User login
Is the US running too many T-cell lymphoma trials?

SAN FRANCISCO—The US currently has 284 open clinical trials enrolling patients with T-cell lymphomas, a fact that is actually detrimental to this patient population, according to an expert in the field.
Anas Younes, MD, of MD Anderson Cancer Center in Houston, presented this perspective at the 4th Annual T-cell Lymphoma Forum, which took place January 26-28.
Dr Younes noted that there are 361 clinical trials worldwide that are currently accruing patients with T-cell lymphomas. Of those, 284 are taking place in the US. Less than half of the US trials are new; 124 of them have been submitted since January 2010.
The new trials are divided pretty evenly between phase 1 and phase 2—66 and 61 trials, respectively. But only 1 of the studies is a phase 3, which suggests that having such a large number of trials may be hindering drug development as well as patient treatment.
“[W]e have too many clinical trials available for a small pool of patients,” Dr Younes said. “I think it’s not a good idea to have that. We’re diluting our efforts, major trials are not able to enroll in a timely manner, and most of them will close before they even enroll [an] adequate [number of] patients.”
As an example, Dr Younes cited lymphoma trials developed at MD Anderson that were open between 2004 and 2011. The center’s accrual of follicular lymphoma patients during this period ranged from roughly 40 to 160 patients. The number of Hodgkin lymphoma patients enrolled ranged from about 25 to 110, and the number of mantle cell lymphoma patients ranged from about 30 to 70.
But the largest number of T-cell lymphoma patients enrolled was about 50 in 2007. And on the whole, the center has not enrolled more than 10 to 15 patients per year.
“And the reason is there are so many competing trials in the United States,” Dr Younes said. “By the time [patients are] referred to us, they’re either not eligible or too sick to be treated . . . . So I think it’s becoming unhealthy competition with such a large number of protocols available for these patients.”
As of right now, MD Anderson is running 5 trials for T-cell lymphoma patients (and planning to open 3 more trials soon), but patient accrual has been slow.
For instance, a trial of vorinostat plus CHOP for untreated T-cell lymphoma has been open since 2008. It has accrued 12 patients but still has 40 slots open.
And a trial of MK-2206 in relapsed or refractory T-cell lymphoma has been open since 2010. It has accrued 1 patient and has 15 slots still open.
“We’re really unable to enroll enough patients in a timely manner anymore,” Dr Younes said. “So we need to prioritize [our trials]. We need to collaborate more.” ![]()
SAN FRANCISCO—The US currently has 284 open clinical trials enrolling patients with T-cell lymphomas, a fact that is actually detrimental to this patient population, according to an expert in the field.
Anas Younes, MD, of MD Anderson Cancer Center in Houston, presented this perspective at the 4th Annual T-cell Lymphoma Forum, which took place January 26-28.
Dr Younes noted that there are 361 clinical trials worldwide that are currently accruing patients with T-cell lymphomas. Of those, 284 are taking place in the US. Less than half of the US trials are new; 124 of them have been submitted since January 2010.
The new trials are divided pretty evenly between phase 1 and phase 2—66 and 61 trials, respectively. But only 1 of the studies is a phase 3, which suggests that having such a large number of trials may be hindering drug development as well as patient treatment.
“[W]e have too many clinical trials available for a small pool of patients,” Dr Younes said. “I think it’s not a good idea to have that. We’re diluting our efforts, major trials are not able to enroll in a timely manner, and most of them will close before they even enroll [an] adequate [number of] patients.”
As an example, Dr Younes cited lymphoma trials developed at MD Anderson that were open between 2004 and 2011. The center’s accrual of follicular lymphoma patients during this period ranged from roughly 40 to 160 patients. The number of Hodgkin lymphoma patients enrolled ranged from about 25 to 110, and the number of mantle cell lymphoma patients ranged from about 30 to 70.
But the largest number of T-cell lymphoma patients enrolled was about 50 in 2007. And on the whole, the center has not enrolled more than 10 to 15 patients per year.
“And the reason is there are so many competing trials in the United States,” Dr Younes said. “By the time [patients are] referred to us, they’re either not eligible or too sick to be treated . . . . So I think it’s becoming unhealthy competition with such a large number of protocols available for these patients.”
As of right now, MD Anderson is running 5 trials for T-cell lymphoma patients (and planning to open 3 more trials soon), but patient accrual has been slow.
For instance, a trial of vorinostat plus CHOP for untreated T-cell lymphoma has been open since 2008. It has accrued 12 patients but still has 40 slots open.
And a trial of MK-2206 in relapsed or refractory T-cell lymphoma has been open since 2010. It has accrued 1 patient and has 15 slots still open.
“We’re really unable to enroll enough patients in a timely manner anymore,” Dr Younes said. “So we need to prioritize [our trials]. We need to collaborate more.” ![]()
SAN FRANCISCO—The US currently has 284 open clinical trials enrolling patients with T-cell lymphomas, a fact that is actually detrimental to this patient population, according to an expert in the field.
Anas Younes, MD, of MD Anderson Cancer Center in Houston, presented this perspective at the 4th Annual T-cell Lymphoma Forum, which took place January 26-28.
Dr Younes noted that there are 361 clinical trials worldwide that are currently accruing patients with T-cell lymphomas. Of those, 284 are taking place in the US. Less than half of the US trials are new; 124 of them have been submitted since January 2010.
The new trials are divided pretty evenly between phase 1 and phase 2—66 and 61 trials, respectively. But only 1 of the studies is a phase 3, which suggests that having such a large number of trials may be hindering drug development as well as patient treatment.
“[W]e have too many clinical trials available for a small pool of patients,” Dr Younes said. “I think it’s not a good idea to have that. We’re diluting our efforts, major trials are not able to enroll in a timely manner, and most of them will close before they even enroll [an] adequate [number of] patients.”
As an example, Dr Younes cited lymphoma trials developed at MD Anderson that were open between 2004 and 2011. The center’s accrual of follicular lymphoma patients during this period ranged from roughly 40 to 160 patients. The number of Hodgkin lymphoma patients enrolled ranged from about 25 to 110, and the number of mantle cell lymphoma patients ranged from about 30 to 70.
But the largest number of T-cell lymphoma patients enrolled was about 50 in 2007. And on the whole, the center has not enrolled more than 10 to 15 patients per year.
“And the reason is there are so many competing trials in the United States,” Dr Younes said. “By the time [patients are] referred to us, they’re either not eligible or too sick to be treated . . . . So I think it’s becoming unhealthy competition with such a large number of protocols available for these patients.”
As of right now, MD Anderson is running 5 trials for T-cell lymphoma patients (and planning to open 3 more trials soon), but patient accrual has been slow.
For instance, a trial of vorinostat plus CHOP for untreated T-cell lymphoma has been open since 2008. It has accrued 12 patients but still has 40 slots open.
And a trial of MK-2206 in relapsed or refractory T-cell lymphoma has been open since 2010. It has accrued 1 patient and has 15 slots still open.
“We’re really unable to enroll enough patients in a timely manner anymore,” Dr Younes said. “So we need to prioritize [our trials]. We need to collaborate more.” ![]()
GAD-alum Antigen Therapy Fails to Halt Progression of Type 1 Diabetes
Antigen therapy with glutamic acid decarboxylase 65 formulated with alum failed to induce immunologic tolerance and stem the loss of stimulated serum C-peptide in a phase III clinical trial of new-onset type 1 diabetes, according to a report in the Feb. 2 issue of the New England Journal of Medicine.
The treatment also failed to improve clinical outcomes during the 15-month study, said Dr. Johnny L. Ludvigsson of the department of clinical and experimental medicine, division of pediatrics, Linkoping (Sweden) University, and his associates.

In a previous phase II study, treatment with the 65-kD isoform of glutamic acid decarboxylase (GAD65) formulated with alum (GAD-alum) had preserved stimulated C-peptide levels and fasting C-peptide levels for 4 years in a subgroup of patients who were treated immediately after diagnosis (Diabetologia 2011;54:634-40). However, a more recent phase II trial of GAD-alum did not show any clinical benefit, the investigators noted.
Dr. Ludvigsson and his colleagues performed their phase III clinical trial at 63 clinics in Finland, France, Germany, Italy, the Netherlands, Slovenia, Spain, Sweden, and the United Kingdom. The 327 study subjects were aged 10-20 years and had been diagnosed as having type 1 diabetes within the preceding 3 months.
The patients were randomly assigned in double-blind fashion to receive one of three regimens of subcutaneous injections: four doses of GAD-alum (on days 1, 30, 90, and 270), two doses of GAD-alum (on days 1 and 30), or four doses of placebo.
The primary outcome was preservation of the stimulated serum C-peptide level after 15 months. Stimulated C-peptide levels showed progressive declines in all three groups throughout the study. The declines were not significantly different among the three groups at any time point, including at the conclusion of the study, the investigators said (N. Engl. J. Med. 2012;366:433-42).
Moreover, there were no differences among the three groups in mean daily insulin dose, glycated hemoglobin levels, or several other clinical outcomes.
The rates of adverse events also were similar among the three study groups.
"Much as treatments for diseases such as childhood cancer and immunotherapy of allergy have developed in a stepwise, gradual manner through the combination of existing therapies, treatment for type 1 diabetes will most likely be based on the knowledge gained from this and other studies, as well as future studies, of single agents or combination therapies for both intervention and prevention," Dr. Ludvigsson and his associates said.
They added that patients who develop stiff person syndrome have been shown in previous studies to carry elevated levels of GAD65 autoantibodies. In this study, all the subjects underwent periodic neurologic assessments, and no symptoms suggestive of stiff person syndrome were seen.
This study was supported by Diamyd Medical and the Swedish Child Diabetes Foundation. Dr. Ludvigsson reported ties to Johnson & Johnson, GlaxoSmithKline, Sanofi-Aventis, and Novo Nordisk; his associates reported ties to Merck Sharp and Dohme, Bristol-Myers Squibb, Eli Lilly, Medtronic, Tolerx, and Andromeda Biotech.
Antigen therapy with glutamic acid decarboxylase 65 formulated with alum failed to induce immunologic tolerance and stem the loss of stimulated serum C-peptide in a phase III clinical trial of new-onset type 1 diabetes, according to a report in the Feb. 2 issue of the New England Journal of Medicine.
The treatment also failed to improve clinical outcomes during the 15-month study, said Dr. Johnny L. Ludvigsson of the department of clinical and experimental medicine, division of pediatrics, Linkoping (Sweden) University, and his associates.
In a previous phase II study, treatment with the 65-kD isoform of glutamic acid decarboxylase (GAD65) formulated with alum (GAD-alum) had preserved stimulated C-peptide levels and fasting C-peptide levels for 4 years in a subgroup of patients who were treated immediately after diagnosis (Diabetologia 2011;54:634-40). However, a more recent phase II trial of GAD-alum did not show any clinical benefit, the investigators noted.
Dr. Ludvigsson and his colleagues performed their phase III clinical trial at 63 clinics in Finland, France, Germany, Italy, the Netherlands, Slovenia, Spain, Sweden, and the United Kingdom. The 327 study subjects were aged 10-20 years and had been diagnosed as having type 1 diabetes within the preceding 3 months.
The patients were randomly assigned in double-blind fashion to receive one of three regimens of subcutaneous injections: four doses of GAD-alum (on days 1, 30, 90, and 270), two doses of GAD-alum (on days 1 and 30), or four doses of placebo.
The primary outcome was preservation of the stimulated serum C-peptide level after 15 months. Stimulated C-peptide levels showed progressive declines in all three groups throughout the study. The declines were not significantly different among the three groups at any time point, including at the conclusion of the study, the investigators said (N. Engl. J. Med. 2012;366:433-42).
Moreover, there were no differences among the three groups in mean daily insulin dose, glycated hemoglobin levels, or several other clinical outcomes.
The rates of adverse events also were similar among the three study groups.
"Much as treatments for diseases such as childhood cancer and immunotherapy of allergy have developed in a stepwise, gradual manner through the combination of existing therapies, treatment for type 1 diabetes will most likely be based on the knowledge gained from this and other studies, as well as future studies, of single agents or combination therapies for both intervention and prevention," Dr. Ludvigsson and his associates said.
They added that patients who develop stiff person syndrome have been shown in previous studies to carry elevated levels of GAD65 autoantibodies. In this study, all the subjects underwent periodic neurologic assessments, and no symptoms suggestive of stiff person syndrome were seen.
This study was supported by Diamyd Medical and the Swedish Child Diabetes Foundation. Dr. Ludvigsson reported ties to Johnson & Johnson, GlaxoSmithKline, Sanofi-Aventis, and Novo Nordisk; his associates reported ties to Merck Sharp and Dohme, Bristol-Myers Squibb, Eli Lilly, Medtronic, Tolerx, and Andromeda Biotech.
Antigen therapy with glutamic acid decarboxylase 65 formulated with alum failed to induce immunologic tolerance and stem the loss of stimulated serum C-peptide in a phase III clinical trial of new-onset type 1 diabetes, according to a report in the Feb. 2 issue of the New England Journal of Medicine.
The treatment also failed to improve clinical outcomes during the 15-month study, said Dr. Johnny L. Ludvigsson of the department of clinical and experimental medicine, division of pediatrics, Linkoping (Sweden) University, and his associates.
In a previous phase II study, treatment with the 65-kD isoform of glutamic acid decarboxylase (GAD65) formulated with alum (GAD-alum) had preserved stimulated C-peptide levels and fasting C-peptide levels for 4 years in a subgroup of patients who were treated immediately after diagnosis (Diabetologia 2011;54:634-40). However, a more recent phase II trial of GAD-alum did not show any clinical benefit, the investigators noted.
Dr. Ludvigsson and his colleagues performed their phase III clinical trial at 63 clinics in Finland, France, Germany, Italy, the Netherlands, Slovenia, Spain, Sweden, and the United Kingdom. The 327 study subjects were aged 10-20 years and had been diagnosed as having type 1 diabetes within the preceding 3 months.
The patients were randomly assigned in double-blind fashion to receive one of three regimens of subcutaneous injections: four doses of GAD-alum (on days 1, 30, 90, and 270), two doses of GAD-alum (on days 1 and 30), or four doses of placebo.
The primary outcome was preservation of the stimulated serum C-peptide level after 15 months. Stimulated C-peptide levels showed progressive declines in all three groups throughout the study. The declines were not significantly different among the three groups at any time point, including at the conclusion of the study, the investigators said (N. Engl. J. Med. 2012;366:433-42).
Moreover, there were no differences among the three groups in mean daily insulin dose, glycated hemoglobin levels, or several other clinical outcomes.
The rates of adverse events also were similar among the three study groups.
"Much as treatments for diseases such as childhood cancer and immunotherapy of allergy have developed in a stepwise, gradual manner through the combination of existing therapies, treatment for type 1 diabetes will most likely be based on the knowledge gained from this and other studies, as well as future studies, of single agents or combination therapies for both intervention and prevention," Dr. Ludvigsson and his associates said.
They added that patients who develop stiff person syndrome have been shown in previous studies to carry elevated levels of GAD65 autoantibodies. In this study, all the subjects underwent periodic neurologic assessments, and no symptoms suggestive of stiff person syndrome were seen.
This study was supported by Diamyd Medical and the Swedish Child Diabetes Foundation. Dr. Ludvigsson reported ties to Johnson & Johnson, GlaxoSmithKline, Sanofi-Aventis, and Novo Nordisk; his associates reported ties to Merck Sharp and Dohme, Bristol-Myers Squibb, Eli Lilly, Medtronic, Tolerx, and Andromeda Biotech.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Major Finding: Antigen therapy with GAD-alum did not preserve serum levels of stimulated C-peptide in new-onset type 1 diabetes or improve clinical outcomes.
Data Source: A 15-month phase III randomized double-blind clinical trial involving 327 patients aged 10-20 years with newly diagnosed type 1 diabetes treated at 63 clinics throughout Europe.
Disclosures: This study was supported by Diamyd Medical and the Swedish Child Diabetes Foundation. Dr. Ludvigsson reported ties to Johnson & Johnson, GlaxoSmithKline, SanofiAventis, and Novo Nordisk; his associates reported ties to Merck Sharp and Dohme, Brystol Myers Squibb, Eli Lilly, Medtronic, Tolerx, and Andromeda Bio.
Comorbidities Up Risk for Thyroidectomy Complications, In-Hospital Deaths
MIAMI BEACH – Cardiac and respiratory comorbidities were "common culprits" and present in more than half of thyroidectomy patients who died in the hospital, according to analysis of a large inpatient database.
Although overall mortality is less than 1% for thyroidectomy patients nationwide, researcher Rishi Vashishta said, "Patient comorbidities can often contribute to perioperative death and should really be considered when discussing treatment options with patients."
Mr. Vashishta and his associates identified 11,862 patients who underwent thyroidectomy using ICD-9 codes from the Healthcare Cost Utilization Project Nationwide Inpatient Sample (NIS) database for 2009. Two-thirds of patients were white and three-fourths were female A total of 73 of these patients died during their hospitalization that year.
"We calculated the mortality rate during hospitalization to be 0.61%," Mr. Vashishta, a medical student at George Washington University, Washington, said at the Triological Society Combined Sections meeting.
Other aims of the study were to assess thyroid surgery complications, length of stay, and total hospital charges. "There are a large number of institutional studies, but there remains a relative paucity of studies examining this procedure on a more macro and socioeconomic level," said Mr. Vashishta.
Among the nearly 12,000 admissions, mean length of stay was 2.97 days and mean total hospital charges accrued was $39,236.
In contrast, a subgroup analysis revealed mean length of stay was 13.8 days and mean increase in total hospital charges was nearly $218,855 among patients who died during hospitalization. "Interestingly, the respiratory status in these patients was markedly worse, with a tracheostomy required in 28%, prolonged mechanical ventilation required in 43%, and endotracheal intubation in 55%," Mr. Vashishta said at the meeting, which was jointly sponsored by the Triological Society and the American College of Surgeons.
Acute cerebrovascular disease was involved in 62% of deaths, he reported.
The mean age of patients who died was 65 years, compared with a mean of 53 years for all thyroidectomy patients in the study.
Approximately 80% of all surgeries in the study were elective. The majority of patients, 55%, underwent total thyroidectomy, 32% underwent unilateral lobectomy, and the remainder had partial thyroidectomy.
When Mr. Vashishta and his colleagues assessed complications, they found hypocalcemia present in 6%, vocal cord paresis in 1.4%, and hypoparathyroidism in 0.77% of patients using bivariate analyses. The incidence of hematoma and hemorrhage were low at 1.43% and 0.67%, respectively. "Our complication rates were generally consistent with those from institutional studies published in the literature."
"We found strong predictors of [these] complications during hospitalization included female gender; hospital location and teaching status; and type of thyroid diagnosis," he said. "Although the majority of cases were conducted at large teaching hospitals in urban centers, no socioeconomic or regional differences were observed," the investigators noted in their abstract but did not offer further explanation.
Admissions data showed that nontoxic nodular goiter was a diagnosis code for 36% of patients. In addition, malignant neoplasm was a code for 31% and benign neoplasm for 11%, "Graves’ disease, which we classified under acquired hypothyroidism, was much less common, around 8%," Mr. Vashishta said. ICD-9 codes for thyrotoxicosis and thyroiditis each were noted on 8% of records.
Errors in coding and sampling are a potential limitation of this and any study based on a large administrative database, Mr. Vashishta said. For example, use of ICD-9 codes "inevitably included patients in our stratified sample admitted for some other problem who underwent incidental thyroidectomies during their hospitalization." Furthermore, thyroidectomy is increasingly being performed as an outpatient procedure and the NIS is an inpatient database. "This effectively skewed our mean total charges and mean length of stay in the hospital upwards."
The study was not funded by industry. Mr. Vashishta said that he had no relevant financial disclosures.
MIAMI BEACH – Cardiac and respiratory comorbidities were "common culprits" and present in more than half of thyroidectomy patients who died in the hospital, according to analysis of a large inpatient database.
Although overall mortality is less than 1% for thyroidectomy patients nationwide, researcher Rishi Vashishta said, "Patient comorbidities can often contribute to perioperative death and should really be considered when discussing treatment options with patients."
Mr. Vashishta and his associates identified 11,862 patients who underwent thyroidectomy using ICD-9 codes from the Healthcare Cost Utilization Project Nationwide Inpatient Sample (NIS) database for 2009. Two-thirds of patients were white and three-fourths were female A total of 73 of these patients died during their hospitalization that year.
"We calculated the mortality rate during hospitalization to be 0.61%," Mr. Vashishta, a medical student at George Washington University, Washington, said at the Triological Society Combined Sections meeting.
Other aims of the study were to assess thyroid surgery complications, length of stay, and total hospital charges. "There are a large number of institutional studies, but there remains a relative paucity of studies examining this procedure on a more macro and socioeconomic level," said Mr. Vashishta.
Among the nearly 12,000 admissions, mean length of stay was 2.97 days and mean total hospital charges accrued was $39,236.
In contrast, a subgroup analysis revealed mean length of stay was 13.8 days and mean increase in total hospital charges was nearly $218,855 among patients who died during hospitalization. "Interestingly, the respiratory status in these patients was markedly worse, with a tracheostomy required in 28%, prolonged mechanical ventilation required in 43%, and endotracheal intubation in 55%," Mr. Vashishta said at the meeting, which was jointly sponsored by the Triological Society and the American College of Surgeons.
Acute cerebrovascular disease was involved in 62% of deaths, he reported.
The mean age of patients who died was 65 years, compared with a mean of 53 years for all thyroidectomy patients in the study.
Approximately 80% of all surgeries in the study were elective. The majority of patients, 55%, underwent total thyroidectomy, 32% underwent unilateral lobectomy, and the remainder had partial thyroidectomy.
When Mr. Vashishta and his colleagues assessed complications, they found hypocalcemia present in 6%, vocal cord paresis in 1.4%, and hypoparathyroidism in 0.77% of patients using bivariate analyses. The incidence of hematoma and hemorrhage were low at 1.43% and 0.67%, respectively. "Our complication rates were generally consistent with those from institutional studies published in the literature."
"We found strong predictors of [these] complications during hospitalization included female gender; hospital location and teaching status; and type of thyroid diagnosis," he said. "Although the majority of cases were conducted at large teaching hospitals in urban centers, no socioeconomic or regional differences were observed," the investigators noted in their abstract but did not offer further explanation.
Admissions data showed that nontoxic nodular goiter was a diagnosis code for 36% of patients. In addition, malignant neoplasm was a code for 31% and benign neoplasm for 11%, "Graves’ disease, which we classified under acquired hypothyroidism, was much less common, around 8%," Mr. Vashishta said. ICD-9 codes for thyrotoxicosis and thyroiditis each were noted on 8% of records.
Errors in coding and sampling are a potential limitation of this and any study based on a large administrative database, Mr. Vashishta said. For example, use of ICD-9 codes "inevitably included patients in our stratified sample admitted for some other problem who underwent incidental thyroidectomies during their hospitalization." Furthermore, thyroidectomy is increasingly being performed as an outpatient procedure and the NIS is an inpatient database. "This effectively skewed our mean total charges and mean length of stay in the hospital upwards."
The study was not funded by industry. Mr. Vashishta said that he had no relevant financial disclosures.
MIAMI BEACH – Cardiac and respiratory comorbidities were "common culprits" and present in more than half of thyroidectomy patients who died in the hospital, according to analysis of a large inpatient database.
Although overall mortality is less than 1% for thyroidectomy patients nationwide, researcher Rishi Vashishta said, "Patient comorbidities can often contribute to perioperative death and should really be considered when discussing treatment options with patients."
Mr. Vashishta and his associates identified 11,862 patients who underwent thyroidectomy using ICD-9 codes from the Healthcare Cost Utilization Project Nationwide Inpatient Sample (NIS) database for 2009. Two-thirds of patients were white and three-fourths were female A total of 73 of these patients died during their hospitalization that year.
"We calculated the mortality rate during hospitalization to be 0.61%," Mr. Vashishta, a medical student at George Washington University, Washington, said at the Triological Society Combined Sections meeting.
Other aims of the study were to assess thyroid surgery complications, length of stay, and total hospital charges. "There are a large number of institutional studies, but there remains a relative paucity of studies examining this procedure on a more macro and socioeconomic level," said Mr. Vashishta.
Among the nearly 12,000 admissions, mean length of stay was 2.97 days and mean total hospital charges accrued was $39,236.
In contrast, a subgroup analysis revealed mean length of stay was 13.8 days and mean increase in total hospital charges was nearly $218,855 among patients who died during hospitalization. "Interestingly, the respiratory status in these patients was markedly worse, with a tracheostomy required in 28%, prolonged mechanical ventilation required in 43%, and endotracheal intubation in 55%," Mr. Vashishta said at the meeting, which was jointly sponsored by the Triological Society and the American College of Surgeons.
Acute cerebrovascular disease was involved in 62% of deaths, he reported.
The mean age of patients who died was 65 years, compared with a mean of 53 years for all thyroidectomy patients in the study.
Approximately 80% of all surgeries in the study were elective. The majority of patients, 55%, underwent total thyroidectomy, 32% underwent unilateral lobectomy, and the remainder had partial thyroidectomy.
When Mr. Vashishta and his colleagues assessed complications, they found hypocalcemia present in 6%, vocal cord paresis in 1.4%, and hypoparathyroidism in 0.77% of patients using bivariate analyses. The incidence of hematoma and hemorrhage were low at 1.43% and 0.67%, respectively. "Our complication rates were generally consistent with those from institutional studies published in the literature."
"We found strong predictors of [these] complications during hospitalization included female gender; hospital location and teaching status; and type of thyroid diagnosis," he said. "Although the majority of cases were conducted at large teaching hospitals in urban centers, no socioeconomic or regional differences were observed," the investigators noted in their abstract but did not offer further explanation.
Admissions data showed that nontoxic nodular goiter was a diagnosis code for 36% of patients. In addition, malignant neoplasm was a code for 31% and benign neoplasm for 11%, "Graves’ disease, which we classified under acquired hypothyroidism, was much less common, around 8%," Mr. Vashishta said. ICD-9 codes for thyrotoxicosis and thyroiditis each were noted on 8% of records.
Errors in coding and sampling are a potential limitation of this and any study based on a large administrative database, Mr. Vashishta said. For example, use of ICD-9 codes "inevitably included patients in our stratified sample admitted for some other problem who underwent incidental thyroidectomies during their hospitalization." Furthermore, thyroidectomy is increasingly being performed as an outpatient procedure and the NIS is an inpatient database. "This effectively skewed our mean total charges and mean length of stay in the hospital upwards."
The study was not funded by industry. Mr. Vashishta said that he had no relevant financial disclosures.
FROM THE TRIOLOGICAL SOCIETY COMBINED SECTIONS MEETING
Major Finding: A total 73 of 11,862 thyroidectomy patients (0.61%) died during hospitalization.
Data Source: Retrospective study of ICD-9 codes for thyroidectomy in 2009 from the Nationwide Inpatient Sample database.
Disclosures: The study was not funded by industry. Mr. Vashishta said that he had no relevant financial disclosures.
Don't Call That Child 'Fat' or 'Obese'
STEAMBOAT SPRINGS, COLO. – In discussing a child’s weight problem with the parents, it’s best for physicians to refrain from using the terms "fat," "extremely obese," and even "obese."
"Parents find those terms undesirable. They’re stigmatizing, blaming, nonmotivating, and condescending," Dr. Paul R. Stricker said at a meeting on practical pediatrics sponsored by the American Academy of Pediatrics.

And that’s not just his personal opinion, either. He cited a recent groundbreaking study in which investigators at Yale University in New Haven, Conn., conducted a national online survey of the parents of 455 children aged 2-18 years. The purpose was to examine parental perceptions of language related to weight in order to improve the quality of physician-parent discussions about their child’s obesity. The underlying idea is that the likelihood of successful long-term weight loss is enhanced if the parents are committed to the proposed lifestyle modifications.
On a 5-point rating scale, most parents ranked "weight" and "unhealthy weight" as terms they preferred physicians to use in describing their child’s extra pounds. Moreover, the parents indicated they found the terms "unhealthy weight," "overweight," and "weight problem" to be the most motivating to lose weight, noted Dr. Stricker, a youth sports medicine specialist at the Scripps Clinic in San Diego.
On the other hand, parents perceived the terms "chubby," "fat," "obese," and "extremely obese" quite negatively, rating them as the least motivating to encourage weight loss (Pediatrics 2011;128:e786-93).
These data cast doubt on the wisdom of the British public health minister's 2010 declaration that U.K. health providers should call their obese patients "fat" to motivate them to lose weight.
As a pediatric sports medicine specialist, Dr. Stricker’s goal is to help overweight kids have a positive sports and exercise experience. He wants it to be "something they’ll want to pass along to their own children." He combines his exercise guidance with dietary instruction in weight loss, with an emphasis placed on eating multiple small meals to keep the metabolic rate revved so more calories are burned.
But lifestyle interventions don’t always work, and Dr. Stricker highlighted a recent German study that’s eye-opening as to why.
The prospective study included 111 overweight and obese 7- to 15-year-olds and their parents. The youths were referred to a 1-year-long best-practice lifestyle intervention program.
Treatment success was defined as at least a 5% weight reduction at follow-up 1 year after completing the year-long intervention. The investigators found – consistent with their study hypothesis – that psychosocial familial characteristics were significantly predictive of long-term success or failure. This was true even after the researchers controlled for familial obesity in order to cancel out the impact of genetic factors.
The strongest predictor of long-term failure for the lifestyle intervention was maternal depression. Maternal attachment insecurity and family adversity also predicted long-term treatment failure (Pediatrics 2011;128: e779-85).
These findings point to the need for further research aimed at developing lifestyle interventions for pediatric weight loss that are tailored to a family’s psychosocial dynamics, Dr. Stricker observed.
He reported having no financial conflicts.
STEAMBOAT SPRINGS, COLO. – In discussing a child’s weight problem with the parents, it’s best for physicians to refrain from using the terms "fat," "extremely obese," and even "obese."
"Parents find those terms undesirable. They’re stigmatizing, blaming, nonmotivating, and condescending," Dr. Paul R. Stricker said at a meeting on practical pediatrics sponsored by the American Academy of Pediatrics.
And that’s not just his personal opinion, either. He cited a recent groundbreaking study in which investigators at Yale University in New Haven, Conn., conducted a national online survey of the parents of 455 children aged 2-18 years. The purpose was to examine parental perceptions of language related to weight in order to improve the quality of physician-parent discussions about their child’s obesity. The underlying idea is that the likelihood of successful long-term weight loss is enhanced if the parents are committed to the proposed lifestyle modifications.
On a 5-point rating scale, most parents ranked "weight" and "unhealthy weight" as terms they preferred physicians to use in describing their child’s extra pounds. Moreover, the parents indicated they found the terms "unhealthy weight," "overweight," and "weight problem" to be the most motivating to lose weight, noted Dr. Stricker, a youth sports medicine specialist at the Scripps Clinic in San Diego.
On the other hand, parents perceived the terms "chubby," "fat," "obese," and "extremely obese" quite negatively, rating them as the least motivating to encourage weight loss (Pediatrics 2011;128:e786-93).
These data cast doubt on the wisdom of the British public health minister's 2010 declaration that U.K. health providers should call their obese patients "fat" to motivate them to lose weight.
As a pediatric sports medicine specialist, Dr. Stricker’s goal is to help overweight kids have a positive sports and exercise experience. He wants it to be "something they’ll want to pass along to their own children." He combines his exercise guidance with dietary instruction in weight loss, with an emphasis placed on eating multiple small meals to keep the metabolic rate revved so more calories are burned.
But lifestyle interventions don’t always work, and Dr. Stricker highlighted a recent German study that’s eye-opening as to why.
The prospective study included 111 overweight and obese 7- to 15-year-olds and their parents. The youths were referred to a 1-year-long best-practice lifestyle intervention program.
Treatment success was defined as at least a 5% weight reduction at follow-up 1 year after completing the year-long intervention. The investigators found – consistent with their study hypothesis – that psychosocial familial characteristics were significantly predictive of long-term success or failure. This was true even after the researchers controlled for familial obesity in order to cancel out the impact of genetic factors.
The strongest predictor of long-term failure for the lifestyle intervention was maternal depression. Maternal attachment insecurity and family adversity also predicted long-term treatment failure (Pediatrics 2011;128: e779-85).
These findings point to the need for further research aimed at developing lifestyle interventions for pediatric weight loss that are tailored to a family’s psychosocial dynamics, Dr. Stricker observed.
He reported having no financial conflicts.
STEAMBOAT SPRINGS, COLO. – In discussing a child’s weight problem with the parents, it’s best for physicians to refrain from using the terms "fat," "extremely obese," and even "obese."
"Parents find those terms undesirable. They’re stigmatizing, blaming, nonmotivating, and condescending," Dr. Paul R. Stricker said at a meeting on practical pediatrics sponsored by the American Academy of Pediatrics.
And that’s not just his personal opinion, either. He cited a recent groundbreaking study in which investigators at Yale University in New Haven, Conn., conducted a national online survey of the parents of 455 children aged 2-18 years. The purpose was to examine parental perceptions of language related to weight in order to improve the quality of physician-parent discussions about their child’s obesity. The underlying idea is that the likelihood of successful long-term weight loss is enhanced if the parents are committed to the proposed lifestyle modifications.
On a 5-point rating scale, most parents ranked "weight" and "unhealthy weight" as terms they preferred physicians to use in describing their child’s extra pounds. Moreover, the parents indicated they found the terms "unhealthy weight," "overweight," and "weight problem" to be the most motivating to lose weight, noted Dr. Stricker, a youth sports medicine specialist at the Scripps Clinic in San Diego.
On the other hand, parents perceived the terms "chubby," "fat," "obese," and "extremely obese" quite negatively, rating them as the least motivating to encourage weight loss (Pediatrics 2011;128:e786-93).
These data cast doubt on the wisdom of the British public health minister's 2010 declaration that U.K. health providers should call their obese patients "fat" to motivate them to lose weight.
As a pediatric sports medicine specialist, Dr. Stricker’s goal is to help overweight kids have a positive sports and exercise experience. He wants it to be "something they’ll want to pass along to their own children." He combines his exercise guidance with dietary instruction in weight loss, with an emphasis placed on eating multiple small meals to keep the metabolic rate revved so more calories are burned.
But lifestyle interventions don’t always work, and Dr. Stricker highlighted a recent German study that’s eye-opening as to why.
The prospective study included 111 overweight and obese 7- to 15-year-olds and their parents. The youths were referred to a 1-year-long best-practice lifestyle intervention program.
Treatment success was defined as at least a 5% weight reduction at follow-up 1 year after completing the year-long intervention. The investigators found – consistent with their study hypothesis – that psychosocial familial characteristics were significantly predictive of long-term success or failure. This was true even after the researchers controlled for familial obesity in order to cancel out the impact of genetic factors.
The strongest predictor of long-term failure for the lifestyle intervention was maternal depression. Maternal attachment insecurity and family adversity also predicted long-term treatment failure (Pediatrics 2011;128: e779-85).
These findings point to the need for further research aimed at developing lifestyle interventions for pediatric weight loss that are tailored to a family’s psychosocial dynamics, Dr. Stricker observed.
He reported having no financial conflicts.
EXPERT ANALYSIS FROM A MEETING ON PRACTICAL PEDIATRICS SPONSORED BY THE AMERICAN ACADEMY OF PEDIATRICS
Transfusion Medicine
Transfusion therapy is an essential part of hematology practice, allowing for curative therapy of diseases such as leukemia, aplastic anemia, and aggressive lymphomas. Nonetheless, transfusions are associated with significant risks, including transfusion-transmitted infections and transfusion-related reactions, and controversy remains about key issues in transfusion therapy, such as triggers for red cell transfusions. This article reviews the available blood products and indications for transfusion along with the associated risks and also discusses specific clinical situations, such as massive transfusion.
To read the full article in PDF:
Transfusion therapy is an essential part of hematology practice, allowing for curative therapy of diseases such as leukemia, aplastic anemia, and aggressive lymphomas. Nonetheless, transfusions are associated with significant risks, including transfusion-transmitted infections and transfusion-related reactions, and controversy remains about key issues in transfusion therapy, such as triggers for red cell transfusions. This article reviews the available blood products and indications for transfusion along with the associated risks and also discusses specific clinical situations, such as massive transfusion.
To read the full article in PDF:
Transfusion therapy is an essential part of hematology practice, allowing for curative therapy of diseases such as leukemia, aplastic anemia, and aggressive lymphomas. Nonetheless, transfusions are associated with significant risks, including transfusion-transmitted infections and transfusion-related reactions, and controversy remains about key issues in transfusion therapy, such as triggers for red cell transfusions. This article reviews the available blood products and indications for transfusion along with the associated risks and also discusses specific clinical situations, such as massive transfusion.
To read the full article in PDF:
A 37-year-old man with a chronic cough
A 37-year-old man presented to the emergency department with an 8-week history of a mildly productive cough and shortness of breath accompanied by high fevers, chills, and night sweats. He also had some nausea but no vomiting.
Four days earlier, he had been evaluated by his primary care physician, who prescribed a 14-day course of one double-strength trimethoprim-sulfamethoxazole tablet (Bactrim DS) every 12 hours for presumed acute bronchitis, but his symptoms did not improve.
He was unemployed, living in Arizona, married with children. He denied any use of tobacco, alcohol, or injection drugs. On further questioning, he disclosed that he had unintentionally lost 30 pounds over the past 2 to 3 months and had been feeling tired.
When asked about his medical history, he revealed that he had been diagnosed with human immunodeficiency virus (HIV) infection in 2008 and that recently he had not been taking his antiretroviral medication, a once-daily combination pill containing efavirenz, emtricitabine, and tenofovir (Atripla). He had no other significant medical history, and the only medication he was currently taking was the trimethoprim-sulfamethoxazole.
On examination, his temperature was 38.7°C (101.7°F), blood pressure 109/68 mm Hg, heart rate 60 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 100% while breathing supplemental oxygen via nasal cannula at 2 L/min. He did not appear seriously ill.
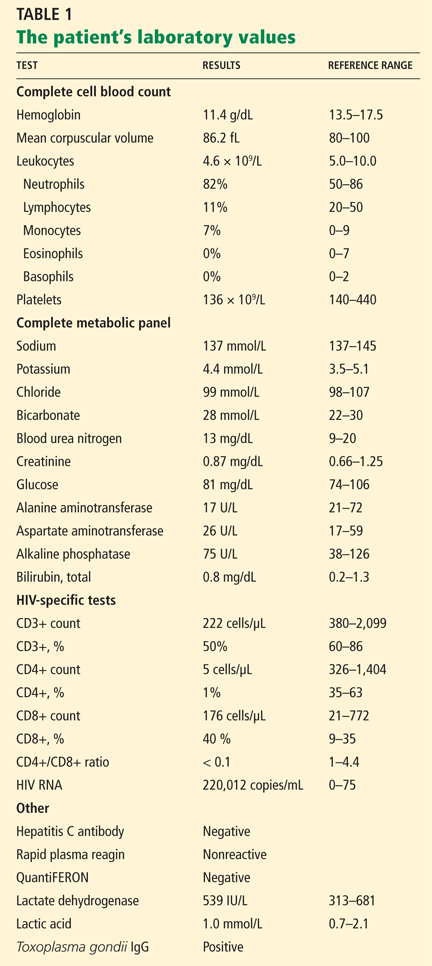
Initial blood tests (Table 1) showed a normal white blood cell count, normal results on a complete metabolic panel, and a lactate dehydrogenase level of 539 IU/L (reference range 313–618). His serum lactate level was within normal limits.
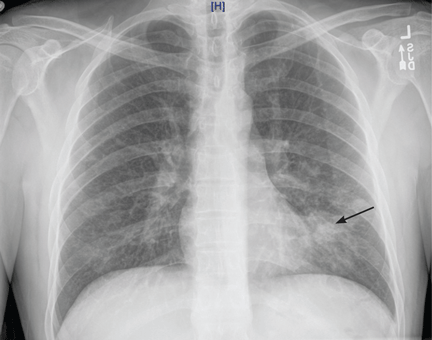
HIV-specific tests performed on the second day of hospitalization showed extreme immunosuppression, with a CD4 count of 5 cells/μL (normal 326–1,404 cells/μL).
WHICH ORGANISM IS CAUSING HIS LUNG INFECTION?
1. Which of the following organisms is the least likely to be associated with this patient’s condition?
- Mycobacterium tuberculosis
- Pneumocystis jirovecii
- Coccidioides immitis
- Candida albicans
- Streptococcus pneumoniae
- Cytomegalovirus
Bacterial, fungal, and viral lung infections are common in HIV-infected patients, especially if they are not on antiretroviral therapy and their CD4 lymphocyte counts are low. Clues to the cause can be derived from the history, physical examination, and general laboratory studies. For instance, knowing where the patient lives and where he has travelled recently provides insight into exposure to endemic infectious agents.
The complete blood cell count with differential white blood cell count can help narrow the differential diagnosis but rarely helps exclude a possibility. Neutrophilia is common in bacterial infections. Lymphocytosis can be seen in tuberculosis, in fungal and viral infections, and, rarely, in hematologic malignancies. Eosinophilia can be seen in acute retroviral syndrome, fungal and helminthic infections, adrenal insufficiency, autoimmune disease, and lymphoma.
A caveat to these clues is that in severely immunocompromised hosts, like this man, diagnoses should not be excluded without firm evidence. This patient has severe, active immunosuppression, and only one of the six answer choices above is not a possible causative agent: C albicans rarely causes lung infection, even in immunocompromised people.
Mycobacterium tuberculosis
Tuberculosis can be the first manifestation of HIV infection. It can occur at any CD4 count, but as the count decreases, the risk of dissemination increases.1 Classic symptoms are fever, night sweats, hemoptysis, and weight loss.
The CD4 count also affects the radiographic presentation. If the count is higher than 350 cells/μL, then infiltration of the upper lobe is likely; if it is lower than 200 cells/μL, then middle, lower, miliary, and extrapulmonary manifestations are likely.1,2 Cavitation is less common in HIV-infected patients, but mediastinal adenopathy is more common.1
Definitive diagnosis is via sputum examination, blood culture, nucleic acid amplification, or microscopic study of biopsy specimens of affected tissues to look for acid-fast bacilli.1
Interferon-gamma-release assays such as the QuantiFERON test (Cellestis, Valencia, CA) or a tuberculin skin test can be used to check for latent tuberculosis infection. These tests can also provide evidence of active infection in the appropriate clinical context.3
Interferon-gamma-release assays have several advantages over skin testing: they are more sensitive (76% to 80%) and specific (97%); they do not give false-positive results in people who previously received bacille Calmette-Guérin vaccine; they react only minimally to previous exposure to nontuberculous mycobacteria; and interpretation is not subject to interreader variability.4,5 However, concordance between skin testing and interferon-gamma-release assays is low. Therefore, either or both tests can be used if tuberculosis is strongly suspected, and a positive result on either test should prompt further workup.6,7
Of note, both tests may be affected by immunosuppression, making both susceptible to false-negative results as the CD4 count declines.3
In any case, a positive acid-fast bacillus smear, radiographic evidence of latent infection, or pulmonary symptoms should be presumed to represent active tuberculosis. In such a situation, directly observed treatment with the typical four-drug regimen—rifampin (Rifadin), isoniazid, pyrazinamide, and ethambutol (Myambutol)—is recommended while awaiting definitive results from culture or polymerase chain reaction (PCR) testing.1
Pneumocystis jirovecii
P jirovecii was previously known as P carinii, and P jirovecii pneumonia is an AIDS-defining illness. Most cases occur when the CD4 count falls below 200 cells/μL.1 Symptoms, including a nonproductive cough, develop insidiously over days to weeks.
