision total hip arthroplasty. J Bone Joint Surg Am. 1999;81(12):1692-1702.
3. Pieringer H, Auersperg V, Böhler N. Reconstruction of severe acetabular bone-deficiency: the Burch-Schneider antiprotrusio cage in primary and revision total hip arthroplasty. J Arthroplasty. 2006;21(4):489-496.
4. Goodman S, Saastamoinen H, Shasha N, Gross A. Complications of ilioischial reconstruction rings in revision total hip arthroplasty. J Arthroplasty. 2004;19(4):436-446.
5. Cates HE, Faris PM, Keating EM, Ritter MA. Polyethylene wear in cemented metal-backed acetabular cups. J Bone Joint Surg Br. 1993;75(2):249-253.
6. Vasu R, Carter DR, Harris WH. Stress distribution in the acetabular region—I. Before and after total joint replacement. J Biomech. 1982;15(3):155-164.
1. Kurtz SM, Ong KL, Schmier J, Zhao K, Mowat F, Lau E. Primary and revision arthroplasty surgery caseloads in the United States from 1990 to 2004. J Arthroplasty. 2009;24(2):195-203.
2. Berry DJ, Lewallen DG, Hanssen AD, Cabanela ME. Pelvic discontinuity in revision total hip arthroplasty. J Bone Joint Surg Am. 1999;81(12):1692-1702.
3. Pieringer H, Auersperg V, Böhler N. Reconstruction of severe acetabular bone-deficiency: the Burch-Schneider antiprotrusio cage in primary and revision total hip arthroplasty. J Arthroplasty. 2006;21(4):489-496.
4. Goodman S, Saastamoinen H, Shasha N, Gross A. Complications of ilioischial reconstruction rings in revision total hip arthroplasty. J Arthroplasty. 2004;19(4):436-446.
5. Cates HE, Faris PM, Keating EM, Ritter MA. Polyethylene wear in cemented metal-backed acetabular cups. J Bone Joint Surg Br. 1993;75(2):249-253.
6. Vasu R, Carter DR, Harris WH. Stress distribution in the acetabular region—I. Before and after total joint replacement. J Biomech. 1982;15(3):155-164.
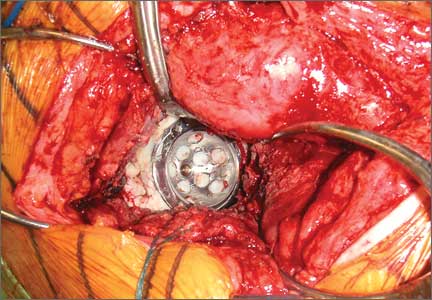
Recorrection Osteotomies and Total Knee Arthroplasties After Failed Bilateral High Tibial Osteotomies
High tibial osteotomy has proved successful in treating unicompartmental arthritis in young, active patients.1-3 However, this procedure fails over time because the other compartments deteriorate.4 The next step is conversion of the osteotomy to total knee arthroplasty (TKA). Conversion results vary, with several authors reporting poor outcomes5-9 and others reporting outcomes equal to those of primary TKA.10-14
The long-term success of TKA depends on proper restoration of the mechanical axis and soft-tissue balancing.15 Preexisting extra-articular deformity may adversely affect outcomes. A deformity of more than 15° may make it difficult to obtain intra-articular correction of an extra-articular deformity through soft-tissue balancing alone.16
In this article, we report the unique case of a patient whose bilateral high tibial osteotomies failed because of excessive extra-articular deformity. TKAs were performed consecutively, in 2 separate settings. Each TKA was combined with a recorrection tibial osteotomy in a single operation, allowing for re-creation of normal knee alignment with ligament balance. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 58-year-old man (weight, 250 pounds; body mass index, 30) underwent staged bilateral medial opening wedge osteotomies using distraction osteogenesis. A uniplanar external fixator was used for fixation on each knee. Before surgery, anatomical axis was 2° (right knee) and –1° (left knee) (Figure 1A), and tibial slope was 9° (right) and 8° (left) (Figures 1B, 1C). The procedures were performed 10 months apart. After surgery, anatomical alignment was 17° valgus (right knee) and 12° valgus (left knee) (Figure 2), and tibial slope was 20° (right) and 13° (left).
The patient received mild relief of his arthritis symptoms. Fifty-six months after the index operation, he decided to undergo conversion of the right high tibial osteotomy to TKA because of progressive painful arthritis of the knee. Excessive valgus alignment caused by the initial osteotomy raised concerns about being able to correct the extra-articular deformity intra-articularly while maintaining kinematic ligament balance. For this reason, a recorrection osteotomy was performed concurrently with the TKA. A posterior cruciate ligament–retaining (PCL-retaining) knee design (NexGen, Zimmer) was selected.
The procedure began with bone cuts for the TKA. Initial cuts were made on the femur. The tibial cut was made in valgus corresponding to the preoperative valgus deformity. The tibial recorrection osteotomy was made at the level of the original osteotomy site. A stemmed tibial component was used to cross the osteotomy site, correcting the valgus deformity and providing stability at the osteotomy site. A 3.5-mm locking compression T-plate (Synthes) was medially placed to prevent loss of correction and control rotation of the osteotomy during healing. The patient began range of motion on postoperative day 1. Continuous passive motion was not used. Protective weight-bearing continued for 6 weeks. After 6 weeks, and once there was radiograph evidence of healing at the osteotomy site, full weight-bearing was allowed.
After 4 months, the patient decided to undergo a similar procedure on the left knee. Postoperative rehabilitation was the same. A year after the bilateral TKAs, the patient maintained a Knee Society Score of 95 and a functional score of 90. After surgery, anatomical alignment was 6° (right knee) and 3° (left knee) (Figure 3), and tibial slope was 6° (right) and 7° (left) (Figures 4A, 4B). In each knee, the PCL was preserved with ligament balance.
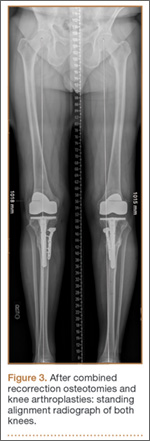
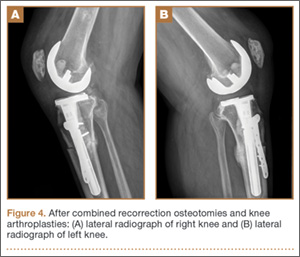
Discussion
Clinical outcomes of TKA after high tibial osteotomy vary. Windsor and colleagues9 reported that knee arthroplasties after tibial osteotomy were less successful than primary TKAs. In small studies, both Staeheli and colleagues17 and Katz and colleagues5 found that TKA outcomes after osteotomy were satisfactory compared with outcomes of primary TKA without previous osteotomy. A meta-analysis by Ramappa and colleagues18 showed no difference in outcomes between TKAs with and without previous osteotomy. In addition, there were no differences in outcomes between TKAs performed after opening wedge versus closing wedge osteotomies.19
An arthritic knee compartment is unloaded when a high tibial osteotomy produces an extra-articular deformity. Neyret and colleagues7 reported difficulties in correcting angulations of 9° or more through soft-tissue release. Cameron and Welsh16 suggested pre–knee arthroplasty correction of the extra-articular deformity for malalignments of more than 15°. In cases of severe malalignment produced by an osteotomy, Katz and colleagues5 also suggested that a second osteotomy be performed to correct alignment before TKA.
For TKA after high tibial osteotomy, a neutral plateau resection removes more bone medially than laterally, creating medial laxity. Without correction of the tibial deformity, lateral release (or, as Krackow and Holtgrewe20 advocated, medial advancement) is required for ligament stability. Both technically demanding options may not provide complete stability throughout the arc of motion. In addition, neither corrects for rotational or sagittal deformities (the concern with correcting an extra-articular deformity with intra-articular ligament balancing).
Another option is valgus tibial resection, which maintains native ligament balance at the cost of excessive valgus alignment. In the low-demand patient, a condylar constrained implant provides a means of correcting the malalignment with knee stability.8,13,17 The increased restraint produces greater forces at the implant–bone interface and may risk early loosening.
The case presented here represents a unique situation of failed bilateral high tibial osteotomies with excessive valgus malalignment. In a similar situation, Papagelopoulos and colleagues21 suggested correcting fracture deformities before or at time of knee arthroplasty. Yoshina and colleagues22 reported using a stemmed tibial component with TKA in treating nonunion of a high tibial osteotomy. As mentioned, Katz and colleagues5 and Neyret and colleagues7 suggested preoperative correction of the osteotomy in cases of severe malalignment. Others have suggested combining recorrection osteotomy and knee arthroplasty in either consecutive operations or a single operation.23-26 Wolff and colleagues27 and Uchinou and colleagues28 described recorrection osteotomy performed concurrent with TKA. The present article is the first to report a case involving concurrent bilateral recorrection osteotomy and TKA.
In one setting, the recorrection osteotomy is performed after the bony cuts are made for the TKA. The initial tibial plateau resection is performed in valgus at the same degree of malalignment as the osteotomy. This allows the plane of the tibial resection to parallel the floor once the recorrection is finished. With use of a tibial stem crossing the osteotomy site and a derotation plate, adequate fixation of the osteotomy is obtained. The recorrection osteotomy prevents the ligaments from overlengthening, allows the native ligament balance of the knee, and preserves the PCL—which lets the surgeon obtain ligament balance for the TKA throughout the arc of motion, avoiding midstance instabilities and achieving knee alignment rotationally and in the coronal and sagittal planes.
The TKA used in the present case was a PCL-retaining design. Both posterior-stabilized and PCL-retaining designs are reasonable options for use in combination with recorrection osteotomy. A stemmed tibial component is needed to cross the osteotomy site. In our patient’s case, use of a PCL-retaining design was based on surgeon preference and experience.
Patella infera has been noted as a problem in studies on converting high tibial osteotomy to TKA.9,12,29 A postulated cause is scarring of the infrapatellar tendon after high tibial osteotomy. In addition, a higher incidence of lateral retinacular release has been identified.9-11 Patella infera did not occur in either knee in the present case, and lateral release was not required.
Our patient’s lateral radiographs (Figures 4A, 4B) showed persistence of the osteotomy plane anterior to the tibia. The osteotomy healed posteriorly but not completely anteriorly. This raises the issue of risk for nonunion when recorrection osteotomy is performed with TKA. Use of a stemmed tibial implant with a derotation plate provides the benefit of intramedullary fixation for the recorrection osteotomy. If the recorrection osteotomy were performed in a separate setting before TKA, plate fixation would be the primary fixation option. Should nonunion occur at the recorrection osteotomy site, revision of the tibial plateau with a new stemmed implant would be required in combination with plate fixation. Madelaine and colleagues30 reported on a series of 15 severe varus knees treated with both osteotomy and TKA. Two nonunions occurred. Fixation was a staple in one case and a cement wedge in the other. Risk for nonunion may be reduced with the combination of stemmed tibial implant and internal fixation with a derotation plate. Protective weight-bearing is recommended for the first 6 postoperative weeks.
Conclusion
Ligament imbalances produced by high tibial osteotomy and exacerbated by conversion to TKA are difficult to address. In this report, we have described successful single-stage high tibial osteotomy recorrection and TKA performed bilaterally in separate settings. With use of a stemmed tibial component and a derotation plate, solid fixation was obtained with an excellent clinical outcome. The malalignment was corrected while ligament balance was maintained for a PCL-retaining TKA design.
1. Billings A, Scott DF, Camargo MP, Hofmann AA. High tibial osteotomy with a calibrated osteotomy guide, rigid internal fixation, and early motion. Long-term follow-up. J Bone Joint Surg Am. 2000;82(1):70-79.
2. Coventry MB, Ilstrup DM, Wallrichs SL. Proximal tibial osteotomy. A critical long-term study of eighty-seven cases. J Bone Joint Surg Am. 1993;75(2):196-201.
3. Rinonapoli E, Mancini GB, Corvaglia A, Musiello S. Tibial osteotomy for varus gonarthrosis. A 10- to 21-year followup study. Clin Orthop Relat Res. 1998;(353):185-193.
4. Ritter MA, Fechtman RA. Proximal tibial osteotomy. A survivorship analysis. J Arthroplasty. 1988;3(4):309-311.
5. Katz MM, Hungerford DS, Krackow KA, Lennox DW. Results of total knee arthroplasty after failed proximal tibial osteotomy for osteoarthritis. J Bone Joint Surg Am. 1987;69(2):225-233.
6. Mont MA, Antonaides S, Krackow KA, Hungerford DS. Total knee arthroplasty after failed high tibial osteotomy. A comparison with a matched group. Clin Orthop Relat Res. 1994;299:125-130.
7. Neyret P, Deroche P, Deschamps G, Dejour H. Total knee replacement after valgus tibial osteotomy. Technical problems [in French]. Rev Chir Orthop Reparatrice Appar Mot. 1992;78(7):438-448.
8. Parvizi J, Hanssen AD, Spangehl MJ. Total knee arthroplasty following proximal tibial osteotomy: risk factors for failure. J Bone Joint Surg Am. 2004;86(3):474-479.
9. Windsor RE, Insall JN, Vince KG. Technical considerations of total knee arthroplasty after proximal tibial osteotomy. J Bone Joint Surg Am. 1988;70(4):547-555.
10. Amendola A, Rorabeck CH, Bourne RB, Apyan PM. Total knee arthroplasty following high tibial osteotomy for osteoarthritis. J Arthroplasty. 1989;(4 suppl):S11-S17.
11. Kazakos KJ, Chatzipapas C, Verettas D, Galanis V, Xarchas KC, Psillakis I. Mid-term results of total knee arthroplasty after high tibial osteotomy. Arch Orthop Trauma Surg. 2008;128(2):167-173.
12. Meding JB, Keating EM, Ritter MA, Faris PM. Total knee arthroplasty after high tibial osteotomy. A comparison study in patients who had bilateral total knee replacement. J Bone Joint Surg Am. 2000;82(9):1252-1259.
13. Niinimaki T, Eskelinen A, Ohtonen P, Puhto AP, Mann BS, Leppilahti J. Total knee arthroplasty after high tibial osteotomy: a registry-based case–control study of 1,036 knees. Arch Orthop Trauma Surg. 2014;134(1):73-77.
14. van Raaij TM, Reijman M, Furlan AD, Verhaar JA. Total knee arthroplasty after high tibial osteotomy. A systematic review. BMC Musculoskelet Disord. 2009;10:88-98.
15. Lotke PA, Ecker ML. Influence of positioning of prosthesis in total knee replacement. J Bone Joint Surg Am. 1977;59(1):77-79.
16. Cameron HU, Welsh RP. Potential complications of total knee replacement following tibial osteotomy. Orthop Rev. 1988;17(1):39-43.
17. Staeheli JW, Cass JR, Morrey BF. Condylar total knee arthroplasty after failed proximal tibial osteotomy. J Bone Joint Surg Am. 1987;69(1):28-31.
