User login
Heavily pretreated myeloma responds to pembrolizumab combo
ORLANDO – The one-two punch of combining the programmed cell death-1 (PD-1) inhibitor pembrolizumab with the immunomodulatory drug lenalidomide and low-dose dexamethasone produced responses in 76% of 17 heavily pretreated patients with relapsed or refractory multiple myeloma in the KEYNOTE-023 study.
This included four very good partial responses (24%) and nine partial responses (53%).
In nine lenalidomide-refractory patients, the overall response rate was 56%, including two very good partial responses (22%) and three partial responses (33%).
The efficacy results are preliminary, but support the continued development of pembrolizumab (Keytruda) in patients with multiple myeloma, Dr. Jesús San Miguel of Clinica Universidad de Navarra, Pamplona, Spain, said at the annual meeting of the American Society of Hematology.

He closed his presentation with two illustrative cases highlighting a rapid response lasting more than a year and a half in a 49-year-old man with myeloma triple-refractory to autologous stem cell transplant (ASCT), lenalidomide (Revlimid), and dexamethasone.
The second case involved a patient with double-refractory myeloma and extramedullary disease who achieved a stringent complete response after two cycles of fourth-line pembrolizumab that was associated with a “striking” reduction in lesion volume on computed tomography scans, he said.
The median duration of response among the 17 evaluable patients was 9.7 months.
The median time to first response was 1.2 months (range 1.0 months to 6.5 months). But some patients require more time and, interestingly, the quality of the response was upgraded in 11% with continued treatment, Dr. San Miguel said.
The rationale for combining PD-1 inhibitors with immunomodulatory drugs (IMiD) lies in recent research showing that lenalidomide reduces PD-ligand 1 and PD-1 expression on multiple myeloma cells as well as on T and myeloid-derived suppressor cells, he explained. In addition, lenalidomide enhances checkpoint blockade–induced effector cytokine production in multiple myeloma bone marrow and induces cytotoxicity against myeloma cells.
“Lenalidomide will increase the number of T cells and the T-cell activation and anti-PD-1 will release the brake in order to allow these activated T cells to interact with the tumor,” Dr. San Miguel said.
Patients enrolled in KEYSTONE-023 were heavily pretreated, with 26% previously exposed to pomalidomide, 76% refractory to lenalidomide, 80% refractory to their last line of therapy, and 86% having undergone prior ASCT. Half the patients were double, triple, or quadruple refractory, he noted.
The study (Abstract 505) was designed to identify the maximum tolerated dose (MTD) of pembrolizumab and to assess its safety and tolerability when given with lenalidomide and dexamethasone in patients with multiple myeloma failing at least two prior lines of therapy including a proteasome inhibitor and an IMiD. Their median age was 62 years; 64% were male.
In the dose-determination stage, three of six patients treated with pembrolizumab 2 mg/kg plus lenalidomide 25 mg and dexamethasone 40 mg experienced dose-limiting toxicities that resolved without treatment discontinuation.
After dose adjustments, a “flat dose” of pembrolizumab 200 mg given every other week in a rapid 30-minute intravenous infusion without premedication with lenalidomide 25 mg on days 1-21 and dexamethasone 40 mg weekly did not cause any dose-limiting toxicities and was identified as the final MTD, Dr. San Miguel said.
The regimen is to continue for 24 months or until tumor progression or excessive side effects and was carried forward into the dose-expansion stage in 33 additional patients with a median follow-up of 48 days.
Among all 50 patients evaluable for safety, 72% experienced at least one treatment-related adverse event of any grade and 46% (23/50 patients) had grade 3/4 adverse events including neutropenia (22%), thrombocytopenia and anemia (8% each), hyperglycemia (6%), and fatigue, muscle spasms, and diarrhea (2% each).
The adverse events were consistent with the individual drug safety profiles, but “the incidence may be underestimated due to the limited drug exposure,” Dr. San Miguel cautioned.
Immune-mediated adverse events included two cases each of hyper- and hypothyroidism, one case of thyroiditis, and one grade 2 adrenal insufficiency. No cases of colitis or pneumonitis were reported. No dose modifications or treatment discontinuations were required to mange the immune-related side effects, he said. No treatment-related deaths occurred.
In a second study reported during the same oral myeloma session, pneumonitis cropped up in 10% of heavily pretreated patients with relapsed multiple myeloma receiving a slightly different regimen of pembrolizumab plus the IMiD pomalidomide (Pomalyst) and dexamethasone. The overall response rate in the phase II study was 60% among 27 evaluable patients and 55% in those double-refractory to IMiDs and proteasome inhibitors.
ORLANDO – The one-two punch of combining the programmed cell death-1 (PD-1) inhibitor pembrolizumab with the immunomodulatory drug lenalidomide and low-dose dexamethasone produced responses in 76% of 17 heavily pretreated patients with relapsed or refractory multiple myeloma in the KEYNOTE-023 study.
This included four very good partial responses (24%) and nine partial responses (53%).
In nine lenalidomide-refractory patients, the overall response rate was 56%, including two very good partial responses (22%) and three partial responses (33%).
The efficacy results are preliminary, but support the continued development of pembrolizumab (Keytruda) in patients with multiple myeloma, Dr. Jesús San Miguel of Clinica Universidad de Navarra, Pamplona, Spain, said at the annual meeting of the American Society of Hematology.
He closed his presentation with two illustrative cases highlighting a rapid response lasting more than a year and a half in a 49-year-old man with myeloma triple-refractory to autologous stem cell transplant (ASCT), lenalidomide (Revlimid), and dexamethasone.
The second case involved a patient with double-refractory myeloma and extramedullary disease who achieved a stringent complete response after two cycles of fourth-line pembrolizumab that was associated with a “striking” reduction in lesion volume on computed tomography scans, he said.
The median duration of response among the 17 evaluable patients was 9.7 months.
The median time to first response was 1.2 months (range 1.0 months to 6.5 months). But some patients require more time and, interestingly, the quality of the response was upgraded in 11% with continued treatment, Dr. San Miguel said.
The rationale for combining PD-1 inhibitors with immunomodulatory drugs (IMiD) lies in recent research showing that lenalidomide reduces PD-ligand 1 and PD-1 expression on multiple myeloma cells as well as on T and myeloid-derived suppressor cells, he explained. In addition, lenalidomide enhances checkpoint blockade–induced effector cytokine production in multiple myeloma bone marrow and induces cytotoxicity against myeloma cells.
“Lenalidomide will increase the number of T cells and the T-cell activation and anti-PD-1 will release the brake in order to allow these activated T cells to interact with the tumor,” Dr. San Miguel said.
Patients enrolled in KEYSTONE-023 were heavily pretreated, with 26% previously exposed to pomalidomide, 76% refractory to lenalidomide, 80% refractory to their last line of therapy, and 86% having undergone prior ASCT. Half the patients were double, triple, or quadruple refractory, he noted.
The study (Abstract 505) was designed to identify the maximum tolerated dose (MTD) of pembrolizumab and to assess its safety and tolerability when given with lenalidomide and dexamethasone in patients with multiple myeloma failing at least two prior lines of therapy including a proteasome inhibitor and an IMiD. Their median age was 62 years; 64% were male.
In the dose-determination stage, three of six patients treated with pembrolizumab 2 mg/kg plus lenalidomide 25 mg and dexamethasone 40 mg experienced dose-limiting toxicities that resolved without treatment discontinuation.
After dose adjustments, a “flat dose” of pembrolizumab 200 mg given every other week in a rapid 30-minute intravenous infusion without premedication with lenalidomide 25 mg on days 1-21 and dexamethasone 40 mg weekly did not cause any dose-limiting toxicities and was identified as the final MTD, Dr. San Miguel said.
The regimen is to continue for 24 months or until tumor progression or excessive side effects and was carried forward into the dose-expansion stage in 33 additional patients with a median follow-up of 48 days.
Among all 50 patients evaluable for safety, 72% experienced at least one treatment-related adverse event of any grade and 46% (23/50 patients) had grade 3/4 adverse events including neutropenia (22%), thrombocytopenia and anemia (8% each), hyperglycemia (6%), and fatigue, muscle spasms, and diarrhea (2% each).
The adverse events were consistent with the individual drug safety profiles, but “the incidence may be underestimated due to the limited drug exposure,” Dr. San Miguel cautioned.
Immune-mediated adverse events included two cases each of hyper- and hypothyroidism, one case of thyroiditis, and one grade 2 adrenal insufficiency. No cases of colitis or pneumonitis were reported. No dose modifications or treatment discontinuations were required to mange the immune-related side effects, he said. No treatment-related deaths occurred.
In a second study reported during the same oral myeloma session, pneumonitis cropped up in 10% of heavily pretreated patients with relapsed multiple myeloma receiving a slightly different regimen of pembrolizumab plus the IMiD pomalidomide (Pomalyst) and dexamethasone. The overall response rate in the phase II study was 60% among 27 evaluable patients and 55% in those double-refractory to IMiDs and proteasome inhibitors.
ORLANDO – The one-two punch of combining the programmed cell death-1 (PD-1) inhibitor pembrolizumab with the immunomodulatory drug lenalidomide and low-dose dexamethasone produced responses in 76% of 17 heavily pretreated patients with relapsed or refractory multiple myeloma in the KEYNOTE-023 study.
This included four very good partial responses (24%) and nine partial responses (53%).
In nine lenalidomide-refractory patients, the overall response rate was 56%, including two very good partial responses (22%) and three partial responses (33%).
The efficacy results are preliminary, but support the continued development of pembrolizumab (Keytruda) in patients with multiple myeloma, Dr. Jesús San Miguel of Clinica Universidad de Navarra, Pamplona, Spain, said at the annual meeting of the American Society of Hematology.
He closed his presentation with two illustrative cases highlighting a rapid response lasting more than a year and a half in a 49-year-old man with myeloma triple-refractory to autologous stem cell transplant (ASCT), lenalidomide (Revlimid), and dexamethasone.
The second case involved a patient with double-refractory myeloma and extramedullary disease who achieved a stringent complete response after two cycles of fourth-line pembrolizumab that was associated with a “striking” reduction in lesion volume on computed tomography scans, he said.
The median duration of response among the 17 evaluable patients was 9.7 months.
The median time to first response was 1.2 months (range 1.0 months to 6.5 months). But some patients require more time and, interestingly, the quality of the response was upgraded in 11% with continued treatment, Dr. San Miguel said.
The rationale for combining PD-1 inhibitors with immunomodulatory drugs (IMiD) lies in recent research showing that lenalidomide reduces PD-ligand 1 and PD-1 expression on multiple myeloma cells as well as on T and myeloid-derived suppressor cells, he explained. In addition, lenalidomide enhances checkpoint blockade–induced effector cytokine production in multiple myeloma bone marrow and induces cytotoxicity against myeloma cells.
“Lenalidomide will increase the number of T cells and the T-cell activation and anti-PD-1 will release the brake in order to allow these activated T cells to interact with the tumor,” Dr. San Miguel said.
Patients enrolled in KEYSTONE-023 were heavily pretreated, with 26% previously exposed to pomalidomide, 76% refractory to lenalidomide, 80% refractory to their last line of therapy, and 86% having undergone prior ASCT. Half the patients were double, triple, or quadruple refractory, he noted.
The study (Abstract 505) was designed to identify the maximum tolerated dose (MTD) of pembrolizumab and to assess its safety and tolerability when given with lenalidomide and dexamethasone in patients with multiple myeloma failing at least two prior lines of therapy including a proteasome inhibitor and an IMiD. Their median age was 62 years; 64% were male.
In the dose-determination stage, three of six patients treated with pembrolizumab 2 mg/kg plus lenalidomide 25 mg and dexamethasone 40 mg experienced dose-limiting toxicities that resolved without treatment discontinuation.
After dose adjustments, a “flat dose” of pembrolizumab 200 mg given every other week in a rapid 30-minute intravenous infusion without premedication with lenalidomide 25 mg on days 1-21 and dexamethasone 40 mg weekly did not cause any dose-limiting toxicities and was identified as the final MTD, Dr. San Miguel said.
The regimen is to continue for 24 months or until tumor progression or excessive side effects and was carried forward into the dose-expansion stage in 33 additional patients with a median follow-up of 48 days.
Among all 50 patients evaluable for safety, 72% experienced at least one treatment-related adverse event of any grade and 46% (23/50 patients) had grade 3/4 adverse events including neutropenia (22%), thrombocytopenia and anemia (8% each), hyperglycemia (6%), and fatigue, muscle spasms, and diarrhea (2% each).
The adverse events were consistent with the individual drug safety profiles, but “the incidence may be underestimated due to the limited drug exposure,” Dr. San Miguel cautioned.
Immune-mediated adverse events included two cases each of hyper- and hypothyroidism, one case of thyroiditis, and one grade 2 adrenal insufficiency. No cases of colitis or pneumonitis were reported. No dose modifications or treatment discontinuations were required to mange the immune-related side effects, he said. No treatment-related deaths occurred.
In a second study reported during the same oral myeloma session, pneumonitis cropped up in 10% of heavily pretreated patients with relapsed multiple myeloma receiving a slightly different regimen of pembrolizumab plus the IMiD pomalidomide (Pomalyst) and dexamethasone. The overall response rate in the phase II study was 60% among 27 evaluable patients and 55% in those double-refractory to IMiDs and proteasome inhibitors.
AT ASH 2015
Key clinical point: Initial results show promising activity for pembrolizumab in combination with lenalidomide and low-dose dexamethasone in heavily pretreated relapsed or refractory multiple myeloma.
Major finding: The overall response rate was 76% (13/17 patients).
Data source: Phase I study in 50 patients with relapsed or refractory multiple myeloma.
Disclosures: The study was supported by Merck. Dr. San Miguel reported consulting for Merck and relationships with Millennium, Janssen, Celgene, Novartis, Onyx, Bristol-Myers Squibb, and Sanofi.

The palliative path: Talking with elderly patients facing emergency surgery
An expert panel has developed a communication framework to improve treatment of older, seriously ill patients who have surgical emergencies, which has been published online in Annals of Surgery.
