User login
Setting Up Your New Physician for Success
Practices and hospitals invest significant time and money in recruiting a new physician. From phone interviews to site visits to contract negotiations, it’s a long and involved process.
Beyond setting up a new physician’s office and appointment schedule, completing human resources paperwork, and ordering business cards, what does your practice do to support new physicians to ensure they are successful? Although a new colleague may arrive with excellent clinical skills, even the most promising surgeon can fall short if not provided with the right expectations, training, and collegial support. Here’s how to fast track your new physician to professional heights.
Credentialing Is Key
At the crux of a new physician’s success is credentialing him or her with hospitals and insurance plans before the official start date to see patients.
“A state medical license is the first domino,” says orthopedic surgeon Michael R. Marks, MD, MBA, consultant and coding educator with KarenZupko & Associates, Inc. Marks has led or participated in physician recruitment in orthopedic and multispecialty groups. The firm has developed a comprehensive New Physician Onboarding Checklist, available at https://www.karenzupko.com/new-physician-onboarding-checklist/.
“Without a medical license,” Marks continues, “you can’t get the new physician hospital privileges and you can’t get him or her credentialed with plans. Without being credentialed, the physician can’t bill for patients treated.” Because commercial carriers won’t allow retrospective billing for services already rendered, “even a 3-month delay in credentialing could cost an orthopedic practice $60,000 to $180,000 in lost revenue.”
And if you think you can bill the new physician’s services under another partner’s name, you are incorrect. “The billing physician will have signed the note, but not have treated the patient,” warns Marks. “This is improper billing. Don’t do it.”
The remedy for ensuring that the new physician is credentialed is simple: get organized and plan ahead.

“When I first started participating in recruitment, I remember telling physicians, ‘I need you tomorrow!’” admits Amon T. Ferry, MD, a practicing orthopedist who leads recruitment efforts at IMS Orthopedics, a division of Integrated Medical Specialists in Phoenix, Arizona. “So they’d get hired before the practice was prepared and before credentialing was completed. Now, I set more realistic expectations,” he says, noting that in Arizona it takes 3 months to get a medical license, 6 months to contract with the hospital, and 9 months to get on insurance plans. And even after a plan has credentialed a new physician, “sometimes it still takes 4 to 6 weeks before the physician’s data is loaded into the plan’s computer systems.”
“The way to do credentialing right is to get all departments communicating,” Marks says. “If you keep everyone siloed, staff don’t understand that a lack of timeliness on their part impacts other areas of the practice.”
Ferry agrees, and says his group learned to organize its multiple departments after making mistakes and missing deadlines. “We now have an 8-page pre-employment application for new physicians,” he explains. “In addition to asking for contact information and everything we need to know in order to get the physician credentialed, we ask questions about malpractice suit history and whether there are issues with the medical board. We also ask about gaps in employment and details about where the physician has practiced in the past.” All of this is done to identify early whether credentialing will require more time and effort. Ferry says that the application has solved a number of processing problems the practice had in the past.
And whether credentialing is done within the practice or outsourced, Ferry says that it pays to be persistent. “Don’t sit back and assume it will get done. Even if you have outsourced credentialing to a company, someone must check with payers and hospitals weekly and provide the practice a status update.”
In one case, when getting a new physician contracted at a hospital was taking forever, Ferry directed the staff to call. “Turns out, they had been trying to reach us and had the wrong phone number,” he says. “When people are processing thousands of physician renewals, things get lost. You have to be proactive and be your own advocate. Don’t be afraid to be the squeaky wheel.”
Staff Relationships and Operational Wisdom
Marks points out that in many practices, the new physician is shown the examination rooms and his or her office, gets electronic health record (EHR) training, and that’s it. To be successful, Marks insists that the new physician must build relationships with personnel and understand operational basics. “In other business industries, successful leaders understand at least the basics of what everyone does. Part of how they do this is by getting to know the employees.”
Ideally, Marks advises that new physicians spend time with each staff member. “The best time to do this is in the first few weeks of employment,” he suggests. “Odds are, the new orthopedist doesn’t have 40 patients a day on the schedule. So schedule conversations within the first few weeks or month, and schedule observation time as well. When a patient complains about check-in, the physician will have an understanding of how things work up there if he or she knows the basic processes.” The new doctor should also spend time in the billing office getting to know the challenges faced by staff, and sit with the surgery coordinator to understand the process of getting cases booked and scheduled.
Plan for an initial and then periodic meetings with the practice administrator and other supervisors. Transparency about business operations, data, and strategy will help the new physician get up to speed faster.
“The executive director of our group was an absolutely invaluable information resource,” says Kathryn J. McCarthy, MD, an orthopedic spine surgeon with Arkansas Specialty Orthopaedics in Little Rock, Arkansas. McCarthy has been with the group for 3 years.
The practice’s executive director developed and presented a PowerPoint (Microsoft) explaining general business procedures, expectations for the coding and billing process, and pertinent compliance and risk issues. She had also developed an interactive model of the compensation formula and buy-in program, using Excel (Microsoft). McCarthy met with the executive director at 3 months, 6 months, and 9 months to review her patient and case volumes and how they were trending against the estimates made about her income, bonus, and buy-in status.
From the new physician’s perspective, McCarthy says having the new physician understand the complexities of certain business systems helps them understand things better. “If you sit in the business meetings long enough, you figure it out,” she says, “but it would have made some of the growing pains less painful if I understood what my overhead charge was going to, or more about the workflow of the clinic.” She adds that an overview of hospital relationships and any overlapping ownership interests will benefit new physicians as well.
“I think it’s useful to provide new physicians with a history of the practice and the vision of where things are going,” McCarthy says. “It’s important to outline the business vision, especially for subspecialties. If you explain to the new physician where you want to grow and when the practice plans on bringing on the next physician, it could really drive someone to grow their practice.”
Don’t Underestimate the Need for Coding Training
“When fellows come out of training, they are comfortable with clinical activity but uncomfortable with business administration,” Marks says. “And we know they don’t get training on coding and billing.”
Marks cites a recent conversation at an American Academy of Orthopaedic Surgeons (AAOS) coding workshop. “A surgeon new in practice told me, ‘I’ve been in practice for 4 months. I understand the clinical side but nobody educated me about coding and billing before this course.’” Practices must provide new physicians with coding and documentation training, and coach them to make sure they feel up to speed and comfortable. “The practice’s future revenue depends on it,” Marks says.
McCarthy agrees. “Having an administrative mentorship for coding is incredibly valuable. They don’t teach it in school.”
So from a practical standpoint, purchase AAOS’ Orthopaedic Code-X, a software tool that will help the new physician navigate and integrate Current Procedural Terminology (CPT), ICD-10 (International Classification of Diseases, Tenth Revision), and other coding data easily and accurately. Send him or her to one of the Academy’s regional coding and reimbursement workshops as well. “It will behoove the practice to send them even before they start seeing patients,” Marks says.
And don’t just stop there. High-performing groups conduct peer reviews of evaluation and management (E/M) and operative notes, blinding the codes billed and discussing which CPT and ICD-10 codes are appropriate for the visit or case. “It will take time for the new physician to completely integrate coding with their clinical care,” says Marks. “Peer review sessions, as well as having a partner review codes before they go to the billing office, can help speed learning.”
Collegial Coaching Counts
The week before her official start day, Mc-Carthy scrubbed in as a first assist with each of her new partners. “It was a great way to start ramping up,” she says. “I could see what kind of equipment was present in the hospitals, and got a touch point for hospital logistics. Plus, as a young surgeon it’s great to see how your skill sets match up with your new partners, and which best practices are being deployed by the group.”
This kind of “collegial coaching” is a vital part of the clinical and cultural integration to the practice. Beyond providing clinical support, it builds relationships and trust among the group, and fosters collaboration.
Arkansas Specialty Orthopaedics organized McCarthy’s clinic and operating room (OR) schedules so that a partner was always present. “There was also someone I could bounce ideas off of,” McCarthy explains. “Every day in the OR, there was a partner there at the same time. If I got into a sticky situation, one of my colleagues was willing to come in and scrub in the OR.”
McCarthy says that patients responded favorably when she told them her plan was developed in conjunction with her partners. “Patients find comfort in knowing that several people’s opinions were considered,” she says. “And as a young surgeon, knowing that you have backup, even if you don’t use it, when caring for high-risk and complex cases really means a lot,” she says.
And although her group didn’t offer a formal mentoring program, McCarthy found that an informal mentorship grew organically when a friendship developed with one of her new partners. “In the first 6 months, every single weekend we sat by the pool and rolled through a ton of cases,” she says. “That was fabulous and it alleviated so much stress for me.” And when it was time for McCarthy to move into board case selection, this colleague and another were instrumental in her board preparation because, “they knew my style and where I would need to focus.”
IMS Orthopedics’ approach is to provide the staff and systems that allow new physicians to step up and take responsibility. “If they want to scrub in with me, that’s great. If they’d like to visit additional facilities and get the lay of the land, we encourage it. But we don’t do a lot of handholding. We set them up for success and make sure people are in place to help them,” says Ferry.
A Marketing Plan Is a Must
“The vast majority of practices do very little when it comes to thinking about how to market and build the practice of their new physician,” Marks says. “Practice-building is more of a challenge for surgical specialists today than it was in the old days when new surgeons could easily meet internists as they were rounding at the hospital. Now, a new physician and the practice must come up with a game plan.”
That game plan starts with the easy things: order business cards, schedule a photo shoot, and update the practice’s Web site pages with the physician’s biography and an introductory video. But with social media, online reviews, and subspecialty competition, Marks says practices must think beyond the basics. Think through each element of marketing, from online to outreach to developing referral relationships.
“I tell practices to draft a written marketing plan,” he says. “Not only does it provide a roadmap for the new physician, but also indicates that the practice has put some thought into how he or she can build a practice. It can make the new physician feel less overwhelmed knowing that he or she doesn’t have to do the marketing alone.” Once you’ve developed a list of actions, Marks suggests creating a spreadsheet with deadlines, and ensuring each action is completed.
McCarthy was scheduled to visit family practice clinics, and joined by the administrator who “handed out cookies and cards while I talked,” she says. Arkansas Specialty Orthopaedics also hired an external marketing firm to develop promotional opportunities for her. For example, “I was scheduled to appear on news channels, where I discussed new and interesting procedures,” she says. “It got my name out into the community.”
If your practice is too small to hire an outside firm, Marks suggests reaching out to agencies such as nursing homes, fitness centers, or the YMCA, which frequently offers educational programs for members. “Contact the administrators or medical directors in these organizations. A few minutes on the phone or a short visit can go a long way to building these relationships and getting your new physician on the map.”
As the old saying goes, an ounce of prevention is worth a pound of cure. Scheduling time for orientation, training, staff integration, and collegial coaching will speed up a new physician’s integration into the practice, and increase his or her opportunity for success.
Practices and hospitals invest significant time and money in recruiting a new physician. From phone interviews to site visits to contract negotiations, it’s a long and involved process.
Beyond setting up a new physician’s office and appointment schedule, completing human resources paperwork, and ordering business cards, what does your practice do to support new physicians to ensure they are successful? Although a new colleague may arrive with excellent clinical skills, even the most promising surgeon can fall short if not provided with the right expectations, training, and collegial support. Here’s how to fast track your new physician to professional heights.
Credentialing Is Key
At the crux of a new physician’s success is credentialing him or her with hospitals and insurance plans before the official start date to see patients.
“A state medical license is the first domino,” says orthopedic surgeon Michael R. Marks, MD, MBA, consultant and coding educator with KarenZupko & Associates, Inc. Marks has led or participated in physician recruitment in orthopedic and multispecialty groups. The firm has developed a comprehensive New Physician Onboarding Checklist, available at https://www.karenzupko.com/new-physician-onboarding-checklist/.
“Without a medical license,” Marks continues, “you can’t get the new physician hospital privileges and you can’t get him or her credentialed with plans. Without being credentialed, the physician can’t bill for patients treated.” Because commercial carriers won’t allow retrospective billing for services already rendered, “even a 3-month delay in credentialing could cost an orthopedic practice $60,000 to $180,000 in lost revenue.”
And if you think you can bill the new physician’s services under another partner’s name, you are incorrect. “The billing physician will have signed the note, but not have treated the patient,” warns Marks. “This is improper billing. Don’t do it.”
The remedy for ensuring that the new physician is credentialed is simple: get organized and plan ahead.
“When I first started participating in recruitment, I remember telling physicians, ‘I need you tomorrow!’” admits Amon T. Ferry, MD, a practicing orthopedist who leads recruitment efforts at IMS Orthopedics, a division of Integrated Medical Specialists in Phoenix, Arizona. “So they’d get hired before the practice was prepared and before credentialing was completed. Now, I set more realistic expectations,” he says, noting that in Arizona it takes 3 months to get a medical license, 6 months to contract with the hospital, and 9 months to get on insurance plans. And even after a plan has credentialed a new physician, “sometimes it still takes 4 to 6 weeks before the physician’s data is loaded into the plan’s computer systems.”
“The way to do credentialing right is to get all departments communicating,” Marks says. “If you keep everyone siloed, staff don’t understand that a lack of timeliness on their part impacts other areas of the practice.”
Ferry agrees, and says his group learned to organize its multiple departments after making mistakes and missing deadlines. “We now have an 8-page pre-employment application for new physicians,” he explains. “In addition to asking for contact information and everything we need to know in order to get the physician credentialed, we ask questions about malpractice suit history and whether there are issues with the medical board. We also ask about gaps in employment and details about where the physician has practiced in the past.” All of this is done to identify early whether credentialing will require more time and effort. Ferry says that the application has solved a number of processing problems the practice had in the past.
And whether credentialing is done within the practice or outsourced, Ferry says that it pays to be persistent. “Don’t sit back and assume it will get done. Even if you have outsourced credentialing to a company, someone must check with payers and hospitals weekly and provide the practice a status update.”
In one case, when getting a new physician contracted at a hospital was taking forever, Ferry directed the staff to call. “Turns out, they had been trying to reach us and had the wrong phone number,” he says. “When people are processing thousands of physician renewals, things get lost. You have to be proactive and be your own advocate. Don’t be afraid to be the squeaky wheel.”
Staff Relationships and Operational Wisdom
Marks points out that in many practices, the new physician is shown the examination rooms and his or her office, gets electronic health record (EHR) training, and that’s it. To be successful, Marks insists that the new physician must build relationships with personnel and understand operational basics. “In other business industries, successful leaders understand at least the basics of what everyone does. Part of how they do this is by getting to know the employees.”
Ideally, Marks advises that new physicians spend time with each staff member. “The best time to do this is in the first few weeks of employment,” he suggests. “Odds are, the new orthopedist doesn’t have 40 patients a day on the schedule. So schedule conversations within the first few weeks or month, and schedule observation time as well. When a patient complains about check-in, the physician will have an understanding of how things work up there if he or she knows the basic processes.” The new doctor should also spend time in the billing office getting to know the challenges faced by staff, and sit with the surgery coordinator to understand the process of getting cases booked and scheduled.
Plan for an initial and then periodic meetings with the practice administrator and other supervisors. Transparency about business operations, data, and strategy will help the new physician get up to speed faster.
“The executive director of our group was an absolutely invaluable information resource,” says Kathryn J. McCarthy, MD, an orthopedic spine surgeon with Arkansas Specialty Orthopaedics in Little Rock, Arkansas. McCarthy has been with the group for 3 years.
The practice’s executive director developed and presented a PowerPoint (Microsoft) explaining general business procedures, expectations for the coding and billing process, and pertinent compliance and risk issues. She had also developed an interactive model of the compensation formula and buy-in program, using Excel (Microsoft). McCarthy met with the executive director at 3 months, 6 months, and 9 months to review her patient and case volumes and how they were trending against the estimates made about her income, bonus, and buy-in status.
From the new physician’s perspective, McCarthy says having the new physician understand the complexities of certain business systems helps them understand things better. “If you sit in the business meetings long enough, you figure it out,” she says, “but it would have made some of the growing pains less painful if I understood what my overhead charge was going to, or more about the workflow of the clinic.” She adds that an overview of hospital relationships and any overlapping ownership interests will benefit new physicians as well.
“I think it’s useful to provide new physicians with a history of the practice and the vision of where things are going,” McCarthy says. “It’s important to outline the business vision, especially for subspecialties. If you explain to the new physician where you want to grow and when the practice plans on bringing on the next physician, it could really drive someone to grow their practice.”
Don’t Underestimate the Need for Coding Training
“When fellows come out of training, they are comfortable with clinical activity but uncomfortable with business administration,” Marks says. “And we know they don’t get training on coding and billing.”
Marks cites a recent conversation at an American Academy of Orthopaedic Surgeons (AAOS) coding workshop. “A surgeon new in practice told me, ‘I’ve been in practice for 4 months. I understand the clinical side but nobody educated me about coding and billing before this course.’” Practices must provide new physicians with coding and documentation training, and coach them to make sure they feel up to speed and comfortable. “The practice’s future revenue depends on it,” Marks says.
McCarthy agrees. “Having an administrative mentorship for coding is incredibly valuable. They don’t teach it in school.”
So from a practical standpoint, purchase AAOS’ Orthopaedic Code-X, a software tool that will help the new physician navigate and integrate Current Procedural Terminology (CPT), ICD-10 (International Classification of Diseases, Tenth Revision), and other coding data easily and accurately. Send him or her to one of the Academy’s regional coding and reimbursement workshops as well. “It will behoove the practice to send them even before they start seeing patients,” Marks says.
And don’t just stop there. High-performing groups conduct peer reviews of evaluation and management (E/M) and operative notes, blinding the codes billed and discussing which CPT and ICD-10 codes are appropriate for the visit or case. “It will take time for the new physician to completely integrate coding with their clinical care,” says Marks. “Peer review sessions, as well as having a partner review codes before they go to the billing office, can help speed learning.”
Collegial Coaching Counts
The week before her official start day, Mc-Carthy scrubbed in as a first assist with each of her new partners. “It was a great way to start ramping up,” she says. “I could see what kind of equipment was present in the hospitals, and got a touch point for hospital logistics. Plus, as a young surgeon it’s great to see how your skill sets match up with your new partners, and which best practices are being deployed by the group.”
This kind of “collegial coaching” is a vital part of the clinical and cultural integration to the practice. Beyond providing clinical support, it builds relationships and trust among the group, and fosters collaboration.
Arkansas Specialty Orthopaedics organized McCarthy’s clinic and operating room (OR) schedules so that a partner was always present. “There was also someone I could bounce ideas off of,” McCarthy explains. “Every day in the OR, there was a partner there at the same time. If I got into a sticky situation, one of my colleagues was willing to come in and scrub in the OR.”
McCarthy says that patients responded favorably when she told them her plan was developed in conjunction with her partners. “Patients find comfort in knowing that several people’s opinions were considered,” she says. “And as a young surgeon, knowing that you have backup, even if you don’t use it, when caring for high-risk and complex cases really means a lot,” she says.
And although her group didn’t offer a formal mentoring program, McCarthy found that an informal mentorship grew organically when a friendship developed with one of her new partners. “In the first 6 months, every single weekend we sat by the pool and rolled through a ton of cases,” she says. “That was fabulous and it alleviated so much stress for me.” And when it was time for McCarthy to move into board case selection, this colleague and another were instrumental in her board preparation because, “they knew my style and where I would need to focus.”
IMS Orthopedics’ approach is to provide the staff and systems that allow new physicians to step up and take responsibility. “If they want to scrub in with me, that’s great. If they’d like to visit additional facilities and get the lay of the land, we encourage it. But we don’t do a lot of handholding. We set them up for success and make sure people are in place to help them,” says Ferry.
A Marketing Plan Is a Must
“The vast majority of practices do very little when it comes to thinking about how to market and build the practice of their new physician,” Marks says. “Practice-building is more of a challenge for surgical specialists today than it was in the old days when new surgeons could easily meet internists as they were rounding at the hospital. Now, a new physician and the practice must come up with a game plan.”
That game plan starts with the easy things: order business cards, schedule a photo shoot, and update the practice’s Web site pages with the physician’s biography and an introductory video. But with social media, online reviews, and subspecialty competition, Marks says practices must think beyond the basics. Think through each element of marketing, from online to outreach to developing referral relationships.
“I tell practices to draft a written marketing plan,” he says. “Not only does it provide a roadmap for the new physician, but also indicates that the practice has put some thought into how he or she can build a practice. It can make the new physician feel less overwhelmed knowing that he or she doesn’t have to do the marketing alone.” Once you’ve developed a list of actions, Marks suggests creating a spreadsheet with deadlines, and ensuring each action is completed.
McCarthy was scheduled to visit family practice clinics, and joined by the administrator who “handed out cookies and cards while I talked,” she says. Arkansas Specialty Orthopaedics also hired an external marketing firm to develop promotional opportunities for her. For example, “I was scheduled to appear on news channels, where I discussed new and interesting procedures,” she says. “It got my name out into the community.”
If your practice is too small to hire an outside firm, Marks suggests reaching out to agencies such as nursing homes, fitness centers, or the YMCA, which frequently offers educational programs for members. “Contact the administrators or medical directors in these organizations. A few minutes on the phone or a short visit can go a long way to building these relationships and getting your new physician on the map.”
As the old saying goes, an ounce of prevention is worth a pound of cure. Scheduling time for orientation, training, staff integration, and collegial coaching will speed up a new physician’s integration into the practice, and increase his or her opportunity for success.
Practices and hospitals invest significant time and money in recruiting a new physician. From phone interviews to site visits to contract negotiations, it’s a long and involved process.
Beyond setting up a new physician’s office and appointment schedule, completing human resources paperwork, and ordering business cards, what does your practice do to support new physicians to ensure they are successful? Although a new colleague may arrive with excellent clinical skills, even the most promising surgeon can fall short if not provided with the right expectations, training, and collegial support. Here’s how to fast track your new physician to professional heights.
Credentialing Is Key
At the crux of a new physician’s success is credentialing him or her with hospitals and insurance plans before the official start date to see patients.
“A state medical license is the first domino,” says orthopedic surgeon Michael R. Marks, MD, MBA, consultant and coding educator with KarenZupko & Associates, Inc. Marks has led or participated in physician recruitment in orthopedic and multispecialty groups. The firm has developed a comprehensive New Physician Onboarding Checklist, available at https://www.karenzupko.com/new-physician-onboarding-checklist/.
“Without a medical license,” Marks continues, “you can’t get the new physician hospital privileges and you can’t get him or her credentialed with plans. Without being credentialed, the physician can’t bill for patients treated.” Because commercial carriers won’t allow retrospective billing for services already rendered, “even a 3-month delay in credentialing could cost an orthopedic practice $60,000 to $180,000 in lost revenue.”
And if you think you can bill the new physician’s services under another partner’s name, you are incorrect. “The billing physician will have signed the note, but not have treated the patient,” warns Marks. “This is improper billing. Don’t do it.”
The remedy for ensuring that the new physician is credentialed is simple: get organized and plan ahead.
“When I first started participating in recruitment, I remember telling physicians, ‘I need you tomorrow!’” admits Amon T. Ferry, MD, a practicing orthopedist who leads recruitment efforts at IMS Orthopedics, a division of Integrated Medical Specialists in Phoenix, Arizona. “So they’d get hired before the practice was prepared and before credentialing was completed. Now, I set more realistic expectations,” he says, noting that in Arizona it takes 3 months to get a medical license, 6 months to contract with the hospital, and 9 months to get on insurance plans. And even after a plan has credentialed a new physician, “sometimes it still takes 4 to 6 weeks before the physician’s data is loaded into the plan’s computer systems.”
“The way to do credentialing right is to get all departments communicating,” Marks says. “If you keep everyone siloed, staff don’t understand that a lack of timeliness on their part impacts other areas of the practice.”
Ferry agrees, and says his group learned to organize its multiple departments after making mistakes and missing deadlines. “We now have an 8-page pre-employment application for new physicians,” he explains. “In addition to asking for contact information and everything we need to know in order to get the physician credentialed, we ask questions about malpractice suit history and whether there are issues with the medical board. We also ask about gaps in employment and details about where the physician has practiced in the past.” All of this is done to identify early whether credentialing will require more time and effort. Ferry says that the application has solved a number of processing problems the practice had in the past.
And whether credentialing is done within the practice or outsourced, Ferry says that it pays to be persistent. “Don’t sit back and assume it will get done. Even if you have outsourced credentialing to a company, someone must check with payers and hospitals weekly and provide the practice a status update.”
In one case, when getting a new physician contracted at a hospital was taking forever, Ferry directed the staff to call. “Turns out, they had been trying to reach us and had the wrong phone number,” he says. “When people are processing thousands of physician renewals, things get lost. You have to be proactive and be your own advocate. Don’t be afraid to be the squeaky wheel.”
Staff Relationships and Operational Wisdom
Marks points out that in many practices, the new physician is shown the examination rooms and his or her office, gets electronic health record (EHR) training, and that’s it. To be successful, Marks insists that the new physician must build relationships with personnel and understand operational basics. “In other business industries, successful leaders understand at least the basics of what everyone does. Part of how they do this is by getting to know the employees.”
Ideally, Marks advises that new physicians spend time with each staff member. “The best time to do this is in the first few weeks of employment,” he suggests. “Odds are, the new orthopedist doesn’t have 40 patients a day on the schedule. So schedule conversations within the first few weeks or month, and schedule observation time as well. When a patient complains about check-in, the physician will have an understanding of how things work up there if he or she knows the basic processes.” The new doctor should also spend time in the billing office getting to know the challenges faced by staff, and sit with the surgery coordinator to understand the process of getting cases booked and scheduled.
Plan for an initial and then periodic meetings with the practice administrator and other supervisors. Transparency about business operations, data, and strategy will help the new physician get up to speed faster.
“The executive director of our group was an absolutely invaluable information resource,” says Kathryn J. McCarthy, MD, an orthopedic spine surgeon with Arkansas Specialty Orthopaedics in Little Rock, Arkansas. McCarthy has been with the group for 3 years.
The practice’s executive director developed and presented a PowerPoint (Microsoft) explaining general business procedures, expectations for the coding and billing process, and pertinent compliance and risk issues. She had also developed an interactive model of the compensation formula and buy-in program, using Excel (Microsoft). McCarthy met with the executive director at 3 months, 6 months, and 9 months to review her patient and case volumes and how they were trending against the estimates made about her income, bonus, and buy-in status.
From the new physician’s perspective, McCarthy says having the new physician understand the complexities of certain business systems helps them understand things better. “If you sit in the business meetings long enough, you figure it out,” she says, “but it would have made some of the growing pains less painful if I understood what my overhead charge was going to, or more about the workflow of the clinic.” She adds that an overview of hospital relationships and any overlapping ownership interests will benefit new physicians as well.
“I think it’s useful to provide new physicians with a history of the practice and the vision of where things are going,” McCarthy says. “It’s important to outline the business vision, especially for subspecialties. If you explain to the new physician where you want to grow and when the practice plans on bringing on the next physician, it could really drive someone to grow their practice.”
Don’t Underestimate the Need for Coding Training
“When fellows come out of training, they are comfortable with clinical activity but uncomfortable with business administration,” Marks says. “And we know they don’t get training on coding and billing.”
Marks cites a recent conversation at an American Academy of Orthopaedic Surgeons (AAOS) coding workshop. “A surgeon new in practice told me, ‘I’ve been in practice for 4 months. I understand the clinical side but nobody educated me about coding and billing before this course.’” Practices must provide new physicians with coding and documentation training, and coach them to make sure they feel up to speed and comfortable. “The practice’s future revenue depends on it,” Marks says.
McCarthy agrees. “Having an administrative mentorship for coding is incredibly valuable. They don’t teach it in school.”
So from a practical standpoint, purchase AAOS’ Orthopaedic Code-X, a software tool that will help the new physician navigate and integrate Current Procedural Terminology (CPT), ICD-10 (International Classification of Diseases, Tenth Revision), and other coding data easily and accurately. Send him or her to one of the Academy’s regional coding and reimbursement workshops as well. “It will behoove the practice to send them even before they start seeing patients,” Marks says.
And don’t just stop there. High-performing groups conduct peer reviews of evaluation and management (E/M) and operative notes, blinding the codes billed and discussing which CPT and ICD-10 codes are appropriate for the visit or case. “It will take time for the new physician to completely integrate coding with their clinical care,” says Marks. “Peer review sessions, as well as having a partner review codes before they go to the billing office, can help speed learning.”
Collegial Coaching Counts
The week before her official start day, Mc-Carthy scrubbed in as a first assist with each of her new partners. “It was a great way to start ramping up,” she says. “I could see what kind of equipment was present in the hospitals, and got a touch point for hospital logistics. Plus, as a young surgeon it’s great to see how your skill sets match up with your new partners, and which best practices are being deployed by the group.”
This kind of “collegial coaching” is a vital part of the clinical and cultural integration to the practice. Beyond providing clinical support, it builds relationships and trust among the group, and fosters collaboration.
Arkansas Specialty Orthopaedics organized McCarthy’s clinic and operating room (OR) schedules so that a partner was always present. “There was also someone I could bounce ideas off of,” McCarthy explains. “Every day in the OR, there was a partner there at the same time. If I got into a sticky situation, one of my colleagues was willing to come in and scrub in the OR.”
McCarthy says that patients responded favorably when she told them her plan was developed in conjunction with her partners. “Patients find comfort in knowing that several people’s opinions were considered,” she says. “And as a young surgeon, knowing that you have backup, even if you don’t use it, when caring for high-risk and complex cases really means a lot,” she says.
And although her group didn’t offer a formal mentoring program, McCarthy found that an informal mentorship grew organically when a friendship developed with one of her new partners. “In the first 6 months, every single weekend we sat by the pool and rolled through a ton of cases,” she says. “That was fabulous and it alleviated so much stress for me.” And when it was time for McCarthy to move into board case selection, this colleague and another were instrumental in her board preparation because, “they knew my style and where I would need to focus.”
IMS Orthopedics’ approach is to provide the staff and systems that allow new physicians to step up and take responsibility. “If they want to scrub in with me, that’s great. If they’d like to visit additional facilities and get the lay of the land, we encourage it. But we don’t do a lot of handholding. We set them up for success and make sure people are in place to help them,” says Ferry.
A Marketing Plan Is a Must
“The vast majority of practices do very little when it comes to thinking about how to market and build the practice of their new physician,” Marks says. “Practice-building is more of a challenge for surgical specialists today than it was in the old days when new surgeons could easily meet internists as they were rounding at the hospital. Now, a new physician and the practice must come up with a game plan.”
That game plan starts with the easy things: order business cards, schedule a photo shoot, and update the practice’s Web site pages with the physician’s biography and an introductory video. But with social media, online reviews, and subspecialty competition, Marks says practices must think beyond the basics. Think through each element of marketing, from online to outreach to developing referral relationships.
“I tell practices to draft a written marketing plan,” he says. “Not only does it provide a roadmap for the new physician, but also indicates that the practice has put some thought into how he or she can build a practice. It can make the new physician feel less overwhelmed knowing that he or she doesn’t have to do the marketing alone.” Once you’ve developed a list of actions, Marks suggests creating a spreadsheet with deadlines, and ensuring each action is completed.
McCarthy was scheduled to visit family practice clinics, and joined by the administrator who “handed out cookies and cards while I talked,” she says. Arkansas Specialty Orthopaedics also hired an external marketing firm to develop promotional opportunities for her. For example, “I was scheduled to appear on news channels, where I discussed new and interesting procedures,” she says. “It got my name out into the community.”
If your practice is too small to hire an outside firm, Marks suggests reaching out to agencies such as nursing homes, fitness centers, or the YMCA, which frequently offers educational programs for members. “Contact the administrators or medical directors in these organizations. A few minutes on the phone or a short visit can go a long way to building these relationships and getting your new physician on the map.”
As the old saying goes, an ounce of prevention is worth a pound of cure. Scheduling time for orientation, training, staff integration, and collegial coaching will speed up a new physician’s integration into the practice, and increase his or her opportunity for success.
Surgical Pearls in Total Knee Arthroplasty: A Lifetime of Lessons Learned
After over 4 decades of experience with total knee arthroplasty (TKA), I have learned many lessons regarding surgical technique. These include exposure issues, alignment methods, bone preparation, correction of deformity, and implantation techniques. Most of these lessons have been self-taught, but some have been suggested by or modified from colleague and student interaction. Attribution is given when possible.
The Incision
The skin incision should be marked in flexion rather than extension because the skin moves approximately 1 cm laterally from extension to flexion.1 This occurs because the tibia internally rotates beneath the skin as the knee is flexed and externally rotates as full extension is achieved. This lateral movement of the skin could bring an incision marked in extension on top of the tibial tubercle when the knee is flexed and may result in pain and dysfunction when the patient attempts to kneel. A review of kneeling ability after TKA showed that most patients are hesitant to kneel initially after their arthroplasty, but gain confidence and improved comfort and ability as their scar matures.2
Exposure
Patellar eversion can be difficult in a markedly obese or ankylosed knee, especially when the patella is difficult to grasp. This is facilitated by the use of a standard patellar clamp that is normally used to compress the patella during component cementation (Figure 1).3

Exposing the Ankylosed Knee and Protecting the Patellar Tendon From Avulsion
A tibial tubercle osteotomy is often recommended in the ankylosed knee but can be avoided by making a short inverted “V” incision in the proximal quadriceps tendon (Figure 2).4


Protecting the Soft Tissues During Surgery
Moist wound towels sewn into the joint capsule protect the underlying soft tissues from debris and desiccation during the procedure and will intuitively lower the chance of wound infection from contamination and tissue injury (Figures 4A, 4B).

Locating and Coagulating the Lateral Inferior Genicular Vessels
The lateral inferior genicular artery and vein can be easily located and coagulated just outside the posterior rim of the lateral meniscus near the popliteus hiatus. This will minimize both intraoperative and postoperative blood loss.
Determining the Entry Point in the Distal Femur for Intramedullary Alignment Devices
Templating the femoral entry point for insertion of an intramedullary alignment device on a preoperative radiograph will help avoid inadvertent excessive distal femoral valgus resection. This is especially important in valgus knees that have a valgus metaphyseal bow (Figure 5).

Avoiding Notching of the Anterior Femoral Cortex
Notching the anterior femoral cortex when in-between femoral sizes or when there is a preexisting dysplastic or shallow trochlea (Figure 6)

Obtaining a Medial Release by Removing Peripheral Medial Tibial Bone
Varus deformities can be corrected without performing a formal medial collateral ligament (MCL) release by a so-called reduction tibial osteotomy.5,6 In mild varus deformity, sufficient medial release can be achieved by removing medial femoral and tibial peripheral osteophytes that tent up the MCL and medial capsule. When this is insufficient, removal of additional peripheral tibial bone further shortens the distance between the origin and insertion of the MCL, effectively lengthening the ligament (Figure 7).
An Inverted Cruciform Lateral Retinacular Release to Correct Severe Valgus Deformity
An inverted cruciform lateral retinacular release effectively corrects a severe valgus deformity and avoids the need for a lateral collateral ligament (LCL) release.7


Relieving Posterior Femoral Impingement
Uncapped posterior condylar bone or retained posterior osteophytes can limit both flexion and extension and cause impingement. Trimming the posterior femoral condyles and removing posterior osteophytes is best accomplished using a trial femoral component as a template.4 A curved osteotome is passed tangential to the metallic condyles to define the bone requiring resection. After removal of the trial, the outlined bone can be easily and accurately resected.
Minimizing Postoperative Posterior Condylar Bone-Cement Radiolucencies
Zone 4 femoral bone-cement radiolucencies8 can be minimized using the “smear” technique.4 These radiolucencies are common because most prosthetic femoral components have posterior condyles that are parallel to the femoral fixation lugs and do not allow for compression of this interface during implantation. Most surgeons put no cement on the posterior condylar bone but place it on the inside of the prosthetic condyle instead. The lack of compression upon insertion leads to a poor interface and the resultant lucencies. In the long term, these lucencies could allow access of wear debris to the posterior condylar bone, with the potential for osteolysis and loosening. To improve this interface, cement can be smeared or packed into the posterior condyles and also placed on the posterior condyles of the prosthesis. This could lead to posterior extrusion of some cement during polymerization, so a removable trial insert should be utilized to allow access posteriorly after polymerization is complete.
Predicting Potential Postoperative Flexion
The best indicator of potential postoperative flexion for any individual patient is not preoperative flexion but is intraoperative flexion against gravity measured after capsular closure.9 Surgeons should measure and record this value for reference if a patient has difficulty regaining flexion during their recovery (Figure 9).

Summary
The short- and long-term success of TKA is highly dependent on surgical technique that allows proper and safe exposure under all circumstances, correction of deformity, and accurate component implantation while minimizing intraoperative and postoperative complications. The surgical pearls shared above will hopefully aid in achieving these goals.
Am J Orthop. 2016;45(6):384-388. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Yacoubian SV, Scott RD. Skin incision translation in total knee arthroplasty: the difference between flexion and extension. J Arthroplasty. 2007;22(3):353-355.
2. Schai PA, Gibbon AJ, Scott RD. Kneeling ability after total knee arthroplasty. Perception and reality. Clin Orthop Relat Res. 1999;367:195-200.
3. Springorum HP, Scott RD. A technique to facilitate everting the patella in stiff or obese knees in total knee arthroplasty. Am J Orthop. 2009;38(10):507-508.
4. Scott RD. Total Knee Arthroplasty. 2nd ed. Philadelphia, PA: Elsevier; 2014.
5. Dixon MC, Parsch D, Brown RR, Scott RD. The correction of severe varus deformity in total knee arthroplasty by tibial component downsizing and resection of uncapped proximal medial bone. J Arthroplasty. 2004;19(1):19-22.
6. Mullaji AB, Padmanabhan V, Jindal G. Total knee arthroplasty for profound varus deformity: technique and radiological results in 173 knees with varus of more than 20 degrees. J Arthroplasty. 2005;20(5):550-561.
7. Politi J, Scott RD. Balancing severe valgus deformity in total knee arthroplasty using a lateral cruciform retinacular release. J Arthroplasty. 2004;19(5):553-557.
8. Huddleston JI, Wiley JW, Scott RD. Zone 4 femoral radiolucent lines in hybrid versus cemented total knee arthroplasties: are they clinically significant? Clin Orthop Relat Res. 2005;441:334-339.
9. Lee DC, Kim DH, Scott RD, Suthers K. Intraoperative flexion against gravity as an indication of ultimate range of motion in individual cases after total knee arthroplasty. J Arthroplasty. 1998;13(5):500-503.
After over 4 decades of experience with total knee arthroplasty (TKA), I have learned many lessons regarding surgical technique. These include exposure issues, alignment methods, bone preparation, correction of deformity, and implantation techniques. Most of these lessons have been self-taught, but some have been suggested by or modified from colleague and student interaction. Attribution is given when possible.
The Incision
The skin incision should be marked in flexion rather than extension because the skin moves approximately 1 cm laterally from extension to flexion.1 This occurs because the tibia internally rotates beneath the skin as the knee is flexed and externally rotates as full extension is achieved. This lateral movement of the skin could bring an incision marked in extension on top of the tibial tubercle when the knee is flexed and may result in pain and dysfunction when the patient attempts to kneel. A review of kneeling ability after TKA showed that most patients are hesitant to kneel initially after their arthroplasty, but gain confidence and improved comfort and ability as their scar matures.2
Exposure
Patellar eversion can be difficult in a markedly obese or ankylosed knee, especially when the patella is difficult to grasp. This is facilitated by the use of a standard patellar clamp that is normally used to compress the patella during component cementation (Figure 1).3
Exposing the Ankylosed Knee and Protecting the Patellar Tendon From Avulsion
A tibial tubercle osteotomy is often recommended in the ankylosed knee but can be avoided by making a short inverted “V” incision in the proximal quadriceps tendon (Figure 2).4
Protecting the Soft Tissues During Surgery
Moist wound towels sewn into the joint capsule protect the underlying soft tissues from debris and desiccation during the procedure and will intuitively lower the chance of wound infection from contamination and tissue injury (Figures 4A, 4B).
Locating and Coagulating the Lateral Inferior Genicular Vessels
The lateral inferior genicular artery and vein can be easily located and coagulated just outside the posterior rim of the lateral meniscus near the popliteus hiatus. This will minimize both intraoperative and postoperative blood loss.
Determining the Entry Point in the Distal Femur for Intramedullary Alignment Devices
Templating the femoral entry point for insertion of an intramedullary alignment device on a preoperative radiograph will help avoid inadvertent excessive distal femoral valgus resection. This is especially important in valgus knees that have a valgus metaphyseal bow (Figure 5).
Avoiding Notching of the Anterior Femoral Cortex
Notching the anterior femoral cortex when in-between femoral sizes or when there is a preexisting dysplastic or shallow trochlea (Figure 6)
Obtaining a Medial Release by Removing Peripheral Medial Tibial Bone
Varus deformities can be corrected without performing a formal medial collateral ligament (MCL) release by a so-called reduction tibial osteotomy.5,6 In mild varus deformity, sufficient medial release can be achieved by removing medial femoral and tibial peripheral osteophytes that tent up the MCL and medial capsule. When this is insufficient, removal of additional peripheral tibial bone further shortens the distance between the origin and insertion of the MCL, effectively lengthening the ligament (Figure 7).
An Inverted Cruciform Lateral Retinacular Release to Correct Severe Valgus Deformity
An inverted cruciform lateral retinacular release effectively corrects a severe valgus deformity and avoids the need for a lateral collateral ligament (LCL) release.7
Relieving Posterior Femoral Impingement
Uncapped posterior condylar bone or retained posterior osteophytes can limit both flexion and extension and cause impingement. Trimming the posterior femoral condyles and removing posterior osteophytes is best accomplished using a trial femoral component as a template.4 A curved osteotome is passed tangential to the metallic condyles to define the bone requiring resection. After removal of the trial, the outlined bone can be easily and accurately resected.
Minimizing Postoperative Posterior Condylar Bone-Cement Radiolucencies
Zone 4 femoral bone-cement radiolucencies8 can be minimized using the “smear” technique.4 These radiolucencies are common because most prosthetic femoral components have posterior condyles that are parallel to the femoral fixation lugs and do not allow for compression of this interface during implantation. Most surgeons put no cement on the posterior condylar bone but place it on the inside of the prosthetic condyle instead. The lack of compression upon insertion leads to a poor interface and the resultant lucencies. In the long term, these lucencies could allow access of wear debris to the posterior condylar bone, with the potential for osteolysis and loosening. To improve this interface, cement can be smeared or packed into the posterior condyles and also placed on the posterior condyles of the prosthesis. This could lead to posterior extrusion of some cement during polymerization, so a removable trial insert should be utilized to allow access posteriorly after polymerization is complete.
Predicting Potential Postoperative Flexion
The best indicator of potential postoperative flexion for any individual patient is not preoperative flexion but is intraoperative flexion against gravity measured after capsular closure.9 Surgeons should measure and record this value for reference if a patient has difficulty regaining flexion during their recovery (Figure 9).
Summary
The short- and long-term success of TKA is highly dependent on surgical technique that allows proper and safe exposure under all circumstances, correction of deformity, and accurate component implantation while minimizing intraoperative and postoperative complications. The surgical pearls shared above will hopefully aid in achieving these goals.
Am J Orthop. 2016;45(6):384-388. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
After over 4 decades of experience with total knee arthroplasty (TKA), I have learned many lessons regarding surgical technique. These include exposure issues, alignment methods, bone preparation, correction of deformity, and implantation techniques. Most of these lessons have been self-taught, but some have been suggested by or modified from colleague and student interaction. Attribution is given when possible.
The Incision
The skin incision should be marked in flexion rather than extension because the skin moves approximately 1 cm laterally from extension to flexion.1 This occurs because the tibia internally rotates beneath the skin as the knee is flexed and externally rotates as full extension is achieved. This lateral movement of the skin could bring an incision marked in extension on top of the tibial tubercle when the knee is flexed and may result in pain and dysfunction when the patient attempts to kneel. A review of kneeling ability after TKA showed that most patients are hesitant to kneel initially after their arthroplasty, but gain confidence and improved comfort and ability as their scar matures.2
Exposure
Patellar eversion can be difficult in a markedly obese or ankylosed knee, especially when the patella is difficult to grasp. This is facilitated by the use of a standard patellar clamp that is normally used to compress the patella during component cementation (Figure 1).3
Exposing the Ankylosed Knee and Protecting the Patellar Tendon From Avulsion
A tibial tubercle osteotomy is often recommended in the ankylosed knee but can be avoided by making a short inverted “V” incision in the proximal quadriceps tendon (Figure 2).4
Protecting the Soft Tissues During Surgery
Moist wound towels sewn into the joint capsule protect the underlying soft tissues from debris and desiccation during the procedure and will intuitively lower the chance of wound infection from contamination and tissue injury (Figures 4A, 4B).
Locating and Coagulating the Lateral Inferior Genicular Vessels
The lateral inferior genicular artery and vein can be easily located and coagulated just outside the posterior rim of the lateral meniscus near the popliteus hiatus. This will minimize both intraoperative and postoperative blood loss.
Determining the Entry Point in the Distal Femur for Intramedullary Alignment Devices
Templating the femoral entry point for insertion of an intramedullary alignment device on a preoperative radiograph will help avoid inadvertent excessive distal femoral valgus resection. This is especially important in valgus knees that have a valgus metaphyseal bow (Figure 5).
Avoiding Notching of the Anterior Femoral Cortex
Notching the anterior femoral cortex when in-between femoral sizes or when there is a preexisting dysplastic or shallow trochlea (Figure 6)
Obtaining a Medial Release by Removing Peripheral Medial Tibial Bone
Varus deformities can be corrected without performing a formal medial collateral ligament (MCL) release by a so-called reduction tibial osteotomy.5,6 In mild varus deformity, sufficient medial release can be achieved by removing medial femoral and tibial peripheral osteophytes that tent up the MCL and medial capsule. When this is insufficient, removal of additional peripheral tibial bone further shortens the distance between the origin and insertion of the MCL, effectively lengthening the ligament (Figure 7).
An Inverted Cruciform Lateral Retinacular Release to Correct Severe Valgus Deformity
An inverted cruciform lateral retinacular release effectively corrects a severe valgus deformity and avoids the need for a lateral collateral ligament (LCL) release.7
Relieving Posterior Femoral Impingement
Uncapped posterior condylar bone or retained posterior osteophytes can limit both flexion and extension and cause impingement. Trimming the posterior femoral condyles and removing posterior osteophytes is best accomplished using a trial femoral component as a template.4 A curved osteotome is passed tangential to the metallic condyles to define the bone requiring resection. After removal of the trial, the outlined bone can be easily and accurately resected.
Minimizing Postoperative Posterior Condylar Bone-Cement Radiolucencies
Zone 4 femoral bone-cement radiolucencies8 can be minimized using the “smear” technique.4 These radiolucencies are common because most prosthetic femoral components have posterior condyles that are parallel to the femoral fixation lugs and do not allow for compression of this interface during implantation. Most surgeons put no cement on the posterior condylar bone but place it on the inside of the prosthetic condyle instead. The lack of compression upon insertion leads to a poor interface and the resultant lucencies. In the long term, these lucencies could allow access of wear debris to the posterior condylar bone, with the potential for osteolysis and loosening. To improve this interface, cement can be smeared or packed into the posterior condyles and also placed on the posterior condyles of the prosthesis. This could lead to posterior extrusion of some cement during polymerization, so a removable trial insert should be utilized to allow access posteriorly after polymerization is complete.
Predicting Potential Postoperative Flexion
The best indicator of potential postoperative flexion for any individual patient is not preoperative flexion but is intraoperative flexion against gravity measured after capsular closure.9 Surgeons should measure and record this value for reference if a patient has difficulty regaining flexion during their recovery (Figure 9).
Summary
The short- and long-term success of TKA is highly dependent on surgical technique that allows proper and safe exposure under all circumstances, correction of deformity, and accurate component implantation while minimizing intraoperative and postoperative complications. The surgical pearls shared above will hopefully aid in achieving these goals.
Am J Orthop. 2016;45(6):384-388. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Yacoubian SV, Scott RD. Skin incision translation in total knee arthroplasty: the difference between flexion and extension. J Arthroplasty. 2007;22(3):353-355.
2. Schai PA, Gibbon AJ, Scott RD. Kneeling ability after total knee arthroplasty. Perception and reality. Clin Orthop Relat Res. 1999;367:195-200.
3. Springorum HP, Scott RD. A technique to facilitate everting the patella in stiff or obese knees in total knee arthroplasty. Am J Orthop. 2009;38(10):507-508.
4. Scott RD. Total Knee Arthroplasty. 2nd ed. Philadelphia, PA: Elsevier; 2014.
5. Dixon MC, Parsch D, Brown RR, Scott RD. The correction of severe varus deformity in total knee arthroplasty by tibial component downsizing and resection of uncapped proximal medial bone. J Arthroplasty. 2004;19(1):19-22.
6. Mullaji AB, Padmanabhan V, Jindal G. Total knee arthroplasty for profound varus deformity: technique and radiological results in 173 knees with varus of more than 20 degrees. J Arthroplasty. 2005;20(5):550-561.
7. Politi J, Scott RD. Balancing severe valgus deformity in total knee arthroplasty using a lateral cruciform retinacular release. J Arthroplasty. 2004;19(5):553-557.
8. Huddleston JI, Wiley JW, Scott RD. Zone 4 femoral radiolucent lines in hybrid versus cemented total knee arthroplasties: are they clinically significant? Clin Orthop Relat Res. 2005;441:334-339.
9. Lee DC, Kim DH, Scott RD, Suthers K. Intraoperative flexion against gravity as an indication of ultimate range of motion in individual cases after total knee arthroplasty. J Arthroplasty. 1998;13(5):500-503.
1. Yacoubian SV, Scott RD. Skin incision translation in total knee arthroplasty: the difference between flexion and extension. J Arthroplasty. 2007;22(3):353-355.
2. Schai PA, Gibbon AJ, Scott RD. Kneeling ability after total knee arthroplasty. Perception and reality. Clin Orthop Relat Res. 1999;367:195-200.
3. Springorum HP, Scott RD. A technique to facilitate everting the patella in stiff or obese knees in total knee arthroplasty. Am J Orthop. 2009;38(10):507-508.
4. Scott RD. Total Knee Arthroplasty. 2nd ed. Philadelphia, PA: Elsevier; 2014.
5. Dixon MC, Parsch D, Brown RR, Scott RD. The correction of severe varus deformity in total knee arthroplasty by tibial component downsizing and resection of uncapped proximal medial bone. J Arthroplasty. 2004;19(1):19-22.
6. Mullaji AB, Padmanabhan V, Jindal G. Total knee arthroplasty for profound varus deformity: technique and radiological results in 173 knees with varus of more than 20 degrees. J Arthroplasty. 2005;20(5):550-561.
7. Politi J, Scott RD. Balancing severe valgus deformity in total knee arthroplasty using a lateral cruciform retinacular release. J Arthroplasty. 2004;19(5):553-557.
8. Huddleston JI, Wiley JW, Scott RD. Zone 4 femoral radiolucent lines in hybrid versus cemented total knee arthroplasties: are they clinically significant? Clin Orthop Relat Res. 2005;441:334-339.
9. Lee DC, Kim DH, Scott RD, Suthers K. Intraoperative flexion against gravity as an indication of ultimate range of motion in individual cases after total knee arthroplasty. J Arthroplasty. 1998;13(5):500-503.
Medical Issues in American Football: Eyes, Teeth, and Skin
Orthopedic conditions are only one of the many medical issues football team physicians may face. In this review, we cover the management of a few of the most common nonorthopedic medical issues football team physicians are likely to encounter, including eye injuries, dental concerns, and skin conditions.
Eye Injuries
More than 2.5 million eye injuries occur each year, with 50,000 people permanently losing part or all of their vision.1 Eye injuries account for over 600,000 yearly emergency department visits; over 30% of these eye injuries were attributed to a sports injury.1 Football is classified as high risk for eye injury, along with baseball, hockey, basketball, and lacrosse.2 Common eye injury mechanisms are categorized as blunt, penetrating, and radiating. Blunt injuries are most common.2 When evaluating an athlete on the sideline, relevant history would include the size of the object, the level of force, and the direction from which the impact occurred. An examination should include best-corrected visual acuity using an eye chart, confrontational visual fields, assessment of extraocular movements, assessment of red reflex, and pupil evaluation with a light source.2
Cornea Injuries
The outermost layer of the eye, the cornea, can be subject to blunt and penetrating injuries. Corneal abrasions often occur from mechanical trauma, such as one from the fingernail of an opposing player, that disrupts the integrity of the corneal epithelium. A corneal abrasion can be identified by applying fluorescein strips after application of a topical anesthetic. Abrasions appear fluorescent green when viewed with a cobalt blue light. If an abrasion is identified, management includes preventing infection and treating pain. Prophylactic topical antibiotics can be applied, particularly for contact lens wearers. Patching has not shown benefit in treatment of pain.3 The physician can consider using topical nonsteroidal anti-inflammatory drugs, such as diclofenac or ketorolac, with a soft contact lens to treat the pain.4 The patient should follow up frequently for monitoring for infection and healing.
Orbital Fractures
Orbital fractures should be considered when an object larger than the orbital opening, such as an elbow or knee, causes blunt trauma to the surrounding bony structures, or a digital poke occurs to the globe.5 The floor of the orbit and medial wall are thin bones that often break sacrificially to protect the globe from rupture. Examination findings may include diplopia, sunken globe, numbness in the distribution of infraorbital nerve, or periorbital emphysema.6 Urgent evaluation should be considered to rule out associated intraocular damage. Imaging and a physical examination can help guide surgical management, if indicated. The most common outcome after this injury is diplopia with upper field gaze.5
Retina Issues
Trauma to the face or head may result in a separation of the retina from the underlying retinal pigment epithelium and allow vitreous fluid to seep in and further separate the layers, causing a retinal detachment. Symptoms may include flashes of light (photopsia), floaters, and visual field defects. Emergent referral is indicated, as the outcomes from this condition are time-sensitive. Consider placing an eye shield to prevent any further pressure on the globe.
Globe Injuries and Rupture
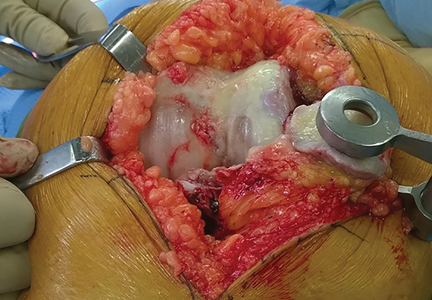
Another emergent ophthalmologic condition that can occur in football is globe rupture. Clinical findings usually prompt the clinician to consider this diagnosis. Hyphema (the collection of blood in the anterior chamber) may be seen in globe injuries. The most common clinical finding of athletes requiring hospitalization after an ocular injury is macroscopic hyphema (Figure 1).7-9

Prompt referral is warranted when there is a sudden decrease or change in vision, pain during movements, photophobia, and floaters and/or flashes.2 Consideration of return to play should take into account the patient’s vision and comfort level, which should not be masked by topical analgesics. Protective eyewear has been mandated in several sports, and has decreased the rate of eye injuries.10 Polycarbonate lenses of 3-mm thickness are recommended due to the significant comparable strength and impact-resistance.2 During the preparticipation physical for high-risk sports, the utilization of protective eyewear should be discussed.
Dental Concerns
Dental injuries may present a challenge for the sports medicine clinician. Contact injuries from elbows, fists, and other nonprojectile objects typically result in low-speed, lower-energy injuries, such as soft tissue lacerations and contusions. On the other hand, high-speed injuries occurring from balls, pucks, and sticks may result in more significant trauma. Baseball accounts for the highest percentage of sports-related dental injuries (40.2%), while basketball was second (20.2%) and football third (12.5%). Over 75% of these injuries occurred in males.11
On-field management of dental injuries should always start with the primary trauma survey, including assessment of the athlete’s airway, breathing, and circulatory function, as well as a targeted cervical spine evaluation. When obtaining a history, one should recognize the mechanism of injury and assess for signs of concomitant injuries, ie, respiratory compromise, concussion, leakage of cerebrospinal fluid, and teeth alignment. Findings from this initial evaluation may reveal critical conditions that will require management in addition to the dental injury.
Of central concern in managing dental trauma is preserving the viability of the injured structures. Therefore, much attention is paid to the pulpal and root vitality of injured teeth. The International Association of Dental Traumology Dental Trauma Guidelines recommend a biological approach to the urgent care of dental injuries:12
1. Stabilize the injury by carefully repositioning displaced entities and suturing soft tissue lacerations.
2. Eliminate or reduce the complications from bacterial contamination by rinsing and flushing with available liquids and use of chlorhexidine when possible.
3. Promote the opportunity for healing by replanting avulsed teeth and repositioning displaced teeth.
4. Make every effort to allow continued development of alveolar ridges in children.
Mouth guards are the single most effective prevention strategy for most contact sport dental injuries. One meta-analysis demonstrated a pooled 86% increased risk of orofacial injuries in nonusers.13
To review the anatomy (and injuries) of the tooth, one must consider the Ellis classification of enamel, dentin, and pulp injuries (Figure 2).