18. Ramappa M, Anand S, Jennings A. Total knee replacement following high tibial osteotomy versus total knee replacement without high tibial osteotomy: a systematic review and meta analysis. Arch Orthop Trauma Surg. 2013;133(11):1587-1593.
19. Preston S, Howard J, Naudie D, Somerville L, McAuley J. Total knee arthroplasty after high tibial osteotomy: no differences between medial and lateral osteotomy approaches. Clin Orthop Relat Res. 2014;472(1):105-110.
20. Krackow KA, Holtgrewe JL. Experience with a new technique for managing severely overcorrected valgus high tibial osteotomy at total knee arthroplasty. Clin Orthop Relat Res. 1990;(258):213-224.
21. Papagelopoulos PJ, Karachalios T, Themistocleous GS, Papadopoulos ECh, Savvidou OD, Rand JA. Total knee arthroplasty in patients with pre-existing fracture deformity. Orthopaedics. 2007;30(5):373-378.
22. Yoshina N, Takai S, Watanabe Y, Nakamura S, Kubo T. Total knee arthroplasty with long stem for treatment of nonunion after high tibial osteotomy. J Arthroplasty. 2004;19(4):528-531.
23. Mont MA, Alexander N, Krackow KA, Hungerford DS. Total knee arthroplasty after failed high tibial osteotomy. Orthop Clin North Am. 1994;25(3):515-525.
24. Scott WN. Insall & Scott’s Surgery of the Knee. Vol 1. 4th ed. Philadelphia, PA: Churchill Livingstone Elsevier; 2006.
25. Gill T, Schemitsch EH, Brick GW, Thornhill TS. Revision total knee arthroplasty after failed unicompartmental knee arthroplasty or high tibial osteotomy. Clin Orthop Relat Res. 1995;(321):10-18.
26. Figgie HE 3rd, Goldberg VM, Heiple KG, Moller HS 3rd, Gordon NH. The influence of tibial-patellofemoral location on function of the knee in patients with the posterior stabilized condylar knee prosthesis. J Bone Joint Surg Am. 1986;68(7):1035-1040.
27. Wolff AM, Hungerford DS, Pepe CL. The effect of extraarticular varus and valgus deformity on total knee arthroplasty. Clin Orthop Relat Res. 1994;(271):35-51.
28. Uchinou S, Yano H, Shimizu K, Masumi S. A severely overcorrected high tibial osteotomy: revision by osteotomy and a long stem component. Acta Orthop Scand. 1996;67(2):193-194.
29. Noda T, Yasuda S, Nagano K, Takahara Y, Namba Y, Inoue H. Clinico-radiological study of total knee arthroplasty after high tibial osteotomy. J Orthop Sci. 2000;5(1):25-36.
30. Madelaine A, Villa V, Yela C, et al. Results and complications of single-stage total knee arthroplasty and high tibial osteotomy. Int Orthop. 2014;38(10):2091-2098.
High tibial osteotomy has proved successful in treating unicompartmental arthritis in young, active patients.1-3 However, this procedure fails over time because the other compartments deteriorate.4 The next step is conversion of the osteotomy to total knee arthroplasty (TKA). Conversion results vary, with several authors reporting poor outcomes5-9 and others reporting outcomes equal to those of primary TKA.10-14
The long-term success of TKA depends on proper restoration of the mechanical axis and soft-tissue balancing.15 Preexisting extra-articular deformity may adversely affect outcomes. A deformity of more than 15° may make it difficult to obtain intra-articular correction of an extra-articular deformity through soft-tissue balancing alone.16
In this article, we report the unique case of a patient whose bilateral high tibial osteotomies failed because of excessive extra-articular deformity. TKAs were performed consecutively, in 2 separate settings. Each TKA was combined with a recorrection tibial osteotomy in a single operation, allowing for re-creation of normal knee alignment with ligament balance. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 58-year-old man (weight, 250 pounds; body mass index, 30) underwent staged bilateral medial opening wedge osteotomies using distraction osteogenesis. A uniplanar external fixator was used for fixation on each knee. Before surgery, anatomical axis was 2° (right knee) and –1° (left knee) (Figure 1A), and tibial slope was 9° (right) and 8° (left) (Figures 1B, 1C). The procedures were performed 10 months apart. After surgery, anatomical alignment was 17° valgus (right knee) and 12° valgus (left knee) (Figure 2), and tibial slope was 20° (right) and 13° (left).
The patient received mild relief of his arthritis symptoms. Fifty-six months after the index operation, he decided to undergo conversion of the right high tibial osteotomy to TKA because of progressive painful arthritis of the knee. Excessive valgus alignment caused by the initial osteotomy raised concerns about being able to correct the extra-articular deformity intra-articularly while maintaining kinematic ligament balance. For this reason, a recorrection osteotomy was performed concurrently with the TKA. A posterior cruciate ligament–retaining (PCL-retaining) knee design (NexGen, Zimmer) was selected.
The procedure began with bone cuts for the TKA. Initial cuts were made on the femur. The tibial cut was made in valgus corresponding to the preoperative valgus deformity. The tibial recorrection osteotomy was made at the level of the original osteotomy site. A stemmed tibial component was used to cross the osteotomy site, correcting the valgus deformity and providing stability at the osteotomy site. A 3.5-mm locking compression T-plate (Synthes) was medially placed to prevent loss of correction and control rotation of the osteotomy during healing. The patient began range of motion on postoperative day 1. Continuous passive motion was not used. Protective weight-bearing continued for 6 weeks. After 6 weeks, and once there was radiograph evidence of healing at the osteotomy site, full weight-bearing was allowed.
After 4 months, the patient decided to undergo a similar procedure on the left knee. Postoperative rehabilitation was the same. A year after the bilateral TKAs, the patient maintained a Knee Society Score of 95 and a functional score of 90. After surgery, anatomical alignment was 6° (right knee) and 3° (left knee) (Figure 3), and tibial slope was 6° (right) and 7° (left) (Figures 4A, 4B). In each knee, the PCL was preserved with ligament balance.
Discussion
Clinical outcomes of TKA after high tibial osteotomy vary. Windsor and colleagues9 reported that knee arthroplasties after tibial osteotomy were less successful than primary TKAs. In small studies, both Staeheli and colleagues17 and Katz and colleagues5 found that TKA outcomes after osteotomy were satisfactory compared with outcomes of primary TKA without previous osteotomy. A meta-analysis by Ramappa and colleagues18 showed no difference in outcomes between TKAs with and without previous osteotomy. In addition, there were no differences in outcomes between TKAs performed after opening wedge versus closing wedge osteotomies.19
An arthritic knee compartment is unloaded when a high tibial osteotomy produces an extra-articular deformity. Neyret and colleagues7 reported difficulties in correcting angulations of 9° or more through soft-tissue release. Cameron and Welsh16 suggested pre–knee arthroplasty correction of the extra-articular deformity for malalignments of more than 15°. In cases of severe malalignment produced by an osteotomy, Katz and colleagues5 also suggested that a second osteotomy be performed to correct alignment before TKA.
For TKA after high tibial osteotomy, a neutral plateau resection removes more bone medially than laterally, creating medial laxity. Without correction of the tibial deformity, lateral release (or, as Krackow and Holtgrewe20 advocated, medial advancement) is required for ligament stability. Both technically demanding options may not provide complete stability throughout the arc of motion. In addition, neither corrects for rotational or sagittal deformities (the concern with correcting an extra-articular deformity with intra-articular ligament balancing).
Another option is valgus tibial resection, which maintains native ligament balance at the cost of excessive valgus alignment. In the low-demand patient, a condylar constrained implant provides a means of correcting the malalignment with knee stability.8,13,17 The increased restraint produces greater forces at the implant–bone interface and may risk early loosening.
The case presented here represents a unique situation of failed bilateral high tibial osteotomies with excessive valgus malalignment. In a similar situation, Papagelopoulos and colleagues21 suggested correcting fracture deformities before or at time of knee arthroplasty. Yoshina and colleagues22 reported using a stemmed tibial component with TKA in treating nonunion of a high tibial osteotomy. As mentioned, Katz and colleagues5 and Neyret and colleagues7 suggested preoperative correction of the osteotomy in cases of severe malalignment. Others have suggested combining recorrection osteotomy and knee arthroplasty in either consecutive operations or a single operation.23-26 Wolff and colleagues27 and Uchinou and colleagues28 described recorrection osteotomy performed concurrent with TKA. The present article is the first to report a case involving concurrent bilateral recorrection osteotomy and TKA.
In one setting, the recorrection osteotomy is performed after the bony cuts are made for the TKA. The initial tibial plateau resection is performed in valgus at the same degree of malalignment as the osteotomy. This allows the plane of the tibial resection to parallel the floor once the recorrection is finished. With use of a tibial stem crossing the osteotomy site and a derotation plate, adequate fixation of the osteotomy is obtained. The recorrection osteotomy prevents the ligaments from overlengthening, allows the native ligament balance of the knee, and preserves the PCL—which lets the surgeon obtain ligament balance for the TKA throughout the arc of motion, avoiding midstance instabilities and achieving knee alignment rotationally and in the coronal and sagittal planes.
The TKA used in the present case was a PCL-retaining design. Both posterior-stabilized and PCL-retaining designs are reasonable options for use in combination with recorrection osteotomy. A stemmed tibial component is needed to cross the osteotomy site. In our patient’s case, use of a PCL-retaining design was based on surgeon preference and experience.
Patella infera has been noted as a problem in studies on converting high tibial osteotomy to TKA.9,12,29 A postulated cause is scarring of the infrapatellar tendon after high tibial osteotomy. In addition, a higher incidence of lateral retinacular release has been identified.9-11 Patella infera did not occur in either knee in the present case, and lateral release was not required.
Our patient’s lateral radiographs (Figures 4A, 4B) showed persistence of the osteotomy plane anterior to the tibia. The osteotomy healed posteriorly but not completely anteriorly. This raises the issue of risk for nonunion when recorrection osteotomy is performed with TKA. Use of a stemmed tibial implant with a derotation plate provides the benefit of intramedullary fixation for the recorrection osteotomy. If the recorrection osteotomy were performed in a separate setting before TKA, plate fixation would be the primary fixation option. Should nonunion occur at the recorrection osteotomy site, revision of the tibial plateau with a new stemmed implant would be required in combination with plate fixation. Madelaine and colleagues30 reported on a series of 15 severe varus knees treated with both osteotomy and TKA. Two nonunions occurred. Fixation was a staple in one case and a cement wedge in the other. Risk for nonunion may be reduced with the combination of stemmed tibial implant and internal fixation with a derotation plate. Protective weight-bearing is recommended for the first 6 postoperative weeks.
Conclusion
Ligament imbalances produced by high tibial osteotomy and exacerbated by conversion to TKA are difficult to address. In this report, we have described successful single-stage high tibial osteotomy recorrection and TKA performed bilaterally in separate settings. With use of a stemmed tibial component and a derotation plate, solid fixation was obtained with an excellent clinical outcome. The malalignment was corrected while ligament balance was maintained for a PCL-retaining TKA design.
High tibial osteotomy has proved successful in treating unicompartmental arthritis in young, active patients.1-3 However, this procedure fails over time because the other compartments deteriorate.4 The next step is conversion of the osteotomy to total knee arthroplasty (TKA). Conversion results vary, with several authors reporting poor outcomes5-9 and others reporting outcomes equal to those of primary TKA.10-14
The long-term success of TKA depends on proper restoration of the mechanical axis and soft-tissue balancing.15 Preexisting extra-articular deformity may adversely affect outcomes. A deformity of more than 15° may make it difficult to obtain intra-articular correction of an extra-articular deformity through soft-tissue balancing alone.16
In this article, we report the unique case of a patient whose bilateral high tibial osteotomies failed because of excessive extra-articular deformity. TKAs were performed consecutively, in 2 separate settings. Each TKA was combined with a recorrection tibial osteotomy in a single operation, allowing for re-creation of normal knee alignment with ligament balance. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 58-year-old man (weight, 250 pounds; body mass index, 30) underwent staged bilateral medial opening wedge osteotomies using distraction osteogenesis. A uniplanar external fixator was used for fixation on each knee. Before surgery, anatomical axis was 2° (right knee) and –1° (left knee) (Figure 1A), and tibial slope was 9° (right) and 8° (left) (Figures 1B, 1C). The procedures were performed 10 months apart. After surgery, anatomical alignment was 17° valgus (right knee) and 12° valgus (left knee) (Figure 2), and tibial slope was 20° (right) and 13° (left).
The patient received mild relief of his arthritis symptoms. Fifty-six months after the index operation, he decided to undergo conversion of the right high tibial osteotomy to TKA because of progressive painful arthritis of the knee. Excessive valgus alignment caused by the initial osteotomy raised concerns about being able to correct the extra-articular deformity intra-articularly while maintaining kinematic ligament balance. For this reason, a recorrection osteotomy was performed concurrently with the TKA. A posterior cruciate ligament–retaining (PCL-retaining) knee design (NexGen, Zimmer) was selected.
The procedure began with bone cuts for the TKA. Initial cuts were made on the femur. The tibial cut was made in valgus corresponding to the preoperative valgus deformity. The tibial recorrection osteotomy was made at the level of the original osteotomy site. A stemmed tibial component was used to cross the osteotomy site, correcting the valgus deformity and providing stability at the osteotomy site. A 3.5-mm locking compression T-plate (Synthes) was medially placed to prevent loss of correction and control rotation of the osteotomy during healing. The patient began range of motion on postoperative day 1. Continuous passive motion was not used. Protective weight-bearing continued for 6 weeks. After 6 weeks, and once there was radiograph evidence of healing at the osteotomy site, full weight-bearing was allowed.
After 4 months, the patient decided to undergo a similar procedure on the left knee. Postoperative rehabilitation was the same. A year after the bilateral TKAs, the patient maintained a Knee Society Score of 95 and a functional score of 90. After surgery, anatomical alignment was 6° (right knee) and 3° (left knee) (Figure 3), and tibial slope was 6° (right) and 7° (left) (Figures 4A, 4B). In each knee, the PCL was preserved with ligament balance.
Discussion
Clinical outcomes of TKA after high tibial osteotomy vary. Windsor and colleagues9 reported that knee arthroplasties after tibial osteotomy were less successful than primary TKAs. In small studies, both Staeheli and colleagues17 and Katz and colleagues5 found that TKA outcomes after osteotomy were satisfactory compared with outcomes of primary TKA without previous osteotomy. A meta-analysis by Ramappa and colleagues18 showed no difference in outcomes between TKAs with and without previous osteotomy. In addition, there were no differences in outcomes between TKAs performed after opening wedge versus closing wedge osteotomies.19
An arthritic knee compartment is unloaded when a high tibial osteotomy produces an extra-articular deformity. Neyret and colleagues7 reported difficulties in correcting angulations of 9° or more through soft-tissue release. Cameron and Welsh16 suggested pre–knee arthroplasty correction of the extra-articular deformity for malalignments of more than 15°. In cases of severe malalignment produced by an osteotomy, Katz and colleagues5 also suggested that a second osteotomy be performed to correct alignment before TKA.
