User login
Residents’ Forum: Docs not at par on post-call days
SAN DIEGO – If you feel sleepy and out of sorts on a post-call day, compared with a normal work-day, you’re not alone.
Anesthesiology faculty reported significant increases in feeling irritable, jittery, and sleepy, along with significant decreases in feeling confident, energetic, and talkative following an on-call period, according to a study presented at the annual meeting of the American Society of Anesthesiologists.
To date, most studies of partial sleep deprivation in health care settings have focused on residents and interns, and less on medical faculty, said lead study author Dr. Haleh Saadat of the department of anesthesiology and pain medicine at Nationwide Children’s Hospital in Columbus, Ohio. “Our call is 17 hours, from 3 p.m. to 7 a.m.; but the call period at most hospitals is 24 hours, and even longer at some private practices,” she said in an interview.
To examine the effects of partial sleep deprivation on reaction time, simple cognitive skills, and mood status in 21 anesthesiologists, Dr. Saadat and her associates obtained verbal consent from the study participants and measured reaction time, mood states, and eight subjective behavioral characteristics at two different time points: between 6:30 a.m. and 8 a.m. on a regular noncall day of work, and between 6:30 a.m. and 8 a.m. after an overnight call (a shift that runs from 3 p.m. to 7 a.m.). The behavioral characteristics included feeling alert, energetic, anxious, confident, irritable, jittery/nervous, sleepy, and talkative, and the researchers used paired t-tests to compare variable means between regular sleep days and post-call days.
Reaction time decreased in all 21 subjects after night call, indicating worse performance (P = .047), while total mood disturbance was significantly higher on post-call days, relative to noncall days (P less than .001).
Of the 21 anesthesiologists, 19 completed all simple cognitive task questions at both time points and reported significant increases in several of these parameters on post-call days, compared with normal work-days.
Post-call observations found participants feeling more irritable, confident, energetic, sleepy (P less than .001), feeling more jittery (P = .003), and feeling less talkative (P less than .001) than on normal work–days.
Coping strategies used to address their sleep deprivation were measured as well, with “most of our subjects using problem solving, followed by seeking social support and avoidance,” Dr. Saadat noted. “People who used avoidance had greater declines in reaction time on post–call days, compared with the rest of the study participants. It didn’t matter whether you were male, female, younger, or older.”
Dr. Saadat called for additional studies to evaluate the neurocognitive impact of partial sleep deprivation on physicians’ on-call duties.
“I would like to see if we can replicate the results in bigger centers,” she said. “If this is what is happening, we may need to pay more attention to faculty’s work hours in both academic and private practice settings – not only among anesthesiologists, but also in other specialties. These observations require a closer look at the potential implications for patients’ and professionals’ safety.”
The researchers reported no financial disclosures.
As a surgical resident, I have experienced firsthand the “drunk-tired” phenomenon, and to be honest, I do not believe it to be such a rare occurrence. “Drunk-tired” may be eloquently defined as being so tired you start behaving like you’re drunk, without actually consuming any alcohol of course.
The first manuscript relating fatigue amongst shift workers to performance impairment was published in 1996 by Dawson et al. demonstrating that moderate levels of fatigue actually produce more impairment than being legally intoxicated (Nature 1997;388:235). It didn’t take much of a leap to translate these observations to health care workers who work long hours, do shift work, and are on-call at times for more than 24 hours at a time. Recently, at the annual meeting of the American Society of Anesthesiologists in San Diego, Dr. Haleh Saadat from Ohio presented her study on the effects of partial sleep deprivation in staff anaesthesiologists leading to significantly decreased reaction times, cognitive skills, and mood disturbances on post-call days, compared with normal work days. No surprise there, as this is in line with what Dawson and his colleagues published nearly two decades ago. This study can certainly be translated to medical students, residents, fellows and staff from the breadth of specialties in medicine. In my opinion, what’s the point? I can already foresee what these studies are going to demonstrate, namely a clean sweep of all forms of cognitive and motor impairments when a subject is sleep deprived. The question becomes how we are translating all of this information into action that changes the lives of health care professionals and more importantly improves patient safety. Understandably, this is a loaded question and I am simply too exhausted to wrap my head around it.
So, next time you’re post call, feeling irritable, discoordinated, and inhibited, just remember: you’re as good as drunk and you should probably sleep it off.
Dr. Laura Drudi is the resident medical editor for Vascular Specialist.
As a surgical resident, I have experienced firsthand the “drunk-tired” phenomenon, and to be honest, I do not believe it to be such a rare occurrence. “Drunk-tired” may be eloquently defined as being so tired you start behaving like you’re drunk, without actually consuming any alcohol of course.
The first manuscript relating fatigue amongst shift workers to performance impairment was published in 1996 by Dawson et al. demonstrating that moderate levels of fatigue actually produce more impairment than being legally intoxicated (Nature 1997;388:235). It didn’t take much of a leap to translate these observations to health care workers who work long hours, do shift work, and are on-call at times for more than 24 hours at a time. Recently, at the annual meeting of the American Society of Anesthesiologists in San Diego, Dr. Haleh Saadat from Ohio presented her study on the effects of partial sleep deprivation in staff anaesthesiologists leading to significantly decreased reaction times, cognitive skills, and mood disturbances on post-call days, compared with normal work days. No surprise there, as this is in line with what Dawson and his colleagues published nearly two decades ago. This study can certainly be translated to medical students, residents, fellows and staff from the breadth of specialties in medicine. In my opinion, what’s the point? I can already foresee what these studies are going to demonstrate, namely a clean sweep of all forms of cognitive and motor impairments when a subject is sleep deprived. The question becomes how we are translating all of this information into action that changes the lives of health care professionals and more importantly improves patient safety. Understandably, this is a loaded question and I am simply too exhausted to wrap my head around it.
So, next time you’re post call, feeling irritable, discoordinated, and inhibited, just remember: you’re as good as drunk and you should probably sleep it off.
Dr. Laura Drudi is the resident medical editor for Vascular Specialist.
As a surgical resident, I have experienced firsthand the “drunk-tired” phenomenon, and to be honest, I do not believe it to be such a rare occurrence. “Drunk-tired” may be eloquently defined as being so tired you start behaving like you’re drunk, without actually consuming any alcohol of course.
The first manuscript relating fatigue amongst shift workers to performance impairment was published in 1996 by Dawson et al. demonstrating that moderate levels of fatigue actually produce more impairment than being legally intoxicated (Nature 1997;388:235). It didn’t take much of a leap to translate these observations to health care workers who work long hours, do shift work, and are on-call at times for more than 24 hours at a time. Recently, at the annual meeting of the American Society of Anesthesiologists in San Diego, Dr. Haleh Saadat from Ohio presented her study on the effects of partial sleep deprivation in staff anaesthesiologists leading to significantly decreased reaction times, cognitive skills, and mood disturbances on post-call days, compared with normal work days. No surprise there, as this is in line with what Dawson and his colleagues published nearly two decades ago. This study can certainly be translated to medical students, residents, fellows and staff from the breadth of specialties in medicine. In my opinion, what’s the point? I can already foresee what these studies are going to demonstrate, namely a clean sweep of all forms of cognitive and motor impairments when a subject is sleep deprived. The question becomes how we are translating all of this information into action that changes the lives of health care professionals and more importantly improves patient safety. Understandably, this is a loaded question and I am simply too exhausted to wrap my head around it.
So, next time you’re post call, feeling irritable, discoordinated, and inhibited, just remember: you’re as good as drunk and you should probably sleep it off.
Dr. Laura Drudi is the resident medical editor for Vascular Specialist.
SAN DIEGO – If you feel sleepy and out of sorts on a post-call day, compared with a normal work-day, you’re not alone.
Anesthesiology faculty reported significant increases in feeling irritable, jittery, and sleepy, along with significant decreases in feeling confident, energetic, and talkative following an on-call period, according to a study presented at the annual meeting of the American Society of Anesthesiologists.
To date, most studies of partial sleep deprivation in health care settings have focused on residents and interns, and less on medical faculty, said lead study author Dr. Haleh Saadat of the department of anesthesiology and pain medicine at Nationwide Children’s Hospital in Columbus, Ohio. “Our call is 17 hours, from 3 p.m. to 7 a.m.; but the call period at most hospitals is 24 hours, and even longer at some private practices,” she said in an interview.
To examine the effects of partial sleep deprivation on reaction time, simple cognitive skills, and mood status in 21 anesthesiologists, Dr. Saadat and her associates obtained verbal consent from the study participants and measured reaction time, mood states, and eight subjective behavioral characteristics at two different time points: between 6:30 a.m. and 8 a.m. on a regular noncall day of work, and between 6:30 a.m. and 8 a.m. after an overnight call (a shift that runs from 3 p.m. to 7 a.m.). The behavioral characteristics included feeling alert, energetic, anxious, confident, irritable, jittery/nervous, sleepy, and talkative, and the researchers used paired t-tests to compare variable means between regular sleep days and post-call days.
Reaction time decreased in all 21 subjects after night call, indicating worse performance (P = .047), while total mood disturbance was significantly higher on post-call days, relative to noncall days (P less than .001).
Of the 21 anesthesiologists, 19 completed all simple cognitive task questions at both time points and reported significant increases in several of these parameters on post-call days, compared with normal work-days.
Post-call observations found participants feeling more irritable, confident, energetic, sleepy (P less than .001), feeling more jittery (P = .003), and feeling less talkative (P less than .001) than on normal work–days.
Coping strategies used to address their sleep deprivation were measured as well, with “most of our subjects using problem solving, followed by seeking social support and avoidance,” Dr. Saadat noted. “People who used avoidance had greater declines in reaction time on post–call days, compared with the rest of the study participants. It didn’t matter whether you were male, female, younger, or older.”
Dr. Saadat called for additional studies to evaluate the neurocognitive impact of partial sleep deprivation on physicians’ on-call duties.
“I would like to see if we can replicate the results in bigger centers,” she said. “If this is what is happening, we may need to pay more attention to faculty’s work hours in both academic and private practice settings – not only among anesthesiologists, but also in other specialties. These observations require a closer look at the potential implications for patients’ and professionals’ safety.”
The researchers reported no financial disclosures.
SAN DIEGO – If you feel sleepy and out of sorts on a post-call day, compared with a normal work-day, you’re not alone.
Anesthesiology faculty reported significant increases in feeling irritable, jittery, and sleepy, along with significant decreases in feeling confident, energetic, and talkative following an on-call period, according to a study presented at the annual meeting of the American Society of Anesthesiologists.
To date, most studies of partial sleep deprivation in health care settings have focused on residents and interns, and less on medical faculty, said lead study author Dr. Haleh Saadat of the department of anesthesiology and pain medicine at Nationwide Children’s Hospital in Columbus, Ohio. “Our call is 17 hours, from 3 p.m. to 7 a.m.; but the call period at most hospitals is 24 hours, and even longer at some private practices,” she said in an interview.
To examine the effects of partial sleep deprivation on reaction time, simple cognitive skills, and mood status in 21 anesthesiologists, Dr. Saadat and her associates obtained verbal consent from the study participants and measured reaction time, mood states, and eight subjective behavioral characteristics at two different time points: between 6:30 a.m. and 8 a.m. on a regular noncall day of work, and between 6:30 a.m. and 8 a.m. after an overnight call (a shift that runs from 3 p.m. to 7 a.m.). The behavioral characteristics included feeling alert, energetic, anxious, confident, irritable, jittery/nervous, sleepy, and talkative, and the researchers used paired t-tests to compare variable means between regular sleep days and post-call days.
Reaction time decreased in all 21 subjects after night call, indicating worse performance (P = .047), while total mood disturbance was significantly higher on post-call days, relative to noncall days (P less than .001).
Of the 21 anesthesiologists, 19 completed all simple cognitive task questions at both time points and reported significant increases in several of these parameters on post-call days, compared with normal work-days.
Post-call observations found participants feeling more irritable, confident, energetic, sleepy (P less than .001), feeling more jittery (P = .003), and feeling less talkative (P less than .001) than on normal work–days.
Coping strategies used to address their sleep deprivation were measured as well, with “most of our subjects using problem solving, followed by seeking social support and avoidance,” Dr. Saadat noted. “People who used avoidance had greater declines in reaction time on post–call days, compared with the rest of the study participants. It didn’t matter whether you were male, female, younger, or older.”
Dr. Saadat called for additional studies to evaluate the neurocognitive impact of partial sleep deprivation on physicians’ on-call duties.
“I would like to see if we can replicate the results in bigger centers,” she said. “If this is what is happening, we may need to pay more attention to faculty’s work hours in both academic and private practice settings – not only among anesthesiologists, but also in other specialties. These observations require a closer look at the potential implications for patients’ and professionals’ safety.”
The researchers reported no financial disclosures.

Antidepressant use associated with subsequent mania diagnosis
Patients with unipolar depression who use antidepressants may increase their risk of subsequently being diagnosed with mania/bipolar disorder, a retrospective cohort study conducted in the United Kingdom showed.
“Our findings demonstrate a significant association between antidepressant therapy in patients with unipolar depression and an increased incidence of mania. This association remained significant after adjusting for age and gender,” wrote Dr. Rashmi Patel of King’s College London and his colleagues.
The study comprised 21,012 adults who were diagnosed with depression and were receiving secondary mental health care for unipolar depression between April 1, 2006, and March 31, 2013. The researchers used electronic health records to determine which patients had used antidepressants prior to being diagnosed with depression and were subsequently diagnosed with mania or bipolar disorder, as well as the dates of the patients’ diagnoses. Patients were followed up to March 31, 2014.
Just under 1,000 (994) of the study participants were diagnosed with mania or bipolar disorder, representing 10.9 per 1,000 person-years. All types of antidepressants taken by the patients were associated with an increased incidence of mania/bipolar disorder (unadjusted hazard ratio greater than 1.0 for all antidepressants), with incidence rates ranging from 13.1 (tricyclic antidepressants) to 19.1 (trazodone) per 1,000 person-years.
“Future research should not only focus on which classes of antidepressants are most associated with mania, but also on other associated factors in order to guide clinicians of the risk of mania in people with depression prior to prescribing antidepressant therapy,” the investigators noted. They disclosed having received research funding from various sources.
Read the full study in BMJ Open (doi: 10.1136/bmjopen-2015-008341).
Patients with unipolar depression who use antidepressants may increase their risk of subsequently being diagnosed with mania/bipolar disorder, a retrospective cohort study conducted in the United Kingdom showed.
“Our findings demonstrate a significant association between antidepressant therapy in patients with unipolar depression and an increased incidence of mania. This association remained significant after adjusting for age and gender,” wrote Dr. Rashmi Patel of King’s College London and his colleagues.
The study comprised 21,012 adults who were diagnosed with depression and were receiving secondary mental health care for unipolar depression between April 1, 2006, and March 31, 2013. The researchers used electronic health records to determine which patients had used antidepressants prior to being diagnosed with depression and were subsequently diagnosed with mania or bipolar disorder, as well as the dates of the patients’ diagnoses. Patients were followed up to March 31, 2014.
Just under 1,000 (994) of the study participants were diagnosed with mania or bipolar disorder, representing 10.9 per 1,000 person-years. All types of antidepressants taken by the patients were associated with an increased incidence of mania/bipolar disorder (unadjusted hazard ratio greater than 1.0 for all antidepressants), with incidence rates ranging from 13.1 (tricyclic antidepressants) to 19.1 (trazodone) per 1,000 person-years.
“Future research should not only focus on which classes of antidepressants are most associated with mania, but also on other associated factors in order to guide clinicians of the risk of mania in people with depression prior to prescribing antidepressant therapy,” the investigators noted. They disclosed having received research funding from various sources.
Read the full study in BMJ Open (doi: 10.1136/bmjopen-2015-008341).
Patients with unipolar depression who use antidepressants may increase their risk of subsequently being diagnosed with mania/bipolar disorder, a retrospective cohort study conducted in the United Kingdom showed.
“Our findings demonstrate a significant association between antidepressant therapy in patients with unipolar depression and an increased incidence of mania. This association remained significant after adjusting for age and gender,” wrote Dr. Rashmi Patel of King’s College London and his colleagues.
The study comprised 21,012 adults who were diagnosed with depression and were receiving secondary mental health care for unipolar depression between April 1, 2006, and March 31, 2013. The researchers used electronic health records to determine which patients had used antidepressants prior to being diagnosed with depression and were subsequently diagnosed with mania or bipolar disorder, as well as the dates of the patients’ diagnoses. Patients were followed up to March 31, 2014.
Just under 1,000 (994) of the study participants were diagnosed with mania or bipolar disorder, representing 10.9 per 1,000 person-years. All types of antidepressants taken by the patients were associated with an increased incidence of mania/bipolar disorder (unadjusted hazard ratio greater than 1.0 for all antidepressants), with incidence rates ranging from 13.1 (tricyclic antidepressants) to 19.1 (trazodone) per 1,000 person-years.
“Future research should not only focus on which classes of antidepressants are most associated with mania, but also on other associated factors in order to guide clinicians of the risk of mania in people with depression prior to prescribing antidepressant therapy,” the investigators noted. They disclosed having received research funding from various sources.
Read the full study in BMJ Open (doi: 10.1136/bmjopen-2015-008341).
FROM BMJ OPEN
Concomitant Ulnar Styloid Fracture and Distal Radius Fracture Portend Poorer Outcome
Distal radius fracture is a common injury treated by orthopedic surgeons. Fifty percent or more of distal radius fractures (DRFs) occur with concomitant ulnar styloid fractures (USFs)1-3 (Figure). The base of the ulnar styloid is the insertion site for portions of the triangular fibrocartilaginous complex (TFCC), which is a primary stabilizer of the distal radioulnar joint (DRUJ).4,5
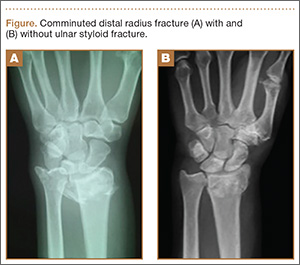
Although the topic has received significant attention in the literature, there remains a lack of consensus on the prognostic and clinical significance of USF occurring with DRF. In a series reported by May and colleagues,6 all patients with DRUJ instability after DRF also had an USF. Some authors have reported USF as a poor prognostic indicator for DRF, as the occurrence of USF was taken as a proxy for DRUJ instability.7,8 Conversely, other authors have reported that USF nonunion has no effect on the outcome of volar plating of DRF.9-11 In a retrospective cohort study of 182 patients, Li and colleagues12 found no clinically significant difference in outcome between presence or absence of USF with DRF. They also reported that the quality of the DRF reduction was the main determinant of clinical outcome in patients with USF.
We examined a large cohort of patients treated for DRF to identify any possible effect of an associated USF on clinical outcome. All patients provided written informed consent for study inclusion.
Materials and Methods
We retrospectively evaluated 315 cases of DRFs treated (184 operatively, 131 nonoperatively) by members of the Trauma and Hand divisions at our institution over a 7-year period. All cases had sufficient follow-up. In each group, patients with concomitant USF were identified.
At presentation, all displaced fractures underwent closed reduction and immobilization with a sugar-tong splint. Baseline demographic data, injury information, and baseline functional scores on the Disabilities of the Arm, Shoulder, and Hand (DASH) questionnaire and the 36-Item Short Form Health Survey (SF-36) were recorded. Complete histories were taken and physical examinations performed. Standard radiographs of the injured and contralateral wrists were obtained at time of initial injury.13
Surgery was indicated in patients with an open fracture and in patients with an inherently unstable fracture pattern, using the instability criteria of Cooney and colleagues.14 According to these criteria, unstable fractures have lost alignment after closed reduction or have more than 20° of dorsal angulation, more than 10 mm of longitudinal shortening, or more than 2 mm of articular displacement.14 Patients were treated with either a volar locked plate or bridging external fixation with supplemental Kirschner-wire fixation (usually 2 or 3 wires). Patients in both groups (operative, nonoperative) participated in a formal outpatient therapy program that emphasized active and passive range of motion (ROM) of the finger, wrist motion (if clinically appropriate), and forearm motion. Mean clinical follow-up was 12 months (range, 8-18 months). At each clinic visit, we used a handheld dynamometer to measure ROM, grip strength, and other parameters and compared them with the same parameters on the uninjured side, along with functional outcome.
Differences in demographic characteristics were evaluated with 2 tests—the χ2 test for categorical variables (eg, USF incidence, sex, hand dominance, fracture pattern) and the Student t test for continuous variables. Mann-Whitney U tests were used to assess differences between groups in DASH and SF-36 scores at long-term follow-up, as well as differences in ROM and radiographic measurements. Statistical significance was set at P < .05.
Results
DRFs occurred in the dominant-side wrist more commonly (P < .05) in the nonoperative group than in the operative group, though there was no difference in hand dominance and presence or absence of USF. There was a significant correlation of intra-articular fractures in the operative group (70%) compared with the nonoperative group (34%), though no association was found between presence of USF and intra-articular fracture location.
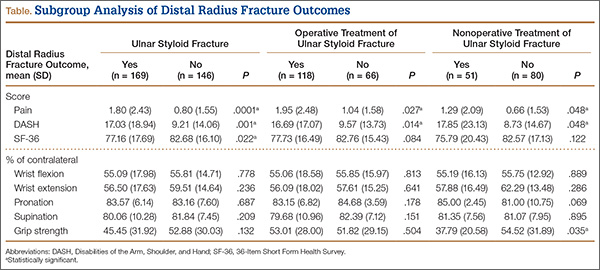
The percentage of concomitant USF was higher (P< .0002) in patients treated operatively (64.1%) than in those treated nonoperatively (38.9%). Mean (SD) pain score was higher (P = .0001) for patients with USF, 1.80 (2.43), than for patients without USF, 0.80 (1.55). This relationship held in both the operative group, 1.95 (2.48) versus 1.04 (1.58) (P = .027), and the nonoperative group, 1.29 (2.09) versus 0.66 (1.53) (P = .048). Similarly, at long-term follow-up for the entire patient cohort, mean (SD) DASH score was negatively affected by presence of USF, 17.03 (18.94) versus 9.21 (14.06) (P = .001), as was mean (SD) SF-36 score, 77.16 (17.69) versus 82.68 (16.10) (P = .022). This relationship also held in the operative and nonoperative groups with respect to pain and DASH scores, though there were only trends in this direction with respect to SF-36 scores. At final follow-up, there was no significant correlation of pain, SF-36, or DASH scores with presence of an intra-articular fracture as compared with an extra-articular fracture.
Time to radiographic healing was not influenced by presence of USF compared with absence of USF (11 vs 10.06 weeks; P > .05). Similarly, healing was no different in intra-articular fractures compared with extra-articular fractures (11 vs 10 weeks; P > .05).
Wrist ROM at final follow-up was not affected by presence of USF; there was no significant difference in wrist flexion, extension, or forearm rotation. In addition, mean (SD) grip strength was unaffected (P = .132) by presence or absence of USF with DRF overall, 45.45% (31.92) of contralateral versus 52.88% (30.03). However, grip strength was negatively affected (P = .035) by presence of USF in the nonoperative group, 37.79% (20.58) versus 54.52% (31.89) (Table).
Discussion
In this study, we determined that presence of USF was a negative predictor for clinical outcomes after DRF. Given the higher incidence of USF in operatively treated DRFs, USF likely represents a higher-energy mechanism of injury. We think these inferior clinical results are attributable to other wrist pathologies that commonly occur with these injuries. These pathologies, identified in the past, include stylocarpal impaction, extensor carpi ulnaris tendinitis, and pain at USF site.6,10,15 In addition, intracarpal ligamentous injuries, including damage to scapholunate and lunotriquetral ligaments, have been shown to occur in roughly 80% of patients who sustain DRFs, with TFCC injuries occurring at a rate of 60%.16
Patient outcome is multifactorial and depends on initial injury characteristics, reduction quality, associated injuries, and patient demographics and lifestyle factors. Li and colleagues12 showed that the quality of the DRF reduction influenced outcomes in these injuries, as the ulnar styloid and its associated TFCC are in turn reduced more anatomically with a restored DRF reduction. This concept applies to injuries treated both operatively and nonoperatively. Similarly, Xarchas and colleagues17 identified malunion of the ulnar styloid as causing chronic wrist pain because of triquetral impingement, which was treated successfully with ulnar styloidectomy. The poor results at final follow-up in their study may reflect severity of the initial injury, as reported by Frykman.18
Additional factors may compromise clinical outcomes after such injuries. For example, the effect of USF fragment size on outcome has been suggested and debated. In a retrospective series, May and colleagues6 identified fractures involving the base of the ulnar styloid or fovea as potentially destabilizing the DRUJ and in turn leading to chronic instability. This mechanism should be considered a potential contributor to protracted clinical recovery. Other studies have shown that, irrespective of USF fragment size, presence of USF with DRF is not a reliable predictor of DRUJ instability.2,10,19 In the present study, we simply identified presence or absence of USF, irrespective of either stability or fragment size. In cases in which there was an USF without instability, we fixed the DRF in isolation, without surgically addressing the USF. Our data demonstrated that, even in the absence of DRUJ instability, presence of USF was a negative prognostic indicator for patient outcome.
This study had several limitations. First, its design was retrospective. A prospective study would have been ideal for eliminating certain inherent bias. Second, USF represents a higher association with DRUJ instability.6 As there are no validated tests for this clinical entity, identification is somewhat subjective. We did not separate patients by presence or absence of DRUJ instability and thus were not able to directly correlate the connection between USF, DRUJ instability, and poor outcomes in association with DRF. In addition, management of an unstable DRUJ after operative fixation of DRF is controversial, with techniques ranging from splinting in supination to pinning the DRUJ. This inconsistency likely contributed to some error between groups of patients in this study. Last, we did not stratify patients by USF fragment size, as previously discussed, which may have affected outcomes within patient groups.
Our data add to the evidence showing that USF in association with DRF portends poorer clinical outcomes. Concomitant USF should alert the treating physician to a higher-energy mechanism of injury and raise the index of suspicion for other associated injuries in the carpus.
1. Richards RS, Bennett JD, Roth JH, Milne K Jr. Arthroscopic diagnosis of intra-articular soft tissue injuries associated with distal radial fractures. J Hand Surg Am. 1997;22(5):772-776.
2. Sammer DM, Shah HM, Shauver MJ, Chung KC. The effect of ulnar styloid fractures on patient-rated outcomes after volar locking plating of distal radius fractures. J Hand Surg Am. 2009;34(9):1595-1602.
3. Villar RN, Marsh D, Rushton N, Greatorex RA. Three years after Colles’ fracture. A prospective review. J Bone Joint Surg Br. 1987;69(4):635-638.
4. Palmer AK, Werner FW. The triangular fibrocartilage complex of the wrist—anatomy and function. J Hand Surg Am. 1981;6(2):153-162.
5. Stuart PR, Berger RA, Linscheid RL, An KN. The dorsopalmar stability of the distal radioulnar joint. J Hand Surg Am. 2000;25(4):689-699.
6. May MM, Lawton JN, Blazar PE. Ulnar styloid fractures associated with distal radius fractures: incidence and implications for distal radioulnar joint instability. J Hand Surg Am. 2002;27(6):965-971.
7. Oskarsson GV, Aaser P, Hjall A. Do we underestimate the predictive value of the ulnar styloid affection in Colles fractures? Arch Orthop Trauma Surg. 1997;116(6-7):341-344.
8. Stoffelen D, De Smet L, Broos P. The importance of the distal radioulnar joint in distal radial fractures. J Hand Surg Br. 1998;23(4):507-511.
9. Buijze GA, Ring D. Clinical impact of united versus nonunited fractures of the proximal half of the ulnar styloid following volar plate fixation of the distal radius. J Hand Surg Am. 2010;35(2):223-227.
10. Kim JK, Yun YH, Kim DJ, Yun GU. Comparison of united and nonunited fractures of the ulnar styloid following volar-plate fixation of distal radius fractures. Injury. 2011;42(4):371-375.
11. Wijffels M, Ring D. The influence of non-union of the ulnar styloid on pain, wrist function and instability after distal radius fracture. J Hand Microsurg. 2011;3(1):11-14.
12. Li S, Chen Y, Lin Z, Fan Q, Cui W, Feng Z. Effect of associated ulnar styloid fracture on wrist function after distal radius [in Chinese]. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2012;26(6):666-670.
13. Egol KA, Walsh M, Romo-Cardoso S, Dorsky S, Paksima N. Distal radial fractures in the elderly: operative compared with nonoperative treatment. J Bone Joint Surg Am. 2010;92(9):1851-1857.
14. Cooney WP 3rd, Linscheid RL, Dobyns JH. External pin fixation for unstable Colles’ fractures. J Bone Joint Surg Am. 1979;61(6):840-845.
15. Cerezal L, del Piñal F, Abascal F, García-Valtuille R, Pereda T, Canga A. Imaging findings in ulnar-sided wrist impaction syndromes. Radiographics. 2002;22(1):105-121.
16. Ogawa T, Tanaka T, Yanai T, Kumagai H, Ochiai N. Analysis of soft tissue injuries associated with distal radius fractures. BMC Sports Sci Med Rehabil. 2013;5(1):19.
17. Xarchas KC, Yfandithis P, Kazakos K. Malunion of the ulnar styloid as a cause of ulnar wrist pain. Clin Anat. 2004;17(5):418-422.
18. Frykman G. Fracture of the distal radius including sequelae—shoulder–hand–finger syndrome, disturbance in the distal radio-ulnar joint and impairment of nerve function. A clinical and experimental study. Acta Orthop Scand. 1967:(suppl 108):3+.
19. Fujitani R, Omokawa S, Akahane M, Iida A, Ono H, Tanaka Y. Predictors of distal radioulnar joint instability in distal radius fractures. J Hand Surg Am. 2011;36(12):1919-1925.
Distal radius fracture is a common injury treated by orthopedic surgeons. Fifty percent or more of distal radius fractures (DRFs) occur with concomitant ulnar styloid fractures (USFs)1-3 (Figure). The base of the ulnar styloid is the insertion site for portions of the triangular fibrocartilaginous complex (TFCC), which is a primary stabilizer of the distal radioulnar joint (DRUJ).4,5
Although the topic has received significant attention in the literature, there remains a lack of consensus on the prognostic and clinical significance of USF occurring with DRF. In a series reported by May and colleagues,6 all patients with DRUJ instability after DRF also had an USF. Some authors have reported USF as a poor prognostic indicator for DRF, as the occurrence of USF was taken as a proxy for DRUJ instability.7,8 Conversely, other authors have reported that USF nonunion has no effect on the outcome of volar plating of DRF.9-11 In a retrospective cohort study of 182 patients, Li and colleagues12 found no clinically significant difference in outcome between presence or absence of USF with DRF. They also reported that the quality of the DRF reduction was the main determinant of clinical outcome in patients with USF.