Tooth Subluxation
Tooth subluxations usually occur secondary to trauma and cause loosening of the tooth in its alveolar socket. A root fracture should be suspected in the setting of a subluxation. On exam, the tooth may be excessively mobile with gentle pressure. If unstable, immobilization with gauze packing or aluminum foil with dental follow-up is recommended.
Fractures
Ellis class I fractures are small chips in the enamel. There should be uniform color at the fracture site. A dental referral may be warranted to smooth rough enamel edges, but if no other injuries are present, these athletes may continue playing with some protection of the fractured surface. A mouth guard may be helpful to avoid mucosal lacerations.
Ellis class II fractures often present with sensitivity to inhaled air and to hot and cold temperatures. Yellow dentin is visible at the fracture site (Figure 3).

Ellis class III fractures may also present with air and temperature sensitivity. Finger pressure may expose a large fracture. Pink or red pulp is visible at the fracture site. Wiping the fracture site with sterile gauze may reveal bleeding from the pulp. This is considered a dental emergency. Immediate restriction from contact sports participation and urgent dental evaluation is indicated for root canal and capping and to prevent abscess formation.
Tooth Avulsion
Tooth avulsions occur when a tooth is completely displaced from the socket (Figure 4).

Skin Issues
Dermatological issues are some of the most common medical conditions faced by a football team physician. Skin infections in particular can pose a significant challenge both diagnostically as well as from a clearance-to-play perspective, given the potential for infections to affect other participants, such as other members of the team. Skin infection rates vary by sport and age group, with one study reporting 28.56 infections per 100,000 athletic exposures in high school wrestlers, which was more than 10 times that of football.14 Still, football players are at a higher risk of skin infections given the contact nature of the sport and close person-to-person proximity. A precise diagnosis may be difficult early in the course of a skin eruption, and with differing guidelines from various professional societies, it may be best suited for medical personnel familiar with these conditions, such as a sports medicine physician or dermatologist, to manage these athletes. A thorough and systematic evaluation is recommended, as athletes are often treated with unnecessary antibiotics, which contributes to antibiotic resistance. Previous antibiotic use may also be a risk factor for developing community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA).15
Two terms sports medicine clinicians must be familiar with are “adequately protected” and “properly covered.” The National Collegiate Athletic Association (NCAA) defines a wound or skin condition as adequately protected when the condition is considered noninfectious, adequately treated by a healthcare provider, and is able to be properly covered. A skin infection is considered properly covered when the lesion is covered by a securely attached bandage or dressing that will contain all drainage and remain intact throughout the sport activity.16
Impetigo
Impetigo is often caused by Staphylococcus and Streptococcus subspecies. The classic presentation is a dry, honey-crusted lesion with an erythematous base. Culture or gram stain may be helpful, but treatment may be initiated on a clinical basis without these studies. Topical antibiotics may be used, but in the setting of multiple lesions or an outbreak, systemic (eg, oral) antibiotics are preferred. Oral antibiotics may also shorten the time to return to play. If not responsive to the initial treatment, MRSA should be considered. No new lesions for 48 hours and a minimum of 72 hours of therapy with no moist, exudative, or draining lesions are required prior to return to play. These lesions cannot be covered as the sole means of return to play.
Methicillin-Resistant Staphylococcus aureus
MRSA is one of the most challenging skin infections for the sports medicine clinician to manage. Several outbreaks have been reported in the high school, college, and professional settings.17-20 Standardized precautions and a proactive approach are key in preventing MRSA outbreaks. It appears that different activities within a given sport may contribute to MRSA risk. One study reported football linemen had the highest attack rate, while another study reported cornerbacks and wide receivers to have the highest rate of MRSA infections.17,20 The elbow area was the most common site infected in both studies.
Abscesses are best initially managed by incision and drainage as well as obtaining wound cultures (Figure 5).

Preventative measures are thought to be useful, especially in the management of teams. The Centers for Disease Control and Prevention has published guidelines for both clinicians and patients. Precautions including hand washing; encouraging good overall hygiene; avoiding whirlpools; discouraging the sharing of towels, razors, and athletic gear; maintaining clean equipment/facilities; and encouraging early reporting of skin lesions.14,17,21,22 Isolated cases of MRSA do not need to be reported, but if more than one athlete is infected, one should notify the athletic training and team coaching staff. In the setting of an outbreak, the physician may need to notify local or state health agencies. No new lesions for 48 hours and a minimum of 72 hours of therapy with no moist, exudative, or draining lesions are required prior to returning to play. These lesions cannot be covered as the sole means of return to play.
Tinea Pedis
Tinea pedis is a common dermatophyte infection involving the feet and is most commonly caused by Trichophyton rubrum. Its distribution is usually interdigital or along the plantar surface of the foot. Topical antifungals with either allylamines or azoles are usually sufficient. Terbinafine has been shown to have a shorter duration of treatment. Athletes with tinea pedis are not restricted from sports participation during treatment, as long as the lesions are properly covered.
Tinea Corporis
Tinea corporis is a common superficial fungal infection of the body. It classically presents as pruritic, annular lesions, with well-demarcated borders and central clearing (

