User login
News Flash! Nocturnists are in high demand
Over 70% of all hospitalist programs have nocturnists, according to the 2016 State of Hospital Medicine Report. For adult-only practices, this has increased to 72.3% from 46.1% in the 2012 State of Hospital Medicine Report.
While one can assert that most hospital medicine practices have nocturnists, not all nights are covered by nocturnists. Thirty-nine percent (39%) of adult practices report that nocturnists cover 100% of nights, and 9.2% report that less than 25% of nights are covered by nocturnists. So, there remains a great deal of variability in the widespread use of nocturnists for nighttime coverage.

Categorically, nocturnists are hospitalists who work primarily at night, providing in-house coverage for hospitalist admissions and coverage for patients cared for by the hospitalist group.
Other clinicians, such as nurses, patient care technicians, medical technologists, and radiology technologists, have worked night shifts for many, many years. The phenomenon of hospital-based physicians and advanced practice clinicians working only at night is reflective of the needs in an acute care environment.
There are many lifestyle benefits to being a nocturnist – raising a family during the day while working at night, working fewer hours for more pay, and being in high demand.
Nocturnists can also allow a hospitalist group to offer more flexible scheduling options and create career longevity within the group. Having a nocturnist can allow a group to offer other hospitalists a “day shift only” option and other flexible scheduling options that many seasoned hospitalists are looking for.
Demand
Because of the increasing demand, it’s becoming more difficult to find long-term nocturnists, and therefore permanent nocturnists are expensive to hire. As reported in the 2016 State of Hospital Medicine Report, groups with nocturnists may offer either a differential in the hours or shifts worked, or compensation, or a combination of both.
About half of nocturnists work fewer shifts, compared with non-nocturnists. Equally stated, about half of nocturnists work the same number of shifts as their day-only counterparts. Of those groups whose nocturnists work fewer shifts than their daytime counterparts, about 60% work 1%-20% fewer shifts.
Nearly 70% of groups with nocturnists pay nocturnists differently. The median pay differential is 15%. While this compensation differential is an increase since the 2014 report, it is on par with the 2012 report.
It should not be construed that every practice with hospitalists offers both fewer shifts and more compensation. In fact, there are many who may offer neither and develop other, more creative ways of recognizing the nocturnist differently, such as evaluating scheduled hours per shift (e.g., 8 vs. 10 vs. 12).
Responsibility
With more adaptation and remuneration for nocturnists comes more responsibility.
Working as a nocturnist can be grueling work. Many times nocturnists may be working alone, and with less support from consultants and fewer hospital resources at night. On the other hand, it’s quieter at night and there can be a strong camaraderie from the smaller team at the hospital at night.
Nocturnists, many times, must be comfortable working alone; they must have strong clinical skills, and may need to seek extra training. In fact, in some hospitals the nocturnists may be the primary, or only, physician covering in-house codes.
Nocturnists must also take responsibility to remain abreast of the quality initiatives of the hospitalist group and hospital, since many of the quality committee meetings and hospitalist group meetings typically occur during daytime hours. Nocturnists may need to make an extra effort to feel a part of the group by voluntarily participating in daytime group activities, so that they don’t feel like an outsider.
Nocturnists should take the lead in receiving hand-off each evening, and handing off each morning to the day shift. This will likely mean handing off valuable patient care information with more than a few of their hospitalist colleagues. This is so immensely important that national patient safety-focused organizations have emphasized it for many years.
Since four out of five hospitalist programs have a hospitalist on site at night, and the majority of those programs have at least some nocturnist coverage, designing hospitalist programs and staffing models that meet the patient care need of 24/7 in-house coverage is a necessity. Also, given the strong demand for nocturnists, more and more program leaders are being challenged to evaluate creative alternatives to provide sustainable hospitalist services.
Some examples of creative solutions for in-house night coverage are implementing telemedicine for admissions, cross cover, or both; expanding coverage by advanced practice clinicians; and staggering shifts to cover late evenings and early mornings.
Perhaps we’ll see more questions about how hospitalist groups are addressing this need in future surveys?
Amanda Trask, MBA, MHA, FACHE, CMPE, SFHM, is national vice president, Hospital Medicine Service Line, at Catholic Health Initiatives, Englewood, Colo.
Over 70% of all hospitalist programs have nocturnists, according to the 2016 State of Hospital Medicine Report. For adult-only practices, this has increased to 72.3% from 46.1% in the 2012 State of Hospital Medicine Report.
While one can assert that most hospital medicine practices have nocturnists, not all nights are covered by nocturnists. Thirty-nine percent (39%) of adult practices report that nocturnists cover 100% of nights, and 9.2% report that less than 25% of nights are covered by nocturnists. So, there remains a great deal of variability in the widespread use of nocturnists for nighttime coverage.
Categorically, nocturnists are hospitalists who work primarily at night, providing in-house coverage for hospitalist admissions and coverage for patients cared for by the hospitalist group.
Other clinicians, such as nurses, patient care technicians, medical technologists, and radiology technologists, have worked night shifts for many, many years. The phenomenon of hospital-based physicians and advanced practice clinicians working only at night is reflective of the needs in an acute care environment.
There are many lifestyle benefits to being a nocturnist – raising a family during the day while working at night, working fewer hours for more pay, and being in high demand.
Nocturnists can also allow a hospitalist group to offer more flexible scheduling options and create career longevity within the group. Having a nocturnist can allow a group to offer other hospitalists a “day shift only” option and other flexible scheduling options that many seasoned hospitalists are looking for.
Demand
Because of the increasing demand, it’s becoming more difficult to find long-term nocturnists, and therefore permanent nocturnists are expensive to hire. As reported in the 2016 State of Hospital Medicine Report, groups with nocturnists may offer either a differential in the hours or shifts worked, or compensation, or a combination of both.
About half of nocturnists work fewer shifts, compared with non-nocturnists. Equally stated, about half of nocturnists work the same number of shifts as their day-only counterparts. Of those groups whose nocturnists work fewer shifts than their daytime counterparts, about 60% work 1%-20% fewer shifts.
Nearly 70% of groups with nocturnists pay nocturnists differently. The median pay differential is 15%. While this compensation differential is an increase since the 2014 report, it is on par with the 2012 report.
It should not be construed that every practice with hospitalists offers both fewer shifts and more compensation. In fact, there are many who may offer neither and develop other, more creative ways of recognizing the nocturnist differently, such as evaluating scheduled hours per shift (e.g., 8 vs. 10 vs. 12).
Responsibility
With more adaptation and remuneration for nocturnists comes more responsibility.
Working as a nocturnist can be grueling work. Many times nocturnists may be working alone, and with less support from consultants and fewer hospital resources at night. On the other hand, it’s quieter at night and there can be a strong camaraderie from the smaller team at the hospital at night.
Nocturnists, many times, must be comfortable working alone; they must have strong clinical skills, and may need to seek extra training. In fact, in some hospitals the nocturnists may be the primary, or only, physician covering in-house codes.
Nocturnists must also take responsibility to remain abreast of the quality initiatives of the hospitalist group and hospital, since many of the quality committee meetings and hospitalist group meetings typically occur during daytime hours. Nocturnists may need to make an extra effort to feel a part of the group by voluntarily participating in daytime group activities, so that they don’t feel like an outsider.
Nocturnists should take the lead in receiving hand-off each evening, and handing off each morning to the day shift. This will likely mean handing off valuable patient care information with more than a few of their hospitalist colleagues. This is so immensely important that national patient safety-focused organizations have emphasized it for many years.
Since four out of five hospitalist programs have a hospitalist on site at night, and the majority of those programs have at least some nocturnist coverage, designing hospitalist programs and staffing models that meet the patient care need of 24/7 in-house coverage is a necessity. Also, given the strong demand for nocturnists, more and more program leaders are being challenged to evaluate creative alternatives to provide sustainable hospitalist services.
Some examples of creative solutions for in-house night coverage are implementing telemedicine for admissions, cross cover, or both; expanding coverage by advanced practice clinicians; and staggering shifts to cover late evenings and early mornings.
Perhaps we’ll see more questions about how hospitalist groups are addressing this need in future surveys?
Amanda Trask, MBA, MHA, FACHE, CMPE, SFHM, is national vice president, Hospital Medicine Service Line, at Catholic Health Initiatives, Englewood, Colo.
Over 70% of all hospitalist programs have nocturnists, according to the 2016 State of Hospital Medicine Report. For adult-only practices, this has increased to 72.3% from 46.1% in the 2012 State of Hospital Medicine Report.
While one can assert that most hospital medicine practices have nocturnists, not all nights are covered by nocturnists. Thirty-nine percent (39%) of adult practices report that nocturnists cover 100% of nights, and 9.2% report that less than 25% of nights are covered by nocturnists. So, there remains a great deal of variability in the widespread use of nocturnists for nighttime coverage.
Categorically, nocturnists are hospitalists who work primarily at night, providing in-house coverage for hospitalist admissions and coverage for patients cared for by the hospitalist group.
Other clinicians, such as nurses, patient care technicians, medical technologists, and radiology technologists, have worked night shifts for many, many years. The phenomenon of hospital-based physicians and advanced practice clinicians working only at night is reflective of the needs in an acute care environment.
There are many lifestyle benefits to being a nocturnist – raising a family during the day while working at night, working fewer hours for more pay, and being in high demand.
Nocturnists can also allow a hospitalist group to offer more flexible scheduling options and create career longevity within the group. Having a nocturnist can allow a group to offer other hospitalists a “day shift only” option and other flexible scheduling options that many seasoned hospitalists are looking for.
Demand
Because of the increasing demand, it’s becoming more difficult to find long-term nocturnists, and therefore permanent nocturnists are expensive to hire. As reported in the 2016 State of Hospital Medicine Report, groups with nocturnists may offer either a differential in the hours or shifts worked, or compensation, or a combination of both.
About half of nocturnists work fewer shifts, compared with non-nocturnists. Equally stated, about half of nocturnists work the same number of shifts as their day-only counterparts. Of those groups whose nocturnists work fewer shifts than their daytime counterparts, about 60% work 1%-20% fewer shifts.
Nearly 70% of groups with nocturnists pay nocturnists differently. The median pay differential is 15%. While this compensation differential is an increase since the 2014 report, it is on par with the 2012 report.
It should not be construed that every practice with hospitalists offers both fewer shifts and more compensation. In fact, there are many who may offer neither and develop other, more creative ways of recognizing the nocturnist differently, such as evaluating scheduled hours per shift (e.g., 8 vs. 10 vs. 12).
Responsibility
With more adaptation and remuneration for nocturnists comes more responsibility.
