User login
Throwing, the Shoulder, and Human Evolution
Charles Darwin once said that apes “...are quite unable, as I have myself seen, to throw a stone with precision”.1 Yet humans can throw with precision and speed, a skill that likely had significant advantages: throwing can affect change at a distance—something few species can do. Throwing can provide protection against predators and can allow for predation for food resources. Throwing would be important in contesting other hominids for scarce resources. As such, throwing has been critically important in human evolution and likely is a skill that has been promoted through natural selection.2-5
In the orthopedic literature, most published work on throwing will ask proximate questions: “how, what, who, when, and where?” Evolutionary biologists are concerned with ultimate questions6,7: “why?” Asking ultimate questions provides insight into how a behavior might offer advantages under natural selection, which can then improve our understanding of the proximate questions for that behavior.
With regard to the shoulder, a number of mysteries exist that, to date, proximate studies have not been able to solve. This article argues that the human shoulder has evolved for throwing and by using this frame of reference, many of the mysteries surrounding the anatomy of the shoulder can be understood.
Pitching Kinematics
The mechanics of pitching have been analyzed extensively. Fleisig and colleagues8 performed kinematic and electromyographic analyses of pitchers to identify the critical moments of pitching (defined as where the forces are highest and injury is most likely going to occur). They found 2 moments where the forces about the shoulder are highest during pitching: the late cocking phase (defined by the point where the humerus reaches maximal external rotation); and the early deceleration phase (defined by the point when the ball is released). If throwing is important in natural selection of humans, then the shoulder anatomy should be optimized to withstand the forces generated in these positions.
Late Cocking Phase of Throwing
The early phases of throwing are attempting to maximize external rotation of the abducted arm as the velocity of the pitched ball correlates to the amount of external rotation achieved.9-11 In this position, kinetic energy in external rotation is stored and converted into kinetic energy in internal rotation.12 The position of the shoulder during late cocking is 94 ± 21° of thoracohumeral abduction, 11 ± 11° of horizontal adduction, and a remarkable 165 ± 11° of thoracohumeral external rotation (Figure 1).8
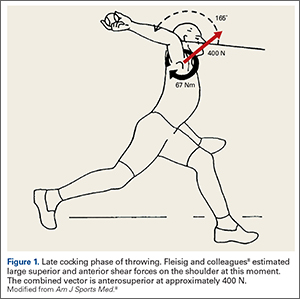
Fleisig and colleagues8 estimated the torque and forces about the shoulder, which are quite high for joint compression (480 ± 130 N). They also analyzed the shear forces and while trying to describe the origin of superior labrum anterior to posterior (SLAP) lesions and anterior labral tears, broke down the major shear vector into an anterior force vector (310 ± 100 N) and a superior force (250 ± 80 N).8 Note that the resulting shear vector is in an anterosuperior direction and is approximately 400 N.
Early Deceleration Phase of Throwing
Interestingly, the position of the humerus during this critical moment of throwing is not much different than the position during the late cocking phase of throwing, with 93 ± 10° of thoracohumeral abduction, 6 ± 8° of horizontal adduction.8 The major difference in the position of the arm is found in the amount of thoracohumeral rotation, which is now 64 ± 35° of external rotation (Figure 2).8
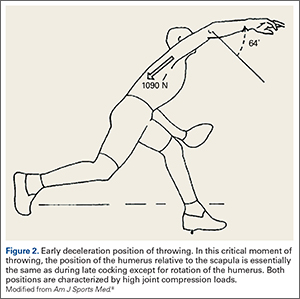
The forces in early deceleration are tremendous, with an estimated 1090 ± 110 N joint compression force, and an anteroinferior shear force of approximately 130 N.8
Clearly, if throwing is an important skill in human evolution, adaptations must exist in the shoulder to withstand the high forces in these 2 critical moments of throwing.
Solving Mysteries of Shoulder Anatomy in the Context of Throwing
There are many anatomic features of the shoulder that remain poorly understood. These include the alignment of the glenohumeral joint, the function of the glenohumeral ligaments, the function of the coracoacromial ligament, the depression of the human greater tuberosity, and the nature and function of the very tendinous subscapularis and long head of the biceps. These mysteries of the human shoulder can be solved if one considers the hypothesis that the shoulder has evolved to throw.
Glenohumeral Joint Alignment
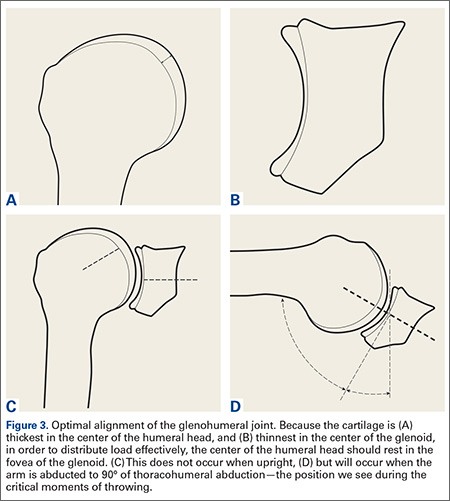
The cartilage of the humeral head is thickest at its center, and thinnest at the periphery (Figure 3A).13,14 Conversely, the cartilage of the glenoid is thinnest at the fovea and thickest in the periphery (Figure 3B).14 It seems obvious that in order to maximally distribute high loads across this joint, the center of the humeral head should rest in the center of the glenoid. Interestingly, this does not occur during most positions of the shoulder. When upright, the center of the humeral head is directed above the glenoid in the coronal plane (Figure 3C). In order to align the glenohumeral joint optimally for the distribution of loads across the joint, the humerus must be abducted approximately 60° relative to the scapula. Assuming a 2:1 glenohumeral to scapulothoracic abduction for arm abduction relative to the thorax,15 this equates to approximately 90° of thoracohumeral abduction—the exact kinematic position of the shoulder during both critical moments of throwing (Figure 3D).
Function of the Glenohumeral Ligaments
The glenohumeral joint capsule has thickenings that help to stabilize the joint. The function of these glenohumeral ligaments has been evaluated biomechanically for their role in preventing translation and instability by a number of authors. The inferior glenohumeral ligament has classically been described as resisting anterior translation of the abducted arm.16 The coracohumeral ligament has been described as important to prevent inferior translation of the adducted arm.17
Interestingly, these ligaments are also the most important ligaments in resisting external rotation of the adducted arm.18 The dominant arm of throwing athletes has been shown to have increased inferior translation19 and increase external rotation.19-22 While the external rotation is partly related to bony adaptation,23,24 the ligamentous restraints to external rotation are likely under tremendous load, which may explain why Dr. Frank Jobe revolutionized the surgical treatment of the throwing athlete by performing an “instability” operation,25,26 as he believed these athletes had “subtle instability” that produced pain, but not symptoms of looseness.27
While these ligaments may exist in part to prevent translation and instability, current thinking suggests that “over-rotation” may lead to internal impingement and may be responsible for symptoms in the thrower’s shoulder,28 as SLAP lesions seem to occur easier with external rotation.29 Again, the importance of maximizing external rotation in throwing and the finding that this position is a critical moment with very high forces suggests that these ligaments may represent an adaptation to restrain external rotation while throwing.
Coracoacromial Ligament
The coracoacromial ligament is unique in that it connects 2 pieces of the same bone, and is only seen in hominids—not other primates.30 Its function has been debated for decades. This ligament is generally thought to limit superior translation of the humeral head,31,32 an effect that is critically important in patients with rotator cuff tears 33,34 Its importance is demonstrated by the fact that it seems to regenerate after it has been resected.35,36 Yet release or resection of this ligament has been a standard treatment for shoulder pain for decades.
Its function becomes clear if one examines the coracoacromial ligament with respect to the kinematics of throwing. As mentioned above, in the late cocking phase of throwing, tremendous shear forces exist in the shoulder. Fleisig and colleagues8 estimated a superior force of 250 ± 80 N, and an anterior shear force of 310 ± 100 N. While Fleisig and colleagues8 analyzed these shear forces with respect to the development of superior and anterior labral tears, it is important to note that these shear forces are vectors that should be combined. When one does this, it becomes apparent that in the late cocking phase of throwing there is shear force in an anterosuperior direction of approximately 400 N (Figure 1). The coracoacromial ligament is positioned to restrain this tremendous force. If throwing is an important adaptation in the evolution of humans, then the function of this ligament and its importance becomes clear.
Depressed Greater Tuberosity and the Pear-Shaped Glenoid
Compared to other primates, the greater tuberosity in humans sits significantly lower (Figure 4). This depression effectively decreases the moment arm of the muscle tendon unit, making the supraspinatus less powerful for raising the arm.37 In addition, by tenting the supraspinatus tendon over the humeral head, a watershed zone is created with decreased vascularity, which is thought to contribute to rotator cuff disease.38 What would be the advantage of the depressed tuberosity?
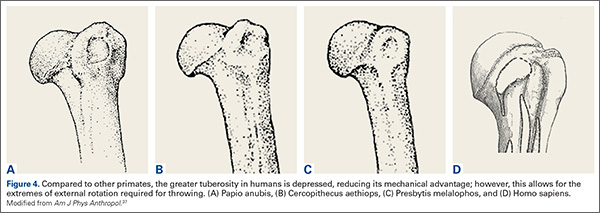
In primates, a lower tuberosity allows for more motion, particularly for arboreal travel.37 In order to throw with velocity, the humerus must achieve extremes of external rotation. A large tuberosity would limit external rotation of the abducted arm. Similarly, the pear-shaped glenoid cavity allows for the depressed tuberosity to achieve maximal external rotation. It is conceivable that a depressed greater tuberosity that allows for throwing would be an adaptation that could be favorable despite its proclivity toward rotator cuff disease in senescence.
Nature of the Subscapularis and the Role of the Long Head of the Biceps
The subscapularis is unique among rotator cuff muscles in that the upper two-thirds of the muscle is surprisingly tendinous.39 Why should this rotator cuff muscle have so much tendon material? Why is the tendon missing from the inferior one-third of the muscle? This situation is not optimal to prevent anterior glenohumeral instability, where inferior tendon material would be preferred.40
The function of the tendon of the long head of the biceps has long been debated and remains unclear.41-43 Cadaver experiments suggest the long head of the biceps provides glenohumeral joint stability in a variety of directions and positions, yet in vivo studies may not show this effect. Electromyography studies show little activity of the long head of the biceps with shoulder motion when the elbow is immobilized, leading some to suggest it is important as a passive restraint.43 This lack of understanding has led some to believe the biceps is not important and can be sacrificed without much concern.42,43
Again, these questions can be answered if one considers them in the context of throwing. At the point of maximal external rotation, the shoulder quickly moves from external rotation to internal rotation. This occurs by converting kinetic energy of external rotation into stored potential energy in the tissues. This energy is then converted into internal rotation. This elastic energy storage is critical for developing the necessary velocities to launch a projectile. While many structures are responsible for storing this energy,12 the subscapularis and long head of the biceps are particularly important. In fact, these 2 structures are important restraints to external rotation of the abducted arm–and become increasingly important with increased external rotation.45,46
One can think of the long head of the biceps as a spring (muscle), a cable (the long tendon), and a pulley (the bicipital groove). Similarly, one can consider the subscapularis as a similar structure, with the coracoid process serving as the pulley. In the late cocking phase of throwing, an interesting alignment occurs such that the pulleys (coracoid process and bicipital groove) are on opposite sides of the joint, providing glenohumeral joint stability. This system, with the inferior glenohumeral ligament (which is the primary restraint to external rotation of the abducted arm18), produces an incredibly stable envelope, preventing the humeral head from over-rotating and translating during the late cocking phase of throwing when the forces about the shoulder are extremely high. Because the muscles serve as springs, this system is also capable of storing kinetic energy during the late cocking phase of throwing and converting it into kinetic energy for internal rotation.
Summary
While throwing is not as critical to survival in today’s culture, the ability to throw was clearly an important adaptation in human evolution. With this in mind, we can approach human anatomy with this perspective, and in fact, many other lines of thinking suggest that throwing was important in the evolution of the hand,47 the brain,48 bipedalism,49 and even human society.50 The shoulder was highly influenced through natural selection to promote the throwing skill. With this perspective, many of the mysteries about the shoulder can be answered.
1. Darwin C. The Descent of Man, and Selection in Relation to Sex. 2nd ed. London, UK: John Murray; 1874:35.
2. Issac B. Throwing and human evolution. African Archeol Record. 1987;5:3-17.
3. Kirschann E. The human throw and a new model of hominid evolution (German). Homo. 1999;50(1):80-85.
4. Knusel CJ. The throwing hypothesis and hominid origins. Human Evolution. 1992;7(1):1-7.
5. Dunsworth H, Challis J, Walker A. The evolution of throwing: a new look at an old idea. Courier Forschungsinstitut Senckenberg. 2003;243:105-110.
6. Mayr E. Animal Species and Evolution. Cambridge, MA: Harvard University Press; 1963.
7. Tinbergen N. On the aims and methods of ethology. Zeitschrift für Tierpsychologie. 1963;20:410-433.
8. Fleisig GS, Andrews JR, Dillman CJ, Escamilla RF. Kinetics of baseball pitching with implications about injury mechanisms. Am J Sports Med. 1995;23(2):233-239.
9. Atwater AE. Biomechanics of overarm throwing movements and of throwing injuries. Exerc Sport Sci Rev. 1975;7:43-85.
10. Matsuo T, Escamila RF, Fleisig GS, Barrentine SW, Andrews JR. Comparison of kinematic and temporal parameters between different pitch velocity groups. J Appl Biomech. 2001;17:1-13.
11. Wang YT, Ford HT III, Ford HT Jr, Shin DM. Three-dimensional kinematic analysis of baseball pitching in acceleration phase. Percept Mot Skills. 1995;80:43-48.
12. Roach NT, Venkadesan M, Rainbow MJ, Lieberman DE. Elastic energy storage in the shoulder and the evolution of high-speed throwing in Homo. Nature. 2013;498(7455):483-486.
13. Fox JA, Cole BJ, Romeo AA, et al. Articular cartilage thickness of the humeral head: an anatomic study. Orthopedics. 2008;31(3):216.
14. Zumstein V, Kraljevic M, Conzen A, Hoechel S, Müller-Gerbl M. Thickness distribution of the glenohumeral joint cartilage: a quantitative study using computed tomography. Surg Radiol Anat. 2014;36(4):327-331.
15. Inman VT, Saunders M, Abbott LC. Observations on the function of the shoulder joint. J Bone Joint Surg Am. 1944;26:1-30.
16. O’Brien SJ, Schwartz RS, Warren RF, Torzilli PA. Capsular restraints to anterior posterior motion of the abducted shoulder: A biomechanical study. J Shoulder Elbow Surg. 1995;4(4):298-308.
17. Warner JJ, Deng XH, Warren RF, Torzilli PA. Static capsuloligamentous restraints to superior-inferior translation of the glenohumeral joint. Am J Sports Med. 1992;20(6):675-685.
18. Kuhn JE, Bey MJ, Huston LJ, Blasier RB, Soslowsky LJ. Ligamentous restraints to external rotation of the humerus in the late-cocking phase of throwing. A cadaveric biomechanical investigation. Am J Sports Med. 2000;28(2):200-205.
19. Bigliani LU, Codd TP, Connor PM, Levine WN, Littlefield MA, Hershon SJ. Shoulder motion and laxity in the professional baseball player. Am J Sports Med. 1997;25(5):609-613.
20. Borsa PA, Dover GC, Wilk KE, Reinold MM. Glenohumeral range of motion and stiffness in professional baseball pitchers. Med Sci Sports Exerc. 2006;38(1):21-26.
21. Hurd WJ, Kaplan KM, Eiattrache NS, Jobe FW, Morrey BF, Kaufman KR. A profile of glenohumeral internal and external rotation motion in the uninjured high school baseball pitcher, part I: motion. J Athl Train. 2011;46(3):282-288.
22. Wilk KE, Macrina LC, Arrigo C. Passive range of motion characteristics in the overhead baseball pitcher and their implications for rehabilitation. Clin Orthop Relat Res. 2012;470(6):1586-1594.
23. Osbahr DC, Cannon DL, Speer KP. Retroversion of the humerus in the throwing shoulder of college baseball pitchers. Am J Sports Med. 2002;30(3):347-353.
24. Greenberg EM, Fernandez-Fernandez A, Lawrence JT, McClure P. The development of humeral retrotorsion and its relationship to throwing sports. Sports Health. 2015;7(6):489-496.
25. Jobe FW, Pink M. The athlete’s shoulder. J Hand Ther. 1994;7(2):107-110.
26. Montgomery WH 3rd, Jobe FW. Functional outcomes in athletes after modified anterior capsulolabral reconstruction. Am J Sports Med. 1994;22(3):352-358.
27. Jobe FW, Kvitne RS, Giangarra CE. Shoulder pain in the overhand or throwing athlete. The relationship of anterior instability and rotator cuff impingement. Orthop Rev. 1989;18(9):963-975.
28. Reinhold MM, Wilk KE, Dugas JR, Andrews JR. Chapter 11. Internal Impingement. In: Wilk K, Reinold MM, Andrews JR, eds. The Athlete’s Shoulder. 2nd ed. Philadelphia, PA: Churchill Livingstone Elsevier; 2009:126.