A substantial portion of older patients who undergo emergency surgeries already have serious life-limiting illnesses such as cardiopulmonary disease, renal failure, liver failure, dementia, severe neurological impairment, or malignancy. The advisory panel based its work on the premise that surgery in these circumstances can lead to significant further morbidity, health care utilization, functional decline, prolonged hospital stay or institutionalization, and death, with attendant physical discomfort and psychological distress at the end of these patients’ lives.

Surgeons consulted in the emergency setting for these patients are hampered by patients unable to communicate well because they are in extremis, by surrogates who are unprepared for their role, and by time constraints, lack of familiarity with the patient, poor understanding of the illness by patients and families, prognostic uncertainty, and inadequate advance care planning. In addition, “many surgeons lack skills to engage in conversations about end-of-life care, or are too unfamiliar with palliative options to discuss them well,” or feel obligated to maintain postoperative life support despite the patient’s wishes, said Dr. Zara Cooper, of Ariadne Labs and the Center for Surgery and Public Health at Brigham and Women’s Hospital, both in Boston, and her associates.
To address these issues and assist surgeons in caring for such patients, an expert panel of 23 national leaders in acute care surgery, general surgery, surgical oncology, palliative medicine, critical care, emergency medicine, anesthesia, and health care innovation was convened at Harvard Medical School, Boston.
The focus of the panel’s recommendations was a structured communications framework prototype to facilitate shared decision-making in these difficult circumstances.
Among the panel’s recommendations for surgeons were the following priorities:
• Review the medical record and consult the treatment team to fully understand the patient’s current condition, comorbidities, expected illness trajectory, and preferences for end-of-life care.
• Assess functional performance as part of the routine history and physical to fully understand the patient’s fitness for surgery.
• Formulate a prognosis regarding the patient’s overall health both with and without surgery.
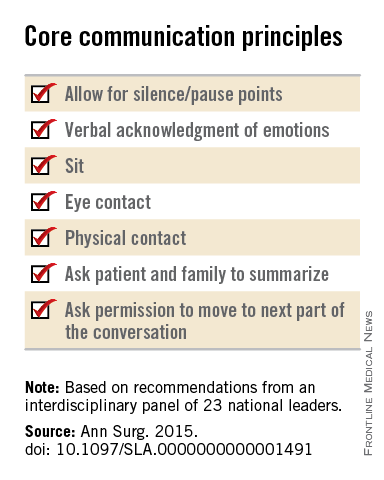
The panel offered a set of principles and specific elements for the meeting with the patient and family:
• The surgeon should begin by introducing himself or herself; according to reports in the literature, physicians fail to do this approximately half of the time.
• Pay attention to nonverbal communication, such as eye contact and physical contact, as this is critical to building rapport. Immediately address pain, anxiety, and other indicators of distress, to maximize the patients’ and the families’ engagement in subsequent medical discussions. “Although adequate analgesia may render a patient unable to make their own decisions, surrogates are more likely to make appropriate decisions when they feel their loved one is comfortable,” the panel noted.
• Allow pauses and silences to occur. Let the patient and the family process information and their own emotions.
• Elicit the patients’ or the surrogates’ understanding of the illness and their views of the patients’ likely trajectory, correcting any inaccuracies. This substantially influences their decisions regarding the aggressiveness of subsequent treatments.
• Inform the patient and family of the life-threatening nature of the patient’s acute condition and its potential impact on the rest of his or her life, including the possibility of prolonged life support, ICU stay, burdensome treatment, and loss of independence. Use accepted techniques for breaking bad news, and check to be sure the patient understands what was conveyed.
• At this point, the surgeon should synthesize and summarize the information from the patient, the family, and the medical record, then pause to give them time to process the information and to assess their emotional state. It is helpful to label and respond to the patient’s emotions at this juncture, and to build empathy with statements such as “I know this is difficult news, and I wish it were different.”
• Describe the benefits, burdens, and range of likely outcomes if surgery is undertaken and if it is not. The surgeon should use nonmedical language to describe symptoms, and should convey his or her expectations regarding length of hospitalization, need for and duration of life support, burdensome symptoms, discharge to an institution, and functional recovery.
• Surgeons should be able to communicate palliative options possible either in combination with surgery or instead of surgery. Palliative care can aid in managing advanced symptoms, providing psychosocial support for patients and caregivers, facilitating interdisciplinary communication, and facilitating medical decisions and care transitions.
• Avoid describing surgical procedures as “doing everything” and palliative care as “doing nothing.” This can make patients and families “feel abandoned, fearful, isolated, and angry, and fails to encompass palliative care’s practices of proactive communication, aggressive symptom management, and timely emotional support to alleviate suffering and affirm quality of life,” the panel said.
• Surgeons should explicitly support the patients’ medical decisions, whether or not they choose surgery.
The panel also cited a few factors that would assist surgeons in following these recommendations. First, surgeons must recognize the importance of communicating well with seriously ill older patients and acknowledge that this is a crucial clinical skill for them to cultivate. They must also recognize that palliative care is vital to delivering high-quality surgical care. Surgeons should consider discharging patients to hospice, which can improve pain and symptom management, improve patient and family satisfaction with care, and avoid unwanted hospitalization or cardiopulmonary resuscitation.
“There are a number of major barriers to introducing palliative care in these situations. One is an education problem - the perception on the part of patients and clinicians, and surgeons in particular, that palliative care is only limited to end-of-life care, which it is not. It is a misperception of what palliative care means in this equation - that palliative care and hospice are the same thing, which they absolutely are not,”said Dr. Cooper in an interview.
”The definition of palliative care has evolved over the past decade and the focus of palliative care is on quality of life and alleviating symptoms. End-of-life palliative care is part of that, and as patients get closer to the end of life, symptom management and quality of life become more focal than life-prolonging treatment... But for patients with chronic and serious illness, there has to be a role for palliative care because we know that when patients feel better, they tend to live longer. And when patients feel their emotional concerns and physical needs are being addressed, they tend to do better. Patients families have improved satisfaction when their loved one receives palliative care,” she noted.”
However, the number of palliative providers is completely inadequate to meet the needs of the number of seriously ill patients, she said. And a lot of hospital-based palliative care is by necessity limited to end-of-life care because of a lack of palliative resources.
Dr. Atul Gawande, a coauthor of the panel recommendations, wrote a best-selling book, Being Mortal (New York: Metropolitan Books, 2014) addressing the shortcomings and potential remaking of medical care in the context of age-related frailty, grave illness, and death. Dr. Cooper noted that there is a growing sentiment among the general public that they want to have their quality of life addressed in the type of medical care they receive. She said that Dr. Gawande’s book tapped into the perception of a lack of recognition of personhood of seriously ill patients.
“We often focus on diagnosis and we don’t have the ‘bandwidth’ to focus on the person carrying that diagnosis, and our patients and focus on the person carrying that diagnosis, but our patients and their families are demanding different types of care. So, ultimately, the patients will be the ones to push us to do better for them.”
The next steps to further developing a widely used and validated communication framework would be to create educational opportunities for clinicians to develop clinical skills in communication with seriously ill patients and palliative care, and to study the impact of these initiatives on improving outcomes most relevant to older patient. This work was supported by the Ariadne Labs, a Joint Center for Health System Innovation at Brigham and Women’s Hospital. Dr. Cooper and her associates reported having no relevant financial disclosures.
An expert panel has developed a communication framework to improve treatment of older, seriously ill patients who have surgical emergencies, which has been published online in Annals of Surgery.
A substantial portion of older patients who undergo emergency surgeries already have serious life-limiting illnesses such as cardiopulmonary disease, renal failure, liver failure, dementia, severe neurological impairment, or malignancy. The advisory panel based its work on the premise that surgery in these circumstances can lead to significant further morbidity, health care utilization, functional decline, prolonged hospital stay or institutionalization, and death, with attendant physical discomfort and psychological distress at the end of these patients’ lives.
Surgeons consulted in the emergency setting for these patients are hampered by patients unable to communicate well because they are in extremis, by surrogates who are unprepared for their role, and by time constraints, lack of familiarity with the patient, poor understanding of the illness by patients and families, prognostic uncertainty, and inadequate advance care planning. In addition, “many surgeons lack skills to engage in conversations about end-of-life care, or are too unfamiliar with palliative options to discuss them well,” or feel obligated to maintain postoperative life support despite the patient’s wishes, said Dr. Zara Cooper, of Ariadne Labs and the Center for Surgery and Public Health at Brigham and Women’s Hospital, both in Boston, and her associates.
To address these issues and assist surgeons in caring for such patients, an expert panel of 23 national leaders in acute care surgery, general surgery, surgical oncology, palliative medicine, critical care, emergency medicine, anesthesia, and health care innovation was convened at Harvard Medical School, Boston.
The focus of the panel’s recommendations was a structured communications framework prototype to facilitate shared decision-making in these difficult circumstances.
Among the panel’s recommendations for surgeons were the following priorities:
• Review the medical record and consult the treatment team to fully understand the patient’s current condition, comorbidities, expected illness trajectory, and preferences for end-of-life care.
• Assess functional performance as part of the routine history and physical to fully understand the patient’s fitness for surgery.
• Formulate a prognosis regarding the patient’s overall health both with and without surgery.
The panel offered a set of principles and specific elements for the meeting with the patient and family:
• The surgeon should begin by introducing himself or herself; according to reports in the literature, physicians fail to do this approximately half of the time.
• Pay attention to nonverbal communication, such as eye contact and physical contact, as this is critical to building rapport. Immediately address pain, anxiety, and other indicators of distress, to maximize the patients’ and the families’ engagement in subsequent medical discussions. “Although adequate analgesia may render a patient unable to make their own decisions, surrogates are more likely to make appropriate decisions when they feel their loved one is comfortable,” the panel noted.
• Allow pauses and silences to occur. Let the patient and the family process information and their own emotions.
• Elicit the patients’ or the surrogates’ understanding of the illness and their views of the patients’ likely trajectory, correcting any inaccuracies. This substantially influences their decisions regarding the aggressiveness of subsequent treatments.
• Inform the patient and family of the life-threatening nature of the patient’s acute condition and its potential impact on the rest of his or her life, including the possibility of prolonged life support, ICU stay, burdensome treatment, and loss of independence. Use accepted techniques for breaking bad news, and check to be sure the patient understands what was conveyed.
• At this point, the surgeon should synthesize and summarize the information from the patient, the family, and the medical record, then pause to give them time to process the information and to assess their emotional state. It is helpful to label and respond to the patient’s emotions at this juncture, and to build empathy with statements such as “I know this is difficult news, and I wish it were different.”
• Describe the benefits, burdens, and range of likely outcomes if surgery is undertaken and if it is not. The surgeon should use nonmedical language to describe symptoms, and should convey his or her expectations regarding length of hospitalization, need for and duration of life support, burdensome symptoms, discharge to an institution, and functional recovery.
• Surgeons should be able to communicate palliative options possible either in combination with surgery or instead of surgery. Palliative care can aid in managing advanced symptoms, providing psychosocial support for patients and caregivers, facilitating interdisciplinary communication, and facilitating medical decisions and care transitions.
• Avoid describing surgical procedures as “doing everything” and palliative care as “doing nothing.” This can make patients and families “feel abandoned, fearful, isolated, and angry, and fails to encompass palliative care’s practices of proactive communication, aggressive symptom management, and timely emotional support to alleviate suffering and affirm quality of life,” the panel said.
• Surgeons should explicitly support the patients’ medical decisions, whether or not they choose surgery.
The panel also cited a few factors that would assist surgeons in following these recommendations. First, surgeons must recognize the importance of communicating well with seriously ill older patients and acknowledge that this is a crucial clinical skill for them to cultivate. They must also recognize that palliative care is vital to delivering high-quality surgical care. Surgeons should consider discharging patients to hospice, which can improve pain and symptom management, improve patient and family satisfaction with care, and avoid unwanted hospitalization or cardiopulmonary resuscitation.
“There are a number of major barriers to introducing palliative care in these situations. One is an education problem - the perception on the part of patients and clinicians, and surgeons in particular, that palliative care is only limited to end-of-life care, which it is not. It is a misperception of what palliative care means in this equation - that palliative care and hospice are the same thing, which they absolutely are not,”said Dr. Cooper in an interview.
”The definition of palliative care has evolved over the past decade and the focus of palliative care is on quality of life and alleviating symptoms. End-of-life palliative care is part of that, and as patients get closer to the end of life, symptom management and quality of life become more focal than life-prolonging treatment... But for patients with chronic and serious illness, there has to be a role for palliative care because we know that when patients feel better, they tend to live longer. And when patients feel their emotional concerns and physical needs are being addressed, they tend to do better. Patients families have improved satisfaction when their loved one receives palliative care,” she noted.”
However, the number of palliative providers is completely inadequate to meet the needs of the number of seriously ill patients, she said. And a lot of hospital-based palliative care is by necessity limited to end-of-life care because of a lack of palliative resources.
Dr. Atul Gawande, a coauthor of the panel recommendations, wrote a best-selling book, Being Mortal (New York: Metropolitan Books, 2014) addressing the shortcomings and potential remaking of medical care in the context of age-related frailty, grave illness, and death. Dr. Cooper noted that there is a growing sentiment among the general public that they want to have their quality of life addressed in the type of medical care they receive. She said that Dr. Gawande’s book tapped into the perception of a lack of recognition of personhood of seriously ill patients.
“We often focus on diagnosis and we don’t have the ‘bandwidth’ to focus on the person carrying that diagnosis, and our patients and focus on the person carrying that diagnosis, but our patients and their families are demanding different types of care. So, ultimately, the patients will be the ones to push us to do better for them.”
The next steps to further developing a widely used and validated communication framework would be to create educational opportunities for clinicians to develop clinical skills in communication with seriously ill patients and palliative care, and to study the impact of these initiatives on improving outcomes most relevant to older patient. This work was supported by the Ariadne Labs, a Joint Center for Health System Innovation at Brigham and Women’s Hospital. Dr. Cooper and her associates reported having no relevant financial disclosures.
An expert panel has developed a communication framework to improve treatment of older, seriously ill patients who have surgical emergencies, which has been published online in Annals of Surgery.
A substantial portion of older patients who undergo emergency surgeries already have serious life-limiting illnesses such as cardiopulmonary disease, renal failure, liver failure, dementia, severe neurological impairment, or malignancy. The advisory panel based its work on the premise that surgery in these circumstances can lead to significant further morbidity, health care utilization, functional decline, prolonged hospital stay or institutionalization, and death, with attendant physical discomfort and psychological distress at the end of these patients’ lives.