Working as a nocturnist can be grueling work. Many times nocturnists may be working alone, and with less support from consultants and fewer hospital resources at night. On the other hand, it’s quieter at night and there can be a strong camaraderie from the smaller team at the hospital at night.
Nocturnists, many times, must be comfortable working alone; they must have strong clinical skills, and may need to seek extra training. In fact, in some hospitals the nocturnists may be the primary, or only, physician covering in-house codes.
Nocturnists must also take responsibility to remain abreast of the quality initiatives of the hospitalist group and hospital, since many of the quality committee meetings and hospitalist group meetings typically occur during daytime hours. Nocturnists may need to make an extra effort to feel a part of the group by voluntarily participating in daytime group activities, so that they don’t feel like an outsider.
Nocturnists should take the lead in receiving hand-off each evening, and handing off each morning to the day shift. This will likely mean handing off valuable patient care information with more than a few of their hospitalist colleagues. This is so immensely important that national patient safety-focused organizations have emphasized it for many years.
Since four out of five hospitalist programs have a hospitalist on site at night, and the majority of those programs have at least some nocturnist coverage, designing hospitalist programs and staffing models that meet the patient care need of 24/7 in-house coverage is a necessity. Also, given the strong demand for nocturnists, more and more program leaders are being challenged to evaluate creative alternatives to provide sustainable hospitalist services.
Some examples of creative solutions for in-house night coverage are implementing telemedicine for admissions, cross cover, or both; expanding coverage by advanced practice clinicians; and staggering shifts to cover late evenings and early mornings.
Perhaps we’ll see more questions about how hospitalist groups are addressing this need in future surveys?
Amanda Trask, MBA, MHA, FACHE, CMPE, SFHM, is national vice president, Hospital Medicine Service Line, at Catholic Health Initiatives, Englewood, Colo.
Diagnosing high-risk keratinocyte carcinomas in the dermatology clinic
SAN FRANCISCO – Patients with high-risk keratinocyte carcinomas sometimes present with neurologic symptoms mimicking Bell’s palsy or trigeminal neuralgia, making the diagnosis of these perineural tumors challenging, Siegrid Yu, MD, said at the annual meeting of the Pacific Dermatologic Association.
Eventually, skin manifestations can land them in a dermatologist’s office. “There is a high incidence of delayed diagnosis and misdiagnosis, which affects the outcome of these patients,” said Dr. Yu of the department of dermatology, University of California, San Francisco.
She presented several cases illustrating the central role that dermatologists can play in the diagnosis and management of high-risk keratinocyte carcinomas. “All of these patients were seen by various doctors, sometimes multiple times, without a diagnosis,” she said.
Perineural invasion occurs in 2.6%-6% of squamous cell carcinoma (SCC) cases and 2% of basal cell carcinoma (BCC) cases. “Perineural invasion presenting with neurologic symptoms is not that common, which is part of why I think it’s easy to misdiagnose these patients,” said Dr. Yu, director of the Mohs Micrographic Surgery and Cutaneous Oncology Fellowship at the UCSF Dermatologic Surgery and Laser Center. In many cases, patients were diagnosed as having Bell’s palsy or trigeminal neuralgia for years before being diagnosed with skin cancer.

Common features of perineural invasion cases include midface location of the tumor, male gender, tumor size larger than 2 cm, recurrence, and poor histologic differentiation. Symptoms often include formication, pain, numbness, and facial weakness. Diagnosis is often delayed by 6 months to 2 years.
One case she described involved a 57-year-old immunosuppressed man who had previously undergone Mohs micrographic surgery for a primary SCC of the nasal sidewall. He experienced delayed numbness and pain of the upper lip and cheek near the surgical site 1 year later. There was no sign of cutaneous recurrence, and MRIs of the head and neck were normal. Examinations by dermatologists, neurologists, and otorhinolaryngologists yielded no diagnosis.
Two years after his initial surgery, the patient developed thickening of the scar from the Mohs surgery, without any overlying skin change. A punch biopsy showed only scar tissue, but a deeper incisional biopsy revealed a recurrence of the SCC. A second head/neck MRI, using a perineural protocol, showed abnormal enhancement at the V2 branch of the trigeminal nerve leading to the foramen rotundum. The patient underwent intensity-modulated radiation, which relies on computer-modeling to deliver doses to the precise location of the tumor. An MRI 2 months later showed a reduction in tumor size and radiographic resolution of trigeminal nerve involvement.
Another case involved a 75-year-old man with progressive right facial droop, who had experienced neurologic symptoms on the right side of his face, including numbness, tingling, oculomotor dysfunction, and radiating pain. He had been diagnosed with shingles on the right side of his face more than 20 years previously, but there was no history of postherpetic neuralgia. He also had hypertension and hypothyroidism, and had been prescribed levothyroxine, amlodipine, losartan, and gabapentin.
He had been evaluated by primary care, dermatology, and ophthalmology with no diagnosis. He then sequentially sought the opinion of four neurologists, and underwent lumbar puncture, serologic evaluation, head CT, and MRI with no findings that correlated with his symptoms. The patient’s neurological symptoms improved transiently with prednisone, and his pain improved slightly with gabapentin.
Finally, a skin biopsy of an ill-defined firmness in the right temple revealed infiltrative SCC. A repeat MRI, this time with perineural protocol, showed perineural spread along the trigeminal nerve, with involvement of the V2 and V3 branches, and possibly the V1 branch.
In another case, complete hemifacial palsy due to perineural spread of SCC was overlooked as having been related to the patient’s history of stroke. However, upon further questioning, the facial palsy involved all branches of the facial nerve, while the patient’s residual stroke symptoms of expressive aphasia and dysphagia were improving. “If you think about head and neck anatomy, an upper motor neuron lesion would not lead to complete facial nerve palsy. It could lead to palsy of the lower two-thirds of the face, sparing the temporal nerve due to cross innervation of the forehead. Only a lower motor neuron can result in progressive palsy of all branches of the facial nerve,” Dr. Yu said. In this case, the facial palsy was due to a large SCC of the external auditory canal.
Dr. Yu highlighted several considerations to keep in mind when examining these patients, including vigilance around prior skin cancer surgeries in cases with neurologic symptoms, the potential need for repeated imaging along with communication with the radiologist regarding suspicion of perineural spread, consideration of anatomy during the clinical exam, and correlation of clinical exam, histopathology, and radiographic findings.
When it comes to imaging, MRI is the most sensitive technique, she noted. It can show increase in nerve diameter, destruction of the nerve-blood barrier, obliteration of the fat below a foramen, nerve enhancement, and denervation atrophy.
Dr. Yu reported having no financial disclosures.
SAN FRANCISCO – Patients with high-risk keratinocyte carcinomas sometimes present with neurologic symptoms mimicking Bell’s palsy or trigeminal neuralgia, making the diagnosis of these perineural tumors challenging, Siegrid Yu, MD, said at the annual meeting of the Pacific Dermatologic Association.
Eventually, skin manifestations can land them in a dermatologist’s office. “There is a high incidence of delayed diagnosis and misdiagnosis, which affects the outcome of these patients,” said Dr. Yu of the department of dermatology, University of California, San Francisco.
She presented several cases illustrating the central role that dermatologists can play in the diagnosis and management of high-risk keratinocyte carcinomas. “All of these patients were seen by various doctors, sometimes multiple times, without a diagnosis,” she said.
Perineural invasion occurs in 2.6%-6% of squamous cell carcinoma (SCC) cases and 2% of basal cell carcinoma (BCC) cases. “Perineural invasion presenting with neurologic symptoms is not that common, which is part of why I think it’s easy to misdiagnose these patients,” said Dr. Yu, director of the Mohs Micrographic Surgery and Cutaneous Oncology Fellowship at the UCSF Dermatologic Surgery and Laser Center. In many cases, patients were diagnosed as having Bell’s palsy or trigeminal neuralgia for years before being diagnosed with skin cancer.
Common features of perineural invasion cases include midface location of the tumor, male gender, tumor size larger than 2 cm, recurrence, and poor histologic differentiation. Symptoms often include formication, pain, numbness, and facial weakness. Diagnosis is often delayed by 6 months to 2 years.
One case she described involved a 57-year-old immunosuppressed man who had previously undergone Mohs micrographic surgery for a primary SCC of the nasal sidewall. He experienced delayed numbness and pain of the upper lip and cheek near the surgical site 1 year later. There was no sign of cutaneous recurrence, and MRIs of the head and neck were normal. Examinations by dermatologists, neurologists, and otorhinolaryngologists yielded no diagnosis.
Two years after his initial surgery, the patient developed thickening of the scar from the Mohs surgery, without any overlying skin change. A punch biopsy showed only scar tissue, but a deeper incisional biopsy revealed a recurrence of the SCC. A second head/neck MRI, using a perineural protocol, showed abnormal enhancement at the V2 branch of the trigeminal nerve leading to the foramen rotundum. The patient underwent intensity-modulated radiation, which relies on computer-modeling to deliver doses to the precise location of the tumor. An MRI 2 months later showed a reduction in tumor size and radiographic resolution of trigeminal nerve involvement.
Another case involved a 75-year-old man with progressive right facial droop, who had experienced neurologic symptoms on the right side of his face, including numbness, tingling, oculomotor dysfunction, and radiating pain. He had been diagnosed with shingles on the right side of his face more than 20 years previously, but there was no history of postherpetic neuralgia. He also had hypertension and hypothyroidism, and had been prescribed levothyroxine, amlodipine, losartan, and gabapentin.
He had been evaluated by primary care, dermatology, and ophthalmology with no diagnosis. He then sequentially sought the opinion of four neurologists, and underwent lumbar puncture, serologic evaluation, head CT, and MRI with no findings that correlated with his symptoms. The patient’s neurological symptoms improved transiently with prednisone, and his pain improved slightly with gabapentin.
Finally, a skin biopsy of an ill-defined firmness in the right temple revealed infiltrative SCC. A repeat MRI, this time with perineural protocol, showed perineural spread along the trigeminal nerve, with involvement of the V2 and V3 branches, and possibly the V1 branch.
In another case, complete hemifacial palsy due to perineural spread of SCC was overlooked as having been related to the patient’s history of stroke. However, upon further questioning, the facial palsy involved all branches of the facial nerve, while the patient’s residual stroke symptoms of expressive aphasia and dysphagia were improving. “If you think about head and neck anatomy, an upper motor neuron lesion would not lead to complete facial nerve palsy. It could lead to palsy of the lower two-thirds of the face, sparing the temporal nerve due to cross innervation of the forehead. Only a lower motor neuron can result in progressive palsy of all branches of the facial nerve,” Dr. Yu said. In this case, the facial palsy was due to a large SCC of the external auditory canal.
