User login
Burnout
My chest and back are sore this week because I was on call last week. It’s my secret to beating burnout. Just keep reading.
The phrase “dermatologist burnout” may seem as oxymoronic as jumbo shrimp, yet both are real. Our work is easier than some other physicians’. Dermatologists don’t sleep in the hospital, and we have many fewer dope-seeking or dying patients. Yet we suffer the same EHR frustrations as any physician. We struggle with an ever-increasing volume of patients and regulations which stultify our ability to care for patients.
[polldaddy:9875293]
According to a recent Mayo Clinic Proceedings study, dermatologists had the highest increase in burnout from 32% to 57% (Mayo Clin Proc. 2015 Dec;90[12]:1600-13). Although some have it worse than others, all physicians today are at high risk. Changing external factors is difficult, but modifying internal aspects of burnout can help.
Challenges
First, I mark difficult weeks on my calendar in red. Do I have extra clinics? Is it post vacation? Am I giving a talk? Then, I set up challenges. For example, I knew last week’s call was going to be tough. So, each morning I challenged myself to do 100 push-ups in 2 minutes, 12 pull-ups, and run 2 miles. I also set goals of plowing through my backlog of journals and upgrading my EHR shortcuts and order sets.
Colleagues
A Navy SEAL training instructor once told me the key to success in BUD/S (the grueling 6-month SEAL training course), is to take care of your teammates:“When you’re focused on the guy to your right and the guy to your left, you find inner strength to endure suffering.” No matter how busy I am, when my phone rings or I get a text, I think to myself, Good, one of my partners needs my help. Framing it that way makes any added work feel lighter.
(Re)Charging
Lastly, I schedule time to recharge and recover. For example, this morning instead of going to the gym, I had a cappuccino and read the entire Sunday New York Times. Later today, my wife and I are going to see Thor: Ragnarok. In reclining seats. With a craft beer.
My call week was sometimes easy and occasionally arduous. Yet, I taught an ER resident how to recognize zoster in its very early stages. I learned the difference between erythema multiforme major and mycoplasma-induced rash with mucositis, and I reassured a family that their hospitalized 9-year-old was going to be just fine. I didn’t miss a workout (however, no SEAL instructor would have credited my pathetic pull-ups #11 and #12).
My next call isn’t long off, and soon, I must work on a big presentation. Medicine is a marathon, punctuated by sprinting. During stressful periods, I challenge myself physically and mentally, focus on helping others, and take the time to rest and recharge after. I think it has helped me beat burnout, I hope it helps you too.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at [email protected]
My chest and back are sore this week because I was on call last week. It’s my secret to beating burnout. Just keep reading.
The phrase “dermatologist burnout” may seem as oxymoronic as jumbo shrimp, yet both are real. Our work is easier than some other physicians’. Dermatologists don’t sleep in the hospital, and we have many fewer dope-seeking or dying patients. Yet we suffer the same EHR frustrations as any physician. We struggle with an ever-increasing volume of patients and regulations which stultify our ability to care for patients.
[polldaddy:9875293]
According to a recent Mayo Clinic Proceedings study, dermatologists had the highest increase in burnout from 32% to 57% (Mayo Clin Proc. 2015 Dec;90[12]:1600-13). Although some have it worse than others, all physicians today are at high risk. Changing external factors is difficult, but modifying internal aspects of burnout can help.
Challenges
First, I mark difficult weeks on my calendar in red. Do I have extra clinics? Is it post vacation? Am I giving a talk? Then, I set up challenges. For example, I knew last week’s call was going to be tough. So, each morning I challenged myself to do 100 push-ups in 2 minutes, 12 pull-ups, and run 2 miles. I also set goals of plowing through my backlog of journals and upgrading my EHR shortcuts and order sets.
Colleagues
A Navy SEAL training instructor once told me the key to success in BUD/S (the grueling 6-month SEAL training course), is to take care of your teammates:“When you’re focused on the guy to your right and the guy to your left, you find inner strength to endure suffering.” No matter how busy I am, when my phone rings or I get a text, I think to myself, Good, one of my partners needs my help. Framing it that way makes any added work feel lighter.
(Re)Charging
Lastly, I schedule time to recharge and recover. For example, this morning instead of going to the gym, I had a cappuccino and read the entire Sunday New York Times. Later today, my wife and I are going to see Thor: Ragnarok. In reclining seats. With a craft beer.
My call week was sometimes easy and occasionally arduous. Yet, I taught an ER resident how to recognize zoster in its very early stages. I learned the difference between erythema multiforme major and mycoplasma-induced rash with mucositis, and I reassured a family that their hospitalized 9-year-old was going to be just fine. I didn’t miss a workout (however, no SEAL instructor would have credited my pathetic pull-ups #11 and #12).
My next call isn’t long off, and soon, I must work on a big presentation. Medicine is a marathon, punctuated by sprinting. During stressful periods, I challenge myself physically and mentally, focus on helping others, and take the time to rest and recharge after. I think it has helped me beat burnout, I hope it helps you too.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at [email protected]
My chest and back are sore this week because I was on call last week. It’s my secret to beating burnout. Just keep reading.
The phrase “dermatologist burnout” may seem as oxymoronic as jumbo shrimp, yet both are real. Our work is easier than some other physicians’. Dermatologists don’t sleep in the hospital, and we have many fewer dope-seeking or dying patients. Yet we suffer the same EHR frustrations as any physician. We struggle with an ever-increasing volume of patients and regulations which stultify our ability to care for patients.
[polldaddy:9875293]
According to a recent Mayo Clinic Proceedings study, dermatologists had the highest increase in burnout from 32% to 57% (Mayo Clin Proc. 2015 Dec;90[12]:1600-13). Although some have it worse than others, all physicians today are at high risk. Changing external factors is difficult, but modifying internal aspects of burnout can help.
Challenges
First, I mark difficult weeks on my calendar in red. Do I have extra clinics? Is it post vacation? Am I giving a talk? Then, I set up challenges. For example, I knew last week’s call was going to be tough. So, each morning I challenged myself to do 100 push-ups in 2 minutes, 12 pull-ups, and run 2 miles. I also set goals of plowing through my backlog of journals and upgrading my EHR shortcuts and order sets.
Colleagues
A Navy SEAL training instructor once told me the key to success in BUD/S (the grueling 6-month SEAL training course), is to take care of your teammates:“When you’re focused on the guy to your right and the guy to your left, you find inner strength to endure suffering.” No matter how busy I am, when my phone rings or I get a text, I think to myself, Good, one of my partners needs my help. Framing it that way makes any added work feel lighter.
(Re)Charging
Lastly, I schedule time to recharge and recover. For example, this morning instead of going to the gym, I had a cappuccino and read the entire Sunday New York Times. Later today, my wife and I are going to see Thor: Ragnarok. In reclining seats. With a craft beer.
My call week was sometimes easy and occasionally arduous. Yet, I taught an ER resident how to recognize zoster in its very early stages. I learned the difference between erythema multiforme major and mycoplasma-induced rash with mucositis, and I reassured a family that their hospitalized 9-year-old was going to be just fine. I didn’t miss a workout (however, no SEAL instructor would have credited my pathetic pull-ups #11 and #12).
My next call isn’t long off, and soon, I must work on a big presentation. Medicine is a marathon, punctuated by sprinting. During stressful periods, I challenge myself physically and mentally, focus on helping others, and take the time to rest and recharge after. I think it has helped me beat burnout, I hope it helps you too.
Dr. Benabio is director of Healthcare Transformation and chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on Twitter. Write to him at [email protected]
Pimavanserin found modestly effective in phase 2 Alzheimer’s psychosis study
BOSTON – Pimavanserin, an atypical antipsychotic approved for use in psychosis associated with Parkinson’s disease, was modestly effective in treating psychosis associated with Alzheimer’s dementia in a phase 2 study.
The study of 181 patients showed that pimavanserin (Nuplazid) was associated with a statistically significant 3.76-point improvement on the Neuropsychiatric Inventory–Nursing Home Version (NPI-NH) psychosis score, Clive Ballard, MD, reported at the Clinical Trials on Alzheimer’s Disease conference. But although pimavanserin was significantly more effective than placebo at 6 weeks, it lost its statistical edge by the trial’s end at 12 weeks, largely because the placebo group improved over the study period.

A key finding was that pimavanserin was more effective in a subset of patients with severe symptoms, reducing those by more than 4 points on the NPI scale, said Dr. Ballard, codirector of the Biomedical Research Unit for Dementia in the Institute for Psychiatry at King’s College London. “A 4-point change is the difference from having moderate symptoms daily to having them weekly. I think this is the most clinically relevant finding.”
The drug seemed to largely spare cognition, which is another notch in its clinical belt, said Richard J. Caselli, MD, professor of neurology at the Mayo Clinic, Scottsdale, Ariz.
“The relative preservation of cognition as seen in an absence of adverse cognitive effects is encouraging, and something pimavanserin may have over its antipsychotic rivals,” Dr. Caselli said in an interview. “The improved scores on the NPI seem modest as does the relative percentage of responders, defined as at least 30% improved NPI-NH score. But at least it is a positive result. One concern is that the company advises it may take 4-6 weeks to see an improvement, which is not the kind of timeline one has with acutely and severely agitated patients. So I suspect antipsychotic drugs, which work more quickly, are likely not going away.”

The 12-week, phase 2 study randomized 181 patients with advanced Alzheimer’s dementia to placebo or 40 mg pimavanserin. They were a mean of 86 years old. The mean baseline NPI-NH psychosis score was 9.8 and the mean Mini–Mental State Exam score was 10.
By 6 weeks, the psychosis score had improved significantly more in the pimavanserin group than in the placebo group (–3.76 vs. –1.93 points; P = .0451). The drug was more effective among patients with severe psychosis at baseline, defined as an NPI-NH psychosis score of at least 12. Among this group, the score improved by 4.43 points. The results were slightly, but not significantly, better in patients who had responded to prior antipsychotic medications and among those who were also taking a selective serotonin reuptake inhibitor. A responder analysis also favored treatment, with 90% of those taking pimavanserin experiencing at least a 30% improvement on the NPI-NH psychosis score, compared with 43% of those taking placebo.
At 12 weeks, however, the overall between-group difference was no longer statistically significant, because the placebo group continued to improve over the treatment period.
Safety and tolerability were important considerations in such an elderly and cognitively compromised group, Dr. Ballard noted. In this respect, pimavanserin performed relatively well. There were more serious adverse events in the treated group (16.7% vs. 11%). These included respiratory infections (5 vs. 2) and urinary tract infection (2 vs. 0). Falls and fractures were similar in both groups. There was one fall in the active group, with one laceration, one hip fracture, and one femoral neck fracture. In the placebo group, there was one fall, one upper limb fracture, one wrist fracture, and one vertebral fracture. Among treated patients, there was also one heart attack and one case of renal failure. Four patients in each group died during the study.
Psychiatric events were more common in the pimavanserin group, most notably agitation (21% vs. 14%). Other psychiatric adverse events included aggression (10% vs. 4%), anxiety (5.6% vs. 2.2%), and dementia-related behavioral symptoms (5.6% vs. 2%). The drug had no effect on Mini–Mental State Exam score.
Pimavanserin was associated with a mean change of 9.4 ms in the heart rate-corrected QT interval, and was more likely to induce a weight loss of 7% or more.
Dr. Ballard had no financial disclosures with regard to pimavanserin or Acadia Pharmaceuticals, which sponsored the trial.
[email protected]
On Twitter @Alz_Gal
BOSTON – Pimavanserin, an atypical antipsychotic approved for use in psychosis associated with Parkinson’s disease, was modestly effective in treating psychosis associated with Alzheimer’s dementia in a phase 2 study.
The study of 181 patients showed that pimavanserin (Nuplazid) was associated with a statistically significant 3.76-point improvement on the Neuropsychiatric Inventory–Nursing Home Version (NPI-NH) psychosis score, Clive Ballard, MD, reported at the Clinical Trials on Alzheimer’s Disease conference. But although pimavanserin was significantly more effective than placebo at 6 weeks, it lost its statistical edge by the trial’s end at 12 weeks, largely because the placebo group improved over the study period.
A key finding was that pimavanserin was more effective in a subset of patients with severe symptoms, reducing those by more than 4 points on the NPI scale, said Dr. Ballard, codirector of the Biomedical Research Unit for Dementia in the Institute for Psychiatry at King’s College London. “A 4-point change is the difference from having moderate symptoms daily to having them weekly. I think this is the most clinically relevant finding.”
The drug seemed to largely spare cognition, which is another notch in its clinical belt, said Richard J. Caselli, MD, professor of neurology at the Mayo Clinic, Scottsdale, Ariz.
“The relative preservation of cognition as seen in an absence of adverse cognitive effects is encouraging, and something pimavanserin may have over its antipsychotic rivals,” Dr. Caselli said in an interview. “The improved scores on the NPI seem modest as does the relative percentage of responders, defined as at least 30% improved NPI-NH score. But at least it is a positive result. One concern is that the company advises it may take 4-6 weeks to see an improvement, which is not the kind of timeline one has with acutely and severely agitated patients. So I suspect antipsychotic drugs, which work more quickly, are likely not going away.”
The 12-week, phase 2 study randomized 181 patients with advanced Alzheimer’s dementia to placebo or 40 mg pimavanserin. They were a mean of 86 years old. The mean baseline NPI-NH psychosis score was 9.8 and the mean Mini–Mental State Exam score was 10.
By 6 weeks, the psychosis score had improved significantly more in the pimavanserin group than in the placebo group (–3.76 vs. –1.93 points; P = .0451). The drug was more effective among patients with severe psychosis at baseline, defined as an NPI-NH psychosis score of at least 12. Among this group, the score improved by 4.43 points. The results were slightly, but not significantly, better in patients who had responded to prior antipsychotic medications and among those who were also taking a selective serotonin reuptake inhibitor. A responder analysis also favored treatment, with 90% of those taking pimavanserin experiencing at least a 30% improvement on the NPI-NH psychosis score, compared with 43% of those taking placebo.
At 12 weeks, however, the overall between-group difference was no longer statistically significant, because the placebo group continued to improve over the treatment period.
Safety and tolerability were important considerations in such an elderly and cognitively compromised group, Dr. Ballard noted. In this respect, pimavanserin performed relatively well. There were more serious adverse events in the treated group (16.7% vs. 11%). These included respiratory infections (5 vs. 2) and urinary tract infection (2 vs. 0). Falls and fractures were similar in both groups. There was one fall in the active group, with one laceration, one hip fracture, and one femoral neck fracture. In the placebo group, there was one fall, one upper limb fracture, one wrist fracture, and one vertebral fracture. Among treated patients, there was also one heart attack and one case of renal failure. Four patients in each group died during the study.
Psychiatric events were more common in the pimavanserin group, most notably agitation (21% vs. 14%). Other psychiatric adverse events included aggression (10% vs. 4%), anxiety (5.6% vs. 2.2%), and dementia-related behavioral symptoms (5.6% vs. 2%). The drug had no effect on Mini–Mental State Exam score.
Pimavanserin was associated with a mean change of 9.4 ms in the heart rate-corrected QT interval, and was more likely to induce a weight loss of 7% or more.
Dr. Ballard had no financial disclosures with regard to pimavanserin or Acadia Pharmaceuticals, which sponsored the trial.
[email protected]
On Twitter @Alz_Gal
BOSTON – Pimavanserin, an atypical antipsychotic approved for use in psychosis associated with Parkinson’s disease, was modestly effective in treating psychosis associated with Alzheimer’s dementia in a phase 2 study.
The study of 181 patients showed that pimavanserin (Nuplazid) was associated with a statistically significant 3.76-point improvement on the Neuropsychiatric Inventory–Nursing Home Version (NPI-NH) psychosis score, Clive Ballard, MD, reported at the Clinical Trials on Alzheimer’s Disease conference. But although pimavanserin was significantly more effective than placebo at 6 weeks, it lost its statistical edge by the trial’s end at 12 weeks, largely because the placebo group improved over the study period.
A key finding was that pimavanserin was more effective in a subset of patients with severe symptoms, reducing those by more than 4 points on the NPI scale, said Dr. Ballard, codirector of the Biomedical Research Unit for Dementia in the Institute for Psychiatry at King’s College London. “A 4-point change is the difference from having moderate symptoms daily to having them weekly. I think this is the most clinically relevant finding.”
The drug seemed to largely spare cognition, which is another notch in its clinical belt, said Richard J. Caselli, MD, professor of neurology at the Mayo Clinic, Scottsdale, Ariz.
“The relative preservation of cognition as seen in an absence of adverse cognitive effects is encouraging, and something pimavanserin may have over its antipsychotic rivals,” Dr. Caselli said in an interview. “The improved scores on the NPI seem modest as does the relative percentage of responders, defined as at least 30% improved NPI-NH score. But at least it is a positive result. One concern is that the company advises it may take 4-6 weeks to see an improvement, which is not the kind of timeline one has with acutely and severely agitated patients. So I suspect antipsychotic drugs, which work more quickly, are likely not going away.”
The 12-week, phase 2 study randomized 181 patients with advanced Alzheimer’s dementia to placebo or 40 mg pimavanserin. They were a mean of 86 years old. The mean baseline NPI-NH psychosis score was 9.8 and the mean Mini–Mental State Exam score was 10.
By 6 weeks, the psychosis score had improved significantly more in the pimavanserin group than in the placebo group (–3.76 vs. –1.93 points; P = .0451). The drug was more effective among patients with severe psychosis at baseline, defined as an NPI-NH psychosis score of at least 12. Among this group, the score improved by 4.43 points. The results were slightly, but not significantly, better in patients who had responded to prior antipsychotic medications and among those who were also taking a selective serotonin reuptake inhibitor. A responder analysis also favored treatment, with 90% of those taking pimavanserin experiencing at least a 30% improvement on the NPI-NH psychosis score, compared with 43% of those taking placebo.
At 12 weeks, however, the overall between-group difference was no longer statistically significant, because the placebo group continued to improve over the treatment period.
Safety and tolerability were important considerations in such an elderly and cognitively compromised group, Dr. Ballard noted. In this respect, pimavanserin performed relatively well. There were more serious adverse events in the treated group (16.7% vs. 11%). These included respiratory infections (5 vs. 2) and urinary tract infection (2 vs. 0). Falls and fractures were similar in both groups. There was one fall in the active group, with one laceration, one hip fracture, and one femoral neck fracture. In the placebo group, there was one fall, one upper limb fracture, one wrist fracture, and one vertebral fracture. Among treated patients, there was also one heart attack and one case of renal failure. Four patients in each group died during the study.
Psychiatric events were more common in the pimavanserin group, most notably agitation (21% vs. 14%). Other psychiatric adverse events included aggression (10% vs. 4%), anxiety (5.6% vs. 2.2%), and dementia-related behavioral symptoms (5.6% vs. 2%). The drug had no effect on Mini–Mental State Exam score.
Pimavanserin was associated with a mean change of 9.4 ms in the heart rate-corrected QT interval, and was more likely to induce a weight loss of 7% or more.
Dr. Ballard had no financial disclosures with regard to pimavanserin or Acadia Pharmaceuticals, which sponsored the trial.
[email protected]
On Twitter @Alz_Gal
AT CTAD
Key clinical point:
Major finding: The psychosis score at 6 weeks improved significantly more in patients taking pimavanserin than in those taking placebo (–3.76 vs. –1.93 points; P = .0451).
Data source: The randomized, placebo-controlled study enrolled 181 patients.
Disclosures: Acadia Pharmaceuticals makes pimavanserin and sponsored the trial. Dr. Ballard has no financial relationship with the company.
Barriers and Facilitators to Adopting Nursing Home Culture Change
From RTI International, Waltham, MA, and Brown University School of Public Health, Providence, RI.
Abstract
- Objective: To review the nursing home culture change literature and identify common barriers to and facilitators of nursing home culture change adoption. Nursing home culture change aims to make nursing homes less institutional by providing more resident-centered care, making environments more homelike, and empowering direct care staff.
- Methods: We reviewed the research literature on nursing home culture change, especially as related to implementation and outcomes.
- Results: Adoption of nursing home culture change practices has been steadily increasing in the past decade, but some practices are more likely to be adopted than others. A commonly reported barrier to culture change adoption is staff resistance to change. Studies suggest that this resistance can be overcome by changes to management practices, including good communication, providing training and education, and leadership support.
- Conclusion: The numerous benefits of nursing home culture change are apparent in the literature. Barriers to its adoption may be overcome by making improvements to nursing home management practices.
Nursing home culture change is a philosophy and combination of diverse practices aimed at making nursing homes less institutional and more resident-centered [1]. Nursing homes have been depicted as dehumanizing “total institutions” [2–4] in which the quality of residents’ lives and the quality of care are generally poor, daily life is medically regimented, only residents’ basic physical needs receive attention [5–8], and direct care workers are subject to poor working conditions for the lowest possible pay [9,10]. Since the 1980s, transforming the culture of nursing homes to be more humanizing, resident-centered, empowering, and homelike has been a primary mission of many stakeholder groups, including nursing home residents and care workers and their advocates [11].
Comprehensive culture change requires transformation of the nursing home environment from that of an institution to that of a home, implementation of more resident-centered care practices, empowerment of direct care staff, and flattening of the traditional organizational hierarchy so that residents and direct-care workers are actively involved in planning and implementing changes that empower them [12,13]. Culture change requires both technical changes, which are relatively straightforward efforts to address issues within a system while fundamentally keeping the system intact, and adaptive changes, which are more complex and entail reforming fundamental values that underlie the system and demand changes to the system itself [14,15].
Over time, nursing home culture change has gained widespread mainstream support. In 2009, the federal government issued new interpretive guidelines for use by nursing home inspectors that call for nursing homes to have more homelike environments and to support more resident-centered care [16]. The Centers for Medicare & Medicaid Services also required state quality improvement organizations to work with nursing homes on culture change efforts [1]. Some states effectively incentivize culture change by tying nursing home reimbursement rates and pay-for-performance policies to the implementation of culture change practices [17]. In addition to federal and state regulations, some nursing home corporations encourage or require facility administrators to implement culture change practices [18]. Overall, nursing homes are pushed to implement culture change practices on many fronts. The promise of beneficial outcomes of culture change also motivates implementation of some culture change practices [19].
In this article, we discuss the key elements of culture change, review the research examining the association between culture change and outcomes, identify key barriers to culture change, and offer suggestions from the literature for overcoming resistance to culture change.
Elements of Culture Change
Changing the Physical Environment
Changing the physical environment of nursing homes to be less institutional and more homelike is a core component of culture change [1]. These include both exterior and interior changes. Exterior changes can include adding walkways, patios, and gardens; interior changes include replacing nurses’ stations with desks, creating resident common areas, introducing the use of linens in dining areas, personalizing mailboxes outside of resident rooms, and adding small kitchens on units [20]. Other ideas for making environments more homelike include providing residents with the choice of colors for painting rooms and the choice of corridor/unit names and replacing public announcement systems with staff pagers [20].
Although changes to the physical environment may be considered cost-prohibitive, many of these changes entail minor and inexpensive enhancements that can help make environments more intimate and reminiscent of home than are traditional nursing homes [21,22]. Additionally, some environmental changes, such as adding raised gardens and walkways, can be designed not only to make the environment more homelike but also to help residents to engage in meaningful activities and connect to former roles, such as those of a homemaker, gardener, or farmer [21–23].
Providing Resident-Centered Care
Making care resident-centered entails enhancing resident choice and decision making and focusing the delivery of services on residents’ needs and preferences. According to Banaszak-Holl and colleagues [24], resident-centered approaches often emphasize the importance of shifting institutional norms and values and drawing employees’ attention to the needs of residents. This cultural shift in values and norms may be signaled by the implementation of practices that strengthen residents’ autonomy regarding everyday decisions. For example, as part of a resident-centered approach, residents would be offered choices and encouraged to make their own decisions about things personally affecting them, such as what to wear or when to go to bed, eating schedules, and menus [1,17,25].
Empowering Care Aides
Nursing home staff empowerment, particularly the empowerment of nursing assistants and other “hands-on” care aides—who are the predominant workforce in nursing homes and provide the vast bulk of care [26]—is a core component of culture change [1]. Such staff empowerment generally entails enhanced participation in decision making and increased autonomy. Staff empowerment practices that were examined in a national survey of nursing home directors [17] included:
- Staff work together to cover shifts when someone cannot come to work
- Staff cross-trained to perform tasks outside of their assigned job duties
- Staff involved in planning social events
- Nursing assistants take part in quality improvement teams
- Nursing assistants know when a resident’s care plan has changed
- Nursing assistants who receive extra training or education receive bonuses or raises
- Nursing assistants can choose which the residents for whom they provide care
We found that the staff empowerment practices most commonly implemented by nursing homes included nursing assistants knowing when a resident’s care plan has changed and staff working together to cover shifts when someone can’t come to work, but it was uncommon for nursing homes to permit nursing assistants to choose which residents they care for [17].
Outcomes of Culture Change
Research over the past 2 decades has examined the outcomes of culture change and the challenges involved in its implementation. Culture change is intended to improve the quality of life for nursing home residents, but the impact of culture change interventions is not clear. Shier and colleagues [27] conducted a comprehensive review of the peer-reviewed and gray literature on culture change published between 2005 and 2012 and found that studies varied widely in scope and evidence was inconsistent. They concluded that there is not yet sufficient evidence to provide specific guidance to nursing homes interested in implementing culture change [27]. The reviewed studies (27 peer-reviewed and 9 gray literature) also were noted to include small sample sizes and restricted geographic coverage, which both limit generalizability.
Although the literature had substantial limitations, Shier and colleagues [27] found numerous beneficial outcomes of culture change. Statistically significant improvements in numerous resident outcome measures were found to be associated with the implementation of culture change practices, including measures of resident quality of life/well-being, engagement and activities, functional status, satisfaction, mood (depression), anxiety/behavior/agitation, and pain/comfort. Two quality of care and services outcome measures also showed significant improvement associated with culture change practices, including increased completion of advance care plans and improved quality of workers’ approach to residents. Various staff outcome measures also showed significant improvement associated with culture change, including improvements in staff turnover/retention, satisfaction/well-being/burnout, absenteeism, knowledge, and attitude. Additionally, studies have shown culture change to be associated with improvements in select organizational outcome measures including operations costs, occupancy rates, revenue/profits, and family satisfaction. Four of the 36 studies reported negative outcomes of culture change. These negative outcomes included increased resident fear/anxiety [28], increased resident incontinence, decreased resident engagement in activities, decreased family engagement [29,30], decreased resident well-being [31], and increased physical incidents [32]. Notably, negative outcomes often co-occurred with positive outcomes [27,28].
To address the limitations of previous culture change research, such as small sample sizes and limited geographic coverage, and to explain some of the previous equivocal findings from quality studies when the extent of culture change practice implementation was not considered or measured, we collaborated on a national study to understand whether nursing home introduction of culture change practices is associated with improved quality [33]. We identified 824 U.S. nursing homes that had implemented some culture change practices, and we classified them by level of culture change practice implementation (high versus low). In nursing homes with high levels of culture change practice implementation, the introduction of nursing home culture change was associated with significant improvements in some care processes (eg, decreased prevalence of restraints, tube feeding, and pressure ulcers; increased proportion of residents on bladder training programs) and improvements in some resident outcomes, including slightly fewer hospitalizations. Among nursing homes with lower levels of culture change practice implementation, the introduction of culture change was associated with fewer health-related and quality-of-life survey deficiencies, but also with a significant increase in the number of resident hospitalizations [33]. Conclusive evidence regarding the impact of nursing homes implementing specific culture change practices or a comprehensive array of culture change practices on resident outcomes and quality of life remains needed, but numerous benefits of culture change are apparent in the literature.
Diffusion of Culture Change Practices
As culture change is widely supported and shows promise for beneficial outcomes, culture change practices are increasingly being implemented in nursing homes nationally. In 2007, a Commonwealth Fund survey found 56% of directors of nursing in U.S. nursing homes reported any culture change implementation or leadership commitment to implementation, but only 5% reported that culture change had completely changed the way the nursing home cared for residents in all areas of the nursing home [34]. In contrast, by 2010, 85% of directors of nursing reported at least partial culture change implementation and 13% reported that culture change had completely changed the way the nursing home cared for residents in all areas [14]. In a more recent survey of nursing home administrators, 16% reported that culture change had completely changed the way the nursing home cared for residents in all areas [35].
Barriers to Culture Change Implementation
Although the growth of culture change in the nursing home industry in the past decade has been impressive, implementation of comprehensive culture change has lagged behind. This is because one notable feature of nursing home culture change is that it is a philosophy that consists of many related practices. As noted above, implementing culture change can involve changes to physical environments, resident-centered care practices, and staff empowerment. This means that facilities can choose to implement as many or as few changes as they would like, and research has shown that there has been a lot of variation in which culture change practices are implemented. For example, in previous research we found that facilities interested in attracting highly reimbursed Medicare rehabilitation patients were more likely to implement hotel-style changes to their physical environments than they were to implement resident-centered care practices or forms of staff empowerment [19]. Sterns and colleagues [36] found that facilities were more likely to implement less complex practices (eg, allowing residents to choose when they go to bed) than more complex practices (eg, involving staff and residents in organizational decision making). The authors suggest that differences in commitment of facility leaders to comprehensive culture change may have contributed to these differences.
Attributes of facility leaders and other aspects of organizational context have been shown to contribute to more and less successful culture change implementation. For example, Scalzi and colleagues [37] found that some important barriers to culture change implementation were not involving all staff in culture change activities and a lack of corporate level support for these efforts. Schuldheis [38] examined differences in organizational context and its role in culture change among 9 Oregon facilities; 3 facilities successfully implemented culture change practices and 6 facilities did not. Results showed that a facility’s existing organizational culture, attention to sustainability, management practices, and staff involvement were important to the success of the initiative. Similarly, Rosemond and colleagues [39] conducted a study involving 8 North Carolina nursing homes. They determined that unsuccessful culture change initiatives could be attributed to the organizations’ readiness for change, a lack of high quality management communications, and unfavorable perceptions of culture change by direct-care workers. A study conducted in 4 nursing homes by Munroe et al [40] found that formal culture change training provided by professional trainers produced better outcomes than informal “train the trainer” sessions provided by other facility managers. Bowers and colleagues [41] also found that unsuccessful implementation of the Green House model of culture change was likely related to a lack of training resources for staff. Similarly, after an in-depth ethnographic study of culture change implementation, Lopez [42] found that it was unrealistic to expect direct-care workers to perform their jobs in radically new ways without being provided with ongoing support from management.
Resistance to Change: A Key Barrier
Our own research sought to understand the barriers and challenges nursing home administrators faced when implementing culture change in their facilities and the strategies they used to overcome them. In interviews conducted with 64 administrators who had participated in a previous nationally representative survey about culture change implementation, administrators reported a wide variety of barriers, including old and outdated physical plants, the costs of some changes, and issues with unions [18]. A key barrier that administrators reported facing was resistance to change on the part of nursing facility staff, residents, and residents’ family members [43]. Administrators reported that residents were resistant to change primarily because they had been institutionalized in their thinking. In other words, nursing homes had essentially trained residents to expect things to be done at certain times and in certain ways. Resistance among staff reportedly included resistance to the overall concept of culture change and to specific culture change practices. Often, staff perceived that changes related to culture change implementation involved additional work or effort on their part without additional resources, but this was not the only reason for resistance. Most often staff, especially longer-term staff, simply were resistant to making any changes to their usual routines or duties.
This type of resistance to change among staff is not unique to culture change implementation and has long been a commonly cited barrier in the organizational change literature. For example, in a 1954 Harvard Business Review article, Lawrence [44] stated that resistance to change was “the most baffling and recalcitrant of the problems which business executives face.” Since that time, resistance to change has been extensively studied as have methods for overcoming such resistance.
Recommendations for Overcoming Resistance to Culture Change
In seminal work on employee resistance to change conducted shortly after World War II, Coch and French [45] challenged the concept that resistance to change was the result of flaws or inadequacies on the part of staff, which would make addressing resistance difficult. Instead, they proposed, and proved through experimental methods, that resistance arose primarily from the context within which the changes were taking place. In other words, they found that managers could ameliorate resistance to change through changes to management and leadership practices. In their experiment, resistance to change in a manufacturing plant was overcome when management effectively communicated to staff the reasons for the change and engaged staff in planning for the desired changes. Studies on the barriers and facilitators of culture change implementation in nursing facilities have similarly found that facility leaders can take steps to address, or even avoid, staff resistance to change.
In our own research, we have found that resistance to change is a common barrier faced by facility leaders. We also found that resistance to change was unique among barriers in that, although strategies used to address other types of barriers varied widely, administrators consistently reported using the same strategies to address and overcome resistance to change. These strategies all involved management and leadership activities, including education and training and improved communication. In addition, administrators discussed in detail the ways they tailored education and communication to their facility’s unique needs. They also indicated that these efforts should be ongoing, communication should be two-way, and that all staff should be included [43].
Good Communication
One important tool for avoiding or overcoming resistance to culture change that facility administrators reported was good communication. They reported that open and bidirectional communication fostered feedback about ongoing culture change efforts and encouraged engagement and buy-in from staff. They also suggested that it is important that this type of communication be ongoing. Good communication about culture change, in particular, included providing a strong rationale for the changes and involved getting input from staff before and during implementation [43].
These findings are similar to other studies of culture change which have found that culture change implementation should involve staff at all levels [37] and that facility leaders should follow through on the plans that have been communicated [39]. Interestingly, the importance of good and open communication has also been identified as important to other forms of nursing facility quality improvement [46].
Training and Education
The facility administrators we interviewed also reported providing education and training for staff about culture change in a variety of ways, including as part of regular in-service training and as a component of new employee orientation. The training materials used were often obtained from the leading culture change organizations. However, importantly, administrators reported tailoring these trainings to the specific needs of their employees or unique context of their facility. For example, administrators reported breaking up long training sessions into shorter segments provided over a longer period of time or organizing trainings to be provided to small groups on the units rather than in more didactic conference-style settings [43]. Administrators explained that providing training in this way was more palatable to staff and helped incorporate learning into everyday care.
Other studies of nursing home culture change have also found training and education to be important to implementation. For example, in a study of a labor-management partnership for culture change implementation, Leutz and colleagues [47] found training of staff from all disciplines by culture change experts to be an important element of successful implementation. Training topics included those that were very general, such as gerontology, and very specific, including person-centered care. Staff were paid for their time participating in training, which took place at their facilities to make participation easier. The trainings were also staggered over the course of several months, so that staff had time to use what they had learned between sessions and could discuss their experiences at the later sessions.
Munroe and colleagues [40] conducted a study of culture change training using pre-post test survey methods and found that formal training had more of an effect on staff than informal training. In the study, staff at 2 facilities received formal education from a consulting group while staff at 2 other facilities then received informal training from the staff of one of the formally trained facilities. An important conclusion of the authors was that the formal training did a better job than the informal training of helping facility leaders and managers view their relationships with staff differently. This suggests that facility leaders and managers may have to alter their management styles to create the supportive context within which culture change efforts can succeed [48].
Leadership Support
Good communication and training/education can be thought of as 2 examples of leadership support, and support from facility leaders and managers has been found, in multiple studies, to be critical to successful culture change efforts. For example, in a recent study of nursing facility culture change in the Netherlands, Snoeren and colleagues [49] found that facility managers can facilitate culture change implementation by supporting a variety of staff needs and promoting the facilities’ new desired values. Another study found that facilities with leaders who are supportive and foster staff flexibility, such as allowing staff to be creative in their problem-solving and have decentralized decision-making, were more likely to report having implemented culture change [24].
In a study focused specifically on facility leadership style and its relation to culture change implementation, Corazzini and colleagues [50] found an adaptive leadership style to be important to culture change implementation. Adaptive leadership styles are ones that acknowledge the importance of staff relationships and recognize that complex changes, like those often implemented in culture change efforts, require complex solutions that will likely evolve over time. These authors conclude that culture change implementation necessitates development of new normative values and behaviors and can, therefore, not be accomplished by simply generating new rules and procedures [50].
Of course, not all nursing facility leaders have the management skills needed to perform in these adaptive and flexible ways. Therefore, management training for facility leaders may be an important first step in a facility’s culture change efforts [51]. This type of training may help improve communication skills and allow facility leaders to perform in more adaptive and flexible ways to better meet the needs of their particular facility and staff. Research also suggests that culture change training for facility leaders may help them to form new and better relationships with staff [40], an important element of culture change.
Conclusion
Nursing home culture change aims to improve care quality and resident satisfaction through changes to physical environments, resident care practices, and staff empowerment. These include both relatively simple technical changes and more complex changes. Nursing home managers and leaders have reported a variety of barriers to implementing nursing home culture change. A common barrier cited is staff resistance to change. Many decades of research in the organizational change literature and more recent research on culture change implementation suggest steps that facility managers and leaders can take to avoid or overcome this resistance. These steps include providing management support, especially in the form of good communication and training and education.
Corresponding author: Denise A. Tyler, PhD, RTI International, 307 Waverly Oaks Rd., Waltham, MA 02452, [email protected].
Financial disclosures: None.
1. Koren MJ. Person-centered care for nursing home residents: The culture-change movement. Health Affairs 2010;29:1–6.
2. Goffman E. Asylums: essays on the social situation of mental patients and other inmates. Garden City, NY: Anchor Books; 1961.
3. Kane RA, Caplan AL. Everyday ethics: resolving dilemmas in nursing home life. New York: Springer; 1990.
4. Mor V, Branco K, Fleishma J, et al. The structure of social engagement among nursing home residents. J Gerontol B Psycholog Sci Soc Sci 1995;50:P1–P8.
5. Foner N. The caregiving dilemma: work in an American nursing home. Berkeley, CA: University of California Press; 1993.
6. Gubrium J. Living and dying at Murray Manor. New York: St. Martins; 1976.
7. Kayser-Jones JS. Old, alone, and neglected: care of the aged in the United States and Scotland. Berkeley, CA: University of California Press; 1990.
8. Vladeck B. Unloving care: the nursing home tragedy. New York: Basic Books; 1980.
9. Diamond T. Social policy and everyday life in nursing homes: a critical ethnography. Soc Sci Med 1986;23:1287–95.
10. Kalleberg A, Reskin BF, Hudson K. Bad jobs in America: standard and nonstandard employment relations and job quality in the United States. Am Sociolog Rev 2000;65:256–78.
11. Rahman AN, Schnelle JF. The nursing home culture-change movement: recent past, present, and future directions for research. Gerontologist 2008;48:142–8.
12. White-Chu EF, Graves WJ, Godfrey SM, et al. Beyond the medical model: the culture change revolution in long-term care. J Am Med Dir Assoc 2009;10:370–8.
13. Misiorski S, Kahn K. Changing the culture of long-term care: Moving beyond programmatic change. J Soc Work Long-Term Care 2006;3:137–46.
14. Anderson RA, Bailey DEJ, Wu B, et al. Adaptive leadership framework for chronic illness: framing a research agenda for transforming care delivery. Adv Nurs Sci 2015;38:83–95.
15. Bailey DE, Docherty S, Adams JA, et al. Studying the clinical encounter with the adaptive leadership framework. J Healthc Leadersh 2012;4:83–91.
16. Centers for Medicare & Medicaid Services Manual System. Revisions to Appendix PP “Guidance to Surveyors of Long Term Care Facilities” Washington, DC: Department of Health and Human Services 2009. Accessed at http://www.cms.gov/Regulations-and-Guidance/Guidance/Transmittals/downloads/R48SOMA.pdf.
17. Miller SC, Looze J, Shield R, et al. Culture change practice in US nursing homes: prevalence and variation by state Medicaid reimbursement policies. Gerontologist 2014;54:434–45.
18. Shield R, Looze J, Tyler D, et al. Why and how do nursing homes implement culture change practices? Insights from qualitative interviews in a mixed methods study. J Appl Gerontol 2014;33:737–63.
19. Lepore MJ, Shield RR, Looze J, et al. Medicare and Medicaid reimbursement rates for nursing homes motivate select culture change practices but not comprehensive culture change. J Aging Soc Pol 2015;27:215–31.
20. Shield RR, Tyler D, Lepore M, et al. Would you do that in your home? Making nursing homes home-like in culture change implementation. J Housing Elderly 2014;28:383–98.
21. Cutler L, Kane RA. As great as all outdoors. J Hous Elderly 2006;19:29–48.
22. Jurkowsky ET. Implementing culture change in long-term care: Benchmarks and strategies for management and practice. New York: Springer; 2013.
23. Wang D , Glicksman A. “Being grounded”: Benefits of gardening for older adults in low-income housing. J Hous Elderly 2013;27:89–104.
24. Banaszak-Holl J, Castle NG, Lin M, Spreitzer G. An assessment of cultural values and resident-centered culture change in US nursing facilities. Healthc Manage Rev 2013;38:295.
25. White-Chu EF, Graves WJ, Godfrey SM, et al. Beyond the medical model: the culture change revolution in long-term care. J Am Med Dir Assoc 2009;10:370–8.
26. Stone RI. Developing a quality direct care workforce: searching for solutions. Pub Pol Aging Rep 2017.
27. Shier V, Khodyakov D, Cohen LW, et al. What does the evidence really say about culture change in nursing homes? Gerontologist 2014;54:S6–S16.
28. Fritsch T, Kwak J, Grant S, et al. Impact of TimeSlips, a creative expression intervention program, on nursing home residents with dementia and their caregivers. Gerontologist 2009;49:117–27.
29. Kane RA, Lum TY, Cutler LJ, et al. Resident outcomes in small-house nursing homes: a longitudinal evaluation of the initial Green House program. J Am Geriatr Soc 2007;55:832-9.
30. Lum TY, Kane RA, Cutler LJ, Yu TC. Effects of Green House nursing homes on residents’ families. Healthc Financ Rev 2008;30:35–51.
31. Brooker DJ, Woolley RJ, Lee D. Enriching opportunities for people living with dementia in nursing homes: an evaluation of a multi-level activity-based model of care. Aging Ment Health 2007;11:361–70.
32. Detweiler MB, Murphy PF, Myers LC, Kim KY. Does a wander garden influence inappropriate behaviors in dementia residents? Am J Alzheimers Dis Other Dement 2008;23:31–45.
33. Miller SC, Lepore M, Lima JC, et al. Does the introduction of nursing home culture change practices improve quality? J Am Geriatr Soc 2014;62:1675–82.
34. Doty MM, Koren MJ, Sturla EL. Culture change in nursing homes: how far have we come? Findings from the Commonweath Fund 2007 National Survey of Nursing Homes; 2008. Accessed at http://www.commonwealthfund.org/Publications/Fund-Reports/2008/May/Culture-Change-in-Nursing-Homes--How-Far-Have-We-Come--Findings-From-The-Commonwealth-Fund-2007-Nati.aspx.
35. Miller SC, Tyler D, Shield R, et al. Nursing home culture change: study framework and survey instrument design. Presentation at the International Association of Gerontology and Geriatrics meeting, San Francisco, CA; 2017.
36. Sterns S, Miller SC, Allen S. The complexity of implementing culture change practices in nursing homes. J Am Med Dir Assoc 2010;11:511–8.
37. Scalzi CC, Evans LK, Barstow A, Hostvedt K. Barriers and enablers to changing organizational culture in nursing homes. Nurs Admin Q 2006;30:368–72.
38. Schuldheis S. Initiating person-centered care practices in long-term care facilities. J Gerontol Nurs 2007;33:47.
39. Rosemond CA, Hanson LC, Ennett ST, et al. Implementing person-centered care in nursing homes. Healthc Manage Rev 2012;37:257–66.
40. Munroe DJ, Kaza PL, Howard D. Culture-change training: Nursing facility staff perceptions of culture change. Geriatr Nurs 2011;32:400–7.
41. Bowers BJ, Nolet K. Developing the Green House nursing care team: Variations on development and implementation. Gerontologist 2014;54:S53–64.
42. Lopez SH. Culture change management in long-term care: a shop-floor view. Pol Soc 2006;34:55–80.
43. Tyler DA, Lepore M, Shield RR, et al. Overcoming resistance to culture change: nursing home administrators’ use of education, training, and communication. Gerontol Geriatr Educ 2014;35:321–36.
44. Lawrence PR. How to deal with resistance to change. Harvard Bus Rev 1954;May/June:49–57.
45. Coch L, French JRP. Overcoming resistance to change. Hum Relat 1948;1:512–32.
46. Scott-Cawiezell J, Schenkman M, Moore L, et al. Exploring nursing home staff’s perceptions of communication and leadership to facilitate quality improvement. J Nurs Care Qual 2004;19:242–52.
47. Leutz W, Bishop CE, Dodson L. Role for a labor–management partnership in nursing home person-centered care. Gerontologist 2009;50:340–51.
48. Tyler DA, Parker VA. Nursing home culture, teamwork, and culture change. J Res Nurs 2011;16:37–49.
49. Snoeren MM, Janssen BM, Niessen TJ, Abma TA. Nurturing cultural change in care for older people: seeing the cherry tree blossom. Health Care Anal 2016;24:349–73.
50. Corazzini K, Twersky J, White HK, et al. Implementing culture change in nursing homes: an adaptive leadership framework. Gerontologist 2014;55:616–27.
51. Morgan JC, Haviland SB, Woodside MA, Konrad TR. Fostering supportive learning environments in long-term care: the case of WIN A STEP UP. Gerontol Geriatr Educ 2007;28:55–75.
From RTI International, Waltham, MA, and Brown University School of Public Health, Providence, RI.
Abstract
- Objective: To review the nursing home culture change literature and identify common barriers to and facilitators of nursing home culture change adoption. Nursing home culture change aims to make nursing homes less institutional by providing more resident-centered care, making environments more homelike, and empowering direct care staff.
- Methods: We reviewed the research literature on nursing home culture change, especially as related to implementation and outcomes.
- Results: Adoption of nursing home culture change practices has been steadily increasing in the past decade, but some practices are more likely to be adopted than others. A commonly reported barrier to culture change adoption is staff resistance to change. Studies suggest that this resistance can be overcome by changes to management practices, including good communication, providing training and education, and leadership support.
- Conclusion: The numerous benefits of nursing home culture change are apparent in the literature. Barriers to its adoption may be overcome by making improvements to nursing home management practices.
Nursing home culture change is a philosophy and combination of diverse practices aimed at making nursing homes less institutional and more resident-centered [1]. Nursing homes have been depicted as dehumanizing “total institutions” [2–4] in which the quality of residents’ lives and the quality of care are generally poor, daily life is medically regimented, only residents’ basic physical needs receive attention [5–8], and direct care workers are subject to poor working conditions for the lowest possible pay [9,10]. Since the 1980s, transforming the culture of nursing homes to be more humanizing, resident-centered, empowering, and homelike has been a primary mission of many stakeholder groups, including nursing home residents and care workers and their advocates [11].
Comprehensive culture change requires transformation of the nursing home environment from that of an institution to that of a home, implementation of more resident-centered care practices, empowerment of direct care staff, and flattening of the traditional organizational hierarchy so that residents and direct-care workers are actively involved in planning and implementing changes that empower them [12,13]. Culture change requires both technical changes, which are relatively straightforward efforts to address issues within a system while fundamentally keeping the system intact, and adaptive changes, which are more complex and entail reforming fundamental values that underlie the system and demand changes to the system itself [14,15].
Over time, nursing home culture change has gained widespread mainstream support. In 2009, the federal government issued new interpretive guidelines for use by nursing home inspectors that call for nursing homes to have more homelike environments and to support more resident-centered care [16]. The Centers for Medicare & Medicaid Services also required state quality improvement organizations to work with nursing homes on culture change efforts [1]. Some states effectively incentivize culture change by tying nursing home reimbursement rates and pay-for-performance policies to the implementation of culture change practices [17]. In addition to federal and state regulations, some nursing home corporations encourage or require facility administrators to implement culture change practices [18]. Overall, nursing homes are pushed to implement culture change practices on many fronts. The promise of beneficial outcomes of culture change also motivates implementation of some culture change practices [19].
In this article, we discuss the key elements of culture change, review the research examining the association between culture change and outcomes, identify key barriers to culture change, and offer suggestions from the literature for overcoming resistance to culture change.
Elements of Culture Change
Changing the Physical Environment
Changing the physical environment of nursing homes to be less institutional and more homelike is a core component of culture change [1]. These include both exterior and interior changes. Exterior changes can include adding walkways, patios, and gardens; interior changes include replacing nurses’ stations with desks, creating resident common areas, introducing the use of linens in dining areas, personalizing mailboxes outside of resident rooms, and adding small kitchens on units [20]. Other ideas for making environments more homelike include providing residents with the choice of colors for painting rooms and the choice of corridor/unit names and replacing public announcement systems with staff pagers [20].
Although changes to the physical environment may be considered cost-prohibitive, many of these changes entail minor and inexpensive enhancements that can help make environments more intimate and reminiscent of home than are traditional nursing homes [21,22]. Additionally, some environmental changes, such as adding raised gardens and walkways, can be designed not only to make the environment more homelike but also to help residents to engage in meaningful activities and connect to former roles, such as those of a homemaker, gardener, or farmer [21–23].
Providing Resident-Centered Care
Making care resident-centered entails enhancing resident choice and decision making and focusing the delivery of services on residents’ needs and preferences. According to Banaszak-Holl and colleagues [24], resident-centered approaches often emphasize the importance of shifting institutional norms and values and drawing employees’ attention to the needs of residents. This cultural shift in values and norms may be signaled by the implementation of practices that strengthen residents’ autonomy regarding everyday decisions. For example, as part of a resident-centered approach, residents would be offered choices and encouraged to make their own decisions about things personally affecting them, such as what to wear or when to go to bed, eating schedules, and menus [1,17,25].
