User login
Idiopathic Eruptive Macular Pigmentation With Papillomatosis
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
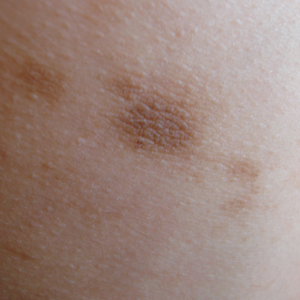
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).

.")
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
Practice Points
- Idiopathic eruptive macular pigmentation with papillomatosis is a rare disorder that most frequently affects children and young adults.
- Idiopathic eruptive macular pigmentation with papillomatosis is characterized by asymptomatic, brownish, hyperpigmented macules involving the neck, trunk, arms, and legs.
- The disorder is important to consider in the differential diagnosis of asymptomatic pigmentary disorders to avoid unnecessary treatment because the disease is self-limiting and resolves over weeks to years.
DOAC’s edge over warfarin fades with low adherence
BOSTON – The direct acting oral anticoagulants boost patient adherence compared with warfarin anticoagulation, but when patients did not adhere to their regimens, those prescribed a new, direct-acting oral anticoagulant had worse outcomes than did patients on warfarin – even patients poorly adherent with warfarin – based on data from more than 80,000 U.S. patients.
Among low-adherence patients, defined as those with adherence rates of 40%-80% based on prescriptions filled, patients with nonvalvular atrial fibrillation and a CHA2DS2-VASc score of at least 2 and treated with warfarin had a 3.37/100 patient-years rate of thromboembolic events–driven hospitalizations or emergency department visits, compared with a 4.05/100 patient-years rate among low-adherence patients receiving a direct-acting oral anticoagulant (DOAC), Dhanunjaya Lakkireddy, MD, said at the annual scientific sessions of the Heart Rhythm Society. The incidence of strokes of any kind was also lower in the low-adherence warfarin patients compared with the low-adherence DOAC patients, although the relationship flipped for hemorrhagic strokes and bleeds, which were more common in the warfarin patients.
In contrast, when patients were adherent, taking more than 80% of their prescribed drug, the performance of the DOACs generally surpassed that of warfarin. In an analysis adjusted for several demographic and clinical confounders, and when compared with patients adherent to a warfarin regimen, those adherent to a DOAC had a 7% lower rate of thromboembolic events, a 36% lower rate of hemorrhagic strokes, an 8% lower rate of any stroke, and a 10% lower rate of bleeds (excluding hemorrhagic strokes), all statistically significant differences, reported Dr. Lakkireddy, medical director of the Kansas City Heart Rhythm Institute in Overland Park, Kan.
The message from this analysis is the importance of maximizing patient adherence, Dr. Lakkireddy said.

“We should not make the false assumption that putting a patient on a DOAC will take care of everything. We need to make it a habit to make sure patients are taking their pills,” he said in an interview. “We were surprised. Our assumption was that the DOACs were more forgiving than warfarin” in poorly compliant patients. “We need to make talking about adherence with patients routine.”
The results also documented that adherence to therapy is better with a DOAC, with a 74% rate of good adherence among the nearly 41,000 patients in the database prescribed a DOAC compared with a 63% rate of good compliance among the more than 42,000 patients prescribed warfarin.
“It’s ironic that we might think low-adherence patients should go on warfarin. That’s sort of backwards,” said Andrew D. Krahn, MD, a cardiac electrophysiologist and professor of medicine at the University of British Columbia in Vancouver. “It’s not biologically plausible” to predict that less adherent patients would do better on warfarin, he noted.

The study run by Dr. Lakkireddy and his associates used data collected by IBM Watson Health Market Scan from about 4 million Medicare patients and 47 million American residents with private insurance during 2012-2016. They focused on the more than 600,000 patients prescribed an anticoagulant during 2014 and 2015, and then narrowed the study group down to just over 83,000 adults with nonvalvular atrial fibrillation, a CHA2DS2-VASc score of 2 or more. The patients’ average age was about 74 years.
Dr. Lakkireddy has been a consultant to or has received research support from Biosense Webster, Boehringer Ingelheim, Bristol Myers Squibb, EstechPharma, Janssen, Pfizer, SentreHeart, and St. Jude. Dr. Krahn has been a consultant to Medtronic and has received research support from Medtronic and Boston Scientific.
SOURCE: Lakkireddy D et al. Heart Rhythm 2018, Abstract B-LBCT02-03.
What is clear from this analysis is that adherence to oral anticoagulant regimens is something we need to address. It would help if we could determine why patients are not well adherent, but regardless of the cause, changing patient behavior and improving adherence will require better patient education and better integration of medical care toward better adherence.
This study has several obvious limitations, including its reliance on an administrative database that does not allow for adjudication of outcomes. The analysis presented so far is also limited by not breaking down the direct-acting oral anticoagulant into individual drugs, and without propensity-score matching of the two treatment subgroups.
We must be very alert to the possibility for confounding by indication in a real-world dataset like this. For example, the stroke rate we see in the patients treated with a DOAC may be affected by clinicians who preferentially prescribed one of the direct-acting drugs to patients who had what they thought was a high stroke risk because they felt these drugs might work better for stroke prevention than warfarin.

What is also notable in the results is that, even among the patients with good adherence, the event and adverse effects rates remained high. Among the highly adherent patients the thromboembolic event rate was greater than 3% per year in both the warfarin and direct-acting drug groups, the stroke rate was also greater than 3% per year in both subgroups, and bleeding events occurred at rates of greater than 4% per year among the patients treated with direct-acting drugs and greater than 5% per year in the warfarin group.
Hein Heidbuchel, MD, professor of medicine and chair of cardiology at the University of Antwerp, Belgium, made these comments as designated discussant for the study. He had no current disclosures.
What is clear from this analysis is that adherence to oral anticoagulant regimens is something we need to address. It would help if we could determine why patients are not well adherent, but regardless of the cause, changing patient behavior and improving adherence will require better patient education and better integration of medical care toward better adherence.
This study has several obvious limitations, including its reliance on an administrative database that does not allow for adjudication of outcomes. The analysis presented so far is also limited by not breaking down the direct-acting oral anticoagulant into individual drugs, and without propensity-score matching of the two treatment subgroups.
We must be very alert to the possibility for confounding by indication in a real-world dataset like this. For example, the stroke rate we see in the patients treated with a DOAC may be affected by clinicians who preferentially prescribed one of the direct-acting drugs to patients who had what they thought was a high stroke risk because they felt these drugs might work better for stroke prevention than warfarin.
What is also notable in the results is that, even among the patients with good adherence, the event and adverse effects rates remained high. Among the highly adherent patients the thromboembolic event rate was greater than 3% per year in both the warfarin and direct-acting drug groups, the stroke rate was also greater than 3% per year in both subgroups, and bleeding events occurred at rates of greater than 4% per year among the patients treated with direct-acting drugs and greater than 5% per year in the warfarin group.
Hein Heidbuchel, MD, professor of medicine and chair of cardiology at the University of Antwerp, Belgium, made these comments as designated discussant for the study. He had no current disclosures.
What is clear from this analysis is that adherence to oral anticoagulant regimens is something we need to address. It would help if we could determine why patients are not well adherent, but regardless of the cause, changing patient behavior and improving adherence will require better patient education and better integration of medical care toward better adherence.
This study has several obvious limitations, including its reliance on an administrative database that does not allow for adjudication of outcomes. The analysis presented so far is also limited by not breaking down the direct-acting oral anticoagulant into individual drugs, and without propensity-score matching of the two treatment subgroups.
We must be very alert to the possibility for confounding by indication in a real-world dataset like this. For example, the stroke rate we see in the patients treated with a DOAC may be affected by clinicians who preferentially prescribed one of the direct-acting drugs to patients who had what they thought was a high stroke risk because they felt these drugs might work better for stroke prevention than warfarin.
What is also notable in the results is that, even among the patients with good adherence, the event and adverse effects rates remained high. Among the highly adherent patients the thromboembolic event rate was greater than 3% per year in both the warfarin and direct-acting drug groups, the stroke rate was also greater than 3% per year in both subgroups, and bleeding events occurred at rates of greater than 4% per year among the patients treated with direct-acting drugs and greater than 5% per year in the warfarin group.
Hein Heidbuchel, MD, professor of medicine and chair of cardiology at the University of Antwerp, Belgium, made these comments as designated discussant for the study. He had no current disclosures.
BOSTON – The direct acting oral anticoagulants boost patient adherence compared with warfarin anticoagulation, but when patients did not adhere to their regimens, those prescribed a new, direct-acting oral anticoagulant had worse outcomes than did patients on warfarin – even patients poorly adherent with warfarin – based on data from more than 80,000 U.S. patients.
Among low-adherence patients, defined as those with adherence rates of 40%-80% based on prescriptions filled, patients with nonvalvular atrial fibrillation and a CHA2DS2-VASc score of at least 2 and treated with warfarin had a 3.37/100 patient-years rate of thromboembolic events–driven hospitalizations or emergency department visits, compared with a 4.05/100 patient-years rate among low-adherence patients receiving a direct-acting oral anticoagulant (DOAC), Dhanunjaya Lakkireddy, MD, said at the annual scientific sessions of the Heart Rhythm Society. The incidence of strokes of any kind was also lower in the low-adherence warfarin patients compared with the low-adherence DOAC patients, although the relationship flipped for hemorrhagic strokes and bleeds, which were more common in the warfarin patients.
In contrast, when patients were adherent, taking more than 80% of their prescribed drug, the performance of the DOACs generally surpassed that of warfarin. In an analysis adjusted for several demographic and clinical confounders, and when compared with patients adherent to a warfarin regimen, those adherent to a DOAC had a 7% lower rate of thromboembolic events, a 36% lower rate of hemorrhagic strokes, an 8% lower rate of any stroke, and a 10% lower rate of bleeds (excluding hemorrhagic strokes), all statistically significant differences, reported Dr. Lakkireddy, medical director of the Kansas City Heart Rhythm Institute in Overland Park, Kan.
The message from this analysis is the importance of maximizing patient adherence, Dr. Lakkireddy said.
“We should not make the false assumption that putting a patient on a DOAC will take care of everything. We need to make it a habit to make sure patients are taking their pills,” he said in an interview. “We were surprised. Our assumption was that the DOACs were more forgiving than warfarin” in poorly compliant patients. “We need to make talking about adherence with patients routine.”