We examined a large cohort of patients treated for DRF to identify any possible effect of an associated USF on clinical outcome. All patients provided written informed consent for study inclusion.
Materials and Methods
We retrospectively evaluated 315 cases of DRFs treated (184 operatively, 131 nonoperatively) by members of the Trauma and Hand divisions at our institution over a 7-year period. All cases had sufficient follow-up. In each group, patients with concomitant USF were identified.
At presentation, all displaced fractures underwent closed reduction and immobilization with a sugar-tong splint. Baseline demographic data, injury information, and baseline functional scores on the Disabilities of the Arm, Shoulder, and Hand (DASH) questionnaire and the 36-Item Short Form Health Survey (SF-36) were recorded. Complete histories were taken and physical examinations performed. Standard radiographs of the injured and contralateral wrists were obtained at time of initial injury.13
Surgery was indicated in patients with an open fracture and in patients with an inherently unstable fracture pattern, using the instability criteria of Cooney and colleagues.14 According to these criteria, unstable fractures have lost alignment after closed reduction or have more than 20° of dorsal angulation, more than 10 mm of longitudinal shortening, or more than 2 mm of articular displacement.14 Patients were treated with either a volar locked plate or bridging external fixation with supplemental Kirschner-wire fixation (usually 2 or 3 wires). Patients in both groups (operative, nonoperative) participated in a formal outpatient therapy program that emphasized active and passive range of motion (ROM) of the finger, wrist motion (if clinically appropriate), and forearm motion. Mean clinical follow-up was 12 months (range, 8-18 months). At each clinic visit, we used a handheld dynamometer to measure ROM, grip strength, and other parameters and compared them with the same parameters on the uninjured side, along with functional outcome.
Differences in demographic characteristics were evaluated with 2 tests—the χ2 test for categorical variables (eg, USF incidence, sex, hand dominance, fracture pattern) and the Student t test for continuous variables. Mann-Whitney U tests were used to assess differences between groups in DASH and SF-36 scores at long-term follow-up, as well as differences in ROM and radiographic measurements. Statistical significance was set at P < .05.
Results
DRFs occurred in the dominant-side wrist more commonly (P < .05) in the nonoperative group than in the operative group, though there was no difference in hand dominance and presence or absence of USF. There was a significant correlation of intra-articular fractures in the operative group (70%) compared with the nonoperative group (34%), though no association was found between presence of USF and intra-articular fracture location.
The percentage of concomitant USF was higher (P< .0002) in patients treated operatively (64.1%) than in those treated nonoperatively (38.9%). Mean (SD) pain score was higher (P = .0001) for patients with USF, 1.80 (2.43), than for patients without USF, 0.80 (1.55). This relationship held in both the operative group, 1.95 (2.48) versus 1.04 (1.58) (P = .027), and the nonoperative group, 1.29 (2.09) versus 0.66 (1.53) (P = .048). Similarly, at long-term follow-up for the entire patient cohort, mean (SD) DASH score was negatively affected by presence of USF, 17.03 (18.94) versus 9.21 (14.06) (P = .001), as was mean (SD) SF-36 score, 77.16 (17.69) versus 82.68 (16.10) (P = .022). This relationship also held in the operative and nonoperative groups with respect to pain and DASH scores, though there were only trends in this direction with respect to SF-36 scores. At final follow-up, there was no significant correlation of pain, SF-36, or DASH scores with presence of an intra-articular fracture as compared with an extra-articular fracture.
Time to radiographic healing was not influenced by presence of USF compared with absence of USF (11 vs 10.06 weeks; P > .05). Similarly, healing was no different in intra-articular fractures compared with extra-articular fractures (11 vs 10 weeks; P > .05).
Wrist ROM at final follow-up was not affected by presence of USF; there was no significant difference in wrist flexion, extension, or forearm rotation. In addition, mean (SD) grip strength was unaffected (P = .132) by presence or absence of USF with DRF overall, 45.45% (31.92) of contralateral versus 52.88% (30.03). However, grip strength was negatively affected (P = .035) by presence of USF in the nonoperative group, 37.79% (20.58) versus 54.52% (31.89) (Table).
Discussion
In this study, we determined that presence of USF was a negative predictor for clinical outcomes after DRF. Given the higher incidence of USF in operatively treated DRFs, USF likely represents a higher-energy mechanism of injury. We think these inferior clinical results are attributable to other wrist pathologies that commonly occur with these injuries. These pathologies, identified in the past, include stylocarpal impaction, extensor carpi ulnaris tendinitis, and pain at USF site.6,10,15 In addition, intracarpal ligamentous injuries, including damage to scapholunate and lunotriquetral ligaments, have been shown to occur in roughly 80% of patients who sustain DRFs, with TFCC injuries occurring at a rate of 60%.16
Patient outcome is multifactorial and depends on initial injury characteristics, reduction quality, associated injuries, and patient demographics and lifestyle factors. Li and colleagues12 showed that the quality of the DRF reduction influenced outcomes in these injuries, as the ulnar styloid and its associated TFCC are in turn reduced more anatomically with a restored DRF reduction. This concept applies to injuries treated both operatively and nonoperatively. Similarly, Xarchas and colleagues17 identified malunion of the ulnar styloid as causing chronic wrist pain because of triquetral impingement, which was treated successfully with ulnar styloidectomy. The poor results at final follow-up in their study may reflect severity of the initial injury, as reported by Frykman.18
Additional factors may compromise clinical outcomes after such injuries. For example, the effect of USF fragment size on outcome has been suggested and debated. In a retrospective series, May and colleagues6 identified fractures involving the base of the ulnar styloid or fovea as potentially destabilizing the DRUJ and in turn leading to chronic instability. This mechanism should be considered a potential contributor to protracted clinical recovery. Other studies have shown that, irrespective of USF fragment size, presence of USF with DRF is not a reliable predictor of DRUJ instability.2,10,19 In the present study, we simply identified presence or absence of USF, irrespective of either stability or fragment size. In cases in which there was an USF without instability, we fixed the DRF in isolation, without surgically addressing the USF. Our data demonstrated that, even in the absence of DRUJ instability, presence of USF was a negative prognostic indicator for patient outcome.
This study had several limitations. First, its design was retrospective. A prospective study would have been ideal for eliminating certain inherent bias. Second, USF represents a higher association with DRUJ instability.6 As there are no validated tests for this clinical entity, identification is somewhat subjective. We did not separate patients by presence or absence of DRUJ instability and thus were not able to directly correlate the connection between USF, DRUJ instability, and poor outcomes in association with DRF. In addition, management of an unstable DRUJ after operative fixation of DRF is controversial, with techniques ranging from splinting in supination to pinning the DRUJ. This inconsistency likely contributed to some error between groups of patients in this study. Last, we did not stratify patients by USF fragment size, as previously discussed, which may have affected outcomes within patient groups.
Our data add to the evidence showing that USF in association with DRF portends poorer clinical outcomes. Concomitant USF should alert the treating physician to a higher-energy mechanism of injury and raise the index of suspicion for other associated injuries in the carpus.
Distal radius fracture is a common injury treated by orthopedic surgeons. Fifty percent or more of distal radius fractures (DRFs) occur with concomitant ulnar styloid fractures (USFs)1-3 (Figure). The base of the ulnar styloid is the insertion site for portions of the triangular fibrocartilaginous complex (TFCC), which is a primary stabilizer of the distal radioulnar joint (DRUJ).4,5
Although the topic has received significant attention in the literature, there remains a lack of consensus on the prognostic and clinical significance of USF occurring with DRF. In a series reported by May and colleagues,6 all patients with DRUJ instability after DRF also had an USF. Some authors have reported USF as a poor prognostic indicator for DRF, as the occurrence of USF was taken as a proxy for DRUJ instability.7,8 Conversely, other authors have reported that USF nonunion has no effect on the outcome of volar plating of DRF.9-11 In a retrospective cohort study of 182 patients, Li and colleagues12 found no clinically significant difference in outcome between presence or absence of USF with DRF. They also reported that the quality of the DRF reduction was the main determinant of clinical outcome in patients with USF.
We examined a large cohort of patients treated for DRF to identify any possible effect of an associated USF on clinical outcome. All patients provided written informed consent for study inclusion.
Materials and Methods
We retrospectively evaluated 315 cases of DRFs treated (184 operatively, 131 nonoperatively) by members of the Trauma and Hand divisions at our institution over a 7-year period. All cases had sufficient follow-up. In each group, patients with concomitant USF were identified.
At presentation, all displaced fractures underwent closed reduction and immobilization with a sugar-tong splint. Baseline demographic data, injury information, and baseline functional scores on the Disabilities of the Arm, Shoulder, and Hand (DASH) questionnaire and the 36-Item Short Form Health Survey (SF-36) were recorded. Complete histories were taken and physical examinations performed. Standard radiographs of the injured and contralateral wrists were obtained at time of initial injury.13
Surgery was indicated in patients with an open fracture and in patients with an inherently unstable fracture pattern, using the instability criteria of Cooney and colleagues.14 According to these criteria, unstable fractures have lost alignment after closed reduction or have more than 20° of dorsal angulation, more than 10 mm of longitudinal shortening, or more than 2 mm of articular displacement.14 Patients were treated with either a volar locked plate or bridging external fixation with supplemental Kirschner-wire fixation (usually 2 or 3 wires). Patients in both groups (operative, nonoperative) participated in a formal outpatient therapy program that emphasized active and passive range of motion (ROM) of the finger, wrist motion (if clinically appropriate), and forearm motion. Mean clinical follow-up was 12 months (range, 8-18 months). At each clinic visit, we used a handheld dynamometer to measure ROM, grip strength, and other parameters and compared them with the same parameters on the uninjured side, along with functional outcome.
Differences in demographic characteristics were evaluated with 2 tests—the χ2 test for categorical variables (eg, USF incidence, sex, hand dominance, fracture pattern) and the Student t test for continuous variables. Mann-Whitney U tests were used to assess differences between groups in DASH and SF-36 scores at long-term follow-up, as well as differences in ROM and radiographic measurements. Statistical significance was set at P < .05.
Results
DRFs occurred in the dominant-side wrist more commonly (P < .05) in the nonoperative group than in the operative group, though there was no difference in hand dominance and presence or absence of USF. There was a significant correlation of intra-articular fractures in the operative group (70%) compared with the nonoperative group (34%), though no association was found between presence of USF and intra-articular fracture location.
The percentage of concomitant USF was higher (P< .0002) in patients treated operatively (64.1%) than in those treated nonoperatively (38.9%). Mean (SD) pain score was higher (P = .0001) for patients with USF, 1.80 (2.43), than for patients without USF, 0.80 (1.55). This relationship held in both the operative group, 1.95 (2.48) versus 1.04 (1.58) (P = .027), and the nonoperative group, 1.29 (2.09) versus 0.66 (1.53) (P = .048). Similarly, at long-term follow-up for the entire patient cohort, mean (SD) DASH score was negatively affected by presence of USF, 17.03 (18.94) versus 9.21 (14.06) (P = .001), as was mean (SD) SF-36 score, 77.16 (17.69) versus 82.68 (16.10) (P = .022). This relationship also held in the operative and nonoperative groups with respect to pain and DASH scores, though there were only trends in this direction with respect to SF-36 scores. At final follow-up, there was no significant correlation of pain, SF-36, or DASH scores with presence of an intra-articular fracture as compared with an extra-articular fracture.
Time to radiographic healing was not influenced by presence of USF compared with absence of USF (11 vs 10.06 weeks; P > .05). Similarly, healing was no different in intra-articular fractures compared with extra-articular fractures (11 vs 10 weeks; P > .05).
Wrist ROM at final follow-up was not affected by presence of USF; there was no significant difference in wrist flexion, extension, or forearm rotation. In addition, mean (SD) grip strength was unaffected (P = .132) by presence or absence of USF with DRF overall, 45.45% (31.92) of contralateral versus 52.88% (30.03). However, grip strength was negatively affected (P = .035) by presence of USF in the nonoperative group, 37.79% (20.58) versus 54.52% (31.89) (Table).
Discussion
In this study, we determined that presence of USF was a negative predictor for clinical outcomes after DRF. Given the higher incidence of USF in operatively treated DRFs, USF likely represents a higher-energy mechanism of injury. We think these inferior clinical results are attributable to other wrist pathologies that commonly occur with these injuries. These pathologies, identified in the past, include stylocarpal impaction, extensor carpi ulnaris tendinitis, and pain at USF site.6,10,15 In addition, intracarpal ligamentous injuries, including damage to scapholunate and lunotriquetral ligaments, have been shown to occur in roughly 80% of patients who sustain DRFs, with TFCC injuries occurring at a rate of 60%.16
Patient outcome is multifactorial and depends on initial injury characteristics, reduction quality, associated injuries, and patient demographics and lifestyle factors. Li and colleagues12 showed that the quality of the DRF reduction influenced outcomes in these injuries, as the ulnar styloid and its associated TFCC are in turn reduced more anatomically with a restored DRF reduction. This concept applies to injuries treated both operatively and nonoperatively. Similarly, Xarchas and colleagues17 identified malunion of the ulnar styloid as causing chronic wrist pain because of triquetral impingement, which was treated successfully with ulnar styloidectomy. The poor results at final follow-up in their study may reflect severity of the initial injury, as reported by Frykman.18
Additional factors may compromise clinical outcomes after such injuries. For example, the effect of USF fragment size on outcome has been suggested and debated. In a retrospective series, May and colleagues6 identified fractures involving the base of the ulnar styloid or fovea as potentially destabilizing the DRUJ and in turn leading to chronic instability. This mechanism should be considered a potential contributor to protracted clinical recovery. Other studies have shown that, irrespective of USF fragment size, presence of USF with DRF is not a reliable predictor of DRUJ instability.2,10,19 In the present study, we simply identified presence or absence of USF, irrespective of either stability or fragment size. In cases in which there was an USF without instability, we fixed the DRF in isolation, without surgically addressing the USF. Our data demonstrated that, even in the absence of DRUJ instability, presence of USF was a negative prognostic indicator for patient outcome.
This study had several limitations. First, its design was retrospective. A prospective study would have been ideal for eliminating certain inherent bias. Second, USF represents a higher association with DRUJ instability.6 As there are no validated tests for this clinical entity, identification is somewhat subjective. We did not separate patients by presence or absence of DRUJ instability and thus were not able to directly correlate the connection between USF, DRUJ instability, and poor outcomes in association with DRF. In addition, management of an unstable DRUJ after operative fixation of DRF is controversial, with techniques ranging from splinting in supination to pinning the DRUJ. This inconsistency likely contributed to some error between groups of patients in this study. Last, we did not stratify patients by USF fragment size, as previously discussed, which may have affected outcomes within patient groups.
Our data add to the evidence showing that USF in association with DRF portends poorer clinical outcomes. Concomitant USF should alert the treating physician to a higher-energy mechanism of injury and raise the index of suspicion for other associated injuries in the carpus.
1. Richards RS, Bennett JD, Roth JH, Milne K Jr. Arthroscopic diagnosis of intra-articular soft tissue injuries associated with distal radial fractures. J Hand Surg Am. 1997;22(5):772-776.
2. Sammer DM, Shah HM, Shauver MJ, Chung KC. The effect of ulnar styloid fractures on patient-rated outcomes after volar locking plating of distal radius fractures. J Hand Surg Am. 2009;34(9):1595-1602.
3. Villar RN, Marsh D, Rushton N, Greatorex RA. Three years after Colles’ fracture. A prospective review. J Bone Joint Surg Br. 1987;69(4):635-638.
4. Palmer AK, Werner FW. The triangular fibrocartilage complex of the wrist—anatomy and function. J Hand Surg Am. 1981;6(2):153-162.
5. Stuart PR, Berger RA, Linscheid RL, An KN. The dorsopalmar stability of the distal radioulnar joint. J Hand Surg Am. 2000;25(4):689-699.
6. May MM, Lawton JN, Blazar PE. Ulnar styloid fractures associated with distal radius fractures: incidence and implications for distal radioulnar joint instability. J Hand Surg Am. 2002;27(6):965-971.
7. Oskarsson GV, Aaser P, Hjall A. Do we underestimate the predictive value of the ulnar styloid affection in Colles fractures? Arch Orthop Trauma Surg. 1997;116(6-7):341-344.
8. Stoffelen D, De Smet L, Broos P. The importance of the distal radioulnar joint in distal radial fractures. J Hand Surg Br. 1998;23(4):507-511.
9. Buijze GA, Ring D. Clinical impact of united versus nonunited fractures of the proximal half of the ulnar styloid following volar plate fixation of the distal radius. J Hand Surg Am. 2010;35(2):223-227.
10. Kim JK, Yun YH, Kim DJ, Yun GU. Comparison of united and nonunited fractures of the ulnar styloid following volar-plate fixation of distal radius fractures. Injury. 2011;42(4):371-375.
11. Wijffels M, Ring D. The influence of non-union of the ulnar styloid on pain, wrist function and instability after distal radius fracture. J Hand Microsurg. 2011;3(1):11-14.
12. Li S, Chen Y, Lin Z, Fan Q, Cui W, Feng Z. Effect of associated ulnar styloid fracture on wrist function after distal radius [in Chinese]. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2012;26(6):666-670.
13. Egol KA, Walsh M, Romo-Cardoso S, Dorsky S, Paksima N. Distal radial fractures in the elderly: operative compared with nonoperative treatment. J Bone Joint Surg Am. 2010;92(9):1851-1857.
14. Cooney WP 3rd, Linscheid RL, Dobyns JH. External pin fixation for unstable Colles’ fractures. J Bone Joint Surg Am. 1979;61(6):840-845.
15. Cerezal L, del Piñal F, Abascal F, García-Valtuille R, Pereda T, Canga A. Imaging findings in ulnar-sided wrist impaction syndromes. Radiographics. 2002;22(1):105-121.
16. Ogawa T, Tanaka T, Yanai T, Kumagai H, Ochiai N. Analysis of soft tissue injuries associated with distal radius fractures. BMC Sports Sci Med Rehabil. 2013;5(1):19.
17. Xarchas KC, Yfandithis P, Kazakos K. Malunion of the ulnar styloid as a cause of ulnar wrist pain. Clin Anat. 2004;17(5):418-422.
18. Frykman G. Fracture of the distal radius including sequelae—shoulder–hand–finger syndrome, disturbance in the distal radio-ulnar joint and impairment of nerve function. A clinical and experimental study. Acta Orthop Scand. 1967:(suppl 108):3+.
19. Fujitani R, Omokawa S, Akahane M, Iida A, Ono H, Tanaka Y. Predictors of distal radioulnar joint instability in distal radius fractures. J Hand Surg Am. 2011;36(12):1919-1925.
1. Richards RS, Bennett JD, Roth JH, Milne K Jr. Arthroscopic diagnosis of intra-articular soft tissue injuries associated with distal radial fractures. J Hand Surg Am. 1997;22(5):772-776.
2. Sammer DM, Shah HM, Shauver MJ, Chung KC. The effect of ulnar styloid fractures on patient-rated outcomes after volar locking plating of distal radius fractures. J Hand Surg Am. 2009;34(9):1595-1602.
3. Villar RN, Marsh D, Rushton N, Greatorex RA. Three years after Colles’ fracture. A prospective review. J Bone Joint Surg Br. 1987;69(4):635-638.
4. Palmer AK, Werner FW. The triangular fibrocartilage complex of the wrist—anatomy and function. J Hand Surg Am. 1981;6(2):153-162.
5. Stuart PR, Berger RA, Linscheid RL, An KN. The dorsopalmar stability of the distal radioulnar joint. J Hand Surg Am. 2000;25(4):689-699.
6. May MM, Lawton JN, Blazar PE. Ulnar styloid fractures associated with distal radius fractures: incidence and implications for distal radioulnar joint instability. J Hand Surg Am. 2002;27(6):965-971.
7. Oskarsson GV, Aaser P, Hjall A. Do we underestimate the predictive value of the ulnar styloid affection in Colles fractures? Arch Orthop Trauma Surg. 1997;116(6-7):341-344.
8. Stoffelen D, De Smet L, Broos P. The importance of the distal radioulnar joint in distal radial fractures. J Hand Surg Br. 1998;23(4):507-511.
9. Buijze GA, Ring D. Clinical impact of united versus nonunited fractures of the proximal half of the ulnar styloid following volar plate fixation of the distal radius. J Hand Surg Am. 2010;35(2):223-227.
10. Kim JK, Yun YH, Kim DJ, Yun GU. Comparison of united and nonunited fractures of the ulnar styloid following volar-plate fixation of distal radius fractures. Injury. 2011;42(4):371-375.
11. Wijffels M, Ring D. The influence of non-union of the ulnar styloid on pain, wrist function and instability after distal radius fracture. J Hand Microsurg. 2011;3(1):11-14.
12. Li S, Chen Y, Lin Z, Fan Q, Cui W, Feng Z. Effect of associated ulnar styloid fracture on wrist function after distal radius [in Chinese]. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2012;26(6):666-670.
13. Egol KA, Walsh M, Romo-Cardoso S, Dorsky S, Paksima N. Distal radial fractures in the elderly: operative compared with nonoperative treatment. J Bone Joint Surg Am. 2010;92(9):1851-1857.
14. Cooney WP 3rd, Linscheid RL, Dobyns JH. External pin fixation for unstable Colles’ fractures. J Bone Joint Surg Am. 1979;61(6):840-845.
15. Cerezal L, del Piñal F, Abascal F, García-Valtuille R, Pereda T, Canga A. Imaging findings in ulnar-sided wrist impaction syndromes. Radiographics. 2002;22(1):105-121.
16. Ogawa T, Tanaka T, Yanai T, Kumagai H, Ochiai N. Analysis of soft tissue injuries associated with distal radius fractures. BMC Sports Sci Med Rehabil. 2013;5(1):19.
17. Xarchas KC, Yfandithis P, Kazakos K. Malunion of the ulnar styloid as a cause of ulnar wrist pain. Clin Anat. 2004;17(5):418-422.
18. Frykman G. Fracture of the distal radius including sequelae—shoulder–hand–finger syndrome, disturbance in the distal radio-ulnar joint and impairment of nerve function. A clinical and experimental study. Acta Orthop Scand. 1967:(suppl 108):3+.
19. Fujitani R, Omokawa S, Akahane M, Iida A, Ono H, Tanaka Y. Predictors of distal radioulnar joint instability in distal radius fractures. J Hand Surg Am. 2011;36(12):1919-1925.
Stewardship at 3 a.m.
The 3-month-old infant presents to the emergency department with a fever of 101° F. The emergency physician decides the infant looks ill enough to warrant some investigation. A urinalysis indicates a urinary tract infection. I am consulted to complete the admission to the hospital. The question arises, “Is a lumbar puncture indicated to rule out meningitis?”
I’ve been down this pathway many times. For years I have relied on a meta-analysis which corrected the bias of an old article from 1972.1 Multiple studies in the 2000-2010 time frame have shown that the risk of concurrent meningitis in a young infant with a UTI is vanishingly small. It is much less than 2%, with many studies finding 0%. So on a typical day, my answer is no tap if there is no clinical suggestion of meningitis.

Hospital medicine has recently focused on reducing overdiagnosis and overtreatment. When you only occasionally admit patients to the hospital, untoward events appear random and uncommon. When you work there day in and day out, you appreciate that all medical interventions have risks and costs.
A recent editorial raised the question of stewardship in medicine.2 It asked why physicians would choose a very expensive drug when there is little evidence of its superiority over a much cheaper predecessor. Physicians, whose actions influence a $3 trillion industry, have not embraced stewardship as a major component of their professional responsibilities. The physician does have a fiduciary duty toward the patient. The physician recommends the best care possible to achieve the patient’s goals of care. Dentistry is distinctly different in this regard. Dentists often have several ways of repairing decayed teeth. Various types of fillings are available. Gold fillings are more expensive. Newer implants are several times more expensive than crowns. Dentists routinely adjust their treatment plan based on what the patient can afford.
While most other industries have market competition and profitability as incentives to avoid extravagance, U.S. health care seems unbridled by fiscal responsibility. The news that a small pharmaceutical company had raised the price of an old generic antibiotic by 5000%3 exposed the irrationality and capriciousness4 of the pricing of medications in the United States. Many politicians decried the behavior but to little effect. Most consumer products, especially computers, become more powerful and cheaper with each decade. Health care does not follow this pattern
There are many factors that influence physician behavior. Concerns about malpractice may bias physicians toward expensive overtreatment. Modern medical research is usually published expounding on the benefits of a new technology over a previous therapy without any acknowledgment that the newer and more expensive treatment may have a downside. This biases people to use the latest and greatest treatment even though it may have only demonstrated noninferiority in its trials.
I try to use evidence-based medicine when it is available. In the clinical case described earlier, I indicate to the emergency doctor that unless there is a clinical impression of coexisting meningitis, the lumbar puncture is not indicated. I cite the meta-analysis as I have many times before. But this night is different. I am simultaneously admitting a teenager whose gastrostomy tube had become dislodged and couldn’t be replaced. This neurologically devastated child had had meningitis as an infant. He is a stark reminder of the consequences of a missed diagnosis.
The parents of that child have provided him wonderful care. His skin is in excellent condition. His moderate contractures are testimony to dedicated stretching regimens at home. It is evident that the parents love the child as he is. But I am sure they would give anything to have avoided this scenario and to reverse the consequences of that meningitis. And so, the best evidence we have, that the risk of meningitis in an infant is low, is not as reassuring to me on this night. At 3 a.m., the juxtaposition of the two patients is unsettling. Is the risk low enough? Would that new test5, serum procalcitonin, help me to make a better decision? How certain must I be that an intervention is unnecessary?
Health care policy, economics, and practice guidelines can be debated with detached objectivity around a conference table in the middle of the day. The trepidation in an emergency room at 3 a.m. is different. This is my patient. I am his doctor. That is the heart of medical ethics.
Dr. Powell is a pediatric hospitalist and clinical ethics consultant living in St. Louis. Dr. Powell said he had no relevant financial disclosures or conflicts of interest. E-mail him at [email protected].
References
1. “How common is co-existing meningitis in infants with urinary tract infection?” on Bestbets.org.
2. “Why do doctors choose a $2,000 cure when a $50 one is just as good?” by Andrew Lam, Washington Post, Dec. 10, 2015.
3. “Drug Goes From $13.50 a Tablet to $750, Overnight” by Andrew Pollack, New York Times, Sept. 20, 2015.
4. “How an $84,000 drug got its price: ‘Let’s hold our position … whatever the headlines’ ” by Carolyn Y. Johnson and Brady Dennis, Washington Post, Dec. 1, 2015.
5. JAMA Pediatr. Published online, 2015 Nov 23. doi:10.1001/jamapediatrics.2015.3267.
The 3-month-old infant presents to the emergency department with a fever of 101° F. The emergency physician decides the infant looks ill enough to warrant some investigation. A urinalysis indicates a urinary tract infection. I am consulted to complete the admission to the hospital. The question arises, “Is a lumbar puncture indicated to rule out meningitis?”
I’ve been down this pathway many times. For years I have relied on a meta-analysis which corrected the bias of an old article from 1972.1 Multiple studies in the 2000-2010 time frame have shown that the risk of concurrent meningitis in a young infant with a UTI is vanishingly small. It is much less than 2%, with many studies finding 0%. So on a typical day, my answer is no tap if there is no clinical suggestion of meningitis.
Hospital medicine has recently focused on reducing overdiagnosis and overtreatment. When you only occasionally admit patients to the hospital, untoward events appear random and uncommon. When you work there day in and day out, you appreciate that all medical interventions have risks and costs.
A recent editorial raised the question of stewardship in medicine.2 It asked why physicians would choose a very expensive drug when there is little evidence of its superiority over a much cheaper predecessor. Physicians, whose actions influence a $3 trillion industry, have not embraced stewardship as a major component of their professional responsibilities. The physician does have a fiduciary duty toward the patient. The physician recommends the best care possible to achieve the patient’s goals of care. Dentistry is distinctly different in this regard. Dentists often have several ways of repairing decayed teeth. Various types of fillings are available. Gold fillings are more expensive. Newer implants are several times more expensive than crowns. Dentists routinely adjust their treatment plan based on what the patient can afford.
While most other industries have market competition and profitability as incentives to avoid extravagance, U.S. health care seems unbridled by fiscal responsibility. The news that a small pharmaceutical company had raised the price of an old generic antibiotic by 5000%3 exposed the irrationality and capriciousness4 of the pricing of medications in the United States. Many politicians decried the behavior but to little effect. Most consumer products, especially computers, become more powerful and cheaper with each decade. Health care does not follow this pattern
There are many factors that influence physician behavior. Concerns about malpractice may bias physicians toward expensive overtreatment. Modern medical research is usually published expounding on the benefits of a new technology over a previous therapy without any acknowledgment that the newer and more expensive treatment may have a downside. This biases people to use the latest and greatest treatment even though it may have only demonstrated noninferiority in its trials.
I try to use evidence-based medicine when it is available. In the clinical case described earlier, I indicate to the emergency doctor that unless there is a clinical impression of coexisting meningitis, the lumbar puncture is not indicated. I cite the meta-analysis as I have many times before. But this night is different. I am simultaneously admitting a teenager whose gastrostomy tube had become dislodged and couldn’t be replaced. This neurologically devastated child had had meningitis as an infant. He is a stark reminder of the consequences of a missed diagnosis.
The parents of that child have provided him wonderful care. His skin is in excellent condition. His moderate contractures are testimony to dedicated stretching regimens at home. It is evident that the parents love the child as he is. But I am sure they would give anything to have avoided this scenario and to reverse the consequences of that meningitis. And so, the best evidence we have, that the risk of meningitis in an infant is low, is not as reassuring to me on this night. At 3 a.m., the juxtaposition of the two patients is unsettling. Is the risk low enough? Would that new test5, serum procalcitonin, help me to make a better decision? How certain must I be that an intervention is unnecessary?
