User login
Blood vessels injured during trocar insertion: $8.7M verdict
Blood vessels injured during trocar insertion: $8.7M verdict
PATIENT’S CLAIM:
The resident was negligent in performing trocar insertion during laparoscopic surgery by inserting the trocar too far into the abdomen. The attending ObGyn did not supervise the resident properly. There is nothing in the patient's medical records to indicate that she had abnormal anatomy. The woman's life is in turmoil after what was supposed to be a routine procedure.
DEFENDANTS' DEFENSE:
There was no negligence. The patient's anatomy was abnormal, making the risk of surgery higher. The injury is a known complication of laparoscopic surgery.
VERDICT:
An $8,718,848 Illinois verdict was returned.
Related article:
How to avoid major vessel injury during gynecologic laparoscopy
Wrong fallopian tube transected: $1.8M award
A 28-year-old woman underwent an appendectomy. During the operation, the surgeon saw an abscess on the patient's right fallopian tube and called in an ObGyn to remove the abscess. While doing so, the ObGyn transected the left fallopian tube. Both fallopian tubes were removed.
PATIENT’S CLAIM:
The surgeon did not tell the ObGyn which fallopian tube was abscessed and therefore the ObGyn operated on the wrong tube. In addition, the surgeon failed to obtain informed consent for bilateral salpingectomy. The patient is now unable to conceive without assisted reproductive treatment.
PHYSICIAN’S DEFENSE:
The surgeon admitted his mistakes but disputed the informed consent claim. The patient probably would not have been able to conceive naturally due to the infection.
VERDICT:
A $1.8 million Connecticut verdict was returned.
Related article:
Elective laparoscopic appendectomy in gynecologic surgery: When, why, and how
Complications after vaginal hysterectomy
A woman underwent laparoscopic vaginal hysterectomy and bilateral salpingo-oophorectomy with anterior and posterior repair using mesh in August 2010. Shortly after surgery, the patient reported vaginal discharge with pain and bleeding. She was treated with antibiotics. Results of a CT scan identified the cause of her symptoms as vaginal cuff granulations.
Her pain continued and in June 2011, she underwent vaginal tissue biopsy. After testing revealed the presence of fecal matter, a small-bowel vaginal fistula was identified. She underwent laparoscopic enterectomy, urethral lysis, an omental pedicle flap, and cystoscopy. The mesh had perforated several loops of the small bowel.
In August 2011, the patient reported spinal pain. Magnetic resonance imaging (MRI) revealed a new fluid abscess in a disc extending through the tract anterior to the soft tissue of the pelvis. She underwent intensive antibiotic therapy.
PATIENT’S CLAIM:
The gynecologic surgeon fell below the standard of care in his treatment of her conditions.
PHYSICIAN’S DEFENSE:
The surgeon denied allegations.
VERDICT:
A Nevada defense verdict was returned.
Related article:
Vaginal hysterectomy with basic instrumentation
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Blood vessels injured during trocar insertion: $8.7M verdict
PATIENT’S CLAIM:
The resident was negligent in performing trocar insertion during laparoscopic surgery by inserting the trocar too far into the abdomen. The attending ObGyn did not supervise the resident properly. There is nothing in the patient's medical records to indicate that she had abnormal anatomy. The woman's life is in turmoil after what was supposed to be a routine procedure.
DEFENDANTS' DEFENSE:
There was no negligence. The patient's anatomy was abnormal, making the risk of surgery higher. The injury is a known complication of laparoscopic surgery.
VERDICT:
An $8,718,848 Illinois verdict was returned.
Related article:
How to avoid major vessel injury during gynecologic laparoscopy
Wrong fallopian tube transected: $1.8M award
A 28-year-old woman underwent an appendectomy. During the operation, the surgeon saw an abscess on the patient's right fallopian tube and called in an ObGyn to remove the abscess. While doing so, the ObGyn transected the left fallopian tube. Both fallopian tubes were removed.
PATIENT’S CLAIM:
The surgeon did not tell the ObGyn which fallopian tube was abscessed and therefore the ObGyn operated on the wrong tube. In addition, the surgeon failed to obtain informed consent for bilateral salpingectomy. The patient is now unable to conceive without assisted reproductive treatment.
PHYSICIAN’S DEFENSE:
The surgeon admitted his mistakes but disputed the informed consent claim. The patient probably would not have been able to conceive naturally due to the infection.
VERDICT:
A $1.8 million Connecticut verdict was returned.
Related article:
Elective laparoscopic appendectomy in gynecologic surgery: When, why, and how
Complications after vaginal hysterectomy
A woman underwent laparoscopic vaginal hysterectomy and bilateral salpingo-oophorectomy with anterior and posterior repair using mesh in August 2010. Shortly after surgery, the patient reported vaginal discharge with pain and bleeding. She was treated with antibiotics. Results of a CT scan identified the cause of her symptoms as vaginal cuff granulations.
Her pain continued and in June 2011, she underwent vaginal tissue biopsy. After testing revealed the presence of fecal matter, a small-bowel vaginal fistula was identified. She underwent laparoscopic enterectomy, urethral lysis, an omental pedicle flap, and cystoscopy. The mesh had perforated several loops of the small bowel.
In August 2011, the patient reported spinal pain. Magnetic resonance imaging (MRI) revealed a new fluid abscess in a disc extending through the tract anterior to the soft tissue of the pelvis. She underwent intensive antibiotic therapy.
PATIENT’S CLAIM:
The gynecologic surgeon fell below the standard of care in his treatment of her conditions.
PHYSICIAN’S DEFENSE:
The surgeon denied allegations.
VERDICT:
A Nevada defense verdict was returned.
Related article:
Vaginal hysterectomy with basic instrumentation
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Blood vessels injured during trocar insertion: $8.7M verdict
PATIENT’S CLAIM:
The resident was negligent in performing trocar insertion during laparoscopic surgery by inserting the trocar too far into the abdomen. The attending ObGyn did not supervise the resident properly. There is nothing in the patient's medical records to indicate that she had abnormal anatomy. The woman's life is in turmoil after what was supposed to be a routine procedure.
DEFENDANTS' DEFENSE:
There was no negligence. The patient's anatomy was abnormal, making the risk of surgery higher. The injury is a known complication of laparoscopic surgery.
VERDICT:
An $8,718,848 Illinois verdict was returned.
Related article:
How to avoid major vessel injury during gynecologic laparoscopy
Wrong fallopian tube transected: $1.8M award
A 28-year-old woman underwent an appendectomy. During the operation, the surgeon saw an abscess on the patient's right fallopian tube and called in an ObGyn to remove the abscess. While doing so, the ObGyn transected the left fallopian tube. Both fallopian tubes were removed.
PATIENT’S CLAIM:
The surgeon did not tell the ObGyn which fallopian tube was abscessed and therefore the ObGyn operated on the wrong tube. In addition, the surgeon failed to obtain informed consent for bilateral salpingectomy. The patient is now unable to conceive without assisted reproductive treatment.
PHYSICIAN’S DEFENSE:
The surgeon admitted his mistakes but disputed the informed consent claim. The patient probably would not have been able to conceive naturally due to the infection.
VERDICT:
A $1.8 million Connecticut verdict was returned.
Related article:
Elective laparoscopic appendectomy in gynecologic surgery: When, why, and how
Complications after vaginal hysterectomy
A woman underwent laparoscopic vaginal hysterectomy and bilateral salpingo-oophorectomy with anterior and posterior repair using mesh in August 2010. Shortly after surgery, the patient reported vaginal discharge with pain and bleeding. She was treated with antibiotics. Results of a CT scan identified the cause of her symptoms as vaginal cuff granulations.
Her pain continued and in June 2011, she underwent vaginal tissue biopsy. After testing revealed the presence of fecal matter, a small-bowel vaginal fistula was identified. She underwent laparoscopic enterectomy, urethral lysis, an omental pedicle flap, and cystoscopy. The mesh had perforated several loops of the small bowel.
In August 2011, the patient reported spinal pain. Magnetic resonance imaging (MRI) revealed a new fluid abscess in a disc extending through the tract anterior to the soft tissue of the pelvis. She underwent intensive antibiotic therapy.
PATIENT’S CLAIM:
The gynecologic surgeon fell below the standard of care in his treatment of her conditions.
PHYSICIAN’S DEFENSE:
The surgeon denied allegations.
VERDICT:
A Nevada defense verdict was returned.
Related article:
Vaginal hysterectomy with basic instrumentation
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Medicaid expansion produced opposing cost effects
States that expanded Medicaid in 2014 both increased and decreased their Medicaid spending that year, compared with the nonexpansion states, according to an analysis from the Centers for Medicare & Medicaid Services.
The 26 states, along with the District of Columbia, that expanded Medicaid eligibility by the end of 2014 had an increase in total Medicaid spending of 12.3% over 2013, compared with an increase of 6.2% in nonexpansion states.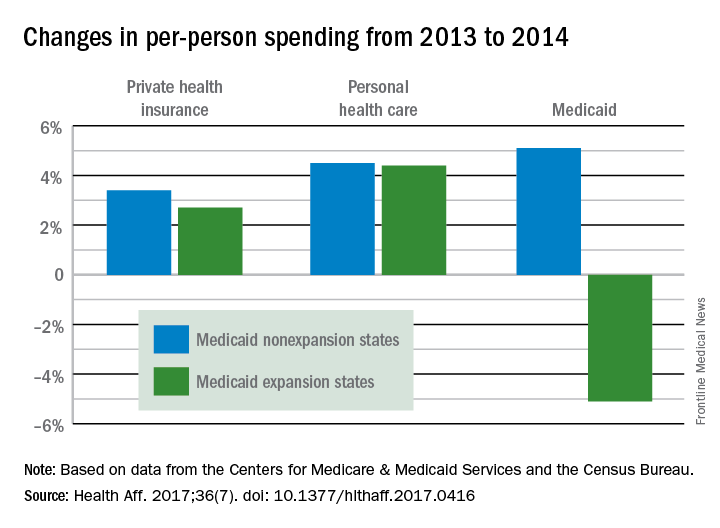
The expansion of coverage “increased the share of ... less expensive enrollees relative to the previous Medicaid beneficiary population mix,” the investigators said. Medicaid expansion brought in more relatively inexpensive adults – 43% of total enrollment in 2014, compared with 32% in 2013 – and reduced the proportion of disabled and aged enrollees, whose cost per person is much higher, they explained.
