User login
App allows monitoring of drug effects in Parkinson’s
BOSTON – The developers of a free app that tests mental and physical symptoms in patients with Parkinson’s disease (PD) report that their software allows the monitoring of responses to treatment.
“We can track markers of medication,” Larsson Omberg, PhD, vice president of systems biology at the nonprofit Sage Bionetworks, said in an interview. “We can statistically predict whether someone has taken their medication or not, and we can even subdivide populations into strong responders to l-dopa and individuals who don’t necessarily have strong responses to medication but have strong fluctuations based on the time of day.”

A total of 10,326 participants (86%) served as a control group of people who do not have PD. Their average age was 32 (interquartile range, 23-38). The other 14% of participants (n = 2,373) disclosed that they have PD. Their average age was 60 (IQR, 54-68), and they reported having the disease for an average of 8 years (IQR, 4-10).
Overall, 80% of the control participants were male, as were 64% of the PD participants.
Researchers found that 96% of the PD participants were taking PD medications, and 10% reported having had deep brain stimulation.
Participants were asked to use the app’s surveys and tests to measure things such as tremor, sleep quality, frequency of gait, tapping ability, and voice jitter.
“We ask them to perform these active tasks, things they might do in the clinic,” Dr. Omberg said. “The difference is that we are capturing measurements using the sensors in the phone. It’s relatively easy so we can track changes over a day and from day to day.”
For example, one of the tests measures how quickly users can tap the screen over a 20-second interval. “It is a two-finger tap between two fingers and two buttons on the screen,” Dr. Omberg said. “Finger tapping is associated with the severity of the disease.”
The researchers found that they could track patient variability throughout the day and connect the data to the times when patients took medication. In half of the PD patients, the data showed a correlation between medication use and performance of at least one of five types of activities – gait, balance, voice, memory, and tapping.
The app and the information it has provided have limitations, he said. While one participant has used the app at least three times a day for 2 years, many users tried it out for a short time and became inactive. And while users can access some of their own data, it’s not very helpful yet: “It’s very raw and probably not the most useful to a clinician,” Dr. Omberg said.
Regarding whether the app could be used to reveal early signs of PD, Dr. Omberg cautioned that it wasn’t designed for that purpose. Still, “I’m pretty sure applications like mPower would be technically able to do this at some point in the future,” he said. “But there is a lot of validation work that will have to be done first.”
For now, the app remains available for free for iOS devices, and researchers are working on a new version. Meanwhile, Dr. Omberg said the app is serving as a research tool to collect baseline and trial data in the Safety of Urate Elevation in Parkinson’s Disease (SURE-PD) study, which is examining the use of oral inosine to boost serum urate in PD patients.
The study was funded by the Robert Wood Johnson Foundation.
BOSTON – The developers of a free app that tests mental and physical symptoms in patients with Parkinson’s disease (PD) report that their software allows the monitoring of responses to treatment.
“We can track markers of medication,” Larsson Omberg, PhD, vice president of systems biology at the nonprofit Sage Bionetworks, said in an interview. “We can statistically predict whether someone has taken their medication or not, and we can even subdivide populations into strong responders to l-dopa and individuals who don’t necessarily have strong responses to medication but have strong fluctuations based on the time of day.”
A total of 10,326 participants (86%) served as a control group of people who do not have PD. Their average age was 32 (interquartile range, 23-38). The other 14% of participants (n = 2,373) disclosed that they have PD. Their average age was 60 (IQR, 54-68), and they reported having the disease for an average of 8 years (IQR, 4-10).
Overall, 80% of the control participants were male, as were 64% of the PD participants.
Researchers found that 96% of the PD participants were taking PD medications, and 10% reported having had deep brain stimulation.
Participants were asked to use the app’s surveys and tests to measure things such as tremor, sleep quality, frequency of gait, tapping ability, and voice jitter.
“We ask them to perform these active tasks, things they might do in the clinic,” Dr. Omberg said. “The difference is that we are capturing measurements using the sensors in the phone. It’s relatively easy so we can track changes over a day and from day to day.”
For example, one of the tests measures how quickly users can tap the screen over a 20-second interval. “It is a two-finger tap between two fingers and two buttons on the screen,” Dr. Omberg said. “Finger tapping is associated with the severity of the disease.”
The researchers found that they could track patient variability throughout the day and connect the data to the times when patients took medication. In half of the PD patients, the data showed a correlation between medication use and performance of at least one of five types of activities – gait, balance, voice, memory, and tapping.
The app and the information it has provided have limitations, he said. While one participant has used the app at least three times a day for 2 years, many users tried it out for a short time and became inactive. And while users can access some of their own data, it’s not very helpful yet: “It’s very raw and probably not the most useful to a clinician,” Dr. Omberg said.
Regarding whether the app could be used to reveal early signs of PD, Dr. Omberg cautioned that it wasn’t designed for that purpose. Still, “I’m pretty sure applications like mPower would be technically able to do this at some point in the future,” he said. “But there is a lot of validation work that will have to be done first.”
For now, the app remains available for free for iOS devices, and researchers are working on a new version. Meanwhile, Dr. Omberg said the app is serving as a research tool to collect baseline and trial data in the Safety of Urate Elevation in Parkinson’s Disease (SURE-PD) study, which is examining the use of oral inosine to boost serum urate in PD patients.
The study was funded by the Robert Wood Johnson Foundation.
BOSTON – The developers of a free app that tests mental and physical symptoms in patients with Parkinson’s disease (PD) report that their software allows the monitoring of responses to treatment.
“We can track markers of medication,” Larsson Omberg, PhD, vice president of systems biology at the nonprofit Sage Bionetworks, said in an interview. “We can statistically predict whether someone has taken their medication or not, and we can even subdivide populations into strong responders to l-dopa and individuals who don’t necessarily have strong responses to medication but have strong fluctuations based on the time of day.”
A total of 10,326 participants (86%) served as a control group of people who do not have PD. Their average age was 32 (interquartile range, 23-38). The other 14% of participants (n = 2,373) disclosed that they have PD. Their average age was 60 (IQR, 54-68), and they reported having the disease for an average of 8 years (IQR, 4-10).
Overall, 80% of the control participants were male, as were 64% of the PD participants.
Researchers found that 96% of the PD participants were taking PD medications, and 10% reported having had deep brain stimulation.
Participants were asked to use the app’s surveys and tests to measure things such as tremor, sleep quality, frequency of gait, tapping ability, and voice jitter.
“We ask them to perform these active tasks, things they might do in the clinic,” Dr. Omberg said. “The difference is that we are capturing measurements using the sensors in the phone. It’s relatively easy so we can track changes over a day and from day to day.”
For example, one of the tests measures how quickly users can tap the screen over a 20-second interval. “It is a two-finger tap between two fingers and two buttons on the screen,” Dr. Omberg said. “Finger tapping is associated with the severity of the disease.”
The researchers found that they could track patient variability throughout the day and connect the data to the times when patients took medication. In half of the PD patients, the data showed a correlation between medication use and performance of at least one of five types of activities – gait, balance, voice, memory, and tapping.
The app and the information it has provided have limitations, he said. While one participant has used the app at least three times a day for 2 years, many users tried it out for a short time and became inactive. And while users can access some of their own data, it’s not very helpful yet: “It’s very raw and probably not the most useful to a clinician,” Dr. Omberg said.
Regarding whether the app could be used to reveal early signs of PD, Dr. Omberg cautioned that it wasn’t designed for that purpose. Still, “I’m pretty sure applications like mPower would be technically able to do this at some point in the future,” he said. “But there is a lot of validation work that will have to be done first.”
For now, the app remains available for free for iOS devices, and researchers are working on a new version. Meanwhile, Dr. Omberg said the app is serving as a research tool to collect baseline and trial data in the Safety of Urate Elevation in Parkinson’s Disease (SURE-PD) study, which is examining the use of oral inosine to boost serum urate in PD patients.
The study was funded by the Robert Wood Johnson Foundation.
AT AAN 2017
Solving stool refusal
When parents bring in their delightful, verbal 3-year-old for refusing to poop on the potty, it may seem laughable. But with impending preschool and costs of diapers, stool refusal can be a major aggravation for families! Fortunately,
Commonly a healthy, typically developing boy stands and urinates in the toilet just fine, but sneaks off behind the sofa to poop. Parent gyrations have gone from cajoling, to punishing, to offering trips to Disney! Flaring tempers can set the stage for stool refusal to be a power play.
There are a number of reasons stool refusal may give clues to child and family tendencies and relevant intervention. We always should be alert to rare medical problems such as Hirschsprung disease or traumas (from slammed toilet lids to sexual abuse). But while learning to use the toilet for urination and defecation generally occur around the same time, there are pitfalls making pooping in the potty different. An impending stool provides stronger sensations and more advance warning than urine and tends to come at regular times, making it logical to start toilet learning with sitting on the potty after meals.
But once seated on the potty, stools can require some waiting – not a typical toddler forte! While running to sit has novelty at first and may be reinforced by celebration, this quickly becomes routine and boring. Very active or very intense children especially hate having their play interrupted by a trip to the bathroom. Oppositional children just won’t perform if they think the parent cares! And unlike for urination, everyone can inhibit defecation long enough for the urge to pass. Repeated stool retention from ignoring the urge makes stools dessicated and harder, with resulting pain when finally passed. One painful stool makes many a young child decide “Never again!” and simply refuse the toilet. A rectal fissure can both start 
During unclogging and establishing a new stool pattern, the toddler should be matter-of-factly put back in diapers (not pull ups) saying “Oh well, you are just not ready for pants yet.” Dramatically placing the treasured Superhero underwear on the top shelf increases motivation (or promised if none have been acquired). Returning to diapers without shaming the child is key, and all caregivers need to buy in. They need to be good “actors,” conveying that they don’t really care about toileting to reduce the power struggle. If controlling poop is a battle, only the child can win!
When the soft stools are occurring several times per day, I suggest “M&M treatment”: 1 for sitting, 2 for peeing, and 3 for pooping = 6 potential M&Ms per episode. The “1 for sitting” (the easiest part), is not painful and restores the habit of complying. Remember, M&Ms are no match for a game on an iPad! By charting the times of stools, the parent can remove electronics ½ hour before the expected poop and restrict the child to one room of the house with a potty nearby. Parents can interact, but should avoid making this a rewarding playtime. When the child uses the potty rather than their pants, the room restriction is removed until the next window for pooping. If they poop outside the toilet, they remain restricted (and no electronics) until the next window (even the next day).
Some parents are especially sensitive to the smell and mess of stools and pass that attitude along to their child by saying “Ugh, you stink!” or “I can’t stand this mess!” or even handing the child to another caregiver in a gesture of rejection. These messages are not missed by the child, who may then not want to deal with the mess, either. I coach parents to stay at least neutral about stools, reminding them that, “Your child is going to have to poop her whole life!”
Demanding a diaper and then getting the special intimacy of bottom cleaning can be reinforcing. If there is a younger sibling, diaper changes may be a desired opportunity for the toddler to regress and retain some “baby privileges.” Other clues to this dynamic include thumb sucking, baby talk, clinginess, or being rough on the sibling. One part of addressing this issue is to prescribe “babying” the toddler by holding in arms, rocking, talking baby talk, offering a pacifier, and feeding him during daily parent-child one-on-one Special Time. This sounds crazy to parents aiming for grown up toileting, but I promise them the child will not go backwards! It addresses the child’s deep fear that the nurturing of infancy is no longer available.
You may have noticed that boys are much more likely to refuse stools than girls. Some of this difference may be that high activity, but learning to urinate standing up also is fun, a Big Boy feat, and a source of pride to fathers. If regular sitting to poop has not been well established before the fun of standing to pee is offered, the little guys are not so interested in sitting again to poop. Plus the wiping and hand washing after poops are further aggravations delaying return to the Legos. But more! By around age 3 years, both genders make the horrifying discovery that boys have a penis and girls don’t. At this age of confusion about potential transformations, the obvious conclusion is that the girl’s penis was lost! And that turd disappearing down the toilet looks a lot like a dismembered body part! Reassurance and education is in order. I address this with my “Penis Talk”: “Boys are made with a penis and girls are made with a vagina. (For boys:) When you get big like your Dad, your penis will be big, too. No one can ever take your penis away. (For girls, a less common concern.) You have always had a vagina. You did not lose a penis.” I recommend that you practice this in front of a mirror before first use!