29. Kuhn JE, Lindholm SR, Huston LJ, Soslowsky LJ, Blasier RB. Failure of the biceps superior labral complex: a cadaveric biomechanical investigation comparing the late cocking and early deceleration positions of throwing. Arthroscopy. 2003;19(4):373-379.
30. Ciochon RL, Corruccini RS. The coraco-acromial ligament and projection index in man and other anthropoid primates. J Anat. 1977;124(Pt 3):627-632.
31. Moorman CT, Warren RF, Deng XH, Wickiewicz TL, Torzilli PA. Role of coracoacromial ligament and related structures in glenohumeral stability: a cadaveric study. J Surg Orthop Adv. 2012;21(4):210-217.
32. Su WR, Budoff JE, Luo ZP. The effect of coracoacromial ligament excision and acromioplasty on superior and anterosuperior glenohumeral stability. Arthroscopy. 2009;25(1):13-18.
33. Wellmann M, Petersen W, Zantop T, Schanz S, Raschke MJ, Hurschler C. Effect of coracoacromial ligament resection on glenohumeral stability under active muscle loading in an in vitro model. Arthroscopy. 2008;24(11):1258-1264.
34. Fagelman M, Sartori M, Freedman KB, Patwardhan AG, Carandang G, Marra G. Biomechanics of coracoacromial arch modification. J Shoulder Elbow Surg. 2007;16(1):101-116.
35. Bak K, Spring IB, Henderson IP. Re-formation of the coracoacromial ligament after open resection or arthroscopic release. J Shoulder Elbow Surg. 2000;9:289-293.
36. Levy O, Copeland SA. Regeneration of the coracoacromial ligament after acromioplasty and arthroscopic subacromial decompression. J Shoulder Elbow Surg. 2001;10(4):317-320.
37. Larson SG, Stern JT Jr. Role of supraspinatus in the quadrupedal locomotion of vervets (Cercopithecus aethiops): Implications for interpretation of humeral morphology. Am J Phys Anthropol. 1989;79(3):369-377.
38. Chansky HA, Iannotti JP. The vascularity of the rotator cuff. Clin Sports Med. 1991;10(4):807-822.
39. Klapper RC, Jobe FW, Matsuura P. Subscapularis muscle and its glenohumeral ligament-like bands. A histomorphologic study. Am J Sports Med. 1992;20(3):307-310.
40. Halder A, Zobitz ME, Schultz E, An KN. Structural properties of the subscapularis tendon. J Orthop Res. 2000;18(5):
829-834.
41. Elser F, Braun S, Dewing CB, Giphart JE, Millett PJ. Anatomy, function, injuries, and treatment of the long head of the biceps brachii tendon. Arthroscopy. 2011;27(4):581-592.
42. Pill SG, Walch G, Hawkins RJ, Kissenberth MJ. The role of the biceps tendon in massive rotator cuff tears. Instr Course Lect. 2012;61:113-120.
43. Krupp RJ, Kevern MA, Gaines MD, Kotara S, Singleton SB. Long head of the biceps tendon pain: differential diagnosis and treatment. J Orthop Sports Phys Ther. 2009;39(2):55-70.
44. Levy AS, Kelly BT, Lintner SA, Osbahr DC, Speer KP. Function of the long head of the biceps at the shoulder: electromyographic analysis. J Shoulder Elbow Surg. 2001;10(3):250-255.
45. Kuhn JE, Huston LJ, Soslowsky LJ, Shyr Y, Blasier RB. External rotation of the glenohumeral joint: ligament restraints and muscle effects in the neutral and abducted positions.
J Shoulder Elbow Surg. 2005;14(1 Suppl S):39S-48S.
46. McGarry MH, Nguyen ML, Quigley RJ, Hanypsiak B, Gupta R, Lee TQ. The effect of long and short head biceps loading on glenohumeral joint rotational range of motion and humeral head postion. Knee Surge Sports Traumatol Arthrosc. 2014 Sep 26. [Epub ahead of print].
47. Young RW. Evolution of the human hand: The role of throwing and clubbing. J Anat. 2003;202:165-174.
48. Calvin WH. Did throwing stones shape hominid brain evolution? Ethology and Sociobiology. 1982;3:115-124.
49. Fifer FC. The adoption of bipedalism by the hominids: A new hypothesis. Human Evolution. 1987;2(2):135-147.
50. Darlington PJ. Group selection, altruism, reinforcement, and throwing in human evolution. Proc Nat Acad Sci. 1973;72(9):3748-3752.
Charles Darwin once said that apes “...are quite unable, as I have myself seen, to throw a stone with precision”.1 Yet humans can throw with precision and speed, a skill that likely had significant advantages: throwing can affect change at a distance—something few species can do. Throwing can provide protection against predators and can allow for predation for food resources. Throwing would be important in contesting other hominids for scarce resources. As such, throwing has been critically important in human evolution and likely is a skill that has been promoted through natural selection.2-5
In the orthopedic literature, most published work on throwing will ask proximate questions: “how, what, who, when, and where?” Evolutionary biologists are concerned with ultimate questions6,7: “why?” Asking ultimate questions provides insight into how a behavior might offer advantages under natural selection, which can then improve our understanding of the proximate questions for that behavior.
With regard to the shoulder, a number of mysteries exist that, to date, proximate studies have not been able to solve. This article argues that the human shoulder has evolved for throwing and by using this frame of reference, many of the mysteries surrounding the anatomy of the shoulder can be understood.
Pitching Kinematics
The mechanics of pitching have been analyzed extensively. Fleisig and colleagues8 performed kinematic and electromyographic analyses of pitchers to identify the critical moments of pitching (defined as where the forces are highest and injury is most likely going to occur). They found 2 moments where the forces about the shoulder are highest during pitching: the late cocking phase (defined by the point where the humerus reaches maximal external rotation); and the early deceleration phase (defined by the point when the ball is released). If throwing is important in natural selection of humans, then the shoulder anatomy should be optimized to withstand the forces generated in these positions.
Late Cocking Phase of Throwing
The early phases of throwing are attempting to maximize external rotation of the abducted arm as the velocity of the pitched ball correlates to the amount of external rotation achieved.9-11 In this position, kinetic energy in external rotation is stored and converted into kinetic energy in internal rotation.12 The position of the shoulder during late cocking is 94 ± 21° of thoracohumeral abduction, 11 ± 11° of horizontal adduction, and a remarkable 165 ± 11° of thoracohumeral external rotation (Figure 1).8
Fleisig and colleagues8 estimated the torque and forces about the shoulder, which are quite high for joint compression (480 ± 130 N). They also analyzed the shear forces and while trying to describe the origin of superior labrum anterior to posterior (SLAP) lesions and anterior labral tears, broke down the major shear vector into an anterior force vector (310 ± 100 N) and a superior force (250 ± 80 N).8 Note that the resulting shear vector is in an anterosuperior direction and is approximately 400 N.
Early Deceleration Phase of Throwing
Interestingly, the position of the humerus during this critical moment of throwing is not much different than the position during the late cocking phase of throwing, with 93 ± 10° of thoracohumeral abduction, 6 ± 8° of horizontal adduction.8 The major difference in the position of the arm is found in the amount of thoracohumeral rotation, which is now 64 ± 35° of external rotation (Figure 2).8
The forces in early deceleration are tremendous, with an estimated 1090 ± 110 N joint compression force, and an anteroinferior shear force of approximately 130 N.8
Clearly, if throwing is an important skill in human evolution, adaptations must exist in the shoulder to withstand the high forces in these 2 critical moments of throwing.
Solving Mysteries of Shoulder Anatomy in the Context of Throwing
There are many anatomic features of the shoulder that remain poorly understood. These include the alignment of the glenohumeral joint, the function of the glenohumeral ligaments, the function of the coracoacromial ligament, the depression of the human greater tuberosity, and the nature and function of the very tendinous subscapularis and long head of the biceps. These mysteries of the human shoulder can be solved if one considers the hypothesis that the shoulder has evolved to throw.
Glenohumeral Joint Alignment
The cartilage of the humeral head is thickest at its center, and thinnest at the periphery (Figure 3A).13,14 Conversely, the cartilage of the glenoid is thinnest at the fovea and thickest in the periphery (Figure 3B).14 It seems obvious that in order to maximally distribute high loads across this joint, the center of the humeral head should rest in the center of the glenoid. Interestingly, this does not occur during most positions of the shoulder. When upright, the center of the humeral head is directed above the glenoid in the coronal plane (Figure 3C). In order to align the glenohumeral joint optimally for the distribution of loads across the joint, the humerus must be abducted approximately 60° relative to the scapula. Assuming a 2:1 glenohumeral to scapulothoracic abduction for arm abduction relative to the thorax,15 this equates to approximately 90° of thoracohumeral abduction—the exact kinematic position of the shoulder during both critical moments of throwing (Figure 3D).
Function of the Glenohumeral Ligaments
The glenohumeral joint capsule has thickenings that help to stabilize the joint. The function of these glenohumeral ligaments has been evaluated biomechanically for their role in preventing translation and instability by a number of authors. The inferior glenohumeral ligament has classically been described as resisting anterior translation of the abducted arm.16 The coracohumeral ligament has been described as important to prevent inferior translation of the adducted arm.17
Interestingly, these ligaments are also the most important ligaments in resisting external rotation of the adducted arm.18 The dominant arm of throwing athletes has been shown to have increased inferior translation19 and increase external rotation.19-22 While the external rotation is partly related to bony adaptation,23,24 the ligamentous restraints to external rotation are likely under tremendous load, which may explain why Dr. Frank Jobe revolutionized the surgical treatment of the throwing athlete by performing an “instability” operation,25,26 as he believed these athletes had “subtle instability” that produced pain, but not symptoms of looseness.27
While these ligaments may exist in part to prevent translation and instability, current thinking suggests that “over-rotation” may lead to internal impingement and may be responsible for symptoms in the thrower’s shoulder,28 as SLAP lesions seem to occur easier with external rotation.29 Again, the importance of maximizing external rotation in throwing and the finding that this position is a critical moment with very high forces suggests that these ligaments may represent an adaptation to restrain external rotation while throwing.
Coracoacromial Ligament
The coracoacromial ligament is unique in that it connects 2 pieces of the same bone, and is only seen in hominids—not other primates.30 Its function has been debated for decades. This ligament is generally thought to limit superior translation of the humeral head,31,32 an effect that is critically important in patients with rotator cuff tears 33,34 Its importance is demonstrated by the fact that it seems to regenerate after it has been resected.35,36 Yet release or resection of this ligament has been a standard treatment for shoulder pain for decades.
Its function becomes clear if one examines the coracoacromial ligament with respect to the kinematics of throwing. As mentioned above, in the late cocking phase of throwing, tremendous shear forces exist in the shoulder. Fleisig and colleagues8 estimated a superior force of 250 ± 80 N, and an anterior shear force of 310 ± 100 N. While Fleisig and colleagues8 analyzed these shear forces with respect to the development of superior and anterior labral tears, it is important to note that these shear forces are vectors that should be combined. When one does this, it becomes apparent that in the late cocking phase of throwing there is shear force in an anterosuperior direction of approximately 400 N (Figure 1). The coracoacromial ligament is positioned to restrain this tremendous force. If throwing is an important adaptation in the evolution of humans, then the function of this ligament and its importance becomes clear.
Depressed Greater Tuberosity and the Pear-Shaped Glenoid
Compared to other primates, the greater tuberosity in humans sits significantly lower (Figure 4). This depression effectively decreases the moment arm of the muscle tendon unit, making the supraspinatus less powerful for raising the arm.37 In addition, by tenting the supraspinatus tendon over the humeral head, a watershed zone is created with decreased vascularity, which is thought to contribute to rotator cuff disease.38 What would be the advantage of the depressed tuberosity?
In primates, a lower tuberosity allows for more motion, particularly for arboreal travel.37 In order to throw with velocity, the humerus must achieve extremes of external rotation. A large tuberosity would limit external rotation of the abducted arm. Similarly, the pear-shaped glenoid cavity allows for the depressed tuberosity to achieve maximal external rotation. It is conceivable that a depressed greater tuberosity that allows for throwing would be an adaptation that could be favorable despite its proclivity toward rotator cuff disease in senescence.
Nature of the Subscapularis and the Role of the Long Head of the Biceps
The subscapularis is unique among rotator cuff muscles in that the upper two-thirds of the muscle is surprisingly tendinous.39 Why should this rotator cuff muscle have so much tendon material? Why is the tendon missing from the inferior one-third of the muscle? This situation is not optimal to prevent anterior glenohumeral instability, where inferior tendon material would be preferred.40
The function of the tendon of the long head of the biceps has long been debated and remains unclear.41-43 Cadaver experiments suggest the long head of the biceps provides glenohumeral joint stability in a variety of directions and positions, yet in vivo studies may not show this effect. Electromyography studies show little activity of the long head of the biceps with shoulder motion when the elbow is immobilized, leading some to suggest it is important as a passive restraint.43 This lack of understanding has led some to believe the biceps is not important and can be sacrificed without much concern.42,43
Again, these questions can be answered if one considers them in the context of throwing. At the point of maximal external rotation, the shoulder quickly moves from external rotation to internal rotation. This occurs by converting kinetic energy of external rotation into stored potential energy in the tissues. This energy is then converted into internal rotation. This elastic energy storage is critical for developing the necessary velocities to launch a projectile. While many structures are responsible for storing this energy,12 the subscapularis and long head of the biceps are particularly important. In fact, these 2 structures are important restraints to external rotation of the abducted arm–and become increasingly important with increased external rotation.45,46
One can think of the long head of the biceps as a spring (muscle), a cable (the long tendon), and a pulley (the bicipital groove). Similarly, one can consider the subscapularis as a similar structure, with the coracoid process serving as the pulley. In the late cocking phase of throwing, an interesting alignment occurs such that the pulleys (coracoid process and bicipital groove) are on opposite sides of the joint, providing glenohumeral joint stability. This system, with the inferior glenohumeral ligament (which is the primary restraint to external rotation of the abducted arm18), produces an incredibly stable envelope, preventing the humeral head from over-rotating and translating during the late cocking phase of throwing when the forces about the shoulder are extremely high. Because the muscles serve as springs, this system is also capable of storing kinetic energy during the late cocking phase of throwing and converting it into kinetic energy for internal rotation.
Summary
While throwing is not as critical to survival in today’s culture, the ability to throw was clearly an important adaptation in human evolution. With this in mind, we can approach human anatomy with this perspective, and in fact, many other lines of thinking suggest that throwing was important in the evolution of the hand,47 the brain,48 bipedalism,49 and even human society.50 The shoulder was highly influenced through natural selection to promote the throwing skill. With this perspective, many of the mysteries about the shoulder can be answered.
Charles Darwin once said that apes “...are quite unable, as I have myself seen, to throw a stone with precision”.1 Yet humans can throw with precision and speed, a skill that likely had significant advantages: throwing can affect change at a distance—something few species can do. Throwing can provide protection against predators and can allow for predation for food resources. Throwing would be important in contesting other hominids for scarce resources. As such, throwing has been critically important in human evolution and likely is a skill that has been promoted through natural selection.2-5
In the orthopedic literature, most published work on throwing will ask proximate questions: “how, what, who, when, and where?” Evolutionary biologists are concerned with ultimate questions6,7: “why?” Asking ultimate questions provides insight into how a behavior might offer advantages under natural selection, which can then improve our understanding of the proximate questions for that behavior.
With regard to the shoulder, a number of mysteries exist that, to date, proximate studies have not been able to solve. This article argues that the human shoulder has evolved for throwing and by using this frame of reference, many of the mysteries surrounding the anatomy of the shoulder can be understood.
Pitching Kinematics
The mechanics of pitching have been analyzed extensively. Fleisig and colleagues8 performed kinematic and electromyographic analyses of pitchers to identify the critical moments of pitching (defined as where the forces are highest and injury is most likely going to occur). They found 2 moments where the forces about the shoulder are highest during pitching: the late cocking phase (defined by the point where the humerus reaches maximal external rotation); and the early deceleration phase (defined by the point when the ball is released). If throwing is important in natural selection of humans, then the shoulder anatomy should be optimized to withstand the forces generated in these positions.
Late Cocking Phase of Throwing
The early phases of throwing are attempting to maximize external rotation of the abducted arm as the velocity of the pitched ball correlates to the amount of external rotation achieved.9-11 In this position, kinetic energy in external rotation is stored and converted into kinetic energy in internal rotation.12 The position of the shoulder during late cocking is 94 ± 21° of thoracohumeral abduction, 11 ± 11° of horizontal adduction, and a remarkable 165 ± 11° of thoracohumeral external rotation (Figure 1).8
Fleisig and colleagues8 estimated the torque and forces about the shoulder, which are quite high for joint compression (480 ± 130 N). They also analyzed the shear forces and while trying to describe the origin of superior labrum anterior to posterior (SLAP) lesions and anterior labral tears, broke down the major shear vector into an anterior force vector (310 ± 100 N) and a superior force (250 ± 80 N).8 Note that the resulting shear vector is in an anterosuperior direction and is approximately 400 N.