Dr. Yu highlighted several considerations to keep in mind when examining these patients, including vigilance around prior skin cancer surgeries in cases with neurologic symptoms, the potential need for repeated imaging along with communication with the radiologist regarding suspicion of perineural spread, consideration of anatomy during the clinical exam, and correlation of clinical exam, histopathology, and radiographic findings.
When it comes to imaging, MRI is the most sensitive technique, she noted. It can show increase in nerve diameter, destruction of the nerve-blood barrier, obliteration of the fat below a foramen, nerve enhancement, and denervation atrophy.
Dr. Yu reported having no financial disclosures.
SAN FRANCISCO – Patients with high-risk keratinocyte carcinomas sometimes present with neurologic symptoms mimicking Bell’s palsy or trigeminal neuralgia, making the diagnosis of these perineural tumors challenging, Siegrid Yu, MD, said at the annual meeting of the Pacific Dermatologic Association.
Eventually, skin manifestations can land them in a dermatologist’s office. “There is a high incidence of delayed diagnosis and misdiagnosis, which affects the outcome of these patients,” said Dr. Yu of the department of dermatology, University of California, San Francisco.
She presented several cases illustrating the central role that dermatologists can play in the diagnosis and management of high-risk keratinocyte carcinomas. “All of these patients were seen by various doctors, sometimes multiple times, without a diagnosis,” she said.
Perineural invasion occurs in 2.6%-6% of squamous cell carcinoma (SCC) cases and 2% of basal cell carcinoma (BCC) cases. “Perineural invasion presenting with neurologic symptoms is not that common, which is part of why I think it’s easy to misdiagnose these patients,” said Dr. Yu, director of the Mohs Micrographic Surgery and Cutaneous Oncology Fellowship at the UCSF Dermatologic Surgery and Laser Center. In many cases, patients were diagnosed as having Bell’s palsy or trigeminal neuralgia for years before being diagnosed with skin cancer.
Common features of perineural invasion cases include midface location of the tumor, male gender, tumor size larger than 2 cm, recurrence, and poor histologic differentiation. Symptoms often include formication, pain, numbness, and facial weakness. Diagnosis is often delayed by 6 months to 2 years.
One case she described involved a 57-year-old immunosuppressed man who had previously undergone Mohs micrographic surgery for a primary SCC of the nasal sidewall. He experienced delayed numbness and pain of the upper lip and cheek near the surgical site 1 year later. There was no sign of cutaneous recurrence, and MRIs of the head and neck were normal. Examinations by dermatologists, neurologists, and otorhinolaryngologists yielded no diagnosis.
Two years after his initial surgery, the patient developed thickening of the scar from the Mohs surgery, without any overlying skin change. A punch biopsy showed only scar tissue, but a deeper incisional biopsy revealed a recurrence of the SCC. A second head/neck MRI, using a perineural protocol, showed abnormal enhancement at the V2 branch of the trigeminal nerve leading to the foramen rotundum. The patient underwent intensity-modulated radiation, which relies on computer-modeling to deliver doses to the precise location of the tumor. An MRI 2 months later showed a reduction in tumor size and radiographic resolution of trigeminal nerve involvement.
Another case involved a 75-year-old man with progressive right facial droop, who had experienced neurologic symptoms on the right side of his face, including numbness, tingling, oculomotor dysfunction, and radiating pain. He had been diagnosed with shingles on the right side of his face more than 20 years previously, but there was no history of postherpetic neuralgia. He also had hypertension and hypothyroidism, and had been prescribed levothyroxine, amlodipine, losartan, and gabapentin.
He had been evaluated by primary care, dermatology, and ophthalmology with no diagnosis. He then sequentially sought the opinion of four neurologists, and underwent lumbar puncture, serologic evaluation, head CT, and MRI with no findings that correlated with his symptoms. The patient’s neurological symptoms improved transiently with prednisone, and his pain improved slightly with gabapentin.
Finally, a skin biopsy of an ill-defined firmness in the right temple revealed infiltrative SCC. A repeat MRI, this time with perineural protocol, showed perineural spread along the trigeminal nerve, with involvement of the V2 and V3 branches, and possibly the V1 branch.
In another case, complete hemifacial palsy due to perineural spread of SCC was overlooked as having been related to the patient’s history of stroke. However, upon further questioning, the facial palsy involved all branches of the facial nerve, while the patient’s residual stroke symptoms of expressive aphasia and dysphagia were improving. “If you think about head and neck anatomy, an upper motor neuron lesion would not lead to complete facial nerve palsy. It could lead to palsy of the lower two-thirds of the face, sparing the temporal nerve due to cross innervation of the forehead. Only a lower motor neuron can result in progressive palsy of all branches of the facial nerve,” Dr. Yu said. In this case, the facial palsy was due to a large SCC of the external auditory canal.
Dr. Yu highlighted several considerations to keep in mind when examining these patients, including vigilance around prior skin cancer surgeries in cases with neurologic symptoms, the potential need for repeated imaging along with communication with the radiologist regarding suspicion of perineural spread, consideration of anatomy during the clinical exam, and correlation of clinical exam, histopathology, and radiographic findings.
When it comes to imaging, MRI is the most sensitive technique, she noted. It can show increase in nerve diameter, destruction of the nerve-blood barrier, obliteration of the fat below a foramen, nerve enhancement, and denervation atrophy.
Dr. Yu reported having no financial disclosures.
AT PDA 2017
Submit VAM Session Topic Proposals
SVS is seeking proposals for invited sessions from internal committees and members alike for the 2018 Vascular Annual Meeting, June 20-23 (exhibits: June 21 to 22; plenaries: June 21 to 23) in Boston, Mass.
Invited sessions consist of postgraduate courses, breakfast sessions, concurrent sessions and workshops/small-group sessions. Submitters will be asked to address educational needs, provide objectives, indicate proposed formats and identify target audiences.
The deadline is 3 p.m. Central Daylight Time, Friday, Sept. 15. Submitters will be notified the week of Sept. 25 if their proposals have been selected for further development. Contact [email protected] or call 312-334-2327 with questions.
SVS is seeking proposals for invited sessions from internal committees and members alike for the 2018 Vascular Annual Meeting, June 20-23 (exhibits: June 21 to 22; plenaries: June 21 to 23) in Boston, Mass.
Invited sessions consist of postgraduate courses, breakfast sessions, concurrent sessions and workshops/small-group sessions. Submitters will be asked to address educational needs, provide objectives, indicate proposed formats and identify target audiences.
The deadline is 3 p.m. Central Daylight Time, Friday, Sept. 15. Submitters will be notified the week of Sept. 25 if their proposals have been selected for further development. Contact [email protected] or call 312-334-2327 with questions.
SVS is seeking proposals for invited sessions from internal committees and members alike for the 2018 Vascular Annual Meeting, June 20-23 (exhibits: June 21 to 22; plenaries: June 21 to 23) in Boston, Mass.
Invited sessions consist of postgraduate courses, breakfast sessions, concurrent sessions and workshops/small-group sessions. Submitters will be asked to address educational needs, provide objectives, indicate proposed formats and identify target audiences.
The deadline is 3 p.m. Central Daylight Time, Friday, Sept. 15. Submitters will be notified the week of Sept. 25 if their proposals have been selected for further development. Contact [email protected] or call 312-334-2327 with questions.
PAD Resources for SVS Members
September is Peripheral Artery Disease Awareness Month. To help SVS members educate patients and to spread awareness about vascular surgeons, we have prepared several things you can share.
1. An infographic for patients and their families. We urge you to print it and post around the office or your institution.
2. A quick resource web page for patients, offering patients a PAD video playlist and links to articles and information on PAD.
3. The latest PAD research information for physicians, along with clinical practice guideline links. If you have contacts among primary care physicians or other referrers, please feel free to send them this link.
4. Two press releases on PAD, to share with your communications people, public relations departments and/or patients
September is Peripheral Artery Disease Awareness Month. To help SVS members educate patients and to spread awareness about vascular surgeons, we have prepared several things you can share.
1. An infographic for patients and their families. We urge you to print it and post around the office or your institution.
2. A quick resource web page for patients, offering patients a PAD video playlist and links to articles and information on PAD.
3. The latest PAD research information for physicians, along with clinical practice guideline links. If you have contacts among primary care physicians or other referrers, please feel free to send them this link.
4. Two press releases on PAD, to share with your communications people, public relations departments and/or patients
September is Peripheral Artery Disease Awareness Month. To help SVS members educate patients and to spread awareness about vascular surgeons, we have prepared several things you can share.
1. An infographic for patients and their families. We urge you to print it and post around the office or your institution.
2. A quick resource web page for patients, offering patients a PAD video playlist and links to articles and information on PAD.
3. The latest PAD research information for physicians, along with clinical practice guideline links. If you have contacts among primary care physicians or other referrers, please feel free to send them this link.
4. Two press releases on PAD, to share with your communications people, public relations departments and/or patients
Three percent of high school seniors report using synthetic cannabinoids
, according to data from the national Monitoring the Future survey.
SCs are more potent than marijuana (as much as 100 times stronger); do not contain the anti-anxiety and antipsychotic constituent of marijuana, cannabidiol; and have a greater risk of a range of adverse effects, such as tachycardia, agitation, nausea, generalized tonic-clonic seizures, psychiatric problems, and death.
Joseph J. Palamar, PhD, MPH, of New York University, and his colleagues set out to examine data about current use of SCs, as previous studies had focused on lifetime or past-year use. Monitoring the Future surveys approximately 15,000 high school seniors annually in the United States. Of the six survey forms (which are distributed randomly), only two asked seniors about their current use of SCs, limiting the current study to a third of the survey sample.
The researchers found that, while 81% of current SC users also reported current use of marijuana, only 9% of current marijuana users reported current use of SCs. SC use was correlated much more highly than marijuana use with the current use of other drugs such as LSD (15% vs. 3%), opioids (14% vs. 6%), cocaine (11% vs. 3%), and heroin (6% vs. 0.1%).
“Current SC use appears to be part of a more extensive polydrug use repertoire involving other illegal drugs that are less prevalent among marijuana-only users. ... These associations suggest the need to target marijuana users who also use other drugs to help prevent initiation of SCs,” concluded Dr. Palamar and his associates.
Read more at (Pediatrics. 2017 Oct;140[4]:e20171330).
, according to data from the national Monitoring the Future survey.