Surgeons consulted in the emergency setting for these patients are hampered by patients unable to communicate well because they are in extremis, by surrogates who are unprepared for their role, and by time constraints, lack of familiarity with the patient, poor understanding of the illness by patients and families, prognostic uncertainty, and inadequate advance care planning. In addition, “many surgeons lack skills to engage in conversations about end-of-life care, or are too unfamiliar with palliative options to discuss them well,” or feel obligated to maintain postoperative life support despite the patient’s wishes, said Dr. Zara Cooper, of Ariadne Labs and the Center for Surgery and Public Health at Brigham and Women’s Hospital, both in Boston, and her associates.
To address these issues and assist surgeons in caring for such patients, an expert panel of 23 national leaders in acute care surgery, general surgery, surgical oncology, palliative medicine, critical care, emergency medicine, anesthesia, and health care innovation was convened at Harvard Medical School, Boston.
The focus of the panel’s recommendations was a structured communications framework prototype to facilitate shared decision-making in these difficult circumstances.
Among the panel’s recommendations for surgeons were the following priorities:
• Review the medical record and consult the treatment team to fully understand the patient’s current condition, comorbidities, expected illness trajectory, and preferences for end-of-life care.
• Assess functional performance as part of the routine history and physical to fully understand the patient’s fitness for surgery.
• Formulate a prognosis regarding the patient’s overall health both with and without surgery.
The panel offered a set of principles and specific elements for the meeting with the patient and family:
• The surgeon should begin by introducing himself or herself; according to reports in the literature, physicians fail to do this approximately half of the time.
• Pay attention to nonverbal communication, such as eye contact and physical contact, as this is critical to building rapport. Immediately address pain, anxiety, and other indicators of distress, to maximize the patients’ and the families’ engagement in subsequent medical discussions. “Although adequate analgesia may render a patient unable to make their own decisions, surrogates are more likely to make appropriate decisions when they feel their loved one is comfortable,” the panel noted.
• Allow pauses and silences to occur. Let the patient and the family process information and their own emotions.
• Elicit the patients’ or the surrogates’ understanding of the illness and their views of the patients’ likely trajectory, correcting any inaccuracies. This substantially influences their decisions regarding the aggressiveness of subsequent treatments.
• Inform the patient and family of the life-threatening nature of the patient’s acute condition and its potential impact on the rest of his or her life, including the possibility of prolonged life support, ICU stay, burdensome treatment, and loss of independence. Use accepted techniques for breaking bad news, and check to be sure the patient understands what was conveyed.
• At this point, the surgeon should synthesize and summarize the information from the patient, the family, and the medical record, then pause to give them time to process the information and to assess their emotional state. It is helpful to label and respond to the patient’s emotions at this juncture, and to build empathy with statements such as “I know this is difficult news, and I wish it were different.”
• Describe the benefits, burdens, and range of likely outcomes if surgery is undertaken and if it is not. The surgeon should use nonmedical language to describe symptoms, and should convey his or her expectations regarding length of hospitalization, need for and duration of life support, burdensome symptoms, discharge to an institution, and functional recovery.
• Surgeons should be able to communicate palliative options possible either in combination with surgery or instead of surgery. Palliative care can aid in managing advanced symptoms, providing psychosocial support for patients and caregivers, facilitating interdisciplinary communication, and facilitating medical decisions and care transitions.
• Avoid describing surgical procedures as “doing everything” and palliative care as “doing nothing.” This can make patients and families “feel abandoned, fearful, isolated, and angry, and fails to encompass palliative care’s practices of proactive communication, aggressive symptom management, and timely emotional support to alleviate suffering and affirm quality of life,” the panel said.
• Surgeons should explicitly support the patients’ medical decisions, whether or not they choose surgery.
The panel also cited a few factors that would assist surgeons in following these recommendations. First, surgeons must recognize the importance of communicating well with seriously ill older patients and acknowledge that this is a crucial clinical skill for them to cultivate. They must also recognize that palliative care is vital to delivering high-quality surgical care. Surgeons should consider discharging patients to hospice, which can improve pain and symptom management, improve patient and family satisfaction with care, and avoid unwanted hospitalization or cardiopulmonary resuscitation.
“There are a number of major barriers to introducing palliative care in these situations. One is an education problem - the perception on the part of patients and clinicians, and surgeons in particular, that palliative care is only limited to end-of-life care, which it is not. It is a misperception of what palliative care means in this equation - that palliative care and hospice are the same thing, which they absolutely are not,”said Dr. Cooper in an interview.
”The definition of palliative care has evolved over the past decade and the focus of palliative care is on quality of life and alleviating symptoms. End-of-life palliative care is part of that, and as patients get closer to the end of life, symptom management and quality of life become more focal than life-prolonging treatment... But for patients with chronic and serious illness, there has to be a role for palliative care because we know that when patients feel better, they tend to live longer. And when patients feel their emotional concerns and physical needs are being addressed, they tend to do better. Patients families have improved satisfaction when their loved one receives palliative care,” she noted.”
However, the number of palliative providers is completely inadequate to meet the needs of the number of seriously ill patients, she said. And a lot of hospital-based palliative care is by necessity limited to end-of-life care because of a lack of palliative resources.
Dr. Atul Gawande, a coauthor of the panel recommendations, wrote a best-selling book, Being Mortal (New York: Metropolitan Books, 2014) addressing the shortcomings and potential remaking of medical care in the context of age-related frailty, grave illness, and death. Dr. Cooper noted that there is a growing sentiment among the general public that they want to have their quality of life addressed in the type of medical care they receive. She said that Dr. Gawande’s book tapped into the perception of a lack of recognition of personhood of seriously ill patients.
“We often focus on diagnosis and we don’t have the ‘bandwidth’ to focus on the person carrying that diagnosis, and our patients and focus on the person carrying that diagnosis, but our patients and their families are demanding different types of care. So, ultimately, the patients will be the ones to push us to do better for them.”
The next steps to further developing a widely used and validated communication framework would be to create educational opportunities for clinicians to develop clinical skills in communication with seriously ill patients and palliative care, and to study the impact of these initiatives on improving outcomes most relevant to older patient. This work was supported by the Ariadne Labs, a Joint Center for Health System Innovation at Brigham and Women’s Hospital. Dr. Cooper and her associates reported having no relevant financial disclosures.
FROM ANNALS OF SURGERY

Private Insurers to Reap Bulk of Spending on Hospitalized Patient Care
Spending on care of hospitalized patients is expected to pass $1 trillion in 2015, a new high. Thomas Selden, PhD, of the Agency for Healthcare Research and Quality recently asked where that money is likely to go. The answer: private insurers.
Dr. Selden and his colleagues report in Health Affairs this month that in 2012, private insurers’ payment rates for inpatient hospital stays were approximately 75% greater than Medicare’s payment rates, a sharp increase from the differential of approximately 10% percent during the period of 1996 to 2001. “We need to understand who’s paying what,” Dr. Selden says. “It’s the first step to a better understanding of public policy.”
The report found that “the predicted percentage difference between the rates of private insurers and those of Medicare has increased substantially over time.” In 1996, private insurers paid 106.1% of Medicare payment rates, a payment rate difference of 6.1% (95% CI: -3.2, 15.5). The difference climbed to 64.1% (95% CI: 48.3, 80.0) in 2011 and 75.3% (95% CI: 52.0, 98.6) in 2012. Medicaid payment rates averaged approximately 90% of Medicare payment rates throughout the study period.
Dr. Selden is hopeful that stakeholders will use the data his team collected to determine the impetus for the widening gap. He also plans to research whether payment differences affect quality metrics.
“Anytime you’re talking about a trillion dollars, it’s really important when a payment difference opens up of this magnitude,” he adds. “The difference is real … what the policy implications are is for the policy makers to decide.” TH
Visit our website for more information on healthcare payment models.
Spending on care of hospitalized patients is expected to pass $1 trillion in 2015, a new high. Thomas Selden, PhD, of the Agency for Healthcare Research and Quality recently asked where that money is likely to go. The answer: private insurers.
Dr. Selden and his colleagues report in Health Affairs this month that in 2012, private insurers’ payment rates for inpatient hospital stays were approximately 75% greater than Medicare’s payment rates, a sharp increase from the differential of approximately 10% percent during the period of 1996 to 2001. “We need to understand who’s paying what,” Dr. Selden says. “It’s the first step to a better understanding of public policy.”
The report found that “the predicted percentage difference between the rates of private insurers and those of Medicare has increased substantially over time.” In 1996, private insurers paid 106.1% of Medicare payment rates, a payment rate difference of 6.1% (95% CI: -3.2, 15.5). The difference climbed to 64.1% (95% CI: 48.3, 80.0) in 2011 and 75.3% (95% CI: 52.0, 98.6) in 2012. Medicaid payment rates averaged approximately 90% of Medicare payment rates throughout the study period.
Dr. Selden is hopeful that stakeholders will use the data his team collected to determine the impetus for the widening gap. He also plans to research whether payment differences affect quality metrics.
“Anytime you’re talking about a trillion dollars, it’s really important when a payment difference opens up of this magnitude,” he adds. “The difference is real … what the policy implications are is for the policy makers to decide.” TH
Visit our website for more information on healthcare payment models.
Spending on care of hospitalized patients is expected to pass $1 trillion in 2015, a new high. Thomas Selden, PhD, of the Agency for Healthcare Research and Quality recently asked where that money is likely to go. The answer: private insurers.
Dr. Selden and his colleagues report in Health Affairs this month that in 2012, private insurers’ payment rates for inpatient hospital stays were approximately 75% greater than Medicare’s payment rates, a sharp increase from the differential of approximately 10% percent during the period of 1996 to 2001. “We need to understand who’s paying what,” Dr. Selden says. “It’s the first step to a better understanding of public policy.”
The report found that “the predicted percentage difference between the rates of private insurers and those of Medicare has increased substantially over time.” In 1996, private insurers paid 106.1% of Medicare payment rates, a payment rate difference of 6.1% (95% CI: -3.2, 15.5). The difference climbed to 64.1% (95% CI: 48.3, 80.0) in 2011 and 75.3% (95% CI: 52.0, 98.6) in 2012. Medicaid payment rates averaged approximately 90% of Medicare payment rates throughout the study period.
Dr. Selden is hopeful that stakeholders will use the data his team collected to determine the impetus for the widening gap. He also plans to research whether payment differences affect quality metrics.
“Anytime you’re talking about a trillion dollars, it’s really important when a payment difference opens up of this magnitude,” he adds. “The difference is real … what the policy implications are is for the policy makers to decide.” TH
Visit our website for more information on healthcare payment models.
New and Noteworthy Information—January 2016
Suicide attempts and recurrent suicide attempts are associated with subsequent epilepsy, suggesting a common underlying biology, according to a study published online ahead of print December 9 in JAMA Psychiatry. The population-based retrospective cohort study in the United Kingdom included patients with incident epilepsy and control patients without a history of epilepsy. For 14,059 patients who later had an onset of epilepsy, versus 56,184 control patients, the risk for a first suicide attempt during the time period before the case patients received a diagnosis of epilepsy was increased 2.9-fold. For 278 case patients who later had an onset of epilepsy, versus 434 control patients, the risk for a recurrent suicide attempt up to and including the day that epilepsy was diagnosed was increased 1.8-fold.
Asthma is associated with an increased risk of new-onset chronic migraine one year later among individuals with episodic migraine, and the highest risk is among people with the greatest number of respiratory symptoms, according to a study published online ahead of print November 19 in Headache. Using the European Community Respiratory Health Survey, researchers defined asthma as a binary variable based on an empirical cut score and developed a Respiratory Symptom Severity Score based on the number of positive responses. This study included 4,446 individuals with episodic migraine in 2008, of whom 17% had asthma. In 2009, new-onset chronic migraine developed in 2.9% of the 2008 episodic migraine cohort, including 5.4% of the asthma subgroup and 2.5% of the non-asthma subgroup.
Vagus nerve stimulation (VNS) paired with rehabilitation is feasible and safe, according to a study published online ahead of print December 8 in Stroke. Twenty-one participants with ischemic stroke more than six months earlier and moderate to severe upper-limb impairment were randomized to VNS plus rehabilitation or rehabilitation alone. Rehabilitation consisted of three two-hour sessions per week for six weeks. There were no serious adverse device effects. One patient had transient vocal cord palsy and dysphagia after implantation. Five patients had minor adverse device effects, including nausea and taste disturbance on the evening of therapy. In the intention-to-treat analysis, the change in Fugl-Meyer Assessment-Upper Extremity scores was not significantly different between groups. In the per-protocol analysis, researchers found a significant difference in change in Fugl-Meyer Assessment-Upper Extremity score between groups.
The Chikungunya virus is a significant cause of CNS disease, according to a study published online ahead of print November 25 in Neurology. During the La Réunion outbreak between September 2005 and June 2006, 57 patients were diagnosed with Chikungunya virus-associated CNS disease, including 24 with Chikungunya virus-associated encephalitis (which corresponded to a cumulative incidence rate of 8.6 per 100,000 people). Patients with encephalitis were observed at both extremes of age categories. The cumulative incidence rates per 100,000 persons were 187 and 37 in patients younger than 1 and patients older than 65, respectively. The case-fatality rate of Chikungunya virus-associated encephalitis was 16.6%, and the proportion of children discharged with persistent disabilities was estimated at between 30% and 45%. Beyond the neonatal period, the clinical presentation and outcomes were less severe in infants than in adults.
Acute stroke is preventable to some extent in most patients, according to a study published online ahead of print December 7 in JAMA Neurology. Researchers evaluated the medical records of 274 consecutive patients discharged with a diagnosis of ischemic stroke between December 2, 2010, and June 11, 2012. Mean patient age was 67.2. Seventy-one patients (25.9%) had scores of 4 or greater on a 10-point scale, indicating that the stroke was highly preventable. Severity of stroke was not related to preventability of stroke. However, 29.6% of patients whose stroke was highly preventable were treated with IV or intra-arterial acute stroke therapy. These treatments were provided for 19.4% of patients with scores of 0, and 14% of patients with scores of 1 to 3.
