User login
Leukocytoclastic Vasculitis Resolution With Topical Dapsone
Leukocytoclastic vasculitis (LCV) is a disease characterized by inflammation of small vessels with characteristic clinical findings of petechiae and palpable purpura.1 Numerous etiologies have been described, but the disease commonly remains idiopathic.2,3 Leukocytoclastic vasculitis often spontaneously resolves within weeks and requires only symptomatic treatment. Chronic or severe disease can require systemic medical treatment with agents such as colchicine, dapsone, and corticosteroids. These agents are effective but carry risks of serious side effects.4,5 These side effects and/or medical contraindications prevent some patients from taking systemic medications for LCV. We present a case of LCV that resolved after treatment with topical dapsone, highlighting a potential new treatment ofLCV with a markedly better side-effect profile.
Case Report
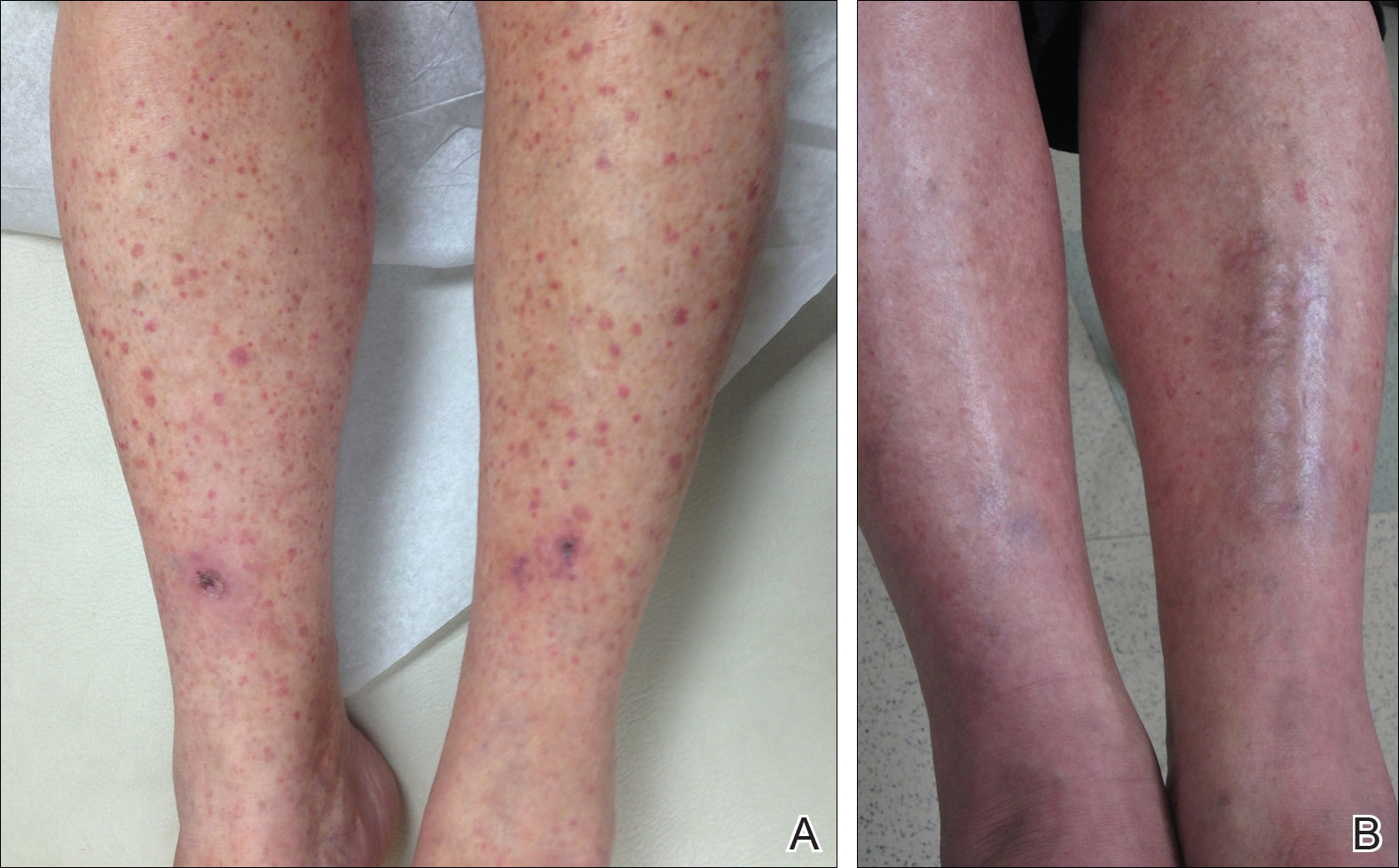
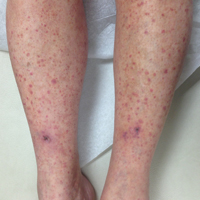
A 60-year-old woman with recent upper respiratory tract and sinus infections presented to our dermatology clinic with painful palpable purpura on the bilateral shins, thighs, and dorsal aspects of the feet of several months’ duration (Figure, A). Her primary care provider initiated treatment with amoxicillin and doxycycline for the infections. When the rash developed approximately 1.5 weeks following initiation of her symptoms, the patient was referred to the dermatology and rheumatology departments at our institution. The treating dermatologist (M.B.T.) obtained a 4-mm punch biopsy from the right lower leg and LCV was shown on histology. The patient completed a 14-day course of doxycycline and amoxicillin without resolution of the eruption. After an extensive investigation, the treating rheumatologist concluded that the LCV was idiopathic or secondary to an infection or drug exposure. The rheumatologist started the patient on oral prednisone for the chronic symptomatic LCV, but she was intolerant of this medication and discontinued it after 1 week. Our dermatology clinic started her on triamcinolone cream 0.1% twice daily, but she continued to experience new and worsening lesions. At her follow-up appointment 1 month later, triamcinolone cream was discontinued and dapsone gel 5% twice daily was started. She experienced resolution of her previously recalcitrant LCV within 3 weeks (Figure, B).
Comment
Established therapies for LCV carry serious side-effect profiles, which can preclude their use.5 Therefore, a topical therapeutic alternative for LCV would be ideal. Systemic prednisone is the first-line therapy for chronic and/or symptomatic LCV, but its side effects include suppression of the hypothalamic-pituitary-adrenal axis, immunosuppression, osteonecrosis, and glucose intolerance.5 Colchicine therapy carries risks for blood dyscrasia, immunosuppression, and gastrointestinal tract upset. Systemic dapsone also is an effective therapy for chronic and/or symptomatic LCV.5,6 However, systemic dapsone requires glucose-6-phosphate dehydrogenase deficiency screening and routine monitoring of blood counts, and it also carries the risk for serious adverse effects including neuropathy, blood dyscrasia, and hypersensitivity syndrome.5,6 Topical dapsone may provide similar efficacy with far fewer adverse effects and has proven to be a safe treatment of acne, even when used in patients with glucose-6-phosphate dehydrogenase deficiency. It displays low systemic absorption and does not accumulate over time once a steady state is reached.7 It also has been shown to be beneficial in other vasculopathies such as erythema elevatum diutinum and in other neutrophilic inflammatory disorders such as pyoderma gangrenosum.8,9 A case of methemoglobinemia due to topical dapsone has been reported.10 Although this effect is rare, clinicians should be aware of such adverse effects when using medications for off-label purposes.
Leukocytoclastic vasculitis can spontaneously resolve; however, our patient’s disease was chronic for several months, and she continued to develop new lesions without signs of resolution. After initiating topical dapsone, she experienced resolution within 3 weeks.
Conclusion
Topical dapsone is a novel approach for treating LCV. Given this drug’s favorable side-effect profile compared to the currently available therapeutic alternatives, we believe it is a reasonable option in select patients. Further investigation is needed to prove its efficacy, but it could be an ideal alternative for patients with contraindications to traditional therapies and/or for those unable to tolerate systemic therapy.
- Koutkia P, Mylonakis E, Rounds S, et al. Leucocytoclastic vasculitis: an update for the clinician. Scand J Rheumatol. 2001;30:315-322.
- Af Ekenstam E, Callen JP. Cutaneous leukocytoclastic vasculitis. clinical and laboratory features of 82 patients seen in private practice. Arch Dermatol. 1984;120:484-489.
- Gyselbrecht L, de Keyser F, Ongenae K, et al. Etiological factors and underlying conditions in patientswith leucocytoclastic vasculitis. Clin Exp Rheumatol. 1996;14:665-668.
- Sais G, Vidaller A, Jucglà A, et al. Colchicine in the treatment of cutaneous leukocytoclastic vasculitis. results of a prospective, randomized controlled trial. Arch Dermatol. 1995;131:1399-1402.
- Sunderkotter C, Bonsmann G, Sindrilaru A, et al. Management of leukocytoclastic vasculitis: clinical review. J Dermatol Treat. 2005;16:193-206.
- Zhu YI, Stiller MJ. Dapsone and sulfones in dermatology: overview and update. J Am Acad Dermatol. 2001;45:420-434.
- Stotland M, Shalita AR, Kissling RF. Dapsone 5% gel: a review of its efficacy and safety in the treatment of acne vulgaris. Am J Clin Dermatol. 2009;10:221-227.
- Frieling GW, Williams NL, Lim SJ, et al. Novel use of topical dapsone 5% gel for erythema elevatum diutinum: safer and effective. J Drugs Dermatol. 2013;12:481-484.
- Handler MZ, Hamilton H, Aires D. Treatment of peristomal pyoderma gangrenosum with topical crushed dapsone. J Drugs Dermatol. 2011;10:1059-1061.
- Swartzentruber GS, Yanta JH, Pizon AF. Methemoglobi-nemia as a complication of topical dapsone. N Engl J Med. 2015;372:491-492.
Leukocytoclastic vasculitis (LCV) is a disease characterized by inflammation of small vessels with characteristic clinical findings of petechiae and palpable purpura.1 Numerous etiologies have been described, but the disease commonly remains idiopathic.2,3 Leukocytoclastic vasculitis often spontaneously resolves within weeks and requires only symptomatic treatment. Chronic or severe disease can require systemic medical treatment with agents such as colchicine, dapsone, and corticosteroids. These agents are effective but carry risks of serious side effects.4,5 These side effects and/or medical contraindications prevent some patients from taking systemic medications for LCV. We present a case of LCV that resolved after treatment with topical dapsone, highlighting a potential new treatment ofLCV with a markedly better side-effect profile.
Case Report
A 60-year-old woman with recent upper respiratory tract and sinus infections presented to our dermatology clinic with painful palpable purpura on the bilateral shins, thighs, and dorsal aspects of the feet of several months’ duration (Figure, A). Her primary care provider initiated treatment with amoxicillin and doxycycline for the infections. When the rash developed approximately 1.5 weeks following initiation of her symptoms, the patient was referred to the dermatology and rheumatology departments at our institution. The treating dermatologist (M.B.T.) obtained a 4-mm punch biopsy from the right lower leg and LCV was shown on histology. The patient completed a 14-day course of doxycycline and amoxicillin without resolution of the eruption. After an extensive investigation, the treating rheumatologist concluded that the LCV was idiopathic or secondary to an infection or drug exposure. The rheumatologist started the patient on oral prednisone for the chronic symptomatic LCV, but she was intolerant of this medication and discontinued it after 1 week. Our dermatology clinic started her on triamcinolone cream 0.1% twice daily, but she continued to experience new and worsening lesions. At her follow-up appointment 1 month later, triamcinolone cream was discontinued and dapsone gel 5% twice daily was started. She experienced resolution of her previously recalcitrant LCV within 3 weeks (Figure, B).
Comment
Established therapies for LCV carry serious side-effect profiles, which can preclude their use.5 Therefore, a topical therapeutic alternative for LCV would be ideal. Systemic prednisone is the first-line therapy for chronic and/or symptomatic LCV, but its side effects include suppression of the hypothalamic-pituitary-adrenal axis, immunosuppression, osteonecrosis, and glucose intolerance.5 Colchicine therapy carries risks for blood dyscrasia, immunosuppression, and gastrointestinal tract upset. Systemic dapsone also is an effective therapy for chronic and/or symptomatic LCV.5,6 However, systemic dapsone requires glucose-6-phosphate dehydrogenase deficiency screening and routine monitoring of blood counts, and it also carries the risk for serious adverse effects including neuropathy, blood dyscrasia, and hypersensitivity syndrome.5,6 Topical dapsone may provide similar efficacy with far fewer adverse effects and has proven to be a safe treatment of acne, even when used in patients with glucose-6-phosphate dehydrogenase deficiency. It displays low systemic absorption and does not accumulate over time once a steady state is reached.7 It also has been shown to be beneficial in other vasculopathies such as erythema elevatum diutinum and in other neutrophilic inflammatory disorders such as pyoderma gangrenosum.8,9 A case of methemoglobinemia due to topical dapsone has been reported.10 Although this effect is rare, clinicians should be aware of such adverse effects when using medications for off-label purposes.
Leukocytoclastic vasculitis can spontaneously resolve; however, our patient’s disease was chronic for several months, and she continued to develop new lesions without signs of resolution. After initiating topical dapsone, she experienced resolution within 3 weeks.
Conclusion
Topical dapsone is a novel approach for treating LCV. Given this drug’s favorable side-effect profile compared to the currently available therapeutic alternatives, we believe it is a reasonable option in select patients. Further investigation is needed to prove its efficacy, but it could be an ideal alternative for patients with contraindications to traditional therapies and/or for those unable to tolerate systemic therapy.
Leukocytoclastic vasculitis (LCV) is a disease characterized by inflammation of small vessels with characteristic clinical findings of petechiae and palpable purpura.1 Numerous etiologies have been described, but the disease commonly remains idiopathic.2,3 Leukocytoclastic vasculitis often spontaneously resolves within weeks and requires only symptomatic treatment. Chronic or severe disease can require systemic medical treatment with agents such as colchicine, dapsone, and corticosteroids. These agents are effective but carry risks of serious side effects.4,5 These side effects and/or medical contraindications prevent some patients from taking systemic medications for LCV. We present a case of LCV that resolved after treatment with topical dapsone, highlighting a potential new treatment ofLCV with a markedly better side-effect profile.
Case Report
A 60-year-old woman with recent upper respiratory tract and sinus infections presented to our dermatology clinic with painful palpable purpura on the bilateral shins, thighs, and dorsal aspects of the feet of several months’ duration (Figure, A). Her primary care provider initiated treatment with amoxicillin and doxycycline for the infections. When the rash developed approximately 1.5 weeks following initiation of her symptoms, the patient was referred to the dermatology and rheumatology departments at our institution. The treating dermatologist (M.B.T.) obtained a 4-mm punch biopsy from the right lower leg and LCV was shown on histology. The patient completed a 14-day course of doxycycline and amoxicillin without resolution of the eruption. After an extensive investigation, the treating rheumatologist concluded that the LCV was idiopathic or secondary to an infection or drug exposure. The rheumatologist started the patient on oral prednisone for the chronic symptomatic LCV, but she was intolerant of this medication and discontinued it after 1 week. Our dermatology clinic started her on triamcinolone cream 0.1% twice daily, but she continued to experience new and worsening lesions. At her follow-up appointment 1 month later, triamcinolone cream was discontinued and dapsone gel 5% twice daily was started. She experienced resolution of her previously recalcitrant LCV within 3 weeks (Figure, B).
Comment
Established therapies for LCV carry serious side-effect profiles, which can preclude their use.5 Therefore, a topical therapeutic alternative for LCV would be ideal. Systemic prednisone is the first-line therapy for chronic and/or symptomatic LCV, but its side effects include suppression of the hypothalamic-pituitary-adrenal axis, immunosuppression, osteonecrosis, and glucose intolerance.5 Colchicine therapy carries risks for blood dyscrasia, immunosuppression, and gastrointestinal tract upset. Systemic dapsone also is an effective therapy for chronic and/or symptomatic LCV.5,6 However, systemic dapsone requires glucose-6-phosphate dehydrogenase deficiency screening and routine monitoring of blood counts, and it also carries the risk for serious adverse effects including neuropathy, blood dyscrasia, and hypersensitivity syndrome.5,6 Topical dapsone may provide similar efficacy with far fewer adverse effects and has proven to be a safe treatment of acne, even when used in patients with glucose-6-phosphate dehydrogenase deficiency. It displays low systemic absorption and does not accumulate over time once a steady state is reached.7 It also has been shown to be beneficial in other vasculopathies such as erythema elevatum diutinum and in other neutrophilic inflammatory disorders such as pyoderma gangrenosum.8,9 A case of methemoglobinemia due to topical dapsone has been reported.10 Although this effect is rare, clinicians should be aware of such adverse effects when using medications for off-label purposes.
Leukocytoclastic vasculitis can spontaneously resolve; however, our patient’s disease was chronic for several months, and she continued to develop new lesions without signs of resolution. After initiating topical dapsone, she experienced resolution within 3 weeks.
Conclusion
Topical dapsone is a novel approach for treating LCV. Given this drug’s favorable side-effect profile compared to the currently available therapeutic alternatives, we believe it is a reasonable option in select patients. Further investigation is needed to prove its efficacy, but it could be an ideal alternative for patients with contraindications to traditional therapies and/or for those unable to tolerate systemic therapy.
- Koutkia P, Mylonakis E, Rounds S, et al. Leucocytoclastic vasculitis: an update for the clinician. Scand J Rheumatol. 2001;30:315-322.
- Af Ekenstam E, Callen JP. Cutaneous leukocytoclastic vasculitis. clinical and laboratory features of 82 patients seen in private practice. Arch Dermatol. 1984;120:484-489.
- Gyselbrecht L, de Keyser F, Ongenae K, et al. Etiological factors and underlying conditions in patientswith leucocytoclastic vasculitis. Clin Exp Rheumatol. 1996;14:665-668.
- Sais G, Vidaller A, Jucglà A, et al. Colchicine in the treatment of cutaneous leukocytoclastic vasculitis. results of a prospective, randomized controlled trial. Arch Dermatol. 1995;131:1399-1402.
- Sunderkotter C, Bonsmann G, Sindrilaru A, et al. Management of leukocytoclastic vasculitis: clinical review. J Dermatol Treat. 2005;16:193-206.
- Zhu YI, Stiller MJ. Dapsone and sulfones in dermatology: overview and update. J Am Acad Dermatol. 2001;45:420-434.
- Stotland M, Shalita AR, Kissling RF. Dapsone 5% gel: a review of its efficacy and safety in the treatment of acne vulgaris. Am J Clin Dermatol. 2009;10:221-227.
- Frieling GW, Williams NL, Lim SJ, et al. Novel use of topical dapsone 5% gel for erythema elevatum diutinum: safer and effective. J Drugs Dermatol. 2013;12:481-484.
- Handler MZ, Hamilton H, Aires D. Treatment of peristomal pyoderma gangrenosum with topical crushed dapsone. J Drugs Dermatol. 2011;10:1059-1061.
- Swartzentruber GS, Yanta JH, Pizon AF. Methemoglobi-nemia as a complication of topical dapsone. N Engl J Med. 2015;372:491-492.
- Koutkia P, Mylonakis E, Rounds S, et al. Leucocytoclastic vasculitis: an update for the clinician. Scand J Rheumatol. 2001;30:315-322.
- Af Ekenstam E, Callen JP. Cutaneous leukocytoclastic vasculitis. clinical and laboratory features of 82 patients seen in private practice. Arch Dermatol. 1984;120:484-489.
- Gyselbrecht L, de Keyser F, Ongenae K, et al. Etiological factors and underlying conditions in patientswith leucocytoclastic vasculitis. Clin Exp Rheumatol. 1996;14:665-668.
- Sais G, Vidaller A, Jucglà A, et al. Colchicine in the treatment of cutaneous leukocytoclastic vasculitis. results of a prospective, randomized controlled trial. Arch Dermatol. 1995;131:1399-1402.
- Sunderkotter C, Bonsmann G, Sindrilaru A, et al. Management of leukocytoclastic vasculitis: clinical review. J Dermatol Treat. 2005;16:193-206.
- Zhu YI, Stiller MJ. Dapsone and sulfones in dermatology: overview and update. J Am Acad Dermatol. 2001;45:420-434.
- Stotland M, Shalita AR, Kissling RF. Dapsone 5% gel: a review of its efficacy and safety in the treatment of acne vulgaris. Am J Clin Dermatol. 2009;10:221-227.
