User login
We can work it out: Should I hire my patient?
Dear Dr. Mossman,
Each month, I see my patient, Mr. R, for a 15-minute medication management appointment. At his latest visit, Mr. R mentioned his financial difficulties. He also observed that our office needed to have some carpentry work done—not a surprise, because he’s known in our area as one of the best carpenters around. He suggested that I hire him as payment for the next 6 appointments. What risks might I encounter if I oblige him?
Submitted by “Dr. Z”
Nearly 29 million Americans are uninsured,1 and even more have trouble accessing mental health care.2 Many psychiatrists struggle to provide affordable services while remaining financially viable.3,4 For outpatients with limited means to pay for care, spacing appointments to fit their budgets might compromise treatment.5 Simply not charging patients poses its own clinical and ethical challenges.6-8
As a result, some mental health professionals make barter arrangements to help their patients enter or continue treatment. To answer Dr. Z’s question on whether exchanging services might be a way to arrange matters with some patients, we explore:
- the idea of bartering for psychiatric treatment
- related ethical and legal considerations
- when and in what situations bartering might be appropriate.
Think of what I’m saying: Bartering for treatment
“Barter” refers to exchanging commodities, products, or services of equivalent value without using money.9 In 2010, Nevada Republican Senate candidate Sue Lowden encouraged barter for health care and harkened back to an earlier time where “they would bring a chicken to the doctor; they would say ‘I’ll paint your house.’”10
Such payment arrangements have been encouraged as health care has become increasingly commoditized.11-13 This happens through both direct barter between physician and patient and barter exchanges. Barter exchange systems have been set up on Web sites (as of 2013, at least 400 such online exchanges were available14), local communities,11,15 and social programs. For example, through the “Swapping Guns for Therapy” program, psychologists in California gave free or reduced-fee care for people who traded in their guns.16
Try to see it my way: A prevailing view of barter
Several psychiatrists recommend against bartering for treatment, for a variety of reasons.7,8,17-19 Simon18 argues that a stable fee policy is part of a proper therapeutic framework, and money is “the only acceptable medium of exchange when receiving payment from patients.” Emotional distress and the power differential inherent in treatment might prevent a patient from making an accurate assessment of the value of the bartered goods or services,7,8,17,18,20 which could lead to future claims of undue influence from trading goods or services below market value.17 To avoid the possibility of exploitating the patient, Simon18 recommends that the psychiatrist’s professional fee be “the only material benefit received from the patient.”
The American Psychiatric Association’s code of ethics states that “it is not ethical to switch a doctor–patient relationship to an employer–employee one … and, in most cases, such an arrangement would be unethical.”21 In some therapeutic settings, employing a patient risks inappropriate self-disclosure and intrusion.16
More than other physicians, psychiatrists pay special attention to professional boundaries, the technical term for the “edge of appropriate behavior,” within which safe, effective care can occur.22,23 Although some boundary crossings can be harmless and even constructive, repeated boundary crossings are the forerunners to improper behavior, including sexual relationships with patients.24-26
Out of concern that bartering could become the first step down a slippery ethical slope toward patient exploitation, mental health clinicians have deemed the practice “ethically troubling,”19 said it did “not usually work out well,”7 and declared it “so fraught with risks for both parties that it seem[ed] illogical to even consider it as an option.”27
While I see it your way: What barter proponents say
Reports of bartering for chickens28 and purchasing fuel from a patient in remote Alaska29 show that not all physicians agree and why they feel that professional codes of ethics reflect an urban bias.28,29 In many rural areas and small towns, access to mental health services is limited, and patients often interact with their doctors outside of clinical encounters.23,29-31
Bartering can benefit a physician’s practice by:
- reducing the need to discount services
- eliminating bureaucratic burdens of traditional insurance arrangements
- facilitating development of a patient base
- allowing patients choice and flexibility in seeking medical care.6,16,32
Bartering could confer certain clinical benefits, such as:
- enhancing trust and empathy32
- encouraging patients to make their needs known constructively6
- modeling financial self-care6
- helping the doctor to feel fairly compensated for providing thoughtful care6
- acknowledging the patient’s cultural values15,33
- affirming that patients and doctors both produce things of value.16
I have always thought: Other ethical models
An ethical approach to bartering that requires careful thought and respect for the patient’s needs appears consistent with a primary goal of treatment: “to increase the capacity of individuals to make more rational choices in their lives and to be relatively freer from disabling conflicts.”20 Some authors criticize slippery-slope arguments and strict-rule ethical approaches as being too rigid, limiting, or risk-averse.22,26,34 In Table 1,6,8,16,18,27,29-31,35-37 we list several factors that might weigh for or against a decision to enter into a barter arrangement as payment for care.
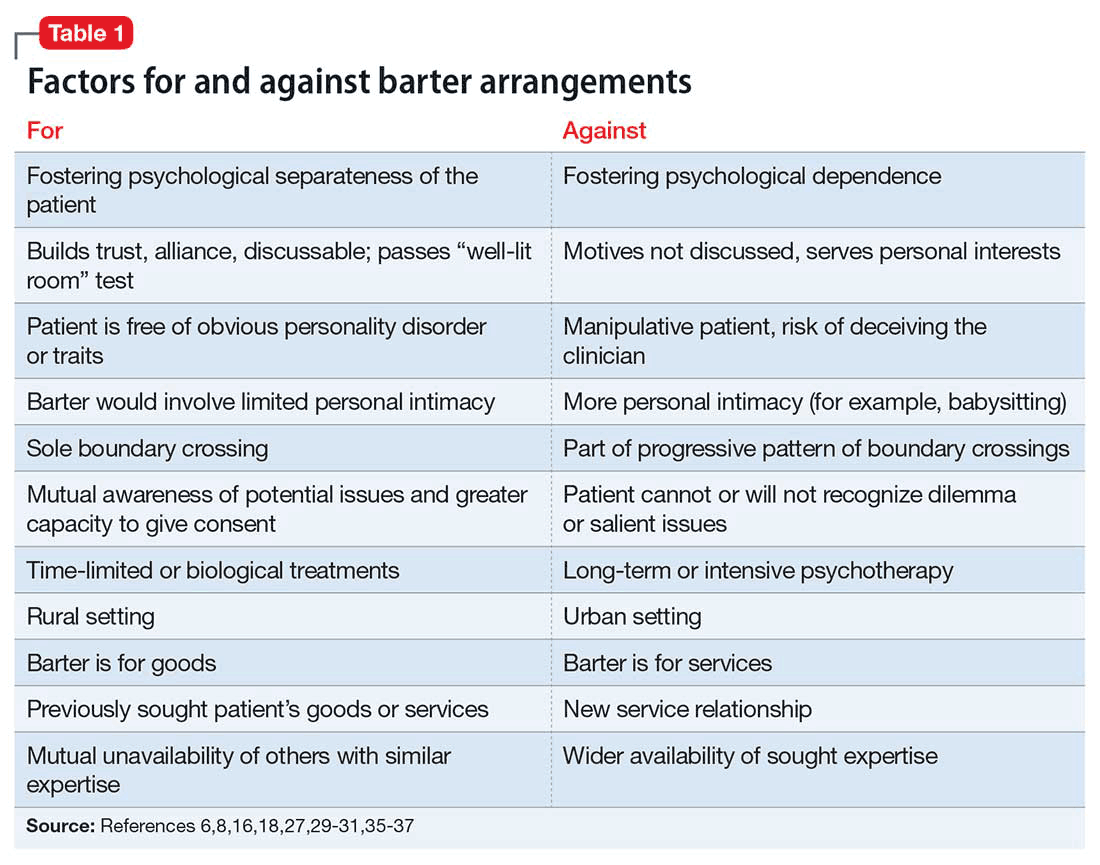
In a similar manner, Martinez33,38 proposed a graded-risk framework that encourages examination of potential harms and benefits of a decision, potential coercive or exploitative elements, the clinician’s intentions and aspiration to professional ideals, and the context of the decision. Within this framework, some bartering arrangements might be encouraged and, perhaps, even obligatory because of the potential benefits to the patient; other arrangements (eg, trading psychotherapy for menial services) might be unjustifiable. Martinez38 argues that this approach fosters mutual decision-making with patients, discourages physician paternalism, and “demands that we struggle with the particulars with each case.”
Gottlieb’s decision-making model35 recognizes that trying to avoid all dual relationships is unrealistic and not all dual relationships are exploitative. Instead, a clinician must assess 3 dimensions of current and proposed relationships:
- the degree of power differential
- the duration of treatment
- the clarity of termination.
The decision-making process also requires involvement of the patient, who if “unable to recognize the dilemma or is unwilling to consider the issues before deciding, should be considered at risk, and the contemplated relationship rejected.”35
So I will ask you once again: Dr. Z’s decision
In the case of Dr. Z and Mr. R, a barter arrangement might work in the sense of permitting and sustaining good care. Mr. R suggested the idea and might not be able to afford care without it. Nothing in Dr. Z’s description suggests that Mr. R has personality characteristics or other conditions that would compromise his ability to give informed consent or to understand the nuances of a barter arrangement. Dr. Z is not providing a treatment (eg, psychodynamic therapy) that a barter arrangement could contaminate. That the arrangement would be circumscribed limits the effect of a power differential, as would its brief duration and defined termination endpoint. Dr. Z’s letter to the authors also shows his willingness to seek consultation.
There’s a chance that we may fall apart: Reasons for caution
Martinez’s graded-risk approach recognizes reasons for caution:
- the risk of harm to the patient or doctor–patient relationship
- the uncertain benefit to the patient
- the blurring of Dr. Z’s self-interest and Mr. R’s needs
- some ambiguity about possible exploitation.
Dr. Z and Mr. R have not discussed the value of Mr. R’s work—which might create a rift between them—and despite Mr. R’s reputation, other carpenters are available. Future med-check appointments will give them little time to explore and discuss the meanings of the barter.
Any proposed barter arrangement creates some clinical perils that can be particularly salient in mental health treatment. Patients could view themselves as “special” or entitled to enhanced access to the doctor because of exchanged services, which could take a toll on the doctor.39 The physician’s objectivity might diminish, and the business aspect of their relationship could make both parties less comfortable when discussing sensitive information relevant to treatment.31,40 Also, the suggested barter is for services to be provided at Dr. Z’s office, where confidentiality may be breeched and transference issues could arise.
A medical malpractice claim states that a doctor has breached a duty of care to a patient such that harm (or “damages”) resulted.41 Should Dr. Z and Mr. R’s barter agreement turn sour and harm follow, Mr. R could sue for recovery of damages based of a claim of duress, undue influence, or other aspects of the doctor–patient power differential.27,42,43 Given the published views we have described, a psychiatrist who barters also may be viewed as violating state regulations that measure the standard of care against generally accepted practice.
Only time will tell if I am right or I am wrong
If you face a situation similar to Dr. Z’s and want to consider a barter arrangement, you can take several steps to mitigate potential risk to your patient and ensure competent care (Table 25,6,15,16,32,35,39,40,44-47). One of the most important steps is to seek ongoing consultation, both before and after a decision to barter. Ideally, the consulting colleague would know you and your circumstances and would have sufficient clinical grasp of the patient to make an informed assessment of risks and benefits.35 This consultation, as well as your own rationale for acting on recommendations, should be thoroughly documented in the patient’s records.26,44,45
Certain types of barter should be off limits, including:
- trading prescription drugs for goods or services
- trading for services that tie into the success of one’s business (eg, business advising or marketing)16
- offering treatment in exchange for illegal or ethically unacceptable services.48
Beyond ethical considerations are some practical issues. The Internal Revenue Service has specific rules regarding taxation of bartered goods and services, which must be included as taxable income.46 If possible, an independent agent should appraise the traded goods or services before the agreement.6 When working in a group practice, the clinician might have to figure out how to allocate the received goods or services such as shared overhead costs.28 Preferably, the patient’s goods or services should be provided before care is delivered.16 If not, the duration of services rendered should be limited, and either party should have the option to disengage from the relationship if one feels dissatisfied.16
A written contract, discussed ahead of time, can be a sound way to summarize the terms of the arrangement. Both sides also should consider what would happen if an injury occurred.16 Finally, you must adhere to any relevant state laws regarding payment for services, particularly if the patient has health insurance.32
If the bartering arrangement does not work, you should take an open and non-defensive approach. If you believe you have made a mistake, consider apologizing.45
1. Kaiser Commission on Medicaid and the Uninsured. Key facts about the uninsured population. http://kff.org/uninsured/fact-sheet/key-facts-about-the-uninsured-population. Published September 29, 2016. Accessed October 7, 2016.
2. National Alliance on Mental Illness. A long road ahead: achieving true parity in mental health and substance use care. https://www.nami.org/About-NAMI/Publications-Reports/Public-Policy-Reports/A-Long-Road-Ahead/2015-ALongRoadAhead.pdf. Published April 2015. Accessed October 7, 2016.
3. Insel T. Director’s blog: the paradox of parity. May 30, 2014. https://www.nimh.nih.gov/about/director/2014/the-paradox-of-parity.shtml. Published May 30, 2014. Accessed October 7, 2016.
4. Bishop TF, Press MJ, Keyhani S, et al. Acceptance of insurance by psychiatrists and the implications for access to mental health care. JAMA Psychiatry. 2014;71(2):176-181.
5. What do you do when patients cannot pay? Psychiatry (Edgmont). 2009;6(5):51-52.
6. Hill M. Barter: ethical considerations in psychotherapy. Women Ther. 2000;22(3):81-91.
7. Simon RI. Commentary: treatment boundaries—flexible guidelines, not rigid standards. J Am Acad Psychiatry Law. 2001;29(3):287-289.
8. Simon RI, Williams IC. Maintaining treatment boundaries in small communities and rural areas. Psychiatr Serv. 1999;50(11):1440-1446.
9. Compact edition of the Oxford English dictionary. New York, NY: Oxford University Press; 1971:171.
10. Coolican JP. Sue Lowden stands by health care plan. Las Vegas Sun. http://lasvegassun.com/news/2010/apr/20/sue-lowden-draws-fire-repeating-health-care-barter. Published April 20, 2010. Accessed September 20, 2016.
11. Consumer Reports. Barter sometimes allow patients to pay for health care they otherwise could not afford. Washington Post. https://www.washingtonpost.com/national/health-science/barter-sometimes-allow-patients-to-pay-for-health-care-they-otherwise-could-not-afford/2013/09/30/e7e5a55e-069d-11e3-88d6-d5795fab4637_story.html. Published September 20, 2013. Accessed September 27, 2016.
12. Ellis B. MediBid auction site lets doctors bid for patients. CNN Money. http://money.cnn.com/2014/01/09/pf/insurance/medibid. Published January 9, 2014. Accessed September 23, 2016.
13. Ambrosino B. Surgery for sale: the ethics of health care bartering in a social media marketplace. http://hub.jhu.edu/2014/01/16/hopkins-ethicist-ponders-medibid. Published January 16, 2014. Accessed September 23, 2016.
14. Thomas C. When patients barter for health care. https://ethicalnag.org/2013/07/30/barter. Published July 30, 2016. Accessed October 9, 2016.
15. Syme G. Fetters or freedom: dual relationships in counselling. Int J Adv Counselling. 2006;28(1):57-69.
16. Zur O. Bartering in psychotherapy and counselling: complexities, case studies and guidelines. New Therapist. 2008;58:18-26.
17. Simon RI. The psychiatrist as a fiduciary: avoiding the double agent role. Psychiatric Annals. 1987;17(9):622-626.
18. Simon RI. Treatment boundary violations: clinical, ethical, and legal considerations. Bull Am Acad Psychiatry Law. 1992;20(3):269-288.
19. Walker R, Clark JJ. Heading off boundary problems: clinical supervision as risk management. Psychiatr Serv. 1999;50(11):1435-1439.
20. Malmquist CP, Norman MT. Psychiatrist-patient boundary issues following treatment termination. Am J Psychiatry. 2001;158(7):1010-1018.
21. American Psychiatric Association. The opinions of the ethics committee on the principles of medical ethics, with annotations especially applicable to psychiatry. https://www.psychiatry.org/psychiatrists/practice/ethics. Published 2016. Accessed October 4, 2016.
22. Gutheil TG, Gabbard GO. Misuses and misunderstandings of boundary theory in clinical and regulatory settings. Am J Psychiatry. 1998;155(3):409-414.
23. Crowden A. Professional boundaries and the ethics of dual and multiple overlapping relationships in psychotherapy. Monash Bioeth Rev. 2008;27(4):10-27.
24. Gabbard GO. Commentary: boundaries, culture, and psychotherapy. J Am Acad Psychiatry Law. 2001;29(3):284-286.
25. Kroll J. Boundary violations: a culture-bound syndrome. J Am Acad Psychiatry Law. 2001;29(3):274-283.
26. Gottlieb MC, Younggren JN. Is there a slippery slope? Considerations regarding multiple relationships and risk management. Professional Psychology: Research and Practice. 2009;40(6):564-557.
27. Woody RH. Bartering for psychological services. Professional Psychology: Research and Practice. 1998;29(2):174-178.
28. Bartering for medical care. MGMA Connex. 2008;8(6):11.
29. Roberts LW, Battaglia J, Epstein RS. Frontier ethics: mental health care needs and ethical dilemmas in rural communities. Psychiatr Serv. 1994;50(4):497-503.
30. Endacott R, Wood A, Judd F, et al. Impact and management of dual relationships in metropolitan, regional and rural mental health practice. Aust N Z J Psychiatry. 2006;40(11-12):987-994.
31. Scopelliti J, Judd F, Grigg M, et al. Dual relationships in mental health practice: issues for clinicians in rural settings. Aust N Z J Psychiatry. 2004;38(11-12):953-959.
32. Ayers AA. Bartering basics for the urgent care operator. http://www.alanayersurgentcare.com/Linked_Files/2013_Articles/Ayers_UCAOA_Bartering_%20Basics_2012_01_09.pdf. Accessed September 23, 2016.
33. Savin D, Martinez R. Cross-cultural boundary dilemmas: a graded-risk assessment approach. Transcult Psychiatry. 2006;42(2):243-258.
34. Glass LL. The gray areas of boundary crossings and violations. Am J Psychother. 2003;57(4):429-444.
35. Gottlieb MC. Avoiding exploitive dual relationships: a decision-making model. Psychotherapy (Chic). 1993;30(1):41-48.
36. Lane JA. The ethical implications of bartering for mental health services: examining interdisciplinary ethical standards. http://pdxscholar.library.pdx.edu/coun_fac/36. Published 2012. Accessed October 17, 2016.
37. Miller RD, Maier GJ. Nonsexual boundary violations: sauce for the gander. J Psychiatry Law. 2002;30(3):309-329.
38. Martinez R. A model for boundary dilemmas: ethical decision-making in the patient-professional relationship. Ethical Hum Sci Serv. 2000;2(1):43-61.
39. Salmon K, Klijnsma M. Boundary issues: employing patients as staff? Br J Gen Pract. 2009;59(558):56-57.
40. College of Physicians and Surgeons Ontario. Hiring patients may compromise physician-patient relationship. Dialogue. 2015;3:47.
41. Bal S. An introduction to medical malpractice in the United States. Clin Orthop Relat Res. 2009;467(2):339-347.
42. What puts a psychiatrist at risk for a malpractice lawsuit? Psychiatry (Edgmont). 2009;6(8):38-39.
43. Geis v Landau, 117 Misc2d 396 (NY Misc 1983).
44. Nisselle P. Danger zone. When boundaries are crossed in the doctor-patient relationship. Aust Fam Physician. 2000;29(6):541-544.
45. Pope KS, Keith-Spiegel P. A practical approach to boundaries in psychotherapy: making decisions, bypassing blunders, and mending fences. J Clin Psychol. 2008;64(5):638-652.
46. IRS Publication 17. https://www.irs.gov/publications/p17/ch12.html. Published 2015. Accessed October 5, 2016.
47. Epstein RS, Simon RI. The exploitation index: an early warning indicator of boundary violations in psychotherapy. Bull Menninger Clin. 1990;54(4):450-465.
48. Skutch J. Savannah doctor accused of trading drugs for sex with strippers. Augusta Chronicle. http://chronicle.augusta.com/news/crime-courts/2013-01-31/savannah-doctor-accused-trading-drugs-sex-strippers. Published January 31, 2013. Accessed October 16, 2016.
Dear Dr. Mossman,
Each month, I see my patient, Mr. R, for a 15-minute medication management appointment. At his latest visit, Mr. R mentioned his financial difficulties. He also observed that our office needed to have some carpentry work done—not a surprise, because he’s known in our area as one of the best carpenters around. He suggested that I hire him as payment for the next 6 appointments. What risks might I encounter if I oblige him?
Submitted by “Dr. Z”
Nearly 29 million Americans are uninsured,1 and even more have trouble accessing mental health care.2 Many psychiatrists struggle to provide affordable services while remaining financially viable.3,4 For outpatients with limited means to pay for care, spacing appointments to fit their budgets might compromise treatment.5 Simply not charging patients poses its own clinical and ethical challenges.6-8
As a result, some mental health professionals make barter arrangements to help their patients enter or continue treatment. To answer Dr. Z’s question on whether exchanging services might be a way to arrange matters with some patients, we explore:
- the idea of bartering for psychiatric treatment
- related ethical and legal considerations
- when and in what situations bartering might be appropriate.
Think of what I’m saying: Bartering for treatment
“Barter” refers to exchanging commodities, products, or services of equivalent value without using money.9 In 2010, Nevada Republican Senate candidate Sue Lowden encouraged barter for health care and harkened back to an earlier time where “they would bring a chicken to the doctor; they would say ‘I’ll paint your house.’”10
Such payment arrangements have been encouraged as health care has become increasingly commoditized.11-13 This happens through both direct barter between physician and patient and barter exchanges. Barter exchange systems have been set up on Web sites (as of 2013, at least 400 such online exchanges were available14), local communities,11,15 and social programs. For example, through the “Swapping Guns for Therapy” program, psychologists in California gave free or reduced-fee care for people who traded in their guns.16
Try to see it my way: A prevailing view of barter
Several psychiatrists recommend against bartering for treatment, for a variety of reasons.7,8,17-19 Simon18 argues that a stable fee policy is part of a proper therapeutic framework, and money is “the only acceptable medium of exchange when receiving payment from patients.” Emotional distress and the power differential inherent in treatment might prevent a patient from making an accurate assessment of the value of the bartered goods or services,7,8,17,18,20 which could lead to future claims of undue influence from trading goods or services below market value.17 To avoid the possibility of exploitating the patient, Simon18 recommends that the psychiatrist’s professional fee be “the only material benefit received from the patient.”
The American Psychiatric Association’s code of ethics states that “it is not ethical to switch a doctor–patient relationship to an employer–employee one … and, in most cases, such an arrangement would be unethical.”21 In some therapeutic settings, employing a patient risks inappropriate self-disclosure and intrusion.16
More than other physicians, psychiatrists pay special attention to professional boundaries, the technical term for the “edge of appropriate behavior,” within which safe, effective care can occur.22,23 Although some boundary crossings can be harmless and even constructive, repeated boundary crossings are the forerunners to improper behavior, including sexual relationships with patients.24-26
Out of concern that bartering could become the first step down a slippery ethical slope toward patient exploitation, mental health clinicians have deemed the practice “ethically troubling,”19 said it did “not usually work out well,”7 and declared it “so fraught with risks for both parties that it seem[ed] illogical to even consider it as an option.”27
While I see it your way: What barter proponents say
Reports of bartering for chickens28 and purchasing fuel from a patient in remote Alaska29 show that not all physicians agree and why they feel that professional codes of ethics reflect an urban bias.28,29 In many rural areas and small towns, access to mental health services is limited, and patients often interact with their doctors outside of clinical encounters.23,29-31
Bartering can benefit a physician’s practice by:
- reducing the need to discount services
- eliminating bureaucratic burdens of traditional insurance arrangements
- facilitating development of a patient base
- allowing patients choice and flexibility in seeking medical care.6,16,32
Bartering could confer certain clinical benefits, such as:
- enhancing trust and empathy32
- encouraging patients to make their needs known constructively6
- modeling financial self-care6
- helping the doctor to feel fairly compensated for providing thoughtful care6
- acknowledging the patient’s cultural values15,33
- affirming that patients and doctors both produce things of value.16
I have always thought: Other ethical models
An ethical approach to bartering that requires careful thought and respect for the patient’s needs appears consistent with a primary goal of treatment: “to increase the capacity of individuals to make more rational choices in their lives and to be relatively freer from disabling conflicts.”20 Some authors criticize slippery-slope arguments and strict-rule ethical approaches as being too rigid, limiting, or risk-averse.22,26,34 In Table 1,6,8,16,18,27,29-31,35-37 we list several factors that might weigh for or against a decision to enter into a barter arrangement as payment for care.
In a similar manner, Martinez33,38 proposed a graded-risk framework that encourages examination of potential harms and benefits of a decision, potential coercive or exploitative elements, the clinician’s intentions and aspiration to professional ideals, and the context of the decision. Within this framework, some bartering arrangements might be encouraged and, perhaps, even obligatory because of the potential benefits to the patient; other arrangements (eg, trading psychotherapy for menial services) might be unjustifiable. Martinez38 argues that this approach fosters mutual decision-making with patients, discourages physician paternalism, and “demands that we struggle with the particulars with each case.”
Gottlieb’s decision-making model35 recognizes that trying to avoid all dual relationships is unrealistic and not all dual relationships are exploitative. Instead, a clinician must assess 3 dimensions of current and proposed relationships:
- the degree of power differential
- the duration of treatment
- the clarity of termination.
The decision-making process also requires involvement of the patient, who if “unable to recognize the dilemma or is unwilling to consider the issues before deciding, should be considered at risk, and the contemplated relationship rejected.”35
So I will ask you once again: Dr. Z’s decision
In the case of Dr. Z and Mr. R, a barter arrangement might work in the sense of permitting and sustaining good care. Mr. R suggested the idea and might not be able to afford care without it. Nothing in Dr. Z’s description suggests that Mr. R has personality characteristics or other conditions that would compromise his ability to give informed consent or to understand the nuances of a barter arrangement. Dr. Z is not providing a treatment (eg, psychodynamic therapy) that a barter arrangement could contaminate. That the arrangement would be circumscribed limits the effect of a power differential, as would its brief duration and defined termination endpoint. Dr. Z’s letter to the authors also shows his willingness to seek consultation.
There’s a chance that we may fall apart: Reasons for caution
Martinez’s graded-risk approach recognizes reasons for caution:
- the risk of harm to the patient or doctor–patient relationship
- the uncertain benefit to the patient
- the blurring of Dr. Z’s self-interest and Mr. R’s needs
- some ambiguity about possible exploitation.
Dr. Z and Mr. R have not discussed the value of Mr. R’s work—which might create a rift between them—and despite Mr. R’s reputation, other carpenters are available. Future med-check appointments will give them little time to explore and discuss the meanings of the barter.
