User login
Twelve medical groups pen letter opposing UHC copay accumulator program
ACR leads outcry against the insurer’s proposed move
Last month, the American College of Rheumatology joined with 11 other medical associations and disease societies asking health insurance giant UnitedHealthcare (UHC) to not proceed with its proposed copay accumulator medical benefit program.

Copay accumulators are policies adopted by insurance companies or their pharmacy benefit managers to exclude patient copayment assistance programs for high-cost drugs, which are promulgated by the drug manufacturers, from being applied to a patient’s annual deductibles or out-of-pocket maximums. The manufacturer’s copay assistance, such as in the form of coupons, is designed to minimize the patient’s out-of-pocket costs. But insurers believe manufacturers will have no pressure to lower the prices of expensive specialty drugs unless patients are unable to afford them. Copay accumulators thus are aimed at giving insurers more leverage in negotiating prices for high-cost drugs.
UHC issued its new copay accumulator protocol for commercial individual and fully insured group plans in early October, effective Jan. 1, 2021, “in order to align employer costs for specialty medications with actual member out of pocket and deductibles,” according to the company’s announcement. In other words, patients will need to pay a higher share of the costs of these medications, said rheumatologist Christopher Phillips, MD, who chairs the Insurance Subcommittee of ACR’s Rheumatologic Care Committee. The annual price of biologic therapies for rheumatologic conditions ranges from $22,000 to $44,000, according to a recent press release from ACR.
The copay accumulator will negate the benefits of manufacturers’ copayment assistance programs for the patient, shifting more of the cost to the patient. With patients being forced to pay a higher share of drug costs for expensive biologic treatments for rheumatoid arthritis, lupus, and other rheumatologic conditions, they’ll stop taking the treatments, Dr. Phillips said.
“In my solo rheumatology practice in Paducah, Kentucky, when I’ve seen this kind of program applied on the pharmacy benefit side, rather than the medical benefit side, almost uniformly patients stop taking the high-cost treatments.” That can lead to disease flares, complications, and permanent disability. The newer rheumatologic drugs can cost $500 to $1,000 per treatment, and in many cases, there’s no generic or lower-cost alternative, he says. “We see policies like this as sacrificing patients to the battle over high drug prices. It’s bad practice, bad for patient outcomes, and nobody – apart from the payer – benefits.”
In ACR’s 2020 Rheumatic Disease Patient Survey, nearly half of 1,109 online survey respondents who had rheumatic diseases reported out-of-pocket costs greater than $1,000 per year for treatment. An IQVIA report from 2016 found that one in four specialty brand prescriptions are abandoned during the deductible phase, three times the rate seen when there is no deductible.
In an Oct. 7 letter to UHC, the 12 groups acknowledged that the drugs targeted by the accumulator policy are expensive. “However, they are also vitally important for our patients.” In addition to the ACR, the organizations involved include the AIDS Institute, American Academy of Dermatology Association, American Academy of Neurology, American College of Gastroenterology, American Gastroenterological Association, American Kidney Fund, Arthritis Foundation, Association for Clinical Oncology, Cancer Support Community, Coalition of State Rheumatology Organizations, and National Multiple Sclerosis Society.
UHC did not reply to questions in time for publication.
First large-scale payer to try copay accumulator program
Under UHC’s proposed policy, providers will be required to use UHC’s portal to report payment information received from drug manufacturer copay assistance programs that are applied to patients’ cost share of these drugs through a complex, 14-step “coupon submission process” involving multiple technology interfaces. “My first oath as a physician is to do no harm to my patient. Many of us are concerned about making these reports, which could harm our patients and undermine the doctor-patient relationship,” Dr. Phillips said.
“If I don’t report, what happens? I don’t think we know the answer to that. Some of us may decide we need to part ways with UHC.” Others may decline to participate in the drug manufacturers’ coupon programs beyond simply informing patients that manufacturer assistance is available.
“We’ve watched these copay accumulator policies for several years,” he said. “Some of them are rather opaque, with names like ‘copay savings programs’ or ‘copay value programs.’ But we had not seen a large-scale payer try to do this until now. Let’s face it: If UHC’s policy goes through, you can count the days until we see it from others.”
The Department of Health & Human Services, in its May 2020 final federal “Notice of Benefit and Payment Parameters for 2021,” indicated that individual states have the responsibility to regulate copay accumulator programs. Five states have banned them or restricted their use for individual and small group health plans. Arizona, Illinois, Virginia, and West Virginia passed such laws in 2019, and Georgia did so earlier this year.
“In next year’s state legislative sessions, we’ll make it a priority to pursue similar laws in other states,” Dr. Phillips said. “I’d encourage rheumatologists to educate their patients on the issues and be active in advocating for them.”
ACR leads outcry against the insurer’s proposed move
ACR leads outcry against the insurer’s proposed move
Last month, the American College of Rheumatology joined with 11 other medical associations and disease societies asking health insurance giant UnitedHealthcare (UHC) to not proceed with its proposed copay accumulator medical benefit program.
Copay accumulators are policies adopted by insurance companies or their pharmacy benefit managers to exclude patient copayment assistance programs for high-cost drugs, which are promulgated by the drug manufacturers, from being applied to a patient’s annual deductibles or out-of-pocket maximums. The manufacturer’s copay assistance, such as in the form of coupons, is designed to minimize the patient’s out-of-pocket costs. But insurers believe manufacturers will have no pressure to lower the prices of expensive specialty drugs unless patients are unable to afford them. Copay accumulators thus are aimed at giving insurers more leverage in negotiating prices for high-cost drugs.
UHC issued its new copay accumulator protocol for commercial individual and fully insured group plans in early October, effective Jan. 1, 2021, “in order to align employer costs for specialty medications with actual member out of pocket and deductibles,” according to the company’s announcement. In other words, patients will need to pay a higher share of the costs of these medications, said rheumatologist Christopher Phillips, MD, who chairs the Insurance Subcommittee of ACR’s Rheumatologic Care Committee. The annual price of biologic therapies for rheumatologic conditions ranges from $22,000 to $44,000, according to a recent press release from ACR.
The copay accumulator will negate the benefits of manufacturers’ copayment assistance programs for the patient, shifting more of the cost to the patient. With patients being forced to pay a higher share of drug costs for expensive biologic treatments for rheumatoid arthritis, lupus, and other rheumatologic conditions, they’ll stop taking the treatments, Dr. Phillips said.
“In my solo rheumatology practice in Paducah, Kentucky, when I’ve seen this kind of program applied on the pharmacy benefit side, rather than the medical benefit side, almost uniformly patients stop taking the high-cost treatments.” That can lead to disease flares, complications, and permanent disability. The newer rheumatologic drugs can cost $500 to $1,000 per treatment, and in many cases, there’s no generic or lower-cost alternative, he says. “We see policies like this as sacrificing patients to the battle over high drug prices. It’s bad practice, bad for patient outcomes, and nobody – apart from the payer – benefits.”
In ACR’s 2020 Rheumatic Disease Patient Survey, nearly half of 1,109 online survey respondents who had rheumatic diseases reported out-of-pocket costs greater than $1,000 per year for treatment. An IQVIA report from 2016 found that one in four specialty brand prescriptions are abandoned during the deductible phase, three times the rate seen when there is no deductible.
In an Oct. 7 letter to UHC, the 12 groups acknowledged that the drugs targeted by the accumulator policy are expensive. “However, they are also vitally important for our patients.” In addition to the ACR, the organizations involved include the AIDS Institute, American Academy of Dermatology Association, American Academy of Neurology, American College of Gastroenterology, American Gastroenterological Association, American Kidney Fund, Arthritis Foundation, Association for Clinical Oncology, Cancer Support Community, Coalition of State Rheumatology Organizations, and National Multiple Sclerosis Society.
UHC did not reply to questions in time for publication.
First large-scale payer to try copay accumulator program
Under UHC’s proposed policy, providers will be required to use UHC’s portal to report payment information received from drug manufacturer copay assistance programs that are applied to patients’ cost share of these drugs through a complex, 14-step “coupon submission process” involving multiple technology interfaces. “My first oath as a physician is to do no harm to my patient. Many of us are concerned about making these reports, which could harm our patients and undermine the doctor-patient relationship,” Dr. Phillips said.
“If I don’t report, what happens? I don’t think we know the answer to that. Some of us may decide we need to part ways with UHC.” Others may decline to participate in the drug manufacturers’ coupon programs beyond simply informing patients that manufacturer assistance is available.
“We’ve watched these copay accumulator policies for several years,” he said. “Some of them are rather opaque, with names like ‘copay savings programs’ or ‘copay value programs.’ But we had not seen a large-scale payer try to do this until now. Let’s face it: If UHC’s policy goes through, you can count the days until we see it from others.”
The Department of Health & Human Services, in its May 2020 final federal “Notice of Benefit and Payment Parameters for 2021,” indicated that individual states have the responsibility to regulate copay accumulator programs. Five states have banned them or restricted their use for individual and small group health plans. Arizona, Illinois, Virginia, and West Virginia passed such laws in 2019, and Georgia did so earlier this year.
“In next year’s state legislative sessions, we’ll make it a priority to pursue similar laws in other states,” Dr. Phillips said. “I’d encourage rheumatologists to educate their patients on the issues and be active in advocating for them.”
Last month, the American College of Rheumatology joined with 11 other medical associations and disease societies asking health insurance giant UnitedHealthcare (UHC) to not proceed with its proposed copay accumulator medical benefit program.
Copay accumulators are policies adopted by insurance companies or their pharmacy benefit managers to exclude patient copayment assistance programs for high-cost drugs, which are promulgated by the drug manufacturers, from being applied to a patient’s annual deductibles or out-of-pocket maximums. The manufacturer’s copay assistance, such as in the form of coupons, is designed to minimize the patient’s out-of-pocket costs. But insurers believe manufacturers will have no pressure to lower the prices of expensive specialty drugs unless patients are unable to afford them. Copay accumulators thus are aimed at giving insurers more leverage in negotiating prices for high-cost drugs.
UHC issued its new copay accumulator protocol for commercial individual and fully insured group plans in early October, effective Jan. 1, 2021, “in order to align employer costs for specialty medications with actual member out of pocket and deductibles,” according to the company’s announcement. In other words, patients will need to pay a higher share of the costs of these medications, said rheumatologist Christopher Phillips, MD, who chairs the Insurance Subcommittee of ACR’s Rheumatologic Care Committee. The annual price of biologic therapies for rheumatologic conditions ranges from $22,000 to $44,000, according to a recent press release from ACR.
The copay accumulator will negate the benefits of manufacturers’ copayment assistance programs for the patient, shifting more of the cost to the patient. With patients being forced to pay a higher share of drug costs for expensive biologic treatments for rheumatoid arthritis, lupus, and other rheumatologic conditions, they’ll stop taking the treatments, Dr. Phillips said.
“In my solo rheumatology practice in Paducah, Kentucky, when I’ve seen this kind of program applied on the pharmacy benefit side, rather than the medical benefit side, almost uniformly patients stop taking the high-cost treatments.” That can lead to disease flares, complications, and permanent disability. The newer rheumatologic drugs can cost $500 to $1,000 per treatment, and in many cases, there’s no generic or lower-cost alternative, he says. “We see policies like this as sacrificing patients to the battle over high drug prices. It’s bad practice, bad for patient outcomes, and nobody – apart from the payer – benefits.”
In ACR’s 2020 Rheumatic Disease Patient Survey, nearly half of 1,109 online survey respondents who had rheumatic diseases reported out-of-pocket costs greater than $1,000 per year for treatment. An IQVIA report from 2016 found that one in four specialty brand prescriptions are abandoned during the deductible phase, three times the rate seen when there is no deductible.
In an Oct. 7 letter to UHC, the 12 groups acknowledged that the drugs targeted by the accumulator policy are expensive. “However, they are also vitally important for our patients.” In addition to the ACR, the organizations involved include the AIDS Institute, American Academy of Dermatology Association, American Academy of Neurology, American College of Gastroenterology, American Gastroenterological Association, American Kidney Fund, Arthritis Foundation, Association for Clinical Oncology, Cancer Support Community, Coalition of State Rheumatology Organizations, and National Multiple Sclerosis Society.
UHC did not reply to questions in time for publication.
First large-scale payer to try copay accumulator program
Under UHC’s proposed policy, providers will be required to use UHC’s portal to report payment information received from drug manufacturer copay assistance programs that are applied to patients’ cost share of these drugs through a complex, 14-step “coupon submission process” involving multiple technology interfaces. “My first oath as a physician is to do no harm to my patient. Many of us are concerned about making these reports, which could harm our patients and undermine the doctor-patient relationship,” Dr. Phillips said.
“If I don’t report, what happens? I don’t think we know the answer to that. Some of us may decide we need to part ways with UHC.” Others may decline to participate in the drug manufacturers’ coupon programs beyond simply informing patients that manufacturer assistance is available.
“We’ve watched these copay accumulator policies for several years,” he said. “Some of them are rather opaque, with names like ‘copay savings programs’ or ‘copay value programs.’ But we had not seen a large-scale payer try to do this until now. Let’s face it: If UHC’s policy goes through, you can count the days until we see it from others.”
The Department of Health & Human Services, in its May 2020 final federal “Notice of Benefit and Payment Parameters for 2021,” indicated that individual states have the responsibility to regulate copay accumulator programs. Five states have banned them or restricted their use for individual and small group health plans. Arizona, Illinois, Virginia, and West Virginia passed such laws in 2019, and Georgia did so earlier this year.
“In next year’s state legislative sessions, we’ll make it a priority to pursue similar laws in other states,” Dr. Phillips said. “I’d encourage rheumatologists to educate their patients on the issues and be active in advocating for them.”
PARTNER registry valve-in-valve outcomes reassuring at 5 years
Transcatheter replacement of a failing surgical bioprosthetic valve showed durably favorable valve hemodynamics coupled with markedly improved patient functional status and excellent quality of life benefits at 5 years of follow-up in the prospective multicenter PARTNER 2 ViV Registry, Rebecca T. Hahn, MD, reported at the Transcatheter Cardiovascular Research Therapeutics virtual annual meeting.
She provided an update on previously reported 3-year outcomes in 365 patients at high to extreme surgical risk who underwent transcatheter aortic valve replacement (TAVR) with a 23-mm or 26-mm Sapien XT valve to address a failing surgical aortic bioprosthesis. The ViV (valve-in-valve) results are quite encouraging, she said at the meeting sponsored by the Cardiovascular Research Foundation.
“I think that this information is changing our algorithm for how we initially make treatment decisions in our patients,” according to the cardiologist.
“We now know that we can salvage a surgical bioprosthetic valve failure with a transcatheter procedure that is relatively safe and has good outcomes out to 5 years – and that’s with a second-generation TAVR valve, not even the third-generation valve,” observed Dr. Hahn, director of interventional echocardiography at New York–Presbyterian/Columbia University Medical Center and professor of clinical medicine at Columbia University, both in New York.
Interventionalists consider the third-generation valve, the Sapien 3, a superior platform compared to the Sapien 2 in use when the PARTNERS 2 ViV Registry started, she added.
At 5 years of follow-up since TAVR valve implantation, the all-cause mortality rate was 50.6%, up significantly from 32.7% at 3 years. However, this high mortality comes as no surprise given that registry participants had a profound comorbidity burden, as reflected in their mean Society of Thoracic Surgeons risk score of 9.1% at the time of TAVR. Of note, the 5-year mortality in surgically high- to extreme-risk patients in the ViV registry was comparable with the 45.9% rate at 5 years following TAVR of a native valve in intermediate-risk patients in the PARTNER 2b trial and superior to the 73% rate with TAVR of a native aortic valve in inoperable patients in PARTNER 2a, the cardiologist said.
The 5-year stroke rate in the ViV registry was 10.1%, up from 6.2% at 3 years. The cumulative incidence of death or stroke through 5 years was 53.8%.
Mortality was significantly lower in recipients of a 26-mm Sapien 2 valve than with the 23-mm version, at 40% at 5 years versus 53%. Recipients of the smaller valve were more often male, had a higher prevalence of coronary artery disease, a higher surgical risk score, a significantly smaller baseline aortic valve area, and a higher mean gradient. Dr. Hahn and her coinvestigators are now examining their data to determine if surgical valve size/patient mismatch was a major driver of adverse outcomes, as has been reported in some other datasets.
At 5 years, the rate of structural valve deterioration–related hemodynamic valve deterioration (SVD-HVD) or bioprosthetic valve failure (BVF) using the soon-to-be-published Valve Academic Research Consortium–3 definitions was 6.6%. The rates of each class of valve deterioration at 5 years in this high- to extreme-risk population were 1.2 per 100 patient-years for SVD-HVD, 0.88 per 100 patient-years for all BVF, and 0.4 per 100 patient-years for SVD-related BVF.
Fully 51% of 5-year survivors were NYHA functional class I, whereas more than 90% of patients were class III or IV at baseline. The mean gradient was 16.8 mm Hg at 5 years, the Doppler velocity index was 0.35, and the mean Kansas City Cardiomyopathy Questionnaire overall summary score was 74.2, all closely similar to the values at 3 years. That dramatic and sustained improvement in the Kansas City Cardiomyopathy Questionnaire from a baseline of 43.1 points is larger than ever seen in any clinical trial of native valve TAVR, Dr. Hahn noted.
For discussant Vinayak N. Bapat, MD a cardiothoracic surgeon at the Minneapolis Heart Institute Foundation, the 5-year PARTNER 2 follow-up data contains a clear take-home message: “These data show that, when we as surgeons are putting in small valves, we ought to put in valves that are expandable.”
Discussant Jeroen J. Bax, MD, had one major caveat regarding the PARTNER 2 ViV Registry findings: They focused on high-surgical-risk patients.
“I think we would all agree that in high-risk patients, valve-in-valve is a better option than redo surgery. But in young, low-risk patients who are getting a bioprosthetic valve – and we’re going to be seeing more and more of them because over 90% of patients in Europe getting aortic valve surgery now are getting a bioprosthetic valve – we really don’t know what the best option is,” said Dr. Bax, professor of cardiology at the University of Leiden (the Netherlands).
