User login
Prostate cancer incidence continues to decrease after recommendation against screening
Incidence rates for localized- and regional-stage prostate cancer continued to decline 2 years following the recommendation by the U.S. Preventive Services Task Force against prostate-specific antigen (PSA) testing in all men.
“Convincing evidence demonstrates that the PSA test often produces false-positive results [and] false-positive PSA test results are associated with negative psychological effects, including persistent worry about prostate cancer,” the task force stated in a recommendation published in October 2011 and finalized in May 2012.

From 2011 to 2012, immediately following the recommendation, there was a significant decline in early-stage cancer incidence rates among men 50 years or older, according to an analysis of data from the Surveillance, Epidemiology, and End Results (SEER) program.
For the current study, Ahmedin Jemal, DVM, PhD, and his associates at the American Cancer Society analyzed incidence data for invasive prostate cancer from 18 SEER registries, which, combined, represented about 28% of the U.S. population.
Investigators reported a continued decline in localized- and regional-stage prostate cancer incidence from 2012 to 2013. Specifically, the incidence rates per 100,000 men decreased from 356.5 to 335.4 in men aged 50-74 years and from 379.2 to 353.6 in men 75 years and older (JAMA Oncol. 2016 Aug 16. doi: 10.1001/jamaoncol.2016.2667).
Incidence rates for distant-stage disease were unchanged during the same time period for men of all ages.
Similar results were reported for non-Hispanic whites and non-Hispanic blacks.
“Whether this pattern will lead to a future increase in the diagnosis of distant-stage disease and prostate cancer mortality requires long-term monitoring because of the slow growing nature of this malignant neoplasm,” the investigators noted.
The American Cancer Society funded the study. The authors had no relevant disclosures to report.
On Twitter @jessnicolecraig
Incidence rates for localized- and regional-stage prostate cancer continued to decline 2 years following the recommendation by the U.S. Preventive Services Task Force against prostate-specific antigen (PSA) testing in all men.
“Convincing evidence demonstrates that the PSA test often produces false-positive results [and] false-positive PSA test results are associated with negative psychological effects, including persistent worry about prostate cancer,” the task force stated in a recommendation published in October 2011 and finalized in May 2012.
From 2011 to 2012, immediately following the recommendation, there was a significant decline in early-stage cancer incidence rates among men 50 years or older, according to an analysis of data from the Surveillance, Epidemiology, and End Results (SEER) program.
For the current study, Ahmedin Jemal, DVM, PhD, and his associates at the American Cancer Society analyzed incidence data for invasive prostate cancer from 18 SEER registries, which, combined, represented about 28% of the U.S. population.
Investigators reported a continued decline in localized- and regional-stage prostate cancer incidence from 2012 to 2013. Specifically, the incidence rates per 100,000 men decreased from 356.5 to 335.4 in men aged 50-74 years and from 379.2 to 353.6 in men 75 years and older (JAMA Oncol. 2016 Aug 16. doi: 10.1001/jamaoncol.2016.2667).
Incidence rates for distant-stage disease were unchanged during the same time period for men of all ages.
Similar results were reported for non-Hispanic whites and non-Hispanic blacks.
“Whether this pattern will lead to a future increase in the diagnosis of distant-stage disease and prostate cancer mortality requires long-term monitoring because of the slow growing nature of this malignant neoplasm,” the investigators noted.
The American Cancer Society funded the study. The authors had no relevant disclosures to report.
On Twitter @jessnicolecraig
Incidence rates for localized- and regional-stage prostate cancer continued to decline 2 years following the recommendation by the U.S. Preventive Services Task Force against prostate-specific antigen (PSA) testing in all men.
“Convincing evidence demonstrates that the PSA test often produces false-positive results [and] false-positive PSA test results are associated with negative psychological effects, including persistent worry about prostate cancer,” the task force stated in a recommendation published in October 2011 and finalized in May 2012.
From 2011 to 2012, immediately following the recommendation, there was a significant decline in early-stage cancer incidence rates among men 50 years or older, according to an analysis of data from the Surveillance, Epidemiology, and End Results (SEER) program.
For the current study, Ahmedin Jemal, DVM, PhD, and his associates at the American Cancer Society analyzed incidence data for invasive prostate cancer from 18 SEER registries, which, combined, represented about 28% of the U.S. population.
Investigators reported a continued decline in localized- and regional-stage prostate cancer incidence from 2012 to 2013. Specifically, the incidence rates per 100,000 men decreased from 356.5 to 335.4 in men aged 50-74 years and from 379.2 to 353.6 in men 75 years and older (JAMA Oncol. 2016 Aug 16. doi: 10.1001/jamaoncol.2016.2667).
Incidence rates for distant-stage disease were unchanged during the same time period for men of all ages.
Similar results were reported for non-Hispanic whites and non-Hispanic blacks.
“Whether this pattern will lead to a future increase in the diagnosis of distant-stage disease and prostate cancer mortality requires long-term monitoring because of the slow growing nature of this malignant neoplasm,” the investigators noted.
The American Cancer Society funded the study. The authors had no relevant disclosures to report.
On Twitter @jessnicolecraig
FROM JAMA ONCOLOGY
Key clinical point: Incidence rates for localized- and regional-stage prostate cancer continue to decline.
Major finding: The incidence rates for localized- and regional-stage prostate cancer per 100,000 men decreased from 356.5 to 335.4 in men aged 50-74 years and from 379.2 to 353.6 in men 75 years and older.
Data source: Meta-analysis from 18 SEER registries.
Disclosures: The American Cancer Society funded the study. The authors had no relevant disclosures to report.

Itch, Scratch, Ad Infinitum, Part 2
1. A 25-year-old woman reports anogenital itching, burning, and redness, present for 3 months. She says she developed a yeast infection after antibiotic therapy for a dental infection. The yeast infection was treated with terconazole, which resulted in immediate severe burning, redness, and swelling. Clobetasol cream used twice daily also caused burning, so she discontinued it. Her symptoms improved when she tried cool soaks and applied topical benzocaine gel as a local anesthetic.
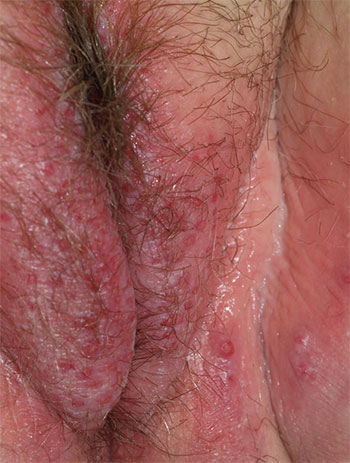
Diagnosis: Irritant contact dermatitis (as opposed to allergic contact dermatitis) associated with the use of terconazole and clobetasol. This was followed by allergic contact dermatitis in association with benzocaine. Treatment consists of withdrawal of benzocaine, reinitiation of cool soaks, and a switch to clobetasol ointment rather than cream. Nighttime sedation enables the patient to sleep through the itching and gradually allows her skin to heal.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
2. This 13-year-old presents with sudden-onset vulvar pain and sores. The child developed a sore throat and low-grade fever 3 days earlier, with vulvar pain and vulvar dysuria the next day. Oral acyclovir was prescribed for herpes simplex virus infection, but the girl’s condition has not improved. She claims sexual abstinence, and her mother believes her.
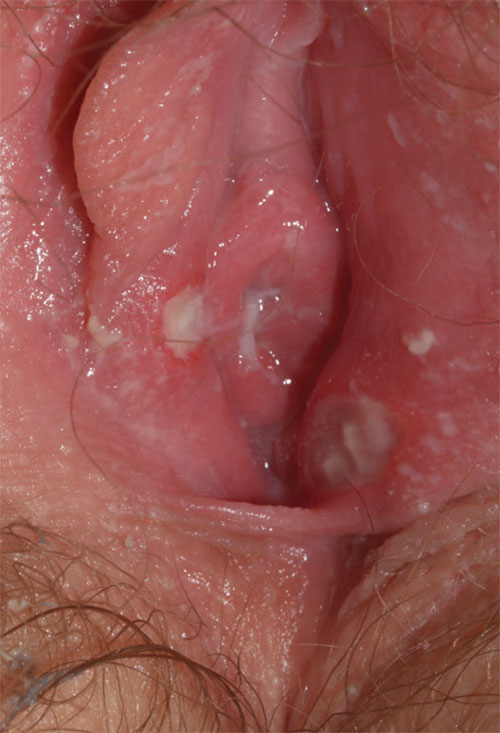
Diagnosis: Vulvar aphthae, believed to be of hyperimmune origin, are often precipitated by a viral syndrome. They are most common in girls aged 9 to 18 years.
