User login
Fecal microbiota transplantation a decolonization treatment option
The use of fecal microbiota transplantation is an option to eradicate highly drug-resistant enteric bacteria carriage, according to results from a small pilot study conducted by French investigators.
“A rapid and dramatic emergence of highly drug-resistant enteric bacteria (HDREB), i.e., carbapenem-resistant Enterobacteriaceae (CRE) and vancomycin-resistant enterococci (VRE), is occurring worldwide,” researchers led by Benjamin Davido, MD, of the infectious diseases unit at Raymond Poincaré Teaching Hospital, Garches, France, wrote in a study published online Feb. 1 in the Journal of Hospital Infection. “Patients carrying these bacteria are at risk of developing severe infections due to these bacteria; these infections are associated with a high mortality rate, partially because of inappropriate antimicrobial treatment.”
Citing recent studies that have demonstrated the efficacy of fecal microbiota transplant (FMT) as an accepted therapy to prevent recurrent Clostridium difficile infection, Dr. Davido and his associates prospectively identified eight different case reports of FMT used in adults for intestinal decolonization from extended-spectrum beta-lactamase (ESBL)–producing Enterobacteriaceae, VRE, or methicillin-resistant Staphylococcus aureus. Patients on immunosuppressive agents were excluded from the study, as were those taking antibiotics at the time of FMT (J Hosp Infect. 2017 Feb. 1. doi: 10.1016/j.jhin.2017.02.001).
The protocol for the procedure involved insertion of a nasoduodenal tube the day before FMT in order to perform a bowel lavage with XPrep solution. FMT was performed using a frozen preparation of fecal microbiota from a universal donor who was previously screened for potential diseases. The main outcome of interest was time to successful decolonization following FMT, which was determined by at least two consecutive negative rectal swabs at a 1-week interval during a follow-up of 3 months.
The mean age of the eight patients was 70 years, their median Charlson comorbidity index score was 5, and the median duration of carriage of HDREB before FMT was 83 days. The researchers observed that 1 month after FMT, two patients were free from CRE colonization, while one more patient was free from VRE after 3 months. Five patients received antibiotic during follow-up. Among them, one patient was decolonized at 1 month, while another was decolonized at 3 months. One patient with cirrhosis and persistent VRE carriage died 3 months after FMT from ascetic fluid infection and VRE bacteremia. No other adverse events were reported.
“In a context where no other efficient strategy is available, our first results show that FMT seems to be safe, with an impact on CRE decolonization at 1 month and on VRE decolonization at 3 months,” the researchers concluded. “Of particular importance, there was no recolonization after the intervention, which is contrary to decolonization with antibiotics. However, our study has important limitations in that the sample size was very small, it was nonrandomized, and follow-up was limited to a 3-month period.” They noted that at least five trials are underway to investigate the impact of FMT on multidrug resistant organism bacterial decolonization. The researchers reported having no financial disclosures.
The use of fecal microbiota transplantation is an option to eradicate highly drug-resistant enteric bacteria carriage, according to results from a small pilot study conducted by French investigators.
“A rapid and dramatic emergence of highly drug-resistant enteric bacteria (HDREB), i.e., carbapenem-resistant Enterobacteriaceae (CRE) and vancomycin-resistant enterococci (VRE), is occurring worldwide,” researchers led by Benjamin Davido, MD, of the infectious diseases unit at Raymond Poincaré Teaching Hospital, Garches, France, wrote in a study published online Feb. 1 in the Journal of Hospital Infection. “Patients carrying these bacteria are at risk of developing severe infections due to these bacteria; these infections are associated with a high mortality rate, partially because of inappropriate antimicrobial treatment.”
Citing recent studies that have demonstrated the efficacy of fecal microbiota transplant (FMT) as an accepted therapy to prevent recurrent Clostridium difficile infection, Dr. Davido and his associates prospectively identified eight different case reports of FMT used in adults for intestinal decolonization from extended-spectrum beta-lactamase (ESBL)–producing Enterobacteriaceae, VRE, or methicillin-resistant Staphylococcus aureus. Patients on immunosuppressive agents were excluded from the study, as were those taking antibiotics at the time of FMT (J Hosp Infect. 2017 Feb. 1. doi: 10.1016/j.jhin.2017.02.001).
The protocol for the procedure involved insertion of a nasoduodenal tube the day before FMT in order to perform a bowel lavage with XPrep solution. FMT was performed using a frozen preparation of fecal microbiota from a universal donor who was previously screened for potential diseases. The main outcome of interest was time to successful decolonization following FMT, which was determined by at least two consecutive negative rectal swabs at a 1-week interval during a follow-up of 3 months.
The mean age of the eight patients was 70 years, their median Charlson comorbidity index score was 5, and the median duration of carriage of HDREB before FMT was 83 days. The researchers observed that 1 month after FMT, two patients were free from CRE colonization, while one more patient was free from VRE after 3 months. Five patients received antibiotic during follow-up. Among them, one patient was decolonized at 1 month, while another was decolonized at 3 months. One patient with cirrhosis and persistent VRE carriage died 3 months after FMT from ascetic fluid infection and VRE bacteremia. No other adverse events were reported.
“In a context where no other efficient strategy is available, our first results show that FMT seems to be safe, with an impact on CRE decolonization at 1 month and on VRE decolonization at 3 months,” the researchers concluded. “Of particular importance, there was no recolonization after the intervention, which is contrary to decolonization with antibiotics. However, our study has important limitations in that the sample size was very small, it was nonrandomized, and follow-up was limited to a 3-month period.” They noted that at least five trials are underway to investigate the impact of FMT on multidrug resistant organism bacterial decolonization. The researchers reported having no financial disclosures.
The use of fecal microbiota transplantation is an option to eradicate highly drug-resistant enteric bacteria carriage, according to results from a small pilot study conducted by French investigators.
“A rapid and dramatic emergence of highly drug-resistant enteric bacteria (HDREB), i.e., carbapenem-resistant Enterobacteriaceae (CRE) and vancomycin-resistant enterococci (VRE), is occurring worldwide,” researchers led by Benjamin Davido, MD, of the infectious diseases unit at Raymond Poincaré Teaching Hospital, Garches, France, wrote in a study published online Feb. 1 in the Journal of Hospital Infection. “Patients carrying these bacteria are at risk of developing severe infections due to these bacteria; these infections are associated with a high mortality rate, partially because of inappropriate antimicrobial treatment.”
Citing recent studies that have demonstrated the efficacy of fecal microbiota transplant (FMT) as an accepted therapy to prevent recurrent Clostridium difficile infection, Dr. Davido and his associates prospectively identified eight different case reports of FMT used in adults for intestinal decolonization from extended-spectrum beta-lactamase (ESBL)–producing Enterobacteriaceae, VRE, or methicillin-resistant Staphylococcus aureus. Patients on immunosuppressive agents were excluded from the study, as were those taking antibiotics at the time of FMT (J Hosp Infect. 2017 Feb. 1. doi: 10.1016/j.jhin.2017.02.001).
The protocol for the procedure involved insertion of a nasoduodenal tube the day before FMT in order to perform a bowel lavage with XPrep solution. FMT was performed using a frozen preparation of fecal microbiota from a universal donor who was previously screened for potential diseases. The main outcome of interest was time to successful decolonization following FMT, which was determined by at least two consecutive negative rectal swabs at a 1-week interval during a follow-up of 3 months.
The mean age of the eight patients was 70 years, their median Charlson comorbidity index score was 5, and the median duration of carriage of HDREB before FMT was 83 days. The researchers observed that 1 month after FMT, two patients were free from CRE colonization, while one more patient was free from VRE after 3 months. Five patients received antibiotic during follow-up. Among them, one patient was decolonized at 1 month, while another was decolonized at 3 months. One patient with cirrhosis and persistent VRE carriage died 3 months after FMT from ascetic fluid infection and VRE bacteremia. No other adverse events were reported.
“In a context where no other efficient strategy is available, our first results show that FMT seems to be safe, with an impact on CRE decolonization at 1 month and on VRE decolonization at 3 months,” the researchers concluded. “Of particular importance, there was no recolonization after the intervention, which is contrary to decolonization with antibiotics. However, our study has important limitations in that the sample size was very small, it was nonrandomized, and follow-up was limited to a 3-month period.” They noted that at least five trials are underway to investigate the impact of FMT on multidrug resistant organism bacterial decolonization. The researchers reported having no financial disclosures.
FROM THE JOURNAL OF HOSPITAL INFECTION
Key clinical point:
Major finding: Of eight patients who underwent fecal microbiota transplantation (FMT) for carbapenem-resistant Enterobacteriaceae (CRE) and vancomycin-resistant enterococci (VRE) digestive tract decolonization, three achieved decolonization after 3 months.
Data source: A prospective study of eight cases of FMT used in adults for intestinal decolonization from drug-resistant enteric bacteria carriage.
Disclosures: The researchers reported having no financial disclosures.
Specific polymorphisms excluded in hemophilic arthropathy
Carriers of a hemochromatosis (HFE) gene mutation or a long (GT)n-repeat length within the HMOX1 promoter regions did not appear to have an increase in hemophilic arthropathy.
In 201 blood samples from patients with severe hemophilia A or B and 37 from patients with moderate disease, neither the presence of an HFE mutation nor a long (GT)n-repeat length was associated with an increase in joint damage. The assessment was based on Pettersson score after adjustment for disease severity, presence of inhibitors, annual joint bleeding rate (AJBR), age at Pettersson score and at clinic entrance, and birth cohort (standardized beta = 0.033 and -0.022, respectively), Lize F. van Vulpen, MD, of University Medical Center Utrecht, the Netherlands, reported at the annual meeting of the European Association of Haemophilia and Allied Disorders.
Study subjects had a median age of 43 years, median AJBRs of 2.5 and 0.5 for severe and moderate disease, respectively, and median Pettersson scores of 22 and 4 in the groups, respectively. An HFE mutation was detected in 91 patients, and their levels of ferritin, iron, and transferrin saturation were significantly increased, but other baseline characteristic were similar in those with and without HFE mutation, and regardless of (GT)n-repeat length.
The marked heterogeneity in joint damage seen among hemophilia patients was hypothesized in this study to be associated with differences in iron handling, but the findings failed to support that hypothesis, Dr. van Vulpen concluded.
Dr. van Vulpen reported having no disclosures.
Carriers of a hemochromatosis (HFE) gene mutation or a long (GT)n-repeat length within the HMOX1 promoter regions did not appear to have an increase in hemophilic arthropathy.
In 201 blood samples from patients with severe hemophilia A or B and 37 from patients with moderate disease, neither the presence of an HFE mutation nor a long (GT)n-repeat length was associated with an increase in joint damage. The assessment was based on Pettersson score after adjustment for disease severity, presence of inhibitors, annual joint bleeding rate (AJBR), age at Pettersson score and at clinic entrance, and birth cohort (standardized beta = 0.033 and -0.022, respectively), Lize F. van Vulpen, MD, of University Medical Center Utrecht, the Netherlands, reported at the annual meeting of the European Association of Haemophilia and Allied Disorders.
Study subjects had a median age of 43 years, median AJBRs of 2.5 and 0.5 for severe and moderate disease, respectively, and median Pettersson scores of 22 and 4 in the groups, respectively. An HFE mutation was detected in 91 patients, and their levels of ferritin, iron, and transferrin saturation were significantly increased, but other baseline characteristic were similar in those with and without HFE mutation, and regardless of (GT)n-repeat length.
The marked heterogeneity in joint damage seen among hemophilia patients was hypothesized in this study to be associated with differences in iron handling, but the findings failed to support that hypothesis, Dr. van Vulpen concluded.
Dr. van Vulpen reported having no disclosures.
Carriers of a hemochromatosis (HFE) gene mutation or a long (GT)n-repeat length within the HMOX1 promoter regions did not appear to have an increase in hemophilic arthropathy.
In 201 blood samples from patients with severe hemophilia A or B and 37 from patients with moderate disease, neither the presence of an HFE mutation nor a long (GT)n-repeat length was associated with an increase in joint damage. The assessment was based on Pettersson score after adjustment for disease severity, presence of inhibitors, annual joint bleeding rate (AJBR), age at Pettersson score and at clinic entrance, and birth cohort (standardized beta = 0.033 and -0.022, respectively), Lize F. van Vulpen, MD, of University Medical Center Utrecht, the Netherlands, reported at the annual meeting of the European Association of Haemophilia and Allied Disorders.
Study subjects had a median age of 43 years, median AJBRs of 2.5 and 0.5 for severe and moderate disease, respectively, and median Pettersson scores of 22 and 4 in the groups, respectively. An HFE mutation was detected in 91 patients, and their levels of ferritin, iron, and transferrin saturation were significantly increased, but other baseline characteristic were similar in those with and without HFE mutation, and regardless of (GT)n-repeat length.
The marked heterogeneity in joint damage seen among hemophilia patients was hypothesized in this study to be associated with differences in iron handling, but the findings failed to support that hypothesis, Dr. van Vulpen concluded.
Dr. van Vulpen reported having no disclosures.
FROM EAHAD 2017
Key clinical point:
Major finding: Neither the presence of an HFE mutation, nor a long (GT)n-repeat length was associated with an increase in joint damage as assessed by Pettersson score (standardized beta = 0.033 and –0.022, respectively).
Data source: An evaluation of blood samples from a cohort of 238 patients.
Disclosures: Dr. van Vulpen reported having no disclosures.
Low inhibitor incidence seen with new generation rhFVIII
Human-cl rhFVIII, a new generation recombinant factor VIII of human origin, appears to be associated with a low inhibitor incidence in patients with severe hemophilia A, according to interim findings from the ongoing NuProtect study.
In 66 evaluable previously untreated patients with at least 20 days of exposure to human-cl rhFVIII, the cumulative incidence of high-titer and low-titer inhibitor development was 12.8% and 8.4%, respectively, Ellis J. Neufeld, MD, reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
The median age at first treatment was 13 months (range of 3-135 months). Of 59 patients with available F8 gene analysis, 44 had high-risk mutations and 47 had null mutations; 1 had no mutation identified. After a median of 11.5 treatment exposure days, 8 of the 66 patients developed high-titer inhibitors and 5 developed low-titer inhibitors; 4 of those were transient, said Dr. Neufeld of Harvard Medical School, Boston.
Only two of the patients developed an inhibitor, including one high- and one low-titer inhibitor, after 20 exposure days. Among the 13 inhibitor patients, 12 had an F8 mutation identified; all were null, and 11 were high risk.
In a prior study of 201 patients with previously treated severe hemophilia A, no inhibitors were reported with human-cl rhFVIII – which is produced in human cells without chemical modification or protein fusion. The NuProtect study is looking at immunogenicity, efficacy, and safety in previously untreated patients. Final data are expected in 2018, Dr. Neufeld noted.
Dr. Neufeld disclosed financial ties to Octapharma, the sponsor of NuProtect, and numerous other pharmaceutical companies.
Human-cl rhFVIII, a new generation recombinant factor VIII of human origin, appears to be associated with a low inhibitor incidence in patients with severe hemophilia A, according to interim findings from the ongoing NuProtect study.
In 66 evaluable previously untreated patients with at least 20 days of exposure to human-cl rhFVIII, the cumulative incidence of high-titer and low-titer inhibitor development was 12.8% and 8.4%, respectively, Ellis J. Neufeld, MD, reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
The median age at first treatment was 13 months (range of 3-135 months). Of 59 patients with available F8 gene analysis, 44 had high-risk mutations and 47 had null mutations; 1 had no mutation identified. After a median of 11.5 treatment exposure days, 8 of the 66 patients developed high-titer inhibitors and 5 developed low-titer inhibitors; 4 of those were transient, said Dr. Neufeld of Harvard Medical School, Boston.
Only two of the patients developed an inhibitor, including one high- and one low-titer inhibitor, after 20 exposure days. Among the 13 inhibitor patients, 12 had an F8 mutation identified; all were null, and 11 were high risk.
In a prior study of 201 patients with previously treated severe hemophilia A, no inhibitors were reported with human-cl rhFVIII – which is produced in human cells without chemical modification or protein fusion. The NuProtect study is looking at immunogenicity, efficacy, and safety in previously untreated patients. Final data are expected in 2018, Dr. Neufeld noted.
Dr. Neufeld disclosed financial ties to Octapharma, the sponsor of NuProtect, and numerous other pharmaceutical companies.
Human-cl rhFVIII, a new generation recombinant factor VIII of human origin, appears to be associated with a low inhibitor incidence in patients with severe hemophilia A, according to interim findings from the ongoing NuProtect study.
In 66 evaluable previously untreated patients with at least 20 days of exposure to human-cl rhFVIII, the cumulative incidence of high-titer and low-titer inhibitor development was 12.8% and 8.4%, respectively, Ellis J. Neufeld, MD, reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
The median age at first treatment was 13 months (range of 3-135 months). Of 59 patients with available F8 gene analysis, 44 had high-risk mutations and 47 had null mutations; 1 had no mutation identified. After a median of 11.5 treatment exposure days, 8 of the 66 patients developed high-titer inhibitors and 5 developed low-titer inhibitors; 4 of those were transient, said Dr. Neufeld of Harvard Medical School, Boston.
Only two of the patients developed an inhibitor, including one high- and one low-titer inhibitor, after 20 exposure days. Among the 13 inhibitor patients, 12 had an F8 mutation identified; all were null, and 11 were high risk.
In a prior study of 201 patients with previously treated severe hemophilia A, no inhibitors were reported with human-cl rhFVIII – which is produced in human cells without chemical modification or protein fusion. The NuProtect study is looking at immunogenicity, efficacy, and safety in previously untreated patients. Final data are expected in 2018, Dr. Neufeld noted.
Dr. Neufeld disclosed financial ties to Octapharma, the sponsor of NuProtect, and numerous other pharmaceutical companies.
Key clinical point:
Major finding: The cumulative incidence of high-titer and low-titer inhibitor development was 12.8% and 8.4%, respectively.