Physical examination may reveal inspiratory crackles; however, half of the time the physical examination is nondiagnostic. Oral candidiasis is a common coinfection. The lactate dehydrogenase level may be elevated.1,8 Radiographs show bilateral interstitial infiltrates, and in 10% to 20% of patients lung cysts develop—hence the name of the organism.1 Pneumothorax in a patient with HIV should prompt a workup for P jirovecii pneumonia.9,10
No consensus exists for the diagnosis. However, if sputum examination is unrevealing but suspicion is high, then bronchoalveolar lavage can help.11–13
Trimethoprim-sulfamethoxazole for 21 days is the first-line treatment, with glucocorticoids added if the Pao2 is less than 70 mm Hg or if the alveolar-arterial oxygen gradient is greater than 35 mm Hg.1
Coccidioides species
Coccidioides infection is typically due to either C immitis or C posadasii.14 People living in or travelling to areas where it is endemic, such as the southwestern United States, Mexico, and Central and South America, are at higher risk.14
Typical signs and symptoms of this fungal infection include an influenza-like illness with fever, cough, adenopathy, and wasting, and when combined with erythema nodosum, erythema multiforme, arthralgia, or ocular involvement, this constellation is colloquially termed “valley fever.”15 Most HIV-infected patients who have CD4 counts higher than 250 cells/μL present with focal pneumonia, while lower counts predispose to disseminated disease.1,2,16
Findings on examination are nonspecific and depend on the various pulmonary manifestations, which include acute, chronic progressive, or diffuse pneumonia, nodules, or cavities.14 Eosinophilia may accompany the infection.15
The diagnosis can be made by finding the organisms on direct microscopic examination of involved tissues or secretions or on culture of clinical specimens.1,2,14 Serologic tests, antigen detection tests, or culture can be helpful if positive, but negative results do not rule out the diagnosis.1,2,14
A caveat about testing: if the pretest probability of infection is low, positive tests for immunoglobulin M (IgM) do not necessarily equal infection, and the IgM test should be followed up with confirmatory testing. Along the same lines, a high pretest probability should not be ignored if initial tests are negative, and patients in this situation should also undergo further evaluation.17
Therapy with an azole drug such as fluconazole (Diflucan) or one of the amphotericin B preparations should be started, depending on the severity of the disease.1,2,14,18
Candida albicans
C albicans is a rare cause of lung infection.19,20 It is, however, a common inhabitant of the upper airway tract, and pulmonary infection is usually the result of aspiration or hematogenous spread from either the gastrointestinal tract or an infected central venous catheter.20
The presentation is relatively nonspecific. Fever despite broad-spectrum antibacterial therapy is a major clue. Radiographic abnormalities usually are due to other causes, such as superimposed infections or pulmonary hemorrhage.21 Sputum culture is unreliable because of colonization. The definitive diagnosis is based on lung biopsy demonstrating organisms within the tissue.19,20,22
Therapy with a systemic antifungal agent is recommended.
Streptococcus pneumoniae
S pneumoniae is one of the most common bacterial causes of community-acquired pneumonia in people with or without HIV.23–25 Moreover, two or more episodes of bacterial pneumonia in 12 months can be an AIDS-defining condition in patients with a positive serologic test for HIV.16 Therefore, in patients with fever, cough, and pulmonary infiltrates on chest radiography, S pneumoniae must always be considered.
Urinary antigen testing has a relatively high positive predictive value (> 89%) and specificity (96%) for diagnosing S pneumoniae pneumonia.26 Blood and sputum cultures should be done not only to confirm the diagnosis, but also because the rates of bacteremia and drug resistance are higher with S pneumoniae infection in the HIV-infected.1
A combination of a beta-lactam and a macrolide or respiratory fluoroquinolone is the treatment of choice.1
Cytomegalovirus
Although influenza is the most common cause of viral pneumonia in HIV-infected people, cytomegalovirus is an opportunistic cause.2 This is usually a reactivation of latent infection rather than new infection.27 Typically, infections occur at CD4 counts lower than 50 cells/μL, with cough, dyspnea, and fever that last for 2 to 4 weeks.2
Crackles may be heard on lung examination. The lactate dehydrogenase level can be elevated, as in P jirovecii pneumonia.2 Radiography can show a wide range of nonspecific findings, from reticular and ground-glass opacities to alveolar or interstitial infiltrates to nodules.
The diagnosis of cytomegalovirus pneumonia is not always clear. Since HIV-infected patients typically shed the virus in their airways, bronchoalveolar lavage is not adequate because a positive finding does not necessarily mean the patient has active viral pneumonitis.27 For this reason, infection should be confirmed by biopsy demonstrating characteristic cytomegalovirus inclusions in lung tissue.2
Importantly, once cytomegalovirus pneumonia is confirmed, the patient should be screened for cytomegalovirus retinitis even if he or she has no visual symptoms, as cytomegalovirus pneumonitis is typically a part of a disseminated infection.1
Treatment with intravenous ganciclovir (Cytovene) is required.1
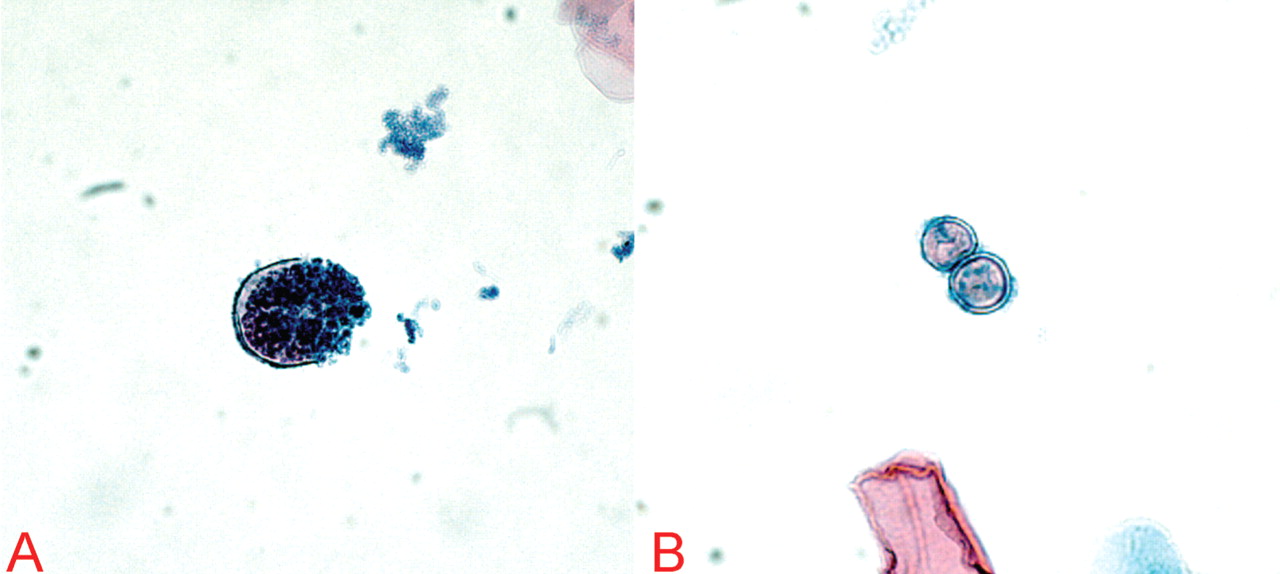
CASE CONTINUED: POSITIVE TESTS FOR COCCIDIOIDES
Our patient began empiric treatment for community-acquired pneumonia with intravenous ceftriaxone (Rocephin) and azithromycin (Zithromax).
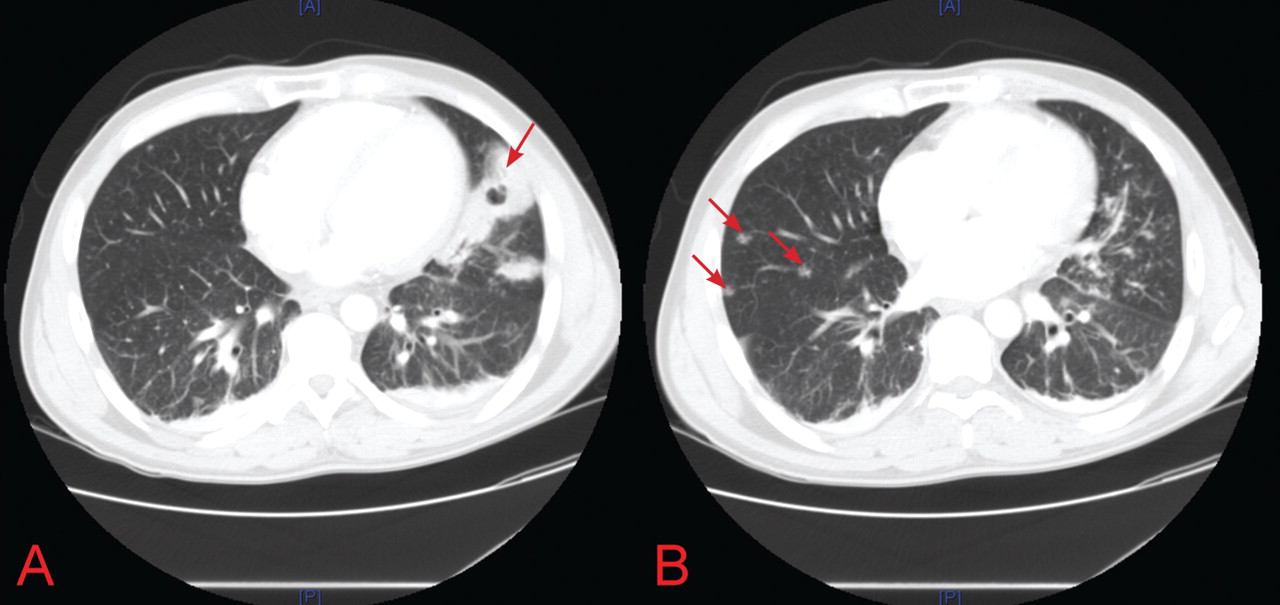
On the basis of these findings, the patient was immediately placed in negative pressure respiratory isolation and underwent induced sputum examinations for tuberculosis. Further tests for S pneumoniae, S aureus, Mycoplasma, Legionella, influenza, Pneumocystis, Cryptococcus, Histoplasma, and Coccidioides species were performed.
QuantiFERON testing was negative, and blood cultures were sterile. The first induced sputum examination was negative for acid-fast bacilli. PCR testing for mycobacterial DNA in the sputum was also negative.
Both silver and direct fluorescent antibody staining of the sputum were negative for Pneumocystis. On the basis of these findings and the patient’s lack of clinical improvement with trimethoprim-sulfamethoxazole, Pneumocystis infection was excluded.
THE PATIENT BEGINS TREATMENT
2. Which treatment is most appropriate for this patient?
- Posaconazole (Noxafil)
- Caspofungin (Cancidas) and surgery
- Fluconazole
- Voriconazole (Vfend) and surgery
- Amphotericin B

Solitary pulmonary cavities tend to be asymptomatic and do not require treatment, even if coccidioidal infection is microbiologically confirmed.
However, if there is pain, hemoptysis, or bacterial superinfection, antifungal therapy may result in improvement but not closure of the cavity.18 Therefore, in all cases of symptomatic coccidioidal pulmonary cavities, surgical resection is the only definitive treatment.
Coccidioidal cavities may rupture and cause pyopneumothorax, but this is an infrequent complication, and antifungal therapy combined with surgical decortication is the treatment of choice.18
Commonly prescribed antifungals include fluconazole and amphotericin B, the latter usually reserved for patients with significant hypoxia or rapid clinical deterioration.18 At this time, there are not enough clinical data to show that voriconazole or posaconazole is effective, and thus neither is approved for the treatment of coccidioidomycosis. Likewise, there have been no human trials of the efficacy of caspofungin against Coccidioides infection, although it has been shown to be active in mouse models.18
Our patient was started on oral fluconazole and observed for clinical improvement or, conversely, for signs of dissemination. After 2 days, he had markedly improved, and within 1 week he was almost back to his baseline level of health. Testing for all other infectious etiologies was unrevealing, and he was removed from negative pressure isolation.
However, as we mentioned above, his CD4 count was 5 cells/μL. We discussed the issue with the patient, and he said he was willing to comply with his treatment for both his Coccidioides and his HIV infection. After much deliberation, he said he was also willing to start and comply with prophylactic treatment for opportunistic infections.
PREVENTING OPPORTUNISTIC INFECTIONS IN HIV PATIENTS
3. Which of the following prophylactic regimens is most appropriate for this patient?
- Trimethoprim-sulfamethoxazole, atovaquone (Mepron), and azithromycin
- Trimethoprim-sulfamethoxazole and azithromycin
- Pentamidine (Nebupent), dapsone, and clarithromycin (Biaxin)
- Dapsone and clarithromycin
- Trimethoprim-sulfamethoxazole by itself
According to guidelines for the prevention of opportunistic diseases in patients with HIV, he needs primary prophylaxis against the following organisms: P jirovecii, Toxoplasma gondii, and Mycobacterium avium complex.1
The CD4 count dictates the appropriate time to start therapy. If the count is lower than 200 cells/μL or if the patient has oropharyngeal candidiasis regardless of the CD4 count, trimethoprim-sulfamethoxazole is indicated to prevent P jirovecii pneumonia. In those who cannot tolerate trimethoprim-sulfamethoxazole or who are allergic to it, dapsone, pentamidine, or atovaquone can be substituted.1
In patients seropositive for T gondii, a CD4 count lower than 100/μL indicates the need for prophylaxis.1 Prophylactic measures are similar to those for Pneumocystis. However, if the patient cannot tolerate trimethoprim-sulfamethoxazole, the recommended alternative is dapsone-pyrimethamine with leucovorin, which is also effective against Pneumocystis.1
Finally, if the CD4 count is lower than 50 cells/μL, prophylaxis against M avium complex is mandatory, with either azithromycin weekly or clarithromycin daily.1
Given our patient’s degree of immunosuppression, trimethoprim-sulfamethoxazole plus azithromycin is his most appropriate option.
Trimethoprim-sulfamethoxazole and azithromycin were added to his antimicrobial regimen before he was discharged. Two weeks later, he noted no side effects from any of the medications, he had no new symptoms, he was feeling well, and his cough had improved greatly. He did not have any signs of dissemination of his coccidioidal infection, and we concluded that the primary and only infection was located in the lungs.
DISSEMINATED COCCIDIOIDOMYCOSIS
4. Which of the following extrapulmonary sites is Coccidioides least likely to infect?
- Brain
- Skin
- Meninges
- Lymph nodes
- Bones
- Joints
Extrapulmonary coccidioidomycosis can involve almost any site. However, the most common sites of dissemination are the skin, lymph nodes, bones, and joints.14 The least likely site is the brain.
Central nervous system involvement
In the central nervous system, involvement is typically with the meninges, rather than frank involvement of the brain parenchyma.18,28,29 Although patients with HIV or those who are otherwise severely immunocompromised are at higher risk for coccidioidal meningitis, it is rare even in this population.30,31 Meningitis most commonly presents as headache, vomiting, meningismus, confusion, or diplopia.32,33
If neurologic findings are absent, experts do not generally recommend lumbar puncture because the incidence of meningeal involvement is low. When cerebrospinal fluid is obtained in an active case of coccidioidal meningitis, fluid analysis typically finds elevated protein, low glucose, and lymphocytic pleocytosis.1,32
Meningeal enhancement on CT or magnetic resonance imaging is common.34 The diagnosis is established by culture or serologic testing of cerebrospinal fluid (IgM titer, IgG titer, immunodiffusion, or complement fixation).14
Of note, cerebral infarction and hydrocephalus are feared complications and pose a serious risk of death in any patient.32,35 In these cases, treatment with antifungals is lifelong, regardless of immune system status.18
Skin involvement
Skin involvement is variable, consisting of nodules, verrucae, abscesses, or ulcerations.15,16 Hemorrhage from the skin is relatively common.36 From the skin, the infection can spread to the lymph nodes, leading to regional lymphadenopathy.14,15 Nodes can ulcerate, drain, or even become necrotic.
Bone and joint involvement
Once integrity of the blood vessels is disrupted, Coccidioides can spread via the blood to the bones or joints,14,15 causing osteomyelitis, septic arthritis, or synovitis. Subcutaneous abscesses or sinus tracts may subsequently develop.14,15
HOW LONG MUST HE BE TREATED?
On follow-up, the patient asked how long he needed to continue his antifungal regimen and if any other testing for his coccidioidal infection was necessary, since he was feeling better.
5. Which is the most appropriate response to the patient’s question?
- He can discontinue his antifungal drugs; no further testing is necessary
- He needs 14 more days of antifungal therapy and periodic serologic tests
- He needs 2.5 more months of antifungal therapy and monthly blood cultures
- He needs lifelong antifungal therapy and periodic urinary antigen levels
- He needs 5.5 more months of antifungal therapy; bronchoscopy with bronchoalveolar lavage at 1 year
How long to treat and how to monitor for coccidioidomycosis vary by patient.
Duration of therapy depends on symptoms and immune status
The severity of infection (Table 2) and the immune status are important factors that must be considered when tailoring a therapeutic regimen.
Immunocompetent patients without symptoms or with mild symptoms usually do not need therapy and are followed periodically for signs of improvement.14,18,29
Immunocompetent patients with severe symptoms typically receive 3 to 6 months of antifungal therapy.18
Immunocompromised patients (especially HIV-infected patients with CD4 counts < 250 cells/μL) need antifungal treatment, regardless of the severity of infection.14,18,29 In many cases, the type of infection will dictate the duration of therapy.
Diffuse pneumonia or extrapulmonary dissemination typically requires treatment for at least 1 year regardless of immune status.14,18 For those with HIV and diffuse pneumonia, dissemination, or meningitis, guidelines dictate that secondary prophylaxis be started after at least 1 year of therapy and improvement in clinical status; it should be continued indefinitely to prevent reactivation of latent infection.18
The guidelines say that in patients with higher CD4 counts (presumably > 250 cells/μL) and nonmeningeal coccidioidomycosis, providers may consider discontinuing secondary prophylaxis, as long as there is clinical evidence of improvement and control of the primary infection.18 However, many experts advocate continuing secondary prophylaxis regardless of the CD4 count, as the rates of relapse and dissemination are high.1,16,37
Monitoring
Regardless of the therapy chosen, disease monitoring every 2 to 4 months with clinical history and examination, radiography, and coccidioidal-specific testing is recommended for at least 1 year, and perhaps longer, to ensure complete resolution and to monitor for signs of dissemination.14,18
Which test to use is not clear. Serologic testing identifies antibodies (IgM or IgG) to coccidioidal antigens. IgM appears during the acute infection, and tests include immunodiffusion, latex agglutination, and enzymelinked immunoassays. The last two are highly sensitive but have a significant false-positive rate, and should be confirmed with the former if found to be positive.17,18 IgG appears weeks after the acute infection and can be evaluated with immunodiffusion or enzyme-linked immunoassay as well.
Keep in mind that these tests provide only qualitative results on the presence of these antibodies, not quantitative information. Furthermore, enzyme-linked immunoassay is not as accurate as immunodiffusion, which has a sensitivity in immunocompromised patients of only approximately 50%.38,39
For that reason, complement fixation titers are extremely helpful because they reflect the severity of infection, can be used to monitor the response to treatment, and can even provide insight into the prognosis.18 The sensitivity of this test in immunocompromised hosts is 60% to 70%.38 Titers can be checked to confirm the diagnosis and can be periodically monitored throughout the treatment course to ensure efficacy of therapy and to watch for reactivation of the infection.1 In fact, an initial complement fixation titer of 1:2 or 1:4 is associated with favorable outcomes, while a titer greater than 1:16 portends dissemination.18
The caveat to any serologic test (immunodiffusion, enzyme-linked immunoassay, and complement fixation) is that severely immunocompromised patients (as in our case) may not mount an immune response and may have falsely low titers even in the face of a severe infection, and therefore these tests may not be reliable.38 In these situations, urinary coccidioidal antigen detection assay (sensitivity 71%) or nucleic acid amplification of coccidioidal DNA (sensitivity 75%) may be of more help.40,41
Therefore, in the setting of HIV infection, an asymptomatic pulmonary cavity, and diffuse pulmonary involvement secondary to coccidioidal infection, lifelong antibiotics (treatment plus secondary prophylaxis) with periodic testing of urinary coccidioidal antigen levels is the best response to the patient’s question, given that his complement fixation titers were initially negative and antigen levels were positive.
CASE CONCLUDED
The patient continues to be followed for his HIV infection. He is undergoing serologic and urinary antigen testing for Coccidioides infection every 3 months in addition to his maintenance HIV testing. He is on chronic suppressive therapy with fluconazole. He has not had a recurrence of his Coccidioides infection, nor have there been any signs of dissemination.
CAVITARY LUNG LESIONS IN HIV PATIENTS
In patients with HIV, cavitary lung lesions on chest radiography can be due to a wide variety of etiologies that range from infection to malignancy. Historical clues, including environmental exposure, occupation, geographic residence, sick contacts, travel, or animal contact can be helpful in ordering subsequent confirmatory testing, especially in the case of infection.
Tuberculosis should be suspected, and appropriate isolation precautions should be taken until it is ruled out.
Laboratory testing, including the complete blood cell count with differential and CD4 count, provide ancillary data to narrow the differential diagnosis. For example, if the CD4 count is greater than 200 cells/μL, mycobacterial infection should be strongly suspected; however, lower CD4 counts should also prompt a search for opportunistic infections. In the appropriate clinical scenario, malignancies including Kaposi sarcoma, non-Hodgkin lymphoma, and bronchogenic carcinoma can be seen and should also be considered.
Nevertheless, the evaluation hinges on the sputum examination and CT scan of the chest to further characterize the cavity, surrounding lung parenchyma, lymph nodes, and potential fluid collections. Usually, further serologic tests and even bronchoscopy with bronchoalveolar lavage and transbronchial biopsy are required. Treatment should begin once the most likely diagnosis is established.
Coccidioidal pneumonia should be considered in all patients with immunodeficiency, including HIV patients, transplant recipients, those undergoing chemotherapy, and those with intrinsic immune system defects, especially if they have a history of exposure or if they are from an endemic region. Antifungal therapy should be initiated early, and dissemination must be ruled out. Suppressive therapy is mandatory for those with a severely compromised immune system, and serologic testing to ensure remission of the infection is needed. Patients who were previously exposed to Coccidioides or who vacationed or live in the southwestern United States (where it is prevalent) are at risk and may present with any number of symptoms.
- Kaplan JE, Benson C, Holmes KH, Brooks JT, Pau A, Masur H; Centers for Disease Control and Prevention (CDC). Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep 2009; 58:1–207.
- Huang L, Crothers K. HIV-associated opportunistic pneumonias. Respirology 2009; 14:474–485.
- Mazurek GH, Jereb J, Lobue P, Iademarco MF, Metchock B, Vernon A; Division of Tuberculosis Elimination, National Center for HIV, STD, and TB Prevention, Centers for Disease Control and Prevention (CDC). Guidelines for using the QuantiFERON-TB Gold test for detecting Mycobacterium tuberculosis infection, United States. MMWR Recomm Rep 2005; 54:49–55.
- Menzies D, Pai M, Comstock G. Meta-analysis: new tests for the diagnosis of latent tuberculosis infection: areas of uncertainty and recommendations for research. Ann Intern Med 2007; 146:340–354.
- Nahid P, Pai M, Hopewell PC. Advances in the diagnosis and treatment of tuberculosis. Proc Am Thorac Soc 2006; 3:103–110.
- Chapman AL, Munkanta M, Wilkinson KA, et al. Rapid detection of active and latent tuberculosis infection in HIV-positive individuals by enumeration of Mycobacterium tuberculosis-specific T cells. AIDS 2002; 16:2285–2293.
- Luetkemeyer AF, Charlebois ED, Flores LL, et al. Comparison of an interferon-gamma release assay with tuberculin skin testing in HIV-infected individuals. Am J Respir Crit Care Med 2007; 175:737–742.
- Zaman MK, White DA. Serum lactate dehydrogenase levels and Pneumocystis carinii pneumonia. Diagnostic and prognostic significance. Am Rev Respir Dis 1988; 137:796–800.
- Metersky ML, Colt HG, Olson LK, Shanks TG. AIDS-related spontaneous pneumothorax. Risk factors and treatment. Chest 1995; 108:946–951.
- Sepkowitz KA, Telzak EE, Gold JW, et al. Pneumothorax in AIDS. Ann Intern Med 1991; 114:455–459.
- Baughman RP, Dohn MN, Frame PT. The continuing utility of bronchoalveolar lavage to diagnose opportunistic infection in AIDS patients. Am J Med 1994; 97:515–522.
- Kovacs JA, Ng VL, Masur H, et al. Diagnosis of Pneumocystis carinii pneumonia: improved detection in sputum with use of monoclonal antibodies. N Engl J Med 1988; 318:589–593.
- Stover DE, Zaman MB, Hajdu SI, Lange M, Gold J, Armstrong D. Bronchoalveolar lavage in the diagnosis of diffuse pulmonary infiltrates in the immunosuppressed host. Ann Intern Med 1984; 101:1–7.
- Parish JM, Blair JE. Coccidioidomycosis. Mayo Clin Proc 2008; 83:343–348.
- Drutz DJ, Catanzaro A. Coccidioidomycosis. Part I. Am Rev Respir Dis 1978; 117:559–585.
- Bartlett JG, Gallant JE, Pham PA. Medical Management of HIV Infection. Durham, NC: Knowledge Source Solutions, LLC; 2009.
- Kuberski T, Herrig J, Pappagianis D. False-positive IgM serology in coccidioidomycosis. J Clin Microbiol 2010; 48:2047–2049.
- Galgiani JN, Ampel NM, Blair JE, et al; Infectious Diseases Society of America. Coccidioidomycosis. Clin Infect Dis 2005; 41:1217–1223.
- Kontoyiannis DP, Reddy BT, Torres HA, et al. Pulmonary candidiasis in patients with cancer: an autopsy study. Clin Infect Dis 2002; 34:400–403.
- Pappas PG, Kauffman CA, Andes D, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:503–535.
- Connolly JE, McAdams HP, Erasmus JJ, Rosado-de-Christenson ML. Opportunistic fungal pneumonia. J Thorac Imaging 1999; 14:51–62.
- Meersseman W, Lagrou K, Spriet I, et al. Significance of the isolation of Candida species from airway samples in critically ill patients: a prospective, autopsy study. Intensive Care Med 2009; 35:1526–1531.
- Miller RF, Foley NM, Kessel D, Jeffrey AA. Community acquired lobar pneumonia in patients with HIV infection and AIDS. Thorax 1994; 49:367–368.
- Polsky B, Gold JW, Whimbey E, et al. Bacterial pneumonia in patients with the acquired immunodeficiency syndrome. Ann Intern Med 1986; 104:38–41.
- Rimland D, Navin TR, Lennox JL, et al; Pulmonary Opportunistic Infection Study Group. Prospective study of etiologic agents of community-acquired pneumonia in patients with HIV infection. AIDS 2002; 16:85–95.
- Boulware DR, Daley CL, Merrifield C, Hopewell PC, Janoff EN. Rapid diagnosis of pneumococcal pneumonia among HIV-infected adults with urine antigen detection. J Infect 2007; 55:300–309.
- Salomon N, Perlman DC. Cytomegalovirus pneumonia. Semin Respir Infect 1999; 14:353–358.
- Chiller TM, Galgiani JN, Stevens DA. Coccidioidomycosis. Infect Dis Clin North Am 2003; 17:41–57.
- Drutz DJ, Catanzaro A. Coccidioidomycosis. Part II. Am Rev Respir Dis 1978; 117:727–771.
- Fish DG, Ampel NM, Galgiani JN, et al. Coccidioidomycosis during human immunodeficiency virus infection. A review of 77 patients. Medicine (Baltimore) 1990; 69:384–391.
- Mischel PS, Vinters HV. Coccidioidomycosis of the central nervous system: neuropathological and vasculopathic manifestations and clinical correlates. Clin Infect Dis 1995; 20:400–405.
- Johnson RH, Einstein HE. Coccidioidal meningitis. Clin Infect Dis 2006; 42:103–107.
- Vincent T, Galgiani JN, Huppert M, Salkin D. The natural history of coccidioidal meningitis: VA-Armed Forces cooperative studies, 1955–1958. Clin Infect Dis 1993; 16:247–254.
- Erly WK, Bellon RJ, Seeger JF, Carmody RF. MR imaging of acute coccidioidal meningitis. AJNR Am J Neuroradiol 1999; 20:509–514.
- Arsura EL, Johnson R, Penrose J, et al. Neuroimaging as a guide to predict outcomes for patients with coccidioidal meningitis. Clin Infect Dis 2005; 40:624–627.
- Tappero JW, Perkins BA, Wenger JD, Berger TG. Cutaneous manifestations of opportunistic infections in patients infected with human immunodeficiency virus. Clin Microbiol Rev 1995; 8:440–450.
- Catanzaro A, Galgiani JN, Levine BE, et al. Fluconazole in the treatment of chronic pulmonary and nonmeningeal disseminated coccidioidomycosis. NIAID Mycoses Study Group. Am J Med 1995; 98:249–256.
- Blair JE, Coakley B, Santelli AC, Hentz JG, Wengenack NL. Serologic testing for symptomatic coccidioidomycosis in immunocompetent and immunosuppressed hosts. Mycopathologia 2006; 162:317–324.
- Martins TB, Jaskowski TD, Mouritsen CL, Hill HR. Comparison of commercially available enzyme immunoassay with traditional serological tests for detection of antibodies to Coccidioides immitis. J Clin Microbiol 1995; 33:940–943.
- Vucicevic D, Blair JE, Binnicker MJ, et al. The utility of Coccidioides polymerase chain reaction testing in the clinical setting. Mycopathologia 2010; 170:345–351.
- Durkin M, Connolly P, Kuberski T, et al. Diagnosis of coccidioidomycosis with use of the Coccidioides antigen enzyme immunoassay. Clin Infect Dis 2008; 47:e69–e73.
A 37-year-old man presented to the emergency department with an 8-week history of a mildly productive cough and shortness of breath accompanied by high fevers, chills, and night sweats. He also had some nausea but no vomiting.
Four days earlier, he had been evaluated by his primary care physician, who prescribed a 14-day course of one double-strength trimethoprim-sulfamethoxazole tablet (Bactrim DS) every 12 hours for presumed acute bronchitis, but his symptoms did not improve.
He was unemployed, living in Arizona, married with children. He denied any use of tobacco, alcohol, or injection drugs. On further questioning, he disclosed that he had unintentionally lost 30 pounds over the past 2 to 3 months and had been feeling tired.
When asked about his medical history, he revealed that he had been diagnosed with human immunodeficiency virus (HIV) infection in 2008 and that recently he had not been taking his antiretroviral medication, a once-daily combination pill containing efavirenz, emtricitabine, and tenofovir (Atripla). He had no other significant medical history, and the only medication he was currently taking was the trimethoprim-sulfamethoxazole.
On examination, his temperature was 38.7°C (101.7°F), blood pressure 109/68 mm Hg, heart rate 60 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 100% while breathing supplemental oxygen via nasal cannula at 2 L/min. He did not appear seriously ill.
Initial blood tests (Table 1) showed a normal white blood cell count, normal results on a complete metabolic panel, and a lactate dehydrogenase level of 539 IU/L (reference range 313–618). His serum lactate level was within normal limits.
HIV-specific tests performed on the second day of hospitalization showed extreme immunosuppression, with a CD4 count of 5 cells/μL (normal 326–1,404 cells/μL).
WHICH ORGANISM IS CAUSING HIS LUNG INFECTION?
1. Which of the following organisms is the least likely to be associated with this patient’s condition?
- Mycobacterium tuberculosis
- Pneumocystis jirovecii
- Coccidioides immitis
- Candida albicans
- Streptococcus pneumoniae
- Cytomegalovirus
Bacterial, fungal, and viral lung infections are common in HIV-infected patients, especially if they are not on antiretroviral therapy and their CD4 lymphocyte counts are low. Clues to the cause can be derived from the history, physical examination, and general laboratory studies. For instance, knowing where the patient lives and where he has travelled recently provides insight into exposure to endemic infectious agents.
The complete blood cell count with differential white blood cell count can help narrow the differential diagnosis but rarely helps exclude a possibility. Neutrophilia is common in bacterial infections. Lymphocytosis can be seen in tuberculosis, in fungal and viral infections, and, rarely, in hematologic malignancies. Eosinophilia can be seen in acute retroviral syndrome, fungal and helminthic infections, adrenal insufficiency, autoimmune disease, and lymphoma.
A caveat to these clues is that in severely immunocompromised hosts, like this man, diagnoses should not be excluded without firm evidence. This patient has severe, active immunosuppression, and only one of the six answer choices above is not a possible causative agent: C albicans rarely causes lung infection, even in immunocompromised people.
Mycobacterium tuberculosis
Tuberculosis can be the first manifestation of HIV infection. It can occur at any CD4 count, but as the count decreases, the risk of dissemination increases.1 Classic symptoms are fever, night sweats, hemoptysis, and weight loss.
The CD4 count also affects the radiographic presentation. If the count is higher than 350 cells/μL, then infiltration of the upper lobe is likely; if it is lower than 200 cells/μL, then middle, lower, miliary, and extrapulmonary manifestations are likely.1,2 Cavitation is less common in HIV-infected patients, but mediastinal adenopathy is more common.1
Definitive diagnosis is via sputum examination, blood culture, nucleic acid amplification, or microscopic study of biopsy specimens of affected tissues to look for acid-fast bacilli.1
Interferon-gamma-release assays such as the QuantiFERON test (Cellestis, Valencia, CA) or a tuberculin skin test can be used to check for latent tuberculosis infection. These tests can also provide evidence of active infection in the appropriate clinical context.3
Interferon-gamma-release assays have several advantages over skin testing: they are more sensitive (76% to 80%) and specific (97%); they do not give false-positive results in people who previously received bacille Calmette-Guérin vaccine; they react only minimally to previous exposure to nontuberculous mycobacteria; and interpretation is not subject to interreader variability.4,5 However, concordance between skin testing and interferon-gamma-release assays is low. Therefore, either or both tests can be used if tuberculosis is strongly suspected, and a positive result on either test should prompt further workup.6,7
Of note, both tests may be affected by immunosuppression, making both susceptible to false-negative results as the CD4 count declines.3
In any case, a positive acid-fast bacillus smear, radiographic evidence of latent infection, or pulmonary symptoms should be presumed to represent active tuberculosis. In such a situation, directly observed treatment with the typical four-drug regimen—rifampin (Rifadin), isoniazid, pyrazinamide, and ethambutol (Myambutol)—is recommended while awaiting definitive results from culture or polymerase chain reaction (PCR) testing.1
Pneumocystis jirovecii
P jirovecii was previously known as P carinii, and P jirovecii pneumonia is an AIDS-defining illness. Most cases occur when the CD4 count falls below 200 cells/μL.1 Symptoms, including a nonproductive cough, develop insidiously over days to weeks.
Physical examination may reveal inspiratory crackles; however, half of the time the physical examination is nondiagnostic. Oral candidiasis is a common coinfection. The lactate dehydrogenase level may be elevated.1,8 Radiographs show bilateral interstitial infiltrates, and in 10% to 20% of patients lung cysts develop—hence the name of the organism.1 Pneumothorax in a patient with HIV should prompt a workup for P jirovecii pneumonia.9,10
No consensus exists for the diagnosis. However, if sputum examination is unrevealing but suspicion is high, then bronchoalveolar lavage can help.11–13
Trimethoprim-sulfamethoxazole for 21 days is the first-line treatment, with glucocorticoids added if the Pao2 is less than 70 mm Hg or if the alveolar-arterial oxygen gradient is greater than 35 mm Hg.1
Coccidioides species
Coccidioides infection is typically due to either C immitis or C posadasii.14 People living in or travelling to areas where it is endemic, such as the southwestern United States, Mexico, and Central and South America, are at higher risk.14
Typical signs and symptoms of this fungal infection include an influenza-like illness with fever, cough, adenopathy, and wasting, and when combined with erythema nodosum, erythema multiforme, arthralgia, or ocular involvement, this constellation is colloquially termed “valley fever.”15 Most HIV-infected patients who have CD4 counts higher than 250 cells/μL present with focal pneumonia, while lower counts predispose to disseminated disease.1,2,16
Findings on examination are nonspecific and depend on the various pulmonary manifestations, which include acute, chronic progressive, or diffuse pneumonia, nodules, or cavities.14 Eosinophilia may accompany the infection.15
The diagnosis can be made by finding the organisms on direct microscopic examination of involved tissues or secretions or on culture of clinical specimens.1,2,14 Serologic tests, antigen detection tests, or culture can be helpful if positive, but negative results do not rule out the diagnosis.1,2,14
A caveat about testing: if the pretest probability of infection is low, positive tests for immunoglobulin M (IgM) do not necessarily equal infection, and the IgM test should be followed up with confirmatory testing. Along the same lines, a high pretest probability should not be ignored if initial tests are negative, and patients in this situation should also undergo further evaluation.17
Therapy with an azole drug such as fluconazole (Diflucan) or one of the amphotericin B preparations should be started, depending on the severity of the disease.1,2,14,18
Candida albicans
C albicans is a rare cause of lung infection.19,20 It is, however, a common inhabitant of the upper airway tract, and pulmonary infection is usually the result of aspiration or hematogenous spread from either the gastrointestinal tract or an infected central venous catheter.20
The presentation is relatively nonspecific. Fever despite broad-spectrum antibacterial therapy is a major clue. Radiographic abnormalities usually are due to other causes, such as superimposed infections or pulmonary hemorrhage.21 Sputum culture is unreliable because of colonization. The definitive diagnosis is based on lung biopsy demonstrating organisms within the tissue.19,20,22
Therapy with a systemic antifungal agent is recommended.
Streptococcus pneumoniae
S pneumoniae is one of the most common bacterial causes of community-acquired pneumonia in people with or without HIV.23–25 Moreover, two or more episodes of bacterial pneumonia in 12 months can be an AIDS-defining condition in patients with a positive serologic test for HIV.16 Therefore, in patients with fever, cough, and pulmonary infiltrates on chest radiography, S pneumoniae must always be considered.
Urinary antigen testing has a relatively high positive predictive value (> 89%) and specificity (96%) for diagnosing S pneumoniae pneumonia.26 Blood and sputum cultures should be done not only to confirm the diagnosis, but also because the rates of bacteremia and drug resistance are higher with S pneumoniae infection in the HIV-infected.1
A combination of a beta-lactam and a macrolide or respiratory fluoroquinolone is the treatment of choice.1
Cytomegalovirus
Although influenza is the most common cause of viral pneumonia in HIV-infected people, cytomegalovirus is an opportunistic cause.2 This is usually a reactivation of latent infection rather than new infection.27 Typically, infections occur at CD4 counts lower than 50 cells/μL, with cough, dyspnea, and fever that last for 2 to 4 weeks.2
Crackles may be heard on lung examination. The lactate dehydrogenase level can be elevated, as in P jirovecii pneumonia.2 Radiography can show a wide range of nonspecific findings, from reticular and ground-glass opacities to alveolar or interstitial infiltrates to nodules.
The diagnosis of cytomegalovirus pneumonia is not always clear. Since HIV-infected patients typically shed the virus in their airways, bronchoalveolar lavage is not adequate because a positive finding does not necessarily mean the patient has active viral pneumonitis.27 For this reason, infection should be confirmed by biopsy demonstrating characteristic cytomegalovirus inclusions in lung tissue.2
Importantly, once cytomegalovirus pneumonia is confirmed, the patient should be screened for cytomegalovirus retinitis even if he or she has no visual symptoms, as cytomegalovirus pneumonitis is typically a part of a disseminated infection.1
Treatment with intravenous ganciclovir (Cytovene) is required.1
CASE CONTINUED: POSITIVE TESTS FOR COCCIDIOIDES
Our patient began empiric treatment for community-acquired pneumonia with intravenous ceftriaxone (Rocephin) and azithromycin (Zithromax).
On the basis of these findings, the patient was immediately placed in negative pressure respiratory isolation and underwent induced sputum examinations for tuberculosis. Further tests for S pneumoniae, S aureus, Mycoplasma, Legionella, influenza, Pneumocystis, Cryptococcus, Histoplasma, and Coccidioides species were performed.
QuantiFERON testing was negative, and blood cultures were sterile. The first induced sputum examination was negative for acid-fast bacilli. PCR testing for mycobacterial DNA in the sputum was also negative.
Both silver and direct fluorescent antibody staining of the sputum were negative for Pneumocystis. On the basis of these findings and the patient’s lack of clinical improvement with trimethoprim-sulfamethoxazole, Pneumocystis infection was excluded.
THE PATIENT BEGINS TREATMENT
2. Which treatment is most appropriate for this patient?
- Posaconazole (Noxafil)
- Caspofungin (Cancidas) and surgery
- Fluconazole
- Voriconazole (Vfend) and surgery
- Amphotericin B
Solitary pulmonary cavities tend to be asymptomatic and do not require treatment, even if coccidioidal infection is microbiologically confirmed.
However, if there is pain, hemoptysis, or bacterial superinfection, antifungal therapy may result in improvement but not closure of the cavity.18 Therefore, in all cases of symptomatic coccidioidal pulmonary cavities, surgical resection is the only definitive treatment.
Coccidioidal cavities may rupture and cause pyopneumothorax, but this is an infrequent complication, and antifungal therapy combined with surgical decortication is the treatment of choice.18
Commonly prescribed antifungals include fluconazole and amphotericin B, the latter usually reserved for patients with significant hypoxia or rapid clinical deterioration.18 At this time, there are not enough clinical data to show that voriconazole or posaconazole is effective, and thus neither is approved for the treatment of coccidioidomycosis. Likewise, there have been no human trials of the efficacy of caspofungin against Coccidioides infection, although it has been shown to be active in mouse models.18
Our patient was started on oral fluconazole and observed for clinical improvement or, conversely, for signs of dissemination. After 2 days, he had markedly improved, and within 1 week he was almost back to his baseline level of health. Testing for all other infectious etiologies was unrevealing, and he was removed from negative pressure isolation.