SCs are more potent than marijuana (as much as 100 times stronger); do not contain the anti-anxiety and antipsychotic constituent of marijuana, cannabidiol; and have a greater risk of a range of adverse effects, such as tachycardia, agitation, nausea, generalized tonic-clonic seizures, psychiatric problems, and death.
Joseph J. Palamar, PhD, MPH, of New York University, and his colleagues set out to examine data about current use of SCs, as previous studies had focused on lifetime or past-year use. Monitoring the Future surveys approximately 15,000 high school seniors annually in the United States. Of the six survey forms (which are distributed randomly), only two asked seniors about their current use of SCs, limiting the current study to a third of the survey sample.
The researchers found that, while 81% of current SC users also reported current use of marijuana, only 9% of current marijuana users reported current use of SCs. SC use was correlated much more highly than marijuana use with the current use of other drugs such as LSD (15% vs. 3%), opioids (14% vs. 6%), cocaine (11% vs. 3%), and heroin (6% vs. 0.1%).
“Current SC use appears to be part of a more extensive polydrug use repertoire involving other illegal drugs that are less prevalent among marijuana-only users. ... These associations suggest the need to target marijuana users who also use other drugs to help prevent initiation of SCs,” concluded Dr. Palamar and his associates.
Read more at (Pediatrics. 2017 Oct;140[4]:e20171330).
, according to data from the national Monitoring the Future survey.
SCs are more potent than marijuana (as much as 100 times stronger); do not contain the anti-anxiety and antipsychotic constituent of marijuana, cannabidiol; and have a greater risk of a range of adverse effects, such as tachycardia, agitation, nausea, generalized tonic-clonic seizures, psychiatric problems, and death.
Joseph J. Palamar, PhD, MPH, of New York University, and his colleagues set out to examine data about current use of SCs, as previous studies had focused on lifetime or past-year use. Monitoring the Future surveys approximately 15,000 high school seniors annually in the United States. Of the six survey forms (which are distributed randomly), only two asked seniors about their current use of SCs, limiting the current study to a third of the survey sample.
The researchers found that, while 81% of current SC users also reported current use of marijuana, only 9% of current marijuana users reported current use of SCs. SC use was correlated much more highly than marijuana use with the current use of other drugs such as LSD (15% vs. 3%), opioids (14% vs. 6%), cocaine (11% vs. 3%), and heroin (6% vs. 0.1%).
“Current SC use appears to be part of a more extensive polydrug use repertoire involving other illegal drugs that are less prevalent among marijuana-only users. ... These associations suggest the need to target marijuana users who also use other drugs to help prevent initiation of SCs,” concluded Dr. Palamar and his associates.
Read more at (Pediatrics. 2017 Oct;140[4]:e20171330).
FROM PEDIATRICS
Reduced fracture risk maintained in abaloparatide extension study
denver – Sequential treatment with abaloparatide and alendronate was associated with reduced vertebral and non-vertebral fractures compared to placebo and alendronate among high-risk women with osteoporosis in a 3.5 year extension study of the ACTIVE trial.
The ACTIVExtend trial included 558 women in the abaloparatide group and 581 women in the placebo group of the original ACTIVE study (NCT01343004). In that double-blind trial, 2,463 postmenopausal women with osteoporosis were randomized to receive daily injections of abaloparatide (80 µg) or placebo, or open-label teriparatide (20 µg). After 18 months of treatment, patients in the placebo and abaloparatide groups were switched to alendronate 70 mg weekly for two years, Henry Bone, MD, director of the Michigan Bone and Mineral Clinic in Detroit, said at the annual meeting of the American Society for Bone and Mineral Research.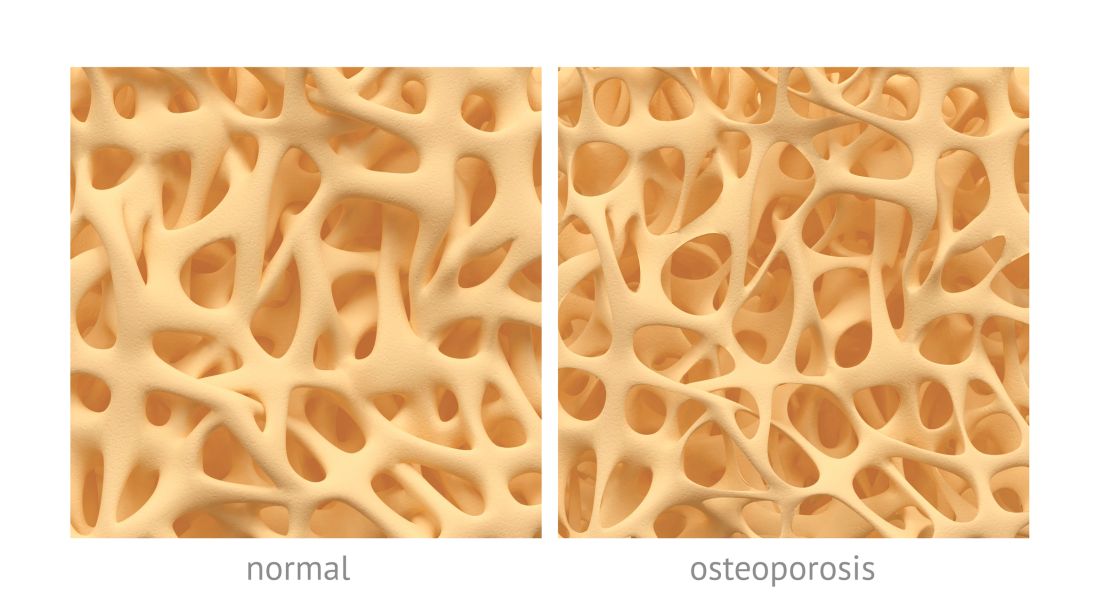
The abaloparatide-alendronate group had a 34% relative risk reduction for all clinical fractures compared to the placebo-alendronate group (P = .045). For major osteoporotic fractures, the abaloparatide-alendronate group had a 50% relative risk reduction compared to the placebo-alendronate group (P = .011).
Among women who had no new vertebral fractures during the first 18 months of the ACTIVE study, 2 women in the abaloparatide-alendronate group and 13 in the placebo-alendronate group had new vertebral fractures during ACTIVExtend.
Adverse events were similar in both arms of the study. There were no cases of atypical femur fracture or osteonecrosis of the jaw.The anabolic agent abaloparatide was approved by the Food and Drug Administration in April for the treatment of osteoporosis in women at high risk of fracture. Sequential treatment with the anti-resorptive agent alendronate aims to preserve the bone density gains from abaloparatide, as previous research has shown that improvements with anabolic agents can be lost once the drug is stopped.
denver – Sequential treatment with abaloparatide and alendronate was associated with reduced vertebral and non-vertebral fractures compared to placebo and alendronate among high-risk women with osteoporosis in a 3.5 year extension study of the ACTIVE trial.
The ACTIVExtend trial included 558 women in the abaloparatide group and 581 women in the placebo group of the original ACTIVE study (NCT01343004). In that double-blind trial, 2,463 postmenopausal women with osteoporosis were randomized to receive daily injections of abaloparatide (80 µg) or placebo, or open-label teriparatide (20 µg). After 18 months of treatment, patients in the placebo and abaloparatide groups were switched to alendronate 70 mg weekly for two years, Henry Bone, MD, director of the Michigan Bone and Mineral Clinic in Detroit, said at the annual meeting of the American Society for Bone and Mineral Research.
The abaloparatide-alendronate group had a 34% relative risk reduction for all clinical fractures compared to the placebo-alendronate group (P = .045). For major osteoporotic fractures, the abaloparatide-alendronate group had a 50% relative risk reduction compared to the placebo-alendronate group (P = .011).
Among women who had no new vertebral fractures during the first 18 months of the ACTIVE study, 2 women in the abaloparatide-alendronate group and 13 in the placebo-alendronate group had new vertebral fractures during ACTIVExtend.
Adverse events were similar in both arms of the study. There were no cases of atypical femur fracture or osteonecrosis of the jaw.The anabolic agent abaloparatide was approved by the Food and Drug Administration in April for the treatment of osteoporosis in women at high risk of fracture. Sequential treatment with the anti-resorptive agent alendronate aims to preserve the bone density gains from abaloparatide, as previous research has shown that improvements with anabolic agents can be lost once the drug is stopped.
denver – Sequential treatment with abaloparatide and alendronate was associated with reduced vertebral and non-vertebral fractures compared to placebo and alendronate among high-risk women with osteoporosis in a 3.5 year extension study of the ACTIVE trial.
The ACTIVExtend trial included 558 women in the abaloparatide group and 581 women in the placebo group of the original ACTIVE study (NCT01343004). In that double-blind trial, 2,463 postmenopausal women with osteoporosis were randomized to receive daily injections of abaloparatide (80 µg) or placebo, or open-label teriparatide (20 µg). After 18 months of treatment, patients in the placebo and abaloparatide groups were switched to alendronate 70 mg weekly for two years, Henry Bone, MD, director of the Michigan Bone and Mineral Clinic in Detroit, said at the annual meeting of the American Society for Bone and Mineral Research.
The abaloparatide-alendronate group had a 34% relative risk reduction for all clinical fractures compared to the placebo-alendronate group (P = .045). For major osteoporotic fractures, the abaloparatide-alendronate group had a 50% relative risk reduction compared to the placebo-alendronate group (P = .011).
Among women who had no new vertebral fractures during the first 18 months of the ACTIVE study, 2 women in the abaloparatide-alendronate group and 13 in the placebo-alendronate group had new vertebral fractures during ACTIVExtend.
Adverse events were similar in both arms of the study. There were no cases of atypical femur fracture or osteonecrosis of the jaw.The anabolic agent abaloparatide was approved by the Food and Drug Administration in April for the treatment of osteoporosis in women at high risk of fracture. Sequential treatment with the anti-resorptive agent alendronate aims to preserve the bone density gains from abaloparatide, as previous research has shown that improvements with anabolic agents can be lost once the drug is stopped.
REPORTING FROM ASBMR 2017
Key clinical point: The reduced fracture risk seen after 18 months of abaloparatide therapy persisted at 43 months with follow up alendronate therapy, and was superior to the results seen in women who received placebo for 18 months followed by alendronate.
Major finding: 0.9% of women who started on abaloparatide experienced at least 1 new vertebral fracture, compared to 5.6% of those who started on placebo.
Data source: The ACTIVExtend trial included 558 women in the abaloparatide group and 581 women in the placebo group.
Disclosures: The study was funded by Radius Health, the maker of abaloparatide. Dr. Bone is a consultant and investigator for Radius Health and Amgen.