Data source: The ongoing NuProtect study of 66 patients.
Disclosures: Dr. Neufeld disclosed financial ties to Octapharma, the sponsor of NuProtect, and numerous other pharmaceutical companies.
ACR guidelines on HBV screening called inadequate
SNOWMASS, COLO. – The ACR guidelines for hepatitis B virus screening prior to starting immunosuppressive therapy are sorely in need of an overhaul, Leonard H. Calabrese, MD, asserted at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
“We need to do better,” declared Dr. Calabrese, professor of medicine and head of the section of clinical immunology at the Cleveland Clinic.
The current ACR guidelines on hepatitis B virus (HBV) screening in rheumatoid arthritis patients (Arthritis Rheumatol. 2016 Jan;68[1]:1-26) are unchanged since 2008. The recommendation is for selective screening confined to patients who are going to go on methotrexate or leflunomide or who state they have an HBV risk factor.

He recommended that his fellow rheumatologists skip the ACR guidance and instead do what he does, which is to follow the American Association for the Study of Liver Diseases (AASLD) guidelines that have been in place for the past 8 years (Hepatology. 2009 Sep;50[3]:661-2).
The AASLD screening algorithm is simple and straightforward: Screen everyone who is going to go on any form of immunosuppressive therapy or cancer therapy by testing for hepatitis B surface antigen (HBsAg) and hepatitis B core antibodies (anti-HBc).
The highest-risk group for HBV reactivation are patients who are HBsAg positive. They have an active infection and should be referred to a hepatologist for antiviral therapy. Depending upon the patient’s HBV DNA level and liver function test results, the antiviral therapy will be started either prior to or simultaneously with the immunosuppressive therapy.
The AASLD guidelines rank immunosuppressive agents in terms of their associated risk of HBV reactivation based upon the best available evidence. It’s a tricky business. Patients with a history of HBV are excluded from clinical trials of immunosuppressants, so the evidence is sketchy.
Nevertheless, the consensus is that among HBsAg-positive patients the risk associated with methotrexate and other antimetabolites is considered low. The tumor necrosis factor inhibitors are deemed to pose a moderate risk, as does cyclosporine. The risk of other cytokine inhibitors, including ustekinumab (Stelara) and abatacept (Orencia), is uncertain, but thought to be moderate to high. The use of corticosteroids at more than 20 mg/day for longer than 4 weeks is considered high-risk therapy. And rituximab (Rituxan), which carries a black box warning regarding HBV reactivation, is rated very-high-risk, as is cancer chemotherapy and hematopoietic stem cell transplantation.
An HBV screen that comes back HBsAg negative but anti-HBc positive indicates past HBV infection. This places a patient at intermediate risk for reactivation during immunosuppressive therapy. If Dr. Calabrese plans to put that patient on rituximab, he’ll make a referral to a hepatologist for consideration of antiviral therapy. If he’s going to use any other form of immunosuppressive therapy in a patient who is HBsAg negative/anti-HBc positive, he tests for HBV DNA every 1-3 months while the patient is on treatment. If HBV DNA becomes detectable, it’s time to start antiviral therapy.
No further action is required if a rheumatology patient’s HBV screen comes back negative for both HBsAg and anti-HBc.
“There are six approved drugs for HBV. They don’t have the power of the direct-acting antiviral agents used for hepatitis C. Cures are very infrequent. But lifelong viral suppression is readily achievable with these drugs, which are well tolerated and have low potential for drug-drug interactions. If patients have undetectable HBV DNA on antiviral therapy, I believe we can treat them with any of our rheumatologic regimens, including rituximab,” Dr. Calabrese said.
He reported having no financial conflicts of interest regarding his presentation.
SNOWMASS, COLO. – The ACR guidelines for hepatitis B virus screening prior to starting immunosuppressive therapy are sorely in need of an overhaul, Leonard H. Calabrese, MD, asserted at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
“We need to do better,” declared Dr. Calabrese, professor of medicine and head of the section of clinical immunology at the Cleveland Clinic.
The current ACR guidelines on hepatitis B virus (HBV) screening in rheumatoid arthritis patients (Arthritis Rheumatol. 2016 Jan;68[1]:1-26) are unchanged since 2008. The recommendation is for selective screening confined to patients who are going to go on methotrexate or leflunomide or who state they have an HBV risk factor.
He recommended that his fellow rheumatologists skip the ACR guidance and instead do what he does, which is to follow the American Association for the Study of Liver Diseases (AASLD) guidelines that have been in place for the past 8 years (Hepatology. 2009 Sep;50[3]:661-2).
The AASLD screening algorithm is simple and straightforward: Screen everyone who is going to go on any form of immunosuppressive therapy or cancer therapy by testing for hepatitis B surface antigen (HBsAg) and hepatitis B core antibodies (anti-HBc).
The highest-risk group for HBV reactivation are patients who are HBsAg positive. They have an active infection and should be referred to a hepatologist for antiviral therapy. Depending upon the patient’s HBV DNA level and liver function test results, the antiviral therapy will be started either prior to or simultaneously with the immunosuppressive therapy.
The AASLD guidelines rank immunosuppressive agents in terms of their associated risk of HBV reactivation based upon the best available evidence. It’s a tricky business. Patients with a history of HBV are excluded from clinical trials of immunosuppressants, so the evidence is sketchy.
Nevertheless, the consensus is that among HBsAg-positive patients the risk associated with methotrexate and other antimetabolites is considered low. The tumor necrosis factor inhibitors are deemed to pose a moderate risk, as does cyclosporine. The risk of other cytokine inhibitors, including ustekinumab (Stelara) and abatacept (Orencia), is uncertain, but thought to be moderate to high. The use of corticosteroids at more than 20 mg/day for longer than 4 weeks is considered high-risk therapy. And rituximab (Rituxan), which carries a black box warning regarding HBV reactivation, is rated very-high-risk, as is cancer chemotherapy and hematopoietic stem cell transplantation.
An HBV screen that comes back HBsAg negative but anti-HBc positive indicates past HBV infection. This places a patient at intermediate risk for reactivation during immunosuppressive therapy. If Dr. Calabrese plans to put that patient on rituximab, he’ll make a referral to a hepatologist for consideration of antiviral therapy. If he’s going to use any other form of immunosuppressive therapy in a patient who is HBsAg negative/anti-HBc positive, he tests for HBV DNA every 1-3 months while the patient is on treatment. If HBV DNA becomes detectable, it’s time to start antiviral therapy.
No further action is required if a rheumatology patient’s HBV screen comes back negative for both HBsAg and anti-HBc.
“There are six approved drugs for HBV. They don’t have the power of the direct-acting antiviral agents used for hepatitis C. Cures are very infrequent. But lifelong viral suppression is readily achievable with these drugs, which are well tolerated and have low potential for drug-drug interactions. If patients have undetectable HBV DNA on antiviral therapy, I believe we can treat them with any of our rheumatologic regimens, including rituximab,” Dr. Calabrese said.
He reported having no financial conflicts of interest regarding his presentation.
SNOWMASS, COLO. – The ACR guidelines for hepatitis B virus screening prior to starting immunosuppressive therapy are sorely in need of an overhaul, Leonard H. Calabrese, MD, asserted at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
“We need to do better,” declared Dr. Calabrese, professor of medicine and head of the section of clinical immunology at the Cleveland Clinic.
The current ACR guidelines on hepatitis B virus (HBV) screening in rheumatoid arthritis patients (Arthritis Rheumatol. 2016 Jan;68[1]:1-26) are unchanged since 2008. The recommendation is for selective screening confined to patients who are going to go on methotrexate or leflunomide or who state they have an HBV risk factor.
He recommended that his fellow rheumatologists skip the ACR guidance and instead do what he does, which is to follow the American Association for the Study of Liver Diseases (AASLD) guidelines that have been in place for the past 8 years (Hepatology. 2009 Sep;50[3]:661-2).
The AASLD screening algorithm is simple and straightforward: Screen everyone who is going to go on any form of immunosuppressive therapy or cancer therapy by testing for hepatitis B surface antigen (HBsAg) and hepatitis B core antibodies (anti-HBc).
The highest-risk group for HBV reactivation are patients who are HBsAg positive. They have an active infection and should be referred to a hepatologist for antiviral therapy. Depending upon the patient’s HBV DNA level and liver function test results, the antiviral therapy will be started either prior to or simultaneously with the immunosuppressive therapy.
The AASLD guidelines rank immunosuppressive agents in terms of their associated risk of HBV reactivation based upon the best available evidence. It’s a tricky business. Patients with a history of HBV are excluded from clinical trials of immunosuppressants, so the evidence is sketchy.
Nevertheless, the consensus is that among HBsAg-positive patients the risk associated with methotrexate and other antimetabolites is considered low. The tumor necrosis factor inhibitors are deemed to pose a moderate risk, as does cyclosporine. The risk of other cytokine inhibitors, including ustekinumab (Stelara) and abatacept (Orencia), is uncertain, but thought to be moderate to high. The use of corticosteroids at more than 20 mg/day for longer than 4 weeks is considered high-risk therapy. And rituximab (Rituxan), which carries a black box warning regarding HBV reactivation, is rated very-high-risk, as is cancer chemotherapy and hematopoietic stem cell transplantation.
An HBV screen that comes back HBsAg negative but anti-HBc positive indicates past HBV infection. This places a patient at intermediate risk for reactivation during immunosuppressive therapy. If Dr. Calabrese plans to put that patient on rituximab, he’ll make a referral to a hepatologist for consideration of antiviral therapy. If he’s going to use any other form of immunosuppressive therapy in a patient who is HBsAg negative/anti-HBc positive, he tests for HBV DNA every 1-3 months while the patient is on treatment. If HBV DNA becomes detectable, it’s time to start antiviral therapy.
No further action is required if a rheumatology patient’s HBV screen comes back negative for both HBsAg and anti-HBc.
“There are six approved drugs for HBV. They don’t have the power of the direct-acting antiviral agents used for hepatitis C. Cures are very infrequent. But lifelong viral suppression is readily achievable with these drugs, which are well tolerated and have low potential for drug-drug interactions. If patients have undetectable HBV DNA on antiviral therapy, I believe we can treat them with any of our rheumatologic regimens, including rituximab,” Dr. Calabrese said.
He reported having no financial conflicts of interest regarding his presentation.
EXPERT ANALYSIS FROM THE WINTER RHEUMATOLOGY SYMPOSIUM
Review of the Long-Term Effects of Proton Pump Inhibitors
Proton pump inhibitors (PPIs) are one of the most frequently used drug classes, given that they are readily accessible over-the-counter as well as via prescription. About 100 million PPI prescriptions dispensed an
The human stomach uses 3 primary neurotransmitters that regulate gastric acid secretion: acetylcholine (ACh), histamine (H), and gastrin (G). The interactions between these neurotransmitters promote and inhibit hydrogen ion (H+) generation. Stimulation of their corresponding receptors draws H+ into parietal cells that line the stomach. Once in the cell, a H+-K+-ATPase (more commonly known as the proton pump) actively transports H+ into the lumen of the stomach. The H+ bind with chlorine ions to form hydrochloric acid, which increases stomach acidity.5 Histamine receptors were thought to be responsible for the greatest degree of stimulation. Hence, histamine type 2-receptor antagonists (H2RAs) became a novel means of therapy to reduce stomach acidity. While utilizing H2RAs was effective, it was theorized the downstream inhibition of the action of all 3 neurotransmitters would serve as a more successful therapy. Therefore, PPIs were developed to target the H+-K+-ATPase Over the past decade, many studies have evaluated the long-term PPI adverse effects (AEs). These include calcium and magnesium malabsorption, vitamin B12 deficiency, Clostridium difficile (C difficile) associated disease (CDAD), and community-acquired pneumonia (CAP). Within the past year, data have become available linking PPI use to dementia and chronic kidney disease (CKD).3,4 The following article reviews literature on the safety of long-term PPI use and proposes recommendations for proper use for their most common indications.
Malabsorption
Calcium & Long-Term Fracture Risk
Calcium is an essential component in bone health and formation. In fact, 99% of all calcium found in the body is stored in bones.6 The primary source of calcium is through diet and oral supplements. After it is ingested, calcium is absorbed from the stomach into the blood in a pH dependent manner. If the pH of the stomach is too high (ie, too basic) calcium is not absorbed into blood and remains in the gastrointestinal (GI) tract for fecal excretion. Without sufficient calcium, the body’s osteoclasts and osteoblasts remain inactive, which hinders proper bone turnover.7
The decrease in acidity leads to calcium malabsorption and increases fracture risk long- term.8 Khalili and colleagues surveyed 80,000 postmenopausal women to measure the incidence of hip fracture in women taking PPIs. The study found that there was a 35% increase in risk of hip fracture among women who regularly used PPIs for at least 2 years (age-adjusted hazard ratio [HR] 1.35; 95% confidence interval [CI], 1.13 -1.62). Adjusted HRs for 4-year and 6- to 8-year use of a PPI was 1.42 (95% CI, 1.05-1.93) and 1.55 (95% CI, 1.03-2.32), respectively, indicating that the longer women were on PPI therapy, the higher the risk of hip fracture. The study also evaluated the time since stopping PPI and the risk of hip fracture. Women who stopped PPI use more than 2 years prior had a similar risk to that of women who never used a PPI, indicating that the effect was reversible.9
Magnesium
Magnesium is an important intracellular ion that has a number of key functions in metabolism and ion transport in the human body. Once ingested, magnesium is absorbed into the bloodstream from the small and large intestines via passive and active transport. Transient receptor potential melastatin 6 (TRPM6) is one of the essential proteins that serve as a transporter for magnesium.10 The high affinity for magnesium of these transporters allows them to maintain adequate levels of magnesium in the blood. In states of low magnesium (hypomagnesemia), the body is at risk for many AEs including seizures, arrhythmias, tetany, and hypotension.11
Proton pump inhibitors have been linked to hypomagnesemia, and recent evaluation has clarified a potential mechanism.12 TRPM6 activity is increased in an acidic environment. When a PPI increases the pH of the stomach, TRPM6 and magnesium levels decrease.12 Luk and colleagues identified 66,102 subjects experiencing AEs while taking a PPI. Hypomagnesemia had a prevalence rate of 1% in these patients. According to the researchers, PPIs were associated with hypomagnesemia and that pantoprazole had the highest incidence among all other PPIs studied (OR, 4.3; 95% CI, 3.3 – 5.7; P < .001).13
Vitamin B12
In recent years, vitamin B12 has been the subject of many studies. An area of concern is vitamin B12’s neurologic effect, as it has been successfully demonstrated that vitamin B12 is essential for proper cognitive function.14 Some data suggest that degeneration is present in parts of the spinal column in patients with cognitive decline or neurologic problems. These lesions are due to improper myelin formation and are specific to vitamin B12 deficiency.15 In 2013 the CDC published the Healthy Brain Initiative, which stated cognitive impairment can be caused by vitamin B12 deficiency.16
Similar to calcium, vitamin B12 needs an acidic environment to be digested and absorbed.17 Vitamin B12 is released from food proteins via gastric acid and pepsin. Once free, the vitamin B12 pairs with R-binders secreted in the stomach. Pancreatic enzymes then degrade this complex into a form that can be absorbed into circulation by the intestine. Given that PPIs reduce the acidity of the stomach, they also reduce the body’s ability to release vitamin B12 from food proteins and be paired with the R-binders.18
In 2013, Lam and colleagues evaluated the association between vitamin B12 deficiency and the use of PPIs and H2RAs. An extensive evaluation was performed on 25,956 patients with a diagnosis of vitamin B12 deficiency and 184,199 patients without. About 12% of patients with vitamin B12 deficiency had received more than a 2-year supply of a PPI, whereas only 7.2% of the patients without vitamin B12 deficiency received a 2-year supply of a PPI. Four point 3 percent of patients with vitamin B12 deficiency received more than a 2-year supply of an H2RA. Only 3.2% of patients without vitamin B12 deficiency received more than a 2-year supply of H2RA. The study concluded that a 2-year or greater history of PPI (OR, 1.65; 95% CI, 1.58-1.73) or H2RA (OR, 1.25; 95% CI, 1.17-1.34) use was associated with vitamin B12 deficiency.19
PPIs and Infections
Clostridium difficile-associated disease
Nationwide CDAD has become a prevalent infection nationwide. In 2011, C difficile caused nearly 500,000 infections and was associated with 29,000 deaths in the U.S.20 One study stated that C difficile is the third most common cause of infectious diarrhea in people aged >75 years.21
C difficile is part of the body’s normal flora in the large intestine. It grows and colonizes in an environment of low acidity. Therefore, in the stomach, where the pH is relatively low, C difficile is unable to colonize.22 When a PPI is introduced, the increased gastric pH increases the risk for CDAD.
