User login
VIDEO: Hone aesthetic technique with upper face first
LAS VEGAS – For those starting to use toxins and fillers, “my first advice is to get a good education,” Christopher B. Zachary, MD, said in a video interview at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
“Know … how to evaluate your patient, know where the problems are, know what the danger zones are, understand your anatomy,” advised Dr. Zachary of the University of California, Irvine.
, he noted. Procedures on the upper face, such as the treatment for crow’s feet or a brow lift, can be “a home run,” while the lower face is much more complicated, he said in the video interview.
Dr. Zachary disclosed relationships with companies including Solta, Zeltiq, Sciton, DUSA, Zimmer, Cutera, Alma, and Amway.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
LAS VEGAS – For those starting to use toxins and fillers, “my first advice is to get a good education,” Christopher B. Zachary, MD, said in a video interview at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
“Know … how to evaluate your patient, know where the problems are, know what the danger zones are, understand your anatomy,” advised Dr. Zachary of the University of California, Irvine.
, he noted. Procedures on the upper face, such as the treatment for crow’s feet or a brow lift, can be “a home run,” while the lower face is much more complicated, he said in the video interview.
Dr. Zachary disclosed relationships with companies including Solta, Zeltiq, Sciton, DUSA, Zimmer, Cutera, Alma, and Amway.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
LAS VEGAS – For those starting to use toxins and fillers, “my first advice is to get a good education,” Christopher B. Zachary, MD, said in a video interview at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
“Know … how to evaluate your patient, know where the problems are, know what the danger zones are, understand your anatomy,” advised Dr. Zachary of the University of California, Irvine.
, he noted. Procedures on the upper face, such as the treatment for crow’s feet or a brow lift, can be “a home run,” while the lower face is much more complicated, he said in the video interview.
Dr. Zachary disclosed relationships with companies including Solta, Zeltiq, Sciton, DUSA, Zimmer, Cutera, Alma, and Amway.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT SDEF LAS VEGAS DERMATOLOGY SEMINAR

VIDEO: U.S. hypertension guidelines reset threshold to 130/80 mm Hg
ANAHEIM, CALIF. – Thirty million Americans became hypertensive overnight on Nov. 13 with the introduction of new high blood pressure guidelines from the American College of Cardiology and American Heart Association.
That happened by resetting the definition of adult hypertension from the long-standing threshold of 140/90 mm Hg to a blood pressure at or above 130/80 mm Hg, a change that jumps the U.S. adult prevalence of hypertension from roughly 32% to 46%. Nearly half of all U.S. adults now have hypertension, bringing the total national hypertensive population to a staggering 103 million.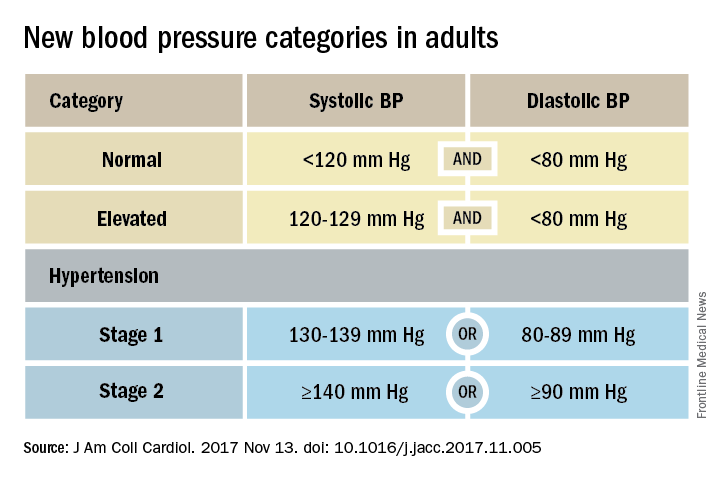
Goal is to transform care
But the new guidelines (J Am Coll Cardiol. 2017 Nov 13. doi: 10.1016/j.jacc.2017.11.005) for preventing, detecting, evaluating, and managing adult hypertension do lots more than just shake up the epidemiology of high blood pressure. With 106 total recommendations, the guidelines seek to transform every aspect of blood pressure in American medical practice, starting with how it’s measured and stretching to redefine applications of medical systems to try to ensure that every person with a blood pressure that truly falls outside the redefined limits gets a comprehensive package of interventions.

Many of these are “seismic changes,” said Lawrence J. Appel, MD. He particularly cited as seismic the new classification of stage 1 hypertension as a pressure at or above 130/80 mm Hg, the emphasis on using some form of out-of-office blood pressure measurement to confirm a diagnosis, the use of risk assessment when deciding whether to treat certain patients with drugs, and the same blood pressure goal of less than 130/80 mm Hg for all hypertensives, regardless of age, as long as they remain ambulatory and community dwelling.
One goal for all adults
“The systolic blood pressure goal for older people has gone from 140 mm Hg to 150 mm Hg and now to 130 mm Hg in the space of 2-3 years,” commented Dr. Appel, professor of epidemiology at Johns Hopkins University in Baltimore and not involved in the guideline-writing process.
In fact, the guidelines simplified the treatment goal all around, to less than 130/80 mm Hg for patients with diabetes, those with chronic kidney disease, and the elderly; that goal remains the same for all adults.
“It will be clearer and easier now that everyone should be less than 130/80 mm Hg. You won’t need to remember a second target,” said Sandra J. Taler, MD, a nephrologist and professor of medicine at the Mayo Clinic in Rochester, Minn., and a member of the guidelines task force.
“Some people may be upset that we changed the rules on them. They had normal blood pressure yesterday, and today it’s high. But it’s a good awakening, especially for using lifestyle interventions,” Dr. Taler said in an interview.
Preferred intervention: Lifestyle, not drugs
Lifestyle optimization is repeatedly cited as the cornerstone of intervention for everyone, including those with elevated blood pressure with a systolic pressure of 120-129 mm Hg, and as the only endorsed intervention for patients with hypertension of 130-139 mm Hg but below a 10% risk for a cardiovascular disease event during the next 10 years on the American College of Cardiology’s online risk calculator. The guidelines list six lifestyle goals: weight loss, following a DASH diet, reducing sodium, enhancing potassium, 90-150 min/wk of physical activity, and moderate alcohol intake.

Team-based care essential
The guidelines also put unprecedented emphasis on using a team-based management approach, which means having nurses, nurse practitioners, pharmacists, dietitians, and other clinicians, allowing for more frequent and focused care. Dr. Whelton and others cited in particular the VA Health System and Kaiser-Permanente as operating team-based and system-driven blood pressure management programs that have resulted in control rates for more than 90% of hypertensive patients. The team-based approach is also a key in the Target:BP program that the American Heart Association and American Medical Association founded. Target:BP will be instrumental in promoting implementation of the new guidelines, Dr. Carey said. Another systems recommendation is that every patient with hypertension should have a “clear, detailed, and current evidence-based plan of care.”
“Using nurse practitioners, physician assistants, and pharmacists has been shown to improve blood pressure levels,” and health systems that use this approach have had “great success,” commented Donald M. Lloyd-Jones, MD, professor and chairman of preventive medicine at Northwestern University in Chicago and not part of the guidelines task force. Some systems have used this approach to achieve high levels of blood pressure control. Now that financial penalties and incentives from payers also exist to push for higher levels of blood pressure control, the alignment of financial and health incentives should result in big changes, Dr. Lloyd-Jones predicted in a video interview.
[email protected]
On Twitter @mitchelzoler
ANAHEIM, CALIF. – Thirty million Americans became hypertensive overnight on Nov. 13 with the introduction of new high blood pressure guidelines from the American College of Cardiology and American Heart Association.
That happened by resetting the definition of adult hypertension from the long-standing threshold of 140/90 mm Hg to a blood pressure at or above 130/80 mm Hg, a change that jumps the U.S. adult prevalence of hypertension from roughly 32% to 46%. Nearly half of all U.S. adults now have hypertension, bringing the total national hypertensive population to a staggering 103 million.
Goal is to transform care
But the new guidelines (J Am Coll Cardiol. 2017 Nov 13. doi: 10.1016/j.jacc.2017.11.005) for preventing, detecting, evaluating, and managing adult hypertension do lots more than just shake up the epidemiology of high blood pressure. With 106 total recommendations, the guidelines seek to transform every aspect of blood pressure in American medical practice, starting with how it’s measured and stretching to redefine applications of medical systems to try to ensure that every person with a blood pressure that truly falls outside the redefined limits gets a comprehensive package of interventions.
Many of these are “seismic changes,” said Lawrence J. Appel, MD. He particularly cited as seismic the new classification of stage 1 hypertension as a pressure at or above 130/80 mm Hg, the emphasis on using some form of out-of-office blood pressure measurement to confirm a diagnosis, the use of risk assessment when deciding whether to treat certain patients with drugs, and the same blood pressure goal of less than 130/80 mm Hg for all hypertensives, regardless of age, as long as they remain ambulatory and community dwelling.
One goal for all adults
“The systolic blood pressure goal for older people has gone from 140 mm Hg to 150 mm Hg and now to 130 mm Hg in the space of 2-3 years,” commented Dr. Appel, professor of epidemiology at Johns Hopkins University in Baltimore and not involved in the guideline-writing process.
In fact, the guidelines simplified the treatment goal all around, to less than 130/80 mm Hg for patients with diabetes, those with chronic kidney disease, and the elderly; that goal remains the same for all adults.
“It will be clearer and easier now that everyone should be less than 130/80 mm Hg. You won’t need to remember a second target,” said Sandra J. Taler, MD, a nephrologist and professor of medicine at the Mayo Clinic in Rochester, Minn., and a member of the guidelines task force.
“Some people may be upset that we changed the rules on them. They had normal blood pressure yesterday, and today it’s high. But it’s a good awakening, especially for using lifestyle interventions,” Dr. Taler said in an interview.
Preferred intervention: Lifestyle, not drugs
Lifestyle optimization is repeatedly cited as the cornerstone of intervention for everyone, including those with elevated blood pressure with a systolic pressure of 120-129 mm Hg, and as the only endorsed intervention for patients with hypertension of 130-139 mm Hg but below a 10% risk for a cardiovascular disease event during the next 10 years on the American College of Cardiology’s online risk calculator. The guidelines list six lifestyle goals: weight loss, following a DASH diet, reducing sodium, enhancing potassium, 90-150 min/wk of physical activity, and moderate alcohol intake.
Team-based care essential
The guidelines also put unprecedented emphasis on using a team-based management approach, which means having nurses, nurse practitioners, pharmacists, dietitians, and other clinicians, allowing for more frequent and focused care. Dr. Whelton and others cited in particular the VA Health System and Kaiser-Permanente as operating team-based and system-driven blood pressure management programs that have resulted in control rates for more than 90% of hypertensive patients. The team-based approach is also a key in the Target:BP program that the American Heart Association and American Medical Association founded. Target:BP will be instrumental in promoting implementation of the new guidelines, Dr. Carey said. Another systems recommendation is that every patient with hypertension should have a “clear, detailed, and current evidence-based plan of care.”
“Using nurse practitioners, physician assistants, and pharmacists has been shown to improve blood pressure levels,” and health systems that use this approach have had “great success,” commented Donald M. Lloyd-Jones, MD, professor and chairman of preventive medicine at Northwestern University in Chicago and not part of the guidelines task force. Some systems have used this approach to achieve high levels of blood pressure control. Now that financial penalties and incentives from payers also exist to push for higher levels of blood pressure control, the alignment of financial and health incentives should result in big changes, Dr. Lloyd-Jones predicted in a video interview.
[email protected]
On Twitter @mitchelzoler
ANAHEIM, CALIF. – Thirty million Americans became hypertensive overnight on Nov. 13 with the introduction of new high blood pressure guidelines from the American College of Cardiology and American Heart Association.
That happened by resetting the definition of adult hypertension from the long-standing threshold of 140/90 mm Hg to a blood pressure at or above 130/80 mm Hg, a change that jumps the U.S. adult prevalence of hypertension from roughly 32% to 46%. Nearly half of all U.S. adults now have hypertension, bringing the total national hypertensive population to a staggering 103 million.