He suggested a randomized trial of TAVR versus redo surgery in low-risk patients with failing bioprosthetic valves is in order, particularly in light of concerns raised by a recent report from a French national patient registry. These were “high-quality, real-world data,” Dr. Bax said, and while they showed better early outcomes for TAVR ViV than with redo surgery, there was a crossing of the curves for heart failure hospitalization already by 2 years.
“We need to look closely at younger, low-risk patients,” he concluded.
The PARTNER 2 ViV Registry is funded by Edwards Lifesciences. Dr. Hahn reported receiving research support from Philips Healthcare and 3Mensio and honoraria from Boston Scientific, Edwards Lifesciences, and Philips Healthcare.
SOURCE: Hahn RT. TCT 2020, Late breaker.
Transcatheter replacement of a failing surgical bioprosthetic valve showed durably favorable valve hemodynamics coupled with markedly improved patient functional status and excellent quality of life benefits at 5 years of follow-up in the prospective multicenter PARTNER 2 ViV Registry, Rebecca T. Hahn, MD, reported at the Transcatheter Cardiovascular Research Therapeutics virtual annual meeting.
She provided an update on previously reported 3-year outcomes in 365 patients at high to extreme surgical risk who underwent transcatheter aortic valve replacement (TAVR) with a 23-mm or 26-mm Sapien XT valve to address a failing surgical aortic bioprosthesis. The ViV (valve-in-valve) results are quite encouraging, she said at the meeting sponsored by the Cardiovascular Research Foundation.
“I think that this information is changing our algorithm for how we initially make treatment decisions in our patients,” according to the cardiologist.
“We now know that we can salvage a surgical bioprosthetic valve failure with a transcatheter procedure that is relatively safe and has good outcomes out to 5 years – and that’s with a second-generation TAVR valve, not even the third-generation valve,” observed Dr. Hahn, director of interventional echocardiography at New York–Presbyterian/Columbia University Medical Center and professor of clinical medicine at Columbia University, both in New York.
Interventionalists consider the third-generation valve, the Sapien 3, a superior platform compared to the Sapien 2 in use when the PARTNERS 2 ViV Registry started, she added.
At 5 years of follow-up since TAVR valve implantation, the all-cause mortality rate was 50.6%, up significantly from 32.7% at 3 years. However, this high mortality comes as no surprise given that registry participants had a profound comorbidity burden, as reflected in their mean Society of Thoracic Surgeons risk score of 9.1% at the time of TAVR. Of note, the 5-year mortality in surgically high- to extreme-risk patients in the ViV registry was comparable with the 45.9% rate at 5 years following TAVR of a native valve in intermediate-risk patients in the PARTNER 2b trial and superior to the 73% rate with TAVR of a native aortic valve in inoperable patients in PARTNER 2a, the cardiologist said.
The 5-year stroke rate in the ViV registry was 10.1%, up from 6.2% at 3 years. The cumulative incidence of death or stroke through 5 years was 53.8%.
Mortality was significantly lower in recipients of a 26-mm Sapien 2 valve than with the 23-mm version, at 40% at 5 years versus 53%. Recipients of the smaller valve were more often male, had a higher prevalence of coronary artery disease, a higher surgical risk score, a significantly smaller baseline aortic valve area, and a higher mean gradient. Dr. Hahn and her coinvestigators are now examining their data to determine if surgical valve size/patient mismatch was a major driver of adverse outcomes, as has been reported in some other datasets.
At 5 years, the rate of structural valve deterioration–related hemodynamic valve deterioration (SVD-HVD) or bioprosthetic valve failure (BVF) using the soon-to-be-published Valve Academic Research Consortium–3 definitions was 6.6%. The rates of each class of valve deterioration at 5 years in this high- to extreme-risk population were 1.2 per 100 patient-years for SVD-HVD, 0.88 per 100 patient-years for all BVF, and 0.4 per 100 patient-years for SVD-related BVF.
Fully 51% of 5-year survivors were NYHA functional class I, whereas more than 90% of patients were class III or IV at baseline. The mean gradient was 16.8 mm Hg at 5 years, the Doppler velocity index was 0.35, and the mean Kansas City Cardiomyopathy Questionnaire overall summary score was 74.2, all closely similar to the values at 3 years. That dramatic and sustained improvement in the Kansas City Cardiomyopathy Questionnaire from a baseline of 43.1 points is larger than ever seen in any clinical trial of native valve TAVR, Dr. Hahn noted.
For discussant Vinayak N. Bapat, MD a cardiothoracic surgeon at the Minneapolis Heart Institute Foundation, the 5-year PARTNER 2 follow-up data contains a clear take-home message: “These data show that, when we as surgeons are putting in small valves, we ought to put in valves that are expandable.”
Discussant Jeroen J. Bax, MD, had one major caveat regarding the PARTNER 2 ViV Registry findings: They focused on high-surgical-risk patients.
“I think we would all agree that in high-risk patients, valve-in-valve is a better option than redo surgery. But in young, low-risk patients who are getting a bioprosthetic valve – and we’re going to be seeing more and more of them because over 90% of patients in Europe getting aortic valve surgery now are getting a bioprosthetic valve – we really don’t know what the best option is,” said Dr. Bax, professor of cardiology at the University of Leiden (the Netherlands).
He suggested a randomized trial of TAVR versus redo surgery in low-risk patients with failing bioprosthetic valves is in order, particularly in light of concerns raised by a recent report from a French national patient registry. These were “high-quality, real-world data,” Dr. Bax said, and while they showed better early outcomes for TAVR ViV than with redo surgery, there was a crossing of the curves for heart failure hospitalization already by 2 years.
“We need to look closely at younger, low-risk patients,” he concluded.
The PARTNER 2 ViV Registry is funded by Edwards Lifesciences. Dr. Hahn reported receiving research support from Philips Healthcare and 3Mensio and honoraria from Boston Scientific, Edwards Lifesciences, and Philips Healthcare.
SOURCE: Hahn RT. TCT 2020, Late breaker.
Transcatheter replacement of a failing surgical bioprosthetic valve showed durably favorable valve hemodynamics coupled with markedly improved patient functional status and excellent quality of life benefits at 5 years of follow-up in the prospective multicenter PARTNER 2 ViV Registry, Rebecca T. Hahn, MD, reported at the Transcatheter Cardiovascular Research Therapeutics virtual annual meeting.
She provided an update on previously reported 3-year outcomes in 365 patients at high to extreme surgical risk who underwent transcatheter aortic valve replacement (TAVR) with a 23-mm or 26-mm Sapien XT valve to address a failing surgical aortic bioprosthesis. The ViV (valve-in-valve) results are quite encouraging, she said at the meeting sponsored by the Cardiovascular Research Foundation.
“I think that this information is changing our algorithm for how we initially make treatment decisions in our patients,” according to the cardiologist.
“We now know that we can salvage a surgical bioprosthetic valve failure with a transcatheter procedure that is relatively safe and has good outcomes out to 5 years – and that’s with a second-generation TAVR valve, not even the third-generation valve,” observed Dr. Hahn, director of interventional echocardiography at New York–Presbyterian/Columbia University Medical Center and professor of clinical medicine at Columbia University, both in New York.
Interventionalists consider the third-generation valve, the Sapien 3, a superior platform compared to the Sapien 2 in use when the PARTNERS 2 ViV Registry started, she added.
At 5 years of follow-up since TAVR valve implantation, the all-cause mortality rate was 50.6%, up significantly from 32.7% at 3 years. However, this high mortality comes as no surprise given that registry participants had a profound comorbidity burden, as reflected in their mean Society of Thoracic Surgeons risk score of 9.1% at the time of TAVR. Of note, the 5-year mortality in surgically high- to extreme-risk patients in the ViV registry was comparable with the 45.9% rate at 5 years following TAVR of a native valve in intermediate-risk patients in the PARTNER 2b trial and superior to the 73% rate with TAVR of a native aortic valve in inoperable patients in PARTNER 2a, the cardiologist said.
The 5-year stroke rate in the ViV registry was 10.1%, up from 6.2% at 3 years. The cumulative incidence of death or stroke through 5 years was 53.8%.
Mortality was significantly lower in recipients of a 26-mm Sapien 2 valve than with the 23-mm version, at 40% at 5 years versus 53%. Recipients of the smaller valve were more often male, had a higher prevalence of coronary artery disease, a higher surgical risk score, a significantly smaller baseline aortic valve area, and a higher mean gradient. Dr. Hahn and her coinvestigators are now examining their data to determine if surgical valve size/patient mismatch was a major driver of adverse outcomes, as has been reported in some other datasets.
At 5 years, the rate of structural valve deterioration–related hemodynamic valve deterioration (SVD-HVD) or bioprosthetic valve failure (BVF) using the soon-to-be-published Valve Academic Research Consortium–3 definitions was 6.6%. The rates of each class of valve deterioration at 5 years in this high- to extreme-risk population were 1.2 per 100 patient-years for SVD-HVD, 0.88 per 100 patient-years for all BVF, and 0.4 per 100 patient-years for SVD-related BVF.
Fully 51% of 5-year survivors were NYHA functional class I, whereas more than 90% of patients were class III or IV at baseline. The mean gradient was 16.8 mm Hg at 5 years, the Doppler velocity index was 0.35, and the mean Kansas City Cardiomyopathy Questionnaire overall summary score was 74.2, all closely similar to the values at 3 years. That dramatic and sustained improvement in the Kansas City Cardiomyopathy Questionnaire from a baseline of 43.1 points is larger than ever seen in any clinical trial of native valve TAVR, Dr. Hahn noted.
For discussant Vinayak N. Bapat, MD a cardiothoracic surgeon at the Minneapolis Heart Institute Foundation, the 5-year PARTNER 2 follow-up data contains a clear take-home message: “These data show that, when we as surgeons are putting in small valves, we ought to put in valves that are expandable.”
Discussant Jeroen J. Bax, MD, had one major caveat regarding the PARTNER 2 ViV Registry findings: They focused on high-surgical-risk patients.
“I think we would all agree that in high-risk patients, valve-in-valve is a better option than redo surgery. But in young, low-risk patients who are getting a bioprosthetic valve – and we’re going to be seeing more and more of them because over 90% of patients in Europe getting aortic valve surgery now are getting a bioprosthetic valve – we really don’t know what the best option is,” said Dr. Bax, professor of cardiology at the University of Leiden (the Netherlands).
He suggested a randomized trial of TAVR versus redo surgery in low-risk patients with failing bioprosthetic valves is in order, particularly in light of concerns raised by a recent report from a French national patient registry. These were “high-quality, real-world data,” Dr. Bax said, and while they showed better early outcomes for TAVR ViV than with redo surgery, there was a crossing of the curves for heart failure hospitalization already by 2 years.
“We need to look closely at younger, low-risk patients,” he concluded.
The PARTNER 2 ViV Registry is funded by Edwards Lifesciences. Dr. Hahn reported receiving research support from Philips Healthcare and 3Mensio and honoraria from Boston Scientific, Edwards Lifesciences, and Philips Healthcare.
SOURCE: Hahn RT. TCT 2020, Late breaker.
FROM TCT 2020
Hemorrhagic Papular Eruption on the Dorsal Hands
The Diagnosis: Heparin-Induced Bullous Hemorrhagic Dermatosis
Results of a punch biopsy of one of the hemorrhagic papules revealed a subcorneal hemorrhagic vesicle without underlying vasculitis, vasculopathy, inflammation, or viral changes (Figure). Tissue and blood cultures were sterile. Heparin and platelet factor 4 antibody testing was negative. The patient was diagnosed with heparin induced bullous hemorrhagic dermatosis (BHD). After chest imaging ruled out a pulmonary embolism, anticoagulation therapy was discontinued. Respiratory symptoms improved on antibiotics, and the skin lesions resolved completely within 2 weeks.
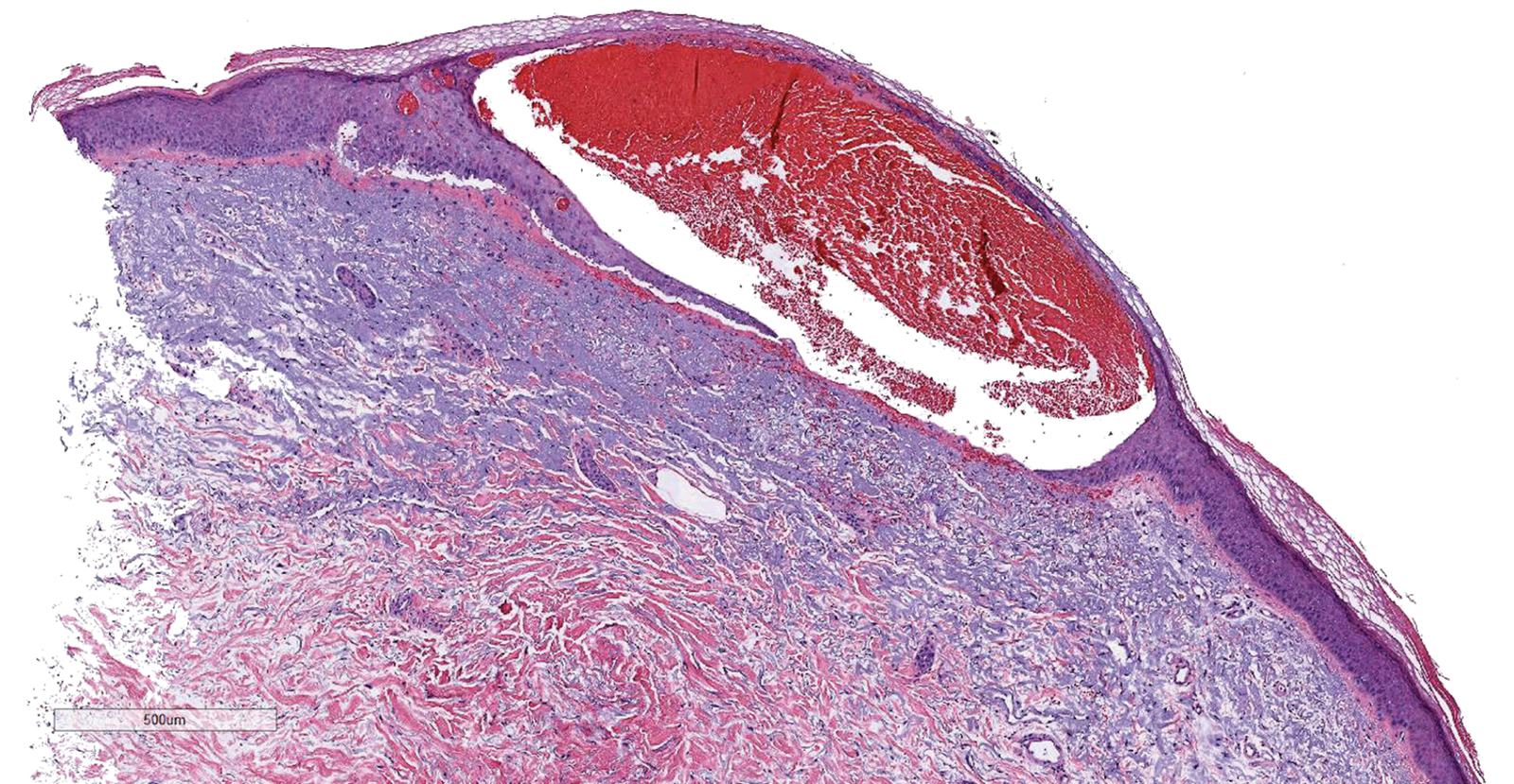
Bullous hemorrhagic dermatosis is an uncommon and underrecognized reaction to various anticoagulants. Bullous hemorrhagic dermatosis presents with painless, noninflammatory, hemorrhagic vesicles and bullae occurring at sites distant from anticoagulant administration. The condition was first characterized in 2006 by Perrinaud et al,1 who presented 3 cases in patients treated with heparin and low-molecular-weight heparin. Since then, there have been at least 90 cases reported in the international literature, with elderly men found to be the more affected demographic (male to female ratio, 1.9:1).2 Typically, BHD presents within 1 week of administration of an anticoagulant, but delayed onset has been reported.2 Bullous hemorrhagic dermatosis is most commonly observed with enoxaparin use but also has been described in association with unfractionated heparin, low-molecular-weight heparin products, and warfarin.2
The noninflammatory-appearing hemorrhagic papules and small plaques of BHD generally are seen on the extremities but can occur anywhere on the body including the oral mucosa.3 The differential diagnosis of BHD may include autoimmune vesiculobullous conditions, bullous drug eruptions, herpetic infection, supratherapeutic anticoagulation, porphyria cutanea tarda, amyloidosis, leukocytoclastic vasculitis, angioinvasive infections, and heparin necrosis. Diagnosis of BHD can be made clinically, but a biopsy is useful to exclude other conditions.
Histologically, BHD is characterized by the presence of intraepidermal hemorrhagic bullae without thrombotic, inflammatory, or vasculitic changes. Although heparinrelated skin lesions have been attributed to various mechanisms, including immune-mediated thrombocytopenia, type IV hypersensitivity reactions, type I allergic hypersensitivity reactions, pustulosis, and skin necrosis, the pathogenesis of BHD remains poorly understood.4 The condition has demonstrated koebnerization in some cases.5
In our patient, the absence of histologic inflammation, viral changes, vasculitis, and amyloid deposition helped rule out the other entities in the differential. The absence of heparin and platelet factor 4 antibodies helped exclude heparin necrosis. Direct immunofluorescence testing was not obtained in our patient but may be used to evaluate for an immunobullous etiology.
Management strategies for BHD are variable, and associated evidence is lacking. Treatment of BHD should be considered in the clinical context based on the necessity for anticoagulation and the severity of the eruption. Discontinuation of anticoagulation therapy, if possible, may prevent morbidity in some cases.6 If it is necessary to continue anticoagulation therapy, changing the drug or decreasing the dose are reasonable options. Skin lesions may resolve even if anticoagulation therapy is continued at the same dose.7,8 Concurrent supportive wound care is beneficial.
