User login
Curb AF recurrences through risk factor modification
SNOWMASS, COLO. – Overlooking the common modifiable risk factors in patients with atrial fibrillation is missing out on an excellent opportunity to help curb the growing global pandemic of the arrhythmia, Patrick T. O’Gara, MD, said at the Annual Cardiovascular Conference at Snowmass.
“My purpose here is a wake up call to improve screening for and treatment of modifiable risk factors in patients with atrial fibrillation,” declared Dr. O’Gara, professor of medicine at Harvard Medical School, Boston.

Overweight/obesity: Investigators at the University of Adelaide (Australia) demonstrated in the LEGACY trial that patients with atrial fibrillation (AF) and a BMI of 27 kg/m2 or more reduced their AF symptom burden in a dose-response fashion as they shed excess pounds as part of an intensive weight management program. Those who shed at least 10% of their baseline body weight had a 46% rate of 5-year freedom from AF without resort to rhythm control medications or ablation procedures of 46%. With 3%-9% weight loss, the rate was 22%. And with 3% weight loss, it was 13%.
The best results came from sustained linear weight loss. Weight fluctuations of greater than 5% – the classic yoyo dieting pattern – partially offset the overall benefit of weight loss with respect to recurrent AF (J Am Coll Cardiol. 2015 May 26;65(20):2159-69).
In a separate study, the same team of Australian investigators offered an opportunity to participate in a risk factor management program to patients with AF and a BMI of 27 kg/m2 or more who were undergoing radiofrequency ablation for their arrhythmia. Participants had significantly fewer repeat ablation procedures during followup and were also less likely to be on antiarrhythmic drugs than the patients who opted for usual care (J Am Coll Cardiol. 2014 Dec 2;64(21):2222-31).
Alcohol: The ‘holiday heart’ syndrome is well known, but alcohol consumption beyond binging can increase risk for AF. Dr. O’Gara noted that in a recent review article entitled “Alcohol and Atrial Fibrillation: A Sobering Review,” investigators at the University of Melbourne showed that while the relationship between the number of standard drinks per week and risk of cardiovascular mortality is J-shaped, with a nadir at 14-21 drinks per week in men and fewer in women, the risk of developing AF is linear over time and appears to increase incrementally with every additional drink per week (J Am Coll Cardiol. 2016 Dec 13;68(23):2567-76).
Also, a prospective study of nearly 80,000 Swedes free from AF at baseline, coupled with a meta-analysis of seven prospective studies found that for each additional drink per day consumed the risk of developing AF rose over time by roughly a further 10% compared to that of teetotalers (J Am Coll Cardiol. 2014; Jul 22;64(3):281-9).
Physical inactivity: In the prospective Tromso Study, in which more than 20,000 Norwegian adults were followed for 20 years, leisure time physical activity displayed a J-shaped relationship with the risk of developing AF. Moderately active subjects were an adjusted 19% less likely to develop AF than those with low physical activity, while the risk in subjects who regularly engaged in vigorous physical activity was 37% higher than in the low-activity group (Eur Heart J. 2016 Aug 1;37(29):2307-13).
“This effect of moderate exercise might be due to the associated weight loss, improved endothelial function, better sleep, perhaps a better balance between the sympathetic and parasympathetic nervous systems,” Dr. O’Gara observed.
How much physical activity is right for patients with AF? Dr. O’Gara said one of the best reviews he’s seen came from the University of Adelaide group (Circulation. 2016 Feb 2;133(5):457-9). They recommended a total of 120-200 minutes of exercise per week spread over three to five sessions. While the research base is strongest for moderate-intensity exercise, the Australians also noted the effectiveness and safety of a novel program of repeated 4-minute intervals of high-intensity exercise at 85%-95% of peak heart rate as demonstrated in a randomized controlled trial by investigators at the Norwegian University of Science and Technology in Trondheim. They showed this approach resulted in reduced time in AF and decreased AF symptoms coupled with improved quality of life and left atrial and ventricular function (Circulation. 2016 Feb 2;133(5):466-73).
“I think you could look at this review and feel very confident that there is some evidence base to substantiate your strong recommendation that patients actively engage in exercise as treatment for their atrial fibrillation,” the cardiologist said.
Sleep apnea: Investigators at Brigham and Women’s Hospital in Boston have demonstrated that effective treatment of sleep apnea with continuous positive airway pressure in patients with atrial fibrillation is associated with smaller atrial size and ventricular mass, lower blood pressure, and a significantly reduced risk of recurrent AF following an AF ablation procedure (J Am Heart Assoc. 2013 Nov 25;2(6):e000421).
“Sleep hygiene is one of the least attended aspects of cardiovascular health,” according to Dr. O’Gara. “We need to ask the partner or spouse, ‘How well does your partner sleep? Do you hear thrashing about, snoring, gagging, or notice restless legs?’ Heart failure folks are really tuned into this, but in the practice of seeing patients come into the emergency room with new-onset atrial fibrillation, it’s tenth on the list of five questions one would ask.”
Dr. O’Gara reported having no financial conflicts.
SNOWMASS, COLO. – Overlooking the common modifiable risk factors in patients with atrial fibrillation is missing out on an excellent opportunity to help curb the growing global pandemic of the arrhythmia, Patrick T. O’Gara, MD, said at the Annual Cardiovascular Conference at Snowmass.
“My purpose here is a wake up call to improve screening for and treatment of modifiable risk factors in patients with atrial fibrillation,” declared Dr. O’Gara, professor of medicine at Harvard Medical School, Boston.
Overweight/obesity: Investigators at the University of Adelaide (Australia) demonstrated in the LEGACY trial that patients with atrial fibrillation (AF) and a BMI of 27 kg/m2 or more reduced their AF symptom burden in a dose-response fashion as they shed excess pounds as part of an intensive weight management program. Those who shed at least 10% of their baseline body weight had a 46% rate of 5-year freedom from AF without resort to rhythm control medications or ablation procedures of 46%. With 3%-9% weight loss, the rate was 22%. And with 3% weight loss, it was 13%.
The best results came from sustained linear weight loss. Weight fluctuations of greater than 5% – the classic yoyo dieting pattern – partially offset the overall benefit of weight loss with respect to recurrent AF (J Am Coll Cardiol. 2015 May 26;65(20):2159-69).
In a separate study, the same team of Australian investigators offered an opportunity to participate in a risk factor management program to patients with AF and a BMI of 27 kg/m2 or more who were undergoing radiofrequency ablation for their arrhythmia. Participants had significantly fewer repeat ablation procedures during followup and were also less likely to be on antiarrhythmic drugs than the patients who opted for usual care (J Am Coll Cardiol. 2014 Dec 2;64(21):2222-31).
Alcohol: The ‘holiday heart’ syndrome is well known, but alcohol consumption beyond binging can increase risk for AF. Dr. O’Gara noted that in a recent review article entitled “Alcohol and Atrial Fibrillation: A Sobering Review,” investigators at the University of Melbourne showed that while the relationship between the number of standard drinks per week and risk of cardiovascular mortality is J-shaped, with a nadir at 14-21 drinks per week in men and fewer in women, the risk of developing AF is linear over time and appears to increase incrementally with every additional drink per week (J Am Coll Cardiol. 2016 Dec 13;68(23):2567-76).
Also, a prospective study of nearly 80,000 Swedes free from AF at baseline, coupled with a meta-analysis of seven prospective studies found that for each additional drink per day consumed the risk of developing AF rose over time by roughly a further 10% compared to that of teetotalers (J Am Coll Cardiol. 2014; Jul 22;64(3):281-9).
Physical inactivity: In the prospective Tromso Study, in which more than 20,000 Norwegian adults were followed for 20 years, leisure time physical activity displayed a J-shaped relationship with the risk of developing AF. Moderately active subjects were an adjusted 19% less likely to develop AF than those with low physical activity, while the risk in subjects who regularly engaged in vigorous physical activity was 37% higher than in the low-activity group (Eur Heart J. 2016 Aug 1;37(29):2307-13).
“This effect of moderate exercise might be due to the associated weight loss, improved endothelial function, better sleep, perhaps a better balance between the sympathetic and parasympathetic nervous systems,” Dr. O’Gara observed.
How much physical activity is right for patients with AF? Dr. O’Gara said one of the best reviews he’s seen came from the University of Adelaide group (Circulation. 2016 Feb 2;133(5):457-9). They recommended a total of 120-200 minutes of exercise per week spread over three to five sessions. While the research base is strongest for moderate-intensity exercise, the Australians also noted the effectiveness and safety of a novel program of repeated 4-minute intervals of high-intensity exercise at 85%-95% of peak heart rate as demonstrated in a randomized controlled trial by investigators at the Norwegian University of Science and Technology in Trondheim. They showed this approach resulted in reduced time in AF and decreased AF symptoms coupled with improved quality of life and left atrial and ventricular function (Circulation. 2016 Feb 2;133(5):466-73).
“I think you could look at this review and feel very confident that there is some evidence base to substantiate your strong recommendation that patients actively engage in exercise as treatment for their atrial fibrillation,” the cardiologist said.
Sleep apnea: Investigators at Brigham and Women’s Hospital in Boston have demonstrated that effective treatment of sleep apnea with continuous positive airway pressure in patients with atrial fibrillation is associated with smaller atrial size and ventricular mass, lower blood pressure, and a significantly reduced risk of recurrent AF following an AF ablation procedure (J Am Heart Assoc. 2013 Nov 25;2(6):e000421).
“Sleep hygiene is one of the least attended aspects of cardiovascular health,” according to Dr. O’Gara. “We need to ask the partner or spouse, ‘How well does your partner sleep? Do you hear thrashing about, snoring, gagging, or notice restless legs?’ Heart failure folks are really tuned into this, but in the practice of seeing patients come into the emergency room with new-onset atrial fibrillation, it’s tenth on the list of five questions one would ask.”
Dr. O’Gara reported having no financial conflicts.
SNOWMASS, COLO. – Overlooking the common modifiable risk factors in patients with atrial fibrillation is missing out on an excellent opportunity to help curb the growing global pandemic of the arrhythmia, Patrick T. O’Gara, MD, said at the Annual Cardiovascular Conference at Snowmass.
“My purpose here is a wake up call to improve screening for and treatment of modifiable risk factors in patients with atrial fibrillation,” declared Dr. O’Gara, professor of medicine at Harvard Medical School, Boston.
Overweight/obesity: Investigators at the University of Adelaide (Australia) demonstrated in the LEGACY trial that patients with atrial fibrillation (AF) and a BMI of 27 kg/m2 or more reduced their AF symptom burden in a dose-response fashion as they shed excess pounds as part of an intensive weight management program. Those who shed at least 10% of their baseline body weight had a 46% rate of 5-year freedom from AF without resort to rhythm control medications or ablation procedures of 46%. With 3%-9% weight loss, the rate was 22%. And with 3% weight loss, it was 13%.
The best results came from sustained linear weight loss. Weight fluctuations of greater than 5% – the classic yoyo dieting pattern – partially offset the overall benefit of weight loss with respect to recurrent AF (J Am Coll Cardiol. 2015 May 26;65(20):2159-69).
In a separate study, the same team of Australian investigators offered an opportunity to participate in a risk factor management program to patients with AF and a BMI of 27 kg/m2 or more who were undergoing radiofrequency ablation for their arrhythmia. Participants had significantly fewer repeat ablation procedures during followup and were also less likely to be on antiarrhythmic drugs than the patients who opted for usual care (J Am Coll Cardiol. 2014 Dec 2;64(21):2222-31).
Alcohol: The ‘holiday heart’ syndrome is well known, but alcohol consumption beyond binging can increase risk for AF. Dr. O’Gara noted that in a recent review article entitled “Alcohol and Atrial Fibrillation: A Sobering Review,” investigators at the University of Melbourne showed that while the relationship between the number of standard drinks per week and risk of cardiovascular mortality is J-shaped, with a nadir at 14-21 drinks per week in men and fewer in women, the risk of developing AF is linear over time and appears to increase incrementally with every additional drink per week (J Am Coll Cardiol. 2016 Dec 13;68(23):2567-76).
Also, a prospective study of nearly 80,000 Swedes free from AF at baseline, coupled with a meta-analysis of seven prospective studies found that for each additional drink per day consumed the risk of developing AF rose over time by roughly a further 10% compared to that of teetotalers (J Am Coll Cardiol. 2014; Jul 22;64(3):281-9).
Physical inactivity: In the prospective Tromso Study, in which more than 20,000 Norwegian adults were followed for 20 years, leisure time physical activity displayed a J-shaped relationship with the risk of developing AF. Moderately active subjects were an adjusted 19% less likely to develop AF than those with low physical activity, while the risk in subjects who regularly engaged in vigorous physical activity was 37% higher than in the low-activity group (Eur Heart J. 2016 Aug 1;37(29):2307-13).
“This effect of moderate exercise might be due to the associated weight loss, improved endothelial function, better sleep, perhaps a better balance between the sympathetic and parasympathetic nervous systems,” Dr. O’Gara observed.