Osimertinib bests PFS achieved with standard care for EGFR-mutated NSCLC
MADRID – The EGFR inhibitor osimertinib (Tagrisso) is being hailed as a new standard of care for first-line therapy of advanced, treatment naïve non-small cell lung cancer (NSCLC) with mutations in the epidermal growth factor receptor (EGFR).
Osimertinib cut the risk of disease progression in these patients by 54% compared gefitinib (Iressa) or erlotinib (Tarceva).
Among 279 patients with EGFR-mutated locally advanced or metastatic NSCLC treated with osemirtinib, the median progression-free survival (PFS) was 18.9 months, compared with 10.2 months for 277 patients treated with the standard of care, which translates into a hazard ratio (HR) of 0.46 (P less than .0001).
“We wanted to see if by shutting down a major escape pathway by giving osemirtinib upfront, whether we would be able to improve patient outcomes,” he said at a briefing at the European Society of Medical Oncology (ESMO) Congress.
Osimertinib is a third-generation EGFR tyrosine kinase inhibitor (TKI) selective for EGFR mutations, including the T790M resistance mutation. It is currently approved in the US for the treatment of patients with EGFR T790M mutation-positive NSCLC whose disease has progressed on or after EGFR TKI therapy.
In the FLAURA trial (NCT02296125) investigators stratified patients with previously untreated NSCLC positive for EGFR resistance mutations according to mutation status (exon 19 deletion or the L858R amino acid substitution in exon 21) and race (Asian or non-Asian).
Patients were randomly assigned to treatment with either oral osimertinib 80 mg daily or an EGFR TKI, either oral gefitinib 250 mg or erlotinib 150 mg daily.
The patients were assessed by Response Evaluation Criteria in Solid Tumors (RECIST) every 6 weeks until objective disease progression.
Patients assigned to the standard-of-care arm who had central confirmation of progression and T790M positivity were allowed to cross over to open-label osimertinib.
PFS, the primary endpoint, was also significantly better with osimertinib than with either of the comparator TKIs in patients with and without central nervous system metastases at study entry (HR 0.47, P = .0009 for patients with CNS metastases, HR 0.46, P less than .0001 for patients with no CNS metastases).
For this interim analysis, with OS data at only 25% maturity, median OS had not been reached in either trial arm. The HR for OS with osimertinib was 0.63, with a P value of .0068, but this was not statistically significant, as the statistical method used required a P of less than .0015 for significance, Dr. Ramalingam explained.
The safety profile of osimertinib was comparable to that of the standard of care, with lower rates of grade 3 or greater adverse events, and a lower discontinuation rate, than with either gefitinib or erlotinib.
Enriqueta Felip, MD, the invited commentator at the briefing, said that “based on these results, osimertinib should be considered a new first-line treatment option for patients with EGFR mutations.”
Tony Mok, MD, from the Chinese University of Hong Kong, the invited discussant at the presidential symposium where Dr. Ramalingam presented the study, commented that “there is little doubt that FLAURA is a positive study demonstrating superiority in PFS with first-line osimertinib.”
He questioned, however whether all patients with EGFR mutation-positve NSCLC should receive osimertinib up front.
“I have confidence that patients with CNS metastases at presentation would benefit the most considering osimertinib’s higher CNS penetration, but the optimal sequence for overall survival benefit is controversial,” he said.
Dr. Mok said that he was reserving judgment about elevating osimertinib to standard-of-care status until there were more answers about the final number of patients who are able to crossover to osimertinib, and about the “mechanism and management of osimertinib resistance upon disease progression.”
The study was funded by AstraZeneca, the maker of osimertinib. Dr Ramalingam is on the advisory boards of AstraZeneca and other companies. Dr Mok disclosed honoraria, fees, and research funding from Astra-Zeneca. Dr. Felip disclosed financial relationships with multiple companies not including AstraZeneca.
MADRID – The EGFR inhibitor osimertinib (Tagrisso) is being hailed as a new standard of care for first-line therapy of advanced, treatment naïve non-small cell lung cancer (NSCLC) with mutations in the epidermal growth factor receptor (EGFR).
Osimertinib cut the risk of disease progression in these patients by 54% compared gefitinib (Iressa) or erlotinib (Tarceva).
Among 279 patients with EGFR-mutated locally advanced or metastatic NSCLC treated with osemirtinib, the median progression-free survival (PFS) was 18.9 months, compared with 10.2 months for 277 patients treated with the standard of care, which translates into a hazard ratio (HR) of 0.46 (P less than .0001).
“We wanted to see if by shutting down a major escape pathway by giving osemirtinib upfront, whether we would be able to improve patient outcomes,” he said at a briefing at the European Society of Medical Oncology (ESMO) Congress.
Osimertinib is a third-generation EGFR tyrosine kinase inhibitor (TKI) selective for EGFR mutations, including the T790M resistance mutation. It is currently approved in the US for the treatment of patients with EGFR T790M mutation-positive NSCLC whose disease has progressed on or after EGFR TKI therapy.
In the FLAURA trial (NCT02296125) investigators stratified patients with previously untreated NSCLC positive for EGFR resistance mutations according to mutation status (exon 19 deletion or the L858R amino acid substitution in exon 21) and race (Asian or non-Asian).
Patients were randomly assigned to treatment with either oral osimertinib 80 mg daily or an EGFR TKI, either oral gefitinib 250 mg or erlotinib 150 mg daily.
The patients were assessed by Response Evaluation Criteria in Solid Tumors (RECIST) every 6 weeks until objective disease progression.
Patients assigned to the standard-of-care arm who had central confirmation of progression and T790M positivity were allowed to cross over to open-label osimertinib.
PFS, the primary endpoint, was also significantly better with osimertinib than with either of the comparator TKIs in patients with and without central nervous system metastases at study entry (HR 0.47, P = .0009 for patients with CNS metastases, HR 0.46, P less than .0001 for patients with no CNS metastases).
For this interim analysis, with OS data at only 25% maturity, median OS had not been reached in either trial arm. The HR for OS with osimertinib was 0.63, with a P value of .0068, but this was not statistically significant, as the statistical method used required a P of less than .0015 for significance, Dr. Ramalingam explained.
The safety profile of osimertinib was comparable to that of the standard of care, with lower rates of grade 3 or greater adverse events, and a lower discontinuation rate, than with either gefitinib or erlotinib.
Enriqueta Felip, MD, the invited commentator at the briefing, said that “based on these results, osimertinib should be considered a new first-line treatment option for patients with EGFR mutations.”
Tony Mok, MD, from the Chinese University of Hong Kong, the invited discussant at the presidential symposium where Dr. Ramalingam presented the study, commented that “there is little doubt that FLAURA is a positive study demonstrating superiority in PFS with first-line osimertinib.”
He questioned, however whether all patients with EGFR mutation-positve NSCLC should receive osimertinib up front.
“I have confidence that patients with CNS metastases at presentation would benefit the most considering osimertinib’s higher CNS penetration, but the optimal sequence for overall survival benefit is controversial,” he said.
Dr. Mok said that he was reserving judgment about elevating osimertinib to standard-of-care status until there were more answers about the final number of patients who are able to crossover to osimertinib, and about the “mechanism and management of osimertinib resistance upon disease progression.”
The study was funded by AstraZeneca, the maker of osimertinib. Dr Ramalingam is on the advisory boards of AstraZeneca and other companies. Dr Mok disclosed honoraria, fees, and research funding from Astra-Zeneca. Dr. Felip disclosed financial relationships with multiple companies not including AstraZeneca.
MADRID – The EGFR inhibitor osimertinib (Tagrisso) is being hailed as a new standard of care for first-line therapy of advanced, treatment naïve non-small cell lung cancer (NSCLC) with mutations in the epidermal growth factor receptor (EGFR).
Osimertinib cut the risk of disease progression in these patients by 54% compared gefitinib (Iressa) or erlotinib (Tarceva).
Among 279 patients with EGFR-mutated locally advanced or metastatic NSCLC treated with osemirtinib, the median progression-free survival (PFS) was 18.9 months, compared with 10.2 months for 277 patients treated with the standard of care, which translates into a hazard ratio (HR) of 0.46 (P less than .0001).
“We wanted to see if by shutting down a major escape pathway by giving osemirtinib upfront, whether we would be able to improve patient outcomes,” he said at a briefing at the European Society of Medical Oncology (ESMO) Congress.
Osimertinib is a third-generation EGFR tyrosine kinase inhibitor (TKI) selective for EGFR mutations, including the T790M resistance mutation. It is currently approved in the US for the treatment of patients with EGFR T790M mutation-positive NSCLC whose disease has progressed on or after EGFR TKI therapy.
In the FLAURA trial (NCT02296125) investigators stratified patients with previously untreated NSCLC positive for EGFR resistance mutations according to mutation status (exon 19 deletion or the L858R amino acid substitution in exon 21) and race (Asian or non-Asian).
Patients were randomly assigned to treatment with either oral osimertinib 80 mg daily or an EGFR TKI, either oral gefitinib 250 mg or erlotinib 150 mg daily.
The patients were assessed by Response Evaluation Criteria in Solid Tumors (RECIST) every 6 weeks until objective disease progression.
Patients assigned to the standard-of-care arm who had central confirmation of progression and T790M positivity were allowed to cross over to open-label osimertinib.
PFS, the primary endpoint, was also significantly better with osimertinib than with either of the comparator TKIs in patients with and without central nervous system metastases at study entry (HR 0.47, P = .0009 for patients with CNS metastases, HR 0.46, P less than .0001 for patients with no CNS metastases).
For this interim analysis, with OS data at only 25% maturity, median OS had not been reached in either trial arm. The HR for OS with osimertinib was 0.63, with a P value of .0068, but this was not statistically significant, as the statistical method used required a P of less than .0015 for significance, Dr. Ramalingam explained.
The safety profile of osimertinib was comparable to that of the standard of care, with lower rates of grade 3 or greater adverse events, and a lower discontinuation rate, than with either gefitinib or erlotinib.
Enriqueta Felip, MD, the invited commentator at the briefing, said that “based on these results, osimertinib should be considered a new first-line treatment option for patients with EGFR mutations.”
Tony Mok, MD, from the Chinese University of Hong Kong, the invited discussant at the presidential symposium where Dr. Ramalingam presented the study, commented that “there is little doubt that FLAURA is a positive study demonstrating superiority in PFS with first-line osimertinib.”
He questioned, however whether all patients with EGFR mutation-positve NSCLC should receive osimertinib up front.
“I have confidence that patients with CNS metastases at presentation would benefit the most considering osimertinib’s higher CNS penetration, but the optimal sequence for overall survival benefit is controversial,” he said.