Empowering Care Aides
Nursing home staff empowerment, particularly the empowerment of nursing assistants and other “hands-on” care aides—who are the predominant workforce in nursing homes and provide the vast bulk of care [26]—is a core component of culture change [1]. Such staff empowerment generally entails enhanced participation in decision making and increased autonomy. Staff empowerment practices that were examined in a national survey of nursing home directors [17] included:
- Staff work together to cover shifts when someone cannot come to work
- Staff cross-trained to perform tasks outside of their assigned job duties
- Staff involved in planning social events
- Nursing assistants take part in quality improvement teams
- Nursing assistants know when a resident’s care plan has changed
- Nursing assistants who receive extra training or education receive bonuses or raises
- Nursing assistants can choose which the residents for whom they provide care
We found that the staff empowerment practices most commonly implemented by nursing homes included nursing assistants knowing when a resident’s care plan has changed and staff working together to cover shifts when someone can’t come to work, but it was uncommon for nursing homes to permit nursing assistants to choose which residents they care for [17].
Outcomes of Culture Change
Research over the past 2 decades has examined the outcomes of culture change and the challenges involved in its implementation. Culture change is intended to improve the quality of life for nursing home residents, but the impact of culture change interventions is not clear. Shier and colleagues [27] conducted a comprehensive review of the peer-reviewed and gray literature on culture change published between 2005 and 2012 and found that studies varied widely in scope and evidence was inconsistent. They concluded that there is not yet sufficient evidence to provide specific guidance to nursing homes interested in implementing culture change [27]. The reviewed studies (27 peer-reviewed and 9 gray literature) also were noted to include small sample sizes and restricted geographic coverage, which both limit generalizability.
Although the literature had substantial limitations, Shier and colleagues [27] found numerous beneficial outcomes of culture change. Statistically significant improvements in numerous resident outcome measures were found to be associated with the implementation of culture change practices, including measures of resident quality of life/well-being, engagement and activities, functional status, satisfaction, mood (depression), anxiety/behavior/agitation, and pain/comfort. Two quality of care and services outcome measures also showed significant improvement associated with culture change practices, including increased completion of advance care plans and improved quality of workers’ approach to residents. Various staff outcome measures also showed significant improvement associated with culture change, including improvements in staff turnover/retention, satisfaction/well-being/burnout, absenteeism, knowledge, and attitude. Additionally, studies have shown culture change to be associated with improvements in select organizational outcome measures including operations costs, occupancy rates, revenue/profits, and family satisfaction. Four of the 36 studies reported negative outcomes of culture change. These negative outcomes included increased resident fear/anxiety [28], increased resident incontinence, decreased resident engagement in activities, decreased family engagement [29,30], decreased resident well-being [31], and increased physical incidents [32]. Notably, negative outcomes often co-occurred with positive outcomes [27,28].
To address the limitations of previous culture change research, such as small sample sizes and limited geographic coverage, and to explain some of the previous equivocal findings from quality studies when the extent of culture change practice implementation was not considered or measured, we collaborated on a national study to understand whether nursing home introduction of culture change practices is associated with improved quality [33]. We identified 824 U.S. nursing homes that had implemented some culture change practices, and we classified them by level of culture change practice implementation (high versus low). In nursing homes with high levels of culture change practice implementation, the introduction of nursing home culture change was associated with significant improvements in some care processes (eg, decreased prevalence of restraints, tube feeding, and pressure ulcers; increased proportion of residents on bladder training programs) and improvements in some resident outcomes, including slightly fewer hospitalizations. Among nursing homes with lower levels of culture change practice implementation, the introduction of culture change was associated with fewer health-related and quality-of-life survey deficiencies, but also with a significant increase in the number of resident hospitalizations [33]. Conclusive evidence regarding the impact of nursing homes implementing specific culture change practices or a comprehensive array of culture change practices on resident outcomes and quality of life remains needed, but numerous benefits of culture change are apparent in the literature.
Diffusion of Culture Change Practices
As culture change is widely supported and shows promise for beneficial outcomes, culture change practices are increasingly being implemented in nursing homes nationally. In 2007, a Commonwealth Fund survey found 56% of directors of nursing in U.S. nursing homes reported any culture change implementation or leadership commitment to implementation, but only 5% reported that culture change had completely changed the way the nursing home cared for residents in all areas of the nursing home [34]. In contrast, by 2010, 85% of directors of nursing reported at least partial culture change implementation and 13% reported that culture change had completely changed the way the nursing home cared for residents in all areas [14]. In a more recent survey of nursing home administrators, 16% reported that culture change had completely changed the way the nursing home cared for residents in all areas [35].
Barriers to Culture Change Implementation
Although the growth of culture change in the nursing home industry in the past decade has been impressive, implementation of comprehensive culture change has lagged behind. This is because one notable feature of nursing home culture change is that it is a philosophy that consists of many related practices. As noted above, implementing culture change can involve changes to physical environments, resident-centered care practices, and staff empowerment. This means that facilities can choose to implement as many or as few changes as they would like, and research has shown that there has been a lot of variation in which culture change practices are implemented. For example, in previous research we found that facilities interested in attracting highly reimbursed Medicare rehabilitation patients were more likely to implement hotel-style changes to their physical environments than they were to implement resident-centered care practices or forms of staff empowerment [19]. Sterns and colleagues [36] found that facilities were more likely to implement less complex practices (eg, allowing residents to choose when they go to bed) than more complex practices (eg, involving staff and residents in organizational decision making). The authors suggest that differences in commitment of facility leaders to comprehensive culture change may have contributed to these differences.
Attributes of facility leaders and other aspects of organizational context have been shown to contribute to more and less successful culture change implementation. For example, Scalzi and colleagues [37] found that some important barriers to culture change implementation were not involving all staff in culture change activities and a lack of corporate level support for these efforts. Schuldheis [38] examined differences in organizational context and its role in culture change among 9 Oregon facilities; 3 facilities successfully implemented culture change practices and 6 facilities did not. Results showed that a facility’s existing organizational culture, attention to sustainability, management practices, and staff involvement were important to the success of the initiative. Similarly, Rosemond and colleagues [39] conducted a study involving 8 North Carolina nursing homes. They determined that unsuccessful culture change initiatives could be attributed to the organizations’ readiness for change, a lack of high quality management communications, and unfavorable perceptions of culture change by direct-care workers. A study conducted in 4 nursing homes by Munroe et al [40] found that formal culture change training provided by professional trainers produced better outcomes than informal “train the trainer” sessions provided by other facility managers. Bowers and colleagues [41] also found that unsuccessful implementation of the Green House model of culture change was likely related to a lack of training resources for staff. Similarly, after an in-depth ethnographic study of culture change implementation, Lopez [42] found that it was unrealistic to expect direct-care workers to perform their jobs in radically new ways without being provided with ongoing support from management.
Resistance to Change: A Key Barrier
Our own research sought to understand the barriers and challenges nursing home administrators faced when implementing culture change in their facilities and the strategies they used to overcome them. In interviews conducted with 64 administrators who had participated in a previous nationally representative survey about culture change implementation, administrators reported a wide variety of barriers, including old and outdated physical plants, the costs of some changes, and issues with unions [18]. A key barrier that administrators reported facing was resistance to change on the part of nursing facility staff, residents, and residents’ family members [43]. Administrators reported that residents were resistant to change primarily because they had been institutionalized in their thinking. In other words, nursing homes had essentially trained residents to expect things to be done at certain times and in certain ways. Resistance among staff reportedly included resistance to the overall concept of culture change and to specific culture change practices. Often, staff perceived that changes related to culture change implementation involved additional work or effort on their part without additional resources, but this was not the only reason for resistance. Most often staff, especially longer-term staff, simply were resistant to making any changes to their usual routines or duties.
This type of resistance to change among staff is not unique to culture change implementation and has long been a commonly cited barrier in the organizational change literature. For example, in a 1954 Harvard Business Review article, Lawrence [44] stated that resistance to change was “the most baffling and recalcitrant of the problems which business executives face.” Since that time, resistance to change has been extensively studied as have methods for overcoming such resistance.
Recommendations for Overcoming Resistance to Culture Change
In seminal work on employee resistance to change conducted shortly after World War II, Coch and French [45] challenged the concept that resistance to change was the result of flaws or inadequacies on the part of staff, which would make addressing resistance difficult. Instead, they proposed, and proved through experimental methods, that resistance arose primarily from the context within which the changes were taking place. In other words, they found that managers could ameliorate resistance to change through changes to management and leadership practices. In their experiment, resistance to change in a manufacturing plant was overcome when management effectively communicated to staff the reasons for the change and engaged staff in planning for the desired changes. Studies on the barriers and facilitators of culture change implementation in nursing facilities have similarly found that facility leaders can take steps to address, or even avoid, staff resistance to change.
In our own research, we have found that resistance to change is a common barrier faced by facility leaders. We also found that resistance to change was unique among barriers in that, although strategies used to address other types of barriers varied widely, administrators consistently reported using the same strategies to address and overcome resistance to change. These strategies all involved management and leadership activities, including education and training and improved communication. In addition, administrators discussed in detail the ways they tailored education and communication to their facility’s unique needs. They also indicated that these efforts should be ongoing, communication should be two-way, and that all staff should be included [43].
Good Communication
One important tool for avoiding or overcoming resistance to culture change that facility administrators reported was good communication. They reported that open and bidirectional communication fostered feedback about ongoing culture change efforts and encouraged engagement and buy-in from staff. They also suggested that it is important that this type of communication be ongoing. Good communication about culture change, in particular, included providing a strong rationale for the changes and involved getting input from staff before and during implementation [43].
These findings are similar to other studies of culture change which have found that culture change implementation should involve staff at all levels [37] and that facility leaders should follow through on the plans that have been communicated [39]. Interestingly, the importance of good and open communication has also been identified as important to other forms of nursing facility quality improvement [46].
Training and Education
The facility administrators we interviewed also reported providing education and training for staff about culture change in a variety of ways, including as part of regular in-service training and as a component of new employee orientation. The training materials used were often obtained from the leading culture change organizations. However, importantly, administrators reported tailoring these trainings to the specific needs of their employees or unique context of their facility. For example, administrators reported breaking up long training sessions into shorter segments provided over a longer period of time or organizing trainings to be provided to small groups on the units rather than in more didactic conference-style settings [43]. Administrators explained that providing training in this way was more palatable to staff and helped incorporate learning into everyday care.
Other studies of nursing home culture change have also found training and education to be important to implementation. For example, in a study of a labor-management partnership for culture change implementation, Leutz and colleagues [47] found training of staff from all disciplines by culture change experts to be an important element of successful implementation. Training topics included those that were very general, such as gerontology, and very specific, including person-centered care. Staff were paid for their time participating in training, which took place at their facilities to make participation easier. The trainings were also staggered over the course of several months, so that staff had time to use what they had learned between sessions and could discuss their experiences at the later sessions.
Munroe and colleagues [40] conducted a study of culture change training using pre-post test survey methods and found that formal training had more of an effect on staff than informal training. In the study, staff at 2 facilities received formal education from a consulting group while staff at 2 other facilities then received informal training from the staff of one of the formally trained facilities. An important conclusion of the authors was that the formal training did a better job than the informal training of helping facility leaders and managers view their relationships with staff differently. This suggests that facility leaders and managers may have to alter their management styles to create the supportive context within which culture change efforts can succeed [48].
Leadership Support
Good communication and training/education can be thought of as 2 examples of leadership support, and support from facility leaders and managers has been found, in multiple studies, to be critical to successful culture change efforts. For example, in a recent study of nursing facility culture change in the Netherlands, Snoeren and colleagues [49] found that facility managers can facilitate culture change implementation by supporting a variety of staff needs and promoting the facilities’ new desired values. Another study found that facilities with leaders who are supportive and foster staff flexibility, such as allowing staff to be creative in their problem-solving and have decentralized decision-making, were more likely to report having implemented culture change [24].
In a study focused specifically on facility leadership style and its relation to culture change implementation, Corazzini and colleagues [50] found an adaptive leadership style to be important to culture change implementation. Adaptive leadership styles are ones that acknowledge the importance of staff relationships and recognize that complex changes, like those often implemented in culture change efforts, require complex solutions that will likely evolve over time. These authors conclude that culture change implementation necessitates development of new normative values and behaviors and can, therefore, not be accomplished by simply generating new rules and procedures [50].
Of course, not all nursing facility leaders have the management skills needed to perform in these adaptive and flexible ways. Therefore, management training for facility leaders may be an important first step in a facility’s culture change efforts [51]. This type of training may help improve communication skills and allow facility leaders to perform in more adaptive and flexible ways to better meet the needs of their particular facility and staff. Research also suggests that culture change training for facility leaders may help them to form new and better relationships with staff [40], an important element of culture change.
Conclusion
Nursing home culture change aims to improve care quality and resident satisfaction through changes to physical environments, resident care practices, and staff empowerment. These include both relatively simple technical changes and more complex changes. Nursing home managers and leaders have reported a variety of barriers to implementing nursing home culture change. A common barrier cited is staff resistance to change. Many decades of research in the organizational change literature and more recent research on culture change implementation suggest steps that facility managers and leaders can take to avoid or overcome this resistance. These steps include providing management support, especially in the form of good communication and training and education.
Corresponding author: Denise A. Tyler, PhD, RTI International, 307 Waverly Oaks Rd., Waltham, MA 02452, [email protected].
Financial disclosures: None.
From RTI International, Waltham, MA, and Brown University School of Public Health, Providence, RI.
Abstract
- Objective: To review the nursing home culture change literature and identify common barriers to and facilitators of nursing home culture change adoption. Nursing home culture change aims to make nursing homes less institutional by providing more resident-centered care, making environments more homelike, and empowering direct care staff.
- Methods: We reviewed the research literature on nursing home culture change, especially as related to implementation and outcomes.
- Results: Adoption of nursing home culture change practices has been steadily increasing in the past decade, but some practices are more likely to be adopted than others. A commonly reported barrier to culture change adoption is staff resistance to change. Studies suggest that this resistance can be overcome by changes to management practices, including good communication, providing training and education, and leadership support.
- Conclusion: The numerous benefits of nursing home culture change are apparent in the literature. Barriers to its adoption may be overcome by making improvements to nursing home management practices.
Nursing home culture change is a philosophy and combination of diverse practices aimed at making nursing homes less institutional and more resident-centered [1]. Nursing homes have been depicted as dehumanizing “total institutions” [2–4] in which the quality of residents’ lives and the quality of care are generally poor, daily life is medically regimented, only residents’ basic physical needs receive attention [5–8], and direct care workers are subject to poor working conditions for the lowest possible pay [9,10]. Since the 1980s, transforming the culture of nursing homes to be more humanizing, resident-centered, empowering, and homelike has been a primary mission of many stakeholder groups, including nursing home residents and care workers and their advocates [11].
Comprehensive culture change requires transformation of the nursing home environment from that of an institution to that of a home, implementation of more resident-centered care practices, empowerment of direct care staff, and flattening of the traditional organizational hierarchy so that residents and direct-care workers are actively involved in planning and implementing changes that empower them [12,13]. Culture change requires both technical changes, which are relatively straightforward efforts to address issues within a system while fundamentally keeping the system intact, and adaptive changes, which are more complex and entail reforming fundamental values that underlie the system and demand changes to the system itself [14,15].
Over time, nursing home culture change has gained widespread mainstream support. In 2009, the federal government issued new interpretive guidelines for use by nursing home inspectors that call for nursing homes to have more homelike environments and to support more resident-centered care [16]. The Centers for Medicare & Medicaid Services also required state quality improvement organizations to work with nursing homes on culture change efforts [1]. Some states effectively incentivize culture change by tying nursing home reimbursement rates and pay-for-performance policies to the implementation of culture change practices [17]. In addition to federal and state regulations, some nursing home corporations encourage or require facility administrators to implement culture change practices [18]. Overall, nursing homes are pushed to implement culture change practices on many fronts. The promise of beneficial outcomes of culture change also motivates implementation of some culture change practices [19].
In this article, we discuss the key elements of culture change, review the research examining the association between culture change and outcomes, identify key barriers to culture change, and offer suggestions from the literature for overcoming resistance to culture change.
Elements of Culture Change
Changing the Physical Environment
Changing the physical environment of nursing homes to be less institutional and more homelike is a core component of culture change [1]. These include both exterior and interior changes. Exterior changes can include adding walkways, patios, and gardens; interior changes include replacing nurses’ stations with desks, creating resident common areas, introducing the use of linens in dining areas, personalizing mailboxes outside of resident rooms, and adding small kitchens on units [20]. Other ideas for making environments more homelike include providing residents with the choice of colors for painting rooms and the choice of corridor/unit names and replacing public announcement systems with staff pagers [20].
Although changes to the physical environment may be considered cost-prohibitive, many of these changes entail minor and inexpensive enhancements that can help make environments more intimate and reminiscent of home than are traditional nursing homes [21,22]. Additionally, some environmental changes, such as adding raised gardens and walkways, can be designed not only to make the environment more homelike but also to help residents to engage in meaningful activities and connect to former roles, such as those of a homemaker, gardener, or farmer [21–23].
Providing Resident-Centered Care
Making care resident-centered entails enhancing resident choice and decision making and focusing the delivery of services on residents’ needs and preferences. According to Banaszak-Holl and colleagues [24], resident-centered approaches often emphasize the importance of shifting institutional norms and values and drawing employees’ attention to the needs of residents. This cultural shift in values and norms may be signaled by the implementation of practices that strengthen residents’ autonomy regarding everyday decisions. For example, as part of a resident-centered approach, residents would be offered choices and encouraged to make their own decisions about things personally affecting them, such as what to wear or when to go to bed, eating schedules, and menus [1,17,25].
Empowering Care Aides
Nursing home staff empowerment, particularly the empowerment of nursing assistants and other “hands-on” care aides—who are the predominant workforce in nursing homes and provide the vast bulk of care [26]—is a core component of culture change [1]. Such staff empowerment generally entails enhanced participation in decision making and increased autonomy. Staff empowerment practices that were examined in a national survey of nursing home directors [17] included:
- Staff work together to cover shifts when someone cannot come to work
- Staff cross-trained to perform tasks outside of their assigned job duties
- Staff involved in planning social events
- Nursing assistants take part in quality improvement teams
- Nursing assistants know when a resident’s care plan has changed
- Nursing assistants who receive extra training or education receive bonuses or raises
- Nursing assistants can choose which the residents for whom they provide care
We found that the staff empowerment practices most commonly implemented by nursing homes included nursing assistants knowing when a resident’s care plan has changed and staff working together to cover shifts when someone can’t come to work, but it was uncommon for nursing homes to permit nursing assistants to choose which residents they care for [17].
Outcomes of Culture Change
Research over the past 2 decades has examined the outcomes of culture change and the challenges involved in its implementation. Culture change is intended to improve the quality of life for nursing home residents, but the impact of culture change interventions is not clear. Shier and colleagues [27] conducted a comprehensive review of the peer-reviewed and gray literature on culture change published between 2005 and 2012 and found that studies varied widely in scope and evidence was inconsistent. They concluded that there is not yet sufficient evidence to provide specific guidance to nursing homes interested in implementing culture change [27]. The reviewed studies (27 peer-reviewed and 9 gray literature) also were noted to include small sample sizes and restricted geographic coverage, which both limit generalizability.
Although the literature had substantial limitations, Shier and colleagues [27] found numerous beneficial outcomes of culture change. Statistically significant improvements in numerous resident outcome measures were found to be associated with the implementation of culture change practices, including measures of resident quality of life/well-being, engagement and activities, functional status, satisfaction, mood (depression), anxiety/behavior/agitation, and pain/comfort. Two quality of care and services outcome measures also showed significant improvement associated with culture change practices, including increased completion of advance care plans and improved quality of workers’ approach to residents. Various staff outcome measures also showed significant improvement associated with culture change, including improvements in staff turnover/retention, satisfaction/well-being/burnout, absenteeism, knowledge, and attitude. Additionally, studies have shown culture change to be associated with improvements in select organizational outcome measures including operations costs, occupancy rates, revenue/profits, and family satisfaction. Four of the 36 studies reported negative outcomes of culture change. These negative outcomes included increased resident fear/anxiety [28], increased resident incontinence, decreased resident engagement in activities, decreased family engagement [29,30], decreased resident well-being [31], and increased physical incidents [32]. Notably, negative outcomes often co-occurred with positive outcomes [27,28].
To address the limitations of previous culture change research, such as small sample sizes and limited geographic coverage, and to explain some of the previous equivocal findings from quality studies when the extent of culture change practice implementation was not considered or measured, we collaborated on a national study to understand whether nursing home introduction of culture change practices is associated with improved quality [33]. We identified 824 U.S. nursing homes that had implemented some culture change practices, and we classified them by level of culture change practice implementation (high versus low). In nursing homes with high levels of culture change practice implementation, the introduction of nursing home culture change was associated with significant improvements in some care processes (eg, decreased prevalence of restraints, tube feeding, and pressure ulcers; increased proportion of residents on bladder training programs) and improvements in some resident outcomes, including slightly fewer hospitalizations. Among nursing homes with lower levels of culture change practice implementation, the introduction of culture change was associated with fewer health-related and quality-of-life survey deficiencies, but also with a significant increase in the number of resident hospitalizations [33]. Conclusive evidence regarding the impact of nursing homes implementing specific culture change practices or a comprehensive array of culture change practices on resident outcomes and quality of life remains needed, but numerous benefits of culture change are apparent in the literature.
Diffusion of Culture Change Practices
As culture change is widely supported and shows promise for beneficial outcomes, culture change practices are increasingly being implemented in nursing homes nationally. In 2007, a Commonwealth Fund survey found 56% of directors of nursing in U.S. nursing homes reported any culture change implementation or leadership commitment to implementation, but only 5% reported that culture change had completely changed the way the nursing home cared for residents in all areas of the nursing home [34]. In contrast, by 2010, 85% of directors of nursing reported at least partial culture change implementation and 13% reported that culture change had completely changed the way the nursing home cared for residents in all areas [14]. In a more recent survey of nursing home administrators, 16% reported that culture change had completely changed the way the nursing home cared for residents in all areas [35].
Barriers to Culture Change Implementation
Although the growth of culture change in the nursing home industry in the past decade has been impressive, implementation of comprehensive culture change has lagged behind. This is because one notable feature of nursing home culture change is that it is a philosophy that consists of many related practices. As noted above, implementing culture change can involve changes to physical environments, resident-centered care practices, and staff empowerment. This means that facilities can choose to implement as many or as few changes as they would like, and research has shown that there has been a lot of variation in which culture change practices are implemented. For example, in previous research we found that facilities interested in attracting highly reimbursed Medicare rehabilitation patients were more likely to implement hotel-style changes to their physical environments than they were to implement resident-centered care practices or forms of staff empowerment [19]. Sterns and colleagues [36] found that facilities were more likely to implement less complex practices (eg, allowing residents to choose when they go to bed) than more complex practices (eg, involving staff and residents in organizational decision making). The authors suggest that differences in commitment of facility leaders to comprehensive culture change may have contributed to these differences.
Attributes of facility leaders and other aspects of organizational context have been shown to contribute to more and less successful culture change implementation. For example, Scalzi and colleagues [37] found that some important barriers to culture change implementation were not involving all staff in culture change activities and a lack of corporate level support for these efforts. Schuldheis [38] examined differences in organizational context and its role in culture change among 9 Oregon facilities; 3 facilities successfully implemented culture change practices and 6 facilities did not. Results showed that a facility’s existing organizational culture, attention to sustainability, management practices, and staff involvement were important to the success of the initiative. Similarly, Rosemond and colleagues [39] conducted a study involving 8 North Carolina nursing homes. They determined that unsuccessful culture change initiatives could be attributed to the organizations’ readiness for change, a lack of high quality management communications, and unfavorable perceptions of culture change by direct-care workers. A study conducted in 4 nursing homes by Munroe et al [40] found that formal culture change training provided by professional trainers produced better outcomes than informal “train the trainer” sessions provided by other facility managers. Bowers and colleagues [41] also found that unsuccessful implementation of the Green House model of culture change was likely related to a lack of training resources for staff. Similarly, after an in-depth ethnographic study of culture change implementation, Lopez [42] found that it was unrealistic to expect direct-care workers to perform their jobs in radically new ways without being provided with ongoing support from management.
Resistance to Change: A Key Barrier
Our own research sought to understand the barriers and challenges nursing home administrators faced when implementing culture change in their facilities and the strategies they used to overcome them. In interviews conducted with 64 administrators who had participated in a previous nationally representative survey about culture change implementation, administrators reported a wide variety of barriers, including old and outdated physical plants, the costs of some changes, and issues with unions [18]. A key barrier that administrators reported facing was resistance to change on the part of nursing facility staff, residents, and residents’ family members [43]. Administrators reported that residents were resistant to change primarily because they had been institutionalized in their thinking. In other words, nursing homes had essentially trained residents to expect things to be done at certain times and in certain ways. Resistance among staff reportedly included resistance to the overall concept of culture change and to specific culture change practices. Often, staff perceived that changes related to culture change implementation involved additional work or effort on their part without additional resources, but this was not the only reason for resistance. Most often staff, especially longer-term staff, simply were resistant to making any changes to their usual routines or duties.
This type of resistance to change among staff is not unique to culture change implementation and has long been a commonly cited barrier in the organizational change literature. For example, in a 1954 Harvard Business Review article, Lawrence [44] stated that resistance to change was “the most baffling and recalcitrant of the problems which business executives face.” Since that time, resistance to change has been extensively studied as have methods for overcoming such resistance.
Recommendations for Overcoming Resistance to Culture Change
In seminal work on employee resistance to change conducted shortly after World War II, Coch and French [45] challenged the concept that resistance to change was the result of flaws or inadequacies on the part of staff, which would make addressing resistance difficult. Instead, they proposed, and proved through experimental methods, that resistance arose primarily from the context within which the changes were taking place. In other words, they found that managers could ameliorate resistance to change through changes to management and leadership practices. In their experiment, resistance to change in a manufacturing plant was overcome when management effectively communicated to staff the reasons for the change and engaged staff in planning for the desired changes. Studies on the barriers and facilitators of culture change implementation in nursing facilities have similarly found that facility leaders can take steps to address, or even avoid, staff resistance to change.
In our own research, we have found that resistance to change is a common barrier faced by facility leaders. We also found that resistance to change was unique among barriers in that, although strategies used to address other types of barriers varied widely, administrators consistently reported using the same strategies to address and overcome resistance to change. These strategies all involved management and leadership activities, including education and training and improved communication. In addition, administrators discussed in detail the ways they tailored education and communication to their facility’s unique needs. They also indicated that these efforts should be ongoing, communication should be two-way, and that all staff should be included [43].
Good Communication
One important tool for avoiding or overcoming resistance to culture change that facility administrators reported was good communication. They reported that open and bidirectional communication fostered feedback about ongoing culture change efforts and encouraged engagement and buy-in from staff. They also suggested that it is important that this type of communication be ongoing. Good communication about culture change, in particular, included providing a strong rationale for the changes and involved getting input from staff before and during implementation [43].
These findings are similar to other studies of culture change which have found that culture change implementation should involve staff at all levels [37] and that facility leaders should follow through on the plans that have been communicated [39]. Interestingly, the importance of good and open communication has also been identified as important to other forms of nursing facility quality improvement [46].
Training and Education
The facility administrators we interviewed also reported providing education and training for staff about culture change in a variety of ways, including as part of regular in-service training and as a component of new employee orientation. The training materials used were often obtained from the leading culture change organizations. However, importantly, administrators reported tailoring these trainings to the specific needs of their employees or unique context of their facility. For example, administrators reported breaking up long training sessions into shorter segments provided over a longer period of time or organizing trainings to be provided to small groups on the units rather than in more didactic conference-style settings [43]. Administrators explained that providing training in this way was more palatable to staff and helped incorporate learning into everyday care.
Other studies of nursing home culture change have also found training and education to be important to implementation. For example, in a study of a labor-management partnership for culture change implementation, Leutz and colleagues [47] found training of staff from all disciplines by culture change experts to be an important element of successful implementation. Training topics included those that were very general, such as gerontology, and very specific, including person-centered care. Staff were paid for their time participating in training, which took place at their facilities to make participation easier. The trainings were also staggered over the course of several months, so that staff had time to use what they had learned between sessions and could discuss their experiences at the later sessions.
Munroe and colleagues [40] conducted a study of culture change training using pre-post test survey methods and found that formal training had more of an effect on staff than informal training. In the study, staff at 2 facilities received formal education from a consulting group while staff at 2 other facilities then received informal training from the staff of one of the formally trained facilities. An important conclusion of the authors was that the formal training did a better job than the informal training of helping facility leaders and managers view their relationships with staff differently. This suggests that facility leaders and managers may have to alter their management styles to create the supportive context within which culture change efforts can succeed [48].
Leadership Support
Good communication and training/education can be thought of as 2 examples of leadership support, and support from facility leaders and managers has been found, in multiple studies, to be critical to successful culture change efforts. For example, in a recent study of nursing facility culture change in the Netherlands, Snoeren and colleagues [49] found that facility managers can facilitate culture change implementation by supporting a variety of staff needs and promoting the facilities’ new desired values. Another study found that facilities with leaders who are supportive and foster staff flexibility, such as allowing staff to be creative in their problem-solving and have decentralized decision-making, were more likely to report having implemented culture change [24].
In a study focused specifically on facility leadership style and its relation to culture change implementation, Corazzini and colleagues [50] found an adaptive leadership style to be important to culture change implementation. Adaptive leadership styles are ones that acknowledge the importance of staff relationships and recognize that complex changes, like those often implemented in culture change efforts, require complex solutions that will likely evolve over time. These authors conclude that culture change implementation necessitates development of new normative values and behaviors and can, therefore, not be accomplished by simply generating new rules and procedures [50].
Of course, not all nursing facility leaders have the management skills needed to perform in these adaptive and flexible ways. Therefore, management training for facility leaders may be an important first step in a facility’s culture change efforts [51]. This type of training may help improve communication skills and allow facility leaders to perform in more adaptive and flexible ways to better meet the needs of their particular facility and staff. Research also suggests that culture change training for facility leaders may help them to form new and better relationships with staff [40], an important element of culture change.
Conclusion
Nursing home culture change aims to improve care quality and resident satisfaction through changes to physical environments, resident care practices, and staff empowerment. These include both relatively simple technical changes and more complex changes. Nursing home managers and leaders have reported a variety of barriers to implementing nursing home culture change. A common barrier cited is staff resistance to change. Many decades of research in the organizational change literature and more recent research on culture change implementation suggest steps that facility managers and leaders can take to avoid or overcome this resistance. These steps include providing management support, especially in the form of good communication and training and education.
Corresponding author: Denise A. Tyler, PhD, RTI International, 307 Waverly Oaks Rd., Waltham, MA 02452, [email protected].
Financial disclosures: None.
1. Koren MJ. Person-centered care for nursing home residents: The culture-change movement. Health Affairs 2010;29:1–6.
2. Goffman E. Asylums: essays on the social situation of mental patients and other inmates. Garden City, NY: Anchor Books; 1961.
3. Kane RA, Caplan AL. Everyday ethics: resolving dilemmas in nursing home life. New York: Springer; 1990.
4. Mor V, Branco K, Fleishma J, et al. The structure of social engagement among nursing home residents. J Gerontol B Psycholog Sci Soc Sci 1995;50:P1–P8.
5. Foner N. The caregiving dilemma: work in an American nursing home. Berkeley, CA: University of California Press; 1993.
6. Gubrium J. Living and dying at Murray Manor. New York: St. Martins; 1976.
7. Kayser-Jones JS. Old, alone, and neglected: care of the aged in the United States and Scotland. Berkeley, CA: University of California Press; 1990.
8. Vladeck B. Unloving care: the nursing home tragedy. New York: Basic Books; 1980.
9. Diamond T. Social policy and everyday life in nursing homes: a critical ethnography. Soc Sci Med 1986;23:1287–95.
10. Kalleberg A, Reskin BF, Hudson K. Bad jobs in America: standard and nonstandard employment relations and job quality in the United States. Am Sociolog Rev 2000;65:256–78.
11. Rahman AN, Schnelle JF. The nursing home culture-change movement: recent past, present, and future directions for research. Gerontologist 2008;48:142–8.
12. White-Chu EF, Graves WJ, Godfrey SM, et al. Beyond the medical model: the culture change revolution in long-term care. J Am Med Dir Assoc 2009;10:370–8.
13. Misiorski S, Kahn K. Changing the culture of long-term care: Moving beyond programmatic change. J Soc Work Long-Term Care 2006;3:137–46.
14. Anderson RA, Bailey DEJ, Wu B, et al. Adaptive leadership framework for chronic illness: framing a research agenda for transforming care delivery. Adv Nurs Sci 2015;38:83–95.
15. Bailey DE, Docherty S, Adams JA, et al. Studying the clinical encounter with the adaptive leadership framework. J Healthc Leadersh 2012;4:83–91.
16. Centers for Medicare & Medicaid Services Manual System. Revisions to Appendix PP “Guidance to Surveyors of Long Term Care Facilities” Washington, DC: Department of Health and Human Services 2009. Accessed at http://www.cms.gov/Regulations-and-Guidance/Guidance/Transmittals/downloads/R48SOMA.pdf.
17. Miller SC, Looze J, Shield R, et al. Culture change practice in US nursing homes: prevalence and variation by state Medicaid reimbursement policies. Gerontologist 2014;54:434–45.
18. Shield R, Looze J, Tyler D, et al. Why and how do nursing homes implement culture change practices? Insights from qualitative interviews in a mixed methods study. J Appl Gerontol 2014;33:737–63.
19. Lepore MJ, Shield RR, Looze J, et al. Medicare and Medicaid reimbursement rates for nursing homes motivate select culture change practices but not comprehensive culture change. J Aging Soc Pol 2015;27:215–31.
20. Shield RR, Tyler D, Lepore M, et al. Would you do that in your home? Making nursing homes home-like in culture change implementation. J Housing Elderly 2014;28:383–98.
21. Cutler L, Kane RA. As great as all outdoors. J Hous Elderly 2006;19:29–48.
22. Jurkowsky ET. Implementing culture change in long-term care: Benchmarks and strategies for management and practice. New York: Springer; 2013.
23. Wang D , Glicksman A. “Being grounded”: Benefits of gardening for older adults in low-income housing. J Hous Elderly 2013;27:89–104.
24. Banaszak-Holl J, Castle NG, Lin M, Spreitzer G. An assessment of cultural values and resident-centered culture change in US nursing facilities. Healthc Manage Rev 2013;38:295.
25. White-Chu EF, Graves WJ, Godfrey SM, et al. Beyond the medical model: the culture change revolution in long-term care. J Am Med Dir Assoc 2009;10:370–8.
26. Stone RI. Developing a quality direct care workforce: searching for solutions. Pub Pol Aging Rep 2017.
27. Shier V, Khodyakov D, Cohen LW, et al. What does the evidence really say about culture change in nursing homes? Gerontologist 2014;54:S6–S16.
28. Fritsch T, Kwak J, Grant S, et al. Impact of TimeSlips, a creative expression intervention program, on nursing home residents with dementia and their caregivers. Gerontologist 2009;49:117–27.
29. Kane RA, Lum TY, Cutler LJ, et al. Resident outcomes in small-house nursing homes: a longitudinal evaluation of the initial Green House program. J Am Geriatr Soc 2007;55:832-9.
30. Lum TY, Kane RA, Cutler LJ, Yu TC. Effects of Green House nursing homes on residents’ families. Healthc Financ Rev 2008;30:35–51.
31. Brooker DJ, Woolley RJ, Lee D. Enriching opportunities for people living with dementia in nursing homes: an evaluation of a multi-level activity-based model of care. Aging Ment Health 2007;11:361–70.
32. Detweiler MB, Murphy PF, Myers LC, Kim KY. Does a wander garden influence inappropriate behaviors in dementia residents? Am J Alzheimers Dis Other Dement 2008;23:31–45.
33. Miller SC, Lepore M, Lima JC, et al. Does the introduction of nursing home culture change practices improve quality? J Am Geriatr Soc 2014;62:1675–82.
34. Doty MM, Koren MJ, Sturla EL. Culture change in nursing homes: how far have we come? Findings from the Commonweath Fund 2007 National Survey of Nursing Homes; 2008. Accessed at http://www.commonwealthfund.org/Publications/Fund-Reports/2008/May/Culture-Change-in-Nursing-Homes--How-Far-Have-We-Come--Findings-From-The-Commonwealth-Fund-2007-Nati.aspx.
35. Miller SC, Tyler D, Shield R, et al. Nursing home culture change: study framework and survey instrument design. Presentation at the International Association of Gerontology and Geriatrics meeting, San Francisco, CA; 2017.
36. Sterns S, Miller SC, Allen S. The complexity of implementing culture change practices in nursing homes. J Am Med Dir Assoc 2010;11:511–8.
37. Scalzi CC, Evans LK, Barstow A, Hostvedt K. Barriers and enablers to changing organizational culture in nursing homes. Nurs Admin Q 2006;30:368–72.
38. Schuldheis S. Initiating person-centered care practices in long-term care facilities. J Gerontol Nurs 2007;33:47.
39. Rosemond CA, Hanson LC, Ennett ST, et al. Implementing person-centered care in nursing homes. Healthc Manage Rev 2012;37:257–66.
40. Munroe DJ, Kaza PL, Howard D. Culture-change training: Nursing facility staff perceptions of culture change. Geriatr Nurs 2011;32:400–7.
41. Bowers BJ, Nolet K. Developing the Green House nursing care team: Variations on development and implementation. Gerontologist 2014;54:S53–64.
42. Lopez SH. Culture change management in long-term care: a shop-floor view. Pol Soc 2006;34:55–80.
43. Tyler DA, Lepore M, Shield RR, et al. Overcoming resistance to culture change: nursing home administrators’ use of education, training, and communication. Gerontol Geriatr Educ 2014;35:321–36.
44. Lawrence PR. How to deal with resistance to change. Harvard Bus Rev 1954;May/June:49–57.
45. Coch L, French JRP. Overcoming resistance to change. Hum Relat 1948;1:512–32.
46. Scott-Cawiezell J, Schenkman M, Moore L, et al. Exploring nursing home staff’s perceptions of communication and leadership to facilitate quality improvement. J Nurs Care Qual 2004;19:242–52.
47. Leutz W, Bishop CE, Dodson L. Role for a labor–management partnership in nursing home person-centered care. Gerontologist 2009;50:340–51.
48. Tyler DA, Parker VA. Nursing home culture, teamwork, and culture change. J Res Nurs 2011;16:37–49.
49. Snoeren MM, Janssen BM, Niessen TJ, Abma TA. Nurturing cultural change in care for older people: seeing the cherry tree blossom. Health Care Anal 2016;24:349–73.
50. Corazzini K, Twersky J, White HK, et al. Implementing culture change in nursing homes: an adaptive leadership framework. Gerontologist 2014;55:616–27.
51. Morgan JC, Haviland SB, Woodside MA, Konrad TR. Fostering supportive learning environments in long-term care: the case of WIN A STEP UP. Gerontol Geriatr Educ 2007;28:55–75.
1. Koren MJ. Person-centered care for nursing home residents: The culture-change movement. Health Affairs 2010;29:1–6.
2. Goffman E. Asylums: essays on the social situation of mental patients and other inmates. Garden City, NY: Anchor Books; 1961.
3. Kane RA, Caplan AL. Everyday ethics: resolving dilemmas in nursing home life. New York: Springer; 1990.
4. Mor V, Branco K, Fleishma J, et al. The structure of social engagement among nursing home residents. J Gerontol B Psycholog Sci Soc Sci 1995;50:P1–P8.
5. Foner N. The caregiving dilemma: work in an American nursing home. Berkeley, CA: University of California Press; 1993.
6. Gubrium J. Living and dying at Murray Manor. New York: St. Martins; 1976.
7. Kayser-Jones JS. Old, alone, and neglected: care of the aged in the United States and Scotland. Berkeley, CA: University of California Press; 1990.
8. Vladeck B. Unloving care: the nursing home tragedy. New York: Basic Books; 1980.
9. Diamond T. Social policy and everyday life in nursing homes: a critical ethnography. Soc Sci Med 1986;23:1287–95.
10. Kalleberg A, Reskin BF, Hudson K. Bad jobs in America: standard and nonstandard employment relations and job quality in the United States. Am Sociolog Rev 2000;65:256–78.
11. Rahman AN, Schnelle JF. The nursing home culture-change movement: recent past, present, and future directions for research. Gerontologist 2008;48:142–8.
12. White-Chu EF, Graves WJ, Godfrey SM, et al. Beyond the medical model: the culture change revolution in long-term care. J Am Med Dir Assoc 2009;10:370–8.
13. Misiorski S, Kahn K. Changing the culture of long-term care: Moving beyond programmatic change. J Soc Work Long-Term Care 2006;3:137–46.
14. Anderson RA, Bailey DEJ, Wu B, et al. Adaptive leadership framework for chronic illness: framing a research agenda for transforming care delivery. Adv Nurs Sci 2015;38:83–95.
15. Bailey DE, Docherty S, Adams JA, et al. Studying the clinical encounter with the adaptive leadership framework. J Healthc Leadersh 2012;4:83–91.
16. Centers for Medicare & Medicaid Services Manual System. Revisions to Appendix PP “Guidance to Surveyors of Long Term Care Facilities” Washington, DC: Department of Health and Human Services 2009. Accessed at http://www.cms.gov/Regulations-and-Guidance/Guidance/Transmittals/downloads/R48SOMA.pdf.
17. Miller SC, Looze J, Shield R, et al. Culture change practice in US nursing homes: prevalence and variation by state Medicaid reimbursement policies. Gerontologist 2014;54:434–45.
18. Shield R, Looze J, Tyler D, et al. Why and how do nursing homes implement culture change practices? Insights from qualitative interviews in a mixed methods study. J Appl Gerontol 2014;33:737–63.
19. Lepore MJ, Shield RR, Looze J, et al. Medicare and Medicaid reimbursement rates for nursing homes motivate select culture change practices but not comprehensive culture change. J Aging Soc Pol 2015;27:215–31.
20. Shield RR, Tyler D, Lepore M, et al. Would you do that in your home? Making nursing homes home-like in culture change implementation. J Housing Elderly 2014;28:383–98.
21. Cutler L, Kane RA. As great as all outdoors. J Hous Elderly 2006;19:29–48.
22. Jurkowsky ET. Implementing culture change in long-term care: Benchmarks and strategies for management and practice. New York: Springer; 2013.
23. Wang D , Glicksman A. “Being grounded”: Benefits of gardening for older adults in low-income housing. J Hous Elderly 2013;27:89–104.
24. Banaszak-Holl J, Castle NG, Lin M, Spreitzer G. An assessment of cultural values and resident-centered culture change in US nursing facilities. Healthc Manage Rev 2013;38:295.
25. White-Chu EF, Graves WJ, Godfrey SM, et al. Beyond the medical model: the culture change revolution in long-term care. J Am Med Dir Assoc 2009;10:370–8.
26. Stone RI. Developing a quality direct care workforce: searching for solutions. Pub Pol Aging Rep 2017.
27. Shier V, Khodyakov D, Cohen LW, et al. What does the evidence really say about culture change in nursing homes? Gerontologist 2014;54:S6–S16.
28. Fritsch T, Kwak J, Grant S, et al. Impact of TimeSlips, a creative expression intervention program, on nursing home residents with dementia and their caregivers. Gerontologist 2009;49:117–27.
29. Kane RA, Lum TY, Cutler LJ, et al. Resident outcomes in small-house nursing homes: a longitudinal evaluation of the initial Green House program. J Am Geriatr Soc 2007;55:832-9.
30. Lum TY, Kane RA, Cutler LJ, Yu TC. Effects of Green House nursing homes on residents’ families. Healthc Financ Rev 2008;30:35–51.
31. Brooker DJ, Woolley RJ, Lee D. Enriching opportunities for people living with dementia in nursing homes: an evaluation of a multi-level activity-based model of care. Aging Ment Health 2007;11:361–70.
32. Detweiler MB, Murphy PF, Myers LC, Kim KY. Does a wander garden influence inappropriate behaviors in dementia residents? Am J Alzheimers Dis Other Dement 2008;23:31–45.
33. Miller SC, Lepore M, Lima JC, et al. Does the introduction of nursing home culture change practices improve quality? J Am Geriatr Soc 2014;62:1675–82.
34. Doty MM, Koren MJ, Sturla EL. Culture change in nursing homes: how far have we come? Findings from the Commonweath Fund 2007 National Survey of Nursing Homes; 2008. Accessed at http://www.commonwealthfund.org/Publications/Fund-Reports/2008/May/Culture-Change-in-Nursing-Homes--How-Far-Have-We-Come--Findings-From-The-Commonwealth-Fund-2007-Nati.aspx.
35. Miller SC, Tyler D, Shield R, et al. Nursing home culture change: study framework and survey instrument design. Presentation at the International Association of Gerontology and Geriatrics meeting, San Francisco, CA; 2017.
36. Sterns S, Miller SC, Allen S. The complexity of implementing culture change practices in nursing homes. J Am Med Dir Assoc 2010;11:511–8.
37. Scalzi CC, Evans LK, Barstow A, Hostvedt K. Barriers and enablers to changing organizational culture in nursing homes. Nurs Admin Q 2006;30:368–72.
38. Schuldheis S. Initiating person-centered care practices in long-term care facilities. J Gerontol Nurs 2007;33:47.
39. Rosemond CA, Hanson LC, Ennett ST, et al. Implementing person-centered care in nursing homes. Healthc Manage Rev 2012;37:257–66.
40. Munroe DJ, Kaza PL, Howard D. Culture-change training: Nursing facility staff perceptions of culture change. Geriatr Nurs 2011;32:400–7.
41. Bowers BJ, Nolet K. Developing the Green House nursing care team: Variations on development and implementation. Gerontologist 2014;54:S53–64.
42. Lopez SH. Culture change management in long-term care: a shop-floor view. Pol Soc 2006;34:55–80.
43. Tyler DA, Lepore M, Shield RR, et al. Overcoming resistance to culture change: nursing home administrators’ use of education, training, and communication. Gerontol Geriatr Educ 2014;35:321–36.
44. Lawrence PR. How to deal with resistance to change. Harvard Bus Rev 1954;May/June:49–57.
45. Coch L, French JRP. Overcoming resistance to change. Hum Relat 1948;1:512–32.
46. Scott-Cawiezell J, Schenkman M, Moore L, et al. Exploring nursing home staff’s perceptions of communication and leadership to facilitate quality improvement. J Nurs Care Qual 2004;19:242–52.
47. Leutz W, Bishop CE, Dodson L. Role for a labor–management partnership in nursing home person-centered care. Gerontologist 2009;50:340–51.
48. Tyler DA, Parker VA. Nursing home culture, teamwork, and culture change. J Res Nurs 2011;16:37–49.
49. Snoeren MM, Janssen BM, Niessen TJ, Abma TA. Nurturing cultural change in care for older people: seeing the cherry tree blossom. Health Care Anal 2016;24:349–73.
50. Corazzini K, Twersky J, White HK, et al. Implementing culture change in nursing homes: an adaptive leadership framework. Gerontologist 2014;55:616–27.
51. Morgan JC, Haviland SB, Woodside MA, Konrad TR. Fostering supportive learning environments in long-term care: the case of WIN A STEP UP. Gerontol Geriatr Educ 2007;28:55–75.
Using Clinical Decision Support to Reduce Inappropriate Imaging: A Health Care Improvement Case Study
From the Office of Science Policy and Communications, National Institute on Drug Abuse, National Institutes of Health, Rockville, MD, and George Washington University, Washington, DC (Dr. Jones), Office of the National Coordinator for Health Information Technology, US Department of Health and Human Services, Washington, DC (Mr. Swain), and Banner Health, Phoenix, AZ (Ms. Burdick).
Abstract
- Objective: Clinical decision support (CDS) can be a useful tool to decrease inappropriate imaging by providing evidence-based information to clinicians at the point of care. The objective of this case study is to highlight lessons from a health care improvement initiative using CDS to encourage use of ultrasound rather than computed tomography (CT) scans as an initial diagnostic tool for suspected appendicitis in pediatric patients.
- Methods: The percentage of suspected pediatric appendicitis cases receiving ultrasounds and CT scans was calculated using electronic health record data. Four steps for implementing health information technology were identified in a literature scan that guided data collection and analysis: planning, software customization and workflow design, training and user support, and optimization.
- Results: During the fourth quarter of 2010, 1 in 7 pediatric patients with suspected appendicitis received an ultrasound and almost half received a CT scan. By the first quarter of 2012, ultrasounds were performed in 40.8% of these cases and the use of CT scans declined to 39.9% of suspected pediatric appendicitis cases.
- Conclusion: Four lessons emerged. First, all levels of staff should be involved in the planning process to make organizational priorities actionable and build buy-in for each healthcare improvement initiative. Second, it takes time to design and test the alert to ensure that clinical guidelines are being properly applied. Third, re-engineering the workflow is critical for usability; in this case, ensuring the availability of ultrasound staff was particularly important. Finally, the effectiveness of CDS depends on applying relevant evidence-based practice guidelines to real-time patient data.
Diagnostic imaging is a useful tool for identifying and guiding the treatment of many health conditions, but evidence indicates that health care providers do not always use imaging appropriately. In fact, a substantial proportion of diagnostic imaging procedures performed in hospital and ambulatory settings are not supported by clinical guideline recommendations [1,2]. Spending on diagnostic imaging is rapidly increasing, and some patients receive unnecessary radiation exposure that can lead to adverse health impacts [3]. Inappropriate imaging falls into 3 broad categories: imaging that does not conform to clinical guidelines, imaging that is contraindicated due to an allergy or implantable medical device, and imaging that might be clinically indicated but is duplicative of prior imaging services.
