User login
Youth tobacco use shows ‘promising declines’
according to the Centers for Disease Control and Prevention.
The prevalence of current tobacco use – defined as use on 1 or more days in the past 30 days – among high schoolers fell from 24.2% in 2011 to 19.6% in 2017, and middle school use decreased from 7.5% to 5.6% over that same time. That means the number of youth tobacco users went from almost 4.6 million in 2011 to slightly more than 3.6 million in 2017, Teresa W. Wang, PhD, and her associates said in the Morbidity and Mortality Weekly Report.
Almost half (47%) of the high school students who used tobacco in 2017 used two or more products, as did two out of five (42%) middle schoolers. That year, black high school students were less likely to use any tobacco product (14.2%) than were whites (22.7%) and Hispanics (16.7%). E-cigarettes were the most popular form of tobacco among white and Hispanic high schoolers, while cigars were the most commonly used form among blacks, they reported based on data from the National Youth Tobacco Surveys, which had sample sizes of 18,766 in 2011 and 17,872 in 2017.
“Despite promising declines in tobacco use, far too many young people continue to use tobacco products, including e-cigarettes,” CDC Director Robert R. Redfield, MD, said in a written statement accompanying the report. “Comprehensive, sustained strategies can help prevent and reduce tobacco use and protect our nation’s youth from this preventable health risk.”
In a separate statement, FDA Commissioner Scott Gottlieb, MD, said, “We are working hard to develop a pathway to put products like e-cigarettes through an appropriate series of regulatory gates to properly evaluate them as an alternative for adults who still want to get access to satisfying levels of nicotine, without all the risks associated with lighting tobacco on fire. And we will continue to encourage the development of potentially less harmful forms of nicotine delivery for currently addicted adult smokers. … But these public health opportunities are put at risk if all we do is hook another generation of kids on nicotine and tobacco products through alternatives like e-cigarettes.”
SOURCE: Wang TW et al. MMWR. 2018;67(22):629-33.
according to the Centers for Disease Control and Prevention.
The prevalence of current tobacco use – defined as use on 1 or more days in the past 30 days – among high schoolers fell from 24.2% in 2011 to 19.6% in 2017, and middle school use decreased from 7.5% to 5.6% over that same time. That means the number of youth tobacco users went from almost 4.6 million in 2011 to slightly more than 3.6 million in 2017, Teresa W. Wang, PhD, and her associates said in the Morbidity and Mortality Weekly Report.
Almost half (47%) of the high school students who used tobacco in 2017 used two or more products, as did two out of five (42%) middle schoolers. That year, black high school students were less likely to use any tobacco product (14.2%) than were whites (22.7%) and Hispanics (16.7%). E-cigarettes were the most popular form of tobacco among white and Hispanic high schoolers, while cigars were the most commonly used form among blacks, they reported based on data from the National Youth Tobacco Surveys, which had sample sizes of 18,766 in 2011 and 17,872 in 2017.
“Despite promising declines in tobacco use, far too many young people continue to use tobacco products, including e-cigarettes,” CDC Director Robert R. Redfield, MD, said in a written statement accompanying the report. “Comprehensive, sustained strategies can help prevent and reduce tobacco use and protect our nation’s youth from this preventable health risk.”
In a separate statement, FDA Commissioner Scott Gottlieb, MD, said, “We are working hard to develop a pathway to put products like e-cigarettes through an appropriate series of regulatory gates to properly evaluate them as an alternative for adults who still want to get access to satisfying levels of nicotine, without all the risks associated with lighting tobacco on fire. And we will continue to encourage the development of potentially less harmful forms of nicotine delivery for currently addicted adult smokers. … But these public health opportunities are put at risk if all we do is hook another generation of kids on nicotine and tobacco products through alternatives like e-cigarettes.”
SOURCE: Wang TW et al. MMWR. 2018;67(22):629-33.
according to the Centers for Disease Control and Prevention.
The prevalence of current tobacco use – defined as use on 1 or more days in the past 30 days – among high schoolers fell from 24.2% in 2011 to 19.6% in 2017, and middle school use decreased from 7.5% to 5.6% over that same time. That means the number of youth tobacco users went from almost 4.6 million in 2011 to slightly more than 3.6 million in 2017, Teresa W. Wang, PhD, and her associates said in the Morbidity and Mortality Weekly Report.
Almost half (47%) of the high school students who used tobacco in 2017 used two or more products, as did two out of five (42%) middle schoolers. That year, black high school students were less likely to use any tobacco product (14.2%) than were whites (22.7%) and Hispanics (16.7%). E-cigarettes were the most popular form of tobacco among white and Hispanic high schoolers, while cigars were the most commonly used form among blacks, they reported based on data from the National Youth Tobacco Surveys, which had sample sizes of 18,766 in 2011 and 17,872 in 2017.
“Despite promising declines in tobacco use, far too many young people continue to use tobacco products, including e-cigarettes,” CDC Director Robert R. Redfield, MD, said in a written statement accompanying the report. “Comprehensive, sustained strategies can help prevent and reduce tobacco use and protect our nation’s youth from this preventable health risk.”
In a separate statement, FDA Commissioner Scott Gottlieb, MD, said, “We are working hard to develop a pathway to put products like e-cigarettes through an appropriate series of regulatory gates to properly evaluate them as an alternative for adults who still want to get access to satisfying levels of nicotine, without all the risks associated with lighting tobacco on fire. And we will continue to encourage the development of potentially less harmful forms of nicotine delivery for currently addicted adult smokers. … But these public health opportunities are put at risk if all we do is hook another generation of kids on nicotine and tobacco products through alternatives like e-cigarettes.”
SOURCE: Wang TW et al. MMWR. 2018;67(22):629-33.
FROM MMWR
HHS to allow insurers’ workaround on 2019 prices
Federal officials will not block insurance companies from again using a workaround to cushion a steep rise in health premiums caused by President Donald Trump’s cancellation of a program established under the Affordable Care Act, Health and Human Services Secretary Alex Azar announced June 6.
The technique – called “silver loading” because it pushed price increases onto the silver-level plans in the ACA marketplaces – was used by many states for 2018 policies. But federal officials had hinted they might bar the practice next year.
At a hearing June 6 before the House Education and Workforce Committee, Mr. Azar said stopping this practice “would require regulations, which simply couldn’t be done in time for the 2019 plan period.”
States moved to silver loading after the president in October cut off federal reimbursement for so-called cost-sharing reduction subsidies that the ACA guaranteed to insurance companies. Those payments offset the cost of discounts that insurers are required by the law to provide to some low-income people to help cover their deductibles and other out-of-pocket costs.
States scrambled to let insurers raise rates so they would stay in the market. And many let them use this technique to recoup the lost funding by adding to the premium costs of midlevel silver plans in the health exchanges.
Because the formula for federal premium subsidies offered to people who purchase through the marketplaces is based on the prices of those silver plans, as those premiums rose so did the subsidies to help people afford them. That meant the federal government ended up paying much of the increase in prices.
At the committee hearing June 6, under questioning from Rep. Joe Courtney (D-Conn.), Mr. Azar declined to say if the department was considering a future ban.
“It’s not an easy question,” Mr. Azar said.
The fact that the federal government ended up effectively making the payments aggravated many Republicans, and there have been rumors over the past several months that HHS might require the premium increases to be applied across all plans, boosting costs for all buyers in the individual market.
Seema Verma, the administrator of the Centers for Medicare & Medicaid Services, told reporters in April that the department was examining the possibility.
Apparently that will not happen, at least not for plan year 2019.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
Federal officials will not block insurance companies from again using a workaround to cushion a steep rise in health premiums caused by President Donald Trump’s cancellation of a program established under the Affordable Care Act, Health and Human Services Secretary Alex Azar announced June 6.
The technique – called “silver loading” because it pushed price increases onto the silver-level plans in the ACA marketplaces – was used by many states for 2018 policies. But federal officials had hinted they might bar the practice next year.
At a hearing June 6 before the House Education and Workforce Committee, Mr. Azar said stopping this practice “would require regulations, which simply couldn’t be done in time for the 2019 plan period.”
States moved to silver loading after the president in October cut off federal reimbursement for so-called cost-sharing reduction subsidies that the ACA guaranteed to insurance companies. Those payments offset the cost of discounts that insurers are required by the law to provide to some low-income people to help cover their deductibles and other out-of-pocket costs.
States scrambled to let insurers raise rates so they would stay in the market. And many let them use this technique to recoup the lost funding by adding to the premium costs of midlevel silver plans in the health exchanges.
Because the formula for federal premium subsidies offered to people who purchase through the marketplaces is based on the prices of those silver plans, as those premiums rose so did the subsidies to help people afford them. That meant the federal government ended up paying much of the increase in prices.
At the committee hearing June 6, under questioning from Rep. Joe Courtney (D-Conn.), Mr. Azar declined to say if the department was considering a future ban.
“It’s not an easy question,” Mr. Azar said.
The fact that the federal government ended up effectively making the payments aggravated many Republicans, and there have been rumors over the past several months that HHS might require the premium increases to be applied across all plans, boosting costs for all buyers in the individual market.
Seema Verma, the administrator of the Centers for Medicare & Medicaid Services, told reporters in April that the department was examining the possibility.
Apparently that will not happen, at least not for plan year 2019.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
Federal officials will not block insurance companies from again using a workaround to cushion a steep rise in health premiums caused by President Donald Trump’s cancellation of a program established under the Affordable Care Act, Health and Human Services Secretary Alex Azar announced June 6.
The technique – called “silver loading” because it pushed price increases onto the silver-level plans in the ACA marketplaces – was used by many states for 2018 policies. But federal officials had hinted they might bar the practice next year.
At a hearing June 6 before the House Education and Workforce Committee, Mr. Azar said stopping this practice “would require regulations, which simply couldn’t be done in time for the 2019 plan period.”
States moved to silver loading after the president in October cut off federal reimbursement for so-called cost-sharing reduction subsidies that the ACA guaranteed to insurance companies. Those payments offset the cost of discounts that insurers are required by the law to provide to some low-income people to help cover their deductibles and other out-of-pocket costs.
States scrambled to let insurers raise rates so they would stay in the market. And many let them use this technique to recoup the lost funding by adding to the premium costs of midlevel silver plans in the health exchanges.
Because the formula for federal premium subsidies offered to people who purchase through the marketplaces is based on the prices of those silver plans, as those premiums rose so did the subsidies to help people afford them. That meant the federal government ended up paying much of the increase in prices.
At the committee hearing June 6, under questioning from Rep. Joe Courtney (D-Conn.), Mr. Azar declined to say if the department was considering a future ban.
“It’s not an easy question,” Mr. Azar said.
The fact that the federal government ended up effectively making the payments aggravated many Republicans, and there have been rumors over the past several months that HHS might require the premium increases to be applied across all plans, boosting costs for all buyers in the individual market.
Seema Verma, the administrator of the Centers for Medicare & Medicaid Services, told reporters in April that the department was examining the possibility.
Apparently that will not happen, at least not for plan year 2019.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
Diabetes places burden on patients
according to a new survey by prescription manager UpWell Health.
The time spent on activities that come with diabetes management – blood glucose monitoring, diet planning, medical appointments – can add up to several hours a week. Among the 5,255 respondents to the online survey, 18% said that such tasks took up 5-10 hours each week, 7% said it was 10-15 hours, and 9% said they spent 15 or more hours a week on diabetes-related tasks, UpWell reported.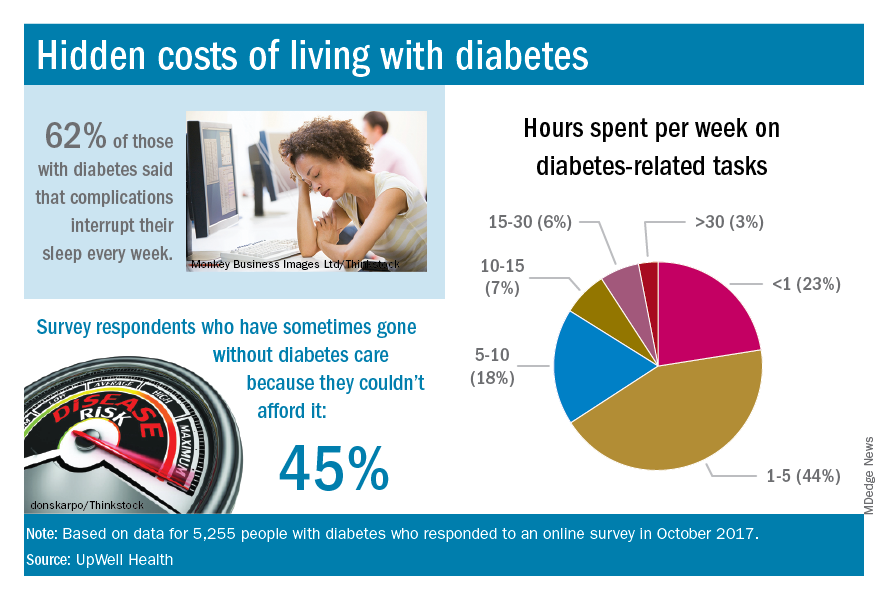
Since medical expenses aren’t always fully covered by insurance, 43% of respondents paid up to $1,000 a year out of pocket to treat diabetes complications, 16% paid $1,000 to $2,000 a year, and 4% paid more than $5,000. Five percent also paid over $5,000 a year out of pocket for diabetes care from a physician and 45% said that they had sometimes gone without diabetes care because they couldn’t afford it, the survey data showed.
“Most people with diabetes are able to manage it successfully and live active, satisfying lives. But doing so requires constant planning, vigilance, and care. They eagerly seek trustworthy resources to help them reduce the burden of living with diabetes,” UpWell said.
according to a new survey by prescription manager UpWell Health.
The time spent on activities that come with diabetes management – blood glucose monitoring, diet planning, medical appointments – can add up to several hours a week. Among the 5,255 respondents to the online survey, 18% said that such tasks took up 5-10 hours each week, 7% said it was 10-15 hours, and 9% said they spent 15 or more hours a week on diabetes-related tasks, UpWell reported.
Since medical expenses aren’t always fully covered by insurance, 43% of respondents paid up to $1,000 a year out of pocket to treat diabetes complications, 16% paid $1,000 to $2,000 a year, and 4% paid more than $5,000. Five percent also paid over $5,000 a year out of pocket for diabetes care from a physician and 45% said that they had sometimes gone without diabetes care because they couldn’t afford it, the survey data showed.
“Most people with diabetes are able to manage it successfully and live active, satisfying lives. But doing so requires constant planning, vigilance, and care. They eagerly seek trustworthy resources to help them reduce the burden of living with diabetes,” UpWell said.
according to a new survey by prescription manager UpWell Health.
The time spent on activities that come with diabetes management – blood glucose monitoring, diet planning, medical appointments – can add up to several hours a week. Among the 5,255 respondents to the online survey, 18% said that such tasks took up 5-10 hours each week, 7% said it was 10-15 hours, and 9% said they spent 15 or more hours a week on diabetes-related tasks, UpWell reported.
Since medical expenses aren’t always fully covered by insurance, 43% of respondents paid up to $1,000 a year out of pocket to treat diabetes complications, 16% paid $1,000 to $2,000 a year, and 4% paid more than $5,000. Five percent also paid over $5,000 a year out of pocket for diabetes care from a physician and 45% said that they had sometimes gone without diabetes care because they couldn’t afford it, the survey data showed.
“Most people with diabetes are able to manage it successfully and live active, satisfying lives. But doing so requires constant planning, vigilance, and care. They eagerly seek trustworthy resources to help them reduce the burden of living with diabetes,” UpWell said.
