User login
50 years in pediatrics: ‘You have to get involved’
A few years before the launch of Pediatric News in 1967, Dr. Henry Kempe and his colleagues at the University of Colorado, Denver, published a groundbreaking report on child abuse, “The Battered Child Syndrome,” in JAMA.
It had a major impact on Calvin C.J. Sia, MD, a now-retired pediatrician in Hawaii, not only because it was pioneering work and one of the milestones of 20th century pediatrics, but also because Dr. Kempe was a close friend and mentor of Dr. Sia, going back to when Dr. Sia was a resident in the 1950s.
Dr. Kempe “alerted me to the battered child syndrome and taught me the importance of looking at the whole child within the context of the family, and he repeatedly reminded me of the importance of preventive care. He taught me how to make preventive change, with the emphasis on the first 3 years” of life, Dr. Sia said in an interview from his home in Honolulu.
To celebrate the 50th anniversary of Pediatric News, it seemed appropriate to turn to Dr. Sia. His practice, launched in 1958, not only spanned our 50 years, but also illustrated one of the major themes in pediatrics over the past half-century. With the old scourges of infectious disease, malnutrition, and infant mortality largely brought under control, pediatrics turned to the broader struggles of childhood, including learning, poverty, and abusive parenting.

Dr. Sia has been president of the Hawaii Medical Association; president of the Hawaii chapter of the American Academy of Pediatrics; chief of staff at the Kauikeolani Children’s Hospital, Honolulu (now the Kapi‘olani Medical Center for Women and Children); chair of the American Medical Association Pediatric Delegation; and founder of the AMA’s Section Council on Pediatrics.
Positions like these eventually led him to contacts with legislators willing to listen and fund child abuse prevention, immunization programs, and other initiatives to help children. It often meant lobbying politicians for money.
“It doesn’t occur overnight; it takes chance and dedication,” said Dr. Sia, whose name, as one of its earliest champions, is now nearly synonymous with the concept of the medical home – an ideal of cradling children in a physician, family, and social services safety net of coordinated care.
Dr. Sia’s career spanned all of the developments that Pediatric News has covered over its 50 years, including the increasing recognition of autism, the earlier survival of premature infants, and phenylketonuria and other screenings at birth. He remembers all of them, and took action on many at the state and federal level.
Taking a step took courage, even when he wasn’t quite sure what to do. “I recognized autism back in the 60s. Children looked normal, were born normal, but then had trouble. I didn’t know what to do with them. That’s how I got involved with learning disabilities. I didn’t know anything, but I got involved,” he said.
With the advice of Robert Cooke, MD, another mentor and an eventual founder of the Head Start program, Dr. Sia cofounded Honolulu’s Variety School for Learning Disabilities in 1967. It’s still in operation; Don Ho and other local “variety hour” entertainers helped with early fundraising.
Dr. Sia also helped launch Healthy Start, one of the nation’s first home visit programs for at-risk kids, and an amendment to the federal Education of the Handicapped Act to extend aid to children with family and developmental challenges.
He convinced Sen. Daniel Inouye, another personal friend, to introduce the Emergency Medical Services for Children Act in the 1980s, which funded states to develop emergency medical services for children. “It was really important because kids need special instruments, IVs, and medications. I had to lobby. It was under Reaganomics,” Dr. Sia said.
At 89 years of age and recovering from a recent heart attack and open heart bypass, he is still trying to help children. Dr. Sia, who was born in Beijing, has been promoting the medical home concept in Asia. Meanwhile, he said he is worried about the fragmentation of health care in the United States, and the likely cutback in federal and state spending on kids.
Even so, “I believe in the future. I see in the generation coming up much more awareness of the whole child. I think they are going to do a much better job than I did,” but they need to learn “how to work the system.” Young physicians also must be mentored to believe in themselves, and take up the torch, he said.
A few years before the launch of Pediatric News in 1967, Dr. Henry Kempe and his colleagues at the University of Colorado, Denver, published a groundbreaking report on child abuse, “The Battered Child Syndrome,” in JAMA.
It had a major impact on Calvin C.J. Sia, MD, a now-retired pediatrician in Hawaii, not only because it was pioneering work and one of the milestones of 20th century pediatrics, but also because Dr. Kempe was a close friend and mentor of Dr. Sia, going back to when Dr. Sia was a resident in the 1950s.
Dr. Kempe “alerted me to the battered child syndrome and taught me the importance of looking at the whole child within the context of the family, and he repeatedly reminded me of the importance of preventive care. He taught me how to make preventive change, with the emphasis on the first 3 years” of life, Dr. Sia said in an interview from his home in Honolulu.
To celebrate the 50th anniversary of Pediatric News, it seemed appropriate to turn to Dr. Sia. His practice, launched in 1958, not only spanned our 50 years, but also illustrated one of the major themes in pediatrics over the past half-century. With the old scourges of infectious disease, malnutrition, and infant mortality largely brought under control, pediatrics turned to the broader struggles of childhood, including learning, poverty, and abusive parenting.
Dr. Sia has been president of the Hawaii Medical Association; president of the Hawaii chapter of the American Academy of Pediatrics; chief of staff at the Kauikeolani Children’s Hospital, Honolulu (now the Kapi‘olani Medical Center for Women and Children); chair of the American Medical Association Pediatric Delegation; and founder of the AMA’s Section Council on Pediatrics.
Positions like these eventually led him to contacts with legislators willing to listen and fund child abuse prevention, immunization programs, and other initiatives to help children. It often meant lobbying politicians for money.
“It doesn’t occur overnight; it takes chance and dedication,” said Dr. Sia, whose name, as one of its earliest champions, is now nearly synonymous with the concept of the medical home – an ideal of cradling children in a physician, family, and social services safety net of coordinated care.
Dr. Sia’s career spanned all of the developments that Pediatric News has covered over its 50 years, including the increasing recognition of autism, the earlier survival of premature infants, and phenylketonuria and other screenings at birth. He remembers all of them, and took action on many at the state and federal level.
Taking a step took courage, even when he wasn’t quite sure what to do. “I recognized autism back in the 60s. Children looked normal, were born normal, but then had trouble. I didn’t know what to do with them. That’s how I got involved with learning disabilities. I didn’t know anything, but I got involved,” he said.
With the advice of Robert Cooke, MD, another mentor and an eventual founder of the Head Start program, Dr. Sia cofounded Honolulu’s Variety School for Learning Disabilities in 1967. It’s still in operation; Don Ho and other local “variety hour” entertainers helped with early fundraising.
Dr. Sia also helped launch Healthy Start, one of the nation’s first home visit programs for at-risk kids, and an amendment to the federal Education of the Handicapped Act to extend aid to children with family and developmental challenges.
He convinced Sen. Daniel Inouye, another personal friend, to introduce the Emergency Medical Services for Children Act in the 1980s, which funded states to develop emergency medical services for children. “It was really important because kids need special instruments, IVs, and medications. I had to lobby. It was under Reaganomics,” Dr. Sia said.
At 89 years of age and recovering from a recent heart attack and open heart bypass, he is still trying to help children. Dr. Sia, who was born in Beijing, has been promoting the medical home concept in Asia. Meanwhile, he said he is worried about the fragmentation of health care in the United States, and the likely cutback in federal and state spending on kids.
Even so, “I believe in the future. I see in the generation coming up much more awareness of the whole child. I think they are going to do a much better job than I did,” but they need to learn “how to work the system.” Young physicians also must be mentored to believe in themselves, and take up the torch, he said.
A few years before the launch of Pediatric News in 1967, Dr. Henry Kempe and his colleagues at the University of Colorado, Denver, published a groundbreaking report on child abuse, “The Battered Child Syndrome,” in JAMA.
It had a major impact on Calvin C.J. Sia, MD, a now-retired pediatrician in Hawaii, not only because it was pioneering work and one of the milestones of 20th century pediatrics, but also because Dr. Kempe was a close friend and mentor of Dr. Sia, going back to when Dr. Sia was a resident in the 1950s.
Dr. Kempe “alerted me to the battered child syndrome and taught me the importance of looking at the whole child within the context of the family, and he repeatedly reminded me of the importance of preventive care. He taught me how to make preventive change, with the emphasis on the first 3 years” of life, Dr. Sia said in an interview from his home in Honolulu.
To celebrate the 50th anniversary of Pediatric News, it seemed appropriate to turn to Dr. Sia. His practice, launched in 1958, not only spanned our 50 years, but also illustrated one of the major themes in pediatrics over the past half-century. With the old scourges of infectious disease, malnutrition, and infant mortality largely brought under control, pediatrics turned to the broader struggles of childhood, including learning, poverty, and abusive parenting.
Dr. Sia has been president of the Hawaii Medical Association; president of the Hawaii chapter of the American Academy of Pediatrics; chief of staff at the Kauikeolani Children’s Hospital, Honolulu (now the Kapi‘olani Medical Center for Women and Children); chair of the American Medical Association Pediatric Delegation; and founder of the AMA’s Section Council on Pediatrics.
Positions like these eventually led him to contacts with legislators willing to listen and fund child abuse prevention, immunization programs, and other initiatives to help children. It often meant lobbying politicians for money.
“It doesn’t occur overnight; it takes chance and dedication,” said Dr. Sia, whose name, as one of its earliest champions, is now nearly synonymous with the concept of the medical home – an ideal of cradling children in a physician, family, and social services safety net of coordinated care.
Dr. Sia’s career spanned all of the developments that Pediatric News has covered over its 50 years, including the increasing recognition of autism, the earlier survival of premature infants, and phenylketonuria and other screenings at birth. He remembers all of them, and took action on many at the state and federal level.
Taking a step took courage, even when he wasn’t quite sure what to do. “I recognized autism back in the 60s. Children looked normal, were born normal, but then had trouble. I didn’t know what to do with them. That’s how I got involved with learning disabilities. I didn’t know anything, but I got involved,” he said.
With the advice of Robert Cooke, MD, another mentor and an eventual founder of the Head Start program, Dr. Sia cofounded Honolulu’s Variety School for Learning Disabilities in 1967. It’s still in operation; Don Ho and other local “variety hour” entertainers helped with early fundraising.
Dr. Sia also helped launch Healthy Start, one of the nation’s first home visit programs for at-risk kids, and an amendment to the federal Education of the Handicapped Act to extend aid to children with family and developmental challenges.
He convinced Sen. Daniel Inouye, another personal friend, to introduce the Emergency Medical Services for Children Act in the 1980s, which funded states to develop emergency medical services for children. “It was really important because kids need special instruments, IVs, and medications. I had to lobby. It was under Reaganomics,” Dr. Sia said.
At 89 years of age and recovering from a recent heart attack and open heart bypass, he is still trying to help children. Dr. Sia, who was born in Beijing, has been promoting the medical home concept in Asia. Meanwhile, he said he is worried about the fragmentation of health care in the United States, and the likely cutback in federal and state spending on kids.
Even so, “I believe in the future. I see in the generation coming up much more awareness of the whole child. I think they are going to do a much better job than I did,” but they need to learn “how to work the system.” Young physicians also must be mentored to believe in themselves, and take up the torch, he said.
Intensifying vedolizumab could counter loss of response in IBD
ORLANDO – In a study of 644 people with moderate to severe inflammatory bowel disease treated with vedolizumab (Entyvio, Takeda Pharmaceuticals), 346 achieved remission or a significant response. This 54% response rate includes 192 people with Crohn’s disease and 154 others with ulcerative colitis.
However, after a median of 143 days, some of the initial responders experienced a loss of response to vedolizumab therapy.
“In our real-world study of vedolizumab use in patients with IBD predominantly refractory to anti-TNF [tumor necrosis factor] therapy, loss of response was observed in about 40% of patients at 12 months,” said Eugenia Shmidt, MD, a gastroenterology fellow at the Icahn School of Medicine at Mount Sinai hospital in New York City.
To counter the loss of response, Dr. Shmidt and her colleagues shortened the dosing interval of vedolizumab to either 4 or 6 weeks in a subgroup of 36 patients who lost response. They also shortened the dosing interval to try to attempt an initial response in a subgroup of 47 people who did not respond to initial induction therapy in the first place. “Dose intensification led to a successful recapture of response in 32% of patients who had initial response and in 19% of patients who did not have an initial response,” Dr. Shmidt said at the Advances in Inflammatory Bowel Diseases meeting.
“We find it interesting that there was greater success in recapturing significant response in patients who responded to vedolizumab initially compared to those who did not initially respond,” she said when asked if she found any of her findings surprising. “This emphasizes a likely need for different management strategies for initial vedolizumab responders and nonresponders, as well as early recognition of the need for dose optimization,” she added.
The study included adults with mild to moderate inflammatory bowel disease, based on clinical factors or confirmed through endoscopy. There were no significant differences in severity of disease between patients who were able to recapture response versus those were not. The mean age among the 374 participants with Crohn’s disease was 39 years; similarly, the mean age in the group of 270 with ulcerative colitis was 41 years. Mean duration of disease was 15 years and 9 years in the two groups, respectively.
To identify risk factors associated with loss of response to vedolizumab, Dr. Shmidt and her colleagues performed univariable and multivariable Cox proportional hazard analyses. They found concomitant use of an immunomodulator for Crohn’s disease was protective against loss of response (hazard ratio, 0.44). In the ulcerative colitis group, a baseline serum albumin level below a normal value achieved lower vedolizumab response rates; a value below 3.2 g/dL was an independent predictor of cumulative loss of response over time (HR, 2.39).
Inflammatory bowel disease patients treated at higher volume centers, which were defined as enrolling at least 100 patients in the study, had higher rates of loss of response to vedolizumab (HR, 1.92 on multivariable analysis).
The multicenter VICTORY consortium coinvestigators in this study are affiliated with Mayo Clinic in Rochester, Minn.; Cleveland Clinic Foundation in Ohio; University of California, San Diego; New York University in New York City; Montefiore Medical Center/Albert Einstein College of Medicine in the Bronx, N.Y.; and Dartmouth-Hitchcock Medical Center in Lebanon, N.H.
Future studies are warranted to evaluate the pharmacodynamics and pharmacokinetics of vedolizumab, the authors noted, as well as to determine an optimal dosing strategy among people with high drug clearance.