Any proposed barter arrangement creates some clinical perils that can be particularly salient in mental health treatment. Patients could view themselves as “special” or entitled to enhanced access to the doctor because of exchanged services, which could take a toll on the doctor.39 The physician’s objectivity might diminish, and the business aspect of their relationship could make both parties less comfortable when discussing sensitive information relevant to treatment.31,40 Also, the suggested barter is for services to be provided at Dr. Z’s office, where confidentiality may be breeched and transference issues could arise.
A medical malpractice claim states that a doctor has breached a duty of care to a patient such that harm (or “damages”) resulted.41 Should Dr. Z and Mr. R’s barter agreement turn sour and harm follow, Mr. R could sue for recovery of damages based of a claim of duress, undue influence, or other aspects of the doctor–patient power differential.27,42,43 Given the published views we have described, a psychiatrist who barters also may be viewed as violating state regulations that measure the standard of care against generally accepted practice.
Only time will tell if I am right or I am wrong
If you face a situation similar to Dr. Z’s and want to consider a barter arrangement, you can take several steps to mitigate potential risk to your patient and ensure competent care (Table 25,6,15,16,32,35,39,40,44-47). One of the most important steps is to seek ongoing consultation, both before and after a decision to barter. Ideally, the consulting colleague would know you and your circumstances and would have sufficient clinical grasp of the patient to make an informed assessment of risks and benefits.35 This consultation, as well as your own rationale for acting on recommendations, should be thoroughly documented in the patient’s records.26,44,45
Certain types of barter should be off limits, including:
- trading prescription drugs for goods or services
- trading for services that tie into the success of one’s business (eg, business advising or marketing)16
- offering treatment in exchange for illegal or ethically unacceptable services.48
Beyond ethical considerations are some practical issues. The Internal Revenue Service has specific rules regarding taxation of bartered goods and services, which must be included as taxable income.46 If possible, an independent agent should appraise the traded goods or services before the agreement.6 When working in a group practice, the clinician might have to figure out how to allocate the received goods or services such as shared overhead costs.28 Preferably, the patient’s goods or services should be provided before care is delivered.16 If not, the duration of services rendered should be limited, and either party should have the option to disengage from the relationship if one feels dissatisfied.16
A written contract, discussed ahead of time, can be a sound way to summarize the terms of the arrangement. Both sides also should consider what would happen if an injury occurred.16 Finally, you must adhere to any relevant state laws regarding payment for services, particularly if the patient has health insurance.32
If the bartering arrangement does not work, you should take an open and non-defensive approach. If you believe you have made a mistake, consider apologizing.45
Dear Dr. Mossman,
Each month, I see my patient, Mr. R, for a 15-minute medication management appointment. At his latest visit, Mr. R mentioned his financial difficulties. He also observed that our office needed to have some carpentry work done—not a surprise, because he’s known in our area as one of the best carpenters around. He suggested that I hire him as payment for the next 6 appointments. What risks might I encounter if I oblige him?
Submitted by “Dr. Z”
Nearly 29 million Americans are uninsured,1 and even more have trouble accessing mental health care.2 Many psychiatrists struggle to provide affordable services while remaining financially viable.3,4 For outpatients with limited means to pay for care, spacing appointments to fit their budgets might compromise treatment.5 Simply not charging patients poses its own clinical and ethical challenges.6-8
As a result, some mental health professionals make barter arrangements to help their patients enter or continue treatment. To answer Dr. Z’s question on whether exchanging services might be a way to arrange matters with some patients, we explore:
- the idea of bartering for psychiatric treatment
- related ethical and legal considerations
- when and in what situations bartering might be appropriate.
Think of what I’m saying: Bartering for treatment
“Barter” refers to exchanging commodities, products, or services of equivalent value without using money.9 In 2010, Nevada Republican Senate candidate Sue Lowden encouraged barter for health care and harkened back to an earlier time where “they would bring a chicken to the doctor; they would say ‘I’ll paint your house.’”10
Such payment arrangements have been encouraged as health care has become increasingly commoditized.11-13 This happens through both direct barter between physician and patient and barter exchanges. Barter exchange systems have been set up on Web sites (as of 2013, at least 400 such online exchanges were available14), local communities,11,15 and social programs. For example, through the “Swapping Guns for Therapy” program, psychologists in California gave free or reduced-fee care for people who traded in their guns.16
Try to see it my way: A prevailing view of barter
Several psychiatrists recommend against bartering for treatment, for a variety of reasons.7,8,17-19 Simon18 argues that a stable fee policy is part of a proper therapeutic framework, and money is “the only acceptable medium of exchange when receiving payment from patients.” Emotional distress and the power differential inherent in treatment might prevent a patient from making an accurate assessment of the value of the bartered goods or services,7,8,17,18,20 which could lead to future claims of undue influence from trading goods or services below market value.17 To avoid the possibility of exploitating the patient, Simon18 recommends that the psychiatrist’s professional fee be “the only material benefit received from the patient.”
The American Psychiatric Association’s code of ethics states that “it is not ethical to switch a doctor–patient relationship to an employer–employee one … and, in most cases, such an arrangement would be unethical.”21 In some therapeutic settings, employing a patient risks inappropriate self-disclosure and intrusion.16
More than other physicians, psychiatrists pay special attention to professional boundaries, the technical term for the “edge of appropriate behavior,” within which safe, effective care can occur.22,23 Although some boundary crossings can be harmless and even constructive, repeated boundary crossings are the forerunners to improper behavior, including sexual relationships with patients.24-26
Out of concern that bartering could become the first step down a slippery ethical slope toward patient exploitation, mental health clinicians have deemed the practice “ethically troubling,”19 said it did “not usually work out well,”7 and declared it “so fraught with risks for both parties that it seem[ed] illogical to even consider it as an option.”27
While I see it your way: What barter proponents say
Reports of bartering for chickens28 and purchasing fuel from a patient in remote Alaska29 show that not all physicians agree and why they feel that professional codes of ethics reflect an urban bias.28,29 In many rural areas and small towns, access to mental health services is limited, and patients often interact with their doctors outside of clinical encounters.23,29-31
Bartering can benefit a physician’s practice by:
- reducing the need to discount services
- eliminating bureaucratic burdens of traditional insurance arrangements
- facilitating development of a patient base
- allowing patients choice and flexibility in seeking medical care.6,16,32
Bartering could confer certain clinical benefits, such as:
- enhancing trust and empathy32
- encouraging patients to make their needs known constructively6
- modeling financial self-care6
- helping the doctor to feel fairly compensated for providing thoughtful care6
- acknowledging the patient’s cultural values15,33
- affirming that patients and doctors both produce things of value.16
I have always thought: Other ethical models
An ethical approach to bartering that requires careful thought and respect for the patient’s needs appears consistent with a primary goal of treatment: “to increase the capacity of individuals to make more rational choices in their lives and to be relatively freer from disabling conflicts.”20 Some authors criticize slippery-slope arguments and strict-rule ethical approaches as being too rigid, limiting, or risk-averse.22,26,34 In Table 1,6,8,16,18,27,29-31,35-37 we list several factors that might weigh for or against a decision to enter into a barter arrangement as payment for care.
In a similar manner, Martinez33,38 proposed a graded-risk framework that encourages examination of potential harms and benefits of a decision, potential coercive or exploitative elements, the clinician’s intentions and aspiration to professional ideals, and the context of the decision. Within this framework, some bartering arrangements might be encouraged and, perhaps, even obligatory because of the potential benefits to the patient; other arrangements (eg, trading psychotherapy for menial services) might be unjustifiable. Martinez38 argues that this approach fosters mutual decision-making with patients, discourages physician paternalism, and “demands that we struggle with the particulars with each case.”
Gottlieb’s decision-making model35 recognizes that trying to avoid all dual relationships is unrealistic and not all dual relationships are exploitative. Instead, a clinician must assess 3 dimensions of current and proposed relationships:
- the degree of power differential
- the duration of treatment
- the clarity of termination.
The decision-making process also requires involvement of the patient, who if “unable to recognize the dilemma or is unwilling to consider the issues before deciding, should be considered at risk, and the contemplated relationship rejected.”35
So I will ask you once again: Dr. Z’s decision
In the case of Dr. Z and Mr. R, a barter arrangement might work in the sense of permitting and sustaining good care. Mr. R suggested the idea and might not be able to afford care without it. Nothing in Dr. Z’s description suggests that Mr. R has personality characteristics or other conditions that would compromise his ability to give informed consent or to understand the nuances of a barter arrangement. Dr. Z is not providing a treatment (eg, psychodynamic therapy) that a barter arrangement could contaminate. That the arrangement would be circumscribed limits the effect of a power differential, as would its brief duration and defined termination endpoint. Dr. Z’s letter to the authors also shows his willingness to seek consultation.
There’s a chance that we may fall apart: Reasons for caution
Martinez’s graded-risk approach recognizes reasons for caution:
- the risk of harm to the patient or doctor–patient relationship
- the uncertain benefit to the patient
- the blurring of Dr. Z’s self-interest and Mr. R’s needs
- some ambiguity about possible exploitation.
Dr. Z and Mr. R have not discussed the value of Mr. R’s work—which might create a rift between them—and despite Mr. R’s reputation, other carpenters are available. Future med-check appointments will give them little time to explore and discuss the meanings of the barter.
Any proposed barter arrangement creates some clinical perils that can be particularly salient in mental health treatment. Patients could view themselves as “special” or entitled to enhanced access to the doctor because of exchanged services, which could take a toll on the doctor.39 The physician’s objectivity might diminish, and the business aspect of their relationship could make both parties less comfortable when discussing sensitive information relevant to treatment.31,40 Also, the suggested barter is for services to be provided at Dr. Z’s office, where confidentiality may be breeched and transference issues could arise.
A medical malpractice claim states that a doctor has breached a duty of care to a patient such that harm (or “damages”) resulted.41 Should Dr. Z and Mr. R’s barter agreement turn sour and harm follow, Mr. R could sue for recovery of damages based of a claim of duress, undue influence, or other aspects of the doctor–patient power differential.27,42,43 Given the published views we have described, a psychiatrist who barters also may be viewed as violating state regulations that measure the standard of care against generally accepted practice.
Only time will tell if I am right or I am wrong
If you face a situation similar to Dr. Z’s and want to consider a barter arrangement, you can take several steps to mitigate potential risk to your patient and ensure competent care (Table 25,6,15,16,32,35,39,40,44-47). One of the most important steps is to seek ongoing consultation, both before and after a decision to barter. Ideally, the consulting colleague would know you and your circumstances and would have sufficient clinical grasp of the patient to make an informed assessment of risks and benefits.35 This consultation, as well as your own rationale for acting on recommendations, should be thoroughly documented in the patient’s records.26,44,45
Certain types of barter should be off limits, including:
- trading prescription drugs for goods or services
- trading for services that tie into the success of one’s business (eg, business advising or marketing)16
- offering treatment in exchange for illegal or ethically unacceptable services.48
Beyond ethical considerations are some practical issues. The Internal Revenue Service has specific rules regarding taxation of bartered goods and services, which must be included as taxable income.46 If possible, an independent agent should appraise the traded goods or services before the agreement.6 When working in a group practice, the clinician might have to figure out how to allocate the received goods or services such as shared overhead costs.28 Preferably, the patient’s goods or services should be provided before care is delivered.16 If not, the duration of services rendered should be limited, and either party should have the option to disengage from the relationship if one feels dissatisfied.16
A written contract, discussed ahead of time, can be a sound way to summarize the terms of the arrangement. Both sides also should consider what would happen if an injury occurred.16 Finally, you must adhere to any relevant state laws regarding payment for services, particularly if the patient has health insurance.32
If the bartering arrangement does not work, you should take an open and non-defensive approach. If you believe you have made a mistake, consider apologizing.45
1. Kaiser Commission on Medicaid and the Uninsured. Key facts about the uninsured population. http://kff.org/uninsured/fact-sheet/key-facts-about-the-uninsured-population. Published September 29, 2016. Accessed October 7, 2016.
2. National Alliance on Mental Illness. A long road ahead: achieving true parity in mental health and substance use care. https://www.nami.org/About-NAMI/Publications-Reports/Public-Policy-Reports/A-Long-Road-Ahead/2015-ALongRoadAhead.pdf. Published April 2015. Accessed October 7, 2016.
3. Insel T. Director’s blog: the paradox of parity. May 30, 2014. https://www.nimh.nih.gov/about/director/2014/the-paradox-of-parity.shtml. Published May 30, 2014. Accessed October 7, 2016.
4. Bishop TF, Press MJ, Keyhani S, et al. Acceptance of insurance by psychiatrists and the implications for access to mental health care. JAMA Psychiatry. 2014;71(2):176-181.
5. What do you do when patients cannot pay? Psychiatry (Edgmont). 2009;6(5):51-52.
6. Hill M. Barter: ethical considerations in psychotherapy. Women Ther. 2000;22(3):81-91.
7. Simon RI. Commentary: treatment boundaries—flexible guidelines, not rigid standards. J Am Acad Psychiatry Law. 2001;29(3):287-289.
8. Simon RI, Williams IC. Maintaining treatment boundaries in small communities and rural areas. Psychiatr Serv. 1999;50(11):1440-1446.
9. Compact edition of the Oxford English dictionary. New York, NY: Oxford University Press; 1971:171.
10. Coolican JP. Sue Lowden stands by health care plan. Las Vegas Sun. http://lasvegassun.com/news/2010/apr/20/sue-lowden-draws-fire-repeating-health-care-barter. Published April 20, 2010. Accessed September 20, 2016.
11. Consumer Reports. Barter sometimes allow patients to pay for health care they otherwise could not afford. Washington Post. https://www.washingtonpost.com/national/health-science/barter-sometimes-allow-patients-to-pay-for-health-care-they-otherwise-could-not-afford/2013/09/30/e7e5a55e-069d-11e3-88d6-d5795fab4637_story.html. Published September 20, 2013. Accessed September 27, 2016.
12. Ellis B. MediBid auction site lets doctors bid for patients. CNN Money. http://money.cnn.com/2014/01/09/pf/insurance/medibid. Published January 9, 2014. Accessed September 23, 2016.
13. Ambrosino B. Surgery for sale: the ethics of health care bartering in a social media marketplace. http://hub.jhu.edu/2014/01/16/hopkins-ethicist-ponders-medibid. Published January 16, 2014. Accessed September 23, 2016.
14. Thomas C. When patients barter for health care. https://ethicalnag.org/2013/07/30/barter. Published July 30, 2016. Accessed October 9, 2016.
15. Syme G. Fetters or freedom: dual relationships in counselling. Int J Adv Counselling. 2006;28(1):57-69.
16. Zur O. Bartering in psychotherapy and counselling: complexities, case studies and guidelines. New Therapist. 2008;58:18-26.
17. Simon RI. The psychiatrist as a fiduciary: avoiding the double agent role. Psychiatric Annals. 1987;17(9):622-626.
18. Simon RI. Treatment boundary violations: clinical, ethical, and legal considerations. Bull Am Acad Psychiatry Law. 1992;20(3):269-288.
19. Walker R, Clark JJ. Heading off boundary problems: clinical supervision as risk management. Psychiatr Serv. 1999;50(11):1435-1439.
20. Malmquist CP, Norman MT. Psychiatrist-patient boundary issues following treatment termination. Am J Psychiatry. 2001;158(7):1010-1018.
21. American Psychiatric Association. The opinions of the ethics committee on the principles of medical ethics, with annotations especially applicable to psychiatry. https://www.psychiatry.org/psychiatrists/practice/ethics. Published 2016. Accessed October 4, 2016.
22. Gutheil TG, Gabbard GO. Misuses and misunderstandings of boundary theory in clinical and regulatory settings. Am J Psychiatry. 1998;155(3):409-414.
23. Crowden A. Professional boundaries and the ethics of dual and multiple overlapping relationships in psychotherapy. Monash Bioeth Rev. 2008;27(4):10-27.
24. Gabbard GO. Commentary: boundaries, culture, and psychotherapy. J Am Acad Psychiatry Law. 2001;29(3):284-286.
25. Kroll J. Boundary violations: a culture-bound syndrome. J Am Acad Psychiatry Law. 2001;29(3):274-283.
26. Gottlieb MC, Younggren JN. Is there a slippery slope? Considerations regarding multiple relationships and risk management. Professional Psychology: Research and Practice. 2009;40(6):564-557.
27. Woody RH. Bartering for psychological services. Professional Psychology: Research and Practice. 1998;29(2):174-178.
28. Bartering for medical care. MGMA Connex. 2008;8(6):11.
29. Roberts LW, Battaglia J, Epstein RS. Frontier ethics: mental health care needs and ethical dilemmas in rural communities. Psychiatr Serv. 1994;50(4):497-503.
30. Endacott R, Wood A, Judd F, et al. Impact and management of dual relationships in metropolitan, regional and rural mental health practice. Aust N Z J Psychiatry. 2006;40(11-12):987-994.
31. Scopelliti J, Judd F, Grigg M, et al. Dual relationships in mental health practice: issues for clinicians in rural settings. Aust N Z J Psychiatry. 2004;38(11-12):953-959.
32. Ayers AA. Bartering basics for the urgent care operator. http://www.alanayersurgentcare.com/Linked_Files/2013_Articles/Ayers_UCAOA_Bartering_%20Basics_2012_01_09.pdf. Accessed September 23, 2016.
33. Savin D, Martinez R. Cross-cultural boundary dilemmas: a graded-risk assessment approach. Transcult Psychiatry. 2006;42(2):243-258.
34. Glass LL. The gray areas of boundary crossings and violations. Am J Psychother. 2003;57(4):429-444.
35. Gottlieb MC. Avoiding exploitive dual relationships: a decision-making model. Psychotherapy (Chic). 1993;30(1):41-48.
36. Lane JA. The ethical implications of bartering for mental health services: examining interdisciplinary ethical standards. http://pdxscholar.library.pdx.edu/coun_fac/36. Published 2012. Accessed October 17, 2016.
37. Miller RD, Maier GJ. Nonsexual boundary violations: sauce for the gander. J Psychiatry Law. 2002;30(3):309-329.
38. Martinez R. A model for boundary dilemmas: ethical decision-making in the patient-professional relationship. Ethical Hum Sci Serv. 2000;2(1):43-61.
39. Salmon K, Klijnsma M. Boundary issues: employing patients as staff? Br J Gen Pract. 2009;59(558):56-57.
40. College of Physicians and Surgeons Ontario. Hiring patients may compromise physician-patient relationship. Dialogue. 2015;3:47.
41. Bal S. An introduction to medical malpractice in the United States. Clin Orthop Relat Res. 2009;467(2):339-347.
42. What puts a psychiatrist at risk for a malpractice lawsuit? Psychiatry (Edgmont). 2009;6(8):38-39.
43. Geis v Landau, 117 Misc2d 396 (NY Misc 1983).
44. Nisselle P. Danger zone. When boundaries are crossed in the doctor-patient relationship. Aust Fam Physician. 2000;29(6):541-544.
45. Pope KS, Keith-Spiegel P. A practical approach to boundaries in psychotherapy: making decisions, bypassing blunders, and mending fences. J Clin Psychol. 2008;64(5):638-652.
46. IRS Publication 17. https://www.irs.gov/publications/p17/ch12.html. Published 2015. Accessed October 5, 2016.
47. Epstein RS, Simon RI. The exploitation index: an early warning indicator of boundary violations in psychotherapy. Bull Menninger Clin. 1990;54(4):450-465.
48. Skutch J. Savannah doctor accused of trading drugs for sex with strippers. Augusta Chronicle. http://chronicle.augusta.com/news/crime-courts/2013-01-31/savannah-doctor-accused-trading-drugs-sex-strippers. Published January 31, 2013. Accessed October 16, 2016.
1. Kaiser Commission on Medicaid and the Uninsured. Key facts about the uninsured population. http://kff.org/uninsured/fact-sheet/key-facts-about-the-uninsured-population. Published September 29, 2016. Accessed October 7, 2016.
2. National Alliance on Mental Illness. A long road ahead: achieving true parity in mental health and substance use care. https://www.nami.org/About-NAMI/Publications-Reports/Public-Policy-Reports/A-Long-Road-Ahead/2015-ALongRoadAhead.pdf. Published April 2015. Accessed October 7, 2016.
3. Insel T. Director’s blog: the paradox of parity. May 30, 2014. https://www.nimh.nih.gov/about/director/2014/the-paradox-of-parity.shtml. Published May 30, 2014. Accessed October 7, 2016.
4. Bishop TF, Press MJ, Keyhani S, et al. Acceptance of insurance by psychiatrists and the implications for access to mental health care. JAMA Psychiatry. 2014;71(2):176-181.
5. What do you do when patients cannot pay? Psychiatry (Edgmont). 2009;6(5):51-52.
6. Hill M. Barter: ethical considerations in psychotherapy. Women Ther. 2000;22(3):81-91.
7. Simon RI. Commentary: treatment boundaries—flexible guidelines, not rigid standards. J Am Acad Psychiatry Law. 2001;29(3):287-289.
8. Simon RI, Williams IC. Maintaining treatment boundaries in small communities and rural areas. Psychiatr Serv. 1999;50(11):1440-1446.
9. Compact edition of the Oxford English dictionary. New York, NY: Oxford University Press; 1971:171.
10. Coolican JP. Sue Lowden stands by health care plan. Las Vegas Sun. http://lasvegassun.com/news/2010/apr/20/sue-lowden-draws-fire-repeating-health-care-barter. Published April 20, 2010. Accessed September 20, 2016.
11. Consumer Reports. Barter sometimes allow patients to pay for health care they otherwise could not afford. Washington Post. https://www.washingtonpost.com/national/health-science/barter-sometimes-allow-patients-to-pay-for-health-care-they-otherwise-could-not-afford/2013/09/30/e7e5a55e-069d-11e3-88d6-d5795fab4637_story.html. Published September 20, 2013. Accessed September 27, 2016.
12. Ellis B. MediBid auction site lets doctors bid for patients. CNN Money. http://money.cnn.com/2014/01/09/pf/insurance/medibid. Published January 9, 2014. Accessed September 23, 2016.
13. Ambrosino B. Surgery for sale: the ethics of health care bartering in a social media marketplace. http://hub.jhu.edu/2014/01/16/hopkins-ethicist-ponders-medibid. Published January 16, 2014. Accessed September 23, 2016.
14. Thomas C. When patients barter for health care. https://ethicalnag.org/2013/07/30/barter. Published July 30, 2016. Accessed October 9, 2016.
15. Syme G. Fetters or freedom: dual relationships in counselling. Int J Adv Counselling. 2006;28(1):57-69.
16. Zur O. Bartering in psychotherapy and counselling: complexities, case studies and guidelines. New Therapist. 2008;58:18-26.
17. Simon RI. The psychiatrist as a fiduciary: avoiding the double agent role. Psychiatric Annals. 1987;17(9):622-626.
18. Simon RI. Treatment boundary violations: clinical, ethical, and legal considerations. Bull Am Acad Psychiatry Law. 1992;20(3):269-288.
19. Walker R, Clark JJ. Heading off boundary problems: clinical supervision as risk management. Psychiatr Serv. 1999;50(11):1435-1439.
20. Malmquist CP, Norman MT. Psychiatrist-patient boundary issues following treatment termination. Am J Psychiatry. 2001;158(7):1010-1018.
21. American Psychiatric Association. The opinions of the ethics committee on the principles of medical ethics, with annotations especially applicable to psychiatry. https://www.psychiatry.org/psychiatrists/practice/ethics. Published 2016. Accessed October 4, 2016.
22. Gutheil TG, Gabbard GO. Misuses and misunderstandings of boundary theory in clinical and regulatory settings. Am J Psychiatry. 1998;155(3):409-414.
23. Crowden A. Professional boundaries and the ethics of dual and multiple overlapping relationships in psychotherapy. Monash Bioeth Rev. 2008;27(4):10-27.
24. Gabbard GO. Commentary: boundaries, culture, and psychotherapy. J Am Acad Psychiatry Law. 2001;29(3):284-286.
25. Kroll J. Boundary violations: a culture-bound syndrome. J Am Acad Psychiatry Law. 2001;29(3):274-283.
26. Gottlieb MC, Younggren JN. Is there a slippery slope? Considerations regarding multiple relationships and risk management. Professional Psychology: Research and Practice. 2009;40(6):564-557.
27. Woody RH. Bartering for psychological services. Professional Psychology: Research and Practice. 1998;29(2):174-178.
28. Bartering for medical care. MGMA Connex. 2008;8(6):11.
29. Roberts LW, Battaglia J, Epstein RS. Frontier ethics: mental health care needs and ethical dilemmas in rural communities. Psychiatr Serv. 1994;50(4):497-503.
30. Endacott R, Wood A, Judd F, et al. Impact and management of dual relationships in metropolitan, regional and rural mental health practice. Aust N Z J Psychiatry. 2006;40(11-12):987-994.
31. Scopelliti J, Judd F, Grigg M, et al. Dual relationships in mental health practice: issues for clinicians in rural settings. Aust N Z J Psychiatry. 2004;38(11-12):953-959.
32. Ayers AA. Bartering basics for the urgent care operator. http://www.alanayersurgentcare.com/Linked_Files/2013_Articles/Ayers_UCAOA_Bartering_%20Basics_2012_01_09.pdf. Accessed September 23, 2016.
33. Savin D, Martinez R. Cross-cultural boundary dilemmas: a graded-risk assessment approach. Transcult Psychiatry. 2006;42(2):243-258.
34. Glass LL. The gray areas of boundary crossings and violations. Am J Psychother. 2003;57(4):429-444.
35. Gottlieb MC. Avoiding exploitive dual relationships: a decision-making model. Psychotherapy (Chic). 1993;30(1):41-48.
36. Lane JA. The ethical implications of bartering for mental health services: examining interdisciplinary ethical standards. http://pdxscholar.library.pdx.edu/coun_fac/36. Published 2012. Accessed October 17, 2016.
37. Miller RD, Maier GJ. Nonsexual boundary violations: sauce for the gander. J Psychiatry Law. 2002;30(3):309-329.
38. Martinez R. A model for boundary dilemmas: ethical decision-making in the patient-professional relationship. Ethical Hum Sci Serv. 2000;2(1):43-61.
39. Salmon K, Klijnsma M. Boundary issues: employing patients as staff? Br J Gen Pract. 2009;59(558):56-57.
40. College of Physicians and Surgeons Ontario. Hiring patients may compromise physician-patient relationship. Dialogue. 2015;3:47.
41. Bal S. An introduction to medical malpractice in the United States. Clin Orthop Relat Res. 2009;467(2):339-347.
42. What puts a psychiatrist at risk for a malpractice lawsuit? Psychiatry (Edgmont). 2009;6(8):38-39.
43. Geis v Landau, 117 Misc2d 396 (NY Misc 1983).
44. Nisselle P. Danger zone. When boundaries are crossed in the doctor-patient relationship. Aust Fam Physician. 2000;29(6):541-544.
45. Pope KS, Keith-Spiegel P. A practical approach to boundaries in psychotherapy: making decisions, bypassing blunders, and mending fences. J Clin Psychol. 2008;64(5):638-652.