The results also documented that adherence to therapy is better with a DOAC, with a 74% rate of good adherence among the nearly 41,000 patients in the database prescribed a DOAC compared with a 63% rate of good compliance among the more than 42,000 patients prescribed warfarin.
“It’s ironic that we might think low-adherence patients should go on warfarin. That’s sort of backwards,” said Andrew D. Krahn, MD, a cardiac electrophysiologist and professor of medicine at the University of British Columbia in Vancouver. “It’s not biologically plausible” to predict that less adherent patients would do better on warfarin, he noted.
The study run by Dr. Lakkireddy and his associates used data collected by IBM Watson Health Market Scan from about 4 million Medicare patients and 47 million American residents with private insurance during 2012-2016. They focused on the more than 600,000 patients prescribed an anticoagulant during 2014 and 2015, and then narrowed the study group down to just over 83,000 adults with nonvalvular atrial fibrillation, a CHA2DS2-VASc score of 2 or more. The patients’ average age was about 74 years.
Dr. Lakkireddy has been a consultant to or has received research support from Biosense Webster, Boehringer Ingelheim, Bristol Myers Squibb, EstechPharma, Janssen, Pfizer, SentreHeart, and St. Jude. Dr. Krahn has been a consultant to Medtronic and has received research support from Medtronic and Boston Scientific.
SOURCE: Lakkireddy D et al. Heart Rhythm 2018, Abstract B-LBCT02-03.
BOSTON – The direct acting oral anticoagulants boost patient adherence compared with warfarin anticoagulation, but when patients did not adhere to their regimens, those prescribed a new, direct-acting oral anticoagulant had worse outcomes than did patients on warfarin – even patients poorly adherent with warfarin – based on data from more than 80,000 U.S. patients.
Among low-adherence patients, defined as those with adherence rates of 40%-80% based on prescriptions filled, patients with nonvalvular atrial fibrillation and a CHA2DS2-VASc score of at least 2 and treated with warfarin had a 3.37/100 patient-years rate of thromboembolic events–driven hospitalizations or emergency department visits, compared with a 4.05/100 patient-years rate among low-adherence patients receiving a direct-acting oral anticoagulant (DOAC), Dhanunjaya Lakkireddy, MD, said at the annual scientific sessions of the Heart Rhythm Society. The incidence of strokes of any kind was also lower in the low-adherence warfarin patients compared with the low-adherence DOAC patients, although the relationship flipped for hemorrhagic strokes and bleeds, which were more common in the warfarin patients.
In contrast, when patients were adherent, taking more than 80% of their prescribed drug, the performance of the DOACs generally surpassed that of warfarin. In an analysis adjusted for several demographic and clinical confounders, and when compared with patients adherent to a warfarin regimen, those adherent to a DOAC had a 7% lower rate of thromboembolic events, a 36% lower rate of hemorrhagic strokes, an 8% lower rate of any stroke, and a 10% lower rate of bleeds (excluding hemorrhagic strokes), all statistically significant differences, reported Dr. Lakkireddy, medical director of the Kansas City Heart Rhythm Institute in Overland Park, Kan.
The message from this analysis is the importance of maximizing patient adherence, Dr. Lakkireddy said.
“We should not make the false assumption that putting a patient on a DOAC will take care of everything. We need to make it a habit to make sure patients are taking their pills,” he said in an interview. “We were surprised. Our assumption was that the DOACs were more forgiving than warfarin” in poorly compliant patients. “We need to make talking about adherence with patients routine.”
The results also documented that adherence to therapy is better with a DOAC, with a 74% rate of good adherence among the nearly 41,000 patients in the database prescribed a DOAC compared with a 63% rate of good compliance among the more than 42,000 patients prescribed warfarin.
“It’s ironic that we might think low-adherence patients should go on warfarin. That’s sort of backwards,” said Andrew D. Krahn, MD, a cardiac electrophysiologist and professor of medicine at the University of British Columbia in Vancouver. “It’s not biologically plausible” to predict that less adherent patients would do better on warfarin, he noted.
The study run by Dr. Lakkireddy and his associates used data collected by IBM Watson Health Market Scan from about 4 million Medicare patients and 47 million American residents with private insurance during 2012-2016. They focused on the more than 600,000 patients prescribed an anticoagulant during 2014 and 2015, and then narrowed the study group down to just over 83,000 adults with nonvalvular atrial fibrillation, a CHA2DS2-VASc score of 2 or more. The patients’ average age was about 74 years.
Dr. Lakkireddy has been a consultant to or has received research support from Biosense Webster, Boehringer Ingelheim, Bristol Myers Squibb, EstechPharma, Janssen, Pfizer, SentreHeart, and St. Jude. Dr. Krahn has been a consultant to Medtronic and has received research support from Medtronic and Boston Scientific.
SOURCE: Lakkireddy D et al. Heart Rhythm 2018, Abstract B-LBCT02-03.
REPORTING FROM HEART RHYTHM 2018
Key clinical point: Good adherence is needed for direct-acting oral anticoagulants to outperform warfarin.
Major finding: Thromboembolic events in low-adherence patients were 4.05/100 patient years with a DOAC and 3.37/100 patient years with warfarin.
Study details: Analysis of 83,168 insured U.S. atrial fibrillation patients treated with an oral anticoagulant in 2014-2015.
Disclosures: Dr. Lakkireddy has been a consultant to or has received research support from Biosense Webster, Boehringer Ingelheim, Bristol Myers Squibb, EstechPharma, Janssen, Pfizer, SentreHeart, and St. Jude. Dr. Krahn has been a consultant to Medtronic and has received research support from Medtronic and Boston Scientific.
Source: Lakkireddy D et al. Heart Rhythm 2018, Abstract B-LBCT02-03.
Four phase 3 studies highlighted at ASCO mark progress in GI cancers
CHICAGO – Findings from four recent, phase 3 gastrointestinal cancer studies mark a step forward toward “the answers we need” for patients with pancreatic, colorectal, or esophageal cancer, according to Andrew S. Epstein, MD.
In this video interview, Dr. Epstein summarizes and provides context for the findings, which were presented at the annual meeting of the American Society of Clinical Oncology and highlighted during a press briefing there. Dr. Epstein, an ASCO Expert and a medical oncologist at Memorial Sloan Kettering Cancer Center, New York, who was invited to discuss each of the studies at the briefing, said the UNICANCER-sponsored Prodige 7 trial addressed an important, long-unanswered question about the value of hyperthermic intraperitoneal chemotherapy (HIPEC) with surgery for colorectal peritoneal carcinomatosis.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
“This randomized study, very importantly, answered that longstanding question and showed us in a less-is-more type of way that the addition of the chemotherapy during surgery actually did not improve the overall survival of these patients,” he said, adding that, at 60 days, HIPEC actually had done more harm than good.
The findings are helpful, as HIPEC has been widely used without a solid data foundation, and now the use of an “additional toxic nonbeneficial treatment” can be avoided in a subset of patients.
Two studies regarding chemotherapy in patients with pancreatic cancer also provided important information about treatment. Preliminary data from one, the PREOPANC-1 trial, suggested that perioperative chemoradiotherapy significantly improves outcomes in resectable and borderline resectable patients, compared with immediate surgery; the other – the Prodige 24/CCTG PA.6 trial – demonstrated that adjuvant mFOLFIRINOX, a four-agent regimen, improved disease-free, metastasis-free, and overall survival, with treated patients living a median of 20 months longer and being cancer free for a median of 9 months longer than those who received gemcitabine therapy.
“We saw a very impressive, encouraging, statistically and clinically significant improvement,” he said regarding survival outcomes in Prodige 24. In patients with good performance status who can tolerate the regimen, mFOLFIRINOX “seems to be the way to go now,” he added, noting that patients receiving the regimen require close monitoring by a medical oncologist.
The fourth study, a prevention trial known as the ASPECT trial, showed that high-dose esomeprazole and low-dose aspirin taken for at least 7 years moderately reduces the risk of high-grade dysplasia and esophageal cancer, and may delay death from any cause in patients with Barrett’s esophagus.
“[It is] obviously of huge importance to be able to prevent a cancer before its onset. ... So with esophagus cancer, which also is a very difficult disease to treat in whatever stage it is, it would be a huge benefit to have a way in which to effectively prevent it,” Dr. Epstein said.
However, more information is needed about the actual benefits in terms of all-cause mortality and the contributors from aspirin versus the proton pump inhibitor versus both, he noted, adding that it is important for the public to know that the findings only apply to those with Barrett’s esophagus and shouldn’t be attempted with over-the-counter treatments as some treatments are associated with complications, and the proton pump inhibitor dose used in this study is not available over the counter.
“So I think it is an intriguing study which needs more clarity and more follow-up, as the author himself said,” he added.
In summing up the findings presented at the briefing, Dr. Epstein said that “collectively we see that the challenge of cancer remains significant and we need high-quality studies like the ones presented today in order to best present ...what the best therapies are for [patients].
“With good sound science like this we continue to inch closer to the answers we need,” he concluded.
Dr. Epstein reported having no disclosures.
CHICAGO – Findings from four recent, phase 3 gastrointestinal cancer studies mark a step forward toward “the answers we need” for patients with pancreatic, colorectal, or esophageal cancer, according to Andrew S. Epstein, MD.
In this video interview, Dr. Epstein summarizes and provides context for the findings, which were presented at the annual meeting of the American Society of Clinical Oncology and highlighted during a press briefing there. Dr. Epstein, an ASCO Expert and a medical oncologist at Memorial Sloan Kettering Cancer Center, New York, who was invited to discuss each of the studies at the briefing, said the UNICANCER-sponsored Prodige 7 trial addressed an important, long-unanswered question about the value of hyperthermic intraperitoneal chemotherapy (HIPEC) with surgery for colorectal peritoneal carcinomatosis.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
“This randomized study, very importantly, answered that longstanding question and showed us in a less-is-more type of way that the addition of the chemotherapy during surgery actually did not improve the overall survival of these patients,” he said, adding that, at 60 days, HIPEC actually had done more harm than good.
The findings are helpful, as HIPEC has been widely used without a solid data foundation, and now the use of an “additional toxic nonbeneficial treatment” can be avoided in a subset of patients.