A U.S. model for Italian hospitals?
In the United States, family physicians (general practitioners) used to manage their patients in the hospital, either as the primary care doctor or in consultation with specialists. Only since the 1990s has a new kind of physician gained widespread acceptance: the hospitalist (“specialist of inpatient care”).1
In Italy the process has not been the same. In our health care system, primary care physicians have always transferred the responsibility of hospital care to an inpatient team. Actually, our hospital-based doctors dedicate their whole working time to inpatient care, and general practitioners are not expected to go to the hospital. The patients were (and are) admitted to one ward or another according to their main clinical problem.
Little by little, a huge number of organ specialty and subspecialty wards have filled Italian hospitals. In this context, the internal medicine specialty was unable to occupy its characteristic role, so that, a few years ago, the medical community wondered if the specialty should have continued to exist.
Anyway, as a result of hyperspecialization, we have many different specialists in inpatient care who are not specialists in global inpatient care.
Nowadays, in our country we are faced with a dramatic epidemiologic change. The Italian population is aging and the majority of patients have not only one clinical problem but multiple comorbidities. When these patients reach the emergency department, it is not easy to identify the main clinical problem and assign him/her to an organ specialty unit. And when he or she eventually arrives there, a considerable number of consultants is frequently required. The vision of organ specialists is not holistic, and they are more prone to maximizing their tools than rationalizing them. So, at present, our traditional hospital model has been generating care fragmentation, overproduction of diagnoses, overprescription of drugs, and increasing costs.
It is obvious that a new model is necessary for the future, and we look with great interest at the American hospitalist model.
We need a new hospital-based clinician who has wide-ranging competencies, and is able to define priorities and appropriateness of care when a patient requires multiple specialists’ interventions; one who is autonomous in performing basic procedures and expert in perioperative medicine; prompt to communicate with primary care doctors at the time of admission and discharge; and prepared to work in managed-care organizations.
We wonder: Are Italian hospital-based internists – the only specialists in global inpatient care – suited to this role?
We think so. However, current Italian training in internal medicine is focused mainly on scientific bases of diseases, pathophysiological, and clinical aspects. Concepts such as complexity or the management of patients with comorbidities are quite difficult to teach to medical school students and therefore often neglected. As a result, internal medicine physicians require a prolonged practical training.
Inspired by the Core Competencies in Hospital Medicine published by the Society of Hospital Medicine, this year in Genoa (the birthplace of Christopher Columbus) we started a 2-year second-level University Master course, called “Hospitalist: Managing complexity in Internal Medicine inpatients” for 35 internal medicine specialists. It is the fruit of collaboration between the main association of Italian hospital-based internists (Federation of Associations of Hospital Doctors on Internal Medicine, or FADOI) and the University of Genoa’s Department of Internal Medicine, Academy of Health Management, and the Center of Simulation and Advanced Training.
In Italy, this is the first concrete initiative to train, and better define, this new type of physician expert in the management of inpatients.
According to SHM’s definition of a hospitalist, we think that the activities of this new physician should also include teaching and research related to hospital medicine. And as Dr. Steven Pantilat wrote, “patient safety, leadership, palliative care and quality improvement are the issues that pertain to all hospitalists.”2
Theoretically, the development of the hospitalist model should be easier in Italy when compared to the United States. Dr. Robert Wachter and Dr. Lee Goldman wrote in 1996 about the objections to the hospitalist model of American primary care physicians (“to preserve continuity”) and specialists (“fewer consultations, lower income”), but in Italy family doctors do not usually follow their patients in the hospital, and specialists have no incentive for in-hospital consultations.3 Moreover, patients with comorbidities, or pathologies on the border between medicine and surgery (e.g. cholecystitis, bowel obstruction, polytrauma, etc.), are already often assigned to internal medicine, and in the smallest hospitals, the internist is most of the time the only specialist doctor continually present.
Nevertheless, the Italian hospitalist model will be a challenge. We know we have to deal with organ specialists, but we strongly believe that this is the most appropriate and the most sustainable model for the future of the Italian hospitals. Our wish is not to become the “bosses” of the hospital, but to ensure global, coordinated, and respectful care to present and future patients.
Published outcomes studies demonstrate that the U.S. hospitalist model has led to consistent and pronounced cost saving with no loss in quality.4 In the United States, the hospitalist field has grown from a few hundred physicians to more than 50,000,5 making it the fastest growing physician specialty in medical history.
Why should the same not occur in Italy?
References
1. Baudendistel TE, Watcher RM. The evolution of the hospitalist movement in USA. Clin Med JRCPL. 2002;2:327-30.
2. Pantilat S. What is a Hospitalist? The Hospitalist 2006 February;2006(2).
3. Wachter RM, Goldman Lee. The emerging role of “Hospitalists” in the American Health Care System. N Engl J Med. 1996;335:514-7.
4. White HL, Glazier RH. Do hospitalist physicians improve the quality of inpatient care delivery? A systematic review of process, efficiency and outcome measures. BMC Medicine. 2011;9:58:1-22. http://www.biomedcentral.com/1741-7015/9/58.
5. Wachter RM, Goldman L. Zero to 50,000 – The 20th Anniversary of the Hospitalist. N Engl J Med. 2016;375:1009-11.
Valerio Verdiani, MD, director of internal medicine, Grosseto, Italy. Francesco Orlandini, MD, internal medicine, health administrator, ASL4 Liguria, Chiavari (GE), Italy. Micaela La Regina, MD, internal medicine, risk management and clinical governance, ASL5 Liguria, La Spezia, Italy. Giovanni Murialdo, MD, department of internal medicine and medical specialty, University of Genoa (Italy). Andrea Fontanella, MD, director of medicine department, president of the Federation of Associations of Hospital Doctors on Internal Medicine (FADOI), Naples, Italy. Mauro Silingardi, MD, director of internal medicine, director of training and refresher of FADOI, Bologna, Italy.
In the United States, family physicians (general practitioners) used to manage their patients in the hospital, either as the primary care doctor or in consultation with specialists. Only since the 1990s has a new kind of physician gained widespread acceptance: the hospitalist (“specialist of inpatient care”).1
In Italy the process has not been the same. In our health care system, primary care physicians have always transferred the responsibility of hospital care to an inpatient team. Actually, our hospital-based doctors dedicate their whole working time to inpatient care, and general practitioners are not expected to go to the hospital. The patients were (and are) admitted to one ward or another according to their main clinical problem.
Little by little, a huge number of organ specialty and subspecialty wards have filled Italian hospitals. In this context, the internal medicine specialty was unable to occupy its characteristic role, so that, a few years ago, the medical community wondered if the specialty should have continued to exist.
Anyway, as a result of hyperspecialization, we have many different specialists in inpatient care who are not specialists in global inpatient care.
Nowadays, in our country we are faced with a dramatic epidemiologic change. The Italian population is aging and the majority of patients have not only one clinical problem but multiple comorbidities. When these patients reach the emergency department, it is not easy to identify the main clinical problem and assign him/her to an organ specialty unit. And when he or she eventually arrives there, a considerable number of consultants is frequently required. The vision of organ specialists is not holistic, and they are more prone to maximizing their tools than rationalizing them. So, at present, our traditional hospital model has been generating care fragmentation, overproduction of diagnoses, overprescription of drugs, and increasing costs.
It is obvious that a new model is necessary for the future, and we look with great interest at the American hospitalist model.
We need a new hospital-based clinician who has wide-ranging competencies, and is able to define priorities and appropriateness of care when a patient requires multiple specialists’ interventions; one who is autonomous in performing basic procedures and expert in perioperative medicine; prompt to communicate with primary care doctors at the time of admission and discharge; and prepared to work in managed-care organizations.
We wonder: Are Italian hospital-based internists – the only specialists in global inpatient care – suited to this role?
We think so. However, current Italian training in internal medicine is focused mainly on scientific bases of diseases, pathophysiological, and clinical aspects. Concepts such as complexity or the management of patients with comorbidities are quite difficult to teach to medical school students and therefore often neglected. As a result, internal medicine physicians require a prolonged practical training.
Inspired by the Core Competencies in Hospital Medicine published by the Society of Hospital Medicine, this year in Genoa (the birthplace of Christopher Columbus) we started a 2-year second-level University Master course, called “Hospitalist: Managing complexity in Internal Medicine inpatients” for 35 internal medicine specialists. It is the fruit of collaboration between the main association of Italian hospital-based internists (Federation of Associations of Hospital Doctors on Internal Medicine, or FADOI) and the University of Genoa’s Department of Internal Medicine, Academy of Health Management, and the Center of Simulation and Advanced Training.
In Italy, this is the first concrete initiative to train, and better define, this new type of physician expert in the management of inpatients.
According to SHM’s definition of a hospitalist, we think that the activities of this new physician should also include teaching and research related to hospital medicine. And as Dr. Steven Pantilat wrote, “patient safety, leadership, palliative care and quality improvement are the issues that pertain to all hospitalists.”2
Theoretically, the development of the hospitalist model should be easier in Italy when compared to the United States. Dr. Robert Wachter and Dr. Lee Goldman wrote in 1996 about the objections to the hospitalist model of American primary care physicians (“to preserve continuity”) and specialists (“fewer consultations, lower income”), but in Italy family doctors do not usually follow their patients in the hospital, and specialists have no incentive for in-hospital consultations.3 Moreover, patients with comorbidities, or pathologies on the border between medicine and surgery (e.g. cholecystitis, bowel obstruction, polytrauma, etc.), are already often assigned to internal medicine, and in the smallest hospitals, the internist is most of the time the only specialist doctor continually present.
Nevertheless, the Italian hospitalist model will be a challenge. We know we have to deal with organ specialists, but we strongly believe that this is the most appropriate and the most sustainable model for the future of the Italian hospitals. Our wish is not to become the “bosses” of the hospital, but to ensure global, coordinated, and respectful care to present and future patients.
Published outcomes studies demonstrate that the U.S. hospitalist model has led to consistent and pronounced cost saving with no loss in quality.4 In the United States, the hospitalist field has grown from a few hundred physicians to more than 50,000,5 making it the fastest growing physician specialty in medical history.
Why should the same not occur in Italy?
References
1. Baudendistel TE, Watcher RM. The evolution of the hospitalist movement in USA. Clin Med JRCPL. 2002;2:327-30.
2. Pantilat S. What is a Hospitalist? The Hospitalist 2006 February;2006(2).
3. Wachter RM, Goldman Lee. The emerging role of “Hospitalists” in the American Health Care System. N Engl J Med. 1996;335:514-7.
4. White HL, Glazier RH. Do hospitalist physicians improve the quality of inpatient care delivery? A systematic review of process, efficiency and outcome measures. BMC Medicine. 2011;9:58:1-22. http://www.biomedcentral.com/1741-7015/9/58.
5. Wachter RM, Goldman L. Zero to 50,000 – The 20th Anniversary of the Hospitalist. N Engl J Med. 2016;375:1009-11.
Valerio Verdiani, MD, director of internal medicine, Grosseto, Italy. Francesco Orlandini, MD, internal medicine, health administrator, ASL4 Liguria, Chiavari (GE), Italy. Micaela La Regina, MD, internal medicine, risk management and clinical governance, ASL5 Liguria, La Spezia, Italy. Giovanni Murialdo, MD, department of internal medicine and medical specialty, University of Genoa (Italy). Andrea Fontanella, MD, director of medicine department, president of the Federation of Associations of Hospital Doctors on Internal Medicine (FADOI), Naples, Italy. Mauro Silingardi, MD, director of internal medicine, director of training and refresher of FADOI, Bologna, Italy.
In the United States, family physicians (general practitioners) used to manage their patients in the hospital, either as the primary care doctor or in consultation with specialists. Only since the 1990s has a new kind of physician gained widespread acceptance: the hospitalist (“specialist of inpatient care”).1
In Italy the process has not been the same. In our health care system, primary care physicians have always transferred the responsibility of hospital care to an inpatient team. Actually, our hospital-based doctors dedicate their whole working time to inpatient care, and general practitioners are not expected to go to the hospital. The patients were (and are) admitted to one ward or another according to their main clinical problem.
Little by little, a huge number of organ specialty and subspecialty wards have filled Italian hospitals. In this context, the internal medicine specialty was unable to occupy its characteristic role, so that, a few years ago, the medical community wondered if the specialty should have continued to exist.
Anyway, as a result of hyperspecialization, we have many different specialists in inpatient care who are not specialists in global inpatient care.
Nowadays, in our country we are faced with a dramatic epidemiologic change. The Italian population is aging and the majority of patients have not only one clinical problem but multiple comorbidities. When these patients reach the emergency department, it is not easy to identify the main clinical problem and assign him/her to an organ specialty unit. And when he or she eventually arrives there, a considerable number of consultants is frequently required. The vision of organ specialists is not holistic, and they are more prone to maximizing their tools than rationalizing them. So, at present, our traditional hospital model has been generating care fragmentation, overproduction of diagnoses, overprescription of drugs, and increasing costs.
It is obvious that a new model is necessary for the future, and we look with great interest at the American hospitalist model.
We need a new hospital-based clinician who has wide-ranging competencies, and is able to define priorities and appropriateness of care when a patient requires multiple specialists’ interventions; one who is autonomous in performing basic procedures and expert in perioperative medicine; prompt to communicate with primary care doctors at the time of admission and discharge; and prepared to work in managed-care organizations.
We wonder: Are Italian hospital-based internists – the only specialists in global inpatient care – suited to this role?
We think so. However, current Italian training in internal medicine is focused mainly on scientific bases of diseases, pathophysiological, and clinical aspects. Concepts such as complexity or the management of patients with comorbidities are quite difficult to teach to medical school students and therefore often neglected. As a result, internal medicine physicians require a prolonged practical training.
Inspired by the Core Competencies in Hospital Medicine published by the Society of Hospital Medicine, this year in Genoa (the birthplace of Christopher Columbus) we started a 2-year second-level University Master course, called “Hospitalist: Managing complexity in Internal Medicine inpatients” for 35 internal medicine specialists. It is the fruit of collaboration between the main association of Italian hospital-based internists (Federation of Associations of Hospital Doctors on Internal Medicine, or FADOI) and the University of Genoa’s Department of Internal Medicine, Academy of Health Management, and the Center of Simulation and Advanced Training.
In Italy, this is the first concrete initiative to train, and better define, this new type of physician expert in the management of inpatients.
According to SHM’s definition of a hospitalist, we think that the activities of this new physician should also include teaching and research related to hospital medicine. And as Dr. Steven Pantilat wrote, “patient safety, leadership, palliative care and quality improvement are the issues that pertain to all hospitalists.”2
Theoretically, the development of the hospitalist model should be easier in Italy when compared to the United States. Dr. Robert Wachter and Dr. Lee Goldman wrote in 1996 about the objections to the hospitalist model of American primary care physicians (“to preserve continuity”) and specialists (“fewer consultations, lower income”), but in Italy family doctors do not usually follow their patients in the hospital, and specialists have no incentive for in-hospital consultations.3 Moreover, patients with comorbidities, or pathologies on the border between medicine and surgery (e.g. cholecystitis, bowel obstruction, polytrauma, etc.), are already often assigned to internal medicine, and in the smallest hospitals, the internist is most of the time the only specialist doctor continually present.
Nevertheless, the Italian hospitalist model will be a challenge. We know we have to deal with organ specialists, but we strongly believe that this is the most appropriate and the most sustainable model for the future of the Italian hospitals. Our wish is not to become the “bosses” of the hospital, but to ensure global, coordinated, and respectful care to present and future patients.