Tinea Cruris
Commonly known as “jock-itch,” this fungal infection is often very pruritic and involves the groin or genital region. The area is also inflamed and scaly. Treatment usually consists of topical allylamines or azoles. Allylamines amines are often preferred, as they require a shorter duration of treatment. There are no specific guidelines on the return to play with these athletes. Clearance is at the team physician’s discretion, but usually there are no restrictions. Athletes with extensive lesions may need to be disqualified from contact sports activities.
Am J Orthop. 2016;45(6):377-382. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Owens PL, Mutter R. Emergency Department Visits Related to Eye Injuries, 2008. Agency for Healthcare Research and Quality Web site. http://www.hcup-us.ahrq.gov/reports/statbriefs/sb112.pdf. Published May 2011. Accessed August 18, 2016.
2. Rodriguez JO, Lavina AM, Agarwai A. Prevention and treatment of common eye injuries in sports. Am Fam Physician. 2003;67(7):1481-1496.
3. Lim CH, Turner A, Lim BX. Patching for corneal abrasion. Cochrane Database Syst Rev. 2016;7:CD004764.
4. Weaver CS, Terrell KM. Evidence-based emergency medicine. Update: do ophthalmic nonsteroidal anti-inflammatory drugs reduce the pain associated with simple corneal abrasion without delaying healing? Ann Emerg Med. 2003;41(1):134-140.
5. Williams RJ 3rd, Marx RG, Barnes R, O’Brien SJ, Warren RF. Fractures about the orbit in professional American football players. Am J Sports Med. 2001;29(1):55-57.
6. Forrest LA, Schuller DE, Strauss RH. Management of orbital blow-out fractures. Case reports and discussion. Am J Sports Med. 1989;17(2):217-220.
7. Barr A, Baines PS, Desai P, MacEwen CJ. Ocular sports injuries: the current picture. Br J Sports Med. 2000;34(6):456-458.
8. Pokhrel PK, Loftus SA. Ocular emergencies. Am Fam Physician. 2007;76(6):829-836.
9. Usatine RP, Smith MA, Mayeaux EJ Jr, Chumley H. Eye Trauma—Hyphema. The Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013.
10. Lincoln AE, Caswell SV, Almquist JL, et al. Effectiveness of the women’s lacrosse protective eyewear mandate in the reduction of eye injuries. Am J Sports Med. 2012;40(3):611-614.
11. Stewart GB, Shields BJ, Fields S, Comstock RD, Smith GA. Consumer products and activities associated with dental injuries to children treated in United States emergency departments, 1990-2003. Dental Traumatol. 2009;25(4):399-405.
12. Bakland LK. Dental trauma guidelines. Pediatric Dent. 2013;35(2):106-108.
13. Knapik J, Marshall SW, Lee RB, et al. Mouthguards in sport activities: history, physical properties and Injury prevention effectiveness. Sports Med. 2007;37(2):117-144.
14. Ashack KA, Burton KA, Johnson TR, Currie DW, Comstock RD, Dellavalle RP. Skin infections among US high school athletes: a national survey. J Am Acad Dermatol. 2016;74(4):679-684.e1.
15. Ellis MW, Hospenthal DR, Dooley DP, Gray PJ, Murray CK. Natural history of community-acquired methicillin-resistant Staphylococcus aureus colonization and infection in soldiers. Clin Infect Dis. 2004;39(7):971-979.
16. The National Collegiate Athletic Association. 2014-15 NCAA Sports Medicine Handbook. http://www.ncaapublications.com/productdownloads/MD15.pdf. Revised June 2008. Accessed August 18, 2016.
17. Anderson BJ. The effectiveness of valacyclovir in preventing reactivation of herpes gladiatorum in wrestlers. Clin J Sport Med. 1999;9(2):86-90.
18. Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18-e55.
19. Jeffords MD, Batts K. Dermatology. In: O’Connor FG, Casa DJ, Davis BA, Pierre PS, Sallis RE, Wilder RP, eds. ACSM’s Sports Medicine: A Comprehensive Review. Riverwoods, IL: Wolters Kluwer; 2016:181-188.
20. Kazakova SV, Hageman JC, Matava M, et al. A clone of methicillin-resistant Staphylococcus aureus among professional football players. N Engl J Med. 2005;352(5):468-475.
21. Begier EM, Frenette K, Barrett NL, et al. A high-morbidity outbreak of methicillin-resistant Staphylococcus aureus among players on a college football team, facilitated by cosmetic body shaving and turf burns. Clin Infect Dis. 2004;39(10):1446-1453.
22. Geissler KE, Borchers JR. More than meets the eye: a rapidly progressive skin infection in a football player. Clin J Sport Med. 2015;25(3):e54-e56.
Orthopedic conditions are only one of the many medical issues football team physicians may face. In this review, we cover the management of a few of the most common nonorthopedic medical issues football team physicians are likely to encounter, including eye injuries, dental concerns, and skin conditions.
Eye Injuries
More than 2.5 million eye injuries occur each year, with 50,000 people permanently losing part or all of their vision.1 Eye injuries account for over 600,000 yearly emergency department visits; over 30% of these eye injuries were attributed to a sports injury.1 Football is classified as high risk for eye injury, along with baseball, hockey, basketball, and lacrosse.2 Common eye injury mechanisms are categorized as blunt, penetrating, and radiating. Blunt injuries are most common.2 When evaluating an athlete on the sideline, relevant history would include the size of the object, the level of force, and the direction from which the impact occurred. An examination should include best-corrected visual acuity using an eye chart, confrontational visual fields, assessment of extraocular movements, assessment of red reflex, and pupil evaluation with a light source.2
Cornea Injuries
The outermost layer of the eye, the cornea, can be subject to blunt and penetrating injuries. Corneal abrasions often occur from mechanical trauma, such as one from the fingernail of an opposing player, that disrupts the integrity of the corneal epithelium. A corneal abrasion can be identified by applying fluorescein strips after application of a topical anesthetic. Abrasions appear fluorescent green when viewed with a cobalt blue light. If an abrasion is identified, management includes preventing infection and treating pain. Prophylactic topical antibiotics can be applied, particularly for contact lens wearers. Patching has not shown benefit in treatment of pain.3 The physician can consider using topical nonsteroidal anti-inflammatory drugs, such as diclofenac or ketorolac, with a soft contact lens to treat the pain.4 The patient should follow up frequently for monitoring for infection and healing.
Orbital Fractures
Orbital fractures should be considered when an object larger than the orbital opening, such as an elbow or knee, causes blunt trauma to the surrounding bony structures, or a digital poke occurs to the globe.5 The floor of the orbit and medial wall are thin bones that often break sacrificially to protect the globe from rupture. Examination findings may include diplopia, sunken globe, numbness in the distribution of infraorbital nerve, or periorbital emphysema.6 Urgent evaluation should be considered to rule out associated intraocular damage. Imaging and a physical examination can help guide surgical management, if indicated. The most common outcome after this injury is diplopia with upper field gaze.5
Retina Issues
Trauma to the face or head may result in a separation of the retina from the underlying retinal pigment epithelium and allow vitreous fluid to seep in and further separate the layers, causing a retinal detachment. Symptoms may include flashes of light (photopsia), floaters, and visual field defects. Emergent referral is indicated, as the outcomes from this condition are time-sensitive. Consider placing an eye shield to prevent any further pressure on the globe.
Globe Injuries and Rupture
Another emergent ophthalmologic condition that can occur in football is globe rupture. Clinical findings usually prompt the clinician to consider this diagnosis. Hyphema (the collection of blood in the anterior chamber) may be seen in globe injuries. The most common clinical finding of athletes requiring hospitalization after an ocular injury is macroscopic hyphema (Figure 1).7-9
Prompt referral is warranted when there is a sudden decrease or change in vision, pain during movements, photophobia, and floaters and/or flashes.2 Consideration of return to play should take into account the patient’s vision and comfort level, which should not be masked by topical analgesics. Protective eyewear has been mandated in several sports, and has decreased the rate of eye injuries.10 Polycarbonate lenses of 3-mm thickness are recommended due to the significant comparable strength and impact-resistance.2 During the preparticipation physical for high-risk sports, the utilization of protective eyewear should be discussed.
Dental Concerns
Dental injuries may present a challenge for the sports medicine clinician. Contact injuries from elbows, fists, and other nonprojectile objects typically result in low-speed, lower-energy injuries, such as soft tissue lacerations and contusions. On the other hand, high-speed injuries occurring from balls, pucks, and sticks may result in more significant trauma. Baseball accounts for the highest percentage of sports-related dental injuries (40.2%), while basketball was second (20.2%) and football third (12.5%). Over 75% of these injuries occurred in males.11
On-field management of dental injuries should always start with the primary trauma survey, including assessment of the athlete’s airway, breathing, and circulatory function, as well as a targeted cervical spine evaluation. When obtaining a history, one should recognize the mechanism of injury and assess for signs of concomitant injuries, ie, respiratory compromise, concussion, leakage of cerebrospinal fluid, and teeth alignment. Findings from this initial evaluation may reveal critical conditions that will require management in addition to the dental injury.
Of central concern in managing dental trauma is preserving the viability of the injured structures. Therefore, much attention is paid to the pulpal and root vitality of injured teeth. The International Association of Dental Traumology Dental Trauma Guidelines recommend a biological approach to the urgent care of dental injuries:12
1. Stabilize the injury by carefully repositioning displaced entities and suturing soft tissue lacerations.
2. Eliminate or reduce the complications from bacterial contamination by rinsing and flushing with available liquids and use of chlorhexidine when possible.
3. Promote the opportunity for healing by replanting avulsed teeth and repositioning displaced teeth.
4. Make every effort to allow continued development of alveolar ridges in children.
Mouth guards are the single most effective prevention strategy for most contact sport dental injuries. One meta-analysis demonstrated a pooled 86% increased risk of orofacial injuries in nonusers.13
To review the anatomy (and injuries) of the tooth, one must consider the Ellis classification of enamel, dentin, and pulp injuries (Figure 2).
Tooth Subluxation
Tooth subluxations usually occur secondary to trauma and cause loosening of the tooth in its alveolar socket. A root fracture should be suspected in the setting of a subluxation. On exam, the tooth may be excessively mobile with gentle pressure. If unstable, immobilization with gauze packing or aluminum foil with dental follow-up is recommended.
Fractures
Ellis class I fractures are small chips in the enamel. There should be uniform color at the fracture site. A dental referral may be warranted to smooth rough enamel edges, but if no other injuries are present, these athletes may continue playing with some protection of the fractured surface. A mouth guard may be helpful to avoid mucosal lacerations.
Ellis class II fractures often present with sensitivity to inhaled air and to hot and cold temperatures. Yellow dentin is visible at the fracture site (Figure 3).
Ellis class III fractures may also present with air and temperature sensitivity. Finger pressure may expose a large fracture. Pink or red pulp is visible at the fracture site. Wiping the fracture site with sterile gauze may reveal bleeding from the pulp. This is considered a dental emergency. Immediate restriction from contact sports participation and urgent dental evaluation is indicated for root canal and capping and to prevent abscess formation.
Tooth Avulsion
Tooth avulsions occur when a tooth is completely displaced from the socket (Figure 4).
Skin Issues
Dermatological issues are some of the most common medical conditions faced by a football team physician. Skin infections in particular can pose a significant challenge both diagnostically as well as from a clearance-to-play perspective, given the potential for infections to affect other participants, such as other members of the team. Skin infection rates vary by sport and age group, with one study reporting 28.56 infections per 100,000 athletic exposures in high school wrestlers, which was more than 10 times that of football.14 Still, football players are at a higher risk of skin infections given the contact nature of the sport and close person-to-person proximity. A precise diagnosis may be difficult early in the course of a skin eruption, and with differing guidelines from various professional societies, it may be best suited for medical personnel familiar with these conditions, such as a sports medicine physician or dermatologist, to manage these athletes. A thorough and systematic evaluation is recommended, as athletes are often treated with unnecessary antibiotics, which contributes to antibiotic resistance. Previous antibiotic use may also be a risk factor for developing community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA).15
Two terms sports medicine clinicians must be familiar with are “adequately protected” and “properly covered.” The National Collegiate Athletic Association (NCAA) defines a wound or skin condition as adequately protected when the condition is considered noninfectious, adequately treated by a healthcare provider, and is able to be properly covered. A skin infection is considered properly covered when the lesion is covered by a securely attached bandage or dressing that will contain all drainage and remain intact throughout the sport activity.16
Impetigo
Impetigo is often caused by Staphylococcus and Streptococcus subspecies. The classic presentation is a dry, honey-crusted lesion with an erythematous base. Culture or gram stain may be helpful, but treatment may be initiated on a clinical basis without these studies. Topical antibiotics may be used, but in the setting of multiple lesions or an outbreak, systemic (eg, oral) antibiotics are preferred. Oral antibiotics may also shorten the time to return to play. If not responsive to the initial treatment, MRSA should be considered. No new lesions for 48 hours and a minimum of 72 hours of therapy with no moist, exudative, or draining lesions are required prior to return to play. These lesions cannot be covered as the sole means of return to play.
Methicillin-Resistant Staphylococcus aureus
MRSA is one of the most challenging skin infections for the sports medicine clinician to manage. Several outbreaks have been reported in the high school, college, and professional settings.17-20 Standardized precautions and a proactive approach are key in preventing MRSA outbreaks. It appears that different activities within a given sport may contribute to MRSA risk. One study reported football linemen had the highest attack rate, while another study reported cornerbacks and wide receivers to have the highest rate of MRSA infections.17,20 The elbow area was the most common site infected in both studies.
Abscesses are best initially managed by incision and drainage as well as obtaining wound cultures (Figure 5).
Preventative measures are thought to be useful, especially in the management of teams. The Centers for Disease Control and Prevention has published guidelines for both clinicians and patients. Precautions including hand washing; encouraging good overall hygiene; avoiding whirlpools; discouraging the sharing of towels, razors, and athletic gear; maintaining clean equipment/facilities; and encouraging early reporting of skin lesions.14,17,21,22 Isolated cases of MRSA do not need to be reported, but if more than one athlete is infected, one should notify the athletic training and team coaching staff. In the setting of an outbreak, the physician may need to notify local or state health agencies. No new lesions for 48 hours and a minimum of 72 hours of therapy with no moist, exudative, or draining lesions are required prior to returning to play. These lesions cannot be covered as the sole means of return to play.
Tinea Pedis
Tinea pedis is a common dermatophyte infection involving the feet and is most commonly caused by Trichophyton rubrum. Its distribution is usually interdigital or along the plantar surface of the foot. Topical antifungals with either allylamines or azoles are usually sufficient. Terbinafine has been shown to have a shorter duration of treatment. Athletes with tinea pedis are not restricted from sports participation during treatment, as long as the lesions are properly covered.
Tinea Corporis
Tinea corporis is a common superficial fungal infection of the body. It classically presents as pruritic, annular lesions, with well-demarcated borders and central clearing (
Tinea Cruris
Commonly known as “jock-itch,” this fungal infection is often very pruritic and involves the groin or genital region. The area is also inflamed and scaly. Treatment usually consists of topical allylamines or azoles. Allylamines amines are often preferred, as they require a shorter duration of treatment. There are no specific guidelines on the return to play with these athletes. Clearance is at the team physician’s discretion, but usually there are no restrictions. Athletes with extensive lesions may need to be disqualified from contact sports activities.
Am J Orthop. 2016;45(6):377-382. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
Orthopedic conditions are only one of the many medical issues football team physicians may face. In this review, we cover the management of a few of the most common nonorthopedic medical issues football team physicians are likely to encounter, including eye injuries, dental concerns, and skin conditions.
Eye Injuries
More than 2.5 million eye injuries occur each year, with 50,000 people permanently losing part or all of their vision.1 Eye injuries account for over 600,000 yearly emergency department visits; over 30% of these eye injuries were attributed to a sports injury.1 Football is classified as high risk for eye injury, along with baseball, hockey, basketball, and lacrosse.2 Common eye injury mechanisms are categorized as blunt, penetrating, and radiating. Blunt injuries are most common.2 When evaluating an athlete on the sideline, relevant history would include the size of the object, the level of force, and the direction from which the impact occurred. An examination should include best-corrected visual acuity using an eye chart, confrontational visual fields, assessment of extraocular movements, assessment of red reflex, and pupil evaluation with a light source.2
Cornea Injuries
The outermost layer of the eye, the cornea, can be subject to blunt and penetrating injuries. Corneal abrasions often occur from mechanical trauma, such as one from the fingernail of an opposing player, that disrupts the integrity of the corneal epithelium. A corneal abrasion can be identified by applying fluorescein strips after application of a topical anesthetic. Abrasions appear fluorescent green when viewed with a cobalt blue light. If an abrasion is identified, management includes preventing infection and treating pain. Prophylactic topical antibiotics can be applied, particularly for contact lens wearers. Patching has not shown benefit in treatment of pain.3 The physician can consider using topical nonsteroidal anti-inflammatory drugs, such as diclofenac or ketorolac, with a soft contact lens to treat the pain.4 The patient should follow up frequently for monitoring for infection and healing.
Orbital Fractures
Orbital fractures should be considered when an object larger than the orbital opening, such as an elbow or knee, causes blunt trauma to the surrounding bony structures, or a digital poke occurs to the globe.5 The floor of the orbit and medial wall are thin bones that often break sacrificially to protect the globe from rupture. Examination findings may include diplopia, sunken globe, numbness in the distribution of infraorbital nerve, or periorbital emphysema.6 Urgent evaluation should be considered to rule out associated intraocular damage. Imaging and a physical examination can help guide surgical management, if indicated. The most common outcome after this injury is diplopia with upper field gaze.5
Retina Issues
Trauma to the face or head may result in a separation of the retina from the underlying retinal pigment epithelium and allow vitreous fluid to seep in and further separate the layers, causing a retinal detachment. Symptoms may include flashes of light (photopsia), floaters, and visual field defects. Emergent referral is indicated, as the outcomes from this condition are time-sensitive. Consider placing an eye shield to prevent any further pressure on the globe.
Globe Injuries and Rupture
Another emergent ophthalmologic condition that can occur in football is globe rupture. Clinical findings usually prompt the clinician to consider this diagnosis. Hyphema (the collection of blood in the anterior chamber) may be seen in globe injuries. The most common clinical finding of athletes requiring hospitalization after an ocular injury is macroscopic hyphema (Figure 1).7-9
Prompt referral is warranted when there is a sudden decrease or change in vision, pain during movements, photophobia, and floaters and/or flashes.2 Consideration of return to play should take into account the patient’s vision and comfort level, which should not be masked by topical analgesics. Protective eyewear has been mandated in several sports, and has decreased the rate of eye injuries.10 Polycarbonate lenses of 3-mm thickness are recommended due to the significant comparable strength and impact-resistance.2 During the preparticipation physical for high-risk sports, the utilization of protective eyewear should be discussed.
Dental Concerns
Dental injuries may present a challenge for the sports medicine clinician. Contact injuries from elbows, fists, and other nonprojectile objects typically result in low-speed, lower-energy injuries, such as soft tissue lacerations and contusions. On the other hand, high-speed injuries occurring from balls, pucks, and sticks may result in more significant trauma. Baseball accounts for the highest percentage of sports-related dental injuries (40.2%), while basketball was second (20.2%) and football third (12.5%). Over 75% of these injuries occurred in males.11
On-field management of dental injuries should always start with the primary trauma survey, including assessment of the athlete’s airway, breathing, and circulatory function, as well as a targeted cervical spine evaluation. When obtaining a history, one should recognize the mechanism of injury and assess for signs of concomitant injuries, ie, respiratory compromise, concussion, leakage of cerebrospinal fluid, and teeth alignment. Findings from this initial evaluation may reveal critical conditions that will require management in addition to the dental injury.
Of central concern in managing dental trauma is preserving the viability of the injured structures. Therefore, much attention is paid to the pulpal and root vitality of injured teeth. The International Association of Dental Traumology Dental Trauma Guidelines recommend a biological approach to the urgent care of dental injuries:12
1. Stabilize the injury by carefully repositioning displaced entities and suturing soft tissue lacerations.
2. Eliminate or reduce the complications from bacterial contamination by rinsing and flushing with available liquids and use of chlorhexidine when possible.
3. Promote the opportunity for healing by replanting avulsed teeth and repositioning displaced teeth.
4. Make every effort to allow continued development of alveolar ridges in children.
Mouth guards are the single most effective prevention strategy for most contact sport dental injuries. One meta-analysis demonstrated a pooled 86% increased risk of orofacial injuries in nonusers.13
To review the anatomy (and injuries) of the tooth, one must consider the Ellis classification of enamel, dentin, and pulp injuries (Figure 2).
Tooth Subluxation
Tooth subluxations usually occur secondary to trauma and cause loosening of the tooth in its alveolar socket. A root fracture should be suspected in the setting of a subluxation. On exam, the tooth may be excessively mobile with gentle pressure. If unstable, immobilization with gauze packing or aluminum foil with dental follow-up is recommended.
Fractures
Ellis class I fractures are small chips in the enamel. There should be uniform color at the fracture site. A dental referral may be warranted to smooth rough enamel edges, but if no other injuries are present, these athletes may continue playing with some protection of the fractured surface. A mouth guard may be helpful to avoid mucosal lacerations.
Ellis class II fractures often present with sensitivity to inhaled air and to hot and cold temperatures. Yellow dentin is visible at the fracture site (Figure 3).
Ellis class III fractures may also present with air and temperature sensitivity. Finger pressure may expose a large fracture. Pink or red pulp is visible at the fracture site. Wiping the fracture site with sterile gauze may reveal bleeding from the pulp. This is considered a dental emergency. Immediate restriction from contact sports participation and urgent dental evaluation is indicated for root canal and capping and to prevent abscess formation.
Tooth Avulsion
Tooth avulsions occur when a tooth is completely displaced from the socket (Figure 4).
Skin Issues
Dermatological issues are some of the most common medical conditions faced by a football team physician. Skin infections in particular can pose a significant challenge both diagnostically as well as from a clearance-to-play perspective, given the potential for infections to affect other participants, such as other members of the team. Skin infection rates vary by sport and age group, with one study reporting 28.56 infections per 100,000 athletic exposures in high school wrestlers, which was more than 10 times that of football.14 Still, football players are at a higher risk of skin infections given the contact nature of the sport and close person-to-person proximity. A precise diagnosis may be difficult early in the course of a skin eruption, and with differing guidelines from various professional societies, it may be best suited for medical personnel familiar with these conditions, such as a sports medicine physician or dermatologist, to manage these athletes. A thorough and systematic evaluation is recommended, as athletes are often treated with unnecessary antibiotics, which contributes to antibiotic resistance. Previous antibiotic use may also be a risk factor for developing community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA).15
Two terms sports medicine clinicians must be familiar with are “adequately protected” and “properly covered.” The National Collegiate Athletic Association (NCAA) defines a wound or skin condition as adequately protected when the condition is considered noninfectious, adequately treated by a healthcare provider, and is able to be properly covered. A skin infection is considered properly covered when the lesion is covered by a securely attached bandage or dressing that will contain all drainage and remain intact throughout the sport activity.16
Impetigo
Impetigo is often caused by Staphylococcus and Streptococcus subspecies. The classic presentation is a dry, honey-crusted lesion with an erythematous base. Culture or gram stain may be helpful, but treatment may be initiated on a clinical basis without these studies. Topical antibiotics may be used, but in the setting of multiple lesions or an outbreak, systemic (eg, oral) antibiotics are preferred. Oral antibiotics may also shorten the time to return to play. If not responsive to the initial treatment, MRSA should be considered. No new lesions for 48 hours and a minimum of 72 hours of therapy with no moist, exudative, or draining lesions are required prior to return to play. These lesions cannot be covered as the sole means of return to play.
Methicillin-Resistant Staphylococcus aureus
MRSA is one of the most challenging skin infections for the sports medicine clinician to manage. Several outbreaks have been reported in the high school, college, and professional settings.17-20 Standardized precautions and a proactive approach are key in preventing MRSA outbreaks. It appears that different activities within a given sport may contribute to MRSA risk. One study reported football linemen had the highest attack rate, while another study reported cornerbacks and wide receivers to have the highest rate of MRSA infections.17,20 The elbow area was the most common site infected in both studies.
Abscesses are best initially managed by incision and drainage as well as obtaining wound cultures (Figure 5).
Preventative measures are thought to be useful, especially in the management of teams. The Centers for Disease Control and Prevention has published guidelines for both clinicians and patients. Precautions including hand washing; encouraging good overall hygiene; avoiding whirlpools; discouraging the sharing of towels, razors, and athletic gear; maintaining clean equipment/facilities; and encouraging early reporting of skin lesions.14,17,21,22 Isolated cases of MRSA do not need to be reported, but if more than one athlete is infected, one should notify the athletic training and team coaching staff. In the setting of an outbreak, the physician may need to notify local or state health agencies. No new lesions for 48 hours and a minimum of 72 hours of therapy with no moist, exudative, or draining lesions are required prior to returning to play. These lesions cannot be covered as the sole means of return to play.
Tinea Pedis
Tinea pedis is a common dermatophyte infection involving the feet and is most commonly caused by Trichophyton rubrum. Its distribution is usually interdigital or along the plantar surface of the foot. Topical antifungals with either allylamines or azoles are usually sufficient. Terbinafine has been shown to have a shorter duration of treatment. Athletes with tinea pedis are not restricted from sports participation during treatment, as long as the lesions are properly covered.
Tinea Corporis
Tinea corporis is a common superficial fungal infection of the body. It classically presents as pruritic, annular lesions, with well-demarcated borders and central clearing (
Tinea Cruris
Commonly known as “jock-itch,” this fungal infection is often very pruritic and involves the groin or genital region. The area is also inflamed and scaly. Treatment usually consists of topical allylamines or azoles. Allylamines amines are often preferred, as they require a shorter duration of treatment. There are no specific guidelines on the return to play with these athletes. Clearance is at the team physician’s discretion, but usually there are no restrictions. Athletes with extensive lesions may need to be disqualified from contact sports activities.
Am J Orthop. 2016;45(6):377-382. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Owens PL, Mutter R. Emergency Department Visits Related to Eye Injuries, 2008. Agency for Healthcare Research and Quality Web site. http://www.hcup-us.ahrq.gov/reports/statbriefs/sb112.pdf. Published May 2011. Accessed August 18, 2016.
2. Rodriguez JO, Lavina AM, Agarwai A. Prevention and treatment of common eye injuries in sports. Am Fam Physician. 2003;67(7):1481-1496.
3. Lim CH, Turner A, Lim BX. Patching for corneal abrasion. Cochrane Database Syst Rev. 2016;7:CD004764.
4. Weaver CS, Terrell KM. Evidence-based emergency medicine. Update: do ophthalmic nonsteroidal anti-inflammatory drugs reduce the pain associated with simple corneal abrasion without delaying healing? Ann Emerg Med. 2003;41(1):134-140.
5. Williams RJ 3rd, Marx RG, Barnes R, O’Brien SJ, Warren RF. Fractures about the orbit in professional American football players. Am J Sports Med. 2001;29(1):55-57.
6. Forrest LA, Schuller DE, Strauss RH. Management of orbital blow-out fractures. Case reports and discussion. Am J Sports Med. 1989;17(2):217-220.
7. Barr A, Baines PS, Desai P, MacEwen CJ. Ocular sports injuries: the current picture. Br J Sports Med. 2000;34(6):456-458.
8. Pokhrel PK, Loftus SA. Ocular emergencies. Am Fam Physician. 2007;76(6):829-836.
9. Usatine RP, Smith MA, Mayeaux EJ Jr, Chumley H. Eye Trauma—Hyphema. The Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013.
10. Lincoln AE, Caswell SV, Almquist JL, et al. Effectiveness of the women’s lacrosse protective eyewear mandate in the reduction of eye injuries. Am J Sports Med. 2012;40(3):611-614.
11. Stewart GB, Shields BJ, Fields S, Comstock RD, Smith GA. Consumer products and activities associated with dental injuries to children treated in United States emergency departments, 1990-2003. Dental Traumatol. 2009;25(4):399-405.
12. Bakland LK. Dental trauma guidelines. Pediatric Dent. 2013;35(2):106-108.
13. Knapik J, Marshall SW, Lee RB, et al. Mouthguards in sport activities: history, physical properties and Injury prevention effectiveness. Sports Med. 2007;37(2):117-144.
14. Ashack KA, Burton KA, Johnson TR, Currie DW, Comstock RD, Dellavalle RP. Skin infections among US high school athletes: a national survey. J Am Acad Dermatol. 2016;74(4):679-684.e1.
15. Ellis MW, Hospenthal DR, Dooley DP, Gray PJ, Murray CK. Natural history of community-acquired methicillin-resistant Staphylococcus aureus colonization and infection in soldiers. Clin Infect Dis. 2004;39(7):971-979.
16. The National Collegiate Athletic Association. 2014-15 NCAA Sports Medicine Handbook. http://www.ncaapublications.com/productdownloads/MD15.pdf. Revised June 2008. Accessed August 18, 2016.
17. Anderson BJ. The effectiveness of valacyclovir in preventing reactivation of herpes gladiatorum in wrestlers. Clin J Sport Med. 1999;9(2):86-90.
18. Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18-e55.
19. Jeffords MD, Batts K. Dermatology. In: O’Connor FG, Casa DJ, Davis BA, Pierre PS, Sallis RE, Wilder RP, eds. ACSM’s Sports Medicine: A Comprehensive Review. Riverwoods, IL: Wolters Kluwer; 2016:181-188.
20. Kazakova SV, Hageman JC, Matava M, et al. A clone of methicillin-resistant Staphylococcus aureus among professional football players. N Engl J Med. 2005;352(5):468-475.
21. Begier EM, Frenette K, Barrett NL, et al. A high-morbidity outbreak of methicillin-resistant Staphylococcus aureus among players on a college football team, facilitated by cosmetic body shaving and turf burns. Clin Infect Dis. 2004;39(10):1446-1453.
22. Geissler KE, Borchers JR. More than meets the eye: a rapidly progressive skin infection in a football player. Clin J Sport Med. 2015;25(3):e54-e56.
1. Owens PL, Mutter R. Emergency Department Visits Related to Eye Injuries, 2008. Agency for Healthcare Research and Quality Web site. http://www.hcup-us.ahrq.gov/reports/statbriefs/sb112.pdf. Published May 2011. Accessed August 18, 2016.
2. Rodriguez JO, Lavina AM, Agarwai A. Prevention and treatment of common eye injuries in sports. Am Fam Physician. 2003;67(7):1481-1496.
3. Lim CH, Turner A, Lim BX. Patching for corneal abrasion. Cochrane Database Syst Rev. 2016;7:CD004764.
4. Weaver CS, Terrell KM. Evidence-based emergency medicine. Update: do ophthalmic nonsteroidal anti-inflammatory drugs reduce the pain associated with simple corneal abrasion without delaying healing? Ann Emerg Med. 2003;41(1):134-140.
5. Williams RJ 3rd, Marx RG, Barnes R, O’Brien SJ, Warren RF. Fractures about the orbit in professional American football players. Am J Sports Med. 2001;29(1):55-57.
6. Forrest LA, Schuller DE, Strauss RH. Management of orbital blow-out fractures. Case reports and discussion. Am J Sports Med. 1989;17(2):217-220.
7. Barr A, Baines PS, Desai P, MacEwen CJ. Ocular sports injuries: the current picture. Br J Sports Med. 2000;34(6):456-458.
8. Pokhrel PK, Loftus SA. Ocular emergencies. Am Fam Physician. 2007;76(6):829-836.
9. Usatine RP, Smith MA, Mayeaux EJ Jr, Chumley H. Eye Trauma—Hyphema. The Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013.
10. Lincoln AE, Caswell SV, Almquist JL, et al. Effectiveness of the women’s lacrosse protective eyewear mandate in the reduction of eye injuries. Am J Sports Med. 2012;40(3):611-614.
11. Stewart GB, Shields BJ, Fields S, Comstock RD, Smith GA. Consumer products and activities associated with dental injuries to children treated in United States emergency departments, 1990-2003. Dental Traumatol. 2009;25(4):399-405.
12. Bakland LK. Dental trauma guidelines. Pediatric Dent. 2013;35(2):106-108.
13. Knapik J, Marshall SW, Lee RB, et al. Mouthguards in sport activities: history, physical properties and Injury prevention effectiveness. Sports Med. 2007;37(2):117-144.
14. Ashack KA, Burton KA, Johnson TR, Currie DW, Comstock RD, Dellavalle RP. Skin infections among US high school athletes: a national survey. J Am Acad Dermatol. 2016;74(4):679-684.e1.
15. Ellis MW, Hospenthal DR, Dooley DP, Gray PJ, Murray CK. Natural history of community-acquired methicillin-resistant Staphylococcus aureus colonization and infection in soldiers. Clin Infect Dis. 2004;39(7):971-979.
16. The National Collegiate Athletic Association. 2014-15 NCAA Sports Medicine Handbook. http://www.ncaapublications.com/productdownloads/MD15.pdf. Revised June 2008. Accessed August 18, 2016.
17. Anderson BJ. The effectiveness of valacyclovir in preventing reactivation of herpes gladiatorum in wrestlers. Clin J Sport Med. 1999;9(2):86-90.
18. Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18-e55.
19. Jeffords MD, Batts K. Dermatology. In: O’Connor FG, Casa DJ, Davis BA, Pierre PS, Sallis RE, Wilder RP, eds. ACSM’s Sports Medicine: A Comprehensive Review. Riverwoods, IL: Wolters Kluwer; 2016:181-188.
20. Kazakova SV, Hageman JC, Matava M, et al. A clone of methicillin-resistant Staphylococcus aureus among professional football players. N Engl J Med. 2005;352(5):468-475.
21. Begier EM, Frenette K, Barrett NL, et al. A high-morbidity outbreak of methicillin-resistant Staphylococcus aureus among players on a college football team, facilitated by cosmetic body shaving and turf burns. Clin Infect Dis. 2004;39(10):1446-1453.
22. Geissler KE, Borchers JR. More than meets the eye: a rapidly progressive skin infection in a football player. Clin J Sport Med. 2015;25(3):e54-e56.
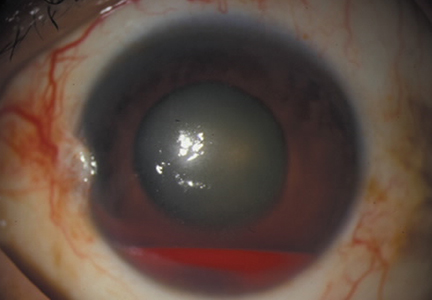
In My Athletic Trainer’s Bag
Editor’s Note: Doug Quon, MAT, ATC, PES, is the Assistant Athletic Trainer for the Washington Redskins. Click the PDF button below to view and download his list of the essential components of an athletic trainer’s bag for high school football
Editor’s Note: Doug Quon, MAT, ATC, PES, is the Assistant Athletic Trainer for the Washington Redskins. Click the PDF button below to view and download his list of the essential components of an athletic trainer’s bag for high school football
Editor’s Note: Doug Quon, MAT, ATC, PES, is the Assistant Athletic Trainer for the Washington Redskins. Click the PDF button below to view and download his list of the essential components of an athletic trainer’s bag for high school football
Knee Injuries in American Football: An Epidemiological Review
Football is one of the most popular sports in the United States. Every year more than 1 million high school males and over 60,000 collegiate males participate in organized football. The number of males who play football is greater than the combined number of males and females who participate in track and field or basketball.1 Football has the highest injury rate amongst popular American sports.2 From 2001 to 2005, there was an estimated 1.1 million emergency room visits as a direct result of football.3 Injuries are more likely to occur during games,1,2,4,5 more likely to require surgery,4 and more likely to end the player’s season or career when compared to other sports.6 Of those injuries that end seasons or careers, the knee is the most common culprit.6 This is of particular concern because knee injuries are most common in football.1,2,5,7 This article reviews the epidemiology of 4 of the most common knee injuries in American football: tears of the anterior cruciate ligament (ACL), medial collateral ligament (MCL), medial patellofemoral ligament (MPFL), and posterior cruciate ligament (PCL).
Anterior Cruciate Ligament
The ACL is the primary structure preventing anterior tibial translation. It is composed of 2 anatomic bundles: the anteromedial and posterolateral bundles. The ACL originates from the posteromedial portion of the lateral femoral condyle and inserts between and slightly anterior to the tibial intercondylar eminence. The bundles are named for their relative insertions onto the tibia.
Injury to the ACL occurs both through noncontact and contact mechanisms. Typical noncontact mechanism is a forceful valgus collapse with the knee close to full extension with combined external or internal rotation of the tibia.8 This is often the result of a sudden deceleration prior to a change in direction.9 Contact injuries to the ACL are the result of a direct blow to the knee causing valgus collapse.9 The majority of ACL injuries amongst all sports are a result of a noncontact mechanism. However, Dragoo and colleagues10 found the majority of football ACL injuries (55%-60%) were from contact. As a result, football players are 4 times more likely to sustain ACL injuries than in other sports.11
ACL injuries are associated with significant time loss from sport. At the high school level, they are the most likely injury to end a season or career.6 Because these are higher-energy injuries, they are frequently associated with damage to additional structures. ACL injuries that occur in football are associated with increased rates of meniscus, chondral, and multi-ligamentous injuries.12,13
The incidence of ACL injuries increases with level of competition. In high school athletes it is 11.1 per 100,000 athlete exposures (AE).1,11 In collegiate football, the rate increases to 14.2 to 18 per 100,000 AE.2,14 Though no incidence data per AE was found in our review of the literature, there were 219 ACL injuries in the National Football League (NFL) from 2010 to 2013.15 In addition, 14.2% of retired NFL athletes in one survey reported a history of ACL injury.16
The most common high-risk positions are running backs and linebackers. Brophy and colleagues17 found that 9.7% of running backs and 8.9% of linebackers participating in the NFL Combine had a history of ACL injury. This may be because both the running back and linebacker are involved in frequent high-energy collisions and often quickly change direction. Other studies have also identified running backs and linebackers as high risk, in addition to tight ends, wide receivers, and interior linemen.13,15,18
Treatment of choice for elite level athletes with ACL injury is reconstruction.19 Of those who undergo ACL reconstruction, the rate of return to play ranges from 63% to 80%.20-22 The average time to return to play is 9 to 13 months. The odds of making a successful return hinges on how successful the athlete was prior to injury. Factors such as prior game experience, position on depth chart, being on scholarship, and draft position for NFL athletes have all been shown to have a positive predictive value on a patient’s chance of returning from ACL reconstruction.20,21
Players who return have variable levels of success afterwards. In a study of NFL quarterbacks who sustained ACL injuries, 12 out of 13 were able to return to game action with no appreciable dropoff in performance based on in-game production.23 Carey and colleagues24 looked specifically at NFL wide receivers and running backs and found an 80% return to play rate but with an approximate decrease in production of one-third upon return. Furthermore, in the Multicenter Orthopaedic Outcomes Network (MOON) cohort study, only 43% of participants felt they returned to their preoperative level.22
Medial Collateral Ligament
The MCL consists of superficial and deep components. The superficial MCL is the primary restraint to valgus laxity at the knee. The superficial MCL has 1 femoral and 2 tibial attachments. The deep MCL is a thickening of the medial joint capsule and runs deep and parallel to the superficial MCL. The amount of medial joint gapping with a valgus force on examination is used to grade severity of MCL injuries. Grade I is a <5-mm opening; Grade II, 5- to 10-mm opening; and grade III, >10-mm opening.
The MCL is the most common knee injury in high school, collegiate, and professional football.1,18,25-28 Injuries are typically due to contact when a valgus force is applied to the knee.29 The annual incidence of MCL injuries amongst high school football players is 24.2 per 100,000 AE.1 The positions that appear to be at greatest risk for MCL injuries are offensive and defensive linemen.18,30-32 In a review of 5047 collegiate athletes participating in the NFL Combine from 1987 to 2000, 23% of offensive linemen had a history of MCL injury, compared to the overall rate of 16%.33 In a similar study, Bradley and colleagues18 performed medical histories on athletes invited to the 2005 NFL Combine and also found offensive linemen had the highest rate of MCL injury at 33%, compared to the overall rate of 22%. They reasonably hypothesized that “chop blocks” and other players “rolling up” on the outside of linemen’s knees were responsible for these injuries. Albright and colleagues32 found that prophylactic knee braces decreased the incidence of MCL injuries in collegiate offensive lineman. However, additional studies have not been able to reproduce these results and the use of prophylactic knee braces remains controversial.26
Treatment of MCL injuries depends upon the grade of injury, associated injuries, and anatomical location of injury. Management of MCL injuries is for the most part nonsurgical. In 1974, Ellsasser and colleagues34 were the first to publish data on nonoperative management of Grade I and Grade II injuries with immediate motion and rehabilitation instead of cast immobilization. They found 93% of patients returned to football in 3 to 8 weeks.34 Derscheid and Garrick27 observed nonoperative treatment of Grade I and II sprains in collegiate football players, with a time loss of 10.6 days and 19.5 days for Grade I and II injuries, respectively. Holden and colleagues35 evaluated nonoperative management of Grade I and II MCL injuries in collegiate football players and found an average return to play of 21 days.
Grade III injury treatment is more controversial. Indelicato and colleagues36 demonstrated successful nonoperative management of Grade III MCL injuries in collegiate football players, with an average return to play of 64.4 days. Jones and colleagues37 had similar success with high school football players, with an average return to play of 34 days. However, isolated Grade III injuries are rare and therefore treatment is likely to be dictated by concomitant injuries. Fetto and Marshall38 found that 78% of Grade III injuries were associated with an additional ligamentous injury. Of those additional injuries, 95% were ACL tears.
Finally, one must consider the location of the MCL injury. Injuries of the distal MCL at its tibial insertion may result in poor healing, as the ligament is displaced away from its insertion. Therefore, some authors recommend surgical management for these injuries.39,40
The patellofemoral joint is a complex structure in which the patella is stabilized within the trochlear groove of the femur by both bony and soft tissue structures. The MPFL is one of the most important soft tissue stabilizers. The MPFL is the primary restraint to lateral patellar translation within the first 20° of knee flexion, contributing to 60% of the total restraining force.41 The MPFL originates on the medial femoral condyle and inserts on the superomedial aspect of the patella.
Patellar instability is the subluxation or dislocation of the patella out of the trochlear groove. Patellar subluxation and dislocation account for approximately 3% of all knee injuries.42 Patella dislocations are more common in younger populations43-45 with the majority (52%-63%) occurring during sports.43,44,46 Mitchell and colleagues47 reported an incidence of 4.1 patellar subluxations/dislocations per 100,000 AE in high school football players.
Dislocation is most commonly the result of knee flexion with the tibia in a valgus position.44,48 The majority of patellar dislocations occur via a noncontact mechanism.44,48 However, the majority of these injuries in football are from contact (63%).47
Acute patellar dislocations are associated with more soft tissue damage than those with recurrent dislocations.46 In acute patella dislocations, the MPFL is almost always ruptured.44 In contrast, Fithian and colleagues46 found only 38% of recurrent dislocators had MPFL injury. As a result, it is thought that those with recurrent instability dislocate without trauma and do not have the same characteristics as those who dislocate from high-energy trauma in sport. Risk factors for atraumatic dislocation are numerous and have been well described in the literature.49 However, traumatic dislocators usually do not have risk factors.50
Traumatic patella dislocations are higher energy and are associated with chondral injury in up to 95%of cases 51 and osteochondral injury 58% to 76% of the time.52,53 In contrast, people with “articular hypermobility” are less likely to sustain articular damage.54 This concept is important when considering risk for recurrent patella dislocation. The literature reports a 17% to 50% rate of recurrent instability after acute patella dislocation.46,55,56 However, most studies do not distinguish between traumatic and atraumatic injuries. Because the majority of patellar dislocations in football occur through contact mechanisms, the rate of recurrent instability in these athletes may in fact be less than what is reported in the literature.
First-time patella dislocations are generally treated nonoperatively. Mitchell and colleagues47 reported that 72.6% of high school athletes with patella subluxation treated conservatively were able to return to sports within 3 weeks, compared to only 34.1% of those with patellar dislocations. In the same study, patellar dislocations were season-ending 37% of the time.47 Atkin and colleagues50 followed 74 patients treated conservatively for first-time patellar dislocation and noted 58% at 6 months still had difficulty in squatting, jumping, or cutting.
Those who have failed conservative management and have an additional dislocation are 7 times more likely to redislocate.46 Therefore, they are usually treated operatively with MPFL reconstruction. Return to sport ranges from 3 to 6 months,57 with 53% to 77.3% reporting return to their previous functionality.57-59 Overall, 84.1% of patients are able to return to sport with 1.2% risk of recurrent dislocation.60
Posterior Cruciate Ligament
The PCL is the primary posterior stabilizer of the knee.61,62 It consists of the anterolateral and posteromedial bundles, named by their insertion on the posterior tibial plateau. The larger, stronger anterolateral bundle is the primary restraint to posterior tibial translation.63
Due to the relative infrequency of PCL injuries, there is a paucity of epidemiological data on sports-related PCL injuries. These injuries in the literature are commonly found due to traffic accidents (45%-57%) or from sports (33%-40%).64,65 According to Swensen and colleagues,1 PCL injuries account for 2.4% of all high school sport knee injuries. In a cohort of 62 knees with PCL injuries, Patel and colleagues66 found football was the most common cause of injury (19.3%).
The most common mechanism of injury in athletes is knee hyperflexion or a direct blow to the tibia in a flexed knee.67 In football, contact mechanisms are the most common. In a 16-year review of the National Collegiate Athletic Association (NCAA) injury surveillance system, the incidence of contact PCL injuries during games were 7.3 times higher than noncontact.68 The most common activity was being tackled, which accounted for 22.9% of all PCL injuries.68
Due to the high energy of these injuries, isolated PCL injuries are rare. In one trauma center’s experience, 96.5% of PCL injuries had an additional ligament injury.64 In that study, injuries to the PCL were associated with posterolateral corner, ACL, and MCL injuries 62%, 46%, and 31% of the time, respectively.64,69
Because isolated PCL injuries are rare, clinicians must rely on a thorough history and physical examination when evaluating athletes with knee injuries. Classification of PCL injuries is based on the amount of posterior tibial translation in relation to the femur with the knee bent to 90°. Grade I is 1 to 5 mm; Grade II, 6 to 10 mm; and Grade III, >10 mm. If there is suspicion of a PCL injury, there should be a very low threshold for magnetic resonance imaging, given the high association with additional injuries.
Natural history of Grade I and II isolated PCL injuries is generally favorable compared to Grade III and multi-ligamentous injuries.70 As a result, isolated Grade I and II PCL injuries are generally treated nonoperatively. Treatment consists of physical therapy with emphasis on quadriceps strengthening. Return to play can be considered as early as 2 to 4 weeks from injury.71 Recent long-term data have shown successful conservative management of Grade I and II injuries with quadriceps strength to 97% of contralateral leg and full range of motion.72 However, there was 11% moderate to severe osteoarthritis in these patients at a mean follow-up of 14.3 years.72 Fowler and Messieh67 managed athletes with 7 isolated complete PCL tears and 5 partial tears nonoperatively, all of whom were able to return to sport without limitation. Parolie and Bergfeld73 managed 25 athletes with isolated PCL tears conservatively. In this study, 80% of athletes reported satisfaction and 68% returned to previous level of play.73 Neither of the aforementioned studies specify the grades of the injuries. Finally, Patel and colleagues66 managed 6 NFL athletes with Grade I and II injuries nonoperatively, and all were able to return to sport.
Treatment of isolated Grade III PCL injuries is more controversial, and no consensus exists in the literature. In an epidemiological study, Dick and colleagues68 found that only 39% of NCAA football athletes underwent surgery for their torn PCLs, compared to 79% of ACL injuries. However, their study makes no mention to the severity of these injuries. Numerous options exist for PCL reconstruction, with no consensus on the preferred method.
Conclusion
Knee injuries are the most common injury in football. Knowledge of the natural history of these injuries, as well as treatment options and expected outcomes, will help treating physicians educate their patients on the optimal treatment and manage return to play expectations.
Am J Orthop. 2016;45(6):368-373. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Swenson DM, Collins CL, Best TM, Flanigan DC, Fields SK, Comstock RD. Epidemiology of knee injuries among U.S. high school athletes, 2005/2006-2010/2011. Med Sci Sports Exerc. 2013;45(3):462-469.
2. Hootman JM, Dick R, Agel J. Epidemiology of collegiate injuries for 15 sports: summary and recommendations for injury prevention initiatives. J Athl Train. 2007;42(2):311-319.
3. Mello MJ, Myers R, Christian JB, Palmisciano L, Linakis JG. Injuries in youth football: national emergency department visits during 2001-2005 for young and adolescent players. Acad Emerg Med. 2009;16(3):243-248.
4. Rechel JA, Collins CL, Comstock RD. Epidemiology of injuries requiring surgery among high school athletes in the United States, 2005 to 2010. J Trauma. 2011;71(4):982-989.
5. Ingram JG, Fields SK, Yard EE, Comstock RD. Epidemiology of knee injuries among boys and girls in US high school athletics. Am J Sports Med. 2008;36(6):1116-1122.
6. Tirabassi J, Brou L, Khodaee M, Lefort R, Fields SK, Comstock RD. Epidemiology of high school sports-related injuries resulting in medical disqualification: 2005-2006 through 2013-2014 academic years. Am J Sports Med. 2016 May 10. [Epub ahead of print]
7. Fernandez WG, Yard EE, Comstock RD. Epidemiology of lower extremity injuries among U.S. high school athletes. Acad Emerg Med. 2007;14(7):641-645.
8. Olsen OE, Myklebust G, Engebretsen L, Bahr R. Injury mechanisms for anterior cruciate ligament injuries in team handball: a systematic video analysis. Am J Sports Med. 2004;32(4):1002-1012.
9. Boden BP, Dean GS, Feagin JA Jr, Garrett WE Jr. Mechanisms of anterior cruciate ligament injury. Orthopedics. 2000;23(6):573-578.
10. Dragoo JL, Braun HJ, Harris AH. The effect of playing surface on the incidence of ACL injuries in National Collegiate Athletic Association American Football. Knee. 2013;20(3):191-195.
11. Joseph AM, Collins CL, Henke NM, Yard EE, Fields SK, Comstock RD. A multisport epidemiologic comparison of anterior cruciate ligament injuries in high school athletics. J Athl Train. 2013;48(6):810-817.
12. Granan LP, Inacio MC, Maletis GB, Funahashi TT, Engebretsen L. Sport-specific injury pattern recorded during anterior cruciate ligament reconstruction. Am J Sports Med. 2013;41(12):2814-2818.
13. Bradley JP, Klimkiewicz JJ, Rytel MJ, Powell JW. Anterior cruciate ligament injuries in the National Football League: epidemiology and current treatment trends among team physicians. Arthroscopy. 2002;18(5):502-509.
14. Dragoo JL, Braun HJ, Durham JL, Chen MR, Harris AH. Incidence and risk factors for injuries to the anterior cruciate ligament in National Collegiate Athletic Association football: data from the 2004-2005 through 2008-2009 National Collegiate Athletic Association Injury Surveillance System. Am J Sports Med. 2012;40(5):990-995.
15. Dodson CC, Secrist ES, Bhat SB, Woods DP, Deluca PF. Anterior cruciate ligamenti in National Football League athletes from 2010 to 2013: a descriptive epidemiology study. Orthop J Sports Med. 2016;4(3):2325967116631949.
16. Golightly YM, Marshall SW, Callahan LF, Guskiewicz K. Early-onset arthritis in retired National Football League players. J Phys Act Health. 2009;6(5):638-643.
17. Brophy RH, Lyman S, Chehab EL, Barnes RP, Rodeo SA, Warren RF. Predictive value of prior injury on career in professional American football is affected by player position. Am J Sports Med. 2009;37(4):768-775.
18. Bradley J, Honkamp NJ, Jost P, West R, Norwig J, Kaplan LD. Incidence and variance of knee injuries in elite college football players. Am J Orthop. 2008;37(6):310-314.
19. Erickson BJ, Harris JD, Fillingham YA, et al. Anterior cruciate ligament reconstruction practice patterns by NFL and NCAA football team physicians. Arthroscopy. 2014;30(6):731-738.
20. Daruawalla JH, Greis PE, Hancock R; ASP Collaborative Group, Xerogeanes JW. Rates and determinants of return to play after anterior cruciate ligament reconstruction in NCAA Division 1 college football athletes: a study of the ACC, SEC, and PAC-12 conferences. Orthop J Sports Med. 2014;2(8):2325967114543901.
21. Shah VM, Andrews JR, Fleisig GS, McMichael CS, Lemak LJ. Return to play after anterior cruciate ligament reconstruction in National Football League athletes. Am J Sports Med. 2010;38(11):2233-2239.
22. McCullough KA, Phelps KD, Spindler KP, et al. Return to high school- and college-level football after anterior cruciate ligament reconstruction: a Multicenter Orthopaedic Outcomes Network (MOON) cohort study. Am J Sports Med. 2012;40(11):2523-2529.
23. Erickson BJ, Harris JD, Heninger JR, et al. Performance and return-to-sport after ACL reconstruction in NFL quarterbacks. Orthopedics. 2014;37(8):e728-e734.
24. Carey JL, Huffman GR, Parekh SG, Sennett BJ. Outcomes of anterior cruciate ligament injuries to running backs and wide receivers in the National Football League. Am J Sports Med. 2006;34(12):1911-1917.
25. Hershman EB, Anderson R, Bergfeld JA, et al. An analysis of specific lower extremity injury rates on grass and FieldTurf playing surfaces in National Football League Games: 2000-2009 seasons. Am J Sports Med. 2012;40(10):2200-2205.
26. Salata MJ, Gibbs AE, Sekiya JK. The effectiveness of prophylactic knee bracing in American football: a systematic review. Sports Health. 2010;2(5):375-379.
27. Derscheid GL, Garrick JG. Medial collateral ligament injuries in football. Nonoperative management of grade I and grade II sprains. Am J Sports Med. 1981;9(6):365-368.
28. Meyers MC, Barnhill BS. Incidence, causes, and severity of high school football injuries on FieldTurf versus natural grass: a 5-year prospective study. Am J Sports Med. 2004;32(7):1626-1638.
29. Lundblad M, Waldén M, Magnusson H, Karlsson J, Ekstrand J. The UEFA injury study: 11-year data concerning 346 MCL injuries and time to return to play. Br J Sports Med. 2013;47(12):759-762.
30. Hewson GF Jr, Mendini RA, Wang JB. Prophylactic knee bracing in college football. Am J Sports Med. 1986;14(4):262-266.
31. Rovere GD, Haupt HA, Yates CS. Prophylactic knee bracing in college football. Am J Sports Med. 1987;15(2):111-116.
32. Albright JP, Powell JW, Smith W, et al. Medial collateral ligament knee sprains in college football. Brace wear preferences and injury risk. Am J Sports Med. 1994;22(1):2-11.
33. Brophy RH, Barnes R, Rodeo SA, Warren RF. Prevalence of musculoskeletal disorders at the NFL Combine--trends from 1987 to 2000. Med Sci Sports Exerc. 2007;39(1):22-27.
34. Ellsasser JC, Reynolds FC, Omohundro JR. The non-operative treatment of collateral ligament injuries of the knee in professional football players. An analysis of seventy-four injuries treated non-operatively and twenty-four injuries treated surgically. J Bone Joint Surg Am. 1974;56(6):1185-1190.
35. Holden DL, Eggert AW, Butler JE. The nonoperative treatment of grade I and II medial collateral ligament injuries to the knee. Am J Sports Med. 1983;11(5):340-344.
36. Indelicato PA, Hermansdorfer J, Huegel M. Nonoperative management of complete tears of the medial collateral ligament of the knee in intercollegiate football players. Clin Orthop Relat Res. 1990;(256):174-177.
37. Jones RE, Henley MB, Francis P. Nonoperative management of isolated grade III collateral ligament injury in high school football players. Clin Orthop Relat Res. 1986;(213):137-140.
38. Fetto JF, Marshall JL. Medial collateral ligament injuries of the knee: a rationale for treatment. Clin Orthop Relat Res. 1978;(132):206-218.
39. Corten K, Hoser C, Fink C, Bellemans J. Case reports: a Stener-like lesion of the medial collateral ligament of the knee. Clin Orthop Relat Res. 2010;468(1):289-293.
40. Marchant MH Jr, Tibor LM, Sekiya JK, Hardaker WT Jr, Garrett WE Jr, Taylor DC. Management of medial-sided knee injuries, part 1: medial collateral ligament. Am J Sports Med. 2011;39(5):1102-1113.
41. Desio SM, Burks RT, Bachus KN. Soft tissue restraints to lateral patellar translation in the human knee. Am J Sports Med. 1998;26(1):59-65.
42. Casteleyn PP, Handelberg F. Arthroscopy in the diagnosis of occult dislocation of the patella. Acta Orthop Belg. 1989;55(3):381-383.
43. Waterman BR, Belmont PJ Jr, Owens BD. Patellar dislocation in the United States: role of sex, age, race, and athletic participation. J Knee Surg. 2012;25(1):51-57.
44. Sillanpää P, Mattila VM, Iivonen T, Visuri T, Pihlajamäki H. Incidence and risk factors of acute traumatic primary patellar dislocation. Med Sci Sports Exerc. 2008;40(4):606-611.
45. Hsiao M, Owens BD, Burks R, Sturdivant RX, Cameron KL. Incidence of acute traumatic patellar dislocation among active-duty United States military service members. Am J Sports Med. 2010;38(10):1997-2004.
46. Fithian DC, Paxton EW, Stone ML, et al. Epidemiology and natural history of acute patellar dislocation. Am J Sports Med. 2004;32(5):1114-1121.
47. Mitchell J, Magnussen RA, Collins CL, et al. Epidemiology of patellofemoral instability injuries among high school athletes in the United States. Am J Sports Med. 2015;43(7):1676-1682.
48. Nikku R, Nietosvaara Y, Aalto K, Kallio PE. The mechanism of primary patellar dislocation: trauma history of 126 patients. Acta Orthop. 2009;80(4):432-434.
49. Tsai CH, Hsu CJ, Hung CH, Hsu HC. Primary traumatic patellar dislocation. J Orthop Surg Res. 2012;7:21.
50. Atkin DM, Fithian DC, Marangi KS, Stone ML, Dobson BE, Mendelsohn C. Characteristics of patients with primary acute lateral patellar dislocation and their recovery within the first 6 months of injury. Am J Sports Med. 2000;28(4):472-479.
51. Nomura E, Inoue M, Kurimura M. Chondral and osteochondral injuries associated with acute patellar dislocation. Arthroscopy. 2003;19(7):717-721.
52. Kirsch MD, Fitzgerald SW, Friedman H, Rogers LF. Transient lateral patellar dislocation: diagnosis with MR imaging. AJR Am J Roentgenol. 1993;161(1):109-113.
53. Virolainen H, Visuri T, Kuusela T. Acute dislocation of the patella: MR findings. Radiology. 1993;189(1):243-246.
54. Stanitski CL. Articular hypermobility and chondral injury in patients with acute patellar dislocation. Am J Sports Med. 1995;23(2):146-150.
55. Mäenpää H, Huhtala H, Lehto MU. Recurrence after patellar dislocation. Redislocation in 37/75 patients followed for 6-24 years. Acta Orthop Scand. 1997;68(5):424-426.
56. Buchner M, Baudendistel B, Sabo D, Schmitt H. Acute traumatic primary patellar dislocation: long-term results comparing conservative and surgical treatment. Clin J Sport Med. 2005;15(2):62-66.
57. Fisher B, Nyland J, Brand E, Curtin B. Medial patellofemoral ligament reconstruction for recurrent patellar dislocation: a systematic review including rehabilitation and return-to-sports efficacy. Arthroscopy. 2010;26(10):1384-1394.
58. Lippacher S, Dreyhaupt J, Williams SR, Reichel H, Nelitz M. Reconstruction of the medial patellofemoral ligament: clinical outcomes and return to sports. Am J Sports Med. 2014;42(7):1661-1668.
59. Panni AS, Alam M, Cerciello S, Vasso M, Maffulli N. Medial patellofemoral ligament reconstruction with a divergent patellar transverse 2-tunnel technique. Am J Sports Med. 2011;39(12):2647-1655.
60. Schneider DK, Grawe B, Magnussen RA, et al. Outcomes after isolated medial patellofemoral ligament reconstruction for the treatment of recurrent lateral patellar dislocations: a systematic review and meta-analysis. Am J Sports Med. 2016 Feb 12. [Epub ahead of print]
61. Amis AA, Bull AM, Gupte CM, Hijazi I, Race A, Robinson JR. Biomechanics of the PCL and related structures: posterolateral, posteromedial and meniscofemoral ligaments. Knee Surg Sports Traumatol Arthrosc. 2003;11(5):271-281.
62. Fu FH, Harner CD, Johnson DL, Miller MD, Woo SL. Biomechanics of knee ligaments: basic concepts and clinical application. Instr Course Lect. 1994;43:137-148.
63. Markolf KL, Feeley BT, Tejwani SG, Martin DE, McAllister DR. Changes in knee laxity and ligament force after sectioning the posteromedial bundle of the posterior cruciate ligament. Arthroscopy. 2006; 22(10):1100-1106.
64. Ganelli GC, Edson CJ. Posterior cruciate ligament injuries in trauma patients: Part II. Arthroscopy. 1995;11(5):526-529.
65. Schulz MS, Russe K, Weiler A, Eichhorn HJ, Strobel MJ. Epidemiology of posterior cruciate ligament injuries. Arch Orthop Trauma Surg. 2003;123(4):186-191.
66. Patel DV, Allen AA, Warren RF, Wickiewicz TL, Simonian PT. The nonoperative treatment of acute, isolated (partial or complete) posterior cruciate ligament-deficient knees: an intermediate-term follow-up study. HSS J. 2007;3(2):137-146.
67. Fowler PJ, Messieh SS. Isolated posterior cruciate ligament injuries in athletes. Am J Sports Med. 1987;15(6):553-557.
68. Dick R, Ferrara MS, Agel J, et al. Descriptive epidemiology of collegiate men’s football injuries: National Collegiate Athletic Association Injury Surveillance System, 1988-1989 through 2003-2004. J Athl Train. 2007;42(2):221-233.
69. LaPrade CM, Civitarese DM, Rasmussen MT, LaPrade RF. Emerging updates on the posterior cruciate ligament: a review of the current literature. Am J Sports Med. 2015;43(12):3077-3092.
70. Torg JS, Barton TM, Pavlov H, Stine R. Natural history of the posterior cruciate ligament-deficient knee. Clin Orthop Relat Res. 1989(246):208-216.
71. Miller MD. Orthopaedic Knowledge Update: Sports Medicine 5. Rosemont, IL; American Academy of Orthopaedic Surgeons; 2016.
72. Shelbourne KD, Clark M, Gray T. Minimum 10-year follow-up of patients after an acute, isolated posterior cruciate ligament injury treated nonoperatively. Am J Sports Med. 2013;41(7):1526-1533.
73. Parolie JM, Bergfeld JA. Long-term results of nonoperative treatment of isolated posterior cruciate ligament injuries in the athlete. Am J Sports Med. 1986;14(1):35-38.
Football is one of the most popular sports in the United States. Every year more than 1 million high school males and over 60,000 collegiate males participate in organized football. The number of males who play football is greater than the combined number of males and females who participate in track and field or basketball.1 Football has the highest injury rate amongst popular American sports.2 From 2001 to 2005, there was an estimated 1.1 million emergency room visits as a direct result of football.3 Injuries are more likely to occur during games,1,2,4,5 more likely to require surgery,4 and more likely to end the player’s season or career when compared to other sports.6 Of those injuries that end seasons or careers, the knee is the most common culprit.6 This is of particular concern because knee injuries are most common in football.1,2,5,7 This article reviews the epidemiology of 4 of the most common knee injuries in American football: tears of the anterior cruciate ligament (ACL), medial collateral ligament (MCL), medial patellofemoral ligament (MPFL), and posterior cruciate ligament (PCL).
Anterior Cruciate Ligament
The ACL is the primary structure preventing anterior tibial translation. It is composed of 2 anatomic bundles: the anteromedial and posterolateral bundles. The ACL originates from the posteromedial portion of the lateral femoral condyle and inserts between and slightly anterior to the tibial intercondylar eminence. The bundles are named for their relative insertions onto the tibia.
Injury to the ACL occurs both through noncontact and contact mechanisms. Typical noncontact mechanism is a forceful valgus collapse with the knee close to full extension with combined external or internal rotation of the tibia.8 This is often the result of a sudden deceleration prior to a change in direction.9 Contact injuries to the ACL are the result of a direct blow to the knee causing valgus collapse.9 The majority of ACL injuries amongst all sports are a result of a noncontact mechanism. However, Dragoo and colleagues10 found the majority of football ACL injuries (55%-60%) were from contact. As a result, football players are 4 times more likely to sustain ACL injuries than in other sports.11
ACL injuries are associated with significant time loss from sport. At the high school level, they are the most likely injury to end a season or career.6 Because these are higher-energy injuries, they are frequently associated with damage to additional structures. ACL injuries that occur in football are associated with increased rates of meniscus, chondral, and multi-ligamentous injuries.12,13
The incidence of ACL injuries increases with level of competition. In high school athletes it is 11.1 per 100,000 athlete exposures (AE).1,11 In collegiate football, the rate increases to 14.2 to 18 per 100,000 AE.2,14 Though no incidence data per AE was found in our review of the literature, there were 219 ACL injuries in the National Football League (NFL) from 2010 to 2013.15 In addition, 14.2% of retired NFL athletes in one survey reported a history of ACL injury.16
The most common high-risk positions are running backs and linebackers. Brophy and colleagues17 found that 9.7% of running backs and 8.9% of linebackers participating in the NFL Combine had a history of ACL injury. This may be because both the running back and linebacker are involved in frequent high-energy collisions and often quickly change direction. Other studies have also identified running backs and linebackers as high risk, in addition to tight ends, wide receivers, and interior linemen.13,15,18
Treatment of choice for elite level athletes with ACL injury is reconstruction.19 Of those who undergo ACL reconstruction, the rate of return to play ranges from 63% to 80%.20-22 The average time to return to play is 9 to 13 months. The odds of making a successful return hinges on how successful the athlete was prior to injury. Factors such as prior game experience, position on depth chart, being on scholarship, and draft position for NFL athletes have all been shown to have a positive predictive value on a patient’s chance of returning from ACL reconstruction.20,21
Players who return have variable levels of success afterwards. In a study of NFL quarterbacks who sustained ACL injuries, 12 out of 13 were able to return to game action with no appreciable dropoff in performance based on in-game production.23 Carey and colleagues24 looked specifically at NFL wide receivers and running backs and found an 80% return to play rate but with an approximate decrease in production of one-third upon return. Furthermore, in the Multicenter Orthopaedic Outcomes Network (MOON) cohort study, only 43% of participants felt they returned to their preoperative level.22
Medial Collateral Ligament
The MCL consists of superficial and deep components. The superficial MCL is the primary restraint to valgus laxity at the knee. The superficial MCL has 1 femoral and 2 tibial attachments. The deep MCL is a thickening of the medial joint capsule and runs deep and parallel to the superficial MCL. The amount of medial joint gapping with a valgus force on examination is used to grade severity of MCL injuries. Grade I is a <5-mm opening; Grade II, 5- to 10-mm opening; and grade III, >10-mm opening.
The MCL is the most common knee injury in high school, collegiate, and professional football.1,18,25-28 Injuries are typically due to contact when a valgus force is applied to the knee.29 The annual incidence of MCL injuries amongst high school football players is 24.2 per 100,000 AE.1 The positions that appear to be at greatest risk for MCL injuries are offensive and defensive linemen.18,30-32 In a review of 5047 collegiate athletes participating in the NFL Combine from 1987 to 2000, 23% of offensive linemen had a history of MCL injury, compared to the overall rate of 16%.33 In a similar study, Bradley and colleagues18 performed medical histories on athletes invited to the 2005 NFL Combine and also found offensive linemen had the highest rate of MCL injury at 33%, compared to the overall rate of 22%. They reasonably hypothesized that “chop blocks” and other players “rolling up” on the outside of linemen’s knees were responsible for these injuries. Albright and colleagues32 found that prophylactic knee braces decreased the incidence of MCL injuries in collegiate offensive lineman. However, additional studies have not been able to reproduce these results and the use of prophylactic knee braces remains controversial.26
Treatment of MCL injuries depends upon the grade of injury, associated injuries, and anatomical location of injury. Management of MCL injuries is for the most part nonsurgical. In 1974, Ellsasser and colleagues34 were the first to publish data on nonoperative management of Grade I and Grade II injuries with immediate motion and rehabilitation instead of cast immobilization. They found 93% of patients returned to football in 3 to 8 weeks.34 Derscheid and Garrick27 observed nonoperative treatment of Grade I and II sprains in collegiate football players, with a time loss of 10.6 days and 19.5 days for Grade I and II injuries, respectively. Holden and colleagues35 evaluated nonoperative management of Grade I and II MCL injuries in collegiate football players and found an average return to play of 21 days.
Grade III injury treatment is more controversial. Indelicato and colleagues36 demonstrated successful nonoperative management of Grade III MCL injuries in collegiate football players, with an average return to play of 64.4 days. Jones and colleagues37 had similar success with high school football players, with an average return to play of 34 days. However, isolated Grade III injuries are rare and therefore treatment is likely to be dictated by concomitant injuries. Fetto and Marshall38 found that 78% of Grade III injuries were associated with an additional ligamentous injury. Of those additional injuries, 95% were ACL tears.
Finally, one must consider the location of the MCL injury. Injuries of the distal MCL at its tibial insertion may result in poor healing, as the ligament is displaced away from its insertion. Therefore, some authors recommend surgical management for these injuries.39,40
The patellofemoral joint is a complex structure in which the patella is stabilized within the trochlear groove of the femur by both bony and soft tissue structures. The MPFL is one of the most important soft tissue stabilizers. The MPFL is the primary restraint to lateral patellar translation within the first 20° of knee flexion, contributing to 60% of the total restraining force.41 The MPFL originates on the medial femoral condyle and inserts on the superomedial aspect of the patella.
Patellar instability is the subluxation or dislocation of the patella out of the trochlear groove. Patellar subluxation and dislocation account for approximately 3% of all knee injuries.42 Patella dislocations are more common in younger populations43-45 with the majority (52%-63%) occurring during sports.43,44,46 Mitchell and colleagues47 reported an incidence of 4.1 patellar subluxations/dislocations per 100,000 AE in high school football players.
Dislocation is most commonly the result of knee flexion with the tibia in a valgus position.44,48 The majority of patellar dislocations occur via a noncontact mechanism.44,48 However, the majority of these injuries in football are from contact (63%).47
Acute patellar dislocations are associated with more soft tissue damage than those with recurrent dislocations.46 In acute patella dislocations, the MPFL is almost always ruptured.44 In contrast, Fithian and colleagues46 found only 38% of recurrent dislocators had MPFL injury. As a result, it is thought that those with recurrent instability dislocate without trauma and do not have the same characteristics as those who dislocate from high-energy trauma in sport. Risk factors for atraumatic dislocation are numerous and have been well described in the literature.49 However, traumatic dislocators usually do not have risk factors.50
Traumatic patella dislocations are higher energy and are associated with chondral injury in up to 95%of cases 51 and osteochondral injury 58% to 76% of the time.52,53 In contrast, people with “articular hypermobility” are less likely to sustain articular damage.54 This concept is important when considering risk for recurrent patella dislocation. The literature reports a 17% to 50% rate of recurrent instability after acute patella dislocation.46,55,56 However, most studies do not distinguish between traumatic and atraumatic injuries. Because the majority of patellar dislocations in football occur through contact mechanisms, the rate of recurrent instability in these athletes may in fact be less than what is reported in the literature.
First-time patella dislocations are generally treated nonoperatively. Mitchell and colleagues47 reported that 72.6% of high school athletes with patella subluxation treated conservatively were able to return to sports within 3 weeks, compared to only 34.1% of those with patellar dislocations. In the same study, patellar dislocations were season-ending 37% of the time.47 Atkin and colleagues50 followed 74 patients treated conservatively for first-time patellar dislocation and noted 58% at 6 months still had difficulty in squatting, jumping, or cutting.
Those who have failed conservative management and have an additional dislocation are 7 times more likely to redislocate.46 Therefore, they are usually treated operatively with MPFL reconstruction. Return to sport ranges from 3 to 6 months,57 with 53% to 77.3% reporting return to their previous functionality.57-59 Overall, 84.1% of patients are able to return to sport with 1.2% risk of recurrent dislocation.60
Posterior Cruciate Ligament
The PCL is the primary posterior stabilizer of the knee.61,62 It consists of the anterolateral and posteromedial bundles, named by their insertion on the posterior tibial plateau. The larger, stronger anterolateral bundle is the primary restraint to posterior tibial translation.63
Due to the relative infrequency of PCL injuries, there is a paucity of epidemiological data on sports-related PCL injuries. These injuries in the literature are commonly found due to traffic accidents (45%-57%) or from sports (33%-40%).64,65 According to Swensen and colleagues,1 PCL injuries account for 2.4% of all high school sport knee injuries. In a cohort of 62 knees with PCL injuries, Patel and colleagues66 found football was the most common cause of injury (19.3%).
The most common mechanism of injury in athletes is knee hyperflexion or a direct blow to the tibia in a flexed knee.67 In football, contact mechanisms are the most common. In a 16-year review of the National Collegiate Athletic Association (NCAA) injury surveillance system, the incidence of contact PCL injuries during games were 7.3 times higher than noncontact.68 The most common activity was being tackled, which accounted for 22.9% of all PCL injuries.68
Due to the high energy of these injuries, isolated PCL injuries are rare. In one trauma center’s experience, 96.5% of PCL injuries had an additional ligament injury.64 In that study, injuries to the PCL were associated with posterolateral corner, ACL, and MCL injuries 62%, 46%, and 31% of the time, respectively.64,69
Because isolated PCL injuries are rare, clinicians must rely on a thorough history and physical examination when evaluating athletes with knee injuries. Classification of PCL injuries is based on the amount of posterior tibial translation in relation to the femur with the knee bent to 90°. Grade I is 1 to 5 mm; Grade II, 6 to 10 mm; and Grade III, >10 mm. If there is suspicion of a PCL injury, there should be a very low threshold for magnetic resonance imaging, given the high association with additional injuries.
Natural history of Grade I and II isolated PCL injuries is generally favorable compared to Grade III and multi-ligamentous injuries.70 As a result, isolated Grade I and II PCL injuries are generally treated nonoperatively. Treatment consists of physical therapy with emphasis on quadriceps strengthening. Return to play can be considered as early as 2 to 4 weeks from injury.71 Recent long-term data have shown successful conservative management of Grade I and II injuries with quadriceps strength to 97% of contralateral leg and full range of motion.72 However, there was 11% moderate to severe osteoarthritis in these patients at a mean follow-up of 14.3 years.72 Fowler and Messieh67 managed athletes with 7 isolated complete PCL tears and 5 partial tears nonoperatively, all of whom were able to return to sport without limitation. Parolie and Bergfeld73 managed 25 athletes with isolated PCL tears conservatively. In this study, 80% of athletes reported satisfaction and 68% returned to previous level of play.73 Neither of the aforementioned studies specify the grades of the injuries. Finally, Patel and colleagues66 managed 6 NFL athletes with Grade I and II injuries nonoperatively, and all were able to return to sport.
Treatment of isolated Grade III PCL injuries is more controversial, and no consensus exists in the literature. In an epidemiological study, Dick and colleagues68 found that only 39% of NCAA football athletes underwent surgery for their torn PCLs, compared to 79% of ACL injuries. However, their study makes no mention to the severity of these injuries. Numerous options exist for PCL reconstruction, with no consensus on the preferred method.
Conclusion
Knee injuries are the most common injury in football. Knowledge of the natural history of these injuries, as well as treatment options and expected outcomes, will help treating physicians educate their patients on the optimal treatment and manage return to play expectations.
Am J Orthop. 2016;45(6):368-373. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
Football is one of the most popular sports in the United States. Every year more than 1 million high school males and over 60,000 collegiate males participate in organized football. The number of males who play football is greater than the combined number of males and females who participate in track and field or basketball.1 Football has the highest injury rate amongst popular American sports.2 From 2001 to 2005, there was an estimated 1.1 million emergency room visits as a direct result of football.3 Injuries are more likely to occur during games,1,2,4,5 more likely to require surgery,4 and more likely to end the player’s season or career when compared to other sports.6 Of those injuries that end seasons or careers, the knee is the most common culprit.6 This is of particular concern because knee injuries are most common in football.1,2,5,7 This article reviews the epidemiology of 4 of the most common knee injuries in American football: tears of the anterior cruciate ligament (ACL), medial collateral ligament (MCL), medial patellofemoral ligament (MPFL), and posterior cruciate ligament (PCL).
Anterior Cruciate Ligament
The ACL is the primary structure preventing anterior tibial translation. It is composed of 2 anatomic bundles: the anteromedial and posterolateral bundles. The ACL originates from the posteromedial portion of the lateral femoral condyle and inserts between and slightly anterior to the tibial intercondylar eminence. The bundles are named for their relative insertions onto the tibia.
Injury to the ACL occurs both through noncontact and contact mechanisms. Typical noncontact mechanism is a forceful valgus collapse with the knee close to full extension with combined external or internal rotation of the tibia.8 This is often the result of a sudden deceleration prior to a change in direction.9 Contact injuries to the ACL are the result of a direct blow to the knee causing valgus collapse.9 The majority of ACL injuries amongst all sports are a result of a noncontact mechanism. However, Dragoo and colleagues10 found the majority of football ACL injuries (55%-60%) were from contact. As a result, football players are 4 times more likely to sustain ACL injuries than in other sports.11
ACL injuries are associated with significant time loss from sport. At the high school level, they are the most likely injury to end a season or career.6 Because these are higher-energy injuries, they are frequently associated with damage to additional structures. ACL injuries that occur in football are associated with increased rates of meniscus, chondral, and multi-ligamentous injuries.12,13
The incidence of ACL injuries increases with level of competition. In high school athletes it is 11.1 per 100,000 athlete exposures (AE).1,11 In collegiate football, the rate increases to 14.2 to 18 per 100,000 AE.2,14 Though no incidence data per AE was found in our review of the literature, there were 219 ACL injuries in the National Football League (NFL) from 2010 to 2013.15 In addition, 14.2% of retired NFL athletes in one survey reported a history of ACL injury.16
The most common high-risk positions are running backs and linebackers. Brophy and colleagues17 found that 9.7% of running backs and 8.9% of linebackers participating in the NFL Combine had a history of ACL injury. This may be because both the running back and linebacker are involved in frequent high-energy collisions and often quickly change direction. Other studies have also identified running backs and linebackers as high risk, in addition to tight ends, wide receivers, and interior linemen.13,15,18
Treatment of choice for elite level athletes with ACL injury is reconstruction.19 Of those who undergo ACL reconstruction, the rate of return to play ranges from 63% to 80%.20-22 The average time to return to play is 9 to 13 months. The odds of making a successful return hinges on how successful the athlete was prior to injury. Factors such as prior game experience, position on depth chart, being on scholarship, and draft position for NFL athletes have all been shown to have a positive predictive value on a patient’s chance of returning from ACL reconstruction.20,21
Players who return have variable levels of success afterwards. In a study of NFL quarterbacks who sustained ACL injuries, 12 out of 13 were able to return to game action with no appreciable dropoff in performance based on in-game production.23 Carey and colleagues24 looked specifically at NFL wide receivers and running backs and found an 80% return to play rate but with an approximate decrease in production of one-third upon return. Furthermore, in the Multicenter Orthopaedic Outcomes Network (MOON) cohort study, only 43% of participants felt they returned to their preoperative level.22
Medial Collateral Ligament
The MCL consists of superficial and deep components. The superficial MCL is the primary restraint to valgus laxity at the knee. The superficial MCL has 1 femoral and 2 tibial attachments. The deep MCL is a thickening of the medial joint capsule and runs deep and parallel to the superficial MCL. The amount of medial joint gapping with a valgus force on examination is used to grade severity of MCL injuries. Grade I is a <5-mm opening; Grade II, 5- to 10-mm opening; and grade III, >10-mm opening.
The MCL is the most common knee injury in high school, collegiate, and professional football.1,18,25-28 Injuries are typically due to contact when a valgus force is applied to the knee.29 The annual incidence of MCL injuries amongst high school football players is 24.2 per 100,000 AE.1 The positions that appear to be at greatest risk for MCL injuries are offensive and defensive linemen.18,30-32 In a review of 5047 collegiate athletes participating in the NFL Combine from 1987 to 2000, 23% of offensive linemen had a history of MCL injury, compared to the overall rate of 16%.33 In a similar study, Bradley and colleagues18 performed medical histories on athletes invited to the 2005 NFL Combine and also found offensive linemen had the highest rate of MCL injury at 33%, compared to the overall rate of 22%. They reasonably hypothesized that “chop blocks” and other players “rolling up” on the outside of linemen’s knees were responsible for these injuries. Albright and colleagues32 found that prophylactic knee braces decreased the incidence of MCL injuries in collegiate offensive lineman. However, additional studies have not been able to reproduce these results and the use of prophylactic knee braces remains controversial.26
Treatment of MCL injuries depends upon the grade of injury, associated injuries, and anatomical location of injury. Management of MCL injuries is for the most part nonsurgical. In 1974, Ellsasser and colleagues34 were the first to publish data on nonoperative management of Grade I and Grade II injuries with immediate motion and rehabilitation instead of cast immobilization. They found 93% of patients returned to football in 3 to 8 weeks.34 Derscheid and Garrick27 observed nonoperative treatment of Grade I and II sprains in collegiate football players, with a time loss of 10.6 days and 19.5 days for Grade I and II injuries, respectively. Holden and colleagues35 evaluated nonoperative management of Grade I and II MCL injuries in collegiate football players and found an average return to play of 21 days.
Grade III injury treatment is more controversial. Indelicato and colleagues36 demonstrated successful nonoperative management of Grade III MCL injuries in collegiate football players, with an average return to play of 64.4 days. Jones and colleagues37 had similar success with high school football players, with an average return to play of 34 days. However, isolated Grade III injuries are rare and therefore treatment is likely to be dictated by concomitant injuries. Fetto and Marshall38 found that 78% of Grade III injuries were associated with an additional ligamentous injury. Of those additional injuries, 95% were ACL tears.
Finally, one must consider the location of the MCL injury. Injuries of the distal MCL at its tibial insertion may result in poor healing, as the ligament is displaced away from its insertion. Therefore, some authors recommend surgical management for these injuries.39,40
The patellofemoral joint is a complex structure in which the patella is stabilized within the trochlear groove of the femur by both bony and soft tissue structures. The MPFL is one of the most important soft tissue stabilizers. The MPFL is the primary restraint to lateral patellar translation within the first 20° of knee flexion, contributing to 60% of the total restraining force.41 The MPFL originates on the medial femoral condyle and inserts on the superomedial aspect of the patella.
Patellar instability is the subluxation or dislocation of the patella out of the trochlear groove. Patellar subluxation and dislocation account for approximately 3% of all knee injuries.42 Patella dislocations are more common in younger populations43-45 with the majority (52%-63%) occurring during sports.43,44,46 Mitchell and colleagues47 reported an incidence of 4.1 patellar subluxations/dislocations per 100,000 AE in high school football players.
Dislocation is most commonly the result of knee flexion with the tibia in a valgus position.44,48 The majority of patellar dislocations occur via a noncontact mechanism.44,48 However, the majority of these injuries in football are from contact (63%).47
Acute patellar dislocations are associated with more soft tissue damage than those with recurrent dislocations.46 In acute patella dislocations, the MPFL is almost always ruptured.44 In contrast, Fithian and colleagues46 found only 38% of recurrent dislocators had MPFL injury. As a result, it is thought that those with recurrent instability dislocate without trauma and do not have the same characteristics as those who dislocate from high-energy trauma in sport. Risk factors for atraumatic dislocation are numerous and have been well described in the literature.49 However, traumatic dislocators usually do not have risk factors.50
Traumatic patella dislocations are higher energy and are associated with chondral injury in up to 95%of cases 51 and osteochondral injury 58% to 76% of the time.52,53 In contrast, people with “articular hypermobility” are less likely to sustain articular damage.54 This concept is important when considering risk for recurrent patella dislocation. The literature reports a 17% to 50% rate of recurrent instability after acute patella dislocation.46,55,56 However, most studies do not distinguish between traumatic and atraumatic injuries. Because the majority of patellar dislocations in football occur through contact mechanisms, the rate of recurrent instability in these athletes may in fact be less than what is reported in the literature.
First-time patella dislocations are generally treated nonoperatively. Mitchell and colleagues47 reported that 72.6% of high school athletes with patella subluxation treated conservatively were able to return to sports within 3 weeks, compared to only 34.1% of those with patellar dislocations. In the same study, patellar dislocations were season-ending 37% of the time.47 Atkin and colleagues50 followed 74 patients treated conservatively for first-time patellar dislocation and noted 58% at 6 months still had difficulty in squatting, jumping, or cutting.
Those who have failed conservative management and have an additional dislocation are 7 times more likely to redislocate.46 Therefore, they are usually treated operatively with MPFL reconstruction. Return to sport ranges from 3 to 6 months,57 with 53% to 77.3% reporting return to their previous functionality.57-59 Overall, 84.1% of patients are able to return to sport with 1.2% risk of recurrent dislocation.60
Posterior Cruciate Ligament
The PCL is the primary posterior stabilizer of the knee.61,62 It consists of the anterolateral and posteromedial bundles, named by their insertion on the posterior tibial plateau. The larger, stronger anterolateral bundle is the primary restraint to posterior tibial translation.63
Due to the relative infrequency of PCL injuries, there is a paucity of epidemiological data on sports-related PCL injuries. These injuries in the literature are commonly found due to traffic accidents (45%-57%) or from sports (33%-40%).64,65 According to Swensen and colleagues,1 PCL injuries account for 2.4% of all high school sport knee injuries. In a cohort of 62 knees with PCL injuries, Patel and colleagues66 found football was the most common cause of injury (19.3%).
The most common mechanism of injury in athletes is knee hyperflexion or a direct blow to the tibia in a flexed knee.67 In football, contact mechanisms are the most common. In a 16-year review of the National Collegiate Athletic Association (NCAA) injury surveillance system, the incidence of contact PCL injuries during games were 7.3 times higher than noncontact.68 The most common activity was being tackled, which accounted for 22.9% of all PCL injuries.68
Due to the high energy of these injuries, isolated PCL injuries are rare. In one trauma center’s experience, 96.5% of PCL injuries had an additional ligament injury.64 In that study, injuries to the PCL were associated with posterolateral corner, ACL, and MCL injuries 62%, 46%, and 31% of the time, respectively.64,69
Because isolated PCL injuries are rare, clinicians must rely on a thorough history and physical examination when evaluating athletes with knee injuries. Classification of PCL injuries is based on the amount of posterior tibial translation in relation to the femur with the knee bent to 90°. Grade I is 1 to 5 mm; Grade II, 6 to 10 mm; and Grade III, >10 mm. If there is suspicion of a PCL injury, there should be a very low threshold for magnetic resonance imaging, given the high association with additional injuries.
Natural history of Grade I and II isolated PCL injuries is generally favorable compared to Grade III and multi-ligamentous injuries.70 As a result, isolated Grade I and II PCL injuries are generally treated nonoperatively. Treatment consists of physical therapy with emphasis on quadriceps strengthening. Return to play can be considered as early as 2 to 4 weeks from injury.71 Recent long-term data have shown successful conservative management of Grade I and II injuries with quadriceps strength to 97% of contralateral leg and full range of motion.72 However, there was 11% moderate to severe osteoarthritis in these patients at a mean follow-up of 14.3 years.72 Fowler and Messieh67 managed athletes with 7 isolated complete PCL tears and 5 partial tears nonoperatively, all of whom were able to return to sport without limitation. Parolie and Bergfeld73 managed 25 athletes with isolated PCL tears conservatively. In this study, 80% of athletes reported satisfaction and 68% returned to previous level of play.73 Neither of the aforementioned studies specify the grades of the injuries. Finally, Patel and colleagues66 managed 6 NFL athletes with Grade I and II injuries nonoperatively, and all were able to return to sport.
Treatment of isolated Grade III PCL injuries is more controversial, and no consensus exists in the literature. In an epidemiological study, Dick and colleagues68 found that only 39% of NCAA football athletes underwent surgery for their torn PCLs, compared to 79% of ACL injuries. However, their study makes no mention to the severity of these injuries. Numerous options exist for PCL reconstruction, with no consensus on the preferred method.
Conclusion
Knee injuries are the most common injury in football. Knowledge of the natural history of these injuries, as well as treatment options and expected outcomes, will help treating physicians educate their patients on the optimal treatment and manage return to play expectations.
Am J Orthop. 2016;45(6):368-373. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Swenson DM, Collins CL, Best TM, Flanigan DC, Fields SK, Comstock RD. Epidemiology of knee injuries among U.S. high school athletes, 2005/2006-2010/2011. Med Sci Sports Exerc. 2013;45(3):462-469.
2. Hootman JM, Dick R, Agel J. Epidemiology of collegiate injuries for 15 sports: summary and recommendations for injury prevention initiatives. J Athl Train. 2007;42(2):311-319.
3. Mello MJ, Myers R, Christian JB, Palmisciano L, Linakis JG. Injuries in youth football: national emergency department visits during 2001-2005 for young and adolescent players. Acad Emerg Med. 2009;16(3):243-248.
4. Rechel JA, Collins CL, Comstock RD. Epidemiology of injuries requiring surgery among high school athletes in the United States, 2005 to 2010. J Trauma. 2011;71(4):982-989.
5. Ingram JG, Fields SK, Yard EE, Comstock RD. Epidemiology of knee injuries among boys and girls in US high school athletics. Am J Sports Med. 2008;36(6):1116-1122.
6. Tirabassi J, Brou L, Khodaee M, Lefort R, Fields SK, Comstock RD. Epidemiology of high school sports-related injuries resulting in medical disqualification: 2005-2006 through 2013-2014 academic years. Am J Sports Med. 2016 May 10. [Epub ahead of print]
7. Fernandez WG, Yard EE, Comstock RD. Epidemiology of lower extremity injuries among U.S. high school athletes. Acad Emerg Med. 2007;14(7):641-645.
8. Olsen OE, Myklebust G, Engebretsen L, Bahr R. Injury mechanisms for anterior cruciate ligament injuries in team handball: a systematic video analysis. Am J Sports Med. 2004;32(4):1002-1012.
9. Boden BP, Dean GS, Feagin JA Jr, Garrett WE Jr. Mechanisms of anterior cruciate ligament injury. Orthopedics. 2000;23(6):573-578.
10. Dragoo JL, Braun HJ, Harris AH. The effect of playing surface on the incidence of ACL injuries in National Collegiate Athletic Association American Football. Knee. 2013;20(3):191-195.
11. Joseph AM, Collins CL, Henke NM, Yard EE, Fields SK, Comstock RD. A multisport epidemiologic comparison of anterior cruciate ligament injuries in high school athletics. J Athl Train. 2013;48(6):810-817.
12. Granan LP, Inacio MC, Maletis GB, Funahashi TT, Engebretsen L. Sport-specific injury pattern recorded during anterior cruciate ligament reconstruction. Am J Sports Med. 2013;41(12):2814-2818.
13. Bradley JP, Klimkiewicz JJ, Rytel MJ, Powell JW. Anterior cruciate ligament injuries in the National Football League: epidemiology and current treatment trends among team physicians. Arthroscopy. 2002;18(5):502-509.
14. Dragoo JL, Braun HJ, Durham JL, Chen MR, Harris AH. Incidence and risk factors for injuries to the anterior cruciate ligament in National Collegiate Athletic Association football: data from the 2004-2005 through 2008-2009 National Collegiate Athletic Association Injury Surveillance System. Am J Sports Med. 2012;40(5):990-995.
15. Dodson CC, Secrist ES, Bhat SB, Woods DP, Deluca PF. Anterior cruciate ligamenti in National Football League athletes from 2010 to 2013: a descriptive epidemiology study. Orthop J Sports Med. 2016;4(3):2325967116631949.
16. Golightly YM, Marshall SW, Callahan LF, Guskiewicz K. Early-onset arthritis in retired National Football League players. J Phys Act Health. 2009;6(5):638-643.
17. Brophy RH, Lyman S, Chehab EL, Barnes RP, Rodeo SA, Warren RF. Predictive value of prior injury on career in professional American football is affected by player position. Am J Sports Med. 2009;37(4):768-775.
18. Bradley J, Honkamp NJ, Jost P, West R, Norwig J, Kaplan LD. Incidence and variance of knee injuries in elite college football players. Am J Orthop. 2008;37(6):310-314.
19. Erickson BJ, Harris JD, Fillingham YA, et al. Anterior cruciate ligament reconstruction practice patterns by NFL and NCAA football team physicians. Arthroscopy. 2014;30(6):731-738.
20. Daruawalla JH, Greis PE, Hancock R; ASP Collaborative Group, Xerogeanes JW. Rates and determinants of return to play after anterior cruciate ligament reconstruction in NCAA Division 1 college football athletes: a study of the ACC, SEC, and PAC-12 conferences. Orthop J Sports Med. 2014;2(8):2325967114543901.
21. Shah VM, Andrews JR, Fleisig GS, McMichael CS, Lemak LJ. Return to play after anterior cruciate ligament reconstruction in National Football League athletes. Am J Sports Med. 2010;38(11):2233-2239.
22. McCullough KA, Phelps KD, Spindler KP, et al. Return to high school- and college-level football after anterior cruciate ligament reconstruction: a Multicenter Orthopaedic Outcomes Network (MOON) cohort study. Am J Sports Med. 2012;40(11):2523-2529.
23. Erickson BJ, Harris JD, Heninger JR, et al. Performance and return-to-sport after ACL reconstruction in NFL quarterbacks. Orthopedics. 2014;37(8):e728-e734.
24. Carey JL, Huffman GR, Parekh SG, Sennett BJ. Outcomes of anterior cruciate ligament injuries to running backs and wide receivers in the National Football League. Am J Sports Med. 2006;34(12):1911-1917.
25. Hershman EB, Anderson R, Bergfeld JA, et al. An analysis of specific lower extremity injury rates on grass and FieldTurf playing surfaces in National Football League Games: 2000-2009 seasons. Am J Sports Med. 2012;40(10):2200-2205.
26. Salata MJ, Gibbs AE, Sekiya JK. The effectiveness of prophylactic knee bracing in American football: a systematic review. Sports Health. 2010;2(5):375-379.
27. Derscheid GL, Garrick JG. Medial collateral ligament injuries in football. Nonoperative management of grade I and grade II sprains. Am J Sports Med. 1981;9(6):365-368.
28. Meyers MC, Barnhill BS. Incidence, causes, and severity of high school football injuries on FieldTurf versus natural grass: a 5-year prospective study. Am J Sports Med. 2004;32(7):1626-1638.
29. Lundblad M, Waldén M, Magnusson H, Karlsson J, Ekstrand J. The UEFA injury study: 11-year data concerning 346 MCL injuries and time to return to play. Br J Sports Med. 2013;47(12):759-762.
30. Hewson GF Jr, Mendini RA, Wang JB. Prophylactic knee bracing in college football. Am J Sports Med. 1986;14(4):262-266.
31. Rovere GD, Haupt HA, Yates CS. Prophylactic knee bracing in college football. Am J Sports Med. 1987;15(2):111-116.
32. Albright JP, Powell JW, Smith W, et al. Medial collateral ligament knee sprains in college football. Brace wear preferences and injury risk. Am J Sports Med. 1994;22(1):2-11.
33. Brophy RH, Barnes R, Rodeo SA, Warren RF. Prevalence of musculoskeletal disorders at the NFL Combine--trends from 1987 to 2000. Med Sci Sports Exerc. 2007;39(1):22-27.
34. Ellsasser JC, Reynolds FC, Omohundro JR. The non-operative treatment of collateral ligament injuries of the knee in professional football players. An analysis of seventy-four injuries treated non-operatively and twenty-four injuries treated surgically. J Bone Joint Surg Am. 1974;56(6):1185-1190.
35. Holden DL, Eggert AW, Butler JE. The nonoperative treatment of grade I and II medial collateral ligament injuries to the knee. Am J Sports Med. 1983;11(5):340-344.
36. Indelicato PA, Hermansdorfer J, Huegel M. Nonoperative management of complete tears of the medial collateral ligament of the knee in intercollegiate football players. Clin Orthop Relat Res. 1990;(256):174-177.
37. Jones RE, Henley MB, Francis P. Nonoperative management of isolated grade III collateral ligament injury in high school football players. Clin Orthop Relat Res. 1986;(213):137-140.
38. Fetto JF, Marshall JL. Medial collateral ligament injuries of the knee: a rationale for treatment. Clin Orthop Relat Res. 1978;(132):206-218.
39. Corten K, Hoser C, Fink C, Bellemans J. Case reports: a Stener-like lesion of the medial collateral ligament of the knee. Clin Orthop Relat Res. 2010;468(1):289-293.
40. Marchant MH Jr, Tibor LM, Sekiya JK, Hardaker WT Jr, Garrett WE Jr, Taylor DC. Management of medial-sided knee injuries, part 1: medial collateral ligament. Am J Sports Med. 2011;39(5):1102-1113.
41. Desio SM, Burks RT, Bachus KN. Soft tissue restraints to lateral patellar translation in the human knee. Am J Sports Med. 1998;26(1):59-65.
42. Casteleyn PP, Handelberg F. Arthroscopy in the diagnosis of occult dislocation of the patella. Acta Orthop Belg. 1989;55(3):381-383.
43. Waterman BR, Belmont PJ Jr, Owens BD. Patellar dislocation in the United States: role of sex, age, race, and athletic participation. J Knee Surg. 2012;25(1):51-57.
44. Sillanpää P, Mattila VM, Iivonen T, Visuri T, Pihlajamäki H. Incidence and risk factors of acute traumatic primary patellar dislocation. Med Sci Sports Exerc. 2008;40(4):606-611.
45. Hsiao M, Owens BD, Burks R, Sturdivant RX, Cameron KL. Incidence of acute traumatic patellar dislocation among active-duty United States military service members. Am J Sports Med. 2010;38(10):1997-2004.
46. Fithian DC, Paxton EW, Stone ML, et al. Epidemiology and natural history of acute patellar dislocation. Am J Sports Med. 2004;32(5):1114-1121.
47. Mitchell J, Magnussen RA, Collins CL, et al. Epidemiology of patellofemoral instability injuries among high school athletes in the United States. Am J Sports Med. 2015;43(7):1676-1682.
48. Nikku R, Nietosvaara Y, Aalto K, Kallio PE. The mechanism of primary patellar dislocation: trauma history of 126 patients. Acta Orthop. 2009;80(4):432-434.
49. Tsai CH, Hsu CJ, Hung CH, Hsu HC. Primary traumatic patellar dislocation. J Orthop Surg Res. 2012;7:21.
50. Atkin DM, Fithian DC, Marangi KS, Stone ML, Dobson BE, Mendelsohn C. Characteristics of patients with primary acute lateral patellar dislocation and their recovery within the first 6 months of injury. Am J Sports Med. 2000;28(4):472-479.
51. Nomura E, Inoue M, Kurimura M. Chondral and osteochondral injuries associated with acute patellar dislocation. Arthroscopy. 2003;19(7):717-721.
52. Kirsch MD, Fitzgerald SW, Friedman H, Rogers LF. Transient lateral patellar dislocation: diagnosis with MR imaging. AJR Am J Roentgenol. 1993;161(1):109-113.
53. Virolainen H, Visuri T, Kuusela T. Acute dislocation of the patella: MR findings. Radiology. 1993;189(1):243-246.
54. Stanitski CL. Articular hypermobility and chondral injury in patients with acute patellar dislocation. Am J Sports Med. 1995;23(2):146-150.
55. Mäenpää H, Huhtala H, Lehto MU. Recurrence after patellar dislocation. Redislocation in 37/75 patients followed for 6-24 years. Acta Orthop Scand. 1997;68(5):424-426.
56. Buchner M, Baudendistel B, Sabo D, Schmitt H. Acute traumatic primary patellar dislocation: long-term results comparing conservative and surgical treatment. Clin J Sport Med. 2005;15(2):62-66.
57. Fisher B, Nyland J, Brand E, Curtin B. Medial patellofemoral ligament reconstruction for recurrent patellar dislocation: a systematic review including rehabilitation and return-to-sports efficacy. Arthroscopy. 2010;26(10):1384-1394.
58. Lippacher S, Dreyhaupt J, Williams SR, Reichel H, Nelitz M. Reconstruction of the medial patellofemoral ligament: clinical outcomes and return to sports. Am J Sports Med. 2014;42(7):1661-1668.
59. Panni AS, Alam M, Cerciello S, Vasso M, Maffulli N. Medial patellofemoral ligament reconstruction with a divergent patellar transverse 2-tunnel technique. Am J Sports Med. 2011;39(12):2647-1655.
60. Schneider DK, Grawe B, Magnussen RA, et al. Outcomes after isolated medial patellofemoral ligament reconstruction for the treatment of recurrent lateral patellar dislocations: a systematic review and meta-analysis. Am J Sports Med. 2016 Feb 12. [Epub ahead of print]
61. Amis AA, Bull AM, Gupte CM, Hijazi I, Race A, Robinson JR. Biomechanics of the PCL and related structures: posterolateral, posteromedial and meniscofemoral ligaments. Knee Surg Sports Traumatol Arthrosc. 2003;11(5):271-281.
62. Fu FH, Harner CD, Johnson DL, Miller MD, Woo SL. Biomechanics of knee ligaments: basic concepts and clinical application. Instr Course Lect. 1994;43:137-148.
63. Markolf KL, Feeley BT, Tejwani SG, Martin DE, McAllister DR. Changes in knee laxity and ligament force after sectioning the posteromedial bundle of the posterior cruciate ligament. Arthroscopy. 2006; 22(10):1100-1106.
64. Ganelli GC, Edson CJ. Posterior cruciate ligament injuries in trauma patients: Part II. Arthroscopy. 1995;11(5):526-529.
65. Schulz MS, Russe K, Weiler A, Eichhorn HJ, Strobel MJ. Epidemiology of posterior cruciate ligament injuries. Arch Orthop Trauma Surg. 2003;123(4):186-191.
66. Patel DV, Allen AA, Warren RF, Wickiewicz TL, Simonian PT. The nonoperative treatment of acute, isolated (partial or complete) posterior cruciate ligament-deficient knees: an intermediate-term follow-up study. HSS J. 2007;3(2):137-146.
67. Fowler PJ, Messieh SS. Isolated posterior cruciate ligament injuries in athletes. Am J Sports Med. 1987;15(6):553-557.
68. Dick R, Ferrara MS, Agel J, et al. Descriptive epidemiology of collegiate men’s football injuries: National Collegiate Athletic Association Injury Surveillance System, 1988-1989 through 2003-2004. J Athl Train. 2007;42(2):221-233.
69. LaPrade CM, Civitarese DM, Rasmussen MT, LaPrade RF. Emerging updates on the posterior cruciate ligament: a review of the current literature. Am J Sports Med. 2015;43(12):3077-3092.
70. Torg JS, Barton TM, Pavlov H, Stine R. Natural history of the posterior cruciate ligament-deficient knee. Clin Orthop Relat Res. 1989(246):208-216.
71. Miller MD. Orthopaedic Knowledge Update: Sports Medicine 5. Rosemont, IL; American Academy of Orthopaedic Surgeons; 2016.
72. Shelbourne KD, Clark M, Gray T. Minimum 10-year follow-up of patients after an acute, isolated posterior cruciate ligament injury treated nonoperatively. Am J Sports Med. 2013;41(7):1526-1533.
73. Parolie JM, Bergfeld JA. Long-term results of nonoperative treatment of isolated posterior cruciate ligament injuries in the athlete. Am J Sports Med. 1986;14(1):35-38.
1. Swenson DM, Collins CL, Best TM, Flanigan DC, Fields SK, Comstock RD. Epidemiology of knee injuries among U.S. high school athletes, 2005/2006-2010/2011. Med Sci Sports Exerc. 2013;45(3):462-469.
2. Hootman JM, Dick R, Agel J. Epidemiology of collegiate injuries for 15 sports: summary and recommendations for injury prevention initiatives. J Athl Train. 2007;42(2):311-319.
3. Mello MJ, Myers R, Christian JB, Palmisciano L, Linakis JG. Injuries in youth football: national emergency department visits during 2001-2005 for young and adolescent players. Acad Emerg Med. 2009;16(3):243-248.
4. Rechel JA, Collins CL, Comstock RD. Epidemiology of injuries requiring surgery among high school athletes in the United States, 2005 to 2010. J Trauma. 2011;71(4):982-989.
5. Ingram JG, Fields SK, Yard EE, Comstock RD. Epidemiology of knee injuries among boys and girls in US high school athletics. Am J Sports Med. 2008;36(6):1116-1122.
6. Tirabassi J, Brou L, Khodaee M, Lefort R, Fields SK, Comstock RD. Epidemiology of high school sports-related injuries resulting in medical disqualification: 2005-2006 through 2013-2014 academic years. Am J Sports Med. 2016 May 10. [Epub ahead of print]
7. Fernandez WG, Yard EE, Comstock RD. Epidemiology of lower extremity injuries among U.S. high school athletes. Acad Emerg Med. 2007;14(7):641-645.
8. Olsen OE, Myklebust G, Engebretsen L, Bahr R. Injury mechanisms for anterior cruciate ligament injuries in team handball: a systematic video analysis. Am J Sports Med. 2004;32(4):1002-1012.
9. Boden BP, Dean GS, Feagin JA Jr, Garrett WE Jr. Mechanisms of anterior cruciate ligament injury. Orthopedics. 2000;23(6):573-578.
10. Dragoo JL, Braun HJ, Harris AH. The effect of playing surface on the incidence of ACL injuries in National Collegiate Athletic Association American Football. Knee. 2013;20(3):191-195.
11. Joseph AM, Collins CL, Henke NM, Yard EE, Fields SK, Comstock RD. A multisport epidemiologic comparison of anterior cruciate ligament injuries in high school athletics. J Athl Train. 2013;48(6):810-817.
12. Granan LP, Inacio MC, Maletis GB, Funahashi TT, Engebretsen L. Sport-specific injury pattern recorded during anterior cruciate ligament reconstruction. Am J Sports Med. 2013;41(12):2814-2818.
13. Bradley JP, Klimkiewicz JJ, Rytel MJ, Powell JW. Anterior cruciate ligament injuries in the National Football League: epidemiology and current treatment trends among team physicians. Arthroscopy. 2002;18(5):502-509.
14. Dragoo JL, Braun HJ, Durham JL, Chen MR, Harris AH. Incidence and risk factors for injuries to the anterior cruciate ligament in National Collegiate Athletic Association football: data from the 2004-2005 through 2008-2009 National Collegiate Athletic Association Injury Surveillance System. Am J Sports Med. 2012;40(5):990-995.
15. Dodson CC, Secrist ES, Bhat SB, Woods DP, Deluca PF. Anterior cruciate ligamenti in National Football League athletes from 2010 to 2013: a descriptive epidemiology study. Orthop J Sports Med. 2016;4(3):2325967116631949.
16. Golightly YM, Marshall SW, Callahan LF, Guskiewicz K. Early-onset arthritis in retired National Football League players. J Phys Act Health. 2009;6(5):638-643.
17. Brophy RH, Lyman S, Chehab EL, Barnes RP, Rodeo SA, Warren RF. Predictive value of prior injury on career in professional American football is affected by player position. Am J Sports Med. 2009;37(4):768-775.
18. Bradley J, Honkamp NJ, Jost P, West R, Norwig J, Kaplan LD. Incidence and variance of knee injuries in elite college football players. Am J Orthop. 2008;37(6):310-314.
19. Erickson BJ, Harris JD, Fillingham YA, et al. Anterior cruciate ligament reconstruction practice patterns by NFL and NCAA football team physicians. Arthroscopy. 2014;30(6):731-738.
20. Daruawalla JH, Greis PE, Hancock R; ASP Collaborative Group, Xerogeanes JW. Rates and determinants of return to play after anterior cruciate ligament reconstruction in NCAA Division 1 college football athletes: a study of the ACC, SEC, and PAC-12 conferences. Orthop J Sports Med. 2014;2(8):2325967114543901.
21. Shah VM, Andrews JR, Fleisig GS, McMichael CS, Lemak LJ. Return to play after anterior cruciate ligament reconstruction in National Football League athletes. Am J Sports Med. 2010;38(11):2233-2239.
22. McCullough KA, Phelps KD, Spindler KP, et al. Return to high school- and college-level football after anterior cruciate ligament reconstruction: a Multicenter Orthopaedic Outcomes Network (MOON) cohort study. Am J Sports Med. 2012;40(11):2523-2529.
23. Erickson BJ, Harris JD, Heninger JR, et al. Performance and return-to-sport after ACL reconstruction in NFL quarterbacks. Orthopedics. 2014;37(8):e728-e734.
24. Carey JL, Huffman GR, Parekh SG, Sennett BJ. Outcomes of anterior cruciate ligament injuries to running backs and wide receivers in the National Football League. Am J Sports Med. 2006;34(12):1911-1917.
25. Hershman EB, Anderson R, Bergfeld JA, et al. An analysis of specific lower extremity injury rates on grass and FieldTurf playing surfaces in National Football League Games: 2000-2009 seasons. Am J Sports Med. 2012;40(10):2200-2205.
26. Salata MJ, Gibbs AE, Sekiya JK. The effectiveness of prophylactic knee bracing in American football: a systematic review. Sports Health. 2010;2(5):375-379.
27. Derscheid GL, Garrick JG. Medial collateral ligament injuries in football. Nonoperative management of grade I and grade II sprains. Am J Sports Med. 1981;9(6):365-368.
28. Meyers MC, Barnhill BS. Incidence, causes, and severity of high school football injuries on FieldTurf versus natural grass: a 5-year prospective study. Am J Sports Med. 2004;32(7):1626-1638.
29. Lundblad M, Waldén M, Magnusson H, Karlsson J, Ekstrand J. The UEFA injury study: 11-year data concerning 346 MCL injuries and time to return to play. Br J Sports Med. 2013;47(12):759-762.
30. Hewson GF Jr, Mendini RA, Wang JB. Prophylactic knee bracing in college football. Am J Sports Med. 1986;14(4):262-266.
31. Rovere GD, Haupt HA, Yates CS. Prophylactic knee bracing in college football. Am J Sports Med. 1987;15(2):111-116.
32. Albright JP, Powell JW, Smith W, et al. Medial collateral ligament knee sprains in college football. Brace wear preferences and injury risk. Am J Sports Med. 1994;22(1):2-11.
33. Brophy RH, Barnes R, Rodeo SA, Warren RF. Prevalence of musculoskeletal disorders at the NFL Combine--trends from 1987 to 2000. Med Sci Sports Exerc. 2007;39(1):22-27.
34. Ellsasser JC, Reynolds FC, Omohundro JR. The non-operative treatment of collateral ligament injuries of the knee in professional football players. An analysis of seventy-four injuries treated non-operatively and twenty-four injuries treated surgically. J Bone Joint Surg Am. 1974;56(6):1185-1190.
35. Holden DL, Eggert AW, Butler JE. The nonoperative treatment of grade I and II medial collateral ligament injuries to the knee. Am J Sports Med. 1983;11(5):340-344.
36. Indelicato PA, Hermansdorfer J, Huegel M. Nonoperative management of complete tears of the medial collateral ligament of the knee in intercollegiate football players. Clin Orthop Relat Res. 1990;(256):174-177.
37. Jones RE, Henley MB, Francis P. Nonoperative management of isolated grade III collateral ligament injury in high school football players. Clin Orthop Relat Res. 1986;(213):137-140.
38. Fetto JF, Marshall JL. Medial collateral ligament injuries of the knee: a rationale for treatment. Clin Orthop Relat Res. 1978;(132):206-218.
39. Corten K, Hoser C, Fink C, Bellemans J. Case reports: a Stener-like lesion of the medial collateral ligament of the knee. Clin Orthop Relat Res. 2010;468(1):289-293.
40. Marchant MH Jr, Tibor LM, Sekiya JK, Hardaker WT Jr, Garrett WE Jr, Taylor DC. Management of medial-sided knee injuries, part 1: medial collateral ligament. Am J Sports Med. 2011;39(5):1102-1113.
41. Desio SM, Burks RT, Bachus KN. Soft tissue restraints to lateral patellar translation in the human knee. Am J Sports Med. 1998;26(1):59-65.
42. Casteleyn PP, Handelberg F. Arthroscopy in the diagnosis of occult dislocation of the patella. Acta Orthop Belg. 1989;55(3):381-383.
43. Waterman BR, Belmont PJ Jr, Owens BD. Patellar dislocation in the United States: role of sex, age, race, and athletic participation. J Knee Surg. 2012;25(1):51-57.
44. Sillanpää P, Mattila VM, Iivonen T, Visuri T, Pihlajamäki H. Incidence and risk factors of acute traumatic primary patellar dislocation. Med Sci Sports Exerc. 2008;40(4):606-611.
45. Hsiao M, Owens BD, Burks R, Sturdivant RX, Cameron KL. Incidence of acute traumatic patellar dislocation among active-duty United States military service members. Am J Sports Med. 2010;38(10):1997-2004.
46. Fithian DC, Paxton EW, Stone ML, et al. Epidemiology and natural history of acute patellar dislocation. Am J Sports Med. 2004;32(5):1114-1121.
47. Mitchell J, Magnussen RA, Collins CL, et al. Epidemiology of patellofemoral instability injuries among high school athletes in the United States. Am J Sports Med. 2015;43(7):1676-1682.
48. Nikku R, Nietosvaara Y, Aalto K, Kallio PE. The mechanism of primary patellar dislocation: trauma history of 126 patients. Acta Orthop. 2009;80(4):432-434.
49. Tsai CH, Hsu CJ, Hung CH, Hsu HC. Primary traumatic patellar dislocation. J Orthop Surg Res. 2012;7:21.
50. Atkin DM, Fithian DC, Marangi KS, Stone ML, Dobson BE, Mendelsohn C. Characteristics of patients with primary acute lateral patellar dislocation and their recovery within the first 6 months of injury. Am J Sports Med. 2000;28(4):472-479.
51. Nomura E, Inoue M, Kurimura M. Chondral and osteochondral injuries associated with acute patellar dislocation. Arthroscopy. 2003;19(7):717-721.
52. Kirsch MD, Fitzgerald SW, Friedman H, Rogers LF. Transient lateral patellar dislocation: diagnosis with MR imaging. AJR Am J Roentgenol. 1993;161(1):109-113.
53. Virolainen H, Visuri T, Kuusela T. Acute dislocation of the patella: MR findings. Radiology. 1993;189(1):243-246.
54. Stanitski CL. Articular hypermobility and chondral injury in patients with acute patellar dislocation. Am J Sports Med. 1995;23(2):146-150.
55. Mäenpää H, Huhtala H, Lehto MU. Recurrence after patellar dislocation. Redislocation in 37/75 patients followed for 6-24 years. Acta Orthop Scand. 1997;68(5):424-426.
56. Buchner M, Baudendistel B, Sabo D, Schmitt H. Acute traumatic primary patellar dislocation: long-term results comparing conservative and surgical treatment. Clin J Sport Med. 2005;15(2):62-66.
57. Fisher B, Nyland J, Brand E, Curtin B. Medial patellofemoral ligament reconstruction for recurrent patellar dislocation: a systematic review including rehabilitation and return-to-sports efficacy. Arthroscopy. 2010;26(10):1384-1394.
58. Lippacher S, Dreyhaupt J, Williams SR, Reichel H, Nelitz M. Reconstruction of the medial patellofemoral ligament: clinical outcomes and return to sports. Am J Sports Med. 2014;42(7):1661-1668.
59. Panni AS, Alam M, Cerciello S, Vasso M, Maffulli N. Medial patellofemoral ligament reconstruction with a divergent patellar transverse 2-tunnel technique. Am J Sports Med. 2011;39(12):2647-1655.
60. Schneider DK, Grawe B, Magnussen RA, et al. Outcomes after isolated medial patellofemoral ligament reconstruction for the treatment of recurrent lateral patellar dislocations: a systematic review and meta-analysis. Am J Sports Med. 2016 Feb 12. [Epub ahead of print]
61. Amis AA, Bull AM, Gupte CM, Hijazi I, Race A, Robinson JR. Biomechanics of the PCL and related structures: posterolateral, posteromedial and meniscofemoral ligaments. Knee Surg Sports Traumatol Arthrosc. 2003;11(5):271-281.
62. Fu FH, Harner CD, Johnson DL, Miller MD, Woo SL. Biomechanics of knee ligaments: basic concepts and clinical application. Instr Course Lect. 1994;43:137-148.
63. Markolf KL, Feeley BT, Tejwani SG, Martin DE, McAllister DR. Changes in knee laxity and ligament force after sectioning the posteromedial bundle of the posterior cruciate ligament. Arthroscopy. 2006; 22(10):1100-1106.
64. Ganelli GC, Edson CJ. Posterior cruciate ligament injuries in trauma patients: Part II. Arthroscopy. 1995;11(5):526-529.
65. Schulz MS, Russe K, Weiler A, Eichhorn HJ, Strobel MJ. Epidemiology of posterior cruciate ligament injuries. Arch Orthop Trauma Surg. 2003;123(4):186-191.
66. Patel DV, Allen AA, Warren RF, Wickiewicz TL, Simonian PT. The nonoperative treatment of acute, isolated (partial or complete) posterior cruciate ligament-deficient knees: an intermediate-term follow-up study. HSS J. 2007;3(2):137-146.
67. Fowler PJ, Messieh SS. Isolated posterior cruciate ligament injuries in athletes. Am J Sports Med. 1987;15(6):553-557.
68. Dick R, Ferrara MS, Agel J, et al. Descriptive epidemiology of collegiate men’s football injuries: National Collegiate Athletic Association Injury Surveillance System, 1988-1989 through 2003-2004. J Athl Train. 2007;42(2):221-233.
69. LaPrade CM, Civitarese DM, Rasmussen MT, LaPrade RF. Emerging updates on the posterior cruciate ligament: a review of the current literature. Am J Sports Med. 2015;43(12):3077-3092.
70. Torg JS, Barton TM, Pavlov H, Stine R. Natural history of the posterior cruciate ligament-deficient knee. Clin Orthop Relat Res. 1989(246):208-216.
71. Miller MD. Orthopaedic Knowledge Update: Sports Medicine 5. Rosemont, IL; American Academy of Orthopaedic Surgeons; 2016.
72. Shelbourne KD, Clark M, Gray T. Minimum 10-year follow-up of patients after an acute, isolated posterior cruciate ligament injury treated nonoperatively. Am J Sports Med. 2013;41(7):1526-1533.
73. Parolie JM, Bergfeld JA. Long-term results of nonoperative treatment of isolated posterior cruciate ligament injuries in the athlete. Am J Sports Med. 1986;14(1):35-38.
Foot and Ankle Injuries in American Football
Foot and ankle injuries are common in American football, with injury rates significantly increasing over the past decade.1-5 Epidemiologic studies of collegiate football players have shown an annual incidence of foot and ankle injuries ranging from 9% to 39%,3,6 with as many as 72% of all collegiate players presenting to the National Football League (NFL) Combine with a history of a foot or ankle injury and 13% undergoing surgical treatment.5 Player position influences the rate and type of foot and ankle injury. Offensive and “skill position” players, including linemen, running backs, and wide receivers, are particularly susceptible to foot and ankle injuries due to high levels of force and torque placed on the distal extremity during running, cutting, and tackling. Shoe wear changes, playing field conditions, increasing player size and speed, and improved reporting of injuries are also contributing to increasing injury rates.
The interaction between player cleats and the playing surface is a central issue of foot and ankle injuries in football. Improved traction relates to performance, but increased subsequent torque on the lower extremity is associated with injury. While lateral ankle sprains are the most common foot and ankle injury experienced by football players,7 numerous other injuries can occur, including turf toe, Jones fractures, Lisfranc injuries, syndesmotic disruption, deltoid complex avulsion, and Achilles ruptures. It is important for physicians to be able to recognize, diagnose, and appropriately treat these injuries in players in order to expedite recovery, restore function, and help prevent future injury and long-term sequelae. This review focuses on updated treatment principles, surgical advances, and rehabilitation protocols for common football foot and ankle injuries.
Turf Toe
The term “turf toe” was first used in 1976 to refer to hyperextension injuries and plantar capsule-ligament sprains of the hallux metatarsophalangeal (MTP) joint that can lead to progressive cock-up deformity.8 While these injuries can occur on any surface and disrupt soft tissue balance with functional implications, predisposing factors include increasing playing surface hardness and decreasing shoe stiffness. In a classic scenario, the foot is fixed in equinus as an axial load is placed on the back of the heel, resulting in forced dorsiflexion of the hallux MTP joint.9 As the proximal phalanx extends, the sesamoids are drawn distally and the more dorsal portion of the metatarsal head articular surface bears the majority of the load, causing partial or complete tearing of the plantar plate with or without hallux MTP dislocation. Osteochondral lesions of the MTP joint and subchondral edema of the metatarsal head can occur concurrently as the proximal phalanx impacts or shears across the metatarsal head articular surface.
Clinical examination should focus on hallux swelling, alignment, and flexor hallucis longus (FHL) function along with vertical instability of the hallux MTP joint using a Lachman test. Radiographs should be evaluated for proximal migration of the sesamoids or diastasis (Figures W1A-W1C).