Dial and colleagues conducted a multicenter case control study to determine whether gastric acid suppression increases the risk of CDAD. Compared with patients who did not take a gastric acid suppressant, those taking a PPI had a 2.9-fold increase in developing CDAD (95% CI, 2.4-3.4). Comparatively, H2RAs had a 2.0-fold increase for CDAD (95% CI, 1.6 to 2.7). These results correlated with the fact that PPIs have a greater impact on gastric pH than do H2RAs.23
Community-Acquired Pneumonia
Community-acquired pneumonia (CAP) has become a growing concern in the U.S. According to the Infectious Disease Society of America (IDSA) and American Thoracic clinical consensus guidelines, CAP remains one of the top reasons for hospitalizations in the U.S., and about 10% of patients admitted to the hospital for CAP end up in the intensive care unit (ICU).24 In the past, PPIs have been linked to patients’ predisposal for developingCAP.25 Although controversial, available evidence suggests a direct association. In 2008 Sarker and colleagues theorized a mechanism that the acid reduction of the gastric lumen allows for increased bacterial colonization in the upper part of the GI tract.26 Since the acidity of the stomach serves as a defense mechanism against many ingested bacteria, many pathogens will be able to survive in the more basic environment.25
Sarkar and colleagues went on to evaluate 80,000 cases over 15 years. The objective was to examine the association between PPI use and the date of diagnosis of the CAP infection, known as the index date. The study demonstrated that PPI use was not associated with increased CAP risk in the long-term (adjusted odds ratio (OR), 1.02; 95% CI, 0.97-1.08). The study did find a strong increase in the risk of CAP if a PPI was started within 2 days (adjusted OR, 6.53; 95% CI, 3.95-10.80), 7 days (adjusted OR, 3.79; 95% CI, 2.66-5.42), and 14 days (adjusted OR, 3.21; 95% CI, 2.46-4.18) of the index date.26
Four years later, de Jagar and colleagues examined the differences in microbial etiology in CAP patients with and without an active PPI. Over a 4-year study period, 463 individuals were selected with clinical suspicion of CAP. The microbial etiology could be determined in 70% of those patients. The remaining 30% were excluded due to an alternative diagnosis. One of the most likely pathogens to cause a CAP infection is Streptococcus pneumoniae (S pneumonia).27 Patients prescribed a PPI were significantly more likely to be infected with S pneumoniae than those not prescribed a PPI (28% vs 11%). The study concluded that the risk of S pneumoniae in patients taking a PPI was 2.23 times more likely (95% CI, 1.28-3.75).28
Dementia
In 2040, it is estimated that more than 80 million people will have from dementia.29 This is expected to become a large fiscal burden on the health care system. In 2010, about $604 billion was spent on therapy for dementia worldwide.30 Although no cure for dementia exists, it is more feasible than in previous years to prevent its occurrence. However, many medications, including PPIs, are associated with the development of dementia; therefore, it is important to minimize their use when possible.
As noted earlier vitamin B12 deficiency may lead to cognitive decline. Due to the malabsorption of vitamin B12 that results from PPI use, it is hypothesized that PPIs may be associated with incidence of dementia. Badiola and colleagues discovered that in the brains of mice given a PPI, levels of β-amyloid increased significantly affecting enzymes responsible for cognition.31 In a February 2016, JAMA article, researchers conducted a prospective cohort study evaluating 73,679 patients aged ≥75 years with no dementia at baseline. They went on to assess regular use of a PPI, defined as at least 1 PPI prescription every 3 months, and the incidence of dementia. Patients with regular use of a PPI (≥ 1 PPI prescription every 3 months) had a 44% increase risk of incident dementia (HR, 1.44; 95% CI, 1.36-1.52; P < .001).3 Therefore, it is theorized that avoiding PPI use in the elderly may prevent the development of dementia.
Chronic Kidney Disease
The prevalence of CKD has drastically increased in recent decades. It is estimated that up to 13% of people in the U.S. are affected by CKD.32 Some studies suggest that dosing errors occur at much higher rates in patients with declined glomerular filtration rate (GFR).33 The correct utilization use of medications becomes especially pertinent to this population. Several studies have already linked PPI use to acute interstitial nephritis (AIN) and acute kidney injury (AKI).34-36
Lazarus and colleagues evaluated the association between PPI use and the incidence of CKD. Their analysis was performed in a long-term running population-based cohort and replicated in a separate health care system. In the running cohort, patients receiving a PPI had a 1.45-fold greater chance of developing CKD (95% CI, 1.11-1.90; P = .006). In that same cohort, patients on a PPI had a 1.72-fold increase risk of AKI (95% CI, 1.28-2.30; P < .001).4 Similar outcomes were seen in the replicated cohort. However, the replicated cohort did observe that twice daily dosing of a PPI (adjusted HR, 1.46; CI, 1.28-1.67; P < .001) had a stronger association with CKD than once- daily dosing (adjusted HR, 1.15; 95% CI, 1.09-1.21; P < .001). H2RAs exhibited no association with CKD in the running cohort (HR, 1.15; 97% CI, 0.98-1.36; P = .10) or the replication cohort (HR, 0.93; 95% CI, 0.88-0.99; P = .03).4
Clinical PPI Recommendations
There are several FDA-approved and unapproved indications that warrant PPI therapy. Proton pump inhibitor indications include gastroesophageal reflux disease (GERD), peptic ulcer disease (PUD), Helicobacter pylori, and ulcers associated with the use of nonsteroidal anti-inflammatory drugs (NSAIDs).
GERD Recommendations
Optimal dosing and duration is important with all medications to maximize efficacy and minimize toxicity. In the case of PPIs, dosing and duration are of particularly concern due to the aforementioned AEs. Table illustrates manufacturer-recommended dosing and duration for the most commonly prescribed PPIs. Although these dosing regimens are based on clinical studies, PPIs are commonly prescribed at higher doses and for longer durations. By extending the duration of therapy, the risk of potential long-term AEs increases dramatically. If durations are limited to the recommended window, risk of AEs can be reduced.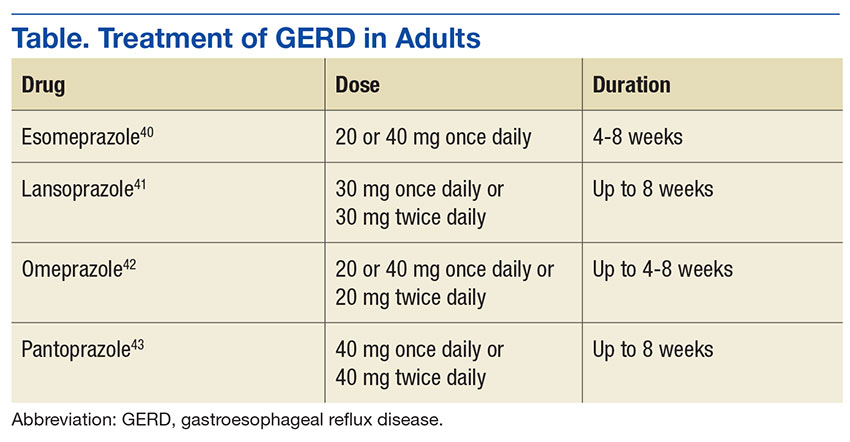
Alternative Therapies
There are several strategies that exist to limit the use of PPIs, including lifestyle modifications to prevent GERD, supplementation of an alternative agent to prevent high doses of the PPI, or discontinuing PPI therapy all together. Lifestyle modifications provide additional benefit as monotherapy or to supplement a pharmacologic regimen.
The American Journal of Gastroenterology promoted lifestyle modifications that include:
- Weight loss for patients with GERD who are overweight and had a recent weight gain;
- Elevation of the head of the bed (if nighttime symptoms present);
- Elimination of dietary triggers;
- Fatty foods, caffeine, chocolate, spicy food, food with high fat content, carbonated beverages, and peppermint;
- Avoiding tight fitting garments to prevent increase in gastric pressure;
- Promote salivation through oral lozenges or chewing gum to neutralize refluxed acid;
- Avoidance of tobacco and alcohol; and
- Abdominal breathing exercise to strengthen the barrier of the lower esophageal sphincter.37
The above modifications may reduce the need for pharmacologic therapy, thereby reducing possible of long-term AEs.
If lifestyle modifications alone are not enough, it is reasonable to use a H2RA for acute symptom relief or reduce high doses and frequencies of a PPI. H2RAs are well studied and effective in the management of GERD. According to the American College of Gastroenterology 2013 clinical practice guidelines, H2RAs can serve as an effective maintenance medication to relieve heartburn in patients without erosive disease. The guideline also states that a bedtime H2RA can be used to supplement a once- daily daytime PPI if nighttime reflux exists. This can eliminate the need to exceed manufacturer-recommended doses.37
One of the final challenges to overcome is a patient that has been maintained on chronic PPI therapy. However, caution should be exercised if choosing to discontinue a PPI. In a study by Niklesson and colleagues, after a 4-week course of pantoprazole given to healthy volunteers, those patients with no preexisting symptoms developed dyspeptic symptoms of GERD, such as heartburn, indigestion, and stomach discomfort. This correlation suggests that a rebound hypersecretion occurs after prolonged suppression of the proton pump, and therefore a gradual taper should be used.38 Although no definitive national recommendations on how to taper a patient off of a PPI exist, one suggestion is a 2- to 3-week taper by using a half-dose once daily or full dose on alternate days.39 This strategy has exhibited moderate success rates when used. Oral and written education on symptom management and the administration of H2RAs for infrequent breakthrough symptoms supplemented the reduction of the PPI.
Conclusion
Proton pump inhibitors have become a popular and effective drug class for a multitude of indications. However, it is crucial to recognize the risk of long-term use. It is important to properly assess the need for a PPI and to use appropriate dosing and duration, since prolonged durations and doses above the manufacturer’s recommendations is a primary contributor to long-term consequences. Both package inserts and clinical guidelines serve as valuable resources to help balance the risks and benefits of this medication class and can help guide therapeutic decisions.
1. U.S. Food and Drug Administration. FDA Drug Safety Communication: Low magnesium levels can be associated with long-term use of Proton Pump Inhibitor drugs (PPIs). http://www.fda.gov/Drugs/DrugSafety/ucm245011.htm. Updated April 7, 2016. Accessed January 12, 2017.
2. Forgacs I. Overprescribing proton pump inhibitors. BMJ. 2008;336(7634):2-3.
3. Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia. JAMA Neurol. 2016;73(4):410-416.
4. Lazarus B, Chen Y, Wilson FP, et al. Proton pump inhibitor use and the risk of chronic kidney disease. JAMA Intern Med. 2016;176(2):238-246.
5. Wolfe MM, Soll AH. The physiology of gastric acid secretion. N Engl J Med. 1988;319(26):1707-1715.
6. Flynn A. The role of dietary calcium in bone health. Proc Nutr Soc. 2003;62(4):851-858.
7. Mizunashi K, Furukawa Y, Katano K, Abe K. Effect of omeprazole, an inhibitor of H+, K(+)-ATPase, on bone resorption in humans. Calcif Tissue Int. 1993;53(1):21-25.
8. O’Connell MB, Darren DM, Murray AM, Heaney RP, Kerzner LJ. Effects of proton pump inhibitors on calcium carbonate absorption in women: a randomized crossover trial. Am J Med. 2005;118(7):778-781.
9. Khalili H, Huang ES, Jacobson BC, Camargo CA Jr, Feskanich D, Chan AT. Use of proton pump inhibitors and risk of hip fracture in relation to dietary and lifestyle factors: a prospective cohort study. BMJ. 2012;344:e372.
10. Schweigel M, Martens H. Magnesium transport in the gastrointestinal tract. Front Biosci. 2000;5:D666-D677.
11. Hess MW, Hoenderop JG, Bindels RJ, Drenth JP. Systematic review: hypomagnesaemia induced by proton pump inhibition. Aliment Pharmacol Ther. 2012;36(5):405-413.
12. William JH, Danziger J. Proton-pump inhibitor-induced hypomagnesemia: current research and proposed mechanisms. World J Nephrol. 2016;5(2):152-157.
13. Luk CP, Parsons R, Lee YP, Hughes JD. Proton pump inhibitor-associated hypomagnesemia: what do FDA data tell us? Ann Pharmacother. 2013;47(6):773-780.
14. Health Quality Ontario. Vitamin B12 and cognitive function: an evidence-based analysis. Ont Health Technol Assess Ser. 2013;13(23):1-45.
15. Green R, Kinsella LJ. Current concepts in the diagnosis of cobalamin deficiency. Neurology. 1995;45(8):1435-1440.
16. Centers for Disease Control and Prevention. The Healthy Brain Initiative. https://www.cdc.gov/aging/pdf/2013-healthy-brain-initiative.pdf. Accessed January 17, 2017.
17. Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med. 1997;337(20):1441-1448.
18. Tefferi A, Pruthi RK. The biochemical basis of cobalamin deficiency. Mayo Clin Proc. 1994;69(2):181-186.
19. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442.
20. Lessa FC, Mu Y, Bamberg WM, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(9):825-834.
21. National Clostridium difficile Standards Group. National Clostridium difficile Standards Group: report to the Department of Health. J Hosp Infect. 2004;56(suppl 1):1-38.
22. Thorens J, Frohlich F, Schwizer W, et al. Bacterial overgrowth during treatment with omeprazole compared with cimetidine. Gut. 1996;39(1):54-59.
23. Dial S, Delaney JAC, Barkun AN, et al. Use of gastric acid-suppressive agents and the risk of community-acquired Clostridium difficile-associated disease. JAMA. 2005;294(23):2989-2995
24. Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; and American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis. 2007;44(suppl 2):S27-S72.
25. Laheij RJ, Sturkenboom MC, Hassing RJ, Dieleman J, Stricker BH, Jansen JB. Risk of community-acquired pneumonia and use of gastric acid-suppressive drugs. JAMA. 2004;292(16):1955-1960.
26. Sarkar M, Hennessy S, Yang Y. Proton-pump inhibitor use and the risk for community-acquired pneumonia. Ann Intern Med. 2008;149(6):391-398.
27. Waterer GW, Wunderink RG. The influence of the severity of community-acquired pneumonia on the usefulness of blood cultures. Respir Med. 2001;95(1):78-82.
28. de Jagar CP, Wever PC, Gemen EF, et al. Proton pump inhibitor therapy predisposes to community-acquired Streptococcus pneumoniae pneumonia. Aliment Pharmacol Ther. 2012;36(10):941-949.
29. Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7(3):137-152.
30. Wimo A, Jönsson L, Bond J, Prince M, Winblad B; Alzheimer Disease International. The worldwide economic impact of dementia 2010. Alzheimers Dement. 2013;9(1):1-11.
31. Badiola N, Alcalde V, Pujol A, et al. The proton-pump inhibitor lansoprazole enhances amyloid beta production. PLoS One. 2013;8(3):e58537.
32. Stevens LA, Li S, Wang C, et al. Prevalence of CKD and comorbid illness in elderly patients in the United States: results from the Kidney Early Evaluation Program (KEEP). Am J Kidney Dis. 2010;55(3)(suppl 2):S23-S33.
33. Weir MR, Fink JC. Safety of medical therapy in patients with chronic kidney disease and end-stage renal disease. Curr Opin Nephrol Hypertens 2014;23(3):306-313.
34. Blank ML, Parkin L, Paul C, Herbison P. A nationwide nested case-control study indicates an increased risk of acute interstitial nephritis with proton pump inhibitor use. Kidney Int. 2014;86(4):837-844.
35. Antoniou T, Macdonald EM, Holland S, et al. Proton pump inhibitors and the risk of acute kidney injury in older patients: a population-based cohort study. CMAJ Open. 2015;3(2):E166-E171.
36. Klepser DG, Collier DS, Cochran GL. Proton pump inhibitors and acute kidney injury: a nested case-control study. BMC Nephrol. 2013;14:150.
37. Katz PO, Gerson LB, Vela MF. Guidelines for the diagnosis and management of gastroesophageal reflux disease. Am J Gastroenterol. 2013;108(3):308-328.
38. Niklasson A, Lindström L, Simrén M, Lindberg G, Björnsson E. Dyspeptic symptom development after discontinuation of a proton pump inhibitor: a double-blind placebo-controlled trial. Am J Gastroenterol. 2010;105(7):1531-1537.
39. Haastrup P, Paulsen MS, Begtrup LM, Hansen JM, Jarbøl DE. Strategies for discontinuation of proton pump inhibitors: a systematic review. Fam Pract. 2014;31(6):625-630.
40. Nexium [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals; 2012.
41. Prevacid [package insert]. Deerfield, IL: Takeda Pharmaceuticals; 2012.
42. Prilosec [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals; 2012.
43. Protonix [package insert]. Konstanz, Germany: Pfizer; 2012.
Proton pump inhibitors (PPIs) are one of the most frequently used drug classes, given that they are readily accessible over-the-counter as well as via prescription. About 100 million PPI prescriptions dispensed an
The human stomach uses 3 primary neurotransmitters that regulate gastric acid secretion: acetylcholine (ACh), histamine (H), and gastrin (G). The interactions between these neurotransmitters promote and inhibit hydrogen ion (H+) generation. Stimulation of their corresponding receptors draws H+ into parietal cells that line the stomach. Once in the cell, a H+-K+-ATPase (more commonly known as the proton pump) actively transports H+ into the lumen of the stomach. The H+ bind with chlorine ions to form hydrochloric acid, which increases stomach acidity.5 Histamine receptors were thought to be responsible for the greatest degree of stimulation. Hence, histamine type 2-receptor antagonists (H2RAs) became a novel means of therapy to reduce stomach acidity. While utilizing H2RAs was effective, it was theorized the downstream inhibition of the action of all 3 neurotransmitters would serve as a more successful therapy. Therefore, PPIs were developed to target the H+-K+-ATPase Over the past decade, many studies have evaluated the long-term PPI adverse effects (AEs). These include calcium and magnesium malabsorption, vitamin B12 deficiency, Clostridium difficile (C difficile) associated disease (CDAD), and community-acquired pneumonia (CAP). Within the past year, data have become available linking PPI use to dementia and chronic kidney disease (CKD).3,4 The following article reviews literature on the safety of long-term PPI use and proposes recommendations for proper use for their most common indications.
