User login
Hundreds of thousands of U.S. troops remain unvaccinated
, according to data analyzed by The Washington Post.
Overall, the military’s vaccination rate has climbed since August, when the Pentagon announced that COVID-19 immunization would become mandatory for the nation’s 2.1 million troops, the newspaper reported. Acting on a directive from President Joe Biden, leaders said exemptions would be rare and unvaccinated service members would face consequences.
But compliance has varied across the services, the newspaper found. About 90% of the active-duty Navy is fully vaccinated, compared with 76% of the active-duty Marine Corps. Both have a November 28 deadline to show proof of full vaccination.
About 81% of the active-duty Air Force is fully vaccinated, leaving more than 60,000 personnel about 3 weeks to get a shot before a November 2 deadline.
Military officials said the variance in vaccination rates is related to the different deadlines, the newspaper reported. As the dates approach, most troops are expected to meet the order.
At the same time, some deadlines are spaced farther out, with the Army Reserve and National Guard required to be fully vaccinated by next summer. About 40% of the Army Reserve and 38% of the National Guard are fully vaccinated. Combined, they account for a quarter of the U.S. military and 40% of the COVID-19 deaths among service members.
“The Army’s policy is incentivizing inaction until the latest possible date,” Katherine Kuzminski, a military policy expert at the Center for a New American Security, told the Post.“The way we’ve seen the virus evolve tells us looking out to June 30 may need to be reconsidered,” she said.
COVID-19 deaths have surged in some of the services in recent months, the newspaper reported. More military personnel died from COVID-19 in September than in all of 2020. None of those who died were fully vaccinated.
Throughout the pandemic, more than 246,000 COVID-19 cases have been reported among service members, according to the latest data from the Defense Department. More than 2,200 have been hospitalized, and 62 personnel died, including 32 in August and September.
For the Army Reserve and National Guard, the June deadline allows “necessary time to update records and process exemption requests,” Lt. Col. Terence Kelley, an Army spokesman, told the Post.
Lt. Col. Kelley noted that the extended date reflects how large the groups are, compared to the other services and their military reserves, as well as the broad geographic distribution of its members. Still, vaccine mandates will apply.
“We expect all unvaccinated soldiers to receive the vaccine as soon as possible,” Lt. Col. Kelley said. “Individual soldiers are required to receive the vaccine when available.”
A version of this article first appeared on Medscape.com.
, according to data analyzed by The Washington Post.
Overall, the military’s vaccination rate has climbed since August, when the Pentagon announced that COVID-19 immunization would become mandatory for the nation’s 2.1 million troops, the newspaper reported. Acting on a directive from President Joe Biden, leaders said exemptions would be rare and unvaccinated service members would face consequences.
But compliance has varied across the services, the newspaper found. About 90% of the active-duty Navy is fully vaccinated, compared with 76% of the active-duty Marine Corps. Both have a November 28 deadline to show proof of full vaccination.
About 81% of the active-duty Air Force is fully vaccinated, leaving more than 60,000 personnel about 3 weeks to get a shot before a November 2 deadline.
Military officials said the variance in vaccination rates is related to the different deadlines, the newspaper reported. As the dates approach, most troops are expected to meet the order.
At the same time, some deadlines are spaced farther out, with the Army Reserve and National Guard required to be fully vaccinated by next summer. About 40% of the Army Reserve and 38% of the National Guard are fully vaccinated. Combined, they account for a quarter of the U.S. military and 40% of the COVID-19 deaths among service members.
“The Army’s policy is incentivizing inaction until the latest possible date,” Katherine Kuzminski, a military policy expert at the Center for a New American Security, told the Post.“The way we’ve seen the virus evolve tells us looking out to June 30 may need to be reconsidered,” she said.
COVID-19 deaths have surged in some of the services in recent months, the newspaper reported. More military personnel died from COVID-19 in September than in all of 2020. None of those who died were fully vaccinated.
Throughout the pandemic, more than 246,000 COVID-19 cases have been reported among service members, according to the latest data from the Defense Department. More than 2,200 have been hospitalized, and 62 personnel died, including 32 in August and September.
For the Army Reserve and National Guard, the June deadline allows “necessary time to update records and process exemption requests,” Lt. Col. Terence Kelley, an Army spokesman, told the Post.
Lt. Col. Kelley noted that the extended date reflects how large the groups are, compared to the other services and their military reserves, as well as the broad geographic distribution of its members. Still, vaccine mandates will apply.
“We expect all unvaccinated soldiers to receive the vaccine as soon as possible,” Lt. Col. Kelley said. “Individual soldiers are required to receive the vaccine when available.”
A version of this article first appeared on Medscape.com.
, according to data analyzed by The Washington Post.
Overall, the military’s vaccination rate has climbed since August, when the Pentagon announced that COVID-19 immunization would become mandatory for the nation’s 2.1 million troops, the newspaper reported. Acting on a directive from President Joe Biden, leaders said exemptions would be rare and unvaccinated service members would face consequences.
But compliance has varied across the services, the newspaper found. About 90% of the active-duty Navy is fully vaccinated, compared with 76% of the active-duty Marine Corps. Both have a November 28 deadline to show proof of full vaccination.
About 81% of the active-duty Air Force is fully vaccinated, leaving more than 60,000 personnel about 3 weeks to get a shot before a November 2 deadline.
Military officials said the variance in vaccination rates is related to the different deadlines, the newspaper reported. As the dates approach, most troops are expected to meet the order.
At the same time, some deadlines are spaced farther out, with the Army Reserve and National Guard required to be fully vaccinated by next summer. About 40% of the Army Reserve and 38% of the National Guard are fully vaccinated. Combined, they account for a quarter of the U.S. military and 40% of the COVID-19 deaths among service members.
“The Army’s policy is incentivizing inaction until the latest possible date,” Katherine Kuzminski, a military policy expert at the Center for a New American Security, told the Post.“The way we’ve seen the virus evolve tells us looking out to June 30 may need to be reconsidered,” she said.
COVID-19 deaths have surged in some of the services in recent months, the newspaper reported. More military personnel died from COVID-19 in September than in all of 2020. None of those who died were fully vaccinated.
Throughout the pandemic, more than 246,000 COVID-19 cases have been reported among service members, according to the latest data from the Defense Department. More than 2,200 have been hospitalized, and 62 personnel died, including 32 in August and September.
For the Army Reserve and National Guard, the June deadline allows “necessary time to update records and process exemption requests,” Lt. Col. Terence Kelley, an Army spokesman, told the Post.
Lt. Col. Kelley noted that the extended date reflects how large the groups are, compared to the other services and their military reserves, as well as the broad geographic distribution of its members. Still, vaccine mandates will apply.
“We expect all unvaccinated soldiers to receive the vaccine as soon as possible,” Lt. Col. Kelley said. “Individual soldiers are required to receive the vaccine when available.”
A version of this article first appeared on Medscape.com.
Women with recurrent UTIs express fear, frustration
Fear of antibiotic overuse and frustration with physicians who prescribe them too freely are key sentiments expressed by women with recurrent urinary tract infections (rUTIs), according to findings from a study involving six focus groups.
“Here in our female pelvic medicine reconstructive urology clinic at Cedars-Sinai and at UCLA, we see many women who are referred for evaluation of rUTIs who are very frustrated with their care,” Victoria Scott, MD, Cedars-Sinai Medical Center, Los Angeles, said in an interview.
“So with these focus groups, we saw an opportunity to explore why women are so frustrated and to try and improve the care delivered,” she added.
Findings from the study were published online Sept. 1 in The Journal of Urology.
“There is a need for physicians to modify management strategies ... and to devote more research efforts to improving nonantibiotic options for the prevention and treatment of recurrent urinary tract infections, as well as management strategies that better empower patients,” the authors wrote.
Six focus groups
Four or five participants were included in each of the six focus groups – a total of 29 women. All participants reported a history of symptomatic, culture-proven UTI episodes. They had experienced two or more infections in 6 months or three or more infections within 1 year. Women were predominantly White. Most were employed part- or full-time and held a college degree.
From a qualitative analysis of all focus group transcripts, two main themes emerged:
- The negative impact of taking antibiotics for the prevention and treatment of rUTIs.
- Resentment of the medical profession for the way it managed rUTIs.
The researchers found that participants had a good understanding of the deleterious effects from inappropriate antibiotic use, largely gleaned from media sources and the Internet. “Numerous women stated that they had reached such a level of concern about antibiotics that they would resist taking them for prevention or treatment of infections,” Dr. Scott and colleagues pointed out.
These concerns centered around the risk of developing resistance to antibiotics and the ill effects that antibiotics can have on the gastrointestinal and genitourinary microbiomes. Several women reported that they had developed Clostridium difficile infections after taking antibiotics; one of the patients required hospitalization for the infection.
Women also reported concerns that they had been given an antibiotic needlessly for symptoms that might have been caused by a genitourinary condition other than a UTI. They also reported feeling resentful toward practitioners, particularly if they felt the practitioner was overprescribing antibiotics. Some had resorted to consultations with alternative practitioners, such as herbalists. “A second concern discussed by participants was the feeling of being ignored by physicians,” the authors observed.
In this regard, the women felt that their physicians underestimated the burden that rUTIs had on their lives and the detrimental effect that repeated infections had on their relationships, work, and overall quality of life. “These perceptions led to a prevalent mistrust of physicians,” the investigators wrote. This prompted many women to insist that the medical community devote more effort to the development of nonantibiotic options for the prevention and treatment of UTIs.
Improved management strategies
Asked how physicians might improve their management of rUTIs, Dr. Scott shared a number of suggestions. Cardinal rule No. 1: Have the patient undergo a urinalysis to make sure she does have a UTI. “There is a subset of patients among women with rUTIs who come in with a diagnosis of an rUTI but who really have not had documentation of more than one positive urine culture,” Dr. Scott noted. Such a history suggests that they do not have an rUTI.
It’s imperative that physicians rule out commonly misdiagnosed disorders, such as overactive bladder, as a cause of the patient’s symptoms. Symptoms of overactive bladder and rUTIs often overlap. While waiting for results from the urinalysis to confirm or rule out a UTI, young and healthy women may be prescribed a nonsteroidal anti-inflammatory drug (NSAID), such as naproxen, which can help ameliorate symptoms.
Because UTIs are frequently self-limiting, Dr. Scott and others have found that for young, otherwise healthy women, NSAIDs alone can often resolve symptoms of the UTI without use of an antibiotic. For relatively severe symptoms, a urinary analgesic, such as phenazopyridine (Pyridium), may soothe the lining of the urinary tract and relieve pain. Cystex is an over-the-counter urinary analgesic that women can procure themselves, Dr. Scott added.
If an antibiotic is indicated, those most commonly prescribed for a single episode of acute cystitis are nitrofurantoin and sulfamethoxazole plus trimethoprim (Bactrim). For recurrent UTIs, “patients are a bit more complicated,” Dr. Scott admitted. “I think the best practice is to look back at a woman’s prior urine culture and select an antibiotic that showed good sensitivity in the last positive urine test,” she said.
Prevention starts with behavioral strategies, such as voiding after sexual intercourse and wiping from front to back following urination to avoid introducing fecal bacteria into the urethra. Evidence suggests that premenopausal women who drink at least 1.5 L of water a day have significantly fewer UTI episodes, Dr. Scott noted. There is also “pretty good” evidence that cranberry supplements (not juice) can prevent rUTIs. Use of cranberry supplements is supported by the American Urological Association (conditional recommendation; evidence level of grade C).
For peri- and postmenopausal women, vaginal estrogen may be effective. It’s use for UTI prevention is well supported by the literature. Although not as well supported by evidence, some women find that a supplement such as D-mannose may prevent or treat UTIs by causing bacteria to bind to it rather than to the bladder wall. Probiotics are another possibility, she noted. Empathy can’t hurt, she added.
“A common theme among satisfied women was the sentiment that their physicians understood their problems and had a system in place to allow rapid diagnosis and treatment for UTI episodes,” the authors emphasized.
“[Such attitudes] highlight the need to investigate each patient’s experience and perceptions to allow for shared decision making regarding the management of rUTIs,” they wrote.
Further commentary
Asked to comment on the findings, editorialist Michelle Van Kuiken, MD, assistant professor of urology, University of California, San Francisco, acknowledged that there is not a lot of good evidence to support many of the strategies recommended by the American Urological Association to prevent and treat rUTIs, but she often follows these recommendations anyway. “The one statement in the guidelines that is the most supported by evidence is the use of cranberry supplements, and I do routinely recommended daily use of some form of concentrated cranberry supplements for all of my patients with rUTIs,” she said in an interview.
Dr. Van Kuiken said that vaginal estrogen is a very good option for all postmenopausal women who suffer from rUTIs and that there is growing acceptance of its use for this and other indications. There is some evidence to support D-mannose as well, although it’s not that robust, she acknowledged.
She said the evidence supporting the use of probiotics for this indication is very thin. She does not routinely recommend them for rUTIs, although they are not inherently harmful. “I think for a lot of women who have rUTIs, it can be pretty debilitating and upsetting for them – it can impact travel plans, work, and social events,” Dr. Van Kuiken said.
“Until we develop better diagnostic and therapeutic strategies, validating women’s experiences and concerns with rUTI while limiting unnecessary antibiotics remains our best option,” she wrote.
Dr. Scott and Dr. Van Kuiken have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Fear of antibiotic overuse and frustration with physicians who prescribe them too freely are key sentiments expressed by women with recurrent urinary tract infections (rUTIs), according to findings from a study involving six focus groups.
“Here in our female pelvic medicine reconstructive urology clinic at Cedars-Sinai and at UCLA, we see many women who are referred for evaluation of rUTIs who are very frustrated with their care,” Victoria Scott, MD, Cedars-Sinai Medical Center, Los Angeles, said in an interview.
“So with these focus groups, we saw an opportunity to explore why women are so frustrated and to try and improve the care delivered,” she added.
Findings from the study were published online Sept. 1 in The Journal of Urology.
“There is a need for physicians to modify management strategies ... and to devote more research efforts to improving nonantibiotic options for the prevention and treatment of recurrent urinary tract infections, as well as management strategies that better empower patients,” the authors wrote.
Six focus groups
Four or five participants were included in each of the six focus groups – a total of 29 women. All participants reported a history of symptomatic, culture-proven UTI episodes. They had experienced two or more infections in 6 months or three or more infections within 1 year. Women were predominantly White. Most were employed part- or full-time and held a college degree.
From a qualitative analysis of all focus group transcripts, two main themes emerged:
- The negative impact of taking antibiotics for the prevention and treatment of rUTIs.
- Resentment of the medical profession for the way it managed rUTIs.
The researchers found that participants had a good understanding of the deleterious effects from inappropriate antibiotic use, largely gleaned from media sources and the Internet. “Numerous women stated that they had reached such a level of concern about antibiotics that they would resist taking them for prevention or treatment of infections,” Dr. Scott and colleagues pointed out.
These concerns centered around the risk of developing resistance to antibiotics and the ill effects that antibiotics can have on the gastrointestinal and genitourinary microbiomes. Several women reported that they had developed Clostridium difficile infections after taking antibiotics; one of the patients required hospitalization for the infection.
Women also reported concerns that they had been given an antibiotic needlessly for symptoms that might have been caused by a genitourinary condition other than a UTI. They also reported feeling resentful toward practitioners, particularly if they felt the practitioner was overprescribing antibiotics. Some had resorted to consultations with alternative practitioners, such as herbalists. “A second concern discussed by participants was the feeling of being ignored by physicians,” the authors observed.
In this regard, the women felt that their physicians underestimated the burden that rUTIs had on their lives and the detrimental effect that repeated infections had on their relationships, work, and overall quality of life. “These perceptions led to a prevalent mistrust of physicians,” the investigators wrote. This prompted many women to insist that the medical community devote more effort to the development of nonantibiotic options for the prevention and treatment of UTIs.
Improved management strategies
Asked how physicians might improve their management of rUTIs, Dr. Scott shared a number of suggestions. Cardinal rule No. 1: Have the patient undergo a urinalysis to make sure she does have a UTI. “There is a subset of patients among women with rUTIs who come in with a diagnosis of an rUTI but who really have not had documentation of more than one positive urine culture,” Dr. Scott noted. Such a history suggests that they do not have an rUTI.
It’s imperative that physicians rule out commonly misdiagnosed disorders, such as overactive bladder, as a cause of the patient’s symptoms. Symptoms of overactive bladder and rUTIs often overlap. While waiting for results from the urinalysis to confirm or rule out a UTI, young and healthy women may be prescribed a nonsteroidal anti-inflammatory drug (NSAID), such as naproxen, which can help ameliorate symptoms.
Because UTIs are frequently self-limiting, Dr. Scott and others have found that for young, otherwise healthy women, NSAIDs alone can often resolve symptoms of the UTI without use of an antibiotic. For relatively severe symptoms, a urinary analgesic, such as phenazopyridine (Pyridium), may soothe the lining of the urinary tract and relieve pain. Cystex is an over-the-counter urinary analgesic that women can procure themselves, Dr. Scott added.
If an antibiotic is indicated, those most commonly prescribed for a single episode of acute cystitis are nitrofurantoin and sulfamethoxazole plus trimethoprim (Bactrim). For recurrent UTIs, “patients are a bit more complicated,” Dr. Scott admitted. “I think the best practice is to look back at a woman’s prior urine culture and select an antibiotic that showed good sensitivity in the last positive urine test,” she said.
Prevention starts with behavioral strategies, such as voiding after sexual intercourse and wiping from front to back following urination to avoid introducing fecal bacteria into the urethra. Evidence suggests that premenopausal women who drink at least 1.5 L of water a day have significantly fewer UTI episodes, Dr. Scott noted. There is also “pretty good” evidence that cranberry supplements (not juice) can prevent rUTIs. Use of cranberry supplements is supported by the American Urological Association (conditional recommendation; evidence level of grade C).
For peri- and postmenopausal women, vaginal estrogen may be effective. It’s use for UTI prevention is well supported by the literature. Although not as well supported by evidence, some women find that a supplement such as D-mannose may prevent or treat UTIs by causing bacteria to bind to it rather than to the bladder wall. Probiotics are another possibility, she noted. Empathy can’t hurt, she added.
“A common theme among satisfied women was the sentiment that their physicians understood their problems and had a system in place to allow rapid diagnosis and treatment for UTI episodes,” the authors emphasized.
“[Such attitudes] highlight the need to investigate each patient’s experience and perceptions to allow for shared decision making regarding the management of rUTIs,” they wrote.
Further commentary
Asked to comment on the findings, editorialist Michelle Van Kuiken, MD, assistant professor of urology, University of California, San Francisco, acknowledged that there is not a lot of good evidence to support many of the strategies recommended by the American Urological Association to prevent and treat rUTIs, but she often follows these recommendations anyway. “The one statement in the guidelines that is the most supported by evidence is the use of cranberry supplements, and I do routinely recommended daily use of some form of concentrated cranberry supplements for all of my patients with rUTIs,” she said in an interview.
Dr. Van Kuiken said that vaginal estrogen is a very good option for all postmenopausal women who suffer from rUTIs and that there is growing acceptance of its use for this and other indications. There is some evidence to support D-mannose as well, although it’s not that robust, she acknowledged.