However, as we mentioned above, his CD4 count was 5 cells/μL. We discussed the issue with the patient, and he said he was willing to comply with his treatment for both his Coccidioides and his HIV infection. After much deliberation, he said he was also willing to start and comply with prophylactic treatment for opportunistic infections.
PREVENTING OPPORTUNISTIC INFECTIONS IN HIV PATIENTS
3. Which of the following prophylactic regimens is most appropriate for this patient?
- Trimethoprim-sulfamethoxazole, atovaquone (Mepron), and azithromycin
- Trimethoprim-sulfamethoxazole and azithromycin
- Pentamidine (Nebupent), dapsone, and clarithromycin (Biaxin)
- Dapsone and clarithromycin
- Trimethoprim-sulfamethoxazole by itself
According to guidelines for the prevention of opportunistic diseases in patients with HIV, he needs primary prophylaxis against the following organisms: P jirovecii, Toxoplasma gondii, and Mycobacterium avium complex.1
The CD4 count dictates the appropriate time to start therapy. If the count is lower than 200 cells/μL or if the patient has oropharyngeal candidiasis regardless of the CD4 count, trimethoprim-sulfamethoxazole is indicated to prevent P jirovecii pneumonia. In those who cannot tolerate trimethoprim-sulfamethoxazole or who are allergic to it, dapsone, pentamidine, or atovaquone can be substituted.1
In patients seropositive for T gondii, a CD4 count lower than 100/μL indicates the need for prophylaxis.1 Prophylactic measures are similar to those for Pneumocystis. However, if the patient cannot tolerate trimethoprim-sulfamethoxazole, the recommended alternative is dapsone-pyrimethamine with leucovorin, which is also effective against Pneumocystis.1
Finally, if the CD4 count is lower than 50 cells/μL, prophylaxis against M avium complex is mandatory, with either azithromycin weekly or clarithromycin daily.1
Given our patient’s degree of immunosuppression, trimethoprim-sulfamethoxazole plus azithromycin is his most appropriate option.
Trimethoprim-sulfamethoxazole and azithromycin were added to his antimicrobial regimen before he was discharged. Two weeks later, he noted no side effects from any of the medications, he had no new symptoms, he was feeling well, and his cough had improved greatly. He did not have any signs of dissemination of his coccidioidal infection, and we concluded that the primary and only infection was located in the lungs.
DISSEMINATED COCCIDIOIDOMYCOSIS
4. Which of the following extrapulmonary sites is Coccidioides least likely to infect?
- Brain
- Skin
- Meninges
- Lymph nodes
- Bones
- Joints
Extrapulmonary coccidioidomycosis can involve almost any site. However, the most common sites of dissemination are the skin, lymph nodes, bones, and joints.14 The least likely site is the brain.
Central nervous system involvement
In the central nervous system, involvement is typically with the meninges, rather than frank involvement of the brain parenchyma.18,28,29 Although patients with HIV or those who are otherwise severely immunocompromised are at higher risk for coccidioidal meningitis, it is rare even in this population.30,31 Meningitis most commonly presents as headache, vomiting, meningismus, confusion, or diplopia.32,33
If neurologic findings are absent, experts do not generally recommend lumbar puncture because the incidence of meningeal involvement is low. When cerebrospinal fluid is obtained in an active case of coccidioidal meningitis, fluid analysis typically finds elevated protein, low glucose, and lymphocytic pleocytosis.1,32
Meningeal enhancement on CT or magnetic resonance imaging is common.34 The diagnosis is established by culture or serologic testing of cerebrospinal fluid (IgM titer, IgG titer, immunodiffusion, or complement fixation).14
Of note, cerebral infarction and hydrocephalus are feared complications and pose a serious risk of death in any patient.32,35 In these cases, treatment with antifungals is lifelong, regardless of immune system status.18
Skin involvement
Skin involvement is variable, consisting of nodules, verrucae, abscesses, or ulcerations.15,16 Hemorrhage from the skin is relatively common.36 From the skin, the infection can spread to the lymph nodes, leading to regional lymphadenopathy.14,15 Nodes can ulcerate, drain, or even become necrotic.
Bone and joint involvement
Once integrity of the blood vessels is disrupted, Coccidioides can spread via the blood to the bones or joints,14,15 causing osteomyelitis, septic arthritis, or synovitis. Subcutaneous abscesses or sinus tracts may subsequently develop.14,15
HOW LONG MUST HE BE TREATED?
On follow-up, the patient asked how long he needed to continue his antifungal regimen and if any other testing for his coccidioidal infection was necessary, since he was feeling better.
5. Which is the most appropriate response to the patient’s question?
- He can discontinue his antifungal drugs; no further testing is necessary
- He needs 14 more days of antifungal therapy and periodic serologic tests
- He needs 2.5 more months of antifungal therapy and monthly blood cultures
- He needs lifelong antifungal therapy and periodic urinary antigen levels
- He needs 5.5 more months of antifungal therapy; bronchoscopy with bronchoalveolar lavage at 1 year
How long to treat and how to monitor for coccidioidomycosis vary by patient.
Duration of therapy depends on symptoms and immune status
The severity of infection (Table 2) and the immune status are important factors that must be considered when tailoring a therapeutic regimen.
Immunocompetent patients without symptoms or with mild symptoms usually do not need therapy and are followed periodically for signs of improvement.14,18,29
Immunocompetent patients with severe symptoms typically receive 3 to 6 months of antifungal therapy.18
Immunocompromised patients (especially HIV-infected patients with CD4 counts < 250 cells/μL) need antifungal treatment, regardless of the severity of infection.14,18,29 In many cases, the type of infection will dictate the duration of therapy.
Diffuse pneumonia or extrapulmonary dissemination typically requires treatment for at least 1 year regardless of immune status.14,18 For those with HIV and diffuse pneumonia, dissemination, or meningitis, guidelines dictate that secondary prophylaxis be started after at least 1 year of therapy and improvement in clinical status; it should be continued indefinitely to prevent reactivation of latent infection.18
The guidelines say that in patients with higher CD4 counts (presumably > 250 cells/μL) and nonmeningeal coccidioidomycosis, providers may consider discontinuing secondary prophylaxis, as long as there is clinical evidence of improvement and control of the primary infection.18 However, many experts advocate continuing secondary prophylaxis regardless of the CD4 count, as the rates of relapse and dissemination are high.1,16,37
Monitoring
Regardless of the therapy chosen, disease monitoring every 2 to 4 months with clinical history and examination, radiography, and coccidioidal-specific testing is recommended for at least 1 year, and perhaps longer, to ensure complete resolution and to monitor for signs of dissemination.14,18
Which test to use is not clear. Serologic testing identifies antibodies (IgM or IgG) to coccidioidal antigens. IgM appears during the acute infection, and tests include immunodiffusion, latex agglutination, and enzymelinked immunoassays. The last two are highly sensitive but have a significant false-positive rate, and should be confirmed with the former if found to be positive.17,18 IgG appears weeks after the acute infection and can be evaluated with immunodiffusion or enzyme-linked immunoassay as well.
Keep in mind that these tests provide only qualitative results on the presence of these antibodies, not quantitative information. Furthermore, enzyme-linked immunoassay is not as accurate as immunodiffusion, which has a sensitivity in immunocompromised patients of only approximately 50%.38,39
For that reason, complement fixation titers are extremely helpful because they reflect the severity of infection, can be used to monitor the response to treatment, and can even provide insight into the prognosis.18 The sensitivity of this test in immunocompromised hosts is 60% to 70%.38 Titers can be checked to confirm the diagnosis and can be periodically monitored throughout the treatment course to ensure efficacy of therapy and to watch for reactivation of the infection.1 In fact, an initial complement fixation titer of 1:2 or 1:4 is associated with favorable outcomes, while a titer greater than 1:16 portends dissemination.18
The caveat to any serologic test (immunodiffusion, enzyme-linked immunoassay, and complement fixation) is that severely immunocompromised patients (as in our case) may not mount an immune response and may have falsely low titers even in the face of a severe infection, and therefore these tests may not be reliable.38 In these situations, urinary coccidioidal antigen detection assay (sensitivity 71%) or nucleic acid amplification of coccidioidal DNA (sensitivity 75%) may be of more help.40,41
Therefore, in the setting of HIV infection, an asymptomatic pulmonary cavity, and diffuse pulmonary involvement secondary to coccidioidal infection, lifelong antibiotics (treatment plus secondary prophylaxis) with periodic testing of urinary coccidioidal antigen levels is the best response to the patient’s question, given that his complement fixation titers were initially negative and antigen levels were positive.
CASE CONCLUDED
The patient continues to be followed for his HIV infection. He is undergoing serologic and urinary antigen testing for Coccidioides infection every 3 months in addition to his maintenance HIV testing. He is on chronic suppressive therapy with fluconazole. He has not had a recurrence of his Coccidioides infection, nor have there been any signs of dissemination.
CAVITARY LUNG LESIONS IN HIV PATIENTS
In patients with HIV, cavitary lung lesions on chest radiography can be due to a wide variety of etiologies that range from infection to malignancy. Historical clues, including environmental exposure, occupation, geographic residence, sick contacts, travel, or animal contact can be helpful in ordering subsequent confirmatory testing, especially in the case of infection.
Tuberculosis should be suspected, and appropriate isolation precautions should be taken until it is ruled out.
Laboratory testing, including the complete blood cell count with differential and CD4 count, provide ancillary data to narrow the differential diagnosis. For example, if the CD4 count is greater than 200 cells/μL, mycobacterial infection should be strongly suspected; however, lower CD4 counts should also prompt a search for opportunistic infections. In the appropriate clinical scenario, malignancies including Kaposi sarcoma, non-Hodgkin lymphoma, and bronchogenic carcinoma can be seen and should also be considered.
Nevertheless, the evaluation hinges on the sputum examination and CT scan of the chest to further characterize the cavity, surrounding lung parenchyma, lymph nodes, and potential fluid collections. Usually, further serologic tests and even bronchoscopy with bronchoalveolar lavage and transbronchial biopsy are required. Treatment should begin once the most likely diagnosis is established.
Coccidioidal pneumonia should be considered in all patients with immunodeficiency, including HIV patients, transplant recipients, those undergoing chemotherapy, and those with intrinsic immune system defects, especially if they have a history of exposure or if they are from an endemic region. Antifungal therapy should be initiated early, and dissemination must be ruled out. Suppressive therapy is mandatory for those with a severely compromised immune system, and serologic testing to ensure remission of the infection is needed. Patients who were previously exposed to Coccidioides or who vacationed or live in the southwestern United States (where it is prevalent) are at risk and may present with any number of symptoms.
A 37-year-old man presented to the emergency department with an 8-week history of a mildly productive cough and shortness of breath accompanied by high fevers, chills, and night sweats. He also had some nausea but no vomiting.
Four days earlier, he had been evaluated by his primary care physician, who prescribed a 14-day course of one double-strength trimethoprim-sulfamethoxazole tablet (Bactrim DS) every 12 hours for presumed acute bronchitis, but his symptoms did not improve.
He was unemployed, living in Arizona, married with children. He denied any use of tobacco, alcohol, or injection drugs. On further questioning, he disclosed that he had unintentionally lost 30 pounds over the past 2 to 3 months and had been feeling tired.
When asked about his medical history, he revealed that he had been diagnosed with human immunodeficiency virus (HIV) infection in 2008 and that recently he had not been taking his antiretroviral medication, a once-daily combination pill containing efavirenz, emtricitabine, and tenofovir (Atripla). He had no other significant medical history, and the only medication he was currently taking was the trimethoprim-sulfamethoxazole.
On examination, his temperature was 38.7°C (101.7°F), blood pressure 109/68 mm Hg, heart rate 60 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 100% while breathing supplemental oxygen via nasal cannula at 2 L/min. He did not appear seriously ill.
Initial blood tests (Table 1) showed a normal white blood cell count, normal results on a complete metabolic panel, and a lactate dehydrogenase level of 539 IU/L (reference range 313–618). His serum lactate level was within normal limits.
HIV-specific tests performed on the second day of hospitalization showed extreme immunosuppression, with a CD4 count of 5 cells/μL (normal 326–1,404 cells/μL).
WHICH ORGANISM IS CAUSING HIS LUNG INFECTION?
1. Which of the following organisms is the least likely to be associated with this patient’s condition?
- Mycobacterium tuberculosis
- Pneumocystis jirovecii
- Coccidioides immitis
- Candida albicans
- Streptococcus pneumoniae
- Cytomegalovirus
Bacterial, fungal, and viral lung infections are common in HIV-infected patients, especially if they are not on antiretroviral therapy and their CD4 lymphocyte counts are low. Clues to the cause can be derived from the history, physical examination, and general laboratory studies. For instance, knowing where the patient lives and where he has travelled recently provides insight into exposure to endemic infectious agents.
The complete blood cell count with differential white blood cell count can help narrow the differential diagnosis but rarely helps exclude a possibility. Neutrophilia is common in bacterial infections. Lymphocytosis can be seen in tuberculosis, in fungal and viral infections, and, rarely, in hematologic malignancies. Eosinophilia can be seen in acute retroviral syndrome, fungal and helminthic infections, adrenal insufficiency, autoimmune disease, and lymphoma.
A caveat to these clues is that in severely immunocompromised hosts, like this man, diagnoses should not be excluded without firm evidence. This patient has severe, active immunosuppression, and only one of the six answer choices above is not a possible causative agent: C albicans rarely causes lung infection, even in immunocompromised people.
Mycobacterium tuberculosis
Tuberculosis can be the first manifestation of HIV infection. It can occur at any CD4 count, but as the count decreases, the risk of dissemination increases.1 Classic symptoms are fever, night sweats, hemoptysis, and weight loss.
The CD4 count also affects the radiographic presentation. If the count is higher than 350 cells/μL, then infiltration of the upper lobe is likely; if it is lower than 200 cells/μL, then middle, lower, miliary, and extrapulmonary manifestations are likely.1,2 Cavitation is less common in HIV-infected patients, but mediastinal adenopathy is more common.1
Definitive diagnosis is via sputum examination, blood culture, nucleic acid amplification, or microscopic study of biopsy specimens of affected tissues to look for acid-fast bacilli.1
Interferon-gamma-release assays such as the QuantiFERON test (Cellestis, Valencia, CA) or a tuberculin skin test can be used to check for latent tuberculosis infection. These tests can also provide evidence of active infection in the appropriate clinical context.3
Interferon-gamma-release assays have several advantages over skin testing: they are more sensitive (76% to 80%) and specific (97%); they do not give false-positive results in people who previously received bacille Calmette-Guérin vaccine; they react only minimally to previous exposure to nontuberculous mycobacteria; and interpretation is not subject to interreader variability.4,5 However, concordance between skin testing and interferon-gamma-release assays is low. Therefore, either or both tests can be used if tuberculosis is strongly suspected, and a positive result on either test should prompt further workup.6,7
Of note, both tests may be affected by immunosuppression, making both susceptible to false-negative results as the CD4 count declines.3
In any case, a positive acid-fast bacillus smear, radiographic evidence of latent infection, or pulmonary symptoms should be presumed to represent active tuberculosis. In such a situation, directly observed treatment with the typical four-drug regimen—rifampin (Rifadin), isoniazid, pyrazinamide, and ethambutol (Myambutol)—is recommended while awaiting definitive results from culture or polymerase chain reaction (PCR) testing.1
Pneumocystis jirovecii
P jirovecii was previously known as P carinii, and P jirovecii pneumonia is an AIDS-defining illness. Most cases occur when the CD4 count falls below 200 cells/μL.1 Symptoms, including a nonproductive cough, develop insidiously over days to weeks.
Physical examination may reveal inspiratory crackles; however, half of the time the physical examination is nondiagnostic. Oral candidiasis is a common coinfection. The lactate dehydrogenase level may be elevated.1,8 Radiographs show bilateral interstitial infiltrates, and in 10% to 20% of patients lung cysts develop—hence the name of the organism.1 Pneumothorax in a patient with HIV should prompt a workup for P jirovecii pneumonia.9,10
No consensus exists for the diagnosis. However, if sputum examination is unrevealing but suspicion is high, then bronchoalveolar lavage can help.11–13
Trimethoprim-sulfamethoxazole for 21 days is the first-line treatment, with glucocorticoids added if the Pao2 is less than 70 mm Hg or if the alveolar-arterial oxygen gradient is greater than 35 mm Hg.1
Coccidioides species
Coccidioides infection is typically due to either C immitis or C posadasii.14 People living in or travelling to areas where it is endemic, such as the southwestern United States, Mexico, and Central and South America, are at higher risk.14
Typical signs and symptoms of this fungal infection include an influenza-like illness with fever, cough, adenopathy, and wasting, and when combined with erythema nodosum, erythema multiforme, arthralgia, or ocular involvement, this constellation is colloquially termed “valley fever.”15 Most HIV-infected patients who have CD4 counts higher than 250 cells/μL present with focal pneumonia, while lower counts predispose to disseminated disease.1,2,16
Findings on examination are nonspecific and depend on the various pulmonary manifestations, which include acute, chronic progressive, or diffuse pneumonia, nodules, or cavities.14 Eosinophilia may accompany the infection.15
The diagnosis can be made by finding the organisms on direct microscopic examination of involved tissues or secretions or on culture of clinical specimens.1,2,14 Serologic tests, antigen detection tests, or culture can be helpful if positive, but negative results do not rule out the diagnosis.1,2,14
A caveat about testing: if the pretest probability of infection is low, positive tests for immunoglobulin M (IgM) do not necessarily equal infection, and the IgM test should be followed up with confirmatory testing. Along the same lines, a high pretest probability should not be ignored if initial tests are negative, and patients in this situation should also undergo further evaluation.17
Therapy with an azole drug such as fluconazole (Diflucan) or one of the amphotericin B preparations should be started, depending on the severity of the disease.1,2,14,18
Candida albicans
C albicans is a rare cause of lung infection.19,20 It is, however, a common inhabitant of the upper airway tract, and pulmonary infection is usually the result of aspiration or hematogenous spread from either the gastrointestinal tract or an infected central venous catheter.20
The presentation is relatively nonspecific. Fever despite broad-spectrum antibacterial therapy is a major clue. Radiographic abnormalities usually are due to other causes, such as superimposed infections or pulmonary hemorrhage.21 Sputum culture is unreliable because of colonization. The definitive diagnosis is based on lung biopsy demonstrating organisms within the tissue.19,20,22
Therapy with a systemic antifungal agent is recommended.
Streptococcus pneumoniae
S pneumoniae is one of the most common bacterial causes of community-acquired pneumonia in people with or without HIV.23–25 Moreover, two or more episodes of bacterial pneumonia in 12 months can be an AIDS-defining condition in patients with a positive serologic test for HIV.16 Therefore, in patients with fever, cough, and pulmonary infiltrates on chest radiography, S pneumoniae must always be considered.
Urinary antigen testing has a relatively high positive predictive value (> 89%) and specificity (96%) for diagnosing S pneumoniae pneumonia.26 Blood and sputum cultures should be done not only to confirm the diagnosis, but also because the rates of bacteremia and drug resistance are higher with S pneumoniae infection in the HIV-infected.1
A combination of a beta-lactam and a macrolide or respiratory fluoroquinolone is the treatment of choice.1
Cytomegalovirus
Although influenza is the most common cause of viral pneumonia in HIV-infected people, cytomegalovirus is an opportunistic cause.2 This is usually a reactivation of latent infection rather than new infection.27 Typically, infections occur at CD4 counts lower than 50 cells/μL, with cough, dyspnea, and fever that last for 2 to 4 weeks.2
Crackles may be heard on lung examination. The lactate dehydrogenase level can be elevated, as in P jirovecii pneumonia.2 Radiography can show a wide range of nonspecific findings, from reticular and ground-glass opacities to alveolar or interstitial infiltrates to nodules.
The diagnosis of cytomegalovirus pneumonia is not always clear. Since HIV-infected patients typically shed the virus in their airways, bronchoalveolar lavage is not adequate because a positive finding does not necessarily mean the patient has active viral pneumonitis.27 For this reason, infection should be confirmed by biopsy demonstrating characteristic cytomegalovirus inclusions in lung tissue.2
Importantly, once cytomegalovirus pneumonia is confirmed, the patient should be screened for cytomegalovirus retinitis even if he or she has no visual symptoms, as cytomegalovirus pneumonitis is typically a part of a disseminated infection.1
Treatment with intravenous ganciclovir (Cytovene) is required.1
CASE CONTINUED: POSITIVE TESTS FOR COCCIDIOIDES
Our patient began empiric treatment for community-acquired pneumonia with intravenous ceftriaxone (Rocephin) and azithromycin (Zithromax).
On the basis of these findings, the patient was immediately placed in negative pressure respiratory isolation and underwent induced sputum examinations for tuberculosis. Further tests for S pneumoniae, S aureus, Mycoplasma, Legionella, influenza, Pneumocystis, Cryptococcus, Histoplasma, and Coccidioides species were performed.
QuantiFERON testing was negative, and blood cultures were sterile. The first induced sputum examination was negative for acid-fast bacilli. PCR testing for mycobacterial DNA in the sputum was also negative.
Both silver and direct fluorescent antibody staining of the sputum were negative for Pneumocystis. On the basis of these findings and the patient’s lack of clinical improvement with trimethoprim-sulfamethoxazole, Pneumocystis infection was excluded.
THE PATIENT BEGINS TREATMENT
2. Which treatment is most appropriate for this patient?
- Posaconazole (Noxafil)
- Caspofungin (Cancidas) and surgery
- Fluconazole
- Voriconazole (Vfend) and surgery
- Amphotericin B
Solitary pulmonary cavities tend to be asymptomatic and do not require treatment, even if coccidioidal infection is microbiologically confirmed.
However, if there is pain, hemoptysis, or bacterial superinfection, antifungal therapy may result in improvement but not closure of the cavity.18 Therefore, in all cases of symptomatic coccidioidal pulmonary cavities, surgical resection is the only definitive treatment.
Coccidioidal cavities may rupture and cause pyopneumothorax, but this is an infrequent complication, and antifungal therapy combined with surgical decortication is the treatment of choice.18
Commonly prescribed antifungals include fluconazole and amphotericin B, the latter usually reserved for patients with significant hypoxia or rapid clinical deterioration.18 At this time, there are not enough clinical data to show that voriconazole or posaconazole is effective, and thus neither is approved for the treatment of coccidioidomycosis. Likewise, there have been no human trials of the efficacy of caspofungin against Coccidioides infection, although it has been shown to be active in mouse models.18
Our patient was started on oral fluconazole and observed for clinical improvement or, conversely, for signs of dissemination. After 2 days, he had markedly improved, and within 1 week he was almost back to his baseline level of health. Testing for all other infectious etiologies was unrevealing, and he was removed from negative pressure isolation.
However, as we mentioned above, his CD4 count was 5 cells/μL. We discussed the issue with the patient, and he said he was willing to comply with his treatment for both his Coccidioides and his HIV infection. After much deliberation, he said he was also willing to start and comply with prophylactic treatment for opportunistic infections.
PREVENTING OPPORTUNISTIC INFECTIONS IN HIV PATIENTS
3. Which of the following prophylactic regimens is most appropriate for this patient?
- Trimethoprim-sulfamethoxazole, atovaquone (Mepron), and azithromycin
- Trimethoprim-sulfamethoxazole and azithromycin
- Pentamidine (Nebupent), dapsone, and clarithromycin (Biaxin)
- Dapsone and clarithromycin
- Trimethoprim-sulfamethoxazole by itself
According to guidelines for the prevention of opportunistic diseases in patients with HIV, he needs primary prophylaxis against the following organisms: P jirovecii, Toxoplasma gondii, and Mycobacterium avium complex.1
The CD4 count dictates the appropriate time to start therapy. If the count is lower than 200 cells/μL or if the patient has oropharyngeal candidiasis regardless of the CD4 count, trimethoprim-sulfamethoxazole is indicated to prevent P jirovecii pneumonia. In those who cannot tolerate trimethoprim-sulfamethoxazole or who are allergic to it, dapsone, pentamidine, or atovaquone can be substituted.1
In patients seropositive for T gondii, a CD4 count lower than 100/μL indicates the need for prophylaxis.1 Prophylactic measures are similar to those for Pneumocystis. However, if the patient cannot tolerate trimethoprim-sulfamethoxazole, the recommended alternative is dapsone-pyrimethamine with leucovorin, which is also effective against Pneumocystis.1
Finally, if the CD4 count is lower than 50 cells/μL, prophylaxis against M avium complex is mandatory, with either azithromycin weekly or clarithromycin daily.1
Given our patient’s degree of immunosuppression, trimethoprim-sulfamethoxazole plus azithromycin is his most appropriate option.
Trimethoprim-sulfamethoxazole and azithromycin were added to his antimicrobial regimen before he was discharged. Two weeks later, he noted no side effects from any of the medications, he had no new symptoms, he was feeling well, and his cough had improved greatly. He did not have any signs of dissemination of his coccidioidal infection, and we concluded that the primary and only infection was located in the lungs.
DISSEMINATED COCCIDIOIDOMYCOSIS
4. Which of the following extrapulmonary sites is Coccidioides least likely to infect?
- Brain
- Skin
- Meninges
- Lymph nodes
- Bones
- Joints
Extrapulmonary coccidioidomycosis can involve almost any site. However, the most common sites of dissemination are the skin, lymph nodes, bones, and joints.14 The least likely site is the brain.
Central nervous system involvement
In the central nervous system, involvement is typically with the meninges, rather than frank involvement of the brain parenchyma.18,28,29 Although patients with HIV or those who are otherwise severely immunocompromised are at higher risk for coccidioidal meningitis, it is rare even in this population.30,31 Meningitis most commonly presents as headache, vomiting, meningismus, confusion, or diplopia.32,33
If neurologic findings are absent, experts do not generally recommend lumbar puncture because the incidence of meningeal involvement is low. When cerebrospinal fluid is obtained in an active case of coccidioidal meningitis, fluid analysis typically finds elevated protein, low glucose, and lymphocytic pleocytosis.1,32
Meningeal enhancement on CT or magnetic resonance imaging is common.34 The diagnosis is established by culture or serologic testing of cerebrospinal fluid (IgM titer, IgG titer, immunodiffusion, or complement fixation).14
Of note, cerebral infarction and hydrocephalus are feared complications and pose a serious risk of death in any patient.32,35 In these cases, treatment with antifungals is lifelong, regardless of immune system status.18
Skin involvement
Skin involvement is variable, consisting of nodules, verrucae, abscesses, or ulcerations.15,16 Hemorrhage from the skin is relatively common.36 From the skin, the infection can spread to the lymph nodes, leading to regional lymphadenopathy.14,15 Nodes can ulcerate, drain, or even become necrotic.
Bone and joint involvement
Once integrity of the blood vessels is disrupted, Coccidioides can spread via the blood to the bones or joints,14,15 causing osteomyelitis, septic arthritis, or synovitis. Subcutaneous abscesses or sinus tracts may subsequently develop.14,15
HOW LONG MUST HE BE TREATED?
On follow-up, the patient asked how long he needed to continue his antifungal regimen and if any other testing for his coccidioidal infection was necessary, since he was feeling better.
5. Which is the most appropriate response to the patient’s question?
- He can discontinue his antifungal drugs; no further testing is necessary
- He needs 14 more days of antifungal therapy and periodic serologic tests
- He needs 2.5 more months of antifungal therapy and monthly blood cultures
- He needs lifelong antifungal therapy and periodic urinary antigen levels
- He needs 5.5 more months of antifungal therapy; bronchoscopy with bronchoalveolar lavage at 1 year
How long to treat and how to monitor for coccidioidomycosis vary by patient.
Duration of therapy depends on symptoms and immune status
The severity of infection (Table 2) and the immune status are important factors that must be considered when tailoring a therapeutic regimen.
Immunocompetent patients without symptoms or with mild symptoms usually do not need therapy and are followed periodically for signs of improvement.14,18,29
Immunocompetent patients with severe symptoms typically receive 3 to 6 months of antifungal therapy.18
Immunocompromised patients (especially HIV-infected patients with CD4 counts < 250 cells/μL) need antifungal treatment, regardless of the severity of infection.14,18,29 In many cases, the type of infection will dictate the duration of therapy.
Diffuse pneumonia or extrapulmonary dissemination typically requires treatment for at least 1 year regardless of immune status.14,18 For those with HIV and diffuse pneumonia, dissemination, or meningitis, guidelines dictate that secondary prophylaxis be started after at least 1 year of therapy and improvement in clinical status; it should be continued indefinitely to prevent reactivation of latent infection.18
The guidelines say that in patients with higher CD4 counts (presumably > 250 cells/μL) and nonmeningeal coccidioidomycosis, providers may consider discontinuing secondary prophylaxis, as long as there is clinical evidence of improvement and control of the primary infection.18 However, many experts advocate continuing secondary prophylaxis regardless of the CD4 count, as the rates of relapse and dissemination are high.1,16,37
Monitoring
Regardless of the therapy chosen, disease monitoring every 2 to 4 months with clinical history and examination, radiography, and coccidioidal-specific testing is recommended for at least 1 year, and perhaps longer, to ensure complete resolution and to monitor for signs of dissemination.14,18
Which test to use is not clear. Serologic testing identifies antibodies (IgM or IgG) to coccidioidal antigens. IgM appears during the acute infection, and tests include immunodiffusion, latex agglutination, and enzymelinked immunoassays. The last two are highly sensitive but have a significant false-positive rate, and should be confirmed with the former if found to be positive.17,18 IgG appears weeks after the acute infection and can be evaluated with immunodiffusion or enzyme-linked immunoassay as well.
Keep in mind that these tests provide only qualitative results on the presence of these antibodies, not quantitative information. Furthermore, enzyme-linked immunoassay is not as accurate as immunodiffusion, which has a sensitivity in immunocompromised patients of only approximately 50%.38,39
For that reason, complement fixation titers are extremely helpful because they reflect the severity of infection, can be used to monitor the response to treatment, and can even provide insight into the prognosis.18 The sensitivity of this test in immunocompromised hosts is 60% to 70%.38 Titers can be checked to confirm the diagnosis and can be periodically monitored throughout the treatment course to ensure efficacy of therapy and to watch for reactivation of the infection.1 In fact, an initial complement fixation titer of 1:2 or 1:4 is associated with favorable outcomes, while a titer greater than 1:16 portends dissemination.18
The caveat to any serologic test (immunodiffusion, enzyme-linked immunoassay, and complement fixation) is that severely immunocompromised patients (as in our case) may not mount an immune response and may have falsely low titers even in the face of a severe infection, and therefore these tests may not be reliable.38 In these situations, urinary coccidioidal antigen detection assay (sensitivity 71%) or nucleic acid amplification of coccidioidal DNA (sensitivity 75%) may be of more help.40,41
Therefore, in the setting of HIV infection, an asymptomatic pulmonary cavity, and diffuse pulmonary involvement secondary to coccidioidal infection, lifelong antibiotics (treatment plus secondary prophylaxis) with periodic testing of urinary coccidioidal antigen levels is the best response to the patient’s question, given that his complement fixation titers were initially negative and antigen levels were positive.
CASE CONCLUDED
The patient continues to be followed for his HIV infection. He is undergoing serologic and urinary antigen testing for Coccidioides infection every 3 months in addition to his maintenance HIV testing. He is on chronic suppressive therapy with fluconazole. He has not had a recurrence of his Coccidioides infection, nor have there been any signs of dissemination.
CAVITARY LUNG LESIONS IN HIV PATIENTS
In patients with HIV, cavitary lung lesions on chest radiography can be due to a wide variety of etiologies that range from infection to malignancy. Historical clues, including environmental exposure, occupation, geographic residence, sick contacts, travel, or animal contact can be helpful in ordering subsequent confirmatory testing, especially in the case of infection.
Tuberculosis should be suspected, and appropriate isolation precautions should be taken until it is ruled out.
Laboratory testing, including the complete blood cell count with differential and CD4 count, provide ancillary data to narrow the differential diagnosis. For example, if the CD4 count is greater than 200 cells/μL, mycobacterial infection should be strongly suspected; however, lower CD4 counts should also prompt a search for opportunistic infections. In the appropriate clinical scenario, malignancies including Kaposi sarcoma, non-Hodgkin lymphoma, and bronchogenic carcinoma can be seen and should also be considered.
Nevertheless, the evaluation hinges on the sputum examination and CT scan of the chest to further characterize the cavity, surrounding lung parenchyma, lymph nodes, and potential fluid collections. Usually, further serologic tests and even bronchoscopy with bronchoalveolar lavage and transbronchial biopsy are required. Treatment should begin once the most likely diagnosis is established.
Coccidioidal pneumonia should be considered in all patients with immunodeficiency, including HIV patients, transplant recipients, those undergoing chemotherapy, and those with intrinsic immune system defects, especially if they have a history of exposure or if they are from an endemic region. Antifungal therapy should be initiated early, and dissemination must be ruled out. Suppressive therapy is mandatory for those with a severely compromised immune system, and serologic testing to ensure remission of the infection is needed. Patients who were previously exposed to Coccidioides or who vacationed or live in the southwestern United States (where it is prevalent) are at risk and may present with any number of symptoms.
- Kaplan JE, Benson C, Holmes KH, Brooks JT, Pau A, Masur H; Centers for Disease Control and Prevention (CDC). Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep 2009; 58:1–207.
- Huang L, Crothers K. HIV-associated opportunistic pneumonias. Respirology 2009; 14:474–485.
- Mazurek GH, Jereb J, Lobue P, Iademarco MF, Metchock B, Vernon A; Division of Tuberculosis Elimination, National Center for HIV, STD, and TB Prevention, Centers for Disease Control and Prevention (CDC). Guidelines for using the QuantiFERON-TB Gold test for detecting Mycobacterium tuberculosis infection, United States. MMWR Recomm Rep 2005; 54:49–55.
- Menzies D, Pai M, Comstock G. Meta-analysis: new tests for the diagnosis of latent tuberculosis infection: areas of uncertainty and recommendations for research. Ann Intern Med 2007; 146:340–354.
- Nahid P, Pai M, Hopewell PC. Advances in the diagnosis and treatment of tuberculosis. Proc Am Thorac Soc 2006; 3:103–110.
- Chapman AL, Munkanta M, Wilkinson KA, et al. Rapid detection of active and latent tuberculosis infection in HIV-positive individuals by enumeration of Mycobacterium tuberculosis-specific T cells. AIDS 2002; 16:2285–2293.
- Luetkemeyer AF, Charlebois ED, Flores LL, et al. Comparison of an interferon-gamma release assay with tuberculin skin testing in HIV-infected individuals. Am J Respir Crit Care Med 2007; 175:737–742.
- Zaman MK, White DA. Serum lactate dehydrogenase levels and Pneumocystis carinii pneumonia. Diagnostic and prognostic significance. Am Rev Respir Dis 1988; 137:796–800.
- Metersky ML, Colt HG, Olson LK, Shanks TG. AIDS-related spontaneous pneumothorax. Risk factors and treatment. Chest 1995; 108:946–951.
- Sepkowitz KA, Telzak EE, Gold JW, et al. Pneumothorax in AIDS. Ann Intern Med 1991; 114:455–459.
- Baughman RP, Dohn MN, Frame PT. The continuing utility of bronchoalveolar lavage to diagnose opportunistic infection in AIDS patients. Am J Med 1994; 97:515–522.
- Kovacs JA, Ng VL, Masur H, et al. Diagnosis of Pneumocystis carinii pneumonia: improved detection in sputum with use of monoclonal antibodies. N Engl J Med 1988; 318:589–593.
- Stover DE, Zaman MB, Hajdu SI, Lange M, Gold J, Armstrong D. Bronchoalveolar lavage in the diagnosis of diffuse pulmonary infiltrates in the immunosuppressed host. Ann Intern Med 1984; 101:1–7.
- Parish JM, Blair JE. Coccidioidomycosis. Mayo Clin Proc 2008; 83:343–348.
- Drutz DJ, Catanzaro A. Coccidioidomycosis. Part I. Am Rev Respir Dis 1978; 117:559–585.
- Bartlett JG, Gallant JE, Pham PA. Medical Management of HIV Infection. Durham, NC: Knowledge Source Solutions, LLC; 2009.
- Kuberski T, Herrig J, Pappagianis D. False-positive IgM serology in coccidioidomycosis. J Clin Microbiol 2010; 48:2047–2049.
- Galgiani JN, Ampel NM, Blair JE, et al; Infectious Diseases Society of America. Coccidioidomycosis. Clin Infect Dis 2005; 41:1217–1223.
- Kontoyiannis DP, Reddy BT, Torres HA, et al. Pulmonary candidiasis in patients with cancer: an autopsy study. Clin Infect Dis 2002; 34:400–403.
- Pappas PG, Kauffman CA, Andes D, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:503–535.
- Connolly JE, McAdams HP, Erasmus JJ, Rosado-de-Christenson ML. Opportunistic fungal pneumonia. J Thorac Imaging 1999; 14:51–62.
- Meersseman W, Lagrou K, Spriet I, et al. Significance of the isolation of Candida species from airway samples in critically ill patients: a prospective, autopsy study. Intensive Care Med 2009; 35:1526–1531.
- Miller RF, Foley NM, Kessel D, Jeffrey AA. Community acquired lobar pneumonia in patients with HIV infection and AIDS. Thorax 1994; 49:367–368.
- Polsky B, Gold JW, Whimbey E, et al. Bacterial pneumonia in patients with the acquired immunodeficiency syndrome. Ann Intern Med 1986; 104:38–41.
- Rimland D, Navin TR, Lennox JL, et al; Pulmonary Opportunistic Infection Study Group. Prospective study of etiologic agents of community-acquired pneumonia in patients with HIV infection. AIDS 2002; 16:85–95.
- Boulware DR, Daley CL, Merrifield C, Hopewell PC, Janoff EN. Rapid diagnosis of pneumococcal pneumonia among HIV-infected adults with urine antigen detection. J Infect 2007; 55:300–309.
- Salomon N, Perlman DC. Cytomegalovirus pneumonia. Semin Respir Infect 1999; 14:353–358.
- Chiller TM, Galgiani JN, Stevens DA. Coccidioidomycosis. Infect Dis Clin North Am 2003; 17:41–57.
- Drutz DJ, Catanzaro A. Coccidioidomycosis. Part II. Am Rev Respir Dis 1978; 117:727–771.
- Fish DG, Ampel NM, Galgiani JN, et al. Coccidioidomycosis during human immunodeficiency virus infection. A review of 77 patients. Medicine (Baltimore) 1990; 69:384–391.
- Mischel PS, Vinters HV. Coccidioidomycosis of the central nervous system: neuropathological and vasculopathic manifestations and clinical correlates. Clin Infect Dis 1995; 20:400–405.
- Johnson RH, Einstein HE. Coccidioidal meningitis. Clin Infect Dis 2006; 42:103–107.
- Vincent T, Galgiani JN, Huppert M, Salkin D. The natural history of coccidioidal meningitis: VA-Armed Forces cooperative studies, 1955–1958. Clin Infect Dis 1993; 16:247–254.
- Erly WK, Bellon RJ, Seeger JF, Carmody RF. MR imaging of acute coccidioidal meningitis. AJNR Am J Neuroradiol 1999; 20:509–514.
- Arsura EL, Johnson R, Penrose J, et al. Neuroimaging as a guide to predict outcomes for patients with coccidioidal meningitis. Clin Infect Dis 2005; 40:624–627.
- Tappero JW, Perkins BA, Wenger JD, Berger TG. Cutaneous manifestations of opportunistic infections in patients infected with human immunodeficiency virus. Clin Microbiol Rev 1995; 8:440–450.
- Catanzaro A, Galgiani JN, Levine BE, et al. Fluconazole in the treatment of chronic pulmonary and nonmeningeal disseminated coccidioidomycosis. NIAID Mycoses Study Group. Am J Med 1995; 98:249–256.
- Blair JE, Coakley B, Santelli AC, Hentz JG, Wengenack NL. Serologic testing for symptomatic coccidioidomycosis in immunocompetent and immunosuppressed hosts. Mycopathologia 2006; 162:317–324.
- Martins TB, Jaskowski TD, Mouritsen CL, Hill HR. Comparison of commercially available enzyme immunoassay with traditional serological tests for detection of antibodies to Coccidioides immitis. J Clin Microbiol 1995; 33:940–943.
- Vucicevic D, Blair JE, Binnicker MJ, et al. The utility of Coccidioides polymerase chain reaction testing in the clinical setting. Mycopathologia 2010; 170:345–351.
- Durkin M, Connolly P, Kuberski T, et al. Diagnosis of coccidioidomycosis with use of the Coccidioides antigen enzyme immunoassay. Clin Infect Dis 2008; 47:e69–e73.
- Kaplan JE, Benson C, Holmes KH, Brooks JT, Pau A, Masur H; Centers for Disease Control and Prevention (CDC). Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep 2009; 58:1–207.
- Huang L, Crothers K. HIV-associated opportunistic pneumonias. Respirology 2009; 14:474–485.
- Mazurek GH, Jereb J, Lobue P, Iademarco MF, Metchock B, Vernon A; Division of Tuberculosis Elimination, National Center for HIV, STD, and TB Prevention, Centers for Disease Control and Prevention (CDC). Guidelines for using the QuantiFERON-TB Gold test for detecting Mycobacterium tuberculosis infection, United States. MMWR Recomm Rep 2005; 54:49–55.
- Menzies D, Pai M, Comstock G. Meta-analysis: new tests for the diagnosis of latent tuberculosis infection: areas of uncertainty and recommendations for research. Ann Intern Med 2007; 146:340–354.
- Nahid P, Pai M, Hopewell PC. Advances in the diagnosis and treatment of tuberculosis. Proc Am Thorac Soc 2006; 3:103–110.
- Chapman AL, Munkanta M, Wilkinson KA, et al. Rapid detection of active and latent tuberculosis infection in HIV-positive individuals by enumeration of Mycobacterium tuberculosis-specific T cells. AIDS 2002; 16:2285–2293.
- Luetkemeyer AF, Charlebois ED, Flores LL, et al. Comparison of an interferon-gamma release assay with tuberculin skin testing in HIV-infected individuals. Am J Respir Crit Care Med 2007; 175:737–742.
- Zaman MK, White DA. Serum lactate dehydrogenase levels and Pneumocystis carinii pneumonia. Diagnostic and prognostic significance. Am Rev Respir Dis 1988; 137:796–800.
- Metersky ML, Colt HG, Olson LK, Shanks TG. AIDS-related spontaneous pneumothorax. Risk factors and treatment. Chest 1995; 108:946–951.
- Sepkowitz KA, Telzak EE, Gold JW, et al. Pneumothorax in AIDS. Ann Intern Med 1991; 114:455–459.
- Baughman RP, Dohn MN, Frame PT. The continuing utility of bronchoalveolar lavage to diagnose opportunistic infection in AIDS patients. Am J Med 1994; 97:515–522.