Another cognitive milestone concerns what sorts of things can disappear dow

Dr. Howard is assistant professor of pediatrics at Johns Hopkins University, Baltimore, and creator of CHADIS (www.CHADIS.com). She had no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to Frontline Medical News. Email her at [email protected].
When parents bring in their delightful, verbal 3-year-old for refusing to poop on the potty, it may seem laughable. But with impending preschool and costs of diapers, stool refusal can be a major aggravation for families! Fortunately,
Commonly a healthy, typically developing boy stands and urinates in the toilet just fine, but sneaks off behind the sofa to poop. Parent gyrations have gone from cajoling, to punishing, to offering trips to Disney! Flaring tempers can set the stage for stool refusal to be a power play.
There are a number of reasons stool refusal may give clues to child and family tendencies and relevant intervention. We always should be alert to rare medical problems such as Hirschsprung disease or traumas (from slammed toilet lids to sexual abuse). But while learning to use the toilet for urination and defecation generally occur around the same time, there are pitfalls making pooping in the potty different. An impending stool provides stronger sensations and more advance warning than urine and tends to come at regular times, making it logical to start toilet learning with sitting on the potty after meals.
But once seated on the potty, stools can require some waiting – not a typical toddler forte! While running to sit has novelty at first and may be reinforced by celebration, this quickly becomes routine and boring. Very active or very intense children especially hate having their play interrupted by a trip to the bathroom. Oppositional children just won’t perform if they think the parent cares! And unlike for urination, everyone can inhibit defecation long enough for the urge to pass. Repeated stool retention from ignoring the urge makes stools dessicated and harder, with resulting pain when finally passed. One painful stool makes many a young child decide “Never again!” and simply refuse the toilet. A rectal fissure can both start
During unclogging and establishing a new stool pattern, the toddler should be matter-of-factly put back in diapers (not pull ups) saying “Oh well, you are just not ready for pants yet.” Dramatically placing the treasured Superhero underwear on the top shelf increases motivation (or promised if none have been acquired). Returning to diapers without shaming the child is key, and all caregivers need to buy in. They need to be good “actors,” conveying that they don’t really care about toileting to reduce the power struggle. If controlling poop is a battle, only the child can win!
When the soft stools are occurring several times per day, I suggest “M&M treatment”: 1 for sitting, 2 for peeing, and 3 for pooping = 6 potential M&Ms per episode. The “1 for sitting” (the easiest part), is not painful and restores the habit of complying. Remember, M&Ms are no match for a game on an iPad! By charting the times of stools, the parent can remove electronics ½ hour before the expected poop and restrict the child to one room of the house with a potty nearby. Parents can interact, but should avoid making this a rewarding playtime. When the child uses the potty rather than their pants, the room restriction is removed until the next window for pooping. If they poop outside the toilet, they remain restricted (and no electronics) until the next window (even the next day).
Some parents are especially sensitive to the smell and mess of stools and pass that attitude along to their child by saying “Ugh, you stink!” or “I can’t stand this mess!” or even handing the child to another caregiver in a gesture of rejection. These messages are not missed by the child, who may then not want to deal with the mess, either. I coach parents to stay at least neutral about stools, reminding them that, “Your child is going to have to poop her whole life!”
Demanding a diaper and then getting the special intimacy of bottom cleaning can be reinforcing. If there is a younger sibling, diaper changes may be a desired opportunity for the toddler to regress and retain some “baby privileges.” Other clues to this dynamic include thumb sucking, baby talk, clinginess, or being rough on the sibling. One part of addressing this issue is to prescribe “babying” the toddler by holding in arms, rocking, talking baby talk, offering a pacifier, and feeding him during daily parent-child one-on-one Special Time. This sounds crazy to parents aiming for grown up toileting, but I promise them the child will not go backwards! It addresses the child’s deep fear that the nurturing of infancy is no longer available.
You may have noticed that boys are much more likely to refuse stools than girls. Some of this difference may be that high activity, but learning to urinate standing up also is fun, a Big Boy feat, and a source of pride to fathers. If regular sitting to poop has not been well established before the fun of standing to pee is offered, the little guys are not so interested in sitting again to poop. Plus the wiping and hand washing after poops are further aggravations delaying return to the Legos. But more! By around age 3 years, both genders make the horrifying discovery that boys have a penis and girls don’t. At this age of confusion about potential transformations, the obvious conclusion is that the girl’s penis was lost! And that turd disappearing down the toilet looks a lot like a dismembered body part! Reassurance and education is in order. I address this with my “Penis Talk”: “Boys are made with a penis and girls are made with a vagina. (For boys:) When you get big like your Dad, your penis will be big, too. No one can ever take your penis away. (For girls, a less common concern.) You have always had a vagina. You did not lose a penis.” I recommend that you practice this in front of a mirror before first use!
Another cognitive milestone concerns what sorts of things can disappear dow
Dr. Howard is assistant professor of pediatrics at Johns Hopkins University, Baltimore, and creator of CHADIS (www.CHADIS.com). She had no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to Frontline Medical News. Email her at [email protected].
When parents bring in their delightful, verbal 3-year-old for refusing to poop on the potty, it may seem laughable. But with impending preschool and costs of diapers, stool refusal can be a major aggravation for families! Fortunately,
Commonly a healthy, typically developing boy stands and urinates in the toilet just fine, but sneaks off behind the sofa to poop. Parent gyrations have gone from cajoling, to punishing, to offering trips to Disney! Flaring tempers can set the stage for stool refusal to be a power play.
There are a number of reasons stool refusal may give clues to child and family tendencies and relevant intervention. We always should be alert to rare medical problems such as Hirschsprung disease or traumas (from slammed toilet lids to sexual abuse). But while learning to use the toilet for urination and defecation generally occur around the same time, there are pitfalls making pooping in the potty different. An impending stool provides stronger sensations and more advance warning than urine and tends to come at regular times, making it logical to start toilet learning with sitting on the potty after meals.
But once seated on the potty, stools can require some waiting – not a typical toddler forte! While running to sit has novelty at first and may be reinforced by celebration, this quickly becomes routine and boring. Very active or very intense children especially hate having their play interrupted by a trip to the bathroom. Oppositional children just won’t perform if they think the parent cares! And unlike for urination, everyone can inhibit defecation long enough for the urge to pass. Repeated stool retention from ignoring the urge makes stools dessicated and harder, with resulting pain when finally passed. One painful stool makes many a young child decide “Never again!” and simply refuse the toilet. A rectal fissure can both start
During unclogging and establishing a new stool pattern, the toddler should be matter-of-factly put back in diapers (not pull ups) saying “Oh well, you are just not ready for pants yet.” Dramatically placing the treasured Superhero underwear on the top shelf increases motivation (or promised if none have been acquired). Returning to diapers without shaming the child is key, and all caregivers need to buy in. They need to be good “actors,” conveying that they don’t really care about toileting to reduce the power struggle. If controlling poop is a battle, only the child can win!
When the soft stools are occurring several times per day, I suggest “M&M treatment”: 1 for sitting, 2 for peeing, and 3 for pooping = 6 potential M&Ms per episode. The “1 for sitting” (the easiest part), is not painful and restores the habit of complying. Remember, M&Ms are no match for a game on an iPad! By charting the times of stools, the parent can remove electronics ½ hour before the expected poop and restrict the child to one room of the house with a potty nearby. Parents can interact, but should avoid making this a rewarding playtime. When the child uses the potty rather than their pants, the room restriction is removed until the next window for pooping. If they poop outside the toilet, they remain restricted (and no electronics) until the next window (even the next day).
Some parents are especially sensitive to the smell and mess of stools and pass that attitude along to their child by saying “Ugh, you stink!” or “I can’t stand this mess!” or even handing the child to another caregiver in a gesture of rejection. These messages are not missed by the child, who may then not want to deal with the mess, either. I coach parents to stay at least neutral about stools, reminding them that, “Your child is going to have to poop her whole life!”
Demanding a diaper and then getting the special intimacy of bottom cleaning can be reinforcing. If there is a younger sibling, diaper changes may be a desired opportunity for the toddler to regress and retain some “baby privileges.” Other clues to this dynamic include thumb sucking, baby talk, clinginess, or being rough on the sibling. One part of addressing this issue is to prescribe “babying” the toddler by holding in arms, rocking, talking baby talk, offering a pacifier, and feeding him during daily parent-child one-on-one Special Time. This sounds crazy to parents aiming for grown up toileting, but I promise them the child will not go backwards! It addresses the child’s deep fear that the nurturing of infancy is no longer available.
You may have noticed that boys are much more likely to refuse stools than girls. Some of this difference may be that high activity, but learning to urinate standing up also is fun, a Big Boy feat, and a source of pride to fathers. If regular sitting to poop has not been well established before the fun of standing to pee is offered, the little guys are not so interested in sitting again to poop. Plus the wiping and hand washing after poops are further aggravations delaying return to the Legos. But more! By around age 3 years, both genders make the horrifying discovery that boys have a penis and girls don’t. At this age of confusion about potential transformations, the obvious conclusion is that the girl’s penis was lost! And that turd disappearing down the toilet looks a lot like a dismembered body part! Reassurance and education is in order. I address this with my “Penis Talk”: “Boys are made with a penis and girls are made with a vagina. (For boys:) When you get big like your Dad, your penis will be big, too. No one can ever take your penis away. (For girls, a less common concern.) You have always had a vagina. You did not lose a penis.” I recommend that you practice this in front of a mirror before first use!
Another cognitive milestone concerns what sorts of things can disappear dow
Dr. Howard is assistant professor of pediatrics at Johns Hopkins University, Baltimore, and creator of CHADIS (www.CHADIS.com). She had no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to Frontline Medical News. Email her at [email protected].
Labor and delivery mismanaged, child has CP: $30.5M award
Labor and delivery mismanaged, child has CP: $30.5M award
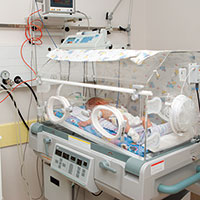
Two days later, the mother reported decreased fetal movement; she was admitted to the hospital for continuous fetal heart-rate (FHR) monitoring, and a maternal-fetal medicine (MFM) specialist was consulted. The mother was not placed on FHR monitoring until 2 hours after admission. Three hours after admission, the MFM, by phone, recommended further testing and later, cesarean delivery.
The child was found to have spastic quadriplegic cerebral palsy, profound developmental delays, a seizure disorder, and cortical blindness.
PARENTS' CLAIM:
The child's injuries were due to mismanagement of labor and delivery. The MFM prescribed ultrasonographic biophysical profiles, but they were not performed until 2 hours after ordered. There were 3 ultrasonography (US) technicians at the hospital when the mother was admitted: 1 was on break, another was performing other tests, and the third was not notified because the hospital's computer system was down. When test results were unfavorable, the MFM recommended emergency cesarean delivery. An earlier delivery could have prevented the child's injuries.
DEFENDANTS' DEFENSE:
The infant's injuries were a result of her mother's failure to keep her GDM under control.
VERDICT:
A $30,545,655 Georgia verdict was returned.
Related article:
10 tips for overcoming common challenges of intrapartum fetal monitoring
Did oxytocin cause child's spastic CP? $14.4M verdict
When a woman went to the hospital in labor, her ObGyn ordered oxytocin to enhance delivery. The FHR monitor showed repetitive decelerations for the next hour, dropping to 60 bpm by 8:00 pm, when the ObGyn expedited delivery but did not stop the oxytocin. By 8:20 pm, the baby's head was crowning, but the ObGyn waited another 10 minutes before performing an episiotomy and delivering the baby.
The child, intubated 5 minutes after birth, was found to have spastic tetraparesis cerebral palsy (CP) with impaired cognition, seizures, and global aphasia.
PARENTS' CLAIM:
The ObGyn and nurses failed to properly monitor labor and delivery. The ObGyn should not have started oxytocin because the patient's labor was progressing normally. He should have taken the mother off oxytocin at 8:00 pm when the FHR dropped to 60 bpm. He should have performed an operative delivery at 8:20 pm when the baby's head crowned. An earlier delivery would have prevented injury.
DEFENDANTS' DEFENSE:
The ObGyn's treatment was within the standard of care. He properly determined that vaginal delivery would be the quickest. It is his practice to stop oxytocin when the FHR slows, though he had no memory of halting oxytocin administration in this case. The baby's CP stemmed from insufficiencies in the placenta, seizures, and meconium aspiration syndrome.
VERDICT:
A $14.4 million Pennsylvania verdict was returned. The ObGyn was found 60% liable for the baby's injuries and the hospital 40% responsible.