Alpha-blocker therapy is associated with an increased risk of ischemic stroke during the early initiation period, especially among patients who are not taking other antihypertensive agents, according to a study published online ahead of print December 7 in Canadian Medical Association Journal. Researchers identified 7,502 men ages 50 and older as of 2007 who were incident users of alpha-blockers and who had a diagnosis of ischemic stroke during the study period, which lasted from 2007 to 2009. Investigators examined the incidence of stroke during risk periods before and after alpha-blocker prescription. Compared with the risk in the unexposed period, the risk of ischemic stroke was increased within 21 days after alpha-blocker initiation among all patients in the study population and among patients without concomitant prescriptions.
In patients with mild Alzheimer’s disease, moderate alcohol consumption (ie, two to three units per day) is associated with a significantly lower mortality over a period of 36 months, according to a study published December 11 in BMJ Open. Investigators examined data collected as part of the Danish Alzheimer’s Intervention Study (DAISY). Information about current daily alcohol consumption was obtained from 321 study participants. In all, 8% abstained from drinking alcohol, 71% drank alcohol occasionally, 17% had two to three units per day, and 4% had more than three units per day. Mortality was not significantly different in abstinent patients or in patients with an alcohol consumption of more than three units per day, compared with patients drinking one or less than one unit per day.
Stress is a potentially remediable risk factor for amnestic mild cognitive impairment (aMCI), according to a study published online ahead of print December 10 in Alzheimer Disease & Associated Disorders. The Perceived Stress Scale (PSS) was administered annually in the Einstein Aging Study to participants age 70 and older who were free of aMCI and dementia at baseline PSS administration and who had at least one subsequent annual follow-up. Cox hazard models were used to examine time to aMCI onset, adjusting for covariates. High levels of perceived stress were associated with a 30% greater risk of incident aMCI, independent of covariates. Overall, understanding the effect that perceived stress has on cognition may lead to intervention strategies that prevent the onset of aMCI and Alzheimer’s-related dementia, said the investigators.
Heptachlor epoxide, a pesticide, is associated with higher risk for signs of Parkinson’s disease, according to a study published online ahead of print December 9 in Neurology. For the study, 449 Japanese-American men with an average age of 54 were followed for more than 30 years and until death in the Honolulu-Asia Aging Study. Tests determined whether participants had lost brain cells in the substantia nigra. In 116 brains, researchers also measured the amount of heptachlor epoxide residue, which was present at high levels in Hawaii’s milk in the early 1980s. Nonsmokers who drank more than two cups of milk per day had 40% fewer brain cells in the substantia nigra than people who drank less than two cups of milk per day.
An in vivo florbetapir PET study confirms previous postmortem evidence showing an association between Alzheimer’s disease pathology and gait speed, and provides additional evidence on potential regional effects of brain β-amyloid on motor function, according to data published online ahead of print December 7 in Neurology. Cross-sectional associations between brain β-amyloid, as measured with [18F]florbetapir PET, and gait speed were examined in 128 elderly participants. Researchers found a significant association between β-amyloid in the posterior and anterior putamen, occipital cortex, precuneus, and anterior cingulate and slow gait speed. A multivariate model emphasized the posterior putamen and the precuneus. The β-amyloid burden explained as much as 9% of the variance in gait speed and significantly improved regression models that contained demographic variables, BMI, and APOE status.
Blast-related injury and loss of consciousness are common in traumatic brain injury (TBI) that is sustained while in the military, according to a study published online ahead of print December 15 in Radiology. Study participants were military service members or dependents recruited between August 2009 and August 2014. There were 834 participants with a history of TBI and 42 participants in a control group without TBI. MRIs were performed at 3 T, primarily with three-dimensional volume imaging at voxels smaller than 1 mm3. In all, 84.2% of participants reported one or more blast-related incidents, and 63.0% reported loss of consciousness at the time of injury. White matter T2-weighted hyperintense areas were the most common pathologic finding and were observed in 51.8% of TBI participants.
Researchers have created a transgenic mouse models of familial amyotrophic lateral sclerosis (ALS), according to research published in the December 2 issue of Neuron. To investigate the pathologic role of C9ORF72 in ALS and frontotemporal dementia (FTD), researchers generated a line of mice carrying a bacterial artificial chromosome containing exons one to six of the human C9ORF72 gene with approximately 500 repeats of the GGGGCC motif. The mice showed no overt behavioral phenotype, but recapitulated distinctive histopathologic features of C9ORF72 ALS/FTD, including sense and antisense intranuclear RNA foci and poly(glycine-proline) dipeptide repeat proteins. Using an artificial microRNA that targets human C9ORF72 in cultures of primary cortical neurons from the C9BAC mice, investigators attenuated expression of the C9BAC transgene and the poly(GP) dipeptides.
Oxidative stress may underlie most of the migraine triggers encountered in clinical practice, according to a study published online ahead of print December 7 in Headache. Investigators searched the literature for studies of common migraine triggers published between 1990 and 2014. The reference lists of the resulting articles were examined for further relevant studies. In all cases except pericranial pain, common migraine triggers are capable of generating oxidative stress. Mechanisms include a high rate of energy production by the mitochondria, toxicity or altered membrane properties of the mitochondria, calcium overload and excitotoxicity, neuroinflammation and activation of microglia, and activation of neuronal nicotinamide adenine dinucleotide phosphate oxidase. For some triggers, oxidants also arise as a byproduct of monoamine oxidase or cytochrome P450 processing, or from uncoupling of nitric oxide synthase.
—Kimberly Williams
Suicide attempts and recurrent suicide attempts are associated with subsequent epilepsy, suggesting a common underlying biology, according to a study published online ahead of print December 9 in JAMA Psychiatry. The population-based retrospective cohort study in the United Kingdom included patients with incident epilepsy and control patients without a history of epilepsy. For 14,059 patients who later had an onset of epilepsy, versus 56,184 control patients, the risk for a first suicide attempt during the time period before the case patients received a diagnosis of epilepsy was increased 2.9-fold. For 278 case patients who later had an onset of epilepsy, versus 434 control patients, the risk for a recurrent suicide attempt up to and including the day that epilepsy was diagnosed was increased 1.8-fold.
Asthma is associated with an increased risk of new-onset chronic migraine one year later among individuals with episodic migraine, and the highest risk is among people with the greatest number of respiratory symptoms, according to a study published online ahead of print November 19 in Headache. Using the European Community Respiratory Health Survey, researchers defined asthma as a binary variable based on an empirical cut score and developed a Respiratory Symptom Severity Score based on the number of positive responses. This study included 4,446 individuals with episodic migraine in 2008, of whom 17% had asthma. In 2009, new-onset chronic migraine developed in 2.9% of the 2008 episodic migraine cohort, including 5.4% of the asthma subgroup and 2.5% of the non-asthma subgroup.
Vagus nerve stimulation (VNS) paired with rehabilitation is feasible and safe, according to a study published online ahead of print December 8 in Stroke. Twenty-one participants with ischemic stroke more than six months earlier and moderate to severe upper-limb impairment were randomized to VNS plus rehabilitation or rehabilitation alone. Rehabilitation consisted of three two-hour sessions per week for six weeks. There were no serious adverse device effects. One patient had transient vocal cord palsy and dysphagia after implantation. Five patients had minor adverse device effects, including nausea and taste disturbance on the evening of therapy. In the intention-to-treat analysis, the change in Fugl-Meyer Assessment-Upper Extremity scores was not significantly different between groups. In the per-protocol analysis, researchers found a significant difference in change in Fugl-Meyer Assessment-Upper Extremity score between groups.
The Chikungunya virus is a significant cause of CNS disease, according to a study published online ahead of print November 25 in Neurology. During the La Réunion outbreak between September 2005 and June 2006, 57 patients were diagnosed with Chikungunya virus-associated CNS disease, including 24 with Chikungunya virus-associated encephalitis (which corresponded to a cumulative incidence rate of 8.6 per 100,000 people). Patients with encephalitis were observed at both extremes of age categories. The cumulative incidence rates per 100,000 persons were 187 and 37 in patients younger than 1 and patients older than 65, respectively. The case-fatality rate of Chikungunya virus-associated encephalitis was 16.6%, and the proportion of children discharged with persistent disabilities was estimated at between 30% and 45%. Beyond the neonatal period, the clinical presentation and outcomes were less severe in infants than in adults.
Acute stroke is preventable to some extent in most patients, according to a study published online ahead of print December 7 in JAMA Neurology. Researchers evaluated the medical records of 274 consecutive patients discharged with a diagnosis of ischemic stroke between December 2, 2010, and June 11, 2012. Mean patient age was 67.2. Seventy-one patients (25.9%) had scores of 4 or greater on a 10-point scale, indicating that the stroke was highly preventable. Severity of stroke was not related to preventability of stroke. However, 29.6% of patients whose stroke was highly preventable were treated with IV or intra-arterial acute stroke therapy. These treatments were provided for 19.4% of patients with scores of 0, and 14% of patients with scores of 1 to 3.
Alpha-blocker therapy is associated with an increased risk of ischemic stroke during the early initiation period, especially among patients who are not taking other antihypertensive agents, according to a study published online ahead of print December 7 in Canadian Medical Association Journal. Researchers identified 7,502 men ages 50 and older as of 2007 who were incident users of alpha-blockers and who had a diagnosis of ischemic stroke during the study period, which lasted from 2007 to 2009. Investigators examined the incidence of stroke during risk periods before and after alpha-blocker prescription. Compared with the risk in the unexposed period, the risk of ischemic stroke was increased within 21 days after alpha-blocker initiation among all patients in the study population and among patients without concomitant prescriptions.
In patients with mild Alzheimer’s disease, moderate alcohol consumption (ie, two to three units per day) is associated with a significantly lower mortality over a period of 36 months, according to a study published December 11 in BMJ Open. Investigators examined data collected as part of the Danish Alzheimer’s Intervention Study (DAISY). Information about current daily alcohol consumption was obtained from 321 study participants. In all, 8% abstained from drinking alcohol, 71% drank alcohol occasionally, 17% had two to three units per day, and 4% had more than three units per day. Mortality was not significantly different in abstinent patients or in patients with an alcohol consumption of more than three units per day, compared with patients drinking one or less than one unit per day.
Stress is a potentially remediable risk factor for amnestic mild cognitive impairment (aMCI), according to a study published online ahead of print December 10 in Alzheimer Disease & Associated Disorders. The Perceived Stress Scale (PSS) was administered annually in the Einstein Aging Study to participants age 70 and older who were free of aMCI and dementia at baseline PSS administration and who had at least one subsequent annual follow-up. Cox hazard models were used to examine time to aMCI onset, adjusting for covariates. High levels of perceived stress were associated with a 30% greater risk of incident aMCI, independent of covariates. Overall, understanding the effect that perceived stress has on cognition may lead to intervention strategies that prevent the onset of aMCI and Alzheimer’s-related dementia, said the investigators.
Heptachlor epoxide, a pesticide, is associated with higher risk for signs of Parkinson’s disease, according to a study published online ahead of print December 9 in Neurology. For the study, 449 Japanese-American men with an average age of 54 were followed for more than 30 years and until death in the Honolulu-Asia Aging Study. Tests determined whether participants had lost brain cells in the substantia nigra. In 116 brains, researchers also measured the amount of heptachlor epoxide residue, which was present at high levels in Hawaii’s milk in the early 1980s. Nonsmokers who drank more than two cups of milk per day had 40% fewer brain cells in the substantia nigra than people who drank less than two cups of milk per day.
An in vivo florbetapir PET study confirms previous postmortem evidence showing an association between Alzheimer’s disease pathology and gait speed, and provides additional evidence on potential regional effects of brain β-amyloid on motor function, according to data published online ahead of print December 7 in Neurology. Cross-sectional associations between brain β-amyloid, as measured with [18F]florbetapir PET, and gait speed were examined in 128 elderly participants. Researchers found a significant association between β-amyloid in the posterior and anterior putamen, occipital cortex, precuneus, and anterior cingulate and slow gait speed. A multivariate model emphasized the posterior putamen and the precuneus. The β-amyloid burden explained as much as 9% of the variance in gait speed and significantly improved regression models that contained demographic variables, BMI, and APOE status.
Blast-related injury and loss of consciousness are common in traumatic brain injury (TBI) that is sustained while in the military, according to a study published online ahead of print December 15 in Radiology. Study participants were military service members or dependents recruited between August 2009 and August 2014. There were 834 participants with a history of TBI and 42 participants in a control group without TBI. MRIs were performed at 3 T, primarily with three-dimensional volume imaging at voxels smaller than 1 mm3. In all, 84.2% of participants reported one or more blast-related incidents, and 63.0% reported loss of consciousness at the time of injury. White matter T2-weighted hyperintense areas were the most common pathologic finding and were observed in 51.8% of TBI participants.
Researchers have created a transgenic mouse models of familial amyotrophic lateral sclerosis (ALS), according to research published in the December 2 issue of Neuron. To investigate the pathologic role of C9ORF72 in ALS and frontotemporal dementia (FTD), researchers generated a line of mice carrying a bacterial artificial chromosome containing exons one to six of the human C9ORF72 gene with approximately 500 repeats of the GGGGCC motif. The mice showed no overt behavioral phenotype, but recapitulated distinctive histopathologic features of C9ORF72 ALS/FTD, including sense and antisense intranuclear RNA foci and poly(glycine-proline) dipeptide repeat proteins. Using an artificial microRNA that targets human C9ORF72 in cultures of primary cortical neurons from the C9BAC mice, investigators attenuated expression of the C9BAC transgene and the poly(GP) dipeptides.
Oxidative stress may underlie most of the migraine triggers encountered in clinical practice, according to a study published online ahead of print December 7 in Headache. Investigators searched the literature for studies of common migraine triggers published between 1990 and 2014. The reference lists of the resulting articles were examined for further relevant studies. In all cases except pericranial pain, common migraine triggers are capable of generating oxidative stress. Mechanisms include a high rate of energy production by the mitochondria, toxicity or altered membrane properties of the mitochondria, calcium overload and excitotoxicity, neuroinflammation and activation of microglia, and activation of neuronal nicotinamide adenine dinucleotide phosphate oxidase. For some triggers, oxidants also arise as a byproduct of monoamine oxidase or cytochrome P450 processing, or from uncoupling of nitric oxide synthase.
