User login
Digital Strategies For Dermatology Patient Education
Technology offers new opportunities that can both enhance and challenge the physician-patient relationship, including the ways in which patients are educated. Ensuring dermatology patients are appropriately educated about their conditions can improve clinical care and treatment adherence, increase patient satisfaction, and potentially decrease medical costs. There are various digital methods by which physicians can deliver information to their patients, and while there are benefits and drawbacks to each, many Americans turn to the Internet for health information—a practice that is only predicted to become more prevalent.1
Dermatologists should strive to keep up with this trend by staying informed about the digital patient education options that are available and which tools they can use to more effectively share their knowledge with patients. Electronic health education has a powerful potential, but it is up to physicians to direct patients to the appropriate resources and education tools that will support their clear understanding of all elements of care.
Effective patient education can transform the role of the patient from passive recipient to active participant in his/her care and subsequently supports the physician-patient relationship. The benefits of patient education are timely and valuable with the new pay-for-performance model instated by the Medicare Access and CHIP Reauthorization Act and the Merit-based Incentive Payment System.2 In dermatology, patient education alone can essentially be a management strategy for numerous conditions (eg, identifying which triggers patients with contact dermatitis should avoid). On the other hand, a lack of patient knowledge can result in perceived noncompliance or treatment failure, when in reality there has simply been a communication gap between the physician and the patient. For example, if a patient notices little to no improvement of a fungal infection after applying ketoconazole shampoo 2% to affected areas and immediately rinsing, this does not necessarily constitute a treatment failure, as the patient should have been educated on the importance of leaving the shampoo on for 5 minutes before rinsing. One study alluded to this communication gap, revealing physicians’ tendency to overestimate how effectively they are communicating with their patients.3
Successful patient education ultimately is dependent on both the content provided and the method of delivery. In one survey of 2636 Internet users, 72% of respondents admitted to searching online for health information within the previous year; however, the same survey showed that physicians remain patients’ most trusted source of information.4 Physicians can use digital education methods to fulfill patient needs by providing them with and directing them to credible up-to-date sources.
Physicians can use electronic medical record (EMR) systems to electronically deliver health information to patients by directly communicating via an online patient portal. Allowing patients to engage with their health care providers electronically has been shown to increase patient satisfaction, promote adherence to preventative and treatment recommendations, improve clinical outcomes, and lower medical costs.5 The online portal can provide direct links for patients to digital resources; for example, MedlinePlus Connect (https://medlineplus.gov/connect/overview.html) is a free service that connects patients to MedlinePlus, an authoritative, up-to-date health information resource for consumer health information; however, many EMR systems lack quality dermatology content, as there is a greater emphasis on primary care, and patient usage of these online portals also is notoriously low.6 Dermatologists can work with EMR vendors to enhance the dermatology content for patient portals, and in some cases, specialty-specific content may be available for an additional fee. Clinicians can make their patients aware of the online portal and incentivize its use by sending an informational email, including a link on their practice’s website, promoting the portal during check-in and check-out at office visits, making tablets or kiosks available in the waiting room for sign-up, hanging posters in the examination rooms, and explaining the portal’s useful features during consultations with patients.
Mobile apps have revolutionized the potential for dermatologists to streamline patient education to a large population. In a 2014 review of 365 dermatology mobile apps, 13% were categorized as educational aids, adding to the realm of possibilities for digital patient education. For example, these apps may provide information on specific dermatologic conditions and medications, help users perform skin cancer checks, and provide reminders for when to administer injections for those on biologics. However, a drawback of medical mobile apps is that, to date, the US Food and Drug Administration has not released formal guidelines for their development.
It would be impractical for busy dermatologists to keep up with the credibility of every mobile app available in a growing market, but one solution could be for physicians to stay informed on only the most popular and most reviewed apps to keep in their digital toolbox. In 2014, the most reviewed dermatology app was the Dermatology app, which provided a guide to common dermatologic conditions and included images and a list of symptoms.7 To help keep physicians up to date on the most reliable dermatology apps, specialty societies, journal task forces, or interested dermatologists, residents, or medical students could publish updated literature on the most popular and most reviewed dermatology apps for patient education annually or biannually.
A practice’s website is a prime place for physicians to direct patients to educational content. Although many dermatology practice websites offer clinical information, the content often is focused on cosmetic procedures or is designed for search engine optimization to support online marketing and therefore may not be helpful to patients trying to understand a specific condition or treatment. Links to trusted resources, such as dermatology journals or medical societies, may be added but also would direct patients away from the practice’s website and would not allow physicians to customize the information he or she would like to share with their patients. Dermatologists should consider investing time and money into customizing educational material for their websites so patients can access health information from the source they trust most: their own physician.
Many of these digital options are useful for patients who want to access education material outside of the physician’s office, but digital opportunities to enhance point-of-care education also are available. In 2016, the American Academy of Dermatology partnered with ContextMedia:Health with the goal of delivering important decision enhancement technologies, educational content, and intelligence to patients and dermatologists for use before and during the consultation.8 ContextMedia:Health’s digital wallboard tablets are an engaging way to visually explain conditions and treatments to patients during the consultation, thus empowering physicians and patients to make decisions together and helping patients to be better advocates of their own health care. The downside is that health care workers must devote time and resources to be trained in using these devices.
The increasing availability of technology for electronic health information can be both beneficial and challenging for dermatologists. Physicians should explore and familiarize themselves with the tools that are available and assess their effectiveness by communicating with patients about their perception and understanding of their conditions. Digital delivery of health information is not meant to replace other methods of patient education but to supplement and reinforce them that which is verbally discussed during the office visit. Electronic health education demonstrates powerful potential, but it is up to the physician to direct patients to the appropriate resources and educational tools that will support a clear understanding of all elements of care.
Acknowledgment
The authors would like to thank Dr. Mark Becker (Berkeley, California) for helpful discussion and reviewing this manuscript.
- Explosive growth in healthcare apps raises oversight questions. Association of American Medical Colleges website. https://www.aamc.org/newsroom/reporter/october2012/308516/health-care-apps.html. Accessed January 16, 2017.
- What’s MACRA? Centers for Medicare and Medicaid Services website. https://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/Value-Based-Programs/MACRA-MIPS-and-APMs/MACRA-MIPS-and-APMs.html. Updated November 16, 2017. Accessed February 26, 2018.
- Duffy FD, Gordon GH, Whelan G, et al. Assessing competence in communication and interpersonal skills: the Kalamazoo II report. Acad Med. 2004;79:495-507.
- Dutta-Bergman M. Trusted online sources of health information: differences in demographics, health beliefs, and health-information orientation [published online September 25, 2003]. J Med Internet Res. 2003;5:E21.
- Griffin A, Skinner A, Thornhill J, et al. Patient portals: who uses them? what features do they use? and do they reduce hospital readmissions? Appl Clin Inform. 2016;7:489-501.
- Lin CT, Wittevrongel L, Moore L, et al. An internet-based patient-provider communication system: randomized controlled trial. J Med Internet Res. 2005;7:E47.
- Patel S, Eluri M, Boyers LN, et al. Update on mobile applications in dermatology [published online November 9, 2014]. Dermatol Online J. 2014;21. pii:13030/qt1zc343js.
- American Academy of Dermatology selects ContextMedia:Health as patient education affinity partner. American Academy of Dermatology website. https://www.aad.org/media/news-releases/aad-selects-patient-education-affinity-partner. Published November 14, 2016. Accessed February 25, 2018.
Technology offers new opportunities that can both enhance and challenge the physician-patient relationship, including the ways in which patients are educated. Ensuring dermatology patients are appropriately educated about their conditions can improve clinical care and treatment adherence, increase patient satisfaction, and potentially decrease medical costs. There are various digital methods by which physicians can deliver information to their patients, and while there are benefits and drawbacks to each, many Americans turn to the Internet for health information—a practice that is only predicted to become more prevalent.1
Dermatologists should strive to keep up with this trend by staying informed about the digital patient education options that are available and which tools they can use to more effectively share their knowledge with patients. Electronic health education has a powerful potential, but it is up to physicians to direct patients to the appropriate resources and education tools that will support their clear understanding of all elements of care.
Effective patient education can transform the role of the patient from passive recipient to active participant in his/her care and subsequently supports the physician-patient relationship. The benefits of patient education are timely and valuable with the new pay-for-performance model instated by the Medicare Access and CHIP Reauthorization Act and the Merit-based Incentive Payment System.2 In dermatology, patient education alone can essentially be a management strategy for numerous conditions (eg, identifying which triggers patients with contact dermatitis should avoid). On the other hand, a lack of patient knowledge can result in perceived noncompliance or treatment failure, when in reality there has simply been a communication gap between the physician and the patient. For example, if a patient notices little to no improvement of a fungal infection after applying ketoconazole shampoo 2% to affected areas and immediately rinsing, this does not necessarily constitute a treatment failure, as the patient should have been educated on the importance of leaving the shampoo on for 5 minutes before rinsing. One study alluded to this communication gap, revealing physicians’ tendency to overestimate how effectively they are communicating with their patients.3
Successful patient education ultimately is dependent on both the content provided and the method of delivery. In one survey of 2636 Internet users, 72% of respondents admitted to searching online for health information within the previous year; however, the same survey showed that physicians remain patients’ most trusted source of information.4 Physicians can use digital education methods to fulfill patient needs by providing them with and directing them to credible up-to-date sources.
Physicians can use electronic medical record (EMR) systems to electronically deliver health information to patients by directly communicating via an online patient portal. Allowing patients to engage with their health care providers electronically has been shown to increase patient satisfaction, promote adherence to preventative and treatment recommendations, improve clinical outcomes, and lower medical costs.5 The online portal can provide direct links for patients to digital resources; for example, MedlinePlus Connect (https://medlineplus.gov/connect/overview.html) is a free service that connects patients to MedlinePlus, an authoritative, up-to-date health information resource for consumer health information; however, many EMR systems lack quality dermatology content, as there is a greater emphasis on primary care, and patient usage of these online portals also is notoriously low.6 Dermatologists can work with EMR vendors to enhance the dermatology content for patient portals, and in some cases, specialty-specific content may be available for an additional fee. Clinicians can make their patients aware of the online portal and incentivize its use by sending an informational email, including a link on their practice’s website, promoting the portal during check-in and check-out at office visits, making tablets or kiosks available in the waiting room for sign-up, hanging posters in the examination rooms, and explaining the portal’s useful features during consultations with patients.
Mobile apps have revolutionized the potential for dermatologists to streamline patient education to a large population. In a 2014 review of 365 dermatology mobile apps, 13% were categorized as educational aids, adding to the realm of possibilities for digital patient education. For example, these apps may provide information on specific dermatologic conditions and medications, help users perform skin cancer checks, and provide reminders for when to administer injections for those on biologics. However, a drawback of medical mobile apps is that, to date, the US Food and Drug Administration has not released formal guidelines for their development.
It would be impractical for busy dermatologists to keep up with the credibility of every mobile app available in a growing market, but one solution could be for physicians to stay informed on only the most popular and most reviewed apps to keep in their digital toolbox. In 2014, the most reviewed dermatology app was the Dermatology app, which provided a guide to common dermatologic conditions and included images and a list of symptoms.7 To help keep physicians up to date on the most reliable dermatology apps, specialty societies, journal task forces, or interested dermatologists, residents, or medical students could publish updated literature on the most popular and most reviewed dermatology apps for patient education annually or biannually.
A practice’s website is a prime place for physicians to direct patients to educational content. Although many dermatology practice websites offer clinical information, the content often is focused on cosmetic procedures or is designed for search engine optimization to support online marketing and therefore may not be helpful to patients trying to understand a specific condition or treatment. Links to trusted resources, such as dermatology journals or medical societies, may be added but also would direct patients away from the practice’s website and would not allow physicians to customize the information he or she would like to share with their patients. Dermatologists should consider investing time and money into customizing educational material for their websites so patients can access health information from the source they trust most: their own physician.
Many of these digital options are useful for patients who want to access education material outside of the physician’s office, but digital opportunities to enhance point-of-care education also are available. In 2016, the American Academy of Dermatology partnered with ContextMedia:Health with the goal of delivering important decision enhancement technologies, educational content, and intelligence to patients and dermatologists for use before and during the consultation.8 ContextMedia:Health’s digital wallboard tablets are an engaging way to visually explain conditions and treatments to patients during the consultation, thus empowering physicians and patients to make decisions together and helping patients to be better advocates of their own health care. The downside is that health care workers must devote time and resources to be trained in using these devices.
The increasing availability of technology for electronic health information can be both beneficial and challenging for dermatologists. Physicians should explore and familiarize themselves with the tools that are available and assess their effectiveness by communicating with patients about their perception and understanding of their conditions. Digital delivery of health information is not meant to replace other methods of patient education but to supplement and reinforce them that which is verbally discussed during the office visit. Electronic health education demonstrates powerful potential, but it is up to the physician to direct patients to the appropriate resources and educational tools that will support a clear understanding of all elements of care.
Acknowledgment
The authors would like to thank Dr. Mark Becker (Berkeley, California) for helpful discussion and reviewing this manuscript.
Technology offers new opportunities that can both enhance and challenge the physician-patient relationship, including the ways in which patients are educated. Ensuring dermatology patients are appropriately educated about their conditions can improve clinical care and treatment adherence, increase patient satisfaction, and potentially decrease medical costs. There are various digital methods by which physicians can deliver information to their patients, and while there are benefits and drawbacks to each, many Americans turn to the Internet for health information—a practice that is only predicted to become more prevalent.1
Dermatologists should strive to keep up with this trend by staying informed about the digital patient education options that are available and which tools they can use to more effectively share their knowledge with patients. Electronic health education has a powerful potential, but it is up to physicians to direct patients to the appropriate resources and education tools that will support their clear understanding of all elements of care.
Effective patient education can transform the role of the patient from passive recipient to active participant in his/her care and subsequently supports the physician-patient relationship. The benefits of patient education are timely and valuable with the new pay-for-performance model instated by the Medicare Access and CHIP Reauthorization Act and the Merit-based Incentive Payment System.2 In dermatology, patient education alone can essentially be a management strategy for numerous conditions (eg, identifying which triggers patients with contact dermatitis should avoid). On the other hand, a lack of patient knowledge can result in perceived noncompliance or treatment failure, when in reality there has simply been a communication gap between the physician and the patient. For example, if a patient notices little to no improvement of a fungal infection after applying ketoconazole shampoo 2% to affected areas and immediately rinsing, this does not necessarily constitute a treatment failure, as the patient should have been educated on the importance of leaving the shampoo on for 5 minutes before rinsing. One study alluded to this communication gap, revealing physicians’ tendency to overestimate how effectively they are communicating with their patients.3
Successful patient education ultimately is dependent on both the content provided and the method of delivery. In one survey of 2636 Internet users, 72% of respondents admitted to searching online for health information within the previous year; however, the same survey showed that physicians remain patients’ most trusted source of information.4 Physicians can use digital education methods to fulfill patient needs by providing them with and directing them to credible up-to-date sources.
Physicians can use electronic medical record (EMR) systems to electronically deliver health information to patients by directly communicating via an online patient portal. Allowing patients to engage with their health care providers electronically has been shown to increase patient satisfaction, promote adherence to preventative and treatment recommendations, improve clinical outcomes, and lower medical costs.5 The online portal can provide direct links for patients to digital resources; for example, MedlinePlus Connect (https://medlineplus.gov/connect/overview.html) is a free service that connects patients to MedlinePlus, an authoritative, up-to-date health information resource for consumer health information; however, many EMR systems lack quality dermatology content, as there is a greater emphasis on primary care, and patient usage of these online portals also is notoriously low.6 Dermatologists can work with EMR vendors to enhance the dermatology content for patient portals, and in some cases, specialty-specific content may be available for an additional fee. Clinicians can make their patients aware of the online portal and incentivize its use by sending an informational email, including a link on their practice’s website, promoting the portal during check-in and check-out at office visits, making tablets or kiosks available in the waiting room for sign-up, hanging posters in the examination rooms, and explaining the portal’s useful features during consultations with patients.
Mobile apps have revolutionized the potential for dermatologists to streamline patient education to a large population. In a 2014 review of 365 dermatology mobile apps, 13% were categorized as educational aids, adding to the realm of possibilities for digital patient education. For example, these apps may provide information on specific dermatologic conditions and medications, help users perform skin cancer checks, and provide reminders for when to administer injections for those on biologics. However, a drawback of medical mobile apps is that, to date, the US Food and Drug Administration has not released formal guidelines for their development.
It would be impractical for busy dermatologists to keep up with the credibility of every mobile app available in a growing market, but one solution could be for physicians to stay informed on only the most popular and most reviewed apps to keep in their digital toolbox. In 2014, the most reviewed dermatology app was the Dermatology app, which provided a guide to common dermatologic conditions and included images and a list of symptoms.7 To help keep physicians up to date on the most reliable dermatology apps, specialty societies, journal task forces, or interested dermatologists, residents, or medical students could publish updated literature on the most popular and most reviewed dermatology apps for patient education annually or biannually.
A practice’s website is a prime place for physicians to direct patients to educational content. Although many dermatology practice websites offer clinical information, the content often is focused on cosmetic procedures or is designed for search engine optimization to support online marketing and therefore may not be helpful to patients trying to understand a specific condition or treatment. Links to trusted resources, such as dermatology journals or medical societies, may be added but also would direct patients away from the practice’s website and would not allow physicians to customize the information he or she would like to share with their patients. Dermatologists should consider investing time and money into customizing educational material for their websites so patients can access health information from the source they trust most: their own physician.
Many of these digital options are useful for patients who want to access education material outside of the physician’s office, but digital opportunities to enhance point-of-care education also are available. In 2016, the American Academy of Dermatology partnered with ContextMedia:Health with the goal of delivering important decision enhancement technologies, educational content, and intelligence to patients and dermatologists for use before and during the consultation.8 ContextMedia:Health’s digital wallboard tablets are an engaging way to visually explain conditions and treatments to patients during the consultation, thus empowering physicians and patients to make decisions together and helping patients to be better advocates of their own health care. The downside is that health care workers must devote time and resources to be trained in using these devices.
The increasing availability of technology for electronic health information can be both beneficial and challenging for dermatologists. Physicians should explore and familiarize themselves with the tools that are available and assess their effectiveness by communicating with patients about their perception and understanding of their conditions. Digital delivery of health information is not meant to replace other methods of patient education but to supplement and reinforce them that which is verbally discussed during the office visit. Electronic health education demonstrates powerful potential, but it is up to the physician to direct patients to the appropriate resources and educational tools that will support a clear understanding of all elements of care.
Acknowledgment
The authors would like to thank Dr. Mark Becker (Berkeley, California) for helpful discussion and reviewing this manuscript.
- Explosive growth in healthcare apps raises oversight questions. Association of American Medical Colleges website. https://www.aamc.org/newsroom/reporter/october2012/308516/health-care-apps.html. Accessed January 16, 2017.
- What’s MACRA? Centers for Medicare and Medicaid Services website. https://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/Value-Based-Programs/MACRA-MIPS-and-APMs/MACRA-MIPS-and-APMs.html. Updated November 16, 2017. Accessed February 26, 2018.
- Duffy FD, Gordon GH, Whelan G, et al. Assessing competence in communication and interpersonal skills: the Kalamazoo II report. Acad Med. 2004;79:495-507.
- Dutta-Bergman M. Trusted online sources of health information: differences in demographics, health beliefs, and health-information orientation [published online September 25, 2003]. J Med Internet Res. 2003;5:E21.
- Griffin A, Skinner A, Thornhill J, et al. Patient portals: who uses them? what features do they use? and do they reduce hospital readmissions? Appl Clin Inform. 2016;7:489-501.
- Lin CT, Wittevrongel L, Moore L, et al. An internet-based patient-provider communication system: randomized controlled trial. J Med Internet Res. 2005;7:E47.
- Patel S, Eluri M, Boyers LN, et al. Update on mobile applications in dermatology [published online November 9, 2014]. Dermatol Online J. 2014;21. pii:13030/qt1zc343js.
- American Academy of Dermatology selects ContextMedia:Health as patient education affinity partner. American Academy of Dermatology website. https://www.aad.org/media/news-releases/aad-selects-patient-education-affinity-partner. Published November 14, 2016. Accessed February 25, 2018.
- Explosive growth in healthcare apps raises oversight questions. Association of American Medical Colleges website. https://www.aamc.org/newsroom/reporter/october2012/308516/health-care-apps.html. Accessed January 16, 2017.
- What’s MACRA? Centers for Medicare and Medicaid Services website. https://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/Value-Based-Programs/MACRA-MIPS-and-APMs/MACRA-MIPS-and-APMs.html. Updated November 16, 2017. Accessed February 26, 2018.
- Duffy FD, Gordon GH, Whelan G, et al. Assessing competence in communication and interpersonal skills: the Kalamazoo II report. Acad Med. 2004;79:495-507.
- Dutta-Bergman M. Trusted online sources of health information: differences in demographics, health beliefs, and health-information orientation [published online September 25, 2003]. J Med Internet Res. 2003;5:E21.
- Griffin A, Skinner A, Thornhill J, et al. Patient portals: who uses them? what features do they use? and do they reduce hospital readmissions? Appl Clin Inform. 2016;7:489-501.
- Lin CT, Wittevrongel L, Moore L, et al. An internet-based patient-provider communication system: randomized controlled trial. J Med Internet Res. 2005;7:E47.
- Patel S, Eluri M, Boyers LN, et al. Update on mobile applications in dermatology [published online November 9, 2014]. Dermatol Online J. 2014;21. pii:13030/qt1zc343js.
- American Academy of Dermatology selects ContextMedia:Health as patient education affinity partner. American Academy of Dermatology website. https://www.aad.org/media/news-releases/aad-selects-patient-education-affinity-partner. Published November 14, 2016. Accessed February 25, 2018.
Linear Terra Firma–Forme Dermatosis of the Midline Back
Terra firma–forme dermatosis (TFFD) was first described by Duncan et al,1 in 1987 and is characterized by brown to black pigmented plaques on the skin that cannot be removed with soap and water but are easily wiped away with isopropyl alcohol. Since that publication, relatively few case reports and case series have been published. We present a case of linear TFFD on the midline back of a 46-year-old woman.
Case Report
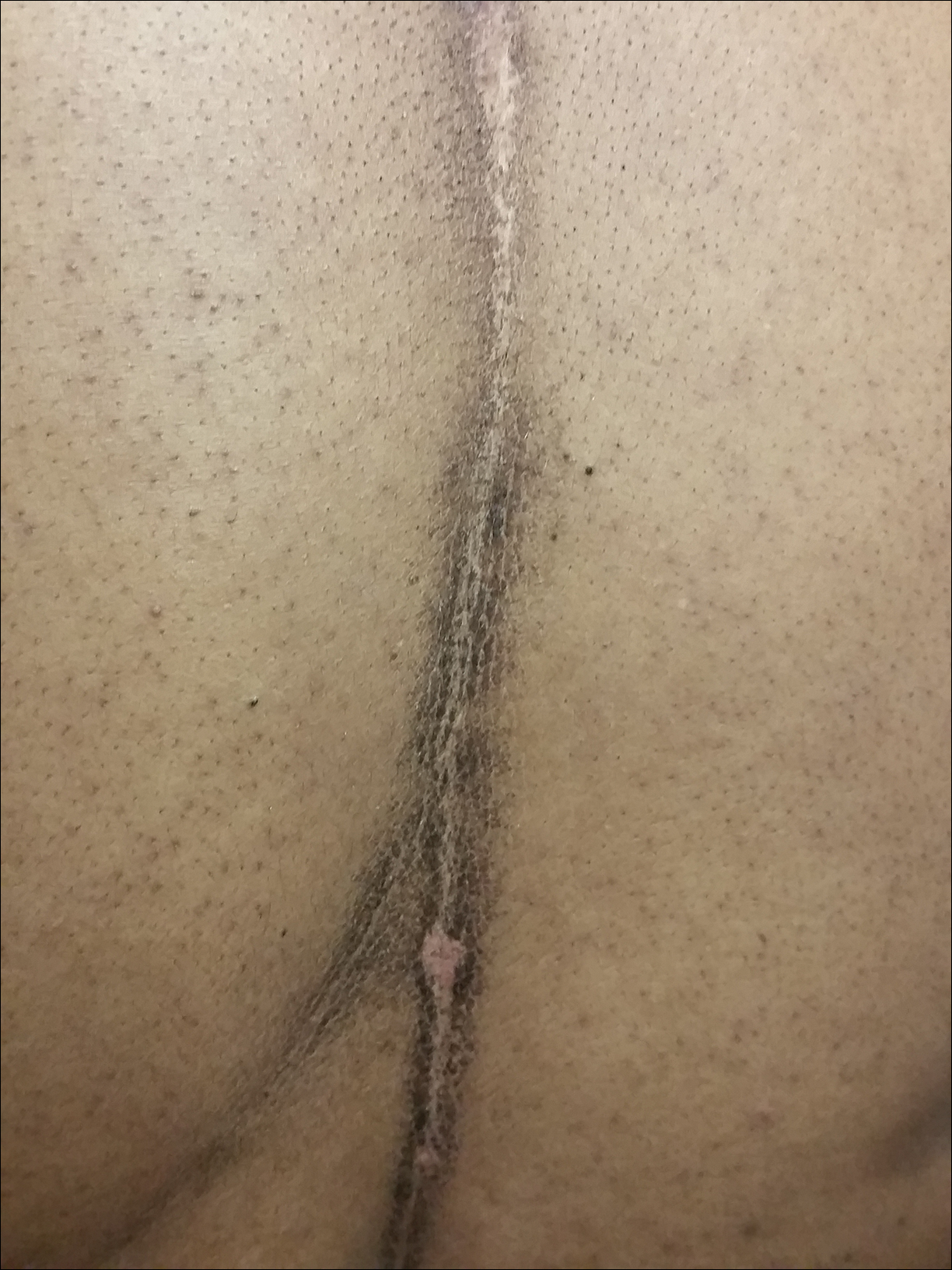
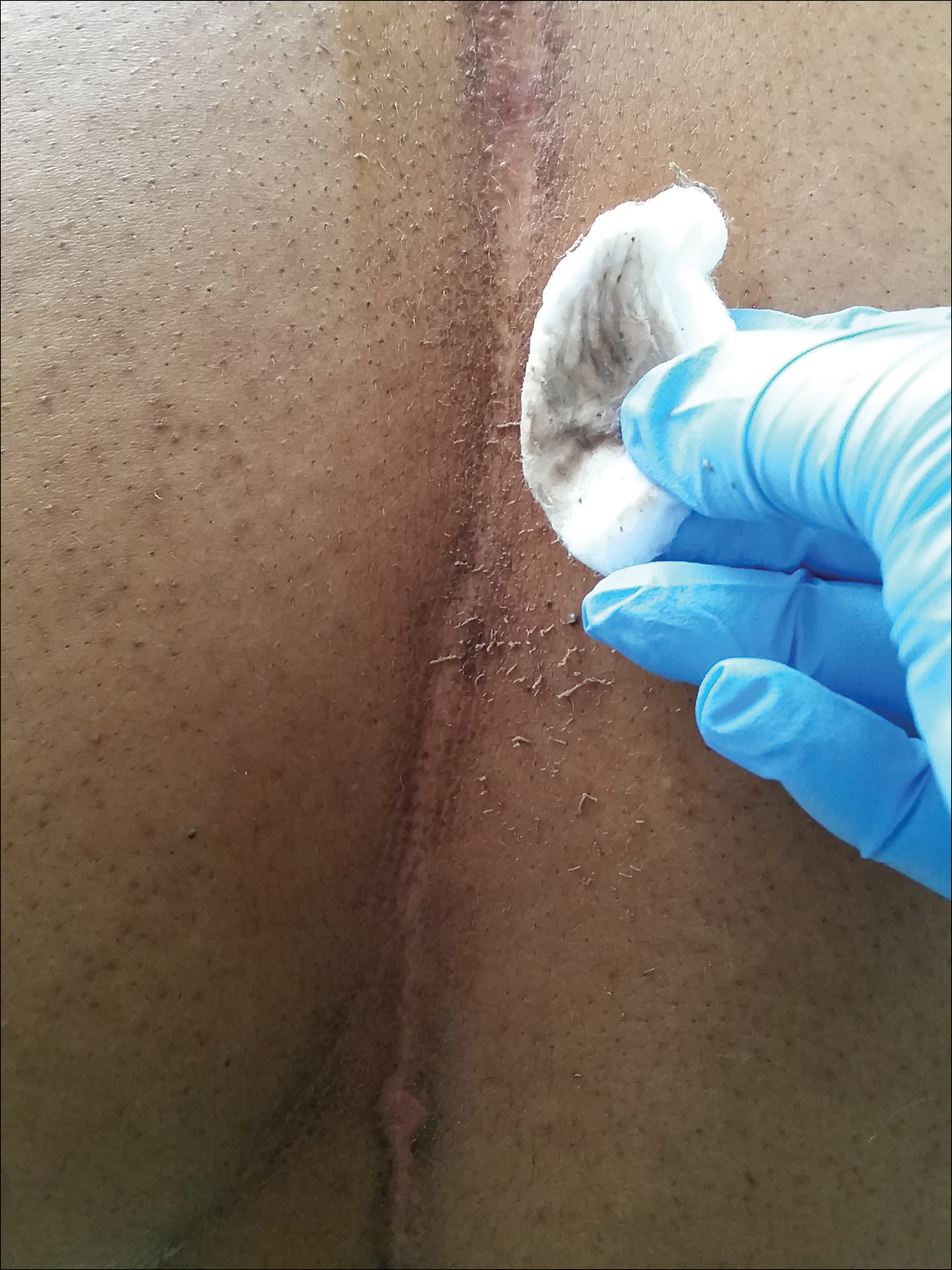
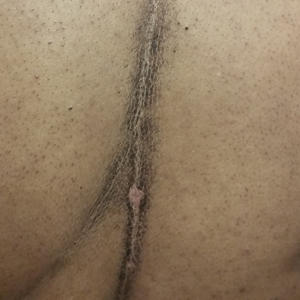
A 46-year-old woman presented to our clinic for evaluation of a lesion on the back that had been present for 3 years. An initial diagnosis of acanthosis nigricans or lichen simplex chronicus was made and treatment with topical triamcinolone cream 0.1% was initiated. However, after 8 months of treatment, no improvement was observed and the patient returned to our clinic. Her medical history was notable for obesity, type 2 diabetes mellitus, and hypertension. The patient stated that she maintained good hygiene, including daily to twice-daily showers with soap. Physical examination revealed a linear, hyperkeratotic, dark-brown plaque on the midline back extending from the top of the sacrum to the upper back (Figure 1). No other areas of skin involvement were noted. The hyperpigmented scales were easily removed with an isopropyl alcohol swab, which confirmed a diagnosis of TFFD (Figure 2). The patient was given ammonium lactate lotion 12% to apply to the lesion once daily using an applicator stick if the lesion recurred. She reported some improvement during this treatment. She occasionally had recurrent lesions, which were removed with isopropyl alcohol on subsequent dermatology visits.
Comment
Terra firma–forme dermatosis is an idiopathic condition that, although benign, can cause notable distress to patients. It presents clinically as asymptomatic, brown or black, hyperpigmented, hyperkeratotic, verrucous, or papillomatous plaques or light scaling in some cases.1-4 It can be readily cleared by rubbing with isopropyl alcohol but is resistant to ordinary soap and water.1
Recent reports have shown that TFFD may be more common than once thought.4-6 Although commonly observed in children, TFFD has been reported over a wide range of ages (4–86 years).2-5 The face, ankles, neck, and trunk are the most commonly affected areas.4,7,8 Areas that are less commonly affected often include surgical incision sites as well as the scalp, axillae, back, umbilical area, pubic area, arms, and legs.2-4,8,9 The lesions may be generalized or localized and are sometimes found to be symmetrical.4,10,11
The exact etiology of TFFD is unknown but is believed to be due to melanin retention and alteration or a delay of keratinization that leads to the buildup and compaction of scales.1,2,12 Poor hygiene generally is considered to exclude the diagnosis of TFFD in favor of dermatitis neglecta.6,12,13 Histopathology typically shows epidermal acanthosis, lamellar hyperkeratosis, and orthokeratotic whorls.3,7 However, biopsies seldom are performed due to the ease of diagnosis by removal by cleaning the lesion with isopropyl alcohol.
The diagnosis is confirmed by resolution of the rash after cleaning with isopropyl alcohol.1 Further confirmation of this diagnosis can be achieved through dermoscopy, as large, polygonal, platelike, brown scales can be found arranged together giving a mosaic pattern.6 In addition to cleaning with isopropyl alcohol,5,8 other treatments have shown efficacy for more resistant cases of TFFD, including topical keratolytic agents (eg, lactic acid, urea lotion).4,14
Conclusion
Terra firma–forme dermatosis is a condition that if recognized early, may provide treatment satisfaction through immediate removal of the lesions. Physicians should keep TFFD in their differential during evaluation of patients with asymptomatic, hyperpigmented, hyperkeratotic plaques. Awareness of TFFD is important, as early diagnosis can prevent unnecessary treatment and diagnostic workup.