Related article:
Q: Following cesarean delivery, what is the optimal oxytocin infusion duration to prevent postpartum bleeding?
Woman with preeclampsia dies: $6M verdict
A 34-year-old woman had been a patient of her family practitioner (FP) for many years. Her blood pressure (BP) averaged 105/63 mm Hg over that time. At a regular prenatal visit on February 26, the patient reported a headache and cough. Her BP was 130/90 mm Hg and she had gained 8.6 lb since her last visit 4 weeks earlier. She was told to return in 2 weeks.
She contacted her FP 2 days later to report acute vaginal bleeding and a severe headache. The FP sent her to the hospital, where potential placental abruption was considered. Two US studies demonstrated oligohydramnios, intrauterine growth restriction, and a grade II placenta. She continued to have repeated high BP readings, headaches, variable and late decelerations, and a dropping platelet count.
She was discharged on the morning of March 3 and sent to another hospital for a specialized US. The FP spoke to the physician who was to perform the US, advising him by phone and in writing to evaluate the oligohydramnios and intrauterine growth restriction. No other information was provided.
At 6:00 pm on March 3, the patient's husband called the FP to report that his wife was vomiting, reporting abdominal pain and intense headache. He was advised to call back in 1 hour, and when he did, he was told to take his wife to the hospital. At the hospital at 8:50 pm on March 3, her BP was 128/103 mm Hg. She reported throbbing headache, vomiting, and facial edema. She was admitted for observation.
At 9:30 pm, when the patient's BP was 155/100 mm Hg, a nurse contacted the FP to report the patient's continued throbbing headache and elevated, labile BP. The FP neither requested a consultation with an attending ObGyn nor went to the hospital until 4:31 am on March 4.
At 3:15 am on March 4, a nurse found the patient with her head hanging over the side of the bed in an obtunded state, having vomited. The rapid response team and an attending ObGyn were called. The ObGyn diagnosed eclampsia, ordered magnesium sulfate and hydralazine and immediately transported her to the operating room for an emergency cesarean delivery. Although the baby was healthy, the mother remained unresponsive. A computed tomography (CT) scan confirmed a massive intracranial hemorrhage. She was pronounced dead at 5:10 pm.
ESTATE'S CLAIM:
The FP negligently deviated from the standard of care, leading to the mother's death. The FP fraudulently misrepresented her experience and training for obstetric conditions. She was negligent for failing to adequately diagnose and react to the patient's condition or refer her to an ObGyn, per hospital policy.
DEFENDANTS' DEFENSE:
The patient's treatment met the standard of care. The FP was credentialed to practice obstetrics at the hospital. The patient's BP never reached or sustained a level that would require the FP to consult an ObGyn until 3:15 am on March 4. When the patient first presented with a headache, the FP had consulted a board-certified ObGyn and an MFM, who suggested continued antepartum testing and induction at 39 weeks. The patient's death was unforeseeable because her BP values were inconsistent; the FP had no knowledge of a family history of stroke. The autopsy reported that a ruptured aneurysm was the cause of death.
VERDICT:
A $6,067,830 Ohio verdict was returned. The award was reduced to $900,000 due to a high/low agreement.
Related article:
Start offering aspirin to pregnant women at high risk for preeclampsia
Fetal abnormalities not diagnosed: Baby has Down syndrome
On September 6, at 10 weeks' gestation, a woman began prenatal care at a clinic with Dr. A, an ObGyn. The mother participated in the California Prenatal Screening Program and received test results on October 23 that showed normal risk for birth defects. On November 1, she saw Dr. B, another ObGyn, who confirmed the negative prenatal screening and ordered an US. A radiologist reported to Dr. Bthat the fetal anatomy was not well visualized. When the mother was at 23 2/7 weeks' gestation (December 6), Dr. B told the parents that the US results were normal.
On January 2, the parents saw Dr. A, who disclosed that the US radiology report indicated an incomplete fetal anatomy scan and ordered a repeat US. The US performed on January 17 showed a cardiac defect. Further testing confirmed that the fetus had Down syndrome. The parents scheduled but did not appear for a late-term abortion because they feared that the procedure was illegal.
PARENTS' CLAIM:
The parents told both ObGyns that they wanted extensive prenatal testing because of a family history of birth defects and that they would terminate the pregnancy if birth defects were discovered. Because Dr. B did not discuss prenatal testing, the parents did not know their child had Down syndrome until it was too late to legally terminate the pregnancy. The mother testified that she had never heard of amniocentesis until mid-January, when a perinatologist confirmed that the baby had Down syndrome.
DEFENDANTS' DEFENSE:
The ObGyns denied having any discussions with the parents about their request for extensive prenatal tests or desire for termination. Difficulty in visualizing the fetus is common in second trimester US, and therefore Dr. B routinely performs another US later in the pregnancy. He also denied responsibility for discussing prenatal testing with the parents, stating that such discussions should happen in the first trimester. Since the parents saw Dr. A during that time, Dr. B believed that those conversations had already taken place. The prenatal screening pamphlet that the mother signed on September 6 discussed amniocentesis. The child's grandmother testified that she had discussed amniocentesis with the parents early in the pregnancy. A clinic employee testified that in January she asked the mother why she had not chosen amniocentesis earlier in the pregnancy; the mother replied that she had decided against it because her prenatal screening test was normal.
VERDICT:
A California defense verdict was returned.
Related article:
When is cell-free DNA best used as a primary screen?
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Labor and delivery mismanaged, child has CP: $30.5M award
Two days later, the mother reported decreased fetal movement; she was admitted to the hospital for continuous fetal heart-rate (FHR) monitoring, and a maternal-fetal medicine (MFM) specialist was consulted. The mother was not placed on FHR monitoring until 2 hours after admission. Three hours after admission, the MFM, by phone, recommended further testing and later, cesarean delivery.
The child was found to have spastic quadriplegic cerebral palsy, profound developmental delays, a seizure disorder, and cortical blindness.
PARENTS' CLAIM:
The child's injuries were due to mismanagement of labor and delivery. The MFM prescribed ultrasonographic biophysical profiles, but they were not performed until 2 hours after ordered. There were 3 ultrasonography (US) technicians at the hospital when the mother was admitted: 1 was on break, another was performing other tests, and the third was not notified because the hospital's computer system was down. When test results were unfavorable, the MFM recommended emergency cesarean delivery. An earlier delivery could have prevented the child's injuries.
DEFENDANTS' DEFENSE:
The infant's injuries were a result of her mother's failure to keep her GDM under control.
VERDICT:
A $30,545,655 Georgia verdict was returned.
Related article:
10 tips for overcoming common challenges of intrapartum fetal monitoring
Did oxytocin cause child's spastic CP? $14.4M verdict
When a woman went to the hospital in labor, her ObGyn ordered oxytocin to enhance delivery. The FHR monitor showed repetitive decelerations for the next hour, dropping to 60 bpm by 8:00 pm, when the ObGyn expedited delivery but did not stop the oxytocin. By 8:20 pm, the baby's head was crowning, but the ObGyn waited another 10 minutes before performing an episiotomy and delivering the baby.
The child, intubated 5 minutes after birth, was found to have spastic tetraparesis cerebral palsy (CP) with impaired cognition, seizures, and global aphasia.
PARENTS' CLAIM:
The ObGyn and nurses failed to properly monitor labor and delivery. The ObGyn should not have started oxytocin because the patient's labor was progressing normally. He should have taken the mother off oxytocin at 8:00 pm when the FHR dropped to 60 bpm. He should have performed an operative delivery at 8:20 pm when the baby's head crowned. An earlier delivery would have prevented injury.
DEFENDANTS' DEFENSE:
The ObGyn's treatment was within the standard of care. He properly determined that vaginal delivery would be the quickest. It is his practice to stop oxytocin when the FHR slows, though he had no memory of halting oxytocin administration in this case. The baby's CP stemmed from insufficiencies in the placenta, seizures, and meconium aspiration syndrome.
VERDICT:
A $14.4 million Pennsylvania verdict was returned. The ObGyn was found 60% liable for the baby's injuries and the hospital 40% responsible.
Related article:
Q: Following cesarean delivery, what is the optimal oxytocin infusion duration to prevent postpartum bleeding?
Woman with preeclampsia dies: $6M verdict
A 34-year-old woman had been a patient of her family practitioner (FP) for many years. Her blood pressure (BP) averaged 105/63 mm Hg over that time. At a regular prenatal visit on February 26, the patient reported a headache and cough. Her BP was 130/90 mm Hg and she had gained 8.6 lb since her last visit 4 weeks earlier. She was told to return in 2 weeks.
She contacted her FP 2 days later to report acute vaginal bleeding and a severe headache. The FP sent her to the hospital, where potential placental abruption was considered. Two US studies demonstrated oligohydramnios, intrauterine growth restriction, and a grade II placenta. She continued to have repeated high BP readings, headaches, variable and late decelerations, and a dropping platelet count.
She was discharged on the morning of March 3 and sent to another hospital for a specialized US. The FP spoke to the physician who was to perform the US, advising him by phone and in writing to evaluate the oligohydramnios and intrauterine growth restriction. No other information was provided.
At 6:00 pm on March 3, the patient's husband called the FP to report that his wife was vomiting, reporting abdominal pain and intense headache. He was advised to call back in 1 hour, and when he did, he was told to take his wife to the hospital. At the hospital at 8:50 pm on March 3, her BP was 128/103 mm Hg. She reported throbbing headache, vomiting, and facial edema. She was admitted for observation.
At 9:30 pm, when the patient's BP was 155/100 mm Hg, a nurse contacted the FP to report the patient's continued throbbing headache and elevated, labile BP. The FP neither requested a consultation with an attending ObGyn nor went to the hospital until 4:31 am on March 4.
At 3:15 am on March 4, a nurse found the patient with her head hanging over the side of the bed in an obtunded state, having vomited. The rapid response team and an attending ObGyn were called. The ObGyn diagnosed eclampsia, ordered magnesium sulfate and hydralazine and immediately transported her to the operating room for an emergency cesarean delivery. Although the baby was healthy, the mother remained unresponsive. A computed tomography (CT) scan confirmed a massive intracranial hemorrhage. She was pronounced dead at 5:10 pm.
ESTATE'S CLAIM:
The FP negligently deviated from the standard of care, leading to the mother's death. The FP fraudulently misrepresented her experience and training for obstetric conditions. She was negligent for failing to adequately diagnose and react to the patient's condition or refer her to an ObGyn, per hospital policy.
DEFENDANTS' DEFENSE:
The patient's treatment met the standard of care. The FP was credentialed to practice obstetrics at the hospital. The patient's BP never reached or sustained a level that would require the FP to consult an ObGyn until 3:15 am on March 4. When the patient first presented with a headache, the FP had consulted a board-certified ObGyn and an MFM, who suggested continued antepartum testing and induction at 39 weeks. The patient's death was unforeseeable because her BP values were inconsistent; the FP had no knowledge of a family history of stroke. The autopsy reported that a ruptured aneurysm was the cause of death.
VERDICT:
A $6,067,830 Ohio verdict was returned. The award was reduced to $900,000 due to a high/low agreement.
Related article:
Start offering aspirin to pregnant women at high risk for preeclampsia
Fetal abnormalities not diagnosed: Baby has Down syndrome
On September 6, at 10 weeks' gestation, a woman began prenatal care at a clinic with Dr. A, an ObGyn. The mother participated in the California Prenatal Screening Program and received test results on October 23 that showed normal risk for birth defects. On November 1, she saw Dr. B, another ObGyn, who confirmed the negative prenatal screening and ordered an US. A radiologist reported to Dr. Bthat the fetal anatomy was not well visualized. When the mother was at 23 2/7 weeks' gestation (December 6), Dr. B told the parents that the US results were normal.
On January 2, the parents saw Dr. A, who disclosed that the US radiology report indicated an incomplete fetal anatomy scan and ordered a repeat US. The US performed on January 17 showed a cardiac defect. Further testing confirmed that the fetus had Down syndrome. The parents scheduled but did not appear for a late-term abortion because they feared that the procedure was illegal.
PARENTS' CLAIM:
The parents told both ObGyns that they wanted extensive prenatal testing because of a family history of birth defects and that they would terminate the pregnancy if birth defects were discovered. Because Dr. B did not discuss prenatal testing, the parents did not know their child had Down syndrome until it was too late to legally terminate the pregnancy. The mother testified that she had never heard of amniocentesis until mid-January, when a perinatologist confirmed that the baby had Down syndrome.