- Perrinaud A, Jacobi D, Machet MC, et al. Bullous hemorrhagic dermatosis occurring at sites distant from subcutaneous injections of heparin: three cases. J Am Acad Dermatol. 2006;54(suppl):S5-S7.
- Russo A, Curtis S, Balbuena-Merle R, et al. Bullous hemorrhagic dermatosis is an under-recognized side effect of full dose low-molecular weight heparin: a case report and review of the literature [published online July 6, 2018]. Exp Hematol. 2018;7:15.
- Harris HB, Kurth BJ, Lam TK, et al. Heparin-induced bullous hemorrhagic dermatosis confined to the oral mucosa. Cutis. 2019;103:365-366, 370.
- Schindewolf M, Schwaner S, Wolter M, et al. Incidence and causes of heparin-induced skin lesions. CMAJ. 2009;181:477-481.
- Gargallo V, Romero FT, Rodríguez-Peralto JL, et al. Heparin induced bullous hemorrhagic dermatosis at a site distant from the injection. a report of five cases. An Bras Dermatol. 2016;91:857-859.
- Choudhry S, Fishman PM, Hernandez C. Heparin-induced bullous hemorrhagic dermatosis. Cutis. 2013;91:93-98.
- Maldonado Cid P, Moreno Alonso de Celada R, Herranz Pinto P, et al. Bullous hemorrhagic dermatosis at sites distant from subcutaneous injections of heparin: a report of 5 cases. J Am Acad Dermatol. 2012;67:E220-E222.
- Snow SC, Pearson DR, Fathi R, et al. Heparin-induced haemorrhagic bullous dermatosis. Clin Exp Dermatol. 2018;43:393-398.
The Diagnosis: Heparin-Induced Bullous Hemorrhagic Dermatosis
Results of a punch biopsy of one of the hemorrhagic papules revealed a subcorneal hemorrhagic vesicle without underlying vasculitis, vasculopathy, inflammation, or viral changes (Figure). Tissue and blood cultures were sterile. Heparin and platelet factor 4 antibody testing was negative. The patient was diagnosed with heparin induced bullous hemorrhagic dermatosis (BHD). After chest imaging ruled out a pulmonary embolism, anticoagulation therapy was discontinued. Respiratory symptoms improved on antibiotics, and the skin lesions resolved completely within 2 weeks.
Bullous hemorrhagic dermatosis is an uncommon and underrecognized reaction to various anticoagulants. Bullous hemorrhagic dermatosis presents with painless, noninflammatory, hemorrhagic vesicles and bullae occurring at sites distant from anticoagulant administration. The condition was first characterized in 2006 by Perrinaud et al,1 who presented 3 cases in patients treated with heparin and low-molecular-weight heparin. Since then, there have been at least 90 cases reported in the international literature, with elderly men found to be the more affected demographic (male to female ratio, 1.9:1).2 Typically, BHD presents within 1 week of administration of an anticoagulant, but delayed onset has been reported.2 Bullous hemorrhagic dermatosis is most commonly observed with enoxaparin use but also has been described in association with unfractionated heparin, low-molecular-weight heparin products, and warfarin.2
The noninflammatory-appearing hemorrhagic papules and small plaques of BHD generally are seen on the extremities but can occur anywhere on the body including the oral mucosa.3 The differential diagnosis of BHD may include autoimmune vesiculobullous conditions, bullous drug eruptions, herpetic infection, supratherapeutic anticoagulation, porphyria cutanea tarda, amyloidosis, leukocytoclastic vasculitis, angioinvasive infections, and heparin necrosis. Diagnosis of BHD can be made clinically, but a biopsy is useful to exclude other conditions.
Histologically, BHD is characterized by the presence of intraepidermal hemorrhagic bullae without thrombotic, inflammatory, or vasculitic changes. Although heparinrelated skin lesions have been attributed to various mechanisms, including immune-mediated thrombocytopenia, type IV hypersensitivity reactions, type I allergic hypersensitivity reactions, pustulosis, and skin necrosis, the pathogenesis of BHD remains poorly understood.4 The condition has demonstrated koebnerization in some cases.5
In our patient, the absence of histologic inflammation, viral changes, vasculitis, and amyloid deposition helped rule out the other entities in the differential. The absence of heparin and platelet factor 4 antibodies helped exclude heparin necrosis. Direct immunofluorescence testing was not obtained in our patient but may be used to evaluate for an immunobullous etiology.
Management strategies for BHD are variable, and associated evidence is lacking. Treatment of BHD should be considered in the clinical context based on the necessity for anticoagulation and the severity of the eruption. Discontinuation of anticoagulation therapy, if possible, may prevent morbidity in some cases.6 If it is necessary to continue anticoagulation therapy, changing the drug or decreasing the dose are reasonable options. Skin lesions may resolve even if anticoagulation therapy is continued at the same dose.7,8 Concurrent supportive wound care is beneficial.
The Diagnosis: Heparin-Induced Bullous Hemorrhagic Dermatosis
Results of a punch biopsy of one of the hemorrhagic papules revealed a subcorneal hemorrhagic vesicle without underlying vasculitis, vasculopathy, inflammation, or viral changes (Figure). Tissue and blood cultures were sterile. Heparin and platelet factor 4 antibody testing was negative. The patient was diagnosed with heparin induced bullous hemorrhagic dermatosis (BHD). After chest imaging ruled out a pulmonary embolism, anticoagulation therapy was discontinued. Respiratory symptoms improved on antibiotics, and the skin lesions resolved completely within 2 weeks.
Bullous hemorrhagic dermatosis is an uncommon and underrecognized reaction to various anticoagulants. Bullous hemorrhagic dermatosis presents with painless, noninflammatory, hemorrhagic vesicles and bullae occurring at sites distant from anticoagulant administration. The condition was first characterized in 2006 by Perrinaud et al,1 who presented 3 cases in patients treated with heparin and low-molecular-weight heparin. Since then, there have been at least 90 cases reported in the international literature, with elderly men found to be the more affected demographic (male to female ratio, 1.9:1).2 Typically, BHD presents within 1 week of administration of an anticoagulant, but delayed onset has been reported.2 Bullous hemorrhagic dermatosis is most commonly observed with enoxaparin use but also has been described in association with unfractionated heparin, low-molecular-weight heparin products, and warfarin.2
The noninflammatory-appearing hemorrhagic papules and small plaques of BHD generally are seen on the extremities but can occur anywhere on the body including the oral mucosa.3 The differential diagnosis of BHD may include autoimmune vesiculobullous conditions, bullous drug eruptions, herpetic infection, supratherapeutic anticoagulation, porphyria cutanea tarda, amyloidosis, leukocytoclastic vasculitis, angioinvasive infections, and heparin necrosis. Diagnosis of BHD can be made clinically, but a biopsy is useful to exclude other conditions.
Histologically, BHD is characterized by the presence of intraepidermal hemorrhagic bullae without thrombotic, inflammatory, or vasculitic changes. Although heparinrelated skin lesions have been attributed to various mechanisms, including immune-mediated thrombocytopenia, type IV hypersensitivity reactions, type I allergic hypersensitivity reactions, pustulosis, and skin necrosis, the pathogenesis of BHD remains poorly understood.4 The condition has demonstrated koebnerization in some cases.5
In our patient, the absence of histologic inflammation, viral changes, vasculitis, and amyloid deposition helped rule out the other entities in the differential. The absence of heparin and platelet factor 4 antibodies helped exclude heparin necrosis. Direct immunofluorescence testing was not obtained in our patient but may be used to evaluate for an immunobullous etiology.
Management strategies for BHD are variable, and associated evidence is lacking. Treatment of BHD should be considered in the clinical context based on the necessity for anticoagulation and the severity of the eruption. Discontinuation of anticoagulation therapy, if possible, may prevent morbidity in some cases.6 If it is necessary to continue anticoagulation therapy, changing the drug or decreasing the dose are reasonable options. Skin lesions may resolve even if anticoagulation therapy is continued at the same dose.7,8 Concurrent supportive wound care is beneficial.
- Perrinaud A, Jacobi D, Machet MC, et al. Bullous hemorrhagic dermatosis occurring at sites distant from subcutaneous injections of heparin: three cases. J Am Acad Dermatol. 2006;54(suppl):S5-S7.
- Russo A, Curtis S, Balbuena-Merle R, et al. Bullous hemorrhagic dermatosis is an under-recognized side effect of full dose low-molecular weight heparin: a case report and review of the literature [published online July 6, 2018]. Exp Hematol. 2018;7:15.
- Harris HB, Kurth BJ, Lam TK, et al. Heparin-induced bullous hemorrhagic dermatosis confined to the oral mucosa. Cutis. 2019;103:365-366, 370.
- Schindewolf M, Schwaner S, Wolter M, et al. Incidence and causes of heparin-induced skin lesions. CMAJ. 2009;181:477-481.
- Gargallo V, Romero FT, Rodríguez-Peralto JL, et al. Heparin induced bullous hemorrhagic dermatosis at a site distant from the injection. a report of five cases. An Bras Dermatol. 2016;91:857-859.
- Choudhry S, Fishman PM, Hernandez C. Heparin-induced bullous hemorrhagic dermatosis. Cutis. 2013;91:93-98.
- Maldonado Cid P, Moreno Alonso de Celada R, Herranz Pinto P, et al. Bullous hemorrhagic dermatosis at sites distant from subcutaneous injections of heparin: a report of 5 cases. J Am Acad Dermatol. 2012;67:E220-E222.
- Snow SC, Pearson DR, Fathi R, et al. Heparin-induced haemorrhagic bullous dermatosis. Clin Exp Dermatol. 2018;43:393-398.
- Perrinaud A, Jacobi D, Machet MC, et al. Bullous hemorrhagic dermatosis occurring at sites distant from subcutaneous injections of heparin: three cases. J Am Acad Dermatol. 2006;54(suppl):S5-S7.
- Russo A, Curtis S, Balbuena-Merle R, et al. Bullous hemorrhagic dermatosis is an under-recognized side effect of full dose low-molecular weight heparin: a case report and review of the literature [published online July 6, 2018]. Exp Hematol. 2018;7:15.
- Harris HB, Kurth BJ, Lam TK, et al. Heparin-induced bullous hemorrhagic dermatosis confined to the oral mucosa. Cutis. 2019;103:365-366, 370.
- Schindewolf M, Schwaner S, Wolter M, et al. Incidence and causes of heparin-induced skin lesions. CMAJ. 2009;181:477-481.
- Gargallo V, Romero FT, Rodríguez-Peralto JL, et al. Heparin induced bullous hemorrhagic dermatosis at a site distant from the injection. a report of five cases. An Bras Dermatol. 2016;91:857-859.
- Choudhry S, Fishman PM, Hernandez C. Heparin-induced bullous hemorrhagic dermatosis. Cutis. 2013;91:93-98.
- Maldonado Cid P, Moreno Alonso de Celada R, Herranz Pinto P, et al. Bullous hemorrhagic dermatosis at sites distant from subcutaneous injections of heparin: a report of 5 cases. J Am Acad Dermatol. 2012;67:E220-E222.
- Snow SC, Pearson DR, Fathi R, et al. Heparin-induced haemorrhagic bullous dermatosis. Clin Exp Dermatol. 2018;43:393-398.
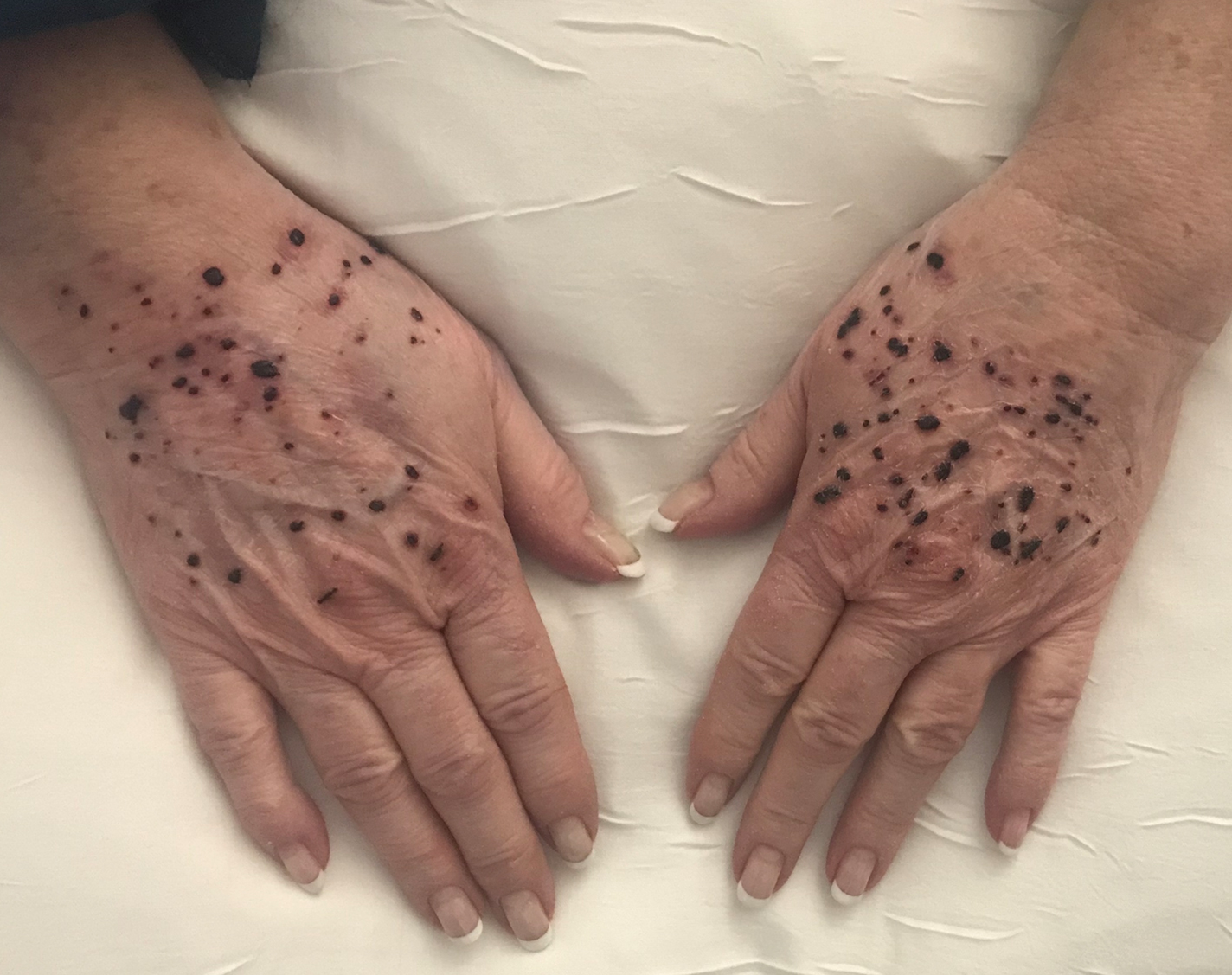
A 66-year-old woman with a history of granulomatous lung disease managed with methotrexate and prednisone, diabetes mellitus, hypertension, and Grave disease was admitted to the hospital for hypoxic respiratory failure. At admission, treatment was empirically initiated for pneumonia with intravenous ceftriaxone and azithromycin. Given the concern of a pulmonary embolism, intravenous heparin also was initiated. Dermatology was consulted for multiple painless blood blisters that erupted on the hands within 24 hours of admission. Physical examination revealed numerous firm hemorrhagic papules on the dorsal hands. Laboratory workup revealed a slightly elevated white blood cell count (11,800/µL [reference range, 4500–11,000/µL]), a normal stable platelet count (231,000/µL [reference range, 150,000– 350,000/µL]), and a normal international normalized ratio.
MADIT-CRT: Resynchronization linked to fewer heart failure hospitalizations
Patients with mild heart failure who received a cardiac resynchronization device had significantly reduced rates of hospitalizations for heart failure during follow-up of 1,820 patients for an average of 5.6 years, identifying in this post hoc analysis another benefit from this device that patients potentially receive in addition to an established survival advantage.
Extended follow-up of patients enrolled in the MADIT-CRT trial showed that patients with either New York Heart Association (NYHA) class I or II cardiomyopathy who received a cardiac resynchronization device with a defibrillator (CRT-D) had a significant reduction in all-cause hospitalization during follow-up, compared with control patients randomized to receive an implantable cardioverter defibrillator (ICD) device. This reduction in all hospitalizations was specifically driven by a significant reduction in cardiovascular hospitalizations, and the drop in cardiovascular hospitalizations was specifically driven by a cut in hospitalizations for heart failure (HHF), Sabu Thomas, MD, said at the annual scientific meeting of the Heart Failure Society of America.
The data showed that during follow-up all-cause hospitalizations occurred in 73% of the CRT-D patients and 83% of those who received an ICD; cardiovascular hospitalizations happened in 29% of the CRT-D patients and in 43% of those with an ICD; and HHF occurred in 12% of the CRT-D patients and in 22% of those with an ICD, reported Dr. Thomas, a heart failure cardiologist at the University of Rochester (N.Y.) Medical Center. All three between-group differences were statistically significant for these post hoc endpoints.
These reduced hospitalizations also linked with better survival. Patients in the trial database with cardiovascular hospitalizations had a nearly fourfold higher rate of death, compared with nonhospitalized patients, Dr. Thomas said.
The findings “suggest that this device [CRT-D] has sustained benefit in these patients for up to 7 years,” said Dr. Thomas and his collaborator, Valentina Kutyifa, MD, in an interview. “However, this was only seen in patients with left bundle branch block [LBBB].” In patients with non-LBBB, CRT-D was not associated with a reduction in [cardiovascular] hospitalizations.
The LBBB connection
In a multivariate analysis, the 1,281 patients with LBBB (70% of the study cohort) who were more than 6 months out from device placement had a significant 43% relative cut in their incidence of cardiovascular hospitalizations, compared with that of control patients who received an ICD, while the 537 patients with non-LBBB showed no benefit from CRT-D treatment, compared with those who received an ICD, for reducing cardiovascular hospitalizations. (Data from two enrolled patients weren’t available for the analyses.) This finding that the HHF benefit focused in patients with LBBB was consistent with many prior observations that CRT-D was most effective in this patient subgroup.
