User login
cTn in Patients Hospitalized with ADHF
Acute decompensated heart failure (ADHF) accounts for over a million hospitalizations per year, with a reported all‐cause mortality rate 11.7% and all‐cause readmission rate 22.5% at 30 days after initial hospitalization.[1]
Risk stratification for accurate identification of ADHF patients at high risk for readmission and mortality may enable clinicians to undertake timely interventions: triage to appropriate level of care and resource allocation for postdischarge care. Further risk stratification may allow the care team to plan and implement a personalized care plan. Several clinical and laboratory variables have been proposed for identification of patients with ADHF who are at increased risk for adverse clinical outcomes. Despite advances in the risk stratification of patients with ADHF, the accurate prediction of individuals at high risk for readmissions and mortality is challenging. Cardiac troponin T (cTnT) and I (cTnI) are highly sensitive and specific biomarkers that are widely used for the risk stratification of patients with acute myocardial infarction and stable heart failure.[2]
In this systematic review and meta‐analysis, we evaluate circulating cardiac troponin in determining risk for increased length of stay (LOS), hospital readmission, and mortality among patients admitted with ADHF.
METHODS
Data Sources and Searches
This systematic review and meta‐analysis was conducted in accordance with the established methods[3] and Preferred Reporting Items for Systematic Review and Meta‐Analysis (PRISMA) guidelines.[4] Risk of bias was evaluated using the Newcastle‐Ottawa Scale for cohort studies.[5] We performed a comprehensive search of several databases from each database's earliest inception to March 2015 without language restrictions. The databases included MEDLINE In‐Process & Other Non‐Indexed Citations, MEDLINE, Embase, Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews, and Scopus. We conducted a manual search for bibliography of pertinent reviews for relevant citations that our electronic searches might have missed. The actual strategy is available from the corresponding author.
Study Selection
Eligibility criteria included: (1) randomized or nonrandomized clinical trials involving adults hospitalized with ADHF, (2) comparator groups stratified by cardiac troponin (cTn) level as defined by individual study investigators, and (3) studies reporting 1 or more of the following clinical outcomes: (1) in‐hospital mortality, (2) hospital LOS, (3) major adverse events during hospitalization (defined as persistent dyspnea,[6] worsening of heart failure,[6, 7, 8, 9] worsening of renal function [creatinine 0.3 mg/dL],[8] or recurrent myocardial ischemia[9] after hospitalization for ADHF), (4) postdischarge readmission, (5) postdischarge mortality rate, and (6) the composite of readmission and mortality. We excluded studies incorporating patients with (1) stable heart failure, (2) acute myocarditis, (3) chemotherapy‐induced cardiomyopathy, (4) postsurgical heart failure, (5) transplanted heart, (6) left ventricular assist device, and (7) hemodialysis.
We incorporated the description of ADHF from national registry for defining ADHF.[10] The lower limit of detection of cTn level in healthy subjects is assay dependent, each with a different cutoff value. To improve uniformity of expression in the present meta‐analysis, we arbitrarily stratified groups by the level of cTn: (1) undetected cTn (cTnT 0.01; cTnI 0.012 g/L), (2) detectable cTn (cTnT 0.010.03; cTnI 0.0120.03 g/L), and (3) elevated cTn (cTnT >0.03; cTnI >0.034 g/L).
Data Extraction and Risk of Bias Assessment
From the results of the initial search, 2 investigators (M.Y. and A.D.A), working independently, reviewed articles for eligibility on the basis of titles and abstracts. Studies that satisfied the inclusion and exclusion criteria were retrieved for full‐text review. Disagreements were resolved by consensus after discussion among investigators, and retained conflicts were adjudicated by a third investigator.
We extracted the following data from each study: type of study, number of participants, age, gender, type of cTn assayed and cut point, comorbidities, length of follow‐up, and outcome measure. Prevalence of detectable or elevated cTn, measure of association with clinical outcomes (hazard ratio [HR], odds ratio [OR], or relative risk) were also abstracted. When HR or OR were not reported for an outcome, based on other provided data, we estimated HR using previously validated methods.[11]
Data Synthesis and Analysis
Studies were stratified by cTn cutoff point and length of follow‐up. To reduce heterogeneity, studies reporting clinical outcomes at multiple time periods after the index hospitalization were grouped in three categories: (1) studies with short‐term follow up (06 months), (2) studies with intermediate‐term follow‐up (up to 1 year), and (3) studies with long‐term follow‐up (up to 3.5 years). We used the DerSimonian and Laird random effects model to combine OR or HR reported by individual studies. The consistency of the results of the studies was assessed by I2 statistics, with values >40% considered as indicators of heterogeneity. We evaluated statistically for publication bias if a sufficient number of studies was available, because such evaluation is unreliable when20 studies are included in a particular analysis.[12]
Sensitivity analyses were performed to investigate the robustness of results to a few assumptions. Analyses were repeated excluding studies reporting unadjusted relative effect measures to assess whether confounding had a large effect on overall results. Similarly, analysis was repeated omitting studies reporting detectable cTn as opposed to elevated cTn level to assess whether these studies influence overall results. All statistical analyses were conducted using Stata 14.0 (StataCorp, College Station, TX).
Quality and Risk of Bias Assessment
The Newcastle‐Ottawa scale was used to assess the quality and risk of bias in cohort studies as suggested by the Cochrane Collaboration.[13] For the assessment of risk of bias, a study was awarded a maximum of 1 star for each of the 7 items from 2 domains: (1) selection of cohort (representativeness of the exposed cohort, selection of the nonexposed cohort, ascertainment of exposure, and demonstration that the outcome of interest was not present at the start of the study) and (2) outcome (assessment of outcome, was the follow‐up long enough, adequacy of the follow‐up of the cohort), and a maximum of 2 stars for comparability of the cohort (comparability on the basis of design and analysis) (Table 1).
| Source | Year | Selection | Compatibility | Outcome | Quality | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 | S2 | S3 | S4 | C1 | C2 | O1 | O2 | O3 | |||
| |||||||||||
| Del Carlo et al.[31] | 2009 | * | * | * | * | * | 5 | ||||
| Felker et al.[6] | 2012 | * | * | * | * | * | 5 | ||||
| Gattis et al.[7] | 2004 | * | * | * | * | * | * | * | 7 | ||
| Guisado Espartero et al.[32] | 2014 | * | * | * | * | * | * | 6 | |||
| Ishii et al.[21] | 2002 | * | * | * | * | * | 5 | ||||
| Kuwabara et al.[22] | 2007 | * | * | * | * | * | 5 | ||||
| La Vecchia et al.[23] | 2000 | * | * | * | * | * | * | * | 7 | ||
| La Corvoisie et al.[17] | 2014 | * | * | * | * | * | * | * | 7 | ||
| Manzano‐Fernandez et al.[33] | 2009 | * | * | * | * | * | 5 | ||||
| Metra et al.[24] | 2007 | * | * | * | * | * | 5 | ||||
| Nakamura et al.[36] | 2014 | * | * | * | * | * | * | * | * | 8 | |
| O'Connor et al.[8] | 2011 | * | * | * | * | * | * | * | * | 8 | |
| Oliveira et al.[34] | 2010 | * | * | * | * | * | * | * | * | 8 | |
| Parissis et al.[20] | 2011 | * | * | * | * | * | * | * | 7 | ||
| Parissis et al.[25] | 2013 | * | * | * | * | * | * | * | * | 8 | |
| Pascual‐Figal et al.[37] | 2012 | * | * | * | * | * | * | 6 | |||
| Peacock et al.[15] | 2008 | * | * | * | * | * | * | * | * | * | 9 |
| Perna et al.[18] | 2005 | * | * | * | * | * | * | * | 7 | ||
| Perna et al.[19] | 2002 | * | * | * | * | * | * | 6 | |||
| Perna et al.[26] | 2012 | * | * | * | * | * | * | 6 | |||
| Rudiger et al.[27] | 2005 | * | * | * | * | * | * | 6 | |||
| Shah et al.[16] | 2007 | * | * | * | * | 4 | |||||
| Wallenborn et al.[28] | 2013 | * | * | * | * | * | * | * | 7 | ||
| Xue et al.[29] | 2011 | * | * | * | * | * | * | * | 7 | ||
| You et al.[35] | 2007 | * | * | * | * | * | * | * | * | * | 9 |
| Zairis et al.[30] | 2010 | * | * | * | * | * | * | * | 7 | ||
RESULTS
Search Results
Figure 1 represents the PRISMA flow diagram for literature search and selection process to identify eligible studies for inclusion.
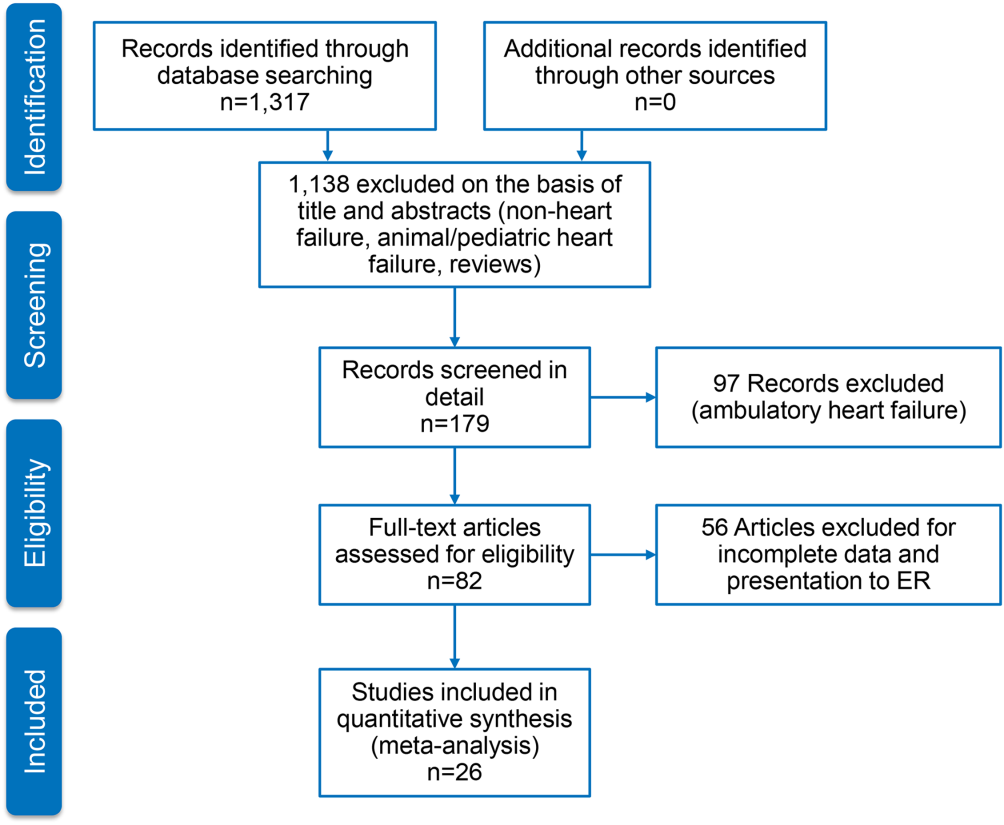
Characteristics of Included Studies
We identified 26 studies, which were all observational cohorts with postdischarge median follow‐up from 30 days to 472 days. Table 2 summarizes the study characteristics. Studies were heterogeneous with regard to prevalence of elevated cTn, cTn assay, and length of follow‐up. Thirteen were single‐center and 5 were multicenter studies, 4 were substudies of large multicenter phase III clinical trials, and 4 were registries. Except for one abstracts, all studies were peer‐reviewed publications. Sample size ranged from 34 to 69,259 patients.
| Source | Year | Design | Patient Population | CAD | HF Type | LVEF, Mean % | Clinical Outcomes | |||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of Patients | Age, y | Men, % | Follow‐up | Endpoints | ||||||
| ||||||||||
| Del Carlo et al.[31] | 2009 | Single | 70 | 54 16 | 69 | 26 | HFrEF | 31 8 | 262 (3393) days | Readmission, mortality |
| Felker et al.[6] | 2012 | Substudy | 808 | 67 | 70 | 61 | Both | 25 | Hosp, 30 days, 6 months | MAE, LOS, readmission, mortality |
| Gattis et al.[7] | 2004 | Substudy | 133 | NR | NR | NR | NR | NR | Hosp | MAE, mortality |
| Guisado Espartero et al.[32] | 2014 | Registry | 406 | 77 (7678) | 42 | 25 | Both | 50 (4456) | 1 year | Readmission, mortality |
| Ishii et al.[21] | 2002 | Single | 98 | 69 9 | 52 | 45 | NR | 42 17 | Hosp, 60 days, >1 year | Readmission, mortality |
| Kuwabara et al.[22] | 2007 | Single | 52 | 72 12 | 59 | 27 | NR | 47 16 | 143 (13540) days | Readmission, mortality |
| La Vecchia et al.[23] | 2000 | Single | 34 | 60 (2885) | 79 | 38 | HFrEF | NR | 90 days | Mortality |
| La Corvoisie et al.[17] | 2014 | Multicenter | 397 | NR | NR | NR | NR | Hosp | Mortality | |
| Manzano‐Fernandez et al.[33] | 2009 | Single | 138 | 74 (67801) | 54 | 35 | NR | NR | 261 (161449) days | Readmission, mortality |
| Metra et al.[24] | 2007 | Single | 116 | NR | NR | NR | NR | NR | 184 (7444) days | Readmission, mortality |
| Nakamura et al.[36] | 2014 | Single | 444 | NR | 63 | 15 | NR | NR | 472 (2001,200) days | Mortality |
| O'Connor et al.[8] | 2011 | Substudy | 288 | 73 (6577) | 59 | 74 | NR | NR | Hosp, 60 days | MAE, readmission, mortality |
| Oliveira et al.[34] | 2010 | Multicenter | 79 | NR | 61 | 18 | HFrEF | 27 | 8 months | MAE, readmission, mortality |
| Parissis et al.[20] | 2011 | Multicenter | 837 | NR | 48 | 20 | HFpEF | NR | Hosp | Mortality |
| Parissis et al.[25] | 2013 | Single | 113 | 73 11 | 68 | 46 | NR | 36 11 | 174 (94728) days | Mortality |
| Pascual‐Figal et al.[37] | 2012 | Single | 202 | 74 (6780) | 55 | 34 | Both | 49 (3260) | 406 (204728) days | Mortality |
| Peacock et al.[15] | 2008 | Registry | 69,259 | 74 14 | 45 | 56 | Both | 34 | Hosp | LOS, mortality |
| Perna et al.[18] | 2005 | Single | 184 | 64 13 | 60 | 38 | Both | NR | Hosp, 3 years | Readmission, mortality |
| Perna et al.[19] | 2002 | Single | 84 | 65 14 | 62 | 55 | NR | NR | Hosp, 1 year | Readmission, mortality |
| Perna et al.[26] | 2012 | Single | 500 | 73 12 | 53 | 38 | HFpEF | 53 11 | 6 months | Readmission, mortality |
| Rudiger et al.[27] | 2005 | Multicenter | 312 | 73 12 | 56 | 70 | Both | NR | 30 days, 1 year | Mortality |
| Shah et al.[16] | 2007 | Substudy | 141 | NR | NR | NR | HFrEF | NR | Hosp, 6 months | LOS, readmission, mortality |
| Wallenborn et al.[28] | 2013 | Registry | 879 | 69 12 | 72 | 50 | NR | 30 8 | 06 months, 618 months | Mortality |
| Xue et al.[29] | 2011 | Single | 144 | 68 13 | 98 | 62 | Both | 43 18 | 90 days | Readmission, mortality |
| You et al.[35] | 2007 | Registry | 2,025 | 76 11 | 50 | 55 | Both | NR | 1 year | Mortality |
| Zairis et al.[30] | 2010 | Multicenter | 577 | 74 8 | 68 | 77 | HFrEF | 23 5 | 31 days | Mortality |
Table 3 stratifies the characteristics of study populations by cTn status. The studies included 77,297 participants hospitalized for ADHF, of whom 7176 (9.3%) had detectable or elevated cTn level. Twenty‐five studies reported data on type of cTn measured (cTnI, cTnT, or both) and reported cutoff values for detectable or elevated cTn (Table 3). The percentages of patients who had detectable or elevated cTn varied widely across the studies (6.2%68%). Most studies utilized standard assays, and the cutoff point for cTn level was chosen arbitrary by study investigators or derived from receiver operating characteristic curve analysis. cTn level is assay dependent. For instance, the 99th centile upper reference limit (URL) is 0.014 ng/mL for cTnT with the Roche high‐sensitivity cTnT assay, and 0.04 ng/mL with the Siemens cTnI‐ultra assay. Few studies of the present meta‐analysis incorporated a cTn cutoff point that defined acute myocardial infarction.[14] Nine studies used a lower threshold cTn level (cTnT >0.01>0.03; cTnI >0.03) for stratification into comparator groups.
| Source | Year | No. of Patients | No. cTn+ (%) | cTn Cutoff | Age | Male | Atrial Fibrillation | CAD | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Tn+ | Tn | Tn+ (%) | No. (%) | Tn+ (%) | Tn+ (%) | |||||
| ||||||||||
| Del Carlo et al.[31] | 2009 | 70 | 12 (17) | cTnT 0.10 | NR | NR | NR | 13 (19) | NR | NR |
| Felker et al.[6] | 2012 | 808 | 404 (50) | cTnT 0.034 | 69 | 65 | 364 (54) | 334 (41) | 170 (42) | 243 (60) |
| Gattis et al.[7] | 2004 | 133 | 91 (68) | cTnT 1.0 | 70 (6180) | 77 (6282) | 46 (50) | NR | NR | NR |
| Guisado Espartero et al.[32] | 2014 | 406 | 241 (60) | cTnT 0.02 | NR | NR | 116 (48) | 236 (58) | 136 (56) | 74 (31) |
| Ishii et al.[21] | 2002 | 98 | NR | cTnT 0.1 | NR | NR | NR | NR | NR | NR |
| Kuwabara et al.[22] | 2007 | 52 | 31 (60) | NR | NR | NR | 31 (59) | 23 (44) | NR | NR |
| La Vecchia et al.[23] | 2000 | 34 | 10 (29) | cTnI 0.4 | 56 13 | 62 12 | 100 | 19 (56) | 5 (50) | 3 (30) |
| La Corvoisie et al.[17] | 2014 | 397 | NR | cTnI0.15 | NR | NR | NR | NR | NR | NR |
| Manzano‐Fernandez et al.[33] | 2009 | 138 | NR | cTnT 0.011 | NR | NR | NR | NR | NR | NR |
| Metra et al.[24] | 2007 | 116 | 41 (38) | cTnT 0.01 | NR | NR | NR | NR | NR | 33 (61) |
| Nakamura et al.[36] | 2014 | 444 | 224 (51) | cTnT 0.028 | 67 14 | 66 14 | 133 (60) | 160 (36) | 72 (32) | 35 (16) |
| O'Connor et al.[8] | 2011 | 288 | 97 (34) | cTnT 0.03 | 71 | 72 | 67 (69) | NR | NR | NR |
| Oliveira et al.[34] | 2010 | 79 | 37 (47) | ctnT 0.02 | 57 18 | 54 17 | 26 (70) | NR | NR | 6 (16) |
| Parissis et al.[20] | 2011 | 837 | 184 (22) | cTnT >0.01 | NR | NR | NR | NR | NR | NR |
| Parissis et al.[25] | 2013 | 113 | 37 (33) | cTnT 0.077 | 74 8 | 72 12 | 22 (59) | 36 (32) | 12 (32) | 18 (49) |
| Pascual‐Figal et al.[37] | 2012 | 202 | NR | cTnT >0.02 | NR | NR | NR | 109 (54) | NR | NR |
| Peacock et al.[15] | 2008 | 69,259 | 4,240 (6.2) | cTnI 1.0; cTnT 0.1 | 73 14 | 73 14 | 2,035 (48) | 207 (30) | 975 (23) | 2,586 (58) |
| Perna et al.[18] | 2005 | 184 | 58 (31) | cTnT 0.1 | 64 13 | 65 13 | 37 (64) | NR | NR | 30 (52) |
| Perna et al.[19] | 2002 | 84 | 46 (55) | cTnT 0.1 | 68 11 | 61 16 | 27 (59) | NR | NR | 33 (72) |
| Perna et al.[26] | 2012 | 500 | 220 (44) | cTnT 0.02 | 74 10 | 72 14 | 125 (59) | 177 (35) | 70 (32) | 110 (50) |
| Rudiger et al.[27] | 2005 | 312 | 88 (28) | cTnT 0.1 | NR | NR | NR | NR | NR | NR |
| Shah et al.[16] | 2007 | 141 | NR | cTnI per 0.1 | NR | NR | NR | NR | NR | NR |
| Wallenborn et al.[28] | 2013 | 879 | 332 (37) | cTnT 0.06 | NR | NR | NR | NR | NR | NR (50) |
| Xue et al.[29] | 2011 | 144 | NR | cTnI 0.023 | NR | NR | NR | NR | NR | NR |
| You et al.[35] | 2007 | 2,025 | 669 (34) | cTnI >0.5 | 77 11 | 75 11 | 364 (53) | NR | NR | 417 (60) |
| Zairis et al.[30] | 2010 | 577 | 114 (20) | cTnI 0.42 | NR | NR | NR | 295 (51) | NR | 443 (77) |
Twenty‐five studies reported performance of cTn as a dichotomized variable. A few studies, additionally, examined clinical outcome in patients grouped by tertiles by cTn and determined the dose‐response relationship using cTn as a continuous variable. The measure of association between cTn and clinical outcome was reported as HR or OR by 16 studies. The remaining 6 studies reported the number of clinical events in the groups by cTn level and therefore provided unadjusted estimates. The results of all meta‐analyses are depicted in Figure 2.

In‐hospital Clinical Outcomes
Three studies examined the association between cTn level and LOS.[6, 15, 16] One study (n = 808) found increased LOS among patients with elevated cTn.[6] Another study (n = 141), which tested the cTn level as a continuous variable, reported no statistically significant association between cTn level and LOS.[16] A large, multicenter ADHF registry (Acute Decompensated Heart Failure National Registry), which reported elevated cTn as a predictor of LOS (mean stay 6.6 vs 5.5 days; P 0.001) but did not provide binary data (OR, confidence interval [CI]), was therefore excluded from the meta‐analysis.[15] The pooled HRs from 2 studies revealed a significant increase in LOS in the cohort with elevated cTn (OR: 1.05, 95% CI: 1.01‐1.10, P = 0.06, I2 = 59.5.0%, n = 949). Six studies assessed in‐hospital mortality,[15, 17, 18, 19, 20, 21] and the meta‐analysis showed a significant increase in the risk of death with no significant heterogeneity (OR: 2.57, 95% CI: 2.27‐2.91, P = 0.744, I2 = 0.0%, n = 69,524). Similarly, 4 clinical studies[6, 7, 8, 9] found detectable or elevated cTn as a predictor of worsened composite clinical outcomes of death and major cardiovascular events (OR: 1.33, 95% CI: 1.03‐1.71, P = 0.473, I2 = 0.0%, n = 1,313).
Short‐term (0 to 6 Months) Clinical Outcomes
Short‐term clinical outcome was assessed in 13 studies.[6, 8, 16, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30] Nine studies addressed mortality,[6, 16, 23, 24, 25, 26, 27, 28, 30] 2 studies readmission,[16, 26] and 7 studies a composite of readmission and mortality during 6 months postdischarge.[6, 8, 21, 22, 24, 26, 29] The meta‐analysis showed increased mortality without significant heterogeneity (OR: 2.11, 95% CI: 1.43‐3.12, P = 0.000, I2 8.5%, 9 studies, n = 3471) and an increase in the composite of readmission and mortality with significant heterogeneity (OR: 2.81, 95%CI: 1.60‐4.92, P = 0.000, I2 89.1%, 7 studies, n = 2028) among ADHF patients with detectable or elevated cTn. The association between cTn level and readmission rate over 6 months post‐discharge did not reach statistical significance (OR: 1.00, 95% CI: 0.37‐2.74, P = 0.034, I2 77.9%, 2 studies, n = 641).
Intermediate‐term (Up to 12 Months) Clinical Outcomes
Intermediate‐term (during the 12 months postdischarge) clinical outcome was assessed in seven studies.[18, 24, 31, 32, 33, 34, 35] Five studies reported an association between cTn level and mortality.[18, 24, 32, 34, 35] The meta‐analysis demonstrated an increase in mortality with significant heterogeneity (OR: 2.21, 95% CI: 1.46‐3.35, P = 0.048, I2 58.4%, 5 studies, n = 2801). The pooled HRs of 2 studies examining the association between cTn and readmission rate[18, 32] did not yield statistical significance (OR: 1.55, 95% CI: 0.96‐2.52, P = 0.233, I2 29.6%, 2 studies, n = 590). A meta‐analysis of 5 studies that assessed an association between cTn and outcome[18, 24, 31, 32, 33] showed a significant increase in the risk of composite of readmission and mortality without significant heterogeneity (OR: 2.30, 95% CI: 1.78‐2.99, P = 0.666, I2 0.0%, 5 studies, n = 905) among patients with a detectable or elevated cTn.