Malabsorption
Calcium & Long-Term Fracture Risk
Calcium is an essential component in bone health and formation. In fact, 99% of all calcium found in the body is stored in bones.6 The primary source of calcium is through diet and oral supplements. After it is ingested, calcium is absorbed from the stomach into the blood in a pH dependent manner. If the pH of the stomach is too high (ie, too basic) calcium is not absorbed into blood and remains in the gastrointestinal (GI) tract for fecal excretion. Without sufficient calcium, the body’s osteoclasts and osteoblasts remain inactive, which hinders proper bone turnover.7
The decrease in acidity leads to calcium malabsorption and increases fracture risk long- term.8 Khalili and colleagues surveyed 80,000 postmenopausal women to measure the incidence of hip fracture in women taking PPIs. The study found that there was a 35% increase in risk of hip fracture among women who regularly used PPIs for at least 2 years (age-adjusted hazard ratio [HR] 1.35; 95% confidence interval [CI], 1.13 -1.62). Adjusted HRs for 4-year and 6- to 8-year use of a PPI was 1.42 (95% CI, 1.05-1.93) and 1.55 (95% CI, 1.03-2.32), respectively, indicating that the longer women were on PPI therapy, the higher the risk of hip fracture. The study also evaluated the time since stopping PPI and the risk of hip fracture. Women who stopped PPI use more than 2 years prior had a similar risk to that of women who never used a PPI, indicating that the effect was reversible.9
Magnesium
Magnesium is an important intracellular ion that has a number of key functions in metabolism and ion transport in the human body. Once ingested, magnesium is absorbed into the bloodstream from the small and large intestines via passive and active transport. Transient receptor potential melastatin 6 (TRPM6) is one of the essential proteins that serve as a transporter for magnesium.10 The high affinity for magnesium of these transporters allows them to maintain adequate levels of magnesium in the blood. In states of low magnesium (hypomagnesemia), the body is at risk for many AEs including seizures, arrhythmias, tetany, and hypotension.11
Proton pump inhibitors have been linked to hypomagnesemia, and recent evaluation has clarified a potential mechanism.12 TRPM6 activity is increased in an acidic environment. When a PPI increases the pH of the stomach, TRPM6 and magnesium levels decrease.12 Luk and colleagues identified 66,102 subjects experiencing AEs while taking a PPI. Hypomagnesemia had a prevalence rate of 1% in these patients. According to the researchers, PPIs were associated with hypomagnesemia and that pantoprazole had the highest incidence among all other PPIs studied (OR, 4.3; 95% CI, 3.3 – 5.7; P < .001).13
Vitamin B12
In recent years, vitamin B12 has been the subject of many studies. An area of concern is vitamin B12’s neurologic effect, as it has been successfully demonstrated that vitamin B12 is essential for proper cognitive function.14 Some data suggest that degeneration is present in parts of the spinal column in patients with cognitive decline or neurologic problems. These lesions are due to improper myelin formation and are specific to vitamin B12 deficiency.15 In 2013 the CDC published the Healthy Brain Initiative, which stated cognitive impairment can be caused by vitamin B12 deficiency.16
Similar to calcium, vitamin B12 needs an acidic environment to be digested and absorbed.17 Vitamin B12 is released from food proteins via gastric acid and pepsin. Once free, the vitamin B12 pairs with R-binders secreted in the stomach. Pancreatic enzymes then degrade this complex into a form that can be absorbed into circulation by the intestine. Given that PPIs reduce the acidity of the stomach, they also reduce the body’s ability to release vitamin B12 from food proteins and be paired with the R-binders.18
In 2013, Lam and colleagues evaluated the association between vitamin B12 deficiency and the use of PPIs and H2RAs. An extensive evaluation was performed on 25,956 patients with a diagnosis of vitamin B12 deficiency and 184,199 patients without. About 12% of patients with vitamin B12 deficiency had received more than a 2-year supply of a PPI, whereas only 7.2% of the patients without vitamin B12 deficiency received a 2-year supply of a PPI. Four point 3 percent of patients with vitamin B12 deficiency received more than a 2-year supply of an H2RA. Only 3.2% of patients without vitamin B12 deficiency received more than a 2-year supply of H2RA. The study concluded that a 2-year or greater history of PPI (OR, 1.65; 95% CI, 1.58-1.73) or H2RA (OR, 1.25; 95% CI, 1.17-1.34) use was associated with vitamin B12 deficiency.19
PPIs and Infections
Clostridium difficile-associated disease
Nationwide CDAD has become a prevalent infection nationwide. In 2011, C difficile caused nearly 500,000 infections and was associated with 29,000 deaths in the U.S.20 One study stated that C difficile is the third most common cause of infectious diarrhea in people aged >75 years.21
C difficile is part of the body’s normal flora in the large intestine. It grows and colonizes in an environment of low acidity. Therefore, in the stomach, where the pH is relatively low, C difficile is unable to colonize.22 When a PPI is introduced, the increased gastric pH increases the risk for CDAD.
Dial and colleagues conducted a multicenter case control study to determine whether gastric acid suppression increases the risk of CDAD. Compared with patients who did not take a gastric acid suppressant, those taking a PPI had a 2.9-fold increase in developing CDAD (95% CI, 2.4-3.4). Comparatively, H2RAs had a 2.0-fold increase for CDAD (95% CI, 1.6 to 2.7). These results correlated with the fact that PPIs have a greater impact on gastric pH than do H2RAs.23
Community-Acquired Pneumonia
Community-acquired pneumonia (CAP) has become a growing concern in the U.S. According to the Infectious Disease Society of America (IDSA) and American Thoracic clinical consensus guidelines, CAP remains one of the top reasons for hospitalizations in the U.S., and about 10% of patients admitted to the hospital for CAP end up in the intensive care unit (ICU).24 In the past, PPIs have been linked to patients’ predisposal for developingCAP.25 Although controversial, available evidence suggests a direct association. In 2008 Sarker and colleagues theorized a mechanism that the acid reduction of the gastric lumen allows for increased bacterial colonization in the upper part of the GI tract.26 Since the acidity of the stomach serves as a defense mechanism against many ingested bacteria, many pathogens will be able to survive in the more basic environment.25
Sarkar and colleagues went on to evaluate 80,000 cases over 15 years. The objective was to examine the association between PPI use and the date of diagnosis of the CAP infection, known as the index date. The study demonstrated that PPI use was not associated with increased CAP risk in the long-term (adjusted odds ratio (OR), 1.02; 95% CI, 0.97-1.08). The study did find a strong increase in the risk of CAP if a PPI was started within 2 days (adjusted OR, 6.53; 95% CI, 3.95-10.80), 7 days (adjusted OR, 3.79; 95% CI, 2.66-5.42), and 14 days (adjusted OR, 3.21; 95% CI, 2.46-4.18) of the index date.26
Four years later, de Jagar and colleagues examined the differences in microbial etiology in CAP patients with and without an active PPI. Over a 4-year study period, 463 individuals were selected with clinical suspicion of CAP. The microbial etiology could be determined in 70% of those patients. The remaining 30% were excluded due to an alternative diagnosis. One of the most likely pathogens to cause a CAP infection is Streptococcus pneumoniae (S pneumonia).27 Patients prescribed a PPI were significantly more likely to be infected with S pneumoniae than those not prescribed a PPI (28% vs 11%). The study concluded that the risk of S pneumoniae in patients taking a PPI was 2.23 times more likely (95% CI, 1.28-3.75).28
Dementia
In 2040, it is estimated that more than 80 million people will have from dementia.29 This is expected to become a large fiscal burden on the health care system. In 2010, about $604 billion was spent on therapy for dementia worldwide.30 Although no cure for dementia exists, it is more feasible than in previous years to prevent its occurrence. However, many medications, including PPIs, are associated with the development of dementia; therefore, it is important to minimize their use when possible.
As noted earlier vitamin B12 deficiency may lead to cognitive decline. Due to the malabsorption of vitamin B12 that results from PPI use, it is hypothesized that PPIs may be associated with incidence of dementia. Badiola and colleagues discovered that in the brains of mice given a PPI, levels of β-amyloid increased significantly affecting enzymes responsible for cognition.31 In a February 2016, JAMA article, researchers conducted a prospective cohort study evaluating 73,679 patients aged ≥75 years with no dementia at baseline. They went on to assess regular use of a PPI, defined as at least 1 PPI prescription every 3 months, and the incidence of dementia. Patients with regular use of a PPI (≥ 1 PPI prescription every 3 months) had a 44% increase risk of incident dementia (HR, 1.44; 95% CI, 1.36-1.52; P < .001).3 Therefore, it is theorized that avoiding PPI use in the elderly may prevent the development of dementia.
Chronic Kidney Disease
The prevalence of CKD has drastically increased in recent decades. It is estimated that up to 13% of people in the U.S. are affected by CKD.32 Some studies suggest that dosing errors occur at much higher rates in patients with declined glomerular filtration rate (GFR).33 The correct utilization use of medications becomes especially pertinent to this population. Several studies have already linked PPI use to acute interstitial nephritis (AIN) and acute kidney injury (AKI).34-36
Lazarus and colleagues evaluated the association between PPI use and the incidence of CKD. Their analysis was performed in a long-term running population-based cohort and replicated in a separate health care system. In the running cohort, patients receiving a PPI had a 1.45-fold greater chance of developing CKD (95% CI, 1.11-1.90; P = .006). In that same cohort, patients on a PPI had a 1.72-fold increase risk of AKI (95% CI, 1.28-2.30; P < .001).4 Similar outcomes were seen in the replicated cohort. However, the replicated cohort did observe that twice daily dosing of a PPI (adjusted HR, 1.46; CI, 1.28-1.67; P < .001) had a stronger association with CKD than once- daily dosing (adjusted HR, 1.15; 95% CI, 1.09-1.21; P < .001). H2RAs exhibited no association with CKD in the running cohort (HR, 1.15; 97% CI, 0.98-1.36; P = .10) or the replication cohort (HR, 0.93; 95% CI, 0.88-0.99; P = .03).4
Clinical PPI Recommendations
There are several FDA-approved and unapproved indications that warrant PPI therapy. Proton pump inhibitor indications include gastroesophageal reflux disease (GERD), peptic ulcer disease (PUD), Helicobacter pylori, and ulcers associated with the use of nonsteroidal anti-inflammatory drugs (NSAIDs).
GERD Recommendations
Optimal dosing and duration is important with all medications to maximize efficacy and minimize toxicity. In the case of PPIs, dosing and duration are of particularly concern due to the aforementioned AEs. Table illustrates manufacturer-recommended dosing and duration for the most commonly prescribed PPIs. Although these dosing regimens are based on clinical studies, PPIs are commonly prescribed at higher doses and for longer durations. By extending the duration of therapy, the risk of potential long-term AEs increases dramatically. If durations are limited to the recommended window, risk of AEs can be reduced.
Alternative Therapies
There are several strategies that exist to limit the use of PPIs, including lifestyle modifications to prevent GERD, supplementation of an alternative agent to prevent high doses of the PPI, or discontinuing PPI therapy all together. Lifestyle modifications provide additional benefit as monotherapy or to supplement a pharmacologic regimen.
The American Journal of Gastroenterology promoted lifestyle modifications that include:
- Weight loss for patients with GERD who are overweight and had a recent weight gain;
- Elevation of the head of the bed (if nighttime symptoms present);
- Elimination of dietary triggers;
- Fatty foods, caffeine, chocolate, spicy food, food with high fat content, carbonated beverages, and peppermint;
- Avoiding tight fitting garments to prevent increase in gastric pressure;
- Promote salivation through oral lozenges or chewing gum to neutralize refluxed acid;
- Avoidance of tobacco and alcohol; and
- Abdominal breathing exercise to strengthen the barrier of the lower esophageal sphincter.37
The above modifications may reduce the need for pharmacologic therapy, thereby reducing possible of long-term AEs.
If lifestyle modifications alone are not enough, it is reasonable to use a H2RA for acute symptom relief or reduce high doses and frequencies of a PPI. H2RAs are well studied and effective in the management of GERD. According to the American College of Gastroenterology 2013 clinical practice guidelines, H2RAs can serve as an effective maintenance medication to relieve heartburn in patients without erosive disease. The guideline also states that a bedtime H2RA can be used to supplement a once- daily daytime PPI if nighttime reflux exists. This can eliminate the need to exceed manufacturer-recommended doses.37
One of the final challenges to overcome is a patient that has been maintained on chronic PPI therapy. However, caution should be exercised if choosing to discontinue a PPI. In a study by Niklesson and colleagues, after a 4-week course of pantoprazole given to healthy volunteers, those patients with no preexisting symptoms developed dyspeptic symptoms of GERD, such as heartburn, indigestion, and stomach discomfort. This correlation suggests that a rebound hypersecretion occurs after prolonged suppression of the proton pump, and therefore a gradual taper should be used.38 Although no definitive national recommendations on how to taper a patient off of a PPI exist, one suggestion is a 2- to 3-week taper by using a half-dose once daily or full dose on alternate days.39 This strategy has exhibited moderate success rates when used. Oral and written education on symptom management and the administration of H2RAs for infrequent breakthrough symptoms supplemented the reduction of the PPI.
Conclusion
Proton pump inhibitors have become a popular and effective drug class for a multitude of indications. However, it is crucial to recognize the risk of long-term use. It is important to properly assess the need for a PPI and to use appropriate dosing and duration, since prolonged durations and doses above the manufacturer’s recommendations is a primary contributor to long-term consequences. Both package inserts and clinical guidelines serve as valuable resources to help balance the risks and benefits of this medication class and can help guide therapeutic decisions.
Proton pump inhibitors (PPIs) are one of the most frequently used drug classes, given that they are readily accessible over-the-counter as well as via prescription. About 100 million PPI prescriptions dispensed an
The human stomach uses 3 primary neurotransmitters that regulate gastric acid secretion: acetylcholine (ACh), histamine (H), and gastrin (G). The interactions between these neurotransmitters promote and inhibit hydrogen ion (H+) generation. Stimulation of their corresponding receptors draws H+ into parietal cells that line the stomach. Once in the cell, a H+-K+-ATPase (more commonly known as the proton pump) actively transports H+ into the lumen of the stomach. The H+ bind with chlorine ions to form hydrochloric acid, which increases stomach acidity.5 Histamine receptors were thought to be responsible for the greatest degree of stimulation. Hence, histamine type 2-receptor antagonists (H2RAs) became a novel means of therapy to reduce stomach acidity. While utilizing H2RAs was effective, it was theorized the downstream inhibition of the action of all 3 neurotransmitters would serve as a more successful therapy. Therefore, PPIs were developed to target the H+-K+-ATPase Over the past decade, many studies have evaluated the long-term PPI adverse effects (AEs). These include calcium and magnesium malabsorption, vitamin B12 deficiency, Clostridium difficile (C difficile) associated disease (CDAD), and community-acquired pneumonia (CAP). Within the past year, data have become available linking PPI use to dementia and chronic kidney disease (CKD).3,4 The following article reviews literature on the safety of long-term PPI use and proposes recommendations for proper use for their most common indications.
Malabsorption
Calcium & Long-Term Fracture Risk
Calcium is an essential component in bone health and formation. In fact, 99% of all calcium found in the body is stored in bones.6 The primary source of calcium is through diet and oral supplements. After it is ingested, calcium is absorbed from the stomach into the blood in a pH dependent manner. If the pH of the stomach is too high (ie, too basic) calcium is not absorbed into blood and remains in the gastrointestinal (GI) tract for fecal excretion. Without sufficient calcium, the body’s osteoclasts and osteoblasts remain inactive, which hinders proper bone turnover.7
The decrease in acidity leads to calcium malabsorption and increases fracture risk long- term.8 Khalili and colleagues surveyed 80,000 postmenopausal women to measure the incidence of hip fracture in women taking PPIs. The study found that there was a 35% increase in risk of hip fracture among women who regularly used PPIs for at least 2 years (age-adjusted hazard ratio [HR] 1.35; 95% confidence interval [CI], 1.13 -1.62). Adjusted HRs for 4-year and 6- to 8-year use of a PPI was 1.42 (95% CI, 1.05-1.93) and 1.55 (95% CI, 1.03-2.32), respectively, indicating that the longer women were on PPI therapy, the higher the risk of hip fracture. The study also evaluated the time since stopping PPI and the risk of hip fracture. Women who stopped PPI use more than 2 years prior had a similar risk to that of women who never used a PPI, indicating that the effect was reversible.9
Magnesium
Magnesium is an important intracellular ion that has a number of key functions in metabolism and ion transport in the human body. Once ingested, magnesium is absorbed into the bloodstream from the small and large intestines via passive and active transport. Transient receptor potential melastatin 6 (TRPM6) is one of the essential proteins that serve as a transporter for magnesium.10 The high affinity for magnesium of these transporters allows them to maintain adequate levels of magnesium in the blood. In states of low magnesium (hypomagnesemia), the body is at risk for many AEs including seizures, arrhythmias, tetany, and hypotension.11
Proton pump inhibitors have been linked to hypomagnesemia, and recent evaluation has clarified a potential mechanism.12 TRPM6 activity is increased in an acidic environment. When a PPI increases the pH of the stomach, TRPM6 and magnesium levels decrease.12 Luk and colleagues identified 66,102 subjects experiencing AEs while taking a PPI. Hypomagnesemia had a prevalence rate of 1% in these patients. According to the researchers, PPIs were associated with hypomagnesemia and that pantoprazole had the highest incidence among all other PPIs studied (OR, 4.3; 95% CI, 3.3 – 5.7; P < .001).13
Vitamin B12
In recent years, vitamin B12 has been the subject of many studies. An area of concern is vitamin B12’s neurologic effect, as it has been successfully demonstrated that vitamin B12 is essential for proper cognitive function.14 Some data suggest that degeneration is present in parts of the spinal column in patients with cognitive decline or neurologic problems. These lesions are due to improper myelin formation and are specific to vitamin B12 deficiency.15 In 2013 the CDC published the Healthy Brain Initiative, which stated cognitive impairment can be caused by vitamin B12 deficiency.16
Similar to calcium, vitamin B12 needs an acidic environment to be digested and absorbed.17 Vitamin B12 is released from food proteins via gastric acid and pepsin. Once free, the vitamin B12 pairs with R-binders secreted in the stomach. Pancreatic enzymes then degrade this complex into a form that can be absorbed into circulation by the intestine. Given that PPIs reduce the acidity of the stomach, they also reduce the body’s ability to release vitamin B12 from food proteins and be paired with the R-binders.18
In 2013, Lam and colleagues evaluated the association between vitamin B12 deficiency and the use of PPIs and H2RAs. An extensive evaluation was performed on 25,956 patients with a diagnosis of vitamin B12 deficiency and 184,199 patients without. About 12% of patients with vitamin B12 deficiency had received more than a 2-year supply of a PPI, whereas only 7.2% of the patients without vitamin B12 deficiency received a 2-year supply of a PPI. Four point 3 percent of patients with vitamin B12 deficiency received more than a 2-year supply of an H2RA. Only 3.2% of patients without vitamin B12 deficiency received more than a 2-year supply of H2RA. The study concluded that a 2-year or greater history of PPI (OR, 1.65; 95% CI, 1.58-1.73) or H2RA (OR, 1.25; 95% CI, 1.17-1.34) use was associated with vitamin B12 deficiency.19
PPIs and Infections
Clostridium difficile-associated disease
Nationwide CDAD has become a prevalent infection nationwide. In 2011, C difficile caused nearly 500,000 infections and was associated with 29,000 deaths in the U.S.20 One study stated that C difficile is the third most common cause of infectious diarrhea in people aged >75 years.21
C difficile is part of the body’s normal flora in the large intestine. It grows and colonizes in an environment of low acidity. Therefore, in the stomach, where the pH is relatively low, C difficile is unable to colonize.22 When a PPI is introduced, the increased gastric pH increases the risk for CDAD.