How much physical activity is right for patients with AF? Dr. O’Gara said one of the best reviews he’s seen came from the University of Adelaide group (Circulation. 2016 Feb 2;133(5):457-9). They recommended a total of 120-200 minutes of exercise per week spread over three to five sessions. While the research base is strongest for moderate-intensity exercise, the Australians also noted the effectiveness and safety of a novel program of repeated 4-minute intervals of high-intensity exercise at 85%-95% of peak heart rate as demonstrated in a randomized controlled trial by investigators at the Norwegian University of Science and Technology in Trondheim. They showed this approach resulted in reduced time in AF and decreased AF symptoms coupled with improved quality of life and left atrial and ventricular function (Circulation. 2016 Feb 2;133(5):466-73).
“I think you could look at this review and feel very confident that there is some evidence base to substantiate your strong recommendation that patients actively engage in exercise as treatment for their atrial fibrillation,” the cardiologist said.
Sleep apnea: Investigators at Brigham and Women’s Hospital in Boston have demonstrated that effective treatment of sleep apnea with continuous positive airway pressure in patients with atrial fibrillation is associated with smaller atrial size and ventricular mass, lower blood pressure, and a significantly reduced risk of recurrent AF following an AF ablation procedure (J Am Heart Assoc. 2013 Nov 25;2(6):e000421).
“Sleep hygiene is one of the least attended aspects of cardiovascular health,” according to Dr. O’Gara. “We need to ask the partner or spouse, ‘How well does your partner sleep? Do you hear thrashing about, snoring, gagging, or notice restless legs?’ Heart failure folks are really tuned into this, but in the practice of seeing patients come into the emergency room with new-onset atrial fibrillation, it’s tenth on the list of five questions one would ask.”
Dr. O’Gara reported having no financial conflicts.
EXPERT ANALYSIS FROM THE CARDIOVASCULAR CONFERENCE AT SNOWMASS
Cardiac procedures tied to higher cancer risk in adult congenital heart disease
NEW ORLEANS – The greater the lifetime number of cardiac procedures entailing exposure to low-dose ionizing radiation, the higher the associated cancer risk in adults with congenital heart disease, Sarah Cohen, MD, reported at the American Heart Association scientific sessions.
This finding takes on added clinical import because the use of such procedures has increased markedly in patients with adult congenital heart disease, added Dr. Cohen of McGill University in Montreal.
She presented analyses of nearly 25,000 patients in the Quebec congenital heart disease database who were ages 18-64 with no history of cancer in January 1995; 602 of them were diagnosed with cancer during follow-up through 2009. Each was matched to four cancer-free controls in the database on the basis of age, sex, congenital heart disease severity, and calendar year.
Patients with a lifetime total of four or five cardiac procedures involving exposure to low-dose ionizing radiation were 1.7-fold more likely to be diagnosed with cancer during follow-up than were those with a history of no such procedures or just one. With a lifetime history of six or more of the procedures, the relative risk climbed to 2.2-fold.
Nearly 90% of the malignancies were genitourinary, respiratory, digestive, breast, or hematologic cancers.
Dr. Cohen and her coinvestigators also conducted a cohort study comparing the relative risk of developing cancer in 1,781 high-exposure Quebec adults in the database who had a lifetime history of four or more cardiac procedures involving radiation exposure to more than 20,000 others with a history of not more than one such procedure. The high-exposure group had a 3.3-fold greater rate of malignancy during follow-up.
In a separate study, Dr. Cohen’s McGill colleagues analyzed the Quebec database and documented the sharp rise over time in low-dose ionizing radiation cardiac procedures, including catheter-based diagnostic procedures, structural heart interventions, nuclear procedures, coronary interventions, computed tomography scans of the chest, coronary interventions, and insertion or repair of pacemakers and implantable cardioverter-defibrillators. In 1990, the rate was 18.5 such procedures per 1,000 patients per year. By 2005, it had nearly tripled to 51.9 per 1,000 per year. The age at first procedure dropped from 5 years to 9 months (Circulation 2016 Jan 5;133[1]:12-20).
Dr. Cohen reported having no financial disclosures.
NEW ORLEANS – The greater the lifetime number of cardiac procedures entailing exposure to low-dose ionizing radiation, the higher the associated cancer risk in adults with congenital heart disease, Sarah Cohen, MD, reported at the American Heart Association scientific sessions.
This finding takes on added clinical import because the use of such procedures has increased markedly in patients with adult congenital heart disease, added Dr. Cohen of McGill University in Montreal.
She presented analyses of nearly 25,000 patients in the Quebec congenital heart disease database who were ages 18-64 with no history of cancer in January 1995; 602 of them were diagnosed with cancer during follow-up through 2009. Each was matched to four cancer-free controls in the database on the basis of age, sex, congenital heart disease severity, and calendar year.
Patients with a lifetime total of four or five cardiac procedures involving exposure to low-dose ionizing radiation were 1.7-fold more likely to be diagnosed with cancer during follow-up than were those with a history of no such procedures or just one. With a lifetime history of six or more of the procedures, the relative risk climbed to 2.2-fold.
Nearly 90% of the malignancies were genitourinary, respiratory, digestive, breast, or hematologic cancers.
Dr. Cohen and her coinvestigators also conducted a cohort study comparing the relative risk of developing cancer in 1,781 high-exposure Quebec adults in the database who had a lifetime history of four or more cardiac procedures involving radiation exposure to more than 20,000 others with a history of not more than one such procedure. The high-exposure group had a 3.3-fold greater rate of malignancy during follow-up.
In a separate study, Dr. Cohen’s McGill colleagues analyzed the Quebec database and documented the sharp rise over time in low-dose ionizing radiation cardiac procedures, including catheter-based diagnostic procedures, structural heart interventions, nuclear procedures, coronary interventions, computed tomography scans of the chest, coronary interventions, and insertion or repair of pacemakers and implantable cardioverter-defibrillators. In 1990, the rate was 18.5 such procedures per 1,000 patients per year. By 2005, it had nearly tripled to 51.9 per 1,000 per year. The age at first procedure dropped from 5 years to 9 months (Circulation 2016 Jan 5;133[1]:12-20).
Dr. Cohen reported having no financial disclosures.
NEW ORLEANS – The greater the lifetime number of cardiac procedures entailing exposure to low-dose ionizing radiation, the higher the associated cancer risk in adults with congenital heart disease, Sarah Cohen, MD, reported at the American Heart Association scientific sessions.
This finding takes on added clinical import because the use of such procedures has increased markedly in patients with adult congenital heart disease, added Dr. Cohen of McGill University in Montreal.
She presented analyses of nearly 25,000 patients in the Quebec congenital heart disease database who were ages 18-64 with no history of cancer in January 1995; 602 of them were diagnosed with cancer during follow-up through 2009. Each was matched to four cancer-free controls in the database on the basis of age, sex, congenital heart disease severity, and calendar year.
Patients with a lifetime total of four or five cardiac procedures involving exposure to low-dose ionizing radiation were 1.7-fold more likely to be diagnosed with cancer during follow-up than were those with a history of no such procedures or just one. With a lifetime history of six or more of the procedures, the relative risk climbed to 2.2-fold.
Nearly 90% of the malignancies were genitourinary, respiratory, digestive, breast, or hematologic cancers.
Dr. Cohen and her coinvestigators also conducted a cohort study comparing the relative risk of developing cancer in 1,781 high-exposure Quebec adults in the database who had a lifetime history of four or more cardiac procedures involving radiation exposure to more than 20,000 others with a history of not more than one such procedure. The high-exposure group had a 3.3-fold greater rate of malignancy during follow-up.
In a separate study, Dr. Cohen’s McGill colleagues analyzed the Quebec database and documented the sharp rise over time in low-dose ionizing radiation cardiac procedures, including catheter-based diagnostic procedures, structural heart interventions, nuclear procedures, coronary interventions, computed tomography scans of the chest, coronary interventions, and insertion or repair of pacemakers and implantable cardioverter-defibrillators. In 1990, the rate was 18.5 such procedures per 1,000 patients per year. By 2005, it had nearly tripled to 51.9 per 1,000 per year. The age at first procedure dropped from 5 years to 9 months (Circulation 2016 Jan 5;133[1]:12-20).
Dr. Cohen reported having no financial disclosures.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point:
Major finding: Adults with congenital heart disease who had a lifetime history of six or more cardiac procedures involving exposure to low-dose ionizing radiation were 2.2-fold more likely to develop cancer during follow-up than were those with not more than one such procedure.
Data source: This nested, case-control study included 602 adults with congenital heart disease who developed cancer, each matched to four controls who did not.
Disclosures: The study presenter reported having no financial disclosures.
Nivolumab safe, effective for salvage in advanced gastric cancer
SAN FRANCISCO – The human monoclonal IgG4 antibody nivolumab was safe and effective as a salvage treatment for patients in a randomized phase III trial who failed standard chemotherapy for advanced gastric or gastroesophageal junction cancer.
Nivolumab, which blocks the human programmed cell death-1 receptor, was superior to placebo with respect to overall survival, progression-free survival, and overall response rate, Yoon-Koo Kang, MD, reported at the symposium sponsored by ASCO, ASTRO, the American Gastroenterological Association, and the Society of Surgical Oncology.
For the study (ONO-4538), 493 patients with ECOG performance status of 0-1 and unresectable advanced or recurrent advanced gastric or gastroesophageal cancer who failed 2 or more prior chemotherapy regimens were randomized to receive placebo or nivolumab in a 2:1 ratio. Nivolumab treatment was associated with significantly improved median overall survival at the time of data cut-off 5.6 months after the last patient was randomized (5.32 months vs. 4.14 months with placebo; hazard ratio, 0.63), said Dr. Kang of Asan Medical Center, University of Ulsan, Seoul, Korea.
The overall survival rate at 12 months was 26.6% vs. 10.9%, respectively, he said.
The overall response rate was 11.2% vs. 0%, and median progression-free survival was 1.61 months vs. 1.45 months (hazard ratio, 0.60) with nivolumab vs. placebo, respectively.
“Median duration of response was 9.53 months,” he said.
Study subjects were adults aged 20 years or older who were enrolled from 49 centers in Japan, Korea, and Taiwan. The 330 patients in the nivolumab group received 3 mg/kg every 2 weeks until toxicity became unacceptable or disease progressed. Treatment-related adverse events occurred in 42.7% vs. 26.7% of nivolumab and placebo group patients, respectively, and grade 3 or greater events occurred in 11.5% vs. 5.5%, respectively.
Grade 3 or 4 events reported in more than 2% of patients were diarrhea, fatigue, decreased appetite, pyrexia, and increased AST and ALT in the treatment group, and fatigue and decreased appetite in the placebo group. The rates of discontinuation of active treatment due to drug-related adverse events were similar at 2.7% and 2.5% in the groups, respectively, he said, noting that the safety profile was consistent with findings in prior studies involving patients with solid tumors.
The findings demonstrate a clinical benefit with nivolumab in pretreated advanced or recurrent gastric cancer, and establish a strong basis for conducting additional studies of the drug in such patients, he concluded.
ONO-4538 was funded by Ono Pharmaceutical Co., Ltd. and Bristol-Myers Squibb. Dr. Kang reported a consulting or advisory role with Lilly/ImClone, Novartis, Ono Pharmaceutical, Roche/Genentech, and Taiho Pharmaceutical, and research funding from Bayer, Novartis, and Roche/Genentech.
SAN FRANCISCO – The human monoclonal IgG4 antibody nivolumab was safe and effective as a salvage treatment for patients in a randomized phase III trial who failed standard chemotherapy for advanced gastric or gastroesophageal junction cancer.
Nivolumab, which blocks the human programmed cell death-1 receptor, was superior to placebo with respect to overall survival, progression-free survival, and overall response rate, Yoon-Koo Kang, MD, reported at the symposium sponsored by ASCO, ASTRO, the American Gastroenterological Association, and the Society of Surgical Oncology.
For the study (ONO-4538), 493 patients with ECOG performance status of 0-1 and unresectable advanced or recurrent advanced gastric or gastroesophageal cancer who failed 2 or more prior chemotherapy regimens were randomized to receive placebo or nivolumab in a 2:1 ratio. Nivolumab treatment was associated with significantly improved median overall survival at the time of data cut-off 5.6 months after the last patient was randomized (5.32 months vs. 4.14 months with placebo; hazard ratio, 0.63), said Dr. Kang of Asan Medical Center, University of Ulsan, Seoul, Korea.
The overall survival rate at 12 months was 26.6% vs. 10.9%, respectively, he said.
The overall response rate was 11.2% vs. 0%, and median progression-free survival was 1.61 months vs. 1.45 months (hazard ratio, 0.60) with nivolumab vs. placebo, respectively.
“Median duration of response was 9.53 months,” he said.
Study subjects were adults aged 20 years or older who were enrolled from 49 centers in Japan, Korea, and Taiwan. The 330 patients in the nivolumab group received 3 mg/kg every 2 weeks until toxicity became unacceptable or disease progressed. Treatment-related adverse events occurred in 42.7% vs. 26.7% of nivolumab and placebo group patients, respectively, and grade 3 or greater events occurred in 11.5% vs. 5.5%, respectively.