Two studies regarding chemotherapy in patients with pancreatic cancer also provided important information about treatment. Preliminary data from one, the PREOPANC-1 trial, suggested that perioperative chemoradiotherapy significantly improves outcomes in resectable and borderline resectable patients, compared with immediate surgery; the other – the Prodige 24/CCTG PA.6 trial – demonstrated that adjuvant mFOLFIRINOX, a four-agent regimen, improved disease-free, metastasis-free, and overall survival, with treated patients living a median of 20 months longer and being cancer free for a median of 9 months longer than those who received gemcitabine therapy.
“We saw a very impressive, encouraging, statistically and clinically significant improvement,” he said regarding survival outcomes in Prodige 24. In patients with good performance status who can tolerate the regimen, mFOLFIRINOX “seems to be the way to go now,” he added, noting that patients receiving the regimen require close monitoring by a medical oncologist.
The fourth study, a prevention trial known as the ASPECT trial, showed that high-dose esomeprazole and low-dose aspirin taken for at least 7 years moderately reduces the risk of high-grade dysplasia and esophageal cancer, and may delay death from any cause in patients with Barrett’s esophagus.
“[It is] obviously of huge importance to be able to prevent a cancer before its onset. ... So with esophagus cancer, which also is a very difficult disease to treat in whatever stage it is, it would be a huge benefit to have a way in which to effectively prevent it,” Dr. Epstein said.
However, more information is needed about the actual benefits in terms of all-cause mortality and the contributors from aspirin versus the proton pump inhibitor versus both, he noted, adding that it is important for the public to know that the findings only apply to those with Barrett’s esophagus and shouldn’t be attempted with over-the-counter treatments as some treatments are associated with complications, and the proton pump inhibitor dose used in this study is not available over the counter.
“So I think it is an intriguing study which needs more clarity and more follow-up, as the author himself said,” he added.
In summing up the findings presented at the briefing, Dr. Epstein said that “collectively we see that the challenge of cancer remains significant and we need high-quality studies like the ones presented today in order to best present ...what the best therapies are for [patients].
“With good sound science like this we continue to inch closer to the answers we need,” he concluded.
Dr. Epstein reported having no disclosures.
CHICAGO – Findings from four recent, phase 3 gastrointestinal cancer studies mark a step forward toward “the answers we need” for patients with pancreatic, colorectal, or esophageal cancer, according to Andrew S. Epstein, MD.
In this video interview, Dr. Epstein summarizes and provides context for the findings, which were presented at the annual meeting of the American Society of Clinical Oncology and highlighted during a press briefing there. Dr. Epstein, an ASCO Expert and a medical oncologist at Memorial Sloan Kettering Cancer Center, New York, who was invited to discuss each of the studies at the briefing, said the UNICANCER-sponsored Prodige 7 trial addressed an important, long-unanswered question about the value of hyperthermic intraperitoneal chemotherapy (HIPEC) with surgery for colorectal peritoneal carcinomatosis.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
“This randomized study, very importantly, answered that longstanding question and showed us in a less-is-more type of way that the addition of the chemotherapy during surgery actually did not improve the overall survival of these patients,” he said, adding that, at 60 days, HIPEC actually had done more harm than good.
The findings are helpful, as HIPEC has been widely used without a solid data foundation, and now the use of an “additional toxic nonbeneficial treatment” can be avoided in a subset of patients.
Two studies regarding chemotherapy in patients with pancreatic cancer also provided important information about treatment. Preliminary data from one, the PREOPANC-1 trial, suggested that perioperative chemoradiotherapy significantly improves outcomes in resectable and borderline resectable patients, compared with immediate surgery; the other – the Prodige 24/CCTG PA.6 trial – demonstrated that adjuvant mFOLFIRINOX, a four-agent regimen, improved disease-free, metastasis-free, and overall survival, with treated patients living a median of 20 months longer and being cancer free for a median of 9 months longer than those who received gemcitabine therapy.
“We saw a very impressive, encouraging, statistically and clinically significant improvement,” he said regarding survival outcomes in Prodige 24. In patients with good performance status who can tolerate the regimen, mFOLFIRINOX “seems to be the way to go now,” he added, noting that patients receiving the regimen require close monitoring by a medical oncologist.
The fourth study, a prevention trial known as the ASPECT trial, showed that high-dose esomeprazole and low-dose aspirin taken for at least 7 years moderately reduces the risk of high-grade dysplasia and esophageal cancer, and may delay death from any cause in patients with Barrett’s esophagus.
“[It is] obviously of huge importance to be able to prevent a cancer before its onset. ... So with esophagus cancer, which also is a very difficult disease to treat in whatever stage it is, it would be a huge benefit to have a way in which to effectively prevent it,” Dr. Epstein said.
However, more information is needed about the actual benefits in terms of all-cause mortality and the contributors from aspirin versus the proton pump inhibitor versus both, he noted, adding that it is important for the public to know that the findings only apply to those with Barrett’s esophagus and shouldn’t be attempted with over-the-counter treatments as some treatments are associated with complications, and the proton pump inhibitor dose used in this study is not available over the counter.
“So I think it is an intriguing study which needs more clarity and more follow-up, as the author himself said,” he added.
In summing up the findings presented at the briefing, Dr. Epstein said that “collectively we see that the challenge of cancer remains significant and we need high-quality studies like the ones presented today in order to best present ...what the best therapies are for [patients].
“With good sound science like this we continue to inch closer to the answers we need,” he concluded.
Dr. Epstein reported having no disclosures.
EXPERT ANALYSIS FROM ASCO 2018
Vaccine nonmedical exemptions creating metro ‘hotspots’
Recent increases in nonmedical exemptions (NMEs) to vaccination have created metropolitan “hotspots” with large numbers of unvaccinated children, according to a report published June 12 in PLoS Medicine.
although rates seem to have plateaued in some states since 2014. As a result of those increases, there were, during the 2016-2017 school year, 15 metro areas with kindergarten NME populations over 400, reported Jacqueline K. Olive, and her associates at Baylor College of Medicine. Their report was based on data from state health departments and the Centers for Disease Control and Prevention.
Leading the way was Maricopa County, Ariz., home of Phoenix and 2,947 unvaccinated kindergartners, which was more than triple the number in county/city No. 2, Salt Lake County/Salt Lake City (NME total: 956). Close behind in third was King County, Wash. (Seattle) at 940, followed by Multnomah County, Ore. (Portland) at 711 and Oakland County, Mich. (Troy) at 686, the investigators said.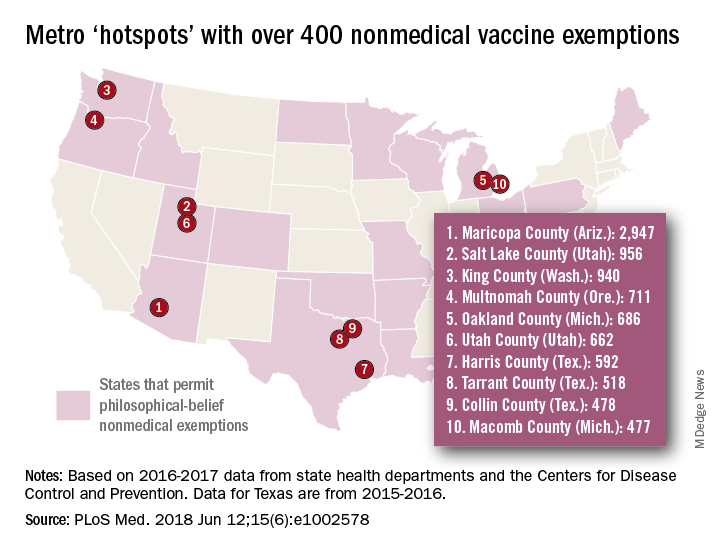
[There was only room for 10 in the map, so here are hotspots 11-15: Wayne County, Mich. (Detroit); Allegheny County, Pa. (Pittsburgh); Travis County, Tex. (Austin); Jackson County, Mo. (Kansas City); and Spokane County, Wash. (Spokane).]
In addition to the large-population hotspots, there are also a number of mainly rural counties with smaller populations but high NME rates. Eight of the 10 highest such rates can be found in Idaho, and at the top of that list is Camas County, which had an NME rate of 27% in 2016-2017, the researchers reported.
Analysis of the relationship between NMEs and MMR vaccination showed that “states with more NME students exhibited lower MMR vaccination rates. In contrast, states that have banned NMEs – Mississippi, California, and West Virginia – exhibit the highest MMR vaccine uptake and lowest incidence of vaccine preventable diseases,” the investigators wrote.
Ms. Olive and her associates said that there was no specific funding for the study and that no conflicts of interest existed.
SOURCE: Olive JK et al. PLoS Med. 2018 Jun 12;15(6): e1002578. doi: 10.1371/journal.pmed.1002578.
Recent increases in nonmedical exemptions (NMEs) to vaccination have created metropolitan “hotspots” with large numbers of unvaccinated children, according to a report published June 12 in PLoS Medicine.
although rates seem to have plateaued in some states since 2014. As a result of those increases, there were, during the 2016-2017 school year, 15 metro areas with kindergarten NME populations over 400, reported Jacqueline K. Olive, and her associates at Baylor College of Medicine. Their report was based on data from state health departments and the Centers for Disease Control and Prevention.
Leading the way was Maricopa County, Ariz., home of Phoenix and 2,947 unvaccinated kindergartners, which was more than triple the number in county/city No. 2, Salt Lake County/Salt Lake City (NME total: 956). Close behind in third was King County, Wash. (Seattle) at 940, followed by Multnomah County, Ore. (Portland) at 711 and Oakland County, Mich. (Troy) at 686, the investigators said.
[There was only room for 10 in the map, so here are hotspots 11-15: Wayne County, Mich. (Detroit); Allegheny County, Pa. (Pittsburgh); Travis County, Tex. (Austin); Jackson County, Mo. (Kansas City); and Spokane County, Wash. (Spokane).]
In addition to the large-population hotspots, there are also a number of mainly rural counties with smaller populations but high NME rates. Eight of the 10 highest such rates can be found in Idaho, and at the top of that list is Camas County, which had an NME rate of 27% in 2016-2017, the researchers reported.
Analysis of the relationship between NMEs and MMR vaccination showed that “states with more NME students exhibited lower MMR vaccination rates. In contrast, states that have banned NMEs – Mississippi, California, and West Virginia – exhibit the highest MMR vaccine uptake and lowest incidence of vaccine preventable diseases,” the investigators wrote.
Ms. Olive and her associates said that there was no specific funding for the study and that no conflicts of interest existed.
