User login
BOS beat placebo for eosinophilic esophagitis
Budesonide oral suspension (BOS) was safe and significantly outperformed placebo on validated measures of eosinophilic esophagitis, according to a first-in-kind, multicenter, randomized, double-blind, phase II trial presented in the March issue of Gastroenterology (doi: 10.1053/j.gastro.2016.11.021).
The novel topical corticosteroid formulation yielded a significant histologic response and was associated with 3 fewer days of dysphagia over 2 weeks compared with placebo, reported Evan S. Dellon, MD, MPH, of University of North Carolina, Chapel Hill, and his associates. “There were no unexpected safety signals, and compliance with medication was high, suggesting that this formulation can be reliably used,” they wrote. Their findings earned BOS (SHP621) an FDA Breakthrough Therapy Designation in June 2016. Although corticosteroids are first-line therapy for eosinophilic esophagitis, symptom response in other studies has been mixed, and the Food and Drug Administration had approved neither fluticasone nor budesonide for this disease, the researchers noted. They formulated BOS to adhere better to the esophageal mucosa in order to enhance esophageal delivery while decreasing unwanted pulmonary deposition.
For the study, they randomly assigned 93 patients aged 11-40 years with eosinophilic esophagitis to receive either placebo or 2 mg BOS twice daily. By week 12, Dysphagia Symptom Questionnaire scores had fallen by 14.3 points with BOS group and by 7.5 points with placebo (P = .001). Endoscopic severity scores dropped by 3.8 points with BOS and rose by 0.4 points with placebo (P less than .0001). Rates of histologic response were 39% and 3%, respectively (P less than .0001). Nonresponders averaged 10 kg more body weight than responders, and had been diagnosed about 21 months earlier (average disease duration, 46 months and 25 months, respectively).
Rates of reported adverse effects were similar with BOS (47%) and placebo (50%). Individual rates of nasopharyngitis, upper respiratory infections, and oropharyngeal pain also were comparable between groups, but one patient stopped BOS after developing dyspnea, nausea, and vomiting that were considered treatment related. Esophageal candidiasis developed in two BOS recipients – a rate similar rate to that in a prior study of BOS (Clin Gastroenterol Hepatol. 2015 Jan 13. doi: 10.1016/j.cgh.2014.05.02), and a lower percentage than in other studies of topical steroids for eosinophilic esophagitis, according to the researchers. Morning cortisol levels were similar between groups, and there were no adverse laboratory effects, they added.
Patients in this trial had severe symptoms and histology and were highly compliant with treatment. They filled out at least 70% of their symptom diary, had at least 15 eosinophils per high-power frame from at least two esophageal levels on screening endoscopy, and reported at least 4 days of dysphagia during the second half of a 4-week, blinded placebo run-in period. Researchers should consider using these strict inclusion criteria in future trials of eosinophilic esophagitis, especially because previous studies have failed to show a treatment benefit for topical steroid therapy, the investigators noted.
Meritage Pharma, which is now a part of the Shire group, makes budesonide oral suspension and sponsored the study. Dr. Dellon disclosed ties to Meritage, Receptos, Regeneron, Aptalis, Banner Life Sciences, Novartis, and Roche. All five coinvestigators disclosed ties to industry, including Meritage, Shire, Receptos, Regeneron, and Biogen Idec.
Budesonide oral suspension (BOS) was safe and significantly outperformed placebo on validated measures of eosinophilic esophagitis, according to a first-in-kind, multicenter, randomized, double-blind, phase II trial presented in the March issue of Gastroenterology (doi: 10.1053/j.gastro.2016.11.021).
The novel topical corticosteroid formulation yielded a significant histologic response and was associated with 3 fewer days of dysphagia over 2 weeks compared with placebo, reported Evan S. Dellon, MD, MPH, of University of North Carolina, Chapel Hill, and his associates. “There were no unexpected safety signals, and compliance with medication was high, suggesting that this formulation can be reliably used,” they wrote. Their findings earned BOS (SHP621) an FDA Breakthrough Therapy Designation in June 2016. Although corticosteroids are first-line therapy for eosinophilic esophagitis, symptom response in other studies has been mixed, and the Food and Drug Administration had approved neither fluticasone nor budesonide for this disease, the researchers noted. They formulated BOS to adhere better to the esophageal mucosa in order to enhance esophageal delivery while decreasing unwanted pulmonary deposition.
For the study, they randomly assigned 93 patients aged 11-40 years with eosinophilic esophagitis to receive either placebo or 2 mg BOS twice daily. By week 12, Dysphagia Symptom Questionnaire scores had fallen by 14.3 points with BOS group and by 7.5 points with placebo (P = .001). Endoscopic severity scores dropped by 3.8 points with BOS and rose by 0.4 points with placebo (P less than .0001). Rates of histologic response were 39% and 3%, respectively (P less than .0001). Nonresponders averaged 10 kg more body weight than responders, and had been diagnosed about 21 months earlier (average disease duration, 46 months and 25 months, respectively).
Rates of reported adverse effects were similar with BOS (47%) and placebo (50%). Individual rates of nasopharyngitis, upper respiratory infections, and oropharyngeal pain also were comparable between groups, but one patient stopped BOS after developing dyspnea, nausea, and vomiting that were considered treatment related. Esophageal candidiasis developed in two BOS recipients – a rate similar rate to that in a prior study of BOS (Clin Gastroenterol Hepatol. 2015 Jan 13. doi: 10.1016/j.cgh.2014.05.02), and a lower percentage than in other studies of topical steroids for eosinophilic esophagitis, according to the researchers. Morning cortisol levels were similar between groups, and there were no adverse laboratory effects, they added.
Patients in this trial had severe symptoms and histology and were highly compliant with treatment. They filled out at least 70% of their symptom diary, had at least 15 eosinophils per high-power frame from at least two esophageal levels on screening endoscopy, and reported at least 4 days of dysphagia during the second half of a 4-week, blinded placebo run-in period. Researchers should consider using these strict inclusion criteria in future trials of eosinophilic esophagitis, especially because previous studies have failed to show a treatment benefit for topical steroid therapy, the investigators noted.
Meritage Pharma, which is now a part of the Shire group, makes budesonide oral suspension and sponsored the study. Dr. Dellon disclosed ties to Meritage, Receptos, Regeneron, Aptalis, Banner Life Sciences, Novartis, and Roche. All five coinvestigators disclosed ties to industry, including Meritage, Shire, Receptos, Regeneron, and Biogen Idec.
Budesonide oral suspension (BOS) was safe and significantly outperformed placebo on validated measures of eosinophilic esophagitis, according to a first-in-kind, multicenter, randomized, double-blind, phase II trial presented in the March issue of Gastroenterology (doi: 10.1053/j.gastro.2016.11.021).
The novel topical corticosteroid formulation yielded a significant histologic response and was associated with 3 fewer days of dysphagia over 2 weeks compared with placebo, reported Evan S. Dellon, MD, MPH, of University of North Carolina, Chapel Hill, and his associates. “There were no unexpected safety signals, and compliance with medication was high, suggesting that this formulation can be reliably used,” they wrote. Their findings earned BOS (SHP621) an FDA Breakthrough Therapy Designation in June 2016. Although corticosteroids are first-line therapy for eosinophilic esophagitis, symptom response in other studies has been mixed, and the Food and Drug Administration had approved neither fluticasone nor budesonide for this disease, the researchers noted. They formulated BOS to adhere better to the esophageal mucosa in order to enhance esophageal delivery while decreasing unwanted pulmonary deposition.
For the study, they randomly assigned 93 patients aged 11-40 years with eosinophilic esophagitis to receive either placebo or 2 mg BOS twice daily. By week 12, Dysphagia Symptom Questionnaire scores had fallen by 14.3 points with BOS group and by 7.5 points with placebo (P = .001). Endoscopic severity scores dropped by 3.8 points with BOS and rose by 0.4 points with placebo (P less than .0001). Rates of histologic response were 39% and 3%, respectively (P less than .0001). Nonresponders averaged 10 kg more body weight than responders, and had been diagnosed about 21 months earlier (average disease duration, 46 months and 25 months, respectively).
Rates of reported adverse effects were similar with BOS (47%) and placebo (50%). Individual rates of nasopharyngitis, upper respiratory infections, and oropharyngeal pain also were comparable between groups, but one patient stopped BOS after developing dyspnea, nausea, and vomiting that were considered treatment related. Esophageal candidiasis developed in two BOS recipients – a rate similar rate to that in a prior study of BOS (Clin Gastroenterol Hepatol. 2015 Jan 13. doi: 10.1016/j.cgh.2014.05.02), and a lower percentage than in other studies of topical steroids for eosinophilic esophagitis, according to the researchers. Morning cortisol levels were similar between groups, and there were no adverse laboratory effects, they added.
Patients in this trial had severe symptoms and histology and were highly compliant with treatment. They filled out at least 70% of their symptom diary, had at least 15 eosinophils per high-power frame from at least two esophageal levels on screening endoscopy, and reported at least 4 days of dysphagia during the second half of a 4-week, blinded placebo run-in period. Researchers should consider using these strict inclusion criteria in future trials of eosinophilic esophagitis, especially because previous studies have failed to show a treatment benefit for topical steroid therapy, the investigators noted.
Meritage Pharma, which is now a part of the Shire group, makes budesonide oral suspension and sponsored the study. Dr. Dellon disclosed ties to Meritage, Receptos, Regeneron, Aptalis, Banner Life Sciences, Novartis, and Roche. All five coinvestigators disclosed ties to industry, including Meritage, Shire, Receptos, Regeneron, and Biogen Idec.
FROM GASTROENTEROLOGY
Key clinical point: Budesonide oral suspension (BOS) (2 mg twice daily) was safe and significantly outperformed placebo on validated measures of eosinophilic esophagitis.
Major finding: Dysphagia Symptom Questionnaire scores decreased by 14.3 points with BOS and by 7.5 points with placebo (P = .001). Endoscopic severity scores decreased by 3.8 points and rose by 0.4 points, respectively (P less than .0001).
Data source: A 12-week, double-blind, placebo-controlled, parallel-group, phase II trial of 93 adolescents and adults with eosinophilic esophagitis.
Disclosures: Meritage Pharma, which is now a part of the Shire group, makes budesonide oral suspension and sponsored the study. Dr. Dellon disclosed ties to Meritage, Receptos, Regeneron, Aptalis, Banner Life Sciences, Novartis, and Roche. All five coinvestigators disclosed ties to industry, including Meritage, Shire, Receptos, Regeneron, and Biogen Idec.
Can a Sigma-1 Agonist Stabilize Cognition and Function in Alzheimer’s Disease?
SAN DIEGO—A novel Alzheimer’s disease drug candidate appeared to stabilize cognition and function over 57 weeks in a small, early-phase, open-label trial.
Patients with mild to moderate Alzheimer’s disease who took ANAVEX 2-73, an agonist of the sigma-1 receptor, experienced virtually no decline on the Mini-Mental State Examination (MMSE) and Alzheimer’s Disease Cooperative
ANAVEX 2-73 (Anavex Life Sciences, New York) also conferred an unexpected benefit upon subjects with insomnia. “Any patient who scored on the insomnia measure [of the Hamilton Depression Rating Scale] at baseline had no sleep disturbance at all by weeks 12 and 26,” Stephen Macfarlane, MBBS, said at the Clinical Trials on Alzheimer’s Disease conference.

The findings must be interpreted cautiously. The phase IIa study was designed to assess safety and tolerability; cognitive and functional end points were secondary. It comprised 32 patients at baseline, 25 of whom completed both a five-week, randomized, dose-finding, crossover trial and a 52-week, open-label, extension study. There was no placebo comparator. Instead, researchers used three different sets of historical control data.
Investigators will continue to treat and follow the extension study cohort, and plan to launch a placebo-controlled study in 2017, said Dr. Macfarlane, Head of Clinical Governance for the Dementia Centre in Melbourne.
The five-week, randomized, dose-finding, crossover trial started one group of patients on 30 mg/day or 50 mg/day oral ANAVEX 2-73 for 11 days after an initial two-day, single-dose, pharmacokinetic analysis, followed by an 11-day washout period, and then 11 days of 3 mg/day or 5 mg/day intravenously. A second group first received 11 days of 3 mg/day or 5 mg/day ANAVEX 2-73 intravenously after an initial 2-day, single-dose, pharmacokinetic analysis, followed by an 11-day washout period, and then 30 mg/day or 50 mg/day oral ANAVEX 2-73 for 11 days. This trial was followed by a 52-week, open-label, extension trial of 10-50 mg/day orally, titrating each patient to the maximum tolerated dose. The extension phase was originally planned to last six months, but patients and caregivers wanted to continue on the medication, so the company extended it to 12 months. It is ongoing.
An Attractive Drug Target
The sigma-1 receptor targeted by ANAVEX 2-73 is found on neurons and glia in many areas of the CNS. It modulates a number of processes implicated in neurodegenerative diseases, including glutamate and calcium activity, reaction to oxidative stress, and mitochondrial function. There is some evidence that sigma-1 receptor activation can induce neuronal regrowth and functional recovery after stroke.
The sigma-1 receptor also appears to play a role in helping cells clear misfolded proteins—a pathway that makes it an attractive drug target in Alzheimer’s disease, as well as other neurodegenerative diseases with aberrant proteins, such as Parkinson’s and Huntington’s diseases.
In preclinical testing, ANAVEX 2-73 seemed to enhance cognition in wild-type and Alzheimer’s disease–model mice.
The mean age of patients in the extension study was 71. The median MMSE score was 20.5. Most patients (78%) were taking a stable dose of an acetylcholinesterase inhibitor. During the extension phase, they were titrated to the maximum tolerated dose; 14 mg was the minimum dose necessary to achieve a therapeutic effect and keep the MMSE stable.
The primary end points were safety, tolerability, and pharmacokinetics. The exploratory measures included the P300 electroencephalogram, MMSE score, the Computerized Cogstate Alzheimer’s Battery, and the ADCS-ADL. The Hamilton Depression (HAM-D) Scale was also employed as a neuropsychiatric symptom measure.
The cohort had low baseline depression scores, with a mean score of 2 on the HAM-D. By the study’s end, it had decreased to a mean of 1 point. Patients also reported improvements in their ability to work or do other activities, and in anxiety, agitation, hypochondriasis, and insight.
Electrophysiologic Measures
The P300 wave amplitude showed a small initial increase from about 6 microvolts to 7 microvolts by four weeks, and then returned to about 6 microvolts until about week 32. Thereafter, it steadily improved to about 8 microvolts by 57 weeks—a level usually seen in healthy age-matched controls. There was a significant separation from the P300 decline seen in a matched historical Alzheimer’s disease cohort, which decreased to about 4 microvolts over a 52-week period while patients were taking donepezil.
Researchers used a second historical control group to assess changes on the Computerized Cogstate Alzheimer’s Battery. All subjects in a large Australian prospective cohort study, called AIBL (Australian Imaging, Biomarkers & Lifestyle Flagship Study of Ageing), were taking standard of care Alzheimer’s drugs. Compared with that cohort, the ANAVEX 2-73 group experienced benefits in processing speed, attention, and working memory, which became statistically significant at week 31 and continued to improve.
At 57 weeks, patients’ mean MMSE score was near the baseline mean score of 20. ADCS-ADL scores declined slightly, from a mean of about 70 to approximately 65.
Finally, the investigators used another historical cohort to assess projected cognitive and functional benefit. Compared with a pooled, placebo-arm, cohort study conducted by the Alzheimer Disease Cooperative Study Group over 12 months, ANAVEX 2-73 would have been associated with 1.8-point benefit on the MMSE and a 4-point benefit on the ADCS-ADL.
“The MMSE declined 45% less and the ADCS-ADL declined 56% less than what we would have expected from the historical control data,” Dr. Macfarlane said. “This is not only statistically significant, but clearly clinically meaningful for patients.”
Nearly all patients (98%) experienced an adverse event. Most events were mild, transitory dizziness or headache; 76% of the events were grade 1, and 2% were grade 2. There were no serious adverse events. Three subjects dropped out of the trial because of adverse events (ie, delirium, dizziness, and a combination of confusion, disorientation, and lethargy). There were no problematic interactions between the study drug and any standard of care Alzheimer’s disease medications.
—Michelle G. Sullivan
SAN DIEGO—A novel Alzheimer’s disease drug candidate appeared to stabilize cognition and function over 57 weeks in a small, early-phase, open-label trial.
Patients with mild to moderate Alzheimer’s disease who took ANAVEX 2-73, an agonist of the sigma-1 receptor, experienced virtually no decline on the Mini-Mental State Examination (MMSE) and Alzheimer’s Disease Cooperative
ANAVEX 2-73 (Anavex Life Sciences, New York) also conferred an unexpected benefit upon subjects with insomnia. “Any patient who scored on the insomnia measure [of the Hamilton Depression Rating Scale] at baseline had no sleep disturbance at all by weeks 12 and 26,” Stephen Macfarlane, MBBS, said at the Clinical Trials on Alzheimer’s Disease conference.
The findings must be interpreted cautiously. The phase IIa study was designed to assess safety and tolerability; cognitive and functional end points were secondary. It comprised 32 patients at baseline, 25 of whom completed both a five-week, randomized, dose-finding, crossover trial and a 52-week, open-label, extension study. There was no placebo comparator. Instead, researchers used three different sets of historical control data.
Investigators will continue to treat and follow the extension study cohort, and plan to launch a placebo-controlled study in 2017, said Dr. Macfarlane, Head of Clinical Governance for the Dementia Centre in Melbourne.
The five-week, randomized, dose-finding, crossover trial started one group of patients on 30 mg/day or 50 mg/day oral ANAVEX 2-73 for 11 days after an initial two-day, single-dose, pharmacokinetic analysis, followed by an 11-day washout period, and then 11 days of 3 mg/day or 5 mg/day intravenously. A second group first received 11 days of 3 mg/day or 5 mg/day ANAVEX 2-73 intravenously after an initial 2-day, single-dose, pharmacokinetic analysis, followed by an 11-day washout period, and then 30 mg/day or 50 mg/day oral ANAVEX 2-73 for 11 days. This trial was followed by a 52-week, open-label, extension trial of 10-50 mg/day orally, titrating each patient to the maximum tolerated dose. The extension phase was originally planned to last six months, but patients and caregivers wanted to continue on the medication, so the company extended it to 12 months. It is ongoing.
An Attractive Drug Target
The sigma-1 receptor targeted by ANAVEX 2-73 is found on neurons and glia in many areas of the CNS. It modulates a number of processes implicated in neurodegenerative diseases, including glutamate and calcium activity, reaction to oxidative stress, and mitochondrial function. There is some evidence that sigma-1 receptor activation can induce neuronal regrowth and functional recovery after stroke.
The sigma-1 receptor also appears to play a role in helping cells clear misfolded proteins—a pathway that makes it an attractive drug target in Alzheimer’s disease, as well as other neurodegenerative diseases with aberrant proteins, such as Parkinson’s and Huntington’s diseases.
In preclinical testing, ANAVEX 2-73 seemed to enhance cognition in wild-type and Alzheimer’s disease–model mice.
The mean age of patients in the extension study was 71. The median MMSE score was 20.5. Most patients (78%) were taking a stable dose of an acetylcholinesterase inhibitor. During the extension phase, they were titrated to the maximum tolerated dose; 14 mg was the minimum dose necessary to achieve a therapeutic effect and keep the MMSE stable.
The primary end points were safety, tolerability, and pharmacokinetics. The exploratory measures included the P300 electroencephalogram, MMSE score, the Computerized Cogstate Alzheimer’s Battery, and the ADCS-ADL. The Hamilton Depression (HAM-D) Scale was also employed as a neuropsychiatric symptom measure.
The cohort had low baseline depression scores, with a mean score of 2 on the HAM-D. By the study’s end, it had decreased to a mean of 1 point. Patients also reported improvements in their ability to work or do other activities, and in anxiety, agitation, hypochondriasis, and insight.
Electrophysiologic Measures
The P300 wave amplitude showed a small initial increase from about 6 microvolts to 7 microvolts by four weeks, and then returned to about 6 microvolts until about week 32. Thereafter, it steadily improved to about 8 microvolts by 57 weeks—a level usually seen in healthy age-matched controls. There was a significant separation from the P300 decline seen in a matched historical Alzheimer’s disease cohort, which decreased to about 4 microvolts over a 52-week period while patients were taking donepezil.
Researchers used a second historical control group to assess changes on the Computerized Cogstate Alzheimer’s Battery. All subjects in a large Australian prospective cohort study, called AIBL (Australian Imaging, Biomarkers & Lifestyle Flagship Study of Ageing), were taking standard of care Alzheimer’s drugs. Compared with that cohort, the ANAVEX 2-73 group experienced benefits in processing speed, attention, and working memory, which became statistically significant at week 31 and continued to improve.
At 57 weeks, patients’ mean MMSE score was near the baseline mean score of 20. ADCS-ADL scores declined slightly, from a mean of about 70 to approximately 65.
Finally, the investigators used another historical cohort to assess projected cognitive and functional benefit. Compared with a pooled, placebo-arm, cohort study conducted by the Alzheimer Disease Cooperative Study Group over 12 months, ANAVEX 2-73 would have been associated with 1.8-point benefit on the MMSE and a 4-point benefit on the ADCS-ADL.
“The MMSE declined 45% less and the ADCS-ADL declined 56% less than what we would have expected from the historical control data,” Dr. Macfarlane said. “This is not only statistically significant, but clearly clinically meaningful for patients.”
Nearly all patients (98%) experienced an adverse event. Most events were mild, transitory dizziness or headache; 76% of the events were grade 1, and 2% were grade 2. There were no serious adverse events. Three subjects dropped out of the trial because of adverse events (ie, delirium, dizziness, and a combination of confusion, disorientation, and lethargy). There were no problematic interactions between the study drug and any standard of care Alzheimer’s disease medications.
—Michelle G. Sullivan
SAN DIEGO—A novel Alzheimer’s disease drug candidate appeared to stabilize cognition and function over 57 weeks in a small, early-phase, open-label trial.
Patients with mild to moderate Alzheimer’s disease who took ANAVEX 2-73, an agonist of the sigma-1 receptor, experienced virtually no decline on the Mini-Mental State Examination (MMSE) and Alzheimer’s Disease Cooperative
ANAVEX 2-73 (Anavex Life Sciences, New York) also conferred an unexpected benefit upon subjects with insomnia. “Any patient who scored on the insomnia measure [of the Hamilton Depression Rating Scale] at baseline had no sleep disturbance at all by weeks 12 and 26,” Stephen Macfarlane, MBBS, said at the Clinical Trials on Alzheimer’s Disease conference.
The findings must be interpreted cautiously. The phase IIa study was designed to assess safety and tolerability; cognitive and functional end points were secondary. It comprised 32 patients at baseline, 25 of whom completed both a five-week, randomized, dose-finding, crossover trial and a 52-week, open-label, extension study. There was no placebo comparator. Instead, researchers used three different sets of historical control data.
Investigators will continue to treat and follow the extension study cohort, and plan to launch a placebo-controlled study in 2017, said Dr. Macfarlane, Head of Clinical Governance for the Dementia Centre in Melbourne.
The five-week, randomized, dose-finding, crossover trial started one group of patients on 30 mg/day or 50 mg/day oral ANAVEX 2-73 for 11 days after an initial two-day, single-dose, pharmacokinetic analysis, followed by an 11-day washout period, and then 11 days of 3 mg/day or 5 mg/day intravenously. A second group first received 11 days of 3 mg/day or 5 mg/day ANAVEX 2-73 intravenously after an initial 2-day, single-dose, pharmacokinetic analysis, followed by an 11-day washout period, and then 30 mg/day or 50 mg/day oral ANAVEX 2-73 for 11 days. This trial was followed by a 52-week, open-label, extension trial of 10-50 mg/day orally, titrating each patient to the maximum tolerated dose. The extension phase was originally planned to last six months, but patients and caregivers wanted to continue on the medication, so the company extended it to 12 months. It is ongoing.
An Attractive Drug Target
The sigma-1 receptor targeted by ANAVEX 2-73 is found on neurons and glia in many areas of the CNS. It modulates a number of processes implicated in neurodegenerative diseases, including glutamate and calcium activity, reaction to oxidative stress, and mitochondrial function. There is some evidence that sigma-1 receptor activation can induce neuronal regrowth and functional recovery after stroke.
The sigma-1 receptor also appears to play a role in helping cells clear misfolded proteins—a pathway that makes it an attractive drug target in Alzheimer’s disease, as well as other neurodegenerative diseases with aberrant proteins, such as Parkinson’s and Huntington’s diseases.
In preclinical testing, ANAVEX 2-73 seemed to enhance cognition in wild-type and Alzheimer’s disease–model mice.
The mean age of patients in the extension study was 71. The median MMSE score was 20.5. Most patients (78%) were taking a stable dose of an acetylcholinesterase inhibitor. During the extension phase, they were titrated to the maximum tolerated dose; 14 mg was the minimum dose necessary to achieve a therapeutic effect and keep the MMSE stable.
The primary end points were safety, tolerability, and pharmacokinetics. The exploratory measures included the P300 electroencephalogram, MMSE score, the Computerized Cogstate Alzheimer’s Battery, and the ADCS-ADL. The Hamilton Depression (HAM-D) Scale was also employed as a neuropsychiatric symptom measure.
The cohort had low baseline depression scores, with a mean score of 2 on the HAM-D. By the study’s end, it had decreased to a mean of 1 point. Patients also reported improvements in their ability to work or do other activities, and in anxiety, agitation, hypochondriasis, and insight.
Electrophysiologic Measures
The P300 wave amplitude showed a small initial increase from about 6 microvolts to 7 microvolts by four weeks, and then returned to about 6 microvolts until about week 32. Thereafter, it steadily improved to about 8 microvolts by 57 weeks—a level usually seen in healthy age-matched controls. There was a significant separation from the P300 decline seen in a matched historical Alzheimer’s disease cohort, which decreased to about 4 microvolts over a 52-week period while patients were taking donepezil.
Researchers used a second historical control group to assess changes on the Computerized Cogstate Alzheimer’s Battery. All subjects in a large Australian prospective cohort study, called AIBL (Australian Imaging, Biomarkers & Lifestyle Flagship Study of Ageing), were taking standard of care Alzheimer’s drugs. Compared with that cohort, the ANAVEX 2-73 group experienced benefits in processing speed, attention, and working memory, which became statistically significant at week 31 and continued to improve.
At 57 weeks, patients’ mean MMSE score was near the baseline mean score of 20. ADCS-ADL scores declined slightly, from a mean of about 70 to approximately 65.
Finally, the investigators used another historical cohort to assess projected cognitive and functional benefit. Compared with a pooled, placebo-arm, cohort study conducted by the Alzheimer Disease Cooperative Study Group over 12 months, ANAVEX 2-73 would have been associated with 1.8-point benefit on the MMSE and a 4-point benefit on the ADCS-ADL.
“The MMSE declined 45% less and the ADCS-ADL declined 56% less than what we would have expected from the historical control data,” Dr. Macfarlane said. “This is not only statistically significant, but clearly clinically meaningful for patients.”
Nearly all patients (98%) experienced an adverse event. Most events were mild, transitory dizziness or headache; 76% of the events were grade 1, and 2% were grade 2. There were no serious adverse events. Three subjects dropped out of the trial because of adverse events (ie, delirium, dizziness, and a combination of confusion, disorientation, and lethargy). There were no problematic interactions between the study drug and any standard of care Alzheimer’s disease medications.
—Michelle G. Sullivan

Clinical Guidelines: ADA 2017 Standards of Medical Care in Diabetes
In 2012, 29.1 million Americans, or 9.3% of the population, had diabetes. Of this number, 21 million were diagnosed, and 8.1 million were undiagnosed. Each year almost 1.5 million Americans receive a new diagnosis of diabetes. The management of diabetes relies upon excellent primary care. Each year the American Diabetes Association reviews new evidence and publishes an updated Standards of Care in the January issue of Diabetes Care. Here we give a short overview of the guidelines with emphasis on fundamentals and changes in the standards over the past year.
Self-management education and support, nutrition therapy, and physical activity
All patients should participate in ongoing diabetes self-management education (DSME) to facilitate the knowledge, skills, and abilities necessary to obtain optimal self-care and incorporate the needs, goals, and life experiences of the person with diabetes as they face new challenges throughout a lifetime of diabetes.
In addition, each patient should receive individualized medical nutrition therapy (MNT) provided by a registered dietitian with knowledge regarding diabetes-specific MNT. Most patients should increase aerobic physical activity to 150 min/week. Providers should encourage patients to reduce the amount of time spent sedentary by briefly standing, walking, or performing other light physical activities every 30 minutes.
Glycemic targets
A reasonable hemoglobin A1c goal for many diabetic nonpregnant adults is less than 7%. A less stringent goal under 8% may be appropriate for patients with a history of severe hypoglycemia, limited life expectancy, advanced microvascular and macrovascular complications, and extensive comorbid conditions. HbA1c measurements should be done at diagnosis and routinely to monitor glycemic control. To aid in achieving glycemic targets, self-monitoring blood glucose (SMBG) allows patients to evaluate their individual response to therapy. Integrating SMBG data into diabetes management can help guide MNT, adjust medications, determine physical activity requirements, and prevent hypoglycemia. Individuals at risk for hypoglycemia should be asked about symptomatic and asymptomatic hypoglycemia at each encounter and counseled regarding treatment of hypoglycemic events.