Aphthae are uncommon and under-recognized on the vulva. Genital aphthae are usually much larger than oral aphthae. Most patients are mistakenly evaluated and treated for sexually transmitted infection, but the large, well-demarcated, painful, nonindurated, deep nature of the ulcer is pathognomonic for an aphthous ulcer.
Recommended treatment is prednisone 40 mg/day plus hydrocodone in usual doses of 5/325, one or two tablets every 4 to 6 hours, as needed; topical petroleum jelly (especially before urination); and sitz baths. When the patient returns one week later, she is much improved.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
3. A 36-year-old woman reports introital itching, vulvar dysuria, and superficial dyspareunia that have lasted 6 months. Apparent on physical examination are redness of the vestibule, medial labia minora, and vaginal walls, with edema of the surrounding skin and yellowish, copious vaginal secretions at the introitus. Lab tests for chlamydia, trichomonas, and gonorrhea are returned as normal.
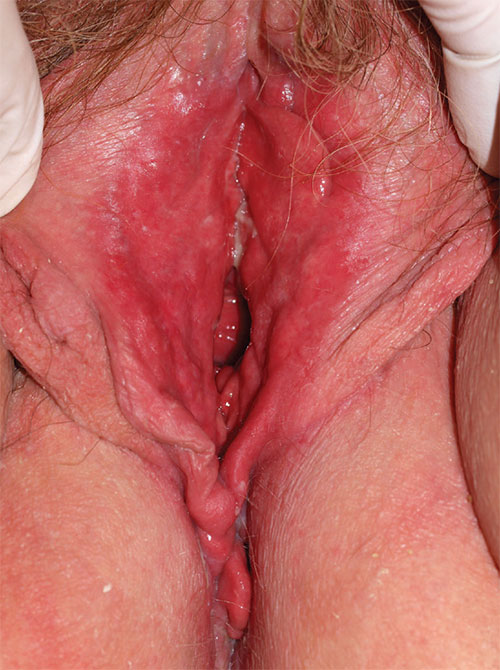
Diagnosis: Desquamative inflammatory vaginitis (DIV) is described as noninfectious inflammatory vaginitis in a setting of normal estrogen and absence of skin disease of the mucous membranes of the vagina. The condition is characterized by an increase in white blood cells and parabasal cells and absent lactobacilli, with relatively high vaginal pH. DIV is thought to represent an inflammatory dermatosis of the vaginal epithelium. Although some clinicians believe that DIV is actually lichen planus, the latter exhibits erosions as well as redness, nearly always affects the mouth and the vulva, and produces remarkable scarring. DIV does not erode, affect any other skin surfaces, or scar.
Treatment for DIV consists of clindamycin vaginal cream, 1/2 to 1 full applicator nightly, with a weekly oral fluconazole tablet (200 mg is more easily covered by insurance) to prevent secondary candidiasis. Schedule a follow-up visit in one month.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
4. A 43-year-old woman reports a “recalcitrant yeast infection” of the vulva, with itching and irritation. She is overweight and diabetic, with mild stress incontinence. Physical examination reveals a fairly well-demarcated red, rough plaque on the vulva and labiocrural folds, with satellite red papules and peripheral peeling. Similar plaques occur in the gluteal cleft, umbilicus, and axillae as well as under the breasts. A fungal preparation of the vagina and skin is negative.

Diagnosis: Of the several morphologic types of psoriasis, anogenital psoriasis is most often of the inverse pattern. Inverse psoriasis preferentially affects skin folds and is frequently mistaken for (and often initially superinfected with) candidiasis. Scale is thin and unapparent, and there often is a shiny, glazed appearance to the skin. Tiny satellite lesions are often visible as well. A skin biopsy of inverse psoriasis often is not diagnostic, showing only nonspecific psoriasiform dermatitis; this does not disprove psoriasis.
Psoriasis is a systemic condition and is associated with metabolic syndrome, carrying an increased risk for overweight, hypertension, diabetes, and cardiovascular disease. Management of these conditions is very important in the overall treatment of the patient.
The recommended treatment is clobetasol ointment applied to the skin folds, along with continuation of the topical miconazole cream. A week later, the patient’s condition is remarkably improved, and her biopsy shows psoriasiform dermatitis. The potency of her corticosteroid was reduced by switching to desonide cream, sparingly applied daily.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
1. A 25-year-old woman reports anogenital itching, burning, and redness, present for 3 months. She says she developed a yeast infection after antibiotic therapy for a dental infection. The yeast infection was treated with terconazole, which resulted in immediate severe burning, redness, and swelling. Clobetasol cream used twice daily also caused burning, so she discontinued it. Her symptoms improved when she tried cool soaks and applied topical benzocaine gel as a local anesthetic.
Diagnosis: Irritant contact dermatitis (as opposed to allergic contact dermatitis) associated with the use of terconazole and clobetasol. This was followed by allergic contact dermatitis in association with benzocaine. Treatment consists of withdrawal of benzocaine, reinitiation of cool soaks, and a switch to clobetasol ointment rather than cream. Nighttime sedation enables the patient to sleep through the itching and gradually allows her skin to heal.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
2. This 13-year-old presents with sudden-onset vulvar pain and sores. The child developed a sore throat and low-grade fever 3 days earlier, with vulvar pain and vulvar dysuria the next day. Oral acyclovir was prescribed for herpes simplex virus infection, but the girl’s condition has not improved. She claims sexual abstinence, and her mother believes her.
Diagnosis: Vulvar aphthae, believed to be of hyperimmune origin, are often precipitated by a viral syndrome. They are most common in girls aged 9 to 18 years.
Aphthae are uncommon and under-recognized on the vulva. Genital aphthae are usually much larger than oral aphthae. Most patients are mistakenly evaluated and treated for sexually transmitted infection, but the large, well-demarcated, painful, nonindurated, deep nature of the ulcer is pathognomonic for an aphthous ulcer.
Recommended treatment is prednisone 40 mg/day plus hydrocodone in usual doses of 5/325, one or two tablets every 4 to 6 hours, as needed; topical petroleum jelly (especially before urination); and sitz baths. When the patient returns one week later, she is much improved.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
3. A 36-year-old woman reports introital itching, vulvar dysuria, and superficial dyspareunia that have lasted 6 months. Apparent on physical examination are redness of the vestibule, medial labia minora, and vaginal walls, with edema of the surrounding skin and yellowish, copious vaginal secretions at the introitus. Lab tests for chlamydia, trichomonas, and gonorrhea are returned as normal.
Diagnosis: Desquamative inflammatory vaginitis (DIV) is described as noninfectious inflammatory vaginitis in a setting of normal estrogen and absence of skin disease of the mucous membranes of the vagina. The condition is characterized by an increase in white blood cells and parabasal cells and absent lactobacilli, with relatively high vaginal pH. DIV is thought to represent an inflammatory dermatosis of the vaginal epithelium. Although some clinicians believe that DIV is actually lichen planus, the latter exhibits erosions as well as redness, nearly always affects the mouth and the vulva, and produces remarkable scarring. DIV does not erode, affect any other skin surfaces, or scar.
Treatment for DIV consists of clindamycin vaginal cream, 1/2 to 1 full applicator nightly, with a weekly oral fluconazole tablet (200 mg is more easily covered by insurance) to prevent secondary candidiasis. Schedule a follow-up visit in one month.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
4. A 43-year-old woman reports a “recalcitrant yeast infection” of the vulva, with itching and irritation. She is overweight and diabetic, with mild stress incontinence. Physical examination reveals a fairly well-demarcated red, rough plaque on the vulva and labiocrural folds, with satellite red papules and peripheral peeling. Similar plaques occur in the gluteal cleft, umbilicus, and axillae as well as under the breasts. A fungal preparation of the vagina and skin is negative.