46. IRS Publication 17. https://www.irs.gov/publications/p17/ch12.html. Published 2015. Accessed October 5, 2016.
47. Epstein RS, Simon RI. The exploitation index: an early warning indicator of boundary violations in psychotherapy. Bull Menninger Clin. 1990;54(4):450-465.
48. Skutch J. Savannah doctor accused of trading drugs for sex with strippers. Augusta Chronicle. http://chronicle.augusta.com/news/crime-courts/2013-01-31/savannah-doctor-accused-trading-drugs-sex-strippers. Published January 31, 2013. Accessed October 16, 2016.

Are you neuroprotecting your patients? 10 Adjunctive therapies to consider
Are you ‘neuroprotecting’ your patients?
Fortunately, many studies have demonstrated that in addition to controlling clinical symptoms, antidepressants, mood stabilizers, and atypical antipsychotics all have neuroprotective effects, including stimulating neurogenesis, preventing apoptosis, and increasing neurotrophins. However, more needs to be done to protect the brain’s gray and white matter and to prevent negative neuroplasticity and disconnectivity of brain circuits that often are documented in patients with psychotic and mood disorders.
There are, in fact, many off-label supplements with strong neuroprotective effects that psychiatrists can use as an adjunct to standard evidence-based pharmacotherapy. These agents generally are safe and well tolerated and often are sold over the counter, but are not covered by insurance. However, considering the disability that often is associated with schizophrenia, treatment-resistant depression, or psychotic mania, it is reasonable to consider using these agents, many of which are supported by studies published in peer-reviewed journals. However, because they are widely available and not proprietary and large, expensive registration trials such as the ones conducted by the pharmaceutical industry are not done, none is likely to receive FDA approval. Therefore, it is up to psychiatrists and nurse practitioners to use them judiciously in patients at risk for neurotoxicity.
Here are 10 agents with neuroprotective effects supported by published data that can be considered as add-on to the standard treatments in an effort to mitigate neurotoxicity and protect the brain from the destructive processes that accompany acute episodes of psychosis, mania, and depression.
Omega-3 fatty acids have been shown in several studies to help reduce psychopathology of psychosis, mania, or depression when used as an adjunctive agent.1 It appears to be more effective in the early stages of psychiatric disorders than in the chronic phase. It has anti-inflammatory, anti-oxidant, and anti-apoptotic effects; activates cell-signaling pathways; and prevents synaptic loss as well as neuronal and glial death.2
N-acetylcysteine (NAC) is a powerful antioxidant that increases glutathione, which is produced in the mitochondria. Schizophrenia is associated with mitochondrial dysfunction with low levels of glutathione, which puts the brain at risk for neurodegeneration caused by high levels of free radicals produced during psychosis. Adding NAC to antipsychotics during acute psychotic episodes—especially the first episode—can significantly reduce the neurotoxic effects of reactive oxygen and nitrogen species, also known as free radicals.3 In studies of traumatic brain injury in rats, NAC reduced brain edema, neuroinflammation, blood–brain barrier permeability, and apoptosis.4
Minocycline. This antibiotic has been studied extensively as an adjunctive treatment in schizophrenia and has proven to have several neuroprotective effects including anti-inflammatory, anti-oxidant, and anti-apoptotic, and reduces glutamate excitotoxicity.5 Several studies have documented its usefulness in acute psychotic episodes.
Vitamin D. Because of its vital role in neurodevelopment (neuronal differentiation, axonal connectivity), vitamin D deficiency has been associated with several psychiatric disorders including autism, schizophrenia, depression, and Alzheimer’s disease.6 Measure serum levels of vitamin D in patients with psychotic and mood disorders and implement supplementation if it is low—and it often is in these patients.
Nicotine is neuroprotective against glutamate excitotoxicity and it also inhibits apoptosis.7 However, it should never be administered via cigarettes, which are loaded with hundreds of toxic substances! It can be administered via patches or nicotine gum, which are usually used to help in smoking cessation. Nicotine also also can have a pro-cognitive effect.
Melatonin. Many people associate melatonin with sleep. However, it has multiple neuroprotective effects by being an antioxidant, protecting mitochondrial integrity, and modulating the immune system, as well as attenuating microglial activation, which triggers neuroinflammation and oxidative stress. It also protects against cellular senescence, which is due to inflammation and reactive oxygen species. Furthermore, melatonin is useful in ameliorating the metabolic syndrome, which is associated with neurotoxic effects on brain tissue caused by the pro-inflammatory effects of peri-omental fat in obesity.8 Adjunctive melatonin could be helpful in patients with schizophrenia or depression who suffer from metabolic syndrome.
Erythropoietin is a hormone produced by the kidneys to promote the formation of red blood cells. It is a potent neuroprotective cytokine that promotes neuronal survival via anti-apoptotic effects. It protects against glutamate and nitrous oxide toxicity9 and haloperidol-induced neuronal death.10 It is clinically used (since FDA approval in 1989) in severe anemia due to chronic kidney disease or chemotherapy, as well as in inflammatory bowel disease. It does have some “black-box” warnings so its use should be limited.
Cox-2 inhibitors. This is a well-known class of anti-inflammatory drugs, which are FDA-approved for pain and inflammation. Studies of adjunctive use of cox-2 inhibitors in acute psychosis show that these drugs accentuate the efficacy of antipsychotic medications.11 The reason is that acute psychosis is associated with neuro-inflammation, which leads to neurotoxicity.
Lithium. Dosages to treat mania are usually 900 to 1500 mg/d. However, in minute (homeopathic) dosages as low as 1 mg/d, lithium has been shown to prevent progression of amnestic mild cognitive impairment to full dementia.12 This interesting observation suggests that lithium not only induces neurogenesis and increases gray matter volume,13 but may be neuroprotective against amyloid neurotoxicity. The effects of very low doses of lithium in depression and schizophrenia have not been studied yet.
Caffeine. Yes, the good old brew people seek all day is neuroprotective and prevents mood and memory dysfunction caused by stress.14 Caffeine should be avoided in patients with anxiety disorders, but it may be helpful for the brains of patients with mood or psychotic disorders. Caffeine reverses synaptic dysfunction in the circuits of the hippocampus caused by chronic unpredictable stress (quite common among our psychiatric patients).
The above interventions may be helpful for some patients but not others. Practitioners should consider using 1 or more of those adjunctive neuroprotective agents in patients who are at risk for neurodegenerative changes secondary to recurrences of acute and severe psychosis or mood episodes. Although clinicians cannot monitor brain structural integrity, they can assess the rate of symptomatic improvement and degree of functional restoration in their patients. Until a cure is found, these little steps—taken cautiously and judiciously—could help alleviate our patients’ suffering and the risk of neurotoxicity associated with their serious psychiatric disorder.
1. Chen AT, Chibnall JT, Nasrallah HA. A meta-analysis of placebo-controlled trials of omega-3 fatty acid augmentation in schizophrenia: possible stage-specific effects. Ann Clin Psychiatry. 2015;27(4):289-296.
2. Calon F, Cole G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: evidence from animal studies. Prostaglandins Leukot Essent Fatty Acids. 2007;7(5-6):287-293.
3. Chen AT, Chibnall JT, Nasrallah HA. Placebo-controlled augmentation trials of the antioxidant NAC in schizophrenia: a review. Ann Clin Psychiatry. 2016;28(3):190-196.
4. Chen G, Shi J, Hu Z, et al. Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of N-acetylcysteine. Mediators Inflamm. 2008;2008:716458. doi: 10.1155/2008/716458
5. Dean OM, Data-Franco J, Giorlando F, et al. Minocycline: therapeutic potential in psychiatry. CNS Drugs. 2012;26(5):391-401.
6. Eyles DW, Burne TH, McGrath JJ. Vitamin D, effects on brain development, adult brain function and the links between low levels of vitamin D and neuropsychiatric disease. Front Neuroendocrinol. 2013;34(1):47-64.
7. Akaike A, Tkada-Takatori Y, Kume T, et al. Mechanisms of neuroprotective effects of nicotine and acetylcholinesterase inhibitors: role of alpha4 and alpha7 receptors in neuroprotection. J Mol Neurosci. 2010;40(1-2):211-216.
8. Cardinali DP, Hardeland R. Inflammaging, metabolic syndrome and melatonin: a call for treatment studies [published online May 11, 2016]. Neuroendocrinology. doi:10.1159/000446543.
9. Yamasaki M, Mishima HK, Yamashita H, et al. Neuroprotective effects of erythropoietin on glutamate and nitric oxide toxicity in primary cultured retinal ganglion cells. Brain Res. 2005;1050(1-2):15-26.
10. Pilllai A, Dhandapani KM, Pillai BA, et al. Erythropoietin prevents haloperidol treatment-induced neuronal apoptosis through regulation of BDNF. Neuropsychopharmacology. 2008;33(8):1942-1951.
11. Müller N, Myint AM, Weidinger E, et al. Anti-inflammatory treatment in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2013;42:146-153.
12. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
13. Dong BT, Tu GJ, Han YX, et al. Lithium enhanced cell proliferation and differentiation of mesenchymal stem cells to neural cells in rat spinal cord. Int J Clin Exp Pathol. 2015;8(3):2473-2483.
14. Kaster MP, Machado NJ, Silva HB, et al. Caffeine acts through neuronal adenosine A24 receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc Natl Acad Sci U S A. 2015;112(25):7833-7838.
Are you ‘neuroprotecting’ your patients?
Fortunately, many studies have demonstrated that in addition to controlling clinical symptoms, antidepressants, mood stabilizers, and atypical antipsychotics all have neuroprotective effects, including stimulating neurogenesis, preventing apoptosis, and increasing neurotrophins. However, more needs to be done to protect the brain’s gray and white matter and to prevent negative neuroplasticity and disconnectivity of brain circuits that often are documented in patients with psychotic and mood disorders.
There are, in fact, many off-label supplements with strong neuroprotective effects that psychiatrists can use as an adjunct to standard evidence-based pharmacotherapy. These agents generally are safe and well tolerated and often are sold over the counter, but are not covered by insurance. However, considering the disability that often is associated with schizophrenia, treatment-resistant depression, or psychotic mania, it is reasonable to consider using these agents, many of which are supported by studies published in peer-reviewed journals. However, because they are widely available and not proprietary and large, expensive registration trials such as the ones conducted by the pharmaceutical industry are not done, none is likely to receive FDA approval. Therefore, it is up to psychiatrists and nurse practitioners to use them judiciously in patients at risk for neurotoxicity.
Here are 10 agents with neuroprotective effects supported by published data that can be considered as add-on to the standard treatments in an effort to mitigate neurotoxicity and protect the brain from the destructive processes that accompany acute episodes of psychosis, mania, and depression.
Omega-3 fatty acids have been shown in several studies to help reduce psychopathology of psychosis, mania, or depression when used as an adjunctive agent.1 It appears to be more effective in the early stages of psychiatric disorders than in the chronic phase. It has anti-inflammatory, anti-oxidant, and anti-apoptotic effects; activates cell-signaling pathways; and prevents synaptic loss as well as neuronal and glial death.2
N-acetylcysteine (NAC) is a powerful antioxidant that increases glutathione, which is produced in the mitochondria. Schizophrenia is associated with mitochondrial dysfunction with low levels of glutathione, which puts the brain at risk for neurodegeneration caused by high levels of free radicals produced during psychosis. Adding NAC to antipsychotics during acute psychotic episodes—especially the first episode—can significantly reduce the neurotoxic effects of reactive oxygen and nitrogen species, also known as free radicals.3 In studies of traumatic brain injury in rats, NAC reduced brain edema, neuroinflammation, blood–brain barrier permeability, and apoptosis.4
Minocycline. This antibiotic has been studied extensively as an adjunctive treatment in schizophrenia and has proven to have several neuroprotective effects including anti-inflammatory, anti-oxidant, and anti-apoptotic, and reduces glutamate excitotoxicity.5 Several studies have documented its usefulness in acute psychotic episodes.
Vitamin D. Because of its vital role in neurodevelopment (neuronal differentiation, axonal connectivity), vitamin D deficiency has been associated with several psychiatric disorders including autism, schizophrenia, depression, and Alzheimer’s disease.6 Measure serum levels of vitamin D in patients with psychotic and mood disorders and implement supplementation if it is low—and it often is in these patients.
Nicotine is neuroprotective against glutamate excitotoxicity and it also inhibits apoptosis.7 However, it should never be administered via cigarettes, which are loaded with hundreds of toxic substances! It can be administered via patches or nicotine gum, which are usually used to help in smoking cessation. Nicotine also also can have a pro-cognitive effect.
Melatonin. Many people associate melatonin with sleep. However, it has multiple neuroprotective effects by being an antioxidant, protecting mitochondrial integrity, and modulating the immune system, as well as attenuating microglial activation, which triggers neuroinflammation and oxidative stress. It also protects against cellular senescence, which is due to inflammation and reactive oxygen species. Furthermore, melatonin is useful in ameliorating the metabolic syndrome, which is associated with neurotoxic effects on brain tissue caused by the pro-inflammatory effects of peri-omental fat in obesity.8 Adjunctive melatonin could be helpful in patients with schizophrenia or depression who suffer from metabolic syndrome.
Erythropoietin is a hormone produced by the kidneys to promote the formation of red blood cells. It is a potent neuroprotective cytokine that promotes neuronal survival via anti-apoptotic effects. It protects against glutamate and nitrous oxide toxicity9 and haloperidol-induced neuronal death.10 It is clinically used (since FDA approval in 1989) in severe anemia due to chronic kidney disease or chemotherapy, as well as in inflammatory bowel disease. It does have some “black-box” warnings so its use should be limited.
Cox-2 inhibitors. This is a well-known class of anti-inflammatory drugs, which are FDA-approved for pain and inflammation. Studies of adjunctive use of cox-2 inhibitors in acute psychosis show that these drugs accentuate the efficacy of antipsychotic medications.11 The reason is that acute psychosis is associated with neuro-inflammation, which leads to neurotoxicity.
Lithium. Dosages to treat mania are usually 900 to 1500 mg/d. However, in minute (homeopathic) dosages as low as 1 mg/d, lithium has been shown to prevent progression of amnestic mild cognitive impairment to full dementia.12 This interesting observation suggests that lithium not only induces neurogenesis and increases gray matter volume,13 but may be neuroprotective against amyloid neurotoxicity. The effects of very low doses of lithium in depression and schizophrenia have not been studied yet.
Caffeine. Yes, the good old brew people seek all day is neuroprotective and prevents mood and memory dysfunction caused by stress.14 Caffeine should be avoided in patients with anxiety disorders, but it may be helpful for the brains of patients with mood or psychotic disorders. Caffeine reverses synaptic dysfunction in the circuits of the hippocampus caused by chronic unpredictable stress (quite common among our psychiatric patients).
The above interventions may be helpful for some patients but not others. Practitioners should consider using 1 or more of those adjunctive neuroprotective agents in patients who are at risk for neurodegenerative changes secondary to recurrences of acute and severe psychosis or mood episodes. Although clinicians cannot monitor brain structural integrity, they can assess the rate of symptomatic improvement and degree of functional restoration in their patients. Until a cure is found, these little steps—taken cautiously and judiciously—could help alleviate our patients’ suffering and the risk of neurotoxicity associated with their serious psychiatric disorder.
Are you ‘neuroprotecting’ your patients?
Fortunately, many studies have demonstrated that in addition to controlling clinical symptoms, antidepressants, mood stabilizers, and atypical antipsychotics all have neuroprotective effects, including stimulating neurogenesis, preventing apoptosis, and increasing neurotrophins. However, more needs to be done to protect the brain’s gray and white matter and to prevent negative neuroplasticity and disconnectivity of brain circuits that often are documented in patients with psychotic and mood disorders.
There are, in fact, many off-label supplements with strong neuroprotective effects that psychiatrists can use as an adjunct to standard evidence-based pharmacotherapy. These agents generally are safe and well tolerated and often are sold over the counter, but are not covered by insurance. However, considering the disability that often is associated with schizophrenia, treatment-resistant depression, or psychotic mania, it is reasonable to consider using these agents, many of which are supported by studies published in peer-reviewed journals. However, because they are widely available and not proprietary and large, expensive registration trials such as the ones conducted by the pharmaceutical industry are not done, none is likely to receive FDA approval. Therefore, it is up to psychiatrists and nurse practitioners to use them judiciously in patients at risk for neurotoxicity.
Here are 10 agents with neuroprotective effects supported by published data that can be considered as add-on to the standard treatments in an effort to mitigate neurotoxicity and protect the brain from the destructive processes that accompany acute episodes of psychosis, mania, and depression.
Omega-3 fatty acids have been shown in several studies to help reduce psychopathology of psychosis, mania, or depression when used as an adjunctive agent.1 It appears to be more effective in the early stages of psychiatric disorders than in the chronic phase. It has anti-inflammatory, anti-oxidant, and anti-apoptotic effects; activates cell-signaling pathways; and prevents synaptic loss as well as neuronal and glial death.2
N-acetylcysteine (NAC) is a powerful antioxidant that increases glutathione, which is produced in the mitochondria. Schizophrenia is associated with mitochondrial dysfunction with low levels of glutathione, which puts the brain at risk for neurodegeneration caused by high levels of free radicals produced during psychosis. Adding NAC to antipsychotics during acute psychotic episodes—especially the first episode—can significantly reduce the neurotoxic effects of reactive oxygen and nitrogen species, also known as free radicals.3 In studies of traumatic brain injury in rats, NAC reduced brain edema, neuroinflammation, blood–brain barrier permeability, and apoptosis.4
Minocycline. This antibiotic has been studied extensively as an adjunctive treatment in schizophrenia and has proven to have several neuroprotective effects including anti-inflammatory, anti-oxidant, and anti-apoptotic, and reduces glutamate excitotoxicity.5 Several studies have documented its usefulness in acute psychotic episodes.
Vitamin D. Because of its vital role in neurodevelopment (neuronal differentiation, axonal connectivity), vitamin D deficiency has been associated with several psychiatric disorders including autism, schizophrenia, depression, and Alzheimer’s disease.6 Measure serum levels of vitamin D in patients with psychotic and mood disorders and implement supplementation if it is low—and it often is in these patients.
Nicotine is neuroprotective against glutamate excitotoxicity and it also inhibits apoptosis.7 However, it should never be administered via cigarettes, which are loaded with hundreds of toxic substances! It can be administered via patches or nicotine gum, which are usually used to help in smoking cessation. Nicotine also also can have a pro-cognitive effect.
Melatonin. Many people associate melatonin with sleep. However, it has multiple neuroprotective effects by being an antioxidant, protecting mitochondrial integrity, and modulating the immune system, as well as attenuating microglial activation, which triggers neuroinflammation and oxidative stress. It also protects against cellular senescence, which is due to inflammation and reactive oxygen species. Furthermore, melatonin is useful in ameliorating the metabolic syndrome, which is associated with neurotoxic effects on brain tissue caused by the pro-inflammatory effects of peri-omental fat in obesity.8 Adjunctive melatonin could be helpful in patients with schizophrenia or depression who suffer from metabolic syndrome.
Erythropoietin is a hormone produced by the kidneys to promote the formation of red blood cells. It is a potent neuroprotective cytokine that promotes neuronal survival via anti-apoptotic effects. It protects against glutamate and nitrous oxide toxicity9 and haloperidol-induced neuronal death.10 It is clinically used (since FDA approval in 1989) in severe anemia due to chronic kidney disease or chemotherapy, as well as in inflammatory bowel disease. It does have some “black-box” warnings so its use should be limited.
Cox-2 inhibitors. This is a well-known class of anti-inflammatory drugs, which are FDA-approved for pain and inflammation. Studies of adjunctive use of cox-2 inhibitors in acute psychosis show that these drugs accentuate the efficacy of antipsychotic medications.11 The reason is that acute psychosis is associated with neuro-inflammation, which leads to neurotoxicity.
Lithium. Dosages to treat mania are usually 900 to 1500 mg/d. However, in minute (homeopathic) dosages as low as 1 mg/d, lithium has been shown to prevent progression of amnestic mild cognitive impairment to full dementia.12 This interesting observation suggests that lithium not only induces neurogenesis and increases gray matter volume,13 but may be neuroprotective against amyloid neurotoxicity. The effects of very low doses of lithium in depression and schizophrenia have not been studied yet.
Caffeine. Yes, the good old brew people seek all day is neuroprotective and prevents mood and memory dysfunction caused by stress.14 Caffeine should be avoided in patients with anxiety disorders, but it may be helpful for the brains of patients with mood or psychotic disorders. Caffeine reverses synaptic dysfunction in the circuits of the hippocampus caused by chronic unpredictable stress (quite common among our psychiatric patients).
The above interventions may be helpful for some patients but not others. Practitioners should consider using 1 or more of those adjunctive neuroprotective agents in patients who are at risk for neurodegenerative changes secondary to recurrences of acute and severe psychosis or mood episodes. Although clinicians cannot monitor brain structural integrity, they can assess the rate of symptomatic improvement and degree of functional restoration in their patients. Until a cure is found, these little steps—taken cautiously and judiciously—could help alleviate our patients’ suffering and the risk of neurotoxicity associated with their serious psychiatric disorder.
1. Chen AT, Chibnall JT, Nasrallah HA. A meta-analysis of placebo-controlled trials of omega-3 fatty acid augmentation in schizophrenia: possible stage-specific effects. Ann Clin Psychiatry. 2015;27(4):289-296.
2. Calon F, Cole G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: evidence from animal studies. Prostaglandins Leukot Essent Fatty Acids. 2007;7(5-6):287-293.
3. Chen AT, Chibnall JT, Nasrallah HA. Placebo-controlled augmentation trials of the antioxidant NAC in schizophrenia: a review. Ann Clin Psychiatry. 2016;28(3):190-196.
4. Chen G, Shi J, Hu Z, et al. Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of N-acetylcysteine. Mediators Inflamm. 2008;2008:716458. doi: 10.1155/2008/716458
5. Dean OM, Data-Franco J, Giorlando F, et al. Minocycline: therapeutic potential in psychiatry. CNS Drugs. 2012;26(5):391-401.
6. Eyles DW, Burne TH, McGrath JJ. Vitamin D, effects on brain development, adult brain function and the links between low levels of vitamin D and neuropsychiatric disease. Front Neuroendocrinol. 2013;34(1):47-64.
7. Akaike A, Tkada-Takatori Y, Kume T, et al. Mechanisms of neuroprotective effects of nicotine and acetylcholinesterase inhibitors: role of alpha4 and alpha7 receptors in neuroprotection. J Mol Neurosci. 2010;40(1-2):211-216.
8. Cardinali DP, Hardeland R. Inflammaging, metabolic syndrome and melatonin: a call for treatment studies [published online May 11, 2016]. Neuroendocrinology. doi:10.1159/000446543.
9. Yamasaki M, Mishima HK, Yamashita H, et al. Neuroprotective effects of erythropoietin on glutamate and nitric oxide toxicity in primary cultured retinal ganglion cells. Brain Res. 2005;1050(1-2):15-26.
10. Pilllai A, Dhandapani KM, Pillai BA, et al. Erythropoietin prevents haloperidol treatment-induced neuronal apoptosis through regulation of BDNF. Neuropsychopharmacology. 2008;33(8):1942-1951.
11. Müller N, Myint AM, Weidinger E, et al. Anti-inflammatory treatment in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2013;42:146-153.
12. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
13. Dong BT, Tu GJ, Han YX, et al. Lithium enhanced cell proliferation and differentiation of mesenchymal stem cells to neural cells in rat spinal cord. Int J Clin Exp Pathol. 2015;8(3):2473-2483.
14. Kaster MP, Machado NJ, Silva HB, et al. Caffeine acts through neuronal adenosine A24 receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc Natl Acad Sci U S A. 2015;112(25):7833-7838.
1. Chen AT, Chibnall JT, Nasrallah HA. A meta-analysis of placebo-controlled trials of omega-3 fatty acid augmentation in schizophrenia: possible stage-specific effects. Ann Clin Psychiatry. 2015;27(4):289-296.
2. Calon F, Cole G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: evidence from animal studies. Prostaglandins Leukot Essent Fatty Acids. 2007;7(5-6):287-293.
3. Chen AT, Chibnall JT, Nasrallah HA. Placebo-controlled augmentation trials of the antioxidant NAC in schizophrenia: a review. Ann Clin Psychiatry. 2016;28(3):190-196.
4. Chen G, Shi J, Hu Z, et al. Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of N-acetylcysteine. Mediators Inflamm. 2008;2008:716458. doi: 10.1155/2008/716458
5. Dean OM, Data-Franco J, Giorlando F, et al. Minocycline: therapeutic potential in psychiatry. CNS Drugs. 2012;26(5):391-401.
6. Eyles DW, Burne TH, McGrath JJ. Vitamin D, effects on brain development, adult brain function and the links between low levels of vitamin D and neuropsychiatric disease. Front Neuroendocrinol. 2013;34(1):47-64.
7. Akaike A, Tkada-Takatori Y, Kume T, et al. Mechanisms of neuroprotective effects of nicotine and acetylcholinesterase inhibitors: role of alpha4 and alpha7 receptors in neuroprotection. J Mol Neurosci. 2010;40(1-2):211-216.
8. Cardinali DP, Hardeland R. Inflammaging, metabolic syndrome and melatonin: a call for treatment studies [published online May 11, 2016]. Neuroendocrinology. doi:10.1159/000446543.
9. Yamasaki M, Mishima HK, Yamashita H, et al. Neuroprotective effects of erythropoietin on glutamate and nitric oxide toxicity in primary cultured retinal ganglion cells. Brain Res. 2005;1050(1-2):15-26.
10. Pilllai A, Dhandapani KM, Pillai BA, et al. Erythropoietin prevents haloperidol treatment-induced neuronal apoptosis through regulation of BDNF. Neuropsychopharmacology. 2008;33(8):1942-1951.
11. Müller N, Myint AM, Weidinger E, et al. Anti-inflammatory treatment in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2013;42:146-153.
12. Forlenza OV, Diniz BS, Radanovic M, et al. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351-356.
13. Dong BT, Tu GJ, Han YX, et al. Lithium enhanced cell proliferation and differentiation of mesenchymal stem cells to neural cells in rat spinal cord. Int J Clin Exp Pathol. 2015;8(3):2473-2483.
14. Kaster MP, Machado NJ, Silva HB, et al. Caffeine acts through neuronal adenosine A24 receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc Natl Acad Sci U S A. 2015;112(25):7833-7838.