- Duncan CW, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Browning J, Rosen T. Terra firmaforme dermatosis revisited. Dermatol Online J. 2005;11:11-13.
- Ashique KT, Kaliyadan F, Goyal T. Terra firma-forme dermatosis: report of a series of 11 cases and a brief review of the literature. Int J Dermatol. 2016;55:769-774.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;29:297-300.
- Greywal T, Cohen PR. Terra firma-forme dermatosis: a report of ten individuals with Duncan’s dirty dermatosis and literature review. Dermatol Pract Concept. 2015;5:29-33.
- Abdel-Razek MM, Fathy H. Terra firm-forme dermatosis: case series and dermoscopic features. Dermatol Online J. 2015;21:4-7.
- Akkash L, Badran D, Al-Omari AQ. Terra firma forme dermatosis. case series and review of the literature. J Dtsch Dermatol Ges. 2009;7:102-107.
- O’Brien TJ, Hall AP. Terra firma-forme dermatosis. Aust J Dermatol. 1997;38:163-164.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
- Santarpia M, Guarneri C. Terra firma-forme dermatosis. Eur J Intern Med. 2016;34:1-2.
- Panchal K, Bhalla N, Salunke P, et al. Extensive terra firma forme dermatosis (TFFD): a rare presentation. Indian Dermatol Online J. 2015;6:458-459.
- Erkek E, Sahin S, Cetin ED, et al. Terra firmaforme dermatosis revisited. Indian J Dermatol Venereol Leprol. 2012;78:358-360.
- Poskitt L, Wayte J, Wojnarowska F, et al. ‘Dermatitis neglecta’: unwashed dermatosis. Br J Dermatol. 1995;132:827-829.
- Unal E, Guarneri C, Chokoeva AA, et al. Terra firma-forme dermatosis [published online October 21, 2016]. Wien Med Wochenschr. 2017;167:66-69.
Terra firma–forme dermatosis (TFFD) was first described by Duncan et al,1 in 1987 and is characterized by brown to black pigmented plaques on the skin that cannot be removed with soap and water but are easily wiped away with isopropyl alcohol. Since that publication, relatively few case reports and case series have been published. We present a case of linear TFFD on the midline back of a 46-year-old woman.
Case Report
A 46-year-old woman presented to our clinic for evaluation of a lesion on the back that had been present for 3 years. An initial diagnosis of acanthosis nigricans or lichen simplex chronicus was made and treatment with topical triamcinolone cream 0.1% was initiated. However, after 8 months of treatment, no improvement was observed and the patient returned to our clinic. Her medical history was notable for obesity, type 2 diabetes mellitus, and hypertension. The patient stated that she maintained good hygiene, including daily to twice-daily showers with soap. Physical examination revealed a linear, hyperkeratotic, dark-brown plaque on the midline back extending from the top of the sacrum to the upper back (Figure 1). No other areas of skin involvement were noted. The hyperpigmented scales were easily removed with an isopropyl alcohol swab, which confirmed a diagnosis of TFFD (Figure 2). The patient was given ammonium lactate lotion 12% to apply to the lesion once daily using an applicator stick if the lesion recurred. She reported some improvement during this treatment. She occasionally had recurrent lesions, which were removed with isopropyl alcohol on subsequent dermatology visits.
Comment
Terra firma–forme dermatosis is an idiopathic condition that, although benign, can cause notable distress to patients. It presents clinically as asymptomatic, brown or black, hyperpigmented, hyperkeratotic, verrucous, or papillomatous plaques or light scaling in some cases.1-4 It can be readily cleared by rubbing with isopropyl alcohol but is resistant to ordinary soap and water.1
Recent reports have shown that TFFD may be more common than once thought.4-6 Although commonly observed in children, TFFD has been reported over a wide range of ages (4–86 years).2-5 The face, ankles, neck, and trunk are the most commonly affected areas.4,7,8 Areas that are less commonly affected often include surgical incision sites as well as the scalp, axillae, back, umbilical area, pubic area, arms, and legs.2-4,8,9 The lesions may be generalized or localized and are sometimes found to be symmetrical.4,10,11
The exact etiology of TFFD is unknown but is believed to be due to melanin retention and alteration or a delay of keratinization that leads to the buildup and compaction of scales.1,2,12 Poor hygiene generally is considered to exclude the diagnosis of TFFD in favor of dermatitis neglecta.6,12,13 Histopathology typically shows epidermal acanthosis, lamellar hyperkeratosis, and orthokeratotic whorls.3,7 However, biopsies seldom are performed due to the ease of diagnosis by removal by cleaning the lesion with isopropyl alcohol.
The diagnosis is confirmed by resolution of the rash after cleaning with isopropyl alcohol.1 Further confirmation of this diagnosis can be achieved through dermoscopy, as large, polygonal, platelike, brown scales can be found arranged together giving a mosaic pattern.6 In addition to cleaning with isopropyl alcohol,5,8 other treatments have shown efficacy for more resistant cases of TFFD, including topical keratolytic agents (eg, lactic acid, urea lotion).4,14
Conclusion
Terra firma–forme dermatosis is a condition that if recognized early, may provide treatment satisfaction through immediate removal of the lesions. Physicians should keep TFFD in their differential during evaluation of patients with asymptomatic, hyperpigmented, hyperkeratotic plaques. Awareness of TFFD is important, as early diagnosis can prevent unnecessary treatment and diagnostic workup.
Terra firma–forme dermatosis (TFFD) was first described by Duncan et al,1 in 1987 and is characterized by brown to black pigmented plaques on the skin that cannot be removed with soap and water but are easily wiped away with isopropyl alcohol. Since that publication, relatively few case reports and case series have been published. We present a case of linear TFFD on the midline back of a 46-year-old woman.
Case Report
A 46-year-old woman presented to our clinic for evaluation of a lesion on the back that had been present for 3 years. An initial diagnosis of acanthosis nigricans or lichen simplex chronicus was made and treatment with topical triamcinolone cream 0.1% was initiated. However, after 8 months of treatment, no improvement was observed and the patient returned to our clinic. Her medical history was notable for obesity, type 2 diabetes mellitus, and hypertension. The patient stated that she maintained good hygiene, including daily to twice-daily showers with soap. Physical examination revealed a linear, hyperkeratotic, dark-brown plaque on the midline back extending from the top of the sacrum to the upper back (Figure 1). No other areas of skin involvement were noted. The hyperpigmented scales were easily removed with an isopropyl alcohol swab, which confirmed a diagnosis of TFFD (Figure 2). The patient was given ammonium lactate lotion 12% to apply to the lesion once daily using an applicator stick if the lesion recurred. She reported some improvement during this treatment. She occasionally had recurrent lesions, which were removed with isopropyl alcohol on subsequent dermatology visits.
Comment
Terra firma–forme dermatosis is an idiopathic condition that, although benign, can cause notable distress to patients. It presents clinically as asymptomatic, brown or black, hyperpigmented, hyperkeratotic, verrucous, or papillomatous plaques or light scaling in some cases.1-4 It can be readily cleared by rubbing with isopropyl alcohol but is resistant to ordinary soap and water.1
Recent reports have shown that TFFD may be more common than once thought.4-6 Although commonly observed in children, TFFD has been reported over a wide range of ages (4–86 years).2-5 The face, ankles, neck, and trunk are the most commonly affected areas.4,7,8 Areas that are less commonly affected often include surgical incision sites as well as the scalp, axillae, back, umbilical area, pubic area, arms, and legs.2-4,8,9 The lesions may be generalized or localized and are sometimes found to be symmetrical.4,10,11
The exact etiology of TFFD is unknown but is believed to be due to melanin retention and alteration or a delay of keratinization that leads to the buildup and compaction of scales.1,2,12 Poor hygiene generally is considered to exclude the diagnosis of TFFD in favor of dermatitis neglecta.6,12,13 Histopathology typically shows epidermal acanthosis, lamellar hyperkeratosis, and orthokeratotic whorls.3,7 However, biopsies seldom are performed due to the ease of diagnosis by removal by cleaning the lesion with isopropyl alcohol.
The diagnosis is confirmed by resolution of the rash after cleaning with isopropyl alcohol.1 Further confirmation of this diagnosis can be achieved through dermoscopy, as large, polygonal, platelike, brown scales can be found arranged together giving a mosaic pattern.6 In addition to cleaning with isopropyl alcohol,5,8 other treatments have shown efficacy for more resistant cases of TFFD, including topical keratolytic agents (eg, lactic acid, urea lotion).4,14
Conclusion
Terra firma–forme dermatosis is a condition that if recognized early, may provide treatment satisfaction through immediate removal of the lesions. Physicians should keep TFFD in their differential during evaluation of patients with asymptomatic, hyperpigmented, hyperkeratotic plaques. Awareness of TFFD is important, as early diagnosis can prevent unnecessary treatment and diagnostic workup.
- Duncan CW, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Browning J, Rosen T. Terra firmaforme dermatosis revisited. Dermatol Online J. 2005;11:11-13.
- Ashique KT, Kaliyadan F, Goyal T. Terra firma-forme dermatosis: report of a series of 11 cases and a brief review of the literature. Int J Dermatol. 2016;55:769-774.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;29:297-300.
- Greywal T, Cohen PR. Terra firma-forme dermatosis: a report of ten individuals with Duncan’s dirty dermatosis and literature review. Dermatol Pract Concept. 2015;5:29-33.
- Abdel-Razek MM, Fathy H. Terra firm-forme dermatosis: case series and dermoscopic features. Dermatol Online J. 2015;21:4-7.
- Akkash L, Badran D, Al-Omari AQ. Terra firma forme dermatosis. case series and review of the literature. J Dtsch Dermatol Ges. 2009;7:102-107.
- O’Brien TJ, Hall AP. Terra firma-forme dermatosis. Aust J Dermatol. 1997;38:163-164.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
- Santarpia M, Guarneri C. Terra firma-forme dermatosis. Eur J Intern Med. 2016;34:1-2.
- Panchal K, Bhalla N, Salunke P, et al. Extensive terra firma forme dermatosis (TFFD): a rare presentation. Indian Dermatol Online J. 2015;6:458-459.
- Erkek E, Sahin S, Cetin ED, et al. Terra firmaforme dermatosis revisited. Indian J Dermatol Venereol Leprol. 2012;78:358-360.
- Poskitt L, Wayte J, Wojnarowska F, et al. ‘Dermatitis neglecta’: unwashed dermatosis. Br J Dermatol. 1995;132:827-829.
- Unal E, Guarneri C, Chokoeva AA, et al. Terra firma-forme dermatosis [published online October 21, 2016]. Wien Med Wochenschr. 2017;167:66-69.
- Duncan CW, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567-569.
- Browning J, Rosen T. Terra firmaforme dermatosis revisited. Dermatol Online J. 2005;11:11-13.
- Ashique KT, Kaliyadan F, Goyal T. Terra firma-forme dermatosis: report of a series of 11 cases and a brief review of the literature. Int J Dermatol. 2016;55:769-774.
- Berk DR. Terra firma-forme dermatosis: a retrospective review of 31 patients. Pediatr Dermatol. 2012;29:297-300.
- Greywal T, Cohen PR. Terra firma-forme dermatosis: a report of ten individuals with Duncan’s dirty dermatosis and literature review. Dermatol Pract Concept. 2015;5:29-33.
- Abdel-Razek MM, Fathy H. Terra firm-forme dermatosis: case series and dermoscopic features. Dermatol Online J. 2015;21:4-7.
- Akkash L, Badran D, Al-Omari AQ. Terra firma forme dermatosis. case series and review of the literature. J Dtsch Dermatol Ges. 2009;7:102-107.
- O’Brien TJ, Hall AP. Terra firma-forme dermatosis. Aust J Dermatol. 1997;38:163-164.
- Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008;47:482-484.
- Santarpia M, Guarneri C. Terra firma-forme dermatosis. Eur J Intern Med. 2016;34:1-2.
- Panchal K, Bhalla N, Salunke P, et al. Extensive terra firma forme dermatosis (TFFD): a rare presentation. Indian Dermatol Online J. 2015;6:458-459.
- Erkek E, Sahin S, Cetin ED, et al. Terra firmaforme dermatosis revisited. Indian J Dermatol Venereol Leprol. 2012;78:358-360.
- Poskitt L, Wayte J, Wojnarowska F, et al. ‘Dermatitis neglecta’: unwashed dermatosis. Br J Dermatol. 1995;132:827-829.
- Unal E, Guarneri C, Chokoeva AA, et al. Terra firma-forme dermatosis [published online October 21, 2016]. Wien Med Wochenschr. 2017;167:66-69.
Practice Points
- Terra firma-forme dermatosis (TFFD) is an idiopathic condition characterized by asymptomatic hyperpigmented and hyperkeratotic plaques that are resistant to removal with soap and water.
- Diagnosis and cure of TFFD can be achieved through removal by rubbing with isopropyl alcohol.
- Increased awareness of the clinical presentation and treatment of TFFD may help patients avoid unnecessary treatment and workup and leads to immediate resolution of the condition.
Postherpetic Isotopic Responses With 3 Simultaneously Occurring Reactions Following Herpes Zoster
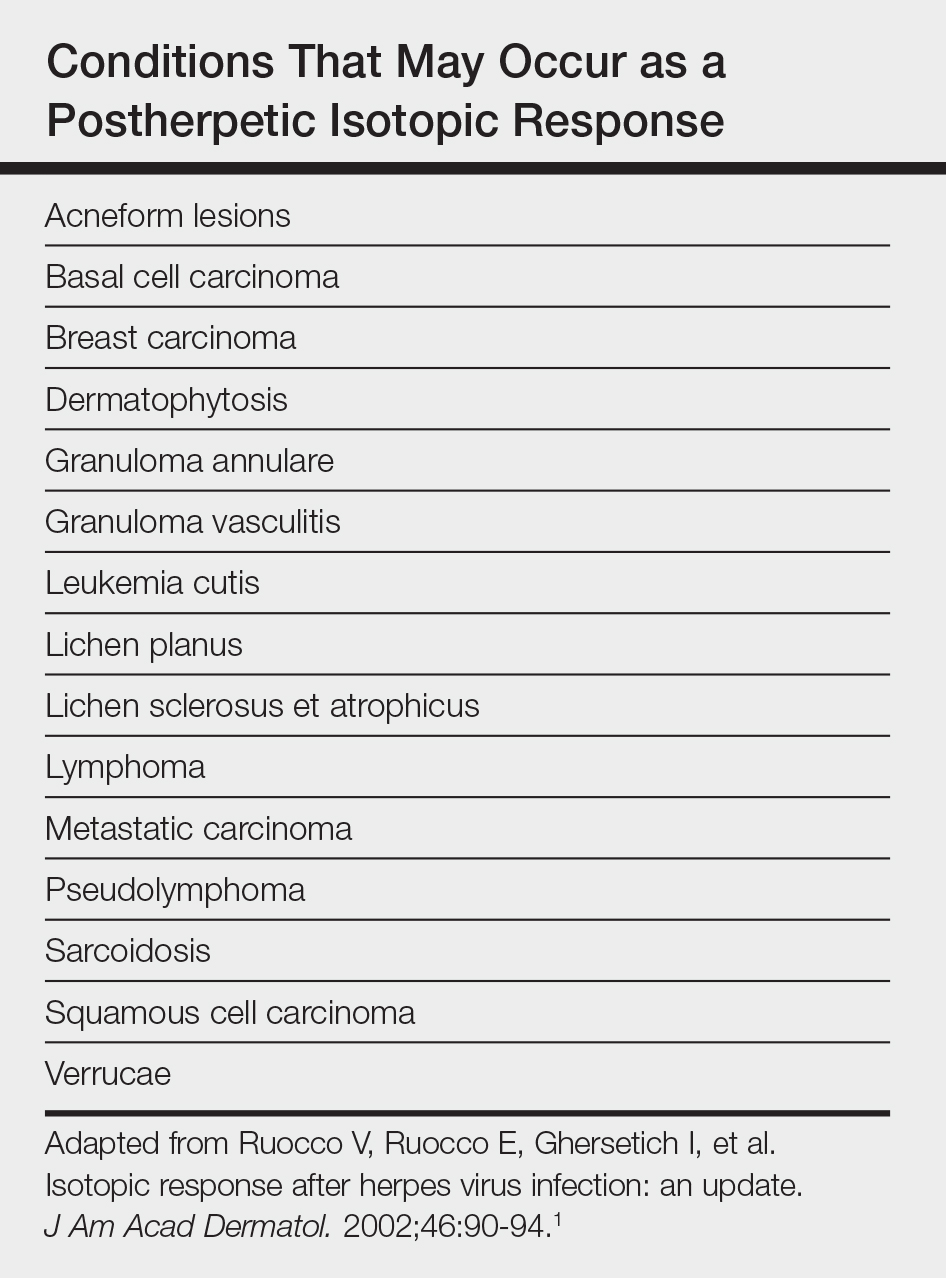
Postherpetic isotopic response (PHIR) refers to the occurrence of a second disease manifesting at the site of prior herpes infection. Many forms of PHIR have been described (Table), with postzoster granulomatous dermatitis (eg, granuloma annulare, sarcoidosis, granulomatous vasculitis) being the most common.1 Both primary and metastatic malignancies also can occur at the site of a prior herpes infection. Rarely, multiple types of PHIRs occur simultaneously. We report a case of 3 simultaneously occurring postzoster isotopic responses--granulomatous dermatitis, vasculitis, and chronic lymphocytic leukemia (CLL)--and review the various types of PHIRs.
Case Report
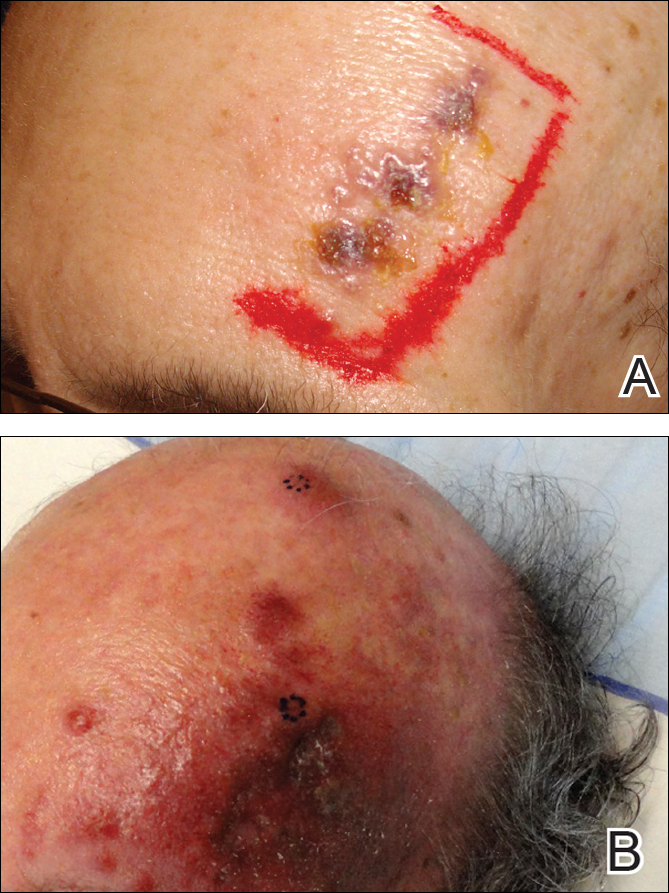
A 55-year-old man with a 4-year history of CLL was admitted to the hospital due to a painful rash on the left side of the face of 2 months' duration. Erythematous to violaceous plaques with surrounding papules and nodules were present on the left side of the forehead and frontal scalp with focal ulceration. Two months prior, the patient had unilateral vesicular lesions in the same distribution (Figure 1A). He initially received a 3-week course of acyclovir for a presumed herpes zoster infection and showed prompt improvement in the vesicular lesions. After resolution of the vesicles, papules and nodules began developing in the prior vesicular areas and he was treated with another course of acyclovir with the addition of clindamycin. When the lesions continued to progress and spread down the left side of the forehead and upper eyelid (Figure 1B), he was admitted to the hospital and assessed by the consultative dermatology team. No fevers, chills, or other systemic symptoms were reported.
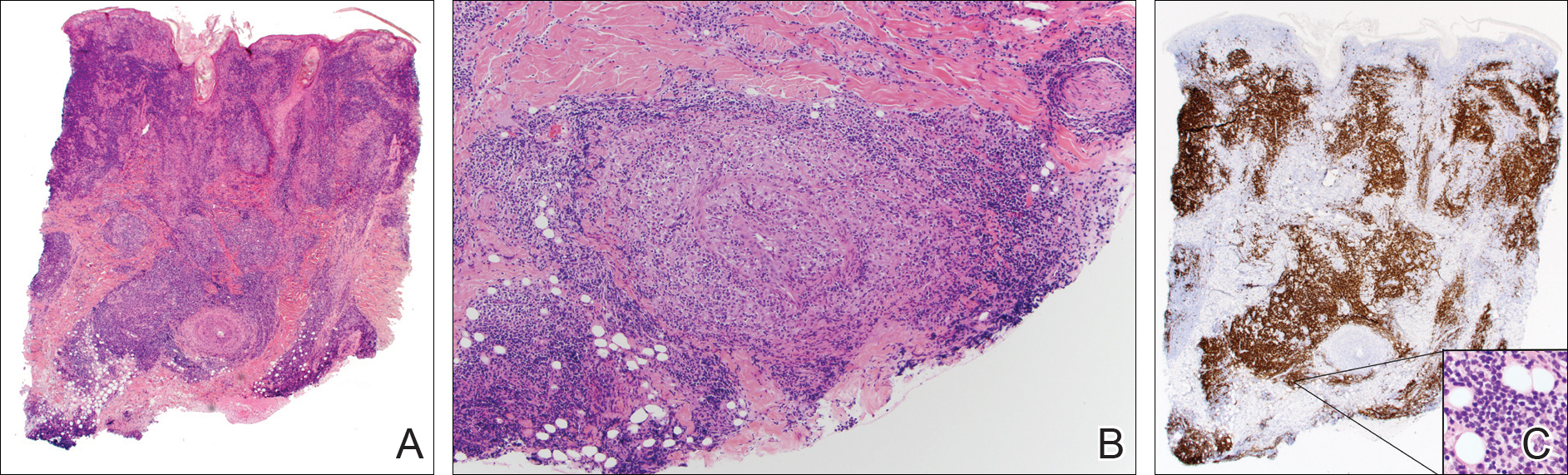
A punch biopsy showed a diffuse lymphocytic infiltrate filling the dermis and extending into the subcutis with nodular collections of histiocytes and some plasma cells scattered throughout (Figure 2A). A medium-vessel vasculitis was present with numerous histiocytes and lymphocytes infiltrating the muscular wall of a blood vessel in the subcutis (Figure 2B). CD3 and CD20 immunostaining showed an overwhelming majority of B cells, some with enlarged atypical nuclei and a smaller number of reactive T lymphocytes (Figure 2C). CD5 and CD43 were diffusely positive in the B cells, confirming the diagnosis of cutaneous CLL. CD23 staining was focally positive. Immunostaining for κ and λ light chains showed a marginal κ predominance. An additional biopsy for tissue culture was negative. A diagnosis of postzoster granulomatous dermatitis with vasculitis and cutaneous CLL was rendered.
Comment
Postherpetic Cutaneous Reactions
Various cutaneous reactions can occur at the site of prior herpes infection. The most frequently reported reactions are granulomatous dermatitides such as granuloma annulare, granulomatous vasculitis, granulomatous folliculitis, sarcoidosis, and nonspecific granulomatous dermatitis.1 Primary cutaneous malignancies and cutaneous metastases, including hematologic malignancies, have also been reported after herpetic infections. In a review of 127 patients with postherpetic cutaneous reactions, 47 had a granulomatous dermatitis, 32 had nonhematologic malignancies, 18 had leukemic or lymphomatous/pseudolymphomatous infiltrates, 10 had acneform lesions, 9 had nongranulomatous dermatitides such as lichen planus and allergic contact dermatitis, and 8 had nonherpetic skin infections; single cases of reactive perforating collagenosis, nodular solar degeneration, and a keloid also were reported.1
Pathogenesis of Cutaneous Reactions
Although postherpetic cutaneous reactions can develop in healthy individuals, they occur more often in immunocompromised patients. Postherpetic isotopic response has been used to describe the development of a nonherpetic disease at the site of prior herpes infection.2 Several different theories have been proposed to explain the pathogenesis of the PHIR, including an unusual delayed-type hypersensitivity reaction to residual viral antigen or host-tissue antigen altered by the virus. This delayed-type hypersensitivity explanation is supported by the presence of helper T cells, activated T lymphocytes, macrophages, varicella major viral envelope glycoproteins, and viral DNA in postherpetic granulomatous lesions3; however, cases that lack detectable virus and viral DNA in these types of lesions also have been reported.4
A second hypothesis proposes that inflammatory or viral-induced alteration of the local microvasculature results in increased site-specific susceptibility to subsequent inflammatory responses and drives these isotopic reactions.2,3 Damage or alteration of local peripheral nerves leading to abnormal release of specific neuromediators involved in regulating cutaneous inflammatory responses also may play a role.5 Varicella-zoster virus utilizes the peripheral nervous system to establish latent infection and can cause destruction of alpha delta and C nerve fibers in the dermis.1 Destruction of nerve fibers may indirectly influence the local immune system by altering the release of neuromediators such as substance P (known to increase blood vessel permeability, increase fibrinolytic activity, and induce mast cell secretion), vasoactive intestinal peptide (enhances monocyte migration, increases histamine release from mast cells, and inhibits natural killer cell activity), calcitonin gene-related peptide (increases vascular permeability, endothelial cell proliferation, and the accumulation of neutrophils), and melanocyte-stimulating hormone (induces anti-inflammatory cytokines). Disruption of the nervous system resulting in an altered local immune response also has been observed in other settings (eg, amputees who develop inflammatory diseases, bacterial and fungal infections, and cutaneous neoplasms confined to stump skin).1
Malignancies in PHIR
The granulomatous inflammation in PHIRs is a nonneoplastic inflammatory reaction with a variable lymphocytic component. Granuloma formation can be seen in both reactive inflammatory infiltrates and in cutaneous involvement of leukemias and lymphomas. Leukemia cutis has been reported in 4% to 20% of patients with CLL/small lymphocytic leukemia.6 In one series of 42 patients with CLL, the malignant cells were confined to the site of postherpetic scars in 14% (6/42) of patients.5 Sixteen percent (7/42) of patients had no prior diagnosis of CLL at the time they developed leukemia cutis, including one patient with leukemia cutis in a postzoster scar. The mechanism involved in the accumulation of neoplastic lymphocytes within postzoster scars has not been fully characterized. The idea that postzoster sites represent a site of least resistance for cutaneous infiltration of CLL due to the changes from prior inflammatory responses has been proposed.7
Combined CLL and granulomatous dermatitis at prior sites of herpes zoster was first reported in 1990.8 In 1995, Cerroni et al9 reported a series of 5 patients with cutaneous CLL following herpes zoster or herpes simplex virus infection. Three of those patients also demonstrated granuloma formation.9 Establishing a new diagnosis of CLL from a biopsy of postzoster granulomatous dermatitis with an associated lymphoid infiltrate also has been reported.10 Cerroni et al9 postulated that cutaneous CLL in post-herpes zoster scars may occur more frequently than reported due to misdiagnoses of CLL as pseudolymphoma. Two additional cases of postherpetic cutaneous CLL and granulomatous dermatitis have been reported since 1995.7,10
Diagnosis of Multiple PHIRs
The presence of 3 concurrent PHIRs is rare. The patient in this report had postzoster cutaneous CLL with an associated granulomatous dermatitis and medium-vessel vasculitis. One other case with these 3 findings was reported by Elgoweini et al.7 Overlooking important diagnoses when multiple findings are present in a biopsy can lead to diagnostic delay and incorrect treatment; we highlighted the importance of careful examination of biopsies in PHIRs to ensure diagnostic accuracy. In cases of postzoster granulomatous dermatitis, assessment of the lymphocytic component should not be overlooked. The presence of a dense lymphocytic infiltrate should raise the possibility of a lymphoproliferative disorder such as CLL, even in patients with no prior history of lymphoma. If initial immunostaining discloses a predominantly B-cell infiltrate, additional immuno-stains (eg, CD5, CD23, CD43) and/or genetic testing for monoclonality should be pursued.
Conclusion
Clinicians and dermatopathologists should be aware of the multiplicity of postherpetic isotopic responses and consider immunohistochemical stains to differentiate between a genuine lymphoma such as CLL and pseudolymphoma in PHIRs with a lymphoid infiltrate.
- Ruocco V, Ruocco E, Ghersetich I, et al. Isotopic response after herpes virus infection: an update. J Am Acad Dermatol. 2002;46:90-94.
- Wolf R, Wolf D, Ruocco E, et al. Wolf's isotopic response. Clin Dermatol. 2011;29:237-240.
- Nikkels AF, Debrus S, Delvenne P, et al. Viral glycoproteins in herpesviridae granulomas. Am J Dermatopathol. 1994;16:588-592.
- Snow J, el-Azhary R, Gibson L, et al. Granulomatous vasculitis associated with herpes virus: a persistent, painful, postherpetic papular eruption. Mayo Clin Proc. 1997;72:851-853.
- Cerroni L, Zenahlik P, Hofler G, et al. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia: a clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol. 1996;20:1000-1010.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Elgoweini M, Blessing K, Jackson R, et al. Coexistent granulomatous vasculitis and leukaemia cutis in a patient with resolving herpes zoster. Clin Exp Dermatol. 2011;36:749-751.
- Pujol RM, Matias-Guiu X, Planaguma M, et al. Chronic lymphocytic leukemia and cutaneous granulomas at sites of herpes zoster scars. Int J Dermatol. 1990;29:652-654.
- Cerroni L, Zenahlik P, Kerl H. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia arising at the site of herpes zoster and herpes simplex scars. Cancer. 1995;76:26-31.
- Trojjet S, Hammami H, Zaraa I, et al. Chronic lymphocytic leukemia revealed by a granulomatous zosteriform eruption. Skinmed. 2012;10:50-52.
Postherpetic isotopic response (PHIR) refers to the occurrence of a second disease manifesting at the site of prior herpes infection. Many forms of PHIR have been described (Table), with postzoster granulomatous dermatitis (eg, granuloma annulare, sarcoidosis, granulomatous vasculitis) being the most common.1 Both primary and metastatic malignancies also can occur at the site of a prior herpes infection. Rarely, multiple types of PHIRs occur simultaneously. We report a case of 3 simultaneously occurring postzoster isotopic responses--granulomatous dermatitis, vasculitis, and chronic lymphocytic leukemia (CLL)--and review the various types of PHIRs.
Case Report
A 55-year-old man with a 4-year history of CLL was admitted to the hospital due to a painful rash on the left side of the face of 2 months' duration. Erythematous to violaceous plaques with surrounding papules and nodules were present on the left side of the forehead and frontal scalp with focal ulceration. Two months prior, the patient had unilateral vesicular lesions in the same distribution (Figure 1A). He initially received a 3-week course of acyclovir for a presumed herpes zoster infection and showed prompt improvement in the vesicular lesions. After resolution of the vesicles, papules and nodules began developing in the prior vesicular areas and he was treated with another course of acyclovir with the addition of clindamycin. When the lesions continued to progress and spread down the left side of the forehead and upper eyelid (Figure 1B), he was admitted to the hospital and assessed by the consultative dermatology team. No fevers, chills, or other systemic symptoms were reported.
A punch biopsy showed a diffuse lymphocytic infiltrate filling the dermis and extending into the subcutis with nodular collections of histiocytes and some plasma cells scattered throughout (Figure 2A). A medium-vessel vasculitis was present with numerous histiocytes and lymphocytes infiltrating the muscular wall of a blood vessel in the subcutis (Figure 2B). CD3 and CD20 immunostaining showed an overwhelming majority of B cells, some with enlarged atypical nuclei and a smaller number of reactive T lymphocytes (Figure 2C). CD5 and CD43 were diffusely positive in the B cells, confirming the diagnosis of cutaneous CLL. CD23 staining was focally positive. Immunostaining for κ and λ light chains showed a marginal κ predominance. An additional biopsy for tissue culture was negative. A diagnosis of postzoster granulomatous dermatitis with vasculitis and cutaneous CLL was rendered.