- Kovacs JA, Ng VL, Masur H, et al. Diagnosis of Pneumocystis carinii pneumonia: improved detection in sputum with use of monoclonal antibodies. N Engl J Med 1988; 318:589–593.
- Stover DE, Zaman MB, Hajdu SI, Lange M, Gold J, Armstrong D. Bronchoalveolar lavage in the diagnosis of diffuse pulmonary infiltrates in the immunosuppressed host. Ann Intern Med 1984; 101:1–7.
- Parish JM, Blair JE. Coccidioidomycosis. Mayo Clin Proc 2008; 83:343–348.
- Drutz DJ, Catanzaro A. Coccidioidomycosis. Part I. Am Rev Respir Dis 1978; 117:559–585.
- Bartlett JG, Gallant JE, Pham PA. Medical Management of HIV Infection. Durham, NC: Knowledge Source Solutions, LLC; 2009.
- Kuberski T, Herrig J, Pappagianis D. False-positive IgM serology in coccidioidomycosis. J Clin Microbiol 2010; 48:2047–2049.
- Galgiani JN, Ampel NM, Blair JE, et al; Infectious Diseases Society of America. Coccidioidomycosis. Clin Infect Dis 2005; 41:1217–1223.
- Kontoyiannis DP, Reddy BT, Torres HA, et al. Pulmonary candidiasis in patients with cancer: an autopsy study. Clin Infect Dis 2002; 34:400–403.
- Pappas PG, Kauffman CA, Andes D, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis 2009; 48:503–535.
- Connolly JE, McAdams HP, Erasmus JJ, Rosado-de-Christenson ML. Opportunistic fungal pneumonia. J Thorac Imaging 1999; 14:51–62.
- Meersseman W, Lagrou K, Spriet I, et al. Significance of the isolation of Candida species from airway samples in critically ill patients: a prospective, autopsy study. Intensive Care Med 2009; 35:1526–1531.
- Miller RF, Foley NM, Kessel D, Jeffrey AA. Community acquired lobar pneumonia in patients with HIV infection and AIDS. Thorax 1994; 49:367–368.
- Polsky B, Gold JW, Whimbey E, et al. Bacterial pneumonia in patients with the acquired immunodeficiency syndrome. Ann Intern Med 1986; 104:38–41.
- Rimland D, Navin TR, Lennox JL, et al; Pulmonary Opportunistic Infection Study Group. Prospective study of etiologic agents of community-acquired pneumonia in patients with HIV infection. AIDS 2002; 16:85–95.
- Boulware DR, Daley CL, Merrifield C, Hopewell PC, Janoff EN. Rapid diagnosis of pneumococcal pneumonia among HIV-infected adults with urine antigen detection. J Infect 2007; 55:300–309.
- Salomon N, Perlman DC. Cytomegalovirus pneumonia. Semin Respir Infect 1999; 14:353–358.
- Chiller TM, Galgiani JN, Stevens DA. Coccidioidomycosis. Infect Dis Clin North Am 2003; 17:41–57.
- Drutz DJ, Catanzaro A. Coccidioidomycosis. Part II. Am Rev Respir Dis 1978; 117:727–771.
- Fish DG, Ampel NM, Galgiani JN, et al. Coccidioidomycosis during human immunodeficiency virus infection. A review of 77 patients. Medicine (Baltimore) 1990; 69:384–391.
- Mischel PS, Vinters HV. Coccidioidomycosis of the central nervous system: neuropathological and vasculopathic manifestations and clinical correlates. Clin Infect Dis 1995; 20:400–405.
- Johnson RH, Einstein HE. Coccidioidal meningitis. Clin Infect Dis 2006; 42:103–107.
- Vincent T, Galgiani JN, Huppert M, Salkin D. The natural history of coccidioidal meningitis: VA-Armed Forces cooperative studies, 1955–1958. Clin Infect Dis 1993; 16:247–254.
- Erly WK, Bellon RJ, Seeger JF, Carmody RF. MR imaging of acute coccidioidal meningitis. AJNR Am J Neuroradiol 1999; 20:509–514.
- Arsura EL, Johnson R, Penrose J, et al. Neuroimaging as a guide to predict outcomes for patients with coccidioidal meningitis. Clin Infect Dis 2005; 40:624–627.
- Tappero JW, Perkins BA, Wenger JD, Berger TG. Cutaneous manifestations of opportunistic infections in patients infected with human immunodeficiency virus. Clin Microbiol Rev 1995; 8:440–450.
- Catanzaro A, Galgiani JN, Levine BE, et al. Fluconazole in the treatment of chronic pulmonary and nonmeningeal disseminated coccidioidomycosis. NIAID Mycoses Study Group. Am J Med 1995; 98:249–256.
- Blair JE, Coakley B, Santelli AC, Hentz JG, Wengenack NL. Serologic testing for symptomatic coccidioidomycosis in immunocompetent and immunosuppressed hosts. Mycopathologia 2006; 162:317–324.
- Martins TB, Jaskowski TD, Mouritsen CL, Hill HR. Comparison of commercially available enzyme immunoassay with traditional serological tests for detection of antibodies to Coccidioides immitis. J Clin Microbiol 1995; 33:940–943.
- Vucicevic D, Blair JE, Binnicker MJ, et al. The utility of Coccidioides polymerase chain reaction testing in the clinical setting. Mycopathologia 2010; 170:345–351.
- Durkin M, Connolly P, Kuberski T, et al. Diagnosis of coccidioidomycosis with use of the Coccidioides antigen enzyme immunoassay. Clin Infect Dis 2008; 47:e69–e73.
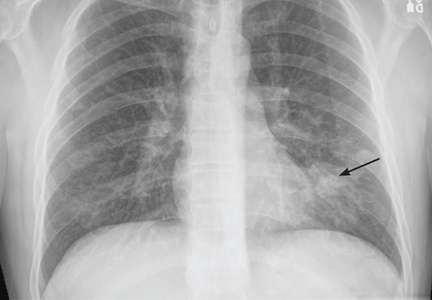
New and future therapies for lupus nephritis
Treatment for lupus nephritis has changed dramatically in recent years. Only 10 years ago, rheumatologists and nephrologists, whether specializing in adult or pediatric medicine, treated lupus nephritis with a similar regimen of monthly intravenous cyclophosphamide (Cytoxan) and glucocorticoids. Although the regimen is effective, side effects such as infection, hair loss, and infertility were extremely common.
Effective but very toxic therapy is common in autoimmune diseases. In the last decade, clinical trials have shown that less toxic drugs are as effective for treating lupus nephritis. This article will review new developments in therapy for lupus nephritis, which can be viewed as a prototype for other fields of medicine.
DEMOGRAPHICS ARE IMPORTANT
Although numerous factors have prognostic value in lupus nephritis (eg, serum creatinine, proteinuria, renal biopsy findings), the most important to consider when designing and interpreting studies are race and socioeconomic variables.
A retrospective study in Miami, FL,1 evaluated 213 patients with lupus nephritis, of whom 47% were Hispanic, 44% African American, and 20% white. At baseline, African Americans had higher blood pressure, higher serum creatinine levels, and lower household income. After 6 years, African Americans fared the worst in terms of doubling of serum creatinine, developing end-stage renal disease, and death; whites had the best outcomes, and Hispanics were in between. Low income was found to be a significant risk factor, independent of racial background.
In a similar retrospective study in New York City in 128 patients (43% white, 40% Hispanic, and 17% African American) with proliferative lupus nephritis,2 disease was much more likely to progress to renal failure over 10 years in patients living in a poor neighborhood, even after adjustment for race.
We need to keep in mind that racial and socioeconomic factors correlate with disease severity when we design and interpret studies of lupus nephritis. Study groups must be carefully balanced with patients of similar racial and socioeconomic profiles. Study findings must be interpreted with caution; for example, whether results from a study from China are applicable to an African American with lupus nephritis in New York City is unclear.
OLDER STANDARD THERAPY: EFFECTIVE BUT TOXIC
The last large National Institutes of Health study that involved only cyclophosphamide and a glucocorticoid was published in 2001,3 with 21 patients receiving cyclophosphamide alone and 20 patients receiving cyclophosphamide plus methylprednisolone. Although lupus nephritis improved, serious side effects occurred in one-third to one-half of patients in each group and included hypertension, hyperlipidemia, valvular heart disease, avascular necrosis, premature menopause, and major infections, including herpes zoster.
Less cyclophosphamide works just as well
The multicenter, prospective Euro-Lupus Nephritis Trial4 randomized 90 patients with proliferative lupus nephritis to receive either standard high-dose intravenous (IV) cyclophosphamide therapy (six monthly pulses and two quarterly pulses, with doses increasing according to the white blood cell count) or low-dose IV cyclophosphamide therapy (six pulses every 2 weeks at a fixed dose of 500 mg). Both regimens were followed by azathioprine (Imuran).
At 4 years, the two treatment groups were not significantly different in terms of treatment failure, remission rates, serum creatinine levels, 24-hour proteinuria, and freedom from renal flares. However, the rates of side effects were significantly different, with more patients in the low-dosage group free of severe infection.
One problem with this study is whether it is applicable to an American lupus nephritis population, since 84% of the patients were white. Since this study, others indicate that this regimen is probably also safe and effective for different racial groups in the United States.
At 10-year follow-up,5 both treatment groups still had identical excellent rates of freedom from end-stage renal disease. Serum creatinine and 24-hour proteinuria were also at excellent levels and identical in both groups. Nearly three quarters of patients still needed glucocorticoid therapy and more than half still needed immunosuppressive therapy, but the rates were not statistically significantly different between the treatment groups.
The cumulative dose of cyclophosphamide was 9.5 g in the standard-treatment group and 5.5 g in the low-dose group. This difference in exposure could make a tremendous difference to patients, not only for immediate side effects such as early menopause and infections, but for the risk of cancer in later decades.
This study showed clearly that low-dose cyclophosphamide is an option for induction therapy. Drawbacks of the study were that the population was mostly white and that patients had only moderately severe disease.
Low-dose cyclophosphamide has largely replaced the older National Institutes of Health regimen, although during the last decade drug therapy has undergone more changes.
MYCOPHENOLATE AND AZATHIOPRINE: ALTERNATIVES TO CYCLOPHOSPHAMIDE
In a Chinese study, mycophenolate was better than cyclophosphamide for induction
In a study in Hong Kong, Chan et al6 randomized 42 patients with severe lupus nephritis to receive either mycophenolate mofetil (available in the United States as CellCept; 2 g/day for 6 months, then 1 g/day for 6 months) or oral cyclophosphamide (2.5 mg/kg per day for 6 months) followed by azathioprine (1.5–2.0 mg/kg per day) for 6 months. Both groups also received prednisolone during the year.
At the end of the first year, the two groups were not significantly different in their rates of complete remission, partial remission, and relapse. The rate of infection, although not significantly different, was higher in the cyclophosphamide group (33% vs 19%). Two patients (10%) died in the cyclophosphamide group, but the difference in mortality rates was not statistically significant.
Nearly 5 years later,7 rates of chronic renal failure and relapse were still statistically the same in the two groups. Infections were fewer in the mycophenolate group (13% vs 40%, P = .013). The rate of amenorrhea was 36% in the cyclophosphamide group and only 4% in the mycophenolate group (P = .004). Four patients in the cyclophosphamide group and none in the mycophenolate group reached the composite end point of end-stage renal failure or death (P = .062).
This study appeared to offer a new option with equal efficacy and fewer side effects than standard therapy. However, its applicability to non-Chinese populations remained to be shown.
In a US study, mycophenolate or azathioprine was better than cyclophosphamide as maintenance
In a study in Miami,8 59 patients with lupus nephritis were given standard induction therapy with IV cyclophosphamide plus glucocorticoids for 6 months, then randomly assigned to one of three maintenance therapies for 1 to 3 years: IV injections of cyclophosphamide every 3 months (standard therapy), oral azathioprine, or oral mycophenolate. The population was 93% female, their average age was 33 years, and nearly half were African American, with many of the others being Hispanic. Patients tended to have severe disease, with nearly two-thirds having nephrotic syndrome.
After 6 years, there had been more deaths in the cyclophosphamide group than in the azathioprine group (P = .02) and in the mycophenolate group, although the latter difference was not statistically significant (P = .11). The combined rate of death and chronic renal failure was significantly higher with cyclophosphamide than with either of the oral agents. The cyclophosphamide group also had the highest relapse rate during the maintenance phase.
The differences in side effects were even more dramatic. Amenorrhea affected 32% of patients in the cyclophosphamide group, and only 7% and 6% in the azathioprine and mycophenolate groups, respectively. Rates of infections were 68% in the cyclophosphamide group and 28% and 21% in the azathioprine and mycophenolate groups, respectively. Patients given cyclophosphamide had 13 hospital days per patient per year, while the other groups each had only 1.
This study showed that maintenance therapy with oral azathioprine or mycophenolate was more effective and had fewer adverse effects than standard IV cyclophosphamide therapy. As a result of this study, oral agents for maintenance therapy became the new standard, but the question remained whether oral agents could safely be used for induction.
In a US study, mycophenolate was better than cyclophosphamide for induction
In a noninferiority study, Ginzler et al9 randomized 140 patients with severe lupus nephritis to receive either monthly IV cyclophosphamide or oral mycophenolate as induction therapy for 6 months. Adjunctive care with glucocorticoids was given in both groups. The study population was from 18 US academic centers and was predominantly female, and more than half were African American.
After 24 weeks, 22.5% of the mycophenolate patients were in complete remission by very strict criteria vs only 4% of those given cyclophosphamide (P = .005). The trend for partial remissions was also in favor of mycophenolate, although the difference was not statistically significant. The rate of complete and partial remissions, a prespecified end point, was significantly higher in the mycophenolate group. Although the study was trying to evaluate equivalency, it actually showed superiority for mycophenolate induction therapy.
Serum creatinine levels declined in both groups, but more in the mycophenolate group by 24 weeks. Urinary protein levels fell the same amount in both groups. At 3 years, the groups were statistically equivalent in terms of renal flares, renal failures, and deaths. However, the study groups were small, and the mycophenolate group did have a better trend for both renal failure (N = 4 vs 7) and deaths (N = 4 vs 8).
Mycophenolate also had fewer side effects, including infection, although again the numbers were too small to show statistical significance. The exception was diarrhea (N = 15 in the mycophenolate group vs 2 in the cyclophosphamide group).
A drawback of the study is that it was designed as a crossover study: a patient for whom therapy was failing after 3 months could switch to the other group, introducing potential confounding. Other problems involved the small population size and the question of whether results from patients in the United States were applicable to others worldwide.
In a worldwide study, mycophenolate was at least equivalent to cyclophosphamide for induction
The Aspreva Lupus Management Study (ALMS)10 used a similar design with 370 patients worldwide (United States, China, South America, and Europe) in one of the largest trials ever conducted in lupus nephritis. Patients were randomized to 6 months of induction therapy with either IV cyclophosphamide or oral mycophenolate but could not cross over.
At 6 months, response rates were identical between the two groups, with response defined as a combination of specific improvement in proteinuria, serum creatinine, and hematuria (50%–55%). In terms of individual renal and nonrenal variables, both groups appeared identical.
However, the side effect profiles differed between the two groups. As expected for mycophenolate, diarrhea was the most common side effect (occurring in 28% vs 12% in the cyclophosphamide group). Nausea and vomiting were more common with cyclophosphamide (45% and 37% respectively vs 14% and 13% in the mycophenolate group). Cyclophosphamide also caused hair loss in 35%, vs 10% in the mycophenolate group.
There were 14 deaths overall, which is a very low number considering the patients’ severity of illness, and it indicates the better results now achieved with therapy. The mortality rate was higher in the mycophenolate group (5% vs 3%), but the difference was not statistically significant. Six of the nine deaths with mycophenolate were from the same center in China, and none were from Europe or the United States. In summary, the study did not show that mycophenolate was superior to IV cyclophosphamide for induction therapy, but that they were equivalent in efficacy with different side effect profiles.
Membranous nephropathy: Mycophenolate vs cyclophosphamide
Less evidence is available about treatment for membranous disease, which is characterized by heavy proteinuria and the nephrotic syndrome but usually does not progress to renal failure. Radhakrishnan et al11 combined data from the trial by Ginzler et al9 and the ALMS trial10 and found 84 patients with pure membranous lupus, who were equally divided between the treatment groups receiving IV cyclophosphamide and mycophenolate. Consistent with the larger group’s data, mycophenolate and cyclophosphamide performed similarly in terms of efficacy, but there was a slightly higher rate of side effects with cyclophosphamide.
Maintenance therapy: Mycophenolate superior to azathioprine
The ALMS Maintenance Trial12 evaluated maintenance therapy in the same worldwide population that was studied for induction therapy. Of the 370 patients involved in the induction phase that compared IV cyclophosphamide and oral mycophenolate, 227 responded sufficiently to be rerandomized in a controlled, double-blinded trial of 36 months of maintenance therapy with corticosteroids and either mycophenolate (1 g twice daily) or azathioprine (2 mg/kg per day).
In intention-to-treat analysis, the time to treatment failure (ie, doubling of the serum creatinine level, progressing to renal failure, or death) was significantly shorter in the azathioprine group (P = .003). Every individual end point—end-stage renal disease, renal flares, doubling of serum creatinine, rescue immunosuppression required—was in favor of mycophenolate maintenance. At 3 years, the completion rate was 63% with mycophenolate and 49% with azathioprine. Serious adverse events and withdrawals because of adverse events were more common in the azathioprine group.
In summary, mycophenolate was superior to azathioprine in maintaining renal response and in preventing relapse in patients with active lupus nephritis who responded to induction therapy with either mycophenolate or IV cyclophosphamide. Mycophenolate was found to be superior regardless of initial induction treatment, race, or region and was confirmed by all key secondary end points.
Only one of the 227 patients died during the 3 years—from an auto accident. Again, this indicates the dramatically improved survival today compared with a decade ago.
RITUXIMAB: PROMISING BUT UNPROVEN
Rituximab (Rituxan) was originally approved to treat tumors, then rheumatoid arthritis, and most recently vasculitis. Evidence thus far is mixed regarding its use as a treatment for lupus nephritis. Although randomized clinical trials have not found it to be superior to standard regimens, there are many signs that it may be effective.
Rituximab in uncontrolled studies
Terrier et al13 analyzed prospective data from 136 patients with systemic lupus erythematosus, most of whom had renal disease, from the French Autoimmunity and Rituximab registry. Response occurred in 71% of patients using rituximab, with no difference found between patients receiving rituximab monotherapy and those concomitantly receiving immunosuppressive agents.
Melander et al14 retrospectively studied 19 women and 1 man who had been treated with rituximab for severe lupus nephritis and followed for at least 1 year. Three patients had concurrent therapy with cyclophosphamide, and 10 patients continued rituximab as maintenance therapy; 12 patients had lupus nephritis that had been refractory to standard treatment, and 6 had relapsing disease.
At a median follow-up of 22 months, 12 patients (60%) had achieved complete or partial renal remission.
Condon et al15 treated 21 patients who had severe lupus nephritis with two doses of rituximab and IV methylprednisolone 2 weeks apart, then maintenance therapy with mycophenolate without any oral steroids. At a mean follow-up of 35 months ( ± 14 months), 16 (76%) were in complete remission, with a mean time to remission of 12 months. Two (9.5%) achieved partial remission. The rate of toxicity was low.
Thus, rituximab appears promising in uncontrolled studies.
Placebo-controlled trials fail to prove rituximab effective
LUNAR trial. On the other hand, the largest placebo-controlled trial to evaluate rituximab in patients with proliferative lupus nephritis, the Lupus Nephritis Assessment With Rituximab (LUNAR) trial16 found differences in favor of rituximab, but none reached statistical significance. The trial randomized 140 patients to receive either mycophenolate plus periodic rituximab infusions or mycophenolate plus placebo infusions for 1 year. All patients received the same dosage of glucocorticoids, which was tapered over the year.
At the end of 1 year, the groups were not statistically different in terms of complete renal response and partial renal response. Rituximab appeared less likely to produce no response, but the difference was not statistically significant.
African Americans appeared to have a higher response rate to rituximab (70% in the rituximab group achieved a response vs 45% in the control group), but again, the difference did not reach statistical significance, and the total study population of African Americans was only 40.
Rituximab did have a statistically significant positive effect on two serologic markers at 1 year: levels of anti-dsDNA fell faster and complement rose faster. In addition, rates of adverse and serious adverse events were similar between the two groups, with no new or unexpected “safety signals.”
This study can be interpreted in a number of ways. The number of patients may have been too small to show significance and the follow-up may have been too short. On the other hand, it may simply not be effective to add rituximab to a full dose of mycophenolate and steroids, an already good treatment.
EXPLORER trial. Similarly, for patients with lupus without nephritis, the Exploratory Phase II/III SLE Evaluation of Rituximab (EXPLORER) trial17 also tested rituximab against a background of an effective therapeutic regimen and found no additional benefit. This study had design problems similar to those of the LUNAR trial.
Rituximab as rescue therapy
The evidence so far indicates that rituximab may have a role as rescue therapy for refractory or relapsing disease. Rituximab must be used with other therapies, but maintenance corticosteroid therapy is not necessary. Its role as a first-line agent in induction therapy for lupus nephritis remains unclear, although it may have an important role for nonwhites. In general, it has been well tolerated. Until a large randomized trial indicates otherwise, it should not be used as a first-line therapy.
The US Food and Drug Administration (FDA) sent out a warning about the danger of progressive multifocal leukoencephalopathy as an adverse effect of rituximab and of mycophenolate, but this does not appear to be a major concern for most patients and is only likely to occur in those who have been over-immunosuppressed for many years.
MULTITARGET THERAPY
The concept of using multiple drugs simultaneously—such as mycophenolate, steroids, and rituximab—is increasingly being tried. Multi-target therapy appears to offer the advantages of combining different modes of action with better results, and it offers fewer side effects because dosages of each individual drug can be lower when combined with other immunosuppressives.
Bao et al18 in China randomly assigned 40 patients with diffuse proliferative and membranous nephritis to 6 to 9 months of induction treatment with either multitarget therapy (mycophenolate, tacrolimus [Prograf], and glucocorticoids) or IV cyclophosphamide. More complete remissions occurred in the multitarget therapy group, both at 6 months (50% vs 5%) and at 9 months (65% vs 15%). Most adverse events were less frequent in the multitarget therapy group, although three patients (15%) in the multitarget therapy group developed new-onset hypertension vs none in the cyclophosphamide group.
NEW MEDICATIONS
Entirely new classes of drugs are being developed with immunomodulatory effects, including tolerance molecules, cytokine blockers, inhibitors of human B lymphocyte stimulator, and costimulatory blockers.
Belimumab offers small improvement for lupus
Belimumab (Benlysta) is a human monoclonal antibody that inhibits the biologic activity of human B lymphocyte stimulator; it has recently been approved by the FDA for lupus nephritis. In a worldwide study,19 867 patients with systemic lupus erythematosus were randomized to receive either belimumab (1 mg/kg or 10 mg/kg) or placebo.
The primary end point was the reduction of disease activity by a scoring system (SELENA-SLEDAI) that incorporated multiple features of lupus, including arthritis, vasculitis, proteinuria, rash, and others. Patients in the belimumab group had better outcomes, but the results were not dramatic. Because the drug is so expensive (about $25,000 per year) and the improvement offered is only incremental, this drug will not likely change the treatment of lupus very much.
Moreover, patients with lupus nephritis were not included in the study, but a new study is being planned to do so. Improvement is harder to demonstrate in lupus nephritis than in rheumatoid arthritis and systemic lupus erythematosus: significant changes in creatinine levels and 24-hour urinary protein must be achieved, rather than more qualitative signs and symptoms of joint pain, rash, and feeling better. Although belimumab is still unproven for lupus nephritis, it might be worth trying for patients failing other therapy.
Laquinimod: A promising experimental drug
Laquinimod is an oral immunomodulatory drug with a number of effects, including down-regulating major histocompatability complex II, chemokines, and adhesion-related molecules related to inflammation. It has been studied in more than 2,500 patients with multiple sclerosis. Pilot studies are now being done for its use for lupus nephritis. If it shows promise, a large randomized, controlled trial will be conducted.
Abatacept is in clinical trials
Abatacept (Orencia), a costimulation blocker, is undergoing clinical trials in lupus nephritis. Results should be available shortly.
INDIVIDUALIZE THERAPY
This past decade has seen such an increase in options to treat lupus nephritis that therapy can now be individualized.
Choosing IV cyclophosphamide vs mycophenolate
As a result of recent trials, doctors in the United States are increasingly using mycophenolate as the first-line drug for lupus nephritis. In Europe, however, many are choosing the shorter regimen of IV cyclophosphamide because of the results of the Euro-Lupus study.
Nowadays, I tend to use IV cyclophosphamide as the first-line drug only for patients with severe crescenteric glomerulonephritis or a very high serum creatinine level. In such cases, there is more experience with cyclophosphamide, and such severe disease does not lend itself to the luxury of trying out different therapies sequentially. If such a severely ill patient insists that a future pregnancy is very important, an alternative therapy of mycophenolate plus rituximab should be considered. I prefer mycophenolate for induction and maintenance therapy in most patients.
Dosing and formulation considerations for mycophenolate
Large dosages of mycophenolate are much better tolerated when broken up throughout the day. A patient who cannot tolerate 1 g twice daily may be able to tolerate 500 mg four times a day. The formulation can also make a difference. Some patients tolerate sustained-release mycophenolate (Myfortic) better than CellCept, and vice versa.
For patients who cannot tolerate mycophenolate, azathioprine is an acceptable alternative. In addition, for a patient who is already doing well on azathioprine, there is no need to change to mycophenolate.
Long maintenance therapy now acceptable
The ALMS Maintenance Trial12 found 3 years of maintenance therapy to be safe and effective. Such a long maintenance period is increasingly viewed as important, especially for patients in their teens and 20s, as it allows them to live a normal life, ie, to finish their education, get married, and become settled socially. Whether 5 years of maintenance therapy or even 10 years is advisable is still unknown.
Treatment during pregnancy
Neither mycophenolate nor azathioprine is recommended during pregnancy, although their effects are unknown. Because we have much more renal transplant experience with azathioprine during pregnancy, I recommend either switching from mycophenolate to azathioprine or trying to stop medication altogether if the patient has been well controlled.
- Contreras G, Lenz O, Pardo V, et al. Outcomes in African Americans and Hispanics with lupus nephritis. Kidney Int 2006; 69:1846–1851.
- Barr RG, Seliger S, Appel GB, et al. Prognosis in proliferative lupus nephritis: the role of socio-economic status and race/ethnicity. Nephrol Dial Transplant 2003; 18:2039–2046.
- Illei GG, Austin HA, Crane M, et al. Combination therapy with pulse cyclophosphamide plus pulse methylprednisolone improves long-term renal outcome without adding toxicity in patients with lupus nephritis. Ann Intern Med 2001; 135:248–257.
- Houssiau FA, Vasconcelos C, D’Cruz D, et al. Immunosuppressive therapy in lupus nephritis: the Euro-Lupus Nephritis Trial, a randomized trial of low-dose versus high-dose intravenous cyclophosphamide. Arthritis Rheum 2002; 46:2121–2131.
- Houssiau FA, Vasconcelos C, D’Cruz D, et al. The 10-year follow-up data of the Euro-Lupus Nephritis Trial comparing low-dose and high-dose intravenous cyclophosphamide. Ann Rheum Dis 2010; 69:61–64.
- Chan TM, Li FK, Tang CS, et al. Efficacy of mycophenolate mofetil in patients with diffuse proliferative lupus nephritis. Hong King-Guangzhou Nephrology Study Group. N Engl J Med 2000; 343:1156–1162.
- Chan TM, Tse KC, Tang CS, Mok MY, Li FK; Hong Kong Nephrology Study Group. Long-term study of mycophenolate mofetil as continuous induction and maintenance treatment for diffuse proliferative lupus nephritis. J Am Soc Nephrol 2005; 16:1076–1084.
- Contreras G, Pardo V, Leclercq B, et al. Sequential therapies for proliferative lupus nephritis. N Engl J Med 2004; 350:971–980.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Appel GB, Contreras G, Dooley MA, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol 2009; 20:1103–1112.
- Radhakrishnan J, Moutzouris DA, Ginzler EM, Solomons N, Siempos II, Appel GB. Mycophenolate mofetil and intravenous cyclophosphamide are similar as induction therapy for class V lupus nephritis. Kidney Int 2010; 77:152–160.
- Dooley MA, Jayne D, Ginzler EM, et al; for the ALMS Group. Mycophenolate versus azathioprine as maintenance therapy for lupus nephritis. N Engl J Med 2011; 365:1886–1895.
- Terrier B, Amoura Z, Ravaud P, et al; Club Rhumatismes et Inflammation. Safety and efficacy of rituximab in systemic lupus erythematosus: results from 136 patients from the French AutoImmunity and Rituximab registry. Arthritis Rheum 2010; 62:2458–2466.
- Melander C, Sallée M, Troillet P, et al. Rituximab in severe lupus nephritis: early B-cell depletion affects long-term renal outcome. Clin J Am Soc Nephrol 2009; 4:579–587.
- Condon MB, Griffith M, Cook HT, Levy J, Lightstone L, Cairns T. Treatment of class IV lupus nephritis with rituximab & mycophenolate mofetil (MMF) with no oral steroids is effective and safe (abstract). J Am Soc Nephrol 2010; 21(suppl):625A–626A.
- Furie RA, Looney RJ, Rovin E, et al. Efficacy and safety of rituximab in subjects with active proliferative lupus nephritis (LN): results from the randomized, double-blind phase III LUNAR study (abstract). Arthritis Rheum 2009; 60(suppl 1):S429.
- Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum 2010; 62:222–233.
- Bao H, Liu ZH, Zie HL, Hu WX, Zhang HT, Li LS. Successful treatment of class V+IV lupus nephritis with multitarget therapy. J Am Soc Nephrol 2008; 19:2001–2010.
- Navarra SV, Guzmán RM, Gallacher AE, et al; BLISS-52 Study Group. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet 2011; 377:721–731.
Treatment for lupus nephritis has changed dramatically in recent years. Only 10 years ago, rheumatologists and nephrologists, whether specializing in adult or pediatric medicine, treated lupus nephritis with a similar regimen of monthly intravenous cyclophosphamide (Cytoxan) and glucocorticoids. Although the regimen is effective, side effects such as infection, hair loss, and infertility were extremely common.
Effective but very toxic therapy is common in autoimmune diseases. In the last decade, clinical trials have shown that less toxic drugs are as effective for treating lupus nephritis. This article will review new developments in therapy for lupus nephritis, which can be viewed as a prototype for other fields of medicine.
DEMOGRAPHICS ARE IMPORTANT
Although numerous factors have prognostic value in lupus nephritis (eg, serum creatinine, proteinuria, renal biopsy findings), the most important to consider when designing and interpreting studies are race and socioeconomic variables.
A retrospective study in Miami, FL,1 evaluated 213 patients with lupus nephritis, of whom 47% were Hispanic, 44% African American, and 20% white. At baseline, African Americans had higher blood pressure, higher serum creatinine levels, and lower household income. After 6 years, African Americans fared the worst in terms of doubling of serum creatinine, developing end-stage renal disease, and death; whites had the best outcomes, and Hispanics were in between. Low income was found to be a significant risk factor, independent of racial background.
In a similar retrospective study in New York City in 128 patients (43% white, 40% Hispanic, and 17% African American) with proliferative lupus nephritis,2 disease was much more likely to progress to renal failure over 10 years in patients living in a poor neighborhood, even after adjustment for race.
We need to keep in mind that racial and socioeconomic factors correlate with disease severity when we design and interpret studies of lupus nephritis. Study groups must be carefully balanced with patients of similar racial and socioeconomic profiles. Study findings must be interpreted with caution; for example, whether results from a study from China are applicable to an African American with lupus nephritis in New York City is unclear.
OLDER STANDARD THERAPY: EFFECTIVE BUT TOXIC
The last large National Institutes of Health study that involved only cyclophosphamide and a glucocorticoid was published in 2001,3 with 21 patients receiving cyclophosphamide alone and 20 patients receiving cyclophosphamide plus methylprednisolone. Although lupus nephritis improved, serious side effects occurred in one-third to one-half of patients in each group and included hypertension, hyperlipidemia, valvular heart disease, avascular necrosis, premature menopause, and major infections, including herpes zoster.
Less cyclophosphamide works just as well
The multicenter, prospective Euro-Lupus Nephritis Trial4 randomized 90 patients with proliferative lupus nephritis to receive either standard high-dose intravenous (IV) cyclophosphamide therapy (six monthly pulses and two quarterly pulses, with doses increasing according to the white blood cell count) or low-dose IV cyclophosphamide therapy (six pulses every 2 weeks at a fixed dose of 500 mg). Both regimens were followed by azathioprine (Imuran).
At 4 years, the two treatment groups were not significantly different in terms of treatment failure, remission rates, serum creatinine levels, 24-hour proteinuria, and freedom from renal flares. However, the rates of side effects were significantly different, with more patients in the low-dosage group free of severe infection.
One problem with this study is whether it is applicable to an American lupus nephritis population, since 84% of the patients were white. Since this study, others indicate that this regimen is probably also safe and effective for different racial groups in the United States.
At 10-year follow-up,5 both treatment groups still had identical excellent rates of freedom from end-stage renal disease. Serum creatinine and 24-hour proteinuria were also at excellent levels and identical in both groups. Nearly three quarters of patients still needed glucocorticoid therapy and more than half still needed immunosuppressive therapy, but the rates were not statistically significantly different between the treatment groups.
The cumulative dose of cyclophosphamide was 9.5 g in the standard-treatment group and 5.5 g in the low-dose group. This difference in exposure could make a tremendous difference to patients, not only for immediate side effects such as early menopause and infections, but for the risk of cancer in later decades.
This study showed clearly that low-dose cyclophosphamide is an option for induction therapy. Drawbacks of the study were that the population was mostly white and that patients had only moderately severe disease.
Low-dose cyclophosphamide has largely replaced the older National Institutes of Health regimen, although during the last decade drug therapy has undergone more changes.
MYCOPHENOLATE AND AZATHIOPRINE: ALTERNATIVES TO CYCLOPHOSPHAMIDE
In a Chinese study, mycophenolate was better than cyclophosphamide for induction
In a study in Hong Kong, Chan et al6 randomized 42 patients with severe lupus nephritis to receive either mycophenolate mofetil (available in the United States as CellCept; 2 g/day for 6 months, then 1 g/day for 6 months) or oral cyclophosphamide (2.5 mg/kg per day for 6 months) followed by azathioprine (1.5–2.0 mg/kg per day) for 6 months. Both groups also received prednisolone during the year.
At the end of the first year, the two groups were not significantly different in their rates of complete remission, partial remission, and relapse. The rate of infection, although not significantly different, was higher in the cyclophosphamide group (33% vs 19%). Two patients (10%) died in the cyclophosphamide group, but the difference in mortality rates was not statistically significant.
Nearly 5 years later,7 rates of chronic renal failure and relapse were still statistically the same in the two groups. Infections were fewer in the mycophenolate group (13% vs 40%, P = .013). The rate of amenorrhea was 36% in the cyclophosphamide group and only 4% in the mycophenolate group (P = .004). Four patients in the cyclophosphamide group and none in the mycophenolate group reached the composite end point of end-stage renal failure or death (P = .062).
This study appeared to offer a new option with equal efficacy and fewer side effects than standard therapy. However, its applicability to non-Chinese populations remained to be shown.
In a US study, mycophenolate or azathioprine was better than cyclophosphamide as maintenance
In a study in Miami,8 59 patients with lupus nephritis were given standard induction therapy with IV cyclophosphamide plus glucocorticoids for 6 months, then randomly assigned to one of three maintenance therapies for 1 to 3 years: IV injections of cyclophosphamide every 3 months (standard therapy), oral azathioprine, or oral mycophenolate. The population was 93% female, their average age was 33 years, and nearly half were African American, with many of the others being Hispanic. Patients tended to have severe disease, with nearly two-thirds having nephrotic syndrome.
After 6 years, there had been more deaths in the cyclophosphamide group than in the azathioprine group (P = .02) and in the mycophenolate group, although the latter difference was not statistically significant (P = .11). The combined rate of death and chronic renal failure was significantly higher with cyclophosphamide than with either of the oral agents. The cyclophosphamide group also had the highest relapse rate during the maintenance phase.
The differences in side effects were even more dramatic. Amenorrhea affected 32% of patients in the cyclophosphamide group, and only 7% and 6% in the azathioprine and mycophenolate groups, respectively. Rates of infections were 68% in the cyclophosphamide group and 28% and 21% in the azathioprine and mycophenolate groups, respectively. Patients given cyclophosphamide had 13 hospital days per patient per year, while the other groups each had only 1.
This study showed that maintenance therapy with oral azathioprine or mycophenolate was more effective and had fewer adverse effects than standard IV cyclophosphamide therapy. As a result of this study, oral agents for maintenance therapy became the new standard, but the question remained whether oral agents could safely be used for induction.
In a US study, mycophenolate was better than cyclophosphamide for induction
In a noninferiority study, Ginzler et al9 randomized 140 patients with severe lupus nephritis to receive either monthly IV cyclophosphamide or oral mycophenolate as induction therapy for 6 months. Adjunctive care with glucocorticoids was given in both groups. The study population was from 18 US academic centers and was predominantly female, and more than half were African American.
After 24 weeks, 22.5% of the mycophenolate patients were in complete remission by very strict criteria vs only 4% of those given cyclophosphamide (P = .005). The trend for partial remissions was also in favor of mycophenolate, although the difference was not statistically significant. The rate of complete and partial remissions, a prespecified end point, was significantly higher in the mycophenolate group. Although the study was trying to evaluate equivalency, it actually showed superiority for mycophenolate induction therapy.
Serum creatinine levels declined in both groups, but more in the mycophenolate group by 24 weeks. Urinary protein levels fell the same amount in both groups. At 3 years, the groups were statistically equivalent in terms of renal flares, renal failures, and deaths. However, the study groups were small, and the mycophenolate group did have a better trend for both renal failure (N = 4 vs 7) and deaths (N = 4 vs 8).
Mycophenolate also had fewer side effects, including infection, although again the numbers were too small to show statistical significance. The exception was diarrhea (N = 15 in the mycophenolate group vs 2 in the cyclophosphamide group).
A drawback of the study is that it was designed as a crossover study: a patient for whom therapy was failing after 3 months could switch to the other group, introducing potential confounding. Other problems involved the small population size and the question of whether results from patients in the United States were applicable to others worldwide.
In a worldwide study, mycophenolate was at least equivalent to cyclophosphamide for induction
The Aspreva Lupus Management Study (ALMS)10 used a similar design with 370 patients worldwide (United States, China, South America, and Europe) in one of the largest trials ever conducted in lupus nephritis. Patients were randomized to 6 months of induction therapy with either IV cyclophosphamide or oral mycophenolate but could not cross over.
At 6 months, response rates were identical between the two groups, with response defined as a combination of specific improvement in proteinuria, serum creatinine, and hematuria (50%–55%). In terms of individual renal and nonrenal variables, both groups appeared identical.
However, the side effect profiles differed between the two groups. As expected for mycophenolate, diarrhea was the most common side effect (occurring in 28% vs 12% in the cyclophosphamide group). Nausea and vomiting were more common with cyclophosphamide (45% and 37% respectively vs 14% and 13% in the mycophenolate group). Cyclophosphamide also caused hair loss in 35%, vs 10% in the mycophenolate group.
There were 14 deaths overall, which is a very low number considering the patients’ severity of illness, and it indicates the better results now achieved with therapy. The mortality rate was higher in the mycophenolate group (5% vs 3%), but the difference was not statistically significant. Six of the nine deaths with mycophenolate were from the same center in China, and none were from Europe or the United States. In summary, the study did not show that mycophenolate was superior to IV cyclophosphamide for induction therapy, but that they were equivalent in efficacy with different side effect profiles.
Membranous nephropathy: Mycophenolate vs cyclophosphamide
Less evidence is available about treatment for membranous disease, which is characterized by heavy proteinuria and the nephrotic syndrome but usually does not progress to renal failure. Radhakrishnan et al11 combined data from the trial by Ginzler et al9 and the ALMS trial10 and found 84 patients with pure membranous lupus, who were equally divided between the treatment groups receiving IV cyclophosphamide and mycophenolate. Consistent with the larger group’s data, mycophenolate and cyclophosphamide performed similarly in terms of efficacy, but there was a slightly higher rate of side effects with cyclophosphamide.
Maintenance therapy: Mycophenolate superior to azathioprine
The ALMS Maintenance Trial12 evaluated maintenance therapy in the same worldwide population that was studied for induction therapy. Of the 370 patients involved in the induction phase that compared IV cyclophosphamide and oral mycophenolate, 227 responded sufficiently to be rerandomized in a controlled, double-blinded trial of 36 months of maintenance therapy with corticosteroids and either mycophenolate (1 g twice daily) or azathioprine (2 mg/kg per day).
In intention-to-treat analysis, the time to treatment failure (ie, doubling of the serum creatinine level, progressing to renal failure, or death) was significantly shorter in the azathioprine group (P = .003). Every individual end point—end-stage renal disease, renal flares, doubling of serum creatinine, rescue immunosuppression required—was in favor of mycophenolate maintenance. At 3 years, the completion rate was 63% with mycophenolate and 49% with azathioprine. Serious adverse events and withdrawals because of adverse events were more common in the azathioprine group.
In summary, mycophenolate was superior to azathioprine in maintaining renal response and in preventing relapse in patients with active lupus nephritis who responded to induction therapy with either mycophenolate or IV cyclophosphamide. Mycophenolate was found to be superior regardless of initial induction treatment, race, or region and was confirmed by all key secondary end points.
Only one of the 227 patients died during the 3 years—from an auto accident. Again, this indicates the dramatically improved survival today compared with a decade ago.
RITUXIMAB: PROMISING BUT UNPROVEN
Rituximab (Rituxan) was originally approved to treat tumors, then rheumatoid arthritis, and most recently vasculitis. Evidence thus far is mixed regarding its use as a treatment for lupus nephritis. Although randomized clinical trials have not found it to be superior to standard regimens, there are many signs that it may be effective.
Rituximab in uncontrolled studies
Terrier et al13 analyzed prospective data from 136 patients with systemic lupus erythematosus, most of whom had renal disease, from the French Autoimmunity and Rituximab registry. Response occurred in 71% of patients using rituximab, with no difference found between patients receiving rituximab monotherapy and those concomitantly receiving immunosuppressive agents.
Melander et al14 retrospectively studied 19 women and 1 man who had been treated with rituximab for severe lupus nephritis and followed for at least 1 year. Three patients had concurrent therapy with cyclophosphamide, and 10 patients continued rituximab as maintenance therapy; 12 patients had lupus nephritis that had been refractory to standard treatment, and 6 had relapsing disease.