- Frieling GW, Williams NL, Lim SJ, et al. Novel use of topical dapsone 5% gel for erythema elevatum diutinum: safer and effective. J Drugs Dermatol. 2013;12:481-484.
- Handler MZ, Hamilton H, Aires D. Treatment of peristomal pyoderma gangrenosum with topical crushed dapsone. J Drugs Dermatol. 2011;10:1059-1061.
- Swartzentruber GS, Yanta JH, Pizon AF. Methemoglobi-nemia as a complication of topical dapsone. N Engl J Med. 2015;372:491-492.
Practice Points
- Leukocytoclastic vasculitis is characterized by inflammation of small vessels with characteristic clinical findings of petechiae and palpable purpura.
- Leukocytoclastic vasculitis often spontaneously resolves within weeks and requires only symptomatic treatment, but chronic or severe disease can require systemic medical treatment with agents such as colchicine, dapsone, and corticosteroids.
Long-acting injectables may be best at preventing relapse in psychosis
SAN DIEGO – A new meta-analysis suggests that second-generation long-acting injectable antipsychotics (LAIs) are slightly better than oral antipsychotics at preventing relapse after a first psychotic incident.
The meta-analysis, released at the annual meeting of the American Psychiatric Association, is limited because it looks at only three studies. Still, study lead author Christine Tran-Boynes, DO, said the findings are useful for psychiatrists.
“For a long time, LAIs were associated with severely ill psychotic patients who were frequently hospitalized and not compliant with their oral meds,” Dr. Tran-Boynes, a resident at the University of Maryland, Baltimore, said in an interview. “The purpose of this paper is to change the perception of LAIs. They are not just a medication of last resort in those with severe, chronic psychosis but, instead, can be used in the early stages of psychosis as prophylaxis against relapse.”
Injectable antipsychotics are more commonly used in Europe, where “there also seemed to be a greater willingness among patients to receive this treatment,” said Peter F. Buckley, MD, dean of the medical school at Virginia Commonwealth University, Richmond.
The APA’s schizophrenia treatment guidelines recommend LAIs for patients with “recurrent relapses related to nonadherence” and patients who prefer the shots. Dr. Tran-Boynes notes that “the most common cause of relapse in patients with schizophrenia is partial adherence or nonadherence to oral antipsychotics. If LAIs can improve adherence in patients and monitoring of adherence for clinicians, they could have a role in preventing relapse during this critical period in psychosis.”
The meta-analysis examines three randomized controlled studies – two from 2015 and one from 2013 – that compare second-generation LAIs to first- and second-generation oral antipsychotics after first episodes of psychosis. Dr. Tran-Boynes said researchers could not find any studies comparing first-generation long-acting antipsychotics to oral antipsychotics.
The largest study had 769 participants; the others had 85 and 86. The subjects, all adults, had diagnoses of schizophrenia, schizoaffective disorder, or schizophreniform disorder. Their diagnoses must have been made within the previous 5 years.
According to the meta-analysis, relapses after first-episode psychosis were more likely (relative risk, 1.078; 95% confidence interval, 1.007-1.154; P = 0.012) in patients taking first- or second-generation oral antipsychotics, compared with those on second-generation LAIs.
“There was an 8% greater efficacy for LAIs preventing relapse after early psychosis, compared to oral antipsychotics,” Dr. Tran-Boynes said. She calculated the number needed to treat as 14.
The percentages of patients who did not relapse while taking second-generation LAIs ranged from 73% (31 of 42 patients randomized to an injectable risperidone arm over 24 months) to 95% (38 of 40 patients over a 12-month study, also of injectable risperidone), Dr. Tran-Boynes said.
When asked about the meta-analysis, Robert Rosenheck, MD, expressed concern.
“While well done, it is based on too few studies to give useful guidance to practice,” said Dr. Rosenheck, professor of psychiatry, epidemiology and public health at Yale University, New Haven, Conn.
Dr. Buckley also noted that the meta-analysis includes a small number of studies. “The effect is sizable for a first-episode population, but other studies to date are more mixed,” he added. “For instance, in a study among a more chronic schizophrenic population, we found no difference between a group receiving long-acting injectable risperidone and oral second-generation antipsychotics” (Schizophr Bull. 2015 Mar;41[2]:449-59).
What should psychiatrists know when they consider prescribing LAIs to prevent psychotic relapse? “If a patient expresses willingness to take an oral antipsychotic on a daily basis and/or has someone to monitor his medication intake, then prescribing an oral antipsychotic would be the ideal route,” Dr. Tran-Boynes said. “However, I would recommend LAIs to patients who have demonstrated poor compliance with previous medications in general, poor awareness of psychosis, poor awareness of need for treatment, poor availability of social support to ensure that the patient will take his/her medication daily, and/or if a patient expresses preference for LAIs.”
She cautioned that LAIs have disadvantages. Compared with oral antipsychotics, it’s harder to adjust patients’ dosages in response to side effects or when they improve, she said. LAIs are also more expensive in the short term, she said.
However, LAIs also may have produced fewer side effects, and there aren’t any questions about compliance, she said. In addition, “there’s less pain at the injection site with second-generation LAIs, compared to first-generation LAIs, due to the water-based solution of the former. The oil-based solutions that are characteristic of first-generation LAIs have been shown in studies to be very painful.”
Dr. Tran-Boynes and Dr. Rosenheck reported no relevant disclosures. Dr. Buckley disclosed that he is a research consultant for the National Institute of Mental Health.
SAN DIEGO – A new meta-analysis suggests that second-generation long-acting injectable antipsychotics (LAIs) are slightly better than oral antipsychotics at preventing relapse after a first psychotic incident.
The meta-analysis, released at the annual meeting of the American Psychiatric Association, is limited because it looks at only three studies. Still, study lead author Christine Tran-Boynes, DO, said the findings are useful for psychiatrists.
“For a long time, LAIs were associated with severely ill psychotic patients who were frequently hospitalized and not compliant with their oral meds,” Dr. Tran-Boynes, a resident at the University of Maryland, Baltimore, said in an interview. “The purpose of this paper is to change the perception of LAIs. They are not just a medication of last resort in those with severe, chronic psychosis but, instead, can be used in the early stages of psychosis as prophylaxis against relapse.”
Injectable antipsychotics are more commonly used in Europe, where “there also seemed to be a greater willingness among patients to receive this treatment,” said Peter F. Buckley, MD, dean of the medical school at Virginia Commonwealth University, Richmond.
The APA’s schizophrenia treatment guidelines recommend LAIs for patients with “recurrent relapses related to nonadherence” and patients who prefer the shots. Dr. Tran-Boynes notes that “the most common cause of relapse in patients with schizophrenia is partial adherence or nonadherence to oral antipsychotics. If LAIs can improve adherence in patients and monitoring of adherence for clinicians, they could have a role in preventing relapse during this critical period in psychosis.”
The meta-analysis examines three randomized controlled studies – two from 2015 and one from 2013 – that compare second-generation LAIs to first- and second-generation oral antipsychotics after first episodes of psychosis. Dr. Tran-Boynes said researchers could not find any studies comparing first-generation long-acting antipsychotics to oral antipsychotics.
The largest study had 769 participants; the others had 85 and 86. The subjects, all adults, had diagnoses of schizophrenia, schizoaffective disorder, or schizophreniform disorder. Their diagnoses must have been made within the previous 5 years.
According to the meta-analysis, relapses after first-episode psychosis were more likely (relative risk, 1.078; 95% confidence interval, 1.007-1.154; P = 0.012) in patients taking first- or second-generation oral antipsychotics, compared with those on second-generation LAIs.
“There was an 8% greater efficacy for LAIs preventing relapse after early psychosis, compared to oral antipsychotics,” Dr. Tran-Boynes said. She calculated the number needed to treat as 14.
The percentages of patients who did not relapse while taking second-generation LAIs ranged from 73% (31 of 42 patients randomized to an injectable risperidone arm over 24 months) to 95% (38 of 40 patients over a 12-month study, also of injectable risperidone), Dr. Tran-Boynes said.
When asked about the meta-analysis, Robert Rosenheck, MD, expressed concern.
“While well done, it is based on too few studies to give useful guidance to practice,” said Dr. Rosenheck, professor of psychiatry, epidemiology and public health at Yale University, New Haven, Conn.
Dr. Buckley also noted that the meta-analysis includes a small number of studies. “The effect is sizable for a first-episode population, but other studies to date are more mixed,” he added. “For instance, in a study among a more chronic schizophrenic population, we found no difference between a group receiving long-acting injectable risperidone and oral second-generation antipsychotics” (Schizophr Bull. 2015 Mar;41[2]:449-59).
What should psychiatrists know when they consider prescribing LAIs to prevent psychotic relapse? “If a patient expresses willingness to take an oral antipsychotic on a daily basis and/or has someone to monitor his medication intake, then prescribing an oral antipsychotic would be the ideal route,” Dr. Tran-Boynes said. “However, I would recommend LAIs to patients who have demonstrated poor compliance with previous medications in general, poor awareness of psychosis, poor awareness of need for treatment, poor availability of social support to ensure that the patient will take his/her medication daily, and/or if a patient expresses preference for LAIs.”
She cautioned that LAIs have disadvantages. Compared with oral antipsychotics, it’s harder to adjust patients’ dosages in response to side effects or when they improve, she said. LAIs are also more expensive in the short term, she said.
However, LAIs also may have produced fewer side effects, and there aren’t any questions about compliance, she said. In addition, “there’s less pain at the injection site with second-generation LAIs, compared to first-generation LAIs, due to the water-based solution of the former. The oil-based solutions that are characteristic of first-generation LAIs have been shown in studies to be very painful.”
Dr. Tran-Boynes and Dr. Rosenheck reported no relevant disclosures. Dr. Buckley disclosed that he is a research consultant for the National Institute of Mental Health.
SAN DIEGO – A new meta-analysis suggests that second-generation long-acting injectable antipsychotics (LAIs) are slightly better than oral antipsychotics at preventing relapse after a first psychotic incident.
The meta-analysis, released at the annual meeting of the American Psychiatric Association, is limited because it looks at only three studies. Still, study lead author Christine Tran-Boynes, DO, said the findings are useful for psychiatrists.
“For a long time, LAIs were associated with severely ill psychotic patients who were frequently hospitalized and not compliant with their oral meds,” Dr. Tran-Boynes, a resident at the University of Maryland, Baltimore, said in an interview. “The purpose of this paper is to change the perception of LAIs. They are not just a medication of last resort in those with severe, chronic psychosis but, instead, can be used in the early stages of psychosis as prophylaxis against relapse.”
Injectable antipsychotics are more commonly used in Europe, where “there also seemed to be a greater willingness among patients to receive this treatment,” said Peter F. Buckley, MD, dean of the medical school at Virginia Commonwealth University, Richmond.
The APA’s schizophrenia treatment guidelines recommend LAIs for patients with “recurrent relapses related to nonadherence” and patients who prefer the shots. Dr. Tran-Boynes notes that “the most common cause of relapse in patients with schizophrenia is partial adherence or nonadherence to oral antipsychotics. If LAIs can improve adherence in patients and monitoring of adherence for clinicians, they could have a role in preventing relapse during this critical period in psychosis.”
The meta-analysis examines three randomized controlled studies – two from 2015 and one from 2013 – that compare second-generation LAIs to first- and second-generation oral antipsychotics after first episodes of psychosis. Dr. Tran-Boynes said researchers could not find any studies comparing first-generation long-acting antipsychotics to oral antipsychotics.
The largest study had 769 participants; the others had 85 and 86. The subjects, all adults, had diagnoses of schizophrenia, schizoaffective disorder, or schizophreniform disorder. Their diagnoses must have been made within the previous 5 years.
According to the meta-analysis, relapses after first-episode psychosis were more likely (relative risk, 1.078; 95% confidence interval, 1.007-1.154; P = 0.012) in patients taking first- or second-generation oral antipsychotics, compared with those on second-generation LAIs.
“There was an 8% greater efficacy for LAIs preventing relapse after early psychosis, compared to oral antipsychotics,” Dr. Tran-Boynes said. She calculated the number needed to treat as 14.
The percentages of patients who did not relapse while taking second-generation LAIs ranged from 73% (31 of 42 patients randomized to an injectable risperidone arm over 24 months) to 95% (38 of 40 patients over a 12-month study, also of injectable risperidone), Dr. Tran-Boynes said.
When asked about the meta-analysis, Robert Rosenheck, MD, expressed concern.
“While well done, it is based on too few studies to give useful guidance to practice,” said Dr. Rosenheck, professor of psychiatry, epidemiology and public health at Yale University, New Haven, Conn.
Dr. Buckley also noted that the meta-analysis includes a small number of studies. “The effect is sizable for a first-episode population, but other studies to date are more mixed,” he added. “For instance, in a study among a more chronic schizophrenic population, we found no difference between a group receiving long-acting injectable risperidone and oral second-generation antipsychotics” (Schizophr Bull. 2015 Mar;41[2]:449-59).
What should psychiatrists know when they consider prescribing LAIs to prevent psychotic relapse? “If a patient expresses willingness to take an oral antipsychotic on a daily basis and/or has someone to monitor his medication intake, then prescribing an oral antipsychotic would be the ideal route,” Dr. Tran-Boynes said. “However, I would recommend LAIs to patients who have demonstrated poor compliance with previous medications in general, poor awareness of psychosis, poor awareness of need for treatment, poor availability of social support to ensure that the patient will take his/her medication daily, and/or if a patient expresses preference for LAIs.”
She cautioned that LAIs have disadvantages. Compared with oral antipsychotics, it’s harder to adjust patients’ dosages in response to side effects or when they improve, she said. LAIs are also more expensive in the short term, she said.
However, LAIs also may have produced fewer side effects, and there aren’t any questions about compliance, she said. In addition, “there’s less pain at the injection site with second-generation LAIs, compared to first-generation LAIs, due to the water-based solution of the former. The oil-based solutions that are characteristic of first-generation LAIs have been shown in studies to be very painful.”
Dr. Tran-Boynes and Dr. Rosenheck reported no relevant disclosures. Dr. Buckley disclosed that he is a research consultant for the National Institute of Mental Health.
AT APA
Key clinical point: Second-generation long-acting injectable antipsychotics (LAIs) may be better than oral antipsychotics at preventing relapse after first episode of psychosis.
Major finding: Relapses after first-episode psychosis were more likely (RR, 1.078; 95% CI, 1.007-1.154; P = 0.012) in patients on first- and second-generation oral antipsychotics, compared with second-generation LAIs.
Data source: Meta-analysis of three randomized controlled trials with 940 total patients.
Disclosures: Dr. Tran-Boynes reported having no relevant disclosures.
Make the Diagnosis - June 2017
Diagnosis: Porokeratotic eccrine ostial and dermal duct nevus (PEODDN)
Porokeratotic eccrine ostial and dermal duct nevus (PEODDN) is a rare, benign adnexal hamartoma first reported by Marsden et al. in 1979 as “comedo nevus of the palm” (Br J Dermatol. 1979 Dec;101[6]:717-22).
To date, 77 cases of PEODDN have been reported in the literature, with 53% having congenital onset. The average age of onset for the acquired lesions was 6 years. PEODDN affects males and females equally. Acral location is the most common (94%), but lesions have been reported less commonly on the trunk and face. A review of the literature found that most cases of PEODDN were independent, not associated with other conditions. Rarely, Bowen’s disease and squamous cell carcinoma have been reported to arise within PEODDN. Interestingly, two cases have been associated with keratitis-ichthyosis-deafness syndrome (KID).
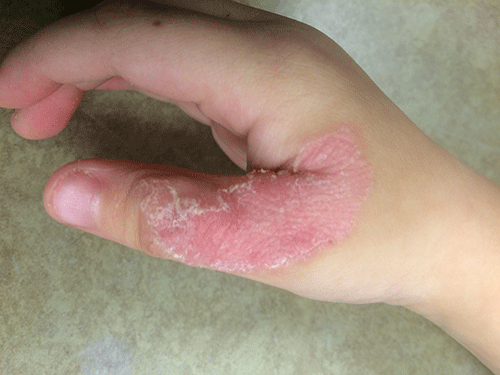
Common differential diagnoses for PEODDN include porokeratosis of Mibelli, linear psoriasis, and linear epidermal nevus. Linear porokeratosis and porokeratosis of Mibelli are characterized by sharply demarcated hyperkeratotic annular lesions with distinct keratotic edges but do not have eccrine gland involvement on histopathology. Linear psoriasis, a rare form of psoriasis, is characterized by late onset linear psoriatic lesions along Blaschko lines. Linear epidermal nevus is a disease characterized by pruritic, erythematous scaly lesions following Blaschko lines that occurs in the first months of life and is slowly progressive. Histopathological features of PEODDN are diagnostic and distinguish it from these other clinical entities. A prominent parakeratotic column within an epidermal invagination that displays loss of the granular layer is found overlaying an eccrine duct with a dilated acrosyringium. Vacuolated and dyskeratotic keratinocytes are also typically present within the epidermal invagination.
The treatment for PEODDN remains elusive. Topical keratolytics, topical retinoids, topical steroids, topical calcipotriol, cryosurgery, phototherapy, and anthralin have been used to treat PEODDN unsuccessfully. Thus far, the most efficacious treatments include CO2 laser and surgical excision for small lesions. Given the young age of this patient, emollient therapy was chosen as treatment until the patient reaches an age at which the lesion is cosmetically more disturbing and other therapies may be safely attempted.
In conclusion, this patient represents a classic case of the rare entity, PEODDN, and draws attention to recent discoveries that a genetic mutation in GJB2 is causative. Because PEODDN shares its pathogenic mutation with KID syndrome, clinicians should be aware that, if the same mutation also affects germline cells, offspring have the potential to express manifestations of KID syndrome.
This case and photo are courtesy of Molly B. Hirt, a medical student at Indiana University, Indianapolis; Carrie L. Davis, MD, of the Dermatology Center of Southern Indiana and Indiana University, Bloomington; and Anita N. Haggstrom, MD, of the departments of dermatology and pediatrics, Indiana University, Indianapolis.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
Diagnosis: Porokeratotic eccrine ostial and dermal duct nevus (PEODDN)
Porokeratotic eccrine ostial and dermal duct nevus (PEODDN) is a rare, benign adnexal hamartoma first reported by Marsden et al. in 1979 as “comedo nevus of the palm” (Br J Dermatol. 1979 Dec;101[6]:717-22).
To date, 77 cases of PEODDN have been reported in the literature, with 53% having congenital onset. The average age of onset for the acquired lesions was 6 years. PEODDN affects males and females equally. Acral location is the most common (94%), but lesions have been reported less commonly on the trunk and face. A review of the literature found that most cases of PEODDN were independent, not associated with other conditions. Rarely, Bowen’s disease and squamous cell carcinoma have been reported to arise within PEODDN. Interestingly, two cases have been associated with keratitis-ichthyosis-deafness syndrome (KID).
Common differential diagnoses for PEODDN include porokeratosis of Mibelli, linear psoriasis, and linear epidermal nevus. Linear porokeratosis and porokeratosis of Mibelli are characterized by sharply demarcated hyperkeratotic annular lesions with distinct keratotic edges but do not have eccrine gland involvement on histopathology. Linear psoriasis, a rare form of psoriasis, is characterized by late onset linear psoriatic lesions along Blaschko lines. Linear epidermal nevus is a disease characterized by pruritic, erythematous scaly lesions following Blaschko lines that occurs in the first months of life and is slowly progressive. Histopathological features of PEODDN are diagnostic and distinguish it from these other clinical entities. A prominent parakeratotic column within an epidermal invagination that displays loss of the granular layer is found overlaying an eccrine duct with a dilated acrosyringium. Vacuolated and dyskeratotic keratinocytes are also typically present within the epidermal invagination.
The treatment for PEODDN remains elusive. Topical keratolytics, topical retinoids, topical steroids, topical calcipotriol, cryosurgery, phototherapy, and anthralin have been used to treat PEODDN unsuccessfully. Thus far, the most efficacious treatments include CO2 laser and surgical excision for small lesions. Given the young age of this patient, emollient therapy was chosen as treatment until the patient reaches an age at which the lesion is cosmetically more disturbing and other therapies may be safely attempted.
In conclusion, this patient represents a classic case of the rare entity, PEODDN, and draws attention to recent discoveries that a genetic mutation in GJB2 is causative. Because PEODDN shares its pathogenic mutation with KID syndrome, clinicians should be aware that, if the same mutation also affects germline cells, offspring have the potential to express manifestations of KID syndrome.