Published outcomes studies demonstrate that the U.S. hospitalist model has led to consistent and pronounced cost saving with no loss in quality.4 In the United States, the hospitalist field has grown from a few hundred physicians to more than 50,000,5 making it the fastest growing physician specialty in medical history.
Why should the same not occur in Italy?
References
1. Baudendistel TE, Watcher RM. The evolution of the hospitalist movement in USA. Clin Med JRCPL. 2002;2:327-30.
2. Pantilat S. What is a Hospitalist? The Hospitalist 2006 February;2006(2).
3. Wachter RM, Goldman Lee. The emerging role of “Hospitalists” in the American Health Care System. N Engl J Med. 1996;335:514-7.
4. White HL, Glazier RH. Do hospitalist physicians improve the quality of inpatient care delivery? A systematic review of process, efficiency and outcome measures. BMC Medicine. 2011;9:58:1-22. http://www.biomedcentral.com/1741-7015/9/58.
5. Wachter RM, Goldman L. Zero to 50,000 – The 20th Anniversary of the Hospitalist. N Engl J Med. 2016;375:1009-11.
Valerio Verdiani, MD, director of internal medicine, Grosseto, Italy. Francesco Orlandini, MD, internal medicine, health administrator, ASL4 Liguria, Chiavari (GE), Italy. Micaela La Regina, MD, internal medicine, risk management and clinical governance, ASL5 Liguria, La Spezia, Italy. Giovanni Murialdo, MD, department of internal medicine and medical specialty, University of Genoa (Italy). Andrea Fontanella, MD, director of medicine department, president of the Federation of Associations of Hospital Doctors on Internal Medicine (FADOI), Naples, Italy. Mauro Silingardi, MD, director of internal medicine, director of training and refresher of FADOI, Bologna, Italy.
ASH urges lawmakers to keep opioids accessible
The American Society of Hematology (ASH) has released a new policy statement in favor of safeguarding access to opioids for hematology patients with chronic, severe pain as policymakers consider restrictions on opioid prescribing.
The statement is a recognition from ASH officials of the large number of opioid overdose deaths that involve prescription medication, and an acknowledgment that hematologists need to be advocates for their patients, said Joseph Alvarnas, MD, of City of Hope, Duarte, Calif.

The scope of the opioid problem is significant and worsening. More than 200,000 people in the United States died from overdoses related to prescription opioids. And in 2016, about 46 people were dying every day from prescription opioid overdoses, according to the Centers for Disease Control and Prevention.
In October 2017, President Trump declared that the opioid crisis was a nationwide “public health emergency” and regulators with the Centers for Medicare % Medicaid Services have already put in place restrictions on opioid dosing through the Medicare Part D program.
In a rule finalized in April 2018, the CMS placed restrictions on the dosage of opioids available for chronic opioid users and limited the days’ supply for first-time opioid users. For chronic users, the CMS set a 90-morphine-milligram-equivalent (MME) per day limit that triggers pharmacist consultation with the prescriber. The agency instructed health plans to implement an “opioid care coordination edit” that would be triggered at 90 MME per day across all opioid prescriptions and would require pharmacists to contact prescribers to override for a higher dosage.
The entire exchange must be documented. The CMS instructed health plans in the Medicare Part D program to implement a “hard safety edit” that limits opioid prescription fills to no more than a 7-day supply for opioid-naive patients being treated for acute pain. The changes are set to take effect in January 2019.
But Diane E. Meier, MD, director of the Center to Advance Palliative Care in New York, said the most current data suggest the risk of addiction and substance use disorder among medically-ill patients taking opioids is less than 10%. “That means that 90% of patients with a serious medical illness can safely take opioids for the relief of pain that is causing functional disorder,” she said.
Policymakers should not conflate the use and prescription of opioids with cases of misuse and abuse, Dr. Alvarnas said. Some patients will require a higher dose of opioids depending on their age or number of pain episodes, or because of their body’s physiological response.

Some policies, such as a prior authorization, create “artificial barriers and delays in getting access to medication” for these patients, Dr. Alvarnas said. “When you create a far more arduous prior [authorization] process or limit prescriptions ... if someone has a severe blood disorder or a bone cancer, then what you’re doing is setting up a system that will fail those patients on a regular basis,” he said.
Patients may also have difficulty finding pharmacies that dispense opioid medications, or doctors who will prescribe them, because of fear of repercussions from the Drug Enforcement Administration or their state licensing boards, Dr. Meier said.
No ‘one-size-fits-all’
Because treatment is individualized, there is no “one-size-fits-all” approach to pain management for patients with hematological diseases.
“One of the concerns is that we care for populations of patients, such as those with sickle cell disease, those with blood cancers that can cause destructive bony lesions like somebody with multiple myeloma might experience, or even pain associated with the complication of a disease like hemophilia, where [patients experience] excruciating pain ... from bleeding into a joint,” Dr. Alvarnas said.
It’s not enough to offer anti-inflammatory medications to these patients – and in some cases doing so may create additional problems, experts said. Contraindications for anti-inflammatory agents tend to be more significant in hematology patients because of low platelet counts, liver disease, and kidney disease. This may prevent them from taking medications such as ibuprofen, acetaminophen, or naproxen sodium. Opioids are the “only option” for patients with these kinds of complications who have severe pain, Dr. Osunkwo said.
Pain management training
While hematologists are trained about the potential risks of common drugs such as steroids, “none of that education and training has occurred” for opioids, Dr. Meier said.

Since hematologists often aren’t trained in pain management, many are uncomfortable with managing pain in their diagnosis and leave the responsibility to a pain specialist, Dr. Osunkwo said. “But the problem is, they know more about the disease than anybody else, and you’ll be safer if [they] are doing the pain management for hematology patients because they know the risk and benefit of the different drugs in light of the diagnosis itself, compared to passing that on to somebody else to manage.”
In the recent policy statement, ASH leaders committed to creating evidence-based guidelines and education activities on pain management.
Finding balance
ASH leaders recognize the longstanding, complex nature of the opioid epidemic and want to be a part of the conversation to ensure their patients’ needs and considerations are met in future legislation, Dr. Alvarnas noted.
Dr. Meier, who is vice chair for public policy at Icahn School of Medicine at Mount Sinai, New York, said the issue of balance is paramount when considering good policymaking.
“No policy at either extreme is the right policy,” Dr. Meier said. “Good policymaking, good public health interventions attempt to achieve some balance ... in preventing harm and maximizing appropriate treatment of vulnerable, seriously ill patients. And the policies we have now don’t achieve that.”
The “Statement on Opioid Use in Patients with Hematologic Diseases and Disorders” is available on the ASH website.
Dr. Alvarnas is chair of the society’s Committee on Practice. Dr. Meier and Dr. Osunkwo reported having no relevant financial disclosures.
The American Society of Hematology (ASH) has released a new policy statement in favor of safeguarding access to opioids for hematology patients with chronic, severe pain as policymakers consider restrictions on opioid prescribing.
The statement is a recognition from ASH officials of the large number of opioid overdose deaths that involve prescription medication, and an acknowledgment that hematologists need to be advocates for their patients, said Joseph Alvarnas, MD, of City of Hope, Duarte, Calif.
The scope of the opioid problem is significant and worsening. More than 200,000 people in the United States died from overdoses related to prescription opioids. And in 2016, about 46 people were dying every day from prescription opioid overdoses, according to the Centers for Disease Control and Prevention.
In October 2017, President Trump declared that the opioid crisis was a nationwide “public health emergency” and regulators with the Centers for Medicare % Medicaid Services have already put in place restrictions on opioid dosing through the Medicare Part D program.
In a rule finalized in April 2018, the CMS placed restrictions on the dosage of opioids available for chronic opioid users and limited the days’ supply for first-time opioid users. For chronic users, the CMS set a 90-morphine-milligram-equivalent (MME) per day limit that triggers pharmacist consultation with the prescriber. The agency instructed health plans to implement an “opioid care coordination edit” that would be triggered at 90 MME per day across all opioid prescriptions and would require pharmacists to contact prescribers to override for a higher dosage.
The entire exchange must be documented. The CMS instructed health plans in the Medicare Part D program to implement a “hard safety edit” that limits opioid prescription fills to no more than a 7-day supply for opioid-naive patients being treated for acute pain. The changes are set to take effect in January 2019.
But Diane E. Meier, MD, director of the Center to Advance Palliative Care in New York, said the most current data suggest the risk of addiction and substance use disorder among medically-ill patients taking opioids is less than 10%. “That means that 90% of patients with a serious medical illness can safely take opioids for the relief of pain that is causing functional disorder,” she said.
Policymakers should not conflate the use and prescription of opioids with cases of misuse and abuse, Dr. Alvarnas said. Some patients will require a higher dose of opioids depending on their age or number of pain episodes, or because of their body’s physiological response.
Some policies, such as a prior authorization, create “artificial barriers and delays in getting access to medication” for these patients, Dr. Alvarnas said. “When you create a far more arduous prior [authorization] process or limit prescriptions ... if someone has a severe blood disorder or a bone cancer, then what you’re doing is setting up a system that will fail those patients on a regular basis,” he said.
Patients may also have difficulty finding pharmacies that dispense opioid medications, or doctors who will prescribe them, because of fear of repercussions from the Drug Enforcement Administration or their state licensing boards, Dr. Meier said.
No ‘one-size-fits-all’
Because treatment is individualized, there is no “one-size-fits-all” approach to pain management for patients with hematological diseases.
“One of the concerns is that we care for populations of patients, such as those with sickle cell disease, those with blood cancers that can cause destructive bony lesions like somebody with multiple myeloma might experience, or even pain associated with the complication of a disease like hemophilia, where [patients experience] excruciating pain ... from bleeding into a joint,” Dr. Alvarnas said.
It’s not enough to offer anti-inflammatory medications to these patients – and in some cases doing so may create additional problems, experts said. Contraindications for anti-inflammatory agents tend to be more significant in hematology patients because of low platelet counts, liver disease, and kidney disease. This may prevent them from taking medications such as ibuprofen, acetaminophen, or naproxen sodium. Opioids are the “only option” for patients with these kinds of complications who have severe pain, Dr. Osunkwo said.
Pain management training
While hematologists are trained about the potential risks of common drugs such as steroids, “none of that education and training has occurred” for opioids, Dr. Meier said.
Since hematologists often aren’t trained in pain management, many are uncomfortable with managing pain in their diagnosis and leave the responsibility to a pain specialist, Dr. Osunkwo said. “But the problem is, they know more about the disease than anybody else, and you’ll be safer if [they] are doing the pain management for hematology patients because they know the risk and benefit of the different drugs in light of the diagnosis itself, compared to passing that on to somebody else to manage.”
In the recent policy statement, ASH leaders committed to creating evidence-based guidelines and education activities on pain management.
Finding balance
ASH leaders recognize the longstanding, complex nature of the opioid epidemic and want to be a part of the conversation to ensure their patients’ needs and considerations are met in future legislation, Dr. Alvarnas noted.
Dr. Meier, who is vice chair for public policy at Icahn School of Medicine at Mount Sinai, New York, said the issue of balance is paramount when considering good policymaking.
“No policy at either extreme is the right policy,” Dr. Meier said. “Good policymaking, good public health interventions attempt to achieve some balance ... in preventing harm and maximizing appropriate treatment of vulnerable, seriously ill patients. And the policies we have now don’t achieve that.”
The “Statement on Opioid Use in Patients with Hematologic Diseases and Disorders” is available on the ASH website.
Dr. Alvarnas is chair of the society’s Committee on Practice. Dr. Meier and Dr. Osunkwo reported having no relevant financial disclosures.
The American Society of Hematology (ASH) has released a new policy statement in favor of safeguarding access to opioids for hematology patients with chronic, severe pain as policymakers consider restrictions on opioid prescribing.
The statement is a recognition from ASH officials of the large number of opioid overdose deaths that involve prescription medication, and an acknowledgment that hematologists need to be advocates for their patients, said Joseph Alvarnas, MD, of City of Hope, Duarte, Calif.
The scope of the opioid problem is significant and worsening. More than 200,000 people in the United States died from overdoses related to prescription opioids. And in 2016, about 46 people were dying every day from prescription opioid overdoses, according to the Centers for Disease Control and Prevention.
In October 2017, President Trump declared that the opioid crisis was a nationwide “public health emergency” and regulators with the Centers for Medicare % Medicaid Services have already put in place restrictions on opioid dosing through the Medicare Part D program.
In a rule finalized in April 2018, the CMS placed restrictions on the dosage of opioids available for chronic opioid users and limited the days’ supply for first-time opioid users. For chronic users, the CMS set a 90-morphine-milligram-equivalent (MME) per day limit that triggers pharmacist consultation with the prescriber. The agency instructed health plans to implement an “opioid care coordination edit” that would be triggered at 90 MME per day across all opioid prescriptions and would require pharmacists to contact prescribers to override for a higher dosage.
The entire exchange must be documented. The CMS instructed health plans in the Medicare Part D program to implement a “hard safety edit” that limits opioid prescription fills to no more than a 7-day supply for opioid-naive patients being treated for acute pain. The changes are set to take effect in January 2019.
But Diane E. Meier, MD, director of the Center to Advance Palliative Care in New York, said the most current data suggest the risk of addiction and substance use disorder among medically-ill patients taking opioids is less than 10%. “That means that 90% of patients with a serious medical illness can safely take opioids for the relief of pain that is causing functional disorder,” she said.
Policymakers should not conflate the use and prescription of opioids with cases of misuse and abuse, Dr. Alvarnas said. Some patients will require a higher dose of opioids depending on their age or number of pain episodes, or because of their body’s physiological response.
Some policies, such as a prior authorization, create “artificial barriers and delays in getting access to medication” for these patients, Dr. Alvarnas said. “When you create a far more arduous prior [authorization] process or limit prescriptions ... if someone has a severe blood disorder or a bone cancer, then what you’re doing is setting up a system that will fail those patients on a regular basis,” he said.
Patients may also have difficulty finding pharmacies that dispense opioid medications, or doctors who will prescribe them, because of fear of repercussions from the Drug Enforcement Administration or their state licensing boards, Dr. Meier said.
No ‘one-size-fits-all’
Because treatment is individualized, there is no “one-size-fits-all” approach to pain management for patients with hematological diseases.
“One of the concerns is that we care for populations of patients, such as those with sickle cell disease, those with blood cancers that can cause destructive bony lesions like somebody with multiple myeloma might experience, or even pain associated with the complication of a disease like hemophilia, where [patients experience] excruciating pain ... from bleeding into a joint,” Dr. Alvarnas said.
It’s not enough to offer anti-inflammatory medications to these patients – and in some cases doing so may create additional problems, experts said. Contraindications for anti-inflammatory agents tend to be more significant in hematology patients because of low platelet counts, liver disease, and kidney disease. This may prevent them from taking medications such as ibuprofen, acetaminophen, or naproxen sodium. Opioids are the “only option” for patients with these kinds of complications who have severe pain, Dr. Osunkwo said.
Pain management training
While hematologists are trained about the potential risks of common drugs such as steroids, “none of that education and training has occurred” for opioids, Dr. Meier said.
Since hematologists often aren’t trained in pain management, many are uncomfortable with managing pain in their diagnosis and leave the responsibility to a pain specialist, Dr. Osunkwo said. “But the problem is, they know more about the disease than anybody else, and you’ll be safer if [they] are doing the pain management for hematology patients because they know the risk and benefit of the different drugs in light of the diagnosis itself, compared to passing that on to somebody else to manage.”