Long‐term (>1 Year) Clinical Outcomes
Long‐term clinical outcome was assessed in 7 studies.[18, 19, 21, 24, 28, 36, 37] The meta‐analysis of 6 studies[18, 19, 21, 28, 36, 37] demonstrated an increase in mortality without significant heterogeneity (OR: 3.69, 95% CI: 2.64‐5.18, P = 0.696, I2 0.0%, 6 studies, n = 1891) among ADHF patients with a detectable or elevated cTn. Likewise, a composite of readmission rate and mortality was also increased (OR: 3.49, 95% CI: 2.08‐5.84, P = 0.070, I2 57.5%, 4 studies, n = 448) in a meta‐analysis of 4 studies.[18, 21, 24, 37] The meta‐analysis of 4 studies[18, 19, 24, 37] that assessed the association between cTn level and readmission rate over long‐term follow‐up showed no significant association (OR: 2.60, 95% CI: 0.80‐8.44, P = 0.000, I2 99.9 %, 4 studies, n = 576).
Confidence in the Estimates
Following the Grading of Recommendations Assessment, Development, and Evaluation approach to evaluate the confidence in the estimates from a systematic review and meta‐analysis[38] (ie, certainty or strength of evidence), we found that the association of a detectable or elevated troponin with mortality and readmission is moderate. This is due to a large effect (ie, relative association measure >2.0) demonstrated in observational studies. The confidence in the estimate of association with hospital LOS is low (smaller magnitude of effect). Analyses of in‐hospital outcomes were not associated with statistical heterogeneity, whereas several posthospital analyses had statistically significant heterogeneity.
DISCUSSION
We conducted a systematic review and meta‐analysis of published studies to assess the association between level of cTn and clinical outcomes including LOS, in‐hospital mortality, and short‐, intermediate‐, and long‐term readmission and death following index hospitalization for ADHF. The results of our meta‐analysis showed that compared with negative or not‐elevated cTn, detectable or elevated cTn was associated with increased LOS and higher rates of in‐hospital death among patients with ADHF. In addition, mortality and composite of mortality and readmission at short‐, intermediate‐ or long‐term after index hospitalization were greater in ADHF patients with a detectable or elevated cTn, as compared with those without elevated cTn, with significant heterogeneity across the studies. Finally, relatively fewer studies examined the association between cTn and readmission rate at multiple time periods after index hospitalization for ADHF, and these associations did not reach statistical significance.
In a review of 67,924 patients with ADHF from the US National Registry, which was limited in assessing inpatient mortality, Peacock et al. reported that a positive cardiac troponin test was associated with higher in‐hospital mortality, independently of other predictive variables.[15] We confirmed this observation in the present meta‐analysis, which also incorporated the study by Peacock et al. Furthermore, our data extended the findings of Peacock et al. to postdischarge readmission and death. The association between cTn and clinical outcomes was adjusted to multiple confounders across most studies included in the present meta‐analysis. Few studies in the present meta‐analysis showed a continuous and graded relationship between cTn level and clinical outcomes in patients with ADHF.[15, 35] Findings of previous studies showed that ADHF patients with persistently elevated cTn, measured at multiple time points during or following hospitalization, had worse clinical outcomes than did patients without similar elevation in cTn.[24, 29] Conversely, a decline in cTnT levels on serial measurements was associated with lower rates of adverse clinical outcomes, potentially through alleviation in ongoing myocardial injury.[39] Additionally, elevated cTn in ambulatory heart failure patients predicted incident hospitalization for acute decompensation.[40] Acute myocardial injury reflected by elevated cTn can be hypothesized to promote ventricular remodeling and thereby heart failure progression and consequent adverse clinical outcomes. Consistent with this hypothesis, a rise in cTn was observed in conjunction with elevated biological markers that characterize extracellular matrix remodeling in patients with heart failure during the acute and postacute phase.[41, 42]cTn is released in blood in direct proportion to myocardial injury.[43] A rise or fall in cTn with 1 value at or above the 99th centile URL in conjunction with clinical evidence of myocardial injury defines acute myocardial infarction.[14] Although patients with chronic stable heart failure often have chronically elevated cTn, those with ADHF may demonstrate an acute rise in cTn, with values reaching above the 99th percentile of URL in the absence of acute myocardial infarction.[15, 44] The pathophysiology of elevated cTn in ADHF is probably multifactorial.[14, 45, 46] The prevalence of elevated cTn in ADHF varied with assay sensitivity and the cutoff point chosen. For instance, in an analysis of >105,000 patients with ADHF, the prevalence of elevated cTn was increased from 6.2% with higher (cTnI >1.0 ng/mL or cTnT >0.1 ng/mL) to 75% with a lower cutoff point for cTn levels (cTnI >0.4 ng/mL and cTnT >0.01 ng/mL).[15]
In the general population with no established coronary artery disease, the prevalence of elevated cTn is contingent on sensitivity of the assay, age, and gender.[47, 48, 49, 50] Elevated cTn, beyond conventional risk factors, identifies a subgroup of individuals from the general population who are at high risk for incident heart failure and death.[51] Furthermore, elevated cTn is an independent predictor of short‐ and long‐term cardiovascular events in patients presenting to an emergency department (ED) for ADHF.[52, 53] In 2 large Canadian registries, an elevated cTn was associated with increased risk of death and cardiovascular readmissions at 30 days after ED visit.[53]
A number of recent studies have identified numerous other biomarkers as independent prognostic indicators in patients with heart failure. cTn, when combined with other biomarkers reflecting different dimensions of heart failure pathophysiology such as brain natriuretic peptide (BNP)/N‐terminal pro‐brain natriuretic peptide, soluble ST2, or cystatin C, enhanced the model's predictive utility beyond individual markers. For instance, patients with elevated cTn who also have increased BNP (840 pg/mL) had in‐hospital mortality of 10.2%, which was significantly greater than the 4.4% in patients with elevated BNP without detectable cTn.[54] Additionally, elevated cTn along with elevated pro‐brain natriuretic peptide and cystatin C has been reported to offer incremental prognostic information in patient with ADHF.[33]
The present systematic review and meta‐analysis is the most comprehensive to date and incorporated many observational cohorts with heterogeneous and unselected patient population. The studies have used various commercially available assays for the measurement of cTnT and cTnI. Therefore, findings of this meta‐analysis are applicable to a wider heart failure patient population. This review has several limitations. The association of elevated cTn and clinical outcome is likely affected by several confounders. Although we used adjusted estimates when possible, we did not have individual participant data. Due to the small number of included studies in each analysis, we could not explore heterogeneity causes using subgroup analysis or metaregression. For the same reasons, we could not statistically evaluate publication bias, which is likely in the setting of observational studies. The meta‐analysis is mainly driven by a few large studies.
In summary, in a broad spectrum of patients with ADHF, a detectable or elevated cTn is an independent predictor of major adverse clinical events not only during acute‐phase hospitalization but also after stabilization during the postdischarge phase. cTn is a widely available and inexpensive biomarker that provides important prognostic information and is likely to have important implications for in‐patient care and postdischarge surveillance of patients hospitalized for ADHF.
Disclosures
The authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers' bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent‐licensing arrangements), or nonfinancial interest (such as personal or professional relationships, affiliations, knowledge, or beliefs) in the subject matter or materials discussed in this article.
- Centers for Medicare 98:1778–1786.
- , , , et al. Meta‐analysis of observational studies in epidemiology: A proposal for reporting. Meta‐analysis of observational studies in epidemiology (moose) group. JAMA. 2000;283:2008–2012.
- , , , et al. The PRISMA statement for reporting systematic reviews and meta‐analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339:b2700.
- . Critical evaluation of the Newcastle‐Ottawa Scale for the assessment of the quality of nonrandomized studies in meta‐analyses. Eur J Epidemiol. 2010;25:603–605.
- , , , et al. Troponin I in acute decompensated heart failure: Insights from the ascend‐hf study. Eur J Heart Fail. 2012;14:1257–1264.
- , , , , , . Usefulness of an elevated troponin‐I in predicting clinical events in patients admitted with acute heart failure and acute coronary syndrome (from the RITZ‐4 trial). Am J Cardiol. 2004;93:1436–1437.
- , , , et al. Impact of serial troponin release on outcomes in patients with acute heart failure: analysis from the protect pilot study. Circulation. 2011;4:724–732.
- , , , et al. Ongoing myocardial injury in stable severe heart failure: value of cardiac troponin t monitoring for high‐risk patient identification. Circulation. 2004;110:2376–2382.
- , , , et al. Characteristics and outcomes of patients hospitalized for heart failure in the united states: rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J. 2005;149:209–216.
- , , . Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Stat Med. 1998;17:2815–2834.
- , , , , . The case of the misleading funnel plot. BMJ. 2006;333:597–600.
- , , , et al. The Newcastle‐Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta‐analyses. Available at: http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp. accessed July 3, 2015.
- , , , et al. Third universal definition of myocardial infarction. J Am Coll Cardiol. 2012;60:1581–1598.
- , , , et al.; ADHERE Investigators. Cardiac troponin and outcome in acute heart failure. N Engl J Med. 2008;358:2117–2126.
- , , , et al. Rapid assay brain natriuretic peptide and troponin I in patients hospitalized with decompensated heart failure (from the Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness trial). Am J Cardiol. 2007;100:1427–1433.
- , , , et al. Functional status and co‐morbidities are associated with in‐hospital mortality among older patients with acute decompensated heart failure: a multicentre prospective cohort study. Age Ageing. 2015;44(2):225–231.
- , , , et al. Minor myocardial damage detected by troponin T is a powerful predictor of long‐term prognosis in patients with acute decompensated heart failure. Int J Cardiol. 2005;99:253–261.
- , , , et al. Cardiac troponin T levels are associated with poor short‐ and long‐term prognosis in patients with acute cardiogenic pulmonary edema. Am Heart J. 2002;143:814–820.
- , , , et al. Clinical characteristics and predictors of in‐hospital mortality in acute heart failure with preserved left ventricular ejection fraction. Am J Cardiol. 2011;107:79–84.
- , , , et al. Risk stratification using a combination of cardiac troponin t and brain natriuretic peptide in patients hospitalized for worsening chronic heart failure. Am J Cardiol. 2002;89:691–695.
- , , , , et al. Persistently increased serum concentrations of cardiac troponin in patients with acutely decompensated heart failure are predictive of adverse outcomes. Circ J. 2007;71:1047–1051.
- , , , et al. Cardiac troponin I as diagnostic and prognostic marker in severe heart failure. J Heart Lung Transplant. 2000;19:644–652.
- , , , et al. The role of plasma biomarkers in acute heart failure. Serial changes and independent prognostic value of NT‐proBNP and cardiac troponin‐T. Eur J Heart Fail. 2007;9:776–786.
- , , , et al. Prognostic value of high sensitivity troponin T in patients with acutely decompensated heart failure and non‐detectable conventional troponin T levels. Int J Cardiol. 2013;168:3609–3612.
- , , , et al. Minor myocardial damage is a prevalent condition in patients with acute heart failure syndromes and preserved systolic function with long‐term prognostic implications: CIAST‐HF (Collaborative Italo‐Argentinean Study on cardiac Troponin T in Heart Failure) study. J Card Fail. 2012;18:822–830.
- , , , et al. Acute heart failure: clinical presentation, one‐year mortality and prognostic factors. Eur J Heart Fail. 2005;7:662–670.
- , , , et al. High‐sensitive troponin I after acute cardiac decompensation‐distribution of baseline values and prognostic significance. Paper presented at: Heart Failure Congress 2013; May 25–28, 2013; Lisbon, Portugal.
- , , , . Serial changes in high‐sensitive troponin I predict outcome in patients with decompensated heart failure. Eur J Heart Fail. 2011;13:37–42.
- , , , et al. Multimarker strategy for the prediction of 31 days cardiac death in patients with acutely decompensated chronic heart failure. Int J Cardiol. 2010;141:284–290.
- , , , , , . Cardiac troponin t for risk stratification in decompensated chronic heart failure. [in Portuguese]. Arq Bras Cardiol. 2009;92(5):404–412.
- , , , et al. Troponin T in acute heart failure: clinical implications and prognosis in the Spanish National Registry on Heart Failure. Eur J Intern Med. 2014;25:739–744.
- , , , et al. Complementary prognostic value of cystatin C, N‐terminal pro‐B‐type natriuretic Peptide and cardiac troponin T in patients with acute heart failure. Am J Cardiol. 2009;103:1753–1759.
- , , . Single cardiac troponin t measurement predicts risk for adverse outcome in decompensated heart failure. Arq Bras Cardiol. 2010;94(4):495–501.
- , , , , . Relation between cardiac troponin i and mortality in acute decompensated heart failure. Am Heart J. 2007;153:462–470.
- , , , et al. High‐sensitivity cardiac troponin t predicts non‐cardiac mortality in heart failure. Circ J. 2014;78:890–895.
- , , , et al. Highly sensitive troponin t for risk stratification of acutely destabilized heart failure. Am Heart J. 2012;163(6):1002–1010.
- , , , et al. How to read a systematic review and meta‐analysis and apply the results to patient care: Users' guides to the medical literature. JAMA. 2014;312:171–179.
- , , , et al. Serial biomarker measurements in ambulatory patients with chronic heart failure: the importance of change over time. Circulation. 2007;116:249–257.
- , , , et al. Prognostic value of very low plasma concentrations of troponin T in patients with stable chronic heart failure. Circulation. 2007;116:1242–1249.
- , , , et al. Episodes of acute heart failure syndrome are associated with increased levels of troponin and extracellular matrix markers. Circ Heart Fail. 2010;3:44–50.
- , , , et al. Cardiac microinjury measured by troponin T predicts collagen metabolism in adults aged >=65 years with heart failure. Circ Heart Fail. 2012;5:406–413.
- . Pathobiology of troponin elevations: so elevations occur with myocardial ischemia as well as necrosis? J Am Coll Cardiol. 2011;57:2406–2408.
- , , . Prognostic value of cardiac troponin in chronic stable heart failure: a systematic review. Heart. 2012;98:1778–1786.
- , , , et al. Vascular endothelial dysfunction and mortality risk in patients with chronic heart failure. Circulation. 2005;111:310–314.
- , , , , . Preload induces troponin I degradation independently of myocardial ischemia. Circulation. 2001;103:2035–2037.
- , , ,et al. Prevalence and determinants of troponin T elevation in the general population. Circulation. 2006;113:1958–1965.
- , , , et al. Cardiac troponin‐I and risk of heart failure: a community‐based cohort study. Eur Heart J. 2009;30:773–781.
- , , , et al. Age‐ and sex‐dependent upper reference limits for the high‐sensitivity cardiac troponin t assay. J Am Coll Cardiol. 2014;63:1441–1448.
- , , , et al. Defining high‐sensitivity cardiac troponin concentrations in the community. Clin Chem. 2013;59:1099–1107.
- , , , et al. Association of serial measures of cardiac troponin T using a sensitive assay with incident heart failure and cardiovascular mortality in older adults. JAMA. 2010;304:2494–2502.
- , , , et al. Sensitive cardiac troponin in the diagnosis and risk stratification of acute heart failure. J Intern Med. 2012;271:598–607.
- , , , et al. Outcomes and care of patients with acute heart failure syndromes and cardiac troponin elevation. Circulation. 2013;6:193–202.
- , , , et al.; ADHERE Scientific Advisory Committee and Investigators. Usefulness of B‐type natriuretic peptide and cardiac troponin levels to predict in‐hospital mortality from ADHERE. Am J Cardiol. 2008;101:231–237.
Acute decompensated heart failure (ADHF) accounts for over a million hospitalizations per year, with a reported all‐cause mortality rate 11.7% and all‐cause readmission rate 22.5% at 30 days after initial hospitalization.[1]
Risk stratification for accurate identification of ADHF patients at high risk for readmission and mortality may enable clinicians to undertake timely interventions: triage to appropriate level of care and resource allocation for postdischarge care. Further risk stratification may allow the care team to plan and implement a personalized care plan. Several clinical and laboratory variables have been proposed for identification of patients with ADHF who are at increased risk for adverse clinical outcomes. Despite advances in the risk stratification of patients with ADHF, the accurate prediction of individuals at high risk for readmissions and mortality is challenging. Cardiac troponin T (cTnT) and I (cTnI) are highly sensitive and specific biomarkers that are widely used for the risk stratification of patients with acute myocardial infarction and stable heart failure.[2]
In this systematic review and meta‐analysis, we evaluate circulating cardiac troponin in determining risk for increased length of stay (LOS), hospital readmission, and mortality among patients admitted with ADHF.
METHODS
Data Sources and Searches
This systematic review and meta‐analysis was conducted in accordance with the established methods[3] and Preferred Reporting Items for Systematic Review and Meta‐Analysis (PRISMA) guidelines.[4] Risk of bias was evaluated using the Newcastle‐Ottawa Scale for cohort studies.[5] We performed a comprehensive search of several databases from each database's earliest inception to March 2015 without language restrictions. The databases included MEDLINE In‐Process & Other Non‐Indexed Citations, MEDLINE, Embase, Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews, and Scopus. We conducted a manual search for bibliography of pertinent reviews for relevant citations that our electronic searches might have missed. The actual strategy is available from the corresponding author.
Study Selection
Eligibility criteria included: (1) randomized or nonrandomized clinical trials involving adults hospitalized with ADHF, (2) comparator groups stratified by cardiac troponin (cTn) level as defined by individual study investigators, and (3) studies reporting 1 or more of the following clinical outcomes: (1) in‐hospital mortality, (2) hospital LOS, (3) major adverse events during hospitalization (defined as persistent dyspnea,[6] worsening of heart failure,[6, 7, 8, 9] worsening of renal function [creatinine 0.3 mg/dL],[8] or recurrent myocardial ischemia[9] after hospitalization for ADHF), (4) postdischarge readmission, (5) postdischarge mortality rate, and (6) the composite of readmission and mortality. We excluded studies incorporating patients with (1) stable heart failure, (2) acute myocarditis, (3) chemotherapy‐induced cardiomyopathy, (4) postsurgical heart failure, (5) transplanted heart, (6) left ventricular assist device, and (7) hemodialysis.
We incorporated the description of ADHF from national registry for defining ADHF.[10] The lower limit of detection of cTn level in healthy subjects is assay dependent, each with a different cutoff value. To improve uniformity of expression in the present meta‐analysis, we arbitrarily stratified groups by the level of cTn: (1) undetected cTn (cTnT 0.01; cTnI 0.012 g/L), (2) detectable cTn (cTnT 0.010.03; cTnI 0.0120.03 g/L), and (3) elevated cTn (cTnT >0.03; cTnI >0.034 g/L).
Data Extraction and Risk of Bias Assessment
From the results of the initial search, 2 investigators (M.Y. and A.D.A), working independently, reviewed articles for eligibility on the basis of titles and abstracts. Studies that satisfied the inclusion and exclusion criteria were retrieved for full‐text review. Disagreements were resolved by consensus after discussion among investigators, and retained conflicts were adjudicated by a third investigator.
We extracted the following data from each study: type of study, number of participants, age, gender, type of cTn assayed and cut point, comorbidities, length of follow‐up, and outcome measure. Prevalence of detectable or elevated cTn, measure of association with clinical outcomes (hazard ratio [HR], odds ratio [OR], or relative risk) were also abstracted. When HR or OR were not reported for an outcome, based on other provided data, we estimated HR using previously validated methods.[11]
Data Synthesis and Analysis
Studies were stratified by cTn cutoff point and length of follow‐up. To reduce heterogeneity, studies reporting clinical outcomes at multiple time periods after the index hospitalization were grouped in three categories: (1) studies with short‐term follow up (06 months), (2) studies with intermediate‐term follow‐up (up to 1 year), and (3) studies with long‐term follow‐up (up to 3.5 years). We used the DerSimonian and Laird random effects model to combine OR or HR reported by individual studies. The consistency of the results of the studies was assessed by I2 statistics, with values >40% considered as indicators of heterogeneity. We evaluated statistically for publication bias if a sufficient number of studies was available, because such evaluation is unreliable when20 studies are included in a particular analysis.[12]
Sensitivity analyses were performed to investigate the robustness of results to a few assumptions. Analyses were repeated excluding studies reporting unadjusted relative effect measures to assess whether confounding had a large effect on overall results. Similarly, analysis was repeated omitting studies reporting detectable cTn as opposed to elevated cTn level to assess whether these studies influence overall results. All statistical analyses were conducted using Stata 14.0 (StataCorp, College Station, TX).
Quality and Risk of Bias Assessment
The Newcastle‐Ottawa scale was used to assess the quality and risk of bias in cohort studies as suggested by the Cochrane Collaboration.[13] For the assessment of risk of bias, a study was awarded a maximum of 1 star for each of the 7 items from 2 domains: (1) selection of cohort (representativeness of the exposed cohort, selection of the nonexposed cohort, ascertainment of exposure, and demonstration that the outcome of interest was not present at the start of the study) and (2) outcome (assessment of outcome, was the follow‐up long enough, adequacy of the follow‐up of the cohort), and a maximum of 2 stars for comparability of the cohort (comparability on the basis of design and analysis) (Table 1).
| Source | Year | Selection | Compatibility | Outcome | Quality | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 | S2 | S3 | S4 | C1 | C2 | O1 | O2 | O3 | |||
| |||||||||||
| Del Carlo et al.[31] | 2009 | * | * | * | * | * | 5 | ||||
| Felker et al.[6] | 2012 | * | * | * | * | * | 5 | ||||
| Gattis et al.[7] | 2004 | * | * | * | * | * | * | * | 7 | ||
| Guisado Espartero et al.[32] | 2014 | * | * | * | * | * | * | 6 | |||
| Ishii et al.[21] | 2002 | * | * | * | * | * | 5 | ||||
| Kuwabara et al.[22] | 2007 | * | * | * | * | * | 5 | ||||
| La Vecchia et al.[23] | 2000 | * | * | * | * | * | * | * | 7 | ||
| La Corvoisie et al.[17] | 2014 | * | * | * | * | * | * | * | 7 | ||
| Manzano‐Fernandez et al.[33] | 2009 | * | * | * | * | * | 5 | ||||
| Metra et al.[24] | 2007 | * | * | * | * | * | 5 | ||||
| Nakamura et al.[36] | 2014 | * | * | * | * | * | * | * | * | 8 | |
| O'Connor et al.[8] | 2011 | * | * | * | * | * | * | * | * | 8 | |
| Oliveira et al.[34] | 2010 | * | * | * | * | * | * | * | * | 8 | |
| Parissis et al.[20] | 2011 | * | * | * | * | * | * | * | 7 | ||
| Parissis et al.[25] | 2013 | * | * | * | * | * | * | * | * | 8 | |
| Pascual‐Figal et al.[37] | 2012 | * | * | * | * | * | * | 6 | |||
| Peacock et al.[15] | 2008 | * | * | * | * | * | * | * | * | * | 9 |
| Perna et al.[18] | 2005 | * | * | * | * | * | * | * | 7 | ||
| Perna et al.[19] | 2002 | * | * | * | * | * | * | 6 | |||
| Perna et al.[26] | 2012 | * | * | * | * | * | * | 6 | |||
| Rudiger et al.[27] | 2005 | * | * | * | * | * | * | 6 | |||
| Shah et al.[16] | 2007 | * | * | * | * | 4 | |||||
| Wallenborn et al.[28] | 2013 | * | * | * | * | * | * | * | 7 | ||
| Xue et al.[29] | 2011 | * | * | * | * | * | * | * | 7 | ||
| You et al.[35] | 2007 | * | * | * | * | * | * | * | * | * | 9 |
| Zairis et al.[30] | 2010 | * | * | * | * | * | * | * | 7 | ||
RESULTS
Search Results
Figure 1 represents the PRISMA flow diagram for literature search and selection process to identify eligible studies for inclusion.