SOURCE: Olive JK et al. PLoS Med. 2018 Jun 12;15(6): e1002578. doi: 10.1371/journal.pmed.1002578.
Recent increases in nonmedical exemptions (NMEs) to vaccination have created metropolitan “hotspots” with large numbers of unvaccinated children, according to a report published June 12 in PLoS Medicine.
although rates seem to have plateaued in some states since 2014. As a result of those increases, there were, during the 2016-2017 school year, 15 metro areas with kindergarten NME populations over 400, reported Jacqueline K. Olive, and her associates at Baylor College of Medicine. Their report was based on data from state health departments and the Centers for Disease Control and Prevention.
Leading the way was Maricopa County, Ariz., home of Phoenix and 2,947 unvaccinated kindergartners, which was more than triple the number in county/city No. 2, Salt Lake County/Salt Lake City (NME total: 956). Close behind in third was King County, Wash. (Seattle) at 940, followed by Multnomah County, Ore. (Portland) at 711 and Oakland County, Mich. (Troy) at 686, the investigators said.
[There was only room for 10 in the map, so here are hotspots 11-15: Wayne County, Mich. (Detroit); Allegheny County, Pa. (Pittsburgh); Travis County, Tex. (Austin); Jackson County, Mo. (Kansas City); and Spokane County, Wash. (Spokane).]
In addition to the large-population hotspots, there are also a number of mainly rural counties with smaller populations but high NME rates. Eight of the 10 highest such rates can be found in Idaho, and at the top of that list is Camas County, which had an NME rate of 27% in 2016-2017, the researchers reported.
Analysis of the relationship between NMEs and MMR vaccination showed that “states with more NME students exhibited lower MMR vaccination rates. In contrast, states that have banned NMEs – Mississippi, California, and West Virginia – exhibit the highest MMR vaccine uptake and lowest incidence of vaccine preventable diseases,” the investigators wrote.
Ms. Olive and her associates said that there was no specific funding for the study and that no conflicts of interest existed.
SOURCE: Olive JK et al. PLoS Med. 2018 Jun 12;15(6): e1002578. doi: 10.1371/journal.pmed.1002578.
FROM PLOS MEDICINE
Cognitive Behavioral Therapy for Pediatric Migraine
Mind and body cognitive behavioral therapy (CBT‐HA) relaxation skills emerged as popular and effective for pediatric migraine sufferers, based on patient and parent reports in a recent study. Qualitative interviews were conducted with 10 patients and 9 of their parents who had undergone CBT‐HA. Interviews were analyzed using an inductive thematic analysis approach based upon modified grounded theory. Patients were ranged in age from 13 to 17.5 years (median=15.4, standard deviation=1.63) and had undergone CBT‐HA about 1 to 2 years prior to participating in the study. Researchers found:
- Overall, patients and their parents reported that CBT‐HA was helpful in reducing headache frequency and related disability.
- Although patients provided mixed reports on the effectiveness of different CBT‐HA skills, the majority of patients indicated that the mind and body relaxation skills of CBT‐HA (deep breathing, progressive muscle relaxation, and activity pacing in particular) were the most helpful and most frequently used skills.
- Patients and parents also generally reported that treatment was easy to learn, and noted at least some aspect of treatment was enjoyable.
CBT for pediatric migraine: A qualitative study of patient and parent experience. Headache. 2018;58(5):661-675. doi:10.1111/head.13285.
Mind and body cognitive behavioral therapy (CBT‐HA) relaxation skills emerged as popular and effective for pediatric migraine sufferers, based on patient and parent reports in a recent study. Qualitative interviews were conducted with 10 patients and 9 of their parents who had undergone CBT‐HA. Interviews were analyzed using an inductive thematic analysis approach based upon modified grounded theory. Patients were ranged in age from 13 to 17.5 years (median=15.4, standard deviation=1.63) and had undergone CBT‐HA about 1 to 2 years prior to participating in the study. Researchers found:
- Overall, patients and their parents reported that CBT‐HA was helpful in reducing headache frequency and related disability.
- Although patients provided mixed reports on the effectiveness of different CBT‐HA skills, the majority of patients indicated that the mind and body relaxation skills of CBT‐HA (deep breathing, progressive muscle relaxation, and activity pacing in particular) were the most helpful and most frequently used skills.
- Patients and parents also generally reported that treatment was easy to learn, and noted at least some aspect of treatment was enjoyable.
CBT for pediatric migraine: A qualitative study of patient and parent experience. Headache. 2018;58(5):661-675. doi:10.1111/head.13285.
Mind and body cognitive behavioral therapy (CBT‐HA) relaxation skills emerged as popular and effective for pediatric migraine sufferers, based on patient and parent reports in a recent study. Qualitative interviews were conducted with 10 patients and 9 of their parents who had undergone CBT‐HA. Interviews were analyzed using an inductive thematic analysis approach based upon modified grounded theory. Patients were ranged in age from 13 to 17.5 years (median=15.4, standard deviation=1.63) and had undergone CBT‐HA about 1 to 2 years prior to participating in the study. Researchers found:
- Overall, patients and their parents reported that CBT‐HA was helpful in reducing headache frequency and related disability.
- Although patients provided mixed reports on the effectiveness of different CBT‐HA skills, the majority of patients indicated that the mind and body relaxation skills of CBT‐HA (deep breathing, progressive muscle relaxation, and activity pacing in particular) were the most helpful and most frequently used skills.
- Patients and parents also generally reported that treatment was easy to learn, and noted at least some aspect of treatment was enjoyable.
CBT for pediatric migraine: A qualitative study of patient and parent experience. Headache. 2018;58(5):661-675. doi:10.1111/head.13285.
Migraineurs Have Reduced Visual Quality of Life
Visual quality of life (QOL) is significantly adversely affected in migraine sufferers, according to a recent study. In fact, patients with chronic migraine may have visual QOL impacts that are as significant as those associated with other common neuro‐ophthalmic disorders. In this cross‐sectional quantitative survey, visual QOL in individuals with chronic and episodic migraine was assessed using the National Eye Institute Visual Function Questionnaire‐25, and the 10‐item National Eye Institute Visual Function Questionnaire‐25 Neuro‐Ophthalmic Supplement. Overall headache severity and impact was assessed using the Migraine‐specific Quality of Life Questionnaire and the Headache Impact Test‐6. Researchers found:
- Among 29 participants with chronic migraine, vision‐specific QOL scores were all statistically significantly decreased compared to disease‐free controls.
- Among 37 participants with episodic migraine, vision‐specific QOL scores were also decreased compared to disease‐free controls.
- Chronic migraineurs had decreased visual QOL scores compared to those with episodic migraines.
- Participants with chronic migraine had visual QOL scores that were as poor as those previously published for patients with other neuro‐ophthalmic disorders, such as multiple sclerosis, myasthenia gravis, and ischemic optic neuropathy.
Patients with migraine have substantial reductions in measures of visual quality of life. [Published online ahead of print June 7, 2018]. Headache. doi:10.1111/head.13330.
Visual quality of life (QOL) is significantly adversely affected in migraine sufferers, according to a recent study. In fact, patients with chronic migraine may have visual QOL impacts that are as significant as those associated with other common neuro‐ophthalmic disorders. In this cross‐sectional quantitative survey, visual QOL in individuals with chronic and episodic migraine was assessed using the National Eye Institute Visual Function Questionnaire‐25, and the 10‐item National Eye Institute Visual Function Questionnaire‐25 Neuro‐Ophthalmic Supplement. Overall headache severity and impact was assessed using the Migraine‐specific Quality of Life Questionnaire and the Headache Impact Test‐6. Researchers found:
- Among 29 participants with chronic migraine, vision‐specific QOL scores were all statistically significantly decreased compared to disease‐free controls.
- Among 37 participants with episodic migraine, vision‐specific QOL scores were also decreased compared to disease‐free controls.
- Chronic migraineurs had decreased visual QOL scores compared to those with episodic migraines.
- Participants with chronic migraine had visual QOL scores that were as poor as those previously published for patients with other neuro‐ophthalmic disorders, such as multiple sclerosis, myasthenia gravis, and ischemic optic neuropathy.
Patients with migraine have substantial reductions in measures of visual quality of life. [Published online ahead of print June 7, 2018]. Headache. doi:10.1111/head.13330.
Visual quality of life (QOL) is significantly adversely affected in migraine sufferers, according to a recent study. In fact, patients with chronic migraine may have visual QOL impacts that are as significant as those associated with other common neuro‐ophthalmic disorders. In this cross‐sectional quantitative survey, visual QOL in individuals with chronic and episodic migraine was assessed using the National Eye Institute Visual Function Questionnaire‐25, and the 10‐item National Eye Institute Visual Function Questionnaire‐25 Neuro‐Ophthalmic Supplement. Overall headache severity and impact was assessed using the Migraine‐specific Quality of Life Questionnaire and the Headache Impact Test‐6. Researchers found:
- Among 29 participants with chronic migraine, vision‐specific QOL scores were all statistically significantly decreased compared to disease‐free controls.
- Among 37 participants with episodic migraine, vision‐specific QOL scores were also decreased compared to disease‐free controls.
- Chronic migraineurs had decreased visual QOL scores compared to those with episodic migraines.
- Participants with chronic migraine had visual QOL scores that were as poor as those previously published for patients with other neuro‐ophthalmic disorders, such as multiple sclerosis, myasthenia gravis, and ischemic optic neuropathy.
Patients with migraine have substantial reductions in measures of visual quality of life. [Published online ahead of print June 7, 2018]. Headache. doi:10.1111/head.13330.
Migraineurs’ Initiation of Behavioral Treatment
Less than one-third of eligible migraineurs were referred for behavioral treatment and only about half initiated behavioral migraine treatment in a recent prospective cohort study. Researchers compared patients who initiated behavioral migraine treatment following a provider recommendation with those who did not (demographics, migraine characteristics, and locus of control) with analysis of variance and chi-square tests. They found:
- Of the 234 eligible patients, 69 (29.5%) were referred for behavioral treatment.
- 53 (76.8%) patients referred for behavioral treatment were reached by phone.
- The mean duration from time of referral to follow-up was 76 (median 76, SD=45) days.
- 30 (56.6%) patients initiated behavioral migraine treatment.
- There was no difference in initiation of behavioral migraine treatment with regard to sex, age, age of diagnosis, years suffered with headaches, health care utilization visits, Migraine Disability Assessment Screen, and locus of control.