Comment
Postherpetic Cutaneous Reactions
Various cutaneous reactions can occur at the site of prior herpes infection. The most frequently reported reactions are granulomatous dermatitides such as granuloma annulare, granulomatous vasculitis, granulomatous folliculitis, sarcoidosis, and nonspecific granulomatous dermatitis.1 Primary cutaneous malignancies and cutaneous metastases, including hematologic malignancies, have also been reported after herpetic infections. In a review of 127 patients with postherpetic cutaneous reactions, 47 had a granulomatous dermatitis, 32 had nonhematologic malignancies, 18 had leukemic or lymphomatous/pseudolymphomatous infiltrates, 10 had acneform lesions, 9 had nongranulomatous dermatitides such as lichen planus and allergic contact dermatitis, and 8 had nonherpetic skin infections; single cases of reactive perforating collagenosis, nodular solar degeneration, and a keloid also were reported.1
Pathogenesis of Cutaneous Reactions
Although postherpetic cutaneous reactions can develop in healthy individuals, they occur more often in immunocompromised patients. Postherpetic isotopic response has been used to describe the development of a nonherpetic disease at the site of prior herpes infection.2 Several different theories have been proposed to explain the pathogenesis of the PHIR, including an unusual delayed-type hypersensitivity reaction to residual viral antigen or host-tissue antigen altered by the virus. This delayed-type hypersensitivity explanation is supported by the presence of helper T cells, activated T lymphocytes, macrophages, varicella major viral envelope glycoproteins, and viral DNA in postherpetic granulomatous lesions3; however, cases that lack detectable virus and viral DNA in these types of lesions also have been reported.4
A second hypothesis proposes that inflammatory or viral-induced alteration of the local microvasculature results in increased site-specific susceptibility to subsequent inflammatory responses and drives these isotopic reactions.2,3 Damage or alteration of local peripheral nerves leading to abnormal release of specific neuromediators involved in regulating cutaneous inflammatory responses also may play a role.5 Varicella-zoster virus utilizes the peripheral nervous system to establish latent infection and can cause destruction of alpha delta and C nerve fibers in the dermis.1 Destruction of nerve fibers may indirectly influence the local immune system by altering the release of neuromediators such as substance P (known to increase blood vessel permeability, increase fibrinolytic activity, and induce mast cell secretion), vasoactive intestinal peptide (enhances monocyte migration, increases histamine release from mast cells, and inhibits natural killer cell activity), calcitonin gene-related peptide (increases vascular permeability, endothelial cell proliferation, and the accumulation of neutrophils), and melanocyte-stimulating hormone (induces anti-inflammatory cytokines). Disruption of the nervous system resulting in an altered local immune response also has been observed in other settings (eg, amputees who develop inflammatory diseases, bacterial and fungal infections, and cutaneous neoplasms confined to stump skin).1
Malignancies in PHIR
The granulomatous inflammation in PHIRs is a nonneoplastic inflammatory reaction with a variable lymphocytic component. Granuloma formation can be seen in both reactive inflammatory infiltrates and in cutaneous involvement of leukemias and lymphomas. Leukemia cutis has been reported in 4% to 20% of patients with CLL/small lymphocytic leukemia.6 In one series of 42 patients with CLL, the malignant cells were confined to the site of postherpetic scars in 14% (6/42) of patients.5 Sixteen percent (7/42) of patients had no prior diagnosis of CLL at the time they developed leukemia cutis, including one patient with leukemia cutis in a postzoster scar. The mechanism involved in the accumulation of neoplastic lymphocytes within postzoster scars has not been fully characterized. The idea that postzoster sites represent a site of least resistance for cutaneous infiltration of CLL due to the changes from prior inflammatory responses has been proposed.7
Combined CLL and granulomatous dermatitis at prior sites of herpes zoster was first reported in 1990.8 In 1995, Cerroni et al9 reported a series of 5 patients with cutaneous CLL following herpes zoster or herpes simplex virus infection. Three of those patients also demonstrated granuloma formation.9 Establishing a new diagnosis of CLL from a biopsy of postzoster granulomatous dermatitis with an associated lymphoid infiltrate also has been reported.10 Cerroni et al9 postulated that cutaneous CLL in post-herpes zoster scars may occur more frequently than reported due to misdiagnoses of CLL as pseudolymphoma. Two additional cases of postherpetic cutaneous CLL and granulomatous dermatitis have been reported since 1995.7,10
Diagnosis of Multiple PHIRs
The presence of 3 concurrent PHIRs is rare. The patient in this report had postzoster cutaneous CLL with an associated granulomatous dermatitis and medium-vessel vasculitis. One other case with these 3 findings was reported by Elgoweini et al.7 Overlooking important diagnoses when multiple findings are present in a biopsy can lead to diagnostic delay and incorrect treatment; we highlighted the importance of careful examination of biopsies in PHIRs to ensure diagnostic accuracy. In cases of postzoster granulomatous dermatitis, assessment of the lymphocytic component should not be overlooked. The presence of a dense lymphocytic infiltrate should raise the possibility of a lymphoproliferative disorder such as CLL, even in patients with no prior history of lymphoma. If initial immunostaining discloses a predominantly B-cell infiltrate, additional immuno-stains (eg, CD5, CD23, CD43) and/or genetic testing for monoclonality should be pursued.
Conclusion
Clinicians and dermatopathologists should be aware of the multiplicity of postherpetic isotopic responses and consider immunohistochemical stains to differentiate between a genuine lymphoma such as CLL and pseudolymphoma in PHIRs with a lymphoid infiltrate.
Postherpetic isotopic response (PHIR) refers to the occurrence of a second disease manifesting at the site of prior herpes infection. Many forms of PHIR have been described (Table), with postzoster granulomatous dermatitis (eg, granuloma annulare, sarcoidosis, granulomatous vasculitis) being the most common.1 Both primary and metastatic malignancies also can occur at the site of a prior herpes infection. Rarely, multiple types of PHIRs occur simultaneously. We report a case of 3 simultaneously occurring postzoster isotopic responses--granulomatous dermatitis, vasculitis, and chronic lymphocytic leukemia (CLL)--and review the various types of PHIRs.
Case Report
A 55-year-old man with a 4-year history of CLL was admitted to the hospital due to a painful rash on the left side of the face of 2 months' duration. Erythematous to violaceous plaques with surrounding papules and nodules were present on the left side of the forehead and frontal scalp with focal ulceration. Two months prior, the patient had unilateral vesicular lesions in the same distribution (Figure 1A). He initially received a 3-week course of acyclovir for a presumed herpes zoster infection and showed prompt improvement in the vesicular lesions. After resolution of the vesicles, papules and nodules began developing in the prior vesicular areas and he was treated with another course of acyclovir with the addition of clindamycin. When the lesions continued to progress and spread down the left side of the forehead and upper eyelid (Figure 1B), he was admitted to the hospital and assessed by the consultative dermatology team. No fevers, chills, or other systemic symptoms were reported.
A punch biopsy showed a diffuse lymphocytic infiltrate filling the dermis and extending into the subcutis with nodular collections of histiocytes and some plasma cells scattered throughout (Figure 2A). A medium-vessel vasculitis was present with numerous histiocytes and lymphocytes infiltrating the muscular wall of a blood vessel in the subcutis (Figure 2B). CD3 and CD20 immunostaining showed an overwhelming majority of B cells, some with enlarged atypical nuclei and a smaller number of reactive T lymphocytes (Figure 2C). CD5 and CD43 were diffusely positive in the B cells, confirming the diagnosis of cutaneous CLL. CD23 staining was focally positive. Immunostaining for κ and λ light chains showed a marginal κ predominance. An additional biopsy for tissue culture was negative. A diagnosis of postzoster granulomatous dermatitis with vasculitis and cutaneous CLL was rendered.
Comment
Postherpetic Cutaneous Reactions
Various cutaneous reactions can occur at the site of prior herpes infection. The most frequently reported reactions are granulomatous dermatitides such as granuloma annulare, granulomatous vasculitis, granulomatous folliculitis, sarcoidosis, and nonspecific granulomatous dermatitis.1 Primary cutaneous malignancies and cutaneous metastases, including hematologic malignancies, have also been reported after herpetic infections. In a review of 127 patients with postherpetic cutaneous reactions, 47 had a granulomatous dermatitis, 32 had nonhematologic malignancies, 18 had leukemic or lymphomatous/pseudolymphomatous infiltrates, 10 had acneform lesions, 9 had nongranulomatous dermatitides such as lichen planus and allergic contact dermatitis, and 8 had nonherpetic skin infections; single cases of reactive perforating collagenosis, nodular solar degeneration, and a keloid also were reported.1
Pathogenesis of Cutaneous Reactions
Although postherpetic cutaneous reactions can develop in healthy individuals, they occur more often in immunocompromised patients. Postherpetic isotopic response has been used to describe the development of a nonherpetic disease at the site of prior herpes infection.2 Several different theories have been proposed to explain the pathogenesis of the PHIR, including an unusual delayed-type hypersensitivity reaction to residual viral antigen or host-tissue antigen altered by the virus. This delayed-type hypersensitivity explanation is supported by the presence of helper T cells, activated T lymphocytes, macrophages, varicella major viral envelope glycoproteins, and viral DNA in postherpetic granulomatous lesions3; however, cases that lack detectable virus and viral DNA in these types of lesions also have been reported.4
A second hypothesis proposes that inflammatory or viral-induced alteration of the local microvasculature results in increased site-specific susceptibility to subsequent inflammatory responses and drives these isotopic reactions.2,3 Damage or alteration of local peripheral nerves leading to abnormal release of specific neuromediators involved in regulating cutaneous inflammatory responses also may play a role.5 Varicella-zoster virus utilizes the peripheral nervous system to establish latent infection and can cause destruction of alpha delta and C nerve fibers in the dermis.1 Destruction of nerve fibers may indirectly influence the local immune system by altering the release of neuromediators such as substance P (known to increase blood vessel permeability, increase fibrinolytic activity, and induce mast cell secretion), vasoactive intestinal peptide (enhances monocyte migration, increases histamine release from mast cells, and inhibits natural killer cell activity), calcitonin gene-related peptide (increases vascular permeability, endothelial cell proliferation, and the accumulation of neutrophils), and melanocyte-stimulating hormone (induces anti-inflammatory cytokines). Disruption of the nervous system resulting in an altered local immune response also has been observed in other settings (eg, amputees who develop inflammatory diseases, bacterial and fungal infections, and cutaneous neoplasms confined to stump skin).1
Malignancies in PHIR
The granulomatous inflammation in PHIRs is a nonneoplastic inflammatory reaction with a variable lymphocytic component. Granuloma formation can be seen in both reactive inflammatory infiltrates and in cutaneous involvement of leukemias and lymphomas. Leukemia cutis has been reported in 4% to 20% of patients with CLL/small lymphocytic leukemia.6 In one series of 42 patients with CLL, the malignant cells were confined to the site of postherpetic scars in 14% (6/42) of patients.5 Sixteen percent (7/42) of patients had no prior diagnosis of CLL at the time they developed leukemia cutis, including one patient with leukemia cutis in a postzoster scar. The mechanism involved in the accumulation of neoplastic lymphocytes within postzoster scars has not been fully characterized. The idea that postzoster sites represent a site of least resistance for cutaneous infiltration of CLL due to the changes from prior inflammatory responses has been proposed.7
Combined CLL and granulomatous dermatitis at prior sites of herpes zoster was first reported in 1990.8 In 1995, Cerroni et al9 reported a series of 5 patients with cutaneous CLL following herpes zoster or herpes simplex virus infection. Three of those patients also demonstrated granuloma formation.9 Establishing a new diagnosis of CLL from a biopsy of postzoster granulomatous dermatitis with an associated lymphoid infiltrate also has been reported.10 Cerroni et al9 postulated that cutaneous CLL in post-herpes zoster scars may occur more frequently than reported due to misdiagnoses of CLL as pseudolymphoma. Two additional cases of postherpetic cutaneous CLL and granulomatous dermatitis have been reported since 1995.7,10
Diagnosis of Multiple PHIRs
The presence of 3 concurrent PHIRs is rare. The patient in this report had postzoster cutaneous CLL with an associated granulomatous dermatitis and medium-vessel vasculitis. One other case with these 3 findings was reported by Elgoweini et al.7 Overlooking important diagnoses when multiple findings are present in a biopsy can lead to diagnostic delay and incorrect treatment; we highlighted the importance of careful examination of biopsies in PHIRs to ensure diagnostic accuracy. In cases of postzoster granulomatous dermatitis, assessment of the lymphocytic component should not be overlooked. The presence of a dense lymphocytic infiltrate should raise the possibility of a lymphoproliferative disorder such as CLL, even in patients with no prior history of lymphoma. If initial immunostaining discloses a predominantly B-cell infiltrate, additional immuno-stains (eg, CD5, CD23, CD43) and/or genetic testing for monoclonality should be pursued.
Conclusion
Clinicians and dermatopathologists should be aware of the multiplicity of postherpetic isotopic responses and consider immunohistochemical stains to differentiate between a genuine lymphoma such as CLL and pseudolymphoma in PHIRs with a lymphoid infiltrate.
- Ruocco V, Ruocco E, Ghersetich I, et al. Isotopic response after herpes virus infection: an update. J Am Acad Dermatol. 2002;46:90-94.
- Wolf R, Wolf D, Ruocco E, et al. Wolf's isotopic response. Clin Dermatol. 2011;29:237-240.
- Nikkels AF, Debrus S, Delvenne P, et al. Viral glycoproteins in herpesviridae granulomas. Am J Dermatopathol. 1994;16:588-592.
- Snow J, el-Azhary R, Gibson L, et al. Granulomatous vasculitis associated with herpes virus: a persistent, painful, postherpetic papular eruption. Mayo Clin Proc. 1997;72:851-853.
- Cerroni L, Zenahlik P, Hofler G, et al. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia: a clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol. 1996;20:1000-1010.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Elgoweini M, Blessing K, Jackson R, et al. Coexistent granulomatous vasculitis and leukaemia cutis in a patient with resolving herpes zoster. Clin Exp Dermatol. 2011;36:749-751.
- Pujol RM, Matias-Guiu X, Planaguma M, et al. Chronic lymphocytic leukemia and cutaneous granulomas at sites of herpes zoster scars. Int J Dermatol. 1990;29:652-654.
- Cerroni L, Zenahlik P, Kerl H. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia arising at the site of herpes zoster and herpes simplex scars. Cancer. 1995;76:26-31.
- Trojjet S, Hammami H, Zaraa I, et al. Chronic lymphocytic leukemia revealed by a granulomatous zosteriform eruption. Skinmed. 2012;10:50-52.
- Ruocco V, Ruocco E, Ghersetich I, et al. Isotopic response after herpes virus infection: an update. J Am Acad Dermatol. 2002;46:90-94.
- Wolf R, Wolf D, Ruocco E, et al. Wolf's isotopic response. Clin Dermatol. 2011;29:237-240.
- Nikkels AF, Debrus S, Delvenne P, et al. Viral glycoproteins in herpesviridae granulomas. Am J Dermatopathol. 1994;16:588-592.
- Snow J, el-Azhary R, Gibson L, et al. Granulomatous vasculitis associated with herpes virus: a persistent, painful, postherpetic papular eruption. Mayo Clin Proc. 1997;72:851-853.
- Cerroni L, Zenahlik P, Hofler G, et al. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia: a clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol. 1996;20:1000-1010.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Elgoweini M, Blessing K, Jackson R, et al. Coexistent granulomatous vasculitis and leukaemia cutis in a patient with resolving herpes zoster. Clin Exp Dermatol. 2011;36:749-751.
- Pujol RM, Matias-Guiu X, Planaguma M, et al. Chronic lymphocytic leukemia and cutaneous granulomas at sites of herpes zoster scars. Int J Dermatol. 1990;29:652-654.
- Cerroni L, Zenahlik P, Kerl H. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia arising at the site of herpes zoster and herpes simplex scars. Cancer. 1995;76:26-31.
- Trojjet S, Hammami H, Zaraa I, et al. Chronic lymphocytic leukemia revealed by a granulomatous zosteriform eruption. Skinmed. 2012;10:50-52.
Practice Points
- Multiple diseases may present in prior sites of herpes infection (postherpetic isotopic response).
- Granulomatous dermatitis is the most common postherpetic isotopic response, but other inflammatory, neoplastic, or infectious conditions also occur.
- Multiple conditions may present simultaneously at sites of herpes infection.
- Cutaneous involvement by chronic lymphocytic leukemia (CLL) can be easily overlooked in this setting.
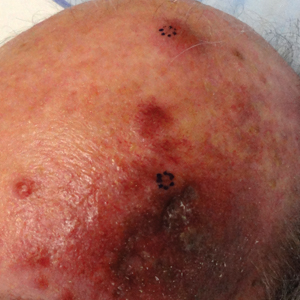
Alemtuzumab-induced autoimmunity: getting closer to answers
SAN DIEGO – The monoclonal antibody alemtuzumab can be an effective treatment for people living with multiple sclerosis, but there’s a catch — the agent is also associated with an increased risk for developing other autoimmune diseases, leaving clinicians with a conundrum.
“This is an efficacious treatment in multiple sclerosis” that can slow the rate of brain atrophy over the long-term, Alasdair Coles, MD, said at ACTRIMS Forum 2018, held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “But 1 or 2 years after each cycle of alemtuzumab [Lemtrada], patients are at very high risk of autoimmune diseases. This is the not-too-worrying thyroid disease, but there are some very troubling and potentially highly threatening complications at lower frequency.”
Subsequent autoimmune thyroid disease can affect up to 40% of patients treated with alemtuzumab, but immune thrombocytopenia (3%) and autoimmune renal disease (0.1%) are also reported. About 1 in 10 people treated with the monoclonal antibody for MS can also develop de novo asymptomatic autoantibodies (10%).
“People ask: ‘Why doesn’t MS come back as part of this generic mechanism?’ and I don’t know the answer to that,” Dr. Coles said.
In the United States, alemtuzumab is indicated for treatment of relapsing multiple sclerosis in adults who have failed to respond adequately to two or more previous therapies. In contrast, “this has become a first-line treatment in the U.K.,” said Dr. Coles, a professor in the department of clinical neurosciences at the University of Cambridge (England).
“Unfortunately, we can offer no proven treatment to prevent this autoimmunity.”
Considering the prospects for different proposed mechanisms
Dr. Coles shared some encouraging news at ACTRIMS Forum 2018. His team and other researchers are getting closer to understanding the cellular mechanism underlying the paradoxical autoimmunity associated with alemtuzumab. Published reports in the literature from others suggest faulty immune B cells could be the culprit, pointing to a similar reconstitution of B cells after bone marrow transplantation. However, he said, “There is no difference in this reconstitution pattern between those who do and don’t get autoimmunity. So we do not think that autoimmunity after alemtuzumab is primarily a B cell problem.”
Other investigators have pointed to possible depletion of a key immune regulatory cell associated with alemtuzumab, such as alterations in CD52-positive T cells that cause depletion in T cells as part of an autoimmune cascade that involve CD52-high expressing cells and sialic acid-binding immunoglobulin-like lectin 10. “I’m not going to describe why we don’t believe any of this,” Dr. Coles said, but added, “We cannot replicate the data in type 1 diabetes or MS about the depletion of T cells.”
Along with his colleague Joanne Jones, PhD, a clinical fellow in the same department at the University of Cambridge, Dr. Coles and his team instead propose that autoimmunity after alemtuzumab therapy is associated with a homeostatic proliferation of T cells in the context of a defective thymus. “We see thymic function reduced after alemtuzumab for a few months. We don’t know if alemtuzumab is having a direct impact on the thymus or if it’s an indirect effect though a cytokine storm at the time of administering alemtuzumab.”
In addition, in contrast to B cells, both CD4-positive and CD8-positive T cells are clonally restricted after alemtuzumab treatment, Dr. Coles explained.
“These are the only changes that distinguish patients who do and do not develop autoimmunity,” he said. “Those who develop autoimmunity have reduced clonality and have impaired thymic function compared to those who don’t.”
As the theory goes, the limited clonal repertoire leads to expansion of the T cells, preferentially expanding autoreactive T cells, leading to B-cell- and antibody-mediated autoimmunity.
The bigger picture
The autoimmune phenomenon is not unique to alemtuzumab or multiple sclerosis. “This turns out to be one of a family of clinical situations where the reconstitution of the depleted lymphocyte repertoire leads to autoimmunity,” Dr. Coles said. A similar effect was seen years ago when very lymphopenic HIV patients were given antiviral therapy, he added, affecting about 10% of treated patients. About 10% of bone marrow transplant patients may experience similar autoimmune concerns.
“What we do think is true is we’ve tapped into a classical expression of autoimmunity,” Dr. Coles said. “Alemtuzumab is a fantastic opportunity to study the mechanisms underlying lymphopenia-associated autoimmunity.”
A ‘tantalizing prospect’
“It’s a tantalizing prospect that susceptible individuals might be identified in the future prior to treatment,” Dr. Coles said. One promising lead, he added, is “we also looked at IL-21. We showed that after treatment, and perhaps more interestingly, before treatment with alemtuzumab, serum IL-21 is greater in those who subsequently develop autoimmune disease. This suggests some individuals are prone to develop autoimmune disease, and could be identified potentially prior to treatment with alemtuzumab.”
More work is needed, including the development of more sensitive IL-21 assays for use in this population, Dr. Coles said. “Please do not attempt to predict the risk of autoimmunity after alemtuzumab using the current commercial assays. This is a source of some frustration for me.”
A potential route of lymphocyte repertoire reconstitution after alemtuzumab is thymic reconstitution, leading to a more diverse immune repertoire, Dr. Coles said. “The obvious corollary of this is if we can direct reconstitution through the thymic reconstitution, we should be able to prevent autoimmunity.”
Dr. Coles disclosed that he receives honoraria for travel and speaking from Sanofi Genzyme, which markets alemtuzumab.
SAN DIEGO – The monoclonal antibody alemtuzumab can be an effective treatment for people living with multiple sclerosis, but there’s a catch — the agent is also associated with an increased risk for developing other autoimmune diseases, leaving clinicians with a conundrum.
“This is an efficacious treatment in multiple sclerosis” that can slow the rate of brain atrophy over the long-term, Alasdair Coles, MD, said at ACTRIMS Forum 2018, held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “But 1 or 2 years after each cycle of alemtuzumab [Lemtrada], patients are at very high risk of autoimmune diseases. This is the not-too-worrying thyroid disease, but there are some very troubling and potentially highly threatening complications at lower frequency.”
Subsequent autoimmune thyroid disease can affect up to 40% of patients treated with alemtuzumab, but immune thrombocytopenia (3%) and autoimmune renal disease (0.1%) are also reported. About 1 in 10 people treated with the monoclonal antibody for MS can also develop de novo asymptomatic autoantibodies (10%).
“People ask: ‘Why doesn’t MS come back as part of this generic mechanism?’ and I don’t know the answer to that,” Dr. Coles said.
In the United States, alemtuzumab is indicated for treatment of relapsing multiple sclerosis in adults who have failed to respond adequately to two or more previous therapies. In contrast, “this has become a first-line treatment in the U.K.,” said Dr. Coles, a professor in the department of clinical neurosciences at the University of Cambridge (England).
“Unfortunately, we can offer no proven treatment to prevent this autoimmunity.”
Considering the prospects for different proposed mechanisms
Dr. Coles shared some encouraging news at ACTRIMS Forum 2018. His team and other researchers are getting closer to understanding the cellular mechanism underlying the paradoxical autoimmunity associated with alemtuzumab. Published reports in the literature from others suggest faulty immune B cells could be the culprit, pointing to a similar reconstitution of B cells after bone marrow transplantation. However, he said, “There is no difference in this reconstitution pattern between those who do and don’t get autoimmunity. So we do not think that autoimmunity after alemtuzumab is primarily a B cell problem.”
Other investigators have pointed to possible depletion of a key immune regulatory cell associated with alemtuzumab, such as alterations in CD52-positive T cells that cause depletion in T cells as part of an autoimmune cascade that involve CD52-high expressing cells and sialic acid-binding immunoglobulin-like lectin 10. “I’m not going to describe why we don’t believe any of this,” Dr. Coles said, but added, “We cannot replicate the data in type 1 diabetes or MS about the depletion of T cells.”
Along with his colleague Joanne Jones, PhD, a clinical fellow in the same department at the University of Cambridge, Dr. Coles and his team instead propose that autoimmunity after alemtuzumab therapy is associated with a homeostatic proliferation of T cells in the context of a defective thymus. “We see thymic function reduced after alemtuzumab for a few months. We don’t know if alemtuzumab is having a direct impact on the thymus or if it’s an indirect effect though a cytokine storm at the time of administering alemtuzumab.”
In addition, in contrast to B cells, both CD4-positive and CD8-positive T cells are clonally restricted after alemtuzumab treatment, Dr. Coles explained.
“These are the only changes that distinguish patients who do and do not develop autoimmunity,” he said. “Those who develop autoimmunity have reduced clonality and have impaired thymic function compared to those who don’t.”
As the theory goes, the limited clonal repertoire leads to expansion of the T cells, preferentially expanding autoreactive T cells, leading to B-cell- and antibody-mediated autoimmunity.
The bigger picture
The autoimmune phenomenon is not unique to alemtuzumab or multiple sclerosis. “This turns out to be one of a family of clinical situations where the reconstitution of the depleted lymphocyte repertoire leads to autoimmunity,” Dr. Coles said. A similar effect was seen years ago when very lymphopenic HIV patients were given antiviral therapy, he added, affecting about 10% of treated patients. About 10% of bone marrow transplant patients may experience similar autoimmune concerns.
“What we do think is true is we’ve tapped into a classical expression of autoimmunity,” Dr. Coles said. “Alemtuzumab is a fantastic opportunity to study the mechanisms underlying lymphopenia-associated autoimmunity.”
A ‘tantalizing prospect’
“It’s a tantalizing prospect that susceptible individuals might be identified in the future prior to treatment,” Dr. Coles said. One promising lead, he added, is “we also looked at IL-21. We showed that after treatment, and perhaps more interestingly, before treatment with alemtuzumab, serum IL-21 is greater in those who subsequently develop autoimmune disease. This suggests some individuals are prone to develop autoimmune disease, and could be identified potentially prior to treatment with alemtuzumab.”
More work is needed, including the development of more sensitive IL-21 assays for use in this population, Dr. Coles said. “Please do not attempt to predict the risk of autoimmunity after alemtuzumab using the current commercial assays. This is a source of some frustration for me.”
A potential route of lymphocyte repertoire reconstitution after alemtuzumab is thymic reconstitution, leading to a more diverse immune repertoire, Dr. Coles said. “The obvious corollary of this is if we can direct reconstitution through the thymic reconstitution, we should be able to prevent autoimmunity.”
Dr. Coles disclosed that he receives honoraria for travel and speaking from Sanofi Genzyme, which markets alemtuzumab.
SAN DIEGO – The monoclonal antibody alemtuzumab can be an effective treatment for people living with multiple sclerosis, but there’s a catch — the agent is also associated with an increased risk for developing other autoimmune diseases, leaving clinicians with a conundrum.
“This is an efficacious treatment in multiple sclerosis” that can slow the rate of brain atrophy over the long-term, Alasdair Coles, MD, said at ACTRIMS Forum 2018, held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “But 1 or 2 years after each cycle of alemtuzumab [Lemtrada], patients are at very high risk of autoimmune diseases. This is the not-too-worrying thyroid disease, but there are some very troubling and potentially highly threatening complications at lower frequency.”
Subsequent autoimmune thyroid disease can affect up to 40% of patients treated with alemtuzumab, but immune thrombocytopenia (3%) and autoimmune renal disease (0.1%) are also reported. About 1 in 10 people treated with the monoclonal antibody for MS can also develop de novo asymptomatic autoantibodies (10%).
“People ask: ‘Why doesn’t MS come back as part of this generic mechanism?’ and I don’t know the answer to that,” Dr. Coles said.
In the United States, alemtuzumab is indicated for treatment of relapsing multiple sclerosis in adults who have failed to respond adequately to two or more previous therapies. In contrast, “this has become a first-line treatment in the U.K.,” said Dr. Coles, a professor in the department of clinical neurosciences at the University of Cambridge (England).
“Unfortunately, we can offer no proven treatment to prevent this autoimmunity.”
Considering the prospects for different proposed mechanisms
Dr. Coles shared some encouraging news at ACTRIMS Forum 2018. His team and other researchers are getting closer to understanding the cellular mechanism underlying the paradoxical autoimmunity associated with alemtuzumab. Published reports in the literature from others suggest faulty immune B cells could be the culprit, pointing to a similar reconstitution of B cells after bone marrow transplantation. However, he said, “There is no difference in this reconstitution pattern between those who do and don’t get autoimmunity. So we do not think that autoimmunity after alemtuzumab is primarily a B cell problem.”
Other investigators have pointed to possible depletion of a key immune regulatory cell associated with alemtuzumab, such as alterations in CD52-positive T cells that cause depletion in T cells as part of an autoimmune cascade that involve CD52-high expressing cells and sialic acid-binding immunoglobulin-like lectin 10. “I’m not going to describe why we don’t believe any of this,” Dr. Coles said, but added, “We cannot replicate the data in type 1 diabetes or MS about the depletion of T cells.”
Along with his colleague Joanne Jones, PhD, a clinical fellow in the same department at the University of Cambridge, Dr. Coles and his team instead propose that autoimmunity after alemtuzumab therapy is associated with a homeostatic proliferation of T cells in the context of a defective thymus. “We see thymic function reduced after alemtuzumab for a few months. We don’t know if alemtuzumab is having a direct impact on the thymus or if it’s an indirect effect though a cytokine storm at the time of administering alemtuzumab.”
In addition, in contrast to B cells, both CD4-positive and CD8-positive T cells are clonally restricted after alemtuzumab treatment, Dr. Coles explained.
“These are the only changes that distinguish patients who do and do not develop autoimmunity,” he said. “Those who develop autoimmunity have reduced clonality and have impaired thymic function compared to those who don’t.”
As the theory goes, the limited clonal repertoire leads to expansion of the T cells, preferentially expanding autoreactive T cells, leading to B-cell- and antibody-mediated autoimmunity.
The bigger picture
The autoimmune phenomenon is not unique to alemtuzumab or multiple sclerosis. “This turns out to be one of a family of clinical situations where the reconstitution of the depleted lymphocyte repertoire leads to autoimmunity,” Dr. Coles said. A similar effect was seen years ago when very lymphopenic HIV patients were given antiviral therapy, he added, affecting about 10% of treated patients. About 10% of bone marrow transplant patients may experience similar autoimmune concerns.
“What we do think is true is we’ve tapped into a classical expression of autoimmunity,” Dr. Coles said. “Alemtuzumab is a fantastic opportunity to study the mechanisms underlying lymphopenia-associated autoimmunity.”
A ‘tantalizing prospect’
“It’s a tantalizing prospect that susceptible individuals might be identified in the future prior to treatment,” Dr. Coles said. One promising lead, he added, is “we also looked at IL-21. We showed that after treatment, and perhaps more interestingly, before treatment with alemtuzumab, serum IL-21 is greater in those who subsequently develop autoimmune disease. This suggests some individuals are prone to develop autoimmune disease, and could be identified potentially prior to treatment with alemtuzumab.”
More work is needed, including the development of more sensitive IL-21 assays for use in this population, Dr. Coles said. “Please do not attempt to predict the risk of autoimmunity after alemtuzumab using the current commercial assays. This is a source of some frustration for me.”
A potential route of lymphocyte repertoire reconstitution after alemtuzumab is thymic reconstitution, leading to a more diverse immune repertoire, Dr. Coles said. “The obvious corollary of this is if we can direct reconstitution through the thymic reconstitution, we should be able to prevent autoimmunity.”
Dr. Coles disclosed that he receives honoraria for travel and speaking from Sanofi Genzyme, which markets alemtuzumab.