Dial and colleagues conducted a multicenter case control study to determine whether gastric acid suppression increases the risk of CDAD. Compared with patients who did not take a gastric acid suppressant, those taking a PPI had a 2.9-fold increase in developing CDAD (95% CI, 2.4-3.4). Comparatively, H2RAs had a 2.0-fold increase for CDAD (95% CI, 1.6 to 2.7). These results correlated with the fact that PPIs have a greater impact on gastric pH than do H2RAs.23
Community-Acquired Pneumonia
Community-acquired pneumonia (CAP) has become a growing concern in the U.S. According to the Infectious Disease Society of America (IDSA) and American Thoracic clinical consensus guidelines, CAP remains one of the top reasons for hospitalizations in the U.S., and about 10% of patients admitted to the hospital for CAP end up in the intensive care unit (ICU).24 In the past, PPIs have been linked to patients’ predisposal for developingCAP.25 Although controversial, available evidence suggests a direct association. In 2008 Sarker and colleagues theorized a mechanism that the acid reduction of the gastric lumen allows for increased bacterial colonization in the upper part of the GI tract.26 Since the acidity of the stomach serves as a defense mechanism against many ingested bacteria, many pathogens will be able to survive in the more basic environment.25
Sarkar and colleagues went on to evaluate 80,000 cases over 15 years. The objective was to examine the association between PPI use and the date of diagnosis of the CAP infection, known as the index date. The study demonstrated that PPI use was not associated with increased CAP risk in the long-term (adjusted odds ratio (OR), 1.02; 95% CI, 0.97-1.08). The study did find a strong increase in the risk of CAP if a PPI was started within 2 days (adjusted OR, 6.53; 95% CI, 3.95-10.80), 7 days (adjusted OR, 3.79; 95% CI, 2.66-5.42), and 14 days (adjusted OR, 3.21; 95% CI, 2.46-4.18) of the index date.26
Four years later, de Jagar and colleagues examined the differences in microbial etiology in CAP patients with and without an active PPI. Over a 4-year study period, 463 individuals were selected with clinical suspicion of CAP. The microbial etiology could be determined in 70% of those patients. The remaining 30% were excluded due to an alternative diagnosis. One of the most likely pathogens to cause a CAP infection is Streptococcus pneumoniae (S pneumonia).27 Patients prescribed a PPI were significantly more likely to be infected with S pneumoniae than those not prescribed a PPI (28% vs 11%). The study concluded that the risk of S pneumoniae in patients taking a PPI was 2.23 times more likely (95% CI, 1.28-3.75).28
Dementia
In 2040, it is estimated that more than 80 million people will have from dementia.29 This is expected to become a large fiscal burden on the health care system. In 2010, about $604 billion was spent on therapy for dementia worldwide.30 Although no cure for dementia exists, it is more feasible than in previous years to prevent its occurrence. However, many medications, including PPIs, are associated with the development of dementia; therefore, it is important to minimize their use when possible.
As noted earlier vitamin B12 deficiency may lead to cognitive decline. Due to the malabsorption of vitamin B12 that results from PPI use, it is hypothesized that PPIs may be associated with incidence of dementia. Badiola and colleagues discovered that in the brains of mice given a PPI, levels of β-amyloid increased significantly affecting enzymes responsible for cognition.31 In a February 2016, JAMA article, researchers conducted a prospective cohort study evaluating 73,679 patients aged ≥75 years with no dementia at baseline. They went on to assess regular use of a PPI, defined as at least 1 PPI prescription every 3 months, and the incidence of dementia. Patients with regular use of a PPI (≥ 1 PPI prescription every 3 months) had a 44% increase risk of incident dementia (HR, 1.44; 95% CI, 1.36-1.52; P < .001).3 Therefore, it is theorized that avoiding PPI use in the elderly may prevent the development of dementia.
Chronic Kidney Disease
The prevalence of CKD has drastically increased in recent decades. It is estimated that up to 13% of people in the U.S. are affected by CKD.32 Some studies suggest that dosing errors occur at much higher rates in patients with declined glomerular filtration rate (GFR).33 The correct utilization use of medications becomes especially pertinent to this population. Several studies have already linked PPI use to acute interstitial nephritis (AIN) and acute kidney injury (AKI).34-36
Lazarus and colleagues evaluated the association between PPI use and the incidence of CKD. Their analysis was performed in a long-term running population-based cohort and replicated in a separate health care system. In the running cohort, patients receiving a PPI had a 1.45-fold greater chance of developing CKD (95% CI, 1.11-1.90; P = .006). In that same cohort, patients on a PPI had a 1.72-fold increase risk of AKI (95% CI, 1.28-2.30; P < .001).4 Similar outcomes were seen in the replicated cohort. However, the replicated cohort did observe that twice daily dosing of a PPI (adjusted HR, 1.46; CI, 1.28-1.67; P < .001) had a stronger association with CKD than once- daily dosing (adjusted HR, 1.15; 95% CI, 1.09-1.21; P < .001). H2RAs exhibited no association with CKD in the running cohort (HR, 1.15; 97% CI, 0.98-1.36; P = .10) or the replication cohort (HR, 0.93; 95% CI, 0.88-0.99; P = .03).4
Clinical PPI Recommendations
There are several FDA-approved and unapproved indications that warrant PPI therapy. Proton pump inhibitor indications include gastroesophageal reflux disease (GERD), peptic ulcer disease (PUD), Helicobacter pylori, and ulcers associated with the use of nonsteroidal anti-inflammatory drugs (NSAIDs).
GERD Recommendations
Optimal dosing and duration is important with all medications to maximize efficacy and minimize toxicity. In the case of PPIs, dosing and duration are of particularly concern due to the aforementioned AEs. Table illustrates manufacturer-recommended dosing and duration for the most commonly prescribed PPIs. Although these dosing regimens are based on clinical studies, PPIs are commonly prescribed at higher doses and for longer durations. By extending the duration of therapy, the risk of potential long-term AEs increases dramatically. If durations are limited to the recommended window, risk of AEs can be reduced.
Alternative Therapies
There are several strategies that exist to limit the use of PPIs, including lifestyle modifications to prevent GERD, supplementation of an alternative agent to prevent high doses of the PPI, or discontinuing PPI therapy all together. Lifestyle modifications provide additional benefit as monotherapy or to supplement a pharmacologic regimen.
The American Journal of Gastroenterology promoted lifestyle modifications that include:
- Weight loss for patients with GERD who are overweight and had a recent weight gain;
- Elevation of the head of the bed (if nighttime symptoms present);
- Elimination of dietary triggers;
- Fatty foods, caffeine, chocolate, spicy food, food with high fat content, carbonated beverages, and peppermint;
- Avoiding tight fitting garments to prevent increase in gastric pressure;
- Promote salivation through oral lozenges or chewing gum to neutralize refluxed acid;
- Avoidance of tobacco and alcohol; and
- Abdominal breathing exercise to strengthen the barrier of the lower esophageal sphincter.37
The above modifications may reduce the need for pharmacologic therapy, thereby reducing possible of long-term AEs.
If lifestyle modifications alone are not enough, it is reasonable to use a H2RA for acute symptom relief or reduce high doses and frequencies of a PPI. H2RAs are well studied and effective in the management of GERD. According to the American College of Gastroenterology 2013 clinical practice guidelines, H2RAs can serve as an effective maintenance medication to relieve heartburn in patients without erosive disease. The guideline also states that a bedtime H2RA can be used to supplement a once- daily daytime PPI if nighttime reflux exists. This can eliminate the need to exceed manufacturer-recommended doses.37
One of the final challenges to overcome is a patient that has been maintained on chronic PPI therapy. However, caution should be exercised if choosing to discontinue a PPI. In a study by Niklesson and colleagues, after a 4-week course of pantoprazole given to healthy volunteers, those patients with no preexisting symptoms developed dyspeptic symptoms of GERD, such as heartburn, indigestion, and stomach discomfort. This correlation suggests that a rebound hypersecretion occurs after prolonged suppression of the proton pump, and therefore a gradual taper should be used.38 Although no definitive national recommendations on how to taper a patient off of a PPI exist, one suggestion is a 2- to 3-week taper by using a half-dose once daily or full dose on alternate days.39 This strategy has exhibited moderate success rates when used. Oral and written education on symptom management and the administration of H2RAs for infrequent breakthrough symptoms supplemented the reduction of the PPI.
Conclusion
Proton pump inhibitors have become a popular and effective drug class for a multitude of indications. However, it is crucial to recognize the risk of long-term use. It is important to properly assess the need for a PPI and to use appropriate dosing and duration, since prolonged durations and doses above the manufacturer’s recommendations is a primary contributor to long-term consequences. Both package inserts and clinical guidelines serve as valuable resources to help balance the risks and benefits of this medication class and can help guide therapeutic decisions.
1. U.S. Food and Drug Administration. FDA Drug Safety Communication: Low magnesium levels can be associated with long-term use of Proton Pump Inhibitor drugs (PPIs). http://www.fda.gov/Drugs/DrugSafety/ucm245011.htm. Updated April 7, 2016. Accessed January 12, 2017.
2. Forgacs I. Overprescribing proton pump inhibitors. BMJ. 2008;336(7634):2-3.
3. Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia. JAMA Neurol. 2016;73(4):410-416.
4. Lazarus B, Chen Y, Wilson FP, et al. Proton pump inhibitor use and the risk of chronic kidney disease. JAMA Intern Med. 2016;176(2):238-246.
5. Wolfe MM, Soll AH. The physiology of gastric acid secretion. N Engl J Med. 1988;319(26):1707-1715.
6. Flynn A. The role of dietary calcium in bone health. Proc Nutr Soc. 2003;62(4):851-858.
7. Mizunashi K, Furukawa Y, Katano K, Abe K. Effect of omeprazole, an inhibitor of H+, K(+)-ATPase, on bone resorption in humans. Calcif Tissue Int. 1993;53(1):21-25.
8. O’Connell MB, Darren DM, Murray AM, Heaney RP, Kerzner LJ. Effects of proton pump inhibitors on calcium carbonate absorption in women: a randomized crossover trial. Am J Med. 2005;118(7):778-781.
9. Khalili H, Huang ES, Jacobson BC, Camargo CA Jr, Feskanich D, Chan AT. Use of proton pump inhibitors and risk of hip fracture in relation to dietary and lifestyle factors: a prospective cohort study. BMJ. 2012;344:e372.
10. Schweigel M, Martens H. Magnesium transport in the gastrointestinal tract. Front Biosci. 2000;5:D666-D677.
11. Hess MW, Hoenderop JG, Bindels RJ, Drenth JP. Systematic review: hypomagnesaemia induced by proton pump inhibition. Aliment Pharmacol Ther. 2012;36(5):405-413.
12. William JH, Danziger J. Proton-pump inhibitor-induced hypomagnesemia: current research and proposed mechanisms. World J Nephrol. 2016;5(2):152-157.
13. Luk CP, Parsons R, Lee YP, Hughes JD. Proton pump inhibitor-associated hypomagnesemia: what do FDA data tell us? Ann Pharmacother. 2013;47(6):773-780.
14. Health Quality Ontario. Vitamin B12 and cognitive function: an evidence-based analysis. Ont Health Technol Assess Ser. 2013;13(23):1-45.
15. Green R, Kinsella LJ. Current concepts in the diagnosis of cobalamin deficiency. Neurology. 1995;45(8):1435-1440.
16. Centers for Disease Control and Prevention. The Healthy Brain Initiative. https://www.cdc.gov/aging/pdf/2013-healthy-brain-initiative.pdf. Accessed January 17, 2017.
17. Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med. 1997;337(20):1441-1448.
18. Tefferi A, Pruthi RK. The biochemical basis of cobalamin deficiency. Mayo Clin Proc. 1994;69(2):181-186.
19. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442.
20. Lessa FC, Mu Y, Bamberg WM, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(9):825-834.
21. National Clostridium difficile Standards Group. National Clostridium difficile Standards Group: report to the Department of Health. J Hosp Infect. 2004;56(suppl 1):1-38.
22. Thorens J, Frohlich F, Schwizer W, et al. Bacterial overgrowth during treatment with omeprazole compared with cimetidine. Gut. 1996;39(1):54-59.
23. Dial S, Delaney JAC, Barkun AN, et al. Use of gastric acid-suppressive agents and the risk of community-acquired Clostridium difficile-associated disease. JAMA. 2005;294(23):2989-2995
24. Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; and American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis. 2007;44(suppl 2):S27-S72.
25. Laheij RJ, Sturkenboom MC, Hassing RJ, Dieleman J, Stricker BH, Jansen JB. Risk of community-acquired pneumonia and use of gastric acid-suppressive drugs. JAMA. 2004;292(16):1955-1960.
26. Sarkar M, Hennessy S, Yang Y. Proton-pump inhibitor use and the risk for community-acquired pneumonia. Ann Intern Med. 2008;149(6):391-398.
27. Waterer GW, Wunderink RG. The influence of the severity of community-acquired pneumonia on the usefulness of blood cultures. Respir Med. 2001;95(1):78-82.
28. de Jagar CP, Wever PC, Gemen EF, et al. Proton pump inhibitor therapy predisposes to community-acquired Streptococcus pneumoniae pneumonia. Aliment Pharmacol Ther. 2012;36(10):941-949.
29. Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7(3):137-152.
30. Wimo A, Jönsson L, Bond J, Prince M, Winblad B; Alzheimer Disease International. The worldwide economic impact of dementia 2010. Alzheimers Dement. 2013;9(1):1-11.
31. Badiola N, Alcalde V, Pujol A, et al. The proton-pump inhibitor lansoprazole enhances amyloid beta production. PLoS One. 2013;8(3):e58537.
32. Stevens LA, Li S, Wang C, et al. Prevalence of CKD and comorbid illness in elderly patients in the United States: results from the Kidney Early Evaluation Program (KEEP). Am J Kidney Dis. 2010;55(3)(suppl 2):S23-S33.
33. Weir MR, Fink JC. Safety of medical therapy in patients with chronic kidney disease and end-stage renal disease. Curr Opin Nephrol Hypertens 2014;23(3):306-313.
34. Blank ML, Parkin L, Paul C, Herbison P. A nationwide nested case-control study indicates an increased risk of acute interstitial nephritis with proton pump inhibitor use. Kidney Int. 2014;86(4):837-844.
35. Antoniou T, Macdonald EM, Holland S, et al. Proton pump inhibitors and the risk of acute kidney injury in older patients: a population-based cohort study. CMAJ Open. 2015;3(2):E166-E171.
36. Klepser DG, Collier DS, Cochran GL. Proton pump inhibitors and acute kidney injury: a nested case-control study. BMC Nephrol. 2013;14:150.
37. Katz PO, Gerson LB, Vela MF. Guidelines for the diagnosis and management of gastroesophageal reflux disease. Am J Gastroenterol. 2013;108(3):308-328.
38. Niklasson A, Lindström L, Simrén M, Lindberg G, Björnsson E. Dyspeptic symptom development after discontinuation of a proton pump inhibitor: a double-blind placebo-controlled trial. Am J Gastroenterol. 2010;105(7):1531-1537.
39. Haastrup P, Paulsen MS, Begtrup LM, Hansen JM, Jarbøl DE. Strategies for discontinuation of proton pump inhibitors: a systematic review. Fam Pract. 2014;31(6):625-630.
40. Nexium [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals; 2012.
41. Prevacid [package insert]. Deerfield, IL: Takeda Pharmaceuticals; 2012.
42. Prilosec [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals; 2012.
43. Protonix [package insert]. Konstanz, Germany: Pfizer; 2012.
1. U.S. Food and Drug Administration. FDA Drug Safety Communication: Low magnesium levels can be associated with long-term use of Proton Pump Inhibitor drugs (PPIs). http://www.fda.gov/Drugs/DrugSafety/ucm245011.htm. Updated April 7, 2016. Accessed January 12, 2017.
2. Forgacs I. Overprescribing proton pump inhibitors. BMJ. 2008;336(7634):2-3.
3. Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia. JAMA Neurol. 2016;73(4):410-416.
4. Lazarus B, Chen Y, Wilson FP, et al. Proton pump inhibitor use and the risk of chronic kidney disease. JAMA Intern Med. 2016;176(2):238-246.
5. Wolfe MM, Soll AH. The physiology of gastric acid secretion. N Engl J Med. 1988;319(26):1707-1715.
6. Flynn A. The role of dietary calcium in bone health. Proc Nutr Soc. 2003;62(4):851-858.
7. Mizunashi K, Furukawa Y, Katano K, Abe K. Effect of omeprazole, an inhibitor of H+, K(+)-ATPase, on bone resorption in humans. Calcif Tissue Int. 1993;53(1):21-25.
8. O’Connell MB, Darren DM, Murray AM, Heaney RP, Kerzner LJ. Effects of proton pump inhibitors on calcium carbonate absorption in women: a randomized crossover trial. Am J Med. 2005;118(7):778-781.