- Patients who had previously seen a psychologist for migraine were more likely to initiate behavioral migraine treatment than patients who had not.
- Time constraints were the most common barrier cited for not initiating behavioral migraine treatment.
Factors related to migraine patients’ decisions to initiate behavioral migraine treatment following a headache specialist’s recommendation: A prospective observational study. [Published online ahead of print June 5, 2018]. Pain Medicine. doi:10.1093/pm/pny028.
Less than one-third of eligible migraineurs were referred for behavioral treatment and only about half initiated behavioral migraine treatment in a recent prospective cohort study. Researchers compared patients who initiated behavioral migraine treatment following a provider recommendation with those who did not (demographics, migraine characteristics, and locus of control) with analysis of variance and chi-square tests. They found:
- Of the 234 eligible patients, 69 (29.5%) were referred for behavioral treatment.
- 53 (76.8%) patients referred for behavioral treatment were reached by phone.
- The mean duration from time of referral to follow-up was 76 (median 76, SD=45) days.
- 30 (56.6%) patients initiated behavioral migraine treatment.
- There was no difference in initiation of behavioral migraine treatment with regard to sex, age, age of diagnosis, years suffered with headaches, health care utilization visits, Migraine Disability Assessment Screen, and locus of control.
- Patients who had previously seen a psychologist for migraine were more likely to initiate behavioral migraine treatment than patients who had not.
- Time constraints were the most common barrier cited for not initiating behavioral migraine treatment.
Factors related to migraine patients’ decisions to initiate behavioral migraine treatment following a headache specialist’s recommendation: A prospective observational study. [Published online ahead of print June 5, 2018]. Pain Medicine. doi:10.1093/pm/pny028.
Less than one-third of eligible migraineurs were referred for behavioral treatment and only about half initiated behavioral migraine treatment in a recent prospective cohort study. Researchers compared patients who initiated behavioral migraine treatment following a provider recommendation with those who did not (demographics, migraine characteristics, and locus of control) with analysis of variance and chi-square tests. They found:
- Of the 234 eligible patients, 69 (29.5%) were referred for behavioral treatment.
- 53 (76.8%) patients referred for behavioral treatment were reached by phone.
- The mean duration from time of referral to follow-up was 76 (median 76, SD=45) days.
- 30 (56.6%) patients initiated behavioral migraine treatment.
- There was no difference in initiation of behavioral migraine treatment with regard to sex, age, age of diagnosis, years suffered with headaches, health care utilization visits, Migraine Disability Assessment Screen, and locus of control.
- Patients who had previously seen a psychologist for migraine were more likely to initiate behavioral migraine treatment than patients who had not.
- Time constraints were the most common barrier cited for not initiating behavioral migraine treatment.
Factors related to migraine patients’ decisions to initiate behavioral migraine treatment following a headache specialist’s recommendation: A prospective observational study. [Published online ahead of print June 5, 2018]. Pain Medicine. doi:10.1093/pm/pny028.
Dismantling the sports-betting ban: A mental health gamble
The Supreme Court decision to overturn the federal law that prohibited state-sanctioned college and professional sports betting is bad news for clinicians who treat patients with addictions.
On May 14, the high court ruled 7-2 that the 1992 law, called the Professional and Amateur Sports Protection Act (PASPA), was unconstitutional. Now every state is free to operate, sponsor, promote, license, advertise, or authorize gambling for any college or professional sport–based event.
Optimistic outlooks on the death of PASPA include the foreseen opportunity by the states to tax and generate revenue on such gambling. Proponents of the ruling also argue that illegal activity that thrived on sports betting will now end.
But to what extent will either of those scenarios benefit the public?
If passage of various state marijuana laws is any example, assumptions that legal avenues will usurp illegal enterprises are flawed. Also, taxation likely will generate a large sum of revenue for each state. But those revenues might be offset by subsequent changes that will be needed in mental health, addiction, and wellness programs – a difficult proposition given the opioid epidemic already overburdening the country. Remember the tobacco cases and promise of state support for education, treatment, and other noble activities? Addiction medicine specialists worry that taxes collected by the states, and promises to prevent and treat gambling problems – and prevent addiction – will not end up in those coffers.
As clinicians, perhaps our most important contribution to the debates on this ruling lies in raising awareness of pathological gambling as an addiction disorder.
Redefining the act of gambling
Breaking down previous barriers to access and increasing convenience to gambling undoubtedly will be associated with increased pathological engagement in gambling. This conclusion is clear, based on past national experiments with substances of addiction (such as alcohol prohibition).
Since the cocaine epidemic of the 1980s, and our increased understanding that addictions need not have prominent withdrawal syndromes, we have focused on addiction as a fatal attraction. Psychiatrists and other clinicians made the case – in some quarters, at least – for sugar, sex, and Internet compulsivity as addictions. Compared with those addictions, the evidence was clearest and most compelling for pathological gambling as an addiction disorder. Indeed, gambling disorder was introduced in 2013 to the DSM-5 as the very first non–substance-based addictive disorder. This was a decisive change, as it recognizes that gambling is more than an environmental hazard for those suffering from dopamine-driven obsessive-compulsive-like dysfunction (the DSM section where it had lived previously). Instead, gambling acts as an agent that can initiate a usurpation of the brain’s reward circuitry. (In addition, this change has reopened the door for other increasingly recognized non–substance-based disorder categories such as video game and pornography addiction, and others.)
Gambling disorder certainly fits well into what many experts view as the essential phenotype of any addiction: Continued use despite harm, waning self-control over engagement, a craving state, and compulsive use. Current research is expanding rapidly and filling in the theoretical framework, strongly supporting gambling disorder based on biological evidence. Much of what we now know about the biology of addiction has been through the efforts of the Yale University–based research group, led by psychiatrist Marc N. Potenza. Dr. Potenza and his colleagues have been investigating gambling disorder in a thorough manner (Harv Rev Psychiatry. 2015 Mar-Apr;23[2]:134-46) and (Curr Treat Options Psychiatry. 2014 Jun 1;1[2]:189-203). Indeed, gambling disorder is much like the other substance-use disorders in which it is grouped, in that it has been found to share some similarities/pathways common to all addictions while also carrying its own specific nuances.
Twin studies have unearthed a wealth of information, such as knowledge that environmental factors seem to be the predominant source of the comorbid development of gambling disorder with the more socially acceptable substances as associated use disorders (alcohol, tobacco, and marijuana) through mechanisms such as peer association and place preference conditioning. Similarly, genetic influences also might be meaningful to treatment. For example, one finding showed that patients with gambling disorder and a family history of alcoholism were found to more preferentially respond to opioid-receptor antagonists as treatment for gambling disorder, compared with individuals without such family history (Psychopharmacology [Berl]. 2008;200[4]:521-7).
Explorations of neurotransmitter involvement and brain connectivity also have been conducted for gambling behaviors. Dopaminergic underpinnings of addiction have been particularly indicated in imaging studies focused on the ventral striatum and other components of reward circuitry. In addition, functional MRI studies have identified both overlapping and discordant brain imaging findings between gambling and many other substance use disorders such as cocaine. All these indicate that gambling seems, like its use-disorder counterparts, to follow a similar but distinct course of hijacking reward systems and priming the brain to seek out further gambling in a pathological manner.
Vulnerable populations
Another key finding of recent research exploring the biological foundations of gambling disorder is gender dimorphism. In numerous studies, women have been found to experience a “telescoping effect” from gambling, compared with their male counterparts, where they seem to more quickly advance from first exposure to problematic use. This phenomenon also is seen in women who use cocaine. Also, functional MRI studies also have found that women appear to have alternative signal changes in regions germane to addiction, compared with their male counterparts. One such example was greater activity in the hippocampus and middle temporal gyrus in women, suggestive of stronger activation of regions key for memory retrieval used in craving/urge-related emotions. These data highlight the need for not only understanding how gambling and other addictions diverge between men and women, but also for how prevention and treatment of these disorders might differ based on sex.
Adolescents also get special consideration: How will they be affected by this expected growth in gambling avenues? Adolescence and young adulthood are periods of development defined by increased impulsivity and risk taking, making this population particularly vulnerable to addiction that can then persist into adulthood. It is expected that age laws will persist and prevent the legal access adults might enjoy, but shifts in opinions of harm and ease of access are likely to contribute to increased gambling exposure. To use another addictive phenomena as an example, data from the Substance Abuse and Mental Health Services Administration show a clear correlation between marijuana use, marijuana legal status, and perceptions of risk. Specifically, areas with unfettered/loosened marijuana regulation have much lower levels of perceived risk among youth and much higher levels of use. Gambling could follow a similar course.
Perhaps the most crucial observation is that the most severe pathological gamblers began gambling before adulthood. Many factors have been identified that seem to increase rates of gambling in youth: Receiving scratch-off lotto cards as gifts, gambling on school grounds, and even smoking status (quite significant given the advent of e-cigarettes now common to many high school students). All of these essentially boil down to the common pathway of proximity and social referencing. As such, the notion that an increased social presence of (what will likely be) large scale, polished, mass televised sports gambling events will be associated with increased gambling behavior (and other mental health comorbidities) among youth is not far-fetched. What also is known for gambling, as well as for other addictive disorders, is that earlier age of onset is correlated to a worse prognosis of gambling disorder in adulthood. In other words, the earlier an addiction strikes, the deeper and more severe it is in the individual – further highlighting the impetus to focus concerns about the PASPA ruling toward the impact on youth.
Prevention and treatment
Lastly, it is important to consider the ground gained in preventing and treating gambling addiction. Many groups focused on treating and preventing gambling already are well established, such as Gamblers Anonymous, and these groups have produced favorable results. More targeted interventions such as cognitive-behavioral therapy adjusted for addiction disorders also have proved effective, as they often not only tackle the gambling disorder but also the collection of conditions it is so often comorbid with (affective illnesses, anxiety disorders).
Pharmacotherapy also has a role, further justifying the view of gambling disorder, and indeed all addiction disorders, as biological processes with biological solutions. Examinations into opiate antagonism and glutamatergic modulation (N-acetylcysteine) also have shown some promise. Prevention programs offer perhaps the best cost-effective ratio in reducing the societal burden of gambling, which is about $7 billion annually, according to 2013 estimates by the National Council on Problem Gambling). These programs have been conducted in schools through parent-teacher groups as well as publicly through distribution of informative psychoeducation via TV and advertising channels.