The merits of buprenorphine for pregnant women with opioid use disorder; A possible solution to the ‘shrinking’ workforce
The merits of buprenorphine for pregnant women with opioid use disorder
Several medication-assisted treatments (MATs) for opioid use disorder were highlighted in “What clinicians need to know about treating opioid use disorder,” (Evidence-Based Reviews,
The landmark study, the Maternal Opioid Treatment: Human Experimental Research, a 2010 multicenter randomized controlled trial compared buprenorphine with methadone in pregnant women with opioid use disorder. The results revealed that neonates exposed to buprenorphine needed 89% less morphine to treat neonatal abstinence syndrome (NAS), 43% shorter hospital stay, and 58% shorter duration of medical treatment for NAS compared with those receiving methadone. Other advantages of buprenorphine over methadone are lower risk of overdose, fewer drug–drug interactions, and the option of receiving treatment in an outpatient setting, rather than a licensed treatment program, such as a methadone maintenance treatment program, which is more tightly controlled.1-3
The previous recommendation was to consider buprenorphine for patients who refused methadone or were unable to take it, or when a methadone treatment program wasn’t available. This study highlighted some clear advantages for treating this subpopulation with methadone instead of buprenorphine: only 18% of patients receiving methadone discontinued treatment, compared with 33% of those receiving buprenorphine,1-3 and methadone had a lower risk of diversion.3 The accepted practice has been to recommend methadone treatment for patients with mental, physical, or social stressors because of the structure of opioid treatment programs (OTP) (also known as methadone maintenance treatment programs). However, buprenorphine can be dispensed through an OTP, following the same stringent rules and regulations.4
The single agent, buprenorphine—not buprenorphine/naloxone—is recommended to prevent prenatal exposure to naloxone. It is thought that exposure to naloxone in utero might produce hormonal changes in the fetus.1,5 O'Connor et al5 noted methadone’s suitability during breast-feeding because of its low concentration in breast milk. Buprenorphine is excreted at breast milk to plasma ratio of 1:1, but because of buprenorphine’s poor oral bioavailability, infant exposure has little impact on the NAS score, therefore it’s suitable for breast-feeding mothers.5
Adegboyega Oyemade, MD, FAPA
Addiction Psychiatrist
Kaiser Permanente
Baltimore, Maryland
References
1. Lori W. Buprenorphine during pregnancy reduces neonate distress. https://www.drugabuse.gov/news-events/nida-notes/2012/07/buprenorphine- during-pregnancy-reduces-neonate-distress. Published July 6, 2016. Accessed September 9, 2016.
2. Jones HE, Kaltenbach K, Heil SH, et al. Neonatal abstinence syndrome after methadone or buprenorphine exposure. N Engl J Med. 2010;363(24):2320-2331.
3. ACOG Committee on Health Care for Underserved Women; American Society of Addiction Medicine. ACOG Committee Opinion No. 524: Opioid abuse, dependence, and addiction in pregnancy. Obest Gynecol. 2012;119(5):1070-1076.
4. Addiction Treatment Forum. Methadone vs. buprenorphine: how do OTPs and patients make the choice? http://atforum.com/2013/11/methadone-vs-buprenorphine-how-do-otps-and-patients-make-the-choice. Published November 15, 2013. Accessed September 9, 2016.
5. O’Connor A, Alto W, Musgrave K, et al. Observational study of buprenorphine treatment of opioid-dependent pregnancy women in a family medicine residency: reports on maternal and infant outcomes. J Am Board Fam Med. 2011;24(2):194-201.
A possible solution to the 'shrinking' workforce
I would like to offer another pragmatic, easy, and quick solution for dealing with the shrinking psychiatrist workforce (The psychiatry workforce pool is shrinking. What are we doing about it? From the Editor.
The United States is short approximately 45,000 psychiatrists.1 Burnout—a silent epidemic among physicians—is prevalent in psychiatry. What consumes time and leads to burn out? “Scut work.”
There are thousands of unmatched residency graduates.2 Most of these graduates have clinical experience in the United States. Psychiatry residency programs should give these unmatched graduates 6 months of training in psychiatry and use them as our primary workforce. These assistant physicians could be paired with 2 to 3 psychiatrists to perform the menial tasks, including, but not limited to, phone calls, prescriptions, prior authorizations, chart review, and other clinical and administrative paper work. This way, psychiatrists can focus on the interview, diagnoses, and treatment, major medical decision-making, and see more patients.
Employing assistant physicians to provide care has been suggested as a solution.3 Arkansas, Kansas, and Missouri have passed laws that allow medical school graduates who did not match with a residency program to work in underserved areas with a collaborating physician.
Because 1 out of 4 individuals have a mental illness and more of them are seeking help because of increasing awareness and the Affordable Care Act, the construct of “assistant physicians” could ease psychiatrists’ workload allowing them to deliver better care to more people.
Maju Mathew Koola, MD
Associate Professor
Department of Psychiatry and Behavioral Sciences
George Washington University
School of Medicine and Health Sciences
Washington, DC
References
1. Carlat D. 45,000 More psychiatrists, anyone? Psychiatric Times. http://www.psychiatrictimes.com/articles/45000-more-psychiatrists-anyone-0.Published August 3, 2010. Accessed November 11, 2016.
2. The Match: National Resident Matching Program. results and data: 2016 main residency match. http://www.nrmp.org/wp-content/uploads/2016/04/Main-Match-Results-and-Data-2016.pdf. Published April 2016. Accessed November 11, 2016.
3. Miller JG, Peterson DJ. Employing nurse practitioners and physician assistants to provide access to care as the psychiatrist shortage continues. Acad Psychiatry. 2015;39(6):685-686.
Concerns in psychiatry
The August 2016 editorial (Unresolved questions about the specialty lurk in the cortext of psychiatrists.
Denis F. Darko, MD
CEO
NeuroSci R&D Consultancy, LLC
Maple Grove, Minnesota
The merits of buprenorphine for pregnant women with opioid use disorder
Several medication-assisted treatments (MATs) for opioid use disorder were highlighted in “What clinicians need to know about treating opioid use disorder,” (Evidence-Based Reviews,
The landmark study, the Maternal Opioid Treatment: Human Experimental Research, a 2010 multicenter randomized controlled trial compared buprenorphine with methadone in pregnant women with opioid use disorder. The results revealed that neonates exposed to buprenorphine needed 89% less morphine to treat neonatal abstinence syndrome (NAS), 43% shorter hospital stay, and 58% shorter duration of medical treatment for NAS compared with those receiving methadone. Other advantages of buprenorphine over methadone are lower risk of overdose, fewer drug–drug interactions, and the option of receiving treatment in an outpatient setting, rather than a licensed treatment program, such as a methadone maintenance treatment program, which is more tightly controlled.1-3
The previous recommendation was to consider buprenorphine for patients who refused methadone or were unable to take it, or when a methadone treatment program wasn’t available. This study highlighted some clear advantages for treating this subpopulation with methadone instead of buprenorphine: only 18% of patients receiving methadone discontinued treatment, compared with 33% of those receiving buprenorphine,1-3 and methadone had a lower risk of diversion.3 The accepted practice has been to recommend methadone treatment for patients with mental, physical, or social stressors because of the structure of opioid treatment programs (OTP) (also known as methadone maintenance treatment programs). However, buprenorphine can be dispensed through an OTP, following the same stringent rules and regulations.4
The single agent, buprenorphine—not buprenorphine/naloxone—is recommended to prevent prenatal exposure to naloxone. It is thought that exposure to naloxone in utero might produce hormonal changes in the fetus.1,5 O'Connor et al5 noted methadone’s suitability during breast-feeding because of its low concentration in breast milk. Buprenorphine is excreted at breast milk to plasma ratio of 1:1, but because of buprenorphine’s poor oral bioavailability, infant exposure has little impact on the NAS score, therefore it’s suitable for breast-feeding mothers.5
Adegboyega Oyemade, MD, FAPA
Addiction Psychiatrist
Kaiser Permanente
Baltimore, Maryland
References
1. Lori W. Buprenorphine during pregnancy reduces neonate distress. https://www.drugabuse.gov/news-events/nida-notes/2012/07/buprenorphine- during-pregnancy-reduces-neonate-distress. Published July 6, 2016. Accessed September 9, 2016.
2. Jones HE, Kaltenbach K, Heil SH, et al. Neonatal abstinence syndrome after methadone or buprenorphine exposure. N Engl J Med. 2010;363(24):2320-2331.
3. ACOG Committee on Health Care for Underserved Women; American Society of Addiction Medicine. ACOG Committee Opinion No. 524: Opioid abuse, dependence, and addiction in pregnancy. Obest Gynecol. 2012;119(5):1070-1076.
4. Addiction Treatment Forum. Methadone vs. buprenorphine: how do OTPs and patients make the choice? http://atforum.com/2013/11/methadone-vs-buprenorphine-how-do-otps-and-patients-make-the-choice. Published November 15, 2013. Accessed September 9, 2016.
5. O’Connor A, Alto W, Musgrave K, et al. Observational study of buprenorphine treatment of opioid-dependent pregnancy women in a family medicine residency: reports on maternal and infant outcomes. J Am Board Fam Med. 2011;24(2):194-201.
A possible solution to the 'shrinking' workforce
I would like to offer another pragmatic, easy, and quick solution for dealing with the shrinking psychiatrist workforce (The psychiatry workforce pool is shrinking. What are we doing about it? From the Editor.
The United States is short approximately 45,000 psychiatrists.1 Burnout—a silent epidemic among physicians—is prevalent in psychiatry. What consumes time and leads to burn out? “Scut work.”
There are thousands of unmatched residency graduates.2 Most of these graduates have clinical experience in the United States. Psychiatry residency programs should give these unmatched graduates 6 months of training in psychiatry and use them as our primary workforce. These assistant physicians could be paired with 2 to 3 psychiatrists to perform the menial tasks, including, but not limited to, phone calls, prescriptions, prior authorizations, chart review, and other clinical and administrative paper work. This way, psychiatrists can focus on the interview, diagnoses, and treatment, major medical decision-making, and see more patients.
Employing assistant physicians to provide care has been suggested as a solution.3 Arkansas, Kansas, and Missouri have passed laws that allow medical school graduates who did not match with a residency program to work in underserved areas with a collaborating physician.
Because 1 out of 4 individuals have a mental illness and more of them are seeking help because of increasing awareness and the Affordable Care Act, the construct of “assistant physicians” could ease psychiatrists’ workload allowing them to deliver better care to more people.
Maju Mathew Koola, MD
Associate Professor
Department of Psychiatry and Behavioral Sciences
George Washington University
School of Medicine and Health Sciences
Washington, DC
References
1. Carlat D. 45,000 More psychiatrists, anyone? Psychiatric Times. http://www.psychiatrictimes.com/articles/45000-more-psychiatrists-anyone-0.Published August 3, 2010. Accessed November 11, 2016.
2. The Match: National Resident Matching Program. results and data: 2016 main residency match. http://www.nrmp.org/wp-content/uploads/2016/04/Main-Match-Results-and-Data-2016.pdf. Published April 2016. Accessed November 11, 2016.
3. Miller JG, Peterson DJ. Employing nurse practitioners and physician assistants to provide access to care as the psychiatrist shortage continues. Acad Psychiatry. 2015;39(6):685-686.
Concerns in psychiatry
The August 2016 editorial (Unresolved questions about the specialty lurk in the cortext of psychiatrists.
Denis F. Darko, MD
CEO
NeuroSci R&D Consultancy, LLC
Maple Grove, Minnesota
The merits of buprenorphine for pregnant women with opioid use disorder
Several medication-assisted treatments (MATs) for opioid use disorder were highlighted in “What clinicians need to know about treating opioid use disorder,” (Evidence-Based Reviews,
The landmark study, the Maternal Opioid Treatment: Human Experimental Research, a 2010 multicenter randomized controlled trial compared buprenorphine with methadone in pregnant women with opioid use disorder. The results revealed that neonates exposed to buprenorphine needed 89% less morphine to treat neonatal abstinence syndrome (NAS), 43% shorter hospital stay, and 58% shorter duration of medical treatment for NAS compared with those receiving methadone. Other advantages of buprenorphine over methadone are lower risk of overdose, fewer drug–drug interactions, and the option of receiving treatment in an outpatient setting, rather than a licensed treatment program, such as a methadone maintenance treatment program, which is more tightly controlled.1-3
The previous recommendation was to consider buprenorphine for patients who refused methadone or were unable to take it, or when a methadone treatment program wasn’t available. This study highlighted some clear advantages for treating this subpopulation with methadone instead of buprenorphine: only 18% of patients receiving methadone discontinued treatment, compared with 33% of those receiving buprenorphine,1-3 and methadone had a lower risk of diversion.3 The accepted practice has been to recommend methadone treatment for patients with mental, physical, or social stressors because of the structure of opioid treatment programs (OTP) (also known as methadone maintenance treatment programs). However, buprenorphine can be dispensed through an OTP, following the same stringent rules and regulations.4
The single agent, buprenorphine—not buprenorphine/naloxone—is recommended to prevent prenatal exposure to naloxone. It is thought that exposure to naloxone in utero might produce hormonal changes in the fetus.1,5 O'Connor et al5 noted methadone’s suitability during breast-feeding because of its low concentration in breast milk. Buprenorphine is excreted at breast milk to plasma ratio of 1:1, but because of buprenorphine’s poor oral bioavailability, infant exposure has little impact on the NAS score, therefore it’s suitable for breast-feeding mothers.5
Adegboyega Oyemade, MD, FAPA
Addiction Psychiatrist
Kaiser Permanente
Baltimore, Maryland
References
1. Lori W. Buprenorphine during pregnancy reduces neonate distress. https://www.drugabuse.gov/news-events/nida-notes/2012/07/buprenorphine- during-pregnancy-reduces-neonate-distress. Published July 6, 2016. Accessed September 9, 2016.
2. Jones HE, Kaltenbach K, Heil SH, et al. Neonatal abstinence syndrome after methadone or buprenorphine exposure. N Engl J Med. 2010;363(24):2320-2331.
3. ACOG Committee on Health Care for Underserved Women; American Society of Addiction Medicine. ACOG Committee Opinion No. 524: Opioid abuse, dependence, and addiction in pregnancy. Obest Gynecol. 2012;119(5):1070-1076.
4. Addiction Treatment Forum. Methadone vs. buprenorphine: how do OTPs and patients make the choice? http://atforum.com/2013/11/methadone-vs-buprenorphine-how-do-otps-and-patients-make-the-choice. Published November 15, 2013. Accessed September 9, 2016.
5. O’Connor A, Alto W, Musgrave K, et al. Observational study of buprenorphine treatment of opioid-dependent pregnancy women in a family medicine residency: reports on maternal and infant outcomes. J Am Board Fam Med. 2011;24(2):194-201.
A possible solution to the 'shrinking' workforce
I would like to offer another pragmatic, easy, and quick solution for dealing with the shrinking psychiatrist workforce (The psychiatry workforce pool is shrinking. What are we doing about it? From the Editor.
The United States is short approximately 45,000 psychiatrists.1 Burnout—a silent epidemic among physicians—is prevalent in psychiatry. What consumes time and leads to burn out? “Scut work.”
There are thousands of unmatched residency graduates.2 Most of these graduates have clinical experience in the United States. Psychiatry residency programs should give these unmatched graduates 6 months of training in psychiatry and use them as our primary workforce. These assistant physicians could be paired with 2 to 3 psychiatrists to perform the menial tasks, including, but not limited to, phone calls, prescriptions, prior authorizations, chart review, and other clinical and administrative paper work. This way, psychiatrists can focus on the interview, diagnoses, and treatment, major medical decision-making, and see more patients.
Employing assistant physicians to provide care has been suggested as a solution.3 Arkansas, Kansas, and Missouri have passed laws that allow medical school graduates who did not match with a residency program to work in underserved areas with a collaborating physician.
Because 1 out of 4 individuals have a mental illness and more of them are seeking help because of increasing awareness and the Affordable Care Act, the construct of “assistant physicians” could ease psychiatrists’ workload allowing them to deliver better care to more people.
Maju Mathew Koola, MD
Associate Professor
Department of Psychiatry and Behavioral Sciences
George Washington University
School of Medicine and Health Sciences
Washington, DC
References
1. Carlat D. 45,000 More psychiatrists, anyone? Psychiatric Times. http://www.psychiatrictimes.com/articles/45000-more-psychiatrists-anyone-0.Published August 3, 2010. Accessed November 11, 2016.
2. The Match: National Resident Matching Program. results and data: 2016 main residency match. http://www.nrmp.org/wp-content/uploads/2016/04/Main-Match-Results-and-Data-2016.pdf. Published April 2016. Accessed November 11, 2016.
3. Miller JG, Peterson DJ. Employing nurse practitioners and physician assistants to provide access to care as the psychiatrist shortage continues. Acad Psychiatry. 2015;39(6):685-686.
Concerns in psychiatry
The August 2016 editorial (Unresolved questions about the specialty lurk in the cortext of psychiatrists.
Denis F. Darko, MD
CEO
NeuroSci R&D Consultancy, LLC
Maple Grove, Minnesota

Wart's That Lesion?
A 10-year-old girl is referred to dermatology for evaluation of an asymptomatic “wart” that manifested on her thigh several years ago. At first, the lesion grew rapidly, but it has since stabilized. It remains unaffected despite multiple treatments with liquid nitrogen.
According to her parents (with confirmation from her medical record), the patient is otherwise quite healthy.
EXAMINATION
A spindle-shaped, pinkish red, 1.2 x 0.7-cm nodule with a smooth surface and rounded borders is located on the patient’s distal left anterior thigh. The lesion itself is quite firm but nontender to the touch, and its long axis is parallel to transverse local skin tension lines. The patient’s type II skin is otherwise unremarkable.
After consultation with the patient and her family, the lesion is excised under local anesthesia with 5-mm margins in an elliptical pattern to yield favorable lines of closure. The sample is sent to pathology.
What is the diagnosis?
DISCUSSION
The pathology report showed both epithelioid and spindle cells with scattered apoptotic cells seen at the dermoepidermal junction, with no atypia. The margins were clear. Such findings are consistent with a diagnosis of Spitz nevus.
Prior to the 1950s, lesions with these clinical and histologic features were believed to be melanoma and therefore termed juvenile melanoma. But their unique morphologic and histologic architecture—and a lack of signs of progressive or metastatic disease in affected patients—prompted suspicion that they were not really malignant. A pathologist named Sophie Spitz conducted a retrospective study that revealed no cancerous activity in any of the patients. Her findings, on a superficial level, eventually led to the elimination of the term “juvenile melanoma”—but more importantly, they eradicated a great deal of unnecessary, and often mutilating, surgery.
The features of the case patient’s lesion—color, texture, shape, and initial rapid growth —are typical of Spitz nevi, as are the location and the age of the patient. However, the range of morphologic presentations includes darker lesions (some almost black) and those of varying vertical depth and appearance (ie, intradermal, compound, junctional).
A note of caution, however: Although Spitz nevi are benign, true melanomas can present with “Spitzoid” morphologic and histologic features (they’re even called “Spitzoid melanomas”). Key distinguishing features can only be identified via microscopic examination of the lesion’s full vertical structure. So, for safety’s sake, the standard of care for Spitz nevi is to excise them totally, with clear, deep, wide margins, whenever possible.
The case patient had her sutures removed 10 days after surgery. Her ongoing care will include a biannual check for signs of recurrence.
TAKE-HOME LEARNING POINTS
• Until the mid-1950s, what we now refer to as Spitz nevi were called “juvenile melanoma” and were treated as such, with radical and often disfiguring surgeries.
• Spitz nevi are usually seen in children. They can be red, pink, or even black, and often have a history of recent growth and an elliptiform shape.
• Excision with totally clear margins is the correct approach, along with pathologic examination.
• Spitz nevi have histologic features suggestive of melanoma, and some melanomas have “Spitzoid” features. Therefore, all cases must be handled with great care.
A 10-year-old girl is referred to dermatology for evaluation of an asymptomatic “wart” that manifested on her thigh several years ago. At first, the lesion grew rapidly, but it has since stabilized. It remains unaffected despite multiple treatments with liquid nitrogen.
According to her parents (with confirmation from her medical record), the patient is otherwise quite healthy.
EXAMINATION
A spindle-shaped, pinkish red, 1.2 x 0.7-cm nodule with a smooth surface and rounded borders is located on the patient’s distal left anterior thigh. The lesion itself is quite firm but nontender to the touch, and its long axis is parallel to transverse local skin tension lines. The patient’s type II skin is otherwise unremarkable.
After consultation with the patient and her family, the lesion is excised under local anesthesia with 5-mm margins in an elliptical pattern to yield favorable lines of closure. The sample is sent to pathology.
What is the diagnosis?
DISCUSSION
The pathology report showed both epithelioid and spindle cells with scattered apoptotic cells seen at the dermoepidermal junction, with no atypia. The margins were clear. Such findings are consistent with a diagnosis of Spitz nevus.
Prior to the 1950s, lesions with these clinical and histologic features were believed to be melanoma and therefore termed juvenile melanoma. But their unique morphologic and histologic architecture—and a lack of signs of progressive or metastatic disease in affected patients—prompted suspicion that they were not really malignant. A pathologist named Sophie Spitz conducted a retrospective study that revealed no cancerous activity in any of the patients. Her findings, on a superficial level, eventually led to the elimination of the term “juvenile melanoma”—but more importantly, they eradicated a great deal of unnecessary, and often mutilating, surgery.
The features of the case patient’s lesion—color, texture, shape, and initial rapid growth —are typical of Spitz nevi, as are the location and the age of the patient. However, the range of morphologic presentations includes darker lesions (some almost black) and those of varying vertical depth and appearance (ie, intradermal, compound, junctional).
A note of caution, however: Although Spitz nevi are benign, true melanomas can present with “Spitzoid” morphologic and histologic features (they’re even called “Spitzoid melanomas”). Key distinguishing features can only be identified via microscopic examination of the lesion’s full vertical structure. So, for safety’s sake, the standard of care for Spitz nevi is to excise them totally, with clear, deep, wide margins, whenever possible.
The case patient had her sutures removed 10 days after surgery. Her ongoing care will include a biannual check for signs of recurrence.
TAKE-HOME LEARNING POINTS
• Until the mid-1950s, what we now refer to as Spitz nevi were called “juvenile melanoma” and were treated as such, with radical and often disfiguring surgeries.
• Spitz nevi are usually seen in children. They can be red, pink, or even black, and often have a history of recent growth and an elliptiform shape.
• Excision with totally clear margins is the correct approach, along with pathologic examination.
• Spitz nevi have histologic features suggestive of melanoma, and some melanomas have “Spitzoid” features. Therefore, all cases must be handled with great care.
A 10-year-old girl is referred to dermatology for evaluation of an asymptomatic “wart” that manifested on her thigh several years ago. At first, the lesion grew rapidly, but it has since stabilized. It remains unaffected despite multiple treatments with liquid nitrogen.
According to her parents (with confirmation from her medical record), the patient is otherwise quite healthy.
EXAMINATION
A spindle-shaped, pinkish red, 1.2 x 0.7-cm nodule with a smooth surface and rounded borders is located on the patient’s distal left anterior thigh. The lesion itself is quite firm but nontender to the touch, and its long axis is parallel to transverse local skin tension lines. The patient’s type II skin is otherwise unremarkable.
After consultation with the patient and her family, the lesion is excised under local anesthesia with 5-mm margins in an elliptical pattern to yield favorable lines of closure. The sample is sent to pathology.
What is the diagnosis?
DISCUSSION
The pathology report showed both epithelioid and spindle cells with scattered apoptotic cells seen at the dermoepidermal junction, with no atypia. The margins were clear. Such findings are consistent with a diagnosis of Spitz nevus.
Prior to the 1950s, lesions with these clinical and histologic features were believed to be melanoma and therefore termed juvenile melanoma. But their unique morphologic and histologic architecture—and a lack of signs of progressive or metastatic disease in affected patients—prompted suspicion that they were not really malignant. A pathologist named Sophie Spitz conducted a retrospective study that revealed no cancerous activity in any of the patients. Her findings, on a superficial level, eventually led to the elimination of the term “juvenile melanoma”—but more importantly, they eradicated a great deal of unnecessary, and often mutilating, surgery.
The features of the case patient’s lesion—color, texture, shape, and initial rapid growth —are typical of Spitz nevi, as are the location and the age of the patient. However, the range of morphologic presentations includes darker lesions (some almost black) and those of varying vertical depth and appearance (ie, intradermal, compound, junctional).
A note of caution, however: Although Spitz nevi are benign, true melanomas can present with “Spitzoid” morphologic and histologic features (they’re even called “Spitzoid melanomas”). Key distinguishing features can only be identified via microscopic examination of the lesion’s full vertical structure. So, for safety’s sake, the standard of care for Spitz nevi is to excise them totally, with clear, deep, wide margins, whenever possible.
The case patient had her sutures removed 10 days after surgery. Her ongoing care will include a biannual check for signs of recurrence.
TAKE-HOME LEARNING POINTS
• Until the mid-1950s, what we now refer to as Spitz nevi were called “juvenile melanoma” and were treated as such, with radical and often disfiguring surgeries.
• Spitz nevi are usually seen in children. They can be red, pink, or even black, and often have a history of recent growth and an elliptiform shape.
• Excision with totally clear margins is the correct approach, along with pathologic examination.
• Spitz nevi have histologic features suggestive of melanoma, and some melanomas have “Spitzoid” features. Therefore, all cases must be handled with great care.
Do progesterone-only contraceptives lead to more mood changes than other types?
Lower, or comparable depression scores compared with other methods
A retrospective cohort trial compared 298 women on progesterone-only contraception with 6356 women on other or no contraception to examine the association between contraception use and depressive symptoms.1
When surveyed with the Center of Epidemiological Studies Depression Scale (using both a 10-question, 30-point questionnaire and a 20-question, 60-point questionnaire), women on progesterone-only contraception demonstrated significantly lower levels of depressive symptoms compared with women using low-efficacy contraception (early withdrawal, spermicides, contraceptive films) or no contraception (mean deviation [MD]=-1.3; 95% confidence interval [CI], -2.4 to -0.2). No significant difference was seen in depression scores when compared with women on other forms of hormonal contraception (MD=-0.3; 95% CI, -1.2 to 0.6).
No significant difference in depression and less anhedonia for nonusers
A cross-sectional, population-based trial conducted by survey in Finland in 1997, 2002, and 2007 investigated the link between contraception and mood symptoms. It included 759 women using the progesterone-only levonorgestrel-releasing intrauterine system (LNG-IUS) and 7036 women on other forms of contraception or none.2
Current LNG-IUS users vs nonusers had no significant difference in diagnosis of depression, as assessed by asking patients if they had been diagnosed with or treated for depression in the previous year of contraception (8.0% vs 7.3%; P>.05); depressive symptoms in the previous year (24% vs 26%; P>.05), or psychological illness (1.9% vs 2.5%; P>.05).
LNG-IUS users reported significantly less anhedonia than nonusers in the previous year (19% vs 22%; P<.05). Moreover, in a partial correlation analysis, LNG-IUS was negatively correlated with anhedonia (r=0.024; P<.05) and symptoms of depression over the previous month (r=0.098; P<.05).
Did relationship satisfaction, rather than contraceptive, influence depression?