Indications for surgical intervention include loss of push-off strength, gross MTP instability, proximal migration of the sesamoids, and progressive hallux malalignment or clawing after immobilization. Cases can involve one or a combination of the following: (1) large capsular avulsion with unstable MTP joint; (2) diastasis of bipartite sesamoid; (3) diastasis of sesamoid fracture; (4) retraction of sesamoid; (5) traumatic hallux valgus deformity; (6) vertical instability (positive Lachman test); (7) loose body in MTP joint; or (8) chondral injury in MTP joint. The goal of surgery is the restoration of anatomy in order to restore normal function of the hallux MTP joint.
We have found that using dual medial and plantar incisions places less traction on the plantar medial cutaneous nerve, improves lateral exposure, and provides better wound healing. The medial capsulotomy extends from the metatarsal neck to the mid-phalanx to provide complete visualization of the sesamoid complex (Figures 1A-1F).


It is important to recognize that not all turf toe injuries involve pure hyperextension on artificial playing surfaces. In recent years, we have found an increasing rate of medial variant turf toe injuries in which a forceful valgus stress on the hallux leads to rupture of the medial collateral ligament, medial or plantar-medial capsule, and/or abductor halluces. Medial variant turf toe can lead to progressive hallux valgus and a traumatic bunion with a significant loss of push-off strength and difficulty with cutting maneuvers. Surgical treatment requires a modified McBride bunionectomy with adductor tenotomy and direct repair of the medial soft tissue defect.
Postoperative management is just as important as proper surgical technique for these injuries and involves a delicate balance between protecting the repair and starting early range of motion (ROM). Patients are immobilized non-weight-bearing (NWB) for 5 to 7 days maximum followed immediately with the initiation of passive hallux plantarflexion to keep the sesamoids moving. Active hallux plantarflexion is started at 4 weeks after surgery with active dorsiflexion from 6 to 8 weeks. Patients are transitioned to an accommodative shoe with stiff hallux insert 8 weeks postoperative with continued therapy focusing on hallux ROM. Running is initiated at 12 weeks and return to play (RTP) is typically allowed 4 months after surgery.
Jones Fractures
Jones fractures are fractures of the 5th metatarsal at the metaphyseal-diaphyseal junction, where there is a watershed area of decreased vascularity between the intramedullary nutrient and metaphyseal arteries. Current thought is that the rising rate of Jones fractures among football players is partially caused by the use of flexible, narrow cleats that do not provide enough stiffness and lateral support for the 5th metatarsal during running and cutting. Additionally, lateral overload from a baseline cavovarus foot posture with or without metatarsus adductus and/or skewfoot is thought to contribute to Jones fractures.10 Preoperative radiographs should be evaluated for fracture location, orientation, amount of cortical thickening, and overall geometry of the foot and 5th metatarsal. In elite athletes, the threshold for surgical intervention is decreasing in order to expedite healing and recovery and decrease re-fracture risk. This rationale is based on delayed union rates of 25% to 66%, nonunion rates of 7% to 28%,11 and re-fracture rates of up to 33% associated with nonoperative treatment.12 Nonoperative management is usually not feasible in the competitive athlete, as it typically involves a period of protected weight-bearing in a tall controlled ankle motion (CAM) boot for 6 to 8 weeks with serial radiographs to evaluate healing.
Our preference for surgical intervention involves percutaneous screw fixation with a “high and inside” starting point on fluoroscopy (Figures 2A-2D).


In career athletes, we augment the fracture site using a mixture of bone marrow aspirate concentrate (BMA) (Magellan, Arteriocyte Medical Systems) and demineralized bone matrix (DBM) (Mini Ignite, Wright Medical Technology) injected percutaneously in and around the fracture site under fluoroscopic guidance. Using this technique in a cohort of 25 NFL players treated operatively for Jones fractures, we found that 100% of athletes were able to RTP in the NFL in an average of 9.5 weeks.14 Two patients (7.5%) suffered re-fractures requiring revision surgery with iliac crest bone graft and repeat screw placement with a RTP after 15 weeks. We did not find an association between RTP and re-fracture rate.
The appropriate rehabilitation protocol for Jones fractures after surgery remains controversial and dependent on individual needs and abilities.15,16 For athletes in-season, we recommend a brief period of NWB for 1 to 2 weeks followed by toe-touch weight-bearing in a tall CAM boot for 2 to 4 weeks. After 4 weeks, patients begin gentle exercises on a stationary bike and pool therapy to reduce impact on the fracture site. Low-intensity pulsed ultrasound bone stimulators (Exogen, Bioventus) are frequently used directly over fracture site throughout the postoperative protocol as an adjuvant therapy. If clinically nontender over the fracture site, patients are allowed to begin running in modified protective shoe wear 4 weeks after surgery with an average RTP of 6 to 8 weeks. RTP is determined clinically, as radiographic union may not be evident for 12 to 16 weeks. Useful custom orthoses include turf toe plates with a cushioned lateral column and lateral heel wedge if hindfoot varus is present preoperatively.10 The solid intramedullary screw is left in place permanently.
In our experience, we have found the average re-fracture and nonunion rate to be approximately 8% across all athletes. Our observation that union rates do not appear to be related to RTP times suggests that underlying biology such as Vitamin D deficiency may play a larger role in union rates than previously thought. We have found that most Jones re-fractures occur in the first year after the initial injury, but can occur up to 2.5 years afterwards as well.14 For the management of symptomatic re-fractures and nonunions, the previous screw must be first removed. This can be difficult if the screw is bent or broken, and may require a lateral corticotomy of the metatarsal.
After hardware removal, we advocate open bone grafting of the fracture site using bone from the iliac crest retrieved with a small, percutaneous trephine. Re-fixation should be achieved using a larger, solid screw and postoperative adjuvants may include bone stimulators, Vitamin D and calcium supplemention, and possible teriparatide use (Forteo, Eli Lilly), depending on individual patient profile. In a cohort of 21 elite athletes treated for Jones fracture revision surgery with screw exchange and bone grafting, we found that 100% of patients had computed tomography (CT) evidence of union, with an average RTP of 12.3 weeks.17
Lisfranc Injuries
Lisfranc injuries include any bony or ligamentous damage that involves the tarsometatarsal (TMT) joints. While axial loading of a fixed, plantarflexed foot has traditionally been thought of as the most common mechanism of Lisfranc injury, we have found that noncontact twisting injuries leading to Lisfranc disruption are actually more common among NFL players. This mechanism is similar to noncontact turf toe and results in a purely ligamentous injury. We have found this to be particularly true in the case of defensive ends engaged with offensive linemen in which no axial loading or contact of the foot occurs. Clinically, patients often have painful weight-bearing, inability to perform a single limb heel rise, plantar ecchymosis, and swelling and point tenderness across the bases of the 1st and 2nd metatarsals.
It is critical to obtain comparison weight-bearing radiographs of both feet during initial work-up to look for evidence of instability. Subtle radiographic findings of Lisfranc injury include a bony “fleck” sign, compression fracture of the cuboid, and diastasis between the base of the 1st and 2nd metatarsals and/or medial and middle cuneiforms (Figures 3A, 3B).

The goal of surgical intervention is to obtain and maintain anatomic reduction of all unstable joints in order to restore a normal foot posture. One of the difficulties with Lisfranc injuries is that there are no exact diastasis parameters and individuals should be treated based on symptoms, functional needs, and degree of instability. It has been shown that 5 mm of displacement can have good long-term clinical results in select cases without surgery.18 For surgery, we recommend open reduction to remove interposed soft tissue debris and directly assess the articular surfaces (Figures 4A-4D).

Proximal-medial column Lisfranc injury variants are increasingly common among football players.20 In these injuries, the force of injury extends through the intercuneiform joint and exits out the naviculocuneiform joint, thus causing instability at multiple joints and an unstable 1st ray. Patients often have minimal clinical findings and normal radiographs and stress radiographs. MRI of the foot often reveals edema at the naviculocuneiform joint. Often patients fail to improve with nonoperative immobilization with continued inability to push off from the hallux. Unrecognized or untreated instability will lead to rapid deterioration of the naviculocuneiform joint. Surgical intervention requires a homerun screw and intercuneiform screw. We do not recommend primary arthrodesis in athletes due to significant risk of malunion and nonunion unless severe articular damage is present.
Patients are typically kept NWB in a splint for 2 weeks after surgery followed by NWB in a tall CAM from 3 to 4 weeks postoperative. Progressive weight-bearing and ROM exercises are initiated from 4 to 8 weeks, followed by return to accommodative shoe wear from 10 to 12 weeks. Hardware removal is performed 4 to 6 months after surgery, typically in the off-season to allow for 6 to 8 weeks or protected recovery afterwards. Premature hardware removal can lead to loss of reduction, particularly at the intercuneiform joints. All hardware crossing the TMT joints should be removed, while the homerun screw can be left in place in addition to the intercuneiform screw. RTP in football typically occurs 6 to 7 months after surgery. Final functional outcome is related to the adequacy of initial reduction and severity of the initial injury.21
Syndesmotic Disruption
Syndesmotic injuries comprise 1% to 18% of ankle sprains in the general population, but occur at much higher rates in football due to the increased rotation forces placed on the ankle during cutting and tackling. RTP after syndesmotic injury often takes twice as long when compared to isolated lateral ankle ligamentous injury.22 Missed injuries are common and if not treated properly can lead to chronic ankle instability and posttraumatic ankle arthritis.23 Syndesmotic injury can occur in isolation or with concomitant adjacent bony, cartilaginous, or ligamentous injuries. Therefore, clinical examination and imaging work-up are critical to successful management.
Syndesmotic injuries often result from an external rotation force applied to a hyperdorsiflexed ankle while the foot is planted. This mechanism causes the fibula to externally rotate while translating posteriorly and laterally, resulting in rupture of the anterior inferior tibiofibular ligament (AITFL) first, followed by the deep deltoid ligament, interosseous ligament (IOL), and lastly posterior talofibular ligament.24 Most syndesmotic injuries involve rupture of only the AITFL and IOL.25 Multiple clinical stress tests have been designed to assess syndesmotic stability, including the squeeze test, external rotation stress test, crossed-leg test, and fibula-translation test.26-29 However, no physical examination maneuver has been shown to reliably predict the presence or degree of syndesmotic injury and therefore imaging studies are necessary.30
Initial imaging should include standing radiographs of the affected ankle. An increase in the medial clear space between the medial malleolus and talus can occur with a combined syndesmotic and deltoid disruption. In the case of subtle syndesmotic injuries, contralateral comparison weight-bearing radiographs are recommended. CT and MRI can provide additional information, but these static imaging tests cannot identify instability. Fluoroscopic stress evaluation is beneficial but has a high false-negative rate in low-grade injuries and may not detect partial rupture of the AITFL and IOL.31 It has been shown that malrotation of as much as 30° of external rotation can occur if relying on intraoperative fluoroscopy alone.32 It has been our practice to recommend surgical reduction and stabilization for any syndesmotic injury with documented diastasis or instability seen on imaging and/or arthroscopy.
Nonoperative treatment is indicated for truly stable grade I syndesmotic injuries. This involves rest and immobilization followed by a progressive rehabilitation program consisting of stretching, strengthening, and proprioceptive exercises.33 After 1 week of protected weight-bearing in a cast or tall CAM boot, progression to full weight-bearing should occur over the following week. Active-assisted ankle ROM exercises and light proprioceptive training should then be initiated followed by sport-specific exercises 2 to 3 weeks after injury.
Arthroscopy can be a valuable diagnostic tool in the setting of subtle syndesmotic injury with negative radiographs, positive MRI for edema, and a protracted recovery course with vague pain (Figures W5A-W5E).

Implants are placed above the true syndesmotic joint (at least 15 mm above the tibial plafond) angled 30° posterior to anterior to follow the normal relationship of the fibula to the distal tibia in the incisura. Typically 2 suture-buttons are used, with the devices placed in a divergent fashion. We highly recommend the use of a fibular buttress plate with button placement in individuals returning to contact activity. This construct increases surface area distribution while preventing stress risers and the risk of fibula fractures. In a cadaver model with deliberate syndesmotic malreduction, suture-button stabilization resulted in decreased postoperative displacement as opposed to conventional screw fixation.34 Therefore, dynamic syndesmotic fixation may help to decrease the negative sequelae of iatrogenic clamp malreduction. Postoperative rehabilitation involves NWB in a cast or tall CAM boot for 4 weeks followed by ankle ROM exercises and progressive weight-bearing and physical therapy. Patients are transitioned to a lace-up ankle brace and athletic shoe from 6 to 12 weeks postoperative with increasing activity. Running and jumping is permitted 4 months after surgery with RTP typically at 6 to 7 months. Athletes who have had surgical stabilization for documented instability without any diastasis may engage in a more rapid recovery and RTP as symptoms and function allow.
Deltoid Complex Avulsion
Missed or neglected deltoid ligament injuries can lead to progressive chondral injury and joint degeneration. These injuries are often subtle and difficult to diagnose. An inability to perform a single limb heel rise, persistent pain with activity, and lack of normal functional improvement despite appropriate care are indicators of subtle ligament instability. These injuries often require an examination under anesthesia with combined ankle arthroscopy. Valgus stress testing of the ankle while directly visualizing the deltoid ligament from the anterolateral portal can reveal medial laxity in addition to potential osteochondral lesions along the anterolateral talar dome.
In American football players, we have observed that infolding and retraction of an avulsed superficial deltoid ligament complex after an ankle fracture, Maisonneuve injury, or severe high ankle sprain can be a source of persistent increased medial clear space, malreduction, and postoperative pain and medial instability. We have found that there is often complete avulsion of the superficial deltoid complex off the proximal aspect of the medial malleolus during high-energy ankle fractures in football players that is amenable to direct repair to bone (Figures W6A-W6E).

During surgical repair, an incision is made along the anterior aspect of the medial malleolus and the superficial deltoid ligament complex can often be found flipped and interposed in the medial gutter. A rongeur is used to create a bleeding cancellous bone surface for soft-tissue healing and 1 to 2 suture anchors are used to repair and imbricate the deltoid ligament complex back to the medial malleolus. The goal of these sutures is to repair the tibionavicular and tibial spring ligaments back to the medial malleolus. We believe that superficial deltoid complex avulsion during high-energy ankle fractures is a distinct injury pattern that should be recognized and may benefit from primary open repair.
We currently open explore every deltoid ligament complex in athletes with unstable syndesmotic injuries, as we believe that deltoid avulsion injuries are underrecognized and do not heal in an anatomic fashion if left alone. Postoperative recovery follows the same immobilization, progressive weight-bearing, and physical therapy protocol as that for syndesmotic disruption.
Achilles Ruptures
Acute midsubstance Achilles tendon ruptures are an increasingly common injury in patients 30 to 50 years of age, with more than 50% of all injuries occurring during basketball.36,37 Among NFL players, we have found that Achilles ruptures tend to occur at a higher rate during training camp, when athletes are deconditioned and quickly returning to explosive push-off activities. Physical examination should include a Thompson test, palpation of a gap within the tendon, and evaluation of resting ankle dorsiflexion in the affected extremity in the prone position with the knees bent. Lateral radiographs should be analyzed for the presence of a bony avulsion fragment indicative of an insertional avulsion injury or midsubstance calcium deposition reflecting chronic Achilles tendinosis, as both of these conditions will change surgical management. MRI is not recommended with acute midsubstance ruptures but may be helpful in the case of chronic ruptures or more proximal tears of the musculotendinous junction.
The management of acute midsubstance Achilles tendon ruptures is controversial, with no general consensus in the literature regarding nonoperative treatment, surgical repair, and ideal repair technique.36,38-42 American Academy of Orthopaedic Surgeons clinical practice guidelines report moderate evidence that nonoperative treatment of Achilles tendon ruptures has lower wound healing complications but higher rates of re-rupture.38,39 Additionally, limited incision approaches have been found to have fewer overall complications compared with traditional open repair. In an effort to reduce the incidence of postoperative wound complications while improving functional recovery, modern repair techniques focus on a limited incision repair using percutaneous suture insertion and management (PARS Achilles Jig System, Arthrex).36 The limited incision technique utilizes a 2-cm transverse incision and non-disposable jig with divergent needle passes and locking suture fixation options to secure and fixate both tendon ends with minimal dissection of skin, subcutaneous tissue, and paratenon. Limited incision repair is ideally performed within 2 weeks of the injury to ensure that both tendon ends are easy to identify, mobilize, and repair. An open repair is generally recommended for midsubstance ruptures more than 4 weeks old and cases of insertional rupture and Achilles tendinopathy.
In a cohort of 9 NFL players treated for midsubstance Achilles ruptures using the PARS technique, we found no re-ruptures, no wound complications, and no sural nerve issues after surgery.43 A comparative review of 270 cases of operatively treated Achilles tendon ruptures (101 PARS, 169 traditional open repair) showed that the PARS group had significantly shorter operative times and a higher number of patients able to return to baseline physical activities by 5 months compared to open repair.36 Although not statistically significant, the overall PARS complication rate was 5% while the open complication rate was 11%. The PARS group had no cases of sural neuritis or deep infection requiring reoperation. We currently use a limited incision technique for all acute midsubstance Achilles ruptures in athletes regardless of sport, patient size, or position played.
During surgery, a 2-cm transverse incision is made over the gap in the Achilles tendon and dissection is carried down to the rupture site with minimal manipulation of the skin (Figures 5A-5F).