The study was funded in part by Takeda Pharmaceuticals. Dr. Eugenia Shmidt did not have any relevant disclosures. The meeting was sponsored by the Crohn’s & Colitis Foundation of America.
ORLANDO – In a study of 644 people with moderate to severe inflammatory bowel disease treated with vedolizumab (Entyvio, Takeda Pharmaceuticals), 346 achieved remission or a significant response. This 54% response rate includes 192 people with Crohn’s disease and 154 others with ulcerative colitis.
However, after a median of 143 days, some of the initial responders experienced a loss of response to vedolizumab therapy.
“In our real-world study of vedolizumab use in patients with IBD predominantly refractory to anti-TNF [tumor necrosis factor] therapy, loss of response was observed in about 40% of patients at 12 months,” said Eugenia Shmidt, MD, a gastroenterology fellow at the Icahn School of Medicine at Mount Sinai hospital in New York City.
To counter the loss of response, Dr. Shmidt and her colleagues shortened the dosing interval of vedolizumab to either 4 or 6 weeks in a subgroup of 36 patients who lost response. They also shortened the dosing interval to try to attempt an initial response in a subgroup of 47 people who did not respond to initial induction therapy in the first place. “Dose intensification led to a successful recapture of response in 32% of patients who had initial response and in 19% of patients who did not have an initial response,” Dr. Shmidt said at the Advances in Inflammatory Bowel Diseases meeting.
“We find it interesting that there was greater success in recapturing significant response in patients who responded to vedolizumab initially compared to those who did not initially respond,” she said when asked if she found any of her findings surprising. “This emphasizes a likely need for different management strategies for initial vedolizumab responders and nonresponders, as well as early recognition of the need for dose optimization,” she added.
The study included adults with mild to moderate inflammatory bowel disease, based on clinical factors or confirmed through endoscopy. There were no significant differences in severity of disease between patients who were able to recapture response versus those were not. The mean age among the 374 participants with Crohn’s disease was 39 years; similarly, the mean age in the group of 270 with ulcerative colitis was 41 years. Mean duration of disease was 15 years and 9 years in the two groups, respectively.
To identify risk factors associated with loss of response to vedolizumab, Dr. Shmidt and her colleagues performed univariable and multivariable Cox proportional hazard analyses. They found concomitant use of an immunomodulator for Crohn’s disease was protective against loss of response (hazard ratio, 0.44). In the ulcerative colitis group, a baseline serum albumin level below a normal value achieved lower vedolizumab response rates; a value below 3.2 g/dL was an independent predictor of cumulative loss of response over time (HR, 2.39).
Inflammatory bowel disease patients treated at higher volume centers, which were defined as enrolling at least 100 patients in the study, had higher rates of loss of response to vedolizumab (HR, 1.92 on multivariable analysis).
The multicenter VICTORY consortium coinvestigators in this study are affiliated with Mayo Clinic in Rochester, Minn.; Cleveland Clinic Foundation in Ohio; University of California, San Diego; New York University in New York City; Montefiore Medical Center/Albert Einstein College of Medicine in the Bronx, N.Y.; and Dartmouth-Hitchcock Medical Center in Lebanon, N.H.
Future studies are warranted to evaluate the pharmacodynamics and pharmacokinetics of vedolizumab, the authors noted, as well as to determine an optimal dosing strategy among people with high drug clearance.
The study was funded in part by Takeda Pharmaceuticals. Dr. Eugenia Shmidt did not have any relevant disclosures. The meeting was sponsored by the Crohn’s & Colitis Foundation of America.
ORLANDO – In a study of 644 people with moderate to severe inflammatory bowel disease treated with vedolizumab (Entyvio, Takeda Pharmaceuticals), 346 achieved remission or a significant response. This 54% response rate includes 192 people with Crohn’s disease and 154 others with ulcerative colitis.
However, after a median of 143 days, some of the initial responders experienced a loss of response to vedolizumab therapy.
“In our real-world study of vedolizumab use in patients with IBD predominantly refractory to anti-TNF [tumor necrosis factor] therapy, loss of response was observed in about 40% of patients at 12 months,” said Eugenia Shmidt, MD, a gastroenterology fellow at the Icahn School of Medicine at Mount Sinai hospital in New York City.
To counter the loss of response, Dr. Shmidt and her colleagues shortened the dosing interval of vedolizumab to either 4 or 6 weeks in a subgroup of 36 patients who lost response. They also shortened the dosing interval to try to attempt an initial response in a subgroup of 47 people who did not respond to initial induction therapy in the first place. “Dose intensification led to a successful recapture of response in 32% of patients who had initial response and in 19% of patients who did not have an initial response,” Dr. Shmidt said at the Advances in Inflammatory Bowel Diseases meeting.
“We find it interesting that there was greater success in recapturing significant response in patients who responded to vedolizumab initially compared to those who did not initially respond,” she said when asked if she found any of her findings surprising. “This emphasizes a likely need for different management strategies for initial vedolizumab responders and nonresponders, as well as early recognition of the need for dose optimization,” she added.
The study included adults with mild to moderate inflammatory bowel disease, based on clinical factors or confirmed through endoscopy. There were no significant differences in severity of disease between patients who were able to recapture response versus those were not. The mean age among the 374 participants with Crohn’s disease was 39 years; similarly, the mean age in the group of 270 with ulcerative colitis was 41 years. Mean duration of disease was 15 years and 9 years in the two groups, respectively.
To identify risk factors associated with loss of response to vedolizumab, Dr. Shmidt and her colleagues performed univariable and multivariable Cox proportional hazard analyses. They found concomitant use of an immunomodulator for Crohn’s disease was protective against loss of response (hazard ratio, 0.44). In the ulcerative colitis group, a baseline serum albumin level below a normal value achieved lower vedolizumab response rates; a value below 3.2 g/dL was an independent predictor of cumulative loss of response over time (HR, 2.39).
Inflammatory bowel disease patients treated at higher volume centers, which were defined as enrolling at least 100 patients in the study, had higher rates of loss of response to vedolizumab (HR, 1.92 on multivariable analysis).
The multicenter VICTORY consortium coinvestigators in this study are affiliated with Mayo Clinic in Rochester, Minn.; Cleveland Clinic Foundation in Ohio; University of California, San Diego; New York University in New York City; Montefiore Medical Center/Albert Einstein College of Medicine in the Bronx, N.Y.; and Dartmouth-Hitchcock Medical Center in Lebanon, N.H.
Future studies are warranted to evaluate the pharmacodynamics and pharmacokinetics of vedolizumab, the authors noted, as well as to determine an optimal dosing strategy among people with high drug clearance.
The study was funded in part by Takeda Pharmaceuticals. Dr. Eugenia Shmidt did not have any relevant disclosures. The meeting was sponsored by the Crohn’s & Colitis Foundation of America.
Key clinical point: Some patients with moderate to severe inflammatory bowel disease experience loss of response to vedolizumab over time.
Major finding: At a median 143 days, 41% of patients with Crohn’s disease and 42% of those with ulcerative colitis who initially had a strong response or experienced remission with vedolizumab experienced subsequent loss of response.
Data source: Study of 644 patients with moderate to severe Crohn’s disease or ulcerative colitis followed for 12 months.
Disclosures: The study was funded in part by Takeda Pharmaceuticals. Dr. Eugenia Shmidt did not have any relevant disclosures.
Immediate postpartum LARC requires cross-disciplinary cooperation in the hospital
Hospitals that aim to offer women long-acting reversible contraception immediately after giving birth require up-front coordination across departments, including early recruitment of nonclinical staff, researchers have found.
One of the advantages to offering LARC postpartum in the hospital, instead of an outpatient clinic, is that patients are not required to present for repeat visits. The American College of Obstetricians and Gynecologists has called the immediate postpartum period an optimal time for LARC placement.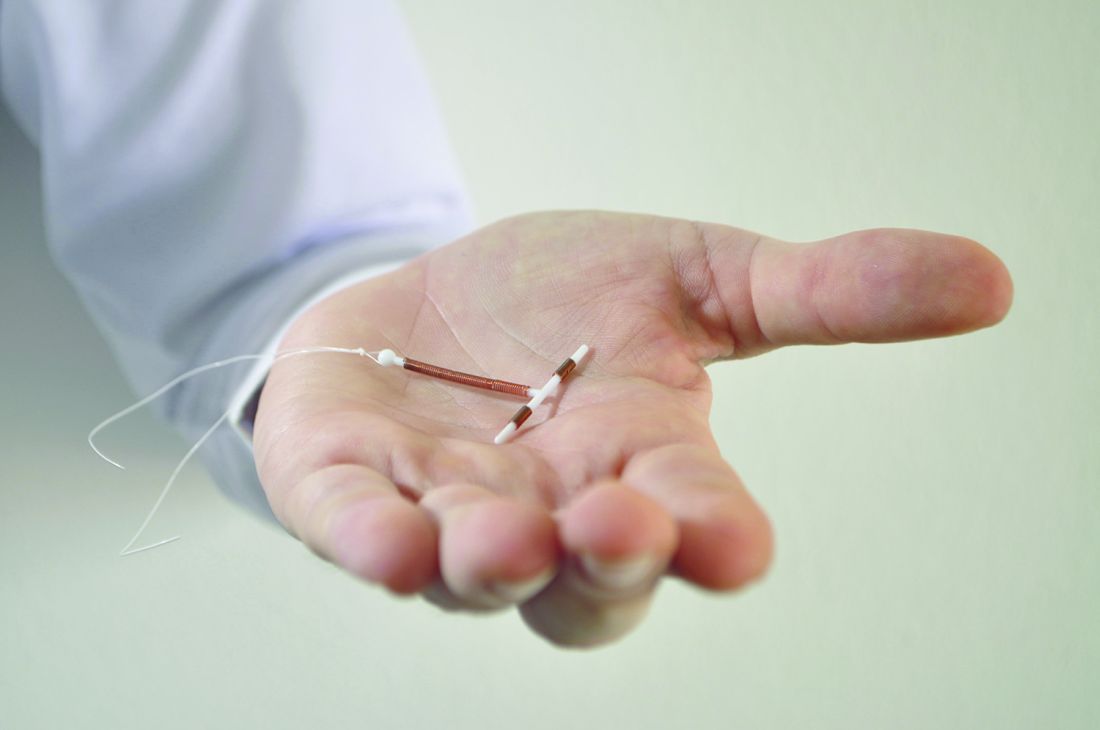
In an effort to fill in this knowledge gap, a team of investigators led by Lisa G. Hofler, MD, MPH, of Emory University in Atlanta, sought to identify barriers to implementation and characteristics of successful efforts among hospitals developing postpartum LARC programs.
Dr. Hofler’s team interviewed clinicians and staff members, including pharmacists and billing employees, at 10 Georgia hospitals starting in March 2015, about a year after the state approved a separate Medicaid-reimbursement protocol for immediate postpartum LARC. Of the hospitals in the study, nine were attempting to launch programs during the study period, and four had active programs by the study endpoint (Obstet Gynecol 2017;129:3–9).
Dr. Hofler and her colleagues found – through interviews conducted separately with 32 employees in clinical or administrative roles – that the hospitals that had succeeded had engaged multidisciplinary teams early in the process.
“We found that implementing an immediate postpartum LARC program in the hospital is initially more complicated than people think, and involves the participation of departments that people might overlook,” Dr. Hofler said in an interview. “It’s about engaging a pharmacy person, a billing person, or a health records expert in addition to the usual nursing and physician staff that one engages when you have some sort of clinical practice change.”
Barriers to successful programs included staff lack of knowledge about LARC, financial concerns, and competing priorities within hospitals, the team found.
“Several participants had little previous exposure to LARC, and clinicians did not always easily appreciate the differences between providing LARC in the inpatient and outpatient settings,” Dr. Hofler and her colleagues reported in their study.
“Early involvement of the necessary members of the implementation team leads to better communication and understanding of the project,” the researchers concluded, noting that implementation cannot move forward without “financial reassurance early in the process.”
Teams should include representation from direct clinical care personnel, pharmacy, or finance and billing, they reported, though the specific team members may vary by hospital.
“Consistent communication and team planning with clear roles and responsibilities are key to navigating the complex and interconnected steps” in launching a program, they wrote.
Though Dr. Hofler stressed that the report was not meant to substitute for formal guidance, it maps the steps needed, and the departments involved, at each stage of the implementation process, from exploration of a program to its eventual launch and maintenance.
The study was supported by a grant from the Society of Family Planning Research Fund. Two of the coauthors disclosed research funding or other relationships with LARC manufacturers.
Hospitals that aim to offer women long-acting reversible contraception immediately after giving birth require up-front coordination across departments, including early recruitment of nonclinical staff, researchers have found.
One of the advantages to offering LARC postpartum in the hospital, instead of an outpatient clinic, is that patients are not required to present for repeat visits. The American College of Obstetricians and Gynecologists has called the immediate postpartum period an optimal time for LARC placement.
In an effort to fill in this knowledge gap, a team of investigators led by Lisa G. Hofler, MD, MPH, of Emory University in Atlanta, sought to identify barriers to implementation and characteristics of successful efforts among hospitals developing postpartum LARC programs.
Dr. Hofler’s team interviewed clinicians and staff members, including pharmacists and billing employees, at 10 Georgia hospitals starting in March 2015, about a year after the state approved a separate Medicaid-reimbursement protocol for immediate postpartum LARC. Of the hospitals in the study, nine were attempting to launch programs during the study period, and four had active programs by the study endpoint (Obstet Gynecol 2017;129:3–9).
Dr. Hofler and her colleagues found – through interviews conducted separately with 32 employees in clinical or administrative roles – that the hospitals that had succeeded had engaged multidisciplinary teams early in the process.
“We found that implementing an immediate postpartum LARC program in the hospital is initially more complicated than people think, and involves the participation of departments that people might overlook,” Dr. Hofler said in an interview. “It’s about engaging a pharmacy person, a billing person, or a health records expert in addition to the usual nursing and physician staff that one engages when you have some sort of clinical practice change.”