At a median follow-up of 22 months, 12 patients (60%) had achieved complete or partial renal remission.
Condon et al15 treated 21 patients who had severe lupus nephritis with two doses of rituximab and IV methylprednisolone 2 weeks apart, then maintenance therapy with mycophenolate without any oral steroids. At a mean follow-up of 35 months ( ± 14 months), 16 (76%) were in complete remission, with a mean time to remission of 12 months. Two (9.5%) achieved partial remission. The rate of toxicity was low.
Thus, rituximab appears promising in uncontrolled studies.
Placebo-controlled trials fail to prove rituximab effective
LUNAR trial. On the other hand, the largest placebo-controlled trial to evaluate rituximab in patients with proliferative lupus nephritis, the Lupus Nephritis Assessment With Rituximab (LUNAR) trial16 found differences in favor of rituximab, but none reached statistical significance. The trial randomized 140 patients to receive either mycophenolate plus periodic rituximab infusions or mycophenolate plus placebo infusions for 1 year. All patients received the same dosage of glucocorticoids, which was tapered over the year.
At the end of 1 year, the groups were not statistically different in terms of complete renal response and partial renal response. Rituximab appeared less likely to produce no response, but the difference was not statistically significant.
African Americans appeared to have a higher response rate to rituximab (70% in the rituximab group achieved a response vs 45% in the control group), but again, the difference did not reach statistical significance, and the total study population of African Americans was only 40.
Rituximab did have a statistically significant positive effect on two serologic markers at 1 year: levels of anti-dsDNA fell faster and complement rose faster. In addition, rates of adverse and serious adverse events were similar between the two groups, with no new or unexpected “safety signals.”
This study can be interpreted in a number of ways. The number of patients may have been too small to show significance and the follow-up may have been too short. On the other hand, it may simply not be effective to add rituximab to a full dose of mycophenolate and steroids, an already good treatment.
EXPLORER trial. Similarly, for patients with lupus without nephritis, the Exploratory Phase II/III SLE Evaluation of Rituximab (EXPLORER) trial17 also tested rituximab against a background of an effective therapeutic regimen and found no additional benefit. This study had design problems similar to those of the LUNAR trial.
Rituximab as rescue therapy
The evidence so far indicates that rituximab may have a role as rescue therapy for refractory or relapsing disease. Rituximab must be used with other therapies, but maintenance corticosteroid therapy is not necessary. Its role as a first-line agent in induction therapy for lupus nephritis remains unclear, although it may have an important role for nonwhites. In general, it has been well tolerated. Until a large randomized trial indicates otherwise, it should not be used as a first-line therapy.
The US Food and Drug Administration (FDA) sent out a warning about the danger of progressive multifocal leukoencephalopathy as an adverse effect of rituximab and of mycophenolate, but this does not appear to be a major concern for most patients and is only likely to occur in those who have been over-immunosuppressed for many years.
MULTITARGET THERAPY
The concept of using multiple drugs simultaneously—such as mycophenolate, steroids, and rituximab—is increasingly being tried. Multi-target therapy appears to offer the advantages of combining different modes of action with better results, and it offers fewer side effects because dosages of each individual drug can be lower when combined with other immunosuppressives.
Bao et al18 in China randomly assigned 40 patients with diffuse proliferative and membranous nephritis to 6 to 9 months of induction treatment with either multitarget therapy (mycophenolate, tacrolimus [Prograf], and glucocorticoids) or IV cyclophosphamide. More complete remissions occurred in the multitarget therapy group, both at 6 months (50% vs 5%) and at 9 months (65% vs 15%). Most adverse events were less frequent in the multitarget therapy group, although three patients (15%) in the multitarget therapy group developed new-onset hypertension vs none in the cyclophosphamide group.
NEW MEDICATIONS
Entirely new classes of drugs are being developed with immunomodulatory effects, including tolerance molecules, cytokine blockers, inhibitors of human B lymphocyte stimulator, and costimulatory blockers.
Belimumab offers small improvement for lupus
Belimumab (Benlysta) is a human monoclonal antibody that inhibits the biologic activity of human B lymphocyte stimulator; it has recently been approved by the FDA for lupus nephritis. In a worldwide study,19 867 patients with systemic lupus erythematosus were randomized to receive either belimumab (1 mg/kg or 10 mg/kg) or placebo.
The primary end point was the reduction of disease activity by a scoring system (SELENA-SLEDAI) that incorporated multiple features of lupus, including arthritis, vasculitis, proteinuria, rash, and others. Patients in the belimumab group had better outcomes, but the results were not dramatic. Because the drug is so expensive (about $25,000 per year) and the improvement offered is only incremental, this drug will not likely change the treatment of lupus very much.
Moreover, patients with lupus nephritis were not included in the study, but a new study is being planned to do so. Improvement is harder to demonstrate in lupus nephritis than in rheumatoid arthritis and systemic lupus erythematosus: significant changes in creatinine levels and 24-hour urinary protein must be achieved, rather than more qualitative signs and symptoms of joint pain, rash, and feeling better. Although belimumab is still unproven for lupus nephritis, it might be worth trying for patients failing other therapy.
Laquinimod: A promising experimental drug
Laquinimod is an oral immunomodulatory drug with a number of effects, including down-regulating major histocompatability complex II, chemokines, and adhesion-related molecules related to inflammation. It has been studied in more than 2,500 patients with multiple sclerosis. Pilot studies are now being done for its use for lupus nephritis. If it shows promise, a large randomized, controlled trial will be conducted.
Abatacept is in clinical trials
Abatacept (Orencia), a costimulation blocker, is undergoing clinical trials in lupus nephritis. Results should be available shortly.
INDIVIDUALIZE THERAPY
This past decade has seen such an increase in options to treat lupus nephritis that therapy can now be individualized.
Choosing IV cyclophosphamide vs mycophenolate
As a result of recent trials, doctors in the United States are increasingly using mycophenolate as the first-line drug for lupus nephritis. In Europe, however, many are choosing the shorter regimen of IV cyclophosphamide because of the results of the Euro-Lupus study.
Nowadays, I tend to use IV cyclophosphamide as the first-line drug only for patients with severe crescenteric glomerulonephritis or a very high serum creatinine level. In such cases, there is more experience with cyclophosphamide, and such severe disease does not lend itself to the luxury of trying out different therapies sequentially. If such a severely ill patient insists that a future pregnancy is very important, an alternative therapy of mycophenolate plus rituximab should be considered. I prefer mycophenolate for induction and maintenance therapy in most patients.
Dosing and formulation considerations for mycophenolate
Large dosages of mycophenolate are much better tolerated when broken up throughout the day. A patient who cannot tolerate 1 g twice daily may be able to tolerate 500 mg four times a day. The formulation can also make a difference. Some patients tolerate sustained-release mycophenolate (Myfortic) better than CellCept, and vice versa.
For patients who cannot tolerate mycophenolate, azathioprine is an acceptable alternative. In addition, for a patient who is already doing well on azathioprine, there is no need to change to mycophenolate.
Long maintenance therapy now acceptable
The ALMS Maintenance Trial12 found 3 years of maintenance therapy to be safe and effective. Such a long maintenance period is increasingly viewed as important, especially for patients in their teens and 20s, as it allows them to live a normal life, ie, to finish their education, get married, and become settled socially. Whether 5 years of maintenance therapy or even 10 years is advisable is still unknown.
Treatment during pregnancy
Neither mycophenolate nor azathioprine is recommended during pregnancy, although their effects are unknown. Because we have much more renal transplant experience with azathioprine during pregnancy, I recommend either switching from mycophenolate to azathioprine or trying to stop medication altogether if the patient has been well controlled.
Treatment for lupus nephritis has changed dramatically in recent years. Only 10 years ago, rheumatologists and nephrologists, whether specializing in adult or pediatric medicine, treated lupus nephritis with a similar regimen of monthly intravenous cyclophosphamide (Cytoxan) and glucocorticoids. Although the regimen is effective, side effects such as infection, hair loss, and infertility were extremely common.
Effective but very toxic therapy is common in autoimmune diseases. In the last decade, clinical trials have shown that less toxic drugs are as effective for treating lupus nephritis. This article will review new developments in therapy for lupus nephritis, which can be viewed as a prototype for other fields of medicine.
DEMOGRAPHICS ARE IMPORTANT
Although numerous factors have prognostic value in lupus nephritis (eg, serum creatinine, proteinuria, renal biopsy findings), the most important to consider when designing and interpreting studies are race and socioeconomic variables.
A retrospective study in Miami, FL,1 evaluated 213 patients with lupus nephritis, of whom 47% were Hispanic, 44% African American, and 20% white. At baseline, African Americans had higher blood pressure, higher serum creatinine levels, and lower household income. After 6 years, African Americans fared the worst in terms of doubling of serum creatinine, developing end-stage renal disease, and death; whites had the best outcomes, and Hispanics were in between. Low income was found to be a significant risk factor, independent of racial background.
In a similar retrospective study in New York City in 128 patients (43% white, 40% Hispanic, and 17% African American) with proliferative lupus nephritis,2 disease was much more likely to progress to renal failure over 10 years in patients living in a poor neighborhood, even after adjustment for race.
We need to keep in mind that racial and socioeconomic factors correlate with disease severity when we design and interpret studies of lupus nephritis. Study groups must be carefully balanced with patients of similar racial and socioeconomic profiles. Study findings must be interpreted with caution; for example, whether results from a study from China are applicable to an African American with lupus nephritis in New York City is unclear.
OLDER STANDARD THERAPY: EFFECTIVE BUT TOXIC
The last large National Institutes of Health study that involved only cyclophosphamide and a glucocorticoid was published in 2001,3 with 21 patients receiving cyclophosphamide alone and 20 patients receiving cyclophosphamide plus methylprednisolone. Although lupus nephritis improved, serious side effects occurred in one-third to one-half of patients in each group and included hypertension, hyperlipidemia, valvular heart disease, avascular necrosis, premature menopause, and major infections, including herpes zoster.
Less cyclophosphamide works just as well
The multicenter, prospective Euro-Lupus Nephritis Trial4 randomized 90 patients with proliferative lupus nephritis to receive either standard high-dose intravenous (IV) cyclophosphamide therapy (six monthly pulses and two quarterly pulses, with doses increasing according to the white blood cell count) or low-dose IV cyclophosphamide therapy (six pulses every 2 weeks at a fixed dose of 500 mg). Both regimens were followed by azathioprine (Imuran).
At 4 years, the two treatment groups were not significantly different in terms of treatment failure, remission rates, serum creatinine levels, 24-hour proteinuria, and freedom from renal flares. However, the rates of side effects were significantly different, with more patients in the low-dosage group free of severe infection.
One problem with this study is whether it is applicable to an American lupus nephritis population, since 84% of the patients were white. Since this study, others indicate that this regimen is probably also safe and effective for different racial groups in the United States.
At 10-year follow-up,5 both treatment groups still had identical excellent rates of freedom from end-stage renal disease. Serum creatinine and 24-hour proteinuria were also at excellent levels and identical in both groups. Nearly three quarters of patients still needed glucocorticoid therapy and more than half still needed immunosuppressive therapy, but the rates were not statistically significantly different between the treatment groups.
The cumulative dose of cyclophosphamide was 9.5 g in the standard-treatment group and 5.5 g in the low-dose group. This difference in exposure could make a tremendous difference to patients, not only for immediate side effects such as early menopause and infections, but for the risk of cancer in later decades.
This study showed clearly that low-dose cyclophosphamide is an option for induction therapy. Drawbacks of the study were that the population was mostly white and that patients had only moderately severe disease.
Low-dose cyclophosphamide has largely replaced the older National Institutes of Health regimen, although during the last decade drug therapy has undergone more changes.
MYCOPHENOLATE AND AZATHIOPRINE: ALTERNATIVES TO CYCLOPHOSPHAMIDE
In a Chinese study, mycophenolate was better than cyclophosphamide for induction
In a study in Hong Kong, Chan et al6 randomized 42 patients with severe lupus nephritis to receive either mycophenolate mofetil (available in the United States as CellCept; 2 g/day for 6 months, then 1 g/day for 6 months) or oral cyclophosphamide (2.5 mg/kg per day for 6 months) followed by azathioprine (1.5–2.0 mg/kg per day) for 6 months. Both groups also received prednisolone during the year.
At the end of the first year, the two groups were not significantly different in their rates of complete remission, partial remission, and relapse. The rate of infection, although not significantly different, was higher in the cyclophosphamide group (33% vs 19%). Two patients (10%) died in the cyclophosphamide group, but the difference in mortality rates was not statistically significant.
Nearly 5 years later,7 rates of chronic renal failure and relapse were still statistically the same in the two groups. Infections were fewer in the mycophenolate group (13% vs 40%, P = .013). The rate of amenorrhea was 36% in the cyclophosphamide group and only 4% in the mycophenolate group (P = .004). Four patients in the cyclophosphamide group and none in the mycophenolate group reached the composite end point of end-stage renal failure or death (P = .062).
This study appeared to offer a new option with equal efficacy and fewer side effects than standard therapy. However, its applicability to non-Chinese populations remained to be shown.
In a US study, mycophenolate or azathioprine was better than cyclophosphamide as maintenance
In a study in Miami,8 59 patients with lupus nephritis were given standard induction therapy with IV cyclophosphamide plus glucocorticoids for 6 months, then randomly assigned to one of three maintenance therapies for 1 to 3 years: IV injections of cyclophosphamide every 3 months (standard therapy), oral azathioprine, or oral mycophenolate. The population was 93% female, their average age was 33 years, and nearly half were African American, with many of the others being Hispanic. Patients tended to have severe disease, with nearly two-thirds having nephrotic syndrome.
After 6 years, there had been more deaths in the cyclophosphamide group than in the azathioprine group (P = .02) and in the mycophenolate group, although the latter difference was not statistically significant (P = .11). The combined rate of death and chronic renal failure was significantly higher with cyclophosphamide than with either of the oral agents. The cyclophosphamide group also had the highest relapse rate during the maintenance phase.
The differences in side effects were even more dramatic. Amenorrhea affected 32% of patients in the cyclophosphamide group, and only 7% and 6% in the azathioprine and mycophenolate groups, respectively. Rates of infections were 68% in the cyclophosphamide group and 28% and 21% in the azathioprine and mycophenolate groups, respectively. Patients given cyclophosphamide had 13 hospital days per patient per year, while the other groups each had only 1.
This study showed that maintenance therapy with oral azathioprine or mycophenolate was more effective and had fewer adverse effects than standard IV cyclophosphamide therapy. As a result of this study, oral agents for maintenance therapy became the new standard, but the question remained whether oral agents could safely be used for induction.
In a US study, mycophenolate was better than cyclophosphamide for induction
In a noninferiority study, Ginzler et al9 randomized 140 patients with severe lupus nephritis to receive either monthly IV cyclophosphamide or oral mycophenolate as induction therapy for 6 months. Adjunctive care with glucocorticoids was given in both groups. The study population was from 18 US academic centers and was predominantly female, and more than half were African American.
After 24 weeks, 22.5% of the mycophenolate patients were in complete remission by very strict criteria vs only 4% of those given cyclophosphamide (P = .005). The trend for partial remissions was also in favor of mycophenolate, although the difference was not statistically significant. The rate of complete and partial remissions, a prespecified end point, was significantly higher in the mycophenolate group. Although the study was trying to evaluate equivalency, it actually showed superiority for mycophenolate induction therapy.
Serum creatinine levels declined in both groups, but more in the mycophenolate group by 24 weeks. Urinary protein levels fell the same amount in both groups. At 3 years, the groups were statistically equivalent in terms of renal flares, renal failures, and deaths. However, the study groups were small, and the mycophenolate group did have a better trend for both renal failure (N = 4 vs 7) and deaths (N = 4 vs 8).
Mycophenolate also had fewer side effects, including infection, although again the numbers were too small to show statistical significance. The exception was diarrhea (N = 15 in the mycophenolate group vs 2 in the cyclophosphamide group).
A drawback of the study is that it was designed as a crossover study: a patient for whom therapy was failing after 3 months could switch to the other group, introducing potential confounding. Other problems involved the small population size and the question of whether results from patients in the United States were applicable to others worldwide.
In a worldwide study, mycophenolate was at least equivalent to cyclophosphamide for induction
The Aspreva Lupus Management Study (ALMS)10 used a similar design with 370 patients worldwide (United States, China, South America, and Europe) in one of the largest trials ever conducted in lupus nephritis. Patients were randomized to 6 months of induction therapy with either IV cyclophosphamide or oral mycophenolate but could not cross over.
At 6 months, response rates were identical between the two groups, with response defined as a combination of specific improvement in proteinuria, serum creatinine, and hematuria (50%–55%). In terms of individual renal and nonrenal variables, both groups appeared identical.
However, the side effect profiles differed between the two groups. As expected for mycophenolate, diarrhea was the most common side effect (occurring in 28% vs 12% in the cyclophosphamide group). Nausea and vomiting were more common with cyclophosphamide (45% and 37% respectively vs 14% and 13% in the mycophenolate group). Cyclophosphamide also caused hair loss in 35%, vs 10% in the mycophenolate group.
There were 14 deaths overall, which is a very low number considering the patients’ severity of illness, and it indicates the better results now achieved with therapy. The mortality rate was higher in the mycophenolate group (5% vs 3%), but the difference was not statistically significant. Six of the nine deaths with mycophenolate were from the same center in China, and none were from Europe or the United States. In summary, the study did not show that mycophenolate was superior to IV cyclophosphamide for induction therapy, but that they were equivalent in efficacy with different side effect profiles.
Membranous nephropathy: Mycophenolate vs cyclophosphamide
Less evidence is available about treatment for membranous disease, which is characterized by heavy proteinuria and the nephrotic syndrome but usually does not progress to renal failure. Radhakrishnan et al11 combined data from the trial by Ginzler et al9 and the ALMS trial10 and found 84 patients with pure membranous lupus, who were equally divided between the treatment groups receiving IV cyclophosphamide and mycophenolate. Consistent with the larger group’s data, mycophenolate and cyclophosphamide performed similarly in terms of efficacy, but there was a slightly higher rate of side effects with cyclophosphamide.
Maintenance therapy: Mycophenolate superior to azathioprine
The ALMS Maintenance Trial12 evaluated maintenance therapy in the same worldwide population that was studied for induction therapy. Of the 370 patients involved in the induction phase that compared IV cyclophosphamide and oral mycophenolate, 227 responded sufficiently to be rerandomized in a controlled, double-blinded trial of 36 months of maintenance therapy with corticosteroids and either mycophenolate (1 g twice daily) or azathioprine (2 mg/kg per day).
In intention-to-treat analysis, the time to treatment failure (ie, doubling of the serum creatinine level, progressing to renal failure, or death) was significantly shorter in the azathioprine group (P = .003). Every individual end point—end-stage renal disease, renal flares, doubling of serum creatinine, rescue immunosuppression required—was in favor of mycophenolate maintenance. At 3 years, the completion rate was 63% with mycophenolate and 49% with azathioprine. Serious adverse events and withdrawals because of adverse events were more common in the azathioprine group.
In summary, mycophenolate was superior to azathioprine in maintaining renal response and in preventing relapse in patients with active lupus nephritis who responded to induction therapy with either mycophenolate or IV cyclophosphamide. Mycophenolate was found to be superior regardless of initial induction treatment, race, or region and was confirmed by all key secondary end points.
Only one of the 227 patients died during the 3 years—from an auto accident. Again, this indicates the dramatically improved survival today compared with a decade ago.
RITUXIMAB: PROMISING BUT UNPROVEN
Rituximab (Rituxan) was originally approved to treat tumors, then rheumatoid arthritis, and most recently vasculitis. Evidence thus far is mixed regarding its use as a treatment for lupus nephritis. Although randomized clinical trials have not found it to be superior to standard regimens, there are many signs that it may be effective.
Rituximab in uncontrolled studies
Terrier et al13 analyzed prospective data from 136 patients with systemic lupus erythematosus, most of whom had renal disease, from the French Autoimmunity and Rituximab registry. Response occurred in 71% of patients using rituximab, with no difference found between patients receiving rituximab monotherapy and those concomitantly receiving immunosuppressive agents.
Melander et al14 retrospectively studied 19 women and 1 man who had been treated with rituximab for severe lupus nephritis and followed for at least 1 year. Three patients had concurrent therapy with cyclophosphamide, and 10 patients continued rituximab as maintenance therapy; 12 patients had lupus nephritis that had been refractory to standard treatment, and 6 had relapsing disease.
At a median follow-up of 22 months, 12 patients (60%) had achieved complete or partial renal remission.
Condon et al15 treated 21 patients who had severe lupus nephritis with two doses of rituximab and IV methylprednisolone 2 weeks apart, then maintenance therapy with mycophenolate without any oral steroids. At a mean follow-up of 35 months ( ± 14 months), 16 (76%) were in complete remission, with a mean time to remission of 12 months. Two (9.5%) achieved partial remission. The rate of toxicity was low.
Thus, rituximab appears promising in uncontrolled studies.
Placebo-controlled trials fail to prove rituximab effective
LUNAR trial. On the other hand, the largest placebo-controlled trial to evaluate rituximab in patients with proliferative lupus nephritis, the Lupus Nephritis Assessment With Rituximab (LUNAR) trial16 found differences in favor of rituximab, but none reached statistical significance. The trial randomized 140 patients to receive either mycophenolate plus periodic rituximab infusions or mycophenolate plus placebo infusions for 1 year. All patients received the same dosage of glucocorticoids, which was tapered over the year.
At the end of 1 year, the groups were not statistically different in terms of complete renal response and partial renal response. Rituximab appeared less likely to produce no response, but the difference was not statistically significant.
African Americans appeared to have a higher response rate to rituximab (70% in the rituximab group achieved a response vs 45% in the control group), but again, the difference did not reach statistical significance, and the total study population of African Americans was only 40.
Rituximab did have a statistically significant positive effect on two serologic markers at 1 year: levels of anti-dsDNA fell faster and complement rose faster. In addition, rates of adverse and serious adverse events were similar between the two groups, with no new or unexpected “safety signals.”
This study can be interpreted in a number of ways. The number of patients may have been too small to show significance and the follow-up may have been too short. On the other hand, it may simply not be effective to add rituximab to a full dose of mycophenolate and steroids, an already good treatment.
EXPLORER trial. Similarly, for patients with lupus without nephritis, the Exploratory Phase II/III SLE Evaluation of Rituximab (EXPLORER) trial17 also tested rituximab against a background of an effective therapeutic regimen and found no additional benefit. This study had design problems similar to those of the LUNAR trial.
Rituximab as rescue therapy
The evidence so far indicates that rituximab may have a role as rescue therapy for refractory or relapsing disease. Rituximab must be used with other therapies, but maintenance corticosteroid therapy is not necessary. Its role as a first-line agent in induction therapy for lupus nephritis remains unclear, although it may have an important role for nonwhites. In general, it has been well tolerated. Until a large randomized trial indicates otherwise, it should not be used as a first-line therapy.
The US Food and Drug Administration (FDA) sent out a warning about the danger of progressive multifocal leukoencephalopathy as an adverse effect of rituximab and of mycophenolate, but this does not appear to be a major concern for most patients and is only likely to occur in those who have been over-immunosuppressed for many years.
MULTITARGET THERAPY
The concept of using multiple drugs simultaneously—such as mycophenolate, steroids, and rituximab—is increasingly being tried. Multi-target therapy appears to offer the advantages of combining different modes of action with better results, and it offers fewer side effects because dosages of each individual drug can be lower when combined with other immunosuppressives.
Bao et al18 in China randomly assigned 40 patients with diffuse proliferative and membranous nephritis to 6 to 9 months of induction treatment with either multitarget therapy (mycophenolate, tacrolimus [Prograf], and glucocorticoids) or IV cyclophosphamide. More complete remissions occurred in the multitarget therapy group, both at 6 months (50% vs 5%) and at 9 months (65% vs 15%). Most adverse events were less frequent in the multitarget therapy group, although three patients (15%) in the multitarget therapy group developed new-onset hypertension vs none in the cyclophosphamide group.
NEW MEDICATIONS
Entirely new classes of drugs are being developed with immunomodulatory effects, including tolerance molecules, cytokine blockers, inhibitors of human B lymphocyte stimulator, and costimulatory blockers.
Belimumab offers small improvement for lupus
Belimumab (Benlysta) is a human monoclonal antibody that inhibits the biologic activity of human B lymphocyte stimulator; it has recently been approved by the FDA for lupus nephritis. In a worldwide study,19 867 patients with systemic lupus erythematosus were randomized to receive either belimumab (1 mg/kg or 10 mg/kg) or placebo.
The primary end point was the reduction of disease activity by a scoring system (SELENA-SLEDAI) that incorporated multiple features of lupus, including arthritis, vasculitis, proteinuria, rash, and others. Patients in the belimumab group had better outcomes, but the results were not dramatic. Because the drug is so expensive (about $25,000 per year) and the improvement offered is only incremental, this drug will not likely change the treatment of lupus very much.
Moreover, patients with lupus nephritis were not included in the study, but a new study is being planned to do so. Improvement is harder to demonstrate in lupus nephritis than in rheumatoid arthritis and systemic lupus erythematosus: significant changes in creatinine levels and 24-hour urinary protein must be achieved, rather than more qualitative signs and symptoms of joint pain, rash, and feeling better. Although belimumab is still unproven for lupus nephritis, it might be worth trying for patients failing other therapy.
Laquinimod: A promising experimental drug
Laquinimod is an oral immunomodulatory drug with a number of effects, including down-regulating major histocompatability complex II, chemokines, and adhesion-related molecules related to inflammation. It has been studied in more than 2,500 patients with multiple sclerosis. Pilot studies are now being done for its use for lupus nephritis. If it shows promise, a large randomized, controlled trial will be conducted.
Abatacept is in clinical trials
Abatacept (Orencia), a costimulation blocker, is undergoing clinical trials in lupus nephritis. Results should be available shortly.
INDIVIDUALIZE THERAPY
This past decade has seen such an increase in options to treat lupus nephritis that therapy can now be individualized.
Choosing IV cyclophosphamide vs mycophenolate
As a result of recent trials, doctors in the United States are increasingly using mycophenolate as the first-line drug for lupus nephritis. In Europe, however, many are choosing the shorter regimen of IV cyclophosphamide because of the results of the Euro-Lupus study.
Nowadays, I tend to use IV cyclophosphamide as the first-line drug only for patients with severe crescenteric glomerulonephritis or a very high serum creatinine level. In such cases, there is more experience with cyclophosphamide, and such severe disease does not lend itself to the luxury of trying out different therapies sequentially. If such a severely ill patient insists that a future pregnancy is very important, an alternative therapy of mycophenolate plus rituximab should be considered. I prefer mycophenolate for induction and maintenance therapy in most patients.
Dosing and formulation considerations for mycophenolate
Large dosages of mycophenolate are much better tolerated when broken up throughout the day. A patient who cannot tolerate 1 g twice daily may be able to tolerate 500 mg four times a day. The formulation can also make a difference. Some patients tolerate sustained-release mycophenolate (Myfortic) better than CellCept, and vice versa.
For patients who cannot tolerate mycophenolate, azathioprine is an acceptable alternative. In addition, for a patient who is already doing well on azathioprine, there is no need to change to mycophenolate.
Long maintenance therapy now acceptable
The ALMS Maintenance Trial12 found 3 years of maintenance therapy to be safe and effective. Such a long maintenance period is increasingly viewed as important, especially for patients in their teens and 20s, as it allows them to live a normal life, ie, to finish their education, get married, and become settled socially. Whether 5 years of maintenance therapy or even 10 years is advisable is still unknown.
Treatment during pregnancy
Neither mycophenolate nor azathioprine is recommended during pregnancy, although their effects are unknown. Because we have much more renal transplant experience with azathioprine during pregnancy, I recommend either switching from mycophenolate to azathioprine or trying to stop medication altogether if the patient has been well controlled.
- Contreras G, Lenz O, Pardo V, et al. Outcomes in African Americans and Hispanics with lupus nephritis. Kidney Int 2006; 69:1846–1851.
- Barr RG, Seliger S, Appel GB, et al. Prognosis in proliferative lupus nephritis: the role of socio-economic status and race/ethnicity. Nephrol Dial Transplant 2003; 18:2039–2046.
- Illei GG, Austin HA, Crane M, et al. Combination therapy with pulse cyclophosphamide plus pulse methylprednisolone improves long-term renal outcome without adding toxicity in patients with lupus nephritis. Ann Intern Med 2001; 135:248–257.
- Houssiau FA, Vasconcelos C, D’Cruz D, et al. Immunosuppressive therapy in lupus nephritis: the Euro-Lupus Nephritis Trial, a randomized trial of low-dose versus high-dose intravenous cyclophosphamide. Arthritis Rheum 2002; 46:2121–2131.
- Houssiau FA, Vasconcelos C, D’Cruz D, et al. The 10-year follow-up data of the Euro-Lupus Nephritis Trial comparing low-dose and high-dose intravenous cyclophosphamide. Ann Rheum Dis 2010; 69:61–64.
- Chan TM, Li FK, Tang CS, et al. Efficacy of mycophenolate mofetil in patients with diffuse proliferative lupus nephritis. Hong King-Guangzhou Nephrology Study Group. N Engl J Med 2000; 343:1156–1162.
- Chan TM, Tse KC, Tang CS, Mok MY, Li FK; Hong Kong Nephrology Study Group. Long-term study of mycophenolate mofetil as continuous induction and maintenance treatment for diffuse proliferative lupus nephritis. J Am Soc Nephrol 2005; 16:1076–1084.
- Contreras G, Pardo V, Leclercq B, et al. Sequential therapies for proliferative lupus nephritis. N Engl J Med 2004; 350:971–980.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Appel GB, Contreras G, Dooley MA, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol 2009; 20:1103–1112.
- Radhakrishnan J, Moutzouris DA, Ginzler EM, Solomons N, Siempos II, Appel GB. Mycophenolate mofetil and intravenous cyclophosphamide are similar as induction therapy for class V lupus nephritis. Kidney Int 2010; 77:152–160.
- Dooley MA, Jayne D, Ginzler EM, et al; for the ALMS Group. Mycophenolate versus azathioprine as maintenance therapy for lupus nephritis. N Engl J Med 2011; 365:1886–1895.
- Terrier B, Amoura Z, Ravaud P, et al; Club Rhumatismes et Inflammation. Safety and efficacy of rituximab in systemic lupus erythematosus: results from 136 patients from the French AutoImmunity and Rituximab registry. Arthritis Rheum 2010; 62:2458–2466.
- Melander C, Sallée M, Troillet P, et al. Rituximab in severe lupus nephritis: early B-cell depletion affects long-term renal outcome. Clin J Am Soc Nephrol 2009; 4:579–587.
- Condon MB, Griffith M, Cook HT, Levy J, Lightstone L, Cairns T. Treatment of class IV lupus nephritis with rituximab & mycophenolate mofetil (MMF) with no oral steroids is effective and safe (abstract). J Am Soc Nephrol 2010; 21(suppl):625A–626A.
- Furie RA, Looney RJ, Rovin E, et al. Efficacy and safety of rituximab in subjects with active proliferative lupus nephritis (LN): results from the randomized, double-blind phase III LUNAR study (abstract). Arthritis Rheum 2009; 60(suppl 1):S429.
- Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum 2010; 62:222–233.
- Bao H, Liu ZH, Zie HL, Hu WX, Zhang HT, Li LS. Successful treatment of class V+IV lupus nephritis with multitarget therapy. J Am Soc Nephrol 2008; 19:2001–2010.
- Navarra SV, Guzmán RM, Gallacher AE, et al; BLISS-52 Study Group. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet 2011; 377:721–731.
- Contreras G, Lenz O, Pardo V, et al. Outcomes in African Americans and Hispanics with lupus nephritis. Kidney Int 2006; 69:1846–1851.
- Barr RG, Seliger S, Appel GB, et al. Prognosis in proliferative lupus nephritis: the role of socio-economic status and race/ethnicity. Nephrol Dial Transplant 2003; 18:2039–2046.
- Illei GG, Austin HA, Crane M, et al. Combination therapy with pulse cyclophosphamide plus pulse methylprednisolone improves long-term renal outcome without adding toxicity in patients with lupus nephritis. Ann Intern Med 2001; 135:248–257.
- Houssiau FA, Vasconcelos C, D’Cruz D, et al. Immunosuppressive therapy in lupus nephritis: the Euro-Lupus Nephritis Trial, a randomized trial of low-dose versus high-dose intravenous cyclophosphamide. Arthritis Rheum 2002; 46:2121–2131.
- Houssiau FA, Vasconcelos C, D’Cruz D, et al. The 10-year follow-up data of the Euro-Lupus Nephritis Trial comparing low-dose and high-dose intravenous cyclophosphamide. Ann Rheum Dis 2010; 69:61–64.
- Chan TM, Li FK, Tang CS, et al. Efficacy of mycophenolate mofetil in patients with diffuse proliferative lupus nephritis. Hong King-Guangzhou Nephrology Study Group. N Engl J Med 2000; 343:1156–1162.
- Chan TM, Tse KC, Tang CS, Mok MY, Li FK; Hong Kong Nephrology Study Group. Long-term study of mycophenolate mofetil as continuous induction and maintenance treatment for diffuse proliferative lupus nephritis. J Am Soc Nephrol 2005; 16:1076–1084.
- Contreras G, Pardo V, Leclercq B, et al. Sequential therapies for proliferative lupus nephritis. N Engl J Med 2004; 350:971–980.
- Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353:2219–2228.
- Appel GB, Contreras G, Dooley MA, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol 2009; 20:1103–1112.
- Radhakrishnan J, Moutzouris DA, Ginzler EM, Solomons N, Siempos II, Appel GB. Mycophenolate mofetil and intravenous cyclophosphamide are similar as induction therapy for class V lupus nephritis. Kidney Int 2010; 77:152–160.
- Dooley MA, Jayne D, Ginzler EM, et al; for the ALMS Group. Mycophenolate versus azathioprine as maintenance therapy for lupus nephritis. N Engl J Med 2011; 365:1886–1895.
- Terrier B, Amoura Z, Ravaud P, et al; Club Rhumatismes et Inflammation. Safety and efficacy of rituximab in systemic lupus erythematosus: results from 136 patients from the French AutoImmunity and Rituximab registry. Arthritis Rheum 2010; 62:2458–2466.
- Melander C, Sallée M, Troillet P, et al. Rituximab in severe lupus nephritis: early B-cell depletion affects long-term renal outcome. Clin J Am Soc Nephrol 2009; 4:579–587.
- Condon MB, Griffith M, Cook HT, Levy J, Lightstone L, Cairns T. Treatment of class IV lupus nephritis with rituximab & mycophenolate mofetil (MMF) with no oral steroids is effective and safe (abstract). J Am Soc Nephrol 2010; 21(suppl):625A–626A.
- Furie RA, Looney RJ, Rovin E, et al. Efficacy and safety of rituximab in subjects with active proliferative lupus nephritis (LN): results from the randomized, double-blind phase III LUNAR study (abstract). Arthritis Rheum 2009; 60(suppl 1):S429.
- Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum 2010; 62:222–233.
- Bao H, Liu ZH, Zie HL, Hu WX, Zhang HT, Li LS. Successful treatment of class V+IV lupus nephritis with multitarget therapy. J Am Soc Nephrol 2008; 19:2001–2010.
- Navarra SV, Guzmán RM, Gallacher AE, et al; BLISS-52 Study Group. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet 2011; 377:721–731.
KEY POINTS
- Mycophenolate is at least equivalent to intravenous cyclophosphamide for induction and maintenance treatment of severe lupus nephritis.
- The role of rituximab is unclear, and for now it should only be used in relapsing patients or patients whose disease is resistant to standard therapy.
- Using combination therapies for induction treatment and maintenance is becoming increasingly common.
- Three-year maintenance therapy is now considered advisable in most patients.
- Entirely new drugs under study include costimulatory blockers, inhibitors of human B lymphocyte stimulator, tolerance molecules, and cytokine blockers.
Finding the cause of acute kidney injury: Which index of fractional excretion is better?
An acute kidney injury can result from a myriad of causes and pathogenic pathways. Of these, the two main categories are prerenal causes (eg, heart failure, volume depletion) and causes that are intrinsic to the kidney (eg, acute tubular necrosis). Together, these categories account for more than 70% of all cases.1–3
While early intervention improves outcomes in both of these categories, the physician in the acute care setting must quickly distinguish between them, as their treatments differ. Similar clinical presentations along with confounding laboratory values make this distinction difficult. Furthermore, prolonged prerenal azotemia can eventually lead to acute tubular necrosis.
Therefore, several methods for distinguishing prerenal from intrinsic causes of acute kidney injury have been developed, including urinalysis, response to fluid challenge, the blood urea nitrogen-to-plasma creatinine ratio, levels of various urine electrolytes and biomarkers, and, the topics of our discussion here, the fractional excretion of sodium (FENa) and the fractional excretion of urea (FEU).4 While each method offers a unique picture of renal function, the validity of each may be affected by specific clinical factors.

In light of the frequent use of diuretics in inpatients and outpatients, a review of the utility of the FEU test is warranted. We will therefore present the theory behind the use of the FENa and the FEU for distinguishing intrinsic from prerenal causes of acute kidney injury, the relevant literature comparing the utility of these investigations, and our suggestions for clinical practice.
ACUTE KIDNEY INJURY DEFINED
Acute kidney injury (formerly called acute renal failure) describes an abrupt decline in renal function. Consensus definitions of it have been published and are gaining more widespread acceptance and use.9,10 The current definition is10:
- An absolute increase in serum creatinine ≥ 0.3 mg/dL (26.4 μmol/L) in 48 hours, or
- A percentage increase in serum creatinine ≥ 50% in 48 hours, or
- Urine output < 0.5 mL/kg/hour for > 6 hours.
These clear criteria allow for earlier recognition and treatment of this condition.
Acute kidney injury is fairly common in hospitalized patients, with 172 to 620 cases per million patients per year.11–14 Furthermore, hospitalized patients with acute kidney injury continue to have high rates of morbidity and death, especially those with more severe cases, in which the mortality rate remains as high as 40%.15
FRACTIONAL EXCRETION OF SODIUM
The FENa is a measure of the extraction of sodium and water from the glomerular filtrate. It is the ratio of the rate of sodium filtration (the urinary sodium concentration times the urinary flow rate, divided by the plasma sodium concentration) to the overall glomerular filtration rate, estimated by the renal filtration of creatinine. It can be calculated as the ratio of plasma creatinine to urine creatinine divided by the ratio of plasma sodium to urine sodium:
A euvolemic person with normal renal function and moderate salt intake in a steady state will have an FENa of approximately 1%.16
In 1976, Espinel17 originally showed that the FENa could be used during the oliguric phase in patients in acute renal failure to differentiate between prerenal acute kidney injury and acute tubular necrosis. Given the kidney’s ability to reabsorb more sodium during times of volume depletion, Espinel suggested that an FENa of less than 1% reflected normal sodium retention, indicating a prerenal cause, ie, diminished effective circulating volume. A value greater than 3% likely represented tubular damage, indicating that the nephrons were unable to properly reabsorb sodium.
The clinical utility of this index was apparent, as the management of prerenal azotemia and acute tubular necrosis differ.18 While both require fluid repletion, the risk of volume overload in acute tubular necrosis is high. Furthermore, acute tubular necrosis secondary to nephrotoxins could require hemodialysis to facilitate clearance of the offending agent.
The FENa test was subsequently validated in a number of studies in different populations and is still widely used.19–21
Limitations to the use of the FENa have been noted in various clinical settings. Notably, it can be falsely depressed in a number of intrinsic renal conditions, such as contrast-induced nephropathy, rhabdomyolysis, and acute glomerulonephritis. Conversely, patients with prerenal acute kidney injury who take diuretics can have a falsely elevated value due to the pharmacologically induced renal excretion of sodium independent of volume status. This is commonly seen in patients on diuretic therapy with baseline low effective circulating volumes, such those with congestive heart failure and hepatic cirrhosis.
FRACTIONAL EXCRETION OF UREA
Urea is continuously produced in the liver as the end product of protein metabolism. It is a small, water-soluble molecule that freely passes across cell membranes and is therefore continuously filtered and excreted by the kidneys. Not merely a waste product, urea is also important in water balance and constitutes approximately half of the normal solute content of urine.22
Urea’s excretion mechanisms are well characterized.22,23 It is absorbed in the proximal tubule, the medullary loop of Henle, and the medullary collecting ducts via facilitated diffusion through specific urea transporters.24 After being absorbed in the loop of Henle, urea is resecreted, a process that creates an osmotic gradient along the medulla that ultimately regulates urea excretion and reabsorption in the medullary collecting duct. Low-volume states are associated with decreased urea excretion due to a physiologic increase in antidiuretic hormone secretion, and the reverse is true for high-volume states.
The FEU has been recognized as a clinically useful tool. The correlation between serum and urine urea concentrations was investigated as early as 1904.25 However, most studies during the ensuing century focused on the serum urea concentration or the creatinine-to-urea ratio as a measure of glomerular failure.26–28 In 1992, Kaplan and Kohn29 proposed that the FEU could be a useful measure for assessing renal dysfunction in acute kidney injury. Conceptually similar to the FENa, the FEU is calculated as:
An FEU less than 35% suggests a prerenal cause of acute kidney injury, while a value greater than 50% suggests an intrinsic one.
FRACTIONAL EXCRETION OF UREA VS FRACTIONAL EXCRETION OF SODIUM
Kaplan and Kohn (1992)
Kaplan and Kohn,29 in their 1992 study, retrospectively analyzed 87 urine samples from 40 patients with renal dysfunction (not specifically acute kidney injury) thought to be secondary to volume depletion in which the FENa was discordant with the FEU.
Findings. Thirty-nine of the 40 patients treated with diuretics had a high FENa value. However, the FEU was low in all of these patients, leading the authors to conclude that the latter may be the more useful of the two indices in evaluating patients receiving diuretics who present with symptoms that suggest prerenal azotemia.
Limitations of the study. On closer inspection, these findings were not generalizable, for several reasons. First, the time that elapsed between administration of diuretics and evaluation of urinary electrolytes varied widely. Additionally, the study was a retrospective analysis of isolated urine specimens without clear correlation to a clinical patient or context. For these reasons, prospective analyses to investigate the utility of the fractional excretion of urea needed to be conducted.
Carvounis et al (2002)
Carvounis et al30 prospectively evaluated the FENa and the FEU in 102 consecutive intensive care patients with acute kidney injury (defined as a serum creatinine concentration > 1.5 mg/dL or an increase of more than 0.5 mg/dL in less than 48 hours). Oliguria was not an inclusion criterion for the study, but patients with acute glomerulonephritis and obstructive nephropathy were excluded. The study grouped subjects into those with prerenal azotemia, prerenal azotemia plus diuretic use, or acute tubular necrosis on the basis of the clinical diagnosis of the attending nephrologist.