Characteristics of Included Studies
We identified 26 studies, which were all observational cohorts with postdischarge median follow‐up from 30 days to 472 days. Table 2 summarizes the study characteristics. Studies were heterogeneous with regard to prevalence of elevated cTn, cTn assay, and length of follow‐up. Thirteen were single‐center and 5 were multicenter studies, 4 were substudies of large multicenter phase III clinical trials, and 4 were registries. Except for one abstracts, all studies were peer‐reviewed publications. Sample size ranged from 34 to 69,259 patients.
| Source | Year | Design | Patient Population | CAD | HF Type | LVEF, Mean % | Clinical Outcomes | |||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of Patients | Age, y | Men, % | Follow‐up | Endpoints | ||||||
| ||||||||||
| Del Carlo et al.[31] | 2009 | Single | 70 | 54 16 | 69 | 26 | HFrEF | 31 8 | 262 (3393) days | Readmission, mortality |
| Felker et al.[6] | 2012 | Substudy | 808 | 67 | 70 | 61 | Both | 25 | Hosp, 30 days, 6 months | MAE, LOS, readmission, mortality |
| Gattis et al.[7] | 2004 | Substudy | 133 | NR | NR | NR | NR | NR | Hosp | MAE, mortality |
| Guisado Espartero et al.[32] | 2014 | Registry | 406 | 77 (7678) | 42 | 25 | Both | 50 (4456) | 1 year | Readmission, mortality |
| Ishii et al.[21] | 2002 | Single | 98 | 69 9 | 52 | 45 | NR | 42 17 | Hosp, 60 days, >1 year | Readmission, mortality |
| Kuwabara et al.[22] | 2007 | Single | 52 | 72 12 | 59 | 27 | NR | 47 16 | 143 (13540) days | Readmission, mortality |
| La Vecchia et al.[23] | 2000 | Single | 34 | 60 (2885) | 79 | 38 | HFrEF | NR | 90 days | Mortality |
| La Corvoisie et al.[17] | 2014 | Multicenter | 397 | NR | NR | NR | NR | Hosp | Mortality | |
| Manzano‐Fernandez et al.[33] | 2009 | Single | 138 | 74 (67801) | 54 | 35 | NR | NR | 261 (161449) days | Readmission, mortality |
| Metra et al.[24] | 2007 | Single | 116 | NR | NR | NR | NR | NR | 184 (7444) days | Readmission, mortality |
| Nakamura et al.[36] | 2014 | Single | 444 | NR | 63 | 15 | NR | NR | 472 (2001,200) days | Mortality |
| O'Connor et al.[8] | 2011 | Substudy | 288 | 73 (6577) | 59 | 74 | NR | NR | Hosp, 60 days | MAE, readmission, mortality |
| Oliveira et al.[34] | 2010 | Multicenter | 79 | NR | 61 | 18 | HFrEF | 27 | 8 months | MAE, readmission, mortality |
| Parissis et al.[20] | 2011 | Multicenter | 837 | NR | 48 | 20 | HFpEF | NR | Hosp | Mortality |
| Parissis et al.[25] | 2013 | Single | 113 | 73 11 | 68 | 46 | NR | 36 11 | 174 (94728) days | Mortality |
| Pascual‐Figal et al.[37] | 2012 | Single | 202 | 74 (6780) | 55 | 34 | Both | 49 (3260) | 406 (204728) days | Mortality |
| Peacock et al.[15] | 2008 | Registry | 69,259 | 74 14 | 45 | 56 | Both | 34 | Hosp | LOS, mortality |
| Perna et al.[18] | 2005 | Single | 184 | 64 13 | 60 | 38 | Both | NR | Hosp, 3 years | Readmission, mortality |
| Perna et al.[19] | 2002 | Single | 84 | 65 14 | 62 | 55 | NR | NR | Hosp, 1 year | Readmission, mortality |
| Perna et al.[26] | 2012 | Single | 500 | 73 12 | 53 | 38 | HFpEF | 53 11 | 6 months | Readmission, mortality |
| Rudiger et al.[27] | 2005 | Multicenter | 312 | 73 12 | 56 | 70 | Both | NR | 30 days, 1 year | Mortality |
| Shah et al.[16] | 2007 | Substudy | 141 | NR | NR | NR | HFrEF | NR | Hosp, 6 months | LOS, readmission, mortality |
| Wallenborn et al.[28] | 2013 | Registry | 879 | 69 12 | 72 | 50 | NR | 30 8 | 06 months, 618 months | Mortality |
| Xue et al.[29] | 2011 | Single | 144 | 68 13 | 98 | 62 | Both | 43 18 | 90 days | Readmission, mortality |
| You et al.[35] | 2007 | Registry | 2,025 | 76 11 | 50 | 55 | Both | NR | 1 year | Mortality |
| Zairis et al.[30] | 2010 | Multicenter | 577 | 74 8 | 68 | 77 | HFrEF | 23 5 | 31 days | Mortality |
Table 3 stratifies the characteristics of study populations by cTn status. The studies included 77,297 participants hospitalized for ADHF, of whom 7176 (9.3%) had detectable or elevated cTn level. Twenty‐five studies reported data on type of cTn measured (cTnI, cTnT, or both) and reported cutoff values for detectable or elevated cTn (Table 3). The percentages of patients who had detectable or elevated cTn varied widely across the studies (6.2%68%). Most studies utilized standard assays, and the cutoff point for cTn level was chosen arbitrary by study investigators or derived from receiver operating characteristic curve analysis. cTn level is assay dependent. For instance, the 99th centile upper reference limit (URL) is 0.014 ng/mL for cTnT with the Roche high‐sensitivity cTnT assay, and 0.04 ng/mL with the Siemens cTnI‐ultra assay. Few studies of the present meta‐analysis incorporated a cTn cutoff point that defined acute myocardial infarction.[14] Nine studies used a lower threshold cTn level (cTnT >0.01>0.03; cTnI >0.03) for stratification into comparator groups.
| Source | Year | No. of Patients | No. cTn+ (%) | cTn Cutoff | Age | Male | Atrial Fibrillation | CAD | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Tn+ | Tn | Tn+ (%) | No. (%) | Tn+ (%) | Tn+ (%) | |||||
| ||||||||||
| Del Carlo et al.[31] | 2009 | 70 | 12 (17) | cTnT 0.10 | NR | NR | NR | 13 (19) | NR | NR |
| Felker et al.[6] | 2012 | 808 | 404 (50) | cTnT 0.034 | 69 | 65 | 364 (54) | 334 (41) | 170 (42) | 243 (60) |
| Gattis et al.[7] | 2004 | 133 | 91 (68) | cTnT 1.0 | 70 (6180) | 77 (6282) | 46 (50) | NR | NR | NR |
| Guisado Espartero et al.[32] | 2014 | 406 | 241 (60) | cTnT 0.02 | NR | NR | 116 (48) | 236 (58) | 136 (56) | 74 (31) |
| Ishii et al.[21] | 2002 | 98 | NR | cTnT 0.1 | NR | NR | NR | NR | NR | NR |
| Kuwabara et al.[22] | 2007 | 52 | 31 (60) | NR | NR | NR | 31 (59) | 23 (44) | NR | NR |
| La Vecchia et al.[23] | 2000 | 34 | 10 (29) | cTnI 0.4 | 56 13 | 62 12 | 100 | 19 (56) | 5 (50) | 3 (30) |
| La Corvoisie et al.[17] | 2014 | 397 | NR | cTnI0.15 | NR | NR | NR | NR | NR | NR |
| Manzano‐Fernandez et al.[33] | 2009 | 138 | NR | cTnT 0.011 | NR | NR | NR | NR | NR | NR |
| Metra et al.[24] | 2007 | 116 | 41 (38) | cTnT 0.01 | NR | NR | NR | NR | NR | 33 (61) |
| Nakamura et al.[36] | 2014 | 444 | 224 (51) | cTnT 0.028 | 67 14 | 66 14 | 133 (60) | 160 (36) | 72 (32) | 35 (16) |
| O'Connor et al.[8] | 2011 | 288 | 97 (34) | cTnT 0.03 | 71 | 72 | 67 (69) | NR | NR | NR |
| Oliveira et al.[34] | 2010 | 79 | 37 (47) | ctnT 0.02 | 57 18 | 54 17 | 26 (70) | NR | NR | 6 (16) |
| Parissis et al.[20] | 2011 | 837 | 184 (22) | cTnT >0.01 | NR | NR | NR | NR | NR | NR |
| Parissis et al.[25] | 2013 | 113 | 37 (33) | cTnT 0.077 | 74 8 | 72 12 | 22 (59) | 36 (32) | 12 (32) | 18 (49) |
| Pascual‐Figal et al.[37] | 2012 | 202 | NR | cTnT >0.02 | NR | NR | NR | 109 (54) | NR | NR |
| Peacock et al.[15] | 2008 | 69,259 | 4,240 (6.2) | cTnI 1.0; cTnT 0.1 | 73 14 | 73 14 | 2,035 (48) | 207 (30) | 975 (23) | 2,586 (58) |
| Perna et al.[18] | 2005 | 184 | 58 (31) | cTnT 0.1 | 64 13 | 65 13 | 37 (64) | NR | NR | 30 (52) |
| Perna et al.[19] | 2002 | 84 | 46 (55) | cTnT 0.1 | 68 11 | 61 16 | 27 (59) | NR | NR | 33 (72) |
| Perna et al.[26] | 2012 | 500 | 220 (44) | cTnT 0.02 | 74 10 | 72 14 | 125 (59) | 177 (35) | 70 (32) | 110 (50) |
| Rudiger et al.[27] | 2005 | 312 | 88 (28) | cTnT 0.1 | NR | NR | NR | NR | NR | NR |
| Shah et al.[16] | 2007 | 141 | NR | cTnI per 0.1 | NR | NR | NR | NR | NR | NR |
| Wallenborn et al.[28] | 2013 | 879 | 332 (37) | cTnT 0.06 | NR | NR | NR | NR | NR | NR (50) |
| Xue et al.[29] | 2011 | 144 | NR | cTnI 0.023 | NR | NR | NR | NR | NR | NR |
| You et al.[35] | 2007 | 2,025 | 669 (34) | cTnI >0.5 | 77 11 | 75 11 | 364 (53) | NR | NR | 417 (60) |
| Zairis et al.[30] | 2010 | 577 | 114 (20) | cTnI 0.42 | NR | NR | NR | 295 (51) | NR | 443 (77) |
Twenty‐five studies reported performance of cTn as a dichotomized variable. A few studies, additionally, examined clinical outcome in patients grouped by tertiles by cTn and determined the dose‐response relationship using cTn as a continuous variable. The measure of association between cTn and clinical outcome was reported as HR or OR by 16 studies. The remaining 6 studies reported the number of clinical events in the groups by cTn level and therefore provided unadjusted estimates. The results of all meta‐analyses are depicted in Figure 2.
In‐hospital Clinical Outcomes
Three studies examined the association between cTn level and LOS.[6, 15, 16] One study (n = 808) found increased LOS among patients with elevated cTn.[6] Another study (n = 141), which tested the cTn level as a continuous variable, reported no statistically significant association between cTn level and LOS.[16] A large, multicenter ADHF registry (Acute Decompensated Heart Failure National Registry), which reported elevated cTn as a predictor of LOS (mean stay 6.6 vs 5.5 days; P 0.001) but did not provide binary data (OR, confidence interval [CI]), was therefore excluded from the meta‐analysis.[15] The pooled HRs from 2 studies revealed a significant increase in LOS in the cohort with elevated cTn (OR: 1.05, 95% CI: 1.01‐1.10, P = 0.06, I2 = 59.5.0%, n = 949). Six studies assessed in‐hospital mortality,[15, 17, 18, 19, 20, 21] and the meta‐analysis showed a significant increase in the risk of death with no significant heterogeneity (OR: 2.57, 95% CI: 2.27‐2.91, P = 0.744, I2 = 0.0%, n = 69,524). Similarly, 4 clinical studies[6, 7, 8, 9] found detectable or elevated cTn as a predictor of worsened composite clinical outcomes of death and major cardiovascular events (OR: 1.33, 95% CI: 1.03‐1.71, P = 0.473, I2 = 0.0%, n = 1,313).
Short‐term (0 to 6 Months) Clinical Outcomes
Short‐term clinical outcome was assessed in 13 studies.[6, 8, 16, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30] Nine studies addressed mortality,[6, 16, 23, 24, 25, 26, 27, 28, 30] 2 studies readmission,[16, 26] and 7 studies a composite of readmission and mortality during 6 months postdischarge.[6, 8, 21, 22, 24, 26, 29] The meta‐analysis showed increased mortality without significant heterogeneity (OR: 2.11, 95% CI: 1.43‐3.12, P = 0.000, I2 8.5%, 9 studies, n = 3471) and an increase in the composite of readmission and mortality with significant heterogeneity (OR: 2.81, 95%CI: 1.60‐4.92, P = 0.000, I2 89.1%, 7 studies, n = 2028) among ADHF patients with detectable or elevated cTn. The association between cTn level and readmission rate over 6 months post‐discharge did not reach statistical significance (OR: 1.00, 95% CI: 0.37‐2.74, P = 0.034, I2 77.9%, 2 studies, n = 641).
Intermediate‐term (Up to 12 Months) Clinical Outcomes
Intermediate‐term (during the 12 months postdischarge) clinical outcome was assessed in seven studies.[18, 24, 31, 32, 33, 34, 35] Five studies reported an association between cTn level and mortality.[18, 24, 32, 34, 35] The meta‐analysis demonstrated an increase in mortality with significant heterogeneity (OR: 2.21, 95% CI: 1.46‐3.35, P = 0.048, I2 58.4%, 5 studies, n = 2801). The pooled HRs of 2 studies examining the association between cTn and readmission rate[18, 32] did not yield statistical significance (OR: 1.55, 95% CI: 0.96‐2.52, P = 0.233, I2 29.6%, 2 studies, n = 590). A meta‐analysis of 5 studies that assessed an association between cTn and outcome[18, 24, 31, 32, 33] showed a significant increase in the risk of composite of readmission and mortality without significant heterogeneity (OR: 2.30, 95% CI: 1.78‐2.99, P = 0.666, I2 0.0%, 5 studies, n = 905) among patients with a detectable or elevated cTn.
Long‐term (>1 Year) Clinical Outcomes
Long‐term clinical outcome was assessed in 7 studies.[18, 19, 21, 24, 28, 36, 37] The meta‐analysis of 6 studies[18, 19, 21, 28, 36, 37] demonstrated an increase in mortality without significant heterogeneity (OR: 3.69, 95% CI: 2.64‐5.18, P = 0.696, I2 0.0%, 6 studies, n = 1891) among ADHF patients with a detectable or elevated cTn. Likewise, a composite of readmission rate and mortality was also increased (OR: 3.49, 95% CI: 2.08‐5.84, P = 0.070, I2 57.5%, 4 studies, n = 448) in a meta‐analysis of 4 studies.[18, 21, 24, 37] The meta‐analysis of 4 studies[18, 19, 24, 37] that assessed the association between cTn level and readmission rate over long‐term follow‐up showed no significant association (OR: 2.60, 95% CI: 0.80‐8.44, P = 0.000, I2 99.9 %, 4 studies, n = 576).
Confidence in the Estimates
Following the Grading of Recommendations Assessment, Development, and Evaluation approach to evaluate the confidence in the estimates from a systematic review and meta‐analysis[38] (ie, certainty or strength of evidence), we found that the association of a detectable or elevated troponin with mortality and readmission is moderate. This is due to a large effect (ie, relative association measure >2.0) demonstrated in observational studies. The confidence in the estimate of association with hospital LOS is low (smaller magnitude of effect). Analyses of in‐hospital outcomes were not associated with statistical heterogeneity, whereas several posthospital analyses had statistically significant heterogeneity.
DISCUSSION
We conducted a systematic review and meta‐analysis of published studies to assess the association between level of cTn and clinical outcomes including LOS, in‐hospital mortality, and short‐, intermediate‐, and long‐term readmission and death following index hospitalization for ADHF. The results of our meta‐analysis showed that compared with negative or not‐elevated cTn, detectable or elevated cTn was associated with increased LOS and higher rates of in‐hospital death among patients with ADHF. In addition, mortality and composite of mortality and readmission at short‐, intermediate‐ or long‐term after index hospitalization were greater in ADHF patients with a detectable or elevated cTn, as compared with those without elevated cTn, with significant heterogeneity across the studies. Finally, relatively fewer studies examined the association between cTn and readmission rate at multiple time periods after index hospitalization for ADHF, and these associations did not reach statistical significance.
In a review of 67,924 patients with ADHF from the US National Registry, which was limited in assessing inpatient mortality, Peacock et al. reported that a positive cardiac troponin test was associated with higher in‐hospital mortality, independently of other predictive variables.[15] We confirmed this observation in the present meta‐analysis, which also incorporated the study by Peacock et al. Furthermore, our data extended the findings of Peacock et al. to postdischarge readmission and death. The association between cTn and clinical outcomes was adjusted to multiple confounders across most studies included in the present meta‐analysis. Few studies in the present meta‐analysis showed a continuous and graded relationship between cTn level and clinical outcomes in patients with ADHF.[15, 35] Findings of previous studies showed that ADHF patients with persistently elevated cTn, measured at multiple time points during or following hospitalization, had worse clinical outcomes than did patients without similar elevation in cTn.[24, 29] Conversely, a decline in cTnT levels on serial measurements was associated with lower rates of adverse clinical outcomes, potentially through alleviation in ongoing myocardial injury.[39] Additionally, elevated cTn in ambulatory heart failure patients predicted incident hospitalization for acute decompensation.[40] Acute myocardial injury reflected by elevated cTn can be hypothesized to promote ventricular remodeling and thereby heart failure progression and consequent adverse clinical outcomes. Consistent with this hypothesis, a rise in cTn was observed in conjunction with elevated biological markers that characterize extracellular matrix remodeling in patients with heart failure during the acute and postacute phase.[41, 42]cTn is released in blood in direct proportion to myocardial injury.[43] A rise or fall in cTn with 1 value at or above the 99th centile URL in conjunction with clinical evidence of myocardial injury defines acute myocardial infarction.[14] Although patients with chronic stable heart failure often have chronically elevated cTn, those with ADHF may demonstrate an acute rise in cTn, with values reaching above the 99th percentile of URL in the absence of acute myocardial infarction.[15, 44] The pathophysiology of elevated cTn in ADHF is probably multifactorial.[14, 45, 46] The prevalence of elevated cTn in ADHF varied with assay sensitivity and the cutoff point chosen. For instance, in an analysis of >105,000 patients with ADHF, the prevalence of elevated cTn was increased from 6.2% with higher (cTnI >1.0 ng/mL or cTnT >0.1 ng/mL) to 75% with a lower cutoff point for cTn levels (cTnI >0.4 ng/mL and cTnT >0.01 ng/mL).[15]
In the general population with no established coronary artery disease, the prevalence of elevated cTn is contingent on sensitivity of the assay, age, and gender.[47, 48, 49, 50] Elevated cTn, beyond conventional risk factors, identifies a subgroup of individuals from the general population who are at high risk for incident heart failure and death.[51] Furthermore, elevated cTn is an independent predictor of short‐ and long‐term cardiovascular events in patients presenting to an emergency department (ED) for ADHF.[52, 53] In 2 large Canadian registries, an elevated cTn was associated with increased risk of death and cardiovascular readmissions at 30 days after ED visit.[53]
A number of recent studies have identified numerous other biomarkers as independent prognostic indicators in patients with heart failure. cTn, when combined with other biomarkers reflecting different dimensions of heart failure pathophysiology such as brain natriuretic peptide (BNP)/N‐terminal pro‐brain natriuretic peptide, soluble ST2, or cystatin C, enhanced the model's predictive utility beyond individual markers. For instance, patients with elevated cTn who also have increased BNP (840 pg/mL) had in‐hospital mortality of 10.2%, which was significantly greater than the 4.4% in patients with elevated BNP without detectable cTn.[54] Additionally, elevated cTn along with elevated pro‐brain natriuretic peptide and cystatin C has been reported to offer incremental prognostic information in patient with ADHF.[33]
The present systematic review and meta‐analysis is the most comprehensive to date and incorporated many observational cohorts with heterogeneous and unselected patient population. The studies have used various commercially available assays for the measurement of cTnT and cTnI. Therefore, findings of this meta‐analysis are applicable to a wider heart failure patient population. This review has several limitations. The association of elevated cTn and clinical outcome is likely affected by several confounders. Although we used adjusted estimates when possible, we did not have individual participant data. Due to the small number of included studies in each analysis, we could not explore heterogeneity causes using subgroup analysis or metaregression. For the same reasons, we could not statistically evaluate publication bias, which is likely in the setting of observational studies. The meta‐analysis is mainly driven by a few large studies.
In summary, in a broad spectrum of patients with ADHF, a detectable or elevated cTn is an independent predictor of major adverse clinical events not only during acute‐phase hospitalization but also after stabilization during the postdischarge phase. cTn is a widely available and inexpensive biomarker that provides important prognostic information and is likely to have important implications for in‐patient care and postdischarge surveillance of patients hospitalized for ADHF.
Disclosures
The authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers' bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent‐licensing arrangements), or nonfinancial interest (such as personal or professional relationships, affiliations, knowledge, or beliefs) in the subject matter or materials discussed in this article.
Acute decompensated heart failure (ADHF) accounts for over a million hospitalizations per year, with a reported all‐cause mortality rate 11.7% and all‐cause readmission rate 22.5% at 30 days after initial hospitalization.[1]
Risk stratification for accurate identification of ADHF patients at high risk for readmission and mortality may enable clinicians to undertake timely interventions: triage to appropriate level of care and resource allocation for postdischarge care. Further risk stratification may allow the care team to plan and implement a personalized care plan. Several clinical and laboratory variables have been proposed for identification of patients with ADHF who are at increased risk for adverse clinical outcomes. Despite advances in the risk stratification of patients with ADHF, the accurate prediction of individuals at high risk for readmissions and mortality is challenging. Cardiac troponin T (cTnT) and I (cTnI) are highly sensitive and specific biomarkers that are widely used for the risk stratification of patients with acute myocardial infarction and stable heart failure.[2]
In this systematic review and meta‐analysis, we evaluate circulating cardiac troponin in determining risk for increased length of stay (LOS), hospital readmission, and mortality among patients admitted with ADHF.
METHODS
Data Sources and Searches
This systematic review and meta‐analysis was conducted in accordance with the established methods[3] and Preferred Reporting Items for Systematic Review and Meta‐Analysis (PRISMA) guidelines.[4] Risk of bias was evaluated using the Newcastle‐Ottawa Scale for cohort studies.[5] We performed a comprehensive search of several databases from each database's earliest inception to March 2015 without language restrictions. The databases included MEDLINE In‐Process & Other Non‐Indexed Citations, MEDLINE, Embase, Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews, and Scopus. We conducted a manual search for bibliography of pertinent reviews for relevant citations that our electronic searches might have missed. The actual strategy is available from the corresponding author.
Study Selection
Eligibility criteria included: (1) randomized or nonrandomized clinical trials involving adults hospitalized with ADHF, (2) comparator groups stratified by cardiac troponin (cTn) level as defined by individual study investigators, and (3) studies reporting 1 or more of the following clinical outcomes: (1) in‐hospital mortality, (2) hospital LOS, (3) major adverse events during hospitalization (defined as persistent dyspnea,[6] worsening of heart failure,[6, 7, 8, 9] worsening of renal function [creatinine 0.3 mg/dL],[8] or recurrent myocardial ischemia[9] after hospitalization for ADHF), (4) postdischarge readmission, (5) postdischarge mortality rate, and (6) the composite of readmission and mortality. We excluded studies incorporating patients with (1) stable heart failure, (2) acute myocarditis, (3) chemotherapy‐induced cardiomyopathy, (4) postsurgical heart failure, (5) transplanted heart, (6) left ventricular assist device, and (7) hemodialysis.
We incorporated the description of ADHF from national registry for defining ADHF.[10] The lower limit of detection of cTn level in healthy subjects is assay dependent, each with a different cutoff value. To improve uniformity of expression in the present meta‐analysis, we arbitrarily stratified groups by the level of cTn: (1) undetected cTn (cTnT 0.01; cTnI 0.012 g/L), (2) detectable cTn (cTnT 0.010.03; cTnI 0.0120.03 g/L), and (3) elevated cTn (cTnT >0.03; cTnI >0.034 g/L).
Data Extraction and Risk of Bias Assessment
From the results of the initial search, 2 investigators (M.Y. and A.D.A), working independently, reviewed articles for eligibility on the basis of titles and abstracts. Studies that satisfied the inclusion and exclusion criteria were retrieved for full‐text review. Disagreements were resolved by consensus after discussion among investigators, and retained conflicts were adjudicated by a third investigator.
We extracted the following data from each study: type of study, number of participants, age, gender, type of cTn assayed and cut point, comorbidities, length of follow‐up, and outcome measure. Prevalence of detectable or elevated cTn, measure of association with clinical outcomes (hazard ratio [HR], odds ratio [OR], or relative risk) were also abstracted. When HR or OR were not reported for an outcome, based on other provided data, we estimated HR using previously validated methods.[11]
Data Synthesis and Analysis
Studies were stratified by cTn cutoff point and length of follow‐up. To reduce heterogeneity, studies reporting clinical outcomes at multiple time periods after the index hospitalization were grouped in three categories: (1) studies with short‐term follow up (06 months), (2) studies with intermediate‐term follow‐up (up to 1 year), and (3) studies with long‐term follow‐up (up to 3.5 years). We used the DerSimonian and Laird random effects model to combine OR or HR reported by individual studies. The consistency of the results of the studies was assessed by I2 statistics, with values >40% considered as indicators of heterogeneity. We evaluated statistically for publication bias if a sufficient number of studies was available, because such evaluation is unreliable when20 studies are included in a particular analysis.[12]
Sensitivity analyses were performed to investigate the robustness of results to a few assumptions. Analyses were repeated excluding studies reporting unadjusted relative effect measures to assess whether confounding had a large effect on overall results. Similarly, analysis was repeated omitting studies reporting detectable cTn as opposed to elevated cTn level to assess whether these studies influence overall results. All statistical analyses were conducted using Stata 14.0 (StataCorp, College Station, TX).