Diagnosis: Of the several morphologic types of psoriasis, anogenital psoriasis is most often of the inverse pattern. Inverse psoriasis preferentially affects skin folds and is frequently mistaken for (and often initially superinfected with) candidiasis. Scale is thin and unapparent, and there often is a shiny, glazed appearance to the skin. Tiny satellite lesions are often visible as well. A skin biopsy of inverse psoriasis often is not diagnostic, showing only nonspecific psoriasiform dermatitis; this does not disprove psoriasis.
Psoriasis is a systemic condition and is associated with metabolic syndrome, carrying an increased risk for overweight, hypertension, diabetes, and cardiovascular disease. Management of these conditions is very important in the overall treatment of the patient.
The recommended treatment is clobetasol ointment applied to the skin folds, along with continuation of the topical miconazole cream. A week later, the patient’s condition is remarkably improved, and her biopsy shows psoriasiform dermatitis. The potency of her corticosteroid was reduced by switching to desonide cream, sparingly applied daily.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
1. A 25-year-old woman reports anogenital itching, burning, and redness, present for 3 months. She says she developed a yeast infection after antibiotic therapy for a dental infection. The yeast infection was treated with terconazole, which resulted in immediate severe burning, redness, and swelling. Clobetasol cream used twice daily also caused burning, so she discontinued it. Her symptoms improved when she tried cool soaks and applied topical benzocaine gel as a local anesthetic.
Diagnosis: Irritant contact dermatitis (as opposed to allergic contact dermatitis) associated with the use of terconazole and clobetasol. This was followed by allergic contact dermatitis in association with benzocaine. Treatment consists of withdrawal of benzocaine, reinitiation of cool soaks, and a switch to clobetasol ointment rather than cream. Nighttime sedation enables the patient to sleep through the itching and gradually allows her skin to heal.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
2. This 13-year-old presents with sudden-onset vulvar pain and sores. The child developed a sore throat and low-grade fever 3 days earlier, with vulvar pain and vulvar dysuria the next day. Oral acyclovir was prescribed for herpes simplex virus infection, but the girl’s condition has not improved. She claims sexual abstinence, and her mother believes her.
Diagnosis: Vulvar aphthae, believed to be of hyperimmune origin, are often precipitated by a viral syndrome. They are most common in girls aged 9 to 18 years.
Aphthae are uncommon and under-recognized on the vulva. Genital aphthae are usually much larger than oral aphthae. Most patients are mistakenly evaluated and treated for sexually transmitted infection, but the large, well-demarcated, painful, nonindurated, deep nature of the ulcer is pathognomonic for an aphthous ulcer.
Recommended treatment is prednisone 40 mg/day plus hydrocodone in usual doses of 5/325, one or two tablets every 4 to 6 hours, as needed; topical petroleum jelly (especially before urination); and sitz baths. When the patient returns one week later, she is much improved.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
3. A 36-year-old woman reports introital itching, vulvar dysuria, and superficial dyspareunia that have lasted 6 months. Apparent on physical examination are redness of the vestibule, medial labia minora, and vaginal walls, with edema of the surrounding skin and yellowish, copious vaginal secretions at the introitus. Lab tests for chlamydia, trichomonas, and gonorrhea are returned as normal.
Diagnosis: Desquamative inflammatory vaginitis (DIV) is described as noninfectious inflammatory vaginitis in a setting of normal estrogen and absence of skin disease of the mucous membranes of the vagina. The condition is characterized by an increase in white blood cells and parabasal cells and absent lactobacilli, with relatively high vaginal pH. DIV is thought to represent an inflammatory dermatosis of the vaginal epithelium. Although some clinicians believe that DIV is actually lichen planus, the latter exhibits erosions as well as redness, nearly always affects the mouth and the vulva, and produces remarkable scarring. DIV does not erode, affect any other skin surfaces, or scar.
Treatment for DIV consists of clindamycin vaginal cream, 1/2 to 1 full applicator nightly, with a weekly oral fluconazole tablet (200 mg is more easily covered by insurance) to prevent secondary candidiasis. Schedule a follow-up visit in one month.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
4. A 43-year-old woman reports a “recalcitrant yeast infection” of the vulva, with itching and irritation. She is overweight and diabetic, with mild stress incontinence. Physical examination reveals a fairly well-demarcated red, rough plaque on the vulva and labiocrural folds, with satellite red papules and peripheral peeling. Similar plaques occur in the gluteal cleft, umbilicus, and axillae as well as under the breasts. A fungal preparation of the vagina and skin is negative.
Diagnosis: Of the several morphologic types of psoriasis, anogenital psoriasis is most often of the inverse pattern. Inverse psoriasis preferentially affects skin folds and is frequently mistaken for (and often initially superinfected with) candidiasis. Scale is thin and unapparent, and there often is a shiny, glazed appearance to the skin. Tiny satellite lesions are often visible as well. A skin biopsy of inverse psoriasis often is not diagnostic, showing only nonspecific psoriasiform dermatitis; this does not disprove psoriasis.
Psoriasis is a systemic condition and is associated with metabolic syndrome, carrying an increased risk for overweight, hypertension, diabetes, and cardiovascular disease. Management of these conditions is very important in the overall treatment of the patient.
The recommended treatment is clobetasol ointment applied to the skin folds, along with continuation of the topical miconazole cream. A week later, the patient’s condition is remarkably improved, and her biopsy shows psoriasiform dermatitis. The potency of her corticosteroid was reduced by switching to desonide cream, sparingly applied daily.
For more information on this case, see “Chronic vulvar irritation, itching, and pain. What is the diagnosis?” OBG Manag. 2014;26(6):30-37.
AATS Week 2017 Call for Abstracts & Videos
AATS welcomes you to submit your abstracts and videos to AATS Week 2017.
AATS Mitral Conclave
April 27-28, 2017
New York, NY
AATS Centennial
April 29-May 3, 2017
Boston, MA
Submission Deadlines:
AATS Centennial: Monday, October 17, 2016 @ 11.59 pm EDT
Mitral Conclave: Sunday, January 8, 2017 @ 11.59 pm EST
AATS welcomes you to submit your abstracts and videos to AATS Week 2017.
AATS Mitral Conclave
April 27-28, 2017
New York, NY
AATS Centennial
April 29-May 3, 2017
Boston, MA
Submission Deadlines:
AATS Centennial: Monday, October 17, 2016 @ 11.59 pm EDT
Mitral Conclave: Sunday, January 8, 2017 @ 11.59 pm EST
AATS welcomes you to submit your abstracts and videos to AATS Week 2017.
AATS Mitral Conclave
April 27-28, 2017
New York, NY
AATS Centennial
April 29-May 3, 2017
Boston, MA
Submission Deadlines:
AATS Centennial: Monday, October 17, 2016 @ 11.59 pm EDT
Mitral Conclave: Sunday, January 8, 2017 @ 11.59 pm EST

“Honoring Our Mentor” Fellowships Open for Submission
The AATS Graham Foundation is calling for submission for its “Honoring Our Mentors” fellowships.
This series is named after prominent CT surgeons who have demonstrated longstanding leadership and dedication over the course of their careers in both their clinical practices and commitment to training the future generation.
Lawrence H. Cohn Clinical Scholar Program
Allows young cardiac surgeons who are in their final year of their residency or have recently completed their residency to obtain advanced experience with valvular surgery or care.
Deadline: December 1, 2016
Denton A. Cooley Fellowship
New! Provides a deserving CT surgeon resident or young postgraduate surgeon the opportunity to enrich his/her education during four weeks of study at the Texas Heart Institute and Baylor St. Luke’s Medical.
Deadline: December 1, 2016
Marc de Leval Fellowship
Offers four to six weeks of congenital heart surgery training at an international center for a North American surgeon.
Deadline: December 1, 2016
F. Griffith Pearson Fellowship
Surgeons who have finished their residency/fellowship in general thoracic surgery advance their clinical techniques at a North American host institute.
Deadline: December 1, 2016
The AATS Graham Foundation is calling for submission for its “Honoring Our Mentors” fellowships.
This series is named after prominent CT surgeons who have demonstrated longstanding leadership and dedication over the course of their careers in both their clinical practices and commitment to training the future generation.