DEFENDANTS' DEFENSE:
The ObGyns denied having any discussions with the parents about their request for extensive prenatal tests or desire for termination. Difficulty in visualizing the fetus is common in second trimester US, and therefore Dr. B routinely performs another US later in the pregnancy. He also denied responsibility for discussing prenatal testing with the parents, stating that such discussions should happen in the first trimester. Since the parents saw Dr. A during that time, Dr. B believed that those conversations had already taken place. The prenatal screening pamphlet that the mother signed on September 6 discussed amniocentesis. The child's grandmother testified that she had discussed amniocentesis with the parents early in the pregnancy. A clinic employee testified that in January she asked the mother why she had not chosen amniocentesis earlier in the pregnancy; the mother replied that she had decided against it because her prenatal screening test was normal.
VERDICT:
A California defense verdict was returned.
Related article:
When is cell-free DNA best used as a primary screen?
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Labor and delivery mismanaged, child has CP: $30.5M award
Two days later, the mother reported decreased fetal movement; she was admitted to the hospital for continuous fetal heart-rate (FHR) monitoring, and a maternal-fetal medicine (MFM) specialist was consulted. The mother was not placed on FHR monitoring until 2 hours after admission. Three hours after admission, the MFM, by phone, recommended further testing and later, cesarean delivery.
The child was found to have spastic quadriplegic cerebral palsy, profound developmental delays, a seizure disorder, and cortical blindness.
PARENTS' CLAIM:
The child's injuries were due to mismanagement of labor and delivery. The MFM prescribed ultrasonographic biophysical profiles, but they were not performed until 2 hours after ordered. There were 3 ultrasonography (US) technicians at the hospital when the mother was admitted: 1 was on break, another was performing other tests, and the third was not notified because the hospital's computer system was down. When test results were unfavorable, the MFM recommended emergency cesarean delivery. An earlier delivery could have prevented the child's injuries.
DEFENDANTS' DEFENSE:
The infant's injuries were a result of her mother's failure to keep her GDM under control.
VERDICT:
A $30,545,655 Georgia verdict was returned.
Related article:
10 tips for overcoming common challenges of intrapartum fetal monitoring
Did oxytocin cause child's spastic CP? $14.4M verdict
When a woman went to the hospital in labor, her ObGyn ordered oxytocin to enhance delivery. The FHR monitor showed repetitive decelerations for the next hour, dropping to 60 bpm by 8:00 pm, when the ObGyn expedited delivery but did not stop the oxytocin. By 8:20 pm, the baby's head was crowning, but the ObGyn waited another 10 minutes before performing an episiotomy and delivering the baby.
The child, intubated 5 minutes after birth, was found to have spastic tetraparesis cerebral palsy (CP) with impaired cognition, seizures, and global aphasia.
PARENTS' CLAIM:
The ObGyn and nurses failed to properly monitor labor and delivery. The ObGyn should not have started oxytocin because the patient's labor was progressing normally. He should have taken the mother off oxytocin at 8:00 pm when the FHR dropped to 60 bpm. He should have performed an operative delivery at 8:20 pm when the baby's head crowned. An earlier delivery would have prevented injury.
DEFENDANTS' DEFENSE:
The ObGyn's treatment was within the standard of care. He properly determined that vaginal delivery would be the quickest. It is his practice to stop oxytocin when the FHR slows, though he had no memory of halting oxytocin administration in this case. The baby's CP stemmed from insufficiencies in the placenta, seizures, and meconium aspiration syndrome.
VERDICT:
A $14.4 million Pennsylvania verdict was returned. The ObGyn was found 60% liable for the baby's injuries and the hospital 40% responsible.
Related article:
Q: Following cesarean delivery, what is the optimal oxytocin infusion duration to prevent postpartum bleeding?
Woman with preeclampsia dies: $6M verdict
A 34-year-old woman had been a patient of her family practitioner (FP) for many years. Her blood pressure (BP) averaged 105/63 mm Hg over that time. At a regular prenatal visit on February 26, the patient reported a headache and cough. Her BP was 130/90 mm Hg and she had gained 8.6 lb since her last visit 4 weeks earlier. She was told to return in 2 weeks.
She contacted her FP 2 days later to report acute vaginal bleeding and a severe headache. The FP sent her to the hospital, where potential placental abruption was considered. Two US studies demonstrated oligohydramnios, intrauterine growth restriction, and a grade II placenta. She continued to have repeated high BP readings, headaches, variable and late decelerations, and a dropping platelet count.
She was discharged on the morning of March 3 and sent to another hospital for a specialized US. The FP spoke to the physician who was to perform the US, advising him by phone and in writing to evaluate the oligohydramnios and intrauterine growth restriction. No other information was provided.
At 6:00 pm on March 3, the patient's husband called the FP to report that his wife was vomiting, reporting abdominal pain and intense headache. He was advised to call back in 1 hour, and when he did, he was told to take his wife to the hospital. At the hospital at 8:50 pm on March 3, her BP was 128/103 mm Hg. She reported throbbing headache, vomiting, and facial edema. She was admitted for observation.
At 9:30 pm, when the patient's BP was 155/100 mm Hg, a nurse contacted the FP to report the patient's continued throbbing headache and elevated, labile BP. The FP neither requested a consultation with an attending ObGyn nor went to the hospital until 4:31 am on March 4.
At 3:15 am on March 4, a nurse found the patient with her head hanging over the side of the bed in an obtunded state, having vomited. The rapid response team and an attending ObGyn were called. The ObGyn diagnosed eclampsia, ordered magnesium sulfate and hydralazine and immediately transported her to the operating room for an emergency cesarean delivery. Although the baby was healthy, the mother remained unresponsive. A computed tomography (CT) scan confirmed a massive intracranial hemorrhage. She was pronounced dead at 5:10 pm.
ESTATE'S CLAIM:
The FP negligently deviated from the standard of care, leading to the mother's death. The FP fraudulently misrepresented her experience and training for obstetric conditions. She was negligent for failing to adequately diagnose and react to the patient's condition or refer her to an ObGyn, per hospital policy.
DEFENDANTS' DEFENSE:
The patient's treatment met the standard of care. The FP was credentialed to practice obstetrics at the hospital. The patient's BP never reached or sustained a level that would require the FP to consult an ObGyn until 3:15 am on March 4. When the patient first presented with a headache, the FP had consulted a board-certified ObGyn and an MFM, who suggested continued antepartum testing and induction at 39 weeks. The patient's death was unforeseeable because her BP values were inconsistent; the FP had no knowledge of a family history of stroke. The autopsy reported that a ruptured aneurysm was the cause of death.
VERDICT:
A $6,067,830 Ohio verdict was returned. The award was reduced to $900,000 due to a high/low agreement.
Related article:
Start offering aspirin to pregnant women at high risk for preeclampsia
Fetal abnormalities not diagnosed: Baby has Down syndrome
On September 6, at 10 weeks' gestation, a woman began prenatal care at a clinic with Dr. A, an ObGyn. The mother participated in the California Prenatal Screening Program and received test results on October 23 that showed normal risk for birth defects. On November 1, she saw Dr. B, another ObGyn, who confirmed the negative prenatal screening and ordered an US. A radiologist reported to Dr. Bthat the fetal anatomy was not well visualized. When the mother was at 23 2/7 weeks' gestation (December 6), Dr. B told the parents that the US results were normal.
On January 2, the parents saw Dr. A, who disclosed that the US radiology report indicated an incomplete fetal anatomy scan and ordered a repeat US. The US performed on January 17 showed a cardiac defect. Further testing confirmed that the fetus had Down syndrome. The parents scheduled but did not appear for a late-term abortion because they feared that the procedure was illegal.
PARENTS' CLAIM:
The parents told both ObGyns that they wanted extensive prenatal testing because of a family history of birth defects and that they would terminate the pregnancy if birth defects were discovered. Because Dr. B did not discuss prenatal testing, the parents did not know their child had Down syndrome until it was too late to legally terminate the pregnancy. The mother testified that she had never heard of amniocentesis until mid-January, when a perinatologist confirmed that the baby had Down syndrome.
DEFENDANTS' DEFENSE:
The ObGyns denied having any discussions with the parents about their request for extensive prenatal tests or desire for termination. Difficulty in visualizing the fetus is common in second trimester US, and therefore Dr. B routinely performs another US later in the pregnancy. He also denied responsibility for discussing prenatal testing with the parents, stating that such discussions should happen in the first trimester. Since the parents saw Dr. A during that time, Dr. B believed that those conversations had already taken place. The prenatal screening pamphlet that the mother signed on September 6 discussed amniocentesis. The child's grandmother testified that she had discussed amniocentesis with the parents early in the pregnancy. A clinic employee testified that in January she asked the mother why she had not chosen amniocentesis earlier in the pregnancy; the mother replied that she had decided against it because her prenatal screening test was normal.
VERDICT:
A California defense verdict was returned.
Related article:
When is cell-free DNA best used as a primary screen?
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Promising phase II results for Rytary reformulation for Parkinson’s
BOSTON – The results of a small phase II trial confirm that IPX203, an investigational reformulation of the extended-release carbidopa-levodopa combination called Rytary, holds the potential to greatly extend “on” time in Parkinson’s disease patients.
While it’s early in the research process, “this is a promising new formulation,” said Mark Stacy, MD, who led the trial that pitted IPX203 against immediate-release (IR) carbidopa-levodopa (CD-LD) and Rytary.
Dr. Stacy, professor of neurology and vice dean for clinical research at Duke University, Durham, N.C., and his colleagues released the phase II results at the annual meeting of the American Academy of Neurology.
Parkinson’s disease patients often aren’t adherent to their drugs despite the risk that they’ll see symptoms return. “It’s been demonstrated that they miss medicines as often as in any other disease,” Dr. Stacy said in an interview.
Rytary, which was approved by the Food and Drug Administration in 2015, has improved adherence in Parkinson’s disease patients by allowing them to reduce the number of doses they need to separately take every day to four. “If this new formulation is able to last 8 hours, you could essentially take that at bedtime and have medication in your system when you awaken,” Dr. Stacy said. “Then you could take it twice more during the day.”
The new, open-label, crossover trial randomly assigned 26 patients on stable IR regimens (at least 400 mg LD/day, at least 4 doses a day) to single doses of IR, Rytary, and IPX203. For 10 hours after dose, raters evaluated every 30 minutes whether the subjects were on (with troublesome dyskinesia) or off (without troublesome dyskinesia).
All but one subject finished the trial. Over the first hour, the patients fared about the same in terms of converting to on status. Overall, PX203 significantly decreased average off time (4.5 hours), compared with IR (7.2 hours; P less than .0001) and Rytary (5.4 hours; P less than .05).
Researchers also found that IPX203 had the highest duration of a 4-point improvement in the Unified Parkinson’s Disease Rating Scale Part III score at 6.1 hours, compared with IR (3.7 hours; P less than .0001) and Rytary (5.2 hours; P less than .05). In addition, 13-point improvements were higher for IPX203 at 4.8 hours, compared with IR (2.2 hours; P less than .0001) and Rytary (3.6 hours; P less than .05).
No serious adverse events were reported. Nausea, dizziness, and hypertension occurred in two or more patients in each treatment group and were more common with IR (28.0%) and IPX203 (19.2%) than with Rytary (8.0%).
The study was funded by Impax Laboratories. Dr. Stacy reported several disclosures, including consulting work for numerous drug makers.
BOSTON – The results of a small phase II trial confirm that IPX203, an investigational reformulation of the extended-release carbidopa-levodopa combination called Rytary, holds the potential to greatly extend “on” time in Parkinson’s disease patients.
While it’s early in the research process, “this is a promising new formulation,” said Mark Stacy, MD, who led the trial that pitted IPX203 against immediate-release (IR) carbidopa-levodopa (CD-LD) and Rytary.
Dr. Stacy, professor of neurology and vice dean for clinical research at Duke University, Durham, N.C., and his colleagues released the phase II results at the annual meeting of the American Academy of Neurology.
Parkinson’s disease patients often aren’t adherent to their drugs despite the risk that they’ll see symptoms return. “It’s been demonstrated that they miss medicines as often as in any other disease,” Dr. Stacy said in an interview.
Rytary, which was approved by the Food and Drug Administration in 2015, has improved adherence in Parkinson’s disease patients by allowing them to reduce the number of doses they need to separately take every day to four. “If this new formulation is able to last 8 hours, you could essentially take that at bedtime and have medication in your system when you awaken,” Dr. Stacy said. “Then you could take it twice more during the day.”