Quality and Risk of Bias Assessment
The Newcastle‐Ottawa scale was used to assess the quality and risk of bias in cohort studies as suggested by the Cochrane Collaboration.[13] For the assessment of risk of bias, a study was awarded a maximum of 1 star for each of the 7 items from 2 domains: (1) selection of cohort (representativeness of the exposed cohort, selection of the nonexposed cohort, ascertainment of exposure, and demonstration that the outcome of interest was not present at the start of the study) and (2) outcome (assessment of outcome, was the follow‐up long enough, adequacy of the follow‐up of the cohort), and a maximum of 2 stars for comparability of the cohort (comparability on the basis of design and analysis) (Table 1).
| Source | Year | Selection | Compatibility | Outcome | Quality | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 | S2 | S3 | S4 | C1 | C2 | O1 | O2 | O3 | |||
| |||||||||||
| Del Carlo et al.[31] | 2009 | * | * | * | * | * | 5 | ||||
| Felker et al.[6] | 2012 | * | * | * | * | * | 5 | ||||
| Gattis et al.[7] | 2004 | * | * | * | * | * | * | * | 7 | ||
| Guisado Espartero et al.[32] | 2014 | * | * | * | * | * | * | 6 | |||
| Ishii et al.[21] | 2002 | * | * | * | * | * | 5 | ||||
| Kuwabara et al.[22] | 2007 | * | * | * | * | * | 5 | ||||
| La Vecchia et al.[23] | 2000 | * | * | * | * | * | * | * | 7 | ||
| La Corvoisie et al.[17] | 2014 | * | * | * | * | * | * | * | 7 | ||
| Manzano‐Fernandez et al.[33] | 2009 | * | * | * | * | * | 5 | ||||
| Metra et al.[24] | 2007 | * | * | * | * | * | 5 | ||||
| Nakamura et al.[36] | 2014 | * | * | * | * | * | * | * | * | 8 | |
| O'Connor et al.[8] | 2011 | * | * | * | * | * | * | * | * | 8 | |
| Oliveira et al.[34] | 2010 | * | * | * | * | * | * | * | * | 8 | |
| Parissis et al.[20] | 2011 | * | * | * | * | * | * | * | 7 | ||
| Parissis et al.[25] | 2013 | * | * | * | * | * | * | * | * | 8 | |
| Pascual‐Figal et al.[37] | 2012 | * | * | * | * | * | * | 6 | |||
| Peacock et al.[15] | 2008 | * | * | * | * | * | * | * | * | * | 9 |
| Perna et al.[18] | 2005 | * | * | * | * | * | * | * | 7 | ||
| Perna et al.[19] | 2002 | * | * | * | * | * | * | 6 | |||
| Perna et al.[26] | 2012 | * | * | * | * | * | * | 6 | |||
| Rudiger et al.[27] | 2005 | * | * | * | * | * | * | 6 | |||
| Shah et al.[16] | 2007 | * | * | * | * | 4 | |||||
| Wallenborn et al.[28] | 2013 | * | * | * | * | * | * | * | 7 | ||
| Xue et al.[29] | 2011 | * | * | * | * | * | * | * | 7 | ||
| You et al.[35] | 2007 | * | * | * | * | * | * | * | * | * | 9 |
| Zairis et al.[30] | 2010 | * | * | * | * | * | * | * | 7 | ||
RESULTS
Search Results
Figure 1 represents the PRISMA flow diagram for literature search and selection process to identify eligible studies for inclusion.
Characteristics of Included Studies
We identified 26 studies, which were all observational cohorts with postdischarge median follow‐up from 30 days to 472 days. Table 2 summarizes the study characteristics. Studies were heterogeneous with regard to prevalence of elevated cTn, cTn assay, and length of follow‐up. Thirteen were single‐center and 5 were multicenter studies, 4 were substudies of large multicenter phase III clinical trials, and 4 were registries. Except for one abstracts, all studies were peer‐reviewed publications. Sample size ranged from 34 to 69,259 patients.
| Source | Year | Design | Patient Population | CAD | HF Type | LVEF, Mean % | Clinical Outcomes | |||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of Patients | Age, y | Men, % | Follow‐up | Endpoints | ||||||
| ||||||||||
| Del Carlo et al.[31] | 2009 | Single | 70 | 54 16 | 69 | 26 | HFrEF | 31 8 | 262 (3393) days | Readmission, mortality |
| Felker et al.[6] | 2012 | Substudy | 808 | 67 | 70 | 61 | Both | 25 | Hosp, 30 days, 6 months | MAE, LOS, readmission, mortality |
| Gattis et al.[7] | 2004 | Substudy | 133 | NR | NR | NR | NR | NR | Hosp | MAE, mortality |
| Guisado Espartero et al.[32] | 2014 | Registry | 406 | 77 (7678) | 42 | 25 | Both | 50 (4456) | 1 year | Readmission, mortality |
| Ishii et al.[21] | 2002 | Single | 98 | 69 9 | 52 | 45 | NR | 42 17 | Hosp, 60 days, >1 year | Readmission, mortality |
| Kuwabara et al.[22] | 2007 | Single | 52 | 72 12 | 59 | 27 | NR | 47 16 | 143 (13540) days | Readmission, mortality |
| La Vecchia et al.[23] | 2000 | Single | 34 | 60 (2885) | 79 | 38 | HFrEF | NR | 90 days | Mortality |
| La Corvoisie et al.[17] | 2014 | Multicenter | 397 | NR | NR | NR | NR | Hosp | Mortality | |
| Manzano‐Fernandez et al.[33] | 2009 | Single | 138 | 74 (67801) | 54 | 35 | NR | NR | 261 (161449) days | Readmission, mortality |
| Metra et al.[24] | 2007 | Single | 116 | NR | NR | NR | NR | NR | 184 (7444) days | Readmission, mortality |
| Nakamura et al.[36] | 2014 | Single | 444 | NR | 63 | 15 | NR | NR | 472 (2001,200) days | Mortality |
| O'Connor et al.[8] | 2011 | Substudy | 288 | 73 (6577) | 59 | 74 | NR | NR | Hosp, 60 days | MAE, readmission, mortality |
| Oliveira et al.[34] | 2010 | Multicenter | 79 | NR | 61 | 18 | HFrEF | 27 | 8 months | MAE, readmission, mortality |
| Parissis et al.[20] | 2011 | Multicenter | 837 | NR | 48 | 20 | HFpEF | NR | Hosp | Mortality |
| Parissis et al.[25] | 2013 | Single | 113 | 73 11 | 68 | 46 | NR | 36 11 | 174 (94728) days | Mortality |
| Pascual‐Figal et al.[37] | 2012 | Single | 202 | 74 (6780) | 55 | 34 | Both | 49 (3260) | 406 (204728) days | Mortality |
| Peacock et al.[15] | 2008 | Registry | 69,259 | 74 14 | 45 | 56 | Both | 34 | Hosp | LOS, mortality |
| Perna et al.[18] | 2005 | Single | 184 | 64 13 | 60 | 38 | Both | NR | Hosp, 3 years | Readmission, mortality |
| Perna et al.[19] | 2002 | Single | 84 | 65 14 | 62 | 55 | NR | NR | Hosp, 1 year | Readmission, mortality |
| Perna et al.[26] | 2012 | Single | 500 | 73 12 | 53 | 38 | HFpEF | 53 11 | 6 months | Readmission, mortality |
| Rudiger et al.[27] | 2005 | Multicenter | 312 | 73 12 | 56 | 70 | Both | NR | 30 days, 1 year | Mortality |
| Shah et al.[16] | 2007 | Substudy | 141 | NR | NR | NR | HFrEF | NR | Hosp, 6 months | LOS, readmission, mortality |
| Wallenborn et al.[28] | 2013 | Registry | 879 | 69 12 | 72 | 50 | NR | 30 8 | 06 months, 618 months | Mortality |
| Xue et al.[29] | 2011 | Single | 144 | 68 13 | 98 | 62 | Both | 43 18 | 90 days | Readmission, mortality |
| You et al.[35] | 2007 | Registry | 2,025 | 76 11 | 50 | 55 | Both | NR | 1 year | Mortality |
| Zairis et al.[30] | 2010 | Multicenter | 577 | 74 8 | 68 | 77 | HFrEF | 23 5 | 31 days | Mortality |
Table 3 stratifies the characteristics of study populations by cTn status. The studies included 77,297 participants hospitalized for ADHF, of whom 7176 (9.3%) had detectable or elevated cTn level. Twenty‐five studies reported data on type of cTn measured (cTnI, cTnT, or both) and reported cutoff values for detectable or elevated cTn (Table 3). The percentages of patients who had detectable or elevated cTn varied widely across the studies (6.2%68%). Most studies utilized standard assays, and the cutoff point for cTn level was chosen arbitrary by study investigators or derived from receiver operating characteristic curve analysis. cTn level is assay dependent. For instance, the 99th centile upper reference limit (URL) is 0.014 ng/mL for cTnT with the Roche high‐sensitivity cTnT assay, and 0.04 ng/mL with the Siemens cTnI‐ultra assay. Few studies of the present meta‐analysis incorporated a cTn cutoff point that defined acute myocardial infarction.[14] Nine studies used a lower threshold cTn level (cTnT >0.01>0.03; cTnI >0.03) for stratification into comparator groups.
| Source | Year | No. of Patients | No. cTn+ (%) | cTn Cutoff | Age | Male | Atrial Fibrillation | CAD | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Tn+ | Tn | Tn+ (%) | No. (%) | Tn+ (%) | Tn+ (%) | |||||
| ||||||||||
| Del Carlo et al.[31] | 2009 | 70 | 12 (17) | cTnT 0.10 | NR | NR | NR | 13 (19) | NR | NR |
| Felker et al.[6] | 2012 | 808 | 404 (50) | cTnT 0.034 | 69 | 65 | 364 (54) | 334 (41) | 170 (42) | 243 (60) |
| Gattis et al.[7] | 2004 | 133 | 91 (68) | cTnT 1.0 | 70 (6180) | 77 (6282) | 46 (50) | NR | NR | NR |
| Guisado Espartero et al.[32] | 2014 | 406 | 241 (60) | cTnT 0.02 | NR | NR | 116 (48) | 236 (58) | 136 (56) | 74 (31) |
| Ishii et al.[21] | 2002 | 98 | NR | cTnT 0.1 | NR | NR | NR | NR | NR | NR |
| Kuwabara et al.[22] | 2007 | 52 | 31 (60) | NR | NR | NR | 31 (59) | 23 (44) | NR | NR |
| La Vecchia et al.[23] | 2000 | 34 | 10 (29) | cTnI 0.4 | 56 13 | 62 12 | 100 | 19 (56) | 5 (50) | 3 (30) |
| La Corvoisie et al.[17] | 2014 | 397 | NR | cTnI0.15 | NR | NR | NR | NR | NR | NR |
| Manzano‐Fernandez et al.[33] | 2009 | 138 | NR | cTnT 0.011 | NR | NR | NR | NR | NR | NR |
| Metra et al.[24] | 2007 | 116 | 41 (38) | cTnT 0.01 | NR | NR | NR | NR | NR | 33 (61) |
| Nakamura et al.[36] | 2014 | 444 | 224 (51) | cTnT 0.028 | 67 14 | 66 14 | 133 (60) | 160 (36) | 72 (32) | 35 (16) |
| O'Connor et al.[8] | 2011 | 288 | 97 (34) | cTnT 0.03 | 71 | 72 | 67 (69) | NR | NR | NR |
| Oliveira et al.[34] | 2010 | 79 | 37 (47) | ctnT 0.02 | 57 18 | 54 17 | 26 (70) | NR | NR | 6 (16) |
| Parissis et al.[20] | 2011 | 837 | 184 (22) | cTnT >0.01 | NR | NR | NR | NR | NR | NR |
| Parissis et al.[25] | 2013 | 113 | 37 (33) | cTnT 0.077 | 74 8 | 72 12 | 22 (59) | 36 (32) | 12 (32) | 18 (49) |
| Pascual‐Figal et al.[37] | 2012 | 202 | NR | cTnT >0.02 | NR | NR | NR | 109 (54) | NR | NR |
| Peacock et al.[15] | 2008 | 69,259 | 4,240 (6.2) | cTnI 1.0; cTnT 0.1 | 73 14 | 73 14 | 2,035 (48) | 207 (30) | 975 (23) | 2,586 (58) |
| Perna et al.[18] | 2005 | 184 | 58 (31) | cTnT 0.1 | 64 13 | 65 13 | 37 (64) | NR | NR | 30 (52) |
| Perna et al.[19] | 2002 | 84 | 46 (55) | cTnT 0.1 | 68 11 | 61 16 | 27 (59) | NR | NR | 33 (72) |
| Perna et al.[26] | 2012 | 500 | 220 (44) | cTnT 0.02 | 74 10 | 72 14 | 125 (59) | 177 (35) | 70 (32) | 110 (50) |
| Rudiger et al.[27] | 2005 | 312 | 88 (28) | cTnT 0.1 | NR | NR | NR | NR | NR | NR |
| Shah et al.[16] | 2007 | 141 | NR | cTnI per 0.1 | NR | NR | NR | NR | NR | NR |
| Wallenborn et al.[28] | 2013 | 879 | 332 (37) | cTnT 0.06 | NR | NR | NR | NR | NR | NR (50) |
| Xue et al.[29] | 2011 | 144 | NR | cTnI 0.023 | NR | NR | NR | NR | NR | NR |
| You et al.[35] | 2007 | 2,025 | 669 (34) | cTnI >0.5 | 77 11 | 75 11 | 364 (53) | NR | NR | 417 (60) |
| Zairis et al.[30] | 2010 | 577 | 114 (20) | cTnI 0.42 | NR | NR | NR | 295 (51) | NR | 443 (77) |
Twenty‐five studies reported performance of cTn as a dichotomized variable. A few studies, additionally, examined clinical outcome in patients grouped by tertiles by cTn and determined the dose‐response relationship using cTn as a continuous variable. The measure of association between cTn and clinical outcome was reported as HR or OR by 16 studies. The remaining 6 studies reported the number of clinical events in the groups by cTn level and therefore provided unadjusted estimates. The results of all meta‐analyses are depicted in Figure 2.
In‐hospital Clinical Outcomes
Three studies examined the association between cTn level and LOS.[6, 15, 16] One study (n = 808) found increased LOS among patients with elevated cTn.[6] Another study (n = 141), which tested the cTn level as a continuous variable, reported no statistically significant association between cTn level and LOS.[16] A large, multicenter ADHF registry (Acute Decompensated Heart Failure National Registry), which reported elevated cTn as a predictor of LOS (mean stay 6.6 vs 5.5 days; P 0.001) but did not provide binary data (OR, confidence interval [CI]), was therefore excluded from the meta‐analysis.[15] The pooled HRs from 2 studies revealed a significant increase in LOS in the cohort with elevated cTn (OR: 1.05, 95% CI: 1.01‐1.10, P = 0.06, I2 = 59.5.0%, n = 949). Six studies assessed in‐hospital mortality,[15, 17, 18, 19, 20, 21] and the meta‐analysis showed a significant increase in the risk of death with no significant heterogeneity (OR: 2.57, 95% CI: 2.27‐2.91, P = 0.744, I2 = 0.0%, n = 69,524). Similarly, 4 clinical studies[6, 7, 8, 9] found detectable or elevated cTn as a predictor of worsened composite clinical outcomes of death and major cardiovascular events (OR: 1.33, 95% CI: 1.03‐1.71, P = 0.473, I2 = 0.0%, n = 1,313).
Short‐term (0 to 6 Months) Clinical Outcomes
Short‐term clinical outcome was assessed in 13 studies.[6, 8, 16, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30] Nine studies addressed mortality,[6, 16, 23, 24, 25, 26, 27, 28, 30] 2 studies readmission,[16, 26] and 7 studies a composite of readmission and mortality during 6 months postdischarge.[6, 8, 21, 22, 24, 26, 29] The meta‐analysis showed increased mortality without significant heterogeneity (OR: 2.11, 95% CI: 1.43‐3.12, P = 0.000, I2 8.5%, 9 studies, n = 3471) and an increase in the composite of readmission and mortality with significant heterogeneity (OR: 2.81, 95%CI: 1.60‐4.92, P = 0.000, I2 89.1%, 7 studies, n = 2028) among ADHF patients with detectable or elevated cTn. The association between cTn level and readmission rate over 6 months post‐discharge did not reach statistical significance (OR: 1.00, 95% CI: 0.37‐2.74, P = 0.034, I2 77.9%, 2 studies, n = 641).
Intermediate‐term (Up to 12 Months) Clinical Outcomes
Intermediate‐term (during the 12 months postdischarge) clinical outcome was assessed in seven studies.[18, 24, 31, 32, 33, 34, 35] Five studies reported an association between cTn level and mortality.[18, 24, 32, 34, 35] The meta‐analysis demonstrated an increase in mortality with significant heterogeneity (OR: 2.21, 95% CI: 1.46‐3.35, P = 0.048, I2 58.4%, 5 studies, n = 2801). The pooled HRs of 2 studies examining the association between cTn and readmission rate[18, 32] did not yield statistical significance (OR: 1.55, 95% CI: 0.96‐2.52, P = 0.233, I2 29.6%, 2 studies, n = 590). A meta‐analysis of 5 studies that assessed an association between cTn and outcome[18, 24, 31, 32, 33] showed a significant increase in the risk of composite of readmission and mortality without significant heterogeneity (OR: 2.30, 95% CI: 1.78‐2.99, P = 0.666, I2 0.0%, 5 studies, n = 905) among patients with a detectable or elevated cTn.
Long‐term (>1 Year) Clinical Outcomes
Long‐term clinical outcome was assessed in 7 studies.[18, 19, 21, 24, 28, 36, 37] The meta‐analysis of 6 studies[18, 19, 21, 28, 36, 37] demonstrated an increase in mortality without significant heterogeneity (OR: 3.69, 95% CI: 2.64‐5.18, P = 0.696, I2 0.0%, 6 studies, n = 1891) among ADHF patients with a detectable or elevated cTn. Likewise, a composite of readmission rate and mortality was also increased (OR: 3.49, 95% CI: 2.08‐5.84, P = 0.070, I2 57.5%, 4 studies, n = 448) in a meta‐analysis of 4 studies.[18, 21, 24, 37] The meta‐analysis of 4 studies[18, 19, 24, 37] that assessed the association between cTn level and readmission rate over long‐term follow‐up showed no significant association (OR: 2.60, 95% CI: 0.80‐8.44, P = 0.000, I2 99.9 %, 4 studies, n = 576).
Confidence in the Estimates
Following the Grading of Recommendations Assessment, Development, and Evaluation approach to evaluate the confidence in the estimates from a systematic review and meta‐analysis[38] (ie, certainty or strength of evidence), we found that the association of a detectable or elevated troponin with mortality and readmission is moderate. This is due to a large effect (ie, relative association measure >2.0) demonstrated in observational studies. The confidence in the estimate of association with hospital LOS is low (smaller magnitude of effect). Analyses of in‐hospital outcomes were not associated with statistical heterogeneity, whereas several posthospital analyses had statistically significant heterogeneity.
DISCUSSION
We conducted a systematic review and meta‐analysis of published studies to assess the association between level of cTn and clinical outcomes including LOS, in‐hospital mortality, and short‐, intermediate‐, and long‐term readmission and death following index hospitalization for ADHF. The results of our meta‐analysis showed that compared with negative or not‐elevated cTn, detectable or elevated cTn was associated with increased LOS and higher rates of in‐hospital death among patients with ADHF. In addition, mortality and composite of mortality and readmission at short‐, intermediate‐ or long‐term after index hospitalization were greater in ADHF patients with a detectable or elevated cTn, as compared with those without elevated cTn, with significant heterogeneity across the studies. Finally, relatively fewer studies examined the association between cTn and readmission rate at multiple time periods after index hospitalization for ADHF, and these associations did not reach statistical significance.
In a review of 67,924 patients with ADHF from the US National Registry, which was limited in assessing inpatient mortality, Peacock et al. reported that a positive cardiac troponin test was associated with higher in‐hospital mortality, independently of other predictive variables.[15] We confirmed this observation in the present meta‐analysis, which also incorporated the study by Peacock et al. Furthermore, our data extended the findings of Peacock et al. to postdischarge readmission and death. The association between cTn and clinical outcomes was adjusted to multiple confounders across most studies included in the present meta‐analysis. Few studies in the present meta‐analysis showed a continuous and graded relationship between cTn level and clinical outcomes in patients with ADHF.[15, 35] Findings of previous studies showed that ADHF patients with persistently elevated cTn, measured at multiple time points during or following hospitalization, had worse clinical outcomes than did patients without similar elevation in cTn.[24, 29] Conversely, a decline in cTnT levels on serial measurements was associated with lower rates of adverse clinical outcomes, potentially through alleviation in ongoing myocardial injury.[39] Additionally, elevated cTn in ambulatory heart failure patients predicted incident hospitalization for acute decompensation.[40] Acute myocardial injury reflected by elevated cTn can be hypothesized to promote ventricular remodeling and thereby heart failure progression and consequent adverse clinical outcomes. Consistent with this hypothesis, a rise in cTn was observed in conjunction with elevated biological markers that characterize extracellular matrix remodeling in patients with heart failure during the acute and postacute phase.[41, 42]cTn is released in blood in direct proportion to myocardial injury.[43] A rise or fall in cTn with 1 value at or above the 99th centile URL in conjunction with clinical evidence of myocardial injury defines acute myocardial infarction.[14] Although patients with chronic stable heart failure often have chronically elevated cTn, those with ADHF may demonstrate an acute rise in cTn, with values reaching above the 99th percentile of URL in the absence of acute myocardial infarction.[15, 44] The pathophysiology of elevated cTn in ADHF is probably multifactorial.[14, 45, 46] The prevalence of elevated cTn in ADHF varied with assay sensitivity and the cutoff point chosen. For instance, in an analysis of >105,000 patients with ADHF, the prevalence of elevated cTn was increased from 6.2% with higher (cTnI >1.0 ng/mL or cTnT >0.1 ng/mL) to 75% with a lower cutoff point for cTn levels (cTnI >0.4 ng/mL and cTnT >0.01 ng/mL).[15]
In the general population with no established coronary artery disease, the prevalence of elevated cTn is contingent on sensitivity of the assay, age, and gender.[47, 48, 49, 50] Elevated cTn, beyond conventional risk factors, identifies a subgroup of individuals from the general population who are at high risk for incident heart failure and death.[51] Furthermore, elevated cTn is an independent predictor of short‐ and long‐term cardiovascular events in patients presenting to an emergency department (ED) for ADHF.[52, 53] In 2 large Canadian registries, an elevated cTn was associated with increased risk of death and cardiovascular readmissions at 30 days after ED visit.[53]
A number of recent studies have identified numerous other biomarkers as independent prognostic indicators in patients with heart failure. cTn, when combined with other biomarkers reflecting different dimensions of heart failure pathophysiology such as brain natriuretic peptide (BNP)/N‐terminal pro‐brain natriuretic peptide, soluble ST2, or cystatin C, enhanced the model's predictive utility beyond individual markers. For instance, patients with elevated cTn who also have increased BNP (840 pg/mL) had in‐hospital mortality of 10.2%, which was significantly greater than the 4.4% in patients with elevated BNP without detectable cTn.[54] Additionally, elevated cTn along with elevated pro‐brain natriuretic peptide and cystatin C has been reported to offer incremental prognostic information in patient with ADHF.[33]
The present systematic review and meta‐analysis is the most comprehensive to date and incorporated many observational cohorts with heterogeneous and unselected patient population. The studies have used various commercially available assays for the measurement of cTnT and cTnI. Therefore, findings of this meta‐analysis are applicable to a wider heart failure patient population. This review has several limitations. The association of elevated cTn and clinical outcome is likely affected by several confounders. Although we used adjusted estimates when possible, we did not have individual participant data. Due to the small number of included studies in each analysis, we could not explore heterogeneity causes using subgroup analysis or metaregression. For the same reasons, we could not statistically evaluate publication bias, which is likely in the setting of observational studies. The meta‐analysis is mainly driven by a few large studies.
In summary, in a broad spectrum of patients with ADHF, a detectable or elevated cTn is an independent predictor of major adverse clinical events not only during acute‐phase hospitalization but also after stabilization during the postdischarge phase. cTn is a widely available and inexpensive biomarker that provides important prognostic information and is likely to have important implications for in‐patient care and postdischarge surveillance of patients hospitalized for ADHF.
Disclosures
The authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers' bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent‐licensing arrangements), or nonfinancial interest (such as personal or professional relationships, affiliations, knowledge, or beliefs) in the subject matter or materials discussed in this article.
- Centers for Medicare 98:1778–1786.
- , , , et al. Meta‐analysis of observational studies in epidemiology: A proposal for reporting. Meta‐analysis of observational studies in epidemiology (moose) group. JAMA. 2000;283:2008–2012.
- , , , et al. The PRISMA statement for reporting systematic reviews and meta‐analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339:b2700.
- . Critical evaluation of the Newcastle‐Ottawa Scale for the assessment of the quality of nonrandomized studies in meta‐analyses. Eur J Epidemiol. 2010;25:603–605.
- , , , et al. Troponin I in acute decompensated heart failure: Insights from the ascend‐hf study. Eur J Heart Fail. 2012;14:1257–1264.
- , , , , , . Usefulness of an elevated troponin‐I in predicting clinical events in patients admitted with acute heart failure and acute coronary syndrome (from the RITZ‐4 trial). Am J Cardiol. 2004;93:1436–1437.
- , , , et al. Impact of serial troponin release on outcomes in patients with acute heart failure: analysis from the protect pilot study. Circulation. 2011;4:724–732.
- , , , et al. Ongoing myocardial injury in stable severe heart failure: value of cardiac troponin t monitoring for high‐risk patient identification. Circulation. 2004;110:2376–2382.
- , , , et al. Characteristics and outcomes of patients hospitalized for heart failure in the united states: rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J. 2005;149:209–216.
- , , . Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Stat Med. 1998;17:2815–2834.
- , , , , . The case of the misleading funnel plot. BMJ. 2006;333:597–600.
- , , , et al. The Newcastle‐Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta‐analyses. Available at: http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp. accessed July 3, 2015.
- , , , et al. Third universal definition of myocardial infarction. J Am Coll Cardiol. 2012;60:1581–1598.
- , , , et al.; ADHERE Investigators. Cardiac troponin and outcome in acute heart failure. N Engl J Med. 2008;358:2117–2126.
- , , , et al. Rapid assay brain natriuretic peptide and troponin I in patients hospitalized with decompensated heart failure (from the Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness trial). Am J Cardiol. 2007;100:1427–1433.
- , , , et al. Functional status and co‐morbidities are associated with in‐hospital mortality among older patients with acute decompensated heart failure: a multicentre prospective cohort study. Age Ageing. 2015;44(2):225–231.
