User login
GOLD: Base COPD treatment on symptoms, exacerbation risk
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) has uncoupled spirometry results from the ABCD treatment algorithm; this move marks the organization’s first announcing of major COPD guidance since 2011.
Spirometry now stands apart from GOLD’s ABCD symptom/exacerbation risk score with its own grade, with possibilities ranging from 1 to 4. A forced expiratory volume in 1 second (FEV1) of 80% or more of the predicted value rates a 1; the score degrades to 4 with an FEV1 below 30%.

“In previous GOLD documents, recommendations for management of COPD were based solely on spirometric category. However, there is considerable evidence that the level of FEV1 is a poor descriptor of disease status, and, for this reason, the management of stable COPD based on … disease impact (determined mainly by symptom burden and activity limitation) and future risk of disease progression (especially of exacerbations) is recommended. ... ABCD groups are now proposed to be derived exclusively from patient symptoms and their history of exacerbations,” GOLD said.
The clear focus on symptoms and exacerbations is “the major accomplishment” of the new report, which has been downloaded more than 45,000 times since it’s release, a testament to GOLD’s importance to clinicians trying to help COPD patients.
“We are trying to do a better job of personalizing treatment,” said GOLD board member Gerard Criner, MD, chair and professor of thoracic medicine and surgery at Temple University in Philadelphia.
The change “allows you to plan treatment based on symptoms [even] if you don’t have immediate access to spirometry, and then refine treatment once you have spirometry results. It also allows you to escalate and deescalate treatment because you are not boxed into a letter grade group” forced by spirometry. “You can also take a better look at pharmacologic versus nonpharmacologic therapy” when deciding what to do, he said.
In short, “we think it gives more freedom” to manage patients based on what seems best, Dr. Criner said.
GOLD included an example of how the new assessment can help. “Consider two patients,” it said, both with an FEV1 less than 30% and a COPD Assessment Test result of 18, but one with no exacerbations in the past year and the other with three. Both would have scored a GOLD D in the old system, and been treated similarly.
“However, with the new proposed scheme, the subject with three exacerbations ... would be labeled GOLD [spirometry] grade 4, group D,” and their treatment would focus on exacerbations. The no-exacerbation patient would be classified as GOLD grade 4, group B. Treatment would focus on symptoms. Drugs are still an option, but also lung volume reduction and lung transplant, GOLD said. Spirometry, in other words, is less important than how the patient is doing.
The group incorporated “every major study up to the first week of November” in the new report, Dr. Criner said, so there’s more to consider.
For instance, it’s clear now that patients benefit from home oxygen if they are severely hypoxemic while sitting on the couch watching TV, but not if they desaturate only when they get up and walk around, or come into the clinic to exercise. “We did not” know that in 2011, he said.
GOLD also recommended pulmonary rehabilitation and palliative care when indicated, as well as ongoing evaluation to make sure patients are able to use their inhalers, a major problem in COPD.
GOLD said that group A patients - those with few symptoms and low exacerbation risk - should be offered a bronchodilator. Initial therapy for group B - more symptoms, but low exacerbation risk - and group C - higher exacerbation risk but fewer symptoms - “should consist of a single long-acting bronchodilator. There is no evidence to recommend one class of long-acting bronchodilator over another.”
For group D - highly symptomatic with frequent exacerbations - “we recommend starting therapy with a [long-acting beta-2 agonist]/[long-acting antimuscarinic antagonist] combination,” the group said.
There was no industry involvement in GOLD’s report, but numerous authors and board members had pharmaceutical company ties, and GOLD’s treatment advice relies on drug company studies. Dr. Criner reported personal payments from Holaria, and research funding and other nonpersonal payments from AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Johnson and Johnson, and others.
As health care moves toward individualized care plans for patients, the updated GOLD recommendations enhance the possibility of personalized COPD treatment. This means more symptom-focused treatment for patients and, as Dr. Criner points out, more freedom for providers to manage patients based on what seems best.
As health care moves toward individualized care plans for patients, the updated GOLD recommendations enhance the possibility of personalized COPD treatment. This means more symptom-focused treatment for patients and, as Dr. Criner points out, more freedom for providers to manage patients based on what seems best.
As health care moves toward individualized care plans for patients, the updated GOLD recommendations enhance the possibility of personalized COPD treatment. This means more symptom-focused treatment for patients and, as Dr. Criner points out, more freedom for providers to manage patients based on what seems best.
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) has uncoupled spirometry results from the ABCD treatment algorithm; this move marks the organization’s first announcing of major COPD guidance since 2011.
Spirometry now stands apart from GOLD’s ABCD symptom/exacerbation risk score with its own grade, with possibilities ranging from 1 to 4. A forced expiratory volume in 1 second (FEV1) of 80% or more of the predicted value rates a 1; the score degrades to 4 with an FEV1 below 30%.
“In previous GOLD documents, recommendations for management of COPD were based solely on spirometric category. However, there is considerable evidence that the level of FEV1 is a poor descriptor of disease status, and, for this reason, the management of stable COPD based on … disease impact (determined mainly by symptom burden and activity limitation) and future risk of disease progression (especially of exacerbations) is recommended. ... ABCD groups are now proposed to be derived exclusively from patient symptoms and their history of exacerbations,” GOLD said.
The clear focus on symptoms and exacerbations is “the major accomplishment” of the new report, which has been downloaded more than 45,000 times since it’s release, a testament to GOLD’s importance to clinicians trying to help COPD patients.
“We are trying to do a better job of personalizing treatment,” said GOLD board member Gerard Criner, MD, chair and professor of thoracic medicine and surgery at Temple University in Philadelphia.
The change “allows you to plan treatment based on symptoms [even] if you don’t have immediate access to spirometry, and then refine treatment once you have spirometry results. It also allows you to escalate and deescalate treatment because you are not boxed into a letter grade group” forced by spirometry. “You can also take a better look at pharmacologic versus nonpharmacologic therapy” when deciding what to do, he said.
In short, “we think it gives more freedom” to manage patients based on what seems best, Dr. Criner said.
GOLD included an example of how the new assessment can help. “Consider two patients,” it said, both with an FEV1 less than 30% and a COPD Assessment Test result of 18, but one with no exacerbations in the past year and the other with three. Both would have scored a GOLD D in the old system, and been treated similarly.
“However, with the new proposed scheme, the subject with three exacerbations ... would be labeled GOLD [spirometry] grade 4, group D,” and their treatment would focus on exacerbations. The no-exacerbation patient would be classified as GOLD grade 4, group B. Treatment would focus on symptoms. Drugs are still an option, but also lung volume reduction and lung transplant, GOLD said. Spirometry, in other words, is less important than how the patient is doing.
The group incorporated “every major study up to the first week of November” in the new report, Dr. Criner said, so there’s more to consider.
For instance, it’s clear now that patients benefit from home oxygen if they are severely hypoxemic while sitting on the couch watching TV, but not if they desaturate only when they get up and walk around, or come into the clinic to exercise. “We did not” know that in 2011, he said.
GOLD also recommended pulmonary rehabilitation and palliative care when indicated, as well as ongoing evaluation to make sure patients are able to use their inhalers, a major problem in COPD.
GOLD said that group A patients - those with few symptoms and low exacerbation risk - should be offered a bronchodilator. Initial therapy for group B - more symptoms, but low exacerbation risk - and group C - higher exacerbation risk but fewer symptoms - “should consist of a single long-acting bronchodilator. There is no evidence to recommend one class of long-acting bronchodilator over another.”
For group D - highly symptomatic with frequent exacerbations - “we recommend starting therapy with a [long-acting beta-2 agonist]/[long-acting antimuscarinic antagonist] combination,” the group said.
There was no industry involvement in GOLD’s report, but numerous authors and board members had pharmaceutical company ties, and GOLD’s treatment advice relies on drug company studies. Dr. Criner reported personal payments from Holaria, and research funding and other nonpersonal payments from AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Johnson and Johnson, and others.
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) has uncoupled spirometry results from the ABCD treatment algorithm; this move marks the organization’s first announcing of major COPD guidance since 2011.
Spirometry now stands apart from GOLD’s ABCD symptom/exacerbation risk score with its own grade, with possibilities ranging from 1 to 4. A forced expiratory volume in 1 second (FEV1) of 80% or more of the predicted value rates a 1; the score degrades to 4 with an FEV1 below 30%.
“In previous GOLD documents, recommendations for management of COPD were based solely on spirometric category. However, there is considerable evidence that the level of FEV1 is a poor descriptor of disease status, and, for this reason, the management of stable COPD based on … disease impact (determined mainly by symptom burden and activity limitation) and future risk of disease progression (especially of exacerbations) is recommended. ... ABCD groups are now proposed to be derived exclusively from patient symptoms and their history of exacerbations,” GOLD said.
The clear focus on symptoms and exacerbations is “the major accomplishment” of the new report, which has been downloaded more than 45,000 times since it’s release, a testament to GOLD’s importance to clinicians trying to help COPD patients.
“We are trying to do a better job of personalizing treatment,” said GOLD board member Gerard Criner, MD, chair and professor of thoracic medicine and surgery at Temple University in Philadelphia.
The change “allows you to plan treatment based on symptoms [even] if you don’t have immediate access to spirometry, and then refine treatment once you have spirometry results. It also allows you to escalate and deescalate treatment because you are not boxed into a letter grade group” forced by spirometry. “You can also take a better look at pharmacologic versus nonpharmacologic therapy” when deciding what to do, he said.
In short, “we think it gives more freedom” to manage patients based on what seems best, Dr. Criner said.
GOLD included an example of how the new assessment can help. “Consider two patients,” it said, both with an FEV1 less than 30% and a COPD Assessment Test result of 18, but one with no exacerbations in the past year and the other with three. Both would have scored a GOLD D in the old system, and been treated similarly.
“However, with the new proposed scheme, the subject with three exacerbations ... would be labeled GOLD [spirometry] grade 4, group D,” and their treatment would focus on exacerbations. The no-exacerbation patient would be classified as GOLD grade 4, group B. Treatment would focus on symptoms. Drugs are still an option, but also lung volume reduction and lung transplant, GOLD said. Spirometry, in other words, is less important than how the patient is doing.
The group incorporated “every major study up to the first week of November” in the new report, Dr. Criner said, so there’s more to consider.
For instance, it’s clear now that patients benefit from home oxygen if they are severely hypoxemic while sitting on the couch watching TV, but not if they desaturate only when they get up and walk around, or come into the clinic to exercise. “We did not” know that in 2011, he said.
GOLD also recommended pulmonary rehabilitation and palliative care when indicated, as well as ongoing evaluation to make sure patients are able to use their inhalers, a major problem in COPD.
GOLD said that group A patients - those with few symptoms and low exacerbation risk - should be offered a bronchodilator. Initial therapy for group B - more symptoms, but low exacerbation risk - and group C - higher exacerbation risk but fewer symptoms - “should consist of a single long-acting bronchodilator. There is no evidence to recommend one class of long-acting bronchodilator over another.”
For group D - highly symptomatic with frequent exacerbations - “we recommend starting therapy with a [long-acting beta-2 agonist]/[long-acting antimuscarinic antagonist] combination,” the group said.
There was no industry involvement in GOLD’s report, but numerous authors and board members had pharmaceutical company ties, and GOLD’s treatment advice relies on drug company studies. Dr. Criner reported personal payments from Holaria, and research funding and other nonpersonal payments from AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Johnson and Johnson, and others.
New guidelines provide standardized hypoglycemia values for clinical evaluation
, joining with the European Association for the Study of Diabetes to specify that a level of less than 3 mmol/L (54 mg/dL) should be considered “clinically important hypoglycemia.”
“A single glucose level should be agreed to that has serious clinical and health-economic consequences,” the ADA and EASD stated in the new guidelines. “This would enable the diabetes and regulatory communities to compare the effectiveness of interventions in reducing hypoglycemia, be they pharmacological, technological, or educational. It would also permit the use of meta-analysis as a statistical tool to increase power when comparing interventions.”
An international, multidisciplinary group – the International Hypoglycemia Study Group – was formed to create distinct definitions of the various levels of severity that hypoglycemia can have. The new guidelines contain three levels, which should be used by clinicians to determine what amounts of blood glucose are significant enough to be clinically reported.

“Currently, there is no uniform agreement to what constitutes reportable hypoglycemia in clinical trials,” she said. “In some studies, it is defined as a blood glucose level of less than 70 mg/dL, whereas in others it is defined as a blood glucose level less than 54 mg/dL.”
The guidelines define first-level hypoglycemia as any glucose level of 3.9 mmol/L (70 mg/dL) or less. This is not considered low enough to be reported on a consistent basis in clinical studies; however, that determination must ultimately be made by the investigators, as the parameters for what is significant often vary from study to study.
The second level is the 3 mmol/L (54 mg/dL), which now is deemed to be a clinically significant level of hypoglycemia. Because it is “sufficiently low to indicate serious, clinically important hypoglycemia,” it should be reported as part of any clinical studies. Finally, the third level, less than 2.8 mmol/L (50 mg/dL), indicates severe hypoglycemia and is classified as any individual with “severe cognitive impairment requiring external assistance for recovery,” according to the guidelines (Diabetes Care. 2016 Dec 1. doi: 10.2337/dc16-2215).

With a new standard of hypoglycemic values that are deemed clinically significant, the ADA and EASD hope that comparing different insulins, medications, technologies, and educational interventions will now become easier and more standardized, leading to better care worldwide.
Although there is general agreement as to where severe hypoglycemia really begins, the newly defined glucose levels are “a step in the right direction,” according to Dr. Rodbard.
The International Hypoglycaemia Study Group developed these guidelines through a grant from Novo Nordisk, awarded to the Six Degrees Academy of Toronto. Dr. Heller has received advisory or consultation fees from Lilly, Novo Nordisk, Takeda, Merck, Sharp & Dohme, and Becton Dickinson; has served as a speaker for AstraZeneca, Lilly, Novo Nordisk, Boehringer Ingelheim, and Takeda; and has received research support from Medtronic U.K. Dr. Rodbard did not report any financial disclosures.
, joining with the European Association for the Study of Diabetes to specify that a level of less than 3 mmol/L (54 mg/dL) should be considered “clinically important hypoglycemia.”
“A single glucose level should be agreed to that has serious clinical and health-economic consequences,” the ADA and EASD stated in the new guidelines. “This would enable the diabetes and regulatory communities to compare the effectiveness of interventions in reducing hypoglycemia, be they pharmacological, technological, or educational. It would also permit the use of meta-analysis as a statistical tool to increase power when comparing interventions.”