Obesity management
There is strong and consistent evidence that obesity management may be beneficial in the treatment of type 2 diabetes. For overweight and obese patients with type 2 diabetes, interventions should be high intensity (more than 16 sessions in 6 months) and focus on diet, physical activity, and behavioral therapy designed to achieve a greater than 5% weight loss (energy deficit of 500-750 kcal/day).
For select patients, weight loss medications may be effective as adjuncts to lifestyle changes. When choosing additional pharmacologic interventions to improve glycemic control in overweight or obese patients, providers should use medications that promote weight loss or are weight neutral including metformin, sodium-glucose cotransporter-2 (SGLT-2) inhibitors, glucagon-like peptide-1 (GLP-1) agonists, and dipeptidyl peptidase-4 inhibitors (DPP-4) versus those that cause weight gain such as insulin secretagogues, thiazolidinediones, and insulin.
Metabolic surgery should be recommended to patients with type 2 diabetes and body mass index above 40 kg/m2 (BMI above 37.5 kg/m2 in Asian Americans), regardless of adequate glycemic control and for patients with BMI above 35 kg/m2 (more than 32.5 kg/m2 in Asian Americans) without adequate glycemic control despite lifestyle modifications and optimal medical therapy. Metabolic surgery should be considered for appropriate candidates with BMIs as low as 30 if hyperglycemia is inadequately controlled despite optical medical control by either oral or injectable medications.
CV disease and risk management: BP, lipids, antiplatelet therapy, and glycemic medication management
Atherosclerotic cardiovascular disease is the leading cause of morbidity and mortality for individuals with diabetes. Screening for atherosclerotic cardiovascular disease is not recommended; rather, the emphasis is on careful risk factor management.
If systolic blood pressure (SBP) is confirmed to be above 140 mm Hg and/or the diastolic blood pressure (DBP) is confirmed to be above 90 mm Hg, pharmacologic therapy should be initiated. A meta-analysis of randomized trials of adults with type 2 diabetes comparing intensive blood pressure targets (upper limit of 130 mm Hg SBP and 80 mm Hg DBP) with standard targets (upper limit of 140-160 mm Hg SBP and 85-100 mm Hg DBP) found no significant reduction in mortality or nonfatal MI. There was a statistically significant, 35% relative risk (RR) reduction in stroke with intensive targets, but intensive targets were associated with an increased risk for adverse events such as hypotension and syncope. Recommendations suggest that antihypertension treatment in adults with diabetes without albuminuria should include any of the classes of medication demonstrated to reduce cardiovascular events in patients with diabetes, such as ACE inhibitors, angiotensin receptor blockers (ARBs), thiazide-like diuretics, or dihydropyridine calcium-channel blockers. ACE inhibitors and ARBs continue to be recommended as first-line medications for the treatment of hypertension in patients with diabetes and elevated urine albumin/creatinine ratios (above 30 mg/g creatinine). The standards also suggest consideration of administering one or more antihypertensive medications at bedtime, which may improve cardiovascular outcomes.
For patients aged 40-75 years who have diabetes without additional atherosclerotic CV disease risk factors, a moderate-intensity statin should be considered. If there are additional cardiovascular risk factors, then a high-intensity statin should be considered. For patients who are younger than 40 years of age and have diabetes with additional atherosclerotic CV disease risk factors, a less strong recommendation is to consider using moderate-intensity or high-intensity statins. For patients older than 75 years with diabetes without additional atherosclerotic CV disease risk factors, consider using moderate-intensity statin therapy; high-intensity statin therapy may be considered in older adults with risk factors for atherosclerotic cardiovascular disease.
Both women and men who are at least 50 years old and have diabetes with at least one additional cardiovascular risk factor should consider taking daily aspirin therapy (75-162 mg/day) if they do not have any risk for excessive bleeding.
In patients with long-standing suboptimally controlled type 2 diabetes and established atherosclerotic CV disease, empagliflozin or liraglutide should be considered as they have been shown to reduce cardiovascular and all-cause mortality when added to standard care.

Microvascular disease and foot care
Large prospective studies have demonstrated that optimized glucose control can reduce the onset and progression of diabetic microvascular complications. Diabetic kidney disease occurs in about 20%-40% of persons with diabetes. Annual screening includes estimated glomerular filtration rate and spot urine albumin-to-creatinine ratio. Treatment includes ACE inhibitors or ARBs in addition to a target blood pressure of under 140/90 mm Hg.
Diabetic retinopathy screening includes a dilated eye exam by an eye care professional. Treatment includes laser photocoagulation therapy for high risk nonproliferative retinopathy or proliferative retinopathy, or intravitreal injections of antivascular endothelial growth factor agents for central-involved diabetic macular edema.
Diabetic peripheral neuropathy screening includes annual 10-g monofilament and 128-HZ tuning fork vibration sensation. Medications for painful diabetic neuropathy may include gabapentin, pregabalin, duloxetine, and other agents.
Neuropathy and vascular disease are contributors to diabetic foot ulcers and amputation. A comprehensive foot examination along with appropriate risk factor oriented history to include neuropathic and vascular components (pulses, claudication) should be performed annually, while all patients with diabetes should have their feet checked at every visit.
Older adults
Prioritizing treatment goals in older adults is important in this heterogeneous population. Cardiovascular risk factor treatment is likely to be beneficial.
In setting HbA1c goals, functional status, and comorbid conditions should be considered. Metformin can still be a first-line agent for many older adults with type 2 diabetes, with consideration to renal status (creatinine clearance above 30 mL/min per 1.73 m2) and heart failure. DPP-4s have few side effects and low risk of hypoglycemia. GLP-1 receptor agonists have a low risk of hypoglycemia but may be associated with GI side effects and weight loss. SGLT-2 inhibitors have a low risk of hypoglycemia, and attention should be paid to renal thresholds for use. Thiazolidinediones should be used cautiously in those with heart failure or at elevated fracture risk. Sulfonylureas should be used cautiously because of their elevated risk of hypoglycemia. When used, a short-acting sulfonylurea – such as glipizide – is preferred, as long-acting sulfonylureas are contraindicated because of even greater hypoglycemic risk. Single-injection basal insulin may be appropriate for many with ease of use and efficacy.
The bottom line
Diabetes is a rapidly changing field and each year the American Diabetes Association updates the Standards of Medical Care document to be consistent with the latest evidence. Highlights of the standards include emphasis on diabetes self-management education, individualized glycemic goal setting, obesity management, setting blood pressure targets to less than 140/90 mm Hg, as well as statins and daily aspirin for most people with diabetes. In addition, ADA now recommends the use of specific antihyperglycemic medications to reduce cardiovascular and all-cause mortality in patients with diabetes and established cardiovascular disease.
Reference
American Diabetes Association Standards of Medical Care in Diabetes – 2017. Diabetes Care 2017; 40 (sup 1):S1-S138
Dr. Skolnik is professor of family and community medicine, Temple University School of Medicine, Philadelphia, and associate director, Family Medicine Residency Program, Abington-Jefferson Health, Abington, Pa. Dr. Johnson is associate professor at the University of North Dakota School of Medicine and Health Sciences, and practices at the Altru Diabetes Center, Grand Forks. Ms. Neuman practices at St. Mark’s Hospital, Salt Lake City.
In 2012, 29.1 million Americans, or 9.3% of the population, had diabetes. Of this number, 21 million were diagnosed, and 8.1 million were undiagnosed. Each year almost 1.5 million Americans receive a new diagnosis of diabetes. The management of diabetes relies upon excellent primary care. Each year the American Diabetes Association reviews new evidence and publishes an updated Standards of Care in the January issue of Diabetes Care. Here we give a short overview of the guidelines with emphasis on fundamentals and changes in the standards over the past year.
Self-management education and support, nutrition therapy, and physical activity
All patients should participate in ongoing diabetes self-management education (DSME) to facilitate the knowledge, skills, and abilities necessary to obtain optimal self-care and incorporate the needs, goals, and life experiences of the person with diabetes as they face new challenges throughout a lifetime of diabetes.
In addition, each patient should receive individualized medical nutrition therapy (MNT) provided by a registered dietitian with knowledge regarding diabetes-specific MNT. Most patients should increase aerobic physical activity to 150 min/week. Providers should encourage patients to reduce the amount of time spent sedentary by briefly standing, walking, or performing other light physical activities every 30 minutes.
Glycemic targets
A reasonable hemoglobin A1c goal for many diabetic nonpregnant adults is less than 7%. A less stringent goal under 8% may be appropriate for patients with a history of severe hypoglycemia, limited life expectancy, advanced microvascular and macrovascular complications, and extensive comorbid conditions. HbA1c measurements should be done at diagnosis and routinely to monitor glycemic control. To aid in achieving glycemic targets, self-monitoring blood glucose (SMBG) allows patients to evaluate their individual response to therapy. Integrating SMBG data into diabetes management can help guide MNT, adjust medications, determine physical activity requirements, and prevent hypoglycemia. Individuals at risk for hypoglycemia should be asked about symptomatic and asymptomatic hypoglycemia at each encounter and counseled regarding treatment of hypoglycemic events.
Obesity management
There is strong and consistent evidence that obesity management may be beneficial in the treatment of type 2 diabetes. For overweight and obese patients with type 2 diabetes, interventions should be high intensity (more than 16 sessions in 6 months) and focus on diet, physical activity, and behavioral therapy designed to achieve a greater than 5% weight loss (energy deficit of 500-750 kcal/day).
For select patients, weight loss medications may be effective as adjuncts to lifestyle changes. When choosing additional pharmacologic interventions to improve glycemic control in overweight or obese patients, providers should use medications that promote weight loss or are weight neutral including metformin, sodium-glucose cotransporter-2 (SGLT-2) inhibitors, glucagon-like peptide-1 (GLP-1) agonists, and dipeptidyl peptidase-4 inhibitors (DPP-4) versus those that cause weight gain such as insulin secretagogues, thiazolidinediones, and insulin.
Metabolic surgery should be recommended to patients with type 2 diabetes and body mass index above 40 kg/m2 (BMI above 37.5 kg/m2 in Asian Americans), regardless of adequate glycemic control and for patients with BMI above 35 kg/m2 (more than 32.5 kg/m2 in Asian Americans) without adequate glycemic control despite lifestyle modifications and optimal medical therapy. Metabolic surgery should be considered for appropriate candidates with BMIs as low as 30 if hyperglycemia is inadequately controlled despite optical medical control by either oral or injectable medications.
CV disease and risk management: BP, lipids, antiplatelet therapy, and glycemic medication management
Atherosclerotic cardiovascular disease is the leading cause of morbidity and mortality for individuals with diabetes. Screening for atherosclerotic cardiovascular disease is not recommended; rather, the emphasis is on careful risk factor management.
If systolic blood pressure (SBP) is confirmed to be above 140 mm Hg and/or the diastolic blood pressure (DBP) is confirmed to be above 90 mm Hg, pharmacologic therapy should be initiated. A meta-analysis of randomized trials of adults with type 2 diabetes comparing intensive blood pressure targets (upper limit of 130 mm Hg SBP and 80 mm Hg DBP) with standard targets (upper limit of 140-160 mm Hg SBP and 85-100 mm Hg DBP) found no significant reduction in mortality or nonfatal MI. There was a statistically significant, 35% relative risk (RR) reduction in stroke with intensive targets, but intensive targets were associated with an increased risk for adverse events such as hypotension and syncope. Recommendations suggest that antihypertension treatment in adults with diabetes without albuminuria should include any of the classes of medication demonstrated to reduce cardiovascular events in patients with diabetes, such as ACE inhibitors, angiotensin receptor blockers (ARBs), thiazide-like diuretics, or dihydropyridine calcium-channel blockers. ACE inhibitors and ARBs continue to be recommended as first-line medications for the treatment of hypertension in patients with diabetes and elevated urine albumin/creatinine ratios (above 30 mg/g creatinine). The standards also suggest consideration of administering one or more antihypertensive medications at bedtime, which may improve cardiovascular outcomes.
For patients aged 40-75 years who have diabetes without additional atherosclerotic CV disease risk factors, a moderate-intensity statin should be considered. If there are additional cardiovascular risk factors, then a high-intensity statin should be considered. For patients who are younger than 40 years of age and have diabetes with additional atherosclerotic CV disease risk factors, a less strong recommendation is to consider using moderate-intensity or high-intensity statins. For patients older than 75 years with diabetes without additional atherosclerotic CV disease risk factors, consider using moderate-intensity statin therapy; high-intensity statin therapy may be considered in older adults with risk factors for atherosclerotic cardiovascular disease.
Both women and men who are at least 50 years old and have diabetes with at least one additional cardiovascular risk factor should consider taking daily aspirin therapy (75-162 mg/day) if they do not have any risk for excessive bleeding.
In patients with long-standing suboptimally controlled type 2 diabetes and established atherosclerotic CV disease, empagliflozin or liraglutide should be considered as they have been shown to reduce cardiovascular and all-cause mortality when added to standard care.
Microvascular disease and foot care
Large prospective studies have demonstrated that optimized glucose control can reduce the onset and progression of diabetic microvascular complications. Diabetic kidney disease occurs in about 20%-40% of persons with diabetes. Annual screening includes estimated glomerular filtration rate and spot urine albumin-to-creatinine ratio. Treatment includes ACE inhibitors or ARBs in addition to a target blood pressure of under 140/90 mm Hg.
Diabetic retinopathy screening includes a dilated eye exam by an eye care professional. Treatment includes laser photocoagulation therapy for high risk nonproliferative retinopathy or proliferative retinopathy, or intravitreal injections of antivascular endothelial growth factor agents for central-involved diabetic macular edema.
Diabetic peripheral neuropathy screening includes annual 10-g monofilament and 128-HZ tuning fork vibration sensation. Medications for painful diabetic neuropathy may include gabapentin, pregabalin, duloxetine, and other agents.
Neuropathy and vascular disease are contributors to diabetic foot ulcers and amputation. A comprehensive foot examination along with appropriate risk factor oriented history to include neuropathic and vascular components (pulses, claudication) should be performed annually, while all patients with diabetes should have their feet checked at every visit.
Older adults
Prioritizing treatment goals in older adults is important in this heterogeneous population. Cardiovascular risk factor treatment is likely to be beneficial.
In setting HbA1c goals, functional status, and comorbid conditions should be considered. Metformin can still be a first-line agent for many older adults with type 2 diabetes, with consideration to renal status (creatinine clearance above 30 mL/min per 1.73 m2) and heart failure. DPP-4s have few side effects and low risk of hypoglycemia. GLP-1 receptor agonists have a low risk of hypoglycemia but may be associated with GI side effects and weight loss. SGLT-2 inhibitors have a low risk of hypoglycemia, and attention should be paid to renal thresholds for use. Thiazolidinediones should be used cautiously in those with heart failure or at elevated fracture risk. Sulfonylureas should be used cautiously because of their elevated risk of hypoglycemia. When used, a short-acting sulfonylurea – such as glipizide – is preferred, as long-acting sulfonylureas are contraindicated because of even greater hypoglycemic risk. Single-injection basal insulin may be appropriate for many with ease of use and efficacy.
The bottom line
Diabetes is a rapidly changing field and each year the American Diabetes Association updates the Standards of Medical Care document to be consistent with the latest evidence. Highlights of the standards include emphasis on diabetes self-management education, individualized glycemic goal setting, obesity management, setting blood pressure targets to less than 140/90 mm Hg, as well as statins and daily aspirin for most people with diabetes. In addition, ADA now recommends the use of specific antihyperglycemic medications to reduce cardiovascular and all-cause mortality in patients with diabetes and established cardiovascular disease.
Reference
American Diabetes Association Standards of Medical Care in Diabetes – 2017. Diabetes Care 2017; 40 (sup 1):S1-S138
Dr. Skolnik is professor of family and community medicine, Temple University School of Medicine, Philadelphia, and associate director, Family Medicine Residency Program, Abington-Jefferson Health, Abington, Pa. Dr. Johnson is associate professor at the University of North Dakota School of Medicine and Health Sciences, and practices at the Altru Diabetes Center, Grand Forks. Ms. Neuman practices at St. Mark’s Hospital, Salt Lake City.
In 2012, 29.1 million Americans, or 9.3% of the population, had diabetes. Of this number, 21 million were diagnosed, and 8.1 million were undiagnosed. Each year almost 1.5 million Americans receive a new diagnosis of diabetes. The management of diabetes relies upon excellent primary care. Each year the American Diabetes Association reviews new evidence and publishes an updated Standards of Care in the January issue of Diabetes Care. Here we give a short overview of the guidelines with emphasis on fundamentals and changes in the standards over the past year.
Self-management education and support, nutrition therapy, and physical activity
All patients should participate in ongoing diabetes self-management education (DSME) to facilitate the knowledge, skills, and abilities necessary to obtain optimal self-care and incorporate the needs, goals, and life experiences of the person with diabetes as they face new challenges throughout a lifetime of diabetes.
In addition, each patient should receive individualized medical nutrition therapy (MNT) provided by a registered dietitian with knowledge regarding diabetes-specific MNT. Most patients should increase aerobic physical activity to 150 min/week. Providers should encourage patients to reduce the amount of time spent sedentary by briefly standing, walking, or performing other light physical activities every 30 minutes.
Glycemic targets
A reasonable hemoglobin A1c goal for many diabetic nonpregnant adults is less than 7%. A less stringent goal under 8% may be appropriate for patients with a history of severe hypoglycemia, limited life expectancy, advanced microvascular and macrovascular complications, and extensive comorbid conditions. HbA1c measurements should be done at diagnosis and routinely to monitor glycemic control. To aid in achieving glycemic targets, self-monitoring blood glucose (SMBG) allows patients to evaluate their individual response to therapy. Integrating SMBG data into diabetes management can help guide MNT, adjust medications, determine physical activity requirements, and prevent hypoglycemia. Individuals at risk for hypoglycemia should be asked about symptomatic and asymptomatic hypoglycemia at each encounter and counseled regarding treatment of hypoglycemic events.
Obesity management
There is strong and consistent evidence that obesity management may be beneficial in the treatment of type 2 diabetes. For overweight and obese patients with type 2 diabetes, interventions should be high intensity (more than 16 sessions in 6 months) and focus on diet, physical activity, and behavioral therapy designed to achieve a greater than 5% weight loss (energy deficit of 500-750 kcal/day).
For select patients, weight loss medications may be effective as adjuncts to lifestyle changes. When choosing additional pharmacologic interventions to improve glycemic control in overweight or obese patients, providers should use medications that promote weight loss or are weight neutral including metformin, sodium-glucose cotransporter-2 (SGLT-2) inhibitors, glucagon-like peptide-1 (GLP-1) agonists, and dipeptidyl peptidase-4 inhibitors (DPP-4) versus those that cause weight gain such as insulin secretagogues, thiazolidinediones, and insulin.
Metabolic surgery should be recommended to patients with type 2 diabetes and body mass index above 40 kg/m2 (BMI above 37.5 kg/m2 in Asian Americans), regardless of adequate glycemic control and for patients with BMI above 35 kg/m2 (more than 32.5 kg/m2 in Asian Americans) without adequate glycemic control despite lifestyle modifications and optimal medical therapy. Metabolic surgery should be considered for appropriate candidates with BMIs as low as 30 if hyperglycemia is inadequately controlled despite optical medical control by either oral or injectable medications.
CV disease and risk management: BP, lipids, antiplatelet therapy, and glycemic medication management
Atherosclerotic cardiovascular disease is the leading cause of morbidity and mortality for individuals with diabetes. Screening for atherosclerotic cardiovascular disease is not recommended; rather, the emphasis is on careful risk factor management.
If systolic blood pressure (SBP) is confirmed to be above 140 mm Hg and/or the diastolic blood pressure (DBP) is confirmed to be above 90 mm Hg, pharmacologic therapy should be initiated. A meta-analysis of randomized trials of adults with type 2 diabetes comparing intensive blood pressure targets (upper limit of 130 mm Hg SBP and 80 mm Hg DBP) with standard targets (upper limit of 140-160 mm Hg SBP and 85-100 mm Hg DBP) found no significant reduction in mortality or nonfatal MI. There was a statistically significant, 35% relative risk (RR) reduction in stroke with intensive targets, but intensive targets were associated with an increased risk for adverse events such as hypotension and syncope. Recommendations suggest that antihypertension treatment in adults with diabetes without albuminuria should include any of the classes of medication demonstrated to reduce cardiovascular events in patients with diabetes, such as ACE inhibitors, angiotensin receptor blockers (ARBs), thiazide-like diuretics, or dihydropyridine calcium-channel blockers. ACE inhibitors and ARBs continue to be recommended as first-line medications for the treatment of hypertension in patients with diabetes and elevated urine albumin/creatinine ratios (above 30 mg/g creatinine). The standards also suggest consideration of administering one or more antihypertensive medications at bedtime, which may improve cardiovascular outcomes.
For patients aged 40-75 years who have diabetes without additional atherosclerotic CV disease risk factors, a moderate-intensity statin should be considered. If there are additional cardiovascular risk factors, then a high-intensity statin should be considered. For patients who are younger than 40 years of age and have diabetes with additional atherosclerotic CV disease risk factors, a less strong recommendation is to consider using moderate-intensity or high-intensity statins. For patients older than 75 years with diabetes without additional atherosclerotic CV disease risk factors, consider using moderate-intensity statin therapy; high-intensity statin therapy may be considered in older adults with risk factors for atherosclerotic cardiovascular disease.
Both women and men who are at least 50 years old and have diabetes with at least one additional cardiovascular risk factor should consider taking daily aspirin therapy (75-162 mg/day) if they do not have any risk for excessive bleeding.
In patients with long-standing suboptimally controlled type 2 diabetes and established atherosclerotic CV disease, empagliflozin or liraglutide should be considered as they have been shown to reduce cardiovascular and all-cause mortality when added to standard care.
Microvascular disease and foot care
Large prospective studies have demonstrated that optimized glucose control can reduce the onset and progression of diabetic microvascular complications. Diabetic kidney disease occurs in about 20%-40% of persons with diabetes. Annual screening includes estimated glomerular filtration rate and spot urine albumin-to-creatinine ratio. Treatment includes ACE inhibitors or ARBs in addition to a target blood pressure of under 140/90 mm Hg.
Diabetic retinopathy screening includes a dilated eye exam by an eye care professional. Treatment includes laser photocoagulation therapy for high risk nonproliferative retinopathy or proliferative retinopathy, or intravitreal injections of antivascular endothelial growth factor agents for central-involved diabetic macular edema.
Diabetic peripheral neuropathy screening includes annual 10-g monofilament and 128-HZ tuning fork vibration sensation. Medications for painful diabetic neuropathy may include gabapentin, pregabalin, duloxetine, and other agents.
Neuropathy and vascular disease are contributors to diabetic foot ulcers and amputation. A comprehensive foot examination along with appropriate risk factor oriented history to include neuropathic and vascular components (pulses, claudication) should be performed annually, while all patients with diabetes should have their feet checked at every visit.
Older adults
Prioritizing treatment goals in older adults is important in this heterogeneous population. Cardiovascular risk factor treatment is likely to be beneficial.
In setting HbA1c goals, functional status, and comorbid conditions should be considered. Metformin can still be a first-line agent for many older adults with type 2 diabetes, with consideration to renal status (creatinine clearance above 30 mL/min per 1.73 m2) and heart failure. DPP-4s have few side effects and low risk of hypoglycemia. GLP-1 receptor agonists have a low risk of hypoglycemia but may be associated with GI side effects and weight loss. SGLT-2 inhibitors have a low risk of hypoglycemia, and attention should be paid to renal thresholds for use. Thiazolidinediones should be used cautiously in those with heart failure or at elevated fracture risk. Sulfonylureas should be used cautiously because of their elevated risk of hypoglycemia. When used, a short-acting sulfonylurea – such as glipizide – is preferred, as long-acting sulfonylureas are contraindicated because of even greater hypoglycemic risk. Single-injection basal insulin may be appropriate for many with ease of use and efficacy.
The bottom line
Diabetes is a rapidly changing field and each year the American Diabetes Association updates the Standards of Medical Care document to be consistent with the latest evidence. Highlights of the standards include emphasis on diabetes self-management education, individualized glycemic goal setting, obesity management, setting blood pressure targets to less than 140/90 mm Hg, as well as statins and daily aspirin for most people with diabetes. In addition, ADA now recommends the use of specific antihyperglycemic medications to reduce cardiovascular and all-cause mortality in patients with diabetes and established cardiovascular disease.
Reference
American Diabetes Association Standards of Medical Care in Diabetes – 2017. Diabetes Care 2017; 40 (sup 1):S1-S138
Dr. Skolnik is professor of family and community medicine, Temple University School of Medicine, Philadelphia, and associate director, Family Medicine Residency Program, Abington-Jefferson Health, Abington, Pa. Dr. Johnson is associate professor at the University of North Dakota School of Medicine and Health Sciences, and practices at the Altru Diabetes Center, Grand Forks. Ms. Neuman practices at St. Mark’s Hospital, Salt Lake City.

Underdosing of Lorazepam in Children Is Associated With Increased Seizure Duration
HOUSTON—The first dose of lorazepam, when administered as a first-line antiepileptic drug (AED) for pediatric refractory status epilepticus, frequently is underdosed, and doses lower than 0.1 mg/kg are associated with increased seizure duration, according to research presented at the 70th Annual Meeting of the American Epilepsy Society (AES).
The results emphasize the importance of following AES status epilepticus guidelines, which call for lorazepam dosing of 0.1 mg/kg, said Dmitry Tchapyjnikov, MD, of Duke University School of Medicine in Durham, North Carolina, and colleagues.