Clinical decision support (CDS) functionality supports health care improvement initiatives to narrow the gap between evidence-based practices and routine care [4]. CDS merges patient-specific clinical information with relevant information about evidence-based practices, providing health care providers with timely information to guide decisions at the point of care [5]. Decision support is most commonly delivered in the form of alerts and reminders [6]. CDS can be effective in reducing adverse drug events [7], sepsis [8,9], and other conditions in hospital [10–12] and ambulatory settings [13,14].
For the evaluation of suspected appendicitis in children, ultrasound is the preferred initial consideration for imaging examination [15]. Evidence suggests that CDS can increase the use of ultrasound for suspected pediatric appendicitis [16,17] and has affirmed the utility of ultrasound as a first-line diagnostic tool for suspected appendicitis [18,19]. In the Choosing Wisely campaign, the American College of Surgeons and the American College of Radiology have both endorsed ultrasound as an option to consider prior to conducting a CT scan to evaluate suspected appendicitis in children [15].
Banner Health, a large health system headquartered in Phoenix, Arizona, implemented a health care improvement initiative using CDS functionality to encourage providers to use ultrasound instead of CT as a first-line diagnostic tool for suspected pediatric appendicitis. We conducted a site visit to Banner Health, an organization who had had attained a high score on the EMR Adoption Model [20] to examine their implementation process. We sought to build on previous research examining the use of health information technology to improve performance in large health systems [21–23].
Methods
Setting
Banner Health is a large not-for-profit health system that is comprised of 24 acute care hospitals across several states, as well as ambulatory medical practices, behavioral health, home care, and ambulatory surgery centers [24,25]. The health system is the largest employer in Arizona and one of the largest in the United States with over 50,000 employees. Banner Health has been nationally recognized for clinical quality [26], an innovative leadership team [27], and using health IT to improve quality [20]. The health system was also selected as one of the Centers for Medicare & Medicaid Services (CMS) Pioneer Accountable Care Organizations.
Site Visit
The first 2 authors conducted a 2-day site visit to the Banner Health headquarters in Phoenix, Arizona in November 2013. The team conducted discussions with over 20 individuals, including health system leadership, frontline clinicians in several units of an acute care hospital, staff members in 2 telehealth hubs—including a tele-ICU hub—and trainers in a simulation facility that is used for staff training. The discussions were conducted with groups of staff or on an individual basis, as appropriate. At the outset of the project, an environmental scan of relevant grey and peer-reviewed literature was conducted under contract on behalf of the authors to guide data collection and analysis [28]. An interview protocol was created to guide the discussions. The protocol contained modules that were used during each discussion, if relevant. The modules addressed topics such as technical issues with designing and deploying health information technology functionalities such as clinical decision support systems, the organizational processes and structures needed to launch health care improvement initiatives, and using health information technology care coordination. Within each module, questions probed about the challenges that arose and the solutions to these challenges, with a focus on the four phases of implementing a health information technology intervention: functionality planning, software customization and workflow design, training and user support, and optimization. To assist with interpreting the qualitative findings, an evolving outline of the findings was maintained. Salient themes and conceptual categories were tracked, which helped the researchers organize, synthesize, and interpret the information collected during the site visit. Once the authors chose to focus on clinical decision support, summary notes from the discussions were reviewed for relevant information, and this information was compiled and organized under the rubric of the four implementation phases. The findings and key themes from the discussion notes were distilled into key lessons for the field.
Data obtained included the percentage of pediatric patients with suspected appendicitis who received ultrasounds and CT scans each month from 1 October 2010 through 31 March 2012. Banner Health staff originally collected the data to support the implementation of health care improvement initiative; the use of these data in this paper is a secondary use [29].
This manuscript was prepared using the SQUIRE 2.0 guidelines [30]. No patient-identifiable data were used, so institutional review board approval was not sought.
Results
The 4 steps of implementing CDS can be described as functionality planning, software customization and workflow design, training and user support, and optimization [31].
Pre-Implementation
The use of computerized provider order entry (CPOE) is a precursor to using clinical decision support, since orders must be entered electronically to be subject to CDS review. Banner Health deployed CPOE to its various facilities starting in 2008. The deployment was staged in a rolling fashion with one or two facilities going live every few months so that the deployment team was available at each facility.
Phase 1: Planning
In contrast to many large health systems, the organization has a single board of directors that oversees the entire system of over 37,000 employees. Activities and relationships to promote the use of evidence-based practices are built into the organizational structure. For example, Banner Health maintains a Care Management Council, a group comprised of clinical and administrative leadership to provide executive oversight of health care improvement projects. The Council convenes on a quarterly basis to review and approve the adoption of new clinical practice guidelines, policies, and standardized standing orders that have been developed by multidisciplinary groups of physicians and other clinicians. A key focus of the Council is ensuring consistent application of evidence-based guidelines to clinical care and disseminating knowledge of clinical best practices across a large and complex enterprise.
Interdisciplinary clinical consensus groups support the Council’s work. These groups are comprised of administrative and program management staff, physicians and other clinicians, and engineers. Each clinical consensus group focuses on emerging issues and improvement opportunities within a specific clinical domain and leads the implementation of health care improvement initiatives in that domain. Providers and staff at all levels of the organization were involved in planning and implementing the health care improvement initiative in inappropriate imaging. This increased buy-in and staff support, which are associated with successful health care improvement initiatives [32]. Banner Health staff rallied around the idea of addressing inappropriate imaging as a key priority initiative. The teams that implement each initiative include an engineer that focuses on redesigning clinical workflows for each initiative. There is also an organizational unit responsible for project management that provides teams with logistical and operational support.
Phase 2: Software Customization and Workflow Redesign
Once the clinical consensus group selected inappropriate imaging as a priority, the next step was to examine the process flow for imaging ordering. In 2011 Banner Health integrated CDS functionality with CPOE into the electronic health record. Before the use of CDS, inpatient and emergency department imaging orders were simply transmitted to imaging staff after the order was entered. After CDS implementation, the process flow begins with an inpatient imaging order and entailed checking the order against clinical guidelines on the proper use of imaging. If the image order did not conform to guidelines, which in this case indicate that ultrasound should be used before CT scans as a diagnostic tool for suspected pediatric appendicitis, the CDS system triggered an alert [15].
Bringing the perspective and skill sets of engineers to the process of redesigning clinical workflows was particularly valuable [33]. While CDS has the potential to reduce inpatient inappropriate imaging, effectiveness depends on adjusting workflows to ensure that the information provided by CDS alerts and reminders is actionable. To reduce alert fatigue among the clinical staff, the team identified the appropriate level of workflow interruption for each alert and reminder (hard stop, workflow interruption, or informational) [5,6].
The design principles that were used to design the alert include intuitive system development to promote ease of use, one set of screen formats and data definitions, and a set of consistent core reports and standard system output formats across facilities. The alert’s appearance was tailored for maximal impact and covered most of the screen. Color contrast was used, but since some people are color-blind, the meaning of the alert did not depend on the color contrast. The alerts included recommendations for changing the treatment plan to encourage using ultrasound as a first-line diagnostic tool. Minimizing the number of clicks to accept the proposed treatment plan change in the alert is desirable.
Phase 3: Training and User Support
Training and support structures and tools were critical to the rollout of the inappropriate imaging alerts. Providers were reminded about clinical best practices and informed during staff meetings about the new CDS rules. In addition, various types of training and support were available to clinicians and staff during the rollout process. Dedicated time for end-user training provided an opportunity to identify and cultivate super-users. These super-users not only helped provide technical support to their colleagues, but also helped create excitement for the initiative. A centralized support desk provided telephone support for providers in facilities throughout the Banner Health system. Site managers were provided toolkits to support providers and staff throughout the implementation process. The toolkits included frequently asked questions and answers, and were maintained as ‘living documents’ that were updated based on emerging issues and questions.
To keep things on track, project managers from the central project management department were involved in the initiative to provide direct project management services to the initiative. They also worked to instill project management competencies throughout the organization, applying a train-the-trainer approach to disseminate best practices for enhancing communication among team members, implementing workflow changes, and monitoring the results.
Phase 4: Optimization
The optimization phase is continuous and continues to the present day. Notably, the success of the CDS rules depends on the availability of current clinical information for each patient, in addition to information about the treatment plan. For this initiative, Banner Health maintained aggregated clinical patient data in the data warehouse that aggregated data from disparate sources, including billing and EHR data from different care settings such as ambulatory offices, inpatient units, the emergency department, home care, and ambulatory surgery centers. The data warehouse is housed in a strategically chosen physical location to minimize the threat of natural disasters, and cloud-based backup is also used. A master patient index and provider credentialing system are integrated with the data warehouse. Query-based health information exchange is used, when possible, to collect information on care received by patients outside of the Banner Health system.
It is important to note that many CDS alerts are over-ridden without changes to clinical care [34]. Previous research indicates that alert fatigue from “false positives” can impede the effectiveness of alerts [35]. Banner Health monitors the rate at which CDS alerts are over-ridden. Figure 1 shows the percentage of all alerts for radiation exposure—including the alert related to using ultrasound as a diagnostic tool for pediatric appendicitis—that led to order cancellations. The percentage of CT orders that generated the alert and were cancelled fell from 18.9% in March 2011 to 13.6% in February 2012. The rate of order cancellations might have declined over time due to a change in provider behavior from the alert. That is, if inappropriate CT scan orders declined over time, then providers would cancel a decreasing percentage of the CT scan orders that prompted an alert. 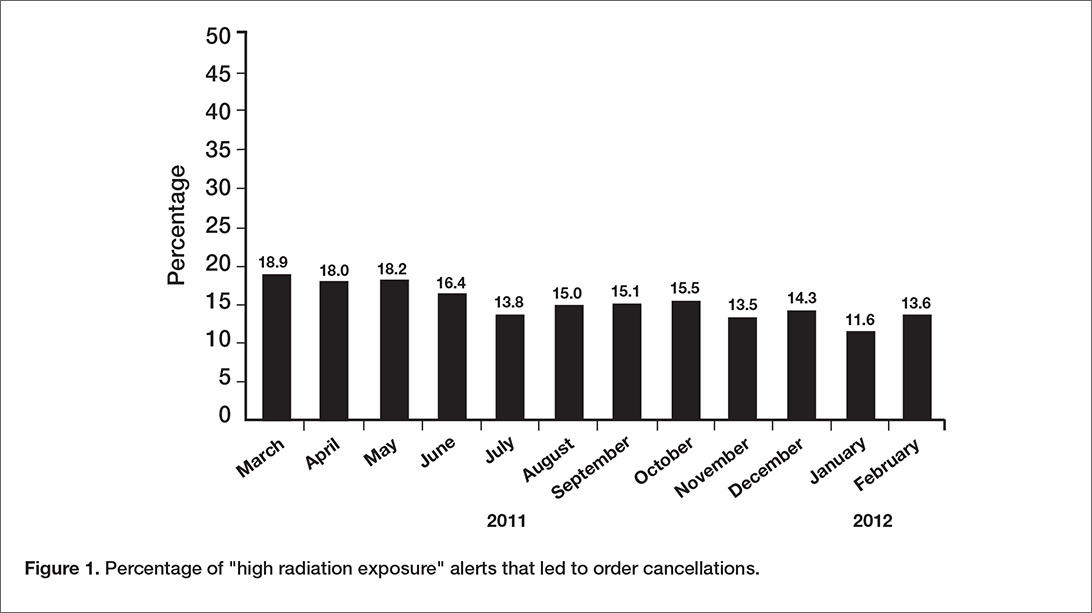
Imaging Use
In Figure 2, data on the use of the 2 imaging procedures for the diagnosis of pediatric appendicitis is presented. During the fourth quarter of 2010, almost half of pediatric patients with suspected appendicitis received a CT scan and only about 1 in 7 received an ultrasound. After the clinical decision support alert was put in place to remind providers to perform an ultrasound as a first-line diagnostic tool, the use of ultrasounds increased sharply. By the first quarter of 2012, ultrasounds were performed in 40.8% of these cases and the use of CT scans declined to 39.9% of suspected pediatric appendicitis cases. 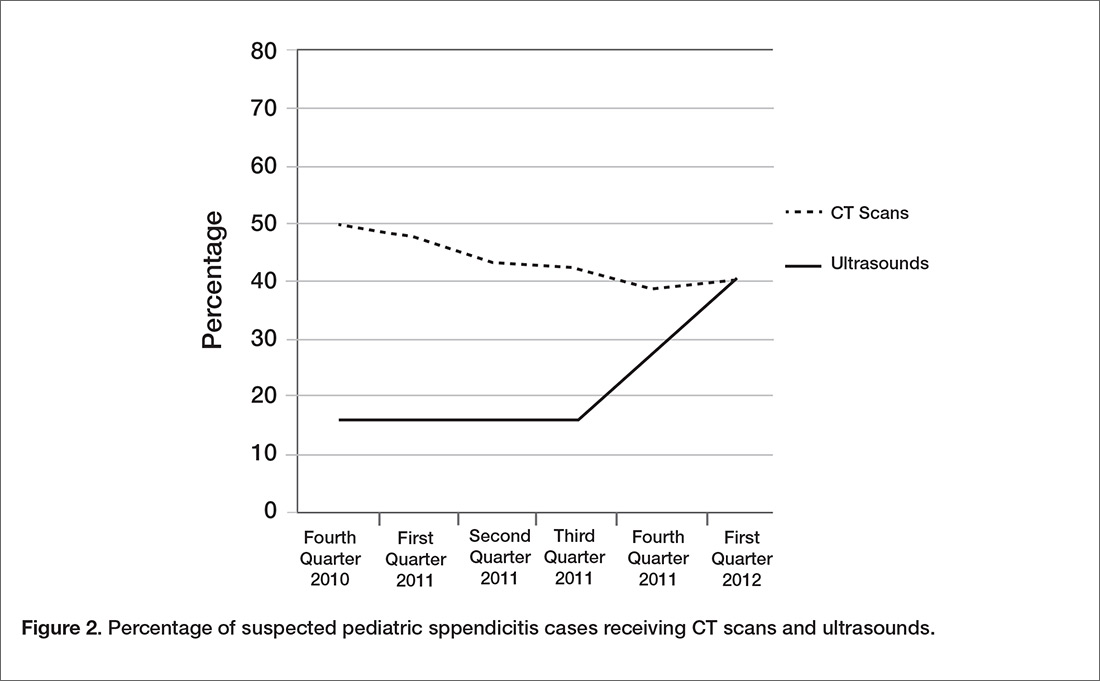
Discussion
This case study discusses the application of CDS functionality in a health care improvement initiative to address inappropriate imaging in a large health system. Four main implementation lessons emerge for the field. First, it is important to involve all levels of staff in the planning process to ensure that health care improvement activities are prioritized correctly and to build buy-in for the priorities addressed with health care improvement activities. Second, it is necessary to allow time to design the alert or reminder, as well as testing it during the implementation process to ensure that clinical guidelines are being properly applied. Third, re-engineering the workflow and ensuring usability of the alert or reminder are important, and using the skills of trained engineers helps in this process. Ensuring the availability of trained ultrasound staff was particularly important to this initiative. Finally, the effectiveness of CDS depends on having complete data for each patient, as well as up-to-date information on the relevant evidence-based practice guidelines.
These results can help guide the implementation of health care improvement initiatives that use CDS functionality to address inappropriate imaging. The adoption of electronic health records with CDS functionality was incentivized and supported by the Medicare and Medicaid Electronic Health Record Incentive Programs; the Medicare program now exists as part of MACRA. Using CDS to reduce inappropriate imaging is required for Medicare fee-for-service patients in the 2014 Protecting Access to Medicare Act (PAMA), highlighting the critical nature of these results, which can guide implementation of CDS to reduce inappropriate imaging [41].
As noted above, the optimization phase is continuous. Banner Health still encourages use of ultrasounds as a first-line diagnostic tool for pediatric appendicitis. Identifying which patients should immediately receive CT scans is difficult, and sometimes the decision depends on the availability of staff to conduct the ultrasound scans. Ways to maximize the productivity of ultrasound technicians have been explored. Another focus area since the original implementation of this health care improvement initiative has been health information exchange, to ensure that complete, up-to-date information is available for each patient.
Banner Health often implements CDS in conjunction with other health IT functionalities. For example, CDS and telehealth are used together to improve care in the intensive care unit (ICU) for patients with sepsis and delirium. An offsite hub of experienced ICU physicians and nurses remotely monitors ICU patients in facilities across Banner Health, using cameras with zoom capability. The intensive care specialists in the tele-hub act as part of the care team; in addition to receiving video feed, they communicate verbally with patients and ICU staff members. Predictive analytics are used to generate clinical decision support alerts and reminders, with a special focus on early intervention if a patient’s clinical indicators are trending downward. The 4 lessons described in this study were also used in the ICU sepsis and delirium initiative; staff were involved in the planning process, alerts and reminders were thoroughly tested, the workflow was adjusted to accommodate the physicians in the tele-ICU hub, and up-to-date and complete clinical information for each patient is maintained. In addition, the design principles for alerts described in this study, such as covering most of the screen and providing recommendations for changing the treatment plan within the alert itself, were also used in the ICU sepsis and delirium initiative.
One limitation of this study is that it was conducted at a single health system. Thus, the findings might not be generalizable to other health systems, particularly if a robust health IT infrastructure is not in place. The culture of Banner Health values quality and involved providers and staff at all levels in selecting and implementing health care improvement initiatives. In addition, engineers assisted with implementation. Finally, the study design does not permit conclusions about the causality of the decline in CT scans and the increase in ultrasounds for suspected pediatric appendicitis cases; unobserved factors might have contributed to the changes in CT and ultrasound use.
Future research should focus on ways to improve the implementation and organization learning process, particularly through engagement of frontline staff by leadership [36] and explore how to operationalize previous findings indicating that innovations in hospital settings are more likely to be sustained when intrinsically rewarding to staff, either by making clinician and staff jobs easier to perform or more gratifying [37]. Future research should focus on facilitating health information exchange between providers in different health systems.
Disclaimer: The views expressed in the article are solely the views of the authors and do not represent those of the National Institutes of Health or the U.S. Government.
Acknowledgments: The authors wish to thank the Banner Health team for taking time to share their insights on how health information technology can be used for health care improvement initiatives, especially John Hensing. We also thank Michael Furukawa of the Agency for Healthcare Research and Quality, formerly of the Office of the National Coordinator for Health Information Technology, who played a key role in the conceptualization of this study and data collection.
Corresponding author: Emily Jones, PhD, MPP, National Institutes of Health, 6001 Executive Blvd., #5232 Rockville, MD 20852, [email protected].
Financial disclosures: None
1. Lehnert B, Bree R. Analysis of appropriateness of outpatient CT and MRI referred from primary care clinics at an academic medical center: how critical is the need for improved decision support? J Am Coll Radiol 2010;7:192–7.
2. Ip I, Schneider L, Hanson R, et al. Adoption and meaningful use of computed physician order entry with an integrated clinical decision support system for radiology: ten-year analysis in an urban teaching hospital. J Am Coll Radiol 2012;9:129–36.
3. Bernardy M, Ullrich C, Rawson J, et al. Strategies for managing imaging utilization. J Am Coll Radiol 2009;6:844–50.
4. Amland R, Dean B, Yu HT et al. Computed clinical decision support to prevent venous thromboembolism among hospitalized patients: proximal outcomes from a multiyear quality improvement project. J Healthcare Qual 2015;37:221–31.
5. Kahn C. Improving outcomes in radiology: bringing computer-based decision support and education to the point of care. Acad Radiology 2005;12:409–14.
6. Phansalkar S, Desai A, Bell D et al. High-priority drug-drug interactions for use in electronic health records. J Am Med Inform Assoc 2012;19:735–43.
7. Wolfstadt J, Gurwitz J, Field T, et al. The effect of computed physician order entry with clinical decision support on the rates of adverse drug events: a systematic review. J Gen Intern Med 2008;23:451–8.
8. Amland R, Hahn-Cover K. Clinical decision support for early recognition of sepsis. Am J Med Qual 2014;1–8.
9. Amland R, Haley J, Lyons J. A multidisciplinary sepsis program enabled by a two-stage clinical decision support system: factors that influence patient outcomes. Am J Med Qual 2015;1–8.
10. Umscheid C, Hanish A, Chittams J, et al. Effectiveness of a novel and scalable clinical decision support intervention to improve venous thromboembolism prophylaxis: a quasi-experimental study. BMC Med Inform Dec Making 2012;12:92–104.
11. Mack EH, Wheeler DS, Embi PJ. Clinical decision support systems in the pediatric intensive care unit. Pediatric Crit Care Med 2009;10:23–8.
12. Kollef M, Heard K, Chen Y, et al. Mortality and length of stay trends following implementation of a rapid response system and real-time automated clinical deterioration alerts. Am J Med Qual 2015; online first.
13. Ali S, Giordano R, Lakhani S, Walker D. A review of randomized controlled trials of medical record powered clinical decision support system to improve quality of diabetes care. Int J Med Informatics 2016;87:91–100.
14. Gill J, Mainous A, Koopman R et al. Impact of EHR-based clinical decision support on adherence to guidelines for patients on NSAIDs: a randomized controlled trial. Ann Fam Med 2011;9:22–30.
15. Choosing Wisely. Accessed 1 May 2017 at http://www.choosingwisely.org/clinician-lists/#keyword=appendicitis.
16. Hendrickson M, Wey A, Gaillard P, Kharbanda A. Implementation of an electronic clinical decision support tool for pediatric appendicitis within a hospital network. Pediatric Emerg Care 2017 (online first).
17. Kharbanda A, Madhok M, Krause E, et al. Implementation of electronic clinical decision support for pediatric appendicitis. Pediatrics 2016;137:e20151745.
18. Schuh S, Chan K, Langer J, et al. Properties of serial ultrasound clinical diagnostic pathway in suspected appendicitis and related computed tomography use. Acad Emerg Med 2015;22:406–14.
19. Ramarajan N, Krishnamoorthi R, Barth R, et al. An interdisciplinary initiative to reduce radiation exposure: evaluation of appendicitis in a pediatric emergency department with clinical assessment supported by a staged ultrasound and computed tomography pathway. Acad Emerg Med 2009;16:1258–65.
20. HIMSS Analytics. Stage 7 Hospitals. Accessed at www.himssanalytics.org/emram/stage7Hospitals.aspx.
21. Rizer M, et al. Top 10 lessons learned from electronic health record implementation in a large academic medical center. Perspectives in Health Information Management. Summer 2015.
22. Cosgrove DM, Fisher M, Gabow P, et al. Ten strategies to lower costs, improve quality, and engage patients: the view from leading health system CEOs. Health Aff (Millwood) 2013;32:321–7.
23. Cresswell KM, Bates DW, Sheikh A. Ten key considerations for the successful implementation and adoption of large-scale health information technology. J Am Med Inform Assoc 2013 Apr 18.
24. Hensing JA. The quest for upper-quartile performance at Banner Health. J Healthc Qual 2008;30:18–24
25. Hensing J, Dahlen D, Warden M, et al. Measuring the benefits of IT-enabled care transformation. Healthc Financ Manage 2008;62:74–80.
26. Truven Health Analytics. 15 Top Health Systems Study. 6th ed. April 2014. Accessed at http://100tophospitals.com/portals/2/assets/15-Top-Health-Systems-Study.pdf.
27. Aiello M. 2011 Top leadership team awards recognize big moves. Health Leaders Media. August 2011. Accessed at www.healthleadersmedia.com/page-2/LED-269808/2011-Top-Leadership-Team-Awards-Recognize-Big-Moves.
 28. Rosenthal D, Stout M. Radiology order entry: features and performance requirements. J Am Coll Radiol 2006;3:554–7.
28. Rosenthal D, Stout M. Radiology order entry: features and performance requirements. J Am Coll Radiol 2006;3:554–7.
29. Kirsh S, Wu WC, Edelman D, Aron D. Research versus quality improvement: distinct or a distinction without a difference? A case study comparison of two studies. Jt Comm J Qual Patient Safety 2014;40:365–75.
30. Ogrinc G, Davies L, Goodman D, et al. SQUIRE 2.0 (Standards for Quality Improvement Reporting Excellence): revised publication guidelines from a detailed consensus process. BMJ Qual Saf 2015;0:1–7.
31. Blavin F, Ramos C, Shah A, Devers K. Lessons from the literature on electronic health record implementation.1 Aug 2013. The Urban Institute. Prepared for the Office of the National Coordinator for Health Information Technology. Available at www.urban.org/research/publication/lessons-literature-electronic-health-record-implementation.
32. Needleman J, Pearson ML, Upenieks VV, et al. Engaging frontline staff in performance improvement: the American Organization of Nurse Executives implementation of transforming care at the beside collaborative. Jt Comm J Qual Patient Safety 2016;42:61–9.
33. Jones E, Swain M, Patel V, Furukawa M. Supporting HITECH implementation and assessing lessons for the future: the role of program evaluation. Healthcare: The Journal of Delivery Science and Innovation 2014;2:4–8.
34. Phansalkar S, Zachariah M, Seidling H, et al. Evaluation of medication alerts in electronic health records for compliance with human factors principles. J Am Med Inform Assoc 2014;21:e332–e340.
35. Handler S, Altman R, Perera S, et al. A systematic review of the performance characteristics of clinical event monitor signals to detect adverse drug events in the hospital setting. J Am Med Inform Assoc 2007;14:451–8.
36. Singer S, Rivard P, Hayes J, et al. Improving patient care through leadership engagement with frontline staff: a Department of Veteran Affairs study. Jt Comm J Qual Patient Safety 2013;39):349–60.
37. Brewster A, Curry L, Cherlin E, et al. Integrating new practices: a qualitative study of how hospital innovations become routine. Implement Sci 2015;5(10):168.
From the Office of Science Policy and Communications, National Institute on Drug Abuse, National Institutes of Health, Rockville, MD, and George Washington University, Washington, DC (Dr. Jones), Office of the National Coordinator for Health Information Technology, US Department of Health and Human Services, Washington, DC (Mr. Swain), and Banner Health, Phoenix, AZ (Ms. Burdick).
Abstract
- Objective: Clinical decision support (CDS) can be a useful tool to decrease inappropriate imaging by providing evidence-based information to clinicians at the point of care. The objective of this case study is to highlight lessons from a health care improvement initiative using CDS to encourage use of ultrasound rather than computed tomography (CT) scans as an initial diagnostic tool for suspected appendicitis in pediatric patients.
- Methods: The percentage of suspected pediatric appendicitis cases receiving ultrasounds and CT scans was calculated using electronic health record data. Four steps for implementing health information technology were identified in a literature scan that guided data collection and analysis: planning, software customization and workflow design, training and user support, and optimization.
- Results: During the fourth quarter of 2010, 1 in 7 pediatric patients with suspected appendicitis received an ultrasound and almost half received a CT scan. By the first quarter of 2012, ultrasounds were performed in 40.8% of these cases and the use of CT scans declined to 39.9% of suspected pediatric appendicitis cases.
- Conclusion: Four lessons emerged. First, all levels of staff should be involved in the planning process to make organizational priorities actionable and build buy-in for each healthcare improvement initiative. Second, it takes time to design and test the alert to ensure that clinical guidelines are being properly applied. Third, re-engineering the workflow is critical for usability; in this case, ensuring the availability of ultrasound staff was particularly important. Finally, the effectiveness of CDS depends on applying relevant evidence-based practice guidelines to real-time patient data.
Diagnostic imaging is a useful tool for identifying and guiding the treatment of many health conditions, but evidence indicates that health care providers do not always use imaging appropriately. In fact, a substantial proportion of diagnostic imaging procedures performed in hospital and ambulatory settings are not supported by clinical guideline recommendations [1,2]. Spending on diagnostic imaging is rapidly increasing, and some patients receive unnecessary radiation exposure that can lead to adverse health impacts [3]. Inappropriate imaging falls into 3 broad categories: imaging that does not conform to clinical guidelines, imaging that is contraindicated due to an allergy or implantable medical device, and imaging that might be clinically indicated but is duplicative of prior imaging services.
Clinical decision support (CDS) functionality supports health care improvement initiatives to narrow the gap between evidence-based practices and routine care [4]. CDS merges patient-specific clinical information with relevant information about evidence-based practices, providing health care providers with timely information to guide decisions at the point of care [5]. Decision support is most commonly delivered in the form of alerts and reminders [6]. CDS can be effective in reducing adverse drug events [7], sepsis [8,9], and other conditions in hospital [10–12] and ambulatory settings [13,14].
For the evaluation of suspected appendicitis in children, ultrasound is the preferred initial consideration for imaging examination [15]. Evidence suggests that CDS can increase the use of ultrasound for suspected pediatric appendicitis [16,17] and has affirmed the utility of ultrasound as a first-line diagnostic tool for suspected appendicitis [18,19]. In the Choosing Wisely campaign, the American College of Surgeons and the American College of Radiology have both endorsed ultrasound as an option to consider prior to conducting a CT scan to evaluate suspected appendicitis in children [15].
Banner Health, a large health system headquartered in Phoenix, Arizona, implemented a health care improvement initiative using CDS functionality to encourage providers to use ultrasound instead of CT as a first-line diagnostic tool for suspected pediatric appendicitis. We conducted a site visit to Banner Health, an organization who had had attained a high score on the EMR Adoption Model [20] to examine their implementation process. We sought to build on previous research examining the use of health information technology to improve performance in large health systems [21–23].
Methods
Setting
Banner Health is a large not-for-profit health system that is comprised of 24 acute care hospitals across several states, as well as ambulatory medical practices, behavioral health, home care, and ambulatory surgery centers [24,25]. The health system is the largest employer in Arizona and one of the largest in the United States with over 50,000 employees. Banner Health has been nationally recognized for clinical quality [26], an innovative leadership team [27], and using health IT to improve quality [20]. The health system was also selected as one of the Centers for Medicare & Medicaid Services (CMS) Pioneer Accountable Care Organizations.
Site Visit
The first 2 authors conducted a 2-day site visit to the Banner Health headquarters in Phoenix, Arizona in November 2013. The team conducted discussions with over 20 individuals, including health system leadership, frontline clinicians in several units of an acute care hospital, staff members in 2 telehealth hubs—including a tele-ICU hub—and trainers in a simulation facility that is used for staff training. The discussions were conducted with groups of staff or on an individual basis, as appropriate. At the outset of the project, an environmental scan of relevant grey and peer-reviewed literature was conducted under contract on behalf of the authors to guide data collection and analysis [28]. An interview protocol was created to guide the discussions. The protocol contained modules that were used during each discussion, if relevant. The modules addressed topics such as technical issues with designing and deploying health information technology functionalities such as clinical decision support systems, the organizational processes and structures needed to launch health care improvement initiatives, and using health information technology care coordination. Within each module, questions probed about the challenges that arose and the solutions to these challenges, with a focus on the four phases of implementing a health information technology intervention: functionality planning, software customization and workflow design, training and user support, and optimization. To assist with interpreting the qualitative findings, an evolving outline of the findings was maintained. Salient themes and conceptual categories were tracked, which helped the researchers organize, synthesize, and interpret the information collected during the site visit. Once the authors chose to focus on clinical decision support, summary notes from the discussions were reviewed for relevant information, and this information was compiled and organized under the rubric of the four implementation phases. The findings and key themes from the discussion notes were distilled into key lessons for the field.
Data obtained included the percentage of pediatric patients with suspected appendicitis who received ultrasounds and CT scans each month from 1 October 2010 through 31 March 2012. Banner Health staff originally collected the data to support the implementation of health care improvement initiative; the use of these data in this paper is a secondary use [29].
This manuscript was prepared using the SQUIRE 2.0 guidelines [30]. No patient-identifiable data were used, so institutional review board approval was not sought.
Results
The 4 steps of implementing CDS can be described as functionality planning, software customization and workflow design, training and user support, and optimization [31].
Pre-Implementation
The use of computerized provider order entry (CPOE) is a precursor to using clinical decision support, since orders must be entered electronically to be subject to CDS review. Banner Health deployed CPOE to its various facilities starting in 2008. The deployment was staged in a rolling fashion with one or two facilities going live every few months so that the deployment team was available at each facility.
Phase 1: Planning
In contrast to many large health systems, the organization has a single board of directors that oversees the entire system of over 37,000 employees. Activities and relationships to promote the use of evidence-based practices are built into the organizational structure. For example, Banner Health maintains a Care Management Council, a group comprised of clinical and administrative leadership to provide executive oversight of health care improvement projects. The Council convenes on a quarterly basis to review and approve the adoption of new clinical practice guidelines, policies, and standardized standing orders that have been developed by multidisciplinary groups of physicians and other clinicians. A key focus of the Council is ensuring consistent application of evidence-based guidelines to clinical care and disseminating knowledge of clinical best practices across a large and complex enterprise.
Interdisciplinary clinical consensus groups support the Council’s work. These groups are comprised of administrative and program management staff, physicians and other clinicians, and engineers. Each clinical consensus group focuses on emerging issues and improvement opportunities within a specific clinical domain and leads the implementation of health care improvement initiatives in that domain. Providers and staff at all levels of the organization were involved in planning and implementing the health care improvement initiative in inappropriate imaging. This increased buy-in and staff support, which are associated with successful health care improvement initiatives [32]. Banner Health staff rallied around the idea of addressing inappropriate imaging as a key priority initiative. The teams that implement each initiative include an engineer that focuses on redesigning clinical workflows for each initiative. There is also an organizational unit responsible for project management that provides teams with logistical and operational support.
Phase 2: Software Customization and Workflow Redesign
Once the clinical consensus group selected inappropriate imaging as a priority, the next step was to examine the process flow for imaging ordering. In 2011 Banner Health integrated CDS functionality with CPOE into the electronic health record. Before the use of CDS, inpatient and emergency department imaging orders were simply transmitted to imaging staff after the order was entered. After CDS implementation, the process flow begins with an inpatient imaging order and entailed checking the order against clinical guidelines on the proper use of imaging. If the image order did not conform to guidelines, which in this case indicate that ultrasound should be used before CT scans as a diagnostic tool for suspected pediatric appendicitis, the CDS system triggered an alert [15].
Bringing the perspective and skill sets of engineers to the process of redesigning clinical workflows was particularly valuable [33]. While CDS has the potential to reduce inpatient inappropriate imaging, effectiveness depends on adjusting workflows to ensure that the information provided by CDS alerts and reminders is actionable. To reduce alert fatigue among the clinical staff, the team identified the appropriate level of workflow interruption for each alert and reminder (hard stop, workflow interruption, or informational) [5,6].
The design principles that were used to design the alert include intuitive system development to promote ease of use, one set of screen formats and data definitions, and a set of consistent core reports and standard system output formats across facilities. The alert’s appearance was tailored for maximal impact and covered most of the screen. Color contrast was used, but since some people are color-blind, the meaning of the alert did not depend on the color contrast. The alerts included recommendations for changing the treatment plan to encourage using ultrasound as a first-line diagnostic tool. Minimizing the number of clicks to accept the proposed treatment plan change in the alert is desirable.
Phase 3: Training and User Support
Training and support structures and tools were critical to the rollout of the inappropriate imaging alerts. Providers were reminded about clinical best practices and informed during staff meetings about the new CDS rules. In addition, various types of training and support were available to clinicians and staff during the rollout process. Dedicated time for end-user training provided an opportunity to identify and cultivate super-users. These super-users not only helped provide technical support to their colleagues, but also helped create excitement for the initiative. A centralized support desk provided telephone support for providers in facilities throughout the Banner Health system. Site managers were provided toolkits to support providers and staff throughout the implementation process. The toolkits included frequently asked questions and answers, and were maintained as ‘living documents’ that were updated based on emerging issues and questions.
To keep things on track, project managers from the central project management department were involved in the initiative to provide direct project management services to the initiative. They also worked to instill project management competencies throughout the organization, applying a train-the-trainer approach to disseminate best practices for enhancing communication among team members, implementing workflow changes, and monitoring the results.
Phase 4: Optimization
The optimization phase is continuous and continues to the present day. Notably, the success of the CDS rules depends on the availability of current clinical information for each patient, in addition to information about the treatment plan. For this initiative, Banner Health maintained aggregated clinical patient data in the data warehouse that aggregated data from disparate sources, including billing and EHR data from different care settings such as ambulatory offices, inpatient units, the emergency department, home care, and ambulatory surgery centers. The data warehouse is housed in a strategically chosen physical location to minimize the threat of natural disasters, and cloud-based backup is also used. A master patient index and provider credentialing system are integrated with the data warehouse. Query-based health information exchange is used, when possible, to collect information on care received by patients outside of the Banner Health system.
It is important to note that many CDS alerts are over-ridden without changes to clinical care [34]. Previous research indicates that alert fatigue from “false positives” can impede the effectiveness of alerts [35]. Banner Health monitors the rate at which CDS alerts are over-ridden. Figure 1 shows the percentage of all alerts for radiation exposure—including the alert related to using ultrasound as a diagnostic tool for pediatric appendicitis—that led to order cancellations. The percentage of CT orders that generated the alert and were cancelled fell from 18.9% in March 2011 to 13.6% in February 2012. The rate of order cancellations might have declined over time due to a change in provider behavior from the alert. That is, if inappropriate CT scan orders declined over time, then providers would cancel a decreasing percentage of the CT scan orders that prompted an alert.
Imaging Use
In Figure 2, data on the use of the 2 imaging procedures for the diagnosis of pediatric appendicitis is presented. During the fourth quarter of 2010, almost half of pediatric patients with suspected appendicitis received a CT scan and only about 1 in 7 received an ultrasound. After the clinical decision support alert was put in place to remind providers to perform an ultrasound as a first-line diagnostic tool, the use of ultrasounds increased sharply. By the first quarter of 2012, ultrasounds were performed in 40.8% of these cases and the use of CT scans declined to 39.9% of suspected pediatric appendicitis cases.
Discussion
This case study discusses the application of CDS functionality in a health care improvement initiative to address inappropriate imaging in a large health system. Four main implementation lessons emerge for the field. First, it is important to involve all levels of staff in the planning process to ensure that health care improvement activities are prioritized correctly and to build buy-in for the priorities addressed with health care improvement activities. Second, it is necessary to allow time to design the alert or reminder, as well as testing it during the implementation process to ensure that clinical guidelines are being properly applied. Third, re-engineering the workflow and ensuring usability of the alert or reminder are important, and using the skills of trained engineers helps in this process. Ensuring the availability of trained ultrasound staff was particularly important to this initiative. Finally, the effectiveness of CDS depends on having complete data for each patient, as well as up-to-date information on the relevant evidence-based practice guidelines.
These results can help guide the implementation of health care improvement initiatives that use CDS functionality to address inappropriate imaging. The adoption of electronic health records with CDS functionality was incentivized and supported by the Medicare and Medicaid Electronic Health Record Incentive Programs; the Medicare program now exists as part of MACRA. Using CDS to reduce inappropriate imaging is required for Medicare fee-for-service patients in the 2014 Protecting Access to Medicare Act (PAMA), highlighting the critical nature of these results, which can guide implementation of CDS to reduce inappropriate imaging [41].
As noted above, the optimization phase is continuous. Banner Health still encourages use of ultrasounds as a first-line diagnostic tool for pediatric appendicitis. Identifying which patients should immediately receive CT scans is difficult, and sometimes the decision depends on the availability of staff to conduct the ultrasound scans. Ways to maximize the productivity of ultrasound technicians have been explored. Another focus area since the original implementation of this health care improvement initiative has been health information exchange, to ensure that complete, up-to-date information is available for each patient.
Banner Health often implements CDS in conjunction with other health IT functionalities. For example, CDS and telehealth are used together to improve care in the intensive care unit (ICU) for patients with sepsis and delirium. An offsite hub of experienced ICU physicians and nurses remotely monitors ICU patients in facilities across Banner Health, using cameras with zoom capability. The intensive care specialists in the tele-hub act as part of the care team; in addition to receiving video feed, they communicate verbally with patients and ICU staff members. Predictive analytics are used to generate clinical decision support alerts and reminders, with a special focus on early intervention if a patient’s clinical indicators are trending downward. The 4 lessons described in this study were also used in the ICU sepsis and delirium initiative; staff were involved in the planning process, alerts and reminders were thoroughly tested, the workflow was adjusted to accommodate the physicians in the tele-ICU hub, and up-to-date and complete clinical information for each patient is maintained. In addition, the design principles for alerts described in this study, such as covering most of the screen and providing recommendations for changing the treatment plan within the alert itself, were also used in the ICU sepsis and delirium initiative.
One limitation of this study is that it was conducted at a single health system. Thus, the findings might not be generalizable to other health systems, particularly if a robust health IT infrastructure is not in place. The culture of Banner Health values quality and involved providers and staff at all levels in selecting and implementing health care improvement initiatives. In addition, engineers assisted with implementation. Finally, the study design does not permit conclusions about the causality of the decline in CT scans and the increase in ultrasounds for suspected pediatric appendicitis cases; unobserved factors might have contributed to the changes in CT and ultrasound use.
Future research should focus on ways to improve the implementation and organization learning process, particularly through engagement of frontline staff by leadership [36] and explore how to operationalize previous findings indicating that innovations in hospital settings are more likely to be sustained when intrinsically rewarding to staff, either by making clinician and staff jobs easier to perform or more gratifying [37]. Future research should focus on facilitating health information exchange between providers in different health systems.
Disclaimer: The views expressed in the article are solely the views of the authors and do not represent those of the National Institutes of Health or the U.S. Government.
Acknowledgments: The authors wish to thank the Banner Health team for taking time to share their insights on how health information technology can be used for health care improvement initiatives, especially John Hensing. We also thank Michael Furukawa of the Agency for Healthcare Research and Quality, formerly of the Office of the National Coordinator for Health Information Technology, who played a key role in the conceptualization of this study and data collection.
Corresponding author: Emily Jones, PhD, MPP, National Institutes of Health, 6001 Executive Blvd., #5232 Rockville, MD 20852, [email protected].
Financial disclosures: None
From the Office of Science Policy and Communications, National Institute on Drug Abuse, National Institutes of Health, Rockville, MD, and George Washington University, Washington, DC (Dr. Jones), Office of the National Coordinator for Health Information Technology, US Department of Health and Human Services, Washington, DC (Mr. Swain), and Banner Health, Phoenix, AZ (Ms. Burdick).
Abstract
- Objective: Clinical decision support (CDS) can be a useful tool to decrease inappropriate imaging by providing evidence-based information to clinicians at the point of care. The objective of this case study is to highlight lessons from a health care improvement initiative using CDS to encourage use of ultrasound rather than computed tomography (CT) scans as an initial diagnostic tool for suspected appendicitis in pediatric patients.
- Methods: The percentage of suspected pediatric appendicitis cases receiving ultrasounds and CT scans was calculated using electronic health record data. Four steps for implementing health information technology were identified in a literature scan that guided data collection and analysis: planning, software customization and workflow design, training and user support, and optimization.
- Results: During the fourth quarter of 2010, 1 in 7 pediatric patients with suspected appendicitis received an ultrasound and almost half received a CT scan. By the first quarter of 2012, ultrasounds were performed in 40.8% of these cases and the use of CT scans declined to 39.9% of suspected pediatric appendicitis cases.
- Conclusion: Four lessons emerged. First, all levels of staff should be involved in the planning process to make organizational priorities actionable and build buy-in for each healthcare improvement initiative. Second, it takes time to design and test the alert to ensure that clinical guidelines are being properly applied. Third, re-engineering the workflow is critical for usability; in this case, ensuring the availability of ultrasound staff was particularly important. Finally, the effectiveness of CDS depends on applying relevant evidence-based practice guidelines to real-time patient data.
Diagnostic imaging is a useful tool for identifying and guiding the treatment of many health conditions, but evidence indicates that health care providers do not always use imaging appropriately. In fact, a substantial proportion of diagnostic imaging procedures performed in hospital and ambulatory settings are not supported by clinical guideline recommendations [1,2]. Spending on diagnostic imaging is rapidly increasing, and some patients receive unnecessary radiation exposure that can lead to adverse health impacts [3]. Inappropriate imaging falls into 3 broad categories: imaging that does not conform to clinical guidelines, imaging that is contraindicated due to an allergy or implantable medical device, and imaging that might be clinically indicated but is duplicative of prior imaging services.
Clinical decision support (CDS) functionality supports health care improvement initiatives to narrow the gap between evidence-based practices and routine care [4]. CDS merges patient-specific clinical information with relevant information about evidence-based practices, providing health care providers with timely information to guide decisions at the point of care [5]. Decision support is most commonly delivered in the form of alerts and reminders [6]. CDS can be effective in reducing adverse drug events [7], sepsis [8,9], and other conditions in hospital [10–12] and ambulatory settings [13,14].
For the evaluation of suspected appendicitis in children, ultrasound is the preferred initial consideration for imaging examination [15]. Evidence suggests that CDS can increase the use of ultrasound for suspected pediatric appendicitis [16,17] and has affirmed the utility of ultrasound as a first-line diagnostic tool for suspected appendicitis [18,19]. In the Choosing Wisely campaign, the American College of Surgeons and the American College of Radiology have both endorsed ultrasound as an option to consider prior to conducting a CT scan to evaluate suspected appendicitis in children [15].
Banner Health, a large health system headquartered in Phoenix, Arizona, implemented a health care improvement initiative using CDS functionality to encourage providers to use ultrasound instead of CT as a first-line diagnostic tool for suspected pediatric appendicitis. We conducted a site visit to Banner Health, an organization who had had attained a high score on the EMR Adoption Model [20] to examine their implementation process. We sought to build on previous research examining the use of health information technology to improve performance in large health systems [21–23].
Methods
Setting
Banner Health is a large not-for-profit health system that is comprised of 24 acute care hospitals across several states, as well as ambulatory medical practices, behavioral health, home care, and ambulatory surgery centers [24,25]. The health system is the largest employer in Arizona and one of the largest in the United States with over 50,000 employees. Banner Health has been nationally recognized for clinical quality [26], an innovative leadership team [27], and using health IT to improve quality [20]. The health system was also selected as one of the Centers for Medicare & Medicaid Services (CMS) Pioneer Accountable Care Organizations.
Site Visit
The first 2 authors conducted a 2-day site visit to the Banner Health headquarters in Phoenix, Arizona in November 2013. The team conducted discussions with over 20 individuals, including health system leadership, frontline clinicians in several units of an acute care hospital, staff members in 2 telehealth hubs—including a tele-ICU hub—and trainers in a simulation facility that is used for staff training. The discussions were conducted with groups of staff or on an individual basis, as appropriate. At the outset of the project, an environmental scan of relevant grey and peer-reviewed literature was conducted under contract on behalf of the authors to guide data collection and analysis [28]. An interview protocol was created to guide the discussions. The protocol contained modules that were used during each discussion, if relevant. The modules addressed topics such as technical issues with designing and deploying health information technology functionalities such as clinical decision support systems, the organizational processes and structures needed to launch health care improvement initiatives, and using health information technology care coordination. Within each module, questions probed about the challenges that arose and the solutions to these challenges, with a focus on the four phases of implementing a health information technology intervention: functionality planning, software customization and workflow design, training and user support, and optimization. To assist with interpreting the qualitative findings, an evolving outline of the findings was maintained. Salient themes and conceptual categories were tracked, which helped the researchers organize, synthesize, and interpret the information collected during the site visit. Once the authors chose to focus on clinical decision support, summary notes from the discussions were reviewed for relevant information, and this information was compiled and organized under the rubric of the four implementation phases. The findings and key themes from the discussion notes were distilled into key lessons for the field.
Data obtained included the percentage of pediatric patients with suspected appendicitis who received ultrasounds and CT scans each month from 1 October 2010 through 31 March 2012. Banner Health staff originally collected the data to support the implementation of health care improvement initiative; the use of these data in this paper is a secondary use [29].
This manuscript was prepared using the SQUIRE 2.0 guidelines [30]. No patient-identifiable data were used, so institutional review board approval was not sought.
Results
The 4 steps of implementing CDS can be described as functionality planning, software customization and workflow design, training and user support, and optimization [31].
Pre-Implementation
The use of computerized provider order entry (CPOE) is a precursor to using clinical decision support, since orders must be entered electronically to be subject to CDS review. Banner Health deployed CPOE to its various facilities starting in 2008. The deployment was staged in a rolling fashion with one or two facilities going live every few months so that the deployment team was available at each facility.
Phase 1: Planning
In contrast to many large health systems, the organization has a single board of directors that oversees the entire system of over 37,000 employees. Activities and relationships to promote the use of evidence-based practices are built into the organizational structure. For example, Banner Health maintains a Care Management Council, a group comprised of clinical and administrative leadership to provide executive oversight of health care improvement projects. The Council convenes on a quarterly basis to review and approve the adoption of new clinical practice guidelines, policies, and standardized standing orders that have been developed by multidisciplinary groups of physicians and other clinicians. A key focus of the Council is ensuring consistent application of evidence-based guidelines to clinical care and disseminating knowledge of clinical best practices across a large and complex enterprise.
Interdisciplinary clinical consensus groups support the Council’s work. These groups are comprised of administrative and program management staff, physicians and other clinicians, and engineers. Each clinical consensus group focuses on emerging issues and improvement opportunities within a specific clinical domain and leads the implementation of health care improvement initiatives in that domain. Providers and staff at all levels of the organization were involved in planning and implementing the health care improvement initiative in inappropriate imaging. This increased buy-in and staff support, which are associated with successful health care improvement initiatives [32]. Banner Health staff rallied around the idea of addressing inappropriate imaging as a key priority initiative. The teams that implement each initiative include an engineer that focuses on redesigning clinical workflows for each initiative. There is also an organizational unit responsible for project management that provides teams with logistical and operational support.