Lawrence H. Cohn Clinical Scholar Program
Allows young cardiac surgeons who are in their final year of their residency or have recently completed their residency to obtain advanced experience with valvular surgery or care.
Deadline: December 1, 2016
Denton A. Cooley Fellowship
New! Provides a deserving CT surgeon resident or young postgraduate surgeon the opportunity to enrich his/her education during four weeks of study at the Texas Heart Institute and Baylor St. Luke’s Medical.
Deadline: December 1, 2016
Marc de Leval Fellowship
Offers four to six weeks of congenital heart surgery training at an international center for a North American surgeon.
Deadline: December 1, 2016
F. Griffith Pearson Fellowship
Surgeons who have finished their residency/fellowship in general thoracic surgery advance their clinical techniques at a North American host institute.
Deadline: December 1, 2016
The AATS Graham Foundation is calling for submission for its “Honoring Our Mentors” fellowships.
This series is named after prominent CT surgeons who have demonstrated longstanding leadership and dedication over the course of their careers in both their clinical practices and commitment to training the future generation.
Lawrence H. Cohn Clinical Scholar Program
Allows young cardiac surgeons who are in their final year of their residency or have recently completed their residency to obtain advanced experience with valvular surgery or care.
Deadline: December 1, 2016
Denton A. Cooley Fellowship
New! Provides a deserving CT surgeon resident or young postgraduate surgeon the opportunity to enrich his/her education during four weeks of study at the Texas Heart Institute and Baylor St. Luke’s Medical.
Deadline: December 1, 2016
Marc de Leval Fellowship
Offers four to six weeks of congenital heart surgery training at an international center for a North American surgeon.
Deadline: December 1, 2016
F. Griffith Pearson Fellowship
Surgeons who have finished their residency/fellowship in general thoracic surgery advance their clinical techniques at a North American host institute.
Deadline: December 1, 2016

Registration and Housing Now Open: AATS Clinical Trials Methods Course 2016
October 20-22, 2016
Hyatt Regency O’Hare
Chicago, IL
Program Directors
David H. Harpole, Jr.
Marco A. Zenati
This course is an intensive, interactive training program for cardiothoracic surgeons across all subspecialties. It will permit them to acquire the critical skills necessary for effective clinical trial design and implementation. The course is particularly suited for professionals who are planning to apply for clinical trial funding, allowing them to better understand the complex nature of preparing and submitting clinical trial proposals.
Invited faculty includes currently funded clinical trial leading investigators and experts in the field of biostatistics and health sciences research. The program will offer a process in which the clinical trial protocol development can be streamlined. Interactive features will include hands-on focus groups and mock study sessions.
Register Today! Space available for only 40 participants.
October 20-22, 2016
Hyatt Regency O’Hare
Chicago, IL
Program Directors
David H. Harpole, Jr.
Marco A. Zenati
This course is an intensive, interactive training program for cardiothoracic surgeons across all subspecialties. It will permit them to acquire the critical skills necessary for effective clinical trial design and implementation. The course is particularly suited for professionals who are planning to apply for clinical trial funding, allowing them to better understand the complex nature of preparing and submitting clinical trial proposals.
Invited faculty includes currently funded clinical trial leading investigators and experts in the field of biostatistics and health sciences research. The program will offer a process in which the clinical trial protocol development can be streamlined. Interactive features will include hands-on focus groups and mock study sessions.
Register Today! Space available for only 40 participants.
October 20-22, 2016
Hyatt Regency O’Hare
Chicago, IL
Program Directors
David H. Harpole, Jr.
Marco A. Zenati
This course is an intensive, interactive training program for cardiothoracic surgeons across all subspecialties. It will permit them to acquire the critical skills necessary for effective clinical trial design and implementation. The course is particularly suited for professionals who are planning to apply for clinical trial funding, allowing them to better understand the complex nature of preparing and submitting clinical trial proposals.
Invited faculty includes currently funded clinical trial leading investigators and experts in the field of biostatistics and health sciences research. The program will offer a process in which the clinical trial protocol development can be streamlined. Interactive features will include hands-on focus groups and mock study sessions.
Register Today! Space available for only 40 participants.

Invest in the Future
Every day, CT surgeons transform the lives of their patients around the world. Your support is essential to ensure the future of our specialty and continue advancing global innovation in CT surgery. Please make a gift to the Graham Foundation or renew your commitment. Together, we can promote our specialty not only for the next generation of surgeons, but also the patients they serve.
Learn more about individual and corporate/organizational giving opportunities.
Contact Development Office: 978-927-8330
Every day, CT surgeons transform the lives of their patients around the world. Your support is essential to ensure the future of our specialty and continue advancing global innovation in CT surgery. Please make a gift to the Graham Foundation or renew your commitment. Together, we can promote our specialty not only for the next generation of surgeons, but also the patients they serve.
Learn more about individual and corporate/organizational giving opportunities.
Contact Development Office: 978-927-8330
Every day, CT surgeons transform the lives of their patients around the world. Your support is essential to ensure the future of our specialty and continue advancing global innovation in CT surgery. Please make a gift to the Graham Foundation or renew your commitment. Together, we can promote our specialty not only for the next generation of surgeons, but also the patients they serve.
Learn more about individual and corporate/organizational giving opportunities.
Contact Development Office: 978-927-8330

PCSK9 Inhibitor Can Nearly Replace Need for Apheresis in HeFH
ROME – Treating patients with heterozygous familial hypercholesterolemia (HeFH) with PCSK9 inhibitors can reduce their need for lipoprotein apheresis and its associated costs, Patrick M. Moriarty, MD, said in a video interview at the annual congress of the European Society of Cardiology.
In the randomized, phase III ODYSSEY ESCAPE trial, HeFH patients who underwent weekly apheresis were treated with either alirocumab, a proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor, or placebo. At the end of the study, the alirocumab-treated patients had a 75% greater reduction in need for apheresis, compared with those on placebo – a statistically significant difference.
With a price tag of $50,000-$75,000 a year for lipoprotein apheresis, compared with the roughly $12,000 cost for a PCSK9 inhibitor, this represents a significant savings for patients with HeFH, which occurs in roughly 1 in 200 people worldwide, Dr. Moriarty of the University of Kansas, Kansas City, told reporter Bruce Jancin.
ROME – Treating patients with heterozygous familial hypercholesterolemia (HeFH) with PCSK9 inhibitors can reduce their need for lipoprotein apheresis and its associated costs, Patrick M. Moriarty, MD, said in a video interview at the annual congress of the European Society of Cardiology.
In the randomized, phase III ODYSSEY ESCAPE trial, HeFH patients who underwent weekly apheresis were treated with either alirocumab, a proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor, or placebo. At the end of the study, the alirocumab-treated patients had a 75% greater reduction in need for apheresis, compared with those on placebo – a statistically significant difference.
With a price tag of $50,000-$75,000 a year for lipoprotein apheresis, compared with the roughly $12,000 cost for a PCSK9 inhibitor, this represents a significant savings for patients with HeFH, which occurs in roughly 1 in 200 people worldwide, Dr. Moriarty of the University of Kansas, Kansas City, told reporter Bruce Jancin.
ROME – Treating patients with heterozygous familial hypercholesterolemia (HeFH) with PCSK9 inhibitors can reduce their need for lipoprotein apheresis and its associated costs, Patrick M. Moriarty, MD, said in a video interview at the annual congress of the European Society of Cardiology.
In the randomized, phase III ODYSSEY ESCAPE trial, HeFH patients who underwent weekly apheresis were treated with either alirocumab, a proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor, or placebo. At the end of the study, the alirocumab-treated patients had a 75% greater reduction in need for apheresis, compared with those on placebo – a statistically significant difference.
With a price tag of $50,000-$75,000 a year for lipoprotein apheresis, compared with the roughly $12,000 cost for a PCSK9 inhibitor, this represents a significant savings for patients with HeFH, which occurs in roughly 1 in 200 people worldwide, Dr. Moriarty of the University of Kansas, Kansas City, told reporter Bruce Jancin.
Guillain-Barré incidence rose with Zika across Americas
Increased incidence of Guillain-Barré syndrome corresponded closely with patterns of Zika virus disease incidence in Central and South America from April 2015 through March 2016, according to results from a new temporal and graphic analysis.
