User login
HIV prevention: A 3-pronged approach
› Screen all pregnant women and individuals ages 15 to 65 for human immunodeficiency virus (HIV) infection. A
› Prescribe tenofovir disoproxil fumarate/emtricitabine (Truvada) for pre-exposure prophylaxis for patients at high risk of acquiring HIV. A
› Offer needle and syringe exchange programs and, when appropriate, opioid substitution therapy to individuals who inject drugs. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Despite advances in human immunodeficiency virus (HIV) screening and treatment over the last 30 years, HIV remains a public health concern. In the United States, after an initial decline, total HIV incidence has failed to significantly decrease in the last 25 years. More than 1.2 million people are living with HIV in the United States, and 12.8% of them (156,300) are unaware they are affected.1 Of those diagnosed with HIV, only 30% are receiving treatment and are virally suppressed.2 Due to structural inequalities and psychosocial factors, African American and Latino patients remain disproportionately affected.3 The incidence of HIV infection among men who have sex with men has increased, and the incidence of HIV infection among people who inject drugs has plateaued after years of progressive decline.4
HIV prevention strategies are highly effective, but in general are underutilized. This article reviews 3 prevention strategies that can be administered by family physicians: HIV screening, pre-exposure prophylaxis (PrEP), and harm reduction.
Who and how to screen for HIV
Early identification of HIV infection generally leads to reduced transmission because diagnosis is associated with decreases in risky behavior.5,6 In addition, antiretroviral therapy (ART) is more effective when initiated early, before the development of advanced immunosuppression.7-9
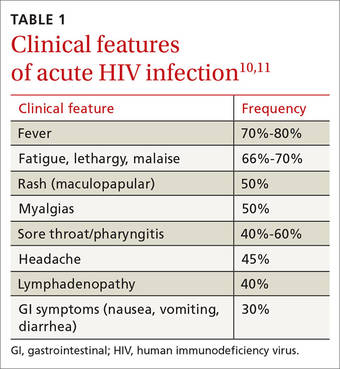
The “window period” of acute HIV infection (AHI) is the time from when the virus is transmitted to when markers of infection can be detected. Because this window period is associated with high viral transmission rates, family physicians must be familiar with symptoms of AHI (TABLE 1)10,11 and associated risk factors (eg, recent condomless sex or sharing of drug injection equipment with someone who is HIV-positive or of unknown HIV status).
Screening for HIV solely based on the presence of risk factors or clinical symptoms is not enough, however. The United States Preventive Services Task Force (USPSTF) recommends screening all pregnant women and individuals ages 15 to 65 for HIV.12 Screening based solely on risk factors or clinical symptoms frequently leads to missed diagnoses and identification of HIV infection at more advanced stages.13,14 Both the USPSTF and the Centers for Disease Control and Prevention (CDC) recommend universal opt-out screening (patients are informed that HIV screening will be performed and that they may decline testing) because such screening identifies HIV earlier and is associated with higher testing rates than opt-in screening, which requires explicit written consent and extensive pre-test counseling.
Which test to use. HIV screening with a fourth-generation antigen/antibody combination immunoassay—which detects both HIV p24 antigen and HIV antibodies—provides greater diagnostic accuracy than older tests.15 These newer tests detect HIV approximately 15 days after initial infection, reducing the window period of AHI.15,16 If you suspect a patient has AHI, consider early repeat HIV testing with a fourth-generation assay, or initial co-testing with a fourth-generation assay and a nucleic acid amplification test for HIV RNA, which makes it possible to detect infection approximately 5 days earlier than fourth-generation assays.15
Offer pre-exposure prophylaxis to high-risk patients
PrEP is the use of ART prior to HIV exposure to prevent transmission of the virus. It should be used with conventional risk reduction strategies, such as providing condoms, counseling patients about reducing risky behaviors, supporting medication adherence, and screening for and treating other sexually transmitted infections.
The US Food and Drug Administration (FDA) has approved only one medication, Truvada (tenofovir disoproxil fumarate/emtricitabine; TDF/FTC), for use as PrEP. Oral tenofovir-based regimens can effectively prevent HIV transmission,17-20 and strong adherence is associated with a risk reduction of 90% to 100%.17-23 The protective effect of oral PrEP is particularly strong in high-risk populations (eg, men who have sex with men, people who inject drugs), where the number needed to treat to prevent one HIV infection ranges from 12 to 100, depending on the patients’ risk profile.24-26 The CDC and Department of Health and Human Services have issued guidelines for using PrEP in high-risk patients.27
Barriers to implementing PrEP. Despite being highly effective, PrEP is not routinely prescribed to high-risk patients; modeling suggests that current use of PrEP is insufficient to significantly impact the incidence of HIV.28 Demand for PrEP is high among target groups,21,29,30 but patients have expressed concerns about adverse effects31 and stigma related to ART, HIV, and being at risk for HIV.32,33 Young age, lack of social support, low perception of risk, and failure to show up for appointments are also barriers to PrEP use.28,30,34
Some physicians have expressed concern that prescribing PrEP may promote high-risk sexual behavior.35 However, because PrEP is most beneficial in individuals who already engage in high-risk sexual behavior, strategic delivery of PrEP remains an effective risk-reducing strategy.17,18,21,26,36,37 Even in instances where PrEP has been associated with higher-risk sexual behavior and higher rates of sexually transmitted infections, it still prevents as much as 100% of new HIV infections.38
Fear of drug resistance also contributes to slow implementation of PrEP. Drug resistance has been observed in clinical trials of PrEP, but it has been exceedingly rare and predominantly limited to patients who had unrecognized AHI when they started PrEP.39 Furthermore, the few cases of drug resistance attributable to PrEP pale in comparison to the large number of estimated HIV infections averted—infections that would require lifelong ART with its own associated risks of drug resistance. By decreasing HIV transmission, PrEP is expected to decrease total drug resistance.40
Cost is another obstacle. Truvada costs approximately $1,540 per month.41 However, analysis has demonstrated that PrEP is cost-effective when targeted to high-risk patients.42 Most insurance plans cover PrEP, but often require high deductibles and copays; fortunately, this financial burden for patients can be mitigated or eliminated by co-pay assistance programs. The manufacturer of Truvada offers assistance programs for both insured and uninsured patients who are candidates for PrEP; details are available at http://www.truvada.com/truvada-patient-assistance.
Stigma has historically burdened individuals who seek to protect their sexual health, including HIV-negative individuals who are candidates for PrEP. Stigma surrounding HIV may decrease ART-based HIV prevention in men who have sex with men,43 while increasing high-risk behaviors44 and all-cause mortality.45
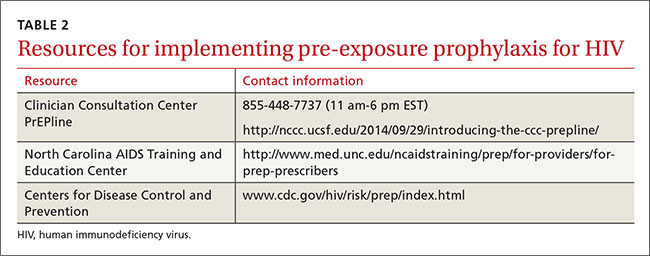
The resources listed in TABLE 2 can help physicians overcome some of the barriers to implementing PrEP.
How to deliver PrEP
Whether HIV specialists or primary care clinicians are best suited to provide PrEP is a subject of debate. HIV specialists are most familiar with ART and routine monitoring of adherence; however, they have less access to HIV-negative patients, who are the target group for PrEP.35 Family physicians tend to work in closer proximity and maintain longitudinal relationships with PrEP target groups, but in general have less experience with ART and evaluating AHI. Some may argue that competing demands may make it impractical to take a detailed sexual history during a primary care visit.46 In truth, both HIV specialists and family physicians can be appropriately equipped to provide PrEP.
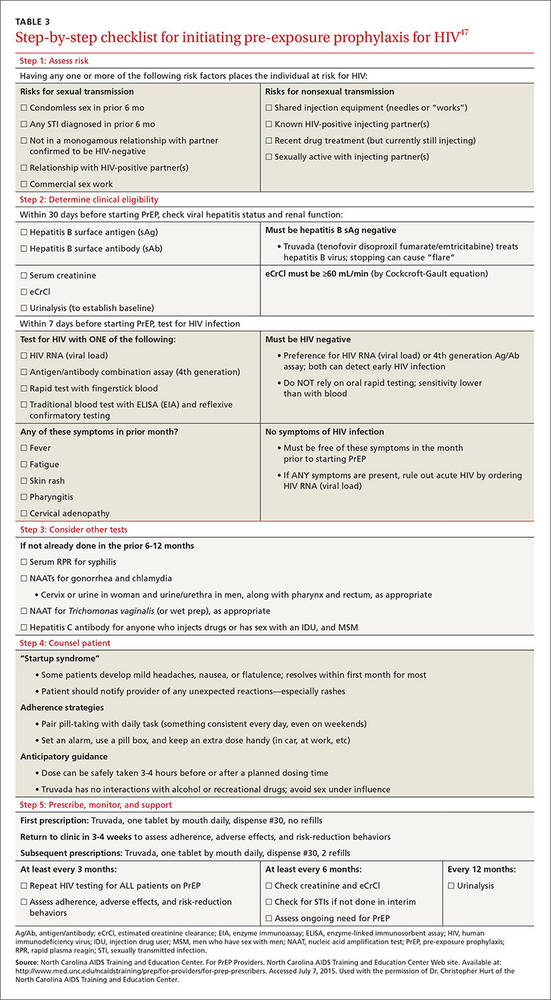
TABLE 3 outlines the steps necessary to provide a patient with PrEP.47 Assessing risk is the initial step; PrEP is beneficial for patients who have one or more risk factors for HIV infection. To be eligible for TDF/FTC, a patient must be HIV-negative, and should be tested for hepatitis B virus (HBV) infection and kidney disease. Because TDF/FTC treats HBV infection, candidates for PrEP who test positive for HBV should be evaluated for treatment of HBV before initiating PrEP. Candidates for PrEP who test negative for HBV infection and immunity should be vaccinated.
Candidates for PrEP should also be screened and monitored for kidney disease. TDF can cause increased serum creatinine due to tubular toxicity. A patient who has an estimated creatinine clearance <60 mL/min should not receive TDF/FTC for PrEP. If a patient’s estimated creatinine clearance falls below 60 mL/min or serum creatinine increases by 0.3 mg/dL above baseline after PrEP is started, TDF/FTC should be discontinued, and the patient should be evaluated for the underlying cause of the kidney disease.27
Before starting PrEP, candidates should be screened for HIV infection and symptoms of AHI. Strongly consider testing for sexually transmitted infections that may increase the risk of HIV transmission, such as syphilis, gonorrhea, or chlamydia.
Candidates who are eligible for PrEP must be counseled on medication adverse effects, adherence strategies, and symptoms of sexually transmitted infections. To initiate PrEP, candidates may be given a one-month supply of TDF/FTC; adherence, adverse effects, and other risk-reduction strategies are assessed at an office visit 3 to 4 weeks later. Subsequent prescriptions are then dispensed as a 3-month supply, with office visits to monitor PrEP scheduled for at least once every 3 months. During these monitoring visits, evaluate the patient’s HIV status, pregnancy status, adherence, adverse effects, risk-reduction behaviors, and indications for continued PrEP. Every 6 months, renal function and sexually transmitted infection status should be reassessed.
Reducing risk of harm among patients who inject drugs
Nonsexual transmission of HIV is a route of high infectivity.48 It includes transfusion of infected blood, sharing of equipment during injection drug use, and percutaneous needle sticks. Sharing of equipment during injection drug use is estimated to account for 8% of new infections in the United States.4
Harm reduction is a collection of strategies meant to reduce complications of illicit drug use, including HIV transmission. These strategies include needle and syringe programs that provide injection drug users with sterile equipment, and opioid substitution therapy.
Needle and syringe programs decrease HIV transmission49 and risky behaviors related to injection drug use,50 but federal funding of such programs is prohibited. Opioid substitution therapy reduces the incidence of HIV,50,51 injection drug use, sharing of drug preparation and injection equipment, and drug-related behaviors associated with a high risk of HIV transmission.50,52 However, in the United States, the quality of these programs varies; a study of opioid substitution therapy delivery found that 22.8% of programs provided doses that were too low to be effective.53
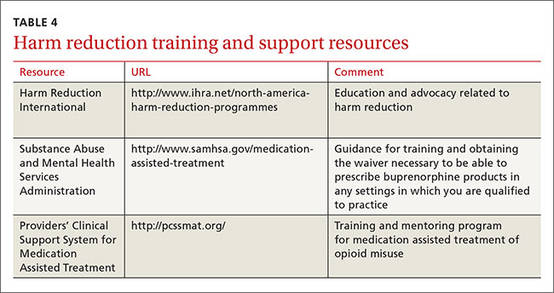
FDA-approved medications for opioid substitution therapy include sublingual buprenorphine, sublingual buprenorphine/naloxone tablets or strips (Suboxone), and oral methadone. Buprenorphine-based regimens can be provided by appropriately trained primary care clinicians; methadone requires a referral to a narcotic treatment program. TABLE 4 provides training and support resources for physicians who want to integrate opioid substitution therapy into their clinical practice.
CORRESPONDENCE
Richard Moore II, MD, 250 Smith Church Road, Roanoke Rapids, NC 27870; [email protected].
1. Hall HI, An Q, Tang T, et al; Centers for Disease Control and Prevention (CDC). Prevalence of diagnosed and undiagnosed HIV infection--United States, 2008-2012. MMWR Morb Mortal Wkly Rep. 2015;64:657-662.
2. Bradley H, Hall HI, Wolitski RJ, et al. Vital signs: HIV diagnosis, care, and treatment among persons living with HIV--United States, 2011. MMWR Morb Mortal Wkly Rep. 2014;63:1113-1117.
3. Maulsby C, Millet G, Lindsey K, et al. HIV among black men who have sex with men (MSM) in the United States: a review of the literature. AIDS Behav. 2014;18:10-25.
4. Centers for Disease Control and Prevention. Estimated HIV incidence among adults and adolescents in the United States, 2007-2010, HIV Surveillance Supplemental Report. 2012. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/hiv/pdf/statistics_hssr_vol_17_no_4.pdf. Accessed October 8, 2015.
5. Cleary PD, Van Devanter N, Rogers TF, et al. Behavior changes after notification of HIV infection. Am J Public Health. 1991;81:1586-1590.
6. Higgins DL, Galavotti C, O’Reilly KR, et al. Evidence for the effects of HIV antibody counseling and testing on risk behaviors. JAMA. 1991;266:2419-2429.
7. Murphy EL, Collier AC, Kalish LA, et al. Highly active antiretroviral therapy decreases mortality and morbidity in patients with advanced HIV disease. Ann Intern Med. 2001;135:17-26.
8. Palella FJ Jr, Deloria-Knoll M, Chmiel JS, et al. Survival benefit of initiating antiretroviral therapy in HIV-infected persons in different CD4 cell strata. Ann Intern Med. 2003;138:620-626.
9. INSIGHT START Study Group, Lundgren JD, Babiker AG, Gordin F, et al. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med. 2015;373:795-807.
10. Daar ES, Pilcher CD, Hecht FM. Clinical presentation and diagnosis of primary HIV-1 infection. Curr Opin HIV AIDS. 2008;3:10-15.
11. Tindall B, Barker S, Donovan B, et al. Characterization of the acute clinical illness associated with human immunodeficiency virus infection. Arch Intern Med. 1988;148:945-949.
12. Moyer V, US Preventative Services Task Force. Screening for HIV: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159:51-60.
13. Jenkins T, Gardner E, Thrun M, et al. Risk-based HIV testing fails to detect the majority of HIV-infected persons in medical care settings. Sex Transm Dis. 2006;33:329-333.
14. Klein D, Hurley LB, Merrill D, et al. Review of medical encounters in the 5 years before a diagnosis of HIV-1 infection: implications for early detection. J Acquir Immune Defic Syndr. 2003;32:143-152.
15. Pandori M, Hackett J Jr, Louie B, et al. Assessment of the ability of a fourth-generation immunoassay for human immunodeficiency virus (HIV) antibody and p24 antigen to detect both acute and recent HIV infections in a high-risk setting. J Clin Microbiol. 2009;47:2639-2642.
16. Branson BM. The future of HIV testing. J Acquir Imm Defic Syndr. 2010;55 Suppl 2:S102-S105.
17. Grant RM, Lama JR, Anderson PL, et al; iPrEx Study Team. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363:2587-2599.
18. Baeten JM, Donnell D, Ndase P, et al; Partners PrEP Study Team. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med. 2012;367:399-410.
19. Thigpen MC, Kebaabetswe PM, Paxton LA, et al. Antiretroviral preexposure prophylaxis for heterosexual HIV transmission in Botswana. N Engl J Med. 2012;367:423-434.
20. Choopanya K, Martin M, Suntharasamai P, et al. Antiretroviral prophylaxis for HIV infection in injecting drug users in Bangkok, Thailand (the Bangkok Tenofovir Study): a randomised, doubleblind, placebo-controlled phase 3 trial. Lancet. 2013;381:2083-2090.
21. Grant RM, Anderson PL, McMahan V, et al. Uptake of pre-exposure prophylaxis, sexual practices, and HIV incidence in men and transgender women who have sex with men: a cohort study. Lancet Infect Dis. 2014;14:820-829.
22. Anderson PL, Glidden DV, Liu A, et al. Emtricitabine-tenofovir concentrations and pre-exposure prophylaxis efficacy in men who have sex with men. Sci Transl Med. 2012;4:151ra125.
23. Henderson FL, Taylor AW, Chirwa LI, et al. Characteristics and oral PrEP adherence in the TDF2 open-label extension in Botswana. Paper presented at International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention; July 1922, 2015; Vancouver, Canada.
24. Murnane PM, Celum C, Mugo N, et al. Efficacy of preexposure prophylaxis for HIV-1 prevention among high-risk heterosexuals: subgroup analyses from a randomized trial. AIDS. 2013;27:2155-2160.
25. Heffron R, Mugo N, Were E, et al. Preexposure prophylaxis is efficacious for HIV-1 prevention among women using depot medroxyprogesterone acetate for contraception. AIDS. 2014;28:2771-2776.
26. Buchbinder SP, Glidden DV, Liu AY, et al. HIV pre-exposure prophylaxis in men who have sex with men and transgender women: a secondary analysis of a phase 3 randomised controlled efficacy trial. Lancet Infect Dis. 2014;14:468-475.
27. Center for Disease Control and Prevention. Preexposure prophylaxis for the prevention of HIV infection in the United States – 2014. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/hiv/pdf/PrEPguidelines2014.pdf. Accessed June 18, 2015.
28. Grant RM. Scale-up of preexposure prophylaxis in San Francisco to impact HIV incidence. Abstract 25. Paper presented at Conference on Retroviruses and Opportunistic Infections; February 23-26, 2015; Seattle, WA.
29. Cohen SE, Vittinghoff E, Bacon O, et al. High interest in preexposure prophylaxis among men who have sex with men at risk for HIV infection: baseline data from the US PrEP demonstration project. J Acquir Immune Defic Syndr. 2015;68:439-448.
30. Haberer JE, Baeten JM, Campbell J, et al. Adherence to antiretroviral prophylaxis for HIV prevention: a substudy cohort within a clinical trial of serodiscordant couples in East Africa. PLoS Med. 2013;10:e1001511.
31. Gilmore H, Koester K, Liu A, et al. To PrEP or not to PrEP: Perspectives from US iPrEx open label extension (OLE) participants. Abstract 440. Paper presented at 9th International Conference on HIV Treatment and Prevention Adherence; June 9, 2014; Miami Beach, FL.
32. Jain S, Gregor C, Krakower D, et al. Attitudes and interest toward HIV pre-exposure prophylaxis (PrEP) among participants using HIV non-occupational post-exposure prophylaxis (NPEP). Poster Abstract 1523. Poster presented at Infectious Disease Society of America Conference; October 8-12, 2014; Philadelphia, PA.
33. van der Straten A, Stadler J, Luecke E, et al; VOICE-C Study Team, Perspectives on use of oral and vaginal antiretrovirals for HIV prevention: the VOICE-C qualitative study in Johannesburg, South Africa. J Int AIDS Soc. 2014;17:19146.
34. Corneli AL, McKenna K, Headley J, et al; FEM-PrEP Study Group. A descriptive analysis of perceptions of HIV risk and worry about acquiring HIV among FEM-PrEP participants who seroconverted in Bondo, Kenya, and Pretoria, South Africa. J Int AIDS Soc. 2014;17:19152.
35. Krakower D, Ware N, Mitty JA, et al. HIV providers’ perceived barriers and facilitators to implementing pre-exposure prophylaxis in care settings: a qualitative study. AIDS Behav. 2014;18:1712-1721.
36. McCormack S, Dunn DT, Desai M, et al. Pre-exposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial. Lancet. 2015. [Epub ahead of print].
37. Mugwanya KK, Donnell D, Celum C, et al. Sexual behaviour of heterosexual men and women receiving antiretroviral pre-exposure prophylaxis for HIV prevention: a longitudinal analysis. Lancet Infect Dis. 2013;13:1021-1028.
38. Volk JE, Marcus JL, Phengrasamy T, et al. No new HIV infections with increasing use of HIV preexposure prophylaxis in a clinical practice setting. Clin Infect Dis. 2015;61:1601-1603.
39. Lehman DA, Baeten JM, McCoy CO, et al. Risk of drug resistance among persons acquiring HIV within a randomized clinical trial of single- or dual-agent preexposure prophylaxis. J Infect Dis. 2015;211:1211-1218.
40. Supervie V, Garcia-Lerma JG, Heneine W, et al. HIV, transmitted drug resistance, and the paradox of preexposure prophylaxis. Proc Natl Acad Sci U S A. 2010;107:12381-12386.
41. AIDSinfo. Cost considerations and antiretroviral therapy. AIDSinfo Web site. Available at: https://aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-arv-guidelines/459/cost-considerations-and-antiretroviral-therapy. Accessed December 14, 2015.
42. Gomez GB, Borquez A, Case KK, et al. The cost and impact of scaling up pre-exposure prophylaxis for HIV prevention: a systematic review of cost-effectiveness modelling studies. PLoS Med. 2013;10:e1001401.
43. Oldenburg CE, Perez-Brumer AG, Hatzenbuehler ML, et al. State-level structural sexual stigma and HIV prevention in a national online sample of HIV-uninfected MSM in the United States. AIDS. 2015;29:837-845.
44. Hatzenbuehler ML, O’Cleirigh C, Mayer KH, et al. Prospective associations between HIV-related stigma, transmission risk behaviors, and adverse mental health outcomes in men who have sex with men. Ann Behav Med. 2011;42:227-234.
45. Hatzenbuehler ML, Bellatorre A, Lee Y, et al. Structural stigma and all-cause mortality in sexual minority populations. Soc Sci Med. 2014;103:33-41.
46. Arnold EA, Hazelton P, Lane T, et al. A qualitative study of provider thoughts on implementing pre-exposure prophylaxis (PrEP) in clinical settings to prevent HIV infection. PLoS One. 2012;7:e40603.
47. North Carolina AIDS Training and Education Center. For PrEP Providers. North Carolina AIDS Training and Education Center Web site. Available at: http://www.med.unc.edu/ncaidstraining/prep/for-providers/for-prep-prescribers. Accessed July 7, 2015.
48. Patel P, Borkowf CB, Brook JT, et al. Estimating per-act HIV transmission risk: a systematic review. AIDS. 2014;28:1509-1519.
49. Aspinall EJ, Nambiar D, Goldberg DJ, et al. Are needle and syringe programmes associated with a reduction in HIV transmission among people who inject drugs: a systematic review and metaanalysis. Int J Epidemiol. 2014;43:235-248.
50. MacArthur GJ, van Velzen E, Palmateer N, et al. Interventions to prevent HIV and Hepatitis C in people who inject drugs: a review of reviews to assess evidence of effectiveness. Int J Drug Policy. 2014;25:34-52.
51. MacArthur GJ, Minozzi S, Martin N, et al. Opiate substitution treatment and HIV transmission in people who inject drugs: systematic review and meta-analysis. BMJ. 2012;345:e5945.
52. Gowing L, Farrell MF, Bornemann R, et al. Oral substitution treatment of injecting opioid users for prevention of HIV infection. Cochrane Database Syst Rev. 2011;(8):CD004145.
53. D’Aunno T, Pollack HA, Frimpong JA, et al. Evidence-based treatment for opioid disorders: a 23-year national study of methadone dose levels. J Subst Abuse Treat. 2014;47:245-250.
› Screen all pregnant women and individuals ages 15 to 65 for human immunodeficiency virus (HIV) infection. A
› Prescribe tenofovir disoproxil fumarate/emtricitabine (Truvada) for pre-exposure prophylaxis for patients at high risk of acquiring HIV. A
› Offer needle and syringe exchange programs and, when appropriate, opioid substitution therapy to individuals who inject drugs. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Despite advances in human immunodeficiency virus (HIV) screening and treatment over the last 30 years, HIV remains a public health concern. In the United States, after an initial decline, total HIV incidence has failed to significantly decrease in the last 25 years. More than 1.2 million people are living with HIV in the United States, and 12.8% of them (156,300) are unaware they are affected.1 Of those diagnosed with HIV, only 30% are receiving treatment and are virally suppressed.2 Due to structural inequalities and psychosocial factors, African American and Latino patients remain disproportionately affected.3 The incidence of HIV infection among men who have sex with men has increased, and the incidence of HIV infection among people who inject drugs has plateaued after years of progressive decline.4
HIV prevention strategies are highly effective, but in general are underutilized. This article reviews 3 prevention strategies that can be administered by family physicians: HIV screening, pre-exposure prophylaxis (PrEP), and harm reduction.
Who and how to screen for HIV
Early identification of HIV infection generally leads to reduced transmission because diagnosis is associated with decreases in risky behavior.5,6 In addition, antiretroviral therapy (ART) is more effective when initiated early, before the development of advanced immunosuppression.7-9
The “window period” of acute HIV infection (AHI) is the time from when the virus is transmitted to when markers of infection can be detected. Because this window period is associated with high viral transmission rates, family physicians must be familiar with symptoms of AHI (TABLE 1)10,11 and associated risk factors (eg, recent condomless sex or sharing of drug injection equipment with someone who is HIV-positive or of unknown HIV status).
Screening for HIV solely based on the presence of risk factors or clinical symptoms is not enough, however. The United States Preventive Services Task Force (USPSTF) recommends screening all pregnant women and individuals ages 15 to 65 for HIV.12 Screening based solely on risk factors or clinical symptoms frequently leads to missed diagnoses and identification of HIV infection at more advanced stages.13,14 Both the USPSTF and the Centers for Disease Control and Prevention (CDC) recommend universal opt-out screening (patients are informed that HIV screening will be performed and that they may decline testing) because such screening identifies HIV earlier and is associated with higher testing rates than opt-in screening, which requires explicit written consent and extensive pre-test counseling.