Early Deceleration Phase of Throwing
Interestingly, the position of the humerus during this critical moment of throwing is not much different than the position during the late cocking phase of throwing, with 93 ± 10° of thoracohumeral abduction, 6 ± 8° of horizontal adduction.8 The major difference in the position of the arm is found in the amount of thoracohumeral rotation, which is now 64 ± 35° of external rotation (Figure 2).8
The forces in early deceleration are tremendous, with an estimated 1090 ± 110 N joint compression force, and an anteroinferior shear force of approximately 130 N.8
Clearly, if throwing is an important skill in human evolution, adaptations must exist in the shoulder to withstand the high forces in these 2 critical moments of throwing.
Solving Mysteries of Shoulder Anatomy in the Context of Throwing
There are many anatomic features of the shoulder that remain poorly understood. These include the alignment of the glenohumeral joint, the function of the glenohumeral ligaments, the function of the coracoacromial ligament, the depression of the human greater tuberosity, and the nature and function of the very tendinous subscapularis and long head of the biceps. These mysteries of the human shoulder can be solved if one considers the hypothesis that the shoulder has evolved to throw.
Glenohumeral Joint Alignment
The cartilage of the humeral head is thickest at its center, and thinnest at the periphery (Figure 3A).13,14 Conversely, the cartilage of the glenoid is thinnest at the fovea and thickest in the periphery (Figure 3B).14 It seems obvious that in order to maximally distribute high loads across this joint, the center of the humeral head should rest in the center of the glenoid. Interestingly, this does not occur during most positions of the shoulder. When upright, the center of the humeral head is directed above the glenoid in the coronal plane (Figure 3C). In order to align the glenohumeral joint optimally for the distribution of loads across the joint, the humerus must be abducted approximately 60° relative to the scapula. Assuming a 2:1 glenohumeral to scapulothoracic abduction for arm abduction relative to the thorax,15 this equates to approximately 90° of thoracohumeral abduction—the exact kinematic position of the shoulder during both critical moments of throwing (Figure 3D).
Function of the Glenohumeral Ligaments
The glenohumeral joint capsule has thickenings that help to stabilize the joint. The function of these glenohumeral ligaments has been evaluated biomechanically for their role in preventing translation and instability by a number of authors. The inferior glenohumeral ligament has classically been described as resisting anterior translation of the abducted arm.16 The coracohumeral ligament has been described as important to prevent inferior translation of the adducted arm.17
Interestingly, these ligaments are also the most important ligaments in resisting external rotation of the adducted arm.18 The dominant arm of throwing athletes has been shown to have increased inferior translation19 and increase external rotation.19-22 While the external rotation is partly related to bony adaptation,23,24 the ligamentous restraints to external rotation are likely under tremendous load, which may explain why Dr. Frank Jobe revolutionized the surgical treatment of the throwing athlete by performing an “instability” operation,25,26 as he believed these athletes had “subtle instability” that produced pain, but not symptoms of looseness.27
While these ligaments may exist in part to prevent translation and instability, current thinking suggests that “over-rotation” may lead to internal impingement and may be responsible for symptoms in the thrower’s shoulder,28 as SLAP lesions seem to occur easier with external rotation.29 Again, the importance of maximizing external rotation in throwing and the finding that this position is a critical moment with very high forces suggests that these ligaments may represent an adaptation to restrain external rotation while throwing.
Coracoacromial Ligament
The coracoacromial ligament is unique in that it connects 2 pieces of the same bone, and is only seen in hominids—not other primates.30 Its function has been debated for decades. This ligament is generally thought to limit superior translation of the humeral head,31,32 an effect that is critically important in patients with rotator cuff tears 33,34 Its importance is demonstrated by the fact that it seems to regenerate after it has been resected.35,36 Yet release or resection of this ligament has been a standard treatment for shoulder pain for decades.
Its function becomes clear if one examines the coracoacromial ligament with respect to the kinematics of throwing. As mentioned above, in the late cocking phase of throwing, tremendous shear forces exist in the shoulder. Fleisig and colleagues8 estimated a superior force of 250 ± 80 N, and an anterior shear force of 310 ± 100 N. While Fleisig and colleagues8 analyzed these shear forces with respect to the development of superior and anterior labral tears, it is important to note that these shear forces are vectors that should be combined. When one does this, it becomes apparent that in the late cocking phase of throwing there is shear force in an anterosuperior direction of approximately 400 N (Figure 1). The coracoacromial ligament is positioned to restrain this tremendous force. If throwing is an important adaptation in the evolution of humans, then the function of this ligament and its importance becomes clear.
Depressed Greater Tuberosity and the Pear-Shaped Glenoid
Compared to other primates, the greater tuberosity in humans sits significantly lower (Figure 4). This depression effectively decreases the moment arm of the muscle tendon unit, making the supraspinatus less powerful for raising the arm.37 In addition, by tenting the supraspinatus tendon over the humeral head, a watershed zone is created with decreased vascularity, which is thought to contribute to rotator cuff disease.38 What would be the advantage of the depressed tuberosity?
In primates, a lower tuberosity allows for more motion, particularly for arboreal travel.37 In order to throw with velocity, the humerus must achieve extremes of external rotation. A large tuberosity would limit external rotation of the abducted arm. Similarly, the pear-shaped glenoid cavity allows for the depressed tuberosity to achieve maximal external rotation. It is conceivable that a depressed greater tuberosity that allows for throwing would be an adaptation that could be favorable despite its proclivity toward rotator cuff disease in senescence.
Nature of the Subscapularis and the Role of the Long Head of the Biceps
The subscapularis is unique among rotator cuff muscles in that the upper two-thirds of the muscle is surprisingly tendinous.39 Why should this rotator cuff muscle have so much tendon material? Why is the tendon missing from the inferior one-third of the muscle? This situation is not optimal to prevent anterior glenohumeral instability, where inferior tendon material would be preferred.40
The function of the tendon of the long head of the biceps has long been debated and remains unclear.41-43 Cadaver experiments suggest the long head of the biceps provides glenohumeral joint stability in a variety of directions and positions, yet in vivo studies may not show this effect. Electromyography studies show little activity of the long head of the biceps with shoulder motion when the elbow is immobilized, leading some to suggest it is important as a passive restraint.43 This lack of understanding has led some to believe the biceps is not important and can be sacrificed without much concern.42,43
Again, these questions can be answered if one considers them in the context of throwing. At the point of maximal external rotation, the shoulder quickly moves from external rotation to internal rotation. This occurs by converting kinetic energy of external rotation into stored potential energy in the tissues. This energy is then converted into internal rotation. This elastic energy storage is critical for developing the necessary velocities to launch a projectile. While many structures are responsible for storing this energy,12 the subscapularis and long head of the biceps are particularly important. In fact, these 2 structures are important restraints to external rotation of the abducted arm–and become increasingly important with increased external rotation.45,46
One can think of the long head of the biceps as a spring (muscle), a cable (the long tendon), and a pulley (the bicipital groove). Similarly, one can consider the subscapularis as a similar structure, with the coracoid process serving as the pulley. In the late cocking phase of throwing, an interesting alignment occurs such that the pulleys (coracoid process and bicipital groove) are on opposite sides of the joint, providing glenohumeral joint stability. This system, with the inferior glenohumeral ligament (which is the primary restraint to external rotation of the abducted arm18), produces an incredibly stable envelope, preventing the humeral head from over-rotating and translating during the late cocking phase of throwing when the forces about the shoulder are extremely high. Because the muscles serve as springs, this system is also capable of storing kinetic energy during the late cocking phase of throwing and converting it into kinetic energy for internal rotation.
Summary
While throwing is not as critical to survival in today’s culture, the ability to throw was clearly an important adaptation in human evolution. With this in mind, we can approach human anatomy with this perspective, and in fact, many other lines of thinking suggest that throwing was important in the evolution of the hand,47 the brain,48 bipedalism,49 and even human society.50 The shoulder was highly influenced through natural selection to promote the throwing skill. With this perspective, many of the mysteries about the shoulder can be answered.
1. Darwin C. The Descent of Man, and Selection in Relation to Sex. 2nd ed. London, UK: John Murray; 1874:35.
2. Issac B. Throwing and human evolution. African Archeol Record. 1987;5:3-17.
3. Kirschann E. The human throw and a new model of hominid evolution (German). Homo. 1999;50(1):80-85.
4. Knusel CJ. The throwing hypothesis and hominid origins. Human Evolution. 1992;7(1):1-7.
5. Dunsworth H, Challis J, Walker A. The evolution of throwing: a new look at an old idea. Courier Forschungsinstitut Senckenberg. 2003;243:105-110.
6. Mayr E. Animal Species and Evolution. Cambridge, MA: Harvard University Press; 1963.
7. Tinbergen N. On the aims and methods of ethology. Zeitschrift für Tierpsychologie. 1963;20:410-433.
8. Fleisig GS, Andrews JR, Dillman CJ, Escamilla RF. Kinetics of baseball pitching with implications about injury mechanisms. Am J Sports Med. 1995;23(2):233-239.
9. Atwater AE. Biomechanics of overarm throwing movements and of throwing injuries. Exerc Sport Sci Rev. 1975;7:43-85.
10. Matsuo T, Escamila RF, Fleisig GS, Barrentine SW, Andrews JR. Comparison of kinematic and temporal parameters between different pitch velocity groups. J Appl Biomech. 2001;17:1-13.
11. Wang YT, Ford HT III, Ford HT Jr, Shin DM. Three-dimensional kinematic analysis of baseball pitching in acceleration phase. Percept Mot Skills. 1995;80:43-48.
12. Roach NT, Venkadesan M, Rainbow MJ, Lieberman DE. Elastic energy storage in the shoulder and the evolution of high-speed throwing in Homo. Nature. 2013;498(7455):483-486.
13. Fox JA, Cole BJ, Romeo AA, et al. Articular cartilage thickness of the humeral head: an anatomic study. Orthopedics. 2008;31(3):216.
14. Zumstein V, Kraljevic M, Conzen A, Hoechel S, Müller-Gerbl M. Thickness distribution of the glenohumeral joint cartilage: a quantitative study using computed tomography. Surg Radiol Anat. 2014;36(4):327-331.
15. Inman VT, Saunders M, Abbott LC. Observations on the function of the shoulder joint. J Bone Joint Surg Am. 1944;26:1-30.
16. O’Brien SJ, Schwartz RS, Warren RF, Torzilli PA. Capsular restraints to anterior posterior motion of the abducted shoulder: A biomechanical study. J Shoulder Elbow Surg. 1995;4(4):298-308.
17. Warner JJ, Deng XH, Warren RF, Torzilli PA. Static capsuloligamentous restraints to superior-inferior translation of the glenohumeral joint. Am J Sports Med. 1992;20(6):675-685.
18. Kuhn JE, Bey MJ, Huston LJ, Blasier RB, Soslowsky LJ. Ligamentous restraints to external rotation of the humerus in the late-cocking phase of throwing. A cadaveric biomechanical investigation. Am J Sports Med. 2000;28(2):200-205.
19. Bigliani LU, Codd TP, Connor PM, Levine WN, Littlefield MA, Hershon SJ. Shoulder motion and laxity in the professional baseball player. Am J Sports Med. 1997;25(5):609-613.
20. Borsa PA, Dover GC, Wilk KE, Reinold MM. Glenohumeral range of motion and stiffness in professional baseball pitchers. Med Sci Sports Exerc. 2006;38(1):21-26.
21. Hurd WJ, Kaplan KM, Eiattrache NS, Jobe FW, Morrey BF, Kaufman KR. A profile of glenohumeral internal and external rotation motion in the uninjured high school baseball pitcher, part I: motion. J Athl Train. 2011;46(3):282-288.
22. Wilk KE, Macrina LC, Arrigo C. Passive range of motion characteristics in the overhead baseball pitcher and their implications for rehabilitation. Clin Orthop Relat Res. 2012;470(6):1586-1594.
23. Osbahr DC, Cannon DL, Speer KP. Retroversion of the humerus in the throwing shoulder of college baseball pitchers. Am J Sports Med. 2002;30(3):347-353.
24. Greenberg EM, Fernandez-Fernandez A, Lawrence JT, McClure P. The development of humeral retrotorsion and its relationship to throwing sports. Sports Health. 2015;7(6):489-496.
25. Jobe FW, Pink M. The athlete’s shoulder. J Hand Ther. 1994;7(2):107-110.
26. Montgomery WH 3rd, Jobe FW. Functional outcomes in athletes after modified anterior capsulolabral reconstruction. Am J Sports Med. 1994;22(3):352-358.
27. Jobe FW, Kvitne RS, Giangarra CE. Shoulder pain in the overhand or throwing athlete. The relationship of anterior instability and rotator cuff impingement. Orthop Rev. 1989;18(9):963-975.
28. Reinhold MM, Wilk KE, Dugas JR, Andrews JR. Chapter 11. Internal Impingement. In: Wilk K, Reinold MM, Andrews JR, eds. The Athlete’s Shoulder. 2nd ed. Philadelphia, PA: Churchill Livingstone Elsevier; 2009:126.
29. Kuhn JE, Lindholm SR, Huston LJ, Soslowsky LJ, Blasier RB. Failure of the biceps superior labral complex: a cadaveric biomechanical investigation comparing the late cocking and early deceleration positions of throwing. Arthroscopy. 2003;19(4):373-379.
30. Ciochon RL, Corruccini RS. The coraco-acromial ligament and projection index in man and other anthropoid primates. J Anat. 1977;124(Pt 3):627-632.
31. Moorman CT, Warren RF, Deng XH, Wickiewicz TL, Torzilli PA. Role of coracoacromial ligament and related structures in glenohumeral stability: a cadaveric study. J Surg Orthop Adv. 2012;21(4):210-217.
32. Su WR, Budoff JE, Luo ZP. The effect of coracoacromial ligament excision and acromioplasty on superior and anterosuperior glenohumeral stability. Arthroscopy. 2009;25(1):13-18.
33. Wellmann M, Petersen W, Zantop T, Schanz S, Raschke MJ, Hurschler C. Effect of coracoacromial ligament resection on glenohumeral stability under active muscle loading in an in vitro model. Arthroscopy. 2008;24(11):1258-1264.
34. Fagelman M, Sartori M, Freedman KB, Patwardhan AG, Carandang G, Marra G. Biomechanics of coracoacromial arch modification. J Shoulder Elbow Surg. 2007;16(1):101-116.
35. Bak K, Spring IB, Henderson IP. Re-formation of the coracoacromial ligament after open resection or arthroscopic release. J Shoulder Elbow Surg. 2000;9:289-293.
36. Levy O, Copeland SA. Regeneration of the coracoacromial ligament after acromioplasty and arthroscopic subacromial decompression. J Shoulder Elbow Surg. 2001;10(4):317-320.
37. Larson SG, Stern JT Jr. Role of supraspinatus in the quadrupedal locomotion of vervets (Cercopithecus aethiops): Implications for interpretation of humeral morphology. Am J Phys Anthropol. 1989;79(3):369-377.
38. Chansky HA, Iannotti JP. The vascularity of the rotator cuff. Clin Sports Med. 1991;10(4):807-822.
39. Klapper RC, Jobe FW, Matsuura P. Subscapularis muscle and its glenohumeral ligament-like bands. A histomorphologic study. Am J Sports Med. 1992;20(3):307-310.
40. Halder A, Zobitz ME, Schultz E, An KN. Structural properties of the subscapularis tendon. J Orthop Res. 2000;18(5):
829-834.
41. Elser F, Braun S, Dewing CB, Giphart JE, Millett PJ. Anatomy, function, injuries, and treatment of the long head of the biceps brachii tendon. Arthroscopy. 2011;27(4):581-592.
42. Pill SG, Walch G, Hawkins RJ, Kissenberth MJ. The role of the biceps tendon in massive rotator cuff tears. Instr Course Lect. 2012;61:113-120.
43. Krupp RJ, Kevern MA, Gaines MD, Kotara S, Singleton SB. Long head of the biceps tendon pain: differential diagnosis and treatment. J Orthop Sports Phys Ther. 2009;39(2):55-70.
44. Levy AS, Kelly BT, Lintner SA, Osbahr DC, Speer KP. Function of the long head of the biceps at the shoulder: electromyographic analysis. J Shoulder Elbow Surg. 2001;10(3):250-255.
45. Kuhn JE, Huston LJ, Soslowsky LJ, Shyr Y, Blasier RB. External rotation of the glenohumeral joint: ligament restraints and muscle effects in the neutral and abducted positions.
J Shoulder Elbow Surg. 2005;14(1 Suppl S):39S-48S.
46. McGarry MH, Nguyen ML, Quigley RJ, Hanypsiak B, Gupta R, Lee TQ. The effect of long and short head biceps loading on glenohumeral joint rotational range of motion and humeral head postion. Knee Surge Sports Traumatol Arthrosc. 2014 Sep 26. [Epub ahead of print].
47. Young RW. Evolution of the human hand: The role of throwing and clubbing. J Anat. 2003;202:165-174.
48. Calvin WH. Did throwing stones shape hominid brain evolution? Ethology and Sociobiology. 1982;3:115-124.
49. Fifer FC. The adoption of bipedalism by the hominids: A new hypothesis. Human Evolution. 1987;2(2):135-147.
50. Darlington PJ. Group selection, altruism, reinforcement, and throwing in human evolution. Proc Nat Acad Sci. 1973;72(9):3748-3752.