Private health insurance spending showed a different pattern: Nonexpansion states had larger increases in both higher per-person and overall costs than did expansion states. Per-person costs were up 3.4% for nonexpanders and 2.7% for expanders, and total costs rose 6.8% in nonexpansion states and 4.6% in the Medicaid expanders, Mr. Lassman and his associates said. Higher per-person spending growth for enrollees in the state and federal marketplaces, compared with nonmarketplace individual coverage, was partially responsible for this trend, they pointed out.
States that expanded Medicaid in 2014 both increased and decreased their Medicaid spending that year, compared with the nonexpansion states, according to an analysis from the Centers for Medicare & Medicaid Services.
The 26 states, along with the District of Columbia, that expanded Medicaid eligibility by the end of 2014 had an increase in total Medicaid spending of 12.3% over 2013, compared with an increase of 6.2% in nonexpansion states.
The expansion of coverage “increased the share of ... less expensive enrollees relative to the previous Medicaid beneficiary population mix,” the investigators said. Medicaid expansion brought in more relatively inexpensive adults – 43% of total enrollment in 2014, compared with 32% in 2013 – and reduced the proportion of disabled and aged enrollees, whose cost per person is much higher, they explained.
Private health insurance spending showed a different pattern: Nonexpansion states had larger increases in both higher per-person and overall costs than did expansion states. Per-person costs were up 3.4% for nonexpanders and 2.7% for expanders, and total costs rose 6.8% in nonexpansion states and 4.6% in the Medicaid expanders, Mr. Lassman and his associates said. Higher per-person spending growth for enrollees in the state and federal marketplaces, compared with nonmarketplace individual coverage, was partially responsible for this trend, they pointed out.
States that expanded Medicaid in 2014 both increased and decreased their Medicaid spending that year, compared with the nonexpansion states, according to an analysis from the Centers for Medicare & Medicaid Services.
The 26 states, along with the District of Columbia, that expanded Medicaid eligibility by the end of 2014 had an increase in total Medicaid spending of 12.3% over 2013, compared with an increase of 6.2% in nonexpansion states.
The expansion of coverage “increased the share of ... less expensive enrollees relative to the previous Medicaid beneficiary population mix,” the investigators said. Medicaid expansion brought in more relatively inexpensive adults – 43% of total enrollment in 2014, compared with 32% in 2013 – and reduced the proportion of disabled and aged enrollees, whose cost per person is much higher, they explained.
Private health insurance spending showed a different pattern: Nonexpansion states had larger increases in both higher per-person and overall costs than did expansion states. Per-person costs were up 3.4% for nonexpanders and 2.7% for expanders, and total costs rose 6.8% in nonexpansion states and 4.6% in the Medicaid expanders, Mr. Lassman and his associates said. Higher per-person spending growth for enrollees in the state and federal marketplaces, compared with nonmarketplace individual coverage, was partially responsible for this trend, they pointed out.
FROM HEALTH AFFAIRS
Patient Handout: Finding the Right Epilepsy Resources


TWEAKing inflammation: Studies reflect potential treatment target for psoriasis, atopic dermatitis
An immunomodulatory pathway that has been linked to cancer, kidney disease, and other disease processes is becoming a focus of dermatologic research.
New evidence suggests that TNF-like weak inducer of apoptosis (TWEAK), a member of the tumor necrosis family (TNF) superfamily, may be involved in both atopic dermatitis (AD) and psoriasis (Nat Commun. 2017 May 22;8:15395. doi: 10.1038/ncomms15395). The research showed that mice engineered to have low TWEAK levels had less severe disease when both AD and psoriasis were induced.

The TWEAK receptor, Fn14, was upregulated in keratinocytes and dermal fibroblasts in mouse disease models of AD and psoriasis, and TWEAK induced production of a range of cytokines associated with both AD and psoriasis. Subcutaneous injection of recombinant TWEAK led to cutaneous inflammation, as well as histological and molecular signals of the two diseases.
The pathophysiology of both AD and psoriasis is nebulously complex, sharing a similar theme of immune dysregulation, but historically polar opposites based on the different branches of the immune response implicated.
The study is not the only recent work tying TWEAK/Fn14 to dermatologic diseases. Other recent papers have shown evidence of their involvement in chronic cutaneous lupus (J Invest Dermatol. 2015;135[8]:1986-95), UVB irradiation-induced cutaneous lupus (Exp Dermatol. 2016 Dec;25[12]:969-76), and bullous pemphigoid (J Invest Dermatol. 2017 Jul;137[7]:1512-22).
The spate of findings hint that TWEAK/Fn14 could be a novel therapeutic pathway to attack inflammatory disease. Many therapies for autoimmune disease focus on immunosuppressive agents, which are associated with an increased risk of infection. But mice engineered to lack either TWEAK or Fn14 appear normal, and a phase I trial of an anti-TWEAK antibody in patients with rheumatoid arthritis did not reveal any worrisome safety concerns. “It doesn’t seem to have the broad immunosuppressive effects which characterize the therapies we currently use,” said Chaim Putterman, MD, chief of the division of rheumatology and professor of medicine and microbiology & immunology at the Albert Einstein College of Medicine, New York.
Instead, TWEAK seems to be regulating inflammation in target organs. It almost certainly plays a role in healthy functions like wound healing and cell survival, but Dr. Putterman believes there are redundant mechanisms that can pick up the slack, as the healthy knockout mice attest. The evidence suggests that the TWEAK pathway may become overactive in some diseases and, if so, a therapeutic antibody might be able to reset it to a more normal balance. “The utopian vision is that you would block this cytokine and bring its downstream effects back to normal levels, rather than totally abrogating its homeostatic functions,” Dr. Putterman noted.
Because blocking TWEAK has no apparent immunosuppressive effects, it might be a candidate for combination therapy with existing cytotoxic drugs. “If you have a disease like psoriasis where some standard of care medications are immunosuppressive, such as methotrexate, you might not get more risk by adding an antibody targeting TWEAK, as opposed to using immunosuppressives in combination. That, I think, has potential,” he said.
Work remains, however. A proof-of-concept study in lupus nephritis, sponsored by Biogen, failed to show a benefit when an anti-TWEAK antibody was combined with the standard of care.
But the potential impact of this approach holds much promise, and the fact that TWEAK has been linked to multiple diseases should make it a more attractive drug target for drug companies. “Now we have a target, that if you knock it out, or its receptor, you can potentially affect both diseases. This may the start of a whole new direction for biologics to treat inflammatory disease, and cancer as well,” Dr. Friedman said.
Dr. Putterman and Dr. Friedman were among the authors of the 2015 JID study on TWEAK/Fn14 signaling in spontaneous lupus and the Experimental Dermatology study. Dr. Putterman has research funding from Biogen Idec. Dr. Friedman had no related disclosures. The authors of the Nature Communications study were from the La Jolla Institute for Allergy and Immunology, and Biogen.
An immunomodulatory pathway that has been linked to cancer, kidney disease, and other disease processes is becoming a focus of dermatologic research.
New evidence suggests that TNF-like weak inducer of apoptosis (TWEAK), a member of the tumor necrosis family (TNF) superfamily, may be involved in both atopic dermatitis (AD) and psoriasis (Nat Commun. 2017 May 22;8:15395. doi: 10.1038/ncomms15395). The research showed that mice engineered to have low TWEAK levels had less severe disease when both AD and psoriasis were induced.
The TWEAK receptor, Fn14, was upregulated in keratinocytes and dermal fibroblasts in mouse disease models of AD and psoriasis, and TWEAK induced production of a range of cytokines associated with both AD and psoriasis. Subcutaneous injection of recombinant TWEAK led to cutaneous inflammation, as well as histological and molecular signals of the two diseases.
The pathophysiology of both AD and psoriasis is nebulously complex, sharing a similar theme of immune dysregulation, but historically polar opposites based on the different branches of the immune response implicated.
The study is not the only recent work tying TWEAK/Fn14 to dermatologic diseases. Other recent papers have shown evidence of their involvement in chronic cutaneous lupus (J Invest Dermatol. 2015;135[8]:1986-95), UVB irradiation-induced cutaneous lupus (Exp Dermatol. 2016 Dec;25[12]:969-76), and bullous pemphigoid (J Invest Dermatol. 2017 Jul;137[7]:1512-22).
The spate of findings hint that TWEAK/Fn14 could be a novel therapeutic pathway to attack inflammatory disease. Many therapies for autoimmune disease focus on immunosuppressive agents, which are associated with an increased risk of infection. But mice engineered to lack either TWEAK or Fn14 appear normal, and a phase I trial of an anti-TWEAK antibody in patients with rheumatoid arthritis did not reveal any worrisome safety concerns. “It doesn’t seem to have the broad immunosuppressive effects which characterize the therapies we currently use,” said Chaim Putterman, MD, chief of the division of rheumatology and professor of medicine and microbiology & immunology at the Albert Einstein College of Medicine, New York.