She said the evidence supporting the use of probiotics for this indication is very thin. She does not routinely recommend them for rUTIs, although they are not inherently harmful. “I think for a lot of women who have rUTIs, it can be pretty debilitating and upsetting for them – it can impact travel plans, work, and social events,” Dr. Van Kuiken said.
“Until we develop better diagnostic and therapeutic strategies, validating women’s experiences and concerns with rUTI while limiting unnecessary antibiotics remains our best option,” she wrote.
Dr. Scott and Dr. Van Kuiken have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Fear of antibiotic overuse and frustration with physicians who prescribe them too freely are key sentiments expressed by women with recurrent urinary tract infections (rUTIs), according to findings from a study involving six focus groups.
“Here in our female pelvic medicine reconstructive urology clinic at Cedars-Sinai and at UCLA, we see many women who are referred for evaluation of rUTIs who are very frustrated with their care,” Victoria Scott, MD, Cedars-Sinai Medical Center, Los Angeles, said in an interview.
“So with these focus groups, we saw an opportunity to explore why women are so frustrated and to try and improve the care delivered,” she added.
Findings from the study were published online Sept. 1 in The Journal of Urology.
“There is a need for physicians to modify management strategies ... and to devote more research efforts to improving nonantibiotic options for the prevention and treatment of recurrent urinary tract infections, as well as management strategies that better empower patients,” the authors wrote.
Six focus groups
Four or five participants were included in each of the six focus groups – a total of 29 women. All participants reported a history of symptomatic, culture-proven UTI episodes. They had experienced two or more infections in 6 months or three or more infections within 1 year. Women were predominantly White. Most were employed part- or full-time and held a college degree.
From a qualitative analysis of all focus group transcripts, two main themes emerged:
- The negative impact of taking antibiotics for the prevention and treatment of rUTIs.
- Resentment of the medical profession for the way it managed rUTIs.
The researchers found that participants had a good understanding of the deleterious effects from inappropriate antibiotic use, largely gleaned from media sources and the Internet. “Numerous women stated that they had reached such a level of concern about antibiotics that they would resist taking them for prevention or treatment of infections,” Dr. Scott and colleagues pointed out.
These concerns centered around the risk of developing resistance to antibiotics and the ill effects that antibiotics can have on the gastrointestinal and genitourinary microbiomes. Several women reported that they had developed Clostridium difficile infections after taking antibiotics; one of the patients required hospitalization for the infection.
Women also reported concerns that they had been given an antibiotic needlessly for symptoms that might have been caused by a genitourinary condition other than a UTI. They also reported feeling resentful toward practitioners, particularly if they felt the practitioner was overprescribing antibiotics. Some had resorted to consultations with alternative practitioners, such as herbalists. “A second concern discussed by participants was the feeling of being ignored by physicians,” the authors observed.
In this regard, the women felt that their physicians underestimated the burden that rUTIs had on their lives and the detrimental effect that repeated infections had on their relationships, work, and overall quality of life. “These perceptions led to a prevalent mistrust of physicians,” the investigators wrote. This prompted many women to insist that the medical community devote more effort to the development of nonantibiotic options for the prevention and treatment of UTIs.
Improved management strategies
Asked how physicians might improve their management of rUTIs, Dr. Scott shared a number of suggestions. Cardinal rule No. 1: Have the patient undergo a urinalysis to make sure she does have a UTI. “There is a subset of patients among women with rUTIs who come in with a diagnosis of an rUTI but who really have not had documentation of more than one positive urine culture,” Dr. Scott noted. Such a history suggests that they do not have an rUTI.
It’s imperative that physicians rule out commonly misdiagnosed disorders, such as overactive bladder, as a cause of the patient’s symptoms. Symptoms of overactive bladder and rUTIs often overlap. While waiting for results from the urinalysis to confirm or rule out a UTI, young and healthy women may be prescribed a nonsteroidal anti-inflammatory drug (NSAID), such as naproxen, which can help ameliorate symptoms.
Because UTIs are frequently self-limiting, Dr. Scott and others have found that for young, otherwise healthy women, NSAIDs alone can often resolve symptoms of the UTI without use of an antibiotic. For relatively severe symptoms, a urinary analgesic, such as phenazopyridine (Pyridium), may soothe the lining of the urinary tract and relieve pain. Cystex is an over-the-counter urinary analgesic that women can procure themselves, Dr. Scott added.
If an antibiotic is indicated, those most commonly prescribed for a single episode of acute cystitis are nitrofurantoin and sulfamethoxazole plus trimethoprim (Bactrim). For recurrent UTIs, “patients are a bit more complicated,” Dr. Scott admitted. “I think the best practice is to look back at a woman’s prior urine culture and select an antibiotic that showed good sensitivity in the last positive urine test,” she said.
Prevention starts with behavioral strategies, such as voiding after sexual intercourse and wiping from front to back following urination to avoid introducing fecal bacteria into the urethra. Evidence suggests that premenopausal women who drink at least 1.5 L of water a day have significantly fewer UTI episodes, Dr. Scott noted. There is also “pretty good” evidence that cranberry supplements (not juice) can prevent rUTIs. Use of cranberry supplements is supported by the American Urological Association (conditional recommendation; evidence level of grade C).
For peri- and postmenopausal women, vaginal estrogen may be effective. It’s use for UTI prevention is well supported by the literature. Although not as well supported by evidence, some women find that a supplement such as D-mannose may prevent or treat UTIs by causing bacteria to bind to it rather than to the bladder wall. Probiotics are another possibility, she noted. Empathy can’t hurt, she added.
“A common theme among satisfied women was the sentiment that their physicians understood their problems and had a system in place to allow rapid diagnosis and treatment for UTI episodes,” the authors emphasized.
“[Such attitudes] highlight the need to investigate each patient’s experience and perceptions to allow for shared decision making regarding the management of rUTIs,” they wrote.
Further commentary
Asked to comment on the findings, editorialist Michelle Van Kuiken, MD, assistant professor of urology, University of California, San Francisco, acknowledged that there is not a lot of good evidence to support many of the strategies recommended by the American Urological Association to prevent and treat rUTIs, but she often follows these recommendations anyway. “The one statement in the guidelines that is the most supported by evidence is the use of cranberry supplements, and I do routinely recommended daily use of some form of concentrated cranberry supplements for all of my patients with rUTIs,” she said in an interview.
Dr. Van Kuiken said that vaginal estrogen is a very good option for all postmenopausal women who suffer from rUTIs and that there is growing acceptance of its use for this and other indications. There is some evidence to support D-mannose as well, although it’s not that robust, she acknowledged.
She said the evidence supporting the use of probiotics for this indication is very thin. She does not routinely recommend them for rUTIs, although they are not inherently harmful. “I think for a lot of women who have rUTIs, it can be pretty debilitating and upsetting for them – it can impact travel plans, work, and social events,” Dr. Van Kuiken said.
“Until we develop better diagnostic and therapeutic strategies, validating women’s experiences and concerns with rUTI while limiting unnecessary antibiotics remains our best option,” she wrote.
Dr. Scott and Dr. Van Kuiken have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Effect of COVID-19 pandemic on respiratory infectious diseases in primary care practice
A secondary consequence of public health measures to prevent the spread of SARS-CoV-2 included a concurrent reduction in risk for children to acquire and spread other respiratory viral infectious diseases. In the Rochester, N.Y., area, we had an ongoing prospective study in primary care pediatric practices that afforded an opportunity to assess the effect of the pandemic control measures on all infectious disease illness visits in young children. Specifically, in children aged 6-36 months old, our study was in place when the pandemic began with a primary objective to evaluate the changing epidemiology of acute otitis media (AOM) and nasopharyngeal colonization by potential bacterial respiratory pathogens in community-based primary care pediatric practices. As the public health measures mandated by New York State Department of Health were implemented, we prospectively quantified their effect on physician-diagnosed infectious disease illness visits. The incidence of infectious disease visits by a cohort of young children during the COVID-19 pandemic period March 15, 2020, through Dec. 31, 2020, was compared with the same time frame in the preceding year, 2019.1

Recommendations of the New York State Department of Health for public health, changes in school and day care attendance, and clinical practice during the study time frame
On March 7, 2020, a state of emergency was declared in New York because of the COVID-19 pandemic. All schools were required to close. A mandated order for public use of masks in adults and children more than 2 years of age was enacted. In the Finger Lakes region of Upstate New York, where the two primary care pediatric practices reside, complete lockdown was partially lifted on May 15, 2020, and further lifted on June 26, 2020. Almost all regional school districts opened to at least hybrid learning models for all students starting Sept. 8, 2020. On March 6, 2020, video telehealth and telephone call visits were introduced as routine practice. Well-child visits were limited to those less than 2 years of age, then gradually expanded to all ages by late May 2020. During the “stay at home” phase of the New York State lockdown, day care services were considered an essential business. Day care child density was limited. All children less than 2 years old were required to wear a mask while in the facility. Upon arrival, children with any respiratory symptoms or fever were excluded. For the school year commencing September 2020, almost all regional school districts opened to virtual, hybrid, or in-person learning models. Exclusion occurred similar to that of the day care facilities.
Incidence of respiratory infectious disease illnesses
Clinical diagnoses and healthy visits of 144 children from March 15 to Dec. 31, 2020 (beginning of the pandemic) were compared to 215 children during the same months in 2019 (prepandemic). Pediatric SARS-CoV-2 positivity rates trended up alongside community spread. Pediatric practice positivity rates rose from 1.9% in October 2020 to 19% in December 2020.
The table shows the incidence of significantly different infectious disease illness visits in the two study cohorts.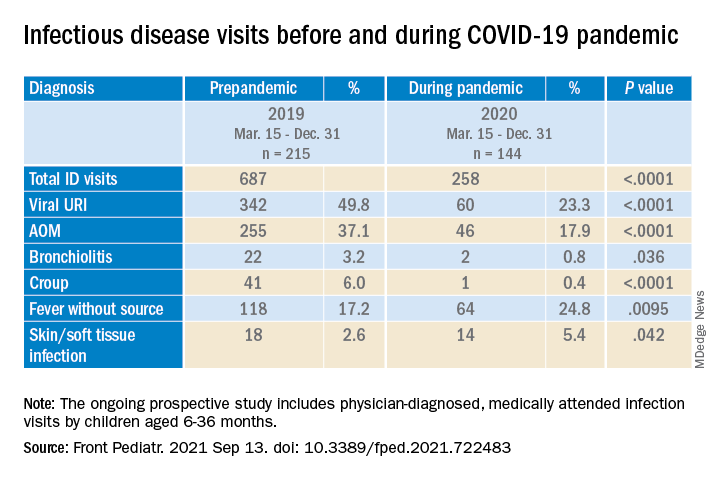
During the pandemic, 258 infection visits occurred among 144 pandemic cohort children, compared with 687 visits among 215 prepandemic cohort children, a 1.8-fold decrease (P < .0001). The proportion of children with visits for AOM (3.7-fold; P < .0001), bronchiolitis (7.4-fold; P = .036), croup (27.5-fold; P < .0001), and viral upper respiratory infection (3.8-fold; P < .0001) decreased significantly. Fever without a source (1.4-fold decrease; P = .009) and skin/soft tissue infection (2.1-fold decrease; P = .042) represented a higher proportion of visits during the pandemic.
Prescription of antibiotics significantly decreased (P < .001) during the pandemic.
Change in care practices
In the prepandemic period, virtual visits, leading to a diagnosis and treatment and referring children to an urgent care or hospital emergency department during regular office hours were rare. During the pandemic, this changed. Significantly increased use of telemedicine visits (P < .0001) and significantly decreased office and urgent care visits (P < .0001) occurred during the pandemic. Telehealth visits peaked the week of April 12, 2020, at 45% of all pediatric visits. In-person illness visits gradually returned to year-to-year volumes in August-September 2020 with school opening. Early in the pandemic, both pediatric offices limited patient encounters to well-child visits in the first 2 years of life to not miss opportunities for childhood vaccinations. However, some parents were reluctant to bring their children to those visits. There was no significant change in frequency of healthy child visits during the pandemic.

To our knowledge, this was the first study from primary care pediatric practices in the United States to analyze the effect on infectious diseases during the first 9 months of the pandemic, including the 6-month time period after the reopening from the first 3 months of lockdown. One prior study from a primary care network in Massachusetts reported significant decreases in respiratory infectious diseases for children aged 0-17 years during the first months of the pandemic during lockdown.2 A study in Tennessee that included hospital emergency department, urgent care, primary care, and retail health clinics also reported respiratory infection diagnoses as well as antibiotic prescription were reduced in the early months of the pandemic.3
Our study shows an overall reduction in frequency of respiratory illness visits in children 6-36 months old during the first 9 months of the COVID-19 pandemic. We learned the value of using technology in the form of virtual visits to render care. Perhaps as the pandemic subsides, many of the hand-washing and sanitizing practices will remain in place and lead to less frequent illness in children in the future. However, there may be temporary negative consequences from the “immune debt” that has occurred from a prolonged time span when children were not becoming infected with respiratory pathogens.4 We will see what unfolds in the future.
Dr. Pichichero is a specialist in pediatric infectious diseases and director of the Research Institute at Rochester (N.Y.) General Hospital. Dr. Schulz is pediatric medical director at Rochester (N.Y.) Regional Health. Dr. Pichichero and Dr. Schulz have no conflicts of interest to disclose. This study was funded in part by the Centers for Disease Control and Prevention.
References
1. Kaur R et al. Front Pediatr. 2021;(9)722483:1-8.
2. Hatoun J et al. Pediatrics. 2020;146(4):e2020006460.
3. Katz SE et al. J Pediatric Infect Dis Soc. 2021;10(1):62-4.
4. Cohen R et al. Infect. Dis Now. 2021; 51(5)418-23.
A secondary consequence of public health measures to prevent the spread of SARS-CoV-2 included a concurrent reduction in risk for children to acquire and spread other respiratory viral infectious diseases. In the Rochester, N.Y., area, we had an ongoing prospective study in primary care pediatric practices that afforded an opportunity to assess the effect of the pandemic control measures on all infectious disease illness visits in young children. Specifically, in children aged 6-36 months old, our study was in place when the pandemic began with a primary objective to evaluate the changing epidemiology of acute otitis media (AOM) and nasopharyngeal colonization by potential bacterial respiratory pathogens in community-based primary care pediatric practices. As the public health measures mandated by New York State Department of Health were implemented, we prospectively quantified their effect on physician-diagnosed infectious disease illness visits. The incidence of infectious disease visits by a cohort of young children during the COVID-19 pandemic period March 15, 2020, through Dec. 31, 2020, was compared with the same time frame in the preceding year, 2019.1
Recommendations of the New York State Department of Health for public health, changes in school and day care attendance, and clinical practice during the study time frame
On March 7, 2020, a state of emergency was declared in New York because of the COVID-19 pandemic. All schools were required to close. A mandated order for public use of masks in adults and children more than 2 years of age was enacted. In the Finger Lakes region of Upstate New York, where the two primary care pediatric practices reside, complete lockdown was partially lifted on May 15, 2020, and further lifted on June 26, 2020. Almost all regional school districts opened to at least hybrid learning models for all students starting Sept. 8, 2020. On March 6, 2020, video telehealth and telephone call visits were introduced as routine practice. Well-child visits were limited to those less than 2 years of age, then gradually expanded to all ages by late May 2020. During the “stay at home” phase of the New York State lockdown, day care services were considered an essential business. Day care child density was limited. All children less than 2 years old were required to wear a mask while in the facility. Upon arrival, children with any respiratory symptoms or fever were excluded. For the school year commencing September 2020, almost all regional school districts opened to virtual, hybrid, or in-person learning models. Exclusion occurred similar to that of the day care facilities.
Incidence of respiratory infectious disease illnesses
Clinical diagnoses and healthy visits of 144 children from March 15 to Dec. 31, 2020 (beginning of the pandemic) were compared to 215 children during the same months in 2019 (prepandemic). Pediatric SARS-CoV-2 positivity rates trended up alongside community spread. Pediatric practice positivity rates rose from 1.9% in October 2020 to 19% in December 2020.
The table shows the incidence of significantly different infectious disease illness visits in the two study cohorts.
During the pandemic, 258 infection visits occurred among 144 pandemic cohort children, compared with 687 visits among 215 prepandemic cohort children, a 1.8-fold decrease (P < .0001). The proportion of children with visits for AOM (3.7-fold; P < .0001), bronchiolitis (7.4-fold; P = .036), croup (27.5-fold; P < .0001), and viral upper respiratory infection (3.8-fold; P < .0001) decreased significantly. Fever without a source (1.4-fold decrease; P = .009) and skin/soft tissue infection (2.1-fold decrease; P = .042) represented a higher proportion of visits during the pandemic.
Prescription of antibiotics significantly decreased (P < .001) during the pandemic.
Change in care practices
In the prepandemic period, virtual visits, leading to a diagnosis and treatment and referring children to an urgent care or hospital emergency department during regular office hours were rare. During the pandemic, this changed. Significantly increased use of telemedicine visits (P < .0001) and significantly decreased office and urgent care visits (P < .0001) occurred during the pandemic. Telehealth visits peaked the week of April 12, 2020, at 45% of all pediatric visits. In-person illness visits gradually returned to year-to-year volumes in August-September 2020 with school opening. Early in the pandemic, both pediatric offices limited patient encounters to well-child visits in the first 2 years of life to not miss opportunities for childhood vaccinations. However, some parents were reluctant to bring their children to those visits. There was no significant change in frequency of healthy child visits during the pandemic.
To our knowledge, this was the first study from primary care pediatric practices in the United States to analyze the effect on infectious diseases during the first 9 months of the pandemic, including the 6-month time period after the reopening from the first 3 months of lockdown. One prior study from a primary care network in Massachusetts reported significant decreases in respiratory infectious diseases for children aged 0-17 years during the first months of the pandemic during lockdown.2 A study in Tennessee that included hospital emergency department, urgent care, primary care, and retail health clinics also reported respiratory infection diagnoses as well as antibiotic prescription were reduced in the early months of the pandemic.3
Our study shows an overall reduction in frequency of respiratory illness visits in children 6-36 months old during the first 9 months of the COVID-19 pandemic. We learned the value of using technology in the form of virtual visits to render care. Perhaps as the pandemic subsides, many of the hand-washing and sanitizing practices will remain in place and lead to less frequent illness in children in the future. However, there may be temporary negative consequences from the “immune debt” that has occurred from a prolonged time span when children were not becoming infected with respiratory pathogens.4 We will see what unfolds in the future.
Dr. Pichichero is a specialist in pediatric infectious diseases and director of the Research Institute at Rochester (N.Y.) General Hospital. Dr. Schulz is pediatric medical director at Rochester (N.Y.) Regional Health. Dr. Pichichero and Dr. Schulz have no conflicts of interest to disclose. This study was funded in part by the Centers for Disease Control and Prevention.