Findings. The FEU was more sensitive than the FENa in detecting prerenal azotemia, especially in those with prerenal azotemia who were receiving diuretics. Overall, the FEU had higher sensitivity and specificity for prerenal azotemia regardless of diuretic usage, and more importantly, the best overall positive and negative predictive value for detecting it (99% and 75% respectively).
These results indicate that, in patients given diuretics, the FENa fails to discriminate between prerenal azotemia and acute tubular necrosis. Conversely, the FEU was excellent in discriminating between all cases of prerenal azotemia and acute tubular necrosis irrespective of the use of diuretics. This has significant practical application, given the frequency of diuretic use in the hospital, particularly in intensive care patients.
Limitations of the study. While the findings supported the utility of the FEU, the study population was limited to intensive care patients. Furthermore, the authors did not report the statistical significance of their findings.30
Pépin et al (2007)
Pépin et al8 performed a similar study, investigating the diagnostic utility of the FENa and the FEU in patients with acute kidney injury, with or without diuretic therapy.
The authors prospectively studied 99 consecutive patients confirmed by an independent nephrologist to have acute kidney injury (defined as an increase in serum creatinine of more than 30% over baseline values within less than 1 week) due to either volume depletion or ischemia. They excluded patients with less common causes of acute kidney injury, such as rhabdomyolysis, obstructive nephropathy, adrenal insufficiency, acute glomerulonephritis, and nephrotoxic acute kidney injury, as well as patients with chronic kidney disease.
Patients were grouped into those with transient acute kidney injury (from decreased kidney perfusion) and persistent acute kidney injury (attributed to acute tubular necrosis), with or without diuretic therapy, according to predefined clinical criteria. They were considered to have diuretic exposure if they had received furosemide (Lasix) within 24 hours or a thiazide within 48 hours of sampling.
Findings. The FENa proved superior to the FEU in patients not taking diuretics and, contrary to the findings of Carvounis et al,30 exhibited diagnostic utility in patients taking diuretics as well. Neither index discriminated between the different etiologies exceptionally well, however.
Of note, the study population was more inclusive than in previous studies, with only 63 intensive care patients, thus making the results more generalizable to all cases of inpatient acute kidney injury. Furthermore, the study included patients with and without oliguria, and the sensitivity and specificity of both the FENa and the FEU were higher in the nonoliguric group (n = 25).
Limitations of the study. The authors admit that a long time may have elapsed between diuretic administration and urine measurements, thereby mitigating the diuretic’s natriuretic effect independent of the patient’s volume status. While this variable may account for the better performance of the FENa than in the other studies, it does not account for the poor performance of the FEU.
Additionally, few of the findings reached statistical significance.
Lastly, a high percentage (30%) of patients had sepsis. The FEU is less effective in patients with infection, as cytokines interfere with the urea transporters in the kidney and colon.31
Lim et al (2009)
Lim et al32 conducted a study similar in design to that of Pépin et al.8
Findings. The FEU was as clinically useful as the FENa at distinguishing transient from persistent acute kidney injury in patients on diuretics. Using a cutoff FEU of less than 30% and a cutoff FENa of less than 1.5% for transient acute kidney injury (based on calculated receiver operating characteristic curves), FENa was more sensitive and specific than FEU in the nondiuretic groups. In patients exposed to diuretics, FEU was more sensitive but less specific than FENa.
FRACTIONAL EXCRETION OF UREA IN OLIGURIA
Diskin et al (2010)
In 2010, Diskin et al33 published a prospective, observational study of 100 consecutive patients with oliguric azotemia referred to a nephrology service. They defined acute kidney injury as serum creatinine concentration greater than 1.9 mg/dL and urine output less than 100 mL in 24 hours. They used a higher FEU cutoff for prerenal azotemia of less than 40% to reflect the known urea secretion rate in oliguric patients (600 mL/24 hours). They used an FENa of less than 1% and greater than 3% to distinguish prerenal azotemia from acute tubular necrosis.
Findings. The FEU was more accurate than the FENa, giving the right diagnosis in 95% vs 54% of cases (P < .0001). The difference was exclusively due to the FEU’s greater utility in the 67 patients who had received diuretics (98% vs 49%, P < .0001). Both the FEU and the FENa accurately detected acute tubular necrosis. As expected, the FENa outperformed FEU in the setting of infection, in which cytokine stimulation interferes with urea excretion.
Limitations of the study. Approximately 80% of the patients had prerenal azotemia, potentially biasing the results toward a test geared toward detecting this condition. However, since prerenal causes are more common than intrinsic causes, the authors argued that their cohort more accurately reflected the population encountered in clinical practice.
Additionally, only patients with oliguria and more advanced kidney injury (serum creatinine > 1.9 mg/dL) were included in the study, potentially limiting the applicability of these results in patients with preserved urine output in the early stages of renal failure.
Table 2 summarizes the findings of the studies discussed above.8,15,30,32,33
FRACTIONAL EXCRETION OF UREA IN CHILDREN AND THE ELDERLY
The FEU has also been validated in populations at the extremes of age.
In children, Fahimi et al34 performed a cross-sectional study in 43 patients referred to a nephrology service because of acute kidney injury.
An FEU less than 35% had greater sensitivity and specificity than an FENa less than 1% for differentiating prerenal from intrinsic causes in pediatric populations. An FEU of less than 30% had an even greater power of distinguishing between the two. Interestingly, 15 of the 26 patients in the group with prerenal azotemia had an FENa greater than 1%, 8 of whom had an obvious cause (diuretic therapy in 5, salt-losing congenital adrenal hyperplasia in 2, and metabolic alkalosis in 1).
In elderly people, urinary indices are less reliable because of reduced sodium and urea reabsorption and urinary concentrating capability. Thus, the FENa and FEU are increased, making the standard cutoff values unreliable and unpredictable for distinguishing prerenal from intrinsic causes of acute kidney injury.35
WHICH TEST SHOULD BE USED?
Both the FENa and the FEU have been validated in prospective trials as useful clinical indices in identifying prerenal azotemia. Results of these studies vary as to which index is superior and when. This may be attributable to the various definitions of acute kidney injury and diagnostic criteria used in the studies as well as the heterogeneity of patients in each study.
However, the preponderance of evidence indicates that the FEU is more useful than the FENa in patients on diuretics. Since diuretics are widely used, particularly in acute care settings in which acute kidney injury is prevalent, the FEU is a useful clinical tool and should be utilized in this context accordingly. Specifically, when there is a history of recent diuretic use, the evidence supports ordering the FEU alone, or at least in conjunction with the FENa. If the two indices yield disparate results, the physician should look for circumstances that would alter each one of them, such as sepsis or an unrecognized dose of diuretic.
In managing acute kidney injury, distinguishing prerenal from intrinsic causes is a difficult task, particularly because prolonged prerenal azotemia can develop into acute tubular necrosis. Therefore, a single index, calculated at a specific time, often is insufficient to properly characterize the pathogenesis of acute kidney injury, and a combination of both of these indices may increase diagnostic sensitivity and specificity.36 Moreover, urine samples collected after acute changes in volume or osmolarity, such as blood loss, administration of intravenous fluids or parenteral nutrition, or dialysis may compromise their diagnostic utility, and care must be taken to interpret the results in the appropriate clinical context.
The clinician must be aware of both the respective applications and limitations of these indices when using them to guide management and navigate the differential diagnosis in the appropriate clinical settings.
- Nolan CR, Anderson RJ. Hospital-acquired acute renal failure. J Am Soc Nephrol 1998; 9:710–718.
- Mehta RL, Pascual MT, Soroko S, et al; Program to Improve Care in Acute Renal Disease. Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney Int 2004; 66:1613–1621.
- Myers BD, Miller DC, Mehigan JT, et al. Nature of the renal injury following total renal ischemia in man. J Clin Invest 1984; 73:329–341.
- Ho E, Fard A, Maisel A. Evolving use of biomarkers for kidney injury in acute care settings. Curr Opin Crit Care 2010; 16:399–407.
- Steiner RW. Low fractional excretion of sodium in myoglobinuric acute renal failure. Arch Intern Med 1982; 142:1216–1217.
- Vaz AJ. Low fractional excretion of urine sodium in acute renal failure due to sepsis. Arch Intern Med 1983; 143:738–739.
- Pru C, Kjellstrand CM. The FENa test is of no prognostic value in acute renal failure. Nephron 1984; 36:20–23.
- Pépin MN, Bouchard J, Legault L, Ethier J. Diagnostic performance of fractional excretion of urea and fractional excretion of sodium in the evaluations of patients with acute kidney injury with or without diuretic treatment. Am J Kidney Dis 2007; 50:566–573.
- Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P; Acute Dialysis Quality Initiative workgroup. Acute renal failure—definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care 2004; 8:R204–R212.
- Mehta RL, Kellum JA, Shah SV, et al; Acute Kidney Injury Network. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care 2007; 11:R31.
- Stevens PE, Tamimi NA, Al-Hasani MK, et al. Non-specialist management of acute renal failure. QJM 2001; 94:533–540.
- Feest TG, Round A, Hamad S. Incidence of severe acute renal failure in adults: results of a community based study. BMJ 1993; 306:481–483.
- Liaño F, Pascual J. Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Madrid Acute Renal Failure Study Group. Kidney Int 1996; 50:811–818.
- Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med 1996; 334:1448–1460.
- Bagshaw SM, George C, Bellomo R; ANZICS Database Management Committee. Changes in the incidence and outcome for early acute kidney injury in a cohort of Australian intensive care units. Crit Care 2007; 11:R68.
- Sodium homeostasis in chronic renal disease. Kidney Int 1982; 21:886–897.
- Espinel CH. The FENa test. Use in the differential diagnosis of acute renal failure. JAMA 1976; 236:579–581.
- Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest 2004; 114:5–14.
- Miller TR, Anderson RJ, Linas SL, et al. Urinary diagnostic indices in acute renal failure: a prospective study. Ann Intern Med 1978; 89:47–50.
- Zarich S, Fang LS, Diamond JR. Fractional excretion of sodium. Exceptions to its diagnostic value. Arch Intern Med 1985; 145:108–112.
- Mandal AK, Baig M, Koutoubi Z. Management of acute renal failure in the elderly. Treatment options. Drugs Aging 1996; 9:226–250.
- Sands JM. Critical role of urea in the urine-concentrating mechanism. J Am Soc Nephrol 2007; 18:670–671.
- Goldstein MH, Lenz PR, Levitt MF. Effect of urine flow rate on urea reabsorption in man: urea as a “tubular marker”. J Appl Physiol 1969; 26:594–599.
- Fenton RA, Knepper MA. Urea and renal function in the 21st century: insights from knockout mice. J Am Soc Nephrol 2007; 18:679–688.
- Gréhant N. Physiologique des reins par le dosage de l’urée dans le sang et dans l’urine. J Physiol Pathol Gen (Paris) 1904; 6:1–8.
- Dossetor JB. Creatininemia versus uremia. The relative significance of blood urea nitrogen and serum creatinine concentrations in azotemia. Ann Intern Med 1966; 65:1287–1299.
- Kahn S, Sagel J, Eales L, Rabkin R. The significance of serum creatinine and the blood urea-serum creatinine ratio in azotaemia. S Afr Med J 1972; 46:1828–1832.
- Kerr DNS, Davison JM. The assessment of renal function. Br J Hosp Med 1975; 14:360–372.
- Kaplan AA, Kohn OF. Fractional excretion of urea as a guide to renal dysfunction. Am J Nephrol 1992; 12:49–54.
- Carvounis CP, Nisar S, Guro-Razuman S. Significance of the fractional excretion of urea in the differential diagnosis of acute renal failure. Kidney Int 2002; 62:2223–2229.
- Schmidt C, Höcherl K, Bucher M. Cytokine-mediated regulation of urea transporters during experimental endotoxemia. Am J Physiol Renal Physiol 2007; 292:F1479–F1489.
- Lim DH, Jeong JM, Oh SH, et al. Diagnostic performance of fractional excretion of urea in evaluating patients with acute kidney injury with diuretics treatment. Korean J Nephrol 2009; 28:190–198.
- Diskin CJ, Stokes TJ, Dansby LM, Radcliff L, Carter TB. The comparative benefits of the fractional excretion of urea and sodium in various azotemic oliguric states. Nephron Clin Pract 2010; 114:c145–c150.
- Fahimi D, Mohajeri S, Hajizadeh N, et al. Comparison between fractional excretions of urea and sodium in children with acute kidney injury. Pediatr Nephrol 2009; 24:2409–2412.
- Musso CG, Liakopoulos V, Ioannidis I, Eleftheriadis T, Stefanidis I. Acute renal failure in the elderly: particular characteristics. Int Urol Nephrol 2006; 38:787–793.
- Schönermarck U, Kehl K, Samtleben W. Diagnostic performance of fractional excretion of urea and sodium in acute kidney injury. Am J Kidney Dis 2008; 51:870–871.
An acute kidney injury can result from a myriad of causes and pathogenic pathways. Of these, the two main categories are prerenal causes (eg, heart failure, volume depletion) and causes that are intrinsic to the kidney (eg, acute tubular necrosis). Together, these categories account for more than 70% of all cases.1–3
While early intervention improves outcomes in both of these categories, the physician in the acute care setting must quickly distinguish between them, as their treatments differ. Similar clinical presentations along with confounding laboratory values make this distinction difficult. Furthermore, prolonged prerenal azotemia can eventually lead to acute tubular necrosis.
Therefore, several methods for distinguishing prerenal from intrinsic causes of acute kidney injury have been developed, including urinalysis, response to fluid challenge, the blood urea nitrogen-to-plasma creatinine ratio, levels of various urine electrolytes and biomarkers, and, the topics of our discussion here, the fractional excretion of sodium (FENa) and the fractional excretion of urea (FEU).4 While each method offers a unique picture of renal function, the validity of each may be affected by specific clinical factors.
In light of the frequent use of diuretics in inpatients and outpatients, a review of the utility of the FEU test is warranted. We will therefore present the theory behind the use of the FENa and the FEU for distinguishing intrinsic from prerenal causes of acute kidney injury, the relevant literature comparing the utility of these investigations, and our suggestions for clinical practice.
ACUTE KIDNEY INJURY DEFINED
Acute kidney injury (formerly called acute renal failure) describes an abrupt decline in renal function. Consensus definitions of it have been published and are gaining more widespread acceptance and use.9,10 The current definition is10:
- An absolute increase in serum creatinine ≥ 0.3 mg/dL (26.4 μmol/L) in 48 hours, or
- A percentage increase in serum creatinine ≥ 50% in 48 hours, or
- Urine output < 0.5 mL/kg/hour for > 6 hours.
These clear criteria allow for earlier recognition and treatment of this condition.
Acute kidney injury is fairly common in hospitalized patients, with 172 to 620 cases per million patients per year.11–14 Furthermore, hospitalized patients with acute kidney injury continue to have high rates of morbidity and death, especially those with more severe cases, in which the mortality rate remains as high as 40%.15
FRACTIONAL EXCRETION OF SODIUM
The FENa is a measure of the extraction of sodium and water from the glomerular filtrate. It is the ratio of the rate of sodium filtration (the urinary sodium concentration times the urinary flow rate, divided by the plasma sodium concentration) to the overall glomerular filtration rate, estimated by the renal filtration of creatinine. It can be calculated as the ratio of plasma creatinine to urine creatinine divided by the ratio of plasma sodium to urine sodium:
A euvolemic person with normal renal function and moderate salt intake in a steady state will have an FENa of approximately 1%.16
In 1976, Espinel17 originally showed that the FENa could be used during the oliguric phase in patients in acute renal failure to differentiate between prerenal acute kidney injury and acute tubular necrosis. Given the kidney’s ability to reabsorb more sodium during times of volume depletion, Espinel suggested that an FENa of less than 1% reflected normal sodium retention, indicating a prerenal cause, ie, diminished effective circulating volume. A value greater than 3% likely represented tubular damage, indicating that the nephrons were unable to properly reabsorb sodium.
The clinical utility of this index was apparent, as the management of prerenal azotemia and acute tubular necrosis differ.18 While both require fluid repletion, the risk of volume overload in acute tubular necrosis is high. Furthermore, acute tubular necrosis secondary to nephrotoxins could require hemodialysis to facilitate clearance of the offending agent.
The FENa test was subsequently validated in a number of studies in different populations and is still widely used.19–21
Limitations to the use of the FENa have been noted in various clinical settings. Notably, it can be falsely depressed in a number of intrinsic renal conditions, such as contrast-induced nephropathy, rhabdomyolysis, and acute glomerulonephritis. Conversely, patients with prerenal acute kidney injury who take diuretics can have a falsely elevated value due to the pharmacologically induced renal excretion of sodium independent of volume status. This is commonly seen in patients on diuretic therapy with baseline low effective circulating volumes, such those with congestive heart failure and hepatic cirrhosis.
FRACTIONAL EXCRETION OF UREA
Urea is continuously produced in the liver as the end product of protein metabolism. It is a small, water-soluble molecule that freely passes across cell membranes and is therefore continuously filtered and excreted by the kidneys. Not merely a waste product, urea is also important in water balance and constitutes approximately half of the normal solute content of urine.22
Urea’s excretion mechanisms are well characterized.22,23 It is absorbed in the proximal tubule, the medullary loop of Henle, and the medullary collecting ducts via facilitated diffusion through specific urea transporters.24 After being absorbed in the loop of Henle, urea is resecreted, a process that creates an osmotic gradient along the medulla that ultimately regulates urea excretion and reabsorption in the medullary collecting duct. Low-volume states are associated with decreased urea excretion due to a physiologic increase in antidiuretic hormone secretion, and the reverse is true for high-volume states.
The FEU has been recognized as a clinically useful tool. The correlation between serum and urine urea concentrations was investigated as early as 1904.25 However, most studies during the ensuing century focused on the serum urea concentration or the creatinine-to-urea ratio as a measure of glomerular failure.26–28 In 1992, Kaplan and Kohn29 proposed that the FEU could be a useful measure for assessing renal dysfunction in acute kidney injury. Conceptually similar to the FENa, the FEU is calculated as:
An FEU less than 35% suggests a prerenal cause of acute kidney injury, while a value greater than 50% suggests an intrinsic one.
FRACTIONAL EXCRETION OF UREA VS FRACTIONAL EXCRETION OF SODIUM
Kaplan and Kohn (1992)
Kaplan and Kohn,29 in their 1992 study, retrospectively analyzed 87 urine samples from 40 patients with renal dysfunction (not specifically acute kidney injury) thought to be secondary to volume depletion in which the FENa was discordant with the FEU.
Findings. Thirty-nine of the 40 patients treated with diuretics had a high FENa value. However, the FEU was low in all of these patients, leading the authors to conclude that the latter may be the more useful of the two indices in evaluating patients receiving diuretics who present with symptoms that suggest prerenal azotemia.
Limitations of the study. On closer inspection, these findings were not generalizable, for several reasons. First, the time that elapsed between administration of diuretics and evaluation of urinary electrolytes varied widely. Additionally, the study was a retrospective analysis of isolated urine specimens without clear correlation to a clinical patient or context. For these reasons, prospective analyses to investigate the utility of the fractional excretion of urea needed to be conducted.
Carvounis et al (2002)
Carvounis et al30 prospectively evaluated the FENa and the FEU in 102 consecutive intensive care patients with acute kidney injury (defined as a serum creatinine concentration > 1.5 mg/dL or an increase of more than 0.5 mg/dL in less than 48 hours). Oliguria was not an inclusion criterion for the study, but patients with acute glomerulonephritis and obstructive nephropathy were excluded. The study grouped subjects into those with prerenal azotemia, prerenal azotemia plus diuretic use, or acute tubular necrosis on the basis of the clinical diagnosis of the attending nephrologist.
Findings. The FEU was more sensitive than the FENa in detecting prerenal azotemia, especially in those with prerenal azotemia who were receiving diuretics. Overall, the FEU had higher sensitivity and specificity for prerenal azotemia regardless of diuretic usage, and more importantly, the best overall positive and negative predictive value for detecting it (99% and 75% respectively).
These results indicate that, in patients given diuretics, the FENa fails to discriminate between prerenal azotemia and acute tubular necrosis. Conversely, the FEU was excellent in discriminating between all cases of prerenal azotemia and acute tubular necrosis irrespective of the use of diuretics. This has significant practical application, given the frequency of diuretic use in the hospital, particularly in intensive care patients.
Limitations of the study. While the findings supported the utility of the FEU, the study population was limited to intensive care patients. Furthermore, the authors did not report the statistical significance of their findings.30
Pépin et al (2007)
Pépin et al8 performed a similar study, investigating the diagnostic utility of the FENa and the FEU in patients with acute kidney injury, with or without diuretic therapy.
The authors prospectively studied 99 consecutive patients confirmed by an independent nephrologist to have acute kidney injury (defined as an increase in serum creatinine of more than 30% over baseline values within less than 1 week) due to either volume depletion or ischemia. They excluded patients with less common causes of acute kidney injury, such as rhabdomyolysis, obstructive nephropathy, adrenal insufficiency, acute glomerulonephritis, and nephrotoxic acute kidney injury, as well as patients with chronic kidney disease.
Patients were grouped into those with transient acute kidney injury (from decreased kidney perfusion) and persistent acute kidney injury (attributed to acute tubular necrosis), with or without diuretic therapy, according to predefined clinical criteria. They were considered to have diuretic exposure if they had received furosemide (Lasix) within 24 hours or a thiazide within 48 hours of sampling.
Findings. The FENa proved superior to the FEU in patients not taking diuretics and, contrary to the findings of Carvounis et al,30 exhibited diagnostic utility in patients taking diuretics as well. Neither index discriminated between the different etiologies exceptionally well, however.
Of note, the study population was more inclusive than in previous studies, with only 63 intensive care patients, thus making the results more generalizable to all cases of inpatient acute kidney injury. Furthermore, the study included patients with and without oliguria, and the sensitivity and specificity of both the FENa and the FEU were higher in the nonoliguric group (n = 25).
Limitations of the study. The authors admit that a long time may have elapsed between diuretic administration and urine measurements, thereby mitigating the diuretic’s natriuretic effect independent of the patient’s volume status. While this variable may account for the better performance of the FENa than in the other studies, it does not account for the poor performance of the FEU.
Additionally, few of the findings reached statistical significance.
Lastly, a high percentage (30%) of patients had sepsis. The FEU is less effective in patients with infection, as cytokines interfere with the urea transporters in the kidney and colon.31
Lim et al (2009)
Lim et al32 conducted a study similar in design to that of Pépin et al.8
Findings. The FEU was as clinically useful as the FENa at distinguishing transient from persistent acute kidney injury in patients on diuretics. Using a cutoff FEU of less than 30% and a cutoff FENa of less than 1.5% for transient acute kidney injury (based on calculated receiver operating characteristic curves), FENa was more sensitive and specific than FEU in the nondiuretic groups. In patients exposed to diuretics, FEU was more sensitive but less specific than FENa.
FRACTIONAL EXCRETION OF UREA IN OLIGURIA
Diskin et al (2010)
In 2010, Diskin et al33 published a prospective, observational study of 100 consecutive patients with oliguric azotemia referred to a nephrology service. They defined acute kidney injury as serum creatinine concentration greater than 1.9 mg/dL and urine output less than 100 mL in 24 hours. They used a higher FEU cutoff for prerenal azotemia of less than 40% to reflect the known urea secretion rate in oliguric patients (600 mL/24 hours). They used an FENa of less than 1% and greater than 3% to distinguish prerenal azotemia from acute tubular necrosis.
Findings. The FEU was more accurate than the FENa, giving the right diagnosis in 95% vs 54% of cases (P < .0001). The difference was exclusively due to the FEU’s greater utility in the 67 patients who had received diuretics (98% vs 49%, P < .0001). Both the FEU and the FENa accurately detected acute tubular necrosis. As expected, the FENa outperformed FEU in the setting of infection, in which cytokine stimulation interferes with urea excretion.
Limitations of the study. Approximately 80% of the patients had prerenal azotemia, potentially biasing the results toward a test geared toward detecting this condition. However, since prerenal causes are more common than intrinsic causes, the authors argued that their cohort more accurately reflected the population encountered in clinical practice.
Additionally, only patients with oliguria and more advanced kidney injury (serum creatinine > 1.9 mg/dL) were included in the study, potentially limiting the applicability of these results in patients with preserved urine output in the early stages of renal failure.
Table 2 summarizes the findings of the studies discussed above.8,15,30,32,33
FRACTIONAL EXCRETION OF UREA IN CHILDREN AND THE ELDERLY
The FEU has also been validated in populations at the extremes of age.
In children, Fahimi et al34 performed a cross-sectional study in 43 patients referred to a nephrology service because of acute kidney injury.
An FEU less than 35% had greater sensitivity and specificity than an FENa less than 1% for differentiating prerenal from intrinsic causes in pediatric populations. An FEU of less than 30% had an even greater power of distinguishing between the two. Interestingly, 15 of the 26 patients in the group with prerenal azotemia had an FENa greater than 1%, 8 of whom had an obvious cause (diuretic therapy in 5, salt-losing congenital adrenal hyperplasia in 2, and metabolic alkalosis in 1).
In elderly people, urinary indices are less reliable because of reduced sodium and urea reabsorption and urinary concentrating capability. Thus, the FENa and FEU are increased, making the standard cutoff values unreliable and unpredictable for distinguishing prerenal from intrinsic causes of acute kidney injury.35
WHICH TEST SHOULD BE USED?
Both the FENa and the FEU have been validated in prospective trials as useful clinical indices in identifying prerenal azotemia. Results of these studies vary as to which index is superior and when. This may be attributable to the various definitions of acute kidney injury and diagnostic criteria used in the studies as well as the heterogeneity of patients in each study.
However, the preponderance of evidence indicates that the FEU is more useful than the FENa in patients on diuretics. Since diuretics are widely used, particularly in acute care settings in which acute kidney injury is prevalent, the FEU is a useful clinical tool and should be utilized in this context accordingly. Specifically, when there is a history of recent diuretic use, the evidence supports ordering the FEU alone, or at least in conjunction with the FENa. If the two indices yield disparate results, the physician should look for circumstances that would alter each one of them, such as sepsis or an unrecognized dose of diuretic.
In managing acute kidney injury, distinguishing prerenal from intrinsic causes is a difficult task, particularly because prolonged prerenal azotemia can develop into acute tubular necrosis. Therefore, a single index, calculated at a specific time, often is insufficient to properly characterize the pathogenesis of acute kidney injury, and a combination of both of these indices may increase diagnostic sensitivity and specificity.36 Moreover, urine samples collected after acute changes in volume or osmolarity, such as blood loss, administration of intravenous fluids or parenteral nutrition, or dialysis may compromise their diagnostic utility, and care must be taken to interpret the results in the appropriate clinical context.
The clinician must be aware of both the respective applications and limitations of these indices when using them to guide management and navigate the differential diagnosis in the appropriate clinical settings.
An acute kidney injury can result from a myriad of causes and pathogenic pathways. Of these, the two main categories are prerenal causes (eg, heart failure, volume depletion) and causes that are intrinsic to the kidney (eg, acute tubular necrosis). Together, these categories account for more than 70% of all cases.1–3
While early intervention improves outcomes in both of these categories, the physician in the acute care setting must quickly distinguish between them, as their treatments differ. Similar clinical presentations along with confounding laboratory values make this distinction difficult. Furthermore, prolonged prerenal azotemia can eventually lead to acute tubular necrosis.
Therefore, several methods for distinguishing prerenal from intrinsic causes of acute kidney injury have been developed, including urinalysis, response to fluid challenge, the blood urea nitrogen-to-plasma creatinine ratio, levels of various urine electrolytes and biomarkers, and, the topics of our discussion here, the fractional excretion of sodium (FENa) and the fractional excretion of urea (FEU).4 While each method offers a unique picture of renal function, the validity of each may be affected by specific clinical factors.
In light of the frequent use of diuretics in inpatients and outpatients, a review of the utility of the FEU test is warranted. We will therefore present the theory behind the use of the FENa and the FEU for distinguishing intrinsic from prerenal causes of acute kidney injury, the relevant literature comparing the utility of these investigations, and our suggestions for clinical practice.
ACUTE KIDNEY INJURY DEFINED
Acute kidney injury (formerly called acute renal failure) describes an abrupt decline in renal function. Consensus definitions of it have been published and are gaining more widespread acceptance and use.9,10 The current definition is10:
- An absolute increase in serum creatinine ≥ 0.3 mg/dL (26.4 μmol/L) in 48 hours, or
- A percentage increase in serum creatinine ≥ 50% in 48 hours, or
- Urine output < 0.5 mL/kg/hour for > 6 hours.
These clear criteria allow for earlier recognition and treatment of this condition.
Acute kidney injury is fairly common in hospitalized patients, with 172 to 620 cases per million patients per year.11–14 Furthermore, hospitalized patients with acute kidney injury continue to have high rates of morbidity and death, especially those with more severe cases, in which the mortality rate remains as high as 40%.15
FRACTIONAL EXCRETION OF SODIUM
The FENa is a measure of the extraction of sodium and water from the glomerular filtrate. It is the ratio of the rate of sodium filtration (the urinary sodium concentration times the urinary flow rate, divided by the plasma sodium concentration) to the overall glomerular filtration rate, estimated by the renal filtration of creatinine. It can be calculated as the ratio of plasma creatinine to urine creatinine divided by the ratio of plasma sodium to urine sodium:
A euvolemic person with normal renal function and moderate salt intake in a steady state will have an FENa of approximately 1%.16
In 1976, Espinel17 originally showed that the FENa could be used during the oliguric phase in patients in acute renal failure to differentiate between prerenal acute kidney injury and acute tubular necrosis. Given the kidney’s ability to reabsorb more sodium during times of volume depletion, Espinel suggested that an FENa of less than 1% reflected normal sodium retention, indicating a prerenal cause, ie, diminished effective circulating volume. A value greater than 3% likely represented tubular damage, indicating that the nephrons were unable to properly reabsorb sodium.
The clinical utility of this index was apparent, as the management of prerenal azotemia and acute tubular necrosis differ.18 While both require fluid repletion, the risk of volume overload in acute tubular necrosis is high. Furthermore, acute tubular necrosis secondary to nephrotoxins could require hemodialysis to facilitate clearance of the offending agent.
The FENa test was subsequently validated in a number of studies in different populations and is still widely used.19–21
Limitations to the use of the FENa have been noted in various clinical settings. Notably, it can be falsely depressed in a number of intrinsic renal conditions, such as contrast-induced nephropathy, rhabdomyolysis, and acute glomerulonephritis. Conversely, patients with prerenal acute kidney injury who take diuretics can have a falsely elevated value due to the pharmacologically induced renal excretion of sodium independent of volume status. This is commonly seen in patients on diuretic therapy with baseline low effective circulating volumes, such those with congestive heart failure and hepatic cirrhosis.
FRACTIONAL EXCRETION OF UREA
Urea is continuously produced in the liver as the end product of protein metabolism. It is a small, water-soluble molecule that freely passes across cell membranes and is therefore continuously filtered and excreted by the kidneys. Not merely a waste product, urea is also important in water balance and constitutes approximately half of the normal solute content of urine.22
Urea’s excretion mechanisms are well characterized.22,23 It is absorbed in the proximal tubule, the medullary loop of Henle, and the medullary collecting ducts via facilitated diffusion through specific urea transporters.24 After being absorbed in the loop of Henle, urea is resecreted, a process that creates an osmotic gradient along the medulla that ultimately regulates urea excretion and reabsorption in the medullary collecting duct. Low-volume states are associated with decreased urea excretion due to a physiologic increase in antidiuretic hormone secretion, and the reverse is true for high-volume states.
The FEU has been recognized as a clinically useful tool. The correlation between serum and urine urea concentrations was investigated as early as 1904.25 However, most studies during the ensuing century focused on the serum urea concentration or the creatinine-to-urea ratio as a measure of glomerular failure.26–28 In 1992, Kaplan and Kohn29 proposed that the FEU could be a useful measure for assessing renal dysfunction in acute kidney injury. Conceptually similar to the FENa, the FEU is calculated as:
An FEU less than 35% suggests a prerenal cause of acute kidney injury, while a value greater than 50% suggests an intrinsic one.
FRACTIONAL EXCRETION OF UREA VS FRACTIONAL EXCRETION OF SODIUM
Kaplan and Kohn (1992)
Kaplan and Kohn,29 in their 1992 study, retrospectively analyzed 87 urine samples from 40 patients with renal dysfunction (not specifically acute kidney injury) thought to be secondary to volume depletion in which the FENa was discordant with the FEU.
Findings. Thirty-nine of the 40 patients treated with diuretics had a high FENa value. However, the FEU was low in all of these patients, leading the authors to conclude that the latter may be the more useful of the two indices in evaluating patients receiving diuretics who present with symptoms that suggest prerenal azotemia.
Limitations of the study. On closer inspection, these findings were not generalizable, for several reasons. First, the time that elapsed between administration of diuretics and evaluation of urinary electrolytes varied widely. Additionally, the study was a retrospective analysis of isolated urine specimens without clear correlation to a clinical patient or context. For these reasons, prospective analyses to investigate the utility of the fractional excretion of urea needed to be conducted.
Carvounis et al (2002)
Carvounis et al30 prospectively evaluated the FENa and the FEU in 102 consecutive intensive care patients with acute kidney injury (defined as a serum creatinine concentration > 1.5 mg/dL or an increase of more than 0.5 mg/dL in less than 48 hours). Oliguria was not an inclusion criterion for the study, but patients with acute glomerulonephritis and obstructive nephropathy were excluded. The study grouped subjects into those with prerenal azotemia, prerenal azotemia plus diuretic use, or acute tubular necrosis on the basis of the clinical diagnosis of the attending nephrologist.
Findings. The FEU was more sensitive than the FENa in detecting prerenal azotemia, especially in those with prerenal azotemia who were receiving diuretics. Overall, the FEU had higher sensitivity and specificity for prerenal azotemia regardless of diuretic usage, and more importantly, the best overall positive and negative predictive value for detecting it (99% and 75% respectively).
These results indicate that, in patients given diuretics, the FENa fails to discriminate between prerenal azotemia and acute tubular necrosis. Conversely, the FEU was excellent in discriminating between all cases of prerenal azotemia and acute tubular necrosis irrespective of the use of diuretics. This has significant practical application, given the frequency of diuretic use in the hospital, particularly in intensive care patients.
Limitations of the study. While the findings supported the utility of the FEU, the study population was limited to intensive care patients. Furthermore, the authors did not report the statistical significance of their findings.30
Pépin et al (2007)
Pépin et al8 performed a similar study, investigating the diagnostic utility of the FENa and the FEU in patients with acute kidney injury, with or without diuretic therapy.
The authors prospectively studied 99 consecutive patients confirmed by an independent nephrologist to have acute kidney injury (defined as an increase in serum creatinine of more than 30% over baseline values within less than 1 week) due to either volume depletion or ischemia. They excluded patients with less common causes of acute kidney injury, such as rhabdomyolysis, obstructive nephropathy, adrenal insufficiency, acute glomerulonephritis, and nephrotoxic acute kidney injury, as well as patients with chronic kidney disease.
Patients were grouped into those with transient acute kidney injury (from decreased kidney perfusion) and persistent acute kidney injury (attributed to acute tubular necrosis), with or without diuretic therapy, according to predefined clinical criteria. They were considered to have diuretic exposure if they had received furosemide (Lasix) within 24 hours or a thiazide within 48 hours of sampling.
Findings. The FENa proved superior to the FEU in patients not taking diuretics and, contrary to the findings of Carvounis et al,30 exhibited diagnostic utility in patients taking diuretics as well. Neither index discriminated between the different etiologies exceptionally well, however.
Of note, the study population was more inclusive than in previous studies, with only 63 intensive care patients, thus making the results more generalizable to all cases of inpatient acute kidney injury. Furthermore, the study included patients with and without oliguria, and the sensitivity and specificity of both the FENa and the FEU were higher in the nonoliguric group (n = 25).
Limitations of the study. The authors admit that a long time may have elapsed between diuretic administration and urine measurements, thereby mitigating the diuretic’s natriuretic effect independent of the patient’s volume status. While this variable may account for the better performance of the FENa than in the other studies, it does not account for the poor performance of the FEU.
Additionally, few of the findings reached statistical significance.
Lastly, a high percentage (30%) of patients had sepsis. The FEU is less effective in patients with infection, as cytokines interfere with the urea transporters in the kidney and colon.31
Lim et al (2009)
Lim et al32 conducted a study similar in design to that of Pépin et al.8
Findings. The FEU was as clinically useful as the FENa at distinguishing transient from persistent acute kidney injury in patients on diuretics. Using a cutoff FEU of less than 30% and a cutoff FENa of less than 1.5% for transient acute kidney injury (based on calculated receiver operating characteristic curves), FENa was more sensitive and specific than FEU in the nondiuretic groups. In patients exposed to diuretics, FEU was more sensitive but less specific than FENa.
FRACTIONAL EXCRETION OF UREA IN OLIGURIA
Diskin et al (2010)
In 2010, Diskin et al33 published a prospective, observational study of 100 consecutive patients with oliguric azotemia referred to a nephrology service. They defined acute kidney injury as serum creatinine concentration greater than 1.9 mg/dL and urine output less than 100 mL in 24 hours. They used a higher FEU cutoff for prerenal azotemia of less than 40% to reflect the known urea secretion rate in oliguric patients (600 mL/24 hours). They used an FENa of less than 1% and greater than 3% to distinguish prerenal azotemia from acute tubular necrosis.
Findings. The FEU was more accurate than the FENa, giving the right diagnosis in 95% vs 54% of cases (P < .0001). The difference was exclusively due to the FEU’s greater utility in the 67 patients who had received diuretics (98% vs 49%, P < .0001). Both the FEU and the FENa accurately detected acute tubular necrosis. As expected, the FENa outperformed FEU in the setting of infection, in which cytokine stimulation interferes with urea excretion.
Limitations of the study. Approximately 80% of the patients had prerenal azotemia, potentially biasing the results toward a test geared toward detecting this condition. However, since prerenal causes are more common than intrinsic causes, the authors argued that their cohort more accurately reflected the population encountered in clinical practice.
Additionally, only patients with oliguria and more advanced kidney injury (serum creatinine > 1.9 mg/dL) were included in the study, potentially limiting the applicability of these results in patients with preserved urine output in the early stages of renal failure.
Table 2 summarizes the findings of the studies discussed above.8,15,30,32,33
FRACTIONAL EXCRETION OF UREA IN CHILDREN AND THE ELDERLY
The FEU has also been validated in populations at the extremes of age.
In children, Fahimi et al34 performed a cross-sectional study in 43 patients referred to a nephrology service because of acute kidney injury.
An FEU less than 35% had greater sensitivity and specificity than an FENa less than 1% for differentiating prerenal from intrinsic causes in pediatric populations. An FEU of less than 30% had an even greater power of distinguishing between the two. Interestingly, 15 of the 26 patients in the group with prerenal azotemia had an FENa greater than 1%, 8 of whom had an obvious cause (diuretic therapy in 5, salt-losing congenital adrenal hyperplasia in 2, and metabolic alkalosis in 1).
In elderly people, urinary indices are less reliable because of reduced sodium and urea reabsorption and urinary concentrating capability. Thus, the FENa and FEU are increased, making the standard cutoff values unreliable and unpredictable for distinguishing prerenal from intrinsic causes of acute kidney injury.35
WHICH TEST SHOULD BE USED?
Both the FENa and the FEU have been validated in prospective trials as useful clinical indices in identifying prerenal azotemia. Results of these studies vary as to which index is superior and when. This may be attributable to the various definitions of acute kidney injury and diagnostic criteria used in the studies as well as the heterogeneity of patients in each study.
However, the preponderance of evidence indicates that the FEU is more useful than the FENa in patients on diuretics. Since diuretics are widely used, particularly in acute care settings in which acute kidney injury is prevalent, the FEU is a useful clinical tool and should be utilized in this context accordingly. Specifically, when there is a history of recent diuretic use, the evidence supports ordering the FEU alone, or at least in conjunction with the FENa. If the two indices yield disparate results, the physician should look for circumstances that would alter each one of them, such as sepsis or an unrecognized dose of diuretic.
In managing acute kidney injury, distinguishing prerenal from intrinsic causes is a difficult task, particularly because prolonged prerenal azotemia can develop into acute tubular necrosis. Therefore, a single index, calculated at a specific time, often is insufficient to properly characterize the pathogenesis of acute kidney injury, and a combination of both of these indices may increase diagnostic sensitivity and specificity.36 Moreover, urine samples collected after acute changes in volume or osmolarity, such as blood loss, administration of intravenous fluids or parenteral nutrition, or dialysis may compromise their diagnostic utility, and care must be taken to interpret the results in the appropriate clinical context.
The clinician must be aware of both the respective applications and limitations of these indices when using them to guide management and navigate the differential diagnosis in the appropriate clinical settings.
- Nolan CR, Anderson RJ. Hospital-acquired acute renal failure. J Am Soc Nephrol 1998; 9:710–718.
- Mehta RL, Pascual MT, Soroko S, et al; Program to Improve Care in Acute Renal Disease. Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney Int 2004; 66:1613–1621.
- Myers BD, Miller DC, Mehigan JT, et al. Nature of the renal injury following total renal ischemia in man. J Clin Invest 1984; 73:329–341.
- Ho E, Fard A, Maisel A. Evolving use of biomarkers for kidney injury in acute care settings. Curr Opin Crit Care 2010; 16:399–407.
- Steiner RW. Low fractional excretion of sodium in myoglobinuric acute renal failure. Arch Intern Med 1982; 142:1216–1217.
- Vaz AJ. Low fractional excretion of urine sodium in acute renal failure due to sepsis. Arch Intern Med 1983; 143:738–739.
- Pru C, Kjellstrand CM. The FENa test is of no prognostic value in acute renal failure. Nephron 1984; 36:20–23.
- Pépin MN, Bouchard J, Legault L, Ethier J. Diagnostic performance of fractional excretion of urea and fractional excretion of sodium in the evaluations of patients with acute kidney injury with or without diuretic treatment. Am J Kidney Dis 2007; 50:566–573.
- Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P; Acute Dialysis Quality Initiative workgroup. Acute renal failure—definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care 2004; 8:R204–R212.
- Mehta RL, Kellum JA, Shah SV, et al; Acute Kidney Injury Network. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care 2007; 11:R31.
- Stevens PE, Tamimi NA, Al-Hasani MK, et al. Non-specialist management of acute renal failure. QJM 2001; 94:533–540.
- Feest TG, Round A, Hamad S. Incidence of severe acute renal failure in adults: results of a community based study. BMJ 1993; 306:481–483.
- Liaño F, Pascual J. Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Madrid Acute Renal Failure Study Group. Kidney Int 1996; 50:811–818.
- Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med 1996; 334:1448–1460.
