User login
Curricular milestones in rheumatology: Is granular better?
A new curricular “road map” attempts to lay out precisely what rheumatology fellows are expected to be able to know and do at different time points in their training.
The 80 “curricular milestones” for rheumatology, developed under the direction of the Accreditation Council for Graduate Medical Education, are meant to complement the 23 internal medicine subspecialty reporting milestones already mandated by the ACGME for use in trainee evaluation.
Unlike the reporting milestones, which came into use in 2014, the curricular milestones are not compulsory. Instead they “offer a guide so that trainees and the people teaching them know what’s expected of them,” said Lisa Criscione-Schreiber, MD, of Duke University, Durham, N.C., the lead author of a recent article in Arthritis Care & Research describing the new milestones (Arthritis Care Res. 2016 Nov 11. doi: 10.1002/acr.23151).
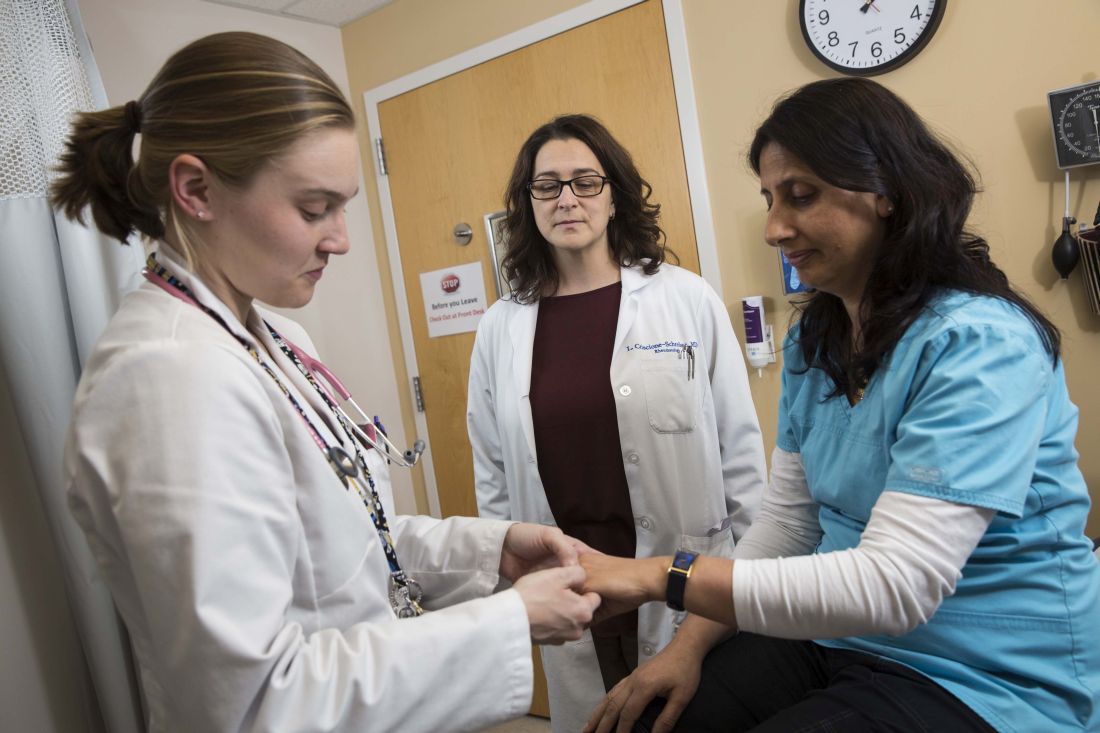
While the reporting milestones are designed to target broader competencies, the curricular milestones are highly granular. For example, a rheumatology fellow at 12 months is expected to be able to “perform compensated polarized microscopy to examine and interpret synovial fluid,” according to one milestone; before 24 months of training, he or she is expected to be able to teach others to do the same.
Authors of an accompanying editorial (Arthritis Care Res. 2016 Nov 11. doi: 10.1002/acr.23150) by Richard Panush, MD, of the University of Southern California in Los Angeles, Eric Hsieh, MD, also of USC, and Nortin Hadler, MD, of the University of North Carolina at Chapel Hill, praised the curricular milestones as meticulous and well conceived, but questioned their complexity and the broader push toward rubric-based training, particularly in the absence of evidence showing it to be superior to established methods.

Ultimately, Dr. Panush acknowledged in an interview, the move to milestones – including the curricular milestones – may prove worthwhile. But it simply “isn’t as sufficiently grounded in science as we would have liked for such a major departure from everything we have done, and with all that implementing the change would imply: the time, the manpower, the cost,” he said.
A leap of faith?
Dr. Criscione-Schreiber, who is Duke’s rheumatology program director, acknowledged that the milestones’ value and utility remain to be determined. “We’re not sure it’s the best way, but it’s a try,” she said. “We’re all in an era of continuous quality improvement in which you change something then test whether it works. The ACGME is collecting data about all this – if they find out it’s burdensome, they have assured program directors that they will change it.”

But Dr. Brown conceded that the editorialists’ concerns were valid. “The ACGME has been the main organization describing parameters by which we train doctors, and it is relatively unopposed. We’ve seen a proliferation of the rubric-based approach, and we are adopting these [curricular milestones] without scientific evidence. I commend the editorial authors in saying we need to find ways to measure this to see if it does improve, but if we wait for that evidence before we move forward, we’ll never get past square one.”
Observation and feedback
A key goal of the milestones is to encourage direct observation of trainees. Dr. Panush and his colleagues, in their editorial, argue that “a wise and astute clinician educator ... can judge when trainees are ready to function independently and have learned how to keep learning and changing without needing menus of evaluations or detailed instruments.”
Dr. Criscione-Schreiber said there’s traditionally been less direct observation in rheumatology training than people would like to think. “It used to be more like what we call the piano analogy. You send them into a room for an hour alone with a piano and some sheet music and they come out saying ‘I can play the piano.’ And you believe them.”
Dr. Brown, meanwhile, compared traditional training approaches to what he called the teabag model. The trainee “was the teabag steeping in the hot water of an academic medical center, and the major criteria for finishing was time,” he said. “I think all of us would recognize that this doesn’t take into consideration different environments, different learning styles and rates of learning.”

Using the milestones
Dr. Panush, in an interview, noted that the implementation of rubric-based education in internal medicine training came with a host of problems resulting in published critiques. He cited a recent article by Ronald Witteles, MD, and Abraham Verghese, MD, both of Stanford (Calif.) University, that criticized the reporting milestones as onerous to implement in large internal medicine programs, leading to excess administrative work and box-ticking (JAMA Intern Med. 2016;176[11]:1599-1600).
But rheumatology fellowship programs, which are generally small, may be less likely to find the curricular milestones burdensome, their champions say. The success of these milestones depends, in large part, on how people choose to integrate them into their training programs.
“The milestones appear a little daunting because they’re new,” Dr. Bolster said, cautioning that they are not designed to be foisted wholesale on trainees. Used judiciously, she said, the curricular milestones can help identify areas of strength and weakness and foster dialogue. “What I do is select a few of them, a couple of times a year, to go over with each fellow,” she said. “I also recommend them for fellows to use as self-reflection, so that they may determine where they are in terms of training.”
Details vs. the big picture
Anne Bass, MD, the rheumatology program director at the Hospital for Special Surgery in New York, who did not take part in either the curricular milestones writing committee or the editorial, said they could be used piecemeal, helping program directors to design portions of their curriculum, or to provide feedback and remediation by pointing out areas of trainee weakness.
But Dr. Bass also said, echoing a concern of the editorialists, that she felt that the milestones’ emphasis on highly specific strengths and weaknesses risked “losing the forest for the trees.”
A trainee who appears weak in one knowledge area is likely to have broader shortcomings, she said.
“If you’re somebody who gets the basic facts, you’re able to interview the patient, do the physical exam, and collect the information, but you’re not really able to apply it yet – that’s probably something global to that trainee that will apply with other areas as well,” not just the milestone you’re measuring, Dr. Bass said.
“The whole point about milestones is it’s showing you where along the continuum you are. But the continuum goes from data collection and description to application, testing, management, teaching – that’s kind of the spectrum, and that development is generally across the board. It’s not usually specific to one area.”
Training rheumatologists like pilots
The authors of the milestones say they’re responding to a sense widely shared in the rheumatology community that training must adapt to the needs of a changing field.
“Medicine is not the same as it was 10 or even 5 years ago, and certainly not as it was 20 years ago. A physician who is going to practice 10 years from now will require a different skill set,” Dr. Criscione-Schreiber said.
This, of course, includes many skills not directly related to patient care but rather successful functioning within current systems of health care delivery.
Dr. Criscione-Schreiber said that amid a coming shortage of rheumatologists, the next generation must be prepared to lead teams.
“We’re going to have clinics with nurses doing much of the education, and a whole team of people taking care of patients, if we have any hope of caring for the population in the U.S. that needs rheumatologists. It’s different from training a one-on-one doctor who sits in the room with a patient for 25 minutes,” she said.
Dr. Brown said he views the milestones effort, whether it proves successful or not, as an attempt to train in a way that ensures all future rheumatologists share the same core skills.
“People ask us all the time which doctor should they see for this or that condition, because they want to see someone really good,” Dr. Brown said. “Contrast that to the last time you got on an airplane. Did you ask a friend which pilot you should fly with? No, because you trusted that the pilot was competent, and had all requisite training, to safely pilot the plane. We want medicine to have many of those qualities and characteristics.”
Human beings are more complicated than airplanes, Dr. Brown acknowledged. “So it’s not perfectly analogous. But if we can more granularly describe training, and what it is we want to achieve, we’d all benefit – to the point where we can confidently tell you to see the rheumatologist trained in an accredited program who’s nearest your home.”
A new curricular “road map” attempts to lay out precisely what rheumatology fellows are expected to be able to know and do at different time points in their training.
The 80 “curricular milestones” for rheumatology, developed under the direction of the Accreditation Council for Graduate Medical Education, are meant to complement the 23 internal medicine subspecialty reporting milestones already mandated by the ACGME for use in trainee evaluation.
Unlike the reporting milestones, which came into use in 2014, the curricular milestones are not compulsory. Instead they “offer a guide so that trainees and the people teaching them know what’s expected of them,” said Lisa Criscione-Schreiber, MD, of Duke University, Durham, N.C., the lead author of a recent article in Arthritis Care & Research describing the new milestones (Arthritis Care Res. 2016 Nov 11. doi: 10.1002/acr.23151).
While the reporting milestones are designed to target broader competencies, the curricular milestones are highly granular. For example, a rheumatology fellow at 12 months is expected to be able to “perform compensated polarized microscopy to examine and interpret synovial fluid,” according to one milestone; before 24 months of training, he or she is expected to be able to teach others to do the same.
Authors of an accompanying editorial (Arthritis Care Res. 2016 Nov 11. doi: 10.1002/acr.23150) by Richard Panush, MD, of the University of Southern California in Los Angeles, Eric Hsieh, MD, also of USC, and Nortin Hadler, MD, of the University of North Carolina at Chapel Hill, praised the curricular milestones as meticulous and well conceived, but questioned their complexity and the broader push toward rubric-based training, particularly in the absence of evidence showing it to be superior to established methods.
Ultimately, Dr. Panush acknowledged in an interview, the move to milestones – including the curricular milestones – may prove worthwhile. But it simply “isn’t as sufficiently grounded in science as we would have liked for such a major departure from everything we have done, and with all that implementing the change would imply: the time, the manpower, the cost,” he said.
A leap of faith?
Dr. Criscione-Schreiber, who is Duke’s rheumatology program director, acknowledged that the milestones’ value and utility remain to be determined. “We’re not sure it’s the best way, but it’s a try,” she said. “We’re all in an era of continuous quality improvement in which you change something then test whether it works. The ACGME is collecting data about all this – if they find out it’s burdensome, they have assured program directors that they will change it.”
But Dr. Brown conceded that the editorialists’ concerns were valid. “The ACGME has been the main organization describing parameters by which we train doctors, and it is relatively unopposed. We’ve seen a proliferation of the rubric-based approach, and we are adopting these [curricular milestones] without scientific evidence. I commend the editorial authors in saying we need to find ways to measure this to see if it does improve, but if we wait for that evidence before we move forward, we’ll never get past square one.”
Observation and feedback
A key goal of the milestones is to encourage direct observation of trainees. Dr. Panush and his colleagues, in their editorial, argue that “a wise and astute clinician educator ... can judge when trainees are ready to function independently and have learned how to keep learning and changing without needing menus of evaluations or detailed instruments.”
Dr. Criscione-Schreiber said there’s traditionally been less direct observation in rheumatology training than people would like to think. “It used to be more like what we call the piano analogy. You send them into a room for an hour alone with a piano and some sheet music and they come out saying ‘I can play the piano.’ And you believe them.”
Dr. Brown, meanwhile, compared traditional training approaches to what he called the teabag model. The trainee “was the teabag steeping in the hot water of an academic medical center, and the major criteria for finishing was time,” he said. “I think all of us would recognize that this doesn’t take into consideration different environments, different learning styles and rates of learning.”
Using the milestones
Dr. Panush, in an interview, noted that the implementation of rubric-based education in internal medicine training came with a host of problems resulting in published critiques. He cited a recent article by Ronald Witteles, MD, and Abraham Verghese, MD, both of Stanford (Calif.) University, that criticized the reporting milestones as onerous to implement in large internal medicine programs, leading to excess administrative work and box-ticking (JAMA Intern Med. 2016;176[11]:1599-1600).
But rheumatology fellowship programs, which are generally small, may be less likely to find the curricular milestones burdensome, their champions say. The success of these milestones depends, in large part, on how people choose to integrate them into their training programs.
“The milestones appear a little daunting because they’re new,” Dr. Bolster said, cautioning that they are not designed to be foisted wholesale on trainees. Used judiciously, she said, the curricular milestones can help identify areas of strength and weakness and foster dialogue. “What I do is select a few of them, a couple of times a year, to go over with each fellow,” she said. “I also recommend them for fellows to use as self-reflection, so that they may determine where they are in terms of training.”
Details vs. the big picture
Anne Bass, MD, the rheumatology program director at the Hospital for Special Surgery in New York, who did not take part in either the curricular milestones writing committee or the editorial, said they could be used piecemeal, helping program directors to design portions of their curriculum, or to provide feedback and remediation by pointing out areas of trainee weakness.
But Dr. Bass also said, echoing a concern of the editorialists, that she felt that the milestones’ emphasis on highly specific strengths and weaknesses risked “losing the forest for the trees.”
A trainee who appears weak in one knowledge area is likely to have broader shortcomings, she said.
“If you’re somebody who gets the basic facts, you’re able to interview the patient, do the physical exam, and collect the information, but you’re not really able to apply it yet – that’s probably something global to that trainee that will apply with other areas as well,” not just the milestone you’re measuring, Dr. Bass said.
“The whole point about milestones is it’s showing you where along the continuum you are. But the continuum goes from data collection and description to application, testing, management, teaching – that’s kind of the spectrum, and that development is generally across the board. It’s not usually specific to one area.”
Training rheumatologists like pilots
The authors of the milestones say they’re responding to a sense widely shared in the rheumatology community that training must adapt to the needs of a changing field.
“Medicine is not the same as it was 10 or even 5 years ago, and certainly not as it was 20 years ago. A physician who is going to practice 10 years from now will require a different skill set,” Dr. Criscione-Schreiber said.
This, of course, includes many skills not directly related to patient care but rather successful functioning within current systems of health care delivery.
Dr. Criscione-Schreiber said that amid a coming shortage of rheumatologists, the next generation must be prepared to lead teams.
“We’re going to have clinics with nurses doing much of the education, and a whole team of people taking care of patients, if we have any hope of caring for the population in the U.S. that needs rheumatologists. It’s different from training a one-on-one doctor who sits in the room with a patient for 25 minutes,” she said.
Dr. Brown said he views the milestones effort, whether it proves successful or not, as an attempt to train in a way that ensures all future rheumatologists share the same core skills.
“People ask us all the time which doctor should they see for this or that condition, because they want to see someone really good,” Dr. Brown said. “Contrast that to the last time you got on an airplane. Did you ask a friend which pilot you should fly with? No, because you trusted that the pilot was competent, and had all requisite training, to safely pilot the plane. We want medicine to have many of those qualities and characteristics.”
Human beings are more complicated than airplanes, Dr. Brown acknowledged. “So it’s not perfectly analogous. But if we can more granularly describe training, and what it is we want to achieve, we’d all benefit – to the point where we can confidently tell you to see the rheumatologist trained in an accredited program who’s nearest your home.”
A new curricular “road map” attempts to lay out precisely what rheumatology fellows are expected to be able to know and do at different time points in their training.
The 80 “curricular milestones” for rheumatology, developed under the direction of the Accreditation Council for Graduate Medical Education, are meant to complement the 23 internal medicine subspecialty reporting milestones already mandated by the ACGME for use in trainee evaluation.
Unlike the reporting milestones, which came into use in 2014, the curricular milestones are not compulsory. Instead they “offer a guide so that trainees and the people teaching them know what’s expected of them,” said Lisa Criscione-Schreiber, MD, of Duke University, Durham, N.C., the lead author of a recent article in Arthritis Care & Research describing the new milestones (Arthritis Care Res. 2016 Nov 11. doi: 10.1002/acr.23151).