Which test to use. HIV screening with a fourth-generation antigen/antibody combination immunoassay—which detects both HIV p24 antigen and HIV antibodies—provides greater diagnostic accuracy than older tests.15 These newer tests detect HIV approximately 15 days after initial infection, reducing the window period of AHI.15,16 If you suspect a patient has AHI, consider early repeat HIV testing with a fourth-generation assay, or initial co-testing with a fourth-generation assay and a nucleic acid amplification test for HIV RNA, which makes it possible to detect infection approximately 5 days earlier than fourth-generation assays.15
Offer pre-exposure prophylaxis to high-risk patients
PrEP is the use of ART prior to HIV exposure to prevent transmission of the virus. It should be used with conventional risk reduction strategies, such as providing condoms, counseling patients about reducing risky behaviors, supporting medication adherence, and screening for and treating other sexually transmitted infections.
The US Food and Drug Administration (FDA) has approved only one medication, Truvada (tenofovir disoproxil fumarate/emtricitabine; TDF/FTC), for use as PrEP. Oral tenofovir-based regimens can effectively prevent HIV transmission,17-20 and strong adherence is associated with a risk reduction of 90% to 100%.17-23 The protective effect of oral PrEP is particularly strong in high-risk populations (eg, men who have sex with men, people who inject drugs), where the number needed to treat to prevent one HIV infection ranges from 12 to 100, depending on the patients’ risk profile.24-26 The CDC and Department of Health and Human Services have issued guidelines for using PrEP in high-risk patients.27
Barriers to implementing PrEP. Despite being highly effective, PrEP is not routinely prescribed to high-risk patients; modeling suggests that current use of PrEP is insufficient to significantly impact the incidence of HIV.28 Demand for PrEP is high among target groups,21,29,30 but patients have expressed concerns about adverse effects31 and stigma related to ART, HIV, and being at risk for HIV.32,33 Young age, lack of social support, low perception of risk, and failure to show up for appointments are also barriers to PrEP use.28,30,34
Some physicians have expressed concern that prescribing PrEP may promote high-risk sexual behavior.35 However, because PrEP is most beneficial in individuals who already engage in high-risk sexual behavior, strategic delivery of PrEP remains an effective risk-reducing strategy.17,18,21,26,36,37 Even in instances where PrEP has been associated with higher-risk sexual behavior and higher rates of sexually transmitted infections, it still prevents as much as 100% of new HIV infections.38
Fear of drug resistance also contributes to slow implementation of PrEP. Drug resistance has been observed in clinical trials of PrEP, but it has been exceedingly rare and predominantly limited to patients who had unrecognized AHI when they started PrEP.39 Furthermore, the few cases of drug resistance attributable to PrEP pale in comparison to the large number of estimated HIV infections averted—infections that would require lifelong ART with its own associated risks of drug resistance. By decreasing HIV transmission, PrEP is expected to decrease total drug resistance.40
Cost is another obstacle. Truvada costs approximately $1,540 per month.41 However, analysis has demonstrated that PrEP is cost-effective when targeted to high-risk patients.42 Most insurance plans cover PrEP, but often require high deductibles and copays; fortunately, this financial burden for patients can be mitigated or eliminated by co-pay assistance programs. The manufacturer of Truvada offers assistance programs for both insured and uninsured patients who are candidates for PrEP; details are available at http://www.truvada.com/truvada-patient-assistance.
Stigma has historically burdened individuals who seek to protect their sexual health, including HIV-negative individuals who are candidates for PrEP. Stigma surrounding HIV may decrease ART-based HIV prevention in men who have sex with men,43 while increasing high-risk behaviors44 and all-cause mortality.45
The resources listed in TABLE 2 can help physicians overcome some of the barriers to implementing PrEP.
How to deliver PrEP
Whether HIV specialists or primary care clinicians are best suited to provide PrEP is a subject of debate. HIV specialists are most familiar with ART and routine monitoring of adherence; however, they have less access to HIV-negative patients, who are the target group for PrEP.35 Family physicians tend to work in closer proximity and maintain longitudinal relationships with PrEP target groups, but in general have less experience with ART and evaluating AHI. Some may argue that competing demands may make it impractical to take a detailed sexual history during a primary care visit.46 In truth, both HIV specialists and family physicians can be appropriately equipped to provide PrEP.
TABLE 3 outlines the steps necessary to provide a patient with PrEP.47 Assessing risk is the initial step; PrEP is beneficial for patients who have one or more risk factors for HIV infection. To be eligible for TDF/FTC, a patient must be HIV-negative, and should be tested for hepatitis B virus (HBV) infection and kidney disease. Because TDF/FTC treats HBV infection, candidates for PrEP who test positive for HBV should be evaluated for treatment of HBV before initiating PrEP. Candidates for PrEP who test negative for HBV infection and immunity should be vaccinated.
Candidates for PrEP should also be screened and monitored for kidney disease. TDF can cause increased serum creatinine due to tubular toxicity. A patient who has an estimated creatinine clearance <60 mL/min should not receive TDF/FTC for PrEP. If a patient’s estimated creatinine clearance falls below 60 mL/min or serum creatinine increases by 0.3 mg/dL above baseline after PrEP is started, TDF/FTC should be discontinued, and the patient should be evaluated for the underlying cause of the kidney disease.27
Before starting PrEP, candidates should be screened for HIV infection and symptoms of AHI. Strongly consider testing for sexually transmitted infections that may increase the risk of HIV transmission, such as syphilis, gonorrhea, or chlamydia.
Candidates who are eligible for PrEP must be counseled on medication adverse effects, adherence strategies, and symptoms of sexually transmitted infections. To initiate PrEP, candidates may be given a one-month supply of TDF/FTC; adherence, adverse effects, and other risk-reduction strategies are assessed at an office visit 3 to 4 weeks later. Subsequent prescriptions are then dispensed as a 3-month supply, with office visits to monitor PrEP scheduled for at least once every 3 months. During these monitoring visits, evaluate the patient’s HIV status, pregnancy status, adherence, adverse effects, risk-reduction behaviors, and indications for continued PrEP. Every 6 months, renal function and sexually transmitted infection status should be reassessed.
Reducing risk of harm among patients who inject drugs
Nonsexual transmission of HIV is a route of high infectivity.48 It includes transfusion of infected blood, sharing of equipment during injection drug use, and percutaneous needle sticks. Sharing of equipment during injection drug use is estimated to account for 8% of new infections in the United States.4
Harm reduction is a collection of strategies meant to reduce complications of illicit drug use, including HIV transmission. These strategies include needle and syringe programs that provide injection drug users with sterile equipment, and opioid substitution therapy.
Needle and syringe programs decrease HIV transmission49 and risky behaviors related to injection drug use,50 but federal funding of such programs is prohibited. Opioid substitution therapy reduces the incidence of HIV,50,51 injection drug use, sharing of drug preparation and injection equipment, and drug-related behaviors associated with a high risk of HIV transmission.50,52 However, in the United States, the quality of these programs varies; a study of opioid substitution therapy delivery found that 22.8% of programs provided doses that were too low to be effective.53
FDA-approved medications for opioid substitution therapy include sublingual buprenorphine, sublingual buprenorphine/naloxone tablets or strips (Suboxone), and oral methadone. Buprenorphine-based regimens can be provided by appropriately trained primary care clinicians; methadone requires a referral to a narcotic treatment program. TABLE 4 provides training and support resources for physicians who want to integrate opioid substitution therapy into their clinical practice.
CORRESPONDENCE
Richard Moore II, MD, 250 Smith Church Road, Roanoke Rapids, NC 27870; [email protected].
› Screen all pregnant women and individuals ages 15 to 65 for human immunodeficiency virus (HIV) infection. A
› Prescribe tenofovir disoproxil fumarate/emtricitabine (Truvada) for pre-exposure prophylaxis for patients at high risk of acquiring HIV. A
› Offer needle and syringe exchange programs and, when appropriate, opioid substitution therapy to individuals who inject drugs. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Despite advances in human immunodeficiency virus (HIV) screening and treatment over the last 30 years, HIV remains a public health concern. In the United States, after an initial decline, total HIV incidence has failed to significantly decrease in the last 25 years. More than 1.2 million people are living with HIV in the United States, and 12.8% of them (156,300) are unaware they are affected.1 Of those diagnosed with HIV, only 30% are receiving treatment and are virally suppressed.2 Due to structural inequalities and psychosocial factors, African American and Latino patients remain disproportionately affected.3 The incidence of HIV infection among men who have sex with men has increased, and the incidence of HIV infection among people who inject drugs has plateaued after years of progressive decline.4
HIV prevention strategies are highly effective, but in general are underutilized. This article reviews 3 prevention strategies that can be administered by family physicians: HIV screening, pre-exposure prophylaxis (PrEP), and harm reduction.
Who and how to screen for HIV
Early identification of HIV infection generally leads to reduced transmission because diagnosis is associated with decreases in risky behavior.5,6 In addition, antiretroviral therapy (ART) is more effective when initiated early, before the development of advanced immunosuppression.7-9
The “window period” of acute HIV infection (AHI) is the time from when the virus is transmitted to when markers of infection can be detected. Because this window period is associated with high viral transmission rates, family physicians must be familiar with symptoms of AHI (TABLE 1)10,11 and associated risk factors (eg, recent condomless sex or sharing of drug injection equipment with someone who is HIV-positive or of unknown HIV status).
Screening for HIV solely based on the presence of risk factors or clinical symptoms is not enough, however. The United States Preventive Services Task Force (USPSTF) recommends screening all pregnant women and individuals ages 15 to 65 for HIV.12 Screening based solely on risk factors or clinical symptoms frequently leads to missed diagnoses and identification of HIV infection at more advanced stages.13,14 Both the USPSTF and the Centers for Disease Control and Prevention (CDC) recommend universal opt-out screening (patients are informed that HIV screening will be performed and that they may decline testing) because such screening identifies HIV earlier and is associated with higher testing rates than opt-in screening, which requires explicit written consent and extensive pre-test counseling.
Which test to use. HIV screening with a fourth-generation antigen/antibody combination immunoassay—which detects both HIV p24 antigen and HIV antibodies—provides greater diagnostic accuracy than older tests.15 These newer tests detect HIV approximately 15 days after initial infection, reducing the window period of AHI.15,16 If you suspect a patient has AHI, consider early repeat HIV testing with a fourth-generation assay, or initial co-testing with a fourth-generation assay and a nucleic acid amplification test for HIV RNA, which makes it possible to detect infection approximately 5 days earlier than fourth-generation assays.15
Offer pre-exposure prophylaxis to high-risk patients
PrEP is the use of ART prior to HIV exposure to prevent transmission of the virus. It should be used with conventional risk reduction strategies, such as providing condoms, counseling patients about reducing risky behaviors, supporting medication adherence, and screening for and treating other sexually transmitted infections.
The US Food and Drug Administration (FDA) has approved only one medication, Truvada (tenofovir disoproxil fumarate/emtricitabine; TDF/FTC), for use as PrEP. Oral tenofovir-based regimens can effectively prevent HIV transmission,17-20 and strong adherence is associated with a risk reduction of 90% to 100%.17-23 The protective effect of oral PrEP is particularly strong in high-risk populations (eg, men who have sex with men, people who inject drugs), where the number needed to treat to prevent one HIV infection ranges from 12 to 100, depending on the patients’ risk profile.24-26 The CDC and Department of Health and Human Services have issued guidelines for using PrEP in high-risk patients.27
Barriers to implementing PrEP. Despite being highly effective, PrEP is not routinely prescribed to high-risk patients; modeling suggests that current use of PrEP is insufficient to significantly impact the incidence of HIV.28 Demand for PrEP is high among target groups,21,29,30 but patients have expressed concerns about adverse effects31 and stigma related to ART, HIV, and being at risk for HIV.32,33 Young age, lack of social support, low perception of risk, and failure to show up for appointments are also barriers to PrEP use.28,30,34
Some physicians have expressed concern that prescribing PrEP may promote high-risk sexual behavior.35 However, because PrEP is most beneficial in individuals who already engage in high-risk sexual behavior, strategic delivery of PrEP remains an effective risk-reducing strategy.17,18,21,26,36,37 Even in instances where PrEP has been associated with higher-risk sexual behavior and higher rates of sexually transmitted infections, it still prevents as much as 100% of new HIV infections.38
Fear of drug resistance also contributes to slow implementation of PrEP. Drug resistance has been observed in clinical trials of PrEP, but it has been exceedingly rare and predominantly limited to patients who had unrecognized AHI when they started PrEP.39 Furthermore, the few cases of drug resistance attributable to PrEP pale in comparison to the large number of estimated HIV infections averted—infections that would require lifelong ART with its own associated risks of drug resistance. By decreasing HIV transmission, PrEP is expected to decrease total drug resistance.40
Cost is another obstacle. Truvada costs approximately $1,540 per month.41 However, analysis has demonstrated that PrEP is cost-effective when targeted to high-risk patients.42 Most insurance plans cover PrEP, but often require high deductibles and copays; fortunately, this financial burden for patients can be mitigated or eliminated by co-pay assistance programs. The manufacturer of Truvada offers assistance programs for both insured and uninsured patients who are candidates for PrEP; details are available at http://www.truvada.com/truvada-patient-assistance.
Stigma has historically burdened individuals who seek to protect their sexual health, including HIV-negative individuals who are candidates for PrEP. Stigma surrounding HIV may decrease ART-based HIV prevention in men who have sex with men,43 while increasing high-risk behaviors44 and all-cause mortality.45
The resources listed in TABLE 2 can help physicians overcome some of the barriers to implementing PrEP.
How to deliver PrEP
Whether HIV specialists or primary care clinicians are best suited to provide PrEP is a subject of debate. HIV specialists are most familiar with ART and routine monitoring of adherence; however, they have less access to HIV-negative patients, who are the target group for PrEP.35 Family physicians tend to work in closer proximity and maintain longitudinal relationships with PrEP target groups, but in general have less experience with ART and evaluating AHI. Some may argue that competing demands may make it impractical to take a detailed sexual history during a primary care visit.46 In truth, both HIV specialists and family physicians can be appropriately equipped to provide PrEP.
TABLE 3 outlines the steps necessary to provide a patient with PrEP.47 Assessing risk is the initial step; PrEP is beneficial for patients who have one or more risk factors for HIV infection. To be eligible for TDF/FTC, a patient must be HIV-negative, and should be tested for hepatitis B virus (HBV) infection and kidney disease. Because TDF/FTC treats HBV infection, candidates for PrEP who test positive for HBV should be evaluated for treatment of HBV before initiating PrEP. Candidates for PrEP who test negative for HBV infection and immunity should be vaccinated.
Candidates for PrEP should also be screened and monitored for kidney disease. TDF can cause increased serum creatinine due to tubular toxicity. A patient who has an estimated creatinine clearance <60 mL/min should not receive TDF/FTC for PrEP. If a patient’s estimated creatinine clearance falls below 60 mL/min or serum creatinine increases by 0.3 mg/dL above baseline after PrEP is started, TDF/FTC should be discontinued, and the patient should be evaluated for the underlying cause of the kidney disease.27
Before starting PrEP, candidates should be screened for HIV infection and symptoms of AHI. Strongly consider testing for sexually transmitted infections that may increase the risk of HIV transmission, such as syphilis, gonorrhea, or chlamydia.
Candidates who are eligible for PrEP must be counseled on medication adverse effects, adherence strategies, and symptoms of sexually transmitted infections. To initiate PrEP, candidates may be given a one-month supply of TDF/FTC; adherence, adverse effects, and other risk-reduction strategies are assessed at an office visit 3 to 4 weeks later. Subsequent prescriptions are then dispensed as a 3-month supply, with office visits to monitor PrEP scheduled for at least once every 3 months. During these monitoring visits, evaluate the patient’s HIV status, pregnancy status, adherence, adverse effects, risk-reduction behaviors, and indications for continued PrEP. Every 6 months, renal function and sexually transmitted infection status should be reassessed.
Reducing risk of harm among patients who inject drugs
Nonsexual transmission of HIV is a route of high infectivity.48 It includes transfusion of infected blood, sharing of equipment during injection drug use, and percutaneous needle sticks. Sharing of equipment during injection drug use is estimated to account for 8% of new infections in the United States.4
Harm reduction is a collection of strategies meant to reduce complications of illicit drug use, including HIV transmission. These strategies include needle and syringe programs that provide injection drug users with sterile equipment, and opioid substitution therapy.
Needle and syringe programs decrease HIV transmission49 and risky behaviors related to injection drug use,50 but federal funding of such programs is prohibited. Opioid substitution therapy reduces the incidence of HIV,50,51 injection drug use, sharing of drug preparation and injection equipment, and drug-related behaviors associated with a high risk of HIV transmission.50,52 However, in the United States, the quality of these programs varies; a study of opioid substitution therapy delivery found that 22.8% of programs provided doses that were too low to be effective.53
FDA-approved medications for opioid substitution therapy include sublingual buprenorphine, sublingual buprenorphine/naloxone tablets or strips (Suboxone), and oral methadone. Buprenorphine-based regimens can be provided by appropriately trained primary care clinicians; methadone requires a referral to a narcotic treatment program. TABLE 4 provides training and support resources for physicians who want to integrate opioid substitution therapy into their clinical practice.
CORRESPONDENCE
Richard Moore II, MD, 250 Smith Church Road, Roanoke Rapids, NC 27870; [email protected].
1. Hall HI, An Q, Tang T, et al; Centers for Disease Control and Prevention (CDC). Prevalence of diagnosed and undiagnosed HIV infection--United States, 2008-2012. MMWR Morb Mortal Wkly Rep. 2015;64:657-662.
2. Bradley H, Hall HI, Wolitski RJ, et al. Vital signs: HIV diagnosis, care, and treatment among persons living with HIV--United States, 2011. MMWR Morb Mortal Wkly Rep. 2014;63:1113-1117.
3. Maulsby C, Millet G, Lindsey K, et al. HIV among black men who have sex with men (MSM) in the United States: a review of the literature. AIDS Behav. 2014;18:10-25.
4. Centers for Disease Control and Prevention. Estimated HIV incidence among adults and adolescents in the United States, 2007-2010, HIV Surveillance Supplemental Report. 2012. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/hiv/pdf/statistics_hssr_vol_17_no_4.pdf. Accessed October 8, 2015.
5. Cleary PD, Van Devanter N, Rogers TF, et al. Behavior changes after notification of HIV infection. Am J Public Health. 1991;81:1586-1590.
6. Higgins DL, Galavotti C, O’Reilly KR, et al. Evidence for the effects of HIV antibody counseling and testing on risk behaviors. JAMA. 1991;266:2419-2429.
7. Murphy EL, Collier AC, Kalish LA, et al. Highly active antiretroviral therapy decreases mortality and morbidity in patients with advanced HIV disease. Ann Intern Med. 2001;135:17-26.
8. Palella FJ Jr, Deloria-Knoll M, Chmiel JS, et al. Survival benefit of initiating antiretroviral therapy in HIV-infected persons in different CD4 cell strata. Ann Intern Med. 2003;138:620-626.
9. INSIGHT START Study Group, Lundgren JD, Babiker AG, Gordin F, et al. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med. 2015;373:795-807.
10. Daar ES, Pilcher CD, Hecht FM. Clinical presentation and diagnosis of primary HIV-1 infection. Curr Opin HIV AIDS. 2008;3:10-15.
11. Tindall B, Barker S, Donovan B, et al. Characterization of the acute clinical illness associated with human immunodeficiency virus infection. Arch Intern Med. 1988;148:945-949.
12. Moyer V, US Preventative Services Task Force. Screening for HIV: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159:51-60.
13. Jenkins T, Gardner E, Thrun M, et al. Risk-based HIV testing fails to detect the majority of HIV-infected persons in medical care settings. Sex Transm Dis. 2006;33:329-333.
14. Klein D, Hurley LB, Merrill D, et al. Review of medical encounters in the 5 years before a diagnosis of HIV-1 infection: implications for early detection. J Acquir Immune Defic Syndr. 2003;32:143-152.
15. Pandori M, Hackett J Jr, Louie B, et al. Assessment of the ability of a fourth-generation immunoassay for human immunodeficiency virus (HIV) antibody and p24 antigen to detect both acute and recent HIV infections in a high-risk setting. J Clin Microbiol. 2009;47:2639-2642.
16. Branson BM. The future of HIV testing. J Acquir Imm Defic Syndr. 2010;55 Suppl 2:S102-S105.
17. Grant RM, Lama JR, Anderson PL, et al; iPrEx Study Team. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363:2587-2599.
18. Baeten JM, Donnell D, Ndase P, et al; Partners PrEP Study Team. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med. 2012;367:399-410.
19. Thigpen MC, Kebaabetswe PM, Paxton LA, et al. Antiretroviral preexposure prophylaxis for heterosexual HIV transmission in Botswana. N Engl J Med. 2012;367:423-434.
20. Choopanya K, Martin M, Suntharasamai P, et al. Antiretroviral prophylaxis for HIV infection in injecting drug users in Bangkok, Thailand (the Bangkok Tenofovir Study): a randomised, doubleblind, placebo-controlled phase 3 trial. Lancet. 2013;381:2083-2090.
21. Grant RM, Anderson PL, McMahan V, et al. Uptake of pre-exposure prophylaxis, sexual practices, and HIV incidence in men and transgender women who have sex with men: a cohort study. Lancet Infect Dis. 2014;14:820-829.
22. Anderson PL, Glidden DV, Liu A, et al. Emtricitabine-tenofovir concentrations and pre-exposure prophylaxis efficacy in men who have sex with men. Sci Transl Med. 2012;4:151ra125.
23. Henderson FL, Taylor AW, Chirwa LI, et al. Characteristics and oral PrEP adherence in the TDF2 open-label extension in Botswana. Paper presented at International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention; July 1922, 2015; Vancouver, Canada.
24. Murnane PM, Celum C, Mugo N, et al. Efficacy of preexposure prophylaxis for HIV-1 prevention among high-risk heterosexuals: subgroup analyses from a randomized trial. AIDS. 2013;27:2155-2160.
25. Heffron R, Mugo N, Were E, et al. Preexposure prophylaxis is efficacious for HIV-1 prevention among women using depot medroxyprogesterone acetate for contraception. AIDS. 2014;28:2771-2776.
26. Buchbinder SP, Glidden DV, Liu AY, et al. HIV pre-exposure prophylaxis in men who have sex with men and transgender women: a secondary analysis of a phase 3 randomised controlled efficacy trial. Lancet Infect Dis. 2014;14:468-475.
27. Center for Disease Control and Prevention. Preexposure prophylaxis for the prevention of HIV infection in the United States – 2014. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/hiv/pdf/PrEPguidelines2014.pdf. Accessed June 18, 2015.
28. Grant RM. Scale-up of preexposure prophylaxis in San Francisco to impact HIV incidence. Abstract 25. Paper presented at Conference on Retroviruses and Opportunistic Infections; February 23-26, 2015; Seattle, WA.
29. Cohen SE, Vittinghoff E, Bacon O, et al. High interest in preexposure prophylaxis among men who have sex with men at risk for HIV infection: baseline data from the US PrEP demonstration project. J Acquir Immune Defic Syndr. 2015;68:439-448.
30. Haberer JE, Baeten JM, Campbell J, et al. Adherence to antiretroviral prophylaxis for HIV prevention: a substudy cohort within a clinical trial of serodiscordant couples in East Africa. PLoS Med. 2013;10:e1001511.
31. Gilmore H, Koester K, Liu A, et al. To PrEP or not to PrEP: Perspectives from US iPrEx open label extension (OLE) participants. Abstract 440. Paper presented at 9th International Conference on HIV Treatment and Prevention Adherence; June 9, 2014; Miami Beach, FL.
32. Jain S, Gregor C, Krakower D, et al. Attitudes and interest toward HIV pre-exposure prophylaxis (PrEP) among participants using HIV non-occupational post-exposure prophylaxis (NPEP). Poster Abstract 1523. Poster presented at Infectious Disease Society of America Conference; October 8-12, 2014; Philadelphia, PA.
33. van der Straten A, Stadler J, Luecke E, et al; VOICE-C Study Team, Perspectives on use of oral and vaginal antiretrovirals for HIV prevention: the VOICE-C qualitative study in Johannesburg, South Africa. J Int AIDS Soc. 2014;17:19146.
34. Corneli AL, McKenna K, Headley J, et al; FEM-PrEP Study Group. A descriptive analysis of perceptions of HIV risk and worry about acquiring HIV among FEM-PrEP participants who seroconverted in Bondo, Kenya, and Pretoria, South Africa. J Int AIDS Soc. 2014;17:19152.
35. Krakower D, Ware N, Mitty JA, et al. HIV providers’ perceived barriers and facilitators to implementing pre-exposure prophylaxis in care settings: a qualitative study. AIDS Behav. 2014;18:1712-1721.
36. McCormack S, Dunn DT, Desai M, et al. Pre-exposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial. Lancet. 2015. [Epub ahead of print].
37. Mugwanya KK, Donnell D, Celum C, et al. Sexual behaviour of heterosexual men and women receiving antiretroviral pre-exposure prophylaxis for HIV prevention: a longitudinal analysis. Lancet Infect Dis. 2013;13:1021-1028.
38. Volk JE, Marcus JL, Phengrasamy T, et al. No new HIV infections with increasing use of HIV preexposure prophylaxis in a clinical practice setting. Clin Infect Dis. 2015;61:1601-1603.
39. Lehman DA, Baeten JM, McCoy CO, et al. Risk of drug resistance among persons acquiring HIV within a randomized clinical trial of single- or dual-agent preexposure prophylaxis. J Infect Dis. 2015;211:1211-1218.
40. Supervie V, Garcia-Lerma JG, Heneine W, et al. HIV, transmitted drug resistance, and the paradox of preexposure prophylaxis. Proc Natl Acad Sci U S A. 2010;107:12381-12386.
41. AIDSinfo. Cost considerations and antiretroviral therapy. AIDSinfo Web site. Available at: https://aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-arv-guidelines/459/cost-considerations-and-antiretroviral-therapy. Accessed December 14, 2015.
42. Gomez GB, Borquez A, Case KK, et al. The cost and impact of scaling up pre-exposure prophylaxis for HIV prevention: a systematic review of cost-effectiveness modelling studies. PLoS Med. 2013;10:e1001401.
