User login
CHMP backs therapy for hemophilia A

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for rurioctocog alfa pegol (Adynovi).
Rurioctocog alfa pegol (formerly BAX 855) is a pegylated, full-length, recombinant factor VIII product built on a licensed recombinant factor VIII product (Advate).
The CHMP is recommending that rurioctocog alfa pegol be approved for the treatment and prophylaxis of bleeding in patients age 12 and older with hemophilia A.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
If approved, rurioctocog alfa pegol would be available as a powder and solvent for solution for injection (250 IU, 500 IU, 1000 IU, and 2000 IU).
Phase 3 trials
Rurioctocog alfa pegol has been studied in 3 phase 3 trials.
One study (phase 2/3) included 137 patients, age 12 and older, with previously treated hemophilia A. Results from this trial were published in Blood in July 2015.
Another study included 15 patients with severe hemophilia A who were undergoing surgical procedures. Results were published in Haemophilia in June 2016.
A third study included 66 patients, age 12 and younger, who had previously treated hemophilia A. Results from this trial were presented at the World Federation of Hemophilia 2016 World Congress in July 2016. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for rurioctocog alfa pegol (Adynovi).
Rurioctocog alfa pegol (formerly BAX 855) is a pegylated, full-length, recombinant factor VIII product built on a licensed recombinant factor VIII product (Advate).
The CHMP is recommending that rurioctocog alfa pegol be approved for the treatment and prophylaxis of bleeding in patients age 12 and older with hemophilia A.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
If approved, rurioctocog alfa pegol would be available as a powder and solvent for solution for injection (250 IU, 500 IU, 1000 IU, and 2000 IU).
Phase 3 trials
Rurioctocog alfa pegol has been studied in 3 phase 3 trials.
One study (phase 2/3) included 137 patients, age 12 and older, with previously treated hemophilia A. Results from this trial were published in Blood in July 2015.
Another study included 15 patients with severe hemophilia A who were undergoing surgical procedures. Results were published in Haemophilia in June 2016.
A third study included 66 patients, age 12 and younger, who had previously treated hemophilia A. Results from this trial were presented at the World Federation of Hemophilia 2016 World Congress in July 2016. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for rurioctocog alfa pegol (Adynovi).
Rurioctocog alfa pegol (formerly BAX 855) is a pegylated, full-length, recombinant factor VIII product built on a licensed recombinant factor VIII product (Advate).
The CHMP is recommending that rurioctocog alfa pegol be approved for the treatment and prophylaxis of bleeding in patients age 12 and older with hemophilia A.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
If approved, rurioctocog alfa pegol would be available as a powder and solvent for solution for injection (250 IU, 500 IU, 1000 IU, and 2000 IU).
Phase 3 trials
Rurioctocog alfa pegol has been studied in 3 phase 3 trials.
One study (phase 2/3) included 137 patients, age 12 and older, with previously treated hemophilia A. Results from this trial were published in Blood in July 2015.
Another study included 15 patients with severe hemophilia A who were undergoing surgical procedures. Results were published in Haemophilia in June 2016.
A third study included 66 patients, age 12 and younger, who had previously treated hemophilia A. Results from this trial were presented at the World Federation of Hemophilia 2016 World Congress in July 2016. ![]()
CHMP wants to expand use of BV to include CTCL

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for brentuximab vedotin (BV, Adcetris).
The CHMP is recommending authorization of BV to treat adults with CD30+ cutaneous T-cell lymphoma (CTCL) who have received at least 1 prior systemic therapy.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The EC previously approved BV to treat:
- Adults with relapsed or refractory CD30+ Hodgkin lymphoma (HL) following autologous stem cell transplant (ASCT) or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- Adults with CD30+ HL at increased risk of relapse or progression following ASCT
- Adults with relapsed or refractory systemic anaplastic large-cell lymphoma.
The CHMP’s recommendation to approve BV for CTCL is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in July 2015 and August 2015.
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for brentuximab vedotin (BV, Adcetris).
The CHMP is recommending authorization of BV to treat adults with CD30+ cutaneous T-cell lymphoma (CTCL) who have received at least 1 prior systemic therapy.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The EC previously approved BV to treat:
- Adults with relapsed or refractory CD30+ Hodgkin lymphoma (HL) following autologous stem cell transplant (ASCT) or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- Adults with CD30+ HL at increased risk of relapse or progression following ASCT
- Adults with relapsed or refractory systemic anaplastic large-cell lymphoma.
The CHMP’s recommendation to approve BV for CTCL is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in July 2015 and August 2015.
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for brentuximab vedotin (BV, Adcetris).
The CHMP is recommending authorization of BV to treat adults with CD30+ cutaneous T-cell lymphoma (CTCL) who have received at least 1 prior systemic therapy.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The EC previously approved BV to treat:
- Adults with relapsed or refractory CD30+ Hodgkin lymphoma (HL) following autologous stem cell transplant (ASCT) or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- Adults with CD30+ HL at increased risk of relapse or progression following ASCT
- Adults with relapsed or refractory systemic anaplastic large-cell lymphoma.
The CHMP’s recommendation to approve BV for CTCL is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in July 2015 and August 2015.
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June. ![]()
CHMP recommends letermovir as CMV prophylaxis
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for letermovir (Prevymis), which belongs to a class of non-nucleoside CMV inhibitors known as 3,4 dihydro-quinazolines.
The CHMP is advocating that letermovir be approved as prophylaxis for cytomegalovirus (CMV) reactivation and disease in patients who receive immunosuppressants after allogeneic hematopoietic stem cell transplant (HSCT).
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
Letermovir previously received an orphan designation from the European Medicines Agency’s Committee for Orphan Medicinal Products in June 2012. Now, the committee will assess whether the orphan designation should be maintained.
Phase 3 trial
The CHMP’s recommendation to authorize use of letermovir is based on data from a phase 3 trial. Results from this trial were presented at the 2017 BMT Tandem Meetings.
The trial enrolled adult recipients of allogeneic HSCTs who were CMV-seropositive. Patients were randomized (2:1) to receive either letermovir (at a dose of 480 mg once-daily, adjusted to 240 mg when co-administered with cyclosporine) or placebo.
Study drug was initiated after HSCT (at any time from day 0 to 28 post-transplant) and continued through week 14 post-transplant. Patients were monitored through week 24 post-HSCT for the primary efficacy endpoint, with continued follow-up through week 48.
Among the 565 treated patients, 34% were engrafted at baseline, and 30% had one or more factors associated with additional risk for CMV reactivation. The most common primary reasons for transplant were acute myeloid leukemia (38%), myelodysplastic syndromes (16%), and lymphoma (12%).
Thirty eight percent of patients in the letermovir arm and 61% in the placebo arm failed prophylaxis.
Reasons for failure (in the letermovir and placebo arms, respectively) included:
- Clinically significant CMV infection—18% vs 42%
- Initiation of PET based on documented CMV viremia—16% vs 40%
- CMV end-organ disease—2% for both
- Study discontinuation before week 24—17% vs 16%
- Missing outcome in week 24 visit window—3% for both.
The stratum-adjusted treatment difference for letermovir vs placebo was -23.5 (95% CI, -32.5, -14.6, P<0.0001).
The Kaplan-Meier event rate for all-cause mortality in the letermovir and placebo arms, respectively, was 12% and 17% at week 24 and 24% and 28% at week 48.
Common adverse events (in the letermovir and placebo arms, respectively) were nausea (27% vs 23%), diarrhea (26% vs 24%), vomiting (19% vs 14%), peripheral edema (14% vs 9%), cough (14% vs 10%), headache (14% vs 9%), fatigue (13% vs 11%), and abdominal pain (12% vs 9%).
The cardiac adverse event rate (regardless of investigator-assessed causality) was 13% in the letermovir arm and 6% in the placebo arm. The most common cardiac adverse events (in the letermovir and placebo arms, respectively) were tachycardia (4% vs 2%) and atrial fibrillation (3% vs 1%). ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for letermovir (Prevymis), which belongs to a class of non-nucleoside CMV inhibitors known as 3,4 dihydro-quinazolines.
The CHMP is advocating that letermovir be approved as prophylaxis for cytomegalovirus (CMV) reactivation and disease in patients who receive immunosuppressants after allogeneic hematopoietic stem cell transplant (HSCT).