1. Darwin C. The Descent of Man, and Selection in Relation to Sex. 2nd ed. London, UK: John Murray; 1874:35.
2. Issac B. Throwing and human evolution. African Archeol Record. 1987;5:3-17.
3. Kirschann E. The human throw and a new model of hominid evolution (German). Homo. 1999;50(1):80-85.
4. Knusel CJ. The throwing hypothesis and hominid origins. Human Evolution. 1992;7(1):1-7.
5. Dunsworth H, Challis J, Walker A. The evolution of throwing: a new look at an old idea. Courier Forschungsinstitut Senckenberg. 2003;243:105-110.
6. Mayr E. Animal Species and Evolution. Cambridge, MA: Harvard University Press; 1963.
7. Tinbergen N. On the aims and methods of ethology. Zeitschrift für Tierpsychologie. 1963;20:410-433.
8. Fleisig GS, Andrews JR, Dillman CJ, Escamilla RF. Kinetics of baseball pitching with implications about injury mechanisms. Am J Sports Med. 1995;23(2):233-239.
9. Atwater AE. Biomechanics of overarm throwing movements and of throwing injuries. Exerc Sport Sci Rev. 1975;7:43-85.
10. Matsuo T, Escamila RF, Fleisig GS, Barrentine SW, Andrews JR. Comparison of kinematic and temporal parameters between different pitch velocity groups. J Appl Biomech. 2001;17:1-13.
11. Wang YT, Ford HT III, Ford HT Jr, Shin DM. Three-dimensional kinematic analysis of baseball pitching in acceleration phase. Percept Mot Skills. 1995;80:43-48.
12. Roach NT, Venkadesan M, Rainbow MJ, Lieberman DE. Elastic energy storage in the shoulder and the evolution of high-speed throwing in Homo. Nature. 2013;498(7455):483-486.
13. Fox JA, Cole BJ, Romeo AA, et al. Articular cartilage thickness of the humeral head: an anatomic study. Orthopedics. 2008;31(3):216.
14. Zumstein V, Kraljevic M, Conzen A, Hoechel S, Müller-Gerbl M. Thickness distribution of the glenohumeral joint cartilage: a quantitative study using computed tomography. Surg Radiol Anat. 2014;36(4):327-331.
15. Inman VT, Saunders M, Abbott LC. Observations on the function of the shoulder joint. J Bone Joint Surg Am. 1944;26:1-30.
16. O’Brien SJ, Schwartz RS, Warren RF, Torzilli PA. Capsular restraints to anterior posterior motion of the abducted shoulder: A biomechanical study. J Shoulder Elbow Surg. 1995;4(4):298-308.
17. Warner JJ, Deng XH, Warren RF, Torzilli PA. Static capsuloligamentous restraints to superior-inferior translation of the glenohumeral joint. Am J Sports Med. 1992;20(6):675-685.
18. Kuhn JE, Bey MJ, Huston LJ, Blasier RB, Soslowsky LJ. Ligamentous restraints to external rotation of the humerus in the late-cocking phase of throwing. A cadaveric biomechanical investigation. Am J Sports Med. 2000;28(2):200-205.
19. Bigliani LU, Codd TP, Connor PM, Levine WN, Littlefield MA, Hershon SJ. Shoulder motion and laxity in the professional baseball player. Am J Sports Med. 1997;25(5):609-613.
20. Borsa PA, Dover GC, Wilk KE, Reinold MM. Glenohumeral range of motion and stiffness in professional baseball pitchers. Med Sci Sports Exerc. 2006;38(1):21-26.
21. Hurd WJ, Kaplan KM, Eiattrache NS, Jobe FW, Morrey BF, Kaufman KR. A profile of glenohumeral internal and external rotation motion in the uninjured high school baseball pitcher, part I: motion. J Athl Train. 2011;46(3):282-288.
22. Wilk KE, Macrina LC, Arrigo C. Passive range of motion characteristics in the overhead baseball pitcher and their implications for rehabilitation. Clin Orthop Relat Res. 2012;470(6):1586-1594.
23. Osbahr DC, Cannon DL, Speer KP. Retroversion of the humerus in the throwing shoulder of college baseball pitchers. Am J Sports Med. 2002;30(3):347-353.
24. Greenberg EM, Fernandez-Fernandez A, Lawrence JT, McClure P. The development of humeral retrotorsion and its relationship to throwing sports. Sports Health. 2015;7(6):489-496.
25. Jobe FW, Pink M. The athlete’s shoulder. J Hand Ther. 1994;7(2):107-110.
26. Montgomery WH 3rd, Jobe FW. Functional outcomes in athletes after modified anterior capsulolabral reconstruction. Am J Sports Med. 1994;22(3):352-358.
27. Jobe FW, Kvitne RS, Giangarra CE. Shoulder pain in the overhand or throwing athlete. The relationship of anterior instability and rotator cuff impingement. Orthop Rev. 1989;18(9):963-975.
28. Reinhold MM, Wilk KE, Dugas JR, Andrews JR. Chapter 11. Internal Impingement. In: Wilk K, Reinold MM, Andrews JR, eds. The Athlete’s Shoulder. 2nd ed. Philadelphia, PA: Churchill Livingstone Elsevier; 2009:126.
29. Kuhn JE, Lindholm SR, Huston LJ, Soslowsky LJ, Blasier RB. Failure of the biceps superior labral complex: a cadaveric biomechanical investigation comparing the late cocking and early deceleration positions of throwing. Arthroscopy. 2003;19(4):373-379.
30. Ciochon RL, Corruccini RS. The coraco-acromial ligament and projection index in man and other anthropoid primates. J Anat. 1977;124(Pt 3):627-632.
31. Moorman CT, Warren RF, Deng XH, Wickiewicz TL, Torzilli PA. Role of coracoacromial ligament and related structures in glenohumeral stability: a cadaveric study. J Surg Orthop Adv. 2012;21(4):210-217.
32. Su WR, Budoff JE, Luo ZP. The effect of coracoacromial ligament excision and acromioplasty on superior and anterosuperior glenohumeral stability. Arthroscopy. 2009;25(1):13-18.
33. Wellmann M, Petersen W, Zantop T, Schanz S, Raschke MJ, Hurschler C. Effect of coracoacromial ligament resection on glenohumeral stability under active muscle loading in an in vitro model. Arthroscopy. 2008;24(11):1258-1264.
34. Fagelman M, Sartori M, Freedman KB, Patwardhan AG, Carandang G, Marra G. Biomechanics of coracoacromial arch modification. J Shoulder Elbow Surg. 2007;16(1):101-116.
35. Bak K, Spring IB, Henderson IP. Re-formation of the coracoacromial ligament after open resection or arthroscopic release. J Shoulder Elbow Surg. 2000;9:289-293.
36. Levy O, Copeland SA. Regeneration of the coracoacromial ligament after acromioplasty and arthroscopic subacromial decompression. J Shoulder Elbow Surg. 2001;10(4):317-320.
37. Larson SG, Stern JT Jr. Role of supraspinatus in the quadrupedal locomotion of vervets (Cercopithecus aethiops): Implications for interpretation of humeral morphology. Am J Phys Anthropol. 1989;79(3):369-377.
38. Chansky HA, Iannotti JP. The vascularity of the rotator cuff. Clin Sports Med. 1991;10(4):807-822.
39. Klapper RC, Jobe FW, Matsuura P. Subscapularis muscle and its glenohumeral ligament-like bands. A histomorphologic study. Am J Sports Med. 1992;20(3):307-310.
40. Halder A, Zobitz ME, Schultz E, An KN. Structural properties of the subscapularis tendon. J Orthop Res. 2000;18(5):
829-834.
41. Elser F, Braun S, Dewing CB, Giphart JE, Millett PJ. Anatomy, function, injuries, and treatment of the long head of the biceps brachii tendon. Arthroscopy. 2011;27(4):581-592.
42. Pill SG, Walch G, Hawkins RJ, Kissenberth MJ. The role of the biceps tendon in massive rotator cuff tears. Instr Course Lect. 2012;61:113-120.
43. Krupp RJ, Kevern MA, Gaines MD, Kotara S, Singleton SB. Long head of the biceps tendon pain: differential diagnosis and treatment. J Orthop Sports Phys Ther. 2009;39(2):55-70.
44. Levy AS, Kelly BT, Lintner SA, Osbahr DC, Speer KP. Function of the long head of the biceps at the shoulder: electromyographic analysis. J Shoulder Elbow Surg. 2001;10(3):250-255.
45. Kuhn JE, Huston LJ, Soslowsky LJ, Shyr Y, Blasier RB. External rotation of the glenohumeral joint: ligament restraints and muscle effects in the neutral and abducted positions.
J Shoulder Elbow Surg. 2005;14(1 Suppl S):39S-48S.
46. McGarry MH, Nguyen ML, Quigley RJ, Hanypsiak B, Gupta R, Lee TQ. The effect of long and short head biceps loading on glenohumeral joint rotational range of motion and humeral head postion. Knee Surge Sports Traumatol Arthrosc. 2014 Sep 26. [Epub ahead of print].
47. Young RW. Evolution of the human hand: The role of throwing and clubbing. J Anat. 2003;202:165-174.
48. Calvin WH. Did throwing stones shape hominid brain evolution? Ethology and Sociobiology. 1982;3:115-124.
49. Fifer FC. The adoption of bipedalism by the hominids: A new hypothesis. Human Evolution. 1987;2(2):135-147.
50. Darlington PJ. Group selection, altruism, reinforcement, and throwing in human evolution. Proc Nat Acad Sci. 1973;72(9):3748-3752.
Why so many pertussis outbreaks despite acellular pertussis vaccine? A call to action
There has been a justified re-examination of acellular pertussis vaccine (aP)1,2 in light of the multiple large outbreaks of pertussis since 2000, particularly the two large California outbreaks in 2010 and 2014.
Lessons learned: aP protection is less durable than originally thought, and much pertussis is not in infants, but in the school-age and adolescent populations.

aP appears to produce reasonable protection (approximately 84% overall) for infants and preschool children, plus a much improved adverse effect profile, compared with whole cell pertussis vaccine (WCP), which provided approximately 94% protection.1 This 10% difference in aP versus WCP, however, means that herd immunity is more difficult to attain. The accepted pertussis immunization rate needed to provide herd immunity is 92%-94%. Because our current tools (DTaP and Tdap) provide only 84% protection at least in infants and preschoolers, even 100% uptake may leave us 6% to 8% short of the threshold for complete herd immunity.
The California outbreak data from school-age and teenage populations show protection rates drop each year post aP booster. That means that by the fourth year after the last dose, protection is less than 10%. So despite a Tdap dose at 11- to 12-years-of-age, protection gaps occur in 8-to 10-year-olds and 14- to 18-year-olds. These vulnerable periods in older children add to aP’s 84% vs. WCP’s 94% protection for those greater than 3 years of age, explaining more frequent pertussis outbreaks as the pool of WCP-immunized children among older populations decreased.
But before we place all blame on switching to aP, consider that we can now confirm more pertussis infections with molecular assays than was possible with culture and fluorescent assay testing in the WCP era. So improved testing sensitivity means more reports of minimally symptomatic cases that may have been missed before. So WCP, if still used today, might not show 94% protection either.
Additionally, aPs rely heavily on pertactin as a target antigen,3 likely less than WCP, given that WCP contained all pertussis antigens rather than just the 3-5 purified antigens in aPs. So the emergence of pertactin-altered pertussis strains could disproportionately affect protection from aP, compared with WCP.
There seem to be no quick fixes to preventing outbreaks using aPs as our vaccine. One suggestion by the authors of the California outbreak report is to use aP mostly to terminate outbreaks rather than routinely in late childhood. My concern is that if we do not continue routine use in 4-to 6-year-olds, 10-to 11-year-olds, and in early adulthood, the vulnerable proportion of the population during outbreaks would be larger, making outbreaks more difficult to terminate. So continuing to produce some protection, albeit short-lived, with current schedules of aP vaccines seems important.
Also remember that T cells, particularly TH 17 pertussis-specific cells, may be as important as pertussis antibody. Therefore, crafting pertussis vaccines that yield improved antibody plus T cell responses is the current goal. Disease and WCP seem to elicit more T-17 response than aP. One method to craft a better vaccine is to use antigen blends that differ from those in the current vaccines, such as antigens derived from circulating pertussis strains instead of the standard laboratory strain. Another option is to use current antigens but with more potent adjuvants. Such vaccines are likely 5 years away.
But we need to have reasonable expectations for pertussis vaccines. Pertussis infection begins in respiratory epithelium. Many of the most obvious signs and symptoms are due to destruction of ciliated respiratory epithelium plus increased tenacity/volume of secretions. Can a parenterally administered vaccine that induces mostly serum antibody protect against infection of epithelium where antibody concentrations are likely 10% or less than in serum? The short answer is – likely not. We should expect neither aP nor WCP to consistently protect against pertussis infection, but it does seem reasonable to expect aP to reduce disease severity. Preventing infections awaits a vaccine that induces surface IgA. Mucosally administered vaccines produce surface IgA – for example, rotavirus vaccine – but no mucosal pertussis vaccine appears imminent.
A key question is whether our most vulnerable populations, young children, have increased morbidity and mortality. Data from the California suggest an increase but mostly in infants under 6 months of age, the group not old enough to benefit from even the most effective of infant vaccines. Protecting young infants depends on vaccine administered prenatally to mothers. The over-representation of the Hispanic infants among fatalities shows a population on which to focus with maternal immunization. Hopefully, the recent universal TdaP recommendation in pregnancy will help when maternal immunization is higher than current approximately 50% rates.4
Despite the problems, it seems clear that we must continue to use current aP vaccines according to the current schedules, attempting to get as close to 100% uptake as possible. While the current, nearly 10% unimmunized rates add to the likelihood that we are losing complete herd immunity, partial herd immunity is better than no herd immunity.
Expectations: There will be ongoing outbreaks. Continue to be alert for signs of pertussis. They are often less obvious in older patients, and may be as subtle as more than 2 weeks of persistent cough. During outbreaks, we may be called upon to give aP doses at intervals shorter than the usual schedule.
Our responsibility: Do not become discouraged or lose enthusiasm for aP, but explain to parents that because aP is less reactogenic, it produces less protection and is less durable, particularly in school-age children. But please emphasize that modest protection is best in the youngest and modest protection of older children is better than none. Emphasize that the adverse effect profile of current aPs puts the harm/benefit balance heavily in favor of aP.
Bottom line: We can hopefully do better than the current 88% to 92% rate of aP vaccine uptake. We need to get as close to 100% uptake as possible until new vaccines or new strategies become available.
1. Clin Infect Dis. 2016 Feb 7; doi: 10.1093/cid/ciw051.
2. Pediatrics. 2016 Feb 5; doi: 10.1542/peds.2015-3326.
3. Expert Rev Vaccines. 2007 Feb;6(1):47-56.
4. Vaccine. 2016 Feb 10;34(7):968-73.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He disclosed that his institution received grant support for a study on hexavalent infant vaccine containing pertussis from GlaxoSmithKline, and he was the local primary investigator.*
*Correction, 2/17/2016: An earlier version of this article incompletely stated Dr. Harrison's disclosure information.
There has been a justified re-examination of acellular pertussis vaccine (aP)1,2 in light of the multiple large outbreaks of pertussis since 2000, particularly the two large California outbreaks in 2010 and 2014.
Lessons learned: aP protection is less durable than originally thought, and much pertussis is not in infants, but in the school-age and adolescent populations.
aP appears to produce reasonable protection (approximately 84% overall) for infants and preschool children, plus a much improved adverse effect profile, compared with whole cell pertussis vaccine (WCP), which provided approximately 94% protection.1 This 10% difference in aP versus WCP, however, means that herd immunity is more difficult to attain. The accepted pertussis immunization rate needed to provide herd immunity is 92%-94%. Because our current tools (DTaP and Tdap) provide only 84% protection at least in infants and preschoolers, even 100% uptake may leave us 6% to 8% short of the threshold for complete herd immunity.
The California outbreak data from school-age and teenage populations show protection rates drop each year post aP booster. That means that by the fourth year after the last dose, protection is less than 10%. So despite a Tdap dose at 11- to 12-years-of-age, protection gaps occur in 8-to 10-year-olds and 14- to 18-year-olds. These vulnerable periods in older children add to aP’s 84% vs. WCP’s 94% protection for those greater than 3 years of age, explaining more frequent pertussis outbreaks as the pool of WCP-immunized children among older populations decreased.
But before we place all blame on switching to aP, consider that we can now confirm more pertussis infections with molecular assays than was possible with culture and fluorescent assay testing in the WCP era. So improved testing sensitivity means more reports of minimally symptomatic cases that may have been missed before. So WCP, if still used today, might not show 94% protection either.
Additionally, aPs rely heavily on pertactin as a target antigen,3 likely less than WCP, given that WCP contained all pertussis antigens rather than just the 3-5 purified antigens in aPs. So the emergence of pertactin-altered pertussis strains could disproportionately affect protection from aP, compared with WCP.