Barriers to successful programs included staff lack of knowledge about LARC, financial concerns, and competing priorities within hospitals, the team found.
“Several participants had little previous exposure to LARC, and clinicians did not always easily appreciate the differences between providing LARC in the inpatient and outpatient settings,” Dr. Hofler and her colleagues reported in their study.
“Early involvement of the necessary members of the implementation team leads to better communication and understanding of the project,” the researchers concluded, noting that implementation cannot move forward without “financial reassurance early in the process.”
Teams should include representation from direct clinical care personnel, pharmacy, or finance and billing, they reported, though the specific team members may vary by hospital.
“Consistent communication and team planning with clear roles and responsibilities are key to navigating the complex and interconnected steps” in launching a program, they wrote.
Though Dr. Hofler stressed that the report was not meant to substitute for formal guidance, it maps the steps needed, and the departments involved, at each stage of the implementation process, from exploration of a program to its eventual launch and maintenance.
The study was supported by a grant from the Society of Family Planning Research Fund. Two of the coauthors disclosed research funding or other relationships with LARC manufacturers.
Hospitals that aim to offer women long-acting reversible contraception immediately after giving birth require up-front coordination across departments, including early recruitment of nonclinical staff, researchers have found.
One of the advantages to offering LARC postpartum in the hospital, instead of an outpatient clinic, is that patients are not required to present for repeat visits. The American College of Obstetricians and Gynecologists has called the immediate postpartum period an optimal time for LARC placement.
In an effort to fill in this knowledge gap, a team of investigators led by Lisa G. Hofler, MD, MPH, of Emory University in Atlanta, sought to identify barriers to implementation and characteristics of successful efforts among hospitals developing postpartum LARC programs.
Dr. Hofler’s team interviewed clinicians and staff members, including pharmacists and billing employees, at 10 Georgia hospitals starting in March 2015, about a year after the state approved a separate Medicaid-reimbursement protocol for immediate postpartum LARC. Of the hospitals in the study, nine were attempting to launch programs during the study period, and four had active programs by the study endpoint (Obstet Gynecol 2017;129:3–9).
Dr. Hofler and her colleagues found – through interviews conducted separately with 32 employees in clinical or administrative roles – that the hospitals that had succeeded had engaged multidisciplinary teams early in the process.
“We found that implementing an immediate postpartum LARC program in the hospital is initially more complicated than people think, and involves the participation of departments that people might overlook,” Dr. Hofler said in an interview. “It’s about engaging a pharmacy person, a billing person, or a health records expert in addition to the usual nursing and physician staff that one engages when you have some sort of clinical practice change.”
Barriers to successful programs included staff lack of knowledge about LARC, financial concerns, and competing priorities within hospitals, the team found.
“Several participants had little previous exposure to LARC, and clinicians did not always easily appreciate the differences between providing LARC in the inpatient and outpatient settings,” Dr. Hofler and her colleagues reported in their study.
“Early involvement of the necessary members of the implementation team leads to better communication and understanding of the project,” the researchers concluded, noting that implementation cannot move forward without “financial reassurance early in the process.”
Teams should include representation from direct clinical care personnel, pharmacy, or finance and billing, they reported, though the specific team members may vary by hospital.
“Consistent communication and team planning with clear roles and responsibilities are key to navigating the complex and interconnected steps” in launching a program, they wrote.
Though Dr. Hofler stressed that the report was not meant to substitute for formal guidance, it maps the steps needed, and the departments involved, at each stage of the implementation process, from exploration of a program to its eventual launch and maintenance.
The study was supported by a grant from the Society of Family Planning Research Fund. Two of the coauthors disclosed research funding or other relationships with LARC manufacturers.
FROM OBSTETRICS & GYNECOLOGY
Key clinical point: Success in establishing an immediate postpartum LARC program involves team-building across hospital disciplines.
Major finding: Factors associated with success included early coordination among financial, administrative, pharmacy, and clinical personnel.
Data source: A qualitative analysis of interviews with 32 employees (clinical and nonclinical) from 10 hospitals in Georgia.
Disclosures: Two authors disclosed relationships with LARC manufacturers.
Ketamine emerging as top treatment for cocaine dependence
VIENNA – The prospect on the horizon of two new effective therapies for chronic cocaine dependence – sustained-release dextroamphetamine and subanesthetic ketamine infusions – was among the top developments of the year in addiction medicine, Wim van den Brink, MD, PhD, said at the annual congress of the European College of Neuropsychopharmacology.
Other highlights on his list included:
• Studies establishing that comorbid attention-deficit/hyperactivity disorder (ADHD) and substance use disorder now can be treated effectively with either extended-release mixed amphetamine salts or high-dose methylphenidate.

• Release of a puzzling array of conflicting studies on the use of high-dose baclofen for treatment of alcohol dependence. It’s tough to reconcile this mishmash of polar opposite results. And that dictates it’s time to declare a moratorium on the use of this therapy in clinical practice, which in many places is now widespread, said Dr. van den Brink, professor of psychiatry and addiction at the University of Amsterdam and director of the Amsterdam Institute for Addiction Research.
“It’s too strange that we have such conflicting evidence out there. Too many people are prescribing crazy-high doses of baclofen with no strong supporting evidence,” Dr. van den Brink said.
Cocaine dependence
Dr. van den Brink was a coinvestigator in a Dutch multicenter randomized, double-blind, placebo-controlled trial of multitreatment-refractory comorbid cocaine dependence in 73 heroin-dependent patients in heroin-assisted treatment. Patients assigned to 60 mg/day of sustained-release dextroamphetamine, in addition to the background methadone and diacetylmorphine all participants were on for their heroin dependence, had significantly fewer days of cocaine use in the 12-week study: a mean of 44.9 days, compared with 60.6 days in placebo-treated controls. Adverse events were transient and well tolerated (Lancet. 2016 May 28;387[10034]:2226-34).
“A lot of medications have been tried for treatment of cocaine dependence, but actually none of them has been shown to be effective with the exception of substitution treatment with stimulants. Ours is one of the most successful trials. These patients were using cocaine an average of 24 days per month along with a lot of other drugs, despite being in heroin treatment for 4 years,” Dr. van den Brink said. “Patients were very willing to take the sustained-release dextroamphetamine. In the last 4 weeks, 84% of them used at least 80% of their medication. And they were blinded to what they were using.
“We saw good effect sizes: 0.6-0.7 for self-report measures and 0.31 for negative urine samples. So this is a very promising approach. But it also means that, like with tobacco dependence or alcohol dependence, we have to start thinking about substitution therapy in stimulant-dependent patients,” he said.
Dr. van den Brink said subanesthetic ketamine as a novel treatment for cocaine dependence is not yet ready for prime time use in clinical practice, because it’s just not practical to bring patients in for a roughly hour-long intravenous infusion on a daily basis, as was done in a highly impressive proof-of-concept study. But new formulations of ketamine are under development that should better lend themselves to use in clinical practice.
In the proof-of-concept study, investigators at the New York State Psychiatric Institute brought into the laboratory cocaine-dependent volunteers not seeking treatment or abstinence and administered 52-minute infusions of ketamine at 0.41 or 71 mg/kg or lorazepam at 2 mg (Biol Psychiatry. 2014 Jul 1;76[1]:40-6). Lorazepam had absolutely no effect on motivation to change, but ketamine was a different story.
“As soon as you give a low dose of ketamine, you see a wonderful effect on motivation to change and on craving ratings in assessments at 24 hours post infusion. This looks like another promising way of treating cocaine dependence,” he said.
Doxazosin for alcoholism
Investigators at the National Institute on Alcohol Abuse and Alcoholism and several U.S. universities hypothesized that the norepinephrine system could be an important treatment target in alcohol dependence. They conducted a double-blind, placebo-controlled randomized trial in which alcohol-dependent patients seeking outpatient treatment were assigned to the alpha1-adrenergic blocker doxazosin (Cardura) titrated to a maximum of 16 mg/day or placebo. They found doxazosin significantly reduced drinks per week and the number of heavy drinking days per week, but only in the subgroup of patients with a strong family history of alcoholism. In patients without such a family history, doxazosin paradoxically increased drinking (Addict Biol. 2016 Jul;21[4]:904-14).
One of the reasons adult ADHD is greatly underrecognized is that it tends to occur in combination with flashier substance use disorders. “Addiction is very comorbid with all kinds of disorders, but especially with externalizing childhood disorders like conduct disorder and ADHD,” Dr. van den Brink said.
It was shown half-a-decade ago that normal doses of methylphenidate have no effect on ADHD symptoms or substance use in comorbid adults. Then Swedish investigators reported that treating criminal offenders with high-dose methylphenidate – roughly three times greater than standard dosing – was effective in reducing both ADHD symptoms and comorbid substance use in criminal offenders. Those findings prompted investigators at the New York State Psychiatric Institute and the University of Minnesota to examine whether prescribing extended-release mixed amphetamine salts in adults with comorbid cocaine use disorder and ADHD would achieve improvement in both conditions. Indeed, it did, Dr. van den Brink said.
One hundred twenty-six affected patients were randomized to 60 or 80 mg/day of extended-release mixed amphetamine salts or placebo for 13 weeks coupled with weekly individual cognitive-behavioral therapy for all in this double-blind, three-arm clinical trial.
“They showed a number-needed-to-treat of about 2.5 in order to achieve a significant reduction in cocaine use and a very nice reduction in ADHD symptoms with a number-needed-to-treat of 3,” Dr. van den Brink said.
The rate of continuous cocaine abstinence in the last 3 weeks of the trial was 30% in the 80-mg group and 17.5% with 60 mg of extended-release mixed amphetamine salts, compared with just 7% with placebo (JAMA Psychiatry. 2015 Jun;72[6]:593-602).
Interpreting baclofen studies
The first high-quality multicenter, randomized, placebo-controlled, double-blind clinical trial, conducted in Germany, showed baclofen (Lioresal) at a mean dose of 180 mg/day was effective in maintaining alcohol abstinence (Eur Neuropsychopharmacol. 2015 Aug;25[8]:1167-77).
“They got wonderful results, with a number-needed-to-treat of 2.3. That is something we’re not used to seeing in the treatment of alcoholism. But there was no dose-response effect, which is a little unusual,” the psychiatrist observed.
Then a multicenter group of Dutch investigators, including Dr. van den Brink, carried out what they believed would be a confirmatory randomized, double-blind, placebo-controlled trial. However, it showed no difference between high- or low-dose baclofen and placebo in time to relapse (Eur Neuropsychopharmacol. 2016 Dec;26[12]:1950-9).
Little further light was shed by the two large French randomized, placebo-controlled clinical trials presented at the 2016 World Congress for Alcohol and Alcoholism in Berlin. One, the BACLOVILLE trial, included 320 patients treated in 60 family practice clinics; it showed strongly positive results for high-dose baclofen. In contrast, the 316-patient ALPADIR study proved negative. These conflicting results were particularly disappointing because France has been at the forefront of using high-dose baclofen to treat alcoholism, Dr. van den Brink said.
“Maybe some 100,000 people have been treated with high-dose baclofen for alcoholism in France,” he said. “What is the conclusion from all these baclofen studies? You can interpret them in many ways. Maybe there are two positive trials and two negative trials, or maybe there are two positive trials and two failed trials. The debate is not closed, even after four randomized trials.”
Dr. van den Brink reported receiving research funding from and/or serving as a consultant to more than half a dozen pharmaceutical companies.
VIENNA – The prospect on the horizon of two new effective therapies for chronic cocaine dependence – sustained-release dextroamphetamine and subanesthetic ketamine infusions – was among the top developments of the year in addiction medicine, Wim van den Brink, MD, PhD, said at the annual congress of the European College of Neuropsychopharmacology.
Other highlights on his list included:
• Studies establishing that comorbid attention-deficit/hyperactivity disorder (ADHD) and substance use disorder now can be treated effectively with either extended-release mixed amphetamine salts or high-dose methylphenidate.
• Release of a puzzling array of conflicting studies on the use of high-dose baclofen for treatment of alcohol dependence. It’s tough to reconcile this mishmash of polar opposite results. And that dictates it’s time to declare a moratorium on the use of this therapy in clinical practice, which in many places is now widespread, said Dr. van den Brink, professor of psychiatry and addiction at the University of Amsterdam and director of the Amsterdam Institute for Addiction Research.
“It’s too strange that we have such conflicting evidence out there. Too many people are prescribing crazy-high doses of baclofen with no strong supporting evidence,” Dr. van den Brink said.
Cocaine dependence
Dr. van den Brink was a coinvestigator in a Dutch multicenter randomized, double-blind, placebo-controlled trial of multitreatment-refractory comorbid cocaine dependence in 73 heroin-dependent patients in heroin-assisted treatment. Patients assigned to 60 mg/day of sustained-release dextroamphetamine, in addition to the background methadone and diacetylmorphine all participants were on for their heroin dependence, had significantly fewer days of cocaine use in the 12-week study: a mean of 44.9 days, compared with 60.6 days in placebo-treated controls. Adverse events were transient and well tolerated (Lancet. 2016 May 28;387[10034]:2226-34).
“A lot of medications have been tried for treatment of cocaine dependence, but actually none of them has been shown to be effective with the exception of substitution treatment with stimulants. Ours is one of the most successful trials. These patients were using cocaine an average of 24 days per month along with a lot of other drugs, despite being in heroin treatment for 4 years,” Dr. van den Brink said. “Patients were very willing to take the sustained-release dextroamphetamine. In the last 4 weeks, 84% of them used at least 80% of their medication. And they were blinded to what they were using.
“We saw good effect sizes: 0.6-0.7 for self-report measures and 0.31 for negative urine samples. So this is a very promising approach. But it also means that, like with tobacco dependence or alcohol dependence, we have to start thinking about substitution therapy in stimulant-dependent patients,” he said.
Dr. van den Brink said subanesthetic ketamine as a novel treatment for cocaine dependence is not yet ready for prime time use in clinical practice, because it’s just not practical to bring patients in for a roughly hour-long intravenous infusion on a daily basis, as was done in a highly impressive proof-of-concept study. But new formulations of ketamine are under development that should better lend themselves to use in clinical practice.