In the recent policy statement, ASH leaders committed to creating evidence-based guidelines and education activities on pain management.
Finding balance
ASH leaders recognize the longstanding, complex nature of the opioid epidemic and want to be a part of the conversation to ensure their patients’ needs and considerations are met in future legislation, Dr. Alvarnas noted.
Dr. Meier, who is vice chair for public policy at Icahn School of Medicine at Mount Sinai, New York, said the issue of balance is paramount when considering good policymaking.
“No policy at either extreme is the right policy,” Dr. Meier said. “Good policymaking, good public health interventions attempt to achieve some balance ... in preventing harm and maximizing appropriate treatment of vulnerable, seriously ill patients. And the policies we have now don’t achieve that.”
The “Statement on Opioid Use in Patients with Hematologic Diseases and Disorders” is available on the ASH website.
Dr. Alvarnas is chair of the society’s Committee on Practice. Dr. Meier and Dr. Osunkwo reported having no relevant financial disclosures.
Antidepressant therapy after MI, stroke cut CVD events
ORLANDO – Although cardiologists and neurologists aren’t typically the physicians who diagnose and treat major depressive disorder that’s newly identified in patients after MI or stroke, they’re the ones who’ll deal with the cardiovascular consequences if the mood disorder isn’t adequately treated.
That was a key message of a study presented by interventional cardiologist Sripal Bangalore, MD, at the annual meeting of the American College of Cardiology.
In his retrospective cohort study of 1,568 patients diagnosed with and treated for major depressive disorder (MDD) following an initial acute MI or stroke, antidepressant therapy deemed inadequate by either of two prespecified measures was associated during a mean follow-up of 2 years with a 20% higher risk of the primary endpoint – a composite of recurrent MI, stroke, angina, or heart failure – than the risk in patients who received what was judged to be adequate antidepressant pharmacotherapy. A precondition for study inclusion was that a patient could not have been taking any antidepressant during the year prior to the index MI or stroke.
The study utilized nationwide claims data from the Truven Health MarketScan Claims Database for 2010-2015. Depression therapy was considered adequate if during the first 90 days following diagnosis of MDD two conditions were met: a patient aged 65 or younger had to be on the equivalent of at least 20 mg of fluoxetine per day, or if older then on a fluoxetine-equivalent dose of at least 10 mg/day, and pharmacy records had to indicate the patient was covered by the antidepressant prescription for at least 72 of those 90 days.
The prevalence of inadequate antidepressant therapy for MDD by these criteria among these patients with known cardiovascular disease was eyebrow-raisingly high: fully 60%, noted Dr. Bangalore of New York University.
In a multivariate logistic regression analysis adjusted for baseline factors that could affect the propensity to receive adequate antidepressant care, Dr. Bangalore and his coinvestigators broke down the risks of insufficient antidepressant therapy associated with each of the individual components of the composite primary endpoint. The 1.2- and 1.95-fold increased risks of stroke and angina, respectively, were statistically significant. However, the 1.37-fold higher risk of MI and 1.14-fold greater risk of heart failure than in adequately treated patients with MDD, while trending in the same direction, didn’t achieve significance.
Dr. Bangalore reported serving as a consultant to Pfizer, which funded the study, as well as to Abbott, Gilead Sciences, and Merck.
ORLANDO – Although cardiologists and neurologists aren’t typically the physicians who diagnose and treat major depressive disorder that’s newly identified in patients after MI or stroke, they’re the ones who’ll deal with the cardiovascular consequences if the mood disorder isn’t adequately treated.
That was a key message of a study presented by interventional cardiologist Sripal Bangalore, MD, at the annual meeting of the American College of Cardiology.
In his retrospective cohort study of 1,568 patients diagnosed with and treated for major depressive disorder (MDD) following an initial acute MI or stroke, antidepressant therapy deemed inadequate by either of two prespecified measures was associated during a mean follow-up of 2 years with a 20% higher risk of the primary endpoint – a composite of recurrent MI, stroke, angina, or heart failure – than the risk in patients who received what was judged to be adequate antidepressant pharmacotherapy. A precondition for study inclusion was that a patient could not have been taking any antidepressant during the year prior to the index MI or stroke.
The study utilized nationwide claims data from the Truven Health MarketScan Claims Database for 2010-2015. Depression therapy was considered adequate if during the first 90 days following diagnosis of MDD two conditions were met: a patient aged 65 or younger had to be on the equivalent of at least 20 mg of fluoxetine per day, or if older then on a fluoxetine-equivalent dose of at least 10 mg/day, and pharmacy records had to indicate the patient was covered by the antidepressant prescription for at least 72 of those 90 days.
The prevalence of inadequate antidepressant therapy for MDD by these criteria among these patients with known cardiovascular disease was eyebrow-raisingly high: fully 60%, noted Dr. Bangalore of New York University.
In a multivariate logistic regression analysis adjusted for baseline factors that could affect the propensity to receive adequate antidepressant care, Dr. Bangalore and his coinvestigators broke down the risks of insufficient antidepressant therapy associated with each of the individual components of the composite primary endpoint. The 1.2- and 1.95-fold increased risks of stroke and angina, respectively, were statistically significant. However, the 1.37-fold higher risk of MI and 1.14-fold greater risk of heart failure than in adequately treated patients with MDD, while trending in the same direction, didn’t achieve significance.
Dr. Bangalore reported serving as a consultant to Pfizer, which funded the study, as well as to Abbott, Gilead Sciences, and Merck.
ORLANDO – Although cardiologists and neurologists aren’t typically the physicians who diagnose and treat major depressive disorder that’s newly identified in patients after MI or stroke, they’re the ones who’ll deal with the cardiovascular consequences if the mood disorder isn’t adequately treated.
That was a key message of a study presented by interventional cardiologist Sripal Bangalore, MD, at the annual meeting of the American College of Cardiology.
In his retrospective cohort study of 1,568 patients diagnosed with and treated for major depressive disorder (MDD) following an initial acute MI or stroke, antidepressant therapy deemed inadequate by either of two prespecified measures was associated during a mean follow-up of 2 years with a 20% higher risk of the primary endpoint – a composite of recurrent MI, stroke, angina, or heart failure – than the risk in patients who received what was judged to be adequate antidepressant pharmacotherapy. A precondition for study inclusion was that a patient could not have been taking any antidepressant during the year prior to the index MI or stroke.
The study utilized nationwide claims data from the Truven Health MarketScan Claims Database for 2010-2015. Depression therapy was considered adequate if during the first 90 days following diagnosis of MDD two conditions were met: a patient aged 65 or younger had to be on the equivalent of at least 20 mg of fluoxetine per day, or if older then on a fluoxetine-equivalent dose of at least 10 mg/day, and pharmacy records had to indicate the patient was covered by the antidepressant prescription for at least 72 of those 90 days.
The prevalence of inadequate antidepressant therapy for MDD by these criteria among these patients with known cardiovascular disease was eyebrow-raisingly high: fully 60%, noted Dr. Bangalore of New York University.
In a multivariate logistic regression analysis adjusted for baseline factors that could affect the propensity to receive adequate antidepressant care, Dr. Bangalore and his coinvestigators broke down the risks of insufficient antidepressant therapy associated with each of the individual components of the composite primary endpoint. The 1.2- and 1.95-fold increased risks of stroke and angina, respectively, were statistically significant. However, the 1.37-fold higher risk of MI and 1.14-fold greater risk of heart failure than in adequately treated patients with MDD, while trending in the same direction, didn’t achieve significance.
Dr. Bangalore reported serving as a consultant to Pfizer, which funded the study, as well as to Abbott, Gilead Sciences, and Merck.
REPORTING FROM ACC 2018
Key clinical point: Make sure patients with newly diagnosed depression post-MI or stroke are getting adequate antidepressant therapy from their primary care physician or psychiatrist.
Major finding: Patients with a first MI or stroke subsequently diagnosed with major depressive disorder were 20% more likely to experience a recurrent cardiovascular event if their antidepressant therapy was judged insufficient than if adequate.
Study details: This was a retrospective cohort study of health insurance claims data for 1,568 patients with an initial diagnosis of MI or stroke who were subsequently diagnosed with and treated for major depressive disorder.
Disclosures: The presenter reported serving as a consultant to Pfizer, which funded the study.
Shingles hospitalization occurs more often among IBD patients
WASHINGTON –

This elevated risk for patients with inflammatory bowel disease (IBD) to develop a herpes zoster virus (HZV) reactivation severe enough to put them in the hospital makes it especially important for IBD patients to receive immunization against shingles, especially now that a more effective vaccine is available, Daniela G. Vinsard, MD, said at the annual Digestive Disease Week®. Ideally, IBD patients should receive the full course of the adjuvanted, recombinant zoster vaccine Shingrix before starting an immunosuppressive regimen, said Dr. Vinsard, a physician at the University of Connecticut, Farmington.
This finding, which underscored the susceptibility of IBD patients to shingles because of their immunosuppressive treatments and the importance of vaccination, recently became even more relevant when the Food and Drug Administration approved tofacitinib (Xeljanz) to treat ulcerative colitis in late May, commented Gil Y. Melmed, MD, director of clinical inflammatory bowel disease at Cedars-Sinai Medical Center, Los Angeles. Tofacitinib, which may be an attractive option to some patients as an oral immunomodulator, carries a black box warning about the added risk for certain serious infections while taking the drug, including HZV. Recent recommendations from the American College of Gastroenterology said that IBD patients aged 51 years or older should “strongly consider” HZV vaccination, including immunosuppressed patients (Am J Gastroenterol. 2017 Feb; 112[2]:241-58). The introduction of a potentially popular drug for ulcerative colitis that’s known to pose a risk for shingles might lead to a stronger recommendation for vaccination in the near future, Dr. Melmed said in an interview.
The study Dr. Vinsard reported used data collected by the National Inpatient Sample from 2012 to September 2015, which represented, with weighting, more than 142 million hospitalized American patients. From this data set she and her associates identified 7,180 IBD patients hospitalized with a primary diagnosis of a vaccine-preventable disease, and about 589,000 weighted patients hospitalized for a vaccine-preventable disease but without IBD. The selection also focused on patients aged 18-65 years. Dr. Vinsard said that she excluded older patients to eliminate advanced age as a cause of immunosuppression.

In a multivariate analysis that controlled for diabetes, HIV infection, cancer, and transplantation, the IBD patients had more than twice the rate of hospitalization for shingles, compared with the patients without IBD, Dr. Vinsard said. When broken down by specific disease type, the rate of HZV infection was 110% higher among ulcerative colitis patients, compared with the general population, and was 140% higher in Crohn’s disease patients, both statistically significant differences.
An additional finding from the analysis was that during the 4 years of study, the rate of hospitalizations of IBD patients for influenza steadily rose, from about 10% in 2012 to nearly 30% in 2015.
Dr. Vinsard reported no disclosures. Dr. Melmed reported consulting with Pfizer, the company that markets tofacitinib, and with several other companies that market biological agents.
WASHINGTON –
This elevated risk for patients with inflammatory bowel disease (IBD) to develop a herpes zoster virus (HZV) reactivation severe enough to put them in the hospital makes it especially important for IBD patients to receive immunization against shingles, especially now that a more effective vaccine is available, Daniela G. Vinsard, MD, said at the annual Digestive Disease Week®. Ideally, IBD patients should receive the full course of the adjuvanted, recombinant zoster vaccine Shingrix before starting an immunosuppressive regimen, said Dr. Vinsard, a physician at the University of Connecticut, Farmington.
This finding, which underscored the susceptibility of IBD patients to shingles because of their immunosuppressive treatments and the importance of vaccination, recently became even more relevant when the Food and Drug Administration approved tofacitinib (Xeljanz) to treat ulcerative colitis in late May, commented Gil Y. Melmed, MD, director of clinical inflammatory bowel disease at Cedars-Sinai Medical Center, Los Angeles. Tofacitinib, which may be an attractive option to some patients as an oral immunomodulator, carries a black box warning about the added risk for certain serious infections while taking the drug, including HZV. Recent recommendations from the American College of Gastroenterology said that IBD patients aged 51 years or older should “strongly consider” HZV vaccination, including immunosuppressed patients (Am J Gastroenterol. 2017 Feb; 112[2]:241-58). The introduction of a potentially popular drug for ulcerative colitis that’s known to pose a risk for shingles might lead to a stronger recommendation for vaccination in the near future, Dr. Melmed said in an interview.
The study Dr. Vinsard reported used data collected by the National Inpatient Sample from 2012 to September 2015, which represented, with weighting, more than 142 million hospitalized American patients. From this data set she and her associates identified 7,180 IBD patients hospitalized with a primary diagnosis of a vaccine-preventable disease, and about 589,000 weighted patients hospitalized for a vaccine-preventable disease but without IBD. The selection also focused on patients aged 18-65 years. Dr. Vinsard said that she excluded older patients to eliminate advanced age as a cause of immunosuppression.
In a multivariate analysis that controlled for diabetes, HIV infection, cancer, and transplantation, the IBD patients had more than twice the rate of hospitalization for shingles, compared with the patients without IBD, Dr. Vinsard said. When broken down by specific disease type, the rate of HZV infection was 110% higher among ulcerative colitis patients, compared with the general population, and was 140% higher in Crohn’s disease patients, both statistically significant differences.
An additional finding from the analysis was that during the 4 years of study, the rate of hospitalizations of IBD patients for influenza steadily rose, from about 10% in 2012 to nearly 30% in 2015.
Dr. Vinsard reported no disclosures. Dr. Melmed reported consulting with Pfizer, the company that markets tofacitinib, and with several other companies that market biological agents.
WASHINGTON –
This elevated risk for patients with inflammatory bowel disease (IBD) to develop a herpes zoster virus (HZV) reactivation severe enough to put them in the hospital makes it especially important for IBD patients to receive immunization against shingles, especially now that a more effective vaccine is available, Daniela G. Vinsard, MD, said at the annual Digestive Disease Week®. Ideally, IBD patients should receive the full course of the adjuvanted, recombinant zoster vaccine Shingrix before starting an immunosuppressive regimen, said Dr. Vinsard, a physician at the University of Connecticut, Farmington.
This finding, which underscored the susceptibility of IBD patients to shingles because of their immunosuppressive treatments and the importance of vaccination, recently became even more relevant when the Food and Drug Administration approved tofacitinib (Xeljanz) to treat ulcerative colitis in late May, commented Gil Y. Melmed, MD, director of clinical inflammatory bowel disease at Cedars-Sinai Medical Center, Los Angeles. Tofacitinib, which may be an attractive option to some patients as an oral immunomodulator, carries a black box warning about the added risk for certain serious infections while taking the drug, including HZV. Recent recommendations from the American College of Gastroenterology said that IBD patients aged 51 years or older should “strongly consider” HZV vaccination, including immunosuppressed patients (Am J Gastroenterol. 2017 Feb; 112[2]:241-58). The introduction of a potentially popular drug for ulcerative colitis that’s known to pose a risk for shingles might lead to a stronger recommendation for vaccination in the near future, Dr. Melmed said in an interview.
The study Dr. Vinsard reported used data collected by the National Inpatient Sample from 2012 to September 2015, which represented, with weighting, more than 142 million hospitalized American patients. From this data set she and her associates identified 7,180 IBD patients hospitalized with a primary diagnosis of a vaccine-preventable disease, and about 589,000 weighted patients hospitalized for a vaccine-preventable disease but without IBD. The selection also focused on patients aged 18-65 years. Dr. Vinsard said that she excluded older patients to eliminate advanced age as a cause of immunosuppression.
