User login
AED Choice Requires an Individualized Approach

New Epilepsy Classification System
A patient’s epilepsy syndrome is the most important factor when choosing an AED, Dr. French said. According to a new epilepsy classification system published by the International League Against Epilepsy, generalized epilepsy may be further categorized as absence, myoclonic, atonic, tonic, and tonic–clonic. Focal epilepsy (formerly called partial epilepsy) may be categorized as focal aware (formerly simple partial), focal impaired awareness (formerly complex partial), and focal to bilateral tonic–clonic (formerly secondary generalized).
Combined generalized and focal epilepsy is a new category associated with epileptic encephalopathies, such as Dravet syndrome and Lennox-Gastaut syndrome. Finally, “seizures of unknown onset” describes cases where the clinician does not have enough information to determine whether the epilepsy is focal or generalized.
Narrow-spectrum drugs, including carbamazepine, oxcarbazepine, tiagabine, gabapentin, and pregabalin, should only be used in patients with focal epilepsy. “Narrow-spectrum drugs might make generalized seizures worse, or they fail to treat generalized seizures,” Dr. French said. “People with generalized epilepsy, generalized and focal epilepsy combined, or epilepsy of unknown onset should be on broad-spectrum agents—valproic acid, topiramate, lamotrigine, levetiracetam, zonisamide, and perampanel.”
Risk of Adverse Effects
“With any AED, the most common adverse events are dose-related,” Dr. French said. Some adverse events occur during titration and eventually resolve. Some drugs are associated with specific adverse events and therefore should be avoided in certain patient populations.
For instance, carbamazepine, oxcarbazepine, and eslicarbazepine may cause hyponatremia and should be given with caution to at-risk patients, such as the elderly. Likewise, drugs that may cause renal calculi, such as topiramate and zonisamide, should be given with caution to patients with that condition. Enzyme-inducing AEDs that increase cholesterol levels (eg, carbamazepine and phenytoin) should be avoided in patients with cardiovascular risk factors. For patients with weight problems or eating disorders, physicians should bear in mind that valproate, gabapentin, carbamazepine, pregabalin, ezogabine, and perampanel have been known to increase weight, while topiramate, zonisamide, and felbamate have been known to decrease weight.
AEDs that may worsen psychiatric function include levetiracetam, topiramate, zonisamide, tiagabine, phenobarbital, and perampanel. “Make sure that you ask patients when they are on levetiracetam whether they are experiencing increased irritability, although not everyone recognizes their mood changes,” Dr. French said. “Sometimes the spouse will complain, but the person will not.” On the other hand, carbamazepine, valproic acid, lamotrigine, and pregabalin have a tendency to improve psychiatric function—but not always. Psychiatric function “can go either way” with any of the AEDs, she said.
Some patients experience adverse drug effects as a result of add-on therapy. In some cases, a pharmacodynamic interaction between the new and old drugs may be responsible. Removing the add-on drug or the background drug may help. “Sometimes it is a better idea to take away the background drugs,” she said.
Older AEDs—carbamazepine, phenytoin, phenobarbital, and valproic acid—are associated with idiosyncratic adverse effects, including serious rash, liver failure, bone marrow failure, and pancreatitis. While some of the newer drugs also have such risks, the overall rate of idiosyncratic adverse reactions with their use is lower. Thus far, levetiracetam, brivaracetam, gabapentin, and pregabalin have not been associated with major idiosyncratic adverse effects, Dr. French noted.
Drug Initiation and Interactions
“The ability to initiate a drug rapidly is sometimes the driving characteristic that causes some doctors to pick one drug over another,” Dr. French said. Drugs that can be initiated at a therapeutic dose in a single day include phenytoin, levetiracetam, valproic acid, gabapentin, pregabalin, lacosamide, and brivaracetam. Other AEDs, such as carbamazepine, lamotrigine, perampanel, and oxcarbazepine, require gradual initiation over one to 10 weeks. Some drugs that take longer to initiate may be better tolerated overall, she said.
Generally, older AEDs are more likely to cause drug interactions, compared with newer drugs. Phenytoin, phenobarbital, and carbamazepine are enzyme inducers, which may cause problems for patients who are on statins. Valproic acid is a hepatic enzyme inhibitor, and phenytoin and valproic acid can have protein-binding interactions. Oral contraceptives reduce lamotrigine’s efficacy by doubling the clearance of the AED, Dr. French explained. “This can be problematic when women do not tell you when they are going on and off the contraceptive pill. They can have a breakthrough seizure, and it is only after the fact that you realize why that happened,” she said.
Interactions between oral contraceptive are common with other AEDs, as well. She cited a recent retrospective study involving 1,144 women with epilepsy ages 18 to 47 who provided demographic, epilepsy, AED, contraceptive, and pregnancy data. Survey results showed that 65% of their pregnancies were unintended. Oral forms of contraception had greater failure rates than nonoral forms, with intrauterine devices having the lowest failure rate.
—Adriene Marshall
Suggested Reading
Herzog AG, Mandle HB, Cahill KE, et al. Predictors of unintended pregnancy in women with epilepsy. Neurology. 2017;88(8):728-733.
Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512-521.
New Epilepsy Classification System
A patient’s epilepsy syndrome is the most important factor when choosing an AED, Dr. French said. According to a new epilepsy classification system published by the International League Against Epilepsy, generalized epilepsy may be further categorized as absence, myoclonic, atonic, tonic, and tonic–clonic. Focal epilepsy (formerly called partial epilepsy) may be categorized as focal aware (formerly simple partial), focal impaired awareness (formerly complex partial), and focal to bilateral tonic–clonic (formerly secondary generalized).
Combined generalized and focal epilepsy is a new category associated with epileptic encephalopathies, such as Dravet syndrome and Lennox-Gastaut syndrome. Finally, “seizures of unknown onset” describes cases where the clinician does not have enough information to determine whether the epilepsy is focal or generalized.
Narrow-spectrum drugs, including carbamazepine, oxcarbazepine, tiagabine, gabapentin, and pregabalin, should only be used in patients with focal epilepsy. “Narrow-spectrum drugs might make generalized seizures worse, or they fail to treat generalized seizures,” Dr. French said. “People with generalized epilepsy, generalized and focal epilepsy combined, or epilepsy of unknown onset should be on broad-spectrum agents—valproic acid, topiramate, lamotrigine, levetiracetam, zonisamide, and perampanel.”
Risk of Adverse Effects
“With any AED, the most common adverse events are dose-related,” Dr. French said. Some adverse events occur during titration and eventually resolve. Some drugs are associated with specific adverse events and therefore should be avoided in certain patient populations.
For instance, carbamazepine, oxcarbazepine, and eslicarbazepine may cause hyponatremia and should be given with caution to at-risk patients, such as the elderly. Likewise, drugs that may cause renal calculi, such as topiramate and zonisamide, should be given with caution to patients with that condition. Enzyme-inducing AEDs that increase cholesterol levels (eg, carbamazepine and phenytoin) should be avoided in patients with cardiovascular risk factors. For patients with weight problems or eating disorders, physicians should bear in mind that valproate, gabapentin, carbamazepine, pregabalin, ezogabine, and perampanel have been known to increase weight, while topiramate, zonisamide, and felbamate have been known to decrease weight.
AEDs that may worsen psychiatric function include levetiracetam, topiramate, zonisamide, tiagabine, phenobarbital, and perampanel. “Make sure that you ask patients when they are on levetiracetam whether they are experiencing increased irritability, although not everyone recognizes their mood changes,” Dr. French said. “Sometimes the spouse will complain, but the person will not.” On the other hand, carbamazepine, valproic acid, lamotrigine, and pregabalin have a tendency to improve psychiatric function—but not always. Psychiatric function “can go either way” with any of the AEDs, she said.
Some patients experience adverse drug effects as a result of add-on therapy. In some cases, a pharmacodynamic interaction between the new and old drugs may be responsible. Removing the add-on drug or the background drug may help. “Sometimes it is a better idea to take away the background drugs,” she said.
Older AEDs—carbamazepine, phenytoin, phenobarbital, and valproic acid—are associated with idiosyncratic adverse effects, including serious rash, liver failure, bone marrow failure, and pancreatitis. While some of the newer drugs also have such risks, the overall rate of idiosyncratic adverse reactions with their use is lower. Thus far, levetiracetam, brivaracetam, gabapentin, and pregabalin have not been associated with major idiosyncratic adverse effects, Dr. French noted.
Drug Initiation and Interactions
“The ability to initiate a drug rapidly is sometimes the driving characteristic that causes some doctors to pick one drug over another,” Dr. French said. Drugs that can be initiated at a therapeutic dose in a single day include phenytoin, levetiracetam, valproic acid, gabapentin, pregabalin, lacosamide, and brivaracetam. Other AEDs, such as carbamazepine, lamotrigine, perampanel, and oxcarbazepine, require gradual initiation over one to 10 weeks. Some drugs that take longer to initiate may be better tolerated overall, she said.
Generally, older AEDs are more likely to cause drug interactions, compared with newer drugs. Phenytoin, phenobarbital, and carbamazepine are enzyme inducers, which may cause problems for patients who are on statins. Valproic acid is a hepatic enzyme inhibitor, and phenytoin and valproic acid can have protein-binding interactions. Oral contraceptives reduce lamotrigine’s efficacy by doubling the clearance of the AED, Dr. French explained. “This can be problematic when women do not tell you when they are going on and off the contraceptive pill. They can have a breakthrough seizure, and it is only after the fact that you realize why that happened,” she said.
Interactions between oral contraceptive are common with other AEDs, as well. She cited a recent retrospective study involving 1,144 women with epilepsy ages 18 to 47 who provided demographic, epilepsy, AED, contraceptive, and pregnancy data. Survey results showed that 65% of their pregnancies were unintended. Oral forms of contraception had greater failure rates than nonoral forms, with intrauterine devices having the lowest failure rate.
—Adriene Marshall
Suggested Reading
Herzog AG, Mandle HB, Cahill KE, et al. Predictors of unintended pregnancy in women with epilepsy. Neurology. 2017;88(8):728-733.
Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512-521.
New Epilepsy Classification System
A patient’s epilepsy syndrome is the most important factor when choosing an AED, Dr. French said. According to a new epilepsy classification system published by the International League Against Epilepsy, generalized epilepsy may be further categorized as absence, myoclonic, atonic, tonic, and tonic–clonic. Focal epilepsy (formerly called partial epilepsy) may be categorized as focal aware (formerly simple partial), focal impaired awareness (formerly complex partial), and focal to bilateral tonic–clonic (formerly secondary generalized).
Combined generalized and focal epilepsy is a new category associated with epileptic encephalopathies, such as Dravet syndrome and Lennox-Gastaut syndrome. Finally, “seizures of unknown onset” describes cases where the clinician does not have enough information to determine whether the epilepsy is focal or generalized.
Narrow-spectrum drugs, including carbamazepine, oxcarbazepine, tiagabine, gabapentin, and pregabalin, should only be used in patients with focal epilepsy. “Narrow-spectrum drugs might make generalized seizures worse, or they fail to treat generalized seizures,” Dr. French said. “People with generalized epilepsy, generalized and focal epilepsy combined, or epilepsy of unknown onset should be on broad-spectrum agents—valproic acid, topiramate, lamotrigine, levetiracetam, zonisamide, and perampanel.”
Risk of Adverse Effects
“With any AED, the most common adverse events are dose-related,” Dr. French said. Some adverse events occur during titration and eventually resolve. Some drugs are associated with specific adverse events and therefore should be avoided in certain patient populations.
For instance, carbamazepine, oxcarbazepine, and eslicarbazepine may cause hyponatremia and should be given with caution to at-risk patients, such as the elderly. Likewise, drugs that may cause renal calculi, such as topiramate and zonisamide, should be given with caution to patients with that condition. Enzyme-inducing AEDs that increase cholesterol levels (eg, carbamazepine and phenytoin) should be avoided in patients with cardiovascular risk factors. For patients with weight problems or eating disorders, physicians should bear in mind that valproate, gabapentin, carbamazepine, pregabalin, ezogabine, and perampanel have been known to increase weight, while topiramate, zonisamide, and felbamate have been known to decrease weight.
AEDs that may worsen psychiatric function include levetiracetam, topiramate, zonisamide, tiagabine, phenobarbital, and perampanel. “Make sure that you ask patients when they are on levetiracetam whether they are experiencing increased irritability, although not everyone recognizes their mood changes,” Dr. French said. “Sometimes the spouse will complain, but the person will not.” On the other hand, carbamazepine, valproic acid, lamotrigine, and pregabalin have a tendency to improve psychiatric function—but not always. Psychiatric function “can go either way” with any of the AEDs, she said.
Some patients experience adverse drug effects as a result of add-on therapy. In some cases, a pharmacodynamic interaction between the new and old drugs may be responsible. Removing the add-on drug or the background drug may help. “Sometimes it is a better idea to take away the background drugs,” she said.
Older AEDs—carbamazepine, phenytoin, phenobarbital, and valproic acid—are associated with idiosyncratic adverse effects, including serious rash, liver failure, bone marrow failure, and pancreatitis. While some of the newer drugs also have such risks, the overall rate of idiosyncratic adverse reactions with their use is lower. Thus far, levetiracetam, brivaracetam, gabapentin, and pregabalin have not been associated with major idiosyncratic adverse effects, Dr. French noted.
Drug Initiation and Interactions
“The ability to initiate a drug rapidly is sometimes the driving characteristic that causes some doctors to pick one drug over another,” Dr. French said. Drugs that can be initiated at a therapeutic dose in a single day include phenytoin, levetiracetam, valproic acid, gabapentin, pregabalin, lacosamide, and brivaracetam. Other AEDs, such as carbamazepine, lamotrigine, perampanel, and oxcarbazepine, require gradual initiation over one to 10 weeks. Some drugs that take longer to initiate may be better tolerated overall, she said.
Generally, older AEDs are more likely to cause drug interactions, compared with newer drugs. Phenytoin, phenobarbital, and carbamazepine are enzyme inducers, which may cause problems for patients who are on statins. Valproic acid is a hepatic enzyme inhibitor, and phenytoin and valproic acid can have protein-binding interactions. Oral contraceptives reduce lamotrigine’s efficacy by doubling the clearance of the AED, Dr. French explained. “This can be problematic when women do not tell you when they are going on and off the contraceptive pill. They can have a breakthrough seizure, and it is only after the fact that you realize why that happened,” she said.
Interactions between oral contraceptive are common with other AEDs, as well. She cited a recent retrospective study involving 1,144 women with epilepsy ages 18 to 47 who provided demographic, epilepsy, AED, contraceptive, and pregnancy data. Survey results showed that 65% of their pregnancies were unintended. Oral forms of contraception had greater failure rates than nonoral forms, with intrauterine devices having the lowest failure rate.
—Adriene Marshall
Suggested Reading
Herzog AG, Mandle HB, Cahill KE, et al. Predictors of unintended pregnancy in women with epilepsy. Neurology. 2017;88(8):728-733.
Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512-521.
New and Noteworthy Information—July 2017
Device Helps Patients Move Paralyzed Hands After Stroke
Patients with stroke who learn to use their minds to open and close a device fitted over their paralyzed hands gain some control over their hands, according to a study published online ahead of print May 26 in Stroke. Ten survivors of chronic hemiparetic stroke with moderate-to-severe upper-limb motor impairment used a powered exoskeleton that opened and closed the affected hand using spectral power from EEG signals from the unaffected hemisphere associated with imagined hand movements of the paretic limb. At 12 weeks, participants had a statistically significant average increase of 6.2 points in the Action Research Arm Test. This behavioral improvement significantly correlated with improvements in brain–computer interface control. Secondary outcomes of grasp strength, Motricity Index, and the Canadian Occupational Performance Measure also significantly improved.
Bundy DT, Souders L, Baranyai K, et al. Contralesional brain-computer interface control of a powered exoskeleton for motor recovery in chronic stroke survivors. Stroke. 2017 May 26 [Epub ahead of print].