Health care policy, economics, and practice guidelines can be debated with detached objectivity around a conference table in the middle of the day. The trepidation in an emergency room at 3 a.m. is different. This is my patient. I am his doctor. That is the heart of medical ethics.
Dr. Powell is a pediatric hospitalist and clinical ethics consultant living in St. Louis. Dr. Powell said he had no relevant financial disclosures or conflicts of interest. E-mail him at [email protected].
References
1. “How common is co-existing meningitis in infants with urinary tract infection?” on Bestbets.org.
2. “Why do doctors choose a $2,000 cure when a $50 one is just as good?” by Andrew Lam, Washington Post, Dec. 10, 2015.
3. “Drug Goes From $13.50 a Tablet to $750, Overnight” by Andrew Pollack, New York Times, Sept. 20, 2015.
4. “How an $84,000 drug got its price: ‘Let’s hold our position … whatever the headlines’ ” by Carolyn Y. Johnson and Brady Dennis, Washington Post, Dec. 1, 2015.
5. JAMA Pediatr. Published online, 2015 Nov 23. doi:10.1001/jamapediatrics.2015.3267.
The 3-month-old infant presents to the emergency department with a fever of 101° F. The emergency physician decides the infant looks ill enough to warrant some investigation. A urinalysis indicates a urinary tract infection. I am consulted to complete the admission to the hospital. The question arises, “Is a lumbar puncture indicated to rule out meningitis?”
I’ve been down this pathway many times. For years I have relied on a meta-analysis which corrected the bias of an old article from 1972.1 Multiple studies in the 2000-2010 time frame have shown that the risk of concurrent meningitis in a young infant with a UTI is vanishingly small. It is much less than 2%, with many studies finding 0%. So on a typical day, my answer is no tap if there is no clinical suggestion of meningitis.
Hospital medicine has recently focused on reducing overdiagnosis and overtreatment. When you only occasionally admit patients to the hospital, untoward events appear random and uncommon. When you work there day in and day out, you appreciate that all medical interventions have risks and costs.
A recent editorial raised the question of stewardship in medicine.2 It asked why physicians would choose a very expensive drug when there is little evidence of its superiority over a much cheaper predecessor. Physicians, whose actions influence a $3 trillion industry, have not embraced stewardship as a major component of their professional responsibilities. The physician does have a fiduciary duty toward the patient. The physician recommends the best care possible to achieve the patient’s goals of care. Dentistry is distinctly different in this regard. Dentists often have several ways of repairing decayed teeth. Various types of fillings are available. Gold fillings are more expensive. Newer implants are several times more expensive than crowns. Dentists routinely adjust their treatment plan based on what the patient can afford.
While most other industries have market competition and profitability as incentives to avoid extravagance, U.S. health care seems unbridled by fiscal responsibility. The news that a small pharmaceutical company had raised the price of an old generic antibiotic by 5000%3 exposed the irrationality and capriciousness4 of the pricing of medications in the United States. Many politicians decried the behavior but to little effect. Most consumer products, especially computers, become more powerful and cheaper with each decade. Health care does not follow this pattern
There are many factors that influence physician behavior. Concerns about malpractice may bias physicians toward expensive overtreatment. Modern medical research is usually published expounding on the benefits of a new technology over a previous therapy without any acknowledgment that the newer and more expensive treatment may have a downside. This biases people to use the latest and greatest treatment even though it may have only demonstrated noninferiority in its trials.
I try to use evidence-based medicine when it is available. In the clinical case described earlier, I indicate to the emergency doctor that unless there is a clinical impression of coexisting meningitis, the lumbar puncture is not indicated. I cite the meta-analysis as I have many times before. But this night is different. I am simultaneously admitting a teenager whose gastrostomy tube had become dislodged and couldn’t be replaced. This neurologically devastated child had had meningitis as an infant. He is a stark reminder of the consequences of a missed diagnosis.
The parents of that child have provided him wonderful care. His skin is in excellent condition. His moderate contractures are testimony to dedicated stretching regimens at home. It is evident that the parents love the child as he is. But I am sure they would give anything to have avoided this scenario and to reverse the consequences of that meningitis. And so, the best evidence we have, that the risk of meningitis in an infant is low, is not as reassuring to me on this night. At 3 a.m., the juxtaposition of the two patients is unsettling. Is the risk low enough? Would that new test5, serum procalcitonin, help me to make a better decision? How certain must I be that an intervention is unnecessary?
Health care policy, economics, and practice guidelines can be debated with detached objectivity around a conference table in the middle of the day. The trepidation in an emergency room at 3 a.m. is different. This is my patient. I am his doctor. That is the heart of medical ethics.
Dr. Powell is a pediatric hospitalist and clinical ethics consultant living in St. Louis. Dr. Powell said he had no relevant financial disclosures or conflicts of interest. E-mail him at [email protected].
References
1. “How common is co-existing meningitis in infants with urinary tract infection?” on Bestbets.org.
2. “Why do doctors choose a $2,000 cure when a $50 one is just as good?” by Andrew Lam, Washington Post, Dec. 10, 2015.
3. “Drug Goes From $13.50 a Tablet to $750, Overnight” by Andrew Pollack, New York Times, Sept. 20, 2015.
4. “How an $84,000 drug got its price: ‘Let’s hold our position … whatever the headlines’ ” by Carolyn Y. Johnson and Brady Dennis, Washington Post, Dec. 1, 2015.
5. JAMA Pediatr. Published online, 2015 Nov 23. doi:10.1001/jamapediatrics.2015.3267.

Radiofrequency Microtenotomy for Elbow Epicondylitis: Midterm Results
Elbow epicondylitis is a painful condition caused by overuse and development of tendon degeneration. It is one of the most common elbow problems in adults, occurring both laterally and medially. “Tennis elbow” or lateral epicondylitis is diagnosed 7 to 10 times more often than the medial form, “golfer’s elbow.”1 Although these injuries are often associated with racquet sports, activities such as bowling and weightlifting and the professions of carpentry, plumbing, and meat-cutting have been described as causes.2,3
Elbow epicondylitis is thought to be the result of multiple microtraumatic events that cause disruption of the internal structure of the tendon and degeneration of the cells and matrix.4 Lesions caused by chronic overuse are now commonly called tendinosis and are not considered inflammatory in nature. Although the term tendinitis is used frequently and indiscriminately, histopathologic studies have shown that specimens of tendon obtained from areas of chronic overuse do not contain large numbers of macrophages, lymphocytes, or neutrophils.5 Rather, tendinosis appears to be a degenerative process that is characterized by the presence of dense populations of fibroblasts, vascular hyperplasia, and disorganized collagen. This constellation of findings has been termed by some authors as angiofibroblastic hyperplasia.6
Conservative care for the treatment of chronic tendinosis has been well described and is often successful. Treatment consists of rest, ice, compression, and elevation in the acute phase. This can be followed with bracing, activity modification, physical therapy, oral nonsteroidal anti-inflammatory drugs, topical applications, and injections of cortisone or platelet-rich plasma. When conservative treatment fails, surgical intervention may be considered. Procedures for the treatment of lateral epicondylitis include open débridement and release, arthroscopic débridement, percutaneous release, and radiofrequency (RF) coblation. The goals of operative treatment are to resect pathological material, to stimulate neovascularization by producing focused local bleeding, and to create a healthy scar while doing the least possible structural damage to surrounding tissues.4
The efficacy of a bipolar RF-based approach for using microtenotomy was first recognized when researchers studied the effects of transmyocardial revascularization for treating congestive heart failure.7 The use of RF- and laser-based transmyocardial revascularization initiated an angiogenic response in degenerated (ischemic) heart tissue. This success led to investigating the use of a RF-based approach for performing microtenotomy. Preclinical studies demonstrated that RF-based microtenotomy was effective for stimulating an angiogenic-healing response in tendon tissue.8 Histologic evaluation of treated tendons showed an early inflammatory response, with new blood-vessel formation by 28 days. In 2005, short-term results of this technique were published.9 This preliminary prospective case series showed that the treatment was safe and effectively improved or eliminated clinical symptoms.9 In the present midterm study, we hypothesized that pain scores would improve after RF microtenotomy and that these favorable results would continue to be observed over a longer term postoperatively.
Materials and Methods
Patients
This was a prospective, nonrandomized, single-center clinical study. After receiving institutional review board approval, patients who were 18 to 65 years of age with a diagnosis of tendinosis were approached for enrollment. For inclusion, patients had to be symptomatic for at least 6 months and had to have failed extensive conservative treatments. Nonoperative treatment included activity modification, enrollment in a facility- or home-based exercise program, bracing, oral nonsteroidal anti-inflammatory medication, and cortisone injection. Candidates with diabetes, confirmed or suspected pregnancy, surgery in the same tendon, implanted hardware adjacent to the target treatment region, or who were receiving care under workers’ compensation or had litigation-related injury were excluded. A single clinician performed a thorough medical history and clinical evaluation. The clinical follow-up and data collection were performed by an independent medical technician.
Clinical Outcomes
Pain status was assessed by using a visual analog scale (VAS). Postoperative clinical assessment was conducted within the first 2 days; at 7 to 10 days; at 4 to 6 weeks; and at 3, 6, 12, and 24 months, up to 9 years postoperatively. The VAS scales were completed annually up to 9 years after the procedure.
The percent improvement of VAS score was calculated. This value represented the difference between the patient’s preoperative and most recent VAS assessments. Failure of the procedure was defined as less than 50% improvement of the VAS score.
The RF-Based Microtenotomy Device
The Topaz Microdebrider (ArthroCare), connected to a System 2000 generator at setting 4 (175 V-RMS), was used to perform the RF-based microtenotomy. The device uses a controlled plasma-mediated RF-based process (coblation). Radiofrequency energy is used to excite the electrolytes in a conductive medium, such as a saline solution, to create precisely focused plasma. The energized particles in the plasma have sufficient energy to break molecular bonds,10,11 excising or dissolving (ie, ablating) soft tissue at relatively low temperatures (typically, 40°-70° C).12,13 The diameter of the active tip of the Topaz device is 0.8 mm.
Surgical Procedure
The senior author performed the majority of procedures in this study. Near the end of the series, the senior author’s associate also performed procedures. The symptomatic area of the tendon was identified and marked while the patient was alert. After the patient was positioned appropriately, light sedation was administered. A tourniquet was placed over the treatment limb and inflated to 250 mm Hg. A small incision, approximately 3 cm in length, was made over the marked treatment site to expose the involved tendon. After initiating sterile isotonic saline flow of 1 drop every 1 to 2 seconds from a line connected to the RF system, the tip of the device was placed on the tendon perpendicular to its surface (Figure 1). Using a light touch, it was activated for 500 milliseconds using a timer accessory for the control box. Five to 8 grams of pressure were applied with the device to penetrate the tendon and achieve successful ablation. The RF applications were performed at 5-mm intervals, to create a grid-like pattern on and throughout the symptomatic tendon area. The tendon was perforated to a depth of several millimeters on every second or third application throughout the treatment grid. After treatment of the symptomatic area, the wound was irrigated with copious amounts of normal saline solution and closed with interrupted nylon suture. Local anesthetic was injected only in the skin and in subcutaneous tissue. Standard wound dressings were applied. In the immediate postoperative period, the patient was advised to begin gentle active and passive range-of-motion exercises. Each patient was evaluated at 1 week postoperatively. At 6 weeks, patients were permitted to increase the intensity of their activities. Return to sports and heavy lifting was allowed once the patient was asymptomatic and had achieved full strength and range of motion; this typically occurred at 6 to 9 weeks after surgery.
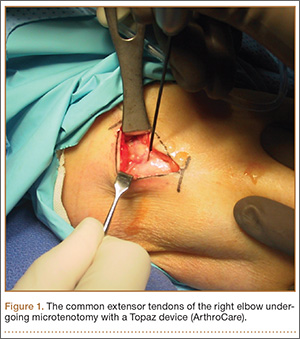
Statistical Analysis
Normally distributed data were described using standard parametric statistics (ie, mean and standard deviation); non-normally distributed data were characterized using nonparametric descriptors (ie, median and quartiles). Statistical evaluation of improvement in pain status was performed by calculating 99% confidence intervals and using the Student t test for change between subsequent time points. Use of confidence intervals provides a descriptive analysis of the observed treatment effect, while permitting determination of statistical relevance. In all statistical testing, confidence bounds not including 0 were considered statistically significant. Probability of P ≤ .01 for committing type I experiment-wise error (rejecting a true null hypothesis) was selected for all statistical testing because of our lack of a control group, small sample size, and evaluation of multiple postoperative time points.
Results
Eighty consecutive patients with tendinosis of the elbow were included in this study. Sixty-nine patients were treated for lateral epicondylitis and 11 for medial epicondylitis. The average age of the patients (33 women, 47 men) was 50 years. The duration of follow-up evaluation ranged from 6 months to 9 years (mean, 2.5 years; median, 2 years). The Table presents the VAS improvement for these patients after the RF microtenotomy.
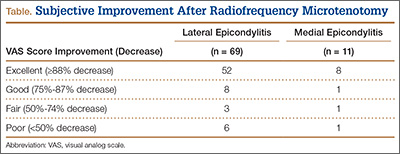
Within the lateral epicondylitis group, 91% (63/69) of the patients reported a successful outcome. The postoperative VAS improved to 1.3 from 6.9, which demonstrated an 81% improvement. Of the 6 patients that did not improve, 2 underwent repeat surgery.
Among the patients treated for medial epicondylitis, 91% (10/11) reported improvement in symptoms. The postoperative VAS improved to 1.3 from 6.1, a 79% improvement. One patient did not improve and did not undergo repeat surgery.
Discussion
For the treatment of medial and lateral elbow epicondylitis, RF microtenotomy is successful in 91% of patients. Symptomatic improvement was observed up to 9 years postoperatively. During this study, no complications were recorded; 7 treatment failures occurred. When compared with other techniques, the results with RF microtenotomy are equivalent or better.
In a retrospective study, Szabo and colleagues14 compared open, arthroscopic, and percutaneous release for lateral elbow tendinosis. They found the 3 methods to be highly effective for the treatment of tendinosis with no significant difference between them. Resection of the epicondyle and transfer of the anconeus muscle was found to be effective (94%) in a retrospective study by Almquist and colleagues.15 Dunn and coauthors16 reported a 97% success rate at 10 to 14 years postoperatively with a mini-open technique. Rubenthaler and colleagues17 showed 88% effectiveness for the open technique and 93% for the arthroscopic technique. With arthroscopic release of the extensor carpi radialis brevis tendon, Lattermann and coauthors18 reported clinical improvement in 94% of patients. In a study by Rose and colleagues,19 denervation of the lateral epicondyle was effective in relieving pain in 80% of patients who had had a positive response to a local anesthetic block. In a recently published study by Koh and coauthors,20 19 of 20 patients experienced a favorable outcome after treatment with ultrasonic microresection.
Regardless of surgical methods and their reported success rate, complications are associated with elbow surgery. Postoperative problems may include restricted function, elbow instability, persistent muscle weakness, and painful neuroma of the posterior cutaneous nerve.10,21,22 The recent introduction of arthroscopic release offers the potential for less morbidity and enables visualization of the elbow joint. However, disadvantages of the arthroscopic approach include violation of the joint for extra-articular pathology, increased operative time and cost, and neurovascular complications. Additionally, it is possible that the entire spectrum of extra-articular tendinosis cannot be effectively identified arthroscopically.23 In a prospective, randomized study, Meknas and colleagues24 compared RF microtenotomy with extensor tendon release and repair. They showed that patients treated with RF-microtenotomy experienced earlier pain relief and improved grip strength over the release group.
Different proposed mechanisms of action have been described to explain the favorable effects of the RF-based microtenotomy procedure, such as induced healing by an angiogenic response in the tendon tissue. In an animal study, Harwood and colleagues8 showed that low-dose RF-based plasma microtenotomy has the ability to stimulate angiogenic growth factors in tendons, such as αv integrin and vascular endothelial growth factor. These factors have been shown to be associated with healing.8 Early inflammatory response with new-vessel formation after 28 days was found in another animal study using the same method.25 Evaluation of RF-based methods in a prospective controlled laboratory study using a rabbit-tendon model showed histologic evidence of early inflammation with development of neovasculature after treatment.8 A later histologic study using an aged Achilles rabbit tendon model was performed to evaluate the effect of RF-based plasma microtenotomy on collagen remodeling.25 The degenerated tendon showed gaps, few normal crimpings, and a lack of reflectivity under polarized light. At 9 days after treatment, the treated tendon showed localized irregular crimpings, and, at 30 days, it showed regular crimping, tightly dense collagen fibers, and hypercellularity with good reflectivity. This was similar in appearance to a normal nondegenerated tendon (Figures 2A-2D). The RF-treated tendon also demonstrated an increase in production of insulin-like growth factor-1, β-fibroblast growth factor-1, αv integrin, and vascular endothelial growth factor.
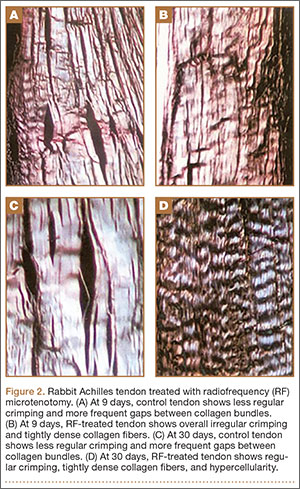
Pathologic nerve ingrowth or nerve irritation in the tendon substance has been considered a possible cause of the pain experienced with tendinosis. Radiofrequency treatment has been shown to induce acute degeneration and ablation of sensory nerve fibers.26 These degenerated nerve fibers were observed to regenerate at 90 days after treatment.27 These findings provide potential evidence for early pain relief that is maintained long term as the nerves regenerate.
This midterm follow-up of patients with elbow epicondylitis has shown that RF-based microtenotomy can produce successful, durable results. Microtenotomy is a technically simple procedure to perform and is associated with a rapid and uncomplicated recovery. It is safe and can effectively eliminate or markedly reduce clinical symptoms.
Limitations
Lateral epicondylitis has been described as a self-limited disease, with resolution of symptoms at 12 to 18 months with conservative treatment. This perspective challenges the indication of any proposed surgical treatment for the condition. Although the results of this research demonstrated the benefits of RF microtenotomy, there are inherent limitations of the study design. The study lacks a control group, and randomization would improve the strength of the study. Additional outcome measures, such as Disabilities of the Arm, Shoulder, and Hand score, and grip strength could complement pain scores to provide more data. These data were collected in a preliminary study.9 Postoperative histologic analysis of treated human tissue would be ideal, but ethical considerations limit study to animal models. An additional limitation is potential examiner bias. Data collection was performed by an independent medical technician; a third-party blinded evaluation could have been performed, but this was not feasible in a clinical setting.
Conclusion
Radiofrequency-based microtenotomy is a safe and effective procedure for elbow epicondylitis. The results are durable with successful outcomes observed 9 years after surgery.
1. Leach RE, Miller JK. Lateral and medial epicondylitis of the elbow. Clin Sports Med. 1987;6(2):259-272.
2. Vangsness CT Jr, Jobe FW. Surgical technique of medial epicondylitis: Results in 35 elbows. J Bone Joint Surg Br. 1991;73(3):409-411.
3. Galloway M, DeMaio M, Mangine R. Rehabilitative techniques in the treatment of medial and lateral epicondylitis. Orthopedics. 1992;15(9):1089-1096.
4. Kraushaar BS, Nirschl RP. Tendinosis of the elbow (tennis elbow). Clinical features and findings of histological, immunohistochemical, and electron microscopy studies. J Bone Joint Surg Am. 1999;81(2):259-278.
5. Leadbetter WB. Cell-matrix response in tendon injury. Clin Sports Med. 1992;11(3):533-578.
6. Nirschl RP. Tennis elbow tendinosis: pathoanatomy, nonsurgical and surgical management. In: Fine LJ, ed. Repetitive Motion Disorders of the Upper Extremity. Rosemont, IL: American Academy of Orthopaedic Surgeons; 1995:467-479.
7. Chu V, Kuang J, Aiaid A, Korkola S, Chiu RC. Angiogenic response induced by mechanical transmyocardial revascularization. J Thorac Cardiovasc Surg 1999;118:849-856.
8. Harwood R, Bowden K, Amiel M, Tasto JP, Amiel D. Structural and angiogenic response to bipolar radiofrequency treatment of normal rabbit achilles tendon: a potential application to the treatment of tendinosis. Trans Orthop Res Soc. 2003;28:819.
9. Tasto JP, Cummings J, Medlock V, Hardesty R, Amiel D. Microtenotomy using a radiofrequency probe to treat lateral epicondylitis. Arthroscopy. 2005;21(7):851-860.
10. Woloszko J, Stalder KR, Brown IG. Plasma characteristics of repetitively-pulsed electrical discharges in saline solutions used for surgical procedures. IEEE Trans Plasma Sci. 2002;30:1376-1383.
11. Stalder KR, Woloszko J, Brown IG, Smith CD. Repetitive plasma discharges in saline solutions. Appl Phys Lett. 2001;79:4503-4505.
12. Woloszko J, Gilbride C. Coblation technology (plasma mediated ablation for otolaryngology applications). Proc SPIE. 2000;3907:306–316.
13. Woloszko J, Kwende MM, Stalder KR. Coblation in otolaryngology. Proc SPIE. 2003;4949:341–352.
14. Szabo SJ, Savoie FH 3rd, Field LD, Ramsey JR, Hosemann CD. Tendinosis of the extensor carpi radialis brevis: an evaluation of three methods of operative treatment. J Shoulder Elbow Surg Am. 2006;15(6):721-727.
15. Almquist EE, Necking L, Bach AW. Epicondylar resection with anconeus transfer for chronic lateral epicondylitis. J Hand Surg Am. 1998;23(4):723-731.
16. Dunn JH, Kim JJ, Davis L, Nirschl RP. Ten- to 14-year follow-up of the Nirschl surgical technique for lateral epicondylitis. Am J Sports Med. 2008;36(2):261-266.
17. Rubenthaler F, Wiese M, Senge A, Keller L, Wittenberg RH. Long-term follow-up of open and endoscopic Hohmann procedures for lateral epicondylitis. Arthroscopy. 2005;21(6):684-690.
18. Lattermann C, Romeo AA, Anbari A, et al. Arthroscopic debridement of the extensor carpi radialis brevis for the treatment of recalcitrant lateral epicondylitis. J Shoulder Elbow Surg. 2010;19(5):651-656.
19. Rose NE, Forman SK, Dellon AL. Denervation of the lateral epicondyle for treatment of chronic lateral epicondylitis. J Hand Surg Am. 2013;38(2):344-349.
20. Koh JS, Mohan PC, Howe TS, et al. Fasciotomy and surgical tenotomy for recalcitrant lateral elbow tendonopathy: early clinical experience with a novel device for minimally invasive percutaneous microresection. Am J Sports Med. 2013;41(3):636-644.
21. Nirschl RP, Ashman ES. Elbow tendonopathy: tennis elbow. Clin Sports Med. 2003;22(4):813-836.
22. Dellon AL, Kim J, Ducic I. Painful neuroma of the posterior cutaneous nerve of the forearm after surgery for lateral humeral epicondylitis. J Hand Surg Am. 2004;29(3):387-390.
23. Cummins CA. Lateral epicondylitis: in-vivo assessment of arthroscopic debridement and correlation with patient outcomes. Am J Sports Med. 2006;34(9):1486-1491.
24. Meknas K, Odden-Miland A, Mercer JB, Castillejo M, Johansen O. Radiofrequency microtenotomy: a promising method for treatment of recalcitrant lateral epicondylitis. Am J Sports Med. 2008;36(10):1960-1965.
25. Takahashi N, Tasto JP, Locke J, et al. The use of radiofrequency (RF) for the treatment of chronic tendinosis. Paper presented at: 6th Biennial Congress of the International Society of Arthroscopy, Knee Surgery, and Orthopaedic Sports Medicine Congress; May 2007; Florence, Italy. Abstract 1433.
26. Takahashi N, Tasto JP, Ritter M, et al. Pain relief through an antinociceptive effect after radiofrequency application. Am J Sports Med. 2007;35(5):805-810.
27. Ochiai N, Tasto JP, Ohtori S, Takahashi N, Moriya H, Amiel D. Nerve regeneration after radiofrequency ablation. Am J Sports Med. 2007;35(11):1940-1944.
Elbow epicondylitis is a painful condition caused by overuse and development of tendon degeneration. It is one of the most common elbow problems in adults, occurring both laterally and medially. “Tennis elbow” or lateral epicondylitis is diagnosed 7 to 10 times more often than the medial form, “golfer’s elbow.”1 Although these injuries are often associated with racquet sports, activities such as bowling and weightlifting and the professions of carpentry, plumbing, and meat-cutting have been described as causes.2,3
Elbow epicondylitis is thought to be the result of multiple microtraumatic events that cause disruption of the internal structure of the tendon and degeneration of the cells and matrix.4 Lesions caused by chronic overuse are now commonly called tendinosis and are not considered inflammatory in nature. Although the term tendinitis is used frequently and indiscriminately, histopathologic studies have shown that specimens of tendon obtained from areas of chronic overuse do not contain large numbers of macrophages, lymphocytes, or neutrophils.5 Rather, tendinosis appears to be a degenerative process that is characterized by the presence of dense populations of fibroblasts, vascular hyperplasia, and disorganized collagen. This constellation of findings has been termed by some authors as angiofibroblastic hyperplasia.6
Conservative care for the treatment of chronic tendinosis has been well described and is often successful. Treatment consists of rest, ice, compression, and elevation in the acute phase. This can be followed with bracing, activity modification, physical therapy, oral nonsteroidal anti-inflammatory drugs, topical applications, and injections of cortisone or platelet-rich plasma. When conservative treatment fails, surgical intervention may be considered. Procedures for the treatment of lateral epicondylitis include open débridement and release, arthroscopic débridement, percutaneous release, and radiofrequency (RF) coblation. The goals of operative treatment are to resect pathological material, to stimulate neovascularization by producing focused local bleeding, and to create a healthy scar while doing the least possible structural damage to surrounding tissues.4
The efficacy of a bipolar RF-based approach for using microtenotomy was first recognized when researchers studied the effects of transmyocardial revascularization for treating congestive heart failure.7 The use of RF- and laser-based transmyocardial revascularization initiated an angiogenic response in degenerated (ischemic) heart tissue. This success led to investigating the use of a RF-based approach for performing microtenotomy. Preclinical studies demonstrated that RF-based microtenotomy was effective for stimulating an angiogenic-healing response in tendon tissue.8 Histologic evaluation of treated tendons showed an early inflammatory response, with new blood-vessel formation by 28 days. In 2005, short-term results of this technique were published.9 This preliminary prospective case series showed that the treatment was safe and effectively improved or eliminated clinical symptoms.9 In the present midterm study, we hypothesized that pain scores would improve after RF microtenotomy and that these favorable results would continue to be observed over a longer term postoperatively.
Materials and Methods
Patients
This was a prospective, nonrandomized, single-center clinical study. After receiving institutional review board approval, patients who were 18 to 65 years of age with a diagnosis of tendinosis were approached for enrollment. For inclusion, patients had to be symptomatic for at least 6 months and had to have failed extensive conservative treatments. Nonoperative treatment included activity modification, enrollment in a facility- or home-based exercise program, bracing, oral nonsteroidal anti-inflammatory medication, and cortisone injection. Candidates with diabetes, confirmed or suspected pregnancy, surgery in the same tendon, implanted hardware adjacent to the target treatment region, or who were receiving care under workers’ compensation or had litigation-related injury were excluded. A single clinician performed a thorough medical history and clinical evaluation. The clinical follow-up and data collection were performed by an independent medical technician.
Clinical Outcomes
Pain status was assessed by using a visual analog scale (VAS). Postoperative clinical assessment was conducted within the first 2 days; at 7 to 10 days; at 4 to 6 weeks; and at 3, 6, 12, and 24 months, up to 9 years postoperatively. The VAS scales were completed annually up to 9 years after the procedure.
The percent improvement of VAS score was calculated. This value represented the difference between the patient’s preoperative and most recent VAS assessments. Failure of the procedure was defined as less than 50% improvement of the VAS score.
The RF-Based Microtenotomy Device
The Topaz Microdebrider (ArthroCare), connected to a System 2000 generator at setting 4 (175 V-RMS), was used to perform the RF-based microtenotomy. The device uses a controlled plasma-mediated RF-based process (coblation). Radiofrequency energy is used to excite the electrolytes in a conductive medium, such as a saline solution, to create precisely focused plasma. The energized particles in the plasma have sufficient energy to break molecular bonds,10,11 excising or dissolving (ie, ablating) soft tissue at relatively low temperatures (typically, 40°-70° C).12,13 The diameter of the active tip of the Topaz device is 0.8 mm.
Surgical Procedure
The senior author performed the majority of procedures in this study. Near the end of the series, the senior author’s associate also performed procedures. The symptomatic area of the tendon was identified and marked while the patient was alert. After the patient was positioned appropriately, light sedation was administered. A tourniquet was placed over the treatment limb and inflated to 250 mm Hg. A small incision, approximately 3 cm in length, was made over the marked treatment site to expose the involved tendon. After initiating sterile isotonic saline flow of 1 drop every 1 to 2 seconds from a line connected to the RF system, the tip of the device was placed on the tendon perpendicular to its surface (Figure 1). Using a light touch, it was activated for 500 milliseconds using a timer accessory for the control box. Five to 8 grams of pressure were applied with the device to penetrate the tendon and achieve successful ablation. The RF applications were performed at 5-mm intervals, to create a grid-like pattern on and throughout the symptomatic tendon area. The tendon was perforated to a depth of several millimeters on every second or third application throughout the treatment grid. After treatment of the symptomatic area, the wound was irrigated with copious amounts of normal saline solution and closed with interrupted nylon suture. Local anesthetic was injected only in the skin and in subcutaneous tissue. Standard wound dressings were applied. In the immediate postoperative period, the patient was advised to begin gentle active and passive range-of-motion exercises. Each patient was evaluated at 1 week postoperatively. At 6 weeks, patients were permitted to increase the intensity of their activities. Return to sports and heavy lifting was allowed once the patient was asymptomatic and had achieved full strength and range of motion; this typically occurred at 6 to 9 weeks after surgery.