Instead, TWEAK seems to be regulating inflammation in target organs. It almost certainly plays a role in healthy functions like wound healing and cell survival, but Dr. Putterman believes there are redundant mechanisms that can pick up the slack, as the healthy knockout mice attest. The evidence suggests that the TWEAK pathway may become overactive in some diseases and, if so, a therapeutic antibody might be able to reset it to a more normal balance. “The utopian vision is that you would block this cytokine and bring its downstream effects back to normal levels, rather than totally abrogating its homeostatic functions,” Dr. Putterman noted.
Because blocking TWEAK has no apparent immunosuppressive effects, it might be a candidate for combination therapy with existing cytotoxic drugs. “If you have a disease like psoriasis where some standard of care medications are immunosuppressive, such as methotrexate, you might not get more risk by adding an antibody targeting TWEAK, as opposed to using immunosuppressives in combination. That, I think, has potential,” he said.
Work remains, however. A proof-of-concept study in lupus nephritis, sponsored by Biogen, failed to show a benefit when an anti-TWEAK antibody was combined with the standard of care.
But the potential impact of this approach holds much promise, and the fact that TWEAK has been linked to multiple diseases should make it a more attractive drug target for drug companies. “Now we have a target, that if you knock it out, or its receptor, you can potentially affect both diseases. This may the start of a whole new direction for biologics to treat inflammatory disease, and cancer as well,” Dr. Friedman said.
Dr. Putterman and Dr. Friedman were among the authors of the 2015 JID study on TWEAK/Fn14 signaling in spontaneous lupus and the Experimental Dermatology study. Dr. Putterman has research funding from Biogen Idec. Dr. Friedman had no related disclosures. The authors of the Nature Communications study were from the La Jolla Institute for Allergy and Immunology, and Biogen.
An immunomodulatory pathway that has been linked to cancer, kidney disease, and other disease processes is becoming a focus of dermatologic research.
New evidence suggests that TNF-like weak inducer of apoptosis (TWEAK), a member of the tumor necrosis family (TNF) superfamily, may be involved in both atopic dermatitis (AD) and psoriasis (Nat Commun. 2017 May 22;8:15395. doi: 10.1038/ncomms15395). The research showed that mice engineered to have low TWEAK levels had less severe disease when both AD and psoriasis were induced.
The TWEAK receptor, Fn14, was upregulated in keratinocytes and dermal fibroblasts in mouse disease models of AD and psoriasis, and TWEAK induced production of a range of cytokines associated with both AD and psoriasis. Subcutaneous injection of recombinant TWEAK led to cutaneous inflammation, as well as histological and molecular signals of the two diseases.
The pathophysiology of both AD and psoriasis is nebulously complex, sharing a similar theme of immune dysregulation, but historically polar opposites based on the different branches of the immune response implicated.
The study is not the only recent work tying TWEAK/Fn14 to dermatologic diseases. Other recent papers have shown evidence of their involvement in chronic cutaneous lupus (J Invest Dermatol. 2015;135[8]:1986-95), UVB irradiation-induced cutaneous lupus (Exp Dermatol. 2016 Dec;25[12]:969-76), and bullous pemphigoid (J Invest Dermatol. 2017 Jul;137[7]:1512-22).
The spate of findings hint that TWEAK/Fn14 could be a novel therapeutic pathway to attack inflammatory disease. Many therapies for autoimmune disease focus on immunosuppressive agents, which are associated with an increased risk of infection. But mice engineered to lack either TWEAK or Fn14 appear normal, and a phase I trial of an anti-TWEAK antibody in patients with rheumatoid arthritis did not reveal any worrisome safety concerns. “It doesn’t seem to have the broad immunosuppressive effects which characterize the therapies we currently use,” said Chaim Putterman, MD, chief of the division of rheumatology and professor of medicine and microbiology & immunology at the Albert Einstein College of Medicine, New York.
Instead, TWEAK seems to be regulating inflammation in target organs. It almost certainly plays a role in healthy functions like wound healing and cell survival, but Dr. Putterman believes there are redundant mechanisms that can pick up the slack, as the healthy knockout mice attest. The evidence suggests that the TWEAK pathway may become overactive in some diseases and, if so, a therapeutic antibody might be able to reset it to a more normal balance. “The utopian vision is that you would block this cytokine and bring its downstream effects back to normal levels, rather than totally abrogating its homeostatic functions,” Dr. Putterman noted.
Because blocking TWEAK has no apparent immunosuppressive effects, it might be a candidate for combination therapy with existing cytotoxic drugs. “If you have a disease like psoriasis where some standard of care medications are immunosuppressive, such as methotrexate, you might not get more risk by adding an antibody targeting TWEAK, as opposed to using immunosuppressives in combination. That, I think, has potential,” he said.
Work remains, however. A proof-of-concept study in lupus nephritis, sponsored by Biogen, failed to show a benefit when an anti-TWEAK antibody was combined with the standard of care.
But the potential impact of this approach holds much promise, and the fact that TWEAK has been linked to multiple diseases should make it a more attractive drug target for drug companies. “Now we have a target, that if you knock it out, or its receptor, you can potentially affect both diseases. This may the start of a whole new direction for biologics to treat inflammatory disease, and cancer as well,” Dr. Friedman said.
Dr. Putterman and Dr. Friedman were among the authors of the 2015 JID study on TWEAK/Fn14 signaling in spontaneous lupus and the Experimental Dermatology study. Dr. Putterman has research funding from Biogen Idec. Dr. Friedman had no related disclosures. The authors of the Nature Communications study were from the La Jolla Institute for Allergy and Immunology, and Biogen.
CBO: Senate health care proposal marginally better than House-passed bill
The Senate health care proposal is only marginally better in terms of the number of uninsured Americans, compared with the House-passed bill it aims to replace, but it still would leave 22 million more Americans without insurance coverage, according to a June 26 analysis by the Congressional Budget Office.
The analysis raised voices of opposition from the medical community.
BCRA would lower the federal deficit by $321 billion between 2017-2026, driven by the dramatic cuts in spending on Medicaid (estimated to be $772 billion), as well as $408 billion saved from reduced tax credits and other subsidies to help people afford health insurance.
The CBO’s estimate also addresses how the bill could impact access to health care.
Initially, patients can expect another short-term spike in insurance premiums, with average premiums in 2018 increasing by 20%, compared with current law, “mainly because the penalty for not having insurance would be eliminated, inducing fewer comparatively healthy people to sign up.” In 2019, premiums are predicted to be about 10% higher than under current law; however, by 2020, premiums for benchmark plans would be 30% lower than with current law.
However, as premiums come down, deductibles would continue to rise for plans that would offer lower levels of coverage, according to the CBO report. Additionally, “starting in 2020, the premium for a silver plan would typically be a relatively high percentage of income for low income people. The deductible for a plan ... would be a significantly higher percentage of income – also making such a plan unattractive but for a different reason. As a result, despite being eligible for premium tax credits, few low-income people would purchase any plan.”
The report also notes that the Senate proposal would not necessarily reverse current concerns regarding consumer choice in the individual markets, stating that “a small fraction of the population resides in areas which – because of this legislation, for at least some of the years after 2019 – no insurers will participate in the nongroup market or insurance would be offered only with very high premiums.” Additionally, removing the employer mandate could result in employers forgoing offering health insurance to their employees.
The bill faces an uphill battle in the Senate as there seemingly are not enough votes to pass the bill at this time. The measure is using the budget reconciliation process, meaning it will need 50 of the 52 Senate Republicans to pass it (all 48 Democrats are expected to vote against it). At least six GOP senators have said they are not ready to start debate. Senate Majority Leader Mitch McConnell (R-Ky) will not present the bill to the chamber for consideration until after the July 4 recess in an effort to tweak the language to garner the 50 votes needed to pass.
Medical societies are pushing back against the bill as well.
The American Medical Association, in a letter to Senate leaders, notes that the first principal that medical professionals operate under is to do no harm. “The draft legislation violates that standard on many levels,” according to the AMA letter.
In a statement from the American Gastroenterological Association, it was pointed out that both the House and Senate bills allow states to opt out of the Affordable Care Act’s essential health benefits package, which includes coverage of colorectal cancer screenings. “A core mission of AGA is to ensure that patients have access to high-quality medical care,“ said Timothy Wang, MD, AGAF, AGA chair. “We have made great strides in the increase in screening and prevention of colorectal cancer, which reduces deaths and downstream health care costs. Erecting barriers to screening will only reverse this process.” The budget cuts and restrictions on patient access under the congressional proposals will create tremendous burdens for the health care system as patients will increasingly rely on practices and academic medical centers to provide uncompensated care, according to the AGA.
The American Osteopathic Association reiterated its objections to BCRA in a statement, citing the CBO’s determination that 22 million would lose coverage.
“As patient advocates, we cannot accept that under [BCRA] patients in need will no longer have the coverage they require to access health care services,” the association said in a statement. “The BCRA does nothing to control health costs but instead focuses on reducing federal health care expenditures by cutting coverage of our nation’s most vulnerable individuals and eliminating policies that promote access to preventive care services that can actually drive down expenses while improving patient outcomes.”
The American College of Cardiology noted that CBO analysis “makes it clear that the [BCRA] would lead to loss of coverage for millions of Americans and limit access to care for our most vulnerable populations. ... The ACC opposes the BCRA as it does not align with our Principles for Health Reform, which stress the need for patient access to meaningful insurance coverage and high-quality care.”