All available research conducted on treatment shows that further research and validation are needed. We should not pretend that increasing access to sports betting and normalizing the activity will not have an effect on gambling prevalence and problems. Prevention, even simple cautionary public warnings, requires time, money, and planning for effective execution.


Dr. Wenzinger is a clinical fellow, PGY-4, in the department of child and adolescent psychiatry at St. Louis Children’s Hospital. Dr. Gold is the 17th Distinguished Alumni Professor at the University of Florida, Gainesville, and professor of psychiatry (adjunct) at Washington University in St. Louis. He also serves as chairman of the scientific advisory boards for RiverMend Health.
The Supreme Court decision to overturn the federal law that prohibited state-sanctioned college and professional sports betting is bad news for clinicians who treat patients with addictions.
On May 14, the high court ruled 7-2 that the 1992 law, called the Professional and Amateur Sports Protection Act (PASPA), was unconstitutional. Now every state is free to operate, sponsor, promote, license, advertise, or authorize gambling for any college or professional sport–based event.
Optimistic outlooks on the death of PASPA include the foreseen opportunity by the states to tax and generate revenue on such gambling. Proponents of the ruling also argue that illegal activity that thrived on sports betting will now end.
But to what extent will either of those scenarios benefit the public?
If passage of various state marijuana laws is any example, assumptions that legal avenues will usurp illegal enterprises are flawed. Also, taxation likely will generate a large sum of revenue for each state. But those revenues might be offset by subsequent changes that will be needed in mental health, addiction, and wellness programs – a difficult proposition given the opioid epidemic already overburdening the country. Remember the tobacco cases and promise of state support for education, treatment, and other noble activities? Addiction medicine specialists worry that taxes collected by the states, and promises to prevent and treat gambling problems – and prevent addiction – will not end up in those coffers.
As clinicians, perhaps our most important contribution to the debates on this ruling lies in raising awareness of pathological gambling as an addiction disorder.
Redefining the act of gambling
Breaking down previous barriers to access and increasing convenience to gambling undoubtedly will be associated with increased pathological engagement in gambling. This conclusion is clear, based on past national experiments with substances of addiction (such as alcohol prohibition).
Since the cocaine epidemic of the 1980s, and our increased understanding that addictions need not have prominent withdrawal syndromes, we have focused on addiction as a fatal attraction. Psychiatrists and other clinicians made the case – in some quarters, at least – for sugar, sex, and Internet compulsivity as addictions. Compared with those addictions, the evidence was clearest and most compelling for pathological gambling as an addiction disorder. Indeed, gambling disorder was introduced in 2013 to the DSM-5 as the very first non–substance-based addictive disorder. This was a decisive change, as it recognizes that gambling is more than an environmental hazard for those suffering from dopamine-driven obsessive-compulsive-like dysfunction (the DSM section where it had lived previously). Instead, gambling acts as an agent that can initiate a usurpation of the brain’s reward circuitry. (In addition, this change has reopened the door for other increasingly recognized non–substance-based disorder categories such as video game and pornography addiction, and others.)
Gambling disorder certainly fits well into what many experts view as the essential phenotype of any addiction: Continued use despite harm, waning self-control over engagement, a craving state, and compulsive use. Current research is expanding rapidly and filling in the theoretical framework, strongly supporting gambling disorder based on biological evidence. Much of what we now know about the biology of addiction has been through the efforts of the Yale University–based research group, led by psychiatrist Marc N. Potenza. Dr. Potenza and his colleagues have been investigating gambling disorder in a thorough manner (Harv Rev Psychiatry. 2015 Mar-Apr;23[2]:134-46) and (Curr Treat Options Psychiatry. 2014 Jun 1;1[2]:189-203). Indeed, gambling disorder is much like the other substance-use disorders in which it is grouped, in that it has been found to share some similarities/pathways common to all addictions while also carrying its own specific nuances.
Twin studies have unearthed a wealth of information, such as knowledge that environmental factors seem to be the predominant source of the comorbid development of gambling disorder with the more socially acceptable substances as associated use disorders (alcohol, tobacco, and marijuana) through mechanisms such as peer association and place preference conditioning. Similarly, genetic influences also might be meaningful to treatment. For example, one finding showed that patients with gambling disorder and a family history of alcoholism were found to more preferentially respond to opioid-receptor antagonists as treatment for gambling disorder, compared with individuals without such family history (Psychopharmacology [Berl]. 2008;200[4]:521-7).
Explorations of neurotransmitter involvement and brain connectivity also have been conducted for gambling behaviors. Dopaminergic underpinnings of addiction have been particularly indicated in imaging studies focused on the ventral striatum and other components of reward circuitry. In addition, functional MRI studies have identified both overlapping and discordant brain imaging findings between gambling and many other substance use disorders such as cocaine. All these indicate that gambling seems, like its use-disorder counterparts, to follow a similar but distinct course of hijacking reward systems and priming the brain to seek out further gambling in a pathological manner.
Vulnerable populations
Another key finding of recent research exploring the biological foundations of gambling disorder is gender dimorphism. In numerous studies, women have been found to experience a “telescoping effect” from gambling, compared with their male counterparts, where they seem to more quickly advance from first exposure to problematic use. This phenomenon also is seen in women who use cocaine. Also, functional MRI studies also have found that women appear to have alternative signal changes in regions germane to addiction, compared with their male counterparts. One such example was greater activity in the hippocampus and middle temporal gyrus in women, suggestive of stronger activation of regions key for memory retrieval used in craving/urge-related emotions. These data highlight the need for not only understanding how gambling and other addictions diverge between men and women, but also for how prevention and treatment of these disorders might differ based on sex.
Adolescents also get special consideration: How will they be affected by this expected growth in gambling avenues? Adolescence and young adulthood are periods of development defined by increased impulsivity and risk taking, making this population particularly vulnerable to addiction that can then persist into adulthood. It is expected that age laws will persist and prevent the legal access adults might enjoy, but shifts in opinions of harm and ease of access are likely to contribute to increased gambling exposure. To use another addictive phenomena as an example, data from the Substance Abuse and Mental Health Services Administration show a clear correlation between marijuana use, marijuana legal status, and perceptions of risk. Specifically, areas with unfettered/loosened marijuana regulation have much lower levels of perceived risk among youth and much higher levels of use. Gambling could follow a similar course.
Perhaps the most crucial observation is that the most severe pathological gamblers began gambling before adulthood. Many factors have been identified that seem to increase rates of gambling in youth: Receiving scratch-off lotto cards as gifts, gambling on school grounds, and even smoking status (quite significant given the advent of e-cigarettes now common to many high school students). All of these essentially boil down to the common pathway of proximity and social referencing. As such, the notion that an increased social presence of (what will likely be) large scale, polished, mass televised sports gambling events will be associated with increased gambling behavior (and other mental health comorbidities) among youth is not far-fetched. What also is known for gambling, as well as for other addictive disorders, is that earlier age of onset is correlated to a worse prognosis of gambling disorder in adulthood. In other words, the earlier an addiction strikes, the deeper and more severe it is in the individual – further highlighting the impetus to focus concerns about the PASPA ruling toward the impact on youth.
Prevention and treatment
Lastly, it is important to consider the ground gained in preventing and treating gambling addiction. Many groups focused on treating and preventing gambling already are well established, such as Gamblers Anonymous, and these groups have produced favorable results. More targeted interventions such as cognitive-behavioral therapy adjusted for addiction disorders also have proved effective, as they often not only tackle the gambling disorder but also the collection of conditions it is so often comorbid with (affective illnesses, anxiety disorders).
Pharmacotherapy also has a role, further justifying the view of gambling disorder, and indeed all addiction disorders, as biological processes with biological solutions. Examinations into opiate antagonism and glutamatergic modulation (N-acetylcysteine) also have shown some promise. Prevention programs offer perhaps the best cost-effective ratio in reducing the societal burden of gambling, which is about $7 billion annually, according to 2013 estimates by the National Council on Problem Gambling). These programs have been conducted in schools through parent-teacher groups as well as publicly through distribution of informative psychoeducation via TV and advertising channels.
All available research conducted on treatment shows that further research and validation are needed. We should not pretend that increasing access to sports betting and normalizing the activity will not have an effect on gambling prevalence and problems. Prevention, even simple cautionary public warnings, requires time, money, and planning for effective execution.
Dr. Wenzinger is a clinical fellow, PGY-4, in the department of child and adolescent psychiatry at St. Louis Children’s Hospital. Dr. Gold is the 17th Distinguished Alumni Professor at the University of Florida, Gainesville, and professor of psychiatry (adjunct) at Washington University in St. Louis. He also serves as chairman of the scientific advisory boards for RiverMend Health.
The Supreme Court decision to overturn the federal law that prohibited state-sanctioned college and professional sports betting is bad news for clinicians who treat patients with addictions.
On May 14, the high court ruled 7-2 that the 1992 law, called the Professional and Amateur Sports Protection Act (PASPA), was unconstitutional. Now every state is free to operate, sponsor, promote, license, advertise, or authorize gambling for any college or professional sport–based event.
Optimistic outlooks on the death of PASPA include the foreseen opportunity by the states to tax and generate revenue on such gambling. Proponents of the ruling also argue that illegal activity that thrived on sports betting will now end.
But to what extent will either of those scenarios benefit the public?
If passage of various state marijuana laws is any example, assumptions that legal avenues will usurp illegal enterprises are flawed. Also, taxation likely will generate a large sum of revenue for each state. But those revenues might be offset by subsequent changes that will be needed in mental health, addiction, and wellness programs – a difficult proposition given the opioid epidemic already overburdening the country. Remember the tobacco cases and promise of state support for education, treatment, and other noble activities? Addiction medicine specialists worry that taxes collected by the states, and promises to prevent and treat gambling problems – and prevent addiction – will not end up in those coffers.
As clinicians, perhaps our most important contribution to the debates on this ruling lies in raising awareness of pathological gambling as an addiction disorder.
Redefining the act of gambling
Breaking down previous barriers to access and increasing convenience to gambling undoubtedly will be associated with increased pathological engagement in gambling. This conclusion is clear, based on past national experiments with substances of addiction (such as alcohol prohibition).
