User login
Second generation ALK inhibitors show good response in second-line NSCLC
GENEVA – Second-generation inhibitors of the anaplastic lymphoma kinase (ALK) are associated with high response rates in patients with non–small cell lung cancer (NSCLC) who have disease progression after treatment with the first-generation ALK inhibitor crizotinib (Xalkori), investigators reported.
Of 117 patients with ALK-positive NSCLC that progressed while on treatment with the ALK-targeting tyrosine-kinase inhibitor (TKI) crizotinib, both ceritinib (Zykadia) and alectinib (Alecensa) were associated with high combined partial and complete response rates, reported Christian Britschgi, MD, PhD, of University Hospital Zürich, and his colleagues.

“We can confirm the high rate of response of ceritinib and alectinib as second TKIs,” Dr. Britschgi said in an interview.
“With ceritinib or alectinib in the first line, however, it’s hard to draw any conclusions because there are very few patients treated with these drugs in the first line who were included in the trials,” he added.
ALK rearrangements occur in 3%-5% of lung adenocarcinomas, and these mutations are potential targets for TKIs directed against ALK.
The investigators analyzed the clinical characteristics of response to, and disease progression on, available ALK-TKIs. They also performed next-generation sequencing on a small group of tumor samples to investigate mechanisms of resistance and to look for new mutations that could be targetable.
They collected data on patients with ALK-positive NSCLC who were receiving palliative care and looked at clinical data and biopsy samples collected before treatment, and where available, post–ALK-TKI therapy.
They collected clinical data on 139 patients, 117 of whom had undergone first-line treatment with chemotherapy (74%), ALK-TKIs (25%), or other therapies (1%). Of this group, 92 received second-line therapy – chemotherapy (17%), ALK-TKIs (74%), or immunotherapies/other (9%).
Third-line treatments were given to 55 patients, 20% of whom received chemotherapy, 75% of whom received ALK-TKIs, and 5% of whom received other therapies.
Twenty-four patients survived long enough to receive fourth-line treatments, which included chemotherapy in 8 patients (33%), ALK-TKIs in 15 (63%), or other therapies in 1 patient.
In all, 102 patients received crizotinib, all but one in the first line and the remaining patient in the second line. The overall response rate was 58.8% (59.4% for the 101 first-line patients). Median progression-free survival (PFS) in these patients was 11.7 months.
Ceritinib was given to 2 patients in the first line, 7 in the second line, and 28 in the third line. The overall response rate was 48.6% (50% in the two patients who received it in the first line, and 50% in patients who received it after crizotinib). The median PFS was 6.5 months.
For the 26 patients who received alectinib – 3 in the first line and 23 after crizotinib – the overall response rate was 100% when the drug was used in the first line, and 60.9% when used in the second and subsequent lines. Median PFS was 12.4 months.
Only a few patients to date have had next-generation sequencing of tumors performed, Dr. Britschgi said. One patient who developed resistance while on alectinib was found to have a point mutation (ALK p.lle117Ser) that was not found in a pretreatment biopsy sample, indicating that it had emerged during therapy. This patient was then started on and had a response to lorlatinib.
A second patient who had received several lines of therapy including chemotherapy and TKIs and was enrolled in the expanded access program for lorlatinib developed hepatic progression while on that drug. A biopsy showed that the tumor carried epidermal growth-factor receptor variant 3, which is known to be highly mutagenic and is associated with glioblastoma. The patient was then put on nivolumab (Opdivo) and had a good response, but had further progression, and was then put on alectinib and again had a good response that is currently ongoing, Dr. Britschgi said.
The study was supported by University Hospital Zurich and Novartis. Dr. Britschgi reported no relevant disclosures. Coauthor Sacha Rothschild, MD, PhD, disclosed institutional compensation for advisory boards from Novartis, Pfizer and Roche.
GENEVA – Second-generation inhibitors of the anaplastic lymphoma kinase (ALK) are associated with high response rates in patients with non–small cell lung cancer (NSCLC) who have disease progression after treatment with the first-generation ALK inhibitor crizotinib (Xalkori), investigators reported.
Of 117 patients with ALK-positive NSCLC that progressed while on treatment with the ALK-targeting tyrosine-kinase inhibitor (TKI) crizotinib, both ceritinib (Zykadia) and alectinib (Alecensa) were associated with high combined partial and complete response rates, reported Christian Britschgi, MD, PhD, of University Hospital Zürich, and his colleagues.
“We can confirm the high rate of response of ceritinib and alectinib as second TKIs,” Dr. Britschgi said in an interview.
“With ceritinib or alectinib in the first line, however, it’s hard to draw any conclusions because there are very few patients treated with these drugs in the first line who were included in the trials,” he added.
ALK rearrangements occur in 3%-5% of lung adenocarcinomas, and these mutations are potential targets for TKIs directed against ALK.
The investigators analyzed the clinical characteristics of response to, and disease progression on, available ALK-TKIs. They also performed next-generation sequencing on a small group of tumor samples to investigate mechanisms of resistance and to look for new mutations that could be targetable.
They collected data on patients with ALK-positive NSCLC who were receiving palliative care and looked at clinical data and biopsy samples collected before treatment, and where available, post–ALK-TKI therapy.
They collected clinical data on 139 patients, 117 of whom had undergone first-line treatment with chemotherapy (74%), ALK-TKIs (25%), or other therapies (1%). Of this group, 92 received second-line therapy – chemotherapy (17%), ALK-TKIs (74%), or immunotherapies/other (9%).
Third-line treatments were given to 55 patients, 20% of whom received chemotherapy, 75% of whom received ALK-TKIs, and 5% of whom received other therapies.
Twenty-four patients survived long enough to receive fourth-line treatments, which included chemotherapy in 8 patients (33%), ALK-TKIs in 15 (63%), or other therapies in 1 patient.
In all, 102 patients received crizotinib, all but one in the first line and the remaining patient in the second line. The overall response rate was 58.8% (59.4% for the 101 first-line patients). Median progression-free survival (PFS) in these patients was 11.7 months.
Ceritinib was given to 2 patients in the first line, 7 in the second line, and 28 in the third line. The overall response rate was 48.6% (50% in the two patients who received it in the first line, and 50% in patients who received it after crizotinib). The median PFS was 6.5 months.
For the 26 patients who received alectinib – 3 in the first line and 23 after crizotinib – the overall response rate was 100% when the drug was used in the first line, and 60.9% when used in the second and subsequent lines. Median PFS was 12.4 months.
Only a few patients to date have had next-generation sequencing of tumors performed, Dr. Britschgi said. One patient who developed resistance while on alectinib was found to have a point mutation (ALK p.lle117Ser) that was not found in a pretreatment biopsy sample, indicating that it had emerged during therapy. This patient was then started on and had a response to lorlatinib.
A second patient who had received several lines of therapy including chemotherapy and TKIs and was enrolled in the expanded access program for lorlatinib developed hepatic progression while on that drug. A biopsy showed that the tumor carried epidermal growth-factor receptor variant 3, which is known to be highly mutagenic and is associated with glioblastoma. The patient was then put on nivolumab (Opdivo) and had a good response, but had further progression, and was then put on alectinib and again had a good response that is currently ongoing, Dr. Britschgi said.
The study was supported by University Hospital Zurich and Novartis. Dr. Britschgi reported no relevant disclosures. Coauthor Sacha Rothschild, MD, PhD, disclosed institutional compensation for advisory boards from Novartis, Pfizer and Roche.
GENEVA – Second-generation inhibitors of the anaplastic lymphoma kinase (ALK) are associated with high response rates in patients with non–small cell lung cancer (NSCLC) who have disease progression after treatment with the first-generation ALK inhibitor crizotinib (Xalkori), investigators reported.
Of 117 patients with ALK-positive NSCLC that progressed while on treatment with the ALK-targeting tyrosine-kinase inhibitor (TKI) crizotinib, both ceritinib (Zykadia) and alectinib (Alecensa) were associated with high combined partial and complete response rates, reported Christian Britschgi, MD, PhD, of University Hospital Zürich, and his colleagues.
“We can confirm the high rate of response of ceritinib and alectinib as second TKIs,” Dr. Britschgi said in an interview.
“With ceritinib or alectinib in the first line, however, it’s hard to draw any conclusions because there are very few patients treated with these drugs in the first line who were included in the trials,” he added.
ALK rearrangements occur in 3%-5% of lung adenocarcinomas, and these mutations are potential targets for TKIs directed against ALK.
The investigators analyzed the clinical characteristics of response to, and disease progression on, available ALK-TKIs. They also performed next-generation sequencing on a small group of tumor samples to investigate mechanisms of resistance and to look for new mutations that could be targetable.
They collected data on patients with ALK-positive NSCLC who were receiving palliative care and looked at clinical data and biopsy samples collected before treatment, and where available, post–ALK-TKI therapy.
They collected clinical data on 139 patients, 117 of whom had undergone first-line treatment with chemotherapy (74%), ALK-TKIs (25%), or other therapies (1%). Of this group, 92 received second-line therapy – chemotherapy (17%), ALK-TKIs (74%), or immunotherapies/other (9%).
Third-line treatments were given to 55 patients, 20% of whom received chemotherapy, 75% of whom received ALK-TKIs, and 5% of whom received other therapies.
Twenty-four patients survived long enough to receive fourth-line treatments, which included chemotherapy in 8 patients (33%), ALK-TKIs in 15 (63%), or other therapies in 1 patient.
In all, 102 patients received crizotinib, all but one in the first line and the remaining patient in the second line. The overall response rate was 58.8% (59.4% for the 101 first-line patients). Median progression-free survival (PFS) in these patients was 11.7 months.
Ceritinib was given to 2 patients in the first line, 7 in the second line, and 28 in the third line. The overall response rate was 48.6% (50% in the two patients who received it in the first line, and 50% in patients who received it after crizotinib). The median PFS was 6.5 months.
For the 26 patients who received alectinib – 3 in the first line and 23 after crizotinib – the overall response rate was 100% when the drug was used in the first line, and 60.9% when used in the second and subsequent lines. Median PFS was 12.4 months.
Only a few patients to date have had next-generation sequencing of tumors performed, Dr. Britschgi said. One patient who developed resistance while on alectinib was found to have a point mutation (ALK p.lle117Ser) that was not found in a pretreatment biopsy sample, indicating that it had emerged during therapy. This patient was then started on and had a response to lorlatinib.
A second patient who had received several lines of therapy including chemotherapy and TKIs and was enrolled in the expanded access program for lorlatinib developed hepatic progression while on that drug. A biopsy showed that the tumor carried epidermal growth-factor receptor variant 3, which is known to be highly mutagenic and is associated with glioblastoma. The patient was then put on nivolumab (Opdivo) and had a good response, but had further progression, and was then put on alectinib and again had a good response that is currently ongoing, Dr. Britschgi said.