A key aspect of postoperative recovery is avoiding excessive ankle dorsiflexion while the tendon is healing during the first 4 weeks after surgery, as this can lead to an elongated tendon with loss of push-off strength. Patients are kept in a plantarflexion splint NWB for 2 weeks after surgery. If the incision is healed at 2 weeks, sutures are removed and patients are transitioned into a NWB tall CAM boot for 2 weeks with gentle ankle ROM exercises. If there is any concern regarding wound healing status, sutures are maintained for an additional 1 to 2 weeks.
From 4 to 8 weeks after surgery, progressive weight-bearing with continued ankle ROM exercises is initiated with peel-away heel lifts (~2 cm thick total, 3 layers). Each layer of the heel lift is gradually removed as pain allows every 2 to 3 days with the goal of being full weight-bearing with the foot flat at 6 weeks postoperative. Physical therapy focusing on ankle ROM and gentle Achilles stretching and strengthening is also started 6 weeks after surgery. From 8 to 12 weeks postoperative, patients are transitioned out of the tall CAM boot into normal, accommodative shoe wear with full weight-bearing. We avoid ankle dorsiflexion past neutral until 12 weeks after surgery, as overlengthening of the Achilles complex and the subsequent loss of push-off power can be devastating to running athletes. Activity levels are increased as tolerated, with no running or jumping from 12 to 16 weeks with full release to all activities after 16 weeks. RTP often takes 5 to 6 months after surgery, depending on the position played.
Am J Orthop. 2016;45(6):358-367. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Canale ST, Cantler ED Jr, Sisk TD, Freeman BL 3rd. A chronicle of injuries of an American intercollegiate football team. Am J Sports Med. 1981;9(6):384-389.2. Robey JM, Blyth CS, Mueller FO. Athletic injuries. Application of epidemiologic methods. JAMA. 1971;217(2):184-189.
3. Saal JA. Common American football injuries. Sports Med. 1991;12(2):132-147.
4. Thompson N, Halpern B, Curl WW, et al. High school football injuries: evaluation. Am J Sports Med. 1987;15(2):117-124.
5. Kaplan LD, Jost PW, Honkamp N, Norwig J, West R, Bradley JP. Incidence and variance of foot and ankle injuries in elite college football players. Am J Orthop. 2011;40(1):40-44.
6. DeLee JC, Farney WC. Incidence of injury in Texas high school football. Am J Sports Med. 1992;20(5):575-580.
7. Brophy RH, Barnes R, Rodeo SA, Warren RF. Prevalence of musculoskeletal disorders at the NFL Combine--trends from 1987 to 2000. Med Sci Sports Exerc. 2007;39(1):22-27.
8. Bowers KD Jr, Martin RB. Turf-toe: a shoe-surface related football injury. Med Sci Sports. 1976;8(2):81-83.
9. McCormick JJ, Anderson RB. Turf toe: anatomy, diagnosis, and treatment. Sports Health. 2010;2(6):487-494.
10. Raikin SM, Slenker N, Ratigan B. The association of a varus hindfoot and fracture of the fifth metatarsal metaphyseal-diaphyseal junction: the Jones fracture. Am J Sports Med. 2008;36(7):1367-1372.
11. Title CI, Katchis SD. Traumatic foot and ankle injuries in the athlete. Orthop Clin North Am. 2002;33(3):587-598.
12. Quill GE Jr. Fractures of the proximal fifth metatarsal. Orthop Clin North Am. 1995;26(2):353-361.
13. Nunley JA, Glisson RR. A new option for intramedullary fixation of Jones fractures: the Charlotte Carolina Jones Fracture System. Foot Ankle Int. 2008;29(12):1216-1221.
14. Lareau CR, Hsu AR, Anderson RB. Return to play in National Football League players after operative Jones fracture treatment. Foot Ankle Int. 2016;37(1):8-16.
15. Larson CM, Almekinders LC, Taft TN, Garrett WE. Intramedullary screw fixation of Jones fractures. Analysis of failure. Am J Sports Med. 2002;30(1):55-60.
16. Portland G, Kelikian A, Kodros S. Acute surgical management of Jones’ fractures. Foot Ankle Int. 2003;24(11):829-833.
17. Hunt KJ, Anderson RB. Treatment of Jones fracture nonunions and refractures in the elite athlete: outcomes of intramedullary screw fixation with bone grafting. Am J Sports Med. 2011;39(9):1948-1954.
18. Nunley JA, Vertullo CJ. Classification, investigation, and management of midfoot sprains: Lisfranc injuries in the athlete. Am J Sports Med. 2002;30(6):871-878.
19. Alberta FG, Aronow MS, Barrero M, Diaz-Doran V, Sullivan RJ, Adams DJ. Ligamentous Lisfranc joint injuries: a biomechanical comparison of dorsal plate and transarticular screw fixation. Foot Ankle Int. 2005;26(6):462-473.
20. Ardoin GT, Anderson RB. Subtle Lisfranc injury. Tech Foot Ankle Surg. 2010;9(3):100-106.
21. Kuo RS, Tejwani NC, Digiovanni CW, et al. Outcome after open reduction and internal fixation of Lisfranc joint injuries. J Bone Joint Surg Am. 2000;82-A(11):1609-1618.
22. Wright RW, Barile RJ, Surprenant DA, Matava MJ. Ankle syndesmosis sprains in national hockey league players. Am J Sports Med. 2004;32(8):1941-1945.
23. Williams GN, Jones MH, Amendola A. Syndesmotic ankle sprains in athletes. Am J Sports Med. 2007;35(7):1197-1207.
24. Beumer A, Valstar ER, Garling EH, et al. Effects of ligament sectioning on the kinematics of the distal tibiofibular syndesmosis: a radiostereometric study of 10 cadaveric specimens based on presumed trauma mechanisms with suggestions for treatment. Acta Orthop. 2006;77(3):531-540.
25. McCollum GA, van den Bekerom MP, Kerkhoffs GM, Calder JD, van Dijk CN. Syndesmosis and deltoid ligament injuries in the athlete. Knee Surg Sports Traumatol Arthrosc. 2013;21(6):1328-1337.
26. Boytim MJ, Fischer DA, Neumann L. Syndesmotic ankle sprains. Am J Sports Med. 1991;19(3):294-298.
27. Nussbaum ED, Hosea TM, Sieler SD, Incremona BR, Kessler DE. Prospective evaluation of syndesmotic ankle sprains without diastasis. Am J Sports Med. 2001;29(1):31-35.
28. Kiter E, Bozkurt M. The crossed-leg test for examination of ankle syndesmosis injuries. Foot Ankle Int. 2005;26(2):187-188.
29. Beumer A, van Hemert WL, Swierstra BA, Jasper LE, Belkoff SM. A biomechanical evaluation of clinical stress tests for syndesmotic ankle instability. Foot Ankle Int. 2003;24(4):358-363.
30. Amendola A, Williams G, Foster D. Evidence-based approach to treatment of acute traumatic syndesmosis (high ankle) sprains. Sports Med Arthrosc. 2006;14(4):232-236.
31. Beumer A, Valstar ER, Garling EH, et al. External rotation stress imaging in syndesmotic injuries of the ankle: comparison of lateral radiography and radiostereometry in a cadaveric model. Acta Orthop Scand. 2003;74(2):201-205.
32. Marmor M, Hansen E, Han HK, Buckley J, Matityahu A. Limitations of standard fluoroscopy in detecting rotational malreduction of the syndesmosis in an ankle fracture model. Foot Ankle Int. 2011;32(6):616-622.
33. Williams GN, Allen EJ. Rehabilitation of syndesmotic (high) ankle sprains. Sports Health. 2010;2(6):460-470.
34. Westermann RW, Rungprai C, Goetz JE, Femino J, Amendola A, Phisitkul P. The effect of suture-button fixation on simulated syndesmotic malreduction: a cadaveric study. J Bone Joint Surg Am. 2014;96(20):1732-1738.
35. Hsu AR, Lareau CR, Anderson RB. Repair of acute superficial deltoid complex avulsion during ankle fracture fixation in National Football League players. Foot Ankle Int. 2015;36(11):1272-1278.
36. Hsu AR, Jones CP, Cohen BE, Davis WH, Ellington JK, Anderson RB. Clinical outcomes and complications of percutaneous Achilles repair system versus open technique for acute achilles tendon ruptures. Foot Ankle Int. 2015;36(11):1279-1286.
37. Raikin SM, Garras DN, Krapchev PV. Achilles tendon injuries in a United States population. Foot Ankle Int. 2013;34(4):475-480.
38. Chiodo CP, Glazebrook M, Bluman EM, et al. American Academy of Orthopaedic Surgeons clinical practice guideline on treatment of achilles tendon rupture. J Bone Joint Surg Am. 2010;92(14):2466-2468.
39. Chiodo CP, Glazebrook M, Bluman EM, et al. Diagnosis and treatment of acute achilles tendon rupture. J Am Acad Orthop Surg. 2010;18(8):503-510.
40. Khan RJ, Fick D, Keogh A, Crawford J, Brammar T, Parker M. Treatment of acute achilles tendon ruptures. A meta-analysis of randomized, controlled trials. J Bone Joint Surg Am. 2005;87(10):2202-2210.
41. Renninger CH, Kuhn K, Fellars T, Youngblood S, Bellamy J. Operative and nonoperative management of achilles tendon ruptures in active duty military population. Foot Ankle Int. 2016;37(3):269-273.
42. Khan RJ, Carey Smith RL. Surgical interventions for treating acute achilles tendon ruptures. Cochrane Database Syst Rev. 2010;(9):CD003674.
43. McCullough KA, Shaw CM, Anderson RB. Mini-open repair of achilles rupture in the national football league. J Surg Orthop Adv. 2014;23(4):179-183.
Foot and ankle injuries are common in American football, with injury rates significantly increasing over the past decade.1-5 Epidemiologic studies of collegiate football players have shown an annual incidence of foot and ankle injuries ranging from 9% to 39%,3,6 with as many as 72% of all collegiate players presenting to the National Football League (NFL) Combine with a history of a foot or ankle injury and 13% undergoing surgical treatment.5 Player position influences the rate and type of foot and ankle injury. Offensive and “skill position” players, including linemen, running backs, and wide receivers, are particularly susceptible to foot and ankle injuries due to high levels of force and torque placed on the distal extremity during running, cutting, and tackling. Shoe wear changes, playing field conditions, increasing player size and speed, and improved reporting of injuries are also contributing to increasing injury rates.
The interaction between player cleats and the playing surface is a central issue of foot and ankle injuries in football. Improved traction relates to performance, but increased subsequent torque on the lower extremity is associated with injury. While lateral ankle sprains are the most common foot and ankle injury experienced by football players,7 numerous other injuries can occur, including turf toe, Jones fractures, Lisfranc injuries, syndesmotic disruption, deltoid complex avulsion, and Achilles ruptures. It is important for physicians to be able to recognize, diagnose, and appropriately treat these injuries in players in order to expedite recovery, restore function, and help prevent future injury and long-term sequelae. This review focuses on updated treatment principles, surgical advances, and rehabilitation protocols for common football foot and ankle injuries.
Turf Toe
The term “turf toe” was first used in 1976 to refer to hyperextension injuries and plantar capsule-ligament sprains of the hallux metatarsophalangeal (MTP) joint that can lead to progressive cock-up deformity.8 While these injuries can occur on any surface and disrupt soft tissue balance with functional implications, predisposing factors include increasing playing surface hardness and decreasing shoe stiffness. In a classic scenario, the foot is fixed in equinus as an axial load is placed on the back of the heel, resulting in forced dorsiflexion of the hallux MTP joint.9 As the proximal phalanx extends, the sesamoids are drawn distally and the more dorsal portion of the metatarsal head articular surface bears the majority of the load, causing partial or complete tearing of the plantar plate with or without hallux MTP dislocation. Osteochondral lesions of the MTP joint and subchondral edema of the metatarsal head can occur concurrently as the proximal phalanx impacts or shears across the metatarsal head articular surface.
Clinical examination should focus on hallux swelling, alignment, and flexor hallucis longus (FHL) function along with vertical instability of the hallux MTP joint using a Lachman test. Radiographs should be evaluated for proximal migration of the sesamoids or diastasis (Figures W1A-W1C).
Indications for surgical intervention include loss of push-off strength, gross MTP instability, proximal migration of the sesamoids, and progressive hallux malalignment or clawing after immobilization. Cases can involve one or a combination of the following: (1) large capsular avulsion with unstable MTP joint; (2) diastasis of bipartite sesamoid; (3) diastasis of sesamoid fracture; (4) retraction of sesamoid; (5) traumatic hallux valgus deformity; (6) vertical instability (positive Lachman test); (7) loose body in MTP joint; or (8) chondral injury in MTP joint. The goal of surgery is the restoration of anatomy in order to restore normal function of the hallux MTP joint.
We have found that using dual medial and plantar incisions places less traction on the plantar medial cutaneous nerve, improves lateral exposure, and provides better wound healing. The medial capsulotomy extends from the metatarsal neck to the mid-phalanx to provide complete visualization of the sesamoid complex (Figures 1A-1F).
It is important to recognize that not all turf toe injuries involve pure hyperextension on artificial playing surfaces. In recent years, we have found an increasing rate of medial variant turf toe injuries in which a forceful valgus stress on the hallux leads to rupture of the medial collateral ligament, medial or plantar-medial capsule, and/or abductor halluces. Medial variant turf toe can lead to progressive hallux valgus and a traumatic bunion with a significant loss of push-off strength and difficulty with cutting maneuvers. Surgical treatment requires a modified McBride bunionectomy with adductor tenotomy and direct repair of the medial soft tissue defect.
Postoperative management is just as important as proper surgical technique for these injuries and involves a delicate balance between protecting the repair and starting early range of motion (ROM). Patients are immobilized non-weight-bearing (NWB) for 5 to 7 days maximum followed immediately with the initiation of passive hallux plantarflexion to keep the sesamoids moving. Active hallux plantarflexion is started at 4 weeks after surgery with active dorsiflexion from 6 to 8 weeks. Patients are transitioned to an accommodative shoe with stiff hallux insert 8 weeks postoperative with continued therapy focusing on hallux ROM. Running is initiated at 12 weeks and return to play (RTP) is typically allowed 4 months after surgery.
Jones Fractures
Jones fractures are fractures of the 5th metatarsal at the metaphyseal-diaphyseal junction, where there is a watershed area of decreased vascularity between the intramedullary nutrient and metaphyseal arteries. Current thought is that the rising rate of Jones fractures among football players is partially caused by the use of flexible, narrow cleats that do not provide enough stiffness and lateral support for the 5th metatarsal during running and cutting. Additionally, lateral overload from a baseline cavovarus foot posture with or without metatarsus adductus and/or skewfoot is thought to contribute to Jones fractures.10 Preoperative radiographs should be evaluated for fracture location, orientation, amount of cortical thickening, and overall geometry of the foot and 5th metatarsal. In elite athletes, the threshold for surgical intervention is decreasing in order to expedite healing and recovery and decrease re-fracture risk. This rationale is based on delayed union rates of 25% to 66%, nonunion rates of 7% to 28%,11 and re-fracture rates of up to 33% associated with nonoperative treatment.12 Nonoperative management is usually not feasible in the competitive athlete, as it typically involves a period of protected weight-bearing in a tall controlled ankle motion (CAM) boot for 6 to 8 weeks with serial radiographs to evaluate healing.
Our preference for surgical intervention involves percutaneous screw fixation with a “high and inside” starting point on fluoroscopy (Figures 2A-2D).
In career athletes, we augment the fracture site using a mixture of bone marrow aspirate concentrate (BMA) (Magellan, Arteriocyte Medical Systems) and demineralized bone matrix (DBM) (Mini Ignite, Wright Medical Technology) injected percutaneously in and around the fracture site under fluoroscopic guidance. Using this technique in a cohort of 25 NFL players treated operatively for Jones fractures, we found that 100% of athletes were able to RTP in the NFL in an average of 9.5 weeks.14 Two patients (7.5%) suffered re-fractures requiring revision surgery with iliac crest bone graft and repeat screw placement with a RTP after 15 weeks. We did not find an association between RTP and re-fracture rate.
The appropriate rehabilitation protocol for Jones fractures after surgery remains controversial and dependent on individual needs and abilities.15,16 For athletes in-season, we recommend a brief period of NWB for 1 to 2 weeks followed by toe-touch weight-bearing in a tall CAM boot for 2 to 4 weeks. After 4 weeks, patients begin gentle exercises on a stationary bike and pool therapy to reduce impact on the fracture site. Low-intensity pulsed ultrasound bone stimulators (Exogen, Bioventus) are frequently used directly over fracture site throughout the postoperative protocol as an adjuvant therapy. If clinically nontender over the fracture site, patients are allowed to begin running in modified protective shoe wear 4 weeks after surgery with an average RTP of 6 to 8 weeks. RTP is determined clinically, as radiographic union may not be evident for 12 to 16 weeks. Useful custom orthoses include turf toe plates with a cushioned lateral column and lateral heel wedge if hindfoot varus is present preoperatively.10 The solid intramedullary screw is left in place permanently.
In our experience, we have found the average re-fracture and nonunion rate to be approximately 8% across all athletes. Our observation that union rates do not appear to be related to RTP times suggests that underlying biology such as Vitamin D deficiency may play a larger role in union rates than previously thought. We have found that most Jones re-fractures occur in the first year after the initial injury, but can occur up to 2.5 years afterwards as well.14 For the management of symptomatic re-fractures and nonunions, the previous screw must be first removed. This can be difficult if the screw is bent or broken, and may require a lateral corticotomy of the metatarsal.
After hardware removal, we advocate open bone grafting of the fracture site using bone from the iliac crest retrieved with a small, percutaneous trephine. Re-fixation should be achieved using a larger, solid screw and postoperative adjuvants may include bone stimulators, Vitamin D and calcium supplemention, and possible teriparatide use (Forteo, Eli Lilly), depending on individual patient profile. In a cohort of 21 elite athletes treated for Jones fracture revision surgery with screw exchange and bone grafting, we found that 100% of patients had computed tomography (CT) evidence of union, with an average RTP of 12.3 weeks.17
Lisfranc Injuries
Lisfranc injuries include any bony or ligamentous damage that involves the tarsometatarsal (TMT) joints. While axial loading of a fixed, plantarflexed foot has traditionally been thought of as the most common mechanism of Lisfranc injury, we have found that noncontact twisting injuries leading to Lisfranc disruption are actually more common among NFL players. This mechanism is similar to noncontact turf toe and results in a purely ligamentous injury. We have found this to be particularly true in the case of defensive ends engaged with offensive linemen in which no axial loading or contact of the foot occurs. Clinically, patients often have painful weight-bearing, inability to perform a single limb heel rise, plantar ecchymosis, and swelling and point tenderness across the bases of the 1st and 2nd metatarsals.
It is critical to obtain comparison weight-bearing radiographs of both feet during initial work-up to look for evidence of instability. Subtle radiographic findings of Lisfranc injury include a bony “fleck” sign, compression fracture of the cuboid, and diastasis between the base of the 1st and 2nd metatarsals and/or medial and middle cuneiforms (Figures 3A, 3B).
The goal of surgical intervention is to obtain and maintain anatomic reduction of all unstable joints in order to restore a normal foot posture. One of the difficulties with Lisfranc injuries is that there are no exact diastasis parameters and individuals should be treated based on symptoms, functional needs, and degree of instability. It has been shown that 5 mm of displacement can have good long-term clinical results in select cases without surgery.18 For surgery, we recommend open reduction to remove interposed soft tissue debris and directly assess the articular surfaces (Figures 4A-4D).
Proximal-medial column Lisfranc injury variants are increasingly common among football players.20 In these injuries, the force of injury extends through the intercuneiform joint and exits out the naviculocuneiform joint, thus causing instability at multiple joints and an unstable 1st ray. Patients often have minimal clinical findings and normal radiographs and stress radiographs. MRI of the foot often reveals edema at the naviculocuneiform joint. Often patients fail to improve with nonoperative immobilization with continued inability to push off from the hallux. Unrecognized or untreated instability will lead to rapid deterioration of the naviculocuneiform joint. Surgical intervention requires a homerun screw and intercuneiform screw. We do not recommend primary arthrodesis in athletes due to significant risk of malunion and nonunion unless severe articular damage is present.
Patients are typically kept NWB in a splint for 2 weeks after surgery followed by NWB in a tall CAM from 3 to 4 weeks postoperative. Progressive weight-bearing and ROM exercises are initiated from 4 to 8 weeks, followed by return to accommodative shoe wear from 10 to 12 weeks. Hardware removal is performed 4 to 6 months after surgery, typically in the off-season to allow for 6 to 8 weeks or protected recovery afterwards. Premature hardware removal can lead to loss of reduction, particularly at the intercuneiform joints. All hardware crossing the TMT joints should be removed, while the homerun screw can be left in place in addition to the intercuneiform screw. RTP in football typically occurs 6 to 7 months after surgery. Final functional outcome is related to the adequacy of initial reduction and severity of the initial injury.21
Syndesmotic Disruption
Syndesmotic injuries comprise 1% to 18% of ankle sprains in the general population, but occur at much higher rates in football due to the increased rotation forces placed on the ankle during cutting and tackling. RTP after syndesmotic injury often takes twice as long when compared to isolated lateral ankle ligamentous injury.22 Missed injuries are common and if not treated properly can lead to chronic ankle instability and posttraumatic ankle arthritis.23 Syndesmotic injury can occur in isolation or with concomitant adjacent bony, cartilaginous, or ligamentous injuries. Therefore, clinical examination and imaging work-up are critical to successful management.
Syndesmotic injuries often result from an external rotation force applied to a hyperdorsiflexed ankle while the foot is planted. This mechanism causes the fibula to externally rotate while translating posteriorly and laterally, resulting in rupture of the anterior inferior tibiofibular ligament (AITFL) first, followed by the deep deltoid ligament, interosseous ligament (IOL), and lastly posterior talofibular ligament.24 Most syndesmotic injuries involve rupture of only the AITFL and IOL.25 Multiple clinical stress tests have been designed to assess syndesmotic stability, including the squeeze test, external rotation stress test, crossed-leg test, and fibula-translation test.26-29 However, no physical examination maneuver has been shown to reliably predict the presence or degree of syndesmotic injury and therefore imaging studies are necessary.30
Initial imaging should include standing radiographs of the affected ankle. An increase in the medial clear space between the medial malleolus and talus can occur with a combined syndesmotic and deltoid disruption. In the case of subtle syndesmotic injuries, contralateral comparison weight-bearing radiographs are recommended. CT and MRI can provide additional information, but these static imaging tests cannot identify instability. Fluoroscopic stress evaluation is beneficial but has a high false-negative rate in low-grade injuries and may not detect partial rupture of the AITFL and IOL.31 It has been shown that malrotation of as much as 30° of external rotation can occur if relying on intraoperative fluoroscopy alone.32 It has been our practice to recommend surgical reduction and stabilization for any syndesmotic injury with documented diastasis or instability seen on imaging and/or arthroscopy.
Nonoperative treatment is indicated for truly stable grade I syndesmotic injuries. This involves rest and immobilization followed by a progressive rehabilitation program consisting of stretching, strengthening, and proprioceptive exercises.33 After 1 week of protected weight-bearing in a cast or tall CAM boot, progression to full weight-bearing should occur over the following week. Active-assisted ankle ROM exercises and light proprioceptive training should then be initiated followed by sport-specific exercises 2 to 3 weeks after injury.
Arthroscopy can be a valuable diagnostic tool in the setting of subtle syndesmotic injury with negative radiographs, positive MRI for edema, and a protracted recovery course with vague pain (Figures W5A-W5E).
Implants are placed above the true syndesmotic joint (at least 15 mm above the tibial plafond) angled 30° posterior to anterior to follow the normal relationship of the fibula to the distal tibia in the incisura. Typically 2 suture-buttons are used, with the devices placed in a divergent fashion. We highly recommend the use of a fibular buttress plate with button placement in individuals returning to contact activity. This construct increases surface area distribution while preventing stress risers and the risk of fibula fractures. In a cadaver model with deliberate syndesmotic malreduction, suture-button stabilization resulted in decreased postoperative displacement as opposed to conventional screw fixation.34 Therefore, dynamic syndesmotic fixation may help to decrease the negative sequelae of iatrogenic clamp malreduction. Postoperative rehabilitation involves NWB in a cast or tall CAM boot for 4 weeks followed by ankle ROM exercises and progressive weight-bearing and physical therapy. Patients are transitioned to a lace-up ankle brace and athletic shoe from 6 to 12 weeks postoperative with increasing activity. Running and jumping is permitted 4 months after surgery with RTP typically at 6 to 7 months. Athletes who have had surgical stabilization for documented instability without any diastasis may engage in a more rapid recovery and RTP as symptoms and function allow.
Deltoid Complex Avulsion
Missed or neglected deltoid ligament injuries can lead to progressive chondral injury and joint degeneration. These injuries are often subtle and difficult to diagnose. An inability to perform a single limb heel rise, persistent pain with activity, and lack of normal functional improvement despite appropriate care are indicators of subtle ligament instability. These injuries often require an examination under anesthesia with combined ankle arthroscopy. Valgus stress testing of the ankle while directly visualizing the deltoid ligament from the anterolateral portal can reveal medial laxity in addition to potential osteochondral lesions along the anterolateral talar dome.
In American football players, we have observed that infolding and retraction of an avulsed superficial deltoid ligament complex after an ankle fracture, Maisonneuve injury, or severe high ankle sprain can be a source of persistent increased medial clear space, malreduction, and postoperative pain and medial instability. We have found that there is often complete avulsion of the superficial deltoid complex off the proximal aspect of the medial malleolus during high-energy ankle fractures in football players that is amenable to direct repair to bone (Figures W6A-W6E).
During surgical repair, an incision is made along the anterior aspect of the medial malleolus and the superficial deltoid ligament complex can often be found flipped and interposed in the medial gutter. A rongeur is used to create a bleeding cancellous bone surface for soft-tissue healing and 1 to 2 suture anchors are used to repair and imbricate the deltoid ligament complex back to the medial malleolus. The goal of these sutures is to repair the tibionavicular and tibial spring ligaments back to the medial malleolus. We believe that superficial deltoid complex avulsion during high-energy ankle fractures is a distinct injury pattern that should be recognized and may benefit from primary open repair.
We currently open explore every deltoid ligament complex in athletes with unstable syndesmotic injuries, as we believe that deltoid avulsion injuries are underrecognized and do not heal in an anatomic fashion if left alone. Postoperative recovery follows the same immobilization, progressive weight-bearing, and physical therapy protocol as that for syndesmotic disruption.
Achilles Ruptures
Acute midsubstance Achilles tendon ruptures are an increasingly common injury in patients 30 to 50 years of age, with more than 50% of all injuries occurring during basketball.36,37 Among NFL players, we have found that Achilles ruptures tend to occur at a higher rate during training camp, when athletes are deconditioned and quickly returning to explosive push-off activities. Physical examination should include a Thompson test, palpation of a gap within the tendon, and evaluation of resting ankle dorsiflexion in the affected extremity in the prone position with the knees bent. Lateral radiographs should be analyzed for the presence of a bony avulsion fragment indicative of an insertional avulsion injury or midsubstance calcium deposition reflecting chronic Achilles tendinosis, as both of these conditions will change surgical management. MRI is not recommended with acute midsubstance ruptures but may be helpful in the case of chronic ruptures or more proximal tears of the musculotendinous junction.
The management of acute midsubstance Achilles tendon ruptures is controversial, with no general consensus in the literature regarding nonoperative treatment, surgical repair, and ideal repair technique.36,38-42 American Academy of Orthopaedic Surgeons clinical practice guidelines report moderate evidence that nonoperative treatment of Achilles tendon ruptures has lower wound healing complications but higher rates of re-rupture.38,39 Additionally, limited incision approaches have been found to have fewer overall complications compared with traditional open repair. In an effort to reduce the incidence of postoperative wound complications while improving functional recovery, modern repair techniques focus on a limited incision repair using percutaneous suture insertion and management (PARS Achilles Jig System, Arthrex).36 The limited incision technique utilizes a 2-cm transverse incision and non-disposable jig with divergent needle passes and locking suture fixation options to secure and fixate both tendon ends with minimal dissection of skin, subcutaneous tissue, and paratenon. Limited incision repair is ideally performed within 2 weeks of the injury to ensure that both tendon ends are easy to identify, mobilize, and repair. An open repair is generally recommended for midsubstance ruptures more than 4 weeks old and cases of insertional rupture and Achilles tendinopathy.
In a cohort of 9 NFL players treated for midsubstance Achilles ruptures using the PARS technique, we found no re-ruptures, no wound complications, and no sural nerve issues after surgery.43 A comparative review of 270 cases of operatively treated Achilles tendon ruptures (101 PARS, 169 traditional open repair) showed that the PARS group had significantly shorter operative times and a higher number of patients able to return to baseline physical activities by 5 months compared to open repair.36 Although not statistically significant, the overall PARS complication rate was 5% while the open complication rate was 11%. The PARS group had no cases of sural neuritis or deep infection requiring reoperation. We currently use a limited incision technique for all acute midsubstance Achilles ruptures in athletes regardless of sport, patient size, or position played.
During surgery, a 2-cm transverse incision is made over the gap in the Achilles tendon and dissection is carried down to the rupture site with minimal manipulation of the skin (Figures 5A-5F).
A key aspect of postoperative recovery is avoiding excessive ankle dorsiflexion while the tendon is healing during the first 4 weeks after surgery, as this can lead to an elongated tendon with loss of push-off strength. Patients are kept in a plantarflexion splint NWB for 2 weeks after surgery. If the incision is healed at 2 weeks, sutures are removed and patients are transitioned into a NWB tall CAM boot for 2 weeks with gentle ankle ROM exercises. If there is any concern regarding wound healing status, sutures are maintained for an additional 1 to 2 weeks.
From 4 to 8 weeks after surgery, progressive weight-bearing with continued ankle ROM exercises is initiated with peel-away heel lifts (~2 cm thick total, 3 layers). Each layer of the heel lift is gradually removed as pain allows every 2 to 3 days with the goal of being full weight-bearing with the foot flat at 6 weeks postoperative. Physical therapy focusing on ankle ROM and gentle Achilles stretching and strengthening is also started 6 weeks after surgery. From 8 to 12 weeks postoperative, patients are transitioned out of the tall CAM boot into normal, accommodative shoe wear with full weight-bearing. We avoid ankle dorsiflexion past neutral until 12 weeks after surgery, as overlengthening of the Achilles complex and the subsequent loss of push-off power can be devastating to running athletes. Activity levels are increased as tolerated, with no running or jumping from 12 to 16 weeks with full release to all activities after 16 weeks. RTP often takes 5 to 6 months after surgery, depending on the position played.
Am J Orthop. 2016;45(6):358-367. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
Foot and ankle injuries are common in American football, with injury rates significantly increasing over the past decade.1-5 Epidemiologic studies of collegiate football players have shown an annual incidence of foot and ankle injuries ranging from 9% to 39%,3,6 with as many as 72% of all collegiate players presenting to the National Football League (NFL) Combine with a history of a foot or ankle injury and 13% undergoing surgical treatment.5 Player position influences the rate and type of foot and ankle injury. Offensive and “skill position” players, including linemen, running backs, and wide receivers, are particularly susceptible to foot and ankle injuries due to high levels of force and torque placed on the distal extremity during running, cutting, and tackling. Shoe wear changes, playing field conditions, increasing player size and speed, and improved reporting of injuries are also contributing to increasing injury rates.
The interaction between player cleats and the playing surface is a central issue of foot and ankle injuries in football. Improved traction relates to performance, but increased subsequent torque on the lower extremity is associated with injury. While lateral ankle sprains are the most common foot and ankle injury experienced by football players,7 numerous other injuries can occur, including turf toe, Jones fractures, Lisfranc injuries, syndesmotic disruption, deltoid complex avulsion, and Achilles ruptures. It is important for physicians to be able to recognize, diagnose, and appropriately treat these injuries in players in order to expedite recovery, restore function, and help prevent future injury and long-term sequelae. This review focuses on updated treatment principles, surgical advances, and rehabilitation protocols for common football foot and ankle injuries.
Turf Toe
The term “turf toe” was first used in 1976 to refer to hyperextension injuries and plantar capsule-ligament sprains of the hallux metatarsophalangeal (MTP) joint that can lead to progressive cock-up deformity.8 While these injuries can occur on any surface and disrupt soft tissue balance with functional implications, predisposing factors include increasing playing surface hardness and decreasing shoe stiffness. In a classic scenario, the foot is fixed in equinus as an axial load is placed on the back of the heel, resulting in forced dorsiflexion of the hallux MTP joint.9 As the proximal phalanx extends, the sesamoids are drawn distally and the more dorsal portion of the metatarsal head articular surface bears the majority of the load, causing partial or complete tearing of the plantar plate with or without hallux MTP dislocation. Osteochondral lesions of the MTP joint and subchondral edema of the metatarsal head can occur concurrently as the proximal phalanx impacts or shears across the metatarsal head articular surface.
Clinical examination should focus on hallux swelling, alignment, and flexor hallucis longus (FHL) function along with vertical instability of the hallux MTP joint using a Lachman test. Radiographs should be evaluated for proximal migration of the sesamoids or diastasis (Figures W1A-W1C).
Indications for surgical intervention include loss of push-off strength, gross MTP instability, proximal migration of the sesamoids, and progressive hallux malalignment or clawing after immobilization. Cases can involve one or a combination of the following: (1) large capsular avulsion with unstable MTP joint; (2) diastasis of bipartite sesamoid; (3) diastasis of sesamoid fracture; (4) retraction of sesamoid; (5) traumatic hallux valgus deformity; (6) vertical instability (positive Lachman test); (7) loose body in MTP joint; or (8) chondral injury in MTP joint. The goal of surgery is the restoration of anatomy in order to restore normal function of the hallux MTP joint.
We have found that using dual medial and plantar incisions places less traction on the plantar medial cutaneous nerve, improves lateral exposure, and provides better wound healing. The medial capsulotomy extends from the metatarsal neck to the mid-phalanx to provide complete visualization of the sesamoid complex (Figures 1A-1F).
It is important to recognize that not all turf toe injuries involve pure hyperextension on artificial playing surfaces. In recent years, we have found an increasing rate of medial variant turf toe injuries in which a forceful valgus stress on the hallux leads to rupture of the medial collateral ligament, medial or plantar-medial capsule, and/or abductor halluces. Medial variant turf toe can lead to progressive hallux valgus and a traumatic bunion with a significant loss of push-off strength and difficulty with cutting maneuvers. Surgical treatment requires a modified McBride bunionectomy with adductor tenotomy and direct repair of the medial soft tissue defect.
Postoperative management is just as important as proper surgical technique for these injuries and involves a delicate balance between protecting the repair and starting early range of motion (ROM). Patients are immobilized non-weight-bearing (NWB) for 5 to 7 days maximum followed immediately with the initiation of passive hallux plantarflexion to keep the sesamoids moving. Active hallux plantarflexion is started at 4 weeks after surgery with active dorsiflexion from 6 to 8 weeks. Patients are transitioned to an accommodative shoe with stiff hallux insert 8 weeks postoperative with continued therapy focusing on hallux ROM. Running is initiated at 12 weeks and return to play (RTP) is typically allowed 4 months after surgery.
Jones Fractures
Jones fractures are fractures of the 5th metatarsal at the metaphyseal-diaphyseal junction, where there is a watershed area of decreased vascularity between the intramedullary nutrient and metaphyseal arteries. Current thought is that the rising rate of Jones fractures among football players is partially caused by the use of flexible, narrow cleats that do not provide enough stiffness and lateral support for the 5th metatarsal during running and cutting. Additionally, lateral overload from a baseline cavovarus foot posture with or without metatarsus adductus and/or skewfoot is thought to contribute to Jones fractures.10 Preoperative radiographs should be evaluated for fracture location, orientation, amount of cortical thickening, and overall geometry of the foot and 5th metatarsal. In elite athletes, the threshold for surgical intervention is decreasing in order to expedite healing and recovery and decrease re-fracture risk. This rationale is based on delayed union rates of 25% to 66%, nonunion rates of 7% to 28%,11 and re-fracture rates of up to 33% associated with nonoperative treatment.12 Nonoperative management is usually not feasible in the competitive athlete, as it typically involves a period of protected weight-bearing in a tall controlled ankle motion (CAM) boot for 6 to 8 weeks with serial radiographs to evaluate healing.
Our preference for surgical intervention involves percutaneous screw fixation with a “high and inside” starting point on fluoroscopy (Figures 2A-2D).
In career athletes, we augment the fracture site using a mixture of bone marrow aspirate concentrate (BMA) (Magellan, Arteriocyte Medical Systems) and demineralized bone matrix (DBM) (Mini Ignite, Wright Medical Technology) injected percutaneously in and around the fracture site under fluoroscopic guidance. Using this technique in a cohort of 25 NFL players treated operatively for Jones fractures, we found that 100% of athletes were able to RTP in the NFL in an average of 9.5 weeks.14 Two patients (7.5%) suffered re-fractures requiring revision surgery with iliac crest bone graft and repeat screw placement with a RTP after 15 weeks. We did not find an association between RTP and re-fracture rate.
The appropriate rehabilitation protocol for Jones fractures after surgery remains controversial and dependent on individual needs and abilities.15,16 For athletes in-season, we recommend a brief period of NWB for 1 to 2 weeks followed by toe-touch weight-bearing in a tall CAM boot for 2 to 4 weeks. After 4 weeks, patients begin gentle exercises on a stationary bike and pool therapy to reduce impact on the fracture site. Low-intensity pulsed ultrasound bone stimulators (Exogen, Bioventus) are frequently used directly over fracture site throughout the postoperative protocol as an adjuvant therapy. If clinically nontender over the fracture site, patients are allowed to begin running in modified protective shoe wear 4 weeks after surgery with an average RTP of 6 to 8 weeks. RTP is determined clinically, as radiographic union may not be evident for 12 to 16 weeks. Useful custom orthoses include turf toe plates with a cushioned lateral column and lateral heel wedge if hindfoot varus is present preoperatively.10 The solid intramedullary screw is left in place permanently.
In our experience, we have found the average re-fracture and nonunion rate to be approximately 8% across all athletes. Our observation that union rates do not appear to be related to RTP times suggests that underlying biology such as Vitamin D deficiency may play a larger role in union rates than previously thought. We have found that most Jones re-fractures occur in the first year after the initial injury, but can occur up to 2.5 years afterwards as well.14 For the management of symptomatic re-fractures and nonunions, the previous screw must be first removed. This can be difficult if the screw is bent or broken, and may require a lateral corticotomy of the metatarsal.
After hardware removal, we advocate open bone grafting of the fracture site using bone from the iliac crest retrieved with a small, percutaneous trephine. Re-fixation should be achieved using a larger, solid screw and postoperative adjuvants may include bone stimulators, Vitamin D and calcium supplemention, and possible teriparatide use (Forteo, Eli Lilly), depending on individual patient profile. In a cohort of 21 elite athletes treated for Jones fracture revision surgery with screw exchange and bone grafting, we found that 100% of patients had computed tomography (CT) evidence of union, with an average RTP of 12.3 weeks.17
Lisfranc Injuries
Lisfranc injuries include any bony or ligamentous damage that involves the tarsometatarsal (TMT) joints. While axial loading of a fixed, plantarflexed foot has traditionally been thought of as the most common mechanism of Lisfranc injury, we have found that noncontact twisting injuries leading to Lisfranc disruption are actually more common among NFL players. This mechanism is similar to noncontact turf toe and results in a purely ligamentous injury. We have found this to be particularly true in the case of defensive ends engaged with offensive linemen in which no axial loading or contact of the foot occurs. Clinically, patients often have painful weight-bearing, inability to perform a single limb heel rise, plantar ecchymosis, and swelling and point tenderness across the bases of the 1st and 2nd metatarsals.
It is critical to obtain comparison weight-bearing radiographs of both feet during initial work-up to look for evidence of instability. Subtle radiographic findings of Lisfranc injury include a bony “fleck” sign, compression fracture of the cuboid, and diastasis between the base of the 1st and 2nd metatarsals and/or medial and middle cuneiforms (Figures 3A, 3B).
The goal of surgical intervention is to obtain and maintain anatomic reduction of all unstable joints in order to restore a normal foot posture. One of the difficulties with Lisfranc injuries is that there are no exact diastasis parameters and individuals should be treated based on symptoms, functional needs, and degree of instability. It has been shown that 5 mm of displacement can have good long-term clinical results in select cases without surgery.18 For surgery, we recommend open reduction to remove interposed soft tissue debris and directly assess the articular surfaces (Figures 4A-4D).
Proximal-medial column Lisfranc injury variants are increasingly common among football players.20 In these injuries, the force of injury extends through the intercuneiform joint and exits out the naviculocuneiform joint, thus causing instability at multiple joints and an unstable 1st ray. Patients often have minimal clinical findings and normal radiographs and stress radiographs. MRI of the foot often reveals edema at the naviculocuneiform joint. Often patients fail to improve with nonoperative immobilization with continued inability to push off from the hallux. Unrecognized or untreated instability will lead to rapid deterioration of the naviculocuneiform joint. Surgical intervention requires a homerun screw and intercuneiform screw. We do not recommend primary arthrodesis in athletes due to significant risk of malunion and nonunion unless severe articular damage is present.
Patients are typically kept NWB in a splint for 2 weeks after surgery followed by NWB in a tall CAM from 3 to 4 weeks postoperative. Progressive weight-bearing and ROM exercises are initiated from 4 to 8 weeks, followed by return to accommodative shoe wear from 10 to 12 weeks. Hardware removal is performed 4 to 6 months after surgery, typically in the off-season to allow for 6 to 8 weeks or protected recovery afterwards. Premature hardware removal can lead to loss of reduction, particularly at the intercuneiform joints. All hardware crossing the TMT joints should be removed, while the homerun screw can be left in place in addition to the intercuneiform screw. RTP in football typically occurs 6 to 7 months after surgery. Final functional outcome is related to the adequacy of initial reduction and severity of the initial injury.21
Syndesmotic Disruption
Syndesmotic injuries comprise 1% to 18% of ankle sprains in the general population, but occur at much higher rates in football due to the increased rotation forces placed on the ankle during cutting and tackling. RTP after syndesmotic injury often takes twice as long when compared to isolated lateral ankle ligamentous injury.22 Missed injuries are common and if not treated properly can lead to chronic ankle instability and posttraumatic ankle arthritis.23 Syndesmotic injury can occur in isolation or with concomitant adjacent bony, cartilaginous, or ligamentous injuries. Therefore, clinical examination and imaging work-up are critical to successful management.
Syndesmotic injuries often result from an external rotation force applied to a hyperdorsiflexed ankle while the foot is planted. This mechanism causes the fibula to externally rotate while translating posteriorly and laterally, resulting in rupture of the anterior inferior tibiofibular ligament (AITFL) first, followed by the deep deltoid ligament, interosseous ligament (IOL), and lastly posterior talofibular ligament.24 Most syndesmotic injuries involve rupture of only the AITFL and IOL.25 Multiple clinical stress tests have been designed to assess syndesmotic stability, including the squeeze test, external rotation stress test, crossed-leg test, and fibula-translation test.26-29 However, no physical examination maneuver has been shown to reliably predict the presence or degree of syndesmotic injury and therefore imaging studies are necessary.30
Initial imaging should include standing radiographs of the affected ankle. An increase in the medial clear space between the medial malleolus and talus can occur with a combined syndesmotic and deltoid disruption. In the case of subtle syndesmotic injuries, contralateral comparison weight-bearing radiographs are recommended. CT and MRI can provide additional information, but these static imaging tests cannot identify instability. Fluoroscopic stress evaluation is beneficial but has a high false-negative rate in low-grade injuries and may not detect partial rupture of the AITFL and IOL.31 It has been shown that malrotation of as much as 30° of external rotation can occur if relying on intraoperative fluoroscopy alone.32 It has been our practice to recommend surgical reduction and stabilization for any syndesmotic injury with documented diastasis or instability seen on imaging and/or arthroscopy.
Nonoperative treatment is indicated for truly stable grade I syndesmotic injuries. This involves rest and immobilization followed by a progressive rehabilitation program consisting of stretching, strengthening, and proprioceptive exercises.33 After 1 week of protected weight-bearing in a cast or tall CAM boot, progression to full weight-bearing should occur over the following week. Active-assisted ankle ROM exercises and light proprioceptive training should then be initiated followed by sport-specific exercises 2 to 3 weeks after injury.
Arthroscopy can be a valuable diagnostic tool in the setting of subtle syndesmotic injury with negative radiographs, positive MRI for edema, and a protracted recovery course with vague pain (Figures W5A-W5E).
Implants are placed above the true syndesmotic joint (at least 15 mm above the tibial plafond) angled 30° posterior to anterior to follow the normal relationship of the fibula to the distal tibia in the incisura. Typically 2 suture-buttons are used, with the devices placed in a divergent fashion. We highly recommend the use of a fibular buttress plate with button placement in individuals returning to contact activity. This construct increases surface area distribution while preventing stress risers and the risk of fibula fractures. In a cadaver model with deliberate syndesmotic malreduction, suture-button stabilization resulted in decreased postoperative displacement as opposed to conventional screw fixation.34 Therefore, dynamic syndesmotic fixation may help to decrease the negative sequelae of iatrogenic clamp malreduction. Postoperative rehabilitation involves NWB in a cast or tall CAM boot for 4 weeks followed by ankle ROM exercises and progressive weight-bearing and physical therapy. Patients are transitioned to a lace-up ankle brace and athletic shoe from 6 to 12 weeks postoperative with increasing activity. Running and jumping is permitted 4 months after surgery with RTP typically at 6 to 7 months. Athletes who have had surgical stabilization for documented instability without any diastasis may engage in a more rapid recovery and RTP as symptoms and function allow.
Deltoid Complex Avulsion
Missed or neglected deltoid ligament injuries can lead to progressive chondral injury and joint degeneration. These injuries are often subtle and difficult to diagnose. An inability to perform a single limb heel rise, persistent pain with activity, and lack of normal functional improvement despite appropriate care are indicators of subtle ligament instability. These injuries often require an examination under anesthesia with combined ankle arthroscopy. Valgus stress testing of the ankle while directly visualizing the deltoid ligament from the anterolateral portal can reveal medial laxity in addition to potential osteochondral lesions along the anterolateral talar dome.
In American football players, we have observed that infolding and retraction of an avulsed superficial deltoid ligament complex after an ankle fracture, Maisonneuve injury, or severe high ankle sprain can be a source of persistent increased medial clear space, malreduction, and postoperative pain and medial instability. We have found that there is often complete avulsion of the superficial deltoid complex off the proximal aspect of the medial malleolus during high-energy ankle fractures in football players that is amenable to direct repair to bone (Figures W6A-W6E).
During surgical repair, an incision is made along the anterior aspect of the medial malleolus and the superficial deltoid ligament complex can often be found flipped and interposed in the medial gutter. A rongeur is used to create a bleeding cancellous bone surface for soft-tissue healing and 1 to 2 suture anchors are used to repair and imbricate the deltoid ligament complex back to the medial malleolus. The goal of these sutures is to repair the tibionavicular and tibial spring ligaments back to the medial malleolus. We believe that superficial deltoid complex avulsion during high-energy ankle fractures is a distinct injury pattern that should be recognized and may benefit from primary open repair.
We currently open explore every deltoid ligament complex in athletes with unstable syndesmotic injuries, as we believe that deltoid avulsion injuries are underrecognized and do not heal in an anatomic fashion if left alone. Postoperative recovery follows the same immobilization, progressive weight-bearing, and physical therapy protocol as that for syndesmotic disruption.
Achilles Ruptures
Acute midsubstance Achilles tendon ruptures are an increasingly common injury in patients 30 to 50 years of age, with more than 50% of all injuries occurring during basketball.36,37 Among NFL players, we have found that Achilles ruptures tend to occur at a higher rate during training camp, when athletes are deconditioned and quickly returning to explosive push-off activities. Physical examination should include a Thompson test, palpation of a gap within the tendon, and evaluation of resting ankle dorsiflexion in the affected extremity in the prone position with the knees bent. Lateral radiographs should be analyzed for the presence of a bony avulsion fragment indicative of an insertional avulsion injury or midsubstance calcium deposition reflecting chronic Achilles tendinosis, as both of these conditions will change surgical management. MRI is not recommended with acute midsubstance ruptures but may be helpful in the case of chronic ruptures or more proximal tears of the musculotendinous junction.
The management of acute midsubstance Achilles tendon ruptures is controversial, with no general consensus in the literature regarding nonoperative treatment, surgical repair, and ideal repair technique.36,38-42 American Academy of Orthopaedic Surgeons clinical practice guidelines report moderate evidence that nonoperative treatment of Achilles tendon ruptures has lower wound healing complications but higher rates of re-rupture.38,39 Additionally, limited incision approaches have been found to have fewer overall complications compared with traditional open repair. In an effort to reduce the incidence of postoperative wound complications while improving functional recovery, modern repair techniques focus on a limited incision repair using percutaneous suture insertion and management (PARS Achilles Jig System, Arthrex).36 The limited incision technique utilizes a 2-cm transverse incision and non-disposable jig with divergent needle passes and locking suture fixation options to secure and fixate both tendon ends with minimal dissection of skin, subcutaneous tissue, and paratenon. Limited incision repair is ideally performed within 2 weeks of the injury to ensure that both tendon ends are easy to identify, mobilize, and repair. An open repair is generally recommended for midsubstance ruptures more than 4 weeks old and cases of insertional rupture and Achilles tendinopathy.
In a cohort of 9 NFL players treated for midsubstance Achilles ruptures using the PARS technique, we found no re-ruptures, no wound complications, and no sural nerve issues after surgery.43 A comparative review of 270 cases of operatively treated Achilles tendon ruptures (101 PARS, 169 traditional open repair) showed that the PARS group had significantly shorter operative times and a higher number of patients able to return to baseline physical activities by 5 months compared to open repair.36 Although not statistically significant, the overall PARS complication rate was 5% while the open complication rate was 11%. The PARS group had no cases of sural neuritis or deep infection requiring reoperation. We currently use a limited incision technique for all acute midsubstance Achilles ruptures in athletes regardless of sport, patient size, or position played.
During surgery, a 2-cm transverse incision is made over the gap in the Achilles tendon and dissection is carried down to the rupture site with minimal manipulation of the skin (Figures 5A-5F).
A key aspect of postoperative recovery is avoiding excessive ankle dorsiflexion while the tendon is healing during the first 4 weeks after surgery, as this can lead to an elongated tendon with loss of push-off strength. Patients are kept in a plantarflexion splint NWB for 2 weeks after surgery. If the incision is healed at 2 weeks, sutures are removed and patients are transitioned into a NWB tall CAM boot for 2 weeks with gentle ankle ROM exercises. If there is any concern regarding wound healing status, sutures are maintained for an additional 1 to 2 weeks.
From 4 to 8 weeks after surgery, progressive weight-bearing with continued ankle ROM exercises is initiated with peel-away heel lifts (~2 cm thick total, 3 layers). Each layer of the heel lift is gradually removed as pain allows every 2 to 3 days with the goal of being full weight-bearing with the foot flat at 6 weeks postoperative. Physical therapy focusing on ankle ROM and gentle Achilles stretching and strengthening is also started 6 weeks after surgery. From 8 to 12 weeks postoperative, patients are transitioned out of the tall CAM boot into normal, accommodative shoe wear with full weight-bearing. We avoid ankle dorsiflexion past neutral until 12 weeks after surgery, as overlengthening of the Achilles complex and the subsequent loss of push-off power can be devastating to running athletes. Activity levels are increased as tolerated, with no running or jumping from 12 to 16 weeks with full release to all activities after 16 weeks. RTP often takes 5 to 6 months after surgery, depending on the position played.
Am J Orthop. 2016;45(6):358-367. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Canale ST, Cantler ED Jr, Sisk TD, Freeman BL 3rd. A chronicle of injuries of an American intercollegiate football team. Am J Sports Med. 1981;9(6):384-389.2. Robey JM, Blyth CS, Mueller FO. Athletic injuries. Application of epidemiologic methods. JAMA. 1971;217(2):184-189.
3. Saal JA. Common American football injuries. Sports Med. 1991;12(2):132-147.
4. Thompson N, Halpern B, Curl WW, et al. High school football injuries: evaluation. Am J Sports Med. 1987;15(2):117-124.
5. Kaplan LD, Jost PW, Honkamp N, Norwig J, West R, Bradley JP. Incidence and variance of foot and ankle injuries in elite college football players. Am J Orthop. 2011;40(1):40-44.
6. DeLee JC, Farney WC. Incidence of injury in Texas high school football. Am J Sports Med. 1992;20(5):575-580.
7. Brophy RH, Barnes R, Rodeo SA, Warren RF. Prevalence of musculoskeletal disorders at the NFL Combine--trends from 1987 to 2000. Med Sci Sports Exerc. 2007;39(1):22-27.
8. Bowers KD Jr, Martin RB. Turf-toe: a shoe-surface related football injury. Med Sci Sports. 1976;8(2):81-83.
9. McCormick JJ, Anderson RB. Turf toe: anatomy, diagnosis, and treatment. Sports Health. 2010;2(6):487-494.
10. Raikin SM, Slenker N, Ratigan B. The association of a varus hindfoot and fracture of the fifth metatarsal metaphyseal-diaphyseal junction: the Jones fracture. Am J Sports Med. 2008;36(7):1367-1372.
11. Title CI, Katchis SD. Traumatic foot and ankle injuries in the athlete. Orthop Clin North Am. 2002;33(3):587-598.
12. Quill GE Jr. Fractures of the proximal fifth metatarsal. Orthop Clin North Am. 1995;26(2):353-361.
13. Nunley JA, Glisson RR. A new option for intramedullary fixation of Jones fractures: the Charlotte Carolina Jones Fracture System. Foot Ankle Int. 2008;29(12):1216-1221.
14. Lareau CR, Hsu AR, Anderson RB. Return to play in National Football League players after operative Jones fracture treatment. Foot Ankle Int. 2016;37(1):8-16.
15. Larson CM, Almekinders LC, Taft TN, Garrett WE. Intramedullary screw fixation of Jones fractures. Analysis of failure. Am J Sports Med. 2002;30(1):55-60.
16. Portland G, Kelikian A, Kodros S. Acute surgical management of Jones’ fractures. Foot Ankle Int. 2003;24(11):829-833.
17. Hunt KJ, Anderson RB. Treatment of Jones fracture nonunions and refractures in the elite athlete: outcomes of intramedullary screw fixation with bone grafting. Am J Sports Med. 2011;39(9):1948-1954.
18. Nunley JA, Vertullo CJ. Classification, investigation, and management of midfoot sprains: Lisfranc injuries in the athlete. Am J Sports Med. 2002;30(6):871-878.
19. Alberta FG, Aronow MS, Barrero M, Diaz-Doran V, Sullivan RJ, Adams DJ. Ligamentous Lisfranc joint injuries: a biomechanical comparison of dorsal plate and transarticular screw fixation. Foot Ankle Int. 2005;26(6):462-473.
20. Ardoin GT, Anderson RB. Subtle Lisfranc injury. Tech Foot Ankle Surg. 2010;9(3):100-106.
21. Kuo RS, Tejwani NC, Digiovanni CW, et al. Outcome after open reduction and internal fixation of Lisfranc joint injuries. J Bone Joint Surg Am. 2000;82-A(11):1609-1618.
22. Wright RW, Barile RJ, Surprenant DA, Matava MJ. Ankle syndesmosis sprains in national hockey league players. Am J Sports Med. 2004;32(8):1941-1945.
23. Williams GN, Jones MH, Amendola A. Syndesmotic ankle sprains in athletes. Am J Sports Med. 2007;35(7):1197-1207.
24. Beumer A, Valstar ER, Garling EH, et al. Effects of ligament sectioning on the kinematics of the distal tibiofibular syndesmosis: a radiostereometric study of 10 cadaveric specimens based on presumed trauma mechanisms with suggestions for treatment. Acta Orthop. 2006;77(3):531-540.
25. McCollum GA, van den Bekerom MP, Kerkhoffs GM, Calder JD, van Dijk CN. Syndesmosis and deltoid ligament injuries in the athlete. Knee Surg Sports Traumatol Arthrosc. 2013;21(6):1328-1337.
26. Boytim MJ, Fischer DA, Neumann L. Syndesmotic ankle sprains. Am J Sports Med. 1991;19(3):294-298.
27. Nussbaum ED, Hosea TM, Sieler SD, Incremona BR, Kessler DE. Prospective evaluation of syndesmotic ankle sprains without diastasis. Am J Sports Med. 2001;29(1):31-35.
28. Kiter E, Bozkurt M. The crossed-leg test for examination of ankle syndesmosis injuries. Foot Ankle Int. 2005;26(2):187-188.
29. Beumer A, van Hemert WL, Swierstra BA, Jasper LE, Belkoff SM. A biomechanical evaluation of clinical stress tests for syndesmotic ankle instability. Foot Ankle Int. 2003;24(4):358-363.
30. Amendola A, Williams G, Foster D. Evidence-based approach to treatment of acute traumatic syndesmosis (high ankle) sprains. Sports Med Arthrosc. 2006;14(4):232-236.
31. Beumer A, Valstar ER, Garling EH, et al. External rotation stress imaging in syndesmotic injuries of the ankle: comparison of lateral radiography and radiostereometry in a cadaveric model. Acta Orthop Scand. 2003;74(2):201-205.
32. Marmor M, Hansen E, Han HK, Buckley J, Matityahu A. Limitations of standard fluoroscopy in detecting rotational malreduction of the syndesmosis in an ankle fracture model. Foot Ankle Int. 2011;32(6):616-622.
33. Williams GN, Allen EJ. Rehabilitation of syndesmotic (high) ankle sprains. Sports Health. 2010;2(6):460-470.
34. Westermann RW, Rungprai C, Goetz JE, Femino J, Amendola A, Phisitkul P. The effect of suture-button fixation on simulated syndesmotic malreduction: a cadaveric study. J Bone Joint Surg Am. 2014;96(20):1732-1738.
35. Hsu AR, Lareau CR, Anderson RB. Repair of acute superficial deltoid complex avulsion during ankle fracture fixation in National Football League players. Foot Ankle Int. 2015;36(11):1272-1278.
36. Hsu AR, Jones CP, Cohen BE, Davis WH, Ellington JK, Anderson RB. Clinical outcomes and complications of percutaneous Achilles repair system versus open technique for acute achilles tendon ruptures. Foot Ankle Int. 2015;36(11):1279-1286.
37. Raikin SM, Garras DN, Krapchev PV. Achilles tendon injuries in a United States population. Foot Ankle Int. 2013;34(4):475-480.
38. Chiodo CP, Glazebrook M, Bluman EM, et al. American Academy of Orthopaedic Surgeons clinical practice guideline on treatment of achilles tendon rupture. J Bone Joint Surg Am. 2010;92(14):2466-2468.
39. Chiodo CP, Glazebrook M, Bluman EM, et al. Diagnosis and treatment of acute achilles tendon rupture. J Am Acad Orthop Surg. 2010;18(8):503-510.
40. Khan RJ, Fick D, Keogh A, Crawford J, Brammar T, Parker M. Treatment of acute achilles tendon ruptures. A meta-analysis of randomized, controlled trials. J Bone Joint Surg Am. 2005;87(10):2202-2210.
41. Renninger CH, Kuhn K, Fellars T, Youngblood S, Bellamy J. Operative and nonoperative management of achilles tendon ruptures in active duty military population. Foot Ankle Int. 2016;37(3):269-273.
42. Khan RJ, Carey Smith RL. Surgical interventions for treating acute achilles tendon ruptures. Cochrane Database Syst Rev. 2010;(9):CD003674.
43. McCullough KA, Shaw CM, Anderson RB. Mini-open repair of achilles rupture in the national football league. J Surg Orthop Adv. 2014;23(4):179-183.
1. Canale ST, Cantler ED Jr, Sisk TD, Freeman BL 3rd. A chronicle of injuries of an American intercollegiate football team. Am J Sports Med. 1981;9(6):384-389.2. Robey JM, Blyth CS, Mueller FO. Athletic injuries. Application of epidemiologic methods. JAMA. 1971;217(2):184-189.
3. Saal JA. Common American football injuries. Sports Med. 1991;12(2):132-147.
4. Thompson N, Halpern B, Curl WW, et al. High school football injuries: evaluation. Am J Sports Med. 1987;15(2):117-124.
5. Kaplan LD, Jost PW, Honkamp N, Norwig J, West R, Bradley JP. Incidence and variance of foot and ankle injuries in elite college football players. Am J Orthop. 2011;40(1):40-44.
6. DeLee JC, Farney WC. Incidence of injury in Texas high school football. Am J Sports Med. 1992;20(5):575-580.
7. Brophy RH, Barnes R, Rodeo SA, Warren RF. Prevalence of musculoskeletal disorders at the NFL Combine--trends from 1987 to 2000. Med Sci Sports Exerc. 2007;39(1):22-27.
8. Bowers KD Jr, Martin RB. Turf-toe: a shoe-surface related football injury. Med Sci Sports. 1976;8(2):81-83.
9. McCormick JJ, Anderson RB. Turf toe: anatomy, diagnosis, and treatment. Sports Health. 2010;2(6):487-494.
10. Raikin SM, Slenker N, Ratigan B. The association of a varus hindfoot and fracture of the fifth metatarsal metaphyseal-diaphyseal junction: the Jones fracture. Am J Sports Med. 2008;36(7):1367-1372.
11. Title CI, Katchis SD. Traumatic foot and ankle injuries in the athlete. Orthop Clin North Am. 2002;33(3):587-598.
12. Quill GE Jr. Fractures of the proximal fifth metatarsal. Orthop Clin North Am. 1995;26(2):353-361.
13. Nunley JA, Glisson RR. A new option for intramedullary fixation of Jones fractures: the Charlotte Carolina Jones Fracture System. Foot Ankle Int. 2008;29(12):1216-1221.
14. Lareau CR, Hsu AR, Anderson RB. Return to play in National Football League players after operative Jones fracture treatment. Foot Ankle Int. 2016;37(1):8-16.
15. Larson CM, Almekinders LC, Taft TN, Garrett WE. Intramedullary screw fixation of Jones fractures. Analysis of failure. Am J Sports Med. 2002;30(1):55-60.
16. Portland G, Kelikian A, Kodros S. Acute surgical management of Jones’ fractures. Foot Ankle Int. 2003;24(11):829-833.
17. Hunt KJ, Anderson RB. Treatment of Jones fracture nonunions and refractures in the elite athlete: outcomes of intramedullary screw fixation with bone grafting. Am J Sports Med. 2011;39(9):1948-1954.
18. Nunley JA, Vertullo CJ. Classification, investigation, and management of midfoot sprains: Lisfranc injuries in the athlete. Am J Sports Med. 2002;30(6):871-878.
19. Alberta FG, Aronow MS, Barrero M, Diaz-Doran V, Sullivan RJ, Adams DJ. Ligamentous Lisfranc joint injuries: a biomechanical comparison of dorsal plate and transarticular screw fixation. Foot Ankle Int. 2005;26(6):462-473.
20. Ardoin GT, Anderson RB. Subtle Lisfranc injury. Tech Foot Ankle Surg. 2010;9(3):100-106.
21. Kuo RS, Tejwani NC, Digiovanni CW, et al. Outcome after open reduction and internal fixation of Lisfranc joint injuries. J Bone Joint Surg Am. 2000;82-A(11):1609-1618.
22. Wright RW, Barile RJ, Surprenant DA, Matava MJ. Ankle syndesmosis sprains in national hockey league players. Am J Sports Med. 2004;32(8):1941-1945.
23. Williams GN, Jones MH, Amendola A. Syndesmotic ankle sprains in athletes. Am J Sports Med. 2007;35(7):1197-1207.
24. Beumer A, Valstar ER, Garling EH, et al. Effects of ligament sectioning on the kinematics of the distal tibiofibular syndesmosis: a radiostereometric study of 10 cadaveric specimens based on presumed trauma mechanisms with suggestions for treatment. Acta Orthop. 2006;77(3):531-540.
25. McCollum GA, van den Bekerom MP, Kerkhoffs GM, Calder JD, van Dijk CN. Syndesmosis and deltoid ligament injuries in the athlete. Knee Surg Sports Traumatol Arthrosc. 2013;21(6):1328-1337.
26. Boytim MJ, Fischer DA, Neumann L. Syndesmotic ankle sprains. Am J Sports Med. 1991;19(3):294-298.
27. Nussbaum ED, Hosea TM, Sieler SD, Incremona BR, Kessler DE. Prospective evaluation of syndesmotic ankle sprains without diastasis. Am J Sports Med. 2001;29(1):31-35.
28. Kiter E, Bozkurt M. The crossed-leg test for examination of ankle syndesmosis injuries. Foot Ankle Int. 2005;26(2):187-188.
29. Beumer A, van Hemert WL, Swierstra BA, Jasper LE, Belkoff SM. A biomechanical evaluation of clinical stress tests for syndesmotic ankle instability. Foot Ankle Int. 2003;24(4):358-363.
30. Amendola A, Williams G, Foster D. Evidence-based approach to treatment of acute traumatic syndesmosis (high ankle) sprains. Sports Med Arthrosc. 2006;14(4):232-236.
31. Beumer A, Valstar ER, Garling EH, et al. External rotation stress imaging in syndesmotic injuries of the ankle: comparison of lateral radiography and radiostereometry in a cadaveric model. Acta Orthop Scand. 2003;74(2):201-205.
32. Marmor M, Hansen E, Han HK, Buckley J, Matityahu A. Limitations of standard fluoroscopy in detecting rotational malreduction of the syndesmosis in an ankle fracture model. Foot Ankle Int. 2011;32(6):616-622.
33. Williams GN, Allen EJ. Rehabilitation of syndesmotic (high) ankle sprains. Sports Health. 2010;2(6):460-470.
34. Westermann RW, Rungprai C, Goetz JE, Femino J, Amendola A, Phisitkul P. The effect of suture-button fixation on simulated syndesmotic malreduction: a cadaveric study. J Bone Joint Surg Am. 2014;96(20):1732-1738.
35. Hsu AR, Lareau CR, Anderson RB. Repair of acute superficial deltoid complex avulsion during ankle fracture fixation in National Football League players. Foot Ankle Int. 2015;36(11):1272-1278.
