User login
Drug granted orphan designation for GVHD

Photo by Linda Bartlett
The European Commission has granted orphan drug designation to ALXN1007 for the treatment of graft-versus-host disease (GVHD).
ALXN1007 is an anti-inflammatory monoclonal antibody targeting complement protein C5a.
The drug is currently under investigation in a phase 2 trial of patients with newly diagnosed acute GVHD of the lower gastrointestinal tract (GI-GVHD).
ALXN1007 is being developed by Alexion Pharmaceuticals, Inc.
About orphan designation
Orphan designation from the European Commission provides regulatory and financial incentives for companies to develop and market therapies that treat a life-threatening or chronically debilitating condition affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity in the European Union if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure.
Phase 2 trial of ALXN1007
Results from the phase 2 trial of ALXN1007 in patients with newly diagnosed, acute GI-GVHD were presented at the 21st Congress of the European Hematology Association (abstract LB2269).
The presentation included 15 patients with biopsy-confirmed acute GI-GVHD. The patients had a median age of 60 (range, 25-69), and 60% were male.
Patients had acute myeloid leukemia/myelodysplastic syndrome (n=8), acute lymphoblastic leukemia (n=2), acute lymphocytic leukemia (n=1), acute myeloblastic leukemia (n=1), aplastic anemia (n=1), cutaneous T-cell lymphoma (n=1), or mantle cell lymphoma (n=1).
Most patients received transplants from matched, unrelated donors (n=11), 3 had matched, related donors, and 1 had a mismatched donor. Ten patients received peripheral blood grafts, 4 received cord blood, and 1 received a bone marrow transplant.
Patients had grade 1 (n=7), grade 2 (n=2), and grade 3 acute GI-GVHD (n=6).
The patients received weekly doses of ALXN1007 at 10 mg/kg, in combination with methylprednisolone at an initial dose of 2 mg/kg, through day 56.
Thirteen patients were evaluable for efficacy. One patient experienced leukemia relapse at day 18, and 1 withdrew from the study early.
The overall acute GVHD response rate was 77% (10/13), both at day 28 and day 56. The complete GI-GVHD response rate was 69% at day 28 and 77% at day 56.
At day 180, the nonrelapse mortality rate was 12.5%, and the overall survival rate was 69.2%.
All of the patients had treatment-emergent adverse events (AEs), and 11 patients (69%) had serious treatment-emergent AEs.
Five patients experienced a total of 12 treatment-related AEs (1 case each)—adenovirus infection, bronchopulmonary aspergillosis, chills, corona virus infection, viral cystitis, Epstein-Barr virus infection, hypersensitivity, influenza, influenza-like illness, infusion-related reaction, respiratory syncytial virus infection, and tremor.
There were 6 deaths, but none were considered treatment-related. ![]()
Photo by Linda Bartlett
The European Commission has granted orphan drug designation to ALXN1007 for the treatment of graft-versus-host disease (GVHD).
ALXN1007 is an anti-inflammatory monoclonal antibody targeting complement protein C5a.
The drug is currently under investigation in a phase 2 trial of patients with newly diagnosed acute GVHD of the lower gastrointestinal tract (GI-GVHD).
ALXN1007 is being developed by Alexion Pharmaceuticals, Inc.
About orphan designation
Orphan designation from the European Commission provides regulatory and financial incentives for companies to develop and market therapies that treat a life-threatening or chronically debilitating condition affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity in the European Union if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure.
Phase 2 trial of ALXN1007
Results from the phase 2 trial of ALXN1007 in patients with newly diagnosed, acute GI-GVHD were presented at the 21st Congress of the European Hematology Association (abstract LB2269).
The presentation included 15 patients with biopsy-confirmed acute GI-GVHD. The patients had a median age of 60 (range, 25-69), and 60% were male.
Patients had acute myeloid leukemia/myelodysplastic syndrome (n=8), acute lymphoblastic leukemia (n=2), acute lymphocytic leukemia (n=1), acute myeloblastic leukemia (n=1), aplastic anemia (n=1), cutaneous T-cell lymphoma (n=1), or mantle cell lymphoma (n=1).
Most patients received transplants from matched, unrelated donors (n=11), 3 had matched, related donors, and 1 had a mismatched donor. Ten patients received peripheral blood grafts, 4 received cord blood, and 1 received a bone marrow transplant.
Patients had grade 1 (n=7), grade 2 (n=2), and grade 3 acute GI-GVHD (n=6).
The patients received weekly doses of ALXN1007 at 10 mg/kg, in combination with methylprednisolone at an initial dose of 2 mg/kg, through day 56.
Thirteen patients were evaluable for efficacy. One patient experienced leukemia relapse at day 18, and 1 withdrew from the study early.
The overall acute GVHD response rate was 77% (10/13), both at day 28 and day 56. The complete GI-GVHD response rate was 69% at day 28 and 77% at day 56.
At day 180, the nonrelapse mortality rate was 12.5%, and the overall survival rate was 69.2%.
All of the patients had treatment-emergent adverse events (AEs), and 11 patients (69%) had serious treatment-emergent AEs.
Five patients experienced a total of 12 treatment-related AEs (1 case each)—adenovirus infection, bronchopulmonary aspergillosis, chills, corona virus infection, viral cystitis, Epstein-Barr virus infection, hypersensitivity, influenza, influenza-like illness, infusion-related reaction, respiratory syncytial virus infection, and tremor.
There were 6 deaths, but none were considered treatment-related. ![]()
Photo by Linda Bartlett
The European Commission has granted orphan drug designation to ALXN1007 for the treatment of graft-versus-host disease (GVHD).
ALXN1007 is an anti-inflammatory monoclonal antibody targeting complement protein C5a.
The drug is currently under investigation in a phase 2 trial of patients with newly diagnosed acute GVHD of the lower gastrointestinal tract (GI-GVHD).
ALXN1007 is being developed by Alexion Pharmaceuticals, Inc.
About orphan designation
Orphan designation from the European Commission provides regulatory and financial incentives for companies to develop and market therapies that treat a life-threatening or chronically debilitating condition affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity in the European Union if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure.
Phase 2 trial of ALXN1007
Results from the phase 2 trial of ALXN1007 in patients with newly diagnosed, acute GI-GVHD were presented at the 21st Congress of the European Hematology Association (abstract LB2269).
The presentation included 15 patients with biopsy-confirmed acute GI-GVHD. The patients had a median age of 60 (range, 25-69), and 60% were male.
Patients had acute myeloid leukemia/myelodysplastic syndrome (n=8), acute lymphoblastic leukemia (n=2), acute lymphocytic leukemia (n=1), acute myeloblastic leukemia (n=1), aplastic anemia (n=1), cutaneous T-cell lymphoma (n=1), or mantle cell lymphoma (n=1).
Most patients received transplants from matched, unrelated donors (n=11), 3 had matched, related donors, and 1 had a mismatched donor. Ten patients received peripheral blood grafts, 4 received cord blood, and 1 received a bone marrow transplant.
Patients had grade 1 (n=7), grade 2 (n=2), and grade 3 acute GI-GVHD (n=6).
The patients received weekly doses of ALXN1007 at 10 mg/kg, in combination with methylprednisolone at an initial dose of 2 mg/kg, through day 56.
Thirteen patients were evaluable for efficacy. One patient experienced leukemia relapse at day 18, and 1 withdrew from the study early.
The overall acute GVHD response rate was 77% (10/13), both at day 28 and day 56. The complete GI-GVHD response rate was 69% at day 28 and 77% at day 56.
At day 180, the nonrelapse mortality rate was 12.5%, and the overall survival rate was 69.2%.
All of the patients had treatment-emergent adverse events (AEs), and 11 patients (69%) had serious treatment-emergent AEs.
Five patients experienced a total of 12 treatment-related AEs (1 case each)—adenovirus infection, bronchopulmonary aspergillosis, chills, corona virus infection, viral cystitis, Epstein-Barr virus infection, hypersensitivity, influenza, influenza-like illness, infusion-related reaction, respiratory syncytial virus infection, and tremor.
There were 6 deaths, but none were considered treatment-related. ![]()
Intake of Vitamins and Minerals Is Inadequate for Most Americans: What Should We Advise Patients About Supplements?


When should primary care physicians prescribe antibiotics to children with respiratory infection symptoms?
Duration of illness, age, and the presence of specific symptoms are key predictors of hospitalization risk due to respiratory infection, according to a study published in The Lancet. These demographic and clinical factors should guide a primary care physician’s decision to prescribe antibiotics.
“More than 80% of all health-service antibiotics [are] prescribed by primary care clinicians,” reported Alastair Hay, MD, of the University of Bristol, England, and his associates.

“Antibiotic prescribing in primary care is increasing and directly affects antimicrobial resistance,” the researchers noted, adding that many primary care clinicians prescribe antibiotics to pediatric patients with respiratory tract infections and/or cough to “mitigate perceived risk of future hospital admission and complications.”
A total of 8,394 pediatric patients who presented with acute cough and one or more other symptoms of respiratory tract infection (such as fever and coryza) were enrolled in the study by primary care physicians at 247 clinical sites in England. All eligible patients were between the ages of 3 months and 16 years; children were excluded if they presented with noninfective exacerbation of asthma, were at high risk of serious infection, or required a throat swab. The study’s primary outcome was hospital admission for any respiratory tract infection within 30 days of enrollment; the data were collected from a review of electronic medical records (Lancet. 2016. Sept 1. doi: 10.1016/S2213-2600[16]30223-5).
The median age of the pediatric cohort was 3 years, 52% were male, and 78% were white. A total of 3,121 patients (37%) were prescribed an antibiotic by their primary care physicians, but only 78 patients (0.9%) were admitted to the hospital, and 27% of discharge diagnoses suggested a possible bacterial cause (lower respiratory tract infection, tonsillitis, and pneumonia).
Multivariate modeling with bootstrap validation demonstrated that duration of illness, age, and the presence or absence of specific respiratory symptoms were the key factors that should be used to identify children at low, normal, and high risk for hospitalization due to respiratory infection. Younger patients with shorter illness durations who presented with wheeze, fever, vomiting, intercostal or subcostal recession, and/or asthma were at higher risk for hospitalization.
“Our data show that 1,846 (33%) of the very-low-risk stratum children received antibiotics. Because these children represent the majority (67%) of all the participants, a 10% overall reduction in antibiotic prescription would be achieved if prescription in this group halved, remained static in the normal risk stratum, and increased to 90% in the high risk stratum, resulting in a similar effect size to other contemporary antimicrobial stewardship interventions,” Dr. Hay and his associates concluded.
This study received funding and sponsorship from the National Institute for Health Research and the University of Bristol. Two investigators reported receiving financial compensation or honoraria from multiple companies including companies with an interest in diagnostic microbiology in respiratory tract infections.
On Twitter @jessnicolecraig
Duration of illness, age, and the presence of specific symptoms are key predictors of hospitalization risk due to respiratory infection, according to a study published in The Lancet. These demographic and clinical factors should guide a primary care physician’s decision to prescribe antibiotics.
“More than 80% of all health-service antibiotics [are] prescribed by primary care clinicians,” reported Alastair Hay, MD, of the University of Bristol, England, and his associates.
“Antibiotic prescribing in primary care is increasing and directly affects antimicrobial resistance,” the researchers noted, adding that many primary care clinicians prescribe antibiotics to pediatric patients with respiratory tract infections and/or cough to “mitigate perceived risk of future hospital admission and complications.”
A total of 8,394 pediatric patients who presented with acute cough and one or more other symptoms of respiratory tract infection (such as fever and coryza) were enrolled in the study by primary care physicians at 247 clinical sites in England. All eligible patients were between the ages of 3 months and 16 years; children were excluded if they presented with noninfective exacerbation of asthma, were at high risk of serious infection, or required a throat swab. The study’s primary outcome was hospital admission for any respiratory tract infection within 30 days of enrollment; the data were collected from a review of electronic medical records (Lancet. 2016. Sept 1. doi: 10.1016/S2213-2600[16]30223-5).
The median age of the pediatric cohort was 3 years, 52% were male, and 78% were white. A total of 3,121 patients (37%) were prescribed an antibiotic by their primary care physicians, but only 78 patients (0.9%) were admitted to the hospital, and 27% of discharge diagnoses suggested a possible bacterial cause (lower respiratory tract infection, tonsillitis, and pneumonia).
Multivariate modeling with bootstrap validation demonstrated that duration of illness, age, and the presence or absence of specific respiratory symptoms were the key factors that should be used to identify children at low, normal, and high risk for hospitalization due to respiratory infection. Younger patients with shorter illness durations who presented with wheeze, fever, vomiting, intercostal or subcostal recession, and/or asthma were at higher risk for hospitalization.
“Our data show that 1,846 (33%) of the very-low-risk stratum children received antibiotics. Because these children represent the majority (67%) of all the participants, a 10% overall reduction in antibiotic prescription would be achieved if prescription in this group halved, remained static in the normal risk stratum, and increased to 90% in the high risk stratum, resulting in a similar effect size to other contemporary antimicrobial stewardship interventions,” Dr. Hay and his associates concluded.
This study received funding and sponsorship from the National Institute for Health Research and the University of Bristol. Two investigators reported receiving financial compensation or honoraria from multiple companies including companies with an interest in diagnostic microbiology in respiratory tract infections.
On Twitter @jessnicolecraig
Duration of illness, age, and the presence of specific symptoms are key predictors of hospitalization risk due to respiratory infection, according to a study published in The Lancet. These demographic and clinical factors should guide a primary care physician’s decision to prescribe antibiotics.
“More than 80% of all health-service antibiotics [are] prescribed by primary care clinicians,” reported Alastair Hay, MD, of the University of Bristol, England, and his associates.
“Antibiotic prescribing in primary care is increasing and directly affects antimicrobial resistance,” the researchers noted, adding that many primary care clinicians prescribe antibiotics to pediatric patients with respiratory tract infections and/or cough to “mitigate perceived risk of future hospital admission and complications.”
A total of 8,394 pediatric patients who presented with acute cough and one or more other symptoms of respiratory tract infection (such as fever and coryza) were enrolled in the study by primary care physicians at 247 clinical sites in England. All eligible patients were between the ages of 3 months and 16 years; children were excluded if they presented with noninfective exacerbation of asthma, were at high risk of serious infection, or required a throat swab. The study’s primary outcome was hospital admission for any respiratory tract infection within 30 days of enrollment; the data were collected from a review of electronic medical records (Lancet. 2016. Sept 1. doi: 10.1016/S2213-2600[16]30223-5).
The median age of the pediatric cohort was 3 years, 52% were male, and 78% were white. A total of 3,121 patients (37%) were prescribed an antibiotic by their primary care physicians, but only 78 patients (0.9%) were admitted to the hospital, and 27% of discharge diagnoses suggested a possible bacterial cause (lower respiratory tract infection, tonsillitis, and pneumonia).
Multivariate modeling with bootstrap validation demonstrated that duration of illness, age, and the presence or absence of specific respiratory symptoms were the key factors that should be used to identify children at low, normal, and high risk for hospitalization due to respiratory infection. Younger patients with shorter illness durations who presented with wheeze, fever, vomiting, intercostal or subcostal recession, and/or asthma were at higher risk for hospitalization.
“Our data show that 1,846 (33%) of the very-low-risk stratum children received antibiotics. Because these children represent the majority (67%) of all the participants, a 10% overall reduction in antibiotic prescription would be achieved if prescription in this group halved, remained static in the normal risk stratum, and increased to 90% in the high risk stratum, resulting in a similar effect size to other contemporary antimicrobial stewardship interventions,” Dr. Hay and his associates concluded.
This study received funding and sponsorship from the National Institute for Health Research and the University of Bristol. Two investigators reported receiving financial compensation or honoraria from multiple companies including companies with an interest in diagnostic microbiology in respiratory tract infections.
On Twitter @jessnicolecraig
FROM THE LANCET
Key clinical point: Duration of illness, age, and the presence of specific symptoms are key predictors of hospitalization risk due to respiratory infection. These factors should guide a primary care physician’s decision to prescribe antibiotics.
Major finding: Younger patients with shorter illness durations who presented with wheeze, fever, vomiting, intercostal or subcostal recession, and/or asthma are at higher risk for hospitalization.
Data source: A prospective, prognostic cohort study of 8,394 children.
Disclosures: This study received funding and sponsorship from the National Institute for Health Research and the University of Bristol. Two investigators reported receiving financial compensation or honoraria from multiple companies, including those with an interest in diagnostic microbiology in respiratory tract infections.

VIDEO: Coronary DES outperform BMS mostly on restenosis
ROME – The difference between contemporary drug-eluting coronary stents and bare-metal stents is not very great, a large Norwegian coronary stent trial showed.
Today’s drug-eluting stents (DES), often called second-generation DES, largely do only what they were designed to do, compared with bare-metal stents (BMS): reduce the rate of stent restenosis and the need for target-lesion revascularization.
“The long-term benefit of contemporary DES over BMS was less that expected,” Kaare H. Bønaa, MD, reported at the annual congress of the European Society of Cardiology.

Results from the Norwegian Coronary Stent Trial (NORSTENT), run with 9,013 patients, showed that patients who received one or more drug-eluting stents had, during nearly 5 years of follow-up, a 5% absolute drop in target-lesion revascularizations (a 53% relative risk reduction), and a 3.3% reduction in all revascularizations (a 24% relative risk reduction), compared with patients who received bare-metal stents, said Dr. Bønaa.
The results also showed that patients who received DES had a 0.4% reduced rate of stent thrombosis (a 36% relative risk reduction), compared with patients treated with BMS during nearly 5 years of follow-up. All three differences were statistically significant.