The researchers also highlighted that their findings apply only to patients with NYHA functional class I or II heart failure with reduced ejection fraction (HFrEF), the only types of patients enrolled in the MADIT-CRT trial (15% had class I disease).
The results also showed that, during the first 6 months on CRT-D treatment, patients with a LBBB showed a significant 43% increase in their cardiovascular hospitalizations, compared with control patients, which may have been driven by device-related events. “We did not investigate this in detail, and it needs more study,” said Dr. Thomas and Dr. Kutyifa, a cardiac electrophysiologist at the University of Rochester.Their new findings extend the initial, prespecified results of the MADIT-CRT (Multicenter Automatic Defibrillator Implantation With Cardiac Resynchronization Therapy) trial, which was designed to examine a primary endpoint of death from any cause or a nonfatal heart failure event. During the initial average follow-up of 2.4 years, patients who received a CRT-D device had a significant relative reduction in this endpoint of 34%, compared with patients on ICD treatment, exclusively in patients with LBBB. Extended follow-up for as long as 7 years of the same cohort showed a continued significant reduction of all-cause death compared with controls, a 41% relative risk reduction, that again was only apparent in patients with LBBB.
The MADIT-CRT findings are generally consistent with prevailing CRT-D recommendations from the American College of Cardiology and American Heart Association from 2013 that give a class I indication (“is indicated”) for using the device in heart failure patients with LBBB, a QRS interval of at least 150 msec, NYHA class II-IV function, and a left ventricular ejection fraction no greater than 35%. A lesser, class IIa recommendation (“can be useful”) exists for patients with a narrower QRS of 120-149 msec with the other class I criteria, and for patients with non-LBBB the recommendation drops to class IIb (“may be considered”).
CRT-D ‘is mysterious,’ especially for non-LBBB patients
“Every time researchers have tried to move beyond the [existing] paradigm of who benefits from CRT-D, it’s never panned out,” commented Jeffrey J. Goldberger, MD, an electrophysiologist, professor, and chief of the cardiovascular division at the University of Miami. “The guidelines are pretty correct on who should get CRT-D. I wouldn’t say that no patients with non-LBBB should get it, but they are less likely to benefit,” although he conceded that responses to CRT-D are highly individualized and hard to predict.
“CRT is mysterious. I’ve had patients who did incredibly well on it,” but “once you start getting outside of where the benefits are proven, you start to run into issues,” Dr. Goldberger said in an interview. “The only solid predictor of a CRT-D response is in patients with LBBB.”
The hospitalizations for heart failure that the University of Rochester investigators assessed as an additional study outcome represent an “important endpoint, but one that is much more subjective than survival,” making its reliability “a bit of a gray area,” he said. The analyses are also limited by being post hoc and, hence, just hypothesis generating.
A recently published analysis of the same dataset by many of the same investigators hinted that CRT-D might reduce HHF in non-LBBB patients when the focus is on recurrent hospitalizations.
Despite the evidence of a survival benefit from CRT-D placement in selected patients, especially those with LBBB, “registry data have shown that use of CRT-D varies widely and has been as low as 27% of eligible patients,” noted Dr. Thomas and Dr. Kutyifa. “There is an opportunity here to understand the barriers to more widespread adoption of CRT-D in appropriate patients,” they said. It is also “possible that CRT-D is overused in non-LBBB patients” given that this subgroup receives about a third of CRT-D devices now. “Future studies should carefully investigate the role of CRT-D in non-LBBB patients.”
MADIT-CRT was funded by Boston Scientific, which markets several CRT-D devices. Dr. Thomas had no disclosures. Dr. Kutyifa has been a consultant to Biotronik and Zoll and has received research funding from Biotronik, Boston Scientific, Spire, and Zoll. Dr Goldberger is director of a not-for-profit think tank on risk stratification for sudden cardiac death that has received unrestricted educational grants from Abbott, Biotronik, Boston Scientific, and Medtronic.
SOURCE: Thomas S et al. HFSA 2020, Abstract 019.
Patients with mild heart failure who received a cardiac resynchronization device had significantly reduced rates of hospitalizations for heart failure during follow-up of 1,820 patients for an average of 5.6 years, identifying in this post hoc analysis another benefit from this device that patients potentially receive in addition to an established survival advantage.
Extended follow-up of patients enrolled in the MADIT-CRT trial showed that patients with either New York Heart Association (NYHA) class I or II cardiomyopathy who received a cardiac resynchronization device with a defibrillator (CRT-D) had a significant reduction in all-cause hospitalization during follow-up, compared with control patients randomized to receive an implantable cardioverter defibrillator (ICD) device. This reduction in all hospitalizations was specifically driven by a significant reduction in cardiovascular hospitalizations, and the drop in cardiovascular hospitalizations was specifically driven by a cut in hospitalizations for heart failure (HHF), Sabu Thomas, MD, said at the annual scientific meeting of the Heart Failure Society of America.
The data showed that during follow-up all-cause hospitalizations occurred in 73% of the CRT-D patients and 83% of those who received an ICD; cardiovascular hospitalizations happened in 29% of the CRT-D patients and in 43% of those with an ICD; and HHF occurred in 12% of the CRT-D patients and in 22% of those with an ICD, reported Dr. Thomas, a heart failure cardiologist at the University of Rochester (N.Y.) Medical Center. All three between-group differences were statistically significant for these post hoc endpoints.
These reduced hospitalizations also linked with better survival. Patients in the trial database with cardiovascular hospitalizations had a nearly fourfold higher rate of death, compared with nonhospitalized patients, Dr. Thomas said.
The findings “suggest that this device [CRT-D] has sustained benefit in these patients for up to 7 years,” said Dr. Thomas and his collaborator, Valentina Kutyifa, MD, in an interview. “However, this was only seen in patients with left bundle branch block [LBBB].” In patients with non-LBBB, CRT-D was not associated with a reduction in [cardiovascular] hospitalizations.
The LBBB connection
In a multivariate analysis, the 1,281 patients with LBBB (70% of the study cohort) who were more than 6 months out from device placement had a significant 43% relative cut in their incidence of cardiovascular hospitalizations, compared with that of control patients who received an ICD, while the 537 patients with non-LBBB showed no benefit from CRT-D treatment, compared with those who received an ICD, for reducing cardiovascular hospitalizations. (Data from two enrolled patients weren’t available for the analyses.) This finding that the HHF benefit focused in patients with LBBB was consistent with many prior observations that CRT-D was most effective in this patient subgroup.
The researchers also highlighted that their findings apply only to patients with NYHA functional class I or II heart failure with reduced ejection fraction (HFrEF), the only types of patients enrolled in the MADIT-CRT trial (15% had class I disease).
The results also showed that, during the first 6 months on CRT-D treatment, patients with a LBBB showed a significant 43% increase in their cardiovascular hospitalizations, compared with control patients, which may have been driven by device-related events. “We did not investigate this in detail, and it needs more study,” said Dr. Thomas and Dr. Kutyifa, a cardiac electrophysiologist at the University of Rochester.Their new findings extend the initial, prespecified results of the MADIT-CRT (Multicenter Automatic Defibrillator Implantation With Cardiac Resynchronization Therapy) trial, which was designed to examine a primary endpoint of death from any cause or a nonfatal heart failure event. During the initial average follow-up of 2.4 years, patients who received a CRT-D device had a significant relative reduction in this endpoint of 34%, compared with patients on ICD treatment, exclusively in patients with LBBB. Extended follow-up for as long as 7 years of the same cohort showed a continued significant reduction of all-cause death compared with controls, a 41% relative risk reduction, that again was only apparent in patients with LBBB.
The MADIT-CRT findings are generally consistent with prevailing CRT-D recommendations from the American College of Cardiology and American Heart Association from 2013 that give a class I indication (“is indicated”) for using the device in heart failure patients with LBBB, a QRS interval of at least 150 msec, NYHA class II-IV function, and a left ventricular ejection fraction no greater than 35%. A lesser, class IIa recommendation (“can be useful”) exists for patients with a narrower QRS of 120-149 msec with the other class I criteria, and for patients with non-LBBB the recommendation drops to class IIb (“may be considered”).
CRT-D ‘is mysterious,’ especially for non-LBBB patients
“Every time researchers have tried to move beyond the [existing] paradigm of who benefits from CRT-D, it’s never panned out,” commented Jeffrey J. Goldberger, MD, an electrophysiologist, professor, and chief of the cardiovascular division at the University of Miami. “The guidelines are pretty correct on who should get CRT-D. I wouldn’t say that no patients with non-LBBB should get it, but they are less likely to benefit,” although he conceded that responses to CRT-D are highly individualized and hard to predict.
“CRT is mysterious. I’ve had patients who did incredibly well on it,” but “once you start getting outside of where the benefits are proven, you start to run into issues,” Dr. Goldberger said in an interview. “The only solid predictor of a CRT-D response is in patients with LBBB.”
The hospitalizations for heart failure that the University of Rochester investigators assessed as an additional study outcome represent an “important endpoint, but one that is much more subjective than survival,” making its reliability “a bit of a gray area,” he said. The analyses are also limited by being post hoc and, hence, just hypothesis generating.
A recently published analysis of the same dataset by many of the same investigators hinted that CRT-D might reduce HHF in non-LBBB patients when the focus is on recurrent hospitalizations.
Despite the evidence of a survival benefit from CRT-D placement in selected patients, especially those with LBBB, “registry data have shown that use of CRT-D varies widely and has been as low as 27% of eligible patients,” noted Dr. Thomas and Dr. Kutyifa. “There is an opportunity here to understand the barriers to more widespread adoption of CRT-D in appropriate patients,” they said. It is also “possible that CRT-D is overused in non-LBBB patients” given that this subgroup receives about a third of CRT-D devices now. “Future studies should carefully investigate the role of CRT-D in non-LBBB patients.”
MADIT-CRT was funded by Boston Scientific, which markets several CRT-D devices. Dr. Thomas had no disclosures. Dr. Kutyifa has been a consultant to Biotronik and Zoll and has received research funding from Biotronik, Boston Scientific, Spire, and Zoll. Dr Goldberger is director of a not-for-profit think tank on risk stratification for sudden cardiac death that has received unrestricted educational grants from Abbott, Biotronik, Boston Scientific, and Medtronic.
SOURCE: Thomas S et al. HFSA 2020, Abstract 019.
Patients with mild heart failure who received a cardiac resynchronization device had significantly reduced rates of hospitalizations for heart failure during follow-up of 1,820 patients for an average of 5.6 years, identifying in this post hoc analysis another benefit from this device that patients potentially receive in addition to an established survival advantage.
Extended follow-up of patients enrolled in the MADIT-CRT trial showed that patients with either New York Heart Association (NYHA) class I or II cardiomyopathy who received a cardiac resynchronization device with a defibrillator (CRT-D) had a significant reduction in all-cause hospitalization during follow-up, compared with control patients randomized to receive an implantable cardioverter defibrillator (ICD) device. This reduction in all hospitalizations was specifically driven by a significant reduction in cardiovascular hospitalizations, and the drop in cardiovascular hospitalizations was specifically driven by a cut in hospitalizations for heart failure (HHF), Sabu Thomas, MD, said at the annual scientific meeting of the Heart Failure Society of America.
The data showed that during follow-up all-cause hospitalizations occurred in 73% of the CRT-D patients and 83% of those who received an ICD; cardiovascular hospitalizations happened in 29% of the CRT-D patients and in 43% of those with an ICD; and HHF occurred in 12% of the CRT-D patients and in 22% of those with an ICD, reported Dr. Thomas, a heart failure cardiologist at the University of Rochester (N.Y.) Medical Center. All three between-group differences were statistically significant for these post hoc endpoints.
These reduced hospitalizations also linked with better survival. Patients in the trial database with cardiovascular hospitalizations had a nearly fourfold higher rate of death, compared with nonhospitalized patients, Dr. Thomas said.
The findings “suggest that this device [CRT-D] has sustained benefit in these patients for up to 7 years,” said Dr. Thomas and his collaborator, Valentina Kutyifa, MD, in an interview. “However, this was only seen in patients with left bundle branch block [LBBB].” In patients with non-LBBB, CRT-D was not associated with a reduction in [cardiovascular] hospitalizations.
The LBBB connection
In a multivariate analysis, the 1,281 patients with LBBB (70% of the study cohort) who were more than 6 months out from device placement had a significant 43% relative cut in their incidence of cardiovascular hospitalizations, compared with that of control patients who received an ICD, while the 537 patients with non-LBBB showed no benefit from CRT-D treatment, compared with those who received an ICD, for reducing cardiovascular hospitalizations. (Data from two enrolled patients weren’t available for the analyses.) This finding that the HHF benefit focused in patients with LBBB was consistent with many prior observations that CRT-D was most effective in this patient subgroup.
The researchers also highlighted that their findings apply only to patients with NYHA functional class I or II heart failure with reduced ejection fraction (HFrEF), the only types of patients enrolled in the MADIT-CRT trial (15% had class I disease).
The results also showed that, during the first 6 months on CRT-D treatment, patients with a LBBB showed a significant 43% increase in their cardiovascular hospitalizations, compared with control patients, which may have been driven by device-related events. “We did not investigate this in detail, and it needs more study,” said Dr. Thomas and Dr. Kutyifa, a cardiac electrophysiologist at the University of Rochester.Their new findings extend the initial, prespecified results of the MADIT-CRT (Multicenter Automatic Defibrillator Implantation With Cardiac Resynchronization Therapy) trial, which was designed to examine a primary endpoint of death from any cause or a nonfatal heart failure event. During the initial average follow-up of 2.4 years, patients who received a CRT-D device had a significant relative reduction in this endpoint of 34%, compared with patients on ICD treatment, exclusively in patients with LBBB. Extended follow-up for as long as 7 years of the same cohort showed a continued significant reduction of all-cause death compared with controls, a 41% relative risk reduction, that again was only apparent in patients with LBBB.
The MADIT-CRT findings are generally consistent with prevailing CRT-D recommendations from the American College of Cardiology and American Heart Association from 2013 that give a class I indication (“is indicated”) for using the device in heart failure patients with LBBB, a QRS interval of at least 150 msec, NYHA class II-IV function, and a left ventricular ejection fraction no greater than 35%. A lesser, class IIa recommendation (“can be useful”) exists for patients with a narrower QRS of 120-149 msec with the other class I criteria, and for patients with non-LBBB the recommendation drops to class IIb (“may be considered”).
CRT-D ‘is mysterious,’ especially for non-LBBB patients
“Every time researchers have tried to move beyond the [existing] paradigm of who benefits from CRT-D, it’s never panned out,” commented Jeffrey J. Goldberger, MD, an electrophysiologist, professor, and chief of the cardiovascular division at the University of Miami. “The guidelines are pretty correct on who should get CRT-D. I wouldn’t say that no patients with non-LBBB should get it, but they are less likely to benefit,” although he conceded that responses to CRT-D are highly individualized and hard to predict.
“CRT is mysterious. I’ve had patients who did incredibly well on it,” but “once you start getting outside of where the benefits are proven, you start to run into issues,” Dr. Goldberger said in an interview. “The only solid predictor of a CRT-D response is in patients with LBBB.”
The hospitalizations for heart failure that the University of Rochester investigators assessed as an additional study outcome represent an “important endpoint, but one that is much more subjective than survival,” making its reliability “a bit of a gray area,” he said. The analyses are also limited by being post hoc and, hence, just hypothesis generating.
A recently published analysis of the same dataset by many of the same investigators hinted that CRT-D might reduce HHF in non-LBBB patients when the focus is on recurrent hospitalizations.
Despite the evidence of a survival benefit from CRT-D placement in selected patients, especially those with LBBB, “registry data have shown that use of CRT-D varies widely and has been as low as 27% of eligible patients,” noted Dr. Thomas and Dr. Kutyifa. “There is an opportunity here to understand the barriers to more widespread adoption of CRT-D in appropriate patients,” they said. It is also “possible that CRT-D is overused in non-LBBB patients” given that this subgroup receives about a third of CRT-D devices now. “Future studies should carefully investigate the role of CRT-D in non-LBBB patients.”
MADIT-CRT was funded by Boston Scientific, which markets several CRT-D devices. Dr. Thomas had no disclosures. Dr. Kutyifa has been a consultant to Biotronik and Zoll and has received research funding from Biotronik, Boston Scientific, Spire, and Zoll. Dr Goldberger is director of a not-for-profit think tank on risk stratification for sudden cardiac death that has received unrestricted educational grants from Abbott, Biotronik, Boston Scientific, and Medtronic.
SOURCE: Thomas S et al. HFSA 2020, Abstract 019.
FROM HFSA 2020
Survey finds European dermatologists unhappy with pandemic teledermatology experience
intensely, according to the findings of a survey presented at the virtual annual congress of the European Academy of Dermatology and Venereology.
“The results of our survey clearly show 7 out of 10 participating dermatologists declared that they were not happy with teledermatology, and most of them declared that they were not at all happy,” according to Mariano Suppa, MD, PhD, of the department of dermatology and venereology, Free University of Brussels.
“It was very interesting: it was not just about the lack of a good quality of consultation, but was also related to some extent to a lack of respect from some patients, and also a lack of empathy. The majority of survey respondents felt [attacked] by their own patients because they were proposing teledermatology. So, yes, we were forced to go to teledermatology, and I think we will be again to some extent, but clearly we’re not happy about it,” he elaborated in response to a question from session chair Brigitte Dreno, MD, professor of dermatology and vice dean of the faculty of medicine at the University of Nantes (France).
The survey, conducted by the EADV communication committee, assessed the pandemic’s impact on European dermatologists’ professional practices and personal lives through 30 brief questions, with space at the end for additional open-ended comments. In the comments section, many dermatologists vented about their income loss, the disorganized response to round one of the pandemic, and most of all about teledermatology. Common complaints were that teledermatology required a huge consumption of energy and constituted a major intrusion upon the physicians’ personal lives. And then there was the common theme of unkind treatment by some patients.