EXPERT ANALYSIS FROM ACTRIMS FORUM 2018
What’s Eating You? Ixodes Tick and Related Diseases, Part 1: Life Cycle, Local Reactions, and Lyme Disease
Ticks are ectoparasitic hemophages that feed on mammals, reptiles, and birds. The Ixodidae family comprises the hard ticks. A hard dorsal plate, scutum, and capitulum that extends outward from the body are features that distinguish the hard tick. 1Ixodes is the largest genus of hard ticks, with more than 250 species localized in temperate climates.2 It has an inornate scutum and lacks festoons (Figure 1).1 The Ixodes ricinus species complex accounts for most species relevant to the spread of human disease (Figure 2), with Ixodes scapularis in the northeastern, north midwestern, and southern United States; Ixodes pacificus in western United States; I ricinus in Europe and North Africa; and Ixodes persulcatus in Russia and Asia. Ixodes holocyclus is endemic to Australia.3,4

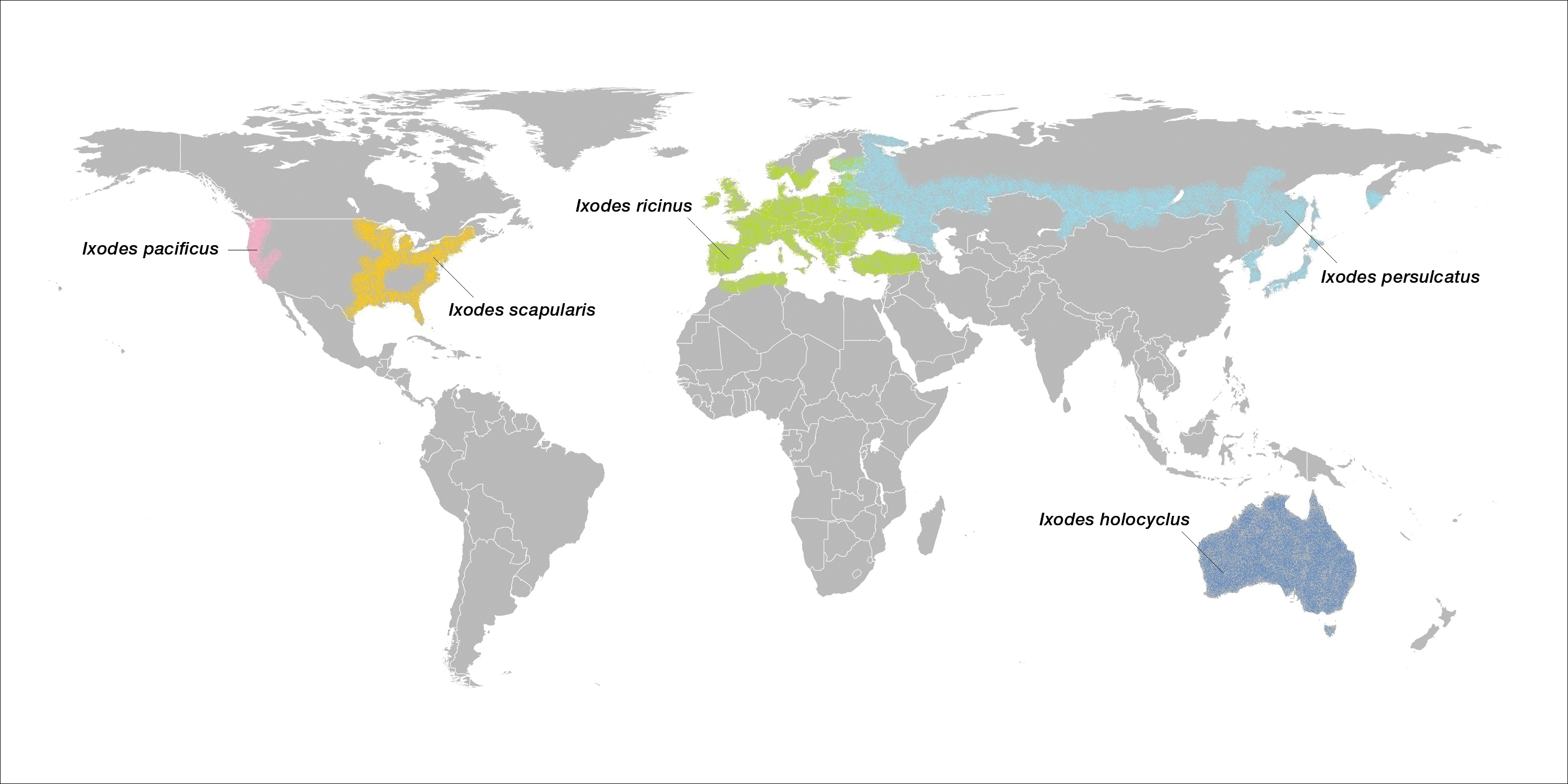
Life Cycle
Ixodes species progress through 4 life stages—egg, larvae, nymph, and adult—during their 3-host life cycle. Lifespan is 2 to 6 years, varying with environmental factors. A blood meal is required between each stage. Female ticks have a small scutum, allowing the abdomen to engorge during meals (Figure 3).

Larvae hatch in the early summer and remain dormant until the spring, emerging as a nymph. Following a blood meal, the nymph molts and reemerges as an adult in autumn. During autumn and winter, the female lays as many as 2000 eggs that emerge in early summer.5 Nymphs are small and easily undetected for the duration required for pathogen transmission, making nymphs the stage most likely to transmit disease.6
The majority of tick-borne diseases present from May to July, corresponding to nymph activity. Fewer cases present in the autumn and early spring because the adult female feeds during cooler months.7
Larvae have 6 legs and are about the size of a sesame seed when engorged. Nymphs are slightly larger with 8 legs. Adults are largest and have 8 legs. Following a blood meal, the tick becomes engorged, increasing in size and lightening in color (Figure 3).1
Ticks are found in low-lying shrubs and tall grass as well as on the forest floor. They search for a host by detecting CO2, warmth, the smell of sweat, and the color white, prompting attachment.8 Habitats hospitable to Ixodes have expanded in the wake of climate, environmental, and socioeconomic changes, potentially contributing to the increasing incidence and expansion of zoonoses associated with this vector.9,10
Local Reactions
A tick bite may induce local hypersensitivity, leading to a red papule or plaque at the bite site, followed by swelling, warmth, and erythema. A cellular immune reaction induces induration and pruritus. Hard ticks are less likely than soft ticks to cause a serious local reaction.11,12
A variety of clinical and histologic features are observed following an arthropod bite. Histologically, acute tick bites show a neutrophilic infiltrate with fibrin deposition. Chronic reactions demonstrate a wedge-shaped, mixed infiltrate with prominent endothelial swelling. Eosinophilic cellulitis, or Wells syndrome, reveals tissue eosinophilia and flame figures.13 Tick mouthparts may be identified in the tissue. B-cell hyperplasia is seen in Borrelia lymphocytoma and is more common in Europe, presenting as erythematous to plum–colored nodules on the ear and areola.14
Lyme Disease
Disease manifestations vary by location. Lyme disease is associated with Borrelia burgdorferi and the recently identified Borrelia mayonii in the United States15; in Europe and Asia, acrodermatitis chronica atrophicans is associated with Borrelia afzelii and neuroborreliosis, with Borrelia garinii. Lyme disease is the most common tick-borne illness in the United States.16 The I ricinus species complex is the most common vector harboring Borrelia species.17 At least 36 hours of tick adherence is required for disease transmission.18 The incubation period is 3 to 20 days (median, 12 days).19
Clinical Findings
Erythema migrans is the most characteristic sign, seen in 80% of cases of Lyme disease. The typical rash is a centrifugally spreading, erythematous, annular patch with central clearing at the site of the tick bite.20 Atypical rashes include vesicular, indurated, ulcerated, and follicular variants.21 Histopathology commonly shows a superficial and deep perivascular lymphocytic infiltrate with plasma cells, histiocytes, and eosinophils.22 Typically, the rash resolves in 3 to 5 weeks.18
Early disseminated Lyme disease can present with any of the following findings: multiple erythema migrans; neurologic involvement, including cranial nerve palsy and meningitis; and Lyme carditis, which may result in atrioventricular block.23,24 Late findings include arthritis, encephalopathy, and polyneuropathy. A late cutaneous manifestation, acrodermatitis chronica atrophicans, is rare in the United States but occurs in as many as 10% of Lyme disease cases in Europe. An initial inflammatory response manifests as blue-red erythema and edema of the extensor surfaces of the extremities, commonly on the dorsal hands, feet, elbows, and knees. Firm fibrotic nodules may develop later over the olecranon and patella.23,24
The term chronic Lyme disease has been used to describe the persistence of symptoms after treatment; however, large clinical trials have not detected a difference in symptom frequency between patients with a history of Lyme disease and matched controls.25,26 Many patients with chronic Lyme disease may instead have posttreatment Lyme disease syndrome, described as nonspecific symptoms including fatigue, arthralgia, and decreased mental acuity following treatment of confirmed Lyme disease. Symptoms generally improve within 1 year.27
Laboratory Testing
The gold standard for laboratory diagnosis of Lyme disease is 2-tiered serologic testing. First, an enzyme immunoassay or immunofluorescence assay is used to screen for antibodies. A Western blot follows if the result of the screen is positive or equivocal. Western blot testing for IgM and IgG is used when illness duration is less than 4 weeks; after 4 weeks, a Western blot for IgG alone is sufficient.27,28 The 2-tiered test has 99% specificity. Sensitivity increases with duration of disease (29%–40% with erythema migrans; 42%–87% in early disseminated disease; 97%–100% in late disease).29,30 A false-positive result can occur in the presence of infectious mononucleosis, an autoimmune disorder, and syphilis. If serologic testing is negative and suspicion remains high, testing should be repeated in 2 to 4 weeks.31 When a patient in a Lyme-endemic area presents with typical erythema migrans, serologic testing is unnecessary prior to treatment.32
Management
Treatment of Lyme disease centers on antibiotic therapy (Table). First-line treatment of early disseminated disease is doxycycline for 14 days (range, 10–21 days).27 In pregnant women, children younger than 8 years, and tetracycline-allergic patients, amoxicillin or cefuroxime axetil for 14 days (range, 14–21 days) may be used.33 For erythema migrans without complications, doxycycline for 10 days is effective. Complications that require hospitalization are treated with intravenous ceftriaxone.27 Re-treatment in patients with posttreatment Lyme disease syndrome is not recommended.34 Prophylaxis with a single dose of doxycycline 200 mg may be indicated when all of the following conditions are met: (1) the patient is in an area where more than 20% of Ixodes ticks are infected with B burgdorferi, (2) the attached tick is I scapularis, (3) the tick has been attached for more than 36 hours, and (4) treatment is begun within 72 hours of tick removal.27
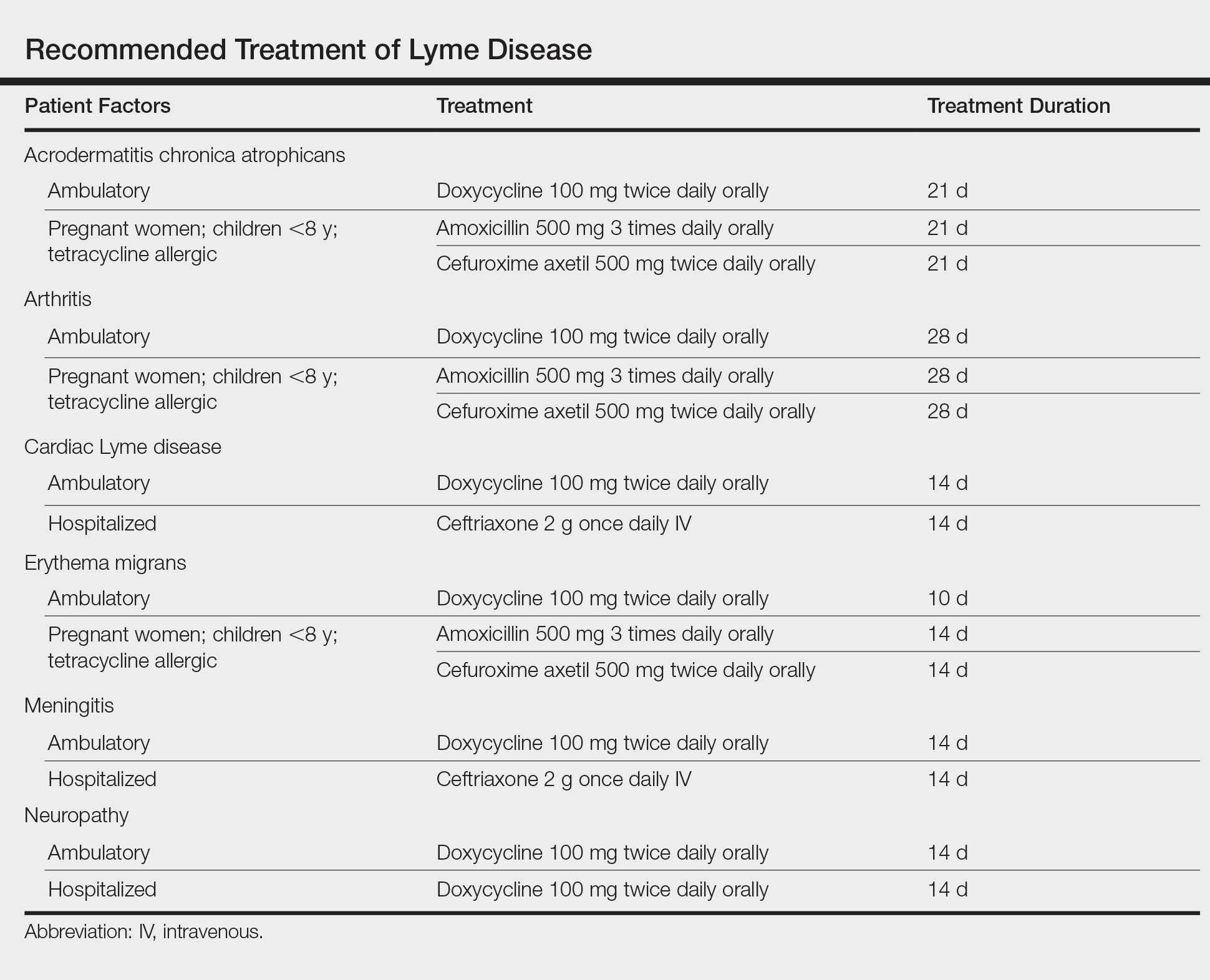
- Anderson JF, Magnarelli LA. Biology of ticks. Infect Dis Clin North Am. 2008;22:195-215.
- Jongejan F, Uilenberg G. The global importance of ticks. Parasitology. 2004;129(suppl):S3-S14.
- Xu G, Fang QQ, Keirans JE, et al. Molecular phylogenetic analyses indicate that the Ixodes ricinus complex is a paraphyletic group. J Parasitol. 2003;89:452-457.
- Swanson SJ, Neitzel D, Reed DK, et al. Coinfections acquired from Ixodes ticks. Clin Microbiol Rev. 2006;19:708-727.
- Mathison BA, Pritt BS. Laboratory identification of arthropod ectoparasites. Clin Microbol Rev. 2014;27:48-67.
- Falco RC, Fish D, Piesman J. Duration of tick bites in a Lyme disease-endemic area. Am J Epidemiol. 1996;143:187-192.
- Centers for Disease Control and Prevention. Lyme disease graphs. http://www.cdc.gov/lyme/stats/graphs.html. Updated November 21, 2016. Accessed November 21, 2017.
- Randolph SE. The impact of tick ecology on pathogen transmission dynamics. In: Bowman AS, Nuttall PA, eds. Ticks: Biology, Disease and Control. Cambridge, UK: Cambridge University Press; 2008:40-72.
- Ostfeld RS, Brunner JL. Climate change and Ixodes tick-borne diseases of humans. Philos Trans R Soc Lond B Biol Sci. 2015;370. pii:20140051. doi:10.1098/rstb.2014.0051.
- Medlock JM, Hansford KM, Bormane A, et al. Driving forces for changes in geographical distribution of Ixodes ricinus ticks in Europe. Parasit Vectors. 2013;6:1.
- McGinley-Smith DE, Tsao SS. Dermatoses from ticks. J Am Acad Dermatol. 2003;49:393-396.
- Middleton DB. Tick-borne infections. What starts as a tiny bite may have a serious outcome. Postgrad Med. 1994;95:131-139.
- Melski JW. Wells’ syndrome, insect bites, and eosinophils. Dermatol Clin. 2015;8:287-293.
- Castelli E, Caputo V, Morello V, et al. Local reactions to tick bites. Am J Dermatopathol. 2008;30:241-248.
- Pritt BS, Mead PS, Johnson DK, et al. Identification of a novel pathogenic Borrelia species causing Lyme borreliosis with unusually high spirochaetaemia: a descriptive study. Lancet Infect Dis. 2016;16:556-564.
- Orloski KA, Hayes EB, Campbell GL, et al. Surveillance for Lyme disease—United States, 1992-1998. MMWR CDC Surveill Summ. 2000;49:1-11.
- Gray JS. The ecology of ticks transmitting Lyme borreliosis. Exp Appl Acarol. 1998;22:249-258.
- Piesman J, Mather TN, Sinsky RJ, et al. Duration of tick attachment and Borrelia burgdorferi transmission. J Clin Microbiol. 1987;25:557-558.
- Richardson M, Elliman D, Maguire H, et al. Evidence base of incubation periods, periods of infectiousness and exclusion policies for the control of communicable diseases in schools and preschools. Pediatr Infect Dis J. 2001;20:380-391.
- Myers SA, Sexton DJ. Dermatologic manifestations of arthropod-borne diseases. Infect Dis Clin North Am. 1994;8:689-712.
- Ducroux E, Debarbieux S, Boibieux A, et al. Follicular borreliosis: an atypical presentation of erythema chronicum migrans. Dermatology. 2009;219:84-85.
- Miraflor AP, Seidel GD, Perry AE, et al. The many masks of cutaneous Lyme disease. J Cutan Pathol. 2016:43:32-40.
- Lenormand C, Jaulhac B, Debarbieux S, et al. Expanding the clinicopathological spectrum of late cutaneous Lyme borreliosis (acrodermatitis chronica atrophicans): a prospective study of 20 culture and/or polymerase chain reaction (PCR) documented cases. J Am Acad Dermatol. 2016;74:685-692.
- Zajkowska J, Czupryna P, Pancewicz SA, et al. Acrodermatitis chronica atrophicans. Lancet Infect Dis. 2011;11:800.
- Seltzer EG, Gerber MA, Cartter ML, et al. Long-term outcomes of persons with Lyme disease. JAMA. 2000;283:609-616.
- Shadick NA, Phillips CB, Sangha O, et al. Musculoskeletal and neurologic outcomes in patients with previously treated Lyme disease. Ann Intern Med. 1999;131:919-926.
- Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43:1089-1134.
- Schriefer ME. Lyme disease diagnosis: serology. Clin Lab Med. 2015;35:797-814.
- Wormser GP, Nowakowski J, Nadelman RB, et al. Impact of clinical variables on Borrelia burgdorferi-specific antibody seropositivity in acute-phase sera from patients in North America with culture-confirmed early Lyme disease. Clin Vaccine Immunol. 2008;15:1519-1522.
- Leeflang MM, Ang CW, Berkhout J, et al. The diagnostic accuracy of serological tests for Lyme borreliosis in Europe: a systematic review and meta-analysis. BMC Infect Dis. 2016;16:140.
- Sanchez E, Vannier E, Wormser GP, et al. Diagnosis, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: a review. JAMA. 2016;315:1767-1777.
- Lantos PM, Brinkerhoff RJ, Wormser GP, et al. Empiric antibiotic treatment of erythema migrans-like skin lesions as a function of geography: a clinical and cost effectiveness modeling study. Vector Borne Zoonotic Dis. 2013;13:877-883.
- Smith GN, Gemmill I, Moore KM. Management of tick bites and Lyme disease during pregnancy. J Obstet Gynaecol Can. 2012;34:1087-1091.
- Berende A, ter Hofstede HJ, Vos FJ, et al. Randomized trial of longer-term therapy for symptoms attributed to Lyme disease. N Engl J Med. 2016;374:1209-1220.
Ticks are ectoparasitic hemophages that feed on mammals, reptiles, and birds. The Ixodidae family comprises the hard ticks. A hard dorsal plate, scutum, and capitulum that extends outward from the body are features that distinguish the hard tick. 1Ixodes is the largest genus of hard ticks, with more than 250 species localized in temperate climates.2 It has an inornate scutum and lacks festoons (Figure 1).1 The Ixodes ricinus species complex accounts for most species relevant to the spread of human disease (Figure 2), with Ixodes scapularis in the northeastern, north midwestern, and southern United States; Ixodes pacificus in western United States; I ricinus in Europe and North Africa; and Ixodes persulcatus in Russia and Asia. Ixodes holocyclus is endemic to Australia.3,4
Life Cycle
Ixodes species progress through 4 life stages—egg, larvae, nymph, and adult—during their 3-host life cycle. Lifespan is 2 to 6 years, varying with environmental factors. A blood meal is required between each stage. Female ticks have a small scutum, allowing the abdomen to engorge during meals (Figure 3).
Larvae hatch in the early summer and remain dormant until the spring, emerging as a nymph. Following a blood meal, the nymph molts and reemerges as an adult in autumn. During autumn and winter, the female lays as many as 2000 eggs that emerge in early summer.5 Nymphs are small and easily undetected for the duration required for pathogen transmission, making nymphs the stage most likely to transmit disease.6
The majority of tick-borne diseases present from May to July, corresponding to nymph activity. Fewer cases present in the autumn and early spring because the adult female feeds during cooler months.7
Larvae have 6 legs and are about the size of a sesame seed when engorged. Nymphs are slightly larger with 8 legs. Adults are largest and have 8 legs. Following a blood meal, the tick becomes engorged, increasing in size and lightening in color (Figure 3).1
Ticks are found in low-lying shrubs and tall grass as well as on the forest floor. They search for a host by detecting CO2, warmth, the smell of sweat, and the color white, prompting attachment.8 Habitats hospitable to Ixodes have expanded in the wake of climate, environmental, and socioeconomic changes, potentially contributing to the increasing incidence and expansion of zoonoses associated with this vector.9,10
Local Reactions
A tick bite may induce local hypersensitivity, leading to a red papule or plaque at the bite site, followed by swelling, warmth, and erythema. A cellular immune reaction induces induration and pruritus. Hard ticks are less likely than soft ticks to cause a serious local reaction.11,12
A variety of clinical and histologic features are observed following an arthropod bite. Histologically, acute tick bites show a neutrophilic infiltrate with fibrin deposition. Chronic reactions demonstrate a wedge-shaped, mixed infiltrate with prominent endothelial swelling. Eosinophilic cellulitis, or Wells syndrome, reveals tissue eosinophilia and flame figures.13 Tick mouthparts may be identified in the tissue. B-cell hyperplasia is seen in Borrelia lymphocytoma and is more common in Europe, presenting as erythematous to plum–colored nodules on the ear and areola.14
Lyme Disease
Disease manifestations vary by location. Lyme disease is associated with Borrelia burgdorferi and the recently identified Borrelia mayonii in the United States15; in Europe and Asia, acrodermatitis chronica atrophicans is associated with Borrelia afzelii and neuroborreliosis, with Borrelia garinii. Lyme disease is the most common tick-borne illness in the United States.16 The I ricinus species complex is the most common vector harboring Borrelia species.17 At least 36 hours of tick adherence is required for disease transmission.18 The incubation period is 3 to 20 days (median, 12 days).19
Clinical Findings
Erythema migrans is the most characteristic sign, seen in 80% of cases of Lyme disease. The typical rash is a centrifugally spreading, erythematous, annular patch with central clearing at the site of the tick bite.20 Atypical rashes include vesicular, indurated, ulcerated, and follicular variants.21 Histopathology commonly shows a superficial and deep perivascular lymphocytic infiltrate with plasma cells, histiocytes, and eosinophils.22 Typically, the rash resolves in 3 to 5 weeks.18
Early disseminated Lyme disease can present with any of the following findings: multiple erythema migrans; neurologic involvement, including cranial nerve palsy and meningitis; and Lyme carditis, which may result in atrioventricular block.23,24 Late findings include arthritis, encephalopathy, and polyneuropathy. A late cutaneous manifestation, acrodermatitis chronica atrophicans, is rare in the United States but occurs in as many as 10% of Lyme disease cases in Europe. An initial inflammatory response manifests as blue-red erythema and edema of the extensor surfaces of the extremities, commonly on the dorsal hands, feet, elbows, and knees. Firm fibrotic nodules may develop later over the olecranon and patella.23,24
The term chronic Lyme disease has been used to describe the persistence of symptoms after treatment; however, large clinical trials have not detected a difference in symptom frequency between patients with a history of Lyme disease and matched controls.25,26 Many patients with chronic Lyme disease may instead have posttreatment Lyme disease syndrome, described as nonspecific symptoms including fatigue, arthralgia, and decreased mental acuity following treatment of confirmed Lyme disease. Symptoms generally improve within 1 year.27
Laboratory Testing
The gold standard for laboratory diagnosis of Lyme disease is 2-tiered serologic testing. First, an enzyme immunoassay or immunofluorescence assay is used to screen for antibodies. A Western blot follows if the result of the screen is positive or equivocal. Western blot testing for IgM and IgG is used when illness duration is less than 4 weeks; after 4 weeks, a Western blot for IgG alone is sufficient.27,28 The 2-tiered test has 99% specificity. Sensitivity increases with duration of disease (29%–40% with erythema migrans; 42%–87% in early disseminated disease; 97%–100% in late disease).29,30 A false-positive result can occur in the presence of infectious mononucleosis, an autoimmune disorder, and syphilis. If serologic testing is negative and suspicion remains high, testing should be repeated in 2 to 4 weeks.31 When a patient in a Lyme-endemic area presents with typical erythema migrans, serologic testing is unnecessary prior to treatment.32
Management
Treatment of Lyme disease centers on antibiotic therapy (Table). First-line treatment of early disseminated disease is doxycycline for 14 days (range, 10–21 days).27 In pregnant women, children younger than 8 years, and tetracycline-allergic patients, amoxicillin or cefuroxime axetil for 14 days (range, 14–21 days) may be used.33 For erythema migrans without complications, doxycycline for 10 days is effective. Complications that require hospitalization are treated with intravenous ceftriaxone.27 Re-treatment in patients with posttreatment Lyme disease syndrome is not recommended.34 Prophylaxis with a single dose of doxycycline 200 mg may be indicated when all of the following conditions are met: (1) the patient is in an area where more than 20% of Ixodes ticks are infected with B burgdorferi, (2) the attached tick is I scapularis, (3) the tick has been attached for more than 36 hours, and (4) treatment is begun within 72 hours of tick removal.27
Ticks are ectoparasitic hemophages that feed on mammals, reptiles, and birds. The Ixodidae family comprises the hard ticks. A hard dorsal plate, scutum, and capitulum that extends outward from the body are features that distinguish the hard tick. 1Ixodes is the largest genus of hard ticks, with more than 250 species localized in temperate climates.2 It has an inornate scutum and lacks festoons (Figure 1).1 The Ixodes ricinus species complex accounts for most species relevant to the spread of human disease (Figure 2), with Ixodes scapularis in the northeastern, north midwestern, and southern United States; Ixodes pacificus in western United States; I ricinus in Europe and North Africa; and Ixodes persulcatus in Russia and Asia. Ixodes holocyclus is endemic to Australia.3,4
Life Cycle
Ixodes species progress through 4 life stages—egg, larvae, nymph, and adult—during their 3-host life cycle. Lifespan is 2 to 6 years, varying with environmental factors. A blood meal is required between each stage. Female ticks have a small scutum, allowing the abdomen to engorge during meals (Figure 3).
Larvae hatch in the early summer and remain dormant until the spring, emerging as a nymph. Following a blood meal, the nymph molts and reemerges as an adult in autumn. During autumn and winter, the female lays as many as 2000 eggs that emerge in early summer.5 Nymphs are small and easily undetected for the duration required for pathogen transmission, making nymphs the stage most likely to transmit disease.6
The majority of tick-borne diseases present from May to July, corresponding to nymph activity. Fewer cases present in the autumn and early spring because the adult female feeds during cooler months.7
Larvae have 6 legs and are about the size of a sesame seed when engorged. Nymphs are slightly larger with 8 legs. Adults are largest and have 8 legs. Following a blood meal, the tick becomes engorged, increasing in size and lightening in color (Figure 3).1
Ticks are found in low-lying shrubs and tall grass as well as on the forest floor. They search for a host by detecting CO2, warmth, the smell of sweat, and the color white, prompting attachment.8 Habitats hospitable to Ixodes have expanded in the wake of climate, environmental, and socioeconomic changes, potentially contributing to the increasing incidence and expansion of zoonoses associated with this vector.9,10
Local Reactions
A tick bite may induce local hypersensitivity, leading to a red papule or plaque at the bite site, followed by swelling, warmth, and erythema. A cellular immune reaction induces induration and pruritus. Hard ticks are less likely than soft ticks to cause a serious local reaction.11,12
A variety of clinical and histologic features are observed following an arthropod bite. Histologically, acute tick bites show a neutrophilic infiltrate with fibrin deposition. Chronic reactions demonstrate a wedge-shaped, mixed infiltrate with prominent endothelial swelling. Eosinophilic cellulitis, or Wells syndrome, reveals tissue eosinophilia and flame figures.13 Tick mouthparts may be identified in the tissue. B-cell hyperplasia is seen in Borrelia lymphocytoma and is more common in Europe, presenting as erythematous to plum–colored nodules on the ear and areola.14
Lyme Disease
Disease manifestations vary by location. Lyme disease is associated with Borrelia burgdorferi and the recently identified Borrelia mayonii in the United States15; in Europe and Asia, acrodermatitis chronica atrophicans is associated with Borrelia afzelii and neuroborreliosis, with Borrelia garinii. Lyme disease is the most common tick-borne illness in the United States.16 The I ricinus species complex is the most common vector harboring Borrelia species.17 At least 36 hours of tick adherence is required for disease transmission.18 The incubation period is 3 to 20 days (median, 12 days).19
Clinical Findings
Erythema migrans is the most characteristic sign, seen in 80% of cases of Lyme disease. The typical rash is a centrifugally spreading, erythematous, annular patch with central clearing at the site of the tick bite.20 Atypical rashes include vesicular, indurated, ulcerated, and follicular variants.21 Histopathology commonly shows a superficial and deep perivascular lymphocytic infiltrate with plasma cells, histiocytes, and eosinophils.22 Typically, the rash resolves in 3 to 5 weeks.18
Early disseminated Lyme disease can present with any of the following findings: multiple erythema migrans; neurologic involvement, including cranial nerve palsy and meningitis; and Lyme carditis, which may result in atrioventricular block.23,24 Late findings include arthritis, encephalopathy, and polyneuropathy. A late cutaneous manifestation, acrodermatitis chronica atrophicans, is rare in the United States but occurs in as many as 10% of Lyme disease cases in Europe. An initial inflammatory response manifests as blue-red erythema and edema of the extensor surfaces of the extremities, commonly on the dorsal hands, feet, elbows, and knees. Firm fibrotic nodules may develop later over the olecranon and patella.23,24
The term chronic Lyme disease has been used to describe the persistence of symptoms after treatment; however, large clinical trials have not detected a difference in symptom frequency between patients with a history of Lyme disease and matched controls.25,26 Many patients with chronic Lyme disease may instead have posttreatment Lyme disease syndrome, described as nonspecific symptoms including fatigue, arthralgia, and decreased mental acuity following treatment of confirmed Lyme disease. Symptoms generally improve within 1 year.27
Laboratory Testing
The gold standard for laboratory diagnosis of Lyme disease is 2-tiered serologic testing. First, an enzyme immunoassay or immunofluorescence assay is used to screen for antibodies. A Western blot follows if the result of the screen is positive or equivocal. Western blot testing for IgM and IgG is used when illness duration is less than 4 weeks; after 4 weeks, a Western blot for IgG alone is sufficient.27,28 The 2-tiered test has 99% specificity. Sensitivity increases with duration of disease (29%–40% with erythema migrans; 42%–87% in early disseminated disease; 97%–100% in late disease).29,30 A false-positive result can occur in the presence of infectious mononucleosis, an autoimmune disorder, and syphilis. If serologic testing is negative and suspicion remains high, testing should be repeated in 2 to 4 weeks.31 When a patient in a Lyme-endemic area presents with typical erythema migrans, serologic testing is unnecessary prior to treatment.32
Management
Treatment of Lyme disease centers on antibiotic therapy (Table). First-line treatment of early disseminated disease is doxycycline for 14 days (range, 10–21 days).27 In pregnant women, children younger than 8 years, and tetracycline-allergic patients, amoxicillin or cefuroxime axetil for 14 days (range, 14–21 days) may be used.33 For erythema migrans without complications, doxycycline for 10 days is effective. Complications that require hospitalization are treated with intravenous ceftriaxone.27 Re-treatment in patients with posttreatment Lyme disease syndrome is not recommended.34 Prophylaxis with a single dose of doxycycline 200 mg may be indicated when all of the following conditions are met: (1) the patient is in an area where more than 20% of Ixodes ticks are infected with B burgdorferi, (2) the attached tick is I scapularis, (3) the tick has been attached for more than 36 hours, and (4) treatment is begun within 72 hours of tick removal.27
- Anderson JF, Magnarelli LA. Biology of ticks. Infect Dis Clin North Am. 2008;22:195-215.
- Jongejan F, Uilenberg G. The global importance of ticks. Parasitology. 2004;129(suppl):S3-S14.
- Xu G, Fang QQ, Keirans JE, et al. Molecular phylogenetic analyses indicate that the Ixodes ricinus complex is a paraphyletic group. J Parasitol. 2003;89:452-457.
- Swanson SJ, Neitzel D, Reed DK, et al. Coinfections acquired from Ixodes ticks. Clin Microbiol Rev. 2006;19:708-727.
- Mathison BA, Pritt BS. Laboratory identification of arthropod ectoparasites. Clin Microbol Rev. 2014;27:48-67.
- Falco RC, Fish D, Piesman J. Duration of tick bites in a Lyme disease-endemic area. Am J Epidemiol. 1996;143:187-192.
- Centers for Disease Control and Prevention. Lyme disease graphs. http://www.cdc.gov/lyme/stats/graphs.html. Updated November 21, 2016. Accessed November 21, 2017.
- Randolph SE. The impact of tick ecology on pathogen transmission dynamics. In: Bowman AS, Nuttall PA, eds. Ticks: Biology, Disease and Control. Cambridge, UK: Cambridge University Press; 2008:40-72.
- Ostfeld RS, Brunner JL. Climate change and Ixodes tick-borne diseases of humans. Philos Trans R Soc Lond B Biol Sci. 2015;370. pii:20140051. doi:10.1098/rstb.2014.0051.