Pyrimethamine Lowers Levels of ALS Biomarker
Pyrimethamine is safe and well tolerated in amyotrophic lateral sclerosis (ALS), according to a study published online ahead of print May 8 in Annals of Neurology. Participants underwent a multicenter, open-label, nine-month dose-ranging study to determine the safety and efficacy of pyrimethamine to lower SOD1 levels in the CSF in patients with SOD1 mutations linked to familial ALS. The study included 32 patients with various SOD1 genetic mutations linked to ALS. Participants had three lumbar punctures, blood studies, and a clinical assessment of strength, motor function, quality of life, and potential adverse effects. A linear mixed effects model showed a significant reduction in CSF SOD1 at visit six, with a mean reduction of 13.5%, and at visit nine, with a mean reduction of 10.5%.
Lange DJ, Shahbazi M, Silani V, et al. Pyrimethamine significantly lowers cerebrospinal fluid Cu/Zn superoxide dismutase in amyotrophic lateral sclerosis patients with SOD1 mutations. Ann Neurol. 2017 May 8 [Epub ahead of print].
Statin Use Linked to Higher Risk of Parkinson’s Disease
Statins, especially lipophilic statins, are associated with higher risk of Parkinson’s disease, according to a study published in the June issue of Movement Disorders. The association is stronger with initial use, which suggests a facilitating effect, said the investigators. Researchers performed a retrospective case–control analysis and identified 2,322 people with incident Parkinson’s disease who had been enrolled in a claims database for at least 2.5 years before diagnosis or prescription of antiparkinson medication. They matched the cases with 2,322 controls by age, gender, and a follow-up window. Statin use was significantly associated with Parkinson’s disease risk. The strongest associations were for lipophilic statins (odds ratio [OR], 1.58) versus hydrophilic statins (OR, 1.19), statins plus nonstatins (OR, 1.95), and for the initial period after starting statins.
Liu G, Sterling NW, Kong L, et al. Statins may facilitate Parkinson’s disease: insight gained from a large, national claims database. Mov Disord. 2017;32(6):913-917.
Is Moderate Drinking Associated With Cognitive Decline?
Moderate alcohol consumption is associated with adverse brain outcomes, including hippocampal atrophy, according to a study published online ahead of print June 6 in BMJ. The study included 550 men and women with a mean age of 43 at study baseline. No patient had alcohol dependence, and all underwent brain MRI at follow-up. Higher alcohol consumption over the 30-year follow-up was associated with increased odds of hippocampal atrophy in a dose-dependent fashion. People consuming more than 30 units/week of alcohol were at the highest risk, compared with abstainers. People who drank moderately had three times the odds of right-sided hippocampal atrophy. There was no protective effect of light drinking over abstinence. Higher alcohol use also was associated with differences in corpus callosum microstructure and faster decline in lexical fluency.
Topiwala A, Allan CL, Valkanova V, et al. Moderate alcohol consumption as risk factor for adverse brain outcomes and cognitive decline: longitudinal cohort study. BMJ. 2017 Jun 6 [Epub ahead of print].
Consuming Low-Fat Dairy May Increase Risk for Parkinson’s Disease
Frequently consuming low-fat dairy products may be associated with an increased risk of Parkinson’s disease, according to a study published online ahead of print June 8 in Neurology. This study is based on data from 80,736 participants in the Nurses’ Health Study and 48,610 participants in the Health Professionals Follow-Up Study, with 26 and 24 years of follow-up, respectively. Both US-based studies were conducted through mailed biennial questionnaires. Dietary intake was assessed with food frequency questionnaires administered repeatedly over the follow-up period. Total dairy intake was not significantly associated with Parkinson’s disease risk, but intake of low-fat dairy foods was associated with Parkinson’s disease risk. This association appeared to result from an increased risk of Parkinson’s disease associated with skim and low-fat milk.
Hughes KC, Gao X, Kim IY, et al. Intake of dairy foods and risk of Parkinson disease. Neurology. 2017 Jun 8 [Epub ahead of print].
Elevated Brain Amyloid Increases Likelihood of Cognitive Decline
Elevated baseline brain amyloid level, compared with normal brain amyloid level, is associated with higher likelihood of cognitive decline, according to a study published June 13 in JAMA. Exploratory analyses were conducted with longitudinal cognitive and biomarker data from 445 cognitively normal people. Participants were classified at baseline as having normal or elevated brain amyloid using PET amyloid imaging or a CSF assay of amyloid β. Outcomes included scores on the Preclinical Alzheimer Cognitive Composite (PACC), Mini-Mental State Examination (MMSE), Clinical Dementia Rating Sum of Boxes (CDR-SOB), and Logical Memory Delayed Recall. Compared with the group with normal amyloid, people with elevated amyloid had worse mean scores at four years on the PACC, MMSE, and CDR-SOB. For Logical Memory Delayed Recall, between-group scores were not significantly different at four years.
Donohue MC, Sperling RA, Petersen R, et al. Association between elevated brain amyloid and subsequent cognitive decline among cognitively normal persons. JAMA. 2017;317(22):2305-2316.
Seven Risk Genes for Insomnia Found
Researchers have found seven risk genes for insomnia, according to a study published online ahead of print June 12 in Nature Genetics. To identify genetic factors for insomnia complaints, investigators performed a genome-wide association study and a genome-wide gene-based association study in 113,006 participants. The authors identified three loci and seven genes, including MEIS1,
Hammerschlag AR, Stringer S, de Leeuw CA, et al. Genome-wide association analysis of insomnia complaints identifies risk genes and genetic overlap with psychiatric and metabolic traits. Nat Genet. 2017 Jun 12 [Epub ahead of print].
Is Telemedicine for Headache as Effective as In-Person Visit?
In people with headache, a video consultation with a neurologist for treatment may be as effective as an in-person visit, according to a study published online ahead of print June 14 in Neurology. Researchers randomized, allocated, and consulted patients with nonacute headache via telemedicine or in a traditional manner in a noninferiority trial. Efficacy end points assessed by questionnaires at three and 12 months included change from baseline in Headache Impact Test-6 (HIT-6) and pain intensity. The primary safety end point was presence of secondary headache within 12 months after consultation. There were no differences between telemedicine and traditional consultations in HIT-6 or pain intensity over three periods. The absolute difference in HIT-6 from baseline was 0.3 at three months and 0.2 at 12 months.
Müller KI, Alstadhaug KB, Bekkelund SI. A randomized trial of telemedicine efficacy and safety for nonacute headaches. Neurology. 2017 Jun 14 [Epub ahead of print].
Minocycline Reduces Risk of Conversion From CIS to MS
The risk of conversion from clinically isolated syndrome (CIS) to multiple sclerosis (MS) is significantly lower with minocycline than with placebo over six months, but not over 24 months, according to a study published June 1 in the New England Journal of Medicine. This study included 142 participants who had had their first demyelinating symptoms within the previous 180 days. At 12 Canadian MS clinics, researchers randomly assigned participants to receive either 100 mg of minocycline, administered orally twice daily, or placebo. Administration of minocycline or placebo was continued until a diagnosis of MS was established or until 24 months after randomization. The unadjusted risk of conversion to MS within six months after randomization was 61.0% in the placebo group and 33.4% in the minocycline group.
Metz LM, Li DKB, Traboulsee AL, et al. Trial of minocycline in a clinically isolated syndrome of multiple sclerosis. N Engl J Med. 2017;376(22):2122-2133.
Can Gene Mutation Speed Memory Loss in Alzheimer’s Disease?
In a middle-aged cohort with Alzheimer’s disease risk, the BDNF Met allele is associated with steeper decline in episodic memory and executive function, according to a study published online ahead of print May 3 in Neurology. One thousand twenty-three adults enrolled in the Wisconsin Registry for Alzheimer’s Prevention underwent BDNF genotyping and cognitive assessment at as many as five time points. A subset of participants underwent Pittsburgh compound B scanning. Compared with BDNF Val/Val homozygotes, Met carriers had steeper decline in verbal learning and memory, and in speed and flexibility. In addition, amyloid β burden moderated the relationship between BDNF and verbal learning and memory, such that Met carriers with greater amyloid β burden showed even steeper cognitive decline.
Boots EA, Schultz SA, Clark LR, et al. BDNF Val66Met predicts cognitive decline in the Wisconsin Registry for Alzheimer’s Prevention. Neurology. 2017 May 3 [Epub ahead of print].
Support From Children Reduces Risk of Dementia
Positive social support from children is associated with reduced risk of dementia, whereas negative social support from children and other immediate family increases the risk, according to a study published in the Journal of Alzheimer’s Disease. Researchers analyzed 10-year follow-up data in 10,055 cognitively normal participants age 50 and older from the English Longitudinal Study of Aging. Incidence of dementia was identified from participant- or informant-reported physician diagnosed dementia or overall score of informant-completed IQCODE questionnaire. Positive social support from children significantly reduced the risk of dementia (hazard ratio, 0.83). Negative support from family and friends was significantly associated with increased risk of dementia. The causal mechanisms that create these associations require further research, said the researchers.
Khondoker M, Rafnsson SB, Morris S, et al. Positive and negative experiences of social support and risk of dementia in later life: an investigation using the English Longitudinal Study of Ageing. J Alzheimers Dis. 2017;58(1):99-108.
—Kimberly Williams
Device Helps Patients Move Paralyzed Hands After Stroke
Patients with stroke who learn to use their minds to open and close a device fitted over their paralyzed hands gain some control over their hands, according to a study published online ahead of print May 26 in Stroke. Ten survivors of chronic hemiparetic stroke with moderate-to-severe upper-limb motor impairment used a powered exoskeleton that opened and closed the affected hand using spectral power from EEG signals from the unaffected hemisphere associated with imagined hand movements of the paretic limb. At 12 weeks, participants had a statistically significant average increase of 6.2 points in the Action Research Arm Test. This behavioral improvement significantly correlated with improvements in brain–computer interface control. Secondary outcomes of grasp strength, Motricity Index, and the Canadian Occupational Performance Measure also significantly improved.
Bundy DT, Souders L, Baranyai K, et al. Contralesional brain-computer interface control of a powered exoskeleton for motor recovery in chronic stroke survivors. Stroke. 2017 May 26 [Epub ahead of print].
Pyrimethamine Lowers Levels of ALS Biomarker
Pyrimethamine is safe and well tolerated in amyotrophic lateral sclerosis (ALS), according to a study published online ahead of print May 8 in Annals of Neurology. Participants underwent a multicenter, open-label, nine-month dose-ranging study to determine the safety and efficacy of pyrimethamine to lower SOD1 levels in the CSF in patients with SOD1 mutations linked to familial ALS. The study included 32 patients with various SOD1 genetic mutations linked to ALS. Participants had three lumbar punctures, blood studies, and a clinical assessment of strength, motor function, quality of life, and potential adverse effects. A linear mixed effects model showed a significant reduction in CSF SOD1 at visit six, with a mean reduction of 13.5%, and at visit nine, with a mean reduction of 10.5%.
Lange DJ, Shahbazi M, Silani V, et al. Pyrimethamine significantly lowers cerebrospinal fluid Cu/Zn superoxide dismutase in amyotrophic lateral sclerosis patients with SOD1 mutations. Ann Neurol. 2017 May 8 [Epub ahead of print].
Statin Use Linked to Higher Risk of Parkinson’s Disease
Statins, especially lipophilic statins, are associated with higher risk of Parkinson’s disease, according to a study published in the June issue of Movement Disorders. The association is stronger with initial use, which suggests a facilitating effect, said the investigators. Researchers performed a retrospective case–control analysis and identified 2,322 people with incident Parkinson’s disease who had been enrolled in a claims database for at least 2.5 years before diagnosis or prescription of antiparkinson medication. They matched the cases with 2,322 controls by age, gender, and a follow-up window. Statin use was significantly associated with Parkinson’s disease risk. The strongest associations were for lipophilic statins (odds ratio [OR], 1.58) versus hydrophilic statins (OR, 1.19), statins plus nonstatins (OR, 1.95), and for the initial period after starting statins.
Liu G, Sterling NW, Kong L, et al. Statins may facilitate Parkinson’s disease: insight gained from a large, national claims database. Mov Disord. 2017;32(6):913-917.
Is Moderate Drinking Associated With Cognitive Decline?
Moderate alcohol consumption is associated with adverse brain outcomes, including hippocampal atrophy, according to a study published online ahead of print June 6 in BMJ. The study included 550 men and women with a mean age of 43 at study baseline. No patient had alcohol dependence, and all underwent brain MRI at follow-up. Higher alcohol consumption over the 30-year follow-up was associated with increased odds of hippocampal atrophy in a dose-dependent fashion. People consuming more than 30 units/week of alcohol were at the highest risk, compared with abstainers. People who drank moderately had three times the odds of right-sided hippocampal atrophy. There was no protective effect of light drinking over abstinence. Higher alcohol use also was associated with differences in corpus callosum microstructure and faster decline in lexical fluency.
Topiwala A, Allan CL, Valkanova V, et al. Moderate alcohol consumption as risk factor for adverse brain outcomes and cognitive decline: longitudinal cohort study. BMJ. 2017 Jun 6 [Epub ahead of print].
Consuming Low-Fat Dairy May Increase Risk for Parkinson’s Disease
Frequently consuming low-fat dairy products may be associated with an increased risk of Parkinson’s disease, according to a study published online ahead of print June 8 in Neurology. This study is based on data from 80,736 participants in the Nurses’ Health Study and 48,610 participants in the Health Professionals Follow-Up Study, with 26 and 24 years of follow-up, respectively. Both US-based studies were conducted through mailed biennial questionnaires. Dietary intake was assessed with food frequency questionnaires administered repeatedly over the follow-up period. Total dairy intake was not significantly associated with Parkinson’s disease risk, but intake of low-fat dairy foods was associated with Parkinson’s disease risk. This association appeared to result from an increased risk of Parkinson’s disease associated with skim and low-fat milk.
Hughes KC, Gao X, Kim IY, et al. Intake of dairy foods and risk of Parkinson disease. Neurology. 2017 Jun 8 [Epub ahead of print].
Elevated Brain Amyloid Increases Likelihood of Cognitive Decline
Elevated baseline brain amyloid level, compared with normal brain amyloid level, is associated with higher likelihood of cognitive decline, according to a study published June 13 in JAMA. Exploratory analyses were conducted with longitudinal cognitive and biomarker data from 445 cognitively normal people. Participants were classified at baseline as having normal or elevated brain amyloid using PET amyloid imaging or a CSF assay of amyloid β. Outcomes included scores on the Preclinical Alzheimer Cognitive Composite (PACC), Mini-Mental State Examination (MMSE), Clinical Dementia Rating Sum of Boxes (CDR-SOB), and Logical Memory Delayed Recall. Compared with the group with normal amyloid, people with elevated amyloid had worse mean scores at four years on the PACC, MMSE, and CDR-SOB. For Logical Memory Delayed Recall, between-group scores were not significantly different at four years.
Donohue MC, Sperling RA, Petersen R, et al. Association between elevated brain amyloid and subsequent cognitive decline among cognitively normal persons. JAMA. 2017;317(22):2305-2316.
Seven Risk Genes for Insomnia Found
Researchers have found seven risk genes for insomnia, according to a study published online ahead of print June 12 in Nature Genetics. To identify genetic factors for insomnia complaints, investigators performed a genome-wide association study and a genome-wide gene-based association study in 113,006 participants. The authors identified three loci and seven genes, including MEIS1,
Hammerschlag AR, Stringer S, de Leeuw CA, et al. Genome-wide association analysis of insomnia complaints identifies risk genes and genetic overlap with psychiatric and metabolic traits. Nat Genet. 2017 Jun 12 [Epub ahead of print].
Is Telemedicine for Headache as Effective as In-Person Visit?
In people with headache, a video consultation with a neurologist for treatment may be as effective as an in-person visit, according to a study published online ahead of print June 14 in Neurology. Researchers randomized, allocated, and consulted patients with nonacute headache via telemedicine or in a traditional manner in a noninferiority trial. Efficacy end points assessed by questionnaires at three and 12 months included change from baseline in Headache Impact Test-6 (HIT-6) and pain intensity. The primary safety end point was presence of secondary headache within 12 months after consultation. There were no differences between telemedicine and traditional consultations in HIT-6 or pain intensity over three periods. The absolute difference in HIT-6 from baseline was 0.3 at three months and 0.2 at 12 months.