The study was supported by University Hospital Zurich and Novartis. Dr. Britschgi reported no relevant disclosures. Coauthor Sacha Rothschild, MD, PhD, disclosed institutional compensation for advisory boards from Novartis, Pfizer and Roche.
FROM ELCC
Key clinical point:
Major finding: Overall response rates after crizotinib were 50% for ceritinib and 60.9% for alectinib.
Data source: A retrospective review of data on 117 patients with ALK-positive NSCLC in Swiss and Italian hospitals.
Disclosures: The study was supported by University Hospital Zurich and Novartis. Dr. Britschgi reported no relevant disclosures. Coauthor Sacha Rothschild, MD, PhD, disclosed institutional compensation for advisory boards from Novartis, Pfizer, and Roche.
FDA: Fluoroquinolone use not linked to retina detachment, aortic problems
The Food and Drug Administration has found no evidence of a link between fluoroquinolone usage and retinal detachment or aortic aneurysm and dissection, according to a new Drug Safety Communication update on potential serious, disabling side effects of oral and injectable fluoroquinolone antibiotics.
Fluoroquinolones are used to treat acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections.
However, after reviewing patient cases and study findings, the FDA said the evidence didn’t support an association between fluoroquinolone usage and potential retinal or aortic dangers, according to its May 10, 2017, Drug Safety Communication update.
Serious side effects associated with fluoroquinolone use include hallucination, depression, suicidal thoughts, tendinitis and tendon rupture, a “pins and needles” feeling in the arms and legs, joint pain and swelling, skin rash, and severe diarrhea.
“We will continue to assess safety issues with fluoroquinolones, and will update the public if additional actions are needed,” the FDA said in a statement.
The Food and Drug Administration has found no evidence of a link between fluoroquinolone usage and retinal detachment or aortic aneurysm and dissection, according to a new Drug Safety Communication update on potential serious, disabling side effects of oral and injectable fluoroquinolone antibiotics.
Fluoroquinolones are used to treat acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections.
However, after reviewing patient cases and study findings, the FDA said the evidence didn’t support an association between fluoroquinolone usage and potential retinal or aortic dangers, according to its May 10, 2017, Drug Safety Communication update.
Serious side effects associated with fluoroquinolone use include hallucination, depression, suicidal thoughts, tendinitis and tendon rupture, a “pins and needles” feeling in the arms and legs, joint pain and swelling, skin rash, and severe diarrhea.
“We will continue to assess safety issues with fluoroquinolones, and will update the public if additional actions are needed,” the FDA said in a statement.
The Food and Drug Administration has found no evidence of a link between fluoroquinolone usage and retinal detachment or aortic aneurysm and dissection, according to a new Drug Safety Communication update on potential serious, disabling side effects of oral and injectable fluoroquinolone antibiotics.
Fluoroquinolones are used to treat acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections.
However, after reviewing patient cases and study findings, the FDA said the evidence didn’t support an association between fluoroquinolone usage and potential retinal or aortic dangers, according to its May 10, 2017, Drug Safety Communication update.
Serious side effects associated with fluoroquinolone use include hallucination, depression, suicidal thoughts, tendinitis and tendon rupture, a “pins and needles” feeling in the arms and legs, joint pain and swelling, skin rash, and severe diarrhea.
“We will continue to assess safety issues with fluoroquinolones, and will update the public if additional actions are needed,” the FDA said in a statement.
SLE linked to subsequent risk of malignant melanoma
PORTLAND, ORE. – A diagnosis of systemic lupus erythematosus (SLE) significantly increases the risk of a subsequent diagnosis of malignant melanoma, according to the results of a large, first-in-kind, single-center longitudinal analysis of electronic medical records.
This finding expands the list of known associations between SLE and cancer, and highlights the need for careful surveillance of this population, Solomiya Grushchak, of the department of dermatology at Northwestern University, Chicago, and her associates, reported in a poster presented at the annual meeting of the Society for Investigative Dermatology.
SLE is increasingly being treated with immune checkpoint inhibitors, which can aggressively disrupt immune reactivity and trigger uncontrolled cellular responses in patients with SLE, Ms. Grushchak noted. “The findings in this large population warrant further exploration of the association between malignant melanoma and SLE to promote optimal patient management, especially in light of recent advances [in the use of] checkpoint inhibitors,” she added.
Past work has linked SLE with several other malignancies, including nonmelanoma skin cancers, non-Hodgkin and Hodgkin lymphomas, and cancers of the larynx, lungs, liver, vulva, vagina, and thyroid gland. Even when patients are not receiving checkpoint inhibitors, SLE causes chronic inflammation and is known to increase cellular dysplasia, which can ultimately trigger uncontrolled proliferation of tumor cells. In 2015, a meta-analysis showed that SLE was associated with a decreased risk of melanoma, but no studies had conclusively evaluated this relationship (PLoS One. 2015;10[4]:e0122964).
Therefore, Ms. Grushchak and her associates analyzed medical records from 2,351 patients from the urban Midwest with SLE diagnosed by a dermatologist or rheumatologist between 2000 and 2016. The data source was the Northwestern Enterprise Data Warehouse, which integrates clinical and research information from more than 50 health data systems used by the Northwestern University Feinberg School of Medicine and its health care partners. To avoid detection bias, the researchers constructed a comparison group from the same database of 1,676 patients diagnosed with systemic sclerosis.
Ten patients (0.4%) with a diagnostic code for SLE were later diagnosed with malignant melanoma, compared with one patient with systemic sclerosis (0.06%), the investigators reported. A Fisher’s exact test confirmed a statistically significant difference between these rates (P = .03). Among the 10 SLE patients with melanoma, 7 were white, 2 were black, and 1 was of Asian ancestry. Nine were females, and one was male. The patient with systemic sclerosis and melanoma was a white male.
The study had several limitations. The investigators did not report how much time elapsed between the diagnoses of SLE and melanoma, or the rates or cumulative exposure to checkpoint inhibitors.
The National Institutes of Health provides support to the Northwestern Enterprise Data Warehouse. The investigators had no relevant financial conflicts.
PORTLAND, ORE. – A diagnosis of systemic lupus erythematosus (SLE) significantly increases the risk of a subsequent diagnosis of malignant melanoma, according to the results of a large, first-in-kind, single-center longitudinal analysis of electronic medical records.
This finding expands the list of known associations between SLE and cancer, and highlights the need for careful surveillance of this population, Solomiya Grushchak, of the department of dermatology at Northwestern University, Chicago, and her associates, reported in a poster presented at the annual meeting of the Society for Investigative Dermatology.
SLE is increasingly being treated with immune checkpoint inhibitors, which can aggressively disrupt immune reactivity and trigger uncontrolled cellular responses in patients with SLE, Ms. Grushchak noted. “The findings in this large population warrant further exploration of the association between malignant melanoma and SLE to promote optimal patient management, especially in light of recent advances [in the use of] checkpoint inhibitors,” she added.
Past work has linked SLE with several other malignancies, including nonmelanoma skin cancers, non-Hodgkin and Hodgkin lymphomas, and cancers of the larynx, lungs, liver, vulva, vagina, and thyroid gland. Even when patients are not receiving checkpoint inhibitors, SLE causes chronic inflammation and is known to increase cellular dysplasia, which can ultimately trigger uncontrolled proliferation of tumor cells. In 2015, a meta-analysis showed that SLE was associated with a decreased risk of melanoma, but no studies had conclusively evaluated this relationship (PLoS One. 2015;10[4]:e0122964).
Therefore, Ms. Grushchak and her associates analyzed medical records from 2,351 patients from the urban Midwest with SLE diagnosed by a dermatologist or rheumatologist between 2000 and 2016. The data source was the Northwestern Enterprise Data Warehouse, which integrates clinical and research information from more than 50 health data systems used by the Northwestern University Feinberg School of Medicine and its health care partners. To avoid detection bias, the researchers constructed a comparison group from the same database of 1,676 patients diagnosed with systemic sclerosis.
Ten patients (0.4%) with a diagnostic code for SLE were later diagnosed with malignant melanoma, compared with one patient with systemic sclerosis (0.06%), the investigators reported. A Fisher’s exact test confirmed a statistically significant difference between these rates (P = .03). Among the 10 SLE patients with melanoma, 7 were white, 2 were black, and 1 was of Asian ancestry. Nine were females, and one was male. The patient with systemic sclerosis and melanoma was a white male.
The study had several limitations. The investigators did not report how much time elapsed between the diagnoses of SLE and melanoma, or the rates or cumulative exposure to checkpoint inhibitors.
The National Institutes of Health provides support to the Northwestern Enterprise Data Warehouse. The investigators had no relevant financial conflicts.
PORTLAND, ORE. – A diagnosis of systemic lupus erythematosus (SLE) significantly increases the risk of a subsequent diagnosis of malignant melanoma, according to the results of a large, first-in-kind, single-center longitudinal analysis of electronic medical records.
This finding expands the list of known associations between SLE and cancer, and highlights the need for careful surveillance of this population, Solomiya Grushchak, of the department of dermatology at Northwestern University, Chicago, and her associates, reported in a poster presented at the annual meeting of the Society for Investigative Dermatology.
SLE is increasingly being treated with immune checkpoint inhibitors, which can aggressively disrupt immune reactivity and trigger uncontrolled cellular responses in patients with SLE, Ms. Grushchak noted. “The findings in this large population warrant further exploration of the association between malignant melanoma and SLE to promote optimal patient management, especially in light of recent advances [in the use of] checkpoint inhibitors,” she added.
Past work has linked SLE with several other malignancies, including nonmelanoma skin cancers, non-Hodgkin and Hodgkin lymphomas, and cancers of the larynx, lungs, liver, vulva, vagina, and thyroid gland. Even when patients are not receiving checkpoint inhibitors, SLE causes chronic inflammation and is known to increase cellular dysplasia, which can ultimately trigger uncontrolled proliferation of tumor cells. In 2015, a meta-analysis showed that SLE was associated with a decreased risk of melanoma, but no studies had conclusively evaluated this relationship (PLoS One. 2015;10[4]:e0122964).
Therefore, Ms. Grushchak and her associates analyzed medical records from 2,351 patients from the urban Midwest with SLE diagnosed by a dermatologist or rheumatologist between 2000 and 2016. The data source was the Northwestern Enterprise Data Warehouse, which integrates clinical and research information from more than 50 health data systems used by the Northwestern University Feinberg School of Medicine and its health care partners. To avoid detection bias, the researchers constructed a comparison group from the same database of 1,676 patients diagnosed with systemic sclerosis.