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
Letermovir previously received an orphan designation from the European Medicines Agency’s Committee for Orphan Medicinal Products in June 2012. Now, the committee will assess whether the orphan designation should be maintained.
Phase 3 trial
The CHMP’s recommendation to authorize use of letermovir is based on data from a phase 3 trial. Results from this trial were presented at the 2017 BMT Tandem Meetings.
The trial enrolled adult recipients of allogeneic HSCTs who were CMV-seropositive. Patients were randomized (2:1) to receive either letermovir (at a dose of 480 mg once-daily, adjusted to 240 mg when co-administered with cyclosporine) or placebo.
Study drug was initiated after HSCT (at any time from day 0 to 28 post-transplant) and continued through week 14 post-transplant. Patients were monitored through week 24 post-HSCT for the primary efficacy endpoint, with continued follow-up through week 48.
Among the 565 treated patients, 34% were engrafted at baseline, and 30% had one or more factors associated with additional risk for CMV reactivation. The most common primary reasons for transplant were acute myeloid leukemia (38%), myelodysplastic syndromes (16%), and lymphoma (12%).
Thirty eight percent of patients in the letermovir arm and 61% in the placebo arm failed prophylaxis.
Reasons for failure (in the letermovir and placebo arms, respectively) included:
- Clinically significant CMV infection—18% vs 42%
- Initiation of PET based on documented CMV viremia—16% vs 40%
- CMV end-organ disease—2% for both
- Study discontinuation before week 24—17% vs 16%
- Missing outcome in week 24 visit window—3% for both.
The stratum-adjusted treatment difference for letermovir vs placebo was -23.5 (95% CI, -32.5, -14.6, P<0.0001).
The Kaplan-Meier event rate for all-cause mortality in the letermovir and placebo arms, respectively, was 12% and 17% at week 24 and 24% and 28% at week 48.
Common adverse events (in the letermovir and placebo arms, respectively) were nausea (27% vs 23%), diarrhea (26% vs 24%), vomiting (19% vs 14%), peripheral edema (14% vs 9%), cough (14% vs 10%), headache (14% vs 9%), fatigue (13% vs 11%), and abdominal pain (12% vs 9%).
The cardiac adverse event rate (regardless of investigator-assessed causality) was 13% in the letermovir arm and 6% in the placebo arm. The most common cardiac adverse events (in the letermovir and placebo arms, respectively) were tachycardia (4% vs 2%) and atrial fibrillation (3% vs 1%). ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for letermovir (Prevymis), which belongs to a class of non-nucleoside CMV inhibitors known as 3,4 dihydro-quinazolines.
The CHMP is advocating that letermovir be approved as prophylaxis for cytomegalovirus (CMV) reactivation and disease in patients who receive immunosuppressants after allogeneic hematopoietic stem cell transplant (HSCT).
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
Letermovir previously received an orphan designation from the European Medicines Agency’s Committee for Orphan Medicinal Products in June 2012. Now, the committee will assess whether the orphan designation should be maintained.
Phase 3 trial
The CHMP’s recommendation to authorize use of letermovir is based on data from a phase 3 trial. Results from this trial were presented at the 2017 BMT Tandem Meetings.
The trial enrolled adult recipients of allogeneic HSCTs who were CMV-seropositive. Patients were randomized (2:1) to receive either letermovir (at a dose of 480 mg once-daily, adjusted to 240 mg when co-administered with cyclosporine) or placebo.
Study drug was initiated after HSCT (at any time from day 0 to 28 post-transplant) and continued through week 14 post-transplant. Patients were monitored through week 24 post-HSCT for the primary efficacy endpoint, with continued follow-up through week 48.
Among the 565 treated patients, 34% were engrafted at baseline, and 30% had one or more factors associated with additional risk for CMV reactivation. The most common primary reasons for transplant were acute myeloid leukemia (38%), myelodysplastic syndromes (16%), and lymphoma (12%).
Thirty eight percent of patients in the letermovir arm and 61% in the placebo arm failed prophylaxis.
Reasons for failure (in the letermovir and placebo arms, respectively) included:
- Clinically significant CMV infection—18% vs 42%
- Initiation of PET based on documented CMV viremia—16% vs 40%
- CMV end-organ disease—2% for both
- Study discontinuation before week 24—17% vs 16%
- Missing outcome in week 24 visit window—3% for both.
The stratum-adjusted treatment difference for letermovir vs placebo was -23.5 (95% CI, -32.5, -14.6, P<0.0001).
The Kaplan-Meier event rate for all-cause mortality in the letermovir and placebo arms, respectively, was 12% and 17% at week 24 and 24% and 28% at week 48.
Common adverse events (in the letermovir and placebo arms, respectively) were nausea (27% vs 23%), diarrhea (26% vs 24%), vomiting (19% vs 14%), peripheral edema (14% vs 9%), cough (14% vs 10%), headache (14% vs 9%), fatigue (13% vs 11%), and abdominal pain (12% vs 9%).
The cardiac adverse event rate (regardless of investigator-assessed causality) was 13% in the letermovir arm and 6% in the placebo arm. The most common cardiac adverse events (in the letermovir and placebo arms, respectively) were tachycardia (4% vs 2%) and atrial fibrillation (3% vs 1%). ![]()
FDA approves cariprazine for schizophrenia maintenance treatment
The Food and Drug Administration has approved a supplemental new drug application for cariprazine (Vraylar) for maintenance treatment of adults with schizophrenia, the drug’s licensor, Allergan, announced Nov. 13. The drug was approved in 2015 for the acute treatment of schizophrenia or for manic or mixed episodes of bipolar I disorder in adults.
The efficacy of the atypical antipsychotic for maintenance treatment of schizophrenia was demonstrated by a 72-week multinational, double-blind, randomized study of a stabilized cariprazine dose of 3, 6, or 9 mg daily, compared with placebo. The daily dose had a significant effect on the study’s primary endpoint – time to relapse. Nearly twice as many placebo-treated patients as cariprazine-treated patients relapsed (49.5% vs. 29.7%).
“The goal of clinicians is to minimize relapses, which can cause significant personal distress and can often have serious implications for a patient’s health,” said Herbert Y. Meltzer, MD, professor of psychiatry and behavioral sciences, pharmacology, and physiology, at Northwestern University, Chicago, in the release. “The approval of Vraylar for the maintenance treatment of schizophrenia provides an important therapy for patients and physicians who are in need of long-term treatment options.”
Cariprazine may cause rash, pruritus, urticaria, and events suggestive of angioedema and is not approved for patients with dementia-related psychosis, as it has an increased mortality risk for elderly patients with dementia. In approved schizophrenia patients, it carries a risk of extrapyramidal symptoms and akathisia.
The Food and Drug Administration has approved a supplemental new drug application for cariprazine (Vraylar) for maintenance treatment of adults with schizophrenia, the drug’s licensor, Allergan, announced Nov. 13. The drug was approved in 2015 for the acute treatment of schizophrenia or for manic or mixed episodes of bipolar I disorder in adults.
The efficacy of the atypical antipsychotic for maintenance treatment of schizophrenia was demonstrated by a 72-week multinational, double-blind, randomized study of a stabilized cariprazine dose of 3, 6, or 9 mg daily, compared with placebo. The daily dose had a significant effect on the study’s primary endpoint – time to relapse. Nearly twice as many placebo-treated patients as cariprazine-treated patients relapsed (49.5% vs. 29.7%).
“The goal of clinicians is to minimize relapses, which can cause significant personal distress and can often have serious implications for a patient’s health,” said Herbert Y. Meltzer, MD, professor of psychiatry and behavioral sciences, pharmacology, and physiology, at Northwestern University, Chicago, in the release. “The approval of Vraylar for the maintenance treatment of schizophrenia provides an important therapy for patients and physicians who are in need of long-term treatment options.”
Cariprazine may cause rash, pruritus, urticaria, and events suggestive of angioedema and is not approved for patients with dementia-related psychosis, as it has an increased mortality risk for elderly patients with dementia. In approved schizophrenia patients, it carries a risk of extrapyramidal symptoms and akathisia.
The Food and Drug Administration has approved a supplemental new drug application for cariprazine (Vraylar) for maintenance treatment of adults with schizophrenia, the drug’s licensor, Allergan, announced Nov. 13. The drug was approved in 2015 for the acute treatment of schizophrenia or for manic or mixed episodes of bipolar I disorder in adults.