9. Khalili H, Huang ES, Jacobson BC, Camargo CA Jr, Feskanich D, Chan AT. Use of proton pump inhibitors and risk of hip fracture in relation to dietary and lifestyle factors: a prospective cohort study. BMJ. 2012;344:e372.
10. Schweigel M, Martens H. Magnesium transport in the gastrointestinal tract. Front Biosci. 2000;5:D666-D677.
11. Hess MW, Hoenderop JG, Bindels RJ, Drenth JP. Systematic review: hypomagnesaemia induced by proton pump inhibition. Aliment Pharmacol Ther. 2012;36(5):405-413.
12. William JH, Danziger J. Proton-pump inhibitor-induced hypomagnesemia: current research and proposed mechanisms. World J Nephrol. 2016;5(2):152-157.
13. Luk CP, Parsons R, Lee YP, Hughes JD. Proton pump inhibitor-associated hypomagnesemia: what do FDA data tell us? Ann Pharmacother. 2013;47(6):773-780.
14. Health Quality Ontario. Vitamin B12 and cognitive function: an evidence-based analysis. Ont Health Technol Assess Ser. 2013;13(23):1-45.
15. Green R, Kinsella LJ. Current concepts in the diagnosis of cobalamin deficiency. Neurology. 1995;45(8):1435-1440.
16. Centers for Disease Control and Prevention. The Healthy Brain Initiative. https://www.cdc.gov/aging/pdf/2013-healthy-brain-initiative.pdf. Accessed January 17, 2017.
17. Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med. 1997;337(20):1441-1448.
18. Tefferi A, Pruthi RK. The biochemical basis of cobalamin deficiency. Mayo Clin Proc. 1994;69(2):181-186.
19. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442.
20. Lessa FC, Mu Y, Bamberg WM, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(9):825-834.
21. National Clostridium difficile Standards Group. National Clostridium difficile Standards Group: report to the Department of Health. J Hosp Infect. 2004;56(suppl 1):1-38.
22. Thorens J, Frohlich F, Schwizer W, et al. Bacterial overgrowth during treatment with omeprazole compared with cimetidine. Gut. 1996;39(1):54-59.
23. Dial S, Delaney JAC, Barkun AN, et al. Use of gastric acid-suppressive agents and the risk of community-acquired Clostridium difficile-associated disease. JAMA. 2005;294(23):2989-2995
24. Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; and American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis. 2007;44(suppl 2):S27-S72.
25. Laheij RJ, Sturkenboom MC, Hassing RJ, Dieleman J, Stricker BH, Jansen JB. Risk of community-acquired pneumonia and use of gastric acid-suppressive drugs. JAMA. 2004;292(16):1955-1960.
26. Sarkar M, Hennessy S, Yang Y. Proton-pump inhibitor use and the risk for community-acquired pneumonia. Ann Intern Med. 2008;149(6):391-398.
27. Waterer GW, Wunderink RG. The influence of the severity of community-acquired pneumonia on the usefulness of blood cultures. Respir Med. 2001;95(1):78-82.
28. de Jagar CP, Wever PC, Gemen EF, et al. Proton pump inhibitor therapy predisposes to community-acquired Streptococcus pneumoniae pneumonia. Aliment Pharmacol Ther. 2012;36(10):941-949.
29. Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7(3):137-152.
30. Wimo A, Jönsson L, Bond J, Prince M, Winblad B; Alzheimer Disease International. The worldwide economic impact of dementia 2010. Alzheimers Dement. 2013;9(1):1-11.
31. Badiola N, Alcalde V, Pujol A, et al. The proton-pump inhibitor lansoprazole enhances amyloid beta production. PLoS One. 2013;8(3):e58537.
32. Stevens LA, Li S, Wang C, et al. Prevalence of CKD and comorbid illness in elderly patients in the United States: results from the Kidney Early Evaluation Program (KEEP). Am J Kidney Dis. 2010;55(3)(suppl 2):S23-S33.
33. Weir MR, Fink JC. Safety of medical therapy in patients with chronic kidney disease and end-stage renal disease. Curr Opin Nephrol Hypertens 2014;23(3):306-313.
34. Blank ML, Parkin L, Paul C, Herbison P. A nationwide nested case-control study indicates an increased risk of acute interstitial nephritis with proton pump inhibitor use. Kidney Int. 2014;86(4):837-844.
35. Antoniou T, Macdonald EM, Holland S, et al. Proton pump inhibitors and the risk of acute kidney injury in older patients: a population-based cohort study. CMAJ Open. 2015;3(2):E166-E171.
36. Klepser DG, Collier DS, Cochran GL. Proton pump inhibitors and acute kidney injury: a nested case-control study. BMC Nephrol. 2013;14:150.
37. Katz PO, Gerson LB, Vela MF. Guidelines for the diagnosis and management of gastroesophageal reflux disease. Am J Gastroenterol. 2013;108(3):308-328.
38. Niklasson A, Lindström L, Simrén M, Lindberg G, Björnsson E. Dyspeptic symptom development after discontinuation of a proton pump inhibitor: a double-blind placebo-controlled trial. Am J Gastroenterol. 2010;105(7):1531-1537.
39. Haastrup P, Paulsen MS, Begtrup LM, Hansen JM, Jarbøl DE. Strategies for discontinuation of proton pump inhibitors: a systematic review. Fam Pract. 2014;31(6):625-630.
40. Nexium [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals; 2012.
41. Prevacid [package insert]. Deerfield, IL: Takeda Pharmaceuticals; 2012.
42. Prilosec [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals; 2012.
43. Protonix [package insert]. Konstanz, Germany: Pfizer; 2012.
Team creates online database of cancer mutations

Image by Spencer Phillips
Researchers have developed an online “knowledgebase” called CIViC, an open access resource for collecting and interpreting information from scientific publications on cancer genetics.
“CIViC” stands for Clinical Interpretations of Variants in Cancer, and the researchers liken it to a Wikipedia of cancer genetics.
Anyone can create an account and contribute information. That information is then curated by editors and moderators who are considered experts in the field.
The researchers described the resource in Nature Genetics.
“It’s relatively easy now to sequence the DNA of tumors—to gather the raw information—but there’s a big interpretation problem,” said study author Obi L. Griffith, PhD, of Washington University School of Medicine in St Louis, Missouri.
“What do these hundreds or thousands of mutations mean for this patient? There are a lot of studies being done to answer these questions. But oncologists trying to interpret the raw data are faced with an overwhelming task of plumbing the literature, reading papers, trying to understand what the latest studies tell them about these mutations and how they may or may not be important.”
The CIViC knowledgebase is an attempt to solve this problem. The researchers said this is one of many efforts to collect and interpret such information, but, to their knowledge, CIViC is the only one that is entirely open access. Anyone is free to contribute and use the content as well as the source code.
“We are committed to keeping this resource open and available to anyone who wants to contribute or make use of the information,” said Malachi Griffith, PhD, of Washington University School of Medicine.
“We would like it to be a community exercise and public resource. The information is in the public domain. There are no restrictions on its use, academic or commercial.”
Though anyone can submit a new piece of information or suggest edits to existing data, at least 2 independent contributors must agree that the new information should be incorporated, and 1 of those users must be an “expert editor.”
Expert editors are not permitted to approve their own submissions. Information on the CIViC website provides details about how new users may be promoted to expert editors and administrators.
To date, the site has seen over 17,500 users from academic institutions, governmental organizations, and commercial entities around the world.
Since CIViC’s launch, 59 users have volunteered their time to contribute their knowledge to CIViC, including descriptions of the clinical relevance of 732 mutations from 285 genes for 203 types of cancer, all gleaned from reviewing 1090 scientific and medical publications.
Despite the fact that there are many groups attempting to collect and interpret genomic variants in cancer, the researchers said the sheer volume of information has resulted in relatively little overlap in data gathered so far.
“While we believe this is the only such open access knowledgebase, there are other large research centers with similar resources,” Malachi Griffith said. “We did an analysis to compare the big ones.”
“Even though we all have access to the same published literature, if you look at the overlap of the information mined by each of these resources, it’s remarkably small. We’re all approaching the same problem, and, just by chance—and probably because of the amount of information out there—we haven’t duplicated our efforts very much yet.”
Obi and Malachi Griffith said finding a way to combine these resources is the primary goal of an international group they are helping lead called the Variant Interpretation for Cancer Consortium, which is a part of the Global Alliance for Genomics and Health (GA4GH).
“We’re just scratching the surface of the potential this holds for precision medicine,” Obi Griffith said. “There’s a lot of work to do.” ![]()
Image by Spencer Phillips
Researchers have developed an online “knowledgebase” called CIViC, an open access resource for collecting and interpreting information from scientific publications on cancer genetics.
“CIViC” stands for Clinical Interpretations of Variants in Cancer, and the researchers liken it to a Wikipedia of cancer genetics.
Anyone can create an account and contribute information. That information is then curated by editors and moderators who are considered experts in the field.
The researchers described the resource in Nature Genetics.
“It’s relatively easy now to sequence the DNA of tumors—to gather the raw information—but there’s a big interpretation problem,” said study author Obi L. Griffith, PhD, of Washington University School of Medicine in St Louis, Missouri.
“What do these hundreds or thousands of mutations mean for this patient? There are a lot of studies being done to answer these questions. But oncologists trying to interpret the raw data are faced with an overwhelming task of plumbing the literature, reading papers, trying to understand what the latest studies tell them about these mutations and how they may or may not be important.”
The CIViC knowledgebase is an attempt to solve this problem. The researchers said this is one of many efforts to collect and interpret such information, but, to their knowledge, CIViC is the only one that is entirely open access. Anyone is free to contribute and use the content as well as the source code.
“We are committed to keeping this resource open and available to anyone who wants to contribute or make use of the information,” said Malachi Griffith, PhD, of Washington University School of Medicine.
“We would like it to be a community exercise and public resource. The information is in the public domain. There are no restrictions on its use, academic or commercial.”
Though anyone can submit a new piece of information or suggest edits to existing data, at least 2 independent contributors must agree that the new information should be incorporated, and 1 of those users must be an “expert editor.”
Expert editors are not permitted to approve their own submissions. Information on the CIViC website provides details about how new users may be promoted to expert editors and administrators.
To date, the site has seen over 17,500 users from academic institutions, governmental organizations, and commercial entities around the world.
Since CIViC’s launch, 59 users have volunteered their time to contribute their knowledge to CIViC, including descriptions of the clinical relevance of 732 mutations from 285 genes for 203 types of cancer, all gleaned from reviewing 1090 scientific and medical publications.
Despite the fact that there are many groups attempting to collect and interpret genomic variants in cancer, the researchers said the sheer volume of information has resulted in relatively little overlap in data gathered so far.
“While we believe this is the only such open access knowledgebase, there are other large research centers with similar resources,” Malachi Griffith said. “We did an analysis to compare the big ones.”
“Even though we all have access to the same published literature, if you look at the overlap of the information mined by each of these resources, it’s remarkably small. We’re all approaching the same problem, and, just by chance—and probably because of the amount of information out there—we haven’t duplicated our efforts very much yet.”
Obi and Malachi Griffith said finding a way to combine these resources is the primary goal of an international group they are helping lead called the Variant Interpretation for Cancer Consortium, which is a part of the Global Alliance for Genomics and Health (GA4GH).
“We’re just scratching the surface of the potential this holds for precision medicine,” Obi Griffith said. “There’s a lot of work to do.” ![]()
Image by Spencer Phillips
Researchers have developed an online “knowledgebase” called CIViC, an open access resource for collecting and interpreting information from scientific publications on cancer genetics.
“CIViC” stands for Clinical Interpretations of Variants in Cancer, and the researchers liken it to a Wikipedia of cancer genetics.
Anyone can create an account and contribute information. That information is then curated by editors and moderators who are considered experts in the field.
The researchers described the resource in Nature Genetics.
“It’s relatively easy now to sequence the DNA of tumors—to gather the raw information—but there’s a big interpretation problem,” said study author Obi L. Griffith, PhD, of Washington University School of Medicine in St Louis, Missouri.
“What do these hundreds or thousands of mutations mean for this patient? There are a lot of studies being done to answer these questions. But oncologists trying to interpret the raw data are faced with an overwhelming task of plumbing the literature, reading papers, trying to understand what the latest studies tell them about these mutations and how they may or may not be important.”
The CIViC knowledgebase is an attempt to solve this problem. The researchers said this is one of many efforts to collect and interpret such information, but, to their knowledge, CIViC is the only one that is entirely open access. Anyone is free to contribute and use the content as well as the source code.
“We are committed to keeping this resource open and available to anyone who wants to contribute or make use of the information,” said Malachi Griffith, PhD, of Washington University School of Medicine.
“We would like it to be a community exercise and public resource. The information is in the public domain. There are no restrictions on its use, academic or commercial.”
Though anyone can submit a new piece of information or suggest edits to existing data, at least 2 independent contributors must agree that the new information should be incorporated, and 1 of those users must be an “expert editor.”
Expert editors are not permitted to approve their own submissions. Information on the CIViC website provides details about how new users may be promoted to expert editors and administrators.
To date, the site has seen over 17,500 users from academic institutions, governmental organizations, and commercial entities around the world.
Since CIViC’s launch, 59 users have volunteered their time to contribute their knowledge to CIViC, including descriptions of the clinical relevance of 732 mutations from 285 genes for 203 types of cancer, all gleaned from reviewing 1090 scientific and medical publications.
Despite the fact that there are many groups attempting to collect and interpret genomic variants in cancer, the researchers said the sheer volume of information has resulted in relatively little overlap in data gathered so far.
“While we believe this is the only such open access knowledgebase, there are other large research centers with similar resources,” Malachi Griffith said. “We did an analysis to compare the big ones.”
“Even though we all have access to the same published literature, if you look at the overlap of the information mined by each of these resources, it’s remarkably small. We’re all approaching the same problem, and, just by chance—and probably because of the amount of information out there—we haven’t duplicated our efforts very much yet.”
Obi and Malachi Griffith said finding a way to combine these resources is the primary goal of an international group they are helping lead called the Variant Interpretation for Cancer Consortium, which is a part of the Global Alliance for Genomics and Health (GA4GH).
“We’re just scratching the surface of the potential this holds for precision medicine,” Obi Griffith said. “There’s a lot of work to do.” ![]()
Noninfectious Penile Lesions
1. A 26-year-old man presents with a penile lesion that has existed at least 10 years without significant change and with no attendant symptoms. The lesion consists of four 1-to-1.5-mm soft, compressible purple papules, in aggregate measuring about 8 mm. No other lesions are seen on the genitals or on the body.
Diagnosis: The lesion proved to be angiokeratoma of Fordyce, a totally benign lesion. This type of angiokeratoma is ectatic, thin-walled vessels in the superficial dermis, with overlying epidermal hyperplasia that forms secondary to normal friction. When they are seen on the scrotum, vulva, or penis, they are usually referred to as angiokeratoma of Fordyce, a type of localized angiokeratoma, other types of which can appear on the legs or hands.
These totally benign lesions must be distinguished from generalized types of angiokeratomata, such as those seen in Fabry disease (angiokeratoma corporis diffusum), an inheritable metabolic disorder. With our patient’s history, his lesion was clearly benign.
Had his lesion been new or changing in any substantive way, additional testing, including a biopsy, might have been necessary to rule out entities such as squamous cell carcinoma (which is almost unknown in circumcised patients), condyloma, melanoma, the aforementioned Kaposi’s sarcoma, or even angiosarcoma.
For more information, see “Skin is skin, no matter the location.” Clinician Reviews. 2013 June;23(6):W5.
2. A 59-year-old uncircumcised man presents with a phimotic foreskin, which cannot be retracted without pain. Only a tiny opening remains through which the patient can urinate (with difficulty). The surface of the foreskin is atrophic, dry, and shiny with focal areas of purpura but little, if any, redness or swelling.
Diagnosis: This patient’s condition is lichen sclerosus, also called balanitis xerotica obliterans (BXO), a diagnosis of sufficient obscurity to almost guarantee initial misdiagnosis as “yeast infection” or “herpes.” One treatment failure after another eventually leads to referral to a provider familiar with BXO, which is the male version of lichen sclerosus et atrophicus (LS&A) and usually affects the glans, foreskin, and distal shaft.
The causes of these conditions are as yet unknown. However, much is known about how they present, how they look under a microscope, and how to treat them.
Treatment entails use of the most powerful topical steroid ointments, which are so effective that they have completely replaced previous treatment options (eg, testosterone ointment). While a cure is unlikely, control is a realistic goal.
BXO, as in this case, can also cause urinary obstruction, both from the overlying foreskin and from actual meatal stenosis. This is why advanced cases need referral to urology for possible circumcision. As with many penile diagnoses (eg, squamous cell carcinoma or condyloma), BXO is far more common in the uncircumcised.
For more information, see “Man has very uncomfortable problem.” Clinician Reviews. 2012 September;22(9):W4.
3. A 31-year-old man has a relatively asymptomatic penile rash that has repeatedly manifested and resolved over a period of months. A round, papulosquamous, bright pink patch on the distal right shaft of his circumcised penis measures > 3 cm in diameter and has a shiny appearance with slightly irregular margins. A similar rash is seen behind both ears, in the umbilicus, and patches of dandruff are noted. Despite good health, he has been under a great deal of stress recently.
Diagnosis: Seborrheic dermatitis (SD), also known as seborrhea, is an extremely common chronic papulosquamous disorder patterned on the sebum-rich areas of the scalp, face, and trunk. Although not directly caused by the highly lipophilic commensal yeast Malassezia furfur, it does appear to be related to increases in the number of those organisms, as well as to immunologic abnormalities and increased production of sebum. It can range from a mild scaly rash to whole-body erythroderma and can affect an astonishing range of areas, including the genitals.
SD, especially in this case, represents the perfect example of the need to “look elsewhere” for clues when confronted with a mysterious rash. Patients can certainly have more than one dermatologic diagnosis at a time, but a single explanation is considerably more likely and should therefore be sought. In this case, corroboration for the diagnosis of SD was readily found by looking for it in its known locations.