- , , , et al. Minor myocardial damage detected by troponin T is a powerful predictor of long‐term prognosis in patients with acute decompensated heart failure. Int J Cardiol. 2005;99:253–261.
- , , , et al. Cardiac troponin T levels are associated with poor short‐ and long‐term prognosis in patients with acute cardiogenic pulmonary edema. Am Heart J. 2002;143:814–820.
- , , , et al. Clinical characteristics and predictors of in‐hospital mortality in acute heart failure with preserved left ventricular ejection fraction. Am J Cardiol. 2011;107:79–84.
- , , , et al. Risk stratification using a combination of cardiac troponin t and brain natriuretic peptide in patients hospitalized for worsening chronic heart failure. Am J Cardiol. 2002;89:691–695.
- , , , , et al. Persistently increased serum concentrations of cardiac troponin in patients with acutely decompensated heart failure are predictive of adverse outcomes. Circ J. 2007;71:1047–1051.
- , , , et al. Cardiac troponin I as diagnostic and prognostic marker in severe heart failure. J Heart Lung Transplant. 2000;19:644–652.
- , , , et al. The role of plasma biomarkers in acute heart failure. Serial changes and independent prognostic value of NT‐proBNP and cardiac troponin‐T. Eur J Heart Fail. 2007;9:776–786.
- , , , et al. Prognostic value of high sensitivity troponin T in patients with acutely decompensated heart failure and non‐detectable conventional troponin T levels. Int J Cardiol. 2013;168:3609–3612.
- , , , et al. Minor myocardial damage is a prevalent condition in patients with acute heart failure syndromes and preserved systolic function with long‐term prognostic implications: CIAST‐HF (Collaborative Italo‐Argentinean Study on cardiac Troponin T in Heart Failure) study. J Card Fail. 2012;18:822–830.
- , , , et al. Acute heart failure: clinical presentation, one‐year mortality and prognostic factors. Eur J Heart Fail. 2005;7:662–670.
- , , , et al. High‐sensitive troponin I after acute cardiac decompensation‐distribution of baseline values and prognostic significance. Paper presented at: Heart Failure Congress 2013; May 25–28, 2013; Lisbon, Portugal.
- , , , . Serial changes in high‐sensitive troponin I predict outcome in patients with decompensated heart failure. Eur J Heart Fail. 2011;13:37–42.
- , , , et al. Multimarker strategy for the prediction of 31 days cardiac death in patients with acutely decompensated chronic heart failure. Int J Cardiol. 2010;141:284–290.
- , , , , , . Cardiac troponin t for risk stratification in decompensated chronic heart failure. [in Portuguese]. Arq Bras Cardiol. 2009;92(5):404–412.
- , , , et al. Troponin T in acute heart failure: clinical implications and prognosis in the Spanish National Registry on Heart Failure. Eur J Intern Med. 2014;25:739–744.
- , , , et al. Complementary prognostic value of cystatin C, N‐terminal pro‐B‐type natriuretic Peptide and cardiac troponin T in patients with acute heart failure. Am J Cardiol. 2009;103:1753–1759.
- , , . Single cardiac troponin t measurement predicts risk for adverse outcome in decompensated heart failure. Arq Bras Cardiol. 2010;94(4):495–501.
- , , , , . Relation between cardiac troponin i and mortality in acute decompensated heart failure. Am Heart J. 2007;153:462–470.
- , , , et al. High‐sensitivity cardiac troponin t predicts non‐cardiac mortality in heart failure. Circ J. 2014;78:890–895.
- , , , et al. Highly sensitive troponin t for risk stratification of acutely destabilized heart failure. Am Heart J. 2012;163(6):1002–1010.
- , , , et al. How to read a systematic review and meta‐analysis and apply the results to patient care: Users' guides to the medical literature. JAMA. 2014;312:171–179.
- , , , et al. Serial biomarker measurements in ambulatory patients with chronic heart failure: the importance of change over time. Circulation. 2007;116:249–257.
- , , , et al. Prognostic value of very low plasma concentrations of troponin T in patients with stable chronic heart failure. Circulation. 2007;116:1242–1249.
- , , , et al. Episodes of acute heart failure syndrome are associated with increased levels of troponin and extracellular matrix markers. Circ Heart Fail. 2010;3:44–50.
- , , , et al. Cardiac microinjury measured by troponin T predicts collagen metabolism in adults aged >=65 years with heart failure. Circ Heart Fail. 2012;5:406–413.
- . Pathobiology of troponin elevations: so elevations occur with myocardial ischemia as well as necrosis? J Am Coll Cardiol. 2011;57:2406–2408.
- , , . Prognostic value of cardiac troponin in chronic stable heart failure: a systematic review. Heart. 2012;98:1778–1786.
- , , , et al. Vascular endothelial dysfunction and mortality risk in patients with chronic heart failure. Circulation. 2005;111:310–314.
- , , , , . Preload induces troponin I degradation independently of myocardial ischemia. Circulation. 2001;103:2035–2037.
- , , ,et al. Prevalence and determinants of troponin T elevation in the general population. Circulation. 2006;113:1958–1965.
- , , , et al. Cardiac troponin‐I and risk of heart failure: a community‐based cohort study. Eur Heart J. 2009;30:773–781.
- , , , et al. Age‐ and sex‐dependent upper reference limits for the high‐sensitivity cardiac troponin t assay. J Am Coll Cardiol. 2014;63:1441–1448.
- , , , et al. Defining high‐sensitivity cardiac troponin concentrations in the community. Clin Chem. 2013;59:1099–1107.
- , , , et al. Association of serial measures of cardiac troponin T using a sensitive assay with incident heart failure and cardiovascular mortality in older adults. JAMA. 2010;304:2494–2502.
- , , , et al. Sensitive cardiac troponin in the diagnosis and risk stratification of acute heart failure. J Intern Med. 2012;271:598–607.
- , , , et al. Outcomes and care of patients with acute heart failure syndromes and cardiac troponin elevation. Circulation. 2013;6:193–202.
- , , , et al.; ADHERE Scientific Advisory Committee and Investigators. Usefulness of B‐type natriuretic peptide and cardiac troponin levels to predict in‐hospital mortality from ADHERE. Am J Cardiol. 2008;101:231–237.
- Centers for Medicare 98:1778–1786.
- , , , et al. Meta‐analysis of observational studies in epidemiology: A proposal for reporting. Meta‐analysis of observational studies in epidemiology (moose) group. JAMA. 2000;283:2008–2012.
- , , , et al. The PRISMA statement for reporting systematic reviews and meta‐analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339:b2700.
- . Critical evaluation of the Newcastle‐Ottawa Scale for the assessment of the quality of nonrandomized studies in meta‐analyses. Eur J Epidemiol. 2010;25:603–605.
- , , , et al. Troponin I in acute decompensated heart failure: Insights from the ascend‐hf study. Eur J Heart Fail. 2012;14:1257–1264.
- , , , , , . Usefulness of an elevated troponin‐I in predicting clinical events in patients admitted with acute heart failure and acute coronary syndrome (from the RITZ‐4 trial). Am J Cardiol. 2004;93:1436–1437.
- , , , et al. Impact of serial troponin release on outcomes in patients with acute heart failure: analysis from the protect pilot study. Circulation. 2011;4:724–732.
- , , , et al. Ongoing myocardial injury in stable severe heart failure: value of cardiac troponin t monitoring for high‐risk patient identification. Circulation. 2004;110:2376–2382.
- , , , et al. Characteristics and outcomes of patients hospitalized for heart failure in the united states: rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J. 2005;149:209–216.
- , , . Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Stat Med. 1998;17:2815–2834.
- , , , , . The case of the misleading funnel plot. BMJ. 2006;333:597–600.
- , , , et al. The Newcastle‐Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta‐analyses. Available at: http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp. accessed July 3, 2015.
- , , , et al. Third universal definition of myocardial infarction. J Am Coll Cardiol. 2012;60:1581–1598.
- , , , et al.; ADHERE Investigators. Cardiac troponin and outcome in acute heart failure. N Engl J Med. 2008;358:2117–2126.
- , , , et al. Rapid assay brain natriuretic peptide and troponin I in patients hospitalized with decompensated heart failure (from the Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness trial). Am J Cardiol. 2007;100:1427–1433.
- , , , et al. Functional status and co‐morbidities are associated with in‐hospital mortality among older patients with acute decompensated heart failure: a multicentre prospective cohort study. Age Ageing. 2015;44(2):225–231.
- , , , et al. Minor myocardial damage detected by troponin T is a powerful predictor of long‐term prognosis in patients with acute decompensated heart failure. Int J Cardiol. 2005;99:253–261.
- , , , et al. Cardiac troponin T levels are associated with poor short‐ and long‐term prognosis in patients with acute cardiogenic pulmonary edema. Am Heart J. 2002;143:814–820.
- , , , et al. Clinical characteristics and predictors of in‐hospital mortality in acute heart failure with preserved left ventricular ejection fraction. Am J Cardiol. 2011;107:79–84.
- , , , et al. Risk stratification using a combination of cardiac troponin t and brain natriuretic peptide in patients hospitalized for worsening chronic heart failure. Am J Cardiol. 2002;89:691–695.
- , , , , et al. Persistently increased serum concentrations of cardiac troponin in patients with acutely decompensated heart failure are predictive of adverse outcomes. Circ J. 2007;71:1047–1051.
- , , , et al. Cardiac troponin I as diagnostic and prognostic marker in severe heart failure. J Heart Lung Transplant. 2000;19:644–652.
- , , , et al. The role of plasma biomarkers in acute heart failure. Serial changes and independent prognostic value of NT‐proBNP and cardiac troponin‐T. Eur J Heart Fail. 2007;9:776–786.
- , , , et al. Prognostic value of high sensitivity troponin T in patients with acutely decompensated heart failure and non‐detectable conventional troponin T levels. Int J Cardiol. 2013;168:3609–3612.
- , , , et al. Minor myocardial damage is a prevalent condition in patients with acute heart failure syndromes and preserved systolic function with long‐term prognostic implications: CIAST‐HF (Collaborative Italo‐Argentinean Study on cardiac Troponin T in Heart Failure) study. J Card Fail. 2012;18:822–830.
- , , , et al. Acute heart failure: clinical presentation, one‐year mortality and prognostic factors. Eur J Heart Fail. 2005;7:662–670.
- , , , et al. High‐sensitive troponin I after acute cardiac decompensation‐distribution of baseline values and prognostic significance. Paper presented at: Heart Failure Congress 2013; May 25–28, 2013; Lisbon, Portugal.
- , , , . Serial changes in high‐sensitive troponin I predict outcome in patients with decompensated heart failure. Eur J Heart Fail. 2011;13:37–42.
- , , , et al. Multimarker strategy for the prediction of 31 days cardiac death in patients with acutely decompensated chronic heart failure. Int J Cardiol. 2010;141:284–290.
- , , , , , . Cardiac troponin t for risk stratification in decompensated chronic heart failure. [in Portuguese]. Arq Bras Cardiol. 2009;92(5):404–412.
- , , , et al. Troponin T in acute heart failure: clinical implications and prognosis in the Spanish National Registry on Heart Failure. Eur J Intern Med. 2014;25:739–744.
- , , , et al. Complementary prognostic value of cystatin C, N‐terminal pro‐B‐type natriuretic Peptide and cardiac troponin T in patients with acute heart failure. Am J Cardiol. 2009;103:1753–1759.
- , , . Single cardiac troponin t measurement predicts risk for adverse outcome in decompensated heart failure. Arq Bras Cardiol. 2010;94(4):495–501.
- , , , , . Relation between cardiac troponin i and mortality in acute decompensated heart failure. Am Heart J. 2007;153:462–470.
- , , , et al. High‐sensitivity cardiac troponin t predicts non‐cardiac mortality in heart failure. Circ J. 2014;78:890–895.
- , , , et al. Highly sensitive troponin t for risk stratification of acutely destabilized heart failure. Am Heart J. 2012;163(6):1002–1010.
- , , , et al. How to read a systematic review and meta‐analysis and apply the results to patient care: Users' guides to the medical literature. JAMA. 2014;312:171–179.
- , , , et al. Serial biomarker measurements in ambulatory patients with chronic heart failure: the importance of change over time. Circulation. 2007;116:249–257.
- , , , et al. Prognostic value of very low plasma concentrations of troponin T in patients with stable chronic heart failure. Circulation. 2007;116:1242–1249.
- , , , et al. Episodes of acute heart failure syndrome are associated with increased levels of troponin and extracellular matrix markers. Circ Heart Fail. 2010;3:44–50.
- , , , et al. Cardiac microinjury measured by troponin T predicts collagen metabolism in adults aged >=65 years with heart failure. Circ Heart Fail. 2012;5:406–413.
- . Pathobiology of troponin elevations: so elevations occur with myocardial ischemia as well as necrosis? J Am Coll Cardiol. 2011;57:2406–2408.
- , , . Prognostic value of cardiac troponin in chronic stable heart failure: a systematic review. Heart. 2012;98:1778–1786.
- , , , et al. Vascular endothelial dysfunction and mortality risk in patients with chronic heart failure. Circulation. 2005;111:310–314.
- , , , , . Preload induces troponin I degradation independently of myocardial ischemia. Circulation. 2001;103:2035–2037.
- , , ,et al. Prevalence and determinants of troponin T elevation in the general population. Circulation. 2006;113:1958–1965.
- , , , et al. Cardiac troponin‐I and risk of heart failure: a community‐based cohort study. Eur Heart J. 2009;30:773–781.
- , , , et al. Age‐ and sex‐dependent upper reference limits for the high‐sensitivity cardiac troponin t assay. J Am Coll Cardiol. 2014;63:1441–1448.
- , , , et al. Defining high‐sensitivity cardiac troponin concentrations in the community. Clin Chem. 2013;59:1099–1107.
- , , , et al. Association of serial measures of cardiac troponin T using a sensitive assay with incident heart failure and cardiovascular mortality in older adults. JAMA. 2010;304:2494–2502.
- , , , et al. Sensitive cardiac troponin in the diagnosis and risk stratification of acute heart failure. J Intern Med. 2012;271:598–607.
- , , , et al. Outcomes and care of patients with acute heart failure syndromes and cardiac troponin elevation. Circulation. 2013;6:193–202.
- , , , et al.; ADHERE Scientific Advisory Committee and Investigators. Usefulness of B‐type natriuretic peptide and cardiac troponin levels to predict in‐hospital mortality from ADHERE. Am J Cardiol. 2008;101:231–237.
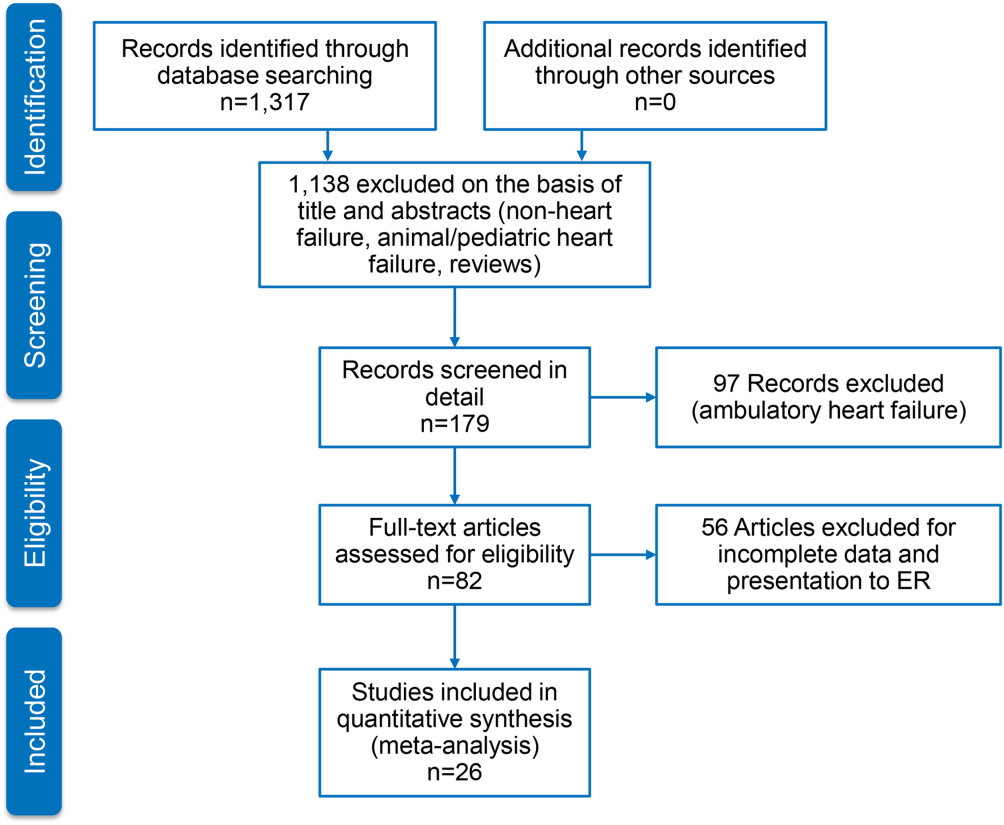
Sex assault by intimate partners as distressing as stranger attack
Sexual assault by an intimate partner was associated with a similar level of psychological distress and a greater likelihood of extragenital traumatic injuries, compared with sexual assaults by a stranger or an acquaintance.
Dr. Patrick Chariot of the department of forensic medicine at Hôpital Jean-Verdier in Bondy, France, and his colleagues conducted an observational and prospective study to compare the psychological and physical symptoms of women assaulted by intimate partners with the symptoms of women who were assaulted by an acquaintance or unknown person.
Participants included girls and women aged 15 years or older who were referred to a sexual assault center and received examination. Data was grouped according to the woman’s reported relationship to their assailant as a current or former intimate partner, stranger, or acquaintance (Obstet Gynecol. 2016 Feb;127:516–26).
A total of 767 patients were included in the study. Assault by an intimate partner was reported by 263 women, assault by an acquaintance by 229 women, and assault by a stranger by 275 women. A 1-month follow-up examination was performed in 38% of participants.
Nearly half of the study participants reported a previous physical or sexual assault. A history of previous assault was more often reported by women in the intimate partner group (71%), compared with those in the acquaintance group (49%) and those in the stranger group (28%).
Extragenital trauma was more common in women assaulted by intimate partners (52% versus 33% and 43%, in acquaintance and strangers, respectively).
The most common psychological symptoms reported at the time of examination included anxiety, fear, shame, and sadness with reports of fear being more common in those with an intimate partner assault (46% versus 30% and 25%). The most common symptoms reported at follow-up included sleep disorders, depression, fear, intrusive thoughts, social withdrawal, shame, and anxiety.
Most of the study participants had scores suggestive of a minor psychiatric disorder (89%) or post-traumatic stress disorder (79%), with similar results in all groups, according to the researchers.
“This study demonstrates that extragenital physical assaults coincident with the sexual assault are more commonly perpetrated by intimate assailants than either strangers or acquaintances,” the researchers wrote. “Additionally, there is no difference in the victim’s psychologic symptoms nor in the reaction to reports of the assault to family and friends.”
The researchers reported having no financial disclosures.
Sexual assault by an intimate partner was associated with a similar level of psychological distress and a greater likelihood of extragenital traumatic injuries, compared with sexual assaults by a stranger or an acquaintance.
Dr. Patrick Chariot of the department of forensic medicine at Hôpital Jean-Verdier in Bondy, France, and his colleagues conducted an observational and prospective study to compare the psychological and physical symptoms of women assaulted by intimate partners with the symptoms of women who were assaulted by an acquaintance or unknown person.
Participants included girls and women aged 15 years or older who were referred to a sexual assault center and received examination. Data was grouped according to the woman’s reported relationship to their assailant as a current or former intimate partner, stranger, or acquaintance (Obstet Gynecol. 2016 Feb;127:516–26).
A total of 767 patients were included in the study. Assault by an intimate partner was reported by 263 women, assault by an acquaintance by 229 women, and assault by a stranger by 275 women. A 1-month follow-up examination was performed in 38% of participants.
Nearly half of the study participants reported a previous physical or sexual assault. A history of previous assault was more often reported by women in the intimate partner group (71%), compared with those in the acquaintance group (49%) and those in the stranger group (28%).
Extragenital trauma was more common in women assaulted by intimate partners (52% versus 33% and 43%, in acquaintance and strangers, respectively).
The most common psychological symptoms reported at the time of examination included anxiety, fear, shame, and sadness with reports of fear being more common in those with an intimate partner assault (46% versus 30% and 25%). The most common symptoms reported at follow-up included sleep disorders, depression, fear, intrusive thoughts, social withdrawal, shame, and anxiety.
Most of the study participants had scores suggestive of a minor psychiatric disorder (89%) or post-traumatic stress disorder (79%), with similar results in all groups, according to the researchers.
“This study demonstrates that extragenital physical assaults coincident with the sexual assault are more commonly perpetrated by intimate assailants than either strangers or acquaintances,” the researchers wrote. “Additionally, there is no difference in the victim’s psychologic symptoms nor in the reaction to reports of the assault to family and friends.”
The researchers reported having no financial disclosures.
Sexual assault by an intimate partner was associated with a similar level of psychological distress and a greater likelihood of extragenital traumatic injuries, compared with sexual assaults by a stranger or an acquaintance.
Dr. Patrick Chariot of the department of forensic medicine at Hôpital Jean-Verdier in Bondy, France, and his colleagues conducted an observational and prospective study to compare the psychological and physical symptoms of women assaulted by intimate partners with the symptoms of women who were assaulted by an acquaintance or unknown person.
Participants included girls and women aged 15 years or older who were referred to a sexual assault center and received examination. Data was grouped according to the woman’s reported relationship to their assailant as a current or former intimate partner, stranger, or acquaintance (Obstet Gynecol. 2016 Feb;127:516–26).
A total of 767 patients were included in the study. Assault by an intimate partner was reported by 263 women, assault by an acquaintance by 229 women, and assault by a stranger by 275 women. A 1-month follow-up examination was performed in 38% of participants.
Nearly half of the study participants reported a previous physical or sexual assault. A history of previous assault was more often reported by women in the intimate partner group (71%), compared with those in the acquaintance group (49%) and those in the stranger group (28%).
Extragenital trauma was more common in women assaulted by intimate partners (52% versus 33% and 43%, in acquaintance and strangers, respectively).
The most common psychological symptoms reported at the time of examination included anxiety, fear, shame, and sadness with reports of fear being more common in those with an intimate partner assault (46% versus 30% and 25%). The most common symptoms reported at follow-up included sleep disorders, depression, fear, intrusive thoughts, social withdrawal, shame, and anxiety.
Most of the study participants had scores suggestive of a minor psychiatric disorder (89%) or post-traumatic stress disorder (79%), with similar results in all groups, according to the researchers.
“This study demonstrates that extragenital physical assaults coincident with the sexual assault are more commonly perpetrated by intimate assailants than either strangers or acquaintances,” the researchers wrote. “Additionally, there is no difference in the victim’s psychologic symptoms nor in the reaction to reports of the assault to family and friends.”
The researchers reported having no financial disclosures.
FROM OBSTETRICS & GYNECOLOGY
Key clinical point: Intimate partner sexual assault had similar psychological distress and lack of support as those assaulted by a stranger or acquaintance.
Major finding: Extragenital trauma was more common in women assaulted by an intimate partner (52% vs. 33% and 43%, in acquaintance and strangers, respectively).
Data source: An observational and prospective study of 767 women aged 15 years or older who were referred to a sexual assault center.
Disclosures: The researchers reported having no financial disclosures.
Treating maternal subclinical hypothyroidism doesn’t improve childhood IQ
ATLANTA – Prenatal treatment of maternal subclinical hypothyroidism or hypothyroxinemia conferred no cognitive, behavioral, or neurodevelopmental benefit to children up to age 5.
The findings of two parallel randomized trials suggest that prenatal screening for these conditions is not necessary, Dr. Brian Casey said at the annual Pregnancy Meeting sponsored by the Society for Maternal-Fetal Medicine. The data also support the American College of Obstetricians and Gynecologists’ 2007 recommendation against routine screening, said Dr. Casey, chief of maternal-fetal medicine and obstetrics at the University of Texas Southwestern Medical Center, Dallas.

The issue of whether to treat pregnant women for subclinical hypothyroidism has been debated for years. Studies in the early 2000s suggested that prenatal levothyroxine did improve child neurocognitive outcomes, but more recent studies, including a 2012 randomized trial, do not.
These data have led both the American College of Obstetricians and Gynecologists and the Endocrine Society to recommend against screening for subclinical hypothyroidism in pregnant women.
Dr. Casey and his colleagues conducted two large parallel randomized studies, one in women with subclinical hypothyroidism and another in women with hypothyroxinemia. Subclinical hypothyroidism was defined as a thyroid stimulating hormone (TSH) level of at least 4 mU/L with normal free T4. Hypothyroxinemia was defined as a normal TSH but a free T4 of less than 0.86 ng/dL.
The primary outcome in each group was child IQ at 5 years. Secondary outcomes were scores on neurodevelopmental and behavioral measures, including the presence of attention deficit hyperactivity disorder, conducted at ages 3, 4, and 5 years.
All women in the trials had a singleton pregnancy of less than 20 weeks’ gestation, with no known history of thyroid disease.