43. Oldenburg CE, Perez-Brumer AG, Hatzenbuehler ML, et al. State-level structural sexual stigma and HIV prevention in a national online sample of HIV-uninfected MSM in the United States. AIDS. 2015;29:837-845.
44. Hatzenbuehler ML, O’Cleirigh C, Mayer KH, et al. Prospective associations between HIV-related stigma, transmission risk behaviors, and adverse mental health outcomes in men who have sex with men. Ann Behav Med. 2011;42:227-234.
45. Hatzenbuehler ML, Bellatorre A, Lee Y, et al. Structural stigma and all-cause mortality in sexual minority populations. Soc Sci Med. 2014;103:33-41.
46. Arnold EA, Hazelton P, Lane T, et al. A qualitative study of provider thoughts on implementing pre-exposure prophylaxis (PrEP) in clinical settings to prevent HIV infection. PLoS One. 2012;7:e40603.
47. North Carolina AIDS Training and Education Center. For PrEP Providers. North Carolina AIDS Training and Education Center Web site. Available at: http://www.med.unc.edu/ncaidstraining/prep/for-providers/for-prep-prescribers. Accessed July 7, 2015.
48. Patel P, Borkowf CB, Brook JT, et al. Estimating per-act HIV transmission risk: a systematic review. AIDS. 2014;28:1509-1519.
49. Aspinall EJ, Nambiar D, Goldberg DJ, et al. Are needle and syringe programmes associated with a reduction in HIV transmission among people who inject drugs: a systematic review and metaanalysis. Int J Epidemiol. 2014;43:235-248.
50. MacArthur GJ, van Velzen E, Palmateer N, et al. Interventions to prevent HIV and Hepatitis C in people who inject drugs: a review of reviews to assess evidence of effectiveness. Int J Drug Policy. 2014;25:34-52.
51. MacArthur GJ, Minozzi S, Martin N, et al. Opiate substitution treatment and HIV transmission in people who inject drugs: systematic review and meta-analysis. BMJ. 2012;345:e5945.
52. Gowing L, Farrell MF, Bornemann R, et al. Oral substitution treatment of injecting opioid users for prevention of HIV infection. Cochrane Database Syst Rev. 2011;(8):CD004145.
53. D’Aunno T, Pollack HA, Frimpong JA, et al. Evidence-based treatment for opioid disorders: a 23-year national study of methadone dose levels. J Subst Abuse Treat. 2014;47:245-250.
1. Hall HI, An Q, Tang T, et al; Centers for Disease Control and Prevention (CDC). Prevalence of diagnosed and undiagnosed HIV infection--United States, 2008-2012. MMWR Morb Mortal Wkly Rep. 2015;64:657-662.
2. Bradley H, Hall HI, Wolitski RJ, et al. Vital signs: HIV diagnosis, care, and treatment among persons living with HIV--United States, 2011. MMWR Morb Mortal Wkly Rep. 2014;63:1113-1117.
3. Maulsby C, Millet G, Lindsey K, et al. HIV among black men who have sex with men (MSM) in the United States: a review of the literature. AIDS Behav. 2014;18:10-25.
4. Centers for Disease Control and Prevention. Estimated HIV incidence among adults and adolescents in the United States, 2007-2010, HIV Surveillance Supplemental Report. 2012. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/hiv/pdf/statistics_hssr_vol_17_no_4.pdf. Accessed October 8, 2015.
5. Cleary PD, Van Devanter N, Rogers TF, et al. Behavior changes after notification of HIV infection. Am J Public Health. 1991;81:1586-1590.
6. Higgins DL, Galavotti C, O’Reilly KR, et al. Evidence for the effects of HIV antibody counseling and testing on risk behaviors. JAMA. 1991;266:2419-2429.
7. Murphy EL, Collier AC, Kalish LA, et al. Highly active antiretroviral therapy decreases mortality and morbidity in patients with advanced HIV disease. Ann Intern Med. 2001;135:17-26.
8. Palella FJ Jr, Deloria-Knoll M, Chmiel JS, et al. Survival benefit of initiating antiretroviral therapy in HIV-infected persons in different CD4 cell strata. Ann Intern Med. 2003;138:620-626.
9. INSIGHT START Study Group, Lundgren JD, Babiker AG, Gordin F, et al. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med. 2015;373:795-807.
10. Daar ES, Pilcher CD, Hecht FM. Clinical presentation and diagnosis of primary HIV-1 infection. Curr Opin HIV AIDS. 2008;3:10-15.
11. Tindall B, Barker S, Donovan B, et al. Characterization of the acute clinical illness associated with human immunodeficiency virus infection. Arch Intern Med. 1988;148:945-949.
12. Moyer V, US Preventative Services Task Force. Screening for HIV: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159:51-60.
13. Jenkins T, Gardner E, Thrun M, et al. Risk-based HIV testing fails to detect the majority of HIV-infected persons in medical care settings. Sex Transm Dis. 2006;33:329-333.
14. Klein D, Hurley LB, Merrill D, et al. Review of medical encounters in the 5 years before a diagnosis of HIV-1 infection: implications for early detection. J Acquir Immune Defic Syndr. 2003;32:143-152.
15. Pandori M, Hackett J Jr, Louie B, et al. Assessment of the ability of a fourth-generation immunoassay for human immunodeficiency virus (HIV) antibody and p24 antigen to detect both acute and recent HIV infections in a high-risk setting. J Clin Microbiol. 2009;47:2639-2642.
16. Branson BM. The future of HIV testing. J Acquir Imm Defic Syndr. 2010;55 Suppl 2:S102-S105.
17. Grant RM, Lama JR, Anderson PL, et al; iPrEx Study Team. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363:2587-2599.
18. Baeten JM, Donnell D, Ndase P, et al; Partners PrEP Study Team. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med. 2012;367:399-410.
19. Thigpen MC, Kebaabetswe PM, Paxton LA, et al. Antiretroviral preexposure prophylaxis for heterosexual HIV transmission in Botswana. N Engl J Med. 2012;367:423-434.
20. Choopanya K, Martin M, Suntharasamai P, et al. Antiretroviral prophylaxis for HIV infection in injecting drug users in Bangkok, Thailand (the Bangkok Tenofovir Study): a randomised, doubleblind, placebo-controlled phase 3 trial. Lancet. 2013;381:2083-2090.
21. Grant RM, Anderson PL, McMahan V, et al. Uptake of pre-exposure prophylaxis, sexual practices, and HIV incidence in men and transgender women who have sex with men: a cohort study. Lancet Infect Dis. 2014;14:820-829.
22. Anderson PL, Glidden DV, Liu A, et al. Emtricitabine-tenofovir concentrations and pre-exposure prophylaxis efficacy in men who have sex with men. Sci Transl Med. 2012;4:151ra125.
23. Henderson FL, Taylor AW, Chirwa LI, et al. Characteristics and oral PrEP adherence in the TDF2 open-label extension in Botswana. Paper presented at International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention; July 1922, 2015; Vancouver, Canada.
24. Murnane PM, Celum C, Mugo N, et al. Efficacy of preexposure prophylaxis for HIV-1 prevention among high-risk heterosexuals: subgroup analyses from a randomized trial. AIDS. 2013;27:2155-2160.
25. Heffron R, Mugo N, Were E, et al. Preexposure prophylaxis is efficacious for HIV-1 prevention among women using depot medroxyprogesterone acetate for contraception. AIDS. 2014;28:2771-2776.
26. Buchbinder SP, Glidden DV, Liu AY, et al. HIV pre-exposure prophylaxis in men who have sex with men and transgender women: a secondary analysis of a phase 3 randomised controlled efficacy trial. Lancet Infect Dis. 2014;14:468-475.
27. Center for Disease Control and Prevention. Preexposure prophylaxis for the prevention of HIV infection in the United States – 2014. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/hiv/pdf/PrEPguidelines2014.pdf. Accessed June 18, 2015.
28. Grant RM. Scale-up of preexposure prophylaxis in San Francisco to impact HIV incidence. Abstract 25. Paper presented at Conference on Retroviruses and Opportunistic Infections; February 23-26, 2015; Seattle, WA.
29. Cohen SE, Vittinghoff E, Bacon O, et al. High interest in preexposure prophylaxis among men who have sex with men at risk for HIV infection: baseline data from the US PrEP demonstration project. J Acquir Immune Defic Syndr. 2015;68:439-448.
30. Haberer JE, Baeten JM, Campbell J, et al. Adherence to antiretroviral prophylaxis for HIV prevention: a substudy cohort within a clinical trial of serodiscordant couples in East Africa. PLoS Med. 2013;10:e1001511.
31. Gilmore H, Koester K, Liu A, et al. To PrEP or not to PrEP: Perspectives from US iPrEx open label extension (OLE) participants. Abstract 440. Paper presented at 9th International Conference on HIV Treatment and Prevention Adherence; June 9, 2014; Miami Beach, FL.
32. Jain S, Gregor C, Krakower D, et al. Attitudes and interest toward HIV pre-exposure prophylaxis (PrEP) among participants using HIV non-occupational post-exposure prophylaxis (NPEP). Poster Abstract 1523. Poster presented at Infectious Disease Society of America Conference; October 8-12, 2014; Philadelphia, PA.
33. van der Straten A, Stadler J, Luecke E, et al; VOICE-C Study Team, Perspectives on use of oral and vaginal antiretrovirals for HIV prevention: the VOICE-C qualitative study in Johannesburg, South Africa. J Int AIDS Soc. 2014;17:19146.
34. Corneli AL, McKenna K, Headley J, et al; FEM-PrEP Study Group. A descriptive analysis of perceptions of HIV risk and worry about acquiring HIV among FEM-PrEP participants who seroconverted in Bondo, Kenya, and Pretoria, South Africa. J Int AIDS Soc. 2014;17:19152.
35. Krakower D, Ware N, Mitty JA, et al. HIV providers’ perceived barriers and facilitators to implementing pre-exposure prophylaxis in care settings: a qualitative study. AIDS Behav. 2014;18:1712-1721.
36. McCormack S, Dunn DT, Desai M, et al. Pre-exposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial. Lancet. 2015. [Epub ahead of print].
37. Mugwanya KK, Donnell D, Celum C, et al. Sexual behaviour of heterosexual men and women receiving antiretroviral pre-exposure prophylaxis for HIV prevention: a longitudinal analysis. Lancet Infect Dis. 2013;13:1021-1028.
38. Volk JE, Marcus JL, Phengrasamy T, et al. No new HIV infections with increasing use of HIV preexposure prophylaxis in a clinical practice setting. Clin Infect Dis. 2015;61:1601-1603.
39. Lehman DA, Baeten JM, McCoy CO, et al. Risk of drug resistance among persons acquiring HIV within a randomized clinical trial of single- or dual-agent preexposure prophylaxis. J Infect Dis. 2015;211:1211-1218.
40. Supervie V, Garcia-Lerma JG, Heneine W, et al. HIV, transmitted drug resistance, and the paradox of preexposure prophylaxis. Proc Natl Acad Sci U S A. 2010;107:12381-12386.
41. AIDSinfo. Cost considerations and antiretroviral therapy. AIDSinfo Web site. Available at: https://aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-arv-guidelines/459/cost-considerations-and-antiretroviral-therapy. Accessed December 14, 2015.
42. Gomez GB, Borquez A, Case KK, et al. The cost and impact of scaling up pre-exposure prophylaxis for HIV prevention: a systematic review of cost-effectiveness modelling studies. PLoS Med. 2013;10:e1001401.
43. Oldenburg CE, Perez-Brumer AG, Hatzenbuehler ML, et al. State-level structural sexual stigma and HIV prevention in a national online sample of HIV-uninfected MSM in the United States. AIDS. 2015;29:837-845.
44. Hatzenbuehler ML, O’Cleirigh C, Mayer KH, et al. Prospective associations between HIV-related stigma, transmission risk behaviors, and adverse mental health outcomes in men who have sex with men. Ann Behav Med. 2011;42:227-234.
45. Hatzenbuehler ML, Bellatorre A, Lee Y, et al. Structural stigma and all-cause mortality in sexual minority populations. Soc Sci Med. 2014;103:33-41.
46. Arnold EA, Hazelton P, Lane T, et al. A qualitative study of provider thoughts on implementing pre-exposure prophylaxis (PrEP) in clinical settings to prevent HIV infection. PLoS One. 2012;7:e40603.
47. North Carolina AIDS Training and Education Center. For PrEP Providers. North Carolina AIDS Training and Education Center Web site. Available at: http://www.med.unc.edu/ncaidstraining/prep/for-providers/for-prep-prescribers. Accessed July 7, 2015.
48. Patel P, Borkowf CB, Brook JT, et al. Estimating per-act HIV transmission risk: a systematic review. AIDS. 2014;28:1509-1519.
49. Aspinall EJ, Nambiar D, Goldberg DJ, et al. Are needle and syringe programmes associated with a reduction in HIV transmission among people who inject drugs: a systematic review and metaanalysis. Int J Epidemiol. 2014;43:235-248.
50. MacArthur GJ, van Velzen E, Palmateer N, et al. Interventions to prevent HIV and Hepatitis C in people who inject drugs: a review of reviews to assess evidence of effectiveness. Int J Drug Policy. 2014;25:34-52.
51. MacArthur GJ, Minozzi S, Martin N, et al. Opiate substitution treatment and HIV transmission in people who inject drugs: systematic review and meta-analysis. BMJ. 2012;345:e5945.
52. Gowing L, Farrell MF, Bornemann R, et al. Oral substitution treatment of injecting opioid users for prevention of HIV infection. Cochrane Database Syst Rev. 2011;(8):CD004145.
53. D’Aunno T, Pollack HA, Frimpong JA, et al. Evidence-based treatment for opioid disorders: a 23-year national study of methadone dose levels. J Subst Abuse Treat. 2014;47:245-250.

Finally, an extra set of hands

In September 2015, the Centers for Medicare & Medicaid Services (CMS) launched a promising 4-year program called the "Transforming Clinical Practice Initiative" to lighten the load for family physicians.1 The central figures in this program are skilled and trained quality improvement advisors (QIA) who will work directly with physicians and their staffs to assist with the heavy lifting of practice improvement. The Oklahoma Physicians Research and Resources Network has used QIAs, which it calls practice enhancement assistants (PEAs), for more than 20 years to help Oklahoma family physicians improve various aspects of their practices, including testing processes, diabetes care, and preventive services. The PEAs have been enormously helpful.2
For this new CMS program, the feds awarded $685 million to 39 national and regional collaborative health care transformation networks and supporting organizations to develop peer-based learning networks to facilitate practice improvements.1 The program is designed to help more than 140,000 primary care physicians improve their practices by providing an extra set of skilled hands.
The American Board of Family Medicine (ABFM) and the American Academy of Family Physicians (AAFP) have teamed up to assist with this national effort. ABFM will cover the cost for the first 6000 family physicians who enroll in one of the regional Practice Transformation Networks to use their newly developed chronic disease registry called PRIME. This registry will extract clinical quality data from diverse electronic health records and report back to practices. The registry will meet the new federal quality measures reporting requirements and will also be a path for maintenance of certification.
The CMS Transforming Clinical Practice Initiative is a great opportunity to get that extra set of skilled hands you need to help meet new quality mandates and make your office more efficient and enjoyable for you, your staff, and your patients. Contact the ABFM (www.theabfm.org) to find out which organization is running the Practice Transformation Network in your area.
1. Centers for Medicare & Medicaid Services (CMS). Transforming clinical practice initiative awards. CMS Web site. Available at: https://www.cms.gov/Newsroom/MediaReleaseDatabase/Fact-sheets/2015-Fact-sheets-items/2015-09-29.html. Accessed December 15, 2015.
2. Nagykaldi Z, Mold JW, Robinson A, et al. Practice facilitators and practice-based research networks. J Am Board Fam Med. 2006;19:506-510.
In September 2015, the Centers for Medicare & Medicaid Services (CMS) launched a promising 4-year program called the "Transforming Clinical Practice Initiative" to lighten the load for family physicians.1 The central figures in this program are skilled and trained quality improvement advisors (QIA) who will work directly with physicians and their staffs to assist with the heavy lifting of practice improvement. The Oklahoma Physicians Research and Resources Network has used QIAs, which it calls practice enhancement assistants (PEAs), for more than 20 years to help Oklahoma family physicians improve various aspects of their practices, including testing processes, diabetes care, and preventive services. The PEAs have been enormously helpful.2
For this new CMS program, the feds awarded $685 million to 39 national and regional collaborative health care transformation networks and supporting organizations to develop peer-based learning networks to facilitate practice improvements.1 The program is designed to help more than 140,000 primary care physicians improve their practices by providing an extra set of skilled hands.
The American Board of Family Medicine (ABFM) and the American Academy of Family Physicians (AAFP) have teamed up to assist with this national effort. ABFM will cover the cost for the first 6000 family physicians who enroll in one of the regional Practice Transformation Networks to use their newly developed chronic disease registry called PRIME. This registry will extract clinical quality data from diverse electronic health records and report back to practices. The registry will meet the new federal quality measures reporting requirements and will also be a path for maintenance of certification.
The CMS Transforming Clinical Practice Initiative is a great opportunity to get that extra set of skilled hands you need to help meet new quality mandates and make your office more efficient and enjoyable for you, your staff, and your patients. Contact the ABFM (www.theabfm.org) to find out which organization is running the Practice Transformation Network in your area.
In September 2015, the Centers for Medicare & Medicaid Services (CMS) launched a promising 4-year program called the "Transforming Clinical Practice Initiative" to lighten the load for family physicians.1 The central figures in this program are skilled and trained quality improvement advisors (QIA) who will work directly with physicians and their staffs to assist with the heavy lifting of practice improvement. The Oklahoma Physicians Research and Resources Network has used QIAs, which it calls practice enhancement assistants (PEAs), for more than 20 years to help Oklahoma family physicians improve various aspects of their practices, including testing processes, diabetes care, and preventive services. The PEAs have been enormously helpful.2
For this new CMS program, the feds awarded $685 million to 39 national and regional collaborative health care transformation networks and supporting organizations to develop peer-based learning networks to facilitate practice improvements.1 The program is designed to help more than 140,000 primary care physicians improve their practices by providing an extra set of skilled hands.
The American Board of Family Medicine (ABFM) and the American Academy of Family Physicians (AAFP) have teamed up to assist with this national effort. ABFM will cover the cost for the first 6000 family physicians who enroll in one of the regional Practice Transformation Networks to use their newly developed chronic disease registry called PRIME. This registry will extract clinical quality data from diverse electronic health records and report back to practices. The registry will meet the new federal quality measures reporting requirements and will also be a path for maintenance of certification.
The CMS Transforming Clinical Practice Initiative is a great opportunity to get that extra set of skilled hands you need to help meet new quality mandates and make your office more efficient and enjoyable for you, your staff, and your patients. Contact the ABFM (www.theabfm.org) to find out which organization is running the Practice Transformation Network in your area.
1. Centers for Medicare & Medicaid Services (CMS). Transforming clinical practice initiative awards. CMS Web site. Available at: https://www.cms.gov/Newsroom/MediaReleaseDatabase/Fact-sheets/2015-Fact-sheets-items/2015-09-29.html. Accessed December 15, 2015.
2. Nagykaldi Z, Mold JW, Robinson A, et al. Practice facilitators and practice-based research networks. J Am Board Fam Med. 2006;19:506-510.
1. Centers for Medicare & Medicaid Services (CMS). Transforming clinical practice initiative awards. CMS Web site. Available at: https://www.cms.gov/Newsroom/MediaReleaseDatabase/Fact-sheets/2015-Fact-sheets-items/2015-09-29.html. Accessed December 15, 2015.
2. Nagykaldi Z, Mold JW, Robinson A, et al. Practice facilitators and practice-based research networks. J Am Board Fam Med. 2006;19:506-510.
Severe anal pain • perianal swelling • no history of injury to the area • Dx?
THE CASE
An 80-year-old man sought medical advice over the phone because he’d had sharp anal pain for 4 days. The pain increased during sitting and defecation. He was told he most likely had an acute anal fissure and was instructed to treat it with sitz baths, a stool-bulking agent, and a topical anesthetic. Despite these treatments, he continued to have intense anal pain.
Two days later, he presented to our practice complaining of severe anal discomfort and perianal swelling that made it almost impossible to sit. The patient led an active lifestyle and was otherwise healthy. He did not recall any injury to the perianal area.
THE DIAGNOSIS
During examination, we noted tender, erythematous swelling over the right perianal region at the 9 o’clock position without fluctuance, discharge, or ulceration. A digital rectal examination revealed a foreign body lying transversely across the anus about 2.5 cm from the anal verge. A pelvic x-ray confirmed the presence of a foreign body—a pin (FIGURE 1).
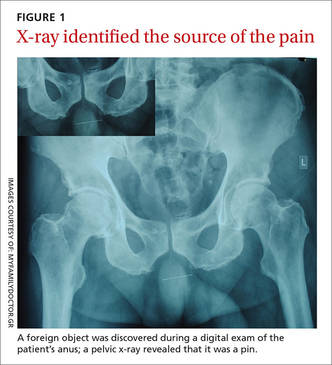
DISCUSSION
Anal pain is a common symptom that is usually caused by hemorrhoids, fissures, fistulas, or abscesses.1 Anal pain is rarely reported to be secondary to the ingestion of sharp foreign bodies, which can produce problems in the lower gastrointestinal tract.
Foreign body ingestion is most common in children ages 6 months to 6 years.2 When it occurs in adults, it tends to involve those who are older, patients without teeth, prisoners, patients under the influence of drugs or alcohol, or those with intellectual disabilities or psychiatric disorders.2,3
The foreign bodies that adults most commonly unintentionally ingest are bones from fish or other animals.2,4 Most of these pass through the alimentary tract uneventfully within a week.2,5,6 Swallowed bones have been known to cause perianal abscesses and anal fistulae, which can cause extreme pain.7
The presence of foreign bodies is not always easy to spot
In our patient’s case, the diagnosis was made by a careful digital rectal examination; however, a foreign body in an abscess cavity can be missed during a digital exam.8 In our patient’s case, a pelvic x-ray confirmed the presence and location of the pin.
Radiography is recommended as an initial screening method and is especially useful for determining the location of radiodense foreign bodies.2 However, most swallowed foreign bodies, such as non-radiodense fish bones, wood, thorns, plastic, small aluminum objects, and glass, cannot be detected by this method.3 Non-radiodense foreign bodies can be identified using computed tomography scanning.9
Removal is typically straightforward
The best method of removing a foreign body in the perianal region is by dilating the anus and then cutting the object in half. Alternatively, the object can be carefully dislodged from the anal canal by freeing one of the impacted ends. Care must be taken while removing the object to avoid accidental injury.
This procedure can be performed in any primary care setting that is adequately equipped for minor surgical services. Consider referral to a secondary care specialist based on the level of risk involved and the physician’s skills and training.
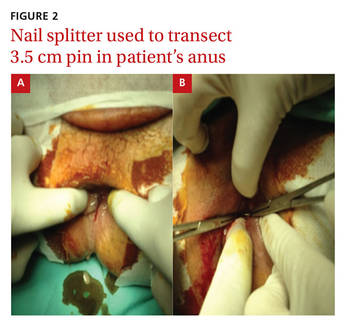
Our patient. After a complete assessment, we prepared the patient for gentle anal dilatation under intravenous conscious sedation. We carefully transected the 3.5 cm pin using a nail splitter (FIGURE 2). Because there was no abscess cavity, no other procedure was needed. We prescribed oral amoxicillin/clavulanic acid 500/125 mg every 8 hours for 7 days to treat a local infection.
After the procedure, we asked the patient about the pin. He said he had no idea how he could have ingested it and didn’t recall any abdominal pain during the previous month. Follow-up was normal, and he recovered without any complications.
THE TAKEAWAY
Consider the possibility of ingested sharp foreign bodies as a cause of severe anal pain, and perform a local and digital rectal examination. Radiography is recommended as an initial screening method. Following anal dilatation, the object can be removed by cutting it in half or by freeing one of the impacted ends.
1. Villalba H, Villalba S, Abbas MA. Anal fissure: a common cause of anal pain. Perm J. 2007;11:62-65.
2. Ambe P, Weber SA, Schauer M, et al. Swallowed foreign bodies in adults. Dtsch Arztebl Int. 2012;109:869-875.
3. Erbil B, Karaca MA, Aslaner MA, et al. Emergency admissions due to swallowed foreign bodies in adults. World J Gastroenterol. 2013;19:6447-6452.
4. Kuo CC, Jen TK, Wen CH, et al. Medical treatment for a fish bone-induced ileal micro-perforation: a case report. World J Gastroenterol. 2012;18:5994-5998.
5. Low VHS, Killius JS. Animal, vegetable, or mineral: A collection of abdominal and alimentary foreign bodies. Appl Radiol. 2000;29:23-30.
6. McCanse DE, Kurchin A, Hinshaw JR. Gastrointestinal foreign bodies. Am J Surg. 1981;142:335-337.
7. Goligher JC, Nixon HH, Duthie HL. Surgery of the anus, rectum and colon. 3rd ed. London: Baillière Tindall;1975:205-255.
8. Doublali M, Chouaib A, Elfassi MJ, et al. Perianal abscesses due to ingested foreign bodies. J Emerg Trauma Shock. 2010;3:395-397.
9. Coulier B, Tancredi MH, Ramboux A. Spiral CT and multidetector-row CT diagnosis of perforation of the small intestine caused by ingested foreign bodies. Eur Radiol. 2004;14:1918-1925.
THE CASE
An 80-year-old man sought medical advice over the phone because he’d had sharp anal pain for 4 days. The pain increased during sitting and defecation. He was told he most likely had an acute anal fissure and was instructed to treat it with sitz baths, a stool-bulking agent, and a topical anesthetic. Despite these treatments, he continued to have intense anal pain.
Two days later, he presented to our practice complaining of severe anal discomfort and perianal swelling that made it almost impossible to sit. The patient led an active lifestyle and was otherwise healthy. He did not recall any injury to the perianal area.
THE DIAGNOSIS
During examination, we noted tender, erythematous swelling over the right perianal region at the 9 o’clock position without fluctuance, discharge, or ulceration. A digital rectal examination revealed a foreign body lying transversely across the anus about 2.5 cm from the anal verge. A pelvic x-ray confirmed the presence of a foreign body—a pin (FIGURE 1).
DISCUSSION
Anal pain is a common symptom that is usually caused by hemorrhoids, fissures, fistulas, or abscesses.1 Anal pain is rarely reported to be secondary to the ingestion of sharp foreign bodies, which can produce problems in the lower gastrointestinal tract.