In this case, treatment comprised a combination of oxiconazole lotion and 2.5% hydrocortisone cream. Many other combinations have been used successfully, including pimecrolimus or tacrolimus combined with ketoconazole cream.
Whatever is used, a cure will not be forthcoming, since the condition is almost always chronic. The main value of an accurate diagnosis in such a case lies in easing the patient’s mind regarding the terrible diseases he doesn’t have.
For more information, see “Relatively Asymptomatic, but Still Problematic." Clinician Reviews. 2014 April;24(4):15-16.
4. This circumscribed inflammatory plaque on the glans penis of allegedly > 20 years' duration was refractory to circumcision and local treatment. Because of unresponsiveness of the lesion to circumcision and focal steroid infiltration, repeated biopsies were performed.
Diagnosis: One biopsy again showed the features of a plasmacellular inflammation, while the other finally revealed the histopathologic features of erythroplasia of Queyrat (carcinoma in situ or Bowen's disease of the glans penis). We assume that either the former biopsy specimens were taken from a plasma cell-rich reactive infiltrate around the neoplastic lesion, or that carcinoma in situ may have arisen due to the chronic inflammation of Zoon's balanitis plasmacellularis. Radiotherapy was performed with good clinical response and subsequent histopathologic proof of complete remission of the lesion.
For more information, see “Bowen's Disease of the Glans Penis (Erythroplasia of Queyrat) in Plasma Cell Balanitis.” Cutis. 2000 June;65(6):395-398.
1. A 26-year-old man presents with a penile lesion that has existed at least 10 years without significant change and with no attendant symptoms. The lesion consists of four 1-to-1.5-mm soft, compressible purple papules, in aggregate measuring about 8 mm. No other lesions are seen on the genitals or on the body.
Diagnosis: The lesion proved to be angiokeratoma of Fordyce, a totally benign lesion. This type of angiokeratoma is ectatic, thin-walled vessels in the superficial dermis, with overlying epidermal hyperplasia that forms secondary to normal friction. When they are seen on the scrotum, vulva, or penis, they are usually referred to as angiokeratoma of Fordyce, a type of localized angiokeratoma, other types of which can appear on the legs or hands.
These totally benign lesions must be distinguished from generalized types of angiokeratomata, such as those seen in Fabry disease (angiokeratoma corporis diffusum), an inheritable metabolic disorder. With our patient’s history, his lesion was clearly benign.
Had his lesion been new or changing in any substantive way, additional testing, including a biopsy, might have been necessary to rule out entities such as squamous cell carcinoma (which is almost unknown in circumcised patients), condyloma, melanoma, the aforementioned Kaposi’s sarcoma, or even angiosarcoma.
For more information, see “Skin is skin, no matter the location.” Clinician Reviews. 2013 June;23(6):W5.
2. A 59-year-old uncircumcised man presents with a phimotic foreskin, which cannot be retracted without pain. Only a tiny opening remains through which the patient can urinate (with difficulty). The surface of the foreskin is atrophic, dry, and shiny with focal areas of purpura but little, if any, redness or swelling.
Diagnosis: This patient’s condition is lichen sclerosus, also called balanitis xerotica obliterans (BXO), a diagnosis of sufficient obscurity to almost guarantee initial misdiagnosis as “yeast infection” or “herpes.” One treatment failure after another eventually leads to referral to a provider familiar with BXO, which is the male version of lichen sclerosus et atrophicus (LS&A) and usually affects the glans, foreskin, and distal shaft.
The causes of these conditions are as yet unknown. However, much is known about how they present, how they look under a microscope, and how to treat them.
Treatment entails use of the most powerful topical steroid ointments, which are so effective that they have completely replaced previous treatment options (eg, testosterone ointment). While a cure is unlikely, control is a realistic goal.
BXO, as in this case, can also cause urinary obstruction, both from the overlying foreskin and from actual meatal stenosis. This is why advanced cases need referral to urology for possible circumcision. As with many penile diagnoses (eg, squamous cell carcinoma or condyloma), BXO is far more common in the uncircumcised.
For more information, see “Man has very uncomfortable problem.” Clinician Reviews. 2012 September;22(9):W4.
3. A 31-year-old man has a relatively asymptomatic penile rash that has repeatedly manifested and resolved over a period of months. A round, papulosquamous, bright pink patch on the distal right shaft of his circumcised penis measures > 3 cm in diameter and has a shiny appearance with slightly irregular margins. A similar rash is seen behind both ears, in the umbilicus, and patches of dandruff are noted. Despite good health, he has been under a great deal of stress recently.
Diagnosis: Seborrheic dermatitis (SD), also known as seborrhea, is an extremely common chronic papulosquamous disorder patterned on the sebum-rich areas of the scalp, face, and trunk. Although not directly caused by the highly lipophilic commensal yeast Malassezia furfur, it does appear to be related to increases in the number of those organisms, as well as to immunologic abnormalities and increased production of sebum. It can range from a mild scaly rash to whole-body erythroderma and can affect an astonishing range of areas, including the genitals.
SD, especially in this case, represents the perfect example of the need to “look elsewhere” for clues when confronted with a mysterious rash. Patients can certainly have more than one dermatologic diagnosis at a time, but a single explanation is considerably more likely and should therefore be sought. In this case, corroboration for the diagnosis of SD was readily found by looking for it in its known locations.
In this case, treatment comprised a combination of oxiconazole lotion and 2.5% hydrocortisone cream. Many other combinations have been used successfully, including pimecrolimus or tacrolimus combined with ketoconazole cream.
Whatever is used, a cure will not be forthcoming, since the condition is almost always chronic. The main value of an accurate diagnosis in such a case lies in easing the patient’s mind regarding the terrible diseases he doesn’t have.
For more information, see “Relatively Asymptomatic, but Still Problematic." Clinician Reviews. 2014 April;24(4):15-16.
4. This circumscribed inflammatory plaque on the glans penis of allegedly > 20 years' duration was refractory to circumcision and local treatment. Because of unresponsiveness of the lesion to circumcision and focal steroid infiltration, repeated biopsies were performed.
Diagnosis: One biopsy again showed the features of a plasmacellular inflammation, while the other finally revealed the histopathologic features of erythroplasia of Queyrat (carcinoma in situ or Bowen's disease of the glans penis). We assume that either the former biopsy specimens were taken from a plasma cell-rich reactive infiltrate around the neoplastic lesion, or that carcinoma in situ may have arisen due to the chronic inflammation of Zoon's balanitis plasmacellularis. Radiotherapy was performed with good clinical response and subsequent histopathologic proof of complete remission of the lesion.
For more information, see “Bowen's Disease of the Glans Penis (Erythroplasia of Queyrat) in Plasma Cell Balanitis.” Cutis. 2000 June;65(6):395-398.
1. A 26-year-old man presents with a penile lesion that has existed at least 10 years without significant change and with no attendant symptoms. The lesion consists of four 1-to-1.5-mm soft, compressible purple papules, in aggregate measuring about 8 mm. No other lesions are seen on the genitals or on the body.
Diagnosis: The lesion proved to be angiokeratoma of Fordyce, a totally benign lesion. This type of angiokeratoma is ectatic, thin-walled vessels in the superficial dermis, with overlying epidermal hyperplasia that forms secondary to normal friction. When they are seen on the scrotum, vulva, or penis, they are usually referred to as angiokeratoma of Fordyce, a type of localized angiokeratoma, other types of which can appear on the legs or hands.
These totally benign lesions must be distinguished from generalized types of angiokeratomata, such as those seen in Fabry disease (angiokeratoma corporis diffusum), an inheritable metabolic disorder. With our patient’s history, his lesion was clearly benign.
Had his lesion been new or changing in any substantive way, additional testing, including a biopsy, might have been necessary to rule out entities such as squamous cell carcinoma (which is almost unknown in circumcised patients), condyloma, melanoma, the aforementioned Kaposi’s sarcoma, or even angiosarcoma.
For more information, see “Skin is skin, no matter the location.” Clinician Reviews. 2013 June;23(6):W5.
2. A 59-year-old uncircumcised man presents with a phimotic foreskin, which cannot be retracted without pain. Only a tiny opening remains through which the patient can urinate (with difficulty). The surface of the foreskin is atrophic, dry, and shiny with focal areas of purpura but little, if any, redness or swelling.
Diagnosis: This patient’s condition is lichen sclerosus, also called balanitis xerotica obliterans (BXO), a diagnosis of sufficient obscurity to almost guarantee initial misdiagnosis as “yeast infection” or “herpes.” One treatment failure after another eventually leads to referral to a provider familiar with BXO, which is the male version of lichen sclerosus et atrophicus (LS&A) and usually affects the glans, foreskin, and distal shaft.
The causes of these conditions are as yet unknown. However, much is known about how they present, how they look under a microscope, and how to treat them.
Treatment entails use of the most powerful topical steroid ointments, which are so effective that they have completely replaced previous treatment options (eg, testosterone ointment). While a cure is unlikely, control is a realistic goal.
BXO, as in this case, can also cause urinary obstruction, both from the overlying foreskin and from actual meatal stenosis. This is why advanced cases need referral to urology for possible circumcision. As with many penile diagnoses (eg, squamous cell carcinoma or condyloma), BXO is far more common in the uncircumcised.
For more information, see “Man has very uncomfortable problem.” Clinician Reviews. 2012 September;22(9):W4.
3. A 31-year-old man has a relatively asymptomatic penile rash that has repeatedly manifested and resolved over a period of months. A round, papulosquamous, bright pink patch on the distal right shaft of his circumcised penis measures > 3 cm in diameter and has a shiny appearance with slightly irregular margins. A similar rash is seen behind both ears, in the umbilicus, and patches of dandruff are noted. Despite good health, he has been under a great deal of stress recently.
Diagnosis: Seborrheic dermatitis (SD), also known as seborrhea, is an extremely common chronic papulosquamous disorder patterned on the sebum-rich areas of the scalp, face, and trunk. Although not directly caused by the highly lipophilic commensal yeast Malassezia furfur, it does appear to be related to increases in the number of those organisms, as well as to immunologic abnormalities and increased production of sebum. It can range from a mild scaly rash to whole-body erythroderma and can affect an astonishing range of areas, including the genitals.
SD, especially in this case, represents the perfect example of the need to “look elsewhere” for clues when confronted with a mysterious rash. Patients can certainly have more than one dermatologic diagnosis at a time, but a single explanation is considerably more likely and should therefore be sought. In this case, corroboration for the diagnosis of SD was readily found by looking for it in its known locations.
In this case, treatment comprised a combination of oxiconazole lotion and 2.5% hydrocortisone cream. Many other combinations have been used successfully, including pimecrolimus or tacrolimus combined with ketoconazole cream.
Whatever is used, a cure will not be forthcoming, since the condition is almost always chronic. The main value of an accurate diagnosis in such a case lies in easing the patient’s mind regarding the terrible diseases he doesn’t have.
For more information, see “Relatively Asymptomatic, but Still Problematic." Clinician Reviews. 2014 April;24(4):15-16.
4. This circumscribed inflammatory plaque on the glans penis of allegedly > 20 years' duration was refractory to circumcision and local treatment. Because of unresponsiveness of the lesion to circumcision and focal steroid infiltration, repeated biopsies were performed.
Diagnosis: One biopsy again showed the features of a plasmacellular inflammation, while the other finally revealed the histopathologic features of erythroplasia of Queyrat (carcinoma in situ or Bowen's disease of the glans penis). We assume that either the former biopsy specimens were taken from a plasma cell-rich reactive infiltrate around the neoplastic lesion, or that carcinoma in situ may have arisen due to the chronic inflammation of Zoon's balanitis plasmacellularis. Radiotherapy was performed with good clinical response and subsequent histopathologic proof of complete remission of the lesion.
For more information, see “Bowen's Disease of the Glans Penis (Erythroplasia of Queyrat) in Plasma Cell Balanitis.” Cutis. 2000 June;65(6):395-398.
NSAIDs and cardiovascular risk
Clinical question: Which NSAID confers the least cardiovascular risk?
Background: Nonsteroidal anti-inflammatory drugs (NSAIDs) reduce pain and inflammation by decreasing prostaglandin production through cyclo-oxygenase (COX) inhibition. However, the COX enzyme stimulates protective prostaglandins for the GI mucosa. The development of cyclo-oxygenase-2 (COX-2) inhibitors did reduce the gastrointestinal side effects of NSAIDs, but subsequent trials pointed to increased cardiovascular events and resulted in rofecoxib being removed from the market. Celecoxib remained on the market as the only COX-2 inhibitor, but the Food and Drug Administration required a cardiovascular safety trial.
Study design: Prospective randomized, double-blind noninferiority study.
Setting: International (926 centers in 13 countries).
Synopsis: The PRECISION trial enrolled 24,081 patients who required NSAIDs for arthritis pain and who either had established cardiovascular disease or an increased risk of CV disease. Patients were randomized to celecoxib 100 mg twice a day, ibuprofen 600 mg three times a day, or naproxen 375 mg twice a day. Patients were allowed to continue taking low-dose aspirin (325 mg or less daily). The primary composite outcome was cardiovascular death (including hemorrhagic death), nonfatal myocardial infarction, or nonfatal stroke. A secondary composite outcome was major adverse CV events (the primary outcome plus coronary revascularization or hospitalization for unstable angina or transient ischemic attack) and significant gastrointestinal events.
The primary outcome occurred in 188 patients in the celecoxib group (2.3%), 201 in the naproxen group (2.5%), and 218 in the ibuprofen group (2.7%). Celecoxib, as compared with either naproxen or ibuprofen, met all four prespecified noninferiority requirements (P less than .01 for noninferiority in both comparisons. Ibuprofen, as compared with naproxen, just met the noninferiority criteria (P = .025).
Bottom line: The PRECISION trial provides statistically strong evidence that the cardiovascular risk associated with moderate doses of celecoxib is not greater than that associated with nonselective NSAIDs (ibuprofen and naproxen).
Citations: Nissen SE, Yeomans ND, Solomon DH, et al. Cardiovascular safety of celecoxib, naproxen, or ibuprofen for arthritis.” N Engl J M. 2016;375(26):2519-2529.
Dr. Cerceo is an assistant professor in the Division of Hospital Medicine, and associate director of the internal medicine residency program at Cooper Medical School of Rowan University, Camden, N.J.
Clinical question: Which NSAID confers the least cardiovascular risk?
Background: Nonsteroidal anti-inflammatory drugs (NSAIDs) reduce pain and inflammation by decreasing prostaglandin production through cyclo-oxygenase (COX) inhibition. However, the COX enzyme stimulates protective prostaglandins for the GI mucosa. The development of cyclo-oxygenase-2 (COX-2) inhibitors did reduce the gastrointestinal side effects of NSAIDs, but subsequent trials pointed to increased cardiovascular events and resulted in rofecoxib being removed from the market. Celecoxib remained on the market as the only COX-2 inhibitor, but the Food and Drug Administration required a cardiovascular safety trial.
Study design: Prospective randomized, double-blind noninferiority study.
Setting: International (926 centers in 13 countries).
Synopsis: The PRECISION trial enrolled 24,081 patients who required NSAIDs for arthritis pain and who either had established cardiovascular disease or an increased risk of CV disease. Patients were randomized to celecoxib 100 mg twice a day, ibuprofen 600 mg three times a day, or naproxen 375 mg twice a day. Patients were allowed to continue taking low-dose aspirin (325 mg or less daily). The primary composite outcome was cardiovascular death (including hemorrhagic death), nonfatal myocardial infarction, or nonfatal stroke. A secondary composite outcome was major adverse CV events (the primary outcome plus coronary revascularization or hospitalization for unstable angina or transient ischemic attack) and significant gastrointestinal events.
The primary outcome occurred in 188 patients in the celecoxib group (2.3%), 201 in the naproxen group (2.5%), and 218 in the ibuprofen group (2.7%). Celecoxib, as compared with either naproxen or ibuprofen, met all four prespecified noninferiority requirements (P less than .01 for noninferiority in both comparisons. Ibuprofen, as compared with naproxen, just met the noninferiority criteria (P = .025).
Bottom line: The PRECISION trial provides statistically strong evidence that the cardiovascular risk associated with moderate doses of celecoxib is not greater than that associated with nonselective NSAIDs (ibuprofen and naproxen).
Citations: Nissen SE, Yeomans ND, Solomon DH, et al. Cardiovascular safety of celecoxib, naproxen, or ibuprofen for arthritis.” N Engl J M. 2016;375(26):2519-2529.
Dr. Cerceo is an assistant professor in the Division of Hospital Medicine, and associate director of the internal medicine residency program at Cooper Medical School of Rowan University, Camden, N.J.
Clinical question: Which NSAID confers the least cardiovascular risk?
Background: Nonsteroidal anti-inflammatory drugs (NSAIDs) reduce pain and inflammation by decreasing prostaglandin production through cyclo-oxygenase (COX) inhibition. However, the COX enzyme stimulates protective prostaglandins for the GI mucosa. The development of cyclo-oxygenase-2 (COX-2) inhibitors did reduce the gastrointestinal side effects of NSAIDs, but subsequent trials pointed to increased cardiovascular events and resulted in rofecoxib being removed from the market. Celecoxib remained on the market as the only COX-2 inhibitor, but the Food and Drug Administration required a cardiovascular safety trial.
Study design: Prospective randomized, double-blind noninferiority study.
Setting: International (926 centers in 13 countries).
Synopsis: The PRECISION trial enrolled 24,081 patients who required NSAIDs for arthritis pain and who either had established cardiovascular disease or an increased risk of CV disease. Patients were randomized to celecoxib 100 mg twice a day, ibuprofen 600 mg three times a day, or naproxen 375 mg twice a day. Patients were allowed to continue taking low-dose aspirin (325 mg or less daily). The primary composite outcome was cardiovascular death (including hemorrhagic death), nonfatal myocardial infarction, or nonfatal stroke. A secondary composite outcome was major adverse CV events (the primary outcome plus coronary revascularization or hospitalization for unstable angina or transient ischemic attack) and significant gastrointestinal events.