The subclinical hypothyroidism group comprised 677 women. They were a mean of 27 years old, with a mean gestational age of 16.5 weeks. Baseline TSH was 4.5 mU/L; baseline free T4 was 1 ng/dL. All had normal urinary iodide. They were randomized to placebo or to daily 100 mcg levothyroxine. The treatment goal was a TSH of 0.1-2.5 mU/L. Most (93%) achieved this by 21 weeks’ gestation.
There was no significant difference in the primary outcome of child IQ at 5 years between the treated and untreated groups (97 vs. 94). On the Bayley Scales of Infant and Toddler Development, scores of cognition, motor skills, and language were similar in each group at both 12 and 24 months. The Differential Ability Scales scores at 3 and 4 years were also similar. The Child Behavior Checklist scores at ages 4 years and 5 years were similar. There was no indication of an increase in ADHD.
The hypothyroxinemia trial comprised 467 women. They were randomized to placebo or to 50 mcg levothyroxine. These women were a mean of 28 years old with a mean gestational age of 18 weeks. Their baseline TSH was 1.5 mU/L, and baseline free T4 was 0.8 ng/dL. All had normal urinary iodide. The treatment goal was a free T4 of between 0.86 and 1.90 ng/dL. Most (83%) achieved this by 23 weeks’ gestation.
There was no difference on the primary outcome of child IQ at 5 years (94 vs. 91). On the Bayley Scales of Infant and Toddler Development, scores of cognition, motor skills, and language were similar in each group at both 12 and 24 months. The Differential Ability Scales scores at 3 years and 4 years were also similar. The Child Behavior Checklist scores at ages 4years and 5 years were similar. There was no indication of an increase in ADHD.
Dr. Casey added that there was no interaction between gestational age at baseline and treatment outcomes, suggesting that there may be little foundation to the argument that treating earlier in pregnancy is key.
He had no financial disclosures.
ATLANTA – Prenatal treatment of maternal subclinical hypothyroidism or hypothyroxinemia conferred no cognitive, behavioral, or neurodevelopmental benefit to children up to age 5.
The findings of two parallel randomized trials suggest that prenatal screening for these conditions is not necessary, Dr. Brian Casey said at the annual Pregnancy Meeting sponsored by the Society for Maternal-Fetal Medicine. The data also support the American College of Obstetricians and Gynecologists’ 2007 recommendation against routine screening, said Dr. Casey, chief of maternal-fetal medicine and obstetrics at the University of Texas Southwestern Medical Center, Dallas.
The issue of whether to treat pregnant women for subclinical hypothyroidism has been debated for years. Studies in the early 2000s suggested that prenatal levothyroxine did improve child neurocognitive outcomes, but more recent studies, including a 2012 randomized trial, do not.
These data have led both the American College of Obstetricians and Gynecologists and the Endocrine Society to recommend against screening for subclinical hypothyroidism in pregnant women.
Dr. Casey and his colleagues conducted two large parallel randomized studies, one in women with subclinical hypothyroidism and another in women with hypothyroxinemia. Subclinical hypothyroidism was defined as a thyroid stimulating hormone (TSH) level of at least 4 mU/L with normal free T4. Hypothyroxinemia was defined as a normal TSH but a free T4 of less than 0.86 ng/dL.
The primary outcome in each group was child IQ at 5 years. Secondary outcomes were scores on neurodevelopmental and behavioral measures, including the presence of attention deficit hyperactivity disorder, conducted at ages 3, 4, and 5 years.
All women in the trials had a singleton pregnancy of less than 20 weeks’ gestation, with no known history of thyroid disease.
The subclinical hypothyroidism group comprised 677 women. They were a mean of 27 years old, with a mean gestational age of 16.5 weeks. Baseline TSH was 4.5 mU/L; baseline free T4 was 1 ng/dL. All had normal urinary iodide. They were randomized to placebo or to daily 100 mcg levothyroxine. The treatment goal was a TSH of 0.1-2.5 mU/L. Most (93%) achieved this by 21 weeks’ gestation.
There was no significant difference in the primary outcome of child IQ at 5 years between the treated and untreated groups (97 vs. 94). On the Bayley Scales of Infant and Toddler Development, scores of cognition, motor skills, and language were similar in each group at both 12 and 24 months. The Differential Ability Scales scores at 3 and 4 years were also similar. The Child Behavior Checklist scores at ages 4 years and 5 years were similar. There was no indication of an increase in ADHD.
The hypothyroxinemia trial comprised 467 women. They were randomized to placebo or to 50 mcg levothyroxine. These women were a mean of 28 years old with a mean gestational age of 18 weeks. Their baseline TSH was 1.5 mU/L, and baseline free T4 was 0.8 ng/dL. All had normal urinary iodide. The treatment goal was a free T4 of between 0.86 and 1.90 ng/dL. Most (83%) achieved this by 23 weeks’ gestation.
There was no difference on the primary outcome of child IQ at 5 years (94 vs. 91). On the Bayley Scales of Infant and Toddler Development, scores of cognition, motor skills, and language were similar in each group at both 12 and 24 months. The Differential Ability Scales scores at 3 years and 4 years were also similar. The Child Behavior Checklist scores at ages 4years and 5 years were similar. There was no indication of an increase in ADHD.
Dr. Casey added that there was no interaction between gestational age at baseline and treatment outcomes, suggesting that there may be little foundation to the argument that treating earlier in pregnancy is key.
He had no financial disclosures.
ATLANTA – Prenatal treatment of maternal subclinical hypothyroidism or hypothyroxinemia conferred no cognitive, behavioral, or neurodevelopmental benefit to children up to age 5.
The findings of two parallel randomized trials suggest that prenatal screening for these conditions is not necessary, Dr. Brian Casey said at the annual Pregnancy Meeting sponsored by the Society for Maternal-Fetal Medicine. The data also support the American College of Obstetricians and Gynecologists’ 2007 recommendation against routine screening, said Dr. Casey, chief of maternal-fetal medicine and obstetrics at the University of Texas Southwestern Medical Center, Dallas.
The issue of whether to treat pregnant women for subclinical hypothyroidism has been debated for years. Studies in the early 2000s suggested that prenatal levothyroxine did improve child neurocognitive outcomes, but more recent studies, including a 2012 randomized trial, do not.
These data have led both the American College of Obstetricians and Gynecologists and the Endocrine Society to recommend against screening for subclinical hypothyroidism in pregnant women.
Dr. Casey and his colleagues conducted two large parallel randomized studies, one in women with subclinical hypothyroidism and another in women with hypothyroxinemia. Subclinical hypothyroidism was defined as a thyroid stimulating hormone (TSH) level of at least 4 mU/L with normal free T4. Hypothyroxinemia was defined as a normal TSH but a free T4 of less than 0.86 ng/dL.
The primary outcome in each group was child IQ at 5 years. Secondary outcomes were scores on neurodevelopmental and behavioral measures, including the presence of attention deficit hyperactivity disorder, conducted at ages 3, 4, and 5 years.
All women in the trials had a singleton pregnancy of less than 20 weeks’ gestation, with no known history of thyroid disease.
The subclinical hypothyroidism group comprised 677 women. They were a mean of 27 years old, with a mean gestational age of 16.5 weeks. Baseline TSH was 4.5 mU/L; baseline free T4 was 1 ng/dL. All had normal urinary iodide. They were randomized to placebo or to daily 100 mcg levothyroxine. The treatment goal was a TSH of 0.1-2.5 mU/L. Most (93%) achieved this by 21 weeks’ gestation.
There was no significant difference in the primary outcome of child IQ at 5 years between the treated and untreated groups (97 vs. 94). On the Bayley Scales of Infant and Toddler Development, scores of cognition, motor skills, and language were similar in each group at both 12 and 24 months. The Differential Ability Scales scores at 3 and 4 years were also similar. The Child Behavior Checklist scores at ages 4 years and 5 years were similar. There was no indication of an increase in ADHD.
The hypothyroxinemia trial comprised 467 women. They were randomized to placebo or to 50 mcg levothyroxine. These women were a mean of 28 years old with a mean gestational age of 18 weeks. Their baseline TSH was 1.5 mU/L, and baseline free T4 was 0.8 ng/dL. All had normal urinary iodide. The treatment goal was a free T4 of between 0.86 and 1.90 ng/dL. Most (83%) achieved this by 23 weeks’ gestation.
There was no difference on the primary outcome of child IQ at 5 years (94 vs. 91). On the Bayley Scales of Infant and Toddler Development, scores of cognition, motor skills, and language were similar in each group at both 12 and 24 months. The Differential Ability Scales scores at 3 years and 4 years were also similar. The Child Behavior Checklist scores at ages 4years and 5 years were similar. There was no indication of an increase in ADHD.
Dr. Casey added that there was no interaction between gestational age at baseline and treatment outcomes, suggesting that there may be little foundation to the argument that treating earlier in pregnancy is key.
He had no financial disclosures.
AT THE PREGNANCY MEETING
Key clinical point: Levothyroxine for women with subclinical hypothyroidism didn’t boost childhood cognition.
Major finding: Child IQ at 5 years old was 97 in the treated group and 94 in the placebo group – not a significant difference.
Data source: The parallel randomized controlled trials comprised a total of 1,144 women.
Disclosures: Dr. Casey had no financial disclosures.

Seven myths about sex and relationships in LGBT youth
Many lesbian, gay, bisexual, and transgender (LGBT) youth face misconceptions about their sexual or gender identity. This is especially true when it comes to sex and relationships. Unfortunately, many clinicians believe these myths, and they can have devastating consequences on the health of LGBT youth.
Here are some common myths about sex and relationships in LGBT youth, and how you, as a provider, can combat them with knowledge and compassion:
Myth No. 1: Bisexual youth are promiscuous. This is a stereotype that even plagues bisexual adults. There is a persistent misconception that just because bisexuals are attracted to both sexes, they are naturally promiscuous. In fact, most bisexuals describe themselves as monogamous.1

Myth No. 2: Youth who are transgender are lesbian/gay/bisexual before transition and are straight after transition. According to the National Transgender Discrimination Survey, regardless of where they are in the transition process, 23% of transgender people identify as heterosexual, 23% identify as gay or lesbian, 25% identify as bisexual, 23% label themselves as queer, 4% describe themselves as asexual and 2% wrote in other answers.2
Myth No. 3: Gay and lesbian teens only have sex or romantic relationships with the same sex. According to the Youth Risk Behavior Survey, although 22% of lesbian and gay teens say they have sex with the same sex only, about 9% say that they have sex with both sexes.3 This shows that sexual identity does not predict sexual behavior and has important implications for the following myths.
Myth No. 4: Lesbian and bisexual girls don’t experience intimate partner violence. Because the majority of those who perpetrate intimate partner violence are men, it is tempting to assume that lesbian and bisexual teenage girls don’t experience abuse in their relationships.
Unfortunately, one study shows that 42% of lesbian and bisexual girls experienced intimate partner violence in the past, compared with 16% of heterosexual girls.4 However, this study and others do not tell us whether they have experienced abuse in their relationships with girls or with boys.
Myth No. 5: Lesbian girls can’t get gonorrhea or chlamydia or pelvic inflammatory disease (PID). About 2% of young lesbians report ever having any sexually transmitted infection (STI). A small percentage of young lesbians report having chlamydia, and this is associated with PID. It is true, however, that gonorrhea is rare among lesbians,5 but don’t forget that young lesbian women may have had sex with men.
Interestingly, the prevalence of bacterial vaginosis, a condition characterized by overgrowth of vaginal anaerobic bacteria, is higher in young women who have sex with women.6 Possible sources of transmission include digital-to-vaginal contact, oral sex, or sex toys.
Myth No. 6: Young women who have sex with women can’t get pregnant, so you don’t have to worry about birth control. Don’t forget that heterosexuals use birth control for other reasons than preventing pregnancy. Some women use birth control to help regulate periods, to ease cramping, or to treat acne. Lesbians and bisexual girls are at the same risk for these problems as are heterosexual girls, so don’t assume that they’re not interested in birth control just because they are not concerned about getting pregnant.
Also, as previously mentioned, lesbian girls may be having sex with boys, so conversations about birth control should be driven by who they are having sex with, not by how they identify.
Myth No. 7: Gay boys can’t get girls pregnant. Lesbian girls can’t get pregnant. A study by the Toronto Teen Sex Survey found that 28% of sexual minority youth report involvement in pregnancy, compared with 7% of heterosexual youth.7
Now many who are reading this may be scratching their heads. If someone finds the same sex attractive, then why are they engaging in heterosexual sex? Some studies suggest that engaging in heterosexual sex is a way to hide their true sexual orientation,8 because we live in a heterosexist and homophobic environment. After all, what better way to prove that you’re heterosexual? Another study suggests that intentionally getting pregnant or getting someone pregnant is the quickest way to parenthood, and becoming a parent can compensate for one’s identity as a sexual minority.9
So how do you overcome these persistent myths? The most important thing to do is not assume. Identity and behaviors are not the same. Always be specific when you’re asking questions about sex and relationships in LGBT youth.
The Centers for Disease Control and Prevention (CDC) recommends the following when obtaining a sexual history:
• Ask, “Are your sexual partner’s male, female, or both?”
• Ask, “When you do have sex with your partner, what do you do?” Here, you have to be very specific. Younger teenagers tend to be concrete thinkers, so don’t just ask “Are you sexually active?” Instead, try asking, “Have you ever had a penis in your mouth, vagina, or anus?” or “Do you use sex toys?”
• In terms of protection from STIs, you might ask, “Do you use condoms or a dental dam?”
• Ask, “Have you ever had an STI, and if so, how was it treated?”
• Ask, “What do you use for birth control?” either hormonal or barrier methods.
In addition to above questions, I would also ask about intimate partner violence. Often, health care providers may ask if their patient has been hit, punch, slapped, or kicked by their partners. But intimate partner violence can go beyond physical violence. It also involves emotional manipulation or birth control sabotage. Sometimes, it is better to ask if a patient has been forced to do something sexual with her partners when she didn’t want to. The patient may deny it, however, even though you highly suspect it. So it is better to remember to build a rapport, and when the patient is ready to get out of an abusive relationship, he or she will come to you for help.
Some clinicians have told me that they have a hard time asking sexual histories in LGBT youth because they’re afraid of offending them, especially when it comes to asking about sex with the opposite sex. This is a valid concern and an area of ongoing research, but I think that by making things normative, just like with any behavior, teens and young adults are more likely to disclose critical pieces of information. It is a good idea, then, to start off with “Because of homophobia, many LGBT youth may engage in heterosexual sex. Tell me, have you ever…”
By not assuming and asking specific questions, LGBT youth are more likely to tell their health care provider important information. With that information, health care providers can prevent many adverse health outcomes like teen pregnancy, STIs, and intimate partner violence. It also will give health care providers an opportunity to address the rampant stigma and discrimination that plagues this vulnerable population.
Here are some resources on sex and relationships in LGBT youth:
• The CDC 2015 STI Guidelines have a special section on STIs in men who have sex with men, women who have sex with women, and transgender men and women.
• Bedsider.org is an excellent website about birth control options and STI prevention for all sexual orientations and gender identities.
• Futures Without Violence provides resources for health care professionals to manage and prevent intimate partner violence.
References
2. National Transgender Discrimination Survey: Full Report. 2012.
3. MMWR Surveill Summ. 2011 Jun 10;60(7):1-133.
4. J Youth Adolesc. 2015 Jan;44(1):211-24.
5. Perspect Sex Reprod Health. 2008 Dec;40(4):212-7.
6. Sex Transm Dis. 2010 May;37(5):335-9.
7. Sexpress: The Toronto teen survey report. 2009.
8. Fletcher RC. Social context and social support: Exploring the lived experiences of LGBTQ youth who have been pregnant. [Master’s Project]: School of Public Health, University of Minnesota; 2011.
9. Can J Hum Sex. 2008;17(3):123-139.
Dr. Montano is an adolescent medicine fellow at Children’s Hospital of Pittsburgh of UPMC and a postdoctoral fellow in the department of pediatrics at the University of Pittsburgh. He has no relevant financial disclosures.
Many lesbian, gay, bisexual, and transgender (LGBT) youth face misconceptions about their sexual or gender identity. This is especially true when it comes to sex and relationships. Unfortunately, many clinicians believe these myths, and they can have devastating consequences on the health of LGBT youth.
Here are some common myths about sex and relationships in LGBT youth, and how you, as a provider, can combat them with knowledge and compassion:
Myth No. 1: Bisexual youth are promiscuous. This is a stereotype that even plagues bisexual adults. There is a persistent misconception that just because bisexuals are attracted to both sexes, they are naturally promiscuous. In fact, most bisexuals describe themselves as monogamous.1
Myth No. 2: Youth who are transgender are lesbian/gay/bisexual before transition and are straight after transition. According to the National Transgender Discrimination Survey, regardless of where they are in the transition process, 23% of transgender people identify as heterosexual, 23% identify as gay or lesbian, 25% identify as bisexual, 23% label themselves as queer, 4% describe themselves as asexual and 2% wrote in other answers.2
Myth No. 3: Gay and lesbian teens only have sex or romantic relationships with the same sex. According to the Youth Risk Behavior Survey, although 22% of lesbian and gay teens say they have sex with the same sex only, about 9% say that they have sex with both sexes.3 This shows that sexual identity does not predict sexual behavior and has important implications for the following myths.
Myth No. 4: Lesbian and bisexual girls don’t experience intimate partner violence. Because the majority of those who perpetrate intimate partner violence are men, it is tempting to assume that lesbian and bisexual teenage girls don’t experience abuse in their relationships.
Unfortunately, one study shows that 42% of lesbian and bisexual girls experienced intimate partner violence in the past, compared with 16% of heterosexual girls.4 However, this study and others do not tell us whether they have experienced abuse in their relationships with girls or with boys.
Myth No. 5: Lesbian girls can’t get gonorrhea or chlamydia or pelvic inflammatory disease (PID). About 2% of young lesbians report ever having any sexually transmitted infection (STI). A small percentage of young lesbians report having chlamydia, and this is associated with PID. It is true, however, that gonorrhea is rare among lesbians,5 but don’t forget that young lesbian women may have had sex with men.
Interestingly, the prevalence of bacterial vaginosis, a condition characterized by overgrowth of vaginal anaerobic bacteria, is higher in young women who have sex with women.6 Possible sources of transmission include digital-to-vaginal contact, oral sex, or sex toys.
Myth No. 6: Young women who have sex with women can’t get pregnant, so you don’t have to worry about birth control. Don’t forget that heterosexuals use birth control for other reasons than preventing pregnancy. Some women use birth control to help regulate periods, to ease cramping, or to treat acne. Lesbians and bisexual girls are at the same risk for these problems as are heterosexual girls, so don’t assume that they’re not interested in birth control just because they are not concerned about getting pregnant.
Also, as previously mentioned, lesbian girls may be having sex with boys, so conversations about birth control should be driven by who they are having sex with, not by how they identify.
Myth No. 7: Gay boys can’t get girls pregnant. Lesbian girls can’t get pregnant. A study by the Toronto Teen Sex Survey found that 28% of sexual minority youth report involvement in pregnancy, compared with 7% of heterosexual youth.7
Now many who are reading this may be scratching their heads. If someone finds the same sex attractive, then why are they engaging in heterosexual sex? Some studies suggest that engaging in heterosexual sex is a way to hide their true sexual orientation,8 because we live in a heterosexist and homophobic environment. After all, what better way to prove that you’re heterosexual? Another study suggests that intentionally getting pregnant or getting someone pregnant is the quickest way to parenthood, and becoming a parent can compensate for one’s identity as a sexual minority.9
So how do you overcome these persistent myths? The most important thing to do is not assume. Identity and behaviors are not the same. Always be specific when you’re asking questions about sex and relationships in LGBT youth.
The Centers for Disease Control and Prevention (CDC) recommends the following when obtaining a sexual history:
• Ask, “Are your sexual partner’s male, female, or both?”
• Ask, “When you do have sex with your partner, what do you do?” Here, you have to be very specific. Younger teenagers tend to be concrete thinkers, so don’t just ask “Are you sexually active?” Instead, try asking, “Have you ever had a penis in your mouth, vagina, or anus?” or “Do you use sex toys?”
• In terms of protection from STIs, you might ask, “Do you use condoms or a dental dam?”
• Ask, “Have you ever had an STI, and if so, how was it treated?”
• Ask, “What do you use for birth control?” either hormonal or barrier methods.
In addition to above questions, I would also ask about intimate partner violence. Often, health care providers may ask if their patient has been hit, punch, slapped, or kicked by their partners. But intimate partner violence can go beyond physical violence. It also involves emotional manipulation or birth control sabotage. Sometimes, it is better to ask if a patient has been forced to do something sexual with her partners when she didn’t want to. The patient may deny it, however, even though you highly suspect it. So it is better to remember to build a rapport, and when the patient is ready to get out of an abusive relationship, he or she will come to you for help.
Some clinicians have told me that they have a hard time asking sexual histories in LGBT youth because they’re afraid of offending them, especially when it comes to asking about sex with the opposite sex. This is a valid concern and an area of ongoing research, but I think that by making things normative, just like with any behavior, teens and young adults are more likely to disclose critical pieces of information. It is a good idea, then, to start off with “Because of homophobia, many LGBT youth may engage in heterosexual sex. Tell me, have you ever…”
By not assuming and asking specific questions, LGBT youth are more likely to tell their health care provider important information. With that information, health care providers can prevent many adverse health outcomes like teen pregnancy, STIs, and intimate partner violence. It also will give health care providers an opportunity to address the rampant stigma and discrimination that plagues this vulnerable population.
Here are some resources on sex and relationships in LGBT youth:
• The CDC 2015 STI Guidelines have a special section on STIs in men who have sex with men, women who have sex with women, and transgender men and women.
• Bedsider.org is an excellent website about birth control options and STI prevention for all sexual orientations and gender identities.
• Futures Without Violence provides resources for health care professionals to manage and prevent intimate partner violence.
References
2. National Transgender Discrimination Survey: Full Report. 2012.
3. MMWR Surveill Summ. 2011 Jun 10;60(7):1-133.
4. J Youth Adolesc. 2015 Jan;44(1):211-24.
5. Perspect Sex Reprod Health. 2008 Dec;40(4):212-7.
6. Sex Transm Dis. 2010 May;37(5):335-9.
7. Sexpress: The Toronto teen survey report. 2009.
8. Fletcher RC. Social context and social support: Exploring the lived experiences of LGBTQ youth who have been pregnant. [Master’s Project]: School of Public Health, University of Minnesota; 2011.
9. Can J Hum Sex. 2008;17(3):123-139.
Dr. Montano is an adolescent medicine fellow at Children’s Hospital of Pittsburgh of UPMC and a postdoctoral fellow in the department of pediatrics at the University of Pittsburgh. He has no relevant financial disclosures.
Many lesbian, gay, bisexual, and transgender (LGBT) youth face misconceptions about their sexual or gender identity. This is especially true when it comes to sex and relationships. Unfortunately, many clinicians believe these myths, and they can have devastating consequences on the health of LGBT youth.
Here are some common myths about sex and relationships in LGBT youth, and how you, as a provider, can combat them with knowledge and compassion:
Myth No. 1: Bisexual youth are promiscuous. This is a stereotype that even plagues bisexual adults. There is a persistent misconception that just because bisexuals are attracted to both sexes, they are naturally promiscuous. In fact, most bisexuals describe themselves as monogamous.1
Myth No. 2: Youth who are transgender are lesbian/gay/bisexual before transition and are straight after transition. According to the National Transgender Discrimination Survey, regardless of where they are in the transition process, 23% of transgender people identify as heterosexual, 23% identify as gay or lesbian, 25% identify as bisexual, 23% label themselves as queer, 4% describe themselves as asexual and 2% wrote in other answers.2
Myth No. 3: Gay and lesbian teens only have sex or romantic relationships with the same sex. According to the Youth Risk Behavior Survey, although 22% of lesbian and gay teens say they have sex with the same sex only, about 9% say that they have sex with both sexes.3 This shows that sexual identity does not predict sexual behavior and has important implications for the following myths.
Myth No. 4: Lesbian and bisexual girls don’t experience intimate partner violence. Because the majority of those who perpetrate intimate partner violence are men, it is tempting to assume that lesbian and bisexual teenage girls don’t experience abuse in their relationships.
Unfortunately, one study shows that 42% of lesbian and bisexual girls experienced intimate partner violence in the past, compared with 16% of heterosexual girls.4 However, this study and others do not tell us whether they have experienced abuse in their relationships with girls or with boys.
Myth No. 5: Lesbian girls can’t get gonorrhea or chlamydia or pelvic inflammatory disease (PID). About 2% of young lesbians report ever having any sexually transmitted infection (STI). A small percentage of young lesbians report having chlamydia, and this is associated with PID. It is true, however, that gonorrhea is rare among lesbians,5 but don’t forget that young lesbian women may have had sex with men.
Interestingly, the prevalence of bacterial vaginosis, a condition characterized by overgrowth of vaginal anaerobic bacteria, is higher in young women who have sex with women.6 Possible sources of transmission include digital-to-vaginal contact, oral sex, or sex toys.
Myth No. 6: Young women who have sex with women can’t get pregnant, so you don’t have to worry about birth control. Don’t forget that heterosexuals use birth control for other reasons than preventing pregnancy. Some women use birth control to help regulate periods, to ease cramping, or to treat acne. Lesbians and bisexual girls are at the same risk for these problems as are heterosexual girls, so don’t assume that they’re not interested in birth control just because they are not concerned about getting pregnant.
Also, as previously mentioned, lesbian girls may be having sex with boys, so conversations about birth control should be driven by who they are having sex with, not by how they identify.