But the NORSTENT findings also documented that the patients who received either DES or BMS had virtually identical rates of all-cause deaths and nonfatal myocardial infarctions. And, on average, the two different types of coronary stents produced identical improvements in patients’ quality of life, reported Dr. Bønaa, a professor and researcher in the Clinic for Heart Disease at St. Olav’s University Hospital in Trondheim, Norway.
The study’s primary endpoint was the combined rate of death or nonfatal MI, and so the nonsignificant difference in that outcome between the two study arms meant that, formally, the NORSTENT trial produced a neutral result. Concurrently with his report, the results appeared in an article online (New Engl J Med. 2016 Aug 30. doi: 10.1056/NEJMoa1607991).
“The difference between the two stent types is not as great as we thought. Patients who get DES do not live longer or better” than those who receive BMS, Dr. Bønaa said. “We suggest that both contemporary DES and BMS can be recommended for contemporary revascularization. The results open up use of BMS for certain patients,” such as those scheduled for surgery or patients who cannot tolerate or afford the drugs used for dual antiplatelet therapy following coronary stent placement.

But the designated discussant for the study, Stefan James, MD, insisted that recent-generation DES “should remain recommended over BMS,” particularly the specific DES that underwent testing in randomized trials that used hard clinical endpoints. The 2014 revascularization guidelines of the European Society of Cardiology recommend new-generation DES over BMS, he noted.
In addition, “BMS should not be specifically recommended for patients at high risk of stent thrombosis or for patients who do not tolerate dual-antiplatelet therapy,” said Dr. James, professor of cardiology at Uppsala University in Sweden.
NORSTENT ran at eight centers in Norway during 2008-2011, and enrolled patients either had acute coronary syndrome (71% of those in the study) or stable coronary disease. Patients averaged 63 years old. The trial excluded patients with prior stents or bifurcated coronary lesions. Enrolled patients received, on average, 1.7 stents. The specific stent in each class that patients received was left to the discretion of each operator, and 95% of patients in the DES arm received a second-generation device. All patients in both arms of the study received dual-antiplatelet therapy for 9 months.
The finding that DES cut the rate of revascularization procedures by 3.3%, compared with patients treated with BMS, means that, on average, clinicians would need to treat 30 patients with DES to avoid the need for one additional repeat revascularization procedure that would occur if BMS were used instead.
That number needed to treat of 30 to avoid one repeat revascularization may seem high, but the money saved that way would still counterbalance the incremental cost of a DES over a BMS, which today in Europe would be about 50-100 euros, noted one cardiologist.
If you multiply 30 procedures by 100 extra euros per stent and by an average of 1.7 stents per patient, you may spend 5,100 euros, less than the cost of a repeat revascularization procedure, commented Carlo Di Mario, MD, a professor of cardiology and an interventional cardiologist at Royal Brompton & Harefield Hospitals in London.
In a video interview, Steen D. Kristensen, MD, of Aarhus University, Denmark, discussed the NORSTENT findings and their implications.
NORSTENT received no commercial support. Dr. Bønaa and Dr. Di Mario had no disclosures. Dr. James has been a consultant to Boston Scientific and has received research support from Boston Scientific and Abbott Vascular.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
NORSTENT was a very well-performed trial. It produced a neutral result for its primary endpoint, but for the secondary endpoint of repeat revascularization, there were significantly more events using bare-metal stents. This is a major finding, and NORSTENT’s design make the results very generalizable.
It may be slightly surprising that the newer drug-eluting stents did not perform better for the primary endpoint of reducing deaths and MIs during 5 years of follow-up, but seeing a difference in the revascularization rate is not surprising; that is what we would expect. We use DES to reduce the problem of restenosis. Results from several earlier studies that had compared DES with BMS had suggested other benefits from DES, and that is also what the European Society of Cardiology guidelines say.
I will not go home now and start using BMS in my own practice. I will continue to use DES, because they have an advantage. I use BMS in patients who cannot tolerate long-term treatment with dual antiplatelet therapy. The results are encouraging for centers where there is a large price difference between DES and BMS, but that is not the case where I practice in Denmark.
Steen D. Kristensen, MD, is a professor of interventional cardiologist at Aarhus University, Denmark. He made these comments in an interview. He had no relevant disclosures.
NORSTENT was a very well-performed trial. It produced a neutral result for its primary endpoint, but for the secondary endpoint of repeat revascularization, there were significantly more events using bare-metal stents. This is a major finding, and NORSTENT’s design make the results very generalizable.
It may be slightly surprising that the newer drug-eluting stents did not perform better for the primary endpoint of reducing deaths and MIs during 5 years of follow-up, but seeing a difference in the revascularization rate is not surprising; that is what we would expect. We use DES to reduce the problem of restenosis. Results from several earlier studies that had compared DES with BMS had suggested other benefits from DES, and that is also what the European Society of Cardiology guidelines say.
I will not go home now and start using BMS in my own practice. I will continue to use DES, because they have an advantage. I use BMS in patients who cannot tolerate long-term treatment with dual antiplatelet therapy. The results are encouraging for centers where there is a large price difference between DES and BMS, but that is not the case where I practice in Denmark.
Steen D. Kristensen, MD, is a professor of interventional cardiologist at Aarhus University, Denmark. He made these comments in an interview. He had no relevant disclosures.
NORSTENT was a very well-performed trial. It produced a neutral result for its primary endpoint, but for the secondary endpoint of repeat revascularization, there were significantly more events using bare-metal stents. This is a major finding, and NORSTENT’s design make the results very generalizable.
It may be slightly surprising that the newer drug-eluting stents did not perform better for the primary endpoint of reducing deaths and MIs during 5 years of follow-up, but seeing a difference in the revascularization rate is not surprising; that is what we would expect. We use DES to reduce the problem of restenosis. Results from several earlier studies that had compared DES with BMS had suggested other benefits from DES, and that is also what the European Society of Cardiology guidelines say.
I will not go home now and start using BMS in my own practice. I will continue to use DES, because they have an advantage. I use BMS in patients who cannot tolerate long-term treatment with dual antiplatelet therapy. The results are encouraging for centers where there is a large price difference between DES and BMS, but that is not the case where I practice in Denmark.
Steen D. Kristensen, MD, is a professor of interventional cardiologist at Aarhus University, Denmark. He made these comments in an interview. He had no relevant disclosures.
ROME – The difference between contemporary drug-eluting coronary stents and bare-metal stents is not very great, a large Norwegian coronary stent trial showed.
Today’s drug-eluting stents (DES), often called second-generation DES, largely do only what they were designed to do, compared with bare-metal stents (BMS): reduce the rate of stent restenosis and the need for target-lesion revascularization.
“The long-term benefit of contemporary DES over BMS was less that expected,” Kaare H. Bønaa, MD, reported at the annual congress of the European Society of Cardiology.
Results from the Norwegian Coronary Stent Trial (NORSTENT), run with 9,013 patients, showed that patients who received one or more drug-eluting stents had, during nearly 5 years of follow-up, a 5% absolute drop in target-lesion revascularizations (a 53% relative risk reduction), and a 3.3% reduction in all revascularizations (a 24% relative risk reduction), compared with patients who received bare-metal stents, said Dr. Bønaa.
The results also showed that patients who received DES had a 0.4% reduced rate of stent thrombosis (a 36% relative risk reduction), compared with patients treated with BMS during nearly 5 years of follow-up. All three differences were statistically significant.
But the NORSTENT findings also documented that the patients who received either DES or BMS had virtually identical rates of all-cause deaths and nonfatal myocardial infarctions. And, on average, the two different types of coronary stents produced identical improvements in patients’ quality of life, reported Dr. Bønaa, a professor and researcher in the Clinic for Heart Disease at St. Olav’s University Hospital in Trondheim, Norway.
The study’s primary endpoint was the combined rate of death or nonfatal MI, and so the nonsignificant difference in that outcome between the two study arms meant that, formally, the NORSTENT trial produced a neutral result. Concurrently with his report, the results appeared in an article online (New Engl J Med. 2016 Aug 30. doi: 10.1056/NEJMoa1607991).
“The difference between the two stent types is not as great as we thought. Patients who get DES do not live longer or better” than those who receive BMS, Dr. Bønaa said. “We suggest that both contemporary DES and BMS can be recommended for contemporary revascularization. The results open up use of BMS for certain patients,” such as those scheduled for surgery or patients who cannot tolerate or afford the drugs used for dual antiplatelet therapy following coronary stent placement.
But the designated discussant for the study, Stefan James, MD, insisted that recent-generation DES “should remain recommended over BMS,” particularly the specific DES that underwent testing in randomized trials that used hard clinical endpoints. The 2014 revascularization guidelines of the European Society of Cardiology recommend new-generation DES over BMS, he noted.
In addition, “BMS should not be specifically recommended for patients at high risk of stent thrombosis or for patients who do not tolerate dual-antiplatelet therapy,” said Dr. James, professor of cardiology at Uppsala University in Sweden.
NORSTENT ran at eight centers in Norway during 2008-2011, and enrolled patients either had acute coronary syndrome (71% of those in the study) or stable coronary disease. Patients averaged 63 years old. The trial excluded patients with prior stents or bifurcated coronary lesions. Enrolled patients received, on average, 1.7 stents. The specific stent in each class that patients received was left to the discretion of each operator, and 95% of patients in the DES arm received a second-generation device. All patients in both arms of the study received dual-antiplatelet therapy for 9 months.
The finding that DES cut the rate of revascularization procedures by 3.3%, compared with patients treated with BMS, means that, on average, clinicians would need to treat 30 patients with DES to avoid the need for one additional repeat revascularization procedure that would occur if BMS were used instead.
That number needed to treat of 30 to avoid one repeat revascularization may seem high, but the money saved that way would still counterbalance the incremental cost of a DES over a BMS, which today in Europe would be about 50-100 euros, noted one cardiologist.
If you multiply 30 procedures by 100 extra euros per stent and by an average of 1.7 stents per patient, you may spend 5,100 euros, less than the cost of a repeat revascularization procedure, commented Carlo Di Mario, MD, a professor of cardiology and an interventional cardiologist at Royal Brompton & Harefield Hospitals in London.
In a video interview, Steen D. Kristensen, MD, of Aarhus University, Denmark, discussed the NORSTENT findings and their implications.
NORSTENT received no commercial support. Dr. Bønaa and Dr. Di Mario had no disclosures. Dr. James has been a consultant to Boston Scientific and has received research support from Boston Scientific and Abbott Vascular.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
ROME – The difference between contemporary drug-eluting coronary stents and bare-metal stents is not very great, a large Norwegian coronary stent trial showed.
Today’s drug-eluting stents (DES), often called second-generation DES, largely do only what they were designed to do, compared with bare-metal stents (BMS): reduce the rate of stent restenosis and the need for target-lesion revascularization.
“The long-term benefit of contemporary DES over BMS was less that expected,” Kaare H. Bønaa, MD, reported at the annual congress of the European Society of Cardiology.
Results from the Norwegian Coronary Stent Trial (NORSTENT), run with 9,013 patients, showed that patients who received one or more drug-eluting stents had, during nearly 5 years of follow-up, a 5% absolute drop in target-lesion revascularizations (a 53% relative risk reduction), and a 3.3% reduction in all revascularizations (a 24% relative risk reduction), compared with patients who received bare-metal stents, said Dr. Bønaa.
The results also showed that patients who received DES had a 0.4% reduced rate of stent thrombosis (a 36% relative risk reduction), compared with patients treated with BMS during nearly 5 years of follow-up. All three differences were statistically significant.
But the NORSTENT findings also documented that the patients who received either DES or BMS had virtually identical rates of all-cause deaths and nonfatal myocardial infarctions. And, on average, the two different types of coronary stents produced identical improvements in patients’ quality of life, reported Dr. Bønaa, a professor and researcher in the Clinic for Heart Disease at St. Olav’s University Hospital in Trondheim, Norway.
The study’s primary endpoint was the combined rate of death or nonfatal MI, and so the nonsignificant difference in that outcome between the two study arms meant that, formally, the NORSTENT trial produced a neutral result. Concurrently with his report, the results appeared in an article online (New Engl J Med. 2016 Aug 30. doi: 10.1056/NEJMoa1607991).
“The difference between the two stent types is not as great as we thought. Patients who get DES do not live longer or better” than those who receive BMS, Dr. Bønaa said. “We suggest that both contemporary DES and BMS can be recommended for contemporary revascularization. The results open up use of BMS for certain patients,” such as those scheduled for surgery or patients who cannot tolerate or afford the drugs used for dual antiplatelet therapy following coronary stent placement.
But the designated discussant for the study, Stefan James, MD, insisted that recent-generation DES “should remain recommended over BMS,” particularly the specific DES that underwent testing in randomized trials that used hard clinical endpoints. The 2014 revascularization guidelines of the European Society of Cardiology recommend new-generation DES over BMS, he noted.
In addition, “BMS should not be specifically recommended for patients at high risk of stent thrombosis or for patients who do not tolerate dual-antiplatelet therapy,” said Dr. James, professor of cardiology at Uppsala University in Sweden.
NORSTENT ran at eight centers in Norway during 2008-2011, and enrolled patients either had acute coronary syndrome (71% of those in the study) or stable coronary disease. Patients averaged 63 years old. The trial excluded patients with prior stents or bifurcated coronary lesions. Enrolled patients received, on average, 1.7 stents. The specific stent in each class that patients received was left to the discretion of each operator, and 95% of patients in the DES arm received a second-generation device. All patients in both arms of the study received dual-antiplatelet therapy for 9 months.
The finding that DES cut the rate of revascularization procedures by 3.3%, compared with patients treated with BMS, means that, on average, clinicians would need to treat 30 patients with DES to avoid the need for one additional repeat revascularization procedure that would occur if BMS were used instead.
That number needed to treat of 30 to avoid one repeat revascularization may seem high, but the money saved that way would still counterbalance the incremental cost of a DES over a BMS, which today in Europe would be about 50-100 euros, noted one cardiologist.
If you multiply 30 procedures by 100 extra euros per stent and by an average of 1.7 stents per patient, you may spend 5,100 euros, less than the cost of a repeat revascularization procedure, commented Carlo Di Mario, MD, a professor of cardiology and an interventional cardiologist at Royal Brompton & Harefield Hospitals in London.
In a video interview, Steen D. Kristensen, MD, of Aarhus University, Denmark, discussed the NORSTENT findings and their implications.
NORSTENT received no commercial support. Dr. Bønaa and Dr. Di Mario had no disclosures. Dr. James has been a consultant to Boston Scientific and has received research support from Boston Scientific and Abbott Vascular.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
AT THE ESC CONGRESS 2016
Key clinical point: The benefit from coronary revascularization with drug-eluting stents, compared with bare-metal stents, was mostly in a reduced need for repeat revascularization, with no difference in mortality or MIs during 5 years of follow-up.
Major finding: Thirty patients need to be treated with drug-eluting stents to prevent one repeat revascularization, compared with bare-metal stents.
Data source: NORSTENT, a randomized, multicenter trial with 9,013 patients.
Disclosures: NORSTENT received no commercial support. Dr. Bønaa and Dr. Di Mario had no disclosures. Dr. James has been a consultant to Boston Scientific and has received research support from Boston Scientific and Abbott Vascular.

VIDEO: Withdrawing antipsychotics is safe and feasible in long-term care
TORONTO – Antipsychotics can be safely withdrawn from many dementia patients in long-term care facilities, two new studies from Australia and Canada have determined.
When the drugs were withdrawn and supplanted with behavior-centered care in the Australian study, 80% of patients experienced no relapse of symptoms, Henry Brodaty, MD, DSc, said at the Alzheimer’s Association International Conference 2016.

“We saw no significant changes at all in agitation, aggression, delusions, or hallucinations,” Dr. Brodaty, the Scientia Professor of Ageing and Mental Health, University of New South Wales, Australia, said in an interview. “Were we surprised at this? No. Because for the majority of these patients, the medications were inappropriately prescribed.”
The 12-month Australian study is still in the process of tracking outcomes after antipsychotic withdrawal. But the Canadian study found great benefits, said Selma Didic, an improvement analyst with the Canadian Foundation for Healthcare Improvement in Ottawa. “We saw falls decrease by 20%. The incidence of verbal abuse and socially disruptive behavior actually decreased as well.”
In fact, she said, patients who discontinued the medications actually started behaving better than the comparator group that stayed on them.
The Australian experience
Dr. Brodaty discussed the HALT (Halting Antipsychotic Use in Long-Term Care) study. HALT is a single-arm, 12-month longitudinal study carried out in 23 nursing homes in New South Wales.
The study team worked with nursing leadership in each facility to identify patients who might be eligible for the program. In order to enroll, each patient’s family and general physician had to agree to a trial of deprescribing. Physicians were instructed to wean patients off the medication by decreasing the dose by half once a week. Most patients were able to stop within a couple of weeks, Dr. Brodaty said.
Getting buy-in wasn’t always easy, he noted. “Some families didn’t want to rock the boat, and some physicians were resistant,” to the idea. Overall, “Families and nurses were very, very worried” about the prospect of dropping drugs that were seen as helpful in everyday patient management.
But getting rid of the medications was just half the picture. Training nurses and care staff to intervene in problematic behaviors without resorting to drugs was just as important. A nurse-leader at each facility received training in person-centered care, and then trained the rest of the staff. This wasn’t always an easy idea to embrace, either, Dr. Brodaty said, especially since nursing staff often leads the discussion about the need for drugs to manage behavioral problems.
“Nursing staff are very task oriented, focused on dressing, bathing, eating, and toileting. They work very hard, and they don’t always have time to sit down and talk to resistant patients. It takes a much different attitude to show that you can actually save time by spending time and engaging the patient.”