- Medlock JM, Hansford KM, Bormane A, et al. Driving forces for changes in geographical distribution of Ixodes ricinus ticks in Europe. Parasit Vectors. 2013;6:1.
- McGinley-Smith DE, Tsao SS. Dermatoses from ticks. J Am Acad Dermatol. 2003;49:393-396.
- Middleton DB. Tick-borne infections. What starts as a tiny bite may have a serious outcome. Postgrad Med. 1994;95:131-139.
- Melski JW. Wells’ syndrome, insect bites, and eosinophils. Dermatol Clin. 2015;8:287-293.
- Castelli E, Caputo V, Morello V, et al. Local reactions to tick bites. Am J Dermatopathol. 2008;30:241-248.
- Pritt BS, Mead PS, Johnson DK, et al. Identification of a novel pathogenic Borrelia species causing Lyme borreliosis with unusually high spirochaetaemia: a descriptive study. Lancet Infect Dis. 2016;16:556-564.
- Orloski KA, Hayes EB, Campbell GL, et al. Surveillance for Lyme disease—United States, 1992-1998. MMWR CDC Surveill Summ. 2000;49:1-11.
- Gray JS. The ecology of ticks transmitting Lyme borreliosis. Exp Appl Acarol. 1998;22:249-258.
- Piesman J, Mather TN, Sinsky RJ, et al. Duration of tick attachment and Borrelia burgdorferi transmission. J Clin Microbiol. 1987;25:557-558.
- Richardson M, Elliman D, Maguire H, et al. Evidence base of incubation periods, periods of infectiousness and exclusion policies for the control of communicable diseases in schools and preschools. Pediatr Infect Dis J. 2001;20:380-391.
- Myers SA, Sexton DJ. Dermatologic manifestations of arthropod-borne diseases. Infect Dis Clin North Am. 1994;8:689-712.
- Ducroux E, Debarbieux S, Boibieux A, et al. Follicular borreliosis: an atypical presentation of erythema chronicum migrans. Dermatology. 2009;219:84-85.
- Miraflor AP, Seidel GD, Perry AE, et al. The many masks of cutaneous Lyme disease. J Cutan Pathol. 2016:43:32-40.
- Lenormand C, Jaulhac B, Debarbieux S, et al. Expanding the clinicopathological spectrum of late cutaneous Lyme borreliosis (acrodermatitis chronica atrophicans): a prospective study of 20 culture and/or polymerase chain reaction (PCR) documented cases. J Am Acad Dermatol. 2016;74:685-692.
- Zajkowska J, Czupryna P, Pancewicz SA, et al. Acrodermatitis chronica atrophicans. Lancet Infect Dis. 2011;11:800.
- Seltzer EG, Gerber MA, Cartter ML, et al. Long-term outcomes of persons with Lyme disease. JAMA. 2000;283:609-616.
- Shadick NA, Phillips CB, Sangha O, et al. Musculoskeletal and neurologic outcomes in patients with previously treated Lyme disease. Ann Intern Med. 1999;131:919-926.
- Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43:1089-1134.
- Schriefer ME. Lyme disease diagnosis: serology. Clin Lab Med. 2015;35:797-814.
- Wormser GP, Nowakowski J, Nadelman RB, et al. Impact of clinical variables on Borrelia burgdorferi-specific antibody seropositivity in acute-phase sera from patients in North America with culture-confirmed early Lyme disease. Clin Vaccine Immunol. 2008;15:1519-1522.
- Leeflang MM, Ang CW, Berkhout J, et al. The diagnostic accuracy of serological tests for Lyme borreliosis in Europe: a systematic review and meta-analysis. BMC Infect Dis. 2016;16:140.
- Sanchez E, Vannier E, Wormser GP, et al. Diagnosis, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: a review. JAMA. 2016;315:1767-1777.
- Lantos PM, Brinkerhoff RJ, Wormser GP, et al. Empiric antibiotic treatment of erythema migrans-like skin lesions as a function of geography: a clinical and cost effectiveness modeling study. Vector Borne Zoonotic Dis. 2013;13:877-883.
- Smith GN, Gemmill I, Moore KM. Management of tick bites and Lyme disease during pregnancy. J Obstet Gynaecol Can. 2012;34:1087-1091.
- Berende A, ter Hofstede HJ, Vos FJ, et al. Randomized trial of longer-term therapy for symptoms attributed to Lyme disease. N Engl J Med. 2016;374:1209-1220.
- Anderson JF, Magnarelli LA. Biology of ticks. Infect Dis Clin North Am. 2008;22:195-215.
- Jongejan F, Uilenberg G. The global importance of ticks. Parasitology. 2004;129(suppl):S3-S14.
- Xu G, Fang QQ, Keirans JE, et al. Molecular phylogenetic analyses indicate that the Ixodes ricinus complex is a paraphyletic group. J Parasitol. 2003;89:452-457.
- Swanson SJ, Neitzel D, Reed DK, et al. Coinfections acquired from Ixodes ticks. Clin Microbiol Rev. 2006;19:708-727.
- Mathison BA, Pritt BS. Laboratory identification of arthropod ectoparasites. Clin Microbol Rev. 2014;27:48-67.
- Falco RC, Fish D, Piesman J. Duration of tick bites in a Lyme disease-endemic area. Am J Epidemiol. 1996;143:187-192.
- Centers for Disease Control and Prevention. Lyme disease graphs. http://www.cdc.gov/lyme/stats/graphs.html. Updated November 21, 2016. Accessed November 21, 2017.
- Randolph SE. The impact of tick ecology on pathogen transmission dynamics. In: Bowman AS, Nuttall PA, eds. Ticks: Biology, Disease and Control. Cambridge, UK: Cambridge University Press; 2008:40-72.
- Ostfeld RS, Brunner JL. Climate change and Ixodes tick-borne diseases of humans. Philos Trans R Soc Lond B Biol Sci. 2015;370. pii:20140051. doi:10.1098/rstb.2014.0051.
- Medlock JM, Hansford KM, Bormane A, et al. Driving forces for changes in geographical distribution of Ixodes ricinus ticks in Europe. Parasit Vectors. 2013;6:1.
- McGinley-Smith DE, Tsao SS. Dermatoses from ticks. J Am Acad Dermatol. 2003;49:393-396.
- Middleton DB. Tick-borne infections. What starts as a tiny bite may have a serious outcome. Postgrad Med. 1994;95:131-139.
- Melski JW. Wells’ syndrome, insect bites, and eosinophils. Dermatol Clin. 2015;8:287-293.
- Castelli E, Caputo V, Morello V, et al. Local reactions to tick bites. Am J Dermatopathol. 2008;30:241-248.
- Pritt BS, Mead PS, Johnson DK, et al. Identification of a novel pathogenic Borrelia species causing Lyme borreliosis with unusually high spirochaetaemia: a descriptive study. Lancet Infect Dis. 2016;16:556-564.
- Orloski KA, Hayes EB, Campbell GL, et al. Surveillance for Lyme disease—United States, 1992-1998. MMWR CDC Surveill Summ. 2000;49:1-11.
- Gray JS. The ecology of ticks transmitting Lyme borreliosis. Exp Appl Acarol. 1998;22:249-258.
- Piesman J, Mather TN, Sinsky RJ, et al. Duration of tick attachment and Borrelia burgdorferi transmission. J Clin Microbiol. 1987;25:557-558.
- Richardson M, Elliman D, Maguire H, et al. Evidence base of incubation periods, periods of infectiousness and exclusion policies for the control of communicable diseases in schools and preschools. Pediatr Infect Dis J. 2001;20:380-391.
- Myers SA, Sexton DJ. Dermatologic manifestations of arthropod-borne diseases. Infect Dis Clin North Am. 1994;8:689-712.
- Ducroux E, Debarbieux S, Boibieux A, et al. Follicular borreliosis: an atypical presentation of erythema chronicum migrans. Dermatology. 2009;219:84-85.
- Miraflor AP, Seidel GD, Perry AE, et al. The many masks of cutaneous Lyme disease. J Cutan Pathol. 2016:43:32-40.
- Lenormand C, Jaulhac B, Debarbieux S, et al. Expanding the clinicopathological spectrum of late cutaneous Lyme borreliosis (acrodermatitis chronica atrophicans): a prospective study of 20 culture and/or polymerase chain reaction (PCR) documented cases. J Am Acad Dermatol. 2016;74:685-692.
- Zajkowska J, Czupryna P, Pancewicz SA, et al. Acrodermatitis chronica atrophicans. Lancet Infect Dis. 2011;11:800.
- Seltzer EG, Gerber MA, Cartter ML, et al. Long-term outcomes of persons with Lyme disease. JAMA. 2000;283:609-616.
- Shadick NA, Phillips CB, Sangha O, et al. Musculoskeletal and neurologic outcomes in patients with previously treated Lyme disease. Ann Intern Med. 1999;131:919-926.
- Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43:1089-1134.
- Schriefer ME. Lyme disease diagnosis: serology. Clin Lab Med. 2015;35:797-814.
- Wormser GP, Nowakowski J, Nadelman RB, et al. Impact of clinical variables on Borrelia burgdorferi-specific antibody seropositivity in acute-phase sera from patients in North America with culture-confirmed early Lyme disease. Clin Vaccine Immunol. 2008;15:1519-1522.
- Leeflang MM, Ang CW, Berkhout J, et al. The diagnostic accuracy of serological tests for Lyme borreliosis in Europe: a systematic review and meta-analysis. BMC Infect Dis. 2016;16:140.
- Sanchez E, Vannier E, Wormser GP, et al. Diagnosis, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: a review. JAMA. 2016;315:1767-1777.
- Lantos PM, Brinkerhoff RJ, Wormser GP, et al. Empiric antibiotic treatment of erythema migrans-like skin lesions as a function of geography: a clinical and cost effectiveness modeling study. Vector Borne Zoonotic Dis. 2013;13:877-883.
- Smith GN, Gemmill I, Moore KM. Management of tick bites and Lyme disease during pregnancy. J Obstet Gynaecol Can. 2012;34:1087-1091.
- Berende A, ter Hofstede HJ, Vos FJ, et al. Randomized trial of longer-term therapy for symptoms attributed to Lyme disease. N Engl J Med. 2016;374:1209-1220.
Practice Points
- Lyme disease is transmitted by Ixodes ticks in the northeastern, midwestern, and far western United States.
- Most tick-borne illnesses, including Lyme disease, respond to treatment with doxycycline.
- Babesiosis, a malarialike illness, can be transmitted concurrently with Lyme disease.

Let There Be Light: Update on Coding for Photodynamic Therapy and Lasers
Winter is the time when many religions celebrate a renewal of the year as the days begin to get longer. On January 1 of each year in the United States we celebrate the official activation of new and revised
Photodynamic Therapy
In 2004, a methyl aminolevulinate cream that was activated with a red light source was brought to market; however, after failing to gain a substantial market share, the product is no longer available in the United States. In May of 2016, a nanoemulsion gel formulation of aminolevulinic acid hydrochloride 10% was approved by the US Food and Drug Administration4 for use with a red light source. Unlike 5-aminolevulinic acid hydrochloride solution, which was approved for application with no prior debridement of the skin,5 the new gel formulation was meant to be applied after degreasing with an ethanol- or isopropanol-soaked cotton pad and removal of any scaling or crusts, followed by roughening of the lesion surfaces (with care taken to avoid bleeding).4 The product must be administered by a health care provider and is reported using CPT codes 96573 and 96574, which are new in 2018 and are discussed in more detail below. Effective January 1, 2018, the Healthcare Common Procedure Coding System supply code for the product is J7345 (aminolevulinic acid hydrochloride gel for topical administration, 10% gel, 10 mg).6 A single tube contains 200 mg, so when an entire tube is used (which is typical), 200 units must be reported. Partial tubes may be used in some patients and should be reported appropriately based on actual usage.
The development of new CPT codes for PDT revealed a middle ground in which many physicians, including myself, have applied the photosensitizing drug themselves instead of a nonphysician provider in order to use their professional judgment to ensure the entire treatment area was covered and also allow for multiple applications of the drug to lesions that in their opinion may have warranted greater dosing, which led to the creation of CPT code 96573. The revision and refinement from one code to 3 (96567, 96573, and 96574) also involved rewording of the preamble for all 3 codes so that the phrase “premalignant and/or malignant lesions” was simplified to “premalignant lesions.” This change was made so that if and when this therapeutic approach is refined enough to be used on malignant lesions, new codes can be created to distinguish between the work performed for both types of lesions.
The new PDT codes include 96573 (photodynamic therapy by external application of light to destroy premalignant lesions of the skin and adjacent mucosa with application and illumination/activation of photosensitizing drug[s] provided by a physician or other qualified healthcare professional, per day) and 96574 (debridement of premalignant hyperkeratotic lesion[s][ie, targeted curettage, abrasion] followed with photodynamic therapy by external application of light to destroy premalignant lesions of the skin and adjacent mucosa with application and illumination/activation of photosensitizing drug[s] provided by a physician or other qualified healthcare professional, per day). According to the 2018 CPT manual,2 these codes should be used to report nonsurgical treatment of cutaneous lesions using PDT (ie, external application of light to destroy premalignant lesions of the skin and adjacent mucosa by activation of photosensitizing drug). A treatment session is defined as an application of a photosensitizer to all lesions within an anatomic area (eg, face, scalp) with or without debridement of all premalignant hyperkeratotic lesions in that area followed by illumination and activation with an appropriate light source. Providers should not report codes for debridement (11000, 11001, 11004, 11005), lesion shaving (11300–11313), biopsy (11100, 11101), or lesion excision (11400–11471) within the treatment area on the same day that PDT is administered.2
With the inclusion of these new PDT codes, the older code 96567 (photodynamic therapy by external application of light to destroy premalignant lesions of the skin and adjacent mucosa with application and illumination/activation of photosensitive drug[s], per day)—which is the base or parent code of the set—should only be used for reporting PDT when a physician or other qualified health care professional is not directly involved in the delivery of the service. Code 96573 is an upgrade to 96567 to account for physician work, while code 96574 captures the extra work of disruption of the skin barrier by debridement.
The novelty here is that old codes often are replaced when new codes come along. The reader should be aware of the distinct differences, as the total value expressed in relative value units for code 96567 is lower than it was in 2017 (3.24 vs 3.80), while the 2 newer codes have higher values (codes 96573 and 96574, 5.37 and 6.92, respectively). Additionally, the reader should note that only one of the 3 codes can be used on a given anatomic area (ie, face and scalp) on a given day. In general, a single-dose package of either of the approved photosensitizing drugs can usually treat an entire anatomic area. The codes themselves are not reserved for specific anatomic areas, but the US Food and Drug Administration clearances are for only face and scalp for both drugs, so the use of more than 2 PDT codes on a given day might raise payer queries.
Whatever you do, be sure your documentation includes an explicit notation about who applied the photosensitizing drug and the technique used for debridement, if performed. Code 96574 explicitly refers to targeted curettage and abrasion but does not include other destructive modalities (eg, chemical peeling), which an auditor may or may not consider an acceptable method of debridement. Personally, I will not be using peels as a justifier for this code.
Lasers
Lasers have played a role in the treatment of severe scarring in wounded warriors and other patient populations.7 Until 2018, there were no CPT codes that allowed precise reporting of these therapies. We now have a series of tracking codes, which are not valued by the Specialty Society Relative Value Scale Update Committee process but are nonetheless reportable, for this valuable treatment.8
The base code for a new pair of codes for reporting fractional ablative laser treatment, which is modeled after the skin graft code series, is 0479T (fractional ablative laser fenestration of burn and traumatic scars for functional improvement; first 100 cm2 or part thereof, or 1% of body surface area of infants and children). The add-on code is 0480T (fractional ablative laser fenestration of burn and traumatic scars for functional improvement; each additional 100 cm2, or each additional 1% of body surface area of infants and children, or part thereof [list separately in addition to code for primary procedure]), which means the code can be reported multiple times in addition to a single unit of 0479T. The aggregate treatment area should only be reported once per day regardless of the number of passes of one or more lasers over the area that day, and codes 0479T and 0480T should not be reported with codes 0491T or 0492T, which are a new family of tracking codes used for ablative laser treatment of chronic open wounds. If the scars are excised in a full-thickness manner, the benign excision codes 11400 to 11446 should be used instead.
For laser treatment of open wounds, 0491T (ablative laser treatment, noncontact, full-field and fractional ablation, open wound, per day, total treatment surface area; first 20 cm2 or less) is the base code for this pair of codes, and 0492T (ablative laser treatment, noncontact, full-field and fractional ablation, open wound, per day, total treatment surface area; each additional 20 cm2, or part thereof [list separately in addition to code for primary procedure]) is the add-on code, similar to the 0479T and 00480T codes described above. Keep in mind that all 4 of these tracking codes do not have defined values, and payment is at the discretion of the payer. If utilization of the procedures increases along with the development of appropriate evidence-based literature to support it, it is possible these will be converted into standard category I CPT codes that will be valued and covered by payers.
Final Thoughts
For more details on the new codes for PDT and lasers, I would strongly suggest obtaining a copy of CPT Changes 2018: An Insider’s View (https://commerce.ama-assn.org/store/catalog/productDetail.jsp?product_id=prod2800018&navAction=push), as well as the 2018 CPT manual for those who are actively practicing. Members of the American Academy of Dermatology also can get the new CPT manual as part of the group’s Coding Value Pack (https://store.aad.org/products/11383) along with Principles of Documentation for Dermatology and 2018 Coding & Billing for Dermatology.
- Daniell MD, Hill JS. A history of photodynamic therapy. Aust N Z J Surg. 1991;61:340-348.
- Current Procedural Terminology 2018, Professional Edition. Chicago, IL: American Medical Association; 2018.
- HCPCS code J7308. HCPCS Complete Reference website. https://hcpcs.codes/j-codes/J7308/. Accessed March 1, 2018.
- Ameluz [package insert]. Wakefield, MA: Biofrontera Inc; 2017.
- Levulan Kerastick [package insert]. Wilmington, MA: Dusa Pharmaceuticals, Inc; 2010.
- Centers for Medicare & Medicaid Services. 2018 Table of drugs. CMS website. https://www.cms.gov/Medicare/Coding/HCPCSReleaseCodeSets/Downloads/2018-Table-of-Drugs.pdf. Updated February 15, 2018. Accessed February 21, 2018.
- Waibel JS, Rudnick A. Current trends and future considerations in scar treatment. Semin Cutan Med Surg. 2015;34:13-16.
- American Medical Association. CPT category III codes. AMA website. https://www.ama-assn.org/sites/default/files/media-browser/public/cpt/cpt-category3-codes-descriptors.pdf. Updated December 21, 2017. Accessed February 21, 2018.
Winter is the time when many religions celebrate a renewal of the year as the days begin to get longer. On January 1 of each year in the United States we celebrate the official activation of new and revised
Photodynamic Therapy
In 2004, a methyl aminolevulinate cream that was activated with a red light source was brought to market; however, after failing to gain a substantial market share, the product is no longer available in the United States. In May of 2016, a nanoemulsion gel formulation of aminolevulinic acid hydrochloride 10% was approved by the US Food and Drug Administration4 for use with a red light source. Unlike 5-aminolevulinic acid hydrochloride solution, which was approved for application with no prior debridement of the skin,5 the new gel formulation was meant to be applied after degreasing with an ethanol- or isopropanol-soaked cotton pad and removal of any scaling or crusts, followed by roughening of the lesion surfaces (with care taken to avoid bleeding).4 The product must be administered by a health care provider and is reported using CPT codes 96573 and 96574, which are new in 2018 and are discussed in more detail below. Effective January 1, 2018, the Healthcare Common Procedure Coding System supply code for the product is J7345 (aminolevulinic acid hydrochloride gel for topical administration, 10% gel, 10 mg).6 A single tube contains 200 mg, so when an entire tube is used (which is typical), 200 units must be reported. Partial tubes may be used in some patients and should be reported appropriately based on actual usage.
The development of new CPT codes for PDT revealed a middle ground in which many physicians, including myself, have applied the photosensitizing drug themselves instead of a nonphysician provider in order to use their professional judgment to ensure the entire treatment area was covered and also allow for multiple applications of the drug to lesions that in their opinion may have warranted greater dosing, which led to the creation of CPT code 96573. The revision and refinement from one code to 3 (96567, 96573, and 96574) also involved rewording of the preamble for all 3 codes so that the phrase “premalignant and/or malignant lesions” was simplified to “premalignant lesions.” This change was made so that if and when this therapeutic approach is refined enough to be used on malignant lesions, new codes can be created to distinguish between the work performed for both types of lesions.
The new PDT codes include 96573 (photodynamic therapy by external application of light to destroy premalignant lesions of the skin and adjacent mucosa with application and illumination/activation of photosensitizing drug[s] provided by a physician or other qualified healthcare professional, per day) and 96574 (debridement of premalignant hyperkeratotic lesion[s][ie, targeted curettage, abrasion] followed with photodynamic therapy by external application of light to destroy premalignant lesions of the skin and adjacent mucosa with application and illumination/activation of photosensitizing drug[s] provided by a physician or other qualified healthcare professional, per day). According to the 2018 CPT manual,2 these codes should be used to report nonsurgical treatment of cutaneous lesions using PDT (ie, external application of light to destroy premalignant lesions of the skin and adjacent mucosa by activation of photosensitizing drug). A treatment session is defined as an application of a photosensitizer to all lesions within an anatomic area (eg, face, scalp) with or without debridement of all premalignant hyperkeratotic lesions in that area followed by illumination and activation with an appropriate light source. Providers should not report codes for debridement (11000, 11001, 11004, 11005), lesion shaving (11300–11313), biopsy (11100, 11101), or lesion excision (11400–11471) within the treatment area on the same day that PDT is administered.2
With the inclusion of these new PDT codes, the older code 96567 (photodynamic therapy by external application of light to destroy premalignant lesions of the skin and adjacent mucosa with application and illumination/activation of photosensitive drug[s], per day)—which is the base or parent code of the set—should only be used for reporting PDT when a physician or other qualified health care professional is not directly involved in the delivery of the service. Code 96573 is an upgrade to 96567 to account for physician work, while code 96574 captures the extra work of disruption of the skin barrier by debridement.
The novelty here is that old codes often are replaced when new codes come along. The reader should be aware of the distinct differences, as the total value expressed in relative value units for code 96567 is lower than it was in 2017 (3.24 vs 3.80), while the 2 newer codes have higher values (codes 96573 and 96574, 5.37 and 6.92, respectively). Additionally, the reader should note that only one of the 3 codes can be used on a given anatomic area (ie, face and scalp) on a given day. In general, a single-dose package of either of the approved photosensitizing drugs can usually treat an entire anatomic area. The codes themselves are not reserved for specific anatomic areas, but the US Food and Drug Administration clearances are for only face and scalp for both drugs, so the use of more than 2 PDT codes on a given day might raise payer queries.
Whatever you do, be sure your documentation includes an explicit notation about who applied the photosensitizing drug and the technique used for debridement, if performed. Code 96574 explicitly refers to targeted curettage and abrasion but does not include other destructive modalities (eg, chemical peeling), which an auditor may or may not consider an acceptable method of debridement. Personally, I will not be using peels as a justifier for this code.
Lasers
Lasers have played a role in the treatment of severe scarring in wounded warriors and other patient populations.7 Until 2018, there were no CPT codes that allowed precise reporting of these therapies. We now have a series of tracking codes, which are not valued by the Specialty Society Relative Value Scale Update Committee process but are nonetheless reportable, for this valuable treatment.8
The base code for a new pair of codes for reporting fractional ablative laser treatment, which is modeled after the skin graft code series, is 0479T (fractional ablative laser fenestration of burn and traumatic scars for functional improvement; first 100 cm2 or part thereof, or 1% of body surface area of infants and children). The add-on code is 0480T (fractional ablative laser fenestration of burn and traumatic scars for functional improvement; each additional 100 cm2, or each additional 1% of body surface area of infants and children, or part thereof [list separately in addition to code for primary procedure]), which means the code can be reported multiple times in addition to a single unit of 0479T. The aggregate treatment area should only be reported once per day regardless of the number of passes of one or more lasers over the area that day, and codes 0479T and 0480T should not be reported with codes 0491T or 0492T, which are a new family of tracking codes used for ablative laser treatment of chronic open wounds. If the scars are excised in a full-thickness manner, the benign excision codes 11400 to 11446 should be used instead.
For laser treatment of open wounds, 0491T (ablative laser treatment, noncontact, full-field and fractional ablation, open wound, per day, total treatment surface area; first 20 cm2 or less) is the base code for this pair of codes, and 0492T (ablative laser treatment, noncontact, full-field and fractional ablation, open wound, per day, total treatment surface area; each additional 20 cm2, or part thereof [list separately in addition to code for primary procedure]) is the add-on code, similar to the 0479T and 00480T codes described above. Keep in mind that all 4 of these tracking codes do not have defined values, and payment is at the discretion of the payer. If utilization of the procedures increases along with the development of appropriate evidence-based literature to support it, it is possible these will be converted into standard category I CPT codes that will be valued and covered by payers.
Final Thoughts
For more details on the new codes for PDT and lasers, I would strongly suggest obtaining a copy of CPT Changes 2018: An Insider’s View (https://commerce.ama-assn.org/store/catalog/productDetail.jsp?product_id=prod2800018&navAction=push), as well as the 2018 CPT manual for those who are actively practicing. Members of the American Academy of Dermatology also can get the new CPT manual as part of the group’s Coding Value Pack (https://store.aad.org/products/11383) along with Principles of Documentation for Dermatology and 2018 Coding & Billing for Dermatology.
Winter is the time when many religions celebrate a renewal of the year as the days begin to get longer. On January 1 of each year in the United States we celebrate the official activation of new and revised
Photodynamic Therapy
In 2004, a methyl aminolevulinate cream that was activated with a red light source was brought to market; however, after failing to gain a substantial market share, the product is no longer available in the United States. In May of 2016, a nanoemulsion gel formulation of aminolevulinic acid hydrochloride 10% was approved by the US Food and Drug Administration4 for use with a red light source. Unlike 5-aminolevulinic acid hydrochloride solution, which was approved for application with no prior debridement of the skin,5 the new gel formulation was meant to be applied after degreasing with an ethanol- or isopropanol-soaked cotton pad and removal of any scaling or crusts, followed by roughening of the lesion surfaces (with care taken to avoid bleeding).4 The product must be administered by a health care provider and is reported using CPT codes 96573 and 96574, which are new in 2018 and are discussed in more detail below. Effective January 1, 2018, the Healthcare Common Procedure Coding System supply code for the product is J7345 (aminolevulinic acid hydrochloride gel for topical administration, 10% gel, 10 mg).6 A single tube contains 200 mg, so when an entire tube is used (which is typical), 200 units must be reported. Partial tubes may be used in some patients and should be reported appropriately based on actual usage.
The development of new CPT codes for PDT revealed a middle ground in which many physicians, including myself, have applied the photosensitizing drug themselves instead of a nonphysician provider in order to use their professional judgment to ensure the entire treatment area was covered and also allow for multiple applications of the drug to lesions that in their opinion may have warranted greater dosing, which led to the creation of CPT code 96573. The revision and refinement from one code to 3 (96567, 96573, and 96574) also involved rewording of the preamble for all 3 codes so that the phrase “premalignant and/or malignant lesions” was simplified to “premalignant lesions.” This change was made so that if and when this therapeutic approach is refined enough to be used on malignant lesions, new codes can be created to distinguish between the work performed for both types of lesions.
The new PDT codes include 96573 (photodynamic therapy by external application of light to destroy premalignant lesions of the skin and adjacent mucosa with application and illumination/activation of photosensitizing drug[s] provided by a physician or other qualified healthcare professional, per day) and 96574 (debridement of premalignant hyperkeratotic lesion[s][ie, targeted curettage, abrasion] followed with photodynamic therapy by external application of light to destroy premalignant lesions of the skin and adjacent mucosa with application and illumination/activation of photosensitizing drug[s] provided by a physician or other qualified healthcare professional, per day). According to the 2018 CPT manual,2 these codes should be used to report nonsurgical treatment of cutaneous lesions using PDT (ie, external application of light to destroy premalignant lesions of the skin and adjacent mucosa by activation of photosensitizing drug). A treatment session is defined as an application of a photosensitizer to all lesions within an anatomic area (eg, face, scalp) with or without debridement of all premalignant hyperkeratotic lesions in that area followed by illumination and activation with an appropriate light source. Providers should not report codes for debridement (11000, 11001, 11004, 11005), lesion shaving (11300–11313), biopsy (11100, 11101), or lesion excision (11400–11471) within the treatment area on the same day that PDT is administered.2
With the inclusion of these new PDT codes, the older code 96567 (photodynamic therapy by external application of light to destroy premalignant lesions of the skin and adjacent mucosa with application and illumination/activation of photosensitive drug[s], per day)—which is the base or parent code of the set—should only be used for reporting PDT when a physician or other qualified health care professional is not directly involved in the delivery of the service. Code 96573 is an upgrade to 96567 to account for physician work, while code 96574 captures the extra work of disruption of the skin barrier by debridement.
The novelty here is that old codes often are replaced when new codes come along. The reader should be aware of the distinct differences, as the total value expressed in relative value units for code 96567 is lower than it was in 2017 (3.24 vs 3.80), while the 2 newer codes have higher values (codes 96573 and 96574, 5.37 and 6.92, respectively). Additionally, the reader should note that only one of the 3 codes can be used on a given anatomic area (ie, face and scalp) on a given day. In general, a single-dose package of either of the approved photosensitizing drugs can usually treat an entire anatomic area. The codes themselves are not reserved for specific anatomic areas, but the US Food and Drug Administration clearances are for only face and scalp for both drugs, so the use of more than 2 PDT codes on a given day might raise payer queries.
Whatever you do, be sure your documentation includes an explicit notation about who applied the photosensitizing drug and the technique used for debridement, if performed. Code 96574 explicitly refers to targeted curettage and abrasion but does not include other destructive modalities (eg, chemical peeling), which an auditor may or may not consider an acceptable method of debridement. Personally, I will not be using peels as a justifier for this code.
Lasers
Lasers have played a role in the treatment of severe scarring in wounded warriors and other patient populations.7 Until 2018, there were no CPT codes that allowed precise reporting of these therapies. We now have a series of tracking codes, which are not valued by the Specialty Society Relative Value Scale Update Committee process but are nonetheless reportable, for this valuable treatment.8
The base code for a new pair of codes for reporting fractional ablative laser treatment, which is modeled after the skin graft code series, is 0479T (fractional ablative laser fenestration of burn and traumatic scars for functional improvement; first 100 cm2 or part thereof, or 1% of body surface area of infants and children). The add-on code is 0480T (fractional ablative laser fenestration of burn and traumatic scars for functional improvement; each additional 100 cm2, or each additional 1% of body surface area of infants and children, or part thereof [list separately in addition to code for primary procedure]), which means the code can be reported multiple times in addition to a single unit of 0479T. The aggregate treatment area should only be reported once per day regardless of the number of passes of one or more lasers over the area that day, and codes 0479T and 0480T should not be reported with codes 0491T or 0492T, which are a new family of tracking codes used for ablative laser treatment of chronic open wounds. If the scars are excised in a full-thickness manner, the benign excision codes 11400 to 11446 should be used instead.
For laser treatment of open wounds, 0491T (ablative laser treatment, noncontact, full-field and fractional ablation, open wound, per day, total treatment surface area; first 20 cm2 or less) is the base code for this pair of codes, and 0492T (ablative laser treatment, noncontact, full-field and fractional ablation, open wound, per day, total treatment surface area; each additional 20 cm2, or part thereof [list separately in addition to code for primary procedure]) is the add-on code, similar to the 0479T and 00480T codes described above. Keep in mind that all 4 of these tracking codes do not have defined values, and payment is at the discretion of the payer. If utilization of the procedures increases along with the development of appropriate evidence-based literature to support it, it is possible these will be converted into standard category I CPT codes that will be valued and covered by payers.
Final Thoughts
For more details on the new codes for PDT and lasers, I would strongly suggest obtaining a copy of CPT Changes 2018: An Insider’s View (https://commerce.ama-assn.org/store/catalog/productDetail.jsp?product_id=prod2800018&navAction=push), as well as the 2018 CPT manual for those who are actively practicing. Members of the American Academy of Dermatology also can get the new CPT manual as part of the group’s Coding Value Pack (https://store.aad.org/products/11383) along with Principles of Documentation for Dermatology and 2018 Coding & Billing for Dermatology.
- Daniell MD, Hill JS. A history of photodynamic therapy. Aust N Z J Surg. 1991;61:340-348.
- Current Procedural Terminology 2018, Professional Edition. Chicago, IL: American Medical Association; 2018.
- HCPCS code J7308. HCPCS Complete Reference website. https://hcpcs.codes/j-codes/J7308/. Accessed March 1, 2018.
- Ameluz [package insert]. Wakefield, MA: Biofrontera Inc; 2017.
- Levulan Kerastick [package insert]. Wilmington, MA: Dusa Pharmaceuticals, Inc; 2010.