For TKA after high tibial osteotomy, a neutral plateau resection removes more bone medially than laterally, creating medial laxity. Without correction of the tibial deformity, lateral release (or, as Krackow and Holtgrewe20 advocated, medial advancement) is required for ligament stability. Both technically demanding options may not provide complete stability throughout the arc of motion. In addition, neither corrects for rotational or sagittal deformities (the concern with correcting an extra-articular deformity with intra-articular ligament balancing).
Another option is valgus tibial resection, which maintains native ligament balance at the cost of excessive valgus alignment. In the low-demand patient, a condylar constrained implant provides a means of correcting the malalignment with knee stability.8,13,17 The increased restraint produces greater forces at the implant–bone interface and may risk early loosening.
The case presented here represents a unique situation of failed bilateral high tibial osteotomies with excessive valgus malalignment. In a similar situation, Papagelopoulos and colleagues21 suggested correcting fracture deformities before or at time of knee arthroplasty. Yoshina and colleagues22 reported using a stemmed tibial component with TKA in treating nonunion of a high tibial osteotomy. As mentioned, Katz and colleagues5 and Neyret and colleagues7 suggested preoperative correction of the osteotomy in cases of severe malalignment. Others have suggested combining recorrection osteotomy and knee arthroplasty in either consecutive operations or a single operation.23-26 Wolff and colleagues27 and Uchinou and colleagues28 described recorrection osteotomy performed concurrent with TKA. The present article is the first to report a case involving concurrent bilateral recorrection osteotomy and TKA.
In one setting, the recorrection osteotomy is performed after the bony cuts are made for the TKA. The initial tibial plateau resection is performed in valgus at the same degree of malalignment as the osteotomy. This allows the plane of the tibial resection to parallel the floor once the recorrection is finished. With use of a tibial stem crossing the osteotomy site and a derotation plate, adequate fixation of the osteotomy is obtained. The recorrection osteotomy prevents the ligaments from overlengthening, allows the native ligament balance of the knee, and preserves the PCL—which lets the surgeon obtain ligament balance for the TKA throughout the arc of motion, avoiding midstance instabilities and achieving knee alignment rotationally and in the coronal and sagittal planes.
The TKA used in the present case was a PCL-retaining design. Both posterior-stabilized and PCL-retaining designs are reasonable options for use in combination with recorrection osteotomy. A stemmed tibial component is needed to cross the osteotomy site. In our patient’s case, use of a PCL-retaining design was based on surgeon preference and experience.
Patella infera has been noted as a problem in studies on converting high tibial osteotomy to TKA.9,12,29 A postulated cause is scarring of the infrapatellar tendon after high tibial osteotomy. In addition, a higher incidence of lateral retinacular release has been identified.9-11 Patella infera did not occur in either knee in the present case, and lateral release was not required.
Our patient’s lateral radiographs (Figures 4A, 4B) showed persistence of the osteotomy plane anterior to the tibia. The osteotomy healed posteriorly but not completely anteriorly. This raises the issue of risk for nonunion when recorrection osteotomy is performed with TKA. Use of a stemmed tibial implant with a derotation plate provides the benefit of intramedullary fixation for the recorrection osteotomy. If the recorrection osteotomy were performed in a separate setting before TKA, plate fixation would be the primary fixation option. Should nonunion occur at the recorrection osteotomy site, revision of the tibial plateau with a new stemmed implant would be required in combination with plate fixation. Madelaine and colleagues30 reported on a series of 15 severe varus knees treated with both osteotomy and TKA. Two nonunions occurred. Fixation was a staple in one case and a cement wedge in the other. Risk for nonunion may be reduced with the combination of stemmed tibial implant and internal fixation with a derotation plate. Protective weight-bearing is recommended for the first 6 postoperative weeks.
Conclusion
Ligament imbalances produced by high tibial osteotomy and exacerbated by conversion to TKA are difficult to address. In this report, we have described successful single-stage high tibial osteotomy recorrection and TKA performed bilaterally in separate settings. With use of a stemmed tibial component and a derotation plate, solid fixation was obtained with an excellent clinical outcome. The malalignment was corrected while ligament balance was maintained for a PCL-retaining TKA design.
1. Billings A, Scott DF, Camargo MP, Hofmann AA. High tibial osteotomy with a calibrated osteotomy guide, rigid internal fixation, and early motion. Long-term follow-up. J Bone Joint Surg Am. 2000;82(1):70-79.
2. Coventry MB, Ilstrup DM, Wallrichs SL. Proximal tibial osteotomy. A critical long-term study of eighty-seven cases. J Bone Joint Surg Am. 1993;75(2):196-201.
3. Rinonapoli E, Mancini GB, Corvaglia A, Musiello S. Tibial osteotomy for varus gonarthrosis. A 10- to 21-year followup study. Clin Orthop Relat Res. 1998;(353):185-193.
4. Ritter MA, Fechtman RA. Proximal tibial osteotomy. A survivorship analysis. J Arthroplasty. 1988;3(4):309-311.
5. Katz MM, Hungerford DS, Krackow KA, Lennox DW. Results of total knee arthroplasty after failed proximal tibial osteotomy for osteoarthritis. J Bone Joint Surg Am. 1987;69(2):225-233.
6. Mont MA, Antonaides S, Krackow KA, Hungerford DS. Total knee arthroplasty after failed high tibial osteotomy. A comparison with a matched group. Clin Orthop Relat Res. 1994;299:125-130.
7. Neyret P, Deroche P, Deschamps G, Dejour H. Total knee replacement after valgus tibial osteotomy. Technical problems [in French]. Rev Chir Orthop Reparatrice Appar Mot. 1992;78(7):438-448.
8. Parvizi J, Hanssen AD, Spangehl MJ. Total knee arthroplasty following proximal tibial osteotomy: risk factors for failure. J Bone Joint Surg Am. 2004;86(3):474-479.
9. Windsor RE, Insall JN, Vince KG. Technical considerations of total knee arthroplasty after proximal tibial osteotomy. J Bone Joint Surg Am. 1988;70(4):547-555.
10. Amendola A, Rorabeck CH, Bourne RB, Apyan PM. Total knee arthroplasty following high tibial osteotomy for osteoarthritis. J Arthroplasty. 1989;(4 suppl):S11-S17.
11. Kazakos KJ, Chatzipapas C, Verettas D, Galanis V, Xarchas KC, Psillakis I. Mid-term results of total knee arthroplasty after high tibial osteotomy. Arch Orthop Trauma Surg. 2008;128(2):167-173.
12. Meding JB, Keating EM, Ritter MA, Faris PM. Total knee arthroplasty after high tibial osteotomy. A comparison study in patients who had bilateral total knee replacement. J Bone Joint Surg Am. 2000;82(9):1252-1259.
13. Niinimaki T, Eskelinen A, Ohtonen P, Puhto AP, Mann BS, Leppilahti J. Total knee arthroplasty after high tibial osteotomy: a registry-based case–control study of 1,036 knees. Arch Orthop Trauma Surg. 2014;134(1):73-77.
14. van Raaij TM, Reijman M, Furlan AD, Verhaar JA. Total knee arthroplasty after high tibial osteotomy. A systematic review. BMC Musculoskelet Disord. 2009;10:88-98.
15. Lotke PA, Ecker ML. Influence of positioning of prosthesis in total knee replacement. J Bone Joint Surg Am. 1977;59(1):77-79.
16. Cameron HU, Welsh RP. Potential complications of total knee replacement following tibial osteotomy. Orthop Rev. 1988;17(1):39-43.
17. Staeheli JW, Cass JR, Morrey BF. Condylar total knee arthroplasty after failed proximal tibial osteotomy. J Bone Joint Surg Am. 1987;69(1):28-31.
18. Ramappa M, Anand S, Jennings A. Total knee replacement following high tibial osteotomy versus total knee replacement without high tibial osteotomy: a systematic review and meta analysis. Arch Orthop Trauma Surg. 2013;133(11):1587-1593.
19. Preston S, Howard J, Naudie D, Somerville L, McAuley J. Total knee arthroplasty after high tibial osteotomy: no differences between medial and lateral osteotomy approaches. Clin Orthop Relat Res. 2014;472(1):105-110.
20. Krackow KA, Holtgrewe JL. Experience with a new technique for managing severely overcorrected valgus high tibial osteotomy at total knee arthroplasty. Clin Orthop Relat Res. 1990;(258):213-224.
21. Papagelopoulos PJ, Karachalios T, Themistocleous GS, Papadopoulos ECh, Savvidou OD, Rand JA. Total knee arthroplasty in patients with pre-existing fracture deformity. Orthopaedics. 2007;30(5):373-378.
22. Yoshina N, Takai S, Watanabe Y, Nakamura S, Kubo T. Total knee arthroplasty with long stem for treatment of nonunion after high tibial osteotomy. J Arthroplasty. 2004;19(4):528-531.
23. Mont MA, Alexander N, Krackow KA, Hungerford DS. Total knee arthroplasty after failed high tibial osteotomy. Orthop Clin North Am. 1994;25(3):515-525.
24. Scott WN. Insall & Scott’s Surgery of the Knee. Vol 1. 4th ed. Philadelphia, PA: Churchill Livingstone Elsevier; 2006.
25. Gill T, Schemitsch EH, Brick GW, Thornhill TS. Revision total knee arthroplasty after failed unicompartmental knee arthroplasty or high tibial osteotomy. Clin Orthop Relat Res. 1995;(321):10-18.
26. Figgie HE 3rd, Goldberg VM, Heiple KG, Moller HS 3rd, Gordon NH. The influence of tibial-patellofemoral location on function of the knee in patients with the posterior stabilized condylar knee prosthesis. J Bone Joint Surg Am. 1986;68(7):1035-1040.
27. Wolff AM, Hungerford DS, Pepe CL. The effect of extraarticular varus and valgus deformity on total knee arthroplasty. Clin Orthop Relat Res. 1994;(271):35-51.
28. Uchinou S, Yano H, Shimizu K, Masumi S. A severely overcorrected high tibial osteotomy: revision by osteotomy and a long stem component. Acta Orthop Scand. 1996;67(2):193-194.
29. Noda T, Yasuda S, Nagano K, Takahara Y, Namba Y, Inoue H. Clinico-radiological study of total knee arthroplasty after high tibial osteotomy. J Orthop Sci. 2000;5(1):25-36.
30. Madelaine A, Villa V, Yela C, et al. Results and complications of single-stage total knee arthroplasty and high tibial osteotomy. Int Orthop. 2014;38(10):2091-2098.
1. Billings A, Scott DF, Camargo MP, Hofmann AA. High tibial osteotomy with a calibrated osteotomy guide, rigid internal fixation, and early motion. Long-term follow-up. J Bone Joint Surg Am. 2000;82(1):70-79.
2. Coventry MB, Ilstrup DM, Wallrichs SL. Proximal tibial osteotomy. A critical long-term study of eighty-seven cases. J Bone Joint Surg Am. 1993;75(2):196-201.
3. Rinonapoli E, Mancini GB, Corvaglia A, Musiello S. Tibial osteotomy for varus gonarthrosis. A 10- to 21-year followup study. Clin Orthop Relat Res. 1998;(353):185-193.
4. Ritter MA, Fechtman RA. Proximal tibial osteotomy. A survivorship analysis. J Arthroplasty. 1988;3(4):309-311.
5. Katz MM, Hungerford DS, Krackow KA, Lennox DW. Results of total knee arthroplasty after failed proximal tibial osteotomy for osteoarthritis. J Bone Joint Surg Am. 1987;69(2):225-233.
6. Mont MA, Antonaides S, Krackow KA, Hungerford DS. Total knee arthroplasty after failed high tibial osteotomy. A comparison with a matched group. Clin Orthop Relat Res. 1994;299:125-130.
7. Neyret P, Deroche P, Deschamps G, Dejour H. Total knee replacement after valgus tibial osteotomy. Technical problems [in French]. Rev Chir Orthop Reparatrice Appar Mot. 1992;78(7):438-448.
8. Parvizi J, Hanssen AD, Spangehl MJ. Total knee arthroplasty following proximal tibial osteotomy: risk factors for failure. J Bone Joint Surg Am. 2004;86(3):474-479.
9. Windsor RE, Insall JN, Vince KG. Technical considerations of total knee arthroplasty after proximal tibial osteotomy. J Bone Joint Surg Am. 1988;70(4):547-555.
10. Amendola A, Rorabeck CH, Bourne RB, Apyan PM. Total knee arthroplasty following high tibial osteotomy for osteoarthritis. J Arthroplasty. 1989;(4 suppl):S11-S17.
11. Kazakos KJ, Chatzipapas C, Verettas D, Galanis V, Xarchas KC, Psillakis I. Mid-term results of total knee arthroplasty after high tibial osteotomy. Arch Orthop Trauma Surg. 2008;128(2):167-173.
12. Meding JB, Keating EM, Ritter MA, Faris PM. Total knee arthroplasty after high tibial osteotomy. A comparison study in patients who had bilateral total knee replacement. J Bone Joint Surg Am. 2000;82(9):1252-1259.
13. Niinimaki T, Eskelinen A, Ohtonen P, Puhto AP, Mann BS, Leppilahti J. Total knee arthroplasty after high tibial osteotomy: a registry-based case–control study of 1,036 knees. Arch Orthop Trauma Surg. 2014;134(1):73-77.
14. van Raaij TM, Reijman M, Furlan AD, Verhaar JA. Total knee arthroplasty after high tibial osteotomy. A systematic review. BMC Musculoskelet Disord. 2009;10:88-98.
15. Lotke PA, Ecker ML. Influence of positioning of prosthesis in total knee replacement. J Bone Joint Surg Am. 1977;59(1):77-79.
16. Cameron HU, Welsh RP. Potential complications of total knee replacement following tibial osteotomy. Orthop Rev. 1988;17(1):39-43.
17. Staeheli JW, Cass JR, Morrey BF. Condylar total knee arthroplasty after failed proximal tibial osteotomy. J Bone Joint Surg Am. 1987;69(1):28-31.
18. Ramappa M, Anand S, Jennings A. Total knee replacement following high tibial osteotomy versus total knee replacement without high tibial osteotomy: a systematic review and meta analysis. Arch Orthop Trauma Surg. 2013;133(11):1587-1593.
19. Preston S, Howard J, Naudie D, Somerville L, McAuley J. Total knee arthroplasty after high tibial osteotomy: no differences between medial and lateral osteotomy approaches. Clin Orthop Relat Res. 2014;472(1):105-110.
20. Krackow KA, Holtgrewe JL. Experience with a new technique for managing severely overcorrected valgus high tibial osteotomy at total knee arthroplasty. Clin Orthop Relat Res. 1990;(258):213-224.