Müller KI, Alstadhaug KB, Bekkelund SI. A randomized trial of telemedicine efficacy and safety for nonacute headaches. Neurology. 2017 Jun 14 [Epub ahead of print].
Minocycline Reduces Risk of Conversion From CIS to MS
The risk of conversion from clinically isolated syndrome (CIS) to multiple sclerosis (MS) is significantly lower with minocycline than with placebo over six months, but not over 24 months, according to a study published June 1 in the New England Journal of Medicine. This study included 142 participants who had had their first demyelinating symptoms within the previous 180 days. At 12 Canadian MS clinics, researchers randomly assigned participants to receive either 100 mg of minocycline, administered orally twice daily, or placebo. Administration of minocycline or placebo was continued until a diagnosis of MS was established or until 24 months after randomization. The unadjusted risk of conversion to MS within six months after randomization was 61.0% in the placebo group and 33.4% in the minocycline group.
Metz LM, Li DKB, Traboulsee AL, et al. Trial of minocycline in a clinically isolated syndrome of multiple sclerosis. N Engl J Med. 2017;376(22):2122-2133.
Can Gene Mutation Speed Memory Loss in Alzheimer’s Disease?
In a middle-aged cohort with Alzheimer’s disease risk, the BDNF Met allele is associated with steeper decline in episodic memory and executive function, according to a study published online ahead of print May 3 in Neurology. One thousand twenty-three adults enrolled in the Wisconsin Registry for Alzheimer’s Prevention underwent BDNF genotyping and cognitive assessment at as many as five time points. A subset of participants underwent Pittsburgh compound B scanning. Compared with BDNF Val/Val homozygotes, Met carriers had steeper decline in verbal learning and memory, and in speed and flexibility. In addition, amyloid β burden moderated the relationship between BDNF and verbal learning and memory, such that Met carriers with greater amyloid β burden showed even steeper cognitive decline.
Boots EA, Schultz SA, Clark LR, et al. BDNF Val66Met predicts cognitive decline in the Wisconsin Registry for Alzheimer’s Prevention. Neurology. 2017 May 3 [Epub ahead of print].
Support From Children Reduces Risk of Dementia
Positive social support from children is associated with reduced risk of dementia, whereas negative social support from children and other immediate family increases the risk, according to a study published in the Journal of Alzheimer’s Disease. Researchers analyzed 10-year follow-up data in 10,055 cognitively normal participants age 50 and older from the English Longitudinal Study of Aging. Incidence of dementia was identified from participant- or informant-reported physician diagnosed dementia or overall score of informant-completed IQCODE questionnaire. Positive social support from children significantly reduced the risk of dementia (hazard ratio, 0.83). Negative support from family and friends was significantly associated with increased risk of dementia. The causal mechanisms that create these associations require further research, said the researchers.
Khondoker M, Rafnsson SB, Morris S, et al. Positive and negative experiences of social support and risk of dementia in later life: an investigation using the English Longitudinal Study of Ageing. J Alzheimers Dis. 2017;58(1):99-108.
—Kimberly Williams
Device Helps Patients Move Paralyzed Hands After Stroke
Patients with stroke who learn to use their minds to open and close a device fitted over their paralyzed hands gain some control over their hands, according to a study published online ahead of print May 26 in Stroke. Ten survivors of chronic hemiparetic stroke with moderate-to-severe upper-limb motor impairment used a powered exoskeleton that opened and closed the affected hand using spectral power from EEG signals from the unaffected hemisphere associated with imagined hand movements of the paretic limb. At 12 weeks, participants had a statistically significant average increase of 6.2 points in the Action Research Arm Test. This behavioral improvement significantly correlated with improvements in brain–computer interface control. Secondary outcomes of grasp strength, Motricity Index, and the Canadian Occupational Performance Measure also significantly improved.
Bundy DT, Souders L, Baranyai K, et al. Contralesional brain-computer interface control of a powered exoskeleton for motor recovery in chronic stroke survivors. Stroke. 2017 May 26 [Epub ahead of print].
Pyrimethamine Lowers Levels of ALS Biomarker
Pyrimethamine is safe and well tolerated in amyotrophic lateral sclerosis (ALS), according to a study published online ahead of print May 8 in Annals of Neurology. Participants underwent a multicenter, open-label, nine-month dose-ranging study to determine the safety and efficacy of pyrimethamine to lower SOD1 levels in the CSF in patients with SOD1 mutations linked to familial ALS. The study included 32 patients with various SOD1 genetic mutations linked to ALS. Participants had three lumbar punctures, blood studies, and a clinical assessment of strength, motor function, quality of life, and potential adverse effects. A linear mixed effects model showed a significant reduction in CSF SOD1 at visit six, with a mean reduction of 13.5%, and at visit nine, with a mean reduction of 10.5%.
Lange DJ, Shahbazi M, Silani V, et al. Pyrimethamine significantly lowers cerebrospinal fluid Cu/Zn superoxide dismutase in amyotrophic lateral sclerosis patients with SOD1 mutations. Ann Neurol. 2017 May 8 [Epub ahead of print].
Statin Use Linked to Higher Risk of Parkinson’s Disease
Statins, especially lipophilic statins, are associated with higher risk of Parkinson’s disease, according to a study published in the June issue of Movement Disorders. The association is stronger with initial use, which suggests a facilitating effect, said the investigators. Researchers performed a retrospective case–control analysis and identified 2,322 people with incident Parkinson’s disease who had been enrolled in a claims database for at least 2.5 years before diagnosis or prescription of antiparkinson medication. They matched the cases with 2,322 controls by age, gender, and a follow-up window. Statin use was significantly associated with Parkinson’s disease risk. The strongest associations were for lipophilic statins (odds ratio [OR], 1.58) versus hydrophilic statins (OR, 1.19), statins plus nonstatins (OR, 1.95), and for the initial period after starting statins.
Liu G, Sterling NW, Kong L, et al. Statins may facilitate Parkinson’s disease: insight gained from a large, national claims database. Mov Disord. 2017;32(6):913-917.
Is Moderate Drinking Associated With Cognitive Decline?
Moderate alcohol consumption is associated with adverse brain outcomes, including hippocampal atrophy, according to a study published online ahead of print June 6 in BMJ. The study included 550 men and women with a mean age of 43 at study baseline. No patient had alcohol dependence, and all underwent brain MRI at follow-up. Higher alcohol consumption over the 30-year follow-up was associated with increased odds of hippocampal atrophy in a dose-dependent fashion. People consuming more than 30 units/week of alcohol were at the highest risk, compared with abstainers. People who drank moderately had three times the odds of right-sided hippocampal atrophy. There was no protective effect of light drinking over abstinence. Higher alcohol use also was associated with differences in corpus callosum microstructure and faster decline in lexical fluency.
Topiwala A, Allan CL, Valkanova V, et al. Moderate alcohol consumption as risk factor for adverse brain outcomes and cognitive decline: longitudinal cohort study. BMJ. 2017 Jun 6 [Epub ahead of print].
Consuming Low-Fat Dairy May Increase Risk for Parkinson’s Disease
Frequently consuming low-fat dairy products may be associated with an increased risk of Parkinson’s disease, according to a study published online ahead of print June 8 in Neurology. This study is based on data from 80,736 participants in the Nurses’ Health Study and 48,610 participants in the Health Professionals Follow-Up Study, with 26 and 24 years of follow-up, respectively. Both US-based studies were conducted through mailed biennial questionnaires. Dietary intake was assessed with food frequency questionnaires administered repeatedly over the follow-up period. Total dairy intake was not significantly associated with Parkinson’s disease risk, but intake of low-fat dairy foods was associated with Parkinson’s disease risk. This association appeared to result from an increased risk of Parkinson’s disease associated with skim and low-fat milk.
Hughes KC, Gao X, Kim IY, et al. Intake of dairy foods and risk of Parkinson disease. Neurology. 2017 Jun 8 [Epub ahead of print].
Elevated Brain Amyloid Increases Likelihood of Cognitive Decline
Elevated baseline brain amyloid level, compared with normal brain amyloid level, is associated with higher likelihood of cognitive decline, according to a study published June 13 in JAMA. Exploratory analyses were conducted with longitudinal cognitive and biomarker data from 445 cognitively normal people. Participants were classified at baseline as having normal or elevated brain amyloid using PET amyloid imaging or a CSF assay of amyloid β. Outcomes included scores on the Preclinical Alzheimer Cognitive Composite (PACC), Mini-Mental State Examination (MMSE), Clinical Dementia Rating Sum of Boxes (CDR-SOB), and Logical Memory Delayed Recall. Compared with the group with normal amyloid, people with elevated amyloid had worse mean scores at four years on the PACC, MMSE, and CDR-SOB. For Logical Memory Delayed Recall, between-group scores were not significantly different at four years.
Donohue MC, Sperling RA, Petersen R, et al. Association between elevated brain amyloid and subsequent cognitive decline among cognitively normal persons. JAMA. 2017;317(22):2305-2316.
Seven Risk Genes for Insomnia Found
Researchers have found seven risk genes for insomnia, according to a study published online ahead of print June 12 in Nature Genetics. To identify genetic factors for insomnia complaints, investigators performed a genome-wide association study and a genome-wide gene-based association study in 113,006 participants. The authors identified three loci and seven genes, including MEIS1,
Hammerschlag AR, Stringer S, de Leeuw CA, et al. Genome-wide association analysis of insomnia complaints identifies risk genes and genetic overlap with psychiatric and metabolic traits. Nat Genet. 2017 Jun 12 [Epub ahead of print].
Is Telemedicine for Headache as Effective as In-Person Visit?
In people with headache, a video consultation with a neurologist for treatment may be as effective as an in-person visit, according to a study published online ahead of print June 14 in Neurology. Researchers randomized, allocated, and consulted patients with nonacute headache via telemedicine or in a traditional manner in a noninferiority trial. Efficacy end points assessed by questionnaires at three and 12 months included change from baseline in Headache Impact Test-6 (HIT-6) and pain intensity. The primary safety end point was presence of secondary headache within 12 months after consultation. There were no differences between telemedicine and traditional consultations in HIT-6 or pain intensity over three periods. The absolute difference in HIT-6 from baseline was 0.3 at three months and 0.2 at 12 months.
Müller KI, Alstadhaug KB, Bekkelund SI. A randomized trial of telemedicine efficacy and safety for nonacute headaches. Neurology. 2017 Jun 14 [Epub ahead of print].
Minocycline Reduces Risk of Conversion From CIS to MS
The risk of conversion from clinically isolated syndrome (CIS) to multiple sclerosis (MS) is significantly lower with minocycline than with placebo over six months, but not over 24 months, according to a study published June 1 in the New England Journal of Medicine. This study included 142 participants who had had their first demyelinating symptoms within the previous 180 days. At 12 Canadian MS clinics, researchers randomly assigned participants to receive either 100 mg of minocycline, administered orally twice daily, or placebo. Administration of minocycline or placebo was continued until a diagnosis of MS was established or until 24 months after randomization. The unadjusted risk of conversion to MS within six months after randomization was 61.0% in the placebo group and 33.4% in the minocycline group.
Metz LM, Li DKB, Traboulsee AL, et al. Trial of minocycline in a clinically isolated syndrome of multiple sclerosis. N Engl J Med. 2017;376(22):2122-2133.
Can Gene Mutation Speed Memory Loss in Alzheimer’s Disease?
In a middle-aged cohort with Alzheimer’s disease risk, the BDNF Met allele is associated with steeper decline in episodic memory and executive function, according to a study published online ahead of print May 3 in Neurology. One thousand twenty-three adults enrolled in the Wisconsin Registry for Alzheimer’s Prevention underwent BDNF genotyping and cognitive assessment at as many as five time points. A subset of participants underwent Pittsburgh compound B scanning. Compared with BDNF Val/Val homozygotes, Met carriers had steeper decline in verbal learning and memory, and in speed and flexibility. In addition, amyloid β burden moderated the relationship between BDNF and verbal learning and memory, such that Met carriers with greater amyloid β burden showed even steeper cognitive decline.
Boots EA, Schultz SA, Clark LR, et al. BDNF Val66Met predicts cognitive decline in the Wisconsin Registry for Alzheimer’s Prevention. Neurology. 2017 May 3 [Epub ahead of print].
Support From Children Reduces Risk of Dementia
Positive social support from children is associated with reduced risk of dementia, whereas negative social support from children and other immediate family increases the risk, according to a study published in the Journal of Alzheimer’s Disease. Researchers analyzed 10-year follow-up data in 10,055 cognitively normal participants age 50 and older from the English Longitudinal Study of Aging. Incidence of dementia was identified from participant- or informant-reported physician diagnosed dementia or overall score of informant-completed IQCODE questionnaire. Positive social support from children significantly reduced the risk of dementia (hazard ratio, 0.83). Negative support from family and friends was significantly associated with increased risk of dementia. The causal mechanisms that create these associations require further research, said the researchers.
Khondoker M, Rafnsson SB, Morris S, et al. Positive and negative experiences of social support and risk of dementia in later life: an investigation using the English Longitudinal Study of Ageing. J Alzheimers Dis. 2017;58(1):99-108.
—Kimberly Williams

T receptor diversity may predict BCP-ALL response to blinatumomab
MADRID – For patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (BCP-ALL), an extensive and diverse T-cell receptor repertoire may be predictive of response to blinatumomab (Blincyto), investigators suggest.
Patients with responses to blinatumomab had a significantly more diverse T-cell receptor–beta (TRB) gene repertoire at the time of screening, compared with patients who would go on to have minimal residual disease after starting on blinatumomab therapy, reported Michaela Kotrova, MD, from the 2nd Faculty of Medicine Charles University and University Hospital Motol in Prague, Czech Republic.

Blinatumomab is a bispecific T-cell engager designed to direct cytotoxic T cells to cancer cells expressing the CD19 receptor. Although it can induce high remission rates in patients with relapsed/refractory BCP-ALL and has been shown to nearly double overall survival among patients with relapsed/refractory BCP-ALL negative for the Philadelphia chromosome, about half of patients do not achieve a minimal residual disease response. This finding prompted the investigators to determine whether differences in the TRB repertoire could have an effect on individual patient responses to blinatumomab.
They performed next-generation sequencing of immunoglobulin and T-cell receptor gene rearrangements to evaluate the diversity of the repertoire, which can have a profound impact on health.
Dr. Kotrova noted that, in young people, there may be as many as 120 million different TRB gene rearrangements, and the more the merrier because a higher diversity repertoire is capable of protecting people from a large variety of pathogens.
They compared the diversity of the TRB repertoire in 114 patients who were either responders to blinatumomab salvage therapy or who had measurable minimal residual disease (persisters).
They found that there was significantly greater probability than mere chance that the TRB repertoire before blinatumomab administration was more diverse in patients with responses, compared with those without responses.
On day 15 of the first cycle of blinatumomab therapy, there was no significant difference in TRB repertoire between responders and persisters, but, by day 29, there was a sharper and statistically significant increase in repertoire diversity but no significant increase among nonresponders.
Their findings raise the intriguing possibility that response to blinatumomab could be predicted by repertoire diversity prior to the start of therapy, but further studies with larger patient cohorts will be necessary to confirm this, Dr. Kotrova said.
The study was supported by Amgen. Dr. Kotrova had no relevant disclosures.
MADRID – For patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (BCP-ALL), an extensive and diverse T-cell receptor repertoire may be predictive of response to blinatumomab (Blincyto), investigators suggest.
Patients with responses to blinatumomab had a significantly more diverse T-cell receptor–beta (TRB) gene repertoire at the time of screening, compared with patients who would go on to have minimal residual disease after starting on blinatumomab therapy, reported Michaela Kotrova, MD, from the 2nd Faculty of Medicine Charles University and University Hospital Motol in Prague, Czech Republic.
Blinatumomab is a bispecific T-cell engager designed to direct cytotoxic T cells to cancer cells expressing the CD19 receptor. Although it can induce high remission rates in patients with relapsed/refractory BCP-ALL and has been shown to nearly double overall survival among patients with relapsed/refractory BCP-ALL negative for the Philadelphia chromosome, about half of patients do not achieve a minimal residual disease response. This finding prompted the investigators to determine whether differences in the TRB repertoire could have an effect on individual patient responses to blinatumomab.