A multicenter prospective cohort trial analyzed the effect of the levonorgestrel implant on mood in 267 women followed for 2 years by evaluating depressive symptoms reported from the Mental Health Inventory, a 6-item questionnaire scored 0 to 24.3
The women demonstrated a significant increase in depressive symptom scores from 7.9 at baseline to 8.8 (P=.01). However, the study authors suggested that relationship satisfaction, not method of birth control, was the cause of depressive symptoms. The 62 women who experienced a decrease in relationship satisfaction exhibited a significant increase in depressive symptoms (6.7-10; P=.001) compared with the 156 women who reported an improvement or no change in relationship satisfaction (7.8-8.2; P=.30).
1. Keyes K, Cheslack-Postava K, Westhoff C, et al. Association of hormonal contraception use with reduced levels of depressive symptoms: a national study of sexually active women in the United States. Am J Epidemiol. 2013;178:1378-1388.
2. Toffol E, Heikinheimo O, Koponen P, et al. Further evidence for lack of negative associations between hormonal contraception and mental health. Contraception. 2012;86:470-480.
3. Westhoff C, Truman C, Kalmuss D, et al. Depressive symptoms and Norplant contraceptive implants. Contraception. 1998;57:241-245.
Lower, or comparable depression scores compared with other methods
A retrospective cohort trial compared 298 women on progesterone-only contraception with 6356 women on other or no contraception to examine the association between contraception use and depressive symptoms.1
When surveyed with the Center of Epidemiological Studies Depression Scale (using both a 10-question, 30-point questionnaire and a 20-question, 60-point questionnaire), women on progesterone-only contraception demonstrated significantly lower levels of depressive symptoms compared with women using low-efficacy contraception (early withdrawal, spermicides, contraceptive films) or no contraception (mean deviation [MD]=-1.3; 95% confidence interval [CI], -2.4 to -0.2). No significant difference was seen in depression scores when compared with women on other forms of hormonal contraception (MD=-0.3; 95% CI, -1.2 to 0.6).
No significant difference in depression and less anhedonia for nonusers
A cross-sectional, population-based trial conducted by survey in Finland in 1997, 2002, and 2007 investigated the link between contraception and mood symptoms. It included 759 women using the progesterone-only levonorgestrel-releasing intrauterine system (LNG-IUS) and 7036 women on other forms of contraception or none.2
Current LNG-IUS users vs nonusers had no significant difference in diagnosis of depression, as assessed by asking patients if they had been diagnosed with or treated for depression in the previous year of contraception (8.0% vs 7.3%; P>.05); depressive symptoms in the previous year (24% vs 26%; P>.05), or psychological illness (1.9% vs 2.5%; P>.05).
LNG-IUS users reported significantly less anhedonia than nonusers in the previous year (19% vs 22%; P<.05). Moreover, in a partial correlation analysis, LNG-IUS was negatively correlated with anhedonia (r=0.024; P<.05) and symptoms of depression over the previous month (r=0.098; P<.05).
Did relationship satisfaction, rather than contraceptive, influence depression?
A multicenter prospective cohort trial analyzed the effect of the levonorgestrel implant on mood in 267 women followed for 2 years by evaluating depressive symptoms reported from the Mental Health Inventory, a 6-item questionnaire scored 0 to 24.3
The women demonstrated a significant increase in depressive symptom scores from 7.9 at baseline to 8.8 (P=.01). However, the study authors suggested that relationship satisfaction, not method of birth control, was the cause of depressive symptoms. The 62 women who experienced a decrease in relationship satisfaction exhibited a significant increase in depressive symptoms (6.7-10; P=.001) compared with the 156 women who reported an improvement or no change in relationship satisfaction (7.8-8.2; P=.30).
Lower, or comparable depression scores compared with other methods
A retrospective cohort trial compared 298 women on progesterone-only contraception with 6356 women on other or no contraception to examine the association between contraception use and depressive symptoms.1
When surveyed with the Center of Epidemiological Studies Depression Scale (using both a 10-question, 30-point questionnaire and a 20-question, 60-point questionnaire), women on progesterone-only contraception demonstrated significantly lower levels of depressive symptoms compared with women using low-efficacy contraception (early withdrawal, spermicides, contraceptive films) or no contraception (mean deviation [MD]=-1.3; 95% confidence interval [CI], -2.4 to -0.2). No significant difference was seen in depression scores when compared with women on other forms of hormonal contraception (MD=-0.3; 95% CI, -1.2 to 0.6).
No significant difference in depression and less anhedonia for nonusers
A cross-sectional, population-based trial conducted by survey in Finland in 1997, 2002, and 2007 investigated the link between contraception and mood symptoms. It included 759 women using the progesterone-only levonorgestrel-releasing intrauterine system (LNG-IUS) and 7036 women on other forms of contraception or none.2
Current LNG-IUS users vs nonusers had no significant difference in diagnosis of depression, as assessed by asking patients if they had been diagnosed with or treated for depression in the previous year of contraception (8.0% vs 7.3%; P>.05); depressive symptoms in the previous year (24% vs 26%; P>.05), or psychological illness (1.9% vs 2.5%; P>.05).
LNG-IUS users reported significantly less anhedonia than nonusers in the previous year (19% vs 22%; P<.05). Moreover, in a partial correlation analysis, LNG-IUS was negatively correlated with anhedonia (r=0.024; P<.05) and symptoms of depression over the previous month (r=0.098; P<.05).
Did relationship satisfaction, rather than contraceptive, influence depression?
A multicenter prospective cohort trial analyzed the effect of the levonorgestrel implant on mood in 267 women followed for 2 years by evaluating depressive symptoms reported from the Mental Health Inventory, a 6-item questionnaire scored 0 to 24.3
The women demonstrated a significant increase in depressive symptom scores from 7.9 at baseline to 8.8 (P=.01). However, the study authors suggested that relationship satisfaction, not method of birth control, was the cause of depressive symptoms. The 62 women who experienced a decrease in relationship satisfaction exhibited a significant increase in depressive symptoms (6.7-10; P=.001) compared with the 156 women who reported an improvement or no change in relationship satisfaction (7.8-8.2; P=.30).
1. Keyes K, Cheslack-Postava K, Westhoff C, et al. Association of hormonal contraception use with reduced levels of depressive symptoms: a national study of sexually active women in the United States. Am J Epidemiol. 2013;178:1378-1388.
2. Toffol E, Heikinheimo O, Koponen P, et al. Further evidence for lack of negative associations between hormonal contraception and mental health. Contraception. 2012;86:470-480.
3. Westhoff C, Truman C, Kalmuss D, et al. Depressive symptoms and Norplant contraceptive implants. Contraception. 1998;57:241-245.
1. Keyes K, Cheslack-Postava K, Westhoff C, et al. Association of hormonal contraception use with reduced levels of depressive symptoms: a national study of sexually active women in the United States. Am J Epidemiol. 2013;178:1378-1388.
2. Toffol E, Heikinheimo O, Koponen P, et al. Further evidence for lack of negative associations between hormonal contraception and mental health. Contraception. 2012;86:470-480.
3. Westhoff C, Truman C, Kalmuss D, et al. Depressive symptoms and Norplant contraceptive implants. Contraception. 1998;57:241-245.
Evidence-based answers from the Family Physicians Inquiries Network
EVIDENCE-BASED ANSWER:
No. Women taking progesterone-only contraceptives don’t appear to experience more depressive symptoms or mood changes than women on other hormonal contraceptives, and they may experience slightly less depression than women using no contraception (strength of recommendation: B, multiple homogeneous cohorts).
Is an intestinal biopsy necessary when the blood work suggests celiac disease?
EVIDENCE SUMMARY
A 2013 Belgian prospective study of 104 non-IgA–deficient adults and children diagnosed with celiac disease and 537 adults and children without celiac disease evaluated the accuracy of 4 manufacturers’ serologic tests for IgA anti-tTG.1 All patients underwent serologic testing followed by a diagnostic biopsy. A Marsh type 3 or greater lesion on duodenal biopsy was considered diagnostic for celiac disease.
Anti-tTG levels greater than 10 times the manufacturer-recommended level for a positive test (cut-off) were associated with a likelihood ratio of 111 to 294 (depending on the manufacturer) of positive biopsy. Post-test probabilities were calculated based on various pretest probabilities using an anti-tTG level of greater than 10 times the cut-off (TABLE1).
Investigators also obtained IgG anti-DGP levels from 2 of the manufacturers.1 Likelihood ratios increased along with antibody levels. Ratios of 80 and 400, depending on the manufacturer, were found at IgG anti-DGP levels 10-fold greater than the cut-off. Pre- and post-test probabilities weren’t calculated.

Positive predictive value rises with antibody levels
A 2013 retrospective study evaluated the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition’s recommendation to forego intestinal biopsy in non-IgA–deficient, symptomatic children and adolescents with positive IgA anti-tTG levels greater than 10 times the cut-off value, positive EMA, and positive HLA-DQ2 or HLA-DQ8.2
Overall, 153 symptomatic patients referred to the gastroenterology unit met these criteria. The age range was 9 months to 14.6 years (mean 4 years). All but 3 of the patients had Marsh 2 or greater lesions with biopsy-confirmed diagnoses of celiac disease. The remaining 3 developed biopsy-positive celiac disease on follow-up. The positive predictive value of combined serologic testing in this small selected patient population was 100%.
A 2013 retrospective study of 2477 symptomatic adults (older than 18 years) who received diagnostic testing for celiac disease at 2 academic institutions in Cleveland, Ohio, evaluated the predictive value of IgA anti-tTG and EMA. Of the patients, 610 (25%) had abnormal serologic tests, and 240 (39%) underwent endoscopy with biopsy.
A total of 50 patients (21%) had biopsy results consistent with celiac disease, defined as a Marsh 3 lesion or greater.3 An IgA anti-tTG level of 118 U/mL (5.9-fold the upper limit of normal on the test) had a positive predictive value of 86.4% with a false-positive value of 2%. An EMA titer greater than 1:160 when IgA anti-tTG was between 21 and 118 U/mL had a positive predictive value of 83%.
Antibody levels 10 times normal show 100% positive predictive value
A 2008 retrospective study of one manufacturer’s IgA anti-tTG serologic test sought to establish the serologic antibody level at which the positive predictive value was 100%.4 Overall, 148 people, 15 years and older, with a positive IgA anti-tTG before biopsy or within 21 days of biopsy were included.
Of the patients biopsied, 139 (93%) had positive biopsies of Marsh 2 or greater and were diagnosed with celiac disease. Using a cut-off of 3.3 and 6.7 times the upper limit of normal, investigators calculated a positive predictive value of 95% and 98%, respectively.
A cut-off of 10 times the upper limit of normal or greater had a positive predictive value of 100%. The highest level of IgA anti-tTG in a patient who didn’t have celiac disease on biopsy was 7.3 times the upper limit of normal.
1. Vermeersch P, Geboes K, Mariën G, et al. Defining thresholds of antibody levels improves diagnosis of celiac disease. Clin Gastroenterol Hepatol. 2013;11:398-403;quiz e32.
2. Klapp G, Masip E, Bolonio M, et al. Celiac disease: The new proposed ESPGHAN diagnostic criteria do work well in a selected population. J Pediatr Gastroenterol Nutr. 2013;56:251-256.
3. Wakim-Fleming J, Pagadala MR, Lemyre MS, et al. Diagnosis of celiac disease in adults based on serology test results, without small-bowel biopsy. Clin Gastroenterol Hepatol. 2013;11:511-516.
4. Hill PG, Holmes GK. Coeliac disease: a biopsy is not always necessary for diagnosis. Aliment Pharmacol Ther. 2008;27:572-577.
EVIDENCE SUMMARY
A 2013 Belgian prospective study of 104 non-IgA–deficient adults and children diagnosed with celiac disease and 537 adults and children without celiac disease evaluated the accuracy of 4 manufacturers’ serologic tests for IgA anti-tTG.1 All patients underwent serologic testing followed by a diagnostic biopsy. A Marsh type 3 or greater lesion on duodenal biopsy was considered diagnostic for celiac disease.
Anti-tTG levels greater than 10 times the manufacturer-recommended level for a positive test (cut-off) were associated with a likelihood ratio of 111 to 294 (depending on the manufacturer) of positive biopsy. Post-test probabilities were calculated based on various pretest probabilities using an anti-tTG level of greater than 10 times the cut-off (TABLE1).
Investigators also obtained IgG anti-DGP levels from 2 of the manufacturers.1 Likelihood ratios increased along with antibody levels. Ratios of 80 and 400, depending on the manufacturer, were found at IgG anti-DGP levels 10-fold greater than the cut-off. Pre- and post-test probabilities weren’t calculated.
Positive predictive value rises with antibody levels
A 2013 retrospective study evaluated the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition’s recommendation to forego intestinal biopsy in non-IgA–deficient, symptomatic children and adolescents with positive IgA anti-tTG levels greater than 10 times the cut-off value, positive EMA, and positive HLA-DQ2 or HLA-DQ8.2
Overall, 153 symptomatic patients referred to the gastroenterology unit met these criteria. The age range was 9 months to 14.6 years (mean 4 years). All but 3 of the patients had Marsh 2 or greater lesions with biopsy-confirmed diagnoses of celiac disease. The remaining 3 developed biopsy-positive celiac disease on follow-up. The positive predictive value of combined serologic testing in this small selected patient population was 100%.
A 2013 retrospective study of 2477 symptomatic adults (older than 18 years) who received diagnostic testing for celiac disease at 2 academic institutions in Cleveland, Ohio, evaluated the predictive value of IgA anti-tTG and EMA. Of the patients, 610 (25%) had abnormal serologic tests, and 240 (39%) underwent endoscopy with biopsy.
A total of 50 patients (21%) had biopsy results consistent with celiac disease, defined as a Marsh 3 lesion or greater.3 An IgA anti-tTG level of 118 U/mL (5.9-fold the upper limit of normal on the test) had a positive predictive value of 86.4% with a false-positive value of 2%. An EMA titer greater than 1:160 when IgA anti-tTG was between 21 and 118 U/mL had a positive predictive value of 83%.
Antibody levels 10 times normal show 100% positive predictive value
A 2008 retrospective study of one manufacturer’s IgA anti-tTG serologic test sought to establish the serologic antibody level at which the positive predictive value was 100%.4 Overall, 148 people, 15 years and older, with a positive IgA anti-tTG before biopsy or within 21 days of biopsy were included.
Of the patients biopsied, 139 (93%) had positive biopsies of Marsh 2 or greater and were diagnosed with celiac disease. Using a cut-off of 3.3 and 6.7 times the upper limit of normal, investigators calculated a positive predictive value of 95% and 98%, respectively.
A cut-off of 10 times the upper limit of normal or greater had a positive predictive value of 100%. The highest level of IgA anti-tTG in a patient who didn’t have celiac disease on biopsy was 7.3 times the upper limit of normal.
EVIDENCE SUMMARY
A 2013 Belgian prospective study of 104 non-IgA–deficient adults and children diagnosed with celiac disease and 537 adults and children without celiac disease evaluated the accuracy of 4 manufacturers’ serologic tests for IgA anti-tTG.1 All patients underwent serologic testing followed by a diagnostic biopsy. A Marsh type 3 or greater lesion on duodenal biopsy was considered diagnostic for celiac disease.
Anti-tTG levels greater than 10 times the manufacturer-recommended level for a positive test (cut-off) were associated with a likelihood ratio of 111 to 294 (depending on the manufacturer) of positive biopsy. Post-test probabilities were calculated based on various pretest probabilities using an anti-tTG level of greater than 10 times the cut-off (TABLE1).
Investigators also obtained IgG anti-DGP levels from 2 of the manufacturers.1 Likelihood ratios increased along with antibody levels. Ratios of 80 and 400, depending on the manufacturer, were found at IgG anti-DGP levels 10-fold greater than the cut-off. Pre- and post-test probabilities weren’t calculated.
Positive predictive value rises with antibody levels
A 2013 retrospective study evaluated the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition’s recommendation to forego intestinal biopsy in non-IgA–deficient, symptomatic children and adolescents with positive IgA anti-tTG levels greater than 10 times the cut-off value, positive EMA, and positive HLA-DQ2 or HLA-DQ8.2
Overall, 153 symptomatic patients referred to the gastroenterology unit met these criteria. The age range was 9 months to 14.6 years (mean 4 years). All but 3 of the patients had Marsh 2 or greater lesions with biopsy-confirmed diagnoses of celiac disease. The remaining 3 developed biopsy-positive celiac disease on follow-up. The positive predictive value of combined serologic testing in this small selected patient population was 100%.
A 2013 retrospective study of 2477 symptomatic adults (older than 18 years) who received diagnostic testing for celiac disease at 2 academic institutions in Cleveland, Ohio, evaluated the predictive value of IgA anti-tTG and EMA. Of the patients, 610 (25%) had abnormal serologic tests, and 240 (39%) underwent endoscopy with biopsy.
A total of 50 patients (21%) had biopsy results consistent with celiac disease, defined as a Marsh 3 lesion or greater.3 An IgA anti-tTG level of 118 U/mL (5.9-fold the upper limit of normal on the test) had a positive predictive value of 86.4% with a false-positive value of 2%. An EMA titer greater than 1:160 when IgA anti-tTG was between 21 and 118 U/mL had a positive predictive value of 83%.
Antibody levels 10 times normal show 100% positive predictive value
A 2008 retrospective study of one manufacturer’s IgA anti-tTG serologic test sought to establish the serologic antibody level at which the positive predictive value was 100%.4 Overall, 148 people, 15 years and older, with a positive IgA anti-tTG before biopsy or within 21 days of biopsy were included.
Of the patients biopsied, 139 (93%) had positive biopsies of Marsh 2 or greater and were diagnosed with celiac disease. Using a cut-off of 3.3 and 6.7 times the upper limit of normal, investigators calculated a positive predictive value of 95% and 98%, respectively.
A cut-off of 10 times the upper limit of normal or greater had a positive predictive value of 100%. The highest level of IgA anti-tTG in a patient who didn’t have celiac disease on biopsy was 7.3 times the upper limit of normal.
1. Vermeersch P, Geboes K, Mariën G, et al. Defining thresholds of antibody levels improves diagnosis of celiac disease. Clin Gastroenterol Hepatol. 2013;11:398-403;quiz e32.
2. Klapp G, Masip E, Bolonio M, et al. Celiac disease: The new proposed ESPGHAN diagnostic criteria do work well in a selected population. J Pediatr Gastroenterol Nutr. 2013;56:251-256.
3. Wakim-Fleming J, Pagadala MR, Lemyre MS, et al. Diagnosis of celiac disease in adults based on serology test results, without small-bowel biopsy. Clin Gastroenterol Hepatol. 2013;11:511-516.
4. Hill PG, Holmes GK. Coeliac disease: a biopsy is not always necessary for diagnosis. Aliment Pharmacol Ther. 2008;27:572-577.
1. Vermeersch P, Geboes K, Mariën G, et al. Defining thresholds of antibody levels improves diagnosis of celiac disease. Clin Gastroenterol Hepatol. 2013;11:398-403;quiz e32.
2. Klapp G, Masip E, Bolonio M, et al. Celiac disease: The new proposed ESPGHAN diagnostic criteria do work well in a selected population. J Pediatr Gastroenterol Nutr. 2013;56:251-256.
3. Wakim-Fleming J, Pagadala MR, Lemyre MS, et al. Diagnosis of celiac disease in adults based on serology test results, without small-bowel biopsy. Clin Gastroenterol Hepatol. 2013;11:511-516.
4. Hill PG, Holmes GK. Coeliac disease: a biopsy is not always necessary for diagnosis. Aliment Pharmacol Ther. 2008;27:572-577.
Evidence-based answers from the Family Physicians Inquiries Network
EVIDENCE-BASED ANSWER:
It depends on the antibody levels in the blood work. Symptomatic patients with serologic levels of immunoglobulin A anti-tissue transglutaminase (IgA anti-tTG) or immunoglobulin G anti-deamidated gliadin peptide antibody (IgG anti-DGP) greater than 10 times the upper limits of normal—especially if they also are positive for endomysial antibodies (EMA) and human leukocyte antigen DQ2 (HLA-DQ2 or HLA-DQ8)—may not need an intestinal biopsy to confirm the diagnosis of celiac disease (strength of recommendation [SOR]: B, inconsistent or limited-quality cohort studies).
Patients with antibody levels lower than 10 times the upper limits of normal or who are asymptomatic most likely need an intestinal biopsy to confirm the diagnosis (SOR: B, inconsistent or limited-quality cohort studies).

Does vitamin D without calcium reduce fracture risk?
EVIDENCE SUMMARY
A 2014 meta-analysis of 15 trials (quasi-random and RCT) with a total of 28,271 patients that compared the effect of vitamin D on fracture risk with placebo or no treatment, found no benefit for vitamin D supplementation (TABLE).1 Patients lived in community and nursing home settings and ranged in age from 50 to 85 years; 24% to 100% were female.
Only 3 trials required patients to have had a previous fracture. Exclusions included: diseases affecting bone metabolism, cognitive impairment, drugs affecting bone metabolism (bisphosphonates, selective estrogen receptor modulators, and corticosteroids), renal failure, hypercalcemia, nephrolithiasis, and decreased mobility (recent stroke recovery and Parkinson’s disease).
Formulations of vitamin D included cholecalciferol (D3) 400 to 2000 IU/d for 4 months to 5 years or 100,000 to 500,000 IU every 3 to 12 months for 1 to 5 years; calcifediol (25(OH)D3) 600 IU/d for 4 years; and ergocalciferol (D2) 400 IU/d for 2 years or 3000 to 300,000 IU every 3 to 12 months for 10 months to 3 years.
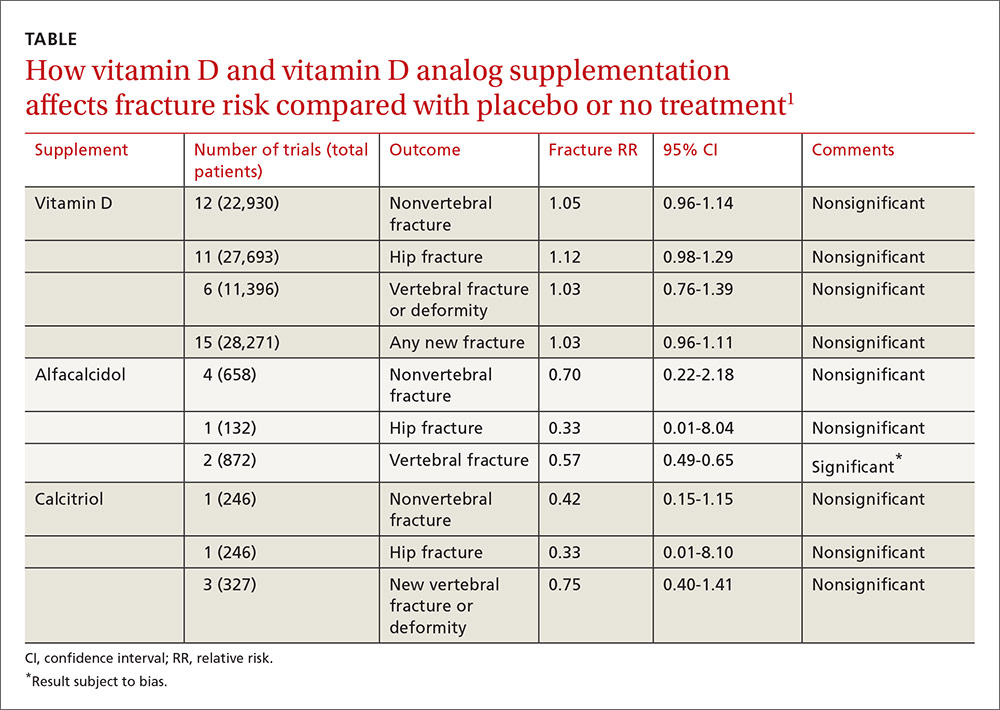
Vitamin D analogs generally have no benefit either
The same meta-analysis compared vitamin D analogs to placebo or no treatment (8 trials, quasi-random and RCT, 1743 patients) on the risk of fracture, again finding no benefit in all but one case. Included patients were mostly by referral to tertiary or university hospitals and outpatient community settings.
Most of the studies included only a small number of patients (about 200), with the largest study having 740 patients. The age range was 50 to 77 years, and 50% to 100% were female. Most of the trials required patients to have osteoporosis or vitamin D deficiency with a previous vertebral deformity on imaging. Study exclusions included osteomalacia, malabsorption, hyperparathyroidism, active kidney stones, history of hypercalciuria, cancer, incurable disease, dementia, severe chronic illness (renal or liver failure), recent stroke or fracture, and drugs that affect bone metabolism.
Vitamin D analogs were given as alfacalcidol (1-alphahydroxyvitamin D3) 0.5 mcg twice daily or 1 mcg/d for 36 weeks to 2 years or calcitriol (1,25-dihydroxyvitamin D3) 0.25 to 1 mcg once or twice daily for one to 3 years. Researchers found a significant reduction in vertebral (but not nonvertebral or hip) fractures with alfacalcidol, but the finding occurred in a single trial that was assessed by the authors of the meta-analysis as subject to bias.
Supplementation doesn’t affect mortality, but does have some side effects
Patients taking vitamin D or an analog with or without calcium showed no difference in risk of death compared with patients taking placebo (29 trials, 71,032 patients; relative risk [RR]=0.97; 95% confidence interval [CI], 0.93-1.01).
Patients taking vitamin D or an analog were more likely than controls to have mild hypercalcemia, with an average increase of 2.7 mmol/L (21 trials, 17,124 patients; RR=2.28; 95% CI, 1.57-3.31). Patients taking calcitriol had the highest risk (4 trials, 988 patients; RR=4.41; 95% CI, 2.14-9.09).
Gastrointestinal adverse effects (4% increase) and renal calculi or mild renal insufficiency (16% increase) were more common with vitamin D and analogs than placebo (GI adverse effects: 15 trials, 47,761 patients; RR=1.04; 95% CI, 1.00-1.08; renal calculi or mild renal insufficiency: 11 trials, 46,548 patients; RR=1.16; 95% CI, 1.02-1.33).
RECOMMENDATIONS
There are no guidelines recommending vitamin D supplementation without calcium to prevent fracture.
1. Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227.
EVIDENCE SUMMARY
A 2014 meta-analysis of 15 trials (quasi-random and RCT) with a total of 28,271 patients that compared the effect of vitamin D on fracture risk with placebo or no treatment, found no benefit for vitamin D supplementation (TABLE).1 Patients lived in community and nursing home settings and ranged in age from 50 to 85 years; 24% to 100% were female.
Only 3 trials required patients to have had a previous fracture. Exclusions included: diseases affecting bone metabolism, cognitive impairment, drugs affecting bone metabolism (bisphosphonates, selective estrogen receptor modulators, and corticosteroids), renal failure, hypercalcemia, nephrolithiasis, and decreased mobility (recent stroke recovery and Parkinson’s disease).
Formulations of vitamin D included cholecalciferol (D3) 400 to 2000 IU/d for 4 months to 5 years or 100,000 to 500,000 IU every 3 to 12 months for 1 to 5 years; calcifediol (25(OH)D3) 600 IU/d for 4 years; and ergocalciferol (D2) 400 IU/d for 2 years or 3000 to 300,000 IU every 3 to 12 months for 10 months to 3 years.