In the proof-of-concept study, investigators at the New York State Psychiatric Institute brought into the laboratory cocaine-dependent volunteers not seeking treatment or abstinence and administered 52-minute infusions of ketamine at 0.41 or 71 mg/kg or lorazepam at 2 mg (Biol Psychiatry. 2014 Jul 1;76[1]:40-6). Lorazepam had absolutely no effect on motivation to change, but ketamine was a different story.
“As soon as you give a low dose of ketamine, you see a wonderful effect on motivation to change and on craving ratings in assessments at 24 hours post infusion. This looks like another promising way of treating cocaine dependence,” he said.
Doxazosin for alcoholism
Investigators at the National Institute on Alcohol Abuse and Alcoholism and several U.S. universities hypothesized that the norepinephrine system could be an important treatment target in alcohol dependence. They conducted a double-blind, placebo-controlled randomized trial in which alcohol-dependent patients seeking outpatient treatment were assigned to the alpha1-adrenergic blocker doxazosin (Cardura) titrated to a maximum of 16 mg/day or placebo. They found doxazosin significantly reduced drinks per week and the number of heavy drinking days per week, but only in the subgroup of patients with a strong family history of alcoholism. In patients without such a family history, doxazosin paradoxically increased drinking (Addict Biol. 2016 Jul;21[4]:904-14).
One of the reasons adult ADHD is greatly underrecognized is that it tends to occur in combination with flashier substance use disorders. “Addiction is very comorbid with all kinds of disorders, but especially with externalizing childhood disorders like conduct disorder and ADHD,” Dr. van den Brink said.
It was shown half-a-decade ago that normal doses of methylphenidate have no effect on ADHD symptoms or substance use in comorbid adults. Then Swedish investigators reported that treating criminal offenders with high-dose methylphenidate – roughly three times greater than standard dosing – was effective in reducing both ADHD symptoms and comorbid substance use in criminal offenders. Those findings prompted investigators at the New York State Psychiatric Institute and the University of Minnesota to examine whether prescribing extended-release mixed amphetamine salts in adults with comorbid cocaine use disorder and ADHD would achieve improvement in both conditions. Indeed, it did, Dr. van den Brink said.
One hundred twenty-six affected patients were randomized to 60 or 80 mg/day of extended-release mixed amphetamine salts or placebo for 13 weeks coupled with weekly individual cognitive-behavioral therapy for all in this double-blind, three-arm clinical trial.
“They showed a number-needed-to-treat of about 2.5 in order to achieve a significant reduction in cocaine use and a very nice reduction in ADHD symptoms with a number-needed-to-treat of 3,” Dr. van den Brink said.
The rate of continuous cocaine abstinence in the last 3 weeks of the trial was 30% in the 80-mg group and 17.5% with 60 mg of extended-release mixed amphetamine salts, compared with just 7% with placebo (JAMA Psychiatry. 2015 Jun;72[6]:593-602).
Interpreting baclofen studies
The first high-quality multicenter, randomized, placebo-controlled, double-blind clinical trial, conducted in Germany, showed baclofen (Lioresal) at a mean dose of 180 mg/day was effective in maintaining alcohol abstinence (Eur Neuropsychopharmacol. 2015 Aug;25[8]:1167-77).
“They got wonderful results, with a number-needed-to-treat of 2.3. That is something we’re not used to seeing in the treatment of alcoholism. But there was no dose-response effect, which is a little unusual,” the psychiatrist observed.
Then a multicenter group of Dutch investigators, including Dr. van den Brink, carried out what they believed would be a confirmatory randomized, double-blind, placebo-controlled trial. However, it showed no difference between high- or low-dose baclofen and placebo in time to relapse (Eur Neuropsychopharmacol. 2016 Dec;26[12]:1950-9).
Little further light was shed by the two large French randomized, placebo-controlled clinical trials presented at the 2016 World Congress for Alcohol and Alcoholism in Berlin. One, the BACLOVILLE trial, included 320 patients treated in 60 family practice clinics; it showed strongly positive results for high-dose baclofen. In contrast, the 316-patient ALPADIR study proved negative. These conflicting results were particularly disappointing because France has been at the forefront of using high-dose baclofen to treat alcoholism, Dr. van den Brink said.
“Maybe some 100,000 people have been treated with high-dose baclofen for alcoholism in France,” he said. “What is the conclusion from all these baclofen studies? You can interpret them in many ways. Maybe there are two positive trials and two negative trials, or maybe there are two positive trials and two failed trials. The debate is not closed, even after four randomized trials.”
Dr. van den Brink reported receiving research funding from and/or serving as a consultant to more than half a dozen pharmaceutical companies.
VIENNA – The prospect on the horizon of two new effective therapies for chronic cocaine dependence – sustained-release dextroamphetamine and subanesthetic ketamine infusions – was among the top developments of the year in addiction medicine, Wim van den Brink, MD, PhD, said at the annual congress of the European College of Neuropsychopharmacology.
Other highlights on his list included:
• Studies establishing that comorbid attention-deficit/hyperactivity disorder (ADHD) and substance use disorder now can be treated effectively with either extended-release mixed amphetamine salts or high-dose methylphenidate.
• Release of a puzzling array of conflicting studies on the use of high-dose baclofen for treatment of alcohol dependence. It’s tough to reconcile this mishmash of polar opposite results. And that dictates it’s time to declare a moratorium on the use of this therapy in clinical practice, which in many places is now widespread, said Dr. van den Brink, professor of psychiatry and addiction at the University of Amsterdam and director of the Amsterdam Institute for Addiction Research.
“It’s too strange that we have such conflicting evidence out there. Too many people are prescribing crazy-high doses of baclofen with no strong supporting evidence,” Dr. van den Brink said.
Cocaine dependence
Dr. van den Brink was a coinvestigator in a Dutch multicenter randomized, double-blind, placebo-controlled trial of multitreatment-refractory comorbid cocaine dependence in 73 heroin-dependent patients in heroin-assisted treatment. Patients assigned to 60 mg/day of sustained-release dextroamphetamine, in addition to the background methadone and diacetylmorphine all participants were on for their heroin dependence, had significantly fewer days of cocaine use in the 12-week study: a mean of 44.9 days, compared with 60.6 days in placebo-treated controls. Adverse events were transient and well tolerated (Lancet. 2016 May 28;387[10034]:2226-34).
“A lot of medications have been tried for treatment of cocaine dependence, but actually none of them has been shown to be effective with the exception of substitution treatment with stimulants. Ours is one of the most successful trials. These patients were using cocaine an average of 24 days per month along with a lot of other drugs, despite being in heroin treatment for 4 years,” Dr. van den Brink said. “Patients were very willing to take the sustained-release dextroamphetamine. In the last 4 weeks, 84% of them used at least 80% of their medication. And they were blinded to what they were using.
“We saw good effect sizes: 0.6-0.7 for self-report measures and 0.31 for negative urine samples. So this is a very promising approach. But it also means that, like with tobacco dependence or alcohol dependence, we have to start thinking about substitution therapy in stimulant-dependent patients,” he said.
Dr. van den Brink said subanesthetic ketamine as a novel treatment for cocaine dependence is not yet ready for prime time use in clinical practice, because it’s just not practical to bring patients in for a roughly hour-long intravenous infusion on a daily basis, as was done in a highly impressive proof-of-concept study. But new formulations of ketamine are under development that should better lend themselves to use in clinical practice.
In the proof-of-concept study, investigators at the New York State Psychiatric Institute brought into the laboratory cocaine-dependent volunteers not seeking treatment or abstinence and administered 52-minute infusions of ketamine at 0.41 or 71 mg/kg or lorazepam at 2 mg (Biol Psychiatry. 2014 Jul 1;76[1]:40-6). Lorazepam had absolutely no effect on motivation to change, but ketamine was a different story.
“As soon as you give a low dose of ketamine, you see a wonderful effect on motivation to change and on craving ratings in assessments at 24 hours post infusion. This looks like another promising way of treating cocaine dependence,” he said.
Doxazosin for alcoholism
Investigators at the National Institute on Alcohol Abuse and Alcoholism and several U.S. universities hypothesized that the norepinephrine system could be an important treatment target in alcohol dependence. They conducted a double-blind, placebo-controlled randomized trial in which alcohol-dependent patients seeking outpatient treatment were assigned to the alpha1-adrenergic blocker doxazosin (Cardura) titrated to a maximum of 16 mg/day or placebo. They found doxazosin significantly reduced drinks per week and the number of heavy drinking days per week, but only in the subgroup of patients with a strong family history of alcoholism. In patients without such a family history, doxazosin paradoxically increased drinking (Addict Biol. 2016 Jul;21[4]:904-14).
One of the reasons adult ADHD is greatly underrecognized is that it tends to occur in combination with flashier substance use disorders. “Addiction is very comorbid with all kinds of disorders, but especially with externalizing childhood disorders like conduct disorder and ADHD,” Dr. van den Brink said.
It was shown half-a-decade ago that normal doses of methylphenidate have no effect on ADHD symptoms or substance use in comorbid adults. Then Swedish investigators reported that treating criminal offenders with high-dose methylphenidate – roughly three times greater than standard dosing – was effective in reducing both ADHD symptoms and comorbid substance use in criminal offenders. Those findings prompted investigators at the New York State Psychiatric Institute and the University of Minnesota to examine whether prescribing extended-release mixed amphetamine salts in adults with comorbid cocaine use disorder and ADHD would achieve improvement in both conditions. Indeed, it did, Dr. van den Brink said.
One hundred twenty-six affected patients were randomized to 60 or 80 mg/day of extended-release mixed amphetamine salts or placebo for 13 weeks coupled with weekly individual cognitive-behavioral therapy for all in this double-blind, three-arm clinical trial.
“They showed a number-needed-to-treat of about 2.5 in order to achieve a significant reduction in cocaine use and a very nice reduction in ADHD symptoms with a number-needed-to-treat of 3,” Dr. van den Brink said.
The rate of continuous cocaine abstinence in the last 3 weeks of the trial was 30% in the 80-mg group and 17.5% with 60 mg of extended-release mixed amphetamine salts, compared with just 7% with placebo (JAMA Psychiatry. 2015 Jun;72[6]:593-602).
Interpreting baclofen studies
The first high-quality multicenter, randomized, placebo-controlled, double-blind clinical trial, conducted in Germany, showed baclofen (Lioresal) at a mean dose of 180 mg/day was effective in maintaining alcohol abstinence (Eur Neuropsychopharmacol. 2015 Aug;25[8]:1167-77).
“They got wonderful results, with a number-needed-to-treat of 2.3. That is something we’re not used to seeing in the treatment of alcoholism. But there was no dose-response effect, which is a little unusual,” the psychiatrist observed.
Then a multicenter group of Dutch investigators, including Dr. van den Brink, carried out what they believed would be a confirmatory randomized, double-blind, placebo-controlled trial. However, it showed no difference between high- or low-dose baclofen and placebo in time to relapse (Eur Neuropsychopharmacol. 2016 Dec;26[12]:1950-9).
Little further light was shed by the two large French randomized, placebo-controlled clinical trials presented at the 2016 World Congress for Alcohol and Alcoholism in Berlin. One, the BACLOVILLE trial, included 320 patients treated in 60 family practice clinics; it showed strongly positive results for high-dose baclofen. In contrast, the 316-patient ALPADIR study proved negative. These conflicting results were particularly disappointing because France has been at the forefront of using high-dose baclofen to treat alcoholism, Dr. van den Brink said.
“Maybe some 100,000 people have been treated with high-dose baclofen for alcoholism in France,” he said. “What is the conclusion from all these baclofen studies? You can interpret them in many ways. Maybe there are two positive trials and two negative trials, or maybe there are two positive trials and two failed trials. The debate is not closed, even after four randomized trials.”
Dr. van den Brink reported receiving research funding from and/or serving as a consultant to more than half a dozen pharmaceutical companies.
EXPERT ANALYSIS FROM THE ECNP CONGRESS

Survey shines new light on weighty comorbidity burden in adult atopic dermatitis
VIENNA – Newly enhanced appreciation of the profound burden of comorbidities associated with adult atopic dermatitis (AD) is provided by the Liberty AD-AWARE study, investigators said at a joint program of the International Eczema Council and the International Psoriasis Council held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
“I think the only reason we thought psoriasis is a systemic disease and atopic dermatitis is not is because people were researching it much more in psoriasis. I think atopic dermatitis will emerge as potentially more systemic than psoriasis, including the comorbidities. It’s just a matter of time before the evidence is put forth for atopic dermatitis,” predicted Emma Guttman-Yassky, MD, PhD, professor and vice chair of the department of dermatology at Mount Sinai School of Medicine in New York.

Dr. Guttman-Yassky noted that 85% of cases of AD begin before 5 years of age. Many cases resolve later in childhood, but for others it becomes a chronic lifelong condition. And while the burden of AD has been well characterized in the pediatric population, that’s not so in affected adults. This was the impetus for the Liberty AD-AWARE (Adults With Atopic Dermatitis Reporting on their Experience) study, an Internet-based cross-sectional survey of more than 1,500 adults with AD receiving their care from dermatologists at eight major U.S. academic medical centers.
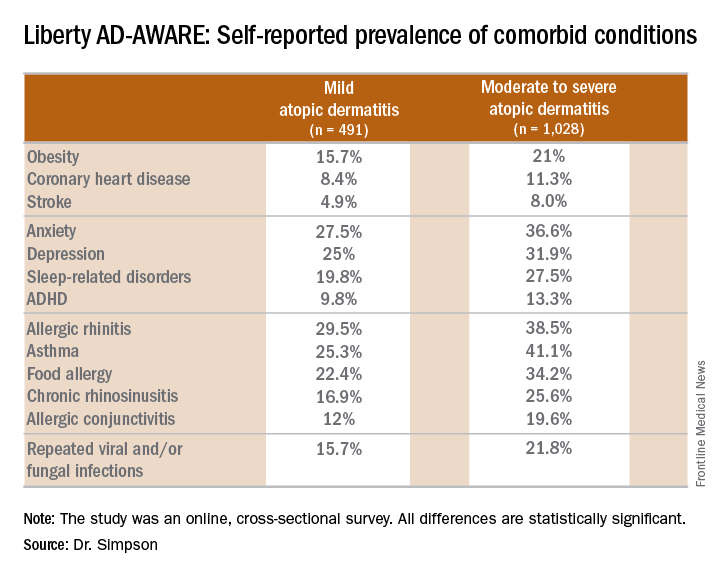
Eric L. Simpson, MD, a coinvestigator with Dr. Guttman-Yassky in Liberty AD-AWARE, observed that the study documented self-reported high rates of a range of psychiatric, cardiovascular, allergic, respiratory, and infectious diseases in participants. And while a cross-sectional study can’t establish causality, it’s important to appreciate that rates of these comorbidities were across the board significantly higher in the 1,028 patients with moderate to severe AD over the prior 12 months than in the 491 classified as having mild AD.