Myth No. 7: Gay boys can’t get girls pregnant. Lesbian girls can’t get pregnant. A study by the Toronto Teen Sex Survey found that 28% of sexual minority youth report involvement in pregnancy, compared with 7% of heterosexual youth.7
Now many who are reading this may be scratching their heads. If someone finds the same sex attractive, then why are they engaging in heterosexual sex? Some studies suggest that engaging in heterosexual sex is a way to hide their true sexual orientation,8 because we live in a heterosexist and homophobic environment. After all, what better way to prove that you’re heterosexual? Another study suggests that intentionally getting pregnant or getting someone pregnant is the quickest way to parenthood, and becoming a parent can compensate for one’s identity as a sexual minority.9
So how do you overcome these persistent myths? The most important thing to do is not assume. Identity and behaviors are not the same. Always be specific when you’re asking questions about sex and relationships in LGBT youth.
The Centers for Disease Control and Prevention (CDC) recommends the following when obtaining a sexual history:
• Ask, “Are your sexual partner’s male, female, or both?”
• Ask, “When you do have sex with your partner, what do you do?” Here, you have to be very specific. Younger teenagers tend to be concrete thinkers, so don’t just ask “Are you sexually active?” Instead, try asking, “Have you ever had a penis in your mouth, vagina, or anus?” or “Do you use sex toys?”
• In terms of protection from STIs, you might ask, “Do you use condoms or a dental dam?”
• Ask, “Have you ever had an STI, and if so, how was it treated?”
• Ask, “What do you use for birth control?” either hormonal or barrier methods.
In addition to above questions, I would also ask about intimate partner violence. Often, health care providers may ask if their patient has been hit, punch, slapped, or kicked by their partners. But intimate partner violence can go beyond physical violence. It also involves emotional manipulation or birth control sabotage. Sometimes, it is better to ask if a patient has been forced to do something sexual with her partners when she didn’t want to. The patient may deny it, however, even though you highly suspect it. So it is better to remember to build a rapport, and when the patient is ready to get out of an abusive relationship, he or she will come to you for help.
Some clinicians have told me that they have a hard time asking sexual histories in LGBT youth because they’re afraid of offending them, especially when it comes to asking about sex with the opposite sex. This is a valid concern and an area of ongoing research, but I think that by making things normative, just like with any behavior, teens and young adults are more likely to disclose critical pieces of information. It is a good idea, then, to start off with “Because of homophobia, many LGBT youth may engage in heterosexual sex. Tell me, have you ever…”
By not assuming and asking specific questions, LGBT youth are more likely to tell their health care provider important information. With that information, health care providers can prevent many adverse health outcomes like teen pregnancy, STIs, and intimate partner violence. It also will give health care providers an opportunity to address the rampant stigma and discrimination that plagues this vulnerable population.
Here are some resources on sex and relationships in LGBT youth:
• The CDC 2015 STI Guidelines have a special section on STIs in men who have sex with men, women who have sex with women, and transgender men and women.
• Bedsider.org is an excellent website about birth control options and STI prevention for all sexual orientations and gender identities.
• Futures Without Violence provides resources for health care professionals to manage and prevent intimate partner violence.
References
2. National Transgender Discrimination Survey: Full Report. 2012.
3. MMWR Surveill Summ. 2011 Jun 10;60(7):1-133.
4. J Youth Adolesc. 2015 Jan;44(1):211-24.
5. Perspect Sex Reprod Health. 2008 Dec;40(4):212-7.
6. Sex Transm Dis. 2010 May;37(5):335-9.
7. Sexpress: The Toronto teen survey report. 2009.
8. Fletcher RC. Social context and social support: Exploring the lived experiences of LGBTQ youth who have been pregnant. [Master’s Project]: School of Public Health, University of Minnesota; 2011.
9. Can J Hum Sex. 2008;17(3):123-139.
Dr. Montano is an adolescent medicine fellow at Children’s Hospital of Pittsburgh of UPMC and a postdoctoral fellow in the department of pediatrics at the University of Pittsburgh. He has no relevant financial disclosures.

Show your FACS pride
The Division of Member Services strives to educate surgical patients about what it means to be treated by a surgeon who is a Fellow of the American College of Surgeons (FACS). As part of this campaign, the College has created a poster of the Fellowship Pledge that is suitable for display in waiting areas, exam rooms, and offices, and is available for purchase or free download. The Fellowship Pledge poster—available in seven languages: English, Arabic, Chinese (both Simplified and Traditional), Japanese, German, Portuguese and Spanish—may be displayed only by surgeons with the FACS designation.
ACS Fellows are committed to providing their patients with the highest standards of surgical care and pledge to protect the welfare and rights of their patients, to respect each patient’s autonomy and individuality, and to advance their knowledge and skills throughout their careers. The Fellowship Pledge poster is available for purchase for $10.00 including standard shipping, or can be downloaded for free. Visit the ACS Online Store at https://goo.gl/sCKiUc to download your poster. Two PDF versions are available; a 22” x 32” poster that may be printed by a professional print shop, or an 11” x 17” version that may be printed on a personal color printer.
The Division of Member Services strives to educate surgical patients about what it means to be treated by a surgeon who is a Fellow of the American College of Surgeons (FACS). As part of this campaign, the College has created a poster of the Fellowship Pledge that is suitable for display in waiting areas, exam rooms, and offices, and is available for purchase or free download. The Fellowship Pledge poster—available in seven languages: English, Arabic, Chinese (both Simplified and Traditional), Japanese, German, Portuguese and Spanish—may be displayed only by surgeons with the FACS designation.
ACS Fellows are committed to providing their patients with the highest standards of surgical care and pledge to protect the welfare and rights of their patients, to respect each patient’s autonomy and individuality, and to advance their knowledge and skills throughout their careers. The Fellowship Pledge poster is available for purchase for $10.00 including standard shipping, or can be downloaded for free. Visit the ACS Online Store at https://goo.gl/sCKiUc to download your poster. Two PDF versions are available; a 22” x 32” poster that may be printed by a professional print shop, or an 11” x 17” version that may be printed on a personal color printer.
The Division of Member Services strives to educate surgical patients about what it means to be treated by a surgeon who is a Fellow of the American College of Surgeons (FACS). As part of this campaign, the College has created a poster of the Fellowship Pledge that is suitable for display in waiting areas, exam rooms, and offices, and is available for purchase or free download. The Fellowship Pledge poster—available in seven languages: English, Arabic, Chinese (both Simplified and Traditional), Japanese, German, Portuguese and Spanish—may be displayed only by surgeons with the FACS designation.
ACS Fellows are committed to providing their patients with the highest standards of surgical care and pledge to protect the welfare and rights of their patients, to respect each patient’s autonomy and individuality, and to advance their knowledge and skills throughout their careers. The Fellowship Pledge poster is available for purchase for $10.00 including standard shipping, or can be downloaded for free. Visit the ACS Online Store at https://goo.gl/sCKiUc to download your poster. Two PDF versions are available; a 22” x 32” poster that may be printed by a professional print shop, or an 11” x 17” version that may be printed on a personal color printer.
Registration opens for 2016 Leadership & Advocacy Summit
Registration is now open for the fifth annual American College of Surgeons (ACS) 2016 Leadership & Advocacy Summit, April 9–12, at the JW Marriott, Washington, DC. The Summit is a dual meeting that offers volunteer leaders and advocates educational sessions fon effective surgeon leadership, as well as interactive advocacy training with coordinated visits to congressional offices.
The 2016 Leadership Summit
(https://www.facs.org/advocacy/participate/summit-2016) will commence the evening of Saturday, April 9, with a Welcome Reception and continue the morning of April 12 with presentations on conflict management, managing difficult or “courageous” conversations, and emotional intelligence. In addition, Adil Haider, MD, MPH, FACS, director, Kessler Center for Surgery and Public Health (CSPH), a joint initiative of Brigham and Women’s Hospital, Harvard Medical School, and the Harvard T.H. Chan School of Public Health, Boston, MA, will discuss unconscious bias and cultural competency in surgical care. During lunch, attendees will meet in small groups by state/region to identify areas for unified efforts in the upcoming year.
The Advocacy Summit will begin the evening of Sunday, April 10, with a keynote address by MSNBC Hardball host, Chris Matthews, who is also a frequent commentator and expert analyst on NBC’s TODAY Show. Monday, April 11, attendees will hear from speakers examining the political environment in Washington, DC, and across the nation, and vital health care issues.
Tuesday morning, attendees will use the knowledge gathered at the Summit when they meet with their senators and representative and/or congressional staff. Tuesday’s meetings provide an opportunity to rally surgery’s collective grassroots advocacy voice on vital issues.
For more information or to register for the 2016 Leadership & Advocacy Summit, go to the ACS website. (https://www.facs.org/advocacy/participate/summit-2016 ) The conference hotel reservation deadline is Friday, March 4.
Registration is now open for the fifth annual American College of Surgeons (ACS) 2016 Leadership & Advocacy Summit, April 9–12, at the JW Marriott, Washington, DC. The Summit is a dual meeting that offers volunteer leaders and advocates educational sessions fon effective surgeon leadership, as well as interactive advocacy training with coordinated visits to congressional offices.
The 2016 Leadership Summit
(https://www.facs.org/advocacy/participate/summit-2016) will commence the evening of Saturday, April 9, with a Welcome Reception and continue the morning of April 12 with presentations on conflict management, managing difficult or “courageous” conversations, and emotional intelligence. In addition, Adil Haider, MD, MPH, FACS, director, Kessler Center for Surgery and Public Health (CSPH), a joint initiative of Brigham and Women’s Hospital, Harvard Medical School, and the Harvard T.H. Chan School of Public Health, Boston, MA, will discuss unconscious bias and cultural competency in surgical care. During lunch, attendees will meet in small groups by state/region to identify areas for unified efforts in the upcoming year.
The Advocacy Summit will begin the evening of Sunday, April 10, with a keynote address by MSNBC Hardball host, Chris Matthews, who is also a frequent commentator and expert analyst on NBC’s TODAY Show. Monday, April 11, attendees will hear from speakers examining the political environment in Washington, DC, and across the nation, and vital health care issues.
Tuesday morning, attendees will use the knowledge gathered at the Summit when they meet with their senators and representative and/or congressional staff. Tuesday’s meetings provide an opportunity to rally surgery’s collective grassroots advocacy voice on vital issues.
For more information or to register for the 2016 Leadership & Advocacy Summit, go to the ACS website. (https://www.facs.org/advocacy/participate/summit-2016 ) The conference hotel reservation deadline is Friday, March 4.
Registration is now open for the fifth annual American College of Surgeons (ACS) 2016 Leadership & Advocacy Summit, April 9–12, at the JW Marriott, Washington, DC. The Summit is a dual meeting that offers volunteer leaders and advocates educational sessions fon effective surgeon leadership, as well as interactive advocacy training with coordinated visits to congressional offices.
The 2016 Leadership Summit
(https://www.facs.org/advocacy/participate/summit-2016) will commence the evening of Saturday, April 9, with a Welcome Reception and continue the morning of April 12 with presentations on conflict management, managing difficult or “courageous” conversations, and emotional intelligence. In addition, Adil Haider, MD, MPH, FACS, director, Kessler Center for Surgery and Public Health (CSPH), a joint initiative of Brigham and Women’s Hospital, Harvard Medical School, and the Harvard T.H. Chan School of Public Health, Boston, MA, will discuss unconscious bias and cultural competency in surgical care. During lunch, attendees will meet in small groups by state/region to identify areas for unified efforts in the upcoming year.
The Advocacy Summit will begin the evening of Sunday, April 10, with a keynote address by MSNBC Hardball host, Chris Matthews, who is also a frequent commentator and expert analyst on NBC’s TODAY Show. Monday, April 11, attendees will hear from speakers examining the political environment in Washington, DC, and across the nation, and vital health care issues.
Tuesday morning, attendees will use the knowledge gathered at the Summit when they meet with their senators and representative and/or congressional staff. Tuesday’s meetings provide an opportunity to rally surgery’s collective grassroots advocacy voice on vital issues.
For more information or to register for the 2016 Leadership & Advocacy Summit, go to the ACS website. (https://www.facs.org/advocacy/participate/summit-2016 ) The conference hotel reservation deadline is Friday, March 4.

Lead poisoning
Lead poisoning is a well-established cause of serious and permanent neurological, cognitive, and behavioral problems, particularly in exposed children.
Children can be exposed to lead from ingesting paint chips in their homes, when old paint is scrapped from the exterior of houses or bridges, and through the water they drink. The damage caused by lead poisoning was first recognized in the United States in the early 20th century, although lead was added to gasoline and paint until the 1970’s. Since then, regulations for lead in consumer products have become increasingly strict, and the Centers for Disease Control and Prevention’s definition of a toxic lead level has shifted from 60 micrograms/deciliter (mcg/dL) in 1970 to 5 mcg/dL in 2012. In many communities, removing lead paint up to the height of a young child is a requirement whenever an older home is sold.
Unfortunately, these regulations did not protect the families in Flint, Michigan from being exposed to high levels of lead when a change in water supply and inadequate water treatment allowed lead to enter the system from decaying water pipes. It is worth reviewing what is known about the short- and long-term consequences of lead exposure, and what lies ahead for the children of Flint.
Lead is a naturally occurring element that is not metabolized, but rather absorbed, distributed to tissues, and excreted. Lead can be inhaled (with 100% absorption) and introduced through the GI tract (with about 70% absorption in children and 20% absorption in adults). GI absorption is enhanced by calcium or iron deficiency, both conditions that are relatively common, especially in poor children and can lead to pica (or eating of non-nutritious materials), further increasing the chances of lead exposure. Absorbed lead is distributed to blood (for 28-36 days), soft tissue, including the nervous system (40 days), and to bone (where it lasts for over 25 years). Blood that is retained in growing bones can be mobilized during periods of physiologic stress (such as illness, injury, or pregnancy), meaning children exposed to lead during a period of rapid bone growth are at long-term risk for acute lead poisoning from their endogenous reservoir without a new exposure. What lead is not retained by tissues is excreted by the kidneys, with adults retaining about 1% of absorbed lead, while children younger than 2 years retain over 30% of absorbed lead. So children, especially toddlers, have a greater likelihood to absorb lead from the GI tract and to retain lead in their tissues, both due to active mineralization of bone and the permeability of the blood brain barrier, primarily in children under 3 years old. This is why we are addressing what will happen to the children of Flint and not to all the residents of Flint.
Lead competitively inhibits interactions between cations and sulfhydryl groups, which are present in most human biochemical reactions. This leads to irreversible cell damage and often cell death, especially within the central nervous system. Lead exposure is associated with particular dysfunction within dopaminergic pathways within the brain, and has been associated in a dose-dependent fashion with decreased prefrontal gray matter volume. Lead poisoning also has hematologic consequences (anemia), renal consequences (interstitial nephritis), gastrointestinal symptoms (vomiting, constipation), and endocrine consequences (reversible inhibition of Vitamin D metabolism and permanently short stature). But the CNS consequences of lead exposure are particularly devastating, as they appear to have no threshold and are permanent. Their incidence is the driving force for the CDC’s lowering of the official toxic lead level and the public health efforts to screen children and educate parents about the risk of lead exposure.

So what do these serious consequences look like? People with severe lead intoxication (blood lead levels greater than 70 mcg/dL) typically present with signs of acute encephalopathy (headache, vomiting, seizures, or coma) and require intensive medical management including chelation therapy. More typically, exposed children have low but accumulating levels of lead and present with nonspecific symptoms, including lost appetite, fatigue, irritability, and insomnia, which gradually worsen.
Behavior
High levels of impulsivity, aggression, and impaired attention are the prototypical sequelae of lead poisoning (following recovery from the acute intoxication). Multiple studies have demonstrated these high levels of aggressive and impulsive behaviors in preschoolers who were exposed to lead, and these behaviors appear to continue into adolescence and adulthood. Indeed, one study found that compared with children with the lowest measurable blood lead levels (0.2-0.7 mcg/dL), those children who were in the next two quartiles had seven and twelve times the odds of meeting diagnostic criteria for conduct disorder.1 There have even been studies which correlated atmospheric lead levels (when leaded gasoline was common) with crime rates 20 years later, which supported an association between childhood lead exposure and adult criminal activity.2-4.
Multiple studies have demonstrated higher rates of inattention, distractibility, and impulsivity in lead-exposed children than would be expected given the prevalence of attention-deficit/hyperactivity disorder (ADHD) in the general population. The incidence of these symptoms goes up in a dose-dependent fashion and appears to have no threshold (so they occur at even the lowest measurable blood lead levels). In a 2006 study of nearly 5,000 children between ages 4-15 years, those with blood lead levels greater than 2 mcg/dL (still below the level the CDC deems toxic) were four times more likely to be carrying a diagnosis of ADHD and be on stimulant medication than their peers with blood lead levels less than 0.8mcg/dL.
Cognition
Closely related to impulse control and attention, the cognitive domains of intelligence and executive function are clearly damaged by lead exposure. Poor performance on tasks requiring focus, cognitive flexibility, and inhibition of automatic responses was directly associated with higher blood lead levels in a group of preschoolers with levels between 0 and 13 mcg/dL.5

IQ has been found to be so consistently diminished by increasing blood lead levels that it is used as an overall index of neurodevelopmental morbidity of lead exposure, leading to the CDC’s adoption of a lower standard definition of toxic lead levels. Even very low blood lead levels are associated with decrements in IQ: children with blood lead levels less than 7.5 mcg/dL lost an average of 3 IQ points for every 1 mcg/dL increase in blood lead levels.6 In a study of 57,000 elementary school students in 2009, Miranda et al. found that those who had a blood lead level of 4 mcg/dL at 3 years old were significantly more likely to be diagnosed with a learning disability in elementary school. Another study of 48,000 children who had a blood lead level of 5 mcg/dL were 30% more likely to fail third grade reading and math tests than their peers without measurable lead levels.
Speech and language
More recent studies have demonstrated that children with higher bone lead concentrations had poorer performance on several language-processing measures, suggesting that childhood lead exposure damages language processing and function as the young people grow. These deficits in language processing can make social development and self-regulation much more challenging in adolescence, and make school and work settings much more challenging. These findings also have implications for the utility of psychotherapy, a language-based treatment, for the other behavioral problems of lead exposure.
Motor skills
Several recent studies have assessed both fine and gross motor skills in lead-exposed children. Findings have demonstrated that balance, coordination, gross motor and fine motor skills all appear to be compromised in a dose-dependent fashion by childhood lead exposure. These findings suggest that not only are children at greater risk for accident and injury through childhood and into adulthood, a risk already increased by their compromised attention and impulse control. But they also are likely to be physically clumsy, compromising an opportunity to cultivate strengths or experience mastery when cognitive tasks may prove frustrating for them.
With deficits in such fundamental cognitive, motor, and behavioral processes, exposed children are clearly vulnerable to more than ADHD, conduct disorder, and learning disabilities. These struggles may lead to secondary vulnerabilities to anxiety or mood symptoms or substance abuse as these children grow into teenagers who face frustration at every turn. In addition to treatment for their deficits in attention and executive function, these children will ideally receive specialized supports in school and at home, to be able to master cognitive tasks, manage new social circumstances and make friends, discover their interests and talents, and generally stay on their best developmental trajectories. Lastly, the specific consequences of lead exposure will vary for any individual child, so parents will have to deal with the uncertainty of their child’s behavior and development over many years. Clearly, the children of Flint face a long road that has been substantially impacted by their lead exposure. The only good that can come from the exposure in Flint is to heighten efforts to ensure that it never happens again.
1. Environ Health Perspect. 2008 Jul;116(7):956-62.
2. Environ Res. 2000 May;83(1):1-22.
3. Environ Res. 2007 Jul;104(3):315-36.
4. Arch Pediatr Adolesc Med. 2001 May;155(5):579-82.
5. Dev Neuropsychol. 2004;26(1):513-40.
6. Environ Health Perspect. 2005 Jul;113(7):894-9.
Dr. Swick is an attending psychiatrist in the division of child psychiatry at Massachusetts General Hospital, Boston, and director of the Parenting at a Challenging Time (PACT) Program at the Vernon Cancer Center at Newton (Mass.) Wellesley Hospital. Dr. Jellinek is professor of psychiatry and of pediatrics at Harvard Medical School, Boston.
Lead poisoning is a well-established cause of serious and permanent neurological, cognitive, and behavioral problems, particularly in exposed children.
Children can be exposed to lead from ingesting paint chips in their homes, when old paint is scrapped from the exterior of houses or bridges, and through the water they drink. The damage caused by lead poisoning was first recognized in the United States in the early 20th century, although lead was added to gasoline and paint until the 1970’s. Since then, regulations for lead in consumer products have become increasingly strict, and the Centers for Disease Control and Prevention’s definition of a toxic lead level has shifted from 60 micrograms/deciliter (mcg/dL) in 1970 to 5 mcg/dL in 2012. In many communities, removing lead paint up to the height of a young child is a requirement whenever an older home is sold.
Unfortunately, these regulations did not protect the families in Flint, Michigan from being exposed to high levels of lead when a change in water supply and inadequate water treatment allowed lead to enter the system from decaying water pipes. It is worth reviewing what is known about the short- and long-term consequences of lead exposure, and what lies ahead for the children of Flint.
Lead is a naturally occurring element that is not metabolized, but rather absorbed, distributed to tissues, and excreted. Lead can be inhaled (with 100% absorption) and introduced through the GI tract (with about 70% absorption in children and 20% absorption in adults). GI absorption is enhanced by calcium or iron deficiency, both conditions that are relatively common, especially in poor children and can lead to pica (or eating of non-nutritious materials), further increasing the chances of lead exposure. Absorbed lead is distributed to blood (for 28-36 days), soft tissue, including the nervous system (40 days), and to bone (where it lasts for over 25 years). Blood that is retained in growing bones can be mobilized during periods of physiologic stress (such as illness, injury, or pregnancy), meaning children exposed to lead during a period of rapid bone growth are at long-term risk for acute lead poisoning from their endogenous reservoir without a new exposure. What lead is not retained by tissues is excreted by the kidneys, with adults retaining about 1% of absorbed lead, while children younger than 2 years retain over 30% of absorbed lead. So children, especially toddlers, have a greater likelihood to absorb lead from the GI tract and to retain lead in their tissues, both due to active mineralization of bone and the permeability of the blood brain barrier, primarily in children under 3 years old. This is why we are addressing what will happen to the children of Flint and not to all the residents of Flint.
Lead competitively inhibits interactions between cations and sulfhydryl groups, which are present in most human biochemical reactions. This leads to irreversible cell damage and often cell death, especially within the central nervous system. Lead exposure is associated with particular dysfunction within dopaminergic pathways within the brain, and has been associated in a dose-dependent fashion with decreased prefrontal gray matter volume. Lead poisoning also has hematologic consequences (anemia), renal consequences (interstitial nephritis), gastrointestinal symptoms (vomiting, constipation), and endocrine consequences (reversible inhibition of Vitamin D metabolism and permanently short stature). But the CNS consequences of lead exposure are particularly devastating, as they appear to have no threshold and are permanent. Their incidence is the driving force for the CDC’s lowering of the official toxic lead level and the public health efforts to screen children and educate parents about the risk of lead exposure.
So what do these serious consequences look like? People with severe lead intoxication (blood lead levels greater than 70 mcg/dL) typically present with signs of acute encephalopathy (headache, vomiting, seizures, or coma) and require intensive medical management including chelation therapy. More typically, exposed children have low but accumulating levels of lead and present with nonspecific symptoms, including lost appetite, fatigue, irritability, and insomnia, which gradually worsen.
Behavior
High levels of impulsivity, aggression, and impaired attention are the prototypical sequelae of lead poisoning (following recovery from the acute intoxication). Multiple studies have demonstrated these high levels of aggressive and impulsive behaviors in preschoolers who were exposed to lead, and these behaviors appear to continue into adolescence and adulthood. Indeed, one study found that compared with children with the lowest measurable blood lead levels (0.2-0.7 mcg/dL), those children who were in the next two quartiles had seven and twelve times the odds of meeting diagnostic criteria for conduct disorder.1 There have even been studies which correlated atmospheric lead levels (when leaded gasoline was common) with crime rates 20 years later, which supported an association between childhood lead exposure and adult criminal activity.2-4.
Multiple studies have demonstrated higher rates of inattention, distractibility, and impulsivity in lead-exposed children than would be expected given the prevalence of attention-deficit/hyperactivity disorder (ADHD) in the general population. The incidence of these symptoms goes up in a dose-dependent fashion and appears to have no threshold (so they occur at even the lowest measurable blood lead levels). In a 2006 study of nearly 5,000 children between ages 4-15 years, those with blood lead levels greater than 2 mcg/dL (still below the level the CDC deems toxic) were four times more likely to be carrying a diagnosis of ADHD and be on stimulant medication than their peers with blood lead levels less than 0.8mcg/dL.
Cognition
Closely related to impulse control and attention, the cognitive domains of intelligence and executive function are clearly damaged by lead exposure. Poor performance on tasks requiring focus, cognitive flexibility, and inhibition of automatic responses was directly associated with higher blood lead levels in a group of preschoolers with levels between 0 and 13 mcg/dL.5
IQ has been found to be so consistently diminished by increasing blood lead levels that it is used as an overall index of neurodevelopmental morbidity of lead exposure, leading to the CDC’s adoption of a lower standard definition of toxic lead levels. Even very low blood lead levels are associated with decrements in IQ: children with blood lead levels less than 7.5 mcg/dL lost an average of 3 IQ points for every 1 mcg/dL increase in blood lead levels.6 In a study of 57,000 elementary school students in 2009, Miranda et al. found that those who had a blood lead level of 4 mcg/dL at 3 years old were significantly more likely to be diagnosed with a learning disability in elementary school. Another study of 48,000 children who had a blood lead level of 5 mcg/dL were 30% more likely to fail third grade reading and math tests than their peers without measurable lead levels.