While the reporting milestones are designed to target broader competencies, the curricular milestones are highly granular. For example, a rheumatology fellow at 12 months is expected to be able to “perform compensated polarized microscopy to examine and interpret synovial fluid,” according to one milestone; before 24 months of training, he or she is expected to be able to teach others to do the same.
Authors of an accompanying editorial (Arthritis Care Res. 2016 Nov 11. doi: 10.1002/acr.23150) by Richard Panush, MD, of the University of Southern California in Los Angeles, Eric Hsieh, MD, also of USC, and Nortin Hadler, MD, of the University of North Carolina at Chapel Hill, praised the curricular milestones as meticulous and well conceived, but questioned their complexity and the broader push toward rubric-based training, particularly in the absence of evidence showing it to be superior to established methods.
Ultimately, Dr. Panush acknowledged in an interview, the move to milestones – including the curricular milestones – may prove worthwhile. But it simply “isn’t as sufficiently grounded in science as we would have liked for such a major departure from everything we have done, and with all that implementing the change would imply: the time, the manpower, the cost,” he said.
A leap of faith?
Dr. Criscione-Schreiber, who is Duke’s rheumatology program director, acknowledged that the milestones’ value and utility remain to be determined. “We’re not sure it’s the best way, but it’s a try,” she said. “We’re all in an era of continuous quality improvement in which you change something then test whether it works. The ACGME is collecting data about all this – if they find out it’s burdensome, they have assured program directors that they will change it.”
But Dr. Brown conceded that the editorialists’ concerns were valid. “The ACGME has been the main organization describing parameters by which we train doctors, and it is relatively unopposed. We’ve seen a proliferation of the rubric-based approach, and we are adopting these [curricular milestones] without scientific evidence. I commend the editorial authors in saying we need to find ways to measure this to see if it does improve, but if we wait for that evidence before we move forward, we’ll never get past square one.”
Observation and feedback
A key goal of the milestones is to encourage direct observation of trainees. Dr. Panush and his colleagues, in their editorial, argue that “a wise and astute clinician educator ... can judge when trainees are ready to function independently and have learned how to keep learning and changing without needing menus of evaluations or detailed instruments.”
Dr. Criscione-Schreiber said there’s traditionally been less direct observation in rheumatology training than people would like to think. “It used to be more like what we call the piano analogy. You send them into a room for an hour alone with a piano and some sheet music and they come out saying ‘I can play the piano.’ And you believe them.”
Dr. Brown, meanwhile, compared traditional training approaches to what he called the teabag model. The trainee “was the teabag steeping in the hot water of an academic medical center, and the major criteria for finishing was time,” he said. “I think all of us would recognize that this doesn’t take into consideration different environments, different learning styles and rates of learning.”
Using the milestones
Dr. Panush, in an interview, noted that the implementation of rubric-based education in internal medicine training came with a host of problems resulting in published critiques. He cited a recent article by Ronald Witteles, MD, and Abraham Verghese, MD, both of Stanford (Calif.) University, that criticized the reporting milestones as onerous to implement in large internal medicine programs, leading to excess administrative work and box-ticking (JAMA Intern Med. 2016;176[11]:1599-1600).
But rheumatology fellowship programs, which are generally small, may be less likely to find the curricular milestones burdensome, their champions say. The success of these milestones depends, in large part, on how people choose to integrate them into their training programs.
“The milestones appear a little daunting because they’re new,” Dr. Bolster said, cautioning that they are not designed to be foisted wholesale on trainees. Used judiciously, she said, the curricular milestones can help identify areas of strength and weakness and foster dialogue. “What I do is select a few of them, a couple of times a year, to go over with each fellow,” she said. “I also recommend them for fellows to use as self-reflection, so that they may determine where they are in terms of training.”
Details vs. the big picture
Anne Bass, MD, the rheumatology program director at the Hospital for Special Surgery in New York, who did not take part in either the curricular milestones writing committee or the editorial, said they could be used piecemeal, helping program directors to design portions of their curriculum, or to provide feedback and remediation by pointing out areas of trainee weakness.
But Dr. Bass also said, echoing a concern of the editorialists, that she felt that the milestones’ emphasis on highly specific strengths and weaknesses risked “losing the forest for the trees.”
A trainee who appears weak in one knowledge area is likely to have broader shortcomings, she said.
“If you’re somebody who gets the basic facts, you’re able to interview the patient, do the physical exam, and collect the information, but you’re not really able to apply it yet – that’s probably something global to that trainee that will apply with other areas as well,” not just the milestone you’re measuring, Dr. Bass said.
“The whole point about milestones is it’s showing you where along the continuum you are. But the continuum goes from data collection and description to application, testing, management, teaching – that’s kind of the spectrum, and that development is generally across the board. It’s not usually specific to one area.”
Training rheumatologists like pilots
The authors of the milestones say they’re responding to a sense widely shared in the rheumatology community that training must adapt to the needs of a changing field.
“Medicine is not the same as it was 10 or even 5 years ago, and certainly not as it was 20 years ago. A physician who is going to practice 10 years from now will require a different skill set,” Dr. Criscione-Schreiber said.
This, of course, includes many skills not directly related to patient care but rather successful functioning within current systems of health care delivery.
Dr. Criscione-Schreiber said that amid a coming shortage of rheumatologists, the next generation must be prepared to lead teams.
“We’re going to have clinics with nurses doing much of the education, and a whole team of people taking care of patients, if we have any hope of caring for the population in the U.S. that needs rheumatologists. It’s different from training a one-on-one doctor who sits in the room with a patient for 25 minutes,” she said.
Dr. Brown said he views the milestones effort, whether it proves successful or not, as an attempt to train in a way that ensures all future rheumatologists share the same core skills.
“People ask us all the time which doctor should they see for this or that condition, because they want to see someone really good,” Dr. Brown said. “Contrast that to the last time you got on an airplane. Did you ask a friend which pilot you should fly with? No, because you trusted that the pilot was competent, and had all requisite training, to safely pilot the plane. We want medicine to have many of those qualities and characteristics.”
Human beings are more complicated than airplanes, Dr. Brown acknowledged. “So it’s not perfectly analogous. But if we can more granularly describe training, and what it is we want to achieve, we’d all benefit – to the point where we can confidently tell you to see the rheumatologist trained in an accredited program who’s nearest your home.”
FROM ARTHRITIS CARE & RESEARCH
Addressing sexuality, gender identity issues is key to positive outcomes
A while ago, I gave a talk on LGBT health to a group of primary care pediatricians. Although I was glad that they invited me to speak, I also sensed some discomfort in the audience. At the end of the lecture, many pediatricians told me that they were uncomfortable with bringing up the topic of sexuality and gender identity with their patients, and others wanted guidance on how to ask questions on sexuality and gender identity.
There are many barriers for primary pediatricians in addressing sexuality and gender identity concerns in their patients. First, pediatricians often will have up to 15 minutes for a visit, so they will have little time to address a complex issue. Second, primary care pediatricians may have known many of their patients since birth, and asking questions on sexuality and gender can feel awkward. Finally, many pediatricians may be working in more conservative areas in the country where asking questions on sexuality and gender identity may be controversial.

Although a very important topic, there is not much empirical evidence on how to ask questions on sexuality and gender appropriately, and most of these recommendations are based on my own experience working with LGBT youth. Regardless, I hope these pointers will help the primary care pediatrician address the needs of LGBT youth efficiently and with sensitivity.
Tip No. 1: The environment counts
I cannot overstate how important it is to make your clinic a welcoming place for LGBT youth. Having various signs and stickers – like rainbow flags or the Human Rights Campaign sticker – will signal to LGBT youth that they are safe in your clinic. Creating a safe and welcoming environment is important because many people in the LGBT community have experienced rejection and discrimination from their primary care doctors.1 Making your clinic a safe space will make it easier and efficient for patients to ask questions about sexuality and gender identity (see Dr. Gaya Chelvakumar’s column “Creating safe spaces for LGBTQ youth, families in health care settings” at pediatricnews.com).
Tip No. 2: Consider the context
Most likely, many presenting complaints – such as colds or sports injuries – of your adolescent patients will not involve sexual orientation or gender identity. There are exceptions. If you suspect an STD, then the risk for certain infections, such as HIV2 or gonorrhea of the anus or of the pharynx3 are higher in gay young men. For the latter, your screening method would be different (that is, obtaining a pharyngeal swab or an anal swab instead of a urine sample). Also, because many LGBT youth have higher rates of mental health problems compared with heterosexual youth,4 you may want to ask questions about sexuality or gender identity to patients complaining of depressive or anxiety symptoms. This is especially important for transgender youth, because the implementation of pubertal blockers or cross-sex hormones can be therapeutic.5 To prevent or reduce many of these health problems, asking about sexuality and gender identity is a good idea during the well visit, when you may have more time.
Tip No. 3: Not all developmental stages are considered equal
Adolescence is a period of rapid and phasic growth. Formation of an identity is one of the major psychosocial tasks for adolescence,6 and sexuality and gender are important identities. In general, in early adolescence identity becomes an issue as the teenager gains autonomy from parents. I typically start asking questions about sexuality and gender when the patient is 11 or 12, because many children may not understand sexuality and gender identity at a younger age. At these ages, I ask these questions with the parents in the room, then I ask them confidentially on subsequent well visits. This approach serves two purposes: it will prepare the adolescent for these complex and thought-provoking questions in future encounters, and it gives the parents an idea of the type of questions you will ask the children when they are old enough for the confidential visit, helping parents feel more comfortable in stepping out of the room during this time.
Tip No. 4: Keep it confidential
Many adolescents are reluctant to see a doctor, even if they are sick. The primary reason adolescents do not seek care is the fear that the provider will tell their parents about their illness.7 Although this should be applicable to all of your adolescent patients, you should make an extra effort to explicitly state to LGBT patients that the clinic visit is confidential (with the exception of risk of suicide, homicide, or child abuse). This is important for LGBT youth who are not out to their parents and may be in danger if they do come out.8
Tip No. 5: Normalize, normalize, normalize
Because of the stigma and discrimination surrounding sexual orientation and gender identity, many LGBT youth will be reluctant to disclose their sexual orientation or gender identity to their health care providers. At the same time, heterosexual youth may think that you’re asking them questions about sexuality or gender identity because you suspect them to be a member of the LGBT community. To avoid this awkward situation, many pediatricians do not ask these questions at all. A good remedy for this is to preface your questions about sexual orientation or gender identity by saying that you ask these questions to all your patients – that way no one feels singled out.
Tip No. 6: Ask for permission
As previously mentioned, members of the LGBT community may experience discrimination from their health care providers after disclosing to them their sexual orientation or gender identity.1 This rejection can be traumatizing for LGBT youth, making them reluctant to discuss any issues related to sexual orientation or gender identity with any medical provider. As part of the trauma-informed approach, asking for permission before delving into issues related to sexual orientation and gender identity will give LGBT patients a sense of control, especially in an environment where there is a significant power differential.
Tip No. 7: Treat this as a skill
Despite the pressures for primary care pediatricians to maintain an efficient and effective clinical practice, many strive to learn new skills to provide the best care for their patients. Asking questions about sexuality and gender identity should be one of those skills. As with any skill, it will feel unnatural at first, and it will require practice. Mastering this skill, however, will help you address the health needs of this vulnerable population.
Asking questions about sexuality and gender identity is difficult for the primary care pediatrician. Hopefully, these tips can help you develop this important skill. It will also help you reach out to a population that is wary of the health care system.
References
1. J Am Board Fam Med. 2016;29(1):156-60.
2. https://www.cdc.gov/hiv/group/msm/index.html.
3. https://www.cdc.gov/std/tg2015/default.htm.
4. J Adolesc Health. 2011;49(2):115-23.
5. Nat Rev Endocrinol. 2011;7(8):466-72.
6. Neinstein LS. Adolescent health care: a practical guide. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2008.
7. J Adolesc Health. 2007;40(3):218-26.
8. Am J Orthopsychiatry. 1998;68(3):361-71.
Dr. Montano is an adolescent medicine fellow at Children’s Hospital of Pittsburgh of the University of Pittsburgh Medical Center and a postdoctoral fellow in the department of pediatrics at the University of Pittsburgh. Email him at [email protected].
A while ago, I gave a talk on LGBT health to a group of primary care pediatricians. Although I was glad that they invited me to speak, I also sensed some discomfort in the audience. At the end of the lecture, many pediatricians told me that they were uncomfortable with bringing up the topic of sexuality and gender identity with their patients, and others wanted guidance on how to ask questions on sexuality and gender identity.
There are many barriers for primary pediatricians in addressing sexuality and gender identity concerns in their patients. First, pediatricians often will have up to 15 minutes for a visit, so they will have little time to address a complex issue. Second, primary care pediatricians may have known many of their patients since birth, and asking questions on sexuality and gender can feel awkward. Finally, many pediatricians may be working in more conservative areas in the country where asking questions on sexuality and gender identity may be controversial.
Although a very important topic, there is not much empirical evidence on how to ask questions on sexuality and gender appropriately, and most of these recommendations are based on my own experience working with LGBT youth. Regardless, I hope these pointers will help the primary care pediatrician address the needs of LGBT youth efficiently and with sensitivity.
Tip No. 1: The environment counts
I cannot overstate how important it is to make your clinic a welcoming place for LGBT youth. Having various signs and stickers – like rainbow flags or the Human Rights Campaign sticker – will signal to LGBT youth that they are safe in your clinic. Creating a safe and welcoming environment is important because many people in the LGBT community have experienced rejection and discrimination from their primary care doctors.1 Making your clinic a safe space will make it easier and efficient for patients to ask questions about sexuality and gender identity (see Dr. Gaya Chelvakumar’s column “Creating safe spaces for LGBTQ youth, families in health care settings” at pediatricnews.com).
Tip No. 2: Consider the context
Most likely, many presenting complaints – such as colds or sports injuries – of your adolescent patients will not involve sexual orientation or gender identity. There are exceptions. If you suspect an STD, then the risk for certain infections, such as HIV2 or gonorrhea of the anus or of the pharynx3 are higher in gay young men. For the latter, your screening method would be different (that is, obtaining a pharyngeal swab or an anal swab instead of a urine sample). Also, because many LGBT youth have higher rates of mental health problems compared with heterosexual youth,4 you may want to ask questions about sexuality or gender identity to patients complaining of depressive or anxiety symptoms. This is especially important for transgender youth, because the implementation of pubertal blockers or cross-sex hormones can be therapeutic.5 To prevent or reduce many of these health problems, asking about sexuality and gender identity is a good idea during the well visit, when you may have more time.
Tip No. 3: Not all developmental stages are considered equal
Adolescence is a period of rapid and phasic growth. Formation of an identity is one of the major psychosocial tasks for adolescence,6 and sexuality and gender are important identities. In general, in early adolescence identity becomes an issue as the teenager gains autonomy from parents. I typically start asking questions about sexuality and gender when the patient is 11 or 12, because many children may not understand sexuality and gender identity at a younger age. At these ages, I ask these questions with the parents in the room, then I ask them confidentially on subsequent well visits. This approach serves two purposes: it will prepare the adolescent for these complex and thought-provoking questions in future encounters, and it gives the parents an idea of the type of questions you will ask the children when they are old enough for the confidential visit, helping parents feel more comfortable in stepping out of the room during this time.
Tip No. 4: Keep it confidential
Many adolescents are reluctant to see a doctor, even if they are sick. The primary reason adolescents do not seek care is the fear that the provider will tell their parents about their illness.7 Although this should be applicable to all of your adolescent patients, you should make an extra effort to explicitly state to LGBT patients that the clinic visit is confidential (with the exception of risk of suicide, homicide, or child abuse). This is important for LGBT youth who are not out to their parents and may be in danger if they do come out.8
Tip No. 5: Normalize, normalize, normalize
Because of the stigma and discrimination surrounding sexual orientation and gender identity, many LGBT youth will be reluctant to disclose their sexual orientation or gender identity to their health care providers. At the same time, heterosexual youth may think that you’re asking them questions about sexuality or gender identity because you suspect them to be a member of the LGBT community. To avoid this awkward situation, many pediatricians do not ask these questions at all. A good remedy for this is to preface your questions about sexual orientation or gender identity by saying that you ask these questions to all your patients – that way no one feels singled out.
Tip No. 6: Ask for permission
As previously mentioned, members of the LGBT community may experience discrimination from their health care providers after disclosing to them their sexual orientation or gender identity.1 This rejection can be traumatizing for LGBT youth, making them reluctant to discuss any issues related to sexual orientation or gender identity with any medical provider. As part of the trauma-informed approach, asking for permission before delving into issues related to sexual orientation and gender identity will give LGBT patients a sense of control, especially in an environment where there is a significant power differential.
Tip No. 7: Treat this as a skill
Despite the pressures for primary care pediatricians to maintain an efficient and effective clinical practice, many strive to learn new skills to provide the best care for their patients. Asking questions about sexuality and gender identity should be one of those skills. As with any skill, it will feel unnatural at first, and it will require practice. Mastering this skill, however, will help you address the health needs of this vulnerable population.
Asking questions about sexuality and gender identity is difficult for the primary care pediatrician. Hopefully, these tips can help you develop this important skill. It will also help you reach out to a population that is wary of the health care system.
References
1. J Am Board Fam Med. 2016;29(1):156-60.
2. https://www.cdc.gov/hiv/group/msm/index.html.
3. https://www.cdc.gov/std/tg2015/default.htm.