The findings show Guillain-Barré syndrome (GBS) cases increasing from 100% to nearly 900% above previously recorded baseline rates during periods of Zika virus transmission in El Salvador, the Dominican Republic, Colombia, Honduras, Suriname, Venezuela, and the Brazilian state of Bahia (N Engl J Med. 2016 Aug 31. doi: 10.1056/NEJMc1609015).

The analysis of the yearlong period also revealed that declines in GBS incidence accompanied declines in Zika virus disease when and where transmission began to wane. The researchers, led by Marcos A. Espinal, MD, DrPH, of the Pan American Health Organization in Washington, did not find significant associations between co-circulation of dengue virus and GBS incidence. The study, which looked at 164,237 confirmed and suspected cases of Zika virus disease and 1,474 cases of GBS, found a 75% higher Zika virus disease incidence rate in women, which Dr. Espinal and colleagues said might be attributable to differences in health care–seeking behavior. GBS incidence, meanwhile, was 28% higher among males. The higher rate of GBS in men, the authors said, was consistent with findings from previous epidemiological studies of GBS.
While the new results did not show that Zika virus causes GBS, Dr. Espinal and colleagues wrote, they argued that they were indicative of a strong association, adding that GBS “could serve as a sentinel for Zika virus disease and other neurological disorders linked to Zika virus,” including microcephaly.
Most of the study authors worked for the Pan American Health Organization or for national health agencies in the data-contributing countries. None declared conflicts of interest.
Increased incidence of Guillain-Barré syndrome corresponded closely with patterns of Zika virus disease incidence in Central and South America from April 2015 through March 2016, according to results from a new temporal and graphic analysis.
The findings show Guillain-Barré syndrome (GBS) cases increasing from 100% to nearly 900% above previously recorded baseline rates during periods of Zika virus transmission in El Salvador, the Dominican Republic, Colombia, Honduras, Suriname, Venezuela, and the Brazilian state of Bahia (N Engl J Med. 2016 Aug 31. doi: 10.1056/NEJMc1609015).
The analysis of the yearlong period also revealed that declines in GBS incidence accompanied declines in Zika virus disease when and where transmission began to wane. The researchers, led by Marcos A. Espinal, MD, DrPH, of the Pan American Health Organization in Washington, did not find significant associations between co-circulation of dengue virus and GBS incidence. The study, which looked at 164,237 confirmed and suspected cases of Zika virus disease and 1,474 cases of GBS, found a 75% higher Zika virus disease incidence rate in women, which Dr. Espinal and colleagues said might be attributable to differences in health care–seeking behavior. GBS incidence, meanwhile, was 28% higher among males. The higher rate of GBS in men, the authors said, was consistent with findings from previous epidemiological studies of GBS.
While the new results did not show that Zika virus causes GBS, Dr. Espinal and colleagues wrote, they argued that they were indicative of a strong association, adding that GBS “could serve as a sentinel for Zika virus disease and other neurological disorders linked to Zika virus,” including microcephaly.
Most of the study authors worked for the Pan American Health Organization or for national health agencies in the data-contributing countries. None declared conflicts of interest.
Increased incidence of Guillain-Barré syndrome corresponded closely with patterns of Zika virus disease incidence in Central and South America from April 2015 through March 2016, according to results from a new temporal and graphic analysis.
The findings show Guillain-Barré syndrome (GBS) cases increasing from 100% to nearly 900% above previously recorded baseline rates during periods of Zika virus transmission in El Salvador, the Dominican Republic, Colombia, Honduras, Suriname, Venezuela, and the Brazilian state of Bahia (N Engl J Med. 2016 Aug 31. doi: 10.1056/NEJMc1609015).
The analysis of the yearlong period also revealed that declines in GBS incidence accompanied declines in Zika virus disease when and where transmission began to wane. The researchers, led by Marcos A. Espinal, MD, DrPH, of the Pan American Health Organization in Washington, did not find significant associations between co-circulation of dengue virus and GBS incidence. The study, which looked at 164,237 confirmed and suspected cases of Zika virus disease and 1,474 cases of GBS, found a 75% higher Zika virus disease incidence rate in women, which Dr. Espinal and colleagues said might be attributable to differences in health care–seeking behavior. GBS incidence, meanwhile, was 28% higher among males. The higher rate of GBS in men, the authors said, was consistent with findings from previous epidemiological studies of GBS.
While the new results did not show that Zika virus causes GBS, Dr. Espinal and colleagues wrote, they argued that they were indicative of a strong association, adding that GBS “could serve as a sentinel for Zika virus disease and other neurological disorders linked to Zika virus,” including microcephaly.
Most of the study authors worked for the Pan American Health Organization or for national health agencies in the data-contributing countries. None declared conflicts of interest.
FROM NEW ENGLAND JOURNAL OF MEDICINE

Back to Basics: The Role of the Team Physician
Editor’s Note: AJO Deputy Editor-in-Chief Robin West, MD, is the Head Team Physician for the Washington Redskins and the Washington Nationals. She has previously served as a team physician for 2 Super Bowl-winning Pittsburgh Steelers teams. I am pleased to “hand off” this issue to her.
—Bryan T. Hanypsiak, MD
The summer is over, football season has begun, and team physicians are busy trying to manage and treat the plethora of injuries that come with the game. Football is one of the most popular sports played by young athletes. Youth participation (ages 6-14 years) in tackle football was 2.169 million in 2015, according to a study conducted by the Physical Activity Council and presented by USA Football. There were 1.084 million boys (and 1500 girls) playing high school football in the 2014-2015 season, nearly twice the number of the next most popular sport, track and field, according to the National Federation of State High School Associations.Due to the sheer volume of athletes and high-impact nature of the game, football leads all other sports in the number of sustained injuries.
Team physicians have the leadership role in the organization, management, and provision of care of the athletes on the team. The roles and responsibilities of the team physician are ever-evolving. The team physician has to meet certain medical qualifications and education requirements, and understand the ethical and medicolegal issues.
The American Academy of Orthopaedic Surgeons and several other medical associations have put together a Team Physician Consensus Statement (available at http://bit.ly/2b8rOzS). All team physicians, coaches, and athletic trainers should read and understand this statement, as it delineates the qualifications, duties, and responsibilities of the team physician.
Our Football Issue focuses on the most common injuries that the team physician will encounter during the season. Our goal is to create a comprehensive guide for the team physician on the acute management of these injuries. As team physicians, we have to make quick return-to-play decisions that are often difficult, as we are dealing with extremely competitive athletes and coaches in the heat of the moment. Since we can’t control the high levels of adrenalin, loud stadium, or rapid speed of the game, we need to be prepared to perform a comprehensive evaluation and diagnosis under these circumstances. This return-to-play decision should be based solely on the severity of the injury and safety of the player. As a team physician, you are responsible for making the “final call” on when the player is safe to return to the game.
This issue includes a section on the most common medical issues (ophthalmology, dental, and dermatology), concussion, exertional heat stroke, knee injuries, and foot and ankle injuries. We also have a special list of the most common items to include in the athletic trainer’s medical bag when covering a high school or collegiate football game (see page 376). Our prominent contributing authors all have extensive experience covering high school, collegiate, and professional teams.
I hope that our Football Issue helps you to keep your athletes safe and injury-free, which is necessary to have a successful season. Remember, as the team physician, your primary focus is the well being of the players. The success of the team only comes when the players are healthy. A cohesive, well-organized medical team, led by the head athletic trainer and team physician, is a key component to the care of the athletes. It truly takes a village to provide top-notch medical care to a football team.
Am J Orthop. 2016;45(6):338. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
Editor’s Note: AJO Deputy Editor-in-Chief Robin West, MD, is the Head Team Physician for the Washington Redskins and the Washington Nationals. She has previously served as a team physician for 2 Super Bowl-winning Pittsburgh Steelers teams. I am pleased to “hand off” this issue to her.
—Bryan T. Hanypsiak, MD
The summer is over, football season has begun, and team physicians are busy trying to manage and treat the plethora of injuries that come with the game. Football is one of the most popular sports played by young athletes. Youth participation (ages 6-14 years) in tackle football was 2.169 million in 2015, according to a study conducted by the Physical Activity Council and presented by USA Football. There were 1.084 million boys (and 1500 girls) playing high school football in the 2014-2015 season, nearly twice the number of the next most popular sport, track and field, according to the National Federation of State High School Associations.Due to the sheer volume of athletes and high-impact nature of the game, football leads all other sports in the number of sustained injuries.