Goal is to transform care
But the new guidelines (J Am Coll Cardiol. 2017 Nov 13. doi: 10.1016/j.jacc.2017.11.005) for preventing, detecting, evaluating, and managing adult hypertension do lots more than just shake up the epidemiology of high blood pressure. With 106 total recommendations, the guidelines seek to transform every aspect of blood pressure in American medical practice, starting with how it’s measured and stretching to redefine applications of medical systems to try to ensure that every person with a blood pressure that truly falls outside the redefined limits gets a comprehensive package of interventions.
Many of these are “seismic changes,” said Lawrence J. Appel, MD. He particularly cited as seismic the new classification of stage 1 hypertension as a pressure at or above 130/80 mm Hg, the emphasis on using some form of out-of-office blood pressure measurement to confirm a diagnosis, the use of risk assessment when deciding whether to treat certain patients with drugs, and the same blood pressure goal of less than 130/80 mm Hg for all hypertensives, regardless of age, as long as they remain ambulatory and community dwelling.
One goal for all adults
“The systolic blood pressure goal for older people has gone from 140 mm Hg to 150 mm Hg and now to 130 mm Hg in the space of 2-3 years,” commented Dr. Appel, professor of epidemiology at Johns Hopkins University in Baltimore and not involved in the guideline-writing process.
In fact, the guidelines simplified the treatment goal all around, to less than 130/80 mm Hg for patients with diabetes, those with chronic kidney disease, and the elderly; that goal remains the same for all adults.
“It will be clearer and easier now that everyone should be less than 130/80 mm Hg. You won’t need to remember a second target,” said Sandra J. Taler, MD, a nephrologist and professor of medicine at the Mayo Clinic in Rochester, Minn., and a member of the guidelines task force.
“Some people may be upset that we changed the rules on them. They had normal blood pressure yesterday, and today it’s high. But it’s a good awakening, especially for using lifestyle interventions,” Dr. Taler said in an interview.
Preferred intervention: Lifestyle, not drugs
Lifestyle optimization is repeatedly cited as the cornerstone of intervention for everyone, including those with elevated blood pressure with a systolic pressure of 120-129 mm Hg, and as the only endorsed intervention for patients with hypertension of 130-139 mm Hg but below a 10% risk for a cardiovascular disease event during the next 10 years on the American College of Cardiology’s online risk calculator. The guidelines list six lifestyle goals: weight loss, following a DASH diet, reducing sodium, enhancing potassium, 90-150 min/wk of physical activity, and moderate alcohol intake.
Team-based care essential
The guidelines also put unprecedented emphasis on using a team-based management approach, which means having nurses, nurse practitioners, pharmacists, dietitians, and other clinicians, allowing for more frequent and focused care. Dr. Whelton and others cited in particular the VA Health System and Kaiser-Permanente as operating team-based and system-driven blood pressure management programs that have resulted in control rates for more than 90% of hypertensive patients. The team-based approach is also a key in the Target:BP program that the American Heart Association and American Medical Association founded. Target:BP will be instrumental in promoting implementation of the new guidelines, Dr. Carey said. Another systems recommendation is that every patient with hypertension should have a “clear, detailed, and current evidence-based plan of care.”
“Using nurse practitioners, physician assistants, and pharmacists has been shown to improve blood pressure levels,” and health systems that use this approach have had “great success,” commented Donald M. Lloyd-Jones, MD, professor and chairman of preventive medicine at Northwestern University in Chicago and not part of the guidelines task force. Some systems have used this approach to achieve high levels of blood pressure control. Now that financial penalties and incentives from payers also exist to push for higher levels of blood pressure control, the alignment of financial and health incentives should result in big changes, Dr. Lloyd-Jones predicted in a video interview.
[email protected]
On Twitter @mitchelzoler
EXPERT ANALYSIS FROM THE AHA SCIENTIFIC SESSIONS
FDA approves dasatinib for pediatric Ph+ CML
(CML).
The tyrosine kinase inhibitor was approved for the treatment of newly diagnosed adult patients with chronic phase Ph+ CML in 2010.
Median follow-up was 4.5 years for newly diagnosed patients and 5.2 years for patients who were resistant to or intolerant of imatinib, the FDA reported. Because more than half of the responding patients had not progressed at the time of data cutoff, the investigators could not estimate median durations of complete cytogenetic response, major cytogenetic response, and major molecular response.
Adverse reactions to dasatinib included headache, nausea, diarrhea, skin rash, vomiting, pain in extremities, abdominal pain, fatigue, and arthralgia; these side effects were reported in approximately 10% of patients.
Dasatinib is marketed as Sprycel by Bristol-Myers Squibb.
The recommended dose of dasatinib for pediatric patients is based on their body weight. Full prescribing information is available here.
[email protected]
On Twitter @nikolaideslaura
(CML).
The tyrosine kinase inhibitor was approved for the treatment of newly diagnosed adult patients with chronic phase Ph+ CML in 2010.
Median follow-up was 4.5 years for newly diagnosed patients and 5.2 years for patients who were resistant to or intolerant of imatinib, the FDA reported. Because more than half of the responding patients had not progressed at the time of data cutoff, the investigators could not estimate median durations of complete cytogenetic response, major cytogenetic response, and major molecular response.
Adverse reactions to dasatinib included headache, nausea, diarrhea, skin rash, vomiting, pain in extremities, abdominal pain, fatigue, and arthralgia; these side effects were reported in approximately 10% of patients.
Dasatinib is marketed as Sprycel by Bristol-Myers Squibb.
The recommended dose of dasatinib for pediatric patients is based on their body weight. Full prescribing information is available here.
[email protected]
On Twitter @nikolaideslaura
(CML).
The tyrosine kinase inhibitor was approved for the treatment of newly diagnosed adult patients with chronic phase Ph+ CML in 2010.
Median follow-up was 4.5 years for newly diagnosed patients and 5.2 years for patients who were resistant to or intolerant of imatinib, the FDA reported. Because more than half of the responding patients had not progressed at the time of data cutoff, the investigators could not estimate median durations of complete cytogenetic response, major cytogenetic response, and major molecular response.
Adverse reactions to dasatinib included headache, nausea, diarrhea, skin rash, vomiting, pain in extremities, abdominal pain, fatigue, and arthralgia; these side effects were reported in approximately 10% of patients.
Dasatinib is marketed as Sprycel by Bristol-Myers Squibb.
The recommended dose of dasatinib for pediatric patients is based on their body weight. Full prescribing information is available here.
[email protected]
On Twitter @nikolaideslaura
The better mammogram: Experts explore sensitivity of new modalities
PHILADELPHIA – Is it time to think about “the better mammogram” as the new standard of care? Can nuclear medicine provide a cost-effective workaround for imaging of women with dense breasts? According to two leading breast imaging researchers,
“Digital breast tomosynthesis is the new kid on the block for screening,” said Emily F. Conant, MD, professor of radiology and chief of breast imaging at the University of Pennsylvania, Philadelphia. “It’s becoming the new standard of care in mammography,” she said, speaking during a plenary session at the annual meeting of the North American Menopause Society.
Digital breast tomosynthesis (DBT) can involve simultaneous acquisition of a conventional 2D mammogram along with a series of images to create a 3D image. Another protocol, which delivers a lower radiation dose, produces a “synthetic” 2D reconstruction of 3D mammography.*

In addition to making visible tumors that otherwise might be obscured by the overlay of dense breast tissue, DBT can help reduce the recall rate, with the 3D images providing immediate clarification at the initial appointment. Studies show that the recall rate can go down by up to 31%, Dr. Conant said.
DBT has been shown to increase detection of invasive cancers, but it does not pick up more ductal carcinoma in situ, Dr. Conant said. This fact helps address the problem of overdiagnosis of small tumors that might regress. Overall, cancer detection is reported to increase by up to 53% with DBT, Dr. Conant said.
When primarily retrospective American studies are taken together with smaller prospective European studies, “the improvement in outcomes achieved with DBT directly addresses the major concerns regarding screening for breast cancer with mammography,” she said.
However, so far the studies have not offered DBT routinely to all comers. Since 2011, DBT has been offered to every woman screened at the University of Pennsylvania, at no additional cost. This created “a sort of natural experiment – there was no bias as to who got it.” Three consecutive years’ worth of outcomes have now been analyzed, Dr. Conant said.
Patient-level data from the University of Pennsylvania experience show statistically significant reductions in recall rate from diagnostic mammography alone. Also, researchers saw a steady increase in the rate of cancers detected per 1,000 patients, from 4.6 with digital mammography alone, to 6.1 by year three of DBT (JAMA Oncol. 2016 Jun 1;2[6]:737-43). This reflected the institutional learning curve with DBT, Dr. Conant said.
She said that the data also showed “a promising trend down in false negatives,” with an early reduction in cancers that were missed by DBT. Time is needed for mature cancer registry data to bear out these early trends, she added.
Other recent data show that DBT has promise to improve detection rates in a population of great interest – younger women, where there are often too many false positives and not enough cancers found, Dr. Conant said. If the risk-benefit ratio for DBT continues to play out as the data pile up, “I would strongly suggest that we should be doing screening in the 40s,” she said.
An important caveat, noted Dr. Conant, is that whether tomosynthesis is used or not, mammography captures anatomy, not physiology, and very dense breast tissue may still obscure a tumor, even when the tomographic slices are peeled back.
Though “DBT is ‘the better mammogram,’ additional outcome data are needed,” she said, including studies that compare modalities, include subgroup analyses, and better delineate the effect of cancer biology.
Molecular breast imaging
Another imaging modality uses nuclear medicine to capture the physiologic changes that accompany cancer. Molecular breast imaging (MBI), or scintimammography, can help “unveil the reservoir of hidden cancers in dense breasts,” said Deborah J. Rhodes, MD, professor of medicine at the Mayo Clinic, Rochester, Minn.
Dr. Rhodes – along with Michael O’Connor, PhD, Connie Hruska, PhD, Katie Hunt, MD, and Amy Conners, MD, her collaborators at the Mayo Clinic – uses a specialized array of gamma cameras to detect uptake of an injected radionuclide that’s preferentially avid for tumor tissue. This technique can unmask smaller tumors not seen on mammogram because it’s not impeded by having to “see” through dense breast tissue.
The radiation dose for an MBI study is a bit more than for DBT, but less than a coronary calcium score scan. The cost is about one-tenth that of breast magnetic resonance imaging (MRI), and interpretation is relatively straightforward, said Dr. Rhodes, who also presented data at the North American Menopause Society plenary.
“The traditional measure of mammography’s performance inflates its effectiveness,” especially in dense breast tissue, said Dr. Rhodes. “What is the sensitivity of mammography in the dense breast? It depends on what you measure it against.”
When cancers detected by MRI or MBI are added, the sensitivity of mammography drops from the 86.9% reported by the Breast Cancer Surveillance Consortium to 21%-31%, according to several published studies.
In one study, Dr. Rhodes and her Mayo colleagues found that the diagnostic yield per 1,000 patients with dense breasts by mammogram alone was 1.9 cancers. When MBI was added, that figure jumped to 8.8 cancers per 1,000 patients, an incremental gain of 363%.
“Tumor size matters profoundly,” she added. “If a tumor is detected above 2 cm, long-term survival drops below 50%.”
That contrasts with the better-than-80% long-term survival rate seen for those with sub-centimeter tumors, even in node-positive disease. “Only a third of tumors are detected when they are less than 1 cm” with regular screening mammography, Dr. Rhodes said.
However, in 2016 the U.S. Preventive Services Task Force concluded that the current evidence was insufficient to assess whether adjunctive screening for breast cancer using breast ultrasonography, MRI, DBT, or other methods should be used in women with dense breasts. The USPSTF noted that there weren’t studies that addressed the effect of supplemental screening on breast cancer morbidity or mortality.
The problem is that it can take 20 years or more to demonstrate mortality reduction, meaning that “no other imaging modality can compete” with mammography when this yardstick is used, Dr. Rhodes said. “This insistence on a mortality endpoint before we change practice” is impeding progress in screening, she said.