In a multivariate analysis that controlled for diabetes, HIV infection, cancer, and transplantation, the IBD patients had more than twice the rate of hospitalization for shingles, compared with the patients without IBD, Dr. Vinsard said. When broken down by specific disease type, the rate of HZV infection was 110% higher among ulcerative colitis patients, compared with the general population, and was 140% higher in Crohn’s disease patients, both statistically significant differences.
An additional finding from the analysis was that during the 4 years of study, the rate of hospitalizations of IBD patients for influenza steadily rose, from about 10% in 2012 to nearly 30% in 2015.
Dr. Vinsard reported no disclosures. Dr. Melmed reported consulting with Pfizer, the company that markets tofacitinib, and with several other companies that market biological agents.
REPORTING FROM DDW 2018
Key clinical point: Patients with inflammatory bowel disease have an increased risk for shingles that results in hospitalization.
Major finding: Patients with IBD hospitalized for a vaccine-preventable infection had twice the rate of shingles as the general population.
Study details: A review of data collected by the U.S. National Inpatient Sample during 2012-2015.
Disclosures: Dr. Vinsard reported no disclosures. Dr. Melmed reported consulting with Pfizer, the company that markets tofacitinib (Xeljanz), and with several other companies that market biological agents.
Imiquimod-Induced Hypopigmentation Following Treatment of Periungual Verruca Vulgaris
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.

 and the right elbow (B).")
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
Practice Points
- Imiquimod commonly is used off label to treat viral and neoplastic processes.
- Clinicians should be aware of the potential for dyspigmentation or depigmentation as a side effect from treatment.
Avelumab does not add punch to ALK inhibitors for ALK+/– NSCLC
CHICAGO – The combination of the immune checkpoint inhibitor avelumab (Bavencio) and the ALK inhibitor lorlatinib in ALK-positive patients was associated with an acceptable safety profile and good activity – albeit not better than lorlatinib alone – in one arm of the phase 1/2b JAVELIN Lung 101 trial.
In contrast, although preclinical data suggested that the combination of an ALK inhibitor and immune checkpoint inhibitor might have synergistic activity in patients with advanced ALK-negative non–small-cell lung cancer (NSCLC), it didn’t pan out in the second arm of the trial, reported Alice T. Shaw, MD, PhD, of Massachusetts General Hospital Cancer Center in Boston.

In the parallel group trial testing combinations of the programmed death ligand-1 (PD-L1) inhibitor avelumab with either of two tyrosine kinase inhibitors (TKIs) – crizotinib (Xalkori) or lorlatinib – the combination of avelumab and crizotinib had an objective response rate (ORR) of just 16.7% in ALK-negative patients, and 5 of 12 patients in this study arm had dose limiting toxicities (DLTs) due to serious adverse events.
“The most common DLTs were increased transaminases, consistent with the recent report of increased hepatotoxicity with the combination of nivolumab and crizotinib in Checkmate 370. While there were two confirmed partial responses, this efficacy would be expected for avelumab alone. No further development of this combination is planned,” Dr. Shaw said in an oral abstract session at the annual meeting of the American Society of Clinical Oncology.
In contrast, in ALK-positive patients, avelumab/lorlatinib was associated with an ORR of 46.4%, although it’s likely that the responses were attributable to lorlatinib alone, and not its anti-PD-L1 partner, she acknowledged.
Synergism sought
The investigators based the study on two hypotheses: The first was that ALK inhibitors, through their immunomodulatory properties, combined with checkpoint inhibitors, could have synergistic activity against non-ALK-driver NSCLC, hence the combination of avelumab and crizotinib in these patients.
Their second hypothesis was that a combination of an ALK inhibitor and checkpoint inhibitor could lead to enhanced efficacy in patients with previously treated ALK-positive NSCLC. To test this combination, they chose to pair avelumab with lorlatinib, a third-generation ALK-targeting TKI with the ability to penetrate the central nervous system. Dr. Shaw and her colleagues had previously shown in a phase 1 trial that this agent has potent activity against ALK-driven tumors with resistance mutations.
Two groups, two TKIs, one PD-L1 inhibitor
Group A in JAVELIN LUNG 101 included 12 patients with ALK-negative NSCLC, no known ROS1 gene rearrangement, c-MET gene amplification, or c-MET exon 14, skipping who had received at least one prior line of systemic therapy, and no prior checkpoint inhibitor. These patients were treated with avelumab 10 mg/kg over a 1-hour IV infusion every 2 weeks, plus 250 mg oral crizotinib twice daily.
Group B included 28 patients with advanced ALK-positive NSCLC, any number of prior regimens (or none), and no prior checkpoint inhibitor therapy. Patients with asymptomatic untreated brain metastases were eligible for treatment. These patients received avelumab at the same dose and schedule as in group A, plus oral lorlatinib 100 mg once daily.
In both arms, patients were assessed for maximum tolerated dose (MTD) and recommended phase 2 doses, DLTs, safety and tolerability, and antitumor activity.
There were no DLTs among 25 patients evaluable for this assessment in the avelumab/lorlatinib arm. In contrast, five patients had DLTs in the avelumab/crizotinib arm, included four transaminases increases and one case each of febrile neutropenia, hepatitis, QT interval prolongation, and rash.
Adverse events of any grade occurred in all patients in group A and in 27 of 28 patients (96.4%) in group B. Grade 3 or greater adverse events occurred in 58.3% and 53.6% of patients, respectively.
In group A, treatment-related serious adverse events were febrile neutropenia, hepatitis, and rash. In group B, these events included pneumonitis, elevated alanine aminotransferase, delirium, fatal dyspnea (one case), and pericardial effusion.
Antitumor activity
In group A (ALK-negative NSCLC), two patients had a partial response (PR), five had stable disease, and five had progressive disease, for an ORR of 16.7. The median time to response was 1.4 months, and the median duration of response was 4.1 months.
In group B (ALK-positive NSCLC). There was one complete response, 12 PRs, six cases of stable disease, and seven of progressive disease. Two patients in this arm were not evaluable for response at the time of the data cutoff in October 2017. The ORR in this arm was 46.4%, median time to response was 1.9 months, and the median duration of response was 7.4 months. Dr. Shaw cautioned, however, that the 95% confidence interval for duration of response in this group was wide (3.7 months to not estimable), because the data were not yet mature and the number of patients was small.
“Longer follow-up will be important to establish the true durability of these responses and to better assess the potential benefit of combined avelumab and lorlatinib in ALK-positive lung cancer,” Dr. Shaw said.

But invited discussant Leora Horn, MD, MSc, of Vanderbilt University Medical Center in Nashville, Tenn., cast doubt on the ALK inhibitor/checkpoint inhibitor combination compared with targeted therapy alone.
“We were hoping to see that combination therapy with ALK tyrosine kinase inhibitors and immune checkpoint inhibitors are superior to therapy with an ALK tyrosine kinase inhibitor alone, that combination therapy is safe with a manageable toxicity profile, and lastly, that combination therapy with an ALK TKI and immune checkpoint inhibitor is superior to immune checkpoint inhibitor alone in ALK wild type patients,” she said.
She noted that previous phase 1 data with lorlatinib monotherapy in NSCLC showed an ORR of 46%, compared with 46.4% seen with the addition of avelumab to lorlatinib.
“So where do we go from here? We’ve seen that combination therapy with ALK tyrosine kinase inhibitors and immune checkpoint inhibitors are not the optimal therapeutic strategy in ALK-positive non–small-cell lung cancer. It is difficult to improve on a 70% response rate [with TKIs]. The progression-free survival, which is more important, was also not improved,” she said.
The mechanisms of additive toxicities between TKIs and checkpoints are not well understood, and she suggested that “further studies with biopsies exploring the tumor microenvironment in ALK or other driver-positive non–small-cell lung cancer prior to and after therapy with a TKI may help us better define the optimal combination strategy going forward.”
In a panel discussion following her talk, Dr. Shaw was asked what proportion of the responses her team saw could be attributed to lorlatinib rather than avelumab.
“As we showed, both the response rates and the duration of response were actually pretty similar to what was seen in our previous phase 1/2 study of lorlatinib alone. So one could say that perhaps all of the response that was seen with lorlatinib/avelumab was due to the lorlatinib,” she replied.
She added that several patients have ongoing responses, and that longer follow-up may reveal a benefit for the combination in terms of duration of response.
SOURCE: Shaw AT et al. ASCO 2018, Abstract 9008.
CHICAGO – The combination of the immune checkpoint inhibitor avelumab (Bavencio) and the ALK inhibitor lorlatinib in ALK-positive patients was associated with an acceptable safety profile and good activity – albeit not better than lorlatinib alone – in one arm of the phase 1/2b JAVELIN Lung 101 trial.
In contrast, although preclinical data suggested that the combination of an ALK inhibitor and immune checkpoint inhibitor might have synergistic activity in patients with advanced ALK-negative non–small-cell lung cancer (NSCLC), it didn’t pan out in the second arm of the trial, reported Alice T. Shaw, MD, PhD, of Massachusetts General Hospital Cancer Center in Boston.
In the parallel group trial testing combinations of the programmed death ligand-1 (PD-L1) inhibitor avelumab with either of two tyrosine kinase inhibitors (TKIs) – crizotinib (Xalkori) or lorlatinib – the combination of avelumab and crizotinib had an objective response rate (ORR) of just 16.7% in ALK-negative patients, and 5 of 12 patients in this study arm had dose limiting toxicities (DLTs) due to serious adverse events.
“The most common DLTs were increased transaminases, consistent with the recent report of increased hepatotoxicity with the combination of nivolumab and crizotinib in Checkmate 370. While there were two confirmed partial responses, this efficacy would be expected for avelumab alone. No further development of this combination is planned,” Dr. Shaw said in an oral abstract session at the annual meeting of the American Society of Clinical Oncology.
In contrast, in ALK-positive patients, avelumab/lorlatinib was associated with an ORR of 46.4%, although it’s likely that the responses were attributable to lorlatinib alone, and not its anti-PD-L1 partner, she acknowledged.
Synergism sought
The investigators based the study on two hypotheses: The first was that ALK inhibitors, through their immunomodulatory properties, combined with checkpoint inhibitors, could have synergistic activity against non-ALK-driver NSCLC, hence the combination of avelumab and crizotinib in these patients.
Their second hypothesis was that a combination of an ALK inhibitor and checkpoint inhibitor could lead to enhanced efficacy in patients with previously treated ALK-positive NSCLC. To test this combination, they chose to pair avelumab with lorlatinib, a third-generation ALK-targeting TKI with the ability to penetrate the central nervous system. Dr. Shaw and her colleagues had previously shown in a phase 1 trial that this agent has potent activity against ALK-driven tumors with resistance mutations.
Two groups, two TKIs, one PD-L1 inhibitor
Group A in JAVELIN LUNG 101 included 12 patients with ALK-negative NSCLC, no known ROS1 gene rearrangement, c-MET gene amplification, or c-MET exon 14, skipping who had received at least one prior line of systemic therapy, and no prior checkpoint inhibitor. These patients were treated with avelumab 10 mg/kg over a 1-hour IV infusion every 2 weeks, plus 250 mg oral crizotinib twice daily.
Group B included 28 patients with advanced ALK-positive NSCLC, any number of prior regimens (or none), and no prior checkpoint inhibitor therapy. Patients with asymptomatic untreated brain metastases were eligible for treatment. These patients received avelumab at the same dose and schedule as in group A, plus oral lorlatinib 100 mg once daily.
In both arms, patients were assessed for maximum tolerated dose (MTD) and recommended phase 2 doses, DLTs, safety and tolerability, and antitumor activity.
There were no DLTs among 25 patients evaluable for this assessment in the avelumab/lorlatinib arm. In contrast, five patients had DLTs in the avelumab/crizotinib arm, included four transaminases increases and one case each of febrile neutropenia, hepatitis, QT interval prolongation, and rash.
Adverse events of any grade occurred in all patients in group A and in 27 of 28 patients (96.4%) in group B. Grade 3 or greater adverse events occurred in 58.3% and 53.6% of patients, respectively.
In group A, treatment-related serious adverse events were febrile neutropenia, hepatitis, and rash. In group B, these events included pneumonitis, elevated alanine aminotransferase, delirium, fatal dyspnea (one case), and pericardial effusion.
Antitumor activity
In group A (ALK-negative NSCLC), two patients had a partial response (PR), five had stable disease, and five had progressive disease, for an ORR of 16.7. The median time to response was 1.4 months, and the median duration of response was 4.1 months.
In group B (ALK-positive NSCLC). There was one complete response, 12 PRs, six cases of stable disease, and seven of progressive disease. Two patients in this arm were not evaluable for response at the time of the data cutoff in October 2017. The ORR in this arm was 46.4%, median time to response was 1.9 months, and the median duration of response was 7.4 months. Dr. Shaw cautioned, however, that the 95% confidence interval for duration of response in this group was wide (3.7 months to not estimable), because the data were not yet mature and the number of patients was small.
“Longer follow-up will be important to establish the true durability of these responses and to better assess the potential benefit of combined avelumab and lorlatinib in ALK-positive lung cancer,” Dr. Shaw said.
But invited discussant Leora Horn, MD, MSc, of Vanderbilt University Medical Center in Nashville, Tenn., cast doubt on the ALK inhibitor/checkpoint inhibitor combination compared with targeted therapy alone.
“We were hoping to see that combination therapy with ALK tyrosine kinase inhibitors and immune checkpoint inhibitors are superior to therapy with an ALK tyrosine kinase inhibitor alone, that combination therapy is safe with a manageable toxicity profile, and lastly, that combination therapy with an ALK TKI and immune checkpoint inhibitor is superior to immune checkpoint inhibitor alone in ALK wild type patients,” she said.
She noted that previous phase 1 data with lorlatinib monotherapy in NSCLC showed an ORR of 46%, compared with 46.4% seen with the addition of avelumab to lorlatinib.
“So where do we go from here? We’ve seen that combination therapy with ALK tyrosine kinase inhibitors and immune checkpoint inhibitors are not the optimal therapeutic strategy in ALK-positive non–small-cell lung cancer. It is difficult to improve on a 70% response rate [with TKIs]. The progression-free survival, which is more important, was also not improved,” she said.
The mechanisms of additive toxicities between TKIs and checkpoints are not well understood, and she suggested that “further studies with biopsies exploring the tumor microenvironment in ALK or other driver-positive non–small-cell lung cancer prior to and after therapy with a TKI may help us better define the optimal combination strategy going forward.”
In a panel discussion following her talk, Dr. Shaw was asked what proportion of the responses her team saw could be attributed to lorlatinib rather than avelumab.
“As we showed, both the response rates and the duration of response were actually pretty similar to what was seen in our previous phase 1/2 study of lorlatinib alone. So one could say that perhaps all of the response that was seen with lorlatinib/avelumab was due to the lorlatinib,” she replied.
She added that several patients have ongoing responses, and that longer follow-up may reveal a benefit for the combination in terms of duration of response.
SOURCE: Shaw AT et al. ASCO 2018, Abstract 9008.
CHICAGO – The combination of the immune checkpoint inhibitor avelumab (Bavencio) and the ALK inhibitor lorlatinib in ALK-positive patients was associated with an acceptable safety profile and good activity – albeit not better than lorlatinib alone – in one arm of the phase 1/2b JAVELIN Lung 101 trial.