Vitamin D analogs generally have no benefit either
The same meta-analysis compared vitamin D analogs to placebo or no treatment (8 trials, quasi-random and RCT, 1743 patients) on the risk of fracture, again finding no benefit in all but one case. Included patients were mostly by referral to tertiary or university hospitals and outpatient community settings.
Most of the studies included only a small number of patients (about 200), with the largest study having 740 patients. The age range was 50 to 77 years, and 50% to 100% were female. Most of the trials required patients to have osteoporosis or vitamin D deficiency with a previous vertebral deformity on imaging. Study exclusions included osteomalacia, malabsorption, hyperparathyroidism, active kidney stones, history of hypercalciuria, cancer, incurable disease, dementia, severe chronic illness (renal or liver failure), recent stroke or fracture, and drugs that affect bone metabolism.
Vitamin D analogs were given as alfacalcidol (1-alphahydroxyvitamin D3) 0.5 mcg twice daily or 1 mcg/d for 36 weeks to 2 years or calcitriol (1,25-dihydroxyvitamin D3) 0.25 to 1 mcg once or twice daily for one to 3 years. Researchers found a significant reduction in vertebral (but not nonvertebral or hip) fractures with alfacalcidol, but the finding occurred in a single trial that was assessed by the authors of the meta-analysis as subject to bias.
Supplementation doesn’t affect mortality, but does have some side effects
Patients taking vitamin D or an analog with or without calcium showed no difference in risk of death compared with patients taking placebo (29 trials, 71,032 patients; relative risk [RR]=0.97; 95% confidence interval [CI], 0.93-1.01).
Patients taking vitamin D or an analog were more likely than controls to have mild hypercalcemia, with an average increase of 2.7 mmol/L (21 trials, 17,124 patients; RR=2.28; 95% CI, 1.57-3.31). Patients taking calcitriol had the highest risk (4 trials, 988 patients; RR=4.41; 95% CI, 2.14-9.09).
Gastrointestinal adverse effects (4% increase) and renal calculi or mild renal insufficiency (16% increase) were more common with vitamin D and analogs than placebo (GI adverse effects: 15 trials, 47,761 patients; RR=1.04; 95% CI, 1.00-1.08; renal calculi or mild renal insufficiency: 11 trials, 46,548 patients; RR=1.16; 95% CI, 1.02-1.33).
RECOMMENDATIONS
There are no guidelines recommending vitamin D supplementation without calcium to prevent fracture.
EVIDENCE SUMMARY
A 2014 meta-analysis of 15 trials (quasi-random and RCT) with a total of 28,271 patients that compared the effect of vitamin D on fracture risk with placebo or no treatment, found no benefit for vitamin D supplementation (TABLE).1 Patients lived in community and nursing home settings and ranged in age from 50 to 85 years; 24% to 100% were female.
Only 3 trials required patients to have had a previous fracture. Exclusions included: diseases affecting bone metabolism, cognitive impairment, drugs affecting bone metabolism (bisphosphonates, selective estrogen receptor modulators, and corticosteroids), renal failure, hypercalcemia, nephrolithiasis, and decreased mobility (recent stroke recovery and Parkinson’s disease).
Formulations of vitamin D included cholecalciferol (D3) 400 to 2000 IU/d for 4 months to 5 years or 100,000 to 500,000 IU every 3 to 12 months for 1 to 5 years; calcifediol (25(OH)D3) 600 IU/d for 4 years; and ergocalciferol (D2) 400 IU/d for 2 years or 3000 to 300,000 IU every 3 to 12 months for 10 months to 3 years.
Vitamin D analogs generally have no benefit either
The same meta-analysis compared vitamin D analogs to placebo or no treatment (8 trials, quasi-random and RCT, 1743 patients) on the risk of fracture, again finding no benefit in all but one case. Included patients were mostly by referral to tertiary or university hospitals and outpatient community settings.
Most of the studies included only a small number of patients (about 200), with the largest study having 740 patients. The age range was 50 to 77 years, and 50% to 100% were female. Most of the trials required patients to have osteoporosis or vitamin D deficiency with a previous vertebral deformity on imaging. Study exclusions included osteomalacia, malabsorption, hyperparathyroidism, active kidney stones, history of hypercalciuria, cancer, incurable disease, dementia, severe chronic illness (renal or liver failure), recent stroke or fracture, and drugs that affect bone metabolism.
Vitamin D analogs were given as alfacalcidol (1-alphahydroxyvitamin D3) 0.5 mcg twice daily or 1 mcg/d for 36 weeks to 2 years or calcitriol (1,25-dihydroxyvitamin D3) 0.25 to 1 mcg once or twice daily for one to 3 years. Researchers found a significant reduction in vertebral (but not nonvertebral or hip) fractures with alfacalcidol, but the finding occurred in a single trial that was assessed by the authors of the meta-analysis as subject to bias.
Supplementation doesn’t affect mortality, but does have some side effects
Patients taking vitamin D or an analog with or without calcium showed no difference in risk of death compared with patients taking placebo (29 trials, 71,032 patients; relative risk [RR]=0.97; 95% confidence interval [CI], 0.93-1.01).
Patients taking vitamin D or an analog were more likely than controls to have mild hypercalcemia, with an average increase of 2.7 mmol/L (21 trials, 17,124 patients; RR=2.28; 95% CI, 1.57-3.31). Patients taking calcitriol had the highest risk (4 trials, 988 patients; RR=4.41; 95% CI, 2.14-9.09).
Gastrointestinal adverse effects (4% increase) and renal calculi or mild renal insufficiency (16% increase) were more common with vitamin D and analogs than placebo (GI adverse effects: 15 trials, 47,761 patients; RR=1.04; 95% CI, 1.00-1.08; renal calculi or mild renal insufficiency: 11 trials, 46,548 patients; RR=1.16; 95% CI, 1.02-1.33).
RECOMMENDATIONS
There are no guidelines recommending vitamin D supplementation without calcium to prevent fracture.
1. Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227.
1. Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227.
Evidence-based answers from the Family Physicians Inquiries Network
EVIDENCE-BASED ANSWER:
No. Supplemental vitamin D without calcium—in doses averaging as much as 800 IU per day—doesn’t reduce the risk of hip, vertebral, or nonvertebral fractures in postmenopausal women and older men (strength of recommendation [SOR]: A, large, high-quality meta-analysis of randomized or quasi-randomized placebo-controlled trials).
The vitamin D analogs alfacalcidol and calcitriol also don’t reduce hip or nonvertebral fractures (SOR: A, multiple randomized, controlled trials [RCTs]), although alfacalcidol (but not calcitriol) does reduce vertebral fractures by 43% (SOR: B, one RCT and one quasi-randomized trial with potential for bias)
Vitamin D supplementation, with or without calcium, doesn’t affect mortality. It does double the risk of mild hypercalcemia (about 2.7 mmol/L increase), raise the risk of renal calculi or mild renal insufficiency by 16%, and slightly increase (4%) gastrointestinal adverse effects (SOR: A, meta-analysis of RCTs or quasi-randomized trials).

Does breastfeeding affect the risk of childhood obesity?
EVIDENCE SUMMARY
A systematic review and meta-analysis of prospective cohort studies evaluating infant risk factors for childhood obesity found that breastfeeding was associated with a lower risk of obesity.1 The authors identified 10 trials (primarily from the United States and Europe) with more than 76,000 infants that compared the effect of some breastfeeding in the first year to no breastfeeding. Follow-up ranged from 2 to 14 years (median 6 years).
Having ever breastfed decreased the odds of future overweight (BMI >85th percentile) or obesity (BMI >95th percentile) by 15% (adjusted odds ratio [AOR]=0.85; 95% confidence interval [CI], 0.74-0.99).
Subsequent studies suggest increased risk with formula feeding
Three large, prospective, longitudinal cohort studies have been published since the meta-analysis. One, which followed 43,367 term infants in Japan, found that formula feeding before 6 months was associated with increased risk of obesity compared with continuous breastfeeding for 6 months.2 Researchers evaluated weight at 7 years and adjusted for child and maternal factors associated with weight gain (AOR for obesity, formula-fed infants=1.8; 95% CI, 1.3-2.6).
A similar prospective longitudinal cohort study of 2868 infants in Australia analyzed maternal breastfeeding diaries and followed children’s weight to age 20 years.3 Introducing a milk other than breast milk before 6 months of age was linked to increased risk of obesity at age 20 (odds ratio [OR]=1.5; 95% CI, 1.1-1.9).
Finally, in a prospective cohort of 568 children in India, 17% of children who breastfed for fewer than 6 months were above the 90th percentile for weight at age 5 years, compared with 10% of children who were breastfed for at least 18 months.4 The result didn’t reach statistical significance, however (P=.08).
Interventions that increase breastfeeding don’t seem to have an impact
An RCT of an intervention to promote breastfeeding didn’t find any effect on subsequent obesity rates. Researchers in Belarus randomized 17,046 mother-infant pairs to breastfeeding promotion, modeled on the UNICEF Baby-Friendly Hospital Initiative, or usual care. The intervention increased the prevalence of exclusive breastfeeding (at 3 months, 43% vs 6%; at 6 months, 7% vs 0.6%; P values not given).
When researchers evaluated 13,879 children at 11 or 12 years by intention-to-treat analysis, however, they found no difference in mean BMI between the children whose mothers received the intervention and those whose mothers didn’t (BMI difference=0.16; 95% CI, -0.02 to 0.35).5
Introduction of solid foods: Later is better
A systematic review investigated the association between the timing of introducing complementary (solid) foods and childhood obesity in 23 primarily cross-sectional and cohort studies (17 from the United States, Canada, and Europe) with more than 33,000 patients. Follow-up ranged from 4 to 19 years.
Eight of the 21 studies that used BMI as an outcome found that early introduction of complementary foods was associated with a higher childhood BMI. In the largest study (a cohort of 17,561 infants), introducing complementary foods before 3 months was associated with higher risk of obesity at age 5 years than introducing them thereafter (OR=1.3; 95% CI, 1.1-1.6).6 Introduction of solids after 4 months was not associated with childhood obesity.
A systematic review of 10 primarily cross-sectional and cohort studies with more than 3000 infants evaluated associations between the types of complementary foods given and the development of childhood obesity.7 Six of the 10 studies were from Europe and none were from the United States. Follow-up ages ranged from 4 to 11 years.
Outcomes were heterogeneous, and no meta-analysis could be performed. The authors cited 3 studies (total 1174 infants) that found various positive associations between total caloric intake during complementary feeding and childhood obesity. No consistent evidence pointed to increased risk from specific foods or food groups.
Scheduled feeding is linked to rapid infant weight gain
A cohort study evaluated the baseline data of an Australian RCT (on an intervention to promote proper nutrition) in 612 infants, mean age 4.3 months.8 Researchers looked at the relationship between feeding on demand vs scheduled feeding (assessed by parental report) and weight gain in infancy. “Rapid weight gain” was defined as >0.67 change in weight-for-age Z-score between birth and enrollment.
Scheduled feeding was associated with rapid weight gain at a higher rate than feeding on demand (OR=2.3; 95% CI, 1.1-4.6). This study didn’t use childhood obesity as an outcome.
1. Weng SF, Redsell SA, Swift JA, et al. Systematic review and meta-analyses of risk factors for childhood overweight identifiable during infancy. Arch Dis Child. 2012;97:1019-1026.
2. Yamakawa M, Yorifuji T, Inoue S, et al. Breastfeeding and obesity among schoolchildren: a national longitudinal survey in Japan. JAMA Pediatr. 2013;167:919-925.
3. Oddy WH, Mari TA, Huang RC, et al. Early infant feeding and adiposity risk: from infancy to adulthood. Ann Nutr Metab. 2014;64:262-270.
4. Caleyachetty A, Krishnaveni GV, Veena SR, et al. Breast-feeding duration, age of starting solids, and high BMI risk and adiposity in Indian children. Matern Child Nutr. 2013;9:199-216.
5. Martin RM, Patel, R, Kramer MS, et al. Effects of promoting longer-term and exclusive breastfeeding on adiposity and insulin-like growth factor-I at age 11.5 years: a randomized trial. JAMA. 2013;309:1005-1013.
6. Pearce J, Taylor MA, Langley-Evans SC. Timing of the introduction of complementary feeding and risk of childhood obesity: a systematic review. Int J Obes (Lond). 2013;37:1295-1306.
7. Pearce J, Langley-Evans. The types of food introduced during complementary feeding and risk of childhood obesity: a systematic review. Int J Obes (Lond). 2013;37:477-485.
8. Mihrshahi S, Battistutta D, Magarey A, et al. Determinants of rapid weight gain during infancy: baseline results from the NOURISH randomised controlled trial. BMC Pediatr. 2011;11:99.
EVIDENCE SUMMARY
A systematic review and meta-analysis of prospective cohort studies evaluating infant risk factors for childhood obesity found that breastfeeding was associated with a lower risk of obesity.1 The authors identified 10 trials (primarily from the United States and Europe) with more than 76,000 infants that compared the effect of some breastfeeding in the first year to no breastfeeding. Follow-up ranged from 2 to 14 years (median 6 years).
Having ever breastfed decreased the odds of future overweight (BMI >85th percentile) or obesity (BMI >95th percentile) by 15% (adjusted odds ratio [AOR]=0.85; 95% confidence interval [CI], 0.74-0.99).
Subsequent studies suggest increased risk with formula feeding
Three large, prospective, longitudinal cohort studies have been published since the meta-analysis. One, which followed 43,367 term infants in Japan, found that formula feeding before 6 months was associated with increased risk of obesity compared with continuous breastfeeding for 6 months.2 Researchers evaluated weight at 7 years and adjusted for child and maternal factors associated with weight gain (AOR for obesity, formula-fed infants=1.8; 95% CI, 1.3-2.6).
A similar prospective longitudinal cohort study of 2868 infants in Australia analyzed maternal breastfeeding diaries and followed children’s weight to age 20 years.3 Introducing a milk other than breast milk before 6 months of age was linked to increased risk of obesity at age 20 (odds ratio [OR]=1.5; 95% CI, 1.1-1.9).
Finally, in a prospective cohort of 568 children in India, 17% of children who breastfed for fewer than 6 months were above the 90th percentile for weight at age 5 years, compared with 10% of children who were breastfed for at least 18 months.4 The result didn’t reach statistical significance, however (P=.08).
Interventions that increase breastfeeding don’t seem to have an impact
An RCT of an intervention to promote breastfeeding didn’t find any effect on subsequent obesity rates. Researchers in Belarus randomized 17,046 mother-infant pairs to breastfeeding promotion, modeled on the UNICEF Baby-Friendly Hospital Initiative, or usual care. The intervention increased the prevalence of exclusive breastfeeding (at 3 months, 43% vs 6%; at 6 months, 7% vs 0.6%; P values not given).
When researchers evaluated 13,879 children at 11 or 12 years by intention-to-treat analysis, however, they found no difference in mean BMI between the children whose mothers received the intervention and those whose mothers didn’t (BMI difference=0.16; 95% CI, -0.02 to 0.35).5
Introduction of solid foods: Later is better
A systematic review investigated the association between the timing of introducing complementary (solid) foods and childhood obesity in 23 primarily cross-sectional and cohort studies (17 from the United States, Canada, and Europe) with more than 33,000 patients. Follow-up ranged from 4 to 19 years.
Eight of the 21 studies that used BMI as an outcome found that early introduction of complementary foods was associated with a higher childhood BMI. In the largest study (a cohort of 17,561 infants), introducing complementary foods before 3 months was associated with higher risk of obesity at age 5 years than introducing them thereafter (OR=1.3; 95% CI, 1.1-1.6).6 Introduction of solids after 4 months was not associated with childhood obesity.
A systematic review of 10 primarily cross-sectional and cohort studies with more than 3000 infants evaluated associations between the types of complementary foods given and the development of childhood obesity.7 Six of the 10 studies were from Europe and none were from the United States. Follow-up ages ranged from 4 to 11 years.
Outcomes were heterogeneous, and no meta-analysis could be performed. The authors cited 3 studies (total 1174 infants) that found various positive associations between total caloric intake during complementary feeding and childhood obesity. No consistent evidence pointed to increased risk from specific foods or food groups.
Scheduled feeding is linked to rapid infant weight gain
A cohort study evaluated the baseline data of an Australian RCT (on an intervention to promote proper nutrition) in 612 infants, mean age 4.3 months.8 Researchers looked at the relationship between feeding on demand vs scheduled feeding (assessed by parental report) and weight gain in infancy. “Rapid weight gain” was defined as >0.67 change in weight-for-age Z-score between birth and enrollment.
Scheduled feeding was associated with rapid weight gain at a higher rate than feeding on demand (OR=2.3; 95% CI, 1.1-4.6). This study didn’t use childhood obesity as an outcome.
EVIDENCE SUMMARY
A systematic review and meta-analysis of prospective cohort studies evaluating infant risk factors for childhood obesity found that breastfeeding was associated with a lower risk of obesity.1 The authors identified 10 trials (primarily from the United States and Europe) with more than 76,000 infants that compared the effect of some breastfeeding in the first year to no breastfeeding. Follow-up ranged from 2 to 14 years (median 6 years).
Having ever breastfed decreased the odds of future overweight (BMI >85th percentile) or obesity (BMI >95th percentile) by 15% (adjusted odds ratio [AOR]=0.85; 95% confidence interval [CI], 0.74-0.99).
Subsequent studies suggest increased risk with formula feeding
Three large, prospective, longitudinal cohort studies have been published since the meta-analysis. One, which followed 43,367 term infants in Japan, found that formula feeding before 6 months was associated with increased risk of obesity compared with continuous breastfeeding for 6 months.2 Researchers evaluated weight at 7 years and adjusted for child and maternal factors associated with weight gain (AOR for obesity, formula-fed infants=1.8; 95% CI, 1.3-2.6).
A similar prospective longitudinal cohort study of 2868 infants in Australia analyzed maternal breastfeeding diaries and followed children’s weight to age 20 years.3 Introducing a milk other than breast milk before 6 months of age was linked to increased risk of obesity at age 20 (odds ratio [OR]=1.5; 95% CI, 1.1-1.9).
Finally, in a prospective cohort of 568 children in India, 17% of children who breastfed for fewer than 6 months were above the 90th percentile for weight at age 5 years, compared with 10% of children who were breastfed for at least 18 months.4 The result didn’t reach statistical significance, however (P=.08).
Interventions that increase breastfeeding don’t seem to have an impact
An RCT of an intervention to promote breastfeeding didn’t find any effect on subsequent obesity rates. Researchers in Belarus randomized 17,046 mother-infant pairs to breastfeeding promotion, modeled on the UNICEF Baby-Friendly Hospital Initiative, or usual care. The intervention increased the prevalence of exclusive breastfeeding (at 3 months, 43% vs 6%; at 6 months, 7% vs 0.6%; P values not given).
When researchers evaluated 13,879 children at 11 or 12 years by intention-to-treat analysis, however, they found no difference in mean BMI between the children whose mothers received the intervention and those whose mothers didn’t (BMI difference=0.16; 95% CI, -0.02 to 0.35).5
Introduction of solid foods: Later is better
A systematic review investigated the association between the timing of introducing complementary (solid) foods and childhood obesity in 23 primarily cross-sectional and cohort studies (17 from the United States, Canada, and Europe) with more than 33,000 patients. Follow-up ranged from 4 to 19 years.
Eight of the 21 studies that used BMI as an outcome found that early introduction of complementary foods was associated with a higher childhood BMI. In the largest study (a cohort of 17,561 infants), introducing complementary foods before 3 months was associated with higher risk of obesity at age 5 years than introducing them thereafter (OR=1.3; 95% CI, 1.1-1.6).6 Introduction of solids after 4 months was not associated with childhood obesity.
A systematic review of 10 primarily cross-sectional and cohort studies with more than 3000 infants evaluated associations between the types of complementary foods given and the development of childhood obesity.7 Six of the 10 studies were from Europe and none were from the United States. Follow-up ages ranged from 4 to 11 years.
Outcomes were heterogeneous, and no meta-analysis could be performed. The authors cited 3 studies (total 1174 infants) that found various positive associations between total caloric intake during complementary feeding and childhood obesity. No consistent evidence pointed to increased risk from specific foods or food groups.
Scheduled feeding is linked to rapid infant weight gain
A cohort study evaluated the baseline data of an Australian RCT (on an intervention to promote proper nutrition) in 612 infants, mean age 4.3 months.8 Researchers looked at the relationship between feeding on demand vs scheduled feeding (assessed by parental report) and weight gain in infancy. “Rapid weight gain” was defined as >0.67 change in weight-for-age Z-score between birth and enrollment.
Scheduled feeding was associated with rapid weight gain at a higher rate than feeding on demand (OR=2.3; 95% CI, 1.1-4.6). This study didn’t use childhood obesity as an outcome.
1. Weng SF, Redsell SA, Swift JA, et al. Systematic review and meta-analyses of risk factors for childhood overweight identifiable during infancy. Arch Dis Child. 2012;97:1019-1026.
2. Yamakawa M, Yorifuji T, Inoue S, et al. Breastfeeding and obesity among schoolchildren: a national longitudinal survey in Japan. JAMA Pediatr. 2013;167:919-925.
3. Oddy WH, Mari TA, Huang RC, et al. Early infant feeding and adiposity risk: from infancy to adulthood. Ann Nutr Metab. 2014;64:262-270.
4. Caleyachetty A, Krishnaveni GV, Veena SR, et al. Breast-feeding duration, age of starting solids, and high BMI risk and adiposity in Indian children. Matern Child Nutr. 2013;9:199-216.
5. Martin RM, Patel, R, Kramer MS, et al. Effects of promoting longer-term and exclusive breastfeeding on adiposity and insulin-like growth factor-I at age 11.5 years: a randomized trial. JAMA. 2013;309:1005-1013.
6. Pearce J, Taylor MA, Langley-Evans SC. Timing of the introduction of complementary feeding and risk of childhood obesity: a systematic review. Int J Obes (Lond). 2013;37:1295-1306.
7. Pearce J, Langley-Evans. The types of food introduced during complementary feeding and risk of childhood obesity: a systematic review. Int J Obes (Lond). 2013;37:477-485.
8. Mihrshahi S, Battistutta D, Magarey A, et al. Determinants of rapid weight gain during infancy: baseline results from the NOURISH randomised controlled trial. BMC Pediatr. 2011;11:99.
1. Weng SF, Redsell SA, Swift JA, et al. Systematic review and meta-analyses of risk factors for childhood overweight identifiable during infancy. Arch Dis Child. 2012;97:1019-1026.
2. Yamakawa M, Yorifuji T, Inoue S, et al. Breastfeeding and obesity among schoolchildren: a national longitudinal survey in Japan. JAMA Pediatr. 2013;167:919-925.
3. Oddy WH, Mari TA, Huang RC, et al. Early infant feeding and adiposity risk: from infancy to adulthood. Ann Nutr Metab. 2014;64:262-270.
4. Caleyachetty A, Krishnaveni GV, Veena SR, et al. Breast-feeding duration, age of starting solids, and high BMI risk and adiposity in Indian children. Matern Child Nutr. 2013;9:199-216.
5. Martin RM, Patel, R, Kramer MS, et al. Effects of promoting longer-term and exclusive breastfeeding on adiposity and insulin-like growth factor-I at age 11.5 years: a randomized trial. JAMA. 2013;309:1005-1013.
6. Pearce J, Taylor MA, Langley-Evans SC. Timing of the introduction of complementary feeding and risk of childhood obesity: a systematic review. Int J Obes (Lond). 2013;37:1295-1306.
7. Pearce J, Langley-Evans. The types of food introduced during complementary feeding and risk of childhood obesity: a systematic review. Int J Obes (Lond). 2013;37:477-485.
8. Mihrshahi S, Battistutta D, Magarey A, et al. Determinants of rapid weight gain during infancy: baseline results from the NOURISH randomised controlled trial. BMC Pediatr. 2011;11:99.
Evidence-based answers from the Family Physicians Inquiries Network
EVIDENCE-BASED ANSWER:
Yes. Ever having breastfed during the first year of life is associated with a 15% lower risk of overweight or obesity over the next 2 to 14 years compared with never having breastfed. Breastfeeding exclusively for 6 months is associated with a 30% to 50% reduction in risk (strength of recommendation [SOR]: B, meta-analysis of cohort studies and subsequent cohort studies). However, interventions that increase breastfeeding rates during the first 3 to 6 months of life don’t appear to alter body mass index (BMI) at 11 to 12 years of age (SOR: B, randomized clinical trial [RCT]).
Introducing complementary (solid) foods before 3 months is associated with a 30% greater risk of childhood obesity than later introduction; starting solid foods after 4 months isn’t linked to increased obesity. High caloric density of complementary feedings may be associated with greater childhood obesity (SOR: C, systematic reviews of heterogeneous cohort studies).
Scheduled feeding doubles the risk of rapid infant weight gain compared with on-demand feeding, although it’s unclear whether a direct relationship exists between rapid infant weight gain and childhood obesity (SOR: B, cohort study).

Worsening of longstanding headaches, dizziness, visual symptoms • Dx?
THE CASE
A 59-year-old woman from the Democratic Republic of the Congo presented to our family medicine clinic with acute worsening of longstanding headaches. Using a Swahili interpreter, the patient reported a 15-year history of recurrent, intermittent headaches that had been previously diagnosed as migraines. Over the prior 2 months, the headaches had intensified with new symptoms of dizziness, ocular pain, and blurred vision with red flashes. She had no hemiplegia, dysarthria, respiratory symptoms, night sweats, or weight loss. A neurologic exam was negative.
Before immigrating to the United States 14 years earlier, the patient lived for 6 months in a refugee camp in the Congo. At the time of her immigration, she was negative for human immunodeficiency virus (HIV), and a tuberculosis (TB) skin test was positive. A chest x-ray was normal and she had no respiratory symptoms. Shortly after her immigration, she completed 6 months of isoniazid treatment for latent TB.
THE DIAGNOSIS
A computed tomography (CT) scan of the patient’s head demonstrated a large right frontal mass. The differential diagnosis included neoplasm, sarcoidosis, or, less likely, an infectious etiology. A contrast-enhanced magnetic resonance image (MRI) of the brain showed multiple heterogeneous enhancing lesions, with the largest measuring 4.4 cm x 4.6 cm x 3 cm (FIGURE 1). Significant surrounding edema caused a 1.6-cm midline shift, subfalcine herniation, and impending uncal herniation. A CT of the abdomen and chest showed no pulmonary masses or metastatic disease, but did reveal a single 1-cm lymph node in the mediastinum and a 1.2-cm right axillary node.