4. J Adolesc Health. 2011;49(2):115-23.
5. Nat Rev Endocrinol. 2011;7(8):466-72.
6. Neinstein LS. Adolescent health care: a practical guide. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2008.
7. J Adolesc Health. 2007;40(3):218-26.
8. Am J Orthopsychiatry. 1998;68(3):361-71.
Dr. Montano is an adolescent medicine fellow at Children’s Hospital of Pittsburgh of the University of Pittsburgh Medical Center and a postdoctoral fellow in the department of pediatrics at the University of Pittsburgh. Email him at [email protected].
A while ago, I gave a talk on LGBT health to a group of primary care pediatricians. Although I was glad that they invited me to speak, I also sensed some discomfort in the audience. At the end of the lecture, many pediatricians told me that they were uncomfortable with bringing up the topic of sexuality and gender identity with their patients, and others wanted guidance on how to ask questions on sexuality and gender identity.
There are many barriers for primary pediatricians in addressing sexuality and gender identity concerns in their patients. First, pediatricians often will have up to 15 minutes for a visit, so they will have little time to address a complex issue. Second, primary care pediatricians may have known many of their patients since birth, and asking questions on sexuality and gender can feel awkward. Finally, many pediatricians may be working in more conservative areas in the country where asking questions on sexuality and gender identity may be controversial.
Although a very important topic, there is not much empirical evidence on how to ask questions on sexuality and gender appropriately, and most of these recommendations are based on my own experience working with LGBT youth. Regardless, I hope these pointers will help the primary care pediatrician address the needs of LGBT youth efficiently and with sensitivity.
Tip No. 1: The environment counts
I cannot overstate how important it is to make your clinic a welcoming place for LGBT youth. Having various signs and stickers – like rainbow flags or the Human Rights Campaign sticker – will signal to LGBT youth that they are safe in your clinic. Creating a safe and welcoming environment is important because many people in the LGBT community have experienced rejection and discrimination from their primary care doctors.1 Making your clinic a safe space will make it easier and efficient for patients to ask questions about sexuality and gender identity (see Dr. Gaya Chelvakumar’s column “Creating safe spaces for LGBTQ youth, families in health care settings” at pediatricnews.com).
Tip No. 2: Consider the context
Most likely, many presenting complaints – such as colds or sports injuries – of your adolescent patients will not involve sexual orientation or gender identity. There are exceptions. If you suspect an STD, then the risk for certain infections, such as HIV2 or gonorrhea of the anus or of the pharynx3 are higher in gay young men. For the latter, your screening method would be different (that is, obtaining a pharyngeal swab or an anal swab instead of a urine sample). Also, because many LGBT youth have higher rates of mental health problems compared with heterosexual youth,4 you may want to ask questions about sexuality or gender identity to patients complaining of depressive or anxiety symptoms. This is especially important for transgender youth, because the implementation of pubertal blockers or cross-sex hormones can be therapeutic.5 To prevent or reduce many of these health problems, asking about sexuality and gender identity is a good idea during the well visit, when you may have more time.
Tip No. 3: Not all developmental stages are considered equal
Adolescence is a period of rapid and phasic growth. Formation of an identity is one of the major psychosocial tasks for adolescence,6 and sexuality and gender are important identities. In general, in early adolescence identity becomes an issue as the teenager gains autonomy from parents. I typically start asking questions about sexuality and gender when the patient is 11 or 12, because many children may not understand sexuality and gender identity at a younger age. At these ages, I ask these questions with the parents in the room, then I ask them confidentially on subsequent well visits. This approach serves two purposes: it will prepare the adolescent for these complex and thought-provoking questions in future encounters, and it gives the parents an idea of the type of questions you will ask the children when they are old enough for the confidential visit, helping parents feel more comfortable in stepping out of the room during this time.
Tip No. 4: Keep it confidential
Many adolescents are reluctant to see a doctor, even if they are sick. The primary reason adolescents do not seek care is the fear that the provider will tell their parents about their illness.7 Although this should be applicable to all of your adolescent patients, you should make an extra effort to explicitly state to LGBT patients that the clinic visit is confidential (with the exception of risk of suicide, homicide, or child abuse). This is important for LGBT youth who are not out to their parents and may be in danger if they do come out.8
Tip No. 5: Normalize, normalize, normalize
Because of the stigma and discrimination surrounding sexual orientation and gender identity, many LGBT youth will be reluctant to disclose their sexual orientation or gender identity to their health care providers. At the same time, heterosexual youth may think that you’re asking them questions about sexuality or gender identity because you suspect them to be a member of the LGBT community. To avoid this awkward situation, many pediatricians do not ask these questions at all. A good remedy for this is to preface your questions about sexual orientation or gender identity by saying that you ask these questions to all your patients – that way no one feels singled out.
Tip No. 6: Ask for permission
As previously mentioned, members of the LGBT community may experience discrimination from their health care providers after disclosing to them their sexual orientation or gender identity.1 This rejection can be traumatizing for LGBT youth, making them reluctant to discuss any issues related to sexual orientation or gender identity with any medical provider. As part of the trauma-informed approach, asking for permission before delving into issues related to sexual orientation and gender identity will give LGBT patients a sense of control, especially in an environment where there is a significant power differential.
Tip No. 7: Treat this as a skill
Despite the pressures for primary care pediatricians to maintain an efficient and effective clinical practice, many strive to learn new skills to provide the best care for their patients. Asking questions about sexuality and gender identity should be one of those skills. As with any skill, it will feel unnatural at first, and it will require practice. Mastering this skill, however, will help you address the health needs of this vulnerable population.
Asking questions about sexuality and gender identity is difficult for the primary care pediatrician. Hopefully, these tips can help you develop this important skill. It will also help you reach out to a population that is wary of the health care system.
References
1. J Am Board Fam Med. 2016;29(1):156-60.
2. https://www.cdc.gov/hiv/group/msm/index.html.
3. https://www.cdc.gov/std/tg2015/default.htm.
4. J Adolesc Health. 2011;49(2):115-23.
5. Nat Rev Endocrinol. 2011;7(8):466-72.
6. Neinstein LS. Adolescent health care: a practical guide. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2008.
7. J Adolesc Health. 2007;40(3):218-26.
8. Am J Orthopsychiatry. 1998;68(3):361-71.
Dr. Montano is an adolescent medicine fellow at Children’s Hospital of Pittsburgh of the University of Pittsburgh Medical Center and a postdoctoral fellow in the department of pediatrics at the University of Pittsburgh. Email him at [email protected].
A Multimodal Strategy to Manage Pain in an In-Patient and Out-Patient Total Joint Program: What Do You Need to Know?

Contents
Opioid-Sparing Pain Control in Outpatient Total Joint Arthroplasty
John W. Barrington, MD
Addressing the Opioid Epidemic With Multimodal Pain Management
Michael A. Kelly, MD
Comprehensive Care for Joint Replacement (CJR) Bundle Expense in Perioperative Pain Management
Susan D. Bear, PharmD, BCPS
The Role of Liposomal Bupivacaine in Value-Based Care
Richard Iorio, MD
Contents
Opioid-Sparing Pain Control in Outpatient Total Joint Arthroplasty
John W. Barrington, MD
Addressing the Opioid Epidemic With Multimodal Pain Management
Michael A. Kelly, MD
Comprehensive Care for Joint Replacement (CJR) Bundle Expense in Perioperative Pain Management
Susan D. Bear, PharmD, BCPS
The Role of Liposomal Bupivacaine in Value-Based Care
Richard Iorio, MD
Contents
Opioid-Sparing Pain Control in Outpatient Total Joint Arthroplasty
John W. Barrington, MD
Addressing the Opioid Epidemic With Multimodal Pain Management
Michael A. Kelly, MD
Comprehensive Care for Joint Replacement (CJR) Bundle Expense in Perioperative Pain Management
Susan D. Bear, PharmD, BCPS
The Role of Liposomal Bupivacaine in Value-Based Care
Richard Iorio, MD

Oral pomalidomide effective against Kaposi’s sarcoma
Oral pomalidomide, a derivative of thalidomide with immunomodulatory, antiangiogenic, and antiproliferative properties, proved to be well tolerated and effective against Kaposi’s sarcoma (KS) in a small phase I/II study, investigators reported in the Journal of Clinical Oncology.
There is a substantial need for oral therapies for KS, which often recurs and requires treatment, at least intermittently, for years. Existing treatments are cytotoxic agents, such as anthracyclines that are poorly tolerated and unsuitable for long-term use, said Mark N. Polizzotto, MD, of the National Cancer Institute, Bethesda, Md., and his associates.
They examined the tolerability and efficacy of pomalidomide against KS in a single-center, open-label trial involving 21 men and 1 transgender woman who had at least five evaluable cutaneous lesions. Fifteen of these participants had HIV infection. All of the patients were heavily pretreated with antiretrovirals and other agents, including thalidomide or lenalidomide, and most (17) had advanced KS with tumor-associated edema. The study participants received 5-mg oral pomalidomide once daily for 21 days in 28-day cycles for up to 12 cycles.
Sixteen patients showed an objective tumor response (overall response rate, 73%), including four who achieved a complete response (ORR, 18%). All 7 of the participants who didn’t have HIV showed an objective tumor response (ORR, 100%), as did 9 of the 15 HIV-infected participants (ORR, 60%). Three more participants, including one who was nonadeherent and lost to follow-up, achieved a partial tumor response.
Tumor responses included a decrease in the number of lesions and a decrease in the number of nodular lesions, as well as complete flattening of lesions, resolution of edema, and improvement in appearance. “The latter is particularly important because visible KS is a major source of stigma,” Dr. Polizzotto and his associates said (J Clin Oncol. 2016 Oct 3;34[34]:4125-32).
Two patients reported substantial subjective improvement in foot pain and appearance; one said he was pleased to be able to resume wearing closed shoes, the investigators noted. Treatment response generally was rapid, often commencing before the first follow-up at 4 weeks.
Pomalidomide was generally well tolerated. Adverse effects were frequent but mild and self-limiting, and they included neutropenia, constipation, anemia, fatigue, and rash. No patients required hospitalization, and there were no thromboembolic events. Three study participants developed malignancies after treatment: an Epstein-Barr-virus–associated Hodgkin lymphoma 1 year after the trial, a primary effusion lymphoma 15 months after, and a squamous-cell skin carcinoma. These probably reflect the patients’ underlying immunodeficiency, but the possibility that pomalidomide could contribute to malignancies in this high-risk population should be carefully explored in future studies, the investigators said.
“In resource-rich regions, pomalidomide may be of particular use in HIV-uninfected patients, in patients who have received substantial cumulative doses of anthracyclines, and in patients with less extensive but symptomatic disease for whom avoidance of cytotoxic chemotherapy would be beneficial. In resource-limited regions, there is an urgent need for effective and tolerable oral agents; with appropriate safeguards and monitoring, pomalidomide could address this,” Dr. Polizzotto and his associates wrote.
“Confirmatory studies of pomalidomide, alone and in combination with cytotoxic chemotherapy, are planned, including in sub-Saharan Africa,” they added.
Oral pomalidomide, a derivative of thalidomide with immunomodulatory, antiangiogenic, and antiproliferative properties, proved to be well tolerated and effective against Kaposi’s sarcoma (KS) in a small phase I/II study, investigators reported in the Journal of Clinical Oncology.
There is a substantial need for oral therapies for KS, which often recurs and requires treatment, at least intermittently, for years. Existing treatments are cytotoxic agents, such as anthracyclines that are poorly tolerated and unsuitable for long-term use, said Mark N. Polizzotto, MD, of the National Cancer Institute, Bethesda, Md., and his associates.
They examined the tolerability and efficacy of pomalidomide against KS in a single-center, open-label trial involving 21 men and 1 transgender woman who had at least five evaluable cutaneous lesions. Fifteen of these participants had HIV infection. All of the patients were heavily pretreated with antiretrovirals and other agents, including thalidomide or lenalidomide, and most (17) had advanced KS with tumor-associated edema. The study participants received 5-mg oral pomalidomide once daily for 21 days in 28-day cycles for up to 12 cycles.
Sixteen patients showed an objective tumor response (overall response rate, 73%), including four who achieved a complete response (ORR, 18%). All 7 of the participants who didn’t have HIV showed an objective tumor response (ORR, 100%), as did 9 of the 15 HIV-infected participants (ORR, 60%). Three more participants, including one who was nonadeherent and lost to follow-up, achieved a partial tumor response.
Tumor responses included a decrease in the number of lesions and a decrease in the number of nodular lesions, as well as complete flattening of lesions, resolution of edema, and improvement in appearance. “The latter is particularly important because visible KS is a major source of stigma,” Dr. Polizzotto and his associates said (J Clin Oncol. 2016 Oct 3;34[34]:4125-32).
Two patients reported substantial subjective improvement in foot pain and appearance; one said he was pleased to be able to resume wearing closed shoes, the investigators noted. Treatment response generally was rapid, often commencing before the first follow-up at 4 weeks.
Pomalidomide was generally well tolerated. Adverse effects were frequent but mild and self-limiting, and they included neutropenia, constipation, anemia, fatigue, and rash. No patients required hospitalization, and there were no thromboembolic events. Three study participants developed malignancies after treatment: an Epstein-Barr-virus–associated Hodgkin lymphoma 1 year after the trial, a primary effusion lymphoma 15 months after, and a squamous-cell skin carcinoma. These probably reflect the patients’ underlying immunodeficiency, but the possibility that pomalidomide could contribute to malignancies in this high-risk population should be carefully explored in future studies, the investigators said.
“In resource-rich regions, pomalidomide may be of particular use in HIV-uninfected patients, in patients who have received substantial cumulative doses of anthracyclines, and in patients with less extensive but symptomatic disease for whom avoidance of cytotoxic chemotherapy would be beneficial. In resource-limited regions, there is an urgent need for effective and tolerable oral agents; with appropriate safeguards and monitoring, pomalidomide could address this,” Dr. Polizzotto and his associates wrote.
“Confirmatory studies of pomalidomide, alone and in combination with cytotoxic chemotherapy, are planned, including in sub-Saharan Africa,” they added.
Oral pomalidomide, a derivative of thalidomide with immunomodulatory, antiangiogenic, and antiproliferative properties, proved to be well tolerated and effective against Kaposi’s sarcoma (KS) in a small phase I/II study, investigators reported in the Journal of Clinical Oncology.
There is a substantial need for oral therapies for KS, which often recurs and requires treatment, at least intermittently, for years. Existing treatments are cytotoxic agents, such as anthracyclines that are poorly tolerated and unsuitable for long-term use, said Mark N. Polizzotto, MD, of the National Cancer Institute, Bethesda, Md., and his associates.
They examined the tolerability and efficacy of pomalidomide against KS in a single-center, open-label trial involving 21 men and 1 transgender woman who had at least five evaluable cutaneous lesions. Fifteen of these participants had HIV infection. All of the patients were heavily pretreated with antiretrovirals and other agents, including thalidomide or lenalidomide, and most (17) had advanced KS with tumor-associated edema. The study participants received 5-mg oral pomalidomide once daily for 21 days in 28-day cycles for up to 12 cycles.
Sixteen patients showed an objective tumor response (overall response rate, 73%), including four who achieved a complete response (ORR, 18%). All 7 of the participants who didn’t have HIV showed an objective tumor response (ORR, 100%), as did 9 of the 15 HIV-infected participants (ORR, 60%). Three more participants, including one who was nonadeherent and lost to follow-up, achieved a partial tumor response.
Tumor responses included a decrease in the number of lesions and a decrease in the number of nodular lesions, as well as complete flattening of lesions, resolution of edema, and improvement in appearance. “The latter is particularly important because visible KS is a major source of stigma,” Dr. Polizzotto and his associates said (J Clin Oncol. 2016 Oct 3;34[34]:4125-32).
Two patients reported substantial subjective improvement in foot pain and appearance; one said he was pleased to be able to resume wearing closed shoes, the investigators noted. Treatment response generally was rapid, often commencing before the first follow-up at 4 weeks.
Pomalidomide was generally well tolerated. Adverse effects were frequent but mild and self-limiting, and they included neutropenia, constipation, anemia, fatigue, and rash. No patients required hospitalization, and there were no thromboembolic events. Three study participants developed malignancies after treatment: an Epstein-Barr-virus–associated Hodgkin lymphoma 1 year after the trial, a primary effusion lymphoma 15 months after, and a squamous-cell skin carcinoma. These probably reflect the patients’ underlying immunodeficiency, but the possibility that pomalidomide could contribute to malignancies in this high-risk population should be carefully explored in future studies, the investigators said.
“In resource-rich regions, pomalidomide may be of particular use in HIV-uninfected patients, in patients who have received substantial cumulative doses of anthracyclines, and in patients with less extensive but symptomatic disease for whom avoidance of cytotoxic chemotherapy would be beneficial. In resource-limited regions, there is an urgent need for effective and tolerable oral agents; with appropriate safeguards and monitoring, pomalidomide could address this,” Dr. Polizzotto and his associates wrote.
“Confirmatory studies of pomalidomide, alone and in combination with cytotoxic chemotherapy, are planned, including in sub-Saharan Africa,” they added.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Oral pomalidomide, a derivative of thalidomide with immunomodulatory, antiangiogenic, and antiproliferative properties, proved to be well tolerated and effective against Kaposi’s sarcoma in a small phase I/II study.