The efficacy of the atypical antipsychotic for maintenance treatment of schizophrenia was demonstrated by a 72-week multinational, double-blind, randomized study of a stabilized cariprazine dose of 3, 6, or 9 mg daily, compared with placebo. The daily dose had a significant effect on the study’s primary endpoint – time to relapse. Nearly twice as many placebo-treated patients as cariprazine-treated patients relapsed (49.5% vs. 29.7%).
“The goal of clinicians is to minimize relapses, which can cause significant personal distress and can often have serious implications for a patient’s health,” said Herbert Y. Meltzer, MD, professor of psychiatry and behavioral sciences, pharmacology, and physiology, at Northwestern University, Chicago, in the release. “The approval of Vraylar for the maintenance treatment of schizophrenia provides an important therapy for patients and physicians who are in need of long-term treatment options.”
Cariprazine may cause rash, pruritus, urticaria, and events suggestive of angioedema and is not approved for patients with dementia-related psychosis, as it has an increased mortality risk for elderly patients with dementia. In approved schizophrenia patients, it carries a risk of extrapyramidal symptoms and akathisia.
Vaccine coverage high among U.S. toddlers in 2016, but gaps remain
, said Holly A. Hill, MD, PhD, and her associates at the National Center for Immunization and Respiratory Diseases, Atlanta.
Coverage still was below 90% for vaccines that needed booster doses during the second year of life (four or more doses of DTaP and pneumococcal conjugate vaccine [PCV] and Haemophilus influenzae type b [Hib] full series) and for other recommended vaccines (hepatitis B [HepB] birth dose, rotavirus, and hepatitis A [HepA]), they reported in Morbidity and Mortality Weekly Report.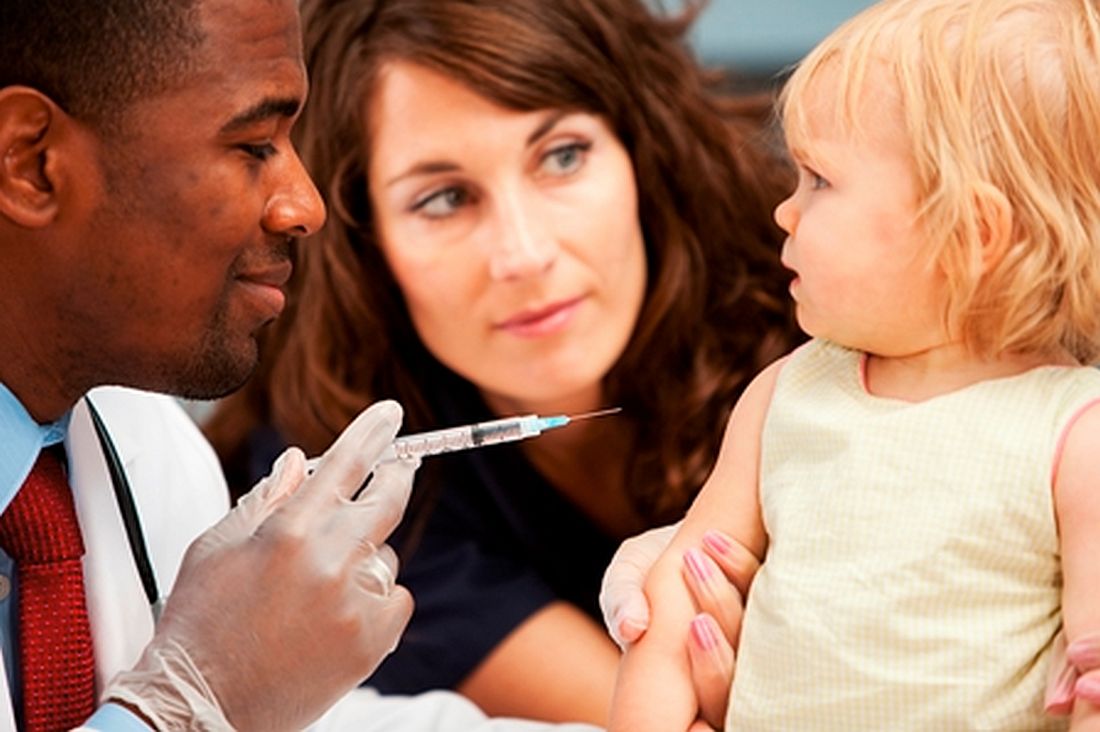
Coverage was estimated to be 61% for two or more doses of HepA vaccine, 71% of the HepB birth dose, 74% of a completed series of rotavirus vaccine, and 71% of the combined seven-vaccine series (four or more doses of DTaP; three or more doses of poliovirus vaccine; one or more doses of measles-containing vaccine; three or four doses of Hib [depending upon product type of vaccine]; three or more doses of HepB; one or more doses of varicella vaccine; and four or more doses of PCV).
Fewer than 1% of children received no vaccinations.
Coverage of most vaccines in 2016 was lower in non-Hispanic black children, compared with non-Hispanic white children. It also was lower for children living below the federal poverty level, compared with children living at or above the poverty level. For Medicaid children, vaccination coverage was lower by 3%-13% than among children who had private insurance; for children with no insurance, vaccination coverage was lower by 12%-25% than among children with private insurance, the investigators reported.
Uninsured children “are eligible for the Vaccines for Children (VFC) program, which was designed to increase access to vaccination among children through age 18 years who might not otherwise be vaccinated because of inability to pay,” the researchers said. “Some families might not be aware of the VFC program, be unable to afford fees associated with visits to a vaccine provider, or might need assistance locating a physician who participates in the VFC program. Children living below poverty and up to a certain percentage above the poverty level are eligible for Medicaid … and are entitled to VFC vaccines.”
The investigators cited language barriers, lack of trust in providers, transportation problems, inconvenient office hours, and other provider- and system-level factors as health care–access barriers among publicly insured children.
“These data indicate that the immunization safety net is not reaching all children early in life,” Dr. Hill and her associates said. “Health care providers can increase vaccination coverage using evidence-based strategies such as provider reminders, standing orders to provide vaccination whenever appropriate, and immunization information systems” such as www.thecommunityguide.org/topic/vaccination.
Read more in MMWR (2017 Nov 3;66[43]:1171-7).
, said Holly A. Hill, MD, PhD, and her associates at the National Center for Immunization and Respiratory Diseases, Atlanta.
Coverage still was below 90% for vaccines that needed booster doses during the second year of life (four or more doses of DTaP and pneumococcal conjugate vaccine [PCV] and Haemophilus influenzae type b [Hib] full series) and for other recommended vaccines (hepatitis B [HepB] birth dose, rotavirus, and hepatitis A [HepA]), they reported in Morbidity and Mortality Weekly Report.
Coverage was estimated to be 61% for two or more doses of HepA vaccine, 71% of the HepB birth dose, 74% of a completed series of rotavirus vaccine, and 71% of the combined seven-vaccine series (four or more doses of DTaP; three or more doses of poliovirus vaccine; one or more doses of measles-containing vaccine; three or four doses of Hib [depending upon product type of vaccine]; three or more doses of HepB; one or more doses of varicella vaccine; and four or more doses of PCV).
Fewer than 1% of children received no vaccinations.
Coverage of most vaccines in 2016 was lower in non-Hispanic black children, compared with non-Hispanic white children. It also was lower for children living below the federal poverty level, compared with children living at or above the poverty level. For Medicaid children, vaccination coverage was lower by 3%-13% than among children who had private insurance; for children with no insurance, vaccination coverage was lower by 12%-25% than among children with private insurance, the investigators reported.
Uninsured children “are eligible for the Vaccines for Children (VFC) program, which was designed to increase access to vaccination among children through age 18 years who might not otherwise be vaccinated because of inability to pay,” the researchers said. “Some families might not be aware of the VFC program, be unable to afford fees associated with visits to a vaccine provider, or might need assistance locating a physician who participates in the VFC program. Children living below poverty and up to a certain percentage above the poverty level are eligible for Medicaid … and are entitled to VFC vaccines.”
The investigators cited language barriers, lack of trust in providers, transportation problems, inconvenient office hours, and other provider- and system-level factors as health care–access barriers among publicly insured children.