They performed next-generation sequencing of immunoglobulin and T-cell receptor gene rearrangements to evaluate the diversity of the repertoire, which can have a profound impact on health.
Dr. Kotrova noted that, in young people, there may be as many as 120 million different TRB gene rearrangements, and the more the merrier because a higher diversity repertoire is capable of protecting people from a large variety of pathogens.
They compared the diversity of the TRB repertoire in 114 patients who were either responders to blinatumomab salvage therapy or who had measurable minimal residual disease (persisters).
They found that there was significantly greater probability than mere chance that the TRB repertoire before blinatumomab administration was more diverse in patients with responses, compared with those without responses.
On day 15 of the first cycle of blinatumomab therapy, there was no significant difference in TRB repertoire between responders and persisters, but, by day 29, there was a sharper and statistically significant increase in repertoire diversity but no significant increase among nonresponders.
Their findings raise the intriguing possibility that response to blinatumomab could be predicted by repertoire diversity prior to the start of therapy, but further studies with larger patient cohorts will be necessary to confirm this, Dr. Kotrova said.
The study was supported by Amgen. Dr. Kotrova had no relevant disclosures.
MADRID – For patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (BCP-ALL), an extensive and diverse T-cell receptor repertoire may be predictive of response to blinatumomab (Blincyto), investigators suggest.
Patients with responses to blinatumomab had a significantly more diverse T-cell receptor–beta (TRB) gene repertoire at the time of screening, compared with patients who would go on to have minimal residual disease after starting on blinatumomab therapy, reported Michaela Kotrova, MD, from the 2nd Faculty of Medicine Charles University and University Hospital Motol in Prague, Czech Republic.
Blinatumomab is a bispecific T-cell engager designed to direct cytotoxic T cells to cancer cells expressing the CD19 receptor. Although it can induce high remission rates in patients with relapsed/refractory BCP-ALL and has been shown to nearly double overall survival among patients with relapsed/refractory BCP-ALL negative for the Philadelphia chromosome, about half of patients do not achieve a minimal residual disease response. This finding prompted the investigators to determine whether differences in the TRB repertoire could have an effect on individual patient responses to blinatumomab.
They performed next-generation sequencing of immunoglobulin and T-cell receptor gene rearrangements to evaluate the diversity of the repertoire, which can have a profound impact on health.
Dr. Kotrova noted that, in young people, there may be as many as 120 million different TRB gene rearrangements, and the more the merrier because a higher diversity repertoire is capable of protecting people from a large variety of pathogens.
They compared the diversity of the TRB repertoire in 114 patients who were either responders to blinatumomab salvage therapy or who had measurable minimal residual disease (persisters).
They found that there was significantly greater probability than mere chance that the TRB repertoire before blinatumomab administration was more diverse in patients with responses, compared with those without responses.
On day 15 of the first cycle of blinatumomab therapy, there was no significant difference in TRB repertoire between responders and persisters, but, by day 29, there was a sharper and statistically significant increase in repertoire diversity but no significant increase among nonresponders.
Their findings raise the intriguing possibility that response to blinatumomab could be predicted by repertoire diversity prior to the start of therapy, but further studies with larger patient cohorts will be necessary to confirm this, Dr. Kotrova said.
The study was supported by Amgen. Dr. Kotrova had no relevant disclosures.
AT EHA 2017
Key clinical point: T-cell receptor–beta diversity is important for protection against a wide variety of pathogens.
Major finding: Patients with responses to blinatumomab had a significantly more diverse TRB gene repertoire at the time of screening, compared with patients who would go on to have minimal residual disease after starting on blinatumomab therapy.
Data source: A next-generation sequencing study of samples from 114 patients with relapsed/refractory B-cell precursor acute lymphocytic leukemia.
Disclosures: The study was supported by Amgen. Dr. Kotrova had no relevant disclosures.
Dermatologist calls for paradigm shift on treating ocular rosacea
A partnership between ophthalmologists and dermatologists holds the potential to produce a better treatment for ocular rosacea, one of the few conditions that brings the two specialties together.
“Hopefully, the paradigm eventually shifts from treating symptoms and signs of ocular rosacea with oral antibiotics or reflexive referral to ophthalmology to approaching cutaneous treatment first,” said dermatologist Kara Capriotti, MD, who has been working with a team of ophthalmologists to develop a treatment that focuses on ocular rosacea as a “lid disease,” instead of as a primary ocular surface problem.

Eye scrubs, artificial tears, stress relief measures, and antibiotics are among the treatments used for the condition, which, however, can be treatment-resistant.
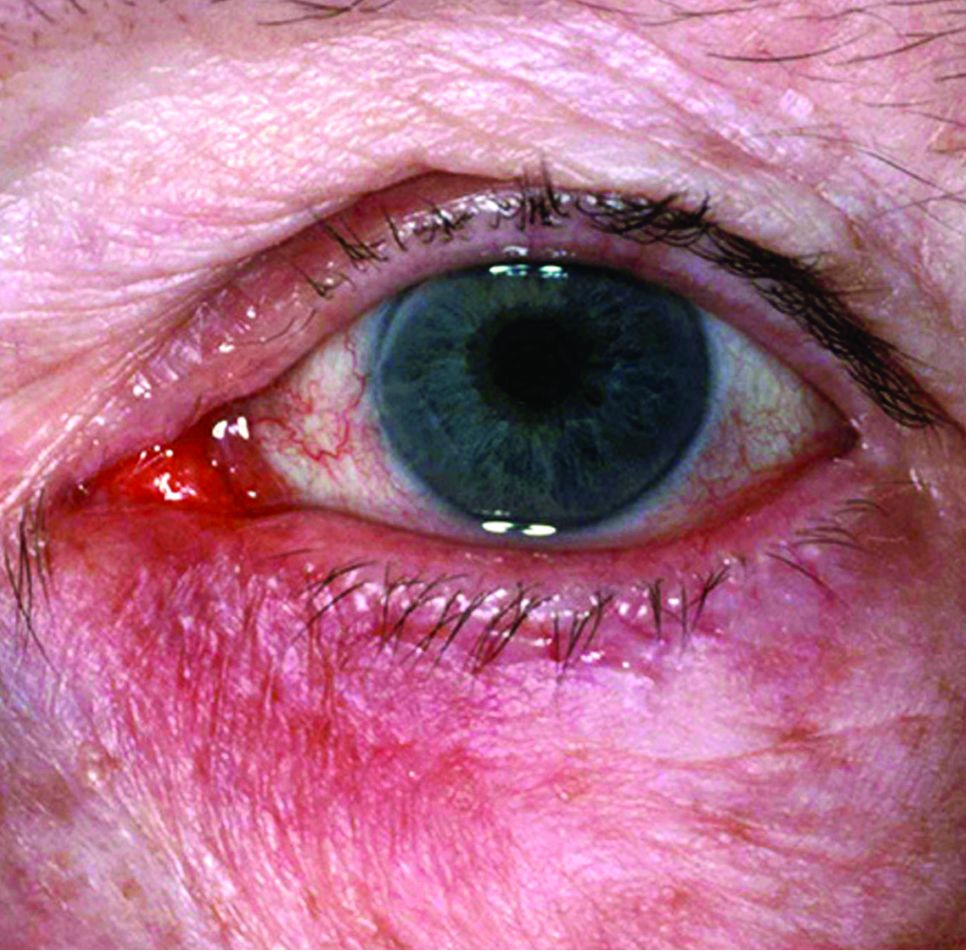
The key, she believes, is to move from a focus on ocular symptoms to an emphasis on lid and skin signs associated with ocular rosacea. “Almost all of the ophthalmology approaches to the disease start at the ocular surface, and these surface treatments are always a little bit [or very] toxic to the ocular surface,” she said. “By treating the lid signs first through a topical approach through the closed lid, we can eliminate a lot of the ocular surface symptoms without ever touching the ocular surface, thus preventing a lot of the associated toxicity and side effects you commonly see with topical ophthalmics.”
Dr. Capriotti and her colleagues have developed a topical treatment that contains dimethyl sulfoxide (DMSO) combined with dilute povidone iodine, which is applied to the lid margin of the closed eye.
In 2015, they published a case report describing the successful use of the treatment for a 78-year-old man with rosacea blepharoconjunctivitis, who had failed oral and topical treatments. He was treated with povidone iodine 10% solution in a DMSO vehicle, which was compounded into a topical gel by a pharmacy and administered twice a day, rubbed onto the lash line and eyelid.
After one week, “remarkable improvements were noted,” with reversal of much of the conjunctivitis, anterior lid erythema, and thickening, they wrote. One month later, after once daily application, “not only were the initial improvements conserved, but the posterior lid margin vessels and telangiectasias had begun to attenuate and involute. Moreover, meibomian capping was no longer present, secretions were less viscous, and tear break-up time normalized.”
DMSO is a skin penetration agent that is rarely used in ophthalmology and povidone iodine is a biocidal agent used in eye care, they pointed out in the case report. “This novel therapy may warrant further investigation in randomized, controlled clinical trials,” they concluded (Ophthalmol Ther. 2015 Dec;4[2]:143-50).
There are plans to start a clinical trial later in 2017, according to Dr. Capriottis, the cofounder of Veloce Biopharma, a company that is developing this product.
Until more specific treatments are available, what can dermatologists do now to improve their care of patients with ocular rosacea? “First and foremost, you have to be comfortable looking at the eyelids and ruling out anything that could be masquerading as blepharitis,” Dr. Capriotti commented. “We have a big disadvantage in dermatology because we don’t use slit-lamp biomicroscopy, but we are pretty good at looking at skin. If the patient has a lot of extraocular rosacea signs, I definitely think dermatologists can manage the prescription of a topical for the lids and evaluate the response.”
She cautioned, however, that referrals are in order in several situations: cases that are severe, those that involve the eye only, and those with which ocular symptoms seem to outweigh the lid signs.
Dr. Capriotti is also the senior vice president of dermatology at Veloce. In the study published in 2015, she and her three coauthors disclosed having financial interest in ALC Therapeutics, a company that was dissolved and restructured into Veloce.
A partnership between ophthalmologists and dermatologists holds the potential to produce a better treatment for ocular rosacea, one of the few conditions that brings the two specialties together.
“Hopefully, the paradigm eventually shifts from treating symptoms and signs of ocular rosacea with oral antibiotics or reflexive referral to ophthalmology to approaching cutaneous treatment first,” said dermatologist Kara Capriotti, MD, who has been working with a team of ophthalmologists to develop a treatment that focuses on ocular rosacea as a “lid disease,” instead of as a primary ocular surface problem.
Eye scrubs, artificial tears, stress relief measures, and antibiotics are among the treatments used for the condition, which, however, can be treatment-resistant.
The key, she believes, is to move from a focus on ocular symptoms to an emphasis on lid and skin signs associated with ocular rosacea. “Almost all of the ophthalmology approaches to the disease start at the ocular surface, and these surface treatments are always a little bit [or very] toxic to the ocular surface,” she said. “By treating the lid signs first through a topical approach through the closed lid, we can eliminate a lot of the ocular surface symptoms without ever touching the ocular surface, thus preventing a lot of the associated toxicity and side effects you commonly see with topical ophthalmics.”
Dr. Capriotti and her colleagues have developed a topical treatment that contains dimethyl sulfoxide (DMSO) combined with dilute povidone iodine, which is applied to the lid margin of the closed eye.
In 2015, they published a case report describing the successful use of the treatment for a 78-year-old man with rosacea blepharoconjunctivitis, who had failed oral and topical treatments. He was treated with povidone iodine 10% solution in a DMSO vehicle, which was compounded into a topical gel by a pharmacy and administered twice a day, rubbed onto the lash line and eyelid.
After one week, “remarkable improvements were noted,” with reversal of much of the conjunctivitis, anterior lid erythema, and thickening, they wrote. One month later, after once daily application, “not only were the initial improvements conserved, but the posterior lid margin vessels and telangiectasias had begun to attenuate and involute. Moreover, meibomian capping was no longer present, secretions were less viscous, and tear break-up time normalized.”
DMSO is a skin penetration agent that is rarely used in ophthalmology and povidone iodine is a biocidal agent used in eye care, they pointed out in the case report. “This novel therapy may warrant further investigation in randomized, controlled clinical trials,” they concluded (Ophthalmol Ther. 2015 Dec;4[2]:143-50).
There are plans to start a clinical trial later in 2017, according to Dr. Capriottis, the cofounder of Veloce Biopharma, a company that is developing this product.
Until more specific treatments are available, what can dermatologists do now to improve their care of patients with ocular rosacea? “First and foremost, you have to be comfortable looking at the eyelids and ruling out anything that could be masquerading as blepharitis,” Dr. Capriotti commented. “We have a big disadvantage in dermatology because we don’t use slit-lamp biomicroscopy, but we are pretty good at looking at skin. If the patient has a lot of extraocular rosacea signs, I definitely think dermatologists can manage the prescription of a topical for the lids and evaluate the response.”
She cautioned, however, that referrals are in order in several situations: cases that are severe, those that involve the eye only, and those with which ocular symptoms seem to outweigh the lid signs.
Dr. Capriotti is also the senior vice president of dermatology at Veloce. In the study published in 2015, she and her three coauthors disclosed having financial interest in ALC Therapeutics, a company that was dissolved and restructured into Veloce.
A partnership between ophthalmologists and dermatologists holds the potential to produce a better treatment for ocular rosacea, one of the few conditions that brings the two specialties together.
“Hopefully, the paradigm eventually shifts from treating symptoms and signs of ocular rosacea with oral antibiotics or reflexive referral to ophthalmology to approaching cutaneous treatment first,” said dermatologist Kara Capriotti, MD, who has been working with a team of ophthalmologists to develop a treatment that focuses on ocular rosacea as a “lid disease,” instead of as a primary ocular surface problem.
Eye scrubs, artificial tears, stress relief measures, and antibiotics are among the treatments used for the condition, which, however, can be treatment-resistant.
The key, she believes, is to move from a focus on ocular symptoms to an emphasis on lid and skin signs associated with ocular rosacea. “Almost all of the ophthalmology approaches to the disease start at the ocular surface, and these surface treatments are always a little bit [or very] toxic to the ocular surface,” she said. “By treating the lid signs first through a topical approach through the closed lid, we can eliminate a lot of the ocular surface symptoms without ever touching the ocular surface, thus preventing a lot of the associated toxicity and side effects you commonly see with topical ophthalmics.”
Dr. Capriotti and her colleagues have developed a topical treatment that contains dimethyl sulfoxide (DMSO) combined with dilute povidone iodine, which is applied to the lid margin of the closed eye.
In 2015, they published a case report describing the successful use of the treatment for a 78-year-old man with rosacea blepharoconjunctivitis, who had failed oral and topical treatments. He was treated with povidone iodine 10% solution in a DMSO vehicle, which was compounded into a topical gel by a pharmacy and administered twice a day, rubbed onto the lash line and eyelid.
After one week, “remarkable improvements were noted,” with reversal of much of the conjunctivitis, anterior lid erythema, and thickening, they wrote. One month later, after once daily application, “not only were the initial improvements conserved, but the posterior lid margin vessels and telangiectasias had begun to attenuate and involute. Moreover, meibomian capping was no longer present, secretions were less viscous, and tear break-up time normalized.”
DMSO is a skin penetration agent that is rarely used in ophthalmology and povidone iodine is a biocidal agent used in eye care, they pointed out in the case report. “This novel therapy may warrant further investigation in randomized, controlled clinical trials,” they concluded (Ophthalmol Ther. 2015 Dec;4[2]:143-50).
There are plans to start a clinical trial later in 2017, according to Dr. Capriottis, the cofounder of Veloce Biopharma, a company that is developing this product.
Until more specific treatments are available, what can dermatologists do now to improve their care of patients with ocular rosacea? “First and foremost, you have to be comfortable looking at the eyelids and ruling out anything that could be masquerading as blepharitis,” Dr. Capriotti commented. “We have a big disadvantage in dermatology because we don’t use slit-lamp biomicroscopy, but we are pretty good at looking at skin. If the patient has a lot of extraocular rosacea signs, I definitely think dermatologists can manage the prescription of a topical for the lids and evaluate the response.”
She cautioned, however, that referrals are in order in several situations: cases that are severe, those that involve the eye only, and those with which ocular symptoms seem to outweigh the lid signs.