An international, multidisciplinary group – the International Hypoglycemia Study Group – was formed to create distinct definitions of the various levels of severity that hypoglycemia can have. The new guidelines contain three levels, which should be used by clinicians to determine what amounts of blood glucose are significant enough to be clinically reported.
“Currently, there is no uniform agreement to what constitutes reportable hypoglycemia in clinical trials,” she said. “In some studies, it is defined as a blood glucose level of less than 70 mg/dL, whereas in others it is defined as a blood glucose level less than 54 mg/dL.”
The guidelines define first-level hypoglycemia as any glucose level of 3.9 mmol/L (70 mg/dL) or less. This is not considered low enough to be reported on a consistent basis in clinical studies; however, that determination must ultimately be made by the investigators, as the parameters for what is significant often vary from study to study.
The second level is the 3 mmol/L (54 mg/dL), which now is deemed to be a clinically significant level of hypoglycemia. Because it is “sufficiently low to indicate serious, clinically important hypoglycemia,” it should be reported as part of any clinical studies. Finally, the third level, less than 2.8 mmol/L (50 mg/dL), indicates severe hypoglycemia and is classified as any individual with “severe cognitive impairment requiring external assistance for recovery,” according to the guidelines (Diabetes Care. 2016 Dec 1. doi: 10.2337/dc16-2215).
With a new standard of hypoglycemic values that are deemed clinically significant, the ADA and EASD hope that comparing different insulins, medications, technologies, and educational interventions will now become easier and more standardized, leading to better care worldwide.
Although there is general agreement as to where severe hypoglycemia really begins, the newly defined glucose levels are “a step in the right direction,” according to Dr. Rodbard.
The International Hypoglycaemia Study Group developed these guidelines through a grant from Novo Nordisk, awarded to the Six Degrees Academy of Toronto. Dr. Heller has received advisory or consultation fees from Lilly, Novo Nordisk, Takeda, Merck, Sharp & Dohme, and Becton Dickinson; has served as a speaker for AstraZeneca, Lilly, Novo Nordisk, Boehringer Ingelheim, and Takeda; and has received research support from Medtronic U.K. Dr. Rodbard did not report any financial disclosures.
, joining with the European Association for the Study of Diabetes to specify that a level of less than 3 mmol/L (54 mg/dL) should be considered “clinically important hypoglycemia.”
“A single glucose level should be agreed to that has serious clinical and health-economic consequences,” the ADA and EASD stated in the new guidelines. “This would enable the diabetes and regulatory communities to compare the effectiveness of interventions in reducing hypoglycemia, be they pharmacological, technological, or educational. It would also permit the use of meta-analysis as a statistical tool to increase power when comparing interventions.”
An international, multidisciplinary group – the International Hypoglycemia Study Group – was formed to create distinct definitions of the various levels of severity that hypoglycemia can have. The new guidelines contain three levels, which should be used by clinicians to determine what amounts of blood glucose are significant enough to be clinically reported.
“Currently, there is no uniform agreement to what constitutes reportable hypoglycemia in clinical trials,” she said. “In some studies, it is defined as a blood glucose level of less than 70 mg/dL, whereas in others it is defined as a blood glucose level less than 54 mg/dL.”
The guidelines define first-level hypoglycemia as any glucose level of 3.9 mmol/L (70 mg/dL) or less. This is not considered low enough to be reported on a consistent basis in clinical studies; however, that determination must ultimately be made by the investigators, as the parameters for what is significant often vary from study to study.
The second level is the 3 mmol/L (54 mg/dL), which now is deemed to be a clinically significant level of hypoglycemia. Because it is “sufficiently low to indicate serious, clinically important hypoglycemia,” it should be reported as part of any clinical studies. Finally, the third level, less than 2.8 mmol/L (50 mg/dL), indicates severe hypoglycemia and is classified as any individual with “severe cognitive impairment requiring external assistance for recovery,” according to the guidelines (Diabetes Care. 2016 Dec 1. doi: 10.2337/dc16-2215).
With a new standard of hypoglycemic values that are deemed clinically significant, the ADA and EASD hope that comparing different insulins, medications, technologies, and educational interventions will now become easier and more standardized, leading to better care worldwide.
Although there is general agreement as to where severe hypoglycemia really begins, the newly defined glucose levels are “a step in the right direction,” according to Dr. Rodbard.
The International Hypoglycaemia Study Group developed these guidelines through a grant from Novo Nordisk, awarded to the Six Degrees Academy of Toronto. Dr. Heller has received advisory or consultation fees from Lilly, Novo Nordisk, Takeda, Merck, Sharp & Dohme, and Becton Dickinson; has served as a speaker for AstraZeneca, Lilly, Novo Nordisk, Boehringer Ingelheim, and Takeda; and has received research support from Medtronic U.K. Dr. Rodbard did not report any financial disclosures.
New PAD guidelines expected to boost awareness and treatment
NEW ORLEANS – The latest revision of U.S. guidelines for diagnosing and managing lower-extremity peripheral artery disease is seen by several experts as primarily a renewed call to action by American physicians to more diligently identify at-risk people in their practices, diagnose the disease with ankle-brachial index measurement, and appropriately treat patients who have the disease.
The new lower-extremity peripheral artery disease (PAD) guidelines “give us a platform to get something done,” said Heather L. Gornik, MD, vice chair of the guidelines panel and medical director of the noninvasive vascular lab at the Cleveland Clinic. Improved PAD identification and care “starts with the fundamentals of recognizing who is at risk for PAD and then doing some clinical investigation to find it, because it’s there. You just need to ask patients if they have leg symptoms, have them take off their socks and examine their feet,” Dr. Gornik said in an interview following a program devoted to the revised guidelines at the American Heart Association scientific sessions.

The new guidelines are “a clarion call to action,” commented Alan T. Hirsch, MD, professor of medicine, epidemiology, and community health and director of the vascular medicine program at the University of Minnesota in Minneapolis. PAD “is the single most morbid and fatal of all cardiovascular diseases, so why in 2016 are we challenged to have every cardiologist trained in basic PAD competency?” asked Dr. Hirsch, who chaired the 2005 guidelines panel.

The AHA wants to “elevate awareness of PAD among the public and health professionals, and we want to better understand how we can be a catalyst for change,” said Terri Wiggins, the organization’s vice president for vascular health programs. AHA publications now stress that clinicians need to have patients “take off their socks and look at the patient’s feet,” she said.
The problem with the way many U.S. physicians handle PAD goes beyond a failure to properly screen and diagnose the disease. Analysis of data collected by the National Ambulatory Medical Care Survey and the National Hospital Ambulatory Medical Care Survey during 2006-2013 showed that, among an average of 3.9 million patients seen each year with PAD, just 38% received treatment with an antiplatelet drug, 35% received a statin, and among those patients who smoked, 36% received counseling for smoking cessation, Jeffrey S. Berger, MD, reported in a separate talk at the meeting. These rates were roughly constant throughout the 8-year period he examined.

“These data are eye-opening. They highlight a clear opportunity to improve the care of patients with PAD with guideline-directed therapy. PAD is problematic because only 10%-15% of patients have typical symptoms, so many physicians have a false perception that these patients are not at high risk,” Dr. Berger said in an interview. “What the AHA is doing is great” for raising awareness, he added.
The revised 2016 PAD guidelines made several changes, compared with what had been on the books from the 2005 guidelines and 2011 update, including classifying vorapaxar (Zontivity) treatment a class IIb recommendation with “uncertain” incremental benefit when used as an add-on agent on top of standard antiplatelet therapy, endorsement of annual influenza vaccination as something every PAD patient should receive and a class I recommendation, and acknowledgment that selected patients with critical limb ischemia are candidates for an “endovascular first” approach to revascularization,
Perhaps just as important was inclusion for the first time in the new guidelines of three advocacy priorities for initiatives by various professional societies with an interest in PAD: easy availability of ankle-brachial index measurement as the initial test to establish a diagnosis of PAD in patients with physical examination findings suggestive of the disease; access to supervised exercise programs for patients diagnosed with PAD; and incorporation of patient-centered outcomes into the regulatory approval process of new medical therapies and revascularization technologies for treating PAD.
The new guidelines “ form the basis for the AHA establishing a Get With The Guidelines program for PAD so that clinicians can be held accountable for delivering these treatments,” said Naomi M. Hamburg, MD, chief of vascular biology at Boston University and a member of the guidelines panel.

Dr. Gornik has an ownership interest in Summit Doppler Systems and Zin Medical and has received research support from AstraZeneca and Theravasc. Dr. Hamburg has been a consultant to Acceleron and has received research support from Everest Genomics, Hershey’s, Unex, and Welch’s. Dr. Hirsch, Ms. Wiggins, and Dr. Berger had no disclosures.
[email protected]
On Twitter @mitchelzoler
NEW ORLEANS – The latest revision of U.S. guidelines for diagnosing and managing lower-extremity peripheral artery disease is seen by several experts as primarily a renewed call to action by American physicians to more diligently identify at-risk people in their practices, diagnose the disease with ankle-brachial index measurement, and appropriately treat patients who have the disease.
The new lower-extremity peripheral artery disease (PAD) guidelines “give us a platform to get something done,” said Heather L. Gornik, MD, vice chair of the guidelines panel and medical director of the noninvasive vascular lab at the Cleveland Clinic. Improved PAD identification and care “starts with the fundamentals of recognizing who is at risk for PAD and then doing some clinical investigation to find it, because it’s there. You just need to ask patients if they have leg symptoms, have them take off their socks and examine their feet,” Dr. Gornik said in an interview following a program devoted to the revised guidelines at the American Heart Association scientific sessions.
The new guidelines are “a clarion call to action,” commented Alan T. Hirsch, MD, professor of medicine, epidemiology, and community health and director of the vascular medicine program at the University of Minnesota in Minneapolis. PAD “is the single most morbid and fatal of all cardiovascular diseases, so why in 2016 are we challenged to have every cardiologist trained in basic PAD competency?” asked Dr. Hirsch, who chaired the 2005 guidelines panel.
The AHA wants to “elevate awareness of PAD among the public and health professionals, and we want to better understand how we can be a catalyst for change,” said Terri Wiggins, the organization’s vice president for vascular health programs. AHA publications now stress that clinicians need to have patients “take off their socks and look at the patient’s feet,” she said.
The problem with the way many U.S. physicians handle PAD goes beyond a failure to properly screen and diagnose the disease. Analysis of data collected by the National Ambulatory Medical Care Survey and the National Hospital Ambulatory Medical Care Survey during 2006-2013 showed that, among an average of 3.9 million patients seen each year with PAD, just 38% received treatment with an antiplatelet drug, 35% received a statin, and among those patients who smoked, 36% received counseling for smoking cessation, Jeffrey S. Berger, MD, reported in a separate talk at the meeting. These rates were roughly constant throughout the 8-year period he examined.
“These data are eye-opening. They highlight a clear opportunity to improve the care of patients with PAD with guideline-directed therapy. PAD is problematic because only 10%-15% of patients have typical symptoms, so many physicians have a false perception that these patients are not at high risk,” Dr. Berger said in an interview. “What the AHA is doing is great” for raising awareness, he added.
The revised 2016 PAD guidelines made several changes, compared with what had been on the books from the 2005 guidelines and 2011 update, including classifying vorapaxar (Zontivity) treatment a class IIb recommendation with “uncertain” incremental benefit when used as an add-on agent on top of standard antiplatelet therapy, endorsement of annual influenza vaccination as something every PAD patient should receive and a class I recommendation, and acknowledgment that selected patients with critical limb ischemia are candidates for an “endovascular first” approach to revascularization,
Perhaps just as important was inclusion for the first time in the new guidelines of three advocacy priorities for initiatives by various professional societies with an interest in PAD: easy availability of ankle-brachial index measurement as the initial test to establish a diagnosis of PAD in patients with physical examination findings suggestive of the disease; access to supervised exercise programs for patients diagnosed with PAD; and incorporation of patient-centered outcomes into the regulatory approval process of new medical therapies and revascularization technologies for treating PAD.
The new guidelines “ form the basis for the AHA establishing a Get With The Guidelines program for PAD so that clinicians can be held accountable for delivering these treatments,” said Naomi M. Hamburg, MD, chief of vascular biology at Boston University and a member of the guidelines panel.
Dr. Gornik has an ownership interest in Summit Doppler Systems and Zin Medical and has received research support from AstraZeneca and Theravasc. Dr. Hamburg has been a consultant to Acceleron and has received research support from Everest Genomics, Hershey’s, Unex, and Welch’s. Dr. Hirsch, Ms. Wiggins, and Dr. Berger had no disclosures.
[email protected]
On Twitter @mitchelzoler
NEW ORLEANS – The latest revision of U.S. guidelines for diagnosing and managing lower-extremity peripheral artery disease is seen by several experts as primarily a renewed call to action by American physicians to more diligently identify at-risk people in their practices, diagnose the disease with ankle-brachial index measurement, and appropriately treat patients who have the disease.
The new lower-extremity peripheral artery disease (PAD) guidelines “give us a platform to get something done,” said Heather L. Gornik, MD, vice chair of the guidelines panel and medical director of the noninvasive vascular lab at the Cleveland Clinic. Improved PAD identification and care “starts with the fundamentals of recognizing who is at risk for PAD and then doing some clinical investigation to find it, because it’s there. You just need to ask patients if they have leg symptoms, have them take off their socks and examine their feet,” Dr. Gornik said in an interview following a program devoted to the revised guidelines at the American Heart Association scientific sessions.
The new guidelines are “a clarion call to action,” commented Alan T. Hirsch, MD, professor of medicine, epidemiology, and community health and director of the vascular medicine program at the University of Minnesota in Minneapolis. PAD “is the single most morbid and fatal of all cardiovascular diseases, so why in 2016 are we challenged to have every cardiologist trained in basic PAD competency?” asked Dr. Hirsch, who chaired the 2005 guidelines panel.
The AHA wants to “elevate awareness of PAD among the public and health professionals, and we want to better understand how we can be a catalyst for change,” said Terri Wiggins, the organization’s vice president for vascular health programs. AHA publications now stress that clinicians need to have patients “take off their socks and look at the patient’s feet,” she said.
The problem with the way many U.S. physicians handle PAD goes beyond a failure to properly screen and diagnose the disease. Analysis of data collected by the National Ambulatory Medical Care Survey and the National Hospital Ambulatory Medical Care Survey during 2006-2013 showed that, among an average of 3.9 million patients seen each year with PAD, just 38% received treatment with an antiplatelet drug, 35% received a statin, and among those patients who smoked, 36% received counseling for smoking cessation, Jeffrey S. Berger, MD, reported in a separate talk at the meeting. These rates were roughly constant throughout the 8-year period he examined.