—Kimberly Williams
Suicide attempts and recurrent suicide attempts are associated with subsequent epilepsy, suggesting a common underlying biology, according to a study published online ahead of print December 9 in JAMA Psychiatry. The population-based retrospective cohort study in the United Kingdom included patients with incident epilepsy and control patients without a history of epilepsy. For 14,059 patients who later had an onset of epilepsy, versus 56,184 control patients, the risk for a first suicide attempt during the time period before the case patients received a diagnosis of epilepsy was increased 2.9-fold. For 278 case patients who later had an onset of epilepsy, versus 434 control patients, the risk for a recurrent suicide attempt up to and including the day that epilepsy was diagnosed was increased 1.8-fold.
Asthma is associated with an increased risk of new-onset chronic migraine one year later among individuals with episodic migraine, and the highest risk is among people with the greatest number of respiratory symptoms, according to a study published online ahead of print November 19 in Headache. Using the European Community Respiratory Health Survey, researchers defined asthma as a binary variable based on an empirical cut score and developed a Respiratory Symptom Severity Score based on the number of positive responses. This study included 4,446 individuals with episodic migraine in 2008, of whom 17% had asthma. In 2009, new-onset chronic migraine developed in 2.9% of the 2008 episodic migraine cohort, including 5.4% of the asthma subgroup and 2.5% of the non-asthma subgroup.
Vagus nerve stimulation (VNS) paired with rehabilitation is feasible and safe, according to a study published online ahead of print December 8 in Stroke. Twenty-one participants with ischemic stroke more than six months earlier and moderate to severe upper-limb impairment were randomized to VNS plus rehabilitation or rehabilitation alone. Rehabilitation consisted of three two-hour sessions per week for six weeks. There were no serious adverse device effects. One patient had transient vocal cord palsy and dysphagia after implantation. Five patients had minor adverse device effects, including nausea and taste disturbance on the evening of therapy. In the intention-to-treat analysis, the change in Fugl-Meyer Assessment-Upper Extremity scores was not significantly different between groups. In the per-protocol analysis, researchers found a significant difference in change in Fugl-Meyer Assessment-Upper Extremity score between groups.
The Chikungunya virus is a significant cause of CNS disease, according to a study published online ahead of print November 25 in Neurology. During the La Réunion outbreak between September 2005 and June 2006, 57 patients were diagnosed with Chikungunya virus-associated CNS disease, including 24 with Chikungunya virus-associated encephalitis (which corresponded to a cumulative incidence rate of 8.6 per 100,000 people). Patients with encephalitis were observed at both extremes of age categories. The cumulative incidence rates per 100,000 persons were 187 and 37 in patients younger than 1 and patients older than 65, respectively. The case-fatality rate of Chikungunya virus-associated encephalitis was 16.6%, and the proportion of children discharged with persistent disabilities was estimated at between 30% and 45%. Beyond the neonatal period, the clinical presentation and outcomes were less severe in infants than in adults.
Acute stroke is preventable to some extent in most patients, according to a study published online ahead of print December 7 in JAMA Neurology. Researchers evaluated the medical records of 274 consecutive patients discharged with a diagnosis of ischemic stroke between December 2, 2010, and June 11, 2012. Mean patient age was 67.2. Seventy-one patients (25.9%) had scores of 4 or greater on a 10-point scale, indicating that the stroke was highly preventable. Severity of stroke was not related to preventability of stroke. However, 29.6% of patients whose stroke was highly preventable were treated with IV or intra-arterial acute stroke therapy. These treatments were provided for 19.4% of patients with scores of 0, and 14% of patients with scores of 1 to 3.
Alpha-blocker therapy is associated with an increased risk of ischemic stroke during the early initiation period, especially among patients who are not taking other antihypertensive agents, according to a study published online ahead of print December 7 in Canadian Medical Association Journal. Researchers identified 7,502 men ages 50 and older as of 2007 who were incident users of alpha-blockers and who had a diagnosis of ischemic stroke during the study period, which lasted from 2007 to 2009. Investigators examined the incidence of stroke during risk periods before and after alpha-blocker prescription. Compared with the risk in the unexposed period, the risk of ischemic stroke was increased within 21 days after alpha-blocker initiation among all patients in the study population and among patients without concomitant prescriptions.
In patients with mild Alzheimer’s disease, moderate alcohol consumption (ie, two to three units per day) is associated with a significantly lower mortality over a period of 36 months, according to a study published December 11 in BMJ Open. Investigators examined data collected as part of the Danish Alzheimer’s Intervention Study (DAISY). Information about current daily alcohol consumption was obtained from 321 study participants. In all, 8% abstained from drinking alcohol, 71% drank alcohol occasionally, 17% had two to three units per day, and 4% had more than three units per day. Mortality was not significantly different in abstinent patients or in patients with an alcohol consumption of more than three units per day, compared with patients drinking one or less than one unit per day.
Stress is a potentially remediable risk factor for amnestic mild cognitive impairment (aMCI), according to a study published online ahead of print December 10 in Alzheimer Disease & Associated Disorders. The Perceived Stress Scale (PSS) was administered annually in the Einstein Aging Study to participants age 70 and older who were free of aMCI and dementia at baseline PSS administration and who had at least one subsequent annual follow-up. Cox hazard models were used to examine time to aMCI onset, adjusting for covariates. High levels of perceived stress were associated with a 30% greater risk of incident aMCI, independent of covariates. Overall, understanding the effect that perceived stress has on cognition may lead to intervention strategies that prevent the onset of aMCI and Alzheimer’s-related dementia, said the investigators.
Heptachlor epoxide, a pesticide, is associated with higher risk for signs of Parkinson’s disease, according to a study published online ahead of print December 9 in Neurology. For the study, 449 Japanese-American men with an average age of 54 were followed for more than 30 years and until death in the Honolulu-Asia Aging Study. Tests determined whether participants had lost brain cells in the substantia nigra. In 116 brains, researchers also measured the amount of heptachlor epoxide residue, which was present at high levels in Hawaii’s milk in the early 1980s. Nonsmokers who drank more than two cups of milk per day had 40% fewer brain cells in the substantia nigra than people who drank less than two cups of milk per day.
An in vivo florbetapir PET study confirms previous postmortem evidence showing an association between Alzheimer’s disease pathology and gait speed, and provides additional evidence on potential regional effects of brain β-amyloid on motor function, according to data published online ahead of print December 7 in Neurology. Cross-sectional associations between brain β-amyloid, as measured with [18F]florbetapir PET, and gait speed were examined in 128 elderly participants. Researchers found a significant association between β-amyloid in the posterior and anterior putamen, occipital cortex, precuneus, and anterior cingulate and slow gait speed. A multivariate model emphasized the posterior putamen and the precuneus. The β-amyloid burden explained as much as 9% of the variance in gait speed and significantly improved regression models that contained demographic variables, BMI, and APOE status.
Blast-related injury and loss of consciousness are common in traumatic brain injury (TBI) that is sustained while in the military, according to a study published online ahead of print December 15 in Radiology. Study participants were military service members or dependents recruited between August 2009 and August 2014. There were 834 participants with a history of TBI and 42 participants in a control group without TBI. MRIs were performed at 3 T, primarily with three-dimensional volume imaging at voxels smaller than 1 mm3. In all, 84.2% of participants reported one or more blast-related incidents, and 63.0% reported loss of consciousness at the time of injury. White matter T2-weighted hyperintense areas were the most common pathologic finding and were observed in 51.8% of TBI participants.
Researchers have created a transgenic mouse models of familial amyotrophic lateral sclerosis (ALS), according to research published in the December 2 issue of Neuron. To investigate the pathologic role of C9ORF72 in ALS and frontotemporal dementia (FTD), researchers generated a line of mice carrying a bacterial artificial chromosome containing exons one to six of the human C9ORF72 gene with approximately 500 repeats of the GGGGCC motif. The mice showed no overt behavioral phenotype, but recapitulated distinctive histopathologic features of C9ORF72 ALS/FTD, including sense and antisense intranuclear RNA foci and poly(glycine-proline) dipeptide repeat proteins. Using an artificial microRNA that targets human C9ORF72 in cultures of primary cortical neurons from the C9BAC mice, investigators attenuated expression of the C9BAC transgene and the poly(GP) dipeptides.
Oxidative stress may underlie most of the migraine triggers encountered in clinical practice, according to a study published online ahead of print December 7 in Headache. Investigators searched the literature for studies of common migraine triggers published between 1990 and 2014. The reference lists of the resulting articles were examined for further relevant studies. In all cases except pericranial pain, common migraine triggers are capable of generating oxidative stress. Mechanisms include a high rate of energy production by the mitochondria, toxicity or altered membrane properties of the mitochondria, calcium overload and excitotoxicity, neuroinflammation and activation of microglia, and activation of neuronal nicotinamide adenine dinucleotide phosphate oxidase. For some triggers, oxidants also arise as a byproduct of monoamine oxidase or cytochrome P450 processing, or from uncoupling of nitric oxide synthase.
—Kimberly Williams

91% who overdose on opioids continue to receive opioid prescriptions
Almost all patients who had nonfatal overdoses while taking long-term opioids for noncancer pain continued to receive opioid prescriptions, usually from the same physicians, in a nationwide cohort study published online Dec. 28 in Annals of Internal Medicine.
Clinical guidelines specify that adverse events related to the misuse of opioids are clear indications to discontinue long-term opioid therapy. But patterns of prescribing after opioid overdoses are not monitored. To examine prescribing trends following nonfatal opioid overdoses, researchers analyzed information in a database of inpatient, outpatient, and pharmacy claims from a large U.S. health insurer covering all 50 states.
They focused on 2,848 insured adults enrolled in 2000-2012 who received hospital or ED treatment for a prescription opioid overdose and were followed in the database for a median of 15 months. The prescribed drugs included codeine, dihydrocodeine, meperidine, morphine, oxycodone, hydrocodone, hydromorphone, fentanyl, oxymorphone, propoxyphene, methadone, tramadol, and levorphanol, said Dr. Marc R. Larochelle of Boston Medical Center and his associates.

A total of 2,597 of these patients (91%) continued to receive opioid prescriptions after their overdose. The primary prescriber was the same person before and after the overdose in 1,198 cases (61%). Two hundred twelve of these patients (7%) had another opioid overdose during follow-up. The likelihood of a second overdose was much higher for patients taking the highest doses of opioids (100 mg or more morphine-equivalent dosage per day), with hazard ratios of 1.13 for patients taking low doses of opioids, 1.89 for those taking mid-range doses, and 2.57 for those taking high doses.
“We could not determine the reason for the treatment patterns after the overdose; however, some prescribers may have been unaware that the opioid overdose had occurred” because there are no procedures in place to ensure provider notification in such cases. Newly introduced prescription monitoring programs may facilitate such communication, but a more rigorous approach would mandate that all overdoses be reported to public health departments, which would then notify providers and pharmacies, and perhaps secure patient referral to substance abuse treatment programs, the investigators said (Ann Intern Med. 2015 Dec 28. doi: 10.7326/M15-0038). It is possible that some overdoses stemmed from therapeutic error rather than opioid misuse, and that providers felt the risk-benefit ratio justified continued opioid treatment. But it also is likely that many providers simply did not have the knowledge and skills to identify and treat opioid misuse, they added.
“Simply eliminating opioid prescribing for patients who had an overdose is not sufficient. … because some [patients] may turn to diverted or illicit opioids. Rather, efforts to identify and treat substance use disorders in these patients are needed,” Dr. Larochelle and his associates said.
Overall, the study findings indicate that nonfatal overdoses provide a meaningful opportunity to improve the safety of opioid prescribing, but that most prescribers at present are missing this opportunity.
It’s tempting to attribute these astonishing findings to poor medical care, bad medical decisions, or sloppy prescribing, but the problem extends well beyond individual prescribers’ practices. These prescribing behaviors take place within a context in which substantial, even deadly, mistakes are inevitable.
Clinicians must be notified when their patients overdose and must know how to act on that notification. They must be taught to recognize pain and addiction as chronic diseases that require team approaches. They must learn how to taper opioid dosages appropriately, how to use buprenorphine, and what other resources in their communities are reliable. Health systems must give physicians the tools and the time they need to identify and coordinate care for affected patients, and would do well to connect overdose patients directly to addiction services at hospital discharge.
This approach would turn an opioid overdose from a devastating event into an opportunity for hope.
Dr. Jessica Gregg is at Central City Concern, a nonprofit agency serving adults and families impacted by homelessness, poverty, and addiction in Portland, Ore. She reported having no relevant financial conflicts of interest. Dr. Gregg made these remarks in an editorial accompanying Dr. Larochelle’s report (Ann Intern Med. 2015 Dec 28. doi: 10.7326/M152687).
It’s tempting to attribute these astonishing findings to poor medical care, bad medical decisions, or sloppy prescribing, but the problem extends well beyond individual prescribers’ practices. These prescribing behaviors take place within a context in which substantial, even deadly, mistakes are inevitable.
Clinicians must be notified when their patients overdose and must know how to act on that notification. They must be taught to recognize pain and addiction as chronic diseases that require team approaches. They must learn how to taper opioid dosages appropriately, how to use buprenorphine, and what other resources in their communities are reliable. Health systems must give physicians the tools and the time they need to identify and coordinate care for affected patients, and would do well to connect overdose patients directly to addiction services at hospital discharge.
This approach would turn an opioid overdose from a devastating event into an opportunity for hope.
Dr. Jessica Gregg is at Central City Concern, a nonprofit agency serving adults and families impacted by homelessness, poverty, and addiction in Portland, Ore. She reported having no relevant financial conflicts of interest. Dr. Gregg made these remarks in an editorial accompanying Dr. Larochelle’s report (Ann Intern Med. 2015 Dec 28. doi: 10.7326/M152687).
It’s tempting to attribute these astonishing findings to poor medical care, bad medical decisions, or sloppy prescribing, but the problem extends well beyond individual prescribers’ practices. These prescribing behaviors take place within a context in which substantial, even deadly, mistakes are inevitable.