Dr. Capriotti is also the senior vice president of dermatology at Veloce. In the study published in 2015, she and her three coauthors disclosed having financial interest in ALC Therapeutics, a company that was dissolved and restructured into Veloce.
Childhood asthma linked to high LV mass later
A history of childhood asthma correlates with high left ventricular mass, a prominent indicator of cardiac damage, in asymptomatic healthy young adults, according to a report published online June 26 in JACC: Heart Failure.
“Our data suggest that aggressive lifestyle modifications or even pharmacologic treatment may be applied to people with a [childhood] history of asthma. … to lower cardiovascular risk,” said Dianjianyi Sun, MD, PhD, of the department of epidemiology, Tulane University, New Orleans, and his associates.
Both mean left ventricular mass (169 g) and mean left ventricular mass index (41 g/m2) were significantly higher in participants who’d had childhood asthma than in those who had not (158 g and 38 g/m2, respectively). This correlation was independent of major cardiovascular risk factors such as race, smoking status, hypertension status, use of antihypertensive medications, C-reactive protein levels, heart rate, and body mass index, the investigators said (JACC: Heart Failure. 2017 Jun 26. doi: 10.1016/j.jchf.2017.03.009).
The association was strongest among young adults who had higher systolic blood pressure (prehypertension or hypertension).
This study was supported by the National Institutes of Health, the Boston Obesity Nutrition Research Center, the United States-Israel Binational Science Foundation, and the American Heart Association. Dr. Sun and his associates reported having no relevant financial disclosures.
Sun et al. report that healthy young adults who had asthma during childhood have a significantly greater left ventricular mass index, by about 8%, than do those who didn’t have the condition.
On an individual patient level, such a difference confers a relatively small incremental risk of incident cardiovascular disease: a roughly 1.08-fold increase. However, at the population level, where there are many major comorbidities, the public health implications could be substantial. Targeting these patients for intensified cardiovascular risk reduction could have important benefits.
John S. Gottdiener, MD, is at the University of Maryland Medical Center, Baltimore. He reported having no relevant financial disclosures. Dr. Gottdiener made these remarks in an editorial comment accompanying Dr. Sun’s report (JACC: Heart Failure. 2017 Jun 26. doi: 10.1016/j.jchf.2017.05.003).
Sun et al. report that healthy young adults who had asthma during childhood have a significantly greater left ventricular mass index, by about 8%, than do those who didn’t have the condition.
On an individual patient level, such a difference confers a relatively small incremental risk of incident cardiovascular disease: a roughly 1.08-fold increase. However, at the population level, where there are many major comorbidities, the public health implications could be substantial. Targeting these patients for intensified cardiovascular risk reduction could have important benefits.
John S. Gottdiener, MD, is at the University of Maryland Medical Center, Baltimore. He reported having no relevant financial disclosures. Dr. Gottdiener made these remarks in an editorial comment accompanying Dr. Sun’s report (JACC: Heart Failure. 2017 Jun 26. doi: 10.1016/j.jchf.2017.05.003).
Sun et al. report that healthy young adults who had asthma during childhood have a significantly greater left ventricular mass index, by about 8%, than do those who didn’t have the condition.
On an individual patient level, such a difference confers a relatively small incremental risk of incident cardiovascular disease: a roughly 1.08-fold increase. However, at the population level, where there are many major comorbidities, the public health implications could be substantial. Targeting these patients for intensified cardiovascular risk reduction could have important benefits.
John S. Gottdiener, MD, is at the University of Maryland Medical Center, Baltimore. He reported having no relevant financial disclosures. Dr. Gottdiener made these remarks in an editorial comment accompanying Dr. Sun’s report (JACC: Heart Failure. 2017 Jun 26. doi: 10.1016/j.jchf.2017.05.003).
A history of childhood asthma correlates with high left ventricular mass, a prominent indicator of cardiac damage, in asymptomatic healthy young adults, according to a report published online June 26 in JACC: Heart Failure.
“Our data suggest that aggressive lifestyle modifications or even pharmacologic treatment may be applied to people with a [childhood] history of asthma. … to lower cardiovascular risk,” said Dianjianyi Sun, MD, PhD, of the department of epidemiology, Tulane University, New Orleans, and his associates.
Both mean left ventricular mass (169 g) and mean left ventricular mass index (41 g/m2) were significantly higher in participants who’d had childhood asthma than in those who had not (158 g and 38 g/m2, respectively). This correlation was independent of major cardiovascular risk factors such as race, smoking status, hypertension status, use of antihypertensive medications, C-reactive protein levels, heart rate, and body mass index, the investigators said (JACC: Heart Failure. 2017 Jun 26. doi: 10.1016/j.jchf.2017.03.009).
The association was strongest among young adults who had higher systolic blood pressure (prehypertension or hypertension).
This study was supported by the National Institutes of Health, the Boston Obesity Nutrition Research Center, the United States-Israel Binational Science Foundation, and the American Heart Association. Dr. Sun and his associates reported having no relevant financial disclosures.
A history of childhood asthma correlates with high left ventricular mass, a prominent indicator of cardiac damage, in asymptomatic healthy young adults, according to a report published online June 26 in JACC: Heart Failure.
“Our data suggest that aggressive lifestyle modifications or even pharmacologic treatment may be applied to people with a [childhood] history of asthma. … to lower cardiovascular risk,” said Dianjianyi Sun, MD, PhD, of the department of epidemiology, Tulane University, New Orleans, and his associates.
Both mean left ventricular mass (169 g) and mean left ventricular mass index (41 g/m2) were significantly higher in participants who’d had childhood asthma than in those who had not (158 g and 38 g/m2, respectively). This correlation was independent of major cardiovascular risk factors such as race, smoking status, hypertension status, use of antihypertensive medications, C-reactive protein levels, heart rate, and body mass index, the investigators said (JACC: Heart Failure. 2017 Jun 26. doi: 10.1016/j.jchf.2017.03.009).
The association was strongest among young adults who had higher systolic blood pressure (prehypertension or hypertension).
This study was supported by the National Institutes of Health, the Boston Obesity Nutrition Research Center, the United States-Israel Binational Science Foundation, and the American Heart Association. Dr. Sun and his associates reported having no relevant financial disclosures.
FROM JACC: HEART FAILURE
Key clinical point: Childhood asthma is associated with high left ventricular mass in asymptomatic young adults.
Major finding: Both mean left ventricular mass (169 g) and mean left ventricular mass index (41 g/m2) were significantly higher in participants who’d had childhood asthma than in those who had not (158 g and 38 g/m2, respectively).
Data source: A secondary analysis of data from the Bogalusa Heart Study, involving 1,118 participants followed for an average of 10 years.
Disclosures: This study was supported by the National Heart, Lung, and Blood Institute, the National Institute of Diabetes and Digestive and Kidney Diseases, the Boston Obesity Nutrition Research Center, the United States-Israel Binational Science Foundation, and the American Heart Association. Dr. Sun and his associates reported having no relevant financial disclosures.
Hospital antibiograms may not apply to emergency department patients
NEW ORLEANS – Patients seen in the emergency department present with a wider range of risk for resistant infections than do hospitalized patients overall, and as a result, hospital-wide antibiograms that dictate empiric therapy may not translate well to the ED setting, a study showed.
Instead, investigators suggested that clinicians choose empiric antimicrobial therapy in the emergency department based on factors associated with differences in susceptibility.
The study researchers compared common antibiotics to treat Escherichia coli between adults treated at their institution overall versus the emergency department, and by sex, patient age, and whether people came to the ED from home versus a long-term care setting. They found some significant differences that could guide empiric treatment in the ED setting.
“Our ED pharmacist observed a lot of broad-spectrum antibiotic prescribing for UTIs,” lead investigator Sarah Jorgensen, PharmD, said at the annual meeting of the American Society for Microbiology. “Also, we’ve had a culture follow-up program in place for the last 5 years, and they had to intervene on a lot of postdischarge antibiotic mismatches.”
E. coli was the most common urinary pathogen detected in this study of 500 randomly selected ED patients with ICD-9/ICD-10 diagnostic codes for urinary tract infection. Investigators found E. coli in 64% of the 226 culture-positive patients presenting to the ED at Huntington Hospital in Pasadena, Calif., between July 2015 and June 2016.
“What was surprising for us, because our enrollment was based on ICD codes for a UTI, is that only about 50% had a positive urine culture,” said Dr. Jorgensen, a pharmacy resident at the University of Southern California in Los Angeles. “Urinalysis was positive in about 99% of the population.”
Dr. Jorgensen and her colleagues found overall low susceptibilities of 71% for ciprofloxacin, 66% for trimethoprim/sulfamethoxazole, and 67% for cefazolin susceptibilities in the ED. They also found that 8% of isolates were positive for extended-spectrum beta-lactamase isolates, as were 1% of E. coli isolates.
The 67% cefazolin susceptibility was significantly lower in the ED, compared with the 86% susceptibility in the institutional antibiogram (P less than .001).
The investigators found E. coli susceptibility to ciprofloxacin was lower in men, at 55%, compared with 74% among women, but the difference was not statistically significant (P = .14). A similar pattern emerged with cefazolin – 55% susceptibility among men and 69% among women (P = .26). In contrast, trimethoprim/sulfamethoxazole susceptibility trended higher in men, at 73%, vs. 64% among women in the ED (P = .63).
When they divided patients by age 50 years and younger versus those older than 50, the investigators found ampicillin susceptibilities were lower in the younger group, at 30%, compared with 51% among those in the older cohort (P = .03). Similarly, gentamicin susceptibilities were 80% in the younger group, compared with 92% in the older group (P = .04).
Ciprofloxacin susceptibility was significantly lower among people coming to the ED from a long-term care facility than among those coming from home – 35% vs. 77% (P less than .001). Differences in ciprofloxacin susceptibility between admitted and discharged patients was less striking – 63% vs. 78% (P = .04).
Nitrofurantoin was the only oral agent with susceptibility greater than 80% in all patient groups, with susceptibility ranging from 88% to 100%.
Because it typically takes 2-3 days to get the susceptibility results back at Huntington Hospital, many patients are discharged on empiric therapy, noted Mira Zurayk, PharmD, a resident at Huntington Hospital. That can present multiple challenges, particularly with homeless patients who are difficult to find and provide follow-up for, Dr. Jorgensen added.
Based partly on the study findings, the investigators developed a clinical algorithm specifically to address UTI antimicrobial prescriptions in the ED. The algorithm incorporates different recommendations for different groups of patients because of their different resistance trends.
“I think that is a good way to tailor empiric therapy when you don’t have culture results up front,” Dr. Jorgensen said. “We just implemented the algorithm, and I’m now analyzing the outcomes.”
Having more data on outcomes will help the clinicians target lowering the rate of “drug-bug mismatches,” as well as UTI-related revisits to the ED. In addition, the work could help expand the antibiotic stewardship program in the hospital to the ED for the first time, Dr. Jorgensen said.
Dr. Jorgensen and Dr. Zurayk had no relevant financial disclosures. One of the study coauthors, Annie Wong-Beringer, PharmD, receives grant funding from Merck.
NEW ORLEANS – Patients seen in the emergency department present with a wider range of risk for resistant infections than do hospitalized patients overall, and as a result, hospital-wide antibiograms that dictate empiric therapy may not translate well to the ED setting, a study showed.
Instead, investigators suggested that clinicians choose empiric antimicrobial therapy in the emergency department based on factors associated with differences in susceptibility.
The study researchers compared common antibiotics to treat Escherichia coli between adults treated at their institution overall versus the emergency department, and by sex, patient age, and whether people came to the ED from home versus a long-term care setting. They found some significant differences that could guide empiric treatment in the ED setting.
“Our ED pharmacist observed a lot of broad-spectrum antibiotic prescribing for UTIs,” lead investigator Sarah Jorgensen, PharmD, said at the annual meeting of the American Society for Microbiology. “Also, we’ve had a culture follow-up program in place for the last 5 years, and they had to intervene on a lot of postdischarge antibiotic mismatches.”
E. coli was the most common urinary pathogen detected in this study of 500 randomly selected ED patients with ICD-9/ICD-10 diagnostic codes for urinary tract infection. Investigators found E. coli in 64% of the 226 culture-positive patients presenting to the ED at Huntington Hospital in Pasadena, Calif., between July 2015 and June 2016.
“What was surprising for us, because our enrollment was based on ICD codes for a UTI, is that only about 50% had a positive urine culture,” said Dr. Jorgensen, a pharmacy resident at the University of Southern California in Los Angeles. “Urinalysis was positive in about 99% of the population.”
Dr. Jorgensen and her colleagues found overall low susceptibilities of 71% for ciprofloxacin, 66% for trimethoprim/sulfamethoxazole, and 67% for cefazolin susceptibilities in the ED. They also found that 8% of isolates were positive for extended-spectrum beta-lactamase isolates, as were 1% of E. coli isolates.
The 67% cefazolin susceptibility was significantly lower in the ED, compared with the 86% susceptibility in the institutional antibiogram (P less than .001).
The investigators found E. coli susceptibility to ciprofloxacin was lower in men, at 55%, compared with 74% among women, but the difference was not statistically significant (P = .14). A similar pattern emerged with cefazolin – 55% susceptibility among men and 69% among women (P = .26). In contrast, trimethoprim/sulfamethoxazole susceptibility trended higher in men, at 73%, vs. 64% among women in the ED (P = .63).
When they divided patients by age 50 years and younger versus those older than 50, the investigators found ampicillin susceptibilities were lower in the younger group, at 30%, compared with 51% among those in the older cohort (P = .03). Similarly, gentamicin susceptibilities were 80% in the younger group, compared with 92% in the older group (P = .04).
Ciprofloxacin susceptibility was significantly lower among people coming to the ED from a long-term care facility than among those coming from home – 35% vs. 77% (P less than .001). Differences in ciprofloxacin susceptibility between admitted and discharged patients was less striking – 63% vs. 78% (P = .04).
Nitrofurantoin was the only oral agent with susceptibility greater than 80% in all patient groups, with susceptibility ranging from 88% to 100%.
Because it typically takes 2-3 days to get the susceptibility results back at Huntington Hospital, many patients are discharged on empiric therapy, noted Mira Zurayk, PharmD, a resident at Huntington Hospital. That can present multiple challenges, particularly with homeless patients who are difficult to find and provide follow-up for, Dr. Jorgensen added.
Based partly on the study findings, the investigators developed a clinical algorithm specifically to address UTI antimicrobial prescriptions in the ED. The algorithm incorporates different recommendations for different groups of patients because of their different resistance trends.
“I think that is a good way to tailor empiric therapy when you don’t have culture results up front,” Dr. Jorgensen said. “We just implemented the algorithm, and I’m now analyzing the outcomes.”
Having more data on outcomes will help the clinicians target lowering the rate of “drug-bug mismatches,” as well as UTI-related revisits to the ED. In addition, the work could help expand the antibiotic stewardship program in the hospital to the ED for the first time, Dr. Jorgensen said.
Dr. Jorgensen and Dr. Zurayk had no relevant financial disclosures. One of the study coauthors, Annie Wong-Beringer, PharmD, receives grant funding from Merck.
NEW ORLEANS – Patients seen in the emergency department present with a wider range of risk for resistant infections than do hospitalized patients overall, and as a result, hospital-wide antibiograms that dictate empiric therapy may not translate well to the ED setting, a study showed.
Instead, investigators suggested that clinicians choose empiric antimicrobial therapy in the emergency department based on factors associated with differences in susceptibility.
The study researchers compared common antibiotics to treat Escherichia coli between adults treated at their institution overall versus the emergency department, and by sex, patient age, and whether people came to the ED from home versus a long-term care setting. They found some significant differences that could guide empiric treatment in the ED setting.
“Our ED pharmacist observed a lot of broad-spectrum antibiotic prescribing for UTIs,” lead investigator Sarah Jorgensen, PharmD, said at the annual meeting of the American Society for Microbiology. “Also, we’ve had a culture follow-up program in place for the last 5 years, and they had to intervene on a lot of postdischarge antibiotic mismatches.”