Foreign body ingestion is most common in children ages 6 months to 6 years.2 When it occurs in adults, it tends to involve those who are older, patients without teeth, prisoners, patients under the influence of drugs or alcohol, or those with intellectual disabilities or psychiatric disorders.2,3
The foreign bodies that adults most commonly unintentionally ingest are bones from fish or other animals.2,4 Most of these pass through the alimentary tract uneventfully within a week.2,5,6 Swallowed bones have been known to cause perianal abscesses and anal fistulae, which can cause extreme pain.7
The presence of foreign bodies is not always easy to spot
In our patient’s case, the diagnosis was made by a careful digital rectal examination; however, a foreign body in an abscess cavity can be missed during a digital exam.8 In our patient’s case, a pelvic x-ray confirmed the presence and location of the pin.
Radiography is recommended as an initial screening method and is especially useful for determining the location of radiodense foreign bodies.2 However, most swallowed foreign bodies, such as non-radiodense fish bones, wood, thorns, plastic, small aluminum objects, and glass, cannot be detected by this method.3 Non-radiodense foreign bodies can be identified using computed tomography scanning.9
Removal is typically straightforward
The best method of removing a foreign body in the perianal region is by dilating the anus and then cutting the object in half. Alternatively, the object can be carefully dislodged from the anal canal by freeing one of the impacted ends. Care must be taken while removing the object to avoid accidental injury.
This procedure can be performed in any primary care setting that is adequately equipped for minor surgical services. Consider referral to a secondary care specialist based on the level of risk involved and the physician’s skills and training.
Our patient. After a complete assessment, we prepared the patient for gentle anal dilatation under intravenous conscious sedation. We carefully transected the 3.5 cm pin using a nail splitter (FIGURE 2). Because there was no abscess cavity, no other procedure was needed. We prescribed oral amoxicillin/clavulanic acid 500/125 mg every 8 hours for 7 days to treat a local infection.
After the procedure, we asked the patient about the pin. He said he had no idea how he could have ingested it and didn’t recall any abdominal pain during the previous month. Follow-up was normal, and he recovered without any complications.
THE TAKEAWAY
Consider the possibility of ingested sharp foreign bodies as a cause of severe anal pain, and perform a local and digital rectal examination. Radiography is recommended as an initial screening method. Following anal dilatation, the object can be removed by cutting it in half or by freeing one of the impacted ends.
THE CASE
An 80-year-old man sought medical advice over the phone because he’d had sharp anal pain for 4 days. The pain increased during sitting and defecation. He was told he most likely had an acute anal fissure and was instructed to treat it with sitz baths, a stool-bulking agent, and a topical anesthetic. Despite these treatments, he continued to have intense anal pain.
Two days later, he presented to our practice complaining of severe anal discomfort and perianal swelling that made it almost impossible to sit. The patient led an active lifestyle and was otherwise healthy. He did not recall any injury to the perianal area.
THE DIAGNOSIS
During examination, we noted tender, erythematous swelling over the right perianal region at the 9 o’clock position without fluctuance, discharge, or ulceration. A digital rectal examination revealed a foreign body lying transversely across the anus about 2.5 cm from the anal verge. A pelvic x-ray confirmed the presence of a foreign body—a pin (FIGURE 1).
DISCUSSION
Anal pain is a common symptom that is usually caused by hemorrhoids, fissures, fistulas, or abscesses.1 Anal pain is rarely reported to be secondary to the ingestion of sharp foreign bodies, which can produce problems in the lower gastrointestinal tract.
Foreign body ingestion is most common in children ages 6 months to 6 years.2 When it occurs in adults, it tends to involve those who are older, patients without teeth, prisoners, patients under the influence of drugs or alcohol, or those with intellectual disabilities or psychiatric disorders.2,3
The foreign bodies that adults most commonly unintentionally ingest are bones from fish or other animals.2,4 Most of these pass through the alimentary tract uneventfully within a week.2,5,6 Swallowed bones have been known to cause perianal abscesses and anal fistulae, which can cause extreme pain.7
The presence of foreign bodies is not always easy to spot
In our patient’s case, the diagnosis was made by a careful digital rectal examination; however, a foreign body in an abscess cavity can be missed during a digital exam.8 In our patient’s case, a pelvic x-ray confirmed the presence and location of the pin.
Radiography is recommended as an initial screening method and is especially useful for determining the location of radiodense foreign bodies.2 However, most swallowed foreign bodies, such as non-radiodense fish bones, wood, thorns, plastic, small aluminum objects, and glass, cannot be detected by this method.3 Non-radiodense foreign bodies can be identified using computed tomography scanning.9
Removal is typically straightforward
The best method of removing a foreign body in the perianal region is by dilating the anus and then cutting the object in half. Alternatively, the object can be carefully dislodged from the anal canal by freeing one of the impacted ends. Care must be taken while removing the object to avoid accidental injury.
This procedure can be performed in any primary care setting that is adequately equipped for minor surgical services. Consider referral to a secondary care specialist based on the level of risk involved and the physician’s skills and training.
Our patient. After a complete assessment, we prepared the patient for gentle anal dilatation under intravenous conscious sedation. We carefully transected the 3.5 cm pin using a nail splitter (FIGURE 2). Because there was no abscess cavity, no other procedure was needed. We prescribed oral amoxicillin/clavulanic acid 500/125 mg every 8 hours for 7 days to treat a local infection.
After the procedure, we asked the patient about the pin. He said he had no idea how he could have ingested it and didn’t recall any abdominal pain during the previous month. Follow-up was normal, and he recovered without any complications.
THE TAKEAWAY
Consider the possibility of ingested sharp foreign bodies as a cause of severe anal pain, and perform a local and digital rectal examination. Radiography is recommended as an initial screening method. Following anal dilatation, the object can be removed by cutting it in half or by freeing one of the impacted ends.
1. Villalba H, Villalba S, Abbas MA. Anal fissure: a common cause of anal pain. Perm J. 2007;11:62-65.
2. Ambe P, Weber SA, Schauer M, et al. Swallowed foreign bodies in adults. Dtsch Arztebl Int. 2012;109:869-875.
3. Erbil B, Karaca MA, Aslaner MA, et al. Emergency admissions due to swallowed foreign bodies in adults. World J Gastroenterol. 2013;19:6447-6452.
4. Kuo CC, Jen TK, Wen CH, et al. Medical treatment for a fish bone-induced ileal micro-perforation: a case report. World J Gastroenterol. 2012;18:5994-5998.
5. Low VHS, Killius JS. Animal, vegetable, or mineral: A collection of abdominal and alimentary foreign bodies. Appl Radiol. 2000;29:23-30.
6. McCanse DE, Kurchin A, Hinshaw JR. Gastrointestinal foreign bodies. Am J Surg. 1981;142:335-337.
7. Goligher JC, Nixon HH, Duthie HL. Surgery of the anus, rectum and colon. 3rd ed. London: Baillière Tindall;1975:205-255.
8. Doublali M, Chouaib A, Elfassi MJ, et al. Perianal abscesses due to ingested foreign bodies. J Emerg Trauma Shock. 2010;3:395-397.
9. Coulier B, Tancredi MH, Ramboux A. Spiral CT and multidetector-row CT diagnosis of perforation of the small intestine caused by ingested foreign bodies. Eur Radiol. 2004;14:1918-1925.
1. Villalba H, Villalba S, Abbas MA. Anal fissure: a common cause of anal pain. Perm J. 2007;11:62-65.
2. Ambe P, Weber SA, Schauer M, et al. Swallowed foreign bodies in adults. Dtsch Arztebl Int. 2012;109:869-875.
3. Erbil B, Karaca MA, Aslaner MA, et al. Emergency admissions due to swallowed foreign bodies in adults. World J Gastroenterol. 2013;19:6447-6452.
4. Kuo CC, Jen TK, Wen CH, et al. Medical treatment for a fish bone-induced ileal micro-perforation: a case report. World J Gastroenterol. 2012;18:5994-5998.
5. Low VHS, Killius JS. Animal, vegetable, or mineral: A collection of abdominal and alimentary foreign bodies. Appl Radiol. 2000;29:23-30.
6. McCanse DE, Kurchin A, Hinshaw JR. Gastrointestinal foreign bodies. Am J Surg. 1981;142:335-337.
7. Goligher JC, Nixon HH, Duthie HL. Surgery of the anus, rectum and colon. 3rd ed. London: Baillière Tindall;1975:205-255.
8. Doublali M, Chouaib A, Elfassi MJ, et al. Perianal abscesses due to ingested foreign bodies. J Emerg Trauma Shock. 2010;3:395-397.
9. Coulier B, Tancredi MH, Ramboux A. Spiral CT and multidetector-row CT diagnosis of perforation of the small intestine caused by ingested foreign bodies. Eur Radiol. 2004;14:1918-1925.

Smoking cessation: What should you recommend?
› Prescribe varenicline, bupropion, or nicotine replacement as first-line single pharmacotherapy for smoking cessation. A
› Provide counseling along with medication, as the combination has proven to be more effective than either option alone. A
› Refer patients to their state Quit Line—a toll-free tobacco cessation coaching service that has been shown to be an effective form of counseling. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
In its 2014 report, “The Health Consequences of Smoking—50 Years of Progress,”1 the US Surgeon General concluded that, while significant improvements have been made since the publication of its landmark 1964 report, cigarette smoking remains a major public health problem. It is the leading cause of preventable death, increasing the risks of such common causes of mortality as cardiovascular disease, pulmonary disease, and malignancy. Cigarette smoking is responsible for an estimated 443,000 deaths annually.2
Overall, 42 million US adults and about 3 million middle and high school students smoke, despite the availability of an array of pharmacologic interventions to help them quit.1 Half of those who continue to smoke will die from a tobacco-related cause. Stopping before the age of 50 years cuts the risk in half, and quitting before age 30 almost completely negates it.3
The most recent comprehensive smoking cessation guideline, sponsored by the US Public Health Service, was published in 2008.4 The US Preventive Services Task Force (USPSTF) recommendation that “clinicians ask all adults about tobacco use and provide tobacco cessation interventions” for those who smoke was issued one year later.5 Since then, multiple studies have assessed the merits of the various medications, forms of nicotine replacement therapy (NRT), and counseling aimed at helping smokers maintain abstinence from tobacco.
This article reviews the guideline and provides family physicians with an evidence-based update.
The guideline: Treating tobacco use and dependence
Prescribing a first-line medication (bupropion SR, varenicline, nicotine gum, nicotine inhaler, nicotine lozenge, nicotine nasal spray, or nicotine patch) for every patient who seeks to quit smoking is a key component of the 2008 guideline (See TABLE W1).4 The only exceptions: patients for whom such agents are medically contraindicated and groups for which there is insufficient evidence of effectiveness, such as pregnant women and adolescents.

The use of any of these medications as a single agent nearly doubles the likelihood of success compared with placebo, with an average cessation rate of 25% (TABLE 1).4
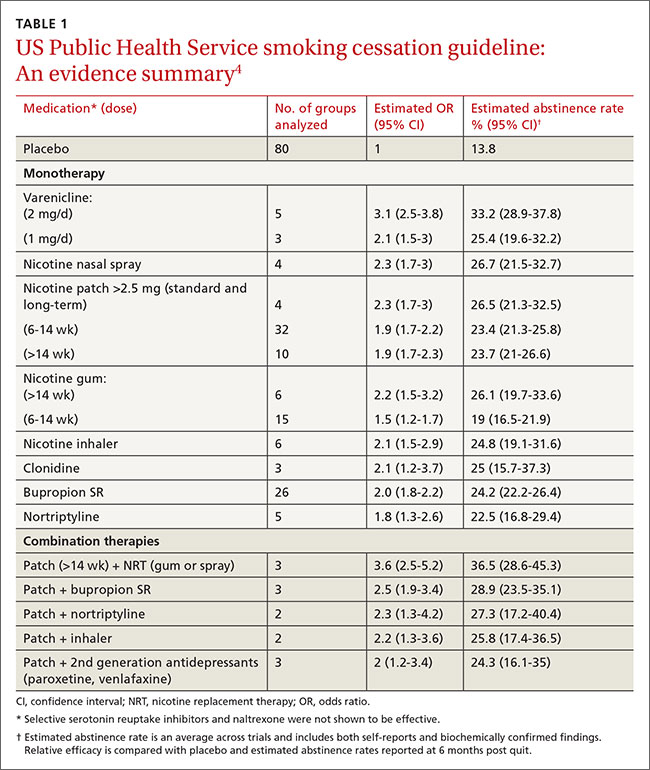
Combination therapy (pairing a nicotine patch and an additional agent) was found to be even more effective, with some combinations attaining success rates as high as 65%.4
Second-line therapies, including clonidine and nortriptyline, were also cited as effective, with an average cessation rate of 24%.4 Although the meta-analyses that these averages were based on did not include head-to-head comparisons, subsequent studies that also showed efficacy did include such comparisons.
Counseling is an essential component
In one of the meta-analyses on which the guideline was based, the combination of counseling and medication proved to be more effective than either intervention alone. Individual, group, and telephone counseling were all effective (odds ratio [OR]=1.7 [1.4-2.0], 1.3 [1.1-1.6], and 1.2 [1.1-1.4], respectively), provided they included practical help that emphasized problem solving and skills training, as well as social support. The benefits of a team-based approach were evident from the finding that counseling provided by more than one type of clinician had higher effect sizes (OR=2.5 [1.9-3.4] when 2 different clinical disciplines were involved and 2.4 [2.1-2.9] for 3 or more disciplines).4
The guideline also found state-sponsored quit lines, accessible at no charge via 800-QUIT-NOW, are an effective option. (For more information about this and other resources, see TABLE W2.)
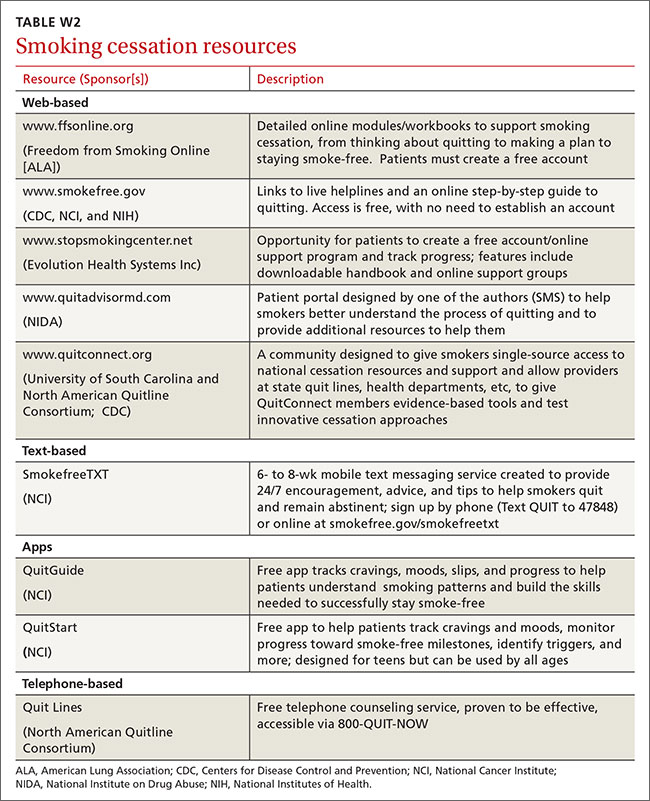
For patients who aren’t ready to stop smoking, the guideline recommends motivational interviewing4—a direct, patient-centered technique used to explore and work through ambivalence. Further information about this method is available at motivational interviewing.org/.
In counseling patients considering a quit attempt, it is important to present all options. A smoking history is needed, too, because factors such as the number of cigarettes smoked per day, how long a patient is typically awake before smoking the first cigarette of the day, and level of dependence are important factors in determining medication and dosage. Consider the advantages and disadvantages of the various medications (TABLE 2)4,6,7 or methods used for prior quit attempts and reasons for relapse, if appropriate; and patient preference.
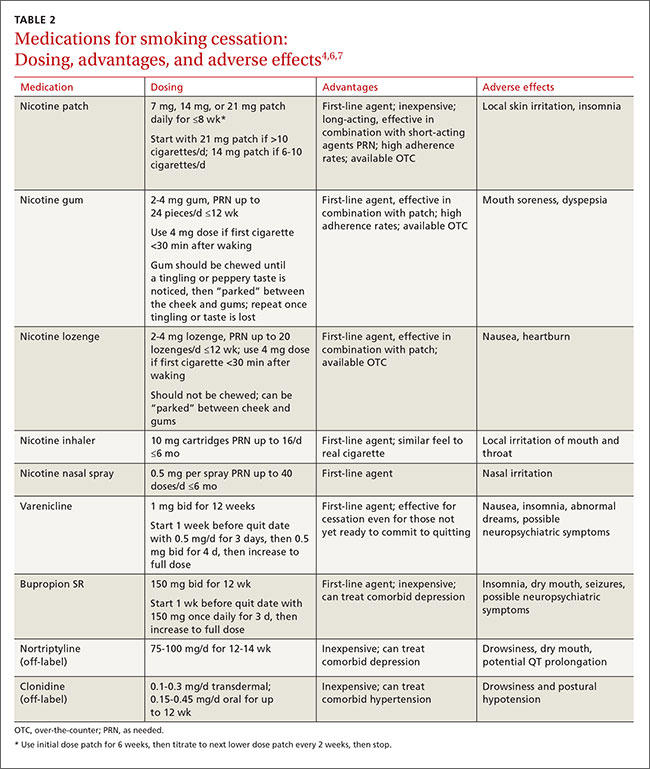
Evidence update: What's best?
Since 2009, many clinical trials have examined the best way to help smokers quit. Here’s a closer look at the latest evidence.
NRT boosts long-term cessation
A 2012 Cochrane review examined 150 trials and found that every type of NRT—gum, lozenge, patch, inhaler, and nasal spray—was associated with long-term cessation (relative risk [RR]=1.60; 95% CI, 1.53-1.68).8 This effect was essentially unchanged regardless of the duration, setting, or intensity of supportive therapy offered, and no single type of NRT was more effective than any other. However, combining a short-acting form like a lozenge with a long-acting patch was found to be more effective than either one alone (RR=1.34; 95% CI, 1.18-1.51).
Starting the NRT before the patient quit did not improve cessation rates over traditional start times (RR=1.18; 95% CI 0.98-1.41). Neither was there an added benefit to using NRT beyond the recommended 24-week prescription period,9 although doing so was found to be safe. Another 2012 Cochrane review looked specifically at the use of pharmacologic smoking cessation interventions during pregnancy and concluded that there was still not sufficient data to document efficacy for this patient population.10
Adherence. In deciding on which type of NRT to prescribe, it is important to consider not only patient preference and previous efforts, but also the latest evidence. A study comparing various NRT formulations found patient adherence to be highest with the patch, followed by nicotine gum, which had a higher compliance rate than either the nicotine inhaler or nasal spray.11
Varenicline is still a first-line agent
Since the 2008 guideline recommended this partial nicotinic receptor agonist/antagonist as a first-line pharmacologic agent, additional meta-analyses have confirmed its long-term efficacy in smokers who are ready to quit.12,13 A 2012 Cochrane review found varenicline to increase long-term cessation compared with placebo (RR=2.27; 95% CI, 2.02-2.55).13 It also showed varenicline to be more effective than bupropion SR (RR=1.52; 95% CI, 1.22-1.88), but about as effective as NRT (RR=1.13; 95% CI, 0.94-1.35).
Newer data suggest that varenicline may also be effective for those who are willing to cut down on smoking but not yet ready to give up cigarettes completely. Used for 24 weeks by those who were initially resistant to quitting, researchers found varenicline nearly tripled the cessation rate at 52 weeks compared with placebo (RR=2.7; 95% CI, 2.1-3.5).14

Latest evidence on safety. Additional concerns about the safety of varenicline have been raised, however, since the 2008 guideline was published. In 2009, the US Food and Drug Administration (FDA) required that black box warnings be added to the labels of both varenicline and bupropion SR based on post-marketing safety reports showing the risk of neuropsychiatric symptoms, including suicidality.15 In 2011, a large case control study by the FDA Adverse Event Reporting System also showed an increased risk of suicidality in patients taking these drugs.16
Follow-up studies, however, including a large prospective cohort study and a large meta-analysis, failed to show an increased association with neuropsychiatric adverse effects.17,18 A smaller randomized controlled trial (RCT) showed that in smokers diagnosed with schizophrenia and bipolar disorder, maintenance therapy with varenicline was effective in preventing smoking relapse for up to 52 weeks. Abstinence rates were 60% for those in the varenicline group vs 19% for those in the placebo group (OR=6.2; 95% CI, 2.2-19.2). Although no increased risk of adverse psychiatric events was found in this study, it was not powered to detect them.19 Also of note: a meta-analysis of 14 RCTs showed an increased rate of cardiovascular events associated with varenicline.20 There are concerns about methodologic flaws in this meta-analysis, however, and 2 subsequent meta-analyses failed to find a cardiovascular risk.21,22
The higher quality studies that have been published since the original concerns about varenicline's safety are reassuring, but it is still essential to carefully weigh the risks and benefits of varenicline. Review cardiac and psychiatric history and conduct a suicidality assessment before prescribing it as a smoking cessation aid, and provide close follow-up.
A closer look at antidepressants
Bupropion SR, an atypical antidepressant, was also listed as a first-line treatment in the 2008 guideline. A 2014 Cochrane review of 90 studies confirmed the evidence for this recommendation.6 Monotherapy with this agent was found to significantly increase rates of long-term cessation (RR=1.62; 95% CI, 1.49-1.76). No increased risk of serious adverse events was identified compared with placebo. As already noted, associations with neuropsychiatric symptoms were found, but this risk must be considered with any antidepressant.
Bupropion’s efficacy was not significantly different from that of NRT, but moderate evidence suggests that it is less effective than varenicline, (RR=0.68; 95% CI, 0.56-0.83). Other classes of antidepressants, including selective serotonin reuptake inhibitors, serotonin norepinephrine reuptake inhibitors, and monoamine oxidase inhibitors, were found to be ineffective for smoking cessation.6
Nortriptyline, a tricyclic antidepressant, was not significantly different from bupropion SR (RR, 1.30; 95% CI, 0.93-1.82) in efficacy for smoking cessation, but it lacks FDA approval for this purpose and is not considered a first-line agent.6
Second-line agents
Clonidine is an alpha-2 adrenergic receptor agonist that was originally used to treat hypertension but found to be effective for smoking cessation in a meta-analysis performed for the 2008 guideline.4 Like nortriptyline, however, clonidine is not FDA-approved for this purpose and is not considered a first-line agent.5 A 2013 Cochrane meta-analysis further showed that clonidine is effective for smoking cessation vs placebo (RR=1.63; 95% CI, 1.22-2.18),7 but suggested that its significant dose-related adverse effects, including postural hypotension and sedation, could limit its usefulness.
Combination therapies are highly effective
Evidence for various combinations of smoking cessation pharmacotherapy continues to mount.23-26 Perhaps the most compelling evidence comes from a comparative effectiveness trial that randomized 1346 patients in 12 primary care clinics to nicotine patches, nicotine lozenges, bupropion SR, a combination of patch plus lozenge, and bupropion SR plus lozenge. The 6-month abstinence rate was 30% for the bupropion SR plus lozenge combination, the most effective option. The combination was superior to either patch or bupropion SR monotherapy (OR, 0.56 and 0.54, respectively).23 Based on data from the 2008 guideline, similar combinations (eg, nicotine patch plus nicotine gum or bupropion SR plus the patch) are likely to be equally effective. The 2008 guideline also supports a nicotine patch and nicotine inhaler combination.
Another study found varenicline combined with the patch to be highly effective, with a 65% abstinence rate at 24 weeks vs 47% for varenicline alone (number needed to treat [NNT]=6; 95% CI, 4-11).24
In heavy smokers—defined as those who smoke ≥20 cigarettes daily—a varenicline and bupropion SR combination was more effective than varenicline alone (NNT= 9; 95% CI, 4.6-71.6), but the combination can lead to increased anxiety and depression.25 A smaller study found triple therapy using nicotine patch plus inhaler plus bupropion SR to be more effective than the nicotine patch alone (35% abstinence vs 19% abstinence at 26 weeks; NNT=6).26 Consider using these combinations in patients who have high nicotine dependency levels or have been unable to quit using a single first-line agent.
What role do e-cigarettes play?
The use of electronic cigarettes or “vapes”—battery-operated devices that deliver nicotine to the user through vapor—has increased significantly since their US introduction in 2007. A recent study found that “ever use” of e-cigarettes increased from 1.8% in 2010 to 13% in 2013; current use increased from 0.3% to 6.8% in the same time frame.27 “Vaping,” as inhaling on an e-cigarette is sometimes known, causes a sensor to detect airflow and initiate the heating element to vaporize the liquid solution within the cartridge, which contains propylene glycol, flavoring, and nicotine.
There is limited evidence of the efficacy of e-cigarettes for smoking cessation, but there is support for their role in reducing the quantity of conventional cigarettes smoked. A 2014 Cochrane review of 2 RCTs evaluating e-cigarette efficacy for smoking cessation or reduction found evidence of increased abstinence at 6 months in users of e-cigarettes containing nicotine compared with placebo e-cigarettes (9% vs 4%; RR=2.29; 95% CI, 1.05-4.96). Additionally, e-cigarette use was associated with >50% decrease in cigarette smoking vs placebo (36% vs 27%; RR=1.31; 95% CI, 1.02-1.68) or patch (61% vs 44%; RR=1.41; 95% CI, 1.20-1.67).28
A survey published after the review also showed a correlation between cigarette reduction (but not cessation) after one year of e-cigarette use.29 A subsequent RCT conducted in a controlled laboratory setting found that e-cigarettes were highly effective in reducing cessation-related cravings.30 And at 8-month follow-up, 44% of those using e-cigarettes were found to have at least a 50% reduction in the use of conventional cigarettes—and complete cessation in some cases.
Concerns about health effects
E-cigarettes have generally been thought to be safer than conventional cigarettes, given that they mainly deliver nicotine and propylene glycol instead of the more toxic chemicals—eg, benzene, carbon monoxide, and formaldehyde—released by conventional cigarettes.31 Carcinogens have also been found in e-cigarettes, but at significantly lower levels.31 However, a systematic review found wide variation in the toxin content of e-cigarettes.32 In addition, recent reports have detailed incidents in which e-cigarette devices were alleged to have exploded, causing severe bodily harm.33
Adverse effects of e-cigarettes include minor irritation of the throat, mouth, and lungs. Among cigarette-naive patients, light-headedness, throat irritation, dizziness, and cough were most commonly reported. No serious adverse events have been reported, but the lack of long-term safety data is a source of concern.32
Additionally, minimal regulatory oversight of the e-cigarette industry exists. Currently, the FDA only has authority to regulate e-cigarettes that are marketed for therapeutic purposes, although the agency is seeking to extend its oversight to all e-cigarettes.