This case and photo are courtesy of Molly B. Hirt, a medical student at Indiana University, Indianapolis; Carrie L. Davis, MD, of the Dermatology Center of Southern Indiana and Indiana University, Bloomington; and Anita N. Haggstrom, MD, of the departments of dermatology and pediatrics, Indiana University, Indianapolis.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
Diagnosis: Porokeratotic eccrine ostial and dermal duct nevus (PEODDN)
Porokeratotic eccrine ostial and dermal duct nevus (PEODDN) is a rare, benign adnexal hamartoma first reported by Marsden et al. in 1979 as “comedo nevus of the palm” (Br J Dermatol. 1979 Dec;101[6]:717-22).
To date, 77 cases of PEODDN have been reported in the literature, with 53% having congenital onset. The average age of onset for the acquired lesions was 6 years. PEODDN affects males and females equally. Acral location is the most common (94%), but lesions have been reported less commonly on the trunk and face. A review of the literature found that most cases of PEODDN were independent, not associated with other conditions. Rarely, Bowen’s disease and squamous cell carcinoma have been reported to arise within PEODDN. Interestingly, two cases have been associated with keratitis-ichthyosis-deafness syndrome (KID).
Common differential diagnoses for PEODDN include porokeratosis of Mibelli, linear psoriasis, and linear epidermal nevus. Linear porokeratosis and porokeratosis of Mibelli are characterized by sharply demarcated hyperkeratotic annular lesions with distinct keratotic edges but do not have eccrine gland involvement on histopathology. Linear psoriasis, a rare form of psoriasis, is characterized by late onset linear psoriatic lesions along Blaschko lines. Linear epidermal nevus is a disease characterized by pruritic, erythematous scaly lesions following Blaschko lines that occurs in the first months of life and is slowly progressive. Histopathological features of PEODDN are diagnostic and distinguish it from these other clinical entities. A prominent parakeratotic column within an epidermal invagination that displays loss of the granular layer is found overlaying an eccrine duct with a dilated acrosyringium. Vacuolated and dyskeratotic keratinocytes are also typically present within the epidermal invagination.
The treatment for PEODDN remains elusive. Topical keratolytics, topical retinoids, topical steroids, topical calcipotriol, cryosurgery, phototherapy, and anthralin have been used to treat PEODDN unsuccessfully. Thus far, the most efficacious treatments include CO2 laser and surgical excision for small lesions. Given the young age of this patient, emollient therapy was chosen as treatment until the patient reaches an age at which the lesion is cosmetically more disturbing and other therapies may be safely attempted.
In conclusion, this patient represents a classic case of the rare entity, PEODDN, and draws attention to recent discoveries that a genetic mutation in GJB2 is causative. Because PEODDN shares its pathogenic mutation with KID syndrome, clinicians should be aware that, if the same mutation also affects germline cells, offspring have the potential to express manifestations of KID syndrome.
This case and photo are courtesy of Molly B. Hirt, a medical student at Indiana University, Indianapolis; Carrie L. Davis, MD, of the Dermatology Center of Southern Indiana and Indiana University, Bloomington; and Anita N. Haggstrom, MD, of the departments of dermatology and pediatrics, Indiana University, Indianapolis.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
Segmental Vitiligo–like Hypopigmentation Associated With Metastatic Melanoma
To the Editor:
Melanoma-associated hypopigmentation frequently has been reported during the disease course and can include different characteristics such as regression of the primary melanoma and/or its metastases as well as common vitiligolike hypopigmentation at sites distant from the melanoma.1,2 Among patients who present with hypopigmentation, the most common clinical presentation is hypopigmented patches in a bilateral symmetric distribution that is similar to vitiligo.1 We report a case of segmental vitiligo–like hypopigmentation associated with melanoma.
RELATED ARTICLE: Novel Melanoma Therapies and Their Side Effects
A 37-year-old man presented with achromic patches on the right side of the neck and lower face of 2 months’ duration. He had a history of melanoma (Breslow thickness, 1.37 mm; mitotic rate, 4/mm2) on the right retroauricular region that was treated by wide local excision 12 years prior; after 10 years, he began to have headaches. At that time, imaging studies including computed tomography, magnetic resonance imaging, and positron emission tomography–computed tomography revealed multiple nodules on the brain, lungs, pancreas, left scapula, and left suprarenal gland. A lung biopsy confirmed metastatic melanoma. Intr
On physical examination using a Wood lamp at the current presentation 2 months later, the achromic patches were linearly distributed on the inferior portion of the right cheek (Figure). A 2×3-cm atrophic scar was present on the right retroauricular region. No regional or distant lymph nodes were enlarged or hard on examination. Although vitiligo is diagnosed using clinical findings,3 a biopsy was performed and showed absence of melanocytes at the dermoepidermal junction (hematoxylin and eosin stain) and complete absence of melanin pigment (Fontana-Masson stain). The patient was treated with topical tacrolimus with poor improvement after 2 months.
The relationship between melanoma and vitiligolike hypopigmentation is a fascinating and controversial topic. Its association is considered to be a consequence of the immune-mediated response against antigens shared by normal melanocytes and melanoma cells.4 Vitiligolike hypopigmentation occurs in 2.8%2 of melanoma patients and is reported in metastatic disease1 as well as those undergoing immunotherapy with or without chemotherapy.5 Its development in patients with stage III or IV melanoma seems to represent an independent positive prognostic factor2 and correlates with a better therapeutic outcome in patients undergoing treatment with biotherapy.5
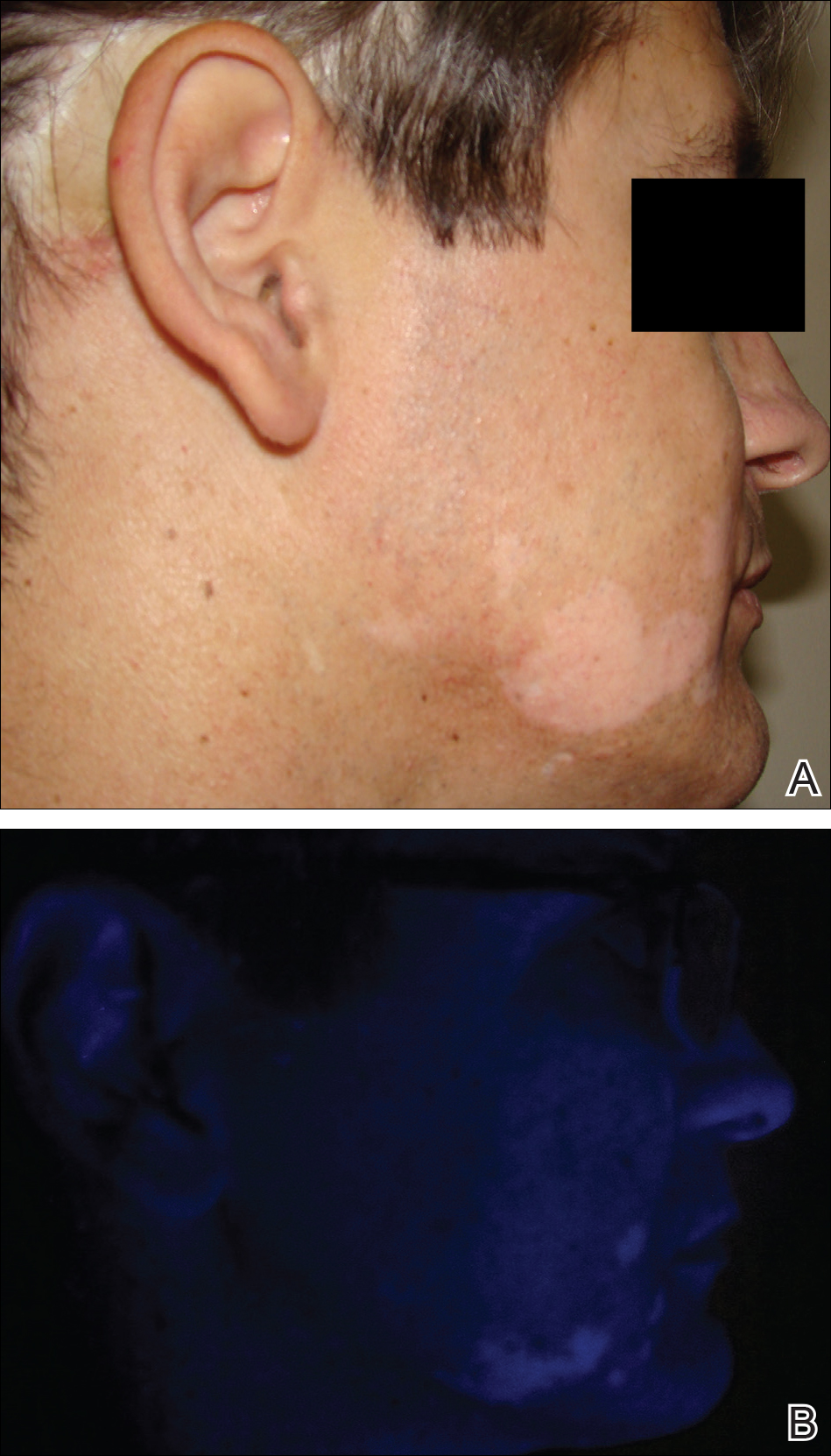
In most cases, the onset of achromic lesions follows the diagnosis of melanoma. Hypopigmentation appears on average 4.8 years after the initial diagnosis and approximately 1 to 2 years after lymph node or distant metastasis.1 In our case, it occurred 12 years after the initial diagnosis and 2 years after metastatic disease was diagnosed.
Despite having widespread metastatic melanoma, our patient only developed achromic patches on the area near the prior melanoma. However, most affected patients present with hypopigmented patches in a bilateral symmetric distribution pattern similar to common vitiligo. No correlation has been found between the hypopigmentation distribution and the location of the primary tumor.1
Because fotemustine is not likely to induce hypopigmentation, we believe that the vitiligolike hypopigmentation in our patient was related to an immune-mediated response associated with melanoma. To help explain our findings, one hypothesis considered was that cutaneous mosaicism may be involved in segmental vitiligo.6 The tumor may have triggered an immune response that affected a close susceptible area of mosaic vitiligo, leading to these clinical findings.
- Hartmann A, Bedenk C, Keikavoussi P, et al. Vitiligo and melanoma-associated hypopigmentation (MAH): shared and discriminative features. J Dtsch Dermatol Ges. 2008;6:1053-1059.
- Quaglino P, Marenco F, Osella-Abate S, et al. Vitiligo is an independent favourable prognostic factor in stage III and IV metastatic melanoma patients: results from a single-institution hospital-based observational cohort study. Ann Oncol. 2010;21:409-414.
- Taïeb A, Picardo M, VETF Members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35.
- Becker JC, Guldberg P, Zeuthen J, et al. Accumulation of identical T cells in melanoma and vitiligo-like leukoderma. J Invest Dermatol. 1999;113:1033-1038.
- Boasberg PD, Hoon DS, Piro LD, et al. Enhanced survival associated with vitiligo expression during maintenance biotherapy for metastatic melanoma. J Invest Dermatol. 2006;126:2658-2663.
- Van Geel N, Speeckaert R, Melsens E, et al. The distribution pattern of segmental vitiligo: clues for somatic mosaicism. Br J Dermatol. 2013;168:56-64.
To the Editor:
Melanoma-associated hypopigmentation frequently has been reported during the disease course and can include different characteristics such as regression of the primary melanoma and/or its metastases as well as common vitiligolike hypopigmentation at sites distant from the melanoma.1,2 Among patients who present with hypopigmentation, the most common clinical presentation is hypopigmented patches in a bilateral symmetric distribution that is similar to vitiligo.1 We report a case of segmental vitiligo–like hypopigmentation associated with melanoma.
RELATED ARTICLE: Novel Melanoma Therapies and Their Side Effects
A 37-year-old man presented with achromic patches on the right side of the neck and lower face of 2 months’ duration. He had a history of melanoma (Breslow thickness, 1.37 mm; mitotic rate, 4/mm2) on the right retroauricular region that was treated by wide local excision 12 years prior; after 10 years, he began to have headaches. At that time, imaging studies including computed tomography, magnetic resonance imaging, and positron emission tomography–computed tomography revealed multiple nodules on the brain, lungs, pancreas, left scapula, and left suprarenal gland. A lung biopsy confirmed metastatic melanoma. Intr
On physical examination using a Wood lamp at the current presentation 2 months later, the achromic patches were linearly distributed on the inferior portion of the right cheek (Figure). A 2×3-cm atrophic scar was present on the right retroauricular region. No regional or distant lymph nodes were enlarged or hard on examination. Although vitiligo is diagnosed using clinical findings,3 a biopsy was performed and showed absence of melanocytes at the dermoepidermal junction (hematoxylin and eosin stain) and complete absence of melanin pigment (Fontana-Masson stain). The patient was treated with topical tacrolimus with poor improvement after 2 months.
The relationship between melanoma and vitiligolike hypopigmentation is a fascinating and controversial topic. Its association is considered to be a consequence of the immune-mediated response against antigens shared by normal melanocytes and melanoma cells.4 Vitiligolike hypopigmentation occurs in 2.8%2 of melanoma patients and is reported in metastatic disease1 as well as those undergoing immunotherapy with or without chemotherapy.5 Its development in patients with stage III or IV melanoma seems to represent an independent positive prognostic factor2 and correlates with a better therapeutic outcome in patients undergoing treatment with biotherapy.5
In most cases, the onset of achromic lesions follows the diagnosis of melanoma. Hypopigmentation appears on average 4.8 years after the initial diagnosis and approximately 1 to 2 years after lymph node or distant metastasis.1 In our case, it occurred 12 years after the initial diagnosis and 2 years after metastatic disease was diagnosed.
Despite having widespread metastatic melanoma, our patient only developed achromic patches on the area near the prior melanoma. However, most affected patients present with hypopigmented patches in a bilateral symmetric distribution pattern similar to common vitiligo. No correlation has been found between the hypopigmentation distribution and the location of the primary tumor.1
Because fotemustine is not likely to induce hypopigmentation, we believe that the vitiligolike hypopigmentation in our patient was related to an immune-mediated response associated with melanoma. To help explain our findings, one hypothesis considered was that cutaneous mosaicism may be involved in segmental vitiligo.6 The tumor may have triggered an immune response that affected a close susceptible area of mosaic vitiligo, leading to these clinical findings.
To the Editor:
Melanoma-associated hypopigmentation frequently has been reported during the disease course and can include different characteristics such as regression of the primary melanoma and/or its metastases as well as common vitiligolike hypopigmentation at sites distant from the melanoma.1,2 Among patients who present with hypopigmentation, the most common clinical presentation is hypopigmented patches in a bilateral symmetric distribution that is similar to vitiligo.1 We report a case of segmental vitiligo–like hypopigmentation associated with melanoma.
RELATED ARTICLE: Novel Melanoma Therapies and Their Side Effects
A 37-year-old man presented with achromic patches on the right side of the neck and lower face of 2 months’ duration. He had a history of melanoma (Breslow thickness, 1.37 mm; mitotic rate, 4/mm2) on the right retroauricular region that was treated by wide local excision 12 years prior; after 10 years, he began to have headaches. At that time, imaging studies including computed tomography, magnetic resonance imaging, and positron emission tomography–computed tomography revealed multiple nodules on the brain, lungs, pancreas, left scapula, and left suprarenal gland. A lung biopsy confirmed metastatic melanoma. Intr
On physical examination using a Wood lamp at the current presentation 2 months later, the achromic patches were linearly distributed on the inferior portion of the right cheek (Figure). A 2×3-cm atrophic scar was present on the right retroauricular region. No regional or distant lymph nodes were enlarged or hard on examination. Although vitiligo is diagnosed using clinical findings,3 a biopsy was performed and showed absence of melanocytes at the dermoepidermal junction (hematoxylin and eosin stain) and complete absence of melanin pigment (Fontana-Masson stain). The patient was treated with topical tacrolimus with poor improvement after 2 months.
The relationship between melanoma and vitiligolike hypopigmentation is a fascinating and controversial topic. Its association is considered to be a consequence of the immune-mediated response against antigens shared by normal melanocytes and melanoma cells.4 Vitiligolike hypopigmentation occurs in 2.8%2 of melanoma patients and is reported in metastatic disease1 as well as those undergoing immunotherapy with or without chemotherapy.5 Its development in patients with stage III or IV melanoma seems to represent an independent positive prognostic factor2 and correlates with a better therapeutic outcome in patients undergoing treatment with biotherapy.5
In most cases, the onset of achromic lesions follows the diagnosis of melanoma. Hypopigmentation appears on average 4.8 years after the initial diagnosis and approximately 1 to 2 years after lymph node or distant metastasis.1 In our case, it occurred 12 years after the initial diagnosis and 2 years after metastatic disease was diagnosed.
Despite having widespread metastatic melanoma, our patient only developed achromic patches on the area near the prior melanoma. However, most affected patients present with hypopigmented patches in a bilateral symmetric distribution pattern similar to common vitiligo. No correlation has been found between the hypopigmentation distribution and the location of the primary tumor.1
Because fotemustine is not likely to induce hypopigmentation, we believe that the vitiligolike hypopigmentation in our patient was related to an immune-mediated response associated with melanoma. To help explain our findings, one hypothesis considered was that cutaneous mosaicism may be involved in segmental vitiligo.6 The tumor may have triggered an immune response that affected a close susceptible area of mosaic vitiligo, leading to these clinical findings.
- Hartmann A, Bedenk C, Keikavoussi P, et al. Vitiligo and melanoma-associated hypopigmentation (MAH): shared and discriminative features. J Dtsch Dermatol Ges. 2008;6:1053-1059.
- Quaglino P, Marenco F, Osella-Abate S, et al. Vitiligo is an independent favourable prognostic factor in stage III and IV metastatic melanoma patients: results from a single-institution hospital-based observational cohort study. Ann Oncol. 2010;21:409-414.
- Taïeb A, Picardo M, VETF Members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35.
- Becker JC, Guldberg P, Zeuthen J, et al. Accumulation of identical T cells in melanoma and vitiligo-like leukoderma. J Invest Dermatol. 1999;113:1033-1038.
- Boasberg PD, Hoon DS, Piro LD, et al. Enhanced survival associated with vitiligo expression during maintenance biotherapy for metastatic melanoma. J Invest Dermatol. 2006;126:2658-2663.
- Van Geel N, Speeckaert R, Melsens E, et al. The distribution pattern of segmental vitiligo: clues for somatic mosaicism. Br J Dermatol. 2013;168:56-64.
- Hartmann A, Bedenk C, Keikavoussi P, et al. Vitiligo and melanoma-associated hypopigmentation (MAH): shared and discriminative features. J Dtsch Dermatol Ges. 2008;6:1053-1059.
- Quaglino P, Marenco F, Osella-Abate S, et al. Vitiligo is an independent favourable prognostic factor in stage III and IV metastatic melanoma patients: results from a single-institution hospital-based observational cohort study. Ann Oncol. 2010;21:409-414.
- Taïeb A, Picardo M, VETF Members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35.
- Becker JC, Guldberg P, Zeuthen J, et al. Accumulation of identical T cells in melanoma and vitiligo-like leukoderma. J Invest Dermatol. 1999;113:1033-1038.
- Boasberg PD, Hoon DS, Piro LD, et al. Enhanced survival associated with vitiligo expression during maintenance biotherapy for metastatic melanoma. J Invest Dermatol. 2006;126:2658-2663.
- Van Geel N, Speeckaert R, Melsens E, et al. The distribution pattern of segmental vitiligo: clues for somatic mosaicism. Br J Dermatol. 2013;168:56-64.
Prac
- Melanoma-associated hypopigmentation usually manifests as common vitiligo; however, little is known about the pathophysiology of segmental vitiligo–like hypopigmentation associated with melanoma.
- This case of segmental vitiligo–like hypopigmentation associated with melanoma sheds light on possible autoimmune and mosaic disease etiology.
Rapid lab test predicts pediatric pneumococcal pneumonia severity
MADRID – Thomsen-Friedenreich antigen activation is useful as a novel early predictor of empyema in pediatric community-acquired pneumonia, Chi-Jung Chang, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
In her retrospective study of 142 Taiwanese children and adolescents hospitalized for community-acquired pneumonia (CAP), Thomsen-Friedenreich antigen (TA) activation had 100% specificity, 100% positive predictive value, and 31% sensitivity for Streptococcus pneumoniae as the causative microorganism.