“There is high variability in lorazepam dosing when used in the treatment of status epilepticus, but little is known about how this dosing variability affects seizure duration,” the researchers said. The investigators analyzed data from a multicenter prospective observational cohort of pediatric patients admitted with refractory status epilepticus (ie, status epilepticus did not resolve after two or more AEDs) between 2011 and 2016. The data were compiled by the Pediatric Status Epilepticus Research Group.
Researchers grouped patients by those who received a lower dose ( < 0.05 mg/kg), medium dose (0.05 mg/kg to < 0.1 mg/kg), and higher dose ( ≥ 0.1 mg/kg) of lorazepam. They used Cox proportional hazards models to assess the association between lorazepam dose and time to seizure resolution, adjusting for age, sex, presumed seizure cause, seizure duration prior to lorazepam administration, home AED use, prior neurologic conditions, and study site.
A total of 103 patients were included in the analysis. Patients had a median age of 4.5, and 48% were female. Lorazepam was administered at a median of 20 minutes after seizure onset. Twenty-eight percent of patients received a lower dose, 43% a medium dose, and 29% a higher dose. Individuals in the higher dose group were significantly more likely to experience seizure resolution sooner than patients in the medium and lower dose groups, with hazard ratios of 1.62 and 2.49, respectively. Median time to total seizure resolution following lorazepam administration was 93 minutes in the higher dose group, 160 minutes in the medium dose group, and 350 minutes in the lower dose group.
Among patients who had convulsive seizures, those in the higher dose group were more likely to experience convulsive seizure resolution sooner than those in the lower dose group (hazard ratio, 1.89). Median time to convulsive seizure resolution was 67 minutes in the higher dose group and 120 minutes in the lower dose group.
—Jake Remaly
HOUSTON—The first dose of lorazepam, when administered as a first-line antiepileptic drug (AED) for pediatric refractory status epilepticus, frequently is underdosed, and doses lower than 0.1 mg/kg are associated with increased seizure duration, according to research presented at the 70th Annual Meeting of the American Epilepsy Society (AES).
The results emphasize the importance of following AES status epilepticus guidelines, which call for lorazepam dosing of 0.1 mg/kg, said Dmitry Tchapyjnikov, MD, of Duke University School of Medicine in Durham, North Carolina, and colleagues.
“There is high variability in lorazepam dosing when used in the treatment of status epilepticus, but little is known about how this dosing variability affects seizure duration,” the researchers said. The investigators analyzed data from a multicenter prospective observational cohort of pediatric patients admitted with refractory status epilepticus (ie, status epilepticus did not resolve after two or more AEDs) between 2011 and 2016. The data were compiled by the Pediatric Status Epilepticus Research Group.
Researchers grouped patients by those who received a lower dose ( < 0.05 mg/kg), medium dose (0.05 mg/kg to < 0.1 mg/kg), and higher dose ( ≥ 0.1 mg/kg) of lorazepam. They used Cox proportional hazards models to assess the association between lorazepam dose and time to seizure resolution, adjusting for age, sex, presumed seizure cause, seizure duration prior to lorazepam administration, home AED use, prior neurologic conditions, and study site.
A total of 103 patients were included in the analysis. Patients had a median age of 4.5, and 48% were female. Lorazepam was administered at a median of 20 minutes after seizure onset. Twenty-eight percent of patients received a lower dose, 43% a medium dose, and 29% a higher dose. Individuals in the higher dose group were significantly more likely to experience seizure resolution sooner than patients in the medium and lower dose groups, with hazard ratios of 1.62 and 2.49, respectively. Median time to total seizure resolution following lorazepam administration was 93 minutes in the higher dose group, 160 minutes in the medium dose group, and 350 minutes in the lower dose group.
Among patients who had convulsive seizures, those in the higher dose group were more likely to experience convulsive seizure resolution sooner than those in the lower dose group (hazard ratio, 1.89). Median time to convulsive seizure resolution was 67 minutes in the higher dose group and 120 minutes in the lower dose group.
—Jake Remaly
HOUSTON—The first dose of lorazepam, when administered as a first-line antiepileptic drug (AED) for pediatric refractory status epilepticus, frequently is underdosed, and doses lower than 0.1 mg/kg are associated with increased seizure duration, according to research presented at the 70th Annual Meeting of the American Epilepsy Society (AES).
The results emphasize the importance of following AES status epilepticus guidelines, which call for lorazepam dosing of 0.1 mg/kg, said Dmitry Tchapyjnikov, MD, of Duke University School of Medicine in Durham, North Carolina, and colleagues.
“There is high variability in lorazepam dosing when used in the treatment of status epilepticus, but little is known about how this dosing variability affects seizure duration,” the researchers said. The investigators analyzed data from a multicenter prospective observational cohort of pediatric patients admitted with refractory status epilepticus (ie, status epilepticus did not resolve after two or more AEDs) between 2011 and 2016. The data were compiled by the Pediatric Status Epilepticus Research Group.
Researchers grouped patients by those who received a lower dose ( < 0.05 mg/kg), medium dose (0.05 mg/kg to < 0.1 mg/kg), and higher dose ( ≥ 0.1 mg/kg) of lorazepam. They used Cox proportional hazards models to assess the association between lorazepam dose and time to seizure resolution, adjusting for age, sex, presumed seizure cause, seizure duration prior to lorazepam administration, home AED use, prior neurologic conditions, and study site.
A total of 103 patients were included in the analysis. Patients had a median age of 4.5, and 48% were female. Lorazepam was administered at a median of 20 minutes after seizure onset. Twenty-eight percent of patients received a lower dose, 43% a medium dose, and 29% a higher dose. Individuals in the higher dose group were significantly more likely to experience seizure resolution sooner than patients in the medium and lower dose groups, with hazard ratios of 1.62 and 2.49, respectively. Median time to total seizure resolution following lorazepam administration was 93 minutes in the higher dose group, 160 minutes in the medium dose group, and 350 minutes in the lower dose group.
Among patients who had convulsive seizures, those in the higher dose group were more likely to experience convulsive seizure resolution sooner than those in the lower dose group (hazard ratio, 1.89). Median time to convulsive seizure resolution was 67 minutes in the higher dose group and 120 minutes in the lower dose group.
—Jake Remaly
7 Myomectomy myths debunked
Fibroids are extremely common and can be detected in 60% of African American women and 40% of white women by age 35. By age 50, more than 80% of African American women and almost 70% of white women have fibroids. Although most women with fibroids are relatively asymptomatic, women who have bothersome symptoms, such as heavy menstrual bleeding, urinary frequency, pelvic or abdominal pressure, or pain, account for nearly 30% of all gynecologic admissions in the United States. The cost of fibroid-related care, including surgery, hospital admissions, outpatient visits, and medications, is estimated at $4 to $9 billion per year.1 In addition, each woman seeking treatment for fibroid-related symptoms incurs an expense of $4,500 to $30,000 for lost work or disability every year.1
Many treatment options, including medical therapy and noninvasive procedures, are now available for women with symptomatic fibroids. For women who require surgical treatment, however, hysterectomy is often recommended. Fibroid-related hysterectomy currently accounts for 45% of all hysterectomies, or approximately 195,700 per year. Although the American College of Obstetricians and Gynecologists (ACOG) clinical management guidelines state that myomectomy is a safe and effective alternative to hysterectomy for treatment of women with symptomatic fibroids, only 30,000 myomectomies (abdominal, laparoscopic, and robotic-assisted approaches) are performed each year.2 Why is this? One reason may be that, although many women wish to have uterus-preserving treatment, they often feel that doctors are too quick to recommend hysterectomy as the first—and sometimes only—treatment option for fibroids.3
CASE: Woman with fibroids seeks alternative to hysterectomy
A 42-year-old woman (G2P2) presents for a third opinion regarding her heavy menstrual bleeding and known uterine fibroids. She does not want to have any more children, but she wishes to avoid a hysterectomy. Both her regular gynecologist and the second gynecologist she consulted recommended hysterectomy as the first, and only, treatment option. Physical examination reveals a 16-week-sized uterus, and ultrasonography shows at least 6 fibroids, 2 of which impinge on the uterine cavity. The patient’s other gynecologists advised her that a myomectomy would be a “bloody operation,” would leave her uterus looking like Swiss cheese, and is not appropriate for women who have completed childbearing.
The patient asks if myomectomy could be considered in her situation. How would you advise her regarding myomectomy as an alternative to hysterectomy?
Organ conservation is important
In 1931, prominent British gynecologic surgeon Victor Bonney said, “Since cure without deformity or loss of function must ever be surgery’s highest ideal, the general proposition that myomectomy is a greater surgical achievement is incontestable.”4 As current hysterectomy and myomectomy rates indicate, however, we are not attempting organ conservation very often.
Other specialties almost never remove an entire organ for benign growths. Using breast cancer surgery as an admirable paradigm, consider that in the early 20th century the standard treatment for breast cancer was a Halsted radical mastectomy with axial lymphadenectomy. By the 1930s, this disfiguring operation was replaced by simple mastectomy and radiation, and by the 1970s, by lumpectomy and lymphadenectomy. Currently, lumpectomy and sentinel node sampling is the standard of care for early stage breast cancer. This is an excellent example of “minimally invasive surgery,” a term fostered by gynecologists. And, these organ-preservingsurgeries are performed for women with cancer, not a benign condition like fibroids.
Although our approach to hysterectomy has evolved with the increasing use of laparoscopic or robotic assistance, removal of the entire uterus nevertheless remains the surgical goal. I think this narrow view of surgical options is a disservice to our patients.
Many of us were taught that myomectomy was associated with more complications and more blood loss than hysterectomy. We were taught that the uterus had no function other than childbearing and that removing the uterus had no adverse health effects. The dogma suggested that myomectomy preserved a uterus that looked like Swiss cheese and would not heal properly and that the risk of fibroid recurrence was high. These beliefs, however, are myths, which are discussed and debunked below. In second and third installments for this series on myomectomy, I present steps for successful abdominal and laparoscopic technique.
Read myths on hysterectomy, myomectomy, and fibroids
MYTH #1: Hysterectomy is safer than myomectomy
Myomectomy is performed within the confines of the uterus and myometrium, with only infrequent occasion to operate near the ureters, uterine vessels, bowel, or bladder. Therefore, it should not be surprising that studies show that fewer complications occur with myomectomy than with hysterectomy.
A retrospective review of 197 women who had myomectomy and 197 women who underwent hysterectomy with similar uterine size (14 vs 15 weeks) reported that 13% (n = 26) of women in the hysterectomy group experienced complications, including 1 bladder injury, 1 ureteral injury, and 3 bowel injuries; 8 women had an ileus and 6 women had a pelvic abscess.5 Only 5% (n = 11) of the myomectomy patients had complications, including 1 bladder injury; 2 women had reoperation for small bowel obstruction, and 6 women had an ileus. The risks of febrile morbidity, unintended surgical procedure, life-threatening events, and rehospitalization were similar for both groups.
Authors of a recent systematic review of 6 studies, which included 1,520 women with uterine size up to 18 weeks, found higher rates of visceral injury and longer hospital stays for women who had a hysterectomy compared with those who had a myomectomy (TABLE 1).6
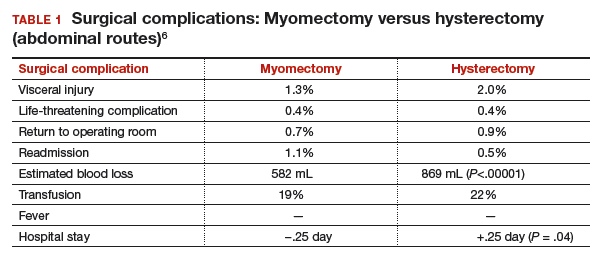
MYTH #2: Myomectomy is associated with more surgical blood loss than hysterectomy
In the previously cited study of 197 women treated with myomectomy and 197 women treated with hysterectomy, the estimated blood loss was greater in the hysterectomy group (484 mL) than in the myomectomy group (227 mL). When uterine size was corrected for, blood loss was no greater for myomectomy than for hysterectomy.5 The risk of hemorrhage (>500 mL blood loss) was greater in the hysterectomy group (14.2% vs 9.6%). Authors of the recent meta-analysis also found that the rate of transfusion was higher in the hysterectomy cohort. Tourniquets, misoprostol, vasopressin, and tranexamic acid all have been shown to significantly decrease surgical blood loss. (These treatments will be discussed in the next installment of this article series.)
MYTH #3: A uterus will look like Swiss cheese after a myomectomy
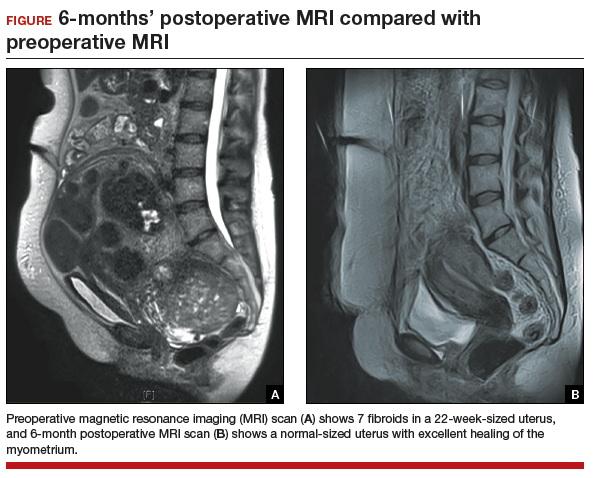
The uterus heals remarkably well after myomectomy. Three months following laparoscopic myomectomy, 3-dimensional Doppler ultrasonography demonstrated complete myometrial healing and normal blood flow to the uterus.7 In a study of women undergoing abdominal myomectomy, follow-up magnetic resonance imaging (MRI) with gadolinium showed complete healing of the myometrium and normal myometrial perfusion by 3 months.8 This study also found that, after removal of 65 g to 380 g of fibroids, the uterine volume 3 months after surgery was 65 mL, essentially equivalent to the normal volume of a uterus without fibroids (57 mL).8 See FIGURE for MRI scans of the uterus before and after myomectomy.
MYTH #4: Fibroids will just grow back after myomectomy
Once a fibroid is completely removed surgically, it does not grow back. The risk of new fibroid growth depends on the number of fibroids originally removed and the amount of time until menopause, when fibroids reduce in size and symptoms usually resolve. Given that the prevalence of fibroids is nearly 80% by age 50, studies measuring the detection of new fibroid growth of 1 cm on ultrasound imaging overstate the problem.9 What is likely a more important consideration for women is whether, following myomectomy, they will need another procedure for new fibroid-related symptoms.
Results of a meta-analysis of 872 women in 7 studies with 10- to 25-year follow-up indicated that 89% of women did not require another surgery.10 In another study, authors found that, over an average follow-up of 7.6 years, a second surgery occurred in 11% of the women who had 1 fibroid initially removed and for 26% of women who had multiple fibroids initially removed.11 In another study of 92 women who had either abdominal or laparoscopic myomectomy after age 45and who were followed for an average of 30 months, only 1 woman (1%) required a hysterectomy for fibroid-related symptoms.12 That patient had growth of a fibroid that was present but was not removed at her initial laparoscopic myomectomy.
Read myths 5–7 on ovarian conservation, fibroid growth, and symptom improvement
MYTH #5: Hysterectomy with ovarian conservation does not change hormone levels
Following hysterectomy with ovarian conservation, some women begin menopause earlier than age-matched women who have not undergone any surgery.13 Hysterectomy with ovarian conservation prior to age 50 has been associated with a significant increase in the risk of coronary heart disease, stroke, and heart failure.14 In a prospective longitudinal study, antimüllerian hormone (AMH) levels were persistently decreased following hysterectomy despite ovarian conservation.15 However, 3 months after myomectomy, no such changes in AMH levels were seen (TABLE 2).15

Early natural menopause has been associated with an increase in cardiovascular disease and death, and bilateral oophorectomy has been associated with increased risks of cardiovascular disease, all-cause mortality, lung cancer, colon cancer, anxiety, and depression. Although taking estrogen might obviate these adverse health effects, the majority of women who receive a prescription for estrogen following surgery are no longer taking it 5 years later.
MYTH #6: Fibroid growth in a premenopausal patient means cancer may be present
While most fibroids grow slowly, rapid growth of benign fibroids is very common. Using computerized analysis of a group of 72 women having serial MRI scans, investigators found that 34% of benign fibroids increased more than 20% in volume over 6 months.16 In premenopausal women, “rapid uterine growth” almost never indicates presence of uterine sarcoma. One study reported only 1 sarcoma among 371 women operated on for rapid growth of presumed fibroids.17 Using current criteria from the World Health Organization to determine the pathologic diagnosis, however, that 1 woman was determined to have had an atypical leiomyoma. Therefore, the prevalence of leiomyosarcoma in that study approached zero. In addition, in the 198 women who had a 6-week increase in uterine size over 1 year (one published definition of rapid growth), no sarcomas were found.17
Because of recent concern about leiomyosarcoma and morcellation of fibroids, some gynecologists have reverted to advising women that growing fibroids might be cancer and that hysterectomy is recommended. However, there is no evidence that fibroid growth is a sign of leiomyosarcoma in premenopausal women. Leiomyosarcoma should strongly be considered in a postmenopausal woman on no hormone therapy who has growth of a presumed fibroid.
MYTH #7: Myomectomy will not improve symptoms
Fibroid-related symptoms can be significant; women who undergo hysterectomy because of fibroid-related symptoms have significantly worse scores on the 36-Item Short-Form Survey (SF-36) quality-of-life questionnaire than women diagnosed with hypertension, heart disease, chronic lung disease, or arthritis.18
For women with fibroid-related symptoms, myomectomy has been shown to improve quality of life. A study of 72 women showed that SF-36 scores improved significantly following myomectomy (TABLE 3, page 48).19 In another study that used the European Quality of Life Five-Dimension Scale and Visual Analog Scale, 95 women had significant improvement in quality of life (P<.001) following laparoscopic myomectomy.20
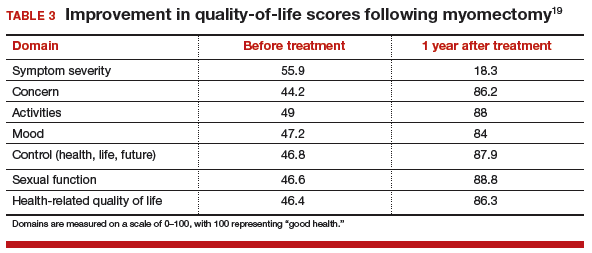
For some women, hysterectomy may have an impact on emotional quality of life. Some women report decreased sexual desire after hysterectomy. They worry that partners will see them as “not whole” and less desirable. Some women expect that hysterectomy will lead to depression, crying, lack of sexual desire, and vaginal dryness.21 No such changes have been reported for women having myomectomy.
CASE Continued: Third consult leads patient to schedule surgical procedure
After reviewing the patient’s symptoms, examination, and ultrasound results, we advise the patient that abdominal myomectomy is indeed appropriate and feasible in her case. She schedules surgery for the following month.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Cardozo ER, Clark AD, Banks NK, Henne MB, Stegmann BJ, Segars JH. The estimated annual cost of leiomyomata in the United States. Am J Obstet Gynecol. 2012;206(3):211.e1–e9.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins–Gynecology. ACOG Practice Bulletin No. 96: alternatives to hysterectomy in the management of leiomyomas. Obstet Gynecol. 2008;112(2 pt 1):387–400.
- Borah BJ, Nicholson WK, Bradley L, Stewart EA. The impact of uterine leiomyomas: a national survey of affected women. Am J Obstet Gynecol. 2013;209(4):319.e1–e20.
- Bonney V. The technique and results of myomectomy. Lancet. 1931;217(5604):171-177.
- Sawin SW, Pilevsky ND, Berlin JA, Barnhart KT. Comparability of perioperative morbidity between abdominal myomectomy and hysterectomy for women with uterine leiomyomas. Am J Obstet Gynecol. 2000;183(6):1448–1455.
- Pundir J, Walawalkar R, Seshadri S, Khalaf Y, El-Toukhy T. Perioperative morbidity associated with abdominal myomectomy compared with total abdominal hysterectomy for uterine fibroids. J Obstet Gynecol. 2013;33(7):655–662.
- Chang WC, Chang DY, Huang SC, et al. Use of three-dimensional ultrasonography in the evaluation of uterine perfusion and healing after laparoscopic myomectomy. Fertil Steril. 2009;92(3):1110–1115.
- Tsuji S, Takahashi K, Imaoka I, Sugimura K, Miyazaki K, Noda Y. MRI evaluation of the uterine structure after myomectomy. Gynecol Obstet Invest. 2006;61(2):106–110.
- Sudik R, Husch K, Steller J, Daume E. Fertility and pregnancy outcome after myomectomy in sterility patients. Eur J Obstet Gynecol Reprod Biol. 1996;65(2):209–214.
- Fauconnier A, Chapron C, Babaki-Fard K, Dubuisson JB. Recurrence of leiomyomata after myomectomy. Hum Reprod Update. 2000;6(6):595–602.
- Malone, LJ. Myomectomy: recurrence after removal of solitary and multiple myomas. Obstet Gynecol. 1969;34(2):200–203.
- Kim DH, Kim ML, Song T, Kim MK, Yoon BS, Seong SJ. Is myomectomy in women aged 45 years and older an effective option? Eur J Obstet Gynecol Reprod Biol. 2014;177:57–60.
- Farquhar CM, Sadler L, Harvey SA, Stewart AW. The association of hysterectomy and menopause: a prospective cohort study. BJOG. 2005;112(7):956–962.
- Ingelsson E, Lundholm C, Johansson AL, Altman D. Hysterectomy and risk of cardiovascular disease: a population-based cohort study. Eur Heart J. 2011;32(6):745–750.
- Wang HY, Quan S, Zhang RL, et al. Comparison of serum anti-Mullerian hormone levels following hysterectomy and myomectomy for benign gynaecological conditions. Eur J Obstet Gynecol Reprod Biol. 2013;171(2):368–371.
- Peddada SD, Laughlin SK, Miner K, et al. Growth of uterine leiomyomata among premenopausal black and white women. Proc Natl Acad Sci. 2008;105(50):19887–19892.
- Parker W, Fu YS, Berek JS. Uterine sarcoma in patients operated on for presumed leiomyoma and rapidly growing leiomyoma. Obstet Gynecol. 1994;83(3):414–418.
- Rowe MK, Kanouse DE, Mittman BS, Bernstein SJ. Quality of life among women undergoing hysterectomies. Obstet Gynecol. 1999;93(6):915–921.
- Dilek S, Ertunc D, Tok EC, Cimen R, Doruk A. The effect of myomectomy on health-related quality of life of women with myoma uteri. J Obstet Gynaecol Res. 2010;36(2):364–369.
- Radosa JC, Radosa CG, Mavrova R, et al. Postoperative quality of life and sexual function in premenopausal women undergoing laparoscopic myomectomy for symptomatic fibroids: a prospective observational cohort study. PLoS One. 2016;29;11(11):e0166659.
- Groff JY, Mullen PD, Byrd T, Shelton AJ, Lees E, Goode J. Decision making, beliefs, and attitudes toward hysterectomy: a focus group study with medically underserved women in Texas. J Womens Health Gend Based Med. 2000;9(suppl 2):39S–50S.