The American College of Obstetricians and Gynecologists “does not recommend adjunctive tests to screening mammography in women with dense breasts who are asymptomatic and have no additional risk factors.” However, the organization “strongly supports additional research to identify more effective screening methods” that will improve outcomes and minimize false positives in women with dense breasts.
Though DBT is becoming more widely available, MBI is still primarily used in research centers. Both Dr. Conant and Dr. Rhodes acknowledged that since these techniques are not required to be covered by insurance, payment – and patient access – may vary. Both physicians said their home institutions have worked hard to keep costs down for their studies.
Dr. Conant is consultant or advisory board member for Hologic. Dr. Rhodes reported having no conflicts of interest.
*Correction, 11/15/2017: An earlier version of this story misstated the synthetic mammography protocol.
[email protected]
On Twitter @karioakes
PHILADELPHIA – Is it time to think about “the better mammogram” as the new standard of care? Can nuclear medicine provide a cost-effective workaround for imaging of women with dense breasts? According to two leading breast imaging researchers,
“Digital breast tomosynthesis is the new kid on the block for screening,” said Emily F. Conant, MD, professor of radiology and chief of breast imaging at the University of Pennsylvania, Philadelphia. “It’s becoming the new standard of care in mammography,” she said, speaking during a plenary session at the annual meeting of the North American Menopause Society.
Digital breast tomosynthesis (DBT) can involve simultaneous acquisition of a conventional 2D mammogram along with a series of images to create a 3D image. Another protocol, which delivers a lower radiation dose, produces a “synthetic” 2D reconstruction of 3D mammography.*
In addition to making visible tumors that otherwise might be obscured by the overlay of dense breast tissue, DBT can help reduce the recall rate, with the 3D images providing immediate clarification at the initial appointment. Studies show that the recall rate can go down by up to 31%, Dr. Conant said.
DBT has been shown to increase detection of invasive cancers, but it does not pick up more ductal carcinoma in situ, Dr. Conant said. This fact helps address the problem of overdiagnosis of small tumors that might regress. Overall, cancer detection is reported to increase by up to 53% with DBT, Dr. Conant said.
When primarily retrospective American studies are taken together with smaller prospective European studies, “the improvement in outcomes achieved with DBT directly addresses the major concerns regarding screening for breast cancer with mammography,” she said.
However, so far the studies have not offered DBT routinely to all comers. Since 2011, DBT has been offered to every woman screened at the University of Pennsylvania, at no additional cost. This created “a sort of natural experiment – there was no bias as to who got it.” Three consecutive years’ worth of outcomes have now been analyzed, Dr. Conant said.
Patient-level data from the University of Pennsylvania experience show statistically significant reductions in recall rate from diagnostic mammography alone. Also, researchers saw a steady increase in the rate of cancers detected per 1,000 patients, from 4.6 with digital mammography alone, to 6.1 by year three of DBT (JAMA Oncol. 2016 Jun 1;2[6]:737-43). This reflected the institutional learning curve with DBT, Dr. Conant said.
She said that the data also showed “a promising trend down in false negatives,” with an early reduction in cancers that were missed by DBT. Time is needed for mature cancer registry data to bear out these early trends, she added.
Other recent data show that DBT has promise to improve detection rates in a population of great interest – younger women, where there are often too many false positives and not enough cancers found, Dr. Conant said. If the risk-benefit ratio for DBT continues to play out as the data pile up, “I would strongly suggest that we should be doing screening in the 40s,” she said.
An important caveat, noted Dr. Conant, is that whether tomosynthesis is used or not, mammography captures anatomy, not physiology, and very dense breast tissue may still obscure a tumor, even when the tomographic slices are peeled back.
Though “DBT is ‘the better mammogram,’ additional outcome data are needed,” she said, including studies that compare modalities, include subgroup analyses, and better delineate the effect of cancer biology.
Molecular breast imaging
Another imaging modality uses nuclear medicine to capture the physiologic changes that accompany cancer. Molecular breast imaging (MBI), or scintimammography, can help “unveil the reservoir of hidden cancers in dense breasts,” said Deborah J. Rhodes, MD, professor of medicine at the Mayo Clinic, Rochester, Minn.
Dr. Rhodes – along with Michael O’Connor, PhD, Connie Hruska, PhD, Katie Hunt, MD, and Amy Conners, MD, her collaborators at the Mayo Clinic – uses a specialized array of gamma cameras to detect uptake of an injected radionuclide that’s preferentially avid for tumor tissue. This technique can unmask smaller tumors not seen on mammogram because it’s not impeded by having to “see” through dense breast tissue.
The radiation dose for an MBI study is a bit more than for DBT, but less than a coronary calcium score scan. The cost is about one-tenth that of breast magnetic resonance imaging (MRI), and interpretation is relatively straightforward, said Dr. Rhodes, who also presented data at the North American Menopause Society plenary.
“The traditional measure of mammography’s performance inflates its effectiveness,” especially in dense breast tissue, said Dr. Rhodes. “What is the sensitivity of mammography in the dense breast? It depends on what you measure it against.”
When cancers detected by MRI or MBI are added, the sensitivity of mammography drops from the 86.9% reported by the Breast Cancer Surveillance Consortium to 21%-31%, according to several published studies.
In one study, Dr. Rhodes and her Mayo colleagues found that the diagnostic yield per 1,000 patients with dense breasts by mammogram alone was 1.9 cancers. When MBI was added, that figure jumped to 8.8 cancers per 1,000 patients, an incremental gain of 363%.
“Tumor size matters profoundly,” she added. “If a tumor is detected above 2 cm, long-term survival drops below 50%.”
That contrasts with the better-than-80% long-term survival rate seen for those with sub-centimeter tumors, even in node-positive disease. “Only a third of tumors are detected when they are less than 1 cm” with regular screening mammography, Dr. Rhodes said.
However, in 2016 the U.S. Preventive Services Task Force concluded that the current evidence was insufficient to assess whether adjunctive screening for breast cancer using breast ultrasonography, MRI, DBT, or other methods should be used in women with dense breasts. The USPSTF noted that there weren’t studies that addressed the effect of supplemental screening on breast cancer morbidity or mortality.
The problem is that it can take 20 years or more to demonstrate mortality reduction, meaning that “no other imaging modality can compete” with mammography when this yardstick is used, Dr. Rhodes said. “This insistence on a mortality endpoint before we change practice” is impeding progress in screening, she said.
The American College of Obstetricians and Gynecologists “does not recommend adjunctive tests to screening mammography in women with dense breasts who are asymptomatic and have no additional risk factors.” However, the organization “strongly supports additional research to identify more effective screening methods” that will improve outcomes and minimize false positives in women with dense breasts.
Though DBT is becoming more widely available, MBI is still primarily used in research centers. Both Dr. Conant and Dr. Rhodes acknowledged that since these techniques are not required to be covered by insurance, payment – and patient access – may vary. Both physicians said their home institutions have worked hard to keep costs down for their studies.
Dr. Conant is consultant or advisory board member for Hologic. Dr. Rhodes reported having no conflicts of interest.
*Correction, 11/15/2017: An earlier version of this story misstated the synthetic mammography protocol.
[email protected]
On Twitter @karioakes
PHILADELPHIA – Is it time to think about “the better mammogram” as the new standard of care? Can nuclear medicine provide a cost-effective workaround for imaging of women with dense breasts? According to two leading breast imaging researchers,
“Digital breast tomosynthesis is the new kid on the block for screening,” said Emily F. Conant, MD, professor of radiology and chief of breast imaging at the University of Pennsylvania, Philadelphia. “It’s becoming the new standard of care in mammography,” she said, speaking during a plenary session at the annual meeting of the North American Menopause Society.
Digital breast tomosynthesis (DBT) can involve simultaneous acquisition of a conventional 2D mammogram along with a series of images to create a 3D image. Another protocol, which delivers a lower radiation dose, produces a “synthetic” 2D reconstruction of 3D mammography.*
In addition to making visible tumors that otherwise might be obscured by the overlay of dense breast tissue, DBT can help reduce the recall rate, with the 3D images providing immediate clarification at the initial appointment. Studies show that the recall rate can go down by up to 31%, Dr. Conant said.
DBT has been shown to increase detection of invasive cancers, but it does not pick up more ductal carcinoma in situ, Dr. Conant said. This fact helps address the problem of overdiagnosis of small tumors that might regress. Overall, cancer detection is reported to increase by up to 53% with DBT, Dr. Conant said.
When primarily retrospective American studies are taken together with smaller prospective European studies, “the improvement in outcomes achieved with DBT directly addresses the major concerns regarding screening for breast cancer with mammography,” she said.
However, so far the studies have not offered DBT routinely to all comers. Since 2011, DBT has been offered to every woman screened at the University of Pennsylvania, at no additional cost. This created “a sort of natural experiment – there was no bias as to who got it.” Three consecutive years’ worth of outcomes have now been analyzed, Dr. Conant said.
Patient-level data from the University of Pennsylvania experience show statistically significant reductions in recall rate from diagnostic mammography alone. Also, researchers saw a steady increase in the rate of cancers detected per 1,000 patients, from 4.6 with digital mammography alone, to 6.1 by year three of DBT (JAMA Oncol. 2016 Jun 1;2[6]:737-43). This reflected the institutional learning curve with DBT, Dr. Conant said.
She said that the data also showed “a promising trend down in false negatives,” with an early reduction in cancers that were missed by DBT. Time is needed for mature cancer registry data to bear out these early trends, she added.
Other recent data show that DBT has promise to improve detection rates in a population of great interest – younger women, where there are often too many false positives and not enough cancers found, Dr. Conant said. If the risk-benefit ratio for DBT continues to play out as the data pile up, “I would strongly suggest that we should be doing screening in the 40s,” she said.
An important caveat, noted Dr. Conant, is that whether tomosynthesis is used or not, mammography captures anatomy, not physiology, and very dense breast tissue may still obscure a tumor, even when the tomographic slices are peeled back.
Though “DBT is ‘the better mammogram,’ additional outcome data are needed,” she said, including studies that compare modalities, include subgroup analyses, and better delineate the effect of cancer biology.
Molecular breast imaging
Another imaging modality uses nuclear medicine to capture the physiologic changes that accompany cancer. Molecular breast imaging (MBI), or scintimammography, can help “unveil the reservoir of hidden cancers in dense breasts,” said Deborah J. Rhodes, MD, professor of medicine at the Mayo Clinic, Rochester, Minn.
Dr. Rhodes – along with Michael O’Connor, PhD, Connie Hruska, PhD, Katie Hunt, MD, and Amy Conners, MD, her collaborators at the Mayo Clinic – uses a specialized array of gamma cameras to detect uptake of an injected radionuclide that’s preferentially avid for tumor tissue. This technique can unmask smaller tumors not seen on mammogram because it’s not impeded by having to “see” through dense breast tissue.
The radiation dose for an MBI study is a bit more than for DBT, but less than a coronary calcium score scan. The cost is about one-tenth that of breast magnetic resonance imaging (MRI), and interpretation is relatively straightforward, said Dr. Rhodes, who also presented data at the North American Menopause Society plenary.
“The traditional measure of mammography’s performance inflates its effectiveness,” especially in dense breast tissue, said Dr. Rhodes. “What is the sensitivity of mammography in the dense breast? It depends on what you measure it against.”
When cancers detected by MRI or MBI are added, the sensitivity of mammography drops from the 86.9% reported by the Breast Cancer Surveillance Consortium to 21%-31%, according to several published studies.
In one study, Dr. Rhodes and her Mayo colleagues found that the diagnostic yield per 1,000 patients with dense breasts by mammogram alone was 1.9 cancers. When MBI was added, that figure jumped to 8.8 cancers per 1,000 patients, an incremental gain of 363%.
“Tumor size matters profoundly,” she added. “If a tumor is detected above 2 cm, long-term survival drops below 50%.”
That contrasts with the better-than-80% long-term survival rate seen for those with sub-centimeter tumors, even in node-positive disease. “Only a third of tumors are detected when they are less than 1 cm” with regular screening mammography, Dr. Rhodes said.