Phase 2: Software Customization and Workflow Redesign
Once the clinical consensus group selected inappropriate imaging as a priority, the next step was to examine the process flow for imaging ordering. In 2011 Banner Health integrated CDS functionality with CPOE into the electronic health record. Before the use of CDS, inpatient and emergency department imaging orders were simply transmitted to imaging staff after the order was entered. After CDS implementation, the process flow begins with an inpatient imaging order and entailed checking the order against clinical guidelines on the proper use of imaging. If the image order did not conform to guidelines, which in this case indicate that ultrasound should be used before CT scans as a diagnostic tool for suspected pediatric appendicitis, the CDS system triggered an alert [15].
Bringing the perspective and skill sets of engineers to the process of redesigning clinical workflows was particularly valuable [33]. While CDS has the potential to reduce inpatient inappropriate imaging, effectiveness depends on adjusting workflows to ensure that the information provided by CDS alerts and reminders is actionable. To reduce alert fatigue among the clinical staff, the team identified the appropriate level of workflow interruption for each alert and reminder (hard stop, workflow interruption, or informational) [5,6].
The design principles that were used to design the alert include intuitive system development to promote ease of use, one set of screen formats and data definitions, and a set of consistent core reports and standard system output formats across facilities. The alert’s appearance was tailored for maximal impact and covered most of the screen. Color contrast was used, but since some people are color-blind, the meaning of the alert did not depend on the color contrast. The alerts included recommendations for changing the treatment plan to encourage using ultrasound as a first-line diagnostic tool. Minimizing the number of clicks to accept the proposed treatment plan change in the alert is desirable.
Phase 3: Training and User Support
Training and support structures and tools were critical to the rollout of the inappropriate imaging alerts. Providers were reminded about clinical best practices and informed during staff meetings about the new CDS rules. In addition, various types of training and support were available to clinicians and staff during the rollout process. Dedicated time for end-user training provided an opportunity to identify and cultivate super-users. These super-users not only helped provide technical support to their colleagues, but also helped create excitement for the initiative. A centralized support desk provided telephone support for providers in facilities throughout the Banner Health system. Site managers were provided toolkits to support providers and staff throughout the implementation process. The toolkits included frequently asked questions and answers, and were maintained as ‘living documents’ that were updated based on emerging issues and questions.
To keep things on track, project managers from the central project management department were involved in the initiative to provide direct project management services to the initiative. They also worked to instill project management competencies throughout the organization, applying a train-the-trainer approach to disseminate best practices for enhancing communication among team members, implementing workflow changes, and monitoring the results.
Phase 4: Optimization
The optimization phase is continuous and continues to the present day. Notably, the success of the CDS rules depends on the availability of current clinical information for each patient, in addition to information about the treatment plan. For this initiative, Banner Health maintained aggregated clinical patient data in the data warehouse that aggregated data from disparate sources, including billing and EHR data from different care settings such as ambulatory offices, inpatient units, the emergency department, home care, and ambulatory surgery centers. The data warehouse is housed in a strategically chosen physical location to minimize the threat of natural disasters, and cloud-based backup is also used. A master patient index and provider credentialing system are integrated with the data warehouse. Query-based health information exchange is used, when possible, to collect information on care received by patients outside of the Banner Health system.
It is important to note that many CDS alerts are over-ridden without changes to clinical care [34]. Previous research indicates that alert fatigue from “false positives” can impede the effectiveness of alerts [35]. Banner Health monitors the rate at which CDS alerts are over-ridden. Figure 1 shows the percentage of all alerts for radiation exposure—including the alert related to using ultrasound as a diagnostic tool for pediatric appendicitis—that led to order cancellations. The percentage of CT orders that generated the alert and were cancelled fell from 18.9% in March 2011 to 13.6% in February 2012. The rate of order cancellations might have declined over time due to a change in provider behavior from the alert. That is, if inappropriate CT scan orders declined over time, then providers would cancel a decreasing percentage of the CT scan orders that prompted an alert.
Imaging Use
In Figure 2, data on the use of the 2 imaging procedures for the diagnosis of pediatric appendicitis is presented. During the fourth quarter of 2010, almost half of pediatric patients with suspected appendicitis received a CT scan and only about 1 in 7 received an ultrasound. After the clinical decision support alert was put in place to remind providers to perform an ultrasound as a first-line diagnostic tool, the use of ultrasounds increased sharply. By the first quarter of 2012, ultrasounds were performed in 40.8% of these cases and the use of CT scans declined to 39.9% of suspected pediatric appendicitis cases.
Discussion
This case study discusses the application of CDS functionality in a health care improvement initiative to address inappropriate imaging in a large health system. Four main implementation lessons emerge for the field. First, it is important to involve all levels of staff in the planning process to ensure that health care improvement activities are prioritized correctly and to build buy-in for the priorities addressed with health care improvement activities. Second, it is necessary to allow time to design the alert or reminder, as well as testing it during the implementation process to ensure that clinical guidelines are being properly applied. Third, re-engineering the workflow and ensuring usability of the alert or reminder are important, and using the skills of trained engineers helps in this process. Ensuring the availability of trained ultrasound staff was particularly important to this initiative. Finally, the effectiveness of CDS depends on having complete data for each patient, as well as up-to-date information on the relevant evidence-based practice guidelines.
These results can help guide the implementation of health care improvement initiatives that use CDS functionality to address inappropriate imaging. The adoption of electronic health records with CDS functionality was incentivized and supported by the Medicare and Medicaid Electronic Health Record Incentive Programs; the Medicare program now exists as part of MACRA. Using CDS to reduce inappropriate imaging is required for Medicare fee-for-service patients in the 2014 Protecting Access to Medicare Act (PAMA), highlighting the critical nature of these results, which can guide implementation of CDS to reduce inappropriate imaging [41].
As noted above, the optimization phase is continuous. Banner Health still encourages use of ultrasounds as a first-line diagnostic tool for pediatric appendicitis. Identifying which patients should immediately receive CT scans is difficult, and sometimes the decision depends on the availability of staff to conduct the ultrasound scans. Ways to maximize the productivity of ultrasound technicians have been explored. Another focus area since the original implementation of this health care improvement initiative has been health information exchange, to ensure that complete, up-to-date information is available for each patient.
Banner Health often implements CDS in conjunction with other health IT functionalities. For example, CDS and telehealth are used together to improve care in the intensive care unit (ICU) for patients with sepsis and delirium. An offsite hub of experienced ICU physicians and nurses remotely monitors ICU patients in facilities across Banner Health, using cameras with zoom capability. The intensive care specialists in the tele-hub act as part of the care team; in addition to receiving video feed, they communicate verbally with patients and ICU staff members. Predictive analytics are used to generate clinical decision support alerts and reminders, with a special focus on early intervention if a patient’s clinical indicators are trending downward. The 4 lessons described in this study were also used in the ICU sepsis and delirium initiative; staff were involved in the planning process, alerts and reminders were thoroughly tested, the workflow was adjusted to accommodate the physicians in the tele-ICU hub, and up-to-date and complete clinical information for each patient is maintained. In addition, the design principles for alerts described in this study, such as covering most of the screen and providing recommendations for changing the treatment plan within the alert itself, were also used in the ICU sepsis and delirium initiative.
One limitation of this study is that it was conducted at a single health system. Thus, the findings might not be generalizable to other health systems, particularly if a robust health IT infrastructure is not in place. The culture of Banner Health values quality and involved providers and staff at all levels in selecting and implementing health care improvement initiatives. In addition, engineers assisted with implementation. Finally, the study design does not permit conclusions about the causality of the decline in CT scans and the increase in ultrasounds for suspected pediatric appendicitis cases; unobserved factors might have contributed to the changes in CT and ultrasound use.
Future research should focus on ways to improve the implementation and organization learning process, particularly through engagement of frontline staff by leadership [36] and explore how to operationalize previous findings indicating that innovations in hospital settings are more likely to be sustained when intrinsically rewarding to staff, either by making clinician and staff jobs easier to perform or more gratifying [37]. Future research should focus on facilitating health information exchange between providers in different health systems.
Disclaimer: The views expressed in the article are solely the views of the authors and do not represent those of the National Institutes of Health or the U.S. Government.
Acknowledgments: The authors wish to thank the Banner Health team for taking time to share their insights on how health information technology can be used for health care improvement initiatives, especially John Hensing. We also thank Michael Furukawa of the Agency for Healthcare Research and Quality, formerly of the Office of the National Coordinator for Health Information Technology, who played a key role in the conceptualization of this study and data collection.
Corresponding author: Emily Jones, PhD, MPP, National Institutes of Health, 6001 Executive Blvd., #5232 Rockville, MD 20852, [email protected].
Financial disclosures: None
1. Lehnert B, Bree R. Analysis of appropriateness of outpatient CT and MRI referred from primary care clinics at an academic medical center: how critical is the need for improved decision support? J Am Coll Radiol 2010;7:192–7.
2. Ip I, Schneider L, Hanson R, et al. Adoption and meaningful use of computed physician order entry with an integrated clinical decision support system for radiology: ten-year analysis in an urban teaching hospital. J Am Coll Radiol 2012;9:129–36.
3. Bernardy M, Ullrich C, Rawson J, et al. Strategies for managing imaging utilization. J Am Coll Radiol 2009;6:844–50.
4. Amland R, Dean B, Yu HT et al. Computed clinical decision support to prevent venous thromboembolism among hospitalized patients: proximal outcomes from a multiyear quality improvement project. J Healthcare Qual 2015;37:221–31.
5. Kahn C. Improving outcomes in radiology: bringing computer-based decision support and education to the point of care. Acad Radiology 2005;12:409–14.
6. Phansalkar S, Desai A, Bell D et al. High-priority drug-drug interactions for use in electronic health records. J Am Med Inform Assoc 2012;19:735–43.
7. Wolfstadt J, Gurwitz J, Field T, et al. The effect of computed physician order entry with clinical decision support on the rates of adverse drug events: a systematic review. J Gen Intern Med 2008;23:451–8.
8. Amland R, Hahn-Cover K. Clinical decision support for early recognition of sepsis. Am J Med Qual 2014;1–8.
9. Amland R, Haley J, Lyons J. A multidisciplinary sepsis program enabled by a two-stage clinical decision support system: factors that influence patient outcomes. Am J Med Qual 2015;1–8.
10. Umscheid C, Hanish A, Chittams J, et al. Effectiveness of a novel and scalable clinical decision support intervention to improve venous thromboembolism prophylaxis: a quasi-experimental study. BMC Med Inform Dec Making 2012;12:92–104.
11. Mack EH, Wheeler DS, Embi PJ. Clinical decision support systems in the pediatric intensive care unit. Pediatric Crit Care Med 2009;10:23–8.
12. Kollef M, Heard K, Chen Y, et al. Mortality and length of stay trends following implementation of a rapid response system and real-time automated clinical deterioration alerts. Am J Med Qual 2015; online first.
13. Ali S, Giordano R, Lakhani S, Walker D. A review of randomized controlled trials of medical record powered clinical decision support system to improve quality of diabetes care. Int J Med Informatics 2016;87:91–100.
14. Gill J, Mainous A, Koopman R et al. Impact of EHR-based clinical decision support on adherence to guidelines for patients on NSAIDs: a randomized controlled trial. Ann Fam Med 2011;9:22–30.
15. Choosing Wisely. Accessed 1 May 2017 at http://www.choosingwisely.org/clinician-lists/#keyword=appendicitis.
16. Hendrickson M, Wey A, Gaillard P, Kharbanda A. Implementation of an electronic clinical decision support tool for pediatric appendicitis within a hospital network. Pediatric Emerg Care 2017 (online first).
17. Kharbanda A, Madhok M, Krause E, et al. Implementation of electronic clinical decision support for pediatric appendicitis. Pediatrics 2016;137:e20151745.
18. Schuh S, Chan K, Langer J, et al. Properties of serial ultrasound clinical diagnostic pathway in suspected appendicitis and related computed tomography use. Acad Emerg Med 2015;22:406–14.
19. Ramarajan N, Krishnamoorthi R, Barth R, et al. An interdisciplinary initiative to reduce radiation exposure: evaluation of appendicitis in a pediatric emergency department with clinical assessment supported by a staged ultrasound and computed tomography pathway. Acad Emerg Med 2009;16:1258–65.
20. HIMSS Analytics. Stage 7 Hospitals. Accessed at www.himssanalytics.org/emram/stage7Hospitals.aspx.
21. Rizer M, et al. Top 10 lessons learned from electronic health record implementation in a large academic medical center. Perspectives in Health Information Management. Summer 2015.
22. Cosgrove DM, Fisher M, Gabow P, et al. Ten strategies to lower costs, improve quality, and engage patients: the view from leading health system CEOs. Health Aff (Millwood) 2013;32:321–7.
23. Cresswell KM, Bates DW, Sheikh A. Ten key considerations for the successful implementation and adoption of large-scale health information technology. J Am Med Inform Assoc 2013 Apr 18.
24. Hensing JA. The quest for upper-quartile performance at Banner Health. J Healthc Qual 2008;30:18–24
25. Hensing J, Dahlen D, Warden M, et al. Measuring the benefits of IT-enabled care transformation. Healthc Financ Manage 2008;62:74–80.
26. Truven Health Analytics. 15 Top Health Systems Study. 6th ed. April 2014. Accessed at http://100tophospitals.com/portals/2/assets/15-Top-Health-Systems-Study.pdf.
27. Aiello M. 2011 Top leadership team awards recognize big moves. Health Leaders Media. August 2011. Accessed at www.healthleadersmedia.com/page-2/LED-269808/2011-Top-Leadership-Team-Awards-Recognize-Big-Moves.
28. Rosenthal D, Stout M. Radiology order entry: features and performance requirements. J Am Coll Radiol 2006;3:554–7.
29. Kirsh S, Wu WC, Edelman D, Aron D. Research versus quality improvement: distinct or a distinction without a difference? A case study comparison of two studies. Jt Comm J Qual Patient Safety 2014;40:365–75.
30. Ogrinc G, Davies L, Goodman D, et al. SQUIRE 2.0 (Standards for Quality Improvement Reporting Excellence): revised publication guidelines from a detailed consensus process. BMJ Qual Saf 2015;0:1–7.
31. Blavin F, Ramos C, Shah A, Devers K. Lessons from the literature on electronic health record implementation.1 Aug 2013. The Urban Institute. Prepared for the Office of the National Coordinator for Health Information Technology. Available at www.urban.org/research/publication/lessons-literature-electronic-health-record-implementation.
32. Needleman J, Pearson ML, Upenieks VV, et al. Engaging frontline staff in performance improvement: the American Organization of Nurse Executives implementation of transforming care at the beside collaborative. Jt Comm J Qual Patient Safety 2016;42:61–9.
33. Jones E, Swain M, Patel V, Furukawa M. Supporting HITECH implementation and assessing lessons for the future: the role of program evaluation. Healthcare: The Journal of Delivery Science and Innovation 2014;2:4–8.
34. Phansalkar S, Zachariah M, Seidling H, et al. Evaluation of medication alerts in electronic health records for compliance with human factors principles. J Am Med Inform Assoc 2014;21:e332–e340.
35. Handler S, Altman R, Perera S, et al. A systematic review of the performance characteristics of clinical event monitor signals to detect adverse drug events in the hospital setting. J Am Med Inform Assoc 2007;14:451–8.
36. Singer S, Rivard P, Hayes J, et al. Improving patient care through leadership engagement with frontline staff: a Department of Veteran Affairs study. Jt Comm J Qual Patient Safety 2013;39):349–60.
37. Brewster A, Curry L, Cherlin E, et al. Integrating new practices: a qualitative study of how hospital innovations become routine. Implement Sci 2015;5(10):168.
1. Lehnert B, Bree R. Analysis of appropriateness of outpatient CT and MRI referred from primary care clinics at an academic medical center: how critical is the need for improved decision support? J Am Coll Radiol 2010;7:192–7.
2. Ip I, Schneider L, Hanson R, et al. Adoption and meaningful use of computed physician order entry with an integrated clinical decision support system for radiology: ten-year analysis in an urban teaching hospital. J Am Coll Radiol 2012;9:129–36.
3. Bernardy M, Ullrich C, Rawson J, et al. Strategies for managing imaging utilization. J Am Coll Radiol 2009;6:844–50.
4. Amland R, Dean B, Yu HT et al. Computed clinical decision support to prevent venous thromboembolism among hospitalized patients: proximal outcomes from a multiyear quality improvement project. J Healthcare Qual 2015;37:221–31.
5. Kahn C. Improving outcomes in radiology: bringing computer-based decision support and education to the point of care. Acad Radiology 2005;12:409–14.
6. Phansalkar S, Desai A, Bell D et al. High-priority drug-drug interactions for use in electronic health records. J Am Med Inform Assoc 2012;19:735–43.
7. Wolfstadt J, Gurwitz J, Field T, et al. The effect of computed physician order entry with clinical decision support on the rates of adverse drug events: a systematic review. J Gen Intern Med 2008;23:451–8.
8. Amland R, Hahn-Cover K. Clinical decision support for early recognition of sepsis. Am J Med Qual 2014;1–8.
9. Amland R, Haley J, Lyons J. A multidisciplinary sepsis program enabled by a two-stage clinical decision support system: factors that influence patient outcomes. Am J Med Qual 2015;1–8.
10. Umscheid C, Hanish A, Chittams J, et al. Effectiveness of a novel and scalable clinical decision support intervention to improve venous thromboembolism prophylaxis: a quasi-experimental study. BMC Med Inform Dec Making 2012;12:92–104.
11. Mack EH, Wheeler DS, Embi PJ. Clinical decision support systems in the pediatric intensive care unit. Pediatric Crit Care Med 2009;10:23–8.
12. Kollef M, Heard K, Chen Y, et al. Mortality and length of stay trends following implementation of a rapid response system and real-time automated clinical deterioration alerts. Am J Med Qual 2015; online first.
13. Ali S, Giordano R, Lakhani S, Walker D. A review of randomized controlled trials of medical record powered clinical decision support system to improve quality of diabetes care. Int J Med Informatics 2016;87:91–100.
14. Gill J, Mainous A, Koopman R et al. Impact of EHR-based clinical decision support on adherence to guidelines for patients on NSAIDs: a randomized controlled trial. Ann Fam Med 2011;9:22–30.
15. Choosing Wisely. Accessed 1 May 2017 at http://www.choosingwisely.org/clinician-lists/#keyword=appendicitis.
16. Hendrickson M, Wey A, Gaillard P, Kharbanda A. Implementation of an electronic clinical decision support tool for pediatric appendicitis within a hospital network. Pediatric Emerg Care 2017 (online first).
17. Kharbanda A, Madhok M, Krause E, et al. Implementation of electronic clinical decision support for pediatric appendicitis. Pediatrics 2016;137:e20151745.
18. Schuh S, Chan K, Langer J, et al. Properties of serial ultrasound clinical diagnostic pathway in suspected appendicitis and related computed tomography use. Acad Emerg Med 2015;22:406–14.
19. Ramarajan N, Krishnamoorthi R, Barth R, et al. An interdisciplinary initiative to reduce radiation exposure: evaluation of appendicitis in a pediatric emergency department with clinical assessment supported by a staged ultrasound and computed tomography pathway. Acad Emerg Med 2009;16:1258–65.
20. HIMSS Analytics. Stage 7 Hospitals. Accessed at www.himssanalytics.org/emram/stage7Hospitals.aspx.
21. Rizer M, et al. Top 10 lessons learned from electronic health record implementation in a large academic medical center. Perspectives in Health Information Management. Summer 2015.
22. Cosgrove DM, Fisher M, Gabow P, et al. Ten strategies to lower costs, improve quality, and engage patients: the view from leading health system CEOs. Health Aff (Millwood) 2013;32:321–7.
23. Cresswell KM, Bates DW, Sheikh A. Ten key considerations for the successful implementation and adoption of large-scale health information technology. J Am Med Inform Assoc 2013 Apr 18.
24. Hensing JA. The quest for upper-quartile performance at Banner Health. J Healthc Qual 2008;30:18–24
25. Hensing J, Dahlen D, Warden M, et al. Measuring the benefits of IT-enabled care transformation. Healthc Financ Manage 2008;62:74–80.
26. Truven Health Analytics. 15 Top Health Systems Study. 6th ed. April 2014. Accessed at http://100tophospitals.com/portals/2/assets/15-Top-Health-Systems-Study.pdf.
27. Aiello M. 2011 Top leadership team awards recognize big moves. Health Leaders Media. August 2011. Accessed at www.healthleadersmedia.com/page-2/LED-269808/2011-Top-Leadership-Team-Awards-Recognize-Big-Moves.
28. Rosenthal D, Stout M. Radiology order entry: features and performance requirements. J Am Coll Radiol 2006;3:554–7.
29. Kirsh S, Wu WC, Edelman D, Aron D. Research versus quality improvement: distinct or a distinction without a difference? A case study comparison of two studies. Jt Comm J Qual Patient Safety 2014;40:365–75.
30. Ogrinc G, Davies L, Goodman D, et al. SQUIRE 2.0 (Standards for Quality Improvement Reporting Excellence): revised publication guidelines from a detailed consensus process. BMJ Qual Saf 2015;0:1–7.
31. Blavin F, Ramos C, Shah A, Devers K. Lessons from the literature on electronic health record implementation.1 Aug 2013. The Urban Institute. Prepared for the Office of the National Coordinator for Health Information Technology. Available at www.urban.org/research/publication/lessons-literature-electronic-health-record-implementation.
32. Needleman J, Pearson ML, Upenieks VV, et al. Engaging frontline staff in performance improvement: the American Organization of Nurse Executives implementation of transforming care at the beside collaborative. Jt Comm J Qual Patient Safety 2016;42:61–9.
33. Jones E, Swain M, Patel V, Furukawa M. Supporting HITECH implementation and assessing lessons for the future: the role of program evaluation. Healthcare: The Journal of Delivery Science and Innovation 2014;2:4–8.
34. Phansalkar S, Zachariah M, Seidling H, et al. Evaluation of medication alerts in electronic health records for compliance with human factors principles. J Am Med Inform Assoc 2014;21:e332–e340.
35. Handler S, Altman R, Perera S, et al. A systematic review of the performance characteristics of clinical event monitor signals to detect adverse drug events in the hospital setting. J Am Med Inform Assoc 2007;14:451–8.
36. Singer S, Rivard P, Hayes J, et al. Improving patient care through leadership engagement with frontline staff: a Department of Veteran Affairs study. Jt Comm J Qual Patient Safety 2013;39):349–60.
37. Brewster A, Curry L, Cherlin E, et al. Integrating new practices: a qualitative study of how hospital innovations become routine. Implement Sci 2015;5(10):168.
Inhaled Corticosteroid Plus Long-Acting Beta-Agonist for Asthma: Real-Life Evidence
Study Overview
Objective. To determine the effectiveness of asthma treatment using fluticasone furoate plus vilanterol in a setting that is closer to usual clinical practice.
Design. Open-label, parallel group, randomised controlled trial.
Setting and participants. The study was conducted at 74 general practice clinics in Salford and South Manchester, UK, between Nov 2012 and Dec 2016. Patients with a general practitioner’s diagnosis of symptomatic asthma and on maintenance inhaler therapy (either inhaled corticosteroid [ICS] alone or in combination with a long-acting bronchodilator [LABA]) were recruited. Patients with recent history of life-threatening asthma, COPD, or concomitant life-threatening disease were excluded. Participants were randomly assigned through a centralized randomization service and stratified by Asthma Control Test (ACT) score and by previous asthma maintenance therapy (ICS or ICS/LABA). Only those with an ACT score < 20 were included in the study.
Intervention. Patients were randomized to receive either a combination of fluticasone furoate and vilanterol (FF/VI) delivered by novel dry powder inhalation (DPI) (Ellipta) or to continue with their maintenance therapy. General practitioners provided care in their usual manner and could continuously optimize therapy according to their clinical opinion. Treatments were dispensed by community pharmacies in the usual way. Patients could modify their treatment and remain in the study. Those in the FF/VI group were allowed to change to other asthma medications and could stop taking FF/VI. Those in the usual care group were also allowed to alter medications, but could not initiate FF/VI.
Main outcome measures. The primary endpoint was ACT score at week 24 (the percentage of patients at week 24 with either an ACT score of 20 or greater or an increase of 3 or greater in the ACT score from baseline, termed responders). Safety endpoints included the incidence of serious pneumonias. The study utilized the Salford electronic medical record system, which allows near to real-time collection and monitoring of safety data. Secondary endpoints included ACT at various weeks, all asthma-related primary and secondary care contacts, annual rate of severe exacerbations, number of salbutamol inhalers dispensed, and time to modification or initial therapy.
Main results. 4233 patients were randomized, with 2119 patients randomized to usual care and 2114 randomized to the FF/VI group. 605 from the usual care group and 602 from the FF/VI group had a baseline ACT score greater than or equal to 20 and were thus excluded from the primary effectiveness analysis population. 306 in the usual care group and 342 in the FF/VI group withdrew for various reasons, including adverse events, or were lost to follow-up or protocol deviations. Mean patient age was 50 years. Within the usual care group, 64% of patients received ICS/LABA combination and 36% received ICS only. Within the FF/VI group, 65% were prescribed 100 μg/25 μg FFI/VI and 35% were prescribed 200 μg/25 μg FF/VI. At week 24, the FF/VI group had 74% responders whereas the usual care group had 60% responders; the odds of being a responder with FF/VI was twice that of being a responder with usual care (OR 1.97; 95% CI 1.71–2.26, P < 0.001). Patients in the FF/VI group had a slightly higher incidence of pneumonia than did the usual care group (23 vs 16; incidence ratio 1.4, 95% CI 0.8–2.7). Also, those in the FF/VI group had an increase in the rate of primary care visits/contacts per year (9.7% increase, 95% CI 4.6%–15.0%).
Conclusion. In patients with a general practitioner’s diagnosis of symptomatic asthma and on maintenance inhaler therapy, initiation of a once-daily treatment regimen of combined FF/VI improved asthma control without increasing the risk of serious adverse events when compared with optimized usual care.
Commentary
Woodcock et al conducted a pragmatic randomized controlled study. This innovative research method prospectively enrolled a large number of patients who were randomized to groups that could involve 1 or more interventions and who were then followed according to the treating physician’s usual practice. The patients’ experience was kept as close to everyday clinical practice care as possible to preserve the real-world nature of the study. The positive aspect of this innovative pragmatic research design is the inclusion of patients with varied disease severity and with comorbidities that are not well represented in conventional double-blind randomized controlled trials, such as patients with smoking history, obesity, or multiple comorbidities. In addition, an electronic health record system was used to track serious adverse events in near real-time and increased the accuracy of the data and minimized data loss.
While the pragmatic study design offers innovation, it also has some limitations. Effectiveness studies using a pragmatic approach are less controlled compared with traditional efficacy RCTs and are more prone to low medication compliance and high rates of follow-up loss. Further, Woodcock et al allowed patients to remain in the FF/VI group even though they may have stopped taking FF/VI. Indeed, in the FF/VI group, 463 (22%) of the 2114 patients changed their medication, and 381 (18%) switched to the usual care group. Patients were analyzed using intention to treat and thus were analyzed in the group to which they were initially randomized. This could have affected results, as a good proportion of patients in the FF/VI group were not actually taking the FF/VI. Within the usual care group, 376 (18%) of 2119 patients altered their medication and 3 (< 1%) switched to FF/VI, though this was prohibited. In routine care, adherence rates are expected to be low (20%–40%) and this is another possible weakness of the study; in closely monitored RCTs, adherence rates are around 80%–90%.
The authors did not include objective measures of the severity or types of asthma, which can be obtained using pulmonary function tests, eosinophil count, or other markers of inflammation. By identifying asthma patients via the general practitioner’s diagnosis, the study is more reflective of real life and primary care–driven; however, one cannot rule out accidental inclusion of patients who do not have asthma (which could include patients with post-infectious cough, vocal cord dysfunction, or anxiety) or patients who would not readily respond to typical asthma therapy (such as those with allergic bronchopulmonary aspergillosis or eosinophilic granulomatosis with polyangitis). In addition, the authors used only subjective measures to define control: ACT score by telephone. Other outcome measures included exacerbation rate, primary care physician visits, and time to exacerbation, which may be insensitive to detecting residual inflammation or severity of asthma. In lieu of objectively measuring the degree of airway obstruction or inflammation, the outcomes measured by the authors may not have comprehensively evaluated efficacy.
The open-label, intention-to-treat, and pragmatic design of the study may have generated major selection bias, despite the randomization. Because general practitioners who directly participated in the recruitment of the patients also monitored their treatment, volunteer or referral bias may have occurred. As the authors admitted, there were differences present in practice and treatment due to variation of training and education of the general practitioners. In addition, the current study was funded by a pharmaceutical company and the trial medication was dispensed free of cost, further generating bias.
Further consideration of the study medication also brings up questions about the study design. Combined therapy with low- to moderate-dose ICS/LABA is currently indicated for asthma patients with moderate persistent or higher severity asthma. The current US insurance system encourages management to begin with low-dose ICS before escalating to a combination of ICS/LABA. Given the previously published evidence of superiority for combined ICS/LABA over ICS alone on asthma control [2,3], inclusion criteria could have been limited only to patients who were already receiving ICS/LABA to more accurately equate the trial medication with the accepted standard medications. By including patients who were on ICS/LABA as well as those only on ICS (in usual care group, 64% were on ICS/LABA and 36% were on ICS) the likelihood of responders in the FF/VI group could have been inflated compared to usual care group. In addition, patients with a low severity of asthma symptoms, such as only intermittent asthma or mild persistent asthma, could have been overtreated by FF/VI per current guidelines. About 30% of the patients initially enrolled in the study had baseline ACT scores greater than 20, and some patients had less severe asthma as indicated by the treatment with ICS alone. The authors also included 2 different doses of fluticasone furoate in their study group.
It is of concern that the incidence of pneumonia with ICS/LABA in this study was slightly higher in the FF/VI than in the usual care group. Although it was not statistically significant in this study, the increased pneumonia risk with ICS has been observed in many other studies [4,5].
Applications for Clinical Practice
Fluticasone furoate plus vilanterol (FF/VI) can be a therapeutic option in patients with asthma, with a small increased risk for pneumonia that is similar to other types of inhaled corticosteroids. However, a stepwise therapeutic approach, following the published asthma treatment strategy [6], should be emphasized when escalating treatment to include FF/VI.
—Minkyung Kwon, MD, Joel Roberson, MD, and Neal Patel, MD, Pulmonary and Critical Care Medicine, Mayo Clinic Florida, Jacksonville, FL (Drs. Kwon and Patel), and Department of Radiology, Oakland University/Beaumont Health, Royal Oak, MI (Dr. Roberson)
1. Chalkidou K, Tunis S, Whicher D, et al. The role for pragmatic randomized controlled trials (pRCTs) in comparative effectiveness research. Clin Trials (London, England) 2012;9:436–46.
2. O’Byrne PM, Bleecker ER, Bateman ED, et al. Once-daily fluticasone furoate alone or combined with vilanterol in persistent asthma. Eur Respir J 2014;43:773–82.
3. Bateman ED, O’Byrne PM, Busse WW, et al. Once-daily fluticasone furoate (FF)/vilanterol reduces risk of severe exacerbations in asthma versus FF alone. Thorax 2014;69:312–9.
4. McKeever T, Harrison TW, Hubbard R, Shaw D. Inhaled corticosteroids and the risk of pneumonia in people with asthma: a case-control study. Chest 2013;144:1788–94.
5. Crim C, Dransfield MT, Bourbeau J, et al. Pneumonia risk with inhaled fluticasone furoate and vilanterol compared with vilanterol alone in patients with COPD. Ann Am Thorac Soc 2015;12:27–34.
6. GINA. Global strategy for asthma management and prevention. 2017. Accessed at ginaasthma.org.
Study Overview
Objective. To determine the effectiveness of asthma treatment using fluticasone furoate plus vilanterol in a setting that is closer to usual clinical practice.
Design. Open-label, parallel group, randomised controlled trial.
Setting and participants. The study was conducted at 74 general practice clinics in Salford and South Manchester, UK, between Nov 2012 and Dec 2016. Patients with a general practitioner’s diagnosis of symptomatic asthma and on maintenance inhaler therapy (either inhaled corticosteroid [ICS] alone or in combination with a long-acting bronchodilator [LABA]) were recruited. Patients with recent history of life-threatening asthma, COPD, or concomitant life-threatening disease were excluded. Participants were randomly assigned through a centralized randomization service and stratified by Asthma Control Test (ACT) score and by previous asthma maintenance therapy (ICS or ICS/LABA). Only those with an ACT score < 20 were included in the study.
Intervention. Patients were randomized to receive either a combination of fluticasone furoate and vilanterol (FF/VI) delivered by novel dry powder inhalation (DPI) (Ellipta) or to continue with their maintenance therapy. General practitioners provided care in their usual manner and could continuously optimize therapy according to their clinical opinion. Treatments were dispensed by community pharmacies in the usual way. Patients could modify their treatment and remain in the study. Those in the FF/VI group were allowed to change to other asthma medications and could stop taking FF/VI. Those in the usual care group were also allowed to alter medications, but could not initiate FF/VI.
Main outcome measures. The primary endpoint was ACT score at week 24 (the percentage of patients at week 24 with either an ACT score of 20 or greater or an increase of 3 or greater in the ACT score from baseline, termed responders). Safety endpoints included the incidence of serious pneumonias. The study utilized the Salford electronic medical record system, which allows near to real-time collection and monitoring of safety data. Secondary endpoints included ACT at various weeks, all asthma-related primary and secondary care contacts, annual rate of severe exacerbations, number of salbutamol inhalers dispensed, and time to modification or initial therapy.
Main results. 4233 patients were randomized, with 2119 patients randomized to usual care and 2114 randomized to the FF/VI group. 605 from the usual care group and 602 from the FF/VI group had a baseline ACT score greater than or equal to 20 and were thus excluded from the primary effectiveness analysis population. 306 in the usual care group and 342 in the FF/VI group withdrew for various reasons, including adverse events, or were lost to follow-up or protocol deviations. Mean patient age was 50 years. Within the usual care group, 64% of patients received ICS/LABA combination and 36% received ICS only. Within the FF/VI group, 65% were prescribed 100 μg/25 μg FFI/VI and 35% were prescribed 200 μg/25 μg FF/VI. At week 24, the FF/VI group had 74% responders whereas the usual care group had 60% responders; the odds of being a responder with FF/VI was twice that of being a responder with usual care (OR 1.97; 95% CI 1.71–2.26, P < 0.001). Patients in the FF/VI group had a slightly higher incidence of pneumonia than did the usual care group (23 vs 16; incidence ratio 1.4, 95% CI 0.8–2.7). Also, those in the FF/VI group had an increase in the rate of primary care visits/contacts per year (9.7% increase, 95% CI 4.6%–15.0%).
Conclusion. In patients with a general practitioner’s diagnosis of symptomatic asthma and on maintenance inhaler therapy, initiation of a once-daily treatment regimen of combined FF/VI improved asthma control without increasing the risk of serious adverse events when compared with optimized usual care.
Commentary
Woodcock et al conducted a pragmatic randomized controlled study. This innovative research method prospectively enrolled a large number of patients who were randomized to groups that could involve 1 or more interventions and who were then followed according to the treating physician’s usual practice. The patients’ experience was kept as close to everyday clinical practice care as possible to preserve the real-world nature of the study. The positive aspect of this innovative pragmatic research design is the inclusion of patients with varied disease severity and with comorbidities that are not well represented in conventional double-blind randomized controlled trials, such as patients with smoking history, obesity, or multiple comorbidities. In addition, an electronic health record system was used to track serious adverse events in near real-time and increased the accuracy of the data and minimized data loss.
While the pragmatic study design offers innovation, it also has some limitations. Effectiveness studies using a pragmatic approach are less controlled compared with traditional efficacy RCTs and are more prone to low medication compliance and high rates of follow-up loss. Further, Woodcock et al allowed patients to remain in the FF/VI group even though they may have stopped taking FF/VI. Indeed, in the FF/VI group, 463 (22%) of the 2114 patients changed their medication, and 381 (18%) switched to the usual care group. Patients were analyzed using intention to treat and thus were analyzed in the group to which they were initially randomized. This could have affected results, as a good proportion of patients in the FF/VI group were not actually taking the FF/VI. Within the usual care group, 376 (18%) of 2119 patients altered their medication and 3 (< 1%) switched to FF/VI, though this was prohibited. In routine care, adherence rates are expected to be low (20%–40%) and this is another possible weakness of the study; in closely monitored RCTs, adherence rates are around 80%–90%.
The authors did not include objective measures of the severity or types of asthma, which can be obtained using pulmonary function tests, eosinophil count, or other markers of inflammation. By identifying asthma patients via the general practitioner’s diagnosis, the study is more reflective of real life and primary care–driven; however, one cannot rule out accidental inclusion of patients who do not have asthma (which could include patients with post-infectious cough, vocal cord dysfunction, or anxiety) or patients who would not readily respond to typical asthma therapy (such as those with allergic bronchopulmonary aspergillosis or eosinophilic granulomatosis with polyangitis). In addition, the authors used only subjective measures to define control: ACT score by telephone. Other outcome measures included exacerbation rate, primary care physician visits, and time to exacerbation, which may be insensitive to detecting residual inflammation or severity of asthma. In lieu of objectively measuring the degree of airway obstruction or inflammation, the outcomes measured by the authors may not have comprehensively evaluated efficacy.
The open-label, intention-to-treat, and pragmatic design of the study may have generated major selection bias, despite the randomization. Because general practitioners who directly participated in the recruitment of the patients also monitored their treatment, volunteer or referral bias may have occurred. As the authors admitted, there were differences present in practice and treatment due to variation of training and education of the general practitioners. In addition, the current study was funded by a pharmaceutical company and the trial medication was dispensed free of cost, further generating bias.
Further consideration of the study medication also brings up questions about the study design. Combined therapy with low- to moderate-dose ICS/LABA is currently indicated for asthma patients with moderate persistent or higher severity asthma. The current US insurance system encourages management to begin with low-dose ICS before escalating to a combination of ICS/LABA. Given the previously published evidence of superiority for combined ICS/LABA over ICS alone on asthma control [2,3], inclusion criteria could have been limited only to patients who were already receiving ICS/LABA to more accurately equate the trial medication with the accepted standard medications. By including patients who were on ICS/LABA as well as those only on ICS (in usual care group, 64% were on ICS/LABA and 36% were on ICS) the likelihood of responders in the FF/VI group could have been inflated compared to usual care group. In addition, patients with a low severity of asthma symptoms, such as only intermittent asthma or mild persistent asthma, could have been overtreated by FF/VI per current guidelines. About 30% of the patients initially enrolled in the study had baseline ACT scores greater than 20, and some patients had less severe asthma as indicated by the treatment with ICS alone. The authors also included 2 different doses of fluticasone furoate in their study group.
It is of concern that the incidence of pneumonia with ICS/LABA in this study was slightly higher in the FF/VI than in the usual care group. Although it was not statistically significant in this study, the increased pneumonia risk with ICS has been observed in many other studies [4,5].
Applications for Clinical Practice
Fluticasone furoate plus vilanterol (FF/VI) can be a therapeutic option in patients with asthma, with a small increased risk for pneumonia that is similar to other types of inhaled corticosteroids. However, a stepwise therapeutic approach, following the published asthma treatment strategy [6], should be emphasized when escalating treatment to include FF/VI.
—Minkyung Kwon, MD, Joel Roberson, MD, and Neal Patel, MD, Pulmonary and Critical Care Medicine, Mayo Clinic Florida, Jacksonville, FL (Drs. Kwon and Patel), and Department of Radiology, Oakland University/Beaumont Health, Royal Oak, MI (Dr. Roberson)
Study Overview
Objective. To determine the effectiveness of asthma treatment using fluticasone furoate plus vilanterol in a setting that is closer to usual clinical practice.
Design. Open-label, parallel group, randomised controlled trial.
Setting and participants. The study was conducted at 74 general practice clinics in Salford and South Manchester, UK, between Nov 2012 and Dec 2016. Patients with a general practitioner’s diagnosis of symptomatic asthma and on maintenance inhaler therapy (either inhaled corticosteroid [ICS] alone or in combination with a long-acting bronchodilator [LABA]) were recruited. Patients with recent history of life-threatening asthma, COPD, or concomitant life-threatening disease were excluded. Participants were randomly assigned through a centralized randomization service and stratified by Asthma Control Test (ACT) score and by previous asthma maintenance therapy (ICS or ICS/LABA). Only those with an ACT score < 20 were included in the study.
Intervention. Patients were randomized to receive either a combination of fluticasone furoate and vilanterol (FF/VI) delivered by novel dry powder inhalation (DPI) (Ellipta) or to continue with their maintenance therapy. General practitioners provided care in their usual manner and could continuously optimize therapy according to their clinical opinion. Treatments were dispensed by community pharmacies in the usual way. Patients could modify their treatment and remain in the study. Those in the FF/VI group were allowed to change to other asthma medications and could stop taking FF/VI. Those in the usual care group were also allowed to alter medications, but could not initiate FF/VI.
Main outcome measures. The primary endpoint was ACT score at week 24 (the percentage of patients at week 24 with either an ACT score of 20 or greater or an increase of 3 or greater in the ACT score from baseline, termed responders). Safety endpoints included the incidence of serious pneumonias. The study utilized the Salford electronic medical record system, which allows near to real-time collection and monitoring of safety data. Secondary endpoints included ACT at various weeks, all asthma-related primary and secondary care contacts, annual rate of severe exacerbations, number of salbutamol inhalers dispensed, and time to modification or initial therapy.
Main results. 4233 patients were randomized, with 2119 patients randomized to usual care and 2114 randomized to the FF/VI group. 605 from the usual care group and 602 from the FF/VI group had a baseline ACT score greater than or equal to 20 and were thus excluded from the primary effectiveness analysis population. 306 in the usual care group and 342 in the FF/VI group withdrew for various reasons, including adverse events, or were lost to follow-up or protocol deviations. Mean patient age was 50 years. Within the usual care group, 64% of patients received ICS/LABA combination and 36% received ICS only. Within the FF/VI group, 65% were prescribed 100 μg/25 μg FFI/VI and 35% were prescribed 200 μg/25 μg FF/VI. At week 24, the FF/VI group had 74% responders whereas the usual care group had 60% responders; the odds of being a responder with FF/VI was twice that of being a responder with usual care (OR 1.97; 95% CI 1.71–2.26, P < 0.001). Patients in the FF/VI group had a slightly higher incidence of pneumonia than did the usual care group (23 vs 16; incidence ratio 1.4, 95% CI 0.8–2.7). Also, those in the FF/VI group had an increase in the rate of primary care visits/contacts per year (9.7% increase, 95% CI 4.6%–15.0%).
Conclusion. In patients with a general practitioner’s diagnosis of symptomatic asthma and on maintenance inhaler therapy, initiation of a once-daily treatment regimen of combined FF/VI improved asthma control without increasing the risk of serious adverse events when compared with optimized usual care.
Commentary
Woodcock et al conducted a pragmatic randomized controlled study. This innovative research method prospectively enrolled a large number of patients who were randomized to groups that could involve 1 or more interventions and who were then followed according to the treating physician’s usual practice. The patients’ experience was kept as close to everyday clinical practice care as possible to preserve the real-world nature of the study. The positive aspect of this innovative pragmatic research design is the inclusion of patients with varied disease severity and with comorbidities that are not well represented in conventional double-blind randomized controlled trials, such as patients with smoking history, obesity, or multiple comorbidities. In addition, an electronic health record system was used to track serious adverse events in near real-time and increased the accuracy of the data and minimized data loss.
While the pragmatic study design offers innovation, it also has some limitations. Effectiveness studies using a pragmatic approach are less controlled compared with traditional efficacy RCTs and are more prone to low medication compliance and high rates of follow-up loss. Further, Woodcock et al allowed patients to remain in the FF/VI group even though they may have stopped taking FF/VI. Indeed, in the FF/VI group, 463 (22%) of the 2114 patients changed their medication, and 381 (18%) switched to the usual care group. Patients were analyzed using intention to treat and thus were analyzed in the group to which they were initially randomized. This could have affected results, as a good proportion of patients in the FF/VI group were not actually taking the FF/VI. Within the usual care group, 376 (18%) of 2119 patients altered their medication and 3 (< 1%) switched to FF/VI, though this was prohibited. In routine care, adherence rates are expected to be low (20%–40%) and this is another possible weakness of the study; in closely monitored RCTs, adherence rates are around 80%–90%.
The authors did not include objective measures of the severity or types of asthma, which can be obtained using pulmonary function tests, eosinophil count, or other markers of inflammation. By identifying asthma patients via the general practitioner’s diagnosis, the study is more reflective of real life and primary care–driven; however, one cannot rule out accidental inclusion of patients who do not have asthma (which could include patients with post-infectious cough, vocal cord dysfunction, or anxiety) or patients who would not readily respond to typical asthma therapy (such as those with allergic bronchopulmonary aspergillosis or eosinophilic granulomatosis with polyangitis). In addition, the authors used only subjective measures to define control: ACT score by telephone. Other outcome measures included exacerbation rate, primary care physician visits, and time to exacerbation, which may be insensitive to detecting residual inflammation or severity of asthma. In lieu of objectively measuring the degree of airway obstruction or inflammation, the outcomes measured by the authors may not have comprehensively evaluated efficacy.
The open-label, intention-to-treat, and pragmatic design of the study may have generated major selection bias, despite the randomization. Because general practitioners who directly participated in the recruitment of the patients also monitored their treatment, volunteer or referral bias may have occurred. As the authors admitted, there were differences present in practice and treatment due to variation of training and education of the general practitioners. In addition, the current study was funded by a pharmaceutical company and the trial medication was dispensed free of cost, further generating bias.
Further consideration of the study medication also brings up questions about the study design. Combined therapy with low- to moderate-dose ICS/LABA is currently indicated for asthma patients with moderate persistent or higher severity asthma. The current US insurance system encourages management to begin with low-dose ICS before escalating to a combination of ICS/LABA. Given the previously published evidence of superiority for combined ICS/LABA over ICS alone on asthma control [2,3], inclusion criteria could have been limited only to patients who were already receiving ICS/LABA to more accurately equate the trial medication with the accepted standard medications. By including patients who were on ICS/LABA as well as those only on ICS (in usual care group, 64% were on ICS/LABA and 36% were on ICS) the likelihood of responders in the FF/VI group could have been inflated compared to usual care group. In addition, patients with a low severity of asthma symptoms, such as only intermittent asthma or mild persistent asthma, could have been overtreated by FF/VI per current guidelines. About 30% of the patients initially enrolled in the study had baseline ACT scores greater than 20, and some patients had less severe asthma as indicated by the treatment with ICS alone. The authors also included 2 different doses of fluticasone furoate in their study group.
It is of concern that the incidence of pneumonia with ICS/LABA in this study was slightly higher in the FF/VI than in the usual care group. Although it was not statistically significant in this study, the increased pneumonia risk with ICS has been observed in many other studies [4,5].
Applications for Clinical Practice
Fluticasone furoate plus vilanterol (FF/VI) can be a therapeutic option in patients with asthma, with a small increased risk for pneumonia that is similar to other types of inhaled corticosteroids. However, a stepwise therapeutic approach, following the published asthma treatment strategy [6], should be emphasized when escalating treatment to include FF/VI.
—Minkyung Kwon, MD, Joel Roberson, MD, and Neal Patel, MD, Pulmonary and Critical Care Medicine, Mayo Clinic Florida, Jacksonville, FL (Drs. Kwon and Patel), and Department of Radiology, Oakland University/Beaumont Health, Royal Oak, MI (Dr. Roberson)
1. Chalkidou K, Tunis S, Whicher D, et al. The role for pragmatic randomized controlled trials (pRCTs) in comparative effectiveness research. Clin Trials (London, England) 2012;9:436–46.
2. O’Byrne PM, Bleecker ER, Bateman ED, et al. Once-daily fluticasone furoate alone or combined with vilanterol in persistent asthma. Eur Respir J 2014;43:773–82.
3. Bateman ED, O’Byrne PM, Busse WW, et al. Once-daily fluticasone furoate (FF)/vilanterol reduces risk of severe exacerbations in asthma versus FF alone. Thorax 2014;69:312–9.
4. McKeever T, Harrison TW, Hubbard R, Shaw D. Inhaled corticosteroids and the risk of pneumonia in people with asthma: a case-control study. Chest 2013;144:1788–94.
5. Crim C, Dransfield MT, Bourbeau J, et al. Pneumonia risk with inhaled fluticasone furoate and vilanterol compared with vilanterol alone in patients with COPD. Ann Am Thorac Soc 2015;12:27–34.
6. GINA. Global strategy for asthma management and prevention. 2017. Accessed at ginaasthma.org.
1. Chalkidou K, Tunis S, Whicher D, et al. The role for pragmatic randomized controlled trials (pRCTs) in comparative effectiveness research. Clin Trials (London, England) 2012;9:436–46.
2. O’Byrne PM, Bleecker ER, Bateman ED, et al. Once-daily fluticasone furoate alone or combined with vilanterol in persistent asthma. Eur Respir J 2014;43:773–82.
3. Bateman ED, O’Byrne PM, Busse WW, et al. Once-daily fluticasone furoate (FF)/vilanterol reduces risk of severe exacerbations in asthma versus FF alone. Thorax 2014;69:312–9.