The survey was sent twice in June 2020 to more than 4,800 EADV members. It was completed by 490 dermatologists from 39 countries. Dr. Suppa attributed the low response rate to physician weariness of the topic due to saturation news media coverage of the pandemic.
Sixty-nine percent of responding dermatologists were women. Fifty-two percent of participants were over age 50, 81% lived in a city, and 53% worked in a university or public hospital or clinic. Twelve percent lived alone.
Impact on professional practice
Many European dermatologists were on the front lines in dealing with the first wave of COVID-19. Twenty-eight percent worked in a COVID-19 unit. Forty-eight percent of dermatologists performed COVID-19 tests, and those who didn’t either had no patient contact or couldn’t get test kits. Thirty-five percent of dermatologists saw patients who presented with skin signs of COVID-19. Four percent of survey respondents became infected.
Seventy percent rescheduled or canceled all or most patient appointments. Clinical care was prioritized: during the peak of the pandemic, 76% of dermatologists saw only urgent cases – mostly potentially serious rashes – and dermato-oncology patients. Seventy-six percent of dermatologists performed teledermatology, although by June 60% of respondents reported seeing at least three-quarters of their patients face-to-face.
Twenty-three percent of dermatologists reported having lost most or all of their income during March through June, and another 26% lost about half.
Impact on dermatologists’ personal lives
About half of survey respondents reported feeling stressed, and a similar percentage checked the box marked ‘anxiety.’ Nine percent reported depressive symptoms, 15% mentioned feeling anger, 17% uselessness, and 2% admitted suicidal ideation. But 30% of dermatologists reported experiencing no negative psychological effects whatsoever stemming from the pandemic.
Sixteen percent of dermatologists reported drinking more alcohol during sequestration.
But respondents cited positive effects as well: a renewed appreciation of the importance of time, and enjoyment of the additional time spent with family and alone. Many dermatologists relished the opportunity to spend more time cooking, reading literature, doing research, listening to or playing music, and practicing yoga or meditation. And dermatologists took solace and pride in being members of the vital medical community.
Dr. Dreno asked if the survey revealed evidence of underdiagnosis and undertreatment of dermatologic diseases during the pandemic. Dr. Suppa replied that the survey didn’t address that issue, but it’s his personal opinion that this was no doubt the case. Roughly one-quarter of dermatologists canceled all appointments, and when dermatology clinics became filled beginning in June, he and his colleagues saw a number of cases of delayed-diagnosis advanced skin cancer.
“I think that the diseases that were really penalized were the chronic inflammatory diseases, such as psoriasis, hidradenitis suppurativa, and also atopic dermatitis. We were doing a lot of telephone consultations for those patients at that time, and we saw in June that for those particular patients there was an unmet need in the pandemic because some of them really needed to have been seen. I think this is a lesson we should learn for the second wave that we’re unfortunately facing right now: We need to adopt restrictive measures to avoid spreading the pandemic, yes, for sure, but we need to keep in mind that there is not just COVID-19, but also other important diseases,” Dr. Suppa said.
A second EADV survey will be performed during the fall/winter wave of the pandemic.
Dr. Suppa reported having no financial conflicts regarding the EADV-funded survey.
SOURCE: Suppa M. EADV 2020. Presentation D3T03.4D
intensely, according to the findings of a survey presented at the virtual annual congress of the European Academy of Dermatology and Venereology.
“The results of our survey clearly show 7 out of 10 participating dermatologists declared that they were not happy with teledermatology, and most of them declared that they were not at all happy,” according to Mariano Suppa, MD, PhD, of the department of dermatology and venereology, Free University of Brussels.
“It was very interesting: it was not just about the lack of a good quality of consultation, but was also related to some extent to a lack of respect from some patients, and also a lack of empathy. The majority of survey respondents felt [attacked] by their own patients because they were proposing teledermatology. So, yes, we were forced to go to teledermatology, and I think we will be again to some extent, but clearly we’re not happy about it,” he elaborated in response to a question from session chair Brigitte Dreno, MD, professor of dermatology and vice dean of the faculty of medicine at the University of Nantes (France).
The survey, conducted by the EADV communication committee, assessed the pandemic’s impact on European dermatologists’ professional practices and personal lives through 30 brief questions, with space at the end for additional open-ended comments. In the comments section, many dermatologists vented about their income loss, the disorganized response to round one of the pandemic, and most of all about teledermatology. Common complaints were that teledermatology required a huge consumption of energy and constituted a major intrusion upon the physicians’ personal lives. And then there was the common theme of unkind treatment by some patients.
The survey was sent twice in June 2020 to more than 4,800 EADV members. It was completed by 490 dermatologists from 39 countries. Dr. Suppa attributed the low response rate to physician weariness of the topic due to saturation news media coverage of the pandemic.
Sixty-nine percent of responding dermatologists were women. Fifty-two percent of participants were over age 50, 81% lived in a city, and 53% worked in a university or public hospital or clinic. Twelve percent lived alone.
Impact on professional practice
Many European dermatologists were on the front lines in dealing with the first wave of COVID-19. Twenty-eight percent worked in a COVID-19 unit. Forty-eight percent of dermatologists performed COVID-19 tests, and those who didn’t either had no patient contact or couldn’t get test kits. Thirty-five percent of dermatologists saw patients who presented with skin signs of COVID-19. Four percent of survey respondents became infected.
Seventy percent rescheduled or canceled all or most patient appointments. Clinical care was prioritized: during the peak of the pandemic, 76% of dermatologists saw only urgent cases – mostly potentially serious rashes – and dermato-oncology patients. Seventy-six percent of dermatologists performed teledermatology, although by June 60% of respondents reported seeing at least three-quarters of their patients face-to-face.
Twenty-three percent of dermatologists reported having lost most or all of their income during March through June, and another 26% lost about half.
Impact on dermatologists’ personal lives
About half of survey respondents reported feeling stressed, and a similar percentage checked the box marked ‘anxiety.’ Nine percent reported depressive symptoms, 15% mentioned feeling anger, 17% uselessness, and 2% admitted suicidal ideation. But 30% of dermatologists reported experiencing no negative psychological effects whatsoever stemming from the pandemic.
Sixteen percent of dermatologists reported drinking more alcohol during sequestration.
But respondents cited positive effects as well: a renewed appreciation of the importance of time, and enjoyment of the additional time spent with family and alone. Many dermatologists relished the opportunity to spend more time cooking, reading literature, doing research, listening to or playing music, and practicing yoga or meditation. And dermatologists took solace and pride in being members of the vital medical community.
Dr. Dreno asked if the survey revealed evidence of underdiagnosis and undertreatment of dermatologic diseases during the pandemic. Dr. Suppa replied that the survey didn’t address that issue, but it’s his personal opinion that this was no doubt the case. Roughly one-quarter of dermatologists canceled all appointments, and when dermatology clinics became filled beginning in June, he and his colleagues saw a number of cases of delayed-diagnosis advanced skin cancer.
“I think that the diseases that were really penalized were the chronic inflammatory diseases, such as psoriasis, hidradenitis suppurativa, and also atopic dermatitis. We were doing a lot of telephone consultations for those patients at that time, and we saw in June that for those particular patients there was an unmet need in the pandemic because some of them really needed to have been seen. I think this is a lesson we should learn for the second wave that we’re unfortunately facing right now: We need to adopt restrictive measures to avoid spreading the pandemic, yes, for sure, but we need to keep in mind that there is not just COVID-19, but also other important diseases,” Dr. Suppa said.
A second EADV survey will be performed during the fall/winter wave of the pandemic.
Dr. Suppa reported having no financial conflicts regarding the EADV-funded survey.
SOURCE: Suppa M. EADV 2020. Presentation D3T03.4D
intensely, according to the findings of a survey presented at the virtual annual congress of the European Academy of Dermatology and Venereology.
“The results of our survey clearly show 7 out of 10 participating dermatologists declared that they were not happy with teledermatology, and most of them declared that they were not at all happy,” according to Mariano Suppa, MD, PhD, of the department of dermatology and venereology, Free University of Brussels.
“It was very interesting: it was not just about the lack of a good quality of consultation, but was also related to some extent to a lack of respect from some patients, and also a lack of empathy. The majority of survey respondents felt [attacked] by their own patients because they were proposing teledermatology. So, yes, we were forced to go to teledermatology, and I think we will be again to some extent, but clearly we’re not happy about it,” he elaborated in response to a question from session chair Brigitte Dreno, MD, professor of dermatology and vice dean of the faculty of medicine at the University of Nantes (France).
The survey, conducted by the EADV communication committee, assessed the pandemic’s impact on European dermatologists’ professional practices and personal lives through 30 brief questions, with space at the end for additional open-ended comments. In the comments section, many dermatologists vented about their income loss, the disorganized response to round one of the pandemic, and most of all about teledermatology. Common complaints were that teledermatology required a huge consumption of energy and constituted a major intrusion upon the physicians’ personal lives. And then there was the common theme of unkind treatment by some patients.
The survey was sent twice in June 2020 to more than 4,800 EADV members. It was completed by 490 dermatologists from 39 countries. Dr. Suppa attributed the low response rate to physician weariness of the topic due to saturation news media coverage of the pandemic.
Sixty-nine percent of responding dermatologists were women. Fifty-two percent of participants were over age 50, 81% lived in a city, and 53% worked in a university or public hospital or clinic. Twelve percent lived alone.
Impact on professional practice
Many European dermatologists were on the front lines in dealing with the first wave of COVID-19. Twenty-eight percent worked in a COVID-19 unit. Forty-eight percent of dermatologists performed COVID-19 tests, and those who didn’t either had no patient contact or couldn’t get test kits. Thirty-five percent of dermatologists saw patients who presented with skin signs of COVID-19. Four percent of survey respondents became infected.
Seventy percent rescheduled or canceled all or most patient appointments. Clinical care was prioritized: during the peak of the pandemic, 76% of dermatologists saw only urgent cases – mostly potentially serious rashes – and dermato-oncology patients. Seventy-six percent of dermatologists performed teledermatology, although by June 60% of respondents reported seeing at least three-quarters of their patients face-to-face.
Twenty-three percent of dermatologists reported having lost most or all of their income during March through June, and another 26% lost about half.
Impact on dermatologists’ personal lives
About half of survey respondents reported feeling stressed, and a similar percentage checked the box marked ‘anxiety.’ Nine percent reported depressive symptoms, 15% mentioned feeling anger, 17% uselessness, and 2% admitted suicidal ideation. But 30% of dermatologists reported experiencing no negative psychological effects whatsoever stemming from the pandemic.
Sixteen percent of dermatologists reported drinking more alcohol during sequestration.
But respondents cited positive effects as well: a renewed appreciation of the importance of time, and enjoyment of the additional time spent with family and alone. Many dermatologists relished the opportunity to spend more time cooking, reading literature, doing research, listening to or playing music, and practicing yoga or meditation. And dermatologists took solace and pride in being members of the vital medical community.
Dr. Dreno asked if the survey revealed evidence of underdiagnosis and undertreatment of dermatologic diseases during the pandemic. Dr. Suppa replied that the survey didn’t address that issue, but it’s his personal opinion that this was no doubt the case. Roughly one-quarter of dermatologists canceled all appointments, and when dermatology clinics became filled beginning in June, he and his colleagues saw a number of cases of delayed-diagnosis advanced skin cancer.
“I think that the diseases that were really penalized were the chronic inflammatory diseases, such as psoriasis, hidradenitis suppurativa, and also atopic dermatitis. We were doing a lot of telephone consultations for those patients at that time, and we saw in June that for those particular patients there was an unmet need in the pandemic because some of them really needed to have been seen. I think this is a lesson we should learn for the second wave that we’re unfortunately facing right now: We need to adopt restrictive measures to avoid spreading the pandemic, yes, for sure, but we need to keep in mind that there is not just COVID-19, but also other important diseases,” Dr. Suppa said.
A second EADV survey will be performed during the fall/winter wave of the pandemic.
Dr. Suppa reported having no financial conflicts regarding the EADV-funded survey.
SOURCE: Suppa M. EADV 2020. Presentation D3T03.4D
FROM THE EADV CONGRESS
Nivolumab Use for First-Line Management of Hepatocellular Carcinoma: Results of a Real-World Cohort of Patients
Hepatocellular carcinoma (HCC) has a poor prognosis and remains an important cause of cancer-related morbidity and mortality.1,2 Potentially curative interventions include surgical resection, radiofrequency ablation, and liver transplantation. However, the majority of patients are not eligible for these procedures because they are diagnosed at an advanced stage, when locoregional therapies are much more limited.3,4 Although the kinase inhibitors sorafenib and lenvatinib are approved as first-line systemic treatment, at the US Department of Veterans Affairs (VA) Kansas City VA Medical Center (KCVAMC) in Missouri, nivolumab was used instead because of concerns for the tolerability of the kinase inhibitors. Locoregional therapies, resection, and transplantation options were either not appropriate or had been exhausted for these patients. The objective of this retrospective study was to determine the outcomes of those veteran patients in a small cohort.
Methods
The KCVAMC Institutional Review Board approved this retrospective chart review. Patients were selected from pharmacy records at KCVAMC. We identified all patients with a diagnosis of HCC who received nivolumab from January 2016 to December 2019. We then included only the patients that had nivolumab in the front-line setting for our final analysis. At the time of initiation of treatment, all patients were informed that immunotherapy was not approved for front-line treatment, but available evidence suggested that it would be easier to tolerate than sorafenib or lenvatinib. These patients were determined to be either ineligible for sorafenib or lenvatinib therapy or expected to tolerate it poorly, and hence they consented to the use of nivolumab. Tumor response and progression were assessed by the investigator according to iRECIST (Immune Response Evaluation Criteria in Solid Tumors) criteria.5 Data were obtained from retrospective health record review.
Results
Fourteen men received nivolumab in the front-line systemic therapy setting from January 2016 to December 2019 at KCVAMC. The median age was 63.5 years (range, 58-72 years), and the median Eastern Cooperative Oncology Group score was 1. The Table highlights patient characteristics.
Of the 14 patients included in the review, 2 patients had a response to nivolumab (14.3%) and 1 patient had a complete response (7.1%). The median duration of immunotherapy was 4.5 months. Immunotherapy was discontinued due to disease progression in 10 patients and toxicity in 3 patients.
The median progression-free survival (PFS) from initiation of immunotherapy was 4 months; median overall survival (OS) was 8 months. The median time from diagnosis to survival was 41 months. Only 1 patient received a second-line treatment.
Incidence of grade 3 or higher toxicity was 35%. Three deaths resulted from auto-immune hepatitis (grade 5 toxicity), as well as 1 grade 3 skin toxicity, and 1 grade 4 liver toxicity.
Discussion
Immunotherapy has shown promise in patients with HCC based on the results of the KEYNOTE-224 and Checkmate-040 studies,6,7 which led to an accelerated US Food and Drug Administration approval of nivolumab and pembrolizumab for HCC following failure of first-line sorafenib.8,9
Several clinical trials are evaluating front-line immunotherapy for HCC. The Checkmate 459 study demonstrated the median OS to be 16.4 months for nivolumab vs 14.7 months for sorafenib, a difference that was not statistically significant. However, tolerability of nivolumab was better than it was for sorafenib, thus positioning it as a potentially attractive first-line option.10 The GO30140 study evaluated
The results from our study differed from the previous studies and raise concern for the applicability of these trials to a real-world population. For example, both the GO30140 and IMbrave150 excluded patients with untreated varices.11,12 Both IMbrave150 and Checkmate 459 limited enrollment only to patients with a Child-Pugh A score for liver disease; 36% of the KCVAMC patients had a Child-Pugh B score. Three patients (21.4%) were homeless, 6 patients (42.8%) had substance abuse history and 5 patients (35.7%) had mental illness. Several psychosocial factors present in our patients, such as substance abuse, mental illness, and homelessness, would have excluded them from clinical trials. Our small cohort of patients, thus, represents a frail real-world population due to multiple medical and psychosocial comorbidities. Real-world experience with immunotherapy as second-line therapy after treatment with sorafenib has been reported, but this is the first reported real-world experience of immunotherapy in the front-line setting for HCC.13,14
Large differences in sociodemographic status and health status exist between the veteran population and typical clinical trial populations. Veterans are predominantly male and older than a clinical trial population. Veterans are more likely to belong to a minority group, more likely to have lower level education and more likely to be poor than a clinical trial population. They are more likely to have poorer health status with higher number of medical conditions and psychosocial conditions.15
Limitations
We acknowledge several limitations to our study, such as the small number of patients and the retrospective single center nature of this study. Patients were older men with multiple psychosocial comorbitities like mental illness, substance abuse, and homelessness. This cohort may not represent the non-VA population, but is an excellent representation of a frail, real-world veteran population.
Conclusions
Despite clinical trials showing the promise of immunotherapy as an attractive front-line systemic treatment option for HCC, our results show poor outcomes in a frail real-world population. In a cohort of patients who received immunotherapy as a front-line systemic treatment for HCC, results were poor with a response rate of 14.3%, a median PFS of 4 months, and a median OS of 8 months. We noted a significantly higher number of adverse effects, including 21% incidence of grade 5 hepatotoxicity. There remains an urgent need to develop more effective and safer therapies for this patient population as well as validation from larger real-world studies.
1. El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118-1127. doi:10.1056/NEJMra1001683
2. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359-E386. doi:10.1002/ijc.29210
3. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362(9399):1907-1917. doi:10.1016/S0140-6736(03)14964-1
4. Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47 Suppl(0):S2-S6. doi:10.1097/MCG.0b013e3182872f29
5. Seymour L, Bogaerts J, Perrone A, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics [published correction appears in Lancet Oncol. 2019 May;20(5):e242]. Lancet Oncol. 2017;18(3):e143-e152. doi:10.1016/S1470-2045(17)30074-8
6. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492-2502.doi:10.1016/S0140-6736(17)31046-2
7. Zhu AX, Finn RS, Edeline J, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial [published correction appears in Lancet Oncol. 2018 Sep;19(9):e440]. Lancet Oncol. 2018;19(7):940-952. doi:10.1016/S1470-2045(18)30351-6
8. US Food and Drug Administration. FDA grants accelerated approval to nivolumab for HCC previously treated with sorafenib. Updated September 25, 2017. Accessed October 7, 2020. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-nivolumab-hcc-previously-treated-sorafenib.