Grade 3 or 4 events reported in more than 2% of patients were diarrhea, fatigue, decreased appetite, pyrexia, and increased AST and ALT in the treatment group, and fatigue and decreased appetite in the placebo group. The rates of discontinuation of active treatment due to drug-related adverse events were similar at 2.7% and 2.5% in the groups, respectively, he said, noting that the safety profile was consistent with findings in prior studies involving patients with solid tumors.
The findings demonstrate a clinical benefit with nivolumab in pretreated advanced or recurrent gastric cancer, and establish a strong basis for conducting additional studies of the drug in such patients, he concluded.
ONO-4538 was funded by Ono Pharmaceutical Co., Ltd. and Bristol-Myers Squibb. Dr. Kang reported a consulting or advisory role with Lilly/ImClone, Novartis, Ono Pharmaceutical, Roche/Genentech, and Taiho Pharmaceutical, and research funding from Bayer, Novartis, and Roche/Genentech.
SAN FRANCISCO – The human monoclonal IgG4 antibody nivolumab was safe and effective as a salvage treatment for patients in a randomized phase III trial who failed standard chemotherapy for advanced gastric or gastroesophageal junction cancer.
Nivolumab, which blocks the human programmed cell death-1 receptor, was superior to placebo with respect to overall survival, progression-free survival, and overall response rate, Yoon-Koo Kang, MD, reported at the symposium sponsored by ASCO, ASTRO, the American Gastroenterological Association, and the Society of Surgical Oncology.
For the study (ONO-4538), 493 patients with ECOG performance status of 0-1 and unresectable advanced or recurrent advanced gastric or gastroesophageal cancer who failed 2 or more prior chemotherapy regimens were randomized to receive placebo or nivolumab in a 2:1 ratio. Nivolumab treatment was associated with significantly improved median overall survival at the time of data cut-off 5.6 months after the last patient was randomized (5.32 months vs. 4.14 months with placebo; hazard ratio, 0.63), said Dr. Kang of Asan Medical Center, University of Ulsan, Seoul, Korea.
The overall survival rate at 12 months was 26.6% vs. 10.9%, respectively, he said.
The overall response rate was 11.2% vs. 0%, and median progression-free survival was 1.61 months vs. 1.45 months (hazard ratio, 0.60) with nivolumab vs. placebo, respectively.
“Median duration of response was 9.53 months,” he said.
Study subjects were adults aged 20 years or older who were enrolled from 49 centers in Japan, Korea, and Taiwan. The 330 patients in the nivolumab group received 3 mg/kg every 2 weeks until toxicity became unacceptable or disease progressed. Treatment-related adverse events occurred in 42.7% vs. 26.7% of nivolumab and placebo group patients, respectively, and grade 3 or greater events occurred in 11.5% vs. 5.5%, respectively.
Grade 3 or 4 events reported in more than 2% of patients were diarrhea, fatigue, decreased appetite, pyrexia, and increased AST and ALT in the treatment group, and fatigue and decreased appetite in the placebo group. The rates of discontinuation of active treatment due to drug-related adverse events were similar at 2.7% and 2.5% in the groups, respectively, he said, noting that the safety profile was consistent with findings in prior studies involving patients with solid tumors.
The findings demonstrate a clinical benefit with nivolumab in pretreated advanced or recurrent gastric cancer, and establish a strong basis for conducting additional studies of the drug in such patients, he concluded.
ONO-4538 was funded by Ono Pharmaceutical Co., Ltd. and Bristol-Myers Squibb. Dr. Kang reported a consulting or advisory role with Lilly/ImClone, Novartis, Ono Pharmaceutical, Roche/Genentech, and Taiho Pharmaceutical, and research funding from Bayer, Novartis, and Roche/Genentech.
AT THE 2017 GASTROINTESTINAL CANCERS SYMPOSIUM
Key clinical point:
Major finding: The overall survival rate at 12 months was 26.6% vs. 10.9% with nivolumab vs. placebo, respectively.
Data source: The randomized phase III ONO-4538 trial.
Disclosures: ONO-4538 was funded by Ono Pharmaceutical Co., Ltd. and Bristol-Myers Squibb. Dr. Kang reported a consulting or advisory role with Lilly/ImClone, Novartis, Ono Pharmaceutical, Roche/Genentech, and Taiho Pharmaceutical, and research funding from Bayer, Novartis, and Roche/Genentech.
New report highlights gaps in knowledge on marijuana use
A new report by the National Academies of Sciences, Engineering and Medicine shines a light on what the existing literature says about the perceived health benefits and dangers of using cannabis and cannabinoids.
“What little we know for certain about the effects of marijuana on human health — and all that we have reason to suspect — justifies serious national concern,” wrote the authors of the report, the evidence and research review of which was chaired by Marie McCormick, MD, of Harvard University in Boston. “The committee’s major recommendation called for an intensification and more comprehensive research effort into the effects of marijuana on the health of the American people.”
The authors concluded current literature shows substantial evidence stating that cannabis is effective at managing chronic pain in adults, while oral cannabinoid use is effective in mitigating nausea or vomiting induced by chemotherapy and improving patient-reported spasticity in patients with multiple sclerosis. Additionally, cannabinoids – specifically, nabiximols – are moderately effective in the short term for improving sleep disturbances brought on by obstructive sleep apnea syndrome, fibromyalgia, chronic pain, and multiple sclerosis.
However, there is limited evidence to support cannabis or cannabinoid use for reducing weight loss or inducing appetite in HIV/AIDS patients, improving clinician-measured spasicity or Tourette syndrome symptoms, reducing anxiety, improving symptoms brought on by post-traumatic stress, and improving outcomes in patients who have suffered traumatic brain injury or intracranial hemorrhage.
Additionally, there is no evidence to support the use of cannabis or cannabinoids in treating cancers or cancer-related anorexia, irritable bowel syndrome symptoms, epilepsy, spinal cord-related spasticity, symptoms of amyotrophic lateral sclerosis, Huntington’s disease, Parkinson’s disease, schizophrenia, and dystonia.
“Present data on drug use progression neither support nor refute the suggestion that medical availability would increase drug abuse,” the authors noted. “However, this question is beyond the issues normally considered for medical uses of drugs and should not be a factor in evaluating the therapeutic potential of marijuana or cannabinoids.”
From a mental health standpoint, suicidal thoughts were found to be more likely in individuals who frequently used cannabis or cannabinoids. Symptoms like depression and anxiety are also more likely in those who smoke marijuana and have bipolar disorder. There is also “limited evidence of a statistical association between sustained abstinence from cannabis use [and] impairments in the cognitive domains of learning, memory, and attention.”
Among the other significant findings of the report, children who live in states where marijuana has been legalized are significantly more likely to ingest cannabis or cannabinoids; so far, marijuana use in some form – either recreational or medical – has been approved in 28 states and Washington, DC. Furthermore, adolescents who use marijuana are more likely to experience difficulties in social and educational development. And individuals of any age who smoke marijuana and drive are more likely to be involved in a car accident.
Also noteworthy is the lack of evidence pointing to marijuana use causing cancer. While chronic marijuana smoking was found to be linked to bronchitis, it was not found to cause cancers that are most commonly associated with chronic smoking of tobacco.
“This report highlights that there are critical gaps in our understanding of the health effects of cannabis,” explained John H. Krystal, MD, of Yale University in New Haven, Connecticut.
A reviewer of the report, Dr. Krystal elaborated on the gaps that exist in the current literature, saying “One reason for these gaps has been regulatory and practical challenges facing those who attempted to conduct this research. For example, what supply of cannabis should they use? Where, in the typical hospital settings where research is conducted, should patients participating in research be permitted to smoke cannabis? What standards should the institutional review committees employ when evaluating studies that involve the administration of cannabis or other cannabinoids?”
Ultimately, Dr. Krystal stated, “what should be evident from this summary is that only a few of the many publicized clinical applications for cannabis are adequately supported by acceptable research standards for determining safety or efficacy.” Specifically, states that have approved cannabis use for managing PTSD symptoms are doing so based off “meager” evidence, and in some cases, are circumventing FDA regulatory processes in a way. This could not only compromise patient care, but muddy the waters for physicians who want to treat their patients safely while also following legal avenues.
“Physicians may face a tension between their roles as physicians [and] their wish to provide a legal path for access to cannabinoids for their patients,” Dr. Krystal said, adding that “the endorsement of particular cannabis prescription practices by the states, even for clinical indications where cannabis has not been shown to be safe and effective, may create pressure for physicians to engage in ineffective or unsafe cannabis prescription practices.”
Ultimately, the report underlines areas of need in terms of understanding and effectively using cannabis and cannabinoid in treating patients. Calling the report a “call to arms” for those in the health care – and, specifically, the public health – arena, Dr. Krystal added that he hopes the findings of the report will be used for educating “legislators, physicians, and consumers [about the] potential benefits and risks of cannabis and thereby help to guide both legislation, clinical practice, and perhaps recreational use.”
A new report by the National Academies of Sciences, Engineering and Medicine shines a light on what the existing literature says about the perceived health benefits and dangers of using cannabis and cannabinoids.
“What little we know for certain about the effects of marijuana on human health — and all that we have reason to suspect — justifies serious national concern,” wrote the authors of the report, the evidence and research review of which was chaired by Marie McCormick, MD, of Harvard University in Boston. “The committee’s major recommendation called for an intensification and more comprehensive research effort into the effects of marijuana on the health of the American people.”
The authors concluded current literature shows substantial evidence stating that cannabis is effective at managing chronic pain in adults, while oral cannabinoid use is effective in mitigating nausea or vomiting induced by chemotherapy and improving patient-reported spasticity in patients with multiple sclerosis. Additionally, cannabinoids – specifically, nabiximols – are moderately effective in the short term for improving sleep disturbances brought on by obstructive sleep apnea syndrome, fibromyalgia, chronic pain, and multiple sclerosis.
However, there is limited evidence to support cannabis or cannabinoid use for reducing weight loss or inducing appetite in HIV/AIDS patients, improving clinician-measured spasicity or Tourette syndrome symptoms, reducing anxiety, improving symptoms brought on by post-traumatic stress, and improving outcomes in patients who have suffered traumatic brain injury or intracranial hemorrhage.
Additionally, there is no evidence to support the use of cannabis or cannabinoids in treating cancers or cancer-related anorexia, irritable bowel syndrome symptoms, epilepsy, spinal cord-related spasticity, symptoms of amyotrophic lateral sclerosis, Huntington’s disease, Parkinson’s disease, schizophrenia, and dystonia.
“Present data on drug use progression neither support nor refute the suggestion that medical availability would increase drug abuse,” the authors noted. “However, this question is beyond the issues normally considered for medical uses of drugs and should not be a factor in evaluating the therapeutic potential of marijuana or cannabinoids.”
From a mental health standpoint, suicidal thoughts were found to be more likely in individuals who frequently used cannabis or cannabinoids. Symptoms like depression and anxiety are also more likely in those who smoke marijuana and have bipolar disorder. There is also “limited evidence of a statistical association between sustained abstinence from cannabis use [and] impairments in the cognitive domains of learning, memory, and attention.”
Among the other significant findings of the report, children who live in states where marijuana has been legalized are significantly more likely to ingest cannabis or cannabinoids; so far, marijuana use in some form – either recreational or medical – has been approved in 28 states and Washington, DC. Furthermore, adolescents who use marijuana are more likely to experience difficulties in social and educational development. And individuals of any age who smoke marijuana and drive are more likely to be involved in a car accident.
Also noteworthy is the lack of evidence pointing to marijuana use causing cancer. While chronic marijuana smoking was found to be linked to bronchitis, it was not found to cause cancers that are most commonly associated with chronic smoking of tobacco.
“This report highlights that there are critical gaps in our understanding of the health effects of cannabis,” explained John H. Krystal, MD, of Yale University in New Haven, Connecticut.
A reviewer of the report, Dr. Krystal elaborated on the gaps that exist in the current literature, saying “One reason for these gaps has been regulatory and practical challenges facing those who attempted to conduct this research. For example, what supply of cannabis should they use? Where, in the typical hospital settings where research is conducted, should patients participating in research be permitted to smoke cannabis? What standards should the institutional review committees employ when evaluating studies that involve the administration of cannabis or other cannabinoids?”
Ultimately, Dr. Krystal stated, “what should be evident from this summary is that only a few of the many publicized clinical applications for cannabis are adequately supported by acceptable research standards for determining safety or efficacy.” Specifically, states that have approved cannabis use for managing PTSD symptoms are doing so based off “meager” evidence, and in some cases, are circumventing FDA regulatory processes in a way. This could not only compromise patient care, but muddy the waters for physicians who want to treat their patients safely while also following legal avenues.
“Physicians may face a tension between their roles as physicians [and] their wish to provide a legal path for access to cannabinoids for their patients,” Dr. Krystal said, adding that “the endorsement of particular cannabis prescription practices by the states, even for clinical indications where cannabis has not been shown to be safe and effective, may create pressure for physicians to engage in ineffective or unsafe cannabis prescription practices.”