- Centers for Medicare & Medicaid Services. 2018 Table of drugs. CMS website. https://www.cms.gov/Medicare/Coding/HCPCSReleaseCodeSets/Downloads/2018-Table-of-Drugs.pdf. Updated February 15, 2018. Accessed February 21, 2018.
- Waibel JS, Rudnick A. Current trends and future considerations in scar treatment. Semin Cutan Med Surg. 2015;34:13-16.
- American Medical Association. CPT category III codes. AMA website. https://www.ama-assn.org/sites/default/files/media-browser/public/cpt/cpt-category3-codes-descriptors.pdf. Updated December 21, 2017. Accessed February 21, 2018.
- Daniell MD, Hill JS. A history of photodynamic therapy. Aust N Z J Surg. 1991;61:340-348.
- Current Procedural Terminology 2018, Professional Edition. Chicago, IL: American Medical Association; 2018.
- HCPCS code J7308. HCPCS Complete Reference website. https://hcpcs.codes/j-codes/J7308/. Accessed March 1, 2018.
- Ameluz [package insert]. Wakefield, MA: Biofrontera Inc; 2017.
- Levulan Kerastick [package insert]. Wilmington, MA: Dusa Pharmaceuticals, Inc; 2010.
- Centers for Medicare & Medicaid Services. 2018 Table of drugs. CMS website. https://www.cms.gov/Medicare/Coding/HCPCSReleaseCodeSets/Downloads/2018-Table-of-Drugs.pdf. Updated February 15, 2018. Accessed February 21, 2018.
- Waibel JS, Rudnick A. Current trends and future considerations in scar treatment. Semin Cutan Med Surg. 2015;34:13-16.
- American Medical Association. CPT category III codes. AMA website. https://www.ama-assn.org/sites/default/files/media-browser/public/cpt/cpt-category3-codes-descriptors.pdf. Updated December 21, 2017. Accessed February 21, 2018.
Practice Points
- In 2018, there are new sets of codes for photodynamic therapy (PDT) and lasers that all dermatologists should be aware of.
- The Current Procedural Terminology (CPT) codes for PDT—96567, 96573, and 96574—can only be used once per patient per day, and only one of the 3 codes can be used on a given anatomic area (ie, face and scalp) on a given day.
- Until 2018, there were no CPT codes that allowed for precise reporting of laser therapies, but there now is a series of tracking codes that are not valued by the Specialty Society Relative Value Scale Update Committee process but are nonetheless reportable.

Onychomycosis Diagnosis and Long-term Treatment
What does your patient need to know at the first visit?
Risk factors for onychomycosis include prior trauma, history of tinea pedis, sports activities, frequenting gyms and pools, hyperhidrosis, advancing age, diabetes mellitus, immunosuppression, smoking, and family history of onychomycosis. Toenails are involved more frequently than fingernails, and typical physical examination findings are distal and lateral nail plate onycholysis with subungual hyperkeratosis. In more severe cases, there may be nail plate thickening, crumbling, yellowing, and involvement of the nail matrix.
Because other nail conditions may resemble onychomycosis, it is imperative to confirm the diagnosis using histopathology, direct microscopy, fungal culture, and/or polymerase chain reaction on nail plate clippings or subungual debris.
What are your go-to treatments? What are the side effects?
After laboratory confirmation, assess the patient for the severity of the infection based on the surface area of nail plate affected, nail plate thickness, involvement of the nail matrix, and number of nails affected. United States Food and Drug Administration-approved oral and topical antifungals are used first line for the treatment of onychomycosis. Devices such as lasers are approved by the US Food and Drug Administration for temporary cosmetic improvement in the appearance of the nail without eradicating the fungus.
Oral antifungals such as terbinafine, itraconazole, and fluconazole (off label) are indicated for patients with severe disease. Patients with mild to moderate disease may benefit from oral or topical antifungals such as efinaconazole, tavaborole, or ciclopirox.
I recommend terbinafine to many of my patients due to its high complete and mycological cure rates, short list of drug-drug interactions, and low incidence of side effects. Adverse reactions are uncommon, with the most common being gastrointestinal upset. While liver injury has been reported, it is exceedingly rare. Itraconazole has many important drug interactions and is contraindicated in patients with congestive heart failure. With topical antifungals, side effects are uncommon, but dermatitis, ingrown nails, and vesicles may occur.
How do you keep patients compliant with treatment?
Patients on a 3-month course of daily oral terbinafine or itraconazole for toenail onychomycosis are typically highly compliant. Compliance for patients on oral fluconazole (off label) is generally more challenging because it is dosed weekly until the nail grows out (1-1.5 years for toenails). To circumvent missed fluconazole doses, I recommend that the patient schedule quarterly visits with me and also to set a cell phone alarm as a weekly reminder to take the medication.
Because topical medications are prescribed for the toenails for a year-long course (with avoidance of nail polish during this period), I prescribe topical antifungals only to highly motivated patients. In addition, because topical antifungals are retained in the nail plate for at least several days after a month-long application, I tell my patients that if they have a big event to attend that they can take a vacation from the topical antifungal, get a pedicure, and then resume treatment after the event.
What do you do if they refuse treatment?
In 2018, we have many options to treat onychomycosis effectively, and therapy is individualized based on the patient's severity of disease, infecting organism(s), comorbidities, concomitant medications, and preferences. If the patient's fungal nail infection is asymptomatic and not aesthetically bothersome, he/she may opt for observation rather than treatment. If the decision is observation, I recommend use of a topical antifungal on the feet and web spaces to prevent worsening of onychomycosis.
Suggested Readings
Gupta AK, Versteeg SG. A critical review of improvement rates for laser therapy used to treat toenail onychomycosis. J Eur Acad Dermatol Venereol. 2017;31:1111-1118.
Lipner SR, Scher RK. Long-standing onychodystrophy in a young woman. JAMA. 2016;316:1915-1916.
Lipner SR, Scher RK. Onychomycosis--a small step for quality of care. Curr Med Res Opin. 2016;32:865-867.
Lipner SR, Scher RK. Onychomycosis: current and investigational therapies. Cutis. 2014;94:E21-E24.
What does your patient need to know at the first visit?
Risk factors for onychomycosis include prior trauma, history of tinea pedis, sports activities, frequenting gyms and pools, hyperhidrosis, advancing age, diabetes mellitus, immunosuppression, smoking, and family history of onychomycosis. Toenails are involved more frequently than fingernails, and typical physical examination findings are distal and lateral nail plate onycholysis with subungual hyperkeratosis. In more severe cases, there may be nail plate thickening, crumbling, yellowing, and involvement of the nail matrix.
Because other nail conditions may resemble onychomycosis, it is imperative to confirm the diagnosis using histopathology, direct microscopy, fungal culture, and/or polymerase chain reaction on nail plate clippings or subungual debris.
What are your go-to treatments? What are the side effects?
After laboratory confirmation, assess the patient for the severity of the infection based on the surface area of nail plate affected, nail plate thickness, involvement of the nail matrix, and number of nails affected. United States Food and Drug Administration-approved oral and topical antifungals are used first line for the treatment of onychomycosis. Devices such as lasers are approved by the US Food and Drug Administration for temporary cosmetic improvement in the appearance of the nail without eradicating the fungus.
Oral antifungals such as terbinafine, itraconazole, and fluconazole (off label) are indicated for patients with severe disease. Patients with mild to moderate disease may benefit from oral or topical antifungals such as efinaconazole, tavaborole, or ciclopirox.
I recommend terbinafine to many of my patients due to its high complete and mycological cure rates, short list of drug-drug interactions, and low incidence of side effects. Adverse reactions are uncommon, with the most common being gastrointestinal upset. While liver injury has been reported, it is exceedingly rare. Itraconazole has many important drug interactions and is contraindicated in patients with congestive heart failure. With topical antifungals, side effects are uncommon, but dermatitis, ingrown nails, and vesicles may occur.
How do you keep patients compliant with treatment?
Patients on a 3-month course of daily oral terbinafine or itraconazole for toenail onychomycosis are typically highly compliant. Compliance for patients on oral fluconazole (off label) is generally more challenging because it is dosed weekly until the nail grows out (1-1.5 years for toenails). To circumvent missed fluconazole doses, I recommend that the patient schedule quarterly visits with me and also to set a cell phone alarm as a weekly reminder to take the medication.
Because topical medications are prescribed for the toenails for a year-long course (with avoidance of nail polish during this period), I prescribe topical antifungals only to highly motivated patients. In addition, because topical antifungals are retained in the nail plate for at least several days after a month-long application, I tell my patients that if they have a big event to attend that they can take a vacation from the topical antifungal, get a pedicure, and then resume treatment after the event.
What do you do if they refuse treatment?
In 2018, we have many options to treat onychomycosis effectively, and therapy is individualized based on the patient's severity of disease, infecting organism(s), comorbidities, concomitant medications, and preferences. If the patient's fungal nail infection is asymptomatic and not aesthetically bothersome, he/she may opt for observation rather than treatment. If the decision is observation, I recommend use of a topical antifungal on the feet and web spaces to prevent worsening of onychomycosis.
Suggested Readings
Gupta AK, Versteeg SG. A critical review of improvement rates for laser therapy used to treat toenail onychomycosis. J Eur Acad Dermatol Venereol. 2017;31:1111-1118.
Lipner SR, Scher RK. Long-standing onychodystrophy in a young woman. JAMA. 2016;316:1915-1916.
Lipner SR, Scher RK. Onychomycosis--a small step for quality of care. Curr Med Res Opin. 2016;32:865-867.
Lipner SR, Scher RK. Onychomycosis: current and investigational therapies. Cutis. 2014;94:E21-E24.
What does your patient need to know at the first visit?
Risk factors for onychomycosis include prior trauma, history of tinea pedis, sports activities, frequenting gyms and pools, hyperhidrosis, advancing age, diabetes mellitus, immunosuppression, smoking, and family history of onychomycosis. Toenails are involved more frequently than fingernails, and typical physical examination findings are distal and lateral nail plate onycholysis with subungual hyperkeratosis. In more severe cases, there may be nail plate thickening, crumbling, yellowing, and involvement of the nail matrix.
Because other nail conditions may resemble onychomycosis, it is imperative to confirm the diagnosis using histopathology, direct microscopy, fungal culture, and/or polymerase chain reaction on nail plate clippings or subungual debris.
What are your go-to treatments? What are the side effects?
After laboratory confirmation, assess the patient for the severity of the infection based on the surface area of nail plate affected, nail plate thickness, involvement of the nail matrix, and number of nails affected. United States Food and Drug Administration-approved oral and topical antifungals are used first line for the treatment of onychomycosis. Devices such as lasers are approved by the US Food and Drug Administration for temporary cosmetic improvement in the appearance of the nail without eradicating the fungus.
Oral antifungals such as terbinafine, itraconazole, and fluconazole (off label) are indicated for patients with severe disease. Patients with mild to moderate disease may benefit from oral or topical antifungals such as efinaconazole, tavaborole, or ciclopirox.
I recommend terbinafine to many of my patients due to its high complete and mycological cure rates, short list of drug-drug interactions, and low incidence of side effects. Adverse reactions are uncommon, with the most common being gastrointestinal upset. While liver injury has been reported, it is exceedingly rare. Itraconazole has many important drug interactions and is contraindicated in patients with congestive heart failure. With topical antifungals, side effects are uncommon, but dermatitis, ingrown nails, and vesicles may occur.
How do you keep patients compliant with treatment?
Patients on a 3-month course of daily oral terbinafine or itraconazole for toenail onychomycosis are typically highly compliant. Compliance for patients on oral fluconazole (off label) is generally more challenging because it is dosed weekly until the nail grows out (1-1.5 years for toenails). To circumvent missed fluconazole doses, I recommend that the patient schedule quarterly visits with me and also to set a cell phone alarm as a weekly reminder to take the medication.
Because topical medications are prescribed for the toenails for a year-long course (with avoidance of nail polish during this period), I prescribe topical antifungals only to highly motivated patients. In addition, because topical antifungals are retained in the nail plate for at least several days after a month-long application, I tell my patients that if they have a big event to attend that they can take a vacation from the topical antifungal, get a pedicure, and then resume treatment after the event.
What do you do if they refuse treatment?
In 2018, we have many options to treat onychomycosis effectively, and therapy is individualized based on the patient's severity of disease, infecting organism(s), comorbidities, concomitant medications, and preferences. If the patient's fungal nail infection is asymptomatic and not aesthetically bothersome, he/she may opt for observation rather than treatment. If the decision is observation, I recommend use of a topical antifungal on the feet and web spaces to prevent worsening of onychomycosis.
Suggested Readings
Gupta AK, Versteeg SG. A critical review of improvement rates for laser therapy used to treat toenail onychomycosis. J Eur Acad Dermatol Venereol. 2017;31:1111-1118.
Lipner SR, Scher RK. Long-standing onychodystrophy in a young woman. JAMA. 2016;316:1915-1916.
Lipner SR, Scher RK. Onychomycosis--a small step for quality of care. Curr Med Res Opin. 2016;32:865-867.
Lipner SR, Scher RK. Onychomycosis: current and investigational therapies. Cutis. 2014;94:E21-E24.

Concurrent Anticytokine Biologics for the Management of Severe Hidradenitis Suppurativa: Are They Safe and Effective?
Dysregulated immune responses including elevations in the inflammatory cytokines tumor necrosis factor (TNF),1-4 IL- 1 β ,3 and IL-12/235-7 have been identified in hidradenitis suppurativa (HS). Targeted biologic agents may offer an opportunity to intervene in specific aberrant inflammatory pathways to effectively treat HS while minimizing a dverse effects (AEs). There is growing evidence, however, that treatment of HS with a single biologic agent is not effective in all patients.6,8-17 The TNF antagonist adalimumab has been shown to achieve clinical response in approximately 50% of patients (N = 633). 18
The administration of concurrent biologics may offer the potential for improved disease control through synergistic targeting of multiple inflammatory pathways, particularly for severe and recalcitrant HS. This approach may be effective given insights from mechanistic studies suggesting the involvement of multiple inflammatory pathways in the disease pathogenesis.3,21 Concurrent anticytokine biologics have been used safely and effectively in other inflammatory diseases; for example, combination therapy with TNF and IL-12/23 antagonists have resulted in near-complete to complete resolution of severe psoriatic skin and joint disease without AEs.22-24
An increased risk for infection without increased efficacy associated with the use of concurrent anticytokine biologics for treatment of rheumatoid arthritis (RA) has raised concerns about the safety of this therapeutic approach. In a study of concurrent etanercept and anakinra therapy for RA (N=244), the combined therapy was not more efficacious than etanercept alone (American College of Rheumatology 50% response at week 24: etanercept 25 mg twice weekly, 41%; etanercept 25 mg twice weekly plus anakinra 100 mg once daily, 31%; etanercept 25 mg once weekly plus anakinra 100 mg once daily, 39% [P=.914]).25 Combination therapy also was associated with a higher overall incidence of serious AEs, serious infections requiring antibiotics or hospitalizations, and serious infections leading to study withdrawal. Reported infections included pneumonia, cellulitis, herpes zoster, pneumonitis, and pyelonephritis, but no opportunistic infections or tuberculosis were reported. A single case of lymphoma was reported in the full-dose etanercept plus anakinra group; however, the association with therapy is unclear, as RA itself is associated with an increased risk of malignancy.25
Although these results are notable, caution must be exercised in extrapolating safety and efficacy data for treatment with concurrent biologics from the RA literature for management of HS for several reasons. First, RA is an autoimmune disease that is associated with an increased risk for genitourinary and bronchopulmonary infections and septic arthritis, even in the absence of treatment with steroids and immunomodulatory drugs.26,27 Increased risk for development of lymphoma, lung cancer, and nonmelanoma skin cancer also has been associated with RA.28,29 The exact etiology of this increased risk is unknown, but it is thought to relate to immunologic disturbances and chronic systemic inflammation associated with RA.29 Furthermore, RA disease characteristics and comorbidities that may contribute to an increased risk for infection and malignancy include advanced age as well as a history of leukopenia, chronic lung disease, diabetes mellitus, alcoholism, and/or smoking.30 Infection and malignancy risk in RA also may be compounded by immunomodulatory therapies.31,32
Conversely, although microbes are believed to play an important role in HS initiation and progression, HS is neither considered an infectious disease nor associated with an increased risk for infection.33 Increased malignancy risk generally is not reported with HS, and systematic therapeutic trials of biologic therapies for HS have been notable for an absence of infectious or malignant AEs compared to placebo.12,14,16,18,19 From a mechanistic standpoint, data suggest that HS may be fundamentally distinct from RA and other autoimmune diseases; therefore, it may not be appropriate to extrapolate safety data from the latter to guide therapeutic strategies for the former.
The concept that different inflammatory diseases harbor distinct risks for comorbidities and AEs associated with medications is further supported by data from patients with PAPA syndrome (pyogenic arthritis, pyoderma gangrenosum, and acne), a monogenic autoinflammatory disease characterized by inflammasome activation and subsequent increased signaling via IL-1.34
We have safely and effectively treated 2 patients with severe HS with extended courses of concurrent TNF and IL-1 antagonists. Both patients had previously failed treatment with multiple therapeutic interventions, including topical and systemic antibiotics, disease-modifying antirheumatic drugs, hormonal therapy, biologic monotherapy with several targeted agents, and wide local excision. In the setting of concurrent certolizumab plus anakinra in the first patient and adalimumab plus anakinra in the second, both patients reported reduced drainage, pain, and number of disease flares. Both patients also were maintained on extended treatment courses (11 months and 2 years, respectively) without evidence of infection or malignancy.
Concurrent biologics may be safe and effective in managing recalcitrant HS; however, large prospective studies are needed to confirm these anecdotal findings. As our understanding of HS pathogenesis expands, novel and more effective therapeutic options will be developed. Until then, concurrent biologics may be a potential option for patients with severe recalcitrant HS.
- Jemec GB. Predicting response to anti-TNF-alpha treatment in hidradenitis suppurativa. Br J Dermatol. 2013;168:233.
- Sbidian E, Hotz C, Seneschal J, et al. Antitumour necrosis factor-α therapy for hidradenitis suppurativa: results from a national cohort study between 2000 and 2013 [published online December 22, 2015]. Br J Dermatol. 2016;174:667-670.
- van der Zee HH, de Ruiter L, van den Broecke DG, et al. Elevated levels of tumour necrosis factor (TNF)-α, interleukin (IL)-1β and IL-10 in hidradenitis suppurativa skin: a rationale for targeting TNF-α and IL-1β [published online May 17, 2011]. Br J Dermatol. 2011;164:1292-1298.
- van Rappard DC, Limpens J, Mekkes JR. The off-label treatment of severe hidradenitis suppurativa with TNF-alpha inhibitors: a systematic review. J Dermatolog Treat. 2013;24:392-404.
- Baerveldt EM, Kappen JH, Thio HB, et al. Successful long-term triple disease control by ustekinumab in a patient with Behcet’s disease, psoriasis and hidradenitis suppurativa. Ann Rheum Dis. 2013;72:626-627.
- Gulliver WP, Jemec GB, Baker KA. Experience with ustekinumab for the treatment of moderate to severe hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2012;26:911-914.
- Santos-Peréz MI, García-Rodicio S, Del Olmo-Revuelto MA, et al. Ustekinumab for hidradenitis suppurativa: a case report [published online December 3, 2013]. Actas Dermosifiliogr. 2014;105:720-722.
- Amano M, Grant A, Kerdel FA. A prospective open-label clinical trial of adalimumab for the treatment of hidradenitis suppurativa. Int J Dermatol. 2010;49:950-955.
- Blanco R, Gonzalez-Lopez MA, Gonzalez-Vela MC, et al. Disparate results in studies of adalimumab in the treatment of hidradenitis suppurativa: comment on the article by Amano et al. Int J Dermatol. 2013;52:380-381.
- Fardet L, Dupuy A, Kerob D, et al. Infliximab for severe hidradenitis suppurativa: transient clinical efficacy in 7 consecutive patients. J Am Acad Dermatol. 2007;56:624-628.
- Grant A, Gonzalez T, Montgomery MO, et al. Infliximab therapy for patients with moderate to severe hidradenitis suppurativa: a randomized, double-blind, placebo-controlled crossover trial. J Am Acad Dermatol. 2010;62:205-217.
- Kimball AB, Kerdel F, Adams D, et al. Adalimumab for the treatment of moderate to severe hidradenitis suppurativa: a parallel randomized trial. Ann Intern Med. 2012;157:846-855.
- Usmani N, Clayton TH, Everett S, et al. Variable response of hidradenitis suppurativa to infliximab in four patients. Clin Exp Dermatol. 2007;32:204-205.
- Leslie KS, Tripathi SV, Nguyen TV, et al. An open-label study of anakinra for the treatment of moderate to severe hidradenitis suppurativa. J Am Acad Dermatol. 2014;70:243-251.
- Menis D, Maronas-Jimenez L, Delgado-Marquez AM, et al. Two cases of severe hidradenitis suppurativa with failure of anakinra therapy [published online January 22, 2015]. Br J Dermatol. 2015;172:810-811.
- Tzanetakou V, Kanni T, Giatrakou S, et al. Safety and efficacy of anakinra in severe hidradenitis suppurativa: a randomized clinical trial. JAMA Dermatol. 2016;152:52-59.
- Zarchi K, Dufour DN, Jemec GB. Successful treatment of severe hidradenitis suppurativa with anakinra. JAMA Dermatol. 2013;149:1192-1194.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Blok JL, Li K, Brodmerkel C, et al. Ustekinumab in hidradenitis suppurativa: clinical results and a search for potential biomarkers in serum. Br J Dermatol. 2016;174:839-846.
- Hoffman LK, Ghias MH, Garg A, et al. Major gaps in understanding and treatment of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:86-92.
- Schlapbach C, Hanni T, Yawalkar N, et al. Expression of the IL-23/Th17 pathway in lesions of hidradenitis suppurativa. J Am Acad Dermatol. 2011;65:790-798.
- Torre KM, Payette MJ. Combination biologic therapy for the treatment of severe palmoplantar pustulosis. JAAD Case Rep. 2017;3:240-242.
- Babalola O, Lakdawala N, Strober BE. Combined biologic therapy for the treatment of psoriasis and psoriatic arthritis: a case report. JAAD Case Rep. 2015;1:3-4.
- Cuchacovich R, Garcia-Valladares I, Espinoza LR. Combination biologic treatment of refractory psoriasis and psoriatic arthritis. J Rheumatol. 2012;39:187-193.
- Genovese MC, Cohen S, Moreland L, et al. Combination therapy with etanercept and anakinra in the treatment of patients with rheumatoid arthritis who have been treated unsuccessfully with methotrexate. Arthritis Rheum. 2004;50:1412-1419.
- Baum J. Infection in rheumatoid arthritis. Arthritis Rheum. 1971;14:135-137.
- Doran MF, Crowson CS, Pond GR, et al. Frequency of infection in patients with rheumatoid arthritis compared with controls: a population-based study. Arthritis Rheum. 2002;46:2287-2293.
- Askling J, Fored CM, Baecklund E, et al. Haematopoietic malignancies in rheumatoid arthritis: lymphoma risk and characteristics after exposure to tumour necrosis factor antagonists. Ann Rheum Dis. 2005;64:1414-1420.
- Smitten AL, Simon TA, Hochberg MC, et al. A meta-analysis of the incidence of malignancy in adult patients with rheumatoid arthritis [published online April 23, 2008]. Arthritis Res Ther. 2008;10:R45.
- Doran MF, Crowson CS, Pond GR, et al. Predictors of infection inrheumatoid arthritis. Arthritis Rheum. 2002;46:2294-2300.
- Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum. 2007;56:2886-2895.
- Raaschou P, Simard JF, Asker Hagelberg C, et al. Rheumatoid arthritis, anti-tumour necrosis factor treatment, and risk of squamous cell and basal cell skin cancer: cohort study based on nationwide prospectively recorded data from Sweden. BMJ. 2016;352:i262.
- Ring HC, Riis Mikkelsen P, Miller IM, et al. The bacteriology of hidradenitis suppurativa: a systematic review. Exp Dermatol. 2015;24:727-731.
- Smith EJ, Allantaz F, Bennett L, et al. Clinical, molecular, and genetic characteristics of PAPA syndrome: a review. Curr Genomics. 2010;11:519-527.
Dysregulated immune responses including elevations in the inflammatory cytokines tumor necrosis factor (TNF),1-4 IL- 1 β ,3 and IL-12/235-7 have been identified in hidradenitis suppurativa (HS). Targeted biologic agents may offer an opportunity to intervene in specific aberrant inflammatory pathways to effectively treat HS while minimizing a dverse effects (AEs). There is growing evidence, however, that treatment of HS with a single biologic agent is not effective in all patients.6,8-17 The TNF antagonist adalimumab has been shown to achieve clinical response in approximately 50% of patients (N = 633). 18
The administration of concurrent biologics may offer the potential for improved disease control through synergistic targeting of multiple inflammatory pathways, particularly for severe and recalcitrant HS. This approach may be effective given insights from mechanistic studies suggesting the involvement of multiple inflammatory pathways in the disease pathogenesis.3,21 Concurrent anticytokine biologics have been used safely and effectively in other inflammatory diseases; for example, combination therapy with TNF and IL-12/23 antagonists have resulted in near-complete to complete resolution of severe psoriatic skin and joint disease without AEs.22-24
An increased risk for infection without increased efficacy associated with the use of concurrent anticytokine biologics for treatment of rheumatoid arthritis (RA) has raised concerns about the safety of this therapeutic approach. In a study of concurrent etanercept and anakinra therapy for RA (N=244), the combined therapy was not more efficacious than etanercept alone (American College of Rheumatology 50% response at week 24: etanercept 25 mg twice weekly, 41%; etanercept 25 mg twice weekly plus anakinra 100 mg once daily, 31%; etanercept 25 mg once weekly plus anakinra 100 mg once daily, 39% [P=.914]).25 Combination therapy also was associated with a higher overall incidence of serious AEs, serious infections requiring antibiotics or hospitalizations, and serious infections leading to study withdrawal. Reported infections included pneumonia, cellulitis, herpes zoster, pneumonitis, and pyelonephritis, but no opportunistic infections or tuberculosis were reported. A single case of lymphoma was reported in the full-dose etanercept plus anakinra group; however, the association with therapy is unclear, as RA itself is associated with an increased risk of malignancy.25
Although these results are notable, caution must be exercised in extrapolating safety and efficacy data for treatment with concurrent biologics from the RA literature for management of HS for several reasons. First, RA is an autoimmune disease that is associated with an increased risk for genitourinary and bronchopulmonary infections and septic arthritis, even in the absence of treatment with steroids and immunomodulatory drugs.26,27 Increased risk for development of lymphoma, lung cancer, and nonmelanoma skin cancer also has been associated with RA.28,29 The exact etiology of this increased risk is unknown, but it is thought to relate to immunologic disturbances and chronic systemic inflammation associated with RA.29 Furthermore, RA disease characteristics and comorbidities that may contribute to an increased risk for infection and malignancy include advanced age as well as a history of leukopenia, chronic lung disease, diabetes mellitus, alcoholism, and/or smoking.30 Infection and malignancy risk in RA also may be compounded by immunomodulatory therapies.31,32
Conversely, although microbes are believed to play an important role in HS initiation and progression, HS is neither considered an infectious disease nor associated with an increased risk for infection.33 Increased malignancy risk generally is not reported with HS, and systematic therapeutic trials of biologic therapies for HS have been notable for an absence of infectious or malignant AEs compared to placebo.12,14,16,18,19 From a mechanistic standpoint, data suggest that HS may be fundamentally distinct from RA and other autoimmune diseases; therefore, it may not be appropriate to extrapolate safety data from the latter to guide therapeutic strategies for the former.
The concept that different inflammatory diseases harbor distinct risks for comorbidities and AEs associated with medications is further supported by data from patients with PAPA syndrome (pyogenic arthritis, pyoderma gangrenosum, and acne), a monogenic autoinflammatory disease characterized by inflammasome activation and subsequent increased signaling via IL-1.34
We have safely and effectively treated 2 patients with severe HS with extended courses of concurrent TNF and IL-1 antagonists. Both patients had previously failed treatment with multiple therapeutic interventions, including topical and systemic antibiotics, disease-modifying antirheumatic drugs, hormonal therapy, biologic monotherapy with several targeted agents, and wide local excision. In the setting of concurrent certolizumab plus anakinra in the first patient and adalimumab plus anakinra in the second, both patients reported reduced drainage, pain, and number of disease flares. Both patients also were maintained on extended treatment courses (11 months and 2 years, respectively) without evidence of infection or malignancy.
Concurrent biologics may be safe and effective in managing recalcitrant HS; however, large prospective studies are needed to confirm these anecdotal findings. As our understanding of HS pathogenesis expands, novel and more effective therapeutic options will be developed. Until then, concurrent biologics may be a potential option for patients with severe recalcitrant HS.
Dysregulated immune responses including elevations in the inflammatory cytokines tumor necrosis factor (TNF),1-4 IL- 1 β ,3 and IL-12/235-7 have been identified in hidradenitis suppurativa (HS). Targeted biologic agents may offer an opportunity to intervene in specific aberrant inflammatory pathways to effectively treat HS while minimizing a dverse effects (AEs). There is growing evidence, however, that treatment of HS with a single biologic agent is not effective in all patients.6,8-17 The TNF antagonist adalimumab has been shown to achieve clinical response in approximately 50% of patients (N = 633). 18
The administration of concurrent biologics may offer the potential for improved disease control through synergistic targeting of multiple inflammatory pathways, particularly for severe and recalcitrant HS. This approach may be effective given insights from mechanistic studies suggesting the involvement of multiple inflammatory pathways in the disease pathogenesis.3,21 Concurrent anticytokine biologics have been used safely and effectively in other inflammatory diseases; for example, combination therapy with TNF and IL-12/23 antagonists have resulted in near-complete to complete resolution of severe psoriatic skin and joint disease without AEs.22-24
An increased risk for infection without increased efficacy associated with the use of concurrent anticytokine biologics for treatment of rheumatoid arthritis (RA) has raised concerns about the safety of this therapeutic approach. In a study of concurrent etanercept and anakinra therapy for RA (N=244), the combined therapy was not more efficacious than etanercept alone (American College of Rheumatology 50% response at week 24: etanercept 25 mg twice weekly, 41%; etanercept 25 mg twice weekly plus anakinra 100 mg once daily, 31%; etanercept 25 mg once weekly plus anakinra 100 mg once daily, 39% [P=.914]).25 Combination therapy also was associated with a higher overall incidence of serious AEs, serious infections requiring antibiotics or hospitalizations, and serious infections leading to study withdrawal. Reported infections included pneumonia, cellulitis, herpes zoster, pneumonitis, and pyelonephritis, but no opportunistic infections or tuberculosis were reported. A single case of lymphoma was reported in the full-dose etanercept plus anakinra group; however, the association with therapy is unclear, as RA itself is associated with an increased risk of malignancy.25
Although these results are notable, caution must be exercised in extrapolating safety and efficacy data for treatment with concurrent biologics from the RA literature for management of HS for several reasons. First, RA is an autoimmune disease that is associated with an increased risk for genitourinary and bronchopulmonary infections and septic arthritis, even in the absence of treatment with steroids and immunomodulatory drugs.26,27 Increased risk for development of lymphoma, lung cancer, and nonmelanoma skin cancer also has been associated with RA.28,29 The exact etiology of this increased risk is unknown, but it is thought to relate to immunologic disturbances and chronic systemic inflammation associated with RA.29 Furthermore, RA disease characteristics and comorbidities that may contribute to an increased risk for infection and malignancy include advanced age as well as a history of leukopenia, chronic lung disease, diabetes mellitus, alcoholism, and/or smoking.30 Infection and malignancy risk in RA also may be compounded by immunomodulatory therapies.31,32
Conversely, although microbes are believed to play an important role in HS initiation and progression, HS is neither considered an infectious disease nor associated with an increased risk for infection.33 Increased malignancy risk generally is not reported with HS, and systematic therapeutic trials of biologic therapies for HS have been notable for an absence of infectious or malignant AEs compared to placebo.12,14,16,18,19 From a mechanistic standpoint, data suggest that HS may be fundamentally distinct from RA and other autoimmune diseases; therefore, it may not be appropriate to extrapolate safety data from the latter to guide therapeutic strategies for the former.
The concept that different inflammatory diseases harbor distinct risks for comorbidities and AEs associated with medications is further supported by data from patients with PAPA syndrome (pyogenic arthritis, pyoderma gangrenosum, and acne), a monogenic autoinflammatory disease characterized by inflammasome activation and subsequent increased signaling via IL-1.34
We have safely and effectively treated 2 patients with severe HS with extended courses of concurrent TNF and IL-1 antagonists. Both patients had previously failed treatment with multiple therapeutic interventions, including topical and systemic antibiotics, disease-modifying antirheumatic drugs, hormonal therapy, biologic monotherapy with several targeted agents, and wide local excision. In the setting of concurrent certolizumab plus anakinra in the first patient and adalimumab plus anakinra in the second, both patients reported reduced drainage, pain, and number of disease flares. Both patients also were maintained on extended treatment courses (11 months and 2 years, respectively) without evidence of infection or malignancy.