4. McKeever T, Harrison TW, Hubbard R, Shaw D. Inhaled corticosteroids and the risk of pneumonia in people with asthma: a case-control study. Chest 2013;144:1788–94.
5. Crim C, Dransfield MT, Bourbeau J, et al. Pneumonia risk with inhaled fluticasone furoate and vilanterol compared with vilanterol alone in patients with COPD. Ann Am Thorac Soc 2015;12:27–34.
6. GINA. Global strategy for asthma management and prevention. 2017. Accessed at ginaasthma.org.
Prolonged Survival in Metastatic Melanoma
Study Overview
Objective. To compare clinical outcomes and toxicities between combined nivolumab plus ipilimumab (N+I) versus ipilimumab alone (I) or nivolumab alone (N) in patients with advanced melanoma.
Design. Randomized controlled trial 1:1:1 of N+I (nivolumab 1 mg/kg + ipilimumab 3 mg/kg every 3 weeks for 4 doses, followed by nivolumab 3 mg/kg every 2 weeks) versus N (3 mg/kg every 2 weeks) versus I plus placebo (3 mg/kg every 3 weeks for four doses).
Setting and participants. Adult patients with previously untreated stage III (unresectable) or stage IV melanoma and ECOG performance status of 0 or 1 (on a scale of 0–5 with higher score indicating greater disability). Patients with active brain metastases, ocular melanoma, or autoimmune disease were excluded. This study took place in academic and community practices across the United States, Europe, and Australia. 945 patients were randomized. If patients progressed, additional therapies were at clinician discretion.
Main outcome measures. Primary end points were progression-free survival and overall survival. Secondary end points were objective response rate, toxicity profile, and evaluation of PD-L1 (programmed death-ligand 1) as a predictive marker for progression-free survival and overall survival.
Main results. Baseline patient characteristics were published previously [1]. There were no significant differences among groups except that the I only group had a higher frequency of brian metastases (4.8%) vs the N only group (2.5%). At censored follow-up of a minimum of 36 months, median overall survival was not reached in the N+I group, was 37.6 months in the N only group and was 19.9 months in the I only group (hazard ratio [HR] for death 0.55 (P < 0.001) for N+I vs. I and 0.65 (P < 0.001) for N vs. I). Overall survival at 3 years was 58% in the N+I group vs. 52% in the N only group vs. 34% in the I only group. The rate of objective response was 58% in the N+I group vs. 44% in the N only group vs. 19% in the I only group. Progression-free survival at 3 years was 39% in the N+I group, 32% in the N only group and 10% in the I only group. The level of PD-L1 expression was not associated with response or overall survival. Grade 3 or 4 treatment-related adverse events occurred in 59% of the N+I group vs. 21% in N vs. 28% in I group. As therapy after progression was left to clinician discretion, crossover was common with 43% of the I only group receiving nivolumab as second-line therapy and 28% of the N only group receiving ipilimumab as second-line therapy.
Treatment-related events that lead to therapy discontinuation occurred much more frequently in those who received N+I (40%) vs. N (12%) vs. I (16%). However, among the N+I patients who discontinued after a median of 3 cycles of treatment, 67% were still alive at 3 years. In addition, when adverse events were treated with safety guidelines, most immune-mediated adverse events resolved within 3 to 4 weeks. The most common grade 3 or 4 adverse events in the N+I group were diarrhea (9%), elevated lipase (11%), and elevated liver transaminases (9%). A total of 2 treatment-related deaths were reported in the N+I group.
Conclusion. Both the combination therapy of nivolumab + ipilimumab and nivolumab alone offer superior 3-year overall survival and progression-free survival compared with ipilimumab alone in advanced melanoma, with acceptable toxicity profiles.
Commentary
Historically, unresectable and metastatic melanoma has had a dismal prognosis, with responses to chemotherapy in about 10% to 15% and rarely were these responses durable [2]. The previous standard of care was high-dose IL-2, a form of immunotherapy which leads to long-term survival in a small minority of patients (~15%) [3]. The encouraging results seen in this small minority lead to optimism for efficacy from additional immune-modifying agents.
The novel immunotherapy agents, known as checkpoint inhibitors, are antibodies directed against PD-1 (nivolumab and pembrolizumab), PD-L1 (atezolizumab, avelumab, and urvalumab), and CTLA-4 (ipilimumab). Each of these antigens are critical in a T cell process known as checkpoint inhibition. When these antigens are activated they inhibit T cells, a process critical for self recognition in the healthy human without cancer. However, many malignancies have developed molecular mechanisms to activate these checkpoint pathways and turn off T cell anti-tumor activity. By implementing checkpoint inhibitor antibodies, as done in this study, these drugs allow the T cells to be disinhibited and therefore exert anti-tumor activity. These drugs have been truly ground-breaking and are now FDA-approved in a number of malignancies, including bladder cancer, non–small cell lung cancer, head and neck squamous cell carcinoma, refractory Hodgkin lymphoma, mismatch repair–affected GI adenocarcinomas, renal cell carcinoma, and Merkel cell carcinoma. They offer the additional advantage of often an improved toxicity profile compared with traditional cytotoxic chemotherapy, as they are not typically associated with cytopenias, nausea, or hair loss, for example [4].
In this study, 3-year data from the CheckMate 067 trial is reported. As reported in this study, checkpoint inhibition has lead to truly remarkable improvements in outcomes for patients with advanced melanoma. In this study, the authors have demonstrated superiority of nivolumab plus ipilimumab and nivolumab alone versus ipilimumab alone. These results are similar to those seen in the KEYNOTE-006 trial which compared pemrolizumab (another anti-PD-1 antibody) to ipilimumab. In the KEYNOTE-006 trial, overall survival at 33 months was 50% in the pembrolizumab group versus 39% in the ipilimumab group.
In this study, the combination therapy was more toxic, requiring more frequent treatment discontinuation, though importantly, 3-year overall survival was 67% even among those who discontinued therapy. Grade 3 or 4 toxicity events seem to be associated with efficacy in this study. This is not surprising as this has been seen in some other tumor types as well [5], though it deserves more dedicated investigation as a prognostic marker in this population.
Applications for Clinical Practice
In this well-designed and -executed multicenter randomized trial, funded by Bristol-Myers Squibb and implemented in a selected population with good performance status, all 3 immunotherapies demonstrated impressive improvements in the management of advanced melanoma. The combination nivolumab and ipilimumab was the most effective, with markedly higher survival and response rates, but also with higher toxicity requiring treatment discontinuation, though this did not decrease the efficacy of the therapy. Both the combination nivolumab plus ipilimumab and nivolimab alone are acceptable treatments for patients with advanced melanoma and good performance status; cost and comorbidities will be critical in personalizing therapy.
—Matthew Painschab, MD, University of North Carolina, Chapel Hill, NC
1. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
2. Hill GJI, Krementz ET, Hill HZ. Dimethyl triazeno imidazole carboxamide and combination therapy for melanoma. Cancer 1984;53:1299–305.
3. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleuken 2 therapy for patiens with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 1999;17:2105–16.
4. Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 2016;54:139–48.
5. Haratani K, Hayashi H, Chiba Y, et al. Association of immune-related adverse events with nivolumab efficacy in non-small-cell lung cancer. JAMA Oncol 2017 Sept 21.
Study Overview
Objective. To compare clinical outcomes and toxicities between combined nivolumab plus ipilimumab (N+I) versus ipilimumab alone (I) or nivolumab alone (N) in patients with advanced melanoma.
Design. Randomized controlled trial 1:1:1 of N+I (nivolumab 1 mg/kg + ipilimumab 3 mg/kg every 3 weeks for 4 doses, followed by nivolumab 3 mg/kg every 2 weeks) versus N (3 mg/kg every 2 weeks) versus I plus placebo (3 mg/kg every 3 weeks for four doses).
Setting and participants. Adult patients with previously untreated stage III (unresectable) or stage IV melanoma and ECOG performance status of 0 or 1 (on a scale of 0–5 with higher score indicating greater disability). Patients with active brain metastases, ocular melanoma, or autoimmune disease were excluded. This study took place in academic and community practices across the United States, Europe, and Australia. 945 patients were randomized. If patients progressed, additional therapies were at clinician discretion.
Main outcome measures. Primary end points were progression-free survival and overall survival. Secondary end points were objective response rate, toxicity profile, and evaluation of PD-L1 (programmed death-ligand 1) as a predictive marker for progression-free survival and overall survival.
Main results. Baseline patient characteristics were published previously [1]. There were no significant differences among groups except that the I only group had a higher frequency of brian metastases (4.8%) vs the N only group (2.5%). At censored follow-up of a minimum of 36 months, median overall survival was not reached in the N+I group, was 37.6 months in the N only group and was 19.9 months in the I only group (hazard ratio [HR] for death 0.55 (P < 0.001) for N+I vs. I and 0.65 (P < 0.001) for N vs. I). Overall survival at 3 years was 58% in the N+I group vs. 52% in the N only group vs. 34% in the I only group. The rate of objective response was 58% in the N+I group vs. 44% in the N only group vs. 19% in the I only group. Progression-free survival at 3 years was 39% in the N+I group, 32% in the N only group and 10% in the I only group. The level of PD-L1 expression was not associated with response or overall survival. Grade 3 or 4 treatment-related adverse events occurred in 59% of the N+I group vs. 21% in N vs. 28% in I group. As therapy after progression was left to clinician discretion, crossover was common with 43% of the I only group receiving nivolumab as second-line therapy and 28% of the N only group receiving ipilimumab as second-line therapy.
Treatment-related events that lead to therapy discontinuation occurred much more frequently in those who received N+I (40%) vs. N (12%) vs. I (16%). However, among the N+I patients who discontinued after a median of 3 cycles of treatment, 67% were still alive at 3 years. In addition, when adverse events were treated with safety guidelines, most immune-mediated adverse events resolved within 3 to 4 weeks. The most common grade 3 or 4 adverse events in the N+I group were diarrhea (9%), elevated lipase (11%), and elevated liver transaminases (9%). A total of 2 treatment-related deaths were reported in the N+I group.
Conclusion. Both the combination therapy of nivolumab + ipilimumab and nivolumab alone offer superior 3-year overall survival and progression-free survival compared with ipilimumab alone in advanced melanoma, with acceptable toxicity profiles.
Commentary
Historically, unresectable and metastatic melanoma has had a dismal prognosis, with responses to chemotherapy in about 10% to 15% and rarely were these responses durable [2]. The previous standard of care was high-dose IL-2, a form of immunotherapy which leads to long-term survival in a small minority of patients (~15%) [3]. The encouraging results seen in this small minority lead to optimism for efficacy from additional immune-modifying agents.
The novel immunotherapy agents, known as checkpoint inhibitors, are antibodies directed against PD-1 (nivolumab and pembrolizumab), PD-L1 (atezolizumab, avelumab, and urvalumab), and CTLA-4 (ipilimumab). Each of these antigens are critical in a T cell process known as checkpoint inhibition. When these antigens are activated they inhibit T cells, a process critical for self recognition in the healthy human without cancer. However, many malignancies have developed molecular mechanisms to activate these checkpoint pathways and turn off T cell anti-tumor activity. By implementing checkpoint inhibitor antibodies, as done in this study, these drugs allow the T cells to be disinhibited and therefore exert anti-tumor activity. These drugs have been truly ground-breaking and are now FDA-approved in a number of malignancies, including bladder cancer, non–small cell lung cancer, head and neck squamous cell carcinoma, refractory Hodgkin lymphoma, mismatch repair–affected GI adenocarcinomas, renal cell carcinoma, and Merkel cell carcinoma. They offer the additional advantage of often an improved toxicity profile compared with traditional cytotoxic chemotherapy, as they are not typically associated with cytopenias, nausea, or hair loss, for example [4].
In this study, 3-year data from the CheckMate 067 trial is reported. As reported in this study, checkpoint inhibition has lead to truly remarkable improvements in outcomes for patients with advanced melanoma. In this study, the authors have demonstrated superiority of nivolumab plus ipilimumab and nivolumab alone versus ipilimumab alone. These results are similar to those seen in the KEYNOTE-006 trial which compared pemrolizumab (another anti-PD-1 antibody) to ipilimumab. In the KEYNOTE-006 trial, overall survival at 33 months was 50% in the pembrolizumab group versus 39% in the ipilimumab group.
In this study, the combination therapy was more toxic, requiring more frequent treatment discontinuation, though importantly, 3-year overall survival was 67% even among those who discontinued therapy. Grade 3 or 4 toxicity events seem to be associated with efficacy in this study. This is not surprising as this has been seen in some other tumor types as well [5], though it deserves more dedicated investigation as a prognostic marker in this population.
Applications for Clinical Practice
In this well-designed and -executed multicenter randomized trial, funded by Bristol-Myers Squibb and implemented in a selected population with good performance status, all 3 immunotherapies demonstrated impressive improvements in the management of advanced melanoma. The combination nivolumab and ipilimumab was the most effective, with markedly higher survival and response rates, but also with higher toxicity requiring treatment discontinuation, though this did not decrease the efficacy of the therapy. Both the combination nivolumab plus ipilimumab and nivolimab alone are acceptable treatments for patients with advanced melanoma and good performance status; cost and comorbidities will be critical in personalizing therapy.
—Matthew Painschab, MD, University of North Carolina, Chapel Hill, NC
Study Overview
Objective. To compare clinical outcomes and toxicities between combined nivolumab plus ipilimumab (N+I) versus ipilimumab alone (I) or nivolumab alone (N) in patients with advanced melanoma.
Design. Randomized controlled trial 1:1:1 of N+I (nivolumab 1 mg/kg + ipilimumab 3 mg/kg every 3 weeks for 4 doses, followed by nivolumab 3 mg/kg every 2 weeks) versus N (3 mg/kg every 2 weeks) versus I plus placebo (3 mg/kg every 3 weeks for four doses).
Setting and participants. Adult patients with previously untreated stage III (unresectable) or stage IV melanoma and ECOG performance status of 0 or 1 (on a scale of 0–5 with higher score indicating greater disability). Patients with active brain metastases, ocular melanoma, or autoimmune disease were excluded. This study took place in academic and community practices across the United States, Europe, and Australia. 945 patients were randomized. If patients progressed, additional therapies were at clinician discretion.
Main outcome measures. Primary end points were progression-free survival and overall survival. Secondary end points were objective response rate, toxicity profile, and evaluation of PD-L1 (programmed death-ligand 1) as a predictive marker for progression-free survival and overall survival.
Main results. Baseline patient characteristics were published previously [1]. There were no significant differences among groups except that the I only group had a higher frequency of brian metastases (4.8%) vs the N only group (2.5%). At censored follow-up of a minimum of 36 months, median overall survival was not reached in the N+I group, was 37.6 months in the N only group and was 19.9 months in the I only group (hazard ratio [HR] for death 0.55 (P < 0.001) for N+I vs. I and 0.65 (P < 0.001) for N vs. I). Overall survival at 3 years was 58% in the N+I group vs. 52% in the N only group vs. 34% in the I only group. The rate of objective response was 58% in the N+I group vs. 44% in the N only group vs. 19% in the I only group. Progression-free survival at 3 years was 39% in the N+I group, 32% in the N only group and 10% in the I only group. The level of PD-L1 expression was not associated with response or overall survival. Grade 3 or 4 treatment-related adverse events occurred in 59% of the N+I group vs. 21% in N vs. 28% in I group. As therapy after progression was left to clinician discretion, crossover was common with 43% of the I only group receiving nivolumab as second-line therapy and 28% of the N only group receiving ipilimumab as second-line therapy.
Treatment-related events that lead to therapy discontinuation occurred much more frequently in those who received N+I (40%) vs. N (12%) vs. I (16%). However, among the N+I patients who discontinued after a median of 3 cycles of treatment, 67% were still alive at 3 years. In addition, when adverse events were treated with safety guidelines, most immune-mediated adverse events resolved within 3 to 4 weeks. The most common grade 3 or 4 adverse events in the N+I group were diarrhea (9%), elevated lipase (11%), and elevated liver transaminases (9%). A total of 2 treatment-related deaths were reported in the N+I group.
Conclusion. Both the combination therapy of nivolumab + ipilimumab and nivolumab alone offer superior 3-year overall survival and progression-free survival compared with ipilimumab alone in advanced melanoma, with acceptable toxicity profiles.
Commentary
Historically, unresectable and metastatic melanoma has had a dismal prognosis, with responses to chemotherapy in about 10% to 15% and rarely were these responses durable [2]. The previous standard of care was high-dose IL-2, a form of immunotherapy which leads to long-term survival in a small minority of patients (~15%) [3]. The encouraging results seen in this small minority lead to optimism for efficacy from additional immune-modifying agents.
The novel immunotherapy agents, known as checkpoint inhibitors, are antibodies directed against PD-1 (nivolumab and pembrolizumab), PD-L1 (atezolizumab, avelumab, and urvalumab), and CTLA-4 (ipilimumab). Each of these antigens are critical in a T cell process known as checkpoint inhibition. When these antigens are activated they inhibit T cells, a process critical for self recognition in the healthy human without cancer. However, many malignancies have developed molecular mechanisms to activate these checkpoint pathways and turn off T cell anti-tumor activity. By implementing checkpoint inhibitor antibodies, as done in this study, these drugs allow the T cells to be disinhibited and therefore exert anti-tumor activity. These drugs have been truly ground-breaking and are now FDA-approved in a number of malignancies, including bladder cancer, non–small cell lung cancer, head and neck squamous cell carcinoma, refractory Hodgkin lymphoma, mismatch repair–affected GI adenocarcinomas, renal cell carcinoma, and Merkel cell carcinoma. They offer the additional advantage of often an improved toxicity profile compared with traditional cytotoxic chemotherapy, as they are not typically associated with cytopenias, nausea, or hair loss, for example [4].
In this study, 3-year data from the CheckMate 067 trial is reported. As reported in this study, checkpoint inhibition has lead to truly remarkable improvements in outcomes for patients with advanced melanoma. In this study, the authors have demonstrated superiority of nivolumab plus ipilimumab and nivolumab alone versus ipilimumab alone. These results are similar to those seen in the KEYNOTE-006 trial which compared pemrolizumab (another anti-PD-1 antibody) to ipilimumab. In the KEYNOTE-006 trial, overall survival at 33 months was 50% in the pembrolizumab group versus 39% in the ipilimumab group.
In this study, the combination therapy was more toxic, requiring more frequent treatment discontinuation, though importantly, 3-year overall survival was 67% even among those who discontinued therapy. Grade 3 or 4 toxicity events seem to be associated with efficacy in this study. This is not surprising as this has been seen in some other tumor types as well [5], though it deserves more dedicated investigation as a prognostic marker in this population.
Applications for Clinical Practice
In this well-designed and -executed multicenter randomized trial, funded by Bristol-Myers Squibb and implemented in a selected population with good performance status, all 3 immunotherapies demonstrated impressive improvements in the management of advanced melanoma. The combination nivolumab and ipilimumab was the most effective, with markedly higher survival and response rates, but also with higher toxicity requiring treatment discontinuation, though this did not decrease the efficacy of the therapy. Both the combination nivolumab plus ipilimumab and nivolimab alone are acceptable treatments for patients with advanced melanoma and good performance status; cost and comorbidities will be critical in personalizing therapy.
—Matthew Painschab, MD, University of North Carolina, Chapel Hill, NC
1. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
2. Hill GJI, Krementz ET, Hill HZ. Dimethyl triazeno imidazole carboxamide and combination therapy for melanoma. Cancer 1984;53:1299–305.
3. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleuken 2 therapy for patiens with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 1999;17:2105–16.
4. Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 2016;54:139–48.
5. Haratani K, Hayashi H, Chiba Y, et al. Association of immune-related adverse events with nivolumab efficacy in non-small-cell lung cancer. JAMA Oncol 2017 Sept 21.
1. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
2. Hill GJI, Krementz ET, Hill HZ. Dimethyl triazeno imidazole carboxamide and combination therapy for melanoma. Cancer 1984;53:1299–305.
3. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleuken 2 therapy for patiens with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 1999;17:2105–16.
4. Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 2016;54:139–48.
5. Haratani K, Hayashi H, Chiba Y, et al. Association of immune-related adverse events with nivolumab efficacy in non-small-cell lung cancer. JAMA Oncol 2017 Sept 21.
Follow-up of Prostatectomy versus Observation for Early Prostate Cancer
Objective. To determine differences in all-cause and prostate cancer–specific mortality between subgroups of patients who underwent watchful waiting versus radical prostactectomy (RP) for early-stage prostate cancer.
Design. Randomized prospective multicenter trial (PIVOT study).
Setting and participants. Study participants were Department of Veterans Affairs (VA) patients younger than age 75 with biopsy-proven local prostate cancer (T1–T2, M0 by TNM staging and centrally confirmed by pathology laboratory in Baylor) between November 1994 and January 2002. They were patients at NCI medical center–associated VA facilities. Patients had to be eligible for RP and not limited by concomitant medical comorbidities. Patients were excluded if they had undergone therapy for prostate cancer other than transurethral resection of prostate cancer (TURP) for diagnostic purposes including radiation, androgen deprivation theory (ADT), chemotherapy, or definitive surgery. They were also excluded if they had a PSA > 50 ng/mL or a bone scan suggestive of metastatic disease.
Main outcome measures. The primary outcome of the study was all-cause mortality. The secondary outcome was prostate cancer–specific mortality. These were measured from date of diagnosis to August 2014 or until the patient died. A third-party end-points committee blinded to patient arm in the trial determined the cause of death from medical record assessment.
Main results. 731 men with a mean age of 67 were randomly assigned to RP or watchful waiting. The median PSA of patients was 7.8 ng/mL with 75% of patients having a Gleason score ≤ 7 and 74% of patients having low- or intermediate-risk prostate cancer. As of August 2014, 468 of 731 men had died; cause of death was unavailable in 7 patients (2 patients in the surgery arm and 5 in the observation arm). Median duration of follow-up to death or end of follow-up was 12.7 years. All-cause mortality was not significantly different between RP and observation arms (hazard ratio 0.84, 95% confidence interval [CI] 0.7–1.01, P = 0.06). The incidence of death at 19.5 years was 61.3% in patients assigned to surgery versus 66.8% in the watchful waiting arm (relative risk 0.92, 95% CI 0.82–1.02). Deaths from prostate cancer or treatment occurred in 69 patients in the study; 65 from prostate cancer and 4 from treatment. Prostate cancer–associated mortality was not significantly lower in the RP arm than in the watchful waiting arm (hazard ratio 0.63, 95% CI 0.39–1.02, P = 0.06). Mortality was not significantly reduced in any examined subgroup (age > or < 65, white or black ethnicity, PSA > 10 ng/mL or < 10 ng/mL, low/high/intermediate grade, Gleason score). Fewer men who underwent surgery (40.9%) had progression compared to those who underwent observation (68.4%). Most of these patients experienced local progression: 34.1% in the surgery arm and 61.9% in the observation arm. Distant progression was seen in 10.7% of patients treated with RP and 14.2% in the untreated arm. Treatment for progression (local, asymptomatic or by PSA rise) occurred in 59.7% of men assigned to observation and in 33.5% of men assigned to surgery. ADT was more frequently utilized as a treatment modality in men who were initially observed (44.4%) than in men who had up-front surgery (21.7%).
With regard to patient-related outcomes (PROs), more men assigned to RP reported bothersome symptoms such as physical discomfort and limitations in performing activities of daily living (ADLs) at 2 years than in men who did not undergo the intervention. This difference did not persist at later time points beyond 2 years. The use of incontinence pads was markedly higher in surgically treated men than in untreated men. 40% of patients in the treatment arm had to use at least 1 incontinence pad per day within 6 months of RP; this number remained unchanged at 10 years. Rates of erectile dysfunction were reported as lower at 2 (80% versus 45%), 5 (80% versus 55%) and 10 (85% versus 70%) years in men who were watched versus those who underwent surgery. Rates of optimal sexual function were reported as lower in resected men at 1 (35% versus 65%), 5 (38% versus 55%) and 10 (50% versus 70%) years than in men who were watched.
Conclusion. Patients with localized prostate cancer who were randomized to observation rather than RP did not experience greater all-cause mortality or prostate cancer–specific mortality than their surgical counterparts. Furthermore, they experienced less erectile dysfunction, less sexual function impairment, and less incontinence than patients who underwent surgery. Patients who underwent surgery had higher rates of ADL dysfunction and physical discomfort although these differences did not persist beyond 2 years.
Commentary
Nearly 162,000 men will be diagnosed with prostate cancer in 2017, and it is anticipated 27,000 will succumb to their disease [1]. This ratio of incident cases to annual mortality represents one of the lowest ratios amongst all cancer sites and suggests most prostate cancers are indolent. Localized prostate cancer is usually defined by low (Gleason score ≤ 6, PSA < 10 ng/mL and ≤ T2 stage) or intermediate (Gleason score ≤ 7, PSA 10–20 ng/mL, and ≤ T2b stage) risk characteristics. 70% of patients present with low-risk disease, which carries a mortality risk of close to 6% at 15 years [2]. Despite this, nearly 90% of these patients are treated with RP, external beam radiation, or brachytherapy. Some published studies suggest up to 60% of low-risk prostate cancer patients may be overtreated [3,4]. The decision to treat low-risk patients is controversial, as morbidities (eg, sexual dysfunction, erectile dysfunction, incontinence) from a radical prostatectomy or focal radiation therapy are significant while the potential gain may be minimal.
Two other trials in addition to current PIVOT follow-up study have sought to answer the question of whether observation (through either watchful waiting or active surveillance) or treatment (surgery or radiation) is the optimal approach in the management of patients with localized prostate cancer. The SPCG-4 trial [5], which began enrollment in the pre-PSA screening era, included Scandinavian patients with biopsy-proven prostate cancer who were < 75, and had life expectancy > 10 years, ≤ T2 lesions, and PSA < 50 ng/mL. Patients began enrollment in 1989 and were watched for more than 20 years. They were seen in clinic every 6 months for the first 2 years and annually thereafter. The primary outcomes of the trial were death from any cause, death from prostate cancer, or risk of bony and visceral metastases. 447 of 695 included men (200 men in the RP group and 247 men in the watchful waiting group) had died by 2012. The cumulative incidence of death from prostate cancer at the 18-year follow-up point was 17.7% in the surgery arm versus 28.7% in the observation arm. The incidence of distant metastases at the 18-year follow-up point was 26.1% in the radical prostatectomy arm and 38.3% in the watchful waiting group. 67.4% of men assigned to watchful waiting utilized ADT while 42.4% of men treated with prostatectomy utilized ADT palliatively post progression [5].

The ProtecT trial was a United Kingdom study that enrolled 1643 men with prostate cancer aged 50–69 years between 1999 and 2009. The trial randomized men to 3 arms: watchful waiting, RP, or radiation therapy. Patients were eligible for the study if they were < 70 and had ≤ T2 stage disease. 97% of patients had a Gleason score ≤ 7. The primary outcome was prostate cancer–associated mortality at 10 years. Secondary outcomes included death from any cause, rates of distant metastases, and clinical progression. At the end of follow-up, prostate cancer–specific survival was 98.8% in all groups with no significant differences between groups. There was no evidence that differences between prostate cancer–associated mortality varied between groups when stratified by Gleason score, age, PSA, or clinical stage. Additionally, all-cause mortality rates were equivalently distributed across groups [6].
One of the primary reasons why PIVOT and ProtecT may have had different outcomes than the SPCG-4 trial may relate to the aggressiveness of tumors in patients in the various studies. Median PSA levels in the PIVOT and ProtecT trials, respectively, were 7.8 ng/mL and 4.2 ng/mL, compared with 13.2 ng/mL in the SPCG-4 trial. 70% and 77% of patients in PIVOT and ProtecT, respectively, had Gleason score ≤ 6 compared with 57% in the SPCG-4 trial. It is possible that SPCG-4 demonstrated the benefit of RP compared to observation because more patients had higher-risk tumors. Other studies have assessed the economic cost of treatment versus observation in low-risk prostate cancer patients using outcomes such as quality-adjusted life events (QALEs). In a 2013 decision analysis, observation was more effective and less costly than up-front treatment with radiation therapy or RP. Specifically, amongst modes of observation, watchful waiting rather than active surveillance (with every-6-months PSA screening) was more effective and less expensive [7].
Some of the strengths of the PIVOT trial include its prospective randomized design, multicenter patient cohorts, central blinded pathology review, and prolonged follow-up time of nearly 20 years. The trial also had several important limitations. First, the trial included a smaller sample size of patients than the investigators originally intended (2000 patients) and was subsequently underpowered to detect the predetermined outcome of mortality difference between the arms. Second, nearly 20% of patients were not adherent with their treatment arm assignments, which could have potentially confounded the results. Finally, the trial included a patient population that was sicker than the average patient diagnosed in the community with prostate cancer. Trial patients were more likely to succumb to diseases other than prostate cancer and thus may not have been alive long enough to demonstrate a difference between the trial arms (20-year mortality rate was close to 50% in trial patients compared with 30% in the general population post prostatectomy).
Applications for Clinical Practice
The NCCN guidelines suggest that patients with low-risk or intermediate-risk prostate cancer with life expectancies < 10 years should proceed with observation alone. In patients with low-risk disease and life expectancies > 10 years, active surveillance, radiation therapy, or RP are all recommended options. In intermediate-risk patients with life expectancies of > 10 years, treatment with surgery or radiation is warranted. Based on the findings from the PIVOT trial and other trials mentioned above, observation seems to be the most reasonable approach in patients with low-risk prostate cancer. The risks of treatment with RP or radiation outweigh the potential benefits from therapy, particularly in the absence of long-term mortality benefit.
—Satya Das, MD, Vanderbilt Ingram Cancer Center, Nashville, TN
1. SEER. https://seer.cancer.gov/statfacts/html/prost.html.
2. Lu-Yao G, Albertsen P, Moore D, et al. Outcomes of localized prostate cancer following conservative management. JAMA 2009;302:1202–9.
3. Cooperberg M, Broering J, Kantoff P, et al. Contemporary trends in low risk prostate cancer: risk assessment and treatment. J Urol 2007;178(3 Pt 2):S14–9.
4. Welch H, Black W. Overdiagnosis in cancer. J Natl Cancer Inst 2010;102:605–13.
5. Bill-Axelson A, Holmberg L, Garmo H, et al. Radical prostatectomy or watchful waiting in early prostate cancer. N Engl J Med 2014;370:932–42.
6. Hamdy F, Donovan J, Lane J, et al. 10-year outcomes after monitoring, surgery, or radiotherapy for localized prostate cancer. N Engl J Med 2016;375:1415–24.
7. Hayes J, Ollendorf D, Pearson S, et al. Observation versus initial treatment for men with localized, low-risk prostate cancer a cost-effectiveness analysis. Ann Intern Med 2013;158:853–60.
Objective. To determine differences in all-cause and prostate cancer–specific mortality between subgroups of patients who underwent watchful waiting versus radical prostactectomy (RP) for early-stage prostate cancer.
Design. Randomized prospective multicenter trial (PIVOT study).
Setting and participants. Study participants were Department of Veterans Affairs (VA) patients younger than age 75 with biopsy-proven local prostate cancer (T1–T2, M0 by TNM staging and centrally confirmed by pathology laboratory in Baylor) between November 1994 and January 2002. They were patients at NCI medical center–associated VA facilities. Patients had to be eligible for RP and not limited by concomitant medical comorbidities. Patients were excluded if they had undergone therapy for prostate cancer other than transurethral resection of prostate cancer (TURP) for diagnostic purposes including radiation, androgen deprivation theory (ADT), chemotherapy, or definitive surgery. They were also excluded if they had a PSA > 50 ng/mL or a bone scan suggestive of metastatic disease.
Main outcome measures. The primary outcome of the study was all-cause mortality. The secondary outcome was prostate cancer–specific mortality. These were measured from date of diagnosis to August 2014 or until the patient died. A third-party end-points committee blinded to patient arm in the trial determined the cause of death from medical record assessment.
Main results. 731 men with a mean age of 67 were randomly assigned to RP or watchful waiting. The median PSA of patients was 7.8 ng/mL with 75% of patients having a Gleason score ≤ 7 and 74% of patients having low- or intermediate-risk prostate cancer. As of August 2014, 468 of 731 men had died; cause of death was unavailable in 7 patients (2 patients in the surgery arm and 5 in the observation arm). Median duration of follow-up to death or end of follow-up was 12.7 years. All-cause mortality was not significantly different between RP and observation arms (hazard ratio 0.84, 95% confidence interval [CI] 0.7–1.01, P = 0.06). The incidence of death at 19.5 years was 61.3% in patients assigned to surgery versus 66.8% in the watchful waiting arm (relative risk 0.92, 95% CI 0.82–1.02). Deaths from prostate cancer or treatment occurred in 69 patients in the study; 65 from prostate cancer and 4 from treatment. Prostate cancer–associated mortality was not significantly lower in the RP arm than in the watchful waiting arm (hazard ratio 0.63, 95% CI 0.39–1.02, P = 0.06). Mortality was not significantly reduced in any examined subgroup (age > or < 65, white or black ethnicity, PSA > 10 ng/mL or < 10 ng/mL, low/high/intermediate grade, Gleason score). Fewer men who underwent surgery (40.9%) had progression compared to those who underwent observation (68.4%). Most of these patients experienced local progression: 34.1% in the surgery arm and 61.9% in the observation arm. Distant progression was seen in 10.7% of patients treated with RP and 14.2% in the untreated arm. Treatment for progression (local, asymptomatic or by PSA rise) occurred in 59.7% of men assigned to observation and in 33.5% of men assigned to surgery. ADT was more frequently utilized as a treatment modality in men who were initially observed (44.4%) than in men who had up-front surgery (21.7%).
With regard to patient-related outcomes (PROs), more men assigned to RP reported bothersome symptoms such as physical discomfort and limitations in performing activities of daily living (ADLs) at 2 years than in men who did not undergo the intervention. This difference did not persist at later time points beyond 2 years. The use of incontinence pads was markedly higher in surgically treated men than in untreated men. 40% of patients in the treatment arm had to use at least 1 incontinence pad per day within 6 months of RP; this number remained unchanged at 10 years. Rates of erectile dysfunction were reported as lower at 2 (80% versus 45%), 5 (80% versus 55%) and 10 (85% versus 70%) years in men who were watched versus those who underwent surgery. Rates of optimal sexual function were reported as lower in resected men at 1 (35% versus 65%), 5 (38% versus 55%) and 10 (50% versus 70%) years than in men who were watched.
Conclusion. Patients with localized prostate cancer who were randomized to observation rather than RP did not experience greater all-cause mortality or prostate cancer–specific mortality than their surgical counterparts. Furthermore, they experienced less erectile dysfunction, less sexual function impairment, and less incontinence than patients who underwent surgery. Patients who underwent surgery had higher rates of ADL dysfunction and physical discomfort although these differences did not persist beyond 2 years.
Commentary
Nearly 162,000 men will be diagnosed with prostate cancer in 2017, and it is anticipated 27,000 will succumb to their disease [1]. This ratio of incident cases to annual mortality represents one of the lowest ratios amongst all cancer sites and suggests most prostate cancers are indolent. Localized prostate cancer is usually defined by low (Gleason score ≤ 6, PSA < 10 ng/mL and ≤ T2 stage) or intermediate (Gleason score ≤ 7, PSA 10–20 ng/mL, and ≤ T2b stage) risk characteristics. 70% of patients present with low-risk disease, which carries a mortality risk of close to 6% at 15 years [2]. Despite this, nearly 90% of these patients are treated with RP, external beam radiation, or brachytherapy. Some published studies suggest up to 60% of low-risk prostate cancer patients may be overtreated [3,4]. The decision to treat low-risk patients is controversial, as morbidities (eg, sexual dysfunction, erectile dysfunction, incontinence) from a radical prostatectomy or focal radiation therapy are significant while the potential gain may be minimal.
Two other trials in addition to current PIVOT follow-up study have sought to answer the question of whether observation (through either watchful waiting or active surveillance) or treatment (surgery or radiation) is the optimal approach in the management of patients with localized prostate cancer. The SPCG-4 trial [5], which began enrollment in the pre-PSA screening era, included Scandinavian patients with biopsy-proven prostate cancer who were < 75, and had life expectancy > 10 years, ≤ T2 lesions, and PSA < 50 ng/mL. Patients began enrollment in 1989 and were watched for more than 20 years. They were seen in clinic every 6 months for the first 2 years and annually thereafter. The primary outcomes of the trial were death from any cause, death from prostate cancer, or risk of bony and visceral metastases. 447 of 695 included men (200 men in the RP group and 247 men in the watchful waiting group) had died by 2012. The cumulative incidence of death from prostate cancer at the 18-year follow-up point was 17.7% in the surgery arm versus 28.7% in the observation arm. The incidence of distant metastases at the 18-year follow-up point was 26.1% in the radical prostatectomy arm and 38.3% in the watchful waiting group. 67.4% of men assigned to watchful waiting utilized ADT while 42.4% of men treated with prostatectomy utilized ADT palliatively post progression [5].
The ProtecT trial was a United Kingdom study that enrolled 1643 men with prostate cancer aged 50–69 years between 1999 and 2009. The trial randomized men to 3 arms: watchful waiting, RP, or radiation therapy. Patients were eligible for the study if they were < 70 and had ≤ T2 stage disease. 97% of patients had a Gleason score ≤ 7. The primary outcome was prostate cancer–associated mortality at 10 years. Secondary outcomes included death from any cause, rates of distant metastases, and clinical progression. At the end of follow-up, prostate cancer–specific survival was 98.8% in all groups with no significant differences between groups. There was no evidence that differences between prostate cancer–associated mortality varied between groups when stratified by Gleason score, age, PSA, or clinical stage. Additionally, all-cause mortality rates were equivalently distributed across groups [6].
One of the primary reasons why PIVOT and ProtecT may have had different outcomes than the SPCG-4 trial may relate to the aggressiveness of tumors in patients in the various studies. Median PSA levels in the PIVOT and ProtecT trials, respectively, were 7.8 ng/mL and 4.2 ng/mL, compared with 13.2 ng/mL in the SPCG-4 trial. 70% and 77% of patients in PIVOT and ProtecT, respectively, had Gleason score ≤ 6 compared with 57% in the SPCG-4 trial. It is possible that SPCG-4 demonstrated the benefit of RP compared to observation because more patients had higher-risk tumors. Other studies have assessed the economic cost of treatment versus observation in low-risk prostate cancer patients using outcomes such as quality-adjusted life events (QALEs). In a 2013 decision analysis, observation was more effective and less costly than up-front treatment with radiation therapy or RP. Specifically, amongst modes of observation, watchful waiting rather than active surveillance (with every-6-months PSA screening) was more effective and less expensive [7].
Some of the strengths of the PIVOT trial include its prospective randomized design, multicenter patient cohorts, central blinded pathology review, and prolonged follow-up time of nearly 20 years. The trial also had several important limitations. First, the trial included a smaller sample size of patients than the investigators originally intended (2000 patients) and was subsequently underpowered to detect the predetermined outcome of mortality difference between the arms. Second, nearly 20% of patients were not adherent with their treatment arm assignments, which could have potentially confounded the results. Finally, the trial included a patient population that was sicker than the average patient diagnosed in the community with prostate cancer. Trial patients were more likely to succumb to diseases other than prostate cancer and thus may not have been alive long enough to demonstrate a difference between the trial arms (20-year mortality rate was close to 50% in trial patients compared with 30% in the general population post prostatectomy).
Applications for Clinical Practice
The NCCN guidelines suggest that patients with low-risk or intermediate-risk prostate cancer with life expectancies < 10 years should proceed with observation alone. In patients with low-risk disease and life expectancies > 10 years, active surveillance, radiation therapy, or RP are all recommended options. In intermediate-risk patients with life expectancies of > 10 years, treatment with surgery or radiation is warranted. Based on the findings from the PIVOT trial and other trials mentioned above, observation seems to be the most reasonable approach in patients with low-risk prostate cancer. The risks of treatment with RP or radiation outweigh the potential benefits from therapy, particularly in the absence of long-term mortality benefit.
—Satya Das, MD, Vanderbilt Ingram Cancer Center, Nashville, TN
Objective. To determine differences in all-cause and prostate cancer–specific mortality between subgroups of patients who underwent watchful waiting versus radical prostactectomy (RP) for early-stage prostate cancer.
Design. Randomized prospective multicenter trial (PIVOT study).
Setting and participants. Study participants were Department of Veterans Affairs (VA) patients younger than age 75 with biopsy-proven local prostate cancer (T1–T2, M0 by TNM staging and centrally confirmed by pathology laboratory in Baylor) between November 1994 and January 2002. They were patients at NCI medical center–associated VA facilities. Patients had to be eligible for RP and not limited by concomitant medical comorbidities. Patients were excluded if they had undergone therapy for prostate cancer other than transurethral resection of prostate cancer (TURP) for diagnostic purposes including radiation, androgen deprivation theory (ADT), chemotherapy, or definitive surgery. They were also excluded if they had a PSA > 50 ng/mL or a bone scan suggestive of metastatic disease.
Main outcome measures. The primary outcome of the study was all-cause mortality. The secondary outcome was prostate cancer–specific mortality. These were measured from date of diagnosis to August 2014 or until the patient died. A third-party end-points committee blinded to patient arm in the trial determined the cause of death from medical record assessment.
Main results. 731 men with a mean age of 67 were randomly assigned to RP or watchful waiting. The median PSA of patients was 7.8 ng/mL with 75% of patients having a Gleason score ≤ 7 and 74% of patients having low- or intermediate-risk prostate cancer. As of August 2014, 468 of 731 men had died; cause of death was unavailable in 7 patients (2 patients in the surgery arm and 5 in the observation arm). Median duration of follow-up to death or end of follow-up was 12.7 years. All-cause mortality was not significantly different between RP and observation arms (hazard ratio 0.84, 95% confidence interval [CI] 0.7–1.01, P = 0.06). The incidence of death at 19.5 years was 61.3% in patients assigned to surgery versus 66.8% in the watchful waiting arm (relative risk 0.92, 95% CI 0.82–1.02). Deaths from prostate cancer or treatment occurred in 69 patients in the study; 65 from prostate cancer and 4 from treatment. Prostate cancer–associated mortality was not significantly lower in the RP arm than in the watchful waiting arm (hazard ratio 0.63, 95% CI 0.39–1.02, P = 0.06). Mortality was not significantly reduced in any examined subgroup (age > or < 65, white or black ethnicity, PSA > 10 ng/mL or < 10 ng/mL, low/high/intermediate grade, Gleason score). Fewer men who underwent surgery (40.9%) had progression compared to those who underwent observation (68.4%). Most of these patients experienced local progression: 34.1% in the surgery arm and 61.9% in the observation arm. Distant progression was seen in 10.7% of patients treated with RP and 14.2% in the untreated arm. Treatment for progression (local, asymptomatic or by PSA rise) occurred in 59.7% of men assigned to observation and in 33.5% of men assigned to surgery. ADT was more frequently utilized as a treatment modality in men who were initially observed (44.4%) than in men who had up-front surgery (21.7%).
With regard to patient-related outcomes (PROs), more men assigned to RP reported bothersome symptoms such as physical discomfort and limitations in performing activities of daily living (ADLs) at 2 years than in men who did not undergo the intervention. This difference did not persist at later time points beyond 2 years. The use of incontinence pads was markedly higher in surgically treated men than in untreated men. 40% of patients in the treatment arm had to use at least 1 incontinence pad per day within 6 months of RP; this number remained unchanged at 10 years. Rates of erectile dysfunction were reported as lower at 2 (80% versus 45%), 5 (80% versus 55%) and 10 (85% versus 70%) years in men who were watched versus those who underwent surgery. Rates of optimal sexual function were reported as lower in resected men at 1 (35% versus 65%), 5 (38% versus 55%) and 10 (50% versus 70%) years than in men who were watched.
Conclusion. Patients with localized prostate cancer who were randomized to observation rather than RP did not experience greater all-cause mortality or prostate cancer–specific mortality than their surgical counterparts. Furthermore, they experienced less erectile dysfunction, less sexual function impairment, and less incontinence than patients who underwent surgery. Patients who underwent surgery had higher rates of ADL dysfunction and physical discomfort although these differences did not persist beyond 2 years.
Commentary
Nearly 162,000 men will be diagnosed with prostate cancer in 2017, and it is anticipated 27,000 will succumb to their disease [1]. This ratio of incident cases to annual mortality represents one of the lowest ratios amongst all cancer sites and suggests most prostate cancers are indolent. Localized prostate cancer is usually defined by low (Gleason score ≤ 6, PSA < 10 ng/mL and ≤ T2 stage) or intermediate (Gleason score ≤ 7, PSA 10–20 ng/mL, and ≤ T2b stage) risk characteristics. 70% of patients present with low-risk disease, which carries a mortality risk of close to 6% at 15 years [2]. Despite this, nearly 90% of these patients are treated with RP, external beam radiation, or brachytherapy. Some published studies suggest up to 60% of low-risk prostate cancer patients may be overtreated [3,4]. The decision to treat low-risk patients is controversial, as morbidities (eg, sexual dysfunction, erectile dysfunction, incontinence) from a radical prostatectomy or focal radiation therapy are significant while the potential gain may be minimal.
Two other trials in addition to current PIVOT follow-up study have sought to answer the question of whether observation (through either watchful waiting or active surveillance) or treatment (surgery or radiation) is the optimal approach in the management of patients with localized prostate cancer. The SPCG-4 trial [5], which began enrollment in the pre-PSA screening era, included Scandinavian patients with biopsy-proven prostate cancer who were < 75, and had life expectancy > 10 years, ≤ T2 lesions, and PSA < 50 ng/mL. Patients began enrollment in 1989 and were watched for more than 20 years. They were seen in clinic every 6 months for the first 2 years and annually thereafter. The primary outcomes of the trial were death from any cause, death from prostate cancer, or risk of bony and visceral metastases. 447 of 695 included men (200 men in the RP group and 247 men in the watchful waiting group) had died by 2012. The cumulative incidence of death from prostate cancer at the 18-year follow-up point was 17.7% in the surgery arm versus 28.7% in the observation arm. The incidence of distant metastases at the 18-year follow-up point was 26.1% in the radical prostatectomy arm and 38.3% in the watchful waiting group. 67.4% of men assigned to watchful waiting utilized ADT while 42.4% of men treated with prostatectomy utilized ADT palliatively post progression [5].
The ProtecT trial was a United Kingdom study that enrolled 1643 men with prostate cancer aged 50–69 years between 1999 and 2009. The trial randomized men to 3 arms: watchful waiting, RP, or radiation therapy. Patients were eligible for the study if they were < 70 and had ≤ T2 stage disease. 97% of patients had a Gleason score ≤ 7. The primary outcome was prostate cancer–associated mortality at 10 years. Secondary outcomes included death from any cause, rates of distant metastases, and clinical progression. At the end of follow-up, prostate cancer–specific survival was 98.8% in all groups with no significant differences between groups. There was no evidence that differences between prostate cancer–associated mortality varied between groups when stratified by Gleason score, age, PSA, or clinical stage. Additionally, all-cause mortality rates were equivalently distributed across groups [6].
One of the primary reasons why PIVOT and ProtecT may have had different outcomes than the SPCG-4 trial may relate to the aggressiveness of tumors in patients in the various studies. Median PSA levels in the PIVOT and ProtecT trials, respectively, were 7.8 ng/mL and 4.2 ng/mL, compared with 13.2 ng/mL in the SPCG-4 trial. 70% and 77% of patients in PIVOT and ProtecT, respectively, had Gleason score ≤ 6 compared with 57% in the SPCG-4 trial. It is possible that SPCG-4 demonstrated the benefit of RP compared to observation because more patients had higher-risk tumors. Other studies have assessed the economic cost of treatment versus observation in low-risk prostate cancer patients using outcomes such as quality-adjusted life events (QALEs). In a 2013 decision analysis, observation was more effective and less costly than up-front treatment with radiation therapy or RP. Specifically, amongst modes of observation, watchful waiting rather than active surveillance (with every-6-months PSA screening) was more effective and less expensive [7].
Some of the strengths of the PIVOT trial include its prospective randomized design, multicenter patient cohorts, central blinded pathology review, and prolonged follow-up time of nearly 20 years. The trial also had several important limitations. First, the trial included a smaller sample size of patients than the investigators originally intended (2000 patients) and was subsequently underpowered to detect the predetermined outcome of mortality difference between the arms. Second, nearly 20% of patients were not adherent with their treatment arm assignments, which could have potentially confounded the results. Finally, the trial included a patient population that was sicker than the average patient diagnosed in the community with prostate cancer. Trial patients were more likely to succumb to diseases other than prostate cancer and thus may not have been alive long enough to demonstrate a difference between the trial arms (20-year mortality rate was close to 50% in trial patients compared with 30% in the general population post prostatectomy).
Applications for Clinical Practice
The NCCN guidelines suggest that patients with low-risk or intermediate-risk prostate cancer with life expectancies < 10 years should proceed with observation alone. In patients with low-risk disease and life expectancies > 10 years, active surveillance, radiation therapy, or RP are all recommended options. In intermediate-risk patients with life expectancies of > 10 years, treatment with surgery or radiation is warranted. Based on the findings from the PIVOT trial and other trials mentioned above, observation seems to be the most reasonable approach in patients with low-risk prostate cancer. The risks of treatment with RP or radiation outweigh the potential benefits from therapy, particularly in the absence of long-term mortality benefit.
—Satya Das, MD, Vanderbilt Ingram Cancer Center, Nashville, TN
1. SEER. https://seer.cancer.gov/statfacts/html/prost.html.
2. Lu-Yao G, Albertsen P, Moore D, et al. Outcomes of localized prostate cancer following conservative management. JAMA 2009;302:1202–9.