21. Papagelopoulos PJ, Karachalios T, Themistocleous GS, Papadopoulos ECh, Savvidou OD, Rand JA. Total knee arthroplasty in patients with pre-existing fracture deformity. Orthopaedics. 2007;30(5):373-378.
22. Yoshina N, Takai S, Watanabe Y, Nakamura S, Kubo T. Total knee arthroplasty with long stem for treatment of nonunion after high tibial osteotomy. J Arthroplasty. 2004;19(4):528-531.
23. Mont MA, Alexander N, Krackow KA, Hungerford DS. Total knee arthroplasty after failed high tibial osteotomy. Orthop Clin North Am. 1994;25(3):515-525.
24. Scott WN. Insall & Scott’s Surgery of the Knee. Vol 1. 4th ed. Philadelphia, PA: Churchill Livingstone Elsevier; 2006.
25. Gill T, Schemitsch EH, Brick GW, Thornhill TS. Revision total knee arthroplasty after failed unicompartmental knee arthroplasty or high tibial osteotomy. Clin Orthop Relat Res. 1995;(321):10-18.
26. Figgie HE 3rd, Goldberg VM, Heiple KG, Moller HS 3rd, Gordon NH. The influence of tibial-patellofemoral location on function of the knee in patients with the posterior stabilized condylar knee prosthesis. J Bone Joint Surg Am. 1986;68(7):1035-1040.
27. Wolff AM, Hungerford DS, Pepe CL. The effect of extraarticular varus and valgus deformity on total knee arthroplasty. Clin Orthop Relat Res. 1994;(271):35-51.
28. Uchinou S, Yano H, Shimizu K, Masumi S. A severely overcorrected high tibial osteotomy: revision by osteotomy and a long stem component. Acta Orthop Scand. 1996;67(2):193-194.
29. Noda T, Yasuda S, Nagano K, Takahara Y, Namba Y, Inoue H. Clinico-radiological study of total knee arthroplasty after high tibial osteotomy. J Orthop Sci. 2000;5(1):25-36.
30. Madelaine A, Villa V, Yela C, et al. Results and complications of single-stage total knee arthroplasty and high tibial osteotomy. Int Orthop. 2014;38(10):2091-2098.

Using a retrojugular approach to the carotid
Carotid endarterectomy is one of the most common operations for vascular surgeons. The standard approach, anteriorly, requires division of the facial vein, frequently additional small tributaries, and sacrifice of the ansa cervicalis.
The facial vein is not infrequently the cause of bleeding on patients returned to the OR who truly have surgical bleeding. The vagus nerve is not well visualized during the standard approach, unless maneuvers are taken to specifically identify it. Although this approach gives good access to the common carotid artery, and to the external carotid, it requires additional maneuvers in order to gain access to the distal internal carotid artery (ICA), which is located more posterolaterally.

This frequently requires ligation of the vascular bundle around the hypoglossal nerve, which can lead to bleeding if not properly addressed. Visualization of the distal ICA is not ideal with this approach.
An alternative: The retrojugular approach
The surgery is performed with the patient prepped in the same fashion as for standard CEA. Skin incision and initial dissection is identical for standard CEA approach, and is made along the sternocleidomastoid, with division of the platysma.
Dissection is carried out just along the border of the SCM until the carotid sheath is encountered. At this point, the dissection differs. Dissection is continued along the lateral border of the internal jugular vein, which is gently dissected from the surrounding tissue.
There are rarely branches along the lateral or posterior border that require ligation. This area is sharply dissected due to the proximity of nervous structures. The vagus nerve is easily visualized utilizing this approach, and left lateral to the carotid artery. Once a sufficient length of jugular has been mobilized, a blunt whietlander is placed, retracting the vein medially. The Carotid artery is then easily visualized, with a long segment of the ICA easily visible.
The hypoglossal nerve and facial vein are not visualized. The ansa cervicalis is not divided. Standard mobilization of the CCA, ICA, and ECA is then performed. With this approach, the ICA is anterior.
Visualization of the ECA is slightly more limited with this approach, but the proximal ECA and superior thyroidal arteries are easily identified and looped. CEA, either with eversion or standard longitudinal incision, is completed in standard fashion. After completion of the CEA, and confirmation of adequate hemostasis, the IJ is simply allowed to return to its normal position. The wound is then closed in standard fashion.
This approach permits significant exposure to a markedly longer segment of the ICA, without division of additional vascular branches, nor mobilization of nerves. There is slightly less visibility of the ECA, but more than adequate for the necessary vascular control. The approach also has less risk of postoperative bleed, and has been reported to be slightly faster than the anterior approach.
Dr. Harris is division chief, Vascular Surgery, at the State University of New York at Buffalo, and an associate medical editor of Vascular Specialist.
Carotid endarterectomy is one of the most common operations for vascular surgeons. The standard approach, anteriorly, requires division of the facial vein, frequently additional small tributaries, and sacrifice of the ansa cervicalis.
The facial vein is not infrequently the cause of bleeding on patients returned to the OR who truly have surgical bleeding. The vagus nerve is not well visualized during the standard approach, unless maneuvers are taken to specifically identify it. Although this approach gives good access to the common carotid artery, and to the external carotid, it requires additional maneuvers in order to gain access to the distal internal carotid artery (ICA), which is located more posterolaterally.
This frequently requires ligation of the vascular bundle around the hypoglossal nerve, which can lead to bleeding if not properly addressed. Visualization of the distal ICA is not ideal with this approach.
An alternative: The retrojugular approach
The surgery is performed with the patient prepped in the same fashion as for standard CEA. Skin incision and initial dissection is identical for standard CEA approach, and is made along the sternocleidomastoid, with division of the platysma.
Dissection is carried out just along the border of the SCM until the carotid sheath is encountered. At this point, the dissection differs. Dissection is continued along the lateral border of the internal jugular vein, which is gently dissected from the surrounding tissue.
There are rarely branches along the lateral or posterior border that require ligation. This area is sharply dissected due to the proximity of nervous structures. The vagus nerve is easily visualized utilizing this approach, and left lateral to the carotid artery. Once a sufficient length of jugular has been mobilized, a blunt whietlander is placed, retracting the vein medially. The Carotid artery is then easily visualized, with a long segment of the ICA easily visible.
The hypoglossal nerve and facial vein are not visualized. The ansa cervicalis is not divided. Standard mobilization of the CCA, ICA, and ECA is then performed. With this approach, the ICA is anterior.
Visualization of the ECA is slightly more limited with this approach, but the proximal ECA and superior thyroidal arteries are easily identified and looped. CEA, either with eversion or standard longitudinal incision, is completed in standard fashion. After completion of the CEA, and confirmation of adequate hemostasis, the IJ is simply allowed to return to its normal position. The wound is then closed in standard fashion.
This approach permits significant exposure to a markedly longer segment of the ICA, without division of additional vascular branches, nor mobilization of nerves. There is slightly less visibility of the ECA, but more than adequate for the necessary vascular control. The approach also has less risk of postoperative bleed, and has been reported to be slightly faster than the anterior approach.
Dr. Harris is division chief, Vascular Surgery, at the State University of New York at Buffalo, and an associate medical editor of Vascular Specialist.
Carotid endarterectomy is one of the most common operations for vascular surgeons. The standard approach, anteriorly, requires division of the facial vein, frequently additional small tributaries, and sacrifice of the ansa cervicalis.
The facial vein is not infrequently the cause of bleeding on patients returned to the OR who truly have surgical bleeding. The vagus nerve is not well visualized during the standard approach, unless maneuvers are taken to specifically identify it. Although this approach gives good access to the common carotid artery, and to the external carotid, it requires additional maneuvers in order to gain access to the distal internal carotid artery (ICA), which is located more posterolaterally.
This frequently requires ligation of the vascular bundle around the hypoglossal nerve, which can lead to bleeding if not properly addressed. Visualization of the distal ICA is not ideal with this approach.
An alternative: The retrojugular approach
The surgery is performed with the patient prepped in the same fashion as for standard CEA. Skin incision and initial dissection is identical for standard CEA approach, and is made along the sternocleidomastoid, with division of the platysma.
Dissection is carried out just along the border of the SCM until the carotid sheath is encountered. At this point, the dissection differs. Dissection is continued along the lateral border of the internal jugular vein, which is gently dissected from the surrounding tissue.
There are rarely branches along the lateral or posterior border that require ligation. This area is sharply dissected due to the proximity of nervous structures. The vagus nerve is easily visualized utilizing this approach, and left lateral to the carotid artery. Once a sufficient length of jugular has been mobilized, a blunt whietlander is placed, retracting the vein medially. The Carotid artery is then easily visualized, with a long segment of the ICA easily visible.
The hypoglossal nerve and facial vein are not visualized. The ansa cervicalis is not divided. Standard mobilization of the CCA, ICA, and ECA is then performed. With this approach, the ICA is anterior.
Visualization of the ECA is slightly more limited with this approach, but the proximal ECA and superior thyroidal arteries are easily identified and looped. CEA, either with eversion or standard longitudinal incision, is completed in standard fashion. After completion of the CEA, and confirmation of adequate hemostasis, the IJ is simply allowed to return to its normal position. The wound is then closed in standard fashion.
This approach permits significant exposure to a markedly longer segment of the ICA, without division of additional vascular branches, nor mobilization of nerves. There is slightly less visibility of the ECA, but more than adequate for the necessary vascular control. The approach also has less risk of postoperative bleed, and has been reported to be slightly faster than the anterior approach.
Dr. Harris is division chief, Vascular Surgery, at the State University of New York at Buffalo, and an associate medical editor of Vascular Specialist.

Role of Surgical Dressings in Total Joint Arthroplasty: A Randomized Controlled Trial
Wound complications (eg, delayed wound healing, blisters, prolonged drainage) have been reported in up to 30% of patients who undergo elective total joint arthroplasty (TJA).1-6 Wound complications increase resource utilization, lengthen hospital stays, and increase costs.7-9 Prolonged wound healing and persistent wound drainage are also harbingers of both superficial and deep surgical site infections.5-11
In several studies, wound complications after TJA were the primary reason for hospital readmissions.12-15 As part of the Patient Protection and Affordable Care Act, hospitals will be penalized by the Centers for Medicaid & Medicare Services for unplanned hospital readmissions within 30 days after TJA. It is imperative, then, to reduce the risk factors and complications associated with surgical site infections to decrease unplanned readmissions.
Historically, little attention has been given to the role of surgical dressings and the effect of dressings on wound healing. Although many subspecialties (eg, cardiothoracic surgery, general surgery) have reported benefits in using occlusive dressings, adoption in TJA has been slow.16-18 At our institution about 5 years ago, we began using an occlusive silver-impregnated barrier dressing based on preliminary data from studies showing benefits of occlusive dressings in TJA.19,20
We conducted a study to determine if use of occlusive antimicrobial barrier dressings decreases rates of wound complications in TJA. We had 3 research questions: Compared with standard surgical dressings, are occlusive dressings associated with decreased rates of wound complications after TJA? Is there a difference in number of dressing changes required between the 2 dressing types? Is satisfaction higher for patients with occlusive dressings than for patients with standard dressings?
Patients and Methods
This randomized controlled trial (RCT) was reviewed and approved by the Institutional Review Board at Carolinas Healthcare. Patients were randomized by the research staff using a parallel, 1:1 allocation method. The randomization table was generated using a random number generator.
An a priori sample size estimate was made using a 2-tailed Fisher exact test with a .05 level of significance. Based on a study by Clarke and colleagues,21 we estimated the incidence of wound problems at 3% in the occlusive dressing (study) group and 13% in the standard dressing (control) group. We determined that 260 participants (130 per group) would be needed to achieve 80% power. We considered a 15% attrition rate for a total enrollment goal of 300 study participants (150 per group).
Between December 2010 and January 2013, patients presenting for either primary total hip arthroplasty (THA) or primary total knee arthroplasty (TKA) were recruited to participate in the study. Eligibility criteria (Table 1) were reviewed, and patients were enrolled by the senior surgeons, Dr. Springer, Dr. Beaver, Dr. Griffin, and Dr. Mason. All eligible participants who provided informed consent were randomized to receive either an occlusive antimicrobial barrier dressing (Aquacel Ag, ConvaTec) or standard surgical dressing (Primapore, Smith & Nephew). The occlusive dressing (Figure 1) consists of an outer barrier layer of hydrocolloid and a central island of hydrofiber, which absorbs and locks in any wound exudate within the fibers and prevents the creation of an overly moist wound environment that can lead to skin maceration and wound breakdown. In addition, the hydrofibers are embedded with ionic silver, which is released only at the site of wound exudate, or drainage; thus, there is no continuous exposure of the entire wound to silver. The standard dressing (Figure 2) consists of a central island of gauze enclosed in low-allergy acrylic adhesive tape.
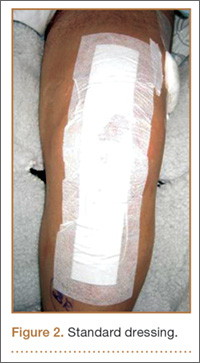
All surgical dressings were placed over a closed incision in a sterile environment in the operating room after the procedure. The groups’ wound closures were identical.
A posterior approach was used for all THAs. The deep fascia was closed with a running barbed suture (Quill, Angiotech), the deep subcutaneous tissue with No. 1 Vicryl suture (Ethicon), and the superficial subcutaneous layer with 2-0 Vicryl suture. A running 3-0 Monocryl stitch (Ethicon) was placed in the subcuticular layer and was followed with a skin adhesive (Dermabond, Ethicon). A closed suction drain, removed on postoperative day (POD) 1, was used for all THAs.
A standard medial parapatellar arthrotomy was used for all TKAs. The arthrotomy was closed with a running barbed suture, the deep subcutaneous tissue with No. 1 Vicryl suture, and the superficial subcutaneous layer with 2-0 Vicryl suture. A running 3-0 Monocryl stitch was placed in the subcuticular layer and was followed with a skin adhesive. A closed suction drain was also used. In addition, a compressive wrap was placed over the dressing in the operating room and was removed the next morning. During the hospital stay, the surgical site was evaluated daily with a standard wound evaluation form.
In the standard dressing group, the bandage was removed for wound evaluation on POD 2, and the dressing was changed every other day during the hospital stay. The dressing was also changed as needed for wound drainage (Figure 3) or other minor wound-healing concerns.
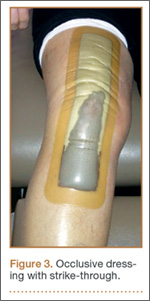
In the occlusive dressing group, the dressing design allowed the dressing to remain in place for about 7 days. It was removed by a home health nurse during a visit closest to but not before the 7-day mark. In addition, it was changed at surgeon discretion if there were concerns about wound drainage or wound healing. For the occlusive barrier, wound drainage was evaluated by strike-through of drainage on the back side of the dressing (Figure 4). If more than 50% of the dressing was saturated, the bandage was changed and the wound evaluated. If there were no immediate concerns about wound complications (eg, infection, blistering), a new occlusive dressing was placed. Because the occlusive dressing was waterproof, patients in the study group were able to shower immediately after surgery. In the control group, patients were allowed to shower if the surgical dressing was kept dry, as the bandage was not waterproof.