E. coli was the most common urinary pathogen detected in this study of 500 randomly selected ED patients with ICD-9/ICD-10 diagnostic codes for urinary tract infection. Investigators found E. coli in 64% of the 226 culture-positive patients presenting to the ED at Huntington Hospital in Pasadena, Calif., between July 2015 and June 2016.
“What was surprising for us, because our enrollment was based on ICD codes for a UTI, is that only about 50% had a positive urine culture,” said Dr. Jorgensen, a pharmacy resident at the University of Southern California in Los Angeles. “Urinalysis was positive in about 99% of the population.”
Dr. Jorgensen and her colleagues found overall low susceptibilities of 71% for ciprofloxacin, 66% for trimethoprim/sulfamethoxazole, and 67% for cefazolin susceptibilities in the ED. They also found that 8% of isolates were positive for extended-spectrum beta-lactamase isolates, as were 1% of E. coli isolates.
The 67% cefazolin susceptibility was significantly lower in the ED, compared with the 86% susceptibility in the institutional antibiogram (P less than .001).
The investigators found E. coli susceptibility to ciprofloxacin was lower in men, at 55%, compared with 74% among women, but the difference was not statistically significant (P = .14). A similar pattern emerged with cefazolin – 55% susceptibility among men and 69% among women (P = .26). In contrast, trimethoprim/sulfamethoxazole susceptibility trended higher in men, at 73%, vs. 64% among women in the ED (P = .63).
When they divided patients by age 50 years and younger versus those older than 50, the investigators found ampicillin susceptibilities were lower in the younger group, at 30%, compared with 51% among those in the older cohort (P = .03). Similarly, gentamicin susceptibilities were 80% in the younger group, compared with 92% in the older group (P = .04).
Ciprofloxacin susceptibility was significantly lower among people coming to the ED from a long-term care facility than among those coming from home – 35% vs. 77% (P less than .001). Differences in ciprofloxacin susceptibility between admitted and discharged patients was less striking – 63% vs. 78% (P = .04).
Nitrofurantoin was the only oral agent with susceptibility greater than 80% in all patient groups, with susceptibility ranging from 88% to 100%.
Because it typically takes 2-3 days to get the susceptibility results back at Huntington Hospital, many patients are discharged on empiric therapy, noted Mira Zurayk, PharmD, a resident at Huntington Hospital. That can present multiple challenges, particularly with homeless patients who are difficult to find and provide follow-up for, Dr. Jorgensen added.
Based partly on the study findings, the investigators developed a clinical algorithm specifically to address UTI antimicrobial prescriptions in the ED. The algorithm incorporates different recommendations for different groups of patients because of their different resistance trends.
“I think that is a good way to tailor empiric therapy when you don’t have culture results up front,” Dr. Jorgensen said. “We just implemented the algorithm, and I’m now analyzing the outcomes.”
Having more data on outcomes will help the clinicians target lowering the rate of “drug-bug mismatches,” as well as UTI-related revisits to the ED. In addition, the work could help expand the antibiotic stewardship program in the hospital to the ED for the first time, Dr. Jorgensen said.
Dr. Jorgensen and Dr. Zurayk had no relevant financial disclosures. One of the study coauthors, Annie Wong-Beringer, PharmD, receives grant funding from Merck.
AT ASM MICROBE 2017
Key clinical point: Escherichia coli susceptibility to common antibiotics in the emergency department varies by age, sex, and admitted versus discharge status.
Major finding: The only oral antimicrobial with greater than 80% susceptibility across all groups was nitrofurantoin.
Data source: A study of 500 randomly selected adults who presented to the ED with a urinary tract infection, based on ICD-9 or ICD-10 codes.
Disclosures: Dr. Jorgensen and Dr. Zurayk had no relevant financial disclosures. One study coauthor, Annie Wong-Beringer, PharmD, receives grant funding from Merck.
Supreme Court allows partial travel ban to proceed
The U.S. Supreme Court has allowed a limited version of President Trump’s travel ban to move forward, prohibiting certain foreign nationals from six majority-Muslim countries from entering the country.
The justices’ June 26 order means that travelers from the affected countries who do not have a bona fide relationship with U.S. nationals or U.S. entities may not enter the United States. Conversely, it means that foreign physicians who have accepted a job at a U.S. institution or students who have been accepted to a U.S. medical school will be allowed to take those positions.
“In practical terms, this means that [the executive order] may not be enforced against foreign nationals who have a credible claim of a bona fide relationship with a person or entity in the United States,” justices wrote in their order. “All other foreign nationals are subject to the provisions. For individuals, a close familial relationship is required. A foreign national who wishes to enter the United States to live with or visit a family member ... clearly has such a relationship. As for entities, the relationship must be formal, documented, and formed in the ordinary course, rather than for the purpose of evading [the executive order].”
In a dissenting statement, Associate Justice Clarence Thomas wrote that the full ban should have gone into effect. Associate Justice Samuel Alito and Associate Justice Neil Gorsuch joined the dissent.
“I agree with the court’s implicit conclusion that the government has made a strong showing that it is likely to succeed on the merits – that is, that the [lower] judgments will be reversed,”Associate Justice Thomas wrote. “The government has also established that failure to stay the injunctions will cause irreparable harm by interfering with its compelling need to provide for the nation’s security. Finally, weighing the government’s interest in preserving national security against the hardships caused to respondents by temporary denials of entry into the country, the balance of the equities favors the government. I would thus grant the government’s applications for a stay in their entirety.”
President Trump’s revised executive order, signed March 6, bars citizens of Iran, Libya, Somalia, Sudan, Syria, and Yemen from obtaining visas for 90 days and blocks refugees from the affected countries from entering the U.S. for 120 days. The executive measure superseded President Trump’s original Jan. 27 travel ban. The revised order clarified that citizens of the six countries who are legal permanent U.S. residents or who have current visas to enter the country are exempt from the travel prohibition. Federal judges in Hawaii and Maryland ruled that the revised order was discriminatory and blocked the order from taking effect, a decision upheld by the 9th U.S. Circuit Court of Appeals.
Justices will hear oral arguments in the case this fall.
[email protected]
On Twitter @legal_med
The U.S. Supreme Court has allowed a limited version of President Trump’s travel ban to move forward, prohibiting certain foreign nationals from six majority-Muslim countries from entering the country.
The justices’ June 26 order means that travelers from the affected countries who do not have a bona fide relationship with U.S. nationals or U.S. entities may not enter the United States. Conversely, it means that foreign physicians who have accepted a job at a U.S. institution or students who have been accepted to a U.S. medical school will be allowed to take those positions.
“In practical terms, this means that [the executive order] may not be enforced against foreign nationals who have a credible claim of a bona fide relationship with a person or entity in the United States,” justices wrote in their order. “All other foreign nationals are subject to the provisions. For individuals, a close familial relationship is required. A foreign national who wishes to enter the United States to live with or visit a family member ... clearly has such a relationship. As for entities, the relationship must be formal, documented, and formed in the ordinary course, rather than for the purpose of evading [the executive order].”
In a dissenting statement, Associate Justice Clarence Thomas wrote that the full ban should have gone into effect. Associate Justice Samuel Alito and Associate Justice Neil Gorsuch joined the dissent.
“I agree with the court’s implicit conclusion that the government has made a strong showing that it is likely to succeed on the merits – that is, that the [lower] judgments will be reversed,”Associate Justice Thomas wrote. “The government has also established that failure to stay the injunctions will cause irreparable harm by interfering with its compelling need to provide for the nation’s security. Finally, weighing the government’s interest in preserving national security against the hardships caused to respondents by temporary denials of entry into the country, the balance of the equities favors the government. I would thus grant the government’s applications for a stay in their entirety.”
President Trump’s revised executive order, signed March 6, bars citizens of Iran, Libya, Somalia, Sudan, Syria, and Yemen from obtaining visas for 90 days and blocks refugees from the affected countries from entering the U.S. for 120 days. The executive measure superseded President Trump’s original Jan. 27 travel ban. The revised order clarified that citizens of the six countries who are legal permanent U.S. residents or who have current visas to enter the country are exempt from the travel prohibition. Federal judges in Hawaii and Maryland ruled that the revised order was discriminatory and blocked the order from taking effect, a decision upheld by the 9th U.S. Circuit Court of Appeals.
Justices will hear oral arguments in the case this fall.
[email protected]
On Twitter @legal_med
The U.S. Supreme Court has allowed a limited version of President Trump’s travel ban to move forward, prohibiting certain foreign nationals from six majority-Muslim countries from entering the country.
The justices’ June 26 order means that travelers from the affected countries who do not have a bona fide relationship with U.S. nationals or U.S. entities may not enter the United States. Conversely, it means that foreign physicians who have accepted a job at a U.S. institution or students who have been accepted to a U.S. medical school will be allowed to take those positions.
“In practical terms, this means that [the executive order] may not be enforced against foreign nationals who have a credible claim of a bona fide relationship with a person or entity in the United States,” justices wrote in their order. “All other foreign nationals are subject to the provisions. For individuals, a close familial relationship is required. A foreign national who wishes to enter the United States to live with or visit a family member ... clearly has such a relationship. As for entities, the relationship must be formal, documented, and formed in the ordinary course, rather than for the purpose of evading [the executive order].”
In a dissenting statement, Associate Justice Clarence Thomas wrote that the full ban should have gone into effect. Associate Justice Samuel Alito and Associate Justice Neil Gorsuch joined the dissent.
“I agree with the court’s implicit conclusion that the government has made a strong showing that it is likely to succeed on the merits – that is, that the [lower] judgments will be reversed,”Associate Justice Thomas wrote. “The government has also established that failure to stay the injunctions will cause irreparable harm by interfering with its compelling need to provide for the nation’s security. Finally, weighing the government’s interest in preserving national security against the hardships caused to respondents by temporary denials of entry into the country, the balance of the equities favors the government. I would thus grant the government’s applications for a stay in their entirety.”
President Trump’s revised executive order, signed March 6, bars citizens of Iran, Libya, Somalia, Sudan, Syria, and Yemen from obtaining visas for 90 days and blocks refugees from the affected countries from entering the U.S. for 120 days. The executive measure superseded President Trump’s original Jan. 27 travel ban. The revised order clarified that citizens of the six countries who are legal permanent U.S. residents or who have current visas to enter the country are exempt from the travel prohibition. Federal judges in Hawaii and Maryland ruled that the revised order was discriminatory and blocked the order from taking effect, a decision upheld by the 9th U.S. Circuit Court of Appeals.
Justices will hear oral arguments in the case this fall.
[email protected]
On Twitter @legal_med
Pembrolizumab + rituximab boost response rates in relapsed follicular lymphoma
LUGANO, SWITZERLAND – A novel combination of the anti-programmed death 1 (PD-1) checkpoint inhibitor pembrolizumab (Keytruda) and the anti-CD20 monoclonal antibody rituximab was associated with a high overall response rate (ORR) in patients with relapsed follicular lymphoma in a phase II clinical trial.
Among 20 patients evaluable for efficacy, the overall response rate to the combination was 65%, including 50% complete responses (CR) reported Loretta J. Nastoupil, MD, of the University of Texas MD Anderson Cancer Center in Houston.
“Follicular lymphoma is probably one of the best examples of targeting the immune system and also one of the earliest examples. Over the last few years we’ve learned a great deal about the different mechanisms of not only negative impact on infiltrating T cells, but also immune escape and T-cell exhaustion,” she said at the International Conference on Malignant Lymphoma.
Although biopsies of follicular lymphoma tumors have demonstrated infiltration of anti-tumor T cells, these cells are typically impeded by immune checkpoints, including PD-1 and its ligand (PD-L1).
The use of anti-PD-1 checkpoint inhibitors such as pembrolizumab has been shown to enhance the function of antitumor T cells in follicular lymphoma, and blocking PD-1 on natural killer cells enhances the antibody-dependent cell-mediated cytotoxicity of the natural killer cells, she said.
Because rituximab, a mainstay of therapy for non-Hodgkin lymphomas, induces antibody-dependent cell-mediated cytotoxicity, the investigators reasoned that combining it with pembrolizumab would simultaneously and synergistically stimulate activation of innate and adaptive immunity.
They designed a phase II, single-arm study in 30 patients with relapsed follicular lymphoma following one or more prior lines of therapy. The patients also had to have rituximab-sensitive disease, defined as a complete response (CR) or partial response lasting for at least 6 months following the most recent rituximab-containing therapy.
The patients were treated with rituximab 375 mg/m2 IV on days 1, 8, 15, and 22 of cycle 1, and pembrolizumab 200 mg IV every 3 weeks for up to 16 cycles starting on day 2 of cycle 1.
The investigators expected that the combination would improve ORR, the primary endpoint, to at least 60%, compared with 40% for historical controls treated with repeat courses of rituximab.
At the data cutoff for the interim analysis, 32 patients had been enrolled, 30 were evaluable for safety, and 20 for efficacy after a median follow-up of 8.2 months.
Among the 20 patients (median age 64) in the efficacy analysis, 10 (50%) had a CR, and 3 (15%) had a partial response, for an ORR of 65%. Three additional patients had stable disease, and four had disease progression as best responses.
Among the patients with CRs, the duration of response ranged from nearly 275 days to more than 600 days.
“This does appear to be durable, and it is time dependent in terms of response. We did see early response, and we also saw deepening of response over time,” Dr. Nastoupil said.
Four patients were discontinued from the study because of immune-related adverse events. All four patients had achieved a CR at the time of study removal, and all four have ongoing CRs.
Among the 30 patients evaluable for safety, there were no grade 4 adverse events, no deaths, and few grade 3 events. Most events were grade 1 or 2, and included fatigue, eye pain/blurred vision/watery eye, nausea and vomiting, diarrhea dyspnea, rash, cough, and lymphopenia.
The investigators also looked at potential biomarkers for response, including PD-L1 expression in tumors prior to treatment. They found in samples from three patients who went on to achieve CRs that PD-L1 expression in tumor cells was low, ranging from 0% to 8%, suggesting that PD-L1 expression may not be necessary to generate a response with the combination.
They then looked at the association between CD8-positive T effector cells and responses in 12 patients, and found that patients with higher levels of expression had better ORR and CR rates.
“These interim results warrant further investigation of this combination in follicular lymphoma, and an expansion to include patients with refractory follicular lymphoma is planned,” Dr. Nastoupil concluded.
The Leukemia & Lymphoma Society supported the study. Dr Nastoupil has disclosed consulting fees from Celgene and contracted research for Abbvie, Janssen, and TG Therapeutics.
LUGANO, SWITZERLAND – A novel combination of the anti-programmed death 1 (PD-1) checkpoint inhibitor pembrolizumab (Keytruda) and the anti-CD20 monoclonal antibody rituximab was associated with a high overall response rate (ORR) in patients with relapsed follicular lymphoma in a phase II clinical trial.
Among 20 patients evaluable for efficacy, the overall response rate to the combination was 65%, including 50% complete responses (CR) reported Loretta J. Nastoupil, MD, of the University of Texas MD Anderson Cancer Center in Houston.
“Follicular lymphoma is probably one of the best examples of targeting the immune system and also one of the earliest examples. Over the last few years we’ve learned a great deal about the different mechanisms of not only negative impact on infiltrating T cells, but also immune escape and T-cell exhaustion,” she said at the International Conference on Malignant Lymphoma.
Although biopsies of follicular lymphoma tumors have demonstrated infiltration of anti-tumor T cells, these cells are typically impeded by immune checkpoints, including PD-1 and its ligand (PD-L1).
The use of anti-PD-1 checkpoint inhibitors such as pembrolizumab has been shown to enhance the function of antitumor T cells in follicular lymphoma, and blocking PD-1 on natural killer cells enhances the antibody-dependent cell-mediated cytotoxicity of the natural killer cells, she said.
Because rituximab, a mainstay of therapy for non-Hodgkin lymphomas, induces antibody-dependent cell-mediated cytotoxicity, the investigators reasoned that combining it with pembrolizumab would simultaneously and synergistically stimulate activation of innate and adaptive immunity.
They designed a phase II, single-arm study in 30 patients with relapsed follicular lymphoma following one or more prior lines of therapy. The patients also had to have rituximab-sensitive disease, defined as a complete response (CR) or partial response lasting for at least 6 months following the most recent rituximab-containing therapy.