Speech and language
More recent studies have demonstrated that children with higher bone lead concentrations had poorer performance on several language-processing measures, suggesting that childhood lead exposure damages language processing and function as the young people grow. These deficits in language processing can make social development and self-regulation much more challenging in adolescence, and make school and work settings much more challenging. These findings also have implications for the utility of psychotherapy, a language-based treatment, for the other behavioral problems of lead exposure.
Motor skills
Several recent studies have assessed both fine and gross motor skills in lead-exposed children. Findings have demonstrated that balance, coordination, gross motor and fine motor skills all appear to be compromised in a dose-dependent fashion by childhood lead exposure. These findings suggest that not only are children at greater risk for accident and injury through childhood and into adulthood, a risk already increased by their compromised attention and impulse control. But they also are likely to be physically clumsy, compromising an opportunity to cultivate strengths or experience mastery when cognitive tasks may prove frustrating for them.
With deficits in such fundamental cognitive, motor, and behavioral processes, exposed children are clearly vulnerable to more than ADHD, conduct disorder, and learning disabilities. These struggles may lead to secondary vulnerabilities to anxiety or mood symptoms or substance abuse as these children grow into teenagers who face frustration at every turn. In addition to treatment for their deficits in attention and executive function, these children will ideally receive specialized supports in school and at home, to be able to master cognitive tasks, manage new social circumstances and make friends, discover their interests and talents, and generally stay on their best developmental trajectories. Lastly, the specific consequences of lead exposure will vary for any individual child, so parents will have to deal with the uncertainty of their child’s behavior and development over many years. Clearly, the children of Flint face a long road that has been substantially impacted by their lead exposure. The only good that can come from the exposure in Flint is to heighten efforts to ensure that it never happens again.
1. Environ Health Perspect. 2008 Jul;116(7):956-62.
2. Environ Res. 2000 May;83(1):1-22.
3. Environ Res. 2007 Jul;104(3):315-36.
4. Arch Pediatr Adolesc Med. 2001 May;155(5):579-82.
5. Dev Neuropsychol. 2004;26(1):513-40.
6. Environ Health Perspect. 2005 Jul;113(7):894-9.
Dr. Swick is an attending psychiatrist in the division of child psychiatry at Massachusetts General Hospital, Boston, and director of the Parenting at a Challenging Time (PACT) Program at the Vernon Cancer Center at Newton (Mass.) Wellesley Hospital. Dr. Jellinek is professor of psychiatry and of pediatrics at Harvard Medical School, Boston.
Lead poisoning is a well-established cause of serious and permanent neurological, cognitive, and behavioral problems, particularly in exposed children.
Children can be exposed to lead from ingesting paint chips in their homes, when old paint is scrapped from the exterior of houses or bridges, and through the water they drink. The damage caused by lead poisoning was first recognized in the United States in the early 20th century, although lead was added to gasoline and paint until the 1970’s. Since then, regulations for lead in consumer products have become increasingly strict, and the Centers for Disease Control and Prevention’s definition of a toxic lead level has shifted from 60 micrograms/deciliter (mcg/dL) in 1970 to 5 mcg/dL in 2012. In many communities, removing lead paint up to the height of a young child is a requirement whenever an older home is sold.
Unfortunately, these regulations did not protect the families in Flint, Michigan from being exposed to high levels of lead when a change in water supply and inadequate water treatment allowed lead to enter the system from decaying water pipes. It is worth reviewing what is known about the short- and long-term consequences of lead exposure, and what lies ahead for the children of Flint.
Lead is a naturally occurring element that is not metabolized, but rather absorbed, distributed to tissues, and excreted. Lead can be inhaled (with 100% absorption) and introduced through the GI tract (with about 70% absorption in children and 20% absorption in adults). GI absorption is enhanced by calcium or iron deficiency, both conditions that are relatively common, especially in poor children and can lead to pica (or eating of non-nutritious materials), further increasing the chances of lead exposure. Absorbed lead is distributed to blood (for 28-36 days), soft tissue, including the nervous system (40 days), and to bone (where it lasts for over 25 years). Blood that is retained in growing bones can be mobilized during periods of physiologic stress (such as illness, injury, or pregnancy), meaning children exposed to lead during a period of rapid bone growth are at long-term risk for acute lead poisoning from their endogenous reservoir without a new exposure. What lead is not retained by tissues is excreted by the kidneys, with adults retaining about 1% of absorbed lead, while children younger than 2 years retain over 30% of absorbed lead. So children, especially toddlers, have a greater likelihood to absorb lead from the GI tract and to retain lead in their tissues, both due to active mineralization of bone and the permeability of the blood brain barrier, primarily in children under 3 years old. This is why we are addressing what will happen to the children of Flint and not to all the residents of Flint.
Lead competitively inhibits interactions between cations and sulfhydryl groups, which are present in most human biochemical reactions. This leads to irreversible cell damage and often cell death, especially within the central nervous system. Lead exposure is associated with particular dysfunction within dopaminergic pathways within the brain, and has been associated in a dose-dependent fashion with decreased prefrontal gray matter volume. Lead poisoning also has hematologic consequences (anemia), renal consequences (interstitial nephritis), gastrointestinal symptoms (vomiting, constipation), and endocrine consequences (reversible inhibition of Vitamin D metabolism and permanently short stature). But the CNS consequences of lead exposure are particularly devastating, as they appear to have no threshold and are permanent. Their incidence is the driving force for the CDC’s lowering of the official toxic lead level and the public health efforts to screen children and educate parents about the risk of lead exposure.
So what do these serious consequences look like? People with severe lead intoxication (blood lead levels greater than 70 mcg/dL) typically present with signs of acute encephalopathy (headache, vomiting, seizures, or coma) and require intensive medical management including chelation therapy. More typically, exposed children have low but accumulating levels of lead and present with nonspecific symptoms, including lost appetite, fatigue, irritability, and insomnia, which gradually worsen.
Behavior
High levels of impulsivity, aggression, and impaired attention are the prototypical sequelae of lead poisoning (following recovery from the acute intoxication). Multiple studies have demonstrated these high levels of aggressive and impulsive behaviors in preschoolers who were exposed to lead, and these behaviors appear to continue into adolescence and adulthood. Indeed, one study found that compared with children with the lowest measurable blood lead levels (0.2-0.7 mcg/dL), those children who were in the next two quartiles had seven and twelve times the odds of meeting diagnostic criteria for conduct disorder.1 There have even been studies which correlated atmospheric lead levels (when leaded gasoline was common) with crime rates 20 years later, which supported an association between childhood lead exposure and adult criminal activity.2-4.
Multiple studies have demonstrated higher rates of inattention, distractibility, and impulsivity in lead-exposed children than would be expected given the prevalence of attention-deficit/hyperactivity disorder (ADHD) in the general population. The incidence of these symptoms goes up in a dose-dependent fashion and appears to have no threshold (so they occur at even the lowest measurable blood lead levels). In a 2006 study of nearly 5,000 children between ages 4-15 years, those with blood lead levels greater than 2 mcg/dL (still below the level the CDC deems toxic) were four times more likely to be carrying a diagnosis of ADHD and be on stimulant medication than their peers with blood lead levels less than 0.8mcg/dL.
Cognition
Closely related to impulse control and attention, the cognitive domains of intelligence and executive function are clearly damaged by lead exposure. Poor performance on tasks requiring focus, cognitive flexibility, and inhibition of automatic responses was directly associated with higher blood lead levels in a group of preschoolers with levels between 0 and 13 mcg/dL.5
IQ has been found to be so consistently diminished by increasing blood lead levels that it is used as an overall index of neurodevelopmental morbidity of lead exposure, leading to the CDC’s adoption of a lower standard definition of toxic lead levels. Even very low blood lead levels are associated with decrements in IQ: children with blood lead levels less than 7.5 mcg/dL lost an average of 3 IQ points for every 1 mcg/dL increase in blood lead levels.6 In a study of 57,000 elementary school students in 2009, Miranda et al. found that those who had a blood lead level of 4 mcg/dL at 3 years old were significantly more likely to be diagnosed with a learning disability in elementary school. Another study of 48,000 children who had a blood lead level of 5 mcg/dL were 30% more likely to fail third grade reading and math tests than their peers without measurable lead levels.
Speech and language
More recent studies have demonstrated that children with higher bone lead concentrations had poorer performance on several language-processing measures, suggesting that childhood lead exposure damages language processing and function as the young people grow. These deficits in language processing can make social development and self-regulation much more challenging in adolescence, and make school and work settings much more challenging. These findings also have implications for the utility of psychotherapy, a language-based treatment, for the other behavioral problems of lead exposure.
Motor skills
Several recent studies have assessed both fine and gross motor skills in lead-exposed children. Findings have demonstrated that balance, coordination, gross motor and fine motor skills all appear to be compromised in a dose-dependent fashion by childhood lead exposure. These findings suggest that not only are children at greater risk for accident and injury through childhood and into adulthood, a risk already increased by their compromised attention and impulse control. But they also are likely to be physically clumsy, compromising an opportunity to cultivate strengths or experience mastery when cognitive tasks may prove frustrating for them.
With deficits in such fundamental cognitive, motor, and behavioral processes, exposed children are clearly vulnerable to more than ADHD, conduct disorder, and learning disabilities. These struggles may lead to secondary vulnerabilities to anxiety or mood symptoms or substance abuse as these children grow into teenagers who face frustration at every turn. In addition to treatment for their deficits in attention and executive function, these children will ideally receive specialized supports in school and at home, to be able to master cognitive tasks, manage new social circumstances and make friends, discover their interests and talents, and generally stay on their best developmental trajectories. Lastly, the specific consequences of lead exposure will vary for any individual child, so parents will have to deal with the uncertainty of their child’s behavior and development over many years. Clearly, the children of Flint face a long road that has been substantially impacted by their lead exposure. The only good that can come from the exposure in Flint is to heighten efforts to ensure that it never happens again.
1. Environ Health Perspect. 2008 Jul;116(7):956-62.
2. Environ Res. 2000 May;83(1):1-22.
3. Environ Res. 2007 Jul;104(3):315-36.
4. Arch Pediatr Adolesc Med. 2001 May;155(5):579-82.
5. Dev Neuropsychol. 2004;26(1):513-40.
6. Environ Health Perspect. 2005 Jul;113(7):894-9.
Dr. Swick is an attending psychiatrist in the division of child psychiatry at Massachusetts General Hospital, Boston, and director of the Parenting at a Challenging Time (PACT) Program at the Vernon Cancer Center at Newton (Mass.) Wellesley Hospital. Dr. Jellinek is professor of psychiatry and of pediatrics at Harvard Medical School, Boston.
From the Washington Office: Medicare audit accountability
The Recovery Audit Contractor (RAC) program was launched in 2010 by the Centers for Medicare and Medicaid Services (CMS) with the intention of identifying and preventing improper payments to Medicare providers. Recovery Audit Contractors are paid on “contingency fee” basis, i.e. a commission on each claim that they deny. Some have thus likened their actions to those of ‘bounty hunters.” Though there is an appeals process, hospitals and physicians bear the cost of audits, denials, and appeals, regardless of the ultimate outcome of the appeals process.

Because of the lack of accountability in the RAC process, concern has been expressed about both the number of inaccurate findings as well as the high volume of appeals. As evidence, the American Hospital Association (AHA) reported that the Office of the Inspector General (OIG) found that 49% of hospital denials are appealed and 72% of the appeals brought before an Administrative Law Judge are overturned in favor of the hospital.
In response to these concerns, Rep. George Holding (R-NC) introduced the H.R. 2568, the Fair Medical Audits Act in May 2015. The bill was jointly referred to the Ways and Means and Energy and Commerce committees in the House of Representatives for further consideration. Currently, H.R. 2568 has 23 cosponsors.
H.R. 2568 addresses many of the concerns in the RAC program by:
• Enhancing transparency in the audit process to improve compliance.
• Improving the claims-review process by mandating that contractors meet appropriate knowledge and experience requirements.
• Promoting provider education while increasing RAC accountability for inaccurate audit findings.
• Ensuring accuracy of those overpayment amounts calculated by contractors using extrapolation methodology.
• Requiring contractors to reimburse certain documentation requests to reduce provider burdens.
• Delaying payment to RACs until after external appeal.
• Reducing the appeals backlog by shortening the “look-back” period.
On Dec. 3, 2015, the American College of Surgeons joined 10 other surgical associations in sending a letter of support to Representative Holding thanking him for introducing the Fair Medical Audits Act. In addition, an ACTION ALERT was posted on the SurgeonsVoice website to facilitate the efforts of Fellows in contacting their individual representatives urging they support the legislation. I would urge all Fellows to log onto www.surgeonsvoice.org, and then click on the “TAKE ACTION” tab on the right side of the screen. It takes only a few moments to send a message to your Member of Congress requesting their assistance in passing this sensible legislation increasing accountability in the Medicare Recovery Audit Contractor program.
Until next month …
Dr. Patrick V. Bailey is an ACS Fellow, a pediatric surgeon, and Medical Director, Advocacy, for the Division of Advocacy and Health Policy, in the ACS offices in Washington, D.C.
The Recovery Audit Contractor (RAC) program was launched in 2010 by the Centers for Medicare and Medicaid Services (CMS) with the intention of identifying and preventing improper payments to Medicare providers. Recovery Audit Contractors are paid on “contingency fee” basis, i.e. a commission on each claim that they deny. Some have thus likened their actions to those of ‘bounty hunters.” Though there is an appeals process, hospitals and physicians bear the cost of audits, denials, and appeals, regardless of the ultimate outcome of the appeals process.
Because of the lack of accountability in the RAC process, concern has been expressed about both the number of inaccurate findings as well as the high volume of appeals. As evidence, the American Hospital Association (AHA) reported that the Office of the Inspector General (OIG) found that 49% of hospital denials are appealed and 72% of the appeals brought before an Administrative Law Judge are overturned in favor of the hospital.
In response to these concerns, Rep. George Holding (R-NC) introduced the H.R. 2568, the Fair Medical Audits Act in May 2015. The bill was jointly referred to the Ways and Means and Energy and Commerce committees in the House of Representatives for further consideration. Currently, H.R. 2568 has 23 cosponsors.
H.R. 2568 addresses many of the concerns in the RAC program by:
• Enhancing transparency in the audit process to improve compliance.
• Improving the claims-review process by mandating that contractors meet appropriate knowledge and experience requirements.
• Promoting provider education while increasing RAC accountability for inaccurate audit findings.
• Ensuring accuracy of those overpayment amounts calculated by contractors using extrapolation methodology.
• Requiring contractors to reimburse certain documentation requests to reduce provider burdens.
• Delaying payment to RACs until after external appeal.
• Reducing the appeals backlog by shortening the “look-back” period.
On Dec. 3, 2015, the American College of Surgeons joined 10 other surgical associations in sending a letter of support to Representative Holding thanking him for introducing the Fair Medical Audits Act. In addition, an ACTION ALERT was posted on the SurgeonsVoice website to facilitate the efforts of Fellows in contacting their individual representatives urging they support the legislation. I would urge all Fellows to log onto www.surgeonsvoice.org, and then click on the “TAKE ACTION” tab on the right side of the screen. It takes only a few moments to send a message to your Member of Congress requesting their assistance in passing this sensible legislation increasing accountability in the Medicare Recovery Audit Contractor program.
Until next month …
Dr. Patrick V. Bailey is an ACS Fellow, a pediatric surgeon, and Medical Director, Advocacy, for the Division of Advocacy and Health Policy, in the ACS offices in Washington, D.C.
The Recovery Audit Contractor (RAC) program was launched in 2010 by the Centers for Medicare and Medicaid Services (CMS) with the intention of identifying and preventing improper payments to Medicare providers. Recovery Audit Contractors are paid on “contingency fee” basis, i.e. a commission on each claim that they deny. Some have thus likened their actions to those of ‘bounty hunters.” Though there is an appeals process, hospitals and physicians bear the cost of audits, denials, and appeals, regardless of the ultimate outcome of the appeals process.
Because of the lack of accountability in the RAC process, concern has been expressed about both the number of inaccurate findings as well as the high volume of appeals. As evidence, the American Hospital Association (AHA) reported that the Office of the Inspector General (OIG) found that 49% of hospital denials are appealed and 72% of the appeals brought before an Administrative Law Judge are overturned in favor of the hospital.
In response to these concerns, Rep. George Holding (R-NC) introduced the H.R. 2568, the Fair Medical Audits Act in May 2015. The bill was jointly referred to the Ways and Means and Energy and Commerce committees in the House of Representatives for further consideration. Currently, H.R. 2568 has 23 cosponsors.
H.R. 2568 addresses many of the concerns in the RAC program by:
• Enhancing transparency in the audit process to improve compliance.
• Improving the claims-review process by mandating that contractors meet appropriate knowledge and experience requirements.
• Promoting provider education while increasing RAC accountability for inaccurate audit findings.
• Ensuring accuracy of those overpayment amounts calculated by contractors using extrapolation methodology.
• Requiring contractors to reimburse certain documentation requests to reduce provider burdens.
• Delaying payment to RACs until after external appeal.
• Reducing the appeals backlog by shortening the “look-back” period.
On Dec. 3, 2015, the American College of Surgeons joined 10 other surgical associations in sending a letter of support to Representative Holding thanking him for introducing the Fair Medical Audits Act. In addition, an ACTION ALERT was posted on the SurgeonsVoice website to facilitate the efforts of Fellows in contacting their individual representatives urging they support the legislation. I would urge all Fellows to log onto www.surgeonsvoice.org, and then click on the “TAKE ACTION” tab on the right side of the screen. It takes only a few moments to send a message to your Member of Congress requesting their assistance in passing this sensible legislation increasing accountability in the Medicare Recovery Audit Contractor program.
Until next month …
Dr. Patrick V. Bailey is an ACS Fellow, a pediatric surgeon, and Medical Director, Advocacy, for the Division of Advocacy and Health Policy, in the ACS offices in Washington, D.C.

Psoriatic Arthritis on the Rise

The primary comorbidity of psoriasis is psoriatic arthritis (PsA). The true incidence of PsA has long been an issue of debate. To estimate the incidence of PsA in patients with psoriasis and to identify risk factors for its development, Eder at al conducted a prospective cohort study involving psoriasis patients without arthritis at study entry that was published online in Arthritis & Rheumatology.
The investigators collected information from patients concerning lifestyle habits, comorbidities, psoriasis activity, and medications. The patients were evaluated at enrollment and annually. A general physical examination, assessment of psoriasis severity, and assessment for the development of musculoskeletal symptoms were conducted at each visit. A diagnosis of PsA was determined by a rheumatologist on the basis of clinical, laboratory, and imaging data; patients also had to fulfill the CASPAR (Classification Criteria for Psoriatic Arthritis) criteria (confirmed cases). The annual incidence of PsA was estimated using an event per person-years analysis.
The results from 464 patients who were followed for 8 years were analyzed. The annual incidence of confirmed PsA was 2.7 per 100 patients with psoriasis (95% CI, 2.1-3.6). Overall, 51 patients developed PsA over the course of the study and an additional 9 were considered suspect cases.
The following baseline variables were associated with the development of PsA in multivariate analysis: severe psoriasis (relative risk [RR], 5.4; P=.006), low level of education (college/university vs high school incomplete: RR, 4.5; P=.005; high school education vs high school incomplete: RR, 3.3; P=.049), and use of retinoid medications (RR, 3.4; P=.02). In addition, psoriatic nail pitting (RR, 2.5; P=.002) and uveitis (RR, 31.5; P<.001) were time-dependent predictors for PsA development.
The authors concluded that the incidence of PsA in patients with psoriasis was higher than previously reported. Possible factors for this finding might include differences in patient recruitment as well as self-reported PsA diagnoses.
What’s the issue?
This prospective analysis is interesting. The incidence of PsA was higher than reported. It reinforces the need for continual evaluation of joint symptoms in patients with psoriasis, even if they have had psoriasis for many years. How will this analysis impact your evaluation of psoriatic patients?
The primary comorbidity of psoriasis is psoriatic arthritis (PsA). The true incidence of PsA has long been an issue of debate. To estimate the incidence of PsA in patients with psoriasis and to identify risk factors for its development, Eder at al conducted a prospective cohort study involving psoriasis patients without arthritis at study entry that was published online in Arthritis & Rheumatology.
The investigators collected information from patients concerning lifestyle habits, comorbidities, psoriasis activity, and medications. The patients were evaluated at enrollment and annually. A general physical examination, assessment of psoriasis severity, and assessment for the development of musculoskeletal symptoms were conducted at each visit. A diagnosis of PsA was determined by a rheumatologist on the basis of clinical, laboratory, and imaging data; patients also had to fulfill the CASPAR (Classification Criteria for Psoriatic Arthritis) criteria (confirmed cases). The annual incidence of PsA was estimated using an event per person-years analysis.
The results from 464 patients who were followed for 8 years were analyzed. The annual incidence of confirmed PsA was 2.7 per 100 patients with psoriasis (95% CI, 2.1-3.6). Overall, 51 patients developed PsA over the course of the study and an additional 9 were considered suspect cases.
The following baseline variables were associated with the development of PsA in multivariate analysis: severe psoriasis (relative risk [RR], 5.4; P=.006), low level of education (college/university vs high school incomplete: RR, 4.5; P=.005; high school education vs high school incomplete: RR, 3.3; P=.049), and use of retinoid medications (RR, 3.4; P=.02). In addition, psoriatic nail pitting (RR, 2.5; P=.002) and uveitis (RR, 31.5; P<.001) were time-dependent predictors for PsA development.
The authors concluded that the incidence of PsA in patients with psoriasis was higher than previously reported. Possible factors for this finding might include differences in patient recruitment as well as self-reported PsA diagnoses.
What’s the issue?
This prospective analysis is interesting. The incidence of PsA was higher than reported. It reinforces the need for continual evaluation of joint symptoms in patients with psoriasis, even if they have had psoriasis for many years. How will this analysis impact your evaluation of psoriatic patients?
The primary comorbidity of psoriasis is psoriatic arthritis (PsA). The true incidence of PsA has long been an issue of debate. To estimate the incidence of PsA in patients with psoriasis and to identify risk factors for its development, Eder at al conducted a prospective cohort study involving psoriasis patients without arthritis at study entry that was published online in Arthritis & Rheumatology.
The investigators collected information from patients concerning lifestyle habits, comorbidities, psoriasis activity, and medications. The patients were evaluated at enrollment and annually. A general physical examination, assessment of psoriasis severity, and assessment for the development of musculoskeletal symptoms were conducted at each visit. A diagnosis of PsA was determined by a rheumatologist on the basis of clinical, laboratory, and imaging data; patients also had to fulfill the CASPAR (Classification Criteria for Psoriatic Arthritis) criteria (confirmed cases). The annual incidence of PsA was estimated using an event per person-years analysis.
The results from 464 patients who were followed for 8 years were analyzed. The annual incidence of confirmed PsA was 2.7 per 100 patients with psoriasis (95% CI, 2.1-3.6). Overall, 51 patients developed PsA over the course of the study and an additional 9 were considered suspect cases.
The following baseline variables were associated with the development of PsA in multivariate analysis: severe psoriasis (relative risk [RR], 5.4; P=.006), low level of education (college/university vs high school incomplete: RR, 4.5; P=.005; high school education vs high school incomplete: RR, 3.3; P=.049), and use of retinoid medications (RR, 3.4; P=.02). In addition, psoriatic nail pitting (RR, 2.5; P=.002) and uveitis (RR, 31.5; P<.001) were time-dependent predictors for PsA development.
The authors concluded that the incidence of PsA in patients with psoriasis was higher than previously reported. Possible factors for this finding might include differences in patient recruitment as well as self-reported PsA diagnoses.
What’s the issue?
This prospective analysis is interesting. The incidence of PsA was higher than reported. It reinforces the need for continual evaluation of joint symptoms in patients with psoriasis, even if they have had psoriasis for many years. How will this analysis impact your evaluation of psoriatic patients?

Erratum (Cutis. 2015;96:E3-E5)
The photo challenge “Sessile Pink Plaque on the Lower Back” (Cutis. 2015;96:E3-E5) contained an error in the byline. The author names should have read:
Robyn Marszalek, MD; Phyu P. Aung, MD, PhD; Deon Wolpowitz, MD, PhD; Amy Yuntzu-Yen Chen, MD
The staff of Cutis® makes every possible effort to ensure accuracy in its articles and apologizes for the mistake.
The photo challenge “Sessile Pink Plaque on the Lower Back” (Cutis. 2015;96:E3-E5) contained an error in the byline. The author names should have read:
Robyn Marszalek, MD; Phyu P. Aung, MD, PhD; Deon Wolpowitz, MD, PhD; Amy Yuntzu-Yen Chen, MD
The staff of Cutis® makes every possible effort to ensure accuracy in its articles and apologizes for the mistake.
The photo challenge “Sessile Pink Plaque on the Lower Back” (Cutis. 2015;96:E3-E5) contained an error in the byline. The author names should have read:
Robyn Marszalek, MD; Phyu P. Aung, MD, PhD; Deon Wolpowitz, MD, PhD; Amy Yuntzu-Yen Chen, MD
The staff of Cutis® makes every possible effort to ensure accuracy in its articles and apologizes for the mistake.