Ten patients (0.4%) with a diagnostic code for SLE were later diagnosed with malignant melanoma, compared with one patient with systemic sclerosis (0.06%), the investigators reported. A Fisher’s exact test confirmed a statistically significant difference between these rates (P = .03). Among the 10 SLE patients with melanoma, 7 were white, 2 were black, and 1 was of Asian ancestry. Nine were females, and one was male. The patient with systemic sclerosis and melanoma was a white male.
The study had several limitations. The investigators did not report how much time elapsed between the diagnoses of SLE and melanoma, or the rates or cumulative exposure to checkpoint inhibitors.
The National Institutes of Health provides support to the Northwestern Enterprise Data Warehouse. The investigators had no relevant financial conflicts.
AT SID 2017
Key clinical point: Compared with controls, patients with systemic lupus erythematosus (SLE) were at significantly increased risk of later being diagnosed with malignant melanoma.
Major finding: Ten patients with SLE (0.4%) were later diagnosed with malignant melanoma, compared with one patient with systemic sclerosis (0.06%), a statistically significant difference (P = .03).
Data source: Electronic medical record reviews of 2,351 patients with SLE and 1,676 patients with systemic sclerosis (controls) between 2000 and 2016.
Disclosures: The National Institutes of Health provides support to the Northwestern Enterprise Data Warehouse. The investigators had no relevant financial conflicts.
Contact dermatitis in children: The top 10 allergens
Contact dermatitis should be suspected in patients with eczematous dermatitis atypical in location, geometric or symmetric in distribution, or unresponsive to or worsened by common therapies, Dr. Catalina Matiz emphasized.
Allergic contact dermatitis is a complex disorder characterized by a type IV delayed-type hypersensitivity reaction that occurs when a patient’s skin is exposed to a substance easily penetrates the skin barrier.

Increasingly recognized in the United States, many pediatric patients are becoming sensitized to contact allergens found in personal care products, including skin care products, topical medications, and clothing.
A study published by Hill et al. in 2016 reviewed pediatric patch test studies to determine the top contact allergens in children (Expert Rev Clin Immunol. 2016;12[5]:551-61).
Dr. Matiz presented the top 10 allergens discovered by this group, and offered practical advice for allergen avoidance.
Topping the list in descending order are the following:
- Tixocortol pivalate (a corticosteroid).
- Propylene glycol.
- Methylchloroisothiazolinone (MCI)/methylisothiazolinone (MI).
- Formaldehyde.
- Cocamidopropyl betaine.
- Lanolin.
- Benzalkonium chloride.
- Fragrance and balsam of peru.
- Neomycin.
- Nickel.
Dr. Matiz educated meeting attendees on allergen-containing products and clinical correlations suggestive of allergic sensitization to common allergens. Tixocortol pivilate is a corticosteroid present in Class A corticosteroids including hydrocortisone acetate. She highlighted the cross reactivity between class A and class D2 corticosteroids including hydrocortisone butyrate and valerate.
“Usually topical corticosteroid–contact sensitivity manifests as a failure to improve or worsening of existing dermatitis,” emphasized Dr. Matiz of departments of dermatology and pediatrics at the university.
Propylene glycol, benzalkonium chloride, and neomycin also represent top contact allergens frequently found in topical medications. Similarly, lanolin contact sensitivity often presents as refractory or worsening atopic dermatitis. “Lanolin, also known as wool alcohol, is commonly found in emollients, medications, and personal care products used by atopic dermatitis patients,” Dr. Matiz stressed.
Methylchloroisothiazolinone/methylisothiazolinone represent preservatives commonly found in wet wipes, hypoallergenic, and sensitive skin products. This was the allergen of the year in 2013 and was subsequently removed from many wet wipe formulations and products in response.
In addition to acute management of allergic contact dermatitis with corticosteroids, Dr. Matiz further emphasized the importance of preemptive avoidance of the top ten allergens.
She recommends patch testing if no clinical improvement is evident after 8 weeks of common allergen avoidance, for definitive allergen identification.
Dr. Matiz said she had no relevant financial disclosures.
Contact dermatitis should be suspected in patients with eczematous dermatitis atypical in location, geometric or symmetric in distribution, or unresponsive to or worsened by common therapies, Dr. Catalina Matiz emphasized.
Allergic contact dermatitis is a complex disorder characterized by a type IV delayed-type hypersensitivity reaction that occurs when a patient’s skin is exposed to a substance easily penetrates the skin barrier.
Increasingly recognized in the United States, many pediatric patients are becoming sensitized to contact allergens found in personal care products, including skin care products, topical medications, and clothing.
A study published by Hill et al. in 2016 reviewed pediatric patch test studies to determine the top contact allergens in children (Expert Rev Clin Immunol. 2016;12[5]:551-61).
Dr. Matiz presented the top 10 allergens discovered by this group, and offered practical advice for allergen avoidance.
Topping the list in descending order are the following:
- Tixocortol pivalate (a corticosteroid).
- Propylene glycol.
- Methylchloroisothiazolinone (MCI)/methylisothiazolinone (MI).
- Formaldehyde.
- Cocamidopropyl betaine.
- Lanolin.
- Benzalkonium chloride.
- Fragrance and balsam of peru.
- Neomycin.
- Nickel.
Dr. Matiz educated meeting attendees on allergen-containing products and clinical correlations suggestive of allergic sensitization to common allergens. Tixocortol pivilate is a corticosteroid present in Class A corticosteroids including hydrocortisone acetate. She highlighted the cross reactivity between class A and class D2 corticosteroids including hydrocortisone butyrate and valerate.
“Usually topical corticosteroid–contact sensitivity manifests as a failure to improve or worsening of existing dermatitis,” emphasized Dr. Matiz of departments of dermatology and pediatrics at the university.
Propylene glycol, benzalkonium chloride, and neomycin also represent top contact allergens frequently found in topical medications. Similarly, lanolin contact sensitivity often presents as refractory or worsening atopic dermatitis. “Lanolin, also known as wool alcohol, is commonly found in emollients, medications, and personal care products used by atopic dermatitis patients,” Dr. Matiz stressed.
Methylchloroisothiazolinone/methylisothiazolinone represent preservatives commonly found in wet wipes, hypoallergenic, and sensitive skin products. This was the allergen of the year in 2013 and was subsequently removed from many wet wipe formulations and products in response.
In addition to acute management of allergic contact dermatitis with corticosteroids, Dr. Matiz further emphasized the importance of preemptive avoidance of the top ten allergens.
She recommends patch testing if no clinical improvement is evident after 8 weeks of common allergen avoidance, for definitive allergen identification.
Dr. Matiz said she had no relevant financial disclosures.
Contact dermatitis should be suspected in patients with eczematous dermatitis atypical in location, geometric or symmetric in distribution, or unresponsive to or worsened by common therapies, Dr. Catalina Matiz emphasized.
Allergic contact dermatitis is a complex disorder characterized by a type IV delayed-type hypersensitivity reaction that occurs when a patient’s skin is exposed to a substance easily penetrates the skin barrier.
Increasingly recognized in the United States, many pediatric patients are becoming sensitized to contact allergens found in personal care products, including skin care products, topical medications, and clothing.
A study published by Hill et al. in 2016 reviewed pediatric patch test studies to determine the top contact allergens in children (Expert Rev Clin Immunol. 2016;12[5]:551-61).
Dr. Matiz presented the top 10 allergens discovered by this group, and offered practical advice for allergen avoidance.
Topping the list in descending order are the following:
- Tixocortol pivalate (a corticosteroid).
- Propylene glycol.
- Methylchloroisothiazolinone (MCI)/methylisothiazolinone (MI).
- Formaldehyde.
- Cocamidopropyl betaine.
- Lanolin.
- Benzalkonium chloride.
- Fragrance and balsam of peru.
- Neomycin.
- Nickel.
Dr. Matiz educated meeting attendees on allergen-containing products and clinical correlations suggestive of allergic sensitization to common allergens. Tixocortol pivilate is a corticosteroid present in Class A corticosteroids including hydrocortisone acetate. She highlighted the cross reactivity between class A and class D2 corticosteroids including hydrocortisone butyrate and valerate.
“Usually topical corticosteroid–contact sensitivity manifests as a failure to improve or worsening of existing dermatitis,” emphasized Dr. Matiz of departments of dermatology and pediatrics at the university.
Propylene glycol, benzalkonium chloride, and neomycin also represent top contact allergens frequently found in topical medications. Similarly, lanolin contact sensitivity often presents as refractory or worsening atopic dermatitis. “Lanolin, also known as wool alcohol, is commonly found in emollients, medications, and personal care products used by atopic dermatitis patients,” Dr. Matiz stressed.
Methylchloroisothiazolinone/methylisothiazolinone represent preservatives commonly found in wet wipes, hypoallergenic, and sensitive skin products. This was the allergen of the year in 2013 and was subsequently removed from many wet wipe formulations and products in response.
In addition to acute management of allergic contact dermatitis with corticosteroids, Dr. Matiz further emphasized the importance of preemptive avoidance of the top ten allergens.
She recommends patch testing if no clinical improvement is evident after 8 weeks of common allergen avoidance, for definitive allergen identification.
Dr. Matiz said she had no relevant financial disclosures.
High-dose oral vitamin D3 significantly reduced effects of sunburn
PORTLAND, ORE. – When given within an hour, a single dose of at least 100,000 IU vitamin D3 rapidly attenuates sunburn, according to the results of a randomized, double-blind, placebo-controlled pilot study of 25 healthy adults.
This is the first in vivo study to evaluate whether vitamin D3 can modulate acute inflammation in target tissues, Jeffrey F. Scott, MD, wrote in a poster presented at the annual meeting of the Society for Investigative Dermatology. The findings “have broad implications for the role of vitamin D in skin homeostasis, and suggest that oral vitamin D may be clinically therapeutic for its immunomodulatory properties,” he and his coauthors concluded.
Higher doses of vitamin D3 also produced significant decreases in skin levels of tumor necrosis factor–alpha (TNF-alpha) (P = .02) and inducible nitric oxide synthase (iNOS) (P = .04) compared with placebo, reported Dr. Scott, a resident in dermatology at University Hospitals Cleveland Medical Center.
Notably, 48 hours after sunburn, hematoxylin and eosin histology of punch biopsies showed that participants who received 200,000 IU vitamin D3 had the least structural damage to the skin, while placebo recipients had the most damage. Expression profiling also linked vitamin D3 treatment with upregulation of genes associated with skin barrier repair.