In contrast, although preclinical data suggested that the combination of an ALK inhibitor and immune checkpoint inhibitor might have synergistic activity in patients with advanced ALK-negative non–small-cell lung cancer (NSCLC), it didn’t pan out in the second arm of the trial, reported Alice T. Shaw, MD, PhD, of Massachusetts General Hospital Cancer Center in Boston.
In the parallel group trial testing combinations of the programmed death ligand-1 (PD-L1) inhibitor avelumab with either of two tyrosine kinase inhibitors (TKIs) – crizotinib (Xalkori) or lorlatinib – the combination of avelumab and crizotinib had an objective response rate (ORR) of just 16.7% in ALK-negative patients, and 5 of 12 patients in this study arm had dose limiting toxicities (DLTs) due to serious adverse events.
“The most common DLTs were increased transaminases, consistent with the recent report of increased hepatotoxicity with the combination of nivolumab and crizotinib in Checkmate 370. While there were two confirmed partial responses, this efficacy would be expected for avelumab alone. No further development of this combination is planned,” Dr. Shaw said in an oral abstract session at the annual meeting of the American Society of Clinical Oncology.
In contrast, in ALK-positive patients, avelumab/lorlatinib was associated with an ORR of 46.4%, although it’s likely that the responses were attributable to lorlatinib alone, and not its anti-PD-L1 partner, she acknowledged.
Synergism sought
The investigators based the study on two hypotheses: The first was that ALK inhibitors, through their immunomodulatory properties, combined with checkpoint inhibitors, could have synergistic activity against non-ALK-driver NSCLC, hence the combination of avelumab and crizotinib in these patients.
Their second hypothesis was that a combination of an ALK inhibitor and checkpoint inhibitor could lead to enhanced efficacy in patients with previously treated ALK-positive NSCLC. To test this combination, they chose to pair avelumab with lorlatinib, a third-generation ALK-targeting TKI with the ability to penetrate the central nervous system. Dr. Shaw and her colleagues had previously shown in a phase 1 trial that this agent has potent activity against ALK-driven tumors with resistance mutations.
Two groups, two TKIs, one PD-L1 inhibitor
Group A in JAVELIN LUNG 101 included 12 patients with ALK-negative NSCLC, no known ROS1 gene rearrangement, c-MET gene amplification, or c-MET exon 14, skipping who had received at least one prior line of systemic therapy, and no prior checkpoint inhibitor. These patients were treated with avelumab 10 mg/kg over a 1-hour IV infusion every 2 weeks, plus 250 mg oral crizotinib twice daily.
Group B included 28 patients with advanced ALK-positive NSCLC, any number of prior regimens (or none), and no prior checkpoint inhibitor therapy. Patients with asymptomatic untreated brain metastases were eligible for treatment. These patients received avelumab at the same dose and schedule as in group A, plus oral lorlatinib 100 mg once daily.
In both arms, patients were assessed for maximum tolerated dose (MTD) and recommended phase 2 doses, DLTs, safety and tolerability, and antitumor activity.
There were no DLTs among 25 patients evaluable for this assessment in the avelumab/lorlatinib arm. In contrast, five patients had DLTs in the avelumab/crizotinib arm, included four transaminases increases and one case each of febrile neutropenia, hepatitis, QT interval prolongation, and rash.
Adverse events of any grade occurred in all patients in group A and in 27 of 28 patients (96.4%) in group B. Grade 3 or greater adverse events occurred in 58.3% and 53.6% of patients, respectively.
In group A, treatment-related serious adverse events were febrile neutropenia, hepatitis, and rash. In group B, these events included pneumonitis, elevated alanine aminotransferase, delirium, fatal dyspnea (one case), and pericardial effusion.
Antitumor activity
In group A (ALK-negative NSCLC), two patients had a partial response (PR), five had stable disease, and five had progressive disease, for an ORR of 16.7. The median time to response was 1.4 months, and the median duration of response was 4.1 months.
In group B (ALK-positive NSCLC). There was one complete response, 12 PRs, six cases of stable disease, and seven of progressive disease. Two patients in this arm were not evaluable for response at the time of the data cutoff in October 2017. The ORR in this arm was 46.4%, median time to response was 1.9 months, and the median duration of response was 7.4 months. Dr. Shaw cautioned, however, that the 95% confidence interval for duration of response in this group was wide (3.7 months to not estimable), because the data were not yet mature and the number of patients was small.
“Longer follow-up will be important to establish the true durability of these responses and to better assess the potential benefit of combined avelumab and lorlatinib in ALK-positive lung cancer,” Dr. Shaw said.
But invited discussant Leora Horn, MD, MSc, of Vanderbilt University Medical Center in Nashville, Tenn., cast doubt on the ALK inhibitor/checkpoint inhibitor combination compared with targeted therapy alone.
“We were hoping to see that combination therapy with ALK tyrosine kinase inhibitors and immune checkpoint inhibitors are superior to therapy with an ALK tyrosine kinase inhibitor alone, that combination therapy is safe with a manageable toxicity profile, and lastly, that combination therapy with an ALK TKI and immune checkpoint inhibitor is superior to immune checkpoint inhibitor alone in ALK wild type patients,” she said.
She noted that previous phase 1 data with lorlatinib monotherapy in NSCLC showed an ORR of 46%, compared with 46.4% seen with the addition of avelumab to lorlatinib.
“So where do we go from here? We’ve seen that combination therapy with ALK tyrosine kinase inhibitors and immune checkpoint inhibitors are not the optimal therapeutic strategy in ALK-positive non–small-cell lung cancer. It is difficult to improve on a 70% response rate [with TKIs]. The progression-free survival, which is more important, was also not improved,” she said.
The mechanisms of additive toxicities between TKIs and checkpoints are not well understood, and she suggested that “further studies with biopsies exploring the tumor microenvironment in ALK or other driver-positive non–small-cell lung cancer prior to and after therapy with a TKI may help us better define the optimal combination strategy going forward.”
In a panel discussion following her talk, Dr. Shaw was asked what proportion of the responses her team saw could be attributed to lorlatinib rather than avelumab.
“As we showed, both the response rates and the duration of response were actually pretty similar to what was seen in our previous phase 1/2 study of lorlatinib alone. So one could say that perhaps all of the response that was seen with lorlatinib/avelumab was due to the lorlatinib,” she replied.
She added that several patients have ongoing responses, and that longer follow-up may reveal a benefit for the combination in terms of duration of response.
SOURCE: Shaw AT et al. ASCO 2018, Abstract 9008.
REPORTING FROM ASCO 2018
Key clinical point: Adding an anti-PD-L1 checkpoint inhibitor to an ALK inhibitor did not add efficacy in patients with either ALK-negative or ALK-positive non–small-cell lung cancer.
Major finding: The ORR in patients treated with avelumab/lorlatinib was 46.4%, the same as ORR with lorlatinib alone.
Study details: Phase 1/2 trial of avelumab/crizotinib in 12 patients with ALK-negative NSCLC, and avelumab/lorlatinib in 28 patients with ALK-positive NSCLC.
Disclosures: Pfizer sponsored the trial. Dr. Shaw disclosed consultancy/advisory board membership, researching funding, and honoraria from Pfizer and other companies. Dr. Horn has previously disclosed serving as a consultant to AbbVie, BMS, Genentech, Merck, and AstraZeneca.
Source: Shaw AT et al. ASCO 2018, Abstract 9008.
Unusual Presentation of Erythema Elevatum Diutinum With Underlying Hepatitis B Infection
Erythema elevatum diutinum (EED) manifests on a clinicopathologic spectrum of chronic cutaneous small vessel vasculitis. The lesions typically present as persistent, symmetric, firm, red to purple papules or nodules on the extensor arms and dorsal hands.1,2 Underlying infectious, malignant, or autoimmune processes are commonly associated with the disease, notably Streptococcus infection and IgA monoclonal gammopathy.2,3 Hepatitis virus also is often implicated in association with EED. Cases of EED have been seen with concomitant human immunodeficiency virus (HIV) infection.4-6 We report a case of EED presenting in various stages of evolution associated with underlying hepatitis B infection alone.
Case Report
A 57-year-old man originally presented to an outpatient dermatology practice with a nodular, painful, episodic rash on the trunk and upper and lower extremities. A biopsy revealed leukocytoclastic vasculitis (LCV) with prominent eosinophils. At the time, the skin findings were believed to be a manifestation of drug hypersensitivity, likely to opioid use. The patient was lost to follow-up.
Seven years later, the patient was admitted to the hospital with new-onset burning and stinging red nodules on the dorsum of the hands and persistence of the original episodic rash over the lower legs and bilateral flanks. In the interim, he was briefly treated with an oral prednisone taper and topical corticosteroids including triamcinolone cream 0.1% and clobetasol cream 0.05% without improvement.
On examination deep red to violaceous discrete nodules and plaques with overlying hyperkeratosis involving all distal and proximal interphalangeal joints of the hands and extensor elbows were seen (Figure 1A). On the bilateral posterior arms (Figure 1B), anterior legs, and periumbilical area were deeply erythematous papules and plaques with background hyperpigmentation. Across his lower back and bilateral flanks were erythematous papules with central hemorrhagic crusting (Figure 1C).
Pertinent laboratory findings included a positive hepatitis B surface antigen with hepatitis B DNA value 4,313,876 IU/mL and a hepatitis B virus quantitative polymerase chain reaction value of 6.64 U. The etiology was suspected to be intravenous drug abuse; however, the patient denied recreational drug use.
An additional infectious workup was negative for hepatitis C, streptococcus, syphilis, tuberculosis, and HIV. A complete blood cell count, complete metabolic panel, urinalysis, complement, cryoglobulins, and serum protein electrophoresis were within reference range. Autoimmune serologies were negative including antinuclear antibody, rheumatoid factor, anti-Sjögren syndrome–related antigen A and B, anticyclic citrullinated peptide, anti-Smith, and antineutrophilic cytoplasmic antibodies. Peripheral blood immunophenotyping, lactate dehydrogenase, quantitative immunoglobulins, and age-appropriate cancer screens did not demonstrate evidence for malignancy underlying the disease. Bilateral hand radiographs showed mild periostitis of the proximal phalanges without obvious erosions.
Three 4-mm punch biopsies were performed from the left fifth digit, left posterior arm, and left flank. Tissue of the left fifth digit showed an intradermal vascular proliferation with a concentric pattern resembling onion skin in a background of increased fibrosis. The blood vessels showed focal fibrinoid necrosis (Figure 2A). The biopsy of the left posterior arm showed an intradermal vascular proliferation with an associated mild acute and chronic perivascular inflammation (Figure 2B). The left flank biopsy showed LCV with focal epidermal necrosis (Figure 2C).
The constellation of clinical findings together with the histopathologic changes represented EED in various stages of evolution. The patient was started on dapsone 100 mg daily and referred to the infectious disease service for treatment of chronic hepatitis B; however, he was subsequently lost to follow-up.
Comment
Overview of EED
Erythema elevatum diutinum represents a rare form of chronic cutaneous small vessel vasculitis. Originally described by Hutchinson7 and Bury8 as symmetric purpuric nodules of the skin, it was later named by Crocker and Williams9 in 1894. The disease classically presents as firm, fixed, red-brown to violaceous papules, plaques, and nodules affecting the extensor upper or lower extremities.1 Lesions are most commonly found symmetrically overlying joints of the hands, feet, elbows, and knees, as well as the Achilles tendon and buttocks.3 Less common locations include the palms and soles, face,10,11 trunk,12 and periauricular region.1 Although they are typically asymptomatic, sensations such as burning, stinging, and pruritus have been noted.1 Our patient was unique because in addition to typical lesions of EED, he presented with crusted papules on the flanks and violaceous papules of the lower legs and periumbilicus.
Etiology
Originally associated with Streptococcus as isolated from EED lesions,3,13 additional infectious etiologies include viral hepatitis,4-6 human herpesvirus 6,14 and rarely HIV.1,15 Hepatitis B and C are well known to be associated with EED, with only rare reports in patients with concomitant HIV infection. Erythema elevatum diutinum also has been described in relationship to myeloproliferative disorders and hematologic malignancies such as IgA myeloma,16 non-Hodgkin lymphoma,17 chronic lymphocytic leukemia,18 and hypergammaglobulinemia.19 In a study of 13 patients with EED, 4 had associated underlying IgA monoclonal gammopathy.2 Autoimmune conditions such as rheumatoid arthritis,20 ulcerative colitis,21 relapsing polychondritis,22 and systemic lupus erythematosus23 also have been implicated.
Pathogenesis
Although the precise pathogenesis of EED remains unknown, it has been suggested that a complement cascade initiated by immune-complex deposition in postcapillary venules induces an LCV.24,25 Chronic antigenic exposure or high antibody levels26 in the face of infections, autoimmune disease, or malignancy may incite this immune-complex reaction. Skin lesions seen in association with hepatitis reflect circulating immune-complex deposition in vessel walls causing destruction. It has been postulated that the duration of immune complexemia may be sufficient to account for the differences in the type of vascular injury seen in acute versus chronic infection.27
Histopathology
Erythema elevatum diutinum may present on a histopathologic spectrum of LCV, as manifested in our patient. Early lesions show predominantly polymorphonuclear cells with nuclear dust pattern in a wedge-shaped infiltrate with fibrin deposition in the superficial and mid dermis.2,3 Later lesions show vasculitis in addition to dermal aggregates of lymphocytes, neutrophils, fibrosis, and areas of granulation tissue. The fibrosis may be dense and comprised of fibroblasts and myofibroblasts.28 Newly formed vessels within the granulation tissue have been postulated to be more susceptible to immune-complex deposition, thus potentiating the process.1,29
Management
Spontaneous resolution of EED may occur, albeit after a prolonged and recurrent course of up to 5 to 10 years.30 Treatment of the underlying cause, when identified, remains paramount. First-line therapy includes dapsone, shown to be effective in reducing lesion size to complete resolution in 80% of the 47 cases reviewed by Momen et al.31 Dapsone monotherapy tends to be less effective in treating nodular lesions associated with HIV-positivity, likely due to the extensive fibrosis.4,31 Combination therapy with dapsone and a sulfonamide,32 niacinamide and tetracycline,33 colchicine,34 or surgical excision35 may be necessary in more resistant cases.
Conclusion
Our case exemplifies the clinical histologic spectrum that EED can present. The constellation of clinical findings was histologically confirmed to be manifestations of the disease in various stages of evolution. When typical lesions of EED present along with cutaneous findings in less common locations, performing multiple biopsies can be helpful. The clinician should retain a high index of suspicion for an underlying etiology and perform a complete workup for infection, malignancy, or autoimmune disease.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Wilkinson SM, English JS, Smith NP, et al. Erythema elevatum diutinum: a clinicopathological study. Clin Exp Dermatol. 1992;17:87-93.
- Fakheri A, Gupta SM, White SM, et al. Erythema elevatum diutinum in a patient with human immunodeficiency virus. Cutis. 2001;68:41-42, 55.
- Kim H. Erythema elevatum diutinum in an HIV-positive patient. J Drugs Dermatol. 2003;2:411-412.
- Revenga F, Vera A, Muñoz A, et al. Erythema elevatum diutinum and AIDS: are they related? Clin Exp Dermatol. 1997;22:250-251.
- Hutchinson J. On two remarkable cases of symmetrieal purple congestion of the skin in patches, with induration. Br J Dermatol. 1888;1:10-15.
- Bury JS. A case of erythema with remarkable nodular thickening and induration of the skin associated with intermittent albuminuria. Illustrated Medical News. 1889;3:145-149.