9. US Food and Drug Administration. FDA grants accelerated approval to pembrolizumab for hepatocellular carcinoma. Updated December 14, 2018. Accessed October 7, 2020. https://www.fda.gov/drugs/fda-grants-accelerated-approval-pembrolizumab-hepatocellular-carcinoma.
10. Yau T, Park JW, Finn RS, et al. CheckMate 459: A randomized, multi-center phase 3 study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Presented at: ESMO 2019 Congress. Barcelona, Spain: September 27, 2019. Ann Onc. 2019;30(suppl_5):v851-v934. doi:10.1093/annonc/mdz394
11. Lee M, Ryoo BY, Hsu CH, et al. Randomised efficacy and safety results for atezolizumab (atezo) + bevacizumab (bev) in patients (pts) with previously untreated, unresectable hepatocellular carcinoma (HCC). Presented at: ESMO 2019 Congress. Barcelona, Spain: September 27, 2019.
12. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894-1905.doi:10.1056/NEJMoa1915745
13. Scheiner B, Kirstein MM, Hucke F, et al. Programmed cell death protein-1 (PD-1)-targeted immunotherapy in advanced hepatocellular carcinoma: efficacy and safety data from an international multicentre real-world cohort. Aliment Pharmacol Ther. 2019;49(10):1323-1333. doi:10.1111/apt.15245
14. Yoon SE, Hur JY, Lee KK, et al. Real-world data on nivolumab treatment in Asian patients with advanced hepatocellular carcinoma. Presented at: ESMO 2018 Congress. Munich, Germany: October 21, 2018. Ann Onc. 2018;29(suppl_8):viii205-viii270. doi:10.1093/annonc/mdy282
15. Agha Z, Lofgren RP, VanRuiswyk JV, Layde PM. Are patients at Veterans Affairs medical centers sicker? A comparative analysis of health status and medical resource use. Arch Intern Med. 2000;160(21):3252-3257. doi:10.1001/archinte.160.21.3252
Hepatocellular carcinoma (HCC) has a poor prognosis and remains an important cause of cancer-related morbidity and mortality.1,2 Potentially curative interventions include surgical resection, radiofrequency ablation, and liver transplantation. However, the majority of patients are not eligible for these procedures because they are diagnosed at an advanced stage, when locoregional therapies are much more limited.3,4 Although the kinase inhibitors sorafenib and lenvatinib are approved as first-line systemic treatment, at the US Department of Veterans Affairs (VA) Kansas City VA Medical Center (KCVAMC) in Missouri, nivolumab was used instead because of concerns for the tolerability of the kinase inhibitors. Locoregional therapies, resection, and transplantation options were either not appropriate or had been exhausted for these patients. The objective of this retrospective study was to determine the outcomes of those veteran patients in a small cohort.
Methods
The KCVAMC Institutional Review Board approved this retrospective chart review. Patients were selected from pharmacy records at KCVAMC. We identified all patients with a diagnosis of HCC who received nivolumab from January 2016 to December 2019. We then included only the patients that had nivolumab in the front-line setting for our final analysis. At the time of initiation of treatment, all patients were informed that immunotherapy was not approved for front-line treatment, but available evidence suggested that it would be easier to tolerate than sorafenib or lenvatinib. These patients were determined to be either ineligible for sorafenib or lenvatinib therapy or expected to tolerate it poorly, and hence they consented to the use of nivolumab. Tumor response and progression were assessed by the investigator according to iRECIST (Immune Response Evaluation Criteria in Solid Tumors) criteria.5 Data were obtained from retrospective health record review.
Results
Fourteen men received nivolumab in the front-line systemic therapy setting from January 2016 to December 2019 at KCVAMC. The median age was 63.5 years (range, 58-72 years), and the median Eastern Cooperative Oncology Group score was 1. The Table highlights patient characteristics.
Of the 14 patients included in the review, 2 patients had a response to nivolumab (14.3%) and 1 patient had a complete response (7.1%). The median duration of immunotherapy was 4.5 months. Immunotherapy was discontinued due to disease progression in 10 patients and toxicity in 3 patients.
The median progression-free survival (PFS) from initiation of immunotherapy was 4 months; median overall survival (OS) was 8 months. The median time from diagnosis to survival was 41 months. Only 1 patient received a second-line treatment.
Incidence of grade 3 or higher toxicity was 35%. Three deaths resulted from auto-immune hepatitis (grade 5 toxicity), as well as 1 grade 3 skin toxicity, and 1 grade 4 liver toxicity.
Discussion
Immunotherapy has shown promise in patients with HCC based on the results of the KEYNOTE-224 and Checkmate-040 studies,6,7 which led to an accelerated US Food and Drug Administration approval of nivolumab and pembrolizumab for HCC following failure of first-line sorafenib.8,9
Several clinical trials are evaluating front-line immunotherapy for HCC. The Checkmate 459 study demonstrated the median OS to be 16.4 months for nivolumab vs 14.7 months for sorafenib, a difference that was not statistically significant. However, tolerability of nivolumab was better than it was for sorafenib, thus positioning it as a potentially attractive first-line option.10 The GO30140 study evaluated
The results from our study differed from the previous studies and raise concern for the applicability of these trials to a real-world population. For example, both the GO30140 and IMbrave150 excluded patients with untreated varices.11,12 Both IMbrave150 and Checkmate 459 limited enrollment only to patients with a Child-Pugh A score for liver disease; 36% of the KCVAMC patients had a Child-Pugh B score. Three patients (21.4%) were homeless, 6 patients (42.8%) had substance abuse history and 5 patients (35.7%) had mental illness. Several psychosocial factors present in our patients, such as substance abuse, mental illness, and homelessness, would have excluded them from clinical trials. Our small cohort of patients, thus, represents a frail real-world population due to multiple medical and psychosocial comorbidities. Real-world experience with immunotherapy as second-line therapy after treatment with sorafenib has been reported, but this is the first reported real-world experience of immunotherapy in the front-line setting for HCC.13,14
Large differences in sociodemographic status and health status exist between the veteran population and typical clinical trial populations. Veterans are predominantly male and older than a clinical trial population. Veterans are more likely to belong to a minority group, more likely to have lower level education and more likely to be poor than a clinical trial population. They are more likely to have poorer health status with higher number of medical conditions and psychosocial conditions.15
Limitations
We acknowledge several limitations to our study, such as the small number of patients and the retrospective single center nature of this study. Patients were older men with multiple psychosocial comorbitities like mental illness, substance abuse, and homelessness. This cohort may not represent the non-VA population, but is an excellent representation of a frail, real-world veteran population.
Conclusions
Despite clinical trials showing the promise of immunotherapy as an attractive front-line systemic treatment option for HCC, our results show poor outcomes in a frail real-world population. In a cohort of patients who received immunotherapy as a front-line systemic treatment for HCC, results were poor with a response rate of 14.3%, a median PFS of 4 months, and a median OS of 8 months. We noted a significantly higher number of adverse effects, including 21% incidence of grade 5 hepatotoxicity. There remains an urgent need to develop more effective and safer therapies for this patient population as well as validation from larger real-world studies.
Hepatocellular carcinoma (HCC) has a poor prognosis and remains an important cause of cancer-related morbidity and mortality.1,2 Potentially curative interventions include surgical resection, radiofrequency ablation, and liver transplantation. However, the majority of patients are not eligible for these procedures because they are diagnosed at an advanced stage, when locoregional therapies are much more limited.3,4 Although the kinase inhibitors sorafenib and lenvatinib are approved as first-line systemic treatment, at the US Department of Veterans Affairs (VA) Kansas City VA Medical Center (KCVAMC) in Missouri, nivolumab was used instead because of concerns for the tolerability of the kinase inhibitors. Locoregional therapies, resection, and transplantation options were either not appropriate or had been exhausted for these patients. The objective of this retrospective study was to determine the outcomes of those veteran patients in a small cohort.
Methods
The KCVAMC Institutional Review Board approved this retrospective chart review. Patients were selected from pharmacy records at KCVAMC. We identified all patients with a diagnosis of HCC who received nivolumab from January 2016 to December 2019. We then included only the patients that had nivolumab in the front-line setting for our final analysis. At the time of initiation of treatment, all patients were informed that immunotherapy was not approved for front-line treatment, but available evidence suggested that it would be easier to tolerate than sorafenib or lenvatinib. These patients were determined to be either ineligible for sorafenib or lenvatinib therapy or expected to tolerate it poorly, and hence they consented to the use of nivolumab. Tumor response and progression were assessed by the investigator according to iRECIST (Immune Response Evaluation Criteria in Solid Tumors) criteria.5 Data were obtained from retrospective health record review.
Results
Fourteen men received nivolumab in the front-line systemic therapy setting from January 2016 to December 2019 at KCVAMC. The median age was 63.5 years (range, 58-72 years), and the median Eastern Cooperative Oncology Group score was 1. The Table highlights patient characteristics.
Of the 14 patients included in the review, 2 patients had a response to nivolumab (14.3%) and 1 patient had a complete response (7.1%). The median duration of immunotherapy was 4.5 months. Immunotherapy was discontinued due to disease progression in 10 patients and toxicity in 3 patients.
The median progression-free survival (PFS) from initiation of immunotherapy was 4 months; median overall survival (OS) was 8 months. The median time from diagnosis to survival was 41 months. Only 1 patient received a second-line treatment.
Incidence of grade 3 or higher toxicity was 35%. Three deaths resulted from auto-immune hepatitis (grade 5 toxicity), as well as 1 grade 3 skin toxicity, and 1 grade 4 liver toxicity.
Discussion
Immunotherapy has shown promise in patients with HCC based on the results of the KEYNOTE-224 and Checkmate-040 studies,6,7 which led to an accelerated US Food and Drug Administration approval of nivolumab and pembrolizumab for HCC following failure of first-line sorafenib.8,9
Several clinical trials are evaluating front-line immunotherapy for HCC. The Checkmate 459 study demonstrated the median OS to be 16.4 months for nivolumab vs 14.7 months for sorafenib, a difference that was not statistically significant. However, tolerability of nivolumab was better than it was for sorafenib, thus positioning it as a potentially attractive first-line option.10 The GO30140 study evaluated
The results from our study differed from the previous studies and raise concern for the applicability of these trials to a real-world population. For example, both the GO30140 and IMbrave150 excluded patients with untreated varices.11,12 Both IMbrave150 and Checkmate 459 limited enrollment only to patients with a Child-Pugh A score for liver disease; 36% of the KCVAMC patients had a Child-Pugh B score. Three patients (21.4%) were homeless, 6 patients (42.8%) had substance abuse history and 5 patients (35.7%) had mental illness. Several psychosocial factors present in our patients, such as substance abuse, mental illness, and homelessness, would have excluded them from clinical trials. Our small cohort of patients, thus, represents a frail real-world population due to multiple medical and psychosocial comorbidities. Real-world experience with immunotherapy as second-line therapy after treatment with sorafenib has been reported, but this is the first reported real-world experience of immunotherapy in the front-line setting for HCC.13,14
Large differences in sociodemographic status and health status exist between the veteran population and typical clinical trial populations. Veterans are predominantly male and older than a clinical trial population. Veterans are more likely to belong to a minority group, more likely to have lower level education and more likely to be poor than a clinical trial population. They are more likely to have poorer health status with higher number of medical conditions and psychosocial conditions.15
Limitations
We acknowledge several limitations to our study, such as the small number of patients and the retrospective single center nature of this study. Patients were older men with multiple psychosocial comorbitities like mental illness, substance abuse, and homelessness. This cohort may not represent the non-VA population, but is an excellent representation of a frail, real-world veteran population.
Conclusions
Despite clinical trials showing the promise of immunotherapy as an attractive front-line systemic treatment option for HCC, our results show poor outcomes in a frail real-world population. In a cohort of patients who received immunotherapy as a front-line systemic treatment for HCC, results were poor with a response rate of 14.3%, a median PFS of 4 months, and a median OS of 8 months. We noted a significantly higher number of adverse effects, including 21% incidence of grade 5 hepatotoxicity. There remains an urgent need to develop more effective and safer therapies for this patient population as well as validation from larger real-world studies.
1. El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118-1127. doi:10.1056/NEJMra1001683
2. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359-E386. doi:10.1002/ijc.29210
3. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362(9399):1907-1917. doi:10.1016/S0140-6736(03)14964-1
4. Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47 Suppl(0):S2-S6. doi:10.1097/MCG.0b013e3182872f29
5. Seymour L, Bogaerts J, Perrone A, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics [published correction appears in Lancet Oncol. 2019 May;20(5):e242]. Lancet Oncol. 2017;18(3):e143-e152. doi:10.1016/S1470-2045(17)30074-8
6. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492-2502.doi:10.1016/S0140-6736(17)31046-2
7. Zhu AX, Finn RS, Edeline J, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial [published correction appears in Lancet Oncol. 2018 Sep;19(9):e440]. Lancet Oncol. 2018;19(7):940-952. doi:10.1016/S1470-2045(18)30351-6
8. US Food and Drug Administration. FDA grants accelerated approval to nivolumab for HCC previously treated with sorafenib. Updated September 25, 2017. Accessed October 7, 2020. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-nivolumab-hcc-previously-treated-sorafenib.
9. US Food and Drug Administration. FDA grants accelerated approval to pembrolizumab for hepatocellular carcinoma. Updated December 14, 2018. Accessed October 7, 2020. https://www.fda.gov/drugs/fda-grants-accelerated-approval-pembrolizumab-hepatocellular-carcinoma.
10. Yau T, Park JW, Finn RS, et al. CheckMate 459: A randomized, multi-center phase 3 study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Presented at: ESMO 2019 Congress. Barcelona, Spain: September 27, 2019. Ann Onc. 2019;30(suppl_5):v851-v934. doi:10.1093/annonc/mdz394
11. Lee M, Ryoo BY, Hsu CH, et al. Randomised efficacy and safety results for atezolizumab (atezo) + bevacizumab (bev) in patients (pts) with previously untreated, unresectable hepatocellular carcinoma (HCC). Presented at: ESMO 2019 Congress. Barcelona, Spain: September 27, 2019.
12. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894-1905.doi:10.1056/NEJMoa1915745
13. Scheiner B, Kirstein MM, Hucke F, et al. Programmed cell death protein-1 (PD-1)-targeted immunotherapy in advanced hepatocellular carcinoma: efficacy and safety data from an international multicentre real-world cohort. Aliment Pharmacol Ther. 2019;49(10):1323-1333. doi:10.1111/apt.15245
14. Yoon SE, Hur JY, Lee KK, et al. Real-world data on nivolumab treatment in Asian patients with advanced hepatocellular carcinoma. Presented at: ESMO 2018 Congress. Munich, Germany: October 21, 2018. Ann Onc. 2018;29(suppl_8):viii205-viii270. doi:10.1093/annonc/mdy282
15. Agha Z, Lofgren RP, VanRuiswyk JV, Layde PM. Are patients at Veterans Affairs medical centers sicker? A comparative analysis of health status and medical resource use. Arch Intern Med. 2000;160(21):3252-3257. doi:10.1001/archinte.160.21.3252
1. El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118-1127. doi:10.1056/NEJMra1001683
2. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359-E386. doi:10.1002/ijc.29210
3. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362(9399):1907-1917. doi:10.1016/S0140-6736(03)14964-1
4. Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47 Suppl(0):S2-S6. doi:10.1097/MCG.0b013e3182872f29
5. Seymour L, Bogaerts J, Perrone A, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics [published correction appears in Lancet Oncol. 2019 May;20(5):e242]. Lancet Oncol. 2017;18(3):e143-e152. doi:10.1016/S1470-2045(17)30074-8
6. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492-2502.doi:10.1016/S0140-6736(17)31046-2
7. Zhu AX, Finn RS, Edeline J, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial [published correction appears in Lancet Oncol. 2018 Sep;19(9):e440]. Lancet Oncol. 2018;19(7):940-952. doi:10.1016/S1470-2045(18)30351-6
8. US Food and Drug Administration. FDA grants accelerated approval to nivolumab for HCC previously treated with sorafenib. Updated September 25, 2017. Accessed October 7, 2020. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-nivolumab-hcc-previously-treated-sorafenib.
9. US Food and Drug Administration. FDA grants accelerated approval to pembrolizumab for hepatocellular carcinoma. Updated December 14, 2018. Accessed October 7, 2020. https://www.fda.gov/drugs/fda-grants-accelerated-approval-pembrolizumab-hepatocellular-carcinoma.
10. Yau T, Park JW, Finn RS, et al. CheckMate 459: A randomized, multi-center phase 3 study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Presented at: ESMO 2019 Congress. Barcelona, Spain: September 27, 2019. Ann Onc. 2019;30(suppl_5):v851-v934. doi:10.1093/annonc/mdz394
11. Lee M, Ryoo BY, Hsu CH, et al. Randomised efficacy and safety results for atezolizumab (atezo) + bevacizumab (bev) in patients (pts) with previously untreated, unresectable hepatocellular carcinoma (HCC). Presented at: ESMO 2019 Congress. Barcelona, Spain: September 27, 2019.
12. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894-1905.doi:10.1056/NEJMoa1915745
13. Scheiner B, Kirstein MM, Hucke F, et al. Programmed cell death protein-1 (PD-1)-targeted immunotherapy in advanced hepatocellular carcinoma: efficacy and safety data from an international multicentre real-world cohort. Aliment Pharmacol Ther. 2019;49(10):1323-1333. doi:10.1111/apt.15245
14. Yoon SE, Hur JY, Lee KK, et al. Real-world data on nivolumab treatment in Asian patients with advanced hepatocellular carcinoma. Presented at: ESMO 2018 Congress. Munich, Germany: October 21, 2018. Ann Onc. 2018;29(suppl_8):viii205-viii270. doi:10.1093/annonc/mdy282
15. Agha Z, Lofgren RP, VanRuiswyk JV, Layde PM. Are patients at Veterans Affairs medical centers sicker? A comparative analysis of health status and medical resource use. Arch Intern Med. 2000;160(21):3252-3257. doi:10.1001/archinte.160.21.3252
COVID-19: U.S. sets new weekly high in children
the American Academy of Pediatrics announced Nov. 2.