These associations between AD and mental health problems have been confirmed in other studies. For example, a recent analysis of data on more than 354,000 children and nearly 35,000 adults in the United States demonstrated that AD was independently associated with a 14% increased likelihood of attention-deficit/hyperactivity disorder in children and a 61% increased risk in adults. Those risks of ADHD rose far higher in individuals with severe AD and sleep disruption (Br J Dermatol. 2016 Nov;175[5]:920-9).
A number of theories have been put forth to explain these associations, including altered brain development stemming from early exposure to inflammatory cytokines or perhaps shared genetic predisposition, but Dr. Simpson proposed a simpler explanation which carries more optimistic implications.
“I suspect the mental health problems associated with adult atopic dermatitis are probably nonspecific sequelae of any chronic skin disorder involving severe itch and sleep disturbances,” said Dr. Simpson, professor of dermatology at Oregon Health & Science University, Portland.
Moreover, there is good reason to believe that novel therapies targeting inflammation more effectively than what’s been available to date may help improve mental health outcomes, as well as asthma in affected adults with AD, he added. He cited a phase IIb, randomized, double-blind, placebo-controlled study for which he was lead investigator. In this trial, 16 weeks of treatment with dupilumab, a first-in-class investigational blocker of the interleukin-4/interleukin-13 signaling pathway, not only resulted in significant reductions in itch and sleep problems, it also decreased anxiety and depression symptoms and improved multiple validated measures of health-related quality of life (J Am Acad Dermatol. 2016 Sep;75[3]:506-15).
Liberty AD-AWARE provides hints of the profound cumulative negative impact moderate to severe AD can have on a patient’s life course. Among the group with moderate to severe disease, 7.5% said AD had a large negative effect on their pursuit of an education, 10.7% said their disease had influenced their career choice “a lot/very much,” 13.3% were unemployed for reasons other than being retired or a student, and 17.1% reported an annual family income of less than $25,000. All these rates were multifold higher than in patients with mild AD in the study, which didn’t include a non-AD control group.
Dr. Guttman-Yassky observed that 42% of the moderate to severe AD group in Liberty AD-AWARE reported their current treatments were ineffective at controlling their disease, even though study participants were presumably receiving high-quality care at academic medical centers. Twenty-eight percent of patients with inadequately controlled AD had used phototherapy or an immunomodulatory drug within the past 7 days, underscoring the limitations of those forms of therapy in patients with more severe AD as well as the need for new and better treatments.
Dr. Guttman-Yassky has played a key role in the paradigm shift regarding understanding of the pathogenesis of AD as involving not just disordered skin barrier function but also immunologic impairment. She was senior author of a study that showed the nonlesional skin of patients with AD is characterized by high-level expression of inflammatory cytokines, whereas the nonlesional skin of psoriasis patients is not, an observation that serves to highlight the need for proactive treatments for AD (J Allergy Clin Immunol. 2011 Apr;127[4]:954-64.e1-4). Later, she and her coworkers demonstrated that AD is characterized by greater levels of T-cell activation among central and effector CD4+ and CD8+CLA+ and CD8+CLA– memory cell subsets (J Allergy Clin Immunol. 2015 Jul;136[1]:208-11).
More recently, she was also senior author of a landmark study that provides a mechanism to account for the reason AD patients would potentially have more comorbid illnesses than psoriasis patients. The investigators demonstrated that AD is accompanied by systemic expansion of transitional and chronically activated memory B cells, plasmablasts, and IgE-expressing memory B cells in both skin and blood. In other words, AD is characterized by a greater level of systemic immune activation, compared with psoriasis, where activated T cells are largely confined to the skin, and activated central memory B cells don’t figure prominently (J Allergy Clin Immunol. 2016 Jan;137[1]:118-29.e5).
The Liberty AD-AWARE study was sponsored by Sanofi and Regeneron. Dr. Simpson and Dr. Guttman-Yassky reported receiving research grants from and serving as consultants to those and other pharmaceutical companies.
VIENNA – Newly enhanced appreciation of the profound burden of comorbidities associated with adult atopic dermatitis (AD) is provided by the Liberty AD-AWARE study, investigators said at a joint program of the International Eczema Council and the International Psoriasis Council held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
“I think the only reason we thought psoriasis is a systemic disease and atopic dermatitis is not is because people were researching it much more in psoriasis. I think atopic dermatitis will emerge as potentially more systemic than psoriasis, including the comorbidities. It’s just a matter of time before the evidence is put forth for atopic dermatitis,” predicted Emma Guttman-Yassky, MD, PhD, professor and vice chair of the department of dermatology at Mount Sinai School of Medicine in New York.
Dr. Guttman-Yassky noted that 85% of cases of AD begin before 5 years of age. Many cases resolve later in childhood, but for others it becomes a chronic lifelong condition. And while the burden of AD has been well characterized in the pediatric population, that’s not so in affected adults. This was the impetus for the Liberty AD-AWARE (Adults With Atopic Dermatitis Reporting on their Experience) study, an Internet-based cross-sectional survey of more than 1,500 adults with AD receiving their care from dermatologists at eight major U.S. academic medical centers.
Eric L. Simpson, MD, a coinvestigator with Dr. Guttman-Yassky in Liberty AD-AWARE, observed that the study documented self-reported high rates of a range of psychiatric, cardiovascular, allergic, respiratory, and infectious diseases in participants. And while a cross-sectional study can’t establish causality, it’s important to appreciate that rates of these comorbidities were across the board significantly higher in the 1,028 patients with moderate to severe AD over the prior 12 months than in the 491 classified as having mild AD.
These associations between AD and mental health problems have been confirmed in other studies. For example, a recent analysis of data on more than 354,000 children and nearly 35,000 adults in the United States demonstrated that AD was independently associated with a 14% increased likelihood of attention-deficit/hyperactivity disorder in children and a 61% increased risk in adults. Those risks of ADHD rose far higher in individuals with severe AD and sleep disruption (Br J Dermatol. 2016 Nov;175[5]:920-9).
A number of theories have been put forth to explain these associations, including altered brain development stemming from early exposure to inflammatory cytokines or perhaps shared genetic predisposition, but Dr. Simpson proposed a simpler explanation which carries more optimistic implications.
“I suspect the mental health problems associated with adult atopic dermatitis are probably nonspecific sequelae of any chronic skin disorder involving severe itch and sleep disturbances,” said Dr. Simpson, professor of dermatology at Oregon Health & Science University, Portland.
Moreover, there is good reason to believe that novel therapies targeting inflammation more effectively than what’s been available to date may help improve mental health outcomes, as well as asthma in affected adults with AD, he added. He cited a phase IIb, randomized, double-blind, placebo-controlled study for which he was lead investigator. In this trial, 16 weeks of treatment with dupilumab, a first-in-class investigational blocker of the interleukin-4/interleukin-13 signaling pathway, not only resulted in significant reductions in itch and sleep problems, it also decreased anxiety and depression symptoms and improved multiple validated measures of health-related quality of life (J Am Acad Dermatol. 2016 Sep;75[3]:506-15).
Liberty AD-AWARE provides hints of the profound cumulative negative impact moderate to severe AD can have on a patient’s life course. Among the group with moderate to severe disease, 7.5% said AD had a large negative effect on their pursuit of an education, 10.7% said their disease had influenced their career choice “a lot/very much,” 13.3% were unemployed for reasons other than being retired or a student, and 17.1% reported an annual family income of less than $25,000. All these rates were multifold higher than in patients with mild AD in the study, which didn’t include a non-AD control group.
Dr. Guttman-Yassky observed that 42% of the moderate to severe AD group in Liberty AD-AWARE reported their current treatments were ineffective at controlling their disease, even though study participants were presumably receiving high-quality care at academic medical centers. Twenty-eight percent of patients with inadequately controlled AD had used phototherapy or an immunomodulatory drug within the past 7 days, underscoring the limitations of those forms of therapy in patients with more severe AD as well as the need for new and better treatments.
Dr. Guttman-Yassky has played a key role in the paradigm shift regarding understanding of the pathogenesis of AD as involving not just disordered skin barrier function but also immunologic impairment. She was senior author of a study that showed the nonlesional skin of patients with AD is characterized by high-level expression of inflammatory cytokines, whereas the nonlesional skin of psoriasis patients is not, an observation that serves to highlight the need for proactive treatments for AD (J Allergy Clin Immunol. 2011 Apr;127[4]:954-64.e1-4). Later, she and her coworkers demonstrated that AD is characterized by greater levels of T-cell activation among central and effector CD4+ and CD8+CLA+ and CD8+CLA– memory cell subsets (J Allergy Clin Immunol. 2015 Jul;136[1]:208-11).
More recently, she was also senior author of a landmark study that provides a mechanism to account for the reason AD patients would potentially have more comorbid illnesses than psoriasis patients. The investigators demonstrated that AD is accompanied by systemic expansion of transitional and chronically activated memory B cells, plasmablasts, and IgE-expressing memory B cells in both skin and blood. In other words, AD is characterized by a greater level of systemic immune activation, compared with psoriasis, where activated T cells are largely confined to the skin, and activated central memory B cells don’t figure prominently (J Allergy Clin Immunol. 2016 Jan;137[1]:118-29.e5).
The Liberty AD-AWARE study was sponsored by Sanofi and Regeneron. Dr. Simpson and Dr. Guttman-Yassky reported receiving research grants from and serving as consultants to those and other pharmaceutical companies.
VIENNA – Newly enhanced appreciation of the profound burden of comorbidities associated with adult atopic dermatitis (AD) is provided by the Liberty AD-AWARE study, investigators said at a joint program of the International Eczema Council and the International Psoriasis Council held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
“I think the only reason we thought psoriasis is a systemic disease and atopic dermatitis is not is because people were researching it much more in psoriasis. I think atopic dermatitis will emerge as potentially more systemic than psoriasis, including the comorbidities. It’s just a matter of time before the evidence is put forth for atopic dermatitis,” predicted Emma Guttman-Yassky, MD, PhD, professor and vice chair of the department of dermatology at Mount Sinai School of Medicine in New York.
Dr. Guttman-Yassky noted that 85% of cases of AD begin before 5 years of age. Many cases resolve later in childhood, but for others it becomes a chronic lifelong condition. And while the burden of AD has been well characterized in the pediatric population, that’s not so in affected adults. This was the impetus for the Liberty AD-AWARE (Adults With Atopic Dermatitis Reporting on their Experience) study, an Internet-based cross-sectional survey of more than 1,500 adults with AD receiving their care from dermatologists at eight major U.S. academic medical centers.
Eric L. Simpson, MD, a coinvestigator with Dr. Guttman-Yassky in Liberty AD-AWARE, observed that the study documented self-reported high rates of a range of psychiatric, cardiovascular, allergic, respiratory, and infectious diseases in participants. And while a cross-sectional study can’t establish causality, it’s important to appreciate that rates of these comorbidities were across the board significantly higher in the 1,028 patients with moderate to severe AD over the prior 12 months than in the 491 classified as having mild AD.
These associations between AD and mental health problems have been confirmed in other studies. For example, a recent analysis of data on more than 354,000 children and nearly 35,000 adults in the United States demonstrated that AD was independently associated with a 14% increased likelihood of attention-deficit/hyperactivity disorder in children and a 61% increased risk in adults. Those risks of ADHD rose far higher in individuals with severe AD and sleep disruption (Br J Dermatol. 2016 Nov;175[5]:920-9).
A number of theories have been put forth to explain these associations, including altered brain development stemming from early exposure to inflammatory cytokines or perhaps shared genetic predisposition, but Dr. Simpson proposed a simpler explanation which carries more optimistic implications.
“I suspect the mental health problems associated with adult atopic dermatitis are probably nonspecific sequelae of any chronic skin disorder involving severe itch and sleep disturbances,” said Dr. Simpson, professor of dermatology at Oregon Health & Science University, Portland.
Moreover, there is good reason to believe that novel therapies targeting inflammation more effectively than what’s been available to date may help improve mental health outcomes, as well as asthma in affected adults with AD, he added. He cited a phase IIb, randomized, double-blind, placebo-controlled study for which he was lead investigator. In this trial, 16 weeks of treatment with dupilumab, a first-in-class investigational blocker of the interleukin-4/interleukin-13 signaling pathway, not only resulted in significant reductions in itch and sleep problems, it also decreased anxiety and depression symptoms and improved multiple validated measures of health-related quality of life (J Am Acad Dermatol. 2016 Sep;75[3]:506-15).
Liberty AD-AWARE provides hints of the profound cumulative negative impact moderate to severe AD can have on a patient’s life course. Among the group with moderate to severe disease, 7.5% said AD had a large negative effect on their pursuit of an education, 10.7% said their disease had influenced their career choice “a lot/very much,” 13.3% were unemployed for reasons other than being retired or a student, and 17.1% reported an annual family income of less than $25,000. All these rates were multifold higher than in patients with mild AD in the study, which didn’t include a non-AD control group.
Dr. Guttman-Yassky observed that 42% of the moderate to severe AD group in Liberty AD-AWARE reported their current treatments were ineffective at controlling their disease, even though study participants were presumably receiving high-quality care at academic medical centers. Twenty-eight percent of patients with inadequately controlled AD had used phototherapy or an immunomodulatory drug within the past 7 days, underscoring the limitations of those forms of therapy in patients with more severe AD as well as the need for new and better treatments.