Statistical Analysis
Normally distributed data were described using standard parametric statistics (ie, mean and standard deviation); non-normally distributed data were characterized using nonparametric descriptors (ie, median and quartiles). Statistical evaluation of improvement in pain status was performed by calculating 99% confidence intervals and using the Student t test for change between subsequent time points. Use of confidence intervals provides a descriptive analysis of the observed treatment effect, while permitting determination of statistical relevance. In all statistical testing, confidence bounds not including 0 were considered statistically significant. Probability of P ≤ .01 for committing type I experiment-wise error (rejecting a true null hypothesis) was selected for all statistical testing because of our lack of a control group, small sample size, and evaluation of multiple postoperative time points.
Results
Eighty consecutive patients with tendinosis of the elbow were included in this study. Sixty-nine patients were treated for lateral epicondylitis and 11 for medial epicondylitis. The average age of the patients (33 women, 47 men) was 50 years. The duration of follow-up evaluation ranged from 6 months to 9 years (mean, 2.5 years; median, 2 years). The Table presents the VAS improvement for these patients after the RF microtenotomy.
Within the lateral epicondylitis group, 91% (63/69) of the patients reported a successful outcome. The postoperative VAS improved to 1.3 from 6.9, which demonstrated an 81% improvement. Of the 6 patients that did not improve, 2 underwent repeat surgery.
Among the patients treated for medial epicondylitis, 91% (10/11) reported improvement in symptoms. The postoperative VAS improved to 1.3 from 6.1, a 79% improvement. One patient did not improve and did not undergo repeat surgery.
Discussion
For the treatment of medial and lateral elbow epicondylitis, RF microtenotomy is successful in 91% of patients. Symptomatic improvement was observed up to 9 years postoperatively. During this study, no complications were recorded; 7 treatment failures occurred. When compared with other techniques, the results with RF microtenotomy are equivalent or better.
In a retrospective study, Szabo and colleagues14 compared open, arthroscopic, and percutaneous release for lateral elbow tendinosis. They found the 3 methods to be highly effective for the treatment of tendinosis with no significant difference between them. Resection of the epicondyle and transfer of the anconeus muscle was found to be effective (94%) in a retrospective study by Almquist and colleagues.15 Dunn and coauthors16 reported a 97% success rate at 10 to 14 years postoperatively with a mini-open technique. Rubenthaler and colleagues17 showed 88% effectiveness for the open technique and 93% for the arthroscopic technique. With arthroscopic release of the extensor carpi radialis brevis tendon, Lattermann and coauthors18 reported clinical improvement in 94% of patients. In a study by Rose and colleagues,19 denervation of the lateral epicondyle was effective in relieving pain in 80% of patients who had had a positive response to a local anesthetic block. In a recently published study by Koh and coauthors,20 19 of 20 patients experienced a favorable outcome after treatment with ultrasonic microresection.
Regardless of surgical methods and their reported success rate, complications are associated with elbow surgery. Postoperative problems may include restricted function, elbow instability, persistent muscle weakness, and painful neuroma of the posterior cutaneous nerve.10,21,22 The recent introduction of arthroscopic release offers the potential for less morbidity and enables visualization of the elbow joint. However, disadvantages of the arthroscopic approach include violation of the joint for extra-articular pathology, increased operative time and cost, and neurovascular complications. Additionally, it is possible that the entire spectrum of extra-articular tendinosis cannot be effectively identified arthroscopically.23 In a prospective, randomized study, Meknas and colleagues24 compared RF microtenotomy with extensor tendon release and repair. They showed that patients treated with RF-microtenotomy experienced earlier pain relief and improved grip strength over the release group.
Different proposed mechanisms of action have been described to explain the favorable effects of the RF-based microtenotomy procedure, such as induced healing by an angiogenic response in the tendon tissue. In an animal study, Harwood and colleagues8 showed that low-dose RF-based plasma microtenotomy has the ability to stimulate angiogenic growth factors in tendons, such as αv integrin and vascular endothelial growth factor. These factors have been shown to be associated with healing.8 Early inflammatory response with new-vessel formation after 28 days was found in another animal study using the same method.25 Evaluation of RF-based methods in a prospective controlled laboratory study using a rabbit-tendon model showed histologic evidence of early inflammation with development of neovasculature after treatment.8 A later histologic study using an aged Achilles rabbit tendon model was performed to evaluate the effect of RF-based plasma microtenotomy on collagen remodeling.25 The degenerated tendon showed gaps, few normal crimpings, and a lack of reflectivity under polarized light. At 9 days after treatment, the treated tendon showed localized irregular crimpings, and, at 30 days, it showed regular crimping, tightly dense collagen fibers, and hypercellularity with good reflectivity. This was similar in appearance to a normal nondegenerated tendon (Figures 2A-2D). The RF-treated tendon also demonstrated an increase in production of insulin-like growth factor-1, β-fibroblast growth factor-1, αv integrin, and vascular endothelial growth factor.
Pathologic nerve ingrowth or nerve irritation in the tendon substance has been considered a possible cause of the pain experienced with tendinosis. Radiofrequency treatment has been shown to induce acute degeneration and ablation of sensory nerve fibers.26 These degenerated nerve fibers were observed to regenerate at 90 days after treatment.27 These findings provide potential evidence for early pain relief that is maintained long term as the nerves regenerate.
This midterm follow-up of patients with elbow epicondylitis has shown that RF-based microtenotomy can produce successful, durable results. Microtenotomy is a technically simple procedure to perform and is associated with a rapid and uncomplicated recovery. It is safe and can effectively eliminate or markedly reduce clinical symptoms.
Limitations
Lateral epicondylitis has been described as a self-limited disease, with resolution of symptoms at 12 to 18 months with conservative treatment. This perspective challenges the indication of any proposed surgical treatment for the condition. Although the results of this research demonstrated the benefits of RF microtenotomy, there are inherent limitations of the study design. The study lacks a control group, and randomization would improve the strength of the study. Additional outcome measures, such as Disabilities of the Arm, Shoulder, and Hand score, and grip strength could complement pain scores to provide more data. These data were collected in a preliminary study.9 Postoperative histologic analysis of treated human tissue would be ideal, but ethical considerations limit study to animal models. An additional limitation is potential examiner bias. Data collection was performed by an independent medical technician; a third-party blinded evaluation could have been performed, but this was not feasible in a clinical setting.
Conclusion
Radiofrequency-based microtenotomy is a safe and effective procedure for elbow epicondylitis. The results are durable with successful outcomes observed 9 years after surgery.
Elbow epicondylitis is a painful condition caused by overuse and development of tendon degeneration. It is one of the most common elbow problems in adults, occurring both laterally and medially. “Tennis elbow” or lateral epicondylitis is diagnosed 7 to 10 times more often than the medial form, “golfer’s elbow.”1 Although these injuries are often associated with racquet sports, activities such as bowling and weightlifting and the professions of carpentry, plumbing, and meat-cutting have been described as causes.2,3
Elbow epicondylitis is thought to be the result of multiple microtraumatic events that cause disruption of the internal structure of the tendon and degeneration of the cells and matrix.4 Lesions caused by chronic overuse are now commonly called tendinosis and are not considered inflammatory in nature. Although the term tendinitis is used frequently and indiscriminately, histopathologic studies have shown that specimens of tendon obtained from areas of chronic overuse do not contain large numbers of macrophages, lymphocytes, or neutrophils.5 Rather, tendinosis appears to be a degenerative process that is characterized by the presence of dense populations of fibroblasts, vascular hyperplasia, and disorganized collagen. This constellation of findings has been termed by some authors as angiofibroblastic hyperplasia.6
Conservative care for the treatment of chronic tendinosis has been well described and is often successful. Treatment consists of rest, ice, compression, and elevation in the acute phase. This can be followed with bracing, activity modification, physical therapy, oral nonsteroidal anti-inflammatory drugs, topical applications, and injections of cortisone or platelet-rich plasma. When conservative treatment fails, surgical intervention may be considered. Procedures for the treatment of lateral epicondylitis include open débridement and release, arthroscopic débridement, percutaneous release, and radiofrequency (RF) coblation. The goals of operative treatment are to resect pathological material, to stimulate neovascularization by producing focused local bleeding, and to create a healthy scar while doing the least possible structural damage to surrounding tissues.4
The efficacy of a bipolar RF-based approach for using microtenotomy was first recognized when researchers studied the effects of transmyocardial revascularization for treating congestive heart failure.7 The use of RF- and laser-based transmyocardial revascularization initiated an angiogenic response in degenerated (ischemic) heart tissue. This success led to investigating the use of a RF-based approach for performing microtenotomy. Preclinical studies demonstrated that RF-based microtenotomy was effective for stimulating an angiogenic-healing response in tendon tissue.8 Histologic evaluation of treated tendons showed an early inflammatory response, with new blood-vessel formation by 28 days. In 2005, short-term results of this technique were published.9 This preliminary prospective case series showed that the treatment was safe and effectively improved or eliminated clinical symptoms.9 In the present midterm study, we hypothesized that pain scores would improve after RF microtenotomy and that these favorable results would continue to be observed over a longer term postoperatively.
Materials and Methods
Patients
This was a prospective, nonrandomized, single-center clinical study. After receiving institutional review board approval, patients who were 18 to 65 years of age with a diagnosis of tendinosis were approached for enrollment. For inclusion, patients had to be symptomatic for at least 6 months and had to have failed extensive conservative treatments. Nonoperative treatment included activity modification, enrollment in a facility- or home-based exercise program, bracing, oral nonsteroidal anti-inflammatory medication, and cortisone injection. Candidates with diabetes, confirmed or suspected pregnancy, surgery in the same tendon, implanted hardware adjacent to the target treatment region, or who were receiving care under workers’ compensation or had litigation-related injury were excluded. A single clinician performed a thorough medical history and clinical evaluation. The clinical follow-up and data collection were performed by an independent medical technician.
Clinical Outcomes
Pain status was assessed by using a visual analog scale (VAS). Postoperative clinical assessment was conducted within the first 2 days; at 7 to 10 days; at 4 to 6 weeks; and at 3, 6, 12, and 24 months, up to 9 years postoperatively. The VAS scales were completed annually up to 9 years after the procedure.
The percent improvement of VAS score was calculated. This value represented the difference between the patient’s preoperative and most recent VAS assessments. Failure of the procedure was defined as less than 50% improvement of the VAS score.
The RF-Based Microtenotomy Device
The Topaz Microdebrider (ArthroCare), connected to a System 2000 generator at setting 4 (175 V-RMS), was used to perform the RF-based microtenotomy. The device uses a controlled plasma-mediated RF-based process (coblation). Radiofrequency energy is used to excite the electrolytes in a conductive medium, such as a saline solution, to create precisely focused plasma. The energized particles in the plasma have sufficient energy to break molecular bonds,10,11 excising or dissolving (ie, ablating) soft tissue at relatively low temperatures (typically, 40°-70° C).12,13 The diameter of the active tip of the Topaz device is 0.8 mm.
Surgical Procedure
The senior author performed the majority of procedures in this study. Near the end of the series, the senior author’s associate also performed procedures. The symptomatic area of the tendon was identified and marked while the patient was alert. After the patient was positioned appropriately, light sedation was administered. A tourniquet was placed over the treatment limb and inflated to 250 mm Hg. A small incision, approximately 3 cm in length, was made over the marked treatment site to expose the involved tendon. After initiating sterile isotonic saline flow of 1 drop every 1 to 2 seconds from a line connected to the RF system, the tip of the device was placed on the tendon perpendicular to its surface (Figure 1). Using a light touch, it was activated for 500 milliseconds using a timer accessory for the control box. Five to 8 grams of pressure were applied with the device to penetrate the tendon and achieve successful ablation. The RF applications were performed at 5-mm intervals, to create a grid-like pattern on and throughout the symptomatic tendon area. The tendon was perforated to a depth of several millimeters on every second or third application throughout the treatment grid. After treatment of the symptomatic area, the wound was irrigated with copious amounts of normal saline solution and closed with interrupted nylon suture. Local anesthetic was injected only in the skin and in subcutaneous tissue. Standard wound dressings were applied. In the immediate postoperative period, the patient was advised to begin gentle active and passive range-of-motion exercises. Each patient was evaluated at 1 week postoperatively. At 6 weeks, patients were permitted to increase the intensity of their activities. Return to sports and heavy lifting was allowed once the patient was asymptomatic and had achieved full strength and range of motion; this typically occurred at 6 to 9 weeks after surgery.
Statistical Analysis
Normally distributed data were described using standard parametric statistics (ie, mean and standard deviation); non-normally distributed data were characterized using nonparametric descriptors (ie, median and quartiles). Statistical evaluation of improvement in pain status was performed by calculating 99% confidence intervals and using the Student t test for change between subsequent time points. Use of confidence intervals provides a descriptive analysis of the observed treatment effect, while permitting determination of statistical relevance. In all statistical testing, confidence bounds not including 0 were considered statistically significant. Probability of P ≤ .01 for committing type I experiment-wise error (rejecting a true null hypothesis) was selected for all statistical testing because of our lack of a control group, small sample size, and evaluation of multiple postoperative time points.
Results
Eighty consecutive patients with tendinosis of the elbow were included in this study. Sixty-nine patients were treated for lateral epicondylitis and 11 for medial epicondylitis. The average age of the patients (33 women, 47 men) was 50 years. The duration of follow-up evaluation ranged from 6 months to 9 years (mean, 2.5 years; median, 2 years). The Table presents the VAS improvement for these patients after the RF microtenotomy.
Within the lateral epicondylitis group, 91% (63/69) of the patients reported a successful outcome. The postoperative VAS improved to 1.3 from 6.9, which demonstrated an 81% improvement. Of the 6 patients that did not improve, 2 underwent repeat surgery.
Among the patients treated for medial epicondylitis, 91% (10/11) reported improvement in symptoms. The postoperative VAS improved to 1.3 from 6.1, a 79% improvement. One patient did not improve and did not undergo repeat surgery.
Discussion
For the treatment of medial and lateral elbow epicondylitis, RF microtenotomy is successful in 91% of patients. Symptomatic improvement was observed up to 9 years postoperatively. During this study, no complications were recorded; 7 treatment failures occurred. When compared with other techniques, the results with RF microtenotomy are equivalent or better.
In a retrospective study, Szabo and colleagues14 compared open, arthroscopic, and percutaneous release for lateral elbow tendinosis. They found the 3 methods to be highly effective for the treatment of tendinosis with no significant difference between them. Resection of the epicondyle and transfer of the anconeus muscle was found to be effective (94%) in a retrospective study by Almquist and colleagues.15 Dunn and coauthors16 reported a 97% success rate at 10 to 14 years postoperatively with a mini-open technique. Rubenthaler and colleagues17 showed 88% effectiveness for the open technique and 93% for the arthroscopic technique. With arthroscopic release of the extensor carpi radialis brevis tendon, Lattermann and coauthors18 reported clinical improvement in 94% of patients. In a study by Rose and colleagues,19 denervation of the lateral epicondyle was effective in relieving pain in 80% of patients who had had a positive response to a local anesthetic block. In a recently published study by Koh and coauthors,20 19 of 20 patients experienced a favorable outcome after treatment with ultrasonic microresection.
Regardless of surgical methods and their reported success rate, complications are associated with elbow surgery. Postoperative problems may include restricted function, elbow instability, persistent muscle weakness, and painful neuroma of the posterior cutaneous nerve.10,21,22 The recent introduction of arthroscopic release offers the potential for less morbidity and enables visualization of the elbow joint. However, disadvantages of the arthroscopic approach include violation of the joint for extra-articular pathology, increased operative time and cost, and neurovascular complications. Additionally, it is possible that the entire spectrum of extra-articular tendinosis cannot be effectively identified arthroscopically.23 In a prospective, randomized study, Meknas and colleagues24 compared RF microtenotomy with extensor tendon release and repair. They showed that patients treated with RF-microtenotomy experienced earlier pain relief and improved grip strength over the release group.
Different proposed mechanisms of action have been described to explain the favorable effects of the RF-based microtenotomy procedure, such as induced healing by an angiogenic response in the tendon tissue. In an animal study, Harwood and colleagues8 showed that low-dose RF-based plasma microtenotomy has the ability to stimulate angiogenic growth factors in tendons, such as αv integrin and vascular endothelial growth factor. These factors have been shown to be associated with healing.8 Early inflammatory response with new-vessel formation after 28 days was found in another animal study using the same method.25 Evaluation of RF-based methods in a prospective controlled laboratory study using a rabbit-tendon model showed histologic evidence of early inflammation with development of neovasculature after treatment.8 A later histologic study using an aged Achilles rabbit tendon model was performed to evaluate the effect of RF-based plasma microtenotomy on collagen remodeling.25 The degenerated tendon showed gaps, few normal crimpings, and a lack of reflectivity under polarized light. At 9 days after treatment, the treated tendon showed localized irregular crimpings, and, at 30 days, it showed regular crimping, tightly dense collagen fibers, and hypercellularity with good reflectivity. This was similar in appearance to a normal nondegenerated tendon (Figures 2A-2D). The RF-treated tendon also demonstrated an increase in production of insulin-like growth factor-1, β-fibroblast growth factor-1, αv integrin, and vascular endothelial growth factor.
Pathologic nerve ingrowth or nerve irritation in the tendon substance has been considered a possible cause of the pain experienced with tendinosis. Radiofrequency treatment has been shown to induce acute degeneration and ablation of sensory nerve fibers.26 These degenerated nerve fibers were observed to regenerate at 90 days after treatment.27 These findings provide potential evidence for early pain relief that is maintained long term as the nerves regenerate.
This midterm follow-up of patients with elbow epicondylitis has shown that RF-based microtenotomy can produce successful, durable results. Microtenotomy is a technically simple procedure to perform and is associated with a rapid and uncomplicated recovery. It is safe and can effectively eliminate or markedly reduce clinical symptoms.
Limitations
Lateral epicondylitis has been described as a self-limited disease, with resolution of symptoms at 12 to 18 months with conservative treatment. This perspective challenges the indication of any proposed surgical treatment for the condition. Although the results of this research demonstrated the benefits of RF microtenotomy, there are inherent limitations of the study design. The study lacks a control group, and randomization would improve the strength of the study. Additional outcome measures, such as Disabilities of the Arm, Shoulder, and Hand score, and grip strength could complement pain scores to provide more data. These data were collected in a preliminary study.9 Postoperative histologic analysis of treated human tissue would be ideal, but ethical considerations limit study to animal models. An additional limitation is potential examiner bias. Data collection was performed by an independent medical technician; a third-party blinded evaluation could have been performed, but this was not feasible in a clinical setting.
Conclusion
Radiofrequency-based microtenotomy is a safe and effective procedure for elbow epicondylitis. The results are durable with successful outcomes observed 9 years after surgery.
1. Leach RE, Miller JK. Lateral and medial epicondylitis of the elbow. Clin Sports Med. 1987;6(2):259-272.
2. Vangsness CT Jr, Jobe FW. Surgical technique of medial epicondylitis: Results in 35 elbows. J Bone Joint Surg Br. 1991;73(3):409-411.
3. Galloway M, DeMaio M, Mangine R. Rehabilitative techniques in the treatment of medial and lateral epicondylitis. Orthopedics. 1992;15(9):1089-1096.
4. Kraushaar BS, Nirschl RP. Tendinosis of the elbow (tennis elbow). Clinical features and findings of histological, immunohistochemical, and electron microscopy studies. J Bone Joint Surg Am. 1999;81(2):259-278.
5. Leadbetter WB. Cell-matrix response in tendon injury. Clin Sports Med. 1992;11(3):533-578.
6. Nirschl RP. Tennis elbow tendinosis: pathoanatomy, nonsurgical and surgical management. In: Fine LJ, ed. Repetitive Motion Disorders of the Upper Extremity. Rosemont, IL: American Academy of Orthopaedic Surgeons; 1995:467-479.
7. Chu V, Kuang J, Aiaid A, Korkola S, Chiu RC. Angiogenic response induced by mechanical transmyocardial revascularization. J Thorac Cardiovasc Surg 1999;118:849-856.
8. Harwood R, Bowden K, Amiel M, Tasto JP, Amiel D. Structural and angiogenic response to bipolar radiofrequency treatment of normal rabbit achilles tendon: a potential application to the treatment of tendinosis. Trans Orthop Res Soc. 2003;28:819.
9. Tasto JP, Cummings J, Medlock V, Hardesty R, Amiel D. Microtenotomy using a radiofrequency probe to treat lateral epicondylitis. Arthroscopy. 2005;21(7):851-860.
10. Woloszko J, Stalder KR, Brown IG. Plasma characteristics of repetitively-pulsed electrical discharges in saline solutions used for surgical procedures. IEEE Trans Plasma Sci. 2002;30:1376-1383.
11. Stalder KR, Woloszko J, Brown IG, Smith CD. Repetitive plasma discharges in saline solutions. Appl Phys Lett. 2001;79:4503-4505.
12. Woloszko J, Gilbride C. Coblation technology (plasma mediated ablation for otolaryngology applications). Proc SPIE. 2000;3907:306–316.
13. Woloszko J, Kwende MM, Stalder KR. Coblation in otolaryngology. Proc SPIE. 2003;4949:341–352.
14. Szabo SJ, Savoie FH 3rd, Field LD, Ramsey JR, Hosemann CD. Tendinosis of the extensor carpi radialis brevis: an evaluation of three methods of operative treatment. J Shoulder Elbow Surg Am. 2006;15(6):721-727.
15. Almquist EE, Necking L, Bach AW. Epicondylar resection with anconeus transfer for chronic lateral epicondylitis. J Hand Surg Am. 1998;23(4):723-731.
16. Dunn JH, Kim JJ, Davis L, Nirschl RP. Ten- to 14-year follow-up of the Nirschl surgical technique for lateral epicondylitis. Am J Sports Med. 2008;36(2):261-266.
17. Rubenthaler F, Wiese M, Senge A, Keller L, Wittenberg RH. Long-term follow-up of open and endoscopic Hohmann procedures for lateral epicondylitis. Arthroscopy. 2005;21(6):684-690.
18. Lattermann C, Romeo AA, Anbari A, et al. Arthroscopic debridement of the extensor carpi radialis brevis for the treatment of recalcitrant lateral epicondylitis. J Shoulder Elbow Surg. 2010;19(5):651-656.
19. Rose NE, Forman SK, Dellon AL. Denervation of the lateral epicondyle for treatment of chronic lateral epicondylitis. J Hand Surg Am. 2013;38(2):344-349.
20. Koh JS, Mohan PC, Howe TS, et al. Fasciotomy and surgical tenotomy for recalcitrant lateral elbow tendonopathy: early clinical experience with a novel device for minimally invasive percutaneous microresection. Am J Sports Med. 2013;41(3):636-644.
21. Nirschl RP, Ashman ES. Elbow tendonopathy: tennis elbow. Clin Sports Med. 2003;22(4):813-836.
22. Dellon AL, Kim J, Ducic I. Painful neuroma of the posterior cutaneous nerve of the forearm after surgery for lateral humeral epicondylitis. J Hand Surg Am. 2004;29(3):387-390.
23. Cummins CA. Lateral epicondylitis: in-vivo assessment of arthroscopic debridement and correlation with patient outcomes. Am J Sports Med. 2006;34(9):1486-1491.
24. Meknas K, Odden-Miland A, Mercer JB, Castillejo M, Johansen O. Radiofrequency microtenotomy: a promising method for treatment of recalcitrant lateral epicondylitis. Am J Sports Med. 2008;36(10):1960-1965.
25. Takahashi N, Tasto JP, Locke J, et al. The use of radiofrequency (RF) for the treatment of chronic tendinosis. Paper presented at: 6th Biennial Congress of the International Society of Arthroscopy, Knee Surgery, and Orthopaedic Sports Medicine Congress; May 2007; Florence, Italy. Abstract 1433.
26. Takahashi N, Tasto JP, Ritter M, et al. Pain relief through an antinociceptive effect after radiofrequency application. Am J Sports Med. 2007;35(5):805-810.
27. Ochiai N, Tasto JP, Ohtori S, Takahashi N, Moriya H, Amiel D. Nerve regeneration after radiofrequency ablation. Am J Sports Med. 2007;35(11):1940-1944.
1. Leach RE, Miller JK. Lateral and medial epicondylitis of the elbow. Clin Sports Med. 1987;6(2):259-272.
2. Vangsness CT Jr, Jobe FW. Surgical technique of medial epicondylitis: Results in 35 elbows. J Bone Joint Surg Br. 1991;73(3):409-411.
3. Galloway M, DeMaio M, Mangine R. Rehabilitative techniques in the treatment of medial and lateral epicondylitis. Orthopedics. 1992;15(9):1089-1096.
4. Kraushaar BS, Nirschl RP. Tendinosis of the elbow (tennis elbow). Clinical features and findings of histological, immunohistochemical, and electron microscopy studies. J Bone Joint Surg Am. 1999;81(2):259-278.
5. Leadbetter WB. Cell-matrix response in tendon injury. Clin Sports Med. 1992;11(3):533-578.
6. Nirschl RP. Tennis elbow tendinosis: pathoanatomy, nonsurgical and surgical management. In: Fine LJ, ed. Repetitive Motion Disorders of the Upper Extremity. Rosemont, IL: American Academy of Orthopaedic Surgeons; 1995:467-479.
7. Chu V, Kuang J, Aiaid A, Korkola S, Chiu RC. Angiogenic response induced by mechanical transmyocardial revascularization. J Thorac Cardiovasc Surg 1999;118:849-856.
8. Harwood R, Bowden K, Amiel M, Tasto JP, Amiel D. Structural and angiogenic response to bipolar radiofrequency treatment of normal rabbit achilles tendon: a potential application to the treatment of tendinosis. Trans Orthop Res Soc. 2003;28:819.
9. Tasto JP, Cummings J, Medlock V, Hardesty R, Amiel D. Microtenotomy using a radiofrequency probe to treat lateral epicondylitis. Arthroscopy. 2005;21(7):851-860.
10. Woloszko J, Stalder KR, Brown IG. Plasma characteristics of repetitively-pulsed electrical discharges in saline solutions used for surgical procedures. IEEE Trans Plasma Sci. 2002;30:1376-1383.
11. Stalder KR, Woloszko J, Brown IG, Smith CD. Repetitive plasma discharges in saline solutions. Appl Phys Lett. 2001;79:4503-4505.
12. Woloszko J, Gilbride C. Coblation technology (plasma mediated ablation for otolaryngology applications). Proc SPIE. 2000;3907:306–316.
13. Woloszko J, Kwende MM, Stalder KR. Coblation in otolaryngology. Proc SPIE. 2003;4949:341–352.
14. Szabo SJ, Savoie FH 3rd, Field LD, Ramsey JR, Hosemann CD. Tendinosis of the extensor carpi radialis brevis: an evaluation of three methods of operative treatment. J Shoulder Elbow Surg Am. 2006;15(6):721-727.
15. Almquist EE, Necking L, Bach AW. Epicondylar resection with anconeus transfer for chronic lateral epicondylitis. J Hand Surg Am. 1998;23(4):723-731.
16. Dunn JH, Kim JJ, Davis L, Nirschl RP. Ten- to 14-year follow-up of the Nirschl surgical technique for lateral epicondylitis. Am J Sports Med. 2008;36(2):261-266.
17. Rubenthaler F, Wiese M, Senge A, Keller L, Wittenberg RH. Long-term follow-up of open and endoscopic Hohmann procedures for lateral epicondylitis. Arthroscopy. 2005;21(6):684-690.
18. Lattermann C, Romeo AA, Anbari A, et al. Arthroscopic debridement of the extensor carpi radialis brevis for the treatment of recalcitrant lateral epicondylitis. J Shoulder Elbow Surg. 2010;19(5):651-656.
19. Rose NE, Forman SK, Dellon AL. Denervation of the lateral epicondyle for treatment of chronic lateral epicondylitis. J Hand Surg Am. 2013;38(2):344-349.
20. Koh JS, Mohan PC, Howe TS, et al. Fasciotomy and surgical tenotomy for recalcitrant lateral elbow tendonopathy: early clinical experience with a novel device for minimally invasive percutaneous microresection. Am J Sports Med. 2013;41(3):636-644.
21. Nirschl RP, Ashman ES. Elbow tendonopathy: tennis elbow. Clin Sports Med. 2003;22(4):813-836.
22. Dellon AL, Kim J, Ducic I. Painful neuroma of the posterior cutaneous nerve of the forearm after surgery for lateral humeral epicondylitis. J Hand Surg Am. 2004;29(3):387-390.
23. Cummins CA. Lateral epicondylitis: in-vivo assessment of arthroscopic debridement and correlation with patient outcomes. Am J Sports Med. 2006;34(9):1486-1491.
24. Meknas K, Odden-Miland A, Mercer JB, Castillejo M, Johansen O. Radiofrequency microtenotomy: a promising method for treatment of recalcitrant lateral epicondylitis. Am J Sports Med. 2008;36(10):1960-1965.
25. Takahashi N, Tasto JP, Locke J, et al. The use of radiofrequency (RF) for the treatment of chronic tendinosis. Paper presented at: 6th Biennial Congress of the International Society of Arthroscopy, Knee Surgery, and Orthopaedic Sports Medicine Congress; May 2007; Florence, Italy. Abstract 1433.
26. Takahashi N, Tasto JP, Ritter M, et al. Pain relief through an antinociceptive effect after radiofrequency application. Am J Sports Med. 2007;35(5):805-810.
27. Ochiai N, Tasto JP, Ohtori S, Takahashi N, Moriya H, Amiel D. Nerve regeneration after radiofrequency ablation. Am J Sports Med. 2007;35(11):1940-1944.
ABIM continues to lighten recertification load
AGA applauds the announcement by the American Board of Internal Medicine that it is extending the suspension of the practice improvement, patient safety, and patient voice requirements until at least December 2018.
Throughout 2015, AGA has pushed ABIM to reconsider the burdensome recertification process. Gastroenterologists need a recertification system that fosters active learning, not high-stakes testing.

Our campaign continues – AGA is communicating with other subspecialty societies to work together to secure the best approach to MOC consistent with the principles we previously published:
• MOC needs to be simpler, less intrusive, and less expensive.
• We support ending the high‐stakes, every-10‐year exam.
• We do not support closed‐book assessments as they do not represent the current realities of medicine in the digital age.
• We support the principles of lifelong learning as evidenced by ongoing CME activities, rather than lifelong testing.
• We support the concept that, for the many diplomates who specialize within certain areas of gastroenterology and hepatology, MOC should not need to include high‐stakes assessments of areas where the diplomate may not practice.