“These data are eye-opening. They highlight a clear opportunity to improve the care of patients with PAD with guideline-directed therapy. PAD is problematic because only 10%-15% of patients have typical symptoms, so many physicians have a false perception that these patients are not at high risk,” Dr. Berger said in an interview. “What the AHA is doing is great” for raising awareness, he added.
The revised 2016 PAD guidelines made several changes, compared with what had been on the books from the 2005 guidelines and 2011 update, including classifying vorapaxar (Zontivity) treatment a class IIb recommendation with “uncertain” incremental benefit when used as an add-on agent on top of standard antiplatelet therapy, endorsement of annual influenza vaccination as something every PAD patient should receive and a class I recommendation, and acknowledgment that selected patients with critical limb ischemia are candidates for an “endovascular first” approach to revascularization,
Perhaps just as important was inclusion for the first time in the new guidelines of three advocacy priorities for initiatives by various professional societies with an interest in PAD: easy availability of ankle-brachial index measurement as the initial test to establish a diagnosis of PAD in patients with physical examination findings suggestive of the disease; access to supervised exercise programs for patients diagnosed with PAD; and incorporation of patient-centered outcomes into the regulatory approval process of new medical therapies and revascularization technologies for treating PAD.
The new guidelines “ form the basis for the AHA establishing a Get With The Guidelines program for PAD so that clinicians can be held accountable for delivering these treatments,” said Naomi M. Hamburg, MD, chief of vascular biology at Boston University and a member of the guidelines panel.
Dr. Gornik has an ownership interest in Summit Doppler Systems and Zin Medical and has received research support from AstraZeneca and Theravasc. Dr. Hamburg has been a consultant to Acceleron and has received research support from Everest Genomics, Hershey’s, Unex, and Welch’s. Dr. Hirsch, Ms. Wiggins, and Dr. Berger had no disclosures.
[email protected]
On Twitter @mitchelzoler
EXPERT ANALYSIS FROM THE AHA SCIENTIFIC SESSIONS
Age correlated with occult cancer in VTE patients
A newly devised risk score may help identify which patients with acute venous thromboembolism (VTE) are most likely to have occult cancer and where they are likely to have it, according to a report published online in CHEST.
Although VTE is known to occur before an occult cancer becomes symptomatic in a minority of patients, clinicians don’t agree on which patients with VTE should be screened for occult cancer or on how extensive that screening should be. Some favor a basic screening with only a clinical history, physical exam, simple lab tests, and a chest X-ray, while others advocate a more thorough work-up to improve the patient’s chance for a cure. “The potential benefits and harms of such screening are controversial, partly because there is little evidence” concerning which patients are at highest risk and which cancer types/sites should be assessed, said Luis Jara-Palomares, MD, PhD, of the medical surgical unit of respiratory diseases, Virgen del Rocio Hospital, Seville, Spain, and his associates.
Dr. Jara-Palomares and his associates devised a prognostic score by assigning points to the variables they found to be most significantly associated with occult cancer. For example, male sex was accorded 1 point, chronic lung disease or a high platelet count was accorded 1 point, age over 70 years or anemia was accorded 2 points, and postoperative or prior VTE was accorded negative 2 points. The incidence of occult cancer was significantly lower among patients whose total score was 2 points or less (5.8%) than among those whose total score was 3 points or higher (12%).
The mean age of these study participants was 63 years, and approximately half were women. A total of 444 (7.6%) were diagnosed as having cancer at 1-24 months after presenting with VTE. Most of these cancers were discovered within 6 months of the VTE.
Patients who had occult cancer were significantly more likely than those who did not to be male and older than 70 years of age; to have chronic lung disease, an elevated platelet count, and/or anemia; and to have a history of recent surgery and/or prior VTE.
The percentage of VTE patients who had occult cancer increased with advancing age, from 2%-3% in the youngest age group (younger than 50 years) to 8%-12% in the oldest age group (older than 70 years). Among men with occult cancer, the most frequently affected sites were the lung (26%), prostate (17%), and colon/rectum (10%). Among women, the most frequent types of cancer were colorectal (19%), breast (12%), uterine (9.1%), hematologic (8.6%), pancreatic (7.6%), and stomach (6.6%).
Overall, more than half of the men who had occult cancer had cancer affecting the lung, prostate, or colon/rectum, while two-thirds of the women who had occult cancer had cancer affecting the colon/rectum, breast, or abdomen. “This is important because [cancer] screening is not necessary in all VTE patients, but any information suggesting [which] patients are at increased risk and [which] sites are more common may be of help to decide the most appropriate work-up for each patient,” the investigators noted.
To determine which patients are most likely to have occult cancer and which cancer types are most likely to develop, the investigators analyzed data from RIETE (the Computerized Registry of Patients With Venous Thromboembolism), an international registry of more than 50,000 consecutive patients with confirmed acute VTE. They focused on 5,863 patients who presented with pulmonary embolism (34%), deep vein thrombosis (48%), or both disorders (18%) and were followed for at least 2 years for the development of cancer.
The accuracy of this risk scoring system must be tested further in a large validation cohort before it can be widely adopted. If it proves to be accurate, then patients with low total risk scores could forgo the expense, discomfort, and psychological stress of extensive cancer screening, Dr. Jara-Palomares and his associates said.
Men whose total score is 3 points or more could benefit from a rectal exam and fecal occult blood test to rule out colorectal cancer, a rectal exam and prostate-specific antigen test to rule out prostate cancer, and a chest CT to rule out lung cancer. Women whose total score is 3 points or higher could benefit from a fecal occult blood test to rule out colorectal cancer, a mammogram to rule out breast cancer, and abdominopelvic CT to rule out uterine, pancreatic, and stomach cancer, they noted.
The RIETE Registry is supported by an unrestricted educational grant from Sanofi Spain and by Bayer Pharma AG. Dr. Jara-Palomares reported having no relevant financial disclosures; his associates reported ties to Sanofi, Bayer, and LEO Pharma.
A newly devised risk score may help identify which patients with acute venous thromboembolism (VTE) are most likely to have occult cancer and where they are likely to have it, according to a report published online in CHEST.
Although VTE is known to occur before an occult cancer becomes symptomatic in a minority of patients, clinicians don’t agree on which patients with VTE should be screened for occult cancer or on how extensive that screening should be. Some favor a basic screening with only a clinical history, physical exam, simple lab tests, and a chest X-ray, while others advocate a more thorough work-up to improve the patient’s chance for a cure. “The potential benefits and harms of such screening are controversial, partly because there is little evidence” concerning which patients are at highest risk and which cancer types/sites should be assessed, said Luis Jara-Palomares, MD, PhD, of the medical surgical unit of respiratory diseases, Virgen del Rocio Hospital, Seville, Spain, and his associates.
Dr. Jara-Palomares and his associates devised a prognostic score by assigning points to the variables they found to be most significantly associated with occult cancer. For example, male sex was accorded 1 point, chronic lung disease or a high platelet count was accorded 1 point, age over 70 years or anemia was accorded 2 points, and postoperative or prior VTE was accorded negative 2 points. The incidence of occult cancer was significantly lower among patients whose total score was 2 points or less (5.8%) than among those whose total score was 3 points or higher (12%).
The mean age of these study participants was 63 years, and approximately half were women. A total of 444 (7.6%) were diagnosed as having cancer at 1-24 months after presenting with VTE. Most of these cancers were discovered within 6 months of the VTE.
Patients who had occult cancer were significantly more likely than those who did not to be male and older than 70 years of age; to have chronic lung disease, an elevated platelet count, and/or anemia; and to have a history of recent surgery and/or prior VTE.
The percentage of VTE patients who had occult cancer increased with advancing age, from 2%-3% in the youngest age group (younger than 50 years) to 8%-12% in the oldest age group (older than 70 years). Among men with occult cancer, the most frequently affected sites were the lung (26%), prostate (17%), and colon/rectum (10%). Among women, the most frequent types of cancer were colorectal (19%), breast (12%), uterine (9.1%), hematologic (8.6%), pancreatic (7.6%), and stomach (6.6%).
Overall, more than half of the men who had occult cancer had cancer affecting the lung, prostate, or colon/rectum, while two-thirds of the women who had occult cancer had cancer affecting the colon/rectum, breast, or abdomen. “This is important because [cancer] screening is not necessary in all VTE patients, but any information suggesting [which] patients are at increased risk and [which] sites are more common may be of help to decide the most appropriate work-up for each patient,” the investigators noted.
To determine which patients are most likely to have occult cancer and which cancer types are most likely to develop, the investigators analyzed data from RIETE (the Computerized Registry of Patients With Venous Thromboembolism), an international registry of more than 50,000 consecutive patients with confirmed acute VTE. They focused on 5,863 patients who presented with pulmonary embolism (34%), deep vein thrombosis (48%), or both disorders (18%) and were followed for at least 2 years for the development of cancer.
The accuracy of this risk scoring system must be tested further in a large validation cohort before it can be widely adopted. If it proves to be accurate, then patients with low total risk scores could forgo the expense, discomfort, and psychological stress of extensive cancer screening, Dr. Jara-Palomares and his associates said.
Men whose total score is 3 points or more could benefit from a rectal exam and fecal occult blood test to rule out colorectal cancer, a rectal exam and prostate-specific antigen test to rule out prostate cancer, and a chest CT to rule out lung cancer. Women whose total score is 3 points or higher could benefit from a fecal occult blood test to rule out colorectal cancer, a mammogram to rule out breast cancer, and abdominopelvic CT to rule out uterine, pancreatic, and stomach cancer, they noted.
The RIETE Registry is supported by an unrestricted educational grant from Sanofi Spain and by Bayer Pharma AG. Dr. Jara-Palomares reported having no relevant financial disclosures; his associates reported ties to Sanofi, Bayer, and LEO Pharma.
A newly devised risk score may help identify which patients with acute venous thromboembolism (VTE) are most likely to have occult cancer and where they are likely to have it, according to a report published online in CHEST.
Although VTE is known to occur before an occult cancer becomes symptomatic in a minority of patients, clinicians don’t agree on which patients with VTE should be screened for occult cancer or on how extensive that screening should be. Some favor a basic screening with only a clinical history, physical exam, simple lab tests, and a chest X-ray, while others advocate a more thorough work-up to improve the patient’s chance for a cure. “The potential benefits and harms of such screening are controversial, partly because there is little evidence” concerning which patients are at highest risk and which cancer types/sites should be assessed, said Luis Jara-Palomares, MD, PhD, of the medical surgical unit of respiratory diseases, Virgen del Rocio Hospital, Seville, Spain, and his associates.
Dr. Jara-Palomares and his associates devised a prognostic score by assigning points to the variables they found to be most significantly associated with occult cancer. For example, male sex was accorded 1 point, chronic lung disease or a high platelet count was accorded 1 point, age over 70 years or anemia was accorded 2 points, and postoperative or prior VTE was accorded negative 2 points. The incidence of occult cancer was significantly lower among patients whose total score was 2 points or less (5.8%) than among those whose total score was 3 points or higher (12%).
The mean age of these study participants was 63 years, and approximately half were women. A total of 444 (7.6%) were diagnosed as having cancer at 1-24 months after presenting with VTE. Most of these cancers were discovered within 6 months of the VTE.
Patients who had occult cancer were significantly more likely than those who did not to be male and older than 70 years of age; to have chronic lung disease, an elevated platelet count, and/or anemia; and to have a history of recent surgery and/or prior VTE.
The percentage of VTE patients who had occult cancer increased with advancing age, from 2%-3% in the youngest age group (younger than 50 years) to 8%-12% in the oldest age group (older than 70 years). Among men with occult cancer, the most frequently affected sites were the lung (26%), prostate (17%), and colon/rectum (10%). Among women, the most frequent types of cancer were colorectal (19%), breast (12%), uterine (9.1%), hematologic (8.6%), pancreatic (7.6%), and stomach (6.6%).
Overall, more than half of the men who had occult cancer had cancer affecting the lung, prostate, or colon/rectum, while two-thirds of the women who had occult cancer had cancer affecting the colon/rectum, breast, or abdomen. “This is important because [cancer] screening is not necessary in all VTE patients, but any information suggesting [which] patients are at increased risk and [which] sites are more common may be of help to decide the most appropriate work-up for each patient,” the investigators noted.
To determine which patients are most likely to have occult cancer and which cancer types are most likely to develop, the investigators analyzed data from RIETE (the Computerized Registry of Patients With Venous Thromboembolism), an international registry of more than 50,000 consecutive patients with confirmed acute VTE. They focused on 5,863 patients who presented with pulmonary embolism (34%), deep vein thrombosis (48%), or both disorders (18%) and were followed for at least 2 years for the development of cancer.
The accuracy of this risk scoring system must be tested further in a large validation cohort before it can be widely adopted. If it proves to be accurate, then patients with low total risk scores could forgo the expense, discomfort, and psychological stress of extensive cancer screening, Dr. Jara-Palomares and his associates said.
Men whose total score is 3 points or more could benefit from a rectal exam and fecal occult blood test to rule out colorectal cancer, a rectal exam and prostate-specific antigen test to rule out prostate cancer, and a chest CT to rule out lung cancer. Women whose total score is 3 points or higher could benefit from a fecal occult blood test to rule out colorectal cancer, a mammogram to rule out breast cancer, and abdominopelvic CT to rule out uterine, pancreatic, and stomach cancer, they noted.
The RIETE Registry is supported by an unrestricted educational grant from Sanofi Spain and by Bayer Pharma AG. Dr. Jara-Palomares reported having no relevant financial disclosures; his associates reported ties to Sanofi, Bayer, and LEO Pharma.
FROM CHEST
Key clinical point:
Major finding: The incidence of occult cancer was significantly lower among patients whose total score was 2 points or less (5.8%) than among those whose total score was 3 points or higher (12%).
Data source: A retrospective cohort study involving 5,863 consecutive VTE patients in an international observational registry.
Disclosures: The RIETE Registry is supported by an unrestricted educational grant from Sanofi Spain and by Bayer Pharma AG. Dr. Jara-Palomares reported having no relevant financial disclosures; his associates reported ties to Sanofi, Bayer, and LEO Pharma.
Biosimilar trastuzumab shows similar efficacy
A trastuzumab biosimilar drug has shown an equivalent response, compared with trastuzumab, in the treatment of ERBB2 (HER2)-positive metastatic breast cancer, according to the results of a randomized double-blind controlled trial.