Dr. Guttman-Yassky has played a key role in the paradigm shift regarding understanding of the pathogenesis of AD as involving not just disordered skin barrier function but also immunologic impairment. She was senior author of a study that showed the nonlesional skin of patients with AD is characterized by high-level expression of inflammatory cytokines, whereas the nonlesional skin of psoriasis patients is not, an observation that serves to highlight the need for proactive treatments for AD (J Allergy Clin Immunol. 2011 Apr;127[4]:954-64.e1-4). Later, she and her coworkers demonstrated that AD is characterized by greater levels of T-cell activation among central and effector CD4+ and CD8+CLA+ and CD8+CLA– memory cell subsets (J Allergy Clin Immunol. 2015 Jul;136[1]:208-11).
More recently, she was also senior author of a landmark study that provides a mechanism to account for the reason AD patients would potentially have more comorbid illnesses than psoriasis patients. The investigators demonstrated that AD is accompanied by systemic expansion of transitional and chronically activated memory B cells, plasmablasts, and IgE-expressing memory B cells in both skin and blood. In other words, AD is characterized by a greater level of systemic immune activation, compared with psoriasis, where activated T cells are largely confined to the skin, and activated central memory B cells don’t figure prominently (J Allergy Clin Immunol. 2016 Jan;137[1]:118-29.e5).
The Liberty AD-AWARE study was sponsored by Sanofi and Regeneron. Dr. Simpson and Dr. Guttman-Yassky reported receiving research grants from and serving as consultants to those and other pharmaceutical companies.
EXPERT ANALYSIS FROM THE EADV CONGRESS
Reasons for noncompliance to ketogenic diet explored
HOUSTON – More than one-third of children discontinued the ketogenic diet prior to completion of a 3-month trial because of reported difficulty, a single-center study showed.
The findings underscore the importance of carefully screening patients and their families prior to initiating the ketogenic diet, lead author Gogi Kumar, MD, said in an interview at the annual meeting of the American Epilepsy Society. “We always talk about how and when the ketogenic diet should be used, but we don’t talk about the barriers to continuing the diet, like the socioeconomic aspects that determine the feasibility of continuing the diet,” said Dr. Kumar, a pediatric neurologist at Dayton (Ohio) Children’s Hospital. “If it’s a single mom who has two jobs and has a kid with intractable epilepsy, she cannot do it because ketogenic diets are complex and very intense. If the child has a gastrostomy tube and you can feed them a formula it might work, but then you have to bring them in for multiple lab tests. The family has to be very committed. They have to have resources.”

The mean age of study participants was 7.4 years, and feeding was accomplished orally in 43%, by tube in 40%, and both routes in 17%. Of the patients who were started on a ketogenic diet, 57% continued on the diet for more than 3 months. Overall, 55% of patients experienced at least a 50% reduction in seizures, while 45% experienced less than a 50% reduction in seizures.
Dr. Kumar went on to report that 15 patients (43%) discontinued the diet before the 3-month trial period. Of these 15 patients, 3 had adverse effects after initiation of the diet and the remaining 12 reported stopping the diet because of difficulty, 1 of whom also reported cost as a barrier. Of the 12 patients who stopped the diet because of difficulty, 8 were on the classic ketogenic diet and 4 were on other diet therapies. All were oral eaters and 50% lived in a single-parent household or had shared parenting in multiple households with poor communication. The remaining 50% had married parents, of whom 25% were teenagers who did not want to commit to the diet and 25% were children with parents who found the diet difficult. Of families who discontinued the diet early, 58% of parents had difficulty learning how to manage the complexity of the ketogenic diet and/or had limited cooking skills.
“Before you try someone on the ketogenic diet, we should evaluate the family’s educational level, their commitment, and their support systems so that we can help them overcome any barriers. It is important to have social workers as part of the ketogenic diet team to help with the process,” Dr. Kumar said. She reported having no financial disclosures.
HOUSTON – More than one-third of children discontinued the ketogenic diet prior to completion of a 3-month trial because of reported difficulty, a single-center study showed.
The findings underscore the importance of carefully screening patients and their families prior to initiating the ketogenic diet, lead author Gogi Kumar, MD, said in an interview at the annual meeting of the American Epilepsy Society. “We always talk about how and when the ketogenic diet should be used, but we don’t talk about the barriers to continuing the diet, like the socioeconomic aspects that determine the feasibility of continuing the diet,” said Dr. Kumar, a pediatric neurologist at Dayton (Ohio) Children’s Hospital. “If it’s a single mom who has two jobs and has a kid with intractable epilepsy, she cannot do it because ketogenic diets are complex and very intense. If the child has a gastrostomy tube and you can feed them a formula it might work, but then you have to bring them in for multiple lab tests. The family has to be very committed. They have to have resources.”
The mean age of study participants was 7.4 years, and feeding was accomplished orally in 43%, by tube in 40%, and both routes in 17%. Of the patients who were started on a ketogenic diet, 57% continued on the diet for more than 3 months. Overall, 55% of patients experienced at least a 50% reduction in seizures, while 45% experienced less than a 50% reduction in seizures.
Dr. Kumar went on to report that 15 patients (43%) discontinued the diet before the 3-month trial period. Of these 15 patients, 3 had adverse effects after initiation of the diet and the remaining 12 reported stopping the diet because of difficulty, 1 of whom also reported cost as a barrier. Of the 12 patients who stopped the diet because of difficulty, 8 were on the classic ketogenic diet and 4 were on other diet therapies. All were oral eaters and 50% lived in a single-parent household or had shared parenting in multiple households with poor communication. The remaining 50% had married parents, of whom 25% were teenagers who did not want to commit to the diet and 25% were children with parents who found the diet difficult. Of families who discontinued the diet early, 58% of parents had difficulty learning how to manage the complexity of the ketogenic diet and/or had limited cooking skills.
“Before you try someone on the ketogenic diet, we should evaluate the family’s educational level, their commitment, and their support systems so that we can help them overcome any barriers. It is important to have social workers as part of the ketogenic diet team to help with the process,” Dr. Kumar said. She reported having no financial disclosures.
HOUSTON – More than one-third of children discontinued the ketogenic diet prior to completion of a 3-month trial because of reported difficulty, a single-center study showed.
The findings underscore the importance of carefully screening patients and their families prior to initiating the ketogenic diet, lead author Gogi Kumar, MD, said in an interview at the annual meeting of the American Epilepsy Society. “We always talk about how and when the ketogenic diet should be used, but we don’t talk about the barriers to continuing the diet, like the socioeconomic aspects that determine the feasibility of continuing the diet,” said Dr. Kumar, a pediatric neurologist at Dayton (Ohio) Children’s Hospital. “If it’s a single mom who has two jobs and has a kid with intractable epilepsy, she cannot do it because ketogenic diets are complex and very intense. If the child has a gastrostomy tube and you can feed them a formula it might work, but then you have to bring them in for multiple lab tests. The family has to be very committed. They have to have resources.”
The mean age of study participants was 7.4 years, and feeding was accomplished orally in 43%, by tube in 40%, and both routes in 17%. Of the patients who were started on a ketogenic diet, 57% continued on the diet for more than 3 months. Overall, 55% of patients experienced at least a 50% reduction in seizures, while 45% experienced less than a 50% reduction in seizures.
Dr. Kumar went on to report that 15 patients (43%) discontinued the diet before the 3-month trial period. Of these 15 patients, 3 had adverse effects after initiation of the diet and the remaining 12 reported stopping the diet because of difficulty, 1 of whom also reported cost as a barrier. Of the 12 patients who stopped the diet because of difficulty, 8 were on the classic ketogenic diet and 4 were on other diet therapies. All were oral eaters and 50% lived in a single-parent household or had shared parenting in multiple households with poor communication. The remaining 50% had married parents, of whom 25% were teenagers who did not want to commit to the diet and 25% were children with parents who found the diet difficult. Of families who discontinued the diet early, 58% of parents had difficulty learning how to manage the complexity of the ketogenic diet and/or had limited cooking skills.
“Before you try someone on the ketogenic diet, we should evaluate the family’s educational level, their commitment, and their support systems so that we can help them overcome any barriers. It is important to have social workers as part of the ketogenic diet team to help with the process,” Dr. Kumar said. She reported having no financial disclosures.
AT AES 2016
Key clinical point:
Major finding: More than one-third of children (43%) discontinued the ketogenic diet before the end of a 3-month trial period.
Data source: A prospective evaluation of 64 patients with intractable epilepsy and their families who were educated about the ketogenic diet at Dayton Children’s Hospital.
Disclosures: Dr. Kumar reported having no financial disclosures.
First visit for tuberous sclerosis complex comes months before diagnosis
HOUSTON – Many patients who eventually receive a diagnosis of tuberous sclerosis complex present with related complaints for months, or even years, before their condition is recognized and correctly diagnosed, according to a retrospective study.
The study, presented at the annual meeting of the American Epilepsy Society, found that patients with tuberous sclerosis complex (TSC) first sought care for TSC-related conditions an average of 7 months before they were diagnosed with the condition. Younger patients received the correct diagnosis sooner than did older patients: Treatment for TSC-related conditions preceded the diagnosis for 3.4 months for those aged 4 years or younger, compared with 5.5 months for those aged 25-29 years, and 21 months for those aged 80 years or older.
Seizures and skin conditions were common initial diagnoses among TSC patients, with 27% of patients aged 0-4 years being diagnosed with seizures prior to receiving their TSC diagnosis. The likelihood of prediagnosis visits for seizures decreased to less than 6% for older age groups. Seizures remained common post-TSC diagnosis among younger patients, with 38% of patients aged 0-4 years having any seizure diagnosis, while the rate fell through the lifespan to zero for those aged 80 or older.
James Wheless, MD, and his associates examined claims and enrollment data records from 2,163 patients diagnosed with TSC between January 2000 and December 2011. In addition to the frequently-diagnosed seizures seen in many TSC patients, skin conditions were diagnosed in 16.3% of patients before their eventual TSC diagnosis.
Other early conditions associated with TSC, according to the study’s multivariable analysis, included bone cysts, anxiety, and ADHD. However, wrote Dr. Wheless, chief of the department of pediatric neurology at the University of Tennessee Health Science Center, Memphis, and his coauthors, “at any point in time, patients with seizures were 2.9 times more likely to receive a TSC diagnosis than patients without seizures.”
The study was drawn from U.S. health plan databases that included both commercial and Medicare Advantage enrollees, and included patients through the lifespan. The date of the first recorded TSC diagnosis was the index date, and patients had to have at least 12 months of prediagnosis health plan enrollment to be included, or 6 months for those aged 2 years or younger. Data were collected for all pre-index visits (some of which stretched back to 1993), and for visits in the 12 months after the index visit.
The proportion of female patients ranged from fewer than half for those under 15 years (0-4 years, 46%; 5-9 years, 43%; 10-14 years, 48%) to 64% for those aged 80 years or older (P less than .001).
Dr. Wheless and his coauthors noted that the claims data used for the analysis “may not adequately capture clinical characteristics such as disease severity,” and that some patient data may have been lost if patients were disenrolled for periods of time during the study period.
The findings of the poster may prompt clinicians to consider TSC as a diagnosis; though rare, occurring in 1-2 per 6,000 live births, it’s thought to be an underrecognized disease entity. “Understanding the initial diagnoses experienced by TSC patients may help lead to earlier diagnosis and treatment of TSC,” Dr. Wheless and his coauthors wrote.
Novartis funded the study. Four study authors are employed by Optum, and one is employed by Novartis.
[email protected]
On Twitter @karioakes
HOUSTON – Many patients who eventually receive a diagnosis of tuberous sclerosis complex present with related complaints for months, or even years, before their condition is recognized and correctly diagnosed, according to a retrospective study.
The study, presented at the annual meeting of the American Epilepsy Society, found that patients with tuberous sclerosis complex (TSC) first sought care for TSC-related conditions an average of 7 months before they were diagnosed with the condition. Younger patients received the correct diagnosis sooner than did older patients: Treatment for TSC-related conditions preceded the diagnosis for 3.4 months for those aged 4 years or younger, compared with 5.5 months for those aged 25-29 years, and 21 months for those aged 80 years or older.
Seizures and skin conditions were common initial diagnoses among TSC patients, with 27% of patients aged 0-4 years being diagnosed with seizures prior to receiving their TSC diagnosis. The likelihood of prediagnosis visits for seizures decreased to less than 6% for older age groups. Seizures remained common post-TSC diagnosis among younger patients, with 38% of patients aged 0-4 years having any seizure diagnosis, while the rate fell through the lifespan to zero for those aged 80 or older.
James Wheless, MD, and his associates examined claims and enrollment data records from 2,163 patients diagnosed with TSC between January 2000 and December 2011. In addition to the frequently-diagnosed seizures seen in many TSC patients, skin conditions were diagnosed in 16.3% of patients before their eventual TSC diagnosis.
Other early conditions associated with TSC, according to the study’s multivariable analysis, included bone cysts, anxiety, and ADHD. However, wrote Dr. Wheless, chief of the department of pediatric neurology at the University of Tennessee Health Science Center, Memphis, and his coauthors, “at any point in time, patients with seizures were 2.9 times more likely to receive a TSC diagnosis than patients without seizures.”
The study was drawn from U.S. health plan databases that included both commercial and Medicare Advantage enrollees, and included patients through the lifespan. The date of the first recorded TSC diagnosis was the index date, and patients had to have at least 12 months of prediagnosis health plan enrollment to be included, or 6 months for those aged 2 years or younger. Data were collected for all pre-index visits (some of which stretched back to 1993), and for visits in the 12 months after the index visit.
The proportion of female patients ranged from fewer than half for those under 15 years (0-4 years, 46%; 5-9 years, 43%; 10-14 years, 48%) to 64% for those aged 80 years or older (P less than .001).
Dr. Wheless and his coauthors noted that the claims data used for the analysis “may not adequately capture clinical characteristics such as disease severity,” and that some patient data may have been lost if patients were disenrolled for periods of time during the study period.