We hear you that MOC is a burden and we will continue to push for the principles of individualization, specialization, and dropping the high-stakes exam – sooner rather than later.
Practicalities for those up for recertification
While we advocate for a new MOC system, there are requirements that still stand. Make sure you are up to date before the end of 2015:
• If you need to complete MOC points by Dec. 31, 2015, AGA has activities you can complete to earn those points. Please visit the MOC section of the AGA website to see how we can help you.
• Each board certified physician’s requirements are slightly different based on the year of your certification or most recent recertification. For details specific to you, we urge you to log in to your ABIM Physician Portal.
For more information, read our paper, The Gastroenterologist-accountable Professionalism in Practice Pathway, and consensus principles for reform that AGA developed with ACG, ASGE, AASLD, ANMS and NASPGHAN.
AGA applauds the announcement by the American Board of Internal Medicine that it is extending the suspension of the practice improvement, patient safety, and patient voice requirements until at least December 2018.
Throughout 2015, AGA has pushed ABIM to reconsider the burdensome recertification process. Gastroenterologists need a recertification system that fosters active learning, not high-stakes testing.
Our campaign continues – AGA is communicating with other subspecialty societies to work together to secure the best approach to MOC consistent with the principles we previously published:
• MOC needs to be simpler, less intrusive, and less expensive.
• We support ending the high‐stakes, every-10‐year exam.
• We do not support closed‐book assessments as they do not represent the current realities of medicine in the digital age.
• We support the principles of lifelong learning as evidenced by ongoing CME activities, rather than lifelong testing.
• We support the concept that, for the many diplomates who specialize within certain areas of gastroenterology and hepatology, MOC should not need to include high‐stakes assessments of areas where the diplomate may not practice.
We hear you that MOC is a burden and we will continue to push for the principles of individualization, specialization, and dropping the high-stakes exam – sooner rather than later.
Practicalities for those up for recertification
While we advocate for a new MOC system, there are requirements that still stand. Make sure you are up to date before the end of 2015:
• If you need to complete MOC points by Dec. 31, 2015, AGA has activities you can complete to earn those points. Please visit the MOC section of the AGA website to see how we can help you.
• Each board certified physician’s requirements are slightly different based on the year of your certification or most recent recertification. For details specific to you, we urge you to log in to your ABIM Physician Portal.
For more information, read our paper, The Gastroenterologist-accountable Professionalism in Practice Pathway, and consensus principles for reform that AGA developed with ACG, ASGE, AASLD, ANMS and NASPGHAN.
AGA applauds the announcement by the American Board of Internal Medicine that it is extending the suspension of the practice improvement, patient safety, and patient voice requirements until at least December 2018.
Throughout 2015, AGA has pushed ABIM to reconsider the burdensome recertification process. Gastroenterologists need a recertification system that fosters active learning, not high-stakes testing.
Our campaign continues – AGA is communicating with other subspecialty societies to work together to secure the best approach to MOC consistent with the principles we previously published:
• MOC needs to be simpler, less intrusive, and less expensive.
• We support ending the high‐stakes, every-10‐year exam.
• We do not support closed‐book assessments as they do not represent the current realities of medicine in the digital age.
• We support the principles of lifelong learning as evidenced by ongoing CME activities, rather than lifelong testing.
• We support the concept that, for the many diplomates who specialize within certain areas of gastroenterology and hepatology, MOC should not need to include high‐stakes assessments of areas where the diplomate may not practice.
We hear you that MOC is a burden and we will continue to push for the principles of individualization, specialization, and dropping the high-stakes exam – sooner rather than later.
Practicalities for those up for recertification
While we advocate for a new MOC system, there are requirements that still stand. Make sure you are up to date before the end of 2015:
• If you need to complete MOC points by Dec. 31, 2015, AGA has activities you can complete to earn those points. Please visit the MOC section of the AGA website to see how we can help you.
• Each board certified physician’s requirements are slightly different based on the year of your certification or most recent recertification. For details specific to you, we urge you to log in to your ABIM Physician Portal.
For more information, read our paper, The Gastroenterologist-accountable Professionalism in Practice Pathway, and consensus principles for reform that AGA developed with ACG, ASGE, AASLD, ANMS and NASPGHAN.

Stewart Tepper, MD
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

A girl refuses to eat solid food because she is afraid of choking
CASE Refusing solid food
Ms. B, age 11, is admitted to a pediatric medical inpatient unit for unintentional weight loss of 14 lb (15% total body weight) over the past month. She reports having 2 traumatic episodes last month: choking on a piece of cheese and having a swab specimen taken for a rapid strep test, which required several people to restrain her (the test was positive). Since then, she has refused to ingest solids, despite hunger and a desire to eat.
Ms. B reports diffuse abdominal pain merely “at the sight of food” and a fear of swallowing solids. She denies difficulty or pain upon swallowing, nausea, vomiting, or any change in bowel habits.
Her mother reports that, on the rare occasion that Ms. B has attempted to eat solid food, she spent as long as an hour cutting it into small pieces before bringing it to her mouth—after which she put the food down without eating. Her mother also witnessed Ms. B holding food in her mouth for “a very long time,” then spitting it out.
Ms. B says she is distressed about the weight loss and recognizes that her fear of solid food is excessive.
What would your diagnosis of Ms. B’s problem be?
a) anorexia nervosa
b) avoidant/restrictive food intake disorder (ARFID)
c) specific phobia (swallowing solids or choking)
d) generalized anxiety disorder (GAD)
The authors’ observations
DSM-5 describes a new eating disorder called ARFID, which replaces the DSM-IV-TR diagnosis of feeding disorder of infancy or early childhood</keyword>. DSM-5 diagnostic criteria define ARFID as:
An eating or feeding disturbance (eg, avoidance based on the sensory characteristics of food…) as manifested by persistent failure to meet appropriate nutritional and/or energy needs associated with at least one of the following: 1. Significant weight loss (or failure to achieve expected weight gain or faltering growth in children). 2. Significant nutritional deficiency. 3. Dependence on enteral feeding or oral nutritional supplements. 4. Marked interference with psychosocial functioning.1
DSM-5 also specifies that the disorder cannot be caused by lack of available food or traditional cultural practices; cannot coexist with anorexia or bulimia nervosa; and is not attributable to a concurrent medical or psychiatric disorder.
Because it is a newly defined diagnosis, the epidemiology of ARFID is unclear. Patients with ARFID have a wide variety of eating symptoms that do not meet diagnostic criteria for anorexia or bulimia nervosa. One study found that, among a cohort of mostly adolescent patients who presented for evaluation of an eating disorder, 14% met diagnostic criteria for ARFID.2 Another retrospective case-control study found a similar prevalence among patients age 8 to 18 (13.8% of 712 patients).3 Because of the variety of maladaptive feeding behaviors seen in ARFID, there is little evidence that pharmacotherapy is effective.4
HISTORY Premature birth
Ms. B’s medical history states that she is twin A of a premature birth at 26 weeks (birth weight, 1,060 g), with a 90-day neonatal intensive care unit hospitalization, during which she required supplemental oxygen and nasogastric tube feeding. She has mild cerebral palsy, and had motor delay of walking at 2.5 years old. Currently, she has no motor difficulties.
Ms. B does not have a psychiatric history and does not take medications. Her mother has a history of major depressive disorder that is well controlled with an unspecified selective serotonin reuptake inhibitor. Ms. B’s maternal uncle has poorly controlled schizophrenia.
During Ms. B’s 6-day hospitalization, her mental status exams are unremarkable. She is shy but cooperative and open. Her mood ranges from “sad” and “nervous” on admission to “fine” with mood-congruent affect. She denies suicidal or homicidal thoughts and hallucinations, and demonstrates good insight and judgment. All laboratory values are within normal limits except for mild hypophosphatemia (3.7 mg/dL) and mild hyperalbuminemia (4.9 g/dL) on admission, which may have been related to her nutritional status.
DIAGNOSIS Not solely psychiatric
The psychiatric differential diagnosis includes:
- ARFID
- specific phobia of swallowing solids or choking (pseudodysphagia)
- GAD
- unspecified feeding disorder.
Ms. B meets diagnostic criteria for ARFID, particularly that of profound acute weight loss due to restrictive eating behaviors. Her presentation also is similar to that of a specific phobia, namely profound anxiety upon even the thought of solid food (phobic stimulus) and recognition that her fear is excessive. However, she fails to meet diagnostic criteria for phobia in children, which require duration of at least 6 months. GAD also is less likely because she has had symptoms for 1 month (also requires 6-month duration) and her anxiety is limited to feeding behaviors.
The treatment team starts exposure therapy, encouraging Ms. B to begin taking small bites of textured foods, such as oatmeal.
On hospital Day 5, barium esophagogram reveals extrinsic compression of the esophagus. To identify the precise cause of compression, chest magnetic resonance angiogram reveals that Ms. B has a right-sided aortic arch with an aberrant left subclavian artery at T4 that, with the ligamentum arteriosum and left pulmonary artery, form a vascular ring impinging around the esophagus.
The authors’ observations
Dysphagia lusoria, coined in 1789 from the root for “natural abnormality,”5 is caused by an aberrant right subclavian artery, which persists because of abnormal involution of the right fourth aortic arch during embryogenesis6 (Figure 1). The condition is diagnosed incidentally in most affected adults, who are asymptomatic throughout life and do not require operative management.6

Symptoms of dysphagia lusoria can include dysphagia and recurrent aspiration if significant esophageal impingement is present.7 It is thought that patients tend to show more symptoms as they age because of sclerosis, aneurysm, or atherosclerosis of the impinging vessel.7 Cough and dyspnea caused by impingement of the trachea also have been reported, and might be more common in children because of tracheal flexibility.5
This case illustrates the complex interrelationship of physical and psychiatric conditions. After the treatment team discovered a physical cause of Ms. B’s symptoms, the initial psychiatric diagnosis became problematic, but remained critically important for long-term treatment of her comorbid eating phobia.
TREATMENT Therapy, surgery
The treatment team is faced with the question of whether dysphagia lusoria fully accounts for Ms. B’s status. The anatomic anomaly might explain the initial choking incident if the food particle was lodged at the site of impingement, but does not account for development of a severe acute eating disorder and subsequent malnutrition.
Because of the presence of dysphagia lusoria, the team concludes that Ms. B does not meet diagnostic criteria for ARFID. She is given a diagnosis of unspecified eating disorder.
Ms. B is discharged and referred to a pediatric cardiothoracic surgeon for operative consult of symptomatic dysphagia lusoria. By discharge, she is successfully eating yogurt with fruit chunks, which she had rejected earlier. She also expresses optimism about eating cake at her birthday party, scheduled for 2 weeks after discharge.
The treatment team strongly recommends outpatient psychiatric follow-up to manage Ms. B’s unspecified eating disorder with cognitive-behavioral therapy and exposure and response prevention, which helped to mildly decrease her anxiety during the hospital stay.
Ms. B’s surgeon deems the case severe enough to warrant surgery. Surgery for dysphagia lusoria can be indicated to prevent progression of symptoms into adulthood8; options include division of the ligamentum arteriosum to loosen the vascular ring and re-implantation of the aberrant subclavian artery.9,10 (On the other hand, dietary modification might be therapeutic in patients whose symptoms are mild.5)
The surgeon elects to divide the ligamentum arteriosum to relieve the pressure of the vascular ring on the esophagus. Intraoperatively, he discovers that Ms. B has a mildly patent ductus arteriosus (PDA) (Figure 2), which is more common in premature infants than in full-term births. The PDA is clipped on both sides and divided. This immediately causes the vascular ring created by the aberrant left subclavian artery, right-sided aorta, and PDA to spring open, releasing pressure on the esophagus.
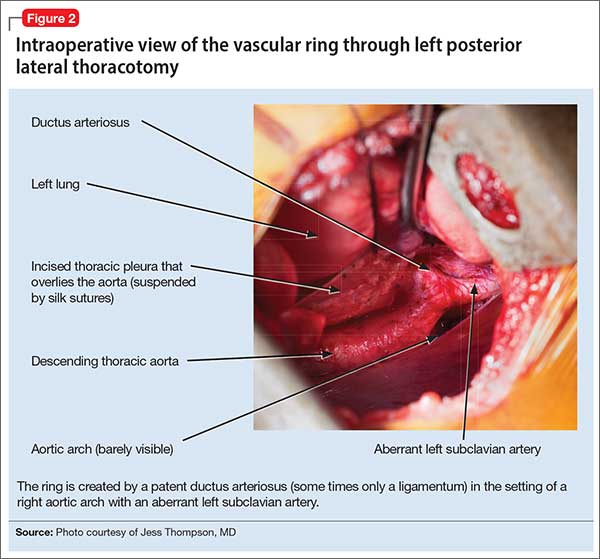
The aberrant left subclavian artery emerges from an abnormal bulging of aorta, known as Kommerell’s diverticulum. After the vascular ring is released, the diverticulum is not observed to impinge on the esophagus; however, as a preventive measure, the surgeon sutures it to the anterior spinous ligament. This will prevent the diverticulum from enlarging and impinging on the esophagus as Ms. B grows to adulthood.
The surgery is completed without complications. Ms. B tolerates the procedure well.
Ten days after surgery, Ms. B is recovering well. She and her mother report satisfaction with the procedure to release the vascular ring. She discontinues her pain medication after 2 days and slowly begins reintroducing solid foods. Her fear of dysphagia and choking rapidly diminish.
Bottom Line
The diagnosis and management of eating disorders, including avoidant/restrictive food intake disorder and choking phobia, can be challenging. Furthermore, if an anatomical or organic anomaly is found, it is important to question how a patient’s eating disorder can be managed best through interdisciplinary collaboration between medical and behavioral specialties.
Related Resources
• Bryant-Waugh R. Feeding and eating disorders in children. Curr Opin Psychiatry. 2013;26(6):537-542.
• Norris ML, Robinson A, Obeid N, et al. Exploring avoidant/restrictive food intake disorder in eating disordered patients: a descriptive study. Int J Eat Disord. 2014;47(5):495-499.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
2. Ornstein RM, Rosen DS, Mammel KA, et al. Distribution of eating disorders in children and adolescents using the proposed DSM-5 criteria for feeding and eating disorders. J Adolesc Health. 2013;53(2):303-305.
3. Fisher MM, Rosen DS, Ornstein RM, et al. Characteristics of avoidant/restrictive food intake disorder in children and adolescents: a “new disorder” in DSM-5. J Adolesc Health. 2014;55(1):49-52.
4. Kelly NR, Shank LM, Bakalar JL, et al. Pediatric feeding and eating disorders: current state of diagnosis and treatment. Curr Psychiatry Rep. 2014;16(5):446.
5. Janssen M, Baggen MG, Veen HF, et al. Dysphagia lusoria: clinical aspects, manometric findings, diagnosis, and therapy. Am J Gastroenterol. 2000;95(6):1411-1416.
6. Abraham V, Mathew A, Cherian V, et al. Aberrant subclavian artery: anatomical curiosity or clinical entity. Int J Surg. 2009;7(2):106-109.
7. Calleja F, Eguaras M, Montero J, et al. Aberrant right subclavian artery associated with common carotid trunk. A rare cause of vascular ring. Eur J Cardiothorac Surg. 1990;4(10):568-570.
8. Jalaie H, Grommes J, Sailer A, et al. Treatment of symptomatic aberrant subclavian arteries. Eur J Vasc Endovasc Surg. 2014;48(5):521-526.
9. Gross RE. Surgical treatment for dysphagia lusoria. Ann Surg. 1946;124:532-534.
10. Morrris CD, Kanter KR, Miller JI Jr. Late-onset dysphagia lusoria. Ann Thorac Surg. 2001;71(2):710-712.
CASE Refusing solid food
Ms. B, age 11, is admitted to a pediatric medical inpatient unit for unintentional weight loss of 14 lb (15% total body weight) over the past month. She reports having 2 traumatic episodes last month: choking on a piece of cheese and having a swab specimen taken for a rapid strep test, which required several people to restrain her (the test was positive). Since then, she has refused to ingest solids, despite hunger and a desire to eat.
Ms. B reports diffuse abdominal pain merely “at the sight of food” and a fear of swallowing solids. She denies difficulty or pain upon swallowing, nausea, vomiting, or any change in bowel habits.
Her mother reports that, on the rare occasion that Ms. B has attempted to eat solid food, she spent as long as an hour cutting it into small pieces before bringing it to her mouth—after which she put the food down without eating. Her mother also witnessed Ms. B holding food in her mouth for “a very long time,” then spitting it out.
Ms. B says she is distressed about the weight loss and recognizes that her fear of solid food is excessive.
What would your diagnosis of Ms. B’s problem be?
a) anorexia nervosa
b) avoidant/restrictive food intake disorder (ARFID)
c) specific phobia (swallowing solids or choking)
d) generalized anxiety disorder (GAD)
The authors’ observations
DSM-5 describes a new eating disorder called ARFID, which replaces the DSM-IV-TR diagnosis of feeding disorder of infancy or early childhood</keyword>. DSM-5 diagnostic criteria define ARFID as:
An eating or feeding disturbance (eg, avoidance based on the sensory characteristics of food…) as manifested by persistent failure to meet appropriate nutritional and/or energy needs associated with at least one of the following: 1. Significant weight loss (or failure to achieve expected weight gain or faltering growth in children). 2. Significant nutritional deficiency. 3. Dependence on enteral feeding or oral nutritional supplements. 4. Marked interference with psychosocial functioning.1
DSM-5 also specifies that the disorder cannot be caused by lack of available food or traditional cultural practices; cannot coexist with anorexia or bulimia nervosa; and is not attributable to a concurrent medical or psychiatric disorder.
Because it is a newly defined diagnosis, the epidemiology of ARFID is unclear. Patients with ARFID have a wide variety of eating symptoms that do not meet diagnostic criteria for anorexia or bulimia nervosa. One study found that, among a cohort of mostly adolescent patients who presented for evaluation of an eating disorder, 14% met diagnostic criteria for ARFID.2 Another retrospective case-control study found a similar prevalence among patients age 8 to 18 (13.8% of 712 patients).3 Because of the variety of maladaptive feeding behaviors seen in ARFID, there is little evidence that pharmacotherapy is effective.4
HISTORY Premature birth
Ms. B’s medical history states that she is twin A of a premature birth at 26 weeks (birth weight, 1,060 g), with a 90-day neonatal intensive care unit hospitalization, during which she required supplemental oxygen and nasogastric tube feeding. She has mild cerebral palsy, and had motor delay of walking at 2.5 years old. Currently, she has no motor difficulties.
Ms. B does not have a psychiatric history and does not take medications. Her mother has a history of major depressive disorder that is well controlled with an unspecified selective serotonin reuptake inhibitor. Ms. B’s maternal uncle has poorly controlled schizophrenia.
During Ms. B’s 6-day hospitalization, her mental status exams are unremarkable. She is shy but cooperative and open. Her mood ranges from “sad” and “nervous” on admission to “fine” with mood-congruent affect. She denies suicidal or homicidal thoughts and hallucinations, and demonstrates good insight and judgment. All laboratory values are within normal limits except for mild hypophosphatemia (3.7 mg/dL) and mild hyperalbuminemia (4.9 g/dL) on admission, which may have been related to her nutritional status.
DIAGNOSIS Not solely psychiatric
The psychiatric differential diagnosis includes:
- ARFID
- specific phobia of swallowing solids or choking (pseudodysphagia)
- GAD
- unspecified feeding disorder.
Ms. B meets diagnostic criteria for ARFID, particularly that of profound acute weight loss due to restrictive eating behaviors. Her presentation also is similar to that of a specific phobia, namely profound anxiety upon even the thought of solid food (phobic stimulus) and recognition that her fear is excessive. However, she fails to meet diagnostic criteria for phobia in children, which require duration of at least 6 months. GAD also is less likely because she has had symptoms for 1 month (also requires 6-month duration) and her anxiety is limited to feeding behaviors.
The treatment team starts exposure therapy, encouraging Ms. B to begin taking small bites of textured foods, such as oatmeal.
On hospital Day 5, barium esophagogram reveals extrinsic compression of the esophagus. To identify the precise cause of compression, chest magnetic resonance angiogram reveals that Ms. B has a right-sided aortic arch with an aberrant left subclavian artery at T4 that, with the ligamentum arteriosum and left pulmonary artery, form a vascular ring impinging around the esophagus.
The authors’ observations
Dysphagia lusoria, coined in 1789 from the root for “natural abnormality,”5 is caused by an aberrant right subclavian artery, which persists because of abnormal involution of the right fourth aortic arch during embryogenesis6 (Figure 1). The condition is diagnosed incidentally in most affected adults, who are asymptomatic throughout life and do not require operative management.6
Symptoms of dysphagia lusoria can include dysphagia and recurrent aspiration if significant esophageal impingement is present.7 It is thought that patients tend to show more symptoms as they age because of sclerosis, aneurysm, or atherosclerosis of the impinging vessel.7 Cough and dyspnea caused by impingement of the trachea also have been reported, and might be more common in children because of tracheal flexibility.5
This case illustrates the complex interrelationship of physical and psychiatric conditions. After the treatment team discovered a physical cause of Ms. B’s symptoms, the initial psychiatric diagnosis became problematic, but remained critically important for long-term treatment of her comorbid eating phobia.
TREATMENT Therapy, surgery
The treatment team is faced with the question of whether dysphagia lusoria fully accounts for Ms. B’s status. The anatomic anomaly might explain the initial choking incident if the food particle was lodged at the site of impingement, but does not account for development of a severe acute eating disorder and subsequent malnutrition.
Because of the presence of dysphagia lusoria, the team concludes that Ms. B does not meet diagnostic criteria for ARFID. She is given a diagnosis of unspecified eating disorder.
Ms. B is discharged and referred to a pediatric cardiothoracic surgeon for operative consult of symptomatic dysphagia lusoria. By discharge, she is successfully eating yogurt with fruit chunks, which she had rejected earlier. She also expresses optimism about eating cake at her birthday party, scheduled for 2 weeks after discharge.
The treatment team strongly recommends outpatient psychiatric follow-up to manage Ms. B’s unspecified eating disorder with cognitive-behavioral therapy and exposure and response prevention, which helped to mildly decrease her anxiety during the hospital stay.
Ms. B’s surgeon deems the case severe enough to warrant surgery. Surgery for dysphagia lusoria can be indicated to prevent progression of symptoms into adulthood8; options include division of the ligamentum arteriosum to loosen the vascular ring and re-implantation of the aberrant subclavian artery.9,10 (On the other hand, dietary modification might be therapeutic in patients whose symptoms are mild.5)
The surgeon elects to divide the ligamentum arteriosum to relieve the pressure of the vascular ring on the esophagus. Intraoperatively, he discovers that Ms. B has a mildly patent ductus arteriosus (PDA) (Figure 2), which is more common in premature infants than in full-term births. The PDA is clipped on both sides and divided. This immediately causes the vascular ring created by the aberrant left subclavian artery, right-sided aorta, and PDA to spring open, releasing pressure on the esophagus.
The aberrant left subclavian artery emerges from an abnormal bulging of aorta, known as Kommerell’s diverticulum. After the vascular ring is released, the diverticulum is not observed to impinge on the esophagus; however, as a preventive measure, the surgeon sutures it to the anterior spinous ligament. This will prevent the diverticulum from enlarging and impinging on the esophagus as Ms. B grows to adulthood.
The surgery is completed without complications. Ms. B tolerates the procedure well.
Ten days after surgery, Ms. B is recovering well. She and her mother report satisfaction with the procedure to release the vascular ring. She discontinues her pain medication after 2 days and slowly begins reintroducing solid foods. Her fear of dysphagia and choking rapidly diminish.
Bottom Line
The diagnosis and management of eating disorders, including avoidant/restrictive food intake disorder and choking phobia, can be challenging. Furthermore, if an anatomical or organic anomaly is found, it is important to question how a patient’s eating disorder can be managed best through interdisciplinary collaboration between medical and behavioral specialties.
Related Resources
• Bryant-Waugh R. Feeding and eating disorders in children. Curr Opin Psychiatry. 2013;26(6):537-542.
• Norris ML, Robinson A, Obeid N, et al. Exploring avoidant/restrictive food intake disorder in eating disordered patients: a descriptive study. Int J Eat Disord. 2014;47(5):495-499.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Refusing solid food
Ms. B, age 11, is admitted to a pediatric medical inpatient unit for unintentional weight loss of 14 lb (15% total body weight) over the past month. She reports having 2 traumatic episodes last month: choking on a piece of cheese and having a swab specimen taken for a rapid strep test, which required several people to restrain her (the test was positive). Since then, she has refused to ingest solids, despite hunger and a desire to eat.
Ms. B reports diffuse abdominal pain merely “at the sight of food” and a fear of swallowing solids. She denies difficulty or pain upon swallowing, nausea, vomiting, or any change in bowel habits.
Her mother reports that, on the rare occasion that Ms. B has attempted to eat solid food, she spent as long as an hour cutting it into small pieces before bringing it to her mouth—after which she put the food down without eating. Her mother also witnessed Ms. B holding food in her mouth for “a very long time,” then spitting it out.
Ms. B says she is distressed about the weight loss and recognizes that her fear of solid food is excessive.
What would your diagnosis of Ms. B’s problem be?
a) anorexia nervosa
b) avoidant/restrictive food intake disorder (ARFID)
c) specific phobia (swallowing solids or choking)
d) generalized anxiety disorder (GAD)
The authors’ observations
DSM-5 describes a new eating disorder called ARFID, which replaces the DSM-IV-TR diagnosis of feeding disorder of infancy or early childhood</keyword>. DSM-5 diagnostic criteria define ARFID as:
An eating or feeding disturbance (eg, avoidance based on the sensory characteristics of food…) as manifested by persistent failure to meet appropriate nutritional and/or energy needs associated with at least one of the following: 1. Significant weight loss (or failure to achieve expected weight gain or faltering growth in children). 2. Significant nutritional deficiency. 3. Dependence on enteral feeding or oral nutritional supplements. 4. Marked interference with psychosocial functioning.1
DSM-5 also specifies that the disorder cannot be caused by lack of available food or traditional cultural practices; cannot coexist with anorexia or bulimia nervosa; and is not attributable to a concurrent medical or psychiatric disorder.
Because it is a newly defined diagnosis, the epidemiology of ARFID is unclear. Patients with ARFID have a wide variety of eating symptoms that do not meet diagnostic criteria for anorexia or bulimia nervosa. One study found that, among a cohort of mostly adolescent patients who presented for evaluation of an eating disorder, 14% met diagnostic criteria for ARFID.2 Another retrospective case-control study found a similar prevalence among patients age 8 to 18 (13.8% of 712 patients).3 Because of the variety of maladaptive feeding behaviors seen in ARFID, there is little evidence that pharmacotherapy is effective.4
HISTORY Premature birth
Ms. B’s medical history states that she is twin A of a premature birth at 26 weeks (birth weight, 1,060 g), with a 90-day neonatal intensive care unit hospitalization, during which she required supplemental oxygen and nasogastric tube feeding. She has mild cerebral palsy, and had motor delay of walking at 2.5 years old. Currently, she has no motor difficulties.
Ms. B does not have a psychiatric history and does not take medications. Her mother has a history of major depressive disorder that is well controlled with an unspecified selective serotonin reuptake inhibitor. Ms. B’s maternal uncle has poorly controlled schizophrenia.
During Ms. B’s 6-day hospitalization, her mental status exams are unremarkable. She is shy but cooperative and open. Her mood ranges from “sad” and “nervous” on admission to “fine” with mood-congruent affect. She denies suicidal or homicidal thoughts and hallucinations, and demonstrates good insight and judgment. All laboratory values are within normal limits except for mild hypophosphatemia (3.7 mg/dL) and mild hyperalbuminemia (4.9 g/dL) on admission, which may have been related to her nutritional status.
DIAGNOSIS Not solely psychiatric
The psychiatric differential diagnosis includes:
- ARFID
- specific phobia of swallowing solids or choking (pseudodysphagia)
- GAD
- unspecified feeding disorder.
Ms. B meets diagnostic criteria for ARFID, particularly that of profound acute weight loss due to restrictive eating behaviors. Her presentation also is similar to that of a specific phobia, namely profound anxiety upon even the thought of solid food (phobic stimulus) and recognition that her fear is excessive. However, she fails to meet diagnostic criteria for phobia in children, which require duration of at least 6 months. GAD also is less likely because she has had symptoms for 1 month (also requires 6-month duration) and her anxiety is limited to feeding behaviors.
The treatment team starts exposure therapy, encouraging Ms. B to begin taking small bites of textured foods, such as oatmeal.
On hospital Day 5, barium esophagogram reveals extrinsic compression of the esophagus. To identify the precise cause of compression, chest magnetic resonance angiogram reveals that Ms. B has a right-sided aortic arch with an aberrant left subclavian artery at T4 that, with the ligamentum arteriosum and left pulmonary artery, form a vascular ring impinging around the esophagus.
The authors’ observations
Dysphagia lusoria, coined in 1789 from the root for “natural abnormality,”5 is caused by an aberrant right subclavian artery, which persists because of abnormal involution of the right fourth aortic arch during embryogenesis6 (Figure 1). The condition is diagnosed incidentally in most affected adults, who are asymptomatic throughout life and do not require operative management.6
Symptoms of dysphagia lusoria can include dysphagia and recurrent aspiration if significant esophageal impingement is present.7 It is thought that patients tend to show more symptoms as they age because of sclerosis, aneurysm, or atherosclerosis of the impinging vessel.7 Cough and dyspnea caused by impingement of the trachea also have been reported, and might be more common in children because of tracheal flexibility.5
This case illustrates the complex interrelationship of physical and psychiatric conditions. After the treatment team discovered a physical cause of Ms. B’s symptoms, the initial psychiatric diagnosis became problematic, but remained critically important for long-term treatment of her comorbid eating phobia.
TREATMENT Therapy, surgery
The treatment team is faced with the question of whether dysphagia lusoria fully accounts for Ms. B’s status. The anatomic anomaly might explain the initial choking incident if the food particle was lodged at the site of impingement, but does not account for development of a severe acute eating disorder and subsequent malnutrition.
Because of the presence of dysphagia lusoria, the team concludes that Ms. B does not meet diagnostic criteria for ARFID. She is given a diagnosis of unspecified eating disorder.