However, in 2016 the U.S. Preventive Services Task Force concluded that the current evidence was insufficient to assess whether adjunctive screening for breast cancer using breast ultrasonography, MRI, DBT, or other methods should be used in women with dense breasts. The USPSTF noted that there weren’t studies that addressed the effect of supplemental screening on breast cancer morbidity or mortality.
The problem is that it can take 20 years or more to demonstrate mortality reduction, meaning that “no other imaging modality can compete” with mammography when this yardstick is used, Dr. Rhodes said. “This insistence on a mortality endpoint before we change practice” is impeding progress in screening, she said.
The American College of Obstetricians and Gynecologists “does not recommend adjunctive tests to screening mammography in women with dense breasts who are asymptomatic and have no additional risk factors.” However, the organization “strongly supports additional research to identify more effective screening methods” that will improve outcomes and minimize false positives in women with dense breasts.
Though DBT is becoming more widely available, MBI is still primarily used in research centers. Both Dr. Conant and Dr. Rhodes acknowledged that since these techniques are not required to be covered by insurance, payment – and patient access – may vary. Both physicians said their home institutions have worked hard to keep costs down for their studies.
Dr. Conant is consultant or advisory board member for Hologic. Dr. Rhodes reported having no conflicts of interest.
*Correction, 11/15/2017: An earlier version of this story misstated the synthetic mammography protocol.
[email protected]
On Twitter @karioakes
EXPERT ANALYSIS FROM NAMS 2017
Statin didn’t slow hepatic steatosis in HIV patients
SAN DIEGO – Statin therapy wasn’t effective against a rising rate of hepatic steatosis in HIV patients, and may increase nonalcoholic fatty liver disease (NAFLD) risk, according to a small study.
The findings are contrary to the limited data currently available, which suggest statins can have a beneficial effect on hepatic steatosis, according to the study’s investigators.
NAFLD is one of the leading causes of mortality and morbidity in people living with HIV, said Vanessa El Kamari, MD, of Case Western Reserve University, Cleveland, and rates are continuing to increase. With a lack of therapeutic interventions for NAFLD in patients with HIV, finding effective treatments is a major concern for providers.
Dr. El Kamari and her colleagues conducted a secondary analysis of the SATURN-HIV trial, a randomized, placebo-controlled trial of 147 patients with HIV who were on stable antiretroviral therapy (ART) with LDL cholesterol levels at or below 130 mg/dL. Patients were either treated with 10 mg rosuvastatin or placebo.
Patients in the treatment and placebo arms were an average age of 45 years, most patients were male (81% and 76%, respectively), and a majority was African American (69% and 67%, respectively). The two groups reported average CD4+ counts of 644 and 636, respectively.
Investigators used validated scores to determine hepatic steatosis in patients, although researchers acknowledged that liver biopsy is the gold standard for hepatic steatosis measurement.
“We understand liver fat score has its limitation,” said Dr. El Kamari in a question and answer session following the presentation at an annual scientific meeting on infectious diseases. “Further studies are needed in this area in order to detect, in noninvasive ways, hepatic steatosis.”
Liver fat scores (LFS) were measured on entry, at week 48, and week 96.
After 96 weeks, investigators saw significant increases in LFS in both the placebo and rosuvastatin groups (P = .01 and P less than .01, respectively). Progression from nonsteatosis at baseline to steatosis over 96 weeks was greater in patients given rosuvastatin than in those given placebo (odds ratio, 4.3; P = .03).
Studying predictors for LFS changes, a trend toward insignificance among the randomization group led investigators to the conclusion that statin may have negatively affected the liver fat score of patients over the 96-week time period.
The study researchers identified 92 patients who did not have hepatic steatosis at baseline to study how statins influenced the development of hepatic steatosis.
Of the 13 patients who developed steatosis during the trial period, 10 were part of the rosuvastatin group, while only 3 were from the placebo group.
Increases in LFS were associated with increases in insulin resistance, detected presence of HIV-1 RNA, and higher interferon-inducible proteins, according to Dr. El Kamari.
While increased homeostatic assessment of insulin resistance was associated with increased hepatic homeostasis, Dr. El Kamari was not able to determine if statins that are not associated with insulin resistance will have different outcomes.
The investigators cautioned that the findings may not be generalizable to the entire HIV population due to the overwhelming majority of African American patients with increased inflammation, as well as the study patients’ LDL cholesterol levels of less than or equal to 130 mg/dL.
The National Institutes of Health funded the study. Dr. El Kamari had no disclosures.
[email protected]
On Twitter @eaztweets
SAN DIEGO – Statin therapy wasn’t effective against a rising rate of hepatic steatosis in HIV patients, and may increase nonalcoholic fatty liver disease (NAFLD) risk, according to a small study.
The findings are contrary to the limited data currently available, which suggest statins can have a beneficial effect on hepatic steatosis, according to the study’s investigators.
NAFLD is one of the leading causes of mortality and morbidity in people living with HIV, said Vanessa El Kamari, MD, of Case Western Reserve University, Cleveland, and rates are continuing to increase. With a lack of therapeutic interventions for NAFLD in patients with HIV, finding effective treatments is a major concern for providers.
Dr. El Kamari and her colleagues conducted a secondary analysis of the SATURN-HIV trial, a randomized, placebo-controlled trial of 147 patients with HIV who were on stable antiretroviral therapy (ART) with LDL cholesterol levels at or below 130 mg/dL. Patients were either treated with 10 mg rosuvastatin or placebo.
Patients in the treatment and placebo arms were an average age of 45 years, most patients were male (81% and 76%, respectively), and a majority was African American (69% and 67%, respectively). The two groups reported average CD4+ counts of 644 and 636, respectively.
Investigators used validated scores to determine hepatic steatosis in patients, although researchers acknowledged that liver biopsy is the gold standard for hepatic steatosis measurement.
“We understand liver fat score has its limitation,” said Dr. El Kamari in a question and answer session following the presentation at an annual scientific meeting on infectious diseases. “Further studies are needed in this area in order to detect, in noninvasive ways, hepatic steatosis.”
Liver fat scores (LFS) were measured on entry, at week 48, and week 96.
After 96 weeks, investigators saw significant increases in LFS in both the placebo and rosuvastatin groups (P = .01 and P less than .01, respectively). Progression from nonsteatosis at baseline to steatosis over 96 weeks was greater in patients given rosuvastatin than in those given placebo (odds ratio, 4.3; P = .03).
Studying predictors for LFS changes, a trend toward insignificance among the randomization group led investigators to the conclusion that statin may have negatively affected the liver fat score of patients over the 96-week time period.
The study researchers identified 92 patients who did not have hepatic steatosis at baseline to study how statins influenced the development of hepatic steatosis.
Of the 13 patients who developed steatosis during the trial period, 10 were part of the rosuvastatin group, while only 3 were from the placebo group.
Increases in LFS were associated with increases in insulin resistance, detected presence of HIV-1 RNA, and higher interferon-inducible proteins, according to Dr. El Kamari.
While increased homeostatic assessment of insulin resistance was associated with increased hepatic homeostasis, Dr. El Kamari was not able to determine if statins that are not associated with insulin resistance will have different outcomes.
The investigators cautioned that the findings may not be generalizable to the entire HIV population due to the overwhelming majority of African American patients with increased inflammation, as well as the study patients’ LDL cholesterol levels of less than or equal to 130 mg/dL.
The National Institutes of Health funded the study. Dr. El Kamari had no disclosures.
[email protected]
On Twitter @eaztweets
SAN DIEGO – Statin therapy wasn’t effective against a rising rate of hepatic steatosis in HIV patients, and may increase nonalcoholic fatty liver disease (NAFLD) risk, according to a small study.
The findings are contrary to the limited data currently available, which suggest statins can have a beneficial effect on hepatic steatosis, according to the study’s investigators.
NAFLD is one of the leading causes of mortality and morbidity in people living with HIV, said Vanessa El Kamari, MD, of Case Western Reserve University, Cleveland, and rates are continuing to increase. With a lack of therapeutic interventions for NAFLD in patients with HIV, finding effective treatments is a major concern for providers.
Dr. El Kamari and her colleagues conducted a secondary analysis of the SATURN-HIV trial, a randomized, placebo-controlled trial of 147 patients with HIV who were on stable antiretroviral therapy (ART) with LDL cholesterol levels at or below 130 mg/dL. Patients were either treated with 10 mg rosuvastatin or placebo.
Patients in the treatment and placebo arms were an average age of 45 years, most patients were male (81% and 76%, respectively), and a majority was African American (69% and 67%, respectively). The two groups reported average CD4+ counts of 644 and 636, respectively.
Investigators used validated scores to determine hepatic steatosis in patients, although researchers acknowledged that liver biopsy is the gold standard for hepatic steatosis measurement.
“We understand liver fat score has its limitation,” said Dr. El Kamari in a question and answer session following the presentation at an annual scientific meeting on infectious diseases. “Further studies are needed in this area in order to detect, in noninvasive ways, hepatic steatosis.”
Liver fat scores (LFS) were measured on entry, at week 48, and week 96.
After 96 weeks, investigators saw significant increases in LFS in both the placebo and rosuvastatin groups (P = .01 and P less than .01, respectively). Progression from nonsteatosis at baseline to steatosis over 96 weeks was greater in patients given rosuvastatin than in those given placebo (odds ratio, 4.3; P = .03).
Studying predictors for LFS changes, a trend toward insignificance among the randomization group led investigators to the conclusion that statin may have negatively affected the liver fat score of patients over the 96-week time period.
The study researchers identified 92 patients who did not have hepatic steatosis at baseline to study how statins influenced the development of hepatic steatosis.
Of the 13 patients who developed steatosis during the trial period, 10 were part of the rosuvastatin group, while only 3 were from the placebo group.
Increases in LFS were associated with increases in insulin resistance, detected presence of HIV-1 RNA, and higher interferon-inducible proteins, according to Dr. El Kamari.
While increased homeostatic assessment of insulin resistance was associated with increased hepatic homeostasis, Dr. El Kamari was not able to determine if statins that are not associated with insulin resistance will have different outcomes.
The investigators cautioned that the findings may not be generalizable to the entire HIV population due to the overwhelming majority of African American patients with increased inflammation, as well as the study patients’ LDL cholesterol levels of less than or equal to 130 mg/dL.
The National Institutes of Health funded the study. Dr. El Kamari had no disclosures.
[email protected]
On Twitter @eaztweets
AT IDWEEK 2017
Key clinical point:
Major finding: Progression from nonsteatosis at baseline to steatosis over 96 weeks was greater in patients given rosuvastatin than in those given placebo (odds ratio, 4.3; P = .03).
Data source: A randomized, placebo-controlled study of 147 patients with HIV on stable ART.
Disclosures: The National Institutes of Health funded the study. Dr. El Kamari had no disclosures.
Large database analysis suggests safety of bariatric surgery in seniors
NATIONAL HARBOR, MD. – despite a slight increase in unadjusted mortality rates, according to an analysis of data from the Metabolic and Bariatric Surgery Accreditation and Quality Improvement Program (MBSAQIP).
Based on data that was collected in 2015 and submitted to MBSAQIP, “bariatric surgery is safe in the elderly, even in those 70 years old and older,” reported Tallal Zeni, MD, director of the Michigan Bariatric Institute in Livonia.

There were 16,568 patients older than age 60 years entered into the MBSAQIP database in 2015. When those were compared with the 117,443 younger patients, the unadjusted rates of morbidity (6.5% vs. 6.0%) and mortality (0.3% vs. 0.1%) were higher for the older patients, but “they are close,” according to Dr. Zeni. Both rates reached significance by the conventional definition (P < .05), but he suggested that they are lower in this study than those in prior studies of MBSAQIP datasets and that they are acceptable relative to the anticipated health benefits.
Above the age of 60 years, no correlation could be made between increasing age and increasing risk of morbidity, mortality, or rate of reoperations, according to Dr. Zeni.