The Senate health care proposal is only marginally better in terms of the number of uninsured Americans, compared with the House-passed bill it aims to replace, but it still would leave 22 million more Americans without insurance coverage, according to a June 26 analysis by the Congressional Budget Office.
The analysis raised voices of opposition from the medical community.
BCRA would lower the federal deficit by $321 billion between 2017-2026, driven by the dramatic cuts in spending on Medicaid (estimated to be $772 billion), as well as $408 billion saved from reduced tax credits and other subsidies to help people afford health insurance.
The CBO’s estimate also addresses how the bill could impact access to health care.
Initially, patients can expect another short-term spike in insurance premiums, with average premiums in 2018 increasing by 20%, compared with current law, “mainly because the penalty for not having insurance would be eliminated, inducing fewer comparatively healthy people to sign up.” In 2019, premiums are predicted to be about 10% higher than under current law; however, by 2020, premiums for benchmark plans would be 30% lower than with current law.
However, as premiums come down, deductibles would continue to rise for plans that would offer lower levels of coverage, according to the CBO report. Additionally, “starting in 2020, the premium for a silver plan would typically be a relatively high percentage of income for low income people. The deductible for a plan ... would be a significantly higher percentage of income – also making such a plan unattractive but for a different reason. As a result, despite being eligible for premium tax credits, few low-income people would purchase any plan.”
The report also notes that the Senate proposal would not necessarily reverse current concerns regarding consumer choice in the individual markets, stating that “a small fraction of the population resides in areas which – because of this legislation, for at least some of the years after 2019 – no insurers will participate in the nongroup market or insurance would be offered only with very high premiums.” Additionally, removing the employer mandate could result in employers forgoing offering health insurance to their employees.
The bill faces an uphill battle in the Senate as there seemingly are not enough votes to pass the bill at this time. The measure is using the budget reconciliation process, meaning it will need 50 of the 52 Senate Republicans to pass it (all 48 Democrats are expected to vote against it). At least six GOP senators have said they are not ready to start debate. Senate Majority Leader Mitch McConnell (R-Ky) will not present the bill to the chamber for consideration until after the July 4 recess in an effort to tweak the language to garner the 50 votes needed to pass.
Medical societies are pushing back against the bill as well.
The American Medical Association, in a letter to Senate leaders, notes that the first principal that medical professionals operate under is to do no harm. “The draft legislation violates that standard on many levels,” according to the AMA letter.
In a statement from the American Gastroenterological Association, it was pointed out that both the House and Senate bills allow states to opt out of the Affordable Care Act’s essential health benefits package, which includes coverage of colorectal cancer screenings. “A core mission of AGA is to ensure that patients have access to high-quality medical care,“ said Timothy Wang, MD, AGAF, AGA chair. “We have made great strides in the increase in screening and prevention of colorectal cancer, which reduces deaths and downstream health care costs. Erecting barriers to screening will only reverse this process.” The budget cuts and restrictions on patient access under the congressional proposals will create tremendous burdens for the health care system as patients will increasingly rely on practices and academic medical centers to provide uncompensated care, according to the AGA.
The American Osteopathic Association reiterated its objections to BCRA in a statement, citing the CBO’s determination that 22 million would lose coverage.
“As patient advocates, we cannot accept that under [BCRA] patients in need will no longer have the coverage they require to access health care services,” the association said in a statement. “The BCRA does nothing to control health costs but instead focuses on reducing federal health care expenditures by cutting coverage of our nation’s most vulnerable individuals and eliminating policies that promote access to preventive care services that can actually drive down expenses while improving patient outcomes.”
The American College of Cardiology noted that CBO analysis “makes it clear that the [BCRA] would lead to loss of coverage for millions of Americans and limit access to care for our most vulnerable populations. ... The ACC opposes the BCRA as it does not align with our Principles for Health Reform, which stress the need for patient access to meaningful insurance coverage and high-quality care.”
The Senate health care proposal is only marginally better in terms of the number of uninsured Americans, compared with the House-passed bill it aims to replace, but it still would leave 22 million more Americans without insurance coverage, according to a June 26 analysis by the Congressional Budget Office.
The analysis raised voices of opposition from the medical community.
BCRA would lower the federal deficit by $321 billion between 2017-2026, driven by the dramatic cuts in spending on Medicaid (estimated to be $772 billion), as well as $408 billion saved from reduced tax credits and other subsidies to help people afford health insurance.
The CBO’s estimate also addresses how the bill could impact access to health care.
Initially, patients can expect another short-term spike in insurance premiums, with average premiums in 2018 increasing by 20%, compared with current law, “mainly because the penalty for not having insurance would be eliminated, inducing fewer comparatively healthy people to sign up.” In 2019, premiums are predicted to be about 10% higher than under current law; however, by 2020, premiums for benchmark plans would be 30% lower than with current law.
However, as premiums come down, deductibles would continue to rise for plans that would offer lower levels of coverage, according to the CBO report. Additionally, “starting in 2020, the premium for a silver plan would typically be a relatively high percentage of income for low income people. The deductible for a plan ... would be a significantly higher percentage of income – also making such a plan unattractive but for a different reason. As a result, despite being eligible for premium tax credits, few low-income people would purchase any plan.”
The report also notes that the Senate proposal would not necessarily reverse current concerns regarding consumer choice in the individual markets, stating that “a small fraction of the population resides in areas which – because of this legislation, for at least some of the years after 2019 – no insurers will participate in the nongroup market or insurance would be offered only with very high premiums.” Additionally, removing the employer mandate could result in employers forgoing offering health insurance to their employees.
The bill faces an uphill battle in the Senate as there seemingly are not enough votes to pass the bill at this time. The measure is using the budget reconciliation process, meaning it will need 50 of the 52 Senate Republicans to pass it (all 48 Democrats are expected to vote against it). At least six GOP senators have said they are not ready to start debate. Senate Majority Leader Mitch McConnell (R-Ky) will not present the bill to the chamber for consideration until after the July 4 recess in an effort to tweak the language to garner the 50 votes needed to pass.
Medical societies are pushing back against the bill as well.
The American Medical Association, in a letter to Senate leaders, notes that the first principal that medical professionals operate under is to do no harm. “The draft legislation violates that standard on many levels,” according to the AMA letter.
In a statement from the American Gastroenterological Association, it was pointed out that both the House and Senate bills allow states to opt out of the Affordable Care Act’s essential health benefits package, which includes coverage of colorectal cancer screenings. “A core mission of AGA is to ensure that patients have access to high-quality medical care,“ said Timothy Wang, MD, AGAF, AGA chair. “We have made great strides in the increase in screening and prevention of colorectal cancer, which reduces deaths and downstream health care costs. Erecting barriers to screening will only reverse this process.” The budget cuts and restrictions on patient access under the congressional proposals will create tremendous burdens for the health care system as patients will increasingly rely on practices and academic medical centers to provide uncompensated care, according to the AGA.
The American Osteopathic Association reiterated its objections to BCRA in a statement, citing the CBO’s determination that 22 million would lose coverage.
“As patient advocates, we cannot accept that under [BCRA] patients in need will no longer have the coverage they require to access health care services,” the association said in a statement. “The BCRA does nothing to control health costs but instead focuses on reducing federal health care expenditures by cutting coverage of our nation’s most vulnerable individuals and eliminating policies that promote access to preventive care services that can actually drive down expenses while improving patient outcomes.”
The American College of Cardiology noted that CBO analysis “makes it clear that the [BCRA] would lead to loss of coverage for millions of Americans and limit access to care for our most vulnerable populations. ... The ACC opposes the BCRA as it does not align with our Principles for Health Reform, which stress the need for patient access to meaningful insurance coverage and high-quality care.”
Senate health care proposal already facing uphill battle
Senate Republican leaders are facing pushback from almost every side on their Affordable Care Act repeal/replace proposal – so much so that the current plan is unlikely to gain enough support to pass.
Sen. Rand Paul (R-Ky.), Sen. Ted Cruz (R-Texas), Sen. Ron Johnson (R-Wis.), and Sen. Mike Lee (R-Utah) said in a joint statement issued June 22, the day the plan was published, “There are provisions in this draft that represent an improvement to our current health care system, but it does not appear this draft as written will accomplish the most important promise that we made to Americans to repeal Obamacare and lower their health care costs.”
Like the House-passed American Health Care Act (H.R. 1628), the proposed BCRA would reduce Medicaid spending and would address rising premiums in the individual health insurance marketplace; however, BCRA would take a slightly different path to the same destination.
Like the House bill, BCRA also targets funding for Planned Parenthood, although because of Senate procedural rules, it is a more indirect funding ban.
A key difference between BCRA and the House bill is how insurance premium support is calculated. The AHCA would base tax credits on age, providing a lesser benefit for older, but pre–Medicare-age adults. In contrast, BCRA would base tax credits on income while limiting eligibility to households at 350% of the federal poverty line. Further, credits would cover only 58% of the actuarial value of health insurance under BCRA.
The draft Senate plan would not allow states to request a waiver from the ACA’s so-called community waiver provisions – the portion of the law that requires health insurance premiums to be the same regardless of age or preexisting condition; the House-passed AHCA would allow those waivers.
Medicaid expansion would be rolled back under the Senate plan, but at a slower pace than the AHCA would require – by 2023 under BCRA vs. 2020 under AHCA.