He related one of his favorite illustrative stories – the milkman who caused a ruckus at bath time. “He got upset and aggressive every night when being put to bed and every morning when being given a shower. The staff spoke to his wife about it. She said that for 40 years, he was accustomed to getting up at 4 a.m. to deliver the milk. He would take a bath at night and get on his track suit and go to bed. Then at 4 a.m., he would get up and be ready to jump in the truck and go.”
When the staff started letting him shower at night and go to bed in his track suit, the milkman’s behavior improved without the need for antipsychotic medications.
“This is what we mean by ‘person-centered care,’ ” Dr. Brodaty said. “We use the ABC paradigm: Addressing the antecedent to the behavior, then the behavior, and then the consequences of the behavior.”
The intervention cohort comprised 139 patients with a mean age of 85 years; most were women. The vast majority (93%) had a diagnosis of dementia. About one-third had Alzheimer’s and one-third vascular dementia. The remainder had other diagnoses, including frontotemporal dementia, Lewy body dementia, and Parkinson’s disease. Common comorbid conditions included depression (56%) and previous stroke (36%). None of the patients had a diagnosis of psychosis.
Risperidone was the most common antipsychotic medication (85%). Other medications were olanzapine, quetiapine, and haloperidol. About 30% had come to the facility on the medication; the others had received it since admission.
Despite the national recommendation to review antipsychotic use every 12 weeks, patients had been on their current antipsychotic for an average of 2 years, and on their current dose for 1 year. In reviewing medications, Dr. Brodaty also found a “concerning” lack of informed consent. In Australia, informed consent for antipsychotic drugs can be given by a family member, but 84% of patients had no documented consent at all.
Of the original group, 125 entered the deprescribing protocol. Of these, 26 (21%) have since resumed their medications, but 79% have done well and are without a relapse of their symptoms or problematic behaviors. An ongoing medication review suggests there has been no concomitant upswing in other psychotropic medications, including benzodiazepines.
Neuropsychiatric symptoms remained stable from baseline. The mean total group score on the Neuropsychiatric Index (NPI) has not changed from its baseline of 30. The mean agitation/aggression NPI subscale has remained about 6, and the mean group score on the Cohen-Mansfield Agitation Inventory about 56. The NPI delusion subscale increased, but the change was nonsignificant, Dr. Brodaty said. The NPI hallucinations subscale decreased slightly, but again the change was nonsignificant.
“Look, we all know antipsychotics are bad for old people, and we all know they are overprescribed,” he said. “Inappropriate use of these medications is an old story, yet we’re still talking about it. Why is this? We have the knowledge now, and we have to build on this knowledge so that we can change practice.”
The Canadian experience
Ms. Didic shared a year-long quality improvement process at 24 long-term care facilities that wanted to improve antipsychotic prescribing for their dementia patients.
The program, which was sponsored by the Canadian Foundation for Healthcare Improvement, used a “train-the-trainer” approach to spread support for antipsychotic deprescribing.

The foundation deployed 15 interdisciplinary teams, which comprised 180 members, including physicians, nurses, pharmacists, recreational therapists, and “clinical champions” who took the methodology directly into participating facilities. Interactive webinars on patient-centered care and deprescribing protocols were part of the process, Ms. Didic said.
In all, 416 patients were included in the outcomes report. Within 12 months, antipsychotics were eliminated in 74 patients (18%) and in 148 (36%), the dosage was reduced.
The benefits of these changes were striking, Ms. Didic said. There were fewer falls and reductions in verbal abuse, care resistance, and socially inappropriate behaviors. These issues either remained the same or got worse in patients who did not decrease antipsychotics. Again, there was no concomitant increase in other psychotropic medications.
The results show that changing the focus from medication-first to behavior-first care is institutionally feasible, Ms. Didic said.
Staff members’ assessments of the program and its personal and institutional impact were positive:
• 91% said they instituted regular medication reviews for every resident.
• 92% said old ways of doing things were adjusted to accommodate the new type of care.
• 94% said the new person-centered care was now a standard way of working.
• 84% said the project improved their ability to lead.
• 80% said it improved their ability to communicate.
“Currently, our teams are now spreading and sharing these resources and tools, serving as advisers, and organizing clinical training and workshops,” for other Canadian nursing homes that want to adopt the strategy.
Dr. Richard Caselli, professor of neurology at the Mayo Clinic, Scottsdale, Ariz., commented on the issues surrounding antipsychotic prescribing in long-term care facilities in a video interview.
Neither Ms. Didic nor Dr. Brodaty had any financial declarations.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @alz_gal
TORONTO – Antipsychotics can be safely withdrawn from many dementia patients in long-term care facilities, two new studies from Australia and Canada have determined.
When the drugs were withdrawn and supplanted with behavior-centered care in the Australian study, 80% of patients experienced no relapse of symptoms, Henry Brodaty, MD, DSc, said at the Alzheimer’s Association International Conference 2016.
“We saw no significant changes at all in agitation, aggression, delusions, or hallucinations,” Dr. Brodaty, the Scientia Professor of Ageing and Mental Health, University of New South Wales, Australia, said in an interview. “Were we surprised at this? No. Because for the majority of these patients, the medications were inappropriately prescribed.”
The 12-month Australian study is still in the process of tracking outcomes after antipsychotic withdrawal. But the Canadian study found great benefits, said Selma Didic, an improvement analyst with the Canadian Foundation for Healthcare Improvement in Ottawa. “We saw falls decrease by 20%. The incidence of verbal abuse and socially disruptive behavior actually decreased as well.”
In fact, she said, patients who discontinued the medications actually started behaving better than the comparator group that stayed on them.
The Australian experience
Dr. Brodaty discussed the HALT (Halting Antipsychotic Use in Long-Term Care) study. HALT is a single-arm, 12-month longitudinal study carried out in 23 nursing homes in New South Wales.
The study team worked with nursing leadership in each facility to identify patients who might be eligible for the program. In order to enroll, each patient’s family and general physician had to agree to a trial of deprescribing. Physicians were instructed to wean patients off the medication by decreasing the dose by half once a week. Most patients were able to stop within a couple of weeks, Dr. Brodaty said.
Getting buy-in wasn’t always easy, he noted. “Some families didn’t want to rock the boat, and some physicians were resistant,” to the idea. Overall, “Families and nurses were very, very worried” about the prospect of dropping drugs that were seen as helpful in everyday patient management.
But getting rid of the medications was just half the picture. Training nurses and care staff to intervene in problematic behaviors without resorting to drugs was just as important. A nurse-leader at each facility received training in person-centered care, and then trained the rest of the staff. This wasn’t always an easy idea to embrace, either, Dr. Brodaty said, especially since nursing staff often leads the discussion about the need for drugs to manage behavioral problems.
“Nursing staff are very task oriented, focused on dressing, bathing, eating, and toileting. They work very hard, and they don’t always have time to sit down and talk to resistant patients. It takes a much different attitude to show that you can actually save time by spending time and engaging the patient.”
He related one of his favorite illustrative stories – the milkman who caused a ruckus at bath time. “He got upset and aggressive every night when being put to bed and every morning when being given a shower. The staff spoke to his wife about it. She said that for 40 years, he was accustomed to getting up at 4 a.m. to deliver the milk. He would take a bath at night and get on his track suit and go to bed. Then at 4 a.m., he would get up and be ready to jump in the truck and go.”
When the staff started letting him shower at night and go to bed in his track suit, the milkman’s behavior improved without the need for antipsychotic medications.
“This is what we mean by ‘person-centered care,’ ” Dr. Brodaty said. “We use the ABC paradigm: Addressing the antecedent to the behavior, then the behavior, and then the consequences of the behavior.”
The intervention cohort comprised 139 patients with a mean age of 85 years; most were women. The vast majority (93%) had a diagnosis of dementia. About one-third had Alzheimer’s and one-third vascular dementia. The remainder had other diagnoses, including frontotemporal dementia, Lewy body dementia, and Parkinson’s disease. Common comorbid conditions included depression (56%) and previous stroke (36%). None of the patients had a diagnosis of psychosis.
Risperidone was the most common antipsychotic medication (85%). Other medications were olanzapine, quetiapine, and haloperidol. About 30% had come to the facility on the medication; the others had received it since admission.
Despite the national recommendation to review antipsychotic use every 12 weeks, patients had been on their current antipsychotic for an average of 2 years, and on their current dose for 1 year. In reviewing medications, Dr. Brodaty also found a “concerning” lack of informed consent. In Australia, informed consent for antipsychotic drugs can be given by a family member, but 84% of patients had no documented consent at all.
Of the original group, 125 entered the deprescribing protocol. Of these, 26 (21%) have since resumed their medications, but 79% have done well and are without a relapse of their symptoms or problematic behaviors. An ongoing medication review suggests there has been no concomitant upswing in other psychotropic medications, including benzodiazepines.
Neuropsychiatric symptoms remained stable from baseline. The mean total group score on the Neuropsychiatric Index (NPI) has not changed from its baseline of 30. The mean agitation/aggression NPI subscale has remained about 6, and the mean group score on the Cohen-Mansfield Agitation Inventory about 56. The NPI delusion subscale increased, but the change was nonsignificant, Dr. Brodaty said. The NPI hallucinations subscale decreased slightly, but again the change was nonsignificant.
“Look, we all know antipsychotics are bad for old people, and we all know they are overprescribed,” he said. “Inappropriate use of these medications is an old story, yet we’re still talking about it. Why is this? We have the knowledge now, and we have to build on this knowledge so that we can change practice.”
The Canadian experience
Ms. Didic shared a year-long quality improvement process at 24 long-term care facilities that wanted to improve antipsychotic prescribing for their dementia patients.
The program, which was sponsored by the Canadian Foundation for Healthcare Improvement, used a “train-the-trainer” approach to spread support for antipsychotic deprescribing.
The foundation deployed 15 interdisciplinary teams, which comprised 180 members, including physicians, nurses, pharmacists, recreational therapists, and “clinical champions” who took the methodology directly into participating facilities. Interactive webinars on patient-centered care and deprescribing protocols were part of the process, Ms. Didic said.
In all, 416 patients were included in the outcomes report. Within 12 months, antipsychotics were eliminated in 74 patients (18%) and in 148 (36%), the dosage was reduced.
The benefits of these changes were striking, Ms. Didic said. There were fewer falls and reductions in verbal abuse, care resistance, and socially inappropriate behaviors. These issues either remained the same or got worse in patients who did not decrease antipsychotics. Again, there was no concomitant increase in other psychotropic medications.
The results show that changing the focus from medication-first to behavior-first care is institutionally feasible, Ms. Didic said.
Staff members’ assessments of the program and its personal and institutional impact were positive:
• 91% said they instituted regular medication reviews for every resident.
• 92% said old ways of doing things were adjusted to accommodate the new type of care.
• 94% said the new person-centered care was now a standard way of working.
• 84% said the project improved their ability to lead.
• 80% said it improved their ability to communicate.
“Currently, our teams are now spreading and sharing these resources and tools, serving as advisers, and organizing clinical training and workshops,” for other Canadian nursing homes that want to adopt the strategy.
Dr. Richard Caselli, professor of neurology at the Mayo Clinic, Scottsdale, Ariz., commented on the issues surrounding antipsychotic prescribing in long-term care facilities in a video interview.
Neither Ms. Didic nor Dr. Brodaty had any financial declarations.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @alz_gal
TORONTO – Antipsychotics can be safely withdrawn from many dementia patients in long-term care facilities, two new studies from Australia and Canada have determined.
When the drugs were withdrawn and supplanted with behavior-centered care in the Australian study, 80% of patients experienced no relapse of symptoms, Henry Brodaty, MD, DSc, said at the Alzheimer’s Association International Conference 2016.
“We saw no significant changes at all in agitation, aggression, delusions, or hallucinations,” Dr. Brodaty, the Scientia Professor of Ageing and Mental Health, University of New South Wales, Australia, said in an interview. “Were we surprised at this? No. Because for the majority of these patients, the medications were inappropriately prescribed.”
The 12-month Australian study is still in the process of tracking outcomes after antipsychotic withdrawal. But the Canadian study found great benefits, said Selma Didic, an improvement analyst with the Canadian Foundation for Healthcare Improvement in Ottawa. “We saw falls decrease by 20%. The incidence of verbal abuse and socially disruptive behavior actually decreased as well.”
In fact, she said, patients who discontinued the medications actually started behaving better than the comparator group that stayed on them.
The Australian experience
Dr. Brodaty discussed the HALT (Halting Antipsychotic Use in Long-Term Care) study. HALT is a single-arm, 12-month longitudinal study carried out in 23 nursing homes in New South Wales.
The study team worked with nursing leadership in each facility to identify patients who might be eligible for the program. In order to enroll, each patient’s family and general physician had to agree to a trial of deprescribing. Physicians were instructed to wean patients off the medication by decreasing the dose by half once a week. Most patients were able to stop within a couple of weeks, Dr. Brodaty said.
Getting buy-in wasn’t always easy, he noted. “Some families didn’t want to rock the boat, and some physicians were resistant,” to the idea. Overall, “Families and nurses were very, very worried” about the prospect of dropping drugs that were seen as helpful in everyday patient management.
But getting rid of the medications was just half the picture. Training nurses and care staff to intervene in problematic behaviors without resorting to drugs was just as important. A nurse-leader at each facility received training in person-centered care, and then trained the rest of the staff. This wasn’t always an easy idea to embrace, either, Dr. Brodaty said, especially since nursing staff often leads the discussion about the need for drugs to manage behavioral problems.
“Nursing staff are very task oriented, focused on dressing, bathing, eating, and toileting. They work very hard, and they don’t always have time to sit down and talk to resistant patients. It takes a much different attitude to show that you can actually save time by spending time and engaging the patient.”
He related one of his favorite illustrative stories – the milkman who caused a ruckus at bath time. “He got upset and aggressive every night when being put to bed and every morning when being given a shower. The staff spoke to his wife about it. She said that for 40 years, he was accustomed to getting up at 4 a.m. to deliver the milk. He would take a bath at night and get on his track suit and go to bed. Then at 4 a.m., he would get up and be ready to jump in the truck and go.”
When the staff started letting him shower at night and go to bed in his track suit, the milkman’s behavior improved without the need for antipsychotic medications.
“This is what we mean by ‘person-centered care,’ ” Dr. Brodaty said. “We use the ABC paradigm: Addressing the antecedent to the behavior, then the behavior, and then the consequences of the behavior.”
The intervention cohort comprised 139 patients with a mean age of 85 years; most were women. The vast majority (93%) had a diagnosis of dementia. About one-third had Alzheimer’s and one-third vascular dementia. The remainder had other diagnoses, including frontotemporal dementia, Lewy body dementia, and Parkinson’s disease. Common comorbid conditions included depression (56%) and previous stroke (36%). None of the patients had a diagnosis of psychosis.
Risperidone was the most common antipsychotic medication (85%). Other medications were olanzapine, quetiapine, and haloperidol. About 30% had come to the facility on the medication; the others had received it since admission.
Despite the national recommendation to review antipsychotic use every 12 weeks, patients had been on their current antipsychotic for an average of 2 years, and on their current dose for 1 year. In reviewing medications, Dr. Brodaty also found a “concerning” lack of informed consent. In Australia, informed consent for antipsychotic drugs can be given by a family member, but 84% of patients had no documented consent at all.
Of the original group, 125 entered the deprescribing protocol. Of these, 26 (21%) have since resumed their medications, but 79% have done well and are without a relapse of their symptoms or problematic behaviors. An ongoing medication review suggests there has been no concomitant upswing in other psychotropic medications, including benzodiazepines.
Neuropsychiatric symptoms remained stable from baseline. The mean total group score on the Neuropsychiatric Index (NPI) has not changed from its baseline of 30. The mean agitation/aggression NPI subscale has remained about 6, and the mean group score on the Cohen-Mansfield Agitation Inventory about 56. The NPI delusion subscale increased, but the change was nonsignificant, Dr. Brodaty said. The NPI hallucinations subscale decreased slightly, but again the change was nonsignificant.
“Look, we all know antipsychotics are bad for old people, and we all know they are overprescribed,” he said. “Inappropriate use of these medications is an old story, yet we’re still talking about it. Why is this? We have the knowledge now, and we have to build on this knowledge so that we can change practice.”
The Canadian experience
Ms. Didic shared a year-long quality improvement process at 24 long-term care facilities that wanted to improve antipsychotic prescribing for their dementia patients.
The program, which was sponsored by the Canadian Foundation for Healthcare Improvement, used a “train-the-trainer” approach to spread support for antipsychotic deprescribing.
The foundation deployed 15 interdisciplinary teams, which comprised 180 members, including physicians, nurses, pharmacists, recreational therapists, and “clinical champions” who took the methodology directly into participating facilities. Interactive webinars on patient-centered care and deprescribing protocols were part of the process, Ms. Didic said.
In all, 416 patients were included in the outcomes report. Within 12 months, antipsychotics were eliminated in 74 patients (18%) and in 148 (36%), the dosage was reduced.
The benefits of these changes were striking, Ms. Didic said. There were fewer falls and reductions in verbal abuse, care resistance, and socially inappropriate behaviors. These issues either remained the same or got worse in patients who did not decrease antipsychotics. Again, there was no concomitant increase in other psychotropic medications.
The results show that changing the focus from medication-first to behavior-first care is institutionally feasible, Ms. Didic said.
Staff members’ assessments of the program and its personal and institutional impact were positive:
• 91% said they instituted regular medication reviews for every resident.
• 92% said old ways of doing things were adjusted to accommodate the new type of care.
• 94% said the new person-centered care was now a standard way of working.
• 84% said the project improved their ability to lead.
• 80% said it improved their ability to communicate.