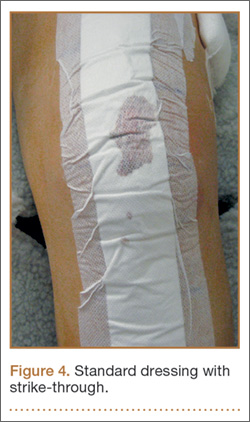
Per the study protocol, all patients were discharged home and followed by a single home health agency. Mean hospital stay was 3 days (range, 0-8 days), which did not differ significantly between groups (P = .133). All home health nurses were trained in evaluation of postsurgical wounds and were aware of the study requirements. The nurses visited all patients 3 days a week until the scheduled 4-week postoperative follow-up with the treating physician or physician assistant. At each visit, the nurse evaluated the wound and surrounding skin using a standard wound document. Dressings were changed based on the criteria we have described. Concerns about wound status (eg, drainage, blistering, erythema) prompted removal of the dressing for further evaluation. The physician was notified of concerns about wound healing, which prompted an office visit for evaluation. The dressing remained in place for a minimum of 7 days but in all cases was removed as close to 7 days as possible, depending on the scheduled nursing visits. Once uneventful wound healing was complete, no further dressing was required. A final wound evaluation was conducted by the surgeon at the 4-week postoperative evaluation.
The primary outcome measure was wound complication (dichotomous variable). Wounds were assessed by describing the amount, type, and color of exudate (Figure 5). The appearance of the wound margins and the surrounding skin was also assessed. Because wounds could not be directly visualized in the occlusive dressing group, drainage (indicated by strike-through) was used as a measure of possible wound complications, prompting removal and full evaluation.

Secondary endpoints included additional wound treatment or surgical procedures for wound complications, number of dressing changes, and patient satisfaction. Patients completed a satisfaction questionnaire at each wound assessment (Figure 6). Using a visual analog scale (VAS), they rated their satisfaction with their ability to perform activities of daily living (personal hygiene, change clothes, sit comfortably, sleep comfortably), drawing a line on the VAS at a point between 0 (totally unsatisfied) and 100 (totally satisfied) for each satisfaction measure. This line was measured and recorded by the study coordinator. The 4 satisfaction measures were averaged for a composite satisfaction measure.

All statistical analyses were conducted using SAS Version 9.2 (SAS Institute). Standard univariate descriptive statistics (means, standard deviations, frequencies, proportions) were calculated and reported. Differences in mean values for continuous data were assessed with independent t test or Wilcoxon rank sum test. Chi-square test and Fisher exact test were used to determine differences between groups for categorical or dichotomous variables. A significance level of .05 was used for all statistical tests.
Results
The 300 patients who consented to participate in the study were randomized to receive either occlusive dressing or standard dressing. After randomization, 38 patients (15 occlusive, 23 standard) were withdrawn from the study (Table 2), leaving a final dataset of 262 patients, 141 in the occlusive group (67 THAs, 74 TKAs) and 121 in the standard group (49 THAs, 72 TKAs). There were no differences in proportion of THAs or TKAs, age, sex, or body mass index between the occlusive and standard groups (Table 3).
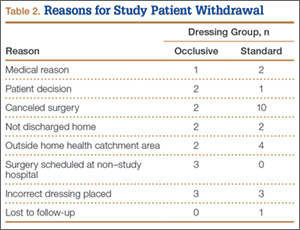
There were statistically significantly (P = .015) fewer wound complications in the occlusive dressing group (10%) than in the standard dressing group (22%). Blisters at or around the wound site were reported in significantly (P = .026) fewer patients with occlusive dressing (1/141, 0.7%) than standard dressing (7/121, 6%). Additional wound care was required in 9 patients (7%) in the standard group and 6 patients (4%) in the occlusive group (P = .27). Two patients (1.7%) in the standard group were readmitted for treatment of wound dehiscence; no one in the occlusive group was readmitted to the hospital or had to return to the operating room for treatment of a wound complication. The difference was not statistically significant (P = .13). There were also no significant (P = .81) differences in rate of wound complications between THA and TKA patients.
There were statistically significantly (P < .0001) fewer dressing changes in the occlusive dressing group. Mean number of dressing changes was 0.14 (median, 0; interquartile range, 0-0) in the occlusive group and 2.8 (median, 2; interquartile range, 1-3) in the standard group.
Compared with patients in the standard dressing group, patients in the occlusive dressing group reported significantly higher satisfaction scores. Mean overall patient satisfaction score was 92 in the occlusive group and 81 in the standard group (P < .0001). Patients in the occlusive group were more satisfied with their ability to take care of their personal hygiene, to change clothes, and to sit and sleep comfortably (Table 4).
Discussion
Wound complications after TJA are common, occurring in up to 30% of patients,1-6 and are associated with development of superficial and deep surgical site infections, increased resource utilization, and longer hospital stays.5-11 Although the role of surgical dressings has received little attention in TJA practice, other subspecialties have found that occlusive barrier dressings can reduce wound complications and promote wound healing.16,17 Mitotic cell division and leukocyte activity, which are critical in wound healing, increase under occlusive dressings. This cellular activity is disrupted with every dressing change, delaying wound healing (biological activity takes 3-4 hours to resume).22 In addition, occlusive dressings increase hypoxia, which promotes angiogenesis and accelerates wound healing.23
Despite being a prospective RCT, this study had several limitations. Because of the need to evaluate wounds and obvious differences between the 2 dressings (eg, color, ability to shower), it was not possible to blind the patient or surgeon to the dressing used. When rating satisfaction, patients were not able to directly compare the 2 dressings. The primary endpoint of the study was the complication rate; however, the deep periprosthetic infection rate may be a superior endpoint and would require a much larger study. Although we assumed that wound complications may be harbingers for periprosthetic infections, no patient in either group developed periprosthetic infection. Therefore, we cannot conclude that surgical dressings play a role in reducing infections. In addition, as the standard dressing was changed on POD 2 (per standard protocol) and the occlusive dressing could remain in place for up to 7 days, there was a selection bias in the evaluation of the number of dressing changes. However, given the characteristics of the standard dressing (eg, tape, gauze, nonocclusive), leaving it in place after POD 2 is not optimal. Therefore, we would expect to see a difference in the number of dressing changes. We think this comparison remains valid, as occlusive dressings were changed when there were indications of wound problems (eg, excessive drainage [strike-through], surrounding erythema, blistering). With an average of less than 1 dressing change in the occlusive group, we think this is a surrogate for uneventful wound healing and decreased wound complication, and our data support this. It is also important to test both dressing durability and patient tolerance for wearing a single dressing for 7 days.
Our RCT results showed that, compared with a standard dressing, an occlusive antimicrobial dressing was associated with a significant decrease in overall wound complications and blisters. These findings are similar to those of other studies of occlusive dressings in a number of surgical subspecialties.16,18 In an RCT of 200 patients who underwent elective and nonelective hip and knee surgery and were randomized to either absorbent perforated dressing with adhesive border (Cutiplast, Smith & Nephew) or Aquacel (ConvaTec) covered with vapor-permeable dressing (Tegaderm, 3M), Ravenscroft and colleagues20 found that Aquacel-plus-Tegaderm was 5.8 times more likely than Cutiplast to produce an uncompromised wound. Similarly, in an RCT of hydrofiber (Aquacel) and central pad (Mepore, Mölnlycke) dressings after primary THA and TKA, Abuzakuk and colleagues19 found significantly fewer dressing changes (43% vs 77%) and blisters (13% vs 26%) in the hydrofiber group than in the pad group.
Hopper and colleagues24 compared 50 consecutive patients treated with modern dressings (Aquacel) with 50 historical control patients treated with traditional surgical dressings (Mepore). Blisters developed in 20% of the patients in the traditional group and 4% of patients in the modern group (P = .028). The authors concluded that adverse outcomes of wound healing can be minimized with modern dressings.
A recent retrospective study by Cai and colleagues25 evaluated the incidence of acute periprosthetic infection (≤3 months after surgery) with use of occlusive (Aquacel) and standard dressings. Incidence of acute periprosthetic infection was 0.44% in the occlusive group and 1.7% in the standard group (P = .005). Incidence of wound-healing problems was not evaluated.
Our second aim in the present study was to evaluate the number of dressing changes required. There were significantly fewer dressing changes in the occlusive dressing group than in the standard dressing group. Therefore, wear time (amount of time a single dressing remains in place) was substantially longer for the occlusive group. In the study by Hopper and colleagues,24 wear time was significantly shorter for the traditional dressing than for the modern dressing (2 vs 7 days; P < .001), and the traditional dressing required more changes (3 vs 0; P < .001).
These findings are important for several reasons. Standard surgical dressings often require frequent changes. If left in place, they create an excessively moist wound environment that promotes blistering and delays wound healing. However, frequent dressing changes expose the wound and increase the risk for surgical site infection.26 A barrier dressing left in place from time of surgery prevents bacteria from entering and contaminating a healing wound. A study by Clarke and colleagues21 demonstrated higher skin colonization rates for patients who had dressings changed on POD 1 than for patients who had their first dressing change on POD 6.
Our third study aim was to evaluate patient satisfaction with surgical dressings. The orthopedic literature has little on this topic.23 Blisters and other wound complications can negatively affect satisfaction.2,3 Our data showed significant improvement in satisfaction, particularly regarding sterility and hygiene.
Other surgical subspecialties have found similar improvement in patient satisfaction with occlusive barrier dressings. In an RCT of 88 pediatric patients, Rasmussen and colleagues27 found that patients reported significantly less pain during changes of an occlusive adhesive dressing (Duoderm, ConvaTec) than during changes of a conventional Steristrip (3M) plus Cutiplast. According to the authors, the occlusive wound dressing seemed to minimize the physical and psychological trauma to the infant or child and lessen disruption of the child’s and the parents’ daily routines, because the children could be bathed immediately after surgery.
Our study did not specifically address cost. Cai and colleagues25 estimated that, if the Aquacel dressing were routinely used in every hip and knee arthroplasty, it would add about $27 million in cost. However, this must be balanced by the cost of managing infection after TJA. In the United States, at an estimated $50,000 to $100,000 per case and an annual incidence of 1% to 2%, the low-end cost for the treatment of periprosthetic infection would be $500 million.28 Cai and colleagues25 found a 4-fold reduction in periprosthetic infection when use of occlusive dressings was implemented. In addition, wound complications remain the number one reason for hospital readmission after TJA.12,13 Cost of hospital readmission, as well as financial penalties to institutions for unplanned readmission for wound complications, must be considered.
Conclusion
Our RCT results demonstrated that use of occlusive antimicrobial barrier dressings (vs standard surgical dressings) significantly reduced wound complications and dressing changes and improved overall patient satisfaction. These findings are similar to those in the literature on TJA and other surgical subspecialties. We conclude that occlusive surgical dressings reduce wound complications after TJA.
1. Cosker T, Elsayed S, Gupta S, Mendonca AD, Tayton KJ. Choice of dressing has a major impact on blistering and healing outcomes in orthopaedic patients. J Wound Care. 2005;14(1):27-29.
2. Koval KJ, Egol KA, Hiebert R, Spratt KF. Tape blisters after hip surgery: can they be eliminated completely? Am J Orthop. 2007;36(5):261-265.
3. Lawrentschuk N, Falkenberg MP, Pirpiris M. Wound blisters post hip surgery: a prospective trial comparing dressings. ANZ J Surg. 2002;72(10):716-719.
4. Mihalko WM, Manaswi A, Brown TE, Parvizi J, Schmalzried TP, Saleh KJ. Infection in primary total knee arthroplasty: contributing factors. Instr Course Lect. 2008;57:317-325.
5. Patel VP, Walsh M, Sehgal B, Preston C, DeWal H, Di Cesare PE. Factors associated with prolonged wound drainage after primary total hip and knee arthroplasty. J Bone Joint Surg Am. 2007;89(1):33-38.
6. Vince KG, Abdeen A. Wound problems in total knee arthroplasty. Clin Orthop Relat Res. 2006;(452):88-90.
7. Galat DD, McGovern SC, Larson DR, Harrington JR, Hanssen AD, Clarke HD. Surgical treatment of early wound complications following primary total knee arthroplasty. J Bone Joint Surg Am. 2009;91(1):48-54.
8. Gordon SM, Culver DH, Simmons BP, Jarvis WR. Risk factors for wound infections after total knee arthroplasty. Am J Epidemiol. 1990;131(5):905-916.
9. Jaberi FM, Parvizi J, Haytmanek CT, Joshi A, Purtill J. Procrastination of wound drainage and malnutrition affect the outcome of joint arthroplasty. Clin Orthop Relat Res. 2008;466(6):1368-1371.
10. Schmalzried TP. The infected hip: telltale signs and treatment options. J Arthroplasty. 2006;21(4 suppl 1):97-100.
11. Weiss AP, Krackow KA. Persistent wound drainage after primary total knee arthroplasty. J Arthroplasty. 1993;8(3):285-289.
12. Avram V, Petruccelli D, Winemaker M, de Beer J. Total joint arthroplasty readmission rates and reasons for 30-day hospital readmission. J Arthroplasty. 2014;29(3):465-468.
13. Dailey EA, Cizik A, Kasten J, Chapman JR, Lee MJ. Risk factors for readmission of orthopaedic surgical patients. J Bone Joint Surg Am. 2013;95(11):1012-1019.
14. Jordan CJ, Goldstein RY, Michels RF, Hutzler L, Slover JD, Bosco JA 3rd. Comprehensive program reduces hospital readmission rates after total joint arthroplasty. Am J Orthop. 2012;41(11):E147-E151.
15. Schairer WW, Sing DC, Vail TP, Bozic KJ. Causes and frequency of unplanned hospital readmission after total hip arthroplasty. Clin Orthop Relat Res. 2014;472(2):464-470.
16. Shinohara T, Yamashita Y, Satoh K, et al. Prospective evaluation of occlusive hydrocolloid dressing versus conventional gauze dressing regarding the healing effect after abdominal operations: randomized controlled trial. Asian J Surg. 2008;31(1):1-5.
17. Siah CJ, Yatim J. Efficacy of a total occlusive ionic silver-containing dressing combination in decreasing risk of surgical site infection: an RCT. J Wound Care. 2011;20(12):561-568.
18. Teshima H, Kawano H, Kashikie H, et al. A new hydrocolloid dressing prevents surgical site infection of median sternotomy wounds. Surg Today. 2009;39(10):848-854.