- Bagshaw SM, George C, Bellomo R; ANZICS Database Management Committee. Changes in the incidence and outcome for early acute kidney injury in a cohort of Australian intensive care units. Crit Care 2007; 11:R68.
- Sodium homeostasis in chronic renal disease. Kidney Int 1982; 21:886–897.
- Espinel CH. The FENa test. Use in the differential diagnosis of acute renal failure. JAMA 1976; 236:579–581.
- Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest 2004; 114:5–14.
- Miller TR, Anderson RJ, Linas SL, et al. Urinary diagnostic indices in acute renal failure: a prospective study. Ann Intern Med 1978; 89:47–50.
- Zarich S, Fang LS, Diamond JR. Fractional excretion of sodium. Exceptions to its diagnostic value. Arch Intern Med 1985; 145:108–112.
- Mandal AK, Baig M, Koutoubi Z. Management of acute renal failure in the elderly. Treatment options. Drugs Aging 1996; 9:226–250.
- Sands JM. Critical role of urea in the urine-concentrating mechanism. J Am Soc Nephrol 2007; 18:670–671.
- Goldstein MH, Lenz PR, Levitt MF. Effect of urine flow rate on urea reabsorption in man: urea as a “tubular marker”. J Appl Physiol 1969; 26:594–599.
- Fenton RA, Knepper MA. Urea and renal function in the 21st century: insights from knockout mice. J Am Soc Nephrol 2007; 18:679–688.
- Gréhant N. Physiologique des reins par le dosage de l’urée dans le sang et dans l’urine. J Physiol Pathol Gen (Paris) 1904; 6:1–8.
- Dossetor JB. Creatininemia versus uremia. The relative significance of blood urea nitrogen and serum creatinine concentrations in azotemia. Ann Intern Med 1966; 65:1287–1299.
- Kahn S, Sagel J, Eales L, Rabkin R. The significance of serum creatinine and the blood urea-serum creatinine ratio in azotaemia. S Afr Med J 1972; 46:1828–1832.
- Kerr DNS, Davison JM. The assessment of renal function. Br J Hosp Med 1975; 14:360–372.
- Kaplan AA, Kohn OF. Fractional excretion of urea as a guide to renal dysfunction. Am J Nephrol 1992; 12:49–54.
- Carvounis CP, Nisar S, Guro-Razuman S. Significance of the fractional excretion of urea in the differential diagnosis of acute renal failure. Kidney Int 2002; 62:2223–2229.
- Schmidt C, Höcherl K, Bucher M. Cytokine-mediated regulation of urea transporters during experimental endotoxemia. Am J Physiol Renal Physiol 2007; 292:F1479–F1489.
- Lim DH, Jeong JM, Oh SH, et al. Diagnostic performance of fractional excretion of urea in evaluating patients with acute kidney injury with diuretics treatment. Korean J Nephrol 2009; 28:190–198.
- Diskin CJ, Stokes TJ, Dansby LM, Radcliff L, Carter TB. The comparative benefits of the fractional excretion of urea and sodium in various azotemic oliguric states. Nephron Clin Pract 2010; 114:c145–c150.
- Fahimi D, Mohajeri S, Hajizadeh N, et al. Comparison between fractional excretions of urea and sodium in children with acute kidney injury. Pediatr Nephrol 2009; 24:2409–2412.
- Musso CG, Liakopoulos V, Ioannidis I, Eleftheriadis T, Stefanidis I. Acute renal failure in the elderly: particular characteristics. Int Urol Nephrol 2006; 38:787–793.
- Schönermarck U, Kehl K, Samtleben W. Diagnostic performance of fractional excretion of urea and sodium in acute kidney injury. Am J Kidney Dis 2008; 51:870–871.
- Nolan CR, Anderson RJ. Hospital-acquired acute renal failure. J Am Soc Nephrol 1998; 9:710–718.
- Mehta RL, Pascual MT, Soroko S, et al; Program to Improve Care in Acute Renal Disease. Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney Int 2004; 66:1613–1621.
- Myers BD, Miller DC, Mehigan JT, et al. Nature of the renal injury following total renal ischemia in man. J Clin Invest 1984; 73:329–341.
- Ho E, Fard A, Maisel A. Evolving use of biomarkers for kidney injury in acute care settings. Curr Opin Crit Care 2010; 16:399–407.
- Steiner RW. Low fractional excretion of sodium in myoglobinuric acute renal failure. Arch Intern Med 1982; 142:1216–1217.
- Vaz AJ. Low fractional excretion of urine sodium in acute renal failure due to sepsis. Arch Intern Med 1983; 143:738–739.
- Pru C, Kjellstrand CM. The FENa test is of no prognostic value in acute renal failure. Nephron 1984; 36:20–23.
- Pépin MN, Bouchard J, Legault L, Ethier J. Diagnostic performance of fractional excretion of urea and fractional excretion of sodium in the evaluations of patients with acute kidney injury with or without diuretic treatment. Am J Kidney Dis 2007; 50:566–573.
- Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P; Acute Dialysis Quality Initiative workgroup. Acute renal failure—definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care 2004; 8:R204–R212.
- Mehta RL, Kellum JA, Shah SV, et al; Acute Kidney Injury Network. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care 2007; 11:R31.
- Stevens PE, Tamimi NA, Al-Hasani MK, et al. Non-specialist management of acute renal failure. QJM 2001; 94:533–540.
- Feest TG, Round A, Hamad S. Incidence of severe acute renal failure in adults: results of a community based study. BMJ 1993; 306:481–483.
- Liaño F, Pascual J. Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Madrid Acute Renal Failure Study Group. Kidney Int 1996; 50:811–818.
- Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med 1996; 334:1448–1460.
- Bagshaw SM, George C, Bellomo R; ANZICS Database Management Committee. Changes in the incidence and outcome for early acute kidney injury in a cohort of Australian intensive care units. Crit Care 2007; 11:R68.
- Sodium homeostasis in chronic renal disease. Kidney Int 1982; 21:886–897.
- Espinel CH. The FENa test. Use in the differential diagnosis of acute renal failure. JAMA 1976; 236:579–581.
- Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest 2004; 114:5–14.
- Miller TR, Anderson RJ, Linas SL, et al. Urinary diagnostic indices in acute renal failure: a prospective study. Ann Intern Med 1978; 89:47–50.
- Zarich S, Fang LS, Diamond JR. Fractional excretion of sodium. Exceptions to its diagnostic value. Arch Intern Med 1985; 145:108–112.
- Mandal AK, Baig M, Koutoubi Z. Management of acute renal failure in the elderly. Treatment options. Drugs Aging 1996; 9:226–250.
- Sands JM. Critical role of urea in the urine-concentrating mechanism. J Am Soc Nephrol 2007; 18:670–671.
- Goldstein MH, Lenz PR, Levitt MF. Effect of urine flow rate on urea reabsorption in man: urea as a “tubular marker”. J Appl Physiol 1969; 26:594–599.
- Fenton RA, Knepper MA. Urea and renal function in the 21st century: insights from knockout mice. J Am Soc Nephrol 2007; 18:679–688.
- Gréhant N. Physiologique des reins par le dosage de l’urée dans le sang et dans l’urine. J Physiol Pathol Gen (Paris) 1904; 6:1–8.
- Dossetor JB. Creatininemia versus uremia. The relative significance of blood urea nitrogen and serum creatinine concentrations in azotemia. Ann Intern Med 1966; 65:1287–1299.
- Kahn S, Sagel J, Eales L, Rabkin R. The significance of serum creatinine and the blood urea-serum creatinine ratio in azotaemia. S Afr Med J 1972; 46:1828–1832.
- Kerr DNS, Davison JM. The assessment of renal function. Br J Hosp Med 1975; 14:360–372.
- Kaplan AA, Kohn OF. Fractional excretion of urea as a guide to renal dysfunction. Am J Nephrol 1992; 12:49–54.
- Carvounis CP, Nisar S, Guro-Razuman S. Significance of the fractional excretion of urea in the differential diagnosis of acute renal failure. Kidney Int 2002; 62:2223–2229.
- Schmidt C, Höcherl K, Bucher M. Cytokine-mediated regulation of urea transporters during experimental endotoxemia. Am J Physiol Renal Physiol 2007; 292:F1479–F1489.
- Lim DH, Jeong JM, Oh SH, et al. Diagnostic performance of fractional excretion of urea in evaluating patients with acute kidney injury with diuretics treatment. Korean J Nephrol 2009; 28:190–198.
- Diskin CJ, Stokes TJ, Dansby LM, Radcliff L, Carter TB. The comparative benefits of the fractional excretion of urea and sodium in various azotemic oliguric states. Nephron Clin Pract 2010; 114:c145–c150.
- Fahimi D, Mohajeri S, Hajizadeh N, et al. Comparison between fractional excretions of urea and sodium in children with acute kidney injury. Pediatr Nephrol 2009; 24:2409–2412.
- Musso CG, Liakopoulos V, Ioannidis I, Eleftheriadis T, Stefanidis I. Acute renal failure in the elderly: particular characteristics. Int Urol Nephrol 2006; 38:787–793.
- Schönermarck U, Kehl K, Samtleben W. Diagnostic performance of fractional excretion of urea and sodium in acute kidney injury. Am J Kidney Dis 2008; 51:870–871.
KEY POINTS
- Finding the cause of acute kidney injury is important, as management strategies differ.
- Although cutoff values differ among studies, in a patient with acute kidney injury, an FENa lower than 1% suggests a prerenal cause, whereas a value higher than 3% suggests an intrinsic cause.
- Similarly, an FEU less than 35% suggests a prerenal cause of acute kidney injury, whereas a value higher than 50% suggests an intrinsic one.
- The FENa can be falsely high in patients taking a diuretic; it can be falsely low in a number of intrinsic renal conditions, such as contrast-induced nephropathy, rhabdomyolysis, and acute glomerulonephritis.
Deep brain stimulation: What can patients expect from it?
Deep brain stimulation is an important therapy for Parkinson disease and other movement disorders. It involves implantation of a pulse generator that can be adjusted by telemetry and can be activated and deactivated by clinicians and patients. It is therefore also a good investigational tool, allowing for double-blind, sham-controlled clinical trials by testing the effects of the stimulation with optimal settings compared with no stimulation.
This article will discuss the approved indications for deep brain stimulation (particularly for managing movement disorders), the benefits that can be expected, the risks, the complications, the maintenance required, how candidates for this treatment are evaluated, and the surgical procedure for implantation of the devices.
DEVICE SIMILAR TO HEART PACEMAKERS
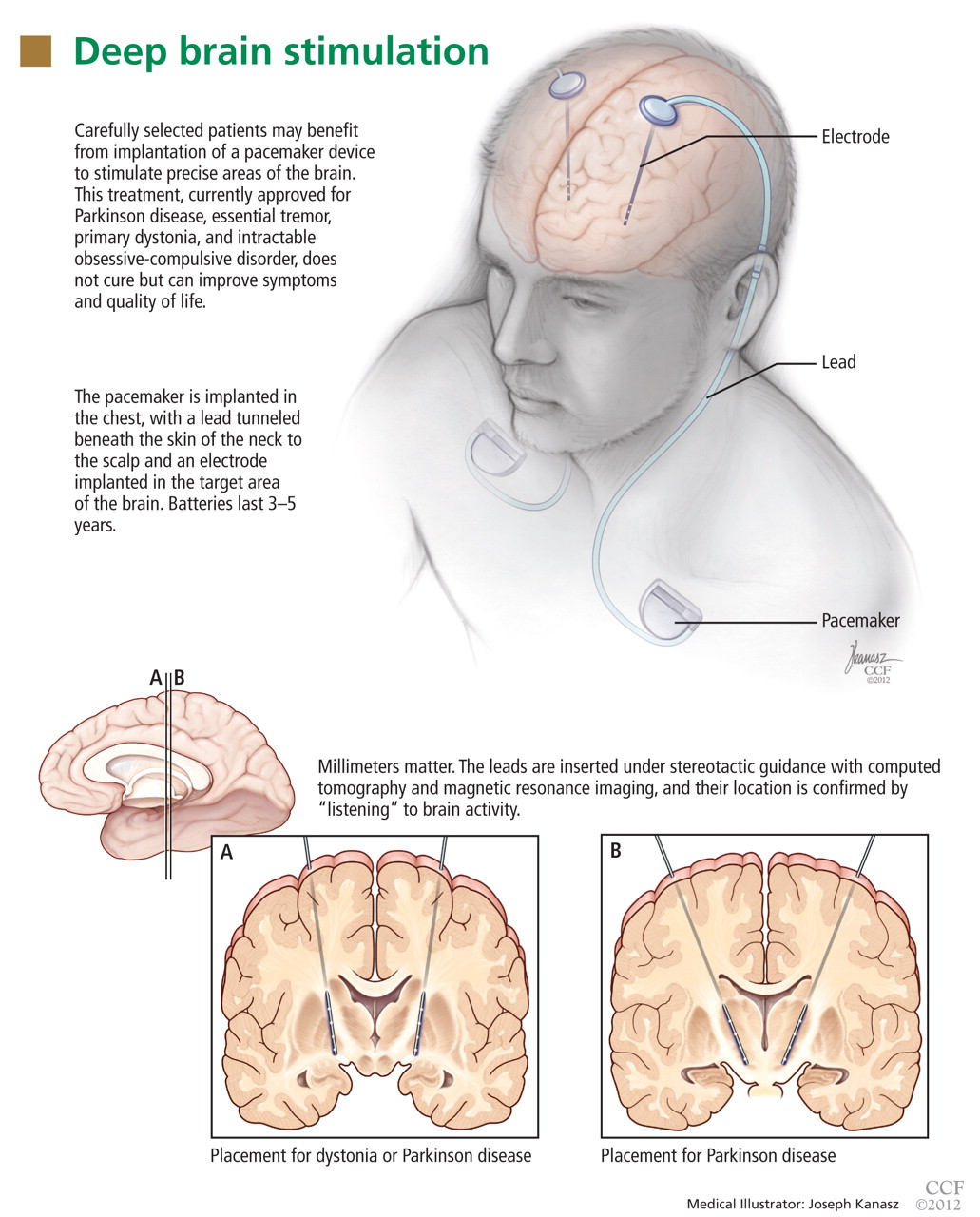
The deep brain stimulation system must be programmed by a physician or midlevel practitioner by observing a symptom and then changing the applied settings to the pulse generator until the symptom improves. This can be a very time-consuming process.
In contrast to heart pacemakers, which run at low frequencies, the brain devices for movement disorders are almost always set to a high frequency, greater than 100 Hz. For this reason, they consume more energy and need larger batteries than those in modern heart pacemakers.
The batteries in these generators typically last 3 to 5 years and are replaced in an outpatient procedure. Newer, smaller, rechargeable devices are expected to last longer but require more maintenance and care by patients, who have to recharge them at home periodically.
INDICATIONS FOR DEEP BRAIN STIMULATION
Deep brain stimulation is approved by the US Food and Drug Administration (FDA) for specific indications:
- Parkinson disease
- Essential tremor
- Primary dystonia (under a humanitarian device exemption)
- Intractable obsessive-compulsive disorder (also under a humanitarian device exemption). We will not discuss this indication further in this paper.
For each of these conditions, deep brain stimulation is considered when nonsurgical management has failed, as is the case for most functional neurosurgical treatments.
Investigations under way in other disorders
Several studies of deep brain stimulation are currently in progress under FDA-approved investigational device exemptions. Some, with funding from industry, are exploring its use in neuropsychiatric conditions other than parkinsonism. Two large clinical trials are evaluating its use for treatment-refractory depression, a common problem and a leading cause of disability in the industrialized world. Multiple investigators are also exploring novel uses of this technology in disorders ranging from obsessive-compulsive disorder to epilepsy.
Investigation is also under way at Cleveland Clinic in a federally funded, prospective, randomized clinical trial of deep brain stimulation for patients with thalamic pain syndrome. The primary hypothesis is that stimulation of the ventral striatal and ventral capsular area will modulate the affective component of this otherwise intractable pain syndrome, reducing pain-related disability and improving quality of life.
DEEP BRAIN STIMULATION VS ABLATION
Before deep brain stimulation became available, the only surgical options for patients with advanced Parkinson disease, tremor, or dystonia were ablative procedures such as pallidotomy (ablation of part of the globus pallidus) and thalamotomy (ablation of part of the thalamus). These procedures had been well known for several decades but fell out of favor when levodopa became available in the 1960s and revolutionized the medical treatment of Parkinson disease.
Surgery for movement disorders, in particular Parkinson disease, had a rebirth in the late 1980s when the limitations and complications associated with the pharmacologic management of Parkinson disease became increasingly evident. Ablative procedures are still used to treat advanced Parkinson disease, but much less commonly in industrialized countries.
Although pallidotomy and thalamotomy can have excellent results, they are not as safe as deep brain stimulation, which has the advantage of being reversible, modulating the function of an area rather than destroying it. Any unwanted effect can be immediately altered or reversed, unlike ablative procedures, in which any change is permanent. In addition, deep brain stimulation is adjustable, and the settings can be optimized as the disease progresses over the years.
Ablative procedures can be risky when performed bilaterally, while deep brain stimulation is routinely done on both hemispheres for patients with bilateral symptoms.
Although deep brain stimulation is today’s surgical treatment of choice, it is not perfect. It has the disadvantage of requiring lifelong maintenance of the hardware, for which the patient remains dependent on a medical center. Patients are usually seen more often at the specialized center in the first few months after surgery for optimization of programming and titration of drugs. (During this time, most patients see a gradual, substantial reduction in medication intake.) They are then followed by their physician and visit the center less often for monitoring of disease status and for further adjustments to the stimulator.
Most patients, to date, receive nonrechargeable pulse generators. As mentioned above, the batteries in these devices typically last 3 to 5 years. Preferably, batteries are replaced before they are completely depleted, to avoid interruption of therapy. Periodic visits to the center allow clinicians to estimate battery expiration ahead of time and plan replacements accordingly.
Rechargeable pulse generators have been recently introduced and are expected to last up to 9 years. They are an option for patients who can comply with the requirements for periodic home recharging of the hardware.
Patients are given a remote control so that they can turn the device on or off and check its status. Most patients keep it turned on all the time, although some turn it off at night to save battery life.
WHAT CAN PARKINSON PATIENTS EXPECT FROM THIS THERAPY?
Typically, some parkinsonian symptoms predominate over others, although some patients with advanced disease present with a severe combination of multiple disabling symptoms. Deep brain stimulation is best suited to address some of the cardinal motor symptoms, particularly tremor, rigidity, and bradykinesia, and motor fluctuations such as “wearing off” and dyskinesia.
Improvement in some motor symptoms
As a general rule, appendicular symptoms such as limb tremor and rigidity are more responsive to this therapy than axial symptoms such as gait and balance problems, but some patients experience improvement in gait as well. Other symptoms, such as swallowing or urinary symptoms, are seldom helped.
Although deep brain stimulation can help manage key motor symptoms and improve quality of life, it does not cure Parkinson disease. Also, there is no evidence to date that it slows disease progression, although this is a topic of ongoing investigation.
Fewer motor fluctuations
A common complaint of patients with advanced Parkinson disease is frequent—and often unpredictable—fluctuations between the “on” state (ie, when the effects of the patient’s levodopa therapy are apparent) and the “off” state (ie, when the levodopa doesn’t seem to be working). Sometimes, in the on state, patients experience involuntary choreic or ballistic movements, called dyskinesias. They also complain that the on time progressively lasts shorter and the day is spent alternating between shorter on states (during which the patient may be dyskinetic) and longer off states, limiting the patient’s independence and quality of life.
Deep brain stimulation can help patients prolong the on time while reducing the amplitude of these fluctuations so that the symptoms are not as severe in the off time and dyskinesias are reduced in the on time.
Some patients undergo deep brain stimulation primarily for managing the adverse effects of levodopa rather than for controlling the symptoms of the disease itself. While these patients need levodopa to address the disabling symptoms of the disease, they also present a greater sensitivity for developing levodopa-induced dyskinesias, quickly fluctuating from a lack of movement (the off state) to a state of uncontrollable movements (during the on state).
Deep brain stimulation typically allows the dosage of levodopa to be significantly reduced and gives patients more on time with fewer side effects and less fluctuation between the on and off states.
Response to levodopa predicts deep brain stimulation’s effects
Whether a patient is likely to be helped by deep brain stimulation can be tested with reasonable predictability by giving a single therapeutic dose of levodopa after the patient has been free of the drug for 12 hours. If there is an obvious difference on objective quantitative testing between the off and on states with a single dose, the patient is likely to benefit from deep brain stimulation. Those who do not respond well or are known to have never been well controlled by levodopa are likely poor candidates.
The test is also used as an indicator of whether the patient’s gait can be improved. Patients whose gait is substantially improved by levodopa, even for only a brief period of time, have a better chance of experiencing improvement in this domain with deep brain stimulation than those who do not show any gait improvement.
An important and notable exception to this rule is tremor control. Even Parkinson patients who do not experience significant improvement in tremor with levodopa (ie, who have medication-resistant tremors) are still likely to benefit from deep brain stimulation. Overall, tremor is the symptom that is most consistently improved with deep brain stimulation.
Results of clinical trials
Several clinical trials have demonstrated that deep brain stimulation plus medication works better than medications alone for advanced Parkinson disease.
Deuschl et al1 conducted a randomized trial in 156 patients with advanced Parkinson disease. Patients receiving subthalamic deep brain stimulation plus medication had significantly greater improvement in motor symptoms as measured by the Unified Parkinson’s Disease Rating Scale as well as in quality-of-life measures than patients receiving medications only.
Krack et al2 reported on the outcomes of 49 patients with advanced Parkinson disease who underwent deep brain stimulation and then were prospectively followed. At 5 years, motor function had improved by approximately 55% from baseline, activities-of-daily-living scores had improved by 49%, and patients continued to need significantly less levodopa and to experience less drug-induced dyskinesia.
Complications related to deep brain stimulation occurred in both studies, including two large intracerebral hemorrhages, one of which was fatal.
Weight gain. During the first 3 months after the device was implanted, patients tended to gain weight (mean 3 kg, maximum 5 kg). Although weight gain is considered an adverse effect, many patients are quite thin by the time they are candidates for deep brain stimulation, and in such cases gaining lean weight can be a benefit.
Patients with poorly controlled Parkinson disease lose weight for several reasons: increased calorie expenditure from shaking and excessive movements; diet modification and protein restriction for some patients who realize that protein competes with levodopa absorption; lack of appetite due to depression or from poor taste sensation (due to anosmia); and decreased overall food consumption due to difficulty swallowing.
DEEP BRAIN STIMULATION FOR ESSENTIAL TREMOR
Essential tremor is more common than Parkinson disease, with a prevalence in the United States estimated at approximately 4,000 per 100,000 people older than 65 years.
The tremor is often bilateral and is characteristically an action tremor, but in many patients it also has a postural, and sometimes a resting, component. It is distinct from parkinsonian tremor, which is usually predominantly a resting tremor. The differential diagnosis includes tremors secondary to central nervous system degenerative disorders as well as psychogenic tremors.
Drinking alcohol tends to relieve essential tremors, a finding that can often be elicited in the patient’s history. Patients whose symptoms improve with an alcoholic beverage are more likely to have essential tremor than another diagnosis.
Response to deep brain stimulation
Most patients with essential tremor respond well to deep brain stimulation of the contralateral ventral intermedius thalamic nucleus.
Treatment is usually started unilaterally, usually aimed at alleviating tremor in the patient’s dominant upper extremity. In selected cases, preference is given to treating the nondominant extremity when it is more severely affected than the dominant extremity.
Implantation of a device on the second side is offered to some patients who continue to be limited in activity and quality of life due to tremor of the untreated extremity. Surgery of the second side can be more complicated than the initial unilateral procedure. In particular, some patients may present with dysarthria, although that seems to be less common in our experience than initially estimated.
In practice, patients with moderate tremors tend to have an excellent response to deep brain stimulation. For this particular indication, if the response is not satisfactory, the treating team tends to consider surgically revising the placement of the lead rather than considering the patient a nonresponder. Patients with very severe tremors may have some residual tremor despite substantial improvement in severity. In our experience, patients with a greater proximal component of tremor tend to have less satisfactory results.
For challenging cases, implantation of additional electrodes in the thalamus or in new targets currently under investigation is sometimes considered, although this is an off-label use.
Treatment of secondary tremors, such as poststroke tremor or tremor due to multiple sclerosis, is sometimes attempted with deep brain stimulation. This is also an off-label option but is considered in selected cases for quality-of-life management.
Patients with axial tremors such as head or voice tremor are less likely to be helped by deep brain stimulation.
DEEP BRAIN STIMULATION FOR PRIMARY DYSTONIA
Generalized dystonia is a less common but severely impairing movement disorder.
Deep brain stimulation is approved for primary dystonia under a humanitarian device exemption, a regulatory mechanism for less common conditions. Deep brain stimulation is an option for patients who have significant impairment related to dystonia and who have not responded to conservative management such as anticholinergic agents, muscle relaxants, benzodiazepines, levodopa, or combinations of these drugs. Surgery has been shown to be effective for patients with primary generalized dystonia, whether or not they tested positive for a dystonia-related gene such as DYT1.
Kupsch et al3 evaluated 40 patients with primary dystonia in a randomized controlled trial of pallidal (globus pallidus pars interna) active deep brain stimulation vs sham stimulation (in which the device was implanted but not activated) for 3 months. Treated patients improved significantly more than controls (39% vs 5%) in the Burke-Fahn- Marsden Dystonia Rating Scale (BFMDRS).4 Similar improvement was noted when those receiving sham stimulation were switched to active stimulation.
During long-term follow-up, the results were generally sustained, with substantial improvement from deep brain stimulation in all movement symptoms evaluated except for speech and swallowing. Unlike improvement in tremor, which is quickly evident during testing in the operating room, the improvement in dystonia occurs gradually, and it may take months for patients to notice a change. Similarly, if stimulation stops because of device malfunction or dead batteries, symptoms sometimes do not recur for weeks or months.
Deep brain stimulation is sometimes offered to patients with dystonia secondary to conditions such as cerebral palsy or trauma (an off-label use). Although benefits are less consistent, deep brain stimulation remains an option for these individuals, aimed at alleviating some of the disabling symptoms. In patients with cerebral palsy or other secondary dystonias, it is sometimes difficult to distinguish how much of the disability is related to spasticity vs dystonia. Deep brain stimulation aims to alleviate the dystonic component; the spasticity may be managed with other options such as intrathecal baclofen (Lioresal).
Patients with tardive dystonia, which is usually secondary to treatment with antipsychotic agents, have been reported to respond well to bilateral deep brain stimulation. Gruber et al5 reported on a series of nine patients with a mean follow-up of 41 months. Patients improved by a mean of approximately 74% on the BFMDRS after 3 to 6 months of deep brain stimulation compared with baseline. None of the patients presented with long-term adverse effects, and quality of life and disability scores also improved significantly.
CANDIDATES ARE EVALUATED BY A MULTIDISCIPLINARY TEAM
Cleveland Clinic conducts a comprehensive 2-day evaluation for patients being considered for deep brain stimulation surgery, including consultations with specialists in neurology, neurosurgery, neuropsychology, and psychiatry.
Patients with significant cognitive deficits—near or meeting the diagnostic criteria for dementia—are usually not recommended to have surgery for Parkinson disease. Deep brain stimulation is not aimed at alleviating cognitive issues related to Parkinson disease or other concomitant dementia. In addition, there is a risk that neurostimulation could further worsen cognitive function in the already compromised brain. Moreover, patients with significant abnormalities detected by neuroimaging may have their diagnosis reconsidered in some cases, and some patients may not be deemed ideal candidates for surgery.
An important part of the process is a discussion with the patient and family about the risks and the potential short-term and long-term benefits. Informed consent requires a good understanding of this equation. Patients are counseled to have realistic expectations about what the procedure can offer. Deep brain stimulation can help some of the symptoms of Parkinson disease but will not cure it, and there is no evidence to date that it reduces its progress. At 5 or 10 years after surgery, patients are expected to be worse overall than they were in the first year after surgery, because of disease progression. However, patients who receive this treatment are expected, in general, to be doing better 5 or 10 years later (or longer) than those who do not receive it.
In addition to the discussion about risks, benefits, and expectations, a careful discussion is also devoted to hardware maintenance, including how to change the batteries. Particularly, younger patients should be informed about the risk of breakage of the leads and the extension wire, as they are likely to outlive their implant. Patients and caregivers should be able to come to the specialized center should hardware malfunction occur.
Patients are also informed that after the system is implanted they cannot undergo magnetic resonance imaging (MRI) except of the head, performed with a specific head coil and under specific parameters. MRI of any other body part and with a body coil is contraindicated.
HOW THE DEVICE IS IMPLANTED
There are several options for implanting a deep brain stimulation device.
Implantation with the patient awake, using a stereotactic headframe
At Cleveland Clinic, we usually prefer implantation with a stereotactic headframe. The base or “halo” of the frame is applied to the head under local anesthesia, followed by imaging via computed tomography (Figure 1). Typically, the tomographic image is fused to a previously acquired MRI image, but the MRI is sometimes either initially performed or repeated on the day of surgery.
Patients are sedated for the beginning of the procedure, while the surgical team is opening the skin and drilling the opening in the skull for placement of the lead. The patient is awakened for placement of the electrodes, which is not painful.
Microelectrode recording is typically performed in order to refine the targeting based on the stereotactic coordinates derived from neuroimaging. Although cadaver atlases exist and provide a guide to the stereotactic localization of subcortical structures, they are not completely accurate in representing the brain anatomy of all patients.
By “listening” to cells and knowing their characteristic signals in specific areas, landmarks can be created, forming an individualized map of the patient’s brain target. Microelectrode recording is invasive and has risks, including the risk of a brain hemorrhage. It is routinely done in most specialized deep brain stimulation centers because it can provide better accuracy and precision in lead placement.
When the target has been located and refined by microelectrode recording, the permanent electrode is inserted. Fluoroscopy is usually used to verify the direction and stability of placement during the procedure.
An intraoperative test of the effects of deep brain stimulation is routinely performed to verify that some benefits can be achieved with the brain lead in its location, to determine the threshold for side effects, or both. For example, the patient may be asked to hold a cup as if trying to drink from it and to write or to draw a spiral on a clipboard to assess for improvements in tremor. Rigidity and bradykinesia can also be tested for improvements.
This intraoperative test is not aimed at assessing the best possible outcome of deep brain stimulation, and not even to see an improvement in all symptoms that burden the patient. Rather, it is to evaluate the likelihood that programming will be feasible with the implanted lead.
Subsequently, implantation of the pulse generator in the chest and connection to the brain lead is completed, usually with the patient under general anesthesia.
Implantation under general anesthesia, with intraoperative MRI
A new alternative to “awake stereotactic surgery” is implantation with the patient under general anesthesia, with intraoperative MRI. We have started to do this procedure in a new operating suite that is attached to an MRI suite. The magnet can be taken in and out of the operating room, allowing the surgeon to verify the location of the implanted leads right at the time of the procedure. In this fashion, intraoperative images are used to guide implantation instead of awake microelectrode recording. This is a new option for patients who cannot tolerate awake surgery and for those who have a contraindication to the regular stereotactic procedure with the patient awake.
Risks of bleeding and infection
The potential complications of implanting a device and leads in the brain can be significant.
Hemorrhage can occur, resulting in a superficial or deep hematoma.
Infection and erosion may require removal of the hardware for antibiotic treatment and possible reimplantation.
Other risks include those related to tunneling the wires from the head to the chest, to implanting the device in the chest, and to serious medical complications after surgery. Hardware failure can occur and requires additional surgery. Finally, environmental risks and risks related to medical devices such as MRI, electrocautery, and cardioversion should also be considered.
Deep brain stimulation is advantageous for its reversibility. If during postoperative programming the brain leads are considered not to be ideally placed, revisions can be done to reposition the leads.
- Deuschl G, Schade-Brittinger C, Krack P, et al; German Parkinson Study Group, Neurostimulation Section. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med 2006; 355:896–908.
- Krack P, Batir A, Van Blercom N, et al. Five-year followup of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med 2003; 349:1925–1934.
- Kupsch A, Benecke R, Müller J, et al; Deep-Brain Stimulation for Dystonia Study Group. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. N Engl J Med 2006; 355:1978–1990.
- Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C, Friedman J. Validity and reliability of a rating scle for the primary torsion dystonias. Neurology 1985; 35:73–77.
- Gruber D, Trottenberg T, Kivi A, et al. Long-term effects of pallidal deep brain stimulation in tardive dystonia. Neurology 2009; 73:53–58.
Deep brain stimulation is an important therapy for Parkinson disease and other movement disorders. It involves implantation of a pulse generator that can be adjusted by telemetry and can be activated and deactivated by clinicians and patients. It is therefore also a good investigational tool, allowing for double-blind, sham-controlled clinical trials by testing the effects of the stimulation with optimal settings compared with no stimulation.
This article will discuss the approved indications for deep brain stimulation (particularly for managing movement disorders), the benefits that can be expected, the risks, the complications, the maintenance required, how candidates for this treatment are evaluated, and the surgical procedure for implantation of the devices.
DEVICE SIMILAR TO HEART PACEMAKERS
The deep brain stimulation system must be programmed by a physician or midlevel practitioner by observing a symptom and then changing the applied settings to the pulse generator until the symptom improves. This can be a very time-consuming process.
In contrast to heart pacemakers, which run at low frequencies, the brain devices for movement disorders are almost always set to a high frequency, greater than 100 Hz. For this reason, they consume more energy and need larger batteries than those in modern heart pacemakers.
The batteries in these generators typically last 3 to 5 years and are replaced in an outpatient procedure. Newer, smaller, rechargeable devices are expected to last longer but require more maintenance and care by patients, who have to recharge them at home periodically.
INDICATIONS FOR DEEP BRAIN STIMULATION
Deep brain stimulation is approved by the US Food and Drug Administration (FDA) for specific indications:
- Parkinson disease
- Essential tremor
- Primary dystonia (under a humanitarian device exemption)
- Intractable obsessive-compulsive disorder (also under a humanitarian device exemption). We will not discuss this indication further in this paper.
For each of these conditions, deep brain stimulation is considered when nonsurgical management has failed, as is the case for most functional neurosurgical treatments.
Investigations under way in other disorders
Several studies of deep brain stimulation are currently in progress under FDA-approved investigational device exemptions. Some, with funding from industry, are exploring its use in neuropsychiatric conditions other than parkinsonism. Two large clinical trials are evaluating its use for treatment-refractory depression, a common problem and a leading cause of disability in the industrialized world. Multiple investigators are also exploring novel uses of this technology in disorders ranging from obsessive-compulsive disorder to epilepsy.
Investigation is also under way at Cleveland Clinic in a federally funded, prospective, randomized clinical trial of deep brain stimulation for patients with thalamic pain syndrome. The primary hypothesis is that stimulation of the ventral striatal and ventral capsular area will modulate the affective component of this otherwise intractable pain syndrome, reducing pain-related disability and improving quality of life.
DEEP BRAIN STIMULATION VS ABLATION
Before deep brain stimulation became available, the only surgical options for patients with advanced Parkinson disease, tremor, or dystonia were ablative procedures such as pallidotomy (ablation of part of the globus pallidus) and thalamotomy (ablation of part of the thalamus). These procedures had been well known for several decades but fell out of favor when levodopa became available in the 1960s and revolutionized the medical treatment of Parkinson disease.
Surgery for movement disorders, in particular Parkinson disease, had a rebirth in the late 1980s when the limitations and complications associated with the pharmacologic management of Parkinson disease became increasingly evident. Ablative procedures are still used to treat advanced Parkinson disease, but much less commonly in industrialized countries.
Although pallidotomy and thalamotomy can have excellent results, they are not as safe as deep brain stimulation, which has the advantage of being reversible, modulating the function of an area rather than destroying it. Any unwanted effect can be immediately altered or reversed, unlike ablative procedures, in which any change is permanent. In addition, deep brain stimulation is adjustable, and the settings can be optimized as the disease progresses over the years.
Ablative procedures can be risky when performed bilaterally, while deep brain stimulation is routinely done on both hemispheres for patients with bilateral symptoms.
Although deep brain stimulation is today’s surgical treatment of choice, it is not perfect. It has the disadvantage of requiring lifelong maintenance of the hardware, for which the patient remains dependent on a medical center. Patients are usually seen more often at the specialized center in the first few months after surgery for optimization of programming and titration of drugs. (During this time, most patients see a gradual, substantial reduction in medication intake.) They are then followed by their physician and visit the center less often for monitoring of disease status and for further adjustments to the stimulator.
Most patients, to date, receive nonrechargeable pulse generators. As mentioned above, the batteries in these devices typically last 3 to 5 years. Preferably, batteries are replaced before they are completely depleted, to avoid interruption of therapy. Periodic visits to the center allow clinicians to estimate battery expiration ahead of time and plan replacements accordingly.
Rechargeable pulse generators have been recently introduced and are expected to last up to 9 years. They are an option for patients who can comply with the requirements for periodic home recharging of the hardware.
Patients are given a remote control so that they can turn the device on or off and check its status. Most patients keep it turned on all the time, although some turn it off at night to save battery life.
WHAT CAN PARKINSON PATIENTS EXPECT FROM THIS THERAPY?
Typically, some parkinsonian symptoms predominate over others, although some patients with advanced disease present with a severe combination of multiple disabling symptoms. Deep brain stimulation is best suited to address some of the cardinal motor symptoms, particularly tremor, rigidity, and bradykinesia, and motor fluctuations such as “wearing off” and dyskinesia.
Improvement in some motor symptoms
As a general rule, appendicular symptoms such as limb tremor and rigidity are more responsive to this therapy than axial symptoms such as gait and balance problems, but some patients experience improvement in gait as well. Other symptoms, such as swallowing or urinary symptoms, are seldom helped.
Although deep brain stimulation can help manage key motor symptoms and improve quality of life, it does not cure Parkinson disease. Also, there is no evidence to date that it slows disease progression, although this is a topic of ongoing investigation.
Fewer motor fluctuations
A common complaint of patients with advanced Parkinson disease is frequent—and often unpredictable—fluctuations between the “on” state (ie, when the effects of the patient’s levodopa therapy are apparent) and the “off” state (ie, when the levodopa doesn’t seem to be working). Sometimes, in the on state, patients experience involuntary choreic or ballistic movements, called dyskinesias. They also complain that the on time progressively lasts shorter and the day is spent alternating between shorter on states (during which the patient may be dyskinetic) and longer off states, limiting the patient’s independence and quality of life.
Deep brain stimulation can help patients prolong the on time while reducing the amplitude of these fluctuations so that the symptoms are not as severe in the off time and dyskinesias are reduced in the on time.
Some patients undergo deep brain stimulation primarily for managing the adverse effects of levodopa rather than for controlling the symptoms of the disease itself. While these patients need levodopa to address the disabling symptoms of the disease, they also present a greater sensitivity for developing levodopa-induced dyskinesias, quickly fluctuating from a lack of movement (the off state) to a state of uncontrollable movements (during the on state).
Deep brain stimulation typically allows the dosage of levodopa to be significantly reduced and gives patients more on time with fewer side effects and less fluctuation between the on and off states.
Response to levodopa predicts deep brain stimulation’s effects
Whether a patient is likely to be helped by deep brain stimulation can be tested with reasonable predictability by giving a single therapeutic dose of levodopa after the patient has been free of the drug for 12 hours. If there is an obvious difference on objective quantitative testing between the off and on states with a single dose, the patient is likely to benefit from deep brain stimulation. Those who do not respond well or are known to have never been well controlled by levodopa are likely poor candidates.
The test is also used as an indicator of whether the patient’s gait can be improved. Patients whose gait is substantially improved by levodopa, even for only a brief period of time, have a better chance of experiencing improvement in this domain with deep brain stimulation than those who do not show any gait improvement.
An important and notable exception to this rule is tremor control. Even Parkinson patients who do not experience significant improvement in tremor with levodopa (ie, who have medication-resistant tremors) are still likely to benefit from deep brain stimulation. Overall, tremor is the symptom that is most consistently improved with deep brain stimulation.
Results of clinical trials
Several clinical trials have demonstrated that deep brain stimulation plus medication works better than medications alone for advanced Parkinson disease.
Deuschl et al1 conducted a randomized trial in 156 patients with advanced Parkinson disease. Patients receiving subthalamic deep brain stimulation plus medication had significantly greater improvement in motor symptoms as measured by the Unified Parkinson’s Disease Rating Scale as well as in quality-of-life measures than patients receiving medications only.
Krack et al2 reported on the outcomes of 49 patients with advanced Parkinson disease who underwent deep brain stimulation and then were prospectively followed. At 5 years, motor function had improved by approximately 55% from baseline, activities-of-daily-living scores had improved by 49%, and patients continued to need significantly less levodopa and to experience less drug-induced dyskinesia.
Complications related to deep brain stimulation occurred in both studies, including two large intracerebral hemorrhages, one of which was fatal.
Weight gain. During the first 3 months after the device was implanted, patients tended to gain weight (mean 3 kg, maximum 5 kg). Although weight gain is considered an adverse effect, many patients are quite thin by the time they are candidates for deep brain stimulation, and in such cases gaining lean weight can be a benefit.
Patients with poorly controlled Parkinson disease lose weight for several reasons: increased calorie expenditure from shaking and excessive movements; diet modification and protein restriction for some patients who realize that protein competes with levodopa absorption; lack of appetite due to depression or from poor taste sensation (due to anosmia); and decreased overall food consumption due to difficulty swallowing.
DEEP BRAIN STIMULATION FOR ESSENTIAL TREMOR
Essential tremor is more common than Parkinson disease, with a prevalence in the United States estimated at approximately 4,000 per 100,000 people older than 65 years.
The tremor is often bilateral and is characteristically an action tremor, but in many patients it also has a postural, and sometimes a resting, component. It is distinct from parkinsonian tremor, which is usually predominantly a resting tremor. The differential diagnosis includes tremors secondary to central nervous system degenerative disorders as well as psychogenic tremors.
Drinking alcohol tends to relieve essential tremors, a finding that can often be elicited in the patient’s history. Patients whose symptoms improve with an alcoholic beverage are more likely to have essential tremor than another diagnosis.
Response to deep brain stimulation
Most patients with essential tremor respond well to deep brain stimulation of the contralateral ventral intermedius thalamic nucleus.
Treatment is usually started unilaterally, usually aimed at alleviating tremor in the patient’s dominant upper extremity. In selected cases, preference is given to treating the nondominant extremity when it is more severely affected than the dominant extremity.
Implantation of a device on the second side is offered to some patients who continue to be limited in activity and quality of life due to tremor of the untreated extremity. Surgery of the second side can be more complicated than the initial unilateral procedure. In particular, some patients may present with dysarthria, although that seems to be less common in our experience than initially estimated.