The BCRA would establish a per capita funding mechanism for Medicaid going forward, which would base funding on historic Medicaid expenditures and uses an economic index to track inflation and adjust payments accordingly.
To address the needs of people with greater health care needs, the Senate proposal would provide $57 billion over the first 4 years, then another $57 billion over the next 8. The funds would be available for programs such as premium support or high-risk pools to help individuals who are expected to be high users of health care. States would be required to match funds starting in 2022.
Experts were quick to weigh in on the Senate plan.
The BCRA needs to do three things, according to Grace-Marie Turner, president of the Galen Institute: Provide a safety net for those covered through the ACA so that they do not lose coverage in the transition, modernize Medicaid, and give states more authority and options to reform their own health insurance markets.
“We have learned that the federal government is not able to regulate something as local as health insurance,” Ms. Turner said. “They cannot create policies and legislation that works for people in downtown Manhattan and rural Montana and southern New Mexico and the panhandle of Florida. There are too many different populations. The states need to do that, and this bill also would give the states more authority to begin to oversee their health insurance markets but with new funding to provide extra help for the people who have difficulty buying progress.”
She said it could be much better if the Senate did not use the reconciliation process, “but within the confines of that, both the House and Senate bills do the same thing.”
Ms. Turner also stressed that there are more reforms coming later, as the Senate and House address other portions of ACA repeal/replacement.
“I hope that [senators] would see moving this forward as beneficial so that then they can move additional pieces of legislation, hopefully, with 60 votes to go through the regular process, to have additional follow-up bills. This is not the end of the story. This is just rescuing us from Obamacare,” she said. “Then we need to go forward and think about what do we need to do to make our health sector work better in the future by putting doctors and patients, rather than government, in charge of choices.”
Doctors, however, did not agree.
“This bill significantly decreases patients’ ability to access high-quality health care, and affordable coverage for millions of Americans will be in jeopardy if the legislation is passed,” Boyd Buser, MD, a doctor of osteopathy and president of the American Osteopathic Association, said in a statement.
He noted that the Medicaid cuts will have a “devastating impact, especially in areas of our country hardest hit by the ongoing opioid epidemic. ... The Senate bill should have prioritized prevention and care coordination, two measures proven to reduce overall health costs by eliminating waste and addressing health problems at the most treatable stage. Decreasing the number of Americans with coverage as it intends does will not lower costs.”
In a statement from the American Gastroenterological Association, it was noted that steep cuts to Medicaid funding are a main driver of loss of coverage. Both bills also allow states to opt out of the Affordable Care Act’s essential health benefits package, which includes coverage of colorectal cancer screenings. “A core mission of AGA is to ensure that patients have access to high-quality medical care,” said Timothy Wang, MD, AGAF, AGA chair. “We have made great strides in the increase in screening and prevention of colorectal cancer, which reduces deaths and downstream health-care costs. Erecting barriers to screening will only reverse this progress.” The budget cuts and restrictions on patient access under the congressional proposals will create tremendous burdens for the health-care system as patients will increasingly rely on practices and academic medical centers to provide uncompensated care, according to the AGA.
Republican lawmakers and the Trump administration have vowed to address the ACA in other ways as well, by reviewing and possibly changing all relevant regulations, then using the regular legislative process, which would need 60 votes, to address issues that cannot be handled by the budget reconciliation process.
Senate Republican leaders are facing pushback from almost every side on their Affordable Care Act repeal/replace proposal – so much so that the current plan is unlikely to gain enough support to pass.
Sen. Rand Paul (R-Ky.), Sen. Ted Cruz (R-Texas), Sen. Ron Johnson (R-Wis.), and Sen. Mike Lee (R-Utah) said in a joint statement issued June 22, the day the plan was published, “There are provisions in this draft that represent an improvement to our current health care system, but it does not appear this draft as written will accomplish the most important promise that we made to Americans to repeal Obamacare and lower their health care costs.”
Like the House-passed American Health Care Act (H.R. 1628), the proposed BCRA would reduce Medicaid spending and would address rising premiums in the individual health insurance marketplace; however, BCRA would take a slightly different path to the same destination.
Like the House bill, BCRA also targets funding for Planned Parenthood, although because of Senate procedural rules, it is a more indirect funding ban.
A key difference between BCRA and the House bill is how insurance premium support is calculated. The AHCA would base tax credits on age, providing a lesser benefit for older, but pre–Medicare-age adults. In contrast, BCRA would base tax credits on income while limiting eligibility to households at 350% of the federal poverty line. Further, credits would cover only 58% of the actuarial value of health insurance under BCRA.
The draft Senate plan would not allow states to request a waiver from the ACA’s so-called community waiver provisions – the portion of the law that requires health insurance premiums to be the same regardless of age or preexisting condition; the House-passed AHCA would allow those waivers.
Medicaid expansion would be rolled back under the Senate plan, but at a slower pace than the AHCA would require – by 2023 under BCRA vs. 2020 under AHCA.
The BCRA would establish a per capita funding mechanism for Medicaid going forward, which would base funding on historic Medicaid expenditures and uses an economic index to track inflation and adjust payments accordingly.
To address the needs of people with greater health care needs, the Senate proposal would provide $57 billion over the first 4 years, then another $57 billion over the next 8. The funds would be available for programs such as premium support or high-risk pools to help individuals who are expected to be high users of health care. States would be required to match funds starting in 2022.
Experts were quick to weigh in on the Senate plan.
The BCRA needs to do three things, according to Grace-Marie Turner, president of the Galen Institute: Provide a safety net for those covered through the ACA so that they do not lose coverage in the transition, modernize Medicaid, and give states more authority and options to reform their own health insurance markets.
“We have learned that the federal government is not able to regulate something as local as health insurance,” Ms. Turner said. “They cannot create policies and legislation that works for people in downtown Manhattan and rural Montana and southern New Mexico and the panhandle of Florida. There are too many different populations. The states need to do that, and this bill also would give the states more authority to begin to oversee their health insurance markets but with new funding to provide extra help for the people who have difficulty buying progress.”
She said it could be much better if the Senate did not use the reconciliation process, “but within the confines of that, both the House and Senate bills do the same thing.”
Ms. Turner also stressed that there are more reforms coming later, as the Senate and House address other portions of ACA repeal/replacement.
“I hope that [senators] would see moving this forward as beneficial so that then they can move additional pieces of legislation, hopefully, with 60 votes to go through the regular process, to have additional follow-up bills. This is not the end of the story. This is just rescuing us from Obamacare,” she said. “Then we need to go forward and think about what do we need to do to make our health sector work better in the future by putting doctors and patients, rather than government, in charge of choices.”
Doctors, however, did not agree.
“This bill significantly decreases patients’ ability to access high-quality health care, and affordable coverage for millions of Americans will be in jeopardy if the legislation is passed,” Boyd Buser, MD, a doctor of osteopathy and president of the American Osteopathic Association, said in a statement.
He noted that the Medicaid cuts will have a “devastating impact, especially in areas of our country hardest hit by the ongoing opioid epidemic. ... The Senate bill should have prioritized prevention and care coordination, two measures proven to reduce overall health costs by eliminating waste and addressing health problems at the most treatable stage. Decreasing the number of Americans with coverage as it intends does will not lower costs.”
In a statement from the American Gastroenterological Association, it was noted that steep cuts to Medicaid funding are a main driver of loss of coverage. Both bills also allow states to opt out of the Affordable Care Act’s essential health benefits package, which includes coverage of colorectal cancer screenings. “A core mission of AGA is to ensure that patients have access to high-quality medical care,” said Timothy Wang, MD, AGAF, AGA chair. “We have made great strides in the increase in screening and prevention of colorectal cancer, which reduces deaths and downstream health-care costs. Erecting barriers to screening will only reverse this progress.” The budget cuts and restrictions on patient access under the congressional proposals will create tremendous burdens for the health-care system as patients will increasingly rely on practices and academic medical centers to provide uncompensated care, according to the AGA.
Republican lawmakers and the Trump administration have vowed to address the ACA in other ways as well, by reviewing and possibly changing all relevant regulations, then using the regular legislative process, which would need 60 votes, to address issues that cannot be handled by the budget reconciliation process.
Senate Republican leaders are facing pushback from almost every side on their Affordable Care Act repeal/replace proposal – so much so that the current plan is unlikely to gain enough support to pass.
Sen. Rand Paul (R-Ky.), Sen. Ted Cruz (R-Texas), Sen. Ron Johnson (R-Wis.), and Sen. Mike Lee (R-Utah) said in a joint statement issued June 22, the day the plan was published, “There are provisions in this draft that represent an improvement to our current health care system, but it does not appear this draft as written will accomplish the most important promise that we made to Americans to repeal Obamacare and lower their health care costs.”
Like the House-passed American Health Care Act (H.R. 1628), the proposed BCRA would reduce Medicaid spending and would address rising premiums in the individual health insurance marketplace; however, BCRA would take a slightly different path to the same destination.
Like the House bill, BCRA also targets funding for Planned Parenthood, although because of Senate procedural rules, it is a more indirect funding ban.
A key difference between BCRA and the House bill is how insurance premium support is calculated. The AHCA would base tax credits on age, providing a lesser benefit for older, but pre–Medicare-age adults. In contrast, BCRA would base tax credits on income while limiting eligibility to households at 350% of the federal poverty line. Further, credits would cover only 58% of the actuarial value of health insurance under BCRA.
The draft Senate plan would not allow states to request a waiver from the ACA’s so-called community waiver provisions – the portion of the law that requires health insurance premiums to be the same regardless of age or preexisting condition; the House-passed AHCA would allow those waivers.