Ultimately, the report underlines areas of need in terms of understanding and effectively using cannabis and cannabinoid in treating patients. Calling the report a “call to arms” for those in the health care – and, specifically, the public health – arena, Dr. Krystal added that he hopes the findings of the report will be used for educating “legislators, physicians, and consumers [about the] potential benefits and risks of cannabis and thereby help to guide both legislation, clinical practice, and perhaps recreational use.”
A new report by the National Academies of Sciences, Engineering and Medicine shines a light on what the existing literature says about the perceived health benefits and dangers of using cannabis and cannabinoids.
“What little we know for certain about the effects of marijuana on human health — and all that we have reason to suspect — justifies serious national concern,” wrote the authors of the report, the evidence and research review of which was chaired by Marie McCormick, MD, of Harvard University in Boston. “The committee’s major recommendation called for an intensification and more comprehensive research effort into the effects of marijuana on the health of the American people.”
The authors concluded current literature shows substantial evidence stating that cannabis is effective at managing chronic pain in adults, while oral cannabinoid use is effective in mitigating nausea or vomiting induced by chemotherapy and improving patient-reported spasticity in patients with multiple sclerosis. Additionally, cannabinoids – specifically, nabiximols – are moderately effective in the short term for improving sleep disturbances brought on by obstructive sleep apnea syndrome, fibromyalgia, chronic pain, and multiple sclerosis.
However, there is limited evidence to support cannabis or cannabinoid use for reducing weight loss or inducing appetite in HIV/AIDS patients, improving clinician-measured spasicity or Tourette syndrome symptoms, reducing anxiety, improving symptoms brought on by post-traumatic stress, and improving outcomes in patients who have suffered traumatic brain injury or intracranial hemorrhage.
Additionally, there is no evidence to support the use of cannabis or cannabinoids in treating cancers or cancer-related anorexia, irritable bowel syndrome symptoms, epilepsy, spinal cord-related spasticity, symptoms of amyotrophic lateral sclerosis, Huntington’s disease, Parkinson’s disease, schizophrenia, and dystonia.
“Present data on drug use progression neither support nor refute the suggestion that medical availability would increase drug abuse,” the authors noted. “However, this question is beyond the issues normally considered for medical uses of drugs and should not be a factor in evaluating the therapeutic potential of marijuana or cannabinoids.”
From a mental health standpoint, suicidal thoughts were found to be more likely in individuals who frequently used cannabis or cannabinoids. Symptoms like depression and anxiety are also more likely in those who smoke marijuana and have bipolar disorder. There is also “limited evidence of a statistical association between sustained abstinence from cannabis use [and] impairments in the cognitive domains of learning, memory, and attention.”
Among the other significant findings of the report, children who live in states where marijuana has been legalized are significantly more likely to ingest cannabis or cannabinoids; so far, marijuana use in some form – either recreational or medical – has been approved in 28 states and Washington, DC. Furthermore, adolescents who use marijuana are more likely to experience difficulties in social and educational development. And individuals of any age who smoke marijuana and drive are more likely to be involved in a car accident.
Also noteworthy is the lack of evidence pointing to marijuana use causing cancer. While chronic marijuana smoking was found to be linked to bronchitis, it was not found to cause cancers that are most commonly associated with chronic smoking of tobacco.
“This report highlights that there are critical gaps in our understanding of the health effects of cannabis,” explained John H. Krystal, MD, of Yale University in New Haven, Connecticut.
A reviewer of the report, Dr. Krystal elaborated on the gaps that exist in the current literature, saying “One reason for these gaps has been regulatory and practical challenges facing those who attempted to conduct this research. For example, what supply of cannabis should they use? Where, in the typical hospital settings where research is conducted, should patients participating in research be permitted to smoke cannabis? What standards should the institutional review committees employ when evaluating studies that involve the administration of cannabis or other cannabinoids?”
Ultimately, Dr. Krystal stated, “what should be evident from this summary is that only a few of the many publicized clinical applications for cannabis are adequately supported by acceptable research standards for determining safety or efficacy.” Specifically, states that have approved cannabis use for managing PTSD symptoms are doing so based off “meager” evidence, and in some cases, are circumventing FDA regulatory processes in a way. This could not only compromise patient care, but muddy the waters for physicians who want to treat their patients safely while also following legal avenues.
“Physicians may face a tension between their roles as physicians [and] their wish to provide a legal path for access to cannabinoids for their patients,” Dr. Krystal said, adding that “the endorsement of particular cannabis prescription practices by the states, even for clinical indications where cannabis has not been shown to be safe and effective, may create pressure for physicians to engage in ineffective or unsafe cannabis prescription practices.”
Ultimately, the report underlines areas of need in terms of understanding and effectively using cannabis and cannabinoid in treating patients. Calling the report a “call to arms” for those in the health care – and, specifically, the public health – arena, Dr. Krystal added that he hopes the findings of the report will be used for educating “legislators, physicians, and consumers [about the] potential benefits and risks of cannabis and thereby help to guide both legislation, clinical practice, and perhaps recreational use.”
FROM THE NATIONAL ACADEMIES OF SCIENCES, ENGINEERING AND MEDICINE
57% drop in central venous catheter–related infections after QI interventions
Quality improvement (QI) interventions related to the use of central venous catheters (CVCs) were, on average, associated with 57% fewer infections and $1.85 million in net savings to hospitals within 1-3 years of implementation, based on the results of a meta-analysis of data from 113 hospitals.
“Hospitals that have already attained very low infection rates (through the use of quality improvement checklists) would likely see smaller clinical benefits and savings than in the studies we have reviewed,” said Dr. Teryl Nuckols of Cedars-Sinai Medical Center, Los Angeles. “Nonetheless, we found that QI interventions can be associated with declines in CLABSI (central line-associated bloodstream infection) and/or CRBSI (catheter-related bloodstream infection) and net savings when checklists are already in use, and when hospitals have CLABSI rates as low as 1.7-3.7 per 1,000 CVC-days.”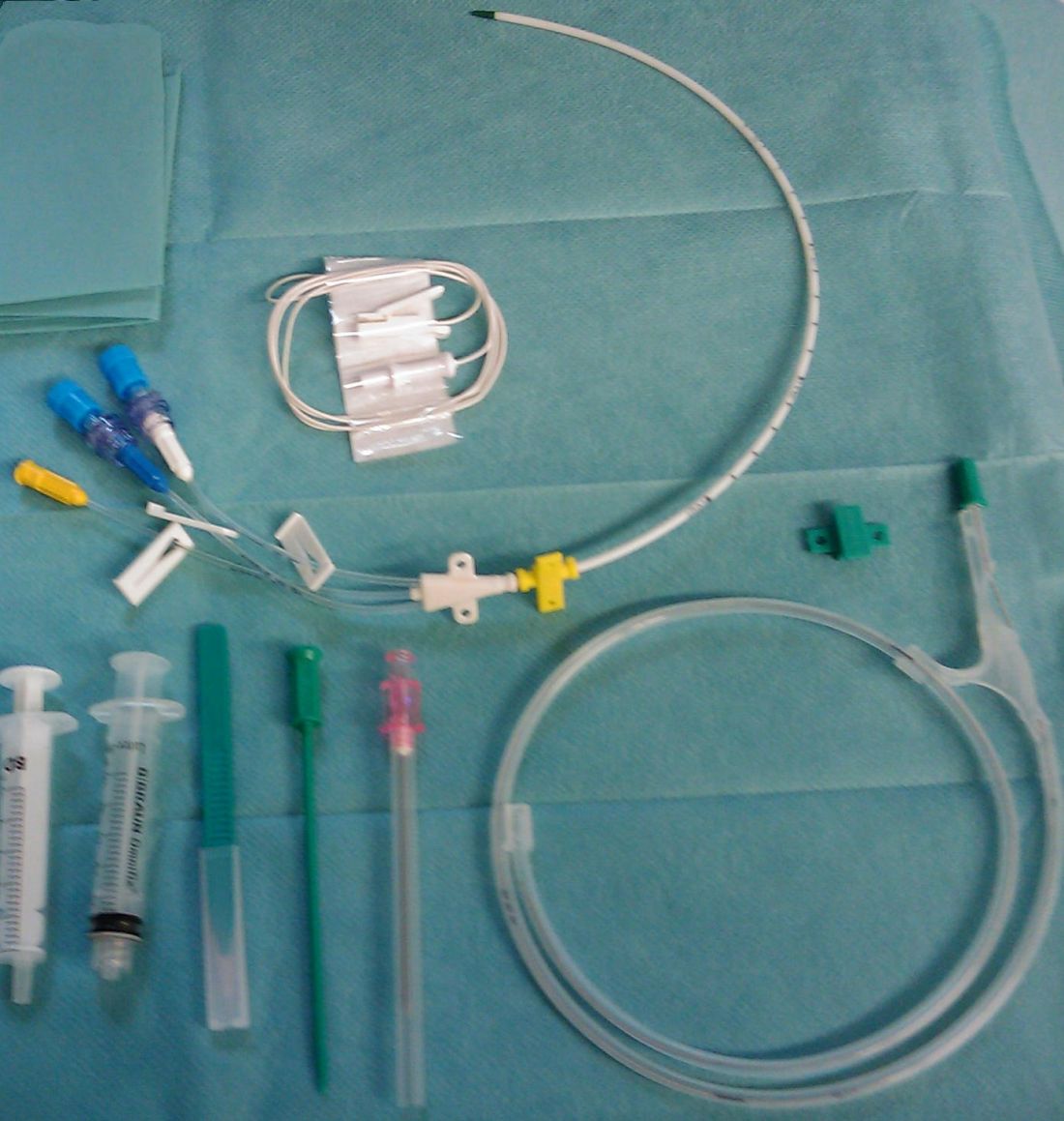
Studies were eligible for the analysis if they reported or estimated the quality improvement intervention’s clinical effectiveness, measured or modeled its costs, compared alternatives to the intervention, and reported both program and infection-related costs.
Insertion checklists were examined in 12 studies, physician education in 11 studies, ultrasound-guided placement of catheters in 3 studies, all-inclusive catheter kits in 5 studies, sterile dressings in 5 studies, chlorhexidine gluconate sponge or antimicrobial dressing in 2 studies, and antimicrobial catheters in 2 studies.
Overall, the weighted mean incidence rate ratio was 0.43 (95% confidence interval, 0.35-0.51) and incremental net savings were $1.85 million (95% CI, $1.30 million to $2.40 million) per hospital over 3 years (2015 U.S. dollars). Each $100,000 increase in program cost was associated with $315,000 greater savings (95% CI, $166 000-$464 000; P less than .001). Infections and net costs declined when hospitals already used checklists or had baseline infection rates of 1.7-3.7 per 1,000 catheter-days. (doi: 10.1001/jamainternmed.2016.6610)
Dr. Nuckols acknowledged that the price tag for achieving these savings “may be burdensome for hospitals with limited financial resources … wages and benefits account for two-thirds of all spending by hospitals, and a quarter of hospitals have had negative operating margins in recent years. We found that, for CLABSI- and CRBSI-prevention interventions, median program costs were about $270,000 per hospital over 3 years – but reached $500,000 to $750,000 in some studies.”
The researchers recommended that “future research should more thoroughly examine the relationships among hospital financial performance, economic investments in QI, and effects on quality of care.”
Quality improvement (QI) interventions related to the use of central venous catheters (CVCs) were, on average, associated with 57% fewer infections and $1.85 million in net savings to hospitals within 1-3 years of implementation, based on the results of a meta-analysis of data from 113 hospitals.
“Hospitals that have already attained very low infection rates (through the use of quality improvement checklists) would likely see smaller clinical benefits and savings than in the studies we have reviewed,” said Dr. Teryl Nuckols of Cedars-Sinai Medical Center, Los Angeles. “Nonetheless, we found that QI interventions can be associated with declines in CLABSI (central line-associated bloodstream infection) and/or CRBSI (catheter-related bloodstream infection) and net savings when checklists are already in use, and when hospitals have CLABSI rates as low as 1.7-3.7 per 1,000 CVC-days.”
Studies were eligible for the analysis if they reported or estimated the quality improvement intervention’s clinical effectiveness, measured or modeled its costs, compared alternatives to the intervention, and reported both program and infection-related costs.
Insertion checklists were examined in 12 studies, physician education in 11 studies, ultrasound-guided placement of catheters in 3 studies, all-inclusive catheter kits in 5 studies, sterile dressings in 5 studies, chlorhexidine gluconate sponge or antimicrobial dressing in 2 studies, and antimicrobial catheters in 2 studies.