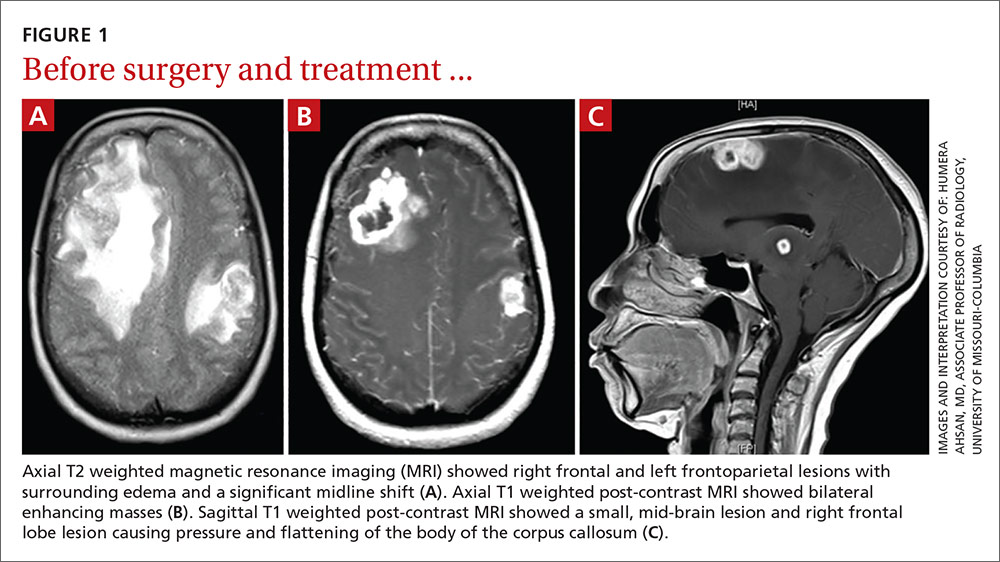
A craniotomy was performed, which confirmed a large mass adhered to the dura. Surgeons removed the mass en bloc; pathology was consistent with a necrotizing granuloma. Acid-fast bacilli (AFB) staining of 3 specimens was negative. Because the tissue was preserved in formalin, mycobacterial cultures could not be obtained. A cerebrospinal fluid analysis showed lymphocytosis and elevated protein, consistent with neurotuberculosis. Blood testing for Mycobacterium tuberculosis with interferon gamma release assay (IGRA) was negative, as was testing for HIV 1 and 2. In addition, induced sputum was AFB-smear negative, as was an M tuberculosis polymerase chain reaction test.
Despite the negative AFB stain and negative IGRA, the patient’s findings were suspicious for TB, so we began to treat her empirically for neurotuberculosis with a 4-drug regimen (isoniazid, rifampin, pyrazinamide, and ethambutol).
In an attempt to confirm the diagnosis of TB and determine sensitivities, we performed a right axillary lymph node biopsy and sent it to the Centers for Disease Control and Prevention (CDC), along with the preserved neural tissue. Using a newly developed technique, the CDC amplified and sequenced mycobacterial DNA from both the central nervous system (CNS) mass and the axillary node, confirming M tuberculosis complex species. Cultures from the axillary node grew pan-sensitive M tuberculosis.
DISCUSSION
About one-third of the world’s population has either active or latent TB.1 In areas where TB is endemic, tuberculomas have accounted for up to 20% of intracranial masses.2 In non-endemic regions, however, they are relatively uncommon. The 3 manifestations of active CNS TB are meningitis, tuberculoma, and abscess.3 The clinical presentation and imaging studies of CNS TB are often indistinguishable from those of patients with malignant neoplasms or metastatic disease. Biopsies may be necessary to distinguish tuberculomas from other intracranial lesions such as pyogenic abscesses or necrotic tumors.4 Mycobacterial cultures were not done on the brain biopsies of our patient because of the high clinical suspicion for neoplasm. Axillary lymph node tissue ultimately confirmed the diagnosis and provided sensitivities.
A diagnosis of CNS tuberculoma without meningitis can be challenging because the clinical presentation is often vague, mild, or even asymptomatic. Constitutional symptoms may include headache, fever, and anorexia.5
In our patient, IGRA testing was also negative. For latent TB, IGRAs are considered to be at least as sensitive as, and considerably more specific than TB skin testing, but their use in CNS TB is less well understood. Studies evaluating IGRA sensitivity for TB meningitis show variable results. In one study, IGRAs were positive in only 50% of culture-confirmed cases of TB meningitis in an HIV-negative population.6
Obtain sputum samples for all patients with extrapulmonary TB
The CDC recommends sputum sampling for all patients with extrapulmonary TB, even in the absence of pulmonary symptoms or radiographic findings, to determine the level of infectivity and potential need for a contact investigation.7
Due to low sensitivity of currently available rapid diagnostic tests and high mortality associated with delayed treatment, initiation of empiric treatment is recommended when the probability of CNS TB is high.5
Treatment duration for CNS tuberculomas is based on one randomized controlled trial,8 a small number of observational studies, a prospective cohort study looking at radiographic resolution,9 and expert opinion. Treatment recommendations often do not distinguish CNS tuberculomas from TB meningitis.10 CNS tuberculomas are commonly treated with a minimum of 12 months of therapy, generally using the same medications and dosages used in the treatment of pulmonary TB, starting with 4 first-line agents: isoniazid, rifampin, pyrazinamide, and ethambutol. Modification of the treatment regimen may be made once sensitivities are available.10
Our patient. After cultures were determined to be pan-sensitive, our patient’s treatment regimen was simplified to rifampin and isoniazid, which she continued for the remainder of her treatment course. Her treatment was discontinued after 18 months when quarterly MRIs showed stabilization of the tuberculomas (FIGURE 2).
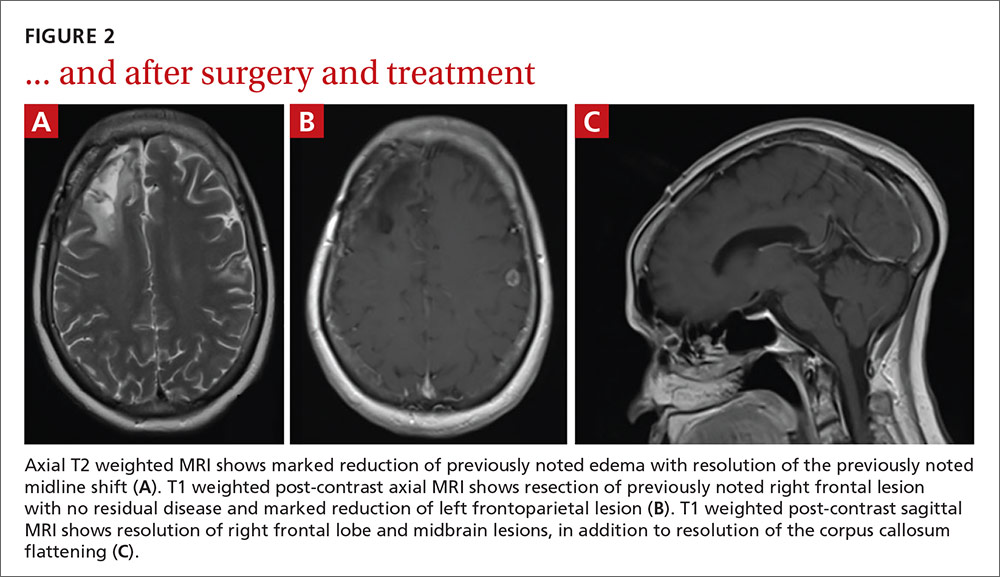
Following her surgery, she was started on levetiractam for seizure prophylaxis. She subsequently had a seizure on 2 occasions when the medication was discontinued or decreased, so we chose to continue it. The patient is asymptomatic from her disease with no residual deficits.
THE TAKEAWAY
A change in headache patterns in a patient over the age of 50 is a red flag that warrants imaging. In patients from countries where TB is endemic,11 consider neurotuberculosis in the differential diagnosis of worsening headaches and progressive neurologic symptoms.
A diagnosis of CNS TB can be difficult and requires a high level of clinical suspicion, but early diagnosis and treatment of neurotuberculosis can minimize the high risk of morbidity and mortality. Treatment for TB shouldn’t be withheld in cases in which there’s a strong clinical suspicion for TB, but for which a definitive diagnosis is still pending.
1. World Health Organization. 10 facts on tuberculosis. Available at: http://www.who.int/features/factfiles/tuberculosis/en/. Accessed September 19, 2014.
2. Dastur DK, Iyer CG. Pathological analysis of 450 intracranial space-occupying lesions. Ind J Cancer. 1966;3:105-115.
3. Chin JH, Mateen FJ. Central nervous system tuberculosis: Challenges and advances in diagnosis and treatment. Curr Infect Dis Rep. 2013;15:631-635.
4. Bayindir C, Mete O, Bilgic B. Retrospective study of 23 pathologically proven cases of central nervous system tuberculomas. Clin Neurol Neurosurg. 2006;108:353-357.
5. Thwaites G, Fisher M, Hemingway C, et al; British Infection Society. British Infection Society guidelines for the diagnosis and treatment of tuberculosis of the central nervous system in adults and children. J Infect. 2009;59:167-187.
6. Simmons CP, Thwaites GE, Quyen NT, et al. Pretreatment intracerebral and peripheral blood immune responses in Vietnamese adults with tuberculous meningitis: diagnostic value and relationship to disease severity and outcome. J Immunol. 2006;176:2007-2014.
7. Centers for Disease Control and Prevention (CDC). Core curriculum on tuberculosis: What the clinician should know. 6th ed. Centers for Disease Control and Prevention, Atlanta, GA; 2013.
8. Rajeswari R, Sivasubramanian S, Balambal R, et al. A controlled clinical trial of short-course chemotherapy for tuberculoma of the brain. Tuber Lung Dis. 1995;76:311-317.
9. Poonnoose SI, Rajshekhar V. Rate of resolution of histologically verified intracranial tuberculomas. Neurosurgery. 2003;53:873-878.
10. American Thoracic Society; CDC; Infectious Diseases Society of America. Treatment of tuberculosis. MMWR Recomm Rep. 2003;52:1-77. Erratum in: MMWR Recomm Rep. 2005;53:1203.
11. Stop TB Partnership. High burden countries. Available at: http://www.stoptb.org/countries/tbdata.asp. Accessed November 7, 2016.
THE CASE
A 59-year-old woman from the Democratic Republic of the Congo presented to our family medicine clinic with acute worsening of longstanding headaches. Using a Swahili interpreter, the patient reported a 15-year history of recurrent, intermittent headaches that had been previously diagnosed as migraines. Over the prior 2 months, the headaches had intensified with new symptoms of dizziness, ocular pain, and blurred vision with red flashes. She had no hemiplegia, dysarthria, respiratory symptoms, night sweats, or weight loss. A neurologic exam was negative.
Before immigrating to the United States 14 years earlier, the patient lived for 6 months in a refugee camp in the Congo. At the time of her immigration, she was negative for human immunodeficiency virus (HIV), and a tuberculosis (TB) skin test was positive. A chest x-ray was normal and she had no respiratory symptoms. Shortly after her immigration, she completed 6 months of isoniazid treatment for latent TB.
THE DIAGNOSIS
A computed tomography (CT) scan of the patient’s head demonstrated a large right frontal mass. The differential diagnosis included neoplasm, sarcoidosis, or, less likely, an infectious etiology. A contrast-enhanced magnetic resonance image (MRI) of the brain showed multiple heterogeneous enhancing lesions, with the largest measuring 4.4 cm x 4.6 cm x 3 cm (FIGURE 1). Significant surrounding edema caused a 1.6-cm midline shift, subfalcine herniation, and impending uncal herniation. A CT of the abdomen and chest showed no pulmonary masses or metastatic disease, but did reveal a single 1-cm lymph node in the mediastinum and a 1.2-cm right axillary node.
A craniotomy was performed, which confirmed a large mass adhered to the dura. Surgeons removed the mass en bloc; pathology was consistent with a necrotizing granuloma. Acid-fast bacilli (AFB) staining of 3 specimens was negative. Because the tissue was preserved in formalin, mycobacterial cultures could not be obtained. A cerebrospinal fluid analysis showed lymphocytosis and elevated protein, consistent with neurotuberculosis. Blood testing for Mycobacterium tuberculosis with interferon gamma release assay (IGRA) was negative, as was testing for HIV 1 and 2. In addition, induced sputum was AFB-smear negative, as was an M tuberculosis polymerase chain reaction test.
Despite the negative AFB stain and negative IGRA, the patient’s findings were suspicious for TB, so we began to treat her empirically for neurotuberculosis with a 4-drug regimen (isoniazid, rifampin, pyrazinamide, and ethambutol).
In an attempt to confirm the diagnosis of TB and determine sensitivities, we performed a right axillary lymph node biopsy and sent it to the Centers for Disease Control and Prevention (CDC), along with the preserved neural tissue. Using a newly developed technique, the CDC amplified and sequenced mycobacterial DNA from both the central nervous system (CNS) mass and the axillary node, confirming M tuberculosis complex species. Cultures from the axillary node grew pan-sensitive M tuberculosis.
DISCUSSION
About one-third of the world’s population has either active or latent TB.1 In areas where TB is endemic, tuberculomas have accounted for up to 20% of intracranial masses.2 In non-endemic regions, however, they are relatively uncommon. The 3 manifestations of active CNS TB are meningitis, tuberculoma, and abscess.3 The clinical presentation and imaging studies of CNS TB are often indistinguishable from those of patients with malignant neoplasms or metastatic disease. Biopsies may be necessary to distinguish tuberculomas from other intracranial lesions such as pyogenic abscesses or necrotic tumors.4 Mycobacterial cultures were not done on the brain biopsies of our patient because of the high clinical suspicion for neoplasm. Axillary lymph node tissue ultimately confirmed the diagnosis and provided sensitivities.
A diagnosis of CNS tuberculoma without meningitis can be challenging because the clinical presentation is often vague, mild, or even asymptomatic. Constitutional symptoms may include headache, fever, and anorexia.5
In our patient, IGRA testing was also negative. For latent TB, IGRAs are considered to be at least as sensitive as, and considerably more specific than TB skin testing, but their use in CNS TB is less well understood. Studies evaluating IGRA sensitivity for TB meningitis show variable results. In one study, IGRAs were positive in only 50% of culture-confirmed cases of TB meningitis in an HIV-negative population.6
Obtain sputum samples for all patients with extrapulmonary TB
The CDC recommends sputum sampling for all patients with extrapulmonary TB, even in the absence of pulmonary symptoms or radiographic findings, to determine the level of infectivity and potential need for a contact investigation.7
Due to low sensitivity of currently available rapid diagnostic tests and high mortality associated with delayed treatment, initiation of empiric treatment is recommended when the probability of CNS TB is high.5
Treatment duration for CNS tuberculomas is based on one randomized controlled trial,8 a small number of observational studies, a prospective cohort study looking at radiographic resolution,9 and expert opinion. Treatment recommendations often do not distinguish CNS tuberculomas from TB meningitis.10 CNS tuberculomas are commonly treated with a minimum of 12 months of therapy, generally using the same medications and dosages used in the treatment of pulmonary TB, starting with 4 first-line agents: isoniazid, rifampin, pyrazinamide, and ethambutol. Modification of the treatment regimen may be made once sensitivities are available.10
Our patient. After cultures were determined to be pan-sensitive, our patient’s treatment regimen was simplified to rifampin and isoniazid, which she continued for the remainder of her treatment course. Her treatment was discontinued after 18 months when quarterly MRIs showed stabilization of the tuberculomas (FIGURE 2).
Following her surgery, she was started on levetiractam for seizure prophylaxis. She subsequently had a seizure on 2 occasions when the medication was discontinued or decreased, so we chose to continue it. The patient is asymptomatic from her disease with no residual deficits.
THE TAKEAWAY
A change in headache patterns in a patient over the age of 50 is a red flag that warrants imaging. In patients from countries where TB is endemic,11 consider neurotuberculosis in the differential diagnosis of worsening headaches and progressive neurologic symptoms.
A diagnosis of CNS TB can be difficult and requires a high level of clinical suspicion, but early diagnosis and treatment of neurotuberculosis can minimize the high risk of morbidity and mortality. Treatment for TB shouldn’t be withheld in cases in which there’s a strong clinical suspicion for TB, but for which a definitive diagnosis is still pending.
THE CASE
A 59-year-old woman from the Democratic Republic of the Congo presented to our family medicine clinic with acute worsening of longstanding headaches. Using a Swahili interpreter, the patient reported a 15-year history of recurrent, intermittent headaches that had been previously diagnosed as migraines. Over the prior 2 months, the headaches had intensified with new symptoms of dizziness, ocular pain, and blurred vision with red flashes. She had no hemiplegia, dysarthria, respiratory symptoms, night sweats, or weight loss. A neurologic exam was negative.
Before immigrating to the United States 14 years earlier, the patient lived for 6 months in a refugee camp in the Congo. At the time of her immigration, she was negative for human immunodeficiency virus (HIV), and a tuberculosis (TB) skin test was positive. A chest x-ray was normal and she had no respiratory symptoms. Shortly after her immigration, she completed 6 months of isoniazid treatment for latent TB.
THE DIAGNOSIS
A computed tomography (CT) scan of the patient’s head demonstrated a large right frontal mass. The differential diagnosis included neoplasm, sarcoidosis, or, less likely, an infectious etiology. A contrast-enhanced magnetic resonance image (MRI) of the brain showed multiple heterogeneous enhancing lesions, with the largest measuring 4.4 cm x 4.6 cm x 3 cm (FIGURE 1). Significant surrounding edema caused a 1.6-cm midline shift, subfalcine herniation, and impending uncal herniation. A CT of the abdomen and chest showed no pulmonary masses or metastatic disease, but did reveal a single 1-cm lymph node in the mediastinum and a 1.2-cm right axillary node.
A craniotomy was performed, which confirmed a large mass adhered to the dura. Surgeons removed the mass en bloc; pathology was consistent with a necrotizing granuloma. Acid-fast bacilli (AFB) staining of 3 specimens was negative. Because the tissue was preserved in formalin, mycobacterial cultures could not be obtained. A cerebrospinal fluid analysis showed lymphocytosis and elevated protein, consistent with neurotuberculosis. Blood testing for Mycobacterium tuberculosis with interferon gamma release assay (IGRA) was negative, as was testing for HIV 1 and 2. In addition, induced sputum was AFB-smear negative, as was an M tuberculosis polymerase chain reaction test.
Despite the negative AFB stain and negative IGRA, the patient’s findings were suspicious for TB, so we began to treat her empirically for neurotuberculosis with a 4-drug regimen (isoniazid, rifampin, pyrazinamide, and ethambutol).
In an attempt to confirm the diagnosis of TB and determine sensitivities, we performed a right axillary lymph node biopsy and sent it to the Centers for Disease Control and Prevention (CDC), along with the preserved neural tissue. Using a newly developed technique, the CDC amplified and sequenced mycobacterial DNA from both the central nervous system (CNS) mass and the axillary node, confirming M tuberculosis complex species. Cultures from the axillary node grew pan-sensitive M tuberculosis.
DISCUSSION
About one-third of the world’s population has either active or latent TB.1 In areas where TB is endemic, tuberculomas have accounted for up to 20% of intracranial masses.2 In non-endemic regions, however, they are relatively uncommon. The 3 manifestations of active CNS TB are meningitis, tuberculoma, and abscess.3 The clinical presentation and imaging studies of CNS TB are often indistinguishable from those of patients with malignant neoplasms or metastatic disease. Biopsies may be necessary to distinguish tuberculomas from other intracranial lesions such as pyogenic abscesses or necrotic tumors.4 Mycobacterial cultures were not done on the brain biopsies of our patient because of the high clinical suspicion for neoplasm. Axillary lymph node tissue ultimately confirmed the diagnosis and provided sensitivities.
A diagnosis of CNS tuberculoma without meningitis can be challenging because the clinical presentation is often vague, mild, or even asymptomatic. Constitutional symptoms may include headache, fever, and anorexia.5
In our patient, IGRA testing was also negative. For latent TB, IGRAs are considered to be at least as sensitive as, and considerably more specific than TB skin testing, but their use in CNS TB is less well understood. Studies evaluating IGRA sensitivity for TB meningitis show variable results. In one study, IGRAs were positive in only 50% of culture-confirmed cases of TB meningitis in an HIV-negative population.6
Obtain sputum samples for all patients with extrapulmonary TB
The CDC recommends sputum sampling for all patients with extrapulmonary TB, even in the absence of pulmonary symptoms or radiographic findings, to determine the level of infectivity and potential need for a contact investigation.7
Due to low sensitivity of currently available rapid diagnostic tests and high mortality associated with delayed treatment, initiation of empiric treatment is recommended when the probability of CNS TB is high.5
Treatment duration for CNS tuberculomas is based on one randomized controlled trial,8 a small number of observational studies, a prospective cohort study looking at radiographic resolution,9 and expert opinion. Treatment recommendations often do not distinguish CNS tuberculomas from TB meningitis.10 CNS tuberculomas are commonly treated with a minimum of 12 months of therapy, generally using the same medications and dosages used in the treatment of pulmonary TB, starting with 4 first-line agents: isoniazid, rifampin, pyrazinamide, and ethambutol. Modification of the treatment regimen may be made once sensitivities are available.10
Our patient. After cultures were determined to be pan-sensitive, our patient’s treatment regimen was simplified to rifampin and isoniazid, which she continued for the remainder of her treatment course. Her treatment was discontinued after 18 months when quarterly MRIs showed stabilization of the tuberculomas (FIGURE 2).
Following her surgery, she was started on levetiractam for seizure prophylaxis. She subsequently had a seizure on 2 occasions when the medication was discontinued or decreased, so we chose to continue it. The patient is asymptomatic from her disease with no residual deficits.
THE TAKEAWAY
A change in headache patterns in a patient over the age of 50 is a red flag that warrants imaging. In patients from countries where TB is endemic,11 consider neurotuberculosis in the differential diagnosis of worsening headaches and progressive neurologic symptoms.
A diagnosis of CNS TB can be difficult and requires a high level of clinical suspicion, but early diagnosis and treatment of neurotuberculosis can minimize the high risk of morbidity and mortality. Treatment for TB shouldn’t be withheld in cases in which there’s a strong clinical suspicion for TB, but for which a definitive diagnosis is still pending.
1. World Health Organization. 10 facts on tuberculosis. Available at: http://www.who.int/features/factfiles/tuberculosis/en/. Accessed September 19, 2014.
2. Dastur DK, Iyer CG. Pathological analysis of 450 intracranial space-occupying lesions. Ind J Cancer. 1966;3:105-115.
3. Chin JH, Mateen FJ. Central nervous system tuberculosis: Challenges and advances in diagnosis and treatment. Curr Infect Dis Rep. 2013;15:631-635.
4. Bayindir C, Mete O, Bilgic B. Retrospective study of 23 pathologically proven cases of central nervous system tuberculomas. Clin Neurol Neurosurg. 2006;108:353-357.
5. Thwaites G, Fisher M, Hemingway C, et al; British Infection Society. British Infection Society guidelines for the diagnosis and treatment of tuberculosis of the central nervous system in adults and children. J Infect. 2009;59:167-187.
6. Simmons CP, Thwaites GE, Quyen NT, et al. Pretreatment intracerebral and peripheral blood immune responses in Vietnamese adults with tuberculous meningitis: diagnostic value and relationship to disease severity and outcome. J Immunol. 2006;176:2007-2014.
7. Centers for Disease Control and Prevention (CDC). Core curriculum on tuberculosis: What the clinician should know. 6th ed. Centers for Disease Control and Prevention, Atlanta, GA; 2013.
8. Rajeswari R, Sivasubramanian S, Balambal R, et al. A controlled clinical trial of short-course chemotherapy for tuberculoma of the brain. Tuber Lung Dis. 1995;76:311-317.
9. Poonnoose SI, Rajshekhar V. Rate of resolution of histologically verified intracranial tuberculomas. Neurosurgery. 2003;53:873-878.
10. American Thoracic Society; CDC; Infectious Diseases Society of America. Treatment of tuberculosis. MMWR Recomm Rep. 2003;52:1-77. Erratum in: MMWR Recomm Rep. 2005;53:1203.
11. Stop TB Partnership. High burden countries. Available at: http://www.stoptb.org/countries/tbdata.asp. Accessed November 7, 2016.
1. World Health Organization. 10 facts on tuberculosis. Available at: http://www.who.int/features/factfiles/tuberculosis/en/. Accessed September 19, 2014.
2. Dastur DK, Iyer CG. Pathological analysis of 450 intracranial space-occupying lesions. Ind J Cancer. 1966;3:105-115.
3. Chin JH, Mateen FJ. Central nervous system tuberculosis: Challenges and advances in diagnosis and treatment. Curr Infect Dis Rep. 2013;15:631-635.
4. Bayindir C, Mete O, Bilgic B. Retrospective study of 23 pathologically proven cases of central nervous system tuberculomas. Clin Neurol Neurosurg. 2006;108:353-357.
5. Thwaites G, Fisher M, Hemingway C, et al; British Infection Society. British Infection Society guidelines for the diagnosis and treatment of tuberculosis of the central nervous system in adults and children. J Infect. 2009;59:167-187.
6. Simmons CP, Thwaites GE, Quyen NT, et al. Pretreatment intracerebral and peripheral blood immune responses in Vietnamese adults with tuberculous meningitis: diagnostic value and relationship to disease severity and outcome. J Immunol. 2006;176:2007-2014.
7. Centers for Disease Control and Prevention (CDC). Core curriculum on tuberculosis: What the clinician should know. 6th ed. Centers for Disease Control and Prevention, Atlanta, GA; 2013.
8. Rajeswari R, Sivasubramanian S, Balambal R, et al. A controlled clinical trial of short-course chemotherapy for tuberculoma of the brain. Tuber Lung Dis. 1995;76:311-317.
9. Poonnoose SI, Rajshekhar V. Rate of resolution of histologically verified intracranial tuberculomas. Neurosurgery. 2003;53:873-878.
10. American Thoracic Society; CDC; Infectious Diseases Society of America. Treatment of tuberculosis. MMWR Recomm Rep. 2003;52:1-77. Erratum in: MMWR Recomm Rep. 2005;53:1203.
11. Stop TB Partnership. High burden countries. Available at: http://www.stoptb.org/countries/tbdata.asp. Accessed November 7, 2016.

Two men with dyspnea, enlarged lymph nodes • Dx?
CASE 1
A 50-year-old man sought care for progressive dyspnea on exertion, abdominal bloating, and bilateral leg edema. He had hypertension that was being treated with atenolol, nifedipine, and enalapril. On examination, his blood pressure was 157/80 mm Hg and his heart rate was 50 beats/min. Jugular venous pressure was grossly elevated with occasional cannon A waves. The patient also had decreased breath sounds in both lower lung zones and moderate pitting edema up to the knees. A chest x-ray showed a small bilateral pleural effusion and no cardiomegaly. An electrocardiogram revealed complete atrioventricular (AV) block with a ventricular response of 50 beats/min. Computed tomography (CT) angiography revealed no evidence of a pulmonary embolus, but did show several enlarged (up to 3.5 cm in diameter) lymph nodes in the upper and middle mediastinum (FIGURE 1). We performed an echocardiogram.