Team physicians have the leadership role in the organization, management, and provision of care of the athletes on the team. The roles and responsibilities of the team physician are ever-evolving. The team physician has to meet certain medical qualifications and education requirements, and understand the ethical and medicolegal issues.
The American Academy of Orthopaedic Surgeons and several other medical associations have put together a Team Physician Consensus Statement (available at http://bit.ly/2b8rOzS). All team physicians, coaches, and athletic trainers should read and understand this statement, as it delineates the qualifications, duties, and responsibilities of the team physician.
Our Football Issue focuses on the most common injuries that the team physician will encounter during the season. Our goal is to create a comprehensive guide for the team physician on the acute management of these injuries. As team physicians, we have to make quick return-to-play decisions that are often difficult, as we are dealing with extremely competitive athletes and coaches in the heat of the moment. Since we can’t control the high levels of adrenalin, loud stadium, or rapid speed of the game, we need to be prepared to perform a comprehensive evaluation and diagnosis under these circumstances. This return-to-play decision should be based solely on the severity of the injury and safety of the player. As a team physician, you are responsible for making the “final call” on when the player is safe to return to the game.
This issue includes a section on the most common medical issues (ophthalmology, dental, and dermatology), concussion, exertional heat stroke, knee injuries, and foot and ankle injuries. We also have a special list of the most common items to include in the athletic trainer’s medical bag when covering a high school or collegiate football game (see page 376). Our prominent contributing authors all have extensive experience covering high school, collegiate, and professional teams.
I hope that our Football Issue helps you to keep your athletes safe and injury-free, which is necessary to have a successful season. Remember, as the team physician, your primary focus is the well being of the players. The success of the team only comes when the players are healthy. A cohesive, well-organized medical team, led by the head athletic trainer and team physician, is a key component to the care of the athletes. It truly takes a village to provide top-notch medical care to a football team.
Am J Orthop. 2016;45(6):338. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.
Editor’s Note: AJO Deputy Editor-in-Chief Robin West, MD, is the Head Team Physician for the Washington Redskins and the Washington Nationals. She has previously served as a team physician for 2 Super Bowl-winning Pittsburgh Steelers teams. I am pleased to “hand off” this issue to her.
—Bryan T. Hanypsiak, MD
The summer is over, football season has begun, and team physicians are busy trying to manage and treat the plethora of injuries that come with the game. Football is one of the most popular sports played by young athletes. Youth participation (ages 6-14 years) in tackle football was 2.169 million in 2015, according to a study conducted by the Physical Activity Council and presented by USA Football. There were 1.084 million boys (and 1500 girls) playing high school football in the 2014-2015 season, nearly twice the number of the next most popular sport, track and field, according to the National Federation of State High School Associations.Due to the sheer volume of athletes and high-impact nature of the game, football leads all other sports in the number of sustained injuries.
Team physicians have the leadership role in the organization, management, and provision of care of the athletes on the team. The roles and responsibilities of the team physician are ever-evolving. The team physician has to meet certain medical qualifications and education requirements, and understand the ethical and medicolegal issues.
The American Academy of Orthopaedic Surgeons and several other medical associations have put together a Team Physician Consensus Statement (available at http://bit.ly/2b8rOzS). All team physicians, coaches, and athletic trainers should read and understand this statement, as it delineates the qualifications, duties, and responsibilities of the team physician.
Our Football Issue focuses on the most common injuries that the team physician will encounter during the season. Our goal is to create a comprehensive guide for the team physician on the acute management of these injuries. As team physicians, we have to make quick return-to-play decisions that are often difficult, as we are dealing with extremely competitive athletes and coaches in the heat of the moment. Since we can’t control the high levels of adrenalin, loud stadium, or rapid speed of the game, we need to be prepared to perform a comprehensive evaluation and diagnosis under these circumstances. This return-to-play decision should be based solely on the severity of the injury and safety of the player. As a team physician, you are responsible for making the “final call” on when the player is safe to return to the game.
This issue includes a section on the most common medical issues (ophthalmology, dental, and dermatology), concussion, exertional heat stroke, knee injuries, and foot and ankle injuries. We also have a special list of the most common items to include in the athletic trainer’s medical bag when covering a high school or collegiate football game (see page 376). Our prominent contributing authors all have extensive experience covering high school, collegiate, and professional teams.
I hope that our Football Issue helps you to keep your athletes safe and injury-free, which is necessary to have a successful season. Remember, as the team physician, your primary focus is the well being of the players. The success of the team only comes when the players are healthy. A cohesive, well-organized medical team, led by the head athletic trainer and team physician, is a key component to the care of the athletes. It truly takes a village to provide top-notch medical care to a football team.
Am J Orthop. 2016;45(6):338. Copyright Frontline Medical Communications Inc. 2016. All rights reserved.

Yeast infection in pregnancy? Think twice about fluconazole
PRACTICE CHANGER
Avoid prescribing oral fluconazole in early pregnancy because it is associated with a higher rate of spontaneous abortion than is topical azole therapy.1
Strength of recommendation
B: Based on large cohort study performed in Denmark.
Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
Illustrative Case
A 25-year-old woman who is 16 weeks pregnant with her first child is experiencing increased vaginal discharge associated with vaginal itching. A microscopic examination of the discharge confirms your suspicions of vaginal candidiasis. Is oral fluconazole or a topical azole your treatment of choice?
Because of the increased production of sex hormones, vaginal candidiasis is common during pregnancy, affecting up to 10% of pregnant women in the United States.1,2 Treatment options include oral fluconazole and a variety of topical azoles. Although topical azoles are recommended as first-line therapy,3 the ease of oral therapy makes it an attractive treatment option.4 The safety of oral fluconazole during pregnancy, however, has recently come under scrutiny.
Case reports have linked high-dose fluconazole use during pregnancy with congenital malformations.5,6 These case reports led to epidemiological studies evaluating fluconazole’s safety, but, in these studies, no association with congenital malformations was found.7,8
A large cohort study involving 1079 fluconazole-exposed pregnancies and 170,453 unexposed pregnancies found no increased risk of congenital malformations or stillbirth; rates of spontaneous abortion and miscarriage were not evaluated.9 A prospective cohort study of 226 pregnant women found no association between fluconazole use during the first trimester and miscarriages.10 However, the validity of both studies’ findings was limited by small numbers of participants. The current study is the largest to date to evaluate whether use of fluconazole compared to that of topical azoles in early pregnancy is associated with increased rates of spontaneous abortion and stillbirth.
Study Summary
Fluconazole significantly increases risk of miscarriage, but not stillbirth
This nationwide cohort study, conducted using the Medical Birth Register in Denmark, evaluated more than 1.4 million pregnancies occurring from 1997 to 2013 for exposure to oral fluconazole between 7 and 22 weeks’ gestation. Each oral fluconazole-exposed pregnancy was matched with up to 4 unexposed pregnancies (based on propensity score, maternal age, calendar year, and gestational age) and to pregnancies exposed to intravaginal formulations of topical azoles. Exposure to fluconazole was documented based on filled prescriptions from the National Prescription Register. Primary outcomes were rates of spontaneous abortion (loss before 22 weeks) and stillbirth (loss after 23 weeks).
Rates of spontaneous abortion. From the total cohort of more than 1.4 million pregnancies, 3315 were exposed to oral fluconazole between 7 and 22 weeks’ gestation. Spontaneous abortions occurred in 147 of the 3315 fluconazole-exposed pregnancies and in 563 of 13,246 unexposed, matched pregnancies (hazard ratio [HR]=1.48; 95% confidence interval [CI], 1.23-1.77).
Rates of stillbirth. Of 5382 pregnancies exposed to fluconazole from week 7 to birth, 21 resulted in stillbirth; 77 stillbirths occurred in the 21,506 unexposed matched pregnancies (HR=1.32; 95% CI, 0.82-2.14). In a sensitivity analysis, however, higher doses of fluconazole (350 mg) were 4 times more likely to be associated with stillbirth (HR=4.10; 95% CI, 1.89-8.90) than lower doses (150 mg) (HR= 0.99; 95% CI, 0.56-1.74).