Overall, the weighted mean incidence rate ratio was 0.43 (95% confidence interval, 0.35-0.51) and incremental net savings were $1.85 million (95% CI, $1.30 million to $2.40 million) per hospital over 3 years (2015 U.S. dollars). Each $100,000 increase in program cost was associated with $315,000 greater savings (95% CI, $166 000-$464 000; P less than .001). Infections and net costs declined when hospitals already used checklists or had baseline infection rates of 1.7-3.7 per 1,000 catheter-days. (doi: 10.1001/jamainternmed.2016.6610)
Dr. Nuckols acknowledged that the price tag for achieving these savings “may be burdensome for hospitals with limited financial resources … wages and benefits account for two-thirds of all spending by hospitals, and a quarter of hospitals have had negative operating margins in recent years. We found that, for CLABSI- and CRBSI-prevention interventions, median program costs were about $270,000 per hospital over 3 years – but reached $500,000 to $750,000 in some studies.”
The researchers recommended that “future research should more thoroughly examine the relationships among hospital financial performance, economic investments in QI, and effects on quality of care.”
Quality improvement (QI) interventions related to the use of central venous catheters (CVCs) were, on average, associated with 57% fewer infections and $1.85 million in net savings to hospitals within 1-3 years of implementation, based on the results of a meta-analysis of data from 113 hospitals.
“Hospitals that have already attained very low infection rates (through the use of quality improvement checklists) would likely see smaller clinical benefits and savings than in the studies we have reviewed,” said Dr. Teryl Nuckols of Cedars-Sinai Medical Center, Los Angeles. “Nonetheless, we found that QI interventions can be associated with declines in CLABSI (central line-associated bloodstream infection) and/or CRBSI (catheter-related bloodstream infection) and net savings when checklists are already in use, and when hospitals have CLABSI rates as low as 1.7-3.7 per 1,000 CVC-days.”
Studies were eligible for the analysis if they reported or estimated the quality improvement intervention’s clinical effectiveness, measured or modeled its costs, compared alternatives to the intervention, and reported both program and infection-related costs.
Insertion checklists were examined in 12 studies, physician education in 11 studies, ultrasound-guided placement of catheters in 3 studies, all-inclusive catheter kits in 5 studies, sterile dressings in 5 studies, chlorhexidine gluconate sponge or antimicrobial dressing in 2 studies, and antimicrobial catheters in 2 studies.
Overall, the weighted mean incidence rate ratio was 0.43 (95% confidence interval, 0.35-0.51) and incremental net savings were $1.85 million (95% CI, $1.30 million to $2.40 million) per hospital over 3 years (2015 U.S. dollars). Each $100,000 increase in program cost was associated with $315,000 greater savings (95% CI, $166 000-$464 000; P less than .001). Infections and net costs declined when hospitals already used checklists or had baseline infection rates of 1.7-3.7 per 1,000 catheter-days. (doi: 10.1001/jamainternmed.2016.6610)
Dr. Nuckols acknowledged that the price tag for achieving these savings “may be burdensome for hospitals with limited financial resources … wages and benefits account for two-thirds of all spending by hospitals, and a quarter of hospitals have had negative operating margins in recent years. We found that, for CLABSI- and CRBSI-prevention interventions, median program costs were about $270,000 per hospital over 3 years – but reached $500,000 to $750,000 in some studies.”
The researchers recommended that “future research should more thoroughly examine the relationships among hospital financial performance, economic investments in QI, and effects on quality of care.”
PET-directed induction therapy improves esophageal cancer outcomes
SAN FRANCISCO – The use of positron emission tomography to assess esophageal tumor response to induction chemotherapy and to guide a regimen change in those who failed to respond was associated with an improved pathologic complete response (pCR) rate in patients in a phase II randomized trial.
Of 257 patients with esophageal and gastroesophageal junction (GEJ) adenocarcinomas who were randomized after a baseline PET scan to receive induction chemotherapy with either modified FOLFOX-6 (oxaliplatin, leucovorin, and 5-fluorouracil) on days 1, 15, and 29, or carboplatin/paclitaxel (CP) on days 1, 8, 22, and 29, 39 patients and 49 patients, respectively, were found on a repeat PET scan performed between days 36 and 42 after initiation of therapy to be nonresponders. Those patients were switched to the alternative regimen during preoperative chemoradiation therapy (CRT). Eligible subjects underwent surgical resection 6 weeks after CRT. The pCR rate after surgery among those who were switched because of initial nonresponse was 18%, compared with an expected rate of 5% based on data from prior studies in which chemotherapy was not changed in nonresponders. The rate was 26% in PET responders, Karyn A. Goodman, MD, reported at the symposium, sponsored by ASCO, ASTRO, the American Gastroenterological Association, and the Society of Surgical Oncology..
“Preoperative chemoradiotherapy is an accepted standard of care for patients with operable esophageal or gastroesophageal junction adenocarcinoma, but even with this aggressive approach, 5-year overall survival rates are on the order of 40%-50%, and most patients die of systemic disease. Optimal systemic therapy for esophageal and gastroesophageal junction cancers remains undefined, so better methods to identify effective therapies are needed,” she said.
Based on findings from the German, phase II, multicenter MUNICON trial, which showed that early PET responders to chemotherapy had significantly better event-free survival than did PET nonresponders (30 vs. 14 months), and that immediate surgical resection among those identified as nonresponders was also associated with improved outcomes, Dr. Goodman and her colleagues “set out to do CALGB 80803 [the current trial] to evaluate the use of early assessment of chemotherapy responsiveness by PET imaging to direct further therapy … with the goal of improving treatment responses,” she said.
Among secondary CALGB 80803 endpoints was the PET response in the treatment arms.
The rate of response to induction chemotherapy was 57% in the FOLFOX group, and 50% in the CP group. The nonresponders were switched to the alternate regimen, and 84% of patients went on to surgery.
The overall pCR rate after surgery was 19% for those who switched from FOLFOX to CP, and 17% for those who switched from CP to FOLFOX.
“The efficacy criteria for changing therapy was met for both induction arms,” she said.
“Of note, patients who were PET responders who had induction and concurrent FOLFOX had a pCR rate of almost 38%. The pCR rate for patients who started on the induction FOLFOX arm was 31%, and for those who started on the induction CP arm it was 14%, and for all patients enrolled on the study, the pCR rate was 22.7%” she said.
PET scans are routinely used to guide therapy decisions in patients with lymphoma, but are only beginning to be explored for this purpose in solid tumors. CALGB 80803 is among the first to show the benefit of PET imaging in directing presurgery treatment decisions for esophageal cancer.
“We’ve shown that a short course of induction chemotherapy followed by early response assessment using PET imaging to determine whether to switch from ineffective therapy to alternative chemotherapy during preoperative chemoradiation for PET nonresponders is feasible for esophageal and GEJ cancers,” she said, adding that the findings demonstrate a benefit with “a new paradigm of using metabolic imaging … to individualize multimodality therapy and improve outcomes in this poor-prognosis population.”
Further, while the study was not powered for a head-to-head comparison of FOLFOX and CP, the “very promising” 38% pCR rate with FOLFOX induction and concurrent therapy is hypothesis generating, she said.
“How these improvements in pCR translate into survival benefit will be determined with longer follow-up,” she added, noting in response to a question about whether these findings will change practice that “this is really the first step in the process. If we can improve outcomes by changing treatment early on, this should be standard of care, but we really do need to get survival outcome information.”
PET at baseline is already standard; adding another scan adds additional cost, but this potentially may be offset by the prevention of costly toxicities and other problems that have to be addressed, she said, concluding that “going forward, early-response assessment using PET imaging can be incorporated into studies to identify more effective new regimens for esophageal and GEJ cancers.”
During a preconference press cast on the findings, press cast moderator Nancy Baxter, MD, of ASCO said that PET scans may prove to be a valuable tool for fine-tuning the use of chemotherapy for esophageal cancer, and for maximizing the benefit of chemotherapy for each patient.
“This is heartening evidence for a new approach to treating a disease where innovation is sorely needed,” she said.
CALGB 80803 was funded by grants from the National Cancer Institute. Dr. Goodman and Dr. Baxter reported having no disclosures.
SAN FRANCISCO – The use of positron emission tomography to assess esophageal tumor response to induction chemotherapy and to guide a regimen change in those who failed to respond was associated with an improved pathologic complete response (pCR) rate in patients in a phase II randomized trial.
Of 257 patients with esophageal and gastroesophageal junction (GEJ) adenocarcinomas who were randomized after a baseline PET scan to receive induction chemotherapy with either modified FOLFOX-6 (oxaliplatin, leucovorin, and 5-fluorouracil) on days 1, 15, and 29, or carboplatin/paclitaxel (CP) on days 1, 8, 22, and 29, 39 patients and 49 patients, respectively, were found on a repeat PET scan performed between days 36 and 42 after initiation of therapy to be nonresponders. Those patients were switched to the alternative regimen during preoperative chemoradiation therapy (CRT). Eligible subjects underwent surgical resection 6 weeks after CRT. The pCR rate after surgery among those who were switched because of initial nonresponse was 18%, compared with an expected rate of 5% based on data from prior studies in which chemotherapy was not changed in nonresponders. The rate was 26% in PET responders, Karyn A. Goodman, MD, reported at the symposium, sponsored by ASCO, ASTRO, the American Gastroenterological Association, and the Society of Surgical Oncology..
“Preoperative chemoradiotherapy is an accepted standard of care for patients with operable esophageal or gastroesophageal junction adenocarcinoma, but even with this aggressive approach, 5-year overall survival rates are on the order of 40%-50%, and most patients die of systemic disease. Optimal systemic therapy for esophageal and gastroesophageal junction cancers remains undefined, so better methods to identify effective therapies are needed,” she said.
Based on findings from the German, phase II, multicenter MUNICON trial, which showed that early PET responders to chemotherapy had significantly better event-free survival than did PET nonresponders (30 vs. 14 months), and that immediate surgical resection among those identified as nonresponders was also associated with improved outcomes, Dr. Goodman and her colleagues “set out to do CALGB 80803 [the current trial] to evaluate the use of early assessment of chemotherapy responsiveness by PET imaging to direct further therapy … with the goal of improving treatment responses,” she said.
Among secondary CALGB 80803 endpoints was the PET response in the treatment arms.
The rate of response to induction chemotherapy was 57% in the FOLFOX group, and 50% in the CP group. The nonresponders were switched to the alternate regimen, and 84% of patients went on to surgery.
The overall pCR rate after surgery was 19% for those who switched from FOLFOX to CP, and 17% for those who switched from CP to FOLFOX.
“The efficacy criteria for changing therapy was met for both induction arms,” she said.
“Of note, patients who were PET responders who had induction and concurrent FOLFOX had a pCR rate of almost 38%. The pCR rate for patients who started on the induction FOLFOX arm was 31%, and for those who started on the induction CP arm it was 14%, and for all patients enrolled on the study, the pCR rate was 22.7%” she said.
PET scans are routinely used to guide therapy decisions in patients with lymphoma, but are only beginning to be explored for this purpose in solid tumors. CALGB 80803 is among the first to show the benefit of PET imaging in directing presurgery treatment decisions for esophageal cancer.
“We’ve shown that a short course of induction chemotherapy followed by early response assessment using PET imaging to determine whether to switch from ineffective therapy to alternative chemotherapy during preoperative chemoradiation for PET nonresponders is feasible for esophageal and GEJ cancers,” she said, adding that the findings demonstrate a benefit with “a new paradigm of using metabolic imaging … to individualize multimodality therapy and improve outcomes in this poor-prognosis population.”
Further, while the study was not powered for a head-to-head comparison of FOLFOX and CP, the “very promising” 38% pCR rate with FOLFOX induction and concurrent therapy is hypothesis generating, she said.
“How these improvements in pCR translate into survival benefit will be determined with longer follow-up,” she added, noting in response to a question about whether these findings will change practice that “this is really the first step in the process. If we can improve outcomes by changing treatment early on, this should be standard of care, but we really do need to get survival outcome information.”
PET at baseline is already standard; adding another scan adds additional cost, but this potentially may be offset by the prevention of costly toxicities and other problems that have to be addressed, she said, concluding that “going forward, early-response assessment using PET imaging can be incorporated into studies to identify more effective new regimens for esophageal and GEJ cancers.”
During a preconference press cast on the findings, press cast moderator Nancy Baxter, MD, of ASCO said that PET scans may prove to be a valuable tool for fine-tuning the use of chemotherapy for esophageal cancer, and for maximizing the benefit of chemotherapy for each patient.
“This is heartening evidence for a new approach to treating a disease where innovation is sorely needed,” she said.
CALGB 80803 was funded by grants from the National Cancer Institute. Dr. Goodman and Dr. Baxter reported having no disclosures.
SAN FRANCISCO – The use of positron emission tomography to assess esophageal tumor response to induction chemotherapy and to guide a regimen change in those who failed to respond was associated with an improved pathologic complete response (pCR) rate in patients in a phase II randomized trial.
Of 257 patients with esophageal and gastroesophageal junction (GEJ) adenocarcinomas who were randomized after a baseline PET scan to receive induction chemotherapy with either modified FOLFOX-6 (oxaliplatin, leucovorin, and 5-fluorouracil) on days 1, 15, and 29, or carboplatin/paclitaxel (CP) on days 1, 8, 22, and 29, 39 patients and 49 patients, respectively, were found on a repeat PET scan performed between days 36 and 42 after initiation of therapy to be nonresponders. Those patients were switched to the alternative regimen during preoperative chemoradiation therapy (CRT). Eligible subjects underwent surgical resection 6 weeks after CRT. The pCR rate after surgery among those who were switched because of initial nonresponse was 18%, compared with an expected rate of 5% based on data from prior studies in which chemotherapy was not changed in nonresponders. The rate was 26% in PET responders, Karyn A. Goodman, MD, reported at the symposium, sponsored by ASCO, ASTRO, the American Gastroenterological Association, and the Society of Surgical Oncology..