Major finding: All 7 of the participants who didn’t have HIV showed an objective tumor response with pomalidomide (ORR, 100%), as did 9 of the 15 HIV-infected participants (ORR, 60%).
Data source: A single-center open-label phase I/II study involving 22 adults with KS, including 15 who had HIV infection.
Disclosures: This study was supported by the National Institutes of Health, the National Cancer Institute, Celgene, and the National Institute of Allergy and Infectious Diseases. Dr. Polizzotto and some of his associates reported that their institutions received research funding from Celgene; one of his associates also reported ties to Ovid, Trek, PTC, Avila, Semorex, and Sorrento.
VIDEO: Watson helps oncologists sleuth out best options in breast cancer
SAN ANTONIO – Every day, practicing oncologists sort through mountains of data from treatment guidelines, clinical trials, protocols, and algorithms to make the best possible therapeutic decisions for their patients.
At Manipal Hospitals in Bangalore, India, those decisions are informed with the aid of Watson for Oncology (WFO), a software platform developed by IBM in concert with clinicians and investigators at Memorial Sloan Kettering Cancer Center in New York City.
In this video interview at the San Antonio Breast Cancer Symposium, S.P. Somashekhar, MBBS, MS, MCH, chairman of the Manipal Comprehensive Cancer Center, discusses how the system is used by the members of the center’s multidisciplinary tumor board to inform decision making. He also describes results of a study he presented at the symposium that shows a relatively high degree of concordance between clinician and WFO choices in the treatment of patients with local breast cancer but less agreement when it comes to the management of metastatic disease, for which there is no universal standard of care.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN ANTONIO – Every day, practicing oncologists sort through mountains of data from treatment guidelines, clinical trials, protocols, and algorithms to make the best possible therapeutic decisions for their patients.
At Manipal Hospitals in Bangalore, India, those decisions are informed with the aid of Watson for Oncology (WFO), a software platform developed by IBM in concert with clinicians and investigators at Memorial Sloan Kettering Cancer Center in New York City.
In this video interview at the San Antonio Breast Cancer Symposium, S.P. Somashekhar, MBBS, MS, MCH, chairman of the Manipal Comprehensive Cancer Center, discusses how the system is used by the members of the center’s multidisciplinary tumor board to inform decision making. He also describes results of a study he presented at the symposium that shows a relatively high degree of concordance between clinician and WFO choices in the treatment of patients with local breast cancer but less agreement when it comes to the management of metastatic disease, for which there is no universal standard of care.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN ANTONIO – Every day, practicing oncologists sort through mountains of data from treatment guidelines, clinical trials, protocols, and algorithms to make the best possible therapeutic decisions for their patients.
At Manipal Hospitals in Bangalore, India, those decisions are informed with the aid of Watson for Oncology (WFO), a software platform developed by IBM in concert with clinicians and investigators at Memorial Sloan Kettering Cancer Center in New York City.
In this video interview at the San Antonio Breast Cancer Symposium, S.P. Somashekhar, MBBS, MS, MCH, chairman of the Manipal Comprehensive Cancer Center, discusses how the system is used by the members of the center’s multidisciplinary tumor board to inform decision making. He also describes results of a study he presented at the symposium that shows a relatively high degree of concordance between clinician and WFO choices in the treatment of patients with local breast cancer but less agreement when it comes to the management of metastatic disease, for which there is no universal standard of care.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT SABCS 2016

History of complex regional pain syndrome increases risk of secondary CRPS
Secondary complex regional pain syndrome is significantly more likely in people currently experiencing CRPS in an unrelated extremity than in the general population, according to Ellen Satteson, MD, and her associates.
In a study of 93 patients with CRPS, 20.4% developed secondary CRPS in another extremity. Twenty patients in the primary CRPS group experienced a secondary inciting event. Of this group, 75% developed secondary CRPS. CRPS in all four extremities occurred in six patients, of whom five had inciting events for each extremity.
The odds ratio for secondary CRPS in the study group compared to the general population was found to be 1,069.6, while the OR for CRPS after an inciting event compared to the general population was 11.7.
“An odds ratio of over 1,000 when comparing the reported population incidence of CRPS to the rate of secondary CRPS documented in this study strongly suggests that patients with a history of CRPS may be at considerable risk of developing secondary CRPS ... Additional, prospective studies with standardized follow-up to assess for subsequent injuries and secondary CRPS, however, are needed to better elucidate the significance of this risk,” the investigators noted.
Find the study in the Scandinavian Journal of Pain (doi: 10.1016/j.sjpain.2016.10.005).
Secondary complex regional pain syndrome is significantly more likely in people currently experiencing CRPS in an unrelated extremity than in the general population, according to Ellen Satteson, MD, and her associates.
In a study of 93 patients with CRPS, 20.4% developed secondary CRPS in another extremity. Twenty patients in the primary CRPS group experienced a secondary inciting event. Of this group, 75% developed secondary CRPS. CRPS in all four extremities occurred in six patients, of whom five had inciting events for each extremity.
The odds ratio for secondary CRPS in the study group compared to the general population was found to be 1,069.6, while the OR for CRPS after an inciting event compared to the general population was 11.7.
“An odds ratio of over 1,000 when comparing the reported population incidence of CRPS to the rate of secondary CRPS documented in this study strongly suggests that patients with a history of CRPS may be at considerable risk of developing secondary CRPS ... Additional, prospective studies with standardized follow-up to assess for subsequent injuries and secondary CRPS, however, are needed to better elucidate the significance of this risk,” the investigators noted.
Find the study in the Scandinavian Journal of Pain (doi: 10.1016/j.sjpain.2016.10.005).
Secondary complex regional pain syndrome is significantly more likely in people currently experiencing CRPS in an unrelated extremity than in the general population, according to Ellen Satteson, MD, and her associates.
In a study of 93 patients with CRPS, 20.4% developed secondary CRPS in another extremity. Twenty patients in the primary CRPS group experienced a secondary inciting event. Of this group, 75% developed secondary CRPS. CRPS in all four extremities occurred in six patients, of whom five had inciting events for each extremity.
The odds ratio for secondary CRPS in the study group compared to the general population was found to be 1,069.6, while the OR for CRPS after an inciting event compared to the general population was 11.7.
“An odds ratio of over 1,000 when comparing the reported population incidence of CRPS to the rate of secondary CRPS documented in this study strongly suggests that patients with a history of CRPS may be at considerable risk of developing secondary CRPS ... Additional, prospective studies with standardized follow-up to assess for subsequent injuries and secondary CRPS, however, are needed to better elucidate the significance of this risk,” the investigators noted.
Find the study in the Scandinavian Journal of Pain (doi: 10.1016/j.sjpain.2016.10.005).
FROM THE SCANDINAVIAN JOURNAL OF PAIN
More evidence shows ties between early inflammation, schizophrenia risk in adulthood
Increases in inflammatory markers during acute psychosis suggest that higher C-reactive protein levels in adolescence may play an important role in subsequent schizophrenia by age 27, but more study is needed, results from a longitudinal study suggest.
“Inflammatory pathways may offer important new prevention and intervention targets for schizophrenia,” wrote Stephen A. Metcalf of the University of Cambridge (England) and his coauthors, most of whom are based in Finland (Brain Behav Immun. 2017 Jan;59:253-9).
The birth cohort consisted of 99% of the live births in two northern Finland provinces between July 1985 and June 1986. Investigators studied 6,362 individuals within the cohort who had gone on to have their blood sampled at age 15 or 16 years and measured for levels of serum C-reactive protein (CRP).
Increased levels of CRP are associated with antipsychotic-naive first-episode psychosis and acute psychotic relapse. However, “it is difficult to ascertain the direction of association between inflammation and schizophrenia from cross-sectional data. Longitudinal studies of inflammatory markers and subsequent psychotic illness are scarce but are necessary to establish whether the increase in circulating inflammatory markers is a cause or consequence of illness,” wrote Mr. Metcalf and his coauthors.
The investigators searched Finnish hospital records for cases and ICD-10 diagnoses of nonaffective psychosis, including schizophrenia, for the years 1994 to 2012 for CRP-measured members of the birth cohort, spanning ages 8-27 years. With the records collected, they sorted the participants for analysis into the following three groups: schizophrenia (n = 22), nonschizophrenia nonaffective psychosis (n = 66), and no psychosis (n = 6,274).
“The adjusted [odds ratio] for schizophrenia by age 27 years for each standard deviation increase in CRP levels at age 15/16 years was 1.25 (95% confidence interval, 1.07-1.46), which was consistent with a linear, dose-response relationship (P = 0.23),” Mr. Metcalf and his coauthors wrote. Participants with high levels of CRP at baseline (greater than 3 mg/L), compared with low levels (less than 1 mg/L), were more likely to develop schizophrenia (adjusted OR, 4.25; 95% CI, 1.30-13.93).
The investigators adjusted their regression models for age, sex, body mass index, maternal education, smoking, and alcohol use. “To exclude reverse causality (i.e., schizophrenia leading to elevated CRP), we repeated the analyses after removing participants who had developed schizophrenia within one year of CRP assay.”
The findings build on those from a longitudinal study in Denmark on increased risks for schizophrenia associated with higher CRP levels (Schizophr Bull. 2014 Sep;40[5]:1117-27). The authors cautioned that the current findings must be replicated elsewhere.
The authors disclosed several individual funding sources, but none played a role in the study.
Increases in inflammatory markers during acute psychosis suggest that higher C-reactive protein levels in adolescence may play an important role in subsequent schizophrenia by age 27, but more study is needed, results from a longitudinal study suggest.
“Inflammatory pathways may offer important new prevention and intervention targets for schizophrenia,” wrote Stephen A. Metcalf of the University of Cambridge (England) and his coauthors, most of whom are based in Finland (Brain Behav Immun. 2017 Jan;59:253-9).
The birth cohort consisted of 99% of the live births in two northern Finland provinces between July 1985 and June 1986. Investigators studied 6,362 individuals within the cohort who had gone on to have their blood sampled at age 15 or 16 years and measured for levels of serum C-reactive protein (CRP).
Increased levels of CRP are associated with antipsychotic-naive first-episode psychosis and acute psychotic relapse. However, “it is difficult to ascertain the direction of association between inflammation and schizophrenia from cross-sectional data. Longitudinal studies of inflammatory markers and subsequent psychotic illness are scarce but are necessary to establish whether the increase in circulating inflammatory markers is a cause or consequence of illness,” wrote Mr. Metcalf and his coauthors.
The investigators searched Finnish hospital records for cases and ICD-10 diagnoses of nonaffective psychosis, including schizophrenia, for the years 1994 to 2012 for CRP-measured members of the birth cohort, spanning ages 8-27 years. With the records collected, they sorted the participants for analysis into the following three groups: schizophrenia (n = 22), nonschizophrenia nonaffective psychosis (n = 66), and no psychosis (n = 6,274).
“The adjusted [odds ratio] for schizophrenia by age 27 years for each standard deviation increase in CRP levels at age 15/16 years was 1.25 (95% confidence interval, 1.07-1.46), which was consistent with a linear, dose-response relationship (P = 0.23),” Mr. Metcalf and his coauthors wrote. Participants with high levels of CRP at baseline (greater than 3 mg/L), compared with low levels (less than 1 mg/L), were more likely to develop schizophrenia (adjusted OR, 4.25; 95% CI, 1.30-13.93).
The investigators adjusted their regression models for age, sex, body mass index, maternal education, smoking, and alcohol use. “To exclude reverse causality (i.e., schizophrenia leading to elevated CRP), we repeated the analyses after removing participants who had developed schizophrenia within one year of CRP assay.”
The findings build on those from a longitudinal study in Denmark on increased risks for schizophrenia associated with higher CRP levels (Schizophr Bull. 2014 Sep;40[5]:1117-27). The authors cautioned that the current findings must be replicated elsewhere.
The authors disclosed several individual funding sources, but none played a role in the study.
Increases in inflammatory markers during acute psychosis suggest that higher C-reactive protein levels in adolescence may play an important role in subsequent schizophrenia by age 27, but more study is needed, results from a longitudinal study suggest.
“Inflammatory pathways may offer important new prevention and intervention targets for schizophrenia,” wrote Stephen A. Metcalf of the University of Cambridge (England) and his coauthors, most of whom are based in Finland (Brain Behav Immun. 2017 Jan;59:253-9).
The birth cohort consisted of 99% of the live births in two northern Finland provinces between July 1985 and June 1986. Investigators studied 6,362 individuals within the cohort who had gone on to have their blood sampled at age 15 or 16 years and measured for levels of serum C-reactive protein (CRP).
Increased levels of CRP are associated with antipsychotic-naive first-episode psychosis and acute psychotic relapse. However, “it is difficult to ascertain the direction of association between inflammation and schizophrenia from cross-sectional data. Longitudinal studies of inflammatory markers and subsequent psychotic illness are scarce but are necessary to establish whether the increase in circulating inflammatory markers is a cause or consequence of illness,” wrote Mr. Metcalf and his coauthors.
The investigators searched Finnish hospital records for cases and ICD-10 diagnoses of nonaffective psychosis, including schizophrenia, for the years 1994 to 2012 for CRP-measured members of the birth cohort, spanning ages 8-27 years. With the records collected, they sorted the participants for analysis into the following three groups: schizophrenia (n = 22), nonschizophrenia nonaffective psychosis (n = 66), and no psychosis (n = 6,274).
“The adjusted [odds ratio] for schizophrenia by age 27 years for each standard deviation increase in CRP levels at age 15/16 years was 1.25 (95% confidence interval, 1.07-1.46), which was consistent with a linear, dose-response relationship (P = 0.23),” Mr. Metcalf and his coauthors wrote. Participants with high levels of CRP at baseline (greater than 3 mg/L), compared with low levels (less than 1 mg/L), were more likely to develop schizophrenia (adjusted OR, 4.25; 95% CI, 1.30-13.93).
The investigators adjusted their regression models for age, sex, body mass index, maternal education, smoking, and alcohol use. “To exclude reverse causality (i.e., schizophrenia leading to elevated CRP), we repeated the analyses after removing participants who had developed schizophrenia within one year of CRP assay.”
The findings build on those from a longitudinal study in Denmark on increased risks for schizophrenia associated with higher CRP levels (Schizophr Bull. 2014 Sep;40[5]:1117-27). The authors cautioned that the current findings must be replicated elsewhere.
The authors disclosed several individual funding sources, but none played a role in the study.
FROM BRAIN, BEHAVIOR, AND IMMUNITY
Phase I results move Alzheimer’s candidate drug aducanumab into phase III trials
SAN DIEGO – The antiamyloid antibody aducanumab significantly reduced amyloid brain plaques in Alzheimer’s disease patients who entered a second year of therapy in an open-label extension trial.
Plaque volume declined in a time- and dose-dependent manner, with a bit of movement even among patients who crossed over to the lowest 1-mg/kg dose after a year of taking placebo in the phase Ib PRIME study, Samantha Haeberlein, PhD, reported at the Clinical Trials on Alzheimer’s Disease conference.
The linear declines in plaque burden were dramatic enough to draw a collective gasp of appreciation from the packed auditorium. But the cognitive and functional data of aducanumab (Biogen), while deemed encouraging, were not as striking. The Clinical Dementia Rating Scale-sum of boxes (CDR-SB) and the Mini-Mental State Examination (MMSE) showed dose-dependent slowing of decline, but the drug’s effect approached statistical significance only among those who finished 2 years on 10 mg/kg aducanumab infused every 4 weeks. This group experienced relative stability of MMSE scores, which dropped only about 1 point from baseline, and also showed the greatest decline in amyloid plaque volume.
Biogen deemed this result on the MMSE, which represented a 3.27-point separation from the 1-mg/kg dose group, as “nominally significant,” with a P value noted as “less than .05.”
However, the finding must be viewed with extreme caution, said Dr. Haeberlein, Biogen’s vice president of clinical development. There were only 15 subjects in this group, and the study was not primarily intended to examine cognition.
“I must emphasize once again that these are exploratory data and these sample sizes are very small for these types of assessments,” she said. “Nonetheless, we find them informative.”
MMSE changes in the other dosing groups of 1, 3, and 6 mg/kg were not statistically significant at the end of the study. There were not any significant findings on the CDR-SB measure.
Alzheimer’s Association reaction
Maria C. Carrillo, PhD, chief science officer of the Alzheimer’s Association, was cautiously optimistic.

“For MMSE, the original placebo group continued to decline even when they switched to the 1-mg/kg treatment, and by a significant number of points. The 1- to 3-mg/kg switching group is interesting, as they do get a little bit of a bump. But we also had the 6-mg/kg arm continuing to decline in MMSE and not improve as much as the others, which is strange. It’s aberrant, but the same thing we saw in the first study report. The 10-mg/kg group, though, stays almost at baseline. That’s pretty amazing. Impressive. Again, small numbers but very encouraging.”
Full 12-month results
Biogen presented two aducanumab abstracts at the meeting, both describing its 12-month phase Ib PRIME study and its 12-month, open-label extension study. The drug is a monoclonal human antibody derived from B cells collected from a cohort of cognitively normal elderly subjects and cognitively impaired elderly subjects who exhibited unusually slow decline, according to the company. It binds to fibrillar and oligomeric amyloid aggregates, thus directly reducing amyloid plaque in the brain.