“Currently, our teams are now spreading and sharing these resources and tools, serving as advisers, and organizing clinical training and workshops,” for other Canadian nursing homes that want to adopt the strategy.
Dr. Richard Caselli, professor of neurology at the Mayo Clinic, Scottsdale, Ariz., commented on the issues surrounding antipsychotic prescribing in long-term care facilities in a video interview.
Neither Ms. Didic nor Dr. Brodaty had any financial declarations.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @alz_gal
EXPERT ANALYSIS FROM AAIC 2016

Thick Scaly Plaques on the Wrists, Knees, and Feet
The Diagnosis: Secondary Syphilis
Syphilis, known as the great mimicker, has a wide-ranging clinical and histologic presentation. There can be overlapping features with many of the entities included in the differential diagnoses. As our patient exemplifies, clinicians and pathologists must have a high index of suspicion, and any concerning features should lead to a more in-depth patient history, spirochete stains, and serologic testing.
Our patient was seen by several dermatologists over the course of 2 years and therapy with topical steroids failed. He was eager to pursue more aggressive therapy with methotrexate, and a punch biopsy was performed to confirm the diagnosis of psoriasis prior to initiating treatment. Hematoxylin and eosin staining results on low power can be seen in Figure 1A. Medium-power view demonstrated vacuolar interface dermatitis (Figure 1B) with psoriasiform epidermal hyperplasia with slender elongation of rete ridges; neutrophils in the stratum corneum; endothelial cell swelling (Figure 1C); and mixed infiltrate with high plasma cells (Figure 1D), lymphocytes, and histiocytes. Although the biopsy results were psoriasiform, there was high suspicion for syphilis in this case. Additional staining for spirochetes was performed with syphilis immunohistochemical stain1 (Figure 2), which revealed spirochetes present on the patient's biopsy, confirming the diagnosis of syphilis. Warthin-Starry stain also can be performed to confirm the diagnosis.
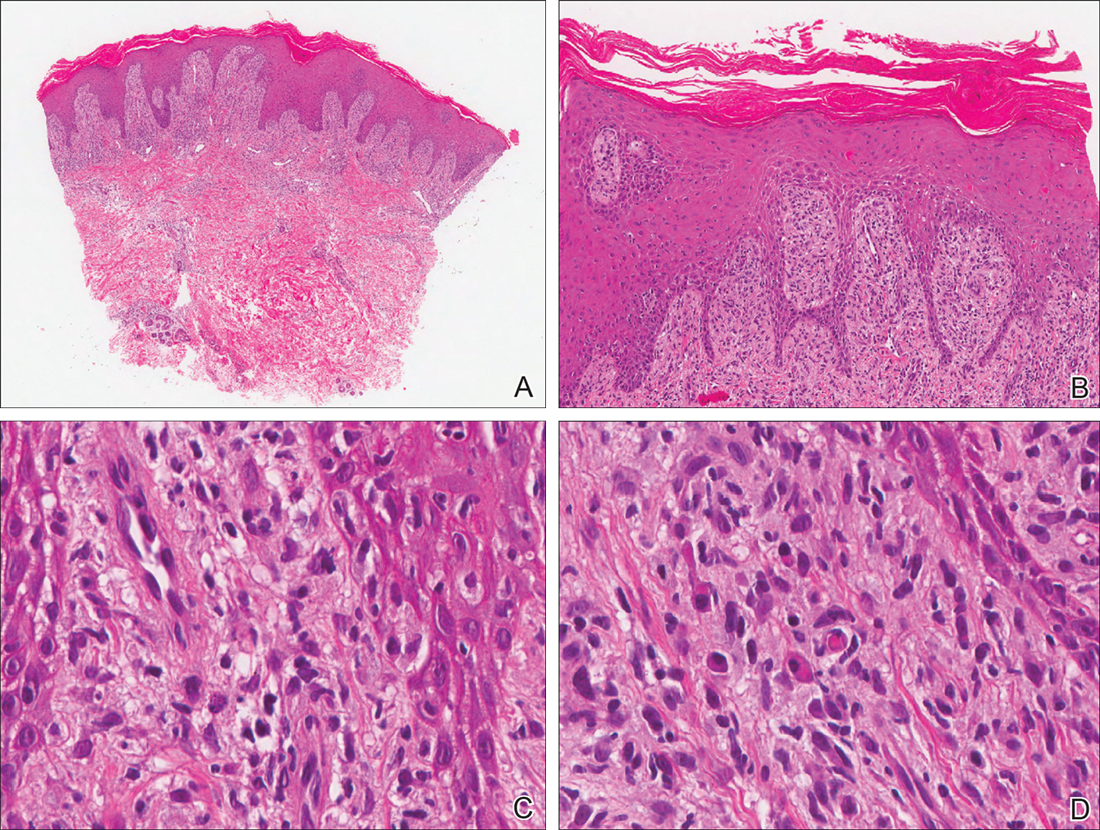
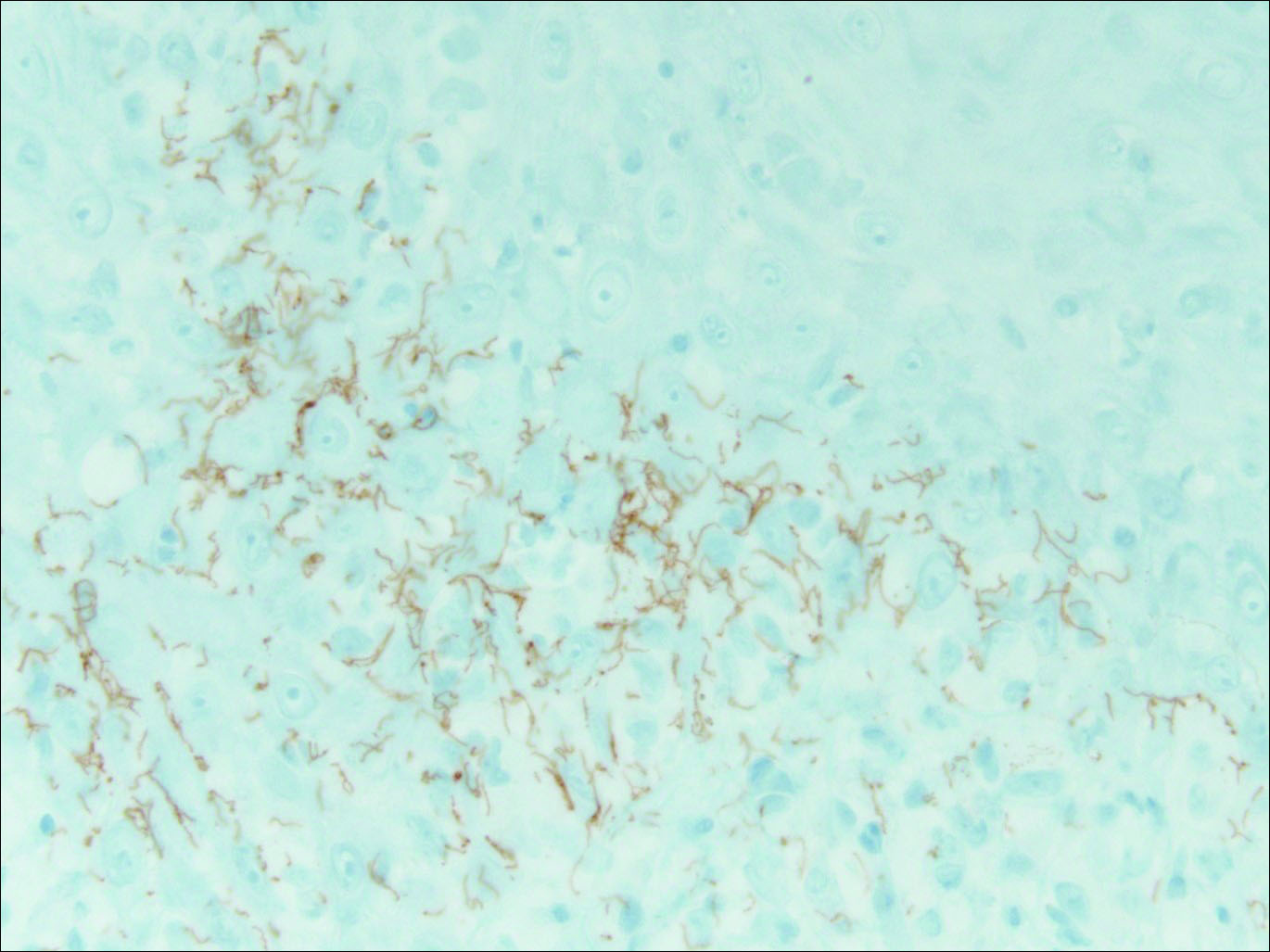
Based on histologic features, the differential diagnosis includes psoriasis vulgaris, eczema, lichen planus, or lichenoid drug eruption. Psoriasis vulgaris displays regular psoriasiform epidermal hyperplasia with hypergranulosis and confluent parakeratosis. The elongated rete pegs are broad rather than slender.2 Neutrophils are present in the stratum corneum. In contrast, eczematous dermatitis is characterized by epidermal hyperplasia, spongiosis, parakeratosis, and eosinophils. Lichen planus classically displays a brisk bandlike lymphocytic infiltrate that closely abuts or obscures the dermoepidermal junction. Parakeratosis, neutrophils, and eosinophils should be absent. The rete pegs taper to a point, similar to a sawtooth, while they are long and slender with syphilis, similar to an ice pick. Although lichenoid drug eruption presents with interface dermatitis, parakeratosis, and eosinophils, the epidermis is hyperplastic without the slender elongation of rete pegs seen in syphilis.
Further workup with serologic testing demonstrated that the patient had a syphilis IgG titer of greater than 8.0 (reactive, >6.0), indicating the patient had been infected.3 Reactive syphilis IgG, a specific treponemal test, should be followed with a nontreponemal assay of either rapid plasma reagin (RPR) or VDRL test to confirm disease activity, according to recommendations from the Centers for Disease Control and Prevention,4 which represents a change to the traditional algorithm that called for screening with a nontreponemal test and confirming with a specific treponemal test. The patient had a positive RPR and quantitative RPR titer was found as 1:2048, indicating that syphilis was active or recently treated. Testing for human immunodeficiency virus (HIV) revealed a quantitative RNA polymerase chain reaction of 145,000 copies/mL and a CD4 count of 18 cells/µL (reference range, 533-1674 cells/µL).
The patient initially was treated for latent syphilis with 3 doses of intramuscular penicillin G benzathine 2.4 million U once weekly for 3 weeks. Due to his high RPR titers and low CD4 count, a lumbar puncture was later pursued, which revealed positive results from a cerebrospinal fluid (CSF)-VDRL test, confirming a diagnosis of neurosyphilis. Although a positive CSF-VDRL test is specific for the diagnosis of neurosyphilis, the sensitivity of the CSF-VDRL test against clinical diagnosis is only 30% to 70%.5 Intravenous aqueous penicillin G 4 million U every 4 hours was started for 14 days for neurosyphilis. One month following the completion of the intravenous penicillin, the rash completely resolved. The patient was in a 10-year monogamous relationship with a man and did not use condoms. Typically, signs and symptoms of secondary syphilis begin 4 to 10 weeks after the appearance of a chancre. However, the classic chancre of primary syphilis among men who have sex with men may go unnoticed in those who may not be able to see anal lesions.6 Also, infection with syphilis increases the likelihood of acquiring and transmitting HIV. All patients diagnosed with syphilis should have additional testing for HIV and other sexually transmitted diseases.
For patients with a history of thick scaly plaques on the wrists, knees, and feet resistant to topical steroid therapy, dermatologists should maintain a high index of clinical suspicion for syphilis.
- Toby M, White J, Van der Walt J. A new test for an old foe... spirochaete immunostaining in the diagnosis of syphilis. Sex Transm Infect. 2013;89:391.
- Nazzaro G, Boneschi V, Coggi A, et al. Syphilis with a lichen planus-like pattern (hypertrophic syphilis). J Cutan Pathol. 2012;39:805-807.
- Yen-Lieberman B, Daniel J, Means C, et al. Identification of false-positive syphilis antibody results using a semiquantitative algorithm. Clin Vaccine Immunol. 2011;18:1038-1040.
- Pope V. Use of syphilis test to screen for syphilis. Infect Med. 2004;21:399-404.
- Larsen S, Kraus S, Whittington W. Diagnostic tests. In: Larsen SA, Hunter E, Kraus S, eds. A Manual of Tests for Syphilis. Washington, DC: American Public Health Association; 1990:2-26.
- Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290:1510-1514.
The Diagnosis: Secondary Syphilis
Syphilis, known as the great mimicker, has a wide-ranging clinical and histologic presentation. There can be overlapping features with many of the entities included in the differential diagnoses. As our patient exemplifies, clinicians and pathologists must have a high index of suspicion, and any concerning features should lead to a more in-depth patient history, spirochete stains, and serologic testing.
Our patient was seen by several dermatologists over the course of 2 years and therapy with topical steroids failed. He was eager to pursue more aggressive therapy with methotrexate, and a punch biopsy was performed to confirm the diagnosis of psoriasis prior to initiating treatment. Hematoxylin and eosin staining results on low power can be seen in Figure 1A. Medium-power view demonstrated vacuolar interface dermatitis (Figure 1B) with psoriasiform epidermal hyperplasia with slender elongation of rete ridges; neutrophils in the stratum corneum; endothelial cell swelling (Figure 1C); and mixed infiltrate with high plasma cells (Figure 1D), lymphocytes, and histiocytes. Although the biopsy results were psoriasiform, there was high suspicion for syphilis in this case. Additional staining for spirochetes was performed with syphilis immunohistochemical stain1 (Figure 2), which revealed spirochetes present on the patient's biopsy, confirming the diagnosis of syphilis. Warthin-Starry stain also can be performed to confirm the diagnosis.
Based on histologic features, the differential diagnosis includes psoriasis vulgaris, eczema, lichen planus, or lichenoid drug eruption. Psoriasis vulgaris displays regular psoriasiform epidermal hyperplasia with hypergranulosis and confluent parakeratosis. The elongated rete pegs are broad rather than slender.2 Neutrophils are present in the stratum corneum. In contrast, eczematous dermatitis is characterized by epidermal hyperplasia, spongiosis, parakeratosis, and eosinophils. Lichen planus classically displays a brisk bandlike lymphocytic infiltrate that closely abuts or obscures the dermoepidermal junction. Parakeratosis, neutrophils, and eosinophils should be absent. The rete pegs taper to a point, similar to a sawtooth, while they are long and slender with syphilis, similar to an ice pick. Although lichenoid drug eruption presents with interface dermatitis, parakeratosis, and eosinophils, the epidermis is hyperplastic without the slender elongation of rete pegs seen in syphilis.
Further workup with serologic testing demonstrated that the patient had a syphilis IgG titer of greater than 8.0 (reactive, >6.0), indicating the patient had been infected.3 Reactive syphilis IgG, a specific treponemal test, should be followed with a nontreponemal assay of either rapid plasma reagin (RPR) or VDRL test to confirm disease activity, according to recommendations from the Centers for Disease Control and Prevention,4 which represents a change to the traditional algorithm that called for screening with a nontreponemal test and confirming with a specific treponemal test. The patient had a positive RPR and quantitative RPR titer was found as 1:2048, indicating that syphilis was active or recently treated. Testing for human immunodeficiency virus (HIV) revealed a quantitative RNA polymerase chain reaction of 145,000 copies/mL and a CD4 count of 18 cells/µL (reference range, 533-1674 cells/µL).
The patient initially was treated for latent syphilis with 3 doses of intramuscular penicillin G benzathine 2.4 million U once weekly for 3 weeks. Due to his high RPR titers and low CD4 count, a lumbar puncture was later pursued, which revealed positive results from a cerebrospinal fluid (CSF)-VDRL test, confirming a diagnosis of neurosyphilis. Although a positive CSF-VDRL test is specific for the diagnosis of neurosyphilis, the sensitivity of the CSF-VDRL test against clinical diagnosis is only 30% to 70%.5 Intravenous aqueous penicillin G 4 million U every 4 hours was started for 14 days for neurosyphilis. One month following the completion of the intravenous penicillin, the rash completely resolved. The patient was in a 10-year monogamous relationship with a man and did not use condoms. Typically, signs and symptoms of secondary syphilis begin 4 to 10 weeks after the appearance of a chancre. However, the classic chancre of primary syphilis among men who have sex with men may go unnoticed in those who may not be able to see anal lesions.6 Also, infection with syphilis increases the likelihood of acquiring and transmitting HIV. All patients diagnosed with syphilis should have additional testing for HIV and other sexually transmitted diseases.
For patients with a history of thick scaly plaques on the wrists, knees, and feet resistant to topical steroid therapy, dermatologists should maintain a high index of clinical suspicion for syphilis.
The Diagnosis: Secondary Syphilis
Syphilis, known as the great mimicker, has a wide-ranging clinical and histologic presentation. There can be overlapping features with many of the entities included in the differential diagnoses. As our patient exemplifies, clinicians and pathologists must have a high index of suspicion, and any concerning features should lead to a more in-depth patient history, spirochete stains, and serologic testing.
Our patient was seen by several dermatologists over the course of 2 years and therapy with topical steroids failed. He was eager to pursue more aggressive therapy with methotrexate, and a punch biopsy was performed to confirm the diagnosis of psoriasis prior to initiating treatment. Hematoxylin and eosin staining results on low power can be seen in Figure 1A. Medium-power view demonstrated vacuolar interface dermatitis (Figure 1B) with psoriasiform epidermal hyperplasia with slender elongation of rete ridges; neutrophils in the stratum corneum; endothelial cell swelling (Figure 1C); and mixed infiltrate with high plasma cells (Figure 1D), lymphocytes, and histiocytes. Although the biopsy results were psoriasiform, there was high suspicion for syphilis in this case. Additional staining for spirochetes was performed with syphilis immunohistochemical stain1 (Figure 2), which revealed spirochetes present on the patient's biopsy, confirming the diagnosis of syphilis. Warthin-Starry stain also can be performed to confirm the diagnosis.