“Preoperative chemoradiotherapy is an accepted standard of care for patients with operable esophageal or gastroesophageal junction adenocarcinoma, but even with this aggressive approach, 5-year overall survival rates are on the order of 40%-50%, and most patients die of systemic disease. Optimal systemic therapy for esophageal and gastroesophageal junction cancers remains undefined, so better methods to identify effective therapies are needed,” she said.
Based on findings from the German, phase II, multicenter MUNICON trial, which showed that early PET responders to chemotherapy had significantly better event-free survival than did PET nonresponders (30 vs. 14 months), and that immediate surgical resection among those identified as nonresponders was also associated with improved outcomes, Dr. Goodman and her colleagues “set out to do CALGB 80803 [the current trial] to evaluate the use of early assessment of chemotherapy responsiveness by PET imaging to direct further therapy … with the goal of improving treatment responses,” she said.
Among secondary CALGB 80803 endpoints was the PET response in the treatment arms.
The rate of response to induction chemotherapy was 57% in the FOLFOX group, and 50% in the CP group. The nonresponders were switched to the alternate regimen, and 84% of patients went on to surgery.
The overall pCR rate after surgery was 19% for those who switched from FOLFOX to CP, and 17% for those who switched from CP to FOLFOX.
“The efficacy criteria for changing therapy was met for both induction arms,” she said.
“Of note, patients who were PET responders who had induction and concurrent FOLFOX had a pCR rate of almost 38%. The pCR rate for patients who started on the induction FOLFOX arm was 31%, and for those who started on the induction CP arm it was 14%, and for all patients enrolled on the study, the pCR rate was 22.7%” she said.
PET scans are routinely used to guide therapy decisions in patients with lymphoma, but are only beginning to be explored for this purpose in solid tumors. CALGB 80803 is among the first to show the benefit of PET imaging in directing presurgery treatment decisions for esophageal cancer.
“We’ve shown that a short course of induction chemotherapy followed by early response assessment using PET imaging to determine whether to switch from ineffective therapy to alternative chemotherapy during preoperative chemoradiation for PET nonresponders is feasible for esophageal and GEJ cancers,” she said, adding that the findings demonstrate a benefit with “a new paradigm of using metabolic imaging … to individualize multimodality therapy and improve outcomes in this poor-prognosis population.”
Further, while the study was not powered for a head-to-head comparison of FOLFOX and CP, the “very promising” 38% pCR rate with FOLFOX induction and concurrent therapy is hypothesis generating, she said.
“How these improvements in pCR translate into survival benefit will be determined with longer follow-up,” she added, noting in response to a question about whether these findings will change practice that “this is really the first step in the process. If we can improve outcomes by changing treatment early on, this should be standard of care, but we really do need to get survival outcome information.”
PET at baseline is already standard; adding another scan adds additional cost, but this potentially may be offset by the prevention of costly toxicities and other problems that have to be addressed, she said, concluding that “going forward, early-response assessment using PET imaging can be incorporated into studies to identify more effective new regimens for esophageal and GEJ cancers.”
During a preconference press cast on the findings, press cast moderator Nancy Baxter, MD, of ASCO said that PET scans may prove to be a valuable tool for fine-tuning the use of chemotherapy for esophageal cancer, and for maximizing the benefit of chemotherapy for each patient.
“This is heartening evidence for a new approach to treating a disease where innovation is sorely needed,” she said.
CALGB 80803 was funded by grants from the National Cancer Institute. Dr. Goodman and Dr. Baxter reported having no disclosures.
AT THE 2017 GASTROINTESTINAL CANCERS SYMPOSIUM
Key clinical point:
Major finding: The pathologic completer response rate among PET nonresponders who switched therapies was 18% vs. an expected rate of 5%.
Data source: The phase II randomized CALGB 80803 of 257 esophageal and GEJ cancer patients.
Disclosures: CALGB 80803 was funded by grants from the National Cancer Institute. Dr. Goodman and Dr. Baxter reported having no disclosures.
Meta-analysis supports statins as VTE prophylaxis

Photo courtesy of the CDC
Results of a meta-analysis support the idea that statins can reduce the risk of venous thromboembolism (VTE).
Researchers analyzed data from 36 studies involving more than 3.2 million people and found an “extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE.”
The team believes these findings could potentially lead to new guidelines and an expansion of the use of statins.
“These findings underscore a potential beneficial role of statin therapy on VTE in addition to its established role in cardiovascular disease prevention,” said Kamlesh Khunti, MD, PhD, of the University of Leicester in the UK.
Dr Khunti and his colleagues reported these findings in The Lancet Haematology.
The researchers analyzed data from 36 studies. This included 13 cohort studies with a total of 3,148,259 subjects and 23 randomized, controlled trials (RCTs) of 118,464 subjects in which statins were compared to placebo or no treatment.
In the observational studies, statin use was associated with a significant reduction in the risk of VTE, when compared to no statin use. The pooled relative risk (RR) was 0.75 (P<0.0001).
Statin use was associated with a significant reduction in the risk of deep vein thrombosis (DVT), with an RR of 0.77 (P<0.0001). (There were only 2 observational studies looking specifically at DVT outcomes.)
However, there was no significant reduction in the risk of pulmonary embolism (PE) with statin use. The RR was 1.02 (P=0.90). (There was only 1 observational study reporting specifically on PE outcomes.)
In the RCTs, statin use was associated with a significant reduction in the risk of VTE, with an RR of 0.85 (P=0.038).
Statin use was also associated with a significant reduction in DVT risk, with an RR of 0.45, but not PE risk, with an RR of 0.77 (no P values available). (There was only 1 RCT that reported specifically on DVT and PE outcomes.)
The RCTs also showed significant differences in VTE risk according to type of statin used. Patients receiving rosuvastatin had the lowest risk of VTE when compared to those receiving other statins, with an RR of 0.57 (P=0.015).
“Currently, statins are only approved for lipid lowering in the primary and secondary prevention of cardiovascular disease,” said study author Setor Kunutsor, MBChB, PhD, of the University of Bristol in the UK.
“But they have shown great promise beyond their established lipid-lowering effects, and these include potential beneficial impact on multiple disease conditions. These results provide an extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE and may support a true protective effect.” ![]()
Photo courtesy of the CDC
Results of a meta-analysis support the idea that statins can reduce the risk of venous thromboembolism (VTE).
Researchers analyzed data from 36 studies involving more than 3.2 million people and found an “extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE.”
The team believes these findings could potentially lead to new guidelines and an expansion of the use of statins.
“These findings underscore a potential beneficial role of statin therapy on VTE in addition to its established role in cardiovascular disease prevention,” said Kamlesh Khunti, MD, PhD, of the University of Leicester in the UK.
Dr Khunti and his colleagues reported these findings in The Lancet Haematology.
The researchers analyzed data from 36 studies. This included 13 cohort studies with a total of 3,148,259 subjects and 23 randomized, controlled trials (RCTs) of 118,464 subjects in which statins were compared to placebo or no treatment.
In the observational studies, statin use was associated with a significant reduction in the risk of VTE, when compared to no statin use. The pooled relative risk (RR) was 0.75 (P<0.0001).
Statin use was associated with a significant reduction in the risk of deep vein thrombosis (DVT), with an RR of 0.77 (P<0.0001). (There were only 2 observational studies looking specifically at DVT outcomes.)
However, there was no significant reduction in the risk of pulmonary embolism (PE) with statin use. The RR was 1.02 (P=0.90). (There was only 1 observational study reporting specifically on PE outcomes.)
In the RCTs, statin use was associated with a significant reduction in the risk of VTE, with an RR of 0.85 (P=0.038).
Statin use was also associated with a significant reduction in DVT risk, with an RR of 0.45, but not PE risk, with an RR of 0.77 (no P values available). (There was only 1 RCT that reported specifically on DVT and PE outcomes.)
The RCTs also showed significant differences in VTE risk according to type of statin used. Patients receiving rosuvastatin had the lowest risk of VTE when compared to those receiving other statins, with an RR of 0.57 (P=0.015).
“Currently, statins are only approved for lipid lowering in the primary and secondary prevention of cardiovascular disease,” said study author Setor Kunutsor, MBChB, PhD, of the University of Bristol in the UK.
“But they have shown great promise beyond their established lipid-lowering effects, and these include potential beneficial impact on multiple disease conditions. These results provide an extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE and may support a true protective effect.” ![]()
Photo courtesy of the CDC
Results of a meta-analysis support the idea that statins can reduce the risk of venous thromboembolism (VTE).
Researchers analyzed data from 36 studies involving more than 3.2 million people and found an “extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE.”
The team believes these findings could potentially lead to new guidelines and an expansion of the use of statins.
“These findings underscore a potential beneficial role of statin therapy on VTE in addition to its established role in cardiovascular disease prevention,” said Kamlesh Khunti, MD, PhD, of the University of Leicester in the UK.
Dr Khunti and his colleagues reported these findings in The Lancet Haematology.
The researchers analyzed data from 36 studies. This included 13 cohort studies with a total of 3,148,259 subjects and 23 randomized, controlled trials (RCTs) of 118,464 subjects in which statins were compared to placebo or no treatment.
In the observational studies, statin use was associated with a significant reduction in the risk of VTE, when compared to no statin use. The pooled relative risk (RR) was 0.75 (P<0.0001).
Statin use was associated with a significant reduction in the risk of deep vein thrombosis (DVT), with an RR of 0.77 (P<0.0001). (There were only 2 observational studies looking specifically at DVT outcomes.)
However, there was no significant reduction in the risk of pulmonary embolism (PE) with statin use. The RR was 1.02 (P=0.90). (There was only 1 observational study reporting specifically on PE outcomes.)
In the RCTs, statin use was associated with a significant reduction in the risk of VTE, with an RR of 0.85 (P=0.038).
Statin use was also associated with a significant reduction in DVT risk, with an RR of 0.45, but not PE risk, with an RR of 0.77 (no P values available). (There was only 1 RCT that reported specifically on DVT and PE outcomes.)
The RCTs also showed significant differences in VTE risk according to type of statin used. Patients receiving rosuvastatin had the lowest risk of VTE when compared to those receiving other statins, with an RR of 0.57 (P=0.015).
“Currently, statins are only approved for lipid lowering in the primary and secondary prevention of cardiovascular disease,” said study author Setor Kunutsor, MBChB, PhD, of the University of Bristol in the UK.
“But they have shown great promise beyond their established lipid-lowering effects, and these include potential beneficial impact on multiple disease conditions. These results provide an extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE and may support a true protective effect.” ![]()
New guidelines for patients requiring red blood cell transfusion
Clinical question: What is a safe target hemoglobin level for patients requiring red blood cell transfusion, and how long can red blood cells be stored prior to transfusion?
Background: The AABB, formerly the American Association of Blood Banks, notes several new, large, rigorous studies on transfusion thresholds were published since their last guideline in 2012. Additionally, there are concerns from initial studies of increased morbidity and mortality with transfusions of red blood cells stored for longer periods of time.
Study design: Systematic review and meta-analysis.
Setting: Summary findings from the AABB clinical transfusion medicine committee.
Synopsis: Thirty-one randomized clinical trials (RCTs) evaluating blood transfusion thresholds were reviewed and analyzed, including 12,587 patients across various clinical scenarios. The authors recommend a restrictive threshold of 7 g/dL for most hospitalized adult patients in the appropriate clinical context. For patients undergoing orthopedic or cardiac surgery, or with cardiovascular disease, a threshold of 8 g/dL is recommended, as it was the threshold used in studies of these patients (though such patients may actually tolerate a lower value).
No recommendations were made for patients with acute coronary syndrome, hematological or oncological disorders, severe thrombocytopenia, or chronic transfusion-dependent anemia given limited data.
To determine a safe period of time for blood storage prior to transfusion, 13 RCTs were reviewed and analyzed. The authors recommend that patients requiring transfusion receive red blood cell units at any period within the standard issue period (less than 42 days), rather than limit transfusion to fresh units (less than 10 days).
Bottom line: A restrictive red blood cell transfusion threshold of 7-8 g/dL is safe in most clinical settings, and there is no advantage to using fresh units as opposed to those stored for the standard period.
Citation: Carson JL, Guyatt G, Heddle NM, et al. Clinical practice guidelines from the AABB. JAMA. 2016;316(19):2025-35.
Dr. Murphy is a clinical instructor at the University of Utah School of Medicine and an academic hospitalist at the University of Utah Hospital.
Clinical question: What is a safe target hemoglobin level for patients requiring red blood cell transfusion, and how long can red blood cells be stored prior to transfusion?