PRIME enrolled 165 patients with prodromal or mild Alzheimer’s disease. Importantly, all of the subjects had brain amyloid proven by PET imaging. PRIME is the first randomized trial of an antiamyloid compound to enroll a pure amyloid-positive cohort. These subjects were randomized to placebo or aducanumab at 1, 3, 6, or 10 mg/kg for 1 year. PRIME’s primary outcomes were safety and tolerability. The cognitive and functional outcomes, not usually assessed in a phase Ib study, were exploratory.
They must also be interpreted in light of the very small numbers, about 30 patients in each dosing group at baseline. In addition, just 69 patients finished the entire 24-month dosing period, leaving only 15-23 patients in each group by the end of the study.
Vissia Viglietta, MD, Biogen’s senior medical director of clinical development, presented the 12-month data. At 52 weeks, all dosing groups, even the 1 mg/kg, saw statistically significant reductions in amyloid plaque, compared with placebo. These changes were dose-dependent; the 10-mg/kg group had the largest reduction, with a P value of less than .001.
There also were dose-dependent changes in the CDR-SB and MMSE, and some of these reached statistical significance.
On the CDR-SB, patients taking placebo declined by an average of 1.89 points. Declines in the 1-, 3-, and 6-mg/kg groups were not significant relative to placebo. However, the 10-mg/kg arm experienced a significant separation from placebo, declining an average of 0.63 points (P less than .05).
The story was similar for the MMSE. Patients taking placebo declined by 2.45 points. The decline was 2.2 points in the 1-mg/kg group; 0.75 in the 3-mg/kg group; and 0.55 in the 10-mg/kg group. The only statistically significant results relative to placebo occurred with the 10-mg/kg group (P less than .05).
The 6-mg/kg group didn’t fit this pattern though, losing an average of 2 points. Biogen has been unable to explain this, but some researchers suggest such an outlying result isn’t surprising, given the small numbers in each group and the exploratory nature of the cognitive analysis.
Amyloid-related imaging abnormalities (ARIA), an inflammatory reaction thought to be related to the removal of amyloid plaque, were the most common adverse event (n = 27). Most of these (22) were in apolipoprotein E4 allele carriers.
Two patients in the placebo arm died, as well as one in the 10-mg/kg arm, but it was not considered related to the study drug. There were no significant changes in hematology, chemistry, urinalysis, electrocardiogram, or vital signs.
Open-label extension results
Dr. Haeberlein focused on the subsequent 12-month, open-label extension trial, which enrolled 117 of the randomized cohort. In this study, patients who had been taking placebo were switched to either 3- or 6-mg/kg aducanumab. Patients who had taken 1 mg/kg were switched to 3 mg/kg. By the end, the remaining patients had taken the antibody for 2 years.
By 24 months, all the dosing groups showed a continued, linear reduction of amyloid plaques. Even those who switched from placebo to 3 mg/kg started to experience plaque reduction, although of a lesser magnitude than with the higher doses.
While still expressing caution, Dr. Haeberlein framed the CDR-SB results as very positive. The placebo and 1-mg/kg switchers continued to progress, but for those who continued on the 3-, 6-, and 10-mg/kg doses, “we saw a saw a numerical slowing of disease progression at both 18 and 24 months.”
However, none of the changes in CDR-SB scores reached statistical significance.
The numbers were somewhat more encouraging in the MMSE analysis. The 3- and 10-mg/kg groups began to separate at 12 months. By 24 months, the 10-mg/kg group had lost about 1 point on the MMSE while there were declines of about 2 points in the 3-mg/kg group and about 3 points in the 1-mg/kg group. Again, the 6-mg/kg group was an outlier, losing about 5 points.
“We already observed that this group behaved differently at 12 months on this endpoint, and we saw that particular cohort continued to follow that trend,” Dr. Haeberlein said.
There were 16 cases of ARIA in the extension trial. Eight were ARIA accompanied by vascular edema (ARIA-E) and these all occurred in the placebo and 1-mg/kg switchers. Three patients discontinued due to ARIA-E.
The remaining eight cases of ARIA were accompanied by microhemorrhage (ARIA-H); these were distributed among all of the dosage groups.
One patient with ARIA-E experienced a seizure and transient loss of pulse. Dr. Haeberlein didn’t elaborate on the possible cause of this event. Two additional patients died, with neither death judged related to the study medication.
The safety data, combined with the reduction of amyloid plaque and hints of cognitive and functional benefit, are enough to continue developing aducanumab, Dr. Haeberlein said. Biogen is recruiting 2,700 subjects with mild cognitive impairment or mild Alzheimer’s for identical phase III studies dubbed ENGAGE and EMERGE.
On Twitter @alz_gal
SAN DIEGO – The antiamyloid antibody aducanumab significantly reduced amyloid brain plaques in Alzheimer’s disease patients who entered a second year of therapy in an open-label extension trial.
Plaque volume declined in a time- and dose-dependent manner, with a bit of movement even among patients who crossed over to the lowest 1-mg/kg dose after a year of taking placebo in the phase Ib PRIME study, Samantha Haeberlein, PhD, reported at the Clinical Trials on Alzheimer’s Disease conference.
The linear declines in plaque burden were dramatic enough to draw a collective gasp of appreciation from the packed auditorium. But the cognitive and functional data of aducanumab (Biogen), while deemed encouraging, were not as striking. The Clinical Dementia Rating Scale-sum of boxes (CDR-SB) and the Mini-Mental State Examination (MMSE) showed dose-dependent slowing of decline, but the drug’s effect approached statistical significance only among those who finished 2 years on 10 mg/kg aducanumab infused every 4 weeks. This group experienced relative stability of MMSE scores, which dropped only about 1 point from baseline, and also showed the greatest decline in amyloid plaque volume.
Biogen deemed this result on the MMSE, which represented a 3.27-point separation from the 1-mg/kg dose group, as “nominally significant,” with a P value noted as “less than .05.”
However, the finding must be viewed with extreme caution, said Dr. Haeberlein, Biogen’s vice president of clinical development. There were only 15 subjects in this group, and the study was not primarily intended to examine cognition.
“I must emphasize once again that these are exploratory data and these sample sizes are very small for these types of assessments,” she said. “Nonetheless, we find them informative.”
MMSE changes in the other dosing groups of 1, 3, and 6 mg/kg were not statistically significant at the end of the study. There were not any significant findings on the CDR-SB measure.
Alzheimer’s Association reaction
Maria C. Carrillo, PhD, chief science officer of the Alzheimer’s Association, was cautiously optimistic.
“For MMSE, the original placebo group continued to decline even when they switched to the 1-mg/kg treatment, and by a significant number of points. The 1- to 3-mg/kg switching group is interesting, as they do get a little bit of a bump. But we also had the 6-mg/kg arm continuing to decline in MMSE and not improve as much as the others, which is strange. It’s aberrant, but the same thing we saw in the first study report. The 10-mg/kg group, though, stays almost at baseline. That’s pretty amazing. Impressive. Again, small numbers but very encouraging.”
Full 12-month results
Biogen presented two aducanumab abstracts at the meeting, both describing its 12-month phase Ib PRIME study and its 12-month, open-label extension study. The drug is a monoclonal human antibody derived from B cells collected from a cohort of cognitively normal elderly subjects and cognitively impaired elderly subjects who exhibited unusually slow decline, according to the company. It binds to fibrillar and oligomeric amyloid aggregates, thus directly reducing amyloid plaque in the brain.
PRIME enrolled 165 patients with prodromal or mild Alzheimer’s disease. Importantly, all of the subjects had brain amyloid proven by PET imaging. PRIME is the first randomized trial of an antiamyloid compound to enroll a pure amyloid-positive cohort. These subjects were randomized to placebo or aducanumab at 1, 3, 6, or 10 mg/kg for 1 year. PRIME’s primary outcomes were safety and tolerability. The cognitive and functional outcomes, not usually assessed in a phase Ib study, were exploratory.
They must also be interpreted in light of the very small numbers, about 30 patients in each dosing group at baseline. In addition, just 69 patients finished the entire 24-month dosing period, leaving only 15-23 patients in each group by the end of the study.
Vissia Viglietta, MD, Biogen’s senior medical director of clinical development, presented the 12-month data. At 52 weeks, all dosing groups, even the 1 mg/kg, saw statistically significant reductions in amyloid plaque, compared with placebo. These changes were dose-dependent; the 10-mg/kg group had the largest reduction, with a P value of less than .001.
There also were dose-dependent changes in the CDR-SB and MMSE, and some of these reached statistical significance.
On the CDR-SB, patients taking placebo declined by an average of 1.89 points. Declines in the 1-, 3-, and 6-mg/kg groups were not significant relative to placebo. However, the 10-mg/kg arm experienced a significant separation from placebo, declining an average of 0.63 points (P less than .05).
The story was similar for the MMSE. Patients taking placebo declined by 2.45 points. The decline was 2.2 points in the 1-mg/kg group; 0.75 in the 3-mg/kg group; and 0.55 in the 10-mg/kg group. The only statistically significant results relative to placebo occurred with the 10-mg/kg group (P less than .05).
The 6-mg/kg group didn’t fit this pattern though, losing an average of 2 points. Biogen has been unable to explain this, but some researchers suggest such an outlying result isn’t surprising, given the small numbers in each group and the exploratory nature of the cognitive analysis.
Amyloid-related imaging abnormalities (ARIA), an inflammatory reaction thought to be related to the removal of amyloid plaque, were the most common adverse event (n = 27). Most of these (22) were in apolipoprotein E4 allele carriers.
Two patients in the placebo arm died, as well as one in the 10-mg/kg arm, but it was not considered related to the study drug. There were no significant changes in hematology, chemistry, urinalysis, electrocardiogram, or vital signs.
Open-label extension results
Dr. Haeberlein focused on the subsequent 12-month, open-label extension trial, which enrolled 117 of the randomized cohort. In this study, patients who had been taking placebo were switched to either 3- or 6-mg/kg aducanumab. Patients who had taken 1 mg/kg were switched to 3 mg/kg. By the end, the remaining patients had taken the antibody for 2 years.
By 24 months, all the dosing groups showed a continued, linear reduction of amyloid plaques. Even those who switched from placebo to 3 mg/kg started to experience plaque reduction, although of a lesser magnitude than with the higher doses.
While still expressing caution, Dr. Haeberlein framed the CDR-SB results as very positive. The placebo and 1-mg/kg switchers continued to progress, but for those who continued on the 3-, 6-, and 10-mg/kg doses, “we saw a saw a numerical slowing of disease progression at both 18 and 24 months.”
However, none of the changes in CDR-SB scores reached statistical significance.
The numbers were somewhat more encouraging in the MMSE analysis. The 3- and 10-mg/kg groups began to separate at 12 months. By 24 months, the 10-mg/kg group had lost about 1 point on the MMSE while there were declines of about 2 points in the 3-mg/kg group and about 3 points in the 1-mg/kg group. Again, the 6-mg/kg group was an outlier, losing about 5 points.
“We already observed that this group behaved differently at 12 months on this endpoint, and we saw that particular cohort continued to follow that trend,” Dr. Haeberlein said.
There were 16 cases of ARIA in the extension trial. Eight were ARIA accompanied by vascular edema (ARIA-E) and these all occurred in the placebo and 1-mg/kg switchers. Three patients discontinued due to ARIA-E.
The remaining eight cases of ARIA were accompanied by microhemorrhage (ARIA-H); these were distributed among all of the dosage groups.
One patient with ARIA-E experienced a seizure and transient loss of pulse. Dr. Haeberlein didn’t elaborate on the possible cause of this event. Two additional patients died, with neither death judged related to the study medication.
The safety data, combined with the reduction of amyloid plaque and hints of cognitive and functional benefit, are enough to continue developing aducanumab, Dr. Haeberlein said. Biogen is recruiting 2,700 subjects with mild cognitive impairment or mild Alzheimer’s for identical phase III studies dubbed ENGAGE and EMERGE.
On Twitter @alz_gal
SAN DIEGO – The antiamyloid antibody aducanumab significantly reduced amyloid brain plaques in Alzheimer’s disease patients who entered a second year of therapy in an open-label extension trial.
Plaque volume declined in a time- and dose-dependent manner, with a bit of movement even among patients who crossed over to the lowest 1-mg/kg dose after a year of taking placebo in the phase Ib PRIME study, Samantha Haeberlein, PhD, reported at the Clinical Trials on Alzheimer’s Disease conference.
The linear declines in plaque burden were dramatic enough to draw a collective gasp of appreciation from the packed auditorium. But the cognitive and functional data of aducanumab (Biogen), while deemed encouraging, were not as striking. The Clinical Dementia Rating Scale-sum of boxes (CDR-SB) and the Mini-Mental State Examination (MMSE) showed dose-dependent slowing of decline, but the drug’s effect approached statistical significance only among those who finished 2 years on 10 mg/kg aducanumab infused every 4 weeks. This group experienced relative stability of MMSE scores, which dropped only about 1 point from baseline, and also showed the greatest decline in amyloid plaque volume.
Biogen deemed this result on the MMSE, which represented a 3.27-point separation from the 1-mg/kg dose group, as “nominally significant,” with a P value noted as “less than .05.”
However, the finding must be viewed with extreme caution, said Dr. Haeberlein, Biogen’s vice president of clinical development. There were only 15 subjects in this group, and the study was not primarily intended to examine cognition.
“I must emphasize once again that these are exploratory data and these sample sizes are very small for these types of assessments,” she said. “Nonetheless, we find them informative.”
MMSE changes in the other dosing groups of 1, 3, and 6 mg/kg were not statistically significant at the end of the study. There were not any significant findings on the CDR-SB measure.
Alzheimer’s Association reaction
Maria C. Carrillo, PhD, chief science officer of the Alzheimer’s Association, was cautiously optimistic.
“For MMSE, the original placebo group continued to decline even when they switched to the 1-mg/kg treatment, and by a significant number of points. The 1- to 3-mg/kg switching group is interesting, as they do get a little bit of a bump. But we also had the 6-mg/kg arm continuing to decline in MMSE and not improve as much as the others, which is strange. It’s aberrant, but the same thing we saw in the first study report. The 10-mg/kg group, though, stays almost at baseline. That’s pretty amazing. Impressive. Again, small numbers but very encouraging.”
Full 12-month results
Biogen presented two aducanumab abstracts at the meeting, both describing its 12-month phase Ib PRIME study and its 12-month, open-label extension study. The drug is a monoclonal human antibody derived from B cells collected from a cohort of cognitively normal elderly subjects and cognitively impaired elderly subjects who exhibited unusually slow decline, according to the company. It binds to fibrillar and oligomeric amyloid aggregates, thus directly reducing amyloid plaque in the brain.
PRIME enrolled 165 patients with prodromal or mild Alzheimer’s disease. Importantly, all of the subjects had brain amyloid proven by PET imaging. PRIME is the first randomized trial of an antiamyloid compound to enroll a pure amyloid-positive cohort. These subjects were randomized to placebo or aducanumab at 1, 3, 6, or 10 mg/kg for 1 year. PRIME’s primary outcomes were safety and tolerability. The cognitive and functional outcomes, not usually assessed in a phase Ib study, were exploratory.
They must also be interpreted in light of the very small numbers, about 30 patients in each dosing group at baseline. In addition, just 69 patients finished the entire 24-month dosing period, leaving only 15-23 patients in each group by the end of the study.
Vissia Viglietta, MD, Biogen’s senior medical director of clinical development, presented the 12-month data. At 52 weeks, all dosing groups, even the 1 mg/kg, saw statistically significant reductions in amyloid plaque, compared with placebo. These changes were dose-dependent; the 10-mg/kg group had the largest reduction, with a P value of less than .001.
There also were dose-dependent changes in the CDR-SB and MMSE, and some of these reached statistical significance.
On the CDR-SB, patients taking placebo declined by an average of 1.89 points. Declines in the 1-, 3-, and 6-mg/kg groups were not significant relative to placebo. However, the 10-mg/kg arm experienced a significant separation from placebo, declining an average of 0.63 points (P less than .05).
The story was similar for the MMSE. Patients taking placebo declined by 2.45 points. The decline was 2.2 points in the 1-mg/kg group; 0.75 in the 3-mg/kg group; and 0.55 in the 10-mg/kg group. The only statistically significant results relative to placebo occurred with the 10-mg/kg group (P less than .05).
The 6-mg/kg group didn’t fit this pattern though, losing an average of 2 points. Biogen has been unable to explain this, but some researchers suggest such an outlying result isn’t surprising, given the small numbers in each group and the exploratory nature of the cognitive analysis.
Amyloid-related imaging abnormalities (ARIA), an inflammatory reaction thought to be related to the removal of amyloid plaque, were the most common adverse event (n = 27). Most of these (22) were in apolipoprotein E4 allele carriers.
Two patients in the placebo arm died, as well as one in the 10-mg/kg arm, but it was not considered related to the study drug. There were no significant changes in hematology, chemistry, urinalysis, electrocardiogram, or vital signs.
Open-label extension results
Dr. Haeberlein focused on the subsequent 12-month, open-label extension trial, which enrolled 117 of the randomized cohort. In this study, patients who had been taking placebo were switched to either 3- or 6-mg/kg aducanumab. Patients who had taken 1 mg/kg were switched to 3 mg/kg. By the end, the remaining patients had taken the antibody for 2 years.
By 24 months, all the dosing groups showed a continued, linear reduction of amyloid plaques. Even those who switched from placebo to 3 mg/kg started to experience plaque reduction, although of a lesser magnitude than with the higher doses.