3. Cooperberg M, Broering J, Kantoff P, et al. Contemporary trends in low risk prostate cancer: risk assessment and treatment. J Urol 2007;178(3 Pt 2):S14–9.
4. Welch H, Black W. Overdiagnosis in cancer. J Natl Cancer Inst 2010;102:605–13.
5. Bill-Axelson A, Holmberg L, Garmo H, et al. Radical prostatectomy or watchful waiting in early prostate cancer. N Engl J Med 2014;370:932–42.
6. Hamdy F, Donovan J, Lane J, et al. 10-year outcomes after monitoring, surgery, or radiotherapy for localized prostate cancer. N Engl J Med 2016;375:1415–24.
7. Hayes J, Ollendorf D, Pearson S, et al. Observation versus initial treatment for men with localized, low-risk prostate cancer a cost-effectiveness analysis. Ann Intern Med 2013;158:853–60.
1. SEER. https://seer.cancer.gov/statfacts/html/prost.html.
2. Lu-Yao G, Albertsen P, Moore D, et al. Outcomes of localized prostate cancer following conservative management. JAMA 2009;302:1202–9.
3. Cooperberg M, Broering J, Kantoff P, et al. Contemporary trends in low risk prostate cancer: risk assessment and treatment. J Urol 2007;178(3 Pt 2):S14–9.
4. Welch H, Black W. Overdiagnosis in cancer. J Natl Cancer Inst 2010;102:605–13.
5. Bill-Axelson A, Holmberg L, Garmo H, et al. Radical prostatectomy or watchful waiting in early prostate cancer. N Engl J Med 2014;370:932–42.
6. Hamdy F, Donovan J, Lane J, et al. 10-year outcomes after monitoring, surgery, or radiotherapy for localized prostate cancer. N Engl J Med 2016;375:1415–24.
7. Hayes J, Ollendorf D, Pearson S, et al. Observation versus initial treatment for men with localized, low-risk prostate cancer a cost-effectiveness analysis. Ann Intern Med 2013;158:853–60.
Session Spotlights Infection in Aneurysms, Grafts, and Endografts
The ongoing discussion over the optimal management of infection in aneurysms and grafts takes center stage in the session, “New Developments in the Treatment of Infected Aneurysms, Prosthetic Arterial Grafts, and Aortic Endografts,” on Friday morning. The session includes two debates: one on mycotic abdominal aortic aneurysms and what to do about them, and the other on the optimal techniques for handling infected aortic grafts and endografts.
“The management of infected aortic grafts is challenging and controversial,” according to Dr. Keith Calligaro, co-moderator of the session. Different aspects of treatment will be discussed, said Dr. Calligaro, chief of the section of vascular surgery and endovascular therapy at Pennsylvania Hospital, and clinical professor of surgery, University of Pennsylvania School of Medicine.

The session begins with a presentation suggesting a change in practice, “With Mycotic AAAs There Has Been a Paradigm Shift in Treatment: A Propensity Matched Multicenter Study Shows That EVAR Is Better than Open Repair as a Durable or Bridge Treatment,” by Dr. Anders Wanhainen, professor of surgery at Uppsala University. Dr. Wanhainen is followed by Dr. Manju Kalra, professor, Mayo Clinic College of Medicine, speaking on “Intraabdominal Extra-Anatomic Bypass for Para- Or Supra-Renal Aortic Infections: Techniques and Results.” Dr. Fred A. Weaver, professor of surgery, Keck School of Medicine at the University of Southern California, then delves into the role of endovascular aortic aneurysm repair (EVAR) for mycotic AAAs.
Next, then the session gears up for a debate, with Dr. Boonprasit Kritpracha, instructor and vascular surgeon, Prince of Songkla University in Thailand, taking the side of “EVAR Should Be the First Choice in Treating Mycotic AAAs: Based on a 10-Year Experience.” Dr. Kritpracha is followed by session co-moderator Dr. Thomas C. Bower, professor of surgery, Mayo Clinic College of Medicine and Science, who takes the view, “Not So: Why Open Repair Should Be the First Choice in Treating Most Mycotic AAAs.”
A talk on the neoaortoiliac system (NAIS) procedure for the treatment of the infected aortic graft, “Technical Tips for Facilitating Deep Vein Grafts for Aortoiliac Arterial and Graft Infections,” by Dr. James H. Black, III, The David Goldfarb, MD Associate Professor of Surgery, The Johns Hopkins University School of Medicine, completes the first half of the session.
The next part of the session focuses on arterial graft and endograft infections. Dr. Max Zegelman, professor of surgery at JWG-University Frankfurt, begins with a review of new techniques for the in situ repair of infected prosthetic arterial grafts and the impact of negative pressure wound therapy.
The presentations are followed by a second debate on the topic of removal vs. saving of infected aortic grafts and endografts. Dr. Colin D. Bicknell, clinical senior lecturer, Imperial College, takes the side of “Definitive Excisional Graft Removal Is a Must for All Infected Aortic Grafts and Endografts,” while co-moderator Dr. Calligaro takes the side of “Not So: More Conservative Graft Saving May Sometimes Be the Best Treatment for infected Aortic Grafts and Endografts if Certain Technical Steps and Adjuncts Are Used.”
“The take-home message is that, in general, total graft excision of infected intracavitary prosthetic and endovascular aortic grafts is recommended, but the surgeon needs to be aware that in certain cases, partial or complete graft preservation may be a better option,” Dr. Calligaro said.
The session continues with more on the topic of treating infected endografts. Dr. Kamphol Laohapensang, professor of vascular surgery, Chiang Mai University Hospital in Thailand, will focus on treating infected endografts after EVAR and under what circumstances endografts are effective for treating mycotic AAAs.
The ongoing discussion over the optimal management of infection in aneurysms and grafts takes center stage in the session, “New Developments in the Treatment of Infected Aneurysms, Prosthetic Arterial Grafts, and Aortic Endografts,” on Friday morning. The session includes two debates: one on mycotic abdominal aortic aneurysms and what to do about them, and the other on the optimal techniques for handling infected aortic grafts and endografts.
“The management of infected aortic grafts is challenging and controversial,” according to Dr. Keith Calligaro, co-moderator of the session. Different aspects of treatment will be discussed, said Dr. Calligaro, chief of the section of vascular surgery and endovascular therapy at Pennsylvania Hospital, and clinical professor of surgery, University of Pennsylvania School of Medicine.
The session begins with a presentation suggesting a change in practice, “With Mycotic AAAs There Has Been a Paradigm Shift in Treatment: A Propensity Matched Multicenter Study Shows That EVAR Is Better than Open Repair as a Durable or Bridge Treatment,” by Dr. Anders Wanhainen, professor of surgery at Uppsala University. Dr. Wanhainen is followed by Dr. Manju Kalra, professor, Mayo Clinic College of Medicine, speaking on “Intraabdominal Extra-Anatomic Bypass for Para- Or Supra-Renal Aortic Infections: Techniques and Results.” Dr. Fred A. Weaver, professor of surgery, Keck School of Medicine at the University of Southern California, then delves into the role of endovascular aortic aneurysm repair (EVAR) for mycotic AAAs.
Next, then the session gears up for a debate, with Dr. Boonprasit Kritpracha, instructor and vascular surgeon, Prince of Songkla University in Thailand, taking the side of “EVAR Should Be the First Choice in Treating Mycotic AAAs: Based on a 10-Year Experience.” Dr. Kritpracha is followed by session co-moderator Dr. Thomas C. Bower, professor of surgery, Mayo Clinic College of Medicine and Science, who takes the view, “Not So: Why Open Repair Should Be the First Choice in Treating Most Mycotic AAAs.”
A talk on the neoaortoiliac system (NAIS) procedure for the treatment of the infected aortic graft, “Technical Tips for Facilitating Deep Vein Grafts for Aortoiliac Arterial and Graft Infections,” by Dr. James H. Black, III, The David Goldfarb, MD Associate Professor of Surgery, The Johns Hopkins University School of Medicine, completes the first half of the session.
The next part of the session focuses on arterial graft and endograft infections. Dr. Max Zegelman, professor of surgery at JWG-University Frankfurt, begins with a review of new techniques for the in situ repair of infected prosthetic arterial grafts and the impact of negative pressure wound therapy.
The presentations are followed by a second debate on the topic of removal vs. saving of infected aortic grafts and endografts. Dr. Colin D. Bicknell, clinical senior lecturer, Imperial College, takes the side of “Definitive Excisional Graft Removal Is a Must for All Infected Aortic Grafts and Endografts,” while co-moderator Dr. Calligaro takes the side of “Not So: More Conservative Graft Saving May Sometimes Be the Best Treatment for infected Aortic Grafts and Endografts if Certain Technical Steps and Adjuncts Are Used.”
“The take-home message is that, in general, total graft excision of infected intracavitary prosthetic and endovascular aortic grafts is recommended, but the surgeon needs to be aware that in certain cases, partial or complete graft preservation may be a better option,” Dr. Calligaro said.
The session continues with more on the topic of treating infected endografts. Dr. Kamphol Laohapensang, professor of vascular surgery, Chiang Mai University Hospital in Thailand, will focus on treating infected endografts after EVAR and under what circumstances endografts are effective for treating mycotic AAAs.
The ongoing discussion over the optimal management of infection in aneurysms and grafts takes center stage in the session, “New Developments in the Treatment of Infected Aneurysms, Prosthetic Arterial Grafts, and Aortic Endografts,” on Friday morning. The session includes two debates: one on mycotic abdominal aortic aneurysms and what to do about them, and the other on the optimal techniques for handling infected aortic grafts and endografts.
“The management of infected aortic grafts is challenging and controversial,” according to Dr. Keith Calligaro, co-moderator of the session. Different aspects of treatment will be discussed, said Dr. Calligaro, chief of the section of vascular surgery and endovascular therapy at Pennsylvania Hospital, and clinical professor of surgery, University of Pennsylvania School of Medicine.
The session begins with a presentation suggesting a change in practice, “With Mycotic AAAs There Has Been a Paradigm Shift in Treatment: A Propensity Matched Multicenter Study Shows That EVAR Is Better than Open Repair as a Durable or Bridge Treatment,” by Dr. Anders Wanhainen, professor of surgery at Uppsala University. Dr. Wanhainen is followed by Dr. Manju Kalra, professor, Mayo Clinic College of Medicine, speaking on “Intraabdominal Extra-Anatomic Bypass for Para- Or Supra-Renal Aortic Infections: Techniques and Results.” Dr. Fred A. Weaver, professor of surgery, Keck School of Medicine at the University of Southern California, then delves into the role of endovascular aortic aneurysm repair (EVAR) for mycotic AAAs.
Next, then the session gears up for a debate, with Dr. Boonprasit Kritpracha, instructor and vascular surgeon, Prince of Songkla University in Thailand, taking the side of “EVAR Should Be the First Choice in Treating Mycotic AAAs: Based on a 10-Year Experience.” Dr. Kritpracha is followed by session co-moderator Dr. Thomas C. Bower, professor of surgery, Mayo Clinic College of Medicine and Science, who takes the view, “Not So: Why Open Repair Should Be the First Choice in Treating Most Mycotic AAAs.”
A talk on the neoaortoiliac system (NAIS) procedure for the treatment of the infected aortic graft, “Technical Tips for Facilitating Deep Vein Grafts for Aortoiliac Arterial and Graft Infections,” by Dr. James H. Black, III, The David Goldfarb, MD Associate Professor of Surgery, The Johns Hopkins University School of Medicine, completes the first half of the session.
The next part of the session focuses on arterial graft and endograft infections. Dr. Max Zegelman, professor of surgery at JWG-University Frankfurt, begins with a review of new techniques for the in situ repair of infected prosthetic arterial grafts and the impact of negative pressure wound therapy.
The presentations are followed by a second debate on the topic of removal vs. saving of infected aortic grafts and endografts. Dr. Colin D. Bicknell, clinical senior lecturer, Imperial College, takes the side of “Definitive Excisional Graft Removal Is a Must for All Infected Aortic Grafts and Endografts,” while co-moderator Dr. Calligaro takes the side of “Not So: More Conservative Graft Saving May Sometimes Be the Best Treatment for infected Aortic Grafts and Endografts if Certain Technical Steps and Adjuncts Are Used.”
“The take-home message is that, in general, total graft excision of infected intracavitary prosthetic and endovascular aortic grafts is recommended, but the surgeon needs to be aware that in certain cases, partial or complete graft preservation may be a better option,” Dr. Calligaro said.
The session continues with more on the topic of treating infected endografts. Dr. Kamphol Laohapensang, professor of vascular surgery, Chiang Mai University Hospital in Thailand, will focus on treating infected endografts after EVAR and under what circumstances endografts are effective for treating mycotic AAAs.
Survival Outcomes in Stage IV Differentiated Thyroid Cancer After Postsurgical RAI versus EBRT
Study Overview
Objective. To evaluate survival trends and differences in a large cohort of patients with stage IV differentiated thyroid cancer treated with radioactive iodine (RAI), external beam radiation therapy (EBRT), or no radiation following surgery.
Design. Multicenter retrospective cohort study using data from the National Cancer Database (NCDB) from 2002–2012.
Setting and participants. The study group consisted of a random sample of all inpatient discharges with a diagnosis of differentiated thyroid cancer (DTC). This yielded a cohort of 11,832 patients with stage IV DTC who underwent primary surgical treatment with thyroidecromy. Patients were stratified by cancer histology into follicular thyroid cancer (FTC) and papillary thyroid cancer (PTC). Patients were additionally stratified into 3 substage groups: IV-A, IV-B, and IV-C. Administrative censoring was implemented at 5 and 10 year marks of survival time.
Main outcome measures. The primary outcome was all-cause mortality. Survival was analyzed at 5 and 10 years. Multivariate analysis was performed on a number of covariates including age, sex, race, socioeconomic status, TNM stage, tumor grade, surgical length of stay, and surgical treatment variables such as neck dissection and lymph node surgery.
Main results. Most patients (91.24%) had PTC and 8.76% had FTC. The average age of patients in the RAI group was younger (FTC, age 66; PTC, age 58) than patients in the EBRT (FTC, age 69; PTC, age 65) or no RT groups. (FTC, age 73; PTC, age 61). In contrast to FTC patients, a large majority of PTC patients underwent surgical neck dissection. There were no significant differences in sex, ethnicity, primary payer, median income quartile, or education level among the 3 groups for patients with FTC. However, in PTC there was a majority of female and ethnically white/Caucasian patients in all 3 groups. In addition, patients with PTC who did not receive RT or received RAI were more likely to have private insurance versus those who underwent EBRT, who were more often covered under Medicare. These differences in primary payer were statistically significant (P < 0.001).
Statistically significant differences in mortality were observed at 5 and 10 years in both papillary and follicular thyroid cancer among the 3 groups. In the PTC groups, patients treated with EBRT had the highest mortality rates (46.6% at 5 years, 50.7% at 10 years), while patients with PTC receiving no RT had lower mortality rates (22.7% at 5 years, 25.5% at 10 years), and PTC patients receiving RAI had the lowest mortality rates (11.0% at 5 years, 14.0% at 10 years). Similar results were seen in patients with FTC, in which patients treated with EBRT had the highest mortality rates (51.4% at 5 years, 59.9% at 10 years), while patient with FTC receiving no RT had lower mortality rates (45.5% at 5 years, 51% at 10 years), and FTC patients receiving RAI had the lowest mortality rates (29.2% at 5 years, 36.8% at 10 years).
Using univariate analysis, EBRT showed a statistically significant increase in 5- and 10-year mortality for patients with PTC stage IV-A and IV-B as compared with no radiation. This was demonstrated in both stage IV-A and IV-B subgroups at 5 years (EBRT 5-year HR PTC stage IV-A = 2.04, 95% confidence interval [CI] 1.74–2.39, P < 0.001; EBRT 5-year HR PTC stage IV-B = 2.23, 95% CI 1.42–3.51, P < 0.001; and 10 years [EBRT 10-year HR PTC stage IV-A = 2.12, 95% CI 1.79-2.52 P < 0.001; EBRT 10-year HR PTC stage IV-B = 2.03, 95% CI 1.33-3.10, P < 0.001). RAI showed a statistically significant decrease in 5- and 10-year mortality in both PTC and FTC compared with no radiation, regardless of pathologic sub-stage. The largest reduction in risk was seen in FTC stage IV-B patients at 5 years [RAI 5 year HR FTC stage IV-B = 0.31, 95% CI 0.12-0.80, P < 0.05). Multivariate analysis was also performed and showed similar results to univariate analysis except that there was no longer a statistically significant difference in EBRT versus no RT in stage IV-A PTC at 5 and 10 years (EBRT 5-year HR PTC stage IV-A = 1.2, 95% CI 0.91–1.59, EBRT 10-year HR PTC stage IV-A = 1.29, 95% CI 0.93–1.79). Reductions in death hazard seen in all groups treated with RAI versus no RT previously observed in univariate analysis remained statistically significant in all groups on multivariate analysis.
Multivariate analysis revealed a number of significant covariates. Increase in age was noted to be associated with higher death hazard in all groups except FTC stage IV-B and stage IV-C. Every additional year of age increased the hazard of death by ~2% to 5%, up to a maximum of 9% per year. Females overall had a lower hazard of death compared with their male counterparts, most notably in PTC. African-American patients had improved survival in FTC (5 years) but lower survival in PTC (5 and 10 years) as compared with white patients. Tumor grade showed a dose response in models studied, with increasing death hazards with worsening tumor differentiation.
Conclusion. RAI was associated with improved survival in patients with stage IV DTC, while EBRT was associated with poorer survival outcomes.
Commentary
Radioiodine therapy has been used for treatment of DTC since the 1940s. Radioactive iodine (I-131) is largely taken up by thyroid follicular cells via their sodium-iodide transporter causing acute thyroid cell death by emission of short path length beta particles [1].
External beam radiation therapy (EBRT) is the most common radiation therapy approach to deliver radiation from a source outside of the patient. EBRT machines produce radiation by either radioactive decay of a nuclide or by acceleration of charged particles such as electrons or protons. Using a linear accelerator, charged particles are accelerated to a high enough energy to allow transmission of particles as an electron beam or x-ray, which is subsequently directed at the tumor [2].
This study by Yang and colleagues aimed to examine survival differences in patients with stage IV DTC who received one of these adjuvant radiation modalities post-thyroidectomy. All treatment groups showed improved survival, with RAI with decreases in death hazard in both univariate and multivariate analysis. Patients with stage IV DTC prolonged their survival by a factor of 1.53–4.66 in multivariate models and 1.63–4.92 in univariate models. This clearly supports the effectiveness of RAI as an adjuvant treatment to DTC following surgical resection.
However, this study has several limitations. As this was a retrospective cohort study, the lack of randomization introduces a potential source of bias. In addition, since data was collected via the National Cancer Database, there was limited information that could be obtained on the subjects studied. Disease-specific survival and recurrence rates were not reported and even histological grades were missing more than 50% of the time. Finally, older age and more advanced stage in the EBRT cohorts were likely confounders in the results of increased death hazard and mortality that were observed. It should be noted, however, that attempts to adjust for these covariates were made by the authors by analyzing the data using multivariate analysis.
There are a number of potential reasons as to why the RAI-treated patients did significantly better than the EBRT-treated patients. Based on the current literature and guidelines, EBRT is mainly recommended as a palliative treatment of locally advanced, unresectable, or metastatic disease in primarily noniodine-avid tumors. Therefore, it is certainly feasible that patients in this study who underwent treatment with EBRT had more aggressive disease and were thus at higher risk to begin with. Perhaps the indications to treat with EBRT inherently confer a poorer prognosis in advanced DTC patients. In addition, RAI is a systemic treatment modality whereas EBRT is only directed locally to the neck and thus may miss micro-metastatic lesions elsewhere in the body.
Applications for Clinical Practice
Current standard practice in thyroid cancer management involve the use of radioiodine therapy in treatment of selected intermediate-risk and all high-risk DTC patients after total thyroidectomy. These patients are treated with 131-I to destroy both remnant normal thyroid tissue and microscopic or subclinical disease remaining after surgery. The decision to administer radioactive iodine post-thyroidectomy in patients with DTC is based on risk stratification of clinicopathologic features of the tumor. The efficacy of RAI is dependent on many factors including sites of disease, patient preparation, tumor characteristics, and dose of radiation administered.
EBRT is currently used much less frequently than RAI in the management of differentiated thyroid cancer. Its main use has been for palliative treatment of locally advanced, unresectable, or metastatic disease in primarily noniodine-avid tumors. It has also been suggested for use in older patients (age 55 years or older) with gross extrathyroidal extension at the time of surgery (T4 disease), or in younger patients with T4b or extensive T4a disease and poor histologic features, with tumors that are strongly suspected to not concentrate iodine. The use of EBRT in other settings is not well established [3,4].
Treatment benefits of RAI in DTC have been extensively studied; however, this is the largest study that has examined long-term survival in a cohort of just under 12,000 patients with stage IV DTC. The results from this large cohort with advanced disease further demonstrates improved overall survival in stage IV DTC patients treated with RAI at 5 and 10 years. It is clear that RAI is the first-line adjuvant radiation therapy of DTC and should remain the standard of care in thyroid cancer management.
—Kayur Bhavsar, MD, University of Maryland School of Medicine
Baltimore, MD
1. Spitzweg C, Harrington KJ, Pinke LA, et al. Clinical review 132: The sodium iodide symporter and its potential role in cancer therapy. J Clin Endocrinol Metab 2001;86:3327–35.
2. Delaney TF, Kooey HM. Protons and charge particle radiotherapy. Philadelphia: Lippincott Williams & Wilkins; 2008.
3. Giuliani M, Brierley J. Indications for the use of external beam radiation in thyroid cancer. Curr Opin Oncol 2014;26:45–50.
4. Cooper DS, et al Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 2009;19:1167–214.
Study Overview
Objective. To evaluate survival trends and differences in a large cohort of patients with stage IV differentiated thyroid cancer treated with radioactive iodine (RAI), external beam radiation therapy (EBRT), or no radiation following surgery.
Design. Multicenter retrospective cohort study using data from the National Cancer Database (NCDB) from 2002–2012.
Setting and participants. The study group consisted of a random sample of all inpatient discharges with a diagnosis of differentiated thyroid cancer (DTC). This yielded a cohort of 11,832 patients with stage IV DTC who underwent primary surgical treatment with thyroidecromy. Patients were stratified by cancer histology into follicular thyroid cancer (FTC) and papillary thyroid cancer (PTC). Patients were additionally stratified into 3 substage groups: IV-A, IV-B, and IV-C. Administrative censoring was implemented at 5 and 10 year marks of survival time.
Main outcome measures. The primary outcome was all-cause mortality. Survival was analyzed at 5 and 10 years. Multivariate analysis was performed on a number of covariates including age, sex, race, socioeconomic status, TNM stage, tumor grade, surgical length of stay, and surgical treatment variables such as neck dissection and lymph node surgery.
Main results. Most patients (91.24%) had PTC and 8.76% had FTC. The average age of patients in the RAI group was younger (FTC, age 66; PTC, age 58) than patients in the EBRT (FTC, age 69; PTC, age 65) or no RT groups. (FTC, age 73; PTC, age 61). In contrast to FTC patients, a large majority of PTC patients underwent surgical neck dissection. There were no significant differences in sex, ethnicity, primary payer, median income quartile, or education level among the 3 groups for patients with FTC. However, in PTC there was a majority of female and ethnically white/Caucasian patients in all 3 groups. In addition, patients with PTC who did not receive RT or received RAI were more likely to have private insurance versus those who underwent EBRT, who were more often covered under Medicare. These differences in primary payer were statistically significant (P < 0.001).
Statistically significant differences in mortality were observed at 5 and 10 years in both papillary and follicular thyroid cancer among the 3 groups. In the PTC groups, patients treated with EBRT had the highest mortality rates (46.6% at 5 years, 50.7% at 10 years), while patients with PTC receiving no RT had lower mortality rates (22.7% at 5 years, 25.5% at 10 years), and PTC patients receiving RAI had the lowest mortality rates (11.0% at 5 years, 14.0% at 10 years). Similar results were seen in patients with FTC, in which patients treated with EBRT had the highest mortality rates (51.4% at 5 years, 59.9% at 10 years), while patient with FTC receiving no RT had lower mortality rates (45.5% at 5 years, 51% at 10 years), and FTC patients receiving RAI had the lowest mortality rates (29.2% at 5 years, 36.8% at 10 years).
Using univariate analysis, EBRT showed a statistically significant increase in 5- and 10-year mortality for patients with PTC stage IV-A and IV-B as compared with no radiation. This was demonstrated in both stage IV-A and IV-B subgroups at 5 years (EBRT 5-year HR PTC stage IV-A = 2.04, 95% confidence interval [CI] 1.74–2.39, P < 0.001; EBRT 5-year HR PTC stage IV-B = 2.23, 95% CI 1.42–3.51, P < 0.001; and 10 years [EBRT 10-year HR PTC stage IV-A = 2.12, 95% CI 1.79-2.52 P < 0.001; EBRT 10-year HR PTC stage IV-B = 2.03, 95% CI 1.33-3.10, P < 0.001). RAI showed a statistically significant decrease in 5- and 10-year mortality in both PTC and FTC compared with no radiation, regardless of pathologic sub-stage. The largest reduction in risk was seen in FTC stage IV-B patients at 5 years [RAI 5 year HR FTC stage IV-B = 0.31, 95% CI 0.12-0.80, P < 0.05). Multivariate analysis was also performed and showed similar results to univariate analysis except that there was no longer a statistically significant difference in EBRT versus no RT in stage IV-A PTC at 5 and 10 years (EBRT 5-year HR PTC stage IV-A = 1.2, 95% CI 0.91–1.59, EBRT 10-year HR PTC stage IV-A = 1.29, 95% CI 0.93–1.79). Reductions in death hazard seen in all groups treated with RAI versus no RT previously observed in univariate analysis remained statistically significant in all groups on multivariate analysis.
Multivariate analysis revealed a number of significant covariates. Increase in age was noted to be associated with higher death hazard in all groups except FTC stage IV-B and stage IV-C. Every additional year of age increased the hazard of death by ~2% to 5%, up to a maximum of 9% per year. Females overall had a lower hazard of death compared with their male counterparts, most notably in PTC. African-American patients had improved survival in FTC (5 years) but lower survival in PTC (5 and 10 years) as compared with white patients. Tumor grade showed a dose response in models studied, with increasing death hazards with worsening tumor differentiation.
Conclusion. RAI was associated with improved survival in patients with stage IV DTC, while EBRT was associated with poorer survival outcomes.
Commentary
Radioiodine therapy has been used for treatment of DTC since the 1940s. Radioactive iodine (I-131) is largely taken up by thyroid follicular cells via their sodium-iodide transporter causing acute thyroid cell death by emission of short path length beta particles [1].
External beam radiation therapy (EBRT) is the most common radiation therapy approach to deliver radiation from a source outside of the patient. EBRT machines produce radiation by either radioactive decay of a nuclide or by acceleration of charged particles such as electrons or protons. Using a linear accelerator, charged particles are accelerated to a high enough energy to allow transmission of particles as an electron beam or x-ray, which is subsequently directed at the tumor [2].
This study by Yang and colleagues aimed to examine survival differences in patients with stage IV DTC who received one of these adjuvant radiation modalities post-thyroidectomy. All treatment groups showed improved survival, with RAI with decreases in death hazard in both univariate and multivariate analysis. Patients with stage IV DTC prolonged their survival by a factor of 1.53–4.66 in multivariate models and 1.63–4.92 in univariate models. This clearly supports the effectiveness of RAI as an adjuvant treatment to DTC following surgical resection.
However, this study has several limitations. As this was a retrospective cohort study, the lack of randomization introduces a potential source of bias. In addition, since data was collected via the National Cancer Database, there was limited information that could be obtained on the subjects studied. Disease-specific survival and recurrence rates were not reported and even histological grades were missing more than 50% of the time. Finally, older age and more advanced stage in the EBRT cohorts were likely confounders in the results of increased death hazard and mortality that were observed. It should be noted, however, that attempts to adjust for these covariates were made by the authors by analyzing the data using multivariate analysis.
There are a number of potential reasons as to why the RAI-treated patients did significantly better than the EBRT-treated patients. Based on the current literature and guidelines, EBRT is mainly recommended as a palliative treatment of locally advanced, unresectable, or metastatic disease in primarily noniodine-avid tumors. Therefore, it is certainly feasible that patients in this study who underwent treatment with EBRT had more aggressive disease and were thus at higher risk to begin with. Perhaps the indications to treat with EBRT inherently confer a poorer prognosis in advanced DTC patients. In addition, RAI is a systemic treatment modality whereas EBRT is only directed locally to the neck and thus may miss micro-metastatic lesions elsewhere in the body.
Applications for Clinical Practice
Current standard practice in thyroid cancer management involve the use of radioiodine therapy in treatment of selected intermediate-risk and all high-risk DTC patients after total thyroidectomy. These patients are treated with 131-I to destroy both remnant normal thyroid tissue and microscopic or subclinical disease remaining after surgery. The decision to administer radioactive iodine post-thyroidectomy in patients with DTC is based on risk stratification of clinicopathologic features of the tumor. The efficacy of RAI is dependent on many factors including sites of disease, patient preparation, tumor characteristics, and dose of radiation administered.
EBRT is currently used much less frequently than RAI in the management of differentiated thyroid cancer. Its main use has been for palliative treatment of locally advanced, unresectable, or metastatic disease in primarily noniodine-avid tumors. It has also been suggested for use in older patients (age 55 years or older) with gross extrathyroidal extension at the time of surgery (T4 disease), or in younger patients with T4b or extensive T4a disease and poor histologic features, with tumors that are strongly suspected to not concentrate iodine. The use of EBRT in other settings is not well established [3,4].
Treatment benefits of RAI in DTC have been extensively studied; however, this is the largest study that has examined long-term survival in a cohort of just under 12,000 patients with stage IV DTC. The results from this large cohort with advanced disease further demonstrates improved overall survival in stage IV DTC patients treated with RAI at 5 and 10 years. It is clear that RAI is the first-line adjuvant radiation therapy of DTC and should remain the standard of care in thyroid cancer management.
—Kayur Bhavsar, MD, University of Maryland School of Medicine
Baltimore, MD
Study Overview
Objective. To evaluate survival trends and differences in a large cohort of patients with stage IV differentiated thyroid cancer treated with radioactive iodine (RAI), external beam radiation therapy (EBRT), or no radiation following surgery.
Design. Multicenter retrospective cohort study using data from the National Cancer Database (NCDB) from 2002–2012.
Setting and participants. The study group consisted of a random sample of all inpatient discharges with a diagnosis of differentiated thyroid cancer (DTC). This yielded a cohort of 11,832 patients with stage IV DTC who underwent primary surgical treatment with thyroidecromy. Patients were stratified by cancer histology into follicular thyroid cancer (FTC) and papillary thyroid cancer (PTC). Patients were additionally stratified into 3 substage groups: IV-A, IV-B, and IV-C. Administrative censoring was implemented at 5 and 10 year marks of survival time.
Main outcome measures. The primary outcome was all-cause mortality. Survival was analyzed at 5 and 10 years. Multivariate analysis was performed on a number of covariates including age, sex, race, socioeconomic status, TNM stage, tumor grade, surgical length of stay, and surgical treatment variables such as neck dissection and lymph node surgery.
Main results. Most patients (91.24%) had PTC and 8.76% had FTC. The average age of patients in the RAI group was younger (FTC, age 66; PTC, age 58) than patients in the EBRT (FTC, age 69; PTC, age 65) or no RT groups. (FTC, age 73; PTC, age 61). In contrast to FTC patients, a large majority of PTC patients underwent surgical neck dissection. There were no significant differences in sex, ethnicity, primary payer, median income quartile, or education level among the 3 groups for patients with FTC. However, in PTC there was a majority of female and ethnically white/Caucasian patients in all 3 groups. In addition, patients with PTC who did not receive RT or received RAI were more likely to have private insurance versus those who underwent EBRT, who were more often covered under Medicare. These differences in primary payer were statistically significant (P < 0.001).
Statistically significant differences in mortality were observed at 5 and 10 years in both papillary and follicular thyroid cancer among the 3 groups. In the PTC groups, patients treated with EBRT had the highest mortality rates (46.6% at 5 years, 50.7% at 10 years), while patients with PTC receiving no RT had lower mortality rates (22.7% at 5 years, 25.5% at 10 years), and PTC patients receiving RAI had the lowest mortality rates (11.0% at 5 years, 14.0% at 10 years). Similar results were seen in patients with FTC, in which patients treated with EBRT had the highest mortality rates (51.4% at 5 years, 59.9% at 10 years), while patient with FTC receiving no RT had lower mortality rates (45.5% at 5 years, 51% at 10 years), and FTC patients receiving RAI had the lowest mortality rates (29.2% at 5 years, 36.8% at 10 years).
Using univariate analysis, EBRT showed a statistically significant increase in 5- and 10-year mortality for patients with PTC stage IV-A and IV-B as compared with no radiation. This was demonstrated in both stage IV-A and IV-B subgroups at 5 years (EBRT 5-year HR PTC stage IV-A = 2.04, 95% confidence interval [CI] 1.74–2.39, P < 0.001; EBRT 5-year HR PTC stage IV-B = 2.23, 95% CI 1.42–3.51, P < 0.001; and 10 years [EBRT 10-year HR PTC stage IV-A = 2.12, 95% CI 1.79-2.52 P < 0.001; EBRT 10-year HR PTC stage IV-B = 2.03, 95% CI 1.33-3.10, P < 0.001). RAI showed a statistically significant decrease in 5- and 10-year mortality in both PTC and FTC compared with no radiation, regardless of pathologic sub-stage. The largest reduction in risk was seen in FTC stage IV-B patients at 5 years [RAI 5 year HR FTC stage IV-B = 0.31, 95% CI 0.12-0.80, P < 0.05). Multivariate analysis was also performed and showed similar results to univariate analysis except that there was no longer a statistically significant difference in EBRT versus no RT in stage IV-A PTC at 5 and 10 years (EBRT 5-year HR PTC stage IV-A = 1.2, 95% CI 0.91–1.59, EBRT 10-year HR PTC stage IV-A = 1.29, 95% CI 0.93–1.79). Reductions in death hazard seen in all groups treated with RAI versus no RT previously observed in univariate analysis remained statistically significant in all groups on multivariate analysis.
Multivariate analysis revealed a number of significant covariates. Increase in age was noted to be associated with higher death hazard in all groups except FTC stage IV-B and stage IV-C. Every additional year of age increased the hazard of death by ~2% to 5%, up to a maximum of 9% per year. Females overall had a lower hazard of death compared with their male counterparts, most notably in PTC. African-American patients had improved survival in FTC (5 years) but lower survival in PTC (5 and 10 years) as compared with white patients. Tumor grade showed a dose response in models studied, with increasing death hazards with worsening tumor differentiation.
Conclusion. RAI was associated with improved survival in patients with stage IV DTC, while EBRT was associated with poorer survival outcomes.
Commentary
Radioiodine therapy has been used for treatment of DTC since the 1940s. Radioactive iodine (I-131) is largely taken up by thyroid follicular cells via their sodium-iodide transporter causing acute thyroid cell death by emission of short path length beta particles [1].
External beam radiation therapy (EBRT) is the most common radiation therapy approach to deliver radiation from a source outside of the patient. EBRT machines produce radiation by either radioactive decay of a nuclide or by acceleration of charged particles such as electrons or protons. Using a linear accelerator, charged particles are accelerated to a high enough energy to allow transmission of particles as an electron beam or x-ray, which is subsequently directed at the tumor [2].
This study by Yang and colleagues aimed to examine survival differences in patients with stage IV DTC who received one of these adjuvant radiation modalities post-thyroidectomy. All treatment groups showed improved survival, with RAI with decreases in death hazard in both univariate and multivariate analysis. Patients with stage IV DTC prolonged their survival by a factor of 1.53–4.66 in multivariate models and 1.63–4.92 in univariate models. This clearly supports the effectiveness of RAI as an adjuvant treatment to DTC following surgical resection.
However, this study has several limitations. As this was a retrospective cohort study, the lack of randomization introduces a potential source of bias. In addition, since data was collected via the National Cancer Database, there was limited information that could be obtained on the subjects studied. Disease-specific survival and recurrence rates were not reported and even histological grades were missing more than 50% of the time. Finally, older age and more advanced stage in the EBRT cohorts were likely confounders in the results of increased death hazard and mortality that were observed. It should be noted, however, that attempts to adjust for these covariates were made by the authors by analyzing the data using multivariate analysis.
There are a number of potential reasons as to why the RAI-treated patients did significantly better than the EBRT-treated patients. Based on the current literature and guidelines, EBRT is mainly recommended as a palliative treatment of locally advanced, unresectable, or metastatic disease in primarily noniodine-avid tumors. Therefore, it is certainly feasible that patients in this study who underwent treatment with EBRT had more aggressive disease and were thus at higher risk to begin with. Perhaps the indications to treat with EBRT inherently confer a poorer prognosis in advanced DTC patients. In addition, RAI is a systemic treatment modality whereas EBRT is only directed locally to the neck and thus may miss micro-metastatic lesions elsewhere in the body.
Applications for Clinical Practice
Current standard practice in thyroid cancer management involve the use of radioiodine therapy in treatment of selected intermediate-risk and all high-risk DTC patients after total thyroidectomy. These patients are treated with 131-I to destroy both remnant normal thyroid tissue and microscopic or subclinical disease remaining after surgery. The decision to administer radioactive iodine post-thyroidectomy in patients with DTC is based on risk stratification of clinicopathologic features of the tumor. The efficacy of RAI is dependent on many factors including sites of disease, patient preparation, tumor characteristics, and dose of radiation administered.
EBRT is currently used much less frequently than RAI in the management of differentiated thyroid cancer. Its main use has been for palliative treatment of locally advanced, unresectable, or metastatic disease in primarily noniodine-avid tumors. It has also been suggested for use in older patients (age 55 years or older) with gross extrathyroidal extension at the time of surgery (T4 disease), or in younger patients with T4b or extensive T4a disease and poor histologic features, with tumors that are strongly suspected to not concentrate iodine. The use of EBRT in other settings is not well established [3,4].
Treatment benefits of RAI in DTC have been extensively studied; however, this is the largest study that has examined long-term survival in a cohort of just under 12,000 patients with stage IV DTC. The results from this large cohort with advanced disease further demonstrates improved overall survival in stage IV DTC patients treated with RAI at 5 and 10 years. It is clear that RAI is the first-line adjuvant radiation therapy of DTC and should remain the standard of care in thyroid cancer management.
—Kayur Bhavsar, MD, University of Maryland School of Medicine
Baltimore, MD
1. Spitzweg C, Harrington KJ, Pinke LA, et al. Clinical review 132: The sodium iodide symporter and its potential role in cancer therapy. J Clin Endocrinol Metab 2001;86:3327–35.
2. Delaney TF, Kooey HM. Protons and charge particle radiotherapy. Philadelphia: Lippincott Williams & Wilkins; 2008.
3. Giuliani M, Brierley J. Indications for the use of external beam radiation in thyroid cancer. Curr Opin Oncol 2014;26:45–50.
4. Cooper DS, et al Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 2009;19:1167–214.
1. Spitzweg C, Harrington KJ, Pinke LA, et al. Clinical review 132: The sodium iodide symporter and its potential role in cancer therapy. J Clin Endocrinol Metab 2001;86:3327–35.
2. Delaney TF, Kooey HM. Protons and charge particle radiotherapy. Philadelphia: Lippincott Williams & Wilkins; 2008.
3. Giuliani M, Brierley J. Indications for the use of external beam radiation in thyroid cancer. Curr Opin Oncol 2014;26:45–50.
4. Cooper DS, et al Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 2009;19:1167–214.
Diagnosis and Treatment of Migraine
From the Department of Neurology, Medstar Georgetown University Hospital, Washington, DC.
Abstract
- Objective: To review the epidemiology, pathophysiology, diagnosis, and treatment of migraine.
- Methods: Review of the literature.
- Results: Migraine is a common disorder associated with significant morbidity. Diagnosis of migraine is performed according to the International Classification of Headache Disorders. Comorbidities are commonly seen with migraine and include mood disorders (depression, anxiety, post-traumatic stress disorder), musculoskeletal disorders (neck pain, fibromyalgia, Ehlors-Danlos syndrome), sleep disorders, asthma, allergies, thyroid dysfunction, obesity, irritable bowel syndrome, epilepsy, stroke, and heart disease. Comorbid conditions can increase migraine disability. Management of migraine with lifestyle modifications, trigger management, and acute and preventive medications can help reduce the frequency, duration, and severity of attacks. Overuse of medications such as opiates, barbiturates, and caffeine-containing medications can increase headache frequency. Educating patients about limiting use of these medications is important.
- Conclusion: Migraine is a common neurologic disease that can be very disabling. Recognizing the condition, making an accurate diagnosis, and starting patients on migraine-specific treatments can help improve patient outcomes.
Migraine is a common neurologic disease that affects 1 in 10 people worldwide [1]. It is 2 to 3 times more prevalent in women than in men [2]. The prevalence of migraine peaks in both sexes during the most productive years of adulthood (age 25 to 55 years) [3]. The Global Burden of Diseases, Injuries, and Risk Factors Study considers it to be the 7th most disabling disease in the world [4]. Over 36 million people in the United States have migraine [5]. However, just 56% of migraineurs have ever been diagnosed [6].
Migraine is associated with a high rate of years lived with disability [7] and the rate has been steadily increasing since 1990. At least 50% of migraine sufferers are severely disabled, many requiring bed rest, during individual migraine attacks lasting hours to days [8]. The total U.S. annual economic costs from headache disorders, including the indirect costs from lost productivity and workplace performance, has been estimated at $31 billion [9,10].
Despite the profound impact of migraine on patients and society, there are numerous barriers to migraine care. Lipton et al [11] identified 3 steps that were minimally necessary to achieve guideline-defined appropriate acute pharmacologic therapy: (1) consulting a prescribing health care professional; (2) receiving a migraine diagnosis; and (3) using migraine-specific or other appropriate acute treatments. In a study they conducted in patients with episodic migraine, 45.5% had consulted health care professional for headache in the preceding year; of these, 86.7% reported receiving a medical diagnosis of migraine, and among the diagnosed consulters, 66.7% currently used acute migraine-specific treatments, resulting in only 26.3% individuals successfully completing all 3 steps. In the recent CaMEO study [12], the proportion patients with chronic migraine that overcame all 3 barriers was less than 5%.
The stigma of migraine often makes it difficult for people to discuss symptoms with their health care providers and family members [13]. When they do discuss their headaches with their provider, often they are not given a diagnosis [14] or do not understand what their diagnosis means [15]. It is important for health care providers to be vigilant about the diagnosis of migraine, discuss treatment goals and strategies, and prescribe appropriate migraine treatment. Migraine is often comorbid with a number of medical, neurological, and psychiatric conditions, and identifying and managing comorbidities is necessary to reduce headache burden and disability. In this article, we provide a review of the diagnosis and treatment of migraine, using a case illustration to highlight key points.
Case Study
Initial Presentation
A 24-year-old woman presents for an evaluation of her headaches.
History and Physical Examination
She initially noted headaches at age 19, which were not memorable and did not cause disability. Her current headaches are a severe throbbing pain over her right forehead. They are associated with light and sound sensitivity and stomach upset. Headaches last 6 to 7 hours without medications and occur 4 to 8 days per month.
She denies vomiting and autonomic symptoms such as runny nose or eye tearing. She also denies preceding aura. She reports headache relief with intake of tablets that contain acetaminophen/aspirin/caffeine and states that she takes between 4 to 15 tablets/month depending on headache frequency. She reports having tried acetaminophen and naproxen with no significant benefit. Aggravating factors include bright lights, strong smells, and soy/ high-sodium foods.
She had no significant past medical problems and denied a history of depression or anxiety. Family history was significant for both her father and sister having a history of headaches. The patient lived alone and denied any major life stressors. She exercises 2 times a week and denies smoking or alcohol use. Review of systems was positive for trouble sleeping, which she described as difficulty falling asleep.
On physical examination, vitals were within normal limits. BMI was 23. Chest, cardiac, abdomen, and general physical examination were all within normal limits. Neurological examination revealed no evidence of papilledema or focal neurological deficits.
What is the pathophysiology of migraine?
Migraine was thought to be a primary vascular disorder of the brain, with the origins of the vascular theory of migraine dating back to 1684 [16]. Trials performed by Wolff concluded that migraine is of vascular origin [17], and this remained the predominant theory over several decades. Current evidence suggests that migraine is unlikely to be a pure vascular disorder and instead may be related to changes in the central or peripheral nervous system [18,19].
Migraine is complex brain network disorder with a strong genetic basis [19]. The trigemino-vascular system, along with neurogenically induced inflammation of the dura mater, mast cell degranulation and release of histamine, are the likely causes of migraine pain. Trigeminal fibers arise from neurons in the trigeminal ganglion that contain substance P and calcitonin gene-related peptide (CGRP) [20]. CGRP is a neuropeptide widely expressed in both peripheral and central neurons. Elevation of CGRP in migraine is linked to diminution of the inhibitory pathways which in turn leads to migraine susceptibility [21]. These findings have led to the development of new drugs that target the CGRP pathway.
In the brainstem, periaqueductal grey matter and the dorsolateral pons have been found to be “migraine generators,” or the driver of changes of cortical activity during migraine [22]. Brainstem nuclei are involved in modulating trigemino-vascular pain transmission and autonomic responses in migraine [23].
The hypothalamus has also been implicated in migraine pathogenesis, particularly its role in nociceptive and autonomic modulation in migraine patients. Schulte and May hypothesized that there is a network change between the hypothalamus and the areas of the brainstem generator leading to the migraine attacks [24].
The thalamus plays a central role for the processing and integration of pain stimuli from the dura mater and cutaneous regions. It maintains complex connections with the somatosensory, motor, visual, auditory, olfactory and limbic regions [25]. The structural and functional alterations in the system play a role in the development of migraine attacks, and also in the sensory hypersensitivity to visual stimuli and mechanical allodynia [26].
Experimental studies in rats show that cortical spreading depression can trigger neurogenic meningeal inflammation and subsequently activate the trigemino-vascular system [27]. It has been observed that between migraine episodes a time-dependent amplitude increase of scalp-evoked potentials to repeated stereotyped stimuli, such as visual, auditory, and somaticstimuli, occurs. This phenomenon is described as “deficient habituation.” In episodic migraine, studies show 2 characteristic changes: a deficient habituation between attacks and sensitization during the attack [28]. Genetic studies have hypothesized an involvement of glutamatergic neurotransmitters and synaptic dysplasticity in causing abnormal cortical excitability in migraine [27].
What are diagnostic criteria for migraine?
Diagnosis of migraine is performed according to the International Classification of Headache Disorders (ICHD) [29]. Based on the number of headache days that the patient reports, migraine is classified into episodic or chronic migraine. Migraines that occur on fewer than 15 days/month are categorized as episodic migraines.
Episodic migraine is divided into 2 categories: migraine with aura (Table 1) and migraine without aura. Migraine without aura is described as recurrent headaches consisting of at least 5 attacks, each lasting 4 to 72 hours if left untreated. At least 2 of the following 4 characteristics must be present: unilateral location, pulsating quality, moderate or severe pain intensity, with aggravation by or causing avoidance of routine physical activity. During headache, at least 1 of nausea and/or vomiting or photophobia and phonophobia should be present. 
In migraine with aura (Table 2), headache characteristics are the same, but in addition there are at least 2 lifetime attacks with fully reversible aura symptoms (visual, sensory, speech/language). In addition, these auras have at least 2 of the following 4 characteristics: at least 1 aura symptom spreads gradually over 5 minutes, and/or 2 or more symptoms occur in succession; each individual aura symptom lasts 5 to 60 minutes; aura symptom is unilateral; and aura is accompanied, or followed within 60 minutes, by headache. Migraine with aura is uncommon, occurring in 20% of patients with migraine [30]. Visual aura is the most common type of aura, occurring in up to 90% of patients [31]. There is also aura without migraine, called typical aura without headache. Patients can present with non-migraine headache with aura, categorized as typical aura with headache [29]. 
Headache occurring on 15 or more days per month for more than 3 months, which has the features of migraine headache on at least 8 days per month, is classified as chronic migraine (Table 3). Evidence indicates that 2.5% of episodic migraine progresses to chronic migraine over 1-year follow-up [32]. There are several risk factors for chronification of migraine. Nonmodifiable factors include female sex, white European heritage, head/neck injury, low education/socioeconomic status, and stressful life events (divorce, moving, work changes, problems with children). Modifiable risk factors are headache frequency, acute medication overuse, caffeine overuse, obesity, comorbid mood disorders, and allodynia. Acute medication use and headache frequency are independent risk factors for development of chronic migraine [33]. The risk of chronic migraine increases exponentially with increased attack frequency, usually when the frequency is ≥ 3 headaches/month. Repetitive episodes of pain may increase central sensitization and result in anatomical changes in the brain and brainstem [34]. 
What information should be elicited during the history?
Specific questions about the headaches can help with making an accurate diagnosis. These include:
- Length of attacks and their frequency
- Pain characteristics (location, quality, intensity)
- Actions that trigger or aggravate headaches (eg, stress, movement, bright lights, menses, certain foods and smells)
- Associated symptoms that accompany headaches (eg, nausea, vomiting)
- How the headaches impact their life (eg, missed days at work or school, missed life events, avoidance of social activities, emergency room visits due to headache)
To assess headache frequency, it is helpful to ask about the number of headache-free days in a month, eg, “how many days a month do you NOT have a headache.” To assist with headache assessment, patients can be asked to keep a calendar in which they mark days of use of medications, including over the counter medications, menses, and headache days. The calendar can be used to assess for migraine patterns, headache frequency, and response to treatment.