“These data indicate that the immunization safety net is not reaching all children early in life,” Dr. Hill and her associates said. “Health care providers can increase vaccination coverage using evidence-based strategies such as provider reminders, standing orders to provide vaccination whenever appropriate, and immunization information systems” such as www.thecommunityguide.org/topic/vaccination.
Read more in MMWR (2017 Nov 3;66[43]:1171-7).
, said Holly A. Hill, MD, PhD, and her associates at the National Center for Immunization and Respiratory Diseases, Atlanta.
Coverage still was below 90% for vaccines that needed booster doses during the second year of life (four or more doses of DTaP and pneumococcal conjugate vaccine [PCV] and Haemophilus influenzae type b [Hib] full series) and for other recommended vaccines (hepatitis B [HepB] birth dose, rotavirus, and hepatitis A [HepA]), they reported in Morbidity and Mortality Weekly Report.
Coverage was estimated to be 61% for two or more doses of HepA vaccine, 71% of the HepB birth dose, 74% of a completed series of rotavirus vaccine, and 71% of the combined seven-vaccine series (four or more doses of DTaP; three or more doses of poliovirus vaccine; one or more doses of measles-containing vaccine; three or four doses of Hib [depending upon product type of vaccine]; three or more doses of HepB; one or more doses of varicella vaccine; and four or more doses of PCV).
Fewer than 1% of children received no vaccinations.
Coverage of most vaccines in 2016 was lower in non-Hispanic black children, compared with non-Hispanic white children. It also was lower for children living below the federal poverty level, compared with children living at or above the poverty level. For Medicaid children, vaccination coverage was lower by 3%-13% than among children who had private insurance; for children with no insurance, vaccination coverage was lower by 12%-25% than among children with private insurance, the investigators reported.
Uninsured children “are eligible for the Vaccines for Children (VFC) program, which was designed to increase access to vaccination among children through age 18 years who might not otherwise be vaccinated because of inability to pay,” the researchers said. “Some families might not be aware of the VFC program, be unable to afford fees associated with visits to a vaccine provider, or might need assistance locating a physician who participates in the VFC program. Children living below poverty and up to a certain percentage above the poverty level are eligible for Medicaid … and are entitled to VFC vaccines.”
The investigators cited language barriers, lack of trust in providers, transportation problems, inconvenient office hours, and other provider- and system-level factors as health care–access barriers among publicly insured children.
“These data indicate that the immunization safety net is not reaching all children early in life,” Dr. Hill and her associates said. “Health care providers can increase vaccination coverage using evidence-based strategies such as provider reminders, standing orders to provide vaccination whenever appropriate, and immunization information systems” such as www.thecommunityguide.org/topic/vaccination.
Read more in MMWR (2017 Nov 3;66[43]:1171-7).
FROM MMWR
Barnacles that come with wisdom
One of the most common reasons for visits to the dermatologist is a brown or flesh-colored lesion on the face or body that is concerning to the patient either because it’s changing; it’s scabbing or bleeding; it feels rough on the surface, and they can’t stand touching it – or because the patient just thinks they’re plain unsightly. After assessing and ruling out a malignant skin cancer or precancerous lesion clinically, the good news is that, in most cases, these turn out to be seborrheic keratoses (SK), benign growths. Patients are often reassured and relieved when we tell them we nickname SKs “barnacles that come with wisdom.” But then they often ask, “can I get rid of them?”
The answer is yes. There are many ways to rid people of these pesky lesions, but the reality is that, even with coding and documentation of an irritated SK, they are rarely covered by insurance. This leaves patients with the choice of whether to pay out of pocket for a cosmetic procedure and puts the dermatologist in a position of either charging the patient for a cosmetic procedure or treating to make the patient happy and not getting compensated for their services. For the cosmetic dermatologist, discussing cosmetic procedures with patients is an easy transition, but for the dermatologist who does not regularly practice cosmetic or fee-for-service dermatology – the majority of dermatologists in the United States – this can put them in an awkward position. According to a 2013 workforce survey, 20% of the dermatology market is cosmetic, while 80% is medical, surgical, and dermatopathology.1

Given the clinically verrucous nature of SKs, a viral etiology, particularly human papilloma virus (HPV), has often been sought. HPV subtypes have been seen in genital “SKs” and HPV-23 has been associated with stucco keratoses, which often resemble the SK family and are found on the legs of aging patients. However, multiple reports have refuted the presence of HPV in nongenital SK lesions.3
Until a potential gene therapy is available, current treatment options for patients who want to have their SKs treated include cryotherapy, electrodesiccation, curettage, or laser therapy with a KTP (potassium titanyl phosphate) laser or an ablative laser, such as a CO2 laser. Cryotherapy, curettage, and electrodesiccation, while effective, run a risk of dyspigmentation, especially hypopigmentation in Fitzpatrick Skin Types III-VI. KTP and ablative lasers can be effective, but are often less cost-effective methods to achieve similar results as cryotherapy or electrodesiccation. Clinical trial data have been published on a topical hydrogen peroxide–based solution, A-101, which is not currently approved by the Food and Drug Administration. In a recently published study, 68% of patients were clear or near clear of SKs on the face with the 40% A-101 solution after up to two treatments.4
SKs are a part of a cosmetic dermatology practice that arises on a daily basis and are often a concern for patients. Discussion of their management, coverage, and treatment options will resonate with every practicing dermatologist.
Dr. Wesley and Dr. Talakoub are cocontributors to this column. Dr. Wesley practices dermatology in Beverly Hills, Calif. Dr. Talakoub is in private practice in McLean, Va. This month’s column is by Dr. Wesley. Dr. Wesley has served on an advisory board panel for Aclaris, which is developing A-101. Write to them at [email protected].
References:
1: www.harriswilliams.com/system/files/industry_update/dermatology_market_overview.pdf
2: Am J Dermatopathol. 2014 Aug;36(8):635-42.
3: Indian J Dermatol. 2013 Jul;58(4):326.
4. Dermatol Surg. 2017 Sep 4. doi: 10.1097/DSS.0000000000001302..
One of the most common reasons for visits to the dermatologist is a brown or flesh-colored lesion on the face or body that is concerning to the patient either because it’s changing; it’s scabbing or bleeding; it feels rough on the surface, and they can’t stand touching it – or because the patient just thinks they’re plain unsightly. After assessing and ruling out a malignant skin cancer or precancerous lesion clinically, the good news is that, in most cases, these turn out to be seborrheic keratoses (SK), benign growths. Patients are often reassured and relieved when we tell them we nickname SKs “barnacles that come with wisdom.” But then they often ask, “can I get rid of them?”
The answer is yes. There are many ways to rid people of these pesky lesions, but the reality is that, even with coding and documentation of an irritated SK, they are rarely covered by insurance. This leaves patients with the choice of whether to pay out of pocket for a cosmetic procedure and puts the dermatologist in a position of either charging the patient for a cosmetic procedure or treating to make the patient happy and not getting compensated for their services. For the cosmetic dermatologist, discussing cosmetic procedures with patients is an easy transition, but for the dermatologist who does not regularly practice cosmetic or fee-for-service dermatology – the majority of dermatologists in the United States – this can put them in an awkward position. According to a 2013 workforce survey, 20% of the dermatology market is cosmetic, while 80% is medical, surgical, and dermatopathology.1
Given the clinically verrucous nature of SKs, a viral etiology, particularly human papilloma virus (HPV), has often been sought. HPV subtypes have been seen in genital “SKs” and HPV-23 has been associated with stucco keratoses, which often resemble the SK family and are found on the legs of aging patients. However, multiple reports have refuted the presence of HPV in nongenital SK lesions.3
Until a potential gene therapy is available, current treatment options for patients who want to have their SKs treated include cryotherapy, electrodesiccation, curettage, or laser therapy with a KTP (potassium titanyl phosphate) laser or an ablative laser, such as a CO2 laser. Cryotherapy, curettage, and electrodesiccation, while effective, run a risk of dyspigmentation, especially hypopigmentation in Fitzpatrick Skin Types III-VI. KTP and ablative lasers can be effective, but are often less cost-effective methods to achieve similar results as cryotherapy or electrodesiccation. Clinical trial data have been published on a topical hydrogen peroxide–based solution, A-101, which is not currently approved by the Food and Drug Administration. In a recently published study, 68% of patients were clear or near clear of SKs on the face with the 40% A-101 solution after up to two treatments.4
SKs are a part of a cosmetic dermatology practice that arises on a daily basis and are often a concern for patients. Discussion of their management, coverage, and treatment options will resonate with every practicing dermatologist.