Based on histologic features, the differential diagnosis includes psoriasis vulgaris, eczema, lichen planus, or lichenoid drug eruption. Psoriasis vulgaris displays regular psoriasiform epidermal hyperplasia with hypergranulosis and confluent parakeratosis. The elongated rete pegs are broad rather than slender.2 Neutrophils are present in the stratum corneum. In contrast, eczematous dermatitis is characterized by epidermal hyperplasia, spongiosis, parakeratosis, and eosinophils. Lichen planus classically displays a brisk bandlike lymphocytic infiltrate that closely abuts or obscures the dermoepidermal junction. Parakeratosis, neutrophils, and eosinophils should be absent. The rete pegs taper to a point, similar to a sawtooth, while they are long and slender with syphilis, similar to an ice pick. Although lichenoid drug eruption presents with interface dermatitis, parakeratosis, and eosinophils, the epidermis is hyperplastic without the slender elongation of rete pegs seen in syphilis.
Further workup with serologic testing demonstrated that the patient had a syphilis IgG titer of greater than 8.0 (reactive, >6.0), indicating the patient had been infected.3 Reactive syphilis IgG, a specific treponemal test, should be followed with a nontreponemal assay of either rapid plasma reagin (RPR) or VDRL test to confirm disease activity, according to recommendations from the Centers for Disease Control and Prevention,4 which represents a change to the traditional algorithm that called for screening with a nontreponemal test and confirming with a specific treponemal test. The patient had a positive RPR and quantitative RPR titer was found as 1:2048, indicating that syphilis was active or recently treated. Testing for human immunodeficiency virus (HIV) revealed a quantitative RNA polymerase chain reaction of 145,000 copies/mL and a CD4 count of 18 cells/µL (reference range, 533-1674 cells/µL).
The patient initially was treated for latent syphilis with 3 doses of intramuscular penicillin G benzathine 2.4 million U once weekly for 3 weeks. Due to his high RPR titers and low CD4 count, a lumbar puncture was later pursued, which revealed positive results from a cerebrospinal fluid (CSF)-VDRL test, confirming a diagnosis of neurosyphilis. Although a positive CSF-VDRL test is specific for the diagnosis of neurosyphilis, the sensitivity of the CSF-VDRL test against clinical diagnosis is only 30% to 70%.5 Intravenous aqueous penicillin G 4 million U every 4 hours was started for 14 days for neurosyphilis. One month following the completion of the intravenous penicillin, the rash completely resolved. The patient was in a 10-year monogamous relationship with a man and did not use condoms. Typically, signs and symptoms of secondary syphilis begin 4 to 10 weeks after the appearance of a chancre. However, the classic chancre of primary syphilis among men who have sex with men may go unnoticed in those who may not be able to see anal lesions.6 Also, infection with syphilis increases the likelihood of acquiring and transmitting HIV. All patients diagnosed with syphilis should have additional testing for HIV and other sexually transmitted diseases.
For patients with a history of thick scaly plaques on the wrists, knees, and feet resistant to topical steroid therapy, dermatologists should maintain a high index of clinical suspicion for syphilis.
- Toby M, White J, Van der Walt J. A new test for an old foe... spirochaete immunostaining in the diagnosis of syphilis. Sex Transm Infect. 2013;89:391.
- Nazzaro G, Boneschi V, Coggi A, et al. Syphilis with a lichen planus-like pattern (hypertrophic syphilis). J Cutan Pathol. 2012;39:805-807.
- Yen-Lieberman B, Daniel J, Means C, et al. Identification of false-positive syphilis antibody results using a semiquantitative algorithm. Clin Vaccine Immunol. 2011;18:1038-1040.
- Pope V. Use of syphilis test to screen for syphilis. Infect Med. 2004;21:399-404.
- Larsen S, Kraus S, Whittington W. Diagnostic tests. In: Larsen SA, Hunter E, Kraus S, eds. A Manual of Tests for Syphilis. Washington, DC: American Public Health Association; 1990:2-26.
- Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290:1510-1514.
- Toby M, White J, Van der Walt J. A new test for an old foe... spirochaete immunostaining in the diagnosis of syphilis. Sex Transm Infect. 2013;89:391.
- Nazzaro G, Boneschi V, Coggi A, et al. Syphilis with a lichen planus-like pattern (hypertrophic syphilis). J Cutan Pathol. 2012;39:805-807.
- Yen-Lieberman B, Daniel J, Means C, et al. Identification of false-positive syphilis antibody results using a semiquantitative algorithm. Clin Vaccine Immunol. 2011;18:1038-1040.
- Pope V. Use of syphilis test to screen for syphilis. Infect Med. 2004;21:399-404.
- Larsen S, Kraus S, Whittington W. Diagnostic tests. In: Larsen SA, Hunter E, Kraus S, eds. A Manual of Tests for Syphilis. Washington, DC: American Public Health Association; 1990:2-26.
- Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290:1510-1514.
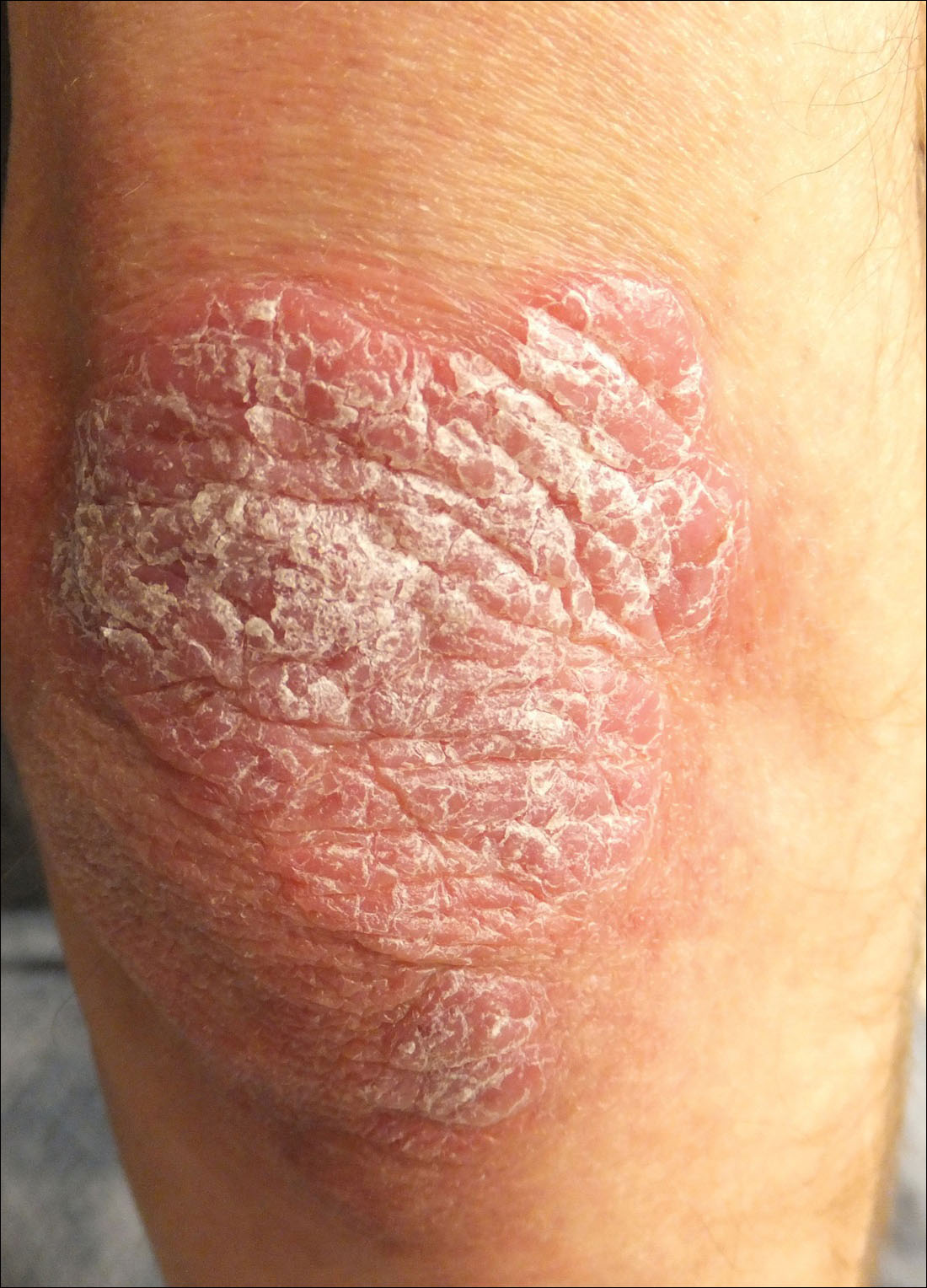
A 34-year-old man presented with thick scaly plaques on the wrists, knees, and feet of 2 years' duration. He had seen several dermatologists, and despite the use of topical steroids, he had no improvement.
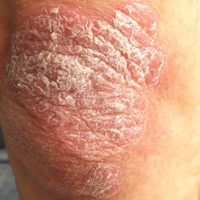
A Boxed Warning for Inadequate Psoriasis Treatment
The US Food and Drug Administration uses the term boxed warning to highlight potentially dangerous situations associated with prescription drugs. A boxed warning is used when “[T]here is an adverse reaction so serious in proportion to the potential benefit from the drug (e.g., a fatal, life-threatening or permanently disabling adverse reaction) that it is essential that it be considered in assessing the risks and benefits of using the drug.”1 However, drugs are not the only potential cause of severe adverse outcomes in patients with psoriasis. Untreated psoriasis also is a well-established cause of serious morbidity and mortality. What are the risks of inadequate psoriasis treatment?
Psoriasis is associated with an increased risk for cardiovascular disease.2-4 Patients with psoriasis also have a higher prevalence of classic cardiovascular risk factors including smoking, diabetes mellitus, hypertension, obesity, and hyperlipidemia.5,6 Psoriasis is a T-cell mediated disease process driven by IL-23 and TH17 helper cell–derived proinflammatory cytokines, sharing certain genetic aspects with metabolic syndrome.6 Cytokine actions on insulin signaling, lipid metabolism, and adipogenesis may underlie the increased prevalence of metabolic syndrome and cardiovascular risk factors in patients with psoriasis. In addition to treating the cutaneous manifestations of psoriasis, reducing inflammation in these patients reduces C-reactive protein and lipid peroxidation and increases high-density lipoprotein levels.6 Tumor necrosis factor α blockers decrease the risk for cardiovascular disease in patients with psoriasis.7,8 Lower than expected rates of cardiovascular disease also have been reported in a large cohort of psoriasis patients (ie, PSOLAR [Psoriasis Longitudinal Assessment and Registry] registry) being treated with either ustekinumab or tumor necrosis factor α blockers.9
Psoriatic arthritis is a chronic inflammatory disease in which active inflammation results in progressive joint destruction.10 Tumor necrosis factor α inhibitors suppress disease progression, preserve function, and delay destruction of the joints. Ustekinumab also helps control psoriatic arthritis and inhibits radiographic progression of joint disease.11
Importantly, untreated moderate to severe psoriasis is associated with several comorbidities that may lead to early death such as heart attacks and strokes.12 Furthermore, patients not taking biologic medications may have higher death rates than patients taking biologic medications.9 Psoriasis also is associated with tremendous suffering and negative psychosocial effects. The mental and physical impact of the disease is comparable to other major medical conditions (eg, cancer, arthritis, hypertension, heart disease, diabetes, depression).13 Patients also may experience physical discomfort from pain and itching.14 Children with psoriasis may experience bullying, which is associated with an increased number of depressive episodes, thereby increasing their risk for developing psychiatric conditions such as depression and anxiety as adults.15 The stigma associated with psoriasis may affect patients’ ability to build relationships. Patients with psoriasis experience higher divorce rates than patients with other chronic medical conditions, and direct involvement of genital regions may negatively impact patients’ sex lives. Patients have noted that the stigma of psoriasis also is associated with the inability to obtain employment.15 Almost one-third of patients with psoriasis who are either not working or are retired base their work status on their skin condition.16 Furthermore, psoriasis may contribute to economic burden for patients due to indirect costs associated with work absenteeism.17
Adequate treatment of psoriasis improves patients’ physical and psychological health as well as their ability to function in the workplace. However, despite the benefits of treatment, 30% of patients with severe psoriasis and 53% of patients with moderate psoriasis receive no treatment or only topical medications instead of systemic therapies.16 The potential adverse events of inadequate psoriasis treatment far outweigh any potential benefits of withholding treatment. Perhaps a boxed warning should be issued for inadequate treatment of psoriasis patients.
- US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. Guidance for industry: warning and precautions, contraindications, and boxed warning sections of labeling for human prescription drug and biological products—content and format. US Food and Drug Administration website. http://www.fda.gov/downloads/Drugs/.../Guidances/ucm075096.pdf. Published October 6, 2011. Accessed August 10, 2016.
- Ogdie A, Yu Y, Haynes K, et al. Risk of major cardiovascular events in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: a population-based cohort study. Ann Rheum Dis. 2015;74:326-332.
- Rose S, Sheth NH, Baker JF, et al. A comparison of vascular inflammation in psoriasis, rheumatoid arthritis, and healthy subjects by FDG-PET/CT: a pilot study. Am J Cardiovasc Dis. 2013;3:273-278.
- Shlyankevich J, Mehta NN, Krueger JG, et al. Accumulating evidence for the association and shared pathogenic mechanisms between psoriasis and cardiovascular-related comorbidities. Am J Med. 2014;127:1148-1153.
- Lee MK, Kim HS, Cho EB, et al. A study of awareness and screening behavior of cardiovascular risk factors in patients with psoriasis and dermatologists. Ann Dermatol. 2015;27:59-65.
- Voiculescu VM, Lupu M, Papagheorghe L, et al. Psoriasis and metabolic syndrome—scientific evidence and therapeutic implications. J Med Life. 2014;7:468-471.
- Wu JJ, Poon KY, Bebchuk JD. Association between the type and length of tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. J Drugs Dermatol. 2013;12:899-903.
- Famenini S, Sako EY, Wu JJ. Effect of treating psoriasis on cardiovascular comorbidities: focus on TNF inhibitors. Am J Clin Dermatol. 2014;15:45-50.
- Gottlieb AB, Kalb RE, Langley RG, et al. Safety observations in 12095 patients with psoriasis enrolled in an international registry (PSOLAR): experience with infliximab and other systemic and biologic therapies. J Drugs Dermatol. 2014;13:1441-1448.
- Chimenti MS, Graceffa D, Perricone R. Anti-TNFα discontinuation in rheumatoid and psoriatic arthritis: is it possible after disease remission [published online Apr 21, 2011]? Autoimmun Rev. 2011;10:636-640.
- Kavanaugh A, Ritchlin C, Rahman P, et al. Ustekinumab, an anti-IL-12/23 p40 monoclonal antibody, inhibits radiographic progression in patients with active psoriatic arthritis: results of an integrated analysis of radiographic data from the phase 3, multicentre, randomised, double-blind, placebo-controlled PSUMMIT-1 and PSUMMIT-2 trials. Ann Rheum Dis. 2014;73:1000-1006.
- Pietrzak A, Bartosinska J, Blaszczyk R, et al. Increased serum level of N-terminal Pro-B-type natriuretic peptide as a possible biomarker of cardiovascular risk in psoriatic patients. J Eur Acad Dermatol Venereol. 2015;29:1010-1014.
- Rapp SR, Feldman SR, Exum ML, et al. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol. 1999;41(3, pt 1):401-407.
- Pettey AA, Balkrishnan R, Rapp SR, et al. Patients with palmoplantar psoriasis have more physical disability and discomfort than patients with other forms of psoriasis: implications for clinical practice. J Am Acad Dermatol. 2003;49:271-275.
- Garshick MK, Kimball AB. Psoriasis and the life cycle of persistent life effects. Dermatol Clin. 2015;33:25-39.
- Feldman SR, Malakouti M, Koo JY. Social impact of the burden of psoriasis: effects on patients and practice. Dermatol Online J. August 17, 2014;20. pii:13030/qt48r4w8h2.
- Brezinski EA, Dhillon JS, Armstrong AW. Economic burden of psoriasis in the United States: a systematic review. JAMA Dermatol. 2015;151:651-658.
The US Food and Drug Administration uses the term boxed warning to highlight potentially dangerous situations associated with prescription drugs. A boxed warning is used when “[T]here is an adverse reaction so serious in proportion to the potential benefit from the drug (e.g., a fatal, life-threatening or permanently disabling adverse reaction) that it is essential that it be considered in assessing the risks and benefits of using the drug.”1 However, drugs are not the only potential cause of severe adverse outcomes in patients with psoriasis. Untreated psoriasis also is a well-established cause of serious morbidity and mortality. What are the risks of inadequate psoriasis treatment?