Studies continue to document diverse biologic effects of vitamin D, including “modulation of immune response, inflammatory disease, cardiovascular health, and carcinogenesis,” the researchers wrote. Vitamin D3 also has been shown to suppress inflammatory mediators and induce autophagy, they added. Previously, their group showed that oral D3 induced similar protective effects in a mouse model of chemical skin injury. Treatment inhibited proinflammatory cytokines and chemokines within the skin, including iNOS and TNF-alpha.
The current study also included a control phase in which participants underwent experimental sunburn on the right arm without any treatment. Forty-eight hours later, punch biopsies revealed high levels of iNOS and TNF-alpha, with increased expression of proinflammatory genes, the researchers wrote.
During the subsequent experimental phase, seven participants were assigned to receive placebo, and six were assigned to receive 50,000 IU, 100,000 IU, or 200,000 IU of vitamin D3. After treatment, participants with the highest serum D3 levels had significantly decreased skin redness compared with participants with lower serum D3 levels (P less than .05). Higher vitamin D3 serum levels were also associated with significant (P less than .05) upregulation of skin barrier repair genes and of arginase-1, a cytosolic enzyme that helps mediate anti-inflammatory activity.
“Arginase-1 may be a clinically useful tissue biomarker for monitoring the immunomodulatory effects of vitamin D3 in humans,” the researchers concluded.
The National Institute of Arthritis Musculoskeletal and Skin Diseases and the National Institutes of Health supported the work. Dr. Scott had no relevant financial disclosures.
PORTLAND, ORE. – When given within an hour, a single dose of at least 100,000 IU vitamin D3 rapidly attenuates sunburn, according to the results of a randomized, double-blind, placebo-controlled pilot study of 25 healthy adults.
This is the first in vivo study to evaluate whether vitamin D3 can modulate acute inflammation in target tissues, Jeffrey F. Scott, MD, wrote in a poster presented at the annual meeting of the Society for Investigative Dermatology. The findings “have broad implications for the role of vitamin D in skin homeostasis, and suggest that oral vitamin D may be clinically therapeutic for its immunomodulatory properties,” he and his coauthors concluded.
Higher doses of vitamin D3 also produced significant decreases in skin levels of tumor necrosis factor–alpha (TNF-alpha) (P = .02) and inducible nitric oxide synthase (iNOS) (P = .04) compared with placebo, reported Dr. Scott, a resident in dermatology at University Hospitals Cleveland Medical Center.
Notably, 48 hours after sunburn, hematoxylin and eosin histology of punch biopsies showed that participants who received 200,000 IU vitamin D3 had the least structural damage to the skin, while placebo recipients had the most damage. Expression profiling also linked vitamin D3 treatment with upregulation of genes associated with skin barrier repair.
Studies continue to document diverse biologic effects of vitamin D, including “modulation of immune response, inflammatory disease, cardiovascular health, and carcinogenesis,” the researchers wrote. Vitamin D3 also has been shown to suppress inflammatory mediators and induce autophagy, they added. Previously, their group showed that oral D3 induced similar protective effects in a mouse model of chemical skin injury. Treatment inhibited proinflammatory cytokines and chemokines within the skin, including iNOS and TNF-alpha.
The current study also included a control phase in which participants underwent experimental sunburn on the right arm without any treatment. Forty-eight hours later, punch biopsies revealed high levels of iNOS and TNF-alpha, with increased expression of proinflammatory genes, the researchers wrote.
During the subsequent experimental phase, seven participants were assigned to receive placebo, and six were assigned to receive 50,000 IU, 100,000 IU, or 200,000 IU of vitamin D3. After treatment, participants with the highest serum D3 levels had significantly decreased skin redness compared with participants with lower serum D3 levels (P less than .05). Higher vitamin D3 serum levels were also associated with significant (P less than .05) upregulation of skin barrier repair genes and of arginase-1, a cytosolic enzyme that helps mediate anti-inflammatory activity.
“Arginase-1 may be a clinically useful tissue biomarker for monitoring the immunomodulatory effects of vitamin D3 in humans,” the researchers concluded.
The National Institute of Arthritis Musculoskeletal and Skin Diseases and the National Institutes of Health supported the work. Dr. Scott had no relevant financial disclosures.
PORTLAND, ORE. – When given within an hour, a single dose of at least 100,000 IU vitamin D3 rapidly attenuates sunburn, according to the results of a randomized, double-blind, placebo-controlled pilot study of 25 healthy adults.
This is the first in vivo study to evaluate whether vitamin D3 can modulate acute inflammation in target tissues, Jeffrey F. Scott, MD, wrote in a poster presented at the annual meeting of the Society for Investigative Dermatology. The findings “have broad implications for the role of vitamin D in skin homeostasis, and suggest that oral vitamin D may be clinically therapeutic for its immunomodulatory properties,” he and his coauthors concluded.
Higher doses of vitamin D3 also produced significant decreases in skin levels of tumor necrosis factor–alpha (TNF-alpha) (P = .02) and inducible nitric oxide synthase (iNOS) (P = .04) compared with placebo, reported Dr. Scott, a resident in dermatology at University Hospitals Cleveland Medical Center.
Notably, 48 hours after sunburn, hematoxylin and eosin histology of punch biopsies showed that participants who received 200,000 IU vitamin D3 had the least structural damage to the skin, while placebo recipients had the most damage. Expression profiling also linked vitamin D3 treatment with upregulation of genes associated with skin barrier repair.
Studies continue to document diverse biologic effects of vitamin D, including “modulation of immune response, inflammatory disease, cardiovascular health, and carcinogenesis,” the researchers wrote. Vitamin D3 also has been shown to suppress inflammatory mediators and induce autophagy, they added. Previously, their group showed that oral D3 induced similar protective effects in a mouse model of chemical skin injury. Treatment inhibited proinflammatory cytokines and chemokines within the skin, including iNOS and TNF-alpha.
The current study also included a control phase in which participants underwent experimental sunburn on the right arm without any treatment. Forty-eight hours later, punch biopsies revealed high levels of iNOS and TNF-alpha, with increased expression of proinflammatory genes, the researchers wrote.
During the subsequent experimental phase, seven participants were assigned to receive placebo, and six were assigned to receive 50,000 IU, 100,000 IU, or 200,000 IU of vitamin D3. After treatment, participants with the highest serum D3 levels had significantly decreased skin redness compared with participants with lower serum D3 levels (P less than .05). Higher vitamin D3 serum levels were also associated with significant (P less than .05) upregulation of skin barrier repair genes and of arginase-1, a cytosolic enzyme that helps mediate anti-inflammatory activity.
“Arginase-1 may be a clinically useful tissue biomarker for monitoring the immunomodulatory effects of vitamin D3 in humans,” the researchers concluded.
The National Institute of Arthritis Musculoskeletal and Skin Diseases and the National Institutes of Health supported the work. Dr. Scott had no relevant financial disclosures.
Key clinical point: A single dose of at least 100,000 IU vitamin D3 rapidly attenuated experimental sunburn.
Major finding: Twenty-four hours after sunburn, recipients of 100,000 IU or 200,000 IU D3 had a marked, sustained reduction in skin redness compared with the placebo and 50,000 IU groups. Higher doses of D3 produced significant decreases in skin levels of tumor necrosis factor–alpha and inducible nitric oxide synthase, compared with placebo.
Data source: A double-blind, randomized, placebo-controlled pilot study of 25 healthy adults.
Disclosures: The National Institute of Arthritis Musculoskeletal and Skin Diseases and the National Institutes of Health supported the work. Dr. Scott had no relevant financial conflicts.

U.S. Zika epidemic could cost billions, or less
A Zika virus epidemic could cost the United States $183 million … or $680 million … or $2.2 billion, according to a new computational model developed to estimate Zika’s economic impact.
The model’s seeming lack of conviction comes from its options for an attack rate – the percentage of the population infected by the virus. An epidemic with a low attack rate of 0.01% would be expected to result in over 7,000 symptomatic cases and cost $183 million in direct medical costs and losses in productivity, said Bruce Y. Lee, MD, of Johns Hopkins Bloomberg School of Public Health, Baltimore, and his associates (PLoS Negl Trop Dis. 2017 Apr 27;11[4]:e0005531).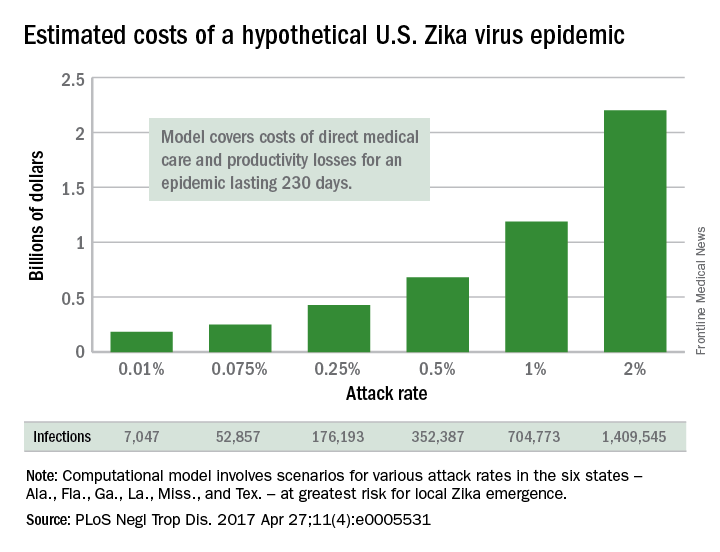
The investigators based their model on the six states at the highest risk for local Zika emergence: Alabama, Florida, Georgia, Louisiana, Mississippi, and Texas. The hypothetical epidemic lasts 230 days, which is equivalent to the Zika-related microcephaly outbreak in Brazil in 2015.
The National Institutes of Health, the Agency for Healthcare Research and Quality, and the United States Agency for International Development funded the study. The authors declared that no competing interests exist.
A Zika virus epidemic could cost the United States $183 million … or $680 million … or $2.2 billion, according to a new computational model developed to estimate Zika’s economic impact.
The model’s seeming lack of conviction comes from its options for an attack rate – the percentage of the population infected by the virus. An epidemic with a low attack rate of 0.01% would be expected to result in over 7,000 symptomatic cases and cost $183 million in direct medical costs and losses in productivity, said Bruce Y. Lee, MD, of Johns Hopkins Bloomberg School of Public Health, Baltimore, and his associates (PLoS Negl Trop Dis. 2017 Apr 27;11[4]:e0005531).
The investigators based their model on the six states at the highest risk for local Zika emergence: Alabama, Florida, Georgia, Louisiana, Mississippi, and Texas. The hypothetical epidemic lasts 230 days, which is equivalent to the Zika-related microcephaly outbreak in Brazil in 2015.
The National Institutes of Health, the Agency for Healthcare Research and Quality, and the United States Agency for International Development funded the study. The authors declared that no competing interests exist.