- Crocker HR, Williams C. Erythema elevatum diutinum. Br J Dermatol. 1894;6:33-38.
- Barzegar M, Davatchi CC, Akhyani M, et al. An atypical presentation of erythema elevatum diutinum involving palms and soles. Int J Dermatol. 2009;48:73-75.
- Futei Y, Konohana I. A case of erythema elevatum diutinum associated with B-cell lymphoma: a rare distribution involving palms, soles and nails. Br J Dermatol. 2000;142:116-119.
- Ben-Zvi GT, Bardsley V, Burrows NP. An atypical distribution of erythema elevatum diutinum. Clin Exp Dermatol. 2014;39:269-270.
- Weidman FD, Besancon JH. Erythema elevatum diutinum. role of streptococci, and relationship to other rheumatic dermatoses. Arch Dermatol Syphilol. 1929;20:593-620.
- Drago F, Semino M, Rampini P, et al. Erythema elevatum diutinum in a patient with human herpesvirus 6 infection. Acta Derm Venereol. 1999;79:91-92.
- Muratori S, Carrera C, Gorani A, et al. Erythema elevatum diutinum and HIV infection: a report of five cases. Br J Dermatol. 1999;141:335-338.
- Archimandritis AJ, Fertakis A, Alegakis G, et al. Erythema elevatum diutinum and IgA myeloma: an interesting association. Br Med J. 1977;2:613-614.
- Hatzitolios A, Tzellos TG, Savopoulos C, et al. Erythema elevatum diutinum with rare distribution as a first clinical sign of non-Hodgkin’s lymphoma: a novel association? J Dermatol. 2008;35:297-300.
- Delaporte E, Alfandari S, Fenaux P, et al. Erythema elevatum diutinum and chronic lymphocytic leukaemia. Clin Exp Dermatol. 1994;19:188-189.
- Miyagawa S, Kitamura W, Morita K, et al. Association of hyperimmunoglobulinaemia D syndrome with erythema elevatum diutinum. Br J Dermatol. 1993;128:572-574.
- Collier PM, Neill SM, Branfoot AC, et al. Erythema elevatum diutinum—a solitary lesion in a patient with rheumatoid arthritis. Clin Exp Dermatol. 1990;15:394-395.
- Buahene K, Hudson M, Mowat A, et al. Erythema elevatum diutinum—an unusual association with ulcerative colitis. Clin Exp Dermatol. 1991;16:204-206.
- Bernard P, Bedane C, Delrous JL, et al. Erythema elevatum diutinum in a patient with relapsing polychondritis. J Am Acad Dermatol. 1992;26:312-315.
- Hancox JG, Wallace CA, Sangueza OP, et al. Erythema elevatum diutinum associated with lupus panniculitis in a patient with discoid lesions of chronic cutaneous lupus erythematosus. J Am Acad Dermatol. 2004;50:652-653.
- Haber H. Erythema elevatum diutinum. Br J Dermatol. 1955;67:121-145.
- Katz SI, Gallin JL, Hertz KC, et al. Erythema elevatum diutinum: skin and systemic manifestations, immunologic studies, and successful treatment with dapsone. Medicine (Baltimore). 1977;56:443-455.
- Walker KD, Badame AJ. Erythema elevatum diutinum in a patient with Crohn’s disease. J Am Acad Dermatol. 1990;22:948-952.
- Popp JW, Harrist T, Dienstag JL, et al. Cutaneous vasculitis associated with acute and chronic hepatitis. Arch Intern Med. 1981;141:623-629.
- Lee AY, Nakagawa H, Nogita T, et al. Erythema elevatum diutinum: an ultrastructural case study. J Cutan Pathol. 1989;16:211-217.
- LeBoit PE, Yen TS, Wintroub B. The evolution of lesions in erythema elevatum diutinum. Am J Dermatopathol. 1986;8:392-402.
- Soubeiran E, Wacker J, Hausser I, et al. Erythema elevatum diutinum with unusual clinical appearance. J Dtsch Dermatol Ges. 2008;6:303-305.
- Momen SE, Jorizzo J, Al-Niaimi F. Erythema elevatum diutinum: a review of presentation and treatment. J Eur Acad Dermatol Venereol. 2014;28:1594-1602.
- Vollum DI. Erythema elevatum diutinum—vesicular lesions and sulfone response. Br J Dermatol. 1968;80:178-183.
- Kohler IK, Lorincz AL. Erythema elevatum diutinum treated with niacinamide and tetracycline. Arch Dermatol. 1980;116:693-695.
- Henriksson R, Hofor PA, Hörngvist R. Erythema elevatum diutinum—a case successfully treated with colchicine. Clin Exp Dermatol. 1989;14:451-453.
- Zacaron LH, Gonçalves JC, Curty VM, et al. Clinical and surgical therapeutic approach in erythema elevatum diutinum—case report. An Bras Dermatol. 2013;88(6, suppl 1):15-18.
Erythema elevatum diutinum (EED) manifests on a clinicopathologic spectrum of chronic cutaneous small vessel vasculitis. The lesions typically present as persistent, symmetric, firm, red to purple papules or nodules on the extensor arms and dorsal hands.1,2 Underlying infectious, malignant, or autoimmune processes are commonly associated with the disease, notably Streptococcus infection and IgA monoclonal gammopathy.2,3 Hepatitis virus also is often implicated in association with EED. Cases of EED have been seen with concomitant human immunodeficiency virus (HIV) infection.4-6 We report a case of EED presenting in various stages of evolution associated with underlying hepatitis B infection alone.
Case Report
A 57-year-old man originally presented to an outpatient dermatology practice with a nodular, painful, episodic rash on the trunk and upper and lower extremities. A biopsy revealed leukocytoclastic vasculitis (LCV) with prominent eosinophils. At the time, the skin findings were believed to be a manifestation of drug hypersensitivity, likely to opioid use. The patient was lost to follow-up.
Seven years later, the patient was admitted to the hospital with new-onset burning and stinging red nodules on the dorsum of the hands and persistence of the original episodic rash over the lower legs and bilateral flanks. In the interim, he was briefly treated with an oral prednisone taper and topical corticosteroids including triamcinolone cream 0.1% and clobetasol cream 0.05% without improvement.
On examination deep red to violaceous discrete nodules and plaques with overlying hyperkeratosis involving all distal and proximal interphalangeal joints of the hands and extensor elbows were seen (Figure 1A). On the bilateral posterior arms (Figure 1B), anterior legs, and periumbilical area were deeply erythematous papules and plaques with background hyperpigmentation. Across his lower back and bilateral flanks were erythematous papules with central hemorrhagic crusting (Figure 1C).
Pertinent laboratory findings included a positive hepatitis B surface antigen with hepatitis B DNA value 4,313,876 IU/mL and a hepatitis B virus quantitative polymerase chain reaction value of 6.64 U. The etiology was suspected to be intravenous drug abuse; however, the patient denied recreational drug use.
An additional infectious workup was negative for hepatitis C, streptococcus, syphilis, tuberculosis, and HIV. A complete blood cell count, complete metabolic panel, urinalysis, complement, cryoglobulins, and serum protein electrophoresis were within reference range. Autoimmune serologies were negative including antinuclear antibody, rheumatoid factor, anti-Sjögren syndrome–related antigen A and B, anticyclic citrullinated peptide, anti-Smith, and antineutrophilic cytoplasmic antibodies. Peripheral blood immunophenotyping, lactate dehydrogenase, quantitative immunoglobulins, and age-appropriate cancer screens did not demonstrate evidence for malignancy underlying the disease. Bilateral hand radiographs showed mild periostitis of the proximal phalanges without obvious erosions.
Three 4-mm punch biopsies were performed from the left fifth digit, left posterior arm, and left flank. Tissue of the left fifth digit showed an intradermal vascular proliferation with a concentric pattern resembling onion skin in a background of increased fibrosis. The blood vessels showed focal fibrinoid necrosis (Figure 2A). The biopsy of the left posterior arm showed an intradermal vascular proliferation with an associated mild acute and chronic perivascular inflammation (Figure 2B). The left flank biopsy showed LCV with focal epidermal necrosis (Figure 2C).
The constellation of clinical findings together with the histopathologic changes represented EED in various stages of evolution. The patient was started on dapsone 100 mg daily and referred to the infectious disease service for treatment of chronic hepatitis B; however, he was subsequently lost to follow-up.
Comment
Overview of EED
Erythema elevatum diutinum represents a rare form of chronic cutaneous small vessel vasculitis. Originally described by Hutchinson7 and Bury8 as symmetric purpuric nodules of the skin, it was later named by Crocker and Williams9 in 1894. The disease classically presents as firm, fixed, red-brown to violaceous papules, plaques, and nodules affecting the extensor upper or lower extremities.1 Lesions are most commonly found symmetrically overlying joints of the hands, feet, elbows, and knees, as well as the Achilles tendon and buttocks.3 Less common locations include the palms and soles, face,10,11 trunk,12 and periauricular region.1 Although they are typically asymptomatic, sensations such as burning, stinging, and pruritus have been noted.1 Our patient was unique because in addition to typical lesions of EED, he presented with crusted papules on the flanks and violaceous papules of the lower legs and periumbilicus.
Etiology
Originally associated with Streptococcus as isolated from EED lesions,3,13 additional infectious etiologies include viral hepatitis,4-6 human herpesvirus 6,14 and rarely HIV.1,15 Hepatitis B and C are well known to be associated with EED, with only rare reports in patients with concomitant HIV infection. Erythema elevatum diutinum also has been described in relationship to myeloproliferative disorders and hematologic malignancies such as IgA myeloma,16 non-Hodgkin lymphoma,17 chronic lymphocytic leukemia,18 and hypergammaglobulinemia.19 In a study of 13 patients with EED, 4 had associated underlying IgA monoclonal gammopathy.2 Autoimmune conditions such as rheumatoid arthritis,20 ulcerative colitis,21 relapsing polychondritis,22 and systemic lupus erythematosus23 also have been implicated.
Pathogenesis
Although the precise pathogenesis of EED remains unknown, it has been suggested that a complement cascade initiated by immune-complex deposition in postcapillary venules induces an LCV.24,25 Chronic antigenic exposure or high antibody levels26 in the face of infections, autoimmune disease, or malignancy may incite this immune-complex reaction. Skin lesions seen in association with hepatitis reflect circulating immune-complex deposition in vessel walls causing destruction. It has been postulated that the duration of immune complexemia may be sufficient to account for the differences in the type of vascular injury seen in acute versus chronic infection.27
Histopathology
Erythema elevatum diutinum may present on a histopathologic spectrum of LCV, as manifested in our patient. Early lesions show predominantly polymorphonuclear cells with nuclear dust pattern in a wedge-shaped infiltrate with fibrin deposition in the superficial and mid dermis.2,3 Later lesions show vasculitis in addition to dermal aggregates of lymphocytes, neutrophils, fibrosis, and areas of granulation tissue. The fibrosis may be dense and comprised of fibroblasts and myofibroblasts.28 Newly formed vessels within the granulation tissue have been postulated to be more susceptible to immune-complex deposition, thus potentiating the process.1,29
Management
Spontaneous resolution of EED may occur, albeit after a prolonged and recurrent course of up to 5 to 10 years.30 Treatment of the underlying cause, when identified, remains paramount. First-line therapy includes dapsone, shown to be effective in reducing lesion size to complete resolution in 80% of the 47 cases reviewed by Momen et al.31 Dapsone monotherapy tends to be less effective in treating nodular lesions associated with HIV-positivity, likely due to the extensive fibrosis.4,31 Combination therapy with dapsone and a sulfonamide,32 niacinamide and tetracycline,33 colchicine,34 or surgical excision35 may be necessary in more resistant cases.
Conclusion
Our case exemplifies the clinical histologic spectrum that EED can present. The constellation of clinical findings was histologically confirmed to be manifestations of the disease in various stages of evolution. When typical lesions of EED present along with cutaneous findings in less common locations, performing multiple biopsies can be helpful. The clinician should retain a high index of suspicion for an underlying etiology and perform a complete workup for infection, malignancy, or autoimmune disease.
Erythema elevatum diutinum (EED) manifests on a clinicopathologic spectrum of chronic cutaneous small vessel vasculitis. The lesions typically present as persistent, symmetric, firm, red to purple papules or nodules on the extensor arms and dorsal hands.1,2 Underlying infectious, malignant, or autoimmune processes are commonly associated with the disease, notably Streptococcus infection and IgA monoclonal gammopathy.2,3 Hepatitis virus also is often implicated in association with EED. Cases of EED have been seen with concomitant human immunodeficiency virus (HIV) infection.4-6 We report a case of EED presenting in various stages of evolution associated with underlying hepatitis B infection alone.
Case Report
A 57-year-old man originally presented to an outpatient dermatology practice with a nodular, painful, episodic rash on the trunk and upper and lower extremities. A biopsy revealed leukocytoclastic vasculitis (LCV) with prominent eosinophils. At the time, the skin findings were believed to be a manifestation of drug hypersensitivity, likely to opioid use. The patient was lost to follow-up.
Seven years later, the patient was admitted to the hospital with new-onset burning and stinging red nodules on the dorsum of the hands and persistence of the original episodic rash over the lower legs and bilateral flanks. In the interim, he was briefly treated with an oral prednisone taper and topical corticosteroids including triamcinolone cream 0.1% and clobetasol cream 0.05% without improvement.
On examination deep red to violaceous discrete nodules and plaques with overlying hyperkeratosis involving all distal and proximal interphalangeal joints of the hands and extensor elbows were seen (Figure 1A). On the bilateral posterior arms (Figure 1B), anterior legs, and periumbilical area were deeply erythematous papules and plaques with background hyperpigmentation. Across his lower back and bilateral flanks were erythematous papules with central hemorrhagic crusting (Figure 1C).
Pertinent laboratory findings included a positive hepatitis B surface antigen with hepatitis B DNA value 4,313,876 IU/mL and a hepatitis B virus quantitative polymerase chain reaction value of 6.64 U. The etiology was suspected to be intravenous drug abuse; however, the patient denied recreational drug use.
An additional infectious workup was negative for hepatitis C, streptococcus, syphilis, tuberculosis, and HIV. A complete blood cell count, complete metabolic panel, urinalysis, complement, cryoglobulins, and serum protein electrophoresis were within reference range. Autoimmune serologies were negative including antinuclear antibody, rheumatoid factor, anti-Sjögren syndrome–related antigen A and B, anticyclic citrullinated peptide, anti-Smith, and antineutrophilic cytoplasmic antibodies. Peripheral blood immunophenotyping, lactate dehydrogenase, quantitative immunoglobulins, and age-appropriate cancer screens did not demonstrate evidence for malignancy underlying the disease. Bilateral hand radiographs showed mild periostitis of the proximal phalanges without obvious erosions.
Three 4-mm punch biopsies were performed from the left fifth digit, left posterior arm, and left flank. Tissue of the left fifth digit showed an intradermal vascular proliferation with a concentric pattern resembling onion skin in a background of increased fibrosis. The blood vessels showed focal fibrinoid necrosis (Figure 2A). The biopsy of the left posterior arm showed an intradermal vascular proliferation with an associated mild acute and chronic perivascular inflammation (Figure 2B). The left flank biopsy showed LCV with focal epidermal necrosis (Figure 2C).
The constellation of clinical findings together with the histopathologic changes represented EED in various stages of evolution. The patient was started on dapsone 100 mg daily and referred to the infectious disease service for treatment of chronic hepatitis B; however, he was subsequently lost to follow-up.