Concurrent biologics may be safe and effective in managing recalcitrant HS; however, large prospective studies are needed to confirm these anecdotal findings. As our understanding of HS pathogenesis expands, novel and more effective therapeutic options will be developed. Until then, concurrent biologics may be a potential option for patients with severe recalcitrant HS.
- Jemec GB. Predicting response to anti-TNF-alpha treatment in hidradenitis suppurativa. Br J Dermatol. 2013;168:233.
- Sbidian E, Hotz C, Seneschal J, et al. Antitumour necrosis factor-α therapy for hidradenitis suppurativa: results from a national cohort study between 2000 and 2013 [published online December 22, 2015]. Br J Dermatol. 2016;174:667-670.
- van der Zee HH, de Ruiter L, van den Broecke DG, et al. Elevated levels of tumour necrosis factor (TNF)-α, interleukin (IL)-1β and IL-10 in hidradenitis suppurativa skin: a rationale for targeting TNF-α and IL-1β [published online May 17, 2011]. Br J Dermatol. 2011;164:1292-1298.
- van Rappard DC, Limpens J, Mekkes JR. The off-label treatment of severe hidradenitis suppurativa with TNF-alpha inhibitors: a systematic review. J Dermatolog Treat. 2013;24:392-404.
- Baerveldt EM, Kappen JH, Thio HB, et al. Successful long-term triple disease control by ustekinumab in a patient with Behcet’s disease, psoriasis and hidradenitis suppurativa. Ann Rheum Dis. 2013;72:626-627.
- Gulliver WP, Jemec GB, Baker KA. Experience with ustekinumab for the treatment of moderate to severe hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2012;26:911-914.
- Santos-Peréz MI, García-Rodicio S, Del Olmo-Revuelto MA, et al. Ustekinumab for hidradenitis suppurativa: a case report [published online December 3, 2013]. Actas Dermosifiliogr. 2014;105:720-722.
- Amano M, Grant A, Kerdel FA. A prospective open-label clinical trial of adalimumab for the treatment of hidradenitis suppurativa. Int J Dermatol. 2010;49:950-955.
- Blanco R, Gonzalez-Lopez MA, Gonzalez-Vela MC, et al. Disparate results in studies of adalimumab in the treatment of hidradenitis suppurativa: comment on the article by Amano et al. Int J Dermatol. 2013;52:380-381.
- Fardet L, Dupuy A, Kerob D, et al. Infliximab for severe hidradenitis suppurativa: transient clinical efficacy in 7 consecutive patients. J Am Acad Dermatol. 2007;56:624-628.
- Grant A, Gonzalez T, Montgomery MO, et al. Infliximab therapy for patients with moderate to severe hidradenitis suppurativa: a randomized, double-blind, placebo-controlled crossover trial. J Am Acad Dermatol. 2010;62:205-217.
- Kimball AB, Kerdel F, Adams D, et al. Adalimumab for the treatment of moderate to severe hidradenitis suppurativa: a parallel randomized trial. Ann Intern Med. 2012;157:846-855.
- Usmani N, Clayton TH, Everett S, et al. Variable response of hidradenitis suppurativa to infliximab in four patients. Clin Exp Dermatol. 2007;32:204-205.
- Leslie KS, Tripathi SV, Nguyen TV, et al. An open-label study of anakinra for the treatment of moderate to severe hidradenitis suppurativa. J Am Acad Dermatol. 2014;70:243-251.
- Menis D, Maronas-Jimenez L, Delgado-Marquez AM, et al. Two cases of severe hidradenitis suppurativa with failure of anakinra therapy [published online January 22, 2015]. Br J Dermatol. 2015;172:810-811.
- Tzanetakou V, Kanni T, Giatrakou S, et al. Safety and efficacy of anakinra in severe hidradenitis suppurativa: a randomized clinical trial. JAMA Dermatol. 2016;152:52-59.
- Zarchi K, Dufour DN, Jemec GB. Successful treatment of severe hidradenitis suppurativa with anakinra. JAMA Dermatol. 2013;149:1192-1194.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Blok JL, Li K, Brodmerkel C, et al. Ustekinumab in hidradenitis suppurativa: clinical results and a search for potential biomarkers in serum. Br J Dermatol. 2016;174:839-846.
- Hoffman LK, Ghias MH, Garg A, et al. Major gaps in understanding and treatment of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:86-92.
- Schlapbach C, Hanni T, Yawalkar N, et al. Expression of the IL-23/Th17 pathway in lesions of hidradenitis suppurativa. J Am Acad Dermatol. 2011;65:790-798.
- Torre KM, Payette MJ. Combination biologic therapy for the treatment of severe palmoplantar pustulosis. JAAD Case Rep. 2017;3:240-242.
- Babalola O, Lakdawala N, Strober BE. Combined biologic therapy for the treatment of psoriasis and psoriatic arthritis: a case report. JAAD Case Rep. 2015;1:3-4.
- Cuchacovich R, Garcia-Valladares I, Espinoza LR. Combination biologic treatment of refractory psoriasis and psoriatic arthritis. J Rheumatol. 2012;39:187-193.
- Genovese MC, Cohen S, Moreland L, et al. Combination therapy with etanercept and anakinra in the treatment of patients with rheumatoid arthritis who have been treated unsuccessfully with methotrexate. Arthritis Rheum. 2004;50:1412-1419.
- Baum J. Infection in rheumatoid arthritis. Arthritis Rheum. 1971;14:135-137.
- Doran MF, Crowson CS, Pond GR, et al. Frequency of infection in patients with rheumatoid arthritis compared with controls: a population-based study. Arthritis Rheum. 2002;46:2287-2293.
- Askling J, Fored CM, Baecklund E, et al. Haematopoietic malignancies in rheumatoid arthritis: lymphoma risk and characteristics after exposure to tumour necrosis factor antagonists. Ann Rheum Dis. 2005;64:1414-1420.
- Smitten AL, Simon TA, Hochberg MC, et al. A meta-analysis of the incidence of malignancy in adult patients with rheumatoid arthritis [published online April 23, 2008]. Arthritis Res Ther. 2008;10:R45.
- Doran MF, Crowson CS, Pond GR, et al. Predictors of infection inrheumatoid arthritis. Arthritis Rheum. 2002;46:2294-2300.
- Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum. 2007;56:2886-2895.
- Raaschou P, Simard JF, Asker Hagelberg C, et al. Rheumatoid arthritis, anti-tumour necrosis factor treatment, and risk of squamous cell and basal cell skin cancer: cohort study based on nationwide prospectively recorded data from Sweden. BMJ. 2016;352:i262.
- Ring HC, Riis Mikkelsen P, Miller IM, et al. The bacteriology of hidradenitis suppurativa: a systematic review. Exp Dermatol. 2015;24:727-731.
- Smith EJ, Allantaz F, Bennett L, et al. Clinical, molecular, and genetic characteristics of PAPA syndrome: a review. Curr Genomics. 2010;11:519-527.
- Jemec GB. Predicting response to anti-TNF-alpha treatment in hidradenitis suppurativa. Br J Dermatol. 2013;168:233.
- Sbidian E, Hotz C, Seneschal J, et al. Antitumour necrosis factor-α therapy for hidradenitis suppurativa: results from a national cohort study between 2000 and 2013 [published online December 22, 2015]. Br J Dermatol. 2016;174:667-670.
- van der Zee HH, de Ruiter L, van den Broecke DG, et al. Elevated levels of tumour necrosis factor (TNF)-α, interleukin (IL)-1β and IL-10 in hidradenitis suppurativa skin: a rationale for targeting TNF-α and IL-1β [published online May 17, 2011]. Br J Dermatol. 2011;164:1292-1298.
- van Rappard DC, Limpens J, Mekkes JR. The off-label treatment of severe hidradenitis suppurativa with TNF-alpha inhibitors: a systematic review. J Dermatolog Treat. 2013;24:392-404.
- Baerveldt EM, Kappen JH, Thio HB, et al. Successful long-term triple disease control by ustekinumab in a patient with Behcet’s disease, psoriasis and hidradenitis suppurativa. Ann Rheum Dis. 2013;72:626-627.
- Gulliver WP, Jemec GB, Baker KA. Experience with ustekinumab for the treatment of moderate to severe hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2012;26:911-914.
- Santos-Peréz MI, García-Rodicio S, Del Olmo-Revuelto MA, et al. Ustekinumab for hidradenitis suppurativa: a case report [published online December 3, 2013]. Actas Dermosifiliogr. 2014;105:720-722.
- Amano M, Grant A, Kerdel FA. A prospective open-label clinical trial of adalimumab for the treatment of hidradenitis suppurativa. Int J Dermatol. 2010;49:950-955.
- Blanco R, Gonzalez-Lopez MA, Gonzalez-Vela MC, et al. Disparate results in studies of adalimumab in the treatment of hidradenitis suppurativa: comment on the article by Amano et al. Int J Dermatol. 2013;52:380-381.
- Fardet L, Dupuy A, Kerob D, et al. Infliximab for severe hidradenitis suppurativa: transient clinical efficacy in 7 consecutive patients. J Am Acad Dermatol. 2007;56:624-628.
- Grant A, Gonzalez T, Montgomery MO, et al. Infliximab therapy for patients with moderate to severe hidradenitis suppurativa: a randomized, double-blind, placebo-controlled crossover trial. J Am Acad Dermatol. 2010;62:205-217.
- Kimball AB, Kerdel F, Adams D, et al. Adalimumab for the treatment of moderate to severe hidradenitis suppurativa: a parallel randomized trial. Ann Intern Med. 2012;157:846-855.
- Usmani N, Clayton TH, Everett S, et al. Variable response of hidradenitis suppurativa to infliximab in four patients. Clin Exp Dermatol. 2007;32:204-205.
- Leslie KS, Tripathi SV, Nguyen TV, et al. An open-label study of anakinra for the treatment of moderate to severe hidradenitis suppurativa. J Am Acad Dermatol. 2014;70:243-251.
- Menis D, Maronas-Jimenez L, Delgado-Marquez AM, et al. Two cases of severe hidradenitis suppurativa with failure of anakinra therapy [published online January 22, 2015]. Br J Dermatol. 2015;172:810-811.
- Tzanetakou V, Kanni T, Giatrakou S, et al. Safety and efficacy of anakinra in severe hidradenitis suppurativa: a randomized clinical trial. JAMA Dermatol. 2016;152:52-59.
- Zarchi K, Dufour DN, Jemec GB. Successful treatment of severe hidradenitis suppurativa with anakinra. JAMA Dermatol. 2013;149:1192-1194.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Blok JL, Li K, Brodmerkel C, et al. Ustekinumab in hidradenitis suppurativa: clinical results and a search for potential biomarkers in serum. Br J Dermatol. 2016;174:839-846.
- Hoffman LK, Ghias MH, Garg A, et al. Major gaps in understanding and treatment of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:86-92.
- Schlapbach C, Hanni T, Yawalkar N, et al. Expression of the IL-23/Th17 pathway in lesions of hidradenitis suppurativa. J Am Acad Dermatol. 2011;65:790-798.
- Torre KM, Payette MJ. Combination biologic therapy for the treatment of severe palmoplantar pustulosis. JAAD Case Rep. 2017;3:240-242.
- Babalola O, Lakdawala N, Strober BE. Combined biologic therapy for the treatment of psoriasis and psoriatic arthritis: a case report. JAAD Case Rep. 2015;1:3-4.
- Cuchacovich R, Garcia-Valladares I, Espinoza LR. Combination biologic treatment of refractory psoriasis and psoriatic arthritis. J Rheumatol. 2012;39:187-193.
- Genovese MC, Cohen S, Moreland L, et al. Combination therapy with etanercept and anakinra in the treatment of patients with rheumatoid arthritis who have been treated unsuccessfully with methotrexate. Arthritis Rheum. 2004;50:1412-1419.
- Baum J. Infection in rheumatoid arthritis. Arthritis Rheum. 1971;14:135-137.
- Doran MF, Crowson CS, Pond GR, et al. Frequency of infection in patients with rheumatoid arthritis compared with controls: a population-based study. Arthritis Rheum. 2002;46:2287-2293.
- Askling J, Fored CM, Baecklund E, et al. Haematopoietic malignancies in rheumatoid arthritis: lymphoma risk and characteristics after exposure to tumour necrosis factor antagonists. Ann Rheum Dis. 2005;64:1414-1420.
- Smitten AL, Simon TA, Hochberg MC, et al. A meta-analysis of the incidence of malignancy in adult patients with rheumatoid arthritis [published online April 23, 2008]. Arthritis Res Ther. 2008;10:R45.
- Doran MF, Crowson CS, Pond GR, et al. Predictors of infection inrheumatoid arthritis. Arthritis Rheum. 2002;46:2294-2300.
- Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum. 2007;56:2886-2895.
- Raaschou P, Simard JF, Asker Hagelberg C, et al. Rheumatoid arthritis, anti-tumour necrosis factor treatment, and risk of squamous cell and basal cell skin cancer: cohort study based on nationwide prospectively recorded data from Sweden. BMJ. 2016;352:i262.
- Ring HC, Riis Mikkelsen P, Miller IM, et al. The bacteriology of hidradenitis suppurativa: a systematic review. Exp Dermatol. 2015;24:727-731.
- Smith EJ, Allantaz F, Bennett L, et al. Clinical, molecular, and genetic characteristics of PAPA syndrome: a review. Curr Genomics. 2010;11:519-527.
MDedge Daily News: Are bigger-dose steroids better at stopping asthma exacerbations?
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Are more inhaled steroids better at blocking asthma exacerbations? A common skin disorder finds its way into more emergency departments, there are new guidelines for handling teen depression in primary care, and learn the five orthopedic tests to avoid in children.
Listen to the MDedge Daily News podcast for all the details on today’s top news.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Are more inhaled steroids better at blocking asthma exacerbations? A common skin disorder finds its way into more emergency departments, there are new guidelines for handling teen depression in primary care, and learn the five orthopedic tests to avoid in children.
Listen to the MDedge Daily News podcast for all the details on today’s top news.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Are more inhaled steroids better at blocking asthma exacerbations? A common skin disorder finds its way into more emergency departments, there are new guidelines for handling teen depression in primary care, and learn the five orthopedic tests to avoid in children.
Listen to the MDedge Daily News podcast for all the details on today’s top news.

A Nationwide Survey and Needs Assessment of Colonoscopy Quality Assurance Programs
Colorectal cancer (CRC) is an important concern for the VA, and colonoscopy is one primary screening, surveillance, and diagnostic modality used. The observed reductions in CRC incidence and mortality over the past decade largely have been attributed to the widespread use of CRC screening options.1,2 Colonoscopy quality is critical to CRC prevention in veterans. However, endoscopy skills to detect and remove colorectal polyps using colonoscopy vary in practice.3-5
Quality benchmarks, linked to patient outcomes, have been established by specialty societies and proposed by the Centers for Medicare and Medicaid Services as reportable quality metrics.6 Colonoscopy quality metrics have been shown to be associated with patient outcomes, such as the risk of developing CRC after colonoscopy. The adenoma detection rate (ADR), defined as the proportion of average-risk screening colonoscopies in which 1 or more adenomas are detected, has the strongest association to interval or “missed” CRC after screening colonoscopy and has been linked to a risk for fatal CRC despite colonoscopy.3
In a landmark study of 314,872 examinations performed by 136 gastroenterologists, the ADR ranged from 7.4% to 52.5%.3 Among patients with ADRs in the highest quintile compared with patients in the lowest, the adjusted hazard ratios (HRs) for any interval cancer was 0.52 (95% confidence interval [CI], 0.39-0.69) and for fatal interval cancers was 0.38 (95% CI, 0.22-0.65).3 Another pooled analysis from 8 surveillance studies that followed more than 800 participants with adenoma(s) after a baseline colonoscopy showed 52% of incident cancers as probable missed lesions, 19% as possibly related to incomplete resection of an earlier, noninvasive lesion, and only 24% as probable new lesions.7 These interval cancers highlight the current imperfections of colonoscopy and the focus on measurement and reporting of quality indicators for colonoscopy.8-12
According to VHA Directive 1015, in December 2014, colonoscopy quality should be monitored as part of an ongoing quality assurance program.13 A recent report from the VA Office of the Inspector General (OIG) highlighted colonoscopy-quality deficiencies.14 The OIG report strongly recommended that the “Acting Under Secretary for Health require standardized documentation of quality indicators based on professional society guidelines and published literature.”14However, no currently standardized and readily available VHA resource measures, reports, and ensures colonoscopy quality.
The authors hypothesized that colonoscopy quality assurance programs vary widely across VHA sites. The objective of this survey was to assess the measurement and reporting practices for colonoscopy quality and identify both strengths and areas for improvement to facilitate implementation of quality assurance programs across the VA health care system.
Methods
The authors performed an online survey of VA sites to assess current colonoscopy quality assurance practices. The institutional review boards (IRBs) at the University of Utah and VA Salt Lake City Health Care System and University of California, San Francisco and San Francisco VA Health Care System classified the study as a quality improvement project that did not qualify for human subjects’ research requiring IRB review.
The authors iteratively developed and refined the questionnaire with a survey methodologist and 2 clinical domain experts. The National Program Director for Gastroenterology, and the National Gastroenterology Field Advisory Committee reviewed the survey content and pretested the survey instrument prior to final data collection. The National Program Office for Gastroenterology provided an e-mail list of all known VA gastroenterology section chiefs. The authors administered the final survey via e-mail, using the Research Electronic Data Capture (REDCap; Vanderbilt University Medical Center) platform beginning January 9, 2017.15
A follow-up reminder e-mail was sent to nonresponders after 2 weeks. After this second invitation, sites were contacted by telephone to verify that the correct contact information had been captured. Subsequently, 50 contacts were updated if e-mails bounced back or the correct contact was obtained. Points of contact received a total of 3 reminder e-mails until the final closeout of the survey on March 28, 2017; 65 of 89 (73%) of the original contacts completed the survey vs 31 of 50 (62%) of the updated contacts.
Analysis
Descriptive statistics of the responses were calculated to determine the overall proportion of VA sites measuring colonoscopy quality metrics and identification of areas in need of quality improvement. The response rate for the survey was defined as the total number of responses obtained as a proportion of the total number of points of contact. This corresponds to the American Association of Public Opinion Research’s RR1, or minimum response rate, formula.16 All categoric responses are presented as proportions. Statistical analyses were performed using STATA SE12.0 (College Station, TX).
Results
Of the 139 points of contact invited, 96 completed the survey (response rate of 69.0%), representing 93 VA facilities (of 141 possible facilities) in 44 different states. Three sites had 2 responses. Sites used various and often a combination of methods to measure quality (Table 1). 
A majority of sites’ (63.5%) quality reports represented individual provider data, whereas fewer provided quality reports for physician groups (22.9%) or for the entire facility (40.6%). Provider quality information was de-identified in 43.8% of reporting sites’ quality reports and identifiable in 37.5% of reporting sites’ quality reports. A majority of sites (74.0%) reported that the local gastroenterology section chief or quality manager has access to the quality reports. Fewer sites reported providing data to individual endoscopists (44.8% for personal and peer data and 32.3% for personal data only). One site (1%) responded that quality reports were available for public access. Survey respondents also were asked to provide the estimated time (hours required per month) to collect the data for quality metrics. Of 75 respondents providing data for this question, 28 (29.2%) and 17 (17.7%), estimated between 1 to 5 and 6 to 10 hours per month, respectively. Ten sites estimated spending between 11 to 20 hours, and 7 sites estimated spending more than 20 hours per month collecting quality metrics. A total of 13 respondents (13.5%) stated uncertainty about the time burden.
As shown in the Figure, numerous quality metrics were collected across sites with more than 80% of sites collecting information on bowel preparation quality (88.5%), cecal intubation rate (87.5%), and complications (83.3%). A majority of sites also reported collecting data on appropriateness of surveillance intervals (62.5%), colonoscopy withdrawal times (62.5%), and ADRs (61.5%). Seven sites (7.3%) did not collect quality metrics.
Information also was collected on colonoscopy procedure documentation to inform future efforts at standardization. A small majority (53.1%) of sites reported using endoscopic software to generate colonoscopy procedure documentation. Within these sites, 6 different types of endoscopic note writing software were used to generate procedure notes (Table 2). 
Most sites (85.4%) were aware of VHA Directive 1015 recommendations for colonoscopy quality assurance programs. A significant majority (89.5%) of respondents also indicated interest in a centralized automatic reporting system to measure and report colonoscopy quality in some form, either with aggregate data, provider data, or both (Table 3).

Discussion
This survey on colonoscopy quality assurance programs is the first assessment of the VHA’s efforts to measure and report colonoscopy quality indicators. The findings indicated that the majority of VA sites are measuring and reporting at least some measures of colonoscopy quality. However, the programs are significantly variable in terms of methods used to collect quality metrics, specific quality measures obtained, and how quality is reported.
The authors’ work is novel in that this is the first report of the status of colonoscopy quality assurance programs in a large U.S. health care system. The VA health care system is the largest integrated health system in the U.S., serving more than 9 million veterans annually. This survey’s high response rate further strengthens the findings. Specifically, the survey found that VA sites are making a strong concerted effort to measure and report colonoscopy quality. However, there is significant variability in documentation, measurement, and reporting practices. Moreover, the majority of VA sites do not have formal performance improvement plans in place for endoscopists who do not meet thresholds for colonoscopy quality.
Screening colonoscopy for CRC offers known mortality benefits to patients.1,17-19 Significant prior work has described and validated the importance of colonoscopy quality metrics, including bowel preparation quality, cecal intubation rate, and ADR and their association with interval colorectal cancer and death.20-23 Gastroenterology professional societies, including the American College of Gastroenterology and the American Society for Gastrointestinal Endoscopy, have recommended and endorsed measurement and reporting of colonoscopy metrics.24 There is general agreement among endoscopists that colonoscopy quality is an important aspect of performing the procedure.
The lack of formal performance improvement programs is a key finding of this survey. Recent studies have shown that improvements in quality metrics, such as the ADR, by individual endoscopists result in reductions in interval colorectal cancer and death.25 Kahi and colleagues previously showed that providing a quarterly report card improves colonoscopy quality.26 Keswani and colleagues studied a combination of a report card and implementation of standards of practice with resultant improvement in colonoscopy quality.27 Most recently, in a large prospective cohort study of individuals who underwent a screening colonoscopy, 294 of the screening endoscopists received annual feedback and quality benchmark indicators to improve colonoscopy performance.25 The majority of the endoscopists (74.5%) increased their annual ADR category over the study period. Moreover, patients examined by endoscopists who reached or maintained the highest ADR quintile (> 24.6%) had significantly lower risk of interval CRC and death. The lack of formal performance improvement programs across the VHA is concerning but reveals a significant opportunity to improve veteran health outcomes on a large scale.
This study’s findings also highlight the intense resources necessary to measure and report colonoscopy quality. The ability to measure and report quality metrics requires having adequate documentation and data to obtain quality metrics. Administrative databases from electronic health records offer some potential for routine monitoring of quality metrics.28 However, most administrative databases, including the VA Corporate Data Warehouse (CDW), contain administrative billing codes (ICD and CPT) linked to limited patient data, including demographics and structured medical record data. The actual data required for quality reporting of important metrics (bowel preparation quality, cecal intubation rates, and ADRs) are usually found in clinical text notes or endoscopic note documentation and not available as structured data. Due to this issue, the majority of VA sites (79.2%) are using manual chart review to collect quality metric data, resulting in widely variable estimates on time burden. A minority of sites in this study (39.6%) reported using automated endoscopic software reporting capability that can help with the time burden. However, even in the VA, an integrated health system, a wide variety of software brands, documentation practices, and photo documentation was found.
Future endoscopy budget and purchase decisions for the individual VA sites should take into account how new technology and software can more easily facilitate accurate quality reporting. A specific policy recommendation would be for the VA to consider a uniform endoscopic note writer for procedure notes. Pathology data, which is necessary for the calculation of ADR, also should be available as structured data in the CDW to more easily measure colonoscopy quality. Continuous measurement and reporting of quality also requires ongoing information technology infrastructure and quality control of the measurement process.
Limitations
This survey was a cross-section of VA sites’ points of contact regarding colonoscopy quality assurance programs, so the results are descriptive in nature. However, the instrument was carefully developed, using both subject matter and survey method expertise. The questionnaire also was refined through pretesting prior to data collection. The initial contact list was found to have errors, and the list had to be updated after launching the survey. Updated information for most of the contacts was available.
Another limitation was the inability to survey nongastroenterologist-run endoscopy centers, because many centers use surgeons or other nongastroenterology providers. The authors speculate that quality monitoring may be less likely to be present at these facilities as they may not be aware of the gastroenterology professional society recommendations. The authors did not require or insist that all questions be answered, so some data were missing from sites. However, 93.7% of respondents completed the entire survey.
Conclusion
The authors have described the status of colonoscopy quality assurance programs across the VA health care system. Many sites are making robust efforts to measure and report quality especially of process measures. However, there are significant time and manual workforce efforts required, and this work is likely associated with the variability in programs. Importantly, ADR, which is the quality metric that has been most strongly associated with risk of colon cancer mortality, is not being measured by 38% of sites.
These results reinforce a critical need for a centralized, automated quality reporting infrastructure to standardize colonoscopy quality reporting, reduce workload, and ensure veterans receive high-quality colonoscopy.
Acknowledgments
The authors acknowledge the support and feedback of the National Gastroenterology Program Field Advisory Committee for survey development and testing. The authors coordinated the survey through the Salt Lake City Specialty Care Center of Innovation in partnership with the National Gastroenterology Program Office and the Quality Enhancement Research Initiative: Quality Enhancement Research Initiative, Measurement Science Program, QUE15-283. The work also was partially supported by the National Center for Advancing Translational Sciences of the National Institutes of Health Award UL1TR001067 and Merit Review Award 1 I01 HX001574-01A1 from the United States Department of Veterans Affairs Health Services Research & Development Service of the VA Office of Research and Development.
1. Brenner H, Stock C, Hoffmeister M. Effect of screening sigmoidoscopy and screening colonoscopy on colorectal cancer incidence and mortality: systematic review and meta-analysis of randomised controlled trials and observational studies. BMJ. 2014;348:g2467.
2. Meester RGS, Doubeni CA, Lansdorp-Vogelaar I, et al. Colorectal cancer deaths attributable to nonuse of screening in the United States. Ann Epidemiol. 2015;25(3):208-213.e1.
3. Corley DA, Jensen CD, Marks AR, et al. Adenoma detection rate and risk of colorectal cancer and death. N Engl J Med. 2014;370(26):1298-1306.
4. Meester RGS, Doubeni CA, Lansdorp-Vogelaar I, et al. Variation in adenoma detection rate and the lifetime benefits and cost of colorectal cancer screening: a microsimulation model. JAMA. 2015;313(23):2349-2358.
5. Boroff ES, Gurudu SR, Hentz JG, Leighton JA, Ramirez FC. Polyp and adenoma detection rates in the proximal and distal colon. Am J Gastroenterol. 2013;108(6):993-999.
6. Center for Medicare and Medicaid Services. Quality measures. https://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/Qual ityMeasures/index.html. Updated December 19, 2017. Accessed January 17, 2018.
7. Robertson DJ, Lieberman DA, Winawer SJ, et al. Colorectal cancers soon after colonoscopy: a pooled multicohort analysis. Gut. 2014;63(6):949-956.
8. Fayad NF, Kahi CJ. Colonoscopy quality assessment. Gastrointest Endosc Clin N Am. 2015;25(2):373-386.
9. de Jonge V, Sint Nicolaas J, Cahen DL, et al; SCoPE Consortium. Quality evaluation of colonoscopy reporting and colonoscopy performance in daily clinical practice. Gastrointest Endosc. 2012;75(1):98-106.
10. Johnson DA. Quality benchmarking for colonoscopy: how do we pick products from the shelf? Gastrointest Endosc. 2012;75(1):107-109.
11. Anderson JC, Butterly LF. Colonoscopy: quality indicators. Clin Transl Gastroenterol. 2015;6(2):e77.
12. Kaminski MF, Regula J, Kraszewska E, et al. Quality indicators for colonoscopy and the risk of interval cancer. N Engl J Med. 2010;362(19):1795-1803.
13. U.S. Department of Veterans Affairs, Veterans Health Administration. Colorectal cancer screening. VHA Directive 1015. Published December 30, 2014.
14. U.S. Department of Veterans Affairs, VA Office of the Inspector General, Office of Healthcare Inspections. Healthcare inspection: alleged access delays and surgery service concerns, VA Roseburg Healthcare System, Roseburg, Oregon. Report No.15-00506-535. https://www.va.gov/oig /pubs/VAOIG-15-00506-535.pdf. Published July 11, 2017. Accessed January 9, 2018.
15. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377-381.
16. The American Association for Public Opinion Research. Standard Definitions: Final Dispositions of Case Codes and Outcome Rates for Surveys. 9th edition. http://www.aapor.org/AAPOR_Main/media/publications/Standard-Definitions20169theditionfinal.pdf. Revised 2016. Accessed January 9, 2018.
17. Kahi CJ, Imperiale TF, Juliar BE, Rex DK. Effect of screening colonoscopy on colorectal cancer incidence and mortality. Clin Gastroenterol Hepatol. 2009;7(7):770-775.
18. Manser CN, Bachmann LM, Brunner J, Hunold F, Bauerfeind P, Marbet UA. Colonoscopy screening markedly reduces the occurrence of colon carcinomas and carcinoma-related death: a closed cohort study. Gastrointest Endosc. 2012;76(1):110-117.
19. Nishihara R, Wu K, Lochhead P, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med. 2013;369(12):1095-1105.
20. Harewood GC, Sharma VK, de Garmo P. Impact of colonoscopy preparation quality on detection of suspected colonic neoplasia. Gastrointest Endosc. 2003;58(1):76-79.
21. Hillyer GC, Lebwohl B, Rosenberg RM, et al. Assessing bowel preparation quality using the mean number of adenomas per colonoscopy. Therap Adv Gastroenterol. 2014;7(6):238-246.
22. Clark BT, Rustagi T, Laine L. What level of bowel prep quality requires early repeat colonoscopy: systematic review and meta-analysis of the impact of preparation quality on adenoma detection rate. Am J Gastroenterol. 2014;109(11):1714-1723; quiz 1724.
23. Johnson DA, Barkun AN, Cohen LB, et al; US Multi-Society Task Force on Colorectal Cancer. Optimizing adequacy of bowel cleansing for colonoscopy: recommendations from the US multi-society task force on colorectal cancer. Gastroenterology. 2014;147(4):903-924.
24. Rex DK, Petrini JL, Baron TH, et al; ASGE/ACG Taskforce on Quality in Endoscopy. Quality indicators for colonoscopy. Am J Gastroenterol. 2006;101(4):873-885.
25. Kaminski MF, Wieszczy P, Rupinski M, et al. Increased rate of adenoma detection associates with reduced risk of colorectal cancer and death. Gastroenterology. 2017;153(1):98-105.
26. Kahi CJ, Ballard D, Shah AS, Mears R, Johnson CS. Impact of a quarterly report card on colonoscopy quality measures. Gastrointest. Endosc. 2013;77(6):925-931.
27. Keswani RN, Yadlapati R, Gleason KM, et al. Physician report cards and implementing standards of practice are both significantly associated with improved screening colonoscopy quality. Am J Gastroenterol. 2015;110(8):1134-1139.
28. Logan JR, Lieberman DA. The use of databases and registries to enhance colonoscopy quality. Gastrointest Endosc Clin N Am. 2010;20(4):717-734.
Colorectal cancer (CRC) is an important concern for the VA, and colonoscopy is one primary screening, surveillance, and diagnostic modality used. The observed reductions in CRC incidence and mortality over the past decade largely have been attributed to the widespread use of CRC screening options.1,2 Colonoscopy quality is critical to CRC prevention in veterans. However, endoscopy skills to detect and remove colorectal polyps using colonoscopy vary in practice.3-5
Quality benchmarks, linked to patient outcomes, have been established by specialty societies and proposed by the Centers for Medicare and Medicaid Services as reportable quality metrics.6 Colonoscopy quality metrics have been shown to be associated with patient outcomes, such as the risk of developing CRC after colonoscopy. The adenoma detection rate (ADR), defined as the proportion of average-risk screening colonoscopies in which 1 or more adenomas are detected, has the strongest association to interval or “missed” CRC after screening colonoscopy and has been linked to a risk for fatal CRC despite colonoscopy.3
In a landmark study of 314,872 examinations performed by 136 gastroenterologists, the ADR ranged from 7.4% to 52.5%.3 Among patients with ADRs in the highest quintile compared with patients in the lowest, the adjusted hazard ratios (HRs) for any interval cancer was 0.52 (95% confidence interval [CI], 0.39-0.69) and for fatal interval cancers was 0.38 (95% CI, 0.22-0.65).3 Another pooled analysis from 8 surveillance studies that followed more than 800 participants with adenoma(s) after a baseline colonoscopy showed 52% of incident cancers as probable missed lesions, 19% as possibly related to incomplete resection of an earlier, noninvasive lesion, and only 24% as probable new lesions.7 These interval cancers highlight the current imperfections of colonoscopy and the focus on measurement and reporting of quality indicators for colonoscopy.8-12
According to VHA Directive 1015, in December 2014, colonoscopy quality should be monitored as part of an ongoing quality assurance program.13 A recent report from the VA Office of the Inspector General (OIG) highlighted colonoscopy-quality deficiencies.14 The OIG report strongly recommended that the “Acting Under Secretary for Health require standardized documentation of quality indicators based on professional society guidelines and published literature.”14However, no currently standardized and readily available VHA resource measures, reports, and ensures colonoscopy quality.