A Zika virus epidemic could cost the United States $183 million … or $680 million … or $2.2 billion, according to a new computational model developed to estimate Zika’s economic impact.
The model’s seeming lack of conviction comes from its options for an attack rate – the percentage of the population infected by the virus. An epidemic with a low attack rate of 0.01% would be expected to result in over 7,000 symptomatic cases and cost $183 million in direct medical costs and losses in productivity, said Bruce Y. Lee, MD, of Johns Hopkins Bloomberg School of Public Health, Baltimore, and his associates (PLoS Negl Trop Dis. 2017 Apr 27;11[4]:e0005531).
The investigators based their model on the six states at the highest risk for local Zika emergence: Alabama, Florida, Georgia, Louisiana, Mississippi, and Texas. The hypothetical epidemic lasts 230 days, which is equivalent to the Zika-related microcephaly outbreak in Brazil in 2015.
The National Institutes of Health, the Agency for Healthcare Research and Quality, and the United States Agency for International Development funded the study. The authors declared that no competing interests exist.
FROM PLOS NEGLECTED TROPICAL DISEASES
A note about OpenNotes
“He is a frequent flyer.” This is a term we reserve for patients who consume a lot of services. In the outpatient clinic, it’s the type of patient who comes for frequent visits, perhaps more often than medically necessary. Oftentimes, more than we’d like. They can be demanding. They can also be an invaluable resource: None of your patients will likely be more forthright with you than those who are so motivated.
I saw one of my frequent flyer patients recently. He, like all patients, has medical problems, but, unlike most, he never misses an opportunity to schedule an appointment to solve them. A once red-, now gray-haired engineer, he has quite a record of skin issues and has meticulously documented all of them himself.

I felt myself stiffening. I added him on to my schedule today because I’m a good guy, yet he wants a piece of me? Bring it.
“So, you can read my notes online?” I asked. “Yes,” he replied, “for some reason I can read all of my charts for dermatology visits.”
“Well, that’s because I volunteered for our OpenNotes program,” I said. As a participant, all of my patients are able to read all of my notes, if they choose to do so. They can access them but cannot make any changes.
Yeah, great idea, Jeff.
“I just want to know, why would you put that if you didn’t do it?” he asked.
“Well, it’s not a lie. We did discuss the risks and benefits of my freezing your AKs previously, right?” “Yes, we did,” he replied. “Did you not want me to freeze them?” I asked. “No, I did,” he answered. “I just wanted you to know that I can see what you write about me, and I don’t want you to say anything you don’t want me to read because I really trust you.”
“I won’t,” I said.
That’s because I understand that you are my patient, and all patients deserve my unmitigated care. It’s what makes me a doctor.
I’ve since added to my EMR template: “Previously discussed risks and benefits.” Not because it really matters. But because it matters to him. And that matters to me.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @dermdoc on Twitter. Write to him at [email protected].
“He is a frequent flyer.” This is a term we reserve for patients who consume a lot of services. In the outpatient clinic, it’s the type of patient who comes for frequent visits, perhaps more often than medically necessary. Oftentimes, more than we’d like. They can be demanding. They can also be an invaluable resource: None of your patients will likely be more forthright with you than those who are so motivated.
I saw one of my frequent flyer patients recently. He, like all patients, has medical problems, but, unlike most, he never misses an opportunity to schedule an appointment to solve them. A once red-, now gray-haired engineer, he has quite a record of skin issues and has meticulously documented all of them himself.
I felt myself stiffening. I added him on to my schedule today because I’m a good guy, yet he wants a piece of me? Bring it.
“So, you can read my notes online?” I asked. “Yes,” he replied, “for some reason I can read all of my charts for dermatology visits.”
“Well, that’s because I volunteered for our OpenNotes program,” I said. As a participant, all of my patients are able to read all of my notes, if they choose to do so. They can access them but cannot make any changes.
Yeah, great idea, Jeff.
“I just want to know, why would you put that if you didn’t do it?” he asked.
“Well, it’s not a lie. We did discuss the risks and benefits of my freezing your AKs previously, right?” “Yes, we did,” he replied. “Did you not want me to freeze them?” I asked. “No, I did,” he answered. “I just wanted you to know that I can see what you write about me, and I don’t want you to say anything you don’t want me to read because I really trust you.”
“I won’t,” I said.
That’s because I understand that you are my patient, and all patients deserve my unmitigated care. It’s what makes me a doctor.
I’ve since added to my EMR template: “Previously discussed risks and benefits.” Not because it really matters. But because it matters to him. And that matters to me.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @dermdoc on Twitter. Write to him at [email protected].
“He is a frequent flyer.” This is a term we reserve for patients who consume a lot of services. In the outpatient clinic, it’s the type of patient who comes for frequent visits, perhaps more often than medically necessary. Oftentimes, more than we’d like. They can be demanding. They can also be an invaluable resource: None of your patients will likely be more forthright with you than those who are so motivated.
I saw one of my frequent flyer patients recently. He, like all patients, has medical problems, but, unlike most, he never misses an opportunity to schedule an appointment to solve them. A once red-, now gray-haired engineer, he has quite a record of skin issues and has meticulously documented all of them himself.
I felt myself stiffening. I added him on to my schedule today because I’m a good guy, yet he wants a piece of me? Bring it.
“So, you can read my notes online?” I asked. “Yes,” he replied, “for some reason I can read all of my charts for dermatology visits.”
“Well, that’s because I volunteered for our OpenNotes program,” I said. As a participant, all of my patients are able to read all of my notes, if they choose to do so. They can access them but cannot make any changes.
Yeah, great idea, Jeff.
“I just want to know, why would you put that if you didn’t do it?” he asked.
“Well, it’s not a lie. We did discuss the risks and benefits of my freezing your AKs previously, right?” “Yes, we did,” he replied. “Did you not want me to freeze them?” I asked. “No, I did,” he answered. “I just wanted you to know that I can see what you write about me, and I don’t want you to say anything you don’t want me to read because I really trust you.”
“I won’t,” I said.
That’s because I understand that you are my patient, and all patients deserve my unmitigated care. It’s what makes me a doctor.
I’ve since added to my EMR template: “Previously discussed risks and benefits.” Not because it really matters. But because it matters to him. And that matters to me.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @dermdoc on Twitter. Write to him at [email protected].
For vertebral osteomyelitis, early switch to oral antibiotics is feasible
VIENNA – A 6-week course of antibiotics, with an early switch from intravenous to oral, appears to be a safe and appropriate option for some patients with pyogenic vertebral osteomyelitis.
A single-center retrospective study of 82 such patients found two treatment failures and two deaths over 1 year (4.8% failure rate). The patients who died were very elderly with serious comorbidities. The two treatment failures occurred in patients with methicillin-resistant coagulase-negative staphylococcal infections of a central catheter.
“Only two of the failures were due to inadequate antibiotic treatment,” Adrien Lemaignen, MD, said at the European Society of Clinical Microbiology and Infectious Diseases annual congress. “Both patients experienced a relapse of bacteremia with the same bacteria a few days after antibiotic cessation in a context of conservative treatment of a catheter-related infection.”
Guidelines recently adopted by the Infectious Diseases Society of America inspired the study, said Dr. Lemaignen of University Hospital of Tours, France. The 2015 document calls for 6-8 weeks of antibiotics, depending upon the infective organism and whether infective endocarditis complicates management. All suggested antibiotic regimens call for initial IV therapy followed by oral, but there are no cut-and-dried recommendations about when to switch. The guideline notes one study in which patients switched to oral after about 2.7 weeks, with a 97% success rate.
Dr. Lemaignen and his colleagues set out to determine cure rates of early oral relay in 82 patients with pyogenic vertebral osteomyelitis (PVO). All patients were treated at a single center from 2011 to 2016. The team defined treatment failure as death, or persistence or relapse of infection in the first year after treatment.
All patients had culture-proven PVO that also was visible on imaging. Patients were excluded if they had any brucellar, fungal, or mycobacterial coinfections, or if they had infected spinal implants.
The mean age of the patients in the cohort was 66 years; 39% had some neuropathology. The mean C-reactive protein level was 115 mg/L. More than half of the cases (56%) involved the lumbar-sacral spine; 30% were thoracic, and the remainder, cervical. About one-fifth had multiple level involvement. There was epidural inflammation in 68%, epidural abscess in 13%, and extradural abscess in 26%.
Staphylococcus aureus was the most common pathogen (34%); two infections were methicillin resistant. Other infective organisms were streptococci (27%), Gram-negative bacilli (15%), and coagulase-negative staph (12%). A few patients had enterococci (5%) or polymicrobial infections (7%).
Infective endocarditis was present in 16 patients; this was associated with enterococcal and streptococcal infections.
Treatment varied by pathogen. Patients with S. aureus received penicillin or cefazolin with an oral relay to fluoroquinolone/rifampicin or clindamycin. Those with streptococci received amoxicillin with or without an aminoglycoside, followed by oral amoxicillin or clindamycin. Those with coagulase-negative streptococci received a glycopeptide with or without blasticidin, followed by fluoroquinolone/rifampicin. Patients with enterococcal infections got a third generation cephalosporin followed by an oral third generation cephalosporin or a fluoroquinolone.
All but six patients received 6 weeks of treatment.
The mean oral relay occurred on day 12, but 30 patients (36%) were able to switch before 7 days elapsed. Thirteen patients had to stay on the IV route for their entire treatment; 25% of this group had infective endocarditis. Six patients, all of whom had motor symptoms, also needed surgery.
The median follow-up was 358 days. During this time, there were two deaths and two treatment failures.
One death was a 93-year-old who had a controlled sepsis, but died at day 79 of a massive hematemesis. The other was an 80-year-old with an amoxicillin-resistant staph infection and decompensated cirrhosis who died at day 49.
There were also two treatment failures. Both of these patients had methicillin-resistant coagulase-negative staph infections of indwelling central catheters. One had a relapse 70 days after the end of IV therapy; the other relapsed on day 26 of treatment, after a 2-week course of oral antibiotics.
Not all patients were able to succeed with 6 weeks of therapy. Three needed prolonged treatment: One of these had an infected vascular prosthesis and two were immunocompromised patients who had cervical osteomyelitis with multiple abscesses.
In light of these results, Dr. Lemaignen said, “We can say confirm the safety of short IV treatment with an early oral relay in pyogenic vertebral osteomyelitis under real-life conditions, with 95% success rate and good functional outcomes at 6 months.”
He had no relevant financial disclosures.
[email protected]
On Twitter @Alz_gal
VIENNA – A 6-week course of antibiotics, with an early switch from intravenous to oral, appears to be a safe and appropriate option for some patients with pyogenic vertebral osteomyelitis.