36. Hsu AR, Jones CP, Cohen BE, Davis WH, Ellington JK, Anderson RB. Clinical outcomes and complications of percutaneous Achilles repair system versus open technique for acute achilles tendon ruptures. Foot Ankle Int. 2015;36(11):1279-1286.
37. Raikin SM, Garras DN, Krapchev PV. Achilles tendon injuries in a United States population. Foot Ankle Int. 2013;34(4):475-480.
38. Chiodo CP, Glazebrook M, Bluman EM, et al. American Academy of Orthopaedic Surgeons clinical practice guideline on treatment of achilles tendon rupture. J Bone Joint Surg Am. 2010;92(14):2466-2468.
39. Chiodo CP, Glazebrook M, Bluman EM, et al. Diagnosis and treatment of acute achilles tendon rupture. J Am Acad Orthop Surg. 2010;18(8):503-510.
40. Khan RJ, Fick D, Keogh A, Crawford J, Brammar T, Parker M. Treatment of acute achilles tendon ruptures. A meta-analysis of randomized, controlled trials. J Bone Joint Surg Am. 2005;87(10):2202-2210.
41. Renninger CH, Kuhn K, Fellars T, Youngblood S, Bellamy J. Operative and nonoperative management of achilles tendon ruptures in active duty military population. Foot Ankle Int. 2016;37(3):269-273.
42. Khan RJ, Carey Smith RL. Surgical interventions for treating acute achilles tendon ruptures. Cochrane Database Syst Rev. 2010;(9):CD003674.
43. McCullough KA, Shaw CM, Anderson RB. Mini-open repair of achilles rupture in the national football league. J Surg Orthop Adv. 2014;23(4):179-183.
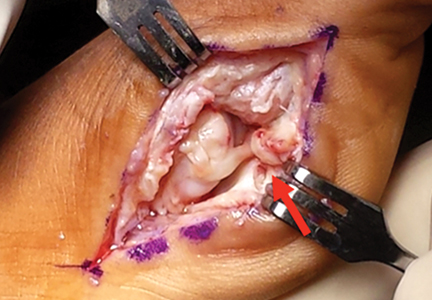
Concussions in American Football
Football is an important component of American culture, with approximately 3 million youth athletes, 1.1 million high school athletes, and 100,000 college athletes participating each year.1 Participation in football provides athletes with physical, social, psychological, and academic benefits. Despite these benefits, widespread focus has been placed on the safety of football due to the risk for sport-related concussion (SRC) and potentially long-term effects; however, little recognition has been given to the advancements in concussion management across time and occurrence of concussions during most life activities. Although it is reasonable for concerns to be presented, it is important to better understand SRC and the current factors leading to prolonged recoveries, increased risk for injury, and potentially long-term effects.
What Is a Concussion?
Concussions occur after sustaining direct or indirect injury to the head or other parts of the body, as long as the injury force is transmitted to the head. Athletes often experience physical, cognitive, emotional, and sleep-related symptoms post-concussion secondary to an “energy crisis” within the brain.2 The energy crisis occurs as the result of transient neurological dysfunction triggered by changes in the brain (eg, release of neurotransmitters, impaired axonal function).2,3 Concussion is undetectable with traditional imaging; however, advanced imaging techniques (eg, diffuse tensor imaging) have shown progress in assessing axonal injury.3 Symptom duration post-concussion is highly variable due to individual differences; a recent study showed recovery took 3 to 4 weeks for memory and symptoms.4,5
Previous Concussion Management
Identification techniques and return-to-play guidelines for concussion have significantly changed across time. In the past, concussion grading scales were utilized for diagnosis and return to play was possible within the same contest.6,7 It has since been recognized that initial concussion severity makes it difficult to predict recovery.3 For example, research revealed memory decline and increased symptoms 36 hours post-injury for athletes with a grade 1 concussion (ie, transient confusion, no loss of consciousness, concussion symptoms or mental status changes that resolve within 15 minutes of injury) compared to baseline.7 Another study found duration of mental status changes to be related to slower symptom resolution and memory impairment 36 hours to 7 days post-injury.6 Consequently, return to play within the same contest was likely too liberal. Guidelines today recommend immediate removal from play with suspected SRC. Nevertheless, the “play through pain” culture has led athletes to continue playing after SRC, contributing to prolonged recoveries and potentially long-term effects.
Current Concussion Management: Continued Concerns and Areas of Improvement
Despite increased awareness of concussions, recent estimates revealed high rates (ie, 27:1 ratio for general players) of underreporting in college football, particularly amongst offensive linemen.8 Researchers have studied recovery implications for remaining in play, with one study revealing a 2.2 times greater risk for prolonged recovery in college athletes with delayed vs immediate removal.9 Another similar study discovered an 8.8 times greater risk for prolonged recovery in adolescent and young adult athletes not removed vs removed from play.10 Further analysis found remaining in play to be the greatest risk factor for prolonged recovery compared to other previously studied risk factors (eg, age, sex, posttraumatic migraine).10 Additionally, significant differences in neurocognitive data were seen between the “removed” and “not removed” groups for verbal memory, visual memory, processing speed, and reaction time at 1 to 7 days and 8 to 30 days.10 The recovery implications of remaining in play and the additional risk for second impact syndrome (SIS), or repeat concussion when recovering from another injury, emphasizes the need for further education efforts amongst athletes to encourage immediate reporting of injury.11
Sideline Assessment
Sideline assessment has become a vital component of concussion management to rule out concussion and/or significant injury other than concussion. Assessment should include observation, cognitive/balance testing, neurologic examination, and possible exertion testing to ensure a comprehensive evaluation of all areas of potential dysfunction.12 Indications for emergency department evaluation include suspicion for cervical spine injury, intracranial hemorrhage, or skull fracture as well as prolonged loss of consciousness, high-risk mechanisms, posttraumatic seizure(s), and/or significant worsening of symptoms.12
Observation
On the sideline, it is important to identify any immediate signs of injury (ie, loss of consciousness, anterograde/retrograde amnesia, and disorientation/confusion). Since immediate signs are not always present, it is important to be aware of the most commonly reported symptoms, including headache, difficulty concentrating, fatigue, drowsiness, and dizziness.13 If symptoms are not reported by the athlete, balance problems, lack of coordination, increased emotionality, and difficulty following instructions may be observed during play.12
On-Field Assessment
Cognitive and balance testing are essential in determining if an athlete has sustained a concussion. Immediate declines in memory, concentration abilities, and balance abilities are common. Given limitations in administering long testing batteries on the sideline, brief standardized tests such as the Standardized Assessment of Concussion (SAC), Balance Error Scoring System (BESS), and Sport Concussion Assessment Tool (SCAT) are commonly utilized. Identification of cognitive and/or balance abnormalities can help the athlete recognize deficits following injury.12 Balance problems are experienced due to abnormalities in sensory organization and generally resolve during the acute recovery period.14,15 Cognitive difficulties typically persist longer than balance problems, though duration varies widely.
Neurologic Evaluation
A neurologic evaluation including cranial nerve testing and evaluation of motor-sensory function (ie, assessment for the strength and sensation of upper and lower extremities) is important to identify focal deficits (ie, sensation changes, loss of fine motor control) indicative of serious intracranial pathology.12 Additionally, clinicians have suggested inclusion of vestibular and oculomotor assessments due to frequent dysfunction post-concussion.12,15,16 Examination of the vestibular/oculomotor systems through tools such as the Vestibular/Ocular Motor Screening (VOMS) assessment (assesses both the vestibular and oculomotor systems) and King-Devick Test (primarily assesses saccadic eye movements) can elicit symptoms that may not present immediately. If assessment appears normal, exertion testing can be utilized to determine if symptoms are provoked through physical exercise that should include cardio, dynamic, and sport-specific activities to stress the vestibular system.12
Risk Factors for Injury and Prolonged Recovery
Medical professionals must consider the presence of risk factors when managing concussion in order to make appropriate treatment recommendations and return-to-play decisions. Research has demonstrated the role of female gender, learning disability, attention-deficit/hyperactivity disorder, psychiatric history, young age, motion sickness, sleep problems, somatization, concussion history, on-field dizziness, posttraumatic migraine, and fogginess in increased risk for injury and/or prolonged recovery.17-25 Additionally, athletes with ongoing symptoms from a previous injury are at risk for sustaining another injury.
Acute Home Concussion Management
Although strict rest has been recommended post-concussion, recent research evaluating strict rest vs usual care for adolescents revealed greater symptom reports and longer recovery periods for the strict rest group.26 Based on these findings and emphasis for regulation within the migraine literature (due to the common pathophysiology between migraine and concussion27), we recommend that athletes follow a regulated daily schedule post-concussion including: 1) regular sleep-wake schedule with avoidance of naps, 2) regular meals, 3) adequate fluid hydration, 4) light noncontact physical activity (ie, walking, with progressions recommended by a physician), and 5) stress management techniques. Use of these strategies immediately can help in preventing against increased symptoms and stress, and decreases the need for medication in select cases. Additionally, over-the-counter medications should be limited to 2 to 3 doses per week to avoid rebound headaches.28
In-Office Concussion Management
Athletes diagnosed with SRC will experience different symptoms based on the injury mechanism, risk factors, and management approach. Comprehensive evaluation should include assessment of risk factors, injury details, symptoms, neurocognitive functioning, vestibular/oculomotor dysfunction, tolerance of physical exertion, balance functioning, and cervical spine integrity (if necessary).29,30 Due to individual differences and the heterogeneous symptom profiles, concussion management must move beyond a “one size fits all” approach to avoid nonspecific treatment strategies and consequently prolonged recoveries.29 Clinicians and researchers at University of Pittsburgh Medical Center have identified 6 concussion clinical profiles (ie, vestibular, ocular, posttraumatic migraine, cervical, anxiety/mood, and cognitive/fatigue) that are generally identifiable 48 hours after injury.29,30 Identification of the clinical profile(s) through a comprehensive evaluation guides the development of individualized treatment plans and targeted rehabilitation strategies.29,30
Vestibular. The vestibular system is responsible for stabilizing vision while the head moves and balance control.15 Athletes can experience central and/or peripheral vestibular dysfunction to include benign paroxysmal positional vertigo (BPPV), visual motion sensitivity, vestibular ocular reflex impairment, and balance impairment.30,31 Symptoms typically include dizziness, impaired balance, blurry vision, difficulty focusing, and environmental sensitivity.15,29,30 Potential treatment options include vestibular rehabilitation, exertion therapy, and school/work accommodations.
Ocular. The oculomotor system is responsible for control of eye movements. Athletes can experience many different posttraumatic vision changes, including convergence problems, eye-tracking difficulties, refractive error, difficulty with pursuits/saccades, and accommodation insufficiency. Symptoms typically include light sensitivity, blurred vision, double vision, headaches, fatigue, and memory difficulties.15,29,30 Potential treatment options include vision therapy, vestibular rehabilitation, and school/work accommodations.32
Posttraumatic Migraine. Headache, the most common post-concussion symptom, can persist and meet criteria for posttraumatic migraine (ie, unilateral headache with accompanying nausea and/or photophobia and phonophobia).29,30,33 Implementation of a routine schedule, daily physical activity, exertion therapy, pharmacologic intervention, and school/work accommodations are potential treatment options.
Cervical. The cervical spine can be injured during whiplash-type injuries. Therefore, determining the location, onset, and typical exacerbations of pain can be helpful in identifying cervical involvement.29,30 Symptoms typically include headaches, neck pain, numbness, and tingling. Evaluation and therapy by a certified physical therapist and pharmacologic intervention (eg, muscle relaxants) are potential treatment options. 29,30
Anxiety/Mood. Anxiety, or worry and fear about everyday situations, is common post-concussion and can sometimes be related to ongoing vestibular impairment. Symptoms typically include ruminative thoughts, avoidance of specific situations, hypervigilance, feelings of being overwhelmed, and difficulty falling asleep.29,30 Potential treatment options include implementation of a routine schedule, exposure to provocative situations, psychotherapy, pharmacologic intervention, and school/work accommodations.34
Cognitive/Fatigue. A global concussion factor (including cognitive, fatigue, and migraine symptoms) has been identified within 1 to 7 days of injury. Although this factor of symptoms generally resolves during the acute recovery period, it persists in select cases.13 Symptoms typically include fatigue, decreased energy levels, nonspecific headaches, potential sleep disruption, increased symptoms towards the end of the day, difficulty concentrating, and increased headache with cognitive activities.29,30,35 Routine schedule, daily physical activity, exertion therapy, pharmacologic intervention (eg, amantadine), and school/work accommodations are potential treatment options.30
Conclusion
Advancements in SRC management warrant change in the conversations regarding concussion in football. Specifically, conversations should address the current understanding of concussion and improvements in the safety of football through stricter concussion guidelines, detailed sideline evaluations, recognition of risk factors, improved acute management, and identification of concussion profiles that help to direct individualized treatment plans and targeted rehabilitation strategies. The biggest concerns related to concussions in football include underreporting of injury, premature return to play, and receiving routine rather than individualized treatment. Therefore, to further improve the safety of football and management of concussion it is essential that future efforts focus on the following 6 areas:
Education: Improved understanding of concussion is imperative to reducing poor outcomes and widespread concerns.
Immediate reporting: Reporting of concussion must be expected and encouraged through consistent responses by coaches to reduce underreporting and fear of reporting in athletes.
Prevention techniques: Athletes must be taught proper form and playing techniques to reduce the risk for concussion. Proper form and technique should be incentivized.
Targeted treatment: Individualized treatment plans and targeted rehabilitation strategies must be developed based on the identified clinical profile(s) to avoid nonspecific treatment recommendations.
Multidisciplinary treatment teams: Given the heterogeneous symptoms profiles and need for care provided by different medical specialties, multidisciplinary teams are essential.
Remain current: With the progress in understanding concussion, providers must remain vigilant of future advances in concussion management to further improve the safety of football.
Am J Orthop. 2016;45(6):352-356. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Dompier TP, Kerr ZY, Marshall SW, et al. Incidence of concussion during practice and games in youth, high school, and collegiate American football players. JAMA Pediatrics. 2015;169(7):659-665.
2. Giza C, Hovda D. The new neurometabolic cascade of concussion
3. Barkhoudarian G, Hovda DA, Giza CC. The molecular pathophysiology of concussive brain injury - an update. Phys Med Rehabil Clin N Am. 2016;27:373-393.
4. Henry L, Elbin R, Collins M, Marchetti G, Kontos A. Examining recovery trajectories after sport-related concussion with a multimodal clinical assessment approach. Neurosurgery. 2016;78(2):232-241.
5. McCrory P, Meeuwisse WH, Aubry M, et al. Consensus statement on concussion in sport: the 4th International Conference on Concussion in Sport held in Zurich, November 2012. Brit J Sports Med. 2013;47(5):250-258.
6. Lovell MR, Collins MW, Iverson GL, et al. Recovery from mild concussion in high school athletes. J Neurosurg. 2003;98(2):296-301.
7. Lovell MR, Collins MW, Iverson GL, Johnston KM, Bradley JP. Grade 1 or “ding” concussions in high school athletes. Am J Sports Med. 2004;32(1):47-54.
8. Baugh CM, Kroshus E, Daneshvar DH, Filali NA, Hiscox MJ, Glantz LH. Concussion management in United States college sports: compliance with National Collegiate Athletic Association concussion policy and areas for improvement. Am J Sports Med. 2015;43(1):47-56.
9. Asken BM, McCrea MA, Clugston JR, Snyder AR, Houck ZM, Bauer RM. “Playing through it”: Delayed reporting and removal from athletic activity after concussion predicts prolonged recovery. J Athl Train. 2016;51(4):329-335.
10. Elbin RJ, Sufrinko A, Schatz P, et al. Athletes that continue to play with concussion demonstrate worse recovery outcomes than athletes immediately removed from play. J Pediatr. In press.
11. Signoretti S, Lazzarino G, Tavazzi B, Vagnozzi R. The pathophysiology of concussion. PM R. 2011;3(10 Suppl 2):S359-S368.
12. Bloom J, Blount JG. Sideline evaluation of concussion. UpToDate. 2016. http://www.uptodate.com/contents/sideline-evaluation-of-concussion. Accessed July 13, 2016.
13. Kontos AP, Elbin RJ, Schatz P, et al. A revised factor structure for the post-concussion symptom scale: baseline and postconcussion factors. Am J Sports Med. 2012;40(10):2375-2384.
14. Guskiewicz KM, Ross SE, Marshall SW. Postural stability and neuropsychological deficits after concussion in collegiate athletes. J Athl Train. 2001;36(3):263.
15. Mucha A, Collins MW, Elbin R, et al. A brief Vestibular/Ocular Motor Screening (VOMS) assessment to evaluate concussions preliminary findings. Am J Sports Med. 2014;42(10):2479-2486.
16. Bloom J. Vestibular and ocular motor assessments: Important pieces to the concussion puzzle. Athletic Training and Sports Health Care. 2013;5(6):246-248.
17. Covassin T, Elbin R, Harris W, Parker T, Kontos A. The role of age and sex in symptoms, neurocognitive performance, and postural stability in athletes after concussion. Am J Sports Med. 2012;40(6):1303-1312.
18. Kontos A, Sufrinko A, Elbin R, Puskar A, Collins M. Reliability and associated risk factors for performance on the Vestibular/Ocular Motor Screening (VOMS) tool in healthy collegiate athletes. Am J Sports Med. 2016;44(6):1400-1406.
19. Guskiewicz KM, McCrea M, Marshall SW, et al. Cumulative effects associated with recurrent concussion in collegiate football players: the NCAA Concussion Study. JAMA. 2003;290(19):2549-2555.
20. Lau B, Lovell MR, Collins MW, Pardini J. Neurocognitive and symptom predictors of recovery in high school athletes. Clin J Sport Med. 2009;19(3):216-221.
21. Lau BC, Kontos AP, Collins MW, Mucha A, Lovell MR. Which on-field signs/symptoms predict protracted recovery from sport-related concussion among high school football players? Am J Sports Med. 2011;39(11):2311-2318.
22. Mihalik JP, Register-Mihalik J, Kerr ZY, Marshall SW, McCrea MC, Guskiewicz KM. Recovery of posttraumatic migraine characteristics in patients after mild traumatic brain injury. Am J Sports Med. 2013;41(7):1490-1496.
23. Covassin T, Moran R, Elbin RJ. Sex differences in reported concussion injury rates and time loss from participation: An update of the National Collegiate Athletic Association injury surveillance program from 2004-2005 through 2008-2009. J Athl Train. 2016;51(3):189-194.
24. Root JM, Zuckerbraun NS, Wang L, et al. History of somatization is associated with prolonged recovery from concussion. J Pediatr. 2016;174:39-44.
25. Sufrinko A, Pearce K, Elbin RJ, et al. The effect of preinjury sleep difficulties on neurocognitive impairment and symptoms after sport-related concussion. Am J Sports Med. 2015;43(4):830-838.
26. Thomas DG, Apps JN, Hoffmann RG, McCrea M, Hammeke T. Benefits of strict rest after acute concussion: a randomized controlled trial. Pediatrics. 2015;135(2):213-223.
27. Choe M, Blume H. Pediatric posttraumatic Headache: a review. J Child Neurol. 2016;31(1):76-85.
28. Tepper SJ, Tepper DE. Breaking the cycle of medication overuse. Cleve Clin J Med. 2010;77(4):236-242.
29. Collins M, Kontos A, Reynolds E, Murawski C, Fu F. A comprehensive, targeted approach to the clinical care of athletes following sport-related concussion. Knee Surg Sports Traumatol Arthrosc. 2014;22(2):235-246.
30. Reynolds E, Collins MW, Mucha A, Troutman-Ensecki C. Establishing a clinical service for the management of sports-related concussions. Neurosurgery. 2014;75 Suppl 4:S71-S81.
31. Broglio SP, Collins MW, Williams RM, Mucha A, Kontos AP. Current and emerging rehabilitation for concussion: a review of the evidence. Clin Sports Med. 2015;34(2):213-231.
32. Master C, Scheiman M, Gallaway M, et al. Vision diagnoses are common after concussion in adolescents. Clin Pediatr (Phila). 2016;55(3):260-267.
33. Headache Classification Committee of the International Headache Society (IHS). The international classification of headache disorders, 3rd edition (beta version). Cephalalgia. 2013;33(9):629-808.
34. Kontos A, Deitrick JM, Reynolds E. Mental health implication and consequences following sport-related concussion. Brit J Sports Med. 2016;50(3):139-140.
35. Kontos AP, Covassin T, Elbin R, Parker T. Depression and neurocognitive performance after concussion among male and female high school and collegiate athletes. Arch Phys Med Rehabil. 2012;93(10):1751-1756.
Football is an important component of American culture, with approximately 3 million youth athletes, 1.1 million high school athletes, and 100,000 college athletes participating each year.1 Participation in football provides athletes with physical, social, psychological, and academic benefits. Despite these benefits, widespread focus has been placed on the safety of football due to the risk for sport-related concussion (SRC) and potentially long-term effects; however, little recognition has been given to the advancements in concussion management across time and occurrence of concussions during most life activities. Although it is reasonable for concerns to be presented, it is important to better understand SRC and the current factors leading to prolonged recoveries, increased risk for injury, and potentially long-term effects.
What Is a Concussion?
Concussions occur after sustaining direct or indirect injury to the head or other parts of the body, as long as the injury force is transmitted to the head. Athletes often experience physical, cognitive, emotional, and sleep-related symptoms post-concussion secondary to an “energy crisis” within the brain.2 The energy crisis occurs as the result of transient neurological dysfunction triggered by changes in the brain (eg, release of neurotransmitters, impaired axonal function).2,3 Concussion is undetectable with traditional imaging; however, advanced imaging techniques (eg, diffuse tensor imaging) have shown progress in assessing axonal injury.3 Symptom duration post-concussion is highly variable due to individual differences; a recent study showed recovery took 3 to 4 weeks for memory and symptoms.4,5
Previous Concussion Management
Identification techniques and return-to-play guidelines for concussion have significantly changed across time. In the past, concussion grading scales were utilized for diagnosis and return to play was possible within the same contest.6,7 It has since been recognized that initial concussion severity makes it difficult to predict recovery.3 For example, research revealed memory decline and increased symptoms 36 hours post-injury for athletes with a grade 1 concussion (ie, transient confusion, no loss of consciousness, concussion symptoms or mental status changes that resolve within 15 minutes of injury) compared to baseline.7 Another study found duration of mental status changes to be related to slower symptom resolution and memory impairment 36 hours to 7 days post-injury.6 Consequently, return to play within the same contest was likely too liberal. Guidelines today recommend immediate removal from play with suspected SRC. Nevertheless, the “play through pain” culture has led athletes to continue playing after SRC, contributing to prolonged recoveries and potentially long-term effects.
Current Concussion Management: Continued Concerns and Areas of Improvement
Despite increased awareness of concussions, recent estimates revealed high rates (ie, 27:1 ratio for general players) of underreporting in college football, particularly amongst offensive linemen.8 Researchers have studied recovery implications for remaining in play, with one study revealing a 2.2 times greater risk for prolonged recovery in college athletes with delayed vs immediate removal.9 Another similar study discovered an 8.8 times greater risk for prolonged recovery in adolescent and young adult athletes not removed vs removed from play.10 Further analysis found remaining in play to be the greatest risk factor for prolonged recovery compared to other previously studied risk factors (eg, age, sex, posttraumatic migraine).10 Additionally, significant differences in neurocognitive data were seen between the “removed” and “not removed” groups for verbal memory, visual memory, processing speed, and reaction time at 1 to 7 days and 8 to 30 days.10 The recovery implications of remaining in play and the additional risk for second impact syndrome (SIS), or repeat concussion when recovering from another injury, emphasizes the need for further education efforts amongst athletes to encourage immediate reporting of injury.11
Sideline Assessment
Sideline assessment has become a vital component of concussion management to rule out concussion and/or significant injury other than concussion. Assessment should include observation, cognitive/balance testing, neurologic examination, and possible exertion testing to ensure a comprehensive evaluation of all areas of potential dysfunction.12 Indications for emergency department evaluation include suspicion for cervical spine injury, intracranial hemorrhage, or skull fracture as well as prolonged loss of consciousness, high-risk mechanisms, posttraumatic seizure(s), and/or significant worsening of symptoms.12
Observation
On the sideline, it is important to identify any immediate signs of injury (ie, loss of consciousness, anterograde/retrograde amnesia, and disorientation/confusion). Since immediate signs are not always present, it is important to be aware of the most commonly reported symptoms, including headache, difficulty concentrating, fatigue, drowsiness, and dizziness.13 If symptoms are not reported by the athlete, balance problems, lack of coordination, increased emotionality, and difficulty following instructions may be observed during play.12
On-Field Assessment
Cognitive and balance testing are essential in determining if an athlete has sustained a concussion. Immediate declines in memory, concentration abilities, and balance abilities are common. Given limitations in administering long testing batteries on the sideline, brief standardized tests such as the Standardized Assessment of Concussion (SAC), Balance Error Scoring System (BESS), and Sport Concussion Assessment Tool (SCAT) are commonly utilized. Identification of cognitive and/or balance abnormalities can help the athlete recognize deficits following injury.12 Balance problems are experienced due to abnormalities in sensory organization and generally resolve during the acute recovery period.14,15 Cognitive difficulties typically persist longer than balance problems, though duration varies widely.
Neurologic Evaluation
A neurologic evaluation including cranial nerve testing and evaluation of motor-sensory function (ie, assessment for the strength and sensation of upper and lower extremities) is important to identify focal deficits (ie, sensation changes, loss of fine motor control) indicative of serious intracranial pathology.12 Additionally, clinicians have suggested inclusion of vestibular and oculomotor assessments due to frequent dysfunction post-concussion.12,15,16 Examination of the vestibular/oculomotor systems through tools such as the Vestibular/Ocular Motor Screening (VOMS) assessment (assesses both the vestibular and oculomotor systems) and King-Devick Test (primarily assesses saccadic eye movements) can elicit symptoms that may not present immediately. If assessment appears normal, exertion testing can be utilized to determine if symptoms are provoked through physical exercise that should include cardio, dynamic, and sport-specific activities to stress the vestibular system.12
Risk Factors for Injury and Prolonged Recovery
Medical professionals must consider the presence of risk factors when managing concussion in order to make appropriate treatment recommendations and return-to-play decisions. Research has demonstrated the role of female gender, learning disability, attention-deficit/hyperactivity disorder, psychiatric history, young age, motion sickness, sleep problems, somatization, concussion history, on-field dizziness, posttraumatic migraine, and fogginess in increased risk for injury and/or prolonged recovery.17-25 Additionally, athletes with ongoing symptoms from a previous injury are at risk for sustaining another injury.
Acute Home Concussion Management
Although strict rest has been recommended post-concussion, recent research evaluating strict rest vs usual care for adolescents revealed greater symptom reports and longer recovery periods for the strict rest group.26 Based on these findings and emphasis for regulation within the migraine literature (due to the common pathophysiology between migraine and concussion27), we recommend that athletes follow a regulated daily schedule post-concussion including: 1) regular sleep-wake schedule with avoidance of naps, 2) regular meals, 3) adequate fluid hydration, 4) light noncontact physical activity (ie, walking, with progressions recommended by a physician), and 5) stress management techniques. Use of these strategies immediately can help in preventing against increased symptoms and stress, and decreases the need for medication in select cases. Additionally, over-the-counter medications should be limited to 2 to 3 doses per week to avoid rebound headaches.28
In-Office Concussion Management
Athletes diagnosed with SRC will experience different symptoms based on the injury mechanism, risk factors, and management approach. Comprehensive evaluation should include assessment of risk factors, injury details, symptoms, neurocognitive functioning, vestibular/oculomotor dysfunction, tolerance of physical exertion, balance functioning, and cervical spine integrity (if necessary).29,30 Due to individual differences and the heterogeneous symptom profiles, concussion management must move beyond a “one size fits all” approach to avoid nonspecific treatment strategies and consequently prolonged recoveries.29 Clinicians and researchers at University of Pittsburgh Medical Center have identified 6 concussion clinical profiles (ie, vestibular, ocular, posttraumatic migraine, cervical, anxiety/mood, and cognitive/fatigue) that are generally identifiable 48 hours after injury.29,30 Identification of the clinical profile(s) through a comprehensive evaluation guides the development of individualized treatment plans and targeted rehabilitation strategies.29,30
Vestibular. The vestibular system is responsible for stabilizing vision while the head moves and balance control.15 Athletes can experience central and/or peripheral vestibular dysfunction to include benign paroxysmal positional vertigo (BPPV), visual motion sensitivity, vestibular ocular reflex impairment, and balance impairment.30,31 Symptoms typically include dizziness, impaired balance, blurry vision, difficulty focusing, and environmental sensitivity.15,29,30 Potential treatment options include vestibular rehabilitation, exertion therapy, and school/work accommodations.
Ocular. The oculomotor system is responsible for control of eye movements. Athletes can experience many different posttraumatic vision changes, including convergence problems, eye-tracking difficulties, refractive error, difficulty with pursuits/saccades, and accommodation insufficiency. Symptoms typically include light sensitivity, blurred vision, double vision, headaches, fatigue, and memory difficulties.15,29,30 Potential treatment options include vision therapy, vestibular rehabilitation, and school/work accommodations.32
Posttraumatic Migraine. Headache, the most common post-concussion symptom, can persist and meet criteria for posttraumatic migraine (ie, unilateral headache with accompanying nausea and/or photophobia and phonophobia).29,30,33 Implementation of a routine schedule, daily physical activity, exertion therapy, pharmacologic intervention, and school/work accommodations are potential treatment options.
Cervical. The cervical spine can be injured during whiplash-type injuries. Therefore, determining the location, onset, and typical exacerbations of pain can be helpful in identifying cervical involvement.29,30 Symptoms typically include headaches, neck pain, numbness, and tingling. Evaluation and therapy by a certified physical therapist and pharmacologic intervention (eg, muscle relaxants) are potential treatment options. 29,30
Anxiety/Mood. Anxiety, or worry and fear about everyday situations, is common post-concussion and can sometimes be related to ongoing vestibular impairment. Symptoms typically include ruminative thoughts, avoidance of specific situations, hypervigilance, feelings of being overwhelmed, and difficulty falling asleep.29,30 Potential treatment options include implementation of a routine schedule, exposure to provocative situations, psychotherapy, pharmacologic intervention, and school/work accommodations.34
Cognitive/Fatigue. A global concussion factor (including cognitive, fatigue, and migraine symptoms) has been identified within 1 to 7 days of injury. Although this factor of symptoms generally resolves during the acute recovery period, it persists in select cases.13 Symptoms typically include fatigue, decreased energy levels, nonspecific headaches, potential sleep disruption, increased symptoms towards the end of the day, difficulty concentrating, and increased headache with cognitive activities.29,30,35 Routine schedule, daily physical activity, exertion therapy, pharmacologic intervention (eg, amantadine), and school/work accommodations are potential treatment options.30
Conclusion
Advancements in SRC management warrant change in the conversations regarding concussion in football. Specifically, conversations should address the current understanding of concussion and improvements in the safety of football through stricter concussion guidelines, detailed sideline evaluations, recognition of risk factors, improved acute management, and identification of concussion profiles that help to direct individualized treatment plans and targeted rehabilitation strategies. The biggest concerns related to concussions in football include underreporting of injury, premature return to play, and receiving routine rather than individualized treatment. Therefore, to further improve the safety of football and management of concussion it is essential that future efforts focus on the following 6 areas:
Education: Improved understanding of concussion is imperative to reducing poor outcomes and widespread concerns.
Immediate reporting: Reporting of concussion must be expected and encouraged through consistent responses by coaches to reduce underreporting and fear of reporting in athletes.
Prevention techniques: Athletes must be taught proper form and playing techniques to reduce the risk for concussion. Proper form and technique should be incentivized.
Targeted treatment: Individualized treatment plans and targeted rehabilitation strategies must be developed based on the identified clinical profile(s) to avoid nonspecific treatment recommendations.
Multidisciplinary treatment teams: Given the heterogeneous symptoms profiles and need for care provided by different medical specialties, multidisciplinary teams are essential.
Remain current: With the progress in understanding concussion, providers must remain vigilant of future advances in concussion management to further improve the safety of football.
Am J Orthop. 2016;45(6):352-356. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
Football is an important component of American culture, with approximately 3 million youth athletes, 1.1 million high school athletes, and 100,000 college athletes participating each year.1 Participation in football provides athletes with physical, social, psychological, and academic benefits. Despite these benefits, widespread focus has been placed on the safety of football due to the risk for sport-related concussion (SRC) and potentially long-term effects; however, little recognition has been given to the advancements in concussion management across time and occurrence of concussions during most life activities. Although it is reasonable for concerns to be presented, it is important to better understand SRC and the current factors leading to prolonged recoveries, increased risk for injury, and potentially long-term effects.
What Is a Concussion?
Concussions occur after sustaining direct or indirect injury to the head or other parts of the body, as long as the injury force is transmitted to the head. Athletes often experience physical, cognitive, emotional, and sleep-related symptoms post-concussion secondary to an “energy crisis” within the brain.2 The energy crisis occurs as the result of transient neurological dysfunction triggered by changes in the brain (eg, release of neurotransmitters, impaired axonal function).2,3 Concussion is undetectable with traditional imaging; however, advanced imaging techniques (eg, diffuse tensor imaging) have shown progress in assessing axonal injury.3 Symptom duration post-concussion is highly variable due to individual differences; a recent study showed recovery took 3 to 4 weeks for memory and symptoms.4,5
Previous Concussion Management
Identification techniques and return-to-play guidelines for concussion have significantly changed across time. In the past, concussion grading scales were utilized for diagnosis and return to play was possible within the same contest.6,7 It has since been recognized that initial concussion severity makes it difficult to predict recovery.3 For example, research revealed memory decline and increased symptoms 36 hours post-injury for athletes with a grade 1 concussion (ie, transient confusion, no loss of consciousness, concussion symptoms or mental status changes that resolve within 15 minutes of injury) compared to baseline.7 Another study found duration of mental status changes to be related to slower symptom resolution and memory impairment 36 hours to 7 days post-injury.6 Consequently, return to play within the same contest was likely too liberal. Guidelines today recommend immediate removal from play with suspected SRC. Nevertheless, the “play through pain” culture has led athletes to continue playing after SRC, contributing to prolonged recoveries and potentially long-term effects.
Current Concussion Management: Continued Concerns and Areas of Improvement
Despite increased awareness of concussions, recent estimates revealed high rates (ie, 27:1 ratio for general players) of underreporting in college football, particularly amongst offensive linemen.8 Researchers have studied recovery implications for remaining in play, with one study revealing a 2.2 times greater risk for prolonged recovery in college athletes with delayed vs immediate removal.9 Another similar study discovered an 8.8 times greater risk for prolonged recovery in adolescent and young adult athletes not removed vs removed from play.10 Further analysis found remaining in play to be the greatest risk factor for prolonged recovery compared to other previously studied risk factors (eg, age, sex, posttraumatic migraine).10 Additionally, significant differences in neurocognitive data were seen between the “removed” and “not removed” groups for verbal memory, visual memory, processing speed, and reaction time at 1 to 7 days and 8 to 30 days.10 The recovery implications of remaining in play and the additional risk for second impact syndrome (SIS), or repeat concussion when recovering from another injury, emphasizes the need for further education efforts amongst athletes to encourage immediate reporting of injury.11
Sideline Assessment
Sideline assessment has become a vital component of concussion management to rule out concussion and/or significant injury other than concussion. Assessment should include observation, cognitive/balance testing, neurologic examination, and possible exertion testing to ensure a comprehensive evaluation of all areas of potential dysfunction.12 Indications for emergency department evaluation include suspicion for cervical spine injury, intracranial hemorrhage, or skull fracture as well as prolonged loss of consciousness, high-risk mechanisms, posttraumatic seizure(s), and/or significant worsening of symptoms.12
Observation
On the sideline, it is important to identify any immediate signs of injury (ie, loss of consciousness, anterograde/retrograde amnesia, and disorientation/confusion). Since immediate signs are not always present, it is important to be aware of the most commonly reported symptoms, including headache, difficulty concentrating, fatigue, drowsiness, and dizziness.13 If symptoms are not reported by the athlete, balance problems, lack of coordination, increased emotionality, and difficulty following instructions may be observed during play.12
On-Field Assessment
Cognitive and balance testing are essential in determining if an athlete has sustained a concussion. Immediate declines in memory, concentration abilities, and balance abilities are common. Given limitations in administering long testing batteries on the sideline, brief standardized tests such as the Standardized Assessment of Concussion (SAC), Balance Error Scoring System (BESS), and Sport Concussion Assessment Tool (SCAT) are commonly utilized. Identification of cognitive and/or balance abnormalities can help the athlete recognize deficits following injury.12 Balance problems are experienced due to abnormalities in sensory organization and generally resolve during the acute recovery period.14,15 Cognitive difficulties typically persist longer than balance problems, though duration varies widely.
Neurologic Evaluation
A neurologic evaluation including cranial nerve testing and evaluation of motor-sensory function (ie, assessment for the strength and sensation of upper and lower extremities) is important to identify focal deficits (ie, sensation changes, loss of fine motor control) indicative of serious intracranial pathology.12 Additionally, clinicians have suggested inclusion of vestibular and oculomotor assessments due to frequent dysfunction post-concussion.12,15,16 Examination of the vestibular/oculomotor systems through tools such as the Vestibular/Ocular Motor Screening (VOMS) assessment (assesses both the vestibular and oculomotor systems) and King-Devick Test (primarily assesses saccadic eye movements) can elicit symptoms that may not present immediately. If assessment appears normal, exertion testing can be utilized to determine if symptoms are provoked through physical exercise that should include cardio, dynamic, and sport-specific activities to stress the vestibular system.12
Risk Factors for Injury and Prolonged Recovery
Medical professionals must consider the presence of risk factors when managing concussion in order to make appropriate treatment recommendations and return-to-play decisions. Research has demonstrated the role of female gender, learning disability, attention-deficit/hyperactivity disorder, psychiatric history, young age, motion sickness, sleep problems, somatization, concussion history, on-field dizziness, posttraumatic migraine, and fogginess in increased risk for injury and/or prolonged recovery.17-25 Additionally, athletes with ongoing symptoms from a previous injury are at risk for sustaining another injury.
Acute Home Concussion Management
Although strict rest has been recommended post-concussion, recent research evaluating strict rest vs usual care for adolescents revealed greater symptom reports and longer recovery periods for the strict rest group.26 Based on these findings and emphasis for regulation within the migraine literature (due to the common pathophysiology between migraine and concussion27), we recommend that athletes follow a regulated daily schedule post-concussion including: 1) regular sleep-wake schedule with avoidance of naps, 2) regular meals, 3) adequate fluid hydration, 4) light noncontact physical activity (ie, walking, with progressions recommended by a physician), and 5) stress management techniques. Use of these strategies immediately can help in preventing against increased symptoms and stress, and decreases the need for medication in select cases. Additionally, over-the-counter medications should be limited to 2 to 3 doses per week to avoid rebound headaches.28
In-Office Concussion Management
Athletes diagnosed with SRC will experience different symptoms based on the injury mechanism, risk factors, and management approach. Comprehensive evaluation should include assessment of risk factors, injury details, symptoms, neurocognitive functioning, vestibular/oculomotor dysfunction, tolerance of physical exertion, balance functioning, and cervical spine integrity (if necessary).29,30 Due to individual differences and the heterogeneous symptom profiles, concussion management must move beyond a “one size fits all” approach to avoid nonspecific treatment strategies and consequently prolonged recoveries.29 Clinicians and researchers at University of Pittsburgh Medical Center have identified 6 concussion clinical profiles (ie, vestibular, ocular, posttraumatic migraine, cervical, anxiety/mood, and cognitive/fatigue) that are generally identifiable 48 hours after injury.29,30 Identification of the clinical profile(s) through a comprehensive evaluation guides the development of individualized treatment plans and targeted rehabilitation strategies.29,30
Vestibular. The vestibular system is responsible for stabilizing vision while the head moves and balance control.15 Athletes can experience central and/or peripheral vestibular dysfunction to include benign paroxysmal positional vertigo (BPPV), visual motion sensitivity, vestibular ocular reflex impairment, and balance impairment.30,31 Symptoms typically include dizziness, impaired balance, blurry vision, difficulty focusing, and environmental sensitivity.15,29,30 Potential treatment options include vestibular rehabilitation, exertion therapy, and school/work accommodations.
Ocular. The oculomotor system is responsible for control of eye movements. Athletes can experience many different posttraumatic vision changes, including convergence problems, eye-tracking difficulties, refractive error, difficulty with pursuits/saccades, and accommodation insufficiency. Symptoms typically include light sensitivity, blurred vision, double vision, headaches, fatigue, and memory difficulties.15,29,30 Potential treatment options include vision therapy, vestibular rehabilitation, and school/work accommodations.32
Posttraumatic Migraine. Headache, the most common post-concussion symptom, can persist and meet criteria for posttraumatic migraine (ie, unilateral headache with accompanying nausea and/or photophobia and phonophobia).29,30,33 Implementation of a routine schedule, daily physical activity, exertion therapy, pharmacologic intervention, and school/work accommodations are potential treatment options.
Cervical. The cervical spine can be injured during whiplash-type injuries. Therefore, determining the location, onset, and typical exacerbations of pain can be helpful in identifying cervical involvement.29,30 Symptoms typically include headaches, neck pain, numbness, and tingling. Evaluation and therapy by a certified physical therapist and pharmacologic intervention (eg, muscle relaxants) are potential treatment options. 29,30
Anxiety/Mood. Anxiety, or worry and fear about everyday situations, is common post-concussion and can sometimes be related to ongoing vestibular impairment. Symptoms typically include ruminative thoughts, avoidance of specific situations, hypervigilance, feelings of being overwhelmed, and difficulty falling asleep.29,30 Potential treatment options include implementation of a routine schedule, exposure to provocative situations, psychotherapy, pharmacologic intervention, and school/work accommodations.34
Cognitive/Fatigue. A global concussion factor (including cognitive, fatigue, and migraine symptoms) has been identified within 1 to 7 days of injury. Although this factor of symptoms generally resolves during the acute recovery period, it persists in select cases.13 Symptoms typically include fatigue, decreased energy levels, nonspecific headaches, potential sleep disruption, increased symptoms towards the end of the day, difficulty concentrating, and increased headache with cognitive activities.29,30,35 Routine schedule, daily physical activity, exertion therapy, pharmacologic intervention (eg, amantadine), and school/work accommodations are potential treatment options.30
Conclusion
Advancements in SRC management warrant change in the conversations regarding concussion in football. Specifically, conversations should address the current understanding of concussion and improvements in the safety of football through stricter concussion guidelines, detailed sideline evaluations, recognition of risk factors, improved acute management, and identification of concussion profiles that help to direct individualized treatment plans and targeted rehabilitation strategies. The biggest concerns related to concussions in football include underreporting of injury, premature return to play, and receiving routine rather than individualized treatment. Therefore, to further improve the safety of football and management of concussion it is essential that future efforts focus on the following 6 areas:
Education: Improved understanding of concussion is imperative to reducing poor outcomes and widespread concerns.
Immediate reporting: Reporting of concussion must be expected and encouraged through consistent responses by coaches to reduce underreporting and fear of reporting in athletes.
Prevention techniques: Athletes must be taught proper form and playing techniques to reduce the risk for concussion. Proper form and technique should be incentivized.
Targeted treatment: Individualized treatment plans and targeted rehabilitation strategies must be developed based on the identified clinical profile(s) to avoid nonspecific treatment recommendations.
Multidisciplinary treatment teams: Given the heterogeneous symptoms profiles and need for care provided by different medical specialties, multidisciplinary teams are essential.
Remain current: With the progress in understanding concussion, providers must remain vigilant of future advances in concussion management to further improve the safety of football.
Am J Orthop. 2016;45(6):352-356. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Dompier TP, Kerr ZY, Marshall SW, et al. Incidence of concussion during practice and games in youth, high school, and collegiate American football players. JAMA Pediatrics. 2015;169(7):659-665.
2. Giza C, Hovda D. The new neurometabolic cascade of concussion
3. Barkhoudarian G, Hovda DA, Giza CC. The molecular pathophysiology of concussive brain injury - an update. Phys Med Rehabil Clin N Am. 2016;27:373-393.
4. Henry L, Elbin R, Collins M, Marchetti G, Kontos A. Examining recovery trajectories after sport-related concussion with a multimodal clinical assessment approach. Neurosurgery. 2016;78(2):232-241.
5. McCrory P, Meeuwisse WH, Aubry M, et al. Consensus statement on concussion in sport: the 4th International Conference on Concussion in Sport held in Zurich, November 2012. Brit J Sports Med. 2013;47(5):250-258.
6. Lovell MR, Collins MW, Iverson GL, et al. Recovery from mild concussion in high school athletes. J Neurosurg. 2003;98(2):296-301.
7. Lovell MR, Collins MW, Iverson GL, Johnston KM, Bradley JP. Grade 1 or “ding” concussions in high school athletes. Am J Sports Med. 2004;32(1):47-54.
8. Baugh CM, Kroshus E, Daneshvar DH, Filali NA, Hiscox MJ, Glantz LH. Concussion management in United States college sports: compliance with National Collegiate Athletic Association concussion policy and areas for improvement. Am J Sports Med. 2015;43(1):47-56.
9. Asken BM, McCrea MA, Clugston JR, Snyder AR, Houck ZM, Bauer RM. “Playing through it”: Delayed reporting and removal from athletic activity after concussion predicts prolonged recovery. J Athl Train. 2016;51(4):329-335.
10. Elbin RJ, Sufrinko A, Schatz P, et al. Athletes that continue to play with concussion demonstrate worse recovery outcomes than athletes immediately removed from play. J Pediatr. In press.
11. Signoretti S, Lazzarino G, Tavazzi B, Vagnozzi R. The pathophysiology of concussion. PM R. 2011;3(10 Suppl 2):S359-S368.
12. Bloom J, Blount JG. Sideline evaluation of concussion. UpToDate. 2016. http://www.uptodate.com/contents/sideline-evaluation-of-concussion. Accessed July 13, 2016.
13. Kontos AP, Elbin RJ, Schatz P, et al. A revised factor structure for the post-concussion symptom scale: baseline and postconcussion factors. Am J Sports Med. 2012;40(10):2375-2384.
14. Guskiewicz KM, Ross SE, Marshall SW. Postural stability and neuropsychological deficits after concussion in collegiate athletes. J Athl Train. 2001;36(3):263.
15. Mucha A, Collins MW, Elbin R, et al. A brief Vestibular/Ocular Motor Screening (VOMS) assessment to evaluate concussions preliminary findings. Am J Sports Med. 2014;42(10):2479-2486.
16. Bloom J. Vestibular and ocular motor assessments: Important pieces to the concussion puzzle. Athletic Training and Sports Health Care. 2013;5(6):246-248.
17. Covassin T, Elbin R, Harris W, Parker T, Kontos A. The role of age and sex in symptoms, neurocognitive performance, and postural stability in athletes after concussion. Am J Sports Med. 2012;40(6):1303-1312.
18. Kontos A, Sufrinko A, Elbin R, Puskar A, Collins M. Reliability and associated risk factors for performance on the Vestibular/Ocular Motor Screening (VOMS) tool in healthy collegiate athletes. Am J Sports Med. 2016;44(6):1400-1406.
19. Guskiewicz KM, McCrea M, Marshall SW, et al. Cumulative effects associated with recurrent concussion in collegiate football players: the NCAA Concussion Study. JAMA. 2003;290(19):2549-2555.
20. Lau B, Lovell MR, Collins MW, Pardini J. Neurocognitive and symptom predictors of recovery in high school athletes. Clin J Sport Med. 2009;19(3):216-221.
21. Lau BC, Kontos AP, Collins MW, Mucha A, Lovell MR. Which on-field signs/symptoms predict protracted recovery from sport-related concussion among high school football players? Am J Sports Med. 2011;39(11):2311-2318.
22. Mihalik JP, Register-Mihalik J, Kerr ZY, Marshall SW, McCrea MC, Guskiewicz KM. Recovery of posttraumatic migraine characteristics in patients after mild traumatic brain injury. Am J Sports Med. 2013;41(7):1490-1496.
23. Covassin T, Moran R, Elbin RJ. Sex differences in reported concussion injury rates and time loss from participation: An update of the National Collegiate Athletic Association injury surveillance program from 2004-2005 through 2008-2009. J Athl Train. 2016;51(3):189-194.
24. Root JM, Zuckerbraun NS, Wang L, et al. History of somatization is associated with prolonged recovery from concussion. J Pediatr. 2016;174:39-44.
25. Sufrinko A, Pearce K, Elbin RJ, et al. The effect of preinjury sleep difficulties on neurocognitive impairment and symptoms after sport-related concussion. Am J Sports Med. 2015;43(4):830-838.
26. Thomas DG, Apps JN, Hoffmann RG, McCrea M, Hammeke T. Benefits of strict rest after acute concussion: a randomized controlled trial. Pediatrics. 2015;135(2):213-223.
27. Choe M, Blume H. Pediatric posttraumatic Headache: a review. J Child Neurol. 2016;31(1):76-85.
28. Tepper SJ, Tepper DE. Breaking the cycle of medication overuse. Cleve Clin J Med. 2010;77(4):236-242.
29. Collins M, Kontos A, Reynolds E, Murawski C, Fu F. A comprehensive, targeted approach to the clinical care of athletes following sport-related concussion. Knee Surg Sports Traumatol Arthrosc. 2014;22(2):235-246.
30. Reynolds E, Collins MW, Mucha A, Troutman-Ensecki C. Establishing a clinical service for the management of sports-related concussions. Neurosurgery. 2014;75 Suppl 4:S71-S81.
31. Broglio SP, Collins MW, Williams RM, Mucha A, Kontos AP. Current and emerging rehabilitation for concussion: a review of the evidence. Clin Sports Med. 2015;34(2):213-231.
32. Master C, Scheiman M, Gallaway M, et al. Vision diagnoses are common after concussion in adolescents. Clin Pediatr (Phila). 2016;55(3):260-267.
33. Headache Classification Committee of the International Headache Society (IHS). The international classification of headache disorders, 3rd edition (beta version). Cephalalgia. 2013;33(9):629-808.
34. Kontos A, Deitrick JM, Reynolds E. Mental health implication and consequences following sport-related concussion. Brit J Sports Med. 2016;50(3):139-140.
35. Kontos AP, Covassin T, Elbin R, Parker T. Depression and neurocognitive performance after concussion among male and female high school and collegiate athletes. Arch Phys Med Rehabil. 2012;93(10):1751-1756.
1. Dompier TP, Kerr ZY, Marshall SW, et al. Incidence of concussion during practice and games in youth, high school, and collegiate American football players. JAMA Pediatrics. 2015;169(7):659-665.
2. Giza C, Hovda D. The new neurometabolic cascade of concussion
3. Barkhoudarian G, Hovda DA, Giza CC. The molecular pathophysiology of concussive brain injury - an update. Phys Med Rehabil Clin N Am. 2016;27:373-393.
4. Henry L, Elbin R, Collins M, Marchetti G, Kontos A. Examining recovery trajectories after sport-related concussion with a multimodal clinical assessment approach. Neurosurgery. 2016;78(2):232-241.
5. McCrory P, Meeuwisse WH, Aubry M, et al. Consensus statement on concussion in sport: the 4th International Conference on Concussion in Sport held in Zurich, November 2012. Brit J Sports Med. 2013;47(5):250-258.
6. Lovell MR, Collins MW, Iverson GL, et al. Recovery from mild concussion in high school athletes. J Neurosurg. 2003;98(2):296-301.
7. Lovell MR, Collins MW, Iverson GL, Johnston KM, Bradley JP. Grade 1 or “ding” concussions in high school athletes. Am J Sports Med. 2004;32(1):47-54.
8. Baugh CM, Kroshus E, Daneshvar DH, Filali NA, Hiscox MJ, Glantz LH. Concussion management in United States college sports: compliance with National Collegiate Athletic Association concussion policy and areas for improvement. Am J Sports Med. 2015;43(1):47-56.
9. Asken BM, McCrea MA, Clugston JR, Snyder AR, Houck ZM, Bauer RM. “Playing through it”: Delayed reporting and removal from athletic activity after concussion predicts prolonged recovery. J Athl Train. 2016;51(4):329-335.
10. Elbin RJ, Sufrinko A, Schatz P, et al. Athletes that continue to play with concussion demonstrate worse recovery outcomes than athletes immediately removed from play. J Pediatr. In press.
11. Signoretti S, Lazzarino G, Tavazzi B, Vagnozzi R. The pathophysiology of concussion. PM R. 2011;3(10 Suppl 2):S359-S368.
12. Bloom J, Blount JG. Sideline evaluation of concussion. UpToDate. 2016. http://www.uptodate.com/contents/sideline-evaluation-of-concussion. Accessed July 13, 2016.
13. Kontos AP, Elbin RJ, Schatz P, et al. A revised factor structure for the post-concussion symptom scale: baseline and postconcussion factors. Am J Sports Med. 2012;40(10):2375-2384.
14. Guskiewicz KM, Ross SE, Marshall SW. Postural stability and neuropsychological deficits after concussion in collegiate athletes. J Athl Train. 2001;36(3):263.
15. Mucha A, Collins MW, Elbin R, et al. A brief Vestibular/Ocular Motor Screening (VOMS) assessment to evaluate concussions preliminary findings. Am J Sports Med. 2014;42(10):2479-2486.
16. Bloom J. Vestibular and ocular motor assessments: Important pieces to the concussion puzzle. Athletic Training and Sports Health Care. 2013;5(6):246-248.
17. Covassin T, Elbin R, Harris W, Parker T, Kontos A. The role of age and sex in symptoms, neurocognitive performance, and postural stability in athletes after concussion. Am J Sports Med. 2012;40(6):1303-1312.
18. Kontos A, Sufrinko A, Elbin R, Puskar A, Collins M. Reliability and associated risk factors for performance on the Vestibular/Ocular Motor Screening (VOMS) tool in healthy collegiate athletes. Am J Sports Med. 2016;44(6):1400-1406.
19. Guskiewicz KM, McCrea M, Marshall SW, et al. Cumulative effects associated with recurrent concussion in collegiate football players: the NCAA Concussion Study. JAMA. 2003;290(19):2549-2555.
20. Lau B, Lovell MR, Collins MW, Pardini J. Neurocognitive and symptom predictors of recovery in high school athletes. Clin J Sport Med. 2009;19(3):216-221.
21. Lau BC, Kontos AP, Collins MW, Mucha A, Lovell MR. Which on-field signs/symptoms predict protracted recovery from sport-related concussion among high school football players? Am J Sports Med. 2011;39(11):2311-2318.
22. Mihalik JP, Register-Mihalik J, Kerr ZY, Marshall SW, McCrea MC, Guskiewicz KM. Recovery of posttraumatic migraine characteristics in patients after mild traumatic brain injury. Am J Sports Med. 2013;41(7):1490-1496.
23. Covassin T, Moran R, Elbin RJ. Sex differences in reported concussion injury rates and time loss from participation: An update of the National Collegiate Athletic Association injury surveillance program from 2004-2005 through 2008-2009. J Athl Train. 2016;51(3):189-194.
24. Root JM, Zuckerbraun NS, Wang L, et al. History of somatization is associated with prolonged recovery from concussion. J Pediatr. 2016;174:39-44.
25. Sufrinko A, Pearce K, Elbin RJ, et al. The effect of preinjury sleep difficulties on neurocognitive impairment and symptoms after sport-related concussion. Am J Sports Med. 2015;43(4):830-838.
26. Thomas DG, Apps JN, Hoffmann RG, McCrea M, Hammeke T. Benefits of strict rest after acute concussion: a randomized controlled trial. Pediatrics. 2015;135(2):213-223.
27. Choe M, Blume H. Pediatric posttraumatic Headache: a review. J Child Neurol. 2016;31(1):76-85.
28. Tepper SJ, Tepper DE. Breaking the cycle of medication overuse. Cleve Clin J Med. 2010;77(4):236-242.
29. Collins M, Kontos A, Reynolds E, Murawski C, Fu F. A comprehensive, targeted approach to the clinical care of athletes following sport-related concussion. Knee Surg Sports Traumatol Arthrosc. 2014;22(2):235-246.
30. Reynolds E, Collins MW, Mucha A, Troutman-Ensecki C. Establishing a clinical service for the management of sports-related concussions. Neurosurgery. 2014;75 Suppl 4:S71-S81.
31. Broglio SP, Collins MW, Williams RM, Mucha A, Kontos AP. Current and emerging rehabilitation for concussion: a review of the evidence. Clin Sports Med. 2015;34(2):213-231.
32. Master C, Scheiman M, Gallaway M, et al. Vision diagnoses are common after concussion in adolescents. Clin Pediatr (Phila). 2016;55(3):260-267.
33. Headache Classification Committee of the International Headache Society (IHS). The international classification of headache disorders, 3rd edition (beta version). Cephalalgia. 2013;33(9):629-808.
34. Kontos A, Deitrick JM, Reynolds E. Mental health implication and consequences following sport-related concussion. Brit J Sports Med. 2016;50(3):139-140.
35. Kontos AP, Covassin T, Elbin R, Parker T. Depression and neurocognitive performance after concussion among male and female high school and collegiate athletes. Arch Phys Med Rehabil. 2012;93(10):1751-1756.
Exertional Heat Stroke and American Football: What the Team Physician Needs to Know
Football, one of the most popular sports in the United States, is additionally recognized as a leading contributor to sports injury secondary to the contact collision nature of the endeavor. There are an estimated 1.1 million high school football players with another 100,000 participants combined in the National Football League (NFL), college, junior college, Arena Football League, and semipro levels of play.1 USA Football estimates that an additional 3 million youth participate in community football leagues.1 The National Center for Catastrophic Sports Injury Research recently calculated a fatality rate of 0.14 per 100,000 participants in 2014 for the 4.2 million who play football at all levels—and 0.45 per 100,000 in high school.1 While direct deaths from head and spine injury remain a significant contributor to the number of catastrophic injuries, indirect deaths (systemic failure) predominate. Exertional heat stroke (EHS) has emerged as one of the leading indirect causes of death in high school and collegiate football. Boden and colleagues2 reported that high school and college football players sustain approximately 12 fatalities annually, with indirect systemic causes being twice as common as direct blunt trauma.2The most common indirect causes identified included cardiac failure, heat illness, and complications of sickle cell trait (SCT). It was also noted that the risk of SCT, heat-related, and cardiac deaths increased during the second decade of the study, indicating these conditions may require a greater emphasis on diagnosis, treatment, and prevention. This review details for the team physician the unique challenge of exercising in the heat to the football player, and the prevention, diagnosis, management and return-to-play issues pertinent to exertional heat illness (EHI).
The Challenge
EHS represents the most severe manifestation of EHI—a gamut of diseases commonly encountered during the hot summer months when American football season begins. The breadth of EHI includes several important clinical diagnoses: exercise-associated muscle cramps (heat cramps); heat exhaustion with and without syncope; heat injury with evidence of end organ injury (eg, rhabdomyolysis); and EHS. EHS is defined as “a form of hyperthermia associated with a systemic inflammatory response leading to a syndrome of multi-organ dysfunction in which encephalopathy predominates.”3 EHS, if left untreated, or even if clinical treatment is delayed, may result in significant end organ morbidity and/or mortality.
During exercise, the human thermoregulatory system mitigates heat gain by increasing skin blood flow and sweating, causing an increased dissipation of heat to the surrounding environment by leveraging conduction, convection, and evaporation.4,5 Elevated environmental temperatures, increased humidity, and dehydration can impede the body’s ability to dissipate heat at a rate needed to maintain thermoregulation. This imbalance can result in hyperthermia secondary to uncompensated heat stress,5 which in turn can lead to EHI. Football players have unique challenges that make them particularly vulnerable to EHI. The summer heat during early-season participation and the requirement for equipment that covers nearly 60% of body surfaces pose increased risk of volume losses and hyperthermia that trigger the onset of EHI.6 Football athletes’ body compositions and physical size are additional contributing risk factors; the relatively high muscle and fat content increase thermogenicity, which require their bodies to dissipate more heat.7
An estimated 9000 cases of EHI occur annually across all high school sports,8 with an incidence of 1.6:100,000 athlete-exposures.8,9 Studies have demonstrated, however, that EHI occurs in football 11.4 times more often than in all other high school sports combined.10 The incidence of nonfatal EHI in all levels of football is 4.42-5:100,000.8,9 Between 2000 and 2014, 41 football players died from EHS.1 In football, approximately 75% of all EHI events occurred during practices, while only 25% of incidents occurred during games.8
Given these potentially deadly consequences, it is important that football team physicians are not only alert to the early symptoms of heat illness and prepared to intervene to prevent the progression to EHS, but are critical leaders in educating coaches and players in evidence-based EHI prevention practices and policies.
Prevention
EHS is a preventable condition, arguably the most common cause of preventable nontraumatic exertional death in young athletes in the United States. Close attention to mitigating risk factors should begin prior to the onset of preseason practice and continue through the early season, where athletes are at the highest risk of developing heat illness.
Primary Prevention
Primary prevention is fundamental to minimizing the occurrences of EHI. It focuses on the following methods: recognition of inherent risk factors, acclimatization, hydration, and avoidance of inciting substances (including supplements).
Pre-Participation Examination. The purpose of the pre-participation examination (PPE) is to maximize an athlete’s safety by identifying medical conditions that place the athlete at risk.11,12 The Preparticipation Physical Evaluation, 4th edition, the most widely used consensus publication, specifically queries if an athlete has a previous history of heat injury. However, it only indirectly addresses intrinsic risk factors that may predispose an athlete to EHI who has never had an EHI before. Therefore, providers should take the opportunity of the PPE to inquire about additional risk factors that may make an athlete high risk for sustaining a heat injury. Common risk factors for EHI are listed in Table 1.
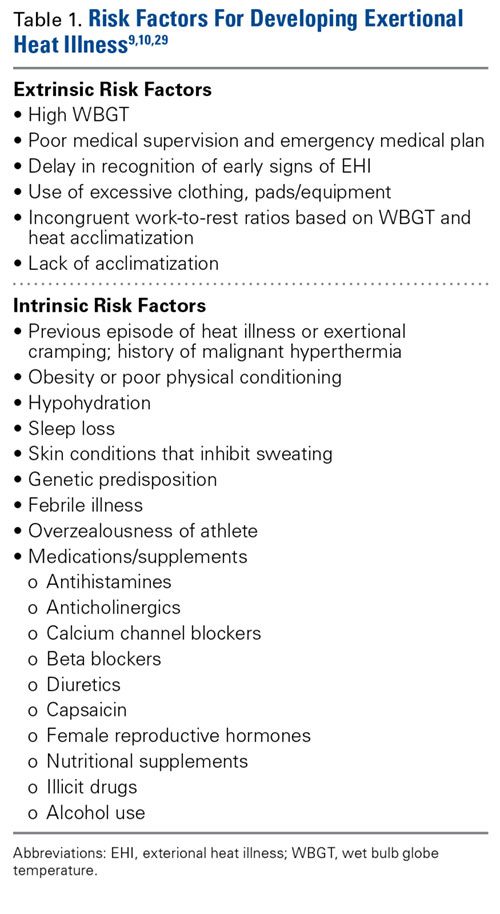
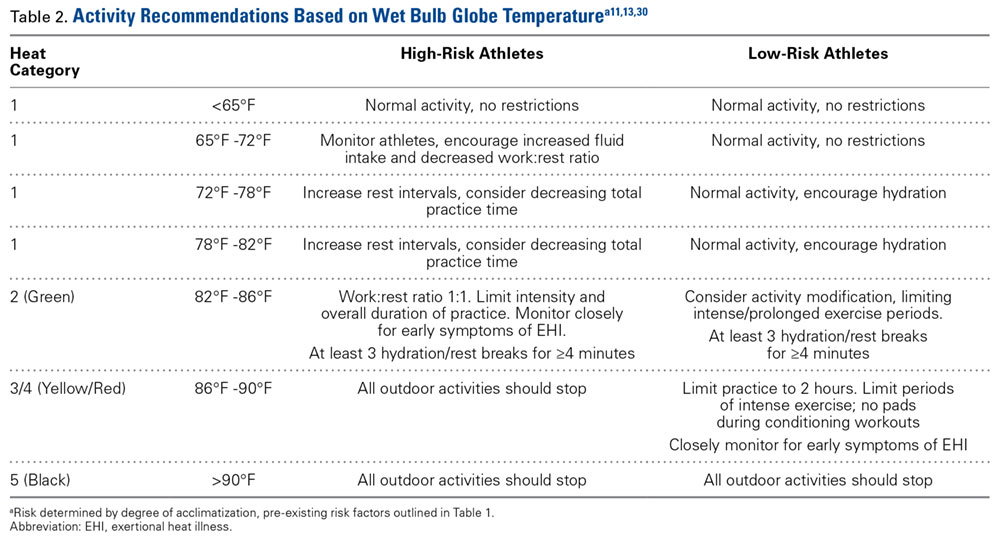
Heat Acclimatization. The risk of EHI escalates significantly when athletes are subjected to multiple stressors during periods of heat exposure, such as sudden increases in intensity or duration of exercise; prolonged new exposures to heat; dehydration; and sleep loss.5 When football season begins in late summer, athletes are least conditioned as temperatures reach their seasonal peak, causing increased risk of EHI.15 Planning for heat acclimatization is vital for all athletes who exercise in hot environments. Acclimatization procedures place progressively mounting physiologic strains on the body to improve athletes’ ability to dissipate heat, diminishing thermoregulatory and cardiovascular exertion.4,5 Acclimatization begins with expansion of plasma volume on days 3 to 6, causing improvements in cardiac efficiency and resulting in an overall decrease in basal internal body temperature.4,5,15 This process results in improvements in heat tolerance and exercise performance, evolving over 10 to 14 days of gradual escalation of exercise intensity and duration.5,10,11,16 However, poor fitness levels and extreme temperatures can prolong this period, requiring up to 2 to 3 months to fully take effect.5,7
The National Athletic Trainers Association (NATA) and National Collegiate Athletic Association (NCAA) have released consensus guidelines regarding heat acclimatization protocols for football athletes at the high school and college levels (Tables 3 and 4). Each of these guidelines involves an initial period without use of protective equipment, followed by a gradual addition of further equipment.11,16