The new, open-label, crossover trial randomly assigned 26 patients on stable IR regimens (at least 400 mg LD/day, at least 4 doses a day) to single doses of IR, Rytary, and IPX203. For 10 hours after dose, raters evaluated every 30 minutes whether the subjects were on (with troublesome dyskinesia) or off (without troublesome dyskinesia).
All but one subject finished the trial. Over the first hour, the patients fared about the same in terms of converting to on status. Overall, PX203 significantly decreased average off time (4.5 hours), compared with IR (7.2 hours; P less than .0001) and Rytary (5.4 hours; P less than .05).
Researchers also found that IPX203 had the highest duration of a 4-point improvement in the Unified Parkinson’s Disease Rating Scale Part III score at 6.1 hours, compared with IR (3.7 hours; P less than .0001) and Rytary (5.2 hours; P less than .05). In addition, 13-point improvements were higher for IPX203 at 4.8 hours, compared with IR (2.2 hours; P less than .0001) and Rytary (3.6 hours; P less than .05).
No serious adverse events were reported. Nausea, dizziness, and hypertension occurred in two or more patients in each treatment group and were more common with IR (28.0%) and IPX203 (19.2%) than with Rytary (8.0%).
The study was funded by Impax Laboratories. Dr. Stacy reported several disclosures, including consulting work for numerous drug makers.
BOSTON – The results of a small phase II trial confirm that IPX203, an investigational reformulation of the extended-release carbidopa-levodopa combination called Rytary, holds the potential to greatly extend “on” time in Parkinson’s disease patients.
While it’s early in the research process, “this is a promising new formulation,” said Mark Stacy, MD, who led the trial that pitted IPX203 against immediate-release (IR) carbidopa-levodopa (CD-LD) and Rytary.
Dr. Stacy, professor of neurology and vice dean for clinical research at Duke University, Durham, N.C., and his colleagues released the phase II results at the annual meeting of the American Academy of Neurology.
Parkinson’s disease patients often aren’t adherent to their drugs despite the risk that they’ll see symptoms return. “It’s been demonstrated that they miss medicines as often as in any other disease,” Dr. Stacy said in an interview.
Rytary, which was approved by the Food and Drug Administration in 2015, has improved adherence in Parkinson’s disease patients by allowing them to reduce the number of doses they need to separately take every day to four. “If this new formulation is able to last 8 hours, you could essentially take that at bedtime and have medication in your system when you awaken,” Dr. Stacy said. “Then you could take it twice more during the day.”
The new, open-label, crossover trial randomly assigned 26 patients on stable IR regimens (at least 400 mg LD/day, at least 4 doses a day) to single doses of IR, Rytary, and IPX203. For 10 hours after dose, raters evaluated every 30 minutes whether the subjects were on (with troublesome dyskinesia) or off (without troublesome dyskinesia).
All but one subject finished the trial. Over the first hour, the patients fared about the same in terms of converting to on status. Overall, PX203 significantly decreased average off time (4.5 hours), compared with IR (7.2 hours; P less than .0001) and Rytary (5.4 hours; P less than .05).
Researchers also found that IPX203 had the highest duration of a 4-point improvement in the Unified Parkinson’s Disease Rating Scale Part III score at 6.1 hours, compared with IR (3.7 hours; P less than .0001) and Rytary (5.2 hours; P less than .05). In addition, 13-point improvements were higher for IPX203 at 4.8 hours, compared with IR (2.2 hours; P less than .0001) and Rytary (3.6 hours; P less than .05).
No serious adverse events were reported. Nausea, dizziness, and hypertension occurred in two or more patients in each treatment group and were more common with IR (28.0%) and IPX203 (19.2%) than with Rytary (8.0%).
The study was funded by Impax Laboratories. Dr. Stacy reported several disclosures, including consulting work for numerous drug makers.
AT AAN 2017
Key clinical point: Investigational drug IPX203, a reformulation of Rytary (extended release carbidopa-levodopa), holds promise as an improved extended-release Parkinson’s disease medication.
Major finding: After one dose, PX203 significantly decreased average off time (4.5 hours), compared with immediate-release carbidopa-levodopa (7.2 hours; P less than .0001) and Rytary (5.4 hours; P less than .05).
Data source: Open-label, rater-blinded, randomized, crossover study of 25 Parkinson’s disease patients
Disclosures: The study was funded by Impax Laboratories. The presenter reported several disclosures, including consulting work for numerous drug makers.
Obtaining coverage for transgender and gender-expansive youth
Transgender and gender-expansive youth face many barriers to health care. (Gender-expansive youth are defined as “youth who do not identify with traditional gender roles but are otherwise not confined to one gender narrative or experience.”) Although some of these youth may be fortunate to have a supportive family and access to health care providers proficient in transgender health care, they still face difficulties in having their insurance cover transgender-related services. This is not an impossible task, but it is a constant struggle for many clinicians.
In this column, I will provide some tips and strategies to help clinicians get insurance companies to cover these critical services. However, keep in mind that there is no one-size-fits-all approach to obtaining insurance coverage. In addition, growing uncertainty over the repeal of the Affordable Care Act (ACA) – which was critical in lifting many of the barriers to insurance coverage for transgender individuals – will make this task challenging.
Health insurance is extraordinarily complex. There are multiple private and public plans that vary in the services they cover. This variation is state dependent. And even within states, there is additional variability. Most health insurance plans are purchased by employers, and employers have a choice of what can be covered in their health plans. So even though an insurance company may state that it covers transgender-related services, the patient’s employer may pay for a plan that doesn’t cover such services. The only way to be sure whether a patient’s insurance will cover transgender-related services or not is to contact the insurance provider directly, but with extremely busy schedules and heavy patient loads, this is easier said than done. It would be helpful to have a social worker perform this task, but even having a social worker can be a luxury for some clinics.
The ACA made it easier for transgender individuals to obtain insurance coverage. Three years ago, the U.S. Department of Health and Human Services stated that Medicare’s longstanding exclusion of “transsexual surgical procedures” was no longer valid.1 Although it did not universally ban transgender exclusion policies, it did allow individual states to do so. Thirteen states have explicit policies that ban exclusions of transgender-related services in both private insurance and in Medicaid, and an additional five states have some policies that discourage such practices.2 This allowed some insurance providers and state Medicaid plans to offer coverage of transgender-related services.
Another challenge in obtaining insurance coverage for transgender and gender-expansive youth is claims denial for sex-specific procedures. For example, if a transwoman is designated as “male” in the electronic medical record and requires a breast ultrasound, the insurance company may automatically reject this claim because this procedure is covered for bodies designated as “female.” If the patient’s insurance plan covers transgender-related services, the clinic can notify the insurance company that the patient is transgender; if the patient’s plan does not, then the clinic will need to appeal to the insurance provider. Alternatively, for clinics associated with federally-funded institutions (e.g., most hospitals), the clinician can use Condition Code 45 in the billing to override the sex mismatch, although not all hospitals have implemented this code.3
1. Patient’s identifying information. Usually the patient’s name and date of birth is sufficient. Clinicians should use the patient’s preferred name in the letter, but provide the insurance or legal name of the patient so that the insurance provider can locate the patient’s records.
2. Result of a psychosocial evaluation and diagnosis (if any). Many insurance providers are looking specifically for the gender dysphoria diagnosis.
3. The duration of the referring health professional’s relationship with the patient, which includes the type of evaluation and therapy or counseling (e.g., cognitive behavior therapy or gender coaching).
4. An explanation that the criteria (usually from the World Professional Association for Transgender Health standard of care4 or the Endocrine Society Guidelines titled Endocrine Treatment of Transsexual Persons5) for hormone therapy have been met, and a brief description of the clinical rationale for supporting the client’s request for hormone therapy.
5. A statement that informed consent has been obtained from the patient (or parental permission if the patient is younger than 18 years).
6. A statement that the referring health professional is available for coordination of care.
If the clinician fails to convince the insurance provider of the necessity of covering transgender-related services, the patient still can pay out of pocket. Some hormones can be affordable to certain patients. In the state of Pennsylvania, for example, a 10-mL vial of testosterone can cost anywhere from $60 to $80, and may generally last anywhere from 10 weeks to a year, depending on dosage. Nevertheless, these costs still may be prohibitive for many transgender youth. Many are chronically unemployed or underemployed, or struggle with homelessness.6 Some transgender youth have to the face the excruciatingly difficult choice between having something to eat for the day or living another day with gender dysphoria.
Clinicians should work very hard to make sure that their transgender and gender-expansive patients obtain the care they need. The above strategies may help navigate the complex insurance system. However, insurance policies vary by state, and anti-trans discrimination creates additional barriers to health care. Therefore, clinicians who take care of transgender youth also should advocate for policies that protect these patients from discrimination, and they should advocate for policies that expand medical coverage for this vulnerable population.

Resources
• The Human Rights Campaign keeps a list of insurance plans that cover transgender-related services, but this list is far from comprehensive.
• Healthcare.gov provides some guidance on how to obtain coverage and navigate the insurance system for transgender individuals.
• UCSF Center of Excellence for Transgender Health provides some excellent resources and guidance on obtaining insurance coverage for transgender individuals.
References
1. LGBT Health 2014;1(4):256-8.
2. Map: State Health Insurance Rules: National Center for Transgender Equality, 2016 [Available from: www.transequality.org/issues/resources/map-state-health-insurance-rules].
3. Health insurance coverage issues for transgender people in the United States: University of California, San Fransisco Center of Excellence for Transgender Health, 2017 [Available from: http://transhealth.ucsf.edu/trans?page=guidelines-insurance].
4. International Journal of Transgenderism 2012;13(4):165-232.
5. J Clin Endocrinol Metab 2009;94(9):3132-54.
6. Injustice at Every Turn: A Report of the National Transgender Discrimination Survey. Washington: National Center for Transgender Equality and National Gay and Lesbian Task Force, 2011.
Transgender and gender-expansive youth face many barriers to health care. (Gender-expansive youth are defined as “youth who do not identify with traditional gender roles but are otherwise not confined to one gender narrative or experience.”) Although some of these youth may be fortunate to have a supportive family and access to health care providers proficient in transgender health care, they still face difficulties in having their insurance cover transgender-related services. This is not an impossible task, but it is a constant struggle for many clinicians.
In this column, I will provide some tips and strategies to help clinicians get insurance companies to cover these critical services. However, keep in mind that there is no one-size-fits-all approach to obtaining insurance coverage. In addition, growing uncertainty over the repeal of the Affordable Care Act (ACA) – which was critical in lifting many of the barriers to insurance coverage for transgender individuals – will make this task challenging.
Health insurance is extraordinarily complex. There are multiple private and public plans that vary in the services they cover. This variation is state dependent. And even within states, there is additional variability. Most health insurance plans are purchased by employers, and employers have a choice of what can be covered in their health plans. So even though an insurance company may state that it covers transgender-related services, the patient’s employer may pay for a plan that doesn’t cover such services. The only way to be sure whether a patient’s insurance will cover transgender-related services or not is to contact the insurance provider directly, but with extremely busy schedules and heavy patient loads, this is easier said than done. It would be helpful to have a social worker perform this task, but even having a social worker can be a luxury for some clinics.
The ACA made it easier for transgender individuals to obtain insurance coverage. Three years ago, the U.S. Department of Health and Human Services stated that Medicare’s longstanding exclusion of “transsexual surgical procedures” was no longer valid.1 Although it did not universally ban transgender exclusion policies, it did allow individual states to do so. Thirteen states have explicit policies that ban exclusions of transgender-related services in both private insurance and in Medicaid, and an additional five states have some policies that discourage such practices.2 This allowed some insurance providers and state Medicaid plans to offer coverage of transgender-related services.
Another challenge in obtaining insurance coverage for transgender and gender-expansive youth is claims denial for sex-specific procedures. For example, if a transwoman is designated as “male” in the electronic medical record and requires a breast ultrasound, the insurance company may automatically reject this claim because this procedure is covered for bodies designated as “female.” If the patient’s insurance plan covers transgender-related services, the clinic can notify the insurance company that the patient is transgender; if the patient’s plan does not, then the clinic will need to appeal to the insurance provider. Alternatively, for clinics associated with federally-funded institutions (e.g., most hospitals), the clinician can use Condition Code 45 in the billing to override the sex mismatch, although not all hospitals have implemented this code.3
1. Patient’s identifying information. Usually the patient’s name and date of birth is sufficient. Clinicians should use the patient’s preferred name in the letter, but provide the insurance or legal name of the patient so that the insurance provider can locate the patient’s records.