The patients were treated with rituximab 375 mg/m2 IV on days 1, 8, 15, and 22 of cycle 1, and pembrolizumab 200 mg IV every 3 weeks for up to 16 cycles starting on day 2 of cycle 1.
The investigators expected that the combination would improve ORR, the primary endpoint, to at least 60%, compared with 40% for historical controls treated with repeat courses of rituximab.
At the data cutoff for the interim analysis, 32 patients had been enrolled, 30 were evaluable for safety, and 20 for efficacy after a median follow-up of 8.2 months.
Among the 20 patients (median age 64) in the efficacy analysis, 10 (50%) had a CR, and 3 (15%) had a partial response, for an ORR of 65%. Three additional patients had stable disease, and four had disease progression as best responses.
Among the patients with CRs, the duration of response ranged from nearly 275 days to more than 600 days.
“This does appear to be durable, and it is time dependent in terms of response. We did see early response, and we also saw deepening of response over time,” Dr. Nastoupil said.
Four patients were discontinued from the study because of immune-related adverse events. All four patients had achieved a CR at the time of study removal, and all four have ongoing CRs.
Among the 30 patients evaluable for safety, there were no grade 4 adverse events, no deaths, and few grade 3 events. Most events were grade 1 or 2, and included fatigue, eye pain/blurred vision/watery eye, nausea and vomiting, diarrhea dyspnea, rash, cough, and lymphopenia.
The investigators also looked at potential biomarkers for response, including PD-L1 expression in tumors prior to treatment. They found in samples from three patients who went on to achieve CRs that PD-L1 expression in tumor cells was low, ranging from 0% to 8%, suggesting that PD-L1 expression may not be necessary to generate a response with the combination.
They then looked at the association between CD8-positive T effector cells and responses in 12 patients, and found that patients with higher levels of expression had better ORR and CR rates.
“These interim results warrant further investigation of this combination in follicular lymphoma, and an expansion to include patients with refractory follicular lymphoma is planned,” Dr. Nastoupil concluded.
The Leukemia & Lymphoma Society supported the study. Dr Nastoupil has disclosed consulting fees from Celgene and contracted research for Abbvie, Janssen, and TG Therapeutics.
LUGANO, SWITZERLAND – A novel combination of the anti-programmed death 1 (PD-1) checkpoint inhibitor pembrolizumab (Keytruda) and the anti-CD20 monoclonal antibody rituximab was associated with a high overall response rate (ORR) in patients with relapsed follicular lymphoma in a phase II clinical trial.
Among 20 patients evaluable for efficacy, the overall response rate to the combination was 65%, including 50% complete responses (CR) reported Loretta J. Nastoupil, MD, of the University of Texas MD Anderson Cancer Center in Houston.
“Follicular lymphoma is probably one of the best examples of targeting the immune system and also one of the earliest examples. Over the last few years we’ve learned a great deal about the different mechanisms of not only negative impact on infiltrating T cells, but also immune escape and T-cell exhaustion,” she said at the International Conference on Malignant Lymphoma.
Although biopsies of follicular lymphoma tumors have demonstrated infiltration of anti-tumor T cells, these cells are typically impeded by immune checkpoints, including PD-1 and its ligand (PD-L1).
The use of anti-PD-1 checkpoint inhibitors such as pembrolizumab has been shown to enhance the function of antitumor T cells in follicular lymphoma, and blocking PD-1 on natural killer cells enhances the antibody-dependent cell-mediated cytotoxicity of the natural killer cells, she said.
Because rituximab, a mainstay of therapy for non-Hodgkin lymphomas, induces antibody-dependent cell-mediated cytotoxicity, the investigators reasoned that combining it with pembrolizumab would simultaneously and synergistically stimulate activation of innate and adaptive immunity.
They designed a phase II, single-arm study in 30 patients with relapsed follicular lymphoma following one or more prior lines of therapy. The patients also had to have rituximab-sensitive disease, defined as a complete response (CR) or partial response lasting for at least 6 months following the most recent rituximab-containing therapy.
The patients were treated with rituximab 375 mg/m2 IV on days 1, 8, 15, and 22 of cycle 1, and pembrolizumab 200 mg IV every 3 weeks for up to 16 cycles starting on day 2 of cycle 1.
The investigators expected that the combination would improve ORR, the primary endpoint, to at least 60%, compared with 40% for historical controls treated with repeat courses of rituximab.
At the data cutoff for the interim analysis, 32 patients had been enrolled, 30 were evaluable for safety, and 20 for efficacy after a median follow-up of 8.2 months.
Among the 20 patients (median age 64) in the efficacy analysis, 10 (50%) had a CR, and 3 (15%) had a partial response, for an ORR of 65%. Three additional patients had stable disease, and four had disease progression as best responses.
Among the patients with CRs, the duration of response ranged from nearly 275 days to more than 600 days.
“This does appear to be durable, and it is time dependent in terms of response. We did see early response, and we also saw deepening of response over time,” Dr. Nastoupil said.
Four patients were discontinued from the study because of immune-related adverse events. All four patients had achieved a CR at the time of study removal, and all four have ongoing CRs.
Among the 30 patients evaluable for safety, there were no grade 4 adverse events, no deaths, and few grade 3 events. Most events were grade 1 or 2, and included fatigue, eye pain/blurred vision/watery eye, nausea and vomiting, diarrhea dyspnea, rash, cough, and lymphopenia.
The investigators also looked at potential biomarkers for response, including PD-L1 expression in tumors prior to treatment. They found in samples from three patients who went on to achieve CRs that PD-L1 expression in tumor cells was low, ranging from 0% to 8%, suggesting that PD-L1 expression may not be necessary to generate a response with the combination.
They then looked at the association between CD8-positive T effector cells and responses in 12 patients, and found that patients with higher levels of expression had better ORR and CR rates.
“These interim results warrant further investigation of this combination in follicular lymphoma, and an expansion to include patients with refractory follicular lymphoma is planned,” Dr. Nastoupil concluded.
The Leukemia & Lymphoma Society supported the study. Dr Nastoupil has disclosed consulting fees from Celgene and contracted research for Abbvie, Janssen, and TG Therapeutics.
AT 14-ICML
Key clinical point: The combination of pembrolizumab and rituximab increased responses compared with repeat rituximab in patients with relapsed follicular lymphoma.
Major finding: The overall response rate with the combination was 65%, including 50% complete responses.
Data source: Open-label, phase II, single-arm study in 32 patients with relapsed follicular lymphoma (20 for efficacy, 30 for safety analysis).
Disclosures: The Leukemia & Lymphoma Society supported the study. Dr Nastoupil has disclosed consulting fees from Celgene and contracted research for Abbvie, Janssen, and TG Therapeutics.
Updated Analysis of Ocrelizumab’s Safety
NEW ORLEANS—The safety profile of ocrelizumab in ongoing open-label extension studies in relapsing multiple sclerosis (MS) and primary progressive MS is generally consistent with that observed in controlled clinical trials, according to a report presented at the 31st Annual Meeting of the Consortium of Multiple Sclerosis Centers (CMSC). “A slight increase in the rate of serious infections was observed in patients with relapsing MS beyond two years, but the rates remained low,” reported Ludwig Kappos, MD, Chair of Neurology at University Hospital Basel in Switzerland, and colleagues. “This was not observed in patients with primary progressive MS, and additional exposure data are needed to determine if the risk increases with further dosing,” the researchers said. “Incidence rates of malignancies and breast cancer observed with ocrelizumab treatment in MS remain within the range of epidemiologic background data.”

The safety and efficacy of ocrelizumab previously have been characterized in a phase II study in patients with relapsing-remitting MS and in phase III studies in patients with relapsing MS (OPERA I and OPERA II) and in patients with primary progressive MS (ORATORIO). Results from these trials showed that ocrelizumab reduced clinical and MRI evidence of disease activity, compared with placebo and interferon beta-1a. The most common adverse events associated with ocrelizumab during the double-blind periods of the phase III trials included infusion-related reactions, nasopharyngitis, upper respiratory tract infections, headache, and urinary tract infections. During the double-blind treatment period in the phase III trials in relapsing MS, serious adverse events, serious infections, and malignancies were reported in 6.9%, 1.3%, and 0.5% of ocrelizumab-treated patients, respectively (vs 8.7%, 2.9%, and 0.2% of patients treated with interferon beta-1a). In the phase III trial in primary progressive MS, these events were reported in 20.4%, 6.2%, and 2.3% of ocrelizumab-treated patients (vs 22.2%, 5.9%, and 0.8% of patients who received placebo). Across studies, few patients who received ocrelizumab (in the range of 2% to 4%) had adverse event–related treatment withdrawals.
At the CMSC annual meeting, data from controlled and open-label extension periods of the clinical trials of ocrelizumab in relapsing MS and primary progressive MS were presented.
Upon completion of a double-blind treatment period, patients from all studies were eligible to enter a long-term open-label extension in which they received ocrelizumab treatment. Safety analyses were based on integrated data from the phase II and phase III studies. The primary analysis was based on the clinical cutoff dates of the individual studies (phase II, January 22, 2015; OPERA I, April 2, 2015; OPERA II, May 12, 2015; ORATORIO, July 24, 2015). The present updated analysis includes all patients who received one or more dose of ocrelizumab during the controlled or open-label periods of the phase II and phase III studies (ie, the ocrelizumab all-exposure population) as of January 20, 2016. Additional data cutoffs of June 30, 2016, and September 15, 2016, were presented for malignancies to demonstrate changes with increasing ocrelizumab exposure.
The ocrelizumab all-exposure group included 2,279 patients. Patients’ mean age was 40.1, and 61.3% were female. Time since MS symptom onset was 6.67 years, and time since MS diagnosis was 3.66 years.
Overall Adverse Events
As of January 20, 2016, the majority (81%) of patients who received ocrelizumab experienced at least one adverse event, corresponding to a rate of 242 events per 100 patient-years. The most common adverse events included nasopharyngitis, upper respiratory tract infection, urinary tract infection, infusion-related reactions, and headache. Serious adverse events were reported in 12% of patients, corresponding to 6.97 events per 100 patient-years, and most commonly included infections. Treatment withdrawal because of an adverse event occurred in 3.3% of patients (rate, 1.40 per 100 patient-years) and primarily included infusion-related reactions (0.9%) and events coded as neoplasms (0.6%) or infections (0.4%).
Infections
As of January 20, 2016, 56.9% of the ocrelizumab all-exposure population reported one or more infection with ocrelizumab, corresponding to a rate of 73.6 per 100 patient-years. “These findings are consistent with the 75.6 rate observed at the primary analysis cutoff date,” the researchers reported. Infections were reported at the highest rate following the first dose and declined with subsequent dosing. Infections reported in 5% or more of patients included nasopharyngitis, upper respiratory tract infection, urinary tract infection, bronchitis, and influenza. In the ocrelizumab all-exposure population, the rate per 100 patient-years of serious infections (1.80) as of January 20, 2016, was comparable with the rate at the primary analysis cutoff date (1.74). Data for the pooled relapsing MS population showed a slight increase in the rate per 100 patient-years of serious infections (1.45) as of January 20, 2016, compared with the rate at the primary analysis cutoff date (1.08); “however, rates remained low, and further exposure is needed to make any interpretation,” the researchers said.
Among patients with primary progressive MS, the rate per 100 patient-years of serious infections remained stable (2.74) as of January 20, 2016, compared with the rate at the primary analysis cutoff date (2.97). The most common serious infection reported was pneumonia. No infections were identified as opportunistic in nature.
Malignancies
In the MS clinical trial program, an imbalance in the crude incidence of malignancies was observed between the ocrelizumab- and comparator-treated patients, which was associated with a cluster of breast cancer events in the ocrelizumab group. Over time, the crude incidence rate of malignancy per 100 patient-years in the ocrelizumab all-exposure population fluctuated but remained within the epidemiologic range of patients with MS. An observed change in the crude incidence rate of malignancy from June to September is attributed to the crude incidence rate of non-melanoma skin cancer, which increased from 0.090 at the primary analysis cutoff date to 0.145 at the September 15, 2016, data cutoff, due to six new cases of basal cell carcinoma. The crude incidence rate of breast cancer remained stable through the September 15, 2016, data cutoff.
More Recent Developments
In late May 2017, Genentech, the maker of ocrelizumab, notified physicians of
—Glenn S. Williams
NEW ORLEANS—The safety profile of ocrelizumab in ongoing open-label extension studies in relapsing multiple sclerosis (MS) and primary progressive MS is generally consistent with that observed in controlled clinical trials, according to a report presented at the 31st Annual Meeting of the Consortium of Multiple Sclerosis Centers (CMSC). “A slight increase in the rate of serious infections was observed in patients with relapsing MS beyond two years, but the rates remained low,” reported Ludwig Kappos, MD, Chair of Neurology at University Hospital Basel in Switzerland, and colleagues. “This was not observed in patients with primary progressive MS, and additional exposure data are needed to determine if the risk increases with further dosing,” the researchers said. “Incidence rates of malignancies and breast cancer observed with ocrelizumab treatment in MS remain within the range of epidemiologic background data.”
The safety and efficacy of ocrelizumab previously have been characterized in a phase II study in patients with relapsing-remitting MS and in phase III studies in patients with relapsing MS (OPERA I and OPERA II) and in patients with primary progressive MS (ORATORIO). Results from these trials showed that ocrelizumab reduced clinical and MRI evidence of disease activity, compared with placebo and interferon beta-1a. The most common adverse events associated with ocrelizumab during the double-blind periods of the phase III trials included infusion-related reactions, nasopharyngitis, upper respiratory tract infections, headache, and urinary tract infections. During the double-blind treatment period in the phase III trials in relapsing MS, serious adverse events, serious infections, and malignancies were reported in 6.9%, 1.3%, and 0.5% of ocrelizumab-treated patients, respectively (vs 8.7%, 2.9%, and 0.2% of patients treated with interferon beta-1a). In the phase III trial in primary progressive MS, these events were reported in 20.4%, 6.2%, and 2.3% of ocrelizumab-treated patients (vs 22.2%, 5.9%, and 0.8% of patients who received placebo). Across studies, few patients who received ocrelizumab (in the range of 2% to 4%) had adverse event–related treatment withdrawals.
At the CMSC annual meeting, data from controlled and open-label extension periods of the clinical trials of ocrelizumab in relapsing MS and primary progressive MS were presented.
Upon completion of a double-blind treatment period, patients from all studies were eligible to enter a long-term open-label extension in which they received ocrelizumab treatment. Safety analyses were based on integrated data from the phase II and phase III studies. The primary analysis was based on the clinical cutoff dates of the individual studies (phase II, January 22, 2015; OPERA I, April 2, 2015; OPERA II, May 12, 2015; ORATORIO, July 24, 2015). The present updated analysis includes all patients who received one or more dose of ocrelizumab during the controlled or open-label periods of the phase II and phase III studies (ie, the ocrelizumab all-exposure population) as of January 20, 2016. Additional data cutoffs of June 30, 2016, and September 15, 2016, were presented for malignancies to demonstrate changes with increasing ocrelizumab exposure.
The ocrelizumab all-exposure group included 2,279 patients. Patients’ mean age was 40.1, and 61.3% were female. Time since MS symptom onset was 6.67 years, and time since MS diagnosis was 3.66 years.
Overall Adverse Events
As of January 20, 2016, the majority (81%) of patients who received ocrelizumab experienced at least one adverse event, corresponding to a rate of 242 events per 100 patient-years. The most common adverse events included nasopharyngitis, upper respiratory tract infection, urinary tract infection, infusion-related reactions, and headache. Serious adverse events were reported in 12% of patients, corresponding to 6.97 events per 100 patient-years, and most commonly included infections. Treatment withdrawal because of an adverse event occurred in 3.3% of patients (rate, 1.40 per 100 patient-years) and primarily included infusion-related reactions (0.9%) and events coded as neoplasms (0.6%) or infections (0.4%).