Dr. Mok said that he was reserving judgment about elevating osimertinib to standard-of-care status until there were more answers about the final number of patients who are able to crossover to osimertinib, and about the “mechanism and management of osimertinib resistance upon disease progression.”
The study was funded by AstraZeneca, the maker of osimertinib. Dr Ramalingam is on the advisory boards of AstraZeneca and other companies. Dr Mok disclosed honoraria, fees, and research funding from Astra-Zeneca. Dr. Felip disclosed financial relationships with multiple companies not including AstraZeneca.
AT ESMO 2017
Key clinical point:
Major finding: The median PFS with osimertinib was 18.9 months, compared with 10.2 months for the standard of care (gefitinib or erlotinib).
Data source: Randomized double-blind study of 556 patients with EGFR-mutated NSCLC.
Disclosures: The study was funded by AstraZeneca. Dr Ramalingam is on the advisory boards of AstraZeneca and other companies. Dr Mok disclosed honoraria, fees, and research funding from Astra-Zeneca. Dr. Felip disclosed financial relationships with multiple companies not including AstraZeneca.
DDSEP® 8 Quick quiz - September 2017 Question 2
Answer B
Objective: Recognize the clinical presentation and imaging features of main duct intraductal papillary mucinous neoplasm (IPMN)
Critique: The patient’s imaging is consistent with main duct IPMN and the mild pancreatitis is likely a consequence of mucin plugging and obstruction. Main duct IPMN is associated with a higher incidence of malignancy, compared with branch duct IPMN and surgical resection is recommended if the patient is a surgical candidate.
While further sampling with endoscopic ultrasound or endoscopic retrograde cholangiopancreatography may be helpful, these tests have a low sensitivity for identifying dysplasia and are unlikely to change management. Surveillance with MRI would be appropriate if the patient does not wish to undergo surgery at this time.
Answer B
Objective: Recognize the clinical presentation and imaging features of main duct intraductal papillary mucinous neoplasm (IPMN)
Critique: The patient’s imaging is consistent with main duct IPMN and the mild pancreatitis is likely a consequence of mucin plugging and obstruction. Main duct IPMN is associated with a higher incidence of malignancy, compared with branch duct IPMN and surgical resection is recommended if the patient is a surgical candidate.
While further sampling with endoscopic ultrasound or endoscopic retrograde cholangiopancreatography may be helpful, these tests have a low sensitivity for identifying dysplasia and are unlikely to change management. Surveillance with MRI would be appropriate if the patient does not wish to undergo surgery at this time.
Answer B
Objective: Recognize the clinical presentation and imaging features of main duct intraductal papillary mucinous neoplasm (IPMN)
Critique: The patient’s imaging is consistent with main duct IPMN and the mild pancreatitis is likely a consequence of mucin plugging and obstruction. Main duct IPMN is associated with a higher incidence of malignancy, compared with branch duct IPMN and surgical resection is recommended if the patient is a surgical candidate.
While further sampling with endoscopic ultrasound or endoscopic retrograde cholangiopancreatography may be helpful, these tests have a low sensitivity for identifying dysplasia and are unlikely to change management. Surveillance with MRI would be appropriate if the patient does not wish to undergo surgery at this time.
A 72-year-old man is admitted to the hospital with mild acute pancreatitis. He reports vague abdominal pain for the past 3 months. He is otherwise healthy and has well-controlled hypertension. He is active and exercises three times a week. CT scan reveals a markedly dilated main pancreatic duct with no stricture as shown below in representative axial and coronal images (Figures 1, 2).
DDSEP® 8 Quick quiz - September 2017 Question 1
Answer D
Objective: Identify the clinical presentation and risk factors for small intestinal bacterial overgrowth.
Rationale: This patient likely has small intestinal bacterial overgrowth (SIBO) based on her symptoms, the steatorrhea with the positive Sudan stain for fat, and a slight anemia with an elevated MCV suggestive of vitamin B12 deficiency secondary to the bacterial overgrowth. She also has scleroderma, a condition commonly associated with SIBO, because it impairs gastrointestinal motility.
While hydrogen breath testing may help establish the diagnosis of SIBO, there is variable sensitivity and specificity of the testing with false-positive and false-negative test results frequently occurring. An alternative strategy is to treat empirically with an accepted antibiotic regimen and assess response after the course is completed.
References
1. Bures J., Cyrany J., Kohoutova D., et al. Small intestinal bacterial overgrowth syndrome. World J Gastroenterol. 2010 Jun 28;16(24):2978-90.
2. Abu-Shanab A., Quigley E.M.. Diagnosis of small intestinal bacterial overgrowth: The challenges persist! Expert Rev Gastroenterol Hepatol. 2009 Feb;3(1):77-87.
3. Khoshini R., Dai S.C., Lezcano S., Pimentel M. A systematic review of diagnostic tests for small intestinal bacterial overgrowth. Dig Dis Sci. 2008 Jun;53(6):1443-54.
Answer D
Objective: Identify the clinical presentation and risk factors for small intestinal bacterial overgrowth.
Rationale: This patient likely has small intestinal bacterial overgrowth (SIBO) based on her symptoms, the steatorrhea with the positive Sudan stain for fat, and a slight anemia with an elevated MCV suggestive of vitamin B12 deficiency secondary to the bacterial overgrowth. She also has scleroderma, a condition commonly associated with SIBO, because it impairs gastrointestinal motility.
While hydrogen breath testing may help establish the diagnosis of SIBO, there is variable sensitivity and specificity of the testing with false-positive and false-negative test results frequently occurring. An alternative strategy is to treat empirically with an accepted antibiotic regimen and assess response after the course is completed.
References
1. Bures J., Cyrany J., Kohoutova D., et al. Small intestinal bacterial overgrowth syndrome. World J Gastroenterol. 2010 Jun 28;16(24):2978-90.
2. Abu-Shanab A., Quigley E.M.. Diagnosis of small intestinal bacterial overgrowth: The challenges persist! Expert Rev Gastroenterol Hepatol. 2009 Feb;3(1):77-87.
3. Khoshini R., Dai S.C., Lezcano S., Pimentel M. A systematic review of diagnostic tests for small intestinal bacterial overgrowth. Dig Dis Sci. 2008 Jun;53(6):1443-54.
Answer D
Objective: Identify the clinical presentation and risk factors for small intestinal bacterial overgrowth.
Rationale: This patient likely has small intestinal bacterial overgrowth (SIBO) based on her symptoms, the steatorrhea with the positive Sudan stain for fat, and a slight anemia with an elevated MCV suggestive of vitamin B12 deficiency secondary to the bacterial overgrowth. She also has scleroderma, a condition commonly associated with SIBO, because it impairs gastrointestinal motility.
While hydrogen breath testing may help establish the diagnosis of SIBO, there is variable sensitivity and specificity of the testing with false-positive and false-negative test results frequently occurring. An alternative strategy is to treat empirically with an accepted antibiotic regimen and assess response after the course is completed.
References
1. Bures J., Cyrany J., Kohoutova D., et al. Small intestinal bacterial overgrowth syndrome. World J Gastroenterol. 2010 Jun 28;16(24):2978-90.
2. Abu-Shanab A., Quigley E.M.. Diagnosis of small intestinal bacterial overgrowth: The challenges persist! Expert Rev Gastroenterol Hepatol. 2009 Feb;3(1):77-87.
3. Khoshini R., Dai S.C., Lezcano S., Pimentel M. A systematic review of diagnostic tests for small intestinal bacterial overgrowth. Dig Dis Sci. 2008 Jun;53(6):1443-54.
A 56-year-old woman with a history of scleroderma presents for evaluation of recurrent episodes of bloating, excess flatulence, mild nausea, and watery diarrhea for the past 5 months without associated weight loss, gastrointestinal bleeding, or fevers.
She had a normal screening colonoscopy 2 years ago, and an upper endoscopy for evaluation of reflux and dyspepsia 5 years ago, which was only notable for a small sliding hiatal hernia. Laboratory testing reveals hemoglobin of 10.9 g/dL with an MCV 106 fL. Stool studies are negative for occult blood, fecal calprotectin is not elevated, but a Sudan stain is positive.
Worsening osteoporosis care for RA patients shows need for action
Osteoporosis care in patients with rheumatoid arthritis is suboptimal, and the relative risk of the application of appropriate osteoporosis care in both RA and osteoarthritis patients has been declining steadily over the past decade, according to findings from a large, prospective, observational study.
The study included 11,669 RA patients and 2,829 OA patients in the National Data Bank for Rheumatic Diseases who were followed from 2003 through 2014 and showed that about half of the RA patients in whom osteoporosis treatment was indicated never received an osteoporosis medication. Further, the decline in the application of appropriate osteoporosis care was apparent even after the release of the 2010 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis, according to the findings, which have been accepted for publication in Arthritis Care & Research (doi: 10.1002/acr.23331).

A more recent study in the general population showed that, regardless of glucocorticoid (GC) use, there was a significant decrease in the use of oral bisphosphonates from 1996 to 2012, noted Dr. Ozen of the University of Nebraska, Omaha, and Marmara University, Istanbul, Turkey.
“The evidence from all these studies suggests that, despite a slight improvement in GIOP care, with a decline in overall OP care in the early 2000s, the OP care gap has always been suboptimal and declined over the last decade significantly,” she said, adding that this is “especially more worrisome in glucocorticoid-receiving RA patients,” in whom bone loss and fracture risk are dramatically increased.
“Additionally, considering that rheumatologists do better regarding comorbidity follow-up and management than other specialists who follow patients with rheumatic diseases, the gap for osteoporosis care might be even higher than we reported,” she said.
Indeed, among physicians caring for patients with osteoporosis, there is concern that “we’re on the precipice of further increase in fracture burden,” according to Kenneth Saag, MD, a rheumatologist and professor at the University of Alabama at Birmingham.
The conclusions of the study by Dr. Ozen and her colleagues are disturbing, but “not tremendously surprising,” said Dr. Saag, who also is president of the National Osteoporosis Foundation.
The findings are consistent with those from previous studies, he explained.
For example, Daniel H. Solomon, MD, and his associates showed that only 23% of 623 RA patients, including 236 patients who were taking glucocorticoids at an index visit in 1999, underwent bone densitometry, and only 42% were prescribed a medication (other than calcium and/or vitamin D), that reduces bone loss (Arthritis Rheum. 2002;46:3136-42). They also showed that, in 2004, despite a slight improvement vs. prior years, only 48% of 193 RA patients included in a chart review had received a bone mineral density test or medication for osteoporosis, and 64% of those taking more than 5 mg of prednisone for more than 3 months were receiving osteoporosis management (Arthritis Care Res. 2006;55:873-7).