Dr. Parker is Director of Minimally Invasive Gynecologic Surgery at Santa Monica-UCLA Medical Center, Santa Monica, California. He is a past president of AAGL.
The author reports no financial relationships relevant to this article.
Dr. Parker is Director of Minimally Invasive Gynecologic Surgery at Santa Monica-UCLA Medical Center, Santa Monica, California. He is a past president of AAGL.
The author reports no financial relationships relevant to this article.
Dr. Parker is Director of Minimally Invasive Gynecologic Surgery at Santa Monica-UCLA Medical Center, Santa Monica, California. He is a past president of AAGL.
The author reports no financial relationships relevant to this article.
Fibroids are extremely common and can be detected in 60% of African American women and 40% of white women by age 35. By age 50, more than 80% of African American women and almost 70% of white women have fibroids. Although most women with fibroids are relatively asymptomatic, women who have bothersome symptoms, such as heavy menstrual bleeding, urinary frequency, pelvic or abdominal pressure, or pain, account for nearly 30% of all gynecologic admissions in the United States. The cost of fibroid-related care, including surgery, hospital admissions, outpatient visits, and medications, is estimated at $4 to $9 billion per year.1 In addition, each woman seeking treatment for fibroid-related symptoms incurs an expense of $4,500 to $30,000 for lost work or disability every year.1
Many treatment options, including medical therapy and noninvasive procedures, are now available for women with symptomatic fibroids. For women who require surgical treatment, however, hysterectomy is often recommended. Fibroid-related hysterectomy currently accounts for 45% of all hysterectomies, or approximately 195,700 per year. Although the American College of Obstetricians and Gynecologists (ACOG) clinical management guidelines state that myomectomy is a safe and effective alternative to hysterectomy for treatment of women with symptomatic fibroids, only 30,000 myomectomies (abdominal, laparoscopic, and robotic-assisted approaches) are performed each year.2 Why is this? One reason may be that, although many women wish to have uterus-preserving treatment, they often feel that doctors are too quick to recommend hysterectomy as the first—and sometimes only—treatment option for fibroids.3
CASE: Woman with fibroids seeks alternative to hysterectomy
A 42-year-old woman (G2P2) presents for a third opinion regarding her heavy menstrual bleeding and known uterine fibroids. She does not want to have any more children, but she wishes to avoid a hysterectomy. Both her regular gynecologist and the second gynecologist she consulted recommended hysterectomy as the first, and only, treatment option. Physical examination reveals a 16-week-sized uterus, and ultrasonography shows at least 6 fibroids, 2 of which impinge on the uterine cavity. The patient’s other gynecologists advised her that a myomectomy would be a “bloody operation,” would leave her uterus looking like Swiss cheese, and is not appropriate for women who have completed childbearing.
The patient asks if myomectomy could be considered in her situation. How would you advise her regarding myomectomy as an alternative to hysterectomy?
Organ conservation is important
In 1931, prominent British gynecologic surgeon Victor Bonney said, “Since cure without deformity or loss of function must ever be surgery’s highest ideal, the general proposition that myomectomy is a greater surgical achievement is incontestable.”4 As current hysterectomy and myomectomy rates indicate, however, we are not attempting organ conservation very often.
Other specialties almost never remove an entire organ for benign growths. Using breast cancer surgery as an admirable paradigm, consider that in the early 20th century the standard treatment for breast cancer was a Halsted radical mastectomy with axial lymphadenectomy. By the 1930s, this disfiguring operation was replaced by simple mastectomy and radiation, and by the 1970s, by lumpectomy and lymphadenectomy. Currently, lumpectomy and sentinel node sampling is the standard of care for early stage breast cancer. This is an excellent example of “minimally invasive surgery,” a term fostered by gynecologists. And, these organ-preservingsurgeries are performed for women with cancer, not a benign condition like fibroids.
Although our approach to hysterectomy has evolved with the increasing use of laparoscopic or robotic assistance, removal of the entire uterus nevertheless remains the surgical goal. I think this narrow view of surgical options is a disservice to our patients.
Many of us were taught that myomectomy was associated with more complications and more blood loss than hysterectomy. We were taught that the uterus had no function other than childbearing and that removing the uterus had no adverse health effects. The dogma suggested that myomectomy preserved a uterus that looked like Swiss cheese and would not heal properly and that the risk of fibroid recurrence was high. These beliefs, however, are myths, which are discussed and debunked below. In second and third installments for this series on myomectomy, I present steps for successful abdominal and laparoscopic technique.
Read myths on hysterectomy, myomectomy, and fibroids
MYTH #1: Hysterectomy is safer than myomectomy
Myomectomy is performed within the confines of the uterus and myometrium, with only infrequent occasion to operate near the ureters, uterine vessels, bowel, or bladder. Therefore, it should not be surprising that studies show that fewer complications occur with myomectomy than with hysterectomy.
A retrospective review of 197 women who had myomectomy and 197 women who underwent hysterectomy with similar uterine size (14 vs 15 weeks) reported that 13% (n = 26) of women in the hysterectomy group experienced complications, including 1 bladder injury, 1 ureteral injury, and 3 bowel injuries; 8 women had an ileus and 6 women had a pelvic abscess.5 Only 5% (n = 11) of the myomectomy patients had complications, including 1 bladder injury; 2 women had reoperation for small bowel obstruction, and 6 women had an ileus. The risks of febrile morbidity, unintended surgical procedure, life-threatening events, and rehospitalization were similar for both groups.
Authors of a recent systematic review of 6 studies, which included 1,520 women with uterine size up to 18 weeks, found higher rates of visceral injury and longer hospital stays for women who had a hysterectomy compared with those who had a myomectomy (TABLE 1).6
MYTH #2: Myomectomy is associated with more surgical blood loss than hysterectomy
In the previously cited study of 197 women treated with myomectomy and 197 women treated with hysterectomy, the estimated blood loss was greater in the hysterectomy group (484 mL) than in the myomectomy group (227 mL). When uterine size was corrected for, blood loss was no greater for myomectomy than for hysterectomy.5 The risk of hemorrhage (>500 mL blood loss) was greater in the hysterectomy group (14.2% vs 9.6%). Authors of the recent meta-analysis also found that the rate of transfusion was higher in the hysterectomy cohort. Tourniquets, misoprostol, vasopressin, and tranexamic acid all have been shown to significantly decrease surgical blood loss. (These treatments will be discussed in the next installment of this article series.)
MYTH #3: A uterus will look like Swiss cheese after a myomectomy
The uterus heals remarkably well after myomectomy. Three months following laparoscopic myomectomy, 3-dimensional Doppler ultrasonography demonstrated complete myometrial healing and normal blood flow to the uterus.7 In a study of women undergoing abdominal myomectomy, follow-up magnetic resonance imaging (MRI) with gadolinium showed complete healing of the myometrium and normal myometrial perfusion by 3 months.8 This study also found that, after removal of 65 g to 380 g of fibroids, the uterine volume 3 months after surgery was 65 mL, essentially equivalent to the normal volume of a uterus without fibroids (57 mL).8 See FIGURE for MRI scans of the uterus before and after myomectomy.
MYTH #4: Fibroids will just grow back after myomectomy
Once a fibroid is completely removed surgically, it does not grow back. The risk of new fibroid growth depends on the number of fibroids originally removed and the amount of time until menopause, when fibroids reduce in size and symptoms usually resolve. Given that the prevalence of fibroids is nearly 80% by age 50, studies measuring the detection of new fibroid growth of 1 cm on ultrasound imaging overstate the problem.9 What is likely a more important consideration for women is whether, following myomectomy, they will need another procedure for new fibroid-related symptoms.
Results of a meta-analysis of 872 women in 7 studies with 10- to 25-year follow-up indicated that 89% of women did not require another surgery.10 In another study, authors found that, over an average follow-up of 7.6 years, a second surgery occurred in 11% of the women who had 1 fibroid initially removed and for 26% of women who had multiple fibroids initially removed.11 In another study of 92 women who had either abdominal or laparoscopic myomectomy after age 45and who were followed for an average of 30 months, only 1 woman (1%) required a hysterectomy for fibroid-related symptoms.12 That patient had growth of a fibroid that was present but was not removed at her initial laparoscopic myomectomy.
Read myths 5–7 on ovarian conservation, fibroid growth, and symptom improvement
MYTH #5: Hysterectomy with ovarian conservation does not change hormone levels
Following hysterectomy with ovarian conservation, some women begin menopause earlier than age-matched women who have not undergone any surgery.13 Hysterectomy with ovarian conservation prior to age 50 has been associated with a significant increase in the risk of coronary heart disease, stroke, and heart failure.14 In a prospective longitudinal study, antimüllerian hormone (AMH) levels were persistently decreased following hysterectomy despite ovarian conservation.15 However, 3 months after myomectomy, no such changes in AMH levels were seen (TABLE 2).15
Early natural menopause has been associated with an increase in cardiovascular disease and death, and bilateral oophorectomy has been associated with increased risks of cardiovascular disease, all-cause mortality, lung cancer, colon cancer, anxiety, and depression. Although taking estrogen might obviate these adverse health effects, the majority of women who receive a prescription for estrogen following surgery are no longer taking it 5 years later.
MYTH #6: Fibroid growth in a premenopausal patient means cancer may be present
While most fibroids grow slowly, rapid growth of benign fibroids is very common. Using computerized analysis of a group of 72 women having serial MRI scans, investigators found that 34% of benign fibroids increased more than 20% in volume over 6 months.16 In premenopausal women, “rapid uterine growth” almost never indicates presence of uterine sarcoma. One study reported only 1 sarcoma among 371 women operated on for rapid growth of presumed fibroids.17 Using current criteria from the World Health Organization to determine the pathologic diagnosis, however, that 1 woman was determined to have had an atypical leiomyoma. Therefore, the prevalence of leiomyosarcoma in that study approached zero. In addition, in the 198 women who had a 6-week increase in uterine size over 1 year (one published definition of rapid growth), no sarcomas were found.17
Because of recent concern about leiomyosarcoma and morcellation of fibroids, some gynecologists have reverted to advising women that growing fibroids might be cancer and that hysterectomy is recommended. However, there is no evidence that fibroid growth is a sign of leiomyosarcoma in premenopausal women. Leiomyosarcoma should strongly be considered in a postmenopausal woman on no hormone therapy who has growth of a presumed fibroid.
MYTH #7: Myomectomy will not improve symptoms
Fibroid-related symptoms can be significant; women who undergo hysterectomy because of fibroid-related symptoms have significantly worse scores on the 36-Item Short-Form Survey (SF-36) quality-of-life questionnaire than women diagnosed with hypertension, heart disease, chronic lung disease, or arthritis.18
For women with fibroid-related symptoms, myomectomy has been shown to improve quality of life. A study of 72 women showed that SF-36 scores improved significantly following myomectomy (TABLE 3, page 48).19 In another study that used the European Quality of Life Five-Dimension Scale and Visual Analog Scale, 95 women had significant improvement in quality of life (P<.001) following laparoscopic myomectomy.20
For some women, hysterectomy may have an impact on emotional quality of life. Some women report decreased sexual desire after hysterectomy. They worry that partners will see them as “not whole” and less desirable. Some women expect that hysterectomy will lead to depression, crying, lack of sexual desire, and vaginal dryness.21 No such changes have been reported for women having myomectomy.
CASE Continued: Third consult leads patient to schedule surgical procedure
After reviewing the patient’s symptoms, examination, and ultrasound results, we advise the patient that abdominal myomectomy is indeed appropriate and feasible in her case. She schedules surgery for the following month.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Fibroids are extremely common and can be detected in 60% of African American women and 40% of white women by age 35. By age 50, more than 80% of African American women and almost 70% of white women have fibroids. Although most women with fibroids are relatively asymptomatic, women who have bothersome symptoms, such as heavy menstrual bleeding, urinary frequency, pelvic or abdominal pressure, or pain, account for nearly 30% of all gynecologic admissions in the United States. The cost of fibroid-related care, including surgery, hospital admissions, outpatient visits, and medications, is estimated at $4 to $9 billion per year.1 In addition, each woman seeking treatment for fibroid-related symptoms incurs an expense of $4,500 to $30,000 for lost work or disability every year.1
Many treatment options, including medical therapy and noninvasive procedures, are now available for women with symptomatic fibroids. For women who require surgical treatment, however, hysterectomy is often recommended. Fibroid-related hysterectomy currently accounts for 45% of all hysterectomies, or approximately 195,700 per year. Although the American College of Obstetricians and Gynecologists (ACOG) clinical management guidelines state that myomectomy is a safe and effective alternative to hysterectomy for treatment of women with symptomatic fibroids, only 30,000 myomectomies (abdominal, laparoscopic, and robotic-assisted approaches) are performed each year.2 Why is this? One reason may be that, although many women wish to have uterus-preserving treatment, they often feel that doctors are too quick to recommend hysterectomy as the first—and sometimes only—treatment option for fibroids.3
CASE: Woman with fibroids seeks alternative to hysterectomy
A 42-year-old woman (G2P2) presents for a third opinion regarding her heavy menstrual bleeding and known uterine fibroids. She does not want to have any more children, but she wishes to avoid a hysterectomy. Both her regular gynecologist and the second gynecologist she consulted recommended hysterectomy as the first, and only, treatment option. Physical examination reveals a 16-week-sized uterus, and ultrasonography shows at least 6 fibroids, 2 of which impinge on the uterine cavity. The patient’s other gynecologists advised her that a myomectomy would be a “bloody operation,” would leave her uterus looking like Swiss cheese, and is not appropriate for women who have completed childbearing.
The patient asks if myomectomy could be considered in her situation. How would you advise her regarding myomectomy as an alternative to hysterectomy?
Organ conservation is important
In 1931, prominent British gynecologic surgeon Victor Bonney said, “Since cure without deformity or loss of function must ever be surgery’s highest ideal, the general proposition that myomectomy is a greater surgical achievement is incontestable.”4 As current hysterectomy and myomectomy rates indicate, however, we are not attempting organ conservation very often.
Other specialties almost never remove an entire organ for benign growths. Using breast cancer surgery as an admirable paradigm, consider that in the early 20th century the standard treatment for breast cancer was a Halsted radical mastectomy with axial lymphadenectomy. By the 1930s, this disfiguring operation was replaced by simple mastectomy and radiation, and by the 1970s, by lumpectomy and lymphadenectomy. Currently, lumpectomy and sentinel node sampling is the standard of care for early stage breast cancer. This is an excellent example of “minimally invasive surgery,” a term fostered by gynecologists. And, these organ-preservingsurgeries are performed for women with cancer, not a benign condition like fibroids.
Although our approach to hysterectomy has evolved with the increasing use of laparoscopic or robotic assistance, removal of the entire uterus nevertheless remains the surgical goal. I think this narrow view of surgical options is a disservice to our patients.
Many of us were taught that myomectomy was associated with more complications and more blood loss than hysterectomy. We were taught that the uterus had no function other than childbearing and that removing the uterus had no adverse health effects. The dogma suggested that myomectomy preserved a uterus that looked like Swiss cheese and would not heal properly and that the risk of fibroid recurrence was high. These beliefs, however, are myths, which are discussed and debunked below. In second and third installments for this series on myomectomy, I present steps for successful abdominal and laparoscopic technique.
Read myths on hysterectomy, myomectomy, and fibroids
MYTH #1: Hysterectomy is safer than myomectomy
Myomectomy is performed within the confines of the uterus and myometrium, with only infrequent occasion to operate near the ureters, uterine vessels, bowel, or bladder. Therefore, it should not be surprising that studies show that fewer complications occur with myomectomy than with hysterectomy.
A retrospective review of 197 women who had myomectomy and 197 women who underwent hysterectomy with similar uterine size (14 vs 15 weeks) reported that 13% (n = 26) of women in the hysterectomy group experienced complications, including 1 bladder injury, 1 ureteral injury, and 3 bowel injuries; 8 women had an ileus and 6 women had a pelvic abscess.5 Only 5% (n = 11) of the myomectomy patients had complications, including 1 bladder injury; 2 women had reoperation for small bowel obstruction, and 6 women had an ileus. The risks of febrile morbidity, unintended surgical procedure, life-threatening events, and rehospitalization were similar for both groups.
Authors of a recent systematic review of 6 studies, which included 1,520 women with uterine size up to 18 weeks, found higher rates of visceral injury and longer hospital stays for women who had a hysterectomy compared with those who had a myomectomy (TABLE 1).6
MYTH #2: Myomectomy is associated with more surgical blood loss than hysterectomy
In the previously cited study of 197 women treated with myomectomy and 197 women treated with hysterectomy, the estimated blood loss was greater in the hysterectomy group (484 mL) than in the myomectomy group (227 mL). When uterine size was corrected for, blood loss was no greater for myomectomy than for hysterectomy.5 The risk of hemorrhage (>500 mL blood loss) was greater in the hysterectomy group (14.2% vs 9.6%). Authors of the recent meta-analysis also found that the rate of transfusion was higher in the hysterectomy cohort. Tourniquets, misoprostol, vasopressin, and tranexamic acid all have been shown to significantly decrease surgical blood loss. (These treatments will be discussed in the next installment of this article series.)
MYTH #3: A uterus will look like Swiss cheese after a myomectomy
The uterus heals remarkably well after myomectomy. Three months following laparoscopic myomectomy, 3-dimensional Doppler ultrasonography demonstrated complete myometrial healing and normal blood flow to the uterus.7 In a study of women undergoing abdominal myomectomy, follow-up magnetic resonance imaging (MRI) with gadolinium showed complete healing of the myometrium and normal myometrial perfusion by 3 months.8 This study also found that, after removal of 65 g to 380 g of fibroids, the uterine volume 3 months after surgery was 65 mL, essentially equivalent to the normal volume of a uterus without fibroids (57 mL).8 See FIGURE for MRI scans of the uterus before and after myomectomy.
MYTH #4: Fibroids will just grow back after myomectomy
Once a fibroid is completely removed surgically, it does not grow back. The risk of new fibroid growth depends on the number of fibroids originally removed and the amount of time until menopause, when fibroids reduce in size and symptoms usually resolve. Given that the prevalence of fibroids is nearly 80% by age 50, studies measuring the detection of new fibroid growth of 1 cm on ultrasound imaging overstate the problem.9 What is likely a more important consideration for women is whether, following myomectomy, they will need another procedure for new fibroid-related symptoms.
Results of a meta-analysis of 872 women in 7 studies with 10- to 25-year follow-up indicated that 89% of women did not require another surgery.10 In another study, authors found that, over an average follow-up of 7.6 years, a second surgery occurred in 11% of the women who had 1 fibroid initially removed and for 26% of women who had multiple fibroids initially removed.11 In another study of 92 women who had either abdominal or laparoscopic myomectomy after age 45and who were followed for an average of 30 months, only 1 woman (1%) required a hysterectomy for fibroid-related symptoms.12 That patient had growth of a fibroid that was present but was not removed at her initial laparoscopic myomectomy.
Read myths 5–7 on ovarian conservation, fibroid growth, and symptom improvement
MYTH #5: Hysterectomy with ovarian conservation does not change hormone levels
Following hysterectomy with ovarian conservation, some women begin menopause earlier than age-matched women who have not undergone any surgery.13 Hysterectomy with ovarian conservation prior to age 50 has been associated with a significant increase in the risk of coronary heart disease, stroke, and heart failure.14 In a prospective longitudinal study, antimüllerian hormone (AMH) levels were persistently decreased following hysterectomy despite ovarian conservation.15 However, 3 months after myomectomy, no such changes in AMH levels were seen (TABLE 2).15
Early natural menopause has been associated with an increase in cardiovascular disease and death, and bilateral oophorectomy has been associated with increased risks of cardiovascular disease, all-cause mortality, lung cancer, colon cancer, anxiety, and depression. Although taking estrogen might obviate these adverse health effects, the majority of women who receive a prescription for estrogen following surgery are no longer taking it 5 years later.
MYTH #6: Fibroid growth in a premenopausal patient means cancer may be present
While most fibroids grow slowly, rapid growth of benign fibroids is very common. Using computerized analysis of a group of 72 women having serial MRI scans, investigators found that 34% of benign fibroids increased more than 20% in volume over 6 months.16 In premenopausal women, “rapid uterine growth” almost never indicates presence of uterine sarcoma. One study reported only 1 sarcoma among 371 women operated on for rapid growth of presumed fibroids.17 Using current criteria from the World Health Organization to determine the pathologic diagnosis, however, that 1 woman was determined to have had an atypical leiomyoma. Therefore, the prevalence of leiomyosarcoma in that study approached zero. In addition, in the 198 women who had a 6-week increase in uterine size over 1 year (one published definition of rapid growth), no sarcomas were found.17
Because of recent concern about leiomyosarcoma and morcellation of fibroids, some gynecologists have reverted to advising women that growing fibroids might be cancer and that hysterectomy is recommended. However, there is no evidence that fibroid growth is a sign of leiomyosarcoma in premenopausal women. Leiomyosarcoma should strongly be considered in a postmenopausal woman on no hormone therapy who has growth of a presumed fibroid.
MYTH #7: Myomectomy will not improve symptoms
Fibroid-related symptoms can be significant; women who undergo hysterectomy because of fibroid-related symptoms have significantly worse scores on the 36-Item Short-Form Survey (SF-36) quality-of-life questionnaire than women diagnosed with hypertension, heart disease, chronic lung disease, or arthritis.18
For women with fibroid-related symptoms, myomectomy has been shown to improve quality of life. A study of 72 women showed that SF-36 scores improved significantly following myomectomy (TABLE 3, page 48).19 In another study that used the European Quality of Life Five-Dimension Scale and Visual Analog Scale, 95 women had significant improvement in quality of life (P<.001) following laparoscopic myomectomy.20
For some women, hysterectomy may have an impact on emotional quality of life. Some women report decreased sexual desire after hysterectomy. They worry that partners will see them as “not whole” and less desirable. Some women expect that hysterectomy will lead to depression, crying, lack of sexual desire, and vaginal dryness.21 No such changes have been reported for women having myomectomy.
CASE Continued: Third consult leads patient to schedule surgical procedure
After reviewing the patient’s symptoms, examination, and ultrasound results, we advise the patient that abdominal myomectomy is indeed appropriate and feasible in her case. She schedules surgery for the following month.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Cardozo ER, Clark AD, Banks NK, Henne MB, Stegmann BJ, Segars JH. The estimated annual cost of leiomyomata in the United States. Am J Obstet Gynecol. 2012;206(3):211.e1–e9.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins–Gynecology. ACOG Practice Bulletin No. 96: alternatives to hysterectomy in the management of leiomyomas. Obstet Gynecol. 2008;112(2 pt 1):387–400.
- Borah BJ, Nicholson WK, Bradley L, Stewart EA. The impact of uterine leiomyomas: a national survey of affected women. Am J Obstet Gynecol. 2013;209(4):319.e1–e20.
- Bonney V. The technique and results of myomectomy. Lancet. 1931;217(5604):171-177.
- Sawin SW, Pilevsky ND, Berlin JA, Barnhart KT. Comparability of perioperative morbidity between abdominal myomectomy and hysterectomy for women with uterine leiomyomas. Am J Obstet Gynecol. 2000;183(6):1448–1455.
- Pundir J, Walawalkar R, Seshadri S, Khalaf Y, El-Toukhy T. Perioperative morbidity associated with abdominal myomectomy compared with total abdominal hysterectomy for uterine fibroids. J Obstet Gynecol. 2013;33(7):655–662.
- Chang WC, Chang DY, Huang SC, et al. Use of three-dimensional ultrasonography in the evaluation of uterine perfusion and healing after laparoscopic myomectomy. Fertil Steril. 2009;92(3):1110–1115.
- Tsuji S, Takahashi K, Imaoka I, Sugimura K, Miyazaki K, Noda Y. MRI evaluation of the uterine structure after myomectomy. Gynecol Obstet Invest. 2006;61(2):106–110.
- Sudik R, Husch K, Steller J, Daume E. Fertility and pregnancy outcome after myomectomy in sterility patients. Eur J Obstet Gynecol Reprod Biol. 1996;65(2):209–214.
- Fauconnier A, Chapron C, Babaki-Fard K, Dubuisson JB. Recurrence of leiomyomata after myomectomy. Hum Reprod Update. 2000;6(6):595–602.
- Malone, LJ. Myomectomy: recurrence after removal of solitary and multiple myomas. Obstet Gynecol. 1969;34(2):200–203.
- Kim DH, Kim ML, Song T, Kim MK, Yoon BS, Seong SJ. Is myomectomy in women aged 45 years and older an effective option? Eur J Obstet Gynecol Reprod Biol. 2014;177:57–60.
- Farquhar CM, Sadler L, Harvey SA, Stewart AW. The association of hysterectomy and menopause: a prospective cohort study. BJOG. 2005;112(7):956–962.
- Ingelsson E, Lundholm C, Johansson AL, Altman D. Hysterectomy and risk of cardiovascular disease: a population-based cohort study. Eur Heart J. 2011;32(6):745–750.
- Wang HY, Quan S, Zhang RL, et al. Comparison of serum anti-Mullerian hormone levels following hysterectomy and myomectomy for benign gynaecological conditions. Eur J Obstet Gynecol Reprod Biol. 2013;171(2):368–371.
- Peddada SD, Laughlin SK, Miner K, et al. Growth of uterine leiomyomata among premenopausal black and white women. Proc Natl Acad Sci. 2008;105(50):19887–19892.
- Parker W, Fu YS, Berek JS. Uterine sarcoma in patients operated on for presumed leiomyoma and rapidly growing leiomyoma. Obstet Gynecol. 1994;83(3):414–418.
- Rowe MK, Kanouse DE, Mittman BS, Bernstein SJ. Quality of life among women undergoing hysterectomies. Obstet Gynecol. 1999;93(6):915–921.
- Dilek S, Ertunc D, Tok EC, Cimen R, Doruk A. The effect of myomectomy on health-related quality of life of women with myoma uteri. J Obstet Gynaecol Res. 2010;36(2):364–369.
- Radosa JC, Radosa CG, Mavrova R, et al. Postoperative quality of life and sexual function in premenopausal women undergoing laparoscopic myomectomy for symptomatic fibroids: a prospective observational cohort study. PLoS One. 2016;29;11(11):e0166659.
- Groff JY, Mullen PD, Byrd T, Shelton AJ, Lees E, Goode J. Decision making, beliefs, and attitudes toward hysterectomy: a focus group study with medically underserved women in Texas. J Womens Health Gend Based Med. 2000;9(suppl 2):39S–50S.
- Cardozo ER, Clark AD, Banks NK, Henne MB, Stegmann BJ, Segars JH. The estimated annual cost of leiomyomata in the United States. Am J Obstet Gynecol. 2012;206(3):211.e1–e9.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins–Gynecology. ACOG Practice Bulletin No. 96: alternatives to hysterectomy in the management of leiomyomas. Obstet Gynecol. 2008;112(2 pt 1):387–400.
- Borah BJ, Nicholson WK, Bradley L, Stewart EA. The impact of uterine leiomyomas: a national survey of affected women. Am J Obstet Gynecol. 2013;209(4):319.e1–e20.
- Bonney V. The technique and results of myomectomy. Lancet. 1931;217(5604):171-177.
- Sawin SW, Pilevsky ND, Berlin JA, Barnhart KT. Comparability of perioperative morbidity between abdominal myomectomy and hysterectomy for women with uterine leiomyomas. Am J Obstet Gynecol. 2000;183(6):1448–1455.
- Pundir J, Walawalkar R, Seshadri S, Khalaf Y, El-Toukhy T. Perioperative morbidity associated with abdominal myomectomy compared with total abdominal hysterectomy for uterine fibroids. J Obstet Gynecol. 2013;33(7):655–662.
- Chang WC, Chang DY, Huang SC, et al. Use of three-dimensional ultrasonography in the evaluation of uterine perfusion and healing after laparoscopic myomectomy. Fertil Steril. 2009;92(3):1110–1115.
- Tsuji S, Takahashi K, Imaoka I, Sugimura K, Miyazaki K, Noda Y. MRI evaluation of the uterine structure after myomectomy. Gynecol Obstet Invest. 2006;61(2):106–110.
- Sudik R, Husch K, Steller J, Daume E. Fertility and pregnancy outcome after myomectomy in sterility patients. Eur J Obstet Gynecol Reprod Biol. 1996;65(2):209–214.
- Fauconnier A, Chapron C, Babaki-Fard K, Dubuisson JB. Recurrence of leiomyomata after myomectomy. Hum Reprod Update. 2000;6(6):595–602.
- Malone, LJ. Myomectomy: recurrence after removal of solitary and multiple myomas. Obstet Gynecol. 1969;34(2):200–203.
- Kim DH, Kim ML, Song T, Kim MK, Yoon BS, Seong SJ. Is myomectomy in women aged 45 years and older an effective option? Eur J Obstet Gynecol Reprod Biol. 2014;177:57–60.
- Farquhar CM, Sadler L, Harvey SA, Stewart AW. The association of hysterectomy and menopause: a prospective cohort study. BJOG. 2005;112(7):956–962.
- Ingelsson E, Lundholm C, Johansson AL, Altman D. Hysterectomy and risk of cardiovascular disease: a population-based cohort study. Eur Heart J. 2011;32(6):745–750.
- Wang HY, Quan S, Zhang RL, et al. Comparison of serum anti-Mullerian hormone levels following hysterectomy and myomectomy for benign gynaecological conditions. Eur J Obstet Gynecol Reprod Biol. 2013;171(2):368–371.
- Peddada SD, Laughlin SK, Miner K, et al. Growth of uterine leiomyomata among premenopausal black and white women. Proc Natl Acad Sci. 2008;105(50):19887–19892.
- Parker W, Fu YS, Berek JS. Uterine sarcoma in patients operated on for presumed leiomyoma and rapidly growing leiomyoma. Obstet Gynecol. 1994;83(3):414–418.
- Rowe MK, Kanouse DE, Mittman BS, Bernstein SJ. Quality of life among women undergoing hysterectomies. Obstet Gynecol. 1999;93(6):915–921.
- Dilek S, Ertunc D, Tok EC, Cimen R, Doruk A. The effect of myomectomy on health-related quality of life of women with myoma uteri. J Obstet Gynaecol Res. 2010;36(2):364–369.
- Radosa JC, Radosa CG, Mavrova R, et al. Postoperative quality of life and sexual function in premenopausal women undergoing laparoscopic myomectomy for symptomatic fibroids: a prospective observational cohort study. PLoS One. 2016;29;11(11):e0166659.
- Groff JY, Mullen PD, Byrd T, Shelton AJ, Lees E, Goode J. Decision making, beliefs, and attitudes toward hysterectomy: a focus group study with medically underserved women in Texas. J Womens Health Gend Based Med. 2000;9(suppl 2):39S–50S.
Acute Plasma Tau Predicts Prolonged Return to Play After Concussion
Among collegiate athletes, elevated plasma tau concentrations within six hours after a sport-related concussion predict a prolonged recovery, according to research published online ahead of print January 6 in Neurology. This finding suggests that tau levels may help to determine when athletes should return to play. Variability of tau concentrations across athletes and the effect of physical exertion on plasma tau may complicate the use of the biomarker for concussion management, however.
Approximately 3.8 million sport-related concussions occur each year in the United States, but no biomarkers are known to predict recovery and an athlete’s readiness to return to play. Postconcussive symptoms typically resolve within 10 days in about half of collegiate athletes, but symptoms are chronic in a subset of patients. Shahim et al found that plasma tau elevations predicted a return to play of more than 10 days in professional ice hockey players in Sweden.
Diagnosing Sport-Related Concussion
Jessica Gill, RN, PhD, an investigator with the National Institute of Nursing Research at the NIH, and colleagues conducted a study to determine whether changes in plasma tau after sport-related concussion relate to return to play in men and women collegiate athletes. The researchers included students with concussion, as well as athlete and nonathlete controls. The athletes participated in various National Collegiate Athletic Association (NCAA) division I and III contact sports (ie, football, soccer, basketball, hockey, and lacrosse).

Between 2009 and 2014, 632 athletes underwent plasma sampling and cognitive testing prior to the sports seasons and were followed prospectively for a diagnosis of sport-related concussion. Sport-related concussions were witnessed by an on-field certified athletic trainer and met the Sport Concussion Assessment Tool 2 definition of concussion.Investigators collected blood samples from athletes with concussion and athlete controls at six hours, 24 hours, 72 hours, and seven days after a concussion. Nonathlete controls had blood draws at an unrelated time point. Investigators measured total tau using an ultrasensitive immunoassay.
Return to play for each athlete was determined by athletic trainers or team physicians. They followed NCAA guidelines, which recommend that athletes be asymptomatic at rest and as they progressively resume activity before returning to play.
A total of 46 athletes were diagnosed with a sport-related concussion. Concussions occurred between 19 days and 218 days after baseline assessments (mean, 92.3 days). Thirty-seven athletes without concussion served as athlete controls. Athletes with and without concussion did not differ significantly in sport played, history of sport-related concussion, or other demographic features. A control group of 21 healthy nonathletes was demographically similar to the athlete groups.Return to play information was available for 41 of the athletes with concussion. Athletes who took more than 10 days to recover were considered to have a long return to play (23 athletes). Those who recovered in less than 10 days had a short return to play (18 athletes). The mean return to play duration was 21.68 days (range, two days to 263 days). Five athletes had a return to play duration of 30 days or more. Approximately 39% returned to play in less than 10 days. There were no significant differences in sport played or history of concussion among those with long return to play versus short return to play. Women made up 61% of the long return to play group and 28% of the short return to play group.
Tau Measurements
Compared with nonathletes, athletes had significantly higher mean tau concentrations at baseline and all other time points. The longitudinal pattern of tau differed significantly between athletes with and without concussion. Athletes with concussion had significantly lower mean total tau at 24 hours (6.06 pg/mL vs 7.89 pg/mL) and 72 hours (5.19 pg/mL vs 6.94 pg/mL), compared with athlete controls.
Athletes with a long return to play had higher tau concentrations overall, after controlling for sex, than those with a short return to play. The differences were statistically significant at six hours (10.98 pg/mL vs 7.02 pg/mL), 24 hours (7.19 pg/mL vs 4.08 pg/mL), and 72 hours (6.29 pg/mL vs 3.94 pg/mL).
Mean change in tau from baseline also significantly differed between the return to play groups. Athletes with long return to play had a mean increase of 2.26 pg/mL at six hours postconcussion, compared with a mean reduction of 1.19 pg/mL in the short return to play group, after controlling for sex. Area under the curve (AUC) analyses revealed that higher total tau six hours post concussion and change in tau from baseline to six hours post concussion predicted long return to play (AUC of 0.81 and 0.80, respectively). Higher total tau at 72 hours postconcussion also was a significant predictor of long return to play (AUC, 0.82).
“These findings suggest that changes in total tau within six hours of a sport-related concussion may provide vital information about return to play decisions, and may serve to mitigate the negative consequences of returning to play prematurely,” Dr. Gill and colleagues said. Preclinical models link insufficient recovery time from a mild traumatic brain injury (mTBI) to greater neuropathology following a subsequent mTBI, including pathology that overlaps with that of chronic traumatic encephalopathy.
Lower levels of tau in athletes with concussion, compared with athletes without concussion, at 24 hours and 72 hours “may be due to the effects of physical exertion on tau,” the researchers said. Limitations of the study include the relatively small sample size within subanalyses of long and short return to play.
More Research Is Needed
“While normally measured in CSF, tau measured in blood could provide the opportunity to assess neurologic injury shortly after concussion, as well as facilitate monitoring of recovery over time,” said Barbara B. Bendlin, PhD, Associate Professor of Medicine and Public Health at the University of Wisconsin–Madison, and Michael Makdissi, MBBS, PhD, research fellow at the Florey Institute of Neuroscience and Mental Health and Adjunct Associate Professor of Rehabilitation, Nutrition, and Sport at the La Trobe Sport and Exercise Medicine Research Centre in Australia, in an accompanying editorial.
However, differences in plasma tau levels between athletes and nonathletes; lower plasma tau levels at 24 hours and 72 hours post concussion in athletes with concussion, compared with nonconcussed teammates; variability across players; and fluctuations in plasma tau levels over time in general may complicate the use of the biomarker in concussion management. In addition, tau in plasma may reflect CNS and peripheral nervous system origins.
“This study and others conducted in the sports setting open the door for further evaluation and possible future implementation of blood-based biomarkers for evaluation of concussion,” they said. “Nevertheless, more work is needed before blood-based biomarkers can be used for management of sport-related concussion.”
—Jake Remaly
Suggested Reading
Bendlin BB, Makdissi M. Blood-based biomarkers for evaluating sport-related concussion: Back in the game. Neurology. 2017 Jan 6 [Epub ahead of print].
Gill J, Merchant-Borna K, Jeromin A, et al. Acute plasma tau relates to prolonged return to play after concussion. Neurology. 2017 Jan 6 [Epub ahead of print].
Shahim P, Tegner Y, Wilson DH, et al. Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol. 2014;71(6):684-692.
Among collegiate athletes, elevated plasma tau concentrations within six hours after a sport-related concussion predict a prolonged recovery, according to research published online ahead of print January 6 in Neurology. This finding suggests that tau levels may help to determine when athletes should return to play. Variability of tau concentrations across athletes and the effect of physical exertion on plasma tau may complicate the use of the biomarker for concussion management, however.
Approximately 3.8 million sport-related concussions occur each year in the United States, but no biomarkers are known to predict recovery and an athlete’s readiness to return to play. Postconcussive symptoms typically resolve within 10 days in about half of collegiate athletes, but symptoms are chronic in a subset of patients. Shahim et al found that plasma tau elevations predicted a return to play of more than 10 days in professional ice hockey players in Sweden.
Diagnosing Sport-Related Concussion
Jessica Gill, RN, PhD, an investigator with the National Institute of Nursing Research at the NIH, and colleagues conducted a study to determine whether changes in plasma tau after sport-related concussion relate to return to play in men and women collegiate athletes. The researchers included students with concussion, as well as athlete and nonathlete controls. The athletes participated in various National Collegiate Athletic Association (NCAA) division I and III contact sports (ie, football, soccer, basketball, hockey, and lacrosse).
Between 2009 and 2014, 632 athletes underwent plasma sampling and cognitive testing prior to the sports seasons and were followed prospectively for a diagnosis of sport-related concussion. Sport-related concussions were witnessed by an on-field certified athletic trainer and met the Sport Concussion Assessment Tool 2 definition of concussion.Investigators collected blood samples from athletes with concussion and athlete controls at six hours, 24 hours, 72 hours, and seven days after a concussion. Nonathlete controls had blood draws at an unrelated time point. Investigators measured total tau using an ultrasensitive immunoassay.
Return to play for each athlete was determined by athletic trainers or team physicians. They followed NCAA guidelines, which recommend that athletes be asymptomatic at rest and as they progressively resume activity before returning to play.
A total of 46 athletes were diagnosed with a sport-related concussion. Concussions occurred between 19 days and 218 days after baseline assessments (mean, 92.3 days). Thirty-seven athletes without concussion served as athlete controls. Athletes with and without concussion did not differ significantly in sport played, history of sport-related concussion, or other demographic features. A control group of 21 healthy nonathletes was demographically similar to the athlete groups.Return to play information was available for 41 of the athletes with concussion. Athletes who took more than 10 days to recover were considered to have a long return to play (23 athletes). Those who recovered in less than 10 days had a short return to play (18 athletes). The mean return to play duration was 21.68 days (range, two days to 263 days). Five athletes had a return to play duration of 30 days or more. Approximately 39% returned to play in less than 10 days. There were no significant differences in sport played or history of concussion among those with long return to play versus short return to play. Women made up 61% of the long return to play group and 28% of the short return to play group.
Tau Measurements
Compared with nonathletes, athletes had significantly higher mean tau concentrations at baseline and all other time points. The longitudinal pattern of tau differed significantly between athletes with and without concussion. Athletes with concussion had significantly lower mean total tau at 24 hours (6.06 pg/mL vs 7.89 pg/mL) and 72 hours (5.19 pg/mL vs 6.94 pg/mL), compared with athlete controls.
Athletes with a long return to play had higher tau concentrations overall, after controlling for sex, than those with a short return to play. The differences were statistically significant at six hours (10.98 pg/mL vs 7.02 pg/mL), 24 hours (7.19 pg/mL vs 4.08 pg/mL), and 72 hours (6.29 pg/mL vs 3.94 pg/mL).
Mean change in tau from baseline also significantly differed between the return to play groups. Athletes with long return to play had a mean increase of 2.26 pg/mL at six hours postconcussion, compared with a mean reduction of 1.19 pg/mL in the short return to play group, after controlling for sex. Area under the curve (AUC) analyses revealed that higher total tau six hours post concussion and change in tau from baseline to six hours post concussion predicted long return to play (AUC of 0.81 and 0.80, respectively). Higher total tau at 72 hours postconcussion also was a significant predictor of long return to play (AUC, 0.82).
“These findings suggest that changes in total tau within six hours of a sport-related concussion may provide vital information about return to play decisions, and may serve to mitigate the negative consequences of returning to play prematurely,” Dr. Gill and colleagues said. Preclinical models link insufficient recovery time from a mild traumatic brain injury (mTBI) to greater neuropathology following a subsequent mTBI, including pathology that overlaps with that of chronic traumatic encephalopathy.
Lower levels of tau in athletes with concussion, compared with athletes without concussion, at 24 hours and 72 hours “may be due to the effects of physical exertion on tau,” the researchers said. Limitations of the study include the relatively small sample size within subanalyses of long and short return to play.
More Research Is Needed
“While normally measured in CSF, tau measured in blood could provide the opportunity to assess neurologic injury shortly after concussion, as well as facilitate monitoring of recovery over time,” said Barbara B. Bendlin, PhD, Associate Professor of Medicine and Public Health at the University of Wisconsin–Madison, and Michael Makdissi, MBBS, PhD, research fellow at the Florey Institute of Neuroscience and Mental Health and Adjunct Associate Professor of Rehabilitation, Nutrition, and Sport at the La Trobe Sport and Exercise Medicine Research Centre in Australia, in an accompanying editorial.
However, differences in plasma tau levels between athletes and nonathletes; lower plasma tau levels at 24 hours and 72 hours post concussion in athletes with concussion, compared with nonconcussed teammates; variability across players; and fluctuations in plasma tau levels over time in general may complicate the use of the biomarker in concussion management. In addition, tau in plasma may reflect CNS and peripheral nervous system origins.
“This study and others conducted in the sports setting open the door for further evaluation and possible future implementation of blood-based biomarkers for evaluation of concussion,” they said. “Nevertheless, more work is needed before blood-based biomarkers can be used for management of sport-related concussion.”
—Jake Remaly
Suggested Reading
Bendlin BB, Makdissi M. Blood-based biomarkers for evaluating sport-related concussion: Back in the game. Neurology. 2017 Jan 6 [Epub ahead of print].
Gill J, Merchant-Borna K, Jeromin A, et al. Acute plasma tau relates to prolonged return to play after concussion. Neurology. 2017 Jan 6 [Epub ahead of print].
Shahim P, Tegner Y, Wilson DH, et al. Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol. 2014;71(6):684-692.
Among collegiate athletes, elevated plasma tau concentrations within six hours after a sport-related concussion predict a prolonged recovery, according to research published online ahead of print January 6 in Neurology. This finding suggests that tau levels may help to determine when athletes should return to play. Variability of tau concentrations across athletes and the effect of physical exertion on plasma tau may complicate the use of the biomarker for concussion management, however.
Approximately 3.8 million sport-related concussions occur each year in the United States, but no biomarkers are known to predict recovery and an athlete’s readiness to return to play. Postconcussive symptoms typically resolve within 10 days in about half of collegiate athletes, but symptoms are chronic in a subset of patients. Shahim et al found that plasma tau elevations predicted a return to play of more than 10 days in professional ice hockey players in Sweden.
Diagnosing Sport-Related Concussion
Jessica Gill, RN, PhD, an investigator with the National Institute of Nursing Research at the NIH, and colleagues conducted a study to determine whether changes in plasma tau after sport-related concussion relate to return to play in men and women collegiate athletes. The researchers included students with concussion, as well as athlete and nonathlete controls. The athletes participated in various National Collegiate Athletic Association (NCAA) division I and III contact sports (ie, football, soccer, basketball, hockey, and lacrosse).
Between 2009 and 2014, 632 athletes underwent plasma sampling and cognitive testing prior to the sports seasons and were followed prospectively for a diagnosis of sport-related concussion. Sport-related concussions were witnessed by an on-field certified athletic trainer and met the Sport Concussion Assessment Tool 2 definition of concussion.Investigators collected blood samples from athletes with concussion and athlete controls at six hours, 24 hours, 72 hours, and seven days after a concussion. Nonathlete controls had blood draws at an unrelated time point. Investigators measured total tau using an ultrasensitive immunoassay.
Return to play for each athlete was determined by athletic trainers or team physicians. They followed NCAA guidelines, which recommend that athletes be asymptomatic at rest and as they progressively resume activity before returning to play.
A total of 46 athletes were diagnosed with a sport-related concussion. Concussions occurred between 19 days and 218 days after baseline assessments (mean, 92.3 days). Thirty-seven athletes without concussion served as athlete controls. Athletes with and without concussion did not differ significantly in sport played, history of sport-related concussion, or other demographic features. A control group of 21 healthy nonathletes was demographically similar to the athlete groups.Return to play information was available for 41 of the athletes with concussion. Athletes who took more than 10 days to recover were considered to have a long return to play (23 athletes). Those who recovered in less than 10 days had a short return to play (18 athletes). The mean return to play duration was 21.68 days (range, two days to 263 days). Five athletes had a return to play duration of 30 days or more. Approximately 39% returned to play in less than 10 days. There were no significant differences in sport played or history of concussion among those with long return to play versus short return to play. Women made up 61% of the long return to play group and 28% of the short return to play group.
Tau Measurements
Compared with nonathletes, athletes had significantly higher mean tau concentrations at baseline and all other time points. The longitudinal pattern of tau differed significantly between athletes with and without concussion. Athletes with concussion had significantly lower mean total tau at 24 hours (6.06 pg/mL vs 7.89 pg/mL) and 72 hours (5.19 pg/mL vs 6.94 pg/mL), compared with athlete controls.
Athletes with a long return to play had higher tau concentrations overall, after controlling for sex, than those with a short return to play. The differences were statistically significant at six hours (10.98 pg/mL vs 7.02 pg/mL), 24 hours (7.19 pg/mL vs 4.08 pg/mL), and 72 hours (6.29 pg/mL vs 3.94 pg/mL).
Mean change in tau from baseline also significantly differed between the return to play groups. Athletes with long return to play had a mean increase of 2.26 pg/mL at six hours postconcussion, compared with a mean reduction of 1.19 pg/mL in the short return to play group, after controlling for sex. Area under the curve (AUC) analyses revealed that higher total tau six hours post concussion and change in tau from baseline to six hours post concussion predicted long return to play (AUC of 0.81 and 0.80, respectively). Higher total tau at 72 hours postconcussion also was a significant predictor of long return to play (AUC, 0.82).
“These findings suggest that changes in total tau within six hours of a sport-related concussion may provide vital information about return to play decisions, and may serve to mitigate the negative consequences of returning to play prematurely,” Dr. Gill and colleagues said. Preclinical models link insufficient recovery time from a mild traumatic brain injury (mTBI) to greater neuropathology following a subsequent mTBI, including pathology that overlaps with that of chronic traumatic encephalopathy.
Lower levels of tau in athletes with concussion, compared with athletes without concussion, at 24 hours and 72 hours “may be due to the effects of physical exertion on tau,” the researchers said. Limitations of the study include the relatively small sample size within subanalyses of long and short return to play.
More Research Is Needed
“While normally measured in CSF, tau measured in blood could provide the opportunity to assess neurologic injury shortly after concussion, as well as facilitate monitoring of recovery over time,” said Barbara B. Bendlin, PhD, Associate Professor of Medicine and Public Health at the University of Wisconsin–Madison, and Michael Makdissi, MBBS, PhD, research fellow at the Florey Institute of Neuroscience and Mental Health and Adjunct Associate Professor of Rehabilitation, Nutrition, and Sport at the La Trobe Sport and Exercise Medicine Research Centre in Australia, in an accompanying editorial.
However, differences in plasma tau levels between athletes and nonathletes; lower plasma tau levels at 24 hours and 72 hours post concussion in athletes with concussion, compared with nonconcussed teammates; variability across players; and fluctuations in plasma tau levels over time in general may complicate the use of the biomarker in concussion management. In addition, tau in plasma may reflect CNS and peripheral nervous system origins.
“This study and others conducted in the sports setting open the door for further evaluation and possible future implementation of blood-based biomarkers for evaluation of concussion,” they said. “Nevertheless, more work is needed before blood-based biomarkers can be used for management of sport-related concussion.”
—Jake Remaly
Suggested Reading
Bendlin BB, Makdissi M. Blood-based biomarkers for evaluating sport-related concussion: Back in the game. Neurology. 2017 Jan 6 [Epub ahead of print].
Gill J, Merchant-Borna K, Jeromin A, et al. Acute plasma tau relates to prolonged return to play after concussion. Neurology. 2017 Jan 6 [Epub ahead of print].
Shahim P, Tegner Y, Wilson DH, et al. Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol. 2014;71(6):684-692.