While still expressing caution, Dr. Haeberlein framed the CDR-SB results as very positive. The placebo and 1-mg/kg switchers continued to progress, but for those who continued on the 3-, 6-, and 10-mg/kg doses, “we saw a saw a numerical slowing of disease progression at both 18 and 24 months.”
However, none of the changes in CDR-SB scores reached statistical significance.
The numbers were somewhat more encouraging in the MMSE analysis. The 3- and 10-mg/kg groups began to separate at 12 months. By 24 months, the 10-mg/kg group had lost about 1 point on the MMSE while there were declines of about 2 points in the 3-mg/kg group and about 3 points in the 1-mg/kg group. Again, the 6-mg/kg group was an outlier, losing about 5 points.
“We already observed that this group behaved differently at 12 months on this endpoint, and we saw that particular cohort continued to follow that trend,” Dr. Haeberlein said.
There were 16 cases of ARIA in the extension trial. Eight were ARIA accompanied by vascular edema (ARIA-E) and these all occurred in the placebo and 1-mg/kg switchers. Three patients discontinued due to ARIA-E.
The remaining eight cases of ARIA were accompanied by microhemorrhage (ARIA-H); these were distributed among all of the dosage groups.
One patient with ARIA-E experienced a seizure and transient loss of pulse. Dr. Haeberlein didn’t elaborate on the possible cause of this event. Two additional patients died, with neither death judged related to the study medication.
The safety data, combined with the reduction of amyloid plaque and hints of cognitive and functional benefit, are enough to continue developing aducanumab, Dr. Haeberlein said. Biogen is recruiting 2,700 subjects with mild cognitive impairment or mild Alzheimer’s for identical phase III studies dubbed ENGAGE and EMERGE.
On Twitter @alz_gal
AT CTAD
Key clinical point:
Major finding: Aducanumab 10 mg/kg was associated with a near-stabilization of the MMSE score over 24 months in 15 patients in an open-label extension of a phase Ib study.
Data source: A 2-year study consisting of a 12-month, randomized, dose-finding, placebo-controlled trial, followed by 12 months of open-label treatment at four different doses.
Disclosures: Dr. Haeberlein and Dr. Viglietta are employees of Biogen, which is developing the molecule.
“Where’s the Music?” Using Music Therapy for Pain Management
Staff at the Malcolm Randall VAMC (MRVAMC) outpatient pain clinic in Gainesville, Florida, found that procedures to reduce a patient’s pain could initially cause pain and anxiety. Typical nursing care plans involved measures to reduce anxiety in patients undergoing interventional procedures expected to produce pain, including identifying and reinforcing coping strategies, providing reassurance and comfort, and giving patients clear explanations slowly and calmly. The MRVAMC nursing staff therefore also advocated to add music therapy to the existing plan.
Background
As part of a quality improvement (QI) project, the authors conducted a literature search to find scientific evidence for the use of music therapy. Multiple medical databases were analyzed to find studies that included total time, dose of sedative medications, pain scores, patient experience, and willingness to repeat the same procedure in the future with use of music vs no music.1 The literature review revealed that music therapy demonstrated effectiveness in decreasing anxiety and pain, supporting relaxation, reducing sedation medication during procedures, and improving patient satisfaction.
As a result of the literature search the authors conducted a prospective, randomized controlled study to investigate music therapy as an adjunct intervention during painful procedures.
Radiofrequency Lesioning
One of the more common (and most painful) procedures performed at MRVAMC is radiofrequency lesioning (RFL).The procedure uses electrical pulses to block nerves for pain relief. Using fluoroscopy, the physician inserts a needle adjacent to the nerve that innervates the facet joint. The sensory and motor nerves are stimulated, causing a tingling or buzzing sensation and tapping. Once the tip of the needle is placed in the correct location, electrical pulses (small radiofrequency currents) are passed through the needle. A lesion is formed that temporarily interrupts the pain messages that the nerve sends to the brain. The procedure can take between30 and 60 minutes, which is longer than most pain clinic procedures.
Radiofrequency lesioning controls pain caused by degenerative disc disease, facet arthropathy, sacroiliac joints, stellate ganglions, and other nerve conditions. Due to the length of the RFL procedure, patients may experience pain and anxiety (as well as other complications, such as vasovagal responses).
The clinic staff anticipated that there would be 20 RFL procedures scheduled per week and selected it as the study procedure for 3 reasons: procedure length, high level of pain, and frequency performed.
After receiving approval from the University of Florida Institutional Review Board and VA Research and Development, the MRVAMC pain clinic initiated the study from September 2013 to April 2014. The purpose of the study was to measure the effects of music on patient’s self-reported anxiety and pain levels before and after nonsedating lumbar RFL.
Methods
Study Design
Veterans aged between 21 and 88 years who were scheduled to return for lumbar RFL and who did not require sedation were invited to participate. Sixty participants consented. The music group had 21 men and 2 women. The no-music group had 19 men and 2 women. Table 1 summarizes descriptive data. Table 2 describes the results of the comparison analysis. Patients were randomly assigned to either the music intervention group or no-music g
The study tools included the global anxiety VAS (GA-VAS) for pain and anxiety and a yes/no self-reported question, “Did music help?” for participants in the music group. Evaluation of the GA-VAS demonstrated reliability and validity and were patient friendly.2,3 Pain was recorded using a Likert scale of 0 for no pain and 10 for severe pain. Anxiety was recorded using a Likert scale of 0 for no anxiety and 4 for extreme anxiety.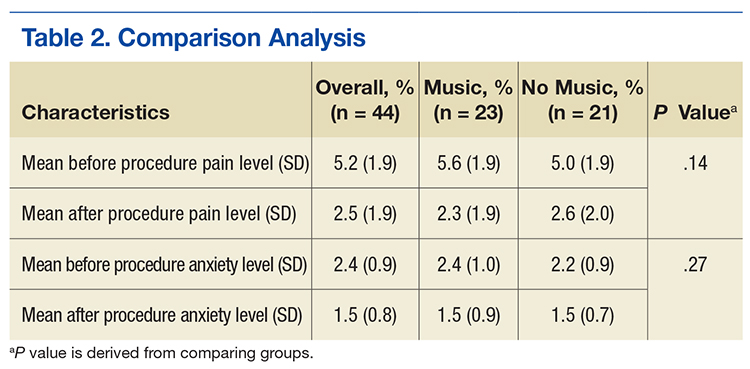
Study subjects were recruited from patients who were on a maintenance lumbar RFL schedule, did not require sedation, and were willing to participate. If sedation was required, the patient was excluded from the study. Returning patients scheduled for RFL were informed about the music study and asked whether they were interested in participating. If they agreed, the study was explained in full, and informed consent was obtained prior to the day of their scheduled procedure. After obtaining informed consent, participants were asked to choose a music genre from 3 options: easy listening, jazz, or classical. Participants received a sealed envelope identifying their group (30 envelopes were created for each group) to be opened by the procedure nurse on the day of the procedure.4,5
Sixty participants consented to participate in the study. Of these 60 patients, 44 were studied. The 16 patients who did not participate had either a change in procedure or did not show for the appointment.
Data Collection
On procedure day, all participants completed the anxiety scale as well as a VAS pain scale (which is the MRVAMC standard of care), preoperatively and postprocedure. Envelopes were opened prior to going into the procedure room to prevent prior knowledge of who was assigned to the music group. Participants in the music intervention group listened to their preselected music on a portable CD player in the procedure room. The music was played softly so the patient could still hear and respond to the physicians instructions during the procedure. The no-music group received everything that the music intervention group received except for music (standard care throughout procedure, which consisted of nurse monitoring, measures to reduce fear and anxiety, and comfort measures). Procedures were performed with local anesthesia; neither group received moderate sedation.
Gender, age, and self-reported pain scores (before and after the lumber RFL procedure) were recorded in the patient’s chart and entered into the study database. Patients in both groups were queried before and after the procedure using the VAS to measure their pain and anxiety levels. Participants in the music intervention group were asked whether they felt that the music helped. They also were asked to provide feedback about their experience. Data were stored in locked filing cabinets, and all forms were de-identified.
Statistical Analysis
SAS version 9.2 (Cary, NC) was used for all analyses. Data were inspected for out-of-range values. The Fisher exact test was used to compare groups on categorical measures. An independent sample t test was used to compare groups on the age variable. Difference scores (formed by subtracting the after score from the before score) were analyzed using paired t tests. Analysis of covariance was used to test for significant group differences on the outcome variables of pain and anxiety with group as the independent variable and the preprocedure measure as the covariate. The level of significance was set at .05, and all testing was 2 sided.
Results
Of the 60 consenting patients, 44 participated in the study.Twenty-three were randomized to the music intervention and 21 to the no-music control group. Both pain and anxiety were significantly reduced (P < .0001) in the total sample (n = 44). The mean (SD) decrease in pain for all participants was 2.80 points (2.31) on a VAS of 0 to 10 and 0.86 points (0.93) decrease in anxiety. In the music intervention group, the mean decrease in pain and anxiety was 3.22 (2.66) and 1.00 (0.85), respectively. In the no-music group, the mean decrease in pain and anxiety was 2.33 (1.80) and 0.69 (1.00), respectively. The magnitude of pain decrease was larger in the music intervention group; however, the difference did not reach statistical significance.
Discussion
Although there was not a statistically significant difference in pain or anxiety reduction due to group assignment, a 2-point reduction in self-reported pain or anxiety may be considered clinically important and has been supported in older studies.6 Importantly, 87% of participants in the music intervention group reported that listening to music was helpful during the procedure (Figure 1).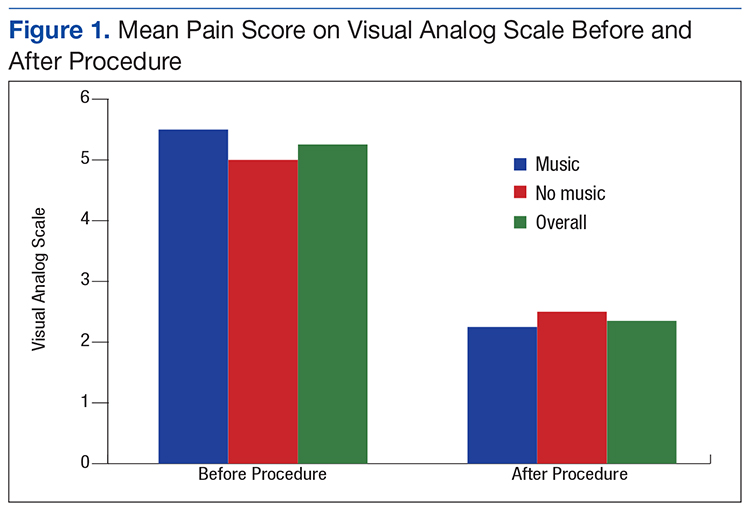
Anxiety levels were not as high as expected when measured before and after the procedure, perhaps due to improvements in patient education and continuity of care (Figure 2). Since all participants were returning patients, they already were familiar with the procedure and the staff. Staff turnover rate is very low at this clinic, which may have contributed to the low anxiety rates among participants at baseline. Other contributing factors included good communication, expert technique, and teamwork.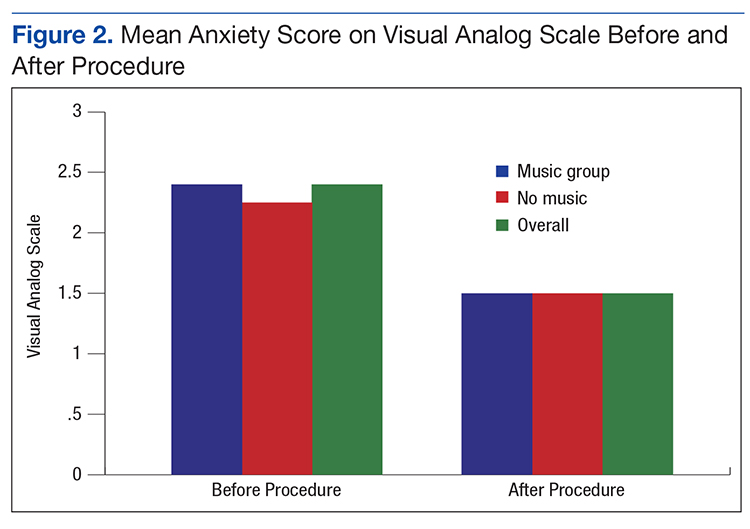
During the study, few negative comments were noted. One participant did not hear the music due to faulty equipment setup. Another participant commented that the physician doing the procedure made negative remarks about the music the patient selected. A third participant commented that the music was too loud, and he was unable to hear the doctor’s instructions, indicating a need for guidelines.
There were many positive comments by participants in the music intervention group. Nurses reported comments such as “The music really helps”; “The music was great, but rock ‘n’ roll would be better”; and “Can I bring my own [music] next time?” Many patients returning for procedures frequently asked, “Where is the music?”
Limitations
Of the 60 consenting patients, only 44 participated, possibly lowering the power of the study to detect significant findings. During the study, the physician staff was reduced, resulting in fewer RFLs performed and causing the study to take longer to conduct and with fewer opportunities to recruit participants.
The CD players used for the study were old, and because earbuds could not be used, volume was difficult to modulate consistently. Earbuds were not used because patient participation was required during the procedure. Also, having only 3 music genres to choose from limited the participant’s choice.
Conclusion
Research supporting the use of music therapy to increase patient comfort is widely accepted and practiced.7 Music therapy is readily available, low risk, inexpensive, and does not require intense training by staff. It may reduce the need for moderate sedation and improve the overall patient experience. During the study, the MRVAMC nursing staff gained a greater appreciation of evidence-based practice; staff are more engaged in QI, based on their personal involvement in research.
Because 87% of the music therapy participants reported that music was helpful, the MRVAMC pain clinic plans to implement music therapy as a standard of care during RFL procedures and all procedure appointments. Music therapy may help reduce pain and anxiety during painful procedures. The goal is to continually increase patient satisfaction and overall procedure experience through integration of evidence-based practice.
Acknowledgments
The authors thank the study team who helped consent participants, perform the experiment, and gather and analyze data. They also acknowledge the pain clinic physicians, Dr. Egle Bavry and Dr. Heidi Goldstein, for their support throughout the study. Special thanks goes to Daniel Prince for his technical support.
1. Bechtold ML, Perez RA, Puli SR, Marshall JB. Effect of music on patients undergoing outpatient colonoscopy. World J Gastroenterol. 2006;12(45):7309-7312.
2. Miller SD, Duncan BL, Brown J, Sparks JA, Claud DA. The outcome rating scale: a preliminary study of the reliability, validity, and feasibility of a brief visual analog measure. J Brief Ther. 2003;2(2):91-100.
3. Williams VS, Morlock RJ, Feltner D. Psychometric evaluation of a visual analog scale for the assessment of anxiety. Health Qual Life Outcomes. 2010;8:57.
4. Dettori J. The random allocation process: two things you need to know. Evid Based Spine Care J. 2010;1(3):7-9.
5. Farrokhyar F, Bajammal S, Kahnamoui K, Bhandari M. Ensuring balanced groups in surgical trials. Can J Surg. 2010;53(6):418-423.
6. Hägg O, Fritzell P, Nordwall A; Swedish Lumbar Spine Study Group. The clinical importance of changes in outcome scores after treatment for chronic low back pain. Eur Spine J. 2003;12(1):12-20.
7. Sonke J. Music and the arts in health: a perspective from the United States. Music Arts Action. 2011;3(2):5-14
Staff at the Malcolm Randall VAMC (MRVAMC) outpatient pain clinic in Gainesville, Florida, found that procedures to reduce a patient’s pain could initially cause pain and anxiety. Typical nursing care plans involved measures to reduce anxiety in patients undergoing interventional procedures expected to produce pain, including identifying and reinforcing coping strategies, providing reassurance and comfort, and giving patients clear explanations slowly and calmly. The MRVAMC nursing staff therefore also advocated to add music therapy to the existing plan.
Background
As part of a quality improvement (QI) project, the authors conducted a literature search to find scientific evidence for the use of music therapy. Multiple medical databases were analyzed to find studies that included total time, dose of sedative medications, pain scores, patient experience, and willingness to repeat the same procedure in the future with use of music vs no music.1 The literature review revealed that music therapy demonstrated effectiveness in decreasing anxiety and pain, supporting relaxation, reducing sedation medication during procedures, and improving patient satisfaction.
As a result of the literature search the authors conducted a prospective, randomized controlled study to investigate music therapy as an adjunct intervention during painful procedures.
Radiofrequency Lesioning
One of the more common (and most painful) procedures performed at MRVAMC is radiofrequency lesioning (RFL).The procedure uses electrical pulses to block nerves for pain relief. Using fluoroscopy, the physician inserts a needle adjacent to the nerve that innervates the facet joint. The sensory and motor nerves are stimulated, causing a tingling or buzzing sensation and tapping. Once the tip of the needle is placed in the correct location, electrical pulses (small radiofrequency currents) are passed through the needle. A lesion is formed that temporarily interrupts the pain messages that the nerve sends to the brain. The procedure can take between30 and 60 minutes, which is longer than most pain clinic procedures.
Radiofrequency lesioning controls pain caused by degenerative disc disease, facet arthropathy, sacroiliac joints, stellate ganglions, and other nerve conditions. Due to the length of the RFL procedure, patients may experience pain and anxiety (as well as other complications, such as vasovagal responses).