The bottom line: More data on safety and regulatory oversight are needed before recommendations on the use of e-cigarettes as a smoking cessation tool can be made.
Looking ahead
Several novel pharmacotherapies have been evaluated for smoking cessation in recent years. Among them is a nicotine vaccine that several drug companies have been pursuing. In theory, such a vaccine would create an immunologic reaction to nicotine in a smoker, thereby preventing the substance from reaching the brain and providing rewarding stimuli. A 2008 Cochrane review of 4 trials assessing the efficacy of nicotine vaccines for tobacco cessation found that none showed efficacy.34
Naltrexone, an opioid antagonist, has shown efficacy in helping those with opioid or alcohol dependence achieve abstinence from these substances, raising the possibility that it might aid in smoking cessation, as well. A 2013 Cochrane review of 8 trials found that this was not the case: Compared with placebo, naltrexone was not beneficial when used alone (RR=1.00; 95% CI, 0.66-1.51) or as an adjunct to NRT compared with NRT alone (RR=0.95; 95% CI, 0.70-1.30).35
Cytisine, an extract from plants in the Faboideae family, has been used in Eastern Europe for decades for smoking cessation. It appears to work as a nicotine receptor partial agonist similar to varenicline. The extract does not have FDA approval, but the National Institutes of Health’s Center for Complementary and Integrative Health is sponsoring early-stage safety trials that could lead to its approval in the United States.36
A 2012 Cochrane review identified 2 recent RCTs evaluating cytisine and found it to be effective in increasing smoking cessation rates vs placebo (RR=3.98; 95% CI, 2.01-7.87).13
CORRESPONDENCE
Paul Bornemann, MD, 3209 Colonial Drive, Columbia, SC 29203; [email protected].
ACKNOWLEDGEMENT
The authors thank Matt Orr, PhD, and Kathryn E. Bornemann for their help with this manuscript.
1. National Center for Chronic Disease Prevention and Health Promotion Office on Smoking and Health. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General. 2014. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24455788. Accessed October 21, 2015.
2. Smoking-attributable mortality, years of potential life lost, and productivity losses—United States, 2000-2004. MMWR Morb Mortal Wkly Rep. 2008;57:1226-1228.
3. Doll R, Peto R, Boreham J, et al. Mortality in relation to smoking: 50 years’ observations on male British doctors. BMJ. 2004;328:1519.
4. US Public Health Service. A clinical practice guideline for treating tobacco use and dependence: 2008 update. A US Public Health Service Report. Am J Prev Med. 2008;35:158-176.
5. US Preventive Services Task Force. Tobacco use in adults and pregnant women: Counseling and interventions. April 2009. Available at: http://www.uspreventiveservicestaskforce.org/Page/Topic/recommendation-summary/tobacco-use-in-adults-and-pregnant-women-counseling-and-interventions. Accessed October 21, 2015.
6. Hughes JR, Stead LF, Hartmann-Boyce J, et al. Antidepressants for smoking cessation. Cochrane Database Syst Rev. 2014;(1):CD000031.
7. Cahill K, Stevens S, Perera R, et al. Pharmacological interventions for smoking cessation: an overview and network meta-analysis. Cochrane Database Syst Rev. 2013;(5):CD009329.
8. Stead LF, Perera R, Bullen C, et al. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2012;(11):CD000146.
9. Schnoll RA, Goelz PM, Veluz-Wilkins A, et al. Long-term nicotine replacement therapy: a randomized clinical trial. JAMA Intern Med. 2015;175:504-511.
10. Coleman T, Chamberlain C, Davey MA, et al. Pharmacological interventions for promoting smoking cessation during pregnancy. Cochrane Database Syst Rev. 2012;(9):CD010078.
11. Hajek P, West R, Foulds J, et al. Randomized comparative trial of nicotine polacrilex, a transdermal patch, nasal spray, and an inhaler. Arch Intern Med. 1999;159:2033-2038.
12. Eisenberg MJ, Filion KB, Yavin D, et al. Pharmacotherapies for smoking cessation: a meta-analysis of randomized controlled trials. CMAJ. 2008;179:135-144.
13. Cahill K, Stead LF, Lancaster T. Nicotine receptor partial agonists for smoking cessation. Cochrane Database Syst Rev. 2012;(4):CD006103.
14. Ebbert JO, Hughes JR, West RJ, et al. Effect of varenicline on smoking cessation through smoking reduction: a randomized clinical trial. JAMA. 2015;313:687-694.
15. US Food and Drug Administration. Reports of suicidality associated with use of varenicline (marketed as CHANTIX) and bupropion (marketed as ZYBAN and GENERICS). FDA Drug Safety News. 2009.
16. Moore TJ, Furberg CD, Glenmullen J, et al. Suicidal behavior and depression in smoking cessation treatments. PLoS One. 2011;6:e27016.
17. Thomas KH, Martin RM, Davies NM, et al. Smoking cessation treatment and risk of depression, suicide, and self harm in the Clinical Practice Research Datalink: prospective cohort study. BMJ. 2013;347:f5704.
18. Thomas KH, Martin RM, Knipe DW, et al. Risk of neuropsychiatric adverse events associated with varenicline: systematic review and meta-analysis. BMJ. 2015;350:h1109.
19. Evins AE, Cather C, Pratt SA, et al. Maintenance treatment with varenicline for smoking cessation in patients with schizophrenia and bipolar disorder: a randomized clinical trial. JAMA. 2014;311:145-154.
20. Singh S, Loke YK, Spangler JG, et al. Risk of serious adverse cardiovascular events associated with varenicline: a systematic review and meta-analysis. CMAJ. 2011;183:1359-1366.
21. Prochaska JJ, Hilton JF. Risk of cardiovascular serious adverse events associated with varenicline use for tobacco cessation: systematic review and meta-analysis. BMJ. 2012;344:e2856.
22. Svanström H, Pasternak B, Hviid A. Use of varenicline for smoking cessation and risk of serious cardiovascular events: nationwide cohort study. BMJ. 2012;345:e7176.
23. Smith SS, McCarthy DE, Japuntich SJ, et al. Comparative effectiveness of five smoking cessation pharmacotherapies in primary care clinics. Arch Intern Med. 2009;169:2148–2155.
24. Koegelenberg CFN, Noor F, Bateman ED, et al. Efficacy of varenicline combined with nicotine replacement therapy vs varenicline alone for smoking cessation. JAMA. 2014;312:155-161.
25. Ebbert JO, Hatsukami DK, Croghan IT, et al. Combination varenicline and bupropion SR for tobacco-dependence treatment in cigarette smokers: a randomized trial. JAMA. 2014;311:155-163.
26. Steinberg MB, Greenhaus S, Schmelzer AC, et al. Triple-combination pharmacotherapy for medically ill smokers: A randomized trial. Ann Intern Med. 2009;150:447-454.
27. McMillen RC, Gottlieb MA, Shaefer RMW, et al. Trends in electronic cigarette use among US. adults: use is increasing in both smokers and nonsmokers. Nicotine Tob Res. 2015;17:1195-1202.
28. McRobbie H, Bullen C, Hartmann-Boyce J, et al. Electronic cigarettes for smoking cessation and reduction. Cochrane Database Syst Rev. 2014;(12):CD010216.
29. Brose LS, Hitchman SC, Brown J, et al. Is the use of electronic cigarettes while smoking associated with smoking cessation attempts, cessation and reduced cigarette consumption? A survey with a 1-year follow-up. Addiction. 2015;110:1160-1168.
30. Adriaens K, Van Gucht D, Declerck P, et al. Effectiveness of the electronic cigarette: an eight-week Flemish study with six-month follow-up on smoking reduction, craving and experienced benefits and complaints. Int J Environ Res Public Health. 2014;11:11220-11248.
31. Goniewicz ML, Knysak J, Gawron M, et al. Levels of selected carcinogens and toxicants in vapour from electronic cigarettes. Tob Control. 2014;23:133-139.
32. Pisinger C, Døssing M. A systematic review of health effects of electronic cigarettes. Prev Med (Baltim). 2014;69C:248-260.
33. Bowerman M. Fla. man hospitalized after e-cigarette explodes in face. USA Today Network. October 29, 2015. Available at: http:// www.usatoday.com/story/news/nation-now/2015/10/29/fla-man-hospitalized-e-cigarette-explodes-face/74791722/. Accessed December 2, 2015.
34. Hatsukami D, Cahill K, Stead LF. Nicotine vaccines for smoking cessation. Cochrane Database Syst Rev. 2008;(2):CD007072.
35. David SP, Lancaster T, Stead LF, et al. Opioid antagonists for smoking cessation. Cochrane Database Syst Rev. 2013;(6):CD003086.
36. Frankel T. Pill that quashes tobacco urge found in plain sight. Washington Post. May 15, 2015. Available at: http://www.washingtonpost.com/business/economy/pill-promises-a-safercheaper-way-than-chantix-to-quit-smoking/2015/05/15/8ce5590c-f830-11e4-9030-b4732caefe81_story.html. Accessed August 3, 2015.
› Prescribe varenicline, bupropion, or nicotine replacement as first-line single pharmacotherapy for smoking cessation. A
› Provide counseling along with medication, as the combination has proven to be more effective than either option alone. A
› Refer patients to their state Quit Line—a toll-free tobacco cessation coaching service that has been shown to be an effective form of counseling. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
In its 2014 report, “The Health Consequences of Smoking—50 Years of Progress,”1 the US Surgeon General concluded that, while significant improvements have been made since the publication of its landmark 1964 report, cigarette smoking remains a major public health problem. It is the leading cause of preventable death, increasing the risks of such common causes of mortality as cardiovascular disease, pulmonary disease, and malignancy. Cigarette smoking is responsible for an estimated 443,000 deaths annually.2
Overall, 42 million US adults and about 3 million middle and high school students smoke, despite the availability of an array of pharmacologic interventions to help them quit.1 Half of those who continue to smoke will die from a tobacco-related cause. Stopping before the age of 50 years cuts the risk in half, and quitting before age 30 almost completely negates it.3
The most recent comprehensive smoking cessation guideline, sponsored by the US Public Health Service, was published in 2008.4 The US Preventive Services Task Force (USPSTF) recommendation that “clinicians ask all adults about tobacco use and provide tobacco cessation interventions” for those who smoke was issued one year later.5 Since then, multiple studies have assessed the merits of the various medications, forms of nicotine replacement therapy (NRT), and counseling aimed at helping smokers maintain abstinence from tobacco.
This article reviews the guideline and provides family physicians with an evidence-based update.
The guideline: Treating tobacco use and dependence
Prescribing a first-line medication (bupropion SR, varenicline, nicotine gum, nicotine inhaler, nicotine lozenge, nicotine nasal spray, or nicotine patch) for every patient who seeks to quit smoking is a key component of the 2008 guideline (See TABLE W1).4 The only exceptions: patients for whom such agents are medically contraindicated and groups for which there is insufficient evidence of effectiveness, such as pregnant women and adolescents.
The use of any of these medications as a single agent nearly doubles the likelihood of success compared with placebo, with an average cessation rate of 25% (TABLE 1).4
Combination therapy (pairing a nicotine patch and an additional agent) was found to be even more effective, with some combinations attaining success rates as high as 65%.4
Second-line therapies, including clonidine and nortriptyline, were also cited as effective, with an average cessation rate of 24%.4 Although the meta-analyses that these averages were based on did not include head-to-head comparisons, subsequent studies that also showed efficacy did include such comparisons.
Counseling is an essential component
In one of the meta-analyses on which the guideline was based, the combination of counseling and medication proved to be more effective than either intervention alone. Individual, group, and telephone counseling were all effective (odds ratio [OR]=1.7 [1.4-2.0], 1.3 [1.1-1.6], and 1.2 [1.1-1.4], respectively), provided they included practical help that emphasized problem solving and skills training, as well as social support. The benefits of a team-based approach were evident from the finding that counseling provided by more than one type of clinician had higher effect sizes (OR=2.5 [1.9-3.4] when 2 different clinical disciplines were involved and 2.4 [2.1-2.9] for 3 or more disciplines).4
The guideline also found state-sponsored quit lines, accessible at no charge via 800-QUIT-NOW, are an effective option. (For more information about this and other resources, see TABLE W2.)
For patients who aren’t ready to stop smoking, the guideline recommends motivational interviewing4—a direct, patient-centered technique used to explore and work through ambivalence. Further information about this method is available at motivational interviewing.org/.
In counseling patients considering a quit attempt, it is important to present all options. A smoking history is needed, too, because factors such as the number of cigarettes smoked per day, how long a patient is typically awake before smoking the first cigarette of the day, and level of dependence are important factors in determining medication and dosage. Consider the advantages and disadvantages of the various medications (TABLE 2)4,6,7 or methods used for prior quit attempts and reasons for relapse, if appropriate; and patient preference.
Evidence update: What's best?
Since 2009, many clinical trials have examined the best way to help smokers quit. Here’s a closer look at the latest evidence.
NRT boosts long-term cessation
A 2012 Cochrane review examined 150 trials and found that every type of NRT—gum, lozenge, patch, inhaler, and nasal spray—was associated with long-term cessation (relative risk [RR]=1.60; 95% CI, 1.53-1.68).8 This effect was essentially unchanged regardless of the duration, setting, or intensity of supportive therapy offered, and no single type of NRT was more effective than any other. However, combining a short-acting form like a lozenge with a long-acting patch was found to be more effective than either one alone (RR=1.34; 95% CI, 1.18-1.51).
Starting the NRT before the patient quit did not improve cessation rates over traditional start times (RR=1.18; 95% CI 0.98-1.41). Neither was there an added benefit to using NRT beyond the recommended 24-week prescription period,9 although doing so was found to be safe. Another 2012 Cochrane review looked specifically at the use of pharmacologic smoking cessation interventions during pregnancy and concluded that there was still not sufficient data to document efficacy for this patient population.10
Adherence. In deciding on which type of NRT to prescribe, it is important to consider not only patient preference and previous efforts, but also the latest evidence. A study comparing various NRT formulations found patient adherence to be highest with the patch, followed by nicotine gum, which had a higher compliance rate than either the nicotine inhaler or nasal spray.11
Varenicline is still a first-line agent
Since the 2008 guideline recommended this partial nicotinic receptor agonist/antagonist as a first-line pharmacologic agent, additional meta-analyses have confirmed its long-term efficacy in smokers who are ready to quit.12,13 A 2012 Cochrane review found varenicline to increase long-term cessation compared with placebo (RR=2.27; 95% CI, 2.02-2.55).13 It also showed varenicline to be more effective than bupropion SR (RR=1.52; 95% CI, 1.22-1.88), but about as effective as NRT (RR=1.13; 95% CI, 0.94-1.35).
Newer data suggest that varenicline may also be effective for those who are willing to cut down on smoking but not yet ready to give up cigarettes completely. Used for 24 weeks by those who were initially resistant to quitting, researchers found varenicline nearly tripled the cessation rate at 52 weeks compared with placebo (RR=2.7; 95% CI, 2.1-3.5).14
Latest evidence on safety. Additional concerns about the safety of varenicline have been raised, however, since the 2008 guideline was published. In 2009, the US Food and Drug Administration (FDA) required that black box warnings be added to the labels of both varenicline and bupropion SR based on post-marketing safety reports showing the risk of neuropsychiatric symptoms, including suicidality.15 In 2011, a large case control study by the FDA Adverse Event Reporting System also showed an increased risk of suicidality in patients taking these drugs.16
Follow-up studies, however, including a large prospective cohort study and a large meta-analysis, failed to show an increased association with neuropsychiatric adverse effects.17,18 A smaller randomized controlled trial (RCT) showed that in smokers diagnosed with schizophrenia and bipolar disorder, maintenance therapy with varenicline was effective in preventing smoking relapse for up to 52 weeks. Abstinence rates were 60% for those in the varenicline group vs 19% for those in the placebo group (OR=6.2; 95% CI, 2.2-19.2). Although no increased risk of adverse psychiatric events was found in this study, it was not powered to detect them.19 Also of note: a meta-analysis of 14 RCTs showed an increased rate of cardiovascular events associated with varenicline.20 There are concerns about methodologic flaws in this meta-analysis, however, and 2 subsequent meta-analyses failed to find a cardiovascular risk.21,22
The higher quality studies that have been published since the original concerns about varenicline's safety are reassuring, but it is still essential to carefully weigh the risks and benefits of varenicline. Review cardiac and psychiatric history and conduct a suicidality assessment before prescribing it as a smoking cessation aid, and provide close follow-up.
A closer look at antidepressants
Bupropion SR, an atypical antidepressant, was also listed as a first-line treatment in the 2008 guideline. A 2014 Cochrane review of 90 studies confirmed the evidence for this recommendation.6 Monotherapy with this agent was found to significantly increase rates of long-term cessation (RR=1.62; 95% CI, 1.49-1.76). No increased risk of serious adverse events was identified compared with placebo. As already noted, associations with neuropsychiatric symptoms were found, but this risk must be considered with any antidepressant.
Bupropion’s efficacy was not significantly different from that of NRT, but moderate evidence suggests that it is less effective than varenicline, (RR=0.68; 95% CI, 0.56-0.83). Other classes of antidepressants, including selective serotonin reuptake inhibitors, serotonin norepinephrine reuptake inhibitors, and monoamine oxidase inhibitors, were found to be ineffective for smoking cessation.6
Nortriptyline, a tricyclic antidepressant, was not significantly different from bupropion SR (RR, 1.30; 95% CI, 0.93-1.82) in efficacy for smoking cessation, but it lacks FDA approval for this purpose and is not considered a first-line agent.6
Second-line agents
Clonidine is an alpha-2 adrenergic receptor agonist that was originally used to treat hypertension but found to be effective for smoking cessation in a meta-analysis performed for the 2008 guideline.4 Like nortriptyline, however, clonidine is not FDA-approved for this purpose and is not considered a first-line agent.5 A 2013 Cochrane meta-analysis further showed that clonidine is effective for smoking cessation vs placebo (RR=1.63; 95% CI, 1.22-2.18),7 but suggested that its significant dose-related adverse effects, including postural hypotension and sedation, could limit its usefulness.
Combination therapies are highly effective
Evidence for various combinations of smoking cessation pharmacotherapy continues to mount.23-26 Perhaps the most compelling evidence comes from a comparative effectiveness trial that randomized 1346 patients in 12 primary care clinics to nicotine patches, nicotine lozenges, bupropion SR, a combination of patch plus lozenge, and bupropion SR plus lozenge. The 6-month abstinence rate was 30% for the bupropion SR plus lozenge combination, the most effective option. The combination was superior to either patch or bupropion SR monotherapy (OR, 0.56 and 0.54, respectively).23 Based on data from the 2008 guideline, similar combinations (eg, nicotine patch plus nicotine gum or bupropion SR plus the patch) are likely to be equally effective. The 2008 guideline also supports a nicotine patch and nicotine inhaler combination.
Another study found varenicline combined with the patch to be highly effective, with a 65% abstinence rate at 24 weeks vs 47% for varenicline alone (number needed to treat [NNT]=6; 95% CI, 4-11).24
In heavy smokers—defined as those who smoke ≥20 cigarettes daily—a varenicline and bupropion SR combination was more effective than varenicline alone (NNT= 9; 95% CI, 4.6-71.6), but the combination can lead to increased anxiety and depression.25 A smaller study found triple therapy using nicotine patch plus inhaler plus bupropion SR to be more effective than the nicotine patch alone (35% abstinence vs 19% abstinence at 26 weeks; NNT=6).26 Consider using these combinations in patients who have high nicotine dependency levels or have been unable to quit using a single first-line agent.
What role do e-cigarettes play?
The use of electronic cigarettes or “vapes”—battery-operated devices that deliver nicotine to the user through vapor—has increased significantly since their US introduction in 2007. A recent study found that “ever use” of e-cigarettes increased from 1.8% in 2010 to 13% in 2013; current use increased from 0.3% to 6.8% in the same time frame.27 “Vaping,” as inhaling on an e-cigarette is sometimes known, causes a sensor to detect airflow and initiate the heating element to vaporize the liquid solution within the cartridge, which contains propylene glycol, flavoring, and nicotine.
There is limited evidence of the efficacy of e-cigarettes for smoking cessation, but there is support for their role in reducing the quantity of conventional cigarettes smoked. A 2014 Cochrane review of 2 RCTs evaluating e-cigarette efficacy for smoking cessation or reduction found evidence of increased abstinence at 6 months in users of e-cigarettes containing nicotine compared with placebo e-cigarettes (9% vs 4%; RR=2.29; 95% CI, 1.05-4.96). Additionally, e-cigarette use was associated with >50% decrease in cigarette smoking vs placebo (36% vs 27%; RR=1.31; 95% CI, 1.02-1.68) or patch (61% vs 44%; RR=1.41; 95% CI, 1.20-1.67).28
A survey published after the review also showed a correlation between cigarette reduction (but not cessation) after one year of e-cigarette use.29 A subsequent RCT conducted in a controlled laboratory setting found that e-cigarettes were highly effective in reducing cessation-related cravings.30 And at 8-month follow-up, 44% of those using e-cigarettes were found to have at least a 50% reduction in the use of conventional cigarettes—and complete cessation in some cases.
Concerns about health effects
E-cigarettes have generally been thought to be safer than conventional cigarettes, given that they mainly deliver nicotine and propylene glycol instead of the more toxic chemicals—eg, benzene, carbon monoxide, and formaldehyde—released by conventional cigarettes.31 Carcinogens have also been found in e-cigarettes, but at significantly lower levels.31 However, a systematic review found wide variation in the toxin content of e-cigarettes.32 In addition, recent reports have detailed incidents in which e-cigarette devices were alleged to have exploded, causing severe bodily harm.33
Adverse effects of e-cigarettes include minor irritation of the throat, mouth, and lungs. Among cigarette-naive patients, light-headedness, throat irritation, dizziness, and cough were most commonly reported. No serious adverse events have been reported, but the lack of long-term safety data is a source of concern.32
Additionally, minimal regulatory oversight of the e-cigarette industry exists. Currently, the FDA only has authority to regulate e-cigarettes that are marketed for therapeutic purposes, although the agency is seeking to extend its oversight to all e-cigarettes.
The bottom line: More data on safety and regulatory oversight are needed before recommendations on the use of e-cigarettes as a smoking cessation tool can be made.
Looking ahead
Several novel pharmacotherapies have been evaluated for smoking cessation in recent years. Among them is a nicotine vaccine that several drug companies have been pursuing. In theory, such a vaccine would create an immunologic reaction to nicotine in a smoker, thereby preventing the substance from reaching the brain and providing rewarding stimuli. A 2008 Cochrane review of 4 trials assessing the efficacy of nicotine vaccines for tobacco cessation found that none showed efficacy.34
Naltrexone, an opioid antagonist, has shown efficacy in helping those with opioid or alcohol dependence achieve abstinence from these substances, raising the possibility that it might aid in smoking cessation, as well. A 2013 Cochrane review of 8 trials found that this was not the case: Compared with placebo, naltrexone was not beneficial when used alone (RR=1.00; 95% CI, 0.66-1.51) or as an adjunct to NRT compared with NRT alone (RR=0.95; 95% CI, 0.70-1.30).35
Cytisine, an extract from plants in the Faboideae family, has been used in Eastern Europe for decades for smoking cessation. It appears to work as a nicotine receptor partial agonist similar to varenicline. The extract does not have FDA approval, but the National Institutes of Health’s Center for Complementary and Integrative Health is sponsoring early-stage safety trials that could lead to its approval in the United States.36
A 2012 Cochrane review identified 2 recent RCTs evaluating cytisine and found it to be effective in increasing smoking cessation rates vs placebo (RR=3.98; 95% CI, 2.01-7.87).13
CORRESPONDENCE
Paul Bornemann, MD, 3209 Colonial Drive, Columbia, SC 29203; [email protected].
ACKNOWLEDGEMENT
The authors thank Matt Orr, PhD, and Kathryn E. Bornemann for their help with this manuscript.
› Prescribe varenicline, bupropion, or nicotine replacement as first-line single pharmacotherapy for smoking cessation. A
› Provide counseling along with medication, as the combination has proven to be more effective than either option alone. A
› Refer patients to their state Quit Line—a toll-free tobacco cessation coaching service that has been shown to be an effective form of counseling. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
In its 2014 report, “The Health Consequences of Smoking—50 Years of Progress,”1 the US Surgeon General concluded that, while significant improvements have been made since the publication of its landmark 1964 report, cigarette smoking remains a major public health problem. It is the leading cause of preventable death, increasing the risks of such common causes of mortality as cardiovascular disease, pulmonary disease, and malignancy. Cigarette smoking is responsible for an estimated 443,000 deaths annually.2
Overall, 42 million US adults and about 3 million middle and high school students smoke, despite the availability of an array of pharmacologic interventions to help them quit.1 Half of those who continue to smoke will die from a tobacco-related cause. Stopping before the age of 50 years cuts the risk in half, and quitting before age 30 almost completely negates it.3
The most recent comprehensive smoking cessation guideline, sponsored by the US Public Health Service, was published in 2008.4 The US Preventive Services Task Force (USPSTF) recommendation that “clinicians ask all adults about tobacco use and provide tobacco cessation interventions” for those who smoke was issued one year later.5 Since then, multiple studies have assessed the merits of the various medications, forms of nicotine replacement therapy (NRT), and counseling aimed at helping smokers maintain abstinence from tobacco.
This article reviews the guideline and provides family physicians with an evidence-based update.
The guideline: Treating tobacco use and dependence
Prescribing a first-line medication (bupropion SR, varenicline, nicotine gum, nicotine inhaler, nicotine lozenge, nicotine nasal spray, or nicotine patch) for every patient who seeks to quit smoking is a key component of the 2008 guideline (See TABLE W1).4 The only exceptions: patients for whom such agents are medically contraindicated and groups for which there is insufficient evidence of effectiveness, such as pregnant women and adolescents.
The use of any of these medications as a single agent nearly doubles the likelihood of success compared with placebo, with an average cessation rate of 25% (TABLE 1).4
Combination therapy (pairing a nicotine patch and an additional agent) was found to be even more effective, with some combinations attaining success rates as high as 65%.4
Second-line therapies, including clonidine and nortriptyline, were also cited as effective, with an average cessation rate of 24%.4 Although the meta-analyses that these averages were based on did not include head-to-head comparisons, subsequent studies that also showed efficacy did include such comparisons.