In practice, patients with moderate tremors tend to have an excellent response to deep brain stimulation. For this particular indication, if the response is not satisfactory, the treating team tends to consider surgically revising the placement of the lead rather than considering the patient a nonresponder. Patients with very severe tremors may have some residual tremor despite substantial improvement in severity. In our experience, patients with a greater proximal component of tremor tend to have less satisfactory results.
For challenging cases, implantation of additional electrodes in the thalamus or in new targets currently under investigation is sometimes considered, although this is an off-label use.
Treatment of secondary tremors, such as poststroke tremor or tremor due to multiple sclerosis, is sometimes attempted with deep brain stimulation. This is also an off-label option but is considered in selected cases for quality-of-life management.
Patients with axial tremors such as head or voice tremor are less likely to be helped by deep brain stimulation.
DEEP BRAIN STIMULATION FOR PRIMARY DYSTONIA
Generalized dystonia is a less common but severely impairing movement disorder.
Deep brain stimulation is approved for primary dystonia under a humanitarian device exemption, a regulatory mechanism for less common conditions. Deep brain stimulation is an option for patients who have significant impairment related to dystonia and who have not responded to conservative management such as anticholinergic agents, muscle relaxants, benzodiazepines, levodopa, or combinations of these drugs. Surgery has been shown to be effective for patients with primary generalized dystonia, whether or not they tested positive for a dystonia-related gene such as DYT1.
Kupsch et al3 evaluated 40 patients with primary dystonia in a randomized controlled trial of pallidal (globus pallidus pars interna) active deep brain stimulation vs sham stimulation (in which the device was implanted but not activated) for 3 months. Treated patients improved significantly more than controls (39% vs 5%) in the Burke-Fahn- Marsden Dystonia Rating Scale (BFMDRS).4 Similar improvement was noted when those receiving sham stimulation were switched to active stimulation.
During long-term follow-up, the results were generally sustained, with substantial improvement from deep brain stimulation in all movement symptoms evaluated except for speech and swallowing. Unlike improvement in tremor, which is quickly evident during testing in the operating room, the improvement in dystonia occurs gradually, and it may take months for patients to notice a change. Similarly, if stimulation stops because of device malfunction or dead batteries, symptoms sometimes do not recur for weeks or months.
Deep brain stimulation is sometimes offered to patients with dystonia secondary to conditions such as cerebral palsy or trauma (an off-label use). Although benefits are less consistent, deep brain stimulation remains an option for these individuals, aimed at alleviating some of the disabling symptoms. In patients with cerebral palsy or other secondary dystonias, it is sometimes difficult to distinguish how much of the disability is related to spasticity vs dystonia. Deep brain stimulation aims to alleviate the dystonic component; the spasticity may be managed with other options such as intrathecal baclofen (Lioresal).
Patients with tardive dystonia, which is usually secondary to treatment with antipsychotic agents, have been reported to respond well to bilateral deep brain stimulation. Gruber et al5 reported on a series of nine patients with a mean follow-up of 41 months. Patients improved by a mean of approximately 74% on the BFMDRS after 3 to 6 months of deep brain stimulation compared with baseline. None of the patients presented with long-term adverse effects, and quality of life and disability scores also improved significantly.
CANDIDATES ARE EVALUATED BY A MULTIDISCIPLINARY TEAM
Cleveland Clinic conducts a comprehensive 2-day evaluation for patients being considered for deep brain stimulation surgery, including consultations with specialists in neurology, neurosurgery, neuropsychology, and psychiatry.
Patients with significant cognitive deficits—near or meeting the diagnostic criteria for dementia—are usually not recommended to have surgery for Parkinson disease. Deep brain stimulation is not aimed at alleviating cognitive issues related to Parkinson disease or other concomitant dementia. In addition, there is a risk that neurostimulation could further worsen cognitive function in the already compromised brain. Moreover, patients with significant abnormalities detected by neuroimaging may have their diagnosis reconsidered in some cases, and some patients may not be deemed ideal candidates for surgery.
An important part of the process is a discussion with the patient and family about the risks and the potential short-term and long-term benefits. Informed consent requires a good understanding of this equation. Patients are counseled to have realistic expectations about what the procedure can offer. Deep brain stimulation can help some of the symptoms of Parkinson disease but will not cure it, and there is no evidence to date that it reduces its progress. At 5 or 10 years after surgery, patients are expected to be worse overall than they were in the first year after surgery, because of disease progression. However, patients who receive this treatment are expected, in general, to be doing better 5 or 10 years later (or longer) than those who do not receive it.
In addition to the discussion about risks, benefits, and expectations, a careful discussion is also devoted to hardware maintenance, including how to change the batteries. Particularly, younger patients should be informed about the risk of breakage of the leads and the extension wire, as they are likely to outlive their implant. Patients and caregivers should be able to come to the specialized center should hardware malfunction occur.
Patients are also informed that after the system is implanted they cannot undergo magnetic resonance imaging (MRI) except of the head, performed with a specific head coil and under specific parameters. MRI of any other body part and with a body coil is contraindicated.
HOW THE DEVICE IS IMPLANTED
There are several options for implanting a deep brain stimulation device.
Implantation with the patient awake, using a stereotactic headframe
At Cleveland Clinic, we usually prefer implantation with a stereotactic headframe. The base or “halo” of the frame is applied to the head under local anesthesia, followed by imaging via computed tomography (Figure 1). Typically, the tomographic image is fused to a previously acquired MRI image, but the MRI is sometimes either initially performed or repeated on the day of surgery.
Patients are sedated for the beginning of the procedure, while the surgical team is opening the skin and drilling the opening in the skull for placement of the lead. The patient is awakened for placement of the electrodes, which is not painful.
Microelectrode recording is typically performed in order to refine the targeting based on the stereotactic coordinates derived from neuroimaging. Although cadaver atlases exist and provide a guide to the stereotactic localization of subcortical structures, they are not completely accurate in representing the brain anatomy of all patients.
By “listening” to cells and knowing their characteristic signals in specific areas, landmarks can be created, forming an individualized map of the patient’s brain target. Microelectrode recording is invasive and has risks, including the risk of a brain hemorrhage. It is routinely done in most specialized deep brain stimulation centers because it can provide better accuracy and precision in lead placement.
When the target has been located and refined by microelectrode recording, the permanent electrode is inserted. Fluoroscopy is usually used to verify the direction and stability of placement during the procedure.
An intraoperative test of the effects of deep brain stimulation is routinely performed to verify that some benefits can be achieved with the brain lead in its location, to determine the threshold for side effects, or both. For example, the patient may be asked to hold a cup as if trying to drink from it and to write or to draw a spiral on a clipboard to assess for improvements in tremor. Rigidity and bradykinesia can also be tested for improvements.
This intraoperative test is not aimed at assessing the best possible outcome of deep brain stimulation, and not even to see an improvement in all symptoms that burden the patient. Rather, it is to evaluate the likelihood that programming will be feasible with the implanted lead.
Subsequently, implantation of the pulse generator in the chest and connection to the brain lead is completed, usually with the patient under general anesthesia.
Implantation under general anesthesia, with intraoperative MRI
A new alternative to “awake stereotactic surgery” is implantation with the patient under general anesthesia, with intraoperative MRI. We have started to do this procedure in a new operating suite that is attached to an MRI suite. The magnet can be taken in and out of the operating room, allowing the surgeon to verify the location of the implanted leads right at the time of the procedure. In this fashion, intraoperative images are used to guide implantation instead of awake microelectrode recording. This is a new option for patients who cannot tolerate awake surgery and for those who have a contraindication to the regular stereotactic procedure with the patient awake.
Risks of bleeding and infection
The potential complications of implanting a device and leads in the brain can be significant.
Hemorrhage can occur, resulting in a superficial or deep hematoma.
Infection and erosion may require removal of the hardware for antibiotic treatment and possible reimplantation.
Other risks include those related to tunneling the wires from the head to the chest, to implanting the device in the chest, and to serious medical complications after surgery. Hardware failure can occur and requires additional surgery. Finally, environmental risks and risks related to medical devices such as MRI, electrocautery, and cardioversion should also be considered.
Deep brain stimulation is advantageous for its reversibility. If during postoperative programming the brain leads are considered not to be ideally placed, revisions can be done to reposition the leads.
Deep brain stimulation is an important therapy for Parkinson disease and other movement disorders. It involves implantation of a pulse generator that can be adjusted by telemetry and can be activated and deactivated by clinicians and patients. It is therefore also a good investigational tool, allowing for double-blind, sham-controlled clinical trials by testing the effects of the stimulation with optimal settings compared with no stimulation.
This article will discuss the approved indications for deep brain stimulation (particularly for managing movement disorders), the benefits that can be expected, the risks, the complications, the maintenance required, how candidates for this treatment are evaluated, and the surgical procedure for implantation of the devices.
DEVICE SIMILAR TO HEART PACEMAKERS
The deep brain stimulation system must be programmed by a physician or midlevel practitioner by observing a symptom and then changing the applied settings to the pulse generator until the symptom improves. This can be a very time-consuming process.
In contrast to heart pacemakers, which run at low frequencies, the brain devices for movement disorders are almost always set to a high frequency, greater than 100 Hz. For this reason, they consume more energy and need larger batteries than those in modern heart pacemakers.
The batteries in these generators typically last 3 to 5 years and are replaced in an outpatient procedure. Newer, smaller, rechargeable devices are expected to last longer but require more maintenance and care by patients, who have to recharge them at home periodically.
INDICATIONS FOR DEEP BRAIN STIMULATION
Deep brain stimulation is approved by the US Food and Drug Administration (FDA) for specific indications:
- Parkinson disease
- Essential tremor
- Primary dystonia (under a humanitarian device exemption)
- Intractable obsessive-compulsive disorder (also under a humanitarian device exemption). We will not discuss this indication further in this paper.
For each of these conditions, deep brain stimulation is considered when nonsurgical management has failed, as is the case for most functional neurosurgical treatments.
Investigations under way in other disorders
Several studies of deep brain stimulation are currently in progress under FDA-approved investigational device exemptions. Some, with funding from industry, are exploring its use in neuropsychiatric conditions other than parkinsonism. Two large clinical trials are evaluating its use for treatment-refractory depression, a common problem and a leading cause of disability in the industrialized world. Multiple investigators are also exploring novel uses of this technology in disorders ranging from obsessive-compulsive disorder to epilepsy.
Investigation is also under way at Cleveland Clinic in a federally funded, prospective, randomized clinical trial of deep brain stimulation for patients with thalamic pain syndrome. The primary hypothesis is that stimulation of the ventral striatal and ventral capsular area will modulate the affective component of this otherwise intractable pain syndrome, reducing pain-related disability and improving quality of life.
DEEP BRAIN STIMULATION VS ABLATION
Before deep brain stimulation became available, the only surgical options for patients with advanced Parkinson disease, tremor, or dystonia were ablative procedures such as pallidotomy (ablation of part of the globus pallidus) and thalamotomy (ablation of part of the thalamus). These procedures had been well known for several decades but fell out of favor when levodopa became available in the 1960s and revolutionized the medical treatment of Parkinson disease.
Surgery for movement disorders, in particular Parkinson disease, had a rebirth in the late 1980s when the limitations and complications associated with the pharmacologic management of Parkinson disease became increasingly evident. Ablative procedures are still used to treat advanced Parkinson disease, but much less commonly in industrialized countries.
Although pallidotomy and thalamotomy can have excellent results, they are not as safe as deep brain stimulation, which has the advantage of being reversible, modulating the function of an area rather than destroying it. Any unwanted effect can be immediately altered or reversed, unlike ablative procedures, in which any change is permanent. In addition, deep brain stimulation is adjustable, and the settings can be optimized as the disease progresses over the years.
Ablative procedures can be risky when performed bilaterally, while deep brain stimulation is routinely done on both hemispheres for patients with bilateral symptoms.
Although deep brain stimulation is today’s surgical treatment of choice, it is not perfect. It has the disadvantage of requiring lifelong maintenance of the hardware, for which the patient remains dependent on a medical center. Patients are usually seen more often at the specialized center in the first few months after surgery for optimization of programming and titration of drugs. (During this time, most patients see a gradual, substantial reduction in medication intake.) They are then followed by their physician and visit the center less often for monitoring of disease status and for further adjustments to the stimulator.
Most patients, to date, receive nonrechargeable pulse generators. As mentioned above, the batteries in these devices typically last 3 to 5 years. Preferably, batteries are replaced before they are completely depleted, to avoid interruption of therapy. Periodic visits to the center allow clinicians to estimate battery expiration ahead of time and plan replacements accordingly.
Rechargeable pulse generators have been recently introduced and are expected to last up to 9 years. They are an option for patients who can comply with the requirements for periodic home recharging of the hardware.
Patients are given a remote control so that they can turn the device on or off and check its status. Most patients keep it turned on all the time, although some turn it off at night to save battery life.
WHAT CAN PARKINSON PATIENTS EXPECT FROM THIS THERAPY?
Typically, some parkinsonian symptoms predominate over others, although some patients with advanced disease present with a severe combination of multiple disabling symptoms. Deep brain stimulation is best suited to address some of the cardinal motor symptoms, particularly tremor, rigidity, and bradykinesia, and motor fluctuations such as “wearing off” and dyskinesia.
Improvement in some motor symptoms
As a general rule, appendicular symptoms such as limb tremor and rigidity are more responsive to this therapy than axial symptoms such as gait and balance problems, but some patients experience improvement in gait as well. Other symptoms, such as swallowing or urinary symptoms, are seldom helped.
Although deep brain stimulation can help manage key motor symptoms and improve quality of life, it does not cure Parkinson disease. Also, there is no evidence to date that it slows disease progression, although this is a topic of ongoing investigation.
Fewer motor fluctuations
A common complaint of patients with advanced Parkinson disease is frequent—and often unpredictable—fluctuations between the “on” state (ie, when the effects of the patient’s levodopa therapy are apparent) and the “off” state (ie, when the levodopa doesn’t seem to be working). Sometimes, in the on state, patients experience involuntary choreic or ballistic movements, called dyskinesias. They also complain that the on time progressively lasts shorter and the day is spent alternating between shorter on states (during which the patient may be dyskinetic) and longer off states, limiting the patient’s independence and quality of life.
Deep brain stimulation can help patients prolong the on time while reducing the amplitude of these fluctuations so that the symptoms are not as severe in the off time and dyskinesias are reduced in the on time.
Some patients undergo deep brain stimulation primarily for managing the adverse effects of levodopa rather than for controlling the symptoms of the disease itself. While these patients need levodopa to address the disabling symptoms of the disease, they also present a greater sensitivity for developing levodopa-induced dyskinesias, quickly fluctuating from a lack of movement (the off state) to a state of uncontrollable movements (during the on state).
Deep brain stimulation typically allows the dosage of levodopa to be significantly reduced and gives patients more on time with fewer side effects and less fluctuation between the on and off states.
Response to levodopa predicts deep brain stimulation’s effects
Whether a patient is likely to be helped by deep brain stimulation can be tested with reasonable predictability by giving a single therapeutic dose of levodopa after the patient has been free of the drug for 12 hours. If there is an obvious difference on objective quantitative testing between the off and on states with a single dose, the patient is likely to benefit from deep brain stimulation. Those who do not respond well or are known to have never been well controlled by levodopa are likely poor candidates.
The test is also used as an indicator of whether the patient’s gait can be improved. Patients whose gait is substantially improved by levodopa, even for only a brief period of time, have a better chance of experiencing improvement in this domain with deep brain stimulation than those who do not show any gait improvement.
An important and notable exception to this rule is tremor control. Even Parkinson patients who do not experience significant improvement in tremor with levodopa (ie, who have medication-resistant tremors) are still likely to benefit from deep brain stimulation. Overall, tremor is the symptom that is most consistently improved with deep brain stimulation.
Results of clinical trials
Several clinical trials have demonstrated that deep brain stimulation plus medication works better than medications alone for advanced Parkinson disease.
Deuschl et al1 conducted a randomized trial in 156 patients with advanced Parkinson disease. Patients receiving subthalamic deep brain stimulation plus medication had significantly greater improvement in motor symptoms as measured by the Unified Parkinson’s Disease Rating Scale as well as in quality-of-life measures than patients receiving medications only.
Krack et al2 reported on the outcomes of 49 patients with advanced Parkinson disease who underwent deep brain stimulation and then were prospectively followed. At 5 years, motor function had improved by approximately 55% from baseline, activities-of-daily-living scores had improved by 49%, and patients continued to need significantly less levodopa and to experience less drug-induced dyskinesia.
Complications related to deep brain stimulation occurred in both studies, including two large intracerebral hemorrhages, one of which was fatal.
Weight gain. During the first 3 months after the device was implanted, patients tended to gain weight (mean 3 kg, maximum 5 kg). Although weight gain is considered an adverse effect, many patients are quite thin by the time they are candidates for deep brain stimulation, and in such cases gaining lean weight can be a benefit.
Patients with poorly controlled Parkinson disease lose weight for several reasons: increased calorie expenditure from shaking and excessive movements; diet modification and protein restriction for some patients who realize that protein competes with levodopa absorption; lack of appetite due to depression or from poor taste sensation (due to anosmia); and decreased overall food consumption due to difficulty swallowing.
DEEP BRAIN STIMULATION FOR ESSENTIAL TREMOR
Essential tremor is more common than Parkinson disease, with a prevalence in the United States estimated at approximately 4,000 per 100,000 people older than 65 years.
The tremor is often bilateral and is characteristically an action tremor, but in many patients it also has a postural, and sometimes a resting, component. It is distinct from parkinsonian tremor, which is usually predominantly a resting tremor. The differential diagnosis includes tremors secondary to central nervous system degenerative disorders as well as psychogenic tremors.
Drinking alcohol tends to relieve essential tremors, a finding that can often be elicited in the patient’s history. Patients whose symptoms improve with an alcoholic beverage are more likely to have essential tremor than another diagnosis.
Response to deep brain stimulation
Most patients with essential tremor respond well to deep brain stimulation of the contralateral ventral intermedius thalamic nucleus.
Treatment is usually started unilaterally, usually aimed at alleviating tremor in the patient’s dominant upper extremity. In selected cases, preference is given to treating the nondominant extremity when it is more severely affected than the dominant extremity.
Implantation of a device on the second side is offered to some patients who continue to be limited in activity and quality of life due to tremor of the untreated extremity. Surgery of the second side can be more complicated than the initial unilateral procedure. In particular, some patients may present with dysarthria, although that seems to be less common in our experience than initially estimated.
In practice, patients with moderate tremors tend to have an excellent response to deep brain stimulation. For this particular indication, if the response is not satisfactory, the treating team tends to consider surgically revising the placement of the lead rather than considering the patient a nonresponder. Patients with very severe tremors may have some residual tremor despite substantial improvement in severity. In our experience, patients with a greater proximal component of tremor tend to have less satisfactory results.
For challenging cases, implantation of additional electrodes in the thalamus or in new targets currently under investigation is sometimes considered, although this is an off-label use.
Treatment of secondary tremors, such as poststroke tremor or tremor due to multiple sclerosis, is sometimes attempted with deep brain stimulation. This is also an off-label option but is considered in selected cases for quality-of-life management.
Patients with axial tremors such as head or voice tremor are less likely to be helped by deep brain stimulation.
DEEP BRAIN STIMULATION FOR PRIMARY DYSTONIA
Generalized dystonia is a less common but severely impairing movement disorder.
Deep brain stimulation is approved for primary dystonia under a humanitarian device exemption, a regulatory mechanism for less common conditions. Deep brain stimulation is an option for patients who have significant impairment related to dystonia and who have not responded to conservative management such as anticholinergic agents, muscle relaxants, benzodiazepines, levodopa, or combinations of these drugs. Surgery has been shown to be effective for patients with primary generalized dystonia, whether or not they tested positive for a dystonia-related gene such as DYT1.
Kupsch et al3 evaluated 40 patients with primary dystonia in a randomized controlled trial of pallidal (globus pallidus pars interna) active deep brain stimulation vs sham stimulation (in which the device was implanted but not activated) for 3 months. Treated patients improved significantly more than controls (39% vs 5%) in the Burke-Fahn- Marsden Dystonia Rating Scale (BFMDRS).4 Similar improvement was noted when those receiving sham stimulation were switched to active stimulation.
During long-term follow-up, the results were generally sustained, with substantial improvement from deep brain stimulation in all movement symptoms evaluated except for speech and swallowing. Unlike improvement in tremor, which is quickly evident during testing in the operating room, the improvement in dystonia occurs gradually, and it may take months for patients to notice a change. Similarly, if stimulation stops because of device malfunction or dead batteries, symptoms sometimes do not recur for weeks or months.
Deep brain stimulation is sometimes offered to patients with dystonia secondary to conditions such as cerebral palsy or trauma (an off-label use). Although benefits are less consistent, deep brain stimulation remains an option for these individuals, aimed at alleviating some of the disabling symptoms. In patients with cerebral palsy or other secondary dystonias, it is sometimes difficult to distinguish how much of the disability is related to spasticity vs dystonia. Deep brain stimulation aims to alleviate the dystonic component; the spasticity may be managed with other options such as intrathecal baclofen (Lioresal).
Patients with tardive dystonia, which is usually secondary to treatment with antipsychotic agents, have been reported to respond well to bilateral deep brain stimulation. Gruber et al5 reported on a series of nine patients with a mean follow-up of 41 months. Patients improved by a mean of approximately 74% on the BFMDRS after 3 to 6 months of deep brain stimulation compared with baseline. None of the patients presented with long-term adverse effects, and quality of life and disability scores also improved significantly.
CANDIDATES ARE EVALUATED BY A MULTIDISCIPLINARY TEAM
Cleveland Clinic conducts a comprehensive 2-day evaluation for patients being considered for deep brain stimulation surgery, including consultations with specialists in neurology, neurosurgery, neuropsychology, and psychiatry.
Patients with significant cognitive deficits—near or meeting the diagnostic criteria for dementia—are usually not recommended to have surgery for Parkinson disease. Deep brain stimulation is not aimed at alleviating cognitive issues related to Parkinson disease or other concomitant dementia. In addition, there is a risk that neurostimulation could further worsen cognitive function in the already compromised brain. Moreover, patients with significant abnormalities detected by neuroimaging may have their diagnosis reconsidered in some cases, and some patients may not be deemed ideal candidates for surgery.
An important part of the process is a discussion with the patient and family about the risks and the potential short-term and long-term benefits. Informed consent requires a good understanding of this equation. Patients are counseled to have realistic expectations about what the procedure can offer. Deep brain stimulation can help some of the symptoms of Parkinson disease but will not cure it, and there is no evidence to date that it reduces its progress. At 5 or 10 years after surgery, patients are expected to be worse overall than they were in the first year after surgery, because of disease progression. However, patients who receive this treatment are expected, in general, to be doing better 5 or 10 years later (or longer) than those who do not receive it.
In addition to the discussion about risks, benefits, and expectations, a careful discussion is also devoted to hardware maintenance, including how to change the batteries. Particularly, younger patients should be informed about the risk of breakage of the leads and the extension wire, as they are likely to outlive their implant. Patients and caregivers should be able to come to the specialized center should hardware malfunction occur.
Patients are also informed that after the system is implanted they cannot undergo magnetic resonance imaging (MRI) except of the head, performed with a specific head coil and under specific parameters. MRI of any other body part and with a body coil is contraindicated.
HOW THE DEVICE IS IMPLANTED
There are several options for implanting a deep brain stimulation device.
Implantation with the patient awake, using a stereotactic headframe
At Cleveland Clinic, we usually prefer implantation with a stereotactic headframe. The base or “halo” of the frame is applied to the head under local anesthesia, followed by imaging via computed tomography (Figure 1). Typically, the tomographic image is fused to a previously acquired MRI image, but the MRI is sometimes either initially performed or repeated on the day of surgery.
Patients are sedated for the beginning of the procedure, while the surgical team is opening the skin and drilling the opening in the skull for placement of the lead. The patient is awakened for placement of the electrodes, which is not painful.
Microelectrode recording is typically performed in order to refine the targeting based on the stereotactic coordinates derived from neuroimaging. Although cadaver atlases exist and provide a guide to the stereotactic localization of subcortical structures, they are not completely accurate in representing the brain anatomy of all patients.
By “listening” to cells and knowing their characteristic signals in specific areas, landmarks can be created, forming an individualized map of the patient’s brain target. Microelectrode recording is invasive and has risks, including the risk of a brain hemorrhage. It is routinely done in most specialized deep brain stimulation centers because it can provide better accuracy and precision in lead placement.
When the target has been located and refined by microelectrode recording, the permanent electrode is inserted. Fluoroscopy is usually used to verify the direction and stability of placement during the procedure.
An intraoperative test of the effects of deep brain stimulation is routinely performed to verify that some benefits can be achieved with the brain lead in its location, to determine the threshold for side effects, or both. For example, the patient may be asked to hold a cup as if trying to drink from it and to write or to draw a spiral on a clipboard to assess for improvements in tremor. Rigidity and bradykinesia can also be tested for improvements.
This intraoperative test is not aimed at assessing the best possible outcome of deep brain stimulation, and not even to see an improvement in all symptoms that burden the patient. Rather, it is to evaluate the likelihood that programming will be feasible with the implanted lead.
Subsequently, implantation of the pulse generator in the chest and connection to the brain lead is completed, usually with the patient under general anesthesia.
Implantation under general anesthesia, with intraoperative MRI
A new alternative to “awake stereotactic surgery” is implantation with the patient under general anesthesia, with intraoperative MRI. We have started to do this procedure in a new operating suite that is attached to an MRI suite. The magnet can be taken in and out of the operating room, allowing the surgeon to verify the location of the implanted leads right at the time of the procedure. In this fashion, intraoperative images are used to guide implantation instead of awake microelectrode recording. This is a new option for patients who cannot tolerate awake surgery and for those who have a contraindication to the regular stereotactic procedure with the patient awake.
Risks of bleeding and infection
The potential complications of implanting a device and leads in the brain can be significant.
Hemorrhage can occur, resulting in a superficial or deep hematoma.
Infection and erosion may require removal of the hardware for antibiotic treatment and possible reimplantation.
Other risks include those related to tunneling the wires from the head to the chest, to implanting the device in the chest, and to serious medical complications after surgery. Hardware failure can occur and requires additional surgery. Finally, environmental risks and risks related to medical devices such as MRI, electrocautery, and cardioversion should also be considered.
Deep brain stimulation is advantageous for its reversibility. If during postoperative programming the brain leads are considered not to be ideally placed, revisions can be done to reposition the leads.
- Deuschl G, Schade-Brittinger C, Krack P, et al; German Parkinson Study Group, Neurostimulation Section. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med 2006; 355:896–908.
- Krack P, Batir A, Van Blercom N, et al. Five-year followup of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med 2003; 349:1925–1934.
- Kupsch A, Benecke R, Müller J, et al; Deep-Brain Stimulation for Dystonia Study Group. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. N Engl J Med 2006; 355:1978–1990.
- Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C, Friedman J. Validity and reliability of a rating scle for the primary torsion dystonias. Neurology 1985; 35:73–77.
- Gruber D, Trottenberg T, Kivi A, et al. Long-term effects of pallidal deep brain stimulation in tardive dystonia. Neurology 2009; 73:53–58.
- Deuschl G, Schade-Brittinger C, Krack P, et al; German Parkinson Study Group, Neurostimulation Section. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med 2006; 355:896–908.
- Krack P, Batir A, Van Blercom N, et al. Five-year followup of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med 2003; 349:1925–1934.
- Kupsch A, Benecke R, Müller J, et al; Deep-Brain Stimulation for Dystonia Study Group. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. N Engl J Med 2006; 355:1978–1990.
- Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C, Friedman J. Validity and reliability of a rating scle for the primary torsion dystonias. Neurology 1985; 35:73–77.
- Gruber D, Trottenberg T, Kivi A, et al. Long-term effects of pallidal deep brain stimulation in tardive dystonia. Neurology 2009; 73:53–58.
KEY POINTS
- Compared with ablative procedures, deep brain stimulation has the advantage of being reversible and adjustable. It is considered safer than ablative surgery, in particular for bilateral procedures, which are often needed for patients with advanced Parkinson disease and other movement disorders.
- For Parkinson disease, deep brain stimulation improves the cardinal motor symptoms, extends medication “on” time, and reduces motor fluctuations during the day.
- In general, patients with Parkinson disease are likely to benefit from this therapy if they show a clear response to levodopa. Patients are therefore asked to stop their Parkinson medications overnight to permit a formal evaluation of their motor response before and after a dose of levodopa.
- Candidates require a thorough evaluation to assess whether they are likely to benefit from deep brain stimulation and if they can comply with the maintenance often required for a successful outcome.
Chest pain followed by sudden collapse
Q: Given what we know so far, what is the most likely cause of the ST segment elevation in leads V1 and V2?
- Brugada syndrome
- Pulmonary embolism
- Right ventricular injury
- Anterior myocardial infarction
A: The correct answer is right ventricular injury (discussed below).
Brugada syndrome is a genetic disorder caused by a mutation in the cardiac sodium channel gene. It is characterized by a pronounced elevation of the J point, a coved-type ST segment elevation in leads V1 and V2, and a propensity to develop malignant ventricular arrhythmias and sudden cardiac death.
In this patient, the pattern of ST segment elevation in leads V1 and V2 may be falsely interpreted as the classic type 1 Brugada electrocardiographic pattern. However, the classic type 1 Brugada electrocardiogram is characterized by a coved ST elevation followed by a negative T wave.1 The absence of T-wave inversion following ST segment elevation in this patient excluded Brugada syndrome. Moreover, the main presentation in patients with Brugada syndrome is either syncopy or sudden cardiac death.
Pulmonary embolism can present with various electrocardiographic patterns. ST segment elevation in the antroseptal leads is an extremely rare sign and has been demonstrated in a few reports.2,3 Pulmonary embolism can also present with abnormal Q waves in leads III and aVF but not in lead II.4 The initial electrocardiographic rhythm in patients who present with cardiac arrest is usually pulseless electrical activity; however, the combination of increased right ventricular oxygen consumption due to increased right ventricular afterload and right ventricular hypoperfusion due to hypotension can lead to right ventricular ischemia and subsequent arrhythmias. Mittal and Arora5 described a case of submassive pulmonary embolism with right ventricular infarction presenting with sustained ventricular tachycardia.
The prognosis is usually poor in patients with cardiac arrest due to pulmonary embolism, which is usually caused by a massive embolus and usually necessitates thrombolytic therapy.
In the patient described here, pulmonary embolism was part of the differential diagnosis, given the presence of ST segment elevation in leads V1 and V2 in the context of the clinical scenario. However, the restoration of spontaneous circulation without any specific treatment for pulmonary embolism and the normal oxygenation after cardiac arrest excluded pulmonary embolism.
Right ventricular myocardial injury is important to recognize for therapeutic and prognostic reasons. It is usually associated with inferior infarction because it is typically secondary to an acute occlusion of the proximal right coronary artery proximal to the take-off of the right ventricular marginal branch. In the described scenario, the presence of ST segment elevation and Q waves in the inferior leads together with reciprocal ST segment depression in leads I and aVL represents an inferior myocardial infarct. ST segment elevation in the right precordial leads V3R and V4R is a marker for right ventricular injury—especially in V4R, in which it is a powerful predictor of right ventricular involvement. ST segment elevation in leads V1 and V2 is not usually demonstrated in patients with right ventricular injury because the electrical current of injury from the left ventricle inferior myocardial infarction dominates the right ventricular electrical forces, blocking the appearance of ST segment elevation in these leads.6 Data from the Hirulog and Early Reperfusion or Occlusion-2 trial showed that ST segment elevation of 1 mm or greater in lead V1 is associated with an increased risk of death in patients with acute inferior myocardial infarction.7 Furthermore, the presence of ST-segment elevation in lead V6 in patients with acute Q-wave inferior myocardial infarction, as evident in the first electrocardiogram, is associated with larger infarct size and a greater incidence of major arrhythmias.8
DETERMINING THE CULPRIT VESSEL
In the scenario described here, differentiating between right ventricular injury and anterior myocardial infarction is important to determine the culprit vessel.
CASE CONCLUDED
- Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005; 111:659–670.
- Livaditis IG, Paraschos M, Dimopoulos K. Massive pulmonary embolism with ST elevation in leads V1–V3 and successful thrombolysis with tenecteplase. Heart 2004; 90:e41.
- Falterman TJ, Martinez JA, Daberkow D, Weiss LD. Pulmonary embolism with ST segment elevation in leads V1 to V4: case report and review of the literature regarding electrocardiographic changes in acute pulmonary embolism. J Emerg Med 2001; 21:255–261.
- Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
- Mittal SR, Arora H. Pulmonary embolism with isolated right ventricular infarction. Indian Heart J 2001; 53:218–220.
- Geft IL, Shah PK, Rodriguez L, et al. ST elevations in leads V1 to V5 may be caused by right coronary artery occlusion and acute right ventricular infarction. Am J Cardiol 1984; 53:991–996.
- Wong CK, Gao W, Stewart RA, et al; Hirulog and Early Reperfusion or Occlusion-2 Investigators. Prognostic value of lead V1 ST elevation during acute inferior myocardial infarction. Circulation 2010; 122:463–469.
- Tsuka Y, Sugiura T, Hatada K, Abe Y, Takahashi N, Iwasaka T. Clinical characteristics of ST-segment elevation in lead V6 in patients with Q-wave acute inferior wall myocardial infarction. Coron Artery Dis 1999; 10:465–469.
Q: Given what we know so far, what is the most likely cause of the ST segment elevation in leads V1 and V2?
- Brugada syndrome
- Pulmonary embolism
- Right ventricular injury
- Anterior myocardial infarction
A: The correct answer is right ventricular injury (discussed below).
Brugada syndrome is a genetic disorder caused by a mutation in the cardiac sodium channel gene. It is characterized by a pronounced elevation of the J point, a coved-type ST segment elevation in leads V1 and V2, and a propensity to develop malignant ventricular arrhythmias and sudden cardiac death.
In this patient, the pattern of ST segment elevation in leads V1 and V2 may be falsely interpreted as the classic type 1 Brugada electrocardiographic pattern. However, the classic type 1 Brugada electrocardiogram is characterized by a coved ST elevation followed by a negative T wave.1 The absence of T-wave inversion following ST segment elevation in this patient excluded Brugada syndrome. Moreover, the main presentation in patients with Brugada syndrome is either syncopy or sudden cardiac death.
Pulmonary embolism can present with various electrocardiographic patterns. ST segment elevation in the antroseptal leads is an extremely rare sign and has been demonstrated in a few reports.2,3 Pulmonary embolism can also present with abnormal Q waves in leads III and aVF but not in lead II.4 The initial electrocardiographic rhythm in patients who present with cardiac arrest is usually pulseless electrical activity; however, the combination of increased right ventricular oxygen consumption due to increased right ventricular afterload and right ventricular hypoperfusion due to hypotension can lead to right ventricular ischemia and subsequent arrhythmias. Mittal and Arora5 described a case of submassive pulmonary embolism with right ventricular infarction presenting with sustained ventricular tachycardia.
The prognosis is usually poor in patients with cardiac arrest due to pulmonary embolism, which is usually caused by a massive embolus and usually necessitates thrombolytic therapy.
In the patient described here, pulmonary embolism was part of the differential diagnosis, given the presence of ST segment elevation in leads V1 and V2 in the context of the clinical scenario. However, the restoration of spontaneous circulation without any specific treatment for pulmonary embolism and the normal oxygenation after cardiac arrest excluded pulmonary embolism.
Right ventricular myocardial injury is important to recognize for therapeutic and prognostic reasons. It is usually associated with inferior infarction because it is typically secondary to an acute occlusion of the proximal right coronary artery proximal to the take-off of the right ventricular marginal branch. In the described scenario, the presence of ST segment elevation and Q waves in the inferior leads together with reciprocal ST segment depression in leads I and aVL represents an inferior myocardial infarct. ST segment elevation in the right precordial leads V3R and V4R is a marker for right ventricular injury—especially in V4R, in which it is a powerful predictor of right ventricular involvement. ST segment elevation in leads V1 and V2 is not usually demonstrated in patients with right ventricular injury because the electrical current of injury from the left ventricle inferior myocardial infarction dominates the right ventricular electrical forces, blocking the appearance of ST segment elevation in these leads.6 Data from the Hirulog and Early Reperfusion or Occlusion-2 trial showed that ST segment elevation of 1 mm or greater in lead V1 is associated with an increased risk of death in patients with acute inferior myocardial infarction.7 Furthermore, the presence of ST-segment elevation in lead V6 in patients with acute Q-wave inferior myocardial infarction, as evident in the first electrocardiogram, is associated with larger infarct size and a greater incidence of major arrhythmias.8
DETERMINING THE CULPRIT VESSEL
In the scenario described here, differentiating between right ventricular injury and anterior myocardial infarction is important to determine the culprit vessel.
CASE CONCLUDED
Q: Given what we know so far, what is the most likely cause of the ST segment elevation in leads V1 and V2?
- Brugada syndrome
- Pulmonary embolism
- Right ventricular injury
- Anterior myocardial infarction
A: The correct answer is right ventricular injury (discussed below).
Brugada syndrome is a genetic disorder caused by a mutation in the cardiac sodium channel gene. It is characterized by a pronounced elevation of the J point, a coved-type ST segment elevation in leads V1 and V2, and a propensity to develop malignant ventricular arrhythmias and sudden cardiac death.
In this patient, the pattern of ST segment elevation in leads V1 and V2 may be falsely interpreted as the classic type 1 Brugada electrocardiographic pattern. However, the classic type 1 Brugada electrocardiogram is characterized by a coved ST elevation followed by a negative T wave.1 The absence of T-wave inversion following ST segment elevation in this patient excluded Brugada syndrome. Moreover, the main presentation in patients with Brugada syndrome is either syncopy or sudden cardiac death.
Pulmonary embolism can present with various electrocardiographic patterns. ST segment elevation in the antroseptal leads is an extremely rare sign and has been demonstrated in a few reports.2,3 Pulmonary embolism can also present with abnormal Q waves in leads III and aVF but not in lead II.4 The initial electrocardiographic rhythm in patients who present with cardiac arrest is usually pulseless electrical activity; however, the combination of increased right ventricular oxygen consumption due to increased right ventricular afterload and right ventricular hypoperfusion due to hypotension can lead to right ventricular ischemia and subsequent arrhythmias. Mittal and Arora5 described a case of submassive pulmonary embolism with right ventricular infarction presenting with sustained ventricular tachycardia.
The prognosis is usually poor in patients with cardiac arrest due to pulmonary embolism, which is usually caused by a massive embolus and usually necessitates thrombolytic therapy.
In the patient described here, pulmonary embolism was part of the differential diagnosis, given the presence of ST segment elevation in leads V1 and V2 in the context of the clinical scenario. However, the restoration of spontaneous circulation without any specific treatment for pulmonary embolism and the normal oxygenation after cardiac arrest excluded pulmonary embolism.
Right ventricular myocardial injury is important to recognize for therapeutic and prognostic reasons. It is usually associated with inferior infarction because it is typically secondary to an acute occlusion of the proximal right coronary artery proximal to the take-off of the right ventricular marginal branch. In the described scenario, the presence of ST segment elevation and Q waves in the inferior leads together with reciprocal ST segment depression in leads I and aVL represents an inferior myocardial infarct. ST segment elevation in the right precordial leads V3R and V4R is a marker for right ventricular injury—especially in V4R, in which it is a powerful predictor of right ventricular involvement. ST segment elevation in leads V1 and V2 is not usually demonstrated in patients with right ventricular injury because the electrical current of injury from the left ventricle inferior myocardial infarction dominates the right ventricular electrical forces, blocking the appearance of ST segment elevation in these leads.6 Data from the Hirulog and Early Reperfusion or Occlusion-2 trial showed that ST segment elevation of 1 mm or greater in lead V1 is associated with an increased risk of death in patients with acute inferior myocardial infarction.7 Furthermore, the presence of ST-segment elevation in lead V6 in patients with acute Q-wave inferior myocardial infarction, as evident in the first electrocardiogram, is associated with larger infarct size and a greater incidence of major arrhythmias.8
DETERMINING THE CULPRIT VESSEL
In the scenario described here, differentiating between right ventricular injury and anterior myocardial infarction is important to determine the culprit vessel.
CASE CONCLUDED
- Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005; 111:659–670.
- Livaditis IG, Paraschos M, Dimopoulos K. Massive pulmonary embolism with ST elevation in leads V1–V3 and successful thrombolysis with tenecteplase. Heart 2004; 90:e41.
- Falterman TJ, Martinez JA, Daberkow D, Weiss LD. Pulmonary embolism with ST segment elevation in leads V1 to V4: case report and review of the literature regarding electrocardiographic changes in acute pulmonary embolism. J Emerg Med 2001; 21:255–261.
- Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
- Mittal SR, Arora H. Pulmonary embolism with isolated right ventricular infarction. Indian Heart J 2001; 53:218–220.
- Geft IL, Shah PK, Rodriguez L, et al. ST elevations in leads V1 to V5 may be caused by right coronary artery occlusion and acute right ventricular infarction. Am J Cardiol 1984; 53:991–996.
- Wong CK, Gao W, Stewart RA, et al; Hirulog and Early Reperfusion or Occlusion-2 Investigators. Prognostic value of lead V1 ST elevation during acute inferior myocardial infarction. Circulation 2010; 122:463–469.
- Tsuka Y, Sugiura T, Hatada K, Abe Y, Takahashi N, Iwasaka T. Clinical characteristics of ST-segment elevation in lead V6 in patients with Q-wave acute inferior wall myocardial infarction. Coron Artery Dis 1999; 10:465–469.
- Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005; 111:659–670.
- Livaditis IG, Paraschos M, Dimopoulos K. Massive pulmonary embolism with ST elevation in leads V1–V3 and successful thrombolysis with tenecteplase. Heart 2004; 90:e41.
- Falterman TJ, Martinez JA, Daberkow D, Weiss LD. Pulmonary embolism with ST segment elevation in leads V1 to V4: case report and review of the literature regarding electrocardiographic changes in acute pulmonary embolism. J Emerg Med 2001; 21:255–261.
- Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
- Mittal SR, Arora H. Pulmonary embolism with isolated right ventricular infarction. Indian Heart J 2001; 53:218–220.
- Geft IL, Shah PK, Rodriguez L, et al. ST elevations in leads V1 to V5 may be caused by right coronary artery occlusion and acute right ventricular infarction. Am J Cardiol 1984; 53:991–996.
- Wong CK, Gao W, Stewart RA, et al; Hirulog and Early Reperfusion or Occlusion-2 Investigators. Prognostic value of lead V1 ST elevation during acute inferior myocardial infarction. Circulation 2010; 122:463–469.
- Tsuka Y, Sugiura T, Hatada K, Abe Y, Takahashi N, Iwasaka T. Clinical characteristics of ST-segment elevation in lead V6 in patients with Q-wave acute inferior wall myocardial infarction. Coron Artery Dis 1999; 10:465–469.