The anti-ERBB2 humanized monoclonal antibody trastuzumab in combination with chemotherapy has been found in numerous trials to significantly improve progression-free survival and overall survival in women with ERBB2-positive metastatic breast, compared with chemotherapy alone.

“With impending patent expiration of some biological agents, development of biosimilars has become a high priority for drug developers and health authorities throughout the world to provide access to high-quality alternatives,” the authors wrote. “A biosimilar drug is a biological product that is highly similar to a licensed biological product, with no clinically meaningful differences in terms of safety, purity, or potency.”
In a phase III multicenter trial, 500 women with ERBB2 (HER2)-positive metastatic breast cancer, recruited from 95 sites in Europe, Africa, South America, and Asia, were randomized 1:1 to intravenous infusions of trastuzumab or a biosimilar labeled MYL-1401O, with both arms also receiving taxane therapy.
At 24 weeks, the overall response rate (ORR) was not significantly different between the biosimilar and trastuzumab groups (69.6% vs. 64.0%; ORR ratio, 1.09; 90% confidence interval, 0.974-1.211) and within the predefined equivalence boundaries, the investigators report (JAMA. 2016 Dec 1. doi: 10.1001/jama.2016.18305).
By week 48, both groups also showed no significant differences in time to tumor progression, progression-free survival (44.3% vs. 44.7%), or overall survival (89.1% vs. 85.1%).
Pharmacokinetic analysis showed the mean concentrations of trastuzumab were similar for the two treatments, and minimum drug concentrations were also comparable at week 16 of treatment.
“This confirmatory efficacy and safety study was the last step in the multistep process to demonstrate similarity of a trastuzumab biosimilar and was adequately powered to demonstrate equivalence with trastuzumab,” the authors wrote. “The results of this study are consistent with the physicochemical and functional similarity shown in vitro and in vivo and with the similar pharmacokinetics shown in healthy participants between the candidate biosimilar and trastuzumab.”
This consistency also extended to adverse events. Almost all participants in both the biosimilar and the trastuzumab groups reported at least one adverse event, which included neutropenia (57.5% vs. 53.3%), peripheral neuropathy (23.1% vs. 24.8%), and diarrhea (20.6% vs. 20.7%).
“A biosimilar treatment option may increase global access to biological cancer therapies, provided, among other issues, that the price of the biosimilar is sufficiently inexpensive to enable women in non–high-income countries to access this therapy,” the authors wrote.
However, they pointed out that the stepwise development program for biosimilar drugs tended to use shorter end-points – 24 weeks for the primary endpoint and 48 weeks for secondary endpoints in this particular study. By 48 weeks, more than 50% of patients had not shown progression, suggesting that the medians for efficacy parameters may have been longer with a longer data cut-off.
“The choice of the 24-week evaluation period for part 1 of this study was related to the ability to analyze the ORR as a short-term measure of clinical activity and safety directly related to the combination of taxanes with trastuzumab and the proposed biosimilar as first-line treatment.”
The study was funded and sponsored by Mylan, which manufactured the biosimilar drug, and Biocon Research Limited. Four authors declared stock in Mylan, two declared consulting fees from Mylan and one also declared stock in Biocon Research Limited. One author declared research and travel support from other pharmaceutical companies.
The availability of a biosimilar agent that achieves equivalent clinical outcomes at lower cost could enable many patients with breast cancer to have access to a therapy that may improve survival. Moreover, given the large number of patients with breast cancer, widespread use of this trastuzumab biosimilar evaluated by Rugo et al. (if approved for use by the U.S. FDA, the European Medicines Agency, and other regulatory agencies) also could have financial implications for the manufacturer of this product.
In announcing their FDA submission for the proposed trastuzumab biosimilar, the sponsors of the trial by Rugo et al. have expressed their “shared commitment to increasing access to these critical medicines worldwide” and indicated that “this advancement in the U.S. will enable us to enhance access to this affordable therapy to larger patient pools.” Ultimately, to fulfill these pledges, the manufacturers must ensure that the pricing of this biosimilar product is responsible and fair and provides access to this important therapy at an affordable price.
Howard Bauchner, MD, is editor in chief of JAMA, Phil B. Fontanarosa, MD, MBA, is executive editor of JAMA, and Robert M. Golub, MD, is deputy editor of JAMA. These comments are taken from an accompanying editorial (JAMA. 2016 Dec 1. doi: 10.1001/jama.2016.18743). No conflicts of interest were declared.
The availability of a biosimilar agent that achieves equivalent clinical outcomes at lower cost could enable many patients with breast cancer to have access to a therapy that may improve survival. Moreover, given the large number of patients with breast cancer, widespread use of this trastuzumab biosimilar evaluated by Rugo et al. (if approved for use by the U.S. FDA, the European Medicines Agency, and other regulatory agencies) also could have financial implications for the manufacturer of this product.
In announcing their FDA submission for the proposed trastuzumab biosimilar, the sponsors of the trial by Rugo et al. have expressed their “shared commitment to increasing access to these critical medicines worldwide” and indicated that “this advancement in the U.S. will enable us to enhance access to this affordable therapy to larger patient pools.” Ultimately, to fulfill these pledges, the manufacturers must ensure that the pricing of this biosimilar product is responsible and fair and provides access to this important therapy at an affordable price.
Howard Bauchner, MD, is editor in chief of JAMA, Phil B. Fontanarosa, MD, MBA, is executive editor of JAMA, and Robert M. Golub, MD, is deputy editor of JAMA. These comments are taken from an accompanying editorial (JAMA. 2016 Dec 1. doi: 10.1001/jama.2016.18743). No conflicts of interest were declared.
The availability of a biosimilar agent that achieves equivalent clinical outcomes at lower cost could enable many patients with breast cancer to have access to a therapy that may improve survival. Moreover, given the large number of patients with breast cancer, widespread use of this trastuzumab biosimilar evaluated by Rugo et al. (if approved for use by the U.S. FDA, the European Medicines Agency, and other regulatory agencies) also could have financial implications for the manufacturer of this product.
In announcing their FDA submission for the proposed trastuzumab biosimilar, the sponsors of the trial by Rugo et al. have expressed their “shared commitment to increasing access to these critical medicines worldwide” and indicated that “this advancement in the U.S. will enable us to enhance access to this affordable therapy to larger patient pools.” Ultimately, to fulfill these pledges, the manufacturers must ensure that the pricing of this biosimilar product is responsible and fair and provides access to this important therapy at an affordable price.
Howard Bauchner, MD, is editor in chief of JAMA, Phil B. Fontanarosa, MD, MBA, is executive editor of JAMA, and Robert M. Golub, MD, is deputy editor of JAMA. These comments are taken from an accompanying editorial (JAMA. 2016 Dec 1. doi: 10.1001/jama.2016.18743). No conflicts of interest were declared.
A trastuzumab biosimilar drug has shown an equivalent response, compared with trastuzumab, in the treatment of ERBB2 (HER2)-positive metastatic breast cancer, according to the results of a randomized double-blind controlled trial.
The anti-ERBB2 humanized monoclonal antibody trastuzumab in combination with chemotherapy has been found in numerous trials to significantly improve progression-free survival and overall survival in women with ERBB2-positive metastatic breast, compared with chemotherapy alone.
“With impending patent expiration of some biological agents, development of biosimilars has become a high priority for drug developers and health authorities throughout the world to provide access to high-quality alternatives,” the authors wrote. “A biosimilar drug is a biological product that is highly similar to a licensed biological product, with no clinically meaningful differences in terms of safety, purity, or potency.”
In a phase III multicenter trial, 500 women with ERBB2 (HER2)-positive metastatic breast cancer, recruited from 95 sites in Europe, Africa, South America, and Asia, were randomized 1:1 to intravenous infusions of trastuzumab or a biosimilar labeled MYL-1401O, with both arms also receiving taxane therapy.
At 24 weeks, the overall response rate (ORR) was not significantly different between the biosimilar and trastuzumab groups (69.6% vs. 64.0%; ORR ratio, 1.09; 90% confidence interval, 0.974-1.211) and within the predefined equivalence boundaries, the investigators report (JAMA. 2016 Dec 1. doi: 10.1001/jama.2016.18305).
By week 48, both groups also showed no significant differences in time to tumor progression, progression-free survival (44.3% vs. 44.7%), or overall survival (89.1% vs. 85.1%).
Pharmacokinetic analysis showed the mean concentrations of trastuzumab were similar for the two treatments, and minimum drug concentrations were also comparable at week 16 of treatment.
“This confirmatory efficacy and safety study was the last step in the multistep process to demonstrate similarity of a trastuzumab biosimilar and was adequately powered to demonstrate equivalence with trastuzumab,” the authors wrote. “The results of this study are consistent with the physicochemical and functional similarity shown in vitro and in vivo and with the similar pharmacokinetics shown in healthy participants between the candidate biosimilar and trastuzumab.”
This consistency also extended to adverse events. Almost all participants in both the biosimilar and the trastuzumab groups reported at least one adverse event, which included neutropenia (57.5% vs. 53.3%), peripheral neuropathy (23.1% vs. 24.8%), and diarrhea (20.6% vs. 20.7%).
“A biosimilar treatment option may increase global access to biological cancer therapies, provided, among other issues, that the price of the biosimilar is sufficiently inexpensive to enable women in non–high-income countries to access this therapy,” the authors wrote.
However, they pointed out that the stepwise development program for biosimilar drugs tended to use shorter end-points – 24 weeks for the primary endpoint and 48 weeks for secondary endpoints in this particular study. By 48 weeks, more than 50% of patients had not shown progression, suggesting that the medians for efficacy parameters may have been longer with a longer data cut-off.
“The choice of the 24-week evaluation period for part 1 of this study was related to the ability to analyze the ORR as a short-term measure of clinical activity and safety directly related to the combination of taxanes with trastuzumab and the proposed biosimilar as first-line treatment.”
The study was funded and sponsored by Mylan, which manufactured the biosimilar drug, and Biocon Research Limited. Four authors declared stock in Mylan, two declared consulting fees from Mylan and one also declared stock in Biocon Research Limited. One author declared research and travel support from other pharmaceutical companies.
A trastuzumab biosimilar drug has shown an equivalent response, compared with trastuzumab, in the treatment of ERBB2 (HER2)-positive metastatic breast cancer, according to the results of a randomized double-blind controlled trial.
The anti-ERBB2 humanized monoclonal antibody trastuzumab in combination with chemotherapy has been found in numerous trials to significantly improve progression-free survival and overall survival in women with ERBB2-positive metastatic breast, compared with chemotherapy alone.
“With impending patent expiration of some biological agents, development of biosimilars has become a high priority for drug developers and health authorities throughout the world to provide access to high-quality alternatives,” the authors wrote. “A biosimilar drug is a biological product that is highly similar to a licensed biological product, with no clinically meaningful differences in terms of safety, purity, or potency.”
In a phase III multicenter trial, 500 women with ERBB2 (HER2)-positive metastatic breast cancer, recruited from 95 sites in Europe, Africa, South America, and Asia, were randomized 1:1 to intravenous infusions of trastuzumab or a biosimilar labeled MYL-1401O, with both arms also receiving taxane therapy.
At 24 weeks, the overall response rate (ORR) was not significantly different between the biosimilar and trastuzumab groups (69.6% vs. 64.0%; ORR ratio, 1.09; 90% confidence interval, 0.974-1.211) and within the predefined equivalence boundaries, the investigators report (JAMA. 2016 Dec 1. doi: 10.1001/jama.2016.18305).
By week 48, both groups also showed no significant differences in time to tumor progression, progression-free survival (44.3% vs. 44.7%), or overall survival (89.1% vs. 85.1%).
Pharmacokinetic analysis showed the mean concentrations of trastuzumab were similar for the two treatments, and minimum drug concentrations were also comparable at week 16 of treatment.
“This confirmatory efficacy and safety study was the last step in the multistep process to demonstrate similarity of a trastuzumab biosimilar and was adequately powered to demonstrate equivalence with trastuzumab,” the authors wrote. “The results of this study are consistent with the physicochemical and functional similarity shown in vitro and in vivo and with the similar pharmacokinetics shown in healthy participants between the candidate biosimilar and trastuzumab.”
This consistency also extended to adverse events. Almost all participants in both the biosimilar and the trastuzumab groups reported at least one adverse event, which included neutropenia (57.5% vs. 53.3%), peripheral neuropathy (23.1% vs. 24.8%), and diarrhea (20.6% vs. 20.7%).
“A biosimilar treatment option may increase global access to biological cancer therapies, provided, among other issues, that the price of the biosimilar is sufficiently inexpensive to enable women in non–high-income countries to access this therapy,” the authors wrote.
However, they pointed out that the stepwise development program for biosimilar drugs tended to use shorter end-points – 24 weeks for the primary endpoint and 48 weeks for secondary endpoints in this particular study. By 48 weeks, more than 50% of patients had not shown progression, suggesting that the medians for efficacy parameters may have been longer with a longer data cut-off.
“The choice of the 24-week evaluation period for part 1 of this study was related to the ability to analyze the ORR as a short-term measure of clinical activity and safety directly related to the combination of taxanes with trastuzumab and the proposed biosimilar as first-line treatment.”
The study was funded and sponsored by Mylan, which manufactured the biosimilar drug, and Biocon Research Limited. Four authors declared stock in Mylan, two declared consulting fees from Mylan and one also declared stock in Biocon Research Limited. One author declared research and travel support from other pharmaceutical companies.
FROM JAMA
Key clinical point: A trastuzumab biosimilar drug has shown an equivalent response, compared with trastuzumab, in the treatment of ERBB2 (HER2)-positive metastatic breast cancer.
Major finding: Patients treated with biosimilar trastuzumab showed no significant differences in response rate, progression, and survival, compared with those treated with trastuzumab.
Data source: Randomized, double-blind phase III controlled trial in 500 women with ERBB2 (HER2)-positive metastatic breast cancer.
Disclosures: The study was funded and sponsored by Mylan, which manufactured the biosimilar drug, and Biocon Research Limited. Four authors declared stock in Mylan, two declared consulting fees from Mylan, and one also declared stock in Biocon Research Limited. One author declared research and travel support from other pharmaceutical companies.
Ferric citrate effective for anemia in non–dialysis-dependent CKD
CHICAGO – Ferric citrate was safe and effective for treatment of iron-deficiency anemia in patients who had non–dialysis-dependent chronic kidney disease (NDD-CKD), based on data from a phase III, randomized, double-blind study.