Ms. B is discharged and referred to a pediatric cardiothoracic surgeon for operative consult of symptomatic dysphagia lusoria. By discharge, she is successfully eating yogurt with fruit chunks, which she had rejected earlier. She also expresses optimism about eating cake at her birthday party, scheduled for 2 weeks after discharge.
The treatment team strongly recommends outpatient psychiatric follow-up to manage Ms. B’s unspecified eating disorder with cognitive-behavioral therapy and exposure and response prevention, which helped to mildly decrease her anxiety during the hospital stay.
Ms. B’s surgeon deems the case severe enough to warrant surgery. Surgery for dysphagia lusoria can be indicated to prevent progression of symptoms into adulthood8; options include division of the ligamentum arteriosum to loosen the vascular ring and re-implantation of the aberrant subclavian artery.9,10 (On the other hand, dietary modification might be therapeutic in patients whose symptoms are mild.5)
The surgeon elects to divide the ligamentum arteriosum to relieve the pressure of the vascular ring on the esophagus. Intraoperatively, he discovers that Ms. B has a mildly patent ductus arteriosus (PDA) (Figure 2), which is more common in premature infants than in full-term births. The PDA is clipped on both sides and divided. This immediately causes the vascular ring created by the aberrant left subclavian artery, right-sided aorta, and PDA to spring open, releasing pressure on the esophagus.
The aberrant left subclavian artery emerges from an abnormal bulging of aorta, known as Kommerell’s diverticulum. After the vascular ring is released, the diverticulum is not observed to impinge on the esophagus; however, as a preventive measure, the surgeon sutures it to the anterior spinous ligament. This will prevent the diverticulum from enlarging and impinging on the esophagus as Ms. B grows to adulthood.
The surgery is completed without complications. Ms. B tolerates the procedure well.
Ten days after surgery, Ms. B is recovering well. She and her mother report satisfaction with the procedure to release the vascular ring. She discontinues her pain medication after 2 days and slowly begins reintroducing solid foods. Her fear of dysphagia and choking rapidly diminish.
Bottom Line
The diagnosis and management of eating disorders, including avoidant/restrictive food intake disorder and choking phobia, can be challenging. Furthermore, if an anatomical or organic anomaly is found, it is important to question how a patient’s eating disorder can be managed best through interdisciplinary collaboration between medical and behavioral specialties.
Related Resources
• Bryant-Waugh R. Feeding and eating disorders in children. Curr Opin Psychiatry. 2013;26(6):537-542.
• Norris ML, Robinson A, Obeid N, et al. Exploring avoidant/restrictive food intake disorder in eating disordered patients: a descriptive study. Int J Eat Disord. 2014;47(5):495-499.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
2. Ornstein RM, Rosen DS, Mammel KA, et al. Distribution of eating disorders in children and adolescents using the proposed DSM-5 criteria for feeding and eating disorders. J Adolesc Health. 2013;53(2):303-305.
3. Fisher MM, Rosen DS, Ornstein RM, et al. Characteristics of avoidant/restrictive food intake disorder in children and adolescents: a “new disorder” in DSM-5. J Adolesc Health. 2014;55(1):49-52.
4. Kelly NR, Shank LM, Bakalar JL, et al. Pediatric feeding and eating disorders: current state of diagnosis and treatment. Curr Psychiatry Rep. 2014;16(5):446.
5. Janssen M, Baggen MG, Veen HF, et al. Dysphagia lusoria: clinical aspects, manometric findings, diagnosis, and therapy. Am J Gastroenterol. 2000;95(6):1411-1416.
6. Abraham V, Mathew A, Cherian V, et al. Aberrant subclavian artery: anatomical curiosity or clinical entity. Int J Surg. 2009;7(2):106-109.
7. Calleja F, Eguaras M, Montero J, et al. Aberrant right subclavian artery associated with common carotid trunk. A rare cause of vascular ring. Eur J Cardiothorac Surg. 1990;4(10):568-570.
8. Jalaie H, Grommes J, Sailer A, et al. Treatment of symptomatic aberrant subclavian arteries. Eur J Vasc Endovasc Surg. 2014;48(5):521-526.
9. Gross RE. Surgical treatment for dysphagia lusoria. Ann Surg. 1946;124:532-534.
10. Morrris CD, Kanter KR, Miller JI Jr. Late-onset dysphagia lusoria. Ann Thorac Surg. 2001;71(2):710-712.
1. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
2. Ornstein RM, Rosen DS, Mammel KA, et al. Distribution of eating disorders in children and adolescents using the proposed DSM-5 criteria for feeding and eating disorders. J Adolesc Health. 2013;53(2):303-305.
3. Fisher MM, Rosen DS, Ornstein RM, et al. Characteristics of avoidant/restrictive food intake disorder in children and adolescents: a “new disorder” in DSM-5. J Adolesc Health. 2014;55(1):49-52.
4. Kelly NR, Shank LM, Bakalar JL, et al. Pediatric feeding and eating disorders: current state of diagnosis and treatment. Curr Psychiatry Rep. 2014;16(5):446.
5. Janssen M, Baggen MG, Veen HF, et al. Dysphagia lusoria: clinical aspects, manometric findings, diagnosis, and therapy. Am J Gastroenterol. 2000;95(6):1411-1416.
6. Abraham V, Mathew A, Cherian V, et al. Aberrant subclavian artery: anatomical curiosity or clinical entity. Int J Surg. 2009;7(2):106-109.
7. Calleja F, Eguaras M, Montero J, et al. Aberrant right subclavian artery associated with common carotid trunk. A rare cause of vascular ring. Eur J Cardiothorac Surg. 1990;4(10):568-570.
8. Jalaie H, Grommes J, Sailer A, et al. Treatment of symptomatic aberrant subclavian arteries. Eur J Vasc Endovasc Surg. 2014;48(5):521-526.
9. Gross RE. Surgical treatment for dysphagia lusoria. Ann Surg. 1946;124:532-534.
10. Morrris CD, Kanter KR, Miller JI Jr. Late-onset dysphagia lusoria. Ann Thorac Surg. 2001;71(2):710-712.

Pigmented Villonodular Synovitis of the Hip: A Systematic Review
Pigmented villonodular synovitis (PVNS) is a rare monoarticular disorder that affects the joints, bursae, or tendon sheaths of 1.8 per million patients.1,2 PVNS is defined by exuberant proliferation of synovial villi and nodules. Although its etiology is unknown, PVNS behaves much as a neoplastic process does, with occasional chromosomal abnormalities, local tissue invasion, and the potential for malignant transformation.3,4 Radiographs show cystic erosions or joint space narrowing, and magnetic resonance imaging shows characteristic low-signal intensity (on T1- and T2-weighted sequences) because of high hemosiderin content. Biopsy remains the gold standard for diagnosis and reveals hemosiderin-laden macrophages, vascularized villi, mononuclear cell infiltration, and sporadic mitotic figures.5 Diffuse PVNS appears as a thickened synovium with matted villi and synovial folds; localized PVNS presents as a pedunculated, firm yellow nodule.6
PVNS has a predilection for large joints, most commonly the knee (up to 80% of cases) and the hip.1,2,7 Treatment strategies for knee PVNS have been well studied and, as an aggregate, show no superiority of arthroscopic or open techniques.8 The literature on hip PVNS is less abundant and more case-based, making it difficult to reach a consensus on effective treatment. Open synovectomy and arthroplasty have been the mainstays of treatment over the past 60 years, but the advent of hip arthroscopy has introduced a new treatment modality.1,9 As arthroscopic management becomes more readily available, it is important to understand and compare the effectiveness of synovectomy and arthroplasty.
We systematically reviewed the treatment modalities for PVNS of the hip to determine how synovectomy and arthroplasty compare with respect to efficacy and revision rates.
Methods
Search Strategy
We systematically reviewed the literature according to PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines using the PRISMA checklist.10 Searches were completed in July 2014 using the PubMed Medline database and the Cochrane Central Register of Clinical Trials. Keyword selection was designed to capture all level I to V evidence English-language studies that reported clinical and/or radiographic outcomes. This was accomplished with a keyword search of all available titles and manuscript abstracts: (pigmented [Title/Abstract] AND villonodular [Title/Abstract] AND synovitis [Title/Abstract]) AND (hip [Title/Abstract]) AND (English [lang])). Abstracts from the 75 resulting studies were reviewed for exclusion criteria, which consisted of any cadaveric, biomechanical, histologic, and/or kinematic results, as well as a lack of any clinical and/or radiographic data (eg, review or technique articles). Studies were also excluded if they did not have clinical follow-up of at least 2 years. Studies not dedicated to hip PVNS specifically were not immediately excluded but were reviewed for outcomes data specific to the hip PVNS subpopulation. If a specific hip PVNS population could be distinguished from other patients, that study was included for review. If a study could not be deconstructed as such or was entirely devoted to one of our exclusion criteria, that study was excluded from our review. This initial search strategy yielded 16 studies.1,6,7,11-28
Bibliographical review of these 16 studies yielded several more for review. To ensure that no patients were counted twice, each study’s authors, data collection period, and ethnic population were reviewed and compared with those of the other studies. If there was any overlap in authorship, period, and place, only the study with the most relevant or comprehensive data was included. After accounting for all inclusion and exclusion criteria, we selected a total of 21 studies with 82 patients (86 hips) for inclusion (Figure 1).
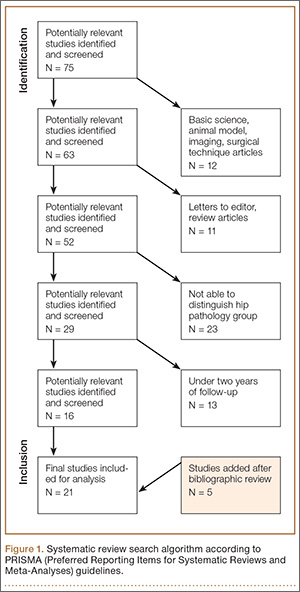
Data Extraction
Details of study design, sample size, and patient demographics, including age, sex, and duration of symptoms, were recorded. Use of diagnostic biopsy, joint space narrowing on radiographs, treatment method, and use of radiation therapy were also abstracted. Some studies described multiple treatment methods. If those methods could not be differentiated into distinct outcomes groups, the study would have been excluded for lack of specific clinical data. Studies with sufficient data were deconstructed such that the patients from each treatment group were isolated.
Fewer than 5 studies reported physical examination findings, validated survey scores, and/or radiographic results. Therefore, the primary outcomes reported and compared between treatment groups were disease recurrence, clinical worsening defined as progressive pain or loss of function, and revision surgery. Revision surgery was subdivided into repeat synovectomy and eventual arthroplasty, arthrodesis, or revision arthroplasty. Time to revision surgery was also documented. Each study’s methodologic quality and bias were evaluated with the Modified Coleman Methodology Score (MCMS), described by Cowan and colleagues.29 MCMS is a 15-item instrument that has been used to assess randomized and nonrandomized patient trials.30,31 It has a scaled potential score ranging from 0 to 100, with scores from 85 through 100 indicating excellent, 70 through 84 good, 55 through 69 fair, and under 55 poor.
Statistical Analysis
We report our data as weighted means (SDs). A mean was calculated for each study reporting on a respective data point, and each mean was then weighted according to the sample size of that study. We multiplied each study’s individual mean by the number of patients enrolled in that study and divided the sum of all the studies’ weighted data points by the number of eligible patients in all relevant studies. The result is that the nonweighted means from studies with a smaller sample size did not carry as much weight as those from larger studies. We then compared 2 groups of patients: those who had only a synovectomy and those who had a combination of synovectomy and arthroplasty. The synovectomy-only group was also compared with a group that underwent total hip arthroplasty (THA) specifically (Figure 2). Groups were compared with Student t test (SPSS Version 18, IBM), and statistical significance was set at α = 0.05.
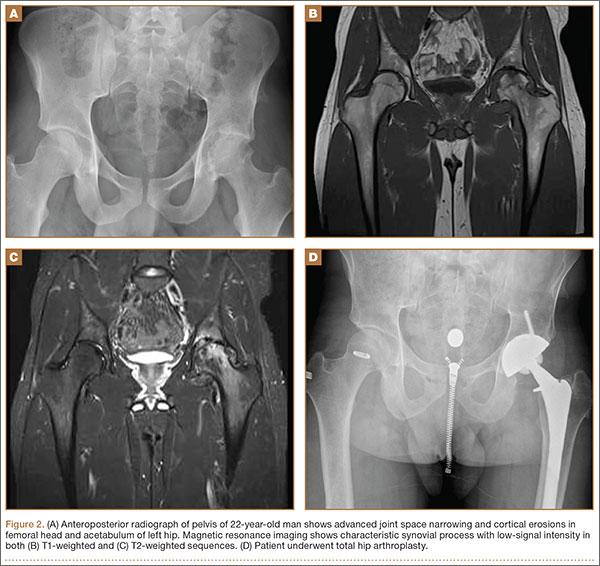
Results
Twenty-one studies (82 patients) were included in the final dataset (Table 1). Of these studies, 19 were retrospective case series (level IV evidence) in which the number of eligible hip PVNS patients ranged from 1 to 15. The other 2 studies were case reports (level V evidence). Mean (SD) MCMS was 25.0 (10.9).
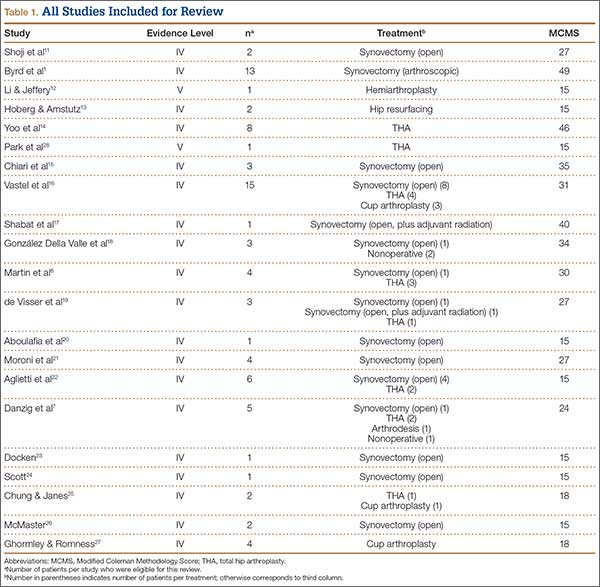
Fifty-one patients (59.3%) were female. Mean (SD) age of all patients was 33.2 (12.6) years. Mean (SD) duration of symptoms was 4.2 (2.7) years. The right hip was affected in 59.5% of patients in whom laterality was documented. Sixty-eight patients (79.1%) had biopsy-proven PVNS; presence or absence of a biopsy was not documented for the other 18 patients.
Of the 82 patients in the study, 45 (54.9%) underwent synovectomy without arthroplasty. Staged radiation was used to augment the synovectomy in 2 of these 45 cases. One series in this group consisted of 15 cases of arthroscopic synovectomy.1 The 37 patients (45.1%) in the other treatment group had arthroplasty at time of synovectomy. These patients underwent 22 THAs, 8 cup arthroplasties, 2 metal-on-metal hip resurfacings, and 1 hemiarthroplasty. The remaining 4 patients were treated nonoperatively (3) or with primary arthrodesis (1).
Comparisons between the synovectomy-only and synovectomy-with-arthroplasty groups are listed in Table 2. Synovectomy patients were younger on average than arthroplasty patients, but the difference was not statistically significant (P = .28). Only 6 studies distinguished between local and diffuse PVNS histology, and the diffuse type was detected in 87.0%, with insufficient data to detect a difference between the synovectomy and arthroplasty groups. In studies with documented radiographic findings, 75.0% of patients had evidence of joint space narrowing, which was significantly (P = .03) more common in the arthroplasty group (96.7% vs 31.3%).
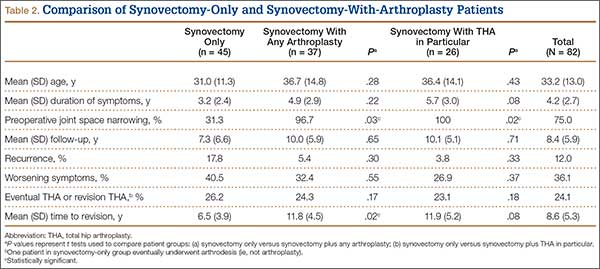
Mean (SD) clinical follow-up was 8.4 (5.9) years for all patients. A larger percentage of synovectomy-only patients experienced recurrence and worsened symptoms, but neither trend achieved statistical significance. The rate of eventual THA or arthrodesis after synovectomy alone was almost identical (P = .17) to the rate of revision THA in the synovectomy-with-arthroplasty group (26.2% vs 24.3%). Time to revision surgery, however, was significantly (P = .02) longer in the arthroplasty group. Two additional patients in the synovectomy-with-arthroplasty group underwent repeat synovectomy alone, but no patients in the synovectomy-only group underwent repeat synovectomy without arthroplasty.
One nonoperatively managed patient experienced symptom progression over the course of 10 years. The other 2 patients were stable after 2- and 4-year follow-up. The arthrodesis patient did not experience recurrence or have a revision operation in the 5 years after the index procedure.
Discussion
PVNS is a proliferative disorder of synovial tissue with a high risk of recurrence.15,32 Metastasis is extremely rare; there is only 1 case report of a fatality, which occurred within 42 months.12 Chiari and colleagues15 suggested that the PVNS recurrence rate is highest in the large joints. Therefore, in hip PVNS, early surgical resection is needed to limit articular destruction and the potential for recurrence. The primary treatment modalities are synovectomy alone and synovectomy with arthroplasty, which includes THA, cup arthroplasty, hip resurfacing, and hemiarthroplasty. According to our systematic review, about one-fourth of all patients in both treatment groups ultimately underwent revision surgery. Mean time to revision was significantly longer for synovectomy-with-arthroplasty patients (almost 12 years) than for synovectomy-only patients (6.5 years). One potential explanation is that arthroplasty component fixation may take longer to loosen than an inadequately synovectomized joint takes to recur. The synovectomy-only group did have a higher recurrence rate, though the difference was not statistically significant.
Open synovectomy is the most widely described technique for addressing hip PVNS. The precise pathophysiology of PVNS remains largely unknown, but most authors agree that aggressive débridement is required to halt its locally invasive course. Scott24 described the invasion of vascular foramina from synovium into bone and thought that radical synovectomy was essential to remove the stalks of these synovial villi. Furthermore, PVNS most commonly affects adults in the third through fifth decades of life,7 and many surgeons want to avoid prosthetic components (which may loosen over time) in this age group. Synovectomy, however, has persistently high recurrence rates, and, without removal of the femoral head and neck, it can be difficult to obtain adequate exposure for complete débridement. Although adjuvant external beam radiation has been used by some authors,17,19,33 its utility is unproven, and other authors have cautioned against unnecessary irradiation of reproductive organs.1,24,34
The high rates of bony involvement, joint destruction, and recurrence after synovectomy have prompted many surgeons to turn to arthroplasty. González Della Valle and colleagues18 theorized that joint space narrowing is more common in hip PVNS because of the poor distensibility of the hip capsule compared with that of the knee and other joints. In turn, bony lesions and arthritis present earlier in hip PVNS.14 Yoo and colleagues14 found a statistically significant increase in Harris Hip Scale (HHS) scores and a high rate of return to athletic activity after THA for PVNS. However, they also reported revisions for component loosening and osteolysis in 2 of 8 patients and periprosthetic osteolysis without loosening in another 2 patients. Vastel and colleagues16 similarly reported aseptic loosening of the acetabular component in half their patient cohort. No studies have determined which condition—PVNS recurrence or debris-related osteolysis—causes the accelerated loosening in this demographic.
Byrd and colleagues1 recently described use of hip arthroscopy in the treatment of PVNS. In a cohort of 13 patients, they found statistically significant improvements in HHS scores, no postoperative complications, and only 1 revision (THA 6 years after surgery). Although there is a prevailing perception that nodular (vs diffuse) PVNS is more appropriately treated with arthroscopic excision, no studies have provided data on this effect, and Byrd and colleagues1 in fact showed a trend of slightly better outcomes in diffuse cases than in nodular cases. The main challenges of hip arthroscopy are the steep learning curve and adequate exposure. Recent innovations include additional arthroscopic portals and enlarged T-capsulotomy, which may be contributing to decreased complication rates in hip arthroscopy in general.35
The limitations of this systematic review were largely imposed by the studies analyzed. The primary limitation was the relative paucity of clinical and radiographic data on hip PVNS. To our knowledge, studies on the treatment of hip PVNS have reported evidence levels no higher than IV. In addition, the studies we reviewed often had only 1 or 2 patient cases satisfying our inclusion criteria. For this reason, we included case reports, which further lowered the level of evidence of studies used. There were no consistently reported physical examination, survey, or radiographic findings that could be used to compare studies. All studies with sufficient data on hip PVNS treatment outcomes were rated poorly with the Modified Coleman Methodology Scoring system.29 Selection bias was minimized by the inclusive nature of studies with level I to V evidence, but this led to a study design bias in that most studies consisted of level IV evidence.
Conclusion
Although the hip PVNS literature is limited, our review provides insight into expected outcomes. No matter which surgery is to be performed, surgeons must counsel patients about the high revision rate. One in 4 patients ultimately undergoes a second surgery, which may be required within 6 or 7 years after synovectomy without arthroplasty. Further development and innovation in hip arthroscopy may transform the treatment of PVNS. We encourage other investigators to conduct prospective, comparative trials with higher evidence levels to assess the utility of arthroscopy and other treatment modalities.
1. Byrd JWT, Jones KS, Maiers GP. Two to 10 years’ follow-up of arthroscopic management of pigmented villonodular synovitis in the hip: a case series. Arthroscopy. 2013;29(11):1783-1787.
2. Myers BW, Masi AT. Pigmented villonodular synovitis and tenosynovitis: a clinical epidemiologic study of 166 cases and literature review. Medicine. 1980;59(3):223-238.
3. Sciot R, Rosai J, Dal Cin P, et al. Analysis of 35 cases of localized and diffuse tenosynovial giant cell tumor: a report from the Chromosomes and Morphology (CHAMP) study group. Mod Pathol. 1999;12(6):576-579.
4. Bertoni F, Unni KK, Beabout JW, Sim FH. Malignant giant cell tumor of the tendon sheaths and joints (malignant pigmented villonodular synovitis). Am J Surg Pathol. 1997;21(2):153-163.
5. Mankin H, Trahan C, Hornicek F. Pigmented villonodular synovitis of joints. J Surg Oncol. 2011;103(5):386-389.
6. Martin RC, Osborne DL, Edwards MJ, Wrightson W, McMasters KM. Giant cell tumor of tendon sheath, tenosynovial giant cell tumor, and pigmented villonodular synovitis: defining the presentation, surgical therapy and recurrence. Oncol Rep. 2000;7(2):413-419.
7. Danzig LA, Gershuni DH, Resnick D. Diagnosis and treatment of diffuse pigmented villonodular synovitis of the hip. Clin Orthop Relat Res. 1982;(168):42-47.
8. Aurégan JC, Klouche S, Bohu Y, Lefèvre N, Herman S, Hardy P. Treatment of pigmented villonodular synovitis of the knee. Arthroscopy. 2014;30(10):1327-1341.
9. Gondolph-Zink B, Puhl W, Noack W. Semiarthroscopic synovectomy of the hip. Int Orthop. 1988;12(1):31-35.
10. Moher D, Liberati A, Tetzlaff J, Altman DG; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J Clin Epidemiol. 2009;62(10):1006-1012.
11. Shoji T, Yasunaga Y, Yamasaki T, et al. Transtrochanteric rotational osteotomy combined with intra-articular procedures for pigmented villonodular synovitis of the hip. J Orthop Sci. 2015;20(5):943-950.
12. Li LM, Jeffery J. Exceptionally aggressive pigmented villonodular synovitis of the hip unresponsive to radiotherapy. J Bone Joint Surg Br. 2011;93(7):995-997.
13. Hoberg M, Amstutz HC. Metal-on-metal hip resurfacing in patients with pigmented villonodular synovitis: a report of two cases. Orthopedics. 2010;33(1):50-53.
14. Yoo JJ, Kwon YS, Koo KH, Yoon KS, Min BW, Kim HJ. Cementless total hip arthroplasty performed in patients with pigmented villonodular synovitis. J Arthroplasty. 2010;25(4):552-557.
15. Chiari C, Pirich C, Brannath W, Kotz R, Trieb K. What affects the recurrence and clinical outcome of pigmented villonodular synovitis? Clin Orthop Relat Res. 2006;(450):172-178.
16. Vastel L, Lambert P, De Pinieux G, Charrois O, Kerboull M, Courpied JP. Surgical treatment of pigmented villonodular synovitis of the hip. J Bone Joint Surg Am. 2005;87(5):1019-1024.
17. Shabat S, Kollender Y, Merimsky O, et al. The use of surgery and yttrium 90 in the management of extensive and diffuse pigmented villonodular synovitis of large joints. Rheumatology. 2002;41(10):1113-1118.
18. González Della Valle A, Piccaluga F, Potter HG, Salvati EA, Pusso R. Pigmented villonodular synovitis of the hip: 2- to 23-year followup study. Clin Orthop Relat Res. 2001;(388):187-199.
19. de Visser E, Veth RP, Pruszczynski M, Wobbes T, Van de Putte LB. Diffuse and localized pigmented villonodular synovitis: evaluation of treatment of 38 patients. Arch Orthop Trauma Surg. 1999;119(7-8):401-404.
20. Aboulafia AJ, Kaplan L, Jelinek J, Benevenia J, Monson DK. Neuropathy secondary to pigmented villonodular synovitis of the hip. Clin Orthop Relat Res. 1996;(325):174-180.
21. Moroni A, Innao V, Picci P. Pigmented villonodular synovitis of the hip. Study of 9 cases. Ital J Orthop Traumatol. 1983;9(3):331-337.
22. Aglietti P, Di Muria GV, Salvati EA, Stringa G. Pigmented villonodular synovitis of the hip joint (review of the literature and report of personal case material). Ital J Orthop Traumatol. 1983;9(4):487-496.
23. Docken WP. Pigmented villonodular synovitis: a review with illustrative case reports. Semin Arthritis Rheum. 1979;9(1):1-22.
24. Scott PM. Bone lesions in pigmented villonodular synovitis. J Bone Joint Surg Br. 1968;50(2):306-311.
25. Chung SM, Janes JM. Diffuse pigmented villonodular synovitis of the hip joint. Review of the literature and report of four cases. J Bone Joint Surg Am. 1965;47:293-303.
26. McMaster PE. Pigmented villonodular synovitis with invasion of bone. Report of six cases. Rheumatology. 1960;42(7):1170-1183.
27. Ghormley RK, Romness JO. Pigmented villonodular synovitis (xanthomatosis) of the hip joint. Proc Staff Meet Mayo Clin. 1954;29(6):171-180.
28. Park KS, Diwanji SR, Yang HK, Yoon TR, Seon JK. Pigmented villonodular synovitis of the hip presenting as a buttock mass treated by total hip arthroplasty. J Arthroplasty. 2010;25(2):333.e9-e12.
29. Cowan J, Lozano-Calderón S, Ring D. Quality of prospective controlled randomized trials. Analysis of trials of treatment for lateral epicondylitis as an example. J Bone Joint Surg Am. 2007;89(8):1693-1699.
30. Harris JD, Siston RA, Pan X, Flanigan DC. Autologous chondrocyte implantation: a systematic review. J Bone Joint Surg Am. 2010;92(12):2220-2233.
31. Harris JD, Siston RA, Brophy RH, Lattermann C, Carey JL, Flanigan DC. Failures, re-operations, and complications after autologous chondrocyte implantation—a systematic review. Osteoarthritis Cartilage. 2011;19(7):779-791.
32. Rao AS, Vigorita VJ. Pigmented villonodular synovitis (giant-cell tumor of the tendon sheath and synovial membrane). A review of eighty-one cases. J Bone Joint Surg Am. 1984;66(1):76-94.
33. Kat S, Kutz R, Elbracht T, Weseloh G, Kuwert T. Radiosynovectomy in pigmented villonodular synovitis. Nuklearmedizin. 2000;39(7):209-213.
34. Gitelis S, Heligman D, Morton T. The treatment of pigmented villonodular synovitis of the hip. A case report and literature review. Clin Orthop Relat Res. 1989;(239):154-160.
35. Harris JD, McCormick FM, Abrams GD, et al. Complications and reoperations during and after hip arthroscopy: a systematic review of 92 studies and more than 6,000 patients. Arthroscopy. 2013;29(3):589-595.
Pigmented villonodular synovitis (PVNS) is a rare monoarticular disorder that affects the joints, bursae, or tendon sheaths of 1.8 per million patients.1,2 PVNS is defined by exuberant proliferation of synovial villi and nodules. Although its etiology is unknown, PVNS behaves much as a neoplastic process does, with occasional chromosomal abnormalities, local tissue invasion, and the potential for malignant transformation.3,4 Radiographs show cystic erosions or joint space narrowing, and magnetic resonance imaging shows characteristic low-signal intensity (on T1- and T2-weighted sequences) because of high hemosiderin content. Biopsy remains the gold standard for diagnosis and reveals hemosiderin-laden macrophages, vascularized villi, mononuclear cell infiltration, and sporadic mitotic figures.5 Diffuse PVNS appears as a thickened synovium with matted villi and synovial folds; localized PVNS presents as a pedunculated, firm yellow nodule.6
PVNS has a predilection for large joints, most commonly the knee (up to 80% of cases) and the hip.1,2,7 Treatment strategies for knee PVNS have been well studied and, as an aggregate, show no superiority of arthroscopic or open techniques.8 The literature on hip PVNS is less abundant and more case-based, making it difficult to reach a consensus on effective treatment. Open synovectomy and arthroplasty have been the mainstays of treatment over the past 60 years, but the advent of hip arthroscopy has introduced a new treatment modality.1,9 As arthroscopic management becomes more readily available, it is important to understand and compare the effectiveness of synovectomy and arthroplasty.
We systematically reviewed the treatment modalities for PVNS of the hip to determine how synovectomy and arthroplasty compare with respect to efficacy and revision rates.