Comment
Overview of EED
Erythema elevatum diutinum represents a rare form of chronic cutaneous small vessel vasculitis. Originally described by Hutchinson7 and Bury8 as symmetric purpuric nodules of the skin, it was later named by Crocker and Williams9 in 1894. The disease classically presents as firm, fixed, red-brown to violaceous papules, plaques, and nodules affecting the extensor upper or lower extremities.1 Lesions are most commonly found symmetrically overlying joints of the hands, feet, elbows, and knees, as well as the Achilles tendon and buttocks.3 Less common locations include the palms and soles, face,10,11 trunk,12 and periauricular region.1 Although they are typically asymptomatic, sensations such as burning, stinging, and pruritus have been noted.1 Our patient was unique because in addition to typical lesions of EED, he presented with crusted papules on the flanks and violaceous papules of the lower legs and periumbilicus.
Etiology
Originally associated with Streptococcus as isolated from EED lesions,3,13 additional infectious etiologies include viral hepatitis,4-6 human herpesvirus 6,14 and rarely HIV.1,15 Hepatitis B and C are well known to be associated with EED, with only rare reports in patients with concomitant HIV infection. Erythema elevatum diutinum also has been described in relationship to myeloproliferative disorders and hematologic malignancies such as IgA myeloma,16 non-Hodgkin lymphoma,17 chronic lymphocytic leukemia,18 and hypergammaglobulinemia.19 In a study of 13 patients with EED, 4 had associated underlying IgA monoclonal gammopathy.2 Autoimmune conditions such as rheumatoid arthritis,20 ulcerative colitis,21 relapsing polychondritis,22 and systemic lupus erythematosus23 also have been implicated.
Pathogenesis
Although the precise pathogenesis of EED remains unknown, it has been suggested that a complement cascade initiated by immune-complex deposition in postcapillary venules induces an LCV.24,25 Chronic antigenic exposure or high antibody levels26 in the face of infections, autoimmune disease, or malignancy may incite this immune-complex reaction. Skin lesions seen in association with hepatitis reflect circulating immune-complex deposition in vessel walls causing destruction. It has been postulated that the duration of immune complexemia may be sufficient to account for the differences in the type of vascular injury seen in acute versus chronic infection.27
Histopathology
Erythema elevatum diutinum may present on a histopathologic spectrum of LCV, as manifested in our patient. Early lesions show predominantly polymorphonuclear cells with nuclear dust pattern in a wedge-shaped infiltrate with fibrin deposition in the superficial and mid dermis.2,3 Later lesions show vasculitis in addition to dermal aggregates of lymphocytes, neutrophils, fibrosis, and areas of granulation tissue. The fibrosis may be dense and comprised of fibroblasts and myofibroblasts.28 Newly formed vessels within the granulation tissue have been postulated to be more susceptible to immune-complex deposition, thus potentiating the process.1,29
Management
Spontaneous resolution of EED may occur, albeit after a prolonged and recurrent course of up to 5 to 10 years.30 Treatment of the underlying cause, when identified, remains paramount. First-line therapy includes dapsone, shown to be effective in reducing lesion size to complete resolution in 80% of the 47 cases reviewed by Momen et al.31 Dapsone monotherapy tends to be less effective in treating nodular lesions associated with HIV-positivity, likely due to the extensive fibrosis.4,31 Combination therapy with dapsone and a sulfonamide,32 niacinamide and tetracycline,33 colchicine,34 or surgical excision35 may be necessary in more resistant cases.
Conclusion
Our case exemplifies the clinical histologic spectrum that EED can present. The constellation of clinical findings was histologically confirmed to be manifestations of the disease in various stages of evolution. When typical lesions of EED present along with cutaneous findings in less common locations, performing multiple biopsies can be helpful. The clinician should retain a high index of suspicion for an underlying etiology and perform a complete workup for infection, malignancy, or autoimmune disease.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Wilkinson SM, English JS, Smith NP, et al. Erythema elevatum diutinum: a clinicopathological study. Clin Exp Dermatol. 1992;17:87-93.
- Fakheri A, Gupta SM, White SM, et al. Erythema elevatum diutinum in a patient with human immunodeficiency virus. Cutis. 2001;68:41-42, 55.
- Kim H. Erythema elevatum diutinum in an HIV-positive patient. J Drugs Dermatol. 2003;2:411-412.
- Revenga F, Vera A, Muñoz A, et al. Erythema elevatum diutinum and AIDS: are they related? Clin Exp Dermatol. 1997;22:250-251.
- Hutchinson J. On two remarkable cases of symmetrieal purple congestion of the skin in patches, with induration. Br J Dermatol. 1888;1:10-15.
- Bury JS. A case of erythema with remarkable nodular thickening and induration of the skin associated with intermittent albuminuria. Illustrated Medical News. 1889;3:145-149.
- Crocker HR, Williams C. Erythema elevatum diutinum. Br J Dermatol. 1894;6:33-38.
- Barzegar M, Davatchi CC, Akhyani M, et al. An atypical presentation of erythema elevatum diutinum involving palms and soles. Int J Dermatol. 2009;48:73-75.
- Futei Y, Konohana I. A case of erythema elevatum diutinum associated with B-cell lymphoma: a rare distribution involving palms, soles and nails. Br J Dermatol. 2000;142:116-119.
- Ben-Zvi GT, Bardsley V, Burrows NP. An atypical distribution of erythema elevatum diutinum. Clin Exp Dermatol. 2014;39:269-270.
- Weidman FD, Besancon JH. Erythema elevatum diutinum. role of streptococci, and relationship to other rheumatic dermatoses. Arch Dermatol Syphilol. 1929;20:593-620.
- Drago F, Semino M, Rampini P, et al. Erythema elevatum diutinum in a patient with human herpesvirus 6 infection. Acta Derm Venereol. 1999;79:91-92.
- Muratori S, Carrera C, Gorani A, et al. Erythema elevatum diutinum and HIV infection: a report of five cases. Br J Dermatol. 1999;141:335-338.
- Archimandritis AJ, Fertakis A, Alegakis G, et al. Erythema elevatum diutinum and IgA myeloma: an interesting association. Br Med J. 1977;2:613-614.
- Hatzitolios A, Tzellos TG, Savopoulos C, et al. Erythema elevatum diutinum with rare distribution as a first clinical sign of non-Hodgkin’s lymphoma: a novel association? J Dermatol. 2008;35:297-300.
- Delaporte E, Alfandari S, Fenaux P, et al. Erythema elevatum diutinum and chronic lymphocytic leukaemia. Clin Exp Dermatol. 1994;19:188-189.
- Miyagawa S, Kitamura W, Morita K, et al. Association of hyperimmunoglobulinaemia D syndrome with erythema elevatum diutinum. Br J Dermatol. 1993;128:572-574.
- Collier PM, Neill SM, Branfoot AC, et al. Erythema elevatum diutinum—a solitary lesion in a patient with rheumatoid arthritis. Clin Exp Dermatol. 1990;15:394-395.
- Buahene K, Hudson M, Mowat A, et al. Erythema elevatum diutinum—an unusual association with ulcerative colitis. Clin Exp Dermatol. 1991;16:204-206.
- Bernard P, Bedane C, Delrous JL, et al. Erythema elevatum diutinum in a patient with relapsing polychondritis. J Am Acad Dermatol. 1992;26:312-315.
- Hancox JG, Wallace CA, Sangueza OP, et al. Erythema elevatum diutinum associated with lupus panniculitis in a patient with discoid lesions of chronic cutaneous lupus erythematosus. J Am Acad Dermatol. 2004;50:652-653.
- Haber H. Erythema elevatum diutinum. Br J Dermatol. 1955;67:121-145.
- Katz SI, Gallin JL, Hertz KC, et al. Erythema elevatum diutinum: skin and systemic manifestations, immunologic studies, and successful treatment with dapsone. Medicine (Baltimore). 1977;56:443-455.
- Walker KD, Badame AJ. Erythema elevatum diutinum in a patient with Crohn’s disease. J Am Acad Dermatol. 1990;22:948-952.
- Popp JW, Harrist T, Dienstag JL, et al. Cutaneous vasculitis associated with acute and chronic hepatitis. Arch Intern Med. 1981;141:623-629.
- Lee AY, Nakagawa H, Nogita T, et al. Erythema elevatum diutinum: an ultrastructural case study. J Cutan Pathol. 1989;16:211-217.
- LeBoit PE, Yen TS, Wintroub B. The evolution of lesions in erythema elevatum diutinum. Am J Dermatopathol. 1986;8:392-402.
- Soubeiran E, Wacker J, Hausser I, et al. Erythema elevatum diutinum with unusual clinical appearance. J Dtsch Dermatol Ges. 2008;6:303-305.
- Momen SE, Jorizzo J, Al-Niaimi F. Erythema elevatum diutinum: a review of presentation and treatment. J Eur Acad Dermatol Venereol. 2014;28:1594-1602.
- Vollum DI. Erythema elevatum diutinum—vesicular lesions and sulfone response. Br J Dermatol. 1968;80:178-183.
- Kohler IK, Lorincz AL. Erythema elevatum diutinum treated with niacinamide and tetracycline. Arch Dermatol. 1980;116:693-695.
- Henriksson R, Hofor PA, Hörngvist R. Erythema elevatum diutinum—a case successfully treated with colchicine. Clin Exp Dermatol. 1989;14:451-453.
- Zacaron LH, Gonçalves JC, Curty VM, et al. Clinical and surgical therapeutic approach in erythema elevatum diutinum—case report. An Bras Dermatol. 2013;88(6, suppl 1):15-18.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Wilkinson SM, English JS, Smith NP, et al. Erythema elevatum diutinum: a clinicopathological study. Clin Exp Dermatol. 1992;17:87-93.
- Fakheri A, Gupta SM, White SM, et al. Erythema elevatum diutinum in a patient with human immunodeficiency virus. Cutis. 2001;68:41-42, 55.
- Kim H. Erythema elevatum diutinum in an HIV-positive patient. J Drugs Dermatol. 2003;2:411-412.
- Revenga F, Vera A, Muñoz A, et al. Erythema elevatum diutinum and AIDS: are they related? Clin Exp Dermatol. 1997;22:250-251.
- Hutchinson J. On two remarkable cases of symmetrieal purple congestion of the skin in patches, with induration. Br J Dermatol. 1888;1:10-15.
- Bury JS. A case of erythema with remarkable nodular thickening and induration of the skin associated with intermittent albuminuria. Illustrated Medical News. 1889;3:145-149.
- Crocker HR, Williams C. Erythema elevatum diutinum. Br J Dermatol. 1894;6:33-38.
- Barzegar M, Davatchi CC, Akhyani M, et al. An atypical presentation of erythema elevatum diutinum involving palms and soles. Int J Dermatol. 2009;48:73-75.
- Futei Y, Konohana I. A case of erythema elevatum diutinum associated with B-cell lymphoma: a rare distribution involving palms, soles and nails. Br J Dermatol. 2000;142:116-119.
- Ben-Zvi GT, Bardsley V, Burrows NP. An atypical distribution of erythema elevatum diutinum. Clin Exp Dermatol. 2014;39:269-270.
- Weidman FD, Besancon JH. Erythema elevatum diutinum. role of streptococci, and relationship to other rheumatic dermatoses. Arch Dermatol Syphilol. 1929;20:593-620.
- Drago F, Semino M, Rampini P, et al. Erythema elevatum diutinum in a patient with human herpesvirus 6 infection. Acta Derm Venereol. 1999;79:91-92.
- Muratori S, Carrera C, Gorani A, et al. Erythema elevatum diutinum and HIV infection: a report of five cases. Br J Dermatol. 1999;141:335-338.
- Archimandritis AJ, Fertakis A, Alegakis G, et al. Erythema elevatum diutinum and IgA myeloma: an interesting association. Br Med J. 1977;2:613-614.
- Hatzitolios A, Tzellos TG, Savopoulos C, et al. Erythema elevatum diutinum with rare distribution as a first clinical sign of non-Hodgkin’s lymphoma: a novel association? J Dermatol. 2008;35:297-300.
- Delaporte E, Alfandari S, Fenaux P, et al. Erythema elevatum diutinum and chronic lymphocytic leukaemia. Clin Exp Dermatol. 1994;19:188-189.
- Miyagawa S, Kitamura W, Morita K, et al. Association of hyperimmunoglobulinaemia D syndrome with erythema elevatum diutinum. Br J Dermatol. 1993;128:572-574.
- Collier PM, Neill SM, Branfoot AC, et al. Erythema elevatum diutinum—a solitary lesion in a patient with rheumatoid arthritis. Clin Exp Dermatol. 1990;15:394-395.
- Buahene K, Hudson M, Mowat A, et al. Erythema elevatum diutinum—an unusual association with ulcerative colitis. Clin Exp Dermatol. 1991;16:204-206.
- Bernard P, Bedane C, Delrous JL, et al. Erythema elevatum diutinum in a patient with relapsing polychondritis. J Am Acad Dermatol. 1992;26:312-315.
- Hancox JG, Wallace CA, Sangueza OP, et al. Erythema elevatum diutinum associated with lupus panniculitis in a patient with discoid lesions of chronic cutaneous lupus erythematosus. J Am Acad Dermatol. 2004;50:652-653.
- Haber H. Erythema elevatum diutinum. Br J Dermatol. 1955;67:121-145.
- Katz SI, Gallin JL, Hertz KC, et al. Erythema elevatum diutinum: skin and systemic manifestations, immunologic studies, and successful treatment with dapsone. Medicine (Baltimore). 1977;56:443-455.
- Walker KD, Badame AJ. Erythema elevatum diutinum in a patient with Crohn’s disease. J Am Acad Dermatol. 1990;22:948-952.
- Popp JW, Harrist T, Dienstag JL, et al. Cutaneous vasculitis associated with acute and chronic hepatitis. Arch Intern Med. 1981;141:623-629.
- Lee AY, Nakagawa H, Nogita T, et al. Erythema elevatum diutinum: an ultrastructural case study. J Cutan Pathol. 1989;16:211-217.
- LeBoit PE, Yen TS, Wintroub B. The evolution of lesions in erythema elevatum diutinum. Am J Dermatopathol. 1986;8:392-402.
- Soubeiran E, Wacker J, Hausser I, et al. Erythema elevatum diutinum with unusual clinical appearance. J Dtsch Dermatol Ges. 2008;6:303-305.
- Momen SE, Jorizzo J, Al-Niaimi F. Erythema elevatum diutinum: a review of presentation and treatment. J Eur Acad Dermatol Venereol. 2014;28:1594-1602.
- Vollum DI. Erythema elevatum diutinum—vesicular lesions and sulfone response. Br J Dermatol. 1968;80:178-183.
- Kohler IK, Lorincz AL. Erythema elevatum diutinum treated with niacinamide and tetracycline. Arch Dermatol. 1980;116:693-695.
- Henriksson R, Hofor PA, Hörngvist R. Erythema elevatum diutinum—a case successfully treated with colchicine. Clin Exp Dermatol. 1989;14:451-453.
- Zacaron LH, Gonçalves JC, Curty VM, et al. Clinical and surgical therapeutic approach in erythema elevatum diutinum—case report. An Bras Dermatol. 2013;88(6, suppl 1):15-18.
Practice Points
- Erythema elevatum diutinum (EED) often is associated with an underlying infectious process, including hepatitis B and hepatitis C, or a hematologic or autoimmune condition.
- If EED is suspected clinically, it may be beneficial to perform multiple biopsies from lesions at different stages of evolution to establish the diagnosis.
- First-line therapy includes treatment of any underlying condition and dapsone.