Since the cocaine epidemic of the 1980s, and our increased understanding that addictions need not have prominent withdrawal syndromes, we have focused on addiction as a fatal attraction. Psychiatrists and other clinicians made the case – in some quarters, at least – for sugar, sex, and Internet compulsivity as addictions. Compared with those addictions, the evidence was clearest and most compelling for pathological gambling as an addiction disorder. Indeed, gambling disorder was introduced in 2013 to the DSM-5 as the very first non–substance-based addictive disorder. This was a decisive change, as it recognizes that gambling is more than an environmental hazard for those suffering from dopamine-driven obsessive-compulsive-like dysfunction (the DSM section where it had lived previously). Instead, gambling acts as an agent that can initiate a usurpation of the brain’s reward circuitry. (In addition, this change has reopened the door for other increasingly recognized non–substance-based disorder categories such as video game and pornography addiction, and others.)
Gambling disorder certainly fits well into what many experts view as the essential phenotype of any addiction: Continued use despite harm, waning self-control over engagement, a craving state, and compulsive use. Current research is expanding rapidly and filling in the theoretical framework, strongly supporting gambling disorder based on biological evidence. Much of what we now know about the biology of addiction has been through the efforts of the Yale University–based research group, led by psychiatrist Marc N. Potenza. Dr. Potenza and his colleagues have been investigating gambling disorder in a thorough manner (Harv Rev Psychiatry. 2015 Mar-Apr;23[2]:134-46) and (Curr Treat Options Psychiatry. 2014 Jun 1;1[2]:189-203). Indeed, gambling disorder is much like the other substance-use disorders in which it is grouped, in that it has been found to share some similarities/pathways common to all addictions while also carrying its own specific nuances.
Twin studies have unearthed a wealth of information, such as knowledge that environmental factors seem to be the predominant source of the comorbid development of gambling disorder with the more socially acceptable substances as associated use disorders (alcohol, tobacco, and marijuana) through mechanisms such as peer association and place preference conditioning. Similarly, genetic influences also might be meaningful to treatment. For example, one finding showed that patients with gambling disorder and a family history of alcoholism were found to more preferentially respond to opioid-receptor antagonists as treatment for gambling disorder, compared with individuals without such family history (Psychopharmacology [Berl]. 2008;200[4]:521-7).
Explorations of neurotransmitter involvement and brain connectivity also have been conducted for gambling behaviors. Dopaminergic underpinnings of addiction have been particularly indicated in imaging studies focused on the ventral striatum and other components of reward circuitry. In addition, functional MRI studies have identified both overlapping and discordant brain imaging findings between gambling and many other substance use disorders such as cocaine. All these indicate that gambling seems, like its use-disorder counterparts, to follow a similar but distinct course of hijacking reward systems and priming the brain to seek out further gambling in a pathological manner.
Vulnerable populations
Another key finding of recent research exploring the biological foundations of gambling disorder is gender dimorphism. In numerous studies, women have been found to experience a “telescoping effect” from gambling, compared with their male counterparts, where they seem to more quickly advance from first exposure to problematic use. This phenomenon also is seen in women who use cocaine. Also, functional MRI studies also have found that women appear to have alternative signal changes in regions germane to addiction, compared with their male counterparts. One such example was greater activity in the hippocampus and middle temporal gyrus in women, suggestive of stronger activation of regions key for memory retrieval used in craving/urge-related emotions. These data highlight the need for not only understanding how gambling and other addictions diverge between men and women, but also for how prevention and treatment of these disorders might differ based on sex.
Adolescents also get special consideration: How will they be affected by this expected growth in gambling avenues? Adolescence and young adulthood are periods of development defined by increased impulsivity and risk taking, making this population particularly vulnerable to addiction that can then persist into adulthood. It is expected that age laws will persist and prevent the legal access adults might enjoy, but shifts in opinions of harm and ease of access are likely to contribute to increased gambling exposure. To use another addictive phenomena as an example, data from the Substance Abuse and Mental Health Services Administration show a clear correlation between marijuana use, marijuana legal status, and perceptions of risk. Specifically, areas with unfettered/loosened marijuana regulation have much lower levels of perceived risk among youth and much higher levels of use. Gambling could follow a similar course.
Perhaps the most crucial observation is that the most severe pathological gamblers began gambling before adulthood. Many factors have been identified that seem to increase rates of gambling in youth: Receiving scratch-off lotto cards as gifts, gambling on school grounds, and even smoking status (quite significant given the advent of e-cigarettes now common to many high school students). All of these essentially boil down to the common pathway of proximity and social referencing. As such, the notion that an increased social presence of (what will likely be) large scale, polished, mass televised sports gambling events will be associated with increased gambling behavior (and other mental health comorbidities) among youth is not far-fetched. What also is known for gambling, as well as for other addictive disorders, is that earlier age of onset is correlated to a worse prognosis of gambling disorder in adulthood. In other words, the earlier an addiction strikes, the deeper and more severe it is in the individual – further highlighting the impetus to focus concerns about the PASPA ruling toward the impact on youth.
Prevention and treatment
Lastly, it is important to consider the ground gained in preventing and treating gambling addiction. Many groups focused on treating and preventing gambling already are well established, such as Gamblers Anonymous, and these groups have produced favorable results. More targeted interventions such as cognitive-behavioral therapy adjusted for addiction disorders also have proved effective, as they often not only tackle the gambling disorder but also the collection of conditions it is so often comorbid with (affective illnesses, anxiety disorders).
Pharmacotherapy also has a role, further justifying the view of gambling disorder, and indeed all addiction disorders, as biological processes with biological solutions. Examinations into opiate antagonism and glutamatergic modulation (N-acetylcysteine) also have shown some promise. Prevention programs offer perhaps the best cost-effective ratio in reducing the societal burden of gambling, which is about $7 billion annually, according to 2013 estimates by the National Council on Problem Gambling). These programs have been conducted in schools through parent-teacher groups as well as publicly through distribution of informative psychoeducation via TV and advertising channels.
All available research conducted on treatment shows that further research and validation are needed. We should not pretend that increasing access to sports betting and normalizing the activity will not have an effect on gambling prevalence and problems. Prevention, even simple cautionary public warnings, requires time, money, and planning for effective execution.
Dr. Wenzinger is a clinical fellow, PGY-4, in the department of child and adolescent psychiatry at St. Louis Children’s Hospital. Dr. Gold is the 17th Distinguished Alumni Professor at the University of Florida, Gainesville, and professor of psychiatry (adjunct) at Washington University in St. Louis. He also serves as chairman of the scientific advisory boards for RiverMend Health.
Debunking Atopic Dermatitis Myths: Should You Use Systemic Therapy?
Myth: Because atopic dermatitis is skin-deep, systemic therapy is unnecessary.
Although atopic dermatitis (AD) primarily is known as a skin condition, recent research has indicated that it may be the start of the “atopic march” leading to the development of 1 or more other atopic conditions with multiorgan involvement. In infancy AD can progress to asthma and allergic rhinitis. Adult AD can be accompanied by systemic diseases such as inflammatory bowel disease, nephritic syndrome, and others. There also is a link between impairment of epidermal barrier function and disturbed skin microbiome in patients with AD. Therefore, systemic therapy may be warranted; the question is when should you use systemic therapy?
Most AD patients have mild to moderate disease that responds well to emollients and avoidance of disease triggers and other skin irritants. However, many AD patients experience a more severe disease course that does not respond adequately to topical therapy. For these patients, systemic therapy is a viable treatment option to improve quality of life (QOL), prevent flares, and control skin inflammation and other AD symptoms.
In 2017 an expert panel of the International Eczema Council proposed an algorithm to be used to determine if systemic therapy is warranted in patients with AD. Dermatologists must consider disease severity, impact on QOL, and risks and benefits of systemic therapies. Before starting systemic therapy, the panel recommends the following:
- Consider alternate or concomitant diagnoses
- Avoid triggers
- Optimize topical therapy
- Ensure adequate patient/caregiver education
- Treat coexistent infection
- Assess QOL
- Consider phototherapy
The American Academy of Dermatology established Guidelines of Care for the Management of AD in 2014, which provide recommendations for the most efficacious systemic agents.
Armed with these guidelines, dermatologists can work with patients to determine the most appropriate treatment course for this condition that is more than skin-deep.
Expert Commentary
Atopic dermatitis is a skin barrier abnormality that causes inflammatory skin disease and an inflammatory disorder triggering abnormal barrier. Whether we choose the outside-in or inside-out approach, it is clear that there is a systemic inflammation associated with skin disease. It is true that children respond well to barrier repair and topical therapy in many settings, as do many adults. However, chronic skin inflammation is not in isolation, triggering mucosal barrier changes allowing for more sensitization to foods and respiratory allergens as well as systemic inflammation in adults. Despite the utility of systemic steroids, the side effects generally outweigh benefit. On the other hand, phototherapy and systemic agents can clear skin and induce remissions and improved QOL. The AAD guidelines were reported before US Food and Drug Administration approval of newer agents such as dupilumab, leaving it up to the dermatologist to find the niche for this first biologic agent for AD.
—Nanette B. Silverberg, MD
Suggested Readings
Darlenski R, Kazandjieva J, Hristakieva E, et al. Atopic dermatitis as a systemic disease [published online November 22, 2013]. Clin Dermatol. 2014;32:409-413.
Sidbury R, Davis DM, Cohen DE, et al; American Academy of Dermatology. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents [published online May 9, 2014]. J Am Acad Dermatol. 2014;71:327-349.
Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? recommendations from an expert panel of the International Eczema Council [published online August 10, 2017]. J Am Acad Dermatol. 2017;77:623-633.
Thomas CL, Fernández-Peñas P. The microbiome and atopic eczema: more than skin deep [published online January 28, 2016]. Australas J Dermatol. 2017;58:18-24.
Myth: Because atopic dermatitis is skin-deep, systemic therapy is unnecessary.
Although atopic dermatitis (AD) primarily is known as a skin condition, recent research has indicated that it may be the start of the “atopic march” leading to the development of 1 or more other atopic conditions with multiorgan involvement. In infancy AD can progress to asthma and allergic rhinitis. Adult AD can be accompanied by systemic diseases such as inflammatory bowel disease, nephritic syndrome, and others. There also is a link between impairment of epidermal barrier function and disturbed skin microbiome in patients with AD. Therefore, systemic therapy may be warranted; the question is when should you use systemic therapy?
Most AD patients have mild to moderate disease that responds well to emollients and avoidance of disease triggers and other skin irritants. However, many AD patients experience a more severe disease course that does not respond adequately to topical therapy. For these patients, systemic therapy is a viable treatment option to improve quality of life (QOL), prevent flares, and control skin inflammation and other AD symptoms.