The responses were durable, and none of the patients received erythropoiesis-stimulating agents (ESAs), presenter Pablo Pergola, MD, PhD, of Renal Associates, San Antonio, said in an interview at a meeting sponsored by the American Society of Nephrology.

The trial involved 234 anemic adults who had NDD-CKD and had not responded to oral iron supplements. The subjects were randomized to receive oral ferric citrate (n = 117) or placebo (n = 115) with meals (one patient did not receive placebo and laboratory data were lacking for one patient). The mean dose in the treatment arm was 5 pills per day.
The primary endpoint was the proportion of patients with hemoglobin (Hgb) greater than or equal to 1.0 g/dL anytime from baseline through week 16. Secondary endpoints included mean changes from baseline in Hgb, transferrin saturation, ferritin, and serum phosphate and evidence of sustained treatment effect based on target changes in Hgb with time.
Both arms were comparable at baseline for demographic and clinical characteristics, including phosphorus and hemoglobin levels and estimated glomerular filtration rate.
The primary endpoint was met by 51.2% of patients receiving ferric citrate and 19.1% of patients receiving placebo (P less than .001). All secondary efficacy endpoints were met, with statistically significant differences between the treatment and placebo arms, Dr. Pergola reported.
Serum phosphate level was significantly reduced from baseline at week 16 (–0.21 mg/dL; 95% confidence interval, –0.39 to –0.03 mg/dL; P equal to .02) in the active treatment group, and the levels remained in the normal range, he said.
During the 16-week treatment period and subsequent 8-week, open-label safety extension period, ferric citrate was well tolerated. Treatment-emergent adverse events (AEs), most commonly diarrhea, occurred in 93 (79.5%) and 75 (64.7%) patients in the treatment and placebo arms, respectively. Serious AEs developed in 14 (12.0%) and 13 (11.2%) of patients in the same respective order. Two deaths occurred, both in the treatment group. The deaths and serious AEs were not considered drug related.
Ferric citrate binds with dietary phosphate in the gastrointestinal tract. The resulting ferric phosphate is insoluble and is excreted. The remaining unbound ferric citrate increases serum iron parameters, including ferritin and transferrin saturation.
The findings potentially extend the therapeutic reach of the drug beyond its Food and Drug Administration–approved use for control of phosphorus levels in CKD patients on dialysis, Dr. Pergola said. The trial data will be used to seek approval for the oral iron medication as a treatment for iron-deficiency anemia in adults with NDD-CKD.
The study was sponsored by Keryx Biopharmaceuticals. Dr. Pergola is supported by honoraria and lecture fees from Akebia Therapeutics, Keryx, Relypsa, Vifor/Fresenius Pharma, and ZS Pharma.
CHICAGO – Ferric citrate was safe and effective for treatment of iron-deficiency anemia in patients who had non–dialysis-dependent chronic kidney disease (NDD-CKD), based on data from a phase III, randomized, double-blind study.
The responses were durable, and none of the patients received erythropoiesis-stimulating agents (ESAs), presenter Pablo Pergola, MD, PhD, of Renal Associates, San Antonio, said in an interview at a meeting sponsored by the American Society of Nephrology.
The trial involved 234 anemic adults who had NDD-CKD and had not responded to oral iron supplements. The subjects were randomized to receive oral ferric citrate (n = 117) or placebo (n = 115) with meals (one patient did not receive placebo and laboratory data were lacking for one patient). The mean dose in the treatment arm was 5 pills per day.
The primary endpoint was the proportion of patients with hemoglobin (Hgb) greater than or equal to 1.0 g/dL anytime from baseline through week 16. Secondary endpoints included mean changes from baseline in Hgb, transferrin saturation, ferritin, and serum phosphate and evidence of sustained treatment effect based on target changes in Hgb with time.
Both arms were comparable at baseline for demographic and clinical characteristics, including phosphorus and hemoglobin levels and estimated glomerular filtration rate.
The primary endpoint was met by 51.2% of patients receiving ferric citrate and 19.1% of patients receiving placebo (P less than .001). All secondary efficacy endpoints were met, with statistically significant differences between the treatment and placebo arms, Dr. Pergola reported.
Serum phosphate level was significantly reduced from baseline at week 16 (–0.21 mg/dL; 95% confidence interval, –0.39 to –0.03 mg/dL; P equal to .02) in the active treatment group, and the levels remained in the normal range, he said.
During the 16-week treatment period and subsequent 8-week, open-label safety extension period, ferric citrate was well tolerated. Treatment-emergent adverse events (AEs), most commonly diarrhea, occurred in 93 (79.5%) and 75 (64.7%) patients in the treatment and placebo arms, respectively. Serious AEs developed in 14 (12.0%) and 13 (11.2%) of patients in the same respective order. Two deaths occurred, both in the treatment group. The deaths and serious AEs were not considered drug related.
Ferric citrate binds with dietary phosphate in the gastrointestinal tract. The resulting ferric phosphate is insoluble and is excreted. The remaining unbound ferric citrate increases serum iron parameters, including ferritin and transferrin saturation.
The findings potentially extend the therapeutic reach of the drug beyond its Food and Drug Administration–approved use for control of phosphorus levels in CKD patients on dialysis, Dr. Pergola said. The trial data will be used to seek approval for the oral iron medication as a treatment for iron-deficiency anemia in adults with NDD-CKD.
The study was sponsored by Keryx Biopharmaceuticals. Dr. Pergola is supported by honoraria and lecture fees from Akebia Therapeutics, Keryx, Relypsa, Vifor/Fresenius Pharma, and ZS Pharma.
CHICAGO – Ferric citrate was safe and effective for treatment of iron-deficiency anemia in patients who had non–dialysis-dependent chronic kidney disease (NDD-CKD), based on data from a phase III, randomized, double-blind study.
The responses were durable, and none of the patients received erythropoiesis-stimulating agents (ESAs), presenter Pablo Pergola, MD, PhD, of Renal Associates, San Antonio, said in an interview at a meeting sponsored by the American Society of Nephrology.
The trial involved 234 anemic adults who had NDD-CKD and had not responded to oral iron supplements. The subjects were randomized to receive oral ferric citrate (n = 117) or placebo (n = 115) with meals (one patient did not receive placebo and laboratory data were lacking for one patient). The mean dose in the treatment arm was 5 pills per day.
The primary endpoint was the proportion of patients with hemoglobin (Hgb) greater than or equal to 1.0 g/dL anytime from baseline through week 16. Secondary endpoints included mean changes from baseline in Hgb, transferrin saturation, ferritin, and serum phosphate and evidence of sustained treatment effect based on target changes in Hgb with time.
Both arms were comparable at baseline for demographic and clinical characteristics, including phosphorus and hemoglobin levels and estimated glomerular filtration rate.
The primary endpoint was met by 51.2% of patients receiving ferric citrate and 19.1% of patients receiving placebo (P less than .001). All secondary efficacy endpoints were met, with statistically significant differences between the treatment and placebo arms, Dr. Pergola reported.
Serum phosphate level was significantly reduced from baseline at week 16 (–0.21 mg/dL; 95% confidence interval, –0.39 to –0.03 mg/dL; P equal to .02) in the active treatment group, and the levels remained in the normal range, he said.
During the 16-week treatment period and subsequent 8-week, open-label safety extension period, ferric citrate was well tolerated. Treatment-emergent adverse events (AEs), most commonly diarrhea, occurred in 93 (79.5%) and 75 (64.7%) patients in the treatment and placebo arms, respectively. Serious AEs developed in 14 (12.0%) and 13 (11.2%) of patients in the same respective order. Two deaths occurred, both in the treatment group. The deaths and serious AEs were not considered drug related.
Ferric citrate binds with dietary phosphate in the gastrointestinal tract. The resulting ferric phosphate is insoluble and is excreted. The remaining unbound ferric citrate increases serum iron parameters, including ferritin and transferrin saturation.
The findings potentially extend the therapeutic reach of the drug beyond its Food and Drug Administration–approved use for control of phosphorus levels in CKD patients on dialysis, Dr. Pergola said. The trial data will be used to seek approval for the oral iron medication as a treatment for iron-deficiency anemia in adults with NDD-CKD.
The study was sponsored by Keryx Biopharmaceuticals. Dr. Pergola is supported by honoraria and lecture fees from Akebia Therapeutics, Keryx, Relypsa, Vifor/Fresenius Pharma, and ZS Pharma.
AT KIDNEY WEEK 2016
Key clinical point: Ferric citrate appears to be safe and effective for treating anemia in non–dialysis-dependent CKD patients.
Major finding: Prevalence of increased hemoglobin was 52.1% in patients receiving the active drug and 19.1% in those given placebo.
Data source: Randomized, double-blind, placebo-controlled, phase III trial with 234 patients.
Disclosures: The study was sponsored by Keryx Biopharmaceuticals. Dr. Pergola is supported by honoraria and lecture fees from Akebia Therapeutics, Keryx, Relypsa, Vifor/Fresenius Pharma, and ZS Pharma.
Flesh-Colored Papular Eruption
Papular Mucinosis/Scleromyxedema
Papular mucinosis/scleromyxedema, also known as generalized lichen myxedematosus, is a rare dermal mucinosis characterized by a papular eruption that can have an associated IgG λ paraproteinemia. The clinical presentation is gradual with the development of firm, flesh-colored, 2- to 3-mm papules often involving the hands, face, and neck that can progress to plaques that cover the entire body. Skin stiffening also can be seen.1 Extracutaneous symptoms are common and include dysphagia, arthralgia, myopathy, and cardiac dysfunction.2 Occasionally, central nervous system involvement can lead to the often fatal dermato-neuro syndrome.3,4
Histologically, papular mucinosis/scleromyxedema demonstrates increased, irregularly arranged fibroblasts in the reticular dermis with increased dermal mucin deposition (quiz image and Figure 1). The epidermis is normal or slightly thinned due to pressure from dermal changes. There may be a mild superficial perivascular lymphocytic infiltrate and atrophy of hair follicles.5 In this case, the clinical and histologic findings best supported a diagnosis of papular mucinosis/scleromyxedema.

Infundibulofolliculitis is a pruritic follicular papular eruption typically involving the neck, trunk, and proximal upper arms and shoulders. It is most common in black men who reside in hot and humid climates. Although infundibulofolliculitis would be included in the clinical differential diagnosis for the current patient, the histopathologic findings were quite distinct for the correct diagnosis of papular mucinosis/scleromyxedema. Infundibulofolliculitis shows widening of the upper part of the hair follicle (infundibulum) and infundibular inflammatory infiltrate with follicular spongiosis (Figure 2). Neither mucin deposition nor fibroblast proliferation is appreciated in infundibulofolliculitis.6,7
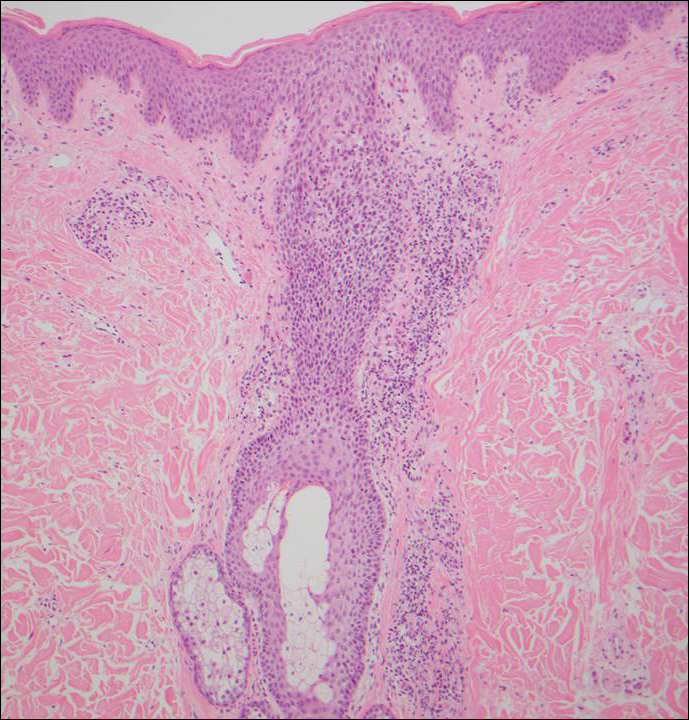
Granuloma annulare (GA) often can be distinguished clinically from papular mucinosis/scleromyxedema due to the annular appearance of papules and plaques in GA and the lack of stiffness of underlying skin. Interstitial granuloma annulare is a histologic variant of GA that can be included in the histologic differential diagnosis of papular mucinosis/scleromyxedema. Histologically, there is an interstitial infiltrate of cytologically bland histiocytes dissecting between collagen bundles in interstitial GA (Figure 3). Necrobiosis and collections of mucin often are inconspicuous. Occasionally, the presence of eosinophils can be a helpful clue.8 A fibroblast proliferation is not a feature of GA.