Moreover, the higher the TA activation titer, the more severe the pneumonia complications that followed, according to Dr. Chang of MacKay Children’s Hospital in Taipei, Taiwan.
The value of this lab test lies in its speed and accuracy for detection of S. pneumoniae CAP. Conventional culture methods are relatively slow and have poor sensitivity, because a child often already has been on empiric antimicrobial therapy and the culture specimen is unwittingly obtained from a sterile site, she explained.
Twenty-two of the 142 children and adolescents hospitalized for lobar CAP were TA activation positive at admission. They were considerably sicker than were the 120 patients who were TA activation negative. Their initial C-reactive protein level was 31.9 mg/dL, twice that of the negative group. Their peak CRP during the hospital stay was significantly higher as well, as was their peak WBC.
Hospital lengths of stay were longer in the TA activation–positive group. Eighteen of 22 TA activation–positive patients (82%) were admitted to the ICU for an average of 8 days, compared with 9% of the negative group.
All TA activation–positive patients had complicated pneumonia with parapneumonic effusions, empyema, necrotizing pneumonia, and/or lung abscesses, as did 36% of the negative group.
S. pneumoniae was the most common pathogen in this study of CAP. It was the responsible microbe in all 22 of the TA activation–positive patients and in 29% of the TA activation–negative ones. The most common serotype in the TA activation group was 19A, which accounted for 12 of the 22 cases. This also was the predominant serotype found in CAP across all Taiwan during the first half of this decade, when the study took place.
In a multivariate logistic regression analysis, TA activation was far and away the strongest independent predictor of empyema, with an associated 15.8-fold increased risk. The other two independent predictors – longer fever duration prior to hospitalization and a higher initial CRP level – were far less robust, according to Dr. Chang.
How TA activation works as a predictor
TA is present on the surface of erythrocytes, platelets, and glomeruli, but ordinarily it is covered by a layer of N-acetylneuraminic acid. Streptococcus pneumoniae produces circulating neuraminidases, which cleave the N-acetylneuraminic acid and expose the underlying TA. The TA then quickly becomes activated through interaction with the anti-TA antibodies, which are normally present in plasma. Once activated, the TA stays so for weeks to months.
Other neuraminidase-producing microorganisms include Clostridium perfringens, Escherichia coli, and Bacteroides.
Dr. Chang and her colleagues used the peanut lectin agglutination method in their TA activation testing.
She reported having no financial conflicts regarding her study.
MADRID – Thomsen-Friedenreich antigen activation is useful as a novel early predictor of empyema in pediatric community-acquired pneumonia, Chi-Jung Chang, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
In her retrospective study of 142 Taiwanese children and adolescents hospitalized for community-acquired pneumonia (CAP), Thomsen-Friedenreich antigen (TA) activation had 100% specificity, 100% positive predictive value, and 31% sensitivity for Streptococcus pneumoniae as the causative microorganism.
Moreover, the higher the TA activation titer, the more severe the pneumonia complications that followed, according to Dr. Chang of MacKay Children’s Hospital in Taipei, Taiwan.
The value of this lab test lies in its speed and accuracy for detection of S. pneumoniae CAP. Conventional culture methods are relatively slow and have poor sensitivity, because a child often already has been on empiric antimicrobial therapy and the culture specimen is unwittingly obtained from a sterile site, she explained.
Twenty-two of the 142 children and adolescents hospitalized for lobar CAP were TA activation positive at admission. They were considerably sicker than were the 120 patients who were TA activation negative. Their initial C-reactive protein level was 31.9 mg/dL, twice that of the negative group. Their peak CRP during the hospital stay was significantly higher as well, as was their peak WBC.
Hospital lengths of stay were longer in the TA activation–positive group. Eighteen of 22 TA activation–positive patients (82%) were admitted to the ICU for an average of 8 days, compared with 9% of the negative group.
All TA activation–positive patients had complicated pneumonia with parapneumonic effusions, empyema, necrotizing pneumonia, and/or lung abscesses, as did 36% of the negative group.
S. pneumoniae was the most common pathogen in this study of CAP. It was the responsible microbe in all 22 of the TA activation–positive patients and in 29% of the TA activation–negative ones. The most common serotype in the TA activation group was 19A, which accounted for 12 of the 22 cases. This also was the predominant serotype found in CAP across all Taiwan during the first half of this decade, when the study took place.
In a multivariate logistic regression analysis, TA activation was far and away the strongest independent predictor of empyema, with an associated 15.8-fold increased risk. The other two independent predictors – longer fever duration prior to hospitalization and a higher initial CRP level – were far less robust, according to Dr. Chang.
How TA activation works as a predictor
TA is present on the surface of erythrocytes, platelets, and glomeruli, but ordinarily it is covered by a layer of N-acetylneuraminic acid. Streptococcus pneumoniae produces circulating neuraminidases, which cleave the N-acetylneuraminic acid and expose the underlying TA. The TA then quickly becomes activated through interaction with the anti-TA antibodies, which are normally present in plasma. Once activated, the TA stays so for weeks to months.
Other neuraminidase-producing microorganisms include Clostridium perfringens, Escherichia coli, and Bacteroides.
Dr. Chang and her colleagues used the peanut lectin agglutination method in their TA activation testing.
She reported having no financial conflicts regarding her study.
MADRID – Thomsen-Friedenreich antigen activation is useful as a novel early predictor of empyema in pediatric community-acquired pneumonia, Chi-Jung Chang, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
In her retrospective study of 142 Taiwanese children and adolescents hospitalized for community-acquired pneumonia (CAP), Thomsen-Friedenreich antigen (TA) activation had 100% specificity, 100% positive predictive value, and 31% sensitivity for Streptococcus pneumoniae as the causative microorganism.
Moreover, the higher the TA activation titer, the more severe the pneumonia complications that followed, according to Dr. Chang of MacKay Children’s Hospital in Taipei, Taiwan.
The value of this lab test lies in its speed and accuracy for detection of S. pneumoniae CAP. Conventional culture methods are relatively slow and have poor sensitivity, because a child often already has been on empiric antimicrobial therapy and the culture specimen is unwittingly obtained from a sterile site, she explained.
Twenty-two of the 142 children and adolescents hospitalized for lobar CAP were TA activation positive at admission. They were considerably sicker than were the 120 patients who were TA activation negative. Their initial C-reactive protein level was 31.9 mg/dL, twice that of the negative group. Their peak CRP during the hospital stay was significantly higher as well, as was their peak WBC.
Hospital lengths of stay were longer in the TA activation–positive group. Eighteen of 22 TA activation–positive patients (82%) were admitted to the ICU for an average of 8 days, compared with 9% of the negative group.
All TA activation–positive patients had complicated pneumonia with parapneumonic effusions, empyema, necrotizing pneumonia, and/or lung abscesses, as did 36% of the negative group.
S. pneumoniae was the most common pathogen in this study of CAP. It was the responsible microbe in all 22 of the TA activation–positive patients and in 29% of the TA activation–negative ones. The most common serotype in the TA activation group was 19A, which accounted for 12 of the 22 cases. This also was the predominant serotype found in CAP across all Taiwan during the first half of this decade, when the study took place.
In a multivariate logistic regression analysis, TA activation was far and away the strongest independent predictor of empyema, with an associated 15.8-fold increased risk. The other two independent predictors – longer fever duration prior to hospitalization and a higher initial CRP level – were far less robust, according to Dr. Chang.
How TA activation works as a predictor
TA is present on the surface of erythrocytes, platelets, and glomeruli, but ordinarily it is covered by a layer of N-acetylneuraminic acid. Streptococcus pneumoniae produces circulating neuraminidases, which cleave the N-acetylneuraminic acid and expose the underlying TA. The TA then quickly becomes activated through interaction with the anti-TA antibodies, which are normally present in plasma. Once activated, the TA stays so for weeks to months.
Other neuraminidase-producing microorganisms include Clostridium perfringens, Escherichia coli, and Bacteroides.
Dr. Chang and her colleagues used the peanut lectin agglutination method in their TA activation testing.
She reported having no financial conflicts regarding her study.
AT ESPID 2017
Key clinical point:
Major finding: A positive Thomsen-Friedenreich antigen activation test in pediatric community-acquired pneumonia had 100% specificity, 100% positive predictive value, and 31% sensitivity for S. pneumoniae as the causative microorganism.
Data source: A retrospective study of 142 Taiwanese children and teens hospitalized for lobular community-acquired pneumonia.
Disclosures: The study presenter reported having no financial conflicts of interest.
Isotretinoin for Acne: Tips for Prescribing and Managing Patient Concerns
What does your patient need to know at the first visit?
Most important is what you need to know before the first visit. As the prescribing physician, you must be familiar with the iPLEDGE program. Because of the complexity of the program, consider identifying a physician in your area to refer patients if you are not going to be a regular prescriber of the medication.
If you are enrolled in iPLEDGE, let your patients (and/or their parents/guardians) know that there is a great deal of misinformation on the Internet. Reiterate that you and your staff are available to discuss their concerns. Also, give them reliable sources of information, such as the American Academy of Dermatology's patient information sheet as well as the Mayo Clinic's acne information. Drugs.com is another resource.
All patients—males, females who cannot become pregnant, and females of childbearing potential (FCBPs)—must be aware that this medication can cause birth defects if taken during pregnancy. They must be informed that the medication is not to be shared with anyone and that they should not give blood while taking this medication.
What treatment course do you recommend?
My evidence-based approach is a course of isotretinoin totaling a minimum of 150 mg per kilogram body weight. Do not give a more abbreviated course unless the patient has cleared early; even then I tend to complete 150 mg when possible. There is published evidence that pushing the course to a total of 220 mg per kilogram body weight results in a longer remission.
Generally, I do few laboratory tests other than pretreatment lipid panels as well as 1 or 2 follow-up lipid panels at monthly intervals. To comply with the iPLEDGE program, FCBP patients must have a monthly pregnancy test, which is reported on the iPLEDGE website before the patient can be prescribed the drug and receive the drug from a pharmacist who is participating in the iPLEDGE program.
One of the defects of the iPLEDGE system is that although only a 30-day supply of pills can be prescribed, it is difficult to always bring a patient back in exactly 30 days; for example, we work on a 4-week cycle and 30 days brings us into the next week or uncommonly the weekend when we do not see patients. Our male patients or females not of childbearing potential are not affected, but for our FCBP patients, it means usually scheduling visits at 35-day intervals because the pregnancy tests must be performed at minimum 28-day intervals and the prescription cannot be written and the pregnancy test recorded until after at least 30 days.
What are the side effects?
The common side effects are what you would expect from a medicine that is supposed to dry up the oil on your skin: dryness of the lips, mouth, and skin, as well as rashes due to the dryness. There also can be minor swelling of the eyelids or lips, nosebleeds, upset stomach, and thinning of the hair; dryness of the scalp may occur. I recommend using a little petroleum jelly inside the nostrils at night to counteract the dryness that leads to nosebleeds, and saline drops or gel for the eyes, especially for contact lens wearers.
Joint aches and pains have been reported, though I rarely see those effects in patients who are physically active such as those participating in competitive sports. Mood changes have been reported, including suicidal ideation.
What do you do if patients refuse treatment?
There is so much false information on the Internet about the dangers of isotretinoin, leaving some patients (and parents/guardians) too afraid to use it. I sympathize with this anxiety, but I do endeavor to point out that the birth defects occur only in women taking the drug while pregnant and have not been reported to occur after the drug is out of the patient's system.
Similarly, I point out that almost all of the evidence-based studies failed to confirm any association between the use of isotretinoin and depression, teenage suicide, and subsequent inflammatory bowel disease. Nonetheless, I mention these issues and recommend that the parents/guardians observe the teenager; in the case of adult patients, they themselves must be sensitive to symptoms.
Suggested Readings
American Academy of Dermatology Association. Position statement on isotretinoin. https://www.aad.org/Forms/Policies/Uploads/PS/PS-Isotretinoin.pdf. Published December 9, 2000. Updated November 13, 2010. Accessed May 18, 2017.
Blasiak RC, Stamey CR, Burkhart CN, et al. High-dose isotretinoin treatment and the rate of retrial, relapse, and adverse effects in patients with acne vulgaris. JAMA Dermatol. 2013;149:1392-1398.
What does your patient need to know at the first visit?
Most important is what you need to know before the first visit. As the prescribing physician, you must be familiar with the iPLEDGE program. Because of the complexity of the program, consider identifying a physician in your area to refer patients if you are not going to be a regular prescriber of the medication.
If you are enrolled in iPLEDGE, let your patients (and/or their parents/guardians) know that there is a great deal of misinformation on the Internet. Reiterate that you and your staff are available to discuss their concerns. Also, give them reliable sources of information, such as the American Academy of Dermatology's patient information sheet as well as the Mayo Clinic's acne information. Drugs.com is another resource.
All patients—males, females who cannot become pregnant, and females of childbearing potential (FCBPs)—must be aware that this medication can cause birth defects if taken during pregnancy. They must be informed that the medication is not to be shared with anyone and that they should not give blood while taking this medication.
What treatment course do you recommend?
My evidence-based approach is a course of isotretinoin totaling a minimum of 150 mg per kilogram body weight. Do not give a more abbreviated course unless the patient has cleared early; even then I tend to complete 150 mg when possible. There is published evidence that pushing the course to a total of 220 mg per kilogram body weight results in a longer remission.
Generally, I do few laboratory tests other than pretreatment lipid panels as well as 1 or 2 follow-up lipid panels at monthly intervals. To comply with the iPLEDGE program, FCBP patients must have a monthly pregnancy test, which is reported on the iPLEDGE website before the patient can be prescribed the drug and receive the drug from a pharmacist who is participating in the iPLEDGE program.
One of the defects of the iPLEDGE system is that although only a 30-day supply of pills can be prescribed, it is difficult to always bring a patient back in exactly 30 days; for example, we work on a 4-week cycle and 30 days brings us into the next week or uncommonly the weekend when we do not see patients. Our male patients or females not of childbearing potential are not affected, but for our FCBP patients, it means usually scheduling visits at 35-day intervals because the pregnancy tests must be performed at minimum 28-day intervals and the prescription cannot be written and the pregnancy test recorded until after at least 30 days.
What are the side effects?
The common side effects are what you would expect from a medicine that is supposed to dry up the oil on your skin: dryness of the lips, mouth, and skin, as well as rashes due to the dryness. There also can be minor swelling of the eyelids or lips, nosebleeds, upset stomach, and thinning of the hair; dryness of the scalp may occur. I recommend using a little petroleum jelly inside the nostrils at night to counteract the dryness that leads to nosebleeds, and saline drops or gel for the eyes, especially for contact lens wearers.
Joint aches and pains have been reported, though I rarely see those effects in patients who are physically active such as those participating in competitive sports. Mood changes have been reported, including suicidal ideation.
What do you do if patients refuse treatment?
There is so much false information on the Internet about the dangers of isotretinoin, leaving some patients (and parents/guardians) too afraid to use it. I sympathize with this anxiety, but I do endeavor to point out that the birth defects occur only in women taking the drug while pregnant and have not been reported to occur after the drug is out of the patient's system.
Similarly, I point out that almost all of the evidence-based studies failed to confirm any association between the use of isotretinoin and depression, teenage suicide, and subsequent inflammatory bowel disease. Nonetheless, I mention these issues and recommend that the parents/guardians observe the teenager; in the case of adult patients, they themselves must be sensitive to symptoms.
Suggested Readings
American Academy of Dermatology Association. Position statement on isotretinoin. https://www.aad.org/Forms/Policies/Uploads/PS/PS-Isotretinoin.pdf. Published December 9, 2000. Updated November 13, 2010. Accessed May 18, 2017.
Blasiak RC, Stamey CR, Burkhart CN, et al. High-dose isotretinoin treatment and the rate of retrial, relapse, and adverse effects in patients with acne vulgaris. JAMA Dermatol. 2013;149:1392-1398.
What does your patient need to know at the first visit?
Most important is what you need to know before the first visit. As the prescribing physician, you must be familiar with the iPLEDGE program. Because of the complexity of the program, consider identifying a physician in your area to refer patients if you are not going to be a regular prescriber of the medication.
If you are enrolled in iPLEDGE, let your patients (and/or their parents/guardians) know that there is a great deal of misinformation on the Internet. Reiterate that you and your staff are available to discuss their concerns. Also, give them reliable sources of information, such as the American Academy of Dermatology's patient information sheet as well as the Mayo Clinic's acne information. Drugs.com is another resource.
All patients—males, females who cannot become pregnant, and females of childbearing potential (FCBPs)—must be aware that this medication can cause birth defects if taken during pregnancy. They must be informed that the medication is not to be shared with anyone and that they should not give blood while taking this medication.
What treatment course do you recommend?
My evidence-based approach is a course of isotretinoin totaling a minimum of 150 mg per kilogram body weight. Do not give a more abbreviated course unless the patient has cleared early; even then I tend to complete 150 mg when possible. There is published evidence that pushing the course to a total of 220 mg per kilogram body weight results in a longer remission.
Generally, I do few laboratory tests other than pretreatment lipid panels as well as 1 or 2 follow-up lipid panels at monthly intervals. To comply with the iPLEDGE program, FCBP patients must have a monthly pregnancy test, which is reported on the iPLEDGE website before the patient can be prescribed the drug and receive the drug from a pharmacist who is participating in the iPLEDGE program.
One of the defects of the iPLEDGE system is that although only a 30-day supply of pills can be prescribed, it is difficult to always bring a patient back in exactly 30 days; for example, we work on a 4-week cycle and 30 days brings us into the next week or uncommonly the weekend when we do not see patients. Our male patients or females not of childbearing potential are not affected, but for our FCBP patients, it means usually scheduling visits at 35-day intervals because the pregnancy tests must be performed at minimum 28-day intervals and the prescription cannot be written and the pregnancy test recorded until after at least 30 days.
What are the side effects?
The common side effects are what you would expect from a medicine that is supposed to dry up the oil on your skin: dryness of the lips, mouth, and skin, as well as rashes due to the dryness. There also can be minor swelling of the eyelids or lips, nosebleeds, upset stomach, and thinning of the hair; dryness of the scalp may occur. I recommend using a little petroleum jelly inside the nostrils at night to counteract the dryness that leads to nosebleeds, and saline drops or gel for the eyes, especially for contact lens wearers.
Joint aches and pains have been reported, though I rarely see those effects in patients who are physically active such as those participating in competitive sports. Mood changes have been reported, including suicidal ideation.
What do you do if patients refuse treatment?
There is so much false information on the Internet about the dangers of isotretinoin, leaving some patients (and parents/guardians) too afraid to use it. I sympathize with this anxiety, but I do endeavor to point out that the birth defects occur only in women taking the drug while pregnant and have not been reported to occur after the drug is out of the patient's system.
Similarly, I point out that almost all of the evidence-based studies failed to confirm any association between the use of isotretinoin and depression, teenage suicide, and subsequent inflammatory bowel disease. Nonetheless, I mention these issues and recommend that the parents/guardians observe the teenager; in the case of adult patients, they themselves must be sensitive to symptoms.
Suggested Readings
American Academy of Dermatology Association. Position statement on isotretinoin. https://www.aad.org/Forms/Policies/Uploads/PS/PS-Isotretinoin.pdf. Published December 9, 2000. Updated November 13, 2010. Accessed May 18, 2017.
Blasiak RC, Stamey CR, Burkhart CN, et al. High-dose isotretinoin treatment and the rate of retrial, relapse, and adverse effects in patients with acne vulgaris. JAMA Dermatol. 2013;149:1392-1398.

In Vivo Reflectance Confocal Microscopy
Reflectance confocal microscopy (RCM) imaging received Category I Current Procedural Terminology (CPT) codes by the Centers for Medicare & Medicaid Services in January 2016 and can now be submitted to insurance companies with reimbursement comparable to a skin biopsy or a global skin pathology service.1 This fairly new technology is a US Food and Drug Administration–cleared noninvasive imaging modality that provides high-resolution in vivo cellular images of the skin. It has been shown to be efficacious in differentiating benign and malignant skin lesions, increasing diagnostic accuracy, and reducing the number of unnecessary skin biopsies that are performed. In addition to skin cancer diagnosis, RCM imaging also can help guide management of malignant lesions by detecting lateral margins prior to surgery as well as monitoring the lesion over time for treatment efficacy or recurrence. The potential impact of RCM imaging is tremendous, and reimbursement may lead to increased use in clinical practice to the benefit of our patients. Herein, we present a brief review of RCM imaging and reimbursement as well as the benefits and limitations of this new technology for dermatologists.