Dr. Wesley and Dr. Talakoub are cocontributors to this column. Dr. Wesley practices dermatology in Beverly Hills, Calif. Dr. Talakoub is in private practice in McLean, Va. This month’s column is by Dr. Wesley. Dr. Wesley has served on an advisory board panel for Aclaris, which is developing A-101. Write to them at [email protected].
References:
1: www.harriswilliams.com/system/files/industry_update/dermatology_market_overview.pdf
2: Am J Dermatopathol. 2014 Aug;36(8):635-42.
3: Indian J Dermatol. 2013 Jul;58(4):326.
4. Dermatol Surg. 2017 Sep 4. doi: 10.1097/DSS.0000000000001302..
One of the most common reasons for visits to the dermatologist is a brown or flesh-colored lesion on the face or body that is concerning to the patient either because it’s changing; it’s scabbing or bleeding; it feels rough on the surface, and they can’t stand touching it – or because the patient just thinks they’re plain unsightly. After assessing and ruling out a malignant skin cancer or precancerous lesion clinically, the good news is that, in most cases, these turn out to be seborrheic keratoses (SK), benign growths. Patients are often reassured and relieved when we tell them we nickname SKs “barnacles that come with wisdom.” But then they often ask, “can I get rid of them?”
The answer is yes. There are many ways to rid people of these pesky lesions, but the reality is that, even with coding and documentation of an irritated SK, they are rarely covered by insurance. This leaves patients with the choice of whether to pay out of pocket for a cosmetic procedure and puts the dermatologist in a position of either charging the patient for a cosmetic procedure or treating to make the patient happy and not getting compensated for their services. For the cosmetic dermatologist, discussing cosmetic procedures with patients is an easy transition, but for the dermatologist who does not regularly practice cosmetic or fee-for-service dermatology – the majority of dermatologists in the United States – this can put them in an awkward position. According to a 2013 workforce survey, 20% of the dermatology market is cosmetic, while 80% is medical, surgical, and dermatopathology.1
Given the clinically verrucous nature of SKs, a viral etiology, particularly human papilloma virus (HPV), has often been sought. HPV subtypes have been seen in genital “SKs” and HPV-23 has been associated with stucco keratoses, which often resemble the SK family and are found on the legs of aging patients. However, multiple reports have refuted the presence of HPV in nongenital SK lesions.3
Until a potential gene therapy is available, current treatment options for patients who want to have their SKs treated include cryotherapy, electrodesiccation, curettage, or laser therapy with a KTP (potassium titanyl phosphate) laser or an ablative laser, such as a CO2 laser. Cryotherapy, curettage, and electrodesiccation, while effective, run a risk of dyspigmentation, especially hypopigmentation in Fitzpatrick Skin Types III-VI. KTP and ablative lasers can be effective, but are often less cost-effective methods to achieve similar results as cryotherapy or electrodesiccation. Clinical trial data have been published on a topical hydrogen peroxide–based solution, A-101, which is not currently approved by the Food and Drug Administration. In a recently published study, 68% of patients were clear or near clear of SKs on the face with the 40% A-101 solution after up to two treatments.4
SKs are a part of a cosmetic dermatology practice that arises on a daily basis and are often a concern for patients. Discussion of their management, coverage, and treatment options will resonate with every practicing dermatologist.
Dr. Wesley and Dr. Talakoub are cocontributors to this column. Dr. Wesley practices dermatology in Beverly Hills, Calif. Dr. Talakoub is in private practice in McLean, Va. This month’s column is by Dr. Wesley. Dr. Wesley has served on an advisory board panel for Aclaris, which is developing A-101. Write to them at [email protected].
References:
1: www.harriswilliams.com/system/files/industry_update/dermatology_market_overview.pdf
2: Am J Dermatopathol. 2014 Aug;36(8):635-42.
3: Indian J Dermatol. 2013 Jul;58(4):326.
4. Dermatol Surg. 2017 Sep 4. doi: 10.1097/DSS.0000000000001302..
CheckMate 214: Updated results for RCC focus on PD-L1 expression, QOL
NATIONAL HARBOR, MD. – The benefits of combined treatment with the immune checkpoint inhibitors nivolumab and ipilimumab (nivo/ipi) vs. the tyrosine kinase inhibitor sunitinib as demonstrated in intermediate- to poor-risk renal cell carcinoma patients in the CheckMate 214 trial were observed across baseline programmed death–ligand 1 (PD-L1) expression levels, according to subgroup analyses from the open-label phase 3 trial.
However, those with PD-L1–positive tumors – defined as tumors with PD-L1 expression in 1% or more of cells – had improved outcomes, compared with those with PD-L1–negative tumors. This was true for all three co-primary endpoints of the study: overall response rate, progression-free survival, and overall survival, Robert J. Motzer, MD, reported at the annual meeting of the Society for Immunotherapy of Cancer.

“For progression-free survival ... [there was] a strong signal in patients who were PD-L1 expression–positive, but not so in those with PD-L1–negative tumors,” he said (P = .003 and .9670, respectively). “In the overall survival endpoint ... patients benefited with longer survival with nivo/ipi, regardless of PD-L1 expression, but the relative benefit seemed higher in patients expressing PD-L1 (P less than .0001 and .0249, respectively).”
The primary efficacy results of CheckMate 214 were reported in September at the European Society of Medical Oncology. The study enrolled 1,096 patients with treatment-naive advanced or metastatic clear-cell renal cell carcinoma with measurable disease and adequate performance status who were stratified by prognostic score and geographical region and randomly assigned to receive either 3 mg/kg nivolumab and 1 mg/kg ipilimumab every 3 weeks for four doses, then 3 mg/kg nivolumab monotherapy every other week, or 50 mg oral sunitinib once daily for 4 weeks in a 6-week cycle. Treatment continued until patients progressed or experienced unacceptable toxicity.
Most of the patients in the study (847 of 1,096) had intermediate- to poor-risk disease and most of those (about 70%) were PD-L1 negative.
Overall, the study met two of the primary endpoints, demonstrating superior overall survival and overall response rates with nivo/ipi vs. sunitinib in intermediate/poor-risk patients with treatment-naive advanced renal cell carcinoma, Dr. Motzer said.
In addition to presenting the subgroup data regarding outcomes across PD-L1 expression levels at the meeting, he also presented new data showing improved self-reported quality of life among patients treated with nivo/ipi vs. sunitinib. Quality of life was measured using the National Comprehensive Cancer Network/ Functional Assessment Of Cancer Therapy–Kidney Symptom Index 19 questionnaire, which “looks at questions particularly relevant to renal cell carcinoma patients,” he said.
The mean change in questionnaire scores from baseline was consistently better in the nivo/ipi arm. At 104 weeks the mean change was about +5 points with nivo/ipi vs. about –7 points with sunitinib.
“These results support the use of nivo/ipi as a new first-line standard of care option for patients with intermediate/poor-risk advanced [renal cell carcinoma],” he concluded.
CheckMate 214 was funded by Bristol-Myers Squibb and Ono Pharmaceutical. Dr. Motzer reported ownership interest in Armo Biosciences.
NATIONAL HARBOR, MD. – The benefits of combined treatment with the immune checkpoint inhibitors nivolumab and ipilimumab (nivo/ipi) vs. the tyrosine kinase inhibitor sunitinib as demonstrated in intermediate- to poor-risk renal cell carcinoma patients in the CheckMate 214 trial were observed across baseline programmed death–ligand 1 (PD-L1) expression levels, according to subgroup analyses from the open-label phase 3 trial.
However, those with PD-L1–positive tumors – defined as tumors with PD-L1 expression in 1% or more of cells – had improved outcomes, compared with those with PD-L1–negative tumors. This was true for all three co-primary endpoints of the study: overall response rate, progression-free survival, and overall survival, Robert J. Motzer, MD, reported at the annual meeting of the Society for Immunotherapy of Cancer.
“For progression-free survival ... [there was] a strong signal in patients who were PD-L1 expression–positive, but not so in those with PD-L1–negative tumors,” he said (P = .003 and .9670, respectively). “In the overall survival endpoint ... patients benefited with longer survival with nivo/ipi, regardless of PD-L1 expression, but the relative benefit seemed higher in patients expressing PD-L1 (P less than .0001 and .0249, respectively).”