Why should bariatric surgery be considered in older patients? He cited data from a study that showed the life expectancy in a 70-year-old without functional limitations is 13 years. As a result, he added, “it behooves us to provide them with the best quality of life we can.”
Relative to prior MBSAQIP evaluations of bariatric surgery in the elderly, the proportion of patients undergoing sleeve gastrectomy relative to gastric bypass has been increasing, Dr. Zeni reported. In the analysis, approximately two-thirds of the bariatric procedures were performed with sleeve gastrectomy, which is higher relative to what previous MBSAQIP analyses have shown.
Based on rates of morbidity for those two surgical approaches in the analysis, that trend makes sense. While the higher 30-day mortality for gastric bypass, compared with sleeve gastrectomy, was not significant (0.38% vs. 0.26%; P = .221), all-cause morbidity was almost two times greater for those undergoing gastric bypass than it was for those undergoing sleeve gastrectomy (10.61% vs. 5.81%; P < .001), Dr. Zeni reported.
However, some of that difference may be explained by baseline disparities between the two groups. In the gastric bypass group, there were higher rates of preoperative diabetes (54% vs. 40%; P < .001), sleep apnea (57% vs. 50%; P < .001) and hyperlipidemia (59% vs. 54%; P < .001). Also, gastric bypass patients were more likely to have a history of a previous bariatric procedure (11% vs. 8.5%; P < .001) and to be in the American Society of Anesthesiologists Physical Status score of 3 (84% vs. 80%; P < .001), according to Dr. Zeni.
The specific complications more common in the gastric bypass group than the sleeve gastrectomy group included anastomotic leak (0.56% vs. 0.3%; P = .017), surgical site infection (1.74% vs. 0.61%; P < .001), pneumonia (0.87% vs. 0.32%; P < .001), and bleeding (1.14% vs. 0.5%; P = .024). Although the average operating time was 40 minutes longer in the bypass group, there were no significant differences in thromboembolic complications.
Overall, despite a modest increase in the risk of complications for bariatric surgery in elderly patients, that risk can be considered acceptable in relation to the potential health benefits, according to Dr. Zeni. He suggested that the data might encourage further growth in the rates of bariatric procedures among patients older than 60 years.
Dr. Zeni reports no relevant financial relationships.
NATIONAL HARBOR, MD. – despite a slight increase in unadjusted mortality rates, according to an analysis of data from the Metabolic and Bariatric Surgery Accreditation and Quality Improvement Program (MBSAQIP).
Based on data that was collected in 2015 and submitted to MBSAQIP, “bariatric surgery is safe in the elderly, even in those 70 years old and older,” reported Tallal Zeni, MD, director of the Michigan Bariatric Institute in Livonia.
There were 16,568 patients older than age 60 years entered into the MBSAQIP database in 2015. When those were compared with the 117,443 younger patients, the unadjusted rates of morbidity (6.5% vs. 6.0%) and mortality (0.3% vs. 0.1%) were higher for the older patients, but “they are close,” according to Dr. Zeni. Both rates reached significance by the conventional definition (P < .05), but he suggested that they are lower in this study than those in prior studies of MBSAQIP datasets and that they are acceptable relative to the anticipated health benefits.
Above the age of 60 years, no correlation could be made between increasing age and increasing risk of morbidity, mortality, or rate of reoperations, according to Dr. Zeni.
Why should bariatric surgery be considered in older patients? He cited data from a study that showed the life expectancy in a 70-year-old without functional limitations is 13 years. As a result, he added, “it behooves us to provide them with the best quality of life we can.”
Relative to prior MBSAQIP evaluations of bariatric surgery in the elderly, the proportion of patients undergoing sleeve gastrectomy relative to gastric bypass has been increasing, Dr. Zeni reported. In the analysis, approximately two-thirds of the bariatric procedures were performed with sleeve gastrectomy, which is higher relative to what previous MBSAQIP analyses have shown.
Based on rates of morbidity for those two surgical approaches in the analysis, that trend makes sense. While the higher 30-day mortality for gastric bypass, compared with sleeve gastrectomy, was not significant (0.38% vs. 0.26%; P = .221), all-cause morbidity was almost two times greater for those undergoing gastric bypass than it was for those undergoing sleeve gastrectomy (10.61% vs. 5.81%; P < .001), Dr. Zeni reported.
However, some of that difference may be explained by baseline disparities between the two groups. In the gastric bypass group, there were higher rates of preoperative diabetes (54% vs. 40%; P < .001), sleep apnea (57% vs. 50%; P < .001) and hyperlipidemia (59% vs. 54%; P < .001). Also, gastric bypass patients were more likely to have a history of a previous bariatric procedure (11% vs. 8.5%; P < .001) and to be in the American Society of Anesthesiologists Physical Status score of 3 (84% vs. 80%; P < .001), according to Dr. Zeni.
The specific complications more common in the gastric bypass group than the sleeve gastrectomy group included anastomotic leak (0.56% vs. 0.3%; P = .017), surgical site infection (1.74% vs. 0.61%; P < .001), pneumonia (0.87% vs. 0.32%; P < .001), and bleeding (1.14% vs. 0.5%; P = .024). Although the average operating time was 40 minutes longer in the bypass group, there were no significant differences in thromboembolic complications.
Overall, despite a modest increase in the risk of complications for bariatric surgery in elderly patients, that risk can be considered acceptable in relation to the potential health benefits, according to Dr. Zeni. He suggested that the data might encourage further growth in the rates of bariatric procedures among patients older than 60 years.
Dr. Zeni reports no relevant financial relationships.
NATIONAL HARBOR, MD. – despite a slight increase in unadjusted mortality rates, according to an analysis of data from the Metabolic and Bariatric Surgery Accreditation and Quality Improvement Program (MBSAQIP).
Based on data that was collected in 2015 and submitted to MBSAQIP, “bariatric surgery is safe in the elderly, even in those 70 years old and older,” reported Tallal Zeni, MD, director of the Michigan Bariatric Institute in Livonia.
There were 16,568 patients older than age 60 years entered into the MBSAQIP database in 2015. When those were compared with the 117,443 younger patients, the unadjusted rates of morbidity (6.5% vs. 6.0%) and mortality (0.3% vs. 0.1%) were higher for the older patients, but “they are close,” according to Dr. Zeni. Both rates reached significance by the conventional definition (P < .05), but he suggested that they are lower in this study than those in prior studies of MBSAQIP datasets and that they are acceptable relative to the anticipated health benefits.
Above the age of 60 years, no correlation could be made between increasing age and increasing risk of morbidity, mortality, or rate of reoperations, according to Dr. Zeni.
Why should bariatric surgery be considered in older patients? He cited data from a study that showed the life expectancy in a 70-year-old without functional limitations is 13 years. As a result, he added, “it behooves us to provide them with the best quality of life we can.”
Relative to prior MBSAQIP evaluations of bariatric surgery in the elderly, the proportion of patients undergoing sleeve gastrectomy relative to gastric bypass has been increasing, Dr. Zeni reported. In the analysis, approximately two-thirds of the bariatric procedures were performed with sleeve gastrectomy, which is higher relative to what previous MBSAQIP analyses have shown.
Based on rates of morbidity for those two surgical approaches in the analysis, that trend makes sense. While the higher 30-day mortality for gastric bypass, compared with sleeve gastrectomy, was not significant (0.38% vs. 0.26%; P = .221), all-cause morbidity was almost two times greater for those undergoing gastric bypass than it was for those undergoing sleeve gastrectomy (10.61% vs. 5.81%; P < .001), Dr. Zeni reported.
However, some of that difference may be explained by baseline disparities between the two groups. In the gastric bypass group, there were higher rates of preoperative diabetes (54% vs. 40%; P < .001), sleep apnea (57% vs. 50%; P < .001) and hyperlipidemia (59% vs. 54%; P < .001). Also, gastric bypass patients were more likely to have a history of a previous bariatric procedure (11% vs. 8.5%; P < .001) and to be in the American Society of Anesthesiologists Physical Status score of 3 (84% vs. 80%; P < .001), according to Dr. Zeni.
The specific complications more common in the gastric bypass group than the sleeve gastrectomy group included anastomotic leak (0.56% vs. 0.3%; P = .017), surgical site infection (1.74% vs. 0.61%; P < .001), pneumonia (0.87% vs. 0.32%; P < .001), and bleeding (1.14% vs. 0.5%; P = .024). Although the average operating time was 40 minutes longer in the bypass group, there were no significant differences in thromboembolic complications.
Overall, despite a modest increase in the risk of complications for bariatric surgery in elderly patients, that risk can be considered acceptable in relation to the potential health benefits, according to Dr. Zeni. He suggested that the data might encourage further growth in the rates of bariatric procedures among patients older than 60 years.
Dr. Zeni reports no relevant financial relationships.
AT OBESITY WEEK 2017
Key clinical point: Based on mortality and morbidity rates, bariatric surgery is acceptably safe in patients older than 60 years of age.
Major finding: Compared with patients younger than 60 years, older patients had only modestly increased rates of morbidity (6.5% vs. 6.0%) and mortality (0.3% vs. 0.1%).
Data source: A retrospective database analysis.
Disclosures: Dr. Zeni reports no relevant financial relationships.
Targeting PCSK9 inhibitors to reap most benefit
ANAHEIM, CALIF. – Patients with symptomatic peripheral artery disease or a high-risk history of MI got the biggest bang for the buck from aggressive LDL cholesterol lowering with evolocumab in two new prespecified subgroup analyses from the landmark FOURIER trial presented at the American Heart Association scientific sessions.
“At the end of the day, not all of our patients with ASCVD [atherosclerotic cardiovascular disease] can have these expensive medications. These subgroup analyses will help clinicians to target use of PCSK9 inhibitors to the patients who will benefit the most,” Lynne T. Braun, PhD, commented in her role as discussant of the two secondary analyses, presented back to back in a late-breaking science session. Dr. Braun is a professor in the department of internal medicine at Rush University, Chicago.
The FOURIER trial included 27,564 high-risk patients with prior MI, stroke, and/or symptomatic peripheral arterial disease (PAD) who had an LDL cholesterol level of 70 mg/dL or more on high- or moderate-intensity statin therapy. They were randomized in double-blind fashion to add-on subcutaneous evolocumab (Repatha) at either 140 mg every 2 weeks or 420 mg/month or to placebo, for a median of 2.5 years of follow-up. The evolocumab group experienced a 59% reduction in LDL cholesterol, compared with the controls on background statin therapy plus placebo, down to a mean LDL cholesterol level of just 30 mg/dL.
As previously reported, the risk of the primary composite endpoint – comprising cardiovascular death, MI, stroke, unstable angina, or coronary revascularization – was reduced by 15% in the evolocumab group at 3 years. The secondary endpoint of cardiovascular death, MI, or stroke was reduced by 20%, from 9.9% to 7.9% (N Engl J Med. 2017;376:1713-22).
Evolocumab tamed PAD
At the AHA scientific sessions, Marc P. Bonaca, MD, presented a secondary analysis restricted to the 3,642 FOURIER participants with symptomatic PAD. The goal was to answer two unresolved questions: Does LDL cholesterol lowering beyond what’s achievable with a statin further reduce PAD patients’ cardiovascular risk? And does it reduce their risk of major adverse leg events (MALE), defined as a composite of acute limb ischemia, major amputation, and urgent revascularization?

The rate of the composite endpoint comprising cardiovascular death, MI, or stroke was 13% over 3 years in PAD patients randomized to placebo, which was 81% greater than the 7.6% rate in placebo-treated participants with a baseline history of stroke or MI but no PAD, in an analysis adjusted for demographics, cardiovascular risk factors, kidney function, body mass index, and prior revascularization.
The event rate was even higher in patients with PAD plus a history of MI or stroke, at 14.9%. Evolocumab reduced that risk by 27%, compared with placebo in patients with PAD, for an absolute risk reduction of 3.5% and a number-needed-to-treat (NNT) of 29 for 2.5 years.
The benefit of evolocumab was even more pronounced in the subgroup of 1,505 patients with baseline PAD but no prior MI or stroke: a 43% relative risk reduction, from 10.3% to 5.5%, for an absolute risk reduction of 4.8% and a NNT of 21.