Common Epilepsies Share Genetic Overlap With Rare Types
Several genes previously implicated only in rare, severe forms of pediatric epilepsy may also contribute to common forms of the disorder, according to a report published online ahead of print January 13 in Lancet Neurology. “Our findings raise hopes that the emerging paradigm for the treatment of rare epilepsies, where therapies are targeted to the precise genetic cause of disease, may also extend to a proportion of common epilepsy syndromes,” said study leader David B. Goldstein, PhD, Director of the Institute for Genomic Medicine and Professor in the Departments of Genetics and Development and Neurology at Columbia University Medical Center in New York City.

In recent years, researchers have identified dozens of genes that, alone or in combination with other factors, cause rare pediatric epilepsies. These discoveries have led to the use of targeted therapies for some seizure disorders, such as the ketogenic diet for patients with Dravet syndrome or GLUT-1 deficiency syndrome. Other therapies such as quinidine, a medication to treat heart arrhythmias, and memantine, an Alzheimer’s disease treatment, have been tried in children with certain gene mutations. These attempts have not proved universally effective for all patients with these mutations, but suggest the potential to repurpose existing medicines to treat rare genetic forms of epilepsy.
“Unlike very rare types of epilepsies, previous studies had shed little light on the genetic underpinnings of common epilepsies, which suggested that this precision medicine paradigm may have a very narrow application,” said Dr. Goldstein.
To learn more about the genetics of epilepsy, Dr. Goldstein and his colleagues conducted a study to identify the genetic contributions to more common forms of epilepsy. Analyses were conducted at Columbia University Medical Center’s Institute for Genomic Medicine, in collaboration with NewYork-Presbyterian, as part of Epi4K, an international consortium of epilepsy clinicians and researchers. Most of the patients were recruited through the Epilepsy Phenome/Genome Project.
The researchers separately compared the sequence data from 640 individuals with familial genetic generalized epilepsy and 525 individuals with familial non-acquired focal epilepsy to the same group of 3,877 controls. The researchers found significantly higher rates of ultra-rare deleterious variation in genes established as causative for dominant epilepsy disorders (familial genetic generalized epilepsy, odd ratio 2.3; familial non-acquired focal epilepsy, odds ratio 3.6). Comparison of an additional cohort of 662 individuals with sporadic non-acquired focal epilepsy to controls did not identify study-wide significant signals.
Five Genes Implicated
For the individuals with familial non-acquired focal epilepsy, the researchers found that five known epilepsy genes—DEPDC5, LG11, PCDH19, SCN1A, and GRIN2A—ranked as the top five genes enriched for ultra-rare deleterious variation. “After accounting for the control carrier rate, we estimate that these five genes contribute to the risk of epilepsy in approximately 8% of individuals with familial non-acquired focal epilepsy,” said Erin Heinzen Cox, PhD, Assistant Professor in the Department of Pathology and Cell Biology and Deputy Director of the Institute for Genomic Medicine at Columbia University Medical Center.

Treatment Targeted to Epilepsy Subtype
The findings have important implications for clinical practice and for research. “At present, all common epilepsies are treated the same way, with the same group of medications,” said Dr. Goldstein. “But as we identify more of these epilepsy genes that span a much wider range of types of epilepsy than previously thought, we can begin to try targeted therapies across these patient populations. As this genetically driven treatment paradigm becomes more established, our field, which is accustomed to undertaking large clinical trials in broad patient populations, will need to take a new approach to clinical research, focusing on patients based on their genetic subtype.”
“This is a very exciting breakthrough in the treatment of epilepsy, in which current treatment is based on whether a child has focal seizures … or generalized seizures,” said James J. Riviello, MD, the Sergievsky Family Professor of Neurology and Pediatrics and Chief of Child Neurology at NewYork-Presbyterian Morgan Stanley Children’s Hospital in New York City. “Genetic testing for epilepsy may allow us to identify the specific anticonvulsant medication that potentially works best for an individual patient. We have already identified children in whom knowing the underlying genetic basis of the epilepsy has guided our treatment choices.”

Additional studies, which will analyze 10,000 to 12,000 samples, are planned for the coming year. “With a larger analysis, we expect to find additional genetic variations that contribute to common epilepsies,” said Dr. Goldstein.
Suggested Reading
Epi4K consortium, Epilepsy Phenome/Genome Project. Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol. 2017;16(2):135-143.
Several genes previously implicated only in rare, severe forms of pediatric epilepsy may also contribute to common forms of the disorder, according to a report published online ahead of print January 13 in Lancet Neurology. “Our findings raise hopes that the emerging paradigm for the treatment of rare epilepsies, where therapies are targeted to the precise genetic cause of disease, may also extend to a proportion of common epilepsy syndromes,” said study leader David B. Goldstein, PhD, Director of the Institute for Genomic Medicine and Professor in the Departments of Genetics and Development and Neurology at Columbia University Medical Center in New York City.
In recent years, researchers have identified dozens of genes that, alone or in combination with other factors, cause rare pediatric epilepsies. These discoveries have led to the use of targeted therapies for some seizure disorders, such as the ketogenic diet for patients with Dravet syndrome or GLUT-1 deficiency syndrome. Other therapies such as quinidine, a medication to treat heart arrhythmias, and memantine, an Alzheimer’s disease treatment, have been tried in children with certain gene mutations. These attempts have not proved universally effective for all patients with these mutations, but suggest the potential to repurpose existing medicines to treat rare genetic forms of epilepsy.
“Unlike very rare types of epilepsies, previous studies had shed little light on the genetic underpinnings of common epilepsies, which suggested that this precision medicine paradigm may have a very narrow application,” said Dr. Goldstein.
To learn more about the genetics of epilepsy, Dr. Goldstein and his colleagues conducted a study to identify the genetic contributions to more common forms of epilepsy. Analyses were conducted at Columbia University Medical Center’s Institute for Genomic Medicine, in collaboration with NewYork-Presbyterian, as part of Epi4K, an international consortium of epilepsy clinicians and researchers. Most of the patients were recruited through the Epilepsy Phenome/Genome Project.
The researchers separately compared the sequence data from 640 individuals with familial genetic generalized epilepsy and 525 individuals with familial non-acquired focal epilepsy to the same group of 3,877 controls. The researchers found significantly higher rates of ultra-rare deleterious variation in genes established as causative for dominant epilepsy disorders (familial genetic generalized epilepsy, odd ratio 2.3; familial non-acquired focal epilepsy, odds ratio 3.6). Comparison of an additional cohort of 662 individuals with sporadic non-acquired focal epilepsy to controls did not identify study-wide significant signals.
Five Genes Implicated
For the individuals with familial non-acquired focal epilepsy, the researchers found that five known epilepsy genes—DEPDC5, LG11, PCDH19, SCN1A, and GRIN2A—ranked as the top five genes enriched for ultra-rare deleterious variation. “After accounting for the control carrier rate, we estimate that these five genes contribute to the risk of epilepsy in approximately 8% of individuals with familial non-acquired focal epilepsy,” said Erin Heinzen Cox, PhD, Assistant Professor in the Department of Pathology and Cell Biology and Deputy Director of the Institute for Genomic Medicine at Columbia University Medical Center.
Treatment Targeted to Epilepsy Subtype
The findings have important implications for clinical practice and for research. “At present, all common epilepsies are treated the same way, with the same group of medications,” said Dr. Goldstein. “But as we identify more of these epilepsy genes that span a much wider range of types of epilepsy than previously thought, we can begin to try targeted therapies across these patient populations. As this genetically driven treatment paradigm becomes more established, our field, which is accustomed to undertaking large clinical trials in broad patient populations, will need to take a new approach to clinical research, focusing on patients based on their genetic subtype.”
“This is a very exciting breakthrough in the treatment of epilepsy, in which current treatment is based on whether a child has focal seizures … or generalized seizures,” said James J. Riviello, MD, the Sergievsky Family Professor of Neurology and Pediatrics and Chief of Child Neurology at NewYork-Presbyterian Morgan Stanley Children’s Hospital in New York City. “Genetic testing for epilepsy may allow us to identify the specific anticonvulsant medication that potentially works best for an individual patient. We have already identified children in whom knowing the underlying genetic basis of the epilepsy has guided our treatment choices.”
Additional studies, which will analyze 10,000 to 12,000 samples, are planned for the coming year. “With a larger analysis, we expect to find additional genetic variations that contribute to common epilepsies,” said Dr. Goldstein.
Suggested Reading
Epi4K consortium, Epilepsy Phenome/Genome Project. Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol. 2017;16(2):135-143.
Several genes previously implicated only in rare, severe forms of pediatric epilepsy may also contribute to common forms of the disorder, according to a report published online ahead of print January 13 in Lancet Neurology. “Our findings raise hopes that the emerging paradigm for the treatment of rare epilepsies, where therapies are targeted to the precise genetic cause of disease, may also extend to a proportion of common epilepsy syndromes,” said study leader David B. Goldstein, PhD, Director of the Institute for Genomic Medicine and Professor in the Departments of Genetics and Development and Neurology at Columbia University Medical Center in New York City.
In recent years, researchers have identified dozens of genes that, alone or in combination with other factors, cause rare pediatric epilepsies. These discoveries have led to the use of targeted therapies for some seizure disorders, such as the ketogenic diet for patients with Dravet syndrome or GLUT-1 deficiency syndrome. Other therapies such as quinidine, a medication to treat heart arrhythmias, and memantine, an Alzheimer’s disease treatment, have been tried in children with certain gene mutations. These attempts have not proved universally effective for all patients with these mutations, but suggest the potential to repurpose existing medicines to treat rare genetic forms of epilepsy.
“Unlike very rare types of epilepsies, previous studies had shed little light on the genetic underpinnings of common epilepsies, which suggested that this precision medicine paradigm may have a very narrow application,” said Dr. Goldstein.
To learn more about the genetics of epilepsy, Dr. Goldstein and his colleagues conducted a study to identify the genetic contributions to more common forms of epilepsy. Analyses were conducted at Columbia University Medical Center’s Institute for Genomic Medicine, in collaboration with NewYork-Presbyterian, as part of Epi4K, an international consortium of epilepsy clinicians and researchers. Most of the patients were recruited through the Epilepsy Phenome/Genome Project.
The researchers separately compared the sequence data from 640 individuals with familial genetic generalized epilepsy and 525 individuals with familial non-acquired focal epilepsy to the same group of 3,877 controls. The researchers found significantly higher rates of ultra-rare deleterious variation in genes established as causative for dominant epilepsy disorders (familial genetic generalized epilepsy, odd ratio 2.3; familial non-acquired focal epilepsy, odds ratio 3.6). Comparison of an additional cohort of 662 individuals with sporadic non-acquired focal epilepsy to controls did not identify study-wide significant signals.
Five Genes Implicated
For the individuals with familial non-acquired focal epilepsy, the researchers found that five known epilepsy genes—DEPDC5, LG11, PCDH19, SCN1A, and GRIN2A—ranked as the top five genes enriched for ultra-rare deleterious variation. “After accounting for the control carrier rate, we estimate that these five genes contribute to the risk of epilepsy in approximately 8% of individuals with familial non-acquired focal epilepsy,” said Erin Heinzen Cox, PhD, Assistant Professor in the Department of Pathology and Cell Biology and Deputy Director of the Institute for Genomic Medicine at Columbia University Medical Center.
Treatment Targeted to Epilepsy Subtype
The findings have important implications for clinical practice and for research. “At present, all common epilepsies are treated the same way, with the same group of medications,” said Dr. Goldstein. “But as we identify more of these epilepsy genes that span a much wider range of types of epilepsy than previously thought, we can begin to try targeted therapies across these patient populations. As this genetically driven treatment paradigm becomes more established, our field, which is accustomed to undertaking large clinical trials in broad patient populations, will need to take a new approach to clinical research, focusing on patients based on their genetic subtype.”
“This is a very exciting breakthrough in the treatment of epilepsy, in which current treatment is based on whether a child has focal seizures … or generalized seizures,” said James J. Riviello, MD, the Sergievsky Family Professor of Neurology and Pediatrics and Chief of Child Neurology at NewYork-Presbyterian Morgan Stanley Children’s Hospital in New York City. “Genetic testing for epilepsy may allow us to identify the specific anticonvulsant medication that potentially works best for an individual patient. We have already identified children in whom knowing the underlying genetic basis of the epilepsy has guided our treatment choices.”
Additional studies, which will analyze 10,000 to 12,000 samples, are planned for the coming year. “With a larger analysis, we expect to find additional genetic variations that contribute to common epilepsies,” said Dr. Goldstein.
Suggested Reading
Epi4K consortium, Epilepsy Phenome/Genome Project. Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol. 2017;16(2):135-143.

Hysteroscopic tubal occlusion: How new product labeling can be a resource for patient counseling
In November 2016, Bayer, the manufacturer of the permanent birth control tubal implant system (Essure), revised the Essure product labeling in accordance with a US Food and Drug Administration (FDA) guidance document.1 The FDA developed its labeling guidance based on its examination of an increasing number of reported adverse events associated with the system’s use (such as persistent pain, perforation of the uterus and/or fallopian tubes, intra-abdominal or pelvic device migration, abnormal or irregular bleeding, and allergy or hypersensitivity reactions) and its evaluation of a trade complaint regarding allegations initially made in a Citizen Petition.
Changes to the new FDA-approved labeling for Essure include:
- the addition of a boxed warning listing adverse events that have been reported either in clinical studies or through postmarket surveillance (see Box)
- updated Instructions for Use document for clinicians and Patient Information Booklet, which contain additional information on safety (contraindications, warnings, and precautions), clinical data, and instructions2,3
- a Patient-Doctor Discussion Checklist (included within the Patient Information Booklet), designed to support appropriate patient counseling, facilitate the patient’s understanding of birth control options, and explain the benefits and risks associated with the device and what to expect during and after the implantation procedure.3
How will these labeling changes impact clinicians and patients? OBG
Reference
1. Essure permanent birth control (Bayer) Instructions for use. http://www.hcp.essure-us.com/assets/pdf/Link%20Essure%20IFU.pdf. Accessed January 5, 2017.
OBG Management: What does the new product labeling mean for clinicians who offer tubal implants as an option for permanent sterilization?
Linda D. Bradley, MD: The FDA-approved revised labeling for the Essure system means that physicians should have a very detailed, in-depth conversation with their patients who are contemplating hysteroscopic tubal insert placement for permanent sterilization. This counseling really should not differ from what doctors were doing before the label was revised. However, physicians can now use the new Patient-Doctor Discussion Checklist as a guide in reviewing the benefits of the device, its known risks and potential risks, outcomes of the insertion procedure, and the possible need for future surgical intervention if device placement–related issues arise.
For clinicians, this counseling adds just a few more minutes to the visit. The Patient-Doctor Discussion Checklist will become an inherent part of the informed consent process, aiding in the review of the device’s benefits, potential risks, and more importantly its permanence.
In the past, there was some concern that perhaps patients did not receive enough guidance for informed consent, so one of the first things listed on the checklist is confirmation—in the form of a printed line where the patient can sign her initials—that she understands that Essure is a permanent form of birth control. The checklist covers additional important issues, including that the doctor has indeed shared with the patient other options for birth control or sterilization, such as laparoscopic sterilization, vasectomy for her male partner, an intrauterine device (IUD), and birth control pills. This is an opportunity to reinforce the fact that tubal implants are a permanent form of birth control, and if the patient is uncertain about ending her fertility, the clinician can inform her about reversible options. The checklist also includes for discussion the pregnancy risk with use of the device, what the patient can expect during the implant insertion procedure and for the days afterwards (such as cramping, mild to moderate pain, nausea and vomiting), and the need for a confirmation test 3 months after device placement.
Other discussion points covered include long-term risks and benefits of the device, the potential for complications, and the possibility (due to pelvic pain) that the hysteroscopically placed devices may need to be removed with a surgical procedure requiring general anesthesia.
Incorporating the checklist into our clinical practice shows that we have listened to patients and complied with recommendations made by the FDA review panel, and we can use this document to have a more complete discussion with our patients.
OBG Management: Do you agree with some clinicians who say that physicians who place the device also should have the skills required to remove it if necessary?
Dr. Bradley: Essure placement—which is a hysteroscopic procedure—is done very differently than a laparoscopic procedure. In the past, among women who needed to have the Essure system removed, most procedures would be done laparoscopically. Since we work collaboratively in teams, someone within the team or division would have the clinical expertise to remove the devices. An ObGyn who does laparoscopy with salpingectomy and/or cornual resection would best be able to remove the devices.
The clinician who does hysteroscopy is not always the same one who does laparoscopy. Someone within the division who is interested in removing the device will develop an expertise and algorithm that suits the practice, so that person in the practice becomes the expert. This is no different from many other things that physicians do. In our clinical practice, for example, we have a pelvic pain specialist, a sexual counselor, someone interested in menopause and management, and someone interested in alternatives to hysterectomies. Those who practice their craft and their art become proficient at it. So if you do not perform a particular procedure such as a tubal implant removal, know the expert to whom you can make a referral.
OBG Management: How do you now advise your colleagues to counsel patients on permanent sterilization?
Dr. Bradley: Hysteroscopic tubal implant sterilization, a minimally invasive procedure, is an excellent and viable option for women who meet the inclusion criteria and who do not have the exclusion criteria for placement. It is overall safe and extremely effective. If a patient has issues after undergoing implant placement—just like with any other surgery or procedure—for example, if she is not feeling better or is not doing as well as anticipated, we must not forget the patient. It is important for our patients to be listened to and to be heard. Postprocedure issues are generally transient and related to pain and discomfort or abnormal bleeding. If they are persistent, then further evaluation is needed.
Tell the patient to contact you if she has questions or issues, and have a tiered approach for working up any problems that she may present with. In addition, reiterate that the patient must use another form of birth control for 3 months until she undergoes the confirmation test and until the results verify that the implants can be relied on for contraception. I am still placing the device. Before I perform the procedure, I speak with my patients—as I did before the checklist was developed—about all of the informed consent issues, the risk−benefit profile, and ruling out contraindications to use. I think this is good medical and surgical practice. The new labeling means we need to have a critical conversation with our patients, and we should be doing that for all procedures.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- US Food and Drug Administration. Labeling for permanent hysteroscopically-placed tubal implants intended for sterilization: guidance for industry and Food and Drug Administration staff. http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM488020.pdf. Published October 31, 2016. Accessed January 5, 2017.
- Essure permanent birth control (Bayer) Instructions for use. http://www.hcp.essure-us.com/assets/pdf/Link_Essure_IFU.pdf. Accessed January 5, 2017.
- Essure patient information booklet. http://labeling.bayerhealthcare.com/html/products/pi/essure_pib_en.pdf. Accessed January 5, 2017.

Dr. Bradley is Professor of Surgery and Vice Chair of Obstetrics and Gynecology at the Women’s Health Institute, and Director, Center for Menstrual Disorders, Fibroids, and Hysteroscopic Services, Cleveland Clinic, Cleveland, Ohio. Dr. Bradley serves on the OBG
Dr. Bradley reports that she has received research or grant support from and is a consultant and speaker for Bayer, is a speaker for Smith & Nephew and Teva, serves on the scientific advisory board for Boston Scientific, is a consultant for Karl Storz, and has received royalties from UpToDate and Elsevier.
Dr. Bradley is Professor of Surgery and Vice Chair of Obstetrics and Gynecology at the Women’s Health Institute, and Director, Center for Menstrual Disorders, Fibroids, and Hysteroscopic Services, Cleveland Clinic, Cleveland, Ohio. Dr. Bradley serves on the OBG
Dr. Bradley reports that she has received research or grant support from and is a consultant and speaker for Bayer, is a speaker for Smith & Nephew and Teva, serves on the scientific advisory board for Boston Scientific, is a consultant for Karl Storz, and has received royalties from UpToDate and Elsevier.
Dr. Bradley is Professor of Surgery and Vice Chair of Obstetrics and Gynecology at the Women’s Health Institute, and Director, Center for Menstrual Disorders, Fibroids, and Hysteroscopic Services, Cleveland Clinic, Cleveland, Ohio. Dr. Bradley serves on the OBG
Dr. Bradley reports that she has received research or grant support from and is a consultant and speaker for Bayer, is a speaker for Smith & Nephew and Teva, serves on the scientific advisory board for Boston Scientific, is a consultant for Karl Storz, and has received royalties from UpToDate and Elsevier.
In November 2016, Bayer, the manufacturer of the permanent birth control tubal implant system (Essure), revised the Essure product labeling in accordance with a US Food and Drug Administration (FDA) guidance document.1 The FDA developed its labeling guidance based on its examination of an increasing number of reported adverse events associated with the system’s use (such as persistent pain, perforation of the uterus and/or fallopian tubes, intra-abdominal or pelvic device migration, abnormal or irregular bleeding, and allergy or hypersensitivity reactions) and its evaluation of a trade complaint regarding allegations initially made in a Citizen Petition.
Changes to the new FDA-approved labeling for Essure include:
- the addition of a boxed warning listing adverse events that have been reported either in clinical studies or through postmarket surveillance (see Box)
- updated Instructions for Use document for clinicians and Patient Information Booklet, which contain additional information on safety (contraindications, warnings, and precautions), clinical data, and instructions2,3
- a Patient-Doctor Discussion Checklist (included within the Patient Information Booklet), designed to support appropriate patient counseling, facilitate the patient’s understanding of birth control options, and explain the benefits and risks associated with the device and what to expect during and after the implantation procedure.3
How will these labeling changes impact clinicians and patients? OBG
Reference
1. Essure permanent birth control (Bayer) Instructions for use. http://www.hcp.essure-us.com/assets/pdf/Link%20Essure%20IFU.pdf. Accessed January 5, 2017.
OBG Management: What does the new product labeling mean for clinicians who offer tubal implants as an option for permanent sterilization?
Linda D. Bradley, MD: The FDA-approved revised labeling for the Essure system means that physicians should have a very detailed, in-depth conversation with their patients who are contemplating hysteroscopic tubal insert placement for permanent sterilization. This counseling really should not differ from what doctors were doing before the label was revised. However, physicians can now use the new Patient-Doctor Discussion Checklist as a guide in reviewing the benefits of the device, its known risks and potential risks, outcomes of the insertion procedure, and the possible need for future surgical intervention if device placement–related issues arise.
For clinicians, this counseling adds just a few more minutes to the visit. The Patient-Doctor Discussion Checklist will become an inherent part of the informed consent process, aiding in the review of the device’s benefits, potential risks, and more importantly its permanence.
In the past, there was some concern that perhaps patients did not receive enough guidance for informed consent, so one of the first things listed on the checklist is confirmation—in the form of a printed line where the patient can sign her initials—that she understands that Essure is a permanent form of birth control. The checklist covers additional important issues, including that the doctor has indeed shared with the patient other options for birth control or sterilization, such as laparoscopic sterilization, vasectomy for her male partner, an intrauterine device (IUD), and birth control pills. This is an opportunity to reinforce the fact that tubal implants are a permanent form of birth control, and if the patient is uncertain about ending her fertility, the clinician can inform her about reversible options. The checklist also includes for discussion the pregnancy risk with use of the device, what the patient can expect during the implant insertion procedure and for the days afterwards (such as cramping, mild to moderate pain, nausea and vomiting), and the need for a confirmation test 3 months after device placement.
Other discussion points covered include long-term risks and benefits of the device, the potential for complications, and the possibility (due to pelvic pain) that the hysteroscopically placed devices may need to be removed with a surgical procedure requiring general anesthesia.
Incorporating the checklist into our clinical practice shows that we have listened to patients and complied with recommendations made by the FDA review panel, and we can use this document to have a more complete discussion with our patients.
OBG Management: Do you agree with some clinicians who say that physicians who place the device also should have the skills required to remove it if necessary?
Dr. Bradley: Essure placement—which is a hysteroscopic procedure—is done very differently than a laparoscopic procedure. In the past, among women who needed to have the Essure system removed, most procedures would be done laparoscopically. Since we work collaboratively in teams, someone within the team or division would have the clinical expertise to remove the devices. An ObGyn who does laparoscopy with salpingectomy and/or cornual resection would best be able to remove the devices.
The clinician who does hysteroscopy is not always the same one who does laparoscopy. Someone within the division who is interested in removing the device will develop an expertise and algorithm that suits the practice, so that person in the practice becomes the expert. This is no different from many other things that physicians do. In our clinical practice, for example, we have a pelvic pain specialist, a sexual counselor, someone interested in menopause and management, and someone interested in alternatives to hysterectomies. Those who practice their craft and their art become proficient at it. So if you do not perform a particular procedure such as a tubal implant removal, know the expert to whom you can make a referral.
OBG Management: How do you now advise your colleagues to counsel patients on permanent sterilization?
Dr. Bradley: Hysteroscopic tubal implant sterilization, a minimally invasive procedure, is an excellent and viable option for women who meet the inclusion criteria and who do not have the exclusion criteria for placement. It is overall safe and extremely effective. If a patient has issues after undergoing implant placement—just like with any other surgery or procedure—for example, if she is not feeling better or is not doing as well as anticipated, we must not forget the patient. It is important for our patients to be listened to and to be heard. Postprocedure issues are generally transient and related to pain and discomfort or abnormal bleeding. If they are persistent, then further evaluation is needed.
Tell the patient to contact you if she has questions or issues, and have a tiered approach for working up any problems that she may present with. In addition, reiterate that the patient must use another form of birth control for 3 months until she undergoes the confirmation test and until the results verify that the implants can be relied on for contraception. I am still placing the device. Before I perform the procedure, I speak with my patients—as I did before the checklist was developed—about all of the informed consent issues, the risk−benefit profile, and ruling out contraindications to use. I think this is good medical and surgical practice. The new labeling means we need to have a critical conversation with our patients, and we should be doing that for all procedures.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
In November 2016, Bayer, the manufacturer of the permanent birth control tubal implant system (Essure), revised the Essure product labeling in accordance with a US Food and Drug Administration (FDA) guidance document.1 The FDA developed its labeling guidance based on its examination of an increasing number of reported adverse events associated with the system’s use (such as persistent pain, perforation of the uterus and/or fallopian tubes, intra-abdominal or pelvic device migration, abnormal or irregular bleeding, and allergy or hypersensitivity reactions) and its evaluation of a trade complaint regarding allegations initially made in a Citizen Petition.
Changes to the new FDA-approved labeling for Essure include:
- the addition of a boxed warning listing adverse events that have been reported either in clinical studies or through postmarket surveillance (see Box)
- updated Instructions for Use document for clinicians and Patient Information Booklet, which contain additional information on safety (contraindications, warnings, and precautions), clinical data, and instructions2,3
- a Patient-Doctor Discussion Checklist (included within the Patient Information Booklet), designed to support appropriate patient counseling, facilitate the patient’s understanding of birth control options, and explain the benefits and risks associated with the device and what to expect during and after the implantation procedure.3
How will these labeling changes impact clinicians and patients? OBG
Reference
1. Essure permanent birth control (Bayer) Instructions for use. http://www.hcp.essure-us.com/assets/pdf/Link%20Essure%20IFU.pdf. Accessed January 5, 2017.
OBG Management: What does the new product labeling mean for clinicians who offer tubal implants as an option for permanent sterilization?
Linda D. Bradley, MD: The FDA-approved revised labeling for the Essure system means that physicians should have a very detailed, in-depth conversation with their patients who are contemplating hysteroscopic tubal insert placement for permanent sterilization. This counseling really should not differ from what doctors were doing before the label was revised. However, physicians can now use the new Patient-Doctor Discussion Checklist as a guide in reviewing the benefits of the device, its known risks and potential risks, outcomes of the insertion procedure, and the possible need for future surgical intervention if device placement–related issues arise.
For clinicians, this counseling adds just a few more minutes to the visit. The Patient-Doctor Discussion Checklist will become an inherent part of the informed consent process, aiding in the review of the device’s benefits, potential risks, and more importantly its permanence.
In the past, there was some concern that perhaps patients did not receive enough guidance for informed consent, so one of the first things listed on the checklist is confirmation—in the form of a printed line where the patient can sign her initials—that she understands that Essure is a permanent form of birth control. The checklist covers additional important issues, including that the doctor has indeed shared with the patient other options for birth control or sterilization, such as laparoscopic sterilization, vasectomy for her male partner, an intrauterine device (IUD), and birth control pills. This is an opportunity to reinforce the fact that tubal implants are a permanent form of birth control, and if the patient is uncertain about ending her fertility, the clinician can inform her about reversible options. The checklist also includes for discussion the pregnancy risk with use of the device, what the patient can expect during the implant insertion procedure and for the days afterwards (such as cramping, mild to moderate pain, nausea and vomiting), and the need for a confirmation test 3 months after device placement.
Other discussion points covered include long-term risks and benefits of the device, the potential for complications, and the possibility (due to pelvic pain) that the hysteroscopically placed devices may need to be removed with a surgical procedure requiring general anesthesia.
Incorporating the checklist into our clinical practice shows that we have listened to patients and complied with recommendations made by the FDA review panel, and we can use this document to have a more complete discussion with our patients.
OBG Management: Do you agree with some clinicians who say that physicians who place the device also should have the skills required to remove it if necessary?
Dr. Bradley: Essure placement—which is a hysteroscopic procedure—is done very differently than a laparoscopic procedure. In the past, among women who needed to have the Essure system removed, most procedures would be done laparoscopically. Since we work collaboratively in teams, someone within the team or division would have the clinical expertise to remove the devices. An ObGyn who does laparoscopy with salpingectomy and/or cornual resection would best be able to remove the devices.
The clinician who does hysteroscopy is not always the same one who does laparoscopy. Someone within the division who is interested in removing the device will develop an expertise and algorithm that suits the practice, so that person in the practice becomes the expert. This is no different from many other things that physicians do. In our clinical practice, for example, we have a pelvic pain specialist, a sexual counselor, someone interested in menopause and management, and someone interested in alternatives to hysterectomies. Those who practice their craft and their art become proficient at it. So if you do not perform a particular procedure such as a tubal implant removal, know the expert to whom you can make a referral.
OBG Management: How do you now advise your colleagues to counsel patients on permanent sterilization?
Dr. Bradley: Hysteroscopic tubal implant sterilization, a minimally invasive procedure, is an excellent and viable option for women who meet the inclusion criteria and who do not have the exclusion criteria for placement. It is overall safe and extremely effective. If a patient has issues after undergoing implant placement—just like with any other surgery or procedure—for example, if she is not feeling better or is not doing as well as anticipated, we must not forget the patient. It is important for our patients to be listened to and to be heard. Postprocedure issues are generally transient and related to pain and discomfort or abnormal bleeding. If they are persistent, then further evaluation is needed.
Tell the patient to contact you if she has questions or issues, and have a tiered approach for working up any problems that she may present with. In addition, reiterate that the patient must use another form of birth control for 3 months until she undergoes the confirmation test and until the results verify that the implants can be relied on for contraception. I am still placing the device. Before I perform the procedure, I speak with my patients—as I did before the checklist was developed—about all of the informed consent issues, the risk−benefit profile, and ruling out contraindications to use. I think this is good medical and surgical practice. The new labeling means we need to have a critical conversation with our patients, and we should be doing that for all procedures.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- US Food and Drug Administration. Labeling for permanent hysteroscopically-placed tubal implants intended for sterilization: guidance for industry and Food and Drug Administration staff. http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM488020.pdf. Published October 31, 2016. Accessed January 5, 2017.
- Essure permanent birth control (Bayer) Instructions for use. http://www.hcp.essure-us.com/assets/pdf/Link_Essure_IFU.pdf. Accessed January 5, 2017.
- Essure patient information booklet. http://labeling.bayerhealthcare.com/html/products/pi/essure_pib_en.pdf. Accessed January 5, 2017.
- US Food and Drug Administration. Labeling for permanent hysteroscopically-placed tubal implants intended for sterilization: guidance for industry and Food and Drug Administration staff. http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM488020.pdf. Published October 31, 2016. Accessed January 5, 2017.
- Essure permanent birth control (Bayer) Instructions for use. http://www.hcp.essure-us.com/assets/pdf/Link_Essure_IFU.pdf. Accessed January 5, 2017.
- Essure patient information booklet. http://labeling.bayerhealthcare.com/html/products/pi/essure_pib_en.pdf. Accessed January 5, 2017.