Oral fluconazole vs topical azole. Use of oral fluconazole in pregnancy was associated with an increased risk of spontaneous abortion when compared to topical azole use: 130 of 2823 pregnancies vs 118 of 2823 pregnancies, respectively (HR=1.62; 95% CI, 1.26-2.07), but not an increased risk of stillbirths: 20 of 4301 pregnancies vs 22 of 4301 pregnancies, respectively (HR=1.18; 95% CI, 0.64-2.16).
What’s New
A sizeable study with a treatment comparison
The authors found that exposure in early pregnancy to oral fluconazole, as compared to topical azoles, increases the risk of spontaneous abortion. By comparing treatments in a sensitivity analysis, the confounder of Candida infections causing spontaneous abortion was removed. In addition, when considering the ease of dosing of fluconazole as compared with topical imidazoles, this study challenges the balance of ease of use with safety.
Caveats
A skewed population and limited generalizability?
This large cohort study using the National Patient Register in Denmark may not be generalizable to a larger, non-Scandinavian population. Since a hospital registry was used, those not seeking care through the hospital were likely missed. If patients
In addition, the study focused on women exposed from 7 to 22 weeks’ gestation; the findings may not be generalizable to fluconazole exposure prior to 7 weeks. Likewise, the registry is unlikely to capture very early spontaneous abortions that are not recognized clinically. In all, given the large sample size and the care taken to match each exposed pregnancy with up to 4 unexposed pregnancies, these limitations are likely to have had little influence on the overall findings of the study.
Challenges to Implementation
Balancing ease of use with safety
Given the ease of using oral fluconazole vs daily topical azole therapy, many physicians and patients may still opt for oral treatment.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
2. Cotch MF, Hillier SL, Gibbs RS, et al. Epidemiology and outcomes associated with moderate to heavy Candida colonization during pregnancy. Vaginal Infections and Prematurity Study Group. Am J Obstet Gynecol. 1998;178:374-380.
3. Workowski KA, Bolan GA, Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
4. Tooley PJ. Patient and doctor preferences in the treatment of vaginal candidosis. Practitioner. 1985;229: 655-660.
5. Aleck KA, Bartley DL. Multiple malformation syndrome following fluconazole use in pregnancy: report of an additional patient. Am J Med Genet. 1997;72:253-256.
6. Lee BE, Feinberg M, Abraham JJ, et al. Congenital malformations in an infant born to a woman treated with fluconazole. Pediatr Infect Dis J. 1992;11:1062-1064.
7. Jick SS. Pregnancy outcomes after maternal exposure to fluconazole. Pharmacotherapy. 1999;19:221-222.
8. Mølgaard-Nielsen D, Pasternak B, Hviid A. Use of oral fluconazole during pregnancy and the risk of birth defects. N Engl J Med. 2013;369:830-839.
9. Nørgaard M, Pedersen L, Gislum M, et al. Maternal use of fluconazole and risk of congenital malformations: a Danish population-based cohort study. J Antimicrob Chemother. 2008;62:172-176.
10. Mastroiacovo P, Mazzone T, Botto LD, et al. Prospective assessment of pregnancy outcomes after first-trimester exposure to fluconazole. Am J Obstet Gynecol. 1996;175:1645-1650.
PRACTICE CHANGER
Avoid prescribing oral fluconazole in early pregnancy because it is associated with a higher rate of spontaneous abortion than is topical azole therapy.1
Strength of recommendation
B: Based on large cohort study performed in Denmark.
Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
Illustrative Case
A 25-year-old woman who is 16 weeks pregnant with her first child is experiencing increased vaginal discharge associated with vaginal itching. A microscopic examination of the discharge confirms your suspicions of vaginal candidiasis. Is oral fluconazole or a topical azole your treatment of choice?
Because of the increased production of sex hormones, vaginal candidiasis is common during pregnancy, affecting up to 10% of pregnant women in the United States.1,2 Treatment options include oral fluconazole and a variety of topical azoles. Although topical azoles are recommended as first-line therapy,3 the ease of oral therapy makes it an attractive treatment option.4 The safety of oral fluconazole during pregnancy, however, has recently come under scrutiny.
Case reports have linked high-dose fluconazole use during pregnancy with congenital malformations.5,6 These case reports led to epidemiological studies evaluating fluconazole’s safety, but, in these studies, no association with congenital malformations was found.7,8
A large cohort study involving 1079 fluconazole-exposed pregnancies and 170,453 unexposed pregnancies found no increased risk of congenital malformations or stillbirth; rates of spontaneous abortion and miscarriage were not evaluated.9 A prospective cohort study of 226 pregnant women found no association between fluconazole use during the first trimester and miscarriages.10 However, the validity of both studies’ findings was limited by small numbers of participants. The current study is the largest to date to evaluate whether use of fluconazole compared to that of topical azoles in early pregnancy is associated with increased rates of spontaneous abortion and stillbirth.
Study Summary
Fluconazole significantly increases risk of miscarriage, but not stillbirth
This nationwide cohort study, conducted using the Medical Birth Register in Denmark, evaluated more than 1.4 million pregnancies occurring from 1997 to 2013 for exposure to oral fluconazole between 7 and 22 weeks’ gestation. Each oral fluconazole-exposed pregnancy was matched with up to 4 unexposed pregnancies (based on propensity score, maternal age, calendar year, and gestational age) and to pregnancies exposed to intravaginal formulations of topical azoles. Exposure to fluconazole was documented based on filled prescriptions from the National Prescription Register. Primary outcomes were rates of spontaneous abortion (loss before 22 weeks) and stillbirth (loss after 23 weeks).
Rates of spontaneous abortion. From the total cohort of more than 1.4 million pregnancies, 3315 were exposed to oral fluconazole between 7 and 22 weeks’ gestation. Spontaneous abortions occurred in 147 of the 3315 fluconazole-exposed pregnancies and in 563 of 13,246 unexposed, matched pregnancies (hazard ratio [HR]=1.48; 95% confidence interval [CI], 1.23-1.77).
Rates of stillbirth. Of 5382 pregnancies exposed to fluconazole from week 7 to birth, 21 resulted in stillbirth; 77 stillbirths occurred in the 21,506 unexposed matched pregnancies (HR=1.32; 95% CI, 0.82-2.14). In a sensitivity analysis, however, higher doses of fluconazole (350 mg) were 4 times more likely to be associated with stillbirth (HR=4.10; 95% CI, 1.89-8.90) than lower doses (150 mg) (HR= 0.99; 95% CI, 0.56-1.74).
Oral fluconazole vs topical azole. Use of oral fluconazole in pregnancy was associated with an increased risk of spontaneous abortion when compared to topical azole use: 130 of 2823 pregnancies vs 118 of 2823 pregnancies, respectively (HR=1.62; 95% CI, 1.26-2.07), but not an increased risk of stillbirths: 20 of 4301 pregnancies vs 22 of 4301 pregnancies, respectively (HR=1.18; 95% CI, 0.64-2.16).
What’s New
A sizeable study with a treatment comparison
The authors found that exposure in early pregnancy to oral fluconazole, as compared to topical azoles, increases the risk of spontaneous abortion. By comparing treatments in a sensitivity analysis, the confounder of Candida infections causing spontaneous abortion was removed. In addition, when considering the ease of dosing of fluconazole as compared with topical imidazoles, this study challenges the balance of ease of use with safety.
Caveats
A skewed population and limited generalizability?
This large cohort study using the National Patient Register in Denmark may not be generalizable to a larger, non-Scandinavian population. Since a hospital registry was used, those not seeking care through the hospital were likely missed. If patients
In addition, the study focused on women exposed from 7 to 22 weeks’ gestation; the findings may not be generalizable to fluconazole exposure prior to 7 weeks. Likewise, the registry is unlikely to capture very early spontaneous abortions that are not recognized clinically. In all, given the large sample size and the care taken to match each exposed pregnancy with up to 4 unexposed pregnancies, these limitations are likely to have had little influence on the overall findings of the study.