Reflectance Confocal Microscopy
In vivo RCM allows us to visualize the epidermis in real time on a cellular level down to the papillary dermis at a high resolution (×30) comparable to histologic examination. With optical sections 3- to 5-µm thick and a lateral resolution of 0.5 to 1.0 µm, RCM produces a stack of 500×500-µm2 images up to a depth of approximately 200 µm.2,3 At any chosen depth, these smaller images are stitched together with sophisticated software into a block, or mosaic, increasing the field of view to up to 8×8 mm2. Imaging is performed in en face planes oriented parallel to the skin surface, similar to dermoscopy.
Current CPT Guidelines and Reimbursement
The CPT codes for RCM imaging provide reimbursement on a per-lesion basis and are similar to those used for skin biopsy and pathology (Table).1 Codes 96931 through 96933 are used for imaging of a single lesion on a patient. The first code—96931—is used when image acquisition, interpretation, and report creation are carried out by a single clinician. The next 2 codes are used when one clinician acquires the image—96932—comparable to the technical component of a pathology code, while another reads it and creates the report—96933—similar to a dermatopathologist billing for the professional component of a pathology report. For patients presenting with multiple lesions, the next 3 codes—96934, 96935, and 96936—are used in conjunction with the applicable first code for each additional lesion with similar global, technical, and professional components. Because these codes are not in the radiology or pathology sections of CPT, a single code cannot be used with modifier -TC (technical component) and modifier -26, as they are in those sections.
The wide-probe VivaScope 1500 (Caliber I.D., Inc) currently is the only confocal device that can be reported with a CPT code and routinely reimbursed. The handheld VivaScope 3000 (Caliber I.D., Inc) can only view a small stack and does not have the ability to acquire a full mosaic image; it is not covered by these codes.
Images can be viewed as a stack captured at the same horizontal position but at sequential depths or as a mosaic, which has a larger field of view but is limited to a single plane. To appropriately assess a lesion, clinicians must obtain a mosaic that needs to be assessed at multiple layers for a diagnosis to be made because it is a cross-section view.
Diagnosis
Studies have demonstrated the usefulness of RCM imaging in the diagnosis of a wide range of skin diseases, including melanoma and nonmelanoma skin cancers, infectious diseases, and inflammatory and autoimmune conditions, as well as wound healing and skin aging. Reflectance confocal microscopy imaging is not limited to the skin; it can be used to evaluate the hair, nails, oral mucosa, and other organs.
According to several studies, RCM imaging notably increases the diagnostic accuracy and detection rate of skin cancers over clinical and dermoscopic examination alone and therefore can act as an aid in differentiating lesions that are benign versus those that are suspicious and should be biopsied.
Reflectance confocal microscopy has been shown to have a mean sensitivity of 94% (range, 92%–96%) and specificity of 83% (range, 81%–84%) for all types of skin cancer when used with dermoscopy.4 In particular, for melanocytic lesions that are ambiguous on dermoscopy, RCM used in addition to dermoscopy increases the mean sensitivity and specificity for melanoma diagnosis to 93% (range, 89%–96%) and 76% (range, 68%–83%), respectively.5 Although these reported sensitivities are comparable to dermoscopy, the specificity is superior, especially for detecting hypomelanotic and amelanotic melanomas, which often lack specific features on dermoscopy.6-8
The combination of RCM with dermoscopy has reduced the number of unnecessary excisions of benign nevi by more than 50% when compared to dermoscopy alone.9 One study showed that the number needed to treat (ie, excise) a melanoma decreased from 14.6 with dermoscopy alone to 6.8 when guided by dermoscopy and RCM imaging.9 In a similar study, the number needed to treat dropped from 19.41 with dermoscopy alone to 6.25 with dermoscopy and RCM.10
These studies were not looking to evaluate RCM as a replacement test but rather as an add-on test to dermoscopy. Reflectance confocal microscopy imaging takes longer than dermoscopy for each lesion; therefore, RCM should only be used as an adjunctive tool to dermoscopy and not as an initial screening test. Consequentially, a dermatologist skilled in dermoscopy is essential in deciding which lesions would be appropriate for subsequent RCM imaging.
In Vivo Margin Mapping as an Adjunct to Surgery
Oftentimes, tumor margins are poorly defined and can be difficult to map clinically and dermoscopically. Studies have demonstrated the use of RCM in delineation of surgical margins prior to surgery or excisional biopsies.11,12 Alternatively, when complete removal at biopsy would be impractical (eg, for extremely large lesions or lesions located in cosmetically sensitive areas such as the face), RCM can be used to pick the best site for an appropriate biopsy, which decreases the chance of sampling error due to skip lesions and increases histologic accuracy.
Nonsurgical Treatment Monitoring
One advantage of RCM over conventional histology is that RCM imaging leaves the tissue intact, allowing dynamic changes to be studied over time, which is useful for monitoring nonmelanoma skin cancers and lentigo maligna being treated with noninvasive therapeutic modalities.13 If not as a definitive treatment, RCM can act as an adjunct for surgery by monitoring reduction in lesion size prior to Mohs micrographic surgery, thereby decreasing the resulting surgical defect.14
Limitations
Imaging Depth
Although RCM is a revolutionary device in the field of dermatology, it has several limitations. With a maximal imaging depth of 350 µm, the imaging resolution decreases substantially with depth, limiting accurate interpretation to 200 µm. Reflectance confocal microscopy can only image the superficial portion of a lesion; therefore, deep tumor margins cannot be assessed. Hypertrophic or hyperkeratotic lesions, including lesions on the palms and soles, also are unable to be imaged with RCM. This limitation in depth penetration makes treatment monitoring impossible for invasive lesions that extend into the dermal layer.
Difficult-to-Reach Areas
Another limitation is the difficulty imaging areas such as the ocular canthi, nasal alae, or helices of the ear due to the wide probe size on the VivaScope 1500. The advent of the smaller handheld VivaScope 3000 device allows for improved imaging of concave services and difficult lesions at the risk of less accurate imaging, low field of view, and no reimbursement at present.
False-Positive Results
Although RCM has been shown to be helpful in reducing unnecessary biopsies, there still is the issue of false-positives on imaging. False-positives most commonly occur in nevi with severe atypia or when Langerhans cells are present that cannot always be differentiated from melanocytic cells.3,15,16 One prospective study found 7 false-positive results from 63 sites using RCM for the diagnosis of lentigo malignas.16 False-negatives can occur in the presence of inflammatory infiltrates and scar tissue that can hide cellular morphology or in sampling errors due to skip lesions.3,16
Time Efficiency
The time required for acquisition of RCM mosaics and stacks followed by reading and interpretation can be substantial depending on the size and complexity of the lesion, which is a major limitation for use of RCM in busy dermatology practices; therefore, RCM should be reserved for lesions selected to undergo biopsy that are clinically equivocal for malignancy prior to RCM examination.17 It would not be cost-effective or time effective to evaluate lesions that either clinically or dermoscopically have a high probability of malignancy; however, patients and physicians may opt for increased specificity at the expense of time, particularly when a lesion is located on a cosmetically sensitive area, as patients can avoid initial histologic biopsy and gain the cosmetic benefit of going straight to surgery versus obtaining an initial diagnostic biopsy.
Cost
Lastly, the high cost involved in purchasing an RCM device and the training involved to use and interpret RCM images currently limits RCM to large academic centers. Reimbursement may make more widespread use feasible. In any event, RCM imaging should be part of the curriculum for both dermatology and pathology trainees.
Future Directions
In vivo RCM is a noninvasive imaging modality that allows for real-time evaluation of the skin. Used in conjunction with dermoscopy, RCM can substantially improve diagnostic accuracy and reduce the number of unnecessary biopsies. Now that RCM has finally gained foundational CPT codes and insurance reimbursement, there may be a growing demand for clinicians to incorporate this technology into their clinical practice.
- Current Procedural Terminology 2017, Professional Edition. Chicago IL: American Medical Association; 2016.
- Que SK, Fraga-Braghiroli N, Grant-Kels JM, et al. Through the looking glass: basics and principles of reflectance confocal microscopy [published online June 4, 2015]. J Am Acad Dermatol. 2015;73:276-284.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside [published online October 27, 2016]. Lasers Surg Med. 2017;49:7-19.
- Xiong YD, Ma S, Li X, et al. A meta-analysis of reflectance confocal microscopy for the diagnosis of malignant skin tumours. J Eur Acad Dermatol Venereol. 2016;30:1295-1302.
- Stevenson AD, Mickan S, Mallett S, et al. Systematic review of diagnostic accuracy of reflectance confocal microscopy for melanoma diagnosis in patients with clinically equivocal skin lesions. Dermatol Pract Concept. 2013;3:19-27.
- Busam KJ, Hester K, Charles C, et al. Detection of clinically amelanotic malignant melanoma and assessment of its margins by in vivo confocal scanning laser microscopy. Arch Dermatol. 2001;137:923-929.
- Losi A, Longo C, Cesinaro AM, et al. Hyporeflective pagetoid cells: a new clue for amelanotic melanoma diagnosis by reflectance confocal microscopy. Br J Dermatol. 2014;171:48-54.
- Guitera P, Menzies SQ, Argenziano G, et al. Dermoscopy and in vivo confocal microscopy are complementary techniques for the diagnosis of difficult amelanotic and light-coloured skin lesions [published online October 12, 2016]. Br J Dermatol. 2016;175:1311-1319.
- Pellacani G, Pepe P, Casari A, et al. Reflectance confocal microscopy as a second-level examination in skin oncology improves diagnostic accuracy and saves unnecessary excisions: a longitudinal prospective study. Br J Dermatol. 2014;171:1044-1051.
- Pellacani G, Witkowski A, Cesinaro AM, et al. Cost-benefit of reflectance confocal microscopy in the diagnostic performance of melanoma. J Eur Acad Dermatol Venereol. 2016;30:413-419.
- Champin J, Perrot JL, Cinotti E, et al. In vivo reflectance confocal microscopy to optimize the spaghetti technique for defining surgical margins of lentigo maligna. Dermatol Surg. 2014;40:247-256.
- Hibler BP, Cordova M, Wong RJ, et al. Intraoperative real-time reflectance confocal microscopy for guiding surgical margins of lentigo maligna melanoma. Dermatol Surg. 2015;41:980-983.
- Ulrich M, Lange-Asschenfeldt S, Gonzalez S. The use of reflectance confocal microscopy for monitoring response to therapy of skin malignancies. Dermatol Pract Concept. 2012;2:202a10.
- Torres A, Niemeyer A, Berkes B, et al. 5% imiquimod cream and reflectance-mode confocal microscopy as adjunct modalities to Mohs micrographic surgery for treatment of basal cell carcinoma. Dermatol Surg. 2004;30(12, pt 1):1462-1469.
- Hashemi P, Pulitzer MP, Scope A, et al. Langerhans cells and melanocytes share similar morphologic features under in vivo reflectance confocal microscopy: a challenge for melanoma diagnosis. J Am Acad Dermatol. 2012;66:452-462.
- Menge TD, Hibler BP, Cordova MA, et al. Concordance of handheld reflectance confocal microscopy (RCM) with histopathology in the diagnosis of lentigo maligna (LM): a prospective study. J Am Acad Dermatol. 2016;74:1114-1120.
- Borsari S, Pampena R, Lallas A, et al. Clinical indications for use of reflectance confocal microscopy for skin cancer diagnosis. JAMA Dermatol. 2016;152:1093-1098.
Reflectance confocal microscopy (RCM) imaging received Category I Current Procedural Terminology (CPT) codes by the Centers for Medicare & Medicaid Services in January 2016 and can now be submitted to insurance companies with reimbursement comparable to a skin biopsy or a global skin pathology service.1 This fairly new technology is a US Food and Drug Administration–cleared noninvasive imaging modality that provides high-resolution in vivo cellular images of the skin. It has been shown to be efficacious in differentiating benign and malignant skin lesions, increasing diagnostic accuracy, and reducing the number of unnecessary skin biopsies that are performed. In addition to skin cancer diagnosis, RCM imaging also can help guide management of malignant lesions by detecting lateral margins prior to surgery as well as monitoring the lesion over time for treatment efficacy or recurrence. The potential impact of RCM imaging is tremendous, and reimbursement may lead to increased use in clinical practice to the benefit of our patients. Herein, we present a brief review of RCM imaging and reimbursement as well as the benefits and limitations of this new technology for dermatologists.
Reflectance Confocal Microscopy
In vivo RCM allows us to visualize the epidermis in real time on a cellular level down to the papillary dermis at a high resolution (×30) comparable to histologic examination. With optical sections 3- to 5-µm thick and a lateral resolution of 0.5 to 1.0 µm, RCM produces a stack of 500×500-µm2 images up to a depth of approximately 200 µm.2,3 At any chosen depth, these smaller images are stitched together with sophisticated software into a block, or mosaic, increasing the field of view to up to 8×8 mm2. Imaging is performed in en face planes oriented parallel to the skin surface, similar to dermoscopy.
Current CPT Guidelines and Reimbursement
The CPT codes for RCM imaging provide reimbursement on a per-lesion basis and are similar to those used for skin biopsy and pathology (Table).1 Codes 96931 through 96933 are used for imaging of a single lesion on a patient. The first code—96931—is used when image acquisition, interpretation, and report creation are carried out by a single clinician. The next 2 codes are used when one clinician acquires the image—96932—comparable to the technical component of a pathology code, while another reads it and creates the report—96933—similar to a dermatopathologist billing for the professional component of a pathology report. For patients presenting with multiple lesions, the next 3 codes—96934, 96935, and 96936—are used in conjunction with the applicable first code for each additional lesion with similar global, technical, and professional components. Because these codes are not in the radiology or pathology sections of CPT, a single code cannot be used with modifier -TC (technical component) and modifier -26, as they are in those sections.
The wide-probe VivaScope 1500 (Caliber I.D., Inc) currently is the only confocal device that can be reported with a CPT code and routinely reimbursed. The handheld VivaScope 3000 (Caliber I.D., Inc) can only view a small stack and does not have the ability to acquire a full mosaic image; it is not covered by these codes.
Images can be viewed as a stack captured at the same horizontal position but at sequential depths or as a mosaic, which has a larger field of view but is limited to a single plane. To appropriately assess a lesion, clinicians must obtain a mosaic that needs to be assessed at multiple layers for a diagnosis to be made because it is a cross-section view.
Diagnosis
Studies have demonstrated the usefulness of RCM imaging in the diagnosis of a wide range of skin diseases, including melanoma and nonmelanoma skin cancers, infectious diseases, and inflammatory and autoimmune conditions, as well as wound healing and skin aging. Reflectance confocal microscopy imaging is not limited to the skin; it can be used to evaluate the hair, nails, oral mucosa, and other organs.
According to several studies, RCM imaging notably increases the diagnostic accuracy and detection rate of skin cancers over clinical and dermoscopic examination alone and therefore can act as an aid in differentiating lesions that are benign versus those that are suspicious and should be biopsied.
Reflectance confocal microscopy has been shown to have a mean sensitivity of 94% (range, 92%–96%) and specificity of 83% (range, 81%–84%) for all types of skin cancer when used with dermoscopy.4 In particular, for melanocytic lesions that are ambiguous on dermoscopy, RCM used in addition to dermoscopy increases the mean sensitivity and specificity for melanoma diagnosis to 93% (range, 89%–96%) and 76% (range, 68%–83%), respectively.5 Although these reported sensitivities are comparable to dermoscopy, the specificity is superior, especially for detecting hypomelanotic and amelanotic melanomas, which often lack specific features on dermoscopy.6-8
The combination of RCM with dermoscopy has reduced the number of unnecessary excisions of benign nevi by more than 50% when compared to dermoscopy alone.9 One study showed that the number needed to treat (ie, excise) a melanoma decreased from 14.6 with dermoscopy alone to 6.8 when guided by dermoscopy and RCM imaging.9 In a similar study, the number needed to treat dropped from 19.41 with dermoscopy alone to 6.25 with dermoscopy and RCM.10
These studies were not looking to evaluate RCM as a replacement test but rather as an add-on test to dermoscopy. Reflectance confocal microscopy imaging takes longer than dermoscopy for each lesion; therefore, RCM should only be used as an adjunctive tool to dermoscopy and not as an initial screening test. Consequentially, a dermatologist skilled in dermoscopy is essential in deciding which lesions would be appropriate for subsequent RCM imaging.
In Vivo Margin Mapping as an Adjunct to Surgery
Oftentimes, tumor margins are poorly defined and can be difficult to map clinically and dermoscopically. Studies have demonstrated the use of RCM in delineation of surgical margins prior to surgery or excisional biopsies.11,12 Alternatively, when complete removal at biopsy would be impractical (eg, for extremely large lesions or lesions located in cosmetically sensitive areas such as the face), RCM can be used to pick the best site for an appropriate biopsy, which decreases the chance of sampling error due to skip lesions and increases histologic accuracy.
Nonsurgical Treatment Monitoring
One advantage of RCM over conventional histology is that RCM imaging leaves the tissue intact, allowing dynamic changes to be studied over time, which is useful for monitoring nonmelanoma skin cancers and lentigo maligna being treated with noninvasive therapeutic modalities.13 If not as a definitive treatment, RCM can act as an adjunct for surgery by monitoring reduction in lesion size prior to Mohs micrographic surgery, thereby decreasing the resulting surgical defect.14
Limitations
Imaging Depth
Although RCM is a revolutionary device in the field of dermatology, it has several limitations. With a maximal imaging depth of 350 µm, the imaging resolution decreases substantially with depth, limiting accurate interpretation to 200 µm. Reflectance confocal microscopy can only image the superficial portion of a lesion; therefore, deep tumor margins cannot be assessed. Hypertrophic or hyperkeratotic lesions, including lesions on the palms and soles, also are unable to be imaged with RCM. This limitation in depth penetration makes treatment monitoring impossible for invasive lesions that extend into the dermal layer.
Difficult-to-Reach Areas
Another limitation is the difficulty imaging areas such as the ocular canthi, nasal alae, or helices of the ear due to the wide probe size on the VivaScope 1500. The advent of the smaller handheld VivaScope 3000 device allows for improved imaging of concave services and difficult lesions at the risk of less accurate imaging, low field of view, and no reimbursement at present.
False-Positive Results
Although RCM has been shown to be helpful in reducing unnecessary biopsies, there still is the issue of false-positives on imaging. False-positives most commonly occur in nevi with severe atypia or when Langerhans cells are present that cannot always be differentiated from melanocytic cells.3,15,16 One prospective study found 7 false-positive results from 63 sites using RCM for the diagnosis of lentigo malignas.16 False-negatives can occur in the presence of inflammatory infiltrates and scar tissue that can hide cellular morphology or in sampling errors due to skip lesions.3,16
Time Efficiency
The time required for acquisition of RCM mosaics and stacks followed by reading and interpretation can be substantial depending on the size and complexity of the lesion, which is a major limitation for use of RCM in busy dermatology practices; therefore, RCM should be reserved for lesions selected to undergo biopsy that are clinically equivocal for malignancy prior to RCM examination.17 It would not be cost-effective or time effective to evaluate lesions that either clinically or dermoscopically have a high probability of malignancy; however, patients and physicians may opt for increased specificity at the expense of time, particularly when a lesion is located on a cosmetically sensitive area, as patients can avoid initial histologic biopsy and gain the cosmetic benefit of going straight to surgery versus obtaining an initial diagnostic biopsy.
Cost
Lastly, the high cost involved in purchasing an RCM device and the training involved to use and interpret RCM images currently limits RCM to large academic centers. Reimbursement may make more widespread use feasible. In any event, RCM imaging should be part of the curriculum for both dermatology and pathology trainees.
Future Directions
In vivo RCM is a noninvasive imaging modality that allows for real-time evaluation of the skin. Used in conjunction with dermoscopy, RCM can substantially improve diagnostic accuracy and reduce the number of unnecessary biopsies. Now that RCM has finally gained foundational CPT codes and insurance reimbursement, there may be a growing demand for clinicians to incorporate this technology into their clinical practice.