The primary efficacy results of CheckMate 214 were reported in September at the European Society of Medical Oncology. The study enrolled 1,096 patients with treatment-naive advanced or metastatic clear-cell renal cell carcinoma with measurable disease and adequate performance status who were stratified by prognostic score and geographical region and randomly assigned to receive either 3 mg/kg nivolumab and 1 mg/kg ipilimumab every 3 weeks for four doses, then 3 mg/kg nivolumab monotherapy every other week, or 50 mg oral sunitinib once daily for 4 weeks in a 6-week cycle. Treatment continued until patients progressed or experienced unacceptable toxicity.
Most of the patients in the study (847 of 1,096) had intermediate- to poor-risk disease and most of those (about 70%) were PD-L1 negative.
Overall, the study met two of the primary endpoints, demonstrating superior overall survival and overall response rates with nivo/ipi vs. sunitinib in intermediate/poor-risk patients with treatment-naive advanced renal cell carcinoma, Dr. Motzer said.
In addition to presenting the subgroup data regarding outcomes across PD-L1 expression levels at the meeting, he also presented new data showing improved self-reported quality of life among patients treated with nivo/ipi vs. sunitinib. Quality of life was measured using the National Comprehensive Cancer Network/ Functional Assessment Of Cancer Therapy–Kidney Symptom Index 19 questionnaire, which “looks at questions particularly relevant to renal cell carcinoma patients,” he said.
The mean change in questionnaire scores from baseline was consistently better in the nivo/ipi arm. At 104 weeks the mean change was about +5 points with nivo/ipi vs. about –7 points with sunitinib.
“These results support the use of nivo/ipi as a new first-line standard of care option for patients with intermediate/poor-risk advanced [renal cell carcinoma],” he concluded.
CheckMate 214 was funded by Bristol-Myers Squibb and Ono Pharmaceutical. Dr. Motzer reported ownership interest in Armo Biosciences.
NATIONAL HARBOR, MD. – The benefits of combined treatment with the immune checkpoint inhibitors nivolumab and ipilimumab (nivo/ipi) vs. the tyrosine kinase inhibitor sunitinib as demonstrated in intermediate- to poor-risk renal cell carcinoma patients in the CheckMate 214 trial were observed across baseline programmed death–ligand 1 (PD-L1) expression levels, according to subgroup analyses from the open-label phase 3 trial.
However, those with PD-L1–positive tumors – defined as tumors with PD-L1 expression in 1% or more of cells – had improved outcomes, compared with those with PD-L1–negative tumors. This was true for all three co-primary endpoints of the study: overall response rate, progression-free survival, and overall survival, Robert J. Motzer, MD, reported at the annual meeting of the Society for Immunotherapy of Cancer.
“For progression-free survival ... [there was] a strong signal in patients who were PD-L1 expression–positive, but not so in those with PD-L1–negative tumors,” he said (P = .003 and .9670, respectively). “In the overall survival endpoint ... patients benefited with longer survival with nivo/ipi, regardless of PD-L1 expression, but the relative benefit seemed higher in patients expressing PD-L1 (P less than .0001 and .0249, respectively).”
The primary efficacy results of CheckMate 214 were reported in September at the European Society of Medical Oncology. The study enrolled 1,096 patients with treatment-naive advanced or metastatic clear-cell renal cell carcinoma with measurable disease and adequate performance status who were stratified by prognostic score and geographical region and randomly assigned to receive either 3 mg/kg nivolumab and 1 mg/kg ipilimumab every 3 weeks for four doses, then 3 mg/kg nivolumab monotherapy every other week, or 50 mg oral sunitinib once daily for 4 weeks in a 6-week cycle. Treatment continued until patients progressed or experienced unacceptable toxicity.
Most of the patients in the study (847 of 1,096) had intermediate- to poor-risk disease and most of those (about 70%) were PD-L1 negative.
Overall, the study met two of the primary endpoints, demonstrating superior overall survival and overall response rates with nivo/ipi vs. sunitinib in intermediate/poor-risk patients with treatment-naive advanced renal cell carcinoma, Dr. Motzer said.
In addition to presenting the subgroup data regarding outcomes across PD-L1 expression levels at the meeting, he also presented new data showing improved self-reported quality of life among patients treated with nivo/ipi vs. sunitinib. Quality of life was measured using the National Comprehensive Cancer Network/ Functional Assessment Of Cancer Therapy–Kidney Symptom Index 19 questionnaire, which “looks at questions particularly relevant to renal cell carcinoma patients,” he said.
The mean change in questionnaire scores from baseline was consistently better in the nivo/ipi arm. At 104 weeks the mean change was about +5 points with nivo/ipi vs. about –7 points with sunitinib.
“These results support the use of nivo/ipi as a new first-line standard of care option for patients with intermediate/poor-risk advanced [renal cell carcinoma],” he concluded.
CheckMate 214 was funded by Bristol-Myers Squibb and Ono Pharmaceutical. Dr. Motzer reported ownership interest in Armo Biosciences.
AT SITC 2017
Key clinical point:
Major finding: Overall response outcomes favored nivo/ipi vs. sunitinib for both PD-L1–positive tumors and PD-L1–negative tumors, but more so for PD-L1–positive tumors (P less than .0001 and P = .252, respectively).
Data source: The 1,096-patient open-label, phase 3 CheckMate 214 trial.
Disclosures: CheckMate 214 was funded by Bristol-Myers Squibb and Ono Pharmaceutical.
In children with ALL, physical and emotional effects persist
Among children with average-risk acute lymphoblastic leukemia (ALL), those with impairments in physical and emotional functioning at 2 months after diagnosis are likely to have continuing difficulties over 2 years later, based on the results of a 594-patient study published online in Cancer (2017 Nov 7. doi: 10.1002/cncr.31085).
Evaluations of quality of life and family functioning as early as 2 months after diagnosis identified those at highest risk of continued impairment, and can be used to target patients and their families for interventions and risk factor modification, the researchers said.
“These results provide a compelling rationale to screen patients for physical and emotional functioning early in therapy to target interventions toward patients at the highest risk of later impairment,” wrote lead author Daniel J. Zheng, MD, of the section of pediatric hematology-oncology at Yale University, New Haven, Conn., and coauthors.
Dr. Zheng and his colleagues measured impairments in children with average-risk ALL using the Pediatric Quality of Life Inventory Generic Core Scales Version 4.0 (PedsQL4.0), a 23-item survey that measures a child’s quality of life, and the 12-question General Functioning subscale of the McMaster Family Assessment Device (FAD-GF). Both are quick and low-cost screening measures that can be conducted in the clinic, they added. Evaluations occurred at approximately 2 months, 8 months, 17 months, 26 months, and 38 months (boys only) after diagnosis. The mean age of participants at diagnosis was 6.0 years (standard deviation, 1.6 years).
At 2 months after diagnosis, 36.5% of the children had impairments in physical functioning, and 26.2% had impairments in emotional functioning. The population norms for these measures are 2.3% for both scales, investigators wrote. At a 26-month evaluation, levels of impairment were still 11.9% for physical and 9.8% for emotional functioning.
Boys had an additional 38-month evaluation, at which time there were no significant differences in quality of life outcomes versus those observed at 26 months in the girls.
Unhealthy family functioning was a significant predictor of emotional impairment (odds ratio, 1.5; 95% confidence interval, 1.1-2.1) in multivariate models controlling for age and sex.
Strategies are needed to “intervene early and help the substantial proportion of children” with quality of life impairments, the researchers said. In particular, family functioning is “potentially modifiable with early intervention,” as suggested by a series of promising studies of techniques such as stress management sessions for parents. Most techniques feature a “targeted family-centered approach to psychosocial needs” that includes “embedded psychologists” as part of the multidisciplinary cancer care team.
Study funding came from the National Institutes of Health, National Cancer Institute, and the St. Baldrick’s Foundation. Dr. Zheng reported funding from a Yale Medical Student research fellowship, while coauthors reported conflict of interest disclosures from entities including Amgen, Takeda Pharmaceuticals International, and Shire Pharmaceuticals.
Among children with average-risk acute lymphoblastic leukemia (ALL), those with impairments in physical and emotional functioning at 2 months after diagnosis are likely to have continuing difficulties over 2 years later, based on the results of a 594-patient study published online in Cancer (2017 Nov 7. doi: 10.1002/cncr.31085).