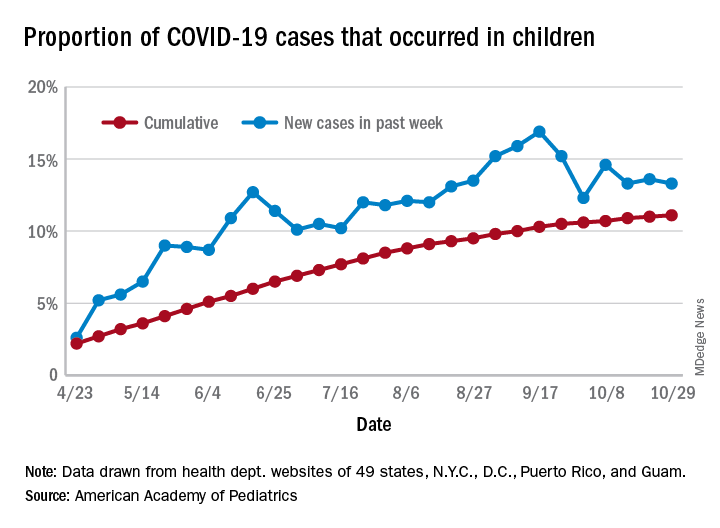
For the week, over 61,000 cases were reported in children, bringing the number of COVID-19 cases for the month of October to nearly 200,000 and the total since the start of the pandemic to over 853,000, the AAP and the Children’s Hospital Association said in their weekly report.
“These numbers reflect a disturbing increase in cases throughout most of the United States in all populations, especially among young adults,” Yvonne Maldonado, MD, chair of the AAP Committee on Infectious Diseases, said in a separate statement. “We are entering a heightened wave of infections around the country. We would encourage family holiday gatherings to be avoided if possible, especially if there are high-risk individuals in the household.”
For the week ending Oct. 29, children represented 13.3% of all cases, possibly constituting a minitrend of stability over the past 3 weeks. For the full length of the pandemic, 11.1% of all COVID-19 cases have occurred in children, although severe illness is much less common: 1.7% of all hospitalizations (data from 24 states and New York City) and 0.06% of all deaths (data from 42 states and New York City), the AAP and CHA report said.
Other data show that 1,134 per 100,000 children in the United States have been infected by the coronavirus, up from 1,053 the previous week, with state rates ranging from 221 per 100,000 in Vermont to 3,321 in North Dakota. In Wyoming, 25.5% of all COVID-19 cases have occurred in children, the highest of any state, while New Jersey has the lowest rate at 4.9%, the AAP/CHA report showed.
In the 10 states making testing data available, children represent the lowest percentage of tests in Iowa (5.0%) and the highest in Indiana (16.9%). Iowa, however, has the highest positivity rate for children at 14.6%, along with Nevada, while West Virginia has the lowest at 3.6%, the AAP and CHA said in the report.
These numbers, however, may not be telling the whole story. “The number of reported COVID-19 cases in children is likely an undercount because children’s symptoms are often mild and they may not be tested for every illness,” the AAP said in its statement.
“We urge policy makers to listen to doctors and public health experts rather than level baseless accusations against them. Physicians, nurses and other health care professionals have put their lives on the line to protect our communities. We can all do our part to protect them, and our communities, by wearing masks, practicing physical distancing, and getting our flu immunizations,” AAP President Sally Goza, MD, said in the AAP statement.
the American Academy of Pediatrics announced Nov. 2.
For the week, over 61,000 cases were reported in children, bringing the number of COVID-19 cases for the month of October to nearly 200,000 and the total since the start of the pandemic to over 853,000, the AAP and the Children’s Hospital Association said in their weekly report.
“These numbers reflect a disturbing increase in cases throughout most of the United States in all populations, especially among young adults,” Yvonne Maldonado, MD, chair of the AAP Committee on Infectious Diseases, said in a separate statement. “We are entering a heightened wave of infections around the country. We would encourage family holiday gatherings to be avoided if possible, especially if there are high-risk individuals in the household.”
For the week ending Oct. 29, children represented 13.3% of all cases, possibly constituting a minitrend of stability over the past 3 weeks. For the full length of the pandemic, 11.1% of all COVID-19 cases have occurred in children, although severe illness is much less common: 1.7% of all hospitalizations (data from 24 states and New York City) and 0.06% of all deaths (data from 42 states and New York City), the AAP and CHA report said.
Other data show that 1,134 per 100,000 children in the United States have been infected by the coronavirus, up from 1,053 the previous week, with state rates ranging from 221 per 100,000 in Vermont to 3,321 in North Dakota. In Wyoming, 25.5% of all COVID-19 cases have occurred in children, the highest of any state, while New Jersey has the lowest rate at 4.9%, the AAP/CHA report showed.
In the 10 states making testing data available, children represent the lowest percentage of tests in Iowa (5.0%) and the highest in Indiana (16.9%). Iowa, however, has the highest positivity rate for children at 14.6%, along with Nevada, while West Virginia has the lowest at 3.6%, the AAP and CHA said in the report.
These numbers, however, may not be telling the whole story. “The number of reported COVID-19 cases in children is likely an undercount because children’s symptoms are often mild and they may not be tested for every illness,” the AAP said in its statement.
“We urge policy makers to listen to doctors and public health experts rather than level baseless accusations against them. Physicians, nurses and other health care professionals have put their lives on the line to protect our communities. We can all do our part to protect them, and our communities, by wearing masks, practicing physical distancing, and getting our flu immunizations,” AAP President Sally Goza, MD, said in the AAP statement.
the American Academy of Pediatrics announced Nov. 2.
For the week, over 61,000 cases were reported in children, bringing the number of COVID-19 cases for the month of October to nearly 200,000 and the total since the start of the pandemic to over 853,000, the AAP and the Children’s Hospital Association said in their weekly report.
“These numbers reflect a disturbing increase in cases throughout most of the United States in all populations, especially among young adults,” Yvonne Maldonado, MD, chair of the AAP Committee on Infectious Diseases, said in a separate statement. “We are entering a heightened wave of infections around the country. We would encourage family holiday gatherings to be avoided if possible, especially if there are high-risk individuals in the household.”
For the week ending Oct. 29, children represented 13.3% of all cases, possibly constituting a minitrend of stability over the past 3 weeks. For the full length of the pandemic, 11.1% of all COVID-19 cases have occurred in children, although severe illness is much less common: 1.7% of all hospitalizations (data from 24 states and New York City) and 0.06% of all deaths (data from 42 states and New York City), the AAP and CHA report said.
Other data show that 1,134 per 100,000 children in the United States have been infected by the coronavirus, up from 1,053 the previous week, with state rates ranging from 221 per 100,000 in Vermont to 3,321 in North Dakota. In Wyoming, 25.5% of all COVID-19 cases have occurred in children, the highest of any state, while New Jersey has the lowest rate at 4.9%, the AAP/CHA report showed.
In the 10 states making testing data available, children represent the lowest percentage of tests in Iowa (5.0%) and the highest in Indiana (16.9%). Iowa, however, has the highest positivity rate for children at 14.6%, along with Nevada, while West Virginia has the lowest at 3.6%, the AAP and CHA said in the report.
These numbers, however, may not be telling the whole story. “The number of reported COVID-19 cases in children is likely an undercount because children’s symptoms are often mild and they may not be tested for every illness,” the AAP said in its statement.
“We urge policy makers to listen to doctors and public health experts rather than level baseless accusations against them. Physicians, nurses and other health care professionals have put their lives on the line to protect our communities. We can all do our part to protect them, and our communities, by wearing masks, practicing physical distancing, and getting our flu immunizations,” AAP President Sally Goza, MD, said in the AAP statement.
IMRT new standard of care for high-risk cervical cancer
“IG-IMRT should represent the new standard of care for postoperative pelvic radiation therapy in women with gynecological cancers,” said study lead author Supriya Chopra, MD, of the Tata Memorial Center in Mumbai, India.
She noted that the study, known as PARCER, is the first in gynecologic cancer to show the impact of advanced technology in reducing long-term morbidity and thus improving the experience of survivors.
At 4 years, rates of late GI toxicity of grade 2 or higher in the IG-IMRT and 3D-CRT arms were 19.2% and 36.2%, respectively (P = .005). Rates of toxicity of grade 3 or higher were 2.0% and 8.7%, respectively (P < .01).
Chopra presented the results at the American Society for Radiation Oncology (ASTRO) 2020 Annual Meeting, which was held online.
Postoperative radiotherapy is indicated for women with cervical and endometrial cancers who have high-risk features, but long-term follow-up has shown an increase in GI symptom burden and toxicity in long-term survivors after adjuvant radiotherapy.
“The uptake of IMRT has been relatively slow in gynecological cancers,” said Chopra. She explained that previous data suggested a benefit with the use of IMRT, but long-term postoperative effects were unclear.
The new data amount to a “practice-change use” of IMRT for this indication, said Sue Yom, MD, PhD, of the University of California, San Francisco, who was not involved with the study. “I see this as having potentially important future impacts on clinical practice.”
Yom explained that, although there have been studies in the United States on the use of postoperative IMRT for pelvic cancer, “this is the first phase 3 study that has shown definite long-term advantages with the use of IMRT, and I would consider it confirmatory.”
In 2015, the preliminary results of PARCER were presented at the plenary session at ASTRO. The results showed that patients treated with IG-IMRT had fewer late GI toxicities at a median follow-up of 20 months. However, the difference between groups was not statistically significant in this earlier analysis.
Now at 49 months’ follow-up
The study was conducted in three clinical sites of Tata Memorial Center and included a total of 300 patients with cervical cancer. The patients had undergone type III hysterectomy and had intermediate- or high-risk features, or they had undergone type I/II hysterectomy necessitating adjuvant chemoradiotherapy. They were randomly assigned to IG-IMRT (n = 151) or 3D-CRT (n = 149). Most patients (117 in the IG-IMRT arm and 114 in the 3D-CRT arm) received concurrent chemotherapy.
The primary endpoint was late GI toxicity of grade 2 or higher. Follow-up included clinical and quality-of-life evaluations, which were conducted every 3 months for 2 years and then every 6 months for years 2 to 5.
Chopra and colleagues evaluated 11 different GI-related side effects. Differences emerged over time between the two groups. Among the group that received IG-IMRT, significantly fewer patients reported moderate to severe acute diarrhea (17% in the IG-IMRT arm vs 27% in the 3D-CRT arm), late abdominal bloating (14% vs 28%), bowel obstruction (1% vs 7%), and anorexia/appetite loss (1% vs 7%).
Overall, for patients treated with IG-IMRT, grade 2 toxicity–free survival rates were significantly higher (78% with IG-IMRT vs 57% with 3D-CRT; P = .0009), as were grade 3 toxicity–free survival rates (97.6% vs 81.6%; P = .001).
As noted above, rates of disease-free survival were similar for both groups (73% with image-guided IMRT vs 68% with 3D-CRT; P = .30).
Funding for the study was provided by the Department of Science and Technology and the Department of Atomic Energy, Clinical Trials Center, in India, and by Varian International and the Terry Fox Foundation. Chopra and Yom have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
“IG-IMRT should represent the new standard of care for postoperative pelvic radiation therapy in women with gynecological cancers,” said study lead author Supriya Chopra, MD, of the Tata Memorial Center in Mumbai, India.
She noted that the study, known as PARCER, is the first in gynecologic cancer to show the impact of advanced technology in reducing long-term morbidity and thus improving the experience of survivors.
At 4 years, rates of late GI toxicity of grade 2 or higher in the IG-IMRT and 3D-CRT arms were 19.2% and 36.2%, respectively (P = .005). Rates of toxicity of grade 3 or higher were 2.0% and 8.7%, respectively (P < .01).
Chopra presented the results at the American Society for Radiation Oncology (ASTRO) 2020 Annual Meeting, which was held online.
Postoperative radiotherapy is indicated for women with cervical and endometrial cancers who have high-risk features, but long-term follow-up has shown an increase in GI symptom burden and toxicity in long-term survivors after adjuvant radiotherapy.
“The uptake of IMRT has been relatively slow in gynecological cancers,” said Chopra. She explained that previous data suggested a benefit with the use of IMRT, but long-term postoperative effects were unclear.
The new data amount to a “practice-change use” of IMRT for this indication, said Sue Yom, MD, PhD, of the University of California, San Francisco, who was not involved with the study. “I see this as having potentially important future impacts on clinical practice.”
Yom explained that, although there have been studies in the United States on the use of postoperative IMRT for pelvic cancer, “this is the first phase 3 study that has shown definite long-term advantages with the use of IMRT, and I would consider it confirmatory.”
In 2015, the preliminary results of PARCER were presented at the plenary session at ASTRO. The results showed that patients treated with IG-IMRT had fewer late GI toxicities at a median follow-up of 20 months. However, the difference between groups was not statistically significant in this earlier analysis.
Now at 49 months’ follow-up
The study was conducted in three clinical sites of Tata Memorial Center and included a total of 300 patients with cervical cancer. The patients had undergone type III hysterectomy and had intermediate- or high-risk features, or they had undergone type I/II hysterectomy necessitating adjuvant chemoradiotherapy. They were randomly assigned to IG-IMRT (n = 151) or 3D-CRT (n = 149). Most patients (117 in the IG-IMRT arm and 114 in the 3D-CRT arm) received concurrent chemotherapy.
The primary endpoint was late GI toxicity of grade 2 or higher. Follow-up included clinical and quality-of-life evaluations, which were conducted every 3 months for 2 years and then every 6 months for years 2 to 5.
Chopra and colleagues evaluated 11 different GI-related side effects. Differences emerged over time between the two groups. Among the group that received IG-IMRT, significantly fewer patients reported moderate to severe acute diarrhea (17% in the IG-IMRT arm vs 27% in the 3D-CRT arm), late abdominal bloating (14% vs 28%), bowel obstruction (1% vs 7%), and anorexia/appetite loss (1% vs 7%).
Overall, for patients treated with IG-IMRT, grade 2 toxicity–free survival rates were significantly higher (78% with IG-IMRT vs 57% with 3D-CRT; P = .0009), as were grade 3 toxicity–free survival rates (97.6% vs 81.6%; P = .001).
As noted above, rates of disease-free survival were similar for both groups (73% with image-guided IMRT vs 68% with 3D-CRT; P = .30).
Funding for the study was provided by the Department of Science and Technology and the Department of Atomic Energy, Clinical Trials Center, in India, and by Varian International and the Terry Fox Foundation. Chopra and Yom have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
“IG-IMRT should represent the new standard of care for postoperative pelvic radiation therapy in women with gynecological cancers,” said study lead author Supriya Chopra, MD, of the Tata Memorial Center in Mumbai, India.
She noted that the study, known as PARCER, is the first in gynecologic cancer to show the impact of advanced technology in reducing long-term morbidity and thus improving the experience of survivors.
At 4 years, rates of late GI toxicity of grade 2 or higher in the IG-IMRT and 3D-CRT arms were 19.2% and 36.2%, respectively (P = .005). Rates of toxicity of grade 3 or higher were 2.0% and 8.7%, respectively (P < .01).
Chopra presented the results at the American Society for Radiation Oncology (ASTRO) 2020 Annual Meeting, which was held online.
Postoperative radiotherapy is indicated for women with cervical and endometrial cancers who have high-risk features, but long-term follow-up has shown an increase in GI symptom burden and toxicity in long-term survivors after adjuvant radiotherapy.
“The uptake of IMRT has been relatively slow in gynecological cancers,” said Chopra. She explained that previous data suggested a benefit with the use of IMRT, but long-term postoperative effects were unclear.
The new data amount to a “practice-change use” of IMRT for this indication, said Sue Yom, MD, PhD, of the University of California, San Francisco, who was not involved with the study. “I see this as having potentially important future impacts on clinical practice.”
Yom explained that, although there have been studies in the United States on the use of postoperative IMRT for pelvic cancer, “this is the first phase 3 study that has shown definite long-term advantages with the use of IMRT, and I would consider it confirmatory.”
In 2015, the preliminary results of PARCER were presented at the plenary session at ASTRO. The results showed that patients treated with IG-IMRT had fewer late GI toxicities at a median follow-up of 20 months. However, the difference between groups was not statistically significant in this earlier analysis.
Now at 49 months’ follow-up
The study was conducted in three clinical sites of Tata Memorial Center and included a total of 300 patients with cervical cancer. The patients had undergone type III hysterectomy and had intermediate- or high-risk features, or they had undergone type I/II hysterectomy necessitating adjuvant chemoradiotherapy. They were randomly assigned to IG-IMRT (n = 151) or 3D-CRT (n = 149). Most patients (117 in the IG-IMRT arm and 114 in the 3D-CRT arm) received concurrent chemotherapy.
The primary endpoint was late GI toxicity of grade 2 or higher. Follow-up included clinical and quality-of-life evaluations, which were conducted every 3 months for 2 years and then every 6 months for years 2 to 5.
Chopra and colleagues evaluated 11 different GI-related side effects. Differences emerged over time between the two groups. Among the group that received IG-IMRT, significantly fewer patients reported moderate to severe acute diarrhea (17% in the IG-IMRT arm vs 27% in the 3D-CRT arm), late abdominal bloating (14% vs 28%), bowel obstruction (1% vs 7%), and anorexia/appetite loss (1% vs 7%).
Overall, for patients treated with IG-IMRT, grade 2 toxicity–free survival rates were significantly higher (78% with IG-IMRT vs 57% with 3D-CRT; P = .0009), as were grade 3 toxicity–free survival rates (97.6% vs 81.6%; P = .001).
As noted above, rates of disease-free survival were similar for both groups (73% with image-guided IMRT vs 68% with 3D-CRT; P = .30).