Counseling is an essential component
In one of the meta-analyses on which the guideline was based, the combination of counseling and medication proved to be more effective than either intervention alone. Individual, group, and telephone counseling were all effective (odds ratio [OR]=1.7 [1.4-2.0], 1.3 [1.1-1.6], and 1.2 [1.1-1.4], respectively), provided they included practical help that emphasized problem solving and skills training, as well as social support. The benefits of a team-based approach were evident from the finding that counseling provided by more than one type of clinician had higher effect sizes (OR=2.5 [1.9-3.4] when 2 different clinical disciplines were involved and 2.4 [2.1-2.9] for 3 or more disciplines).4
The guideline also found state-sponsored quit lines, accessible at no charge via 800-QUIT-NOW, are an effective option. (For more information about this and other resources, see TABLE W2.)
For patients who aren’t ready to stop smoking, the guideline recommends motivational interviewing4—a direct, patient-centered technique used to explore and work through ambivalence. Further information about this method is available at motivational interviewing.org/.
In counseling patients considering a quit attempt, it is important to present all options. A smoking history is needed, too, because factors such as the number of cigarettes smoked per day, how long a patient is typically awake before smoking the first cigarette of the day, and level of dependence are important factors in determining medication and dosage. Consider the advantages and disadvantages of the various medications (TABLE 2)4,6,7 or methods used for prior quit attempts and reasons for relapse, if appropriate; and patient preference.
Evidence update: What's best?
Since 2009, many clinical trials have examined the best way to help smokers quit. Here’s a closer look at the latest evidence.
NRT boosts long-term cessation
A 2012 Cochrane review examined 150 trials and found that every type of NRT—gum, lozenge, patch, inhaler, and nasal spray—was associated with long-term cessation (relative risk [RR]=1.60; 95% CI, 1.53-1.68).8 This effect was essentially unchanged regardless of the duration, setting, or intensity of supportive therapy offered, and no single type of NRT was more effective than any other. However, combining a short-acting form like a lozenge with a long-acting patch was found to be more effective than either one alone (RR=1.34; 95% CI, 1.18-1.51).
Starting the NRT before the patient quit did not improve cessation rates over traditional start times (RR=1.18; 95% CI 0.98-1.41). Neither was there an added benefit to using NRT beyond the recommended 24-week prescription period,9 although doing so was found to be safe. Another 2012 Cochrane review looked specifically at the use of pharmacologic smoking cessation interventions during pregnancy and concluded that there was still not sufficient data to document efficacy for this patient population.10
Adherence. In deciding on which type of NRT to prescribe, it is important to consider not only patient preference and previous efforts, but also the latest evidence. A study comparing various NRT formulations found patient adherence to be highest with the patch, followed by nicotine gum, which had a higher compliance rate than either the nicotine inhaler or nasal spray.11
Varenicline is still a first-line agent
Since the 2008 guideline recommended this partial nicotinic receptor agonist/antagonist as a first-line pharmacologic agent, additional meta-analyses have confirmed its long-term efficacy in smokers who are ready to quit.12,13 A 2012 Cochrane review found varenicline to increase long-term cessation compared with placebo (RR=2.27; 95% CI, 2.02-2.55).13 It also showed varenicline to be more effective than bupropion SR (RR=1.52; 95% CI, 1.22-1.88), but about as effective as NRT (RR=1.13; 95% CI, 0.94-1.35).
Newer data suggest that varenicline may also be effective for those who are willing to cut down on smoking but not yet ready to give up cigarettes completely. Used for 24 weeks by those who were initially resistant to quitting, researchers found varenicline nearly tripled the cessation rate at 52 weeks compared with placebo (RR=2.7; 95% CI, 2.1-3.5).14
Latest evidence on safety. Additional concerns about the safety of varenicline have been raised, however, since the 2008 guideline was published. In 2009, the US Food and Drug Administration (FDA) required that black box warnings be added to the labels of both varenicline and bupropion SR based on post-marketing safety reports showing the risk of neuropsychiatric symptoms, including suicidality.15 In 2011, a large case control study by the FDA Adverse Event Reporting System also showed an increased risk of suicidality in patients taking these drugs.16
Follow-up studies, however, including a large prospective cohort study and a large meta-analysis, failed to show an increased association with neuropsychiatric adverse effects.17,18 A smaller randomized controlled trial (RCT) showed that in smokers diagnosed with schizophrenia and bipolar disorder, maintenance therapy with varenicline was effective in preventing smoking relapse for up to 52 weeks. Abstinence rates were 60% for those in the varenicline group vs 19% for those in the placebo group (OR=6.2; 95% CI, 2.2-19.2). Although no increased risk of adverse psychiatric events was found in this study, it was not powered to detect them.19 Also of note: a meta-analysis of 14 RCTs showed an increased rate of cardiovascular events associated with varenicline.20 There are concerns about methodologic flaws in this meta-analysis, however, and 2 subsequent meta-analyses failed to find a cardiovascular risk.21,22
The higher quality studies that have been published since the original concerns about varenicline's safety are reassuring, but it is still essential to carefully weigh the risks and benefits of varenicline. Review cardiac and psychiatric history and conduct a suicidality assessment before prescribing it as a smoking cessation aid, and provide close follow-up.
A closer look at antidepressants
Bupropion SR, an atypical antidepressant, was also listed as a first-line treatment in the 2008 guideline. A 2014 Cochrane review of 90 studies confirmed the evidence for this recommendation.6 Monotherapy with this agent was found to significantly increase rates of long-term cessation (RR=1.62; 95% CI, 1.49-1.76). No increased risk of serious adverse events was identified compared with placebo. As already noted, associations with neuropsychiatric symptoms were found, but this risk must be considered with any antidepressant.
Bupropion’s efficacy was not significantly different from that of NRT, but moderate evidence suggests that it is less effective than varenicline, (RR=0.68; 95% CI, 0.56-0.83). Other classes of antidepressants, including selective serotonin reuptake inhibitors, serotonin norepinephrine reuptake inhibitors, and monoamine oxidase inhibitors, were found to be ineffective for smoking cessation.6
Nortriptyline, a tricyclic antidepressant, was not significantly different from bupropion SR (RR, 1.30; 95% CI, 0.93-1.82) in efficacy for smoking cessation, but it lacks FDA approval for this purpose and is not considered a first-line agent.6
Second-line agents
Clonidine is an alpha-2 adrenergic receptor agonist that was originally used to treat hypertension but found to be effective for smoking cessation in a meta-analysis performed for the 2008 guideline.4 Like nortriptyline, however, clonidine is not FDA-approved for this purpose and is not considered a first-line agent.5 A 2013 Cochrane meta-analysis further showed that clonidine is effective for smoking cessation vs placebo (RR=1.63; 95% CI, 1.22-2.18),7 but suggested that its significant dose-related adverse effects, including postural hypotension and sedation, could limit its usefulness.
Combination therapies are highly effective
Evidence for various combinations of smoking cessation pharmacotherapy continues to mount.23-26 Perhaps the most compelling evidence comes from a comparative effectiveness trial that randomized 1346 patients in 12 primary care clinics to nicotine patches, nicotine lozenges, bupropion SR, a combination of patch plus lozenge, and bupropion SR plus lozenge. The 6-month abstinence rate was 30% for the bupropion SR plus lozenge combination, the most effective option. The combination was superior to either patch or bupropion SR monotherapy (OR, 0.56 and 0.54, respectively).23 Based on data from the 2008 guideline, similar combinations (eg, nicotine patch plus nicotine gum or bupropion SR plus the patch) are likely to be equally effective. The 2008 guideline also supports a nicotine patch and nicotine inhaler combination.
Another study found varenicline combined with the patch to be highly effective, with a 65% abstinence rate at 24 weeks vs 47% for varenicline alone (number needed to treat [NNT]=6; 95% CI, 4-11).24
In heavy smokers—defined as those who smoke ≥20 cigarettes daily—a varenicline and bupropion SR combination was more effective than varenicline alone (NNT= 9; 95% CI, 4.6-71.6), but the combination can lead to increased anxiety and depression.25 A smaller study found triple therapy using nicotine patch plus inhaler plus bupropion SR to be more effective than the nicotine patch alone (35% abstinence vs 19% abstinence at 26 weeks; NNT=6).26 Consider using these combinations in patients who have high nicotine dependency levels or have been unable to quit using a single first-line agent.
What role do e-cigarettes play?
The use of electronic cigarettes or “vapes”—battery-operated devices that deliver nicotine to the user through vapor—has increased significantly since their US introduction in 2007. A recent study found that “ever use” of e-cigarettes increased from 1.8% in 2010 to 13% in 2013; current use increased from 0.3% to 6.8% in the same time frame.27 “Vaping,” as inhaling on an e-cigarette is sometimes known, causes a sensor to detect airflow and initiate the heating element to vaporize the liquid solution within the cartridge, which contains propylene glycol, flavoring, and nicotine.
There is limited evidence of the efficacy of e-cigarettes for smoking cessation, but there is support for their role in reducing the quantity of conventional cigarettes smoked. A 2014 Cochrane review of 2 RCTs evaluating e-cigarette efficacy for smoking cessation or reduction found evidence of increased abstinence at 6 months in users of e-cigarettes containing nicotine compared with placebo e-cigarettes (9% vs 4%; RR=2.29; 95% CI, 1.05-4.96). Additionally, e-cigarette use was associated with >50% decrease in cigarette smoking vs placebo (36% vs 27%; RR=1.31; 95% CI, 1.02-1.68) or patch (61% vs 44%; RR=1.41; 95% CI, 1.20-1.67).28
A survey published after the review also showed a correlation between cigarette reduction (but not cessation) after one year of e-cigarette use.29 A subsequent RCT conducted in a controlled laboratory setting found that e-cigarettes were highly effective in reducing cessation-related cravings.30 And at 8-month follow-up, 44% of those using e-cigarettes were found to have at least a 50% reduction in the use of conventional cigarettes—and complete cessation in some cases.
Concerns about health effects
E-cigarettes have generally been thought to be safer than conventional cigarettes, given that they mainly deliver nicotine and propylene glycol instead of the more toxic chemicals—eg, benzene, carbon monoxide, and formaldehyde—released by conventional cigarettes.31 Carcinogens have also been found in e-cigarettes, but at significantly lower levels.31 However, a systematic review found wide variation in the toxin content of e-cigarettes.32 In addition, recent reports have detailed incidents in which e-cigarette devices were alleged to have exploded, causing severe bodily harm.33
Adverse effects of e-cigarettes include minor irritation of the throat, mouth, and lungs. Among cigarette-naive patients, light-headedness, throat irritation, dizziness, and cough were most commonly reported. No serious adverse events have been reported, but the lack of long-term safety data is a source of concern.32
Additionally, minimal regulatory oversight of the e-cigarette industry exists. Currently, the FDA only has authority to regulate e-cigarettes that are marketed for therapeutic purposes, although the agency is seeking to extend its oversight to all e-cigarettes.
The bottom line: More data on safety and regulatory oversight are needed before recommendations on the use of e-cigarettes as a smoking cessation tool can be made.
Looking ahead
Several novel pharmacotherapies have been evaluated for smoking cessation in recent years. Among them is a nicotine vaccine that several drug companies have been pursuing. In theory, such a vaccine would create an immunologic reaction to nicotine in a smoker, thereby preventing the substance from reaching the brain and providing rewarding stimuli. A 2008 Cochrane review of 4 trials assessing the efficacy of nicotine vaccines for tobacco cessation found that none showed efficacy.34
Naltrexone, an opioid antagonist, has shown efficacy in helping those with opioid or alcohol dependence achieve abstinence from these substances, raising the possibility that it might aid in smoking cessation, as well. A 2013 Cochrane review of 8 trials found that this was not the case: Compared with placebo, naltrexone was not beneficial when used alone (RR=1.00; 95% CI, 0.66-1.51) or as an adjunct to NRT compared with NRT alone (RR=0.95; 95% CI, 0.70-1.30).35
Cytisine, an extract from plants in the Faboideae family, has been used in Eastern Europe for decades for smoking cessation. It appears to work as a nicotine receptor partial agonist similar to varenicline. The extract does not have FDA approval, but the National Institutes of Health’s Center for Complementary and Integrative Health is sponsoring early-stage safety trials that could lead to its approval in the United States.36
A 2012 Cochrane review identified 2 recent RCTs evaluating cytisine and found it to be effective in increasing smoking cessation rates vs placebo (RR=3.98; 95% CI, 2.01-7.87).13
CORRESPONDENCE
Paul Bornemann, MD, 3209 Colonial Drive, Columbia, SC 29203; [email protected].
ACKNOWLEDGEMENT
The authors thank Matt Orr, PhD, and Kathryn E. Bornemann for their help with this manuscript.
1. National Center for Chronic Disease Prevention and Health Promotion Office on Smoking and Health. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General. 2014. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24455788. Accessed October 21, 2015.
2. Smoking-attributable mortality, years of potential life lost, and productivity losses—United States, 2000-2004. MMWR Morb Mortal Wkly Rep. 2008;57:1226-1228.
3. Doll R, Peto R, Boreham J, et al. Mortality in relation to smoking: 50 years’ observations on male British doctors. BMJ. 2004;328:1519.
4. US Public Health Service. A clinical practice guideline for treating tobacco use and dependence: 2008 update. A US Public Health Service Report. Am J Prev Med. 2008;35:158-176.
5. US Preventive Services Task Force. Tobacco use in adults and pregnant women: Counseling and interventions. April 2009. Available at: http://www.uspreventiveservicestaskforce.org/Page/Topic/recommendation-summary/tobacco-use-in-adults-and-pregnant-women-counseling-and-interventions. Accessed October 21, 2015.
6. Hughes JR, Stead LF, Hartmann-Boyce J, et al. Antidepressants for smoking cessation. Cochrane Database Syst Rev. 2014;(1):CD000031.
7. Cahill K, Stevens S, Perera R, et al. Pharmacological interventions for smoking cessation: an overview and network meta-analysis. Cochrane Database Syst Rev. 2013;(5):CD009329.
8. Stead LF, Perera R, Bullen C, et al. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2012;(11):CD000146.
9. Schnoll RA, Goelz PM, Veluz-Wilkins A, et al. Long-term nicotine replacement therapy: a randomized clinical trial. JAMA Intern Med. 2015;175:504-511.
10. Coleman T, Chamberlain C, Davey MA, et al. Pharmacological interventions for promoting smoking cessation during pregnancy. Cochrane Database Syst Rev. 2012;(9):CD010078.
11. Hajek P, West R, Foulds J, et al. Randomized comparative trial of nicotine polacrilex, a transdermal patch, nasal spray, and an inhaler. Arch Intern Med. 1999;159:2033-2038.
12. Eisenberg MJ, Filion KB, Yavin D, et al. Pharmacotherapies for smoking cessation: a meta-analysis of randomized controlled trials. CMAJ. 2008;179:135-144.
13. Cahill K, Stead LF, Lancaster T. Nicotine receptor partial agonists for smoking cessation. Cochrane Database Syst Rev. 2012;(4):CD006103.
14. Ebbert JO, Hughes JR, West RJ, et al. Effect of varenicline on smoking cessation through smoking reduction: a randomized clinical trial. JAMA. 2015;313:687-694.
15. US Food and Drug Administration. Reports of suicidality associated with use of varenicline (marketed as CHANTIX) and bupropion (marketed as ZYBAN and GENERICS). FDA Drug Safety News. 2009.
16. Moore TJ, Furberg CD, Glenmullen J, et al. Suicidal behavior and depression in smoking cessation treatments. PLoS One. 2011;6:e27016.
17. Thomas KH, Martin RM, Davies NM, et al. Smoking cessation treatment and risk of depression, suicide, and self harm in the Clinical Practice Research Datalink: prospective cohort study. BMJ. 2013;347:f5704.
18. Thomas KH, Martin RM, Knipe DW, et al. Risk of neuropsychiatric adverse events associated with varenicline: systematic review and meta-analysis. BMJ. 2015;350:h1109.
19. Evins AE, Cather C, Pratt SA, et al. Maintenance treatment with varenicline for smoking cessation in patients with schizophrenia and bipolar disorder: a randomized clinical trial. JAMA. 2014;311:145-154.
20. Singh S, Loke YK, Spangler JG, et al. Risk of serious adverse cardiovascular events associated with varenicline: a systematic review and meta-analysis. CMAJ. 2011;183:1359-1366.
21. Prochaska JJ, Hilton JF. Risk of cardiovascular serious adverse events associated with varenicline use for tobacco cessation: systematic review and meta-analysis. BMJ. 2012;344:e2856.
22. Svanström H, Pasternak B, Hviid A. Use of varenicline for smoking cessation and risk of serious cardiovascular events: nationwide cohort study. BMJ. 2012;345:e7176.
23. Smith SS, McCarthy DE, Japuntich SJ, et al. Comparative effectiveness of five smoking cessation pharmacotherapies in primary care clinics. Arch Intern Med. 2009;169:2148–2155.
24. Koegelenberg CFN, Noor F, Bateman ED, et al. Efficacy of varenicline combined with nicotine replacement therapy vs varenicline alone for smoking cessation. JAMA. 2014;312:155-161.
25. Ebbert JO, Hatsukami DK, Croghan IT, et al. Combination varenicline and bupropion SR for tobacco-dependence treatment in cigarette smokers: a randomized trial. JAMA. 2014;311:155-163.
26. Steinberg MB, Greenhaus S, Schmelzer AC, et al. Triple-combination pharmacotherapy for medically ill smokers: A randomized trial. Ann Intern Med. 2009;150:447-454.
27. McMillen RC, Gottlieb MA, Shaefer RMW, et al. Trends in electronic cigarette use among US. adults: use is increasing in both smokers and nonsmokers. Nicotine Tob Res. 2015;17:1195-1202.
28. McRobbie H, Bullen C, Hartmann-Boyce J, et al. Electronic cigarettes for smoking cessation and reduction. Cochrane Database Syst Rev. 2014;(12):CD010216.
29. Brose LS, Hitchman SC, Brown J, et al. Is the use of electronic cigarettes while smoking associated with smoking cessation attempts, cessation and reduced cigarette consumption? A survey with a 1-year follow-up. Addiction. 2015;110:1160-1168.
30. Adriaens K, Van Gucht D, Declerck P, et al. Effectiveness of the electronic cigarette: an eight-week Flemish study with six-month follow-up on smoking reduction, craving and experienced benefits and complaints. Int J Environ Res Public Health. 2014;11:11220-11248.
31. Goniewicz ML, Knysak J, Gawron M, et al. Levels of selected carcinogens and toxicants in vapour from electronic cigarettes. Tob Control. 2014;23:133-139.
32. Pisinger C, Døssing M. A systematic review of health effects of electronic cigarettes. Prev Med (Baltim). 2014;69C:248-260.
33. Bowerman M. Fla. man hospitalized after e-cigarette explodes in face. USA Today Network. October 29, 2015. Available at: http:// www.usatoday.com/story/news/nation-now/2015/10/29/fla-man-hospitalized-e-cigarette-explodes-face/74791722/. Accessed December 2, 2015.
34. Hatsukami D, Cahill K, Stead LF. Nicotine vaccines for smoking cessation. Cochrane Database Syst Rev. 2008;(2):CD007072.
35. David SP, Lancaster T, Stead LF, et al. Opioid antagonists for smoking cessation. Cochrane Database Syst Rev. 2013;(6):CD003086.
36. Frankel T. Pill that quashes tobacco urge found in plain sight. Washington Post. May 15, 2015. Available at: http://www.washingtonpost.com/business/economy/pill-promises-a-safercheaper-way-than-chantix-to-quit-smoking/2015/05/15/8ce5590c-f830-11e4-9030-b4732caefe81_story.html. Accessed August 3, 2015.
1. National Center for Chronic Disease Prevention and Health Promotion Office on Smoking and Health. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General. 2014. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24455788. Accessed October 21, 2015.
2. Smoking-attributable mortality, years of potential life lost, and productivity losses—United States, 2000-2004. MMWR Morb Mortal Wkly Rep. 2008;57:1226-1228.
3. Doll R, Peto R, Boreham J, et al. Mortality in relation to smoking: 50 years’ observations on male British doctors. BMJ. 2004;328:1519.
4. US Public Health Service. A clinical practice guideline for treating tobacco use and dependence: 2008 update. A US Public Health Service Report. Am J Prev Med. 2008;35:158-176.
5. US Preventive Services Task Force. Tobacco use in adults and pregnant women: Counseling and interventions. April 2009. Available at: http://www.uspreventiveservicestaskforce.org/Page/Topic/recommendation-summary/tobacco-use-in-adults-and-pregnant-women-counseling-and-interventions. Accessed October 21, 2015.
6. Hughes JR, Stead LF, Hartmann-Boyce J, et al. Antidepressants for smoking cessation. Cochrane Database Syst Rev. 2014;(1):CD000031.
7. Cahill K, Stevens S, Perera R, et al. Pharmacological interventions for smoking cessation: an overview and network meta-analysis. Cochrane Database Syst Rev. 2013;(5):CD009329.
8. Stead LF, Perera R, Bullen C, et al. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2012;(11):CD000146.
9. Schnoll RA, Goelz PM, Veluz-Wilkins A, et al. Long-term nicotine replacement therapy: a randomized clinical trial. JAMA Intern Med. 2015;175:504-511.
10. Coleman T, Chamberlain C, Davey MA, et al. Pharmacological interventions for promoting smoking cessation during pregnancy. Cochrane Database Syst Rev. 2012;(9):CD010078.
11. Hajek P, West R, Foulds J, et al. Randomized comparative trial of nicotine polacrilex, a transdermal patch, nasal spray, and an inhaler. Arch Intern Med. 1999;159:2033-2038.
12. Eisenberg MJ, Filion KB, Yavin D, et al. Pharmacotherapies for smoking cessation: a meta-analysis of randomized controlled trials. CMAJ. 2008;179:135-144.
13. Cahill K, Stead LF, Lancaster T. Nicotine receptor partial agonists for smoking cessation. Cochrane Database Syst Rev. 2012;(4):CD006103.
14. Ebbert JO, Hughes JR, West RJ, et al. Effect of varenicline on smoking cessation through smoking reduction: a randomized clinical trial. JAMA. 2015;313:687-694.
15. US Food and Drug Administration. Reports of suicidality associated with use of varenicline (marketed as CHANTIX) and bupropion (marketed as ZYBAN and GENERICS). FDA Drug Safety News. 2009.
16. Moore TJ, Furberg CD, Glenmullen J, et al. Suicidal behavior and depression in smoking cessation treatments. PLoS One. 2011;6:e27016.
17. Thomas KH, Martin RM, Davies NM, et al. Smoking cessation treatment and risk of depression, suicide, and self harm in the Clinical Practice Research Datalink: prospective cohort study. BMJ. 2013;347:f5704.
18. Thomas KH, Martin RM, Knipe DW, et al. Risk of neuropsychiatric adverse events associated with varenicline: systematic review and meta-analysis. BMJ. 2015;350:h1109.
19. Evins AE, Cather C, Pratt SA, et al. Maintenance treatment with varenicline for smoking cessation in patients with schizophrenia and bipolar disorder: a randomized clinical trial. JAMA. 2014;311:145-154.
20. Singh S, Loke YK, Spangler JG, et al. Risk of serious adverse cardiovascular events associated with varenicline: a systematic review and meta-analysis. CMAJ. 2011;183:1359-1366.
21. Prochaska JJ, Hilton JF. Risk of cardiovascular serious adverse events associated with varenicline use for tobacco cessation: systematic review and meta-analysis. BMJ. 2012;344:e2856.
22. Svanström H, Pasternak B, Hviid A. Use of varenicline for smoking cessation and risk of serious cardiovascular events: nationwide cohort study. BMJ. 2012;345:e7176.
23. Smith SS, McCarthy DE, Japuntich SJ, et al. Comparative effectiveness of five smoking cessation pharmacotherapies in primary care clinics. Arch Intern Med. 2009;169:2148–2155.
24. Koegelenberg CFN, Noor F, Bateman ED, et al. Efficacy of varenicline combined with nicotine replacement therapy vs varenicline alone for smoking cessation. JAMA. 2014;312:155-161.
25. Ebbert JO, Hatsukami DK, Croghan IT, et al. Combination varenicline and bupropion SR for tobacco-dependence treatment in cigarette smokers: a randomized trial. JAMA. 2014;311:155-163.
26. Steinberg MB, Greenhaus S, Schmelzer AC, et al. Triple-combination pharmacotherapy for medically ill smokers: A randomized trial. Ann Intern Med. 2009;150:447-454.
27. McMillen RC, Gottlieb MA, Shaefer RMW, et al. Trends in electronic cigarette use among US. adults: use is increasing in both smokers and nonsmokers. Nicotine Tob Res. 2015;17:1195-1202.
28. McRobbie H, Bullen C, Hartmann-Boyce J, et al. Electronic cigarettes for smoking cessation and reduction. Cochrane Database Syst Rev. 2014;(12):CD010216.
29. Brose LS, Hitchman SC, Brown J, et al. Is the use of electronic cigarettes while smoking associated with smoking cessation attempts, cessation and reduced cigarette consumption? A survey with a 1-year follow-up. Addiction. 2015;110:1160-1168.
30. Adriaens K, Van Gucht D, Declerck P, et al. Effectiveness of the electronic cigarette: an eight-week Flemish study with six-month follow-up on smoking reduction, craving and experienced benefits and complaints. Int J Environ Res Public Health. 2014;11:11220-11248.
31. Goniewicz ML, Knysak J, Gawron M, et al. Levels of selected carcinogens and toxicants in vapour from electronic cigarettes. Tob Control. 2014;23:133-139.
32. Pisinger C, Døssing M. A systematic review of health effects of electronic cigarettes. Prev Med (Baltim). 2014;69C:248-260.
33. Bowerman M. Fla. man hospitalized after e-cigarette explodes in face. USA Today Network. October 29, 2015. Available at: http:// www.usatoday.com/story/news/nation-now/2015/10/29/fla-man-hospitalized-e-cigarette-explodes-face/74791722/. Accessed December 2, 2015.
34. Hatsukami D, Cahill K, Stead LF. Nicotine vaccines for smoking cessation. Cochrane Database Syst Rev. 2008;(2):CD007072.
35. David SP, Lancaster T, Stead LF, et al. Opioid antagonists for smoking cessation. Cochrane Database Syst Rev. 2013;(6):CD003086.
36. Frankel T. Pill that quashes tobacco urge found in plain sight. Washington Post. May 15, 2015. Available at: http://www.washingtonpost.com/business/economy/pill-promises-a-safercheaper-way-than-chantix-to-quit-smoking/2015/05/15/8ce5590c-f830-11e4-9030-b4732caefe81_story.html. Accessed August 3, 2015.

Botanical over-the-counter regimen reduces acne lesions
An over-the-counter botanical acne regimen outperformed a conventional acne regimen, with improved skin appearance and fewer lesions after 12 weeks of treatment, in a double-blind randomized controlled trial.