In the current study, Dr. Ozen and her colleagues looked at both RA and OA patients and found that overall, osteoporosis treatment or screening was reported in 67.4% of study subjects over a mean of 5.5 years follow-up. Of those eligible for osteoporosis treatment based on the 2010 ACR guidelines, including 48.4% of RA patients and 17.6% of OA patients, 55% reported osteoporosis medication use. Despite their increased risk of osteoporosis, particularly in the setting of glucocorticoid use, RA patients were not more likely than OA patients to undergo screening or receive treatment (hazard ratio, 1.04).

“This suboptimal and decreasing trend for osteoporosis treatment and screening in RA patients is important as the prevalence of osteoporosis leading to fractures in RA, regardless of GC use, is still high, despite aggressive management strategies and the use of [biologic disease-modifying antirheumatic agents],” they wrote.
Factors contributing to the decline
The reasons for the decline after 2008 are not fully known, but data suggest that both patient and clinician factors play a role.
Lack of knowledge, unwillingness to take additional drugs, and cost are among patient factors that may interfere with screening and treatment, and lack of experience and time, and a focus more on disease activity and other comorbid conditions may be among clinician factors, they said.
Other factors that may have affected patient and clinician willingness to pursue care include 2007 cuts in Medicare reimbursement for dual-energy x-ray absorptiometry (DXA), a 2006 warning regarding jaw osteonecrosis with bisphosphonate use, and 2007 and 2010 publications about atrial fibrillation and atypical femoral fractures associated with long-term bisphosphonate treatment, they noted.
Dr. Saag agreed that it is difficult to pinpoint exact reasons for the decline, but said the DXA reimbursement cuts have had a significant effect.
“Declining reimbursement is likely one of the major factors, if not the major factor in the decline in testing that has been observed nationally,” he said, explaining that reimbursement was below the break-even point for many physicians in freestanding medical practices, and that slight adjustments have applied only to facilities located in hospitals. As a result, the availability of DXA screening has declined.
The National Osteoporosis Foundation and other organizations such as the International Society for Clinical Densitometry are lobbying for changes, as there is a very strong link between testing and treatment.
“If we can’t get more people tested, we’re unlikely to get more people treated,” Dr. Saag said.
The availability of new agents for the treatment of osteoporosis could also lead to improvements in screening and care, he added.
In July, Amgen submitted to the Food and Drug Administration a supplemental new Biologics License Application for Prolia (denosumab) for the treatment of patients with GIOP. The application is based on a phase 3 study evaluating the safety and efficacy of Prolia, compared with risedronate, in patients receiving glucocorticoid treatment. Denosumab was also shown in a phase 3 study presented at the 2016 ACR meeting to result in greater improvement in bone mineral density vs. bisphosphonates across multiple sites, said Dr. Saag, an investigator for the trial.
“So we’re pleased about that finding, and we’re hopeful that eventually denosumab may become another treatment option,” he said, adding that having more choices leads to more personalized care for patients.
“We hope that, if it does receive approval and is utilized, that it may partially help address the gap in treatment that has been identified,” he said, adding that abaloparatide (Tymlos), which is approved for use in postmenopausal women with osteoporosis and high fracture risk, and romosozumab, currently in late-phase development, could also eventually help bridge the gap.
Recent guideline update
There is also optimism that a recent update to the 2010 guideline, published in August in Arthritis Care & Research, will help address the problem (Arthritis Care Res. 2017;69:1095-1110).
“We hope that one the most important impacts of the new 2017 ACR GIOP guideline will be increasing the awareness of GIOP care,” Dr. Ozen said, noting that changes in the new guideline will also be helpful for guiding management of certain patients not addressed in the 2010 guideline, including adults under age 40 years and children.
The new guideline also incorporates FRAX hip fracture scores into the risk-assessment recommendations .
“Lastly, the addition of other oral bisphosphonates and denosumab to the guideline has broadened the potential therapeutic options for physicians and patients,” she said.

Efforts by the ACR to make these guidelines widely known among clinicians could also help promote improved screening and treatment, said Dr. Buckley, a professor of internal medicine and pediatrics at Yale University, New Haven, Conn.
And that awareness is what is needed most, Dr. Ozen said.
“What we need in osteoporosis care of RA or GIOP patients is more awareness and education about risk-benefit ratios of antiosteoporotic medications instead of focusing on the medication type. We hope that our paper showing the worsening osteoporosis care and the new 2017 ACR GIOP guideline provide more awareness and guidance for the physicians managing these patients,” she said, adding that overcoming obstacles in osteoporosis care requires identification and attention to all potential causes for the suboptimal management of osteoporosis from both patients’ and physicians’ perspectives.
“With the improvement in management strategies of rheumatic diseases ... we anticipate improvement in life expectancy and cardiovascular outcomes in RA and inflammatory rheumatic diseases. In this regard, with aging and decreased but ongoing clinical or subclinical chronic inflammation, it is highly likely that we might be dealing with much older patients with higher osteoporosis and fracture risks. Considering the significant contribution of fractures on cost, disability, morbidity, and mortality of these diseases, rheumatologists and other specialists should work together more vigilantly to overcome the obstacles and improve osteoporosis care,” she said.
Dr. Ozen and Dr. Buckley reported having no conflicts of interest. Dr. Saag is an investigator and consultant for Amgen, Merck, and Radius Health, and is a consultant for Lilly.
Osteoporosis care in patients with rheumatoid arthritis is suboptimal, and the relative risk of the application of appropriate osteoporosis care in both RA and osteoarthritis patients has been declining steadily over the past decade, according to findings from a large, prospective, observational study.
The study included 11,669 RA patients and 2,829 OA patients in the National Data Bank for Rheumatic Diseases who were followed from 2003 through 2014 and showed that about half of the RA patients in whom osteoporosis treatment was indicated never received an osteoporosis medication. Further, the decline in the application of appropriate osteoporosis care was apparent even after the release of the 2010 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis, according to the findings, which have been accepted for publication in Arthritis Care & Research (doi: 10.1002/acr.23331).
A more recent study in the general population showed that, regardless of glucocorticoid (GC) use, there was a significant decrease in the use of oral bisphosphonates from 1996 to 2012, noted Dr. Ozen of the University of Nebraska, Omaha, and Marmara University, Istanbul, Turkey.
“The evidence from all these studies suggests that, despite a slight improvement in GIOP care, with a decline in overall OP care in the early 2000s, the OP care gap has always been suboptimal and declined over the last decade significantly,” she said, adding that this is “especially more worrisome in glucocorticoid-receiving RA patients,” in whom bone loss and fracture risk are dramatically increased.
“Additionally, considering that rheumatologists do better regarding comorbidity follow-up and management than other specialists who follow patients with rheumatic diseases, the gap for osteoporosis care might be even higher than we reported,” she said.
Indeed, among physicians caring for patients with osteoporosis, there is concern that “we’re on the precipice of further increase in fracture burden,” according to Kenneth Saag, MD, a rheumatologist and professor at the University of Alabama at Birmingham.
The conclusions of the study by Dr. Ozen and her colleagues are disturbing, but “not tremendously surprising,” said Dr. Saag, who also is president of the National Osteoporosis Foundation.
The findings are consistent with those from previous studies, he explained.
For example, Daniel H. Solomon, MD, and his associates showed that only 23% of 623 RA patients, including 236 patients who were taking glucocorticoids at an index visit in 1999, underwent bone densitometry, and only 42% were prescribed a medication (other than calcium and/or vitamin D), that reduces bone loss (Arthritis Rheum. 2002;46:3136-42). They also showed that, in 2004, despite a slight improvement vs. prior years, only 48% of 193 RA patients included in a chart review had received a bone mineral density test or medication for osteoporosis, and 64% of those taking more than 5 mg of prednisone for more than 3 months were receiving osteoporosis management (Arthritis Care Res. 2006;55:873-7).
In the current study, Dr. Ozen and her colleagues looked at both RA and OA patients and found that overall, osteoporosis treatment or screening was reported in 67.4% of study subjects over a mean of 5.5 years follow-up. Of those eligible for osteoporosis treatment based on the 2010 ACR guidelines, including 48.4% of RA patients and 17.6% of OA patients, 55% reported osteoporosis medication use. Despite their increased risk of osteoporosis, particularly in the setting of glucocorticoid use, RA patients were not more likely than OA patients to undergo screening or receive treatment (hazard ratio, 1.04).
“This suboptimal and decreasing trend for osteoporosis treatment and screening in RA patients is important as the prevalence of osteoporosis leading to fractures in RA, regardless of GC use, is still high, despite aggressive management strategies and the use of [biologic disease-modifying antirheumatic agents],” they wrote.
Factors contributing to the decline
The reasons for the decline after 2008 are not fully known, but data suggest that both patient and clinician factors play a role.
Lack of knowledge, unwillingness to take additional drugs, and cost are among patient factors that may interfere with screening and treatment, and lack of experience and time, and a focus more on disease activity and other comorbid conditions may be among clinician factors, they said.
Other factors that may have affected patient and clinician willingness to pursue care include 2007 cuts in Medicare reimbursement for dual-energy x-ray absorptiometry (DXA), a 2006 warning regarding jaw osteonecrosis with bisphosphonate use, and 2007 and 2010 publications about atrial fibrillation and atypical femoral fractures associated with long-term bisphosphonate treatment, they noted.
Dr. Saag agreed that it is difficult to pinpoint exact reasons for the decline, but said the DXA reimbursement cuts have had a significant effect.
“Declining reimbursement is likely one of the major factors, if not the major factor in the decline in testing that has been observed nationally,” he said, explaining that reimbursement was below the break-even point for many physicians in freestanding medical practices, and that slight adjustments have applied only to facilities located in hospitals. As a result, the availability of DXA screening has declined.
The National Osteoporosis Foundation and other organizations such as the International Society for Clinical Densitometry are lobbying for changes, as there is a very strong link between testing and treatment.
“If we can’t get more people tested, we’re unlikely to get more people treated,” Dr. Saag said.
The availability of new agents for the treatment of osteoporosis could also lead to improvements in screening and care, he added.
In July, Amgen submitted to the Food and Drug Administration a supplemental new Biologics License Application for Prolia (denosumab) for the treatment of patients with GIOP. The application is based on a phase 3 study evaluating the safety and efficacy of Prolia, compared with risedronate, in patients receiving glucocorticoid treatment. Denosumab was also shown in a phase 3 study presented at the 2016 ACR meeting to result in greater improvement in bone mineral density vs. bisphosphonates across multiple sites, said Dr. Saag, an investigator for the trial.