The primary outcome occurred in 188 patients in the celecoxib group (2.3%), 201 in the naproxen group (2.5%), and 218 in the ibuprofen group (2.7%). Celecoxib, as compared with either naproxen or ibuprofen, met all four prespecified noninferiority requirements (P less than .01 for noninferiority in both comparisons. Ibuprofen, as compared with naproxen, just met the noninferiority criteria (P = .025).
Bottom line: The PRECISION trial provides statistically strong evidence that the cardiovascular risk associated with moderate doses of celecoxib is not greater than that associated with nonselective NSAIDs (ibuprofen and naproxen).
Citations: Nissen SE, Yeomans ND, Solomon DH, et al. Cardiovascular safety of celecoxib, naproxen, or ibuprofen for arthritis.” N Engl J M. 2016;375(26):2519-2529.
Dr. Cerceo is an assistant professor in the Division of Hospital Medicine, and associate director of the internal medicine residency program at Cooper Medical School of Rowan University, Camden, N.J.
Palliative care: a systematic review for patients and their caregivers
Clinical question: What is the association of palliative care programs on quality of life, symptoms, and survival for patients and their caregivers?
Background: Palliative care programs have expanded across the country: More than 65% of U.S. hospitals have such a program. Efforts have been made to assess their effectiveness for terminally ill patients and their caregivers.
Study design: Systematic review and meta-analysis of 43 randomized controlled trials.
Setting: Not applicable
Synopsis: Two reviewers independently assessed 43 trials (12,731 patients and 2,479 caregivers) with the main outcomes being quality of life, symptom burden, survival, mood, advance care planning, site of death, health care satisfaction, resource utilization, and health care expenditures. Estimates of QOL were translated to units of the Functional Assessment of Chronic Illness Therapy–palliative care scale (FACIT-Pal) instrument and symptom burden was translated into the Edmonton Symptom Assessment Scale (ESAS). Palliative care was associated with statistically and clinically significant improvements in patient QOL at the 1- to 3-month follow-up (standardized mean difference, 0.46; 95% CI, 0.08-0.83; FACIT-Pal mean difference, 11.36) and symptom burden at the 1- to 3-month follow-up (standardized mean difference, −0.66; 95% CI, −1.25 to −0.07; ESAS mean difference, −10.30). 
When analyses were limited to trials at low risk of bias (n = 5), the association between palliative care and QOL was attenuated but remained statistically significant (standardized mean difference, 0.20; 95% CI, 0.06-0.34; FACIT-Pal mean difference, 4.94), whereas the association with symptom burden was no longer statistically significant (standardized mean difference, −0.21; 95% CI, −0.42-0.00; ESAS mean difference, −3.28). Caregiver outcomes were mixed but with limited quality of evidence.
Bottom line: Although there was no significant association between palliative care and survival, palliative interventions were associated with improved patient QOL, patient and caregiver satisfaction, lower health care utilization, and symptom burden.
Citations: Kavalieratos D, Corbelli J, Zhang D, et al. Association between palliative care and patient and caregiver outcomes: A systematic review and meta-analysis. JAMA. 2016;316(20):2104-2114. doi: 10.1001/jama.2016.16840
Dr. Cerceo is an assistant professor in the Division of Hospital Medicine, and associate director of the internal medicine residency program at Cooper Medical School of Rowan University, Camden, N.J.
Clinical question: What is the association of palliative care programs on quality of life, symptoms, and survival for patients and their caregivers?
Background: Palliative care programs have expanded across the country: More than 65% of U.S. hospitals have such a program. Efforts have been made to assess their effectiveness for terminally ill patients and their caregivers.
Study design: Systematic review and meta-analysis of 43 randomized controlled trials.
Setting: Not applicable
Synopsis: Two reviewers independently assessed 43 trials (12,731 patients and 2,479 caregivers) with the main outcomes being quality of life, symptom burden, survival, mood, advance care planning, site of death, health care satisfaction, resource utilization, and health care expenditures. Estimates of QOL were translated to units of the Functional Assessment of Chronic Illness Therapy–palliative care scale (FACIT-Pal) instrument and symptom burden was translated into the Edmonton Symptom Assessment Scale (ESAS). Palliative care was associated with statistically and clinically significant improvements in patient QOL at the 1- to 3-month follow-up (standardized mean difference, 0.46; 95% CI, 0.08-0.83; FACIT-Pal mean difference, 11.36) and symptom burden at the 1- to 3-month follow-up (standardized mean difference, −0.66; 95% CI, −1.25 to −0.07; ESAS mean difference, −10.30).
When analyses were limited to trials at low risk of bias (n = 5), the association between palliative care and QOL was attenuated but remained statistically significant (standardized mean difference, 0.20; 95% CI, 0.06-0.34; FACIT-Pal mean difference, 4.94), whereas the association with symptom burden was no longer statistically significant (standardized mean difference, −0.21; 95% CI, −0.42-0.00; ESAS mean difference, −3.28). Caregiver outcomes were mixed but with limited quality of evidence.
Bottom line: Although there was no significant association between palliative care and survival, palliative interventions were associated with improved patient QOL, patient and caregiver satisfaction, lower health care utilization, and symptom burden.
Citations: Kavalieratos D, Corbelli J, Zhang D, et al. Association between palliative care and patient and caregiver outcomes: A systematic review and meta-analysis. JAMA. 2016;316(20):2104-2114. doi: 10.1001/jama.2016.16840
Dr. Cerceo is an assistant professor in the Division of Hospital Medicine, and associate director of the internal medicine residency program at Cooper Medical School of Rowan University, Camden, N.J.
Clinical question: What is the association of palliative care programs on quality of life, symptoms, and survival for patients and their caregivers?
Background: Palliative care programs have expanded across the country: More than 65% of U.S. hospitals have such a program. Efforts have been made to assess their effectiveness for terminally ill patients and their caregivers.
Study design: Systematic review and meta-analysis of 43 randomized controlled trials.
Setting: Not applicable
Synopsis: Two reviewers independently assessed 43 trials (12,731 patients and 2,479 caregivers) with the main outcomes being quality of life, symptom burden, survival, mood, advance care planning, site of death, health care satisfaction, resource utilization, and health care expenditures. Estimates of QOL were translated to units of the Functional Assessment of Chronic Illness Therapy–palliative care scale (FACIT-Pal) instrument and symptom burden was translated into the Edmonton Symptom Assessment Scale (ESAS). Palliative care was associated with statistically and clinically significant improvements in patient QOL at the 1- to 3-month follow-up (standardized mean difference, 0.46; 95% CI, 0.08-0.83; FACIT-Pal mean difference, 11.36) and symptom burden at the 1- to 3-month follow-up (standardized mean difference, −0.66; 95% CI, −1.25 to −0.07; ESAS mean difference, −10.30).
When analyses were limited to trials at low risk of bias (n = 5), the association between palliative care and QOL was attenuated but remained statistically significant (standardized mean difference, 0.20; 95% CI, 0.06-0.34; FACIT-Pal mean difference, 4.94), whereas the association with symptom burden was no longer statistically significant (standardized mean difference, −0.21; 95% CI, −0.42-0.00; ESAS mean difference, −3.28). Caregiver outcomes were mixed but with limited quality of evidence.
Bottom line: Although there was no significant association between palliative care and survival, palliative interventions were associated with improved patient QOL, patient and caregiver satisfaction, lower health care utilization, and symptom burden.
Citations: Kavalieratos D, Corbelli J, Zhang D, et al. Association between palliative care and patient and caregiver outcomes: A systematic review and meta-analysis. JAMA. 2016;316(20):2104-2114. doi: 10.1001/jama.2016.16840
Dr. Cerceo is an assistant professor in the Division of Hospital Medicine, and associate director of the internal medicine residency program at Cooper Medical School of Rowan University, Camden, N.J.
Esophageal retractor found handy tool in AF ablation
ORLANDO – A relatively simple mechanical tool to move the esophagus away from the energy delivered during ablation of atrial fibrillation (AF) does what it is supposed to do, according to data from a multicenter observational study presented at the AF Symposium 2017.
When esophageal temperature during the ablation procedure was monitored in 101 consecutive cases, no recording exceeded 38° C, according to Valay Parikh, MD, a clinical cardiac electrophysiology fellow working under Dhanunjaya Lakkireddy, MD, at the Kansas University Medical Center, Kansas City.
The tool is a stylet constructed from a nickel-titanium (nitinol) alloy. Malleable at room temperature, the stylet is inserted into an 18 Fr orogastric (OG) tube. Firmer at body temperature, the stylet within the OG tube is maneuvered to displace the esophagus away from the adjacent left atrium when radiofrequency ablation (RFA) is being administered.
The tool, marketed under the brand name EsoSure, was first made available almost 2 years ago, but the recently completed multicenter observational study was conducted to provide a more systematic evaluation of its safety and efficacy in routine use. In this study, 101 consecutive patients scheduled for RFA for AF had their esophagus displaced by the stylet during the procedure. The temperature of the esophagus as well as any adverse events involving the upper gastrointestinal tract were evaluated during the procedure. Patients were then followed for at least 6 months.
“The principal finding of our study is that mechanical displacement of the esophagus with the help of the EsoSure device is safe and provides sufficient room to deliver the intended energy at the site of ablation without any rise in temperature over 38° C,” Dr. Parikh reported.
The mean age of the 101 patients who participated in this study was 65 years. About half were female. The mean body mass index (BMI) was 32 kg/m2. The mean CHA2DS2-VASc score was 2.4. Barium x-rays were used to confirm esophageal displacement.
After the procedure, patients were discharged on a proton pump inhibitor and sucralfate, which inhibits pepsin activity and protects against ulceration. Follow-up endoscopy was performed only when medically indicated, but all patients were evaluated over the course of follow-up for odynophagia, dysphagia, hematemesis, dyspepsia, and other GI symptoms.
Pulmonary vein isolation (PVI) was achieved successfully in all cases. Although there was a rapid temperature rise at the site of ablation over the course of RFA, the esophagus was adequately displaced from the source of energy, as confirmed with the absence of significant temperature rises in this tissue. The mean esophageal displacement was 2.53 cm.
The only complication in this series, occurring in 7% of patients, was dysphagia. All cases of dysphagia developed immediately or soon after the procedure. All were mild, and all resolved within several days. There were no late GI complications observed, although Dr. Parikh acknowledged that no follow-up endoscopy was performed to rule out any esophageal injury.
In contrast, without the deviation permitted by the esophageal retractor, the rapid rise in temperature “could have precluded PVI,” Dr. Parikh maintained. He said that the retractor permitted the esophagus to be cleared from potential injury, as indicated by the lack of a temperature rise in esophageal tissue, in 100% of the cases. He noted that the tool is now in routine use at his center.
According to Dr. Lakkireddy, this tool is already in routine use at several centers across the country. The goal of this study was to provide an objective documentation of the ability of the device to enable successful posterior wall isolation during PVI without esophageal injury.
“It helped us get to the endpoint without a problem 100% of the time,” Dr. Lakkireddy reported.
Dr. Parikh has no industry relationships relevant to this study.
ORLANDO – A relatively simple mechanical tool to move the esophagus away from the energy delivered during ablation of atrial fibrillation (AF) does what it is supposed to do, according to data from a multicenter observational study presented at the AF Symposium 2017.
When esophageal temperature during the ablation procedure was monitored in 101 consecutive cases, no recording exceeded 38° C, according to Valay Parikh, MD, a clinical cardiac electrophysiology fellow working under Dhanunjaya Lakkireddy, MD, at the Kansas University Medical Center, Kansas City.
The tool is a stylet constructed from a nickel-titanium (nitinol) alloy. Malleable at room temperature, the stylet is inserted into an 18 Fr orogastric (OG) tube. Firmer at body temperature, the stylet within the OG tube is maneuvered to displace the esophagus away from the adjacent left atrium when radiofrequency ablation (RFA) is being administered.
The tool, marketed under the brand name EsoSure, was first made available almost 2 years ago, but the recently completed multicenter observational study was conducted to provide a more systematic evaluation of its safety and efficacy in routine use. In this study, 101 consecutive patients scheduled for RFA for AF had their esophagus displaced by the stylet during the procedure. The temperature of the esophagus as well as any adverse events involving the upper gastrointestinal tract were evaluated during the procedure. Patients were then followed for at least 6 months.
“The principal finding of our study is that mechanical displacement of the esophagus with the help of the EsoSure device is safe and provides sufficient room to deliver the intended energy at the site of ablation without any rise in temperature over 38° C,” Dr. Parikh reported.
The mean age of the 101 patients who participated in this study was 65 years. About half were female. The mean body mass index (BMI) was 32 kg/m2. The mean CHA2DS2-VASc score was 2.4. Barium x-rays were used to confirm esophageal displacement.
After the procedure, patients were discharged on a proton pump inhibitor and sucralfate, which inhibits pepsin activity and protects against ulceration. Follow-up endoscopy was performed only when medically indicated, but all patients were evaluated over the course of follow-up for odynophagia, dysphagia, hematemesis, dyspepsia, and other GI symptoms.
Pulmonary vein isolation (PVI) was achieved successfully in all cases. Although there was a rapid temperature rise at the site of ablation over the course of RFA, the esophagus was adequately displaced from the source of energy, as confirmed with the absence of significant temperature rises in this tissue. The mean esophageal displacement was 2.53 cm.
The only complication in this series, occurring in 7% of patients, was dysphagia. All cases of dysphagia developed immediately or soon after the procedure. All were mild, and all resolved within several days. There were no late GI complications observed, although Dr. Parikh acknowledged that no follow-up endoscopy was performed to rule out any esophageal injury.
In contrast, without the deviation permitted by the esophageal retractor, the rapid rise in temperature “could have precluded PVI,” Dr. Parikh maintained. He said that the retractor permitted the esophagus to be cleared from potential injury, as indicated by the lack of a temperature rise in esophageal tissue, in 100% of the cases. He noted that the tool is now in routine use at his center.
According to Dr. Lakkireddy, this tool is already in routine use at several centers across the country. The goal of this study was to provide an objective documentation of the ability of the device to enable successful posterior wall isolation during PVI without esophageal injury.
“It helped us get to the endpoint without a problem 100% of the time,” Dr. Lakkireddy reported.
Dr. Parikh has no industry relationships relevant to this study.
ORLANDO – A relatively simple mechanical tool to move the esophagus away from the energy delivered during ablation of atrial fibrillation (AF) does what it is supposed to do, according to data from a multicenter observational study presented at the AF Symposium 2017.
When esophageal temperature during the ablation procedure was monitored in 101 consecutive cases, no recording exceeded 38° C, according to Valay Parikh, MD, a clinical cardiac electrophysiology fellow working under Dhanunjaya Lakkireddy, MD, at the Kansas University Medical Center, Kansas City.
The tool is a stylet constructed from a nickel-titanium (nitinol) alloy. Malleable at room temperature, the stylet is inserted into an 18 Fr orogastric (OG) tube. Firmer at body temperature, the stylet within the OG tube is maneuvered to displace the esophagus away from the adjacent left atrium when radiofrequency ablation (RFA) is being administered.
The tool, marketed under the brand name EsoSure, was first made available almost 2 years ago, but the recently completed multicenter observational study was conducted to provide a more systematic evaluation of its safety and efficacy in routine use. In this study, 101 consecutive patients scheduled for RFA for AF had their esophagus displaced by the stylet during the procedure. The temperature of the esophagus as well as any adverse events involving the upper gastrointestinal tract were evaluated during the procedure. Patients were then followed for at least 6 months.
“The principal finding of our study is that mechanical displacement of the esophagus with the help of the EsoSure device is safe and provides sufficient room to deliver the intended energy at the site of ablation without any rise in temperature over 38° C,” Dr. Parikh reported.
The mean age of the 101 patients who participated in this study was 65 years. About half were female. The mean body mass index (BMI) was 32 kg/m2. The mean CHA2DS2-VASc score was 2.4. Barium x-rays were used to confirm esophageal displacement.
After the procedure, patients were discharged on a proton pump inhibitor and sucralfate, which inhibits pepsin activity and protects against ulceration. Follow-up endoscopy was performed only when medically indicated, but all patients were evaluated over the course of follow-up for odynophagia, dysphagia, hematemesis, dyspepsia, and other GI symptoms.
Pulmonary vein isolation (PVI) was achieved successfully in all cases. Although there was a rapid temperature rise at the site of ablation over the course of RFA, the esophagus was adequately displaced from the source of energy, as confirmed with the absence of significant temperature rises in this tissue. The mean esophageal displacement was 2.53 cm.
The only complication in this series, occurring in 7% of patients, was dysphagia. All cases of dysphagia developed immediately or soon after the procedure. All were mild, and all resolved within several days. There were no late GI complications observed, although Dr. Parikh acknowledged that no follow-up endoscopy was performed to rule out any esophageal injury.
In contrast, without the deviation permitted by the esophageal retractor, the rapid rise in temperature “could have precluded PVI,” Dr. Parikh maintained. He said that the retractor permitted the esophagus to be cleared from potential injury, as indicated by the lack of a temperature rise in esophageal tissue, in 100% of the cases. He noted that the tool is now in routine use at his center.
According to Dr. Lakkireddy, this tool is already in routine use at several centers across the country. The goal of this study was to provide an objective documentation of the ability of the device to enable successful posterior wall isolation during PVI without esophageal injury.
“It helped us get to the endpoint without a problem 100% of the time,” Dr. Lakkireddy reported.
Dr. Parikh has no industry relationships relevant to this study.
AT THE INTERNATIONAL AF SYMPOSIUM
Key clinical point: The efficacy of a tool to move the esophagus out of the way when performing ablation for atrial fibrillation is supported by a multicenter study.
Major finding: In 101 consecutive cases at four centers, all ablations were completed successfully with no esophageal temperature rise.
Data source: Prospective observational study.
Disclosures: Dr. Parikh has no industry relationships relevant to this study.