Methods
Search Strategy
We systematically reviewed the literature according to PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines using the PRISMA checklist.10 Searches were completed in July 2014 using the PubMed Medline database and the Cochrane Central Register of Clinical Trials. Keyword selection was designed to capture all level I to V evidence English-language studies that reported clinical and/or radiographic outcomes. This was accomplished with a keyword search of all available titles and manuscript abstracts: (pigmented [Title/Abstract] AND villonodular [Title/Abstract] AND synovitis [Title/Abstract]) AND (hip [Title/Abstract]) AND (English [lang])). Abstracts from the 75 resulting studies were reviewed for exclusion criteria, which consisted of any cadaveric, biomechanical, histologic, and/or kinematic results, as well as a lack of any clinical and/or radiographic data (eg, review or technique articles). Studies were also excluded if they did not have clinical follow-up of at least 2 years. Studies not dedicated to hip PVNS specifically were not immediately excluded but were reviewed for outcomes data specific to the hip PVNS subpopulation. If a specific hip PVNS population could be distinguished from other patients, that study was included for review. If a study could not be deconstructed as such or was entirely devoted to one of our exclusion criteria, that study was excluded from our review. This initial search strategy yielded 16 studies.1,6,7,11-28
Bibliographical review of these 16 studies yielded several more for review. To ensure that no patients were counted twice, each study’s authors, data collection period, and ethnic population were reviewed and compared with those of the other studies. If there was any overlap in authorship, period, and place, only the study with the most relevant or comprehensive data was included. After accounting for all inclusion and exclusion criteria, we selected a total of 21 studies with 82 patients (86 hips) for inclusion (Figure 1).
Data Extraction
Details of study design, sample size, and patient demographics, including age, sex, and duration of symptoms, were recorded. Use of diagnostic biopsy, joint space narrowing on radiographs, treatment method, and use of radiation therapy were also abstracted. Some studies described multiple treatment methods. If those methods could not be differentiated into distinct outcomes groups, the study would have been excluded for lack of specific clinical data. Studies with sufficient data were deconstructed such that the patients from each treatment group were isolated.
Fewer than 5 studies reported physical examination findings, validated survey scores, and/or radiographic results. Therefore, the primary outcomes reported and compared between treatment groups were disease recurrence, clinical worsening defined as progressive pain or loss of function, and revision surgery. Revision surgery was subdivided into repeat synovectomy and eventual arthroplasty, arthrodesis, or revision arthroplasty. Time to revision surgery was also documented. Each study’s methodologic quality and bias were evaluated with the Modified Coleman Methodology Score (MCMS), described by Cowan and colleagues.29 MCMS is a 15-item instrument that has been used to assess randomized and nonrandomized patient trials.30,31 It has a scaled potential score ranging from 0 to 100, with scores from 85 through 100 indicating excellent, 70 through 84 good, 55 through 69 fair, and under 55 poor.
Statistical Analysis
We report our data as weighted means (SDs). A mean was calculated for each study reporting on a respective data point, and each mean was then weighted according to the sample size of that study. We multiplied each study’s individual mean by the number of patients enrolled in that study and divided the sum of all the studies’ weighted data points by the number of eligible patients in all relevant studies. The result is that the nonweighted means from studies with a smaller sample size did not carry as much weight as those from larger studies. We then compared 2 groups of patients: those who had only a synovectomy and those who had a combination of synovectomy and arthroplasty. The synovectomy-only group was also compared with a group that underwent total hip arthroplasty (THA) specifically (Figure 2). Groups were compared with Student t test (SPSS Version 18, IBM), and statistical significance was set at α = 0.05.
Results
Twenty-one studies (82 patients) were included in the final dataset (Table 1). Of these studies, 19 were retrospective case series (level IV evidence) in which the number of eligible hip PVNS patients ranged from 1 to 15. The other 2 studies were case reports (level V evidence). Mean (SD) MCMS was 25.0 (10.9).
Fifty-one patients (59.3%) were female. Mean (SD) age of all patients was 33.2 (12.6) years. Mean (SD) duration of symptoms was 4.2 (2.7) years. The right hip was affected in 59.5% of patients in whom laterality was documented. Sixty-eight patients (79.1%) had biopsy-proven PVNS; presence or absence of a biopsy was not documented for the other 18 patients.
Of the 82 patients in the study, 45 (54.9%) underwent synovectomy without arthroplasty. Staged radiation was used to augment the synovectomy in 2 of these 45 cases. One series in this group consisted of 15 cases of arthroscopic synovectomy.1 The 37 patients (45.1%) in the other treatment group had arthroplasty at time of synovectomy. These patients underwent 22 THAs, 8 cup arthroplasties, 2 metal-on-metal hip resurfacings, and 1 hemiarthroplasty. The remaining 4 patients were treated nonoperatively (3) or with primary arthrodesis (1).
Comparisons between the synovectomy-only and synovectomy-with-arthroplasty groups are listed in Table 2. Synovectomy patients were younger on average than arthroplasty patients, but the difference was not statistically significant (P = .28). Only 6 studies distinguished between local and diffuse PVNS histology, and the diffuse type was detected in 87.0%, with insufficient data to detect a difference between the synovectomy and arthroplasty groups. In studies with documented radiographic findings, 75.0% of patients had evidence of joint space narrowing, which was significantly (P = .03) more common in the arthroplasty group (96.7% vs 31.3%).
Mean (SD) clinical follow-up was 8.4 (5.9) years for all patients. A larger percentage of synovectomy-only patients experienced recurrence and worsened symptoms, but neither trend achieved statistical significance. The rate of eventual THA or arthrodesis after synovectomy alone was almost identical (P = .17) to the rate of revision THA in the synovectomy-with-arthroplasty group (26.2% vs 24.3%). Time to revision surgery, however, was significantly (P = .02) longer in the arthroplasty group. Two additional patients in the synovectomy-with-arthroplasty group underwent repeat synovectomy alone, but no patients in the synovectomy-only group underwent repeat synovectomy without arthroplasty.
One nonoperatively managed patient experienced symptom progression over the course of 10 years. The other 2 patients were stable after 2- and 4-year follow-up. The arthrodesis patient did not experience recurrence or have a revision operation in the 5 years after the index procedure.
Discussion
PVNS is a proliferative disorder of synovial tissue with a high risk of recurrence.15,32 Metastasis is extremely rare; there is only 1 case report of a fatality, which occurred within 42 months.12 Chiari and colleagues15 suggested that the PVNS recurrence rate is highest in the large joints. Therefore, in hip PVNS, early surgical resection is needed to limit articular destruction and the potential for recurrence. The primary treatment modalities are synovectomy alone and synovectomy with arthroplasty, which includes THA, cup arthroplasty, hip resurfacing, and hemiarthroplasty. According to our systematic review, about one-fourth of all patients in both treatment groups ultimately underwent revision surgery. Mean time to revision was significantly longer for synovectomy-with-arthroplasty patients (almost 12 years) than for synovectomy-only patients (6.5 years). One potential explanation is that arthroplasty component fixation may take longer to loosen than an inadequately synovectomized joint takes to recur. The synovectomy-only group did have a higher recurrence rate, though the difference was not statistically significant.
Open synovectomy is the most widely described technique for addressing hip PVNS. The precise pathophysiology of PVNS remains largely unknown, but most authors agree that aggressive débridement is required to halt its locally invasive course. Scott24 described the invasion of vascular foramina from synovium into bone and thought that radical synovectomy was essential to remove the stalks of these synovial villi. Furthermore, PVNS most commonly affects adults in the third through fifth decades of life,7 and many surgeons want to avoid prosthetic components (which may loosen over time) in this age group. Synovectomy, however, has persistently high recurrence rates, and, without removal of the femoral head and neck, it can be difficult to obtain adequate exposure for complete débridement. Although adjuvant external beam radiation has been used by some authors,17,19,33 its utility is unproven, and other authors have cautioned against unnecessary irradiation of reproductive organs.1,24,34
The high rates of bony involvement, joint destruction, and recurrence after synovectomy have prompted many surgeons to turn to arthroplasty. González Della Valle and colleagues18 theorized that joint space narrowing is more common in hip PVNS because of the poor distensibility of the hip capsule compared with that of the knee and other joints. In turn, bony lesions and arthritis present earlier in hip PVNS.14 Yoo and colleagues14 found a statistically significant increase in Harris Hip Scale (HHS) scores and a high rate of return to athletic activity after THA for PVNS. However, they also reported revisions for component loosening and osteolysis in 2 of 8 patients and periprosthetic osteolysis without loosening in another 2 patients. Vastel and colleagues16 similarly reported aseptic loosening of the acetabular component in half their patient cohort. No studies have determined which condition—PVNS recurrence or debris-related osteolysis—causes the accelerated loosening in this demographic.
Byrd and colleagues1 recently described use of hip arthroscopy in the treatment of PVNS. In a cohort of 13 patients, they found statistically significant improvements in HHS scores, no postoperative complications, and only 1 revision (THA 6 years after surgery). Although there is a prevailing perception that nodular (vs diffuse) PVNS is more appropriately treated with arthroscopic excision, no studies have provided data on this effect, and Byrd and colleagues1 in fact showed a trend of slightly better outcomes in diffuse cases than in nodular cases. The main challenges of hip arthroscopy are the steep learning curve and adequate exposure. Recent innovations include additional arthroscopic portals and enlarged T-capsulotomy, which may be contributing to decreased complication rates in hip arthroscopy in general.35
The limitations of this systematic review were largely imposed by the studies analyzed. The primary limitation was the relative paucity of clinical and radiographic data on hip PVNS. To our knowledge, studies on the treatment of hip PVNS have reported evidence levels no higher than IV. In addition, the studies we reviewed often had only 1 or 2 patient cases satisfying our inclusion criteria. For this reason, we included case reports, which further lowered the level of evidence of studies used. There were no consistently reported physical examination, survey, or radiographic findings that could be used to compare studies. All studies with sufficient data on hip PVNS treatment outcomes were rated poorly with the Modified Coleman Methodology Scoring system.29 Selection bias was minimized by the inclusive nature of studies with level I to V evidence, but this led to a study design bias in that most studies consisted of level IV evidence.
Conclusion
Although the hip PVNS literature is limited, our review provides insight into expected outcomes. No matter which surgery is to be performed, surgeons must counsel patients about the high revision rate. One in 4 patients ultimately undergoes a second surgery, which may be required within 6 or 7 years after synovectomy without arthroplasty. Further development and innovation in hip arthroscopy may transform the treatment of PVNS. We encourage other investigators to conduct prospective, comparative trials with higher evidence levels to assess the utility of arthroscopy and other treatment modalities.
Pigmented villonodular synovitis (PVNS) is a rare monoarticular disorder that affects the joints, bursae, or tendon sheaths of 1.8 per million patients.1,2 PVNS is defined by exuberant proliferation of synovial villi and nodules. Although its etiology is unknown, PVNS behaves much as a neoplastic process does, with occasional chromosomal abnormalities, local tissue invasion, and the potential for malignant transformation.3,4 Radiographs show cystic erosions or joint space narrowing, and magnetic resonance imaging shows characteristic low-signal intensity (on T1- and T2-weighted sequences) because of high hemosiderin content. Biopsy remains the gold standard for diagnosis and reveals hemosiderin-laden macrophages, vascularized villi, mononuclear cell infiltration, and sporadic mitotic figures.5 Diffuse PVNS appears as a thickened synovium with matted villi and synovial folds; localized PVNS presents as a pedunculated, firm yellow nodule.6
PVNS has a predilection for large joints, most commonly the knee (up to 80% of cases) and the hip.1,2,7 Treatment strategies for knee PVNS have been well studied and, as an aggregate, show no superiority of arthroscopic or open techniques.8 The literature on hip PVNS is less abundant and more case-based, making it difficult to reach a consensus on effective treatment. Open synovectomy and arthroplasty have been the mainstays of treatment over the past 60 years, but the advent of hip arthroscopy has introduced a new treatment modality.1,9 As arthroscopic management becomes more readily available, it is important to understand and compare the effectiveness of synovectomy and arthroplasty.
We systematically reviewed the treatment modalities for PVNS of the hip to determine how synovectomy and arthroplasty compare with respect to efficacy and revision rates.
Methods
Search Strategy
We systematically reviewed the literature according to PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines using the PRISMA checklist.10 Searches were completed in July 2014 using the PubMed Medline database and the Cochrane Central Register of Clinical Trials. Keyword selection was designed to capture all level I to V evidence English-language studies that reported clinical and/or radiographic outcomes. This was accomplished with a keyword search of all available titles and manuscript abstracts: (pigmented [Title/Abstract] AND villonodular [Title/Abstract] AND synovitis [Title/Abstract]) AND (hip [Title/Abstract]) AND (English [lang])). Abstracts from the 75 resulting studies were reviewed for exclusion criteria, which consisted of any cadaveric, biomechanical, histologic, and/or kinematic results, as well as a lack of any clinical and/or radiographic data (eg, review or technique articles). Studies were also excluded if they did not have clinical follow-up of at least 2 years. Studies not dedicated to hip PVNS specifically were not immediately excluded but were reviewed for outcomes data specific to the hip PVNS subpopulation. If a specific hip PVNS population could be distinguished from other patients, that study was included for review. If a study could not be deconstructed as such or was entirely devoted to one of our exclusion criteria, that study was excluded from our review. This initial search strategy yielded 16 studies.1,6,7,11-28
Bibliographical review of these 16 studies yielded several more for review. To ensure that no patients were counted twice, each study’s authors, data collection period, and ethnic population were reviewed and compared with those of the other studies. If there was any overlap in authorship, period, and place, only the study with the most relevant or comprehensive data was included. After accounting for all inclusion and exclusion criteria, we selected a total of 21 studies with 82 patients (86 hips) for inclusion (Figure 1).
Data Extraction
Details of study design, sample size, and patient demographics, including age, sex, and duration of symptoms, were recorded. Use of diagnostic biopsy, joint space narrowing on radiographs, treatment method, and use of radiation therapy were also abstracted. Some studies described multiple treatment methods. If those methods could not be differentiated into distinct outcomes groups, the study would have been excluded for lack of specific clinical data. Studies with sufficient data were deconstructed such that the patients from each treatment group were isolated.
Fewer than 5 studies reported physical examination findings, validated survey scores, and/or radiographic results. Therefore, the primary outcomes reported and compared between treatment groups were disease recurrence, clinical worsening defined as progressive pain or loss of function, and revision surgery. Revision surgery was subdivided into repeat synovectomy and eventual arthroplasty, arthrodesis, or revision arthroplasty. Time to revision surgery was also documented. Each study’s methodologic quality and bias were evaluated with the Modified Coleman Methodology Score (MCMS), described by Cowan and colleagues.29 MCMS is a 15-item instrument that has been used to assess randomized and nonrandomized patient trials.30,31 It has a scaled potential score ranging from 0 to 100, with scores from 85 through 100 indicating excellent, 70 through 84 good, 55 through 69 fair, and under 55 poor.
Statistical Analysis
We report our data as weighted means (SDs). A mean was calculated for each study reporting on a respective data point, and each mean was then weighted according to the sample size of that study. We multiplied each study’s individual mean by the number of patients enrolled in that study and divided the sum of all the studies’ weighted data points by the number of eligible patients in all relevant studies. The result is that the nonweighted means from studies with a smaller sample size did not carry as much weight as those from larger studies. We then compared 2 groups of patients: those who had only a synovectomy and those who had a combination of synovectomy and arthroplasty. The synovectomy-only group was also compared with a group that underwent total hip arthroplasty (THA) specifically (Figure 2). Groups were compared with Student t test (SPSS Version 18, IBM), and statistical significance was set at α = 0.05.
Results
Twenty-one studies (82 patients) were included in the final dataset (Table 1). Of these studies, 19 were retrospective case series (level IV evidence) in which the number of eligible hip PVNS patients ranged from 1 to 15. The other 2 studies were case reports (level V evidence). Mean (SD) MCMS was 25.0 (10.9).
Fifty-one patients (59.3%) were female. Mean (SD) age of all patients was 33.2 (12.6) years. Mean (SD) duration of symptoms was 4.2 (2.7) years. The right hip was affected in 59.5% of patients in whom laterality was documented. Sixty-eight patients (79.1%) had biopsy-proven PVNS; presence or absence of a biopsy was not documented for the other 18 patients.
Of the 82 patients in the study, 45 (54.9%) underwent synovectomy without arthroplasty. Staged radiation was used to augment the synovectomy in 2 of these 45 cases. One series in this group consisted of 15 cases of arthroscopic synovectomy.1 The 37 patients (45.1%) in the other treatment group had arthroplasty at time of synovectomy. These patients underwent 22 THAs, 8 cup arthroplasties, 2 metal-on-metal hip resurfacings, and 1 hemiarthroplasty. The remaining 4 patients were treated nonoperatively (3) or with primary arthrodesis (1).
Comparisons between the synovectomy-only and synovectomy-with-arthroplasty groups are listed in Table 2. Synovectomy patients were younger on average than arthroplasty patients, but the difference was not statistically significant (P = .28). Only 6 studies distinguished between local and diffuse PVNS histology, and the diffuse type was detected in 87.0%, with insufficient data to detect a difference between the synovectomy and arthroplasty groups. In studies with documented radiographic findings, 75.0% of patients had evidence of joint space narrowing, which was significantly (P = .03) more common in the arthroplasty group (96.7% vs 31.3%).
Mean (SD) clinical follow-up was 8.4 (5.9) years for all patients. A larger percentage of synovectomy-only patients experienced recurrence and worsened symptoms, but neither trend achieved statistical significance. The rate of eventual THA or arthrodesis after synovectomy alone was almost identical (P = .17) to the rate of revision THA in the synovectomy-with-arthroplasty group (26.2% vs 24.3%). Time to revision surgery, however, was significantly (P = .02) longer in the arthroplasty group. Two additional patients in the synovectomy-with-arthroplasty group underwent repeat synovectomy alone, but no patients in the synovectomy-only group underwent repeat synovectomy without arthroplasty.
One nonoperatively managed patient experienced symptom progression over the course of 10 years. The other 2 patients were stable after 2- and 4-year follow-up. The arthrodesis patient did not experience recurrence or have a revision operation in the 5 years after the index procedure.
Discussion
PVNS is a proliferative disorder of synovial tissue with a high risk of recurrence.15,32 Metastasis is extremely rare; there is only 1 case report of a fatality, which occurred within 42 months.12 Chiari and colleagues15 suggested that the PVNS recurrence rate is highest in the large joints. Therefore, in hip PVNS, early surgical resection is needed to limit articular destruction and the potential for recurrence. The primary treatment modalities are synovectomy alone and synovectomy with arthroplasty, which includes THA, cup arthroplasty, hip resurfacing, and hemiarthroplasty. According to our systematic review, about one-fourth of all patients in both treatment groups ultimately underwent revision surgery. Mean time to revision was significantly longer for synovectomy-with-arthroplasty patients (almost 12 years) than for synovectomy-only patients (6.5 years). One potential explanation is that arthroplasty component fixation may take longer to loosen than an inadequately synovectomized joint takes to recur. The synovectomy-only group did have a higher recurrence rate, though the difference was not statistically significant.
Open synovectomy is the most widely described technique for addressing hip PVNS. The precise pathophysiology of PVNS remains largely unknown, but most authors agree that aggressive débridement is required to halt its locally invasive course. Scott24 described the invasion of vascular foramina from synovium into bone and thought that radical synovectomy was essential to remove the stalks of these synovial villi. Furthermore, PVNS most commonly affects adults in the third through fifth decades of life,7 and many surgeons want to avoid prosthetic components (which may loosen over time) in this age group. Synovectomy, however, has persistently high recurrence rates, and, without removal of the femoral head and neck, it can be difficult to obtain adequate exposure for complete débridement. Although adjuvant external beam radiation has been used by some authors,17,19,33 its utility is unproven, and other authors have cautioned against unnecessary irradiation of reproductive organs.1,24,34
The high rates of bony involvement, joint destruction, and recurrence after synovectomy have prompted many surgeons to turn to arthroplasty. González Della Valle and colleagues18 theorized that joint space narrowing is more common in hip PVNS because of the poor distensibility of the hip capsule compared with that of the knee and other joints. In turn, bony lesions and arthritis present earlier in hip PVNS.14 Yoo and colleagues14 found a statistically significant increase in Harris Hip Scale (HHS) scores and a high rate of return to athletic activity after THA for PVNS. However, they also reported revisions for component loosening and osteolysis in 2 of 8 patients and periprosthetic osteolysis without loosening in another 2 patients. Vastel and colleagues16 similarly reported aseptic loosening of the acetabular component in half their patient cohort. No studies have determined which condition—PVNS recurrence or debris-related osteolysis—causes the accelerated loosening in this demographic.
Byrd and colleagues1 recently described use of hip arthroscopy in the treatment of PVNS. In a cohort of 13 patients, they found statistically significant improvements in HHS scores, no postoperative complications, and only 1 revision (THA 6 years after surgery). Although there is a prevailing perception that nodular (vs diffuse) PVNS is more appropriately treated with arthroscopic excision, no studies have provided data on this effect, and Byrd and colleagues1 in fact showed a trend of slightly better outcomes in diffuse cases than in nodular cases. The main challenges of hip arthroscopy are the steep learning curve and adequate exposure. Recent innovations include additional arthroscopic portals and enlarged T-capsulotomy, which may be contributing to decreased complication rates in hip arthroscopy in general.35
The limitations of this systematic review were largely imposed by the studies analyzed. The primary limitation was the relative paucity of clinical and radiographic data on hip PVNS. To our knowledge, studies on the treatment of hip PVNS have reported evidence levels no higher than IV. In addition, the studies we reviewed often had only 1 or 2 patient cases satisfying our inclusion criteria. For this reason, we included case reports, which further lowered the level of evidence of studies used. There were no consistently reported physical examination, survey, or radiographic findings that could be used to compare studies. All studies with sufficient data on hip PVNS treatment outcomes were rated poorly with the Modified Coleman Methodology Scoring system.29 Selection bias was minimized by the inclusive nature of studies with level I to V evidence, but this led to a study design bias in that most studies consisted of level IV evidence.
Conclusion
Although the hip PVNS literature is limited, our review provides insight into expected outcomes. No matter which surgery is to be performed, surgeons must counsel patients about the high revision rate. One in 4 patients ultimately undergoes a second surgery, which may be required within 6 or 7 years after synovectomy without arthroplasty. Further development and innovation in hip arthroscopy may transform the treatment of PVNS. We encourage other investigators to conduct prospective, comparative trials with higher evidence levels to assess the utility of arthroscopy and other treatment modalities.
1. Byrd JWT, Jones KS, Maiers GP. Two to 10 years’ follow-up of arthroscopic management of pigmented villonodular synovitis in the hip: a case series. Arthroscopy. 2013;29(11):1783-1787.
2. Myers BW, Masi AT. Pigmented villonodular synovitis and tenosynovitis: a clinical epidemiologic study of 166 cases and literature review. Medicine. 1980;59(3):223-238.
3. Sciot R, Rosai J, Dal Cin P, et al. Analysis of 35 cases of localized and diffuse tenosynovial giant cell tumor: a report from the Chromosomes and Morphology (CHAMP) study group. Mod Pathol. 1999;12(6):576-579.
4. Bertoni F, Unni KK, Beabout JW, Sim FH. Malignant giant cell tumor of the tendon sheaths and joints (malignant pigmented villonodular synovitis). Am J Surg Pathol. 1997;21(2):153-163.
5. Mankin H, Trahan C, Hornicek F. Pigmented villonodular synovitis of joints. J Surg Oncol. 2011;103(5):386-389.
6. Martin RC, Osborne DL, Edwards MJ, Wrightson W, McMasters KM. Giant cell tumor of tendon sheath, tenosynovial giant cell tumor, and pigmented villonodular synovitis: defining the presentation, surgical therapy and recurrence. Oncol Rep. 2000;7(2):413-419.
7. Danzig LA, Gershuni DH, Resnick D. Diagnosis and treatment of diffuse pigmented villonodular synovitis of the hip. Clin Orthop Relat Res. 1982;(168):42-47.
8. Aurégan JC, Klouche S, Bohu Y, Lefèvre N, Herman S, Hardy P. Treatment of pigmented villonodular synovitis of the knee. Arthroscopy. 2014;30(10):1327-1341.
9. Gondolph-Zink B, Puhl W, Noack W. Semiarthroscopic synovectomy of the hip. Int Orthop. 1988;12(1):31-35.
10. Moher D, Liberati A, Tetzlaff J, Altman DG; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J Clin Epidemiol. 2009;62(10):1006-1012.
11. Shoji T, Yasunaga Y, Yamasaki T, et al. Transtrochanteric rotational osteotomy combined with intra-articular procedures for pigmented villonodular synovitis of the hip. J Orthop Sci. 2015;20(5):943-950.
12. Li LM, Jeffery J. Exceptionally aggressive pigmented villonodular synovitis of the hip unresponsive to radiotherapy. J Bone Joint Surg Br. 2011;93(7):995-997.
13. Hoberg M, Amstutz HC. Metal-on-metal hip resurfacing in patients with pigmented villonodular synovitis: a report of two cases. Orthopedics. 2010;33(1):50-53.
14. Yoo JJ, Kwon YS, Koo KH, Yoon KS, Min BW, Kim HJ. Cementless total hip arthroplasty performed in patients with pigmented villonodular synovitis. J Arthroplasty. 2010;25(4):552-557.
15. Chiari C, Pirich C, Brannath W, Kotz R, Trieb K. What affects the recurrence and clinical outcome of pigmented villonodular synovitis? Clin Orthop Relat Res. 2006;(450):172-178.
16. Vastel L, Lambert P, De Pinieux G, Charrois O, Kerboull M, Courpied JP. Surgical treatment of pigmented villonodular synovitis of the hip. J Bone Joint Surg Am. 2005;87(5):1019-1024.
17. Shabat S, Kollender Y, Merimsky O, et al. The use of surgery and yttrium 90 in the management of extensive and diffuse pigmented villonodular synovitis of large joints. Rheumatology. 2002;41(10):1113-1118.
18. González Della Valle A, Piccaluga F, Potter HG, Salvati EA, Pusso R. Pigmented villonodular synovitis of the hip: 2- to 23-year followup study. Clin Orthop Relat Res. 2001;(388):187-199.
19. de Visser E, Veth RP, Pruszczynski M, Wobbes T, Van de Putte LB. Diffuse and localized pigmented villonodular synovitis: evaluation of treatment of 38 patients. Arch Orthop Trauma Surg. 1999;119(7-8):401-404.
20. Aboulafia AJ, Kaplan L, Jelinek J, Benevenia J, Monson DK. Neuropathy secondary to pigmented villonodular synovitis of the hip. Clin Orthop Relat Res. 1996;(325):174-180.
21. Moroni A, Innao V, Picci P. Pigmented villonodular synovitis of the hip. Study of 9 cases. Ital J Orthop Traumatol. 1983;9(3):331-337.
22. Aglietti P, Di Muria GV, Salvati EA, Stringa G. Pigmented villonodular synovitis of the hip joint (review of the literature and report of personal case material). Ital J Orthop Traumatol. 1983;9(4):487-496.
23. Docken WP. Pigmented villonodular synovitis: a review with illustrative case reports. Semin Arthritis Rheum. 1979;9(1):1-22.
24. Scott PM. Bone lesions in pigmented villonodular synovitis. J Bone Joint Surg Br. 1968;50(2):306-311.
25. Chung SM, Janes JM. Diffuse pigmented villonodular synovitis of the hip joint. Review of the literature and report of four cases. J Bone Joint Surg Am. 1965;47:293-303.
26. McMaster PE. Pigmented villonodular synovitis with invasion of bone. Report of six cases. Rheumatology. 1960;42(7):1170-1183.
27. Ghormley RK, Romness JO. Pigmented villonodular synovitis (xanthomatosis) of the hip joint. Proc Staff Meet Mayo Clin. 1954;29(6):171-180.
28. Park KS, Diwanji SR, Yang HK, Yoon TR, Seon JK. Pigmented villonodular synovitis of the hip presenting as a buttock mass treated by total hip arthroplasty. J Arthroplasty. 2010;25(2):333.e9-e12.
29. Cowan J, Lozano-Calderón S, Ring D. Quality of prospective controlled randomized trials. Analysis of trials of treatment for lateral epicondylitis as an example. J Bone Joint Surg Am. 2007;89(8):1693-1699.
30. Harris JD, Siston RA, Pan X, Flanigan DC. Autologous chondrocyte implantation: a systematic review. J Bone Joint Surg Am. 2010;92(12):2220-2233.
31. Harris JD, Siston RA, Brophy RH, Lattermann C, Carey JL, Flanigan DC. Failures, re-operations, and complications after autologous chondrocyte implantation—a systematic review. Osteoarthritis Cartilage. 2011;19(7):779-791.
32. Rao AS, Vigorita VJ. Pigmented villonodular synovitis (giant-cell tumor of the tendon sheath and synovial membrane). A review of eighty-one cases. J Bone Joint Surg Am. 1984;66(1):76-94.
33. Kat S, Kutz R, Elbracht T, Weseloh G, Kuwert T. Radiosynovectomy in pigmented villonodular synovitis. Nuklearmedizin. 2000;39(7):209-213.
34. Gitelis S, Heligman D, Morton T. The treatment of pigmented villonodular synovitis of the hip. A case report and literature review. Clin Orthop Relat Res. 1989;(239):154-160.
35. Harris JD, McCormick FM, Abrams GD, et al. Complications and reoperations during and after hip arthroscopy: a systematic review of 92 studies and more than 6,000 patients. Arthroscopy. 2013;29(3):589-595.
1. Byrd JWT, Jones KS, Maiers GP. Two to 10 years’ follow-up of arthroscopic management of pigmented villonodular synovitis in the hip: a case series. Arthroscopy. 2013;29(11):1783-1787.
2. Myers BW, Masi AT. Pigmented villonodular synovitis and tenosynovitis: a clinical epidemiologic study of 166 cases and literature review. Medicine. 1980;59(3):223-238.
3. Sciot R, Rosai J, Dal Cin P, et al. Analysis of 35 cases of localized and diffuse tenosynovial giant cell tumor: a report from the Chromosomes and Morphology (CHAMP) study group. Mod Pathol. 1999;12(6):576-579.
4. Bertoni F, Unni KK, Beabout JW, Sim FH. Malignant giant cell tumor of the tendon sheaths and joints (malignant pigmented villonodular synovitis). Am J Surg Pathol. 1997;21(2):153-163.