A linear relationship was seen between the MALE rate during follow-up and LDL cholesterol level after 1 month of therapy, down to an LDL cholesterol level of less than 10 mg/dL. The clinically relevant composite endpoint of MACE (major adverse cardiovascular events – a composite of cardiovascular death, MI, and stroke) or MALE in patients with baseline PAD but no history of MI or stroke occurred in 12.8% of controls and 6.5% of the evolocumab group. This translated to a 48% relative risk reduction, a 6.3% absolute risk reduction, and a NNT of 16. The event curves in the evolocumab and control arms separated quite early, within the first 90 days of treatment.
The take home message: “LDL reduction to very low levels should be considered in patients with PAD, regardless of their history of MI or stroke, to reduce the risk of MACE [major adverse cardiovascular event] and MALE,” declared Dr. Bonaca of Brigham and Women’s Hospital and Harvard Medical School, both in Boston.
Spotting the patients with a history of MI who’re at highest risk
Marc S. Sabatine, MD, presented the subanalysis involving the 22,351 FOURIER patients with a prior MI. He and his coinvestigators identified three high-risk features within this group: an MI within the past 2 years, a history of two or more MIs, and residual multivessel CAD. Each of these three features was individually associated with a 34%-90% increased risk of MACE during follow-up. All told, 63% of FOURIER participants with prior MI had one or more of the high-risk features.

The use of evolocumab in patients with at least one of the three high-risk features was associated with a 22% relative risk reduction and an absolute 2.5% risk reduction, compared with placebo. The event curves diverged at about 6 months, and the gap between them steadily widened during follow-up. Extrapolating from this pattern, it’s likely that evolocumab would achieve an absolute 5% risk reduction in MACE, compared with placebo over 5 years, with an NNT of 20, according to Dr. Sabatine, professor of medicine at Harvard Medical School and chairman of the Thrombolysis in Myocardial Infarction (TIMI) Study Group.
Lingering questions
Dr. Braun was particularly impressed that the absolute risk reduction in MACE was even larger in patients with baseline PAD but no history of stroke or MI than in PAD patients with such a history. She added that, while she recognizes the value of selecting objectively assessable hard clinical MACE as the primary endpoint in FOURIER, her own patients care even more about other outcomes.
“What my patients with PAD care most about is whether profound LDL lowering translates to less claudication, improved quality of life, and greater physical activity tolerance. These were prespecified secondary outcomes in FOURIER, and I look forward to future reports addressing those issues,” she said.
Another unanswered question involves the mechanism by which intensive LDL cholesterol lowering results in fewer MACE and MALE events in high-risk subgroups. The possibilities include the plaque regression that was documented in the GLAGOV trial, an anti-inflammatory plaque-stabilizing effect being exerted through PCSK9 inhibition, or perhaps the PCSK9 inhibitors’ ability to moderately lower lipoprotein(a) cholesterol levels.
Simultaneous with Dr. Bonaca’s presentation at the AHA, the FOURIER PAD analysis was published online in Circulation (2017 Nov 13; doi: 10.1161/CIRCULATIONAHA.117.032235).
The FOURIER trial was sponsored by Amgen. Dr. Bonaca and Dr. Sabatine reported receiving research grants from and serving as consultants to Amgen and other companies.
ANAHEIM, CALIF. – Patients with symptomatic peripheral artery disease or a high-risk history of MI got the biggest bang for the buck from aggressive LDL cholesterol lowering with evolocumab in two new prespecified subgroup analyses from the landmark FOURIER trial presented at the American Heart Association scientific sessions.
“At the end of the day, not all of our patients with ASCVD [atherosclerotic cardiovascular disease] can have these expensive medications. These subgroup analyses will help clinicians to target use of PCSK9 inhibitors to the patients who will benefit the most,” Lynne T. Braun, PhD, commented in her role as discussant of the two secondary analyses, presented back to back in a late-breaking science session. Dr. Braun is a professor in the department of internal medicine at Rush University, Chicago.
The FOURIER trial included 27,564 high-risk patients with prior MI, stroke, and/or symptomatic peripheral arterial disease (PAD) who had an LDL cholesterol level of 70 mg/dL or more on high- or moderate-intensity statin therapy. They were randomized in double-blind fashion to add-on subcutaneous evolocumab (Repatha) at either 140 mg every 2 weeks or 420 mg/month or to placebo, for a median of 2.5 years of follow-up. The evolocumab group experienced a 59% reduction in LDL cholesterol, compared with the controls on background statin therapy plus placebo, down to a mean LDL cholesterol level of just 30 mg/dL.
As previously reported, the risk of the primary composite endpoint – comprising cardiovascular death, MI, stroke, unstable angina, or coronary revascularization – was reduced by 15% in the evolocumab group at 3 years. The secondary endpoint of cardiovascular death, MI, or stroke was reduced by 20%, from 9.9% to 7.9% (N Engl J Med. 2017;376:1713-22).
Evolocumab tamed PAD
At the AHA scientific sessions, Marc P. Bonaca, MD, presented a secondary analysis restricted to the 3,642 FOURIER participants with symptomatic PAD. The goal was to answer two unresolved questions: Does LDL cholesterol lowering beyond what’s achievable with a statin further reduce PAD patients’ cardiovascular risk? And does it reduce their risk of major adverse leg events (MALE), defined as a composite of acute limb ischemia, major amputation, and urgent revascularization?
The rate of the composite endpoint comprising cardiovascular death, MI, or stroke was 13% over 3 years in PAD patients randomized to placebo, which was 81% greater than the 7.6% rate in placebo-treated participants with a baseline history of stroke or MI but no PAD, in an analysis adjusted for demographics, cardiovascular risk factors, kidney function, body mass index, and prior revascularization.
The event rate was even higher in patients with PAD plus a history of MI or stroke, at 14.9%. Evolocumab reduced that risk by 27%, compared with placebo in patients with PAD, for an absolute risk reduction of 3.5% and a number-needed-to-treat (NNT) of 29 for 2.5 years.
The benefit of evolocumab was even more pronounced in the subgroup of 1,505 patients with baseline PAD but no prior MI or stroke: a 43% relative risk reduction, from 10.3% to 5.5%, for an absolute risk reduction of 4.8% and a NNT of 21.
A linear relationship was seen between the MALE rate during follow-up and LDL cholesterol level after 1 month of therapy, down to an LDL cholesterol level of less than 10 mg/dL. The clinically relevant composite endpoint of MACE (major adverse cardiovascular events – a composite of cardiovascular death, MI, and stroke) or MALE in patients with baseline PAD but no history of MI or stroke occurred in 12.8% of controls and 6.5% of the evolocumab group. This translated to a 48% relative risk reduction, a 6.3% absolute risk reduction, and a NNT of 16. The event curves in the evolocumab and control arms separated quite early, within the first 90 days of treatment.
The take home message: “LDL reduction to very low levels should be considered in patients with PAD, regardless of their history of MI or stroke, to reduce the risk of MACE [major adverse cardiovascular event] and MALE,” declared Dr. Bonaca of Brigham and Women’s Hospital and Harvard Medical School, both in Boston.
Spotting the patients with a history of MI who’re at highest risk
Marc S. Sabatine, MD, presented the subanalysis involving the 22,351 FOURIER patients with a prior MI. He and his coinvestigators identified three high-risk features within this group: an MI within the past 2 years, a history of two or more MIs, and residual multivessel CAD. Each of these three features was individually associated with a 34%-90% increased risk of MACE during follow-up. All told, 63% of FOURIER participants with prior MI had one or more of the high-risk features.
The use of evolocumab in patients with at least one of the three high-risk features was associated with a 22% relative risk reduction and an absolute 2.5% risk reduction, compared with placebo. The event curves diverged at about 6 months, and the gap between them steadily widened during follow-up. Extrapolating from this pattern, it’s likely that evolocumab would achieve an absolute 5% risk reduction in MACE, compared with placebo over 5 years, with an NNT of 20, according to Dr. Sabatine, professor of medicine at Harvard Medical School and chairman of the Thrombolysis in Myocardial Infarction (TIMI) Study Group.
Lingering questions
Dr. Braun was particularly impressed that the absolute risk reduction in MACE was even larger in patients with baseline PAD but no history of stroke or MI than in PAD patients with such a history. She added that, while she recognizes the value of selecting objectively assessable hard clinical MACE as the primary endpoint in FOURIER, her own patients care even more about other outcomes.
“What my patients with PAD care most about is whether profound LDL lowering translates to less claudication, improved quality of life, and greater physical activity tolerance. These were prespecified secondary outcomes in FOURIER, and I look forward to future reports addressing those issues,” she said.
Another unanswered question involves the mechanism by which intensive LDL cholesterol lowering results in fewer MACE and MALE events in high-risk subgroups. The possibilities include the plaque regression that was documented in the GLAGOV trial, an anti-inflammatory plaque-stabilizing effect being exerted through PCSK9 inhibition, or perhaps the PCSK9 inhibitors’ ability to moderately lower lipoprotein(a) cholesterol levels.
Simultaneous with Dr. Bonaca’s presentation at the AHA, the FOURIER PAD analysis was published online in Circulation (2017 Nov 13; doi: 10.1161/CIRCULATIONAHA.117.032235).
The FOURIER trial was sponsored by Amgen. Dr. Bonaca and Dr. Sabatine reported receiving research grants from and serving as consultants to Amgen and other companies.
ANAHEIM, CALIF. – Patients with symptomatic peripheral artery disease or a high-risk history of MI got the biggest bang for the buck from aggressive LDL cholesterol lowering with evolocumab in two new prespecified subgroup analyses from the landmark FOURIER trial presented at the American Heart Association scientific sessions.
“At the end of the day, not all of our patients with ASCVD [atherosclerotic cardiovascular disease] can have these expensive medications. These subgroup analyses will help clinicians to target use of PCSK9 inhibitors to the patients who will benefit the most,” Lynne T. Braun, PhD, commented in her role as discussant of the two secondary analyses, presented back to back in a late-breaking science session. Dr. Braun is a professor in the department of internal medicine at Rush University, Chicago.
The FOURIER trial included 27,564 high-risk patients with prior MI, stroke, and/or symptomatic peripheral arterial disease (PAD) who had an LDL cholesterol level of 70 mg/dL or more on high- or moderate-intensity statin therapy. They were randomized in double-blind fashion to add-on subcutaneous evolocumab (Repatha) at either 140 mg every 2 weeks or 420 mg/month or to placebo, for a median of 2.5 years of follow-up. The evolocumab group experienced a 59% reduction in LDL cholesterol, compared with the controls on background statin therapy plus placebo, down to a mean LDL cholesterol level of just 30 mg/dL.
As previously reported, the risk of the primary composite endpoint – comprising cardiovascular death, MI, stroke, unstable angina, or coronary revascularization – was reduced by 15% in the evolocumab group at 3 years. The secondary endpoint of cardiovascular death, MI, or stroke was reduced by 20%, from 9.9% to 7.9% (N Engl J Med. 2017;376:1713-22).
Evolocumab tamed PAD
At the AHA scientific sessions, Marc P. Bonaca, MD, presented a secondary analysis restricted to the 3,642 FOURIER participants with symptomatic PAD. The goal was to answer two unresolved questions: Does LDL cholesterol lowering beyond what’s achievable with a statin further reduce PAD patients’ cardiovascular risk? And does it reduce their risk of major adverse leg events (MALE), defined as a composite of acute limb ischemia, major amputation, and urgent revascularization?
The rate of the composite endpoint comprising cardiovascular death, MI, or stroke was 13% over 3 years in PAD patients randomized to placebo, which was 81% greater than the 7.6% rate in placebo-treated participants with a baseline history of stroke or MI but no PAD, in an analysis adjusted for demographics, cardiovascular risk factors, kidney function, body mass index, and prior revascularization.
The event rate was even higher in patients with PAD plus a history of MI or stroke, at 14.9%. Evolocumab reduced that risk by 27%, compared with placebo in patients with PAD, for an absolute risk reduction of 3.5% and a number-needed-to-treat (NNT) of 29 for 2.5 years.