2017 Update on fertility
Zika virus is a serious problem. Education and infection prevention are critical to effective management, and why we chose to include Zika virus as a topic for this year’s Update. We also discuss obesity’s effects on reproduction—a very relevant concern for all ObGyns and patients alike as about half of reproductive-age women are obese. Finally, subclinical hypothyroidism can present unique management challenges, such as determining when it is present and when treatment is indicated.
Read about counseling patients about Zika virus
Managing attempted pregnancy in the era of Zika virus
Oduyebo T, Igbinosa I, Petersen EE, et al. Update: interim guidance for health care providers caring for pregnant women with possible Zika virus exposure--United States, July 2016. MMWR Morb Mortal Wkly Rep. 2016;65(29):739-744.
Petersen EE, Meaney-Delman D, Neblett-Fanfair R, et al. Update: interim guidance for preconception counseling and prevention of sexual transmission of Zika virus for persons with possible Zika virus exposure--United States, September 2016. MMWR Morb Mortal Wkly Rep. 2016;65(39):1077-1081.
US Food and Drug Administration. Donor Screening Recommendations to Reduce the Risk of Transmission of Zika Virus by Human Cells, Tissues, and Cellular and Tissue-Based Products. http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Tissue/UCM488582.pdf. Published March 2016. Accessed January 12, 2017.
National Institutes of Health. Zika: Overview. https://www.nichd.nih.gov/health/topics/zika/Pages/default.aspx. Accessed January 12, 2017.
World Health Organization. Prevention of sexual transmission of Zika virus interim guidance. WHO reference number: WHO/ZIKV/MOC/16. 1 Rev. 3, September 6, 2016.
Zika Virus Guidance Task Force of the American Society for Reproductive Medicine. Rev. 13, September 2016.
Zika virus presents unique challenges to physicians managing the care of patients attempting pregnancy, with or without fertility treatment. Neonatal Zika virus infection sequelae only recently have been appreciated; microcephaly was associated with Zika virus in October 2015, followed by other neurologic conditions including brain abnormalities, neural tube defects, and eye abnormalities. Results of recent studies involving the US Zika Pregnancy Registry show that 6% of women with Zika at any time in pregnancy had affected babies, but 11% of those who contracted the disease in the first trimester were affected.
Diagnosis is difficult because symptoms are generally mild, with 80% of affected patients asymptomatic. Possible Zika virus exposure is defined as travel to or residence in an area of active Zika virus transmission, or sex without a condom with a partner who traveled to or lived in an area of active transmission. Much is unknown about the interval from exposure to symptoms. Testing availability is limited and variable, and much is unknown about sensitivity and specificity of direct viral RNA testing, appearance and disappearance of detectable immunoglobulin (Ig) M and IgG antibodies that affect false positive and false negative test results, duration of infectious phase, risk of transmission, and numerous other factors.
Positive serum viral testing likely indicates virus in semen or other bodily fluids, but a negative serum viral test cannot definitively preclude virus in other bodily fluids. Zika virus likely can be passed from any combination of semen and vaginal and cervical fluids, but validating tests for these fluids are not yet available. It is not known if sperm preparation and assisted reproductive technology (ART) procedures that minimize risk of HIV transmission are effective against Zika virus or whether or not cryopreservation can destroy the virus.
Pregnancy timing
The Centers for Disease Control and Prevention now recommends that all men with possible Zika virus exposure who are considering attempting pregnancy with their partner wait to get pregnant until at least 6 months after symptom onset (if symptomatic) or last possible Zika virus exposure (if asymptomatic). Women with possible Zika virus exposure are recommended to wait to get pregnant until at least 8 weeks after symptom onset (if symptomatic) or last possible Zika virus exposure (if asymptomatic).
Women and men with possible exposure to Zika virus but without clinical symptoms of illness should consider testing for Zika viral RNA within 2 weeks of suspected exposure and wait at least 8 weeks after the last date of exposure before being re-tested. If direct viral testing (using rRT-PCR) results initially are negative, ideally, antibody testing would be obtained, if available, at 8 weeks. However, no testing paradigm will absolutely guarantee lack of Zika virus infectivity.
Virus management problems are dramatically compounded in areas endemic for Zika. Women and men who have had Zika virus disease should wait at least 6 months after illness onset to attempt reproduction. The temporal relationship between the presence of viral RNA and infectivity is not known definitively, and so the absolute duration of time to wait before attempting pregnancy is unknown. Male and female partners who become infected should avoid all forms of intimate sexual conduct or use condoms for the same 6 months. There is no evidence Zika will cause congenital infection in pregnancies initiated after resolution of maternal Zika viremia. However, any testing performed at a time other than the time of treatment might not reflect true viral status, particularly in areas of active Zika virus transmission.
Prevention
Women and men, especially those residing in areas of active Zika virus transmission, should talk with their physicians regarding pregnancy plans and avoid mosquito bites using the usual precautions: avoid mosquito areas, drain standing water, use mosquito repellent containing DEET, and use mosquito netting. Some people have gone so far as to relocate to nonendemic areas.
Those contemplating pregnancy should be advised to consider what they would do if they become exposed to or have suspected or confirmed Zika virus during pregnancy. Additional considerations are gamete or embryo cryopreservation and quarantine until a subsequent rRT-PCR test result is negative in both the male and female and at least 8 weeks have passed from gamete collection.

Patient counseling essentials
Counsel patients considering reproduction about:
- Zika virus as a new reproductive hazard
- the significance of the hazard to the fetus if infected
- the areas of active transmission, and that they are constantly changing
- avoidance of Zika areas if possible
- methods of transmission through mosquito bites or sex
- avoidance of mosquito bites
- symptoms of Zika infection
- safe sex practices
- testing limitations and knowledge deficiency about Zika.
Not uncommonly, clinical situations require complex individualized management decisions regarding trade-offs of risks, especially in older patients with decreased ovarian reserve. Consultation with infectious disease and reproductive specialists should be obtained when complicated and consequential decisions have to be made.
All practitioners should inform their patients, especially those undergoing fertility treatments, about Zika, and develop language in their informed consent that conveys the gap in knowledge to these patients.
Read how obesity specifically affects reproduction in an adverse way
Obesity adversely affects reproduction, but how specifically?
Practice Committee of the American Society for Reproductive Medicine. Obesity and Reproduction: A committee opinion. Fertil Steril. 2015;104(5):1116-1126.
The prevalence of obesity has increased substantially over the past 2 decades. Almost two-thirds of women and three-fourths of men in the United States are overweight or obese (defined as a body mass index [BMI] ≥25 kg/m2 and BMI ≥30 kg/m2, respectively; TABLE). Nearly 50% of reproductive-age women are obese.
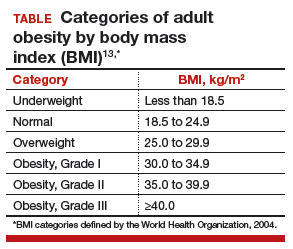
A disease of excess body fat and insulin resistance, obesity increases the risks of hypertension, diabetes, dyslipidemia, cardiovascular disease, sleep apnea, respiratory problems, and cancer as well as other serious health problems. While not all individuals with obesity will have infertility, obesity is associated with impaired reproduction in both women and men, adverse obstetric outcomes, and health problems in offspring. The American Society for Reproductive Medicine (ASRM) reviewed this important issue in a recent practice committee opinion.
Menstrual cycle and ovulatory dysfunction
Menstrual cycle abnormalities are more common in women with obesity. Elevated levels of insulin in obese women suppress sex hormone−binding globulin (SHBG) which in turn reduces gonadotropin secretion due to increased production of estrogen from conversion of androgens by adipose aromatase.1 Adipose tissue produces adipokines, which directly can suppress ovarian function.2
Ovulatory dysfunction is common among obese women; the relative risk of such dysfunction is 3.1 (95% confidence interval [CI], 2.2−4.4) among women with BMI levels >27 kg/m2 versus BMI levels 20.0 to 24.9 kg/m2.3,4 Obesity decreases fecundity even in women with normal menstrual cycles.5 This may in part be due to altered ovulatory dynamics with reduced early follicular luteinizing hormone pulse amplitude accompanied by prolonged folliculogenesis and reduced luteal progesterone levels.6
Compared with normal-weight women, obese women have a lower chance of conception within 1 year of stopping contraception; about 66% of obese women conceive within 1 year of stopping contraception, compared with about 81% of women with normal weight.7 Results of a Dutch study of 3,029 women with regular ovulation, at least one patent tube, and a partner with a normal semen analysis indicated a direct correlation between obesity and delayed conception, with a 4% lower spontaneous pregnancy rate per kg/m2 increase in women with a BMI >29 kg/m2 versus a BMI of 21 to 29 kg/m2 (hazard ratio, 0.96; 95% CI, 0.91−0.99).8
Assisted reproduction
Assisted reproduction in women with obesity is associated with lower success rates than in women with normal weight. A systematic review of 27 in vitro fertilization (IVF) studies (23 of which were retrospective) reveals 10% lower live-birth rate in overweight (BMI >25 kg/m2) versus normal-weight women (BMI <25 kg/m2) undergoing IVF (odds ratio [OR], 0.90; 95% CI, 0.82−1.0).9 Data from a meta-analysis of 33 IVF studies, including 47,967 cycles, show that, compared with women with a BMI <25 kg/m2, overweight or obese women have significantly reduced rates of clinical pregnancy (relative risk [RR], 0.90; P<.0001) and live birth (RR, 0.84; P = .0002).10
Results of a retrospective study of 4,609 women undergoing first IVF or IVF/intracytoplasmic sperm injection cycles revealed impaired embryo implantation (controlling for embryo quality and transfer day), reducing the age-adjusted odds of live birth in a BMI-dependent manner by 37% (BMI, 30.0−34.9 kg/m2), 61% (BMI, 35.0−39.9 kg/m2), and 68% (BMI, >40 kg/m2) compared with women with a BMI of 18.5 to 24.9 kg/m2.11 In a study of 12,566 Danish couples undergoing assisted reproduction, overweight and obese ovulatory women had a 12% (95% CI, 0.79−0.99) and 25% (95% CI, 0.63−0.90) reduction in IVF-related live birth rate, respectively (referent BMI, 18.5−24.9 kg/m2), with a 2% (95% CI, 0.97−0.99) decrease in live-birth rate for every one-unit increase in BMI.12 Putative mechanisms for these findings include altered oocyte morphology and reduced fertilization in eggs from obese women,13 and impaired embryo quality in women less than age 35.14 Oocytes from women with a BMI >25 kg/m2 are smaller and less likely to complete development postfertilization, with embryos arrested prior to blastulation containing more triglyceride than those forming blastocysts.15
Blastocysts developed from oocytes of high-BMI women are smaller, contain fewer cells and have a higher content of triglycerides, lower glucose consumption, and altered amino acid metabolism compared with embryos of normal-weight women (BMI <24.9 kg/m2).15 Obesity may alter endometrial receptivity during IVF given the finding that third-party surrogate women with a BMI >35 kg/m2 have a lower live-birth rate (25%) compared with women with a BMI <35 kg/m2 (49%; P<.05).16
Pregnancy outcomes
Obesity is linked to an increased risk of miscarriage. Results of a meta-analysis of 33 IVF studies including 47,967 cycles indicated that overweight or obese women have a higher rate of miscarriage (RR, 1.31; P<.0001) than normal-weight women (BMI <25 kg/m2).17 Maternal and perinatal morbid obesity are strongly associated with obstetric and perinatal complications, including gestational diabetes, hypertension, preeclampsia, preterm delivery, shoulder dystocia, fetal distress, early neonatal death, and small- as well as large-for-gestational age infants.
Obese women who conceive by IVF are at increased risk for preeclampsia, gestational diabetes, preterm delivery, and cesarean delivery.13 Authors of a meta-analysis of 18 observational studies concluded that obese mothers were at increased odds of pregnancies affected by such birth defects as neural tube defects, cardiovascular anomalies, and cleft lip and palate, among others.18
In addition to being the cause of these fetal abnormalities, maternal metabolic dysfunction is linked to promoting obesity in offspring, thereby perpetuating a cycle of obesity and adverse health outcomes that include an increased risk of premature death in adult offspring in subsequent generations.13
Treatment for obesity
Lifestyle modification is the first-line treatment for obesity.
Pre-fertility therapy and pregnancy goals. Targets for pregnancy should include:
- preconception weight loss to a BMI of 35 kg/m2
- prevention of excess weight gain in pregnancy
- long-term reduction in weight.
For all obese individuals, lifestyle modifications should include a weight loss of 7% of body weight and increased physical activity to at least 150 minutes of moderate activity, such as walking, per week. Calorie restriction should be emphasized. A 500 to 1,000 kcal/day decrease from usual dietary intake is expected to result in a 1- to 2-lb weight loss per week. A low-calorie diet of 1,000 to 1,200 kcal/day can lead to an average 10% decrease in total body weight over 6 months.
Adjunct supervised medical therapy or bariatric surgery can play an important role in successful weight loss prepregnancy but are not appropriate for women actively attempting conception. Importantly, pregnancy should be deferred for a minimum of 1 year after bariatric surgery. The decision to postpone pregnancy to achieve weight loss must be balanced against the risk of declining fertility with advancing age of the woman.
Read about when to treat subclinical hypothyroidism
Optimal management of subclinical hypothyroidism in women with infertility
Practice Committee of the American Society for Reproductive Medicine. Subclinical hypothyroidism in the infertile female population: a guideline. Fertil Steril. 2015;104(3):545-553.
Thyroid disorders long have been associated with the potential for adverse reproductive outcomes. While overt hypothyroidism has been linked to infertility, increased miscarriage risk, and poor maternal and fetal outcomes, controversy has existed regarding the association between subclinical hypothyroidism (SCH) and reproductive problems. The ASRM recently published a guideline on the role of SCH in the infertile female population.
How is subclinical hypothyroidism defined?
SCH is classically defined as a thyrotropin (TSH) level above the upper limit of normal range (4.5−5.0 mIU/L) with normal free thyroxine (FT4) levels. The National Health and Nutrition Examination Survey (NHANES III) population has been used to establish normative data for TSH for a disease-free population. These include a median serum level for TSH of 1.5 mIU/L, with the corresponding 2.5 and 97.5 percentiles of 0.41 and 6.10, respectively.19 Data from the National Academy of Clinical Biochemistry, however, reveal that 95% of individuals without evidence of thyroid disease have a TSH level <2.5 mIU/L, and that the normal reference range is skewed to the right.20 Adjusting the upper limit of the normal range to 2.5 mIU/L would result in an additional 11.8% to 14.2% of the United States population (22 to 28 million individuals) being diagnosed with hypothyroidism.
This information raises several important questions.
1. Should nonpregnant women be treated for SCH?
No. There is no benefit from the standpoint of lipid profile or alteration of cardiovascular risk in the treatment of TSH levels between 5 and 10 mIU/L and, therefore, treatment of individuals with TSH <5 mIU/L is questionable. Furthermore, the risk of overtreatment resulting in bone loss is a concern. The Endocrine Society does not recommend changing the current normal TSH range for nonpregnant women.
2. What are normal TSH levels in pregnant women?
Because human chorionic gonadotropin (hCG) can bind to and affect the TSH receptor, thereby influencing TSH values, the normal range for TSH is modified in pregnancy. The Endocrine Society recommends the following pregnancy trimester guidelines for TSH levels: 2.5 mIU/L is the recommended upper limit of normal in the first trimester, 3.0 mIU/L in the second trimester, and 3.5 mIU/L in the third trimester.
3. Is untreated SCH associated with miscarriage?
There is fair evidence that SCH, defined as a TSH level >4 mIU/L during pregnancy, is associated with miscarriage, but there is insufficient evidence that TSH levels between 2.5 and 4 mIU/L are associated with miscarriage.
4. Is untreated SCH associated with infertility?
Limited data are available to assess the effect of SCH on infertility. While a few studies show an association between SCH on unexplained infertility and ovulatory disorders, SCH does not appear to be increased in other causes of infertility.
5. Is SCH associated with adverse obstetric outcomes?
Available data reveal that SCH with TSH levels outside the normal pregnancy range are associated with an increased risk of such obstetric complications as placental abruption, preterm birth, fetal death, and preterm premature rupture of membranes (PPROM). However, it is unclear if prepregnancy TSH levels between 2.5 and 4 mIU/L are associated with adverse obstetric outcomes.
6. Does untreated SCH affect developmental outcomes in children?
The fetus is solely dependent on maternal thyroid hormone in early pregnancy because the fetal thyroid does not produce thyroid hormone before 10 to 13 weeks of gestation. Significant evidence has associated untreated maternal hypothyroidism with delayed fetal neurologic development, impaired school performance, and lower intelligence quotient (IQ) among offspring.21 There is fair evidence that SCH diagnosed in pregnancy is associated with adverse neurologic development. There is no evidence that SCH prior to pregnancy is associated with adverse neurodevelopmental outcomes. It should be noted that only one study has examined whether treatment of SCH improves developmental outcomes (measured by IQ scored at age 3 years) and no significant differences were observed in women with SCH who were treated with levothyroxine versus those who were not.22
7. Does treatment of SCH improve miscarriage rates, live-birth rates, and/or clinical pregnancy rates?
Small randomized controlled studies of women undergoing infertility treatment and a few observational studies in the general population yield good evidence that levothyroxine treatment in women with SCH defined as TSH >4.0 mIU/L is associated with improvement in pregnancy, live birth, and miscarriage rates. There are no randomized trials assessing whether levothyroxine treatment in women with TSH levels between 2.5 and 4 mIU/L would yield similar benefits to those observed in women with TSH levels above 4 mIU/L.
8. Are thyroid antibodies associated with infertility or adverse reproductive outcomes?
There is good evidence that the thyroid autoimmunity, or the presence of TPO-Ab, is associated with miscarriage and fair evidence that it is associated with infertility. Treatment with levothyroxine may improve pregnancy outcomes especially if the TSH level is above 2.5 mIU/L.
9. Should there be universal screening for hypothyroidism in the first trimester of pregnancy?
Current evidence does not reveal a benefit of universal screening at this time. The American College of Obstetricians and Gynecologists does not recommend routine screening for hypothyroidism in pregnancy unless women have risk factors for thyroid disease, including a personal or family history of thyroid disease, physical findings or symptoms of goiter or hypothyroidism, type 1 diabetes mellitus, infertility, history of miscarriage or preterm delivery, and/or personal or family history of autoimmune disease.
The bottom line
SCH, defined as a TSH level greater than the upper limit of normal range (4.5−5.0 mIU/L)with normal FT4 levels, is associated with adverse reproductive outcomes including miscarriage, pregnancy complications, and delayed fetal neurodevelopment. Thyroid supplementation is beneficial; however, treatment has not been shown to improve long-term neurologic developmental outcomes in offspring. Data are limited on whether TSH values between 2.5 mIU/L and the upper range of normal are associated with adverse pregnancy outcomes and therefore treatment in this group remains controversial. Although available evidence is weak, there may be a benefit in some subgroups, and because risk is minimal, it may be reasonable to treat or to monitor levels and treat above nonpregnant and pregnancy ranges. There is fair evidence that thyroid autoimmunity (positive thyroid antibody) is associated with miscarriage and infertility. Levothyroxine therapy may improve pregnancy outcomes especially if the TSH level is above 2.5 mIU/L. While universal screening of thyroid function in pregnancy is not recommended, women at high risk for thyroid disease should be screened.23
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Pasquali R, Pelusi C, Genghini S, Cacciari M, Gambineri A. Obesity and reproductive disorders in women. Hum Reprod Update. 2003;9(4):359-372.
- Greisen S, Ledet T, Møller N, et al. Effects of leptin on basal and FSH stimulated steroidogenesis in human granulosa luteal cells. Acta Obstet Gynecol Scand. 2000;79(11):931-935.
- Rich-Edwards JW, Goldman MB, Willett WC, et al. Adolescent body mass index and infertility caused by ovulatory disorder. Am J Obstet Gynecol. 1994;171(1):171-177.
- Grodstein F, Goldman MB, Cramer DW. Body mass index and ovulatory infertility. Epidemiology. 1994;5(2):247-250.
- Gesink Law DC, Maclehose RF, Longnecker MP. Obesity and time to pregnancy. Hum Reprod. 2007;22(2):414-420.
- Jain A, Polotsky AJ, Rochester D, et al. Pulsatile luteinizing hormone amplitude and progesterone metabolite excretion are reduced in obese women. J Clin Endocrinol Metab. 2007;92(7):2468-2473.
- Lake JK, Power C, Cole TJ. Women's reproductive health: the role of body mass index in early and adult life. Int J Obes Relat Metab Disord. 1997;21(6):432-438.
- van der Steeg JW, Steures P, Eijkemans MJ, et al. Obesity affects spontaneous pregnancy chances in subfertile, ovulatory women. Hum Reprod. 2008;23(2):324-328.
- Koning AM, Mutsaerts MA, Kuchenbecker WK, et al. Complications and outcome of assisted reproduction technologies in overweight and obese women [Published correction appears in Hum Reprod. 2012;27(8):2570.] Hum Reprod. 2012;27(2):457-467.
- Rittenberg V, Seshadri S, Sunkara SK, Sobaleva S, Oteng-Ntim E, El-Toukhy T. Effect of body mass index on IVF treatment outcome: an updated systematic review and meta-analysis. Reprod Biomed Online. 2011;23(4):421-439.
- Moragianni VA, Jones SM, Ryley DA. The effect of body mass index on the outcomes of first assisted reproductive technology cycles. Fertil Steril. 2012;98(1):102-108.
- Petersen GL, Schmidt L, Pinborg A, Kamper-Jørgensen M. The influence of female and male body mass index on live births after assisted reproductive technology treatment: a nationwide register-based cohort study. Fertil Steril. 2013;99(6):1654-1662.
- Practice Committee of the American Society for Reproductive Medicine. Obesity and Reproduction: A committee opinion. Fertil Steril. 2015;104(5):1116-1126.
- Metwally M, Cutting R, Tipton A, Skull J, Ledger WL, Li TC. Effect of increased body mass index on oocyte and embryo quality in IVF patients. Reprod Biomed Online. 2007;15(5):532-538.
- Leary C, Leese HJ, Sturmey RG. Human embryos from overweight and obese women display phenotypic and metabolic abnormalities. Hum Reprod. 2015;30(1):122-132.
- Deugarte D, Deugarte C, Sahakian V. Surrogate obesity negatively impacts pregnancy rates in third-party reproduction. Fertil Steril. 2010;93(3):1008-1010.
- Rittenberg V, Seshadri S, Sunkara SK, Sobaleva S, Oteng-Ntim E, El-Toukhy T. Effect of body mass index on IVF treatment outcome: an updated systematic review and meta-analysis. Reprod Biomed Online. 2011;23(4):421-439.
- Stothard KJ, Tennant PWG, Bell R, Rankin J. Maternal overweight and obesity and the risk of congenital anomalies: a systematic review and meta-analysis. JAMA. 2009;301(6):636-650.
- Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87(2):489-499.
- Baloch Z, Carayon P, Conte-Devolx B, et al. Laboratory medicine practice guidelines. Laboratory support for the diagnosis and monitoring of thyroid disease. Thyroid. 2003;13(1):3-126.
- Pop VJ, Kuijpens JL, van Baar AL, et al. Low maternal free thyroxine concentrations during early pregnancy are associated with impaired psychomotor development in infancy. Clin Endocrinol (Oxf). 1999;50(2):149-155.
- Lazarus JH, Bestwick JP, Channon S, et al. Antenatal thyroid screening and childhood cognitive function. N Engl J Med. 2012;366(17):493-501.
- Practice Committee of the American Society for Reproductive Medicine. Subclinical hypothyroidism in the infertile female population: a guideline. Fertil Steril. 2015;104(3):545-553.

Dr. Adamson is Founder/Executive Chairman of Advanced Reproductive Care, Inc; Adjunct Clinical Professor at Stanford University; and Associate Clinical Professor at the University of California, San Francisco. He is also Medical Director, Assisted Reproductive Technologies Program, Palo Alto Medical Foundation Fertility Physicians of Northern California in Palo Alto and San Jose.