Funding for the study was provided by the Department of Science and Technology and the Department of Atomic Energy, Clinical Trials Center, in India, and by Varian International and the Terry Fox Foundation. Chopra and Yom have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Birch bark derivative gel found effective for EB, in phase 3 study
A . The results come from the largest double-blind, randomized trial performed in this patient population.

More than 41% of EB target wounds that were treated with Oleogel-S10 healed within 45 days, compared with about 29% of target wounds treated with placebo, in the EASE phase 3 trial, conducted at 58 sites in 28 countries.
A group of rare genetic disorders, EB “is described as the worst disease you’ve never heard of,” explained lead investigator Dedee Murrell, MD, director of dermatology, St. George Hospital at the University of New South Wales, Sydney. “It starts in children and is like having burns that heal with scars, and no treatment has been approved for it” by the Food and Drug Administration.
“This is the first large clinical trial with placebo of a topical treatment that’s worked for this terrible disease,” Dr. Murrell said in an interview. She noted that standard EB treatment currently consists of applying nonstick dressings to wounds to protect skin from trauma and infection.
Dr. Murrell, who has focused her work on EB patients since 1990, presented the findings at the virtual annual Congress of the European Academy of Dermatology and Venereology.
The trial enrolled 223 patients (average age, 12 years, but ages ranged to 81 years) with three types of EB, including dystrophic and junctional EB and Kindler syndrome. For each participant, a target wound was selected for use as the primary efficacy endpoint. Those wounds had a partial thickness of between 10 cm2 and 50 cm2 and lasted between 21 days and 9 months. Patients were stratified into groups depending on type of EB and size of target wound.
Participants were randomly assigned to receive either Oleogel-S10 (n = 109) or placebo (n = 114). All applied the blinded-study gel to all their wounds at least every 4 days at the time dressings were changed.
The primary endpoint was the percentage of patients whose target wounds completely closed within 45 days. Key secondary endpoints included time to wound healing and percentage of target wounds that healed within 90 days of treatment; incidence and severity of target wound infection; change in total body wound burden, as measured by the Epidermolysis Bullosa Disease Activity and Scarring Index skin activity subscore; change in itching, as measured by the Itch Man Scale and the Leuven Itch Scale; and adverse events.
Nearly 92% of patients who were treated with Oleogel-S10 completed the double-blind phase of the trial, compared with nearly 87% who received placebo. As noted, the primary endpoint was met, with 41.3% of Oleogel-S10 patients achieving target wound closure within 45 days, compared with 28.9% of the patients who received placebo (P = .013).
But the difference in time to wound healing by day 90 between the two patient groups was not statistically significant (P = .302), with 50.5% of Oleogel-S10 patients achieving wound closure vs. 43.9% of control patients.
Target-wound infection occurred in eight participants, including three who used Oleogel-S10 and five who received placebo; all moderate or severe infections occurred in patients who received placebo. Total wound burden was reduced to a greater extent among Oleogel-S10 patients by day 60, but there was no apparent difference at day 90.
Both treatment groups reported qualitative improvements in itch, with no significant differences between groups. The prevalence of adverse events was also similar between groups (Oleogel-S10, 81.7%; placebo, 80.7%). The most frequently reported adverse events among Oleogel-S10 patients, compared with patients who received placebo, were wound complications, pyrexia, wound infection, pruritus, and anemia; only 4.5% of adverse events were deemed severe.
Dr. Murrell said that, on the basis of the trial results, she expects the FDA to fast-track approval of Oleogel-S10, which contains triterpene extract and sunflower oil.
The gel is “a treatment patients will be able to put under their dressings, added to normal treatment, which will accelerate their wound healing, with no significant increase in any side effects,” she added.
Jemima Mellerio, MD, of St. Thomas’ Hospital in London who sees about 400 EB patients each year, agreed with Dr. Murrell that the results are “very exciting.” Dr. Mellerio was not involved in the study.
“Practicing dermatologists seeing people with EB will have something to offer that appears to speed up wound healing in chronic wounds,” Dr. Mellerio said in an interview. “It’s a positive option rather than just supportive treatment, something that makes a difference to the natural history of wounds.”
She said the trial’s biggest strength was including “such a large cohort of patients.
“It’s extremely difficult to do that kind of study, especially with a placebo-controlled arm and especially in a rare disease,” Dr. Mellerio said. “If you think about the product itself, it’s easy to apply, so it’s not particularly onerous for people to add to their daily regimen of dressings.”
The study was funded by Amryt Pharma. Dr. Murrell is an advisory board member for Amryt Pharma. Dr. Mellerio is a consultant for Amryt Pharma.
A version of this article originally appeared on Medscape.com.
A . The results come from the largest double-blind, randomized trial performed in this patient population.
More than 41% of EB target wounds that were treated with Oleogel-S10 healed within 45 days, compared with about 29% of target wounds treated with placebo, in the EASE phase 3 trial, conducted at 58 sites in 28 countries.
A group of rare genetic disorders, EB “is described as the worst disease you’ve never heard of,” explained lead investigator Dedee Murrell, MD, director of dermatology, St. George Hospital at the University of New South Wales, Sydney. “It starts in children and is like having burns that heal with scars, and no treatment has been approved for it” by the Food and Drug Administration.
“This is the first large clinical trial with placebo of a topical treatment that’s worked for this terrible disease,” Dr. Murrell said in an interview. She noted that standard EB treatment currently consists of applying nonstick dressings to wounds to protect skin from trauma and infection.
Dr. Murrell, who has focused her work on EB patients since 1990, presented the findings at the virtual annual Congress of the European Academy of Dermatology and Venereology.
The trial enrolled 223 patients (average age, 12 years, but ages ranged to 81 years) with three types of EB, including dystrophic and junctional EB and Kindler syndrome. For each participant, a target wound was selected for use as the primary efficacy endpoint. Those wounds had a partial thickness of between 10 cm2 and 50 cm2 and lasted between 21 days and 9 months. Patients were stratified into groups depending on type of EB and size of target wound.
Participants were randomly assigned to receive either Oleogel-S10 (n = 109) or placebo (n = 114). All applied the blinded-study gel to all their wounds at least every 4 days at the time dressings were changed.
The primary endpoint was the percentage of patients whose target wounds completely closed within 45 days. Key secondary endpoints included time to wound healing and percentage of target wounds that healed within 90 days of treatment; incidence and severity of target wound infection; change in total body wound burden, as measured by the Epidermolysis Bullosa Disease Activity and Scarring Index skin activity subscore; change in itching, as measured by the Itch Man Scale and the Leuven Itch Scale; and adverse events.
Nearly 92% of patients who were treated with Oleogel-S10 completed the double-blind phase of the trial, compared with nearly 87% who received placebo. As noted, the primary endpoint was met, with 41.3% of Oleogel-S10 patients achieving target wound closure within 45 days, compared with 28.9% of the patients who received placebo (P = .013).
But the difference in time to wound healing by day 90 between the two patient groups was not statistically significant (P = .302), with 50.5% of Oleogel-S10 patients achieving wound closure vs. 43.9% of control patients.
Target-wound infection occurred in eight participants, including three who used Oleogel-S10 and five who received placebo; all moderate or severe infections occurred in patients who received placebo. Total wound burden was reduced to a greater extent among Oleogel-S10 patients by day 60, but there was no apparent difference at day 90.
Both treatment groups reported qualitative improvements in itch, with no significant differences between groups. The prevalence of adverse events was also similar between groups (Oleogel-S10, 81.7%; placebo, 80.7%). The most frequently reported adverse events among Oleogel-S10 patients, compared with patients who received placebo, were wound complications, pyrexia, wound infection, pruritus, and anemia; only 4.5% of adverse events were deemed severe.
Dr. Murrell said that, on the basis of the trial results, she expects the FDA to fast-track approval of Oleogel-S10, which contains triterpene extract and sunflower oil.
The gel is “a treatment patients will be able to put under their dressings, added to normal treatment, which will accelerate their wound healing, with no significant increase in any side effects,” she added.
Jemima Mellerio, MD, of St. Thomas’ Hospital in London who sees about 400 EB patients each year, agreed with Dr. Murrell that the results are “very exciting.” Dr. Mellerio was not involved in the study.
“Practicing dermatologists seeing people with EB will have something to offer that appears to speed up wound healing in chronic wounds,” Dr. Mellerio said in an interview. “It’s a positive option rather than just supportive treatment, something that makes a difference to the natural history of wounds.”
She said the trial’s biggest strength was including “such a large cohort of patients.
“It’s extremely difficult to do that kind of study, especially with a placebo-controlled arm and especially in a rare disease,” Dr. Mellerio said. “If you think about the product itself, it’s easy to apply, so it’s not particularly onerous for people to add to their daily regimen of dressings.”
The study was funded by Amryt Pharma. Dr. Murrell is an advisory board member for Amryt Pharma. Dr. Mellerio is a consultant for Amryt Pharma.
A version of this article originally appeared on Medscape.com.
A . The results come from the largest double-blind, randomized trial performed in this patient population.
More than 41% of EB target wounds that were treated with Oleogel-S10 healed within 45 days, compared with about 29% of target wounds treated with placebo, in the EASE phase 3 trial, conducted at 58 sites in 28 countries.
A group of rare genetic disorders, EB “is described as the worst disease you’ve never heard of,” explained lead investigator Dedee Murrell, MD, director of dermatology, St. George Hospital at the University of New South Wales, Sydney. “It starts in children and is like having burns that heal with scars, and no treatment has been approved for it” by the Food and Drug Administration.
“This is the first large clinical trial with placebo of a topical treatment that’s worked for this terrible disease,” Dr. Murrell said in an interview. She noted that standard EB treatment currently consists of applying nonstick dressings to wounds to protect skin from trauma and infection.
Dr. Murrell, who has focused her work on EB patients since 1990, presented the findings at the virtual annual Congress of the European Academy of Dermatology and Venereology.
The trial enrolled 223 patients (average age, 12 years, but ages ranged to 81 years) with three types of EB, including dystrophic and junctional EB and Kindler syndrome. For each participant, a target wound was selected for use as the primary efficacy endpoint. Those wounds had a partial thickness of between 10 cm2 and 50 cm2 and lasted between 21 days and 9 months. Patients were stratified into groups depending on type of EB and size of target wound.
Participants were randomly assigned to receive either Oleogel-S10 (n = 109) or placebo (n = 114). All applied the blinded-study gel to all their wounds at least every 4 days at the time dressings were changed.
The primary endpoint was the percentage of patients whose target wounds completely closed within 45 days. Key secondary endpoints included time to wound healing and percentage of target wounds that healed within 90 days of treatment; incidence and severity of target wound infection; change in total body wound burden, as measured by the Epidermolysis Bullosa Disease Activity and Scarring Index skin activity subscore; change in itching, as measured by the Itch Man Scale and the Leuven Itch Scale; and adverse events.
Nearly 92% of patients who were treated with Oleogel-S10 completed the double-blind phase of the trial, compared with nearly 87% who received placebo. As noted, the primary endpoint was met, with 41.3% of Oleogel-S10 patients achieving target wound closure within 45 days, compared with 28.9% of the patients who received placebo (P = .013).
But the difference in time to wound healing by day 90 between the two patient groups was not statistically significant (P = .302), with 50.5% of Oleogel-S10 patients achieving wound closure vs. 43.9% of control patients.
Target-wound infection occurred in eight participants, including three who used Oleogel-S10 and five who received placebo; all moderate or severe infections occurred in patients who received placebo. Total wound burden was reduced to a greater extent among Oleogel-S10 patients by day 60, but there was no apparent difference at day 90.
Both treatment groups reported qualitative improvements in itch, with no significant differences between groups. The prevalence of adverse events was also similar between groups (Oleogel-S10, 81.7%; placebo, 80.7%). The most frequently reported adverse events among Oleogel-S10 patients, compared with patients who received placebo, were wound complications, pyrexia, wound infection, pruritus, and anemia; only 4.5% of adverse events were deemed severe.
Dr. Murrell said that, on the basis of the trial results, she expects the FDA to fast-track approval of Oleogel-S10, which contains triterpene extract and sunflower oil.
The gel is “a treatment patients will be able to put under their dressings, added to normal treatment, which will accelerate their wound healing, with no significant increase in any side effects,” she added.
Jemima Mellerio, MD, of St. Thomas’ Hospital in London who sees about 400 EB patients each year, agreed with Dr. Murrell that the results are “very exciting.” Dr. Mellerio was not involved in the study.
“Practicing dermatologists seeing people with EB will have something to offer that appears to speed up wound healing in chronic wounds,” Dr. Mellerio said in an interview. “It’s a positive option rather than just supportive treatment, something that makes a difference to the natural history of wounds.”
She said the trial’s biggest strength was including “such a large cohort of patients.
“It’s extremely difficult to do that kind of study, especially with a placebo-controlled arm and especially in a rare disease,” Dr. Mellerio said. “If you think about the product itself, it’s easy to apply, so it’s not particularly onerous for people to add to their daily regimen of dressings.”
The study was funded by Amryt Pharma. Dr. Murrell is an advisory board member for Amryt Pharma. Dr. Mellerio is a consultant for Amryt Pharma.
A version of this article originally appeared on Medscape.com.
AMA discharge linked to increased readmissions, discontinuity of care
Background: AMA discharges are common (1%-2% of all U.S. discharges) and disproportionately affect vulnerable patient populations, specifically those of lower socioeconomic status and the uninsured. Previous studies have been insufficiently powered to assess the effects of AMA discharge on 30-day readmission rates at a national level.

Study design: Retrospective cohort.
Setting: Community and teaching hospitals in 22 states.
Synopsis: With use of the 2014 Nationwide Readmissions Database of 23,110,641 index hospitalizations of patients 18 years or older, this study found that AMA discharge occurred with 1.3% of admissions. AMA discharge was associated with greater than twice the odds of 30-day readmission, compared with routine discharge. Of patients discharged AMA, 20.2% had an unplanned readmission within 30 days, compared with 10.1% of patients discharged routinely (OR, 2.25; 95% CI, 2.20-2.30; P less than .001).
Patients who were discharged AMA had almost 20 times the odds of undergoing repeat AMA discharge at readmission (OR, 18.41; 95% CI, 17.46-19.41; P less than .001) and twice the odds of presenting to a different hospital (OR, 2.35; 95% CI, 2.22-2.49; P less than .001). The study did not capture readmissions in a different state than that of the index hospital and was limited to the 22 states participating in the 2014 Readmissions Database.
Bottom line: Discharge AMA is associated with significantly higher odds of 30-day readmission, subsequent AMA discharge and presentation to another hospital, compared with routine discharge.
Citation: Kumar N. Burden of 30-day readmissions associated with discharge against medical advice among inpatients in the United States. Am J Med. 2019 Jun;132(6):708-17.
Dr. Webber is a hospitalist at Vanderbilt University Medical Center, Nashville, Tenn.
Background: AMA discharges are common (1%-2% of all U.S. discharges) and disproportionately affect vulnerable patient populations, specifically those of lower socioeconomic status and the uninsured. Previous studies have been insufficiently powered to assess the effects of AMA discharge on 30-day readmission rates at a national level.
Study design: Retrospective cohort.
Setting: Community and teaching hospitals in 22 states.
Synopsis: With use of the 2014 Nationwide Readmissions Database of 23,110,641 index hospitalizations of patients 18 years or older, this study found that AMA discharge occurred with 1.3% of admissions. AMA discharge was associated with greater than twice the odds of 30-day readmission, compared with routine discharge. Of patients discharged AMA, 20.2% had an unplanned readmission within 30 days, compared with 10.1% of patients discharged routinely (OR, 2.25; 95% CI, 2.20-2.30; P less than .001).
Patients who were discharged AMA had almost 20 times the odds of undergoing repeat AMA discharge at readmission (OR, 18.41; 95% CI, 17.46-19.41; P less than .001) and twice the odds of presenting to a different hospital (OR, 2.35; 95% CI, 2.22-2.49; P less than .001). The study did not capture readmissions in a different state than that of the index hospital and was limited to the 22 states participating in the 2014 Readmissions Database.
Bottom line: Discharge AMA is associated with significantly higher odds of 30-day readmission, subsequent AMA discharge and presentation to another hospital, compared with routine discharge.
Citation: Kumar N. Burden of 30-day readmissions associated with discharge against medical advice among inpatients in the United States. Am J Med. 2019 Jun;132(6):708-17.
Dr. Webber is a hospitalist at Vanderbilt University Medical Center, Nashville, Tenn.
Background: AMA discharges are common (1%-2% of all U.S. discharges) and disproportionately affect vulnerable patient populations, specifically those of lower socioeconomic status and the uninsured. Previous studies have been insufficiently powered to assess the effects of AMA discharge on 30-day readmission rates at a national level.
Study design: Retrospective cohort.
Setting: Community and teaching hospitals in 22 states.
Synopsis: With use of the 2014 Nationwide Readmissions Database of 23,110,641 index hospitalizations of patients 18 years or older, this study found that AMA discharge occurred with 1.3% of admissions. AMA discharge was associated with greater than twice the odds of 30-day readmission, compared with routine discharge. Of patients discharged AMA, 20.2% had an unplanned readmission within 30 days, compared with 10.1% of patients discharged routinely (OR, 2.25; 95% CI, 2.20-2.30; P less than .001).
Patients who were discharged AMA had almost 20 times the odds of undergoing repeat AMA discharge at readmission (OR, 18.41; 95% CI, 17.46-19.41; P less than .001) and twice the odds of presenting to a different hospital (OR, 2.35; 95% CI, 2.22-2.49; P less than .001). The study did not capture readmissions in a different state than that of the index hospital and was limited to the 22 states participating in the 2014 Readmissions Database.
Bottom line: Discharge AMA is associated with significantly higher odds of 30-day readmission, subsequent AMA discharge and presentation to another hospital, compared with routine discharge.
Citation: Kumar N. Burden of 30-day readmissions associated with discharge against medical advice among inpatients in the United States. Am J Med. 2019 Jun;132(6):708-17.
Dr. Webber is a hospitalist at Vanderbilt University Medical Center, Nashville, Tenn.