Secondary Prevention
Despite physicians’ best efforts to prevent all cases of EHI, athletes will still experience the effects of exercise-induced hyperthermia. The goal of secondary prevention is to slow the progression of this hyperthermia so that it does not progress to more dangerous EHI.

Hydration. Dehydration is an important risk factor for EHI. Sweat maintains thermoregulation by dissipating heat generated during exercise; however, it also contributes to body water losses. Furthermore, intravascular depletion decreases stroke volume, thereby increasing cardiovascular strain. It is estimated that for every 1% loss in body mass from dehydration, body temperature rises 0.22°C in comparison to a euhydrated state.6 Dehydration occurs more rapidly in hot environments, as fluid is lost through increased sweat production.7 After approximately 6% to 10% body weight volume loss, cardiac output cannot be maintained, diminishing sweat production and blood flow to both skin and muscle and causing diminished performance and a significant risk of heat exhaustion.7 If left unchecked, these physiologic changes result in further elevations in body temperature and increased cardiovascular strain, ultimately placing the athlete at significant risk for development of EHS.
Adequate hydration to maintain euvolemia is an important step in avoiding possible EHI. Multiple studies have shown that football players experience a baseline hypovolemia during their competition season,6 a deficit that is most marked during the first week of practices.17 This deficit is multifactorial, as football players expend a significant amount of fluid through sweat, are not able to adequately replace these losses during practice, and do not appropriately hydrate off the field.6,18 Some players, especially linemen, sweat at a higher rate than their teammates, posing a possible risk of significant dehydration.6 Coaches and players alike should be educated on the importance of adequate hydration to meet their fluid needs.
The goal of hydration during exercise is to prevent large fluid losses that can adversely affect performance and increase risk of EHI;6 it may be unrealistic to replace all fluid losses during the practice period. Instead, athletes should target complete volume replacement over the post-exercise period.6 Some recommend hydrating based upon thirst drive; however, thirst is activated following a volume loss of approximately 2% body mass, the same degree of losses that place athletes at an increased risk for performance impairment and EHI.4,6,11,12 Individuals should have access to fluids throughout practice and competition and be encouraged to hydrate as needed.6,12,15 Furthermore, staff should modify their practices based upon WBGT and acclimatization status to provide more frequent hydration breaks.
Hyperhydration and Salt Intake. Of note, there are inherent risks to hyperhydration. Athletes with low sweat rates have an increased risk of overhydration and the development of exercise-associated hyponatremia (EAH),6 a condition whose presentation is very similar to EHS. In addition, inadequate sodium intake and excessive sweating can also contribute to the development of EAH. EAH has been implicated in the deaths of 2 football players in 2014.1,6 Establishing team hydration guidelines and educating players and staff on appropriate hydration and dietary salt intake is essential to reduce the risk of both dehydration and hyperhydration and their complications.6Intra-Event Cooling. During exercise, team physicians can employ strategies for cooling athletes during exertion to mitigate their risk of EHI by decreasing thermal and cardiovascular strain.4,19 Cooling during exercise is hypothesized to allow for accelerated heat dissipation, where heat is lost from the body more effectively. This accelerated loss enables athletes to maintain a higher heat storage capacity over the duration of exercise, avoiding uncompensated heat stresses that ultimately cause EHI.19
Some intra-event cooling strategies include the use of cooling garments, cooling packs, and cold water/slurry ingestion. Cooling garments lower skin temperature, which in turn can decrease thermoregulatory strains;4 a recent meta-analysis of intra-event cooling modalities revealed that wearing an ice vest during exercise resulted in the greatest decrease in thermal heat strain.19 Internal cooling strategies—namely ingestion of cold fluids/ice slurry—have shown some mild benefit in decreasing internal temperatures; however, some studies have demonstrated some decrease in sweat production associated with cold oral intake used in isolation.19 Overall, studies have shown that combining external (cooling clothing, ice packs, fanning) and internal (cold water, ice slurry) cooling methods result in a greater cooling effect than a use of a single method.4
Tertiary Prevention
The goal of tertiary prevention is to mitigate the risk of long-term adverse outcomes following an EHS event. The most effective means of reducing risk for morbidity and mortality is rapid identification and treatment of EHS as well as close evaluation of an athlete’s return to activity in heat. This process is spearheaded by an effective and well-rehearsed emergency action plan.
Diagnosis and Management
Rapid identification and treatment of EHS is crucial to minimizing the risk of poor outcomes.7 Any delay in the treatment of EHS can dramatically increase the likelihood of associated morbidity and mortality.20
EHS is diagnosed by an elevated rectal temperature ≥40°C (104°F) and associated central nervous system (CNS) dysfunction.21 EHS should be strongly suspected in any athlete exercising in heat who exhibits signs of CNS dysfunction, including disorientation, confusion, dizziness, erratic behavior, irritability, headache, loss of coordination, delirium, collapse, or seizures.7,12,15 EHS may also present with symptoms of heat exhaustion, including fatigue, hyperventilation, tachycardia, vomiting, diarrhea, and hypotension.7,12,15
Rectal temperature should be taken for any athlete with suspected EHS, as other modalities—oral, skin, axillary, and aural—can be inaccurate and easily modified by ambient confounders such as ambient and skin temperature, athlete hyperventilation, and consumption of liquids.7,11,12 Athletes exhibiting CNS symptoms with moderately elevated rectal temperatures that do not exceed 40°C should also be assumed to be suffering from EHS and treated with rapid cooling.11 On the other hand, athletes with CNS symptoms who are normothermic should be assumed to have EAH until ruled out by electrolyte assessment; IV fluids should be at no more than keep vein open (KVO) pending this determination.11 In some cases, an athlete may initially present with altered mental status but return to “normal.” However, this improvement may represent a “lucid period”; evaluation should continue with rectal temperature and treatment, as EHS in these cases may progress quickly.15
Treatment is centered on rapid, whole body cooling initiated at the first sign of heat illness.7,22 The goal of treatment is to achieve a rectal temperature <38.9°C within 30 minutes of the onset of EHS.15 Upon diagnosis, the athlete should be quickly placed in a tub of ice water to facilitate cold water immersion (CWI) therapy. Some guidelines suggest the athlete’s clothing be removed to potentiate evaporative cooling during CWI;12 however, cooling should not be delayed due to difficulties in removing equipment. CWI, where a heat stroke victim is submerged in ice water up to their neck while water is continuously circulated, is generally considered to be the gold standard treatment as it is the modality with the highest recorded cooling rates and the lowest rate of morbidity and mortality.7,20,21 Multiple studies of CWI have shown that survival nears 100% when aggressive cooling starts within 5 minutes of collapse or identification of EHS.20,21,22
If whole body CWI is unavailable, alternative methods of rapid cooling should be employed. Partial CWI, with torso immersion being preferable to the extremities, has been shown to achieve an acceptable rate of cooling to achieve sufficient drops in internal body temperature.20,23 However, one popular treatment—applying ice packs to the whole body, in particular to the groin and axillae—has not been shown to be sufficient to achieve standard cooling goals.20 None of these methods have been shown to be as effective as CWI.23
Intravenous access should be initiated with fluid resuscitation dictated by the provider’s assessment. Normal saline is recommended as the resuscitative fluid of choice, with the rate dictated by clinical judgment and adjusted as guided by electrolyte determination and clinical response. It cannot be overstated that in normothermic patients with confusion, EAH is the diagnosis of exclusion and aggressive fluid resuscitation should be withheld until electrolyte determination.
Once rectal temperature descends appropriately (~38.9°C), the cooling process should stop and the individual should be transported to a hospital for further observation20 and evaluation of possible sequelae, including rhabdomyolysis and renal injury, cardiac dysfunction and arrhythmia, severe electrolyte abnormalities, acute respiratory distress syndrome, lactic acidosis, and other forms of end-organ failure (Figure).

Rapid cooling is more crucial than transport; transport poses a risk of delayed cooling, which can dramatically increase an individual’s risk of morbidity and mortality.20,23 In situations where a patient can be cooled on-site, physicians should pursue cooling before transporting the patient to a medical treatment facility.
Emergency Action Plan
Team physicians should be proactive in developing an emergency action plan to address possible EHS events. These plans should be site-specific, addressing procedures for all practice and home competition locations.12 All competition venues should have a CWI tub on-site in events where there is an increased risk of EHS.12,15,20 This tub should be set up and functional for all high-risk activities, including practices.12
Following recognition of a potential case of EHS, treatment teams should have procedures in place to transport athletes to the treatment area, obtain rectal temperature, initiate rapid cooling, and stabilize the athlete for transport to an emergency department (ED) for further evaluation.12,15 A written record of treatments and medications provided during athlete stabilization should be maintained and transported with the athlete to the ED.15 A list of helpful equipment and supplies for treatment of EHS can be found in Table 5.

EHS is a unique life-threatening situation where it is best to treat the patient on the sideline before transport.15 Those athletes transported before cooling risk spending an increased amount of time above critical temperatures for cell damage, which has been associated with increased morbidity and mortality. This mantra of “cool first, transport second” cannot be overemphasized, as those individuals with EHS who present to the ED with a persisting rectal temperature >41°F may risk up to an 80% mortality rate.24 Conversely, a recent large, retrospective study of 274 EHS events sustained during the Falmouth Road Race found a 100% survival rate when athletes were rapidly identified via rectal thermometry and treated with aggressive, rapid cooling through CWI.21
Return to Play
Perhaps the most challenging and important role the team physician has is determining an athlete’s return to play following EHI, as there currently are no evidence-based guidelines for return to activity for these athletes.7 The decisions surrounding return to play are highly individualized, as recovery from EHS and heat injury is associated with the duration of internal body temperature elevation above the critical level (40°C).7,20 Guidelines for return to activity following recovery from EHI differ among experts and institutions.7,25 The general consensus from these guidelines is that, at minimum, athletes should not participate in any physical activity until they are asymptomatic and all blood tests have normalized.11 Following this asymptomatic period, most guidelines advocate for a slow, deliberate return to activity.11 The American College of Sports Medicine (ACSM) offers one reasonable approach to the returning athlete following EHS:7
- No exercise for at least 7 days following release from medical care.
- Follow-up with a physician 1 week after release from medical care for physical examination and any warranted lab or radiologic studies (based upon organ systems affected during EHS).
- Once cleared to return to activity, the athlete begins exercise in a cool environment, gradually increasing the duration, intensity, and heat exposure over 2 weeks to demonstrate heat tolerance and acclimatization.
- Athletes who cannot resume vigorous activity due to recurrent symptoms (eg, excessive fatigue) should be reevaluated after 4 weeks. Laboratory exercise-heat tolerance testing may be useful in this setting.
- The athlete may resume full competition once they are able to participate in full training in the heat for 2 to 4 weeks without adverse effects.
Heat tolerance testing (HTT) in these athletes remains controversial.5 26 The ACSM recommends that HTT be considered only for those unable to return to vigorous activity after a suitable period (approximately 4 weeks). In contrast, the Israeli Defense Force (IDF) uses HTT to evaluate soldiers following EHS to guide decision-making about return to duty.27 The IDF HTT assumes that individuals will respond differently to heat stresses. They identify individuals who are “heat intolerant” as being unable to tolerate specific heat challenges, indicated by increases in body temperature occurring more rapidly than normal responders under identical environmental and exercise conditions. However, despite being used for more than 30 years, there is no clear evidence that HTT adequately predicts who will experience subsequent episodes of EHS.
Conclusion
While the recognized cornerstone of being a team physician is the provision of medical care, the ACSM Team Physician Consensus Statement28 further delineates the medical and administrative responsibilities as both (1) understanding medical management and prevention of injury and illness in athletes; and (2) awareness of or involvement in the development and rehearsal of an emergency action plan. These tenets are critical for the team physician who accepts the responsibility to cover sports at the high school level or higher. Football team physicians play an essential role in mitigating risk of EHI in their athletes. Through development and execution of both comprehensive prevention strategies and emergency action plans, physicians can work to minimize athletes’ risk of both developing and experiencing significant adverse outcomes from an EHI.
Am J Orthop. 2016;45(6):340-348. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Kucera KL, Klossner D, Colgate B, Cantu RC. Annual Survey of Football Injury Research: 1931-2014. National Center for Catastrophic Sport Injury Research Web site. https://nccsir.unc.edu/files/2013/10/Annual-Football-2014-Fatalities-Final.pdf. Accessed May 31, 2016.
2. Boden BP, Breit I, Beachler JA, Williams A, Mueller FO. Fatalities in high school and college football players. Am J Sports Med. 2013;41(5):1108-1116.
3. Bouchama A, Knochel JP. Heat stroke. N Engl J Med. 2002;346(25):1978-1988.
4. Racinais S, Alonso JM, Coutts AJ, et al. Consensus recommendations on training and competing in the heat. Scand J Med Sci Sports. 2015;25 Suppl 1:6-19.
5. Pryor RR, Casa DJ, Adams WM, et al. Maximizing athletic performance in the heat. Strength Cond J. 2013;35(6):24-33.
6. Adams WM, Casa DJ. Hydration for football athletes. Sports Sci Exchange. 2015;28(141):1-5.
7. American College of Sports Medicine, Armstrong LE, Casa DJ, et al. American College of Sports Medicine position stand. Exertional heat illness during training and competition. Med Sci Sports Exerc. 2007;39(3):556-572.
8. Yard EE, Gilchrist J, Haileyesus T, et al. Heat illness among high school athletes--United States, 2005-2009. J Safety Res. 2010;41(6):471-474.
9. Huffman EA, Yard EE, Fields SK, Collins CL, Comstock RD. Epidemiology of rare injuries and conditions among United States high school athletes during the 2005-2006 and 2006-2007 school years. J Athl Train. 2008;43(6):624-630.
10. Kerr ZY, Casa DJ, Marshall SW, Comstock RD. Epidemiology of exertional heat illness among U.S. high school athletes. Am J Prev Med. 2013;44(1):8-14.
11. Casa DJ, DeMartini JK, Bergeron MF, et al. National Athletic Trainers’ Association position statement: exertional heat illnesses. J Athl Train. 2015;50(9):986-1000.
12. Casa DJ, Almquist J, Anderson SA. The inter-association task force for preventing sudden death in secondary school athletics programs: best-practices recommendations. J Athl Train. 2013;48(4):546-553.
13. Gardner JW, Kark JA, Karnei K, et al. Risk factors predicting exertional heat illness in male Marine Corps recruits. Med Sci Sports Exerc. 1996;28(8):939-944.
14. Gundstein AJ, Ramseyer C, Zhao F, et al. A retrospective analysis of American football hyperthermia deaths in the United States. Int J Biometerol. 2012;56(1):11-20.
15. Armstrong LE, Johnson EC, Casa DJ, et al. The American football uniform: uncompensable heat stress and hyperthermic exhaustion. J Athl Train. 2010;45(2):117-127.
16. Casa DJ, Csillan D; Inter-Association Task Force for Preseason Secondary School Athletics Participants, et al. Preseason heat-acclimatization guidelines for secondary school athletics. J Athl Train. 2009;44(3):332-333.
17. Godek SF, Godek JJ, Bartolozzi AR. Hydration status in college football players during consecutive days of twice-a-day preseason practices. Am J Sports Med. 2005;33(6):843-851.
18. Stover EA, Zachwieja J, Stofan J, Murray R, Horswill CA. Consistently high urine specific gravity in adolescent American football players and the impact of an acute drinking strategy. Int J Sports Med. 2006;27(4):330-335.
19. Bongers CC, Thijssen DH, Veltmeijer MTW, Hopman MT, Eijsvogels TM. Precooling and percooling (cooling during exercise) both improve performance in the heat: a meta-analytical review. Br J Sports Med. 2015;49(6):377-384.
20. Casa DJ, McDermott BP, Lee EC, Yeargin SW, Armstrong LE, Maresh CM. Cold water immersion: the gold standard for exertional heatstroke treatment. Exerc Sport Sci Rev. 2007;35(3):141-149.
21. DeMartini JK, Casa DJ, Stearns R, et al. Effectiveness of cold water immersion in the treatment of exertional heat stroke at the Falmouth Road Race. Med Sci Sports Exerc. 2015;47(2):240-245.
22. Casa DJ, Kenny GP, Taylor NA. Immersion treatment for exertional hyperthermia: cold or temperate water? Med Sci Sports Exerc. 2010;42(7):1246-1252.
23. Casa DJ, Armstrong LE, Kenny GP, O’Connor FG, Huggins RA. Exertional heat stroke: new concepts regarding cause and care. Curr Sports Med Rep. 2012;11(3):115-122.
24. Argaud L, Ferry T, Le QH, et al. Short- and long-term outcomes of heat stroke following the 2003 heat wave in Lyon, France. Arch Intern Med. 2007;167(20):2177-2183.
25. O’Connor FG, Casa DJ, Bergeron MF, et al. American College of Sports Medicine Roundtable on exertional heat stroke--return to duty/return to play: conference proceedings. Curr Sports Med Rep. 2010;9(5):314-321.
26. Kazman JB, Heled Y, Lisman PJ, Druyan A, Deuster PA, O’Connor FG. Exertional heat illness: the role of heat tolerance testing. Curr Sports Med Rep. 2013;12(2):101-105.
27. Moran DS, Heled Y, Still L, Laor A, Shapiro Y. Assessment of heat tolerance for post exertional heat stroke individuals. Med Sci Monit. 2004;10(6):CR252-CR257.
28. Herring SA, Kibler WB, Putukian M. Team Physician Consensus Statement: 2013 update. Med Sci Sports Exerc. 2013;45(8):1618-1622.
29. Heat stroke treatment. Korey Stringer Institute University of Connecticut Web site. http://ksi.uconn.edu/emergency-conditions/heat-illnesses/exertional-heat-stroke/heat-stroke-treatment/. Accessed June 14, 2016.
30. Headquarters, Department of the Army and the Air Force. Heat Stress Control and Heat Casualty Management. Technical Bulletin Medical 507. http://www.dir.ca.gov/oshsb/documents/Heat_illness_prevention_tbmed507.pdf. Published March 7, 2003. Accessed June 14, 2016.
Football, one of the most popular sports in the United States, is additionally recognized as a leading contributor to sports injury secondary to the contact collision nature of the endeavor. There are an estimated 1.1 million high school football players with another 100,000 participants combined in the National Football League (NFL), college, junior college, Arena Football League, and semipro levels of play.1 USA Football estimates that an additional 3 million youth participate in community football leagues.1 The National Center for Catastrophic Sports Injury Research recently calculated a fatality rate of 0.14 per 100,000 participants in 2014 for the 4.2 million who play football at all levels—and 0.45 per 100,000 in high school.1 While direct deaths from head and spine injury remain a significant contributor to the number of catastrophic injuries, indirect deaths (systemic failure) predominate. Exertional heat stroke (EHS) has emerged as one of the leading indirect causes of death in high school and collegiate football. Boden and colleagues2 reported that high school and college football players sustain approximately 12 fatalities annually, with indirect systemic causes being twice as common as direct blunt trauma.2The most common indirect causes identified included cardiac failure, heat illness, and complications of sickle cell trait (SCT). It was also noted that the risk of SCT, heat-related, and cardiac deaths increased during the second decade of the study, indicating these conditions may require a greater emphasis on diagnosis, treatment, and prevention. This review details for the team physician the unique challenge of exercising in the heat to the football player, and the prevention, diagnosis, management and return-to-play issues pertinent to exertional heat illness (EHI).
The Challenge
EHS represents the most severe manifestation of EHI—a gamut of diseases commonly encountered during the hot summer months when American football season begins. The breadth of EHI includes several important clinical diagnoses: exercise-associated muscle cramps (heat cramps); heat exhaustion with and without syncope; heat injury with evidence of end organ injury (eg, rhabdomyolysis); and EHS. EHS is defined as “a form of hyperthermia associated with a systemic inflammatory response leading to a syndrome of multi-organ dysfunction in which encephalopathy predominates.”3 EHS, if left untreated, or even if clinical treatment is delayed, may result in significant end organ morbidity and/or mortality.
During exercise, the human thermoregulatory system mitigates heat gain by increasing skin blood flow and sweating, causing an increased dissipation of heat to the surrounding environment by leveraging conduction, convection, and evaporation.4,5 Elevated environmental temperatures, increased humidity, and dehydration can impede the body’s ability to dissipate heat at a rate needed to maintain thermoregulation. This imbalance can result in hyperthermia secondary to uncompensated heat stress,5 which in turn can lead to EHI. Football players have unique challenges that make them particularly vulnerable to EHI. The summer heat during early-season participation and the requirement for equipment that covers nearly 60% of body surfaces pose increased risk of volume losses and hyperthermia that trigger the onset of EHI.6 Football athletes’ body compositions and physical size are additional contributing risk factors; the relatively high muscle and fat content increase thermogenicity, which require their bodies to dissipate more heat.7
An estimated 9000 cases of EHI occur annually across all high school sports,8 with an incidence of 1.6:100,000 athlete-exposures.8,9 Studies have demonstrated, however, that EHI occurs in football 11.4 times more often than in all other high school sports combined.10 The incidence of nonfatal EHI in all levels of football is 4.42-5:100,000.8,9 Between 2000 and 2014, 41 football players died from EHS.1 In football, approximately 75% of all EHI events occurred during practices, while only 25% of incidents occurred during games.8
Given these potentially deadly consequences, it is important that football team physicians are not only alert to the early symptoms of heat illness and prepared to intervene to prevent the progression to EHS, but are critical leaders in educating coaches and players in evidence-based EHI prevention practices and policies.
Prevention
EHS is a preventable condition, arguably the most common cause of preventable nontraumatic exertional death in young athletes in the United States. Close attention to mitigating risk factors should begin prior to the onset of preseason practice and continue through the early season, where athletes are at the highest risk of developing heat illness.
Primary Prevention
Primary prevention is fundamental to minimizing the occurrences of EHI. It focuses on the following methods: recognition of inherent risk factors, acclimatization, hydration, and avoidance of inciting substances (including supplements).
Pre-Participation Examination. The purpose of the pre-participation examination (PPE) is to maximize an athlete’s safety by identifying medical conditions that place the athlete at risk.11,12 The Preparticipation Physical Evaluation, 4th edition, the most widely used consensus publication, specifically queries if an athlete has a previous history of heat injury. However, it only indirectly addresses intrinsic risk factors that may predispose an athlete to EHI who has never had an EHI before. Therefore, providers should take the opportunity of the PPE to inquire about additional risk factors that may make an athlete high risk for sustaining a heat injury. Common risk factors for EHI are listed in Table 1.
Heat Acclimatization. The risk of EHI escalates significantly when athletes are subjected to multiple stressors during periods of heat exposure, such as sudden increases in intensity or duration of exercise; prolonged new exposures to heat; dehydration; and sleep loss.5 When football season begins in late summer, athletes are least conditioned as temperatures reach their seasonal peak, causing increased risk of EHI.15 Planning for heat acclimatization is vital for all athletes who exercise in hot environments. Acclimatization procedures place progressively mounting physiologic strains on the body to improve athletes’ ability to dissipate heat, diminishing thermoregulatory and cardiovascular exertion.4,5 Acclimatization begins with expansion of plasma volume on days 3 to 6, causing improvements in cardiac efficiency and resulting in an overall decrease in basal internal body temperature.4,5,15 This process results in improvements in heat tolerance and exercise performance, evolving over 10 to 14 days of gradual escalation of exercise intensity and duration.5,10,11,16 However, poor fitness levels and extreme temperatures can prolong this period, requiring up to 2 to 3 months to fully take effect.5,7
The National Athletic Trainers Association (NATA) and National Collegiate Athletic Association (NCAA) have released consensus guidelines regarding heat acclimatization protocols for football athletes at the high school and college levels (Tables 3 and 4). Each of these guidelines involves an initial period without use of protective equipment, followed by a gradual addition of further equipment.11,16
Secondary Prevention
Despite physicians’ best efforts to prevent all cases of EHI, athletes will still experience the effects of exercise-induced hyperthermia. The goal of secondary prevention is to slow the progression of this hyperthermia so that it does not progress to more dangerous EHI.
Hydration. Dehydration is an important risk factor for EHI. Sweat maintains thermoregulation by dissipating heat generated during exercise; however, it also contributes to body water losses. Furthermore, intravascular depletion decreases stroke volume, thereby increasing cardiovascular strain. It is estimated that for every 1% loss in body mass from dehydration, body temperature rises 0.22°C in comparison to a euhydrated state.6 Dehydration occurs more rapidly in hot environments, as fluid is lost through increased sweat production.7 After approximately 6% to 10% body weight volume loss, cardiac output cannot be maintained, diminishing sweat production and blood flow to both skin and muscle and causing diminished performance and a significant risk of heat exhaustion.7 If left unchecked, these physiologic changes result in further elevations in body temperature and increased cardiovascular strain, ultimately placing the athlete at significant risk for development of EHS.
Adequate hydration to maintain euvolemia is an important step in avoiding possible EHI. Multiple studies have shown that football players experience a baseline hypovolemia during their competition season,6 a deficit that is most marked during the first week of practices.17 This deficit is multifactorial, as football players expend a significant amount of fluid through sweat, are not able to adequately replace these losses during practice, and do not appropriately hydrate off the field.6,18 Some players, especially linemen, sweat at a higher rate than their teammates, posing a possible risk of significant dehydration.6 Coaches and players alike should be educated on the importance of adequate hydration to meet their fluid needs.
The goal of hydration during exercise is to prevent large fluid losses that can adversely affect performance and increase risk of EHI;6 it may be unrealistic to replace all fluid losses during the practice period. Instead, athletes should target complete volume replacement over the post-exercise period.6 Some recommend hydrating based upon thirst drive; however, thirst is activated following a volume loss of approximately 2% body mass, the same degree of losses that place athletes at an increased risk for performance impairment and EHI.4,6,11,12 Individuals should have access to fluids throughout practice and competition and be encouraged to hydrate as needed.6,12,15 Furthermore, staff should modify their practices based upon WBGT and acclimatization status to provide more frequent hydration breaks.
Hyperhydration and Salt Intake. Of note, there are inherent risks to hyperhydration. Athletes with low sweat rates have an increased risk of overhydration and the development of exercise-associated hyponatremia (EAH),6 a condition whose presentation is very similar to EHS. In addition, inadequate sodium intake and excessive sweating can also contribute to the development of EAH. EAH has been implicated in the deaths of 2 football players in 2014.1,6 Establishing team hydration guidelines and educating players and staff on appropriate hydration and dietary salt intake is essential to reduce the risk of both dehydration and hyperhydration and their complications.6Intra-Event Cooling. During exercise, team physicians can employ strategies for cooling athletes during exertion to mitigate their risk of EHI by decreasing thermal and cardiovascular strain.4,19 Cooling during exercise is hypothesized to allow for accelerated heat dissipation, where heat is lost from the body more effectively. This accelerated loss enables athletes to maintain a higher heat storage capacity over the duration of exercise, avoiding uncompensated heat stresses that ultimately cause EHI.19
Some intra-event cooling strategies include the use of cooling garments, cooling packs, and cold water/slurry ingestion. Cooling garments lower skin temperature, which in turn can decrease thermoregulatory strains;4 a recent meta-analysis of intra-event cooling modalities revealed that wearing an ice vest during exercise resulted in the greatest decrease in thermal heat strain.19 Internal cooling strategies—namely ingestion of cold fluids/ice slurry—have shown some mild benefit in decreasing internal temperatures; however, some studies have demonstrated some decrease in sweat production associated with cold oral intake used in isolation.19 Overall, studies have shown that combining external (cooling clothing, ice packs, fanning) and internal (cold water, ice slurry) cooling methods result in a greater cooling effect than a use of a single method.4
Tertiary Prevention
The goal of tertiary prevention is to mitigate the risk of long-term adverse outcomes following an EHS event. The most effective means of reducing risk for morbidity and mortality is rapid identification and treatment of EHS as well as close evaluation of an athlete’s return to activity in heat. This process is spearheaded by an effective and well-rehearsed emergency action plan.
Diagnosis and Management
Rapid identification and treatment of EHS is crucial to minimizing the risk of poor outcomes.7 Any delay in the treatment of EHS can dramatically increase the likelihood of associated morbidity and mortality.20
EHS is diagnosed by an elevated rectal temperature ≥40°C (104°F) and associated central nervous system (CNS) dysfunction.21 EHS should be strongly suspected in any athlete exercising in heat who exhibits signs of CNS dysfunction, including disorientation, confusion, dizziness, erratic behavior, irritability, headache, loss of coordination, delirium, collapse, or seizures.7,12,15 EHS may also present with symptoms of heat exhaustion, including fatigue, hyperventilation, tachycardia, vomiting, diarrhea, and hypotension.7,12,15
Rectal temperature should be taken for any athlete with suspected EHS, as other modalities—oral, skin, axillary, and aural—can be inaccurate and easily modified by ambient confounders such as ambient and skin temperature, athlete hyperventilation, and consumption of liquids.7,11,12 Athletes exhibiting CNS symptoms with moderately elevated rectal temperatures that do not exceed 40°C should also be assumed to be suffering from EHS and treated with rapid cooling.11 On the other hand, athletes with CNS symptoms who are normothermic should be assumed to have EAH until ruled out by electrolyte assessment; IV fluids should be at no more than keep vein open (KVO) pending this determination.11 In some cases, an athlete may initially present with altered mental status but return to “normal.” However, this improvement may represent a “lucid period”; evaluation should continue with rectal temperature and treatment, as EHS in these cases may progress quickly.15
Treatment is centered on rapid, whole body cooling initiated at the first sign of heat illness.7,22 The goal of treatment is to achieve a rectal temperature <38.9°C within 30 minutes of the onset of EHS.15 Upon diagnosis, the athlete should be quickly placed in a tub of ice water to facilitate cold water immersion (CWI) therapy. Some guidelines suggest the athlete’s clothing be removed to potentiate evaporative cooling during CWI;12 however, cooling should not be delayed due to difficulties in removing equipment. CWI, where a heat stroke victim is submerged in ice water up to their neck while water is continuously circulated, is generally considered to be the gold standard treatment as it is the modality with the highest recorded cooling rates and the lowest rate of morbidity and mortality.7,20,21 Multiple studies of CWI have shown that survival nears 100% when aggressive cooling starts within 5 minutes of collapse or identification of EHS.20,21,22
If whole body CWI is unavailable, alternative methods of rapid cooling should be employed. Partial CWI, with torso immersion being preferable to the extremities, has been shown to achieve an acceptable rate of cooling to achieve sufficient drops in internal body temperature.20,23 However, one popular treatment—applying ice packs to the whole body, in particular to the groin and axillae—has not been shown to be sufficient to achieve standard cooling goals.20 None of these methods have been shown to be as effective as CWI.23
Intravenous access should be initiated with fluid resuscitation dictated by the provider’s assessment. Normal saline is recommended as the resuscitative fluid of choice, with the rate dictated by clinical judgment and adjusted as guided by electrolyte determination and clinical response. It cannot be overstated that in normothermic patients with confusion, EAH is the diagnosis of exclusion and aggressive fluid resuscitation should be withheld until electrolyte determination.
Once rectal temperature descends appropriately (~38.9°C), the cooling process should stop and the individual should be transported to a hospital for further observation20 and evaluation of possible sequelae, including rhabdomyolysis and renal injury, cardiac dysfunction and arrhythmia, severe electrolyte abnormalities, acute respiratory distress syndrome, lactic acidosis, and other forms of end-organ failure (Figure).
Rapid cooling is more crucial than transport; transport poses a risk of delayed cooling, which can dramatically increase an individual’s risk of morbidity and mortality.20,23 In situations where a patient can be cooled on-site, physicians should pursue cooling before transporting the patient to a medical treatment facility.
Emergency Action Plan
Team physicians should be proactive in developing an emergency action plan to address possible EHS events. These plans should be site-specific, addressing procedures for all practice and home competition locations.12 All competition venues should have a CWI tub on-site in events where there is an increased risk of EHS.12,15,20 This tub should be set up and functional for all high-risk activities, including practices.12
Following recognition of a potential case of EHS, treatment teams should have procedures in place to transport athletes to the treatment area, obtain rectal temperature, initiate rapid cooling, and stabilize the athlete for transport to an emergency department (ED) for further evaluation.12,15 A written record of treatments and medications provided during athlete stabilization should be maintained and transported with the athlete to the ED.15 A list of helpful equipment and supplies for treatment of EHS can be found in Table 5.
EHS is a unique life-threatening situation where it is best to treat the patient on the sideline before transport.15 Those athletes transported before cooling risk spending an increased amount of time above critical temperatures for cell damage, which has been associated with increased morbidity and mortality. This mantra of “cool first, transport second” cannot be overemphasized, as those individuals with EHS who present to the ED with a persisting rectal temperature >41°F may risk up to an 80% mortality rate.24 Conversely, a recent large, retrospective study of 274 EHS events sustained during the Falmouth Road Race found a 100% survival rate when athletes were rapidly identified via rectal thermometry and treated with aggressive, rapid cooling through CWI.21
Return to Play
Perhaps the most challenging and important role the team physician has is determining an athlete’s return to play following EHI, as there currently are no evidence-based guidelines for return to activity for these athletes.7 The decisions surrounding return to play are highly individualized, as recovery from EHS and heat injury is associated with the duration of internal body temperature elevation above the critical level (40°C).7,20 Guidelines for return to activity following recovery from EHI differ among experts and institutions.7,25 The general consensus from these guidelines is that, at minimum, athletes should not participate in any physical activity until they are asymptomatic and all blood tests have normalized.11 Following this asymptomatic period, most guidelines advocate for a slow, deliberate return to activity.11 The American College of Sports Medicine (ACSM) offers one reasonable approach to the returning athlete following EHS:7
- No exercise for at least 7 days following release from medical care.
- Follow-up with a physician 1 week after release from medical care for physical examination and any warranted lab or radiologic studies (based upon organ systems affected during EHS).
- Once cleared to return to activity, the athlete begins exercise in a cool environment, gradually increasing the duration, intensity, and heat exposure over 2 weeks to demonstrate heat tolerance and acclimatization.
- Athletes who cannot resume vigorous activity due to recurrent symptoms (eg, excessive fatigue) should be reevaluated after 4 weeks. Laboratory exercise-heat tolerance testing may be useful in this setting.
- The athlete may resume full competition once they are able to participate in full training in the heat for 2 to 4 weeks without adverse effects.
Heat tolerance testing (HTT) in these athletes remains controversial.5 26 The ACSM recommends that HTT be considered only for those unable to return to vigorous activity after a suitable period (approximately 4 weeks). In contrast, the Israeli Defense Force (IDF) uses HTT to evaluate soldiers following EHS to guide decision-making about return to duty.27 The IDF HTT assumes that individuals will respond differently to heat stresses. They identify individuals who are “heat intolerant” as being unable to tolerate specific heat challenges, indicated by increases in body temperature occurring more rapidly than normal responders under identical environmental and exercise conditions. However, despite being used for more than 30 years, there is no clear evidence that HTT adequately predicts who will experience subsequent episodes of EHS.
Conclusion
While the recognized cornerstone of being a team physician is the provision of medical care, the ACSM Team Physician Consensus Statement28 further delineates the medical and administrative responsibilities as both (1) understanding medical management and prevention of injury and illness in athletes; and (2) awareness of or involvement in the development and rehearsal of an emergency action plan. These tenets are critical for the team physician who accepts the responsibility to cover sports at the high school level or higher. Football team physicians play an essential role in mitigating risk of EHI in their athletes. Through development and execution of both comprehensive prevention strategies and emergency action plans, physicians can work to minimize athletes’ risk of both developing and experiencing significant adverse outcomes from an EHI.
Am J Orthop. 2016;45(6):340-348. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
Football, one of the most popular sports in the United States, is additionally recognized as a leading contributor to sports injury secondary to the contact collision nature of the endeavor. There are an estimated 1.1 million high school football players with another 100,000 participants combined in the National Football League (NFL), college, junior college, Arena Football League, and semipro levels of play.1 USA Football estimates that an additional 3 million youth participate in community football leagues.1 The National Center for Catastrophic Sports Injury Research recently calculated a fatality rate of 0.14 per 100,000 participants in 2014 for the 4.2 million who play football at all levels—and 0.45 per 100,000 in high school.1 While direct deaths from head and spine injury remain a significant contributor to the number of catastrophic injuries, indirect deaths (systemic failure) predominate. Exertional heat stroke (EHS) has emerged as one of the leading indirect causes of death in high school and collegiate football. Boden and colleagues2 reported that high school and college football players sustain approximately 12 fatalities annually, with indirect systemic causes being twice as common as direct blunt trauma.2The most common indirect causes identified included cardiac failure, heat illness, and complications of sickle cell trait (SCT). It was also noted that the risk of SCT, heat-related, and cardiac deaths increased during the second decade of the study, indicating these conditions may require a greater emphasis on diagnosis, treatment, and prevention. This review details for the team physician the unique challenge of exercising in the heat to the football player, and the prevention, diagnosis, management and return-to-play issues pertinent to exertional heat illness (EHI).
The Challenge
EHS represents the most severe manifestation of EHI—a gamut of diseases commonly encountered during the hot summer months when American football season begins. The breadth of EHI includes several important clinical diagnoses: exercise-associated muscle cramps (heat cramps); heat exhaustion with and without syncope; heat injury with evidence of end organ injury (eg, rhabdomyolysis); and EHS. EHS is defined as “a form of hyperthermia associated with a systemic inflammatory response leading to a syndrome of multi-organ dysfunction in which encephalopathy predominates.”3 EHS, if left untreated, or even if clinical treatment is delayed, may result in significant end organ morbidity and/or mortality.
During exercise, the human thermoregulatory system mitigates heat gain by increasing skin blood flow and sweating, causing an increased dissipation of heat to the surrounding environment by leveraging conduction, convection, and evaporation.4,5 Elevated environmental temperatures, increased humidity, and dehydration can impede the body’s ability to dissipate heat at a rate needed to maintain thermoregulation. This imbalance can result in hyperthermia secondary to uncompensated heat stress,5 which in turn can lead to EHI. Football players have unique challenges that make them particularly vulnerable to EHI. The summer heat during early-season participation and the requirement for equipment that covers nearly 60% of body surfaces pose increased risk of volume losses and hyperthermia that trigger the onset of EHI.6 Football athletes’ body compositions and physical size are additional contributing risk factors; the relatively high muscle and fat content increase thermogenicity, which require their bodies to dissipate more heat.7
An estimated 9000 cases of EHI occur annually across all high school sports,8 with an incidence of 1.6:100,000 athlete-exposures.8,9 Studies have demonstrated, however, that EHI occurs in football 11.4 times more often than in all other high school sports combined.10 The incidence of nonfatal EHI in all levels of football is 4.42-5:100,000.8,9 Between 2000 and 2014, 41 football players died from EHS.1 In football, approximately 75% of all EHI events occurred during practices, while only 25% of incidents occurred during games.8
Given these potentially deadly consequences, it is important that football team physicians are not only alert to the early symptoms of heat illness and prepared to intervene to prevent the progression to EHS, but are critical leaders in educating coaches and players in evidence-based EHI prevention practices and policies.
Prevention
EHS is a preventable condition, arguably the most common cause of preventable nontraumatic exertional death in young athletes in the United States. Close attention to mitigating risk factors should begin prior to the onset of preseason practice and continue through the early season, where athletes are at the highest risk of developing heat illness.
Primary Prevention
Primary prevention is fundamental to minimizing the occurrences of EHI. It focuses on the following methods: recognition of inherent risk factors, acclimatization, hydration, and avoidance of inciting substances (including supplements).
Pre-Participation Examination. The purpose of the pre-participation examination (PPE) is to maximize an athlete’s safety by identifying medical conditions that place the athlete at risk.11,12 The Preparticipation Physical Evaluation, 4th edition, the most widely used consensus publication, specifically queries if an athlete has a previous history of heat injury. However, it only indirectly addresses intrinsic risk factors that may predispose an athlete to EHI who has never had an EHI before. Therefore, providers should take the opportunity of the PPE to inquire about additional risk factors that may make an athlete high risk for sustaining a heat injury. Common risk factors for EHI are listed in Table 1.
Heat Acclimatization. The risk of EHI escalates significantly when athletes are subjected to multiple stressors during periods of heat exposure, such as sudden increases in intensity or duration of exercise; prolonged new exposures to heat; dehydration; and sleep loss.5 When football season begins in late summer, athletes are least conditioned as temperatures reach their seasonal peak, causing increased risk of EHI.15 Planning for heat acclimatization is vital for all athletes who exercise in hot environments. Acclimatization procedures place progressively mounting physiologic strains on the body to improve athletes’ ability to dissipate heat, diminishing thermoregulatory and cardiovascular exertion.4,5 Acclimatization begins with expansion of plasma volume on days 3 to 6, causing improvements in cardiac efficiency and resulting in an overall decrease in basal internal body temperature.4,5,15 This process results in improvements in heat tolerance and exercise performance, evolving over 10 to 14 days of gradual escalation of exercise intensity and duration.5,10,11,16 However, poor fitness levels and extreme temperatures can prolong this period, requiring up to 2 to 3 months to fully take effect.5,7
The National Athletic Trainers Association (NATA) and National Collegiate Athletic Association (NCAA) have released consensus guidelines regarding heat acclimatization protocols for football athletes at the high school and college levels (Tables 3 and 4). Each of these guidelines involves an initial period without use of protective equipment, followed by a gradual addition of further equipment.11,16
Secondary Prevention
Despite physicians’ best efforts to prevent all cases of EHI, athletes will still experience the effects of exercise-induced hyperthermia. The goal of secondary prevention is to slow the progression of this hyperthermia so that it does not progress to more dangerous EHI.
Hydration. Dehydration is an important risk factor for EHI. Sweat maintains thermoregulation by dissipating heat generated during exercise; however, it also contributes to body water losses. Furthermore, intravascular depletion decreases stroke volume, thereby increasing cardiovascular strain. It is estimated that for every 1% loss in body mass from dehydration, body temperature rises 0.22°C in comparison to a euhydrated state.6 Dehydration occurs more rapidly in hot environments, as fluid is lost through increased sweat production.7 After approximately 6% to 10% body weight volume loss, cardiac output cannot be maintained, diminishing sweat production and blood flow to both skin and muscle and causing diminished performance and a significant risk of heat exhaustion.7 If left unchecked, these physiologic changes result in further elevations in body temperature and increased cardiovascular strain, ultimately placing the athlete at significant risk for development of EHS.
Adequate hydration to maintain euvolemia is an important step in avoiding possible EHI. Multiple studies have shown that football players experience a baseline hypovolemia during their competition season,6 a deficit that is most marked during the first week of practices.17 This deficit is multifactorial, as football players expend a significant amount of fluid through sweat, are not able to adequately replace these losses during practice, and do not appropriately hydrate off the field.6,18 Some players, especially linemen, sweat at a higher rate than their teammates, posing a possible risk of significant dehydration.6 Coaches and players alike should be educated on the importance of adequate hydration to meet their fluid needs.
The goal of hydration during exercise is to prevent large fluid losses that can adversely affect performance and increase risk of EHI;6 it may be unrealistic to replace all fluid losses during the practice period. Instead, athletes should target complete volume replacement over the post-exercise period.6 Some recommend hydrating based upon thirst drive; however, thirst is activated following a volume loss of approximately 2% body mass, the same degree of losses that place athletes at an increased risk for performance impairment and EHI.4,6,11,12 Individuals should have access to fluids throughout practice and competition and be encouraged to hydrate as needed.6,12,15 Furthermore, staff should modify their practices based upon WBGT and acclimatization status to provide more frequent hydration breaks.
Hyperhydration and Salt Intake. Of note, there are inherent risks to hyperhydration. Athletes with low sweat rates have an increased risk of overhydration and the development of exercise-associated hyponatremia (EAH),6 a condition whose presentation is very similar to EHS. In addition, inadequate sodium intake and excessive sweating can also contribute to the development of EAH. EAH has been implicated in the deaths of 2 football players in 2014.1,6 Establishing team hydration guidelines and educating players and staff on appropriate hydration and dietary salt intake is essential to reduce the risk of both dehydration and hyperhydration and their complications.6Intra-Event Cooling. During exercise, team physicians can employ strategies for cooling athletes during exertion to mitigate their risk of EHI by decreasing thermal and cardiovascular strain.4,19 Cooling during exercise is hypothesized to allow for accelerated heat dissipation, where heat is lost from the body more effectively. This accelerated loss enables athletes to maintain a higher heat storage capacity over the duration of exercise, avoiding uncompensated heat stresses that ultimately cause EHI.19
Some intra-event cooling strategies include the use of cooling garments, cooling packs, and cold water/slurry ingestion. Cooling garments lower skin temperature, which in turn can decrease thermoregulatory strains;4 a recent meta-analysis of intra-event cooling modalities revealed that wearing an ice vest during exercise resulted in the greatest decrease in thermal heat strain.19 Internal cooling strategies—namely ingestion of cold fluids/ice slurry—have shown some mild benefit in decreasing internal temperatures; however, some studies have demonstrated some decrease in sweat production associated with cold oral intake used in isolation.19 Overall, studies have shown that combining external (cooling clothing, ice packs, fanning) and internal (cold water, ice slurry) cooling methods result in a greater cooling effect than a use of a single method.4
Tertiary Prevention
The goal of tertiary prevention is to mitigate the risk of long-term adverse outcomes following an EHS event. The most effective means of reducing risk for morbidity and mortality is rapid identification and treatment of EHS as well as close evaluation of an athlete’s return to activity in heat. This process is spearheaded by an effective and well-rehearsed emergency action plan.
Diagnosis and Management
Rapid identification and treatment of EHS is crucial to minimizing the risk of poor outcomes.7 Any delay in the treatment of EHS can dramatically increase the likelihood of associated morbidity and mortality.20
EHS is diagnosed by an elevated rectal temperature ≥40°C (104°F) and associated central nervous system (CNS) dysfunction.21 EHS should be strongly suspected in any athlete exercising in heat who exhibits signs of CNS dysfunction, including disorientation, confusion, dizziness, erratic behavior, irritability, headache, loss of coordination, delirium, collapse, or seizures.7,12,15 EHS may also present with symptoms of heat exhaustion, including fatigue, hyperventilation, tachycardia, vomiting, diarrhea, and hypotension.7,12,15
Rectal temperature should be taken for any athlete with suspected EHS, as other modalities—oral, skin, axillary, and aural—can be inaccurate and easily modified by ambient confounders such as ambient and skin temperature, athlete hyperventilation, and consumption of liquids.7,11,12 Athletes exhibiting CNS symptoms with moderately elevated rectal temperatures that do not exceed 40°C should also be assumed to be suffering from EHS and treated with rapid cooling.11 On the other hand, athletes with CNS symptoms who are normothermic should be assumed to have EAH until ruled out by electrolyte assessment; IV fluids should be at no more than keep vein open (KVO) pending this determination.11 In some cases, an athlete may initially present with altered mental status but return to “normal.” However, this improvement may represent a “lucid period”; evaluation should continue with rectal temperature and treatment, as EHS in these cases may progress quickly.15
Treatment is centered on rapid, whole body cooling initiated at the first sign of heat illness.7,22 The goal of treatment is to achieve a rectal temperature <38.9°C within 30 minutes of the onset of EHS.15 Upon diagnosis, the athlete should be quickly placed in a tub of ice water to facilitate cold water immersion (CWI) therapy. Some guidelines suggest the athlete’s clothing be removed to potentiate evaporative cooling during CWI;12 however, cooling should not be delayed due to difficulties in removing equipment. CWI, where a heat stroke victim is submerged in ice water up to their neck while water is continuously circulated, is generally considered to be the gold standard treatment as it is the modality with the highest recorded cooling rates and the lowest rate of morbidity and mortality.7,20,21 Multiple studies of CWI have shown that survival nears 100% when aggressive cooling starts within 5 minutes of collapse or identification of EHS.20,21,22
If whole body CWI is unavailable, alternative methods of rapid cooling should be employed. Partial CWI, with torso immersion being preferable to the extremities, has been shown to achieve an acceptable rate of cooling to achieve sufficient drops in internal body temperature.20,23 However, one popular treatment—applying ice packs to the whole body, in particular to the groin and axillae—has not been shown to be sufficient to achieve standard cooling goals.20 None of these methods have been shown to be as effective as CWI.23
Intravenous access should be initiated with fluid resuscitation dictated by the provider’s assessment. Normal saline is recommended as the resuscitative fluid of choice, with the rate dictated by clinical judgment and adjusted as guided by electrolyte determination and clinical response. It cannot be overstated that in normothermic patients with confusion, EAH is the diagnosis of exclusion and aggressive fluid resuscitation should be withheld until electrolyte determination.
Once rectal temperature descends appropriately (~38.9°C), the cooling process should stop and the individual should be transported to a hospital for further observation20 and evaluation of possible sequelae, including rhabdomyolysis and renal injury, cardiac dysfunction and arrhythmia, severe electrolyte abnormalities, acute respiratory distress syndrome, lactic acidosis, and other forms of end-organ failure (Figure).
Rapid cooling is more crucial than transport; transport poses a risk of delayed cooling, which can dramatically increase an individual’s risk of morbidity and mortality.20,23 In situations where a patient can be cooled on-site, physicians should pursue cooling before transporting the patient to a medical treatment facility.
Emergency Action Plan
Team physicians should be proactive in developing an emergency action plan to address possible EHS events. These plans should be site-specific, addressing procedures for all practice and home competition locations.12 All competition venues should have a CWI tub on-site in events where there is an increased risk of EHS.12,15,20 This tub should be set up and functional for all high-risk activities, including practices.12
Following recognition of a potential case of EHS, treatment teams should have procedures in place to transport athletes to the treatment area, obtain rectal temperature, initiate rapid cooling, and stabilize the athlete for transport to an emergency department (ED) for further evaluation.12,15 A written record of treatments and medications provided during athlete stabilization should be maintained and transported with the athlete to the ED.15 A list of helpful equipment and supplies for treatment of EHS can be found in Table 5.
EHS is a unique life-threatening situation where it is best to treat the patient on the sideline before transport.15 Those athletes transported before cooling risk spending an increased amount of time above critical temperatures for cell damage, which has been associated with increased morbidity and mortality. This mantra of “cool first, transport second” cannot be overemphasized, as those individuals with EHS who present to the ED with a persisting rectal temperature >41°F may risk up to an 80% mortality rate.24 Conversely, a recent large, retrospective study of 274 EHS events sustained during the Falmouth Road Race found a 100% survival rate when athletes were rapidly identified via rectal thermometry and treated with aggressive, rapid cooling through CWI.21
Return to Play
Perhaps the most challenging and important role the team physician has is determining an athlete’s return to play following EHI, as there currently are no evidence-based guidelines for return to activity for these athletes.7 The decisions surrounding return to play are highly individualized, as recovery from EHS and heat injury is associated with the duration of internal body temperature elevation above the critical level (40°C).7,20 Guidelines for return to activity following recovery from EHI differ among experts and institutions.7,25 The general consensus from these guidelines is that, at minimum, athletes should not participate in any physical activity until they are asymptomatic and all blood tests have normalized.11 Following this asymptomatic period, most guidelines advocate for a slow, deliberate return to activity.11 The American College of Sports Medicine (ACSM) offers one reasonable approach to the returning athlete following EHS:7
- No exercise for at least 7 days following release from medical care.
- Follow-up with a physician 1 week after release from medical care for physical examination and any warranted lab or radiologic studies (based upon organ systems affected during EHS).
- Once cleared to return to activity, the athlete begins exercise in a cool environment, gradually increasing the duration, intensity, and heat exposure over 2 weeks to demonstrate heat tolerance and acclimatization.
- Athletes who cannot resume vigorous activity due to recurrent symptoms (eg, excessive fatigue) should be reevaluated after 4 weeks. Laboratory exercise-heat tolerance testing may be useful in this setting.
- The athlete may resume full competition once they are able to participate in full training in the heat for 2 to 4 weeks without adverse effects.
Heat tolerance testing (HTT) in these athletes remains controversial.5 26 The ACSM recommends that HTT be considered only for those unable to return to vigorous activity after a suitable period (approximately 4 weeks). In contrast, the Israeli Defense Force (IDF) uses HTT to evaluate soldiers following EHS to guide decision-making about return to duty.27 The IDF HTT assumes that individuals will respond differently to heat stresses. They identify individuals who are “heat intolerant” as being unable to tolerate specific heat challenges, indicated by increases in body temperature occurring more rapidly than normal responders under identical environmental and exercise conditions. However, despite being used for more than 30 years, there is no clear evidence that HTT adequately predicts who will experience subsequent episodes of EHS.
Conclusion
While the recognized cornerstone of being a team physician is the provision of medical care, the ACSM Team Physician Consensus Statement28 further delineates the medical and administrative responsibilities as both (1) understanding medical management and prevention of injury and illness in athletes; and (2) awareness of or involvement in the development and rehearsal of an emergency action plan. These tenets are critical for the team physician who accepts the responsibility to cover sports at the high school level or higher. Football team physicians play an essential role in mitigating risk of EHI in their athletes. Through development and execution of both comprehensive prevention strategies and emergency action plans, physicians can work to minimize athletes’ risk of both developing and experiencing significant adverse outcomes from an EHI.
Am J Orthop. 2016;45(6):340-348. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
1. Kucera KL, Klossner D, Colgate B, Cantu RC. Annual Survey of Football Injury Research: 1931-2014. National Center for Catastrophic Sport Injury Research Web site. https://nccsir.unc.edu/files/2013/10/Annual-Football-2014-Fatalities-Final.pdf. Accessed May 31, 2016.
2. Boden BP, Breit I, Beachler JA, Williams A, Mueller FO. Fatalities in high school and college football players. Am J Sports Med. 2013;41(5):1108-1116.
3. Bouchama A, Knochel JP. Heat stroke. N Engl J Med. 2002;346(25):1978-1988.
4. Racinais S, Alonso JM, Coutts AJ, et al. Consensus recommendations on training and competing in the heat. Scand J Med Sci Sports. 2015;25 Suppl 1:6-19.
5. Pryor RR, Casa DJ, Adams WM, et al. Maximizing athletic performance in the heat. Strength Cond J. 2013;35(6):24-33.
6. Adams WM, Casa DJ. Hydration for football athletes. Sports Sci Exchange. 2015;28(141):1-5.
7. American College of Sports Medicine, Armstrong LE, Casa DJ, et al. American College of Sports Medicine position stand. Exertional heat illness during training and competition. Med Sci Sports Exerc. 2007;39(3):556-572.
8. Yard EE, Gilchrist J, Haileyesus T, et al. Heat illness among high school athletes--United States, 2005-2009. J Safety Res. 2010;41(6):471-474.
9. Huffman EA, Yard EE, Fields SK, Collins CL, Comstock RD. Epidemiology of rare injuries and conditions among United States high school athletes during the 2005-2006 and 2006-2007 school years. J Athl Train. 2008;43(6):624-630.
10. Kerr ZY, Casa DJ, Marshall SW, Comstock RD. Epidemiology of exertional heat illness among U.S. high school athletes. Am J Prev Med. 2013;44(1):8-14.
11. Casa DJ, DeMartini JK, Bergeron MF, et al. National Athletic Trainers’ Association position statement: exertional heat illnesses. J Athl Train. 2015;50(9):986-1000.
12. Casa DJ, Almquist J, Anderson SA. The inter-association task force for preventing sudden death in secondary school athletics programs: best-practices recommendations. J Athl Train. 2013;48(4):546-553.
13. Gardner JW, Kark JA, Karnei K, et al. Risk factors predicting exertional heat illness in male Marine Corps recruits. Med Sci Sports Exerc. 1996;28(8):939-944.
14. Gundstein AJ, Ramseyer C, Zhao F, et al. A retrospective analysis of American football hyperthermia deaths in the United States. Int J Biometerol. 2012;56(1):11-20.
15. Armstrong LE, Johnson EC, Casa DJ, et al. The American football uniform: uncompensable heat stress and hyperthermic exhaustion. J Athl Train. 2010;45(2):117-127.
16. Casa DJ, Csillan D; Inter-Association Task Force for Preseason Secondary School Athletics Participants, et al. Preseason heat-acclimatization guidelines for secondary school athletics. J Athl Train. 2009;44(3):332-333.
17. Godek SF, Godek JJ, Bartolozzi AR. Hydration status in college football players during consecutive days of twice-a-day preseason practices. Am J Sports Med. 2005;33(6):843-851.
18. Stover EA, Zachwieja J, Stofan J, Murray R, Horswill CA. Consistently high urine specific gravity in adolescent American football players and the impact of an acute drinking strategy. Int J Sports Med. 2006;27(4):330-335.
19. Bongers CC, Thijssen DH, Veltmeijer MTW, Hopman MT, Eijsvogels TM. Precooling and percooling (cooling during exercise) both improve performance in the heat: a meta-analytical review. Br J Sports Med. 2015;49(6):377-384.
20. Casa DJ, McDermott BP, Lee EC, Yeargin SW, Armstrong LE, Maresh CM. Cold water immersion: the gold standard for exertional heatstroke treatment. Exerc Sport Sci Rev. 2007;35(3):141-149.
21. DeMartini JK, Casa DJ, Stearns R, et al. Effectiveness of cold water immersion in the treatment of exertional heat stroke at the Falmouth Road Race. Med Sci Sports Exerc. 2015;47(2):240-245.
22. Casa DJ, Kenny GP, Taylor NA. Immersion treatment for exertional hyperthermia: cold or temperate water? Med Sci Sports Exerc. 2010;42(7):1246-1252.
23. Casa DJ, Armstrong LE, Kenny GP, O’Connor FG, Huggins RA. Exertional heat stroke: new concepts regarding cause and care. Curr Sports Med Rep. 2012;11(3):115-122.
24. Argaud L, Ferry T, Le QH, et al. Short- and long-term outcomes of heat stroke following the 2003 heat wave in Lyon, France. Arch Intern Med. 2007;167(20):2177-2183.
25. O’Connor FG, Casa DJ, Bergeron MF, et al. American College of Sports Medicine Roundtable on exertional heat stroke--return to duty/return to play: conference proceedings. Curr Sports Med Rep. 2010;9(5):314-321.
26. Kazman JB, Heled Y, Lisman PJ, Druyan A, Deuster PA, O’Connor FG. Exertional heat illness: the role of heat tolerance testing. Curr Sports Med Rep. 2013;12(2):101-105.
27. Moran DS, Heled Y, Still L, Laor A, Shapiro Y. Assessment of heat tolerance for post exertional heat stroke individuals. Med Sci Monit. 2004;10(6):CR252-CR257.
28. Herring SA, Kibler WB, Putukian M. Team Physician Consensus Statement: 2013 update. Med Sci Sports Exerc. 2013;45(8):1618-1622.
29. Heat stroke treatment. Korey Stringer Institute University of Connecticut Web site. http://ksi.uconn.edu/emergency-conditions/heat-illnesses/exertional-heat-stroke/heat-stroke-treatment/. Accessed June 14, 2016.
30. Headquarters, Department of the Army and the Air Force. Heat Stress Control and Heat Casualty Management. Technical Bulletin Medical 507. http://www.dir.ca.gov/oshsb/documents/Heat_illness_prevention_tbmed507.pdf. Published March 7, 2003. Accessed June 14, 2016.
1. Kucera KL, Klossner D, Colgate B, Cantu RC. Annual Survey of Football Injury Research: 1931-2014. National Center for Catastrophic Sport Injury Research Web site. https://nccsir.unc.edu/files/2013/10/Annual-Football-2014-Fatalities-Final.pdf. Accessed May 31, 2016.
2. Boden BP, Breit I, Beachler JA, Williams A, Mueller FO. Fatalities in high school and college football players. Am J Sports Med. 2013;41(5):1108-1116.
3. Bouchama A, Knochel JP. Heat stroke. N Engl J Med. 2002;346(25):1978-1988.
4. Racinais S, Alonso JM, Coutts AJ, et al. Consensus recommendations on training and competing in the heat. Scand J Med Sci Sports. 2015;25 Suppl 1:6-19.
5. Pryor RR, Casa DJ, Adams WM, et al. Maximizing athletic performance in the heat. Strength Cond J. 2013;35(6):24-33.
6. Adams WM, Casa DJ. Hydration for football athletes. Sports Sci Exchange. 2015;28(141):1-5.
7. American College of Sports Medicine, Armstrong LE, Casa DJ, et al. American College of Sports Medicine position stand. Exertional heat illness during training and competition. Med Sci Sports Exerc. 2007;39(3):556-572.
8. Yard EE, Gilchrist J, Haileyesus T, et al. Heat illness among high school athletes--United States, 2005-2009. J Safety Res. 2010;41(6):471-474.
9. Huffman EA, Yard EE, Fields SK, Collins CL, Comstock RD. Epidemiology of rare injuries and conditions among United States high school athletes during the 2005-2006 and 2006-2007 school years. J Athl Train. 2008;43(6):624-630.
10. Kerr ZY, Casa DJ, Marshall SW, Comstock RD. Epidemiology of exertional heat illness among U.S. high school athletes. Am J Prev Med. 2013;44(1):8-14.
11. Casa DJ, DeMartini JK, Bergeron MF, et al. National Athletic Trainers’ Association position statement: exertional heat illnesses. J Athl Train. 2015;50(9):986-1000.
12. Casa DJ, Almquist J, Anderson SA. The inter-association task force for preventing sudden death in secondary school athletics programs: best-practices recommendations. J Athl Train. 2013;48(4):546-553.
13. Gardner JW, Kark JA, Karnei K, et al. Risk factors predicting exertional heat illness in male Marine Corps recruits. Med Sci Sports Exerc. 1996;28(8):939-944.
14. Gundstein AJ, Ramseyer C, Zhao F, et al. A retrospective analysis of American football hyperthermia deaths in the United States. Int J Biometerol. 2012;56(1):11-20.
15. Armstrong LE, Johnson EC, Casa DJ, et al. The American football uniform: uncompensable heat stress and hyperthermic exhaustion. J Athl Train. 2010;45(2):117-127.
16. Casa DJ, Csillan D; Inter-Association Task Force for Preseason Secondary School Athletics Participants, et al. Preseason heat-acclimatization guidelines for secondary school athletics. J Athl Train. 2009;44(3):332-333.
17. Godek SF, Godek JJ, Bartolozzi AR. Hydration status in college football players during consecutive days of twice-a-day preseason practices. Am J Sports Med. 2005;33(6):843-851.
18. Stover EA, Zachwieja J, Stofan J, Murray R, Horswill CA. Consistently high urine specific gravity in adolescent American football players and the impact of an acute drinking strategy. Int J Sports Med. 2006;27(4):330-335.
19. Bongers CC, Thijssen DH, Veltmeijer MTW, Hopman MT, Eijsvogels TM. Precooling and percooling (cooling during exercise) both improve performance in the heat: a meta-analytical review. Br J Sports Med. 2015;49(6):377-384.
20. Casa DJ, McDermott BP, Lee EC, Yeargin SW, Armstrong LE, Maresh CM. Cold water immersion: the gold standard for exertional heatstroke treatment. Exerc Sport Sci Rev. 2007;35(3):141-149.
21. DeMartini JK, Casa DJ, Stearns R, et al. Effectiveness of cold water immersion in the treatment of exertional heat stroke at the Falmouth Road Race. Med Sci Sports Exerc. 2015;47(2):240-245.
22. Casa DJ, Kenny GP, Taylor NA. Immersion treatment for exertional hyperthermia: cold or temperate water? Med Sci Sports Exerc. 2010;42(7):1246-1252.
23. Casa DJ, Armstrong LE, Kenny GP, O’Connor FG, Huggins RA. Exertional heat stroke: new concepts regarding cause and care. Curr Sports Med Rep. 2012;11(3):115-122.
24. Argaud L, Ferry T, Le QH, et al. Short- and long-term outcomes of heat stroke following the 2003 heat wave in Lyon, France. Arch Intern Med. 2007;167(20):2177-2183.
25. O’Connor FG, Casa DJ, Bergeron MF, et al. American College of Sports Medicine Roundtable on exertional heat stroke--return to duty/return to play: conference proceedings. Curr Sports Med Rep. 2010;9(5):314-321.
26. Kazman JB, Heled Y, Lisman PJ, Druyan A, Deuster PA, O’Connor FG. Exertional heat illness: the role of heat tolerance testing. Curr Sports Med Rep. 2013;12(2):101-105.
27. Moran DS, Heled Y, Still L, Laor A, Shapiro Y. Assessment of heat tolerance for post exertional heat stroke individuals. Med Sci Monit. 2004;10(6):CR252-CR257.
28. Herring SA, Kibler WB, Putukian M. Team Physician Consensus Statement: 2013 update. Med Sci Sports Exerc. 2013;45(8):1618-1622.
29. Heat stroke treatment. Korey Stringer Institute University of Connecticut Web site. http://ksi.uconn.edu/emergency-conditions/heat-illnesses/exertional-heat-stroke/heat-stroke-treatment/. Accessed June 14, 2016.
30. Headquarters, Department of the Army and the Air Force. Heat Stress Control and Heat Casualty Management. Technical Bulletin Medical 507. http://www.dir.ca.gov/oshsb/documents/Heat_illness_prevention_tbmed507.pdf. Published March 7, 2003. Accessed June 14, 2016.