Eighty individuals aged 12 years and older with mild to moderate acne were randomized either to a three-step botanical-based acne treatment regimen (Receutics) consisting of a skin cleanser, breakout treatment, and tone and complexion corrector twice daily, or the currently marketed acne treatment kit, Proactiv (Guthy-Renker).

The botanical-based acne treatment contains a range of botanical ingredients, including algae and lentil seed extracts; cranberry seed, grape seed, and pumpkin seed oils; and allantoin; with 3.4% benzoyl peroxide as the active ingredient. The active ingredient in the cleanser is 2% salicylic acid, and niacinamide is the active ingredient in the tone and complexion corrector; both also contain botanical ingredients.
In the study, published in December, the investigator, Dr. Zoe Diana Draelos, reported that the botanical regimen achieved a significantly greater reduction in lesion count, in terms of closed comedones and inflammatory lesions, by week four, compared with the control treatment (J Drugs Dermatol. 2015 Dec; 14 [12]:1418-21).
This effect persisted at 12 weeks, with fewer closed comedones (P = .006). The botanical regimen also achieved greater reductions in pus, erythema, lesion height, and inflammation at weeks 2 and 4; although this difference disappeared by week 12. By week four and onwards, the botanical regimen also outperformed the conventional treatment on all blinded, investigator-assessed cosmetic appearance parameters, including skin tone, blemishes, erythema, and overall appearance.
While both treatments were effective at improving acne, Dr. Draelos, of Dermatology Consulting Services, High Point, N.C., said the botanical three-step regimen had the advantage of cosmetic ingredients such as emollients, anti-inflammatory/antioxidants, and “sensitive skin modulators.”
“This study demonstrates the value of combining monographed acne ingredients with advanced cosmeceutical technology,” she wrote.
The author received a grant from manufacturer Receutics to conduct the study.
An over-the-counter botanical acne regimen outperformed a conventional acne regimen, with improved skin appearance and fewer lesions after 12 weeks of treatment, in a double-blind randomized controlled trial.
Eighty individuals aged 12 years and older with mild to moderate acne were randomized either to a three-step botanical-based acne treatment regimen (Receutics) consisting of a skin cleanser, breakout treatment, and tone and complexion corrector twice daily, or the currently marketed acne treatment kit, Proactiv (Guthy-Renker).
The botanical-based acne treatment contains a range of botanical ingredients, including algae and lentil seed extracts; cranberry seed, grape seed, and pumpkin seed oils; and allantoin; with 3.4% benzoyl peroxide as the active ingredient. The active ingredient in the cleanser is 2% salicylic acid, and niacinamide is the active ingredient in the tone and complexion corrector; both also contain botanical ingredients.
In the study, published in December, the investigator, Dr. Zoe Diana Draelos, reported that the botanical regimen achieved a significantly greater reduction in lesion count, in terms of closed comedones and inflammatory lesions, by week four, compared with the control treatment (J Drugs Dermatol. 2015 Dec; 14 [12]:1418-21).
This effect persisted at 12 weeks, with fewer closed comedones (P = .006). The botanical regimen also achieved greater reductions in pus, erythema, lesion height, and inflammation at weeks 2 and 4; although this difference disappeared by week 12. By week four and onwards, the botanical regimen also outperformed the conventional treatment on all blinded, investigator-assessed cosmetic appearance parameters, including skin tone, blemishes, erythema, and overall appearance.
While both treatments were effective at improving acne, Dr. Draelos, of Dermatology Consulting Services, High Point, N.C., said the botanical three-step regimen had the advantage of cosmetic ingredients such as emollients, anti-inflammatory/antioxidants, and “sensitive skin modulators.”
“This study demonstrates the value of combining monographed acne ingredients with advanced cosmeceutical technology,” she wrote.
The author received a grant from manufacturer Receutics to conduct the study.
An over-the-counter botanical acne regimen outperformed a conventional acne regimen, with improved skin appearance and fewer lesions after 12 weeks of treatment, in a double-blind randomized controlled trial.
Eighty individuals aged 12 years and older with mild to moderate acne were randomized either to a three-step botanical-based acne treatment regimen (Receutics) consisting of a skin cleanser, breakout treatment, and tone and complexion corrector twice daily, or the currently marketed acne treatment kit, Proactiv (Guthy-Renker).
The botanical-based acne treatment contains a range of botanical ingredients, including algae and lentil seed extracts; cranberry seed, grape seed, and pumpkin seed oils; and allantoin; with 3.4% benzoyl peroxide as the active ingredient. The active ingredient in the cleanser is 2% salicylic acid, and niacinamide is the active ingredient in the tone and complexion corrector; both also contain botanical ingredients.
In the study, published in December, the investigator, Dr. Zoe Diana Draelos, reported that the botanical regimen achieved a significantly greater reduction in lesion count, in terms of closed comedones and inflammatory lesions, by week four, compared with the control treatment (J Drugs Dermatol. 2015 Dec; 14 [12]:1418-21).
This effect persisted at 12 weeks, with fewer closed comedones (P = .006). The botanical regimen also achieved greater reductions in pus, erythema, lesion height, and inflammation at weeks 2 and 4; although this difference disappeared by week 12. By week four and onwards, the botanical regimen also outperformed the conventional treatment on all blinded, investigator-assessed cosmetic appearance parameters, including skin tone, blemishes, erythema, and overall appearance.
While both treatments were effective at improving acne, Dr. Draelos, of Dermatology Consulting Services, High Point, N.C., said the botanical three-step regimen had the advantage of cosmetic ingredients such as emollients, anti-inflammatory/antioxidants, and “sensitive skin modulators.”
“This study demonstrates the value of combining monographed acne ingredients with advanced cosmeceutical technology,” she wrote.
The author received a grant from manufacturer Receutics to conduct the study.
FROM THE JOURNAL OF DRUGS IN DERMATOLOGY
Key clinical point: A botanical over-the-counter acne regimen achieved greater reductions in lesions than a currently marketed acne treatment.
Major finding: A three-step botanical-based acne regimen achieved significantly greater reduction in lesion counts and improvements in skin appearance than a conventional acne treatment.
Data source: A randomized, double-blind, controlled trial of 80 patients with mild to moderate acne.
Disclosures: The author received a grant from treatment manufacturer Receutics to conduct the study.

Breast cancer

Should You Bypass Anticoagulant “Bridging” Before and After Surgery?
PRACTICE CHANGER
Stop using low molecular weight heparin (LMWH) for surgical procedures to “bridge” low- to moderate-risk patients with atrial fibrillation (CHADS2 score ≤ 4) who are receiving warfarin. The risks outweigh the benefits.1
STRENGTH OF RECOMMENDATION
B: Based on a single good-quality randomized controlled trial.1
CASE A 75-year-old man comes to your office for surgical clearance before right knee replacement surgery. He has diabetes and high blood pressure and is taking warfarin for atrial fibrillation. He is scheduled for surgery in a week. What is the safest way to manage his warfarin in the perioperative period?
More than 2 million people are being treated with oral anticoagulation in North America to prevent stroke or to prevent or treat venous thromboembolism.2 Since 2010, several new oral anticoagulants have been approved, including dabigatran, apixaban, and rivaroxaban. These new medications have a shorter half-life than older anticoagulants, which enables them to be stopped one to two days before surgery.
On the other hand, warfarin—which remains a common choice for anticoagulation—has a three- to seven-day onset and elimination.3,4 This long clinical half-life presents a special challenge during the perioperative period. To reduce the risk for operative bleeding, the warfarin must be stopped days prior to the procedure, but clinicians often worry that this will increase the risk for arterial or venous thromboembolism, including stroke.
An estimated 250,000 patients need perioperative management of their anticoagulation each year.5 As the US population continues to age and the incidence of conditions requiring anticoagulation (particularly atrial fibrillation) increases, this number is only going to rise.6
Current guidelines on bridging. American College of Chest Physicians (ACCP) guidelines recommend transition to “a short-acting anticoagulant, consisting of subcutaneous low molecular weight heparin (LMWH) or intravenous unfractionated heparin, for a 10- to 12-day period during interruption of vitamin K antagonist (VKA) therapy.”5Furthermore, for an appropriate bridging regimen, the ACCP guidelines recommend stopping VKA therapy five days prior to the procedure and utilizing LMWH from within 24 to 48 hours of stopping VKA therapy until up to 24 hours before surgery.5 Postoperatively, VKA or LMWH therapy should be reinitiated within 24 hours and 24 to 72 hours, respectively, depending on the patient’s risk for bleeding during surgery.5
These guidelines recommend using CHADS2 scoring (see the table) to determine arterial thromboembolism (ATE) risk in atrial fibrillation.3,5 Patients at low risk for ATE (CHADS2 score, 0-2) should not be bridged, and patients at high risk (CHADS2 score, 5-6) should always be bridged.5 These guidelines are less clear about bridging recommendations for patients considered to be at moderate risk (CHADS2 score, 3-4).
Previous evidence on bridging. A 2012 meta-analysis of 34 studies evaluated the safety and efficacy of perioperative bridging with heparin in patients receiving VKA.7Researchers found no difference in ATE events in eight studies that compared groups that received bridging vs groups that simply stopped anticoagulation (odds ratio [OR], 0.80).7 The group that received bridging had an increased risk for overall bleeding in 13 studies and of major bleeding in five studies.7This meta-analysis was limited by poor study quality and variation in the indication for VKA therapy.
A 2015 subgroup analysis of a larger cohort study of patients receiving anticoagulants for atrial fibrillation found an increased risk for bleeding when their anticoagulation was interrupted for procedures (OR for major bleeding, 3.84).8
Douketis et al1 conducted a randomized trial to clarify the need for and safety of bridging anticoagulation for ATE in patients with atrial fibrillation who were receiving warfarin.
Continue for study summary >>
STUDY SUMMARY
When it comes to stroke/TIA, there’s no advantage to bridging
This double-blind, placebo-controlled trial compared bridging with dalteparin, a form of LMWH, to placebo among 1,884 patients with atrial fibrillation who were taking warfarin and whose anticoagulation therapy needed to be interrupted for an elective procedure. Patients were included if they were receiving warfarin to prevent stroke and had been taking it for at least 12 weeks, with a goal International Normalized Ratio (INR) of 2.0 to 3.0. Exclusion criteria included having a mechanical heart valve or having a stroke/transient ischemic attack (TIA; 12 weeks prior) or major bleeding (six weeks prior). Patients undergoing cardiac, intracranial, and intraspinal surgeries were also excluded from the study.
The mean CHADS2 score was 2.3; 38.3% of patients had a CHADS2 score ≥ 3, and 9.4% of patients had a history of stroke. Forty-four percent of patients underwent a gastrointestinal procedure, 17.2% underwent a cardiothoracic procedure, and 9.2% underwent an orthopedic procedure.
Patients stopped taking warfarin five days before their procedure and began subcutaneous dalteparin (100 IU/kg) or an identical placebo three days before the procedure. The dalteparin/placebo was stopped 24 hours before the procedure and restarted after the procedure, until the patient’s INR was in the therapeutic range. Warfarin was resumed on the evening of the procedure or the following day.
The primary efficacy outcome was ATE, including stroke, TIA, or systemic embolism. The primary safety endpoint was major bleeding (defined as bleeding at a critical anatomic site, symptomatic or clinically overt bleeding, or a decrease in hemoglobin > 2 g/dL). Secondary efficacy and safety outcomes included minor bleeding, acute myocardial infarction, deep vein thrombosis, pulmonary embolism, and death. Outcomes were assessed within 37 days of the procedure.
The incidence of ATE was 0.4% (four events) in the no-bridging group vs 0.3% (three events) in the bridging group. Major bleeding occurred in 1.3% of the no-bridging group (12 events) and in 3.2% of the bridging group (29 events), indicating that no bridging was superior in terms of the major bleeding outcome (number needed to harm [NNH], 53; relative risk [RR], 0.41).
The no-bridging group also had significantly fewer minor bleeds in comparison to the bridging group (NNH, 11; 12% vs 20.9%). There were no differences between groups in other secondary outcomes.
Continue for what's new >>
WHAT’S NEW
High-quality evidence suggests it’s OK to stop warfarin before surgery
This is the largest good-quality study to evaluate perioperative bridging in patients with atrial fibrillation who were at low or moderate risk for ATE (CHADS2 score, 0-4). Previous studies suggested bridging increased bleeding and offered limited benefit for reducing the risk for ATE. However, this is the first study to include a large group of moderate-risk patients (CHADS2 score, 3-4). This trial provides high-quality evidence to support the practice of simply stopping warfarin in the perioperative period, rather than bridging with LMWH.
CAVEATS
Findings might not apply to patients at highest risk
Most patients in this study had a CHADS2 score ≤ 3. About 3% had a CHADS2 score ≥ 5. It’s not clear whether these findings apply to patients with a CHADS2 score of 5 or 6.
This trial categorized ATE risk using the CHADS2 score, rather than the CHA2DS2-VASc, which includes additional risk factors and may more accurately predict stroke risk. Both patients who received bridging therapy and those who did not had a lower rate of stroke than predicted by CHADS2. This may reflect a limit of the predictive value of CHADS2 but should not have affected the rate of bleeding or ATE outcomes in this study.
Continue for challenges to implementation >>
CHALLENGES TO IMPLEMENTATION
Providers may hesitate to disregard current guidelines
Strokes are devastating events for patients, families, and clinicians, and they pose a greater risk for morbidity and mortality compared to bleeding. However, this study suggests patients who receive bridging have a higher risk for bleeding than stroke, which is in contrast to some providers’ experience and current recommendations.
A clinician caring for a patient who’s had a stroke may be inclined to recommend bridging despite the lack of efficacy and evidence of bleeding risk. Additionally, until guidelines reflect the most current research, clinicians may be reluctant to provide care in contrast to these recommendations.
REFERENCES
1. Douketis JD, Spyropoulos AC, Kaatz S, et al. Perioperative bridging anticoagulation in patients with atrial fibrillation. N Engl J Med. 2015;373:823-833.
2. Guyatt GH, Akl EA, Crowther M, et al; American College of Chest Physicians Antithrombotic Therapy and Prevention of Thrombosis Panel. Executive summary: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141:7S-47S.
3. Clark NP, Witt DM, Davies LE, et al. Bleeding, recurrent venous thromboembolism and mortality risks during warfarin interruption for invasive procedures. JAMA Intern Med. 2015; 175:1163-1168.
4. Lip GY, Lane DA. Stroke prevention in atrial fibrillation: a systematic review.JAMA. 2015;313:1950-1962.
5. Douketis JD, Spyropoulos AC, Spencer FA, et al; American College of Chest Physicians. Perioperative management of antithrombotic therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141:e326S-e350S.
6. Miyasaka Y, Barnes ME, Gersh BJ, et al. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence.Circulation. 2006;114:119-125.
7. Siegal D, Yudin J, Kaatz S, et al. Periprocedural heparin bridging in patients receiving vitamin K antagonists: systematic review and meta-analysis of bleeding and thromboembolic rates.Circulation. 2012;126:1630-1639.
8. Steinberg B, Peterson E, Kim S, et al; Outcomes Registry for Better Informed Treatment of Atrial Fibrillation Investigators and Patients. Use and outcomes associated with bridging during anticoagulation interruptions in patients with atrial fibrillation: findings from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF).Circulation. 2015;131:488-494.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2015. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2015;64(12):794-795, 800.
PRACTICE CHANGER
Stop using low molecular weight heparin (LMWH) for surgical procedures to “bridge” low- to moderate-risk patients with atrial fibrillation (CHADS2 score ≤ 4) who are receiving warfarin. The risks outweigh the benefits.1
STRENGTH OF RECOMMENDATION
B: Based on a single good-quality randomized controlled trial.1
CASE A 75-year-old man comes to your office for surgical clearance before right knee replacement surgery. He has diabetes and high blood pressure and is taking warfarin for atrial fibrillation. He is scheduled for surgery in a week. What is the safest way to manage his warfarin in the perioperative period?
More than 2 million people are being treated with oral anticoagulation in North America to prevent stroke or to prevent or treat venous thromboembolism.2 Since 2010, several new oral anticoagulants have been approved, including dabigatran, apixaban, and rivaroxaban. These new medications have a shorter half-life than older anticoagulants, which enables them to be stopped one to two days before surgery.
On the other hand, warfarin—which remains a common choice for anticoagulation—has a three- to seven-day onset and elimination.3,4 This long clinical half-life presents a special challenge during the perioperative period. To reduce the risk for operative bleeding, the warfarin must be stopped days prior to the procedure, but clinicians often worry that this will increase the risk for arterial or venous thromboembolism, including stroke.
An estimated 250,000 patients need perioperative management of their anticoagulation each year.5 As the US population continues to age and the incidence of conditions requiring anticoagulation (particularly atrial fibrillation) increases, this number is only going to rise.6
Current guidelines on bridging. American College of Chest Physicians (ACCP) guidelines recommend transition to “a short-acting anticoagulant, consisting of subcutaneous low molecular weight heparin (LMWH) or intravenous unfractionated heparin, for a 10- to 12-day period during interruption of vitamin K antagonist (VKA) therapy.”5Furthermore, for an appropriate bridging regimen, the ACCP guidelines recommend stopping VKA therapy five days prior to the procedure and utilizing LMWH from within 24 to 48 hours of stopping VKA therapy until up to 24 hours before surgery.5 Postoperatively, VKA or LMWH therapy should be reinitiated within 24 hours and 24 to 72 hours, respectively, depending on the patient’s risk for bleeding during surgery.5
These guidelines recommend using CHADS2 scoring (see the table) to determine arterial thromboembolism (ATE) risk in atrial fibrillation.3,5 Patients at low risk for ATE (CHADS2 score, 0-2) should not be bridged, and patients at high risk (CHADS2 score, 5-6) should always be bridged.5 These guidelines are less clear about bridging recommendations for patients considered to be at moderate risk (CHADS2 score, 3-4).
Previous evidence on bridging. A 2012 meta-analysis of 34 studies evaluated the safety and efficacy of perioperative bridging with heparin in patients receiving VKA.7Researchers found no difference in ATE events in eight studies that compared groups that received bridging vs groups that simply stopped anticoagulation (odds ratio [OR], 0.80).7 The group that received bridging had an increased risk for overall bleeding in 13 studies and of major bleeding in five studies.7This meta-analysis was limited by poor study quality and variation in the indication for VKA therapy.
A 2015 subgroup analysis of a larger cohort study of patients receiving anticoagulants for atrial fibrillation found an increased risk for bleeding when their anticoagulation was interrupted for procedures (OR for major bleeding, 3.84).8
Douketis et al1 conducted a randomized trial to clarify the need for and safety of bridging anticoagulation for ATE in patients with atrial fibrillation who were receiving warfarin.
Continue for study summary >>
STUDY SUMMARY
When it comes to stroke/TIA, there’s no advantage to bridging
This double-blind, placebo-controlled trial compared bridging with dalteparin, a form of LMWH, to placebo among 1,884 patients with atrial fibrillation who were taking warfarin and whose anticoagulation therapy needed to be interrupted for an elective procedure. Patients were included if they were receiving warfarin to prevent stroke and had been taking it for at least 12 weeks, with a goal International Normalized Ratio (INR) of 2.0 to 3.0. Exclusion criteria included having a mechanical heart valve or having a stroke/transient ischemic attack (TIA; 12 weeks prior) or major bleeding (six weeks prior). Patients undergoing cardiac, intracranial, and intraspinal surgeries were also excluded from the study.
The mean CHADS2 score was 2.3; 38.3% of patients had a CHADS2 score ≥ 3, and 9.4% of patients had a history of stroke. Forty-four percent of patients underwent a gastrointestinal procedure, 17.2% underwent a cardiothoracic procedure, and 9.2% underwent an orthopedic procedure.
Patients stopped taking warfarin five days before their procedure and began subcutaneous dalteparin (100 IU/kg) or an identical placebo three days before the procedure. The dalteparin/placebo was stopped 24 hours before the procedure and restarted after the procedure, until the patient’s INR was in the therapeutic range. Warfarin was resumed on the evening of the procedure or the following day.
The primary efficacy outcome was ATE, including stroke, TIA, or systemic embolism. The primary safety endpoint was major bleeding (defined as bleeding at a critical anatomic site, symptomatic or clinically overt bleeding, or a decrease in hemoglobin > 2 g/dL). Secondary efficacy and safety outcomes included minor bleeding, acute myocardial infarction, deep vein thrombosis, pulmonary embolism, and death. Outcomes were assessed within 37 days of the procedure.
The incidence of ATE was 0.4% (four events) in the no-bridging group vs 0.3% (three events) in the bridging group. Major bleeding occurred in 1.3% of the no-bridging group (12 events) and in 3.2% of the bridging group (29 events), indicating that no bridging was superior in terms of the major bleeding outcome (number needed to harm [NNH], 53; relative risk [RR], 0.41).
The no-bridging group also had significantly fewer minor bleeds in comparison to the bridging group (NNH, 11; 12% vs 20.9%). There were no differences between groups in other secondary outcomes.
Continue for what's new >>
WHAT’S NEW
High-quality evidence suggests it’s OK to stop warfarin before surgery
This is the largest good-quality study to evaluate perioperative bridging in patients with atrial fibrillation who were at low or moderate risk for ATE (CHADS2 score, 0-4). Previous studies suggested bridging increased bleeding and offered limited benefit for reducing the risk for ATE. However, this is the first study to include a large group of moderate-risk patients (CHADS2 score, 3-4). This trial provides high-quality evidence to support the practice of simply stopping warfarin in the perioperative period, rather than bridging with LMWH.
CAVEATS
Findings might not apply to patients at highest risk
Most patients in this study had a CHADS2 score ≤ 3. About 3% had a CHADS2 score ≥ 5. It’s not clear whether these findings apply to patients with a CHADS2 score of 5 or 6.
This trial categorized ATE risk using the CHADS2 score, rather than the CHA2DS2-VASc, which includes additional risk factors and may more accurately predict stroke risk. Both patients who received bridging therapy and those who did not had a lower rate of stroke than predicted by CHADS2. This may reflect a limit of the predictive value of CHADS2 but should not have affected the rate of bleeding or ATE outcomes in this study.
Continue for challenges to implementation >>
CHALLENGES TO IMPLEMENTATION
Providers may hesitate to disregard current guidelines
Strokes are devastating events for patients, families, and clinicians, and they pose a greater risk for morbidity and mortality compared to bleeding. However, this study suggests patients who receive bridging have a higher risk for bleeding than stroke, which is in contrast to some providers’ experience and current recommendations.
A clinician caring for a patient who’s had a stroke may be inclined to recommend bridging despite the lack of efficacy and evidence of bleeding risk. Additionally, until guidelines reflect the most current research, clinicians may be reluctant to provide care in contrast to these recommendations.
REFERENCES
1. Douketis JD, Spyropoulos AC, Kaatz S, et al. Perioperative bridging anticoagulation in patients with atrial fibrillation. N Engl J Med. 2015;373:823-833.
2. Guyatt GH, Akl EA, Crowther M, et al; American College of Chest Physicians Antithrombotic Therapy and Prevention of Thrombosis Panel. Executive summary: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141:7S-47S.
3. Clark NP, Witt DM, Davies LE, et al. Bleeding, recurrent venous thromboembolism and mortality risks during warfarin interruption for invasive procedures. JAMA Intern Med. 2015; 175:1163-1168.
4. Lip GY, Lane DA. Stroke prevention in atrial fibrillation: a systematic review.JAMA. 2015;313:1950-1962.
5. Douketis JD, Spyropoulos AC, Spencer FA, et al; American College of Chest Physicians. Perioperative management of antithrombotic therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141:e326S-e350S.
6. Miyasaka Y, Barnes ME, Gersh BJ, et al. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence.Circulation. 2006;114:119-125.
7. Siegal D, Yudin J, Kaatz S, et al. Periprocedural heparin bridging in patients receiving vitamin K antagonists: systematic review and meta-analysis of bleeding and thromboembolic rates.Circulation. 2012;126:1630-1639.
8. Steinberg B, Peterson E, Kim S, et al; Outcomes Registry for Better Informed Treatment of Atrial Fibrillation Investigators and Patients. Use and outcomes associated with bridging during anticoagulation interruptions in patients with atrial fibrillation: findings from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF).Circulation. 2015;131:488-494.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2015. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2015;64(12):794-795, 800.
PRACTICE CHANGER
Stop using low molecular weight heparin (LMWH) for surgical procedures to “bridge” low- to moderate-risk patients with atrial fibrillation (CHADS2 score ≤ 4) who are receiving warfarin. The risks outweigh the benefits.1
STRENGTH OF RECOMMENDATION
B: Based on a single good-quality randomized controlled trial.1
CASE A 75-year-old man comes to your office for surgical clearance before right knee replacement surgery. He has diabetes and high blood pressure and is taking warfarin for atrial fibrillation. He is scheduled for surgery in a week. What is the safest way to manage his warfarin in the perioperative period?
More than 2 million people are being treated with oral anticoagulation in North America to prevent stroke or to prevent or treat venous thromboembolism.2 Since 2010, several new oral anticoagulants have been approved, including dabigatran, apixaban, and rivaroxaban. These new medications have a shorter half-life than older anticoagulants, which enables them to be stopped one to two days before surgery.
On the other hand, warfarin—which remains a common choice for anticoagulation—has a three- to seven-day onset and elimination.3,4 This long clinical half-life presents a special challenge during the perioperative period. To reduce the risk for operative bleeding, the warfarin must be stopped days prior to the procedure, but clinicians often worry that this will increase the risk for arterial or venous thromboembolism, including stroke.
An estimated 250,000 patients need perioperative management of their anticoagulation each year.5 As the US population continues to age and the incidence of conditions requiring anticoagulation (particularly atrial fibrillation) increases, this number is only going to rise.6
Current guidelines on bridging. American College of Chest Physicians (ACCP) guidelines recommend transition to “a short-acting anticoagulant, consisting of subcutaneous low molecular weight heparin (LMWH) or intravenous unfractionated heparin, for a 10- to 12-day period during interruption of vitamin K antagonist (VKA) therapy.”5Furthermore, for an appropriate bridging regimen, the ACCP guidelines recommend stopping VKA therapy five days prior to the procedure and utilizing LMWH from within 24 to 48 hours of stopping VKA therapy until up to 24 hours before surgery.5 Postoperatively, VKA or LMWH therapy should be reinitiated within 24 hours and 24 to 72 hours, respectively, depending on the patient’s risk for bleeding during surgery.5
These guidelines recommend using CHADS2 scoring (see the table) to determine arterial thromboembolism (ATE) risk in atrial fibrillation.3,5 Patients at low risk for ATE (CHADS2 score, 0-2) should not be bridged, and patients at high risk (CHADS2 score, 5-6) should always be bridged.5 These guidelines are less clear about bridging recommendations for patients considered to be at moderate risk (CHADS2 score, 3-4).