19. Abuzakuk TM, Coward P, Shenava Y, Kumar VS, Skinner JA. The management of wounds following primary lower limb arthroplasty: a prospective, randomised study comparing hydrofibre and central pad dressings. Int Wound J. 2006;3(2):133-137.
20. Ravenscroft MJ, Harker J, Buch KA. A prospective, randomised, controlled trial comparing wound dressings used in hip and knee surgery: Aquacel and Tegaderm versus Cutiplast. Ann R Coll Surg Engl. 2006;88(1):18-22.
21. Clarke JV, Deakin AH, Dillon JM, Emmerson S, Kinninmonth AW. A prospective clinical audit of a new dressing design for lower limb arthroplasty wounds. J Wound Care. 2009;18(1):5-8, 10-11.
22. Kloeters O. The use of a semi-occlusive dressing reduces epidermal inflammatory cytokine expression and mitigates dermal proliferation and inflammation in a rat incisional model. Wound Repair Regen. 2008;16(4):568-575.
23. Michie DD, Hugill JV. Influence of occlusive and impregnated gauze dressings on incisional healing: a prospective, randomized, controlled study. Ann Plast Surg. 1994;32(1):57-64.
24. Hopper GP, Deakin AH, Crane EO, Clarke JV. Enhancing patient recovery following lower limb arthroplasty with a modern wound dressing: a prospective, comparative audit. J Wound Care. 2012;21(4):200-203.
25. Cai J, Karam JA, Parvizi J, Smith EB, Sharkey PF. Aquacel surgical dressing reduces the rate of acute PJI following total joint arthroplasty: a case–control study. J Arthroplasty. 2014;29(6):1098-1100.
26. Berg A, Fleischer S, Kuss O, Unverzagt S, Langer G. Timing of dressing removal in the healing of surgical wounds by primary intention: quantitative systematic review protocol. J Adv Nurs. 2012;68(2):264-270.
27. Rasmussen H, Larsen MJ, Skeie E. Surgical wound dressing in outpatient paediatric surgery. A randomised study. Dan Med Bull. 1993;40(2):252-254.
28. Kurtz SM, Lau E, Schmier J, Ong KL, Zhao K, Parvizi J. Infection burden for hip and knee arthroplasty in the United States. J Arthroplasty. 2008;23(7):984-991.
Wound complications (eg, delayed wound healing, blisters, prolonged drainage) have been reported in up to 30% of patients who undergo elective total joint arthroplasty (TJA).1-6 Wound complications increase resource utilization, lengthen hospital stays, and increase costs.7-9 Prolonged wound healing and persistent wound drainage are also harbingers of both superficial and deep surgical site infections.5-11
In several studies, wound complications after TJA were the primary reason for hospital readmissions.12-15 As part of the Patient Protection and Affordable Care Act, hospitals will be penalized by the Centers for Medicaid & Medicare Services for unplanned hospital readmissions within 30 days after TJA. It is imperative, then, to reduce the risk factors and complications associated with surgical site infections to decrease unplanned readmissions.
Historically, little attention has been given to the role of surgical dressings and the effect of dressings on wound healing. Although many subspecialties (eg, cardiothoracic surgery, general surgery) have reported benefits in using occlusive dressings, adoption in TJA has been slow.16-18 At our institution about 5 years ago, we began using an occlusive silver-impregnated barrier dressing based on preliminary data from studies showing benefits of occlusive dressings in TJA.19,20
We conducted a study to determine if use of occlusive antimicrobial barrier dressings decreases rates of wound complications in TJA. We had 3 research questions: Compared with standard surgical dressings, are occlusive dressings associated with decreased rates of wound complications after TJA? Is there a difference in number of dressing changes required between the 2 dressing types? Is satisfaction higher for patients with occlusive dressings than for patients with standard dressings?
Patients and Methods
This randomized controlled trial (RCT) was reviewed and approved by the Institutional Review Board at Carolinas Healthcare. Patients were randomized by the research staff using a parallel, 1:1 allocation method. The randomization table was generated using a random number generator.
An a priori sample size estimate was made using a 2-tailed Fisher exact test with a .05 level of significance. Based on a study by Clarke and colleagues,21 we estimated the incidence of wound problems at 3% in the occlusive dressing (study) group and 13% in the standard dressing (control) group. We determined that 260 participants (130 per group) would be needed to achieve 80% power. We considered a 15% attrition rate for a total enrollment goal of 300 study participants (150 per group).
Between December 2010 and January 2013, patients presenting for either primary total hip arthroplasty (THA) or primary total knee arthroplasty (TKA) were recruited to participate in the study. Eligibility criteria (Table 1) were reviewed, and patients were enrolled by the senior surgeons, Dr. Springer, Dr. Beaver, Dr. Griffin, and Dr. Mason. All eligible participants who provided informed consent were randomized to receive either an occlusive antimicrobial barrier dressing (Aquacel Ag, ConvaTec) or standard surgical dressing (Primapore, Smith & Nephew). The occlusive dressing (Figure 1) consists of an outer barrier layer of hydrocolloid and a central island of hydrofiber, which absorbs and locks in any wound exudate within the fibers and prevents the creation of an overly moist wound environment that can lead to skin maceration and wound breakdown. In addition, the hydrofibers are embedded with ionic silver, which is released only at the site of wound exudate, or drainage; thus, there is no continuous exposure of the entire wound to silver. The standard dressing (Figure 2) consists of a central island of gauze enclosed in low-allergy acrylic adhesive tape.
All surgical dressings were placed over a closed incision in a sterile environment in the operating room after the procedure. The groups’ wound closures were identical.
A posterior approach was used for all THAs. The deep fascia was closed with a running barbed suture (Quill, Angiotech), the deep subcutaneous tissue with No. 1 Vicryl suture (Ethicon), and the superficial subcutaneous layer with 2-0 Vicryl suture. A running 3-0 Monocryl stitch (Ethicon) was placed in the subcuticular layer and was followed with a skin adhesive (Dermabond, Ethicon). A closed suction drain, removed on postoperative day (POD) 1, was used for all THAs.
A standard medial parapatellar arthrotomy was used for all TKAs. The arthrotomy was closed with a running barbed suture, the deep subcutaneous tissue with No. 1 Vicryl suture, and the superficial subcutaneous layer with 2-0 Vicryl suture. A running 3-0 Monocryl stitch was placed in the subcuticular layer and was followed with a skin adhesive. A closed suction drain was also used. In addition, a compressive wrap was placed over the dressing in the operating room and was removed the next morning. During the hospital stay, the surgical site was evaluated daily with a standard wound evaluation form.
In the standard dressing group, the bandage was removed for wound evaluation on POD 2, and the dressing was changed every other day during the hospital stay. The dressing was also changed as needed for wound drainage (Figure 3) or other minor wound-healing concerns.
In the occlusive dressing group, the dressing design allowed the dressing to remain in place for about 7 days. It was removed by a home health nurse during a visit closest to but not before the 7-day mark. In addition, it was changed at surgeon discretion if there were concerns about wound drainage or wound healing. For the occlusive barrier, wound drainage was evaluated by strike-through of drainage on the back side of the dressing (Figure 4). If more than 50% of the dressing was saturated, the bandage was changed and the wound evaluated. If there were no immediate concerns about wound complications (eg, infection, blistering), a new occlusive dressing was placed. Because the occlusive dressing was waterproof, patients in the study group were able to shower immediately after surgery. In the control group, patients were allowed to shower if the surgical dressing was kept dry, as the bandage was not waterproof.
Per the study protocol, all patients were discharged home and followed by a single home health agency. Mean hospital stay was 3 days (range, 0-8 days), which did not differ significantly between groups (P = .133). All home health nurses were trained in evaluation of postsurgical wounds and were aware of the study requirements. The nurses visited all patients 3 days a week until the scheduled 4-week postoperative follow-up with the treating physician or physician assistant. At each visit, the nurse evaluated the wound and surrounding skin using a standard wound document. Dressings were changed based on the criteria we have described. Concerns about wound status (eg, drainage, blistering, erythema) prompted removal of the dressing for further evaluation. The physician was notified of concerns about wound healing, which prompted an office visit for evaluation. The dressing remained in place for a minimum of 7 days but in all cases was removed as close to 7 days as possible, depending on the scheduled nursing visits. Once uneventful wound healing was complete, no further dressing was required. A final wound evaluation was conducted by the surgeon at the 4-week postoperative evaluation.
The primary outcome measure was wound complication (dichotomous variable). Wounds were assessed by describing the amount, type, and color of exudate (Figure 5). The appearance of the wound margins and the surrounding skin was also assessed. Because wounds could not be directly visualized in the occlusive dressing group, drainage (indicated by strike-through) was used as a measure of possible wound complications, prompting removal and full evaluation.
Secondary endpoints included additional wound treatment or surgical procedures for wound complications, number of dressing changes, and patient satisfaction. Patients completed a satisfaction questionnaire at each wound assessment (Figure 6). Using a visual analog scale (VAS), they rated their satisfaction with their ability to perform activities of daily living (personal hygiene, change clothes, sit comfortably, sleep comfortably), drawing a line on the VAS at a point between 0 (totally unsatisfied) and 100 (totally satisfied) for each satisfaction measure. This line was measured and recorded by the study coordinator. The 4 satisfaction measures were averaged for a composite satisfaction measure.
All statistical analyses were conducted using SAS Version 9.2 (SAS Institute). Standard univariate descriptive statistics (means, standard deviations, frequencies, proportions) were calculated and reported. Differences in mean values for continuous data were assessed with independent t test or Wilcoxon rank sum test. Chi-square test and Fisher exact test were used to determine differences between groups for categorical or dichotomous variables. A significance level of .05 was used for all statistical tests.
Results
The 300 patients who consented to participate in the study were randomized to receive either occlusive dressing or standard dressing. After randomization, 38 patients (15 occlusive, 23 standard) were withdrawn from the study (Table 2), leaving a final dataset of 262 patients, 141 in the occlusive group (67 THAs, 74 TKAs) and 121 in the standard group (49 THAs, 72 TKAs). There were no differences in proportion of THAs or TKAs, age, sex, or body mass index between the occlusive and standard groups (Table 3).
There were statistically significantly (P = .015) fewer wound complications in the occlusive dressing group (10%) than in the standard dressing group (22%). Blisters at or around the wound site were reported in significantly (P = .026) fewer patients with occlusive dressing (1/141, 0.7%) than standard dressing (7/121, 6%). Additional wound care was required in 9 patients (7%) in the standard group and 6 patients (4%) in the occlusive group (P = .27). Two patients (1.7%) in the standard group were readmitted for treatment of wound dehiscence; no one in the occlusive group was readmitted to the hospital or had to return to the operating room for treatment of a wound complication. The difference was not statistically significant (P = .13). There were also no significant (P = .81) differences in rate of wound complications between THA and TKA patients.
There were statistically significantly (P < .0001) fewer dressing changes in the occlusive dressing group. Mean number of dressing changes was 0.14 (median, 0; interquartile range, 0-0) in the occlusive group and 2.8 (median, 2; interquartile range, 1-3) in the standard group.
Compared with patients in the standard dressing group, patients in the occlusive dressing group reported significantly higher satisfaction scores. Mean overall patient satisfaction score was 92 in the occlusive group and 81 in the standard group (P < .0001). Patients in the occlusive group were more satisfied with their ability to take care of their personal hygiene, to change clothes, and to sit and sleep comfortably (Table 4).
Discussion
Wound complications after TJA are common, occurring in up to 30% of patients,1-6 and are associated with development of superficial and deep surgical site infections, increased resource utilization, and longer hospital stays.5-11 Although the role of surgical dressings has received little attention in TJA practice, other subspecialties have found that occlusive barrier dressings can reduce wound complications and promote wound healing.16,17 Mitotic cell division and leukocyte activity, which are critical in wound healing, increase under occlusive dressings. This cellular activity is disrupted with every dressing change, delaying wound healing (biological activity takes 3-4 hours to resume).22 In addition, occlusive dressings increase hypoxia, which promotes angiogenesis and accelerates wound healing.23
Despite being a prospective RCT, this study had several limitations. Because of the need to evaluate wounds and obvious differences between the 2 dressings (eg, color, ability to shower), it was not possible to blind the patient or surgeon to the dressing used. When rating satisfaction, patients were not able to directly compare the 2 dressings. The primary endpoint of the study was the complication rate; however, the deep periprosthetic infection rate may be a superior endpoint and would require a much larger study. Although we assumed that wound complications may be harbingers for periprosthetic infections, no patient in either group developed periprosthetic infection. Therefore, we cannot conclude that surgical dressings play a role in reducing infections. In addition, as the standard dressing was changed on POD 2 (per standard protocol) and the occlusive dressing could remain in place for up to 7 days, there was a selection bias in the evaluation of the number of dressing changes. However, given the characteristics of the standard dressing (eg, tape, gauze, nonocclusive), leaving it in place after POD 2 is not optimal. Therefore, we would expect to see a difference in the number of dressing changes. We think this comparison remains valid, as occlusive dressings were changed when there were indications of wound problems (eg, excessive drainage [strike-through], surrounding erythema, blistering). With an average of less than 1 dressing change in the occlusive group, we think this is a surrogate for uneventful wound healing and decreased wound complication, and our data support this. It is also important to test both dressing durability and patient tolerance for wearing a single dressing for 7 days.
Our RCT results showed that, compared with a standard dressing, an occlusive antimicrobial dressing was associated with a significant decrease in overall wound complications and blisters. These findings are similar to those of other studies of occlusive dressings in a number of surgical subspecialties.16,18 In an RCT of 200 patients who underwent elective and nonelective hip and knee surgery and were randomized to either absorbent perforated dressing with adhesive border (Cutiplast, Smith & Nephew) or Aquacel (ConvaTec) covered with vapor-permeable dressing (Tegaderm, 3M), Ravenscroft and colleagues20 found that Aquacel-plus-Tegaderm was 5.8 times more likely than Cutiplast to produce an uncompromised wound. Similarly, in an RCT of hydrofiber (Aquacel) and central pad (Mepore, Mölnlycke) dressings after primary THA and TKA, Abuzakuk and colleagues19 found significantly fewer dressing changes (43% vs 77%) and blisters (13% vs 26%) in the hydrofiber group than in the pad group.
Hopper and colleagues24 compared 50 consecutive patients treated with modern dressings (Aquacel) with 50 historical control patients treated with traditional surgical dressings (Mepore). Blisters developed in 20% of the patients in the traditional group and 4% of patients in the modern group (P = .028). The authors concluded that adverse outcomes of wound healing can be minimized with modern dressings.
A recent retrospective study by Cai and colleagues25 evaluated the incidence of acute periprosthetic infection (≤3 months after surgery) with use of occlusive (Aquacel) and standard dressings. Incidence of acute periprosthetic infection was 0.44% in the occlusive group and 1.7% in the standard group (P = .005). Incidence of wound-healing problems was not evaluated.