Reflectance confocal microscopy (RCM) imaging received Category I Current Procedural Terminology (CPT) codes by the Centers for Medicare & Medicaid Services in January 2016 and can now be submitted to insurance companies with reimbursement comparable to a skin biopsy or a global skin pathology service.1 This fairly new technology is a US Food and Drug Administration–cleared noninvasive imaging modality that provides high-resolution in vivo cellular images of the skin. It has been shown to be efficacious in differentiating benign and malignant skin lesions, increasing diagnostic accuracy, and reducing the number of unnecessary skin biopsies that are performed. In addition to skin cancer diagnosis, RCM imaging also can help guide management of malignant lesions by detecting lateral margins prior to surgery as well as monitoring the lesion over time for treatment efficacy or recurrence. The potential impact of RCM imaging is tremendous, and reimbursement may lead to increased use in clinical practice to the benefit of our patients. Herein, we present a brief review of RCM imaging and reimbursement as well as the benefits and limitations of this new technology for dermatologists.
Reflectance Confocal Microscopy
In vivo RCM allows us to visualize the epidermis in real time on a cellular level down to the papillary dermis at a high resolution (×30) comparable to histologic examination. With optical sections 3- to 5-µm thick and a lateral resolution of 0.5 to 1.0 µm, RCM produces a stack of 500×500-µm2 images up to a depth of approximately 200 µm.2,3 At any chosen depth, these smaller images are stitched together with sophisticated software into a block, or mosaic, increasing the field of view to up to 8×8 mm2. Imaging is performed in en face planes oriented parallel to the skin surface, similar to dermoscopy.
Current CPT Guidelines and Reimbursement
The CPT codes for RCM imaging provide reimbursement on a per-lesion basis and are similar to those used for skin biopsy and pathology (Table).1 Codes 96931 through 96933 are used for imaging of a single lesion on a patient. The first code—96931—is used when image acquisition, interpretation, and report creation are carried out by a single clinician. The next 2 codes are used when one clinician acquires the image—96932—comparable to the technical component of a pathology code, while another reads it and creates the report—96933—similar to a dermatopathologist billing for the professional component of a pathology report. For patients presenting with multiple lesions, the next 3 codes—96934, 96935, and 96936—are used in conjunction with the applicable first code for each additional lesion with similar global, technical, and professional components. Because these codes are not in the radiology or pathology sections of CPT, a single code cannot be used with modifier -TC (technical component) and modifier -26, as they are in those sections.
The wide-probe VivaScope 1500 (Caliber I.D., Inc) currently is the only confocal device that can be reported with a CPT code and routinely reimbursed. The handheld VivaScope 3000 (Caliber I.D., Inc) can only view a small stack and does not have the ability to acquire a full mosaic image; it is not covered by these codes.
Images can be viewed as a stack captured at the same horizontal position but at sequential depths or as a mosaic, which has a larger field of view but is limited to a single plane. To appropriately assess a lesion, clinicians must obtain a mosaic that needs to be assessed at multiple layers for a diagnosis to be made because it is a cross-section view.
Diagnosis
Studies have demonstrated the usefulness of RCM imaging in the diagnosis of a wide range of skin diseases, including melanoma and nonmelanoma skin cancers, infectious diseases, and inflammatory and autoimmune conditions, as well as wound healing and skin aging. Reflectance confocal microscopy imaging is not limited to the skin; it can be used to evaluate the hair, nails, oral mucosa, and other organs.
According to several studies, RCM imaging notably increases the diagnostic accuracy and detection rate of skin cancers over clinical and dermoscopic examination alone and therefore can act as an aid in differentiating lesions that are benign versus those that are suspicious and should be biopsied.
Reflectance confocal microscopy has been shown to have a mean sensitivity of 94% (range, 92%–96%) and specificity of 83% (range, 81%–84%) for all types of skin cancer when used with dermoscopy.4 In particular, for melanocytic lesions that are ambiguous on dermoscopy, RCM used in addition to dermoscopy increases the mean sensitivity and specificity for melanoma diagnosis to 93% (range, 89%–96%) and 76% (range, 68%–83%), respectively.5 Although these reported sensitivities are comparable to dermoscopy, the specificity is superior, especially for detecting hypomelanotic and amelanotic melanomas, which often lack specific features on dermoscopy.6-8
The combination of RCM with dermoscopy has reduced the number of unnecessary excisions of benign nevi by more than 50% when compared to dermoscopy alone.9 One study showed that the number needed to treat (ie, excise) a melanoma decreased from 14.6 with dermoscopy alone to 6.8 when guided by dermoscopy and RCM imaging.9 In a similar study, the number needed to treat dropped from 19.41 with dermoscopy alone to 6.25 with dermoscopy and RCM.10
These studies were not looking to evaluate RCM as a replacement test but rather as an add-on test to dermoscopy. Reflectance confocal microscopy imaging takes longer than dermoscopy for each lesion; therefore, RCM should only be used as an adjunctive tool to dermoscopy and not as an initial screening test. Consequentially, a dermatologist skilled in dermoscopy is essential in deciding which lesions would be appropriate for subsequent RCM imaging.
In Vivo Margin Mapping as an Adjunct to Surgery
Oftentimes, tumor margins are poorly defined and can be difficult to map clinically and dermoscopically. Studies have demonstrated the use of RCM in delineation of surgical margins prior to surgery or excisional biopsies.11,12 Alternatively, when complete removal at biopsy would be impractical (eg, for extremely large lesions or lesions located in cosmetically sensitive areas such as the face), RCM can be used to pick the best site for an appropriate biopsy, which decreases the chance of sampling error due to skip lesions and increases histologic accuracy.
Nonsurgical Treatment Monitoring
One advantage of RCM over conventional histology is that RCM imaging leaves the tissue intact, allowing dynamic changes to be studied over time, which is useful for monitoring nonmelanoma skin cancers and lentigo maligna being treated with noninvasive therapeutic modalities.13 If not as a definitive treatment, RCM can act as an adjunct for surgery by monitoring reduction in lesion size prior to Mohs micrographic surgery, thereby decreasing the resulting surgical defect.14
Limitations
Imaging Depth
Although RCM is a revolutionary device in the field of dermatology, it has several limitations. With a maximal imaging depth of 350 µm, the imaging resolution decreases substantially with depth, limiting accurate interpretation to 200 µm. Reflectance confocal microscopy can only image the superficial portion of a lesion; therefore, deep tumor margins cannot be assessed. Hypertrophic or hyperkeratotic lesions, including lesions on the palms and soles, also are unable to be imaged with RCM. This limitation in depth penetration makes treatment monitoring impossible for invasive lesions that extend into the dermal layer.
Difficult-to-Reach Areas
Another limitation is the difficulty imaging areas such as the ocular canthi, nasal alae, or helices of the ear due to the wide probe size on the VivaScope 1500. The advent of the smaller handheld VivaScope 3000 device allows for improved imaging of concave services and difficult lesions at the risk of less accurate imaging, low field of view, and no reimbursement at present.
False-Positive Results
Although RCM has been shown to be helpful in reducing unnecessary biopsies, there still is the issue of false-positives on imaging. False-positives most commonly occur in nevi with severe atypia or when Langerhans cells are present that cannot always be differentiated from melanocytic cells.3,15,16 One prospective study found 7 false-positive results from 63 sites using RCM for the diagnosis of lentigo malignas.16 False-negatives can occur in the presence of inflammatory infiltrates and scar tissue that can hide cellular morphology or in sampling errors due to skip lesions.3,16
Time Efficiency
The time required for acquisition of RCM mosaics and stacks followed by reading and interpretation can be substantial depending on the size and complexity of the lesion, which is a major limitation for use of RCM in busy dermatology practices; therefore, RCM should be reserved for lesions selected to undergo biopsy that are clinically equivocal for malignancy prior to RCM examination.17 It would not be cost-effective or time effective to evaluate lesions that either clinically or dermoscopically have a high probability of malignancy; however, patients and physicians may opt for increased specificity at the expense of time, particularly when a lesion is located on a cosmetically sensitive area, as patients can avoid initial histologic biopsy and gain the cosmetic benefit of going straight to surgery versus obtaining an initial diagnostic biopsy.
Cost
Lastly, the high cost involved in purchasing an RCM device and the training involved to use and interpret RCM images currently limits RCM to large academic centers. Reimbursement may make more widespread use feasible. In any event, RCM imaging should be part of the curriculum for both dermatology and pathology trainees.
Future Directions
In vivo RCM is a noninvasive imaging modality that allows for real-time evaluation of the skin. Used in conjunction with dermoscopy, RCM can substantially improve diagnostic accuracy and reduce the number of unnecessary biopsies. Now that RCM has finally gained foundational CPT codes and insurance reimbursement, there may be a growing demand for clinicians to incorporate this technology into their clinical practice.
- Current Procedural Terminology 2017, Professional Edition. Chicago IL: American Medical Association; 2016.
- Que SK, Fraga-Braghiroli N, Grant-Kels JM, et al. Through the looking glass: basics and principles of reflectance confocal microscopy [published online June 4, 2015]. J Am Acad Dermatol. 2015;73:276-284.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside [published online October 27, 2016]. Lasers Surg Med. 2017;49:7-19.
- Xiong YD, Ma S, Li X, et al. A meta-analysis of reflectance confocal microscopy for the diagnosis of malignant skin tumours. J Eur Acad Dermatol Venereol. 2016;30:1295-1302.
- Stevenson AD, Mickan S, Mallett S, et al. Systematic review of diagnostic accuracy of reflectance confocal microscopy for melanoma diagnosis in patients with clinically equivocal skin lesions. Dermatol Pract Concept. 2013;3:19-27.
- Busam KJ, Hester K, Charles C, et al. Detection of clinically amelanotic malignant melanoma and assessment of its margins by in vivo confocal scanning laser microscopy. Arch Dermatol. 2001;137:923-929.
- Losi A, Longo C, Cesinaro AM, et al. Hyporeflective pagetoid cells: a new clue for amelanotic melanoma diagnosis by reflectance confocal microscopy. Br J Dermatol. 2014;171:48-54.
- Guitera P, Menzies SQ, Argenziano G, et al. Dermoscopy and in vivo confocal microscopy are complementary techniques for the diagnosis of difficult amelanotic and light-coloured skin lesions [published online October 12, 2016]. Br J Dermatol. 2016;175:1311-1319.
- Pellacani G, Pepe P, Casari A, et al. Reflectance confocal microscopy as a second-level examination in skin oncology improves diagnostic accuracy and saves unnecessary excisions: a longitudinal prospective study. Br J Dermatol. 2014;171:1044-1051.
- Pellacani G, Witkowski A, Cesinaro AM, et al. Cost-benefit of reflectance confocal microscopy in the diagnostic performance of melanoma. J Eur Acad Dermatol Venereol. 2016;30:413-419.
- Champin J, Perrot JL, Cinotti E, et al. In vivo reflectance confocal microscopy to optimize the spaghetti technique for defining surgical margins of lentigo maligna. Dermatol Surg. 2014;40:247-256.
- Hibler BP, Cordova M, Wong RJ, et al. Intraoperative real-time reflectance confocal microscopy for guiding surgical margins of lentigo maligna melanoma. Dermatol Surg. 2015;41:980-983.
- Ulrich M, Lange-Asschenfeldt S, Gonzalez S. The use of reflectance confocal microscopy for monitoring response to therapy of skin malignancies. Dermatol Pract Concept. 2012;2:202a10.
- Torres A, Niemeyer A, Berkes B, et al. 5% imiquimod cream and reflectance-mode confocal microscopy as adjunct modalities to Mohs micrographic surgery for treatment of basal cell carcinoma. Dermatol Surg. 2004;30(12, pt 1):1462-1469.
- Hashemi P, Pulitzer MP, Scope A, et al. Langerhans cells and melanocytes share similar morphologic features under in vivo reflectance confocal microscopy: a challenge for melanoma diagnosis. J Am Acad Dermatol. 2012;66:452-462.
- Menge TD, Hibler BP, Cordova MA, et al. Concordance of handheld reflectance confocal microscopy (RCM) with histopathology in the diagnosis of lentigo maligna (LM): a prospective study. J Am Acad Dermatol. 2016;74:1114-1120.
- Borsari S, Pampena R, Lallas A, et al. Clinical indications for use of reflectance confocal microscopy for skin cancer diagnosis. JAMA Dermatol. 2016;152:1093-1098.
- Current Procedural Terminology 2017, Professional Edition. Chicago IL: American Medical Association; 2016.
- Que SK, Fraga-Braghiroli N, Grant-Kels JM, et al. Through the looking glass: basics and principles of reflectance confocal microscopy [published online June 4, 2015]. J Am Acad Dermatol. 2015;73:276-284.
- Rajadhyaksha M, Marghoob A, Rossi A, et al. Reflectance confocal microscopy of skin in vivo: from bench to bedside [published online October 27, 2016]. Lasers Surg Med. 2017;49:7-19.
- Xiong YD, Ma S, Li X, et al. A meta-analysis of reflectance confocal microscopy for the diagnosis of malignant skin tumours. J Eur Acad Dermatol Venereol. 2016;30:1295-1302.
- Stevenson AD, Mickan S, Mallett S, et al. Systematic review of diagnostic accuracy of reflectance confocal microscopy for melanoma diagnosis in patients with clinically equivocal skin lesions. Dermatol Pract Concept. 2013;3:19-27.
- Busam KJ, Hester K, Charles C, et al. Detection of clinically amelanotic malignant melanoma and assessment of its margins by in vivo confocal scanning laser microscopy. Arch Dermatol. 2001;137:923-929.
- Losi A, Longo C, Cesinaro AM, et al. Hyporeflective pagetoid cells: a new clue for amelanotic melanoma diagnosis by reflectance confocal microscopy. Br J Dermatol. 2014;171:48-54.
- Guitera P, Menzies SQ, Argenziano G, et al. Dermoscopy and in vivo confocal microscopy are complementary techniques for the diagnosis of difficult amelanotic and light-coloured skin lesions [published online October 12, 2016]. Br J Dermatol. 2016;175:1311-1319.
- Pellacani G, Pepe P, Casari A, et al. Reflectance confocal microscopy as a second-level examination in skin oncology improves diagnostic accuracy and saves unnecessary excisions: a longitudinal prospective study. Br J Dermatol. 2014;171:1044-1051.
- Pellacani G, Witkowski A, Cesinaro AM, et al. Cost-benefit of reflectance confocal microscopy in the diagnostic performance of melanoma. J Eur Acad Dermatol Venereol. 2016;30:413-419.
- Champin J, Perrot JL, Cinotti E, et al. In vivo reflectance confocal microscopy to optimize the spaghetti technique for defining surgical margins of lentigo maligna. Dermatol Surg. 2014;40:247-256.
- Hibler BP, Cordova M, Wong RJ, et al. Intraoperative real-time reflectance confocal microscopy for guiding surgical margins of lentigo maligna melanoma. Dermatol Surg. 2015;41:980-983.
- Ulrich M, Lange-Asschenfeldt S, Gonzalez S. The use of reflectance confocal microscopy for monitoring response to therapy of skin malignancies. Dermatol Pract Concept. 2012;2:202a10.
- Torres A, Niemeyer A, Berkes B, et al. 5% imiquimod cream and reflectance-mode confocal microscopy as adjunct modalities to Mohs micrographic surgery for treatment of basal cell carcinoma. Dermatol Surg. 2004;30(12, pt 1):1462-1469.
- Hashemi P, Pulitzer MP, Scope A, et al. Langerhans cells and melanocytes share similar morphologic features under in vivo reflectance confocal microscopy: a challenge for melanoma diagnosis. J Am Acad Dermatol. 2012;66:452-462.
- Menge TD, Hibler BP, Cordova MA, et al. Concordance of handheld reflectance confocal microscopy (RCM) with histopathology in the diagnosis of lentigo maligna (LM): a prospective study. J Am Acad Dermatol. 2016;74:1114-1120.
- Borsari S, Pampena R, Lallas A, et al. Clinical indications for use of reflectance confocal microscopy for skin cancer diagnosis. JAMA Dermatol. 2016;152:1093-1098.
Practice Points
- Reflectance confocal microscopy (RCM) recently received Category I Current Procedural Terminology codes for reimbursement comparable to a skin biopsy.
- When used in combination with dermoscopy, RCM has been shown to increase diagnostic accuracy of skin cancer.
- Reflectance confocal microscopy also is useful in surgical treatment planning and monitoring nonsurgical treatments over time.
- Limitations of RCM imaging include low imaging depth, difficulty in imaging certain areas of the skin, learning curve for interpreting these images, and the cost of equipment.

Mitigating Burnout - Part 3
It is easy to look at the changes required to mitigate burnout and improve compassionate care and see the burden being placed mainly on the physician. Many of the proposed modifications seem to require the one commodity surgeons lack most, time. Any widespread effort to mitigate the burnout crisis must involve decreasing the barriers to patient care and reducing the physicians’ time constraints.
This may seem daunting, but broad changes in our healthcare system have been implemented in the name of quality, reducing errors, and alleviating trainee fatigue. Burnout can be a similar force for change. Much like the resident’s 80-hour workweek, however, in what manner this change is applied will be the ultimate determinant of the movement’s success.

This series of articles dealt with the adverse consequences of surgeon burnout on both clinicians and their patients, and then presented a conceptual framework to promote workforce well-being. Strategies that the SVS might adopt have been suggested. The central proposal is that helping physicians to deliver compassionate, collaborative care will not only mitigate burnout but also will enhance provider engagement, patient experience, and clinical outcomes, as well as improve the quality and safety of healthcare delivery. It seems reasonable to question if such broad strategic proposals are scalable to individual clinical settings.
The characteristics of compassionate care have been well described by patients, and, not surprisingly, only 53% reported that their last encounter with the health care system was compassionate. In 2014, a multidisciplinary consortium published recommendations for advancing compassionate person- and family-centered care. They detailed the attributes, values, and behaviors of such care, including focusing one’s attention, recognizing nonverbal clues, active listening, demonstrating nonjudgmental interest in the whole person, understanding the context of a person’s disease, and asking about the patient’s chief concerns in addition to their chief complaints. Most significantly, the authors outlined how these attributes could be integrated into existing competency documents such as those provided by the Association of American Medical Colleges (AAMC) Entrustable Professional Activities, or the milestones programs of the Accreditation Council of Graduate Medical Education (ACGME) and the American Board of Internal Medicine (ABIM).
It is but a small step to enhance current criteria for certification, clinical appointment, and privileging by including these professional attributes. Bear in mind that these skills are teachable and easily incorporated into health professional education and clinical care. As for metrics, the Schwartz Compassionate Care Scale is a validated patient-rated questionnaire that reliably measures physicians’ compassion and overall patient satisfaction and is available in the public domain. It also provides a metric that is important to clinicians and, when placed on the hospital dashboard, highlights that compassion is an organizational priority. Such priorities become the fabric of the workforce when organizational leadership installs programs such as values-based recruitment, retention, and promotion as Vivian Lee describes at the University of Utah Health System. Mitigating burnout requires changing the culture in which clinicians work and with whom they work.
Vascular surgeons and their professional surgical societies have a leadership opportunity to design high performing teams. Most patient care models have been structured around traditional medical and surgical departments. This paradigm overlooks the fact that patients do not “get sick” within traditional teaching disciplines but do so across varied medical and surgical specialties. Changes in organizational hierarchy are needed so that team-based care is supported. In addition to physician and nurse clinicians, the new teams would do well to expand to all “caregivers,” i.e. everyone who touches the patient (technologists, interpreters, pharmacists, transport workers, support staff, and administrators).
On the front line
On April 15, 2013, at 2:49 pm, two homemade bombs detonated near the finish line of the Boston Marathon killing three people at the scene and injuring 264 others; the most severe sustaining mutilating lower extremity injuries. Much has been written about the preparedness, the emergency response, and the fact that all those who made it to the hospital survived. Jeffrey Kalish, MD, a vascular surgeon at Boston Medical Center and SVS member, was on the front line that day. We asked him to share his personal experience of caring for the victims through the lens of compassionate collaborative care.
“It has been over four years since I went from being a spectator near the Boston Marathon finish line to rushing directly to the operating rooms at Boston Medical Center to help our teams perform lifesaving procedures on critically ill patients, including amputations and complex vascular repairs. While I have learned a tremendous amount since that experience with regard to limb salvage and amputation, reconstructive techniques, and prosthetics, I will focus here on the care the patients and their families received, the lessons our hospital learned from the weeks and months that followed, and how we modeled this care going forward for all amputation patients at Boston Medical Center.
"Based on lessons from the Boston Marathon bombings, I aligned a multidisciplinary team of health care providers in order to formalize and standardize best practices to benefit our amputation patients. STRONG (Surgery To Rehab Ongoing Needs Group) continues to strive toward the ultimate goal of improving and coordinating care for amputation patients and their families as they transition from the hospital setting to rehabilitation. Some of our guiding principles, along with their positive impacts on patient care and physician well-being, are highlighted below:
1. Sustaining hope with a new mindset: Although surgeons have historically considered amputation as a treatment failure, a more appropriate mindset is that amputation can often be a reconstructive procedure in the surgical armamentarium designed to restore a patient back to full function.