There seem to be no quick fixes to preventing outbreaks using aPs as our vaccine. One suggestion by the authors of the California outbreak report is to use aP mostly to terminate outbreaks rather than routinely in late childhood. My concern is that if we do not continue routine use in 4-to 6-year-olds, 10-to 11-year-olds, and in early adulthood, the vulnerable proportion of the population during outbreaks would be larger, making outbreaks more difficult to terminate. So continuing to produce some protection, albeit short-lived, with current schedules of aP vaccines seems important.
Also remember that T cells, particularly TH 17 pertussis-specific cells, may be as important as pertussis antibody. Therefore, crafting pertussis vaccines that yield improved antibody plus T cell responses is the current goal. Disease and WCP seem to elicit more T-17 response than aP. One method to craft a better vaccine is to use antigen blends that differ from those in the current vaccines, such as antigens derived from circulating pertussis strains instead of the standard laboratory strain. Another option is to use current antigens but with more potent adjuvants. Such vaccines are likely 5 years away.
But we need to have reasonable expectations for pertussis vaccines. Pertussis infection begins in respiratory epithelium. Many of the most obvious signs and symptoms are due to destruction of ciliated respiratory epithelium plus increased tenacity/volume of secretions. Can a parenterally administered vaccine that induces mostly serum antibody protect against infection of epithelium where antibody concentrations are likely 10% or less than in serum? The short answer is – likely not. We should expect neither aP nor WCP to consistently protect against pertussis infection, but it does seem reasonable to expect aP to reduce disease severity. Preventing infections awaits a vaccine that induces surface IgA. Mucosally administered vaccines produce surface IgA – for example, rotavirus vaccine – but no mucosal pertussis vaccine appears imminent.
A key question is whether our most vulnerable populations, young children, have increased morbidity and mortality. Data from the California suggest an increase but mostly in infants under 6 months of age, the group not old enough to benefit from even the most effective of infant vaccines. Protecting young infants depends on vaccine administered prenatally to mothers. The over-representation of the Hispanic infants among fatalities shows a population on which to focus with maternal immunization. Hopefully, the recent universal TdaP recommendation in pregnancy will help when maternal immunization is higher than current approximately 50% rates.4
Despite the problems, it seems clear that we must continue to use current aP vaccines according to the current schedules, attempting to get as close to 100% uptake as possible. While the current, nearly 10% unimmunized rates add to the likelihood that we are losing complete herd immunity, partial herd immunity is better than no herd immunity.
Expectations: There will be ongoing outbreaks. Continue to be alert for signs of pertussis. They are often less obvious in older patients, and may be as subtle as more than 2 weeks of persistent cough. During outbreaks, we may be called upon to give aP doses at intervals shorter than the usual schedule.
Our responsibility: Do not become discouraged or lose enthusiasm for aP, but explain to parents that because aP is less reactogenic, it produces less protection and is less durable, particularly in school-age children. But please emphasize that modest protection is best in the youngest and modest protection of older children is better than none. Emphasize that the adverse effect profile of current aPs puts the harm/benefit balance heavily in favor of aP.
Bottom line: We can hopefully do better than the current 88% to 92% rate of aP vaccine uptake. We need to get as close to 100% uptake as possible until new vaccines or new strategies become available.
1. Clin Infect Dis. 2016 Feb 7; doi: 10.1093/cid/ciw051.
2. Pediatrics. 2016 Feb 5; doi: 10.1542/peds.2015-3326.
3. Expert Rev Vaccines. 2007 Feb;6(1):47-56.
4. Vaccine. 2016 Feb 10;34(7):968-73.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He disclosed that his institution received grant support for a study on hexavalent infant vaccine containing pertussis from GlaxoSmithKline, and he was the local primary investigator.*
*Correction, 2/17/2016: An earlier version of this article incompletely stated Dr. Harrison's disclosure information.
There has been a justified re-examination of acellular pertussis vaccine (aP)1,2 in light of the multiple large outbreaks of pertussis since 2000, particularly the two large California outbreaks in 2010 and 2014.
Lessons learned: aP protection is less durable than originally thought, and much pertussis is not in infants, but in the school-age and adolescent populations.
aP appears to produce reasonable protection (approximately 84% overall) for infants and preschool children, plus a much improved adverse effect profile, compared with whole cell pertussis vaccine (WCP), which provided approximately 94% protection.1 This 10% difference in aP versus WCP, however, means that herd immunity is more difficult to attain. The accepted pertussis immunization rate needed to provide herd immunity is 92%-94%. Because our current tools (DTaP and Tdap) provide only 84% protection at least in infants and preschoolers, even 100% uptake may leave us 6% to 8% short of the threshold for complete herd immunity.
The California outbreak data from school-age and teenage populations show protection rates drop each year post aP booster. That means that by the fourth year after the last dose, protection is less than 10%. So despite a Tdap dose at 11- to 12-years-of-age, protection gaps occur in 8-to 10-year-olds and 14- to 18-year-olds. These vulnerable periods in older children add to aP’s 84% vs. WCP’s 94% protection for those greater than 3 years of age, explaining more frequent pertussis outbreaks as the pool of WCP-immunized children among older populations decreased.
But before we place all blame on switching to aP, consider that we can now confirm more pertussis infections with molecular assays than was possible with culture and fluorescent assay testing in the WCP era. So improved testing sensitivity means more reports of minimally symptomatic cases that may have been missed before. So WCP, if still used today, might not show 94% protection either.
Additionally, aPs rely heavily on pertactin as a target antigen,3 likely less than WCP, given that WCP contained all pertussis antigens rather than just the 3-5 purified antigens in aPs. So the emergence of pertactin-altered pertussis strains could disproportionately affect protection from aP, compared with WCP.
There seem to be no quick fixes to preventing outbreaks using aPs as our vaccine. One suggestion by the authors of the California outbreak report is to use aP mostly to terminate outbreaks rather than routinely in late childhood. My concern is that if we do not continue routine use in 4-to 6-year-olds, 10-to 11-year-olds, and in early adulthood, the vulnerable proportion of the population during outbreaks would be larger, making outbreaks more difficult to terminate. So continuing to produce some protection, albeit short-lived, with current schedules of aP vaccines seems important.
Also remember that T cells, particularly TH 17 pertussis-specific cells, may be as important as pertussis antibody. Therefore, crafting pertussis vaccines that yield improved antibody plus T cell responses is the current goal. Disease and WCP seem to elicit more T-17 response than aP. One method to craft a better vaccine is to use antigen blends that differ from those in the current vaccines, such as antigens derived from circulating pertussis strains instead of the standard laboratory strain. Another option is to use current antigens but with more potent adjuvants. Such vaccines are likely 5 years away.
But we need to have reasonable expectations for pertussis vaccines. Pertussis infection begins in respiratory epithelium. Many of the most obvious signs and symptoms are due to destruction of ciliated respiratory epithelium plus increased tenacity/volume of secretions. Can a parenterally administered vaccine that induces mostly serum antibody protect against infection of epithelium where antibody concentrations are likely 10% or less than in serum? The short answer is – likely not. We should expect neither aP nor WCP to consistently protect against pertussis infection, but it does seem reasonable to expect aP to reduce disease severity. Preventing infections awaits a vaccine that induces surface IgA. Mucosally administered vaccines produce surface IgA – for example, rotavirus vaccine – but no mucosal pertussis vaccine appears imminent.
A key question is whether our most vulnerable populations, young children, have increased morbidity and mortality. Data from the California suggest an increase but mostly in infants under 6 months of age, the group not old enough to benefit from even the most effective of infant vaccines. Protecting young infants depends on vaccine administered prenatally to mothers. The over-representation of the Hispanic infants among fatalities shows a population on which to focus with maternal immunization. Hopefully, the recent universal TdaP recommendation in pregnancy will help when maternal immunization is higher than current approximately 50% rates.4
Despite the problems, it seems clear that we must continue to use current aP vaccines according to the current schedules, attempting to get as close to 100% uptake as possible. While the current, nearly 10% unimmunized rates add to the likelihood that we are losing complete herd immunity, partial herd immunity is better than no herd immunity.
Expectations: There will be ongoing outbreaks. Continue to be alert for signs of pertussis. They are often less obvious in older patients, and may be as subtle as more than 2 weeks of persistent cough. During outbreaks, we may be called upon to give aP doses at intervals shorter than the usual schedule.
Our responsibility: Do not become discouraged or lose enthusiasm for aP, but explain to parents that because aP is less reactogenic, it produces less protection and is less durable, particularly in school-age children. But please emphasize that modest protection is best in the youngest and modest protection of older children is better than none. Emphasize that the adverse effect profile of current aPs puts the harm/benefit balance heavily in favor of aP.
Bottom line: We can hopefully do better than the current 88% to 92% rate of aP vaccine uptake. We need to get as close to 100% uptake as possible until new vaccines or new strategies become available.
1. Clin Infect Dis. 2016 Feb 7; doi: 10.1093/cid/ciw051.
2. Pediatrics. 2016 Feb 5; doi: 10.1542/peds.2015-3326.
3. Expert Rev Vaccines. 2007 Feb;6(1):47-56.
4. Vaccine. 2016 Feb 10;34(7):968-73.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He disclosed that his institution received grant support for a study on hexavalent infant vaccine containing pertussis from GlaxoSmithKline, and he was the local primary investigator.*
*Correction, 2/17/2016: An earlier version of this article incompletely stated Dr. Harrison's disclosure information.

HIPAA enforcement in 2016: Is your practice ready?
Two reports from the Office of the Inspector General (OIG) have attracted a lot of attention in recent weeks: The Office for Civil Rights (OCR), OIG said, needs to improve and expand its enforcement of the Health Insurance Portability and Accountability Act (HIPAA). In response, the OCR announced that it plans to identify a pool of potential audit targets and launch a permanent audit program this year. That, combined with the substantial fine levied against a dermatology group last year for violating one of the new rules, signals the importance of reviewing your practice’s HIPAA compliance as soon as possible.
You can compare your office’s compliance status against the recommendations listed on the OCR website, but pay particular attention to your agreements with Business Associates (BAs). Those are the individuals or businesses, other than your employees, who perform “functions or activities” on behalf of your practice that involve “creating, receiving, maintaining, or transmitting” personal health information.

First, make sure that all individuals and enterprises fitting that definition have a signed agreement in place. Typical BAs include answering and billing services, independent transcriptionists, hardware and software companies, and any other vendors involved in creating or maintaining your medical records. Practice management consultants, attorneys, specialty pharmacies, and record storage, microfilming, and shredding services are BAs if they must have direct access to confidential information in order to do their job.
The revised rules place additional onus on physicians for confidentiality breaches committed by their BAs. It’s not enough to simply have a BA contract; you are expected to use “reasonable diligence” in monitoring their work. BAs and their subcontractors are directly responsible for their own actions, but the primary responsibility is yours. Furthermore, you must now assume the worst-case scenario: Previously, when protected health information (PHI) was compromised, you would have to notify only affected patients (and the government) if there was a “significant risk of financial or reputational harm,” but now, any incident involving patient records is assumed to be a breach, and must be reported.
Failure to do so could subject your practice, as well as the contractor, to significant fines. That is where the Massachusetts dermatology group ran into trouble: It lost a thumb drive containing unencrypted patient records, and was forced to pay a $150,000 fine, even though there was no evidence that the information was found or exploited.
Had the lost drive been encrypted, the incident would not have been considered a breach, according to the Centers for Medicare & Medicaid Services, because its contents would not have been viewable by the finder. The biggest vulnerability in most practices is probably mobile devices carrying patient data. There is no longer any excuse for not encrypting HIPAA-protected information; encryption software is cheap, readily available, and easy to use.
Patients have new rights under the new rules as well; they may now restrict any PHI shared with third-party insurers and health plans, if they pay for the services themselves. They also have the right to request copies of their electronic health records (EHRs). You can bill the costs of responding to such requests. If you have EHRs, work out a system for doing this, because the response time has been decreased from 90 days to 30 days – even shorter in some states.
If you haven’t yet revised your Notice of Privacy Practices (NPP) to explain your relationships with BAs, and their status under the new rules, do it now. (You should have done it last year.) You need to explain the breach notification process too, as well as the new patient rights mentioned above. You must post your revised NPP in your office, and make copies available there, but you need not mail a copy to every patient.
You also should examine every part of your office where patient information is handled to identify potential violations. Examples include computer screens in your reception area that are visible to patients; laptops not locked up after hours; unencrypted emails or texts that might reveal confidential information; and documents designated for shredding that sit, unshredded, in the “to shred” bin for days.
And make sure you correct any problems you find before the OCR auditors come calling.
To view the recommendations at the OCR website so you can check your office’s compliance status, go to: www.hhs.gov/hipaa/index.html.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News.
Two reports from the Office of the Inspector General (OIG) have attracted a lot of attention in recent weeks: The Office for Civil Rights (OCR), OIG said, needs to improve and expand its enforcement of the Health Insurance Portability and Accountability Act (HIPAA). In response, the OCR announced that it plans to identify a pool of potential audit targets and launch a permanent audit program this year. That, combined with the substantial fine levied against a dermatology group last year for violating one of the new rules, signals the importance of reviewing your practice’s HIPAA compliance as soon as possible.
You can compare your office’s compliance status against the recommendations listed on the OCR website, but pay particular attention to your agreements with Business Associates (BAs). Those are the individuals or businesses, other than your employees, who perform “functions or activities” on behalf of your practice that involve “creating, receiving, maintaining, or transmitting” personal health information.
First, make sure that all individuals and enterprises fitting that definition have a signed agreement in place. Typical BAs include answering and billing services, independent transcriptionists, hardware and software companies, and any other vendors involved in creating or maintaining your medical records. Practice management consultants, attorneys, specialty pharmacies, and record storage, microfilming, and shredding services are BAs if they must have direct access to confidential information in order to do their job.
The revised rules place additional onus on physicians for confidentiality breaches committed by their BAs. It’s not enough to simply have a BA contract; you are expected to use “reasonable diligence” in monitoring their work. BAs and their subcontractors are directly responsible for their own actions, but the primary responsibility is yours. Furthermore, you must now assume the worst-case scenario: Previously, when protected health information (PHI) was compromised, you would have to notify only affected patients (and the government) if there was a “significant risk of financial or reputational harm,” but now, any incident involving patient records is assumed to be a breach, and must be reported.
Failure to do so could subject your practice, as well as the contractor, to significant fines. That is where the Massachusetts dermatology group ran into trouble: It lost a thumb drive containing unencrypted patient records, and was forced to pay a $150,000 fine, even though there was no evidence that the information was found or exploited.
Had the lost drive been encrypted, the incident would not have been considered a breach, according to the Centers for Medicare & Medicaid Services, because its contents would not have been viewable by the finder. The biggest vulnerability in most practices is probably mobile devices carrying patient data. There is no longer any excuse for not encrypting HIPAA-protected information; encryption software is cheap, readily available, and easy to use.
Patients have new rights under the new rules as well; they may now restrict any PHI shared with third-party insurers and health plans, if they pay for the services themselves. They also have the right to request copies of their electronic health records (EHRs). You can bill the costs of responding to such requests. If you have EHRs, work out a system for doing this, because the response time has been decreased from 90 days to 30 days – even shorter in some states.
If you haven’t yet revised your Notice of Privacy Practices (NPP) to explain your relationships with BAs, and their status under the new rules, do it now. (You should have done it last year.) You need to explain the breach notification process too, as well as the new patient rights mentioned above. You must post your revised NPP in your office, and make copies available there, but you need not mail a copy to every patient.
You also should examine every part of your office where patient information is handled to identify potential violations. Examples include computer screens in your reception area that are visible to patients; laptops not locked up after hours; unencrypted emails or texts that might reveal confidential information; and documents designated for shredding that sit, unshredded, in the “to shred” bin for days.
And make sure you correct any problems you find before the OCR auditors come calling.
To view the recommendations at the OCR website so you can check your office’s compliance status, go to: www.hhs.gov/hipaa/index.html.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News.
Two reports from the Office of the Inspector General (OIG) have attracted a lot of attention in recent weeks: The Office for Civil Rights (OCR), OIG said, needs to improve and expand its enforcement of the Health Insurance Portability and Accountability Act (HIPAA). In response, the OCR announced that it plans to identify a pool of potential audit targets and launch a permanent audit program this year. That, combined with the substantial fine levied against a dermatology group last year for violating one of the new rules, signals the importance of reviewing your practice’s HIPAA compliance as soon as possible.
You can compare your office’s compliance status against the recommendations listed on the OCR website, but pay particular attention to your agreements with Business Associates (BAs). Those are the individuals or businesses, other than your employees, who perform “functions or activities” on behalf of your practice that involve “creating, receiving, maintaining, or transmitting” personal health information.
First, make sure that all individuals and enterprises fitting that definition have a signed agreement in place. Typical BAs include answering and billing services, independent transcriptionists, hardware and software companies, and any other vendors involved in creating or maintaining your medical records. Practice management consultants, attorneys, specialty pharmacies, and record storage, microfilming, and shredding services are BAs if they must have direct access to confidential information in order to do their job.