Challenges to Implementation
Balancing ease of use with safety
Given the ease of using oral fluconazole vs daily topical azole therapy, many physicians and patients may still opt for oral treatment.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
PRACTICE CHANGER
Avoid prescribing oral fluconazole in early pregnancy because it is associated with a higher rate of spontaneous abortion than is topical azole therapy.1
Strength of recommendation
B: Based on large cohort study performed in Denmark.
Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
Illustrative Case
A 25-year-old woman who is 16 weeks pregnant with her first child is experiencing increased vaginal discharge associated with vaginal itching. A microscopic examination of the discharge confirms your suspicions of vaginal candidiasis. Is oral fluconazole or a topical azole your treatment of choice?
Because of the increased production of sex hormones, vaginal candidiasis is common during pregnancy, affecting up to 10% of pregnant women in the United States.1,2 Treatment options include oral fluconazole and a variety of topical azoles. Although topical azoles are recommended as first-line therapy,3 the ease of oral therapy makes it an attractive treatment option.4 The safety of oral fluconazole during pregnancy, however, has recently come under scrutiny.
Case reports have linked high-dose fluconazole use during pregnancy with congenital malformations.5,6 These case reports led to epidemiological studies evaluating fluconazole’s safety, but, in these studies, no association with congenital malformations was found.7,8
A large cohort study involving 1079 fluconazole-exposed pregnancies and 170,453 unexposed pregnancies found no increased risk of congenital malformations or stillbirth; rates of spontaneous abortion and miscarriage were not evaluated.9 A prospective cohort study of 226 pregnant women found no association between fluconazole use during the first trimester and miscarriages.10 However, the validity of both studies’ findings was limited by small numbers of participants. The current study is the largest to date to evaluate whether use of fluconazole compared to that of topical azoles in early pregnancy is associated with increased rates of spontaneous abortion and stillbirth.
Study Summary
Fluconazole significantly increases risk of miscarriage, but not stillbirth
This nationwide cohort study, conducted using the Medical Birth Register in Denmark, evaluated more than 1.4 million pregnancies occurring from 1997 to 2013 for exposure to oral fluconazole between 7 and 22 weeks’ gestation. Each oral fluconazole-exposed pregnancy was matched with up to 4 unexposed pregnancies (based on propensity score, maternal age, calendar year, and gestational age) and to pregnancies exposed to intravaginal formulations of topical azoles. Exposure to fluconazole was documented based on filled prescriptions from the National Prescription Register. Primary outcomes were rates of spontaneous abortion (loss before 22 weeks) and stillbirth (loss after 23 weeks).
Rates of spontaneous abortion. From the total cohort of more than 1.4 million pregnancies, 3315 were exposed to oral fluconazole between 7 and 22 weeks’ gestation. Spontaneous abortions occurred in 147 of the 3315 fluconazole-exposed pregnancies and in 563 of 13,246 unexposed, matched pregnancies (hazard ratio [HR]=1.48; 95% confidence interval [CI], 1.23-1.77).
Rates of stillbirth. Of 5382 pregnancies exposed to fluconazole from week 7 to birth, 21 resulted in stillbirth; 77 stillbirths occurred in the 21,506 unexposed matched pregnancies (HR=1.32; 95% CI, 0.82-2.14). In a sensitivity analysis, however, higher doses of fluconazole (350 mg) were 4 times more likely to be associated with stillbirth (HR=4.10; 95% CI, 1.89-8.90) than lower doses (150 mg) (HR= 0.99; 95% CI, 0.56-1.74).
Oral fluconazole vs topical azole. Use of oral fluconazole in pregnancy was associated with an increased risk of spontaneous abortion when compared to topical azole use: 130 of 2823 pregnancies vs 118 of 2823 pregnancies, respectively (HR=1.62; 95% CI, 1.26-2.07), but not an increased risk of stillbirths: 20 of 4301 pregnancies vs 22 of 4301 pregnancies, respectively (HR=1.18; 95% CI, 0.64-2.16).
What’s New
A sizeable study with a treatment comparison
The authors found that exposure in early pregnancy to oral fluconazole, as compared to topical azoles, increases the risk of spontaneous abortion. By comparing treatments in a sensitivity analysis, the confounder of Candida infections causing spontaneous abortion was removed. In addition, when considering the ease of dosing of fluconazole as compared with topical imidazoles, this study challenges the balance of ease of use with safety.
Caveats
A skewed population and limited generalizability?
This large cohort study using the National Patient Register in Denmark may not be generalizable to a larger, non-Scandinavian population. Since a hospital registry was used, those not seeking care through the hospital were likely missed. If patients
In addition, the study focused on women exposed from 7 to 22 weeks’ gestation; the findings may not be generalizable to fluconazole exposure prior to 7 weeks. Likewise, the registry is unlikely to capture very early spontaneous abortions that are not recognized clinically. In all, given the large sample size and the care taken to match each exposed pregnancy with up to 4 unexposed pregnancies, these limitations are likely to have had little influence on the overall findings of the study.
Challenges to Implementation
Balancing ease of use with safety
Given the ease of using oral fluconazole vs daily topical azole therapy, many physicians and patients may still opt for oral treatment.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
2. Cotch MF, Hillier SL, Gibbs RS, et al. Epidemiology and outcomes associated with moderate to heavy Candida colonization during pregnancy. Vaginal Infections and Prematurity Study Group. Am J Obstet Gynecol. 1998;178:374-380.
3. Workowski KA, Bolan GA, Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
4. Tooley PJ. Patient and doctor preferences in the treatment of vaginal candidosis. Practitioner. 1985;229: 655-660.
5. Aleck KA, Bartley DL. Multiple malformation syndrome following fluconazole use in pregnancy: report of an additional patient. Am J Med Genet. 1997;72:253-256.
6. Lee BE, Feinberg M, Abraham JJ, et al. Congenital malformations in an infant born to a woman treated with fluconazole. Pediatr Infect Dis J. 1992;11:1062-1064.
7. Jick SS. Pregnancy outcomes after maternal exposure to fluconazole. Pharmacotherapy. 1999;19:221-222.
8. Mølgaard-Nielsen D, Pasternak B, Hviid A. Use of oral fluconazole during pregnancy and the risk of birth defects. N Engl J Med. 2013;369:830-839.
9. Nørgaard M, Pedersen L, Gislum M, et al. Maternal use of fluconazole and risk of congenital malformations: a Danish population-based cohort study. J Antimicrob Chemother. 2008;62:172-176.
10. Mastroiacovo P, Mazzone T, Botto LD, et al. Prospective assessment of pregnancy outcomes after first-trimester exposure to fluconazole. Am J Obstet Gynecol. 1996;175:1645-1650.
1. Mølgaard-Nielsen D, Svanström H, Melbye M, et al. Association between use of oral fluconazole during pregnancy and risk of spontaneous abortion and stillbirth. JAMA. 2016;315:58-67.
2. Cotch MF, Hillier SL, Gibbs RS, et al. Epidemiology and outcomes associated with moderate to heavy Candida colonization during pregnancy. Vaginal Infections and Prematurity Study Group. Am J Obstet Gynecol. 1998;178:374-380.
3. Workowski KA, Bolan GA, Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
4. Tooley PJ. Patient and doctor preferences in the treatment of vaginal candidosis. Practitioner. 1985;229: 655-660.
5. Aleck KA, Bartley DL. Multiple malformation syndrome following fluconazole use in pregnancy: report of an additional patient. Am J Med Genet. 1997;72:253-256.
6. Lee BE, Feinberg M, Abraham JJ, et al. Congenital malformations in an infant born to a woman treated with fluconazole. Pediatr Infect Dis J. 1992;11:1062-1064.
7. Jick SS. Pregnancy outcomes after maternal exposure to fluconazole. Pharmacotherapy. 1999;19:221-222.
8. Mølgaard-Nielsen D, Pasternak B, Hviid A. Use of oral fluconazole during pregnancy and the risk of birth defects. N Engl J Med. 2013;369:830-839.
9. Nørgaard M, Pedersen L, Gislum M, et al. Maternal use of fluconazole and risk of congenital malformations: a Danish population-based cohort study. J Antimicrob Chemother. 2008;62:172-176.
10. Mastroiacovo P, Mazzone T, Botto LD, et al. Prospective assessment of pregnancy outcomes after first-trimester exposure to fluconazole. Am J Obstet Gynecol. 1996;175:1645-1650.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.