Our second aim in the present study was to evaluate the number of dressing changes required. There were significantly fewer dressing changes in the occlusive dressing group than in the standard dressing group. Therefore, wear time (amount of time a single dressing remains in place) was substantially longer for the occlusive group. In the study by Hopper and colleagues,24 wear time was significantly shorter for the traditional dressing than for the modern dressing (2 vs 7 days; P < .001), and the traditional dressing required more changes (3 vs 0; P < .001).
These findings are important for several reasons. Standard surgical dressings often require frequent changes. If left in place, they create an excessively moist wound environment that promotes blistering and delays wound healing. However, frequent dressing changes expose the wound and increase the risk for surgical site infection.26 A barrier dressing left in place from time of surgery prevents bacteria from entering and contaminating a healing wound. A study by Clarke and colleagues21 demonstrated higher skin colonization rates for patients who had dressings changed on POD 1 than for patients who had their first dressing change on POD 6.
Our third study aim was to evaluate patient satisfaction with surgical dressings. The orthopedic literature has little on this topic.23 Blisters and other wound complications can negatively affect satisfaction.2,3 Our data showed significant improvement in satisfaction, particularly regarding sterility and hygiene.
Other surgical subspecialties have found similar improvement in patient satisfaction with occlusive barrier dressings. In an RCT of 88 pediatric patients, Rasmussen and colleagues27 found that patients reported significantly less pain during changes of an occlusive adhesive dressing (Duoderm, ConvaTec) than during changes of a conventional Steristrip (3M) plus Cutiplast. According to the authors, the occlusive wound dressing seemed to minimize the physical and psychological trauma to the infant or child and lessen disruption of the child’s and the parents’ daily routines, because the children could be bathed immediately after surgery.
Our study did not specifically address cost. Cai and colleagues25 estimated that, if the Aquacel dressing were routinely used in every hip and knee arthroplasty, it would add about $27 million in cost. However, this must be balanced by the cost of managing infection after TJA. In the United States, at an estimated $50,000 to $100,000 per case and an annual incidence of 1% to 2%, the low-end cost for the treatment of periprosthetic infection would be $500 million.28 Cai and colleagues25 found a 4-fold reduction in periprosthetic infection when use of occlusive dressings was implemented. In addition, wound complications remain the number one reason for hospital readmission after TJA.12,13 Cost of hospital readmission, as well as financial penalties to institutions for unplanned readmission for wound complications, must be considered.
Conclusion
Our RCT results demonstrated that use of occlusive antimicrobial barrier dressings (vs standard surgical dressings) significantly reduced wound complications and dressing changes and improved overall patient satisfaction. These findings are similar to those in the literature on TJA and other surgical subspecialties. We conclude that occlusive surgical dressings reduce wound complications after TJA.
Wound complications (eg, delayed wound healing, blisters, prolonged drainage) have been reported in up to 30% of patients who undergo elective total joint arthroplasty (TJA).1-6 Wound complications increase resource utilization, lengthen hospital stays, and increase costs.7-9 Prolonged wound healing and persistent wound drainage are also harbingers of both superficial and deep surgical site infections.5-11
In several studies, wound complications after TJA were the primary reason for hospital readmissions.12-15 As part of the Patient Protection and Affordable Care Act, hospitals will be penalized by the Centers for Medicaid & Medicare Services for unplanned hospital readmissions within 30 days after TJA. It is imperative, then, to reduce the risk factors and complications associated with surgical site infections to decrease unplanned readmissions.
Historically, little attention has been given to the role of surgical dressings and the effect of dressings on wound healing. Although many subspecialties (eg, cardiothoracic surgery, general surgery) have reported benefits in using occlusive dressings, adoption in TJA has been slow.16-18 At our institution about 5 years ago, we began using an occlusive silver-impregnated barrier dressing based on preliminary data from studies showing benefits of occlusive dressings in TJA.19,20
We conducted a study to determine if use of occlusive antimicrobial barrier dressings decreases rates of wound complications in TJA. We had 3 research questions: Compared with standard surgical dressings, are occlusive dressings associated with decreased rates of wound complications after TJA? Is there a difference in number of dressing changes required between the 2 dressing types? Is satisfaction higher for patients with occlusive dressings than for patients with standard dressings?
Patients and Methods
This randomized controlled trial (RCT) was reviewed and approved by the Institutional Review Board at Carolinas Healthcare. Patients were randomized by the research staff using a parallel, 1:1 allocation method. The randomization table was generated using a random number generator.
An a priori sample size estimate was made using a 2-tailed Fisher exact test with a .05 level of significance. Based on a study by Clarke and colleagues,21 we estimated the incidence of wound problems at 3% in the occlusive dressing (study) group and 13% in the standard dressing (control) group. We determined that 260 participants (130 per group) would be needed to achieve 80% power. We considered a 15% attrition rate for a total enrollment goal of 300 study participants (150 per group).
Between December 2010 and January 2013, patients presenting for either primary total hip arthroplasty (THA) or primary total knee arthroplasty (TKA) were recruited to participate in the study. Eligibility criteria (Table 1) were reviewed, and patients were enrolled by the senior surgeons, Dr. Springer, Dr. Beaver, Dr. Griffin, and Dr. Mason. All eligible participants who provided informed consent were randomized to receive either an occlusive antimicrobial barrier dressing (Aquacel Ag, ConvaTec) or standard surgical dressing (Primapore, Smith & Nephew). The occlusive dressing (Figure 1) consists of an outer barrier layer of hydrocolloid and a central island of hydrofiber, which absorbs and locks in any wound exudate within the fibers and prevents the creation of an overly moist wound environment that can lead to skin maceration and wound breakdown. In addition, the hydrofibers are embedded with ionic silver, which is released only at the site of wound exudate, or drainage; thus, there is no continuous exposure of the entire wound to silver. The standard dressing (Figure 2) consists of a central island of gauze enclosed in low-allergy acrylic adhesive tape.
All surgical dressings were placed over a closed incision in a sterile environment in the operating room after the procedure. The groups’ wound closures were identical.
A posterior approach was used for all THAs. The deep fascia was closed with a running barbed suture (Quill, Angiotech), the deep subcutaneous tissue with No. 1 Vicryl suture (Ethicon), and the superficial subcutaneous layer with 2-0 Vicryl suture. A running 3-0 Monocryl stitch (Ethicon) was placed in the subcuticular layer and was followed with a skin adhesive (Dermabond, Ethicon). A closed suction drain, removed on postoperative day (POD) 1, was used for all THAs.
A standard medial parapatellar arthrotomy was used for all TKAs. The arthrotomy was closed with a running barbed suture, the deep subcutaneous tissue with No. 1 Vicryl suture, and the superficial subcutaneous layer with 2-0 Vicryl suture. A running 3-0 Monocryl stitch was placed in the subcuticular layer and was followed with a skin adhesive. A closed suction drain was also used. In addition, a compressive wrap was placed over the dressing in the operating room and was removed the next morning. During the hospital stay, the surgical site was evaluated daily with a standard wound evaluation form.
In the standard dressing group, the bandage was removed for wound evaluation on POD 2, and the dressing was changed every other day during the hospital stay. The dressing was also changed as needed for wound drainage (Figure 3) or other minor wound-healing concerns.
In the occlusive dressing group, the dressing design allowed the dressing to remain in place for about 7 days. It was removed by a home health nurse during a visit closest to but not before the 7-day mark. In addition, it was changed at surgeon discretion if there were concerns about wound drainage or wound healing. For the occlusive barrier, wound drainage was evaluated by strike-through of drainage on the back side of the dressing (Figure 4). If more than 50% of the dressing was saturated, the bandage was changed and the wound evaluated. If there were no immediate concerns about wound complications (eg, infection, blistering), a new occlusive dressing was placed. Because the occlusive dressing was waterproof, patients in the study group were able to shower immediately after surgery. In the control group, patients were allowed to shower if the surgical dressing was kept dry, as the bandage was not waterproof.
Per the study protocol, all patients were discharged home and followed by a single home health agency. Mean hospital stay was 3 days (range, 0-8 days), which did not differ significantly between groups (P = .133). All home health nurses were trained in evaluation of postsurgical wounds and were aware of the study requirements. The nurses visited all patients 3 days a week until the scheduled 4-week postoperative follow-up with the treating physician or physician assistant. At each visit, the nurse evaluated the wound and surrounding skin using a standard wound document. Dressings were changed based on the criteria we have described. Concerns about wound status (eg, drainage, blistering, erythema) prompted removal of the dressing for further evaluation. The physician was notified of concerns about wound healing, which prompted an office visit for evaluation. The dressing remained in place for a minimum of 7 days but in all cases was removed as close to 7 days as possible, depending on the scheduled nursing visits. Once uneventful wound healing was complete, no further dressing was required. A final wound evaluation was conducted by the surgeon at the 4-week postoperative evaluation.
The primary outcome measure was wound complication (dichotomous variable). Wounds were assessed by describing the amount, type, and color of exudate (Figure 5). The appearance of the wound margins and the surrounding skin was also assessed. Because wounds could not be directly visualized in the occlusive dressing group, drainage (indicated by strike-through) was used as a measure of possible wound complications, prompting removal and full evaluation.
Secondary endpoints included additional wound treatment or surgical procedures for wound complications, number of dressing changes, and patient satisfaction. Patients completed a satisfaction questionnaire at each wound assessment (Figure 6). Using a visual analog scale (VAS), they rated their satisfaction with their ability to perform activities of daily living (personal hygiene, change clothes, sit comfortably, sleep comfortably), drawing a line on the VAS at a point between 0 (totally unsatisfied) and 100 (totally satisfied) for each satisfaction measure. This line was measured and recorded by the study coordinator. The 4 satisfaction measures were averaged for a composite satisfaction measure.
All statistical analyses were conducted using SAS Version 9.2 (SAS Institute). Standard univariate descriptive statistics (means, standard deviations, frequencies, proportions) were calculated and reported. Differences in mean values for continuous data were assessed with independent t test or Wilcoxon rank sum test. Chi-square test and Fisher exact test were used to determine differences between groups for categorical or dichotomous variables. A significance level of .05 was used for all statistical tests.
Results
The 300 patients who consented to participate in the study were randomized to receive either occlusive dressing or standard dressing. After randomization, 38 patients (15 occlusive, 23 standard) were withdrawn from the study (Table 2), leaving a final dataset of 262 patients, 141 in the occlusive group (67 THAs, 74 TKAs) and 121 in the standard group (49 THAs, 72 TKAs). There were no differences in proportion of THAs or TKAs, age, sex, or body mass index between the occlusive and standard groups (Table 3).
There were statistically significantly (P = .015) fewer wound complications in the occlusive dressing group (10%) than in the standard dressing group (22%). Blisters at or around the wound site were reported in significantly (P = .026) fewer patients with occlusive dressing (1/141, 0.7%) than standard dressing (7/121, 6%). Additional wound care was required in 9 patients (7%) in the standard group and 6 patients (4%) in the occlusive group (P = .27). Two patients (1.7%) in the standard group were readmitted for treatment of wound dehiscence; no one in the occlusive group was readmitted to the hospital or had to return to the operating room for treatment of a wound complication. The difference was not statistically significant (P = .13). There were also no significant (P = .81) differences in rate of wound complications between THA and TKA patients.
There were statistically significantly (P < .0001) fewer dressing changes in the occlusive dressing group. Mean number of dressing changes was 0.14 (median, 0; interquartile range, 0-0) in the occlusive group and 2.8 (median, 2; interquartile range, 1-3) in the standard group.
Compared with patients in the standard dressing group, patients in the occlusive dressing group reported significantly higher satisfaction scores. Mean overall patient satisfaction score was 92 in the occlusive group and 81 in the standard group (P < .0001). Patients in the occlusive group were more satisfied with their ability to take care of their personal hygiene, to change clothes, and to sit and sleep comfortably (Table 4).
Discussion
Wound complications after TJA are common, occurring in up to 30% of patients,1-6 and are associated with development of superficial and deep surgical site infections, increased resource utilization, and longer hospital stays.5-11 Although the role of surgical dressings has received little attention in TJA practice, other subspecialties have found that occlusive barrier dressings can reduce wound complications and promote wound healing.16,17 Mitotic cell division and leukocyte activity, which are critical in wound healing, increase under occlusive dressings. This cellular activity is disrupted with every dressing change, delaying wound healing (biological activity takes 3-4 hours to resume).22 In addition, occlusive dressings increase hypoxia, which promotes angiogenesis and accelerates wound healing.23
Despite being a prospective RCT, this study had several limitations. Because of the need to evaluate wounds and obvious differences between the 2 dressings (eg, color, ability to shower), it was not possible to blind the patient or surgeon to the dressing used. When rating satisfaction, patients were not able to directly compare the 2 dressings. The primary endpoint of the study was the complication rate; however, the deep periprosthetic infection rate may be a superior endpoint and would require a much larger study. Although we assumed that wound complications may be harbingers for periprosthetic infections, no patient in either group developed periprosthetic infection. Therefore, we cannot conclude that surgical dressings play a role in reducing infections. In addition, as the standard dressing was changed on POD 2 (per standard protocol) and the occlusive dressing could remain in place for up to 7 days, there was a selection bias in the evaluation of the number of dressing changes. However, given the characteristics of the standard dressing (eg, tape, gauze, nonocclusive), leaving it in place after POD 2 is not optimal. Therefore, we would expect to see a difference in the number of dressing changes. We think this comparison remains valid, as occlusive dressings were changed when there were indications of wound problems (eg, excessive drainage [strike-through], surrounding erythema, blistering). With an average of less than 1 dressing change in the occlusive group, we think this is a surrogate for uneventful wound healing and decreased wound complication, and our data support this. It is also important to test both dressing durability and patient tolerance for wearing a single dressing for 7 days.
Our RCT results showed that, compared with a standard dressing, an occlusive antimicrobial dressing was associated with a significant decrease in overall wound complications and blisters. These findings are similar to those of other studies of occlusive dressings in a number of surgical subspecialties.16,18 In an RCT of 200 patients who underwent elective and nonelective hip and knee surgery and were randomized to either absorbent perforated dressing with adhesive border (Cutiplast, Smith & Nephew) or Aquacel (ConvaTec) covered with vapor-permeable dressing (Tegaderm, 3M), Ravenscroft and colleagues20 found that Aquacel-plus-Tegaderm was 5.8 times more likely than Cutiplast to produce an uncompromised wound. Similarly, in an RCT of hydrofiber (Aquacel) and central pad (Mepore, Mölnlycke) dressings after primary THA and TKA, Abuzakuk and colleagues19 found significantly fewer dressing changes (43% vs 77%) and blisters (13% vs 26%) in the hydrofiber group than in the pad group.
Hopper and colleagues24 compared 50 consecutive patients treated with modern dressings (Aquacel) with 50 historical control patients treated with traditional surgical dressings (Mepore). Blisters developed in 20% of the patients in the traditional group and 4% of patients in the modern group (P = .028). The authors concluded that adverse outcomes of wound healing can be minimized with modern dressings.