In 2017 an expert panel of the International Eczema Council proposed an algorithm to be used to determine if systemic therapy is warranted in patients with AD. Dermatologists must consider disease severity, impact on QOL, and risks and benefits of systemic therapies. Before starting systemic therapy, the panel recommends the following:
- Consider alternate or concomitant diagnoses
- Avoid triggers
- Optimize topical therapy
- Ensure adequate patient/caregiver education
- Treat coexistent infection
- Assess QOL
- Consider phototherapy
The American Academy of Dermatology established Guidelines of Care for the Management of AD in 2014, which provide recommendations for the most efficacious systemic agents.
Armed with these guidelines, dermatologists can work with patients to determine the most appropriate treatment course for this condition that is more than skin-deep.
Expert Commentary
Atopic dermatitis is a skin barrier abnormality that causes inflammatory skin disease and an inflammatory disorder triggering abnormal barrier. Whether we choose the outside-in or inside-out approach, it is clear that there is a systemic inflammation associated with skin disease. It is true that children respond well to barrier repair and topical therapy in many settings, as do many adults. However, chronic skin inflammation is not in isolation, triggering mucosal barrier changes allowing for more sensitization to foods and respiratory allergens as well as systemic inflammation in adults. Despite the utility of systemic steroids, the side effects generally outweigh benefit. On the other hand, phototherapy and systemic agents can clear skin and induce remissions and improved QOL. The AAD guidelines were reported before US Food and Drug Administration approval of newer agents such as dupilumab, leaving it up to the dermatologist to find the niche for this first biologic agent for AD.
—Nanette B. Silverberg, MD
Suggested Readings
Darlenski R, Kazandjieva J, Hristakieva E, et al. Atopic dermatitis as a systemic disease [published online November 22, 2013]. Clin Dermatol. 2014;32:409-413.
Sidbury R, Davis DM, Cohen DE, et al; American Academy of Dermatology. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents [published online May 9, 2014]. J Am Acad Dermatol. 2014;71:327-349.
Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? recommendations from an expert panel of the International Eczema Council [published online August 10, 2017]. J Am Acad Dermatol. 2017;77:623-633.
Thomas CL, Fernández-Peñas P. The microbiome and atopic eczema: more than skin deep [published online January 28, 2016]. Australas J Dermatol. 2017;58:18-24.
Myth: Because atopic dermatitis is skin-deep, systemic therapy is unnecessary.
Although atopic dermatitis (AD) primarily is known as a skin condition, recent research has indicated that it may be the start of the “atopic march” leading to the development of 1 or more other atopic conditions with multiorgan involvement. In infancy AD can progress to asthma and allergic rhinitis. Adult AD can be accompanied by systemic diseases such as inflammatory bowel disease, nephritic syndrome, and others. There also is a link between impairment of epidermal barrier function and disturbed skin microbiome in patients with AD. Therefore, systemic therapy may be warranted; the question is when should you use systemic therapy?
Most AD patients have mild to moderate disease that responds well to emollients and avoidance of disease triggers and other skin irritants. However, many AD patients experience a more severe disease course that does not respond adequately to topical therapy. For these patients, systemic therapy is a viable treatment option to improve quality of life (QOL), prevent flares, and control skin inflammation and other AD symptoms.
In 2017 an expert panel of the International Eczema Council proposed an algorithm to be used to determine if systemic therapy is warranted in patients with AD. Dermatologists must consider disease severity, impact on QOL, and risks and benefits of systemic therapies. Before starting systemic therapy, the panel recommends the following:
- Consider alternate or concomitant diagnoses
- Avoid triggers
- Optimize topical therapy
- Ensure adequate patient/caregiver education
- Treat coexistent infection
- Assess QOL
- Consider phototherapy
The American Academy of Dermatology established Guidelines of Care for the Management of AD in 2014, which provide recommendations for the most efficacious systemic agents.
Armed with these guidelines, dermatologists can work with patients to determine the most appropriate treatment course for this condition that is more than skin-deep.
Expert Commentary
Atopic dermatitis is a skin barrier abnormality that causes inflammatory skin disease and an inflammatory disorder triggering abnormal barrier. Whether we choose the outside-in or inside-out approach, it is clear that there is a systemic inflammation associated with skin disease. It is true that children respond well to barrier repair and topical therapy in many settings, as do many adults. However, chronic skin inflammation is not in isolation, triggering mucosal barrier changes allowing for more sensitization to foods and respiratory allergens as well as systemic inflammation in adults. Despite the utility of systemic steroids, the side effects generally outweigh benefit. On the other hand, phototherapy and systemic agents can clear skin and induce remissions and improved QOL. The AAD guidelines were reported before US Food and Drug Administration approval of newer agents such as dupilumab, leaving it up to the dermatologist to find the niche for this first biologic agent for AD.
—Nanette B. Silverberg, MD
Suggested Readings
Darlenski R, Kazandjieva J, Hristakieva E, et al. Atopic dermatitis as a systemic disease [published online November 22, 2013]. Clin Dermatol. 2014;32:409-413.
Sidbury R, Davis DM, Cohen DE, et al; American Academy of Dermatology. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents [published online May 9, 2014]. J Am Acad Dermatol. 2014;71:327-349.
Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? recommendations from an expert panel of the International Eczema Council [published online August 10, 2017]. J Am Acad Dermatol. 2017;77:623-633.
Thomas CL, Fernández-Peñas P. The microbiome and atopic eczema: more than skin deep [published online January 28, 2016]. Australas J Dermatol. 2017;58:18-24.

The demise of family dinners may play role in picky eating
My wife and I have dinner together almost every evening. There is a candle on the table regardless of the menu. And the meal begins with a toast, usually “To this chance to be together.” I can hear you muttering to yourself, “They must be one of those sappy, sweet hand-holding couples that appear to be joined at the hip.” Far from it, we lead very busy, active, but separate lives that only rarely intersect. But we make it a priority that one of those intersections occurs at a meal. For us, an evening dinner works best.
Listening to our friends, we have learned that an increasing number of them have drifted away from sharing a meal together. This phenomenon is surprising because most of them are retired, and time is not an issue. Of course, it is no secret that, for young overscheduled families, sitting down for a shared dining experience is becoming increasingly less frequent. Like some of you, I would like to claim that a return to family meal times would solve all of society’s ills. But some of the literature supporting this claim suggests shared family experiences in general, not particularly those associated with eating, may be just as important in supporting emotional health. But because everyone needs to eat, meals seem to me to be the easy target, low-hanging fruit if you will.
It is interesting that parents’ reports of their children’s eating patterns were validated by the behaviors videotaped by the researchers. But what is really interesting is that children who grew up in households where mealtimes followed a routine were more likely to be in the low–picky eating group. Routines included things like having a specific place times for eating, a habitual way of serving food, and other rituals such as saying grace.

Unfortunately, because this Michigan study began at age 4 years it doesn’t tell us if the worst picky eaters were that way from the beginning. I suspect that some were. But my hunch is that picky eaters who are managed in a home environment that includes mealtime rituals and puts dining together as a high priority are more likely to outgrow their pickiness.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “Coping with a Picky Eater: A Guide for the Perplexed Parent.” Email him at [email protected].
My wife and I have dinner together almost every evening. There is a candle on the table regardless of the menu. And the meal begins with a toast, usually “To this chance to be together.” I can hear you muttering to yourself, “They must be one of those sappy, sweet hand-holding couples that appear to be joined at the hip.” Far from it, we lead very busy, active, but separate lives that only rarely intersect. But we make it a priority that one of those intersections occurs at a meal. For us, an evening dinner works best.
Listening to our friends, we have learned that an increasing number of them have drifted away from sharing a meal together. This phenomenon is surprising because most of them are retired, and time is not an issue. Of course, it is no secret that, for young overscheduled families, sitting down for a shared dining experience is becoming increasingly less frequent. Like some of you, I would like to claim that a return to family meal times would solve all of society’s ills. But some of the literature supporting this claim suggests shared family experiences in general, not particularly those associated with eating, may be just as important in supporting emotional health. But because everyone needs to eat, meals seem to me to be the easy target, low-hanging fruit if you will.
It is interesting that parents’ reports of their children’s eating patterns were validated by the behaviors videotaped by the researchers. But what is really interesting is that children who grew up in households where mealtimes followed a routine were more likely to be in the low–picky eating group. Routines included things like having a specific place times for eating, a habitual way of serving food, and other rituals such as saying grace.
Unfortunately, because this Michigan study began at age 4 years it doesn’t tell us if the worst picky eaters were that way from the beginning. I suspect that some were. But my hunch is that picky eaters who are managed in a home environment that includes mealtime rituals and puts dining together as a high priority are more likely to outgrow their pickiness.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “Coping with a Picky Eater: A Guide for the Perplexed Parent.” Email him at [email protected].
My wife and I have dinner together almost every evening. There is a candle on the table regardless of the menu. And the meal begins with a toast, usually “To this chance to be together.” I can hear you muttering to yourself, “They must be one of those sappy, sweet hand-holding couples that appear to be joined at the hip.” Far from it, we lead very busy, active, but separate lives that only rarely intersect. But we make it a priority that one of those intersections occurs at a meal. For us, an evening dinner works best.
Listening to our friends, we have learned that an increasing number of them have drifted away from sharing a meal together. This phenomenon is surprising because most of them are retired, and time is not an issue. Of course, it is no secret that, for young overscheduled families, sitting down for a shared dining experience is becoming increasingly less frequent. Like some of you, I would like to claim that a return to family meal times would solve all of society’s ills. But some of the literature supporting this claim suggests shared family experiences in general, not particularly those associated with eating, may be just as important in supporting emotional health. But because everyone needs to eat, meals seem to me to be the easy target, low-hanging fruit if you will.
It is interesting that parents’ reports of their children’s eating patterns were validated by the behaviors videotaped by the researchers. But what is really interesting is that children who grew up in households where mealtimes followed a routine were more likely to be in the low–picky eating group. Routines included things like having a specific place times for eating, a habitual way of serving food, and other rituals such as saying grace.
Unfortunately, because this Michigan study began at age 4 years it doesn’t tell us if the worst picky eaters were that way from the beginning. I suspect that some were. But my hunch is that picky eaters who are managed in a home environment that includes mealtime rituals and puts dining together as a high priority are more likely to outgrow their pickiness.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “Coping with a Picky Eater: A Guide for the Perplexed Parent.” Email him at [email protected].