The revised rules place additional onus on physicians for confidentiality breaches committed by their BAs. It’s not enough to simply have a BA contract; you are expected to use “reasonable diligence” in monitoring their work. BAs and their subcontractors are directly responsible for their own actions, but the primary responsibility is yours. Furthermore, you must now assume the worst-case scenario: Previously, when protected health information (PHI) was compromised, you would have to notify only affected patients (and the government) if there was a “significant risk of financial or reputational harm,” but now, any incident involving patient records is assumed to be a breach, and must be reported.
Failure to do so could subject your practice, as well as the contractor, to significant fines. That is where the Massachusetts dermatology group ran into trouble: It lost a thumb drive containing unencrypted patient records, and was forced to pay a $150,000 fine, even though there was no evidence that the information was found or exploited.
Had the lost drive been encrypted, the incident would not have been considered a breach, according to the Centers for Medicare & Medicaid Services, because its contents would not have been viewable by the finder. The biggest vulnerability in most practices is probably mobile devices carrying patient data. There is no longer any excuse for not encrypting HIPAA-protected information; encryption software is cheap, readily available, and easy to use.
Patients have new rights under the new rules as well; they may now restrict any PHI shared with third-party insurers and health plans, if they pay for the services themselves. They also have the right to request copies of their electronic health records (EHRs). You can bill the costs of responding to such requests. If you have EHRs, work out a system for doing this, because the response time has been decreased from 90 days to 30 days – even shorter in some states.
If you haven’t yet revised your Notice of Privacy Practices (NPP) to explain your relationships with BAs, and their status under the new rules, do it now. (You should have done it last year.) You need to explain the breach notification process too, as well as the new patient rights mentioned above. You must post your revised NPP in your office, and make copies available there, but you need not mail a copy to every patient.
You also should examine every part of your office where patient information is handled to identify potential violations. Examples include computer screens in your reception area that are visible to patients; laptops not locked up after hours; unencrypted emails or texts that might reveal confidential information; and documents designated for shredding that sit, unshredded, in the “to shred” bin for days.
And make sure you correct any problems you find before the OCR auditors come calling.
To view the recommendations at the OCR website so you can check your office’s compliance status, go to: www.hhs.gov/hipaa/index.html.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News.

Obesity
Emily is a 15-year-old girl who was referred by her pediatrician because of cutting behavior and conflict with her parents. Her parents reported that she has had a high body weight in the obese range since early in life. She had tried various diets without success, and her parents were frustrated with the pediatrician’s emphasis on weight over the years.
Mood problems had begun when she was in the sixth grade when she began to be severely bullied about her weight. Emily said this time was so difficult that she did not have clear memories of it. She described feeling numb. She began experiencing intense anxiety about school, and she was sometimes reluctant to attend and started cutting herself as a means of managing her emotions. In middle school, she began to fight back and associated herself with a group of “mean girls” who drank. She began having increasing conflict with her parents over the drinking and the cutting.

Discussion
Obesity is an extremely complex issue without simple answers. Severe obesity is correlated with numerous health risks including not only cardiovascular disease, type 2 diabetes, hypertension, and cancer, but also psychiatric problems such as depression, anxiety, body dissatisfaction, eating disorders, and unhealthy weight control behaviors. While some of these issues relate directly to the weight itself, many of the psychiatric concerns stem from society’s extremely harsh response to obesity.
We are all aware that the percentage of overweight and obese children, teens, and adults has increased in the past 50 years, although with some recent stabilization.1 The rise in obesity is related to societal factors – the prevalence and advertising of nutrient-poor/high-calorie processed foods in the marketplace, the rise of technologies that have decreased the need for movement, increases in portion sizes in restaurants, especially fast food settings, as well as the subsidizing of unhealthy foods, limited access to and greater cost of more nutritious foods, and limited access to exercise opportunities in poorer areas. This is the “obesogenic environment.” As in numerous aspects of health, weight is also influenced by genetics. Those who are genetically more likely to gain weight are the ones who suffer most from these social changes.
The problem is that, except for bariatric surgery, the interventions prescribed for individuals with obesity don’t work for the vast majority of people in the long run. There is an assumption that if the obese would just eat and exercise the way a thin person does, then they would be thin. While there is evidence that lifestyle strategies that induce a negative energy balance through cutting calories (often by 500-1,000) and “programmed exercise” can help some people lose weight over the course of 6 months to a year, longer-term follow-up suggests that most people regain this weight in the long run, at 5 years out. Even the most optimistic estimates suggest that only about one out of five people can maintain weight losses of 10% in the long term with current standard lifestyle interventions.2

There is evidence that someone attaining a particular body mass index (BMI) through dieting is not able to consume as many calories as another person who has always been at that BMI, requiring constant dietary restraint and a very high level of exercise to maintain the weight loss.3 The great majority of people who are unable to lose the weight, or briefly succeed and then gain the weight back or more, are seen as failing by society, by many medical professionals, and by themselves. There is clearly a need to focus more of our efforts on making changes on a societal level.
There also are alternative individual approaches that take the emphasis away from dieting and weight loss and instead focus on body acceptance and self-care. These interventions go by several names including mindful eating, intuitive eating, weight neutral, and “Health at Every Size.” This approach acknowledges the environmental and genetic factors beyond personal control and discusses how society pressures people to be thin. Instead of emphasizing repeated restrictive dieting, these programs stress maximizing health through making sustainable changes to increase activity and nutrition. These programs encourage people to care for themselves now rather than focusing on dieting toward a future weight where one can start enjoying life. Enjoyment of food, taking time to savor food, and being aware of when one is hungry and when not are central. For physical activity, the emphasis is on discovering something that is pleasurable and sustainable, rather than an onerous duty, as a means to an end of weight loss.4
Management
For Emily, struggling on the individual level, there is not a neat resolution. Psychotherapy to address anxiety, trauma, and substance abuse is indicated. Psychotherapy also should address Emily’s relationship with her body, as this is at the heart of many of these issues. Acknowledging the powerful stigma that society places on the obese while tolerating and even promoting an obesogenic environment, and the reality that weight loss is in fact extremely difficult, would open the door to a discussion with Emily and her family about what she wants and all her options to find the healthiest and most enjoyable way for her to live her life.
1. Pediatr Clin North Am. 2015 Oct;62(5):1241-61.
2. Annu Rev Nutr. 2001;21:323-41.
3. Am J Clin Nutr. 2005 Jul;82(1 Suppl):222S-225S.
4. Tylka TL, Annunziato RA, Burgard D, et al. “The Weight-Inclusive versus Weight-Normative Approach to Health: Evaluating the Evidence for Prioritizing Well-Being over Weight Loss.” J Obes. 2014;2014:983495. doi: 10.1155/2014/983495.
Dr. Hall is an assistant professor of psychiatry and pediatrics at the University of Vermont, Burlington.
Emily is a 15-year-old girl who was referred by her pediatrician because of cutting behavior and conflict with her parents. Her parents reported that she has had a high body weight in the obese range since early in life. She had tried various diets without success, and her parents were frustrated with the pediatrician’s emphasis on weight over the years.
Mood problems had begun when she was in the sixth grade when she began to be severely bullied about her weight. Emily said this time was so difficult that she did not have clear memories of it. She described feeling numb. She began experiencing intense anxiety about school, and she was sometimes reluctant to attend and started cutting herself as a means of managing her emotions. In middle school, she began to fight back and associated herself with a group of “mean girls” who drank. She began having increasing conflict with her parents over the drinking and the cutting.
Discussion
Obesity is an extremely complex issue without simple answers. Severe obesity is correlated with numerous health risks including not only cardiovascular disease, type 2 diabetes, hypertension, and cancer, but also psychiatric problems such as depression, anxiety, body dissatisfaction, eating disorders, and unhealthy weight control behaviors. While some of these issues relate directly to the weight itself, many of the psychiatric concerns stem from society’s extremely harsh response to obesity.
We are all aware that the percentage of overweight and obese children, teens, and adults has increased in the past 50 years, although with some recent stabilization.1 The rise in obesity is related to societal factors – the prevalence and advertising of nutrient-poor/high-calorie processed foods in the marketplace, the rise of technologies that have decreased the need for movement, increases in portion sizes in restaurants, especially fast food settings, as well as the subsidizing of unhealthy foods, limited access to and greater cost of more nutritious foods, and limited access to exercise opportunities in poorer areas. This is the “obesogenic environment.” As in numerous aspects of health, weight is also influenced by genetics. Those who are genetically more likely to gain weight are the ones who suffer most from these social changes.
The problem is that, except for bariatric surgery, the interventions prescribed for individuals with obesity don’t work for the vast majority of people in the long run. There is an assumption that if the obese would just eat and exercise the way a thin person does, then they would be thin. While there is evidence that lifestyle strategies that induce a negative energy balance through cutting calories (often by 500-1,000) and “programmed exercise” can help some people lose weight over the course of 6 months to a year, longer-term follow-up suggests that most people regain this weight in the long run, at 5 years out. Even the most optimistic estimates suggest that only about one out of five people can maintain weight losses of 10% in the long term with current standard lifestyle interventions.2
There is evidence that someone attaining a particular body mass index (BMI) through dieting is not able to consume as many calories as another person who has always been at that BMI, requiring constant dietary restraint and a very high level of exercise to maintain the weight loss.3 The great majority of people who are unable to lose the weight, or briefly succeed and then gain the weight back or more, are seen as failing by society, by many medical professionals, and by themselves. There is clearly a need to focus more of our efforts on making changes on a societal level.
There also are alternative individual approaches that take the emphasis away from dieting and weight loss and instead focus on body acceptance and self-care. These interventions go by several names including mindful eating, intuitive eating, weight neutral, and “Health at Every Size.” This approach acknowledges the environmental and genetic factors beyond personal control and discusses how society pressures people to be thin. Instead of emphasizing repeated restrictive dieting, these programs stress maximizing health through making sustainable changes to increase activity and nutrition. These programs encourage people to care for themselves now rather than focusing on dieting toward a future weight where one can start enjoying life. Enjoyment of food, taking time to savor food, and being aware of when one is hungry and when not are central. For physical activity, the emphasis is on discovering something that is pleasurable and sustainable, rather than an onerous duty, as a means to an end of weight loss.4
Management
For Emily, struggling on the individual level, there is not a neat resolution. Psychotherapy to address anxiety, trauma, and substance abuse is indicated. Psychotherapy also should address Emily’s relationship with her body, as this is at the heart of many of these issues. Acknowledging the powerful stigma that society places on the obese while tolerating and even promoting an obesogenic environment, and the reality that weight loss is in fact extremely difficult, would open the door to a discussion with Emily and her family about what she wants and all her options to find the healthiest and most enjoyable way for her to live her life.
1. Pediatr Clin North Am. 2015 Oct;62(5):1241-61.
2. Annu Rev Nutr. 2001;21:323-41.
3. Am J Clin Nutr. 2005 Jul;82(1 Suppl):222S-225S.
4. Tylka TL, Annunziato RA, Burgard D, et al. “The Weight-Inclusive versus Weight-Normative Approach to Health: Evaluating the Evidence for Prioritizing Well-Being over Weight Loss.” J Obes. 2014;2014:983495. doi: 10.1155/2014/983495.
Dr. Hall is an assistant professor of psychiatry and pediatrics at the University of Vermont, Burlington.
Emily is a 15-year-old girl who was referred by her pediatrician because of cutting behavior and conflict with her parents. Her parents reported that she has had a high body weight in the obese range since early in life. She had tried various diets without success, and her parents were frustrated with the pediatrician’s emphasis on weight over the years.
Mood problems had begun when she was in the sixth grade when she began to be severely bullied about her weight. Emily said this time was so difficult that she did not have clear memories of it. She described feeling numb. She began experiencing intense anxiety about school, and she was sometimes reluctant to attend and started cutting herself as a means of managing her emotions. In middle school, she began to fight back and associated herself with a group of “mean girls” who drank. She began having increasing conflict with her parents over the drinking and the cutting.
Discussion
Obesity is an extremely complex issue without simple answers. Severe obesity is correlated with numerous health risks including not only cardiovascular disease, type 2 diabetes, hypertension, and cancer, but also psychiatric problems such as depression, anxiety, body dissatisfaction, eating disorders, and unhealthy weight control behaviors. While some of these issues relate directly to the weight itself, many of the psychiatric concerns stem from society’s extremely harsh response to obesity.
We are all aware that the percentage of overweight and obese children, teens, and adults has increased in the past 50 years, although with some recent stabilization.1 The rise in obesity is related to societal factors – the prevalence and advertising of nutrient-poor/high-calorie processed foods in the marketplace, the rise of technologies that have decreased the need for movement, increases in portion sizes in restaurants, especially fast food settings, as well as the subsidizing of unhealthy foods, limited access to and greater cost of more nutritious foods, and limited access to exercise opportunities in poorer areas. This is the “obesogenic environment.” As in numerous aspects of health, weight is also influenced by genetics. Those who are genetically more likely to gain weight are the ones who suffer most from these social changes.
The problem is that, except for bariatric surgery, the interventions prescribed for individuals with obesity don’t work for the vast majority of people in the long run. There is an assumption that if the obese would just eat and exercise the way a thin person does, then they would be thin. While there is evidence that lifestyle strategies that induce a negative energy balance through cutting calories (often by 500-1,000) and “programmed exercise” can help some people lose weight over the course of 6 months to a year, longer-term follow-up suggests that most people regain this weight in the long run, at 5 years out. Even the most optimistic estimates suggest that only about one out of five people can maintain weight losses of 10% in the long term with current standard lifestyle interventions.2
There is evidence that someone attaining a particular body mass index (BMI) through dieting is not able to consume as many calories as another person who has always been at that BMI, requiring constant dietary restraint and a very high level of exercise to maintain the weight loss.3 The great majority of people who are unable to lose the weight, or briefly succeed and then gain the weight back or more, are seen as failing by society, by many medical professionals, and by themselves. There is clearly a need to focus more of our efforts on making changes on a societal level.
There also are alternative individual approaches that take the emphasis away from dieting and weight loss and instead focus on body acceptance and self-care. These interventions go by several names including mindful eating, intuitive eating, weight neutral, and “Health at Every Size.” This approach acknowledges the environmental and genetic factors beyond personal control and discusses how society pressures people to be thin. Instead of emphasizing repeated restrictive dieting, these programs stress maximizing health through making sustainable changes to increase activity and nutrition. These programs encourage people to care for themselves now rather than focusing on dieting toward a future weight where one can start enjoying life. Enjoyment of food, taking time to savor food, and being aware of when one is hungry and when not are central. For physical activity, the emphasis is on discovering something that is pleasurable and sustainable, rather than an onerous duty, as a means to an end of weight loss.4
Management
For Emily, struggling on the individual level, there is not a neat resolution. Psychotherapy to address anxiety, trauma, and substance abuse is indicated. Psychotherapy also should address Emily’s relationship with her body, as this is at the heart of many of these issues. Acknowledging the powerful stigma that society places on the obese while tolerating and even promoting an obesogenic environment, and the reality that weight loss is in fact extremely difficult, would open the door to a discussion with Emily and her family about what she wants and all her options to find the healthiest and most enjoyable way for her to live her life.
1. Pediatr Clin North Am. 2015 Oct;62(5):1241-61.
2. Annu Rev Nutr. 2001;21:323-41.
3. Am J Clin Nutr. 2005 Jul;82(1 Suppl):222S-225S.
4. Tylka TL, Annunziato RA, Burgard D, et al. “The Weight-Inclusive versus Weight-Normative Approach to Health: Evaluating the Evidence for Prioritizing Well-Being over Weight Loss.” J Obes. 2014;2014:983495. doi: 10.1155/2014/983495.
Dr. Hall is an assistant professor of psychiatry and pediatrics at the University of Vermont, Burlington.

Make Room on Your Shelves
As orthopedic surgeons, we’ve made a commitment to lifelong learning. I can’t think of a single surgery that I perform the same way I did when I was in training. With rapidly evolving technology, continuously advancing procedures, and ever-increasing documentation requirements, it’s hard to stay on top of it all. We know your time is precious and that you have less of it than ever before. What little time you have that is not dedicated to work is reserved for your family or your hobbies. There’s no time to read every orthopedic journal, many filled with articles that have no practical value to your practice. That’s why we’ve created the new AJO. Our goal, as an editorial staff, is to provide a journal where every article, column, and feature contains information that directly benefits your practice, your patients, or your bottom line, and keeps you informed of the latest techniques, procedures, and products. We will help surgeons “work smarter, not harder,” implement new technologies into their practices, and find creative revenue streams that are both legal and compliant.
We’ve assembled a team of talented editors to accomplish this task, and will introduce them throughout the coming year. In this issue, you will meet our Deputy Editors-in-Chief and some of our new Associate Editors who’ve collaborated to bring you the “new AJO”.
At this year’s Academy, the AJO launched an extensive rebranding. We have a new look, a new logo, and a new creative directive. The journal will now feature new columns, invited articles, and innovative surgical techniques. We will publish 5 issues for the remainder of 2016. Our March/April issue is a special edition dedicated to baseball. In time for Spring Training/Opening Day, this issue includes articles from Major League Baseball’s physicians and trainers, a “Codes to Know” segment, “Tips of the Trade,” and a “Tools of the Trade” feature. “The Baseball Issue” will set the tone for what readers can expect from the “new AJO”.