2. Seeing the patient in context: Shared decision making can occur more readily once a surgeon and the care team seek to understand the whole person and their family, including what that person does for work and leisure.
3. Communication with colleagues, patients, and families: Patient and family fear and confusion can often be reduced after establishment of a multi-disciplinary team with daily care coordination and consistent messaging. Breaking down traditional hospital silos to allow for improved coordination of care benefits both the patients and the practitioners.
4. Managing emotional and physical suffering: Introducing social workers, mental health professionals, or pastoral care advocates into the care team as soon as a patient is ready can help manage the emotional and psychosocial needs of patients and their families.
5. Sustaining long term surgeon/patient relationships: As clinicians, we can feel rewarded after restoring functional performance in our patients and by meeting the needs of our patients and families. This can occur both in the short-term during the acute hospital stay and in the long-term as we follow our patients’ progress towards achievement of their ultimate goals.
6. Attend to one’s own well-being and foster resilience. There will never be a substitute in our profession for the human connection between our patients and ourselves as their caregivers, and this connection should be one of the most treasured aspects of our work life. We should seek daily reminders of these positive interactions to nurture our ability to cope with a grueling and challenging field, where the outcomes are not always as ideal as we hope.
For me, STRONG is now a consistent and powerful reminder of why I originally became a doctor in the first place, and I strive to propagate this model of compassionate care to benefit future patients as we move forward together.”
We are at an inflexion point in health care. No amount of individual resilience can withstand a toxic or nonsupportive environment. It is unreasonable to think that simply by taking better care of ourselves we are going to resolve the issue of burnout. We need to rethink our current systems of care and focus our energy on developing those that support our ability to deliver the kind of care we know our patients need and deserve. We have an opportunity to alleviate the suffering many providers are experiencing as they strive to heal their patients. We have an opportunity to improve both physician and patient engagement, develop care delivery systems we know our patients deserve, and restore the deep sense of satisfaction that comes from practicing medicine and surgery. There is abundant expertise within our professional medical and surgical societies. The SVS has the courage and the duty to lead.
Drs. Colman, Kalish, and Sheahan extend their thanks and appreciation for the guidance, resources and support of Michael Goldberg, M.D., Scholar in Residence, Schwartz Center for Compassionate Care, Boston, Mass., and Clinical Professor of Orthopedics at Seattle Children’s Hospital.
Bibliography
1. Acad Med (2016) 91:338-344
2. Health Aff (2011) 30:1772-1778
3. Patient Educ Couns (2015) 98:1005-10
4. Acad Med (2016) 91:310-316
5. http://www.theschwartzcenter.org/media/Triple-C-Conference-Framework-Tables_FINAL.pdf
6. http://www.theschwartzcenter.org/media/Triple-C-Conference-Recommendations-Report_FINAL1.pdf
7. JAMA (2017) 317: 901-2
8. Clin Orthop Relat Res. (2017) 475:1309-14.
It is easy to look at the changes required to mitigate burnout and improve compassionate care and see the burden being placed mainly on the physician. Many of the proposed modifications seem to require the one commodity surgeons lack most, time. Any widespread effort to mitigate the burnout crisis must involve decreasing the barriers to patient care and reducing the physicians’ time constraints.
This may seem daunting, but broad changes in our healthcare system have been implemented in the name of quality, reducing errors, and alleviating trainee fatigue. Burnout can be a similar force for change. Much like the resident’s 80-hour workweek, however, in what manner this change is applied will be the ultimate determinant of the movement’s success.
This series of articles dealt with the adverse consequences of surgeon burnout on both clinicians and their patients, and then presented a conceptual framework to promote workforce well-being. Strategies that the SVS might adopt have been suggested. The central proposal is that helping physicians to deliver compassionate, collaborative care will not only mitigate burnout but also will enhance provider engagement, patient experience, and clinical outcomes, as well as improve the quality and safety of healthcare delivery. It seems reasonable to question if such broad strategic proposals are scalable to individual clinical settings.
The characteristics of compassionate care have been well described by patients, and, not surprisingly, only 53% reported that their last encounter with the health care system was compassionate. In 2014, a multidisciplinary consortium published recommendations for advancing compassionate person- and family-centered care. They detailed the attributes, values, and behaviors of such care, including focusing one’s attention, recognizing nonverbal clues, active listening, demonstrating nonjudgmental interest in the whole person, understanding the context of a person’s disease, and asking about the patient’s chief concerns in addition to their chief complaints. Most significantly, the authors outlined how these attributes could be integrated into existing competency documents such as those provided by the Association of American Medical Colleges (AAMC) Entrustable Professional Activities, or the milestones programs of the Accreditation Council of Graduate Medical Education (ACGME) and the American Board of Internal Medicine (ABIM).
It is but a small step to enhance current criteria for certification, clinical appointment, and privileging by including these professional attributes. Bear in mind that these skills are teachable and easily incorporated into health professional education and clinical care. As for metrics, the Schwartz Compassionate Care Scale is a validated patient-rated questionnaire that reliably measures physicians’ compassion and overall patient satisfaction and is available in the public domain. It also provides a metric that is important to clinicians and, when placed on the hospital dashboard, highlights that compassion is an organizational priority. Such priorities become the fabric of the workforce when organizational leadership installs programs such as values-based recruitment, retention, and promotion as Vivian Lee describes at the University of Utah Health System. Mitigating burnout requires changing the culture in which clinicians work and with whom they work.
Vascular surgeons and their professional surgical societies have a leadership opportunity to design high performing teams. Most patient care models have been structured around traditional medical and surgical departments. This paradigm overlooks the fact that patients do not “get sick” within traditional teaching disciplines but do so across varied medical and surgical specialties. Changes in organizational hierarchy are needed so that team-based care is supported. In addition to physician and nurse clinicians, the new teams would do well to expand to all “caregivers,” i.e. everyone who touches the patient (technologists, interpreters, pharmacists, transport workers, support staff, and administrators).
On the front line
On April 15, 2013, at 2:49 pm, two homemade bombs detonated near the finish line of the Boston Marathon killing three people at the scene and injuring 264 others; the most severe sustaining mutilating lower extremity injuries. Much has been written about the preparedness, the emergency response, and the fact that all those who made it to the hospital survived. Jeffrey Kalish, MD, a vascular surgeon at Boston Medical Center and SVS member, was on the front line that day. We asked him to share his personal experience of caring for the victims through the lens of compassionate collaborative care.
“It has been over four years since I went from being a spectator near the Boston Marathon finish line to rushing directly to the operating rooms at Boston Medical Center to help our teams perform lifesaving procedures on critically ill patients, including amputations and complex vascular repairs. While I have learned a tremendous amount since that experience with regard to limb salvage and amputation, reconstructive techniques, and prosthetics, I will focus here on the care the patients and their families received, the lessons our hospital learned from the weeks and months that followed, and how we modeled this care going forward for all amputation patients at Boston Medical Center.
"Based on lessons from the Boston Marathon bombings, I aligned a multidisciplinary team of health care providers in order to formalize and standardize best practices to benefit our amputation patients. STRONG (Surgery To Rehab Ongoing Needs Group) continues to strive toward the ultimate goal of improving and coordinating care for amputation patients and their families as they transition from the hospital setting to rehabilitation. Some of our guiding principles, along with their positive impacts on patient care and physician well-being, are highlighted below:
1. Sustaining hope with a new mindset: Although surgeons have historically considered amputation as a treatment failure, a more appropriate mindset is that amputation can often be a reconstructive procedure in the surgical armamentarium designed to restore a patient back to full function.
2. Seeing the patient in context: Shared decision making can occur more readily once a surgeon and the care team seek to understand the whole person and their family, including what that person does for work and leisure.
3. Communication with colleagues, patients, and families: Patient and family fear and confusion can often be reduced after establishment of a multi-disciplinary team with daily care coordination and consistent messaging. Breaking down traditional hospital silos to allow for improved coordination of care benefits both the patients and the practitioners.
4. Managing emotional and physical suffering: Introducing social workers, mental health professionals, or pastoral care advocates into the care team as soon as a patient is ready can help manage the emotional and psychosocial needs of patients and their families.
5. Sustaining long term surgeon/patient relationships: As clinicians, we can feel rewarded after restoring functional performance in our patients and by meeting the needs of our patients and families. This can occur both in the short-term during the acute hospital stay and in the long-term as we follow our patients’ progress towards achievement of their ultimate goals.
6. Attend to one’s own well-being and foster resilience. There will never be a substitute in our profession for the human connection between our patients and ourselves as their caregivers, and this connection should be one of the most treasured aspects of our work life. We should seek daily reminders of these positive interactions to nurture our ability to cope with a grueling and challenging field, where the outcomes are not always as ideal as we hope.
For me, STRONG is now a consistent and powerful reminder of why I originally became a doctor in the first place, and I strive to propagate this model of compassionate care to benefit future patients as we move forward together.”
We are at an inflexion point in health care. No amount of individual resilience can withstand a toxic or nonsupportive environment. It is unreasonable to think that simply by taking better care of ourselves we are going to resolve the issue of burnout. We need to rethink our current systems of care and focus our energy on developing those that support our ability to deliver the kind of care we know our patients need and deserve. We have an opportunity to alleviate the suffering many providers are experiencing as they strive to heal their patients. We have an opportunity to improve both physician and patient engagement, develop care delivery systems we know our patients deserve, and restore the deep sense of satisfaction that comes from practicing medicine and surgery. There is abundant expertise within our professional medical and surgical societies. The SVS has the courage and the duty to lead.
Drs. Colman, Kalish, and Sheahan extend their thanks and appreciation for the guidance, resources and support of Michael Goldberg, M.D., Scholar in Residence, Schwartz Center for Compassionate Care, Boston, Mass., and Clinical Professor of Orthopedics at Seattle Children’s Hospital.
Bibliography
1. Acad Med (2016) 91:338-344
2. Health Aff (2011) 30:1772-1778
3. Patient Educ Couns (2015) 98:1005-10
4. Acad Med (2016) 91:310-316
5. http://www.theschwartzcenter.org/media/Triple-C-Conference-Framework-Tables_FINAL.pdf
6. http://www.theschwartzcenter.org/media/Triple-C-Conference-Recommendations-Report_FINAL1.pdf
7. JAMA (2017) 317: 901-2
8. Clin Orthop Relat Res. (2017) 475:1309-14.
It is easy to look at the changes required to mitigate burnout and improve compassionate care and see the burden being placed mainly on the physician. Many of the proposed modifications seem to require the one commodity surgeons lack most, time. Any widespread effort to mitigate the burnout crisis must involve decreasing the barriers to patient care and reducing the physicians’ time constraints.
This may seem daunting, but broad changes in our healthcare system have been implemented in the name of quality, reducing errors, and alleviating trainee fatigue. Burnout can be a similar force for change. Much like the resident’s 80-hour workweek, however, in what manner this change is applied will be the ultimate determinant of the movement’s success.
This series of articles dealt with the adverse consequences of surgeon burnout on both clinicians and their patients, and then presented a conceptual framework to promote workforce well-being. Strategies that the SVS might adopt have been suggested. The central proposal is that helping physicians to deliver compassionate, collaborative care will not only mitigate burnout but also will enhance provider engagement, patient experience, and clinical outcomes, as well as improve the quality and safety of healthcare delivery. It seems reasonable to question if such broad strategic proposals are scalable to individual clinical settings.
The characteristics of compassionate care have been well described by patients, and, not surprisingly, only 53% reported that their last encounter with the health care system was compassionate. In 2014, a multidisciplinary consortium published recommendations for advancing compassionate person- and family-centered care. They detailed the attributes, values, and behaviors of such care, including focusing one’s attention, recognizing nonverbal clues, active listening, demonstrating nonjudgmental interest in the whole person, understanding the context of a person’s disease, and asking about the patient’s chief concerns in addition to their chief complaints. Most significantly, the authors outlined how these attributes could be integrated into existing competency documents such as those provided by the Association of American Medical Colleges (AAMC) Entrustable Professional Activities, or the milestones programs of the Accreditation Council of Graduate Medical Education (ACGME) and the American Board of Internal Medicine (ABIM).
It is but a small step to enhance current criteria for certification, clinical appointment, and privileging by including these professional attributes. Bear in mind that these skills are teachable and easily incorporated into health professional education and clinical care. As for metrics, the Schwartz Compassionate Care Scale is a validated patient-rated questionnaire that reliably measures physicians’ compassion and overall patient satisfaction and is available in the public domain. It also provides a metric that is important to clinicians and, when placed on the hospital dashboard, highlights that compassion is an organizational priority. Such priorities become the fabric of the workforce when organizational leadership installs programs such as values-based recruitment, retention, and promotion as Vivian Lee describes at the University of Utah Health System. Mitigating burnout requires changing the culture in which clinicians work and with whom they work.
Vascular surgeons and their professional surgical societies have a leadership opportunity to design high performing teams. Most patient care models have been structured around traditional medical and surgical departments. This paradigm overlooks the fact that patients do not “get sick” within traditional teaching disciplines but do so across varied medical and surgical specialties. Changes in organizational hierarchy are needed so that team-based care is supported. In addition to physician and nurse clinicians, the new teams would do well to expand to all “caregivers,” i.e. everyone who touches the patient (technologists, interpreters, pharmacists, transport workers, support staff, and administrators).
On the front line
On April 15, 2013, at 2:49 pm, two homemade bombs detonated near the finish line of the Boston Marathon killing three people at the scene and injuring 264 others; the most severe sustaining mutilating lower extremity injuries. Much has been written about the preparedness, the emergency response, and the fact that all those who made it to the hospital survived. Jeffrey Kalish, MD, a vascular surgeon at Boston Medical Center and SVS member, was on the front line that day. We asked him to share his personal experience of caring for the victims through the lens of compassionate collaborative care.
“It has been over four years since I went from being a spectator near the Boston Marathon finish line to rushing directly to the operating rooms at Boston Medical Center to help our teams perform lifesaving procedures on critically ill patients, including amputations and complex vascular repairs. While I have learned a tremendous amount since that experience with regard to limb salvage and amputation, reconstructive techniques, and prosthetics, I will focus here on the care the patients and their families received, the lessons our hospital learned from the weeks and months that followed, and how we modeled this care going forward for all amputation patients at Boston Medical Center.
"Based on lessons from the Boston Marathon bombings, I aligned a multidisciplinary team of health care providers in order to formalize and standardize best practices to benefit our amputation patients. STRONG (Surgery To Rehab Ongoing Needs Group) continues to strive toward the ultimate goal of improving and coordinating care for amputation patients and their families as they transition from the hospital setting to rehabilitation. Some of our guiding principles, along with their positive impacts on patient care and physician well-being, are highlighted below:
1. Sustaining hope with a new mindset: Although surgeons have historically considered amputation as a treatment failure, a more appropriate mindset is that amputation can often be a reconstructive procedure in the surgical armamentarium designed to restore a patient back to full function.
2. Seeing the patient in context: Shared decision making can occur more readily once a surgeon and the care team seek to understand the whole person and their family, including what that person does for work and leisure.
3. Communication with colleagues, patients, and families: Patient and family fear and confusion can often be reduced after establishment of a multi-disciplinary team with daily care coordination and consistent messaging. Breaking down traditional hospital silos to allow for improved coordination of care benefits both the patients and the practitioners.
4. Managing emotional and physical suffering: Introducing social workers, mental health professionals, or pastoral care advocates into the care team as soon as a patient is ready can help manage the emotional and psychosocial needs of patients and their families.
5. Sustaining long term surgeon/patient relationships: As clinicians, we can feel rewarded after restoring functional performance in our patients and by meeting the needs of our patients and families. This can occur both in the short-term during the acute hospital stay and in the long-term as we follow our patients’ progress towards achievement of their ultimate goals.
6. Attend to one’s own well-being and foster resilience. There will never be a substitute in our profession for the human connection between our patients and ourselves as their caregivers, and this connection should be one of the most treasured aspects of our work life. We should seek daily reminders of these positive interactions to nurture our ability to cope with a grueling and challenging field, where the outcomes are not always as ideal as we hope.
For me, STRONG is now a consistent and powerful reminder of why I originally became a doctor in the first place, and I strive to propagate this model of compassionate care to benefit future patients as we move forward together.”
We are at an inflexion point in health care. No amount of individual resilience can withstand a toxic or nonsupportive environment. It is unreasonable to think that simply by taking better care of ourselves we are going to resolve the issue of burnout. We need to rethink our current systems of care and focus our energy on developing those that support our ability to deliver the kind of care we know our patients need and deserve. We have an opportunity to alleviate the suffering many providers are experiencing as they strive to heal their patients. We have an opportunity to improve both physician and patient engagement, develop care delivery systems we know our patients deserve, and restore the deep sense of satisfaction that comes from practicing medicine and surgery. There is abundant expertise within our professional medical and surgical societies. The SVS has the courage and the duty to lead.
Drs. Colman, Kalish, and Sheahan extend their thanks and appreciation for the guidance, resources and support of Michael Goldberg, M.D., Scholar in Residence, Schwartz Center for Compassionate Care, Boston, Mass., and Clinical Professor of Orthopedics at Seattle Children’s Hospital.
Bibliography
1. Acad Med (2016) 91:338-344
2. Health Aff (2011) 30:1772-1778
3. Patient Educ Couns (2015) 98:1005-10
4. Acad Med (2016) 91:310-316
5. http://www.theschwartzcenter.org/media/Triple-C-Conference-Framework-Tables_FINAL.pdf
6. http://www.theschwartzcenter.org/media/Triple-C-Conference-Recommendations-Report_FINAL1.pdf
7. JAMA (2017) 317: 901-2
8. Clin Orthop Relat Res. (2017) 475:1309-14.
Cutaneous laser surgery: Basic caution isn’t enough to prevent lawsuits
SAN DIEGO – Injuries and lawsuits related to laser cosmetic surgery are increasing and potential legal threats are not always easy to predict, according to two dermatologists who spoke at the annual meeting of the American Society for Laser Medicine and Surgery (ASLMS).
A laser procedure could go smoothly, for example, but the patient might be able to successfully sue if he or she is allowed to drive home after receiving a sedative. Or a physician might get sued because his or her nurse set a laser at the wrong setting and singed a patient.
The risk of a lawsuit is high, H. Ray Jalian, MD, a dermatologist in Los Angeles, said at the meeting. “The reality is that we’re all at some point going to face this.”

The most common procedure litigated was laser hair removal, making up almost 40% of the cases, which is not an indication that this particular procedure is dangerous, Dr. Jalian said. “It’s quite safe, and the complication rate is quite low,” but more of these procedures are being done, he noted. Rejuvenation procedures followed, accounting for 25% of cases.
The alleged injuries sustained from laser surgery included burns (47%), scars (39%), and pigmentation problems (24%). Deaths occurred in just over 2% of the cases. In the study, almost a third of plaintiffs alleged that they were not provided informed consent. Plaintiffs also alleged fraud (9%) and assault/battery (5%), and a family member occasionally sued for loss of consortium (8% of cases). The specialty with the largest percentage of the cases was plastic surgery (26%), followed by dermatology (21%).
Dr. Jalian and his copresenter, Mathew Avram, MD, JD, director of the Dermatology Laser & Cosmetic Center, and director of dermatologic surgery at Massachusetts General Hospital, Boston, offered these lessons about the legal risks associated with laser procedures:
• You may have a duty to protect your patient from bad choices.
Physicians aren’t expected to keep patients from making certain bad decisions such as sunbathing after a traditional resurfacing procedure, said Dr. Avram, of the department of dermatology, Harvard Medical School, Boston, and the ASLMS president. But in some cases, he said, the law may expect the physician to step in to prevent harm. For example, he said, a patient who has undergone a fractional ablative laser procedure and has received a sedative should not be allowed to drive home.
• You may get sued even if your employee is at fault.
The 2013 study found physicians were often sued even when they did not perform the laser procedure in question. Nonphysicians such as physician assistants and nurses often perform laser operations, and many states allow them to do so. “Nonphysicians were less likely to be sued even if they were the operators,” Dr. Jalian said. In the study, almost 38% of the 174 analyzed cases involved nonphysician operators, but they were sued in just 26% of the cases. In 33 of the 174 cases in the study, plaintiffs alleged failure to properly hire, train, or supervise staff.