“So we’re pleased about that finding, and we’re hopeful that eventually denosumab may become another treatment option,” he said, adding that having more choices leads to more personalized care for patients.
“We hope that, if it does receive approval and is utilized, that it may partially help address the gap in treatment that has been identified,” he said, adding that abaloparatide (Tymlos), which is approved for use in postmenopausal women with osteoporosis and high fracture risk, and romosozumab, currently in late-phase development, could also eventually help bridge the gap.
Recent guideline update
There is also optimism that a recent update to the 2010 guideline, published in August in Arthritis Care & Research, will help address the problem (Arthritis Care Res. 2017;69:1095-1110).
“We hope that one the most important impacts of the new 2017 ACR GIOP guideline will be increasing the awareness of GIOP care,” Dr. Ozen said, noting that changes in the new guideline will also be helpful for guiding management of certain patients not addressed in the 2010 guideline, including adults under age 40 years and children.
The new guideline also incorporates FRAX hip fracture scores into the risk-assessment recommendations .
“Lastly, the addition of other oral bisphosphonates and denosumab to the guideline has broadened the potential therapeutic options for physicians and patients,” she said.
Efforts by the ACR to make these guidelines widely known among clinicians could also help promote improved screening and treatment, said Dr. Buckley, a professor of internal medicine and pediatrics at Yale University, New Haven, Conn.
And that awareness is what is needed most, Dr. Ozen said.
“What we need in osteoporosis care of RA or GIOP patients is more awareness and education about risk-benefit ratios of antiosteoporotic medications instead of focusing on the medication type. We hope that our paper showing the worsening osteoporosis care and the new 2017 ACR GIOP guideline provide more awareness and guidance for the physicians managing these patients,” she said, adding that overcoming obstacles in osteoporosis care requires identification and attention to all potential causes for the suboptimal management of osteoporosis from both patients’ and physicians’ perspectives.
“With the improvement in management strategies of rheumatic diseases ... we anticipate improvement in life expectancy and cardiovascular outcomes in RA and inflammatory rheumatic diseases. In this regard, with aging and decreased but ongoing clinical or subclinical chronic inflammation, it is highly likely that we might be dealing with much older patients with higher osteoporosis and fracture risks. Considering the significant contribution of fractures on cost, disability, morbidity, and mortality of these diseases, rheumatologists and other specialists should work together more vigilantly to overcome the obstacles and improve osteoporosis care,” she said.
Dr. Ozen and Dr. Buckley reported having no conflicts of interest. Dr. Saag is an investigator and consultant for Amgen, Merck, and Radius Health, and is a consultant for Lilly.
Osteoporosis care in patients with rheumatoid arthritis is suboptimal, and the relative risk of the application of appropriate osteoporosis care in both RA and osteoarthritis patients has been declining steadily over the past decade, according to findings from a large, prospective, observational study.
The study included 11,669 RA patients and 2,829 OA patients in the National Data Bank for Rheumatic Diseases who were followed from 2003 through 2014 and showed that about half of the RA patients in whom osteoporosis treatment was indicated never received an osteoporosis medication. Further, the decline in the application of appropriate osteoporosis care was apparent even after the release of the 2010 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis, according to the findings, which have been accepted for publication in Arthritis Care & Research (doi: 10.1002/acr.23331).
A more recent study in the general population showed that, regardless of glucocorticoid (GC) use, there was a significant decrease in the use of oral bisphosphonates from 1996 to 2012, noted Dr. Ozen of the University of Nebraska, Omaha, and Marmara University, Istanbul, Turkey.
“The evidence from all these studies suggests that, despite a slight improvement in GIOP care, with a decline in overall OP care in the early 2000s, the OP care gap has always been suboptimal and declined over the last decade significantly,” she said, adding that this is “especially more worrisome in glucocorticoid-receiving RA patients,” in whom bone loss and fracture risk are dramatically increased.
“Additionally, considering that rheumatologists do better regarding comorbidity follow-up and management than other specialists who follow patients with rheumatic diseases, the gap for osteoporosis care might be even higher than we reported,” she said.
Indeed, among physicians caring for patients with osteoporosis, there is concern that “we’re on the precipice of further increase in fracture burden,” according to Kenneth Saag, MD, a rheumatologist and professor at the University of Alabama at Birmingham.
The conclusions of the study by Dr. Ozen and her colleagues are disturbing, but “not tremendously surprising,” said Dr. Saag, who also is president of the National Osteoporosis Foundation.
The findings are consistent with those from previous studies, he explained.
For example, Daniel H. Solomon, MD, and his associates showed that only 23% of 623 RA patients, including 236 patients who were taking glucocorticoids at an index visit in 1999, underwent bone densitometry, and only 42% were prescribed a medication (other than calcium and/or vitamin D), that reduces bone loss (Arthritis Rheum. 2002;46:3136-42). They also showed that, in 2004, despite a slight improvement vs. prior years, only 48% of 193 RA patients included in a chart review had received a bone mineral density test or medication for osteoporosis, and 64% of those taking more than 5 mg of prednisone for more than 3 months were receiving osteoporosis management (Arthritis Care Res. 2006;55:873-7).
In the current study, Dr. Ozen and her colleagues looked at both RA and OA patients and found that overall, osteoporosis treatment or screening was reported in 67.4% of study subjects over a mean of 5.5 years follow-up. Of those eligible for osteoporosis treatment based on the 2010 ACR guidelines, including 48.4% of RA patients and 17.6% of OA patients, 55% reported osteoporosis medication use. Despite their increased risk of osteoporosis, particularly in the setting of glucocorticoid use, RA patients were not more likely than OA patients to undergo screening or receive treatment (hazard ratio, 1.04).
“This suboptimal and decreasing trend for osteoporosis treatment and screening in RA patients is important as the prevalence of osteoporosis leading to fractures in RA, regardless of GC use, is still high, despite aggressive management strategies and the use of [biologic disease-modifying antirheumatic agents],” they wrote.
Factors contributing to the decline
The reasons for the decline after 2008 are not fully known, but data suggest that both patient and clinician factors play a role.
Lack of knowledge, unwillingness to take additional drugs, and cost are among patient factors that may interfere with screening and treatment, and lack of experience and time, and a focus more on disease activity and other comorbid conditions may be among clinician factors, they said.
Other factors that may have affected patient and clinician willingness to pursue care include 2007 cuts in Medicare reimbursement for dual-energy x-ray absorptiometry (DXA), a 2006 warning regarding jaw osteonecrosis with bisphosphonate use, and 2007 and 2010 publications about atrial fibrillation and atypical femoral fractures associated with long-term bisphosphonate treatment, they noted.
Dr. Saag agreed that it is difficult to pinpoint exact reasons for the decline, but said the DXA reimbursement cuts have had a significant effect.
“Declining reimbursement is likely one of the major factors, if not the major factor in the decline in testing that has been observed nationally,” he said, explaining that reimbursement was below the break-even point for many physicians in freestanding medical practices, and that slight adjustments have applied only to facilities located in hospitals. As a result, the availability of DXA screening has declined.
The National Osteoporosis Foundation and other organizations such as the International Society for Clinical Densitometry are lobbying for changes, as there is a very strong link between testing and treatment.
“If we can’t get more people tested, we’re unlikely to get more people treated,” Dr. Saag said.
The availability of new agents for the treatment of osteoporosis could also lead to improvements in screening and care, he added.
In July, Amgen submitted to the Food and Drug Administration a supplemental new Biologics License Application for Prolia (denosumab) for the treatment of patients with GIOP. The application is based on a phase 3 study evaluating the safety and efficacy of Prolia, compared with risedronate, in patients receiving glucocorticoid treatment. Denosumab was also shown in a phase 3 study presented at the 2016 ACR meeting to result in greater improvement in bone mineral density vs. bisphosphonates across multiple sites, said Dr. Saag, an investigator for the trial.
“So we’re pleased about that finding, and we’re hopeful that eventually denosumab may become another treatment option,” he said, adding that having more choices leads to more personalized care for patients.
“We hope that, if it does receive approval and is utilized, that it may partially help address the gap in treatment that has been identified,” he said, adding that abaloparatide (Tymlos), which is approved for use in postmenopausal women with osteoporosis and high fracture risk, and romosozumab, currently in late-phase development, could also eventually help bridge the gap.
Recent guideline update
There is also optimism that a recent update to the 2010 guideline, published in August in Arthritis Care & Research, will help address the problem (Arthritis Care Res. 2017;69:1095-1110).
“We hope that one the most important impacts of the new 2017 ACR GIOP guideline will be increasing the awareness of GIOP care,” Dr. Ozen said, noting that changes in the new guideline will also be helpful for guiding management of certain patients not addressed in the 2010 guideline, including adults under age 40 years and children.
The new guideline also incorporates FRAX hip fracture scores into the risk-assessment recommendations .
“Lastly, the addition of other oral bisphosphonates and denosumab to the guideline has broadened the potential therapeutic options for physicians and patients,” she said.
Efforts by the ACR to make these guidelines widely known among clinicians could also help promote improved screening and treatment, said Dr. Buckley, a professor of internal medicine and pediatrics at Yale University, New Haven, Conn.
And that awareness is what is needed most, Dr. Ozen said.
“What we need in osteoporosis care of RA or GIOP patients is more awareness and education about risk-benefit ratios of antiosteoporotic medications instead of focusing on the medication type. We hope that our paper showing the worsening osteoporosis care and the new 2017 ACR GIOP guideline provide more awareness and guidance for the physicians managing these patients,” she said, adding that overcoming obstacles in osteoporosis care requires identification and attention to all potential causes for the suboptimal management of osteoporosis from both patients’ and physicians’ perspectives.
“With the improvement in management strategies of rheumatic diseases ... we anticipate improvement in life expectancy and cardiovascular outcomes in RA and inflammatory rheumatic diseases. In this regard, with aging and decreased but ongoing clinical or subclinical chronic inflammation, it is highly likely that we might be dealing with much older patients with higher osteoporosis and fracture risks. Considering the significant contribution of fractures on cost, disability, morbidity, and mortality of these diseases, rheumatologists and other specialists should work together more vigilantly to overcome the obstacles and improve osteoporosis care,” she said.
Dr. Ozen and Dr. Buckley reported having no conflicts of interest. Dr. Saag is an investigator and consultant for Amgen, Merck, and Radius Health, and is a consultant for Lilly.
FROM ARTHRITIS CARE & RESEARCH