Clinicians must be notified when their patients overdose and must know how to act on that notification. They must be taught to recognize pain and addiction as chronic diseases that require team approaches. They must learn how to taper opioid dosages appropriately, how to use buprenorphine, and what other resources in their communities are reliable. Health systems must give physicians the tools and the time they need to identify and coordinate care for affected patients, and would do well to connect overdose patients directly to addiction services at hospital discharge.
This approach would turn an opioid overdose from a devastating event into an opportunity for hope.
Dr. Jessica Gregg is at Central City Concern, a nonprofit agency serving adults and families impacted by homelessness, poverty, and addiction in Portland, Ore. She reported having no relevant financial conflicts of interest. Dr. Gregg made these remarks in an editorial accompanying Dr. Larochelle’s report (Ann Intern Med. 2015 Dec 28. doi: 10.7326/M152687).
Almost all patients who had nonfatal overdoses while taking long-term opioids for noncancer pain continued to receive opioid prescriptions, usually from the same physicians, in a nationwide cohort study published online Dec. 28 in Annals of Internal Medicine.
Clinical guidelines specify that adverse events related to the misuse of opioids are clear indications to discontinue long-term opioid therapy. But patterns of prescribing after opioid overdoses are not monitored. To examine prescribing trends following nonfatal opioid overdoses, researchers analyzed information in a database of inpatient, outpatient, and pharmacy claims from a large U.S. health insurer covering all 50 states.
They focused on 2,848 insured adults enrolled in 2000-2012 who received hospital or ED treatment for a prescription opioid overdose and were followed in the database for a median of 15 months. The prescribed drugs included codeine, dihydrocodeine, meperidine, morphine, oxycodone, hydrocodone, hydromorphone, fentanyl, oxymorphone, propoxyphene, methadone, tramadol, and levorphanol, said Dr. Marc R. Larochelle of Boston Medical Center and his associates.
A total of 2,597 of these patients (91%) continued to receive opioid prescriptions after their overdose. The primary prescriber was the same person before and after the overdose in 1,198 cases (61%). Two hundred twelve of these patients (7%) had another opioid overdose during follow-up. The likelihood of a second overdose was much higher for patients taking the highest doses of opioids (100 mg or more morphine-equivalent dosage per day), with hazard ratios of 1.13 for patients taking low doses of opioids, 1.89 for those taking mid-range doses, and 2.57 for those taking high doses.
“We could not determine the reason for the treatment patterns after the overdose; however, some prescribers may have been unaware that the opioid overdose had occurred” because there are no procedures in place to ensure provider notification in such cases. Newly introduced prescription monitoring programs may facilitate such communication, but a more rigorous approach would mandate that all overdoses be reported to public health departments, which would then notify providers and pharmacies, and perhaps secure patient referral to substance abuse treatment programs, the investigators said (Ann Intern Med. 2015 Dec 28. doi: 10.7326/M15-0038). It is possible that some overdoses stemmed from therapeutic error rather than opioid misuse, and that providers felt the risk-benefit ratio justified continued opioid treatment. But it also is likely that many providers simply did not have the knowledge and skills to identify and treat opioid misuse, they added.
“Simply eliminating opioid prescribing for patients who had an overdose is not sufficient. … because some [patients] may turn to diverted or illicit opioids. Rather, efforts to identify and treat substance use disorders in these patients are needed,” Dr. Larochelle and his associates said.
Overall, the study findings indicate that nonfatal overdoses provide a meaningful opportunity to improve the safety of opioid prescribing, but that most prescribers at present are missing this opportunity.
Almost all patients who had nonfatal overdoses while taking long-term opioids for noncancer pain continued to receive opioid prescriptions, usually from the same physicians, in a nationwide cohort study published online Dec. 28 in Annals of Internal Medicine.
Clinical guidelines specify that adverse events related to the misuse of opioids are clear indications to discontinue long-term opioid therapy. But patterns of prescribing after opioid overdoses are not monitored. To examine prescribing trends following nonfatal opioid overdoses, researchers analyzed information in a database of inpatient, outpatient, and pharmacy claims from a large U.S. health insurer covering all 50 states.
They focused on 2,848 insured adults enrolled in 2000-2012 who received hospital or ED treatment for a prescription opioid overdose and were followed in the database for a median of 15 months. The prescribed drugs included codeine, dihydrocodeine, meperidine, morphine, oxycodone, hydrocodone, hydromorphone, fentanyl, oxymorphone, propoxyphene, methadone, tramadol, and levorphanol, said Dr. Marc R. Larochelle of Boston Medical Center and his associates.
A total of 2,597 of these patients (91%) continued to receive opioid prescriptions after their overdose. The primary prescriber was the same person before and after the overdose in 1,198 cases (61%). Two hundred twelve of these patients (7%) had another opioid overdose during follow-up. The likelihood of a second overdose was much higher for patients taking the highest doses of opioids (100 mg or more morphine-equivalent dosage per day), with hazard ratios of 1.13 for patients taking low doses of opioids, 1.89 for those taking mid-range doses, and 2.57 for those taking high doses.
“We could not determine the reason for the treatment patterns after the overdose; however, some prescribers may have been unaware that the opioid overdose had occurred” because there are no procedures in place to ensure provider notification in such cases. Newly introduced prescription monitoring programs may facilitate such communication, but a more rigorous approach would mandate that all overdoses be reported to public health departments, which would then notify providers and pharmacies, and perhaps secure patient referral to substance abuse treatment programs, the investigators said (Ann Intern Med. 2015 Dec 28. doi: 10.7326/M15-0038). It is possible that some overdoses stemmed from therapeutic error rather than opioid misuse, and that providers felt the risk-benefit ratio justified continued opioid treatment. But it also is likely that many providers simply did not have the knowledge and skills to identify and treat opioid misuse, they added.
“Simply eliminating opioid prescribing for patients who had an overdose is not sufficient. … because some [patients] may turn to diverted or illicit opioids. Rather, efforts to identify and treat substance use disorders in these patients are needed,” Dr. Larochelle and his associates said.
Overall, the study findings indicate that nonfatal overdoses provide a meaningful opportunity to improve the safety of opioid prescribing, but that most prescribers at present are missing this opportunity.
FROM ANNALS OF INTERNAL MEDICINE
Key clinical point: Almost all patients treated at EDs for nonfatal opioid overdose continue to receive opioid prescriptions.
Major finding: 2,597 patients (91%) continued to receive opioid prescriptions after their overdose; the primary prescriber was the same person before and after the overdose in 1,198 cases (61%).
Data source: A retrospective cohort study involving 2,848 adults taking opioids for noncancer pain who overdosed and were followed for up to 2 years.
Disclosures: This study was funded by the U.S. Health Resources and Services Administration, which had no role in the design or conduct of the study. Dr. Larochelle reported also receiving support from the Ryoichi Sasakawa Fellowship Fund and Harvard Pilgrim Health Care Institute. His associates reported having no relevant financial disclosures.

My mental health hopes for 2016
The world has become a very complicated place. I suppose it always was, but today’s complexities are new and different and constantly changing with technology. It’s easy to think of ways our professional lives could be simpler and the treatment of our patients more attentive. Reflections, predictions, and all things “Happy New Year” tend to come as lists, and I don’t want to break with tradition. A list of my mental health hopes for 2016 follows:
1. I hope … that as our legislators rethink the Affordable Care Act (Obamacare), they don’t throw out the baby with the bath water, and the gains that were made remain in place. Among the good that’s been done has been the ability to keep adult children on a family policy to age 26, the elimination of “preexisting conditions” as exclusions from health insurance coverage, and the overall expansion of who is able to get some form of health insurance.

2. I hope … psychiatric treatment comes to be about so much more than medications, and that psychiatry resumes the priority of listening to patients on so many different levels. People are about more than checklists of symptoms and side effects, and their problems occur within the context of their lives. This is often more than a psychiatrist can piece together when seeing four or five patients an hour. In addition, there has been the added burden of attending to screens, data collection that is irrelevant to treatment, and paperwork burdens. I’d like to see the end of “meaningful use,” irrelevant maintenance of certification exams, and demands to use electronic medical records that divert psychiatrist time and attention without improving patient care. Technology should facilitate excellent care, not detract from it. And in my continued hope for our Internet-based world, I’ll wish for efficiency and innovation in how we use technology to learn, to communicate with one another, and to offer care to our patients, without compromising patient privacy.
3. I hope … discussions about involuntary treatment come to be about reducing the suffering of our patients, and not about preventing mass murders. It’s an expectation psychiatry simply cannot meet.
4. I hope … our legislators will come to understand that “mental illness” does not cause gun violence, and that a better predictor of violent behavior is a past history of violence, substance abuse, anger, and impulse control problems, and not the presence of a particular diagnosis or the catchall category of mental illness.
5. I hope ... we stop political discussions calling for an end to stigma while simultaneously stigmatizing those with psychiatric disorders.
6. I hope … that insurers and pharmacy benefit managers are required to limit preauthorization requirements to the most expensive and controversial forms of care. They must be required to standardize, simplify, and streamline any preauthorization procedures, and to be held to a level of accountability for the care that is denied.
7. I hope … that Medicare and Medicaid become user friendly entities that are easy to navigate and welcoming to psychiatrists so that our patients with limited incomes have access to treatment.
8. I hope … we come to appreciate the need for “housing-first” options for people who live and sleep on our streets. In a civilized society, this is a disgrace, and it benefits no one. It shouldn’t matter whether people with nowhere else to sleep do so because they are mentally ill, addicted, or simply impoverished: We need to provide better housing options. It is the humane thing to do, and it is more cost effective than paying for the correctional and medical care that result from homelessness. For those who are homeless because of untreated mental illness, it is so much easier to take medications when they have a shelf to put the bottles on.
9. I hope … for an understanding that our world is not black and white: We don’t neatly divide into those who are mentally ill and those who are not any more than we neatly divide into those who are good guys and those who are bad guys. People are complex, and mentally healthy people can behave in very disordered ways given the wrong set of circumstances. And, quite obviously, we all have a bit of both the good guy and the bad guy in us, and to believe otherwise is to be naive.
10. I hope … for both mental health and all of medicine, that all our treatments become available to all of our patients. It doesn’t matter how wonderful new treatments are if they are available only to those patients who are wealthy enough to pay for their own care, or who have Cadillac insurance policies that will reimburse for a given therapy.
Convention (or at least David Letterman) would suggest that I stop at 10 hopes. I never was one to follow arbitrary rules, so please allow me to express more mental health wishes for 2016.
11. I hope … we find some way of addressing the shortage of psychiatrists and services. We need to make it easier for patients to link with psychiatrists they find to be helpful. Our current system is difficult to negotiate for those who are healthy and have resources. Linking to care is nearly impossible for someone with limited resources who is psychotic or severely depressed.
12. And finally, I hope ... for better treatments for our patients, ones that cure mental illnesses without causing side effects, adverse reactions, or trade-offs that sometime make the cures worse than the diseases.
Thank you for bearing with me. I’m sure you have your own mental health hopes for the coming year, and perhaps some of our wishes might even come true. Wishing you a Healthy and Happy New Year!
Dr. Miller is a coauthor of “Shrink Rap: Three Psychiatrists Explain Their Work” (Baltimore: Johns Hopkins University Press, 2011).
The world has become a very complicated place. I suppose it always was, but today’s complexities are new and different and constantly changing with technology. It’s easy to think of ways our professional lives could be simpler and the treatment of our patients more attentive. Reflections, predictions, and all things “Happy New Year” tend to come as lists, and I don’t want to break with tradition. A list of my mental health hopes for 2016 follows:
1. I hope … that as our legislators rethink the Affordable Care Act (Obamacare), they don’t throw out the baby with the bath water, and the gains that were made remain in place. Among the good that’s been done has been the ability to keep adult children on a family policy to age 26, the elimination of “preexisting conditions” as exclusions from health insurance coverage, and the overall expansion of who is able to get some form of health insurance.
2. I hope … psychiatric treatment comes to be about so much more than medications, and that psychiatry resumes the priority of listening to patients on so many different levels. People are about more than checklists of symptoms and side effects, and their problems occur within the context of their lives. This is often more than a psychiatrist can piece together when seeing four or five patients an hour. In addition, there has been the added burden of attending to screens, data collection that is irrelevant to treatment, and paperwork burdens. I’d like to see the end of “meaningful use,” irrelevant maintenance of certification exams, and demands to use electronic medical records that divert psychiatrist time and attention without improving patient care. Technology should facilitate excellent care, not detract from it. And in my continued hope for our Internet-based world, I’ll wish for efficiency and innovation in how we use technology to learn, to communicate with one another, and to offer care to our patients, without compromising patient privacy.
3. I hope … discussions about involuntary treatment come to be about reducing the suffering of our patients, and not about preventing mass murders. It’s an expectation psychiatry simply cannot meet.
4. I hope … our legislators will come to understand that “mental illness” does not cause gun violence, and that a better predictor of violent behavior is a past history of violence, substance abuse, anger, and impulse control problems, and not the presence of a particular diagnosis or the catchall category of mental illness.
5. I hope ... we stop political discussions calling for an end to stigma while simultaneously stigmatizing those with psychiatric disorders.
6. I hope … that insurers and pharmacy benefit managers are required to limit preauthorization requirements to the most expensive and controversial forms of care. They must be required to standardize, simplify, and streamline any preauthorization procedures, and to be held to a level of accountability for the care that is denied.
7. I hope … that Medicare and Medicaid become user friendly entities that are easy to navigate and welcoming to psychiatrists so that our patients with limited incomes have access to treatment.
8. I hope … we come to appreciate the need for “housing-first” options for people who live and sleep on our streets. In a civilized society, this is a disgrace, and it benefits no one. It shouldn’t matter whether people with nowhere else to sleep do so because they are mentally ill, addicted, or simply impoverished: We need to provide better housing options. It is the humane thing to do, and it is more cost effective than paying for the correctional and medical care that result from homelessness. For those who are homeless because of untreated mental illness, it is so much easier to take medications when they have a shelf to put the bottles on.
9. I hope … for an understanding that our world is not black and white: We don’t neatly divide into those who are mentally ill and those who are not any more than we neatly divide into those who are good guys and those who are bad guys. People are complex, and mentally healthy people can behave in very disordered ways given the wrong set of circumstances. And, quite obviously, we all have a bit of both the good guy and the bad guy in us, and to believe otherwise is to be naive.