Background: The AABB, formerly the American Association of Blood Banks, notes several new, large, rigorous studies on transfusion thresholds were published since their last guideline in 2012. Additionally, there are concerns from initial studies of increased morbidity and mortality with transfusions of red blood cells stored for longer periods of time.
Study design: Systematic review and meta-analysis.
Setting: Summary findings from the AABB clinical transfusion medicine committee.
Synopsis: Thirty-one randomized clinical trials (RCTs) evaluating blood transfusion thresholds were reviewed and analyzed, including 12,587 patients across various clinical scenarios. The authors recommend a restrictive threshold of 7 g/dL for most hospitalized adult patients in the appropriate clinical context. For patients undergoing orthopedic or cardiac surgery, or with cardiovascular disease, a threshold of 8 g/dL is recommended, as it was the threshold used in studies of these patients (though such patients may actually tolerate a lower value).
No recommendations were made for patients with acute coronary syndrome, hematological or oncological disorders, severe thrombocytopenia, or chronic transfusion-dependent anemia given limited data.
To determine a safe period of time for blood storage prior to transfusion, 13 RCTs were reviewed and analyzed. The authors recommend that patients requiring transfusion receive red blood cell units at any period within the standard issue period (less than 42 days), rather than limit transfusion to fresh units (less than 10 days).
Bottom line: A restrictive red blood cell transfusion threshold of 7-8 g/dL is safe in most clinical settings, and there is no advantage to using fresh units as opposed to those stored for the standard period.
Citation: Carson JL, Guyatt G, Heddle NM, et al. Clinical practice guidelines from the AABB. JAMA. 2016;316(19):2025-35.
Dr. Murphy is a clinical instructor at the University of Utah School of Medicine and an academic hospitalist at the University of Utah Hospital.
Clinical question: What is a safe target hemoglobin level for patients requiring red blood cell transfusion, and how long can red blood cells be stored prior to transfusion?
Background: The AABB, formerly the American Association of Blood Banks, notes several new, large, rigorous studies on transfusion thresholds were published since their last guideline in 2012. Additionally, there are concerns from initial studies of increased morbidity and mortality with transfusions of red blood cells stored for longer periods of time.
Study design: Systematic review and meta-analysis.
Setting: Summary findings from the AABB clinical transfusion medicine committee.
Synopsis: Thirty-one randomized clinical trials (RCTs) evaluating blood transfusion thresholds were reviewed and analyzed, including 12,587 patients across various clinical scenarios. The authors recommend a restrictive threshold of 7 g/dL for most hospitalized adult patients in the appropriate clinical context. For patients undergoing orthopedic or cardiac surgery, or with cardiovascular disease, a threshold of 8 g/dL is recommended, as it was the threshold used in studies of these patients (though such patients may actually tolerate a lower value).
No recommendations were made for patients with acute coronary syndrome, hematological or oncological disorders, severe thrombocytopenia, or chronic transfusion-dependent anemia given limited data.
To determine a safe period of time for blood storage prior to transfusion, 13 RCTs were reviewed and analyzed. The authors recommend that patients requiring transfusion receive red blood cell units at any period within the standard issue period (less than 42 days), rather than limit transfusion to fresh units (less than 10 days).
Bottom line: A restrictive red blood cell transfusion threshold of 7-8 g/dL is safe in most clinical settings, and there is no advantage to using fresh units as opposed to those stored for the standard period.
Citation: Carson JL, Guyatt G, Heddle NM, et al. Clinical practice guidelines from the AABB. JAMA. 2016;316(19):2025-35.
Dr. Murphy is a clinical instructor at the University of Utah School of Medicine and an academic hospitalist at the University of Utah Hospital.
Eight things hospitalists need to know about post-acute care
Whether you’re a hospitalist who works only in a hospital, a hospitalist who works only in a post-acute care (PAC) setting, or a hospitalist who works in both types of facilities, knowing about current trends at PAC facilities and what the future may hold can help you excel in your current capacity and, ultimately, improve patient care.
The Hospitalist tapped experts in the post-acute space to tell us what they thought HM should know about working in PAC – which, in many ways, is quite different from the hospital setting. Here’s a compilation of their top eight must-knows.
1. PAC settings rely more on mid-level medical staff than hospitals do.
PAC facilities employ more mid-level providers, such as nurse practitioners and physician assistants, because they can support the level of medical complexity and decision making 95% of the time, says James D. Tollman, MD, FHM, president of Essex Inpatient Physicians in Boxford, Mass. Further, they are more heavily staffed by licensed practical nurses than are acute-care settings.

Usually, there is no physician or nurse practitioner presence at night. Clinicians rely on nursing staff’s assessment to make decisions regarding changes in patient status during off-hours, says Virginia Cummings, MD, director of long-term care, gerontology division, at Boston-based Beth Israel Deaconess Medical Center.

2. Testing takes longer, and options are limited.
Access to some acute urgent resources such as laboratory testing, imaging tests, and pharmacy products is more challenging at PAC facilities because most of these resources are not on-site. Consequently, there is a time lag between ordering tests and new medications and implementing these orders.
“If a patient needs something performed diagnostically immediately, they usually have to be transported to the emergency room or a facility with the necessary testing equipment,” Dr. Tollman says.

However, Paul T. Liistro, managing partner, Arbors of Hop Brook Limited Partnership in Manchester, Conn., and Vernon Manor Health Care Center in Vernon, Conn., and administrator, Manchester Manor Health Care Center, notes that it’s possible for a laboratory service or mobile diagnostic unit to provide laboratory testing or certain imaging at a PAC facility. More-involved diagnostics, such as an MRI or a PET scan, typically require testing at a remote location.

3. Patient populations mainly include rehab and terminally ill patients.
Patients are typically sent to a PAC facility either to recover from an illness or injury or because they are chronically ill and have exhausted treatment options. Regarding the latter, “They are mostly there for palliation; we don’t perform daily tests or prescribe aggressive medications on these patients,” Dr. Nazir says.
Dr. Cummings explains that PAC clinicians go through “the dying process with the patient.”
“They may or may not have assistance from hospice organizations,” she says, “and when they don’t, [hospitalists] take on the role of palliative-care providers.”
Dr. Cummings has seen an increase in psychiatric patients entering PAC facilities.
“Many patients with chronic psychological problems are aging, and there are fewer inpatient psychiatric beds available to those with concurrent medical and psychiatric problems,” she says. Much of this work is now being done in PAC settings. 
4. You can build a relationship with your patient.
Because the pace of a PAC facility is slower and a patient typically stays in a PAC facility longer than at a hospital, there’s time for a hospitalist to have more in-depth conversations with patients and their families.
“Building a deeper relationship with a patient may give the hospitalist an opportunity to discover the cause of an acute problem,” Dr. Nazir says. “They can go in-depth into the psychosocial aspect of medicine and may be able to find out what led to the initial problem and the real root cause, which can help prevent future recurrences, such as repeat falls or forgetting to take a medication.”
5. Using EHRs can improve transitions.
Care transitions between a hospital and PAC facility can be compromised by a lack of information sharing, and they can affect the quality and safety of patient care, says Dori Cross, a doctoral candidate in health services organization and policy at the University of Michigan School of Public Health in Ann Arbor. Handoffs between providers require information continuity – information that is complete, timely, and in a usable format – to ensure appropriate medical decisions and to provide high-quality care during and after transition.
Electronic health records (EHRs) as well as health information exchanges (HIEs) allow providers to communicate and share patient information. For example, hospitals can send information electronically to PAC facilities (“push” exchange) or make information available online securely for PAC providers to log in and access (“pull” exchange). According to a 2014 survey data by the American Hospital Association, more than 50% of hospitals report sending structured summary-of-care records electronically to long-term care settings; a little less than half of those hospitals (23% of the total sample of hospitals) were also receiving information electronically from long-term care sites.1
“This bidirectional exchange, in particular, can make it easier to share information across provider organizations electronically and, in turn, improve care delivery,” says Ms. Cross, who authored an accepted paper on the subject in the Journal of Post-Acute and Long-Term Medicine.
6. Hospitalists can work with providers in PAC settings to improve transitions.
Despite improvements in the electronic transfer of medical information, gaps still exist and can cause problems. One chasm when discharging patients to a PAC facility, is when a hospital IT system is incapable of communicating with the PAC facility system. In this instance, Dr. Nazir says, the hospitalist “can help bridge the gap.”
“[We] can verbally relay relevant information to physicians at PAC facilities so they understand the patient’s status, needs, and expectations,” he says. “Furthermore, hospitalists and a PAC facility’s administration can brainstorm methods to improve the systems of care so the patient receives more effective and timely care.”
7. Hospitalists switching to the PAC setting should have formal training.
The two main obstacles for hospitalists who change from working in a hospital to a PAC facility are the lack of exposure to PAC work in training and the assumption that it requires the same skills sets of a typical hospitalist, according to Manoj K. Mathew, MD, SFHM, national medical director of Los Angeles–based Agilon Health. The PAC setting has quite a number of differences compared with a hospital setting. For example, some regulations apply specifically to PAC facilities. In addition to formal training, hospitalists can benefit from using SHM’s Post-Acute Care Transitions Toolkit, having a mentor, or using resources from other organizations that function in this space such as The Society for Post-Acute and Long-Term Care Medicine, Dr. Nazir says.

8. A variety of payors and payment models are in play.
Commercial insurers continue to be major payors for PAC, especially for individuals younger than 65 years. Medicare and Medicaid, administered by the Centers for Medicare & Medicaid Services, are the primary payors for patients aged 65 years and older.

“These scorecards are using a variety of criteria to rank providers, such as length of stay, cost, readmissions to hospitals, and quality.”
Because Medicare Part A covers many patients discharged to a PAC setting, any changes in payment incentives or benefit structures by the Medicare program will drive changes in PAC.
“For example, as Medicare implements payment adjustments for hospitals that have high rates of readmissions, hospitals have a new incentive to work closely with SNFs and other providers of PAC to ensure patients can avoid unnecessary readmissions,” says Tiffany A. Radcliff, PhD, a health economist and associate professor in the department of health policy and management at Texas A&M University School of Public Health in College Station.
Providers must follow the billing rules for each payor. The rules for Medicare payments are outlined on CMS’ website. Bundled payments for PAC under the Medicare Part A program are scheduled to be implemented by 2018.
Reference
Cross DA, Adler-Milstein J. Investing in post-acute care transitions: electronic information exchange between hospitals and long-term care facilities [published online ahead of print Sept. 14, 2016]. JAMDA. doi: http://dx.doi.org/10.1016/j.jamda.2016.07.024.
Whether you’re a hospitalist who works only in a hospital, a hospitalist who works only in a post-acute care (PAC) setting, or a hospitalist who works in both types of facilities, knowing about current trends at PAC facilities and what the future may hold can help you excel in your current capacity and, ultimately, improve patient care.
The Hospitalist tapped experts in the post-acute space to tell us what they thought HM should know about working in PAC – which, in many ways, is quite different from the hospital setting. Here’s a compilation of their top eight must-knows.
1. PAC settings rely more on mid-level medical staff than hospitals do.
PAC facilities employ more mid-level providers, such as nurse practitioners and physician assistants, because they can support the level of medical complexity and decision making 95% of the time, says James D. Tollman, MD, FHM, president of Essex Inpatient Physicians in Boxford, Mass. Further, they are more heavily staffed by licensed practical nurses than are acute-care settings.
Usually, there is no physician or nurse practitioner presence at night. Clinicians rely on nursing staff’s assessment to make decisions regarding changes in patient status during off-hours, says Virginia Cummings, MD, director of long-term care, gerontology division, at Boston-based Beth Israel Deaconess Medical Center.
2. Testing takes longer, and options are limited.
Access to some acute urgent resources such as laboratory testing, imaging tests, and pharmacy products is more challenging at PAC facilities because most of these resources are not on-site. Consequently, there is a time lag between ordering tests and new medications and implementing these orders.
“If a patient needs something performed diagnostically immediately, they usually have to be transported to the emergency room or a facility with the necessary testing equipment,” Dr. Tollman says.
However, Paul T. Liistro, managing partner, Arbors of Hop Brook Limited Partnership in Manchester, Conn., and Vernon Manor Health Care Center in Vernon, Conn., and administrator, Manchester Manor Health Care Center, notes that it’s possible for a laboratory service or mobile diagnostic unit to provide laboratory testing or certain imaging at a PAC facility. More-involved diagnostics, such as an MRI or a PET scan, typically require testing at a remote location.
3. Patient populations mainly include rehab and terminally ill patients.
Patients are typically sent to a PAC facility either to recover from an illness or injury or because they are chronically ill and have exhausted treatment options. Regarding the latter, “They are mostly there for palliation; we don’t perform daily tests or prescribe aggressive medications on these patients,” Dr. Nazir says.
Dr. Cummings explains that PAC clinicians go through “the dying process with the patient.”
“They may or may not have assistance from hospice organizations,” she says, “and when they don’t, [hospitalists] take on the role of palliative-care providers.”