The clinic staff anticipated that there would be 20 RFL procedures scheduled per week and selected it as the study procedure for 3 reasons: procedure length, high level of pain, and frequency performed.
After receiving approval from the University of Florida Institutional Review Board and VA Research and Development, the MRVAMC pain clinic initiated the study from September 2013 to April 2014. The purpose of the study was to measure the effects of music on patient’s self-reported anxiety and pain levels before and after nonsedating lumbar RFL.
Methods
Study Design
Veterans aged between 21 and 88 years who were scheduled to return for lumbar RFL and who did not require sedation were invited to participate. Sixty participants consented. The music group had 21 men and 2 women. The no-music group had 19 men and 2 women. Table 1 summarizes descriptive data. Table 2 describes the results of the comparison analysis. Patients were randomly assigned to either the music intervention group or no-music g
The study tools included the global anxiety VAS (GA-VAS) for pain and anxiety and a yes/no self-reported question, “Did music help?” for participants in the music group. Evaluation of the GA-VAS demonstrated reliability and validity and were patient friendly.2,3 Pain was recorded using a Likert scale of 0 for no pain and 10 for severe pain. Anxiety was recorded using a Likert scale of 0 for no anxiety and 4 for extreme anxiety.
Study subjects were recruited from patients who were on a maintenance lumbar RFL schedule, did not require sedation, and were willing to participate. If sedation was required, the patient was excluded from the study. Returning patients scheduled for RFL were informed about the music study and asked whether they were interested in participating. If they agreed, the study was explained in full, and informed consent was obtained prior to the day of their scheduled procedure. After obtaining informed consent, participants were asked to choose a music genre from 3 options: easy listening, jazz, or classical. Participants received a sealed envelope identifying their group (30 envelopes were created for each group) to be opened by the procedure nurse on the day of the procedure.4,5
Sixty participants consented to participate in the study. Of these 60 patients, 44 were studied. The 16 patients who did not participate had either a change in procedure or did not show for the appointment.
Data Collection
On procedure day, all participants completed the anxiety scale as well as a VAS pain scale (which is the MRVAMC standard of care), preoperatively and postprocedure. Envelopes were opened prior to going into the procedure room to prevent prior knowledge of who was assigned to the music group. Participants in the music intervention group listened to their preselected music on a portable CD player in the procedure room. The music was played softly so the patient could still hear and respond to the physicians instructions during the procedure. The no-music group received everything that the music intervention group received except for music (standard care throughout procedure, which consisted of nurse monitoring, measures to reduce fear and anxiety, and comfort measures). Procedures were performed with local anesthesia; neither group received moderate sedation.
Gender, age, and self-reported pain scores (before and after the lumber RFL procedure) were recorded in the patient’s chart and entered into the study database. Patients in both groups were queried before and after the procedure using the VAS to measure their pain and anxiety levels. Participants in the music intervention group were asked whether they felt that the music helped. They also were asked to provide feedback about their experience. Data were stored in locked filing cabinets, and all forms were de-identified.
Statistical Analysis
SAS version 9.2 (Cary, NC) was used for all analyses. Data were inspected for out-of-range values. The Fisher exact test was used to compare groups on categorical measures. An independent sample t test was used to compare groups on the age variable. Difference scores (formed by subtracting the after score from the before score) were analyzed using paired t tests. Analysis of covariance was used to test for significant group differences on the outcome variables of pain and anxiety with group as the independent variable and the preprocedure measure as the covariate. The level of significance was set at .05, and all testing was 2 sided.
Results
Of the 60 consenting patients, 44 participated in the study.Twenty-three were randomized to the music intervention and 21 to the no-music control group. Both pain and anxiety were significantly reduced (P < .0001) in the total sample (n = 44). The mean (SD) decrease in pain for all participants was 2.80 points (2.31) on a VAS of 0 to 10 and 0.86 points (0.93) decrease in anxiety. In the music intervention group, the mean decrease in pain and anxiety was 3.22 (2.66) and 1.00 (0.85), respectively. In the no-music group, the mean decrease in pain and anxiety was 2.33 (1.80) and 0.69 (1.00), respectively. The magnitude of pain decrease was larger in the music intervention group; however, the difference did not reach statistical significance.
Discussion
Although there was not a statistically significant difference in pain or anxiety reduction due to group assignment, a 2-point reduction in self-reported pain or anxiety may be considered clinically important and has been supported in older studies.6 Importantly, 87% of participants in the music intervention group reported that listening to music was helpful during the procedure (Figure 1).
Anxiety levels were not as high as expected when measured before and after the procedure, perhaps due to improvements in patient education and continuity of care (Figure 2). Since all participants were returning patients, they already were familiar with the procedure and the staff. Staff turnover rate is very low at this clinic, which may have contributed to the low anxiety rates among participants at baseline. Other contributing factors included good communication, expert technique, and teamwork.
During the study, few negative comments were noted. One participant did not hear the music due to faulty equipment setup. Another participant commented that the physician doing the procedure made negative remarks about the music the patient selected. A third participant commented that the music was too loud, and he was unable to hear the doctor’s instructions, indicating a need for guidelines.
There were many positive comments by participants in the music intervention group. Nurses reported comments such as “The music really helps”; “The music was great, but rock ‘n’ roll would be better”; and “Can I bring my own [music] next time?” Many patients returning for procedures frequently asked, “Where is the music?”
Limitations
Of the 60 consenting patients, only 44 participated, possibly lowering the power of the study to detect significant findings. During the study, the physician staff was reduced, resulting in fewer RFLs performed and causing the study to take longer to conduct and with fewer opportunities to recruit participants.
The CD players used for the study were old, and because earbuds could not be used, volume was difficult to modulate consistently. Earbuds were not used because patient participation was required during the procedure. Also, having only 3 music genres to choose from limited the participant’s choice.
Conclusion
Research supporting the use of music therapy to increase patient comfort is widely accepted and practiced.7 Music therapy is readily available, low risk, inexpensive, and does not require intense training by staff. It may reduce the need for moderate sedation and improve the overall patient experience. During the study, the MRVAMC nursing staff gained a greater appreciation of evidence-based practice; staff are more engaged in QI, based on their personal involvement in research.
Because 87% of the music therapy participants reported that music was helpful, the MRVAMC pain clinic plans to implement music therapy as a standard of care during RFL procedures and all procedure appointments. Music therapy may help reduce pain and anxiety during painful procedures. The goal is to continually increase patient satisfaction and overall procedure experience through integration of evidence-based practice.
Acknowledgments
The authors thank the study team who helped consent participants, perform the experiment, and gather and analyze data. They also acknowledge the pain clinic physicians, Dr. Egle Bavry and Dr. Heidi Goldstein, for their support throughout the study. Special thanks goes to Daniel Prince for his technical support.
Staff at the Malcolm Randall VAMC (MRVAMC) outpatient pain clinic in Gainesville, Florida, found that procedures to reduce a patient’s pain could initially cause pain and anxiety. Typical nursing care plans involved measures to reduce anxiety in patients undergoing interventional procedures expected to produce pain, including identifying and reinforcing coping strategies, providing reassurance and comfort, and giving patients clear explanations slowly and calmly. The MRVAMC nursing staff therefore also advocated to add music therapy to the existing plan.
Background
As part of a quality improvement (QI) project, the authors conducted a literature search to find scientific evidence for the use of music therapy. Multiple medical databases were analyzed to find studies that included total time, dose of sedative medications, pain scores, patient experience, and willingness to repeat the same procedure in the future with use of music vs no music.1 The literature review revealed that music therapy demonstrated effectiveness in decreasing anxiety and pain, supporting relaxation, reducing sedation medication during procedures, and improving patient satisfaction.
As a result of the literature search the authors conducted a prospective, randomized controlled study to investigate music therapy as an adjunct intervention during painful procedures.
Radiofrequency Lesioning
One of the more common (and most painful) procedures performed at MRVAMC is radiofrequency lesioning (RFL).The procedure uses electrical pulses to block nerves for pain relief. Using fluoroscopy, the physician inserts a needle adjacent to the nerve that innervates the facet joint. The sensory and motor nerves are stimulated, causing a tingling or buzzing sensation and tapping. Once the tip of the needle is placed in the correct location, electrical pulses (small radiofrequency currents) are passed through the needle. A lesion is formed that temporarily interrupts the pain messages that the nerve sends to the brain. The procedure can take between30 and 60 minutes, which is longer than most pain clinic procedures.
Radiofrequency lesioning controls pain caused by degenerative disc disease, facet arthropathy, sacroiliac joints, stellate ganglions, and other nerve conditions. Due to the length of the RFL procedure, patients may experience pain and anxiety (as well as other complications, such as vasovagal responses).
The clinic staff anticipated that there would be 20 RFL procedures scheduled per week and selected it as the study procedure for 3 reasons: procedure length, high level of pain, and frequency performed.
After receiving approval from the University of Florida Institutional Review Board and VA Research and Development, the MRVAMC pain clinic initiated the study from September 2013 to April 2014. The purpose of the study was to measure the effects of music on patient’s self-reported anxiety and pain levels before and after nonsedating lumbar RFL.
Methods
Study Design
Veterans aged between 21 and 88 years who were scheduled to return for lumbar RFL and who did not require sedation were invited to participate. Sixty participants consented. The music group had 21 men and 2 women. The no-music group had 19 men and 2 women. Table 1 summarizes descriptive data. Table 2 describes the results of the comparison analysis. Patients were randomly assigned to either the music intervention group or no-music g
The study tools included the global anxiety VAS (GA-VAS) for pain and anxiety and a yes/no self-reported question, “Did music help?” for participants in the music group. Evaluation of the GA-VAS demonstrated reliability and validity and were patient friendly.2,3 Pain was recorded using a Likert scale of 0 for no pain and 10 for severe pain. Anxiety was recorded using a Likert scale of 0 for no anxiety and 4 for extreme anxiety.
Study subjects were recruited from patients who were on a maintenance lumbar RFL schedule, did not require sedation, and were willing to participate. If sedation was required, the patient was excluded from the study. Returning patients scheduled for RFL were informed about the music study and asked whether they were interested in participating. If they agreed, the study was explained in full, and informed consent was obtained prior to the day of their scheduled procedure. After obtaining informed consent, participants were asked to choose a music genre from 3 options: easy listening, jazz, or classical. Participants received a sealed envelope identifying their group (30 envelopes were created for each group) to be opened by the procedure nurse on the day of the procedure.4,5
Sixty participants consented to participate in the study. Of these 60 patients, 44 were studied. The 16 patients who did not participate had either a change in procedure or did not show for the appointment.
Data Collection
On procedure day, all participants completed the anxiety scale as well as a VAS pain scale (which is the MRVAMC standard of care), preoperatively and postprocedure. Envelopes were opened prior to going into the procedure room to prevent prior knowledge of who was assigned to the music group. Participants in the music intervention group listened to their preselected music on a portable CD player in the procedure room. The music was played softly so the patient could still hear and respond to the physicians instructions during the procedure. The no-music group received everything that the music intervention group received except for music (standard care throughout procedure, which consisted of nurse monitoring, measures to reduce fear and anxiety, and comfort measures). Procedures were performed with local anesthesia; neither group received moderate sedation.
Gender, age, and self-reported pain scores (before and after the lumber RFL procedure) were recorded in the patient’s chart and entered into the study database. Patients in both groups were queried before and after the procedure using the VAS to measure their pain and anxiety levels. Participants in the music intervention group were asked whether they felt that the music helped. They also were asked to provide feedback about their experience. Data were stored in locked filing cabinets, and all forms were de-identified.
Statistical Analysis
SAS version 9.2 (Cary, NC) was used for all analyses. Data were inspected for out-of-range values. The Fisher exact test was used to compare groups on categorical measures. An independent sample t test was used to compare groups on the age variable. Difference scores (formed by subtracting the after score from the before score) were analyzed using paired t tests. Analysis of covariance was used to test for significant group differences on the outcome variables of pain and anxiety with group as the independent variable and the preprocedure measure as the covariate. The level of significance was set at .05, and all testing was 2 sided.
Results
Of the 60 consenting patients, 44 participated in the study.Twenty-three were randomized to the music intervention and 21 to the no-music control group. Both pain and anxiety were significantly reduced (P < .0001) in the total sample (n = 44). The mean (SD) decrease in pain for all participants was 2.80 points (2.31) on a VAS of 0 to 10 and 0.86 points (0.93) decrease in anxiety. In the music intervention group, the mean decrease in pain and anxiety was 3.22 (2.66) and 1.00 (0.85), respectively. In the no-music group, the mean decrease in pain and anxiety was 2.33 (1.80) and 0.69 (1.00), respectively. The magnitude of pain decrease was larger in the music intervention group; however, the difference did not reach statistical significance.
Discussion
Although there was not a statistically significant difference in pain or anxiety reduction due to group assignment, a 2-point reduction in self-reported pain or anxiety may be considered clinically important and has been supported in older studies.6 Importantly, 87% of participants in the music intervention group reported that listening to music was helpful during the procedure (Figure 1).
Anxiety levels were not as high as expected when measured before and after the procedure, perhaps due to improvements in patient education and continuity of care (Figure 2). Since all participants were returning patients, they already were familiar with the procedure and the staff. Staff turnover rate is very low at this clinic, which may have contributed to the low anxiety rates among participants at baseline. Other contributing factors included good communication, expert technique, and teamwork.
During the study, few negative comments were noted. One participant did not hear the music due to faulty equipment setup. Another participant commented that the physician doing the procedure made negative remarks about the music the patient selected. A third participant commented that the music was too loud, and he was unable to hear the doctor’s instructions, indicating a need for guidelines.
There were many positive comments by participants in the music intervention group. Nurses reported comments such as “The music really helps”; “The music was great, but rock ‘n’ roll would be better”; and “Can I bring my own [music] next time?” Many patients returning for procedures frequently asked, “Where is the music?”
Limitations
Of the 60 consenting patients, only 44 participated, possibly lowering the power of the study to detect significant findings. During the study, the physician staff was reduced, resulting in fewer RFLs performed and causing the study to take longer to conduct and with fewer opportunities to recruit participants.
The CD players used for the study were old, and because earbuds could not be used, volume was difficult to modulate consistently. Earbuds were not used because patient participation was required during the procedure. Also, having only 3 music genres to choose from limited the participant’s choice.
Conclusion
Research supporting the use of music therapy to increase patient comfort is widely accepted and practiced.7 Music therapy is readily available, low risk, inexpensive, and does not require intense training by staff. It may reduce the need for moderate sedation and improve the overall patient experience. During the study, the MRVAMC nursing staff gained a greater appreciation of evidence-based practice; staff are more engaged in QI, based on their personal involvement in research.
Because 87% of the music therapy participants reported that music was helpful, the MRVAMC pain clinic plans to implement music therapy as a standard of care during RFL procedures and all procedure appointments. Music therapy may help reduce pain and anxiety during painful procedures. The goal is to continually increase patient satisfaction and overall procedure experience through integration of evidence-based practice.
Acknowledgments
The authors thank the study team who helped consent participants, perform the experiment, and gather and analyze data. They also acknowledge the pain clinic physicians, Dr. Egle Bavry and Dr. Heidi Goldstein, for their support throughout the study. Special thanks goes to Daniel Prince for his technical support.
1. Bechtold ML, Perez RA, Puli SR, Marshall JB. Effect of music on patients undergoing outpatient colonoscopy. World J Gastroenterol. 2006;12(45):7309-7312.
2. Miller SD, Duncan BL, Brown J, Sparks JA, Claud DA. The outcome rating scale: a preliminary study of the reliability, validity, and feasibility of a brief visual analog measure. J Brief Ther. 2003;2(2):91-100.
3. Williams VS, Morlock RJ, Feltner D. Psychometric evaluation of a visual analog scale for the assessment of anxiety. Health Qual Life Outcomes. 2010;8:57.
4. Dettori J. The random allocation process: two things you need to know. Evid Based Spine Care J. 2010;1(3):7-9.
5. Farrokhyar F, Bajammal S, Kahnamoui K, Bhandari M. Ensuring balanced groups in surgical trials. Can J Surg. 2010;53(6):418-423.
6. Hägg O, Fritzell P, Nordwall A; Swedish Lumbar Spine Study Group. The clinical importance of changes in outcome scores after treatment for chronic low back pain. Eur Spine J. 2003;12(1):12-20.
7. Sonke J. Music and the arts in health: a perspective from the United States. Music Arts Action. 2011;3(2):5-14
1. Bechtold ML, Perez RA, Puli SR, Marshall JB. Effect of music on patients undergoing outpatient colonoscopy. World J Gastroenterol. 2006;12(45):7309-7312.
2. Miller SD, Duncan BL, Brown J, Sparks JA, Claud DA. The outcome rating scale: a preliminary study of the reliability, validity, and feasibility of a brief visual analog measure. J Brief Ther. 2003;2(2):91-100.
3. Williams VS, Morlock RJ, Feltner D. Psychometric evaluation of a visual analog scale for the assessment of anxiety. Health Qual Life Outcomes. 2010;8:57.
4. Dettori J. The random allocation process: two things you need to know. Evid Based Spine Care J. 2010;1(3):7-9.
5. Farrokhyar F, Bajammal S, Kahnamoui K, Bhandari M. Ensuring balanced groups in surgical trials. Can J Surg. 2010;53(6):418-423.