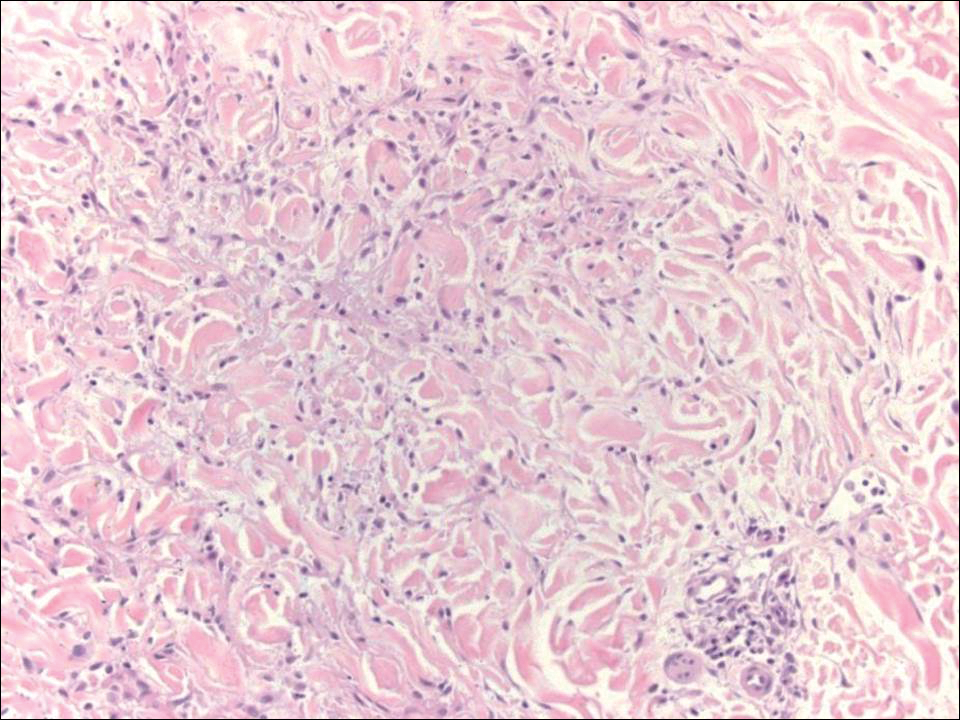
Reticular erythematous mucinosis also is a type of cutaneous mucinosis but with a classic clinical appearance of a reticulated erythematous plaque on the chest or back, making it clinically distinct from papular mucinosis/scleromyxedema and the presentation described in the current patient. Reticular erythematous mucinosis can be histologically distinguished from papular mucinosis/scleromyxedema by the presence of a superficial and deep perivascular lymphocytic infiltrate with increased dermal mucin deposition (Figure 4). It often shows a positive IgM deposition on the basement membrane on direct immunofluorescence.9

Similar to papular mucinosis/scleromyxedema, scleredema shows thickening of the skin with decreased movement of involved areas. Scleredema often involves the upper back, shoulders, and neck where affected areas often have a peau d'orange appearance. Scleredema is classified into 3 clinical forms based on clinical associations. Type 1 often is preceded by an infection, classically Streptococcus pyogenes. Type 2 is associated with a hematologic dyscrasia such as multiple myeloma, or it can have an associated paraproteinemia that is typically of the IgA κ type, which is distinct from papular mucinosis/scleromyxedema where IgG λ paraproteinemia typically is seen. Type 3 is associated with diabetes mellitus. Histologically, scleredema also is distinct from papular mucinosis/scleromyxedema. Although increased mucin is seen in the dermis, the mucin is classically more prominent in the deep reticular dermis as compared with papular mucinosis/scleromyxedema (Figure 5). Additionally, collagen bundles are thickened with clear separation between them. Hyperplasia of fibroblasts in the dermis that is a characteristic feature of papular mucinosis/scleromyxedema is not observed in scleredema.10
- Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Fleming KE, Virmani D, Sutton E, et al. Scleromyxedema and the dermato-neuro syndrome: case report and review of the literature. J Cutan Pathol. 2012;39:508-517.
- Hummers LK. Scleromyxedema. Curr Opin Rheumatol. 2014;26:658-662.
- Rongioleti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;5:174-175.
- Soyinka F. Recurrent disseminated infundibulofolliculitis. Int J Dermatol. 1973;12:314-317.
- Keimig EL. Granuloma annulare. Dermatol Clin. 2015;33:315-329.
- Thareja S, Paghdal K, Lein MH, et al. Reticular erythematous mucinosis--a review. Int J Dermatol. 2012;51:903-909.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
Papular Mucinosis/Scleromyxedema
Papular mucinosis/scleromyxedema, also known as generalized lichen myxedematosus, is a rare dermal mucinosis characterized by a papular eruption that can have an associated IgG λ paraproteinemia. The clinical presentation is gradual with the development of firm, flesh-colored, 2- to 3-mm papules often involving the hands, face, and neck that can progress to plaques that cover the entire body. Skin stiffening also can be seen.1 Extracutaneous symptoms are common and include dysphagia, arthralgia, myopathy, and cardiac dysfunction.2 Occasionally, central nervous system involvement can lead to the often fatal dermato-neuro syndrome.3,4
Histologically, papular mucinosis/scleromyxedema demonstrates increased, irregularly arranged fibroblasts in the reticular dermis with increased dermal mucin deposition (quiz image and Figure 1). The epidermis is normal or slightly thinned due to pressure from dermal changes. There may be a mild superficial perivascular lymphocytic infiltrate and atrophy of hair follicles.5 In this case, the clinical and histologic findings best supported a diagnosis of papular mucinosis/scleromyxedema.
Infundibulofolliculitis is a pruritic follicular papular eruption typically involving the neck, trunk, and proximal upper arms and shoulders. It is most common in black men who reside in hot and humid climates. Although infundibulofolliculitis would be included in the clinical differential diagnosis for the current patient, the histopathologic findings were quite distinct for the correct diagnosis of papular mucinosis/scleromyxedema. Infundibulofolliculitis shows widening of the upper part of the hair follicle (infundibulum) and infundibular inflammatory infiltrate with follicular spongiosis (Figure 2). Neither mucin deposition nor fibroblast proliferation is appreciated in infundibulofolliculitis.6,7
Granuloma annulare (GA) often can be distinguished clinically from papular mucinosis/scleromyxedema due to the annular appearance of papules and plaques in GA and the lack of stiffness of underlying skin. Interstitial granuloma annulare is a histologic variant of GA that can be included in the histologic differential diagnosis of papular mucinosis/scleromyxedema. Histologically, there is an interstitial infiltrate of cytologically bland histiocytes dissecting between collagen bundles in interstitial GA (Figure 3). Necrobiosis and collections of mucin often are inconspicuous. Occasionally, the presence of eosinophils can be a helpful clue.8 A fibroblast proliferation is not a feature of GA.
Reticular erythematous mucinosis also is a type of cutaneous mucinosis but with a classic clinical appearance of a reticulated erythematous plaque on the chest or back, making it clinically distinct from papular mucinosis/scleromyxedema and the presentation described in the current patient. Reticular erythematous mucinosis can be histologically distinguished from papular mucinosis/scleromyxedema by the presence of a superficial and deep perivascular lymphocytic infiltrate with increased dermal mucin deposition (Figure 4). It often shows a positive IgM deposition on the basement membrane on direct immunofluorescence.9
Similar to papular mucinosis/scleromyxedema, scleredema shows thickening of the skin with decreased movement of involved areas. Scleredema often involves the upper back, shoulders, and neck where affected areas often have a peau d'orange appearance. Scleredema is classified into 3 clinical forms based on clinical associations. Type 1 often is preceded by an infection, classically Streptococcus pyogenes. Type 2 is associated with a hematologic dyscrasia such as multiple myeloma, or it can have an associated paraproteinemia that is typically of the IgA κ type, which is distinct from papular mucinosis/scleromyxedema where IgG λ paraproteinemia typically is seen. Type 3 is associated with diabetes mellitus. Histologically, scleredema also is distinct from papular mucinosis/scleromyxedema. Although increased mucin is seen in the dermis, the mucin is classically more prominent in the deep reticular dermis as compared with papular mucinosis/scleromyxedema (Figure 5). Additionally, collagen bundles are thickened with clear separation between them. Hyperplasia of fibroblasts in the dermis that is a characteristic feature of papular mucinosis/scleromyxedema is not observed in scleredema.10
Papular Mucinosis/Scleromyxedema
Papular mucinosis/scleromyxedema, also known as generalized lichen myxedematosus, is a rare dermal mucinosis characterized by a papular eruption that can have an associated IgG λ paraproteinemia. The clinical presentation is gradual with the development of firm, flesh-colored, 2- to 3-mm papules often involving the hands, face, and neck that can progress to plaques that cover the entire body. Skin stiffening also can be seen.1 Extracutaneous symptoms are common and include dysphagia, arthralgia, myopathy, and cardiac dysfunction.2 Occasionally, central nervous system involvement can lead to the often fatal dermato-neuro syndrome.3,4
Histologically, papular mucinosis/scleromyxedema demonstrates increased, irregularly arranged fibroblasts in the reticular dermis with increased dermal mucin deposition (quiz image and Figure 1). The epidermis is normal or slightly thinned due to pressure from dermal changes. There may be a mild superficial perivascular lymphocytic infiltrate and atrophy of hair follicles.5 In this case, the clinical and histologic findings best supported a diagnosis of papular mucinosis/scleromyxedema.
Infundibulofolliculitis is a pruritic follicular papular eruption typically involving the neck, trunk, and proximal upper arms and shoulders. It is most common in black men who reside in hot and humid climates. Although infundibulofolliculitis would be included in the clinical differential diagnosis for the current patient, the histopathologic findings were quite distinct for the correct diagnosis of papular mucinosis/scleromyxedema. Infundibulofolliculitis shows widening of the upper part of the hair follicle (infundibulum) and infundibular inflammatory infiltrate with follicular spongiosis (Figure 2). Neither mucin deposition nor fibroblast proliferation is appreciated in infundibulofolliculitis.6,7
Granuloma annulare (GA) often can be distinguished clinically from papular mucinosis/scleromyxedema due to the annular appearance of papules and plaques in GA and the lack of stiffness of underlying skin. Interstitial granuloma annulare is a histologic variant of GA that can be included in the histologic differential diagnosis of papular mucinosis/scleromyxedema. Histologically, there is an interstitial infiltrate of cytologically bland histiocytes dissecting between collagen bundles in interstitial GA (Figure 3). Necrobiosis and collections of mucin often are inconspicuous. Occasionally, the presence of eosinophils can be a helpful clue.8 A fibroblast proliferation is not a feature of GA.
Reticular erythematous mucinosis also is a type of cutaneous mucinosis but with a classic clinical appearance of a reticulated erythematous plaque on the chest or back, making it clinically distinct from papular mucinosis/scleromyxedema and the presentation described in the current patient. Reticular erythematous mucinosis can be histologically distinguished from papular mucinosis/scleromyxedema by the presence of a superficial and deep perivascular lymphocytic infiltrate with increased dermal mucin deposition (Figure 4). It often shows a positive IgM deposition on the basement membrane on direct immunofluorescence.9
Similar to papular mucinosis/scleromyxedema, scleredema shows thickening of the skin with decreased movement of involved areas. Scleredema often involves the upper back, shoulders, and neck where affected areas often have a peau d'orange appearance. Scleredema is classified into 3 clinical forms based on clinical associations. Type 1 often is preceded by an infection, classically Streptococcus pyogenes. Type 2 is associated with a hematologic dyscrasia such as multiple myeloma, or it can have an associated paraproteinemia that is typically of the IgA κ type, which is distinct from papular mucinosis/scleromyxedema where IgG λ paraproteinemia typically is seen. Type 3 is associated with diabetes mellitus. Histologically, scleredema also is distinct from papular mucinosis/scleromyxedema. Although increased mucin is seen in the dermis, the mucin is classically more prominent in the deep reticular dermis as compared with papular mucinosis/scleromyxedema (Figure 5). Additionally, collagen bundles are thickened with clear separation between them. Hyperplasia of fibroblasts in the dermis that is a characteristic feature of papular mucinosis/scleromyxedema is not observed in scleredema.10
- Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Fleming KE, Virmani D, Sutton E, et al. Scleromyxedema and the dermato-neuro syndrome: case report and review of the literature. J Cutan Pathol. 2012;39:508-517.
- Hummers LK. Scleromyxedema. Curr Opin Rheumatol. 2014;26:658-662.
- Rongioleti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;5:174-175.
- Soyinka F. Recurrent disseminated infundibulofolliculitis. Int J Dermatol. 1973;12:314-317.
- Keimig EL. Granuloma annulare. Dermatol Clin. 2015;33:315-329.
- Thareja S, Paghdal K, Lein MH, et al. Reticular erythematous mucinosis--a review. Int J Dermatol. 2012;51:903-909.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Fleming KE, Virmani D, Sutton E, et al. Scleromyxedema and the dermato-neuro syndrome: case report and review of the literature. J Cutan Pathol. 2012;39:508-517.
- Hummers LK. Scleromyxedema. Curr Opin Rheumatol. 2014;26:658-662.
- Rongioleti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;5:174-175.
- Soyinka F. Recurrent disseminated infundibulofolliculitis. Int J Dermatol. 1973;12:314-317.
- Keimig EL. Granuloma annulare. Dermatol Clin. 2015;33:315-329.
- Thareja S, Paghdal K, Lein MH, et al. Reticular erythematous mucinosis--a review. Int J Dermatol. 2012;51:903-909.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
A 48-year-old black man presented with a rash of 7 months' duration that started on the face and spread to the body. He had extreme pruritus, increased stiffness in the hands and joints, and paresthesia. Physical examination revealed an eruption of 2- to 4-mm, flesh-colored papules with follicular accentuation on the face, neck, bilateral upper extremities, back, and thighs.
Parental online sharing involves balancing risks, benefits
SAN FRANCISCO – More than two-thirds of parents worry about their children’s privacy online and/or that photos of their children might be reshared on the wider Web, according to a survey conducted by C.S. Mott Children’s Hospital.
Those fears are not baseless, and they need to be considered more often by parents themselves in posting about their children online, presenters agreed at a symposium on the media at the annual meeting of the American Academy of Pediatrics.
“The first children of social media are just now entering adulthood, entering the job market,” said Stacey Steinberg, JD, a legal skills professor at the University of Florida Levin College of Law, Gainesville. She is also with the law school’s center on children and families.
She and Bahareh Keith, DO, a pediatrician at the University of Florida, discussed the challenges and risks of “sharenting” – parents’ sharing information and photos of their children online – and pediatricians’ role in advising parents and looking out for children’s best interests.
“The dearth of discussion on this topic leaves even the most well-intentioned parents without enough information to thoroughly analyze this,” Ms. Steinberg said. “We’re not sitting here saying we know what the answers are. But we’re saying this is an important issue that affects families, and these children require a voice in this discussion.”
The way social media and blogging have changed the landscape for children coming of age today means that they often have a digital footprint shaped by their parents long before they create their own first account. This reality means it’s necessary to consider how to balance children’s right to privacy with parents’ right to free speech and expression.
The 2015 C.S. Mott survey asked 569 parents of children aged 4 years and younger about how they use social media as parents, and reported that more than half of mothers (56%) and a third of fathers (34%) discuss parenting and child health topics on Facebook, Twitter, blogs, online forums, and other online platforms.
The risks of this sharenting can range from embarrassment of the child to significantly more sinister repercussions. Just over half of the parents (52%) in the Mott survey reported that they are concerned their child might feel embarrassed when they grow older and discover what their parents shared online. But that embarrassment also can lead to bullying or determent of psychosocial development, Ms. Steinberg and Dr. Keith explained.
More serious, if less common, risks include the possibility that data brokers could access and use information about the children or that online child pornographers could repurpose the photos inappropriately. One worst case scenario of the former is digital kidnapping, a disturbing practice in which a stranger uses baby photos and information that is not their own to pass off the child as their own or to invite others to “invent” identities for the child. The Children’s Online Privacy Protection Rule of the Federal Trade Commission addresses only online use by those under age 13 years, not others’ use of those images.