Medicaid expansion would be rolled back under the Senate plan, but at a slower pace than the AHCA would require – by 2023 under BCRA vs. 2020 under AHCA.
The BCRA would establish a per capita funding mechanism for Medicaid going forward, which would base funding on historic Medicaid expenditures and uses an economic index to track inflation and adjust payments accordingly.
To address the needs of people with greater health care needs, the Senate proposal would provide $57 billion over the first 4 years, then another $57 billion over the next 8. The funds would be available for programs such as premium support or high-risk pools to help individuals who are expected to be high users of health care. States would be required to match funds starting in 2022.
Experts were quick to weigh in on the Senate plan.
The BCRA needs to do three things, according to Grace-Marie Turner, president of the Galen Institute: Provide a safety net for those covered through the ACA so that they do not lose coverage in the transition, modernize Medicaid, and give states more authority and options to reform their own health insurance markets.
“We have learned that the federal government is not able to regulate something as local as health insurance,” Ms. Turner said. “They cannot create policies and legislation that works for people in downtown Manhattan and rural Montana and southern New Mexico and the panhandle of Florida. There are too many different populations. The states need to do that, and this bill also would give the states more authority to begin to oversee their health insurance markets but with new funding to provide extra help for the people who have difficulty buying progress.”
She said it could be much better if the Senate did not use the reconciliation process, “but within the confines of that, both the House and Senate bills do the same thing.”
Ms. Turner also stressed that there are more reforms coming later, as the Senate and House address other portions of ACA repeal/replacement.
“I hope that [senators] would see moving this forward as beneficial so that then they can move additional pieces of legislation, hopefully, with 60 votes to go through the regular process, to have additional follow-up bills. This is not the end of the story. This is just rescuing us from Obamacare,” she said. “Then we need to go forward and think about what do we need to do to make our health sector work better in the future by putting doctors and patients, rather than government, in charge of choices.”
Doctors, however, did not agree.
“This bill significantly decreases patients’ ability to access high-quality health care, and affordable coverage for millions of Americans will be in jeopardy if the legislation is passed,” Boyd Buser, MD, a doctor of osteopathy and president of the American Osteopathic Association, said in a statement.
He noted that the Medicaid cuts will have a “devastating impact, especially in areas of our country hardest hit by the ongoing opioid epidemic. ... The Senate bill should have prioritized prevention and care coordination, two measures proven to reduce overall health costs by eliminating waste and addressing health problems at the most treatable stage. Decreasing the number of Americans with coverage as it intends does will not lower costs.”
In a statement from the American Gastroenterological Association, it was noted that steep cuts to Medicaid funding are a main driver of loss of coverage. Both bills also allow states to opt out of the Affordable Care Act’s essential health benefits package, which includes coverage of colorectal cancer screenings. “A core mission of AGA is to ensure that patients have access to high-quality medical care,” said Timothy Wang, MD, AGAF, AGA chair. “We have made great strides in the increase in screening and prevention of colorectal cancer, which reduces deaths and downstream health-care costs. Erecting barriers to screening will only reverse this progress.” The budget cuts and restrictions on patient access under the congressional proposals will create tremendous burdens for the health-care system as patients will increasingly rely on practices and academic medical centers to provide uncompensated care, according to the AGA.
Republican lawmakers and the Trump administration have vowed to address the ACA in other ways as well, by reviewing and possibly changing all relevant regulations, then using the regular legislative process, which would need 60 votes, to address issues that cannot be handled by the budget reconciliation process.
How would you handle predictions of Alzheimer’s disease?
We love to try and predict the future. Some of it is scientific, like checking the weather forecast to see what we’re in for. (Here in Phoenix, it’s always hot, hotter, or melting.)
On the other hand, some of it is just for entertainment, like checking a horoscope or seeing what a fortune cookie says.
The breakthroughs in biomarkers for Alzheimer’s disease are accelerating. Although still experimental, we’re getting pretty close to predicting the disease many years before it develops. At the same time, we aren’t nearly as close to a treatment that will have a meaningful impact on the course of the disease.
In 1993, the genetic marker for Huntington’s disease was identified, quickly leading to a blood test with high accuracy to know if you were – or were not – going to develop the fatal disorder down the road.
Some wanted to know and used the information to decide if they wanted to have families. Others, understandably fearful, decided not to and let their lives play out as they will. Sadly, either way we have nothing close to a cure for the disease.
Now, we come to Alzheimer’s disease, many times more common than Huntington’s. Close to predicting its coming and not really close to a cure.
What would you do?
[polldaddy:9778279]
In “Back to the Future,” Doc Brown said “no man should know too much about their own destiny” (though later changed his mind). But, for Doc Brown, a bulletproof vest was all he needed. In Alzheimer’s disease, it’s not that simple.
I’m sure some would see it as a way to have their affairs in order long in advance, to spare themselves and their loved ones the frantic scramble that often comes after a diagnosis. Others would be afraid to know what the future holds, with every misplaced set of keys or iPhone becoming a reason to panic.
Obviously, if we had a true cure for the disorder, the decision would be easy. Then, it becomes a preventive measure in the same category as mammograms and colonoscopies. Early detection saves lives.

What would you do? And how will you guide the patients who ask your opinion?
For better or worse, these questions are coming. All of us need to think about how we’ll handle them.
Dr. Block has a solo neurology private practice in Scottsdale, Ariz.
We love to try and predict the future. Some of it is scientific, like checking the weather forecast to see what we’re in for. (Here in Phoenix, it’s always hot, hotter, or melting.)
On the other hand, some of it is just for entertainment, like checking a horoscope or seeing what a fortune cookie says.
The breakthroughs in biomarkers for Alzheimer’s disease are accelerating. Although still experimental, we’re getting pretty close to predicting the disease many years before it develops. At the same time, we aren’t nearly as close to a treatment that will have a meaningful impact on the course of the disease.
In 1993, the genetic marker for Huntington’s disease was identified, quickly leading to a blood test with high accuracy to know if you were – or were not – going to develop the fatal disorder down the road.
Some wanted to know and used the information to decide if they wanted to have families. Others, understandably fearful, decided not to and let their lives play out as they will. Sadly, either way we have nothing close to a cure for the disease.
Now, we come to Alzheimer’s disease, many times more common than Huntington’s. Close to predicting its coming and not really close to a cure.
What would you do?
[polldaddy:9778279]
In “Back to the Future,” Doc Brown said “no man should know too much about their own destiny” (though later changed his mind). But, for Doc Brown, a bulletproof vest was all he needed. In Alzheimer’s disease, it’s not that simple.
I’m sure some would see it as a way to have their affairs in order long in advance, to spare themselves and their loved ones the frantic scramble that often comes after a diagnosis. Others would be afraid to know what the future holds, with every misplaced set of keys or iPhone becoming a reason to panic.
Obviously, if we had a true cure for the disorder, the decision would be easy. Then, it becomes a preventive measure in the same category as mammograms and colonoscopies. Early detection saves lives.
What would you do? And how will you guide the patients who ask your opinion?
For better or worse, these questions are coming. All of us need to think about how we’ll handle them.
Dr. Block has a solo neurology private practice in Scottsdale, Ariz.
We love to try and predict the future. Some of it is scientific, like checking the weather forecast to see what we’re in for. (Here in Phoenix, it’s always hot, hotter, or melting.)
On the other hand, some of it is just for entertainment, like checking a horoscope or seeing what a fortune cookie says.
The breakthroughs in biomarkers for Alzheimer’s disease are accelerating. Although still experimental, we’re getting pretty close to predicting the disease many years before it develops. At the same time, we aren’t nearly as close to a treatment that will have a meaningful impact on the course of the disease.
In 1993, the genetic marker for Huntington’s disease was identified, quickly leading to a blood test with high accuracy to know if you were – or were not – going to develop the fatal disorder down the road.
Some wanted to know and used the information to decide if they wanted to have families. Others, understandably fearful, decided not to and let their lives play out as they will. Sadly, either way we have nothing close to a cure for the disease.
Now, we come to Alzheimer’s disease, many times more common than Huntington’s. Close to predicting its coming and not really close to a cure.
What would you do?
[polldaddy:9778279]
In “Back to the Future,” Doc Brown said “no man should know too much about their own destiny” (though later changed his mind). But, for Doc Brown, a bulletproof vest was all he needed. In Alzheimer’s disease, it’s not that simple.
I’m sure some would see it as a way to have their affairs in order long in advance, to spare themselves and their loved ones the frantic scramble that often comes after a diagnosis. Others would be afraid to know what the future holds, with every misplaced set of keys or iPhone becoming a reason to panic.
Obviously, if we had a true cure for the disorder, the decision would be easy. Then, it becomes a preventive measure in the same category as mammograms and colonoscopies. Early detection saves lives.
What would you do? And how will you guide the patients who ask your opinion?
For better or worse, these questions are coming. All of us need to think about how we’ll handle them.
Dr. Block has a solo neurology private practice in Scottsdale, Ariz.
Nocturia and sleep apnea
Author’s note: I have been writing “Myth of the Month” columns for the last several years. I will try to continue to write about myths when possible, but I would like to introduce a new column, “Pearl of the Month.” I want to share with you pearls that I have found really helpful in medical practice. Some of these will be new news, while some may be old news that may not be well known.
A 65-year-old man comes to a clinic concerned about frequent nocturia. He is getting up four times a night to urinate, and he has been urinating about every 5 hours during the day. He has been seen twice for this problem and was diagnosed with benign prostatic hyperplasia and started on tamsulosin.