Our first feature article, written by Jed Kuhn, takes a philosophical look at the evolution of the throwing shoulder, and invites the reader to help unlock some of the great shoulder anatomy mysteries by viewing them from a time when throwing was an activity of daily living. In ancient times, if you couldn’t throw, you couldn’t eat. We know that children who play baseball remodel their shoulder to allow for increased external rotation. Read Dr. Kuhn’s article and imagine when a shoulder optimized for throwing was a competitive advantage for survival.
Our second feature article is written by Stan Conte, a legend of the game and longtime trainer for the Los Angeles Dodgers. Dr. Conte studied injury trends in baseball over the past 18 seasons and provides an analysis of the staggering cost of placing players on the disabled list.
A baseball issue could not be complete without an article on Tommy John surgery. In this issue, AJO shares a revolutionary new technique for treating players with MUCL tears by author Jeffrey Dugas. Named the “Internal Brace”, Dr. Dugas shares his technique for augmenting the injured MUCL and we are proud to bring it to you first.
A recurring feature in the new AJO will be a section we refer to as “Codes to Know.” In partnership with Karen Zupko, AJO will present little-known coding secrets and proper coding techniques to help you get reimbursed appropriately for your work. This month, in the first article of a 3-part series, Alan Hirahara teaches us how to properly code for a diagnostic ultrasound examination of the shoulder. The article includes templates available for download to assist you with proper documentation. Parts 2 and 3 will provide a tutorial on the proper technique for examinations and injections.
While shoulder and elbow injuries get more attention, Major League Baseball’s Injury Panel has produced a look at the staggering amount of knee injuries over the 2011-2014 seasons, inspiring us to feature the knee in our 2 “Trade” Columns.
The “Tips of the Trade” column will continue, featuring this month a guide to identifying and treating meniscal root tears. A new segment, referred to as “Tools of the Trade,” reviews the latest products for all-inside meniscal repair. Our “Tools” section will feature announcements and reviews of the hottest new products, with a buying guide and surgical pearls from the surgeons who know them best.
While we are discussing the lower extremity, we should point out that we plan to do the “leg work” for you. Each AJO issue will have handouts that can be downloaded from our website and utilized in your practice. Read Robin West’s article entitled “Interval Throwing and Hitting Programs in Baseball: Biomechanics and Rehabilitation,” and download Return to Throwing and Hitting programs your patients and therapists can use.
Finally, I’d like to thank our previous Editor-in-Chief Dr. Peter McCann for his stewardship the last 10 years and recognize him for his dedication to the journal.
Thank you for reading AJO and for continuing to do so in the future. I know that collectively, we can turn AJO into a product worthy of its title. We know our past reputation. We are no longer that journal. Spend some time to get to know the “new AJO”, and make some room on your shelves, because the information between the covers will provide a template to implement new technologies and revenue streams into your practice and help fulfill your commitment to learning.
As orthopedic surgeons, we’ve made a commitment to lifelong learning. I can’t think of a single surgery that I perform the same way I did when I was in training. With rapidly evolving technology, continuously advancing procedures, and ever-increasing documentation requirements, it’s hard to stay on top of it all. We know your time is precious and that you have less of it than ever before. What little time you have that is not dedicated to work is reserved for your family or your hobbies. There’s no time to read every orthopedic journal, many filled with articles that have no practical value to your practice. That’s why we’ve created the new AJO. Our goal, as an editorial staff, is to provide a journal where every article, column, and feature contains information that directly benefits your practice, your patients, or your bottom line, and keeps you informed of the latest techniques, procedures, and products. We will help surgeons “work smarter, not harder,” implement new technologies into their practices, and find creative revenue streams that are both legal and compliant.
We’ve assembled a team of talented editors to accomplish this task, and will introduce them throughout the coming year. In this issue, you will meet our Deputy Editors-in-Chief and some of our new Associate Editors who’ve collaborated to bring you the “new AJO”.
At this year’s Academy, the AJO launched an extensive rebranding. We have a new look, a new logo, and a new creative directive. The journal will now feature new columns, invited articles, and innovative surgical techniques. We will publish 5 issues for the remainder of 2016. Our March/April issue is a special edition dedicated to baseball. In time for Spring Training/Opening Day, this issue includes articles from Major League Baseball’s physicians and trainers, a “Codes to Know” segment, “Tips of the Trade,” and a “Tools of the Trade” feature. “The Baseball Issue” will set the tone for what readers can expect from the “new AJO”.
Our first feature article, written by Jed Kuhn, takes a philosophical look at the evolution of the throwing shoulder, and invites the reader to help unlock some of the great shoulder anatomy mysteries by viewing them from a time when throwing was an activity of daily living. In ancient times, if you couldn’t throw, you couldn’t eat. We know that children who play baseball remodel their shoulder to allow for increased external rotation. Read Dr. Kuhn’s article and imagine when a shoulder optimized for throwing was a competitive advantage for survival.
Our second feature article is written by Stan Conte, a legend of the game and longtime trainer for the Los Angeles Dodgers. Dr. Conte studied injury trends in baseball over the past 18 seasons and provides an analysis of the staggering cost of placing players on the disabled list.
A baseball issue could not be complete without an article on Tommy John surgery. In this issue, AJO shares a revolutionary new technique for treating players with MUCL tears by author Jeffrey Dugas. Named the “Internal Brace”, Dr. Dugas shares his technique for augmenting the injured MUCL and we are proud to bring it to you first.
A recurring feature in the new AJO will be a section we refer to as “Codes to Know.” In partnership with Karen Zupko, AJO will present little-known coding secrets and proper coding techniques to help you get reimbursed appropriately for your work. This month, in the first article of a 3-part series, Alan Hirahara teaches us how to properly code for a diagnostic ultrasound examination of the shoulder. The article includes templates available for download to assist you with proper documentation. Parts 2 and 3 will provide a tutorial on the proper technique for examinations and injections.
While shoulder and elbow injuries get more attention, Major League Baseball’s Injury Panel has produced a look at the staggering amount of knee injuries over the 2011-2014 seasons, inspiring us to feature the knee in our 2 “Trade” Columns.
The “Tips of the Trade” column will continue, featuring this month a guide to identifying and treating meniscal root tears. A new segment, referred to as “Tools of the Trade,” reviews the latest products for all-inside meniscal repair. Our “Tools” section will feature announcements and reviews of the hottest new products, with a buying guide and surgical pearls from the surgeons who know them best.
While we are discussing the lower extremity, we should point out that we plan to do the “leg work” for you. Each AJO issue will have handouts that can be downloaded from our website and utilized in your practice. Read Robin West’s article entitled “Interval Throwing and Hitting Programs in Baseball: Biomechanics and Rehabilitation,” and download Return to Throwing and Hitting programs your patients and therapists can use.
Finally, I’d like to thank our previous Editor-in-Chief Dr. Peter McCann for his stewardship the last 10 years and recognize him for his dedication to the journal.
Thank you for reading AJO and for continuing to do so in the future. I know that collectively, we can turn AJO into a product worthy of its title. We know our past reputation. We are no longer that journal. Spend some time to get to know the “new AJO”, and make some room on your shelves, because the information between the covers will provide a template to implement new technologies and revenue streams into your practice and help fulfill your commitment to learning.
As orthopedic surgeons, we’ve made a commitment to lifelong learning. I can’t think of a single surgery that I perform the same way I did when I was in training. With rapidly evolving technology, continuously advancing procedures, and ever-increasing documentation requirements, it’s hard to stay on top of it all. We know your time is precious and that you have less of it than ever before. What little time you have that is not dedicated to work is reserved for your family or your hobbies. There’s no time to read every orthopedic journal, many filled with articles that have no practical value to your practice. That’s why we’ve created the new AJO. Our goal, as an editorial staff, is to provide a journal where every article, column, and feature contains information that directly benefits your practice, your patients, or your bottom line, and keeps you informed of the latest techniques, procedures, and products. We will help surgeons “work smarter, not harder,” implement new technologies into their practices, and find creative revenue streams that are both legal and compliant.
We’ve assembled a team of talented editors to accomplish this task, and will introduce them throughout the coming year. In this issue, you will meet our Deputy Editors-in-Chief and some of our new Associate Editors who’ve collaborated to bring you the “new AJO”.
At this year’s Academy, the AJO launched an extensive rebranding. We have a new look, a new logo, and a new creative directive. The journal will now feature new columns, invited articles, and innovative surgical techniques. We will publish 5 issues for the remainder of 2016. Our March/April issue is a special edition dedicated to baseball. In time for Spring Training/Opening Day, this issue includes articles from Major League Baseball’s physicians and trainers, a “Codes to Know” segment, “Tips of the Trade,” and a “Tools of the Trade” feature. “The Baseball Issue” will set the tone for what readers can expect from the “new AJO”.
Our first feature article, written by Jed Kuhn, takes a philosophical look at the evolution of the throwing shoulder, and invites the reader to help unlock some of the great shoulder anatomy mysteries by viewing them from a time when throwing was an activity of daily living. In ancient times, if you couldn’t throw, you couldn’t eat. We know that children who play baseball remodel their shoulder to allow for increased external rotation. Read Dr. Kuhn’s article and imagine when a shoulder optimized for throwing was a competitive advantage for survival.
Our second feature article is written by Stan Conte, a legend of the game and longtime trainer for the Los Angeles Dodgers. Dr. Conte studied injury trends in baseball over the past 18 seasons and provides an analysis of the staggering cost of placing players on the disabled list.
A baseball issue could not be complete without an article on Tommy John surgery. In this issue, AJO shares a revolutionary new technique for treating players with MUCL tears by author Jeffrey Dugas. Named the “Internal Brace”, Dr. Dugas shares his technique for augmenting the injured MUCL and we are proud to bring it to you first.
A recurring feature in the new AJO will be a section we refer to as “Codes to Know.” In partnership with Karen Zupko, AJO will present little-known coding secrets and proper coding techniques to help you get reimbursed appropriately for your work. This month, in the first article of a 3-part series, Alan Hirahara teaches us how to properly code for a diagnostic ultrasound examination of the shoulder. The article includes templates available for download to assist you with proper documentation. Parts 2 and 3 will provide a tutorial on the proper technique for examinations and injections.
While shoulder and elbow injuries get more attention, Major League Baseball’s Injury Panel has produced a look at the staggering amount of knee injuries over the 2011-2014 seasons, inspiring us to feature the knee in our 2 “Trade” Columns.
The “Tips of the Trade” column will continue, featuring this month a guide to identifying and treating meniscal root tears. A new segment, referred to as “Tools of the Trade,” reviews the latest products for all-inside meniscal repair. Our “Tools” section will feature announcements and reviews of the hottest new products, with a buying guide and surgical pearls from the surgeons who know them best.
While we are discussing the lower extremity, we should point out that we plan to do the “leg work” for you. Each AJO issue will have handouts that can be downloaded from our website and utilized in your practice. Read Robin West’s article entitled “Interval Throwing and Hitting Programs in Baseball: Biomechanics and Rehabilitation,” and download Return to Throwing and Hitting programs your patients and therapists can use.
Finally, I’d like to thank our previous Editor-in-Chief Dr. Peter McCann for his stewardship the last 10 years and recognize him for his dedication to the journal.
Thank you for reading AJO and for continuing to do so in the future. I know that collectively, we can turn AJO into a product worthy of its title. We know our past reputation. We are no longer that journal. Spend some time to get to know the “new AJO”, and make some room on your shelves, because the information between the covers will provide a template to implement new technologies and revenue streams into your practice and help fulfill your commitment to learning.

Piebaldism in Children
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
Practice Points
- Poliosis circumscripta (or white forelock) is commonly the only manifestation of piebaldism in children.
- Affected areas of leukoderma in piebaldism are classically distributed on the central forehead, anterior trunk, and mid extremities.
- The presence of congenital leukoderma should prompt a thorough skin examination and review of the patient’s medical history for evidence of ocular, auditory, and/or neurologic abnormalities.
Poverty promotes flu hospitalizations
Influenza-related hospitalizations were approximately twice as high among residents of areas where at least 20% of the population lived below the federal poverty level, compared with areas of less poverty, based on data from more than 27 million individuals in the United States.
Census and hospitalization data included 14 states and spanned two flu seasons (2010-2011 and 2011-2012). Overall, the incidence of flu-related hospitalizations was approximately 21 per 100,000 person-years in high-poverty areas, compared with approximately 11 per 100,000 person-years in census areas where less than 5% of the population lived below the poverty level. The data were consistent across all age groups and ethnicities.
Flu vaccination rates were inversely associated with poverty level, ranging from a high of 48% in high-income areas to a low of 35% in areas with the most poverty. This finding, however, was probably caused by lower vaccination rates among adults aged 65 years and older in higher-poverty areas, compared with high-income areas (80% vs. 94%) noted Dr. James L. Hadler of the Yale School of Public Health in New Haven, Conn., and colleagues.
“Enhanced influenza outreach to improve influenza vaccination coverage for persons living in poorer neighborhoods and efforts to increase use of antivirals by clinicians serving these neighborhoods could reduce poverty-related disparities in severe influenza outcomes,” the researchers noted.
The findings were published in the Morbidity and Mortality Weekly Report (MMWR 2016;65[5]:101-5). Read the full article here.
Influenza-related hospitalizations were approximately twice as high among residents of areas where at least 20% of the population lived below the federal poverty level, compared with areas of less poverty, based on data from more than 27 million individuals in the United States.
Census and hospitalization data included 14 states and spanned two flu seasons (2010-2011 and 2011-2012). Overall, the incidence of flu-related hospitalizations was approximately 21 per 100,000 person-years in high-poverty areas, compared with approximately 11 per 100,000 person-years in census areas where less than 5% of the population lived below the poverty level. The data were consistent across all age groups and ethnicities.
Flu vaccination rates were inversely associated with poverty level, ranging from a high of 48% in high-income areas to a low of 35% in areas with the most poverty. This finding, however, was probably caused by lower vaccination rates among adults aged 65 years and older in higher-poverty areas, compared with high-income areas (80% vs. 94%) noted Dr. James L. Hadler of the Yale School of Public Health in New Haven, Conn., and colleagues.
“Enhanced influenza outreach to improve influenza vaccination coverage for persons living in poorer neighborhoods and efforts to increase use of antivirals by clinicians serving these neighborhoods could reduce poverty-related disparities in severe influenza outcomes,” the researchers noted.
The findings were published in the Morbidity and Mortality Weekly Report (MMWR 2016;65[5]:101-5). Read the full article here.
Influenza-related hospitalizations were approximately twice as high among residents of areas where at least 20% of the population lived below the federal poverty level, compared with areas of less poverty, based on data from more than 27 million individuals in the United States.
Census and hospitalization data included 14 states and spanned two flu seasons (2010-2011 and 2011-2012). Overall, the incidence of flu-related hospitalizations was approximately 21 per 100,000 person-years in high-poverty areas, compared with approximately 11 per 100,000 person-years in census areas where less than 5% of the population lived below the poverty level. The data were consistent across all age groups and ethnicities.
Flu vaccination rates were inversely associated with poverty level, ranging from a high of 48% in high-income areas to a low of 35% in areas with the most poverty. This finding, however, was probably caused by lower vaccination rates among adults aged 65 years and older in higher-poverty areas, compared with high-income areas (80% vs. 94%) noted Dr. James L. Hadler of the Yale School of Public Health in New Haven, Conn., and colleagues.
“Enhanced influenza outreach to improve influenza vaccination coverage for persons living in poorer neighborhoods and efforts to increase use of antivirals by clinicians serving these neighborhoods could reduce poverty-related disparities in severe influenza outcomes,” the researchers noted.
The findings were published in the Morbidity and Mortality Weekly Report (MMWR 2016;65[5]:101-5). Read the full article here.
FROM MMWR
What Is Your Diagnosis? Stinkbug Staining
The Diagnosis: Stinkbug Staining
After discussing management options with the patient including biopsy, we decided that we would photograph the lesion and follow-up in clinic. While dressing, the patient discovered the source of the pigment, a stinkbug, stuck to the corresponding area of the sock.
The brown marmorated stinkbug (Halyomorpha halys)(Figure) is a member of the Pentatomidae family. This insect is native to East Asia and has become an invasive species in the United States. Their presence has recently increased in the eastern United States and they have become an important agricultural pest as well as a household nuisance. Stinkbugs most commonly interact with humans during the fall and winter months when they enter homes because of cooler temperatures outdoors. They can fit into many unexpected places because of their thin profile.1

Stinkbugs earned their name because of their defensive release of a malodorous chemical. This chemical is comprised of trans-2-decenal and trans-2-octenal, which are both aldehydes and are chemically related to formaldehyde. Based on the material safety data sheet, trans-2-decenal also may be responsible for the orange-brown color seen on the patient’s skin.2 Contact dermatitis caused by direct excretion of this chemical onto human skin has been reported3; anecdotal reports of irritation in agricultural workers have been noted. Stinkbugs are becoming a more common household and agricultural pest and should be recognized as possible causes of some presentations in the dermatology clinic.
- Nielsen AL, Hamilton GC. Seasonal occurrence and impact of Halyomorpha halys (Hemiptera: Pentatomidae) in tree fruit. J Econ Entomol. 2009;102:1133-1140.