Psoriasis is associated with an increased risk for cardiovascular disease.2-4 Patients with psoriasis also have a higher prevalence of classic cardiovascular risk factors including smoking, diabetes mellitus, hypertension, obesity, and hyperlipidemia.5,6 Psoriasis is a T-cell mediated disease process driven by IL-23 and TH17 helper cell–derived proinflammatory cytokines, sharing certain genetic aspects with metabolic syndrome.6 Cytokine actions on insulin signaling, lipid metabolism, and adipogenesis may underlie the increased prevalence of metabolic syndrome and cardiovascular risk factors in patients with psoriasis. In addition to treating the cutaneous manifestations of psoriasis, reducing inflammation in these patients reduces C-reactive protein and lipid peroxidation and increases high-density lipoprotein levels.6 Tumor necrosis factor α blockers decrease the risk for cardiovascular disease in patients with psoriasis.7,8 Lower than expected rates of cardiovascular disease also have been reported in a large cohort of psoriasis patients (ie, PSOLAR [Psoriasis Longitudinal Assessment and Registry] registry) being treated with either ustekinumab or tumor necrosis factor α blockers.9
Psoriatic arthritis is a chronic inflammatory disease in which active inflammation results in progressive joint destruction.10 Tumor necrosis factor α inhibitors suppress disease progression, preserve function, and delay destruction of the joints. Ustekinumab also helps control psoriatic arthritis and inhibits radiographic progression of joint disease.11
Importantly, untreated moderate to severe psoriasis is associated with several comorbidities that may lead to early death such as heart attacks and strokes.12 Furthermore, patients not taking biologic medications may have higher death rates than patients taking biologic medications.9 Psoriasis also is associated with tremendous suffering and negative psychosocial effects. The mental and physical impact of the disease is comparable to other major medical conditions (eg, cancer, arthritis, hypertension, heart disease, diabetes, depression).13 Patients also may experience physical discomfort from pain and itching.14 Children with psoriasis may experience bullying, which is associated with an increased number of depressive episodes, thereby increasing their risk for developing psychiatric conditions such as depression and anxiety as adults.15 The stigma associated with psoriasis may affect patients’ ability to build relationships. Patients with psoriasis experience higher divorce rates than patients with other chronic medical conditions, and direct involvement of genital regions may negatively impact patients’ sex lives. Patients have noted that the stigma of psoriasis also is associated with the inability to obtain employment.15 Almost one-third of patients with psoriasis who are either not working or are retired base their work status on their skin condition.16 Furthermore, psoriasis may contribute to economic burden for patients due to indirect costs associated with work absenteeism.17
Adequate treatment of psoriasis improves patients’ physical and psychological health as well as their ability to function in the workplace. However, despite the benefits of treatment, 30% of patients with severe psoriasis and 53% of patients with moderate psoriasis receive no treatment or only topical medications instead of systemic therapies.16 The potential adverse events of inadequate psoriasis treatment far outweigh any potential benefits of withholding treatment. Perhaps a boxed warning should be issued for inadequate treatment of psoriasis patients.
The US Food and Drug Administration uses the term boxed warning to highlight potentially dangerous situations associated with prescription drugs. A boxed warning is used when “[T]here is an adverse reaction so serious in proportion to the potential benefit from the drug (e.g., a fatal, life-threatening or permanently disabling adverse reaction) that it is essential that it be considered in assessing the risks and benefits of using the drug.”1 However, drugs are not the only potential cause of severe adverse outcomes in patients with psoriasis. Untreated psoriasis also is a well-established cause of serious morbidity and mortality. What are the risks of inadequate psoriasis treatment?
Psoriasis is associated with an increased risk for cardiovascular disease.2-4 Patients with psoriasis also have a higher prevalence of classic cardiovascular risk factors including smoking, diabetes mellitus, hypertension, obesity, and hyperlipidemia.5,6 Psoriasis is a T-cell mediated disease process driven by IL-23 and TH17 helper cell–derived proinflammatory cytokines, sharing certain genetic aspects with metabolic syndrome.6 Cytokine actions on insulin signaling, lipid metabolism, and adipogenesis may underlie the increased prevalence of metabolic syndrome and cardiovascular risk factors in patients with psoriasis. In addition to treating the cutaneous manifestations of psoriasis, reducing inflammation in these patients reduces C-reactive protein and lipid peroxidation and increases high-density lipoprotein levels.6 Tumor necrosis factor α blockers decrease the risk for cardiovascular disease in patients with psoriasis.7,8 Lower than expected rates of cardiovascular disease also have been reported in a large cohort of psoriasis patients (ie, PSOLAR [Psoriasis Longitudinal Assessment and Registry] registry) being treated with either ustekinumab or tumor necrosis factor α blockers.9
Psoriatic arthritis is a chronic inflammatory disease in which active inflammation results in progressive joint destruction.10 Tumor necrosis factor α inhibitors suppress disease progression, preserve function, and delay destruction of the joints. Ustekinumab also helps control psoriatic arthritis and inhibits radiographic progression of joint disease.11
Importantly, untreated moderate to severe psoriasis is associated with several comorbidities that may lead to early death such as heart attacks and strokes.12 Furthermore, patients not taking biologic medications may have higher death rates than patients taking biologic medications.9 Psoriasis also is associated with tremendous suffering and negative psychosocial effects. The mental and physical impact of the disease is comparable to other major medical conditions (eg, cancer, arthritis, hypertension, heart disease, diabetes, depression).13 Patients also may experience physical discomfort from pain and itching.14 Children with psoriasis may experience bullying, which is associated with an increased number of depressive episodes, thereby increasing their risk for developing psychiatric conditions such as depression and anxiety as adults.15 The stigma associated with psoriasis may affect patients’ ability to build relationships. Patients with psoriasis experience higher divorce rates than patients with other chronic medical conditions, and direct involvement of genital regions may negatively impact patients’ sex lives. Patients have noted that the stigma of psoriasis also is associated with the inability to obtain employment.15 Almost one-third of patients with psoriasis who are either not working or are retired base their work status on their skin condition.16 Furthermore, psoriasis may contribute to economic burden for patients due to indirect costs associated with work absenteeism.17
Adequate treatment of psoriasis improves patients’ physical and psychological health as well as their ability to function in the workplace. However, despite the benefits of treatment, 30% of patients with severe psoriasis and 53% of patients with moderate psoriasis receive no treatment or only topical medications instead of systemic therapies.16 The potential adverse events of inadequate psoriasis treatment far outweigh any potential benefits of withholding treatment. Perhaps a boxed warning should be issued for inadequate treatment of psoriasis patients.
- US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. Guidance for industry: warning and precautions, contraindications, and boxed warning sections of labeling for human prescription drug and biological products—content and format. US Food and Drug Administration website. http://www.fda.gov/downloads/Drugs/.../Guidances/ucm075096.pdf. Published October 6, 2011. Accessed August 10, 2016.
- Ogdie A, Yu Y, Haynes K, et al. Risk of major cardiovascular events in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: a population-based cohort study. Ann Rheum Dis. 2015;74:326-332.
- Rose S, Sheth NH, Baker JF, et al. A comparison of vascular inflammation in psoriasis, rheumatoid arthritis, and healthy subjects by FDG-PET/CT: a pilot study. Am J Cardiovasc Dis. 2013;3:273-278.
- Shlyankevich J, Mehta NN, Krueger JG, et al. Accumulating evidence for the association and shared pathogenic mechanisms between psoriasis and cardiovascular-related comorbidities. Am J Med. 2014;127:1148-1153.
- Lee MK, Kim HS, Cho EB, et al. A study of awareness and screening behavior of cardiovascular risk factors in patients with psoriasis and dermatologists. Ann Dermatol. 2015;27:59-65.
- Voiculescu VM, Lupu M, Papagheorghe L, et al. Psoriasis and metabolic syndrome—scientific evidence and therapeutic implications. J Med Life. 2014;7:468-471.
- Wu JJ, Poon KY, Bebchuk JD. Association between the type and length of tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. J Drugs Dermatol. 2013;12:899-903.
- Famenini S, Sako EY, Wu JJ. Effect of treating psoriasis on cardiovascular comorbidities: focus on TNF inhibitors. Am J Clin Dermatol. 2014;15:45-50.
- Gottlieb AB, Kalb RE, Langley RG, et al. Safety observations in 12095 patients with psoriasis enrolled in an international registry (PSOLAR): experience with infliximab and other systemic and biologic therapies. J Drugs Dermatol. 2014;13:1441-1448.
- Chimenti MS, Graceffa D, Perricone R. Anti-TNFα discontinuation in rheumatoid and psoriatic arthritis: is it possible after disease remission [published online Apr 21, 2011]? Autoimmun Rev. 2011;10:636-640.
- Kavanaugh A, Ritchlin C, Rahman P, et al. Ustekinumab, an anti-IL-12/23 p40 monoclonal antibody, inhibits radiographic progression in patients with active psoriatic arthritis: results of an integrated analysis of radiographic data from the phase 3, multicentre, randomised, double-blind, placebo-controlled PSUMMIT-1 and PSUMMIT-2 trials. Ann Rheum Dis. 2014;73:1000-1006.
- Pietrzak A, Bartosinska J, Blaszczyk R, et al. Increased serum level of N-terminal Pro-B-type natriuretic peptide as a possible biomarker of cardiovascular risk in psoriatic patients. J Eur Acad Dermatol Venereol. 2015;29:1010-1014.
- Rapp SR, Feldman SR, Exum ML, et al. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol. 1999;41(3, pt 1):401-407.
- Pettey AA, Balkrishnan R, Rapp SR, et al. Patients with palmoplantar psoriasis have more physical disability and discomfort than patients with other forms of psoriasis: implications for clinical practice. J Am Acad Dermatol. 2003;49:271-275.
- Garshick MK, Kimball AB. Psoriasis and the life cycle of persistent life effects. Dermatol Clin. 2015;33:25-39.
- Feldman SR, Malakouti M, Koo JY. Social impact of the burden of psoriasis: effects on patients and practice. Dermatol Online J. August 17, 2014;20. pii:13030/qt48r4w8h2.
- Brezinski EA, Dhillon JS, Armstrong AW. Economic burden of psoriasis in the United States: a systematic review. JAMA Dermatol. 2015;151:651-658.
- US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. Guidance for industry: warning and precautions, contraindications, and boxed warning sections of labeling for human prescription drug and biological products—content and format. US Food and Drug Administration website. http://www.fda.gov/downloads/Drugs/.../Guidances/ucm075096.pdf. Published October 6, 2011. Accessed August 10, 2016.
- Ogdie A, Yu Y, Haynes K, et al. Risk of major cardiovascular events in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: a population-based cohort study. Ann Rheum Dis. 2015;74:326-332.
- Rose S, Sheth NH, Baker JF, et al. A comparison of vascular inflammation in psoriasis, rheumatoid arthritis, and healthy subjects by FDG-PET/CT: a pilot study. Am J Cardiovasc Dis. 2013;3:273-278.
- Shlyankevich J, Mehta NN, Krueger JG, et al. Accumulating evidence for the association and shared pathogenic mechanisms between psoriasis and cardiovascular-related comorbidities. Am J Med. 2014;127:1148-1153.
- Lee MK, Kim HS, Cho EB, et al. A study of awareness and screening behavior of cardiovascular risk factors in patients with psoriasis and dermatologists. Ann Dermatol. 2015;27:59-65.
- Voiculescu VM, Lupu M, Papagheorghe L, et al. Psoriasis and metabolic syndrome—scientific evidence and therapeutic implications. J Med Life. 2014;7:468-471.
- Wu JJ, Poon KY, Bebchuk JD. Association between the type and length of tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. J Drugs Dermatol. 2013;12:899-903.
- Famenini S, Sako EY, Wu JJ. Effect of treating psoriasis on cardiovascular comorbidities: focus on TNF inhibitors. Am J Clin Dermatol. 2014;15:45-50.
- Gottlieb AB, Kalb RE, Langley RG, et al. Safety observations in 12095 patients with psoriasis enrolled in an international registry (PSOLAR): experience with infliximab and other systemic and biologic therapies. J Drugs Dermatol. 2014;13:1441-1448.
- Chimenti MS, Graceffa D, Perricone R. Anti-TNFα discontinuation in rheumatoid and psoriatic arthritis: is it possible after disease remission [published online Apr 21, 2011]? Autoimmun Rev. 2011;10:636-640.
- Kavanaugh A, Ritchlin C, Rahman P, et al. Ustekinumab, an anti-IL-12/23 p40 monoclonal antibody, inhibits radiographic progression in patients with active psoriatic arthritis: results of an integrated analysis of radiographic data from the phase 3, multicentre, randomised, double-blind, placebo-controlled PSUMMIT-1 and PSUMMIT-2 trials. Ann Rheum Dis. 2014;73:1000-1006.
- Pietrzak A, Bartosinska J, Blaszczyk R, et al. Increased serum level of N-terminal Pro-B-type natriuretic peptide as a possible biomarker of cardiovascular risk in psoriatic patients. J Eur Acad Dermatol Venereol. 2015;29:1010-1014.
- Rapp SR, Feldman SR, Exum ML, et al. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol. 1999;41(3, pt 1):401-407.
- Pettey AA, Balkrishnan R, Rapp SR, et al. Patients with palmoplantar psoriasis have more physical disability and discomfort than patients with other forms of psoriasis: implications for clinical practice. J Am Acad Dermatol. 2003;49:271-275.
- Garshick MK, Kimball AB. Psoriasis and the life cycle of persistent life effects. Dermatol Clin. 2015;33:25-39.
- Feldman SR, Malakouti M, Koo JY. Social impact of the burden of psoriasis: effects on patients and practice. Dermatol Online J. August 17, 2014;20. pii:13030/qt48r4w8h2.
- Brezinski EA, Dhillon JS, Armstrong AW. Economic burden of psoriasis in the United States: a systematic review. JAMA Dermatol. 2015;151:651-658.

Desmoplastic Hairless Hypopigmented Nevus
To the Editor:
We report 2 cases of desmoplastic hairless hypopigmented nevi (DHHN), which are giant congenital melanocytic nevi (GCMN) that show sclerosis with progressive loss of pigment and hair. These changes in GCMN could be considered signs of regression.
A 6-year-old boy presented in the dermatology department with an asymptomatic skin lesion on the right buttock since birth. The parents claimed that the lesion was darkly pigmented at birth and gradually increased in size, with progressive reduction in color in the last 2 years. Physical examination revealed a 10×6-cm, well-defined, raised plaque on the upper medial side of the right buttock (Figure 1). The plaque was firm with a shiny smooth surface and was devoid of hair. The surface was flesh colored with scattered pigmented spots. A punch biopsy of the lesion showed increased melanin content in the basal cell layer. The upper dermis showed small nests of epithelioid nevus cells, most of them containing melanin pigment (Figure 2). In the lower two-thirds of the dermis, nevus cells were both epithelioid and spindle shaped and were arranged in between thick sclerotic collagen bundles with an increased number of fibroblasts. There was a marked reduction in the number of hair follicles. Immunohistochemical staining results were S-100 positive and CD34 negative.
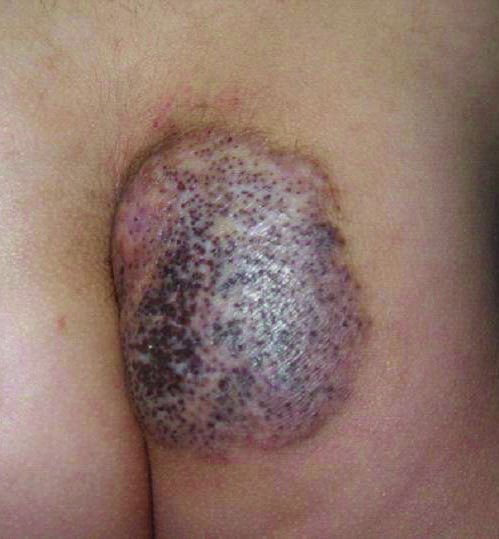
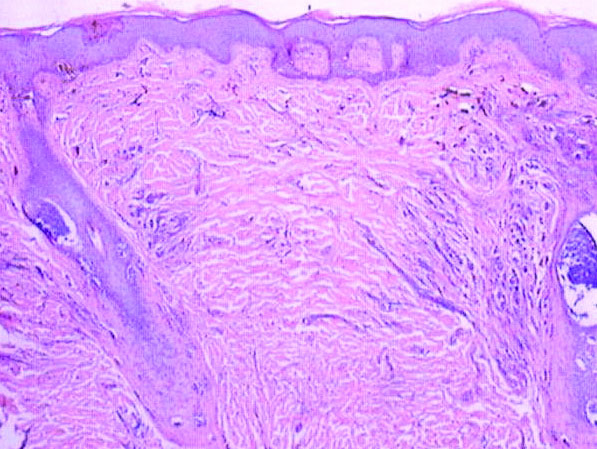
A 5-year-old boy presented in the dermatology department with a large hairy GCMN covering most of the trunk since birth. In the last 1.5 years the parents noted gradual fading of color, decreased hair density, and increased induration of the nevus. Physical examination revealed a large plaque covering the anterior aspect of the trunk (Figure 3) and the back extending down to the buttocks. The lesion formed large skin folds that were more pronounced on the back. The nevus was darkly pigmented with large areas of lighter color that were indurated, devoid of hair, and showed small spots of dark pigmentation. A punch biopsy from the lesion showed small nests of nevus cells in the upper part of the reticular dermis. In the lower part of the dermis, nevus cells were arranged in single units in between thick collagen bundles.