2. Result of a psychosocial evaluation and diagnosis (if any). Many insurance providers are looking specifically for the gender dysphoria diagnosis.
3. The duration of the referring health professional’s relationship with the patient, which includes the type of evaluation and therapy or counseling (e.g., cognitive behavior therapy or gender coaching).
4. An explanation that the criteria (usually from the World Professional Association for Transgender Health standard of care4 or the Endocrine Society Guidelines titled Endocrine Treatment of Transsexual Persons5) for hormone therapy have been met, and a brief description of the clinical rationale for supporting the client’s request for hormone therapy.
5. A statement that informed consent has been obtained from the patient (or parental permission if the patient is younger than 18 years).
6. A statement that the referring health professional is available for coordination of care.
If the clinician fails to convince the insurance provider of the necessity of covering transgender-related services, the patient still can pay out of pocket. Some hormones can be affordable to certain patients. In the state of Pennsylvania, for example, a 10-mL vial of testosterone can cost anywhere from $60 to $80, and may generally last anywhere from 10 weeks to a year, depending on dosage. Nevertheless, these costs still may be prohibitive for many transgender youth. Many are chronically unemployed or underemployed, or struggle with homelessness.6 Some transgender youth have to the face the excruciatingly difficult choice between having something to eat for the day or living another day with gender dysphoria.
Clinicians should work very hard to make sure that their transgender and gender-expansive patients obtain the care they need. The above strategies may help navigate the complex insurance system. However, insurance policies vary by state, and anti-trans discrimination creates additional barriers to health care. Therefore, clinicians who take care of transgender youth also should advocate for policies that protect these patients from discrimination, and they should advocate for policies that expand medical coverage for this vulnerable population.
Resources
• The Human Rights Campaign keeps a list of insurance plans that cover transgender-related services, but this list is far from comprehensive.
• Healthcare.gov provides some guidance on how to obtain coverage and navigate the insurance system for transgender individuals.
• UCSF Center of Excellence for Transgender Health provides some excellent resources and guidance on obtaining insurance coverage for transgender individuals.
References
1. LGBT Health 2014;1(4):256-8.
2. Map: State Health Insurance Rules: National Center for Transgender Equality, 2016 [Available from: www.transequality.org/issues/resources/map-state-health-insurance-rules].
3. Health insurance coverage issues for transgender people in the United States: University of California, San Fransisco Center of Excellence for Transgender Health, 2017 [Available from: http://transhealth.ucsf.edu/trans?page=guidelines-insurance].
4. International Journal of Transgenderism 2012;13(4):165-232.
5. J Clin Endocrinol Metab 2009;94(9):3132-54.
6. Injustice at Every Turn: A Report of the National Transgender Discrimination Survey. Washington: National Center for Transgender Equality and National Gay and Lesbian Task Force, 2011.
Transgender and gender-expansive youth face many barriers to health care. (Gender-expansive youth are defined as “youth who do not identify with traditional gender roles but are otherwise not confined to one gender narrative or experience.”) Although some of these youth may be fortunate to have a supportive family and access to health care providers proficient in transgender health care, they still face difficulties in having their insurance cover transgender-related services. This is not an impossible task, but it is a constant struggle for many clinicians.
In this column, I will provide some tips and strategies to help clinicians get insurance companies to cover these critical services. However, keep in mind that there is no one-size-fits-all approach to obtaining insurance coverage. In addition, growing uncertainty over the repeal of the Affordable Care Act (ACA) – which was critical in lifting many of the barriers to insurance coverage for transgender individuals – will make this task challenging.
Health insurance is extraordinarily complex. There are multiple private and public plans that vary in the services they cover. This variation is state dependent. And even within states, there is additional variability. Most health insurance plans are purchased by employers, and employers have a choice of what can be covered in their health plans. So even though an insurance company may state that it covers transgender-related services, the patient’s employer may pay for a plan that doesn’t cover such services. The only way to be sure whether a patient’s insurance will cover transgender-related services or not is to contact the insurance provider directly, but with extremely busy schedules and heavy patient loads, this is easier said than done. It would be helpful to have a social worker perform this task, but even having a social worker can be a luxury for some clinics.
The ACA made it easier for transgender individuals to obtain insurance coverage. Three years ago, the U.S. Department of Health and Human Services stated that Medicare’s longstanding exclusion of “transsexual surgical procedures” was no longer valid.1 Although it did not universally ban transgender exclusion policies, it did allow individual states to do so. Thirteen states have explicit policies that ban exclusions of transgender-related services in both private insurance and in Medicaid, and an additional five states have some policies that discourage such practices.2 This allowed some insurance providers and state Medicaid plans to offer coverage of transgender-related services.
Another challenge in obtaining insurance coverage for transgender and gender-expansive youth is claims denial for sex-specific procedures. For example, if a transwoman is designated as “male” in the electronic medical record and requires a breast ultrasound, the insurance company may automatically reject this claim because this procedure is covered for bodies designated as “female.” If the patient’s insurance plan covers transgender-related services, the clinic can notify the insurance company that the patient is transgender; if the patient’s plan does not, then the clinic will need to appeal to the insurance provider. Alternatively, for clinics associated with federally-funded institutions (e.g., most hospitals), the clinician can use Condition Code 45 in the billing to override the sex mismatch, although not all hospitals have implemented this code.3
1. Patient’s identifying information. Usually the patient’s name and date of birth is sufficient. Clinicians should use the patient’s preferred name in the letter, but provide the insurance or legal name of the patient so that the insurance provider can locate the patient’s records.
2. Result of a psychosocial evaluation and diagnosis (if any). Many insurance providers are looking specifically for the gender dysphoria diagnosis.
3. The duration of the referring health professional’s relationship with the patient, which includes the type of evaluation and therapy or counseling (e.g., cognitive behavior therapy or gender coaching).
4. An explanation that the criteria (usually from the World Professional Association for Transgender Health standard of care4 or the Endocrine Society Guidelines titled Endocrine Treatment of Transsexual Persons5) for hormone therapy have been met, and a brief description of the clinical rationale for supporting the client’s request for hormone therapy.
5. A statement that informed consent has been obtained from the patient (or parental permission if the patient is younger than 18 years).
6. A statement that the referring health professional is available for coordination of care.
If the clinician fails to convince the insurance provider of the necessity of covering transgender-related services, the patient still can pay out of pocket. Some hormones can be affordable to certain patients. In the state of Pennsylvania, for example, a 10-mL vial of testosterone can cost anywhere from $60 to $80, and may generally last anywhere from 10 weeks to a year, depending on dosage. Nevertheless, these costs still may be prohibitive for many transgender youth. Many are chronically unemployed or underemployed, or struggle with homelessness.6 Some transgender youth have to the face the excruciatingly difficult choice between having something to eat for the day or living another day with gender dysphoria.
Clinicians should work very hard to make sure that their transgender and gender-expansive patients obtain the care they need. The above strategies may help navigate the complex insurance system. However, insurance policies vary by state, and anti-trans discrimination creates additional barriers to health care. Therefore, clinicians who take care of transgender youth also should advocate for policies that protect these patients from discrimination, and they should advocate for policies that expand medical coverage for this vulnerable population.
Resources
• The Human Rights Campaign keeps a list of insurance plans that cover transgender-related services, but this list is far from comprehensive.
• Healthcare.gov provides some guidance on how to obtain coverage and navigate the insurance system for transgender individuals.
• UCSF Center of Excellence for Transgender Health provides some excellent resources and guidance on obtaining insurance coverage for transgender individuals.
References
1. LGBT Health 2014;1(4):256-8.
2. Map: State Health Insurance Rules: National Center for Transgender Equality, 2016 [Available from: www.transequality.org/issues/resources/map-state-health-insurance-rules].
3. Health insurance coverage issues for transgender people in the United States: University of California, San Fransisco Center of Excellence for Transgender Health, 2017 [Available from: http://transhealth.ucsf.edu/trans?page=guidelines-insurance].
4. International Journal of Transgenderism 2012;13(4):165-232.
5. J Clin Endocrinol Metab 2009;94(9):3132-54.
6. Injustice at Every Turn: A Report of the National Transgender Discrimination Survey. Washington: National Center for Transgender Equality and National Gay and Lesbian Task Force, 2011.
Why extended release metformin?
“TREATING POLYCYSTIC OVARY SYNDROME: START USING DUAL MEDICAL THERAPY”
ROBERT L. BARBIERI, MD (EDITORIAL; APRIL 2017)
Why extended release metformin?
I read with interest Dr. Barbieri’s editorial on polycystic ovary syndrome. It left me wondering: Is there a metabolic or pharmacologic reason why you give metformin XR 1,500 mg with dinner instead of 750 mg orally twice per day?
Marcelo Andreoli, MD
Vienna, Virginia
Dr. Barbieri responds
I thank Dr. Andreoli for the important clinical question about one-time or multiple dosing of metformin. To improve patient adherence with metformin treatment, I think once-daily dosing at dinner with an extended-release formulation is more convenient than twice-daily dosing with immediate-release metformin. Following ingestion of immediate- or extended-release metformin, peak metformin blood concentrations are achieved after 2 and 7 hours, respectively.1 There is some evidence that extended-release metformin has fewer gastrointestinal (GI) adverse effects than immediate-release metformin.2 In one study, the reported rates of GI adverse effects were 29% versus 39% with extended-release and immediate-release formulations, respectively.2
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Ali S, Fonseca V. Overview of metformin: special focus on metformin extended release. Expert Opin Pharmacother. 2012;13(12):1797–1805.
- Fujioka K, Pans M, Joyal S. Glycemic control in patients with type 2 diabetes mellitus switched from twice-daily immediate-release metformin to a once-daily extended-release formulation. Clin Ther. 2003;25(2):515–529.
“TREATING POLYCYSTIC OVARY SYNDROME: START USING DUAL MEDICAL THERAPY”
ROBERT L. BARBIERI, MD (EDITORIAL; APRIL 2017)
Why extended release metformin?
I read with interest Dr. Barbieri’s editorial on polycystic ovary syndrome. It left me wondering: Is there a metabolic or pharmacologic reason why you give metformin XR 1,500 mg with dinner instead of 750 mg orally twice per day?
Marcelo Andreoli, MD
Vienna, Virginia
Dr. Barbieri responds
I thank Dr. Andreoli for the important clinical question about one-time or multiple dosing of metformin. To improve patient adherence with metformin treatment, I think once-daily dosing at dinner with an extended-release formulation is more convenient than twice-daily dosing with immediate-release metformin. Following ingestion of immediate- or extended-release metformin, peak metformin blood concentrations are achieved after 2 and 7 hours, respectively.1 There is some evidence that extended-release metformin has fewer gastrointestinal (GI) adverse effects than immediate-release metformin.2 In one study, the reported rates of GI adverse effects were 29% versus 39% with extended-release and immediate-release formulations, respectively.2
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
“TREATING POLYCYSTIC OVARY SYNDROME: START USING DUAL MEDICAL THERAPY”
ROBERT L. BARBIERI, MD (EDITORIAL; APRIL 2017)
Why extended release metformin?
I read with interest Dr. Barbieri’s editorial on polycystic ovary syndrome. It left me wondering: Is there a metabolic or pharmacologic reason why you give metformin XR 1,500 mg with dinner instead of 750 mg orally twice per day?
Marcelo Andreoli, MD
Vienna, Virginia
Dr. Barbieri responds
I thank Dr. Andreoli for the important clinical question about one-time or multiple dosing of metformin. To improve patient adherence with metformin treatment, I think once-daily dosing at dinner with an extended-release formulation is more convenient than twice-daily dosing with immediate-release metformin. Following ingestion of immediate- or extended-release metformin, peak metformin blood concentrations are achieved after 2 and 7 hours, respectively.1 There is some evidence that extended-release metformin has fewer gastrointestinal (GI) adverse effects than immediate-release metformin.2 In one study, the reported rates of GI adverse effects were 29% versus 39% with extended-release and immediate-release formulations, respectively.2
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Ali S, Fonseca V. Overview of metformin: special focus on metformin extended release. Expert Opin Pharmacother. 2012;13(12):1797–1805.
- Fujioka K, Pans M, Joyal S. Glycemic control in patients with type 2 diabetes mellitus switched from twice-daily immediate-release metformin to a once-daily extended-release formulation. Clin Ther. 2003;25(2):515–529.