The authors hypothesized that colonoscopy quality assurance programs vary widely across VHA sites. The objective of this survey was to assess the measurement and reporting practices for colonoscopy quality and identify both strengths and areas for improvement to facilitate implementation of quality assurance programs across the VA health care system.
Methods
The authors performed an online survey of VA sites to assess current colonoscopy quality assurance practices. The institutional review boards (IRBs) at the University of Utah and VA Salt Lake City Health Care System and University of California, San Francisco and San Francisco VA Health Care System classified the study as a quality improvement project that did not qualify for human subjects’ research requiring IRB review.
The authors iteratively developed and refined the questionnaire with a survey methodologist and 2 clinical domain experts. The National Program Director for Gastroenterology, and the National Gastroenterology Field Advisory Committee reviewed the survey content and pretested the survey instrument prior to final data collection. The National Program Office for Gastroenterology provided an e-mail list of all known VA gastroenterology section chiefs. The authors administered the final survey via e-mail, using the Research Electronic Data Capture (REDCap; Vanderbilt University Medical Center) platform beginning January 9, 2017.15
A follow-up reminder e-mail was sent to nonresponders after 2 weeks. After this second invitation, sites were contacted by telephone to verify that the correct contact information had been captured. Subsequently, 50 contacts were updated if e-mails bounced back or the correct contact was obtained. Points of contact received a total of 3 reminder e-mails until the final closeout of the survey on March 28, 2017; 65 of 89 (73%) of the original contacts completed the survey vs 31 of 50 (62%) of the updated contacts.
Analysis
Descriptive statistics of the responses were calculated to determine the overall proportion of VA sites measuring colonoscopy quality metrics and identification of areas in need of quality improvement. The response rate for the survey was defined as the total number of responses obtained as a proportion of the total number of points of contact. This corresponds to the American Association of Public Opinion Research’s RR1, or minimum response rate, formula.16 All categoric responses are presented as proportions. Statistical analyses were performed using STATA SE12.0 (College Station, TX).
Results
Of the 139 points of contact invited, 96 completed the survey (response rate of 69.0%), representing 93 VA facilities (of 141 possible facilities) in 44 different states. Three sites had 2 responses. Sites used various and often a combination of methods to measure quality (Table 1).
A majority of sites’ (63.5%) quality reports represented individual provider data, whereas fewer provided quality reports for physician groups (22.9%) or for the entire facility (40.6%). Provider quality information was de-identified in 43.8% of reporting sites’ quality reports and identifiable in 37.5% of reporting sites’ quality reports. A majority of sites (74.0%) reported that the local gastroenterology section chief or quality manager has access to the quality reports. Fewer sites reported providing data to individual endoscopists (44.8% for personal and peer data and 32.3% for personal data only). One site (1%) responded that quality reports were available for public access. Survey respondents also were asked to provide the estimated time (hours required per month) to collect the data for quality metrics. Of 75 respondents providing data for this question, 28 (29.2%) and 17 (17.7%), estimated between 1 to 5 and 6 to 10 hours per month, respectively. Ten sites estimated spending between 11 to 20 hours, and 7 sites estimated spending more than 20 hours per month collecting quality metrics. A total of 13 respondents (13.5%) stated uncertainty about the time burden.
As shown in the Figure, numerous quality metrics were collected across sites with more than 80% of sites collecting information on bowel preparation quality (88.5%), cecal intubation rate (87.5%), and complications (83.3%). A majority of sites also reported collecting data on appropriateness of surveillance intervals (62.5%), colonoscopy withdrawal times (62.5%), and ADRs (61.5%). Seven sites (7.3%) did not collect quality metrics.
Information also was collected on colonoscopy procedure documentation to inform future efforts at standardization. A small majority (53.1%) of sites reported using endoscopic software to generate colonoscopy procedure documentation. Within these sites, 6 different types of endoscopic note writing software were used to generate procedure notes (Table 2).
Most sites (85.4%) were aware of VHA Directive 1015 recommendations for colonoscopy quality assurance programs. A significant majority (89.5%) of respondents also indicated interest in a centralized automatic reporting system to measure and report colonoscopy quality in some form, either with aggregate data, provider data, or both (Table 3).
Discussion
This survey on colonoscopy quality assurance programs is the first assessment of the VHA’s efforts to measure and report colonoscopy quality indicators. The findings indicated that the majority of VA sites are measuring and reporting at least some measures of colonoscopy quality. However, the programs are significantly variable in terms of methods used to collect quality metrics, specific quality measures obtained, and how quality is reported.
The authors’ work is novel in that this is the first report of the status of colonoscopy quality assurance programs in a large U.S. health care system. The VA health care system is the largest integrated health system in the U.S., serving more than 9 million veterans annually. This survey’s high response rate further strengthens the findings. Specifically, the survey found that VA sites are making a strong concerted effort to measure and report colonoscopy quality. However, there is significant variability in documentation, measurement, and reporting practices. Moreover, the majority of VA sites do not have formal performance improvement plans in place for endoscopists who do not meet thresholds for colonoscopy quality.
Screening colonoscopy for CRC offers known mortality benefits to patients.1,17-19 Significant prior work has described and validated the importance of colonoscopy quality metrics, including bowel preparation quality, cecal intubation rate, and ADR and their association with interval colorectal cancer and death.20-23 Gastroenterology professional societies, including the American College of Gastroenterology and the American Society for Gastrointestinal Endoscopy, have recommended and endorsed measurement and reporting of colonoscopy metrics.24 There is general agreement among endoscopists that colonoscopy quality is an important aspect of performing the procedure.
The lack of formal performance improvement programs is a key finding of this survey. Recent studies have shown that improvements in quality metrics, such as the ADR, by individual endoscopists result in reductions in interval colorectal cancer and death.25 Kahi and colleagues previously showed that providing a quarterly report card improves colonoscopy quality.26 Keswani and colleagues studied a combination of a report card and implementation of standards of practice with resultant improvement in colonoscopy quality.27 Most recently, in a large prospective cohort study of individuals who underwent a screening colonoscopy, 294 of the screening endoscopists received annual feedback and quality benchmark indicators to improve colonoscopy performance.25 The majority of the endoscopists (74.5%) increased their annual ADR category over the study period. Moreover, patients examined by endoscopists who reached or maintained the highest ADR quintile (> 24.6%) had significantly lower risk of interval CRC and death. The lack of formal performance improvement programs across the VHA is concerning but reveals a significant opportunity to improve veteran health outcomes on a large scale.
This study’s findings also highlight the intense resources necessary to measure and report colonoscopy quality. The ability to measure and report quality metrics requires having adequate documentation and data to obtain quality metrics. Administrative databases from electronic health records offer some potential for routine monitoring of quality metrics.28 However, most administrative databases, including the VA Corporate Data Warehouse (CDW), contain administrative billing codes (ICD and CPT) linked to limited patient data, including demographics and structured medical record data. The actual data required for quality reporting of important metrics (bowel preparation quality, cecal intubation rates, and ADRs) are usually found in clinical text notes or endoscopic note documentation and not available as structured data. Due to this issue, the majority of VA sites (79.2%) are using manual chart review to collect quality metric data, resulting in widely variable estimates on time burden. A minority of sites in this study (39.6%) reported using automated endoscopic software reporting capability that can help with the time burden. However, even in the VA, an integrated health system, a wide variety of software brands, documentation practices, and photo documentation was found.
Future endoscopy budget and purchase decisions for the individual VA sites should take into account how new technology and software can more easily facilitate accurate quality reporting. A specific policy recommendation would be for the VA to consider a uniform endoscopic note writer for procedure notes. Pathology data, which is necessary for the calculation of ADR, also should be available as structured data in the CDW to more easily measure colonoscopy quality. Continuous measurement and reporting of quality also requires ongoing information technology infrastructure and quality control of the measurement process.
Limitations
This survey was a cross-section of VA sites’ points of contact regarding colonoscopy quality assurance programs, so the results are descriptive in nature. However, the instrument was carefully developed, using both subject matter and survey method expertise. The questionnaire also was refined through pretesting prior to data collection. The initial contact list was found to have errors, and the list had to be updated after launching the survey. Updated information for most of the contacts was available.
Another limitation was the inability to survey nongastroenterologist-run endoscopy centers, because many centers use surgeons or other nongastroenterology providers. The authors speculate that quality monitoring may be less likely to be present at these facilities as they may not be aware of the gastroenterology professional society recommendations. The authors did not require or insist that all questions be answered, so some data were missing from sites. However, 93.7% of respondents completed the entire survey.
Conclusion
The authors have described the status of colonoscopy quality assurance programs across the VA health care system. Many sites are making robust efforts to measure and report quality especially of process measures. However, there are significant time and manual workforce efforts required, and this work is likely associated with the variability in programs. Importantly, ADR, which is the quality metric that has been most strongly associated with risk of colon cancer mortality, is not being measured by 38% of sites.
These results reinforce a critical need for a centralized, automated quality reporting infrastructure to standardize colonoscopy quality reporting, reduce workload, and ensure veterans receive high-quality colonoscopy.
Acknowledgments
The authors acknowledge the support and feedback of the National Gastroenterology Program Field Advisory Committee for survey development and testing. The authors coordinated the survey through the Salt Lake City Specialty Care Center of Innovation in partnership with the National Gastroenterology Program Office and the Quality Enhancement Research Initiative: Quality Enhancement Research Initiative, Measurement Science Program, QUE15-283. The work also was partially supported by the National Center for Advancing Translational Sciences of the National Institutes of Health Award UL1TR001067 and Merit Review Award 1 I01 HX001574-01A1 from the United States Department of Veterans Affairs Health Services Research & Development Service of the VA Office of Research and Development.
Colorectal cancer (CRC) is an important concern for the VA, and colonoscopy is one primary screening, surveillance, and diagnostic modality used. The observed reductions in CRC incidence and mortality over the past decade largely have been attributed to the widespread use of CRC screening options.1,2 Colonoscopy quality is critical to CRC prevention in veterans. However, endoscopy skills to detect and remove colorectal polyps using colonoscopy vary in practice.3-5
Quality benchmarks, linked to patient outcomes, have been established by specialty societies and proposed by the Centers for Medicare and Medicaid Services as reportable quality metrics.6 Colonoscopy quality metrics have been shown to be associated with patient outcomes, such as the risk of developing CRC after colonoscopy. The adenoma detection rate (ADR), defined as the proportion of average-risk screening colonoscopies in which 1 or more adenomas are detected, has the strongest association to interval or “missed” CRC after screening colonoscopy and has been linked to a risk for fatal CRC despite colonoscopy.3
In a landmark study of 314,872 examinations performed by 136 gastroenterologists, the ADR ranged from 7.4% to 52.5%.3 Among patients with ADRs in the highest quintile compared with patients in the lowest, the adjusted hazard ratios (HRs) for any interval cancer was 0.52 (95% confidence interval [CI], 0.39-0.69) and for fatal interval cancers was 0.38 (95% CI, 0.22-0.65).3 Another pooled analysis from 8 surveillance studies that followed more than 800 participants with adenoma(s) after a baseline colonoscopy showed 52% of incident cancers as probable missed lesions, 19% as possibly related to incomplete resection of an earlier, noninvasive lesion, and only 24% as probable new lesions.7 These interval cancers highlight the current imperfections of colonoscopy and the focus on measurement and reporting of quality indicators for colonoscopy.8-12
According to VHA Directive 1015, in December 2014, colonoscopy quality should be monitored as part of an ongoing quality assurance program.13 A recent report from the VA Office of the Inspector General (OIG) highlighted colonoscopy-quality deficiencies.14 The OIG report strongly recommended that the “Acting Under Secretary for Health require standardized documentation of quality indicators based on professional society guidelines and published literature.”14However, no currently standardized and readily available VHA resource measures, reports, and ensures colonoscopy quality.
The authors hypothesized that colonoscopy quality assurance programs vary widely across VHA sites. The objective of this survey was to assess the measurement and reporting practices for colonoscopy quality and identify both strengths and areas for improvement to facilitate implementation of quality assurance programs across the VA health care system.
Methods
The authors performed an online survey of VA sites to assess current colonoscopy quality assurance practices. The institutional review boards (IRBs) at the University of Utah and VA Salt Lake City Health Care System and University of California, San Francisco and San Francisco VA Health Care System classified the study as a quality improvement project that did not qualify for human subjects’ research requiring IRB review.
The authors iteratively developed and refined the questionnaire with a survey methodologist and 2 clinical domain experts. The National Program Director for Gastroenterology, and the National Gastroenterology Field Advisory Committee reviewed the survey content and pretested the survey instrument prior to final data collection. The National Program Office for Gastroenterology provided an e-mail list of all known VA gastroenterology section chiefs. The authors administered the final survey via e-mail, using the Research Electronic Data Capture (REDCap; Vanderbilt University Medical Center) platform beginning January 9, 2017.15
A follow-up reminder e-mail was sent to nonresponders after 2 weeks. After this second invitation, sites were contacted by telephone to verify that the correct contact information had been captured. Subsequently, 50 contacts were updated if e-mails bounced back or the correct contact was obtained. Points of contact received a total of 3 reminder e-mails until the final closeout of the survey on March 28, 2017; 65 of 89 (73%) of the original contacts completed the survey vs 31 of 50 (62%) of the updated contacts.
Analysis
Descriptive statistics of the responses were calculated to determine the overall proportion of VA sites measuring colonoscopy quality metrics and identification of areas in need of quality improvement. The response rate for the survey was defined as the total number of responses obtained as a proportion of the total number of points of contact. This corresponds to the American Association of Public Opinion Research’s RR1, or minimum response rate, formula.16 All categoric responses are presented as proportions. Statistical analyses were performed using STATA SE12.0 (College Station, TX).
Results
Of the 139 points of contact invited, 96 completed the survey (response rate of 69.0%), representing 93 VA facilities (of 141 possible facilities) in 44 different states. Three sites had 2 responses. Sites used various and often a combination of methods to measure quality (Table 1).
A majority of sites’ (63.5%) quality reports represented individual provider data, whereas fewer provided quality reports for physician groups (22.9%) or for the entire facility (40.6%). Provider quality information was de-identified in 43.8% of reporting sites’ quality reports and identifiable in 37.5% of reporting sites’ quality reports. A majority of sites (74.0%) reported that the local gastroenterology section chief or quality manager has access to the quality reports. Fewer sites reported providing data to individual endoscopists (44.8% for personal and peer data and 32.3% for personal data only). One site (1%) responded that quality reports were available for public access. Survey respondents also were asked to provide the estimated time (hours required per month) to collect the data for quality metrics. Of 75 respondents providing data for this question, 28 (29.2%) and 17 (17.7%), estimated between 1 to 5 and 6 to 10 hours per month, respectively. Ten sites estimated spending between 11 to 20 hours, and 7 sites estimated spending more than 20 hours per month collecting quality metrics. A total of 13 respondents (13.5%) stated uncertainty about the time burden.
As shown in the Figure, numerous quality metrics were collected across sites with more than 80% of sites collecting information on bowel preparation quality (88.5%), cecal intubation rate (87.5%), and complications (83.3%). A majority of sites also reported collecting data on appropriateness of surveillance intervals (62.5%), colonoscopy withdrawal times (62.5%), and ADRs (61.5%). Seven sites (7.3%) did not collect quality metrics.
Information also was collected on colonoscopy procedure documentation to inform future efforts at standardization. A small majority (53.1%) of sites reported using endoscopic software to generate colonoscopy procedure documentation. Within these sites, 6 different types of endoscopic note writing software were used to generate procedure notes (Table 2).
Most sites (85.4%) were aware of VHA Directive 1015 recommendations for colonoscopy quality assurance programs. A significant majority (89.5%) of respondents also indicated interest in a centralized automatic reporting system to measure and report colonoscopy quality in some form, either with aggregate data, provider data, or both (Table 3).
Discussion
This survey on colonoscopy quality assurance programs is the first assessment of the VHA’s efforts to measure and report colonoscopy quality indicators. The findings indicated that the majority of VA sites are measuring and reporting at least some measures of colonoscopy quality. However, the programs are significantly variable in terms of methods used to collect quality metrics, specific quality measures obtained, and how quality is reported.
The authors’ work is novel in that this is the first report of the status of colonoscopy quality assurance programs in a large U.S. health care system. The VA health care system is the largest integrated health system in the U.S., serving more than 9 million veterans annually. This survey’s high response rate further strengthens the findings. Specifically, the survey found that VA sites are making a strong concerted effort to measure and report colonoscopy quality. However, there is significant variability in documentation, measurement, and reporting practices. Moreover, the majority of VA sites do not have formal performance improvement plans in place for endoscopists who do not meet thresholds for colonoscopy quality.
Screening colonoscopy for CRC offers known mortality benefits to patients.1,17-19 Significant prior work has described and validated the importance of colonoscopy quality metrics, including bowel preparation quality, cecal intubation rate, and ADR and their association with interval colorectal cancer and death.20-23 Gastroenterology professional societies, including the American College of Gastroenterology and the American Society for Gastrointestinal Endoscopy, have recommended and endorsed measurement and reporting of colonoscopy metrics.24 There is general agreement among endoscopists that colonoscopy quality is an important aspect of performing the procedure.
The lack of formal performance improvement programs is a key finding of this survey. Recent studies have shown that improvements in quality metrics, such as the ADR, by individual endoscopists result in reductions in interval colorectal cancer and death.25 Kahi and colleagues previously showed that providing a quarterly report card improves colonoscopy quality.26 Keswani and colleagues studied a combination of a report card and implementation of standards of practice with resultant improvement in colonoscopy quality.27 Most recently, in a large prospective cohort study of individuals who underwent a screening colonoscopy, 294 of the screening endoscopists received annual feedback and quality benchmark indicators to improve colonoscopy performance.25 The majority of the endoscopists (74.5%) increased their annual ADR category over the study period. Moreover, patients examined by endoscopists who reached or maintained the highest ADR quintile (> 24.6%) had significantly lower risk of interval CRC and death. The lack of formal performance improvement programs across the VHA is concerning but reveals a significant opportunity to improve veteran health outcomes on a large scale.
This study’s findings also highlight the intense resources necessary to measure and report colonoscopy quality. The ability to measure and report quality metrics requires having adequate documentation and data to obtain quality metrics. Administrative databases from electronic health records offer some potential for routine monitoring of quality metrics.28 However, most administrative databases, including the VA Corporate Data Warehouse (CDW), contain administrative billing codes (ICD and CPT) linked to limited patient data, including demographics and structured medical record data. The actual data required for quality reporting of important metrics (bowel preparation quality, cecal intubation rates, and ADRs) are usually found in clinical text notes or endoscopic note documentation and not available as structured data. Due to this issue, the majority of VA sites (79.2%) are using manual chart review to collect quality metric data, resulting in widely variable estimates on time burden. A minority of sites in this study (39.6%) reported using automated endoscopic software reporting capability that can help with the time burden. However, even in the VA, an integrated health system, a wide variety of software brands, documentation practices, and photo documentation was found.
Future endoscopy budget and purchase decisions for the individual VA sites should take into account how new technology and software can more easily facilitate accurate quality reporting. A specific policy recommendation would be for the VA to consider a uniform endoscopic note writer for procedure notes. Pathology data, which is necessary for the calculation of ADR, also should be available as structured data in the CDW to more easily measure colonoscopy quality. Continuous measurement and reporting of quality also requires ongoing information technology infrastructure and quality control of the measurement process.
Limitations
This survey was a cross-section of VA sites’ points of contact regarding colonoscopy quality assurance programs, so the results are descriptive in nature. However, the instrument was carefully developed, using both subject matter and survey method expertise. The questionnaire also was refined through pretesting prior to data collection. The initial contact list was found to have errors, and the list had to be updated after launching the survey. Updated information for most of the contacts was available.
Another limitation was the inability to survey nongastroenterologist-run endoscopy centers, because many centers use surgeons or other nongastroenterology providers. The authors speculate that quality monitoring may be less likely to be present at these facilities as they may not be aware of the gastroenterology professional society recommendations. The authors did not require or insist that all questions be answered, so some data were missing from sites. However, 93.7% of respondents completed the entire survey.
Conclusion
The authors have described the status of colonoscopy quality assurance programs across the VA health care system. Many sites are making robust efforts to measure and report quality especially of process measures. However, there are significant time and manual workforce efforts required, and this work is likely associated with the variability in programs. Importantly, ADR, which is the quality metric that has been most strongly associated with risk of colon cancer mortality, is not being measured by 38% of sites.
These results reinforce a critical need for a centralized, automated quality reporting infrastructure to standardize colonoscopy quality reporting, reduce workload, and ensure veterans receive high-quality colonoscopy.
Acknowledgments
The authors acknowledge the support and feedback of the National Gastroenterology Program Field Advisory Committee for survey development and testing. The authors coordinated the survey through the Salt Lake City Specialty Care Center of Innovation in partnership with the National Gastroenterology Program Office and the Quality Enhancement Research Initiative: Quality Enhancement Research Initiative, Measurement Science Program, QUE15-283. The work also was partially supported by the National Center for Advancing Translational Sciences of the National Institutes of Health Award UL1TR001067 and Merit Review Award 1 I01 HX001574-01A1 from the United States Department of Veterans Affairs Health Services Research & Development Service of the VA Office of Research and Development.
1. Brenner H, Stock C, Hoffmeister M. Effect of screening sigmoidoscopy and screening colonoscopy on colorectal cancer incidence and mortality: systematic review and meta-analysis of randomised controlled trials and observational studies. BMJ. 2014;348:g2467.
2. Meester RGS, Doubeni CA, Lansdorp-Vogelaar I, et al. Colorectal cancer deaths attributable to nonuse of screening in the United States. Ann Epidemiol. 2015;25(3):208-213.e1.
3. Corley DA, Jensen CD, Marks AR, et al. Adenoma detection rate and risk of colorectal cancer and death. N Engl J Med. 2014;370(26):1298-1306.
4. Meester RGS, Doubeni CA, Lansdorp-Vogelaar I, et al. Variation in adenoma detection rate and the lifetime benefits and cost of colorectal cancer screening: a microsimulation model. JAMA. 2015;313(23):2349-2358.
5. Boroff ES, Gurudu SR, Hentz JG, Leighton JA, Ramirez FC. Polyp and adenoma detection rates in the proximal and distal colon. Am J Gastroenterol. 2013;108(6):993-999.
6. Center for Medicare and Medicaid Services. Quality measures. https://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/Qual ityMeasures/index.html. Updated December 19, 2017. Accessed January 17, 2018.
7. Robertson DJ, Lieberman DA, Winawer SJ, et al. Colorectal cancers soon after colonoscopy: a pooled multicohort analysis. Gut. 2014;63(6):949-956.
8. Fayad NF, Kahi CJ. Colonoscopy quality assessment. Gastrointest Endosc Clin N Am. 2015;25(2):373-386.
9. de Jonge V, Sint Nicolaas J, Cahen DL, et al; SCoPE Consortium. Quality evaluation of colonoscopy reporting and colonoscopy performance in daily clinical practice. Gastrointest Endosc. 2012;75(1):98-106.
10. Johnson DA. Quality benchmarking for colonoscopy: how do we pick products from the shelf? Gastrointest Endosc. 2012;75(1):107-109.
11. Anderson JC, Butterly LF. Colonoscopy: quality indicators. Clin Transl Gastroenterol. 2015;6(2):e77.
12. Kaminski MF, Regula J, Kraszewska E, et al. Quality indicators for colonoscopy and the risk of interval cancer. N Engl J Med. 2010;362(19):1795-1803.
13. U.S. Department of Veterans Affairs, Veterans Health Administration. Colorectal cancer screening. VHA Directive 1015. Published December 30, 2014.
14. U.S. Department of Veterans Affairs, VA Office of the Inspector General, Office of Healthcare Inspections. Healthcare inspection: alleged access delays and surgery service concerns, VA Roseburg Healthcare System, Roseburg, Oregon. Report No.15-00506-535. https://www.va.gov/oig /pubs/VAOIG-15-00506-535.pdf. Published July 11, 2017. Accessed January 9, 2018.
15. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377-381.
16. The American Association for Public Opinion Research. Standard Definitions: Final Dispositions of Case Codes and Outcome Rates for Surveys. 9th edition. http://www.aapor.org/AAPOR_Main/media/publications/Standard-Definitions20169theditionfinal.pdf. Revised 2016. Accessed January 9, 2018.
17. Kahi CJ, Imperiale TF, Juliar BE, Rex DK. Effect of screening colonoscopy on colorectal cancer incidence and mortality. Clin Gastroenterol Hepatol. 2009;7(7):770-775.
18. Manser CN, Bachmann LM, Brunner J, Hunold F, Bauerfeind P, Marbet UA. Colonoscopy screening markedly reduces the occurrence of colon carcinomas and carcinoma-related death: a closed cohort study. Gastrointest Endosc. 2012;76(1):110-117.
19. Nishihara R, Wu K, Lochhead P, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med. 2013;369(12):1095-1105.
20. Harewood GC, Sharma VK, de Garmo P. Impact of colonoscopy preparation quality on detection of suspected colonic neoplasia. Gastrointest Endosc. 2003;58(1):76-79.
21. Hillyer GC, Lebwohl B, Rosenberg RM, et al. Assessing bowel preparation quality using the mean number of adenomas per colonoscopy. Therap Adv Gastroenterol. 2014;7(6):238-246.
22. Clark BT, Rustagi T, Laine L. What level of bowel prep quality requires early repeat colonoscopy: systematic review and meta-analysis of the impact of preparation quality on adenoma detection rate. Am J Gastroenterol. 2014;109(11):1714-1723; quiz 1724.
23. Johnson DA, Barkun AN, Cohen LB, et al; US Multi-Society Task Force on Colorectal Cancer. Optimizing adequacy of bowel cleansing for colonoscopy: recommendations from the US multi-society task force on colorectal cancer. Gastroenterology. 2014;147(4):903-924.
24. Rex DK, Petrini JL, Baron TH, et al; ASGE/ACG Taskforce on Quality in Endoscopy. Quality indicators for colonoscopy. Am J Gastroenterol. 2006;101(4):873-885.
25. Kaminski MF, Wieszczy P, Rupinski M, et al. Increased rate of adenoma detection associates with reduced risk of colorectal cancer and death. Gastroenterology. 2017;153(1):98-105.
26. Kahi CJ, Ballard D, Shah AS, Mears R, Johnson CS. Impact of a quarterly report card on colonoscopy quality measures. Gastrointest. Endosc. 2013;77(6):925-931.
27. Keswani RN, Yadlapati R, Gleason KM, et al. Physician report cards and implementing standards of practice are both significantly associated with improved screening colonoscopy quality. Am J Gastroenterol. 2015;110(8):1134-1139.
28. Logan JR, Lieberman DA. The use of databases and registries to enhance colonoscopy quality. Gastrointest Endosc Clin N Am. 2010;20(4):717-734.
1. Brenner H, Stock C, Hoffmeister M. Effect of screening sigmoidoscopy and screening colonoscopy on colorectal cancer incidence and mortality: systematic review and meta-analysis of randomised controlled trials and observational studies. BMJ. 2014;348:g2467.
2. Meester RGS, Doubeni CA, Lansdorp-Vogelaar I, et al. Colorectal cancer deaths attributable to nonuse of screening in the United States. Ann Epidemiol. 2015;25(3):208-213.e1.
3. Corley DA, Jensen CD, Marks AR, et al. Adenoma detection rate and risk of colorectal cancer and death. N Engl J Med. 2014;370(26):1298-1306.
4. Meester RGS, Doubeni CA, Lansdorp-Vogelaar I, et al. Variation in adenoma detection rate and the lifetime benefits and cost of colorectal cancer screening: a microsimulation model. JAMA. 2015;313(23):2349-2358.
5. Boroff ES, Gurudu SR, Hentz JG, Leighton JA, Ramirez FC. Polyp and adenoma detection rates in the proximal and distal colon. Am J Gastroenterol. 2013;108(6):993-999.
6. Center for Medicare and Medicaid Services. Quality measures. https://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/Qual ityMeasures/index.html. Updated December 19, 2017. Accessed January 17, 2018.
7. Robertson DJ, Lieberman DA, Winawer SJ, et al. Colorectal cancers soon after colonoscopy: a pooled multicohort analysis. Gut. 2014;63(6):949-956.
8. Fayad NF, Kahi CJ. Colonoscopy quality assessment. Gastrointest Endosc Clin N Am. 2015;25(2):373-386.
9. de Jonge V, Sint Nicolaas J, Cahen DL, et al; SCoPE Consortium. Quality evaluation of colonoscopy reporting and colonoscopy performance in daily clinical practice. Gastrointest Endosc. 2012;75(1):98-106.
10. Johnson DA. Quality benchmarking for colonoscopy: how do we pick products from the shelf? Gastrointest Endosc. 2012;75(1):107-109.
11. Anderson JC, Butterly LF. Colonoscopy: quality indicators. Clin Transl Gastroenterol. 2015;6(2):e77.
12. Kaminski MF, Regula J, Kraszewska E, et al. Quality indicators for colonoscopy and the risk of interval cancer. N Engl J Med. 2010;362(19):1795-1803.
13. U.S. Department of Veterans Affairs, Veterans Health Administration. Colorectal cancer screening. VHA Directive 1015. Published December 30, 2014.
14. U.S. Department of Veterans Affairs, VA Office of the Inspector General, Office of Healthcare Inspections. Healthcare inspection: alleged access delays and surgery service concerns, VA Roseburg Healthcare System, Roseburg, Oregon. Report No.15-00506-535. https://www.va.gov/oig /pubs/VAOIG-15-00506-535.pdf. Published July 11, 2017. Accessed January 9, 2018.
15. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377-381.
16. The American Association for Public Opinion Research. Standard Definitions: Final Dispositions of Case Codes and Outcome Rates for Surveys. 9th edition. http://www.aapor.org/AAPOR_Main/media/publications/Standard-Definitions20169theditionfinal.pdf. Revised 2016. Accessed January 9, 2018.
17. Kahi CJ, Imperiale TF, Juliar BE, Rex DK. Effect of screening colonoscopy on colorectal cancer incidence and mortality. Clin Gastroenterol Hepatol. 2009;7(7):770-775.
18. Manser CN, Bachmann LM, Brunner J, Hunold F, Bauerfeind P, Marbet UA. Colonoscopy screening markedly reduces the occurrence of colon carcinomas and carcinoma-related death: a closed cohort study. Gastrointest Endosc. 2012;76(1):110-117.
19. Nishihara R, Wu K, Lochhead P, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med. 2013;369(12):1095-1105.
20. Harewood GC, Sharma VK, de Garmo P. Impact of colonoscopy preparation quality on detection of suspected colonic neoplasia. Gastrointest Endosc. 2003;58(1):76-79.
21. Hillyer GC, Lebwohl B, Rosenberg RM, et al. Assessing bowel preparation quality using the mean number of adenomas per colonoscopy. Therap Adv Gastroenterol. 2014;7(6):238-246.
22. Clark BT, Rustagi T, Laine L. What level of bowel prep quality requires early repeat colonoscopy: systematic review and meta-analysis of the impact of preparation quality on adenoma detection rate. Am J Gastroenterol. 2014;109(11):1714-1723; quiz 1724.
23. Johnson DA, Barkun AN, Cohen LB, et al; US Multi-Society Task Force on Colorectal Cancer. Optimizing adequacy of bowel cleansing for colonoscopy: recommendations from the US multi-society task force on colorectal cancer. Gastroenterology. 2014;147(4):903-924.
24. Rex DK, Petrini JL, Baron TH, et al; ASGE/ACG Taskforce on Quality in Endoscopy. Quality indicators for colonoscopy. Am J Gastroenterol. 2006;101(4):873-885.
25. Kaminski MF, Wieszczy P, Rupinski M, et al. Increased rate of adenoma detection associates with reduced risk of colorectal cancer and death. Gastroenterology. 2017;153(1):98-105.
26. Kahi CJ, Ballard D, Shah AS, Mears R, Johnson CS. Impact of a quarterly report card on colonoscopy quality measures. Gastrointest. Endosc. 2013;77(6):925-931.
27. Keswani RN, Yadlapati R, Gleason KM, et al. Physician report cards and implementing standards of practice are both significantly associated with improved screening colonoscopy quality. Am J Gastroenterol. 2015;110(8):1134-1139.
28. Logan JR, Lieberman DA. The use of databases and registries to enhance colonoscopy quality. Gastrointest Endosc Clin N Am. 2010;20(4):717-734.