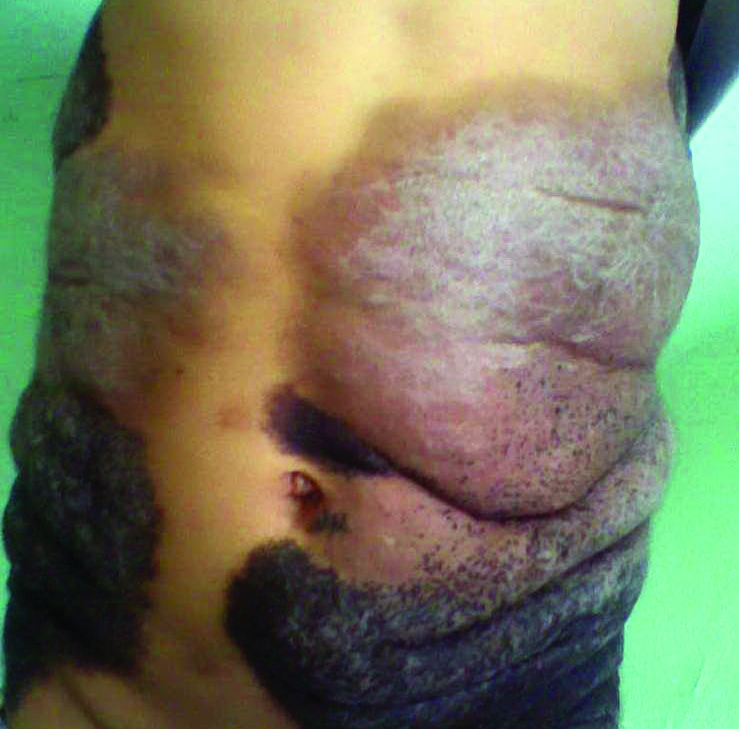
In 2003, Ruiz-Maldonado et al1 described 4 cases of GCMN that showed progressive loss of pigmentation, sclerosis, and hair loss. They proposed the term desmoplastic hairless hypopigmented nevus for their cases and considered it as a variant of GCMN.1 Prior to these reported cases, 2 similar cases were described. The first was a report by Hogan et al2 in 1988 of a 7-month-old girl with a GCMN involving the occipital area and the upper back that became indurated and ulcerated with progressive involution that led to complete disappearance of the nevus. The second was a report by Pattee et al3 in 2001 of a newborn with a GCMN located on the trunk with progressive sclerodermiform reaction. After surgical excision of the nevus, the sclerotic margin disappeared.3
Following the report by Ruiz-Maldonaldo et al,1 5 more cases of DHHN were described.4-8 All cases of DHHN share the same clinical and histopathological features. The clinical features include a GCMN present since birth with progressive sclerosis over time and loss of both pigmentation and hair. Histologically, DHHN shows the typical changes of a congenital melanocytic nevus with decreased numbers of nevus cells, thick sclerotic collagen bundles of the reticular dermis, increased number of fibroblasts, and decreased number of hair follicles. The progressive reduction in the number of nevus cells in melanocytic nevi is considered a sign of regression. Spontaneous regression was rarely described in GCMN, and all the reported cases of regression were associated with desmoplasia.4 Desmoplasia is thought to be induced by either melanocytes that function as adaptive fibroblasts or by fibroblasts themselves, as fibroblasts can show multifunctional differentiation capabilities.9 The direct correlation between the increased induration of DHHN and pigment depletion supports the former hypothesis. The absence of inflammatory cells within the sections of DHHN lesions is against the possibility of an immune-mediated reaction as a cause for the clinical and histological changes seen in this rare form of GCMN. The progressive hair loss in DHHN may be explained by the progressive fibrotic changes in the reticular dermis that affect the blood supply to follicles, leading to atrophy or even absence of the follicles. The progressive reduction in the number of nevus cells in DHHN reduces the potential for malignant transformation and hence following a watchful waiting strategy is a reasonable way to manage these nevi.
We present 2 patients with DHHN, which is a rare form of GCMN that shows signs of regression. The cause of these changes is still unclear.
- Ruiz-Maldonado R, Orozco-Covarrubias L, Ridaura-Sanz C, et al. Desmoplastic hairless hypopigmented naevus: a variant of giant congenital melanocytic naevus. Br J Dermatol. 2003;148:1253-1257.
- Hogan DJ, Murphy F, Bremner RM. Spontaneous resolution of a giant congenital melanocytic nevus. Pediatr Dermatol. 1988;5:170-172.
- Pattee SF, Hansen RC, Bangert JL, et al. Giant congenital nevus with progressive sclerodermoid reaction in a newborn. Pediatr Dermatol. 2001;18:321-324.
- Boente MC, Asial RA. Desmoplastic hairless hypopigmented nevus (DHHN). a distinct variant of giant melanocytic nevus. Eur J Dermatol. 2005;15:451-453.
- Bushby SA, Rajan NJ, Shehade SA. Spontaneous resolution of a giant melanocytic naevus involving a desmoplastic process. Br J Dermatol. 2005;153(suppl 1):13-19.
- Martin JM, Jorda E, Monteagudo C, et al. Desmoplastic giant congenital nevus with progressive depigmentation. J Am Acad Dermatol. 2007;56(suppl 2):S10-S14.
- Hermandez-Martin A, Torrelo A, Echevarria C, et al. Ulcerated sclerotic giant congenital melanocytic naevus: case report and review of the literature. Clin Exp Dermatol. 2007;32:529-532.
- Werner B, Carvalho VO, Nacif SB, et al. Desmoplastic hypopigmented hairless nevus: a variant with progressive depigmentation, induration and overgrowth [published online May 16, 2011]. Pediatr Dermatol. 2012;29:336-340.
- Fearns C, Dowdle EB. The desmoplastic response: induction of collagen synthesis by melanoma cells in vitro. Int J Cancer. 1992;50:621-627.
To the Editor:
We report 2 cases of desmoplastic hairless hypopigmented nevi (DHHN), which are giant congenital melanocytic nevi (GCMN) that show sclerosis with progressive loss of pigment and hair. These changes in GCMN could be considered signs of regression.
A 6-year-old boy presented in the dermatology department with an asymptomatic skin lesion on the right buttock since birth. The parents claimed that the lesion was darkly pigmented at birth and gradually increased in size, with progressive reduction in color in the last 2 years. Physical examination revealed a 10×6-cm, well-defined, raised plaque on the upper medial side of the right buttock (Figure 1). The plaque was firm with a shiny smooth surface and was devoid of hair. The surface was flesh colored with scattered pigmented spots. A punch biopsy of the lesion showed increased melanin content in the basal cell layer. The upper dermis showed small nests of epithelioid nevus cells, most of them containing melanin pigment (Figure 2). In the lower two-thirds of the dermis, nevus cells were both epithelioid and spindle shaped and were arranged in between thick sclerotic collagen bundles with an increased number of fibroblasts. There was a marked reduction in the number of hair follicles. Immunohistochemical staining results were S-100 positive and CD34 negative.
A 5-year-old boy presented in the dermatology department with a large hairy GCMN covering most of the trunk since birth. In the last 1.5 years the parents noted gradual fading of color, decreased hair density, and increased induration of the nevus. Physical examination revealed a large plaque covering the anterior aspect of the trunk (Figure 3) and the back extending down to the buttocks. The lesion formed large skin folds that were more pronounced on the back. The nevus was darkly pigmented with large areas of lighter color that were indurated, devoid of hair, and showed small spots of dark pigmentation. A punch biopsy from the lesion showed small nests of nevus cells in the upper part of the reticular dermis. In the lower part of the dermis, nevus cells were arranged in single units in between thick collagen bundles.
In 2003, Ruiz-Maldonado et al1 described 4 cases of GCMN that showed progressive loss of pigmentation, sclerosis, and hair loss. They proposed the term desmoplastic hairless hypopigmented nevus for their cases and considered it as a variant of GCMN.1 Prior to these reported cases, 2 similar cases were described. The first was a report by Hogan et al2 in 1988 of a 7-month-old girl with a GCMN involving the occipital area and the upper back that became indurated and ulcerated with progressive involution that led to complete disappearance of the nevus. The second was a report by Pattee et al3 in 2001 of a newborn with a GCMN located on the trunk with progressive sclerodermiform reaction. After surgical excision of the nevus, the sclerotic margin disappeared.3
Following the report by Ruiz-Maldonaldo et al,1 5 more cases of DHHN were described.4-8 All cases of DHHN share the same clinical and histopathological features. The clinical features include a GCMN present since birth with progressive sclerosis over time and loss of both pigmentation and hair. Histologically, DHHN shows the typical changes of a congenital melanocytic nevus with decreased numbers of nevus cells, thick sclerotic collagen bundles of the reticular dermis, increased number of fibroblasts, and decreased number of hair follicles. The progressive reduction in the number of nevus cells in melanocytic nevi is considered a sign of regression. Spontaneous regression was rarely described in GCMN, and all the reported cases of regression were associated with desmoplasia.4 Desmoplasia is thought to be induced by either melanocytes that function as adaptive fibroblasts or by fibroblasts themselves, as fibroblasts can show multifunctional differentiation capabilities.9 The direct correlation between the increased induration of DHHN and pigment depletion supports the former hypothesis. The absence of inflammatory cells within the sections of DHHN lesions is against the possibility of an immune-mediated reaction as a cause for the clinical and histological changes seen in this rare form of GCMN. The progressive hair loss in DHHN may be explained by the progressive fibrotic changes in the reticular dermis that affect the blood supply to follicles, leading to atrophy or even absence of the follicles. The progressive reduction in the number of nevus cells in DHHN reduces the potential for malignant transformation and hence following a watchful waiting strategy is a reasonable way to manage these nevi.
We present 2 patients with DHHN, which is a rare form of GCMN that shows signs of regression. The cause of these changes is still unclear.
To the Editor:
We report 2 cases of desmoplastic hairless hypopigmented nevi (DHHN), which are giant congenital melanocytic nevi (GCMN) that show sclerosis with progressive loss of pigment and hair. These changes in GCMN could be considered signs of regression.
A 6-year-old boy presented in the dermatology department with an asymptomatic skin lesion on the right buttock since birth. The parents claimed that the lesion was darkly pigmented at birth and gradually increased in size, with progressive reduction in color in the last 2 years. Physical examination revealed a 10×6-cm, well-defined, raised plaque on the upper medial side of the right buttock (Figure 1). The plaque was firm with a shiny smooth surface and was devoid of hair. The surface was flesh colored with scattered pigmented spots. A punch biopsy of the lesion showed increased melanin content in the basal cell layer. The upper dermis showed small nests of epithelioid nevus cells, most of them containing melanin pigment (Figure 2). In the lower two-thirds of the dermis, nevus cells were both epithelioid and spindle shaped and were arranged in between thick sclerotic collagen bundles with an increased number of fibroblasts. There was a marked reduction in the number of hair follicles. Immunohistochemical staining results were S-100 positive and CD34 negative.
A 5-year-old boy presented in the dermatology department with a large hairy GCMN covering most of the trunk since birth. In the last 1.5 years the parents noted gradual fading of color, decreased hair density, and increased induration of the nevus. Physical examination revealed a large plaque covering the anterior aspect of the trunk (Figure 3) and the back extending down to the buttocks. The lesion formed large skin folds that were more pronounced on the back. The nevus was darkly pigmented with large areas of lighter color that were indurated, devoid of hair, and showed small spots of dark pigmentation. A punch biopsy from the lesion showed small nests of nevus cells in the upper part of the reticular dermis. In the lower part of the dermis, nevus cells were arranged in single units in between thick collagen bundles.
In 2003, Ruiz-Maldonado et al1 described 4 cases of GCMN that showed progressive loss of pigmentation, sclerosis, and hair loss. They proposed the term desmoplastic hairless hypopigmented nevus for their cases and considered it as a variant of GCMN.1 Prior to these reported cases, 2 similar cases were described. The first was a report by Hogan et al2 in 1988 of a 7-month-old girl with a GCMN involving the occipital area and the upper back that became indurated and ulcerated with progressive involution that led to complete disappearance of the nevus. The second was a report by Pattee et al3 in 2001 of a newborn with a GCMN located on the trunk with progressive sclerodermiform reaction. After surgical excision of the nevus, the sclerotic margin disappeared.3
Following the report by Ruiz-Maldonaldo et al,1 5 more cases of DHHN were described.4-8 All cases of DHHN share the same clinical and histopathological features. The clinical features include a GCMN present since birth with progressive sclerosis over time and loss of both pigmentation and hair. Histologically, DHHN shows the typical changes of a congenital melanocytic nevus with decreased numbers of nevus cells, thick sclerotic collagen bundles of the reticular dermis, increased number of fibroblasts, and decreased number of hair follicles. The progressive reduction in the number of nevus cells in melanocytic nevi is considered a sign of regression. Spontaneous regression was rarely described in GCMN, and all the reported cases of regression were associated with desmoplasia.4 Desmoplasia is thought to be induced by either melanocytes that function as adaptive fibroblasts or by fibroblasts themselves, as fibroblasts can show multifunctional differentiation capabilities.9 The direct correlation between the increased induration of DHHN and pigment depletion supports the former hypothesis. The absence of inflammatory cells within the sections of DHHN lesions is against the possibility of an immune-mediated reaction as a cause for the clinical and histological changes seen in this rare form of GCMN. The progressive hair loss in DHHN may be explained by the progressive fibrotic changes in the reticular dermis that affect the blood supply to follicles, leading to atrophy or even absence of the follicles. The progressive reduction in the number of nevus cells in DHHN reduces the potential for malignant transformation and hence following a watchful waiting strategy is a reasonable way to manage these nevi.
We present 2 patients with DHHN, which is a rare form of GCMN that shows signs of regression. The cause of these changes is still unclear.
- Ruiz-Maldonado R, Orozco-Covarrubias L, Ridaura-Sanz C, et al. Desmoplastic hairless hypopigmented naevus: a variant of giant congenital melanocytic naevus. Br J Dermatol. 2003;148:1253-1257.
- Hogan DJ, Murphy F, Bremner RM. Spontaneous resolution of a giant congenital melanocytic nevus. Pediatr Dermatol. 1988;5:170-172.
- Pattee SF, Hansen RC, Bangert JL, et al. Giant congenital nevus with progressive sclerodermoid reaction in a newborn. Pediatr Dermatol. 2001;18:321-324.
- Boente MC, Asial RA. Desmoplastic hairless hypopigmented nevus (DHHN). a distinct variant of giant melanocytic nevus. Eur J Dermatol. 2005;15:451-453.
- Bushby SA, Rajan NJ, Shehade SA. Spontaneous resolution of a giant melanocytic naevus involving a desmoplastic process. Br J Dermatol. 2005;153(suppl 1):13-19.
- Martin JM, Jorda E, Monteagudo C, et al. Desmoplastic giant congenital nevus with progressive depigmentation. J Am Acad Dermatol. 2007;56(suppl 2):S10-S14.
- Hermandez-Martin A, Torrelo A, Echevarria C, et al. Ulcerated sclerotic giant congenital melanocytic naevus: case report and review of the literature. Clin Exp Dermatol. 2007;32:529-532.
- Werner B, Carvalho VO, Nacif SB, et al. Desmoplastic hypopigmented hairless nevus: a variant with progressive depigmentation, induration and overgrowth [published online May 16, 2011]. Pediatr Dermatol. 2012;29:336-340.
- Fearns C, Dowdle EB. The desmoplastic response: induction of collagen synthesis by melanoma cells in vitro. Int J Cancer. 1992;50:621-627.
- Ruiz-Maldonado R, Orozco-Covarrubias L, Ridaura-Sanz C, et al. Desmoplastic hairless hypopigmented naevus: a variant of giant congenital melanocytic naevus. Br J Dermatol. 2003;148:1253-1257.
- Hogan DJ, Murphy F, Bremner RM. Spontaneous resolution of a giant congenital melanocytic nevus. Pediatr Dermatol. 1988;5:170-172.
- Pattee SF, Hansen RC, Bangert JL, et al. Giant congenital nevus with progressive sclerodermoid reaction in a newborn. Pediatr Dermatol. 2001;18:321-324.
- Boente MC, Asial RA. Desmoplastic hairless hypopigmented nevus (DHHN). a distinct variant of giant melanocytic nevus. Eur J Dermatol. 2005;15:451-453.
- Bushby SA, Rajan NJ, Shehade SA. Spontaneous resolution of a giant melanocytic naevus involving a desmoplastic process. Br J Dermatol. 2005;153(suppl 1):13-19.
- Martin JM, Jorda E, Monteagudo C, et al. Desmoplastic giant congenital nevus with progressive depigmentation. J Am Acad Dermatol. 2007;56(suppl 2):S10-S14.
- Hermandez-Martin A, Torrelo A, Echevarria C, et al. Ulcerated sclerotic giant congenital melanocytic naevus: case report and review of the literature. Clin Exp Dermatol. 2007;32:529-532.
- Werner B, Carvalho VO, Nacif SB, et al. Desmoplastic hypopigmented hairless nevus: a variant with progressive depigmentation, induration and overgrowth [published online May 16, 2011]. Pediatr Dermatol. 2012;29:336-340.
- Fearns C, Dowdle EB. The desmoplastic response: induction of collagen synthesis by melanoma cells in vitro. Int J Cancer. 1992;50:621-627.
Cutaneous Artifactual Disease Represented as Recurrent Toxic Epidermal Necrolysis
To the Editor:
Lyell1 coined the term cutaneous artifactual disease to describe the spectrum of factitious disorders associated with skin presentations. Interestingly, Lyell was the first to name toxic epidermal necrolysis (TEN).2,3 We present a rare case of factitial TEN, a dangerous and life-threatening manifestation of factitial disease.