Regarding the latter, Ms. Steinberg and Dr. Keith showed an example of a bare-bottomed baby standing in front of a bathtub that had been reshared hundreds of times, but other images that have been shared on child pornography sites depict children in everyday situations such as playing on a playground, running at the beach, or doing gymnastics.
“These are images that many of us would think are innocent, but pornographers would categorize these into folders,” Dr. Keith said. “It’s not even naked or half-naked pictures.”
A study conducted by an e-safety commission in Australia, for example, found that half of the thousands of photos shared on a sample of child pornography sites had originated from parental sharing.
But Ms. Steinberg and Dr. Keith pointed out that benefits of parents’ online sharing exist as well, as the Mott survey found. In that survey, 72% of parents who discuss parenting and/or their children on social media reported that doing so helps them feel less alone. Similarly, 70% said they learn what not to do through those experiences, 67% said they receive advice from more experienced parents, and 62% said they consequently worry less. Common topics they discussed included sleep, nutrition, discipline, day care, and behavior management.
Other benefits, Ms. Steinberg pointed out, are that families geographically spread apart can stay connected, and communities can grow stronger with shared communal experiences of parents meeting others online.
“For some parents, it gives them an opportunity for advocacy work and raises awareness for important social issues,” Ms. Steinberg said, although she added, “If you’re going to share your children’s behavioral problems, consider sharing anonymously.”
Neither Ms. Steinberg and Dr. Keith said they had simple solutions to these challenges. Rather, they recommended using the public health model of raising awareness and encouraging open dialogue among pediatricians, parents, and their children to look for ways to balance competing interests.
“Social media offers many positive benefits, and we don’t want to silence the many voices of parents who take part in online sharing,” Ms. Steinberg explained. But she and Ms. Keith said it’s also worth considering children’s potential interest in controlling what their digital footprint is as they become adults.
For example, one study they cited found that, among 249 pairs of parents and their children, three times more children than parents wanted the parents to have and follow rules regarding what they could share on social media about their children.
Although guidance for parents on monitoring children’s social media use is a part of the AAP policy statement on media, only one recommendation obliquely addresses how parents should or shouldn’t use social media by advising them to model appropriate use for their children.
“It’s just like any medical decision: What is the benefit, and what is the risk, and does the benefit outweigh the risk?” said Wendy Sue Swanson, MD, executive director of digital health at Seattle Children’s Hospital. She recommended that parents ask their child for permission before posting a story or photo if their kids are aged 6 or older.
Ms. Steinberg and Dr. Keith recommended that pediatricians broach this subject with parents to help them think about risks they simply might not have considered before.
“When we looked at what sorts of best practices could be encouraged or doctors could talk to parents about – the tangible harms, such as whether data brokers or people interested in child pornography could access the information – we didn’t want to create any unnecessary panic,” Ms. Steinberg said. “But we did find some concerns that were troublesome, and we thought that parents or at least physicians [should] be aware of those potential risks.”
Ms. Steinberg and Dr. Keith reported that they had no relevant financial disclosures.
SAN FRANCISCO – More than two-thirds of parents worry about their children’s privacy online and/or that photos of their children might be reshared on the wider Web, according to a survey conducted by C.S. Mott Children’s Hospital.
Those fears are not baseless, and they need to be considered more often by parents themselves in posting about their children online, presenters agreed at a symposium on the media at the annual meeting of the American Academy of Pediatrics.
“The first children of social media are just now entering adulthood, entering the job market,” said Stacey Steinberg, JD, a legal skills professor at the University of Florida Levin College of Law, Gainesville. She is also with the law school’s center on children and families.
She and Bahareh Keith, DO, a pediatrician at the University of Florida, discussed the challenges and risks of “sharenting” – parents’ sharing information and photos of their children online – and pediatricians’ role in advising parents and looking out for children’s best interests.
“The dearth of discussion on this topic leaves even the most well-intentioned parents without enough information to thoroughly analyze this,” Ms. Steinberg said. “We’re not sitting here saying we know what the answers are. But we’re saying this is an important issue that affects families, and these children require a voice in this discussion.”
The way social media and blogging have changed the landscape for children coming of age today means that they often have a digital footprint shaped by their parents long before they create their own first account. This reality means it’s necessary to consider how to balance children’s right to privacy with parents’ right to free speech and expression.
The 2015 C.S. Mott survey asked 569 parents of children aged 4 years and younger about how they use social media as parents, and reported that more than half of mothers (56%) and a third of fathers (34%) discuss parenting and child health topics on Facebook, Twitter, blogs, online forums, and other online platforms.
The risks of this sharenting can range from embarrassment of the child to significantly more sinister repercussions. Just over half of the parents (52%) in the Mott survey reported that they are concerned their child might feel embarrassed when they grow older and discover what their parents shared online. But that embarrassment also can lead to bullying or determent of psychosocial development, Ms. Steinberg and Dr. Keith explained.
More serious, if less common, risks include the possibility that data brokers could access and use information about the children or that online child pornographers could repurpose the photos inappropriately. One worst case scenario of the former is digital kidnapping, a disturbing practice in which a stranger uses baby photos and information that is not their own to pass off the child as their own or to invite others to “invent” identities for the child. The Children’s Online Privacy Protection Rule of the Federal Trade Commission addresses only online use by those under age 13 years, not others’ use of those images.
Regarding the latter, Ms. Steinberg and Dr. Keith showed an example of a bare-bottomed baby standing in front of a bathtub that had been reshared hundreds of times, but other images that have been shared on child pornography sites depict children in everyday situations such as playing on a playground, running at the beach, or doing gymnastics.
“These are images that many of us would think are innocent, but pornographers would categorize these into folders,” Dr. Keith said. “It’s not even naked or half-naked pictures.”
A study conducted by an e-safety commission in Australia, for example, found that half of the thousands of photos shared on a sample of child pornography sites had originated from parental sharing.
But Ms. Steinberg and Dr. Keith pointed out that benefits of parents’ online sharing exist as well, as the Mott survey found. In that survey, 72% of parents who discuss parenting and/or their children on social media reported that doing so helps them feel less alone. Similarly, 70% said they learn what not to do through those experiences, 67% said they receive advice from more experienced parents, and 62% said they consequently worry less. Common topics they discussed included sleep, nutrition, discipline, day care, and behavior management.
Other benefits, Ms. Steinberg pointed out, are that families geographically spread apart can stay connected, and communities can grow stronger with shared communal experiences of parents meeting others online.
“For some parents, it gives them an opportunity for advocacy work and raises awareness for important social issues,” Ms. Steinberg said, although she added, “If you’re going to share your children’s behavioral problems, consider sharing anonymously.”
Neither Ms. Steinberg and Dr. Keith said they had simple solutions to these challenges. Rather, they recommended using the public health model of raising awareness and encouraging open dialogue among pediatricians, parents, and their children to look for ways to balance competing interests.
“Social media offers many positive benefits, and we don’t want to silence the many voices of parents who take part in online sharing,” Ms. Steinberg explained. But she and Ms. Keith said it’s also worth considering children’s potential interest in controlling what their digital footprint is as they become adults.
For example, one study they cited found that, among 249 pairs of parents and their children, three times more children than parents wanted the parents to have and follow rules regarding what they could share on social media about their children.
Although guidance for parents on monitoring children’s social media use is a part of the AAP policy statement on media, only one recommendation obliquely addresses how parents should or shouldn’t use social media by advising them to model appropriate use for their children.
“It’s just like any medical decision: What is the benefit, and what is the risk, and does the benefit outweigh the risk?” said Wendy Sue Swanson, MD, executive director of digital health at Seattle Children’s Hospital. She recommended that parents ask their child for permission before posting a story or photo if their kids are aged 6 or older.
Ms. Steinberg and Dr. Keith recommended that pediatricians broach this subject with parents to help them think about risks they simply might not have considered before.
“When we looked at what sorts of best practices could be encouraged or doctors could talk to parents about – the tangible harms, such as whether data brokers or people interested in child pornography could access the information – we didn’t want to create any unnecessary panic,” Ms. Steinberg said. “But we did find some concerns that were troublesome, and we thought that parents or at least physicians [should] be aware of those potential risks.”
Ms. Steinberg and Dr. Keith reported that they had no relevant financial disclosures.
SAN FRANCISCO – More than two-thirds of parents worry about their children’s privacy online and/or that photos of their children might be reshared on the wider Web, according to a survey conducted by C.S. Mott Children’s Hospital.
Those fears are not baseless, and they need to be considered more often by parents themselves in posting about their children online, presenters agreed at a symposium on the media at the annual meeting of the American Academy of Pediatrics.
“The first children of social media are just now entering adulthood, entering the job market,” said Stacey Steinberg, JD, a legal skills professor at the University of Florida Levin College of Law, Gainesville. She is also with the law school’s center on children and families.
She and Bahareh Keith, DO, a pediatrician at the University of Florida, discussed the challenges and risks of “sharenting” – parents’ sharing information and photos of their children online – and pediatricians’ role in advising parents and looking out for children’s best interests.
“The dearth of discussion on this topic leaves even the most well-intentioned parents without enough information to thoroughly analyze this,” Ms. Steinberg said. “We’re not sitting here saying we know what the answers are. But we’re saying this is an important issue that affects families, and these children require a voice in this discussion.”
The way social media and blogging have changed the landscape for children coming of age today means that they often have a digital footprint shaped by their parents long before they create their own first account. This reality means it’s necessary to consider how to balance children’s right to privacy with parents’ right to free speech and expression.
The 2015 C.S. Mott survey asked 569 parents of children aged 4 years and younger about how they use social media as parents, and reported that more than half of mothers (56%) and a third of fathers (34%) discuss parenting and child health topics on Facebook, Twitter, blogs, online forums, and other online platforms.
The risks of this sharenting can range from embarrassment of the child to significantly more sinister repercussions. Just over half of the parents (52%) in the Mott survey reported that they are concerned their child might feel embarrassed when they grow older and discover what their parents shared online. But that embarrassment also can lead to bullying or determent of psychosocial development, Ms. Steinberg and Dr. Keith explained.
More serious, if less common, risks include the possibility that data brokers could access and use information about the children or that online child pornographers could repurpose the photos inappropriately. One worst case scenario of the former is digital kidnapping, a disturbing practice in which a stranger uses baby photos and information that is not their own to pass off the child as their own or to invite others to “invent” identities for the child. The Children’s Online Privacy Protection Rule of the Federal Trade Commission addresses only online use by those under age 13 years, not others’ use of those images.
Regarding the latter, Ms. Steinberg and Dr. Keith showed an example of a bare-bottomed baby standing in front of a bathtub that had been reshared hundreds of times, but other images that have been shared on child pornography sites depict children in everyday situations such as playing on a playground, running at the beach, or doing gymnastics.
“These are images that many of us would think are innocent, but pornographers would categorize these into folders,” Dr. Keith said. “It’s not even naked or half-naked pictures.”
A study conducted by an e-safety commission in Australia, for example, found that half of the thousands of photos shared on a sample of child pornography sites had originated from parental sharing.
But Ms. Steinberg and Dr. Keith pointed out that benefits of parents’ online sharing exist as well, as the Mott survey found. In that survey, 72% of parents who discuss parenting and/or their children on social media reported that doing so helps them feel less alone. Similarly, 70% said they learn what not to do through those experiences, 67% said they receive advice from more experienced parents, and 62% said they consequently worry less. Common topics they discussed included sleep, nutrition, discipline, day care, and behavior management.
Other benefits, Ms. Steinberg pointed out, are that families geographically spread apart can stay connected, and communities can grow stronger with shared communal experiences of parents meeting others online.
“For some parents, it gives them an opportunity for advocacy work and raises awareness for important social issues,” Ms. Steinberg said, although she added, “If you’re going to share your children’s behavioral problems, consider sharing anonymously.”
Neither Ms. Steinberg and Dr. Keith said they had simple solutions to these challenges. Rather, they recommended using the public health model of raising awareness and encouraging open dialogue among pediatricians, parents, and their children to look for ways to balance competing interests.
“Social media offers many positive benefits, and we don’t want to silence the many voices of parents who take part in online sharing,” Ms. Steinberg explained. But she and Ms. Keith said it’s also worth considering children’s potential interest in controlling what their digital footprint is as they become adults.
For example, one study they cited found that, among 249 pairs of parents and their children, three times more children than parents wanted the parents to have and follow rules regarding what they could share on social media about their children.
Although guidance for parents on monitoring children’s social media use is a part of the AAP policy statement on media, only one recommendation obliquely addresses how parents should or shouldn’t use social media by advising them to model appropriate use for their children.
“It’s just like any medical decision: What is the benefit, and what is the risk, and does the benefit outweigh the risk?” said Wendy Sue Swanson, MD, executive director of digital health at Seattle Children’s Hospital. She recommended that parents ask their child for permission before posting a story or photo if their kids are aged 6 or older.
Ms. Steinberg and Dr. Keith recommended that pediatricians broach this subject with parents to help them think about risks they simply might not have considered before.
“When we looked at what sorts of best practices could be encouraged or doctors could talk to parents about – the tangible harms, such as whether data brokers or people interested in child pornography could access the information – we didn’t want to create any unnecessary panic,” Ms. Steinberg said. “But we did find some concerns that were troublesome, and we thought that parents or at least physicians [should] be aware of those potential risks.”
Ms. Steinberg and Dr. Keith reported that they had no relevant financial disclosures.
AT AAP 2016
SABCS 2016: Top picks from Dr. Hope S. Rugo and Dr. William J. Gradishar
Oncology Practice associate editors Dr. Hope S. Rugo and Dr. William J. Gradishar reveal several anticipated studies from the 39th annual San Antonio Breast Cancer Symposium, set to begin on Wednesday, Dec. 7:
• S1-02. PrECOG 0102. A randomized, double-blind, phase II trial of fulvestrant plus everolimus or placebo in postmenopausal women with hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative metastatic breast cancer resistant to aromatase inhibitor therapy
• S1-03. First results from the multicenter phase III DATA study comparing 3 vs. 6 years of anastrozole after 2-3 years of tamoxifen in postmenopausal women with HR–positive early breast cancer.
• S1-04. Optimal duration of extended letrozole treatment after 5 years of adjuvant endocrine therapy; results of the randomized phase III IDEAL trial (BOOG 2006-05).
• S1-05. A randomized, double-blind, placebo-controlled clinical trial to evaluate extended adjuvant endocrine therapy (5 years of letrozole) in postmenopausal women with HR–positive breast cancer who have completed previous adjuvant endocrine therapy: initial results of NRG oncology/NSABP B-42.