Dr. Cummings has seen an increase in psychiatric patients entering PAC facilities.
“Many patients with chronic psychological problems are aging, and there are fewer inpatient psychiatric beds available to those with concurrent medical and psychiatric problems,” she says. Much of this work is now being done in PAC settings.
4. You can build a relationship with your patient.
Because the pace of a PAC facility is slower and a patient typically stays in a PAC facility longer than at a hospital, there’s time for a hospitalist to have more in-depth conversations with patients and their families.
“Building a deeper relationship with a patient may give the hospitalist an opportunity to discover the cause of an acute problem,” Dr. Nazir says. “They can go in-depth into the psychosocial aspect of medicine and may be able to find out what led to the initial problem and the real root cause, which can help prevent future recurrences, such as repeat falls or forgetting to take a medication.”
5. Using EHRs can improve transitions.
Care transitions between a hospital and PAC facility can be compromised by a lack of information sharing, and they can affect the quality and safety of patient care, says Dori Cross, a doctoral candidate in health services organization and policy at the University of Michigan School of Public Health in Ann Arbor. Handoffs between providers require information continuity – information that is complete, timely, and in a usable format – to ensure appropriate medical decisions and to provide high-quality care during and after transition.
Electronic health records (EHRs) as well as health information exchanges (HIEs) allow providers to communicate and share patient information. For example, hospitals can send information electronically to PAC facilities (“push” exchange) or make information available online securely for PAC providers to log in and access (“pull” exchange). According to a 2014 survey data by the American Hospital Association, more than 50% of hospitals report sending structured summary-of-care records electronically to long-term care settings; a little less than half of those hospitals (23% of the total sample of hospitals) were also receiving information electronically from long-term care sites.1
“This bidirectional exchange, in particular, can make it easier to share information across provider organizations electronically and, in turn, improve care delivery,” says Ms. Cross, who authored an accepted paper on the subject in the Journal of Post-Acute and Long-Term Medicine.
6. Hospitalists can work with providers in PAC settings to improve transitions.
Despite improvements in the electronic transfer of medical information, gaps still exist and can cause problems. One chasm when discharging patients to a PAC facility, is when a hospital IT system is incapable of communicating with the PAC facility system. In this instance, Dr. Nazir says, the hospitalist “can help bridge the gap.”
“[We] can verbally relay relevant information to physicians at PAC facilities so they understand the patient’s status, needs, and expectations,” he says. “Furthermore, hospitalists and a PAC facility’s administration can brainstorm methods to improve the systems of care so the patient receives more effective and timely care.”
7. Hospitalists switching to the PAC setting should have formal training.
The two main obstacles for hospitalists who change from working in a hospital to a PAC facility are the lack of exposure to PAC work in training and the assumption that it requires the same skills sets of a typical hospitalist, according to Manoj K. Mathew, MD, SFHM, national medical director of Los Angeles–based Agilon Health. The PAC setting has quite a number of differences compared with a hospital setting. For example, some regulations apply specifically to PAC facilities. In addition to formal training, hospitalists can benefit from using SHM’s Post-Acute Care Transitions Toolkit, having a mentor, or using resources from other organizations that function in this space such as The Society for Post-Acute and Long-Term Care Medicine, Dr. Nazir says.
8. A variety of payors and payment models are in play.
Commercial insurers continue to be major payors for PAC, especially for individuals younger than 65 years. Medicare and Medicaid, administered by the Centers for Medicare & Medicaid Services, are the primary payors for patients aged 65 years and older.
“These scorecards are using a variety of criteria to rank providers, such as length of stay, cost, readmissions to hospitals, and quality.”
Because Medicare Part A covers many patients discharged to a PAC setting, any changes in payment incentives or benefit structures by the Medicare program will drive changes in PAC.
“For example, as Medicare implements payment adjustments for hospitals that have high rates of readmissions, hospitals have a new incentive to work closely with SNFs and other providers of PAC to ensure patients can avoid unnecessary readmissions,” says Tiffany A. Radcliff, PhD, a health economist and associate professor in the department of health policy and management at Texas A&M University School of Public Health in College Station.
Providers must follow the billing rules for each payor. The rules for Medicare payments are outlined on CMS’ website. Bundled payments for PAC under the Medicare Part A program are scheduled to be implemented by 2018.
Reference
Cross DA, Adler-Milstein J. Investing in post-acute care transitions: electronic information exchange between hospitals and long-term care facilities [published online ahead of print Sept. 14, 2016]. JAMDA. doi: http://dx.doi.org/10.1016/j.jamda.2016.07.024.
Whether you’re a hospitalist who works only in a hospital, a hospitalist who works only in a post-acute care (PAC) setting, or a hospitalist who works in both types of facilities, knowing about current trends at PAC facilities and what the future may hold can help you excel in your current capacity and, ultimately, improve patient care.
The Hospitalist tapped experts in the post-acute space to tell us what they thought HM should know about working in PAC – which, in many ways, is quite different from the hospital setting. Here’s a compilation of their top eight must-knows.
1. PAC settings rely more on mid-level medical staff than hospitals do.
PAC facilities employ more mid-level providers, such as nurse practitioners and physician assistants, because they can support the level of medical complexity and decision making 95% of the time, says James D. Tollman, MD, FHM, president of Essex Inpatient Physicians in Boxford, Mass. Further, they are more heavily staffed by licensed practical nurses than are acute-care settings.
Usually, there is no physician or nurse practitioner presence at night. Clinicians rely on nursing staff’s assessment to make decisions regarding changes in patient status during off-hours, says Virginia Cummings, MD, director of long-term care, gerontology division, at Boston-based Beth Israel Deaconess Medical Center.
2. Testing takes longer, and options are limited.
Access to some acute urgent resources such as laboratory testing, imaging tests, and pharmacy products is more challenging at PAC facilities because most of these resources are not on-site. Consequently, there is a time lag between ordering tests and new medications and implementing these orders.
“If a patient needs something performed diagnostically immediately, they usually have to be transported to the emergency room or a facility with the necessary testing equipment,” Dr. Tollman says.
However, Paul T. Liistro, managing partner, Arbors of Hop Brook Limited Partnership in Manchester, Conn., and Vernon Manor Health Care Center in Vernon, Conn., and administrator, Manchester Manor Health Care Center, notes that it’s possible for a laboratory service or mobile diagnostic unit to provide laboratory testing or certain imaging at a PAC facility. More-involved diagnostics, such as an MRI or a PET scan, typically require testing at a remote location.
3. Patient populations mainly include rehab and terminally ill patients.
Patients are typically sent to a PAC facility either to recover from an illness or injury or because they are chronically ill and have exhausted treatment options. Regarding the latter, “They are mostly there for palliation; we don’t perform daily tests or prescribe aggressive medications on these patients,” Dr. Nazir says.
Dr. Cummings explains that PAC clinicians go through “the dying process with the patient.”
“They may or may not have assistance from hospice organizations,” she says, “and when they don’t, [hospitalists] take on the role of palliative-care providers.”
Dr. Cummings has seen an increase in psychiatric patients entering PAC facilities.
“Many patients with chronic psychological problems are aging, and there are fewer inpatient psychiatric beds available to those with concurrent medical and psychiatric problems,” she says. Much of this work is now being done in PAC settings.
4. You can build a relationship with your patient.
Because the pace of a PAC facility is slower and a patient typically stays in a PAC facility longer than at a hospital, there’s time for a hospitalist to have more in-depth conversations with patients and their families.
“Building a deeper relationship with a patient may give the hospitalist an opportunity to discover the cause of an acute problem,” Dr. Nazir says. “They can go in-depth into the psychosocial aspect of medicine and may be able to find out what led to the initial problem and the real root cause, which can help prevent future recurrences, such as repeat falls or forgetting to take a medication.”
5. Using EHRs can improve transitions.
Care transitions between a hospital and PAC facility can be compromised by a lack of information sharing, and they can affect the quality and safety of patient care, says Dori Cross, a doctoral candidate in health services organization and policy at the University of Michigan School of Public Health in Ann Arbor. Handoffs between providers require information continuity – information that is complete, timely, and in a usable format – to ensure appropriate medical decisions and to provide high-quality care during and after transition.
Electronic health records (EHRs) as well as health information exchanges (HIEs) allow providers to communicate and share patient information. For example, hospitals can send information electronically to PAC facilities (“push” exchange) or make information available online securely for PAC providers to log in and access (“pull” exchange). According to a 2014 survey data by the American Hospital Association, more than 50% of hospitals report sending structured summary-of-care records electronically to long-term care settings; a little less than half of those hospitals (23% of the total sample of hospitals) were also receiving information electronically from long-term care sites.1
“This bidirectional exchange, in particular, can make it easier to share information across provider organizations electronically and, in turn, improve care delivery,” says Ms. Cross, who authored an accepted paper on the subject in the Journal of Post-Acute and Long-Term Medicine.
6. Hospitalists can work with providers in PAC settings to improve transitions.
Despite improvements in the electronic transfer of medical information, gaps still exist and can cause problems. One chasm when discharging patients to a PAC facility, is when a hospital IT system is incapable of communicating with the PAC facility system. In this instance, Dr. Nazir says, the hospitalist “can help bridge the gap.”
“[We] can verbally relay relevant information to physicians at PAC facilities so they understand the patient’s status, needs, and expectations,” he says. “Furthermore, hospitalists and a PAC facility’s administration can brainstorm methods to improve the systems of care so the patient receives more effective and timely care.”
7. Hospitalists switching to the PAC setting should have formal training.
The two main obstacles for hospitalists who change from working in a hospital to a PAC facility are the lack of exposure to PAC work in training and the assumption that it requires the same skills sets of a typical hospitalist, according to Manoj K. Mathew, MD, SFHM, national medical director of Los Angeles–based Agilon Health. The PAC setting has quite a number of differences compared with a hospital setting. For example, some regulations apply specifically to PAC facilities. In addition to formal training, hospitalists can benefit from using SHM’s Post-Acute Care Transitions Toolkit, having a mentor, or using resources from other organizations that function in this space such as The Society for Post-Acute and Long-Term Care Medicine, Dr. Nazir says.
8. A variety of payors and payment models are in play.
Commercial insurers continue to be major payors for PAC, especially for individuals younger than 65 years. Medicare and Medicaid, administered by the Centers for Medicare & Medicaid Services, are the primary payors for patients aged 65 years and older.
“These scorecards are using a variety of criteria to rank providers, such as length of stay, cost, readmissions to hospitals, and quality.”
Because Medicare Part A covers many patients discharged to a PAC setting, any changes in payment incentives or benefit structures by the Medicare program will drive changes in PAC.
“For example, as Medicare implements payment adjustments for hospitals that have high rates of readmissions, hospitals have a new incentive to work closely with SNFs and other providers of PAC to ensure patients can avoid unnecessary readmissions,” says Tiffany A. Radcliff, PhD, a health economist and associate professor in the department of health policy and management at Texas A&M University School of Public Health in College Station.
Providers must follow the billing rules for each payor. The rules for Medicare payments are outlined on CMS’ website. Bundled payments for PAC under the Medicare Part A program are scheduled to be implemented by 2018.
Reference
Cross DA, Adler-Milstein J. Investing in post-acute care transitions: electronic information exchange between hospitals and long-term care facilities [published online ahead of print Sept. 14, 2016]. JAMDA. doi: http://dx.doi.org/10.1016/j.jamda.2016.07.024.
VIDEO: Health law changes under new administration
WASHINGTON – A new president is taking office along with new staffers to lead the country’s top health care agencies.
In this video, Joyce Hall, chair of the American Bar Association Health Law Section, discusses what changes she foresees in health law issues under the new administration and what to expect from the leadership transition. Ms. Hall also speaks on potential alterations to the Medicare Access and CHIP Reauthorization Act of 2015 and whether the potential appointment of Rep. Tom Price (R-Ga.) as U.S. Department of Health and Human Services Secretary will be positive or negative for health care providers.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
[email protected]
On Twitter @legal_med
WASHINGTON – A new president is taking office along with new staffers to lead the country’s top health care agencies.
In this video, Joyce Hall, chair of the American Bar Association Health Law Section, discusses what changes she foresees in health law issues under the new administration and what to expect from the leadership transition. Ms. Hall also speaks on potential alterations to the Medicare Access and CHIP Reauthorization Act of 2015 and whether the potential appointment of Rep. Tom Price (R-Ga.) as U.S. Department of Health and Human Services Secretary will be positive or negative for health care providers.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
[email protected]
On Twitter @legal_med
WASHINGTON – A new president is taking office along with new staffers to lead the country’s top health care agencies.
In this video, Joyce Hall, chair of the American Bar Association Health Law Section, discusses what changes she foresees in health law issues under the new administration and what to expect from the leadership transition. Ms. Hall also speaks on potential alterations to the Medicare Access and CHIP Reauthorization Act of 2015 and whether the potential appointment of Rep. Tom Price (R-Ga.) as U.S. Department of Health and Human Services Secretary will be positive or negative for health care providers.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
[email protected]
On Twitter @legal_med
AT THE WASHINGTON HEALTH LAW SUMMIT