References
1. Kaur R et al. Front Pediatr. 2021;(9)722483:1-8.
2. Hatoun J et al. Pediatrics. 2020;146(4):e2020006460.
3. Katz SE et al. J Pediatric Infect Dis Soc. 2021;10(1):62-4.
4. Cohen R et al. Infect. Dis Now. 2021; 51(5)418-23.
A secondary consequence of public health measures to prevent the spread of SARS-CoV-2 included a concurrent reduction in risk for children to acquire and spread other respiratory viral infectious diseases. In the Rochester, N.Y., area, we had an ongoing prospective study in primary care pediatric practices that afforded an opportunity to assess the effect of the pandemic control measures on all infectious disease illness visits in young children. Specifically, in children aged 6-36 months old, our study was in place when the pandemic began with a primary objective to evaluate the changing epidemiology of acute otitis media (AOM) and nasopharyngeal colonization by potential bacterial respiratory pathogens in community-based primary care pediatric practices. As the public health measures mandated by New York State Department of Health were implemented, we prospectively quantified their effect on physician-diagnosed infectious disease illness visits. The incidence of infectious disease visits by a cohort of young children during the COVID-19 pandemic period March 15, 2020, through Dec. 31, 2020, was compared with the same time frame in the preceding year, 2019.1
Recommendations of the New York State Department of Health for public health, changes in school and day care attendance, and clinical practice during the study time frame
On March 7, 2020, a state of emergency was declared in New York because of the COVID-19 pandemic. All schools were required to close. A mandated order for public use of masks in adults and children more than 2 years of age was enacted. In the Finger Lakes region of Upstate New York, where the two primary care pediatric practices reside, complete lockdown was partially lifted on May 15, 2020, and further lifted on June 26, 2020. Almost all regional school districts opened to at least hybrid learning models for all students starting Sept. 8, 2020. On March 6, 2020, video telehealth and telephone call visits were introduced as routine practice. Well-child visits were limited to those less than 2 years of age, then gradually expanded to all ages by late May 2020. During the “stay at home” phase of the New York State lockdown, day care services were considered an essential business. Day care child density was limited. All children less than 2 years old were required to wear a mask while in the facility. Upon arrival, children with any respiratory symptoms or fever were excluded. For the school year commencing September 2020, almost all regional school districts opened to virtual, hybrid, or in-person learning models. Exclusion occurred similar to that of the day care facilities.
Incidence of respiratory infectious disease illnesses
Clinical diagnoses and healthy visits of 144 children from March 15 to Dec. 31, 2020 (beginning of the pandemic) were compared to 215 children during the same months in 2019 (prepandemic). Pediatric SARS-CoV-2 positivity rates trended up alongside community spread. Pediatric practice positivity rates rose from 1.9% in October 2020 to 19% in December 2020.
The table shows the incidence of significantly different infectious disease illness visits in the two study cohorts.
During the pandemic, 258 infection visits occurred among 144 pandemic cohort children, compared with 687 visits among 215 prepandemic cohort children, a 1.8-fold decrease (P < .0001). The proportion of children with visits for AOM (3.7-fold; P < .0001), bronchiolitis (7.4-fold; P = .036), croup (27.5-fold; P < .0001), and viral upper respiratory infection (3.8-fold; P < .0001) decreased significantly. Fever without a source (1.4-fold decrease; P = .009) and skin/soft tissue infection (2.1-fold decrease; P = .042) represented a higher proportion of visits during the pandemic.
Prescription of antibiotics significantly decreased (P < .001) during the pandemic.
Change in care practices
In the prepandemic period, virtual visits, leading to a diagnosis and treatment and referring children to an urgent care or hospital emergency department during regular office hours were rare. During the pandemic, this changed. Significantly increased use of telemedicine visits (P < .0001) and significantly decreased office and urgent care visits (P < .0001) occurred during the pandemic. Telehealth visits peaked the week of April 12, 2020, at 45% of all pediatric visits. In-person illness visits gradually returned to year-to-year volumes in August-September 2020 with school opening. Early in the pandemic, both pediatric offices limited patient encounters to well-child visits in the first 2 years of life to not miss opportunities for childhood vaccinations. However, some parents were reluctant to bring their children to those visits. There was no significant change in frequency of healthy child visits during the pandemic.
To our knowledge, this was the first study from primary care pediatric practices in the United States to analyze the effect on infectious diseases during the first 9 months of the pandemic, including the 6-month time period after the reopening from the first 3 months of lockdown. One prior study from a primary care network in Massachusetts reported significant decreases in respiratory infectious diseases for children aged 0-17 years during the first months of the pandemic during lockdown.2 A study in Tennessee that included hospital emergency department, urgent care, primary care, and retail health clinics also reported respiratory infection diagnoses as well as antibiotic prescription were reduced in the early months of the pandemic.3
Our study shows an overall reduction in frequency of respiratory illness visits in children 6-36 months old during the first 9 months of the COVID-19 pandemic. We learned the value of using technology in the form of virtual visits to render care. Perhaps as the pandemic subsides, many of the hand-washing and sanitizing practices will remain in place and lead to less frequent illness in children in the future. However, there may be temporary negative consequences from the “immune debt” that has occurred from a prolonged time span when children were not becoming infected with respiratory pathogens.4 We will see what unfolds in the future.
Dr. Pichichero is a specialist in pediatric infectious diseases and director of the Research Institute at Rochester (N.Y.) General Hospital. Dr. Schulz is pediatric medical director at Rochester (N.Y.) Regional Health. Dr. Pichichero and Dr. Schulz have no conflicts of interest to disclose. This study was funded in part by the Centers for Disease Control and Prevention.
References
1. Kaur R et al. Front Pediatr. 2021;(9)722483:1-8.
2. Hatoun J et al. Pediatrics. 2020;146(4):e2020006460.
3. Katz SE et al. J Pediatric Infect Dis Soc. 2021;10(1):62-4.
4. Cohen R et al. Infect. Dis Now. 2021; 51(5)418-23.
Staff education cuts psychotropic drug use in long-term care
The effect of the intervention was transient, possibly because of high staff turnover, according to the investigators in the new randomized, controlled trial.
The findings were presented by Ulla Aalto, MD, PhD, during a session at the European Geriatric Medicine Society annual congress, a hybrid live and online meeting.
There was a significant reduction in the use of psychotropic agents at 6 months in long-term care wards where the nursing staff had undergone a short training session on drug therapy for older patients, but there was no improvement in wards that were randomly assigned to serve as controls, Dr. Aalto, from Helsinki Hospital, reported during the session.
“Future research would be investigating how we could maintain the positive effects that were gained at 6 months but not seen any more at 1 year, and how to implement the good practice in nursing homes by this kind of staff training,” she said.
Heavy drug use
Psychotropic medications are widely used in long-term care settings, but their indiscriminate use or use of the wrong drug for the wrong patient can be harmful. Inappropriate drug use in long-term care settings is also associated with higher costs, Dr. Aalto said.
To see whether a staff-training intervention could reduce drugs use and lower costs, the investigators conducted a randomized clinical trial in assisted living facilities in Helsinki in 2011, with a total of 227 patients 65 years and older.
Long-term care wards were randomly assigned to either an intervention for nursing staff consisting of two 4-hour sessions on good drug-therapy practice for older adults, or to serve as controls (10 wards in each group).
Drug use and costs were monitored at both 6 and 12 months after randomization. Psychotropic drugs included antipsychotics, antidepressants, anxiolytics, and hypnotics as classified by the World Health Organization. For the purposes of comparison, actual doses were counted and converted into relative proportions of defined daily doses.
The baseline characteristics of patients in each group were generally similar, with a mean age of around 83 years. In each study arm, nearly two-thirds of patients were on at least one psychotropic drug, and of this group, a third had been prescribed 2 or more psychotropic agents.
Nearly half of the patients were on at least one antipsychotic agent and/or antidepressant.
Short-term benefit
As noted before, in the wards randomized to staff training, there was a significant reduction in use of all psychotropics from baseline at 6 months after randomization (P = .045), but there was no change among the control wards.
By 12 months, however, the differences between the intervention and control arms narrowed, and drug use in the intervention arm was no longer significantly lower over baseline.
Drugs costs significantly decreased in the intervention group at 6 months (P = .027) and were numerically but not statistically lower over baseline at 12 months.
In contrast, drug costs in the control arm were numerically (but not statistically) higher at both 6 and 12 months of follow-up.
Annual drug costs in the intervention group decreased by mean of 12.3 euros ($14.22) whereas costs in the control group increased by a mean of 20.6 euros ($23.81).
“This quite light and feasible intervention succeeded in reducing overall defined daily doses of psychotropics in the short term,” Dr. Aalto said.
The waning of the intervention’s effect on drug use and costs may be caused partly by the high employee turnover rate in long-term care facilities and to the dilution effect, she said, referring to a form of judgment bias in which people tend to devalue diagnostic information when other, nondiagnostic information is also available.
Randomized design
In the question-and-answer session following her presentation, audience member Jesper Ryg, MD, PhD from Odense (Denmark) University Hospital and the University of Southern Denmark, also in Odense, commented: “It’s a great study, doing a [randomized, controlled trial] on deprescribing, we need more of those.”
“But what we know now is that a lot of studies show it is possible to deprescribe and get less drugs, but do we have any clinical data? Does this deprescribing lead to less falls, did it lead to lower mortality?” he asked.
Dr. Aalto replied that, in an earlier report from this study, investigators showed that harmful medication use was reduced and negative outcomes were reduced.
Another audience member asked why nursing staff were the target of the intervention, given that physicians do the actual drug prescribing.
Dr. Aalto responded: “It is the physician of course who prescribes, but in nursing homes and long-term care, nursing staff is there all the time, and the physicians are kind of consultants who just come there once in a while, so it’s important that the nurses also know about these harmful medications and can bring them to the doctor when he or she arrives there.”
Dr. Aalto and Dr. Ryg had no disclosures.
The effect of the intervention was transient, possibly because of high staff turnover, according to the investigators in the new randomized, controlled trial.
The findings were presented by Ulla Aalto, MD, PhD, during a session at the European Geriatric Medicine Society annual congress, a hybrid live and online meeting.
There was a significant reduction in the use of psychotropic agents at 6 months in long-term care wards where the nursing staff had undergone a short training session on drug therapy for older patients, but there was no improvement in wards that were randomly assigned to serve as controls, Dr. Aalto, from Helsinki Hospital, reported during the session.
“Future research would be investigating how we could maintain the positive effects that were gained at 6 months but not seen any more at 1 year, and how to implement the good practice in nursing homes by this kind of staff training,” she said.
Heavy drug use
Psychotropic medications are widely used in long-term care settings, but their indiscriminate use or use of the wrong drug for the wrong patient can be harmful. Inappropriate drug use in long-term care settings is also associated with higher costs, Dr. Aalto said.
To see whether a staff-training intervention could reduce drugs use and lower costs, the investigators conducted a randomized clinical trial in assisted living facilities in Helsinki in 2011, with a total of 227 patients 65 years and older.
Long-term care wards were randomly assigned to either an intervention for nursing staff consisting of two 4-hour sessions on good drug-therapy practice for older adults, or to serve as controls (10 wards in each group).
Drug use and costs were monitored at both 6 and 12 months after randomization. Psychotropic drugs included antipsychotics, antidepressants, anxiolytics, and hypnotics as classified by the World Health Organization. For the purposes of comparison, actual doses were counted and converted into relative proportions of defined daily doses.
The baseline characteristics of patients in each group were generally similar, with a mean age of around 83 years. In each study arm, nearly two-thirds of patients were on at least one psychotropic drug, and of this group, a third had been prescribed 2 or more psychotropic agents.
Nearly half of the patients were on at least one antipsychotic agent and/or antidepressant.
Short-term benefit
As noted before, in the wards randomized to staff training, there was a significant reduction in use of all psychotropics from baseline at 6 months after randomization (P = .045), but there was no change among the control wards.
By 12 months, however, the differences between the intervention and control arms narrowed, and drug use in the intervention arm was no longer significantly lower over baseline.
Drugs costs significantly decreased in the intervention group at 6 months (P = .027) and were numerically but not statistically lower over baseline at 12 months.
In contrast, drug costs in the control arm were numerically (but not statistically) higher at both 6 and 12 months of follow-up.
Annual drug costs in the intervention group decreased by mean of 12.3 euros ($14.22) whereas costs in the control group increased by a mean of 20.6 euros ($23.81).
“This quite light and feasible intervention succeeded in reducing overall defined daily doses of psychotropics in the short term,” Dr. Aalto said.
The waning of the intervention’s effect on drug use and costs may be caused partly by the high employee turnover rate in long-term care facilities and to the dilution effect, she said, referring to a form of judgment bias in which people tend to devalue diagnostic information when other, nondiagnostic information is also available.
Randomized design
In the question-and-answer session following her presentation, audience member Jesper Ryg, MD, PhD from Odense (Denmark) University Hospital and the University of Southern Denmark, also in Odense, commented: “It’s a great study, doing a [randomized, controlled trial] on deprescribing, we need more of those.”
“But what we know now is that a lot of studies show it is possible to deprescribe and get less drugs, but do we have any clinical data? Does this deprescribing lead to less falls, did it lead to lower mortality?” he asked.
Dr. Aalto replied that, in an earlier report from this study, investigators showed that harmful medication use was reduced and negative outcomes were reduced.
Another audience member asked why nursing staff were the target of the intervention, given that physicians do the actual drug prescribing.
Dr. Aalto responded: “It is the physician of course who prescribes, but in nursing homes and long-term care, nursing staff is there all the time, and the physicians are kind of consultants who just come there once in a while, so it’s important that the nurses also know about these harmful medications and can bring them to the doctor when he or she arrives there.”
Dr. Aalto and Dr. Ryg had no disclosures.
The effect of the intervention was transient, possibly because of high staff turnover, according to the investigators in the new randomized, controlled trial.
The findings were presented by Ulla Aalto, MD, PhD, during a session at the European Geriatric Medicine Society annual congress, a hybrid live and online meeting.
There was a significant reduction in the use of psychotropic agents at 6 months in long-term care wards where the nursing staff had undergone a short training session on drug therapy for older patients, but there was no improvement in wards that were randomly assigned to serve as controls, Dr. Aalto, from Helsinki Hospital, reported during the session.
“Future research would be investigating how we could maintain the positive effects that were gained at 6 months but not seen any more at 1 year, and how to implement the good practice in nursing homes by this kind of staff training,” she said.
Heavy drug use
Psychotropic medications are widely used in long-term care settings, but their indiscriminate use or use of the wrong drug for the wrong patient can be harmful. Inappropriate drug use in long-term care settings is also associated with higher costs, Dr. Aalto said.
To see whether a staff-training intervention could reduce drugs use and lower costs, the investigators conducted a randomized clinical trial in assisted living facilities in Helsinki in 2011, with a total of 227 patients 65 years and older.
Long-term care wards were randomly assigned to either an intervention for nursing staff consisting of two 4-hour sessions on good drug-therapy practice for older adults, or to serve as controls (10 wards in each group).
Drug use and costs were monitored at both 6 and 12 months after randomization. Psychotropic drugs included antipsychotics, antidepressants, anxiolytics, and hypnotics as classified by the World Health Organization. For the purposes of comparison, actual doses were counted and converted into relative proportions of defined daily doses.
The baseline characteristics of patients in each group were generally similar, with a mean age of around 83 years. In each study arm, nearly two-thirds of patients were on at least one psychotropic drug, and of this group, a third had been prescribed 2 or more psychotropic agents.
Nearly half of the patients were on at least one antipsychotic agent and/or antidepressant.
Short-term benefit
As noted before, in the wards randomized to staff training, there was a significant reduction in use of all psychotropics from baseline at 6 months after randomization (P = .045), but there was no change among the control wards.
By 12 months, however, the differences between the intervention and control arms narrowed, and drug use in the intervention arm was no longer significantly lower over baseline.
Drugs costs significantly decreased in the intervention group at 6 months (P = .027) and were numerically but not statistically lower over baseline at 12 months.
In contrast, drug costs in the control arm were numerically (but not statistically) higher at both 6 and 12 months of follow-up.
Annual drug costs in the intervention group decreased by mean of 12.3 euros ($14.22) whereas costs in the control group increased by a mean of 20.6 euros ($23.81).
“This quite light and feasible intervention succeeded in reducing overall defined daily doses of psychotropics in the short term,” Dr. Aalto said.
The waning of the intervention’s effect on drug use and costs may be caused partly by the high employee turnover rate in long-term care facilities and to the dilution effect, she said, referring to a form of judgment bias in which people tend to devalue diagnostic information when other, nondiagnostic information is also available.
Randomized design
In the question-and-answer session following her presentation, audience member Jesper Ryg, MD, PhD from Odense (Denmark) University Hospital and the University of Southern Denmark, also in Odense, commented: “It’s a great study, doing a [randomized, controlled trial] on deprescribing, we need more of those.”
“But what we know now is that a lot of studies show it is possible to deprescribe and get less drugs, but do we have any clinical data? Does this deprescribing lead to less falls, did it lead to lower mortality?” he asked.
Dr. Aalto replied that, in an earlier report from this study, investigators showed that harmful medication use was reduced and negative outcomes were reduced.
Another audience member asked why nursing staff were the target of the intervention, given that physicians do the actual drug prescribing.
Dr. Aalto responded: “It is the physician of course who prescribes, but in nursing homes and long-term care, nursing staff is there all the time, and the physicians are kind of consultants who just come there once in a while, so it’s important that the nurses also know about these harmful medications and can bring them to the doctor when he or she arrives there.”
Dr. Aalto and Dr. Ryg had no disclosures.
FROM EUGMS 2021
Low preconception complement levels linked to adverse pregnancy outcomes in antiphospholipid syndrome
Low serum levels of two complement proteins are linked to worse pregnancy outcomes in women with antiphospholipid syndrome (APS), the results of a multicenter study appear to confirm.
The study evaluated preconception complement levels in 260 pregnancies in 197 women who had APS or carried antiphospholipid antibodies (aPL), and found that low levels of C3 and C4 in the 6 months prior to pregnancy were associated with several gestational complications and resulted in pregnancy losses.
“This study has validated, on large scale, the possible utility of preconception measurement of C3 and C4 levels to predict pregnancy loss in patients with aPL, even at a high-risk profile,” said study investigator Daniele Lini, MD, of ASST Spedali Civili and the University of Brescia (Italy).
“The tests are easy and cheap to be routinely performed, and they could therefore represent a valid aid to identify women that need particular monitoring and management,” he said at the 14th International Congress on Systemic Lupus Erythematosus held together with the 6th International Congress on Controversies in Rheumatology and Autoimmunity.
aPL and adverse obstetric outcomes
aPL, which include lupus anticoagulant, anti–beta2-glycoprotein 1, and anticardiolipin antibodies, have been shown to induce fetal loss in animal models. Their influence on the outcome of human pregnancies, however, has been less clear, with several studies failing to prove a link between their presence and obstetric complications.
Dr. Lini and coinvestigators conducted a multicenter study involving 11 Italian centers and one Russian center, retrospectively looking for women with primary APS or women who had persistently high levels of aPL but no symptoms who had become pregnant. Of 503 pregnancies, information on complement levels before conception was available for 260, of which 184 had occurred in women with APS and 76 in women with persistently high aPL.