• S1-08. Prognostic associations of tumor-infiltrating lymphocytes in metastatic HER2-positive breast cancer treated with trastuzumab and pertuzumab: a secondary analysis of the CLEOPATRA study.

• S2-03. Does BRCA status affect outcome in young breast cancer patients? Results from the Prospective Study of Outcomes in Sporadic versus Hereditary Breast Cancer (POSH).
• S2-05. Efficacy and tolerability of veliparib (ABT-888) in combination with carboplatin (C) and paclitaxel (P) vs. placebo (Plc)+C/P in patients with BRCA1 or BRCA2 mutations and metastatic breast cancer: a randomized, phase II study.
• S2-06. DNA repair deficiency biomarkers and MammaPrint High1/(ultra)High2 risk as predictors of veliparib/carboplatin response: results from the neoadjuvant I-SPY 2 trial for high-risk breast cancer.

• S3-02. Plasma microRNA levels for predicting therapeutic response to neoadjuvant treatment in HER2-positive breast cancer: results from Neo-ALTTO.
• S3-04. Primary analysis of PERTAIN: a randomized, two-arm, open-label, multicenter phase II trial assessing the efficacy and safety of pertuzumab given in combination with trastuzumab plus an aromatase inhibitor in first-line patients with HER2-positive and HR–positive metastatic or locally advanced breast cancer.
• S4-06. Biologic effects of abemaciclib in a phase II neoadjuvant study for postmenopausal patients with HR–positive, HER2-negative breast cancer.
• S4-07. BELLE-3: a phase III study of buparlisib + fulvestrant in postmenopausal women with HR-positive, HER2-negative, aromatase inhibitor–treated locally advanced or metastatic breast cancer, who progressed on or after mTOR inhibitor–based treatment.
• S5-02. Scalp Cooling Alopecia Prevention (SCALP) trial for patients with early-stage breast cancer.
Dr. Rugo is professor of medicine, University of California, San Francisco, and director, breast oncology and clinical trials education, UCSF Helen Diller Family Comprehensive Cancer Center. Dr. Gradishar is the Betsy Bramsen Professor of Breast Oncology, professor of medicine, and deputy chief in the division of hematology/oncology, Feinberg School of Medicine, Chicago; and deputy director for the Clinical Network and director of the Maggie Daley Center for Women’s Cancer Care, Robert H. Lurie Comprehensive Cancer Center Network of Northwestern University, Chicago.
Oncology Practice associate editors Dr. Hope S. Rugo and Dr. William J. Gradishar reveal several anticipated studies from the 39th annual San Antonio Breast Cancer Symposium, set to begin on Wednesday, Dec. 7:
• S1-02. PrECOG 0102. A randomized, double-blind, phase II trial of fulvestrant plus everolimus or placebo in postmenopausal women with hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative metastatic breast cancer resistant to aromatase inhibitor therapy
• S1-03. First results from the multicenter phase III DATA study comparing 3 vs. 6 years of anastrozole after 2-3 years of tamoxifen in postmenopausal women with HR–positive early breast cancer.
• S1-04. Optimal duration of extended letrozole treatment after 5 years of adjuvant endocrine therapy; results of the randomized phase III IDEAL trial (BOOG 2006-05).
• S1-05. A randomized, double-blind, placebo-controlled clinical trial to evaluate extended adjuvant endocrine therapy (5 years of letrozole) in postmenopausal women with HR–positive breast cancer who have completed previous adjuvant endocrine therapy: initial results of NRG oncology/NSABP B-42.
• S1-08. Prognostic associations of tumor-infiltrating lymphocytes in metastatic HER2-positive breast cancer treated with trastuzumab and pertuzumab: a secondary analysis of the CLEOPATRA study.
• S2-03. Does BRCA status affect outcome in young breast cancer patients? Results from the Prospective Study of Outcomes in Sporadic versus Hereditary Breast Cancer (POSH).
• S2-05. Efficacy and tolerability of veliparib (ABT-888) in combination with carboplatin (C) and paclitaxel (P) vs. placebo (Plc)+C/P in patients with BRCA1 or BRCA2 mutations and metastatic breast cancer: a randomized, phase II study.
• S2-06. DNA repair deficiency biomarkers and MammaPrint High1/(ultra)High2 risk as predictors of veliparib/carboplatin response: results from the neoadjuvant I-SPY 2 trial for high-risk breast cancer.
• S3-02. Plasma microRNA levels for predicting therapeutic response to neoadjuvant treatment in HER2-positive breast cancer: results from Neo-ALTTO.
• S3-04. Primary analysis of PERTAIN: a randomized, two-arm, open-label, multicenter phase II trial assessing the efficacy and safety of pertuzumab given in combination with trastuzumab plus an aromatase inhibitor in first-line patients with HER2-positive and HR–positive metastatic or locally advanced breast cancer.
• S4-06. Biologic effects of abemaciclib in a phase II neoadjuvant study for postmenopausal patients with HR–positive, HER2-negative breast cancer.
• S4-07. BELLE-3: a phase III study of buparlisib + fulvestrant in postmenopausal women with HR-positive, HER2-negative, aromatase inhibitor–treated locally advanced or metastatic breast cancer, who progressed on or after mTOR inhibitor–based treatment.
• S5-02. Scalp Cooling Alopecia Prevention (SCALP) trial for patients with early-stage breast cancer.
Dr. Rugo is professor of medicine, University of California, San Francisco, and director, breast oncology and clinical trials education, UCSF Helen Diller Family Comprehensive Cancer Center. Dr. Gradishar is the Betsy Bramsen Professor of Breast Oncology, professor of medicine, and deputy chief in the division of hematology/oncology, Feinberg School of Medicine, Chicago; and deputy director for the Clinical Network and director of the Maggie Daley Center for Women’s Cancer Care, Robert H. Lurie Comprehensive Cancer Center Network of Northwestern University, Chicago.
Oncology Practice associate editors Dr. Hope S. Rugo and Dr. William J. Gradishar reveal several anticipated studies from the 39th annual San Antonio Breast Cancer Symposium, set to begin on Wednesday, Dec. 7:
• S1-02. PrECOG 0102. A randomized, double-blind, phase II trial of fulvestrant plus everolimus or placebo in postmenopausal women with hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative metastatic breast cancer resistant to aromatase inhibitor therapy
• S1-03. First results from the multicenter phase III DATA study comparing 3 vs. 6 years of anastrozole after 2-3 years of tamoxifen in postmenopausal women with HR–positive early breast cancer.
• S1-04. Optimal duration of extended letrozole treatment after 5 years of adjuvant endocrine therapy; results of the randomized phase III IDEAL trial (BOOG 2006-05).
• S1-05. A randomized, double-blind, placebo-controlled clinical trial to evaluate extended adjuvant endocrine therapy (5 years of letrozole) in postmenopausal women with HR–positive breast cancer who have completed previous adjuvant endocrine therapy: initial results of NRG oncology/NSABP B-42.
• S1-08. Prognostic associations of tumor-infiltrating lymphocytes in metastatic HER2-positive breast cancer treated with trastuzumab and pertuzumab: a secondary analysis of the CLEOPATRA study.
• S2-03. Does BRCA status affect outcome in young breast cancer patients? Results from the Prospective Study of Outcomes in Sporadic versus Hereditary Breast Cancer (POSH).
• S2-05. Efficacy and tolerability of veliparib (ABT-888) in combination with carboplatin (C) and paclitaxel (P) vs. placebo (Plc)+C/P in patients with BRCA1 or BRCA2 mutations and metastatic breast cancer: a randomized, phase II study.
• S2-06. DNA repair deficiency biomarkers and MammaPrint High1/(ultra)High2 risk as predictors of veliparib/carboplatin response: results from the neoadjuvant I-SPY 2 trial for high-risk breast cancer.
• S3-02. Plasma microRNA levels for predicting therapeutic response to neoadjuvant treatment in HER2-positive breast cancer: results from Neo-ALTTO.
• S3-04. Primary analysis of PERTAIN: a randomized, two-arm, open-label, multicenter phase II trial assessing the efficacy and safety of pertuzumab given in combination with trastuzumab plus an aromatase inhibitor in first-line patients with HER2-positive and HR–positive metastatic or locally advanced breast cancer.
• S4-06. Biologic effects of abemaciclib in a phase II neoadjuvant study for postmenopausal patients with HR–positive, HER2-negative breast cancer.
• S4-07. BELLE-3: a phase III study of buparlisib + fulvestrant in postmenopausal women with HR-positive, HER2-negative, aromatase inhibitor–treated locally advanced or metastatic breast cancer, who progressed on or after mTOR inhibitor–based treatment.
• S5-02. Scalp Cooling Alopecia Prevention (SCALP) trial for patients with early-stage breast cancer.
Dr. Rugo is professor of medicine, University of California, San Francisco, and director, breast oncology and clinical trials education, UCSF Helen Diller Family Comprehensive Cancer Center. Dr. Gradishar is the Betsy Bramsen Professor of Breast Oncology, professor of medicine, and deputy chief in the division of hematology/oncology, Feinberg School of Medicine, Chicago; and deputy director for the Clinical Network and director of the Maggie Daley Center for Women’s Cancer Care, Robert H. Lurie Comprehensive Cancer Center Network of Northwestern University, Chicago.
FROM SABCS 2016
Clinical Challenges - December 2016
What is the most plausible diagnosis and what would be the next step?
The diagnosis
To clarify the diagnosis, endoscopic resection of the smaller lesion was performed and deeper biopsies of the other lesions were taken. Histology revealed lymphoid, centroblast, and centrocyte-like cell proliferation with follicular pattern (Figure D). Immunohistochemically, the follicles stained for bcl-6, CD20, and bcl-2 (Figure E), but not CD3, CD5, CD10, or cyclin D1.
Malignant lymphomas of the colon represent about 0.2% of all colonic neoplasms and most frequently are diffuse large B-cell, mucosa-associated lymphoid tissue, and mantle cell lymphomas.2 This phenotypic presentation, as multiple lymphomatous polyposis, has been reported in colon follicular lymphomas but is more typical of mantle cell lymphoma.3 Treatment usually consists of chemotherapy containing rituximab (anti-CD20) and should be decided on a case-by-case basis owing to possible relapse and the often indolent course.1
References
1. Damaj, G., Verkarre, V., Delmer, A. et al. Primary follicular lymphoma of the gastrointestinal tract: A study of 25 cases and a literature review. Ann Oncol. 2003;14:623-9.
2. Muller-Hermelink, H.K., Chott, A., Gascoyne, R.D. et al. B-cell lymphoma of the colon and rectum. In: S.R. Hamilton, L.A. Asltonen, eds. WHO Classification of Tumours. Lyon, France: IARC Press;2001:139-41.
3. Hiraide, T., Shoji, T., Higashi, Y. et al. Extranodal multiple polypoid follicular lymphoma of the sigmoid colon. Gastrointest Endosc. 2011;73(1):182-4.
The diagnosis
To clarify the diagnosis, endoscopic resection of the smaller lesion was performed and deeper biopsies of the other lesions were taken. Histology revealed lymphoid, centroblast, and centrocyte-like cell proliferation with follicular pattern (Figure D). Immunohistochemically, the follicles stained for bcl-6, CD20, and bcl-2 (Figure E), but not CD3, CD5, CD10, or cyclin D1.
Malignant lymphomas of the colon represent about 0.2% of all colonic neoplasms and most frequently are diffuse large B-cell, mucosa-associated lymphoid tissue, and mantle cell lymphomas.2 This phenotypic presentation, as multiple lymphomatous polyposis, has been reported in colon follicular lymphomas but is more typical of mantle cell lymphoma.3 Treatment usually consists of chemotherapy containing rituximab (anti-CD20) and should be decided on a case-by-case basis owing to possible relapse and the often indolent course.1
References
1. Damaj, G., Verkarre, V., Delmer, A. et al. Primary follicular lymphoma of the gastrointestinal tract: A study of 25 cases and a literature review. Ann Oncol. 2003;14:623-9.
2. Muller-Hermelink, H.K., Chott, A., Gascoyne, R.D. et al. B-cell lymphoma of the colon and rectum. In: S.R. Hamilton, L.A. Asltonen, eds. WHO Classification of Tumours. Lyon, France: IARC Press;2001:139-41.
3. Hiraide, T., Shoji, T., Higashi, Y. et al. Extranodal multiple polypoid follicular lymphoma of the sigmoid colon. Gastrointest Endosc. 2011;73(1):182-4.
The diagnosis
To clarify the diagnosis, endoscopic resection of the smaller lesion was performed and deeper biopsies of the other lesions were taken. Histology revealed lymphoid, centroblast, and centrocyte-like cell proliferation with follicular pattern (Figure D). Immunohistochemically, the follicles stained for bcl-6, CD20, and bcl-2 (Figure E), but not CD3, CD5, CD10, or cyclin D1.
Malignant lymphomas of the colon represent about 0.2% of all colonic neoplasms and most frequently are diffuse large B-cell, mucosa-associated lymphoid tissue, and mantle cell lymphomas.2 This phenotypic presentation, as multiple lymphomatous polyposis, has been reported in colon follicular lymphomas but is more typical of mantle cell lymphoma.3 Treatment usually consists of chemotherapy containing rituximab (anti-CD20) and should be decided on a case-by-case basis owing to possible relapse and the often indolent course.1
References
1. Damaj, G., Verkarre, V., Delmer, A. et al. Primary follicular lymphoma of the gastrointestinal tract: A study of 25 cases and a literature review. Ann Oncol. 2003;14:623-9.
2. Muller-Hermelink, H.K., Chott, A., Gascoyne, R.D. et al. B-cell lymphoma of the colon and rectum. In: S.R. Hamilton, L.A. Asltonen, eds. WHO Classification of Tumours. Lyon, France: IARC Press;2001:139-41.
3. Hiraide, T., Shoji, T., Higashi, Y. et al. Extranodal multiple polypoid follicular lymphoma of the sigmoid colon. Gastrointest Endosc. 2011;73(1):182-4.
What is the most plausible diagnosis and what would be the next step?
What is the most plausible diagnosis and what would be the next step?
What’s your diagnosis?
By Aníbal Ferreira, MD, PhD , Raquel Gonçalves, MD, and Carla Rolanda, MD. Published previously in Gastroenterology (2012 Dec;143[6]:1440, 1693-4). 
An asymptomatic, 74-year-old woman with type 2 diabetes was referred for endoscopic colorectal cancer screening. Colonoscopy revealed a 30-mm, polypoid, firm lesion in the transverse colon (Figure A), a 20-mm similar lesion in the cecum (Figure B),