Evaluations of quality of life and family functioning as early as 2 months after diagnosis identified those at highest risk of continued impairment, and can be used to target patients and their families for interventions and risk factor modification, the researchers said.
“These results provide a compelling rationale to screen patients for physical and emotional functioning early in therapy to target interventions toward patients at the highest risk of later impairment,” wrote lead author Daniel J. Zheng, MD, of the section of pediatric hematology-oncology at Yale University, New Haven, Conn., and coauthors.
Dr. Zheng and his colleagues measured impairments in children with average-risk ALL using the Pediatric Quality of Life Inventory Generic Core Scales Version 4.0 (PedsQL4.0), a 23-item survey that measures a child’s quality of life, and the 12-question General Functioning subscale of the McMaster Family Assessment Device (FAD-GF). Both are quick and low-cost screening measures that can be conducted in the clinic, they added. Evaluations occurred at approximately 2 months, 8 months, 17 months, 26 months, and 38 months (boys only) after diagnosis. The mean age of participants at diagnosis was 6.0 years (standard deviation, 1.6 years).
At 2 months after diagnosis, 36.5% of the children had impairments in physical functioning, and 26.2% had impairments in emotional functioning. The population norms for these measures are 2.3% for both scales, investigators wrote. At a 26-month evaluation, levels of impairment were still 11.9% for physical and 9.8% for emotional functioning.
Boys had an additional 38-month evaluation, at which time there were no significant differences in quality of life outcomes versus those observed at 26 months in the girls.
Unhealthy family functioning was a significant predictor of emotional impairment (odds ratio, 1.5; 95% confidence interval, 1.1-2.1) in multivariate models controlling for age and sex.
Strategies are needed to “intervene early and help the substantial proportion of children” with quality of life impairments, the researchers said. In particular, family functioning is “potentially modifiable with early intervention,” as suggested by a series of promising studies of techniques such as stress management sessions for parents. Most techniques feature a “targeted family-centered approach to psychosocial needs” that includes “embedded psychologists” as part of the multidisciplinary cancer care team.
Study funding came from the National Institutes of Health, National Cancer Institute, and the St. Baldrick’s Foundation. Dr. Zheng reported funding from a Yale Medical Student research fellowship, while coauthors reported conflict of interest disclosures from entities including Amgen, Takeda Pharmaceuticals International, and Shire Pharmaceuticals.
Among children with average-risk acute lymphoblastic leukemia (ALL), those with impairments in physical and emotional functioning at 2 months after diagnosis are likely to have continuing difficulties over 2 years later, based on the results of a 594-patient study published online in Cancer (2017 Nov 7. doi: 10.1002/cncr.31085).
Evaluations of quality of life and family functioning as early as 2 months after diagnosis identified those at highest risk of continued impairment, and can be used to target patients and their families for interventions and risk factor modification, the researchers said.
“These results provide a compelling rationale to screen patients for physical and emotional functioning early in therapy to target interventions toward patients at the highest risk of later impairment,” wrote lead author Daniel J. Zheng, MD, of the section of pediatric hematology-oncology at Yale University, New Haven, Conn., and coauthors.
Dr. Zheng and his colleagues measured impairments in children with average-risk ALL using the Pediatric Quality of Life Inventory Generic Core Scales Version 4.0 (PedsQL4.0), a 23-item survey that measures a child’s quality of life, and the 12-question General Functioning subscale of the McMaster Family Assessment Device (FAD-GF). Both are quick and low-cost screening measures that can be conducted in the clinic, they added. Evaluations occurred at approximately 2 months, 8 months, 17 months, 26 months, and 38 months (boys only) after diagnosis. The mean age of participants at diagnosis was 6.0 years (standard deviation, 1.6 years).
At 2 months after diagnosis, 36.5% of the children had impairments in physical functioning, and 26.2% had impairments in emotional functioning. The population norms for these measures are 2.3% for both scales, investigators wrote. At a 26-month evaluation, levels of impairment were still 11.9% for physical and 9.8% for emotional functioning.
Boys had an additional 38-month evaluation, at which time there were no significant differences in quality of life outcomes versus those observed at 26 months in the girls.
Unhealthy family functioning was a significant predictor of emotional impairment (odds ratio, 1.5; 95% confidence interval, 1.1-2.1) in multivariate models controlling for age and sex.
Strategies are needed to “intervene early and help the substantial proportion of children” with quality of life impairments, the researchers said. In particular, family functioning is “potentially modifiable with early intervention,” as suggested by a series of promising studies of techniques such as stress management sessions for parents. Most techniques feature a “targeted family-centered approach to psychosocial needs” that includes “embedded psychologists” as part of the multidisciplinary cancer care team.
Study funding came from the National Institutes of Health, National Cancer Institute, and the St. Baldrick’s Foundation. Dr. Zheng reported funding from a Yale Medical Student research fellowship, while coauthors reported conflict of interest disclosures from entities including Amgen, Takeda Pharmaceuticals International, and Shire Pharmaceuticals.
FROM CANCER
Key clinical point: Simple
Major finding: At 26 months after diagnosis, a considerable proportion of children identified at 2 months after diagnosis still had impairments in physical functioning (11.9%) and emotional functioning (9.8%).
Data source: A prospective cohort study of 594 participants with average-risk ALL in the Children’s Oncology Group AALL0932 trial.
Disclosures: The National Institutes of Health, National Cancer Institute, and St. Baldrick’s Foundation provided funding for the study.
10 Ways You Championed Lung Health in 2017
1. You funded more than a half-million dollars in community service and clinical research grants awarded to the next generation of CHEST leaders.
2. You improved patient outcomes by supporting more than 65 patient education resources focused on procedures and disease states—easily accessed for free at chestfoundation.org/patienteducation.![]()
3. You brought the Lung Health Experience to local communities, where more than 1,000 people received free COPD screening and spirometry testing.
4. You raised nearly $200,000 at the Irv Feldman Texas Hold’em and Casino Night to create new patient resources for pulmonary fibrosis.
5. You influenced the careers of 40 young professionals through travel grants and mentorship programs for CHEST Annual Meeting 2017.
6. You educated millions by supporting nationwide disease awareness campaigns for COPD, asthma, sarcoidosis, and lung cancer.
7. You funded asthma training sessions for community-based asthma educators.
8. You supplied physicians in Tanzania pulmonary reference textbooks so that they can learn to do bronchoscopy for the first time.
9. You translated nearly 50 critical care course manuals into French to help Haitian pediatricians save children’s lives.
10. You’re supporting an asthma app that will teach patients how to use their asthma devices.
Thank you for being a champion for lung health. You make this and so much more possible.
1. You funded more than a half-million dollars in community service and clinical research grants awarded to the next generation of CHEST leaders.
2. You improved patient outcomes by supporting more than 65 patient education resources focused on procedures and disease states—easily accessed for free at chestfoundation.org/patienteducation.![]()
3. You brought the Lung Health Experience to local communities, where more than 1,000 people received free COPD screening and spirometry testing.
4. You raised nearly $200,000 at the Irv Feldman Texas Hold’em and Casino Night to create new patient resources for pulmonary fibrosis.
5. You influenced the careers of 40 young professionals through travel grants and mentorship programs for CHEST Annual Meeting 2017.
6. You educated millions by supporting nationwide disease awareness campaigns for COPD, asthma, sarcoidosis, and lung cancer.
7. You funded asthma training sessions for community-based asthma educators.
8. You supplied physicians in Tanzania pulmonary reference textbooks so that they can learn to do bronchoscopy for the first time.
9. You translated nearly 50 critical care course manuals into French to help Haitian pediatricians save children’s lives.
10. You’re supporting an asthma app that will teach patients how to use their asthma devices.
Thank you for being a champion for lung health. You make this and so much more possible.
1. You funded more than a half-million dollars in community service and clinical research grants awarded to the next generation of CHEST leaders.
2. You improved patient outcomes by supporting more than 65 patient education resources focused on procedures and disease states—easily accessed for free at chestfoundation.org/patienteducation.![]()
3. You brought the Lung Health Experience to local communities, where more than 1,000 people received free COPD screening and spirometry testing.