The benefit of evolocumab was even more pronounced in the subgroup of 1,505 patients with baseline PAD but no prior MI or stroke: a 43% relative risk reduction, from 10.3% to 5.5%, for an absolute risk reduction of 4.8% and a NNT of 21.
A linear relationship was seen between the MALE rate during follow-up and LDL cholesterol level after 1 month of therapy, down to an LDL cholesterol level of less than 10 mg/dL. The clinically relevant composite endpoint of MACE (major adverse cardiovascular events – a composite of cardiovascular death, MI, and stroke) or MALE in patients with baseline PAD but no history of MI or stroke occurred in 12.8% of controls and 6.5% of the evolocumab group. This translated to a 48% relative risk reduction, a 6.3% absolute risk reduction, and a NNT of 16. The event curves in the evolocumab and control arms separated quite early, within the first 90 days of treatment.
The take home message: “LDL reduction to very low levels should be considered in patients with PAD, regardless of their history of MI or stroke, to reduce the risk of MACE [major adverse cardiovascular event] and MALE,” declared Dr. Bonaca of Brigham and Women’s Hospital and Harvard Medical School, both in Boston.
Spotting the patients with a history of MI who’re at highest risk
Marc S. Sabatine, MD, presented the subanalysis involving the 22,351 FOURIER patients with a prior MI. He and his coinvestigators identified three high-risk features within this group: an MI within the past 2 years, a history of two or more MIs, and residual multivessel CAD. Each of these three features was individually associated with a 34%-90% increased risk of MACE during follow-up. All told, 63% of FOURIER participants with prior MI had one or more of the high-risk features.
The use of evolocumab in patients with at least one of the three high-risk features was associated with a 22% relative risk reduction and an absolute 2.5% risk reduction, compared with placebo. The event curves diverged at about 6 months, and the gap between them steadily widened during follow-up. Extrapolating from this pattern, it’s likely that evolocumab would achieve an absolute 5% risk reduction in MACE, compared with placebo over 5 years, with an NNT of 20, according to Dr. Sabatine, professor of medicine at Harvard Medical School and chairman of the Thrombolysis in Myocardial Infarction (TIMI) Study Group.
Lingering questions
Dr. Braun was particularly impressed that the absolute risk reduction in MACE was even larger in patients with baseline PAD but no history of stroke or MI than in PAD patients with such a history. She added that, while she recognizes the value of selecting objectively assessable hard clinical MACE as the primary endpoint in FOURIER, her own patients care even more about other outcomes.
“What my patients with PAD care most about is whether profound LDL lowering translates to less claudication, improved quality of life, and greater physical activity tolerance. These were prespecified secondary outcomes in FOURIER, and I look forward to future reports addressing those issues,” she said.
Another unanswered question involves the mechanism by which intensive LDL cholesterol lowering results in fewer MACE and MALE events in high-risk subgroups. The possibilities include the plaque regression that was documented in the GLAGOV trial, an anti-inflammatory plaque-stabilizing effect being exerted through PCSK9 inhibition, or perhaps the PCSK9 inhibitors’ ability to moderately lower lipoprotein(a) cholesterol levels.
Simultaneous with Dr. Bonaca’s presentation at the AHA, the FOURIER PAD analysis was published online in Circulation (2017 Nov 13; doi: 10.1161/CIRCULATIONAHA.117.032235).
The FOURIER trial was sponsored by Amgen. Dr. Bonaca and Dr. Sabatine reported receiving research grants from and serving as consultants to Amgen and other companies.
EXPERT ANALYSIS FROM THE AHA SCIENTIFIC SESSIONS
Adherence boon, or Big Brother loom?
The Food and Drug Administration has approved the first drug in the United States with a digital ingestion tracking system. Abilify MyCite (aripiprazole tablets with sensor) has an ingestible sensor embedded in the pill that records that the medication was taken. The product is approved for the treatment of schizophrenia, acute treatment of manic and mixed episodes associated with bipolar I disorder, and for use as an add-on treatment for depression in adults.
The system works by sending a message from the pill’s sensor to a wearable patch, according to a statement issued by the FDA. The patch transmits the information to a mobile application so that patients can track the ingestion of the medication on their smartphones. Patients can also permit their caregivers and physician to access the information through a web-based portal.
[polldaddy:9874958]
The Food and Drug Administration has approved the first drug in the United States with a digital ingestion tracking system. Abilify MyCite (aripiprazole tablets with sensor) has an ingestible sensor embedded in the pill that records that the medication was taken. The product is approved for the treatment of schizophrenia, acute treatment of manic and mixed episodes associated with bipolar I disorder, and for use as an add-on treatment for depression in adults.
The system works by sending a message from the pill’s sensor to a wearable patch, according to a statement issued by the FDA. The patch transmits the information to a mobile application so that patients can track the ingestion of the medication on their smartphones. Patients can also permit their caregivers and physician to access the information through a web-based portal.
[polldaddy:9874958]
The Food and Drug Administration has approved the first drug in the United States with a digital ingestion tracking system. Abilify MyCite (aripiprazole tablets with sensor) has an ingestible sensor embedded in the pill that records that the medication was taken. The product is approved for the treatment of schizophrenia, acute treatment of manic and mixed episodes associated with bipolar I disorder, and for use as an add-on treatment for depression in adults.
The system works by sending a message from the pill’s sensor to a wearable patch, according to a statement issued by the FDA. The patch transmits the information to a mobile application so that patients can track the ingestion of the medication on their smartphones. Patients can also permit their caregivers and physician to access the information through a web-based portal.
[polldaddy:9874958]
VIDEO: Beware of over-relying on MRI findings in axSpA
SAN DIEGO – Healthy individuals can show signs of spinal and pelvic inflammation on MRI, but these scans can be misleading if relied on to make a diagnosis of axial spondyloarthritis, according to findings from three separate studies at the annual meeting of the American College of Rheumatology.
“Don’t rely on MRI alone is our message,” said Robert Landewé, MD, PhD, of the University of Amsterdam, who was a coauthor of one of the three studies. “A positive MRI may occur in individuals that are completely healthy. We need to make sure that not too many patients with chronic lower back pain are diagnosed with a disease they don’t have.”
The axial form of spondyloarthritis (axSpA) affects the spinal and pelvic joints of an estimated 1.4% of the U.S. population, and the term encompasses the diagnosis of ankylosing spondylitis (0.5% of U.S. population) in which advanced sacroiliitis is seen on conventional radiography, according to the ACR. Axial SpA is particularly common in young people, especially males, in their teens and 20s.
Researchers believe that MRI scans can misleadingly suggest that patients have the condition. “We know that MRI is a sensitive method, but there’s a lack of data regarding its specificity,” Thomas Renson, MD, of Ghent (Belgium) University, said at a press conference during the meeting.
Dr. Landewé led a study that compared MRIs of sacroiliac joints in 47 healthy people, 47 axSpA patients matched for gender and age, 47 chronic back pain patients, 7 women with postpartum back pain, and 24 frequent runners. Positive MRIs were common in the axSpA patients (43 of 47), but they were also found in healthy people (11 of 47), chronic back pain patients (3 of 47), frequent runners (3 of 24), and women with postpartum back pain (4 of 7).
In another study, Dr. Renson and his colleagues sought to understand whether a sustained period of intense physical activity affected spinal findings in 22 healthy military recruits who did not have SpA.

However, there was a statistically significant increase of combined structural and inflammatory lesions (P = .038) from baseline to post training.
The findings underscore “the importance of interpretation of imaging in the right clinical context,” Dr. Renson said, since they point to the possibility of an incorrect diagnosis “even in a young, active population.”
Another study, led by Ulrich Weber, MD, of King Christian 10th Hospital for Rheumatic Diseases, Gråsten, Denmark, sought to understand levels of normal low-grade BME in 20 amateur runners (8 men) and 22 professional Danish hockey players (all men). On average, the researchers found signs of BME in 3.1 sacroiliac joint quadrants in the runners before and after they ran a race. Hockey players were scanned at the end of the competitive season and showed signs of BME in an average of 3.6 sacroiliac joint quadrants.
In an interview, Dr. Landewé said the studies point to how common positive MRIs are in healthy people. “It was far higher than we would have thought 10 years ago,” he said.
Are MRIs still useful then? Dr. Weber said MRI scans are still helpful in axSpA diagnoses even though they have major limitations. “The imaging method is the only one that’s halfway reliable,” he said. “These joints are deep in the body, so we have virtually no clinical ways to diagnose this.”
However, Dr. Landewé said, “you should do it only when you have sufficient suspicion of spondyloarthritis” – due to accompanying conditions such as positive family history, acute anterior uveitis, psoriasis, or peripheral arthritis – and not just when a patient has chronic back pain.
Dr. Renson reported having no relevant disclosures; two of his coauthors reported extensive disclosures. Dr. Weber and his coauthors reported having no relevant disclosures. Dr. Landewé reported having no relevant disclosures; several of his coauthors reported various disclosures. Funding for the studies was not reported.
SAN DIEGO – Healthy individuals can show signs of spinal and pelvic inflammation on MRI, but these scans can be misleading if relied on to make a diagnosis of axial spondyloarthritis, according to findings from three separate studies at the annual meeting of the American College of Rheumatology.
“Don’t rely on MRI alone is our message,” said Robert Landewé, MD, PhD, of the University of Amsterdam, who was a coauthor of one of the three studies. “A positive MRI may occur in individuals that are completely healthy. We need to make sure that not too many patients with chronic lower back pain are diagnosed with a disease they don’t have.”
The axial form of spondyloarthritis (axSpA) affects the spinal and pelvic joints of an estimated 1.4% of the U.S. population, and the term encompasses the diagnosis of ankylosing spondylitis (0.5% of U.S. population) in which advanced sacroiliitis is seen on conventional radiography, according to the ACR. Axial SpA is particularly common in young people, especially males, in their teens and 20s.
Researchers believe that MRI scans can misleadingly suggest that patients have the condition. “We know that MRI is a sensitive method, but there’s a lack of data regarding its specificity,” Thomas Renson, MD, of Ghent (Belgium) University, said at a press conference during the meeting.
Dr. Landewé led a study that compared MRIs of sacroiliac joints in 47 healthy people, 47 axSpA patients matched for gender and age, 47 chronic back pain patients, 7 women with postpartum back pain, and 24 frequent runners. Positive MRIs were common in the axSpA patients (43 of 47), but they were also found in healthy people (11 of 47), chronic back pain patients (3 of 47), frequent runners (3 of 24), and women with postpartum back pain (4 of 7).
In another study, Dr. Renson and his colleagues sought to understand whether a sustained period of intense physical activity affected spinal findings in 22 healthy military recruits who did not have SpA.
However, there was a statistically significant increase of combined structural and inflammatory lesions (P = .038) from baseline to post training.
The findings underscore “the importance of interpretation of imaging in the right clinical context,” Dr. Renson said, since they point to the possibility of an incorrect diagnosis “even in a young, active population.”
Another study, led by Ulrich Weber, MD, of King Christian 10th Hospital for Rheumatic Diseases, Gråsten, Denmark, sought to understand levels of normal low-grade BME in 20 amateur runners (8 men) and 22 professional Danish hockey players (all men). On average, the researchers found signs of BME in 3.1 sacroiliac joint quadrants in the runners before and after they ran a race. Hockey players were scanned at the end of the competitive season and showed signs of BME in an average of 3.6 sacroiliac joint quadrants.
In an interview, Dr. Landewé said the studies point to how common positive MRIs are in healthy people. “It was far higher than we would have thought 10 years ago,” he said.
Are MRIs still useful then? Dr. Weber said MRI scans are still helpful in axSpA diagnoses even though they have major limitations. “The imaging method is the only one that’s halfway reliable,” he said. “These joints are deep in the body, so we have virtually no clinical ways to diagnose this.”