Dr. Abusief is a Board-Certified Specialist in Reproductive Endocrinology and Infertility and Chair, Department of Reproductive Endocrine Fertility at Palo Alto Medical Foundation Fertility Physicians of Northern California.
Dr. Adamson reports being a consultant to Abbvie, Bayer, and Ferring and that he has equity in ARC Fertility. Dr. Abusief reports no financial relationships relevant to this article.
Dr. Adamson is Founder/Executive Chairman of Advanced Reproductive Care, Inc; Adjunct Clinical Professor at Stanford University; and Associate Clinical Professor at the University of California, San Francisco. He is also Medical Director, Assisted Reproductive Technologies Program, Palo Alto Medical Foundation Fertility Physicians of Northern California in Palo Alto and San Jose.
Dr. Abusief is a Board-Certified Specialist in Reproductive Endocrinology and Infertility and Chair, Department of Reproductive Endocrine Fertility at Palo Alto Medical Foundation Fertility Physicians of Northern California.
Dr. Adamson reports being a consultant to Abbvie, Bayer, and Ferring and that he has equity in ARC Fertility. Dr. Abusief reports no financial relationships relevant to this article.
Dr. Adamson is Founder/Executive Chairman of Advanced Reproductive Care, Inc; Adjunct Clinical Professor at Stanford University; and Associate Clinical Professor at the University of California, San Francisco. He is also Medical Director, Assisted Reproductive Technologies Program, Palo Alto Medical Foundation Fertility Physicians of Northern California in Palo Alto and San Jose.
Dr. Abusief is a Board-Certified Specialist in Reproductive Endocrinology and Infertility and Chair, Department of Reproductive Endocrine Fertility at Palo Alto Medical Foundation Fertility Physicians of Northern California.
Dr. Adamson reports being a consultant to Abbvie, Bayer, and Ferring and that he has equity in ARC Fertility. Dr. Abusief reports no financial relationships relevant to this article.
Zika virus is a serious problem. Education and infection prevention are critical to effective management, and why we chose to include Zika virus as a topic for this year’s Update. We also discuss obesity’s effects on reproduction—a very relevant concern for all ObGyns and patients alike as about half of reproductive-age women are obese. Finally, subclinical hypothyroidism can present unique management challenges, such as determining when it is present and when treatment is indicated.
Read about counseling patients about Zika virus
Managing attempted pregnancy in the era of Zika virus
Oduyebo T, Igbinosa I, Petersen EE, et al. Update: interim guidance for health care providers caring for pregnant women with possible Zika virus exposure--United States, July 2016. MMWR Morb Mortal Wkly Rep. 2016;65(29):739-744.
Petersen EE, Meaney-Delman D, Neblett-Fanfair R, et al. Update: interim guidance for preconception counseling and prevention of sexual transmission of Zika virus for persons with possible Zika virus exposure--United States, September 2016. MMWR Morb Mortal Wkly Rep. 2016;65(39):1077-1081.
US Food and Drug Administration. Donor Screening Recommendations to Reduce the Risk of Transmission of Zika Virus by Human Cells, Tissues, and Cellular and Tissue-Based Products. http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Tissue/UCM488582.pdf. Published March 2016. Accessed January 12, 2017.
National Institutes of Health. Zika: Overview. https://www.nichd.nih.gov/health/topics/zika/Pages/default.aspx. Accessed January 12, 2017.
World Health Organization. Prevention of sexual transmission of Zika virus interim guidance. WHO reference number: WHO/ZIKV/MOC/16. 1 Rev. 3, September 6, 2016.
Zika Virus Guidance Task Force of the American Society for Reproductive Medicine. Rev. 13, September 2016.
Zika virus presents unique challenges to physicians managing the care of patients attempting pregnancy, with or without fertility treatment. Neonatal Zika virus infection sequelae only recently have been appreciated; microcephaly was associated with Zika virus in October 2015, followed by other neurologic conditions including brain abnormalities, neural tube defects, and eye abnormalities. Results of recent studies involving the US Zika Pregnancy Registry show that 6% of women with Zika at any time in pregnancy had affected babies, but 11% of those who contracted the disease in the first trimester were affected.
Diagnosis is difficult because symptoms are generally mild, with 80% of affected patients asymptomatic. Possible Zika virus exposure is defined as travel to or residence in an area of active Zika virus transmission, or sex without a condom with a partner who traveled to or lived in an area of active transmission. Much is unknown about the interval from exposure to symptoms. Testing availability is limited and variable, and much is unknown about sensitivity and specificity of direct viral RNA testing, appearance and disappearance of detectable immunoglobulin (Ig) M and IgG antibodies that affect false positive and false negative test results, duration of infectious phase, risk of transmission, and numerous other factors.
Positive serum viral testing likely indicates virus in semen or other bodily fluids, but a negative serum viral test cannot definitively preclude virus in other bodily fluids. Zika virus likely can be passed from any combination of semen and vaginal and cervical fluids, but validating tests for these fluids are not yet available. It is not known if sperm preparation and assisted reproductive technology (ART) procedures that minimize risk of HIV transmission are effective against Zika virus or whether or not cryopreservation can destroy the virus.
Pregnancy timing
The Centers for Disease Control and Prevention now recommends that all men with possible Zika virus exposure who are considering attempting pregnancy with their partner wait to get pregnant until at least 6 months after symptom onset (if symptomatic) or last possible Zika virus exposure (if asymptomatic). Women with possible Zika virus exposure are recommended to wait to get pregnant until at least 8 weeks after symptom onset (if symptomatic) or last possible Zika virus exposure (if asymptomatic).
Women and men with possible exposure to Zika virus but without clinical symptoms of illness should consider testing for Zika viral RNA within 2 weeks of suspected exposure and wait at least 8 weeks after the last date of exposure before being re-tested. If direct viral testing (using rRT-PCR) results initially are negative, ideally, antibody testing would be obtained, if available, at 8 weeks. However, no testing paradigm will absolutely guarantee lack of Zika virus infectivity.
Virus management problems are dramatically compounded in areas endemic for Zika. Women and men who have had Zika virus disease should wait at least 6 months after illness onset to attempt reproduction. The temporal relationship between the presence of viral RNA and infectivity is not known definitively, and so the absolute duration of time to wait before attempting pregnancy is unknown. Male and female partners who become infected should avoid all forms of intimate sexual conduct or use condoms for the same 6 months. There is no evidence Zika will cause congenital infection in pregnancies initiated after resolution of maternal Zika viremia. However, any testing performed at a time other than the time of treatment might not reflect true viral status, particularly in areas of active Zika virus transmission.
Prevention
Women and men, especially those residing in areas of active Zika virus transmission, should talk with their physicians regarding pregnancy plans and avoid mosquito bites using the usual precautions: avoid mosquito areas, drain standing water, use mosquito repellent containing DEET, and use mosquito netting. Some people have gone so far as to relocate to nonendemic areas.
Those contemplating pregnancy should be advised to consider what they would do if they become exposed to or have suspected or confirmed Zika virus during pregnancy. Additional considerations are gamete or embryo cryopreservation and quarantine until a subsequent rRT-PCR test result is negative in both the male and female and at least 8 weeks have passed from gamete collection.
Patient counseling essentials
Counsel patients considering reproduction about:
- Zika virus as a new reproductive hazard
- the significance of the hazard to the fetus if infected
- the areas of active transmission, and that they are constantly changing
- avoidance of Zika areas if possible
- methods of transmission through mosquito bites or sex
- avoidance of mosquito bites
- symptoms of Zika infection
- safe sex practices
- testing limitations and knowledge deficiency about Zika.
Not uncommonly, clinical situations require complex individualized management decisions regarding trade-offs of risks, especially in older patients with decreased ovarian reserve. Consultation with infectious disease and reproductive specialists should be obtained when complicated and consequential decisions have to be made.
All practitioners should inform their patients, especially those undergoing fertility treatments, about Zika, and develop language in their informed consent that conveys the gap in knowledge to these patients.
Read how obesity specifically affects reproduction in an adverse way
Obesity adversely affects reproduction, but how specifically?
Practice Committee of the American Society for Reproductive Medicine. Obesity and Reproduction: A committee opinion. Fertil Steril. 2015;104(5):1116-1126.
The prevalence of obesity has increased substantially over the past 2 decades. Almost two-thirds of women and three-fourths of men in the United States are overweight or obese (defined as a body mass index [BMI] ≥25 kg/m2 and BMI ≥30 kg/m2, respectively; TABLE). Nearly 50% of reproductive-age women are obese.
A disease of excess body fat and insulin resistance, obesity increases the risks of hypertension, diabetes, dyslipidemia, cardiovascular disease, sleep apnea, respiratory problems, and cancer as well as other serious health problems. While not all individuals with obesity will have infertility, obesity is associated with impaired reproduction in both women and men, adverse obstetric outcomes, and health problems in offspring. The American Society for Reproductive Medicine (ASRM) reviewed this important issue in a recent practice committee opinion.
Menstrual cycle and ovulatory dysfunction
Menstrual cycle abnormalities are more common in women with obesity. Elevated levels of insulin in obese women suppress sex hormone−binding globulin (SHBG) which in turn reduces gonadotropin secretion due to increased production of estrogen from conversion of androgens by adipose aromatase.1 Adipose tissue produces adipokines, which directly can suppress ovarian function.2
Ovulatory dysfunction is common among obese women; the relative risk of such dysfunction is 3.1 (95% confidence interval [CI], 2.2−4.4) among women with BMI levels >27 kg/m2 versus BMI levels 20.0 to 24.9 kg/m2.3,4 Obesity decreases fecundity even in women with normal menstrual cycles.5 This may in part be due to altered ovulatory dynamics with reduced early follicular luteinizing hormone pulse amplitude accompanied by prolonged folliculogenesis and reduced luteal progesterone levels.6
Compared with normal-weight women, obese women have a lower chance of conception within 1 year of stopping contraception; about 66% of obese women conceive within 1 year of stopping contraception, compared with about 81% of women with normal weight.7 Results of a Dutch study of 3,029 women with regular ovulation, at least one patent tube, and a partner with a normal semen analysis indicated a direct correlation between obesity and delayed conception, with a 4% lower spontaneous pregnancy rate per kg/m2 increase in women with a BMI >29 kg/m2 versus a BMI of 21 to 29 kg/m2 (hazard ratio, 0.96; 95% CI, 0.91−0.99).8
Assisted reproduction
Assisted reproduction in women with obesity is associated with lower success rates than in women with normal weight. A systematic review of 27 in vitro fertilization (IVF) studies (23 of which were retrospective) reveals 10% lower live-birth rate in overweight (BMI >25 kg/m2) versus normal-weight women (BMI <25 kg/m2) undergoing IVF (odds ratio [OR], 0.90; 95% CI, 0.82−1.0).9 Data from a meta-analysis of 33 IVF studies, including 47,967 cycles, show that, compared with women with a BMI <25 kg/m2, overweight or obese women have significantly reduced rates of clinical pregnancy (relative risk [RR], 0.90; P<.0001) and live birth (RR, 0.84; P = .0002).10
Results of a retrospective study of 4,609 women undergoing first IVF or IVF/intracytoplasmic sperm injection cycles revealed impaired embryo implantation (controlling for embryo quality and transfer day), reducing the age-adjusted odds of live birth in a BMI-dependent manner by 37% (BMI, 30.0−34.9 kg/m2), 61% (BMI, 35.0−39.9 kg/m2), and 68% (BMI, >40 kg/m2) compared with women with a BMI of 18.5 to 24.9 kg/m2.11 In a study of 12,566 Danish couples undergoing assisted reproduction, overweight and obese ovulatory women had a 12% (95% CI, 0.79−0.99) and 25% (95% CI, 0.63−0.90) reduction in IVF-related live birth rate, respectively (referent BMI, 18.5−24.9 kg/m2), with a 2% (95% CI, 0.97−0.99) decrease in live-birth rate for every one-unit increase in BMI.12 Putative mechanisms for these findings include altered oocyte morphology and reduced fertilization in eggs from obese women,13 and impaired embryo quality in women less than age 35.14 Oocytes from women with a BMI >25 kg/m2 are smaller and less likely to complete development postfertilization, with embryos arrested prior to blastulation containing more triglyceride than those forming blastocysts.15
Blastocysts developed from oocytes of high-BMI women are smaller, contain fewer cells and have a higher content of triglycerides, lower glucose consumption, and altered amino acid metabolism compared with embryos of normal-weight women (BMI <24.9 kg/m2).15 Obesity may alter endometrial receptivity during IVF given the finding that third-party surrogate women with a BMI >35 kg/m2 have a lower live-birth rate (25%) compared with women with a BMI <35 kg/m2 (49%; P<.05).16
Pregnancy outcomes
Obesity is linked to an increased risk of miscarriage. Results of a meta-analysis of 33 IVF studies including 47,967 cycles indicated that overweight or obese women have a higher rate of miscarriage (RR, 1.31; P<.0001) than normal-weight women (BMI <25 kg/m2).17 Maternal and perinatal morbid obesity are strongly associated with obstetric and perinatal complications, including gestational diabetes, hypertension, preeclampsia, preterm delivery, shoulder dystocia, fetal distress, early neonatal death, and small- as well as large-for-gestational age infants.
Obese women who conceive by IVF are at increased risk for preeclampsia, gestational diabetes, preterm delivery, and cesarean delivery.13 Authors of a meta-analysis of 18 observational studies concluded that obese mothers were at increased odds of pregnancies affected by such birth defects as neural tube defects, cardiovascular anomalies, and cleft lip and palate, among others.18
In addition to being the cause of these fetal abnormalities, maternal metabolic dysfunction is linked to promoting obesity in offspring, thereby perpetuating a cycle of obesity and adverse health outcomes that include an increased risk of premature death in adult offspring in subsequent generations.13
Treatment for obesity
Lifestyle modification is the first-line treatment for obesity.
Pre-fertility therapy and pregnancy goals. Targets for pregnancy should include:
- preconception weight loss to a BMI of 35 kg/m2
- prevention of excess weight gain in pregnancy
- long-term reduction in weight.
For all obese individuals, lifestyle modifications should include a weight loss of 7% of body weight and increased physical activity to at least 150 minutes of moderate activity, such as walking, per week. Calorie restriction should be emphasized. A 500 to 1,000 kcal/day decrease from usual dietary intake is expected to result in a 1- to 2-lb weight loss per week. A low-calorie diet of 1,000 to 1,200 kcal/day can lead to an average 10% decrease in total body weight over 6 months.
Adjunct supervised medical therapy or bariatric surgery can play an important role in successful weight loss prepregnancy but are not appropriate for women actively attempting conception. Importantly, pregnancy should be deferred for a minimum of 1 year after bariatric surgery. The decision to postpone pregnancy to achieve weight loss must be balanced against the risk of declining fertility with advancing age of the woman.
Read about when to treat subclinical hypothyroidism
Optimal management of subclinical hypothyroidism in women with infertility
Practice Committee of the American Society for Reproductive Medicine. Subclinical hypothyroidism in the infertile female population: a guideline. Fertil Steril. 2015;104(3):545-553.
Thyroid disorders long have been associated with the potential for adverse reproductive outcomes. While overt hypothyroidism has been linked to infertility, increased miscarriage risk, and poor maternal and fetal outcomes, controversy has existed regarding the association between subclinical hypothyroidism (SCH) and reproductive problems. The ASRM recently published a guideline on the role of SCH in the infertile female population.
How is subclinical hypothyroidism defined?
SCH is classically defined as a thyrotropin (TSH) level above the upper limit of normal range (4.5−5.0 mIU/L) with normal free thyroxine (FT4) levels. The National Health and Nutrition Examination Survey (NHANES III) population has been used to establish normative data for TSH for a disease-free population. These include a median serum level for TSH of 1.5 mIU/L, with the corresponding 2.5 and 97.5 percentiles of 0.41 and 6.10, respectively.19 Data from the National Academy of Clinical Biochemistry, however, reveal that 95% of individuals without evidence of thyroid disease have a TSH level <2.5 mIU/L, and that the normal reference range is skewed to the right.20 Adjusting the upper limit of the normal range to 2.5 mIU/L would result in an additional 11.8% to 14.2% of the United States population (22 to 28 million individuals) being diagnosed with hypothyroidism.
This information raises several important questions.
1. Should nonpregnant women be treated for SCH?
No. There is no benefit from the standpoint of lipid profile or alteration of cardiovascular risk in the treatment of TSH levels between 5 and 10 mIU/L and, therefore, treatment of individuals with TSH <5 mIU/L is questionable. Furthermore, the risk of overtreatment resulting in bone loss is a concern. The Endocrine Society does not recommend changing the current normal TSH range for nonpregnant women.
2. What are normal TSH levels in pregnant women?
Because human chorionic gonadotropin (hCG) can bind to and affect the TSH receptor, thereby influencing TSH values, the normal range for TSH is modified in pregnancy. The Endocrine Society recommends the following pregnancy trimester guidelines for TSH levels: 2.5 mIU/L is the recommended upper limit of normal in the first trimester, 3.0 mIU/L in the second trimester, and 3.5 mIU/L in the third trimester.
3. Is untreated SCH associated with miscarriage?
There is fair evidence that SCH, defined as a TSH level >4 mIU/L during pregnancy, is associated with miscarriage, but there is insufficient evidence that TSH levels between 2.5 and 4 mIU/L are associated with miscarriage.
4. Is untreated SCH associated with infertility?
Limited data are available to assess the effect of SCH on infertility. While a few studies show an association between SCH on unexplained infertility and ovulatory disorders, SCH does not appear to be increased in other causes of infertility.
5. Is SCH associated with adverse obstetric outcomes?
Available data reveal that SCH with TSH levels outside the normal pregnancy range are associated with an increased risk of such obstetric complications as placental abruption, preterm birth, fetal death, and preterm premature rupture of membranes (PPROM). However, it is unclear if prepregnancy TSH levels between 2.5 and 4 mIU/L are associated with adverse obstetric outcomes.
6. Does untreated SCH affect developmental outcomes in children?
The fetus is solely dependent on maternal thyroid hormone in early pregnancy because the fetal thyroid does not produce thyroid hormone before 10 to 13 weeks of gestation. Significant evidence has associated untreated maternal hypothyroidism with delayed fetal neurologic development, impaired school performance, and lower intelligence quotient (IQ) among offspring.21 There is fair evidence that SCH diagnosed in pregnancy is associated with adverse neurologic development. There is no evidence that SCH prior to pregnancy is associated with adverse neurodevelopmental outcomes. It should be noted that only one study has examined whether treatment of SCH improves developmental outcomes (measured by IQ scored at age 3 years) and no significant differences were observed in women with SCH who were treated with levothyroxine versus those who were not.22
7. Does treatment of SCH improve miscarriage rates, live-birth rates, and/or clinical pregnancy rates?
Small randomized controlled studies of women undergoing infertility treatment and a few observational studies in the general population yield good evidence that levothyroxine treatment in women with SCH defined as TSH >4.0 mIU/L is associated with improvement in pregnancy, live birth, and miscarriage rates. There are no randomized trials assessing whether levothyroxine treatment in women with TSH levels between 2.5 and 4 mIU/L would yield similar benefits to those observed in women with TSH levels above 4 mIU/L.
8. Are thyroid antibodies associated with infertility or adverse reproductive outcomes?
There is good evidence that the thyroid autoimmunity, or the presence of TPO-Ab, is associated with miscarriage and fair evidence that it is associated with infertility. Treatment with levothyroxine may improve pregnancy outcomes especially if the TSH level is above 2.5 mIU/L.
9. Should there be universal screening for hypothyroidism in the first trimester of pregnancy?
Current evidence does not reveal a benefit of universal screening at this time. The American College of Obstetricians and Gynecologists does not recommend routine screening for hypothyroidism in pregnancy unless women have risk factors for thyroid disease, including a personal or family history of thyroid disease, physical findings or symptoms of goiter or hypothyroidism, type 1 diabetes mellitus, infertility, history of miscarriage or preterm delivery, and/or personal or family history of autoimmune disease.
The bottom line
SCH, defined as a TSH level greater than the upper limit of normal range (4.5−5.0 mIU/L)with normal FT4 levels, is associated with adverse reproductive outcomes including miscarriage, pregnancy complications, and delayed fetal neurodevelopment. Thyroid supplementation is beneficial; however, treatment has not been shown to improve long-term neurologic developmental outcomes in offspring. Data are limited on whether TSH values between 2.5 mIU/L and the upper range of normal are associated with adverse pregnancy outcomes and therefore treatment in this group remains controversial. Although available evidence is weak, there may be a benefit in some subgroups, and because risk is minimal, it may be reasonable to treat or to monitor levels and treat above nonpregnant and pregnancy ranges. There is fair evidence that thyroid autoimmunity (positive thyroid antibody) is associated with miscarriage and infertility. Levothyroxine therapy may improve pregnancy outcomes especially if the TSH level is above 2.5 mIU/L. While universal screening of thyroid function in pregnancy is not recommended, women at high risk for thyroid disease should be screened.23
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Zika virus is a serious problem. Education and infection prevention are critical to effective management, and why we chose to include Zika virus as a topic for this year’s Update. We also discuss obesity’s effects on reproduction—a very relevant concern for all ObGyns and patients alike as about half of reproductive-age women are obese. Finally, subclinical hypothyroidism can present unique management challenges, such as determining when it is present and when treatment is indicated.
Read about counseling patients about Zika virus
Managing attempted pregnancy in the era of Zika virus
Oduyebo T, Igbinosa I, Petersen EE, et al. Update: interim guidance for health care providers caring for pregnant women with possible Zika virus exposure--United States, July 2016. MMWR Morb Mortal Wkly Rep. 2016;65(29):739-744.
Petersen EE, Meaney-Delman D, Neblett-Fanfair R, et al. Update: interim guidance for preconception counseling and prevention of sexual transmission of Zika virus for persons with possible Zika virus exposure--United States, September 2016. MMWR Morb Mortal Wkly Rep. 2016;65(39):1077-1081.
US Food and Drug Administration. Donor Screening Recommendations to Reduce the Risk of Transmission of Zika Virus by Human Cells, Tissues, and Cellular and Tissue-Based Products. http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Tissue/UCM488582.pdf. Published March 2016. Accessed January 12, 2017.
National Institutes of Health. Zika: Overview. https://www.nichd.nih.gov/health/topics/zika/Pages/default.aspx. Accessed January 12, 2017.
World Health Organization. Prevention of sexual transmission of Zika virus interim guidance. WHO reference number: WHO/ZIKV/MOC/16. 1 Rev. 3, September 6, 2016.
Zika Virus Guidance Task Force of the American Society for Reproductive Medicine. Rev. 13, September 2016.
Zika virus presents unique challenges to physicians managing the care of patients attempting pregnancy, with or without fertility treatment. Neonatal Zika virus infection sequelae only recently have been appreciated; microcephaly was associated with Zika virus in October 2015, followed by other neurologic conditions including brain abnormalities, neural tube defects, and eye abnormalities. Results of recent studies involving the US Zika Pregnancy Registry show that 6% of women with Zika at any time in pregnancy had affected babies, but 11% of those who contracted the disease in the first trimester were affected.
Diagnosis is difficult because symptoms are generally mild, with 80% of affected patients asymptomatic. Possible Zika virus exposure is defined as travel to or residence in an area of active Zika virus transmission, or sex without a condom with a partner who traveled to or lived in an area of active transmission. Much is unknown about the interval from exposure to symptoms. Testing availability is limited and variable, and much is unknown about sensitivity and specificity of direct viral RNA testing, appearance and disappearance of detectable immunoglobulin (Ig) M and IgG antibodies that affect false positive and false negative test results, duration of infectious phase, risk of transmission, and numerous other factors.
Positive serum viral testing likely indicates virus in semen or other bodily fluids, but a negative serum viral test cannot definitively preclude virus in other bodily fluids. Zika virus likely can be passed from any combination of semen and vaginal and cervical fluids, but validating tests for these fluids are not yet available. It is not known if sperm preparation and assisted reproductive technology (ART) procedures that minimize risk of HIV transmission are effective against Zika virus or whether or not cryopreservation can destroy the virus.
Pregnancy timing
The Centers for Disease Control and Prevention now recommends that all men with possible Zika virus exposure who are considering attempting pregnancy with their partner wait to get pregnant until at least 6 months after symptom onset (if symptomatic) or last possible Zika virus exposure (if asymptomatic). Women with possible Zika virus exposure are recommended to wait to get pregnant until at least 8 weeks after symptom onset (if symptomatic) or last possible Zika virus exposure (if asymptomatic).
Women and men with possible exposure to Zika virus but without clinical symptoms of illness should consider testing for Zika viral RNA within 2 weeks of suspected exposure and wait at least 8 weeks after the last date of exposure before being re-tested. If direct viral testing (using rRT-PCR) results initially are negative, ideally, antibody testing would be obtained, if available, at 8 weeks. However, no testing paradigm will absolutely guarantee lack of Zika virus infectivity.
Virus management problems are dramatically compounded in areas endemic for Zika. Women and men who have had Zika virus disease should wait at least 6 months after illness onset to attempt reproduction. The temporal relationship between the presence of viral RNA and infectivity is not known definitively, and so the absolute duration of time to wait before attempting pregnancy is unknown. Male and female partners who become infected should avoid all forms of intimate sexual conduct or use condoms for the same 6 months. There is no evidence Zika will cause congenital infection in pregnancies initiated after resolution of maternal Zika viremia. However, any testing performed at a time other than the time of treatment might not reflect true viral status, particularly in areas of active Zika virus transmission.
Prevention
Women and men, especially those residing in areas of active Zika virus transmission, should talk with their physicians regarding pregnancy plans and avoid mosquito bites using the usual precautions: avoid mosquito areas, drain standing water, use mosquito repellent containing DEET, and use mosquito netting. Some people have gone so far as to relocate to nonendemic areas.
Those contemplating pregnancy should be advised to consider what they would do if they become exposed to or have suspected or confirmed Zika virus during pregnancy. Additional considerations are gamete or embryo cryopreservation and quarantine until a subsequent rRT-PCR test result is negative in both the male and female and at least 8 weeks have passed from gamete collection.
Patient counseling essentials
Counsel patients considering reproduction about:
- Zika virus as a new reproductive hazard
- the significance of the hazard to the fetus if infected
- the areas of active transmission, and that they are constantly changing
- avoidance of Zika areas if possible
- methods of transmission through mosquito bites or sex
- avoidance of mosquito bites
- symptoms of Zika infection
- safe sex practices
- testing limitations and knowledge deficiency about Zika.
Not uncommonly, clinical situations require complex individualized management decisions regarding trade-offs of risks, especially in older patients with decreased ovarian reserve. Consultation with infectious disease and reproductive specialists should be obtained when complicated and consequential decisions have to be made.
All practitioners should inform their patients, especially those undergoing fertility treatments, about Zika, and develop language in their informed consent that conveys the gap in knowledge to these patients.
Read how obesity specifically affects reproduction in an adverse way
Obesity adversely affects reproduction, but how specifically?
Practice Committee of the American Society for Reproductive Medicine. Obesity and Reproduction: A committee opinion. Fertil Steril. 2015;104(5):1116-1126.
The prevalence of obesity has increased substantially over the past 2 decades. Almost two-thirds of women and three-fourths of men in the United States are overweight or obese (defined as a body mass index [BMI] ≥25 kg/m2 and BMI ≥30 kg/m2, respectively; TABLE). Nearly 50% of reproductive-age women are obese.
A disease of excess body fat and insulin resistance, obesity increases the risks of hypertension, diabetes, dyslipidemia, cardiovascular disease, sleep apnea, respiratory problems, and cancer as well as other serious health problems. While not all individuals with obesity will have infertility, obesity is associated with impaired reproduction in both women and men, adverse obstetric outcomes, and health problems in offspring. The American Society for Reproductive Medicine (ASRM) reviewed this important issue in a recent practice committee opinion.
Menstrual cycle and ovulatory dysfunction
Menstrual cycle abnormalities are more common in women with obesity. Elevated levels of insulin in obese women suppress sex hormone−binding globulin (SHBG) which in turn reduces gonadotropin secretion due to increased production of estrogen from conversion of androgens by adipose aromatase.1 Adipose tissue produces adipokines, which directly can suppress ovarian function.2
Ovulatory dysfunction is common among obese women; the relative risk of such dysfunction is 3.1 (95% confidence interval [CI], 2.2−4.4) among women with BMI levels >27 kg/m2 versus BMI levels 20.0 to 24.9 kg/m2.3,4 Obesity decreases fecundity even in women with normal menstrual cycles.5 This may in part be due to altered ovulatory dynamics with reduced early follicular luteinizing hormone pulse amplitude accompanied by prolonged folliculogenesis and reduced luteal progesterone levels.6
Compared with normal-weight women, obese women have a lower chance of conception within 1 year of stopping contraception; about 66% of obese women conceive within 1 year of stopping contraception, compared with about 81% of women with normal weight.7 Results of a Dutch study of 3,029 women with regular ovulation, at least one patent tube, and a partner with a normal semen analysis indicated a direct correlation between obesity and delayed conception, with a 4% lower spontaneous pregnancy rate per kg/m2 increase in women with a BMI >29 kg/m2 versus a BMI of 21 to 29 kg/m2 (hazard ratio, 0.96; 95% CI, 0.91−0.99).8
Assisted reproduction
Assisted reproduction in women with obesity is associated with lower success rates than in women with normal weight. A systematic review of 27 in vitro fertilization (IVF) studies (23 of which were retrospective) reveals 10% lower live-birth rate in overweight (BMI >25 kg/m2) versus normal-weight women (BMI <25 kg/m2) undergoing IVF (odds ratio [OR], 0.90; 95% CI, 0.82−1.0).9 Data from a meta-analysis of 33 IVF studies, including 47,967 cycles, show that, compared with women with a BMI <25 kg/m2, overweight or obese women have significantly reduced rates of clinical pregnancy (relative risk [RR], 0.90; P<.0001) and live birth (RR, 0.84; P = .0002).10