A single-center retrospective study of 82 such patients found two treatment failures and two deaths over 1 year (4.8% failure rate). The patients who died were very elderly with serious comorbidities. The two treatment failures occurred in patients with methicillin-resistant coagulase-negative staphylococcal infections of a central catheter.
“Only two of the failures were due to inadequate antibiotic treatment,” Adrien Lemaignen, MD, said at the European Society of Clinical Microbiology and Infectious Diseases annual congress. “Both patients experienced a relapse of bacteremia with the same bacteria a few days after antibiotic cessation in a context of conservative treatment of a catheter-related infection.”
Guidelines recently adopted by the Infectious Diseases Society of America inspired the study, said Dr. Lemaignen of University Hospital of Tours, France. The 2015 document calls for 6-8 weeks of antibiotics, depending upon the infective organism and whether infective endocarditis complicates management. All suggested antibiotic regimens call for initial IV therapy followed by oral, but there are no cut-and-dried recommendations about when to switch. The guideline notes one study in which patients switched to oral after about 2.7 weeks, with a 97% success rate.
Dr. Lemaignen and his colleagues set out to determine cure rates of early oral relay in 82 patients with pyogenic vertebral osteomyelitis (PVO). All patients were treated at a single center from 2011 to 2016. The team defined treatment failure as death, or persistence or relapse of infection in the first year after treatment.
All patients had culture-proven PVO that also was visible on imaging. Patients were excluded if they had any brucellar, fungal, or mycobacterial coinfections, or if they had infected spinal implants.
The mean age of the patients in the cohort was 66 years; 39% had some neuropathology. The mean C-reactive protein level was 115 mg/L. More than half of the cases (56%) involved the lumbar-sacral spine; 30% were thoracic, and the remainder, cervical. About one-fifth had multiple level involvement. There was epidural inflammation in 68%, epidural abscess in 13%, and extradural abscess in 26%.
Staphylococcus aureus was the most common pathogen (34%); two infections were methicillin resistant. Other infective organisms were streptococci (27%), Gram-negative bacilli (15%), and coagulase-negative staph (12%). A few patients had enterococci (5%) or polymicrobial infections (7%).
Infective endocarditis was present in 16 patients; this was associated with enterococcal and streptococcal infections.
Treatment varied by pathogen. Patients with S. aureus received penicillin or cefazolin with an oral relay to fluoroquinolone/rifampicin or clindamycin. Those with streptococci received amoxicillin with or without an aminoglycoside, followed by oral amoxicillin or clindamycin. Those with coagulase-negative streptococci received a glycopeptide with or without blasticidin, followed by fluoroquinolone/rifampicin. Patients with enterococcal infections got a third generation cephalosporin followed by an oral third generation cephalosporin or a fluoroquinolone.
All but six patients received 6 weeks of treatment.
The mean oral relay occurred on day 12, but 30 patients (36%) were able to switch before 7 days elapsed. Thirteen patients had to stay on the IV route for their entire treatment; 25% of this group had infective endocarditis. Six patients, all of whom had motor symptoms, also needed surgery.
The median follow-up was 358 days. During this time, there were two deaths and two treatment failures.
One death was a 93-year-old who had a controlled sepsis, but died at day 79 of a massive hematemesis. The other was an 80-year-old with an amoxicillin-resistant staph infection and decompensated cirrhosis who died at day 49.
There were also two treatment failures. Both of these patients had methicillin-resistant coagulase-negative staph infections of indwelling central catheters. One had a relapse 70 days after the end of IV therapy; the other relapsed on day 26 of treatment, after a 2-week course of oral antibiotics.
Not all patients were able to succeed with 6 weeks of therapy. Three needed prolonged treatment: One of these had an infected vascular prosthesis and two were immunocompromised patients who had cervical osteomyelitis with multiple abscesses.
In light of these results, Dr. Lemaignen said, “We can say confirm the safety of short IV treatment with an early oral relay in pyogenic vertebral osteomyelitis under real-life conditions, with 95% success rate and good functional outcomes at 6 months.”
He had no relevant financial disclosures.
[email protected]
On Twitter @Alz_gal
VIENNA – A 6-week course of antibiotics, with an early switch from intravenous to oral, appears to be a safe and appropriate option for some patients with pyogenic vertebral osteomyelitis.
A single-center retrospective study of 82 such patients found two treatment failures and two deaths over 1 year (4.8% failure rate). The patients who died were very elderly with serious comorbidities. The two treatment failures occurred in patients with methicillin-resistant coagulase-negative staphylococcal infections of a central catheter.
“Only two of the failures were due to inadequate antibiotic treatment,” Adrien Lemaignen, MD, said at the European Society of Clinical Microbiology and Infectious Diseases annual congress. “Both patients experienced a relapse of bacteremia with the same bacteria a few days after antibiotic cessation in a context of conservative treatment of a catheter-related infection.”
Guidelines recently adopted by the Infectious Diseases Society of America inspired the study, said Dr. Lemaignen of University Hospital of Tours, France. The 2015 document calls for 6-8 weeks of antibiotics, depending upon the infective organism and whether infective endocarditis complicates management. All suggested antibiotic regimens call for initial IV therapy followed by oral, but there are no cut-and-dried recommendations about when to switch. The guideline notes one study in which patients switched to oral after about 2.7 weeks, with a 97% success rate.
Dr. Lemaignen and his colleagues set out to determine cure rates of early oral relay in 82 patients with pyogenic vertebral osteomyelitis (PVO). All patients were treated at a single center from 2011 to 2016. The team defined treatment failure as death, or persistence or relapse of infection in the first year after treatment.
All patients had culture-proven PVO that also was visible on imaging. Patients were excluded if they had any brucellar, fungal, or mycobacterial coinfections, or if they had infected spinal implants.
The mean age of the patients in the cohort was 66 years; 39% had some neuropathology. The mean C-reactive protein level was 115 mg/L. More than half of the cases (56%) involved the lumbar-sacral spine; 30% were thoracic, and the remainder, cervical. About one-fifth had multiple level involvement. There was epidural inflammation in 68%, epidural abscess in 13%, and extradural abscess in 26%.
Staphylococcus aureus was the most common pathogen (34%); two infections were methicillin resistant. Other infective organisms were streptococci (27%), Gram-negative bacilli (15%), and coagulase-negative staph (12%). A few patients had enterococci (5%) or polymicrobial infections (7%).
Infective endocarditis was present in 16 patients; this was associated with enterococcal and streptococcal infections.
Treatment varied by pathogen. Patients with S. aureus received penicillin or cefazolin with an oral relay to fluoroquinolone/rifampicin or clindamycin. Those with streptococci received amoxicillin with or without an aminoglycoside, followed by oral amoxicillin or clindamycin. Those with coagulase-negative streptococci received a glycopeptide with or without blasticidin, followed by fluoroquinolone/rifampicin. Patients with enterococcal infections got a third generation cephalosporin followed by an oral third generation cephalosporin or a fluoroquinolone.
All but six patients received 6 weeks of treatment.
The mean oral relay occurred on day 12, but 30 patients (36%) were able to switch before 7 days elapsed. Thirteen patients had to stay on the IV route for their entire treatment; 25% of this group had infective endocarditis. Six patients, all of whom had motor symptoms, also needed surgery.
The median follow-up was 358 days. During this time, there were two deaths and two treatment failures.
One death was a 93-year-old who had a controlled sepsis, but died at day 79 of a massive hematemesis. The other was an 80-year-old with an amoxicillin-resistant staph infection and decompensated cirrhosis who died at day 49.
There were also two treatment failures. Both of these patients had methicillin-resistant coagulase-negative staph infections of indwelling central catheters. One had a relapse 70 days after the end of IV therapy; the other relapsed on day 26 of treatment, after a 2-week course of oral antibiotics.
Not all patients were able to succeed with 6 weeks of therapy. Three needed prolonged treatment: One of these had an infected vascular prosthesis and two were immunocompromised patients who had cervical osteomyelitis with multiple abscesses.
In light of these results, Dr. Lemaignen said, “We can say confirm the safety of short IV treatment with an early oral relay in pyogenic vertebral osteomyelitis under real-life conditions, with 95% success rate and good functional outcomes at 6 months.”
He had no relevant financial disclosures.
[email protected]
On Twitter @Alz_gal
AT ECCMID 2017
Key clinical point:
Major finding: There were two treatment failures attributable to the antibiotic regimen, and two deaths that were not, for a total treatment success rate of 95%.
Data source: A retrospective cohort comprising 82 patients.
Disclosures: Dr. Lemaignen had no financial disclosures.
Ribaxamase prevented C. difficile infections by protecting microbiome
VIENNA – An investigational beta-lactamase reduced Clostridium difficile infections by 71% in patients receiving extended antibiotic therapy for respiratory infections but not by killing the opportunistic bacteria.
Rather, ribaxamase prevented C. difficile infections (CDI) by breaking down excess therapeutic antibiotics in the gut before they could injure an otherwise healthy microbiome, John Kokai-Kun, PhD, said at the European Society of Clinical Microbiology and Infectious Diseases annual congress.

Ribaxamase is an oral enzyme that breaks the lactam ring in penicillins and cephalosporins. It’s formulated to release at a pH of 5.5 or higher, an environment that begins to develop in the upper small intestine near the bile duct – the same place that excess antibiotics are excreted.
“The drug is intended to be administered during, and for a short time after, intravenous administration of specific beta-lactam–containing antibiotics,” Dr. Kokai-Kun said. Ribaxamase doesn’t work on carbapenem-type antibiotics, he noted, and Synthetic Biologics is working on an effective enzyme for those as well.
In early human studies, ribaxamase was well tolerated and didn’t interfere with the pharmacokinetics of therapeutic antibiotics (Antimicrob Agents Chemother. 2017 Mar;61[3]:e02197-16). It’s also effective in patients who are taking a proton pump inhibitor, he said.
Dr. Kokai-Kun reported the results of a phase IIb study of 412 patients who received IV ceftriaxone for lower respiratory infections. They were assigned 1:1 to either 150 mg ribaxamase daily or placebo throughout the IV treatment and for 3 days after.
The primary endpoint was prevention of C. difficile infection. The secondary endpoint was prevention of non–C. difficile antibiotic-associated diarrhea. An exploratory endpoint examined the drug’s ability to protect the microbiome. Patients were monitored for 6 weeks after treatment stopped.
The cohort was a mean 70 years old. One-third of patients also received a macrolide during their hospitalization, and one-third were taking proton pump inhibitors. The respiratory infection cure rate was about 99% in both groups at both 72 hours and 4 weeks.