4. You raised nearly $200,000 at the Irv Feldman Texas Hold’em and Casino Night to create new patient resources for pulmonary fibrosis.
5. You influenced the careers of 40 young professionals through travel grants and mentorship programs for CHEST Annual Meeting 2017.
6. You educated millions by supporting nationwide disease awareness campaigns for COPD, asthma, sarcoidosis, and lung cancer.
7. You funded asthma training sessions for community-based asthma educators.
8. You supplied physicians in Tanzania pulmonary reference textbooks so that they can learn to do bronchoscopy for the first time.
9. You translated nearly 50 critical care course manuals into French to help Haitian pediatricians save children’s lives.
10. You’re supporting an asthma app that will teach patients how to use their asthma devices.
Thank you for being a champion for lung health. You make this and so much more possible.
Salivary biomarker identified for Huntington’s disease
SAN DIEGO – Huntingtin protein – the key biomarker for Huntington’s disease – can be detected in saliva, which might prove to be an easy and inexpensive way to diagnose and monitor Huntington’s, and perhaps even predict clinical onset, according to investigators at the University of California, San Diego.
In a study of 178 subjects, they found that salivary total huntingtin protein (Htt) was significantly increased in saliva from individuals with Huntington’s disease (HD), compared with controls without HD (mean, 0.775 ng/mL vs. 0.359 ng/mL; P = .0012). Levels remained consistent throughout the day and from day to day, and were not affected by age or sex.

Meanwhile, salivary mutant Htt levels were higher in gene-positive, premanifest HD subjects than in normal controls (P less than .05). Salivary C-reactive protein level was also significantly elevated in premanifest HD subjects (9,548 pg/mL vs. 3,399 pg/mL, P = .025), indicating a pathologic inflammatory or metabolic state. When considered together, the two measurements might herald the onset of symptoms.
There’s an acute need for a convenient, inexpensive HD biomarker. Htt isn’t often measured in clinical practice, and when it is, it’s assessed from blood or cerebrospinal fluid. With salivary Htt, “you don’t need any specialized personnel, [and samples] are easy to obtain and process. They keep well and are very stable. We don’t have to rush to get them somewhere,” said lead investigator Jody Corey-Bloom, MD, PhD, professor emeritus of neurosciences at the university.
“We are really excited about the potential of salivary Htt. We think this is going to be an easy way to follow patients,” she said at the annual meeting of the American Neurological Association.
The next step is to see how salivary Htt correlates with cerebrospinal fluid and blood levels. The team will also investigate it as a diagnostic tool; perhaps there’s a cut point that diagnoses HD. “It’s an intriguing idea,” Dr. Corey-Bloom said.
Perhaps the greatest potential is for predicting disease onset so treatments can be started before symptoms emerge. There’s nothing on the market yet that can delay or prevent progression, but trials are in the works for therapeutics that lower levels of mutant Htt in the brain. “If we can use something simple like salivary Htt [to start preemptive treatment] that would be phenomenal. That’s the hope,” she said.
Subjects refrained from smoking, eating, drinking, and brushing their teeth for at least an hour before saliva samples were taken. Testing was done by Western blot and enzyme-linked immunosorbent assay.
The work was funded by the university. Dr. Corey-Bloom said she had no relevant disclosures.
SAN DIEGO – Huntingtin protein – the key biomarker for Huntington’s disease – can be detected in saliva, which might prove to be an easy and inexpensive way to diagnose and monitor Huntington’s, and perhaps even predict clinical onset, according to investigators at the University of California, San Diego.
In a study of 178 subjects, they found that salivary total huntingtin protein (Htt) was significantly increased in saliva from individuals with Huntington’s disease (HD), compared with controls without HD (mean, 0.775 ng/mL vs. 0.359 ng/mL; P = .0012). Levels remained consistent throughout the day and from day to day, and were not affected by age or sex.
Meanwhile, salivary mutant Htt levels were higher in gene-positive, premanifest HD subjects than in normal controls (P less than .05). Salivary C-reactive protein level was also significantly elevated in premanifest HD subjects (9,548 pg/mL vs. 3,399 pg/mL, P = .025), indicating a pathologic inflammatory or metabolic state. When considered together, the two measurements might herald the onset of symptoms.
There’s an acute need for a convenient, inexpensive HD biomarker. Htt isn’t often measured in clinical practice, and when it is, it’s assessed from blood or cerebrospinal fluid. With salivary Htt, “you don’t need any specialized personnel, [and samples] are easy to obtain and process. They keep well and are very stable. We don’t have to rush to get them somewhere,” said lead investigator Jody Corey-Bloom, MD, PhD, professor emeritus of neurosciences at the university.
“We are really excited about the potential of salivary Htt. We think this is going to be an easy way to follow patients,” she said at the annual meeting of the American Neurological Association.
The next step is to see how salivary Htt correlates with cerebrospinal fluid and blood levels. The team will also investigate it as a diagnostic tool; perhaps there’s a cut point that diagnoses HD. “It’s an intriguing idea,” Dr. Corey-Bloom said.
Perhaps the greatest potential is for predicting disease onset so treatments can be started before symptoms emerge. There’s nothing on the market yet that can delay or prevent progression, but trials are in the works for therapeutics that lower levels of mutant Htt in the brain. “If we can use something simple like salivary Htt [to start preemptive treatment] that would be phenomenal. That’s the hope,” she said.
Subjects refrained from smoking, eating, drinking, and brushing their teeth for at least an hour before saliva samples were taken. Testing was done by Western blot and enzyme-linked immunosorbent assay.
The work was funded by the university. Dr. Corey-Bloom said she had no relevant disclosures.
SAN DIEGO – Huntingtin protein – the key biomarker for Huntington’s disease – can be detected in saliva, which might prove to be an easy and inexpensive way to diagnose and monitor Huntington’s, and perhaps even predict clinical onset, according to investigators at the University of California, San Diego.
In a study of 178 subjects, they found that salivary total huntingtin protein (Htt) was significantly increased in saliva from individuals with Huntington’s disease (HD), compared with controls without HD (mean, 0.775 ng/mL vs. 0.359 ng/mL; P = .0012). Levels remained consistent throughout the day and from day to day, and were not affected by age or sex.
Meanwhile, salivary mutant Htt levels were higher in gene-positive, premanifest HD subjects than in normal controls (P less than .05). Salivary C-reactive protein level was also significantly elevated in premanifest HD subjects (9,548 pg/mL vs. 3,399 pg/mL, P = .025), indicating a pathologic inflammatory or metabolic state. When considered together, the two measurements might herald the onset of symptoms.
There’s an acute need for a convenient, inexpensive HD biomarker. Htt isn’t often measured in clinical practice, and when it is, it’s assessed from blood or cerebrospinal fluid. With salivary Htt, “you don’t need any specialized personnel, [and samples] are easy to obtain and process. They keep well and are very stable. We don’t have to rush to get them somewhere,” said lead investigator Jody Corey-Bloom, MD, PhD, professor emeritus of neurosciences at the university.
“We are really excited about the potential of salivary Htt. We think this is going to be an easy way to follow patients,” she said at the annual meeting of the American Neurological Association.
The next step is to see how salivary Htt correlates with cerebrospinal fluid and blood levels. The team will also investigate it as a diagnostic tool; perhaps there’s a cut point that diagnoses HD. “It’s an intriguing idea,” Dr. Corey-Bloom said.
Perhaps the greatest potential is for predicting disease onset so treatments can be started before symptoms emerge. There’s nothing on the market yet that can delay or prevent progression, but trials are in the works for therapeutics that lower levels of mutant Htt in the brain. “If we can use something simple like salivary Htt [to start preemptive treatment] that would be phenomenal. That’s the hope,” she said.
Subjects refrained from smoking, eating, drinking, and brushing their teeth for at least an hour before saliva samples were taken. Testing was done by Western blot and enzyme-linked immunosorbent assay.
The work was funded by the university. Dr. Corey-Bloom said she had no relevant disclosures.
AT ANA 2017
Key clinical point:
Major finding: Salivary total huntingtin protein was significantly increased in saliva from individuals with Huntington’s disease, compared with controls (mean, 0.775 ng/mL vs. 0.359 ng/mL, P = .0012).
Data source: Measurement of salivary Htt and other analytes in 178 subjects.
Disclosures: The work was funded by the University of California, San Diego. The lead investigator had no relevant financial disclosures.