He found a slight improvement when he started on 0.4 mg qhs, reducing his nocturia episodes from four to three. His dose was increased to 0.8 mg qhs, with no improvement in nocturia.
Exam today: BP, 140/94; pulse, 70. Rectal exam: Prostate is twice normal size without nodules. Labs: Na, 140; K, 4.0; glucose, 80; Ca, 9.6.
He is frustrated because he feels tired and sleepy from having to get up so often to urinate every night.
What is the best treatment/advice at this point?
A. Check hemoglobin A1C.
B. Start finasteride.
C. Switch tamsulosin to terazosin.
D. Evaluate for sleep apnea.
Umpei Yamamoto, MD, of Kyushu University Hospital, Japan, and colleagues studied the prevalence of sleep-disordered breathing among patients who presented to a urology clinic with nocturia and in those who visited a sleep apnea clinic with symptoms of excessive daytime sleepiness.1 Sleep-disordered breathing was found in 91% of the patients from the sleep apnea clinic and 70% of the patients from the urology clinic. The frequency of nocturia was reduced with continuous positive airway pressure (CPAP) in both groups in the patients who had not responded to conventional therapy or nocturia.
The symptom of nocturia as a symptom of sleep apnea might be even more common in women.2 Ozen K. Basoglu, MD, and Mehmet Sezai Tasbakan, MD, of Ege University, Izmir, Turkey, described clinical similarities and differences based on gender in a large group of patients with sleep apnea. Both men and women with sleep apnea had similar rates of excessive daytime sleepiness, snoring, and impaired concentration. Women had more frequent nocturia.
Nocturia especially should be considered a possible clue for the presence of sleep apnea in younger patients who have fewer other reasons to have nocturia. Takahiro Maeda, MD, of Keio University, Tokyo, and colleagues found that men younger than 50 years had more nocturnal urinations the worse their apnea-hypopnea index was.3 Overall in the study, 85% of the patients had a reduction in nighttime urination after CPAP therapy.
Treatment of sleep apnea has been shown in several studies to improve the nocturia that occurs in patients with sleep apnea. Hyoung Keun Park, MD, of Konkuk University, Seoul, and colleagues studied whether surgical intervention with uvulopalatopharyngoplasty (UPPP) reduced nocturia in patients with sleep apnea.4 In the study, there was a 73% success rate in treatment for sleep apnea with the UPPP surgery, and, among those who had successful surgeries, nocturia episodes decreased from 1.9 preoperatively to 0.7 postoperatively (P less than .001).
Minoru Miyazato, MD, PhD, of University of the Ryukyus, Okinawa, Japan, and colleagues looked at the effect of CPAP treatment on nighttime urine production in patients with obstructive sleep apnea.5 In this small study of 40 patients, mean nighttime voiding episodes decreased from 2.1 to 1.2 (P less than .01).

Pearl: Sleep apnea should be considered in the differential diagnosis of patients with nocturia, and treatment of sleep apnea may decrease nocturia.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at [email protected].
References
1. Intern Med. 2016;55(8):901-5.
2. Sleep Breath. 2017 Feb 14. doi: 10.1007/s11325-017-1482-9.
3. Can Urol Assoc J. 2016 Jul-Aug;10(7-8):E241-5.
4. Int Neurourol J. 2016 Dec;20(4):329-34.
5. Neurourol Urodyn. 2017 Feb;36(2):376-9.
Author’s note: I have been writing “Myth of the Month” columns for the last several years. I will try to continue to write about myths when possible, but I would like to introduce a new column, “Pearl of the Month.” I want to share with you pearls that I have found really helpful in medical practice. Some of these will be new news, while some may be old news that may not be well known.
A 65-year-old man comes to a clinic concerned about frequent nocturia. He is getting up four times a night to urinate, and he has been urinating about every 5 hours during the day. He has been seen twice for this problem and was diagnosed with benign prostatic hyperplasia and started on tamsulosin.
He found a slight improvement when he started on 0.4 mg qhs, reducing his nocturia episodes from four to three. His dose was increased to 0.8 mg qhs, with no improvement in nocturia.
Exam today: BP, 140/94; pulse, 70. Rectal exam: Prostate is twice normal size without nodules. Labs: Na, 140; K, 4.0; glucose, 80; Ca, 9.6.
He is frustrated because he feels tired and sleepy from having to get up so often to urinate every night.
What is the best treatment/advice at this point?
A. Check hemoglobin A1C.
B. Start finasteride.
C. Switch tamsulosin to terazosin.
D. Evaluate for sleep apnea.
Umpei Yamamoto, MD, of Kyushu University Hospital, Japan, and colleagues studied the prevalence of sleep-disordered breathing among patients who presented to a urology clinic with nocturia and in those who visited a sleep apnea clinic with symptoms of excessive daytime sleepiness.1 Sleep-disordered breathing was found in 91% of the patients from the sleep apnea clinic and 70% of the patients from the urology clinic. The frequency of nocturia was reduced with continuous positive airway pressure (CPAP) in both groups in the patients who had not responded to conventional therapy or nocturia.
The symptom of nocturia as a symptom of sleep apnea might be even more common in women.2 Ozen K. Basoglu, MD, and Mehmet Sezai Tasbakan, MD, of Ege University, Izmir, Turkey, described clinical similarities and differences based on gender in a large group of patients with sleep apnea. Both men and women with sleep apnea had similar rates of excessive daytime sleepiness, snoring, and impaired concentration. Women had more frequent nocturia.
Nocturia especially should be considered a possible clue for the presence of sleep apnea in younger patients who have fewer other reasons to have nocturia. Takahiro Maeda, MD, of Keio University, Tokyo, and colleagues found that men younger than 50 years had more nocturnal urinations the worse their apnea-hypopnea index was.3 Overall in the study, 85% of the patients had a reduction in nighttime urination after CPAP therapy.
Treatment of sleep apnea has been shown in several studies to improve the nocturia that occurs in patients with sleep apnea. Hyoung Keun Park, MD, of Konkuk University, Seoul, and colleagues studied whether surgical intervention with uvulopalatopharyngoplasty (UPPP) reduced nocturia in patients with sleep apnea.4 In the study, there was a 73% success rate in treatment for sleep apnea with the UPPP surgery, and, among those who had successful surgeries, nocturia episodes decreased from 1.9 preoperatively to 0.7 postoperatively (P less than .001).
Minoru Miyazato, MD, PhD, of University of the Ryukyus, Okinawa, Japan, and colleagues looked at the effect of CPAP treatment on nighttime urine production in patients with obstructive sleep apnea.5 In this small study of 40 patients, mean nighttime voiding episodes decreased from 2.1 to 1.2 (P less than .01).
Pearl: Sleep apnea should be considered in the differential diagnosis of patients with nocturia, and treatment of sleep apnea may decrease nocturia.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at [email protected].
References
1. Intern Med. 2016;55(8):901-5.
2. Sleep Breath. 2017 Feb 14. doi: 10.1007/s11325-017-1482-9.
3. Can Urol Assoc J. 2016 Jul-Aug;10(7-8):E241-5.
4. Int Neurourol J. 2016 Dec;20(4):329-34.
5. Neurourol Urodyn. 2017 Feb;36(2):376-9.
Author’s note: I have been writing “Myth of the Month” columns for the last several years. I will try to continue to write about myths when possible, but I would like to introduce a new column, “Pearl of the Month.” I want to share with you pearls that I have found really helpful in medical practice. Some of these will be new news, while some may be old news that may not be well known.
A 65-year-old man comes to a clinic concerned about frequent nocturia. He is getting up four times a night to urinate, and he has been urinating about every 5 hours during the day. He has been seen twice for this problem and was diagnosed with benign prostatic hyperplasia and started on tamsulosin.
He found a slight improvement when he started on 0.4 mg qhs, reducing his nocturia episodes from four to three. His dose was increased to 0.8 mg qhs, with no improvement in nocturia.
Exam today: BP, 140/94; pulse, 70. Rectal exam: Prostate is twice normal size without nodules. Labs: Na, 140; K, 4.0; glucose, 80; Ca, 9.6.
He is frustrated because he feels tired and sleepy from having to get up so often to urinate every night.
What is the best treatment/advice at this point?
A. Check hemoglobin A1C.
B. Start finasteride.
C. Switch tamsulosin to terazosin.
D. Evaluate for sleep apnea.
Umpei Yamamoto, MD, of Kyushu University Hospital, Japan, and colleagues studied the prevalence of sleep-disordered breathing among patients who presented to a urology clinic with nocturia and in those who visited a sleep apnea clinic with symptoms of excessive daytime sleepiness.1 Sleep-disordered breathing was found in 91% of the patients from the sleep apnea clinic and 70% of the patients from the urology clinic. The frequency of nocturia was reduced with continuous positive airway pressure (CPAP) in both groups in the patients who had not responded to conventional therapy or nocturia.
The symptom of nocturia as a symptom of sleep apnea might be even more common in women.2 Ozen K. Basoglu, MD, and Mehmet Sezai Tasbakan, MD, of Ege University, Izmir, Turkey, described clinical similarities and differences based on gender in a large group of patients with sleep apnea. Both men and women with sleep apnea had similar rates of excessive daytime sleepiness, snoring, and impaired concentration. Women had more frequent nocturia.
Nocturia especially should be considered a possible clue for the presence of sleep apnea in younger patients who have fewer other reasons to have nocturia. Takahiro Maeda, MD, of Keio University, Tokyo, and colleagues found that men younger than 50 years had more nocturnal urinations the worse their apnea-hypopnea index was.3 Overall in the study, 85% of the patients had a reduction in nighttime urination after CPAP therapy.