The pregnancies were grouped according to whether there were low (n = 93) or normal (n = 167) levels of C3 and C4 in the last 6 months.
“Women with adverse pregnancy outcomes showed significantly lower preconception complement levels than those with successful pregnancies, without any difference between APS and aPL carriers,” Dr. Lini reported.
Comparing those with low to those with high complement levels, the preterm live birth rate (before 37 weeks’ gestation) was 37% versus 18% (P < .0001).
The full-term live birth rates were a respective 42% and 72% (P < .0001).
The rate of pregnancy loss, which included both abortion and miscarriage, was a respective 21% and 10% (P = .008).
A subgroup analysis focusing on where there was triple aPL positivity found that preconception low C3 and/or C4 levels was associated with an increased rate of pregnancy loss (P = .05). This association disappeared if there was just one or two aPL present.
The researchers found no correlation between complement levels and rates of venous thromboembolism or thrombocytopenia.
Study highlights ‘impact and importance’ of complement in APS
The study indicates “the impact and the importance of complement” in APS, said Yehuda Shoenfeld, MD, the founder and head of the Zabludowicz Center for Autoimmune Diseases at the Sheba Medical Center in Tel Hashomer, Israel.
In the early days of understanding APS, said Dr. Shoenfeld, it was thought that complement was not as important as it was in systemic lupus erythematosus (SLE). The importance of raised complement seen in studies of APS would often be discounted or neglected in comparison to SLE.
However, “slowly, slowly” it has been found that “complement [in APS] is activated very similarly to SLE,” Dr. Shoenfeld noted.
“I think that it’s important to assess the component levels,” Dr. Lini said in discussion. “This is needed to be done in the preconception counseling for APS and aPL carrier patients.”
Determining whether there is single, double, or even triple aPL positivity could be useful in guiding clinical decisions.
“If we have triple positivity, that could mean that there may be a more immunologic activation of the system and that it could be useful to administrate hydroxychloroquine [to] those patients who would like to have a pregnancy,” Dr. Lini suggested.
Plus, in those with decreased complement levels, “this could be a very useful tool” to identify where something could go wrong during their pregnancy.
The study had no outside funding. Dr. Lini and Dr. Shoenfeld disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Low serum levels of two complement proteins are linked to worse pregnancy outcomes in women with antiphospholipid syndrome (APS), the results of a multicenter study appear to confirm.
The study evaluated preconception complement levels in 260 pregnancies in 197 women who had APS or carried antiphospholipid antibodies (aPL), and found that low levels of C3 and C4 in the 6 months prior to pregnancy were associated with several gestational complications and resulted in pregnancy losses.
“This study has validated, on large scale, the possible utility of preconception measurement of C3 and C4 levels to predict pregnancy loss in patients with aPL, even at a high-risk profile,” said study investigator Daniele Lini, MD, of ASST Spedali Civili and the University of Brescia (Italy).
“The tests are easy and cheap to be routinely performed, and they could therefore represent a valid aid to identify women that need particular monitoring and management,” he said at the 14th International Congress on Systemic Lupus Erythematosus held together with the 6th International Congress on Controversies in Rheumatology and Autoimmunity.
aPL and adverse obstetric outcomes
aPL, which include lupus anticoagulant, anti–beta2-glycoprotein 1, and anticardiolipin antibodies, have been shown to induce fetal loss in animal models. Their influence on the outcome of human pregnancies, however, has been less clear, with several studies failing to prove a link between their presence and obstetric complications.
Dr. Lini and coinvestigators conducted a multicenter study involving 11 Italian centers and one Russian center, retrospectively looking for women with primary APS or women who had persistently high levels of aPL but no symptoms who had become pregnant. Of 503 pregnancies, information on complement levels before conception was available for 260, of which 184 had occurred in women with APS and 76 in women with persistently high aPL.
The pregnancies were grouped according to whether there were low (n = 93) or normal (n = 167) levels of C3 and C4 in the last 6 months.
“Women with adverse pregnancy outcomes showed significantly lower preconception complement levels than those with successful pregnancies, without any difference between APS and aPL carriers,” Dr. Lini reported.
Comparing those with low to those with high complement levels, the preterm live birth rate (before 37 weeks’ gestation) was 37% versus 18% (P < .0001).
The full-term live birth rates were a respective 42% and 72% (P < .0001).
The rate of pregnancy loss, which included both abortion and miscarriage, was a respective 21% and 10% (P = .008).
A subgroup analysis focusing on where there was triple aPL positivity found that preconception low C3 and/or C4 levels was associated with an increased rate of pregnancy loss (P = .05). This association disappeared if there was just one or two aPL present.
The researchers found no correlation between complement levels and rates of venous thromboembolism or thrombocytopenia.
Study highlights ‘impact and importance’ of complement in APS
The study indicates “the impact and the importance of complement” in APS, said Yehuda Shoenfeld, MD, the founder and head of the Zabludowicz Center for Autoimmune Diseases at the Sheba Medical Center in Tel Hashomer, Israel.
In the early days of understanding APS, said Dr. Shoenfeld, it was thought that complement was not as important as it was in systemic lupus erythematosus (SLE). The importance of raised complement seen in studies of APS would often be discounted or neglected in comparison to SLE.
However, “slowly, slowly” it has been found that “complement [in APS] is activated very similarly to SLE,” Dr. Shoenfeld noted.
“I think that it’s important to assess the component levels,” Dr. Lini said in discussion. “This is needed to be done in the preconception counseling for APS and aPL carrier patients.”
Determining whether there is single, double, or even triple aPL positivity could be useful in guiding clinical decisions.
“If we have triple positivity, that could mean that there may be a more immunologic activation of the system and that it could be useful to administrate hydroxychloroquine [to] those patients who would like to have a pregnancy,” Dr. Lini suggested.
Plus, in those with decreased complement levels, “this could be a very useful tool” to identify where something could go wrong during their pregnancy.
The study had no outside funding. Dr. Lini and Dr. Shoenfeld disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Low serum levels of two complement proteins are linked to worse pregnancy outcomes in women with antiphospholipid syndrome (APS), the results of a multicenter study appear to confirm.
The study evaluated preconception complement levels in 260 pregnancies in 197 women who had APS or carried antiphospholipid antibodies (aPL), and found that low levels of C3 and C4 in the 6 months prior to pregnancy were associated with several gestational complications and resulted in pregnancy losses.
“This study has validated, on large scale, the possible utility of preconception measurement of C3 and C4 levels to predict pregnancy loss in patients with aPL, even at a high-risk profile,” said study investigator Daniele Lini, MD, of ASST Spedali Civili and the University of Brescia (Italy).
“The tests are easy and cheap to be routinely performed, and they could therefore represent a valid aid to identify women that need particular monitoring and management,” he said at the 14th International Congress on Systemic Lupus Erythematosus held together with the 6th International Congress on Controversies in Rheumatology and Autoimmunity.
aPL and adverse obstetric outcomes
aPL, which include lupus anticoagulant, anti–beta2-glycoprotein 1, and anticardiolipin antibodies, have been shown to induce fetal loss in animal models. Their influence on the outcome of human pregnancies, however, has been less clear, with several studies failing to prove a link between their presence and obstetric complications.
Dr. Lini and coinvestigators conducted a multicenter study involving 11 Italian centers and one Russian center, retrospectively looking for women with primary APS or women who had persistently high levels of aPL but no symptoms who had become pregnant. Of 503 pregnancies, information on complement levels before conception was available for 260, of which 184 had occurred in women with APS and 76 in women with persistently high aPL.
The pregnancies were grouped according to whether there were low (n = 93) or normal (n = 167) levels of C3 and C4 in the last 6 months.
“Women with adverse pregnancy outcomes showed significantly lower preconception complement levels than those with successful pregnancies, without any difference between APS and aPL carriers,” Dr. Lini reported.
Comparing those with low to those with high complement levels, the preterm live birth rate (before 37 weeks’ gestation) was 37% versus 18% (P < .0001).
The full-term live birth rates were a respective 42% and 72% (P < .0001).
The rate of pregnancy loss, which included both abortion and miscarriage, was a respective 21% and 10% (P = .008).
A subgroup analysis focusing on where there was triple aPL positivity found that preconception low C3 and/or C4 levels was associated with an increased rate of pregnancy loss (P = .05). This association disappeared if there was just one or two aPL present.
The researchers found no correlation between complement levels and rates of venous thromboembolism or thrombocytopenia.
Study highlights ‘impact and importance’ of complement in APS
The study indicates “the impact and the importance of complement” in APS, said Yehuda Shoenfeld, MD, the founder and head of the Zabludowicz Center for Autoimmune Diseases at the Sheba Medical Center in Tel Hashomer, Israel.
In the early days of understanding APS, said Dr. Shoenfeld, it was thought that complement was not as important as it was in systemic lupus erythematosus (SLE). The importance of raised complement seen in studies of APS would often be discounted or neglected in comparison to SLE.
However, “slowly, slowly” it has been found that “complement [in APS] is activated very similarly to SLE,” Dr. Shoenfeld noted.
“I think that it’s important to assess the component levels,” Dr. Lini said in discussion. “This is needed to be done in the preconception counseling for APS and aPL carrier patients.”
Determining whether there is single, double, or even triple aPL positivity could be useful in guiding clinical decisions.
“If we have triple positivity, that could mean that there may be a more immunologic activation of the system and that it could be useful to administrate hydroxychloroquine [to] those patients who would like to have a pregnancy,” Dr. Lini suggested.
Plus, in those with decreased complement levels, “this could be a very useful tool” to identify where something could go wrong during their pregnancy.
The study had no outside funding. Dr. Lini and Dr. Shoenfeld disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Is the end near for surgical and transbronchial biopsies? Challenges in the pediatric workforce; Cascade testing in PAH; and more ...
Interventional chest/diagnostic procedures
Endobronchial optical coherence tomography and interstitial lung diseases: Is the end near for surgical and transbronchial lung biopsies?
The early diagnosis of interstitial lung diseases (ILD) is paramount to initiating appropriate treatment and preventing irreversible pulmonary damage. Specific ILD subtypes may be diagnosed based on clinical evaluation, high resolution chest CT (HRCT) patterns, and serologic testing, but many patients require invasive procedures for histopathologic evaluation of lung tissue. Current modalities for obtaining tissue include transbronchial lung cryobiopsy (TBLC) and surgical lung biopsy (SLB), both of which carry a risk of potential complications (Troy LK, et al. Lancet Respir Med. 2020;8:171-81; Hutchinson JP, et al. Am J Respir Crit Care Med. 2016;193[10]:1161-7).

Recently, genomic classifiers applied to transbronchial biopsies have been proposed to facilitate the diagnosis of usual interstitial pneumonia (UIP), but the limited information provided still does not obviate the need for tissue diagnosis when needed (Raghu G, et al. Lancet Respir Med. 2019;7[6]:487-96). It is in this context that endobronchial optical coherence tomography (EB-OCT) was proposed as a real-time, in vivo, optical biopsy method for ILD.

EB-OCT uses near infrared light to generate large volumes of in-vivo three-dimensional tissue imaging with microscopic resolution (Goorsenberg A, et al. Respiration. 2020;99:190-205; Nandy S, et al. Am J Respir Crit Care Med. 2021;article in press). The OCT catheter is advanced through the bronchoscope working channel and can be used during outpatient procedures under conscious sedation. Available data suggests that minimal training is necessary, both for proceduralists and interpreting pathologists, but this will need to be confirmed in larger studies and various practice settings. Early studies suggest that OCT can identify microscopic honeycombing and other abnormalities even before they are evident on HRCT scans (Goorsenberg A, et al. Respiration. 2020;99:190-205). Newer research comparing ILD diagnosis from EB-OCT cross-sectional images with that obtained from SLB specimens revealed EB-OCT can distinguish UIP from non-UIP ILD with high sensitivity and specificity (Nandy S, et al. Am J Respir Crit Care Med. 2021;article in press). Could this mean the end of SLB and TBLC for the diagnosis of ILD? While the ability to diagnose ILD subtypes with high reliability and low risk of complications is certainly promising, studies remain admittedly small and the technique itself is only available to highly select individuals and specialized ILD centers. Let’s not pack up the cryoprobe just yet.
Audra J. Schwalk, MD, MBA: Steering Committee Member
Fabien Maldonado, MD, FCCP: Steering Committee Member
Pediatric chest medicine
Challenges in the pediatric pulmonary workforce
The future of the pediatric workforce has been the source of extensive discussion within the pediatric community and resulted in a considerable body of medical literature (Vinci RJ. Pediatrics. 2021;147[6]:e2020013292). In pediatric pulmonology, there is growing concern that current trends will lead to a workforce shortage resulting in patients having difficulty accessing subspecialty care (Harris C, et al. Pediatric Pulmonol. 2019;54[4]:444-50). The etiology of this shortage is multifactorial. Duration of fellowship training and subsequent financial implications are reported potential barriers to pursuing a fellowship (Nelson BA, et al. Pediatric Pulmonol. 2020;1-7). Discrepancies between pediatric and adult compensation may be another barrier. Insightful recruitment strategies based on the results of a recent study included maximizing resident interaction with pulmonary faculty, early identification and support of interested trainees, and consideration of flexible training models (Nelson BA, et al. ATS Sch. 2020;1:372-83). Lifestyle has also been a factor that contributes to a trainee’s decision to go into pediatric pulmonology (Freed GL, et al. Pediatrics. 2009;123(suppl 1):S31‐S37).

As our field addresses the critical need to recruit more trainees in light of the unfilled fellowship positions and the increasing average age of members of the field, we should not underestimate the prevalence of systemic racism and bias in medicine (Chiel L, et al. ATS Sch. 2020;1[4]:337-39) nor gender discrimination. Instead, we should seize the opportunity to understand and knock down barriers that trainees who are underrepresented in medicine face in pursuing pediatric subspecialty careers and build upon the excellent recent body of literature in this field to help recruit, support , and grow a robust, diverse workforce to provide the best pediatric care to all.
Anne C. Coates, MD – Steering Committee Member
Pulmonary vascular disease
Cascade testing in PAH: Is there a role?
Pediatric guidelines for pulmonary arterial hypertension (PAH) recommends genetic screening as a part of the evaluation for the newly diagnosed, with expansion to first-degree relatives as indicated. Currently, this is not mandated, and implementation is variable. In adults, genetic screening is not routinely offered, and family screening is rare. This reflects a lack of definitive guidelines (Abman SH, et al. Circulation. 2015:24;132[21]:2037-99). However, it is intuitive that if carriers are not identified by screening, they will come to attention after pulmonary vascular disease burden causes symptoms and affects outcomes.

Cascade testing is a screening methodology that is used in heritable cancers (George RM, et al. Genet Couns. 2015;24[3]:388-99). In cascade testing, identification of an index case prompts screening of at-risk family members. If these relatives are positive for mutations, the cycle is repeated (cascaded) to their immediate relatives, allowing for targeted screening. This approach is especially effective in genetic mutations that are inherited in an autosomal dominant fashion, such as in BMPR2 gene mutation. Cascade testing is an effective way to capture relatives who would otherwise be overlooked.

Unfortunately, in the United States, the cost of genetic testing is a significant obstacle to universal implementation. A new diagnosis of heritable pulmonary arterial hypertension (HPAH) is often followed by a multigene panel with costs exceeding $1000 and may prompt subsequent targeted testing resulting in additional expense (Chung WK, et al. Can J Cardiol. 2015;31[4]:544-47). Furthermore, a positive mutation detected on screening is not definitively associated with disease due to variable penetrance (Morrell NW, et al. Eur Respir J. 2019;53[1]:1801899]. As such, mass screening strategies are not recommended. The recent DELPHI-2 study [Montani D, et al. Eur Respir J. 2021;58[1]:2004229) have demonstrated that genetic screening is impactful in families with HPAH. A genetic screening algorithm should be considered, and cascade testing could be a cost-effective targeted approach.
Sandeep Sahay, MD, MSc, FCCP: Steering Committee Member
Jean M. Elwing, MD, FCCP: Chair
Pulmonary physiology, function, and rehabilitation network
Physiological benefits of awake proning: Its role and relevance in the COVID-19 pandemic
The advent of the COVID-19 pandemic has put a significant strain on the health care systems and critical care services across several countries, including the United States. Amidst this, several concerted efforts to reduce the need for mechanical ventilation has resulted in the emergence of awake proning as a strategy to improve oxygenation, which has been instituted in critical care units, in-patient settings, as well as in EDs. Although the evidence on this strategy has been vastly limited to case series and observational studies, several societies have incorporated awake proning as an initial management strategy in hypoxemic respiratory failure within their clinical guidelines (Chalmers JD, et al. Eur Respir J. 2021;57:2100048; Koeckerling D, et al. Thorax. 2020;75:833-4) and consensus statements (Nasa P, et al. Crit Care. 2021;25:106).

Physiological benefits of awake proning include improvement in ventilation-perfusion matching secondary to relative increase in ventilation in dorsal nondependent areas in the setting of higher density of perfusion within these units, thus reducing shunt and, hence, improving oxygenation. Other physiological mechanisms include homogenization of transpulmonary pressures, reduction of ventilator-induced lung injury (VILI) or patient self-inflicted lung injury (P-SILI), and possibly lung injury from pendelluft (Telias I, et al. JAMA. 2020;323[22]:2265-67).
A recent meta-trial involving randomized controlled trials done across six countries compared prone positioning with standard care in patients with hypoxemic respiratory failure (defined as SpO2/ FiO2 < 315 and on high flow oxygen therapy) showed a reduced incidence of treatment failure and need for intubation without any signal of harm; although no mortality benefit was reported (Ehrmann S, et al. Lancet Respir Med. 2021 Aug 20;S2213-2600(21)00356-8). The number needed to treat to prevent one intubation was 14. While promising and reinforcing the safety of this relatively easy maneuver, several questions remain—which patients would benefit the most? Can it be applied within general wards safely? Does institution of awake proning delay intubation rates with consequent worse outcomes? Several ongoing (NCT 04402879) and completed studies (NCT 04383613 and NCT 04350723) may shed light on these important questions (Weatherald J, et al. Lancet Respir Med. 2021 Aug 20;S2213-2600[21]00368-4).
Sujith Cherian, MD, FCCP: Steering Committee Member
Interventional chest/diagnostic procedures
Endobronchial optical coherence tomography and interstitial lung diseases: Is the end near for surgical and transbronchial lung biopsies?
The early diagnosis of interstitial lung diseases (ILD) is paramount to initiating appropriate treatment and preventing irreversible pulmonary damage. Specific ILD subtypes may be diagnosed based on clinical evaluation, high resolution chest CT (HRCT) patterns, and serologic testing, but many patients require invasive procedures for histopathologic evaluation of lung tissue. Current modalities for obtaining tissue include transbronchial lung cryobiopsy (TBLC) and surgical lung biopsy (SLB), both of which carry a risk of potential complications (Troy LK, et al. Lancet Respir Med. 2020;8:171-81; Hutchinson JP, et al. Am J Respir Crit Care Med. 2016;193[10]:1161-7).