Eight patients in the placebo group (3.8%) and two in the active group (less than 1%) developed C. difficile infection. That translated to a statistically significant 71% risk reduction, with a P value of .027, Dr. Kokai-Kun said. Ribaxamase did not hit its secondary endpoint of preventing all-cause diarrhea or antibiotic-associated diarrhea that was not caused by C. difficile infection.
Although not a primary finding, ribaxamase also inhibited colonization by vancomycin-resistant enterococci, which occurred in about 70 (40%) patients in the placebo group and 40 (20%) in the ribaxamase group at both 72 hours and 4 weeks.
All patients contributed stool samples at baseline and after treatment for microbiome analysis. That portion of the study is still ongoing, Dr. Kokai-Kun said.
Synthetic Biologics sponsored the study and is developing ribaxamase. Dr. Kokai-Kun is the company’s vice president of nonclinical affairs.
[email protected]
On Twitter @alz_gal

VIENNA – An investigational beta-lactamase reduced Clostridium difficile infections by 71% in patients receiving extended antibiotic therapy for respiratory infections but not by killing the opportunistic bacteria.
Rather, ribaxamase prevented C. difficile infections (CDI) by breaking down excess therapeutic antibiotics in the gut before they could injure an otherwise healthy microbiome, John Kokai-Kun, PhD, said at the European Society of Clinical Microbiology and Infectious Diseases annual congress.
Ribaxamase is an oral enzyme that breaks the lactam ring in penicillins and cephalosporins. It’s formulated to release at a pH of 5.5 or higher, an environment that begins to develop in the upper small intestine near the bile duct – the same place that excess antibiotics are excreted.
“The drug is intended to be administered during, and for a short time after, intravenous administration of specific beta-lactam–containing antibiotics,” Dr. Kokai-Kun said. Ribaxamase doesn’t work on carbapenem-type antibiotics, he noted, and Synthetic Biologics is working on an effective enzyme for those as well.
In early human studies, ribaxamase was well tolerated and didn’t interfere with the pharmacokinetics of therapeutic antibiotics (Antimicrob Agents Chemother. 2017 Mar;61[3]:e02197-16). It’s also effective in patients who are taking a proton pump inhibitor, he said.
Dr. Kokai-Kun reported the results of a phase IIb study of 412 patients who received IV ceftriaxone for lower respiratory infections. They were assigned 1:1 to either 150 mg ribaxamase daily or placebo throughout the IV treatment and for 3 days after.
The primary endpoint was prevention of C. difficile infection. The secondary endpoint was prevention of non–C. difficile antibiotic-associated diarrhea. An exploratory endpoint examined the drug’s ability to protect the microbiome. Patients were monitored for 6 weeks after treatment stopped.
The cohort was a mean 70 years old. One-third of patients also received a macrolide during their hospitalization, and one-third were taking proton pump inhibitors. The respiratory infection cure rate was about 99% in both groups at both 72 hours and 4 weeks.
Eight patients in the placebo group (3.8%) and two in the active group (less than 1%) developed C. difficile infection. That translated to a statistically significant 71% risk reduction, with a P value of .027, Dr. Kokai-Kun said. Ribaxamase did not hit its secondary endpoint of preventing all-cause diarrhea or antibiotic-associated diarrhea that was not caused by C. difficile infection.
Although not a primary finding, ribaxamase also inhibited colonization by vancomycin-resistant enterococci, which occurred in about 70 (40%) patients in the placebo group and 40 (20%) in the ribaxamase group at both 72 hours and 4 weeks.
All patients contributed stool samples at baseline and after treatment for microbiome analysis. That portion of the study is still ongoing, Dr. Kokai-Kun said.
Synthetic Biologics sponsored the study and is developing ribaxamase. Dr. Kokai-Kun is the company’s vice president of nonclinical affairs.
[email protected]
On Twitter @alz_gal
VIENNA – An investigational beta-lactamase reduced Clostridium difficile infections by 71% in patients receiving extended antibiotic therapy for respiratory infections but not by killing the opportunistic bacteria.
Rather, ribaxamase prevented C. difficile infections (CDI) by breaking down excess therapeutic antibiotics in the gut before they could injure an otherwise healthy microbiome, John Kokai-Kun, PhD, said at the European Society of Clinical Microbiology and Infectious Diseases annual congress.
Ribaxamase is an oral enzyme that breaks the lactam ring in penicillins and cephalosporins. It’s formulated to release at a pH of 5.5 or higher, an environment that begins to develop in the upper small intestine near the bile duct – the same place that excess antibiotics are excreted.
“The drug is intended to be administered during, and for a short time after, intravenous administration of specific beta-lactam–containing antibiotics,” Dr. Kokai-Kun said. Ribaxamase doesn’t work on carbapenem-type antibiotics, he noted, and Synthetic Biologics is working on an effective enzyme for those as well.
In early human studies, ribaxamase was well tolerated and didn’t interfere with the pharmacokinetics of therapeutic antibiotics (Antimicrob Agents Chemother. 2017 Mar;61[3]:e02197-16). It’s also effective in patients who are taking a proton pump inhibitor, he said.
Dr. Kokai-Kun reported the results of a phase IIb study of 412 patients who received IV ceftriaxone for lower respiratory infections. They were assigned 1:1 to either 150 mg ribaxamase daily or placebo throughout the IV treatment and for 3 days after.
The primary endpoint was prevention of C. difficile infection. The secondary endpoint was prevention of non–C. difficile antibiotic-associated diarrhea. An exploratory endpoint examined the drug’s ability to protect the microbiome. Patients were monitored for 6 weeks after treatment stopped.
The cohort was a mean 70 years old. One-third of patients also received a macrolide during their hospitalization, and one-third were taking proton pump inhibitors. The respiratory infection cure rate was about 99% in both groups at both 72 hours and 4 weeks.
Eight patients in the placebo group (3.8%) and two in the active group (less than 1%) developed C. difficile infection. That translated to a statistically significant 71% risk reduction, with a P value of .027, Dr. Kokai-Kun said. Ribaxamase did not hit its secondary endpoint of preventing all-cause diarrhea or antibiotic-associated diarrhea that was not caused by C. difficile infection.
Although not a primary finding, ribaxamase also inhibited colonization by vancomycin-resistant enterococci, which occurred in about 70 (40%) patients in the placebo group and 40 (20%) in the ribaxamase group at both 72 hours and 4 weeks.
All patients contributed stool samples at baseline and after treatment for microbiome analysis. That portion of the study is still ongoing, Dr. Kokai-Kun said.
Synthetic Biologics sponsored the study and is developing ribaxamase. Dr. Kokai-Kun is the company’s vice president of nonclinical affairs.
[email protected]
On Twitter @alz_gal
AT ECCMID 2017
Key clinical point:
Major finding: Ribaxamase reduced C. difficile infections by 71%, relative to a placebo.
Data source: The study randomized 412 patients to either placebo or ribaxamase in addition to their therapeutic antibiotics.
Disclosures: Synthetic Biologics sponsored the study and is developing ribaxamase. Dr. Kokai-Kun is the company’s vice president of nonclinical affairs.
Topical imiquimod boosted response to intradermal hepatitis B vaccine
VIENNA – Topical imiquimod appeared to enhance the immunogenicity of an intradermal hepatitis B vaccine in patients on renal replacement therapy.
Patients on hemodialysis or peritoneal dialysis who got the combination developed significantly higher seroprotection and antibody levels than those who got either the typical intramuscular vaccination or an intradermal vaccination on unprepared skin, Ivan Fan-Ngai Hung, MD, said at the European Conference on Clinical Microbiology and Infectious Diseases. By 1 year, the protection and titers did begin to fall, but they still remained significantly higher than in the two comparator groups, said Dr. Hung, a clinical professor at the University of Hong Kong.

Dr. Hung and his colleagues have been investigating imiquimod’s immunogenicity-boosting potential for several years. Their initial murine work with an H1N1 influenza virus appeared in 2014 (Clin Vaccine Immunol. 2014 Apr;21[4]: 570-9). The investigators intraperitoneally immunized mice with a monovalent A(H1N1) vaccine combined with imiquimod (VIC) then intranasally inoculated them with a lethal dose of the virus. When compared with mice who received only vaccine, only imiquimod, or only placebo, the VIC group showed significantly greater and significantly longer survival. Virus-specific serum immunoglobulin M, IgG, and neutralizing antibodies were all significantly higher.
The investigators theorized that imiquimod, a Toll-like receptor 7 agonist, plays several key roles in boosting immune response, including inducing the differentiation and migration of dendritic cells, enhancing B cell differentiation, and increasing long-term B cell memory.
Within the past 2 years, the group has advanced to human influenza trials in healthy young adults and elders with comorbidities.
Both studies employed a 5% imiquimod cream delivering 250 mg of the drug. It was applied at the injection site 5 minutes before vaccination. In the elder study, 90% of the 91 subjects who got the combination achieved seroconversion, compared with 13% of those who got an intramuscular injection and 39% of those who got an intradermal injection plus placebo cream. The geometric mean titers went up faster and stayed elevated longer. The better immunogenicity was associated with fewer hospitalizations for influenza or pneumonia (Clin Infect Dis. 2014;59[9]:1246-55).
The immunogenicity findings were similar in the study of 160 healthy young people. This study had a surprising twist too, Dr. Hung said in his talk. Not only did the combination significantly improve immunogenicity against the vaccine influenza strains, it increased immunogenicity against the nonvaccine strains, especially the antigenically drifted H3N2 strain of 2015, which was not included in the 2013-2014 recommended vaccine (Lancet Inf Dis. 2016 Feb;16(2):209-18).
The study Dr. Hung presented in Vienna was an interim analysis of the first to apply this technique to a hepatitis B vaccine. It enrolled 69 patients (51 on peritoneal dialysis and 18 on hemodialysis). They were a mean 65 years old. All received 10 mcg of the Sci-B-Vac at baseline, 1 month, and 6 months. Vaccine was delivered in a trineedle unit designed for shallow intradermal penetration (MicronJet600; NanoPass Technologies) Group IQ received topical imiquimod along with the intradermal vaccine. Group ID received a placebo cream and the intradermal vaccine. Group IM received a placebo cream and an intramuscular vaccination.
Anti–hepatitis B titers were measured at baseline and at 1, 3, 6, and 12 months. The primary outcome was seroprotection at 1 month. The secondary outcomes were seroprotection at 3, 6, and 12 months; anti–hepatitis B antibody titer; and safety.
By 1 month, seroprotection was already significantly higher in the IQ group than in the ID and IM groups (60% vs. 50% and 38%, respectively).
By 3 months, the seroprotection rate in group IQ had risen to 85%. It remained elevated there at 6 months then tailed off to about 70% by 12 months. The ID and IM groups followed this same rising and falling curve but remained significantly lower at all time points. At 12 months, seroprotection was similar in both these groups – about 40%.