The findings of the poster may prompt clinicians to consider TSC as a diagnosis; though rare, occurring in 1-2 per 6,000 live births, it’s thought to be an underrecognized disease entity. “Understanding the initial diagnoses experienced by TSC patients may help lead to earlier diagnosis and treatment of TSC,” Dr. Wheless and his coauthors wrote.
Novartis funded the study. Four study authors are employed by Optum, and one is employed by Novartis.
[email protected]
On Twitter @karioakes
HOUSTON – Many patients who eventually receive a diagnosis of tuberous sclerosis complex present with related complaints for months, or even years, before their condition is recognized and correctly diagnosed, according to a retrospective study.
The study, presented at the annual meeting of the American Epilepsy Society, found that patients with tuberous sclerosis complex (TSC) first sought care for TSC-related conditions an average of 7 months before they were diagnosed with the condition. Younger patients received the correct diagnosis sooner than did older patients: Treatment for TSC-related conditions preceded the diagnosis for 3.4 months for those aged 4 years or younger, compared with 5.5 months for those aged 25-29 years, and 21 months for those aged 80 years or older.
Seizures and skin conditions were common initial diagnoses among TSC patients, with 27% of patients aged 0-4 years being diagnosed with seizures prior to receiving their TSC diagnosis. The likelihood of prediagnosis visits for seizures decreased to less than 6% for older age groups. Seizures remained common post-TSC diagnosis among younger patients, with 38% of patients aged 0-4 years having any seizure diagnosis, while the rate fell through the lifespan to zero for those aged 80 or older.
James Wheless, MD, and his associates examined claims and enrollment data records from 2,163 patients diagnosed with TSC between January 2000 and December 2011. In addition to the frequently-diagnosed seizures seen in many TSC patients, skin conditions were diagnosed in 16.3% of patients before their eventual TSC diagnosis.
Other early conditions associated with TSC, according to the study’s multivariable analysis, included bone cysts, anxiety, and ADHD. However, wrote Dr. Wheless, chief of the department of pediatric neurology at the University of Tennessee Health Science Center, Memphis, and his coauthors, “at any point in time, patients with seizures were 2.9 times more likely to receive a TSC diagnosis than patients without seizures.”
The study was drawn from U.S. health plan databases that included both commercial and Medicare Advantage enrollees, and included patients through the lifespan. The date of the first recorded TSC diagnosis was the index date, and patients had to have at least 12 months of prediagnosis health plan enrollment to be included, or 6 months for those aged 2 years or younger. Data were collected for all pre-index visits (some of which stretched back to 1993), and for visits in the 12 months after the index visit.
The proportion of female patients ranged from fewer than half for those under 15 years (0-4 years, 46%; 5-9 years, 43%; 10-14 years, 48%) to 64% for those aged 80 years or older (P less than .001).
Dr. Wheless and his coauthors noted that the claims data used for the analysis “may not adequately capture clinical characteristics such as disease severity,” and that some patient data may have been lost if patients were disenrolled for periods of time during the study period.
The findings of the poster may prompt clinicians to consider TSC as a diagnosis; though rare, occurring in 1-2 per 6,000 live births, it’s thought to be an underrecognized disease entity. “Understanding the initial diagnoses experienced by TSC patients may help lead to earlier diagnosis and treatment of TSC,” Dr. Wheless and his coauthors wrote.
Novartis funded the study. Four study authors are employed by Optum, and one is employed by Novartis.
[email protected]
On Twitter @karioakes
FROM AES 2016
Key clinical point:
Major finding: Patients were seen an average of 6.9 months before they received their TSC diagnosis.
Data source: Retrospective review of claims and enrollment data from 2,163 patients with tuberous sclerosis.
Disclosures: Novartis funded the study. Four study authors are employed by Optum; one is employed by Novartis.
Physicians and EHR time
Clinical question: How much time do ambulatory-care physicians spend on electronic health records (EHRs)?
Background: There is growing concern about physicians’ increased time and effort allocated to the EHR and decreased clinical face time and meaningful interaction with patients. Prior studies have shown that increased physician EHR task load is associated with increased physician stress and dissatisfaction.
Setting: Ambulatory-care practices.
Synopsis: Fifty-seven physicians from 16 practices in four U.S. states participated and were observed for more than 430 office hours. Additionally, 21 physicians completed a self-reported after-hours diary. During office hours, physicians spent 49.2% of their total time on the EHR and desk work and only 27% on face time with patients. While in the exam room, physicians spent 52.9% of the time on direct clinical face time and 37% on the EHR and desk work. Self-reported diaries showed an additional 1-2 hours of follow-up work on the EHR. These observations might not be generalizable to other practices. No formal statistical comparisons by physicians, practice, or EHR characteristics were done.
Bottom line: Ambulatory-care physicians appear to spend more time with EHR tasks and desk work than clinical face time with patients.
Citation: Sinsky C, Colligan L, Li L, et al. Allocation of physician time in ambulatory practice: a time and motion studies in 4 specialties [published online ahead of print Sept. 6, 2016]. Ann Intern Med. 165(11):753-760.
Dr. Briones is an assistant professor at the University of Miami Miller School of Medicine and medical director of the hospitalist service at the University of Miami Hospital.
Clinical question: How much time do ambulatory-care physicians spend on electronic health records (EHRs)?
Background: There is growing concern about physicians’ increased time and effort allocated to the EHR and decreased clinical face time and meaningful interaction with patients. Prior studies have shown that increased physician EHR task load is associated with increased physician stress and dissatisfaction.
Setting: Ambulatory-care practices.
Synopsis: Fifty-seven physicians from 16 practices in four U.S. states participated and were observed for more than 430 office hours. Additionally, 21 physicians completed a self-reported after-hours diary. During office hours, physicians spent 49.2% of their total time on the EHR and desk work and only 27% on face time with patients. While in the exam room, physicians spent 52.9% of the time on direct clinical face time and 37% on the EHR and desk work. Self-reported diaries showed an additional 1-2 hours of follow-up work on the EHR. These observations might not be generalizable to other practices. No formal statistical comparisons by physicians, practice, or EHR characteristics were done.
Bottom line: Ambulatory-care physicians appear to spend more time with EHR tasks and desk work than clinical face time with patients.
Citation: Sinsky C, Colligan L, Li L, et al. Allocation of physician time in ambulatory practice: a time and motion studies in 4 specialties [published online ahead of print Sept. 6, 2016]. Ann Intern Med. 165(11):753-760.
Dr. Briones is an assistant professor at the University of Miami Miller School of Medicine and medical director of the hospitalist service at the University of Miami Hospital.
Clinical question: How much time do ambulatory-care physicians spend on electronic health records (EHRs)?
Background: There is growing concern about physicians’ increased time and effort allocated to the EHR and decreased clinical face time and meaningful interaction with patients. Prior studies have shown that increased physician EHR task load is associated with increased physician stress and dissatisfaction.
Setting: Ambulatory-care practices.
Synopsis: Fifty-seven physicians from 16 practices in four U.S. states participated and were observed for more than 430 office hours. Additionally, 21 physicians completed a self-reported after-hours diary. During office hours, physicians spent 49.2% of their total time on the EHR and desk work and only 27% on face time with patients. While in the exam room, physicians spent 52.9% of the time on direct clinical face time and 37% on the EHR and desk work. Self-reported diaries showed an additional 1-2 hours of follow-up work on the EHR. These observations might not be generalizable to other practices. No formal statistical comparisons by physicians, practice, or EHR characteristics were done.
Bottom line: Ambulatory-care physicians appear to spend more time with EHR tasks and desk work than clinical face time with patients.
Citation: Sinsky C, Colligan L, Li L, et al. Allocation of physician time in ambulatory practice: a time and motion studies in 4 specialties [published online ahead of print Sept. 6, 2016]. Ann Intern Med. 165(11):753-760.
Dr. Briones is an assistant professor at the University of Miami Miller School of Medicine and medical director of the hospitalist service at the University of Miami Hospital.

Machine learning beats clinical prediction of temporal lobe epilepsy surgery outcomes
HOUSTON – A machine learning interpretation of presurgical MRI studies did a better job of predicting which patients would have a successful outcome after anterior temporal lobectomy for temporal lobe epilepsy than did commonly-used clinical indicators in a prospective cohort study.
Xiaosong He, PhD, and his associates used two different machine learning classification methods to find two markers for thalamocortical connectedness that best predicted a good surgical outcome for temporal lobe epilepsy (TLE) in a small sample of patients. They presented their findings during a poster session of the annual meeting of the American Epilepsy Society.
After selecting a variety of possible predictors and building a model using resting state functional MRI (rsfMRI) data from 48 patients, the investigators then validated the prediction accuracies with rsfMRI data from 8 patients.
In predicting which TLE patients would have a good surgical outcome, models built with machine learning techniques using rsfMRI functional connectivity values had sensitivity ranging from 80% to 89% and specificity ranging from 52% to 57%. By comparison, models using clinical predictors only had sensitivity of 66% to 83% and specificity of 29% to 33%.
Dr. He and his coauthors dichotomized the surgical outcome for 56 patients who underwent TLE surgery into good outcome (n = 35) for those achieving and Engel class I and poor outcome (n = 21, class II-IV) at 1 year post surgery. All patients had a 5-minute rsfMRI scan before surgery.
MRI has been helpful in elucidating the importance of thalamocortical network pathology in TLE. Dr. He and his associates had previously used rsfMRI to examine the strength of functional connectivity between thalamic regions and their corresponding cortical regions in patients with TLE. Analysis of rsfMRI data of “both the left and right TLE groups showed that compared to controls there was a pattern of decreased thalamocortical [functional connectivity] in multiple thalamic segments,” wrote Dr. He and his collaborators (Epilepsia. 2015;56[10]:1571-9).
For the validation cohort, the two measures of connectedness found most predictive of a good surgical outcome were degree centrality and eigenvector centrality. In the graph theory and network analysis used in mapping functional connectivity, centrality refers to how highly connected one node, or data point, is to other data.
In the present study, the investigators used the Automated Anatomical Labeling cortical parcellation map to identify 45 cortical regions of interest per hemisphere, for a total of 90 cortical regions. They built a matrix with five topological parameters (global efficiency, global clustering coefficient, degree centrality, betweenness centrality, and eigenvector centrality) and the 90 cortical regions, yielding 272 variables. When nine commonly-used clinical predictors of surgical outcome (age, gender, handedness, laterality of TLE, epilepsy onset age and duration, seizure focality, interictal-spike type, and the presence of hippocampal sclerosis) were included, the model was made up of 281 variables.
The investigators used two different machine learning classification methods, called support vector machine and random forest, to build models that included various combinations of the 281 variables based on data from the initial 48 patients. The models were then tested with data from the remaining 8 patients.
Of the 35 patients with a good outcome, 18 had a left-sided epileptogenic temporal lobe; for the 21 patients with a poor outcome, the left temporal lobe was epileptogenic in 8. The mean age was similar for both groups: 41.25 years in those with good outcome, and 38.58 years in those with a poor outcome. Age at epilepsy onset also was similar, with each group having had epilepsy for about 17 years at the time of surgery. A total of 15 of the 20 patients with good outcome had seizure focality, compared with 10 of the 11 with poor outcome. Of those with a good outcome, 29 had an ipsilateral interactive spike, while 15 of those with poor outcomes had an ipsilateral interactive spike.
Since the random forest model best predicted surgical outcomes in the small sample size tested, the investigators plan to further fine-tune the random forest parameters to increase the robustness of their model.
Dr. He reported no conflicts of interest.
[email protected]
On Twitter @karioakes
HOUSTON – A machine learning interpretation of presurgical MRI studies did a better job of predicting which patients would have a successful outcome after anterior temporal lobectomy for temporal lobe epilepsy than did commonly-used clinical indicators in a prospective cohort study.
Xiaosong He, PhD, and his associates used two different machine learning classification methods to find two markers for thalamocortical connectedness that best predicted a good surgical outcome for temporal lobe epilepsy (TLE) in a small sample of patients. They presented their findings during a poster session of the annual meeting of the American Epilepsy Society.
After selecting a variety of possible predictors and building a model using resting state functional MRI (rsfMRI) data from 48 patients, the investigators then validated the prediction accuracies with rsfMRI data from 8 patients.
In predicting which TLE patients would have a good surgical outcome, models built with machine learning techniques using rsfMRI functional connectivity values had sensitivity ranging from 80% to 89% and specificity ranging from 52% to 57%. By comparison, models using clinical predictors only had sensitivity of 66% to 83% and specificity of 29% to 33%.
Dr. He and his coauthors dichotomized the surgical outcome for 56 patients who underwent TLE surgery into good outcome (n = 35) for those achieving and Engel class I and poor outcome (n = 21, class II-IV) at 1 year post surgery. All patients had a 5-minute rsfMRI scan before surgery.
MRI has been helpful in elucidating the importance of thalamocortical network pathology in TLE. Dr. He and his associates had previously used rsfMRI to examine the strength of functional connectivity between thalamic regions and their corresponding cortical regions in patients with TLE. Analysis of rsfMRI data of “both the left and right TLE groups showed that compared to controls there was a pattern of decreased thalamocortical [functional connectivity] in multiple thalamic segments,” wrote Dr. He and his collaborators (Epilepsia. 2015;56[10]:1571-9).
For the validation cohort, the two measures of connectedness found most predictive of a good surgical outcome were degree centrality and eigenvector centrality. In the graph theory and network analysis used in mapping functional connectivity, centrality refers to how highly connected one node, or data point, is to other data.
In the present study, the investigators used the Automated Anatomical Labeling cortical parcellation map to identify 45 cortical regions of interest per hemisphere, for a total of 90 cortical regions. They built a matrix with five topological parameters (global efficiency, global clustering coefficient, degree centrality, betweenness centrality, and eigenvector centrality) and the 90 cortical regions, yielding 272 variables. When nine commonly-used clinical predictors of surgical outcome (age, gender, handedness, laterality of TLE, epilepsy onset age and duration, seizure focality, interictal-spike type, and the presence of hippocampal sclerosis) were included, the model was made up of 281 variables.