Recently, genomic classifiers applied to transbronchial biopsies have been proposed to facilitate the diagnosis of usual interstitial pneumonia (UIP), but the limited information provided still does not obviate the need for tissue diagnosis when needed (Raghu G, et al. Lancet Respir Med. 2019;7[6]:487-96). It is in this context that endobronchial optical coherence tomography (EB-OCT) was proposed as a real-time, in vivo, optical biopsy method for ILD.
EB-OCT uses near infrared light to generate large volumes of in-vivo three-dimensional tissue imaging with microscopic resolution (Goorsenberg A, et al. Respiration. 2020;99:190-205; Nandy S, et al. Am J Respir Crit Care Med. 2021;article in press). The OCT catheter is advanced through the bronchoscope working channel and can be used during outpatient procedures under conscious sedation. Available data suggests that minimal training is necessary, both for proceduralists and interpreting pathologists, but this will need to be confirmed in larger studies and various practice settings. Early studies suggest that OCT can identify microscopic honeycombing and other abnormalities even before they are evident on HRCT scans (Goorsenberg A, et al. Respiration. 2020;99:190-205). Newer research comparing ILD diagnosis from EB-OCT cross-sectional images with that obtained from SLB specimens revealed EB-OCT can distinguish UIP from non-UIP ILD with high sensitivity and specificity (Nandy S, et al. Am J Respir Crit Care Med. 2021;article in press). Could this mean the end of SLB and TBLC for the diagnosis of ILD? While the ability to diagnose ILD subtypes with high reliability and low risk of complications is certainly promising, studies remain admittedly small and the technique itself is only available to highly select individuals and specialized ILD centers. Let’s not pack up the cryoprobe just yet.
Audra J. Schwalk, MD, MBA: Steering Committee Member
Fabien Maldonado, MD, FCCP: Steering Committee Member
Pediatric chest medicine
Challenges in the pediatric pulmonary workforce
The future of the pediatric workforce has been the source of extensive discussion within the pediatric community and resulted in a considerable body of medical literature (Vinci RJ. Pediatrics. 2021;147[6]:e2020013292). In pediatric pulmonology, there is growing concern that current trends will lead to a workforce shortage resulting in patients having difficulty accessing subspecialty care (Harris C, et al. Pediatric Pulmonol. 2019;54[4]:444-50). The etiology of this shortage is multifactorial. Duration of fellowship training and subsequent financial implications are reported potential barriers to pursuing a fellowship (Nelson BA, et al. Pediatric Pulmonol. 2020;1-7). Discrepancies between pediatric and adult compensation may be another barrier. Insightful recruitment strategies based on the results of a recent study included maximizing resident interaction with pulmonary faculty, early identification and support of interested trainees, and consideration of flexible training models (Nelson BA, et al. ATS Sch. 2020;1:372-83). Lifestyle has also been a factor that contributes to a trainee’s decision to go into pediatric pulmonology (Freed GL, et al. Pediatrics. 2009;123(suppl 1):S31‐S37).
As our field addresses the critical need to recruit more trainees in light of the unfilled fellowship positions and the increasing average age of members of the field, we should not underestimate the prevalence of systemic racism and bias in medicine (Chiel L, et al. ATS Sch. 2020;1[4]:337-39) nor gender discrimination. Instead, we should seize the opportunity to understand and knock down barriers that trainees who are underrepresented in medicine face in pursuing pediatric subspecialty careers and build upon the excellent recent body of literature in this field to help recruit, support , and grow a robust, diverse workforce to provide the best pediatric care to all.
Anne C. Coates, MD – Steering Committee Member
Pulmonary vascular disease
Cascade testing in PAH: Is there a role?
Pediatric guidelines for pulmonary arterial hypertension (PAH) recommends genetic screening as a part of the evaluation for the newly diagnosed, with expansion to first-degree relatives as indicated. Currently, this is not mandated, and implementation is variable. In adults, genetic screening is not routinely offered, and family screening is rare. This reflects a lack of definitive guidelines (Abman SH, et al. Circulation. 2015:24;132[21]:2037-99). However, it is intuitive that if carriers are not identified by screening, they will come to attention after pulmonary vascular disease burden causes symptoms and affects outcomes.
Cascade testing is a screening methodology that is used in heritable cancers (George RM, et al. Genet Couns. 2015;24[3]:388-99). In cascade testing, identification of an index case prompts screening of at-risk family members. If these relatives are positive for mutations, the cycle is repeated (cascaded) to their immediate relatives, allowing for targeted screening. This approach is especially effective in genetic mutations that are inherited in an autosomal dominant fashion, such as in BMPR2 gene mutation. Cascade testing is an effective way to capture relatives who would otherwise be overlooked.
Unfortunately, in the United States, the cost of genetic testing is a significant obstacle to universal implementation. A new diagnosis of heritable pulmonary arterial hypertension (HPAH) is often followed by a multigene panel with costs exceeding $1000 and may prompt subsequent targeted testing resulting in additional expense (Chung WK, et al. Can J Cardiol. 2015;31[4]:544-47). Furthermore, a positive mutation detected on screening is not definitively associated with disease due to variable penetrance (Morrell NW, et al. Eur Respir J. 2019;53[1]:1801899]. As such, mass screening strategies are not recommended. The recent DELPHI-2 study [Montani D, et al. Eur Respir J. 2021;58[1]:2004229) have demonstrated that genetic screening is impactful in families with HPAH. A genetic screening algorithm should be considered, and cascade testing could be a cost-effective targeted approach.
Sandeep Sahay, MD, MSc, FCCP: Steering Committee Member
Jean M. Elwing, MD, FCCP: Chair
Pulmonary physiology, function, and rehabilitation network
Physiological benefits of awake proning: Its role and relevance in the COVID-19 pandemic
The advent of the COVID-19 pandemic has put a significant strain on the health care systems and critical care services across several countries, including the United States. Amidst this, several concerted efforts to reduce the need for mechanical ventilation has resulted in the emergence of awake proning as a strategy to improve oxygenation, which has been instituted in critical care units, in-patient settings, as well as in EDs. Although the evidence on this strategy has been vastly limited to case series and observational studies, several societies have incorporated awake proning as an initial management strategy in hypoxemic respiratory failure within their clinical guidelines (Chalmers JD, et al. Eur Respir J. 2021;57:2100048; Koeckerling D, et al. Thorax. 2020;75:833-4) and consensus statements (Nasa P, et al. Crit Care. 2021;25:106).
Physiological benefits of awake proning include improvement in ventilation-perfusion matching secondary to relative increase in ventilation in dorsal nondependent areas in the setting of higher density of perfusion within these units, thus reducing shunt and, hence, improving oxygenation. Other physiological mechanisms include homogenization of transpulmonary pressures, reduction of ventilator-induced lung injury (VILI) or patient self-inflicted lung injury (P-SILI), and possibly lung injury from pendelluft (Telias I, et al. JAMA. 2020;323[22]:2265-67).
A recent meta-trial involving randomized controlled trials done across six countries compared prone positioning with standard care in patients with hypoxemic respiratory failure (defined as SpO2/ FiO2 < 315 and on high flow oxygen therapy) showed a reduced incidence of treatment failure and need for intubation without any signal of harm; although no mortality benefit was reported (Ehrmann S, et al. Lancet Respir Med. 2021 Aug 20;S2213-2600(21)00356-8). The number needed to treat to prevent one intubation was 14. While promising and reinforcing the safety of this relatively easy maneuver, several questions remain—which patients would benefit the most? Can it be applied within general wards safely? Does institution of awake proning delay intubation rates with consequent worse outcomes? Several ongoing (NCT 04402879) and completed studies (NCT 04383613 and NCT 04350723) may shed light on these important questions (Weatherald J, et al. Lancet Respir Med. 2021 Aug 20;S2213-2600[21]00368-4).
Sujith Cherian, MD, FCCP: Steering Committee Member
Interventional chest/diagnostic procedures
Endobronchial optical coherence tomography and interstitial lung diseases: Is the end near for surgical and transbronchial lung biopsies?
The early diagnosis of interstitial lung diseases (ILD) is paramount to initiating appropriate treatment and preventing irreversible pulmonary damage. Specific ILD subtypes may be diagnosed based on clinical evaluation, high resolution chest CT (HRCT) patterns, and serologic testing, but many patients require invasive procedures for histopathologic evaluation of lung tissue. Current modalities for obtaining tissue include transbronchial lung cryobiopsy (TBLC) and surgical lung biopsy (SLB), both of which carry a risk of potential complications (Troy LK, et al. Lancet Respir Med. 2020;8:171-81; Hutchinson JP, et al. Am J Respir Crit Care Med. 2016;193[10]:1161-7).
Recently, genomic classifiers applied to transbronchial biopsies have been proposed to facilitate the diagnosis of usual interstitial pneumonia (UIP), but the limited information provided still does not obviate the need for tissue diagnosis when needed (Raghu G, et al. Lancet Respir Med. 2019;7[6]:487-96). It is in this context that endobronchial optical coherence tomography (EB-OCT) was proposed as a real-time, in vivo, optical biopsy method for ILD.
EB-OCT uses near infrared light to generate large volumes of in-vivo three-dimensional tissue imaging with microscopic resolution (Goorsenberg A, et al. Respiration. 2020;99:190-205; Nandy S, et al. Am J Respir Crit Care Med. 2021;article in press). The OCT catheter is advanced through the bronchoscope working channel and can be used during outpatient procedures under conscious sedation. Available data suggests that minimal training is necessary, both for proceduralists and interpreting pathologists, but this will need to be confirmed in larger studies and various practice settings. Early studies suggest that OCT can identify microscopic honeycombing and other abnormalities even before they are evident on HRCT scans (Goorsenberg A, et al. Respiration. 2020;99:190-205). Newer research comparing ILD diagnosis from EB-OCT cross-sectional images with that obtained from SLB specimens revealed EB-OCT can distinguish UIP from non-UIP ILD with high sensitivity and specificity (Nandy S, et al. Am J Respir Crit Care Med. 2021;article in press). Could this mean the end of SLB and TBLC for the diagnosis of ILD? While the ability to diagnose ILD subtypes with high reliability and low risk of complications is certainly promising, studies remain admittedly small and the technique itself is only available to highly select individuals and specialized ILD centers. Let’s not pack up the cryoprobe just yet.
Audra J. Schwalk, MD, MBA: Steering Committee Member
Fabien Maldonado, MD, FCCP: Steering Committee Member
Pediatric chest medicine
Challenges in the pediatric pulmonary workforce
The future of the pediatric workforce has been the source of extensive discussion within the pediatric community and resulted in a considerable body of medical literature (Vinci RJ. Pediatrics. 2021;147[6]:e2020013292). In pediatric pulmonology, there is growing concern that current trends will lead to a workforce shortage resulting in patients having difficulty accessing subspecialty care (Harris C, et al. Pediatric Pulmonol. 2019;54[4]:444-50). The etiology of this shortage is multifactorial. Duration of fellowship training and subsequent financial implications are reported potential barriers to pursuing a fellowship (Nelson BA, et al. Pediatric Pulmonol. 2020;1-7). Discrepancies between pediatric and adult compensation may be another barrier. Insightful recruitment strategies based on the results of a recent study included maximizing resident interaction with pulmonary faculty, early identification and support of interested trainees, and consideration of flexible training models (Nelson BA, et al. ATS Sch. 2020;1:372-83). Lifestyle has also been a factor that contributes to a trainee’s decision to go into pediatric pulmonology (Freed GL, et al. Pediatrics. 2009;123(suppl 1):S31‐S37).
As our field addresses the critical need to recruit more trainees in light of the unfilled fellowship positions and the increasing average age of members of the field, we should not underestimate the prevalence of systemic racism and bias in medicine (Chiel L, et al. ATS Sch. 2020;1[4]:337-39) nor gender discrimination. Instead, we should seize the opportunity to understand and knock down barriers that trainees who are underrepresented in medicine face in pursuing pediatric subspecialty careers and build upon the excellent recent body of literature in this field to help recruit, support , and grow a robust, diverse workforce to provide the best pediatric care to all.
Anne C. Coates, MD – Steering Committee Member
Pulmonary vascular disease
Cascade testing in PAH: Is there a role?
Pediatric guidelines for pulmonary arterial hypertension (PAH) recommends genetic screening as a part of the evaluation for the newly diagnosed, with expansion to first-degree relatives as indicated. Currently, this is not mandated, and implementation is variable. In adults, genetic screening is not routinely offered, and family screening is rare. This reflects a lack of definitive guidelines (Abman SH, et al. Circulation. 2015:24;132[21]:2037-99). However, it is intuitive that if carriers are not identified by screening, they will come to attention after pulmonary vascular disease burden causes symptoms and affects outcomes.
Cascade testing is a screening methodology that is used in heritable cancers (George RM, et al. Genet Couns. 2015;24[3]:388-99). In cascade testing, identification of an index case prompts screening of at-risk family members. If these relatives are positive for mutations, the cycle is repeated (cascaded) to their immediate relatives, allowing for targeted screening. This approach is especially effective in genetic mutations that are inherited in an autosomal dominant fashion, such as in BMPR2 gene mutation. Cascade testing is an effective way to capture relatives who would otherwise be overlooked.
Unfortunately, in the United States, the cost of genetic testing is a significant obstacle to universal implementation. A new diagnosis of heritable pulmonary arterial hypertension (HPAH) is often followed by a multigene panel with costs exceeding $1000 and may prompt subsequent targeted testing resulting in additional expense (Chung WK, et al. Can J Cardiol. 2015;31[4]:544-47). Furthermore, a positive mutation detected on screening is not definitively associated with disease due to variable penetrance (Morrell NW, et al. Eur Respir J. 2019;53[1]:1801899]. As such, mass screening strategies are not recommended. The recent DELPHI-2 study [Montani D, et al. Eur Respir J. 2021;58[1]:2004229) have demonstrated that genetic screening is impactful in families with HPAH. A genetic screening algorithm should be considered, and cascade testing could be a cost-effective targeted approach.
Sandeep Sahay, MD, MSc, FCCP: Steering Committee Member
Jean M. Elwing, MD, FCCP: Chair
Pulmonary physiology, function, and rehabilitation network
Physiological benefits of awake proning: Its role and relevance in the COVID-19 pandemic
The advent of the COVID-19 pandemic has put a significant strain on the health care systems and critical care services across several countries, including the United States. Amidst this, several concerted efforts to reduce the need for mechanical ventilation has resulted in the emergence of awake proning as a strategy to improve oxygenation, which has been instituted in critical care units, in-patient settings, as well as in EDs. Although the evidence on this strategy has been vastly limited to case series and observational studies, several societies have incorporated awake proning as an initial management strategy in hypoxemic respiratory failure within their clinical guidelines (Chalmers JD, et al. Eur Respir J. 2021;57:2100048; Koeckerling D, et al. Thorax. 2020;75:833-4) and consensus statements (Nasa P, et al. Crit Care. 2021;25:106).
Physiological benefits of awake proning include improvement in ventilation-perfusion matching secondary to relative increase in ventilation in dorsal nondependent areas in the setting of higher density of perfusion within these units, thus reducing shunt and, hence, improving oxygenation. Other physiological mechanisms include homogenization of transpulmonary pressures, reduction of ventilator-induced lung injury (VILI) or patient self-inflicted lung injury (P-SILI), and possibly lung injury from pendelluft (Telias I, et al. JAMA. 2020;323[22]:2265-67).
A recent meta-trial involving randomized controlled trials done across six countries compared prone positioning with standard care in patients with hypoxemic respiratory failure (defined as SpO2/ FiO2 < 315 and on high flow oxygen therapy) showed a reduced incidence of treatment failure and need for intubation without any signal of harm; although no mortality benefit was reported (Ehrmann S, et al. Lancet Respir Med. 2021 Aug 20;S2213-2600(21)00356-8). The number needed to treat to prevent one intubation was 14. While promising and reinforcing the safety of this relatively easy maneuver, several questions remain—which patients would benefit the most? Can it be applied within general wards safely? Does institution of awake proning delay intubation rates with consequent worse outcomes? Several ongoing (NCT 04402879) and completed studies (NCT 04383613 and NCT 04350723) may shed light on these important questions (Weatherald J, et al. Lancet Respir Med. 2021 Aug 20;S2213-2600[21]00368-4).
Sujith Cherian, MD, FCCP: Steering Committee Member
Thoughts on becoming CHEST President
I am honored to have the privilege of serving as the 84th President of the American College of Chest Physicians. When I attended my first CHEST meeting, I sat in the opening plenary session with thousands of other members, never imagining that I would have the opportunity to lead the organization just two decades later. And while I don’t recall many sessions from that meeting, I vividly remember the way it made an emotional impact. I never felt like one of a drove of nameless learners; both faculty and staff made it a collegial experience, much like attending pulmonary grand rounds at my own institution. Speakers would stay after their presentations to answer questions from even the most junior members. Leadership made themselves available over coffee or in the hallways between sessions. And that experience was the first of a great many memorable interactions I have had with CHEST.

CHEST has meant a great deal to me personally; it served as my first professional home away from home. I had the opportunity to grow in a number of different areas through my service to CHEST, in ways that I would not have been able to do easily at my own institution. I’ve worked with incredible staff and volunteers in my service on a number of our committees, including the Council of NetWorks, the Training and Transitions Committee, the Education Committee, and the Program Committee, to name a few. While I’ve had a chance to learn what role each of these component parts of the College serves during my tenure on those committees, it wasn’t until far more recently that I better understood the role of the President. Before I get into what I’d like to achieve during my year as President, I’d like to briefly review what that role entails.
Contrary to popular belief, the President does not set the organizational goals for CHEST; those are set by the Board of Regents. While I will have the privilege of running the Board meetings, it is the seventeen incredibly talented folks who serve as voting members of the Board that set the College’s direction. Once the organizational goals are set, it is our committees that take charge of designing and implementing plans to work toward those goals. Concomitantly, Dr. Robert Musacchio (CHEST chief executive officer and executive vice president) meets with his own executive leadership team to design a structure that lets the CHEST staff work, both on their own and in tandem with our members, to achieve these goals. One of the President’s main roles, as I see it, is to serve as a liaison. When the Board makes decisions that affect the membership, it will be my job to communicate changes and why they are being made. When our members have challenges that the College might be able to help solve, it is my role to work with the Board and the CEO to see what we can do about them. And when there is need to interface with other organizations, the President (or their designee) can speak on behalf of the College in those interactions.
In the context of those duties, what are the things that I would like to accomplish during my tenure as CHEST president? First, I want to spend more time with our committees and you, our members. CHEST is a member-focused organization; I believe that this is the main thing that sets our professional society apart from its sister societies. I have always found CHEST to be very collegial and welcoming. But I am aware that some of our members haven’t always found it accessible. And I get that; our structure is complex. That’s the reason I provided a description of my role, and the reason that I intend to spend time making CHEST more accessible to all of you. We’ve already developed dedicated social media channels for a number of our NetWorks in order to make you all more aware of their activities. In the coming year, I’ll provide regular updates to membership about ongoing CHEST activities. I’ll work to provide more member awareness of what role each of our committees plays in forwarding the College’s goals. And I’ll provide you with more information about the type of qualifications that each committee seeks in its nominees, in an effort to encourage you to run for a leadership position that best suits your interests and skill set.