When asking about headache history, it is important for patients to describe their untreated headaches. Patients taking medications may have pain that is less severe or disabling or have reduced associated symptoms. Understanding what the headaches were like when they did not treat is important in making a diagnosis.
Other important questions include when was the first time they recall ever experiencing a headache. Migraine is often present early in life, and understanding the change in headache over time is important. Also ask patients about what they want to do when they have a headache. Often patients want to lie down in a cool dark room. Ask what they would prefer to do if they didn’t have any pending responsibilities.
Comorbidities
Comorbidities are commonly seen with migraine. Common comorbidities are mood disorders (depression, anxiety, post-traumatic stress disorder), musculoskeletal disorders (neck pain, fibromyalgia, Ehlors-Danlos syndrome), sleep disorders, asthma, allergies, thyroid dysfunction, obesity, irritable bowel syndrome, epilepsy, stroke, and heart disease.
Comorbid conditions can increase migraine disability and also can provide information about the pathophysiology of migraine and guide treatment. Management of the underlying comorbidity often leads to improved migraine outcomes. For example, serotonergic dysfunction is a possible pathway involved in both migraine and mood disorders. Treatment with medications that alter the serotonin system may help both migraine and coexisting mood disorders. Bigal et al proposed that activation of the HPA axis with reduced serotonin synthesis is a main pathway involved in affective disorders, migraine, and obesity [35].
In the early 1950s, Wolff conceptualized migraine as a psychophysiologic disorder [36]. The relationship between migraine and psychiatric conditions is complex, and comorbid psychiatric disorders are risk factors for headache progression and chronicity. Psychiatric conditions also play a role in nonadherence to headache medication, which contributes to poor outcome in these patients. Hence, there is a need for assessment and treatment of psychiatric disorders in people with migraine. A study by Guidetti et al found that headache patients with multiple psychiatric conditions have poor outcomes, with 86 % of these headache patients having no improvement and even deterioration in their headache [37]. Another study by Mongini et al concluded that psychiatric disorder appears to influence the result of treatment on a long-term basis [38].
In addition, migraine has been shown to impact mood disorders. Worsening headache was found to be associated with poorer prognosis for depression. Patients with active migraine not on medications with comorbid major depressive disorder (MDD) had more severe anxiety and somatic symptoms as compared with MDD patients without migraine [39].
Case Continued
Our patient has a normal neurologic examination and classic migraine headache history and stable frequency. The physician tells her she meets criteria for episodic migraine without aura. The patient asks if she needs a “brain scan” to see if something more serious may be causing her symptoms.
What workup is recommended for patients with migraine?
If patient symptoms fit the criteria for migraine and there is a normal neurologic examination, the differential is often limited. When there are neurologic abnormalities on examination (eg, papilledema), or if the patient has concerning signs or symptoms (see below), then neuroimaging should be obtained to rule out secondary causes of headache.
In 2014, the American Academy of Neurology (AAN) published practice parameters on the evaluation of adults with recurrent headache based on guidelines published by the US Headache Consortium [40]. As per AAN guidelines, routine laboratory studies, lumbar puncture, and electroencephalogram are not recommended in the evaluation of non-acute migraines. Neuroimaging is not warranted in patients with migraine and a normal neurologic examination (grade B recommendation). Imaging may need to be considered in patients with non-acute headache and an unexplained abnormal finding on the neurologic examination (grade B recommendation).
When patients exhibit particular warning signs, or headache “red flags,” it is recommended that neuroimaging be considered. Red flags include patients with recurrent headaches and systemic symptoms (fever, weight loss), neurologic symptoms or abnormal signs (confusion, impaired alertness or consciousness), sudden onset, abrupt, or split second in nature, patients age > 50 with new onset or progressive headache, previous headache history with new or different headache (change in frequency, severity, or clinical features) and if there are secondary risk factors (HIV, cancer) [41].
Case Continued
Our patient has no red flags and can be reassured that given her normal physical examination and history suggestive of a migraine, a secondary cause of her headache is unlikely. The physician describes the treatments available, including implementing lifestyles changes and preventive and abortive medications. The patient expresses apprehension about being on prescription medications. She is concerned about side effects as well as the need to take daily medication over a long period of time. She reports that these were the main reasons she did not take the rizatriptan and propranolol that was prescribed by her previous doctor.
How is migraine treated?
Migraine is managed with a combination of lifestyle changes and pharmacologic therapy. Pharmacologic management targets treating an attack when it occurs (abortive medication), as well as reducing the frequency and severity of future attacks (preventive medication).
Lifestyle Changes
Patients should be advised that making healthy lifestyle choices, eg, regular sleep, balanced meals, proper hydration, and regular exercise, can mitigate migraine [42–44]. Other lifestyle changes that can be helpful include weight loss in the obese population, as weight loss appears to result in migraine improvement. People who are obese also are at higher risk for the progression to chronic migraine.
Acute Therapy
There are varieties of abortive therapies [45] (Table 4) that are commonly used in clinical practice. Abortive therapy can be taken as needed and is most effective if used within the first 2 hours of headache. For patients with daily or frequent headache, these medications need to be restricted to 8 to 12 days a month of use and their use should be restricted to when headache is worsening. This usually works well in patients with moderate level pain, and especially in patients with no associated nausea. Selective migraine treatments, like triptans and ergots, are used when nonspecific treatments fail, or when headache is more severe. It is preferable that patients avoid opioids, butalbital, and caffeine-containing medications. In the real world, it is difficult to convince patient to stop these medications; it is more realistic to discuss use limitation with patients, who often run out their weekly limit for triptans. 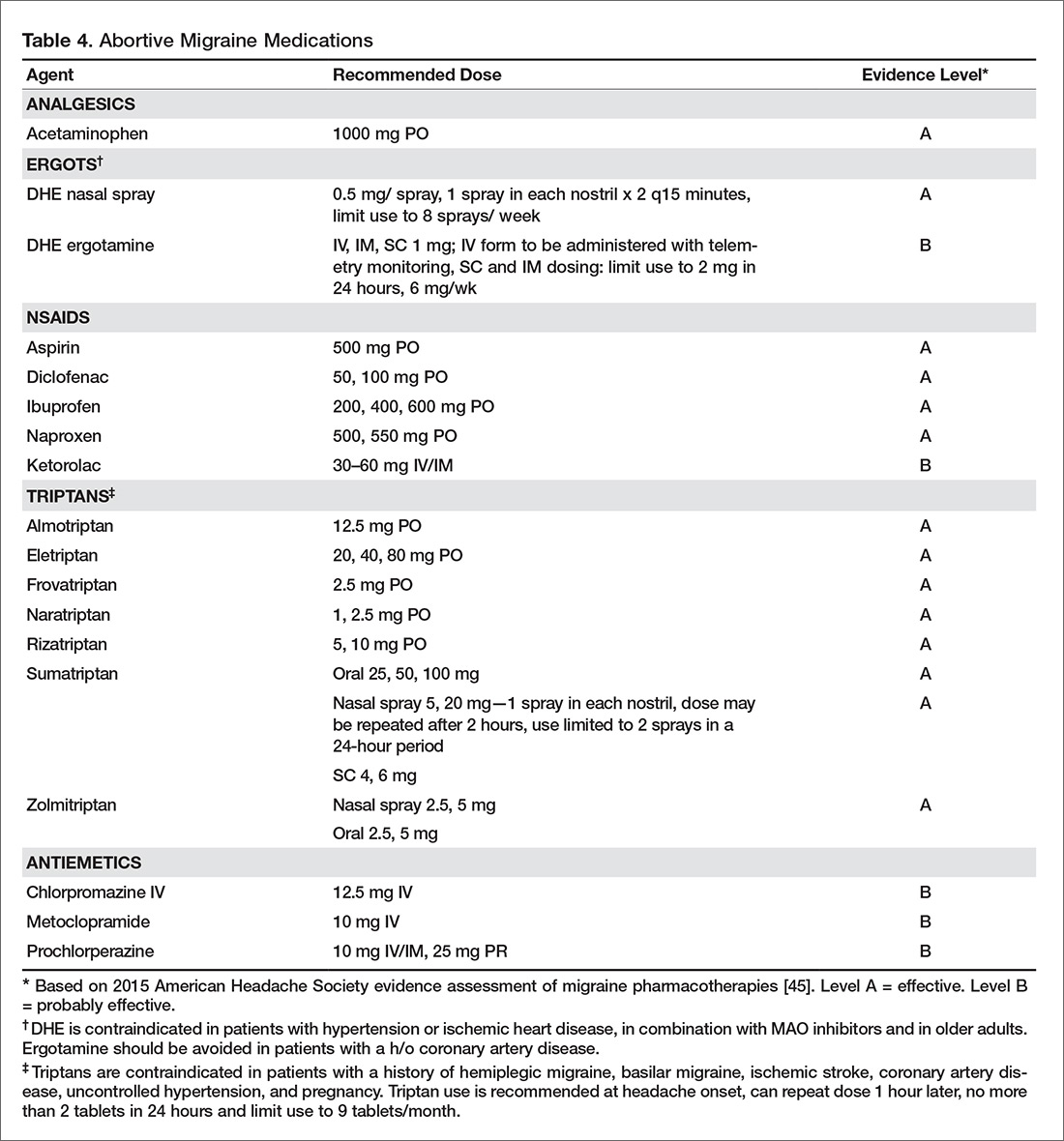
Triptans are effective medications for acute management of migraine but headache recurrence rate is high, occurring in 15% to 40 % of patients taking oral triptans. It is difficult to predict the response to a triptan [46]. The choice of an abortive agent is often directed partially by patient preference (side effect profile, cost, non-sedating vs. prefers to sleep, long vs short half-life), comorbid conditions (avoid triptans and ergots in uncontrolled hypertension, cardiovascular disease, or peripheral vascular disease or stroke/aneurysm; avoid NSAIDS in patients with cardiovascular disease), and migraine-associated symptoms (nausea and/or vomiting). Consider non-oral formulations via subcutaneous or nasal routes in patients who have nausea or vomiting with their migraine attacks. Some patients may require more than one type of abortive medication. The high recurrence rate is similar across different triptans and so switching from one triptan to another has not been found to be useful. Adding NSAIDS to triptans has been found to be more useful than switching between triptans.Overuse of acute medications has been associated with transformation of headache from episodic to chronic (medication overuse headache or rebound headache). The risk of transformation appears to be greatest with medications containing caffeine, opiates, or barbiturates [47]. Use of acute medications should be limited based on the type of medication. Patients should take triptans for no more than 10 days a month. Combined medications and opioids should be used fewer than 8 days a month, and butalbital-containing medications should be avoided or used fewer than 5 days a month [48]. Use of acute therapy should be monitored with headache calendars. It is unclear if and to what degree NSAIDS and acetaminophen cause overuse headaches.
Medication overuse headache can be difficult to treat as patients have to stop using the medication causing rebound. Further, headaches often resemble migraine and it can be difficult to differentiate them from the patients’ routine headache. Vigilance with medication use in patients with frequent headache is an essential part of migraine management, and patients should receive clear instructions regarding how to use acute medications.
Prevention
Patients presenting with more than 4 headaches per month, or headaches that last longer than 12 hours, require preventive therapy. The goals of preventive therapy is to reduce attack frequency, severity, and duration, to improve responsiveness to treatment of acute attacks, to improve function and reduce disability, and to prevent progression or transformation of episodic migraine to chronic migraine. Preventive medications usually need to be taken daily to reduce frequency or severity of the headache. The goal in this approach is 50% reduction of headache frequency and severity. Migraine preventive medications usually belong to 1 of 3 categories of drugs: antihypertensives, antiepileptics, and antidepressants. At present there are many medications for migraine prevention with different levels of evidence [49] (Table 5). Onabotulinuma toxin is the only approved medication for chronic migraine based on promising results of the PREEMPT trial [50]. 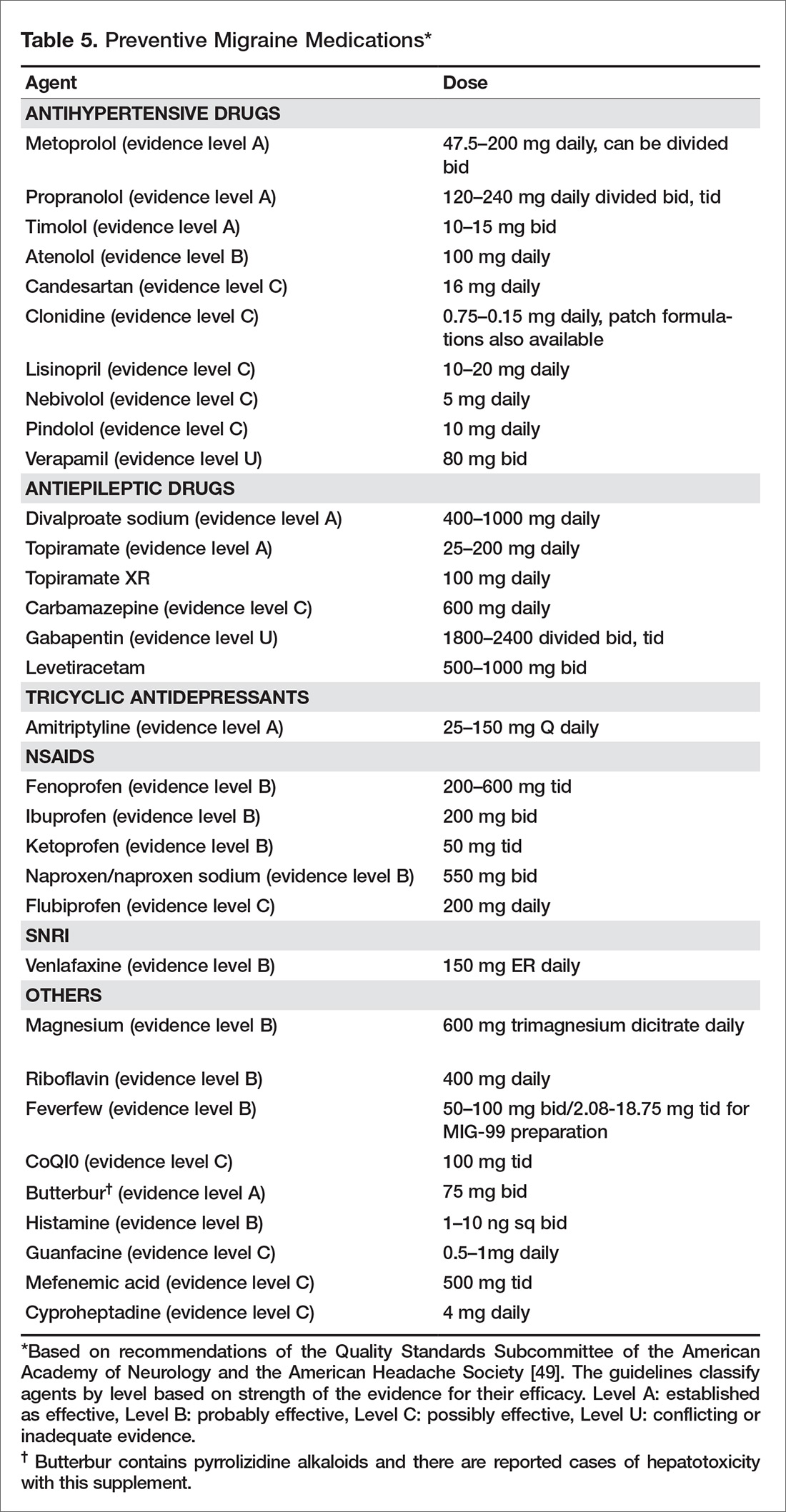
Other Considerations
A multidisciplinary approach to treatment may be warranted. Psychiatric evaluation and management of underlying depression and mood disorders can help reduce headache frequency and severity. Physical therapy should be prescribed for neck and shoulder pain. Sleep specialists should be consulted if ongoing sleep issues continue despite behavioral management.
How common is nonadherence with migraine medication?
One third of patients who are prescribed triptans discontinue the medication within a year. Lack of efficacy and concerns over medication side effects are 2 of the most common reasons for poor adherence [51]. In addition, age plays a significant role in discontinuing medication, with the elderly population more likely to stop taking triptans [52]. Seng et al reported that among patients with migraine, being male, being single, having frequent headache, and having mild pain are all associated with medication nonadherence [53]. Formulary restrictions and type of insurance coverage also were associated with nonadherence. Among adherent patients, some individuals were found to be hoarding their tablets and waiting until they were sure it was a migraine. Delaying administration of abortive medications increases the chance of incomplete treatment response, leading to patients taking more medication and in turn have more side effects [53].
Educating patients about their medications and how they need to be taken (preventive vs. abortive, when to administer) can help with adherence (Table 6). Monitoring medication use and headache frequency is an essential part of continued care for migraine patients. Maintain follow up with patients to review how they are doing with the medication and avoid providing refills without visits. The patient may not be taking medication consistently or may be using more medication than prescribed. 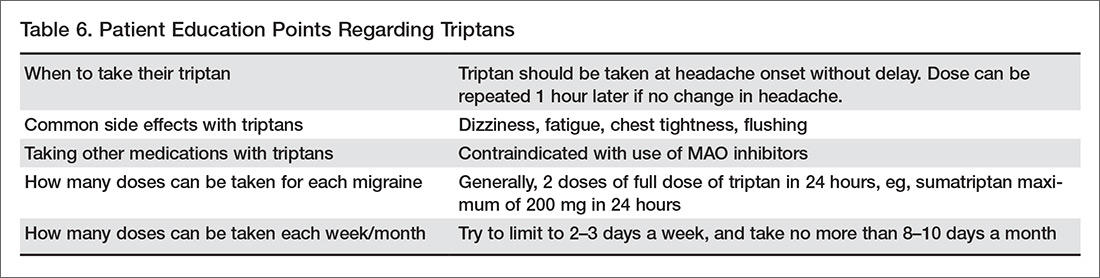
What is the role of nonpharmacologic therapy?
Most patients respond to pharmacologic treatment, but some patients with mood disorder, anxiety, difficulties or disability associated with headache, and patients with difficulty managing stress or other triggers may benefit from the addition of behavioral treatments (eg, relaxation, biofeedback, cognitive behavioral therapy, stress management) [54].
Cognitive behavioral therapy and mindfulness are techniques that have been found to be effective in decreasing intensity of pain and associated disability. The goal of these techniques is to manage the cognitive, affective, and behavioral precipitants of headache. In this process, patients are helped to identify the thoughts and behavior that play a role in generating headache. These techniques have been found to improve many headache-related outcomes like pain intensity, headache-related disability, measures of quality of life, mood and medication consumption [55]. A multidisciplinary intervention that included group exercise, stress management and relaxation lectures, and massage therapy was found to reduce self-perceived pain intensity, frequency, and duration of the headache, and improve functional status and quality of life in migraineurs [56]. A randomized controlled trial of yoga therapy compared with self care showed that yoga led to significant reduction in migraine headache frequency and improved overall outcome [57].
Overall, results from studies of nonpharmacologic techniques have been mixed [58,59]. A systematic review by Sullivan et al found a large range in the efficacy of psychological interventions for migraine [60]. A 2015 systematic review that examined if cognitive behavioral therapy (CBT) can reduce the physical symptoms of chronic headache and migraines obtained mixed results [58]. Holryod et al’s study [61] found that behavioral management combined with a ß blocker is useful in improving outcomes, but neither the ß blocker alone or behavioral migraine management alone was. Also, a trial by Penzien et al showed that nonpharmacological management helped reduce migraines by 40% to 50% and this was similar to results seen with preventive drugs [62].
Patient education may be helpful in improving outcomes. Smith et al reported a 50% reduction in headache frequency at 12 months in 46% of patients who received migraine education [63]. A randomized controlled trial by Rothrock et al involving 100 migraine patients found that patients who attended a “headache school” consisting of three 90-minute educational sessions focused on topics such as acute treatment and prevention of migraine had a significant reduction in mean migraine disability assessment score (MIDAS) than the group randomized to routine medical management only. The patients also experienced a reduction in functionally incapacitating headache days per month, less need for abortive therapy and were more compliant with prophylactic therapy [64].
Case Conclusion
Our patient is a young woman with a history of headaches suggestive of migraine without aura. Since her headache frequency ranges from 4-8 headaches month, she has episodic migraines. She also has a strong family history of headaches. She denies any other medical or psychiatric comorbidity. She reports an intake of a caffeine-containing medication of 4 to 15 tablets per month.
The physician recommended that she limit her intake of the caffeine-containing medication to 5 days or less per month given the risk of migraine transformation. The physician also recommended maintaining a good sleep schedule, limiting excessive caffeine intake, a stress reduction program, regular cardiovascular exercise, and avoiding skipping or delaying meals. The patient was educated about migraine and its underlying mechanisms and the benefits of taking medications, and her fears regarding medication use and side effects were allayed. Sumatriptan 100 mg oral tablets were prescribed to be taken at headache onset. She was hesitant to be started on an antihypertensive or antiseizure medication, so she was prescribed amitriptyline 30 mg at night for headache prevention. She was also asked to maintain a headache diary. The patient was agreeable with this plan.
Summary
Migraine is often underdiagnosed and undertreated. Primary care providers are often the first point of contact for these patients. Identifying the type and frequency of migraine and comorbidities is necessary to guide appropriate management in terms of medications and lifestyle modifications. Often no testing or imaging is required. Educating patients about this chronic disease, treatment expectations, and limiting intake of medication is essential.
Corresponding author: Pooja Mohan Rao, MBBS, MD, Georgetown University Hospital, 3800 Reservoir Rd. NW, 7 PHC, Washington, DC 20007, [email protected].
Financial disclosures: Dr. Ailani reports receiving honoraria for speaking and consulting for Allergan, Avanir, and Eli Lilly.
1. Woldeamanuel YW, Cowan RP. Migraine affects 1 in 10 people worldwide featuring recent rise: A systematic review and meta-analysis of community-based studies involving 6 million participants, J Neurol Sci 2017;372:307–15.
2. Vetvik KG, MacGregor EA. Sex differences in the epidemiology, clinical features, and pathophysiology of migraine. Lancet Neurol 2017;16:76–87.
3. Lipton RB, Bigal ME. Migraine: epidemiology, impact, and risk factors for progression. Headache 2005;45 Suppl 1:S3–S13.
4. GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016;388:1545–602.
5. Lipton RB, Silberstein SD. Episodic and chronic migraine headache: breaking down barriers to optimal treatment and prevention Headache 2015;55 Suppl 2:103–22.
6. Diamond S, Bigal ME, Silberstein S, et al. Patterns of diagnosis and acute and preventive treatment for migraine in the United States: results from the American Migraine Prevalence and Prevention study. Headache 2007;47:355–63.
7. Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study. Lancet 2012;380:2163–96.
8. Lipton RB, Stewart WF, Diamond S, et al. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache 2001;41:646–57.
9. Stewart WF, Ricci JA, Chee E, et al. Lost productive time and cost due to common pain in the US workforce. JAMA 2003;290:2443–54.
10. Hawkins K, Wang S, Rupnow M. Direct cost burden among insured US employees with migraine. Headache 2008;48:553–63.
11. Lipton RB, Serrano D, Holland S, et al. Barriers to the diagnosis and treatment of migraine: Effects of sex, income, and headache features. Headache 2013;53:81–92.
12. Dodick DW, Loder EW, Manack Adams A, et al. Assessing barriers to chronic migraine consultation, diagnosis, and treatment: Results from the chronic migraine epidemiology and outcomes (CaMEO) study. Headache 2016;56:821–34.
13. Young WB, Park JE, Tian IX, Kempner J. The stigma of migraine. PLoS One 2013;8(1):e54074.
14. Mia M, Ashna S, Audrey H. A migraine management training program for primary care providers: an overview of a survey and pilot study findings, lessons learned, and considerations for further research. Headache 2016;56:725–40.
15. Lipton RB, Amatniek JC, Ferrari MD, Gross M. Migraine: identifying and removing barriers to care. Neurology 1994;44(6 Suppl 4):S63–8.
16. Knapp RD Jr. Reports from the past 2. Headache 1963;3:112–22.
17. Levine M, Wolff HG. Cerebral circulation: afferent impulses from the blood vessels of the pia. Arch Neurol Psychiat 1932;28:140.
18. Amin FM, Asghar MS, Hougaard A, et al. Magnetic resonance angiography of intracranial and extracranial arteries in patients with spontaneous migraine without aura: a cross sectional study. Lancet Neurol 2013;12:454–61.
19. Goadsby PJ, Holland PR, Martins-Oliveira M, et al. Pathophysiology of migraine—a disorder of sensory processing. Physiol Rev 2017;97:553–622.
20. Goadsby PJ. Pathophysiology of migraine. Ann Indian Acad Neurol 2012;15(Suppl 1):S15–S22.
21. Puledda F, Messina R, Goadsby PJ, et al. An update on migraine: current understanding and future J Neurol 2017 Mar 20.
22. Vinogradova LV. Comparative potency of sensory-induced brainstem activation to trigger spreading depression and seizures in the cortex of awake rats: implications for the pathophysiology of migraine aura. Cephalalgia 2015;35:979–86.
23. Bahra A, Matharu MS, Buchel C, et al. Brainstem activation specific to migraine headache. Lancet 2001;357:1016–7.
24. Schulte LH, May A. The migraine generator revisited: continuous scanning of the migraine cycle over 30 days and three spontaneous attacks. Brain 2016;139:1987–93.
25. Noseda R, Jakubowski M, Kainz V, et al. Cortical projections of functionally identified thalamic trigeminovascular neurons: implications for migraine headache and its associated symptoms. J Neurosci 2011;31:14204–17.
26. Noseda R, Kainz V, Jakubowski M, et al. A neural mechanism for exacerbation of headache by light. Nat Neurosci 2010;13:239–45.
27. Puledda F, Messina R, Goadsby PJ. An update on migraine: current understanding and future directions. J Neurol 2017 Mar 20.
28. Coppola G, Di Lorenzo C, Schoenen J, Pierelli F. Habituation and sensitization in primary headaches. J Headache Pain 2013;14:65.
29. The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia 2013;33:629–808.
30. Yusheng H, Y Li. Typical aura without headache: a case report and review of the literature J Med Case Rep 2015;9:40.
31. Buture A, Khalil M, Ahmed F. Iatrogenic visual aura: a case report and a brief review of the literature Ther Clin Risk Manag 2017;13:643–6.
32. Lipton RB. Tracing transformation: Chronic migraine classification, progression, and epidemiology. Neurology 2009;72:S3–7.
33. Lipton RB. Headache 2011;51;S2:77–83.
34. Scher AI, Stewart WF, et al. Factors associated with the onset and remission of chronic daily headache in a population-based study. Pain 2003;106:81–9.
35. Bigal ME, Lipton RB, Holland PR, Goadsby PJ. Obesity, migraine, and chronic migraine: possible mechanisms of interaction. Neurology 2007;68:1851–61.
36. Wolff HG. Stress and disease. Springfield, IL: Charles C. Thomas; 1953.
37. Guidetti V, Galli F, Fabrizi P, et al. Headache and psychiatric comorbidity: clinical aspects and outcome in an 8-year follow-up study. Cephalalgia 1998;18:455–62.
38. Mongini F, Keller R, Deregibus A, et al. Personality traits, depression and migraine in women: a longitudinal study. Cephalalgia 2003;23:186–92.
39. Hung CI, Liu CY, Yang CH, Wang SJ. The impacts of migraine among outpatients with major depressive disorder at a two-year follow-up. PLoS One 2015;10:e0128087.
40. Frishberg BM, Rosenberg JH, Matchar DB, et al. Evidence-based guidelines in the primary care setting: neuroimaging in patients with nonacute headache. St Paul: US Headache Consortium; 2000.
41. Headache Measurement Set 2014 Revised. American Academy of Neurology. Accessed at www.aan.com/uploadedFiles/Website_Library_Assets/Documents/3.Practice_Management/2.Quality_Improvement/1.Quality_Measures/1.All_Measures/2014.
42. Taylor FR. Lifestyle changes, dietary restrictions, and nutraceuticals in migraine prevention. Techn Reg Anesth Pain Manage 2009;13:28–37.
43. Varkey E, Cider A, Carlsson J, Linde M. Exercise as migraine prophylaxis: A randomized study using relaxation and topiramate as controls. Cephalalgia 2011;14:1428–38.
44. Ahn AH. Why does increased exercise decrease migraine? Curr Pain Headache Rep 2013;17:379.
45. Marmura MJ, Silberstein SD, Schwedt TJ. The acute treatment of migraine in adults: The American Headache Society evidence assessment of migraine pharmacotherapies. Headache 2015;55:3–20.
46. Belvis R, Mas N, Aceituno A. Migraine attack treatment : a tailor-made suit, not one size fits all. Recent Pat CNS Drug Discov 2014;9:26–40.
47. Bigal ME, Serrano D, Buse D, et al. Acute migraine medications and evolution from episodic to chronic migraine: A longitudinal population-based study. Headache 2008;48:1157–68.
48. Bigal ME, Rapoport AM, Sheftell FD, et al. Transformed migraine and medication overuse in a tertiary headache centre--clinical characteristics and treatment outcomes Cephalalgia 2004;24:483–90.
49. Silberstein SD, Holland S, Freitag F, et al; Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Evidence-based guideline update: pharmacologic treatment for episodic migraine prevention in adults: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology 2012;78:1337–45.
50. Diener HC, Dodick DW, Aurora SK, et al. OnabotulinumtoxinA for treatment of chronic migraine: Results from the double-blind, randomized, placebo-controlled phase of the PREEMPT 2 trial Cephalagia year;30:804–14.
51. Wells RE, Markowitz SY, Baron EP, et al. Identifying the factors underlying discontinuation of triptans. Headache 2014;54:278–89.
52. Holland S, Fanning KM, Serrano D, et al. Rates and reasons for discontinuation of triptans and opioids in episodic migraine: results from the American Migraine Prevalence and Prevention (AMPP) study. J Neurol Sci 2013;326:10–7.
53. Seng EK, Rains JA, Nicholson RA, Lipton RB. Improving medication adherence in migraine treatment. Curr Pain Headache Rep 2015;19:24.
54. Nicholson RA, Buse DC, Andrasik F, Lipton RB. Nonpharmacologic treatments for migraine and tension-type headache: how to choose and when to use. Curr Treatment Opt Neurol 2011;13:28–40.
55. Probyn K, Bowers H, Mistry D, et al. Non-pharmacological self-management for people living with migraine or tension-type headache: a systematic review including analysis of intervention components BMJ Open 2017;7:e016670.
56. Lemstra M, Stewart B, Olszynski WP. Effectiveness of multidisciplinary intervention in the treatment of migraine: a randomized clinical trial. Headache 2002;42:845–54.
57. John PJ, Sharma N, Sharma CM, Kankane A. Effectiveness of yoga therapy in the treatment of migraine without aura: a randomized controlled trial. Headache 2007;47:654–61.
58. Harris P, Loveman E, Clegg A, et al. Systematic review of cognitive behavioural therapy for the management of headaches and migraines in adults Br J Pain 2015;9:213–24.
59. Kropp P, Meyer B, Meyer W, Dresler T. An update on behavioral treatments in migraine - current knowledge and future options. Expert Rev Neurother 2017:1–10.
60. Sullivan A, Cousins S, Ridsdale L. Psychological interventions for migraine: a systematic review. J Neurol 2016;263:2369–77.
61. Holroyd KA, Cottrell CK, O’Donnell FJ, et al. Effect of preventive (beta blocker) treatment, behavioural migraine management, or their combination on outcomes of optimised acute treatment in frequent migraine: randomised controlled trial. BMJ 2010;341:c4871.
62. Penzien DB, Rains JC, Andrasik F. Behavioral management of recurrent headache: three decades of experience and empiricism. Appl Psychophysiol Biofeedback 2002;27:163–81.
63. Smith TR, Nicholson RA, Banks JW. A primary care migraine education program has benefit on headache impact and quality of life: results from the mercy migraine management program. Headache 2010;50:600–12.
64. Rothrock JF, Parada VA, Sims C, et al.The impact of intensive patient education on clinical outcome in a clinic-based migraine population. Headache 2006;46:726–31.
From the Department of Neurology, Medstar Georgetown University Hospital, Washington, DC.
Abstract
- Objective: To review the epidemiology, pathophysiology, diagnosis, and treatment of migraine.
- Methods: Review of the literature.
- Results: Migraine is a common disorder associated with significant morbidity. Diagnosis of migraine is performed according to the International Classification of Headache Disorders. Comorbidities are commonly seen with migraine and include mood disorders (depression, anxiety, post-traumatic stress disorder), musculoskeletal disorders (neck pain, fibromyalgia, Ehlors-Danlos syndrome), sleep disorders, asthma, allergies, thyroid dysfunction, obesity, irritable bowel syndrome, epilepsy, stroke, and heart disease. Comorbid conditions can increase migraine disability. Management of migraine with lifestyle modifications, trigger management, and acute and preventive medications can help reduce the frequency, duration, and severity of attacks. Overuse of medications such as opiates, barbiturates, and caffeine-containing medications can increase headache frequency. Educating patients about limiting use of these medications is important.
- Conclusion: Migraine is a common neurologic disease that can be very disabling. Recognizing the condition, making an accurate diagnosis, and starting patients on migraine-specific treatments can help improve patient outcomes.
Migraine is a common neurologic disease that affects 1 in 10 people worldwide [1]. It is 2 to 3 times more prevalent in women than in men [2]. The prevalence of migraine peaks in both sexes during the most productive years of adulthood (age 25 to 55 years) [3]. The Global Burden of Diseases, Injuries, and Risk Factors Study considers it to be the 7th most disabling disease in the world [4]. Over 36 million people in the United States have migraine [5]. However, just 56% of migraineurs have ever been diagnosed [6].
Migraine is associated with a high rate of years lived with disability [7] and the rate has been steadily increasing since 1990. At least 50% of migraine sufferers are severely disabled, many requiring bed rest, during individual migraine attacks lasting hours to days [8]. The total U.S. annual economic costs from headache disorders, including the indirect costs from lost productivity and workplace performance, has been estimated at $31 billion [9,10].
Despite the profound impact of migraine on patients and society, there are numerous barriers to migraine care. Lipton et al [11] identified 3 steps that were minimally necessary to achieve guideline-defined appropriate acute pharmacologic therapy: (1) consulting a prescribing health care professional; (2) receiving a migraine diagnosis; and (3) using migraine-specific or other appropriate acute treatments. In a study they conducted in patients with episodic migraine, 45.5% had consulted health care professional for headache in the preceding year; of these, 86.7% reported receiving a medical diagnosis of migraine, and among the diagnosed consulters, 66.7% currently used acute migraine-specific treatments, resulting in only 26.3% individuals successfully completing all 3 steps. In the recent CaMEO study [12], the proportion patients with chronic migraine that overcame all 3 barriers was less than 5%.
The stigma of migraine often makes it difficult for people to discuss symptoms with their health care providers and family members [13]. When they do discuss their headaches with their provider, often they are not given a diagnosis [14] or do not understand what their diagnosis means [15]. It is important for health care providers to be vigilant about the diagnosis of migraine, discuss treatment goals and strategies, and prescribe appropriate migraine treatment. Migraine is often comorbid with a number of medical, neurological, and psychiatric conditions, and identifying and managing comorbidities is necessary to reduce headache burden and disability. In this article, we provide a review of the diagnosis and treatment of migraine, using a case illustration to highlight key points.
Case Study
Initial Presentation
A 24-year-old woman presents for an evaluation of her headaches.
History and Physical Examination
She initially noted headaches at age 19, which were not memorable and did not cause disability. Her current headaches are a severe throbbing pain over her right forehead. They are associated with light and sound sensitivity and stomach upset. Headaches last 6 to 7 hours without medications and occur 4 to 8 days per month.
She denies vomiting and autonomic symptoms such as runny nose or eye tearing. She also denies preceding aura. She reports headache relief with intake of tablets that contain acetaminophen/aspirin/caffeine and states that she takes between 4 to 15 tablets/month depending on headache frequency. She reports having tried acetaminophen and naproxen with no significant benefit. Aggravating factors include bright lights, strong smells, and soy/ high-sodium foods.
She had no significant past medical problems and denied a history of depression or anxiety. Family history was significant for both her father and sister having a history of headaches. The patient lived alone and denied any major life stressors. She exercises 2 times a week and denies smoking or alcohol use. Review of systems was positive for trouble sleeping, which she described as difficulty falling asleep.
On physical examination, vitals were within normal limits. BMI was 23. Chest, cardiac, abdomen, and general physical examination were all within normal limits. Neurological examination revealed no evidence of papilledema or focal neurological deficits.
What is the pathophysiology of migraine?
Migraine was thought to be a primary vascular disorder of the brain, with the origins of the vascular theory of migraine dating back to 1684 [16]. Trials performed by Wolff concluded that migraine is of vascular origin [17], and this remained the predominant theory over several decades. Current evidence suggests that migraine is unlikely to be a pure vascular disorder and instead may be related to changes in the central or peripheral nervous system [18,19].
Migraine is complex brain network disorder with a strong genetic basis [19]. The trigemino-vascular system, along with neurogenically induced inflammation of the dura mater, mast cell degranulation and release of histamine, are the likely causes of migraine pain. Trigeminal fibers arise from neurons in the trigeminal ganglion that contain substance P and calcitonin gene-related peptide (CGRP) [20]. CGRP is a neuropeptide widely expressed in both peripheral and central neurons. Elevation of CGRP in migraine is linked to diminution of the inhibitory pathways which in turn leads to migraine susceptibility [21]. These findings have led to the development of new drugs that target the CGRP pathway.
In the brainstem, periaqueductal grey matter and the dorsolateral pons have been found to be “migraine generators,” or the driver of changes of cortical activity during migraine [22]. Brainstem nuclei are involved in modulating trigemino-vascular pain transmission and autonomic responses in migraine [23].
The hypothalamus has also been implicated in migraine pathogenesis, particularly its role in nociceptive and autonomic modulation in migraine patients. Schulte and May hypothesized that there is a network change between the hypothalamus and the areas of the brainstem generator leading to the migraine attacks [24].
The thalamus plays a central role for the processing and integration of pain stimuli from the dura mater and cutaneous regions. It maintains complex connections with the somatosensory, motor, visual, auditory, olfactory and limbic regions [25]. The structural and functional alterations in the system play a role in the development of migraine attacks, and also in the sensory hypersensitivity to visual stimuli and mechanical allodynia [26].
Experimental studies in rats show that cortical spreading depression can trigger neurogenic meningeal inflammation and subsequently activate the trigemino-vascular system [27]. It has been observed that between migraine episodes a time-dependent amplitude increase of scalp-evoked potentials to repeated stereotyped stimuli, such as visual, auditory, and somaticstimuli, occurs. This phenomenon is described as “deficient habituation.” In episodic migraine, studies show 2 characteristic changes: a deficient habituation between attacks and sensitization during the attack [28]. Genetic studies have hypothesized an involvement of glutamatergic neurotransmitters and synaptic dysplasticity in causing abnormal cortical excitability in migraine [27].
What are diagnostic criteria for migraine?
Diagnosis of migraine is performed according to the International Classification of Headache Disorders (ICHD) [29]. Based on the number of headache days that the patient reports, migraine is classified into episodic or chronic migraine. Migraines that occur on fewer than 15 days/month are categorized as episodic migraines.
Episodic migraine is divided into 2 categories: migraine with aura (Table 1) and migraine without aura. Migraine without aura is described as recurrent headaches consisting of at least 5 attacks, each lasting 4 to 72 hours if left untreated. At least 2 of the following 4 characteristics must be present: unilateral location, pulsating quality, moderate or severe pain intensity, with aggravation by or causing avoidance of routine physical activity. During headache, at least 1 of nausea and/or vomiting or photophobia and phonophobia should be present.
In migraine with aura (Table 2), headache characteristics are the same, but in addition there are at least 2 lifetime attacks with fully reversible aura symptoms (visual, sensory, speech/language). In addition, these auras have at least 2 of the following 4 characteristics: at least 1 aura symptom spreads gradually over 5 minutes, and/or 2 or more symptoms occur in succession; each individual aura symptom lasts 5 to 60 minutes; aura symptom is unilateral; and aura is accompanied, or followed within 60 minutes, by headache. Migraine with aura is uncommon, occurring in 20% of patients with migraine [30]. Visual aura is the most common type of aura, occurring in up to 90% of patients [31]. There is also aura without migraine, called typical aura without headache. Patients can present with non-migraine headache with aura, categorized as typical aura with headache [29].
Headache occurring on 15 or more days per month for more than 3 months, which has the features of migraine headache on at least 8 days per month, is classified as chronic migraine (Table 3). Evidence indicates that 2.5% of episodic migraine progresses to chronic migraine over 1-year follow-up [32]. There are several risk factors for chronification of migraine. Nonmodifiable factors include female sex, white European heritage, head/neck injury, low education/socioeconomic status, and stressful life events (divorce, moving, work changes, problems with children). Modifiable risk factors are headache frequency, acute medication overuse, caffeine overuse, obesity, comorbid mood disorders, and allodynia. Acute medication use and headache frequency are independent risk factors for development of chronic migraine [33]. The risk of chronic migraine increases exponentially with increased attack frequency, usually when the frequency is ≥ 3 headaches/month. Repetitive episodes of pain may increase central sensitization and result in anatomical changes in the brain and brainstem [34].
What information should be elicited during the history?
Specific questions about the headaches can help with making an accurate diagnosis. These include:
- Length of attacks and their frequency
- Pain characteristics (location, quality, intensity)
- Actions that trigger or aggravate headaches (eg, stress, movement, bright lights, menses, certain foods and smells)
- Associated symptoms that accompany headaches (eg, nausea, vomiting)
- How the headaches impact their life (eg, missed days at work or school, missed life events, avoidance of social activities, emergency room visits due to headache)
To assess headache frequency, it is helpful to ask about the number of headache-free days in a month, eg, “how many days a month do you NOT have a headache.” To assist with headache assessment, patients can be asked to keep a calendar in which they mark days of use of medications, including over the counter medications, menses, and headache days. The calendar can be used to assess for migraine patterns, headache frequency, and response to treatment.
When asking about headache history, it is important for patients to describe their untreated headaches. Patients taking medications may have pain that is less severe or disabling or have reduced associated symptoms. Understanding what the headaches were like when they did not treat is important in making a diagnosis.
Other important questions include when was the first time they recall ever experiencing a headache. Migraine is often present early in life, and understanding the change in headache over time is important. Also ask patients about what they want to do when they have a headache. Often patients want to lie down in a cool dark room. Ask what they would prefer to do if they didn’t have any pending responsibilities.
Comorbidities
Comorbidities are commonly seen with migraine. Common comorbidities are mood disorders (depression, anxiety, post-traumatic stress disorder), musculoskeletal disorders (neck pain, fibromyalgia, Ehlors-Danlos syndrome), sleep disorders, asthma, allergies, thyroid dysfunction, obesity, irritable bowel syndrome, epilepsy, stroke, and heart disease.
Comorbid conditions can increase migraine disability and also can provide information about the pathophysiology of migraine and guide treatment. Management of the underlying comorbidity often leads to improved migraine outcomes. For example, serotonergic dysfunction is a possible pathway involved in both migraine and mood disorders. Treatment with medications that alter the serotonin system may help both migraine and coexisting mood disorders. Bigal et al proposed that activation of the HPA axis with reduced serotonin synthesis is a main pathway involved in affective disorders, migraine, and obesity [35].
In the early 1950s, Wolff conceptualized migraine as a psychophysiologic disorder [36]. The relationship between migraine and psychiatric conditions is complex, and comorbid psychiatric disorders are risk factors for headache progression and chronicity. Psychiatric conditions also play a role in nonadherence to headache medication, which contributes to poor outcome in these patients. Hence, there is a need for assessment and treatment of psychiatric disorders in people with migraine. A study by Guidetti et al found that headache patients with multiple psychiatric conditions have poor outcomes, with 86 % of these headache patients having no improvement and even deterioration in their headache [37]. Another study by Mongini et al concluded that psychiatric disorder appears to influence the result of treatment on a long-term basis [38].
In addition, migraine has been shown to impact mood disorders. Worsening headache was found to be associated with poorer prognosis for depression. Patients with active migraine not on medications with comorbid major depressive disorder (MDD) had more severe anxiety and somatic symptoms as compared with MDD patients without migraine [39].
Case Continued
Our patient has a normal neurologic examination and classic migraine headache history and stable frequency. The physician tells her she meets criteria for episodic migraine without aura. The patient asks if she needs a “brain scan” to see if something more serious may be causing her symptoms.
What workup is recommended for patients with migraine?
If patient symptoms fit the criteria for migraine and there is a normal neurologic examination, the differential is often limited. When there are neurologic abnormalities on examination (eg, papilledema), or if the patient has concerning signs or symptoms (see below), then neuroimaging should be obtained to rule out secondary causes of headache.
In 2014, the American Academy of Neurology (AAN) published practice parameters on the evaluation of adults with recurrent headache based on guidelines published by the US Headache Consortium [40]. As per AAN guidelines, routine laboratory studies, lumbar puncture, and electroencephalogram are not recommended in the evaluation of non-acute migraines. Neuroimaging is not warranted in patients with migraine and a normal neurologic examination (grade B recommendation). Imaging may need to be considered in patients with non-acute headache and an unexplained abnormal finding on the neurologic examination (grade B recommendation).
When patients exhibit particular warning signs, or headache “red flags,” it is recommended that neuroimaging be considered. Red flags include patients with recurrent headaches and systemic symptoms (fever, weight loss), neurologic symptoms or abnormal signs (confusion, impaired alertness or consciousness), sudden onset, abrupt, or split second in nature, patients age > 50 with new onset or progressive headache, previous headache history with new or different headache (change in frequency, severity, or clinical features) and if there are secondary risk factors (HIV, cancer) [41].
Case Continued
Our patient has no red flags and can be reassured that given her normal physical examination and history suggestive of a migraine, a secondary cause of her headache is unlikely. The physician describes the treatments available, including implementing lifestyles changes and preventive and abortive medications. The patient expresses apprehension about being on prescription medications. She is concerned about side effects as well as the need to take daily medication over a long period of time. She reports that these were the main reasons she did not take the rizatriptan and propranolol that was prescribed by her previous doctor.
How is migraine treated?
Migraine is managed with a combination of lifestyle changes and pharmacologic therapy. Pharmacologic management targets treating an attack when it occurs (abortive medication), as well as reducing the frequency and severity of future attacks (preventive medication).
Lifestyle Changes
Patients should be advised that making healthy lifestyle choices, eg, regular sleep, balanced meals, proper hydration, and regular exercise, can mitigate migraine [42–44]. Other lifestyle changes that can be helpful include weight loss in the obese population, as weight loss appears to result in migraine improvement. People who are obese also are at higher risk for the progression to chronic migraine.
Acute Therapy
There are varieties of abortive therapies [45] (Table 4) that are commonly used in clinical practice. Abortive therapy can be taken as needed and is most effective if used within the first 2 hours of headache. For patients with daily or frequent headache, these medications need to be restricted to 8 to 12 days a month of use and their use should be restricted to when headache is worsening. This usually works well in patients with moderate level pain, and especially in patients with no associated nausea. Selective migraine treatments, like triptans and ergots, are used when nonspecific treatments fail, or when headache is more severe. It is preferable that patients avoid opioids, butalbital, and caffeine-containing medications. In the real world, it is difficult to convince patient to stop these medications; it is more realistic to discuss use limitation with patients, who often run out their weekly limit for triptans.
Triptans are effective medications for acute management of migraine but headache recurrence rate is high, occurring in 15% to 40 % of patients taking oral triptans. It is difficult to predict the response to a triptan [46]. The choice of an abortive agent is often directed partially by patient preference (side effect profile, cost, non-sedating vs. prefers to sleep, long vs short half-life), comorbid conditions (avoid triptans and ergots in uncontrolled hypertension, cardiovascular disease, or peripheral vascular disease or stroke/aneurysm; avoid NSAIDS in patients with cardiovascular disease), and migraine-associated symptoms (nausea and/or vomiting). Consider non-oral formulations via subcutaneous or nasal routes in patients who have nausea or vomiting with their migraine attacks. Some patients may require more than one type of abortive medication. The high recurrence rate is similar across different triptans and so switching from one triptan to another has not been found to be useful. Adding NSAIDS to triptans has been found to be more useful than switching between triptans.Overuse of acute medications has been associated with transformation of headache from episodic to chronic (medication overuse headache or rebound headache). The risk of transformation appears to be greatest with medications containing caffeine, opiates, or barbiturates [47]. Use of acute medications should be limited based on the type of medication. Patients should take triptans for no more than 10 days a month. Combined medications and opioids should be used fewer than 8 days a month, and butalbital-containing medications should be avoided or used fewer than 5 days a month [48]. Use of acute therapy should be monitored with headache calendars. It is unclear if and to what degree NSAIDS and acetaminophen cause overuse headaches.
Medication overuse headache can be difficult to treat as patients have to stop using the medication causing rebound. Further, headaches often resemble migraine and it can be difficult to differentiate them from the patients’ routine headache. Vigilance with medication use in patients with frequent headache is an essential part of migraine management, and patients should receive clear instructions regarding how to use acute medications.