Woman, 36, With Fever and Malaise
IN THIS ARTICLE
- Clinical presentation and evaluation
- Terminology table
- Outcome for the case patient
A 36-year-old Bengali woman with a history of well-controlled diabetes presents to the emergency department with complaints of feeling “unwell” for about two weeks. She does not speak English, and a hospital-provided phone translator is used to obtain history and explain hospital course. The patient is vague regarding symptomatology, describing general malaise and tiredness. She says she became “much worse” two days ago and has shaking chills, sore throat, headache, and nonproductive cough, but she denies shortness of breath or chest pain. She also developed nausea and vomiting, stating, “I can’t keep anything down.”
She has not recently traveled out of the country and has no known sick contacts. Influenza activity is high in the area, and the patient has not received immunization. She had a “normal” menstrual period two weeks ago and firmly states, “There is no way I can be pregnant.” She admits to vaginal “spotting” off and on for the past two weeks without abdominal pain. She is married with six children and has no history of miscarriage, ectopic pregnancy, or induced abortion; she is not taking any form of birth control.
On exam, the patient is tachycardic, with a heart rate of 127 beats/min, and has a fever of 103.3°F. Blood pressure, respiratory rate, and pulse oximetry are normal. She appears unwell and dehydrated. Her mucous membranes are dry, but no skin rash is noted. There is no tonsillar swelling or exudate and no meningismus; the lung exam is clear, with no adventitious sounds. Abdominal exam demonstrates mild, generalized tenderness in the lower abdomen without peritoneal signs. No costovertebral angle tenderness is noted. Initial diagnostic considerations include sources of fever (eg, influenza, pneumonia, urinary tract infection, viral illness), or abdominal sources, such as appendicitis.
An upright anteroposterior chest x-ray shows no infiltrate, pleural effusion, or cardiomegaly. Laboratory results include a high white blood cell (WBC) count (16.9 k/mm3) with bandemia and normal electrolytes without anion gap. Rapid influenza A and B testing is negative. A urine pregnancy test is positive, and the urinalysis shows no infection but +2 ketones. Rh factor is positive. A serum quantitative β-hCG is 130,581 mIU/mL. Blood cultures are obtained, but results are not available.
Due to cultural differences, the patient is very reluctant to consent to a pelvic exam. After extensive counseling, she agrees to a bimanual exam only. The uterus is boggy and enlarged to about 12 weeks. There is exquisite uterine tenderness and purulent discharge on the gloved finger. The cervical os is closed, and there is scant bleeding.
A transvaginal ultrasound is obtained; it reveals a thickened endometrium with echogenicity, without increased vascularity, and no identifiable intrauterine pregnancy. The adnexa have no masses, and there is no free fluid in the endometrium (see Figures 1 and 2).
The patient is given broad-spectrum antibiotics and urgently transported to the operating room by Ob-Gyn for uterine evacuation. She is found to have a septic abortion due to retained products of conception (RPOC) from an incomplete miscarriage.
Continue for discussion >>
DISCUSSIONIt is not uncommon for a woman to miscarry a very early pregnancy and not realize she had been pregnant.1 Many attribute it to a “heavy” or unusual period. In one study, 11% of patients who denied the possibility of pregnancy were, in fact, pregnant.2
Miscarriage is a frequent outcome of early pregnancy; it is estimated that 11% to 20% of early pregnancies result in a spontaneous miscarriage.3-5 Most resolve without complications, but risk increases with gestational age. When they do occur, complications include RPOC, heavy prolonged bleeding, and endometritis. RPOC refers to placental or fetal tissue that remains in the uterus after a miscarriage, surgical abortion, or preterm/term delivery (see Table for additional terminology related to miscarriage and abortion). Because of increased morbidity, it is important to suspect RPOC after a known miscarriage or an induced abortion, or in a pregnant patient with bleeding.
Incidence and pathophysiologySeptic abortion is a relatively rare complication of miscarriage. It can refer to a spontaneous miscarriage complicated by a subsequent intrauterine infection, often caused by RPOC. Septic abortion is much more common after an induced abortion, in which there is instrumentation of the uterus.
The infection after a spontaneous miscarriage usually begins as endometritis. It involves the necrotic RPOC, which are prone to infection by the cervical and vaginal flora. It may spread further into the parametrium/myometrium and the peritoneal cavity. The infection may then progress to bacteremia and sepsis. Typical causative organisms include Escherichia coli, Enterobacter aerogenes, Proteus vulgaris, hemolytic streptococci, staphylococci, and some anaerobic organisms, including Clostridium perfringens.3
Death, although rare in developed countries, is usually secondary to the sequela of sepsis, including septic shock, renal failure, adult respiratory distress syndrome, and disseminated intravascular coagulation.3,6,7 Pelvic adhesions and hysterectomy are also possible outcomes of a septic abortion.
Continue for clinical presentation and evaluation >>
Clinical presentation and evaluationMany findings suggestive of septic abortion are nonspecific, such as bleeding, pain, uterine tenderness, and fever. A combination of historical risk, physical exam, and laboratory and ultrasound findings will often be needed to confirm the diagnosis.
Fever is never to be expected in an uncomplicated miscarriage. Vaginal bleeding and some cramping are common after miscarriage; women will bleed, on average, between eight and 11 days afterward.5 Women who fall outside the normal range and experience prolonged bleeding, heavy bleeding, or severe abdominal pain should be evaluated.
A workup for patients with a possible septic abortion should include a complete blood count, blood culture with additional laboratory investigation if there is concern for bacteremia/sepsis, and type and screen for Rh factor and for possible blood transfusion, if needed.
All patients with postabortion complications should be screened for Rh factor; Rho(D) immune globulin (RhoGAM) should be administered if results indicate that the patient is Rh-negative and unsensitized. A quantitative β-hCG level can be obtained to confirm pregnancy. A single measurement will not be helpful; β-hCG can remain positive for weeks after an uncomplicated miscarriage. On the other hand, a low level does not exclude RPOC—the RPOC, if necrotic, may remain in the uterus without secreting hormone. The trend of β-hCG over time can be helpful if the diagnosis is unclear.
A careful physical exam, including a pelvic exam, should be performed. Assess for uterine tenderness, peritoneal signs, and purulent discharge from the cervix. An open cervical os is suggestive of RPOC, as the cervix closes quickly after a complete miscarriage, but a closed cervical os does not exclude the possibility of RPOC or septic abortion. The amount of bleeding should be noted, along with any tissue or clots within the vaginal vault or cervix.
A pelvic ultrasound should be obtained in all patients concerning for a septic incomplete miscarriage. Ultrasound findings can be nonspecific, because small amounts of retained tissue can look like blood (a common finding after miscarriage). Ultrasound findings of heterogeneous, echogenic material within the uterus or a thick, irregular endometrium support a diagnosis of RPOC in patients considered at risk.8,9 Increased color Doppler flow is often seen with RPOC, but there may be decreased flow in the case of necrotic RPOC. Ultrasound findings consistent with RPOC in a febrile, ill patient suggest a septic abortion.
Continue for treatment and prognosis >>
Treatment and prognosisPatients with a septic abortion require immediate evacuation of the uterus to prevent deadly complications; antibiotics may not be able to perfuse to the necrotic source of infection.10 Suction curettage is less likely than sharp curettage to cause perforation.
Broad-spectrum antibiotics should be administered. The bacteria associated with a septic incomplete miscarriage are usually polymicrobial and represent the normal flora of the vagina and cervix. The choice of agents recommended is usually the same as for pelvic inflammatory disease.11
The treatment regimen typically includes clindamycin (900 mg IV q8h), plus gentamicin (5 mg/kg IV once a day), with or without ampicillin (2 g IV q4h).11,12 Alternatively, a combination of ampicillin, gentamicin, and metronidazole (500 mg IV q8h) can be used.
Further surgery, including laparotomy and possible hysterectomy, is indicated in patients who do not respond to uterine evacuation and parenteral antibiotics. Other possible complications requiring surgery include pelvic abscess, necrotizing Clostridium infections in the myometrium, and uterine perforation.
OUTCOME FOR THE CASE PATIENTThe patient was started on IV ampicillin, gentamicin, and clindamycin and taken promptly for a suction dilation and curettage. Pathology later showed a gestational sac with severe acute necrotizing chorioamnionitis and extensive bacterial growth. This confirmed the diagnosis of a septic, incomplete miscarriage.
Blood cultures remained without any growth, and the patient was afebrile on the second postop day. The WBC count and β-hCG level trended downward.
The patient was discharged on a 14-day course of oral doxycycline and metronidazole. She was then lost to further follow-up.
CONCLUSIONThe differential diagnosis in this ill, febrile patient was initially very broad. The importance of suspecting pregnancy in all women of childbearing age, especially those not using contraception, cannot be underestimated. The accuracy of patient history and recall of last menstrual period in determining the possibility of pregnancy is not sufficiently reliable.
1. Promislow JH, Baird DD, Wilcox AJ, et al. Bleeding following pregnancy loss prior to six weeks gestation. Hum Reprod. 2007;22(3):853-857.
2. Ramoska EA, Sacchetti AD, Nepp M. Reliability of patient history in determining the possibility of pregnancy. Ann Emerg Med. 1989;18(1):48-50.
3. Osazuwa H, Aziken M. Septic abortion: a review of social and demographic characteristics. Arch Gynecol Obstet. 2007;275(2):117-119.
4. Hure AJ, Powers JR, Mishra GD, et al. Miscarriage, preterm delivery, and stillbirth: large variations in rates within a cohort of Australian women. PLoS One. 2012;7(5):e37109.
5. Nielsen S, Hahlin M. Expectant management of first-trimester spontaneous abortion. Lancet. 1995;345(8942):84-86.
6. Eschenbach DA. Treating spontaneous and induced septic abortions. Obstet Gynecol. 2015;125(5):1042-1048.
7. Rana A, Pradhan N, Gurung G, Singh M. Induced septic abortion: a major factor in maternal mortality and morbidity. J Obstet Gynaecol Res. 2004;30(1):3-8.
8. Abbasi S, Jamal A, Eslamian L, Marsousi V. Role of clinical and ultrasound findings in the diagnosis of retained products of conception. Ultrasound Obstet Gynecol. 2008;32(5):704-707.
9. Esmaeillou H, Jamal A, Eslamian L, et al. Accurate detection of retained products of conception after first- and second-trimester abortion by color doppler sonography. J Med Ultrasound. 2015;23(7):34-38.
10. Finkielman JD, De Feo FD, Heller PG, Afessa B. The clinical course of patients with septic abortion admitted to an intensive care unit. Intensive Care Med. 2004;30(6):1097-1102.
11. CDC. Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep. 2010;59(RR-12):1-110.
12. Mackeen AD, Packard RE, Ota E, Speer L. Antibiotic regimens for postpartum endometritis. Cochrane Database Syst Rev. 2015;2:CD001067.
IN THIS ARTICLE
- Clinical presentation and evaluation
- Terminology table
- Outcome for the case patient
A 36-year-old Bengali woman with a history of well-controlled diabetes presents to the emergency department with complaints of feeling “unwell” for about two weeks. She does not speak English, and a hospital-provided phone translator is used to obtain history and explain hospital course. The patient is vague regarding symptomatology, describing general malaise and tiredness. She says she became “much worse” two days ago and has shaking chills, sore throat, headache, and nonproductive cough, but she denies shortness of breath or chest pain. She also developed nausea and vomiting, stating, “I can’t keep anything down.”
She has not recently traveled out of the country and has no known sick contacts. Influenza activity is high in the area, and the patient has not received immunization. She had a “normal” menstrual period two weeks ago and firmly states, “There is no way I can be pregnant.” She admits to vaginal “spotting” off and on for the past two weeks without abdominal pain. She is married with six children and has no history of miscarriage, ectopic pregnancy, or induced abortion; she is not taking any form of birth control.
On exam, the patient is tachycardic, with a heart rate of 127 beats/min, and has a fever of 103.3°F. Blood pressure, respiratory rate, and pulse oximetry are normal. She appears unwell and dehydrated. Her mucous membranes are dry, but no skin rash is noted. There is no tonsillar swelling or exudate and no meningismus; the lung exam is clear, with no adventitious sounds. Abdominal exam demonstrates mild, generalized tenderness in the lower abdomen without peritoneal signs. No costovertebral angle tenderness is noted. Initial diagnostic considerations include sources of fever (eg, influenza, pneumonia, urinary tract infection, viral illness), or abdominal sources, such as appendicitis.
An upright anteroposterior chest x-ray shows no infiltrate, pleural effusion, or cardiomegaly. Laboratory results include a high white blood cell (WBC) count (16.9 k/mm3) with bandemia and normal electrolytes without anion gap. Rapid influenza A and B testing is negative. A urine pregnancy test is positive, and the urinalysis shows no infection but +2 ketones. Rh factor is positive. A serum quantitative β-hCG is 130,581 mIU/mL. Blood cultures are obtained, but results are not available.
Due to cultural differences, the patient is very reluctant to consent to a pelvic exam. After extensive counseling, she agrees to a bimanual exam only. The uterus is boggy and enlarged to about 12 weeks. There is exquisite uterine tenderness and purulent discharge on the gloved finger. The cervical os is closed, and there is scant bleeding.
A transvaginal ultrasound is obtained; it reveals a thickened endometrium with echogenicity, without increased vascularity, and no identifiable intrauterine pregnancy. The adnexa have no masses, and there is no free fluid in the endometrium (see Figures 1 and 2).
The patient is given broad-spectrum antibiotics and urgently transported to the operating room by Ob-Gyn for uterine evacuation. She is found to have a septic abortion due to retained products of conception (RPOC) from an incomplete miscarriage.
Continue for discussion >>
DISCUSSIONIt is not uncommon for a woman to miscarry a very early pregnancy and not realize she had been pregnant.1 Many attribute it to a “heavy” or unusual period. In one study, 11% of patients who denied the possibility of pregnancy were, in fact, pregnant.2
Miscarriage is a frequent outcome of early pregnancy; it is estimated that 11% to 20% of early pregnancies result in a spontaneous miscarriage.3-5 Most resolve without complications, but risk increases with gestational age. When they do occur, complications include RPOC, heavy prolonged bleeding, and endometritis. RPOC refers to placental or fetal tissue that remains in the uterus after a miscarriage, surgical abortion, or preterm/term delivery (see Table for additional terminology related to miscarriage and abortion). Because of increased morbidity, it is important to suspect RPOC after a known miscarriage or an induced abortion, or in a pregnant patient with bleeding.
Incidence and pathophysiologySeptic abortion is a relatively rare complication of miscarriage. It can refer to a spontaneous miscarriage complicated by a subsequent intrauterine infection, often caused by RPOC. Septic abortion is much more common after an induced abortion, in which there is instrumentation of the uterus.
The infection after a spontaneous miscarriage usually begins as endometritis. It involves the necrotic RPOC, which are prone to infection by the cervical and vaginal flora. It may spread further into the parametrium/myometrium and the peritoneal cavity. The infection may then progress to bacteremia and sepsis. Typical causative organisms include Escherichia coli, Enterobacter aerogenes, Proteus vulgaris, hemolytic streptococci, staphylococci, and some anaerobic organisms, including Clostridium perfringens.3
Death, although rare in developed countries, is usually secondary to the sequela of sepsis, including septic shock, renal failure, adult respiratory distress syndrome, and disseminated intravascular coagulation.3,6,7 Pelvic adhesions and hysterectomy are also possible outcomes of a septic abortion.
Continue for clinical presentation and evaluation >>
Clinical presentation and evaluationMany findings suggestive of septic abortion are nonspecific, such as bleeding, pain, uterine tenderness, and fever. A combination of historical risk, physical exam, and laboratory and ultrasound findings will often be needed to confirm the diagnosis.
Fever is never to be expected in an uncomplicated miscarriage. Vaginal bleeding and some cramping are common after miscarriage; women will bleed, on average, between eight and 11 days afterward.5 Women who fall outside the normal range and experience prolonged bleeding, heavy bleeding, or severe abdominal pain should be evaluated.
A workup for patients with a possible septic abortion should include a complete blood count, blood culture with additional laboratory investigation if there is concern for bacteremia/sepsis, and type and screen for Rh factor and for possible blood transfusion, if needed.
All patients with postabortion complications should be screened for Rh factor; Rho(D) immune globulin (RhoGAM) should be administered if results indicate that the patient is Rh-negative and unsensitized. A quantitative β-hCG level can be obtained to confirm pregnancy. A single measurement will not be helpful; β-hCG can remain positive for weeks after an uncomplicated miscarriage. On the other hand, a low level does not exclude RPOC—the RPOC, if necrotic, may remain in the uterus without secreting hormone. The trend of β-hCG over time can be helpful if the diagnosis is unclear.
A careful physical exam, including a pelvic exam, should be performed. Assess for uterine tenderness, peritoneal signs, and purulent discharge from the cervix. An open cervical os is suggestive of RPOC, as the cervix closes quickly after a complete miscarriage, but a closed cervical os does not exclude the possibility of RPOC or septic abortion. The amount of bleeding should be noted, along with any tissue or clots within the vaginal vault or cervix.
A pelvic ultrasound should be obtained in all patients concerning for a septic incomplete miscarriage. Ultrasound findings can be nonspecific, because small amounts of retained tissue can look like blood (a common finding after miscarriage). Ultrasound findings of heterogeneous, echogenic material within the uterus or a thick, irregular endometrium support a diagnosis of RPOC in patients considered at risk.8,9 Increased color Doppler flow is often seen with RPOC, but there may be decreased flow in the case of necrotic RPOC. Ultrasound findings consistent with RPOC in a febrile, ill patient suggest a septic abortion.
Continue for treatment and prognosis >>
Treatment and prognosisPatients with a septic abortion require immediate evacuation of the uterus to prevent deadly complications; antibiotics may not be able to perfuse to the necrotic source of infection.10 Suction curettage is less likely than sharp curettage to cause perforation.
Broad-spectrum antibiotics should be administered. The bacteria associated with a septic incomplete miscarriage are usually polymicrobial and represent the normal flora of the vagina and cervix. The choice of agents recommended is usually the same as for pelvic inflammatory disease.11
The treatment regimen typically includes clindamycin (900 mg IV q8h), plus gentamicin (5 mg/kg IV once a day), with or without ampicillin (2 g IV q4h).11,12 Alternatively, a combination of ampicillin, gentamicin, and metronidazole (500 mg IV q8h) can be used.
Further surgery, including laparotomy and possible hysterectomy, is indicated in patients who do not respond to uterine evacuation and parenteral antibiotics. Other possible complications requiring surgery include pelvic abscess, necrotizing Clostridium infections in the myometrium, and uterine perforation.
OUTCOME FOR THE CASE PATIENTThe patient was started on IV ampicillin, gentamicin, and clindamycin and taken promptly for a suction dilation and curettage. Pathology later showed a gestational sac with severe acute necrotizing chorioamnionitis and extensive bacterial growth. This confirmed the diagnosis of a septic, incomplete miscarriage.
Blood cultures remained without any growth, and the patient was afebrile on the second postop day. The WBC count and β-hCG level trended downward.
The patient was discharged on a 14-day course of oral doxycycline and metronidazole. She was then lost to further follow-up.
CONCLUSIONThe differential diagnosis in this ill, febrile patient was initially very broad. The importance of suspecting pregnancy in all women of childbearing age, especially those not using contraception, cannot be underestimated. The accuracy of patient history and recall of last menstrual period in determining the possibility of pregnancy is not sufficiently reliable.
IN THIS ARTICLE
- Clinical presentation and evaluation
- Terminology table
- Outcome for the case patient
A 36-year-old Bengali woman with a history of well-controlled diabetes presents to the emergency department with complaints of feeling “unwell” for about two weeks. She does not speak English, and a hospital-provided phone translator is used to obtain history and explain hospital course. The patient is vague regarding symptomatology, describing general malaise and tiredness. She says she became “much worse” two days ago and has shaking chills, sore throat, headache, and nonproductive cough, but she denies shortness of breath or chest pain. She also developed nausea and vomiting, stating, “I can’t keep anything down.”
She has not recently traveled out of the country and has no known sick contacts. Influenza activity is high in the area, and the patient has not received immunization. She had a “normal” menstrual period two weeks ago and firmly states, “There is no way I can be pregnant.” She admits to vaginal “spotting” off and on for the past two weeks without abdominal pain. She is married with six children and has no history of miscarriage, ectopic pregnancy, or induced abortion; she is not taking any form of birth control.
On exam, the patient is tachycardic, with a heart rate of 127 beats/min, and has a fever of 103.3°F. Blood pressure, respiratory rate, and pulse oximetry are normal. She appears unwell and dehydrated. Her mucous membranes are dry, but no skin rash is noted. There is no tonsillar swelling or exudate and no meningismus; the lung exam is clear, with no adventitious sounds. Abdominal exam demonstrates mild, generalized tenderness in the lower abdomen without peritoneal signs. No costovertebral angle tenderness is noted. Initial diagnostic considerations include sources of fever (eg, influenza, pneumonia, urinary tract infection, viral illness), or abdominal sources, such as appendicitis.
An upright anteroposterior chest x-ray shows no infiltrate, pleural effusion, or cardiomegaly. Laboratory results include a high white blood cell (WBC) count (16.9 k/mm3) with bandemia and normal electrolytes without anion gap. Rapid influenza A and B testing is negative. A urine pregnancy test is positive, and the urinalysis shows no infection but +2 ketones. Rh factor is positive. A serum quantitative β-hCG is 130,581 mIU/mL. Blood cultures are obtained, but results are not available.
Due to cultural differences, the patient is very reluctant to consent to a pelvic exam. After extensive counseling, she agrees to a bimanual exam only. The uterus is boggy and enlarged to about 12 weeks. There is exquisite uterine tenderness and purulent discharge on the gloved finger. The cervical os is closed, and there is scant bleeding.
A transvaginal ultrasound is obtained; it reveals a thickened endometrium with echogenicity, without increased vascularity, and no identifiable intrauterine pregnancy. The adnexa have no masses, and there is no free fluid in the endometrium (see Figures 1 and 2).
The patient is given broad-spectrum antibiotics and urgently transported to the operating room by Ob-Gyn for uterine evacuation. She is found to have a septic abortion due to retained products of conception (RPOC) from an incomplete miscarriage.
Continue for discussion >>
DISCUSSIONIt is not uncommon for a woman to miscarry a very early pregnancy and not realize she had been pregnant.1 Many attribute it to a “heavy” or unusual period. In one study, 11% of patients who denied the possibility of pregnancy were, in fact, pregnant.2
Miscarriage is a frequent outcome of early pregnancy; it is estimated that 11% to 20% of early pregnancies result in a spontaneous miscarriage.3-5 Most resolve without complications, but risk increases with gestational age. When they do occur, complications include RPOC, heavy prolonged bleeding, and endometritis. RPOC refers to placental or fetal tissue that remains in the uterus after a miscarriage, surgical abortion, or preterm/term delivery (see Table for additional terminology related to miscarriage and abortion). Because of increased morbidity, it is important to suspect RPOC after a known miscarriage or an induced abortion, or in a pregnant patient with bleeding.
Incidence and pathophysiologySeptic abortion is a relatively rare complication of miscarriage. It can refer to a spontaneous miscarriage complicated by a subsequent intrauterine infection, often caused by RPOC. Septic abortion is much more common after an induced abortion, in which there is instrumentation of the uterus.
The infection after a spontaneous miscarriage usually begins as endometritis. It involves the necrotic RPOC, which are prone to infection by the cervical and vaginal flora. It may spread further into the parametrium/myometrium and the peritoneal cavity. The infection may then progress to bacteremia and sepsis. Typical causative organisms include Escherichia coli, Enterobacter aerogenes, Proteus vulgaris, hemolytic streptococci, staphylococci, and some anaerobic organisms, including Clostridium perfringens.3
Death, although rare in developed countries, is usually secondary to the sequela of sepsis, including septic shock, renal failure, adult respiratory distress syndrome, and disseminated intravascular coagulation.3,6,7 Pelvic adhesions and hysterectomy are also possible outcomes of a septic abortion.
Continue for clinical presentation and evaluation >>
Clinical presentation and evaluationMany findings suggestive of septic abortion are nonspecific, such as bleeding, pain, uterine tenderness, and fever. A combination of historical risk, physical exam, and laboratory and ultrasound findings will often be needed to confirm the diagnosis.
Fever is never to be expected in an uncomplicated miscarriage. Vaginal bleeding and some cramping are common after miscarriage; women will bleed, on average, between eight and 11 days afterward.5 Women who fall outside the normal range and experience prolonged bleeding, heavy bleeding, or severe abdominal pain should be evaluated.
A workup for patients with a possible septic abortion should include a complete blood count, blood culture with additional laboratory investigation if there is concern for bacteremia/sepsis, and type and screen for Rh factor and for possible blood transfusion, if needed.
All patients with postabortion complications should be screened for Rh factor; Rho(D) immune globulin (RhoGAM) should be administered if results indicate that the patient is Rh-negative and unsensitized. A quantitative β-hCG level can be obtained to confirm pregnancy. A single measurement will not be helpful; β-hCG can remain positive for weeks after an uncomplicated miscarriage. On the other hand, a low level does not exclude RPOC—the RPOC, if necrotic, may remain in the uterus without secreting hormone. The trend of β-hCG over time can be helpful if the diagnosis is unclear.
A careful physical exam, including a pelvic exam, should be performed. Assess for uterine tenderness, peritoneal signs, and purulent discharge from the cervix. An open cervical os is suggestive of RPOC, as the cervix closes quickly after a complete miscarriage, but a closed cervical os does not exclude the possibility of RPOC or septic abortion. The amount of bleeding should be noted, along with any tissue or clots within the vaginal vault or cervix.
A pelvic ultrasound should be obtained in all patients concerning for a septic incomplete miscarriage. Ultrasound findings can be nonspecific, because small amounts of retained tissue can look like blood (a common finding after miscarriage). Ultrasound findings of heterogeneous, echogenic material within the uterus or a thick, irregular endometrium support a diagnosis of RPOC in patients considered at risk.8,9 Increased color Doppler flow is often seen with RPOC, but there may be decreased flow in the case of necrotic RPOC. Ultrasound findings consistent with RPOC in a febrile, ill patient suggest a septic abortion.
Continue for treatment and prognosis >>
Treatment and prognosisPatients with a septic abortion require immediate evacuation of the uterus to prevent deadly complications; antibiotics may not be able to perfuse to the necrotic source of infection.10 Suction curettage is less likely than sharp curettage to cause perforation.
Broad-spectrum antibiotics should be administered. The bacteria associated with a septic incomplete miscarriage are usually polymicrobial and represent the normal flora of the vagina and cervix. The choice of agents recommended is usually the same as for pelvic inflammatory disease.11
The treatment regimen typically includes clindamycin (900 mg IV q8h), plus gentamicin (5 mg/kg IV once a day), with or without ampicillin (2 g IV q4h).11,12 Alternatively, a combination of ampicillin, gentamicin, and metronidazole (500 mg IV q8h) can be used.
Further surgery, including laparotomy and possible hysterectomy, is indicated in patients who do not respond to uterine evacuation and parenteral antibiotics. Other possible complications requiring surgery include pelvic abscess, necrotizing Clostridium infections in the myometrium, and uterine perforation.
OUTCOME FOR THE CASE PATIENTThe patient was started on IV ampicillin, gentamicin, and clindamycin and taken promptly for a suction dilation and curettage. Pathology later showed a gestational sac with severe acute necrotizing chorioamnionitis and extensive bacterial growth. This confirmed the diagnosis of a septic, incomplete miscarriage.
Blood cultures remained without any growth, and the patient was afebrile on the second postop day. The WBC count and β-hCG level trended downward.
The patient was discharged on a 14-day course of oral doxycycline and metronidazole. She was then lost to further follow-up.
CONCLUSIONThe differential diagnosis in this ill, febrile patient was initially very broad. The importance of suspecting pregnancy in all women of childbearing age, especially those not using contraception, cannot be underestimated. The accuracy of patient history and recall of last menstrual period in determining the possibility of pregnancy is not sufficiently reliable.
1. Promislow JH, Baird DD, Wilcox AJ, et al. Bleeding following pregnancy loss prior to six weeks gestation. Hum Reprod. 2007;22(3):853-857.
2. Ramoska EA, Sacchetti AD, Nepp M. Reliability of patient history in determining the possibility of pregnancy. Ann Emerg Med. 1989;18(1):48-50.
3. Osazuwa H, Aziken M. Septic abortion: a review of social and demographic characteristics. Arch Gynecol Obstet. 2007;275(2):117-119.
4. Hure AJ, Powers JR, Mishra GD, et al. Miscarriage, preterm delivery, and stillbirth: large variations in rates within a cohort of Australian women. PLoS One. 2012;7(5):e37109.
5. Nielsen S, Hahlin M. Expectant management of first-trimester spontaneous abortion. Lancet. 1995;345(8942):84-86.
6. Eschenbach DA. Treating spontaneous and induced septic abortions. Obstet Gynecol. 2015;125(5):1042-1048.
7. Rana A, Pradhan N, Gurung G, Singh M. Induced septic abortion: a major factor in maternal mortality and morbidity. J Obstet Gynaecol Res. 2004;30(1):3-8.
8. Abbasi S, Jamal A, Eslamian L, Marsousi V. Role of clinical and ultrasound findings in the diagnosis of retained products of conception. Ultrasound Obstet Gynecol. 2008;32(5):704-707.
9. Esmaeillou H, Jamal A, Eslamian L, et al. Accurate detection of retained products of conception after first- and second-trimester abortion by color doppler sonography. J Med Ultrasound. 2015;23(7):34-38.
10. Finkielman JD, De Feo FD, Heller PG, Afessa B. The clinical course of patients with septic abortion admitted to an intensive care unit. Intensive Care Med. 2004;30(6):1097-1102.
11. CDC. Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep. 2010;59(RR-12):1-110.
12. Mackeen AD, Packard RE, Ota E, Speer L. Antibiotic regimens for postpartum endometritis. Cochrane Database Syst Rev. 2015;2:CD001067.
1. Promislow JH, Baird DD, Wilcox AJ, et al. Bleeding following pregnancy loss prior to six weeks gestation. Hum Reprod. 2007;22(3):853-857.
2. Ramoska EA, Sacchetti AD, Nepp M. Reliability of patient history in determining the possibility of pregnancy. Ann Emerg Med. 1989;18(1):48-50.
3. Osazuwa H, Aziken M. Septic abortion: a review of social and demographic characteristics. Arch Gynecol Obstet. 2007;275(2):117-119.
4. Hure AJ, Powers JR, Mishra GD, et al. Miscarriage, preterm delivery, and stillbirth: large variations in rates within a cohort of Australian women. PLoS One. 2012;7(5):e37109.
5. Nielsen S, Hahlin M. Expectant management of first-trimester spontaneous abortion. Lancet. 1995;345(8942):84-86.
6. Eschenbach DA. Treating spontaneous and induced septic abortions. Obstet Gynecol. 2015;125(5):1042-1048.
7. Rana A, Pradhan N, Gurung G, Singh M. Induced septic abortion: a major factor in maternal mortality and morbidity. J Obstet Gynaecol Res. 2004;30(1):3-8.
8. Abbasi S, Jamal A, Eslamian L, Marsousi V. Role of clinical and ultrasound findings in the diagnosis of retained products of conception. Ultrasound Obstet Gynecol. 2008;32(5):704-707.
9. Esmaeillou H, Jamal A, Eslamian L, et al. Accurate detection of retained products of conception after first- and second-trimester abortion by color doppler sonography. J Med Ultrasound. 2015;23(7):34-38.
10. Finkielman JD, De Feo FD, Heller PG, Afessa B. The clinical course of patients with septic abortion admitted to an intensive care unit. Intensive Care Med. 2004;30(6):1097-1102.
11. CDC. Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep. 2010;59(RR-12):1-110.
12. Mackeen AD, Packard RE, Ota E, Speer L. Antibiotic regimens for postpartum endometritis. Cochrane Database Syst Rev. 2015;2:CD001067.