While improving our members’ understanding of the inner workings at CHEST will help each of you better see how the College can meet your needs, my hope is that this increase in organizational accessibility will motivate each of you to engage more actively with us. This is my second goal as President. For some of you, that engagement may take the form of joining our Twitter chats; for others, it could mean attending one of our live learning courses in Chicago for the first time. But I hope that some of you will consider submitting session proposals to our annual meeting for the first time, or running for an available leadership position within the College when nominations open in the Spring.
As our organization grows (now almost twenty thousand members strong!), I want to provide a second home for all our members, spanning the range from medical students to full professors, from lifelong academic physicians to those just starting out in community practices, from busy clinicians to physician scientists, and including all members of the healthcare team. Although the makeup of our volunteer leadership is becoming more representative of the full breadth of our membership, we are not fully there yet. Until we get to that intended target, I would like to ask each of you to reach out to me with any thoughts about how CHEST can better meet your professional needs. Creating greater access to leadership to let each of your opinions be heard is my third goal as President of CHEST. I’ll provide more details about how I’m hoping to achieve this in the coming months.
The world has been a crazy place over the last eighteen months, filled with challenges that we could never have foreseen even a year prior. Our members have been on the front lines of the pandemic; in addition to the professional stresses related to caring for innumerable critically ill patients, many of us have suffered personal losses. Although none of us knows what 2022 holds, I look forward to a brighter future, knowing that regardless of what the coming year brings, we will face it together.
I am honored to have the privilege of serving as the 84th President of the American College of Chest Physicians. When I attended my first CHEST meeting, I sat in the opening plenary session with thousands of other members, never imagining that I would have the opportunity to lead the organization just two decades later. And while I don’t recall many sessions from that meeting, I vividly remember the way it made an emotional impact. I never felt like one of a drove of nameless learners; both faculty and staff made it a collegial experience, much like attending pulmonary grand rounds at my own institution. Speakers would stay after their presentations to answer questions from even the most junior members. Leadership made themselves available over coffee or in the hallways between sessions. And that experience was the first of a great many memorable interactions I have had with CHEST.
CHEST has meant a great deal to me personally; it served as my first professional home away from home. I had the opportunity to grow in a number of different areas through my service to CHEST, in ways that I would not have been able to do easily at my own institution. I’ve worked with incredible staff and volunteers in my service on a number of our committees, including the Council of NetWorks, the Training and Transitions Committee, the Education Committee, and the Program Committee, to name a few. While I’ve had a chance to learn what role each of these component parts of the College serves during my tenure on those committees, it wasn’t until far more recently that I better understood the role of the President. Before I get into what I’d like to achieve during my year as President, I’d like to briefly review what that role entails.
Contrary to popular belief, the President does not set the organizational goals for CHEST; those are set by the Board of Regents. While I will have the privilege of running the Board meetings, it is the seventeen incredibly talented folks who serve as voting members of the Board that set the College’s direction. Once the organizational goals are set, it is our committees that take charge of designing and implementing plans to work toward those goals. Concomitantly, Dr. Robert Musacchio (CHEST chief executive officer and executive vice president) meets with his own executive leadership team to design a structure that lets the CHEST staff work, both on their own and in tandem with our members, to achieve these goals. One of the President’s main roles, as I see it, is to serve as a liaison. When the Board makes decisions that affect the membership, it will be my job to communicate changes and why they are being made. When our members have challenges that the College might be able to help solve, it is my role to work with the Board and the CEO to see what we can do about them. And when there is need to interface with other organizations, the President (or their designee) can speak on behalf of the College in those interactions.
In the context of those duties, what are the things that I would like to accomplish during my tenure as CHEST president? First, I want to spend more time with our committees and you, our members. CHEST is a member-focused organization; I believe that this is the main thing that sets our professional society apart from its sister societies. I have always found CHEST to be very collegial and welcoming. But I am aware that some of our members haven’t always found it accessible. And I get that; our structure is complex. That’s the reason I provided a description of my role, and the reason that I intend to spend time making CHEST more accessible to all of you. We’ve already developed dedicated social media channels for a number of our NetWorks in order to make you all more aware of their activities. In the coming year, I’ll provide regular updates to membership about ongoing CHEST activities. I’ll work to provide more member awareness of what role each of our committees plays in forwarding the College’s goals. And I’ll provide you with more information about the type of qualifications that each committee seeks in its nominees, in an effort to encourage you to run for a leadership position that best suits your interests and skill set.
While improving our members’ understanding of the inner workings at CHEST will help each of you better see how the College can meet your needs, my hope is that this increase in organizational accessibility will motivate each of you to engage more actively with us. This is my second goal as President. For some of you, that engagement may take the form of joining our Twitter chats; for others, it could mean attending one of our live learning courses in Chicago for the first time. But I hope that some of you will consider submitting session proposals to our annual meeting for the first time, or running for an available leadership position within the College when nominations open in the Spring.
As our organization grows (now almost twenty thousand members strong!), I want to provide a second home for all our members, spanning the range from medical students to full professors, from lifelong academic physicians to those just starting out in community practices, from busy clinicians to physician scientists, and including all members of the healthcare team. Although the makeup of our volunteer leadership is becoming more representative of the full breadth of our membership, we are not fully there yet. Until we get to that intended target, I would like to ask each of you to reach out to me with any thoughts about how CHEST can better meet your professional needs. Creating greater access to leadership to let each of your opinions be heard is my third goal as President of CHEST. I’ll provide more details about how I’m hoping to achieve this in the coming months.
The world has been a crazy place over the last eighteen months, filled with challenges that we could never have foreseen even a year prior. Our members have been on the front lines of the pandemic; in addition to the professional stresses related to caring for innumerable critically ill patients, many of us have suffered personal losses. Although none of us knows what 2022 holds, I look forward to a brighter future, knowing that regardless of what the coming year brings, we will face it together.
I am honored to have the privilege of serving as the 84th President of the American College of Chest Physicians. When I attended my first CHEST meeting, I sat in the opening plenary session with thousands of other members, never imagining that I would have the opportunity to lead the organization just two decades later. And while I don’t recall many sessions from that meeting, I vividly remember the way it made an emotional impact. I never felt like one of a drove of nameless learners; both faculty and staff made it a collegial experience, much like attending pulmonary grand rounds at my own institution. Speakers would stay after their presentations to answer questions from even the most junior members. Leadership made themselves available over coffee or in the hallways between sessions. And that experience was the first of a great many memorable interactions I have had with CHEST.
CHEST has meant a great deal to me personally; it served as my first professional home away from home. I had the opportunity to grow in a number of different areas through my service to CHEST, in ways that I would not have been able to do easily at my own institution. I’ve worked with incredible staff and volunteers in my service on a number of our committees, including the Council of NetWorks, the Training and Transitions Committee, the Education Committee, and the Program Committee, to name a few. While I’ve had a chance to learn what role each of these component parts of the College serves during my tenure on those committees, it wasn’t until far more recently that I better understood the role of the President. Before I get into what I’d like to achieve during my year as President, I’d like to briefly review what that role entails.
Contrary to popular belief, the President does not set the organizational goals for CHEST; those are set by the Board of Regents. While I will have the privilege of running the Board meetings, it is the seventeen incredibly talented folks who serve as voting members of the Board that set the College’s direction. Once the organizational goals are set, it is our committees that take charge of designing and implementing plans to work toward those goals. Concomitantly, Dr. Robert Musacchio (CHEST chief executive officer and executive vice president) meets with his own executive leadership team to design a structure that lets the CHEST staff work, both on their own and in tandem with our members, to achieve these goals. One of the President’s main roles, as I see it, is to serve as a liaison. When the Board makes decisions that affect the membership, it will be my job to communicate changes and why they are being made. When our members have challenges that the College might be able to help solve, it is my role to work with the Board and the CEO to see what we can do about them. And when there is need to interface with other organizations, the President (or their designee) can speak on behalf of the College in those interactions.
In the context of those duties, what are the things that I would like to accomplish during my tenure as CHEST president? First, I want to spend more time with our committees and you, our members. CHEST is a member-focused organization; I believe that this is the main thing that sets our professional society apart from its sister societies. I have always found CHEST to be very collegial and welcoming. But I am aware that some of our members haven’t always found it accessible. And I get that; our structure is complex. That’s the reason I provided a description of my role, and the reason that I intend to spend time making CHEST more accessible to all of you. We’ve already developed dedicated social media channels for a number of our NetWorks in order to make you all more aware of their activities. In the coming year, I’ll provide regular updates to membership about ongoing CHEST activities. I’ll work to provide more member awareness of what role each of our committees plays in forwarding the College’s goals. And I’ll provide you with more information about the type of qualifications that each committee seeks in its nominees, in an effort to encourage you to run for a leadership position that best suits your interests and skill set.
While improving our members’ understanding of the inner workings at CHEST will help each of you better see how the College can meet your needs, my hope is that this increase in organizational accessibility will motivate each of you to engage more actively with us. This is my second goal as President. For some of you, that engagement may take the form of joining our Twitter chats; for others, it could mean attending one of our live learning courses in Chicago for the first time. But I hope that some of you will consider submitting session proposals to our annual meeting for the first time, or running for an available leadership position within the College when nominations open in the Spring.
As our organization grows (now almost twenty thousand members strong!), I want to provide a second home for all our members, spanning the range from medical students to full professors, from lifelong academic physicians to those just starting out in community practices, from busy clinicians to physician scientists, and including all members of the healthcare team. Although the makeup of our volunteer leadership is becoming more representative of the full breadth of our membership, we are not fully there yet. Until we get to that intended target, I would like to ask each of you to reach out to me with any thoughts about how CHEST can better meet your professional needs. Creating greater access to leadership to let each of your opinions be heard is my third goal as President of CHEST. I’ll provide more details about how I’m hoping to achieve this in the coming months.
The world has been a crazy place over the last eighteen months, filled with challenges that we could never have foreseen even a year prior. Our members have been on the front lines of the pandemic; in addition to the professional stresses related to caring for innumerable critically ill patients, many of us have suffered personal losses. Although none of us knows what 2022 holds, I look forward to a brighter future, knowing that regardless of what the coming year brings, we will face it together.
TTM2: Is there anything therapeutic about therapeutic hypothermia?
Animal and human models of the effects of therapeutic hypothermia, now called targeted temperature management (TTM), began to surface in the late 1980s. The first randomized clinical trial employing TTM as a neuroprotective strategy following cardiac arrest did not appear until the early 2000s. When compared with normothermia, the HACA trial (Holzer M, et al. N Engl J Med. 2002;346[8]:549-56) demonstrated a 14% reduction in mortality and improved neurologic outcomes following out of hospital cardiac arrest (OHCA) due to ventricular fibrillation (VF) or ventricular tachycardia (VT) when maintaining body temperature between 32˚C and 34˚C post-arrest. Following the results of this trial, TTM in comatose patients following cardiac arrest was recommended by international guidelines and became the standard of care. It was not until the publication of the TTM1 trial (Nielsen N, et al. N Engl J Med. 2013;369[23]:2197-206) about a decade later, that serious questions regarding the efficacy of TTM were raised. The TTM1 trial showed no difference in mortality or neurologic outcomes when comparing TTM at 33˚C vs 36˚C for OHCA. The results of this trial heralded widespread practice change, with many abandoning deep cooling, and often active cooling measures, in favor of fever avoidance. The HYPERION trial (Lascarrou J, et al. N Engl J Med. 2019;381:2327-37) came next, comparing TTM at 33˚C to normothermia (<37.5˚C) for cardiac arrest with non-hockable rhythm. This study did not identify any improvement in mortality with utilization of TTM but suggested it may be associated with more favorable neurologic outcomes, albeit in a small number of patients.

The TTM2 trial (Dankiewicz J, et al. N Engl J Med. 2021;384:2283-94) is the most recent trial to address the question of TTM post-cardiac arrest. The TTM2 trial was an international, randomized controlled superiority trial of TTM at 33˚C vs normothermia (≤37.8˚C) for patients with coma following OHCA with any initial rhythm. It was conducted by the same group as the TTM1 trial and, to date, represents the largest (N= 1,850) and most robust trial conducted in this area. The trial spanned 61 institutions across 14 countries and had nearly complete follow-up at 6 months. Once again, there was no significant difference in all-cause mortality at 6 months in the TTM group when compared with the normothermia group. Equally important, there were no differences observed in secondary outcomes, including functional neurologic status and health-related quality of life at 6 months. With the results of the TTM1 and TTM2 trials failing to show any neurologic or mortality benefit to TTM, we are left wondering, is there anything therapeutic about “therapeutic hypothermia”?
Both the 2020 American Heart Association (AHA) and 2021 European Resuscitation Council (ERC) guidelines predate this trial; they recommend cooling any OHCA or in-hospital cardiac arrest (IHCA) patient who remains unresponsive after return of spontaneous circulation (ROSC) regardless of initial rhythm. They further suggest maintaining a target temperature between 32˚C and 36˚C for at least 24 hours, followed by avoidance of fever (>37.7˚C) for at least 72 hours after ROSC in patients who remain comatose. While it will be interesting to see what future iterations of the guidelines recommend, the results from the TTM1 and TTM2 trials support a shift in clinical practice away from TTM and toward more active fever avoidance. Additionally, careful review of adverse events in the TTM2 trial suggests that induced hypothermia is not without risk of harm. When compared with the normothermia group in the TTM2 trial, the hypothermia group experienced higher rates of arrhythmias with hemodynamic instability (16% vs 24%), increased exposure to sedation, increased use of neuromuscular blockade, and increased duration of mechanical ventilation.
While the results of the TTM2 trial move the needle away from therapeutic hypothermia for OHCA patients, there is some nuance that warrants further discussion. First, the initial HACA trial, upon which the standard of TTM was based, included only patients with an initial shockable rhythm (VT/VF). Inherently, the etiology of these arrests is likely to be cardiac and more reversible in nature. Most subsequent landmark trials on TTM, including the TTM2 trial, have included OHCA patients with both shockable and nonshockable initial rhythms. Still, the majority of patients in the TTM2 trial had an initial shockable rhythm on presentation (72% hypothermia vs 75% normothermia). This may limit broad generalizability of study findings as an increasing number of OHCA patients are presenting with nonshockable initial rhythms. Next, it is well known that bystander CPR improves outcomes following OHCA. Impressively, over 75% of patients in both groups in the TTM2 trial received bystander CPR compared with an average of 46% of arrest patients in the US according to AHA data. Finally, like most of its predecessors, the TTM2 trial only included OHCA patients meaning no real conclusions can be drawn regarding application of TTM to IHCA patients. Of the major trials to date, only the HYPERION trial included IHCA patients – representing about 25% of the study population. Thus, the utility of TTM in the setting of IHCA remains largely unknown.
Taken in summation, recent trials, including TTM2, suggest that fever-avoidance post-cardiac arrest is likely the best option for improving mortality and neurologic outcomes while mitigating risk to the patient. We must remain vigilant in our enforcement of normothermia though as worse neurologic outcomes have been observed with hyperthermia in the early post-arrest period (Zeiner A, et al. Arch Intern Med. 2001;161[16]:2007-12). A key takeaway from recent trials is that maintaining normothermia without active temperature control measures is likely to be difficult to achieve. A criticism of the HYPERION trial was that a “substantial proportion” of patients in the normothermia group had temperatures above 38˚C. Similarly, 10% to15% of patients in the TTM2 trial had body temperatures above 37.7˚C, 40 to 72 hours after randomization and, ultimately, 46% of patients in the normothermia group required cooling with a temperature management device. Thus, we can conclude that maintenance of strict normothermia will likely continue to require active control with a temperature management device.
Despite an increasing number of well conducted studies in this area, there are several questions that remain unanswered. The first is whether cooling patients even earlier post-arrest is felt to increase the likelihood of survival with improved neurologic outcomes. Like HACA and HYPERION, the rate of cooling in the TTM2 trial was relatively quick with a time to randomization after onset of cardiac arrest of about 2 hours in both groups and a median time from intervention until reaching target temperature of 3 hours. While some retrospective data suggest ultra-early cooling may be beneficial, neither induction of therapeutic hypothermia during OHCA using a rapid infusion of cold saline (Bernard SA, et al. Circulation. 2016;134[11]:797-805) nor transnasal evaporative cooling in the pre-hospital setting (Nordeberg P, et al. JAMA. 2019;321(17):1677-85) has shown improvement in survival with good neurologic outcomes. Next, if we are going to continue TTM, the TTM2 trial does not provide guidance on optimal duration of cooling. Although the current guidelines are to cool for at least 24 hours after ROSC, it is unclear for how long strict temperature control should be continued. The currently enrolling ICECAP study aims to further elucidate the optimal duration of TTM for OHCA patients with both shockable and non-shockable initial rhythms.
Post-cardiac arrest management continues to be a significant area of interest in clinical research and for good reason. Although steady improvement has occurred with regards to survival and neurologic function for IHCA, of the approximately 350,000 nontraumatic OHCA that occur in a year in the United States, only about 10.2% of those patients will survive their initial hospitalization, and only about 8.2% of those who survive will have good functional status (American Heart Association. Circulation. 2020;142(suppl 2):S366-S468). There remains much room for continued study and improvement.
Dr. Capp is a Pulmonary and Critical Care Fellow; and Dr. Pendleton is Assistant Professor of Medicine; Division of Pulmonary, Allergy, Critical Care, and Sleep Medicine; University of Minnesota, Minneapolis, Minnesota.
Animal and human models of the effects of therapeutic hypothermia, now called targeted temperature management (TTM), began to surface in the late 1980s. The first randomized clinical trial employing TTM as a neuroprotective strategy following cardiac arrest did not appear until the early 2000s. When compared with normothermia, the HACA trial (Holzer M, et al. N Engl J Med. 2002;346[8]:549-56) demonstrated a 14% reduction in mortality and improved neurologic outcomes following out of hospital cardiac arrest (OHCA) due to ventricular fibrillation (VF) or ventricular tachycardia (VT) when maintaining body temperature between 32˚C and 34˚C post-arrest. Following the results of this trial, TTM in comatose patients following cardiac arrest was recommended by international guidelines and became the standard of care. It was not until the publication of the TTM1 trial (Nielsen N, et al. N Engl J Med. 2013;369[23]:2197-206) about a decade later, that serious questions regarding the efficacy of TTM were raised. The TTM1 trial showed no difference in mortality or neurologic outcomes when comparing TTM at 33˚C vs 36˚C for OHCA. The results of this trial heralded widespread practice change, with many abandoning deep cooling, and often active cooling measures, in favor of fever avoidance. The HYPERION trial (Lascarrou J, et al. N Engl J Med. 2019;381:2327-37) came next, comparing TTM at 33˚C to normothermia (<37.5˚C) for cardiac arrest with non-hockable rhythm. This study did not identify any improvement in mortality with utilization of TTM but suggested it may be associated with more favorable neurologic outcomes, albeit in a small number of patients.