A 49-year-old homeless man with a history of Stevens-Johnson syndrome (SJS)/TEN from trimethoprim-sulfamethoxazole (TMP-SMX) was admitted on 4 separate occasions over an 18-month period for recurrent exposure to the medication producing SJS/TEN. Originally, this patient was given TMP-SMX for a skin infection and 10 days later presented with 15% body surface area (BSA) involvement of SJS/TEN. He was successfully treated with intravenous immunoglobulin (IVIg) in the burn intensive care unit (BICU) and discharged. Several months later, the patient was given TMP-SMX for a leg infection by a different clinic. He was admitted to the BICU with 40% BSA, treated with IVIg, and survived. Eight months later, the patient was again admitted to the BICU with 30% BSA and treated with IVIg; however, this admission required intubation due to complications secondary to volume resuscitation. He was evaluated by psychiatry and confessed to purposely seeking TMP-SMX, stating that he “liked the food and care in the hospital.” He was diagnosed with factitial disorder and given a referral for further treatment at an outpatient facility. Two months later, the patient was again admitted to the BICU after taking a single dose of TMP-SMX obtained from a “friend.” He had 10% BSA with conjunctival involvement and was again successfully treated with IVIg. He was discharged with the state psychiatric system for further treatment and evaluation.
Factitial disease in dermatology is difficult to diagnose. Its incidence is unknown, as only case reports exist in the literature. In factitial disease, patients “perform self-mutilating and clinically relevant damage to themselves without the direct intention of suicide.”4 Harth et al4 described 3 subcategories of factitious disorders: dermatitis artefacta syndrome, dermatitis para-artefacta syndrome, or malingering. Dermatitis artefacta syndrome is “a dissociated self-injury or behavior where the patient unconsciously simulated disease with intention to be cared for as a patient.”4 Dermatitis para-artefacta syndrome was described as a disorder of impulse control in which a patient will produce or manipulate a specific dermatosis presentation. The patient usually admits to doing it in a semiconscious state. Dermatitis artefacta and dermatitis para-artefacta differ from malingering in that malingering patients knowingly fake symptoms for external gain, which can be monetary or the avoidance of responsibility.4 More familiar examples to dermatologists of factitial disease include factitial panniculitis,1 direct applications of caustic agents to the skin, and excoriations from instruments or fingernails.4,5
This case illustrates the difficult and potentially dangerous nature of factitial disorders, specifically dermatitis para-artefacta syndrome. Our patient was intensely preoccupied with the outcome of being a patient in a hospital. Our patient sought out a medication from multiple providers to produce a deadly and severe life-threatening reaction. If his main intentions were solely to obtain a bed and 3 square meals a day, then malingering would have represented his factitial disease. However, his main intent was to be seen as a patient, and then doted on and cared for by medical professionals in a hospital setting. From this assessment, the patient’s behavior would fall under the factitial disorder of dermatitis para-artefacta syndrome.
Factitious disorders pose immense challenges for diagnosis and treatment. It is prudent for physicians to learn to recognize patterns of history and examination that do not coincide. The first step in treatment is the recognition and early involvement of psychiatry to aid in curbing this behavior. Remission of factitial disorders can be induced with proper diagnosis and treatment. Patients with the highest chance of remission are those with treatment centered on behavioral therapy in conjunction with psychotropic medications.1
- Lyell A. Cutaneous artifactual disease. J Am Acad Dermatol. 1979;1:391-402.
- Palmieri TL, Greenhalgh, DG, Saffle JR, et al. A multicenter review of toxic epidermal necrolysis treated in U.S. burn centers at the end of the twentieth century. J Burn Care Rehab. 2002;23:87-96.
- Lyell A. Toxic epidermal necrolysis: an eruption resembling scalding of the skin. Br J Dermatol. 1956;68:355-361.
- Harth W, Gieler U, Kusnir D, et al. Primarily psychogenic dermatoses. In: Clinical Management in Psychodermatology. 1st ed. Leipzog, Germany: Springer-Verlag Berlin Heidelberg; 2009:11-19.
- Sanmartin O, Requena C, Requena L. Factitial panniculitis. Dermatol Clin. 2008;26:519-527.
To the Editor:
Lyell1 coined the term cutaneous artifactual disease to describe the spectrum of factitious disorders associated with skin presentations. Interestingly, Lyell was the first to name toxic epidermal necrolysis (TEN).2,3 We present a rare case of factitial TEN, a dangerous and life-threatening manifestation of factitial disease.
A 49-year-old homeless man with a history of Stevens-Johnson syndrome (SJS)/TEN from trimethoprim-sulfamethoxazole (TMP-SMX) was admitted on 4 separate occasions over an 18-month period for recurrent exposure to the medication producing SJS/TEN. Originally, this patient was given TMP-SMX for a skin infection and 10 days later presented with 15% body surface area (BSA) involvement of SJS/TEN. He was successfully treated with intravenous immunoglobulin (IVIg) in the burn intensive care unit (BICU) and discharged. Several months later, the patient was given TMP-SMX for a leg infection by a different clinic. He was admitted to the BICU with 40% BSA, treated with IVIg, and survived. Eight months later, the patient was again admitted to the BICU with 30% BSA and treated with IVIg; however, this admission required intubation due to complications secondary to volume resuscitation. He was evaluated by psychiatry and confessed to purposely seeking TMP-SMX, stating that he “liked the food and care in the hospital.” He was diagnosed with factitial disorder and given a referral for further treatment at an outpatient facility. Two months later, the patient was again admitted to the BICU after taking a single dose of TMP-SMX obtained from a “friend.” He had 10% BSA with conjunctival involvement and was again successfully treated with IVIg. He was discharged with the state psychiatric system for further treatment and evaluation.
Factitial disease in dermatology is difficult to diagnose. Its incidence is unknown, as only case reports exist in the literature. In factitial disease, patients “perform self-mutilating and clinically relevant damage to themselves without the direct intention of suicide.”4 Harth et al4 described 3 subcategories of factitious disorders: dermatitis artefacta syndrome, dermatitis para-artefacta syndrome, or malingering. Dermatitis artefacta syndrome is “a dissociated self-injury or behavior where the patient unconsciously simulated disease with intention to be cared for as a patient.”4 Dermatitis para-artefacta syndrome was described as a disorder of impulse control in which a patient will produce or manipulate a specific dermatosis presentation. The patient usually admits to doing it in a semiconscious state. Dermatitis artefacta and dermatitis para-artefacta differ from malingering in that malingering patients knowingly fake symptoms for external gain, which can be monetary or the avoidance of responsibility.4 More familiar examples to dermatologists of factitial disease include factitial panniculitis,1 direct applications of caustic agents to the skin, and excoriations from instruments or fingernails.4,5
This case illustrates the difficult and potentially dangerous nature of factitial disorders, specifically dermatitis para-artefacta syndrome. Our patient was intensely preoccupied with the outcome of being a patient in a hospital. Our patient sought out a medication from multiple providers to produce a deadly and severe life-threatening reaction. If his main intentions were solely to obtain a bed and 3 square meals a day, then malingering would have represented his factitial disease. However, his main intent was to be seen as a patient, and then doted on and cared for by medical professionals in a hospital setting. From this assessment, the patient’s behavior would fall under the factitial disorder of dermatitis para-artefacta syndrome.
Factitious disorders pose immense challenges for diagnosis and treatment. It is prudent for physicians to learn to recognize patterns of history and examination that do not coincide. The first step in treatment is the recognition and early involvement of psychiatry to aid in curbing this behavior. Remission of factitial disorders can be induced with proper diagnosis and treatment. Patients with the highest chance of remission are those with treatment centered on behavioral therapy in conjunction with psychotropic medications.1
To the Editor:
Lyell1 coined the term cutaneous artifactual disease to describe the spectrum of factitious disorders associated with skin presentations. Interestingly, Lyell was the first to name toxic epidermal necrolysis (TEN).2,3 We present a rare case of factitial TEN, a dangerous and life-threatening manifestation of factitial disease.
A 49-year-old homeless man with a history of Stevens-Johnson syndrome (SJS)/TEN from trimethoprim-sulfamethoxazole (TMP-SMX) was admitted on 4 separate occasions over an 18-month period for recurrent exposure to the medication producing SJS/TEN. Originally, this patient was given TMP-SMX for a skin infection and 10 days later presented with 15% body surface area (BSA) involvement of SJS/TEN. He was successfully treated with intravenous immunoglobulin (IVIg) in the burn intensive care unit (BICU) and discharged. Several months later, the patient was given TMP-SMX for a leg infection by a different clinic. He was admitted to the BICU with 40% BSA, treated with IVIg, and survived. Eight months later, the patient was again admitted to the BICU with 30% BSA and treated with IVIg; however, this admission required intubation due to complications secondary to volume resuscitation. He was evaluated by psychiatry and confessed to purposely seeking TMP-SMX, stating that he “liked the food and care in the hospital.” He was diagnosed with factitial disorder and given a referral for further treatment at an outpatient facility. Two months later, the patient was again admitted to the BICU after taking a single dose of TMP-SMX obtained from a “friend.” He had 10% BSA with conjunctival involvement and was again successfully treated with IVIg. He was discharged with the state psychiatric system for further treatment and evaluation.
Factitial disease in dermatology is difficult to diagnose. Its incidence is unknown, as only case reports exist in the literature. In factitial disease, patients “perform self-mutilating and clinically relevant damage to themselves without the direct intention of suicide.”4 Harth et al4 described 3 subcategories of factitious disorders: dermatitis artefacta syndrome, dermatitis para-artefacta syndrome, or malingering. Dermatitis artefacta syndrome is “a dissociated self-injury or behavior where the patient unconsciously simulated disease with intention to be cared for as a patient.”4 Dermatitis para-artefacta syndrome was described as a disorder of impulse control in which a patient will produce or manipulate a specific dermatosis presentation. The patient usually admits to doing it in a semiconscious state. Dermatitis artefacta and dermatitis para-artefacta differ from malingering in that malingering patients knowingly fake symptoms for external gain, which can be monetary or the avoidance of responsibility.4 More familiar examples to dermatologists of factitial disease include factitial panniculitis,1 direct applications of caustic agents to the skin, and excoriations from instruments or fingernails.4,5
This case illustrates the difficult and potentially dangerous nature of factitial disorders, specifically dermatitis para-artefacta syndrome. Our patient was intensely preoccupied with the outcome of being a patient in a hospital. Our patient sought out a medication from multiple providers to produce a deadly and severe life-threatening reaction. If his main intentions were solely to obtain a bed and 3 square meals a day, then malingering would have represented his factitial disease. However, his main intent was to be seen as a patient, and then doted on and cared for by medical professionals in a hospital setting. From this assessment, the patient’s behavior would fall under the factitial disorder of dermatitis para-artefacta syndrome.
Factitious disorders pose immense challenges for diagnosis and treatment. It is prudent for physicians to learn to recognize patterns of history and examination that do not coincide. The first step in treatment is the recognition and early involvement of psychiatry to aid in curbing this behavior. Remission of factitial disorders can be induced with proper diagnosis and treatment. Patients with the highest chance of remission are those with treatment centered on behavioral therapy in conjunction with psychotropic medications.1
- Lyell A. Cutaneous artifactual disease. J Am Acad Dermatol. 1979;1:391-402.
- Palmieri TL, Greenhalgh, DG, Saffle JR, et al. A multicenter review of toxic epidermal necrolysis treated in U.S. burn centers at the end of the twentieth century. J Burn Care Rehab. 2002;23:87-96.
- Lyell A. Toxic epidermal necrolysis: an eruption resembling scalding of the skin. Br J Dermatol. 1956;68:355-361.
- Harth W, Gieler U, Kusnir D, et al. Primarily psychogenic dermatoses. In: Clinical Management in Psychodermatology. 1st ed. Leipzog, Germany: Springer-Verlag Berlin Heidelberg; 2009:11-19.
- Sanmartin O, Requena C, Requena L. Factitial panniculitis. Dermatol Clin. 2008;26:519-527.
- Lyell A. Cutaneous artifactual disease. J Am Acad Dermatol. 1979;1:391-402.
- Palmieri TL, Greenhalgh, DG, Saffle JR, et al. A multicenter review of toxic epidermal necrolysis treated in U.S. burn centers at the end of the twentieth century. J Burn Care Rehab. 2002;23:87-96.
- Lyell A. Toxic epidermal necrolysis: an eruption resembling scalding of the skin. Br J Dermatol. 1956;68:355-361.
- Harth W, Gieler U, Kusnir D, et al. Primarily psychogenic dermatoses. In: Clinical Management in Psychodermatology. 1st ed. Leipzog, Germany: Springer-Verlag Berlin Heidelberg; 2009:11-19.
- Sanmartin O, Requena C, Requena L. Factitial panniculitis. Dermatol Clin. 2008;26:519-527.
Practice Points
- It is important to consider an underlying psychiatric disorder (eg, factitial disorders) in dermatologic patients, even when an exogenous cause can be identified.
- On occasion, dermatologic disease is best treated and prevented with routine psychiatric care and psychotropic therapy.

PCV vaccines less prominent in children with meningitis
Pneumococcal conjugate vaccines 7-valent and 13-valent (PCV7/PCV13) in children younger than 5 years of age in Israel were less prominent in meningitis than in nonmeningitis invasive pneumococcal disease (nm-IPD), according to S. Ben-Shimol, MD, and associates.
Between July 2000 and June 2015, 4,168 IPD episodes were reported; 426 (10.2%) were meningitis. The PCV13 serotype (13VT) meningitis rates significantly declined by 93% (incidence rate ratio = 0.07), from 3.6 ± 1.3 in the pre-PCV period to 0.3 in the last year of the study. Also, the 13VT nm-IPD rates significantly declined by 95% (IRR = 0.05), from a rate of 40.0 ± 5.4 in the pre-PCV period to 1.9. The non-13VT meningitis rates significantly increased by 273% (IRR = 3.73), from 0.8 ± 0.3 in the pre-PCV period to 3.0. And the non-13VT nm-IPD rates also significantly increased by 162% (IRR = 2.62), from 4.5 ± 0.8 in the pre-PCV period to 11.8.

The researchers noted that the increase in non-13VT meningitis was partially driven by a sharp and significant increase of serotype 12F, along with the other predominant non-13VT serotypes that caused meningitis: 15B/C, 24F, and 27. The serotypes also were predominant in non-13VT nm-IPD, as were additional serotypes 8, 10A, 33F, 7B and 10B.
“This finding may be attributed to the younger age of children with meningitis and differences in causative serotypes between the two groups, as the decline of the incidence of meningitis and nm-IPD caused by vaccine-serotypes is similar,” researchers concluded. “Continuous monitoring of meningitis and nm-IPD is warranted.”
Find the full study in Vaccine (doi: 10.1016/j.vaccine.2016.07.038).
Pneumococcal conjugate vaccines 7-valent and 13-valent (PCV7/PCV13) in children younger than 5 years of age in Israel were less prominent in meningitis than in nonmeningitis invasive pneumococcal disease (nm-IPD), according to S. Ben-Shimol, MD, and associates.
Between July 2000 and June 2015, 4,168 IPD episodes were reported; 426 (10.2%) were meningitis. The PCV13 serotype (13VT) meningitis rates significantly declined by 93% (incidence rate ratio = 0.07), from 3.6 ± 1.3 in the pre-PCV period to 0.3 in the last year of the study. Also, the 13VT nm-IPD rates significantly declined by 95% (IRR = 0.05), from a rate of 40.0 ± 5.4 in the pre-PCV period to 1.9. The non-13VT meningitis rates significantly increased by 273% (IRR = 3.73), from 0.8 ± 0.3 in the pre-PCV period to 3.0. And the non-13VT nm-IPD rates also significantly increased by 162% (IRR = 2.62), from 4.5 ± 0.8 in the pre-PCV period to 11.8.
The researchers noted that the increase in non-13VT meningitis was partially driven by a sharp and significant increase of serotype 12F, along with the other predominant non-13VT serotypes that caused meningitis: 15B/C, 24F, and 27. The serotypes also were predominant in non-13VT nm-IPD, as were additional serotypes 8, 10A, 33F, 7B and 10B.
“This finding may be attributed to the younger age of children with meningitis and differences in causative serotypes between the two groups, as the decline of the incidence of meningitis and nm-IPD caused by vaccine-serotypes is similar,” researchers concluded. “Continuous monitoring of meningitis and nm-IPD is warranted.”
Find the full study in Vaccine (doi: 10.1016/j.vaccine.2016.07.038).
Pneumococcal conjugate vaccines 7-valent and 13-valent (PCV7/PCV13) in children younger than 5 years of age in Israel were less prominent in meningitis than in nonmeningitis invasive pneumococcal disease (nm-IPD), according to S. Ben-Shimol, MD, and associates.
Between July 2000 and June 2015, 4,168 IPD episodes were reported; 426 (10.2%) were meningitis. The PCV13 serotype (13VT) meningitis rates significantly declined by 93% (incidence rate ratio = 0.07), from 3.6 ± 1.3 in the pre-PCV period to 0.3 in the last year of the study. Also, the 13VT nm-IPD rates significantly declined by 95% (IRR = 0.05), from a rate of 40.0 ± 5.4 in the pre-PCV period to 1.9. The non-13VT meningitis rates significantly increased by 273% (IRR = 3.73), from 0.8 ± 0.3 in the pre-PCV period to 3.0. And the non-13VT nm-IPD rates also significantly increased by 162% (IRR = 2.62), from 4.5 ± 0.8 in the pre-PCV period to 11.8.
The researchers noted that the increase in non-13VT meningitis was partially driven by a sharp and significant increase of serotype 12F, along with the other predominant non-13VT serotypes that caused meningitis: 15B/C, 24F, and 27. The serotypes also were predominant in non-13VT nm-IPD, as were additional serotypes 8, 10A, 33F, 7B and 10B.
“This finding may be attributed to the younger age of children with meningitis and differences in causative serotypes between the two groups, as the decline of the incidence of meningitis and nm-IPD caused by vaccine-serotypes is similar,” researchers concluded. “Continuous monitoring of meningitis and nm-IPD is warranted.”
Find the full study in Vaccine (doi: 10.1016/j.vaccine.2016.07.038).
FROM VACCINE