Results of a retrospective study of 4,609 women undergoing first IVF or IVF/intracytoplasmic sperm injection cycles revealed impaired embryo implantation (controlling for embryo quality and transfer day), reducing the age-adjusted odds of live birth in a BMI-dependent manner by 37% (BMI, 30.0−34.9 kg/m2), 61% (BMI, 35.0−39.9 kg/m2), and 68% (BMI, >40 kg/m2) compared with women with a BMI of 18.5 to 24.9 kg/m2.11 In a study of 12,566 Danish couples undergoing assisted reproduction, overweight and obese ovulatory women had a 12% (95% CI, 0.79−0.99) and 25% (95% CI, 0.63−0.90) reduction in IVF-related live birth rate, respectively (referent BMI, 18.5−24.9 kg/m2), with a 2% (95% CI, 0.97−0.99) decrease in live-birth rate for every one-unit increase in BMI.12 Putative mechanisms for these findings include altered oocyte morphology and reduced fertilization in eggs from obese women,13 and impaired embryo quality in women less than age 35.14 Oocytes from women with a BMI >25 kg/m2 are smaller and less likely to complete development postfertilization, with embryos arrested prior to blastulation containing more triglyceride than those forming blastocysts.15
Blastocysts developed from oocytes of high-BMI women are smaller, contain fewer cells and have a higher content of triglycerides, lower glucose consumption, and altered amino acid metabolism compared with embryos of normal-weight women (BMI <24.9 kg/m2).15 Obesity may alter endometrial receptivity during IVF given the finding that third-party surrogate women with a BMI >35 kg/m2 have a lower live-birth rate (25%) compared with women with a BMI <35 kg/m2 (49%; P<.05).16
Pregnancy outcomes
Obesity is linked to an increased risk of miscarriage. Results of a meta-analysis of 33 IVF studies including 47,967 cycles indicated that overweight or obese women have a higher rate of miscarriage (RR, 1.31; P<.0001) than normal-weight women (BMI <25 kg/m2).17 Maternal and perinatal morbid obesity are strongly associated with obstetric and perinatal complications, including gestational diabetes, hypertension, preeclampsia, preterm delivery, shoulder dystocia, fetal distress, early neonatal death, and small- as well as large-for-gestational age infants.
Obese women who conceive by IVF are at increased risk for preeclampsia, gestational diabetes, preterm delivery, and cesarean delivery.13 Authors of a meta-analysis of 18 observational studies concluded that obese mothers were at increased odds of pregnancies affected by such birth defects as neural tube defects, cardiovascular anomalies, and cleft lip and palate, among others.18
In addition to being the cause of these fetal abnormalities, maternal metabolic dysfunction is linked to promoting obesity in offspring, thereby perpetuating a cycle of obesity and adverse health outcomes that include an increased risk of premature death in adult offspring in subsequent generations.13
Treatment for obesity
Lifestyle modification is the first-line treatment for obesity.
Pre-fertility therapy and pregnancy goals. Targets for pregnancy should include:
- preconception weight loss to a BMI of 35 kg/m2
- prevention of excess weight gain in pregnancy
- long-term reduction in weight.
For all obese individuals, lifestyle modifications should include a weight loss of 7% of body weight and increased physical activity to at least 150 minutes of moderate activity, such as walking, per week. Calorie restriction should be emphasized. A 500 to 1,000 kcal/day decrease from usual dietary intake is expected to result in a 1- to 2-lb weight loss per week. A low-calorie diet of 1,000 to 1,200 kcal/day can lead to an average 10% decrease in total body weight over 6 months.
Adjunct supervised medical therapy or bariatric surgery can play an important role in successful weight loss prepregnancy but are not appropriate for women actively attempting conception. Importantly, pregnancy should be deferred for a minimum of 1 year after bariatric surgery. The decision to postpone pregnancy to achieve weight loss must be balanced against the risk of declining fertility with advancing age of the woman.
Read about when to treat subclinical hypothyroidism
Optimal management of subclinical hypothyroidism in women with infertility
Practice Committee of the American Society for Reproductive Medicine. Subclinical hypothyroidism in the infertile female population: a guideline. Fertil Steril. 2015;104(3):545-553.
Thyroid disorders long have been associated with the potential for adverse reproductive outcomes. While overt hypothyroidism has been linked to infertility, increased miscarriage risk, and poor maternal and fetal outcomes, controversy has existed regarding the association between subclinical hypothyroidism (SCH) and reproductive problems. The ASRM recently published a guideline on the role of SCH in the infertile female population.
How is subclinical hypothyroidism defined?
SCH is classically defined as a thyrotropin (TSH) level above the upper limit of normal range (4.5−5.0 mIU/L) with normal free thyroxine (FT4) levels. The National Health and Nutrition Examination Survey (NHANES III) population has been used to establish normative data for TSH for a disease-free population. These include a median serum level for TSH of 1.5 mIU/L, with the corresponding 2.5 and 97.5 percentiles of 0.41 and 6.10, respectively.19 Data from the National Academy of Clinical Biochemistry, however, reveal that 95% of individuals without evidence of thyroid disease have a TSH level <2.5 mIU/L, and that the normal reference range is skewed to the right.20 Adjusting the upper limit of the normal range to 2.5 mIU/L would result in an additional 11.8% to 14.2% of the United States population (22 to 28 million individuals) being diagnosed with hypothyroidism.
This information raises several important questions.
1. Should nonpregnant women be treated for SCH?
No. There is no benefit from the standpoint of lipid profile or alteration of cardiovascular risk in the treatment of TSH levels between 5 and 10 mIU/L and, therefore, treatment of individuals with TSH <5 mIU/L is questionable. Furthermore, the risk of overtreatment resulting in bone loss is a concern. The Endocrine Society does not recommend changing the current normal TSH range for nonpregnant women.
2. What are normal TSH levels in pregnant women?
Because human chorionic gonadotropin (hCG) can bind to and affect the TSH receptor, thereby influencing TSH values, the normal range for TSH is modified in pregnancy. The Endocrine Society recommends the following pregnancy trimester guidelines for TSH levels: 2.5 mIU/L is the recommended upper limit of normal in the first trimester, 3.0 mIU/L in the second trimester, and 3.5 mIU/L in the third trimester.
3. Is untreated SCH associated with miscarriage?
There is fair evidence that SCH, defined as a TSH level >4 mIU/L during pregnancy, is associated with miscarriage, but there is insufficient evidence that TSH levels between 2.5 and 4 mIU/L are associated with miscarriage.
4. Is untreated SCH associated with infertility?
Limited data are available to assess the effect of SCH on infertility. While a few studies show an association between SCH on unexplained infertility and ovulatory disorders, SCH does not appear to be increased in other causes of infertility.
5. Is SCH associated with adverse obstetric outcomes?
Available data reveal that SCH with TSH levels outside the normal pregnancy range are associated with an increased risk of such obstetric complications as placental abruption, preterm birth, fetal death, and preterm premature rupture of membranes (PPROM). However, it is unclear if prepregnancy TSH levels between 2.5 and 4 mIU/L are associated with adverse obstetric outcomes.
6. Does untreated SCH affect developmental outcomes in children?
The fetus is solely dependent on maternal thyroid hormone in early pregnancy because the fetal thyroid does not produce thyroid hormone before 10 to 13 weeks of gestation. Significant evidence has associated untreated maternal hypothyroidism with delayed fetal neurologic development, impaired school performance, and lower intelligence quotient (IQ) among offspring.21 There is fair evidence that SCH diagnosed in pregnancy is associated with adverse neurologic development. There is no evidence that SCH prior to pregnancy is associated with adverse neurodevelopmental outcomes. It should be noted that only one study has examined whether treatment of SCH improves developmental outcomes (measured by IQ scored at age 3 years) and no significant differences were observed in women with SCH who were treated with levothyroxine versus those who were not.22
7. Does treatment of SCH improve miscarriage rates, live-birth rates, and/or clinical pregnancy rates?
Small randomized controlled studies of women undergoing infertility treatment and a few observational studies in the general population yield good evidence that levothyroxine treatment in women with SCH defined as TSH >4.0 mIU/L is associated with improvement in pregnancy, live birth, and miscarriage rates. There are no randomized trials assessing whether levothyroxine treatment in women with TSH levels between 2.5 and 4 mIU/L would yield similar benefits to those observed in women with TSH levels above 4 mIU/L.
8. Are thyroid antibodies associated with infertility or adverse reproductive outcomes?
There is good evidence that the thyroid autoimmunity, or the presence of TPO-Ab, is associated with miscarriage and fair evidence that it is associated with infertility. Treatment with levothyroxine may improve pregnancy outcomes especially if the TSH level is above 2.5 mIU/L.
9. Should there be universal screening for hypothyroidism in the first trimester of pregnancy?
Current evidence does not reveal a benefit of universal screening at this time. The American College of Obstetricians and Gynecologists does not recommend routine screening for hypothyroidism in pregnancy unless women have risk factors for thyroid disease, including a personal or family history of thyroid disease, physical findings or symptoms of goiter or hypothyroidism, type 1 diabetes mellitus, infertility, history of miscarriage or preterm delivery, and/or personal or family history of autoimmune disease.
The bottom line
SCH, defined as a TSH level greater than the upper limit of normal range (4.5−5.0 mIU/L)with normal FT4 levels, is associated with adverse reproductive outcomes including miscarriage, pregnancy complications, and delayed fetal neurodevelopment. Thyroid supplementation is beneficial; however, treatment has not been shown to improve long-term neurologic developmental outcomes in offspring. Data are limited on whether TSH values between 2.5 mIU/L and the upper range of normal are associated with adverse pregnancy outcomes and therefore treatment in this group remains controversial. Although available evidence is weak, there may be a benefit in some subgroups, and because risk is minimal, it may be reasonable to treat or to monitor levels and treat above nonpregnant and pregnancy ranges. There is fair evidence that thyroid autoimmunity (positive thyroid antibody) is associated with miscarriage and infertility. Levothyroxine therapy may improve pregnancy outcomes especially if the TSH level is above 2.5 mIU/L. While universal screening of thyroid function in pregnancy is not recommended, women at high risk for thyroid disease should be screened.23
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Pasquali R, Pelusi C, Genghini S, Cacciari M, Gambineri A. Obesity and reproductive disorders in women. Hum Reprod Update. 2003;9(4):359-372.
- Greisen S, Ledet T, Møller N, et al. Effects of leptin on basal and FSH stimulated steroidogenesis in human granulosa luteal cells. Acta Obstet Gynecol Scand. 2000;79(11):931-935.
- Rich-Edwards JW, Goldman MB, Willett WC, et al. Adolescent body mass index and infertility caused by ovulatory disorder. Am J Obstet Gynecol. 1994;171(1):171-177.
- Grodstein F, Goldman MB, Cramer DW. Body mass index and ovulatory infertility. Epidemiology. 1994;5(2):247-250.
- Gesink Law DC, Maclehose RF, Longnecker MP. Obesity and time to pregnancy. Hum Reprod. 2007;22(2):414-420.
- Jain A, Polotsky AJ, Rochester D, et al. Pulsatile luteinizing hormone amplitude and progesterone metabolite excretion are reduced in obese women. J Clin Endocrinol Metab. 2007;92(7):2468-2473.
- Lake JK, Power C, Cole TJ. Women's reproductive health: the role of body mass index in early and adult life. Int J Obes Relat Metab Disord. 1997;21(6):432-438.
- van der Steeg JW, Steures P, Eijkemans MJ, et al. Obesity affects spontaneous pregnancy chances in subfertile, ovulatory women. Hum Reprod. 2008;23(2):324-328.
- Koning AM, Mutsaerts MA, Kuchenbecker WK, et al. Complications and outcome of assisted reproduction technologies in overweight and obese women [Published correction appears in Hum Reprod. 2012;27(8):2570.] Hum Reprod. 2012;27(2):457-467.
- Rittenberg V, Seshadri S, Sunkara SK, Sobaleva S, Oteng-Ntim E, El-Toukhy T. Effect of body mass index on IVF treatment outcome: an updated systematic review and meta-analysis. Reprod Biomed Online. 2011;23(4):421-439.
- Moragianni VA, Jones SM, Ryley DA. The effect of body mass index on the outcomes of first assisted reproductive technology cycles. Fertil Steril. 2012;98(1):102-108.
- Petersen GL, Schmidt L, Pinborg A, Kamper-Jørgensen M. The influence of female and male body mass index on live births after assisted reproductive technology treatment: a nationwide register-based cohort study. Fertil Steril. 2013;99(6):1654-1662.
- Practice Committee of the American Society for Reproductive Medicine. Obesity and Reproduction: A committee opinion. Fertil Steril. 2015;104(5):1116-1126.
- Metwally M, Cutting R, Tipton A, Skull J, Ledger WL, Li TC. Effect of increased body mass index on oocyte and embryo quality in IVF patients. Reprod Biomed Online. 2007;15(5):532-538.
- Leary C, Leese HJ, Sturmey RG. Human embryos from overweight and obese women display phenotypic and metabolic abnormalities. Hum Reprod. 2015;30(1):122-132.
- Deugarte D, Deugarte C, Sahakian V. Surrogate obesity negatively impacts pregnancy rates in third-party reproduction. Fertil Steril. 2010;93(3):1008-1010.
- Rittenberg V, Seshadri S, Sunkara SK, Sobaleva S, Oteng-Ntim E, El-Toukhy T. Effect of body mass index on IVF treatment outcome: an updated systematic review and meta-analysis. Reprod Biomed Online. 2011;23(4):421-439.
- Stothard KJ, Tennant PWG, Bell R, Rankin J. Maternal overweight and obesity and the risk of congenital anomalies: a systematic review and meta-analysis. JAMA. 2009;301(6):636-650.
- Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87(2):489-499.
- Baloch Z, Carayon P, Conte-Devolx B, et al. Laboratory medicine practice guidelines. Laboratory support for the diagnosis and monitoring of thyroid disease. Thyroid. 2003;13(1):3-126.
- Pop VJ, Kuijpens JL, van Baar AL, et al. Low maternal free thyroxine concentrations during early pregnancy are associated with impaired psychomotor development in infancy. Clin Endocrinol (Oxf). 1999;50(2):149-155.
- Lazarus JH, Bestwick JP, Channon S, et al. Antenatal thyroid screening and childhood cognitive function. N Engl J Med. 2012;366(17):493-501.
- Practice Committee of the American Society for Reproductive Medicine. Subclinical hypothyroidism in the infertile female population: a guideline. Fertil Steril. 2015;104(3):545-553.
- Pasquali R, Pelusi C, Genghini S, Cacciari M, Gambineri A. Obesity and reproductive disorders in women. Hum Reprod Update. 2003;9(4):359-372.
- Greisen S, Ledet T, Møller N, et al. Effects of leptin on basal and FSH stimulated steroidogenesis in human granulosa luteal cells. Acta Obstet Gynecol Scand. 2000;79(11):931-935.
- Rich-Edwards JW, Goldman MB, Willett WC, et al. Adolescent body mass index and infertility caused by ovulatory disorder. Am J Obstet Gynecol. 1994;171(1):171-177.
- Grodstein F, Goldman MB, Cramer DW. Body mass index and ovulatory infertility. Epidemiology. 1994;5(2):247-250.
- Gesink Law DC, Maclehose RF, Longnecker MP. Obesity and time to pregnancy. Hum Reprod. 2007;22(2):414-420.
- Jain A, Polotsky AJ, Rochester D, et al. Pulsatile luteinizing hormone amplitude and progesterone metabolite excretion are reduced in obese women. J Clin Endocrinol Metab. 2007;92(7):2468-2473.
- Lake JK, Power C, Cole TJ. Women's reproductive health: the role of body mass index in early and adult life. Int J Obes Relat Metab Disord. 1997;21(6):432-438.
- van der Steeg JW, Steures P, Eijkemans MJ, et al. Obesity affects spontaneous pregnancy chances in subfertile, ovulatory women. Hum Reprod. 2008;23(2):324-328.
- Koning AM, Mutsaerts MA, Kuchenbecker WK, et al. Complications and outcome of assisted reproduction technologies in overweight and obese women [Published correction appears in Hum Reprod. 2012;27(8):2570.] Hum Reprod. 2012;27(2):457-467.
- Rittenberg V, Seshadri S, Sunkara SK, Sobaleva S, Oteng-Ntim E, El-Toukhy T. Effect of body mass index on IVF treatment outcome: an updated systematic review and meta-analysis. Reprod Biomed Online. 2011;23(4):421-439.
- Moragianni VA, Jones SM, Ryley DA. The effect of body mass index on the outcomes of first assisted reproductive technology cycles. Fertil Steril. 2012;98(1):102-108.
- Petersen GL, Schmidt L, Pinborg A, Kamper-Jørgensen M. The influence of female and male body mass index on live births after assisted reproductive technology treatment: a nationwide register-based cohort study. Fertil Steril. 2013;99(6):1654-1662.
- Practice Committee of the American Society for Reproductive Medicine. Obesity and Reproduction: A committee opinion. Fertil Steril. 2015;104(5):1116-1126.
- Metwally M, Cutting R, Tipton A, Skull J, Ledger WL, Li TC. Effect of increased body mass index on oocyte and embryo quality in IVF patients. Reprod Biomed Online. 2007;15(5):532-538.
- Leary C, Leese HJ, Sturmey RG. Human embryos from overweight and obese women display phenotypic and metabolic abnormalities. Hum Reprod. 2015;30(1):122-132.
- Deugarte D, Deugarte C, Sahakian V. Surrogate obesity negatively impacts pregnancy rates in third-party reproduction. Fertil Steril. 2010;93(3):1008-1010.
- Rittenberg V, Seshadri S, Sunkara SK, Sobaleva S, Oteng-Ntim E, El-Toukhy T. Effect of body mass index on IVF treatment outcome: an updated systematic review and meta-analysis. Reprod Biomed Online. 2011;23(4):421-439.
- Stothard KJ, Tennant PWG, Bell R, Rankin J. Maternal overweight and obesity and the risk of congenital anomalies: a systematic review and meta-analysis. JAMA. 2009;301(6):636-650.
- Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87(2):489-499.
- Baloch Z, Carayon P, Conte-Devolx B, et al. Laboratory medicine practice guidelines. Laboratory support for the diagnosis and monitoring of thyroid disease. Thyroid. 2003;13(1):3-126.
- Pop VJ, Kuijpens JL, van Baar AL, et al. Low maternal free thyroxine concentrations during early pregnancy are associated with impaired psychomotor development in infancy. Clin Endocrinol (Oxf). 1999;50(2):149-155.
- Lazarus JH, Bestwick JP, Channon S, et al. Antenatal thyroid screening and childhood cognitive function. N Engl J Med. 2012;366(17):493-501.
- Practice Committee of the American Society for Reproductive Medicine. Subclinical hypothyroidism in the infertile female population: a guideline. Fertil Steril. 2015;104(3):545-553.
As-needed anticoagulation for intermittent Afib raises concerns
ORLANDO – A pilot study that suggested as-needed anticoagulation could be effective in preventing stroke in at least some patients after successful ablation of atrial fibrillation (AF) was received with caution at the annual International AF Symposium.
The positive findings, originally reported at the 2016 annual meeting of the Heart Rhythm Society (HRS), were updated at AF Symposium 2017 by Francis Marchlinski, MD, director of cardiac electrophysiology at the University of Pennsylvania. When delivering the data, he provided several caveats before other AF experts added their own.
The study was conducted in response to the substantial number of patients who request discontinuing anticoagulation therapy after a successful ablation for atrial fibrillation, according to Dr. Marchlinski. Current guidelines recommend anticoagulation in AF patients following ablation if they have risk factors for stroke even if their AF is controlled. However, according to Dr. Marchlinski, who cited five observational studies, the risk of stroke in patients with a negative electrocardiogram after ablation appears to be “in the neighborhood of 0.1%.”
“There are no randomized prospective trials that have assessed the safety of stopping anticoagulants, but the fact is that this is a pretty low event rate if the observational studies are accurate, and even if they are off by severalfold, it is likely that we would be unable to show the benefit of continuing anticoagulants in these patients,” Dr. Marchlinski observed.
A strategy of as-needed anticoagulants has been made practical by the introduction of novel oral anticoagulants (NOACs), which have a rapid onset of action relative to warfarin and would, therefore, be expected to provide rapid protection against AF-related stroke risk if initiated upon AF onset, according to Dr. Marchlinski. To test this approach, 105 “highly motivated” AF patients were selected for the pilot study.
In addition to 3 weeks of ECG monitoring to confirm the absence of AF, patients participating in the trial were required to demonstrate skill in pulse assessment, which they agreed to perform on a twice-daily basis. Use of a smartphone app that can detect AF was encouraged but not required. All patients were required to fill a prescription for a NOAC and told to initiate therapy for any AF episode of more than 1 hour.
Of the 105 patients, four were noncompliant with AF monitoring and removed from the study. Another two patients voluntarily requested to return to daily NOAC treatment. The remaining 99 were followed for 30 months. Of these, 18 had multiple episodes of AF and were transitioned back to daily NOAC therapy, 15 used NOAC on an as-needed basis at least once but remained off daily therapy, and the remaining 66 did not have an episode of AF that triggered a course of NOAC therapy.
In 263 patient years of follow-up, there was a single cerebrovascular accident (CVA). This occurred in an 81-year-old patient with a history of hypertrophic cardiomyopathy and an atherosclerotic aortic arch on imaging. The patient presented with neurologic symptoms but had a negative ECG. The CVA symptoms resolved with treatment.
In presenting these data, Dr. Marchlinski said, “PRN use of NOACs may be safe and effective to maintain a low risk of stroke when patients are adherent to diligent pulse monitoring.” However, he reiterated that the study group consisted of “a select group of motivated patients,” and he emphasized the patients must be followed closely.
In a discussion that followed this presentation, several experts expressed the usual caution about drawing conclusions from a single uncontrolled study, but Elaine M. Hylek, MD, professor of medicine, Boston University, added additional reservations to the “pill in a pocket” strategy. In particular, she noted an imperfect correlation between onset of AF and stroke risk. “I think this makes us [reluctant] to stop oral anticoagulation,” she said.
According to Daniel Singer, MD, chief of epidemiology, Harvard School of Public Health, Boston, the available data suggest that “once the AF is gone, the risk of stroke recedes,” but he indicated that all the variables of risk may not be fully understood. He said more “hard data” are needed to endorse a wider application of on-demand anticoagulation in patients like those entered into this study.
The fact that patients without AF following ablation remain at substantial risk of AF recurrences, including asymptomatic episodes, is a liability of as-needed anticoagulation, conceded Dr. Marchlinski. However, these initial results provide promise for the substantial proportion of patients without AF after ablation that wish to avoid anticoagulants and are willing to consider risks and benefits.
Dr. Marchlinski reports financial relationships with Abbott, Biosense Webster, Biotronik, Boston Scientific, St. Jude Medical, and Medtronic.
ORLANDO – A pilot study that suggested as-needed anticoagulation could be effective in preventing stroke in at least some patients after successful ablation of atrial fibrillation (AF) was received with caution at the annual International AF Symposium.
The positive findings, originally reported at the 2016 annual meeting of the Heart Rhythm Society (HRS), were updated at AF Symposium 2017 by Francis Marchlinski, MD, director of cardiac electrophysiology at the University of Pennsylvania. When delivering the data, he provided several caveats before other AF experts added their own.
The study was conducted in response to the substantial number of patients who request discontinuing anticoagulation therapy after a successful ablation for atrial fibrillation, according to Dr. Marchlinski. Current guidelines recommend anticoagulation in AF patients following ablation if they have risk factors for stroke even if their AF is controlled. However, according to Dr. Marchlinski, who cited five observational studies, the risk of stroke in patients with a negative electrocardiogram after ablation appears to be “in the neighborhood of 0.1%.”
“There are no randomized prospective trials that have assessed the safety of stopping anticoagulants, but the fact is that this is a pretty low event rate if the observational studies are accurate, and even if they are off by severalfold, it is likely that we would be unable to show the benefit of continuing anticoagulants in these patients,” Dr. Marchlinski observed.
A strategy of as-needed anticoagulants has been made practical by the introduction of novel oral anticoagulants (NOACs), which have a rapid onset of action relative to warfarin and would, therefore, be expected to provide rapid protection against AF-related stroke risk if initiated upon AF onset, according to Dr. Marchlinski. To test this approach, 105 “highly motivated” AF patients were selected for the pilot study.
In addition to 3 weeks of ECG monitoring to confirm the absence of AF, patients participating in the trial were required to demonstrate skill in pulse assessment, which they agreed to perform on a twice-daily basis. Use of a smartphone app that can detect AF was encouraged but not required. All patients were required to fill a prescription for a NOAC and told to initiate therapy for any AF episode of more than 1 hour.
Of the 105 patients, four were noncompliant with AF monitoring and removed from the study. Another two patients voluntarily requested to return to daily NOAC treatment. The remaining 99 were followed for 30 months. Of these, 18 had multiple episodes of AF and were transitioned back to daily NOAC therapy, 15 used NOAC on an as-needed basis at least once but remained off daily therapy, and the remaining 66 did not have an episode of AF that triggered a course of NOAC therapy.
In 263 patient years of follow-up, there was a single cerebrovascular accident (CVA). This occurred in an 81-year-old patient with a history of hypertrophic cardiomyopathy and an atherosclerotic aortic arch on imaging. The patient presented with neurologic symptoms but had a negative ECG. The CVA symptoms resolved with treatment.
In presenting these data, Dr. Marchlinski said, “PRN use of NOACs may be safe and effective to maintain a low risk of stroke when patients are adherent to diligent pulse monitoring.” However, he reiterated that the study group consisted of “a select group of motivated patients,” and he emphasized the patients must be followed closely.
In a discussion that followed this presentation, several experts expressed the usual caution about drawing conclusions from a single uncontrolled study, but Elaine M. Hylek, MD, professor of medicine, Boston University, added additional reservations to the “pill in a pocket” strategy. In particular, she noted an imperfect correlation between onset of AF and stroke risk. “I think this makes us [reluctant] to stop oral anticoagulation,” she said.
According to Daniel Singer, MD, chief of epidemiology, Harvard School of Public Health, Boston, the available data suggest that “once the AF is gone, the risk of stroke recedes,” but he indicated that all the variables of risk may not be fully understood. He said more “hard data” are needed to endorse a wider application of on-demand anticoagulation in patients like those entered into this study.
The fact that patients without AF following ablation remain at substantial risk of AF recurrences, including asymptomatic episodes, is a liability of as-needed anticoagulation, conceded Dr. Marchlinski. However, these initial results provide promise for the substantial proportion of patients without AF after ablation that wish to avoid anticoagulants and are willing to consider risks and benefits.
Dr. Marchlinski reports financial relationships with Abbott, Biosense Webster, Biotronik, Boston Scientific, St. Jude Medical, and Medtronic.
ORLANDO – A pilot study that suggested as-needed anticoagulation could be effective in preventing stroke in at least some patients after successful ablation of atrial fibrillation (AF) was received with caution at the annual International AF Symposium.
The positive findings, originally reported at the 2016 annual meeting of the Heart Rhythm Society (HRS), were updated at AF Symposium 2017 by Francis Marchlinski, MD, director of cardiac electrophysiology at the University of Pennsylvania. When delivering the data, he provided several caveats before other AF experts added their own.
The study was conducted in response to the substantial number of patients who request discontinuing anticoagulation therapy after a successful ablation for atrial fibrillation, according to Dr. Marchlinski. Current guidelines recommend anticoagulation in AF patients following ablation if they have risk factors for stroke even if their AF is controlled. However, according to Dr. Marchlinski, who cited five observational studies, the risk of stroke in patients with a negative electrocardiogram after ablation appears to be “in the neighborhood of 0.1%.”
“There are no randomized prospective trials that have assessed the safety of stopping anticoagulants, but the fact is that this is a pretty low event rate if the observational studies are accurate, and even if they are off by severalfold, it is likely that we would be unable to show the benefit of continuing anticoagulants in these patients,” Dr. Marchlinski observed.
A strategy of as-needed anticoagulants has been made practical by the introduction of novel oral anticoagulants (NOACs), which have a rapid onset of action relative to warfarin and would, therefore, be expected to provide rapid protection against AF-related stroke risk if initiated upon AF onset, according to Dr. Marchlinski. To test this approach, 105 “highly motivated” AF patients were selected for the pilot study.
In addition to 3 weeks of ECG monitoring to confirm the absence of AF, patients participating in the trial were required to demonstrate skill in pulse assessment, which they agreed to perform on a twice-daily basis. Use of a smartphone app that can detect AF was encouraged but not required. All patients were required to fill a prescription for a NOAC and told to initiate therapy for any AF episode of more than 1 hour.
Of the 105 patients, four were noncompliant with AF monitoring and removed from the study. Another two patients voluntarily requested to return to daily NOAC treatment. The remaining 99 were followed for 30 months. Of these, 18 had multiple episodes of AF and were transitioned back to daily NOAC therapy, 15 used NOAC on an as-needed basis at least once but remained off daily therapy, and the remaining 66 did not have an episode of AF that triggered a course of NOAC therapy.
In 263 patient years of follow-up, there was a single cerebrovascular accident (CVA). This occurred in an 81-year-old patient with a history of hypertrophic cardiomyopathy and an atherosclerotic aortic arch on imaging. The patient presented with neurologic symptoms but had a negative ECG. The CVA symptoms resolved with treatment.
In presenting these data, Dr. Marchlinski said, “PRN use of NOACs may be safe and effective to maintain a low risk of stroke when patients are adherent to diligent pulse monitoring.” However, he reiterated that the study group consisted of “a select group of motivated patients,” and he emphasized the patients must be followed closely.
In a discussion that followed this presentation, several experts expressed the usual caution about drawing conclusions from a single uncontrolled study, but Elaine M. Hylek, MD, professor of medicine, Boston University, added additional reservations to the “pill in a pocket” strategy. In particular, she noted an imperfect correlation between onset of AF and stroke risk. “I think this makes us [reluctant] to stop oral anticoagulation,” she said.
According to Daniel Singer, MD, chief of epidemiology, Harvard School of Public Health, Boston, the available data suggest that “once the AF is gone, the risk of stroke recedes,” but he indicated that all the variables of risk may not be fully understood. He said more “hard data” are needed to endorse a wider application of on-demand anticoagulation in patients like those entered into this study.
The fact that patients without AF following ablation remain at substantial risk of AF recurrences, including asymptomatic episodes, is a liability of as-needed anticoagulation, conceded Dr. Marchlinski. However, these initial results provide promise for the substantial proportion of patients without AF after ablation that wish to avoid anticoagulants and are willing to consider risks and benefits.
Dr. Marchlinski reports financial relationships with Abbott, Biosense Webster, Biotronik, Boston Scientific, St. Jude Medical, and Medtronic.
Key clinical point: Despite a positive pilot study, the efficacy of intermittent anticoagulation for stroke prevention after ablation for atrial fibrillation will be difficult to validate in a definitive fashion.
Major finding: Sixty-six percent of atrial fibrillation patients entirely avoided anticoagulation over 30 months of follow-up, but there are at least theoretical concerns.
Data source: A prospective, nonrandomized study.
Disclosures: Dr. Marchlinski reported financial relationships with Abbott, Biosense Webster, Biotronik, Boston Scientific, St. Jude Medical, and Medtronic.