The TTM2 trial (Dankiewicz J, et al. N Engl J Med. 2021;384:2283-94) is the most recent trial to address the question of TTM post-cardiac arrest. The TTM2 trial was an international, randomized controlled superiority trial of TTM at 33˚C vs normothermia (≤37.8˚C) for patients with coma following OHCA with any initial rhythm. It was conducted by the same group as the TTM1 trial and, to date, represents the largest (N= 1,850) and most robust trial conducted in this area. The trial spanned 61 institutions across 14 countries and had nearly complete follow-up at 6 months. Once again, there was no significant difference in all-cause mortality at 6 months in the TTM group when compared with the normothermia group. Equally important, there were no differences observed in secondary outcomes, including functional neurologic status and health-related quality of life at 6 months. With the results of the TTM1 and TTM2 trials failing to show any neurologic or mortality benefit to TTM, we are left wondering, is there anything therapeutic about “therapeutic hypothermia”?
Both the 2020 American Heart Association (AHA) and 2021 European Resuscitation Council (ERC) guidelines predate this trial; they recommend cooling any OHCA or in-hospital cardiac arrest (IHCA) patient who remains unresponsive after return of spontaneous circulation (ROSC) regardless of initial rhythm. They further suggest maintaining a target temperature between 32˚C and 36˚C for at least 24 hours, followed by avoidance of fever (>37.7˚C) for at least 72 hours after ROSC in patients who remain comatose. While it will be interesting to see what future iterations of the guidelines recommend, the results from the TTM1 and TTM2 trials support a shift in clinical practice away from TTM and toward more active fever avoidance. Additionally, careful review of adverse events in the TTM2 trial suggests that induced hypothermia is not without risk of harm. When compared with the normothermia group in the TTM2 trial, the hypothermia group experienced higher rates of arrhythmias with hemodynamic instability (16% vs 24%), increased exposure to sedation, increased use of neuromuscular blockade, and increased duration of mechanical ventilation.
While the results of the TTM2 trial move the needle away from therapeutic hypothermia for OHCA patients, there is some nuance that warrants further discussion. First, the initial HACA trial, upon which the standard of TTM was based, included only patients with an initial shockable rhythm (VT/VF). Inherently, the etiology of these arrests is likely to be cardiac and more reversible in nature. Most subsequent landmark trials on TTM, including the TTM2 trial, have included OHCA patients with both shockable and nonshockable initial rhythms. Still, the majority of patients in the TTM2 trial had an initial shockable rhythm on presentation (72% hypothermia vs 75% normothermia). This may limit broad generalizability of study findings as an increasing number of OHCA patients are presenting with nonshockable initial rhythms. Next, it is well known that bystander CPR improves outcomes following OHCA. Impressively, over 75% of patients in both groups in the TTM2 trial received bystander CPR compared with an average of 46% of arrest patients in the US according to AHA data. Finally, like most of its predecessors, the TTM2 trial only included OHCA patients meaning no real conclusions can be drawn regarding application of TTM to IHCA patients. Of the major trials to date, only the HYPERION trial included IHCA patients – representing about 25% of the study population. Thus, the utility of TTM in the setting of IHCA remains largely unknown.
Taken in summation, recent trials, including TTM2, suggest that fever-avoidance post-cardiac arrest is likely the best option for improving mortality and neurologic outcomes while mitigating risk to the patient. We must remain vigilant in our enforcement of normothermia though as worse neurologic outcomes have been observed with hyperthermia in the early post-arrest period (Zeiner A, et al. Arch Intern Med. 2001;161[16]:2007-12). A key takeaway from recent trials is that maintaining normothermia without active temperature control measures is likely to be difficult to achieve. A criticism of the HYPERION trial was that a “substantial proportion” of patients in the normothermia group had temperatures above 38˚C. Similarly, 10% to15% of patients in the TTM2 trial had body temperatures above 37.7˚C, 40 to 72 hours after randomization and, ultimately, 46% of patients in the normothermia group required cooling with a temperature management device. Thus, we can conclude that maintenance of strict normothermia will likely continue to require active control with a temperature management device.
Despite an increasing number of well conducted studies in this area, there are several questions that remain unanswered. The first is whether cooling patients even earlier post-arrest is felt to increase the likelihood of survival with improved neurologic outcomes. Like HACA and HYPERION, the rate of cooling in the TTM2 trial was relatively quick with a time to randomization after onset of cardiac arrest of about 2 hours in both groups and a median time from intervention until reaching target temperature of 3 hours. While some retrospective data suggest ultra-early cooling may be beneficial, neither induction of therapeutic hypothermia during OHCA using a rapid infusion of cold saline (Bernard SA, et al. Circulation. 2016;134[11]:797-805) nor transnasal evaporative cooling in the pre-hospital setting (Nordeberg P, et al. JAMA. 2019;321(17):1677-85) has shown improvement in survival with good neurologic outcomes. Next, if we are going to continue TTM, the TTM2 trial does not provide guidance on optimal duration of cooling. Although the current guidelines are to cool for at least 24 hours after ROSC, it is unclear for how long strict temperature control should be continued. The currently enrolling ICECAP study aims to further elucidate the optimal duration of TTM for OHCA patients with both shockable and non-shockable initial rhythms.
Post-cardiac arrest management continues to be a significant area of interest in clinical research and for good reason. Although steady improvement has occurred with regards to survival and neurologic function for IHCA, of the approximately 350,000 nontraumatic OHCA that occur in a year in the United States, only about 10.2% of those patients will survive their initial hospitalization, and only about 8.2% of those who survive will have good functional status (American Heart Association. Circulation. 2020;142(suppl 2):S366-S468). There remains much room for continued study and improvement.
Dr. Capp is a Pulmonary and Critical Care Fellow; and Dr. Pendleton is Assistant Professor of Medicine; Division of Pulmonary, Allergy, Critical Care, and Sleep Medicine; University of Minnesota, Minneapolis, Minnesota.
Animal and human models of the effects of therapeutic hypothermia, now called targeted temperature management (TTM), began to surface in the late 1980s. The first randomized clinical trial employing TTM as a neuroprotective strategy following cardiac arrest did not appear until the early 2000s. When compared with normothermia, the HACA trial (Holzer M, et al. N Engl J Med. 2002;346[8]:549-56) demonstrated a 14% reduction in mortality and improved neurologic outcomes following out of hospital cardiac arrest (OHCA) due to ventricular fibrillation (VF) or ventricular tachycardia (VT) when maintaining body temperature between 32˚C and 34˚C post-arrest. Following the results of this trial, TTM in comatose patients following cardiac arrest was recommended by international guidelines and became the standard of care. It was not until the publication of the TTM1 trial (Nielsen N, et al. N Engl J Med. 2013;369[23]:2197-206) about a decade later, that serious questions regarding the efficacy of TTM were raised. The TTM1 trial showed no difference in mortality or neurologic outcomes when comparing TTM at 33˚C vs 36˚C for OHCA. The results of this trial heralded widespread practice change, with many abandoning deep cooling, and often active cooling measures, in favor of fever avoidance. The HYPERION trial (Lascarrou J, et al. N Engl J Med. 2019;381:2327-37) came next, comparing TTM at 33˚C to normothermia (<37.5˚C) for cardiac arrest with non-hockable rhythm. This study did not identify any improvement in mortality with utilization of TTM but suggested it may be associated with more favorable neurologic outcomes, albeit in a small number of patients.
The TTM2 trial (Dankiewicz J, et al. N Engl J Med. 2021;384:2283-94) is the most recent trial to address the question of TTM post-cardiac arrest. The TTM2 trial was an international, randomized controlled superiority trial of TTM at 33˚C vs normothermia (≤37.8˚C) for patients with coma following OHCA with any initial rhythm. It was conducted by the same group as the TTM1 trial and, to date, represents the largest (N= 1,850) and most robust trial conducted in this area. The trial spanned 61 institutions across 14 countries and had nearly complete follow-up at 6 months. Once again, there was no significant difference in all-cause mortality at 6 months in the TTM group when compared with the normothermia group. Equally important, there were no differences observed in secondary outcomes, including functional neurologic status and health-related quality of life at 6 months. With the results of the TTM1 and TTM2 trials failing to show any neurologic or mortality benefit to TTM, we are left wondering, is there anything therapeutic about “therapeutic hypothermia”?
Both the 2020 American Heart Association (AHA) and 2021 European Resuscitation Council (ERC) guidelines predate this trial; they recommend cooling any OHCA or in-hospital cardiac arrest (IHCA) patient who remains unresponsive after return of spontaneous circulation (ROSC) regardless of initial rhythm. They further suggest maintaining a target temperature between 32˚C and 36˚C for at least 24 hours, followed by avoidance of fever (>37.7˚C) for at least 72 hours after ROSC in patients who remain comatose. While it will be interesting to see what future iterations of the guidelines recommend, the results from the TTM1 and TTM2 trials support a shift in clinical practice away from TTM and toward more active fever avoidance. Additionally, careful review of adverse events in the TTM2 trial suggests that induced hypothermia is not without risk of harm. When compared with the normothermia group in the TTM2 trial, the hypothermia group experienced higher rates of arrhythmias with hemodynamic instability (16% vs 24%), increased exposure to sedation, increased use of neuromuscular blockade, and increased duration of mechanical ventilation.
While the results of the TTM2 trial move the needle away from therapeutic hypothermia for OHCA patients, there is some nuance that warrants further discussion. First, the initial HACA trial, upon which the standard of TTM was based, included only patients with an initial shockable rhythm (VT/VF). Inherently, the etiology of these arrests is likely to be cardiac and more reversible in nature. Most subsequent landmark trials on TTM, including the TTM2 trial, have included OHCA patients with both shockable and nonshockable initial rhythms. Still, the majority of patients in the TTM2 trial had an initial shockable rhythm on presentation (72% hypothermia vs 75% normothermia). This may limit broad generalizability of study findings as an increasing number of OHCA patients are presenting with nonshockable initial rhythms. Next, it is well known that bystander CPR improves outcomes following OHCA. Impressively, over 75% of patients in both groups in the TTM2 trial received bystander CPR compared with an average of 46% of arrest patients in the US according to AHA data. Finally, like most of its predecessors, the TTM2 trial only included OHCA patients meaning no real conclusions can be drawn regarding application of TTM to IHCA patients. Of the major trials to date, only the HYPERION trial included IHCA patients – representing about 25% of the study population. Thus, the utility of TTM in the setting of IHCA remains largely unknown.
Taken in summation, recent trials, including TTM2, suggest that fever-avoidance post-cardiac arrest is likely the best option for improving mortality and neurologic outcomes while mitigating risk to the patient. We must remain vigilant in our enforcement of normothermia though as worse neurologic outcomes have been observed with hyperthermia in the early post-arrest period (Zeiner A, et al. Arch Intern Med. 2001;161[16]:2007-12). A key takeaway from recent trials is that maintaining normothermia without active temperature control measures is likely to be difficult to achieve. A criticism of the HYPERION trial was that a “substantial proportion” of patients in the normothermia group had temperatures above 38˚C. Similarly, 10% to15% of patients in the TTM2 trial had body temperatures above 37.7˚C, 40 to 72 hours after randomization and, ultimately, 46% of patients in the normothermia group required cooling with a temperature management device. Thus, we can conclude that maintenance of strict normothermia will likely continue to require active control with a temperature management device.
Despite an increasing number of well conducted studies in this area, there are several questions that remain unanswered. The first is whether cooling patients even earlier post-arrest is felt to increase the likelihood of survival with improved neurologic outcomes. Like HACA and HYPERION, the rate of cooling in the TTM2 trial was relatively quick with a time to randomization after onset of cardiac arrest of about 2 hours in both groups and a median time from intervention until reaching target temperature of 3 hours. While some retrospective data suggest ultra-early cooling may be beneficial, neither induction of therapeutic hypothermia during OHCA using a rapid infusion of cold saline (Bernard SA, et al. Circulation. 2016;134[11]:797-805) nor transnasal evaporative cooling in the pre-hospital setting (Nordeberg P, et al. JAMA. 2019;321(17):1677-85) has shown improvement in survival with good neurologic outcomes. Next, if we are going to continue TTM, the TTM2 trial does not provide guidance on optimal duration of cooling. Although the current guidelines are to cool for at least 24 hours after ROSC, it is unclear for how long strict temperature control should be continued. The currently enrolling ICECAP study aims to further elucidate the optimal duration of TTM for OHCA patients with both shockable and non-shockable initial rhythms.
Post-cardiac arrest management continues to be a significant area of interest in clinical research and for good reason. Although steady improvement has occurred with regards to survival and neurologic function for IHCA, of the approximately 350,000 nontraumatic OHCA that occur in a year in the United States, only about 10.2% of those patients will survive their initial hospitalization, and only about 8.2% of those who survive will have good functional status (American Heart Association. Circulation. 2020;142(suppl 2):S366-S468). There remains much room for continued study and improvement.
Dr. Capp is a Pulmonary and Critical Care Fellow; and Dr. Pendleton is Assistant Professor of Medicine; Division of Pulmonary, Allergy, Critical Care, and Sleep Medicine; University of Minnesota, Minneapolis, Minnesota.
Lung transplantation for patients with severe COVID-19
As of September 2021, over 222 million people worldwide (WHO, 2021) and 40 million Americans (CDC, 2021) have been infected with the novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). The total number of infections in the United States began climbing again this summer with the persistence of vaccine reluctance among a significant proportion of the population and the emergence of the much more infectious B.1.617.2 (Delta) variant. While the clinical illness caused by the SARS-CoV-2 virus, referred to as the Coronavirus disease 2019 (COVID-19), is mostly mild, approximately 10% of cases develop acute respiratory distress syndrome (ARDS) (Remuzzi A, et al. Lancet. 2020;395[10231]:1225-8). A small but substantial proportion of patients with COVID-19 ARDS fails to respond to the various supportive measures and requires extracorporeal membrane oxygenation (ECMO) support. The overarching goal of the different support strategies, including ECMO, is to provide time for the lungs to recover from ARDS. ECMO has the theoretical advantage over other strategies in facilitating recovery by allowing the injured lungs to ‘rest’ as the oxygenation and ventilation needs are met in an extracorporeal fashion. Regardless, a small number of patients with COVID-19 ARDS will not recover enough pulmonary function to allow them to be weaned from the various respiratory support strategies.

For patients with irreversible lung injury, lung transplantation (LT) is a potential consideration. Earlier in the pandemic, older patients with significant comorbid illnesses were more vulnerable to severe COVID-19, often precluding consideration for transplantation. However, the emergence of the Delta variant may have altered this dynamic via a substantial increase in the incidence of COVID-19 ARDS among younger and healthier patients. A handful of patients with COVID-19 ARDS have already had successful transplantation. However, the overall number is still small (Bharat A, et al. Sci Translat Med. 2020 Dec 16;12[574]:eabe4282. doi: 10.1126/scitranslmed.abe4282. Epub 2020 Nov 30; and Hawkins R, et al. Transplantation. 2021;6:1381-7), and there is a lack of long-term outcomes data among these patients.

There is currently little guidance regarding criteria for patient selection and consideration for LT among patients with COVID-19 ARDS. Given that the SARS-CoV-2 virus is a novel pathogen that leads to an illness that is unique from other forms of viral pneumonia, specific considerations regarding LT should be made among these patients. In the current article, we discuss some of the pertinent issues related to the consideration of LT among patients with COVID-19 ARDS.
The timing for considering LT is one of the most important aspects. First, patients with COVID-19 ARDS must not be actively infected at the time of transplantation consideration. It has been suggested that LT should only be considered in patients with two separate negative polymerase chain reaction (PCR) test results for SARS-CoV-2 from bronchoalveolar lavage fluid 24 hours apart and at least 4 weeks after the onset of COVID-19 symptoms (Bharat A, et al. Sci Translat Med. 2020 Dec 16;12[574]:eabe4282. doi: 10.1126/scitranslmed.abe4282. Epub 2020 Nov 30). Among patients with persistently positive SARS-CoV-2 PCR 4 to 6 weeks after symptom onset, a negative viral culture from a bronchoalveolar lavage (BAL) can be used to confirm viral inactivity (Lang C, et al. Lancet Respir Med. 2020;8[10]:1057-60).
Despite the sparse data in this domain, there seems to be a consensus in the literature that LT could be considered once 4 to 6 weeks have elapsed since the onset of the respiratory failure (Cypel M, et al. Lancet Respir Med. 2020;8[10]:944-6). This timeline is felt to be long enough to alleviate the concerns regarding ongoing inflammatory processes that may be reversible while not so long to risk the development of non-pulmonary complications or severe debility that may become significant barriers to transplant candidacy. An exception may be made in patients with medically unmanageable complications such as recalcitrant bronchopleural fistulae in the background of fibrotic changes or right ventricular failure from severe pulmonary hypertension. Regardless, this timeline is borrowed from the approach to irreversible ARDS from other forms of viral pneumonia. It is not clear if it is appropriate to extrapolate past experience to COVID-19, which is a disease unlike any other seen during the LT era: a profound inflammatory phase mediated by a cytokine storm as the etiologic basis for the organ dysfunction, activation of coagulation pathways in pulmonary circulation leading to immunothrombosis contributing to the refractory hypoxemia, favorable effects of anticoagulants, diverse pulmonary physiologic phenotypes of ARDS, an increased risk of pleural complications, and utilization of novel anti-inflammatory therapies with consequent risks ofsecondary infections are all unique to COVID-19. A recent study found that patients requiring ECMO for COVID-19 ARDS took longer to recover lung function but had similar survival rates to patients on ECMO with other virus-induced ARDS (Raff LA, et al. Am J Surg. 2021;S0002-9610[21]00233-6. doi: 10.1016/j.amjsurg.2021.04.004. Online ahead of print).These data support pursuing a more conservative timeline for consideration of LT.
Determining the reversibility of pulmonary impairment in COVID-19 ARDS is another challenge. The nature of the pulmonary opacities should be assessed on CT scan imaging as close as possible to the time of LT consideration. Differentiating the extent of irreversible parenchymal scarring vs salvageability during acute illness can be challenging. The presence of extensive architectural distortion with or without bullous changes, while being the best indicator of irreversibility, may not be sensitive enough. The standard of care in such situations remains serial assessments, often weekly, by a dedicated multidisciplinary group. We have found it useful to augment the imaging data with pulmonary physiologic assessments, including the extent of ventilator and ECMO support as well as dynamic and static compliance trends. Improvement in physiologic data often precedes radiologic improvement. Nonetheless, an important area of future research is to identify objective markers for determining reversibility, which could include novel biomarkers in serum or bronchoalveolar lavage fluid.
When a determination is made regarding the irreversibility of pulmonary impairment, the LT evaluation should begin promptly. Pre-transplant deconditioning and debility is associated with worse post-transplant outcomes. In this regard, patients managed using an ambulatory ECMO strategy may have superior rehabilitation potential. Furthermore, an attempt should be made during the evaluation to wean sedation in order to facilitate discussions regarding the rigors of LT with the patient alongside present family members. An additional consideration, given the use of immunomodulatory medications for COVID-19 and prolonged intubation, is the dramatically increased risk of multi-drug resistant infections in this population; these must be aggressively managed for patients to remain eligible for LT.