10. I hope … for both mental health and all of medicine, that all our treatments become available to all of our patients. It doesn’t matter how wonderful new treatments are if they are available only to those patients who are wealthy enough to pay for their own care, or who have Cadillac insurance policies that will reimburse for a given therapy.
Convention (or at least David Letterman) would suggest that I stop at 10 hopes. I never was one to follow arbitrary rules, so please allow me to express more mental health wishes for 2016.
11. I hope … we find some way of addressing the shortage of psychiatrists and services. We need to make it easier for patients to link with psychiatrists they find to be helpful. Our current system is difficult to negotiate for those who are healthy and have resources. Linking to care is nearly impossible for someone with limited resources who is psychotic or severely depressed.
12. And finally, I hope ... for better treatments for our patients, ones that cure mental illnesses without causing side effects, adverse reactions, or trade-offs that sometime make the cures worse than the diseases.
Thank you for bearing with me. I’m sure you have your own mental health hopes for the coming year, and perhaps some of our wishes might even come true. Wishing you a Healthy and Happy New Year!
Dr. Miller is a coauthor of “Shrink Rap: Three Psychiatrists Explain Their Work” (Baltimore: Johns Hopkins University Press, 2011).
The world has become a very complicated place. I suppose it always was, but today’s complexities are new and different and constantly changing with technology. It’s easy to think of ways our professional lives could be simpler and the treatment of our patients more attentive. Reflections, predictions, and all things “Happy New Year” tend to come as lists, and I don’t want to break with tradition. A list of my mental health hopes for 2016 follows:
1. I hope … that as our legislators rethink the Affordable Care Act (Obamacare), they don’t throw out the baby with the bath water, and the gains that were made remain in place. Among the good that’s been done has been the ability to keep adult children on a family policy to age 26, the elimination of “preexisting conditions” as exclusions from health insurance coverage, and the overall expansion of who is able to get some form of health insurance.
2. I hope … psychiatric treatment comes to be about so much more than medications, and that psychiatry resumes the priority of listening to patients on so many different levels. People are about more than checklists of symptoms and side effects, and their problems occur within the context of their lives. This is often more than a psychiatrist can piece together when seeing four or five patients an hour. In addition, there has been the added burden of attending to screens, data collection that is irrelevant to treatment, and paperwork burdens. I’d like to see the end of “meaningful use,” irrelevant maintenance of certification exams, and demands to use electronic medical records that divert psychiatrist time and attention without improving patient care. Technology should facilitate excellent care, not detract from it. And in my continued hope for our Internet-based world, I’ll wish for efficiency and innovation in how we use technology to learn, to communicate with one another, and to offer care to our patients, without compromising patient privacy.
3. I hope … discussions about involuntary treatment come to be about reducing the suffering of our patients, and not about preventing mass murders. It’s an expectation psychiatry simply cannot meet.
4. I hope … our legislators will come to understand that “mental illness” does not cause gun violence, and that a better predictor of violent behavior is a past history of violence, substance abuse, anger, and impulse control problems, and not the presence of a particular diagnosis or the catchall category of mental illness.
5. I hope ... we stop political discussions calling for an end to stigma while simultaneously stigmatizing those with psychiatric disorders.
6. I hope … that insurers and pharmacy benefit managers are required to limit preauthorization requirements to the most expensive and controversial forms of care. They must be required to standardize, simplify, and streamline any preauthorization procedures, and to be held to a level of accountability for the care that is denied.
7. I hope … that Medicare and Medicaid become user friendly entities that are easy to navigate and welcoming to psychiatrists so that our patients with limited incomes have access to treatment.
8. I hope … we come to appreciate the need for “housing-first” options for people who live and sleep on our streets. In a civilized society, this is a disgrace, and it benefits no one. It shouldn’t matter whether people with nowhere else to sleep do so because they are mentally ill, addicted, or simply impoverished: We need to provide better housing options. It is the humane thing to do, and it is more cost effective than paying for the correctional and medical care that result from homelessness. For those who are homeless because of untreated mental illness, it is so much easier to take medications when they have a shelf to put the bottles on.
9. I hope … for an understanding that our world is not black and white: We don’t neatly divide into those who are mentally ill and those who are not any more than we neatly divide into those who are good guys and those who are bad guys. People are complex, and mentally healthy people can behave in very disordered ways given the wrong set of circumstances. And, quite obviously, we all have a bit of both the good guy and the bad guy in us, and to believe otherwise is to be naive.
10. I hope … for both mental health and all of medicine, that all our treatments become available to all of our patients. It doesn’t matter how wonderful new treatments are if they are available only to those patients who are wealthy enough to pay for their own care, or who have Cadillac insurance policies that will reimburse for a given therapy.
Convention (or at least David Letterman) would suggest that I stop at 10 hopes. I never was one to follow arbitrary rules, so please allow me to express more mental health wishes for 2016.
11. I hope … we find some way of addressing the shortage of psychiatrists and services. We need to make it easier for patients to link with psychiatrists they find to be helpful. Our current system is difficult to negotiate for those who are healthy and have resources. Linking to care is nearly impossible for someone with limited resources who is psychotic or severely depressed.
12. And finally, I hope ... for better treatments for our patients, ones that cure mental illnesses without causing side effects, adverse reactions, or trade-offs that sometime make the cures worse than the diseases.
Thank you for bearing with me. I’m sure you have your own mental health hopes for the coming year, and perhaps some of our wishes might even come true. Wishing you a Healthy and Happy New Year!
Dr. Miller is a coauthor of “Shrink Rap: Three Psychiatrists Explain Their Work” (Baltimore: Johns Hopkins University Press, 2011).

U.S. influenza cases rise above baseline level
For the first time this flu season, the proportion of outpatient visits for influenza-like illness (ILI) was higher than the national baseline level of 2.1%, the Centers for Disease Control and Prevention reported.
According to data from the U.S. Outpatient Influenza-like Illness Surveillance Network, for the week ending Dec. 19 (week 10 of the 2015-2016 season), 2.2% of outpatient visits nationwide involved ILI, the CDC said.
South Carolina remained the only state in the “high” range of activity. New Jersey went up to level 7 – the high end of the “moderate” range – to remain the second most affected state, and Texas jumped from level 2 last week to level 6 this week to move into “moderate” territory. Alabama and Georgia both moved up to “low” status for the first time with ILI activity at level 5, and Virginia stayed at level 4 – the only other state in the “low” range, according to the CDC.
A total of 17 states were at level 2 or higher during week 10, up from 15 states the week before. Arizona, Illinois, Louisiana, Mississippi, and Pennsylvania were at level 3, and Colorado, Hawaii, Minnesota, New Mexico, and Utah were at level 2, data from the Outpatient Influenza-like Illness Surveillance Network revealed.
One influenza-related pediatric death was reported during week 10, bringing the total for the season to four.
For the first time this flu season, the proportion of outpatient visits for influenza-like illness (ILI) was higher than the national baseline level of 2.1%, the Centers for Disease Control and Prevention reported.
According to data from the U.S. Outpatient Influenza-like Illness Surveillance Network, for the week ending Dec. 19 (week 10 of the 2015-2016 season), 2.2% of outpatient visits nationwide involved ILI, the CDC said.
South Carolina remained the only state in the “high” range of activity. New Jersey went up to level 7 – the high end of the “moderate” range – to remain the second most affected state, and Texas jumped from level 2 last week to level 6 this week to move into “moderate” territory. Alabama and Georgia both moved up to “low” status for the first time with ILI activity at level 5, and Virginia stayed at level 4 – the only other state in the “low” range, according to the CDC.
A total of 17 states were at level 2 or higher during week 10, up from 15 states the week before. Arizona, Illinois, Louisiana, Mississippi, and Pennsylvania were at level 3, and Colorado, Hawaii, Minnesota, New Mexico, and Utah were at level 2, data from the Outpatient Influenza-like Illness Surveillance Network revealed.
One influenza-related pediatric death was reported during week 10, bringing the total for the season to four.
For the first time this flu season, the proportion of outpatient visits for influenza-like illness (ILI) was higher than the national baseline level of 2.1%, the Centers for Disease Control and Prevention reported.
According to data from the U.S. Outpatient Influenza-like Illness Surveillance Network, for the week ending Dec. 19 (week 10 of the 2015-2016 season), 2.2% of outpatient visits nationwide involved ILI, the CDC said.
South Carolina remained the only state in the “high” range of activity. New Jersey went up to level 7 – the high end of the “moderate” range – to remain the second most affected state, and Texas jumped from level 2 last week to level 6 this week to move into “moderate” territory. Alabama and Georgia both moved up to “low” status for the first time with ILI activity at level 5, and Virginia stayed at level 4 – the only other state in the “low” range, according to the CDC.
A total of 17 states were at level 2 or higher during week 10, up from 15 states the week before. Arizona, Illinois, Louisiana, Mississippi, and Pennsylvania were at level 3, and Colorado, Hawaii, Minnesota, New Mexico, and Utah were at level 2, data from the Outpatient Influenza-like Illness Surveillance Network revealed.
One influenza-related pediatric death was reported during week 10, bringing the total for the season to four.
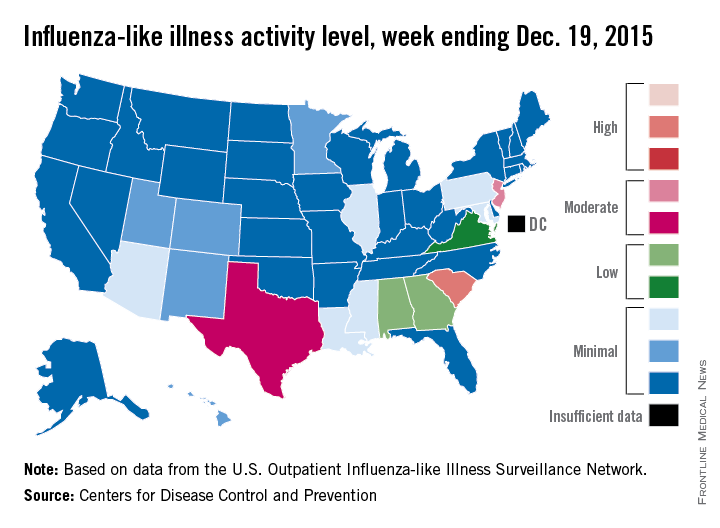
SHM Members Can Share Their Success Stories on Social Media
From care transitions to antibiotic stewardship

What does that mean? When you experience a success related to being a member, attend an SHM event, or use an SHM resource or program, tweet about it using the hashtag #SHeMpowered and mention SHM, @SHMLive. We’ll retweet and share your fantastic work with our thousands of followers.

What kinds of things can you tweet about? Pretty much anything! Attended Leadership Academy? Finished a course in the Learning Portal? Implemented a checklist from Project BOOST in your hospital? Tweet about it! Just remember to use the hashtag #SHeMpowered to be a part of the larger conversation on social media, and follow the hashtag to see how others are using their SHM membership to enhance patient care.

For more information, visit www.hospitalmedicine.org/getinvolved, and be sure to follow SHM on Twitter @SHMLive. While you’re online, check out our Facebook page, YouTube channel, and LinkedIn group, and get #SHeMpowered!
From care transitions to antibiotic stewardship
What does that mean? When you experience a success related to being a member, attend an SHM event, or use an SHM resource or program, tweet about it using the hashtag #SHeMpowered and mention SHM, @SHMLive. We’ll retweet and share your fantastic work with our thousands of followers.
What kinds of things can you tweet about? Pretty much anything! Attended Leadership Academy? Finished a course in the Learning Portal? Implemented a checklist from Project BOOST in your hospital? Tweet about it! Just remember to use the hashtag #SHeMpowered to be a part of the larger conversation on social media, and follow the hashtag to see how others are using their SHM membership to enhance patient care.
For more information, visit www.hospitalmedicine.org/getinvolved, and be sure to follow SHM on Twitter @SHMLive. While you’re online, check out our Facebook page, YouTube channel, and LinkedIn group, and get #SHeMpowered!
From care transitions to antibiotic stewardship
What does that mean? When you experience a success related to being a member, attend an SHM event, or use an SHM resource or program, tweet about it using the hashtag #SHeMpowered and mention SHM, @SHMLive. We’ll retweet and share your fantastic work with our thousands of followers.
What kinds of things can you tweet about? Pretty much anything! Attended Leadership Academy? Finished a course in the Learning Portal? Implemented a checklist from Project BOOST in your hospital? Tweet about it! Just remember to use the hashtag #SHeMpowered to be a part of the larger conversation on social media, and follow the hashtag to see how others are using their SHM membership to enhance patient care.
For more information, visit www.hospitalmedicine.org/getinvolved, and be sure to follow SHM on Twitter @SHMLive. While you’re online, check out our Facebook page, YouTube channel, and LinkedIn group, and get #SHeMpowered!
What if we are all they have?
I recently attended a CME conference at Johns Hopkins University titled “Infectious Diseases Update for Primary Care and Hospital Medicine.” As one would assume, some things were highly germane to my practice as a hospitalist, while others were, well, not relevant at all.
Don’t get me wrong, the conference was excellent and very thought provoking. It not only taught me clinically useful information, it challenged me to do more than I am used to doing for my patients; thus, I pass this challenge along to you.
One expert presented a case of an otherwise healthy patient who was found to have mildly elevated liver function tests on routine lab work done for life insurance purposes. His ALT and AST were 73 and 36, respectively, numbers that many of us would simply defer to the primary care provider to follow. But what if there is no primary care doctor? What if we are all they have?
Upon further evaluation, this patient was found to have hepatitis C. A more detailed history revealed that he had injected drugs with friends a few times over 20 years ago. The conference presenter shared statistics showing there are 2.7 million to 5 million people living with chronic HCV in America, and an estimated 45% to 60% of them are unaware of their disease – a disease that responds so well to treatment that simply screening baby boomers has the potential to prevent over 120,000 HCV-related deaths! It’s mind boggling to imagine how many people of all ages will die from this disease alone, completely oblivious to its existence.
Many people have obtained health insurance as a direct result of the Affordable Care Act, yet there are still many Americans who remain uninsured. When they are hospitalized for an acute illness, it may be the only encounter they have had with a medical professional in years. So, I ask the question again: What if we are all they have?