Treatment of sleep apnea has been shown in several studies to improve the nocturia that occurs in patients with sleep apnea. Hyoung Keun Park, MD, of Konkuk University, Seoul, and colleagues studied whether surgical intervention with uvulopalatopharyngoplasty (UPPP) reduced nocturia in patients with sleep apnea.4 In the study, there was a 73% success rate in treatment for sleep apnea with the UPPP surgery, and, among those who had successful surgeries, nocturia episodes decreased from 1.9 preoperatively to 0.7 postoperatively (P less than .001).
Minoru Miyazato, MD, PhD, of University of the Ryukyus, Okinawa, Japan, and colleagues looked at the effect of CPAP treatment on nighttime urine production in patients with obstructive sleep apnea.5 In this small study of 40 patients, mean nighttime voiding episodes decreased from 2.1 to 1.2 (P less than .01).
Pearl: Sleep apnea should be considered in the differential diagnosis of patients with nocturia, and treatment of sleep apnea may decrease nocturia.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at [email protected].
References
1. Intern Med. 2016;55(8):901-5.
2. Sleep Breath. 2017 Feb 14. doi: 10.1007/s11325-017-1482-9.
3. Can Urol Assoc J. 2016 Jul-Aug;10(7-8):E241-5.
4. Int Neurourol J. 2016 Dec;20(4):329-34.
5. Neurourol Urodyn. 2017 Feb;36(2):376-9.
Preliminary results promising from ublituximab phase II study for MS
NEW ORLEANS – The investigative agent ublituximab was well tolerated and demonstrated rapid B-cell depletion in patients with relapsing multiple sclerosis, results from an ongoing phase II trial showed.
“Ublituximab is a novel, chimeric monoclonal antibody targeting a unique epitope on the CD20 antigen, and glycoengineered to enhance affinity for all variants of FcyRIlla receptors, thereby demonstrating greater antibody-dependent cellular toxicity activity than rituximab and ofatumumab,” Amy Lovett-Racke, PhD, said at the annual meeting of the Consortium of Multiple Sclerosis Centers.

In a trial supported by TG Therapeutics, which is developing the agent, Dr. Lovett-Racke of the department of microbial infection and immunity at the Ohio State University Medical Center, Columbus, and her associates reported data from 24 patients who received ublituximab at doses markedly less than those used in ongoing phase III oncology studies, and at a range of infusion times, with a goal of rapid infusions.
The study ran for 48 weeks, and the primary endpoint was responders rate, defined as a percent of patients with at least a 95% reduction in peripheral CD19-positive B-cells within 2 weeks after the second infusion. All patients received the same total dose of 600 mg; only infusion times differed. (Ublituximab was administered on day 1, day 15, and week 24. Placebo IV infusion dose was administered on day 1 and day 15 only.)
The mean age of the study participants was 40 years, and the distribution of time from diagnosis was less than 5 years in 11 patients, 5-10 years in 7 patients, and greater than 10 years in 6 patients.
To date, ublituximab has been well tolerated, with only mild infusion reactions being observed, even with infusion times reduced to 1 hour. The researchers also found that ublituximab resulted in 99% B-cell depletion, meeting the study endpoint of greater than 95% depletion within 2 weeks of the second dose, which is comparable to ocrelizumab.
“Every patient has met the endpoint so far,” Dr. Lovett-Racke said. “Although there is a transient decrease in T cells after the initial dose of ublituximab, T-cell numbers are fairly stable over time. Memory B cells seem slightly more resistant to depletion, but are efficiently depleted in all patients. A comprehensive analysis of B- and T-cell profiles is being performed to understand how B-cell depletion influences T-cell profiles, and to characterize the B-cell repletion.”
TG Therapeutics funded the study. Dr. Lovett-Racke disclosed having received research grants from the National Institutes of Health, the National Multiple Sclerosis Society, and the Strategic Pharmaceutical Academic Research Consortium.
NEW ORLEANS – The investigative agent ublituximab was well tolerated and demonstrated rapid B-cell depletion in patients with relapsing multiple sclerosis, results from an ongoing phase II trial showed.
“Ublituximab is a novel, chimeric monoclonal antibody targeting a unique epitope on the CD20 antigen, and glycoengineered to enhance affinity for all variants of FcyRIlla receptors, thereby demonstrating greater antibody-dependent cellular toxicity activity than rituximab and ofatumumab,” Amy Lovett-Racke, PhD, said at the annual meeting of the Consortium of Multiple Sclerosis Centers.
In a trial supported by TG Therapeutics, which is developing the agent, Dr. Lovett-Racke of the department of microbial infection and immunity at the Ohio State University Medical Center, Columbus, and her associates reported data from 24 patients who received ublituximab at doses markedly less than those used in ongoing phase III oncology studies, and at a range of infusion times, with a goal of rapid infusions.
The study ran for 48 weeks, and the primary endpoint was responders rate, defined as a percent of patients with at least a 95% reduction in peripheral CD19-positive B-cells within 2 weeks after the second infusion. All patients received the same total dose of 600 mg; only infusion times differed. (Ublituximab was administered on day 1, day 15, and week 24. Placebo IV infusion dose was administered on day 1 and day 15 only.)
The mean age of the study participants was 40 years, and the distribution of time from diagnosis was less than 5 years in 11 patients, 5-10 years in 7 patients, and greater than 10 years in 6 patients.
To date, ublituximab has been well tolerated, with only mild infusion reactions being observed, even with infusion times reduced to 1 hour. The researchers also found that ublituximab resulted in 99% B-cell depletion, meeting the study endpoint of greater than 95% depletion within 2 weeks of the second dose, which is comparable to ocrelizumab.
“Every patient has met the endpoint so far,” Dr. Lovett-Racke said. “Although there is a transient decrease in T cells after the initial dose of ublituximab, T-cell numbers are fairly stable over time. Memory B cells seem slightly more resistant to depletion, but are efficiently depleted in all patients. A comprehensive analysis of B- and T-cell profiles is being performed to understand how B-cell depletion influences T-cell profiles, and to characterize the B-cell repletion.”
TG Therapeutics funded the study. Dr. Lovett-Racke disclosed having received research grants from the National Institutes of Health, the National Multiple Sclerosis Society, and the Strategic Pharmaceutical Academic Research Consortium.
NEW ORLEANS – The investigative agent ublituximab was well tolerated and demonstrated rapid B-cell depletion in patients with relapsing multiple sclerosis, results from an ongoing phase II trial showed.
“Ublituximab is a novel, chimeric monoclonal antibody targeting a unique epitope on the CD20 antigen, and glycoengineered to enhance affinity for all variants of FcyRIlla receptors, thereby demonstrating greater antibody-dependent cellular toxicity activity than rituximab and ofatumumab,” Amy Lovett-Racke, PhD, said at the annual meeting of the Consortium of Multiple Sclerosis Centers.
In a trial supported by TG Therapeutics, which is developing the agent, Dr. Lovett-Racke of the department of microbial infection and immunity at the Ohio State University Medical Center, Columbus, and her associates reported data from 24 patients who received ublituximab at doses markedly less than those used in ongoing phase III oncology studies, and at a range of infusion times, with a goal of rapid infusions.
The study ran for 48 weeks, and the primary endpoint was responders rate, defined as a percent of patients with at least a 95% reduction in peripheral CD19-positive B-cells within 2 weeks after the second infusion. All patients received the same total dose of 600 mg; only infusion times differed. (Ublituximab was administered on day 1, day 15, and week 24. Placebo IV infusion dose was administered on day 1 and day 15 only.)
The mean age of the study participants was 40 years, and the distribution of time from diagnosis was less than 5 years in 11 patients, 5-10 years in 7 patients, and greater than 10 years in 6 patients.
To date, ublituximab has been well tolerated, with only mild infusion reactions being observed, even with infusion times reduced to 1 hour. The researchers also found that ublituximab resulted in 99% B-cell depletion, meeting the study endpoint of greater than 95% depletion within 2 weeks of the second dose, which is comparable to ocrelizumab.
“Every patient has met the endpoint so far,” Dr. Lovett-Racke said. “Although there is a transient decrease in T cells after the initial dose of ublituximab, T-cell numbers are fairly stable over time. Memory B cells seem slightly more resistant to depletion, but are efficiently depleted in all patients. A comprehensive analysis of B- and T-cell profiles is being performed to understand how B-cell depletion influences T-cell profiles, and to characterize the B-cell repletion.”
TG Therapeutics funded the study. Dr. Lovett-Racke disclosed having received research grants from the National Institutes of Health, the National Multiple Sclerosis Society, and the Strategic Pharmaceutical Academic Research Consortium.
AT THE CMSC ANNUAL MEETING
Key clinical point:
Major finding: Use of ublituximab resulted in 99% B-cell depletion among patients with relapsing MS.
Data source: Preliminary results from a phase II placebo-controlled study of 24 MS patients who received ublituximab.
Disclosures: TG Therapeutics funded the study. Dr. Lovett-Racke disclosed having received research grants from the National Institutes of Health, the National Multiple Sclerosis Society, and the Strategic Pharmaceutical Academic Research Consortium.
Laparoscopic myomectomy technique
Read the accompanying article by Dr. Parker: “Laparoscopic myomectomy: Tips for patient selection and technique”
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Read the accompanying article by Dr. Parker: “Laparoscopic myomectomy: Tips for patient selection and technique”
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Read the accompanying article by Dr. Parker: “Laparoscopic myomectomy: Tips for patient selection and technique”
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.