Prevention
Patients presenting with more than 4 headaches per month, or headaches that last longer than 12 hours, require preventive therapy. The goals of preventive therapy is to reduce attack frequency, severity, and duration, to improve responsiveness to treatment of acute attacks, to improve function and reduce disability, and to prevent progression or transformation of episodic migraine to chronic migraine. Preventive medications usually need to be taken daily to reduce frequency or severity of the headache. The goal in this approach is 50% reduction of headache frequency and severity. Migraine preventive medications usually belong to 1 of 3 categories of drugs: antihypertensives, antiepileptics, and antidepressants. At present there are many medications for migraine prevention with different levels of evidence [49] (Table 5). Onabotulinuma toxin is the only approved medication for chronic migraine based on promising results of the PREEMPT trial [50].
Other Considerations
A multidisciplinary approach to treatment may be warranted. Psychiatric evaluation and management of underlying depression and mood disorders can help reduce headache frequency and severity. Physical therapy should be prescribed for neck and shoulder pain. Sleep specialists should be consulted if ongoing sleep issues continue despite behavioral management.
How common is nonadherence with migraine medication?
One third of patients who are prescribed triptans discontinue the medication within a year. Lack of efficacy and concerns over medication side effects are 2 of the most common reasons for poor adherence [51]. In addition, age plays a significant role in discontinuing medication, with the elderly population more likely to stop taking triptans [52]. Seng et al reported that among patients with migraine, being male, being single, having frequent headache, and having mild pain are all associated with medication nonadherence [53]. Formulary restrictions and type of insurance coverage also were associated with nonadherence. Among adherent patients, some individuals were found to be hoarding their tablets and waiting until they were sure it was a migraine. Delaying administration of abortive medications increases the chance of incomplete treatment response, leading to patients taking more medication and in turn have more side effects [53].
Educating patients about their medications and how they need to be taken (preventive vs. abortive, when to administer) can help with adherence (Table 6). Monitoring medication use and headache frequency is an essential part of continued care for migraine patients. Maintain follow up with patients to review how they are doing with the medication and avoid providing refills without visits. The patient may not be taking medication consistently or may be using more medication than prescribed.
What is the role of nonpharmacologic therapy?
Most patients respond to pharmacologic treatment, but some patients with mood disorder, anxiety, difficulties or disability associated with headache, and patients with difficulty managing stress or other triggers may benefit from the addition of behavioral treatments (eg, relaxation, biofeedback, cognitive behavioral therapy, stress management) [54].
Cognitive behavioral therapy and mindfulness are techniques that have been found to be effective in decreasing intensity of pain and associated disability. The goal of these techniques is to manage the cognitive, affective, and behavioral precipitants of headache. In this process, patients are helped to identify the thoughts and behavior that play a role in generating headache. These techniques have been found to improve many headache-related outcomes like pain intensity, headache-related disability, measures of quality of life, mood and medication consumption [55]. A multidisciplinary intervention that included group exercise, stress management and relaxation lectures, and massage therapy was found to reduce self-perceived pain intensity, frequency, and duration of the headache, and improve functional status and quality of life in migraineurs [56]. A randomized controlled trial of yoga therapy compared with self care showed that yoga led to significant reduction in migraine headache frequency and improved overall outcome [57].
Overall, results from studies of nonpharmacologic techniques have been mixed [58,59]. A systematic review by Sullivan et al found a large range in the efficacy of psychological interventions for migraine [60]. A 2015 systematic review that examined if cognitive behavioral therapy (CBT) can reduce the physical symptoms of chronic headache and migraines obtained mixed results [58]. Holryod et al’s study [61] found that behavioral management combined with a ß blocker is useful in improving outcomes, but neither the ß blocker alone or behavioral migraine management alone was. Also, a trial by Penzien et al showed that nonpharmacological management helped reduce migraines by 40% to 50% and this was similar to results seen with preventive drugs [62].
Patient education may be helpful in improving outcomes. Smith et al reported a 50% reduction in headache frequency at 12 months in 46% of patients who received migraine education [63]. A randomized controlled trial by Rothrock et al involving 100 migraine patients found that patients who attended a “headache school” consisting of three 90-minute educational sessions focused on topics such as acute treatment and prevention of migraine had a significant reduction in mean migraine disability assessment score (MIDAS) than the group randomized to routine medical management only. The patients also experienced a reduction in functionally incapacitating headache days per month, less need for abortive therapy and were more compliant with prophylactic therapy [64].
Case Conclusion
Our patient is a young woman with a history of headaches suggestive of migraine without aura. Since her headache frequency ranges from 4-8 headaches month, she has episodic migraines. She also has a strong family history of headaches. She denies any other medical or psychiatric comorbidity. She reports an intake of a caffeine-containing medication of 4 to 15 tablets per month.
The physician recommended that she limit her intake of the caffeine-containing medication to 5 days or less per month given the risk of migraine transformation. The physician also recommended maintaining a good sleep schedule, limiting excessive caffeine intake, a stress reduction program, regular cardiovascular exercise, and avoiding skipping or delaying meals. The patient was educated about migraine and its underlying mechanisms and the benefits of taking medications, and her fears regarding medication use and side effects were allayed. Sumatriptan 100 mg oral tablets were prescribed to be taken at headache onset. She was hesitant to be started on an antihypertensive or antiseizure medication, so she was prescribed amitriptyline 30 mg at night for headache prevention. She was also asked to maintain a headache diary. The patient was agreeable with this plan.
Summary
Migraine is often underdiagnosed and undertreated. Primary care providers are often the first point of contact for these patients. Identifying the type and frequency of migraine and comorbidities is necessary to guide appropriate management in terms of medications and lifestyle modifications. Often no testing or imaging is required. Educating patients about this chronic disease, treatment expectations, and limiting intake of medication is essential.
Corresponding author: Pooja Mohan Rao, MBBS, MD, Georgetown University Hospital, 3800 Reservoir Rd. NW, 7 PHC, Washington, DC 20007, [email protected].
Financial disclosures: Dr. Ailani reports receiving honoraria for speaking and consulting for Allergan, Avanir, and Eli Lilly.
From the Department of Neurology, Medstar Georgetown University Hospital, Washington, DC.
Abstract
- Objective: To review the epidemiology, pathophysiology, diagnosis, and treatment of migraine.
- Methods: Review of the literature.
- Results: Migraine is a common disorder associated with significant morbidity. Diagnosis of migraine is performed according to the International Classification of Headache Disorders. Comorbidities are commonly seen with migraine and include mood disorders (depression, anxiety, post-traumatic stress disorder), musculoskeletal disorders (neck pain, fibromyalgia, Ehlors-Danlos syndrome), sleep disorders, asthma, allergies, thyroid dysfunction, obesity, irritable bowel syndrome, epilepsy, stroke, and heart disease. Comorbid conditions can increase migraine disability. Management of migraine with lifestyle modifications, trigger management, and acute and preventive medications can help reduce the frequency, duration, and severity of attacks. Overuse of medications such as opiates, barbiturates, and caffeine-containing medications can increase headache frequency. Educating patients about limiting use of these medications is important.
- Conclusion: Migraine is a common neurologic disease that can be very disabling. Recognizing the condition, making an accurate diagnosis, and starting patients on migraine-specific treatments can help improve patient outcomes.
Migraine is a common neurologic disease that affects 1 in 10 people worldwide [1]. It is 2 to 3 times more prevalent in women than in men [2]. The prevalence of migraine peaks in both sexes during the most productive years of adulthood (age 25 to 55 years) [3]. The Global Burden of Diseases, Injuries, and Risk Factors Study considers it to be the 7th most disabling disease in the world [4]. Over 36 million people in the United States have migraine [5]. However, just 56% of migraineurs have ever been diagnosed [6].
Migraine is associated with a high rate of years lived with disability [7] and the rate has been steadily increasing since 1990. At least 50% of migraine sufferers are severely disabled, many requiring bed rest, during individual migraine attacks lasting hours to days [8]. The total U.S. annual economic costs from headache disorders, including the indirect costs from lost productivity and workplace performance, has been estimated at $31 billion [9,10].
Despite the profound impact of migraine on patients and society, there are numerous barriers to migraine care. Lipton et al [11] identified 3 steps that were minimally necessary to achieve guideline-defined appropriate acute pharmacologic therapy: (1) consulting a prescribing health care professional; (2) receiving a migraine diagnosis; and (3) using migraine-specific or other appropriate acute treatments. In a study they conducted in patients with episodic migraine, 45.5% had consulted health care professional for headache in the preceding year; of these, 86.7% reported receiving a medical diagnosis of migraine, and among the diagnosed consulters, 66.7% currently used acute migraine-specific treatments, resulting in only 26.3% individuals successfully completing all 3 steps. In the recent CaMEO study [12], the proportion patients with chronic migraine that overcame all 3 barriers was less than 5%.
The stigma of migraine often makes it difficult for people to discuss symptoms with their health care providers and family members [13]. When they do discuss their headaches with their provider, often they are not given a diagnosis [14] or do not understand what their diagnosis means [15]. It is important for health care providers to be vigilant about the diagnosis of migraine, discuss treatment goals and strategies, and prescribe appropriate migraine treatment. Migraine is often comorbid with a number of medical, neurological, and psychiatric conditions, and identifying and managing comorbidities is necessary to reduce headache burden and disability. In this article, we provide a review of the diagnosis and treatment of migraine, using a case illustration to highlight key points.
Case Study
Initial Presentation
A 24-year-old woman presents for an evaluation of her headaches.
History and Physical Examination
She initially noted headaches at age 19, which were not memorable and did not cause disability. Her current headaches are a severe throbbing pain over her right forehead. They are associated with light and sound sensitivity and stomach upset. Headaches last 6 to 7 hours without medications and occur 4 to 8 days per month.
She denies vomiting and autonomic symptoms such as runny nose or eye tearing. She also denies preceding aura. She reports headache relief with intake of tablets that contain acetaminophen/aspirin/caffeine and states that she takes between 4 to 15 tablets/month depending on headache frequency. She reports having tried acetaminophen and naproxen with no significant benefit. Aggravating factors include bright lights, strong smells, and soy/ high-sodium foods.
She had no significant past medical problems and denied a history of depression or anxiety. Family history was significant for both her father and sister having a history of headaches. The patient lived alone and denied any major life stressors. She exercises 2 times a week and denies smoking or alcohol use. Review of systems was positive for trouble sleeping, which she described as difficulty falling asleep.
On physical examination, vitals were within normal limits. BMI was 23. Chest, cardiac, abdomen, and general physical examination were all within normal limits. Neurological examination revealed no evidence of papilledema or focal neurological deficits.
What is the pathophysiology of migraine?
Migraine was thought to be a primary vascular disorder of the brain, with the origins of the vascular theory of migraine dating back to 1684 [16]. Trials performed by Wolff concluded that migraine is of vascular origin [17], and this remained the predominant theory over several decades. Current evidence suggests that migraine is unlikely to be a pure vascular disorder and instead may be related to changes in the central or peripheral nervous system [18,19].
Migraine is complex brain network disorder with a strong genetic basis [19]. The trigemino-vascular system, along with neurogenically induced inflammation of the dura mater, mast cell degranulation and release of histamine, are the likely causes of migraine pain. Trigeminal fibers arise from neurons in the trigeminal ganglion that contain substance P and calcitonin gene-related peptide (CGRP) [20]. CGRP is a neuropeptide widely expressed in both peripheral and central neurons. Elevation of CGRP in migraine is linked to diminution of the inhibitory pathways which in turn leads to migraine susceptibility [21]. These findings have led to the development of new drugs that target the CGRP pathway.
In the brainstem, periaqueductal grey matter and the dorsolateral pons have been found to be “migraine generators,” or the driver of changes of cortical activity during migraine [22]. Brainstem nuclei are involved in modulating trigemino-vascular pain transmission and autonomic responses in migraine [23].
The hypothalamus has also been implicated in migraine pathogenesis, particularly its role in nociceptive and autonomic modulation in migraine patients. Schulte and May hypothesized that there is a network change between the hypothalamus and the areas of the brainstem generator leading to the migraine attacks [24].
The thalamus plays a central role for the processing and integration of pain stimuli from the dura mater and cutaneous regions. It maintains complex connections with the somatosensory, motor, visual, auditory, olfactory and limbic regions [25]. The structural and functional alterations in the system play a role in the development of migraine attacks, and also in the sensory hypersensitivity to visual stimuli and mechanical allodynia [26].
Experimental studies in rats show that cortical spreading depression can trigger neurogenic meningeal inflammation and subsequently activate the trigemino-vascular system [27]. It has been observed that between migraine episodes a time-dependent amplitude increase of scalp-evoked potentials to repeated stereotyped stimuli, such as visual, auditory, and somaticstimuli, occurs. This phenomenon is described as “deficient habituation.” In episodic migraine, studies show 2 characteristic changes: a deficient habituation between attacks and sensitization during the attack [28]. Genetic studies have hypothesized an involvement of glutamatergic neurotransmitters and synaptic dysplasticity in causing abnormal cortical excitability in migraine [27].
What are diagnostic criteria for migraine?
Diagnosis of migraine is performed according to the International Classification of Headache Disorders (ICHD) [29]. Based on the number of headache days that the patient reports, migraine is classified into episodic or chronic migraine. Migraines that occur on fewer than 15 days/month are categorized as episodic migraines.
Episodic migraine is divided into 2 categories: migraine with aura (Table 1) and migraine without aura. Migraine without aura is described as recurrent headaches consisting of at least 5 attacks, each lasting 4 to 72 hours if left untreated. At least 2 of the following 4 characteristics must be present: unilateral location, pulsating quality, moderate or severe pain intensity, with aggravation by or causing avoidance of routine physical activity. During headache, at least 1 of nausea and/or vomiting or photophobia and phonophobia should be present.
In migraine with aura (Table 2), headache characteristics are the same, but in addition there are at least 2 lifetime attacks with fully reversible aura symptoms (visual, sensory, speech/language). In addition, these auras have at least 2 of the following 4 characteristics: at least 1 aura symptom spreads gradually over 5 minutes, and/or 2 or more symptoms occur in succession; each individual aura symptom lasts 5 to 60 minutes; aura symptom is unilateral; and aura is accompanied, or followed within 60 minutes, by headache. Migraine with aura is uncommon, occurring in 20% of patients with migraine [30]. Visual aura is the most common type of aura, occurring in up to 90% of patients [31]. There is also aura without migraine, called typical aura without headache. Patients can present with non-migraine headache with aura, categorized as typical aura with headache [29].
Headache occurring on 15 or more days per month for more than 3 months, which has the features of migraine headache on at least 8 days per month, is classified as chronic migraine (Table 3). Evidence indicates that 2.5% of episodic migraine progresses to chronic migraine over 1-year follow-up [32]. There are several risk factors for chronification of migraine. Nonmodifiable factors include female sex, white European heritage, head/neck injury, low education/socioeconomic status, and stressful life events (divorce, moving, work changes, problems with children). Modifiable risk factors are headache frequency, acute medication overuse, caffeine overuse, obesity, comorbid mood disorders, and allodynia. Acute medication use and headache frequency are independent risk factors for development of chronic migraine [33]. The risk of chronic migraine increases exponentially with increased attack frequency, usually when the frequency is ≥ 3 headaches/month. Repetitive episodes of pain may increase central sensitization and result in anatomical changes in the brain and brainstem [34].
What information should be elicited during the history?
Specific questions about the headaches can help with making an accurate diagnosis. These include:
- Length of attacks and their frequency
- Pain characteristics (location, quality, intensity)
- Actions that trigger or aggravate headaches (eg, stress, movement, bright lights, menses, certain foods and smells)
- Associated symptoms that accompany headaches (eg, nausea, vomiting)
- How the headaches impact their life (eg, missed days at work or school, missed life events, avoidance of social activities, emergency room visits due to headache)
To assess headache frequency, it is helpful to ask about the number of headache-free days in a month, eg, “how many days a month do you NOT have a headache.” To assist with headache assessment, patients can be asked to keep a calendar in which they mark days of use of medications, including over the counter medications, menses, and headache days. The calendar can be used to assess for migraine patterns, headache frequency, and response to treatment.
When asking about headache history, it is important for patients to describe their untreated headaches. Patients taking medications may have pain that is less severe or disabling or have reduced associated symptoms. Understanding what the headaches were like when they did not treat is important in making a diagnosis.
Other important questions include when was the first time they recall ever experiencing a headache. Migraine is often present early in life, and understanding the change in headache over time is important. Also ask patients about what they want to do when they have a headache. Often patients want to lie down in a cool dark room. Ask what they would prefer to do if they didn’t have any pending responsibilities.
Comorbidities
Comorbidities are commonly seen with migraine. Common comorbidities are mood disorders (depression, anxiety, post-traumatic stress disorder), musculoskeletal disorders (neck pain, fibromyalgia, Ehlors-Danlos syndrome), sleep disorders, asthma, allergies, thyroid dysfunction, obesity, irritable bowel syndrome, epilepsy, stroke, and heart disease.
Comorbid conditions can increase migraine disability and also can provide information about the pathophysiology of migraine and guide treatment. Management of the underlying comorbidity often leads to improved migraine outcomes. For example, serotonergic dysfunction is a possible pathway involved in both migraine and mood disorders. Treatment with medications that alter the serotonin system may help both migraine and coexisting mood disorders. Bigal et al proposed that activation of the HPA axis with reduced serotonin synthesis is a main pathway involved in affective disorders, migraine, and obesity [35].
In the early 1950s, Wolff conceptualized migraine as a psychophysiologic disorder [36]. The relationship between migraine and psychiatric conditions is complex, and comorbid psychiatric disorders are risk factors for headache progression and chronicity. Psychiatric conditions also play a role in nonadherence to headache medication, which contributes to poor outcome in these patients. Hence, there is a need for assessment and treatment of psychiatric disorders in people with migraine. A study by Guidetti et al found that headache patients with multiple psychiatric conditions have poor outcomes, with 86 % of these headache patients having no improvement and even deterioration in their headache [37]. Another study by Mongini et al concluded that psychiatric disorder appears to influence the result of treatment on a long-term basis [38].
In addition, migraine has been shown to impact mood disorders. Worsening headache was found to be associated with poorer prognosis for depression. Patients with active migraine not on medications with comorbid major depressive disorder (MDD) had more severe anxiety and somatic symptoms as compared with MDD patients without migraine [39].
Case Continued
Our patient has a normal neurologic examination and classic migraine headache history and stable frequency. The physician tells her she meets criteria for episodic migraine without aura. The patient asks if she needs a “brain scan” to see if something more serious may be causing her symptoms.
What workup is recommended for patients with migraine?
If patient symptoms fit the criteria for migraine and there is a normal neurologic examination, the differential is often limited. When there are neurologic abnormalities on examination (eg, papilledema), or if the patient has concerning signs or symptoms (see below), then neuroimaging should be obtained to rule out secondary causes of headache.
In 2014, the American Academy of Neurology (AAN) published practice parameters on the evaluation of adults with recurrent headache based on guidelines published by the US Headache Consortium [40]. As per AAN guidelines, routine laboratory studies, lumbar puncture, and electroencephalogram are not recommended in the evaluation of non-acute migraines. Neuroimaging is not warranted in patients with migraine and a normal neurologic examination (grade B recommendation). Imaging may need to be considered in patients with non-acute headache and an unexplained abnormal finding on the neurologic examination (grade B recommendation).
When patients exhibit particular warning signs, or headache “red flags,” it is recommended that neuroimaging be considered. Red flags include patients with recurrent headaches and systemic symptoms (fever, weight loss), neurologic symptoms or abnormal signs (confusion, impaired alertness or consciousness), sudden onset, abrupt, or split second in nature, patients age > 50 with new onset or progressive headache, previous headache history with new or different headache (change in frequency, severity, or clinical features) and if there are secondary risk factors (HIV, cancer) [41].
Case Continued
Our patient has no red flags and can be reassured that given her normal physical examination and history suggestive of a migraine, a secondary cause of her headache is unlikely. The physician describes the treatments available, including implementing lifestyles changes and preventive and abortive medications. The patient expresses apprehension about being on prescription medications. She is concerned about side effects as well as the need to take daily medication over a long period of time. She reports that these were the main reasons she did not take the rizatriptan and propranolol that was prescribed by her previous doctor.
How is migraine treated?
Migraine is managed with a combination of lifestyle changes and pharmacologic therapy. Pharmacologic management targets treating an attack when it occurs (abortive medication), as well as reducing the frequency and severity of future attacks (preventive medication).
Lifestyle Changes
Patients should be advised that making healthy lifestyle choices, eg, regular sleep, balanced meals, proper hydration, and regular exercise, can mitigate migraine [42–44]. Other lifestyle changes that can be helpful include weight loss in the obese population, as weight loss appears to result in migraine improvement. People who are obese also are at higher risk for the progression to chronic migraine.
Acute Therapy
There are varieties of abortive therapies [45] (Table 4) that are commonly used in clinical practice. Abortive therapy can be taken as needed and is most effective if used within the first 2 hours of headache. For patients with daily or frequent headache, these medications need to be restricted to 8 to 12 days a month of use and their use should be restricted to when headache is worsening. This usually works well in patients with moderate level pain, and especially in patients with no associated nausea. Selective migraine treatments, like triptans and ergots, are used when nonspecific treatments fail, or when headache is more severe. It is preferable that patients avoid opioids, butalbital, and caffeine-containing medications. In the real world, it is difficult to convince patient to stop these medications; it is more realistic to discuss use limitation with patients, who often run out their weekly limit for triptans.
Triptans are effective medications for acute management of migraine but headache recurrence rate is high, occurring in 15% to 40 % of patients taking oral triptans. It is difficult to predict the response to a triptan [46]. The choice of an abortive agent is often directed partially by patient preference (side effect profile, cost, non-sedating vs. prefers to sleep, long vs short half-life), comorbid conditions (avoid triptans and ergots in uncontrolled hypertension, cardiovascular disease, or peripheral vascular disease or stroke/aneurysm; avoid NSAIDS in patients with cardiovascular disease), and migraine-associated symptoms (nausea and/or vomiting). Consider non-oral formulations via subcutaneous or nasal routes in patients who have nausea or vomiting with their migraine attacks. Some patients may require more than one type of abortive medication. The high recurrence rate is similar across different triptans and so switching from one triptan to another has not been found to be useful. Adding NSAIDS to triptans has been found to be more useful than switching between triptans.Overuse of acute medications has been associated with transformation of headache from episodic to chronic (medication overuse headache or rebound headache). The risk of transformation appears to be greatest with medications containing caffeine, opiates, or barbiturates [47]. Use of acute medications should be limited based on the type of medication. Patients should take triptans for no more than 10 days a month. Combined medications and opioids should be used fewer than 8 days a month, and butalbital-containing medications should be avoided or used fewer than 5 days a month [48]. Use of acute therapy should be monitored with headache calendars. It is unclear if and to what degree NSAIDS and acetaminophen cause overuse headaches.
Medication overuse headache can be difficult to treat as patients have to stop using the medication causing rebound. Further, headaches often resemble migraine and it can be difficult to differentiate them from the patients’ routine headache. Vigilance with medication use in patients with frequent headache is an essential part of migraine management, and patients should receive clear instructions regarding how to use acute medications.
Prevention
Patients presenting with more than 4 headaches per month, or headaches that last longer than 12 hours, require preventive therapy. The goals of preventive therapy is to reduce attack frequency, severity, and duration, to improve responsiveness to treatment of acute attacks, to improve function and reduce disability, and to prevent progression or transformation of episodic migraine to chronic migraine. Preventive medications usually need to be taken daily to reduce frequency or severity of the headache. The goal in this approach is 50% reduction of headache frequency and severity. Migraine preventive medications usually belong to 1 of 3 categories of drugs: antihypertensives, antiepileptics, and antidepressants. At present there are many medications for migraine prevention with different levels of evidence [49] (Table 5). Onabotulinuma toxin is the only approved medication for chronic migraine based on promising results of the PREEMPT trial [50].
Other Considerations
A multidisciplinary approach to treatment may be warranted. Psychiatric evaluation and management of underlying depression and mood disorders can help reduce headache frequency and severity. Physical therapy should be prescribed for neck and shoulder pain. Sleep specialists should be consulted if ongoing sleep issues continue despite behavioral management.
How common is nonadherence with migraine medication?
One third of patients who are prescribed triptans discontinue the medication within a year. Lack of efficacy and concerns over medication side effects are 2 of the most common reasons for poor adherence [51]. In addition, age plays a significant role in discontinuing medication, with the elderly population more likely to stop taking triptans [52]. Seng et al reported that among patients with migraine, being male, being single, having frequent headache, and having mild pain are all associated with medication nonadherence [53]. Formulary restrictions and type of insurance coverage also were associated with nonadherence. Among adherent patients, some individuals were found to be hoarding their tablets and waiting until they were sure it was a migraine. Delaying administration of abortive medications increases the chance of incomplete treatment response, leading to patients taking more medication and in turn have more side effects [53].
Educating patients about their medications and how they need to be taken (preventive vs. abortive, when to administer) can help with adherence (Table 6). Monitoring medication use and headache frequency is an essential part of continued care for migraine patients. Maintain follow up with patients to review how they are doing with the medication and avoid providing refills without visits. The patient may not be taking medication consistently or may be using more medication than prescribed.
What is the role of nonpharmacologic therapy?
Most patients respond to pharmacologic treatment, but some patients with mood disorder, anxiety, difficulties or disability associated with headache, and patients with difficulty managing stress or other triggers may benefit from the addition of behavioral treatments (eg, relaxation, biofeedback, cognitive behavioral therapy, stress management) [54].
Cognitive behavioral therapy and mindfulness are techniques that have been found to be effective in decreasing intensity of pain and associated disability. The goal of these techniques is to manage the cognitive, affective, and behavioral precipitants of headache. In this process, patients are helped to identify the thoughts and behavior that play a role in generating headache. These techniques have been found to improve many headache-related outcomes like pain intensity, headache-related disability, measures of quality of life, mood and medication consumption [55]. A multidisciplinary intervention that included group exercise, stress management and relaxation lectures, and massage therapy was found to reduce self-perceived pain intensity, frequency, and duration of the headache, and improve functional status and quality of life in migraineurs [56]. A randomized controlled trial of yoga therapy compared with self care showed that yoga led to significant reduction in migraine headache frequency and improved overall outcome [57].
Overall, results from studies of nonpharmacologic techniques have been mixed [58,59]. A systematic review by Sullivan et al found a large range in the efficacy of psychological interventions for migraine [60]. A 2015 systematic review that examined if cognitive behavioral therapy (CBT) can reduce the physical symptoms of chronic headache and migraines obtained mixed results [58]. Holryod et al’s study [61] found that behavioral management combined with a ß blocker is useful in improving outcomes, but neither the ß blocker alone or behavioral migraine management alone was. Also, a trial by Penzien et al showed that nonpharmacological management helped reduce migraines by 40% to 50% and this was similar to results seen with preventive drugs [62].
Patient education may be helpful in improving outcomes. Smith et al reported a 50% reduction in headache frequency at 12 months in 46% of patients who received migraine education [63]. A randomized controlled trial by Rothrock et al involving 100 migraine patients found that patients who attended a “headache school” consisting of three 90-minute educational sessions focused on topics such as acute treatment and prevention of migraine had a significant reduction in mean migraine disability assessment score (MIDAS) than the group randomized to routine medical management only. The patients also experienced a reduction in functionally incapacitating headache days per month, less need for abortive therapy and were more compliant with prophylactic therapy [64].
Case Conclusion
Our patient is a young woman with a history of headaches suggestive of migraine without aura. Since her headache frequency ranges from 4-8 headaches month, she has episodic migraines. She also has a strong family history of headaches. She denies any other medical or psychiatric comorbidity. She reports an intake of a caffeine-containing medication of 4 to 15 tablets per month.
The physician recommended that she limit her intake of the caffeine-containing medication to 5 days or less per month given the risk of migraine transformation. The physician also recommended maintaining a good sleep schedule, limiting excessive caffeine intake, a stress reduction program, regular cardiovascular exercise, and avoiding skipping or delaying meals. The patient was educated about migraine and its underlying mechanisms and the benefits of taking medications, and her fears regarding medication use and side effects were allayed. Sumatriptan 100 mg oral tablets were prescribed to be taken at headache onset. She was hesitant to be started on an antihypertensive or antiseizure medication, so she was prescribed amitriptyline 30 mg at night for headache prevention. She was also asked to maintain a headache diary. The patient was agreeable with this plan.
Summary
Migraine is often underdiagnosed and undertreated. Primary care providers are often the first point of contact for these patients. Identifying the type and frequency of migraine and comorbidities is necessary to guide appropriate management in terms of medications and lifestyle modifications. Often no testing or imaging is required. Educating patients about this chronic disease, treatment expectations, and limiting intake of medication is essential.
Corresponding author: Pooja Mohan Rao, MBBS, MD, Georgetown University Hospital, 3800 Reservoir Rd. NW, 7 PHC, Washington, DC 20007, [email protected].
Financial disclosures: Dr. Ailani reports receiving honoraria for speaking and consulting for Allergan, Avanir, and Eli Lilly.
1. Woldeamanuel YW, Cowan RP. Migraine affects 1 in 10 people worldwide featuring recent rise: A systematic review and meta-analysis of community-based studies involving 6 million participants, J Neurol Sci 2017;372:307–15.
2. Vetvik KG, MacGregor EA. Sex differences in the epidemiology, clinical features, and pathophysiology of migraine. Lancet Neurol 2017;16:76–87.
3. Lipton RB, Bigal ME. Migraine: epidemiology, impact, and risk factors for progression. Headache 2005;45 Suppl 1:S3–S13.
4. GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016;388:1545–602.
5. Lipton RB, Silberstein SD. Episodic and chronic migraine headache: breaking down barriers to optimal treatment and prevention Headache 2015;55 Suppl 2:103–22.
6. Diamond S, Bigal ME, Silberstein S, et al. Patterns of diagnosis and acute and preventive treatment for migraine in the United States: results from the American Migraine Prevalence and Prevention study. Headache 2007;47:355–63.
7. Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study. Lancet 2012;380:2163–96.
8. Lipton RB, Stewart WF, Diamond S, et al. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache 2001;41:646–57.
9. Stewart WF, Ricci JA, Chee E, et al. Lost productive time and cost due to common pain in the US workforce. JAMA 2003;290:2443–54.
10. Hawkins K, Wang S, Rupnow M. Direct cost burden among insured US employees with migraine. Headache 2008;48:553–63.
11. Lipton RB, Serrano D, Holland S, et al. Barriers to the diagnosis and treatment of migraine: Effects of sex, income, and headache features. Headache 2013;53:81–92.
12. Dodick DW, Loder EW, Manack Adams A, et al. Assessing barriers to chronic migraine consultation, diagnosis, and treatment: Results from the chronic migraine epidemiology and outcomes (CaMEO) study. Headache 2016;56:821–34.
13. Young WB, Park JE, Tian IX, Kempner J. The stigma of migraine. PLoS One 2013;8(1):e54074.
14. Mia M, Ashna S, Audrey H. A migraine management training program for primary care providers: an overview of a survey and pilot study findings, lessons learned, and considerations for further research. Headache 2016;56:725–40.
15. Lipton RB, Amatniek JC, Ferrari MD, Gross M. Migraine: identifying and removing barriers to care. Neurology 1994;44(6 Suppl 4):S63–8.
16. Knapp RD Jr. Reports from the past 2. Headache 1963;3:112–22.
17. Levine M, Wolff HG. Cerebral circulation: afferent impulses from the blood vessels of the pia. Arch Neurol Psychiat 1932;28:140.
18. Amin FM, Asghar MS, Hougaard A, et al. Magnetic resonance angiography of intracranial and extracranial arteries in patients with spontaneous migraine without aura: a cross sectional study. Lancet Neurol 2013;12:454–61.
19. Goadsby PJ, Holland PR, Martins-Oliveira M, et al. Pathophysiology of migraine—a disorder of sensory processing. Physiol Rev 2017;97:553–622.
20. Goadsby PJ. Pathophysiology of migraine. Ann Indian Acad Neurol 2012;15(Suppl 1):S15–S22.
21. Puledda F, Messina R, Goadsby PJ, et al. An update on migraine: current understanding and future J Neurol 2017 Mar 20.
22. Vinogradova LV. Comparative potency of sensory-induced brainstem activation to trigger spreading depression and seizures in the cortex of awake rats: implications for the pathophysiology of migraine aura. Cephalalgia 2015;35:979–86.
23. Bahra A, Matharu MS, Buchel C, et al. Brainstem activation specific to migraine headache. Lancet 2001;357:1016–7.
24. Schulte LH, May A. The migraine generator revisited: continuous scanning of the migraine cycle over 30 days and three spontaneous attacks. Brain 2016;139:1987–93.
25. Noseda R, Jakubowski M, Kainz V, et al. Cortical projections of functionally identified thalamic trigeminovascular neurons: implications for migraine headache and its associated symptoms. J Neurosci 2011;31:14204–17.
26. Noseda R, Kainz V, Jakubowski M, et al. A neural mechanism for exacerbation of headache by light. Nat Neurosci 2010;13:239–45.
27. Puledda F, Messina R, Goadsby PJ. An update on migraine: current understanding and future directions. J Neurol 2017 Mar 20.
28. Coppola G, Di Lorenzo C, Schoenen J, Pierelli F. Habituation and sensitization in primary headaches. J Headache Pain 2013;14:65.
29. The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia 2013;33:629–808.
30. Yusheng H, Y Li. Typical aura without headache: a case report and review of the literature J Med Case Rep 2015;9:40.
31. Buture A, Khalil M, Ahmed F. Iatrogenic visual aura: a case report and a brief review of the literature Ther Clin Risk Manag 2017;13:643–6.
32. Lipton RB. Tracing transformation: Chronic migraine classification, progression, and epidemiology. Neurology 2009;72:S3–7.
33. Lipton RB. Headache 2011;51;S2:77–83.
34. Scher AI, Stewart WF, et al. Factors associated with the onset and remission of chronic daily headache in a population-based study. Pain 2003;106:81–9.
35. Bigal ME, Lipton RB, Holland PR, Goadsby PJ. Obesity, migraine, and chronic migraine: possible mechanisms of interaction. Neurology 2007;68:1851–61.
36. Wolff HG. Stress and disease. Springfield, IL: Charles C. Thomas; 1953.
37. Guidetti V, Galli F, Fabrizi P, et al. Headache and psychiatric comorbidity: clinical aspects and outcome in an 8-year follow-up study. Cephalalgia 1998;18:455–62.
38. Mongini F, Keller R, Deregibus A, et al. Personality traits, depression and migraine in women: a longitudinal study. Cephalalgia 2003;23:186–92.
39. Hung CI, Liu CY, Yang CH, Wang SJ. The impacts of migraine among outpatients with major depressive disorder at a two-year follow-up. PLoS One 2015;10:e0128087.
40. Frishberg BM, Rosenberg JH, Matchar DB, et al. Evidence-based guidelines in the primary care setting: neuroimaging in patients with nonacute headache. St Paul: US Headache Consortium; 2000.
41. Headache Measurement Set 2014 Revised. American Academy of Neurology. Accessed at www.aan.com/uploadedFiles/Website_Library_Assets/Documents/3.Practice_Management/2.Quality_Improvement/1.Quality_Measures/1.All_Measures/2014.
42. Taylor FR. Lifestyle changes, dietary restrictions, and nutraceuticals in migraine prevention. Techn Reg Anesth Pain Manage 2009;13:28–37.
43. Varkey E, Cider A, Carlsson J, Linde M. Exercise as migraine prophylaxis: A randomized study using relaxation and topiramate as controls. Cephalalgia 2011;14:1428–38.
44. Ahn AH. Why does increased exercise decrease migraine? Curr Pain Headache Rep 2013;17:379.
45. Marmura MJ, Silberstein SD, Schwedt TJ. The acute treatment of migraine in adults: The American Headache Society evidence assessment of migraine pharmacotherapies. Headache 2015;55:3–20.
46. Belvis R, Mas N, Aceituno A. Migraine attack treatment : a tailor-made suit, not one size fits all. Recent Pat CNS Drug Discov 2014;9:26–40.
47. Bigal ME, Serrano D, Buse D, et al. Acute migraine medications and evolution from episodic to chronic migraine: A longitudinal population-based study. Headache 2008;48:1157–68.
48. Bigal ME, Rapoport AM, Sheftell FD, et al. Transformed migraine and medication overuse in a tertiary headache centre--clinical characteristics and treatment outcomes Cephalalgia 2004;24:483–90.
49. Silberstein SD, Holland S, Freitag F, et al; Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Evidence-based guideline update: pharmacologic treatment for episodic migraine prevention in adults: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology 2012;78:1337–45.
50. Diener HC, Dodick DW, Aurora SK, et al. OnabotulinumtoxinA for treatment of chronic migraine: Results from the double-blind, randomized, placebo-controlled phase of the PREEMPT 2 trial Cephalagia year;30:804–14.
51. Wells RE, Markowitz SY, Baron EP, et al. Identifying the factors underlying discontinuation of triptans. Headache 2014;54:278–89.
52. Holland S, Fanning KM, Serrano D, et al. Rates and reasons for discontinuation of triptans and opioids in episodic migraine: results from the American Migraine Prevalence and Prevention (AMPP) study. J Neurol Sci 2013;326:10–7.
53. Seng EK, Rains JA, Nicholson RA, Lipton RB. Improving medication adherence in migraine treatment. Curr Pain Headache Rep 2015;19:24.
54. Nicholson RA, Buse DC, Andrasik F, Lipton RB. Nonpharmacologic treatments for migraine and tension-type headache: how to choose and when to use. Curr Treatment Opt Neurol 2011;13:28–40.
55. Probyn K, Bowers H, Mistry D, et al. Non-pharmacological self-management for people living with migraine or tension-type headache: a systematic review including analysis of intervention components BMJ Open 2017;7:e016670.
56. Lemstra M, Stewart B, Olszynski WP. Effectiveness of multidisciplinary intervention in the treatment of migraine: a randomized clinical trial. Headache 2002;42:845–54.
57. John PJ, Sharma N, Sharma CM, Kankane A. Effectiveness of yoga therapy in the treatment of migraine without aura: a randomized controlled trial. Headache 2007;47:654–61.
58. Harris P, Loveman E, Clegg A, et al. Systematic review of cognitive behavioural therapy for the management of headaches and migraines in adults Br J Pain 2015;9:213–24.
59. Kropp P, Meyer B, Meyer W, Dresler T. An update on behavioral treatments in migraine - current knowledge and future options. Expert Rev Neurother 2017:1–10.
60. Sullivan A, Cousins S, Ridsdale L. Psychological interventions for migraine: a systematic review. J Neurol 2016;263:2369–77.
61. Holroyd KA, Cottrell CK, O’Donnell FJ, et al. Effect of preventive (beta blocker) treatment, behavioural migraine management, or their combination on outcomes of optimised acute treatment in frequent migraine: randomised controlled trial. BMJ 2010;341:c4871.
62. Penzien DB, Rains JC, Andrasik F. Behavioral management of recurrent headache: three decades of experience and empiricism. Appl Psychophysiol Biofeedback 2002;27:163–81.
63. Smith TR, Nicholson RA, Banks JW. A primary care migraine education program has benefit on headache impact and quality of life: results from the mercy migraine management program. Headache 2010;50:600–12.
64. Rothrock JF, Parada VA, Sims C, et al.The impact of intensive patient education on clinical outcome in a clinic-based migraine population. Headache 2006;46:726–31.
1. Woldeamanuel YW, Cowan RP. Migraine affects 1 in 10 people worldwide featuring recent rise: A systematic review and meta-analysis of community-based studies involving 6 million participants, J Neurol Sci 2017;372:307–15.
2. Vetvik KG, MacGregor EA. Sex differences in the epidemiology, clinical features, and pathophysiology of migraine. Lancet Neurol 2017;16:76–87.
3. Lipton RB, Bigal ME. Migraine: epidemiology, impact, and risk factors for progression. Headache 2005;45 Suppl 1:S3–S13.
4. GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016;388:1545–602.
5. Lipton RB, Silberstein SD. Episodic and chronic migraine headache: breaking down barriers to optimal treatment and prevention Headache 2015;55 Suppl 2:103–22.
6. Diamond S, Bigal ME, Silberstein S, et al. Patterns of diagnosis and acute and preventive treatment for migraine in the United States: results from the American Migraine Prevalence and Prevention study. Headache 2007;47:355–63.
7. Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study. Lancet 2012;380:2163–96.
8. Lipton RB, Stewart WF, Diamond S, et al. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache 2001;41:646–57.
9. Stewart WF, Ricci JA, Chee E, et al. Lost productive time and cost due to common pain in the US workforce. JAMA 2003;290:2443–54.
10. Hawkins K, Wang S, Rupnow M. Direct cost burden among insured US employees with migraine. Headache 2008;48:553–63.
11. Lipton RB, Serrano D, Holland S, et al. Barriers to the diagnosis and treatment of migraine: Effects of sex, income, and headache features. Headache 2013;53:81–92.
12. Dodick DW, Loder EW, Manack Adams A, et al. Assessing barriers to chronic migraine consultation, diagnosis, and treatment: Results from the chronic migraine epidemiology and outcomes (CaMEO) study. Headache 2016;56:821–34.
13. Young WB, Park JE, Tian IX, Kempner J. The stigma of migraine. PLoS One 2013;8(1):e54074.
14. Mia M, Ashna S, Audrey H. A migraine management training program for primary care providers: an overview of a survey and pilot study findings, lessons learned, and considerations for further research. Headache 2016;56:725–40.
15. Lipton RB, Amatniek JC, Ferrari MD, Gross M. Migraine: identifying and removing barriers to care. Neurology 1994;44(6 Suppl 4):S63–8.
16. Knapp RD Jr. Reports from the past 2. Headache 1963;3:112–22.
17. Levine M, Wolff HG. Cerebral circulation: afferent impulses from the blood vessels of the pia. Arch Neurol Psychiat 1932;28:140.
18. Amin FM, Asghar MS, Hougaard A, et al. Magnetic resonance angiography of intracranial and extracranial arteries in patients with spontaneous migraine without aura: a cross sectional study. Lancet Neurol 2013;12:454–61.
19. Goadsby PJ, Holland PR, Martins-Oliveira M, et al. Pathophysiology of migraine—a disorder of sensory processing. Physiol Rev 2017;97:553–622.
20. Goadsby PJ. Pathophysiology of migraine. Ann Indian Acad Neurol 2012;15(Suppl 1):S15–S22.
21. Puledda F, Messina R, Goadsby PJ, et al. An update on migraine: current understanding and future J Neurol 2017 Mar 20.
22. Vinogradova LV. Comparative potency of sensory-induced brainstem activation to trigger spreading depression and seizures in the cortex of awake rats: implications for the pathophysiology of migraine aura. Cephalalgia 2015;35:979–86.
23. Bahra A, Matharu MS, Buchel C, et al. Brainstem activation specific to migraine headache. Lancet 2001;357:1016–7.
24. Schulte LH, May A. The migraine generator revisited: continuous scanning of the migraine cycle over 30 days and three spontaneous attacks. Brain 2016;139:1987–93.
25. Noseda R, Jakubowski M, Kainz V, et al. Cortical projections of functionally identified thalamic trigeminovascular neurons: implications for migraine headache and its associated symptoms. J Neurosci 2011;31:14204–17.
26. Noseda R, Kainz V, Jakubowski M, et al. A neural mechanism for exacerbation of headache by light. Nat Neurosci 2010;13:239–45.
27. Puledda F, Messina R, Goadsby PJ. An update on migraine: current understanding and future directions. J Neurol 2017 Mar 20.
28. Coppola G, Di Lorenzo C, Schoenen J, Pierelli F. Habituation and sensitization in primary headaches. J Headache Pain 2013;14:65.
29. The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia 2013;33:629–808.
30. Yusheng H, Y Li. Typical aura without headache: a case report and review of the literature J Med Case Rep 2015;9:40.
31. Buture A, Khalil M, Ahmed F. Iatrogenic visual aura: a case report and a brief review of the literature Ther Clin Risk Manag 2017;13:643–6.
32. Lipton RB. Tracing transformation: Chronic migraine classification, progression, and epidemiology. Neurology 2009;72:S3–7.
33. Lipton RB. Headache 2011;51;S2:77–83.
34. Scher AI, Stewart WF, et al. Factors associated with the onset and remission of chronic daily headache in a population-based study. Pain 2003;106:81–9.
35. Bigal ME, Lipton RB, Holland PR, Goadsby PJ. Obesity, migraine, and chronic migraine: possible mechanisms of interaction. Neurology 2007;68:1851–61.
36. Wolff HG. Stress and disease. Springfield, IL: Charles C. Thomas; 1953.
37. Guidetti V, Galli F, Fabrizi P, et al. Headache and psychiatric comorbidity: clinical aspects and outcome in an 8-year follow-up study. Cephalalgia 1998;18:455–62.
38. Mongini F, Keller R, Deregibus A, et al. Personality traits, depression and migraine in women: a longitudinal study. Cephalalgia 2003;23:186–92.
39. Hung CI, Liu CY, Yang CH, Wang SJ. The impacts of migraine among outpatients with major depressive disorder at a two-year follow-up. PLoS One 2015;10:e0128087.
40. Frishberg BM, Rosenberg JH, Matchar DB, et al. Evidence-based guidelines in the primary care setting: neuroimaging in patients with nonacute headache. St Paul: US Headache Consortium; 2000.
41. Headache Measurement Set 2014 Revised. American Academy of Neurology. Accessed at www.aan.com/uploadedFiles/Website_Library_Assets/Documents/3.Practice_Management/2.Quality_Improvement/1.Quality_Measures/1.All_Measures/2014.
42. Taylor FR. Lifestyle changes, dietary restrictions, and nutraceuticals in migraine prevention. Techn Reg Anesth Pain Manage 2009;13:28–37.
43. Varkey E, Cider A, Carlsson J, Linde M. Exercise as migraine prophylaxis: A randomized study using relaxation and topiramate as controls. Cephalalgia 2011;14:1428–38.
44. Ahn AH. Why does increased exercise decrease migraine? Curr Pain Headache Rep 2013;17:379.
45. Marmura MJ, Silberstein SD, Schwedt TJ. The acute treatment of migraine in adults: The American Headache Society evidence assessment of migraine pharmacotherapies. Headache 2015;55:3–20.
46. Belvis R, Mas N, Aceituno A. Migraine attack treatment : a tailor-made suit, not one size fits all. Recent Pat CNS Drug Discov 2014;9:26–40.
47. Bigal ME, Serrano D, Buse D, et al. Acute migraine medications and evolution from episodic to chronic migraine: A longitudinal population-based study. Headache 2008;48:1157–68.
48. Bigal ME, Rapoport AM, Sheftell FD, et al. Transformed migraine and medication overuse in a tertiary headache centre--clinical characteristics and treatment outcomes Cephalalgia 2004;24:483–90.
49. Silberstein SD, Holland S, Freitag F, et al; Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Evidence-based guideline update: pharmacologic treatment for episodic migraine prevention in adults: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology 2012;78:1337–45.
50. Diener HC, Dodick DW, Aurora SK, et al. OnabotulinumtoxinA for treatment of chronic migraine: Results from the double-blind, randomized, placebo-controlled phase of the PREEMPT 2 trial Cephalagia year;30:804–14.
51. Wells RE, Markowitz SY, Baron EP, et al. Identifying the factors underlying discontinuation of triptans. Headache 2014;54:278–89.
52. Holland S, Fanning KM, Serrano D, et al. Rates and reasons for discontinuation of triptans and opioids in episodic migraine: results from the American Migraine Prevalence and Prevention (AMPP) study. J Neurol Sci 2013;326:10–7.
53. Seng EK, Rains JA, Nicholson RA, Lipton RB. Improving medication adherence in migraine treatment. Curr Pain Headache Rep 2015;19:24.
54. Nicholson RA, Buse DC, Andrasik F, Lipton RB. Nonpharmacologic treatments for migraine and tension-type headache: how to choose and when to use. Curr Treatment Opt Neurol 2011;13:28–40.
55. Probyn K, Bowers H, Mistry D, et al. Non-pharmacological self-management for people living with migraine or tension-type headache: a systematic review including analysis of intervention components BMJ Open 2017;7:e016670.
56. Lemstra M, Stewart B, Olszynski WP. Effectiveness of multidisciplinary intervention in the treatment of migraine: a randomized clinical trial. Headache 2002;42:845–54.
57. John PJ, Sharma N, Sharma CM, Kankane A. Effectiveness of yoga therapy in the treatment of migraine without aura: a randomized controlled trial. Headache 2007;47:654–61.
58. Harris P, Loveman E, Clegg A, et al. Systematic review of cognitive behavioural therapy for the management of headaches and migraines in adults Br J Pain 2015;9:213–24.
59. Kropp P, Meyer B, Meyer W, Dresler T. An update on behavioral treatments in migraine - current knowledge and future options. Expert Rev Neurother 2017:1–10.
60. Sullivan A, Cousins S, Ridsdale L. Psychological interventions for migraine: a systematic review. J Neurol 2016;263:2369–77.
61. Holroyd KA, Cottrell CK, O’Donnell FJ, et al. Effect of preventive (beta blocker) treatment, behavioural migraine management, or their combination on outcomes of optimised acute treatment in frequent migraine: randomised controlled trial. BMJ 2010;341:c4871.
62. Penzien DB, Rains JC, Andrasik F. Behavioral management of recurrent headache: three decades of experience and empiricism. Appl Psychophysiol Biofeedback 2002;27:163–81.
63. Smith TR, Nicholson RA, Banks JW. A primary care migraine education program has benefit on headache impact and quality of life: results from the mercy migraine management program. Headache 2010;50:600–12.
64. Rothrock JF, Parada VA, Sims C, et al.The impact of intensive patient education on clinical outcome in a clinic-based migraine population. Headache 2006;46:726–31.