- Ali S, Fonseca V. Overview of metformin: special focus on metformin extended release. Expert Opin Pharmacother. 2012;13(12):1797–1805.
- Fujioka K, Pans M, Joyal S. Glycemic control in patients with type 2 diabetes mellitus switched from twice-daily immediate-release metformin to a once-daily extended-release formulation. Clin Ther. 2003;25(2):515–529.

Look for symptoms of IBS, PCOS, and PMS
“WHY ARE THERE DELAYS IN THE DIAGNOSIS OF ENDOMETRIOSIS?”
ROBERT L. BARBIERI, MD (EDITORIAL; MARCH 2017)
Look for symptoms of IBS, PCOS, and PMS
I practiced reproductive endocrinology for 40 years and saw too many patients whose endometriosis had been ignored or undertreated. I found that the initial suspicion for the disease could be discovered by looking for symptoms of 3 comorbidities: irritable bowel syndrome, polycystic ovary syndrome, and premenstrual syndrome.
Wilbur (Dub) Howard, MD
Dallas, Texas
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
“WHY ARE THERE DELAYS IN THE DIAGNOSIS OF ENDOMETRIOSIS?”
ROBERT L. BARBIERI, MD (EDITORIAL; MARCH 2017)
Look for symptoms of IBS, PCOS, and PMS
I practiced reproductive endocrinology for 40 years and saw too many patients whose endometriosis had been ignored or undertreated. I found that the initial suspicion for the disease could be discovered by looking for symptoms of 3 comorbidities: irritable bowel syndrome, polycystic ovary syndrome, and premenstrual syndrome.
Wilbur (Dub) Howard, MD
Dallas, Texas
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
“WHY ARE THERE DELAYS IN THE DIAGNOSIS OF ENDOMETRIOSIS?”
ROBERT L. BARBIERI, MD (EDITORIAL; MARCH 2017)
Look for symptoms of IBS, PCOS, and PMS
I practiced reproductive endocrinology for 40 years and saw too many patients whose endometriosis had been ignored or undertreated. I found that the initial suspicion for the disease could be discovered by looking for symptoms of 3 comorbidities: irritable bowel syndrome, polycystic ovary syndrome, and premenstrual syndrome.
Wilbur (Dub) Howard, MD
Dallas, Texas
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
The fallopian tube should have been removed
OBIANUJU SANDRA MADUEKE-LAVEAUX, MD, MPH; BETH W. RACKOW, MD; AND ARNOLD P. ADVINCULA, MD (VIDEO; JANUARY 2017)
The fallopian tube should have been removed
I watched the video by Dr. Advincula and colleagues and as always was impressed with the surgical skills demonstrated. While the robot-assisted approach is quite nice, this case could have been accomplished with only three 5-mm lower abdominal port sites and traditional straight-stick laparoscopic methods. The cosmetic benefit to a 15-year-old patient of this alternative should have been considered.
More importantly, the fallopian tube separated from the rudimentary horn should have been removed. Leaving the right tube in situ exposes the patient to the possibility of a future ectopic pregnancy in that tube and provides no benefit to the patient.
David L. Zisow, MD
Baltimore, Maryland
Dr. Advincula and team respond
We appreciate Dr. Zisow’s perspective. As is known, tool selection is based on surgeon preference. Inherent to this point, a discussion about route of surgery, and any implications it would have, such as cosmesis, was had. Cosmesis was not an issue with this patient, and she was quite pleased with her cosmetic outcome.
We also discussed preoperatively, among our team and with the patient, the right fallopian tube. Although removal would have been optimal, there was concern intraoperatively of possible compromise to the ovary. Hence, a decision was made to forego removal particularly in light of the extremely rare risk of transperitoneal migration of spermatozoa weighed against the risk of compromising a perfectly healthy ovary in a 15-year-old woman.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
OBIANUJU SANDRA MADUEKE-LAVEAUX, MD, MPH; BETH W. RACKOW, MD; AND ARNOLD P. ADVINCULA, MD (VIDEO; JANUARY 2017)
The fallopian tube should have been removed
I watched the video by Dr. Advincula and colleagues and as always was impressed with the surgical skills demonstrated. While the robot-assisted approach is quite nice, this case could have been accomplished with only three 5-mm lower abdominal port sites and traditional straight-stick laparoscopic methods. The cosmetic benefit to a 15-year-old patient of this alternative should have been considered.
More importantly, the fallopian tube separated from the rudimentary horn should have been removed. Leaving the right tube in situ exposes the patient to the possibility of a future ectopic pregnancy in that tube and provides no benefit to the patient.
David L. Zisow, MD
Baltimore, Maryland
Dr. Advincula and team respond
We appreciate Dr. Zisow’s perspective. As is known, tool selection is based on surgeon preference. Inherent to this point, a discussion about route of surgery, and any implications it would have, such as cosmesis, was had. Cosmesis was not an issue with this patient, and she was quite pleased with her cosmetic outcome.
We also discussed preoperatively, among our team and with the patient, the right fallopian tube. Although removal would have been optimal, there was concern intraoperatively of possible compromise to the ovary. Hence, a decision was made to forego removal particularly in light of the extremely rare risk of transperitoneal migration of spermatozoa weighed against the risk of compromising a perfectly healthy ovary in a 15-year-old woman.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
OBIANUJU SANDRA MADUEKE-LAVEAUX, MD, MPH; BETH W. RACKOW, MD; AND ARNOLD P. ADVINCULA, MD (VIDEO; JANUARY 2017)
The fallopian tube should have been removed
I watched the video by Dr. Advincula and colleagues and as always was impressed with the surgical skills demonstrated. While the robot-assisted approach is quite nice, this case could have been accomplished with only three 5-mm lower abdominal port sites and traditional straight-stick laparoscopic methods. The cosmetic benefit to a 15-year-old patient of this alternative should have been considered.
More importantly, the fallopian tube separated from the rudimentary horn should have been removed. Leaving the right tube in situ exposes the patient to the possibility of a future ectopic pregnancy in that tube and provides no benefit to the patient.
David L. Zisow, MD
Baltimore, Maryland
Dr. Advincula and team respond
We appreciate Dr. Zisow’s perspective. As is known, tool selection is based on surgeon preference. Inherent to this point, a discussion about route of surgery, and any implications it would have, such as cosmesis, was had. Cosmesis was not an issue with this patient, and she was quite pleased with her cosmetic outcome.
We also discussed preoperatively, among our team and with the patient, the right fallopian tube. Although removal would have been optimal, there was concern intraoperatively of possible compromise to the ovary. Hence, a decision was made to forego removal particularly in light of the extremely rare risk of transperitoneal migration of spermatozoa weighed against the risk of compromising a perfectly healthy ovary in a 15-year-old woman.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
VIDEO: Does biologic immunogenicity matter in daily practice?
MADRID – Measuring the formation of antibodies against biologic agents has no real value in daily practice as their presence or absence does not really change how patients are likely to be treated, Johannes W.J. Bijlsma, MD, observed at the European Congress of Rheumatology.
Consider a female patient who is 59 years old, diagnosed with rheumatoid arthritis (RA) in 2014, he said. She was being treated with methotrexate at a dose of 20 mg with additional glucocorticoids, initially given at a dose of 10 mg, reduced to 5 mg after 2 years, and then stopped. The patient experiences a disease flare, however, and for various other reasons is given a tumor necrosis factor inhibitor (TNFi). She does well initially but then has another flare, so would there be any point of measuring anti-drug antibodies (ADAbs) as this point? Not really, Dr. Bijlsma suggested, as the same decision to change the biologic agent would probably result if ADAbs were detected or not.
“If I do not measure them, I decide to change the biological. If I measure them and they are present, I change the biological, and if they are absent, I still change the biological,” said Dr. Bijlsma, professor and head of the department of rheumatology and clinical immunology at University Medical Center Utrecht (the Netherlands).
Following the European League Against Rheumatism (EULAR) recommendations for biologic disease-modifying antirheumatic drug (bDMARD) use (Ann Rheum Dis. 2017;76:960-77) would then mean that the first bDMARD, in this case adalimumab (Humira), would be replaced by another biologic with a different mechanism of action or a second TNFi.
“The immune response is always there,” Dr. Bijlsma said. It does not matter what or how it is administered, introducing any foreign protein, humanized or not, will instigate some kind of immune reaction, he said. The extent to which an immune reaction is raised might vary between biologic agents, but it will be there. He cited a review paper (Rheumatology [Oxford]. 2016;55:210-20) showing that the mean estimated occurrence of ADAbs in patients with RA ranges from 0.6% with the interleukin-6-targeting agent tocilizumab (Actemra) to 30% with infliximab (Remicade).
Measuring the level of ADAbs becomes problematic when considering that different biologics will induce different levels of immune response. The level of detection also will be dependent on which of three current types of assays are used. In addition, “humanization of biological agents is not the key point in preventing anti-drug antibodies,” Dr. Bijlsma said, pointing out that the prevalence of ADAbs against adalimumab did not appear to by any lower than ADAbs against infliximab.
Preventing ADAbs can be achieved by co-administering methotrexate or alternating the treatment schedule, Dr. Bijlsma said. Treatment with methotrexate, which is usually continued when patients start a biologic, “diminishes the immune response,” he noted. Indeed, while 50% of patients who are not treated with this conventional DMARD develop ADAbs, only 14%-35% develop them while taking methotrexate, depending on the dose used.
It is likely to be more useful in clinical practice to measure individual patients’ drug trough levels than to measure ADAb levels, he suggested, with dosing continued or adjusted accordingly for each patient. Using drug trough levels to personalize adalimumab treatment has been tested (Ann Rheum Dis. 2015;74:361-8) using a theoretical algorithm based on whether patients achieve a EULAR response at 6 months. If they do achieve a EULAR response and drug trough levels are between 5 and 12 mg/L or greater than 12 mg/L, then adalimumab treatment should continue. However, if the trough levels fall below 5 mg/L, there is probably no point in continuing treatment and this TNFi should be stopped. If patients do not respond and drug testing shows a trough level above 5 mg/L, then a switch to infliximab might be advantageous, while a trough level below this threshold could indicated that a TNFi with a lower immunogenic potential such as etanercept might be a better choice.
Using drug trough levels is still very much research based right now and is not ready for clinical practice just yet, but the theory is that it could help decide if patients should continue, stop, or perhaps switch their biologic, Dr. Bijlsma said.
Dr. Bijlsma spoke about these issues in a video interview at the congress.
Dr. Bijlsma has worked with many of the pharmaceutical companies that produce biologic agents for the management of rheumatic diseases but had no specific disclosures in relation to his comments.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
MADRID – Measuring the formation of antibodies against biologic agents has no real value in daily practice as their presence or absence does not really change how patients are likely to be treated, Johannes W.J. Bijlsma, MD, observed at the European Congress of Rheumatology.
Consider a female patient who is 59 years old, diagnosed with rheumatoid arthritis (RA) in 2014, he said. She was being treated with methotrexate at a dose of 20 mg with additional glucocorticoids, initially given at a dose of 10 mg, reduced to 5 mg after 2 years, and then stopped. The patient experiences a disease flare, however, and for various other reasons is given a tumor necrosis factor inhibitor (TNFi). She does well initially but then has another flare, so would there be any point of measuring anti-drug antibodies (ADAbs) as this point? Not really, Dr. Bijlsma suggested, as the same decision to change the biologic agent would probably result if ADAbs were detected or not.
“If I do not measure them, I decide to change the biological. If I measure them and they are present, I change the biological, and if they are absent, I still change the biological,” said Dr. Bijlsma, professor and head of the department of rheumatology and clinical immunology at University Medical Center Utrecht (the Netherlands).
Following the European League Against Rheumatism (EULAR) recommendations for biologic disease-modifying antirheumatic drug (bDMARD) use (Ann Rheum Dis. 2017;76:960-77) would then mean that the first bDMARD, in this case adalimumab (Humira), would be replaced by another biologic with a different mechanism of action or a second TNFi.
“The immune response is always there,” Dr. Bijlsma said. It does not matter what or how it is administered, introducing any foreign protein, humanized or not, will instigate some kind of immune reaction, he said. The extent to which an immune reaction is raised might vary between biologic agents, but it will be there. He cited a review paper (Rheumatology [Oxford]. 2016;55:210-20) showing that the mean estimated occurrence of ADAbs in patients with RA ranges from 0.6% with the interleukin-6-targeting agent tocilizumab (Actemra) to 30% with infliximab (Remicade).