The investigators used two different machine learning classification methods, called support vector machine and random forest, to build models that included various combinations of the 281 variables based on data from the initial 48 patients. The models were then tested with data from the remaining 8 patients.
Of the 35 patients with a good outcome, 18 had a left-sided epileptogenic temporal lobe; for the 21 patients with a poor outcome, the left temporal lobe was epileptogenic in 8. The mean age was similar for both groups: 41.25 years in those with good outcome, and 38.58 years in those with a poor outcome. Age at epilepsy onset also was similar, with each group having had epilepsy for about 17 years at the time of surgery. A total of 15 of the 20 patients with good outcome had seizure focality, compared with 10 of the 11 with poor outcome. Of those with a good outcome, 29 had an ipsilateral interactive spike, while 15 of those with poor outcomes had an ipsilateral interactive spike.
Since the random forest model best predicted surgical outcomes in the small sample size tested, the investigators plan to further fine-tune the random forest parameters to increase the robustness of their model.
Dr. He reported no conflicts of interest.
[email protected]
On Twitter @karioakes
HOUSTON – A machine learning interpretation of presurgical MRI studies did a better job of predicting which patients would have a successful outcome after anterior temporal lobectomy for temporal lobe epilepsy than did commonly-used clinical indicators in a prospective cohort study.
Xiaosong He, PhD, and his associates used two different machine learning classification methods to find two markers for thalamocortical connectedness that best predicted a good surgical outcome for temporal lobe epilepsy (TLE) in a small sample of patients. They presented their findings during a poster session of the annual meeting of the American Epilepsy Society.
After selecting a variety of possible predictors and building a model using resting state functional MRI (rsfMRI) data from 48 patients, the investigators then validated the prediction accuracies with rsfMRI data from 8 patients.
In predicting which TLE patients would have a good surgical outcome, models built with machine learning techniques using rsfMRI functional connectivity values had sensitivity ranging from 80% to 89% and specificity ranging from 52% to 57%. By comparison, models using clinical predictors only had sensitivity of 66% to 83% and specificity of 29% to 33%.
Dr. He and his coauthors dichotomized the surgical outcome for 56 patients who underwent TLE surgery into good outcome (n = 35) for those achieving and Engel class I and poor outcome (n = 21, class II-IV) at 1 year post surgery. All patients had a 5-minute rsfMRI scan before surgery.
MRI has been helpful in elucidating the importance of thalamocortical network pathology in TLE. Dr. He and his associates had previously used rsfMRI to examine the strength of functional connectivity between thalamic regions and their corresponding cortical regions in patients with TLE. Analysis of rsfMRI data of “both the left and right TLE groups showed that compared to controls there was a pattern of decreased thalamocortical [functional connectivity] in multiple thalamic segments,” wrote Dr. He and his collaborators (Epilepsia. 2015;56[10]:1571-9).
For the validation cohort, the two measures of connectedness found most predictive of a good surgical outcome were degree centrality and eigenvector centrality. In the graph theory and network analysis used in mapping functional connectivity, centrality refers to how highly connected one node, or data point, is to other data.
In the present study, the investigators used the Automated Anatomical Labeling cortical parcellation map to identify 45 cortical regions of interest per hemisphere, for a total of 90 cortical regions. They built a matrix with five topological parameters (global efficiency, global clustering coefficient, degree centrality, betweenness centrality, and eigenvector centrality) and the 90 cortical regions, yielding 272 variables. When nine commonly-used clinical predictors of surgical outcome (age, gender, handedness, laterality of TLE, epilepsy onset age and duration, seizure focality, interictal-spike type, and the presence of hippocampal sclerosis) were included, the model was made up of 281 variables.
The investigators used two different machine learning classification methods, called support vector machine and random forest, to build models that included various combinations of the 281 variables based on data from the initial 48 patients. The models were then tested with data from the remaining 8 patients.
Of the 35 patients with a good outcome, 18 had a left-sided epileptogenic temporal lobe; for the 21 patients with a poor outcome, the left temporal lobe was epileptogenic in 8. The mean age was similar for both groups: 41.25 years in those with good outcome, and 38.58 years in those with a poor outcome. Age at epilepsy onset also was similar, with each group having had epilepsy for about 17 years at the time of surgery. A total of 15 of the 20 patients with good outcome had seizure focality, compared with 10 of the 11 with poor outcome. Of those with a good outcome, 29 had an ipsilateral interactive spike, while 15 of those with poor outcomes had an ipsilateral interactive spike.
Since the random forest model best predicted surgical outcomes in the small sample size tested, the investigators plan to further fine-tune the random forest parameters to increase the robustness of their model.
Dr. He reported no conflicts of interest.
[email protected]
On Twitter @karioakes
AT AES 2016
Key clinical point:
Major finding: Models built with machine learning techniques using resting state fMRI functional connectivity values had sensitivity ranging from 80% to 89% and specificity ranging from 52% to 57%.
Data source: A prospective study of 56 patients with temporal lobe epilepsy.
Disclosures: Dr. He reported no conflicts of interest.
USPSTF nixes routine genital herpes screening
Asymptomatic adolescents and adults, including pregnant women, should not undergo routine serologic screening for genital herpes because the harms of this approach outweigh the benefits, according to an updated US Preventive Services Task Force (USPSTF) Recommendation Statement published online in JAMA.
The USPSTF last addressed screening for herpes simplex virus (HSV) in a 2005 Recommendation Statement, which advised against routine screening of asymptomatic patients at that time. “A small number” of clinical trials examined the issue since then, so the group commissioned a review of the literature through October 2016 to incorporate any new findings into the update, said Kirsten Bibbins-Domingo, MD, PhD, chair of the USPSTF and lead author of the update, and her associates.
Dr. Feltner and her colleagues concluded that current serologic screening tests produce a high rate of false-positive results – as much as 50% – and that those in turn lead to psychosocial harms such as distress and disruption of personal relationships, as well as increased costs and potential medical harm associated with confirmatory testing and unnecessary treatment.
Given the false-positive rates of the most widely used screening tests and the 15% prevalence of genital herpes in the general population, “screening 10,000 persons would result in approximately 1,485 true-positive and 1,445 false-positive results.” Moreover, confirmatory testing is offered only at a single research laboratory, said Dr. Bibbins-Domingo, professor of medicine at the University of California, San Francisco, and her associates on the Task Force.
Based on these findings and given the natural history and epidemiology of genital herpes, they characterized the potential benefits of routine screening as “no greater than small,” and the harms as “at least moderate” (JAMA. 2016 Dec 20;316[23]:2525-30. doi: 10.1001/jama.2016.16776).
The American Academy of Family Physicians, the American College of Obstetrics and Gynecology, and the Centers for Disease Control and Prevention also recommend against routine HSV screening of adolescents and adults, including pregnant women, the investigators noted.
The USPSTF is an independent, voluntary group that evaluates preventive health care services and is funded by the Agency for Healthcare Research and Quality by mandate of the U.S. Congress. The authors’ financial disclosures are available at www.uspreventiveservicestaskforce.org.
[email protected]
On Twitter @idpractitioner
It’s disappointing that the performance of screening tests hasn’t improved during the 10 years since the last USPSTF Recommendation Statement.
These findings should serve as a call to action for federal health agencies and their partners in industry to prioritize the development of better tests and stem the continuing epidemic of genital herpes.
Another important step would be to work vigorously to reduce the pervasive stigma of this disease, which also hinders management and control efforts. The public continues to harbor misperceptions about the severity of genital herpes and the availability of effective treatment, which adds to disproportionate stigmatization of those infected.
Edward W. Hook III, MD, is in the department of microbiology at the University of Alabama at Birmingham. He reported ties to Hologic, Roche Molecular, Becton Dickinson, and Cepheid. Dr. Hook made these remarks in an editorial accompanying the USPSTF Recommendation Statement (JAMA. 2016 Dec 20;316[23]:2493-4).
It’s disappointing that the performance of screening tests hasn’t improved during the 10 years since the last USPSTF Recommendation Statement.
These findings should serve as a call to action for federal health agencies and their partners in industry to prioritize the development of better tests and stem the continuing epidemic of genital herpes.
Another important step would be to work vigorously to reduce the pervasive stigma of this disease, which also hinders management and control efforts. The public continues to harbor misperceptions about the severity of genital herpes and the availability of effective treatment, which adds to disproportionate stigmatization of those infected.
Edward W. Hook III, MD, is in the department of microbiology at the University of Alabama at Birmingham. He reported ties to Hologic, Roche Molecular, Becton Dickinson, and Cepheid. Dr. Hook made these remarks in an editorial accompanying the USPSTF Recommendation Statement (JAMA. 2016 Dec 20;316[23]:2493-4).
It’s disappointing that the performance of screening tests hasn’t improved during the 10 years since the last USPSTF Recommendation Statement.
These findings should serve as a call to action for federal health agencies and their partners in industry to prioritize the development of better tests and stem the continuing epidemic of genital herpes.
Another important step would be to work vigorously to reduce the pervasive stigma of this disease, which also hinders management and control efforts. The public continues to harbor misperceptions about the severity of genital herpes and the availability of effective treatment, which adds to disproportionate stigmatization of those infected.
Edward W. Hook III, MD, is in the department of microbiology at the University of Alabama at Birmingham. He reported ties to Hologic, Roche Molecular, Becton Dickinson, and Cepheid. Dr. Hook made these remarks in an editorial accompanying the USPSTF Recommendation Statement (JAMA. 2016 Dec 20;316[23]:2493-4).
Asymptomatic adolescents and adults, including pregnant women, should not undergo routine serologic screening for genital herpes because the harms of this approach outweigh the benefits, according to an updated US Preventive Services Task Force (USPSTF) Recommendation Statement published online in JAMA.
The USPSTF last addressed screening for herpes simplex virus (HSV) in a 2005 Recommendation Statement, which advised against routine screening of asymptomatic patients at that time. “A small number” of clinical trials examined the issue since then, so the group commissioned a review of the literature through October 2016 to incorporate any new findings into the update, said Kirsten Bibbins-Domingo, MD, PhD, chair of the USPSTF and lead author of the update, and her associates.
Dr. Feltner and her colleagues concluded that current serologic screening tests produce a high rate of false-positive results – as much as 50% – and that those in turn lead to psychosocial harms such as distress and disruption of personal relationships, as well as increased costs and potential medical harm associated with confirmatory testing and unnecessary treatment.
Given the false-positive rates of the most widely used screening tests and the 15% prevalence of genital herpes in the general population, “screening 10,000 persons would result in approximately 1,485 true-positive and 1,445 false-positive results.” Moreover, confirmatory testing is offered only at a single research laboratory, said Dr. Bibbins-Domingo, professor of medicine at the University of California, San Francisco, and her associates on the Task Force.
Based on these findings and given the natural history and epidemiology of genital herpes, they characterized the potential benefits of routine screening as “no greater than small,” and the harms as “at least moderate” (JAMA. 2016 Dec 20;316[23]:2525-30. doi: 10.1001/jama.2016.16776).
The American Academy of Family Physicians, the American College of Obstetrics and Gynecology, and the Centers for Disease Control and Prevention also recommend against routine HSV screening of adolescents and adults, including pregnant women, the investigators noted.
The USPSTF is an independent, voluntary group that evaluates preventive health care services and is funded by the Agency for Healthcare Research and Quality by mandate of the U.S. Congress. The authors’ financial disclosures are available at www.uspreventiveservicestaskforce.org.
[email protected]
On Twitter @idpractitioner
Asymptomatic adolescents and adults, including pregnant women, should not undergo routine serologic screening for genital herpes because the harms of this approach outweigh the benefits, according to an updated US Preventive Services Task Force (USPSTF) Recommendation Statement published online in JAMA.
The USPSTF last addressed screening for herpes simplex virus (HSV) in a 2005 Recommendation Statement, which advised against routine screening of asymptomatic patients at that time. “A small number” of clinical trials examined the issue since then, so the group commissioned a review of the literature through October 2016 to incorporate any new findings into the update, said Kirsten Bibbins-Domingo, MD, PhD, chair of the USPSTF and lead author of the update, and her associates.
Dr. Feltner and her colleagues concluded that current serologic screening tests produce a high rate of false-positive results – as much as 50% – and that those in turn lead to psychosocial harms such as distress and disruption of personal relationships, as well as increased costs and potential medical harm associated with confirmatory testing and unnecessary treatment.
Given the false-positive rates of the most widely used screening tests and the 15% prevalence of genital herpes in the general population, “screening 10,000 persons would result in approximately 1,485 true-positive and 1,445 false-positive results.” Moreover, confirmatory testing is offered only at a single research laboratory, said Dr. Bibbins-Domingo, professor of medicine at the University of California, San Francisco, and her associates on the Task Force.
Based on these findings and given the natural history and epidemiology of genital herpes, they characterized the potential benefits of routine screening as “no greater than small,” and the harms as “at least moderate” (JAMA. 2016 Dec 20;316[23]:2525-30. doi: 10.1001/jama.2016.16776).
The American Academy of Family Physicians, the American College of Obstetrics and Gynecology, and the Centers for Disease Control and Prevention also recommend against routine HSV screening of adolescents and adults, including pregnant women, the investigators noted.
The USPSTF is an independent, voluntary group that evaluates preventive health care services and is funded by the Agency for Healthcare Research and Quality by mandate of the U.S. Congress. The authors’ financial disclosures are available at www.uspreventiveservicestaskforce.org.
[email protected]
On Twitter @idpractitioner
FROM JAMA
Key clinical point: Asymptomatic adults and adolescents should not undergo routine serologic screening for genital herpes because the harms outweigh the benefits, according to an updated USPSTF recommendation statement.
Major finding: Given the nearly 50% false-positive rate of current screening tests, screening 10,000 members of the general population would result in approximately 1,485 true-positive and 1,445 false-positive results.
Data source: A systematic review of 17 studies involving 9,736 participants regarding the accuracy, benefits, and harms of HSV screening.
Disclosures: The USPSTF is an independent, voluntary group that evaluates preventive health care services and is funded by the Agency for Healthcare Research and Quality by mandate of the U.S. Congress. The authors’ financial disclosures are available at www.uspreventiveservicestaskforce.org.