Previous evidence on bridging. A 2012 meta-analysis of 34 studies evaluated the safety and efficacy of perioperative bridging with heparin in patients receiving VKA.7Researchers found no difference in ATE events in eight studies that compared groups that received bridging vs groups that simply stopped anticoagulation (odds ratio [OR], 0.80).7 The group that received bridging had an increased risk for overall bleeding in 13 studies and of major bleeding in five studies.7This meta-analysis was limited by poor study quality and variation in the indication for VKA therapy.
A 2015 subgroup analysis of a larger cohort study of patients receiving anticoagulants for atrial fibrillation found an increased risk for bleeding when their anticoagulation was interrupted for procedures (OR for major bleeding, 3.84).8
Douketis et al1 conducted a randomized trial to clarify the need for and safety of bridging anticoagulation for ATE in patients with atrial fibrillation who were receiving warfarin.
Continue for study summary >>
STUDY SUMMARY
When it comes to stroke/TIA, there’s no advantage to bridging
This double-blind, placebo-controlled trial compared bridging with dalteparin, a form of LMWH, to placebo among 1,884 patients with atrial fibrillation who were taking warfarin and whose anticoagulation therapy needed to be interrupted for an elective procedure. Patients were included if they were receiving warfarin to prevent stroke and had been taking it for at least 12 weeks, with a goal International Normalized Ratio (INR) of 2.0 to 3.0. Exclusion criteria included having a mechanical heart valve or having a stroke/transient ischemic attack (TIA; 12 weeks prior) or major bleeding (six weeks prior). Patients undergoing cardiac, intracranial, and intraspinal surgeries were also excluded from the study.
The mean CHADS2 score was 2.3; 38.3% of patients had a CHADS2 score ≥ 3, and 9.4% of patients had a history of stroke. Forty-four percent of patients underwent a gastrointestinal procedure, 17.2% underwent a cardiothoracic procedure, and 9.2% underwent an orthopedic procedure.
Patients stopped taking warfarin five days before their procedure and began subcutaneous dalteparin (100 IU/kg) or an identical placebo three days before the procedure. The dalteparin/placebo was stopped 24 hours before the procedure and restarted after the procedure, until the patient’s INR was in the therapeutic range. Warfarin was resumed on the evening of the procedure or the following day.
The primary efficacy outcome was ATE, including stroke, TIA, or systemic embolism. The primary safety endpoint was major bleeding (defined as bleeding at a critical anatomic site, symptomatic or clinically overt bleeding, or a decrease in hemoglobin > 2 g/dL). Secondary efficacy and safety outcomes included minor bleeding, acute myocardial infarction, deep vein thrombosis, pulmonary embolism, and death. Outcomes were assessed within 37 days of the procedure.
The incidence of ATE was 0.4% (four events) in the no-bridging group vs 0.3% (three events) in the bridging group. Major bleeding occurred in 1.3% of the no-bridging group (12 events) and in 3.2% of the bridging group (29 events), indicating that no bridging was superior in terms of the major bleeding outcome (number needed to harm [NNH], 53; relative risk [RR], 0.41).
The no-bridging group also had significantly fewer minor bleeds in comparison to the bridging group (NNH, 11; 12% vs 20.9%). There were no differences between groups in other secondary outcomes.
Continue for what's new >>
WHAT’S NEW
High-quality evidence suggests it’s OK to stop warfarin before surgery
This is the largest good-quality study to evaluate perioperative bridging in patients with atrial fibrillation who were at low or moderate risk for ATE (CHADS2 score, 0-4). Previous studies suggested bridging increased bleeding and offered limited benefit for reducing the risk for ATE. However, this is the first study to include a large group of moderate-risk patients (CHADS2 score, 3-4). This trial provides high-quality evidence to support the practice of simply stopping warfarin in the perioperative period, rather than bridging with LMWH.
CAVEATS
Findings might not apply to patients at highest risk
Most patients in this study had a CHADS2 score ≤ 3. About 3% had a CHADS2 score ≥ 5. It’s not clear whether these findings apply to patients with a CHADS2 score of 5 or 6.
This trial categorized ATE risk using the CHADS2 score, rather than the CHA2DS2-VASc, which includes additional risk factors and may more accurately predict stroke risk. Both patients who received bridging therapy and those who did not had a lower rate of stroke than predicted by CHADS2. This may reflect a limit of the predictive value of CHADS2 but should not have affected the rate of bleeding or ATE outcomes in this study.
Continue for challenges to implementation >>
CHALLENGES TO IMPLEMENTATION
Providers may hesitate to disregard current guidelines
Strokes are devastating events for patients, families, and clinicians, and they pose a greater risk for morbidity and mortality compared to bleeding. However, this study suggests patients who receive bridging have a higher risk for bleeding than stroke, which is in contrast to some providers’ experience and current recommendations.
A clinician caring for a patient who’s had a stroke may be inclined to recommend bridging despite the lack of efficacy and evidence of bleeding risk. Additionally, until guidelines reflect the most current research, clinicians may be reluctant to provide care in contrast to these recommendations.
REFERENCES
1. Douketis JD, Spyropoulos AC, Kaatz S, et al. Perioperative bridging anticoagulation in patients with atrial fibrillation. N Engl J Med. 2015;373:823-833.
2. Guyatt GH, Akl EA, Crowther M, et al; American College of Chest Physicians Antithrombotic Therapy and Prevention of Thrombosis Panel. Executive summary: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141:7S-47S.
3. Clark NP, Witt DM, Davies LE, et al. Bleeding, recurrent venous thromboembolism and mortality risks during warfarin interruption for invasive procedures. JAMA Intern Med. 2015; 175:1163-1168.
4. Lip GY, Lane DA. Stroke prevention in atrial fibrillation: a systematic review.JAMA. 2015;313:1950-1962.
5. Douketis JD, Spyropoulos AC, Spencer FA, et al; American College of Chest Physicians. Perioperative management of antithrombotic therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141:e326S-e350S.
6. Miyasaka Y, Barnes ME, Gersh BJ, et al. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence.Circulation. 2006;114:119-125.
7. Siegal D, Yudin J, Kaatz S, et al. Periprocedural heparin bridging in patients receiving vitamin K antagonists: systematic review and meta-analysis of bleeding and thromboembolic rates.Circulation. 2012;126:1630-1639.
8. Steinberg B, Peterson E, Kim S, et al; Outcomes Registry for Better Informed Treatment of Atrial Fibrillation Investigators and Patients. Use and outcomes associated with bridging during anticoagulation interruptions in patients with atrial fibrillation: findings from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF).Circulation. 2015;131:488-494.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2015. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice. 2015;64(12):794-795, 800.
Value of Ultra-Brief Cognitive Assessments in Predicting Negative Hospital Outcomes
Clinical question: What is the value of ultra-brief cognitive assessments in predicting hospital outcomes?

Background: Cognitive assessment tools can be used to predict patient outcomes in the hospital setting. Physician time constraints limit use of longer traditional cognitive testing, and little is known about the effectiveness of ultra-brief (less than one minute) assessments and their predictive value.
Study design: Secondary data analysis of a quality improvement project.
Setting: Tertiary, Veterans Administration hospital.
Synopsis: Using data from a prior inpatient database, 3,232 patients over the age of 60 were screened on admission using the modified Richmond Agitation and Sedation Scale (mRASS) for arousal and the months of the year backwards (MOTYB) for attention. Abnormal mRASS and incorrect MOTYB predicted negative hospital outcomes: increased length of stay (incident rate ratio 1.23, 95% CI 1.17-1.3); increased restraint use (risk ratio 5.05, 95% CI); increased hospital mortality (RR 3.46, 95% CI 1.24-9.63); and decreased rates of being discharged home (RR 2.97, 95% CI: 2.42-3.64).
This study highlights the value of two ultra-brief cognitive assessment tools in the prediction of potential poor outcomes during inpatient admission. Hospitalists need to identify high-risk patients, and these tools allow for rapid assessment at the time of admission, without a significant time constraint for the busy hospitalist.
Bottom Line: The use of ultra-brief cognitive assessment tools in patients over age 60 can predict negative inpatient outcomes.
Citation: Yevchak AM, Doherty K, Archambault EG, Kelly B, Fonda JR, Rudolph JL. The association between an ultra-brief cognitive screening in older adults and hospital outcomes. J Hosp Med. 2015;10(10):651-657.
Clinical question: What is the value of ultra-brief cognitive assessments in predicting hospital outcomes?
Background: Cognitive assessment tools can be used to predict patient outcomes in the hospital setting. Physician time constraints limit use of longer traditional cognitive testing, and little is known about the effectiveness of ultra-brief (less than one minute) assessments and their predictive value.
Study design: Secondary data analysis of a quality improvement project.
Setting: Tertiary, Veterans Administration hospital.
Synopsis: Using data from a prior inpatient database, 3,232 patients over the age of 60 were screened on admission using the modified Richmond Agitation and Sedation Scale (mRASS) for arousal and the months of the year backwards (MOTYB) for attention. Abnormal mRASS and incorrect MOTYB predicted negative hospital outcomes: increased length of stay (incident rate ratio 1.23, 95% CI 1.17-1.3); increased restraint use (risk ratio 5.05, 95% CI); increased hospital mortality (RR 3.46, 95% CI 1.24-9.63); and decreased rates of being discharged home (RR 2.97, 95% CI: 2.42-3.64).
This study highlights the value of two ultra-brief cognitive assessment tools in the prediction of potential poor outcomes during inpatient admission. Hospitalists need to identify high-risk patients, and these tools allow for rapid assessment at the time of admission, without a significant time constraint for the busy hospitalist.
Bottom Line: The use of ultra-brief cognitive assessment tools in patients over age 60 can predict negative inpatient outcomes.
Citation: Yevchak AM, Doherty K, Archambault EG, Kelly B, Fonda JR, Rudolph JL. The association between an ultra-brief cognitive screening in older adults and hospital outcomes. J Hosp Med. 2015;10(10):651-657.
Clinical question: What is the value of ultra-brief cognitive assessments in predicting hospital outcomes?
Background: Cognitive assessment tools can be used to predict patient outcomes in the hospital setting. Physician time constraints limit use of longer traditional cognitive testing, and little is known about the effectiveness of ultra-brief (less than one minute) assessments and their predictive value.
Study design: Secondary data analysis of a quality improvement project.
Setting: Tertiary, Veterans Administration hospital.
Synopsis: Using data from a prior inpatient database, 3,232 patients over the age of 60 were screened on admission using the modified Richmond Agitation and Sedation Scale (mRASS) for arousal and the months of the year backwards (MOTYB) for attention. Abnormal mRASS and incorrect MOTYB predicted negative hospital outcomes: increased length of stay (incident rate ratio 1.23, 95% CI 1.17-1.3); increased restraint use (risk ratio 5.05, 95% CI); increased hospital mortality (RR 3.46, 95% CI 1.24-9.63); and decreased rates of being discharged home (RR 2.97, 95% CI: 2.42-3.64).
This study highlights the value of two ultra-brief cognitive assessment tools in the prediction of potential poor outcomes during inpatient admission. Hospitalists need to identify high-risk patients, and these tools allow for rapid assessment at the time of admission, without a significant time constraint for the busy hospitalist.
Bottom Line: The use of ultra-brief cognitive assessment tools in patients over age 60 can predict negative inpatient outcomes.
Citation: Yevchak AM, Doherty K, Archambault EG, Kelly B, Fonda JR, Rudolph JL. The association between an ultra-brief cognitive screening in older adults and hospital outcomes. J Hosp Med. 2015;10(10):651-657.

Criteria for Appropriate Use of Peripherally Inserted Central Catheters
Clinical question: What are criteria for appropriate and inappropriate use of PICCs?
Background: PICCs are commonly used in medical care in a variety of clinical contexts; however, criteria defining the appropriate use of PICCs and practices related to PICC placement have not been previously established.
Study design: A multispecialty panel classified indications for PICC use as appropriate or inappropriate using the RAND/UCLA Appropriateness Method.
Synopsis: Selected appropriate PICC uses include:
- Infusion of peripherally compatible infusates, intermittent infusions, or infrequent phlebotomy in patients with poor or difficult venous access when the expected duration of use is at least six days;
- Phlebotomy at least every eight hours when the expected duration of use is at least six days; and
- Invasive hemodynamic monitoring in a critically ill patient only if the duration of use is expected to exceed 15 days.
Selected appropriate PICC-related practices:
- Verify PICC tip position using a chest radiograph only after non-ECG or non-fluoroscopically guided PICC insertion;
- Provide an interval without a PICC to allow resolution of bacteremia when managing PICC-related bloodstream infections; and
- For PICC-related DVT, provide at least three months of systemic anticoagulation if not otherwise contraindicated.
Selected inappropriate PICC-related practices:
- Adjustment of PICC tips that reside in the lower third of the superior vena cava, cavoatrial junction, or right atrium; and
- Removal or replacement of PICCs that are clinically necessary, well positioned, and functional in the setting of PICC-related DVT or without evidence of catheter-associated bloodstream infection.
Bottom line: A multispecialty expert panel provides guidance for appropriate use of PICCs and PICC-related practices.
Citation: Chopra V, Flanders SA, Saint S, et al. The Michigan appropriateness guide for intravenous catheters (MAGIC): results from a multispecialty panel using the RAND/UCLA appropriateness method. Ann Intern Med. 2015;163(6):S1-S40.
Clinical question: What are criteria for appropriate and inappropriate use of PICCs?
Background: PICCs are commonly used in medical care in a variety of clinical contexts; however, criteria defining the appropriate use of PICCs and practices related to PICC placement have not been previously established.
Study design: A multispecialty panel classified indications for PICC use as appropriate or inappropriate using the RAND/UCLA Appropriateness Method.
Synopsis: Selected appropriate PICC uses include:
- Infusion of peripherally compatible infusates, intermittent infusions, or infrequent phlebotomy in patients with poor or difficult venous access when the expected duration of use is at least six days;
- Phlebotomy at least every eight hours when the expected duration of use is at least six days; and
- Invasive hemodynamic monitoring in a critically ill patient only if the duration of use is expected to exceed 15 days.
Selected appropriate PICC-related practices:
- Verify PICC tip position using a chest radiograph only after non-ECG or non-fluoroscopically guided PICC insertion;
- Provide an interval without a PICC to allow resolution of bacteremia when managing PICC-related bloodstream infections; and
- For PICC-related DVT, provide at least three months of systemic anticoagulation if not otherwise contraindicated.
Selected inappropriate PICC-related practices:
- Adjustment of PICC tips that reside in the lower third of the superior vena cava, cavoatrial junction, or right atrium; and
- Removal or replacement of PICCs that are clinically necessary, well positioned, and functional in the setting of PICC-related DVT or without evidence of catheter-associated bloodstream infection.
Bottom line: A multispecialty expert panel provides guidance for appropriate use of PICCs and PICC-related practices.
Citation: Chopra V, Flanders SA, Saint S, et al. The Michigan appropriateness guide for intravenous catheters (MAGIC): results from a multispecialty panel using the RAND/UCLA appropriateness method. Ann Intern Med. 2015;163(6):S1-S40.
Clinical question: What are criteria for appropriate and inappropriate use of PICCs?
Background: PICCs are commonly used in medical care in a variety of clinical contexts; however, criteria defining the appropriate use of PICCs and practices related to PICC placement have not been previously established.
Study design: A multispecialty panel classified indications for PICC use as appropriate or inappropriate using the RAND/UCLA Appropriateness Method.
Synopsis: Selected appropriate PICC uses include:
- Infusion of peripherally compatible infusates, intermittent infusions, or infrequent phlebotomy in patients with poor or difficult venous access when the expected duration of use is at least six days;
- Phlebotomy at least every eight hours when the expected duration of use is at least six days; and
- Invasive hemodynamic monitoring in a critically ill patient only if the duration of use is expected to exceed 15 days.
Selected appropriate PICC-related practices:
- Verify PICC tip position using a chest radiograph only after non-ECG or non-fluoroscopically guided PICC insertion;
- Provide an interval without a PICC to allow resolution of bacteremia when managing PICC-related bloodstream infections; and
- For PICC-related DVT, provide at least three months of systemic anticoagulation if not otherwise contraindicated.
Selected inappropriate PICC-related practices:
- Adjustment of PICC tips that reside in the lower third of the superior vena cava, cavoatrial junction, or right atrium; and
- Removal or replacement of PICCs that are clinically necessary, well positioned, and functional in the setting of PICC-related DVT or without evidence of catheter-associated bloodstream infection.
Bottom line: A multispecialty expert panel provides guidance for appropriate use of PICCs and PICC-related practices.
Citation: Chopra V, Flanders SA, Saint S, et al. The Michigan appropriateness guide for intravenous catheters (MAGIC): results from a multispecialty panel using the RAND/UCLA appropriateness method. Ann Intern Med. 2015;163(6):S1-S40.
Kidney Transplants
Q) All I hear about is the shortage of kidneys for transplantation. A friend of mine is on the local transplant list, and it is eight years long! Are there any ideas out there to grow your own kidneys?
Eight years is a long time for people dealing with the physical and emotional effects of kidney disease coupled with hemodialysis or peritoneal dialysis. Your friend is one of 110,000 patients (as of January 2015) in the United States on the United Network for Organ Sharing (UNOS) kidney transplant waiting list.1 The UNOS/Organ Procurement and Transplant Network (OPTN) implemented new polices in 2014 to shorten the wait.
Among them: For pediatric patients (those younger than 18), the wait list time starts when the glomerular filtration rate (GFR) is ≤ 20 mL/min or the child starts dialysis. UNOS also has attempted to match posttransplant survival time of the graft with posttransplant survival time of the recipient through use of calculations that classify kidneys on the basis of how long they are likely to function once transplanted. Priority is now given to candidates with high immune system sensitivity or uncommon blood types, as they are less likely to obtain a kidney otherwise.2
The million-dollar question is how to obtain a kidney transplant in a timely fashion. Grave robbing, in case you are wondering, is not a viable option! Nor is transplant tourism (traveling outside the US to obtain an organ transplant), which confers a higher risk for severe infectious complications and acute rejection, possibly related to less extensive donor screening.3
There are other possibilities: Living donors can donate one kidney. Or, as is becoming increasingly common, paired organ transplants can be arranged. These occur when a patient in need of a kidney has a willing but incompatible donor; if a different match can be found, a “swap” is orchestrated, in which Donor A’s kidney is transplanted into Recipient B and Donor B’s kidney is given to Recipient A. This can be and has been done with multiple donors and recipients—in some cases, dozens—allowing the gift of donation to go on and on. (See “Trading Kidneys: Innovative Program Could Save Thousands of Lives” for an overview of how this concept started.)
Some exciting research is going on with regard to 3D printing of kidneys; they are miniature for now but showing survival of the printed cells. Another area of exploration is regenerative medicine; researchers in the field are investigating the bioengineering of organs by taking the “scaffolding” of an organ and implanting a patient’s own cells to “grow” a new organ (which, if successful, would also eliminate the need for immunosuppressants). Other signs of progress include recent news that scientists are getting lab-grown kidneys to work in animals.
It will be several years before any of these options will be viable. In the meantime, it never hurts to ask loved ones if they are willing to donate a kidney. Best wishes to your friend. —DC
Della Connor, PhD, RN, FNP-BC
East Texas Nephrology Associates, Lufkin, Texas
REFERENCES
1. Organ Procurement and Transplantation Network. http://optn.transplant.hrsa.gov. Accessed December 10, 2015.
2. Organ Procurement and Transplantation Network. New OPTN requirements and resources for the living donor kidney transplant programs. Prog Transplant. 2013;23(2):117.
3. Gill J, Madhira BR, Gjertson D, et al. Transplant tourism in the United States: a single-center experience. Clin J Am Soc Nephrol. 2008;3(6):1820-1828.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a retired physician assistant who works with the American Academy of Nephrology PAs and is also past chair of the NKF-CAP. This month’s responses were authored by Della Connor, PhD, RN, FNP-BC, who is an Assistant Professor at Stephen F. Austin State University in Nacogdoches, Texas, and practices at East Texas Nephrology Associates in Lufkin, and the department editors.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a retired physician assistant who works with the American Academy of Nephrology PAs and is also past chair of the NKF-CAP. This month’s responses were authored by Della Connor, PhD, RN, FNP-BC, who is an Assistant Professor at Stephen F. Austin State University in Nacogdoches, Texas, and practices at East Texas Nephrology Associates in Lufkin, and the department editors.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a retired physician assistant who works with the American Academy of Nephrology PAs and is also past chair of the NKF-CAP. This month’s responses were authored by Della Connor, PhD, RN, FNP-BC, who is an Assistant Professor at Stephen F. Austin State University in Nacogdoches, Texas, and practices at East Texas Nephrology Associates in Lufkin, and the department editors.
Q) All I hear about is the shortage of kidneys for transplantation. A friend of mine is on the local transplant list, and it is eight years long! Are there any ideas out there to grow your own kidneys?
Eight years is a long time for people dealing with the physical and emotional effects of kidney disease coupled with hemodialysis or peritoneal dialysis. Your friend is one of 110,000 patients (as of January 2015) in the United States on the United Network for Organ Sharing (UNOS) kidney transplant waiting list.1 The UNOS/Organ Procurement and Transplant Network (OPTN) implemented new polices in 2014 to shorten the wait.
Among them: For pediatric patients (those younger than 18), the wait list time starts when the glomerular filtration rate (GFR) is ≤ 20 mL/min or the child starts dialysis. UNOS also has attempted to match posttransplant survival time of the graft with posttransplant survival time of the recipient through use of calculations that classify kidneys on the basis of how long they are likely to function once transplanted. Priority is now given to candidates with high immune system sensitivity or uncommon blood types, as they are less likely to obtain a kidney otherwise.2
The million-dollar question is how to obtain a kidney transplant in a timely fashion. Grave robbing, in case you are wondering, is not a viable option! Nor is transplant tourism (traveling outside the US to obtain an organ transplant), which confers a higher risk for severe infectious complications and acute rejection, possibly related to less extensive donor screening.3
There are other possibilities: Living donors can donate one kidney. Or, as is becoming increasingly common, paired organ transplants can be arranged. These occur when a patient in need of a kidney has a willing but incompatible donor; if a different match can be found, a “swap” is orchestrated, in which Donor A’s kidney is transplanted into Recipient B and Donor B’s kidney is given to Recipient A. This can be and has been done with multiple donors and recipients—in some cases, dozens—allowing the gift of donation to go on and on. (See “Trading Kidneys: Innovative Program Could Save Thousands of Lives” for an overview of how this concept started.)
Some exciting research is going on with regard to 3D printing of kidneys; they are miniature for now but showing survival of the printed cells. Another area of exploration is regenerative medicine; researchers in the field are investigating the bioengineering of organs by taking the “scaffolding” of an organ and implanting a patient’s own cells to “grow” a new organ (which, if successful, would also eliminate the need for immunosuppressants). Other signs of progress include recent news that scientists are getting lab-grown kidneys to work in animals.
It will be several years before any of these options will be viable. In the meantime, it never hurts to ask loved ones if they are willing to donate a kidney. Best wishes to your friend. —DC
Della Connor, PhD, RN, FNP-BC
East Texas Nephrology Associates, Lufkin, Texas
REFERENCES
1. Organ Procurement and Transplantation Network. http://optn.transplant.hrsa.gov. Accessed December 10, 2015.
2. Organ Procurement and Transplantation Network. New OPTN requirements and resources for the living donor kidney transplant programs. Prog Transplant. 2013;23(2):117.
3. Gill J, Madhira BR, Gjertson D, et al. Transplant tourism in the United States: a single-center experience. Clin J Am Soc Nephrol. 2008;3(6):1820-1828.
Q) All I hear about is the shortage of kidneys for transplantation. A friend of mine is on the local transplant list, and it is eight years long! Are there any ideas out there to grow your own kidneys?
Eight years is a long time for people dealing with the physical and emotional effects of kidney disease coupled with hemodialysis or peritoneal dialysis. Your friend is one of 110,000 patients (as of January 2015) in the United States on the United Network for Organ Sharing (UNOS) kidney transplant waiting list.1 The UNOS/Organ Procurement and Transplant Network (OPTN) implemented new polices in 2014 to shorten the wait.
Among them: For pediatric patients (those younger than 18), the wait list time starts when the glomerular filtration rate (GFR) is ≤ 20 mL/min or the child starts dialysis. UNOS also has attempted to match posttransplant survival time of the graft with posttransplant survival time of the recipient through use of calculations that classify kidneys on the basis of how long they are likely to function once transplanted. Priority is now given to candidates with high immune system sensitivity or uncommon blood types, as they are less likely to obtain a kidney otherwise.2
The million-dollar question is how to obtain a kidney transplant in a timely fashion. Grave robbing, in case you are wondering, is not a viable option! Nor is transplant tourism (traveling outside the US to obtain an organ transplant), which confers a higher risk for severe infectious complications and acute rejection, possibly related to less extensive donor screening.3
There are other possibilities: Living donors can donate one kidney. Or, as is becoming increasingly common, paired organ transplants can be arranged. These occur when a patient in need of a kidney has a willing but incompatible donor; if a different match can be found, a “swap” is orchestrated, in which Donor A’s kidney is transplanted into Recipient B and Donor B’s kidney is given to Recipient A. This can be and has been done with multiple donors and recipients—in some cases, dozens—allowing the gift of donation to go on and on. (See “Trading Kidneys: Innovative Program Could Save Thousands of Lives” for an overview of how this concept started.)
Some exciting research is going on with regard to 3D printing of kidneys; they are miniature for now but showing survival of the printed cells. Another area of exploration is regenerative medicine; researchers in the field are investigating the bioengineering of organs by taking the “scaffolding” of an organ and implanting a patient’s own cells to “grow” a new organ (which, if successful, would also eliminate the need for immunosuppressants). Other signs of progress include recent news that scientists are getting lab-grown kidneys to work in animals.
It will be several years before any of these options will be viable. In the meantime, it never hurts to ask loved ones if they are willing to donate a kidney. Best wishes to your friend. —DC
Della Connor, PhD, RN, FNP-BC
East Texas Nephrology Associates, Lufkin, Texas
REFERENCES
1. Organ Procurement and Transplantation Network. http://optn.transplant.hrsa.gov. Accessed December 10, 2015.
2. Organ Procurement and Transplantation Network. New OPTN requirements and resources for the living donor kidney transplant programs. Prog Transplant. 2013;23(2):117.
3. Gill J, Madhira BR, Gjertson D, et al. Transplant tourism in the United States: a single-center experience. Clin J Am Soc Nephrol. 2008;3(6):1820-1828.