A recent retrospective study by Cai and colleagues25 evaluated the incidence of acute periprosthetic infection (≤3 months after surgery) with use of occlusive (Aquacel) and standard dressings. Incidence of acute periprosthetic infection was 0.44% in the occlusive group and 1.7% in the standard group (P = .005). Incidence of wound-healing problems was not evaluated.
Our second aim in the present study was to evaluate the number of dressing changes required. There were significantly fewer dressing changes in the occlusive dressing group than in the standard dressing group. Therefore, wear time (amount of time a single dressing remains in place) was substantially longer for the occlusive group. In the study by Hopper and colleagues,24 wear time was significantly shorter for the traditional dressing than for the modern dressing (2 vs 7 days; P < .001), and the traditional dressing required more changes (3 vs 0; P < .001).
These findings are important for several reasons. Standard surgical dressings often require frequent changes. If left in place, they create an excessively moist wound environment that promotes blistering and delays wound healing. However, frequent dressing changes expose the wound and increase the risk for surgical site infection.26 A barrier dressing left in place from time of surgery prevents bacteria from entering and contaminating a healing wound. A study by Clarke and colleagues21 demonstrated higher skin colonization rates for patients who had dressings changed on POD 1 than for patients who had their first dressing change on POD 6.
Our third study aim was to evaluate patient satisfaction with surgical dressings. The orthopedic literature has little on this topic.23 Blisters and other wound complications can negatively affect satisfaction.2,3 Our data showed significant improvement in satisfaction, particularly regarding sterility and hygiene.
Other surgical subspecialties have found similar improvement in patient satisfaction with occlusive barrier dressings. In an RCT of 88 pediatric patients, Rasmussen and colleagues27 found that patients reported significantly less pain during changes of an occlusive adhesive dressing (Duoderm, ConvaTec) than during changes of a conventional Steristrip (3M) plus Cutiplast. According to the authors, the occlusive wound dressing seemed to minimize the physical and psychological trauma to the infant or child and lessen disruption of the child’s and the parents’ daily routines, because the children could be bathed immediately after surgery.
Our study did not specifically address cost. Cai and colleagues25 estimated that, if the Aquacel dressing were routinely used in every hip and knee arthroplasty, it would add about $27 million in cost. However, this must be balanced by the cost of managing infection after TJA. In the United States, at an estimated $50,000 to $100,000 per case and an annual incidence of 1% to 2%, the low-end cost for the treatment of periprosthetic infection would be $500 million.28 Cai and colleagues25 found a 4-fold reduction in periprosthetic infection when use of occlusive dressings was implemented. In addition, wound complications remain the number one reason for hospital readmission after TJA.12,13 Cost of hospital readmission, as well as financial penalties to institutions for unplanned readmission for wound complications, must be considered.
Conclusion
Our RCT results demonstrated that use of occlusive antimicrobial barrier dressings (vs standard surgical dressings) significantly reduced wound complications and dressing changes and improved overall patient satisfaction. These findings are similar to those in the literature on TJA and other surgical subspecialties. We conclude that occlusive surgical dressings reduce wound complications after TJA.
1. Cosker T, Elsayed S, Gupta S, Mendonca AD, Tayton KJ. Choice of dressing has a major impact on blistering and healing outcomes in orthopaedic patients. J Wound Care. 2005;14(1):27-29.
2. Koval KJ, Egol KA, Hiebert R, Spratt KF. Tape blisters after hip surgery: can they be eliminated completely? Am J Orthop. 2007;36(5):261-265.
3. Lawrentschuk N, Falkenberg MP, Pirpiris M. Wound blisters post hip surgery: a prospective trial comparing dressings. ANZ J Surg. 2002;72(10):716-719.
4. Mihalko WM, Manaswi A, Brown TE, Parvizi J, Schmalzried TP, Saleh KJ. Infection in primary total knee arthroplasty: contributing factors. Instr Course Lect. 2008;57:317-325.
5. Patel VP, Walsh M, Sehgal B, Preston C, DeWal H, Di Cesare PE. Factors associated with prolonged wound drainage after primary total hip and knee arthroplasty. J Bone Joint Surg Am. 2007;89(1):33-38.
6. Vince KG, Abdeen A. Wound problems in total knee arthroplasty. Clin Orthop Relat Res. 2006;(452):88-90.
7. Galat DD, McGovern SC, Larson DR, Harrington JR, Hanssen AD, Clarke HD. Surgical treatment of early wound complications following primary total knee arthroplasty. J Bone Joint Surg Am. 2009;91(1):48-54.
8. Gordon SM, Culver DH, Simmons BP, Jarvis WR. Risk factors for wound infections after total knee arthroplasty. Am J Epidemiol. 1990;131(5):905-916.
9. Jaberi FM, Parvizi J, Haytmanek CT, Joshi A, Purtill J. Procrastination of wound drainage and malnutrition affect the outcome of joint arthroplasty. Clin Orthop Relat Res. 2008;466(6):1368-1371.
10. Schmalzried TP. The infected hip: telltale signs and treatment options. J Arthroplasty. 2006;21(4 suppl 1):97-100.
11. Weiss AP, Krackow KA. Persistent wound drainage after primary total knee arthroplasty. J Arthroplasty. 1993;8(3):285-289.
12. Avram V, Petruccelli D, Winemaker M, de Beer J. Total joint arthroplasty readmission rates and reasons for 30-day hospital readmission. J Arthroplasty. 2014;29(3):465-468.
13. Dailey EA, Cizik A, Kasten J, Chapman JR, Lee MJ. Risk factors for readmission of orthopaedic surgical patients. J Bone Joint Surg Am. 2013;95(11):1012-1019.
14. Jordan CJ, Goldstein RY, Michels RF, Hutzler L, Slover JD, Bosco JA 3rd. Comprehensive program reduces hospital readmission rates after total joint arthroplasty. Am J Orthop. 2012;41(11):E147-E151.
15. Schairer WW, Sing DC, Vail TP, Bozic KJ. Causes and frequency of unplanned hospital readmission after total hip arthroplasty. Clin Orthop Relat Res. 2014;472(2):464-470.
16. Shinohara T, Yamashita Y, Satoh K, et al. Prospective evaluation of occlusive hydrocolloid dressing versus conventional gauze dressing regarding the healing effect after abdominal operations: randomized controlled trial. Asian J Surg. 2008;31(1):1-5.
17. Siah CJ, Yatim J. Efficacy of a total occlusive ionic silver-containing dressing combination in decreasing risk of surgical site infection: an RCT. J Wound Care. 2011;20(12):561-568.
18. Teshima H, Kawano H, Kashikie H, et al. A new hydrocolloid dressing prevents surgical site infection of median sternotomy wounds. Surg Today. 2009;39(10):848-854.
19. Abuzakuk TM, Coward P, Shenava Y, Kumar VS, Skinner JA. The management of wounds following primary lower limb arthroplasty: a prospective, randomised study comparing hydrofibre and central pad dressings. Int Wound J. 2006;3(2):133-137.
20. Ravenscroft MJ, Harker J, Buch KA. A prospective, randomised, controlled trial comparing wound dressings used in hip and knee surgery: Aquacel and Tegaderm versus Cutiplast. Ann R Coll Surg Engl. 2006;88(1):18-22.
21. Clarke JV, Deakin AH, Dillon JM, Emmerson S, Kinninmonth AW. A prospective clinical audit of a new dressing design for lower limb arthroplasty wounds. J Wound Care. 2009;18(1):5-8, 10-11.
22. Kloeters O. The use of a semi-occlusive dressing reduces epidermal inflammatory cytokine expression and mitigates dermal proliferation and inflammation in a rat incisional model. Wound Repair Regen. 2008;16(4):568-575.
23. Michie DD, Hugill JV. Influence of occlusive and impregnated gauze dressings on incisional healing: a prospective, randomized, controlled study. Ann Plast Surg. 1994;32(1):57-64.
24. Hopper GP, Deakin AH, Crane EO, Clarke JV. Enhancing patient recovery following lower limb arthroplasty with a modern wound dressing: a prospective, comparative audit. J Wound Care. 2012;21(4):200-203.
25. Cai J, Karam JA, Parvizi J, Smith EB, Sharkey PF. Aquacel surgical dressing reduces the rate of acute PJI following total joint arthroplasty: a case–control study. J Arthroplasty. 2014;29(6):1098-1100.
26. Berg A, Fleischer S, Kuss O, Unverzagt S, Langer G. Timing of dressing removal in the healing of surgical wounds by primary intention: quantitative systematic review protocol. J Adv Nurs. 2012;68(2):264-270.
27. Rasmussen H, Larsen MJ, Skeie E. Surgical wound dressing in outpatient paediatric surgery. A randomised study. Dan Med Bull. 1993;40(2):252-254.
28. Kurtz SM, Lau E, Schmier J, Ong KL, Zhao K, Parvizi J. Infection burden for hip and knee arthroplasty in the United States. J Arthroplasty. 2008;23(7):984-991.
1. Cosker T, Elsayed S, Gupta S, Mendonca AD, Tayton KJ. Choice of dressing has a major impact on blistering and healing outcomes in orthopaedic patients. J Wound Care. 2005;14(1):27-29.
2. Koval KJ, Egol KA, Hiebert R, Spratt KF. Tape blisters after hip surgery: can they be eliminated completely? Am J Orthop. 2007;36(5):261-265.
3. Lawrentschuk N, Falkenberg MP, Pirpiris M. Wound blisters post hip surgery: a prospective trial comparing dressings. ANZ J Surg. 2002;72(10):716-719.
4. Mihalko WM, Manaswi A, Brown TE, Parvizi J, Schmalzried TP, Saleh KJ. Infection in primary total knee arthroplasty: contributing factors. Instr Course Lect. 2008;57:317-325.
5. Patel VP, Walsh M, Sehgal B, Preston C, DeWal H, Di Cesare PE. Factors associated with prolonged wound drainage after primary total hip and knee arthroplasty. J Bone Joint Surg Am. 2007;89(1):33-38.
6. Vince KG, Abdeen A. Wound problems in total knee arthroplasty. Clin Orthop Relat Res. 2006;(452):88-90.
7. Galat DD, McGovern SC, Larson DR, Harrington JR, Hanssen AD, Clarke HD. Surgical treatment of early wound complications following primary total knee arthroplasty. J Bone Joint Surg Am. 2009;91(1):48-54.
8. Gordon SM, Culver DH, Simmons BP, Jarvis WR. Risk factors for wound infections after total knee arthroplasty. Am J Epidemiol. 1990;131(5):905-916.
9. Jaberi FM, Parvizi J, Haytmanek CT, Joshi A, Purtill J. Procrastination of wound drainage and malnutrition affect the outcome of joint arthroplasty. Clin Orthop Relat Res. 2008;466(6):1368-1371.
10. Schmalzried TP. The infected hip: telltale signs and treatment options. J Arthroplasty. 2006;21(4 suppl 1):97-100.
11. Weiss AP, Krackow KA. Persistent wound drainage after primary total knee arthroplasty. J Arthroplasty. 1993;8(3):285-289.
12. Avram V, Petruccelli D, Winemaker M, de Beer J. Total joint arthroplasty readmission rates and reasons for 30-day hospital readmission. J Arthroplasty. 2014;29(3):465-468.
13. Dailey EA, Cizik A, Kasten J, Chapman JR, Lee MJ. Risk factors for readmission of orthopaedic surgical patients. J Bone Joint Surg Am. 2013;95(11):1012-1019.
14. Jordan CJ, Goldstein RY, Michels RF, Hutzler L, Slover JD, Bosco JA 3rd. Comprehensive program reduces hospital readmission rates after total joint arthroplasty. Am J Orthop. 2012;41(11):E147-E151.
15. Schairer WW, Sing DC, Vail TP, Bozic KJ. Causes and frequency of unplanned hospital readmission after total hip arthroplasty. Clin Orthop Relat Res. 2014;472(2):464-470.
16. Shinohara T, Yamashita Y, Satoh K, et al. Prospective evaluation of occlusive hydrocolloid dressing versus conventional gauze dressing regarding the healing effect after abdominal operations: randomized controlled trial. Asian J Surg. 2008;31(1):1-5.
17. Siah CJ, Yatim J. Efficacy of a total occlusive ionic silver-containing dressing combination in decreasing risk of surgical site infection: an RCT. J Wound Care. 2011;20(12):561-568.
18. Teshima H, Kawano H, Kashikie H, et al. A new hydrocolloid dressing prevents surgical site infection of median sternotomy wounds. Surg Today. 2009;39(10):848-854.
19. Abuzakuk TM, Coward P, Shenava Y, Kumar VS, Skinner JA. The management of wounds following primary lower limb arthroplasty: a prospective, randomised study comparing hydrofibre and central pad dressings. Int Wound J. 2006;3(2):133-137.
20. Ravenscroft MJ, Harker J, Buch KA. A prospective, randomised, controlled trial comparing wound dressings used in hip and knee surgery: Aquacel and Tegaderm versus Cutiplast. Ann R Coll Surg Engl. 2006;88(1):18-22.
21. Clarke JV, Deakin AH, Dillon JM, Emmerson S, Kinninmonth AW. A prospective clinical audit of a new dressing design for lower limb arthroplasty wounds. J Wound Care. 2009;18(1):5-8, 10-11.
22. Kloeters O. The use of a semi-occlusive dressing reduces epidermal inflammatory cytokine expression and mitigates dermal proliferation and inflammation in a rat incisional model. Wound Repair Regen. 2008;16(4):568-575.
23. Michie DD, Hugill JV. Influence of occlusive and impregnated gauze dressings on incisional healing: a prospective, randomized, controlled study. Ann Plast Surg. 1994;32(1):57-64.
24. Hopper GP, Deakin AH, Crane EO, Clarke JV. Enhancing patient recovery following lower limb arthroplasty with a modern wound dressing: a prospective, comparative audit. J Wound Care. 2012;21(4):200-203.
25. Cai J, Karam JA, Parvizi J, Smith EB, Sharkey PF. Aquacel surgical dressing reduces the rate of acute PJI following total joint arthroplasty: a case–control study. J Arthroplasty. 2014;29(6):1098-1100.
26. Berg A, Fleischer S, Kuss O, Unverzagt S, Langer G. Timing of dressing removal in the healing of surgical wounds by primary intention: quantitative systematic review protocol. J Adv Nurs. 2012;68(2):264-270.
27. Rasmussen H, Larsen MJ, Skeie E. Surgical wound dressing in outpatient paediatric surgery. A randomised study. Dan Med Bull. 1993;40(2):252-254.
28. Kurtz SM, Lau E, Schmier J, Ong KL, Zhao K, Parvizi J. Infection burden for hip and knee arthroplasty in the United States. J Arthroplasty. 2008;23(7):984-991.