Measuring the level of ADAbs becomes problematic when considering that different biologics will induce different levels of immune response. The level of detection also will be dependent on which of three current types of assays are used. In addition, “humanization of biological agents is not the key point in preventing anti-drug antibodies,” Dr. Bijlsma said, pointing out that the prevalence of ADAbs against adalimumab did not appear to by any lower than ADAbs against infliximab.
Preventing ADAbs can be achieved by co-administering methotrexate or alternating the treatment schedule, Dr. Bijlsma said. Treatment with methotrexate, which is usually continued when patients start a biologic, “diminishes the immune response,” he noted. Indeed, while 50% of patients who are not treated with this conventional DMARD develop ADAbs, only 14%-35% develop them while taking methotrexate, depending on the dose used.
It is likely to be more useful in clinical practice to measure individual patients’ drug trough levels than to measure ADAb levels, he suggested, with dosing continued or adjusted accordingly for each patient. Using drug trough levels to personalize adalimumab treatment has been tested (Ann Rheum Dis. 2015;74:361-8) using a theoretical algorithm based on whether patients achieve a EULAR response at 6 months. If they do achieve a EULAR response and drug trough levels are between 5 and 12 mg/L or greater than 12 mg/L, then adalimumab treatment should continue. However, if the trough levels fall below 5 mg/L, there is probably no point in continuing treatment and this TNFi should be stopped. If patients do not respond and drug testing shows a trough level above 5 mg/L, then a switch to infliximab might be advantageous, while a trough level below this threshold could indicated that a TNFi with a lower immunogenic potential such as etanercept might be a better choice.
Using drug trough levels is still very much research based right now and is not ready for clinical practice just yet, but the theory is that it could help decide if patients should continue, stop, or perhaps switch their biologic, Dr. Bijlsma said.
Dr. Bijlsma spoke about these issues in a video interview at the congress.
Dr. Bijlsma has worked with many of the pharmaceutical companies that produce biologic agents for the management of rheumatic diseases but had no specific disclosures in relation to his comments.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
MADRID – Measuring the formation of antibodies against biologic agents has no real value in daily practice as their presence or absence does not really change how patients are likely to be treated, Johannes W.J. Bijlsma, MD, observed at the European Congress of Rheumatology.
Consider a female patient who is 59 years old, diagnosed with rheumatoid arthritis (RA) in 2014, he said. She was being treated with methotrexate at a dose of 20 mg with additional glucocorticoids, initially given at a dose of 10 mg, reduced to 5 mg after 2 years, and then stopped. The patient experiences a disease flare, however, and for various other reasons is given a tumor necrosis factor inhibitor (TNFi). She does well initially but then has another flare, so would there be any point of measuring anti-drug antibodies (ADAbs) as this point? Not really, Dr. Bijlsma suggested, as the same decision to change the biologic agent would probably result if ADAbs were detected or not.
“If I do not measure them, I decide to change the biological. If I measure them and they are present, I change the biological, and if they are absent, I still change the biological,” said Dr. Bijlsma, professor and head of the department of rheumatology and clinical immunology at University Medical Center Utrecht (the Netherlands).
Following the European League Against Rheumatism (EULAR) recommendations for biologic disease-modifying antirheumatic drug (bDMARD) use (Ann Rheum Dis. 2017;76:960-77) would then mean that the first bDMARD, in this case adalimumab (Humira), would be replaced by another biologic with a different mechanism of action or a second TNFi.
“The immune response is always there,” Dr. Bijlsma said. It does not matter what or how it is administered, introducing any foreign protein, humanized or not, will instigate some kind of immune reaction, he said. The extent to which an immune reaction is raised might vary between biologic agents, but it will be there. He cited a review paper (Rheumatology [Oxford]. 2016;55:210-20) showing that the mean estimated occurrence of ADAbs in patients with RA ranges from 0.6% with the interleukin-6-targeting agent tocilizumab (Actemra) to 30% with infliximab (Remicade).
Measuring the level of ADAbs becomes problematic when considering that different biologics will induce different levels of immune response. The level of detection also will be dependent on which of three current types of assays are used. In addition, “humanization of biological agents is not the key point in preventing anti-drug antibodies,” Dr. Bijlsma said, pointing out that the prevalence of ADAbs against adalimumab did not appear to by any lower than ADAbs against infliximab.
Preventing ADAbs can be achieved by co-administering methotrexate or alternating the treatment schedule, Dr. Bijlsma said. Treatment with methotrexate, which is usually continued when patients start a biologic, “diminishes the immune response,” he noted. Indeed, while 50% of patients who are not treated with this conventional DMARD develop ADAbs, only 14%-35% develop them while taking methotrexate, depending on the dose used.
It is likely to be more useful in clinical practice to measure individual patients’ drug trough levels than to measure ADAb levels, he suggested, with dosing continued or adjusted accordingly for each patient. Using drug trough levels to personalize adalimumab treatment has been tested (Ann Rheum Dis. 2015;74:361-8) using a theoretical algorithm based on whether patients achieve a EULAR response at 6 months. If they do achieve a EULAR response and drug trough levels are between 5 and 12 mg/L or greater than 12 mg/L, then adalimumab treatment should continue. However, if the trough levels fall below 5 mg/L, there is probably no point in continuing treatment and this TNFi should be stopped. If patients do not respond and drug testing shows a trough level above 5 mg/L, then a switch to infliximab might be advantageous, while a trough level below this threshold could indicated that a TNFi with a lower immunogenic potential such as etanercept might be a better choice.
Using drug trough levels is still very much research based right now and is not ready for clinical practice just yet, but the theory is that it could help decide if patients should continue, stop, or perhaps switch their biologic, Dr. Bijlsma said.
Dr. Bijlsma spoke about these issues in a video interview at the congress.
Dr. Bijlsma has worked with many of the pharmaceutical companies that produce biologic agents for the management of rheumatic diseases but had no specific disclosures in relation to his comments.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
EXPERT ANALYSIS FROM THE EULAR 2017 CONGRESS

Hysteroscopy equipment too expensive for employed or small-group practitioners
“2017 UPDATE ON ABNORMAL UTERINE BLEEDING”
HOWARD T. SHARP, MD, AND MARISSA ADELMAN, MD (APRIL 2017)
Hysteroscopy equipment too expensive for employed or small-group practitioners
I could not agree more with Drs. Sharp and Adelman that diagnostic hysteroscopy should be performed in the office whenever possible. However, as a solo gynecologist in private practice, I could not afford or justify the cost of purchasing the equipment as well as its care and maintenance. Sometimes I was able to bring a third-party vendor to provide the equipment and a technician so that I could perform a diagnostic hysteroscopy in my office when I did an ablation with my own Thermachoice equipment and balloon system.
The hysteroscopy was bundled/required for the Current Procedural Terminology (CPT) code to work in the office. Most of these patients already had undergone an ultrasonography, endometrial biopsy, and some had an outpatient hysteroscopic dilation and curettage under general anesthesia, which did not resolve their bleeding. All of this adds to the cost and increased patient discomfort and inconvenience. Reimbursement for the office procedure was better than when performed at the hospital, and patients avoided $500 to $1,000 copays to the hospital and anesthesiologist.
When I closed my private practice and became employed by the hospital, I proposed that they purchase office hysteroscopy equipment for the other gynecologist and me to share. I continued to perform uterine ablations with my own equipment. Together we performed more than 100 outpatient diagnostic hysteroscopies per year, some with global endometrial ablation. Since there were only 2 gyns, the 2 new hysteroscopy sets they purchased sat in the closet most of the time.
I suggested they “lease” the equipment back to us on a case-by-case basis for office use since they owned and managed our practices. The hospital administration basically saw office procedures as taking away revenue from the hospital and decreasing operating room volume. The patients I treated in the office setting did well, preferred to avoid general anesthesia, and enjoyed the cost savings.
Large ObGyn groups with multiple providers and high volumes can justify the expenses of the equipment, but for those in solo practice or employed by a hospital, it may not be feasible. I sincerely hope that articles focusing on in-office hysteroscopy will open up the discussion to enable and encourage more physicians and hospital administrators to see the advantages of office-based procedures.
Steven R. Moffett, MD
Knoxville, Tennessee
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
“2017 UPDATE ON ABNORMAL UTERINE BLEEDING”
HOWARD T. SHARP, MD, AND MARISSA ADELMAN, MD (APRIL 2017)
Hysteroscopy equipment too expensive for employed or small-group practitioners
I could not agree more with Drs. Sharp and Adelman that diagnostic hysteroscopy should be performed in the office whenever possible. However, as a solo gynecologist in private practice, I could not afford or justify the cost of purchasing the equipment as well as its care and maintenance. Sometimes I was able to bring a third-party vendor to provide the equipment and a technician so that I could perform a diagnostic hysteroscopy in my office when I did an ablation with my own Thermachoice equipment and balloon system.
The hysteroscopy was bundled/required for the Current Procedural Terminology (CPT) code to work in the office. Most of these patients already had undergone an ultrasonography, endometrial biopsy, and some had an outpatient hysteroscopic dilation and curettage under general anesthesia, which did not resolve their bleeding. All of this adds to the cost and increased patient discomfort and inconvenience. Reimbursement for the office procedure was better than when performed at the hospital, and patients avoided $500 to $1,000 copays to the hospital and anesthesiologist.
When I closed my private practice and became employed by the hospital, I proposed that they purchase office hysteroscopy equipment for the other gynecologist and me to share. I continued to perform uterine ablations with my own equipment. Together we performed more than 100 outpatient diagnostic hysteroscopies per year, some with global endometrial ablation. Since there were only 2 gyns, the 2 new hysteroscopy sets they purchased sat in the closet most of the time.
I suggested they “lease” the equipment back to us on a case-by-case basis for office use since they owned and managed our practices. The hospital administration basically saw office procedures as taking away revenue from the hospital and decreasing operating room volume. The patients I treated in the office setting did well, preferred to avoid general anesthesia, and enjoyed the cost savings.
Large ObGyn groups with multiple providers and high volumes can justify the expenses of the equipment, but for those in solo practice or employed by a hospital, it may not be feasible. I sincerely hope that articles focusing on in-office hysteroscopy will open up the discussion to enable and encourage more physicians and hospital administrators to see the advantages of office-based procedures.
Steven R. Moffett, MD
Knoxville, Tennessee
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
“2017 UPDATE ON ABNORMAL UTERINE BLEEDING”
HOWARD T. SHARP, MD, AND MARISSA ADELMAN, MD (APRIL 2017)
Hysteroscopy equipment too expensive for employed or small-group practitioners
I could not agree more with Drs. Sharp and Adelman that diagnostic hysteroscopy should be performed in the office whenever possible. However, as a solo gynecologist in private practice, I could not afford or justify the cost of purchasing the equipment as well as its care and maintenance. Sometimes I was able to bring a third-party vendor to provide the equipment and a technician so that I could perform a diagnostic hysteroscopy in my office when I did an ablation with my own Thermachoice equipment and balloon system.
The hysteroscopy was bundled/required for the Current Procedural Terminology (CPT) code to work in the office. Most of these patients already had undergone an ultrasonography, endometrial biopsy, and some had an outpatient hysteroscopic dilation and curettage under general anesthesia, which did not resolve their bleeding. All of this adds to the cost and increased patient discomfort and inconvenience. Reimbursement for the office procedure was better than when performed at the hospital, and patients avoided $500 to $1,000 copays to the hospital and anesthesiologist.
When I closed my private practice and became employed by the hospital, I proposed that they purchase office hysteroscopy equipment for the other gynecologist and me to share. I continued to perform uterine ablations with my own equipment. Together we performed more than 100 outpatient diagnostic hysteroscopies per year, some with global endometrial ablation. Since there were only 2 gyns, the 2 new hysteroscopy sets they purchased sat in the closet most of the time.
I suggested they “lease” the equipment back to us on a case-by-case basis for office use since they owned and managed our practices. The hospital administration basically saw office procedures as taking away revenue from the hospital and decreasing operating room volume. The patients I treated in the office setting did well, preferred to avoid general anesthesia, and enjoyed the cost savings.
Large ObGyn groups with multiple providers and high volumes can justify the expenses of the equipment, but for those in solo practice or employed by a hospital, it may not be feasible. I sincerely hope that articles focusing on in-office hysteroscopy will open up the discussion to enable and encourage more physicians and hospital administrators to see the advantages of office-based procedures.
Steven R. Moffett, MD
Knoxville, Tennessee
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.