Infections
As of January 20, 2016, 56.9% of the ocrelizumab all-exposure population reported one or more infection with ocrelizumab, corresponding to a rate of 73.6 per 100 patient-years. “These findings are consistent with the 75.6 rate observed at the primary analysis cutoff date,” the researchers reported. Infections were reported at the highest rate following the first dose and declined with subsequent dosing. Infections reported in 5% or more of patients included nasopharyngitis, upper respiratory tract infection, urinary tract infection, bronchitis, and influenza. In the ocrelizumab all-exposure population, the rate per 100 patient-years of serious infections (1.80) as of January 20, 2016, was comparable with the rate at the primary analysis cutoff date (1.74). Data for the pooled relapsing MS population showed a slight increase in the rate per 100 patient-years of serious infections (1.45) as of January 20, 2016, compared with the rate at the primary analysis cutoff date (1.08); “however, rates remained low, and further exposure is needed to make any interpretation,” the researchers said.
Among patients with primary progressive MS, the rate per 100 patient-years of serious infections remained stable (2.74) as of January 20, 2016, compared with the rate at the primary analysis cutoff date (2.97). The most common serious infection reported was pneumonia. No infections were identified as opportunistic in nature.
Malignancies
In the MS clinical trial program, an imbalance in the crude incidence of malignancies was observed between the ocrelizumab- and comparator-treated patients, which was associated with a cluster of breast cancer events in the ocrelizumab group. Over time, the crude incidence rate of malignancy per 100 patient-years in the ocrelizumab all-exposure population fluctuated but remained within the epidemiologic range of patients with MS. An observed change in the crude incidence rate of malignancy from June to September is attributed to the crude incidence rate of non-melanoma skin cancer, which increased from 0.090 at the primary analysis cutoff date to 0.145 at the September 15, 2016, data cutoff, due to six new cases of basal cell carcinoma. The crude incidence rate of breast cancer remained stable through the September 15, 2016, data cutoff.
More Recent Developments
In late May 2017, Genentech, the maker of ocrelizumab, notified physicians of
—Glenn S. Williams
NEW ORLEANS—The safety profile of ocrelizumab in ongoing open-label extension studies in relapsing multiple sclerosis (MS) and primary progressive MS is generally consistent with that observed in controlled clinical trials, according to a report presented at the 31st Annual Meeting of the Consortium of Multiple Sclerosis Centers (CMSC). “A slight increase in the rate of serious infections was observed in patients with relapsing MS beyond two years, but the rates remained low,” reported Ludwig Kappos, MD, Chair of Neurology at University Hospital Basel in Switzerland, and colleagues. “This was not observed in patients with primary progressive MS, and additional exposure data are needed to determine if the risk increases with further dosing,” the researchers said. “Incidence rates of malignancies and breast cancer observed with ocrelizumab treatment in MS remain within the range of epidemiologic background data.”
The safety and efficacy of ocrelizumab previously have been characterized in a phase II study in patients with relapsing-remitting MS and in phase III studies in patients with relapsing MS (OPERA I and OPERA II) and in patients with primary progressive MS (ORATORIO). Results from these trials showed that ocrelizumab reduced clinical and MRI evidence of disease activity, compared with placebo and interferon beta-1a. The most common adverse events associated with ocrelizumab during the double-blind periods of the phase III trials included infusion-related reactions, nasopharyngitis, upper respiratory tract infections, headache, and urinary tract infections. During the double-blind treatment period in the phase III trials in relapsing MS, serious adverse events, serious infections, and malignancies were reported in 6.9%, 1.3%, and 0.5% of ocrelizumab-treated patients, respectively (vs 8.7%, 2.9%, and 0.2% of patients treated with interferon beta-1a). In the phase III trial in primary progressive MS, these events were reported in 20.4%, 6.2%, and 2.3% of ocrelizumab-treated patients (vs 22.2%, 5.9%, and 0.8% of patients who received placebo). Across studies, few patients who received ocrelizumab (in the range of 2% to 4%) had adverse event–related treatment withdrawals.
At the CMSC annual meeting, data from controlled and open-label extension periods of the clinical trials of ocrelizumab in relapsing MS and primary progressive MS were presented.
Upon completion of a double-blind treatment period, patients from all studies were eligible to enter a long-term open-label extension in which they received ocrelizumab treatment. Safety analyses were based on integrated data from the phase II and phase III studies. The primary analysis was based on the clinical cutoff dates of the individual studies (phase II, January 22, 2015; OPERA I, April 2, 2015; OPERA II, May 12, 2015; ORATORIO, July 24, 2015). The present updated analysis includes all patients who received one or more dose of ocrelizumab during the controlled or open-label periods of the phase II and phase III studies (ie, the ocrelizumab all-exposure population) as of January 20, 2016. Additional data cutoffs of June 30, 2016, and September 15, 2016, were presented for malignancies to demonstrate changes with increasing ocrelizumab exposure.
The ocrelizumab all-exposure group included 2,279 patients. Patients’ mean age was 40.1, and 61.3% were female. Time since MS symptom onset was 6.67 years, and time since MS diagnosis was 3.66 years.
Overall Adverse Events
As of January 20, 2016, the majority (81%) of patients who received ocrelizumab experienced at least one adverse event, corresponding to a rate of 242 events per 100 patient-years. The most common adverse events included nasopharyngitis, upper respiratory tract infection, urinary tract infection, infusion-related reactions, and headache. Serious adverse events were reported in 12% of patients, corresponding to 6.97 events per 100 patient-years, and most commonly included infections. Treatment withdrawal because of an adverse event occurred in 3.3% of patients (rate, 1.40 per 100 patient-years) and primarily included infusion-related reactions (0.9%) and events coded as neoplasms (0.6%) or infections (0.4%).
Infections
As of January 20, 2016, 56.9% of the ocrelizumab all-exposure population reported one or more infection with ocrelizumab, corresponding to a rate of 73.6 per 100 patient-years. “These findings are consistent with the 75.6 rate observed at the primary analysis cutoff date,” the researchers reported. Infections were reported at the highest rate following the first dose and declined with subsequent dosing. Infections reported in 5% or more of patients included nasopharyngitis, upper respiratory tract infection, urinary tract infection, bronchitis, and influenza. In the ocrelizumab all-exposure population, the rate per 100 patient-years of serious infections (1.80) as of January 20, 2016, was comparable with the rate at the primary analysis cutoff date (1.74). Data for the pooled relapsing MS population showed a slight increase in the rate per 100 patient-years of serious infections (1.45) as of January 20, 2016, compared with the rate at the primary analysis cutoff date (1.08); “however, rates remained low, and further exposure is needed to make any interpretation,” the researchers said.
Among patients with primary progressive MS, the rate per 100 patient-years of serious infections remained stable (2.74) as of January 20, 2016, compared with the rate at the primary analysis cutoff date (2.97). The most common serious infection reported was pneumonia. No infections were identified as opportunistic in nature.
Malignancies
In the MS clinical trial program, an imbalance in the crude incidence of malignancies was observed between the ocrelizumab- and comparator-treated patients, which was associated with a cluster of breast cancer events in the ocrelizumab group. Over time, the crude incidence rate of malignancy per 100 patient-years in the ocrelizumab all-exposure population fluctuated but remained within the epidemiologic range of patients with MS. An observed change in the crude incidence rate of malignancy from June to September is attributed to the crude incidence rate of non-melanoma skin cancer, which increased from 0.090 at the primary analysis cutoff date to 0.145 at the September 15, 2016, data cutoff, due to six new cases of basal cell carcinoma. The crude incidence rate of breast cancer remained stable through the September 15, 2016, data cutoff.
More Recent Developments
In late May 2017, Genentech, the maker of ocrelizumab, notified physicians of
—Glenn S. Williams
New Tool Can Distinguish Between Tremor Syndromes
A new tool called the tremor stability index (TSI) can aid the differential diagnosis of essential tremor and Parkinson’s disease, according to research published online ahead of print April 27 in Brain. The TSI has a diagnostic accuracy of approximately 90% for these syndromes, and neurologists can derive it from brief, inexpensive, and noninvasive tremor recordings.
Misdiagnosis of tremor syndromes is common and affects clinical care and research. The current gold standard for diagnosis is clinical evaluation by a specialist in movement disorders. In 2015, Brittain et al found that the frequency of tremor remains stable over a narrow range of frequencies in essential tremor, but remains stable over a broader range in Parkinson’s disease.

Lazzaro di Biase, PhD, a postdoctoral researcher at Università Campus Bio-Medico in Rome, and colleagues analyzed the overall tremor stability characteristics of essential tremor and Parkinson’s disease to develop the TSI, an interquartile range of the change in tremor frequency. The investigators performed kinematic measurements on a test cohort of 16 patients with tremor-dominant Parkinson’s disease and 20 patients with essential tremor. Data were collected after overnight withdrawal of medications. A validation cohort included new and original data taken from published studies and encompassed 42 rest tremor recordings of patients with Parkinson’s disease and eight postural tremor recordings of patients with essential tremor.
In the test cohort, Dr. di Biase and colleagues found no difference in mean instantaneous frequency between patient groups, but saw a difference in TSI between groups. For every unit increase in TSI, the odds of a patient having a diagnosis of essential tremor increased by a factor of 14.8. The optimal TSI threshold in the test cohort was 1.05; TSI values greater than 1.05 indicated a diagnosis of essential tremor, and TSI values of 1.05 or less indicated a diagnosis of Parkinson’s disease. Follow-up recordings made one year and seven months later confirmed the stability of the TSI over time for patients with Parkinson’s disease.
The investigators found a difference in mean instantaneous frequency between groups in the validation cohort, as well as a significant difference in TSI between essential tremor and Parkinson’s disease. For every unit increase in TSI, the odds of a patient having a diagnosis of essential tremor increased by a factor of 5.7. When the researchers applied the threshold of 1.05, they found that the TSI had excellent discriminatory ability. In addition, bootstrap analysis revealed that the TSI outperformed the mean harmonic power of postural tremor harmonics 72% of the time.
The TSI was robust when applied to various patient groups with differing demographics and clinical characteristics. The devices used to record tremor did not affect the TSI’s reliability, nor did postural context. According to the researchers, the TSI can be rapidly estimated from 10-second recordings of tremor.
“The TSI provides a promising diagnostic aid in clinical practice and could be a useful tool to avoid selection errors in clinical trials,” said Dr. di Biase. “It will be important to see if this index can also distinguish dystonic tremor, which may masquerade as Parkinson’s disease or essential tremor,” he added.
—Erik Greb
Suggested Reading
di Biase L, Brittain JS, Shah SA, et al. Tremor stability index: a new tool for differential diagnosis in tremor syndromes. Brain. 2017 Apr 27 [Epub ahead of print].
A new tool called the tremor stability index (TSI) can aid the differential diagnosis of essential tremor and Parkinson’s disease, according to research published online ahead of print April 27 in Brain. The TSI has a diagnostic accuracy of approximately 90% for these syndromes, and neurologists can derive it from brief, inexpensive, and noninvasive tremor recordings.
Misdiagnosis of tremor syndromes is common and affects clinical care and research. The current gold standard for diagnosis is clinical evaluation by a specialist in movement disorders. In 2015, Brittain et al found that the frequency of tremor remains stable over a narrow range of frequencies in essential tremor, but remains stable over a broader range in Parkinson’s disease.
Lazzaro di Biase, PhD, a postdoctoral researcher at Università Campus Bio-Medico in Rome, and colleagues analyzed the overall tremor stability characteristics of essential tremor and Parkinson’s disease to develop the TSI, an interquartile range of the change in tremor frequency. The investigators performed kinematic measurements on a test cohort of 16 patients with tremor-dominant Parkinson’s disease and 20 patients with essential tremor. Data were collected after overnight withdrawal of medications. A validation cohort included new and original data taken from published studies and encompassed 42 rest tremor recordings of patients with Parkinson’s disease and eight postural tremor recordings of patients with essential tremor.
In the test cohort, Dr. di Biase and colleagues found no difference in mean instantaneous frequency between patient groups, but saw a difference in TSI between groups. For every unit increase in TSI, the odds of a patient having a diagnosis of essential tremor increased by a factor of 14.8. The optimal TSI threshold in the test cohort was 1.05; TSI values greater than 1.05 indicated a diagnosis of essential tremor, and TSI values of 1.05 or less indicated a diagnosis of Parkinson’s disease. Follow-up recordings made one year and seven months later confirmed the stability of the TSI over time for patients with Parkinson’s disease.
The investigators found a difference in mean instantaneous frequency between groups in the validation cohort, as well as a significant difference in TSI between essential tremor and Parkinson’s disease. For every unit increase in TSI, the odds of a patient having a diagnosis of essential tremor increased by a factor of 5.7. When the researchers applied the threshold of 1.05, they found that the TSI had excellent discriminatory ability. In addition, bootstrap analysis revealed that the TSI outperformed the mean harmonic power of postural tremor harmonics 72% of the time.
The TSI was robust when applied to various patient groups with differing demographics and clinical characteristics. The devices used to record tremor did not affect the TSI’s reliability, nor did postural context. According to the researchers, the TSI can be rapidly estimated from 10-second recordings of tremor.
“The TSI provides a promising diagnostic aid in clinical practice and could be a useful tool to avoid selection errors in clinical trials,” said Dr. di Biase. “It will be important to see if this index can also distinguish dystonic tremor, which may masquerade as Parkinson’s disease or essential tremor,” he added.
—Erik Greb
Suggested Reading
di Biase L, Brittain JS, Shah SA, et al. Tremor stability index: a new tool for differential diagnosis in tremor syndromes. Brain. 2017 Apr 27 [Epub ahead of print].
A new tool called the tremor stability index (TSI) can aid the differential diagnosis of essential tremor and Parkinson’s disease, according to research published online ahead of print April 27 in Brain. The TSI has a diagnostic accuracy of approximately 90% for these syndromes, and neurologists can derive it from brief, inexpensive, and noninvasive tremor recordings.
Misdiagnosis of tremor syndromes is common and affects clinical care and research. The current gold standard for diagnosis is clinical evaluation by a specialist in movement disorders. In 2015, Brittain et al found that the frequency of tremor remains stable over a narrow range of frequencies in essential tremor, but remains stable over a broader range in Parkinson’s disease.
Lazzaro di Biase, PhD, a postdoctoral researcher at Università Campus Bio-Medico in Rome, and colleagues analyzed the overall tremor stability characteristics of essential tremor and Parkinson’s disease to develop the TSI, an interquartile range of the change in tremor frequency. The investigators performed kinematic measurements on a test cohort of 16 patients with tremor-dominant Parkinson’s disease and 20 patients with essential tremor. Data were collected after overnight withdrawal of medications. A validation cohort included new and original data taken from published studies and encompassed 42 rest tremor recordings of patients with Parkinson’s disease and eight postural tremor recordings of patients with essential tremor.
In the test cohort, Dr. di Biase and colleagues found no difference in mean instantaneous frequency between patient groups, but saw a difference in TSI between groups. For every unit increase in TSI, the odds of a patient having a diagnosis of essential tremor increased by a factor of 14.8. The optimal TSI threshold in the test cohort was 1.05; TSI values greater than 1.05 indicated a diagnosis of essential tremor, and TSI values of 1.05 or less indicated a diagnosis of Parkinson’s disease. Follow-up recordings made one year and seven months later confirmed the stability of the TSI over time for patients with Parkinson’s disease.
The investigators found a difference in mean instantaneous frequency between groups in the validation cohort, as well as a significant difference in TSI between essential tremor and Parkinson’s disease. For every unit increase in TSI, the odds of a patient having a diagnosis of essential tremor increased by a factor of 5.7. When the researchers applied the threshold of 1.05, they found that the TSI had excellent discriminatory ability. In addition, bootstrap analysis revealed that the TSI outperformed the mean harmonic power of postural tremor harmonics 72% of the time.
The TSI was robust when applied to various patient groups with differing demographics and clinical characteristics. The devices used to record tremor did not affect the TSI’s reliability, nor did postural context. According to the researchers, the TSI can be rapidly estimated from 10-second recordings of tremor.
“The TSI provides a promising diagnostic aid in clinical practice and could be a useful tool to avoid selection errors in clinical trials,” said Dr. di Biase. “It will be important to see if this index can also distinguish dystonic tremor, which may masquerade as Parkinson’s disease or essential tremor,” he added.
—Erik Greb
Suggested Reading
di Biase L, Brittain JS, Shah SA, et al. Tremor stability index: a new tool for differential diagnosis in tremor syndromes. Brain. 2017 Apr 27 [Epub ahead of print].