He recommended looking at state laws, which differ greatly in their regulations – or lack of them – regarding the operation of medical lasers. In some cases, physicians must supervise laser use, he said. “But what are the requirements? Can you be available by phone down the street or in the Caribbean?”
Dr. Jalian, Dr. Avram, and a colleague followed up the 2013 study with another study that tracked 175 legal cases from 1999 to 2012 involving alleged injuries from cutaneous laser surgery. During this time period, 75 (43%) involved a nonphysician operating a laser, increasing from 36% in 2008 to 78% in 2012.
In almost two-thirds of cases, the procedures in question were done by nonphysicians outside a “traditional medical setting” such as a salon or spa (JAMA Dermatol. 2014 Apr;150[4]:407-11).
• Delayed side effects could mean delayed lawsuits.
According to Dr. Avram, statutes of limitations – the length of time in which a patient can file a lawsuit – typically last for 2-3 years in malpractice cases. But he said that the period begins when the physician is alleged to have made a mistake or when the patient becomes aware of – or should reasonably be aware of – an injury. Therefore, physicians could face legal trouble over delayed hypopigmentation that appears 6 months after a laser resurfacing treatment, or granulomas that appear years after a filler treatment, he said.
• A signed form is not a cure-all.
It is wise to make patients sign an extensive informed consent form, but this will not protect a physician against a claim of negligence, Dr. Avram said. And the reverse is also true: If a patient did not sign a proper consent form, he or she could still sue even if the procedure went perfectly, he noted.
• Your instincts are worth trusting.
When it comes to lawsuit prevention, Dr. Avram said, “by far the most important thing you can do happens within a minute of when you see the patient. Assess and trust your own intuition and your staff’s intuition. For elective, cosmetic treatments, don’t be afraid to say no. There’s no legal obligation to perform a cosmetic treatment on a patient.”
If you do choose to treat a patient, he advised, be open about the procedure and “maybe even tell them some of the tougher, worse-case scenarios.” If a procedure goes poorly, he said, consider how to fix it. “Many complications can be significantly improved or cleared with timely and appropriate intervention,” he said.
In some cases, refunding the patient’s money can be considered, with the patient signing a release, he said. “Document that you are refunding the money in order to preserve the doctor-patient relationship, not to avoid negligence.”
Dr. Jalian and Dr. Avram reported no relevant disclosures.
SAN DIEGO – Injuries and lawsuits related to laser cosmetic surgery are increasing and potential legal threats are not always easy to predict, according to two dermatologists who spoke at the annual meeting of the American Society for Laser Medicine and Surgery (ASLMS).
A laser procedure could go smoothly, for example, but the patient might be able to successfully sue if he or she is allowed to drive home after receiving a sedative. Or a physician might get sued because his or her nurse set a laser at the wrong setting and singed a patient.
The risk of a lawsuit is high, H. Ray Jalian, MD, a dermatologist in Los Angeles, said at the meeting. “The reality is that we’re all at some point going to face this.”
The most common procedure litigated was laser hair removal, making up almost 40% of the cases, which is not an indication that this particular procedure is dangerous, Dr. Jalian said. “It’s quite safe, and the complication rate is quite low,” but more of these procedures are being done, he noted. Rejuvenation procedures followed, accounting for 25% of cases.
The alleged injuries sustained from laser surgery included burns (47%), scars (39%), and pigmentation problems (24%). Deaths occurred in just over 2% of the cases. In the study, almost a third of plaintiffs alleged that they were not provided informed consent. Plaintiffs also alleged fraud (9%) and assault/battery (5%), and a family member occasionally sued for loss of consortium (8% of cases). The specialty with the largest percentage of the cases was plastic surgery (26%), followed by dermatology (21%).
Dr. Jalian and his copresenter, Mathew Avram, MD, JD, director of the Dermatology Laser & Cosmetic Center, and director of dermatologic surgery at Massachusetts General Hospital, Boston, offered these lessons about the legal risks associated with laser procedures:
• You may have a duty to protect your patient from bad choices.
Physicians aren’t expected to keep patients from making certain bad decisions such as sunbathing after a traditional resurfacing procedure, said Dr. Avram, of the department of dermatology, Harvard Medical School, Boston, and the ASLMS president. But in some cases, he said, the law may expect the physician to step in to prevent harm. For example, he said, a patient who has undergone a fractional ablative laser procedure and has received a sedative should not be allowed to drive home.
• You may get sued even if your employee is at fault.
The 2013 study found physicians were often sued even when they did not perform the laser procedure in question. Nonphysicians such as physician assistants and nurses often perform laser operations, and many states allow them to do so. “Nonphysicians were less likely to be sued even if they were the operators,” Dr. Jalian said. In the study, almost 38% of the 174 analyzed cases involved nonphysician operators, but they were sued in just 26% of the cases. In 33 of the 174 cases in the study, plaintiffs alleged failure to properly hire, train, or supervise staff.
He recommended looking at state laws, which differ greatly in their regulations – or lack of them – regarding the operation of medical lasers. In some cases, physicians must supervise laser use, he said. “But what are the requirements? Can you be available by phone down the street or in the Caribbean?”
Dr. Jalian, Dr. Avram, and a colleague followed up the 2013 study with another study that tracked 175 legal cases from 1999 to 2012 involving alleged injuries from cutaneous laser surgery. During this time period, 75 (43%) involved a nonphysician operating a laser, increasing from 36% in 2008 to 78% in 2012.
In almost two-thirds of cases, the procedures in question were done by nonphysicians outside a “traditional medical setting” such as a salon or spa (JAMA Dermatol. 2014 Apr;150[4]:407-11).
• Delayed side effects could mean delayed lawsuits.
According to Dr. Avram, statutes of limitations – the length of time in which a patient can file a lawsuit – typically last for 2-3 years in malpractice cases. But he said that the period begins when the physician is alleged to have made a mistake or when the patient becomes aware of – or should reasonably be aware of – an injury. Therefore, physicians could face legal trouble over delayed hypopigmentation that appears 6 months after a laser resurfacing treatment, or granulomas that appear years after a filler treatment, he said.
• A signed form is not a cure-all.
It is wise to make patients sign an extensive informed consent form, but this will not protect a physician against a claim of negligence, Dr. Avram said. And the reverse is also true: If a patient did not sign a proper consent form, he or she could still sue even if the procedure went perfectly, he noted.
• Your instincts are worth trusting.
When it comes to lawsuit prevention, Dr. Avram said, “by far the most important thing you can do happens within a minute of when you see the patient. Assess and trust your own intuition and your staff’s intuition. For elective, cosmetic treatments, don’t be afraid to say no. There’s no legal obligation to perform a cosmetic treatment on a patient.”
If you do choose to treat a patient, he advised, be open about the procedure and “maybe even tell them some of the tougher, worse-case scenarios.” If a procedure goes poorly, he said, consider how to fix it. “Many complications can be significantly improved or cleared with timely and appropriate intervention,” he said.
In some cases, refunding the patient’s money can be considered, with the patient signing a release, he said. “Document that you are refunding the money in order to preserve the doctor-patient relationship, not to avoid negligence.”
Dr. Jalian and Dr. Avram reported no relevant disclosures.
SAN DIEGO – Injuries and lawsuits related to laser cosmetic surgery are increasing and potential legal threats are not always easy to predict, according to two dermatologists who spoke at the annual meeting of the American Society for Laser Medicine and Surgery (ASLMS).
A laser procedure could go smoothly, for example, but the patient might be able to successfully sue if he or she is allowed to drive home after receiving a sedative. Or a physician might get sued because his or her nurse set a laser at the wrong setting and singed a patient.
The risk of a lawsuit is high, H. Ray Jalian, MD, a dermatologist in Los Angeles, said at the meeting. “The reality is that we’re all at some point going to face this.”
The most common procedure litigated was laser hair removal, making up almost 40% of the cases, which is not an indication that this particular procedure is dangerous, Dr. Jalian said. “It’s quite safe, and the complication rate is quite low,” but more of these procedures are being done, he noted. Rejuvenation procedures followed, accounting for 25% of cases.
The alleged injuries sustained from laser surgery included burns (47%), scars (39%), and pigmentation problems (24%). Deaths occurred in just over 2% of the cases. In the study, almost a third of plaintiffs alleged that they were not provided informed consent. Plaintiffs also alleged fraud (9%) and assault/battery (5%), and a family member occasionally sued for loss of consortium (8% of cases). The specialty with the largest percentage of the cases was plastic surgery (26%), followed by dermatology (21%).
Dr. Jalian and his copresenter, Mathew Avram, MD, JD, director of the Dermatology Laser & Cosmetic Center, and director of dermatologic surgery at Massachusetts General Hospital, Boston, offered these lessons about the legal risks associated with laser procedures:
• You may have a duty to protect your patient from bad choices.
Physicians aren’t expected to keep patients from making certain bad decisions such as sunbathing after a traditional resurfacing procedure, said Dr. Avram, of the department of dermatology, Harvard Medical School, Boston, and the ASLMS president. But in some cases, he said, the law may expect the physician to step in to prevent harm. For example, he said, a patient who has undergone a fractional ablative laser procedure and has received a sedative should not be allowed to drive home.
• You may get sued even if your employee is at fault.
The 2013 study found physicians were often sued even when they did not perform the laser procedure in question. Nonphysicians such as physician assistants and nurses often perform laser operations, and many states allow them to do so. “Nonphysicians were less likely to be sued even if they were the operators,” Dr. Jalian said. In the study, almost 38% of the 174 analyzed cases involved nonphysician operators, but they were sued in just 26% of the cases. In 33 of the 174 cases in the study, plaintiffs alleged failure to properly hire, train, or supervise staff.
He recommended looking at state laws, which differ greatly in their regulations – or lack of them – regarding the operation of medical lasers. In some cases, physicians must supervise laser use, he said. “But what are the requirements? Can you be available by phone down the street or in the Caribbean?”
Dr. Jalian, Dr. Avram, and a colleague followed up the 2013 study with another study that tracked 175 legal cases from 1999 to 2012 involving alleged injuries from cutaneous laser surgery. During this time period, 75 (43%) involved a nonphysician operating a laser, increasing from 36% in 2008 to 78% in 2012.
In almost two-thirds of cases, the procedures in question were done by nonphysicians outside a “traditional medical setting” such as a salon or spa (JAMA Dermatol. 2014 Apr;150[4]:407-11).
• Delayed side effects could mean delayed lawsuits.
According to Dr. Avram, statutes of limitations – the length of time in which a patient can file a lawsuit – typically last for 2-3 years in malpractice cases. But he said that the period begins when the physician is alleged to have made a mistake or when the patient becomes aware of – or should reasonably be aware of – an injury. Therefore, physicians could face legal trouble over delayed hypopigmentation that appears 6 months after a laser resurfacing treatment, or granulomas that appear years after a filler treatment, he said.
• A signed form is not a cure-all.
It is wise to make patients sign an extensive informed consent form, but this will not protect a physician against a claim of negligence, Dr. Avram said. And the reverse is also true: If a patient did not sign a proper consent form, he or she could still sue even if the procedure went perfectly, he noted.
• Your instincts are worth trusting.
When it comes to lawsuit prevention, Dr. Avram said, “by far the most important thing you can do happens within a minute of when you see the patient. Assess and trust your own intuition and your staff’s intuition. For elective, cosmetic treatments, don’t be afraid to say no. There’s no legal obligation to perform a cosmetic treatment on a patient.”
If you do choose to treat a patient, he advised, be open about the procedure and “maybe even tell them some of the tougher, worse-case scenarios.” If a procedure goes poorly, he said, consider how to fix it. “Many complications can be significantly improved or cleared with timely and appropriate intervention,” he said.
In some cases, refunding the patient’s money can be considered, with the patient signing a release, he said. “Document that you are refunding the money in order to preserve the doctor-patient relationship, not to avoid negligence.”
Dr. Jalian and Dr. Avram reported no relevant disclosures.
AT LASER 2017
Biomarker predicts prolonged depression in breast cancer patients
SAN FRANCISCO – Nearly 40% of breast cancer patients experience prolonged depression lasting for at least 16 months after diagnosis of their malignancy; those at increased risk may be identifiable in a timely way by their exaggerated cortisol awakening response when measured after surgery but before adjuvant therapy, according to Kate R. Kuhlman, PhD.
“There are several psychological interventions that mitigate depressive symptoms and psychologic distress in women with breast cancer. This time period immediately following cancer diagnosis and surgery may be the optimal time to intervene,” said Dr. Kuhlman, a psychologist at the University of California, Los Angeles.

She presented a prospective study of 135 women with breast cancer who collected saliva samples for analysis of hypothalamic-pituitary-adrenal axis functioning on 3 consecutive days after their primary surgery but prior to starting adjuvant therapy. Samples were obtained on each of the 3 days upon awakening, 30 minutes later, 8 hours later, and at bedtime. The women also completed the Center for Epidemiologic Studies Depression Scale (CES-D) then and again 6 months after completing their breast cancer treatment.
At baseline, 45 of the 135 women scored 16 points or higher out of a possible 60 on the 20-question CES-D, indicative of clinically significant depression. Hypothalamic-pituitary-adrenal axis functioning wasn’t associated with depressive symptoms at that time. Importantly, however, one measure of baseline HPA axis functioning – the cortisol awakening response – proved to be associated with an increase in depressive symptoms over time, Dr. Kuhlman reported.
In a multivariate analysis adjusted for age, breast cancer stage, type of surgery, and forms of adjuvant therapy, a 1-standard-deviation increase above the mean in baseline cortisol awakening response was associated with a 6-point increase in CES-D score at follow-up 6 months after completion of breast cancer therapy. This association was seen only in the 90 women without significant depressive symptoms at baseline. And that’s exactly the population where a predictive biologic marker for future depression is most needed, Dr. Kuhlman said at the annual conference of the Anxiety and Depression Association of America.
“The people at highest risk of depressive symptoms in the future are the ones who have the most symptoms now. They’re easy to identify. We have good reliable measures. But then there are also people at risk whom we would miss by using those measures because they don’t have high symptoms right now,” the psychologist explained.
She and her coinvestigators zeroed in on cortisol awakening response as a potential biomarker of increased future risk of depression because it reflects the adrenal gland’s sensitivity to adrenocorticotropic hormone and the gland’s ability to signal the pituitary to produce cortisol. This action is triggered when people go from sleep to awakening.
The next steps in this research are to confirm these novel findings and hunt for an alternative marker of adrenal sensitivity to adrenocorticotropic hormone that’s simpler than sending a waking saliva sample off to a laboratory.
This ongoing longitudinal study is funded by the National Cancer Institute. Dr. Kuhlman reported having no relevant financial conflicts.
SAN FRANCISCO – Nearly 40% of breast cancer patients experience prolonged depression lasting for at least 16 months after diagnosis of their malignancy; those at increased risk may be identifiable in a timely way by their exaggerated cortisol awakening response when measured after surgery but before adjuvant therapy, according to Kate R. Kuhlman, PhD.
“There are several psychological interventions that mitigate depressive symptoms and psychologic distress in women with breast cancer. This time period immediately following cancer diagnosis and surgery may be the optimal time to intervene,” said Dr. Kuhlman, a psychologist at the University of California, Los Angeles.
She presented a prospective study of 135 women with breast cancer who collected saliva samples for analysis of hypothalamic-pituitary-adrenal axis functioning on 3 consecutive days after their primary surgery but prior to starting adjuvant therapy. Samples were obtained on each of the 3 days upon awakening, 30 minutes later, 8 hours later, and at bedtime. The women also completed the Center for Epidemiologic Studies Depression Scale (CES-D) then and again 6 months after completing their breast cancer treatment.
At baseline, 45 of the 135 women scored 16 points or higher out of a possible 60 on the 20-question CES-D, indicative of clinically significant depression. Hypothalamic-pituitary-adrenal axis functioning wasn’t associated with depressive symptoms at that time. Importantly, however, one measure of baseline HPA axis functioning – the cortisol awakening response – proved to be associated with an increase in depressive symptoms over time, Dr. Kuhlman reported.
In a multivariate analysis adjusted for age, breast cancer stage, type of surgery, and forms of adjuvant therapy, a 1-standard-deviation increase above the mean in baseline cortisol awakening response was associated with a 6-point increase in CES-D score at follow-up 6 months after completion of breast cancer therapy. This association was seen only in the 90 women without significant depressive symptoms at baseline. And that’s exactly the population where a predictive biologic marker for future depression is most needed, Dr. Kuhlman said at the annual conference of the Anxiety and Depression Association of America.
“The people at highest risk of depressive symptoms in the future are the ones who have the most symptoms now. They’re easy to identify. We have good reliable measures. But then there are also people at risk whom we would miss by using those measures because they don’t have high symptoms right now,” the psychologist explained.
She and her coinvestigators zeroed in on cortisol awakening response as a potential biomarker of increased future risk of depression because it reflects the adrenal gland’s sensitivity to adrenocorticotropic hormone and the gland’s ability to signal the pituitary to produce cortisol. This action is triggered when people go from sleep to awakening.
The next steps in this research are to confirm these novel findings and hunt for an alternative marker of adrenal sensitivity to adrenocorticotropic hormone that’s simpler than sending a waking saliva sample off to a laboratory.
This ongoing longitudinal study is funded by the National Cancer Institute. Dr. Kuhlman reported having no relevant financial conflicts.
SAN FRANCISCO – Nearly 40% of breast cancer patients experience prolonged depression lasting for at least 16 months after diagnosis of their malignancy; those at increased risk may be identifiable in a timely way by their exaggerated cortisol awakening response when measured after surgery but before adjuvant therapy, according to Kate R. Kuhlman, PhD.
“There are several psychological interventions that mitigate depressive symptoms and psychologic distress in women with breast cancer. This time period immediately following cancer diagnosis and surgery may be the optimal time to intervene,” said Dr. Kuhlman, a psychologist at the University of California, Los Angeles.
She presented a prospective study of 135 women with breast cancer who collected saliva samples for analysis of hypothalamic-pituitary-adrenal axis functioning on 3 consecutive days after their primary surgery but prior to starting adjuvant therapy. Samples were obtained on each of the 3 days upon awakening, 30 minutes later, 8 hours later, and at bedtime. The women also completed the Center for Epidemiologic Studies Depression Scale (CES-D) then and again 6 months after completing their breast cancer treatment.
At baseline, 45 of the 135 women scored 16 points or higher out of a possible 60 on the 20-question CES-D, indicative of clinically significant depression. Hypothalamic-pituitary-adrenal axis functioning wasn’t associated with depressive symptoms at that time. Importantly, however, one measure of baseline HPA axis functioning – the cortisol awakening response – proved to be associated with an increase in depressive symptoms over time, Dr. Kuhlman reported.
In a multivariate analysis adjusted for age, breast cancer stage, type of surgery, and forms of adjuvant therapy, a 1-standard-deviation increase above the mean in baseline cortisol awakening response was associated with a 6-point increase in CES-D score at follow-up 6 months after completion of breast cancer therapy. This association was seen only in the 90 women without significant depressive symptoms at baseline. And that’s exactly the population where a predictive biologic marker for future depression is most needed, Dr. Kuhlman said at the annual conference of the Anxiety and Depression Association of America.
“The people at highest risk of depressive symptoms in the future are the ones who have the most symptoms now. They’re easy to identify. We have good reliable measures. But then there are also people at risk whom we would miss by using those measures because they don’t have high symptoms right now,” the psychologist explained.
She and her coinvestigators zeroed in on cortisol awakening response as a potential biomarker of increased future risk of depression because it reflects the adrenal gland’s sensitivity to adrenocorticotropic hormone and the gland’s ability to signal the pituitary to produce cortisol. This action is triggered when people go from sleep to awakening.
The next steps in this research are to confirm these novel findings and hunt for an alternative marker of adrenal sensitivity to adrenocorticotropic hormone that’s simpler than sending a waking saliva sample off to a laboratory.
This ongoing longitudinal study is funded by the National Cancer Institute. Dr. Kuhlman reported having no relevant financial conflicts.
AT ANXIETY AND DEPRESSION CONFERENCE 2017
Key clinical point:
Major finding: Nondepressed breast cancer patients whose saliva samples show an exaggerated cortisol awakening response when measured after surgery but before adjuvant therapy are at increased risk for developing prolonged depression as treatment progresses.
Data source: A prospective longitudinal study of 135 women with breast cancer.
Disclosures: This ongoing study is funded by the National Cancer Institute. The presenter reported having no relevant financial conflicts.