The anti–hepatitis B antibody titers told a similar story. Titers in the IQ group rose more rapidly and sharply, to 544 mIU/mL at 6 months and 566 mIU/mL at 12 months. The ID group also experienced a strong response, rising to 489 mIU/mL at 6 months. However, by 12 months, titer levels had dropped to 170 mIU/mL.
Titers in the IM group barely moved at all during the entire follow-up period, never rising above 21 mIU/mL.
There were no differences in systemic reactions among the three groups, but those who got the intradermal vaccines reported slightly more swelling and induration at the injection site.
“Since this is an interim analysis, we cannot determine long-term protection or antibody titers,” Dr. Hung cautioned. “However, we are starting a similar study in elderly patients and also one for those who are on low-dose immunosuppressants. We believe this regimen will also work for them.”
Dr. Hung has been on advisory boards for Pfizer and Gilead Sciences.
[email protected]
On Twitter @alz_gal
VIENNA – Topical imiquimod appeared to enhance the immunogenicity of an intradermal hepatitis B vaccine in patients on renal replacement therapy.
Patients on hemodialysis or peritoneal dialysis who got the combination developed significantly higher seroprotection and antibody levels than those who got either the typical intramuscular vaccination or an intradermal vaccination on unprepared skin, Ivan Fan-Ngai Hung, MD, said at the European Conference on Clinical Microbiology and Infectious Diseases. By 1 year, the protection and titers did begin to fall, but they still remained significantly higher than in the two comparator groups, said Dr. Hung, a clinical professor at the University of Hong Kong.
Dr. Hung and his colleagues have been investigating imiquimod’s immunogenicity-boosting potential for several years. Their initial murine work with an H1N1 influenza virus appeared in 2014 (Clin Vaccine Immunol. 2014 Apr;21[4]: 570-9). The investigators intraperitoneally immunized mice with a monovalent A(H1N1) vaccine combined with imiquimod (VIC) then intranasally inoculated them with a lethal dose of the virus. When compared with mice who received only vaccine, only imiquimod, or only placebo, the VIC group showed significantly greater and significantly longer survival. Virus-specific serum immunoglobulin M, IgG, and neutralizing antibodies were all significantly higher.
The investigators theorized that imiquimod, a Toll-like receptor 7 agonist, plays several key roles in boosting immune response, including inducing the differentiation and migration of dendritic cells, enhancing B cell differentiation, and increasing long-term B cell memory.
Within the past 2 years, the group has advanced to human influenza trials in healthy young adults and elders with comorbidities.
Both studies employed a 5% imiquimod cream delivering 250 mg of the drug. It was applied at the injection site 5 minutes before vaccination. In the elder study, 90% of the 91 subjects who got the combination achieved seroconversion, compared with 13% of those who got an intramuscular injection and 39% of those who got an intradermal injection plus placebo cream. The geometric mean titers went up faster and stayed elevated longer. The better immunogenicity was associated with fewer hospitalizations for influenza or pneumonia (Clin Infect Dis. 2014;59[9]:1246-55).
The immunogenicity findings were similar in the study of 160 healthy young people. This study had a surprising twist too, Dr. Hung said in his talk. Not only did the combination significantly improve immunogenicity against the vaccine influenza strains, it increased immunogenicity against the nonvaccine strains, especially the antigenically drifted H3N2 strain of 2015, which was not included in the 2013-2014 recommended vaccine (Lancet Inf Dis. 2016 Feb;16(2):209-18).
The study Dr. Hung presented in Vienna was an interim analysis of the first to apply this technique to a hepatitis B vaccine. It enrolled 69 patients (51 on peritoneal dialysis and 18 on hemodialysis). They were a mean 65 years old. All received 10 mcg of the Sci-B-Vac at baseline, 1 month, and 6 months. Vaccine was delivered in a trineedle unit designed for shallow intradermal penetration (MicronJet600; NanoPass Technologies) Group IQ received topical imiquimod along with the intradermal vaccine. Group ID received a placebo cream and the intradermal vaccine. Group IM received a placebo cream and an intramuscular vaccination.
Anti–hepatitis B titers were measured at baseline and at 1, 3, 6, and 12 months. The primary outcome was seroprotection at 1 month. The secondary outcomes were seroprotection at 3, 6, and 12 months; anti–hepatitis B antibody titer; and safety.
By 1 month, seroprotection was already significantly higher in the IQ group than in the ID and IM groups (60% vs. 50% and 38%, respectively).
By 3 months, the seroprotection rate in group IQ had risen to 85%. It remained elevated there at 6 months then tailed off to about 70% by 12 months. The ID and IM groups followed this same rising and falling curve but remained significantly lower at all time points. At 12 months, seroprotection was similar in both these groups – about 40%.
The anti–hepatitis B antibody titers told a similar story. Titers in the IQ group rose more rapidly and sharply, to 544 mIU/mL at 6 months and 566 mIU/mL at 12 months. The ID group also experienced a strong response, rising to 489 mIU/mL at 6 months. However, by 12 months, titer levels had dropped to 170 mIU/mL.
Titers in the IM group barely moved at all during the entire follow-up period, never rising above 21 mIU/mL.
There were no differences in systemic reactions among the three groups, but those who got the intradermal vaccines reported slightly more swelling and induration at the injection site.
“Since this is an interim analysis, we cannot determine long-term protection or antibody titers,” Dr. Hung cautioned. “However, we are starting a similar study in elderly patients and also one for those who are on low-dose immunosuppressants. We believe this regimen will also work for them.”
Dr. Hung has been on advisory boards for Pfizer and Gilead Sciences.
[email protected]
On Twitter @alz_gal
VIENNA – Topical imiquimod appeared to enhance the immunogenicity of an intradermal hepatitis B vaccine in patients on renal replacement therapy.
Patients on hemodialysis or peritoneal dialysis who got the combination developed significantly higher seroprotection and antibody levels than those who got either the typical intramuscular vaccination or an intradermal vaccination on unprepared skin, Ivan Fan-Ngai Hung, MD, said at the European Conference on Clinical Microbiology and Infectious Diseases. By 1 year, the protection and titers did begin to fall, but they still remained significantly higher than in the two comparator groups, said Dr. Hung, a clinical professor at the University of Hong Kong.
Dr. Hung and his colleagues have been investigating imiquimod’s immunogenicity-boosting potential for several years. Their initial murine work with an H1N1 influenza virus appeared in 2014 (Clin Vaccine Immunol. 2014 Apr;21[4]: 570-9). The investigators intraperitoneally immunized mice with a monovalent A(H1N1) vaccine combined with imiquimod (VIC) then intranasally inoculated them with a lethal dose of the virus. When compared with mice who received only vaccine, only imiquimod, or only placebo, the VIC group showed significantly greater and significantly longer survival. Virus-specific serum immunoglobulin M, IgG, and neutralizing antibodies were all significantly higher.
The investigators theorized that imiquimod, a Toll-like receptor 7 agonist, plays several key roles in boosting immune response, including inducing the differentiation and migration of dendritic cells, enhancing B cell differentiation, and increasing long-term B cell memory.
Within the past 2 years, the group has advanced to human influenza trials in healthy young adults and elders with comorbidities.
Both studies employed a 5% imiquimod cream delivering 250 mg of the drug. It was applied at the injection site 5 minutes before vaccination. In the elder study, 90% of the 91 subjects who got the combination achieved seroconversion, compared with 13% of those who got an intramuscular injection and 39% of those who got an intradermal injection plus placebo cream. The geometric mean titers went up faster and stayed elevated longer. The better immunogenicity was associated with fewer hospitalizations for influenza or pneumonia (Clin Infect Dis. 2014;59[9]:1246-55).
The immunogenicity findings were similar in the study of 160 healthy young people. This study had a surprising twist too, Dr. Hung said in his talk. Not only did the combination significantly improve immunogenicity against the vaccine influenza strains, it increased immunogenicity against the nonvaccine strains, especially the antigenically drifted H3N2 strain of 2015, which was not included in the 2013-2014 recommended vaccine (Lancet Inf Dis. 2016 Feb;16(2):209-18).
The study Dr. Hung presented in Vienna was an interim analysis of the first to apply this technique to a hepatitis B vaccine. It enrolled 69 patients (51 on peritoneal dialysis and 18 on hemodialysis). They were a mean 65 years old. All received 10 mcg of the Sci-B-Vac at baseline, 1 month, and 6 months. Vaccine was delivered in a trineedle unit designed for shallow intradermal penetration (MicronJet600; NanoPass Technologies) Group IQ received topical imiquimod along with the intradermal vaccine. Group ID received a placebo cream and the intradermal vaccine. Group IM received a placebo cream and an intramuscular vaccination.
Anti–hepatitis B titers were measured at baseline and at 1, 3, 6, and 12 months. The primary outcome was seroprotection at 1 month. The secondary outcomes were seroprotection at 3, 6, and 12 months; anti–hepatitis B antibody titer; and safety.
By 1 month, seroprotection was already significantly higher in the IQ group than in the ID and IM groups (60% vs. 50% and 38%, respectively).
By 3 months, the seroprotection rate in group IQ had risen to 85%. It remained elevated there at 6 months then tailed off to about 70% by 12 months. The ID and IM groups followed this same rising and falling curve but remained significantly lower at all time points. At 12 months, seroprotection was similar in both these groups – about 40%.
The anti–hepatitis B antibody titers told a similar story. Titers in the IQ group rose more rapidly and sharply, to 544 mIU/mL at 6 months and 566 mIU/mL at 12 months. The ID group also experienced a strong response, rising to 489 mIU/mL at 6 months. However, by 12 months, titer levels had dropped to 170 mIU/mL.
Titers in the IM group barely moved at all during the entire follow-up period, never rising above 21 mIU/mL.
There were no differences in systemic reactions among the three groups, but those who got the intradermal vaccines reported slightly more swelling and induration at the injection site.
“Since this is an interim analysis, we cannot determine long-term protection or antibody titers,” Dr. Hung cautioned. “However, we are starting a similar study in elderly patients and also one for those who are on low-dose immunosuppressants. We believe this regimen will also work for them.”
Dr. Hung has been on advisory boards for Pfizer and Gilead Sciences.
[email protected]
On Twitter @alz_gal
AT ECCMID 2017
Key clinical point:
Major finding: By 1 month, seroprotection was significantly higher in the imiquimod group than in those who got an intradermal vaccination without imiquimod and those who had an intramuscular injection only (60% vs. 50% and 38%, respectively).
Data source: The four-armed randomized study comprised 69 patients on hemodialysis or peritoneal dialysis.
Disclosures: Dr. Hung has served on advisory boards for Pfizer and Gilead Sciences.