6. Hägg O, Fritzell P, Nordwall A; Swedish Lumbar Spine Study Group. The clinical importance of changes in outcome scores after treatment for chronic low back pain. Eur Spine J. 2003;12(1):12-20.
7. Sonke J. Music and the arts in health: a perspective from the United States. Music Arts Action. 2011;3(2):5-14
MDS patients with mutated IDH2 benefit from enasidenib

Photo courtesy of ASH
SAN DIEGO—Daily treatment with enasidenib monotherapy in patients with mutated IDH2-positive myelodysplastic syndromes (MDS) induced responses in the majority of patients treated, according to a presentation at the 2016 ASH Annual Meeting.
The study was a portion of a larger phase 1/2 trial of the agent in patients with acute myeloid leukemia (AML) and other hematologic malignancies, so the subset was relatively small, numbering 17 patients.
Nevertheless, enasidenib was well tolerated and induced responses in these predominantly higher-risk patients.
Enasidenib (AG-221/CC-9007) is a selective, oral, potent inhibitor of mutant IDH2 (mIDH2), which produces 2-HG and thus alters DNA methylation and blocks cellular differentiation of hematopoietic progenitor cells.
Approximately 15% of AML patients and 5% of MDS patients have mIDH2. So investigators undertook the study to evaluate the safety and efficacy of enasidenib monotherapy in these diseases.
Eytan Stein, MD, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the analysis of enasidenib in mIDH2-positive MDS patients as abstract 343.*
Study design
MDS patients were allowed to enroll during the dose-escalation and expansion phase of the study, Dr Stein explained.
Patients had to be 18 or older and have an advanced hematologic malignancy with mutated IDH2—relapsed or refractory AML, relapsed or refractory MDS, untreated AML, or other hematologic malignancy with mIDH2.
MDS patients could not be candidates for other therapies, had to be IPSS-R high risk, and had to have relapsed or refractory RAEB-1/RAEB-2 disease.
Investigators also performed co-molecular profiling using next-generation sequencing with a FoundationOne® Heme Panel.
All patients received daily oral enasidenib at 100 mg daily in 28-day cycles.
Patient characteristics
The study accrued a total of 239 patients—176 with relapsed or refractory AML, 37 with untreated AML, 9 with another hematologic malignancy, and 17 with MDS.
The median age of the MDS patients was 67 (range, 45-78), and 71% were male. All had the IDH2 mutation, 88% had R140 mutations, and 12% had R172.
Thirteen patients (76%) had an ECOG performance status of 0-1, and 4 (24%) had a performance status of 2.
A little more than a third (35%) of patients had 2 or more prior anti-cancer regimens.
Two patients (12%) received prior lenalidomide therapy, 8 (47%) received other treatments, including sorafenib (n=2) and vosaroxin, epoetin alfa, pracinostat, cytarabine plus clofarabine, ruxolitinib, and rigosertib (n=1 each). Four patients (24%) were untreated.
“I want to make the point,” Dr Stein said, “that, of those patients, three quarters of them, 76% [n=13], had received a prior hypomethylating agent, really understanding that this is a very poor-risk group of patients that we are studying here.”
About half of patients (47%) had intermediate-2/high IPSS risk status, good MDS cytogenetic risk, and high/very high IPSS-R risk status.
Adverse events
Grade 3-4 treatment-emergent adverse events (AEs) occurring in 2 or more patients were hyperbilirubinemia (n=5), pneumonia (n=4), thrombocytopenia (n=4), anemia (n=3), hypokalemia (n=3), dyspnea (n=2), and tumor lysis syndrome (n=2).
“As I’ve said in a number of meetings where I’ve talked about IDH2 inhibitors, and specifically enasidenib, the hyperbilirubinemia that is seen with this drug is an indirect hyperbilirubinemia,” Dr Stein said.
“A known off-target effect of this drug is inhibition of the UGT1a1 enzyme, which conjugates bilirubin, and this indirect hyperbilirubinemia, which is typically relatively mild [and] does not appear to have any clinical sequelae.”
Investigators considered 9 of the AEs reported for 6 patients to be drug-related.
Four serious enasidenib-related AEs included tumor lysis syndrome (n=2), increased blood bilirubin (n=1), and transaminitis (n=1).
There were no treatment-related deaths.
Response and survival
Ten of 17 patients (59%) achieved an overall response, defined as complete response (CR) plus partial response, plus marrow CR, plus hematologic improvement (HI).
One patient achieved CR, 1 had a partial response, 3 had marrow CR, and 5 had HI.
Dr Stein noted that, of the 13 patients who had received prior hypomethylating agent therapy, 7 (54%) had a response with enasidenib.
Of the patients who attained HI, 2 had trilineage and 2 had bilineage improvement.
The median time to response was 21 days (range, 10-87), and the median number of treatment cycles was 3.0.
Patients remained on study for up to 24 months. Four patients in hematologic remission are still on study, and 3 patients proceeded to stem cell or bone marrow transplant, Dr Stein said.
Two patients discontinued because of investigator decision, and 8 discontinued therapy due to death or disease progression.
The limitation of the study regarding overall survival, Dr Stein said, is that the number is very small.
At a median follow-up of 7.5 months, the median overall survival was not reached.
“So I’m not arguing that this is the end word of this,” he said. “This is going to be dynamic as more patients come on these studies. But I think it’s a nice indication that this treatment appears to be well-tolerated and doing good for a large subset of patients.”
Co-occurring mutations
The investigators also analyzed co-occurring mutations in 13 MDS patients.
The small patient population prevented the investigators from making definitive conclusions regarding potential correlations between response and co-mutations.
Nevertheless, Dr Stein said the analysis revealed something “very, very intriguing.”
He noted that 7 patients had ASXL1 mutations, and “those are typically patients who are bad actors.”
“Five of those 7 patients had a response,” Dr Stein said. “And of those 5, 3 of them had received a prior hypomethylating agent. I think it’s at least food for thought that you can salvage a patient who has failed a hypomethylating agent, with poor risk disease. I think this is very, very exciting.”
Celgene Corporation and its collaborator, Agios Pharmaceuticals, sponsored the trial. ![]()
*Information in the abstract differs from the presentation.
Photo courtesy of ASH
SAN DIEGO—Daily treatment with enasidenib monotherapy in patients with mutated IDH2-positive myelodysplastic syndromes (MDS) induced responses in the majority of patients treated, according to a presentation at the 2016 ASH Annual Meeting.
The study was a portion of a larger phase 1/2 trial of the agent in patients with acute myeloid leukemia (AML) and other hematologic malignancies, so the subset was relatively small, numbering 17 patients.
Nevertheless, enasidenib was well tolerated and induced responses in these predominantly higher-risk patients.
Enasidenib (AG-221/CC-9007) is a selective, oral, potent inhibitor of mutant IDH2 (mIDH2), which produces 2-HG and thus alters DNA methylation and blocks cellular differentiation of hematopoietic progenitor cells.
Approximately 15% of AML patients and 5% of MDS patients have mIDH2. So investigators undertook the study to evaluate the safety and efficacy of enasidenib monotherapy in these diseases.
Eytan Stein, MD, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the analysis of enasidenib in mIDH2-positive MDS patients as abstract 343.*
Study design
MDS patients were allowed to enroll during the dose-escalation and expansion phase of the study, Dr Stein explained.
Patients had to be 18 or older and have an advanced hematologic malignancy with mutated IDH2—relapsed or refractory AML, relapsed or refractory MDS, untreated AML, or other hematologic malignancy with mIDH2.
MDS patients could not be candidates for other therapies, had to be IPSS-R high risk, and had to have relapsed or refractory RAEB-1/RAEB-2 disease.
Investigators also performed co-molecular profiling using next-generation sequencing with a FoundationOne® Heme Panel.
All patients received daily oral enasidenib at 100 mg daily in 28-day cycles.
Patient characteristics
The study accrued a total of 239 patients—176 with relapsed or refractory AML, 37 with untreated AML, 9 with another hematologic malignancy, and 17 with MDS.
The median age of the MDS patients was 67 (range, 45-78), and 71% were male. All had the IDH2 mutation, 88% had R140 mutations, and 12% had R172.
Thirteen patients (76%) had an ECOG performance status of 0-1, and 4 (24%) had a performance status of 2.
A little more than a third (35%) of patients had 2 or more prior anti-cancer regimens.
Two patients (12%) received prior lenalidomide therapy, 8 (47%) received other treatments, including sorafenib (n=2) and vosaroxin, epoetin alfa, pracinostat, cytarabine plus clofarabine, ruxolitinib, and rigosertib (n=1 each). Four patients (24%) were untreated.
“I want to make the point,” Dr Stein said, “that, of those patients, three quarters of them, 76% [n=13], had received a prior hypomethylating agent, really understanding that this is a very poor-risk group of patients that we are studying here.”
About half of patients (47%) had intermediate-2/high IPSS risk status, good MDS cytogenetic risk, and high/very high IPSS-R risk status.
Adverse events
Grade 3-4 treatment-emergent adverse events (AEs) occurring in 2 or more patients were hyperbilirubinemia (n=5), pneumonia (n=4), thrombocytopenia (n=4), anemia (n=3), hypokalemia (n=3), dyspnea (n=2), and tumor lysis syndrome (n=2).
“As I’ve said in a number of meetings where I’ve talked about IDH2 inhibitors, and specifically enasidenib, the hyperbilirubinemia that is seen with this drug is an indirect hyperbilirubinemia,” Dr Stein said.
“A known off-target effect of this drug is inhibition of the UGT1a1 enzyme, which conjugates bilirubin, and this indirect hyperbilirubinemia, which is typically relatively mild [and] does not appear to have any clinical sequelae.”
Investigators considered 9 of the AEs reported for 6 patients to be drug-related.
Four serious enasidenib-related AEs included tumor lysis syndrome (n=2), increased blood bilirubin (n=1), and transaminitis (n=1).
There were no treatment-related deaths.
Response and survival
Ten of 17 patients (59%) achieved an overall response, defined as complete response (CR) plus partial response, plus marrow CR, plus hematologic improvement (HI).
One patient achieved CR, 1 had a partial response, 3 had marrow CR, and 5 had HI.
Dr Stein noted that, of the 13 patients who had received prior hypomethylating agent therapy, 7 (54%) had a response with enasidenib.
Of the patients who attained HI, 2 had trilineage and 2 had bilineage improvement.
The median time to response was 21 days (range, 10-87), and the median number of treatment cycles was 3.0.
Patients remained on study for up to 24 months. Four patients in hematologic remission are still on study, and 3 patients proceeded to stem cell or bone marrow transplant, Dr Stein said.
Two patients discontinued because of investigator decision, and 8 discontinued therapy due to death or disease progression.
The limitation of the study regarding overall survival, Dr Stein said, is that the number is very small.
At a median follow-up of 7.5 months, the median overall survival was not reached.
“So I’m not arguing that this is the end word of this,” he said. “This is going to be dynamic as more patients come on these studies. But I think it’s a nice indication that this treatment appears to be well-tolerated and doing good for a large subset of patients.”
Co-occurring mutations
The investigators also analyzed co-occurring mutations in 13 MDS patients.
The small patient population prevented the investigators from making definitive conclusions regarding potential correlations between response and co-mutations.
Nevertheless, Dr Stein said the analysis revealed something “very, very intriguing.”
He noted that 7 patients had ASXL1 mutations, and “those are typically patients who are bad actors.”
“Five of those 7 patients had a response,” Dr Stein said. “And of those 5, 3 of them had received a prior hypomethylating agent. I think it’s at least food for thought that you can salvage a patient who has failed a hypomethylating agent, with poor risk disease. I think this is very, very exciting.”
Celgene Corporation and its collaborator, Agios Pharmaceuticals, sponsored the trial. ![]()
*Information in the abstract differs from the presentation.
Photo courtesy of ASH
SAN DIEGO—Daily treatment with enasidenib monotherapy in patients with mutated IDH2-positive myelodysplastic syndromes (MDS) induced responses in the majority of patients treated, according to a presentation at the 2016 ASH Annual Meeting.
The study was a portion of a larger phase 1/2 trial of the agent in patients with acute myeloid leukemia (AML) and other hematologic malignancies, so the subset was relatively small, numbering 17 patients.
Nevertheless, enasidenib was well tolerated and induced responses in these predominantly higher-risk patients.
Enasidenib (AG-221/CC-9007) is a selective, oral, potent inhibitor of mutant IDH2 (mIDH2), which produces 2-HG and thus alters DNA methylation and blocks cellular differentiation of hematopoietic progenitor cells.
Approximately 15% of AML patients and 5% of MDS patients have mIDH2. So investigators undertook the study to evaluate the safety and efficacy of enasidenib monotherapy in these diseases.
Eytan Stein, MD, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the analysis of enasidenib in mIDH2-positive MDS patients as abstract 343.*
Study design
MDS patients were allowed to enroll during the dose-escalation and expansion phase of the study, Dr Stein explained.
Patients had to be 18 or older and have an advanced hematologic malignancy with mutated IDH2—relapsed or refractory AML, relapsed or refractory MDS, untreated AML, or other hematologic malignancy with mIDH2.
MDS patients could not be candidates for other therapies, had to be IPSS-R high risk, and had to have relapsed or refractory RAEB-1/RAEB-2 disease.
Investigators also performed co-molecular profiling using next-generation sequencing with a FoundationOne® Heme Panel.
All patients received daily oral enasidenib at 100 mg daily in 28-day cycles.
Patient characteristics
The study accrued a total of 239 patients—176 with relapsed or refractory AML, 37 with untreated AML, 9 with another hematologic malignancy, and 17 with MDS.
The median age of the MDS patients was 67 (range, 45-78), and 71% were male. All had the IDH2 mutation, 88% had R140 mutations, and 12% had R172.
Thirteen patients (76%) had an ECOG performance status of 0-1, and 4 (24%) had a performance status of 2.
A little more than a third (35%) of patients had 2 or more prior anti-cancer regimens.
Two patients (12%) received prior lenalidomide therapy, 8 (47%) received other treatments, including sorafenib (n=2) and vosaroxin, epoetin alfa, pracinostat, cytarabine plus clofarabine, ruxolitinib, and rigosertib (n=1 each). Four patients (24%) were untreated.
“I want to make the point,” Dr Stein said, “that, of those patients, three quarters of them, 76% [n=13], had received a prior hypomethylating agent, really understanding that this is a very poor-risk group of patients that we are studying here.”
About half of patients (47%) had intermediate-2/high IPSS risk status, good MDS cytogenetic risk, and high/very high IPSS-R risk status.
Adverse events
Grade 3-4 treatment-emergent adverse events (AEs) occurring in 2 or more patients were hyperbilirubinemia (n=5), pneumonia (n=4), thrombocytopenia (n=4), anemia (n=3), hypokalemia (n=3), dyspnea (n=2), and tumor lysis syndrome (n=2).
“As I’ve said in a number of meetings where I’ve talked about IDH2 inhibitors, and specifically enasidenib, the hyperbilirubinemia that is seen with this drug is an indirect hyperbilirubinemia,” Dr Stein said.
“A known off-target effect of this drug is inhibition of the UGT1a1 enzyme, which conjugates bilirubin, and this indirect hyperbilirubinemia, which is typically relatively mild [and] does not appear to have any clinical sequelae.”
Investigators considered 9 of the AEs reported for 6 patients to be drug-related.
Four serious enasidenib-related AEs included tumor lysis syndrome (n=2), increased blood bilirubin (n=1), and transaminitis (n=1).
There were no treatment-related deaths.
Response and survival
Ten of 17 patients (59%) achieved an overall response, defined as complete response (CR) plus partial response, plus marrow CR, plus hematologic improvement (HI).
One patient achieved CR, 1 had a partial response, 3 had marrow CR, and 5 had HI.
Dr Stein noted that, of the 13 patients who had received prior hypomethylating agent therapy, 7 (54%) had a response with enasidenib.
Of the patients who attained HI, 2 had trilineage and 2 had bilineage improvement.
The median time to response was 21 days (range, 10-87), and the median number of treatment cycles was 3.0.
Patients remained on study for up to 24 months. Four patients in hematologic remission are still on study, and 3 patients proceeded to stem cell or bone marrow transplant, Dr Stein said.
Two patients discontinued because of investigator decision, and 8 discontinued therapy due to death or disease progression.
The limitation of the study regarding overall survival, Dr Stein said, is that the number is very small.
At a median follow-up of 7.5 months, the median overall survival was not reached.
“So I’m not arguing that this is the end word of this,” he said. “This is going to be dynamic as more patients come on these studies. But I think it’s a nice indication that this treatment appears to be well-tolerated and doing good for a large subset of patients.”
Co-occurring mutations
The investigators also analyzed co-occurring mutations in 13 MDS patients.
The small patient population prevented the investigators from making definitive conclusions regarding potential correlations between response and co-mutations.
Nevertheless, Dr Stein said the analysis revealed something “very, very intriguing.”
He noted that 7 patients had ASXL1 mutations, and “those are typically patients who are bad actors.”
“Five of those 7 patients had a response,” Dr Stein said. “And of those 5, 3 of them had received a prior hypomethylating agent. I think it’s at least food for thought that you can salvage a patient who has failed a hypomethylating agent, with poor risk disease. I think this is very, very exciting.”
Celgene Corporation and its collaborator, Agios Pharmaceuticals, sponsored the trial. ![]()
*Information in the abstract differs from the presentation.