The degree of pulmonary impairment and frequent colonization of the airways will likely dictate bilateral LT as the preferred strategy, although surgical feasibility may, at times, be the overriding determinant. Regardless of the type of transplant, certain unique aspects should be anticipated. The inflammatory responses during COVID-19 that often spill outside the confines of the pulmonary parenchyma, along with potentially frequent thoracic interventions prior to transplant, create significant technical challenges during the operation. Native pneumonectomy can take longer than usual leading to prolonged ischemic time, increased need for intra-operative blood products, and raised risk for primary graft dysfunction. All of these factors have a significant impact on early and late outcomes. Finally, the long-term immunologic consequences of severe infection from a novel virus remain unknown, and it is unclear if COVID-19 ARDS patients bridged to transplant will enjoy comparable survival. It is pertinent to acknowledge that the high-risk nature of such transplants is substantially accentuated due to several unique characteristics of the illness related to COVID-19.
The emergence of the COVID-19 pandemic has led to an increase in the number of urgent inpatient lung transplant consultations for refractory ARDS. While the basic principles of LT candidate selection should continue to guide us, the unique characteristics of this illness merit using a customized approach. There are few validated predictors to guide decision-making, and longitudinal assessments by a dedicated multidisciplinary group remain the best strategy. Finally, in the absence of systemic studies and lack of longitudinal outcomes data, there is an emergent need to establish consensus guidelines regarding the approach to LT consideration in these patients.
Dr. Quinn and Dr. Banga are with the Lung Transplant Program, Divisions of Pulmonary and Critical Care Medicine, University of Texas Southwestern Medical Center, Dallas.
As of September 2021, over 222 million people worldwide (WHO, 2021) and 40 million Americans (CDC, 2021) have been infected with the novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). The total number of infections in the United States began climbing again this summer with the persistence of vaccine reluctance among a significant proportion of the population and the emergence of the much more infectious B.1.617.2 (Delta) variant. While the clinical illness caused by the SARS-CoV-2 virus, referred to as the Coronavirus disease 2019 (COVID-19), is mostly mild, approximately 10% of cases develop acute respiratory distress syndrome (ARDS) (Remuzzi A, et al. Lancet. 2020;395[10231]:1225-8). A small but substantial proportion of patients with COVID-19 ARDS fails to respond to the various supportive measures and requires extracorporeal membrane oxygenation (ECMO) support. The overarching goal of the different support strategies, including ECMO, is to provide time for the lungs to recover from ARDS. ECMO has the theoretical advantage over other strategies in facilitating recovery by allowing the injured lungs to ‘rest’ as the oxygenation and ventilation needs are met in an extracorporeal fashion. Regardless, a small number of patients with COVID-19 ARDS will not recover enough pulmonary function to allow them to be weaned from the various respiratory support strategies.
For patients with irreversible lung injury, lung transplantation (LT) is a potential consideration. Earlier in the pandemic, older patients with significant comorbid illnesses were more vulnerable to severe COVID-19, often precluding consideration for transplantation. However, the emergence of the Delta variant may have altered this dynamic via a substantial increase in the incidence of COVID-19 ARDS among younger and healthier patients. A handful of patients with COVID-19 ARDS have already had successful transplantation. However, the overall number is still small (Bharat A, et al. Sci Translat Med. 2020 Dec 16;12[574]:eabe4282. doi: 10.1126/scitranslmed.abe4282. Epub 2020 Nov 30; and Hawkins R, et al. Transplantation. 2021;6:1381-7), and there is a lack of long-term outcomes data among these patients.
There is currently little guidance regarding criteria for patient selection and consideration for LT among patients with COVID-19 ARDS. Given that the SARS-CoV-2 virus is a novel pathogen that leads to an illness that is unique from other forms of viral pneumonia, specific considerations regarding LT should be made among these patients. In the current article, we discuss some of the pertinent issues related to the consideration of LT among patients with COVID-19 ARDS.
The timing for considering LT is one of the most important aspects. First, patients with COVID-19 ARDS must not be actively infected at the time of transplantation consideration. It has been suggested that LT should only be considered in patients with two separate negative polymerase chain reaction (PCR) test results for SARS-CoV-2 from bronchoalveolar lavage fluid 24 hours apart and at least 4 weeks after the onset of COVID-19 symptoms (Bharat A, et al. Sci Translat Med. 2020 Dec 16;12[574]:eabe4282. doi: 10.1126/scitranslmed.abe4282. Epub 2020 Nov 30). Among patients with persistently positive SARS-CoV-2 PCR 4 to 6 weeks after symptom onset, a negative viral culture from a bronchoalveolar lavage (BAL) can be used to confirm viral inactivity (Lang C, et al. Lancet Respir Med. 2020;8[10]:1057-60).
Despite the sparse data in this domain, there seems to be a consensus in the literature that LT could be considered once 4 to 6 weeks have elapsed since the onset of the respiratory failure (Cypel M, et al. Lancet Respir Med. 2020;8[10]:944-6). This timeline is felt to be long enough to alleviate the concerns regarding ongoing inflammatory processes that may be reversible while not so long to risk the development of non-pulmonary complications or severe debility that may become significant barriers to transplant candidacy. An exception may be made in patients with medically unmanageable complications such as recalcitrant bronchopleural fistulae in the background of fibrotic changes or right ventricular failure from severe pulmonary hypertension. Regardless, this timeline is borrowed from the approach to irreversible ARDS from other forms of viral pneumonia. It is not clear if it is appropriate to extrapolate past experience to COVID-19, which is a disease unlike any other seen during the LT era: a profound inflammatory phase mediated by a cytokine storm as the etiologic basis for the organ dysfunction, activation of coagulation pathways in pulmonary circulation leading to immunothrombosis contributing to the refractory hypoxemia, favorable effects of anticoagulants, diverse pulmonary physiologic phenotypes of ARDS, an increased risk of pleural complications, and utilization of novel anti-inflammatory therapies with consequent risks ofsecondary infections are all unique to COVID-19. A recent study found that patients requiring ECMO for COVID-19 ARDS took longer to recover lung function but had similar survival rates to patients on ECMO with other virus-induced ARDS (Raff LA, et al. Am J Surg. 2021;S0002-9610[21]00233-6. doi: 10.1016/j.amjsurg.2021.04.004. Online ahead of print).These data support pursuing a more conservative timeline for consideration of LT.
Determining the reversibility of pulmonary impairment in COVID-19 ARDS is another challenge. The nature of the pulmonary opacities should be assessed on CT scan imaging as close as possible to the time of LT consideration. Differentiating the extent of irreversible parenchymal scarring vs salvageability during acute illness can be challenging. The presence of extensive architectural distortion with or without bullous changes, while being the best indicator of irreversibility, may not be sensitive enough. The standard of care in such situations remains serial assessments, often weekly, by a dedicated multidisciplinary group. We have found it useful to augment the imaging data with pulmonary physiologic assessments, including the extent of ventilator and ECMO support as well as dynamic and static compliance trends. Improvement in physiologic data often precedes radiologic improvement. Nonetheless, an important area of future research is to identify objective markers for determining reversibility, which could include novel biomarkers in serum or bronchoalveolar lavage fluid.
When a determination is made regarding the irreversibility of pulmonary impairment, the LT evaluation should begin promptly. Pre-transplant deconditioning and debility is associated with worse post-transplant outcomes. In this regard, patients managed using an ambulatory ECMO strategy may have superior rehabilitation potential. Furthermore, an attempt should be made during the evaluation to wean sedation in order to facilitate discussions regarding the rigors of LT with the patient alongside present family members. An additional consideration, given the use of immunomodulatory medications for COVID-19 and prolonged intubation, is the dramatically increased risk of multi-drug resistant infections in this population; these must be aggressively managed for patients to remain eligible for LT.
The degree of pulmonary impairment and frequent colonization of the airways will likely dictate bilateral LT as the preferred strategy, although surgical feasibility may, at times, be the overriding determinant. Regardless of the type of transplant, certain unique aspects should be anticipated. The inflammatory responses during COVID-19 that often spill outside the confines of the pulmonary parenchyma, along with potentially frequent thoracic interventions prior to transplant, create significant technical challenges during the operation. Native pneumonectomy can take longer than usual leading to prolonged ischemic time, increased need for intra-operative blood products, and raised risk for primary graft dysfunction. All of these factors have a significant impact on early and late outcomes. Finally, the long-term immunologic consequences of severe infection from a novel virus remain unknown, and it is unclear if COVID-19 ARDS patients bridged to transplant will enjoy comparable survival. It is pertinent to acknowledge that the high-risk nature of such transplants is substantially accentuated due to several unique characteristics of the illness related to COVID-19.
The emergence of the COVID-19 pandemic has led to an increase in the number of urgent inpatient lung transplant consultations for refractory ARDS. While the basic principles of LT candidate selection should continue to guide us, the unique characteristics of this illness merit using a customized approach. There are few validated predictors to guide decision-making, and longitudinal assessments by a dedicated multidisciplinary group remain the best strategy. Finally, in the absence of systemic studies and lack of longitudinal outcomes data, there is an emergent need to establish consensus guidelines regarding the approach to LT consideration in these patients.
Dr. Quinn and Dr. Banga are with the Lung Transplant Program, Divisions of Pulmonary and Critical Care Medicine, University of Texas Southwestern Medical Center, Dallas.
As of September 2021, over 222 million people worldwide (WHO, 2021) and 40 million Americans (CDC, 2021) have been infected with the novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). The total number of infections in the United States began climbing again this summer with the persistence of vaccine reluctance among a significant proportion of the population and the emergence of the much more infectious B.1.617.2 (Delta) variant. While the clinical illness caused by the SARS-CoV-2 virus, referred to as the Coronavirus disease 2019 (COVID-19), is mostly mild, approximately 10% of cases develop acute respiratory distress syndrome (ARDS) (Remuzzi A, et al. Lancet. 2020;395[10231]:1225-8). A small but substantial proportion of patients with COVID-19 ARDS fails to respond to the various supportive measures and requires extracorporeal membrane oxygenation (ECMO) support. The overarching goal of the different support strategies, including ECMO, is to provide time for the lungs to recover from ARDS. ECMO has the theoretical advantage over other strategies in facilitating recovery by allowing the injured lungs to ‘rest’ as the oxygenation and ventilation needs are met in an extracorporeal fashion. Regardless, a small number of patients with COVID-19 ARDS will not recover enough pulmonary function to allow them to be weaned from the various respiratory support strategies.
For patients with irreversible lung injury, lung transplantation (LT) is a potential consideration. Earlier in the pandemic, older patients with significant comorbid illnesses were more vulnerable to severe COVID-19, often precluding consideration for transplantation. However, the emergence of the Delta variant may have altered this dynamic via a substantial increase in the incidence of COVID-19 ARDS among younger and healthier patients. A handful of patients with COVID-19 ARDS have already had successful transplantation. However, the overall number is still small (Bharat A, et al. Sci Translat Med. 2020 Dec 16;12[574]:eabe4282. doi: 10.1126/scitranslmed.abe4282. Epub 2020 Nov 30; and Hawkins R, et al. Transplantation. 2021;6:1381-7), and there is a lack of long-term outcomes data among these patients.
There is currently little guidance regarding criteria for patient selection and consideration for LT among patients with COVID-19 ARDS. Given that the SARS-CoV-2 virus is a novel pathogen that leads to an illness that is unique from other forms of viral pneumonia, specific considerations regarding LT should be made among these patients. In the current article, we discuss some of the pertinent issues related to the consideration of LT among patients with COVID-19 ARDS.
The timing for considering LT is one of the most important aspects. First, patients with COVID-19 ARDS must not be actively infected at the time of transplantation consideration. It has been suggested that LT should only be considered in patients with two separate negative polymerase chain reaction (PCR) test results for SARS-CoV-2 from bronchoalveolar lavage fluid 24 hours apart and at least 4 weeks after the onset of COVID-19 symptoms (Bharat A, et al. Sci Translat Med. 2020 Dec 16;12[574]:eabe4282. doi: 10.1126/scitranslmed.abe4282. Epub 2020 Nov 30). Among patients with persistently positive SARS-CoV-2 PCR 4 to 6 weeks after symptom onset, a negative viral culture from a bronchoalveolar lavage (BAL) can be used to confirm viral inactivity (Lang C, et al. Lancet Respir Med. 2020;8[10]:1057-60).
Despite the sparse data in this domain, there seems to be a consensus in the literature that LT could be considered once 4 to 6 weeks have elapsed since the onset of the respiratory failure (Cypel M, et al. Lancet Respir Med. 2020;8[10]:944-6). This timeline is felt to be long enough to alleviate the concerns regarding ongoing inflammatory processes that may be reversible while not so long to risk the development of non-pulmonary complications or severe debility that may become significant barriers to transplant candidacy. An exception may be made in patients with medically unmanageable complications such as recalcitrant bronchopleural fistulae in the background of fibrotic changes or right ventricular failure from severe pulmonary hypertension. Regardless, this timeline is borrowed from the approach to irreversible ARDS from other forms of viral pneumonia. It is not clear if it is appropriate to extrapolate past experience to COVID-19, which is a disease unlike any other seen during the LT era: a profound inflammatory phase mediated by a cytokine storm as the etiologic basis for the organ dysfunction, activation of coagulation pathways in pulmonary circulation leading to immunothrombosis contributing to the refractory hypoxemia, favorable effects of anticoagulants, diverse pulmonary physiologic phenotypes of ARDS, an increased risk of pleural complications, and utilization of novel anti-inflammatory therapies with consequent risks ofsecondary infections are all unique to COVID-19. A recent study found that patients requiring ECMO for COVID-19 ARDS took longer to recover lung function but had similar survival rates to patients on ECMO with other virus-induced ARDS (Raff LA, et al. Am J Surg. 2021;S0002-9610[21]00233-6. doi: 10.1016/j.amjsurg.2021.04.004. Online ahead of print).These data support pursuing a more conservative timeline for consideration of LT.
Determining the reversibility of pulmonary impairment in COVID-19 ARDS is another challenge. The nature of the pulmonary opacities should be assessed on CT scan imaging as close as possible to the time of LT consideration. Differentiating the extent of irreversible parenchymal scarring vs salvageability during acute illness can be challenging. The presence of extensive architectural distortion with or without bullous changes, while being the best indicator of irreversibility, may not be sensitive enough. The standard of care in such situations remains serial assessments, often weekly, by a dedicated multidisciplinary group. We have found it useful to augment the imaging data with pulmonary physiologic assessments, including the extent of ventilator and ECMO support as well as dynamic and static compliance trends. Improvement in physiologic data often precedes radiologic improvement. Nonetheless, an important area of future research is to identify objective markers for determining reversibility, which could include novel biomarkers in serum or bronchoalveolar lavage fluid.
When a determination is made regarding the irreversibility of pulmonary impairment, the LT evaluation should begin promptly. Pre-transplant deconditioning and debility is associated with worse post-transplant outcomes. In this regard, patients managed using an ambulatory ECMO strategy may have superior rehabilitation potential. Furthermore, an attempt should be made during the evaluation to wean sedation in order to facilitate discussions regarding the rigors of LT with the patient alongside present family members. An additional consideration, given the use of immunomodulatory medications for COVID-19 and prolonged intubation, is the dramatically increased risk of multi-drug resistant infections in this population; these must be aggressively managed for patients to remain eligible for LT.
The degree of pulmonary impairment and frequent colonization of the airways will likely dictate bilateral LT as the preferred strategy, although surgical feasibility may, at times, be the overriding determinant. Regardless of the type of transplant, certain unique aspects should be anticipated. The inflammatory responses during COVID-19 that often spill outside the confines of the pulmonary parenchyma, along with potentially frequent thoracic interventions prior to transplant, create significant technical challenges during the operation. Native pneumonectomy can take longer than usual leading to prolonged ischemic time, increased need for intra-operative blood products, and raised risk for primary graft dysfunction. All of these factors have a significant impact on early and late outcomes. Finally, the long-term immunologic consequences of severe infection from a novel virus remain unknown, and it is unclear if COVID-19 ARDS patients bridged to transplant will enjoy comparable survival. It is pertinent to acknowledge that the high-risk nature of such transplants is substantially accentuated due to several unique characteristics of the illness related to COVID-19.
The emergence of the COVID-19 pandemic has led to an increase in the number of urgent inpatient lung transplant consultations for refractory ARDS. While the basic principles of LT candidate selection should continue to guide us, the unique characteristics of this illness merit using a customized approach. There are few validated predictors to guide decision-making, and longitudinal assessments by a dedicated multidisciplinary group remain the best strategy. Finally, in the absence of systemic studies and lack of longitudinal outcomes data, there is an emergent need to establish consensus guidelines regarding the approach to LT consideration in these patients.
Dr. Quinn and Dr. Banga are with the Lung Transplant Program, Divisions of Pulmonary and Critical Care Medicine, University of Texas Southwestern Medical Center, Dallas.
This month in the journal CHEST®
Editor’s picks
How I do it: Transitioning asthma care from adolescents to adults: Severe Asthma Series. By Dr. A. Nanzer.
Outpatient management of patients with COVID-19: Multicenter prospective validation of the HOME-CoV Rule to safely discharge patients. By Dr. D. Douillet, et al.
Emphysema progression and lung function decline among angiotensin converting enzyme inhibitors (ACEi) and angiotensin-receptor blockade (ARB) users in the COPDGene Cohort. By Dr. V. Tejwani, et al.
Sarcoidosis: An occupational disease? By Dr. C.L. Oliver, et al.
Pulmonary thrombosis and thromboembolism in COVID-19. By Dr. H. Poor.
How I do it: Mediastinal staging for lung cancer. By Dr. F. Farjah, et al.
Editor’s picks
Editor’s picks
How I do it: Transitioning asthma care from adolescents to adults: Severe Asthma Series. By Dr. A. Nanzer.
Outpatient management of patients with COVID-19: Multicenter prospective validation of the HOME-CoV Rule to safely discharge patients. By Dr. D. Douillet, et al.
Emphysema progression and lung function decline among angiotensin converting enzyme inhibitors (ACEi) and angiotensin-receptor blockade (ARB) users in the COPDGene Cohort. By Dr. V. Tejwani, et al.
Sarcoidosis: An occupational disease? By Dr. C.L. Oliver, et al.
Pulmonary thrombosis and thromboembolism in COVID-19. By Dr. H. Poor.
How I do it: Mediastinal staging for lung cancer. By Dr. F. Farjah, et al.
How I do it: Transitioning asthma care from adolescents to adults: Severe Asthma Series. By Dr. A. Nanzer.
Outpatient management of patients with COVID-19: Multicenter prospective validation of the HOME-CoV Rule to safely discharge patients. By Dr. D. Douillet, et al.
Emphysema progression and lung function decline among angiotensin converting enzyme inhibitors (ACEi) and angiotensin-receptor blockade (ARB) users in the COPDGene Cohort. By Dr. V. Tejwani, et al.
Sarcoidosis: An occupational disease? By Dr. C.L. Oliver, et al.
Pulmonary thrombosis and thromboembolism in COVID-19. By Dr. H. Poor.
How I do it: Mediastinal staging for lung cancer. By Dr. F. Farjah, et al.