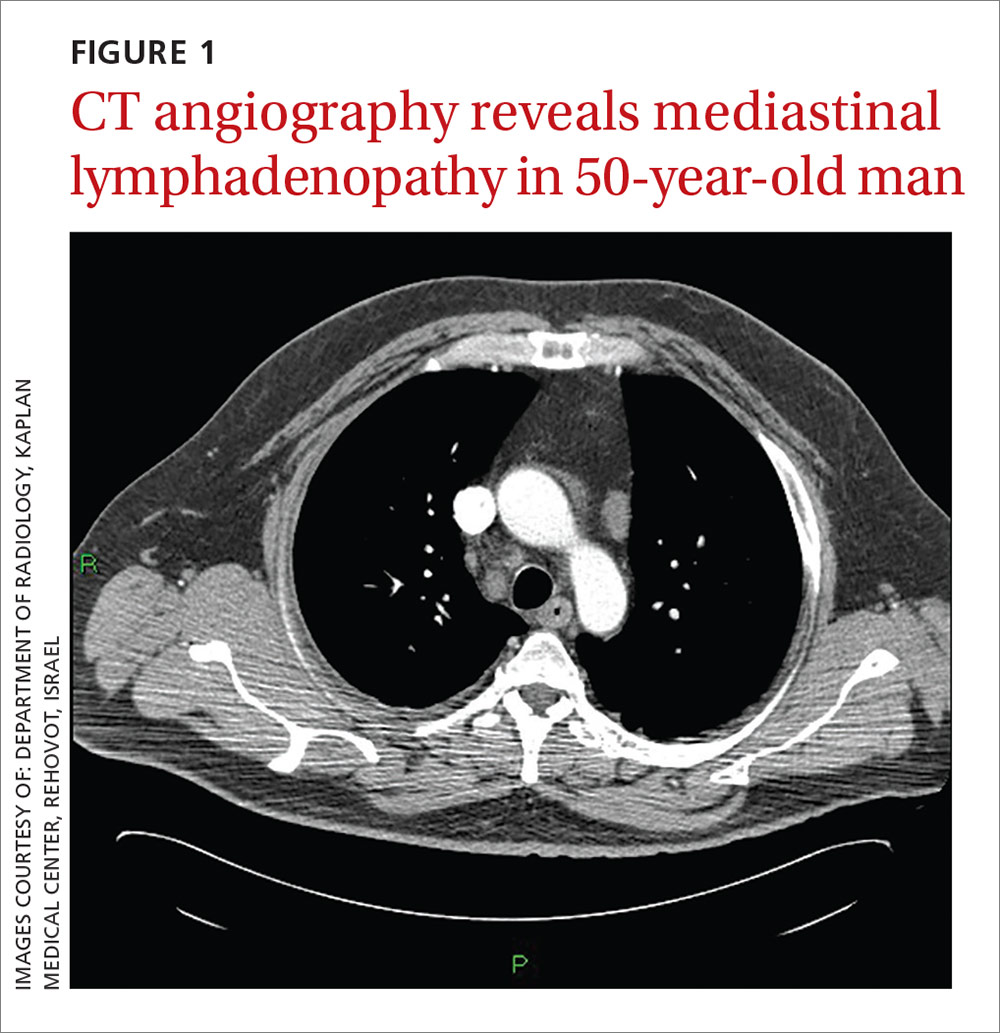
CASE 2
A 79-year-old man with hypertension and diabetes presented to our medical center with acute dyspnea. During the physical examination, we noted bilateral diminished breath sounds with expiratory wheezes and an irregular pulse. Chest x-ray showed mild pulmonary congestion. A chest CT demonstrated bilateral small pleural effusions and multiple enlarged mediastinal lymph nodes with a maximal diameter of 2.4 cm (FIGURE 2A). One week later, the patient’s shortness of breath increased and he was hospitalized. A chest x-ray at that time showed moderate pulmonary congestion, so we performed an echocardiogram.
THE DIAGNOSIS
The echocardiogram for the 50-year-old patient in Case 1 revealed a mildly dilated left ventricle with normal systolic function, diastolic left ventricular (LV) dysfunction, moderate tricuspid regurgitation, and mild pulmonary hypertension. Extensive testing for malignancy and tuberculosis was negative.
For the 79-year-old patient in Case 2, echocardiography demonstrated concentric LV hypertrophy, mild dilatation of the left ventricle, normal LV systolic function, LV diastolic dysfunction with elevated LV diastolic filling pressure, and mild-to-moderate pulmonary hypertension.
Based on these results, we diagnosed both patients with diastolic heart failure. The patient in the second case had features of cardiac asthma, as well. Both patients had also developed reversible mediastinal lymphadenopathy (MLN), of which the diastolic heart failure was the only apparent cause. In both cases, radiologists did not note any suspicious findings for malignancy beyond the MLN.
DISCUSSION
Systolic heart failure has been previously recognized as a cause of MLN.1,2 Other causes of MLN include sarcoidosis, various malignancies, pulmonary infections, and occupational lung diseases. There are, however, no reports of MLN in patients with diastolic heart failure.
Heart failure and MLN. Slanetz et al reported one series of 46 patients who had undergone CT of the chest during periods of congestive heart failure (CHF).1 There was mediastinal lymph node enlargement in 55% of these patients. In a subset of 17 patients who had elevated capillary wedge pressure, 82% had some degree of lymphadenopathy.
Erly et al2 retrospectively studied 44 patients who had a thoracic CT performed before cardiac transplantation. Twenty-nine (66%) had at least one enlarged mediastinal lymph node (>1 cm). Eighty-one percent of patients with an ejection fraction <35% had lymphadenopathy, while none of the patients with an ejection fraction >35% had lymphadenopathy. Most enlarged lymph nodes were pretracheal, with a mean short axis diameter of 1.3 cm.
However, Storto et al reported that an association between CHF and MLN was not found in 7 patients undergoing high-resolution CT imaging.3 There are also cases of MLN in patients with pulmonary hypertension without systolic dysfunction.4
Chabbert et al studied 31 consecutive patients with subacute left heart failure (mean ejection fraction, 39%).5 Enlarged mediastinal lymph nodes were present in 13 patients (42%). Other radiographic features included blurred contour of the lymph nodes in 5 patients (16%) and hazy mediastinal fat in one patient (3%). Follow-up CT showed a significant decrease in the size of the lymph nodes in 8 of 13 patients (62%) following initiation of treatment.
Heart failure and malignancy. A PubMed search with the keywords “diastolic dysfunction” and “lymphoma” found 7 references in the English language. There is a report of 125 survivors of childhood lymphomas treated with mediastinal radiotherapy and anthracyclines,6 another of 44 children treated for acute lymphoblastic leukemia and Hodgkin’s lymphoma7, a report of 294 patients who had received mediastinal irradiation for the treatment of Hodgkin’s disease,8 and another of 106 survivors of non-Hodgkin’s and Hodgkin’s lymphomas.9 None of these reports, however, made any mention of mediastinal lymphadenopathy.
What caused the lymphadenopathy in our patients?
Our 2 patients had volume overload due to diastolic dysfunction with elevated LV end diastolic pressure. Our first patient also had a loss of AV synchronization—which was reversible upon pacemaker insertion—that probably exacerbated the heart failure.
The mechanism for the lymphadenopathy is not clear, but may be due to cardiogenic pulmonary edema causing distension of the pulmonary lymphatic vessels and pulmonary hypertension. In a study of patients with severe systolic dysfunction undergoing evaluation for cardiac transplant, there was a relationship (albeit weak), between MLN and mitral regurgitation, tricuspid regurgitation, elevated mean pulmonary artery pressure, elevated pulmonary capillary wedge pressure, and elevated right atrial pressure.10
How to accurately detect and treat MLN
MLN may be detected by chest x-ray, CT, magnetic resonance imaging, or endoscopic ultrasound examinations. The clinical situation will dictate the imaging modality used. Keep in mind that it is difficult to make a comparison between a finding of lymphadenopathy on one modality and another, especially if one is looking for a change in size.
If clinically appropriate, a trial of diuretics, such as intravenous (IV) furosemide 80 mg,
Our patients. The 50-year-old man in Case 1 responded well to 80 mg of IV furosemide after one hour and improved further upon receipt of a pacemaker the next day. A repeat thoracic CT one month later showed complete resolution of the MLN.
The 79-year-old man in Case 2 also received 80 mg of IV furosemide and improved within 3 hours. A month later, a repeat thoracic CT showed a significant reduction in the size of all the enlarged lymph nodes (FIGURE 2B).
THE TAKEAWAY
The importance of these 2 cases is that they show that heart failure—even diastolic alone—can produce enlarged mediastinal lymph nodes. In patients with heart failure in whom unexpected MLN is detected, consideration should be given to performing a repeat imaging examination after the administration of diuretics.
1. Slanetz PJ, Truong M, Shepard JA, et al. Mediastinal lymphadenopathy and hazy mediastinal fat: new CT findings of congestive heart failure. AJR Am J Roentgenol. 1998;171:1307-1309.
2. Erly WK, Borders RJ, Outwater EK, et al. Location, size, and distribution of mediastinal lymph node enlargement in chronic congestive heart failure. J Comput Assist Tomogr. 2003;27:485-489.
3. Storto ML, Kee ST, Golden JA, et al. Hydrostatic pulmonary edema: high-resolution CT findings. AJR Am J Roentgenol. 1995;165:817-820.
4. Moua T, Levin DL, Carmona EM, et al. Frequency of mediastinal lymphadenopathy in patients with idiopathic pulmonary arterial hypertension. Chest. 2013;143:344-348.
5. Chabbert V, Canevet G, Baixas C, et al. Mediastinal lymphadenopathy in congestive heart failure: a sequential CT evaluation with clinical and echocardiographic correlations. Eur Radiol. 2004;14:881-889.
6. Christiansen JR, Hamre H, Massey R, et al. Left ventricular function in long-term survivors of childhood lymphoma. Am J Cardiol. 2014;114:483-490.
7. Krawczuk-Rybak M, Dakowicz L, Hryniewicz A, et al. Cardiac function in survivors of acute lymphoblastic leukaemia and Hodgkin’s lymphoma. J Paediatr Child Health. 2011;47:455-459.
8. Heidenreich PA, Hancock SL, Vagelos RH, et al. Diastolic dysfunction after mediastinal irradiation. Am Heart J. 2005;150:977-982.
9. Elbl L, Vasova I, Tomaskova I, et al. Cardiopulmonary exercise testing in the evaluation of functional capacity after treatment of lymphomas in adults. Leuk Lymphoma. 2006;47:843-851.
10. Pastis NJ Jr, Van Bakel AB, Brand TM, et al. Mediastinal lymphadenopathy in patients undergoing cardiac transplant evaluation. Chest. 2011;139:1451-1457.
CASE 1
A 50-year-old man sought care for progressive dyspnea on exertion, abdominal bloating, and bilateral leg edema. He had hypertension that was being treated with atenolol, nifedipine, and enalapril. On examination, his blood pressure was 157/80 mm Hg and his heart rate was 50 beats/min. Jugular venous pressure was grossly elevated with occasional cannon A waves. The patient also had decreased breath sounds in both lower lung zones and moderate pitting edema up to the knees. A chest x-ray showed a small bilateral pleural effusion and no cardiomegaly. An electrocardiogram revealed complete atrioventricular (AV) block with a ventricular response of 50 beats/min. Computed tomography (CT) angiography revealed no evidence of a pulmonary embolus, but did show several enlarged (up to 3.5 cm in diameter) lymph nodes in the upper and middle mediastinum (FIGURE 1). We performed an echocardiogram.
CASE 2
A 79-year-old man with hypertension and diabetes presented to our medical center with acute dyspnea. During the physical examination, we noted bilateral diminished breath sounds with expiratory wheezes and an irregular pulse. Chest x-ray showed mild pulmonary congestion. A chest CT demonstrated bilateral small pleural effusions and multiple enlarged mediastinal lymph nodes with a maximal diameter of 2.4 cm (FIGURE 2A). One week later, the patient’s shortness of breath increased and he was hospitalized. A chest x-ray at that time showed moderate pulmonary congestion, so we performed an echocardiogram.
THE DIAGNOSIS
The echocardiogram for the 50-year-old patient in Case 1 revealed a mildly dilated left ventricle with normal systolic function, diastolic left ventricular (LV) dysfunction, moderate tricuspid regurgitation, and mild pulmonary hypertension. Extensive testing for malignancy and tuberculosis was negative.
For the 79-year-old patient in Case 2, echocardiography demonstrated concentric LV hypertrophy, mild dilatation of the left ventricle, normal LV systolic function, LV diastolic dysfunction with elevated LV diastolic filling pressure, and mild-to-moderate pulmonary hypertension.
Based on these results, we diagnosed both patients with diastolic heart failure. The patient in the second case had features of cardiac asthma, as well. Both patients had also developed reversible mediastinal lymphadenopathy (MLN), of which the diastolic heart failure was the only apparent cause. In both cases, radiologists did not note any suspicious findings for malignancy beyond the MLN.
DISCUSSION
Systolic heart failure has been previously recognized as a cause of MLN.1,2 Other causes of MLN include sarcoidosis, various malignancies, pulmonary infections, and occupational lung diseases. There are, however, no reports of MLN in patients with diastolic heart failure.
Heart failure and MLN. Slanetz et al reported one series of 46 patients who had undergone CT of the chest during periods of congestive heart failure (CHF).1 There was mediastinal lymph node enlargement in 55% of these patients. In a subset of 17 patients who had elevated capillary wedge pressure, 82% had some degree of lymphadenopathy.
Erly et al2 retrospectively studied 44 patients who had a thoracic CT performed before cardiac transplantation. Twenty-nine (66%) had at least one enlarged mediastinal lymph node (>1 cm). Eighty-one percent of patients with an ejection fraction <35% had lymphadenopathy, while none of the patients with an ejection fraction >35% had lymphadenopathy. Most enlarged lymph nodes were pretracheal, with a mean short axis diameter of 1.3 cm.
However, Storto et al reported that an association between CHF and MLN was not found in 7 patients undergoing high-resolution CT imaging.3 There are also cases of MLN in patients with pulmonary hypertension without systolic dysfunction.4
Chabbert et al studied 31 consecutive patients with subacute left heart failure (mean ejection fraction, 39%).5 Enlarged mediastinal lymph nodes were present in 13 patients (42%). Other radiographic features included blurred contour of the lymph nodes in 5 patients (16%) and hazy mediastinal fat in one patient (3%). Follow-up CT showed a significant decrease in the size of the lymph nodes in 8 of 13 patients (62%) following initiation of treatment.
Heart failure and malignancy. A PubMed search with the keywords “diastolic dysfunction” and “lymphoma” found 7 references in the English language. There is a report of 125 survivors of childhood lymphomas treated with mediastinal radiotherapy and anthracyclines,6 another of 44 children treated for acute lymphoblastic leukemia and Hodgkin’s lymphoma7, a report of 294 patients who had received mediastinal irradiation for the treatment of Hodgkin’s disease,8 and another of 106 survivors of non-Hodgkin’s and Hodgkin’s lymphomas.9 None of these reports, however, made any mention of mediastinal lymphadenopathy.
What caused the lymphadenopathy in our patients?
Our 2 patients had volume overload due to diastolic dysfunction with elevated LV end diastolic pressure. Our first patient also had a loss of AV synchronization—which was reversible upon pacemaker insertion—that probably exacerbated the heart failure.
The mechanism for the lymphadenopathy is not clear, but may be due to cardiogenic pulmonary edema causing distension of the pulmonary lymphatic vessels and pulmonary hypertension. In a study of patients with severe systolic dysfunction undergoing evaluation for cardiac transplant, there was a relationship (albeit weak), between MLN and mitral regurgitation, tricuspid regurgitation, elevated mean pulmonary artery pressure, elevated pulmonary capillary wedge pressure, and elevated right atrial pressure.10
How to accurately detect and treat MLN
MLN may be detected by chest x-ray, CT, magnetic resonance imaging, or endoscopic ultrasound examinations. The clinical situation will dictate the imaging modality used. Keep in mind that it is difficult to make a comparison between a finding of lymphadenopathy on one modality and another, especially if one is looking for a change in size.
If clinically appropriate, a trial of diuretics, such as intravenous (IV) furosemide 80 mg,
Our patients. The 50-year-old man in Case 1 responded well to 80 mg of IV furosemide after one hour and improved further upon receipt of a pacemaker the next day. A repeat thoracic CT one month later showed complete resolution of the MLN.
The 79-year-old man in Case 2 also received 80 mg of IV furosemide and improved within 3 hours. A month later, a repeat thoracic CT showed a significant reduction in the size of all the enlarged lymph nodes (FIGURE 2B).
THE TAKEAWAY
The importance of these 2 cases is that they show that heart failure—even diastolic alone—can produce enlarged mediastinal lymph nodes. In patients with heart failure in whom unexpected MLN is detected, consideration should be given to performing a repeat imaging examination after the administration of diuretics.
CASE 1
A 50-year-old man sought care for progressive dyspnea on exertion, abdominal bloating, and bilateral leg edema. He had hypertension that was being treated with atenolol, nifedipine, and enalapril. On examination, his blood pressure was 157/80 mm Hg and his heart rate was 50 beats/min. Jugular venous pressure was grossly elevated with occasional cannon A waves. The patient also had decreased breath sounds in both lower lung zones and moderate pitting edema up to the knees. A chest x-ray showed a small bilateral pleural effusion and no cardiomegaly. An electrocardiogram revealed complete atrioventricular (AV) block with a ventricular response of 50 beats/min. Computed tomography (CT) angiography revealed no evidence of a pulmonary embolus, but did show several enlarged (up to 3.5 cm in diameter) lymph nodes in the upper and middle mediastinum (FIGURE 1). We performed an echocardiogram.
CASE 2
A 79-year-old man with hypertension and diabetes presented to our medical center with acute dyspnea. During the physical examination, we noted bilateral diminished breath sounds with expiratory wheezes and an irregular pulse. Chest x-ray showed mild pulmonary congestion. A chest CT demonstrated bilateral small pleural effusions and multiple enlarged mediastinal lymph nodes with a maximal diameter of 2.4 cm (FIGURE 2A). One week later, the patient’s shortness of breath increased and he was hospitalized. A chest x-ray at that time showed moderate pulmonary congestion, so we performed an echocardiogram.
THE DIAGNOSIS
The echocardiogram for the 50-year-old patient in Case 1 revealed a mildly dilated left ventricle with normal systolic function, diastolic left ventricular (LV) dysfunction, moderate tricuspid regurgitation, and mild pulmonary hypertension. Extensive testing for malignancy and tuberculosis was negative.
For the 79-year-old patient in Case 2, echocardiography demonstrated concentric LV hypertrophy, mild dilatation of the left ventricle, normal LV systolic function, LV diastolic dysfunction with elevated LV diastolic filling pressure, and mild-to-moderate pulmonary hypertension.
Based on these results, we diagnosed both patients with diastolic heart failure. The patient in the second case had features of cardiac asthma, as well. Both patients had also developed reversible mediastinal lymphadenopathy (MLN), of which the diastolic heart failure was the only apparent cause. In both cases, radiologists did not note any suspicious findings for malignancy beyond the MLN.
DISCUSSION
Systolic heart failure has been previously recognized as a cause of MLN.1,2 Other causes of MLN include sarcoidosis, various malignancies, pulmonary infections, and occupational lung diseases. There are, however, no reports of MLN in patients with diastolic heart failure.
Heart failure and MLN. Slanetz et al reported one series of 46 patients who had undergone CT of the chest during periods of congestive heart failure (CHF).1 There was mediastinal lymph node enlargement in 55% of these patients. In a subset of 17 patients who had elevated capillary wedge pressure, 82% had some degree of lymphadenopathy.
Erly et al2 retrospectively studied 44 patients who had a thoracic CT performed before cardiac transplantation. Twenty-nine (66%) had at least one enlarged mediastinal lymph node (>1 cm). Eighty-one percent of patients with an ejection fraction <35% had lymphadenopathy, while none of the patients with an ejection fraction >35% had lymphadenopathy. Most enlarged lymph nodes were pretracheal, with a mean short axis diameter of 1.3 cm.
However, Storto et al reported that an association between CHF and MLN was not found in 7 patients undergoing high-resolution CT imaging.3 There are also cases of MLN in patients with pulmonary hypertension without systolic dysfunction.4
Chabbert et al studied 31 consecutive patients with subacute left heart failure (mean ejection fraction, 39%).5 Enlarged mediastinal lymph nodes were present in 13 patients (42%). Other radiographic features included blurred contour of the lymph nodes in 5 patients (16%) and hazy mediastinal fat in one patient (3%). Follow-up CT showed a significant decrease in the size of the lymph nodes in 8 of 13 patients (62%) following initiation of treatment.
Heart failure and malignancy. A PubMed search with the keywords “diastolic dysfunction” and “lymphoma” found 7 references in the English language. There is a report of 125 survivors of childhood lymphomas treated with mediastinal radiotherapy and anthracyclines,6 another of 44 children treated for acute lymphoblastic leukemia and Hodgkin’s lymphoma7, a report of 294 patients who had received mediastinal irradiation for the treatment of Hodgkin’s disease,8 and another of 106 survivors of non-Hodgkin’s and Hodgkin’s lymphomas.9 None of these reports, however, made any mention of mediastinal lymphadenopathy.
What caused the lymphadenopathy in our patients?
Our 2 patients had volume overload due to diastolic dysfunction with elevated LV end diastolic pressure. Our first patient also had a loss of AV synchronization—which was reversible upon pacemaker insertion—that probably exacerbated the heart failure.
The mechanism for the lymphadenopathy is not clear, but may be due to cardiogenic pulmonary edema causing distension of the pulmonary lymphatic vessels and pulmonary hypertension. In a study of patients with severe systolic dysfunction undergoing evaluation for cardiac transplant, there was a relationship (albeit weak), between MLN and mitral regurgitation, tricuspid regurgitation, elevated mean pulmonary artery pressure, elevated pulmonary capillary wedge pressure, and elevated right atrial pressure.10
How to accurately detect and treat MLN
MLN may be detected by chest x-ray, CT, magnetic resonance imaging, or endoscopic ultrasound examinations. The clinical situation will dictate the imaging modality used. Keep in mind that it is difficult to make a comparison between a finding of lymphadenopathy on one modality and another, especially if one is looking for a change in size.
If clinically appropriate, a trial of diuretics, such as intravenous (IV) furosemide 80 mg,
Our patients. The 50-year-old man in Case 1 responded well to 80 mg of IV furosemide after one hour and improved further upon receipt of a pacemaker the next day. A repeat thoracic CT one month later showed complete resolution of the MLN.
The 79-year-old man in Case 2 also received 80 mg of IV furosemide and improved within 3 hours. A month later, a repeat thoracic CT showed a significant reduction in the size of all the enlarged lymph nodes (FIGURE 2B).
THE TAKEAWAY
The importance of these 2 cases is that they show that heart failure—even diastolic alone—can produce enlarged mediastinal lymph nodes. In patients with heart failure in whom unexpected MLN is detected, consideration should be given to performing a repeat imaging examination after the administration of diuretics.
1. Slanetz PJ, Truong M, Shepard JA, et al. Mediastinal lymphadenopathy and hazy mediastinal fat: new CT findings of congestive heart failure. AJR Am J Roentgenol. 1998;171:1307-1309.
2. Erly WK, Borders RJ, Outwater EK, et al. Location, size, and distribution of mediastinal lymph node enlargement in chronic congestive heart failure. J Comput Assist Tomogr. 2003;27:485-489.
3. Storto ML, Kee ST, Golden JA, et al. Hydrostatic pulmonary edema: high-resolution CT findings. AJR Am J Roentgenol. 1995;165:817-820.
4. Moua T, Levin DL, Carmona EM, et al. Frequency of mediastinal lymphadenopathy in patients with idiopathic pulmonary arterial hypertension. Chest. 2013;143:344-348.
5. Chabbert V, Canevet G, Baixas C, et al. Mediastinal lymphadenopathy in congestive heart failure: a sequential CT evaluation with clinical and echocardiographic correlations. Eur Radiol. 2004;14:881-889.
6. Christiansen JR, Hamre H, Massey R, et al. Left ventricular function in long-term survivors of childhood lymphoma. Am J Cardiol. 2014;114:483-490.
7. Krawczuk-Rybak M, Dakowicz L, Hryniewicz A, et al. Cardiac function in survivors of acute lymphoblastic leukaemia and Hodgkin’s lymphoma. J Paediatr Child Health. 2011;47:455-459.
8. Heidenreich PA, Hancock SL, Vagelos RH, et al. Diastolic dysfunction after mediastinal irradiation. Am Heart J. 2005;150:977-982.
9. Elbl L, Vasova I, Tomaskova I, et al. Cardiopulmonary exercise testing in the evaluation of functional capacity after treatment of lymphomas in adults. Leuk Lymphoma. 2006;47:843-851.
10. Pastis NJ Jr, Van Bakel AB, Brand TM, et al. Mediastinal lymphadenopathy in patients undergoing cardiac transplant evaluation. Chest. 2011;139:1451-1457.
1. Slanetz PJ, Truong M, Shepard JA, et al. Mediastinal lymphadenopathy and hazy mediastinal fat: new CT findings of congestive heart failure. AJR Am J Roentgenol. 1998;171:1307-1309.
2. Erly WK, Borders RJ, Outwater EK, et al. Location, size, and distribution of mediastinal lymph node enlargement in chronic congestive heart failure. J Comput Assist Tomogr. 2003;27:485-489.
3. Storto ML, Kee ST, Golden JA, et al. Hydrostatic pulmonary edema: high-resolution CT findings. AJR Am J Roentgenol. 1995;165:817-820.
4. Moua T, Levin DL, Carmona EM, et al. Frequency of mediastinal lymphadenopathy in patients with idiopathic pulmonary arterial hypertension. Chest. 2013;143:344-348.
5. Chabbert V, Canevet G, Baixas C, et al. Mediastinal lymphadenopathy in congestive heart failure: a sequential CT evaluation with clinical and echocardiographic correlations. Eur Radiol. 2004;14:881-889.
6. Christiansen JR, Hamre H, Massey R, et al. Left ventricular function in long-term survivors of childhood lymphoma. Am J Cardiol. 2014;114:483-490.
7. Krawczuk-Rybak M, Dakowicz L, Hryniewicz A, et al. Cardiac function in survivors of acute lymphoblastic leukaemia and Hodgkin’s lymphoma. J Paediatr Child Health. 2011;47:455-459.
8. Heidenreich PA, Hancock SL, Vagelos RH, et al. Diastolic dysfunction after mediastinal irradiation. Am Heart J. 2005;150:977-982.
9. Elbl L, Vasova I, Tomaskova I, et al. Cardiopulmonary exercise testing in the evaluation of functional capacity after treatment of lymphomas in adults. Leuk Lymphoma. 2006;47:843-851.
10. Pastis NJ Jr, Van Bakel AB, Brand TM, et al. Mediastinal lymphadenopathy in patients undergoing cardiac transplant evaluation. Chest. 2011;139:1451-1457.