5. Mankin H, Trahan C, Hornicek F. Pigmented villonodular synovitis of joints. J Surg Oncol. 2011;103(5):386-389.
6. Martin RC, Osborne DL, Edwards MJ, Wrightson W, McMasters KM. Giant cell tumor of tendon sheath, tenosynovial giant cell tumor, and pigmented villonodular synovitis: defining the presentation, surgical therapy and recurrence. Oncol Rep. 2000;7(2):413-419.
7. Danzig LA, Gershuni DH, Resnick D. Diagnosis and treatment of diffuse pigmented villonodular synovitis of the hip. Clin Orthop Relat Res. 1982;(168):42-47.
8. Aurégan JC, Klouche S, Bohu Y, Lefèvre N, Herman S, Hardy P. Treatment of pigmented villonodular synovitis of the knee. Arthroscopy. 2014;30(10):1327-1341.
9. Gondolph-Zink B, Puhl W, Noack W. Semiarthroscopic synovectomy of the hip. Int Orthop. 1988;12(1):31-35.
10. Moher D, Liberati A, Tetzlaff J, Altman DG; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J Clin Epidemiol. 2009;62(10):1006-1012.
11. Shoji T, Yasunaga Y, Yamasaki T, et al. Transtrochanteric rotational osteotomy combined with intra-articular procedures for pigmented villonodular synovitis of the hip. J Orthop Sci. 2015;20(5):943-950.
12. Li LM, Jeffery J. Exceptionally aggressive pigmented villonodular synovitis of the hip unresponsive to radiotherapy. J Bone Joint Surg Br. 2011;93(7):995-997.
13. Hoberg M, Amstutz HC. Metal-on-metal hip resurfacing in patients with pigmented villonodular synovitis: a report of two cases. Orthopedics. 2010;33(1):50-53.
14. Yoo JJ, Kwon YS, Koo KH, Yoon KS, Min BW, Kim HJ. Cementless total hip arthroplasty performed in patients with pigmented villonodular synovitis. J Arthroplasty. 2010;25(4):552-557.
15. Chiari C, Pirich C, Brannath W, Kotz R, Trieb K. What affects the recurrence and clinical outcome of pigmented villonodular synovitis? Clin Orthop Relat Res. 2006;(450):172-178.
16. Vastel L, Lambert P, De Pinieux G, Charrois O, Kerboull M, Courpied JP. Surgical treatment of pigmented villonodular synovitis of the hip. J Bone Joint Surg Am. 2005;87(5):1019-1024.
17. Shabat S, Kollender Y, Merimsky O, et al. The use of surgery and yttrium 90 in the management of extensive and diffuse pigmented villonodular synovitis of large joints. Rheumatology. 2002;41(10):1113-1118.
18. González Della Valle A, Piccaluga F, Potter HG, Salvati EA, Pusso R. Pigmented villonodular synovitis of the hip: 2- to 23-year followup study. Clin Orthop Relat Res. 2001;(388):187-199.
19. de Visser E, Veth RP, Pruszczynski M, Wobbes T, Van de Putte LB. Diffuse and localized pigmented villonodular synovitis: evaluation of treatment of 38 patients. Arch Orthop Trauma Surg. 1999;119(7-8):401-404.
20. Aboulafia AJ, Kaplan L, Jelinek J, Benevenia J, Monson DK. Neuropathy secondary to pigmented villonodular synovitis of the hip. Clin Orthop Relat Res. 1996;(325):174-180.
21. Moroni A, Innao V, Picci P. Pigmented villonodular synovitis of the hip. Study of 9 cases. Ital J Orthop Traumatol. 1983;9(3):331-337.
22. Aglietti P, Di Muria GV, Salvati EA, Stringa G. Pigmented villonodular synovitis of the hip joint (review of the literature and report of personal case material). Ital J Orthop Traumatol. 1983;9(4):487-496.
23. Docken WP. Pigmented villonodular synovitis: a review with illustrative case reports. Semin Arthritis Rheum. 1979;9(1):1-22.
24. Scott PM. Bone lesions in pigmented villonodular synovitis. J Bone Joint Surg Br. 1968;50(2):306-311.
25. Chung SM, Janes JM. Diffuse pigmented villonodular synovitis of the hip joint. Review of the literature and report of four cases. J Bone Joint Surg Am. 1965;47:293-303.
26. McMaster PE. Pigmented villonodular synovitis with invasion of bone. Report of six cases. Rheumatology. 1960;42(7):1170-1183.
27. Ghormley RK, Romness JO. Pigmented villonodular synovitis (xanthomatosis) of the hip joint. Proc Staff Meet Mayo Clin. 1954;29(6):171-180.
28. Park KS, Diwanji SR, Yang HK, Yoon TR, Seon JK. Pigmented villonodular synovitis of the hip presenting as a buttock mass treated by total hip arthroplasty. J Arthroplasty. 2010;25(2):333.e9-e12.
29. Cowan J, Lozano-Calderón S, Ring D. Quality of prospective controlled randomized trials. Analysis of trials of treatment for lateral epicondylitis as an example. J Bone Joint Surg Am. 2007;89(8):1693-1699.
30. Harris JD, Siston RA, Pan X, Flanigan DC. Autologous chondrocyte implantation: a systematic review. J Bone Joint Surg Am. 2010;92(12):2220-2233.
31. Harris JD, Siston RA, Brophy RH, Lattermann C, Carey JL, Flanigan DC. Failures, re-operations, and complications after autologous chondrocyte implantation—a systematic review. Osteoarthritis Cartilage. 2011;19(7):779-791.
32. Rao AS, Vigorita VJ. Pigmented villonodular synovitis (giant-cell tumor of the tendon sheath and synovial membrane). A review of eighty-one cases. J Bone Joint Surg Am. 1984;66(1):76-94.
33. Kat S, Kutz R, Elbracht T, Weseloh G, Kuwert T. Radiosynovectomy in pigmented villonodular synovitis. Nuklearmedizin. 2000;39(7):209-213.
34. Gitelis S, Heligman D, Morton T. The treatment of pigmented villonodular synovitis of the hip. A case report and literature review. Clin Orthop Relat Res. 1989;(239):154-160.
35. Harris JD, McCormick FM, Abrams GD, et al. Complications and reoperations during and after hip arthroscopy: a systematic review of 92 studies and more than 6,000 patients. Arthroscopy. 2013;29(3):589-595.
Debunking marijuana myths for teens
The annual checkup has long provided an opportunity for early adolescents to learn about the risks of alcohol and drug use from a trusted source who may be less biased than parents, teachers, or police. Parents also turn to their child’s pediatrician for guidance on how to broach this important topic with their children, or they may come with concerns about their children’s use of drugs or alcohol.
Marijuana has become an increasingly complex topic, as its legal status has rapidly changed: It’s legal to purchase marijuana in four states (Alaska, Colorado, Oregon, and Washington, as well as the District of Columbia); it is decriminalized in 20 states and the District of Columbia for certain marijuana possession offenses; and it is legal to use medical marijuana in 23 states. As its legal status changes, attitudes about its use also have shifted, and its availability, form, and potency all have changed dramatically in just the past decade. Further, we ourselves may have mixed feelings about marijuana use based on our own experience as adolescents and sampling bias. We may have seen its low-level use and minimal effects in young or mature adults, or we may have seen substantial use of marijuana have a major deleterious impact on a friend or become a gateway drug for addiction to dangerous substances.
Before addressing marijuana use with adolescent patients and dealing with their potential skepticism concerning any harm, it is worth spending a little time looking in the mirror to consider your perspective on marijuana use and your response to disbelief.

According to the National Institute on Drug Abuse’s Monitoring the Future (MTF) survey, almost 12% of 8th graders, 27% of 10th graders, and 35% of 12th graders in the United States reported having used marijuana in the past year. Among the 12th graders in that 2014 survey, almost 20% were current users of marijuana and 6% were daily users. Many surveys, including the MTF, have demonstrated that attitudes of teenagers have shifted about marijuana’s dangerousness, with a steep and steady decline in the number of teenagers believing that regular marijuana use poses a risk to their health and well-being. In 2014, less than 40% of 12th graders in the MTF survey agreed that regular use of marijuana would pose a risk to their well-being, compared with a peak of almost 80% of 12th graders in the early 1990’s.
Pediatricians have an opportunity to change their patient’s thinking about marijuana. At the checkup when you routinely ask about alcohol and drug use, ask about marijuana use in particular. You might start by asking if they have heard their friends talking about marijuana? What have they heard? Are other kids using it? Have they ever seen anyone use it? Have their friends invited them to try? You should find out if they think it is safe or dangerous, and how it compares with cigarettes, alcohol, and other drugs on this score. Then you may be able to debunk some myths you hear from them.
Myth No. 1: Marijuana is medicine
Although 23 states allow the legal sale of marijuana for “medicinal purposes,” it is important to note that there are currently no Food and Drug Administration–approved indications for medical marijuana. There is modest evidence that the active compounds in marijuana (delta-9-tetra-hydrocannabinol [THC] and other cannabinoids) can be effective in the management of the muscle spasticity associated with multiple sclerosis, the treatment of nausea associated with chemotherapy, and increasing the appetite of patients with wasting due to AIDS, and there are FDA-approved synthetic cannabinoids that can be prescribed for these symptoms. It is also important to note that there is no evidence that THC or other cannabinoids are useful in the treatment of mood or anxiety symptoms, even though these are often used as reasons for seeking medicinal marijuana. Indeed, marijuana may cause or worsen several psychiatric problems.
Myth No. 2: Marijuana is safe

Although there is consensus that moderate marijuana use in adulthood poses only limited health risks (including the known risks of smoking), there is robust evidence that marijuana use during youth (through the early 20s) causes several serious and permanent effects on the developing brain. One 2012 study showed that for youth who are dependent on marijuana before they are 18 years, there is an 8-point drop in IQ in adulthood (Proc Natl Acad Sci USA. 2012 Oct 2;109[40]:E2657-64). This IQ drop persists even if they quit smoking, and does not occur for those who first become dependent on marijuana in adulthood. A 2015 study demonstrated that even for adolescents who are light smokers (one to two times weekly) with no evidence of marijuana dependence, there are significant abnormalities in the size and shape of their amygdala and nucleus accumbens, with associated changes in their motivation, decision making, attention, functional memory, and processing of emotions(J Neurosci. 2014 Apr 16;34[16]:5529-38). These abnormalities increase with increased frequency of use, and are not seen in those who begin smoking in adulthood (mid-20s and later).
Beyond these findings of cognitive deficits, evidence is growing that adolescent marijuana use is associated with several psychiatric illnesses, including depression and anxiety. There is especially strong evidence for a causal link between marijuana use and psychotic illnesses in (genetically) vulnerable young people. Any marijuana user can experience a brief psychotic reaction if the amount ingested or smoked is great enough, but for those young people who carry a specific variant of the gene for catechol-o-methyltransferase (COMT, an enzyme that degrades neurotransmitters), smoking marijuana in adolescence nearly triples their risk of developing schizophrenia in adulthood. For youth with a variant of the AKT gene (another enzyme affecting dopamine signaling in the brain), daily use of marijuana raises their risk of developing schizophrenia sevenfold. Clearly, marijuana can be the critical environmental trigger for schizophrenia in genetically vulnerable youth. Until we have a comprehensive knowledge of the relevant genes, and routinely check every patient’s complete genetic profile, it is reasonable to assume that any young person using marijuana is significantly increasing the risk of developing schizophrenia, a chronic and disabling condition.
Myth No. 3: Marijuana has no effect on driving

Marijuana intoxication significantly affects motor coordination, reaction time, and judgment, and multiple studies have demonstrated a direct relationship between blood THC concentration and impaired driving ability. A recent meta-analysis demonstrated that the risk of being in a car accident doubled after marijuana use (Drug Alcohol Depend. 2004 Feb 7;73[2]:109-19). These studies usually involved adults, and it is reasonable to assume that the risks may be more pronounced in adolescents, particularly ones who are new to driving or have other problems that could affect their attention or reaction time (such as attention-deficit/hyperactivity disorder). Beyond letting patients know about the increased risks of accidents, it may be worth reminding them that driving while intoxicated – even with legal use marijuana – is a criminal offense.
Myth No. 4: Marijuana has no effect on schoolwork
Aside from the risks of causing long-term cognitive changes and psychiatric problems that can affect school performance, the direct effects of marijuana intoxication can linger and affect school performance well after its use. The “high” from marijuana typically lasts from 1 to 3 hours, but the drug’s effects on higher-level cognitive processes (mediated by the neocortex and hippocampus) can last for days. So a teenager who smokes on Saturday night may have lingering impairment of motivation, the ability to shift attention, the ability to learn complex tasks, and working memory. These are all critical cognitive abilities for learning, and can make studying on Sunday and performing well on a test on Monday much more difficult.
Myth No. 5: Marijuana is not addictive
Marijuana is addictive, with studies suggesting that nearly 9% of marijuana users will become addicted. Again, the risks are far greater for young people. Among people who begin using marijuana during adolescence, the rate of addiction climbs to 17%, and can be as high as 50% in daily users. Remember that addiction describes a pattern of continued use despite that use causing significant legal, social, or school and work problems. Users also may develop physical dependence, with a withdrawal syndrome that includes irritability, restlessness, insomnia, and appetite changes; these can last as long as 2 weeks.
Currently available forms of marijuana are much more potent than those that were studied and used in prior decades. On average, the potency of smoked marijuana has tripled, and there are concentrates (in oil form, for example) and hybrids with much higher potency still. More potent marijuana increases the high from even a small dose, and increases the likelihood of addiction and of other immediate and lingering complications of its use. So, parents who think they know what marijuana does to adolescents based on their own youthful experiences are significantly underestimating the risks.
When asking your patients explicitly about marijuana use, be curious and nonjudgmental, but also be frank and forthright about what is known about the risks associated with its use. Although the current legal and political changes around marijuana use may have given them the impression that marijuana use is safe, you want them to have the facts they need to make informed decisions. Even if you only discuss one of these myths with your patients, you will have equipped them with powerful information that they may use and share with their friends.
Dr. Swick is an attending psychiatrist in the division of child psychiatry at Massachusetts General Hospital, Boston, and director of the Parenting at a Challenging Time (PACT) Program at the Vernon Cancer Center at Newton (Mass.) Wellesley Hospital. Dr. Jellinek is professor of psychiatry and of pediatrics at Harvard Medical School, Boston. Email them at [email protected].
The annual checkup has long provided an opportunity for early adolescents to learn about the risks of alcohol and drug use from a trusted source who may be less biased than parents, teachers, or police. Parents also turn to their child’s pediatrician for guidance on how to broach this important topic with their children, or they may come with concerns about their children’s use of drugs or alcohol.
Marijuana has become an increasingly complex topic, as its legal status has rapidly changed: It’s legal to purchase marijuana in four states (Alaska, Colorado, Oregon, and Washington, as well as the District of Columbia); it is decriminalized in 20 states and the District of Columbia for certain marijuana possession offenses; and it is legal to use medical marijuana in 23 states. As its legal status changes, attitudes about its use also have shifted, and its availability, form, and potency all have changed dramatically in just the past decade. Further, we ourselves may have mixed feelings about marijuana use based on our own experience as adolescents and sampling bias. We may have seen its low-level use and minimal effects in young or mature adults, or we may have seen substantial use of marijuana have a major deleterious impact on a friend or become a gateway drug for addiction to dangerous substances.
Before addressing marijuana use with adolescent patients and dealing with their potential skepticism concerning any harm, it is worth spending a little time looking in the mirror to consider your perspective on marijuana use and your response to disbelief.
According to the National Institute on Drug Abuse’s Monitoring the Future (MTF) survey, almost 12% of 8th graders, 27% of 10th graders, and 35% of 12th graders in the United States reported having used marijuana in the past year. Among the 12th graders in that 2014 survey, almost 20% were current users of marijuana and 6% were daily users. Many surveys, including the MTF, have demonstrated that attitudes of teenagers have shifted about marijuana’s dangerousness, with a steep and steady decline in the number of teenagers believing that regular marijuana use poses a risk to their health and well-being. In 2014, less than 40% of 12th graders in the MTF survey agreed that regular use of marijuana would pose a risk to their well-being, compared with a peak of almost 80% of 12th graders in the early 1990’s.
Pediatricians have an opportunity to change their patient’s thinking about marijuana. At the checkup when you routinely ask about alcohol and drug use, ask about marijuana use in particular. You might start by asking if they have heard their friends talking about marijuana? What have they heard? Are other kids using it? Have they ever seen anyone use it? Have their friends invited them to try? You should find out if they think it is safe or dangerous, and how it compares with cigarettes, alcohol, and other drugs on this score. Then you may be able to debunk some myths you hear from them.
Myth No. 1: Marijuana is medicine
Although 23 states allow the legal sale of marijuana for “medicinal purposes,” it is important to note that there are currently no Food and Drug Administration–approved indications for medical marijuana. There is modest evidence that the active compounds in marijuana (delta-9-tetra-hydrocannabinol [THC] and other cannabinoids) can be effective in the management of the muscle spasticity associated with multiple sclerosis, the treatment of nausea associated with chemotherapy, and increasing the appetite of patients with wasting due to AIDS, and there are FDA-approved synthetic cannabinoids that can be prescribed for these symptoms. It is also important to note that there is no evidence that THC or other cannabinoids are useful in the treatment of mood or anxiety symptoms, even though these are often used as reasons for seeking medicinal marijuana. Indeed, marijuana may cause or worsen several psychiatric problems.
Myth No. 2: Marijuana is safe
Although there is consensus that moderate marijuana use in adulthood poses only limited health risks (including the known risks of smoking), there is robust evidence that marijuana use during youth (through the early 20s) causes several serious and permanent effects on the developing brain. One 2012 study showed that for youth who are dependent on marijuana before they are 18 years, there is an 8-point drop in IQ in adulthood (Proc Natl Acad Sci USA. 2012 Oct 2;109[40]:E2657-64). This IQ drop persists even if they quit smoking, and does not occur for those who first become dependent on marijuana in adulthood. A 2015 study demonstrated that even for adolescents who are light smokers (one to two times weekly) with no evidence of marijuana dependence, there are significant abnormalities in the size and shape of their amygdala and nucleus accumbens, with associated changes in their motivation, decision making, attention, functional memory, and processing of emotions(J Neurosci. 2014 Apr 16;34[16]:5529-38). These abnormalities increase with increased frequency of use, and are not seen in those who begin smoking in adulthood (mid-20s and later).
Beyond these findings of cognitive deficits, evidence is growing that adolescent marijuana use is associated with several psychiatric illnesses, including depression and anxiety. There is especially strong evidence for a causal link between marijuana use and psychotic illnesses in (genetically) vulnerable young people. Any marijuana user can experience a brief psychotic reaction if the amount ingested or smoked is great enough, but for those young people who carry a specific variant of the gene for catechol-o-methyltransferase (COMT, an enzyme that degrades neurotransmitters), smoking marijuana in adolescence nearly triples their risk of developing schizophrenia in adulthood. For youth with a variant of the AKT gene (another enzyme affecting dopamine signaling in the brain), daily use of marijuana raises their risk of developing schizophrenia sevenfold. Clearly, marijuana can be the critical environmental trigger for schizophrenia in genetically vulnerable youth. Until we have a comprehensive knowledge of the relevant genes, and routinely check every patient’s complete genetic profile, it is reasonable to assume that any young person using marijuana is significantly increasing the risk of developing schizophrenia, a chronic and disabling condition.
Myth No. 3: Marijuana has no effect on driving
Marijuana intoxication significantly affects motor coordination, reaction time, and judgment, and multiple studies have demonstrated a direct relationship between blood THC concentration and impaired driving ability. A recent meta-analysis demonstrated that the risk of being in a car accident doubled after marijuana use (Drug Alcohol Depend. 2004 Feb 7;73[2]:109-19). These studies usually involved adults, and it is reasonable to assume that the risks may be more pronounced in adolescents, particularly ones who are new to driving or have other problems that could affect their attention or reaction time (such as attention-deficit/hyperactivity disorder). Beyond letting patients know about the increased risks of accidents, it may be worth reminding them that driving while intoxicated – even with legal use marijuana – is a criminal offense.
Myth No. 4: Marijuana has no effect on schoolwork
Aside from the risks of causing long-term cognitive changes and psychiatric problems that can affect school performance, the direct effects of marijuana intoxication can linger and affect school performance well after its use. The “high” from marijuana typically lasts from 1 to 3 hours, but the drug’s effects on higher-level cognitive processes (mediated by the neocortex and hippocampus) can last for days. So a teenager who smokes on Saturday night may have lingering impairment of motivation, the ability to shift attention, the ability to learn complex tasks, and working memory. These are all critical cognitive abilities for learning, and can make studying on Sunday and performing well on a test on Monday much more difficult.
Myth No. 5: Marijuana is not addictive
Marijuana is addictive, with studies suggesting that nearly 9% of marijuana users will become addicted. Again, the risks are far greater for young people. Among people who begin using marijuana during adolescence, the rate of addiction climbs to 17%, and can be as high as 50% in daily users. Remember that addiction describes a pattern of continued use despite that use causing significant legal, social, or school and work problems. Users also may develop physical dependence, with a withdrawal syndrome that includes irritability, restlessness, insomnia, and appetite changes; these can last as long as 2 weeks.
Currently available forms of marijuana are much more potent than those that were studied and used in prior decades. On average, the potency of smoked marijuana has tripled, and there are concentrates (in oil form, for example) and hybrids with much higher potency still. More potent marijuana increases the high from even a small dose, and increases the likelihood of addiction and of other immediate and lingering complications of its use. So, parents who think they know what marijuana does to adolescents based on their own youthful experiences are significantly underestimating the risks.
When asking your patients explicitly about marijuana use, be curious and nonjudgmental, but also be frank and forthright about what is known about the risks associated with its use. Although the current legal and political changes around marijuana use may have given them the impression that marijuana use is safe, you want them to have the facts they need to make informed decisions. Even if you only discuss one of these myths with your patients, you will have equipped them with powerful information that they may use and share with their friends.
Dr. Swick is an attending psychiatrist in the division of child psychiatry at Massachusetts General Hospital, Boston, and director of the Parenting at a Challenging Time (PACT) Program at the Vernon Cancer Center at Newton (Mass.) Wellesley Hospital. Dr. Jellinek is professor of psychiatry and of pediatrics at Harvard Medical School, Boston. Email them at [email protected].
The annual checkup has long provided an opportunity for early adolescents to learn about the risks of alcohol and drug use from a trusted source who may be less biased than parents, teachers, or police. Parents also turn to their child’s pediatrician for guidance on how to broach this important topic with their children, or they may come with concerns about their children’s use of drugs or alcohol.
Marijuana has become an increasingly complex topic, as its legal status has rapidly changed: It’s legal to purchase marijuana in four states (Alaska, Colorado, Oregon, and Washington, as well as the District of Columbia); it is decriminalized in 20 states and the District of Columbia for certain marijuana possession offenses; and it is legal to use medical marijuana in 23 states. As its legal status changes, attitudes about its use also have shifted, and its availability, form, and potency all have changed dramatically in just the past decade. Further, we ourselves may have mixed feelings about marijuana use based on our own experience as adolescents and sampling bias. We may have seen its low-level use and minimal effects in young or mature adults, or we may have seen substantial use of marijuana have a major deleterious impact on a friend or become a gateway drug for addiction to dangerous substances.
Before addressing marijuana use with adolescent patients and dealing with their potential skepticism concerning any harm, it is worth spending a little time looking in the mirror to consider your perspective on marijuana use and your response to disbelief.
According to the National Institute on Drug Abuse’s Monitoring the Future (MTF) survey, almost 12% of 8th graders, 27% of 10th graders, and 35% of 12th graders in the United States reported having used marijuana in the past year. Among the 12th graders in that 2014 survey, almost 20% were current users of marijuana and 6% were daily users. Many surveys, including the MTF, have demonstrated that attitudes of teenagers have shifted about marijuana’s dangerousness, with a steep and steady decline in the number of teenagers believing that regular marijuana use poses a risk to their health and well-being. In 2014, less than 40% of 12th graders in the MTF survey agreed that regular use of marijuana would pose a risk to their well-being, compared with a peak of almost 80% of 12th graders in the early 1990’s.
Pediatricians have an opportunity to change their patient’s thinking about marijuana. At the checkup when you routinely ask about alcohol and drug use, ask about marijuana use in particular. You might start by asking if they have heard their friends talking about marijuana? What have they heard? Are other kids using it? Have they ever seen anyone use it? Have their friends invited them to try? You should find out if they think it is safe or dangerous, and how it compares with cigarettes, alcohol, and other drugs on this score. Then you may be able to debunk some myths you hear from them.
Myth No. 1: Marijuana is medicine
Although 23 states allow the legal sale of marijuana for “medicinal purposes,” it is important to note that there are currently no Food and Drug Administration–approved indications for medical marijuana. There is modest evidence that the active compounds in marijuana (delta-9-tetra-hydrocannabinol [THC] and other cannabinoids) can be effective in the management of the muscle spasticity associated with multiple sclerosis, the treatment of nausea associated with chemotherapy, and increasing the appetite of patients with wasting due to AIDS, and there are FDA-approved synthetic cannabinoids that can be prescribed for these symptoms. It is also important to note that there is no evidence that THC or other cannabinoids are useful in the treatment of mood or anxiety symptoms, even though these are often used as reasons for seeking medicinal marijuana. Indeed, marijuana may cause or worsen several psychiatric problems.
Myth No. 2: Marijuana is safe
Although there is consensus that moderate marijuana use in adulthood poses only limited health risks (including the known risks of smoking), there is robust evidence that marijuana use during youth (through the early 20s) causes several serious and permanent effects on the developing brain. One 2012 study showed that for youth who are dependent on marijuana before they are 18 years, there is an 8-point drop in IQ in adulthood (Proc Natl Acad Sci USA. 2012 Oct 2;109[40]:E2657-64). This IQ drop persists even if they quit smoking, and does not occur for those who first become dependent on marijuana in adulthood. A 2015 study demonstrated that even for adolescents who are light smokers (one to two times weekly) with no evidence of marijuana dependence, there are significant abnormalities in the size and shape of their amygdala and nucleus accumbens, with associated changes in their motivation, decision making, attention, functional memory, and processing of emotions(J Neurosci. 2014 Apr 16;34[16]:5529-38). These abnormalities increase with increased frequency of use, and are not seen in those who begin smoking in adulthood (mid-20s and later).
Beyond these findings of cognitive deficits, evidence is growing that adolescent marijuana use is associated with several psychiatric illnesses, including depression and anxiety. There is especially strong evidence for a causal link between marijuana use and psychotic illnesses in (genetically) vulnerable young people. Any marijuana user can experience a brief psychotic reaction if the amount ingested or smoked is great enough, but for those young people who carry a specific variant of the gene for catechol-o-methyltransferase (COMT, an enzyme that degrades neurotransmitters), smoking marijuana in adolescence nearly triples their risk of developing schizophrenia in adulthood. For youth with a variant of the AKT gene (another enzyme affecting dopamine signaling in the brain), daily use of marijuana raises their risk of developing schizophrenia sevenfold. Clearly, marijuana can be the critical environmental trigger for schizophrenia in genetically vulnerable youth. Until we have a comprehensive knowledge of the relevant genes, and routinely check every patient’s complete genetic profile, it is reasonable to assume that any young person using marijuana is significantly increasing the risk of developing schizophrenia, a chronic and disabling condition.
Myth No. 3: Marijuana has no effect on driving
Marijuana intoxication significantly affects motor coordination, reaction time, and judgment, and multiple studies have demonstrated a direct relationship between blood THC concentration and impaired driving ability. A recent meta-analysis demonstrated that the risk of being in a car accident doubled after marijuana use (Drug Alcohol Depend. 2004 Feb 7;73[2]:109-19). These studies usually involved adults, and it is reasonable to assume that the risks may be more pronounced in adolescents, particularly ones who are new to driving or have other problems that could affect their attention or reaction time (such as attention-deficit/hyperactivity disorder). Beyond letting patients know about the increased risks of accidents, it may be worth reminding them that driving while intoxicated – even with legal use marijuana – is a criminal offense.
Myth No. 4: Marijuana has no effect on schoolwork
Aside from the risks of causing long-term cognitive changes and psychiatric problems that can affect school performance, the direct effects of marijuana intoxication can linger and affect school performance well after its use. The “high” from marijuana typically lasts from 1 to 3 hours, but the drug’s effects on higher-level cognitive processes (mediated by the neocortex and hippocampus) can last for days. So a teenager who smokes on Saturday night may have lingering impairment of motivation, the ability to shift attention, the ability to learn complex tasks, and working memory. These are all critical cognitive abilities for learning, and can make studying on Sunday and performing well on a test on Monday much more difficult.
Myth No. 5: Marijuana is not addictive
Marijuana is addictive, with studies suggesting that nearly 9% of marijuana users will become addicted. Again, the risks are far greater for young people. Among people who begin using marijuana during adolescence, the rate of addiction climbs to 17%, and can be as high as 50% in daily users. Remember that addiction describes a pattern of continued use despite that use causing significant legal, social, or school and work problems. Users also may develop physical dependence, with a withdrawal syndrome that includes irritability, restlessness, insomnia, and appetite changes; these can last as long as 2 weeks.
Currently available forms of marijuana are much more potent than those that were studied and used in prior decades. On average, the potency of smoked marijuana has tripled, and there are concentrates (in oil form, for example) and hybrids with much higher potency still. More potent marijuana increases the high from even a small dose, and increases the likelihood of addiction and of other immediate and lingering complications of its use. So, parents who think they know what marijuana does to adolescents based on their own youthful experiences are significantly underestimating the risks.
When asking your patients explicitly about marijuana use, be curious and nonjudgmental, but also be frank and forthright about what is known about the risks associated with its use. Although the current legal and political changes around marijuana use may have given them the impression that marijuana use is safe, you want them to have the facts they need to make informed decisions. Even if you only discuss one of these myths with your patients, you will have equipped them with powerful information that they may use and share with their friends.
Dr. Swick is an attending psychiatrist in the division of child psychiatry at Massachusetts General Hospital, Boston, and director of the Parenting at a Challenging Time (PACT) Program at the Vernon Cancer Center at Newton (Mass.) Wellesley Hospital. Dr. Jellinek is professor of psychiatry and of pediatrics at Harvard Medical School, Boston. Email them at [email protected].