However, Dr. Landewé said, “you should do it only when you have sufficient suspicion of spondyloarthritis” – due to accompanying conditions such as positive family history, acute anterior uveitis, psoriasis, or peripheral arthritis – and not just when a patient has chronic back pain.
Dr. Renson reported having no relevant disclosures; two of his coauthors reported extensive disclosures. Dr. Weber and his coauthors reported having no relevant disclosures. Dr. Landewé reported having no relevant disclosures; several of his coauthors reported various disclosures. Funding for the studies was not reported.
SAN DIEGO – Healthy individuals can show signs of spinal and pelvic inflammation on MRI, but these scans can be misleading if relied on to make a diagnosis of axial spondyloarthritis, according to findings from three separate studies at the annual meeting of the American College of Rheumatology.
“Don’t rely on MRI alone is our message,” said Robert Landewé, MD, PhD, of the University of Amsterdam, who was a coauthor of one of the three studies. “A positive MRI may occur in individuals that are completely healthy. We need to make sure that not too many patients with chronic lower back pain are diagnosed with a disease they don’t have.”
The axial form of spondyloarthritis (axSpA) affects the spinal and pelvic joints of an estimated 1.4% of the U.S. population, and the term encompasses the diagnosis of ankylosing spondylitis (0.5% of U.S. population) in which advanced sacroiliitis is seen on conventional radiography, according to the ACR. Axial SpA is particularly common in young people, especially males, in their teens and 20s.
Researchers believe that MRI scans can misleadingly suggest that patients have the condition. “We know that MRI is a sensitive method, but there’s a lack of data regarding its specificity,” Thomas Renson, MD, of Ghent (Belgium) University, said at a press conference during the meeting.
Dr. Landewé led a study that compared MRIs of sacroiliac joints in 47 healthy people, 47 axSpA patients matched for gender and age, 47 chronic back pain patients, 7 women with postpartum back pain, and 24 frequent runners. Positive MRIs were common in the axSpA patients (43 of 47), but they were also found in healthy people (11 of 47), chronic back pain patients (3 of 47), frequent runners (3 of 24), and women with postpartum back pain (4 of 7).
In another study, Dr. Renson and his colleagues sought to understand whether a sustained period of intense physical activity affected spinal findings in 22 healthy military recruits who did not have SpA.
However, there was a statistically significant increase of combined structural and inflammatory lesions (P = .038) from baseline to post training.
The findings underscore “the importance of interpretation of imaging in the right clinical context,” Dr. Renson said, since they point to the possibility of an incorrect diagnosis “even in a young, active population.”
Another study, led by Ulrich Weber, MD, of King Christian 10th Hospital for Rheumatic Diseases, Gråsten, Denmark, sought to understand levels of normal low-grade BME in 20 amateur runners (8 men) and 22 professional Danish hockey players (all men). On average, the researchers found signs of BME in 3.1 sacroiliac joint quadrants in the runners before and after they ran a race. Hockey players were scanned at the end of the competitive season and showed signs of BME in an average of 3.6 sacroiliac joint quadrants.
In an interview, Dr. Landewé said the studies point to how common positive MRIs are in healthy people. “It was far higher than we would have thought 10 years ago,” he said.
Are MRIs still useful then? Dr. Weber said MRI scans are still helpful in axSpA diagnoses even though they have major limitations. “The imaging method is the only one that’s halfway reliable,” he said. “These joints are deep in the body, so we have virtually no clinical ways to diagnose this.”
However, Dr. Landewé said, “you should do it only when you have sufficient suspicion of spondyloarthritis” – due to accompanying conditions such as positive family history, acute anterior uveitis, psoriasis, or peripheral arthritis – and not just when a patient has chronic back pain.
Dr. Renson reported having no relevant disclosures; two of his coauthors reported extensive disclosures. Dr. Weber and his coauthors reported having no relevant disclosures. Dr. Landewé reported having no relevant disclosures; several of his coauthors reported various disclosures. Funding for the studies was not reported.
AT ACR 2017
Fibroids associated with lower chance of unsuspected malignancy
NATIONAL HARBOR, MD. – Women undergoing hysterectomy or myomectomy for benign indications, who also had fibroids, were less likely to have a malignant diagnosis, according to a study presented at the AAGL Global Congress.
These findings could change the conversation when it comes to counseling patients about the risks associated with morcellation, a procedure that was strongly discouraged by the FDA in 2014 due to the concern that it might have the potential to spread malignancy.
“There’s a lot of things going on in the media about morcellation and risk of malignancy at the time of benign fibroid surgery, but this research actually makes apparent the higher risk of malignancy when fibroids are not present,” Farah Alvi, MD, a second-year fellow at Northwestern University, Chicago, said in an interview. Despite the concerns regarding morcellation and malignancy, this research suggests that patients who have fibroids at time of surgery may have a lower chance of malignancy, compared with patients who have other indications for surgery, she explained.
Dr. Alvi and her colleagues studied 2,987 hysterectomy or myomectomy patients with benign indications between January 2005 and December 2014.
Among patients studied, researchers found 33 confirmed malignant or borderline tumors, 16 of 1,790 (0.89%) in the leiomyoma group and 17 of 1,197 (1.42%) in the group with other indications (P = 0.04). The malignancies/borderline tumors included three leiomyosarcomas, two endometrial sarcomas, two endometrioid adenocarcinomas, one granulose cell tumor, three smooth muscle tumors of uncertain malignant potential, three atypical leiomyoma, and one serous papillary borderline ovarian tumor.
Of those with leiomyomata, 1 in 600 patients were diagnosed with leiomyosarcoma, compared with a risk of 1 in 350 for unanticipated malignancy in general.
Patients with surgical indications of symptomatic leiomyoma had an odds ratio of 0.63 (P = .18) for diagnosis of an unanticipated malignancy, compared with those without leiomyoma, according to Dr. Alvi. The odds of malignancy were also reduced in patients with uterine sizes of 15-20 weeks (OR, 0.65; P = .43) and those with specimen sizes of 250-500 grams (OR, 0.68; P = .64).
These findings will have implications for how physicians counsel women undergoing minimally invasive hysterectomy or myomectomy, Dr. Alvi said.
“In counseling patients about morcellation, we often have quoted them an estimated risk of 1 in 458 for leiomyosarcoma, based on the FDA morcellation warnings, and one thing we can learn is that risk is actually much lower than we think it is,” Dr. Alvi said.
The findings also suggest a shift in focus toward identifying the factors that put women at higher risk for malignancy. For example, older age is one of the most significant risk factors identified in the study, she added.
Dr. Alvi reported having no relevant financial disclosures.
[email protected]
On Twitter @eaztweets
NATIONAL HARBOR, MD. – Women undergoing hysterectomy or myomectomy for benign indications, who also had fibroids, were less likely to have a malignant diagnosis, according to a study presented at the AAGL Global Congress.
These findings could change the conversation when it comes to counseling patients about the risks associated with morcellation, a procedure that was strongly discouraged by the FDA in 2014 due to the concern that it might have the potential to spread malignancy.
“There’s a lot of things going on in the media about morcellation and risk of malignancy at the time of benign fibroid surgery, but this research actually makes apparent the higher risk of malignancy when fibroids are not present,” Farah Alvi, MD, a second-year fellow at Northwestern University, Chicago, said in an interview. Despite the concerns regarding morcellation and malignancy, this research suggests that patients who have fibroids at time of surgery may have a lower chance of malignancy, compared with patients who have other indications for surgery, she explained.
Dr. Alvi and her colleagues studied 2,987 hysterectomy or myomectomy patients with benign indications between January 2005 and December 2014.
Among patients studied, researchers found 33 confirmed malignant or borderline tumors, 16 of 1,790 (0.89%) in the leiomyoma group and 17 of 1,197 (1.42%) in the group with other indications (P = 0.04). The malignancies/borderline tumors included three leiomyosarcomas, two endometrial sarcomas, two endometrioid adenocarcinomas, one granulose cell tumor, three smooth muscle tumors of uncertain malignant potential, three atypical leiomyoma, and one serous papillary borderline ovarian tumor.
Of those with leiomyomata, 1 in 600 patients were diagnosed with leiomyosarcoma, compared with a risk of 1 in 350 for unanticipated malignancy in general.
Patients with surgical indications of symptomatic leiomyoma had an odds ratio of 0.63 (P = .18) for diagnosis of an unanticipated malignancy, compared with those without leiomyoma, according to Dr. Alvi. The odds of malignancy were also reduced in patients with uterine sizes of 15-20 weeks (OR, 0.65; P = .43) and those with specimen sizes of 250-500 grams (OR, 0.68; P = .64).
These findings will have implications for how physicians counsel women undergoing minimally invasive hysterectomy or myomectomy, Dr. Alvi said.
“In counseling patients about morcellation, we often have quoted them an estimated risk of 1 in 458 for leiomyosarcoma, based on the FDA morcellation warnings, and one thing we can learn is that risk is actually much lower than we think it is,” Dr. Alvi said.
The findings also suggest a shift in focus toward identifying the factors that put women at higher risk for malignancy. For example, older age is one of the most significant risk factors identified in the study, she added.
Dr. Alvi reported having no relevant financial disclosures.
[email protected]
On Twitter @eaztweets
NATIONAL HARBOR, MD. – Women undergoing hysterectomy or myomectomy for benign indications, who also had fibroids, were less likely to have a malignant diagnosis, according to a study presented at the AAGL Global Congress.
These findings could change the conversation when it comes to counseling patients about the risks associated with morcellation, a procedure that was strongly discouraged by the FDA in 2014 due to the concern that it might have the potential to spread malignancy.
“There’s a lot of things going on in the media about morcellation and risk of malignancy at the time of benign fibroid surgery, but this research actually makes apparent the higher risk of malignancy when fibroids are not present,” Farah Alvi, MD, a second-year fellow at Northwestern University, Chicago, said in an interview. Despite the concerns regarding morcellation and malignancy, this research suggests that patients who have fibroids at time of surgery may have a lower chance of malignancy, compared with patients who have other indications for surgery, she explained.
Dr. Alvi and her colleagues studied 2,987 hysterectomy or myomectomy patients with benign indications between January 2005 and December 2014.
Among patients studied, researchers found 33 confirmed malignant or borderline tumors, 16 of 1,790 (0.89%) in the leiomyoma group and 17 of 1,197 (1.42%) in the group with other indications (P = 0.04). The malignancies/borderline tumors included three leiomyosarcomas, two endometrial sarcomas, two endometrioid adenocarcinomas, one granulose cell tumor, three smooth muscle tumors of uncertain malignant potential, three atypical leiomyoma, and one serous papillary borderline ovarian tumor.
Of those with leiomyomata, 1 in 600 patients were diagnosed with leiomyosarcoma, compared with a risk of 1 in 350 for unanticipated malignancy in general.
Patients with surgical indications of symptomatic leiomyoma had an odds ratio of 0.63 (P = .18) for diagnosis of an unanticipated malignancy, compared with those without leiomyoma, according to Dr. Alvi. The odds of malignancy were also reduced in patients with uterine sizes of 15-20 weeks (OR, 0.65; P = .43) and those with specimen sizes of 250-500 grams (OR, 0.68; P = .64).
These findings will have implications for how physicians counsel women undergoing minimally invasive hysterectomy or myomectomy, Dr. Alvi said.
“In counseling patients about morcellation, we often have quoted them an estimated risk of 1 in 458 for leiomyosarcoma, based on the FDA morcellation warnings, and one thing we can learn is that risk is actually much lower than we think it is,” Dr. Alvi said.
The findings also suggest a shift in focus toward identifying the factors that put women at higher risk for malignancy. For example, older age is one of the most significant risk factors identified in the study, she added.
Dr. Alvi reported having no relevant financial disclosures.
[email protected]
On Twitter @eaztweets
AT AAGL 2017
Key clinical point:
Major finding: Patients with preoperative indication of symptomatic leiomyoma had an odds ratio of 0.63 (P = .18) of having a diagnosis of malignancy.
Data source: Retrospective study of 2,987 hysterectomies or myomectomies between January 2005 and December 2014.
Disclosures: Dr. Alvi reported having no relevant financial disclosures.