- Material safety data sheet: trans-2-Decenal. https://fscimage.fishersci.com/msds/45077.htm. Published October 24, 1998. Updated November 20, 2008. Accessed January 11, 2016.
- Anderson BE, Miller JJ, Adams DR. Irritant contact dermatitis to the brown marmorated stink bug, Halyomorpha halys. Dermatitis. 2012;23:170-172.
The Diagnosis: Stinkbug Staining
After discussing management options with the patient including biopsy, we decided that we would photograph the lesion and follow-up in clinic. While dressing, the patient discovered the source of the pigment, a stinkbug, stuck to the corresponding area of the sock.
The brown marmorated stinkbug (Halyomorpha halys)(Figure) is a member of the Pentatomidae family. This insect is native to East Asia and has become an invasive species in the United States. Their presence has recently increased in the eastern United States and they have become an important agricultural pest as well as a household nuisance. Stinkbugs most commonly interact with humans during the fall and winter months when they enter homes because of cooler temperatures outdoors. They can fit into many unexpected places because of their thin profile.1
Stinkbugs earned their name because of their defensive release of a malodorous chemical. This chemical is comprised of trans-2-decenal and trans-2-octenal, which are both aldehydes and are chemically related to formaldehyde. Based on the material safety data sheet, trans-2-decenal also may be responsible for the orange-brown color seen on the patient’s skin.2 Contact dermatitis caused by direct excretion of this chemical onto human skin has been reported3; anecdotal reports of irritation in agricultural workers have been noted. Stinkbugs are becoming a more common household and agricultural pest and should be recognized as possible causes of some presentations in the dermatology clinic.
The Diagnosis: Stinkbug Staining
After discussing management options with the patient including biopsy, we decided that we would photograph the lesion and follow-up in clinic. While dressing, the patient discovered the source of the pigment, a stinkbug, stuck to the corresponding area of the sock.
The brown marmorated stinkbug (Halyomorpha halys)(Figure) is a member of the Pentatomidae family. This insect is native to East Asia and has become an invasive species in the United States. Their presence has recently increased in the eastern United States and they have become an important agricultural pest as well as a household nuisance. Stinkbugs most commonly interact with humans during the fall and winter months when they enter homes because of cooler temperatures outdoors. They can fit into many unexpected places because of their thin profile.1
Stinkbugs earned their name because of their defensive release of a malodorous chemical. This chemical is comprised of trans-2-decenal and trans-2-octenal, which are both aldehydes and are chemically related to formaldehyde. Based on the material safety data sheet, trans-2-decenal also may be responsible for the orange-brown color seen on the patient’s skin.2 Contact dermatitis caused by direct excretion of this chemical onto human skin has been reported3; anecdotal reports of irritation in agricultural workers have been noted. Stinkbugs are becoming a more common household and agricultural pest and should be recognized as possible causes of some presentations in the dermatology clinic.
- Nielsen AL, Hamilton GC. Seasonal occurrence and impact of Halyomorpha halys (Hemiptera: Pentatomidae) in tree fruit. J Econ Entomol. 2009;102:1133-1140.
- Material safety data sheet: trans-2-Decenal. https://fscimage.fishersci.com/msds/45077.htm. Published October 24, 1998. Updated November 20, 2008. Accessed January 11, 2016.
- Anderson BE, Miller JJ, Adams DR. Irritant contact dermatitis to the brown marmorated stink bug, Halyomorpha halys. Dermatitis. 2012;23:170-172.
- Nielsen AL, Hamilton GC. Seasonal occurrence and impact of Halyomorpha halys (Hemiptera: Pentatomidae) in tree fruit. J Econ Entomol. 2009;102:1133-1140.
- Material safety data sheet: trans-2-Decenal. https://fscimage.fishersci.com/msds/45077.htm. Published October 24, 1998. Updated November 20, 2008. Accessed January 11, 2016.
- Anderson BE, Miller JJ, Adams DR. Irritant contact dermatitis to the brown marmorated stink bug, Halyomorpha halys. Dermatitis. 2012;23:170-172.
A 56-year-old woman presented at the clinic for a total-body skin examination. A pigmented lesion was found on the medial aspect of the left first toe during the examination. The patient did not recognize this spot as a long-standing nevus. The area was scrubbed vigorously with an alcohol swab, which did not change the pigment. Clinically the lesion was concerning for an atypical nevus. Dermoscopic examination showed an unusual pattern with pigment deposition in ridges and on furrows.
Rare Neurological Disease Special Report
Click here to visit the digital edition.

Click here to visit the digital edition.
Click here to visit the digital edition.

Overuse of Antibiotics for Acne Vulgaris: Too Much of a Good Thing
In recent years, resistance to antimicrobial drugs has become increasingly widespread, resulting in a health threat of epidemic proportions. The long list of drug-resistant bacteria continues to expand at an accelerated pace. What does this mean in the dermatology world? We are not the only problem but are certainly part of the problem, representing 5% of all antibiotic prescriptions annually even though we represent only 1% of all physicians in the United States. These prescriptions certainly do not just include skin and soft tissue functions, as a survey-based study by Chouake et al (J Drugs Dermatol. 2014;13:119-124.) showed that dermatologists are overusing antibiotics in the treatment of simple skin abscesses such as acne vulgaris, one of the most common inflammatory skin diseases.
Although the inappropriate utilization of antibiotics for acne has been a subject of great discourse for years, it recently reentered the limelight in a study by Nagler et al published online in October 2015 in the Journal of the American Academy of Dermatology. They showed that patients who ultimately were treated with isotretinoin had been receiving antibiotics for months without any sign of therapeutic life or course end in sight. This retrospective chart review evaluated the duration of systemic antibiotic use prior to starting isotretinoin in 137 patients with inflammatory/nodulocystic acne. Antibiotic use continued for a mean of 331.3 days (median, 238 days). Duration of antibiotic use was divided into categories: 3 months or less (15.3%), 6 months or more (64.2%), or 1 year or more (33.6%).
Let’s take a broad look at antimicrobial resistance. Bacterial drug resistance has numerous negative effects on medicine and society. Drug-resistant bacterial infections result in higher doses of drugs, the addition of treatments with higher toxicity, longer hospital stays, and increased mortality. In the United States, infections due to antibiotic-resistant bacteria add $20 billion to total health care costs plus $35 billion in costs to society.
Unfortunately, it is relatively easy for bacterium to develop drug resistance through 3 simple steps: acquisition by microbes of resistance genes, expression of those resistance genes, and selection for pathogens expressing those resistance genes. The selective pressure in favor of resistance occurs whenever microbes are exposed to a drug but not eradicated, either by the killing effects of the drug itself or by inhibitory effects of the drug followed by killing by the host’s immune system. In any setting that creates this selective pressure in favor of drug resistance, such as poor patient compliance (ie, infrequent dosing, taking an antibiotic for too long as we see with the use of antibiotics for the treatment of inflammatory skin diseases such as acne), the likelihood of that resistance actually developing is increased. In addition, drugs that inhibit but do not kill microbes are more likely to allow some microbial cells to live and therefore develop resistance when exposed to a drug, which accounts for the majority of antibiotics in our armament. Lastly, abuse of broad-spectrum antibiotics has further spurred the emergence of many antibiotic-resistant strains. For instance, Pseudomonas aeruginosa is one of many evolving multidrug-resistant microorganisms that have been collectively coined the “ESKAPE” pathogens (Enterococcus faecalis, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, P aeruginosa, Enterobacter species) to emphasize the fact that they “escape” the effects of many antibacterial agents.
All of the above does not take into account the environmental factors that play a role in this resistance. The close quarters, mass/public transportation, and stressful pace of life of urban living not only bring these organisms together to share resistance genes but also increase our susceptibility.
What’s the issue?
We can all do our part in the fight against microbial resistance and join the antimicrobial stewardship. Here are a couple tips for dermatologists:
- Stop using over-the-counter antibiotic ointment for every biopsy or minor procedure, which is one of the recommendations of the American Academy of Dermatology based on the ABIM Foundation’s Choosing Wisely campaign.
- Oral and topical antibiotics for inflammatory skin diseases such as acne, rosacea, and hidradenitis suppurativa should only be used temporarily or at subantimicrobial dosing. Always combine a benzoyl peroxide–containing wash with a topical or oral antibiotic to hit the bacteria with multiple mechanisms of antibacterial activity to limit resistance. Don’t use benzoyl peroxide stronger than 2.5% for the face; make sure to wash it off completely to avoid staining your towels, sheets, and clothing.
We can all play our part in the fight against antimicrobial resistance. How do you fight the resistance?
Suggested Readings
Boucher HW. Challenges in anti-infective development in the era of bad bugs, no drugs: a regulatory perspective using the example of bloodstream infection as an indication. Clin Infect Dis. 2010;50(suppl 1):S4-S9.
Spellberg B, Guidos R, Gilbert D, et al. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:155-164.
In recent years, resistance to antimicrobial drugs has become increasingly widespread, resulting in a health threat of epidemic proportions. The long list of drug-resistant bacteria continues to expand at an accelerated pace. What does this mean in the dermatology world? We are not the only problem but are certainly part of the problem, representing 5% of all antibiotic prescriptions annually even though we represent only 1% of all physicians in the United States. These prescriptions certainly do not just include skin and soft tissue functions, as a survey-based study by Chouake et al (J Drugs Dermatol. 2014;13:119-124.) showed that dermatologists are overusing antibiotics in the treatment of simple skin abscesses such as acne vulgaris, one of the most common inflammatory skin diseases.
Although the inappropriate utilization of antibiotics for acne has been a subject of great discourse for years, it recently reentered the limelight in a study by Nagler et al published online in October 2015 in the Journal of the American Academy of Dermatology. They showed that patients who ultimately were treated with isotretinoin had been receiving antibiotics for months without any sign of therapeutic life or course end in sight. This retrospective chart review evaluated the duration of systemic antibiotic use prior to starting isotretinoin in 137 patients with inflammatory/nodulocystic acne. Antibiotic use continued for a mean of 331.3 days (median, 238 days). Duration of antibiotic use was divided into categories: 3 months or less (15.3%), 6 months or more (64.2%), or 1 year or more (33.6%).
Let’s take a broad look at antimicrobial resistance. Bacterial drug resistance has numerous negative effects on medicine and society. Drug-resistant bacterial infections result in higher doses of drugs, the addition of treatments with higher toxicity, longer hospital stays, and increased mortality. In the United States, infections due to antibiotic-resistant bacteria add $20 billion to total health care costs plus $35 billion in costs to society.
Unfortunately, it is relatively easy for bacterium to develop drug resistance through 3 simple steps: acquisition by microbes of resistance genes, expression of those resistance genes, and selection for pathogens expressing those resistance genes. The selective pressure in favor of resistance occurs whenever microbes are exposed to a drug but not eradicated, either by the killing effects of the drug itself or by inhibitory effects of the drug followed by killing by the host’s immune system. In any setting that creates this selective pressure in favor of drug resistance, such as poor patient compliance (ie, infrequent dosing, taking an antibiotic for too long as we see with the use of antibiotics for the treatment of inflammatory skin diseases such as acne), the likelihood of that resistance actually developing is increased. In addition, drugs that inhibit but do not kill microbes are more likely to allow some microbial cells to live and therefore develop resistance when exposed to a drug, which accounts for the majority of antibiotics in our armament. Lastly, abuse of broad-spectrum antibiotics has further spurred the emergence of many antibiotic-resistant strains. For instance, Pseudomonas aeruginosa is one of many evolving multidrug-resistant microorganisms that have been collectively coined the “ESKAPE” pathogens (Enterococcus faecalis, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, P aeruginosa, Enterobacter species) to emphasize the fact that they “escape” the effects of many antibacterial agents.
All of the above does not take into account the environmental factors that play a role in this resistance. The close quarters, mass/public transportation, and stressful pace of life of urban living not only bring these organisms together to share resistance genes but also increase our susceptibility.
What’s the issue?
We can all do our part in the fight against microbial resistance and join the antimicrobial stewardship. Here are a couple tips for dermatologists:
- Stop using over-the-counter antibiotic ointment for every biopsy or minor procedure, which is one of the recommendations of the American Academy of Dermatology based on the ABIM Foundation’s Choosing Wisely campaign.
- Oral and topical antibiotics for inflammatory skin diseases such as acne, rosacea, and hidradenitis suppurativa should only be used temporarily or at subantimicrobial dosing. Always combine a benzoyl peroxide–containing wash with a topical or oral antibiotic to hit the bacteria with multiple mechanisms of antibacterial activity to limit resistance. Don’t use benzoyl peroxide stronger than 2.5% for the face; make sure to wash it off completely to avoid staining your towels, sheets, and clothing.
We can all play our part in the fight against antimicrobial resistance. How do you fight the resistance?
In recent years, resistance to antimicrobial drugs has become increasingly widespread, resulting in a health threat of epidemic proportions. The long list of drug-resistant bacteria continues to expand at an accelerated pace. What does this mean in the dermatology world? We are not the only problem but are certainly part of the problem, representing 5% of all antibiotic prescriptions annually even though we represent only 1% of all physicians in the United States. These prescriptions certainly do not just include skin and soft tissue functions, as a survey-based study by Chouake et al (J Drugs Dermatol. 2014;13:119-124.) showed that dermatologists are overusing antibiotics in the treatment of simple skin abscesses such as acne vulgaris, one of the most common inflammatory skin diseases.
Although the inappropriate utilization of antibiotics for acne has been a subject of great discourse for years, it recently reentered the limelight in a study by Nagler et al published online in October 2015 in the Journal of the American Academy of Dermatology. They showed that patients who ultimately were treated with isotretinoin had been receiving antibiotics for months without any sign of therapeutic life or course end in sight. This retrospective chart review evaluated the duration of systemic antibiotic use prior to starting isotretinoin in 137 patients with inflammatory/nodulocystic acne. Antibiotic use continued for a mean of 331.3 days (median, 238 days). Duration of antibiotic use was divided into categories: 3 months or less (15.3%), 6 months or more (64.2%), or 1 year or more (33.6%).
Let’s take a broad look at antimicrobial resistance. Bacterial drug resistance has numerous negative effects on medicine and society. Drug-resistant bacterial infections result in higher doses of drugs, the addition of treatments with higher toxicity, longer hospital stays, and increased mortality. In the United States, infections due to antibiotic-resistant bacteria add $20 billion to total health care costs plus $35 billion in costs to society.
Unfortunately, it is relatively easy for bacterium to develop drug resistance through 3 simple steps: acquisition by microbes of resistance genes, expression of those resistance genes, and selection for pathogens expressing those resistance genes. The selective pressure in favor of resistance occurs whenever microbes are exposed to a drug but not eradicated, either by the killing effects of the drug itself or by inhibitory effects of the drug followed by killing by the host’s immune system. In any setting that creates this selective pressure in favor of drug resistance, such as poor patient compliance (ie, infrequent dosing, taking an antibiotic for too long as we see with the use of antibiotics for the treatment of inflammatory skin diseases such as acne), the likelihood of that resistance actually developing is increased. In addition, drugs that inhibit but do not kill microbes are more likely to allow some microbial cells to live and therefore develop resistance when exposed to a drug, which accounts for the majority of antibiotics in our armament. Lastly, abuse of broad-spectrum antibiotics has further spurred the emergence of many antibiotic-resistant strains. For instance, Pseudomonas aeruginosa is one of many evolving multidrug-resistant microorganisms that have been collectively coined the “ESKAPE” pathogens (Enterococcus faecalis, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, P aeruginosa, Enterobacter species) to emphasize the fact that they “escape” the effects of many antibacterial agents.
All of the above does not take into account the environmental factors that play a role in this resistance. The close quarters, mass/public transportation, and stressful pace of life of urban living not only bring these organisms together to share resistance genes but also increase our susceptibility.
What’s the issue?
We can all do our part in the fight against microbial resistance and join the antimicrobial stewardship. Here are a couple tips for dermatologists:
- Stop using over-the-counter antibiotic ointment for every biopsy or minor procedure, which is one of the recommendations of the American Academy of Dermatology based on the ABIM Foundation’s Choosing Wisely campaign.
- Oral and topical antibiotics for inflammatory skin diseases such as acne, rosacea, and hidradenitis suppurativa should only be used temporarily or at subantimicrobial dosing. Always combine a benzoyl peroxide–containing wash with a topical or oral antibiotic to hit the bacteria with multiple mechanisms of antibacterial activity to limit resistance. Don’t use benzoyl peroxide stronger than 2.5% for the face; make sure to wash it off completely to avoid staining your towels, sheets, and clothing.
We can all play our part in the fight against antimicrobial resistance. How do you fight the resistance?
Suggested Readings
Boucher HW. Challenges in anti-infective development in the era of bad bugs, no drugs: a regulatory perspective using the example of bloodstream infection as an indication. Clin Infect Dis. 2010;50(suppl 1):S4-S9.
Spellberg B, Guidos R, Gilbert D, et al. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:155-164.
Suggested Readings
Boucher HW. Challenges in anti-infective development in the era of bad bugs, no drugs: a regulatory perspective using the example of bloodstream infection as an indication. Clin Infect Dis. 2010;50(suppl 1):S4-S9.
Spellberg B, Guidos R, Gilbert D, et al. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:155-164.