We can design all the elaborate hand-offs, discharge summaries, and patient instruction forms we want, but what if patients are unable to actually act on our “easy-to-understand” recommendations? Many of our patients will, out of embarrassment, nod their heads in agreement when we stress the extreme importance of following up with a primary care doctor and getting their prescriptions filled, knowing all the while that they simply don’t have the means to do so. I don’t think I will ever forget how out of touch I felt after giving a patient my spiel about taking his medication as prescribed to decrease his risk of a heart attack. He looked straight into my eyes and frankly, yet ever so respectively said, “Dr. Hester, I can either buy my medicine or I can eat.”
Sometimes it’s just that simple.
We all feel the urgency to provide high-quality care while keeping that care cost effective and time efficient, but hospitalists have a unique opportunity to not only serve our patients’ acute needs when they present via EMS to the ED, but to protect them from unforeseen catastrophes in the future. An extra (needed) test here and there, a little more time spent counseling on lifestyle changes, a few more minutes spent trying to help coordinate affordable (or free) follow-up care can all pay big dividends, and you may never have to see those patients in the hospital again. Isn’t that the goal?
Dr. Hester is a hospitalist at Baltimore-Washington Medical Center in Glen Burnie, Md. She is the creator of the Patient Whiz, a patient-engagement app for iOS. Reach her at [email protected].
I recently attended a CME conference at Johns Hopkins University titled “Infectious Diseases Update for Primary Care and Hospital Medicine.” As one would assume, some things were highly germane to my practice as a hospitalist, while others were, well, not relevant at all.
Don’t get me wrong, the conference was excellent and very thought provoking. It not only taught me clinically useful information, it challenged me to do more than I am used to doing for my patients; thus, I pass this challenge along to you.
One expert presented a case of an otherwise healthy patient who was found to have mildly elevated liver function tests on routine lab work done for life insurance purposes. His ALT and AST were 73 and 36, respectively, numbers that many of us would simply defer to the primary care provider to follow. But what if there is no primary care doctor? What if we are all they have?
Upon further evaluation, this patient was found to have hepatitis C. A more detailed history revealed that he had injected drugs with friends a few times over 20 years ago. The conference presenter shared statistics showing there are 2.7 million to 5 million people living with chronic HCV in America, and an estimated 45% to 60% of them are unaware of their disease – a disease that responds so well to treatment that simply screening baby boomers has the potential to prevent over 120,000 HCV-related deaths! It’s mind boggling to imagine how many people of all ages will die from this disease alone, completely oblivious to its existence.
Many people have obtained health insurance as a direct result of the Affordable Care Act, yet there are still many Americans who remain uninsured. When they are hospitalized for an acute illness, it may be the only encounter they have had with a medical professional in years. So, I ask the question again: What if we are all they have?
We can design all the elaborate hand-offs, discharge summaries, and patient instruction forms we want, but what if patients are unable to actually act on our “easy-to-understand” recommendations? Many of our patients will, out of embarrassment, nod their heads in agreement when we stress the extreme importance of following up with a primary care doctor and getting their prescriptions filled, knowing all the while that they simply don’t have the means to do so. I don’t think I will ever forget how out of touch I felt after giving a patient my spiel about taking his medication as prescribed to decrease his risk of a heart attack. He looked straight into my eyes and frankly, yet ever so respectively said, “Dr. Hester, I can either buy my medicine or I can eat.”
Sometimes it’s just that simple.
We all feel the urgency to provide high-quality care while keeping that care cost effective and time efficient, but hospitalists have a unique opportunity to not only serve our patients’ acute needs when they present via EMS to the ED, but to protect them from unforeseen catastrophes in the future. An extra (needed) test here and there, a little more time spent counseling on lifestyle changes, a few more minutes spent trying to help coordinate affordable (or free) follow-up care can all pay big dividends, and you may never have to see those patients in the hospital again. Isn’t that the goal?
Dr. Hester is a hospitalist at Baltimore-Washington Medical Center in Glen Burnie, Md. She is the creator of the Patient Whiz, a patient-engagement app for iOS. Reach her at [email protected].
I recently attended a CME conference at Johns Hopkins University titled “Infectious Diseases Update for Primary Care and Hospital Medicine.” As one would assume, some things were highly germane to my practice as a hospitalist, while others were, well, not relevant at all.
Don’t get me wrong, the conference was excellent and very thought provoking. It not only taught me clinically useful information, it challenged me to do more than I am used to doing for my patients; thus, I pass this challenge along to you.
One expert presented a case of an otherwise healthy patient who was found to have mildly elevated liver function tests on routine lab work done for life insurance purposes. His ALT and AST were 73 and 36, respectively, numbers that many of us would simply defer to the primary care provider to follow. But what if there is no primary care doctor? What if we are all they have?
Upon further evaluation, this patient was found to have hepatitis C. A more detailed history revealed that he had injected drugs with friends a few times over 20 years ago. The conference presenter shared statistics showing there are 2.7 million to 5 million people living with chronic HCV in America, and an estimated 45% to 60% of them are unaware of their disease – a disease that responds so well to treatment that simply screening baby boomers has the potential to prevent over 120,000 HCV-related deaths! It’s mind boggling to imagine how many people of all ages will die from this disease alone, completely oblivious to its existence.
Many people have obtained health insurance as a direct result of the Affordable Care Act, yet there are still many Americans who remain uninsured. When they are hospitalized for an acute illness, it may be the only encounter they have had with a medical professional in years. So, I ask the question again: What if we are all they have?
We can design all the elaborate hand-offs, discharge summaries, and patient instruction forms we want, but what if patients are unable to actually act on our “easy-to-understand” recommendations? Many of our patients will, out of embarrassment, nod their heads in agreement when we stress the extreme importance of following up with a primary care doctor and getting their prescriptions filled, knowing all the while that they simply don’t have the means to do so. I don’t think I will ever forget how out of touch I felt after giving a patient my spiel about taking his medication as prescribed to decrease his risk of a heart attack. He looked straight into my eyes and frankly, yet ever so respectively said, “Dr. Hester, I can either buy my medicine or I can eat.”
Sometimes it’s just that simple.
We all feel the urgency to provide high-quality care while keeping that care cost effective and time efficient, but hospitalists have a unique opportunity to not only serve our patients’ acute needs when they present via EMS to the ED, but to protect them from unforeseen catastrophes in the future. An extra (needed) test here and there, a little more time spent counseling on lifestyle changes, a few more minutes spent trying to help coordinate affordable (or free) follow-up care can all pay big dividends, and you may never have to see those patients in the hospital again. Isn’t that the goal?
Dr. Hester is a hospitalist at Baltimore-Washington Medical Center in Glen Burnie, Md. She is the creator of the Patient Whiz, a patient-engagement app for iOS. Reach her at [email protected].
Genetic Pathways, Part 2

After, test your knowledge by answering the 5 practice questions.
Practice Questions
1. A 6-month-old male infant presented to your dermatology clinic with an ash-leaf macule on the right back. What is the most common gene defect seen in this condition?
a. tuberin
b. merlin
c. neurofibromin
d. smoothened
e. hamartin
2. Bilateral acoustic neuromas are associated with what gene mutation?
a. NF1 (neurofibromin 1)
b. NF2 (neurofibromin 2)
c. PTCH1 (patched 1)
d. TSC1 (tuberous sclerosis 1)
e. TSC2 (tuberous sclerosis 2)
3. Which of the following would least likely be seen in neurofibromatosis types 1 or 2?
a. angiofibromas
b. café au lait macules
c. gliomas
d. Lisch nodules
e. neurofibromas
4. What protein is the patched 1 gene a receptor for?
a. fused
b. glioma-associated oncogene
c. smoothened
d. sonic hedgehog
e. suppressor of fused
5. A 20-year-old woman presented to your dermatology clinic with a history of numerous basal cell carcinomas. On physical examination, it is noted that she has numerous palmar pits. What finding could you find from radiograph of the head?
a. calcification of the dura
b. calcifications of the temporal lobe
c. cysts of the mandible
d. thickening of the corpus callosum
e. tumor of the cerebellum
1. A 6-month-old male infant presented to your dermatology clinic with an ash-leaf macule on the right back. What is the most common gene defect seen in this condition?
a. tuberin
b. merlin
c. neurofibromin
d. smoothened
e. hamartin
2. Bilateral acoustic neuromas are associated with what gene mutation?
a. NF1 (neurofibromin 1)
b. NF2 (neurofibromin 2)
c. PTCH1 (patched 1)
d. TSC1 (tuberous sclerosis 1)
e. TSC2 (tuberous sclerosis 2)
3. Which of the following would least likely be seen in neurofibromatosis types 1 or 2?
a. angiofibromas
b. café au lait macules
c. gliomas
d. Lisch nodules
e. neurofibromas
4. What protein is the patched 1 gene a receptor for?
a. fused
b. glioma-associated oncogene
c. smoothened
d. sonic hedgehog
e. suppressor of fused
5. A 20-year-old woman presented to your dermatology clinic with a history of numerous basal cell carcinomas. On physical examination, it is noted that she has numerous palmar pits. What finding could you find from radiograph of the head?
a. calcification of the dura
b. calcifications of the temporal lobe
c. cysts of the mandible
d. thickening of the corpus callosum
e. tumor of the cerebellum
After, test your knowledge by answering the 5 practice questions.
Practice Questions
1. A 6-month-old male infant presented to your dermatology clinic with an ash-leaf macule on the right back. What is the most common gene defect seen in this condition?
a. tuberin
b. merlin
c. neurofibromin
d. smoothened
e. hamartin
2. Bilateral acoustic neuromas are associated with what gene mutation?
a. NF1 (neurofibromin 1)
b. NF2 (neurofibromin 2)
c. PTCH1 (patched 1)
d. TSC1 (tuberous sclerosis 1)
e. TSC2 (tuberous sclerosis 2)
3. Which of the following would least likely be seen in neurofibromatosis types 1 or 2?
a. angiofibromas
b. café au lait macules
c. gliomas
d. Lisch nodules
e. neurofibromas
4. What protein is the patched 1 gene a receptor for?
a. fused
b. glioma-associated oncogene
c. smoothened
d. sonic hedgehog
e. suppressor of fused
5. A 20-year-old woman presented to your dermatology clinic with a history of numerous basal cell carcinomas. On physical examination, it is noted that she has numerous palmar pits. What finding could you find from radiograph of the head?
a. calcification of the dura
b. calcifications of the temporal lobe
c. cysts of the mandible
d. thickening of the corpus callosum
e. tumor of the cerebellum
1. A 6-month-old male infant presented to your dermatology clinic with an ash-leaf macule on the right back. What is the most common gene defect seen in this condition?
a. tuberin
b. merlin
c. neurofibromin
d. smoothened
e. hamartin
2. Bilateral acoustic neuromas are associated with what gene mutation?
a. NF1 (neurofibromin 1)
b. NF2 (neurofibromin 2)
c. PTCH1 (patched 1)
d. TSC1 (tuberous sclerosis 1)
e. TSC2 (tuberous sclerosis 2)
3. Which of the following would least likely be seen in neurofibromatosis types 1 or 2?
a. angiofibromas
b. café au lait macules
c. gliomas
d. Lisch nodules
e. neurofibromas
4. What protein is the patched 1 gene a receptor for?
a. fused
b. glioma-associated oncogene
c. smoothened
d. sonic hedgehog
e. suppressor of fused
5. A 20-year-old woman presented to your dermatology clinic with a history of numerous basal cell carcinomas. On physical examination, it is noted that she has numerous palmar pits. What finding could you find from radiograph of the head?
a. calcification of the dura
b. calcifications of the temporal lobe
c. cysts of the mandible
d. thickening of the corpus callosum
e. tumor of the cerebellum
After, test your knowledge by answering the 5 practice questions.
Practice Questions
1. A 6-month-old male infant presented to your dermatology clinic with an ash-leaf macule on the right back. What is the most common gene defect seen in this condition?
a. tuberin
b. merlin
c. neurofibromin
d. smoothened
e. hamartin
2. Bilateral acoustic neuromas are associated with what gene mutation?
a. NF1 (neurofibromin 1)
b. NF2 (neurofibromin 2)
c. PTCH1 (patched 1)
d. TSC1 (tuberous sclerosis 1)
e. TSC2 (tuberous sclerosis 2)
3. Which of the following would least likely be seen in neurofibromatosis types 1 or 2?
a. angiofibromas
b. café au lait macules
c. gliomas
d. Lisch nodules
e. neurofibromas
4. What protein is the patched 1 gene a receptor for?
a. fused
b. glioma-associated oncogene
c. smoothened
d. sonic hedgehog
e. suppressor of fused
5. A 20-year-old woman presented to your dermatology clinic with a history of numerous basal cell carcinomas. On physical examination, it is noted that she has numerous palmar pits. What finding could you find from radiograph of the head?
a. calcification of the dura
b. calcifications of the temporal lobe
c. cysts of the mandible
d. thickening of the corpus callosum
e. tumor of the cerebellum
1. A 6-month-old male infant presented to your dermatology clinic with an ash-leaf macule on the right back. What is the most common gene defect seen in this condition?
a. tuberin
b. merlin
c. neurofibromin
d. smoothened
e. hamartin
2. Bilateral acoustic neuromas are associated with what gene mutation?
a. NF1 (neurofibromin 1)
b. NF2 (neurofibromin 2)
c. PTCH1 (patched 1)
d. TSC1 (tuberous sclerosis 1)
e. TSC2 (tuberous sclerosis 2)
3. Which of the following would least likely be seen in neurofibromatosis types 1 or 2?
a. angiofibromas
b. café au lait macules
c. gliomas
d. Lisch nodules
e. neurofibromas
4. What protein is the patched 1 gene a receptor for?
a. fused
b. glioma-associated oncogene
c. smoothened
d. sonic hedgehog
e. suppressor of fused
5. A 20-year-old woman presented to your dermatology clinic with a history of numerous basal cell carcinomas. On physical examination, it is noted that she has numerous palmar pits. What finding could you find from radiograph of the head?
a. calcification of the dura
b. calcifications of the temporal lobe
c. cysts of the mandible
d. thickening of the corpus callosum
e. tumor of the cerebellum
