User login
A Novel Pharmaceutical Care Model for High-Risk Patients
Nonadherence is a significant problem that has a negative impact on both patients and public health. Patients with multiple diseases often have complicated medication regimens, which can be difficult for them to manage. Unfortunately, nonadherence in these high-risk patients can have drastic consequences, including disease progression, hospitalization, and death, resulting in billions of dollars in unnecessary costs nationwide.1,2 The Wheel Model of Pharmaceutical Care (Figure) is a novel care model developed at the Gallup Indian Medical Center (GIMC) in New Mexico to address these problems by positioning pharmacy as a proactive service. The Wheel Model of Pharmaceutical Care was designed to improve adherence and patient outcomes and to encourage communication among the patient, pharmacists, prescribers, and other health care team members.
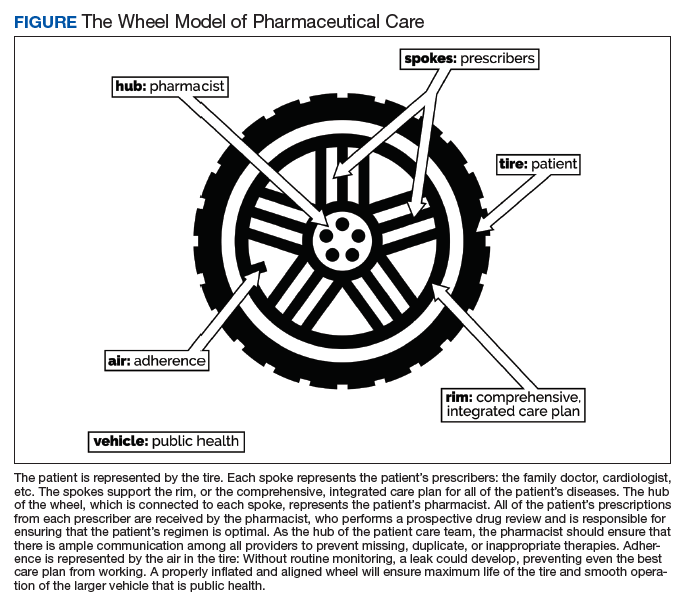
Pharmacists are central to managing patients’ medication therapies and coordinating communication among the health care providers (HCPs).1,3 Medication therapy management (MTM), a required component of Medicare Part D plans, helps ensure appropriate drug use and reduce the risk of adverse events.3 Since pharmacists receive prescriptions from all of the patient’s HCPs, patients may see pharmacists more often than they see any other HCP. GIMC is currently piloting a new clinic, the Medication Optimization, Synchronization, and Adherence Improvement Clinic (MOSAIC), that was created to implement the Wheel Model of Pharmaceutical Care. MOSAIC aims to provide proactive pharmacy services and continuous MTM to high-risk patients and will enable the effectiveness of this new pharmaceutical care model to be assessed.
Methods
Studies have identified certain populations who are at an increased risk for nonadherence: the elderly, patients with complex or extensive medication regimens, patients with multiple chronic medical conditions, substance misusers, certain ethnicities, patients of lower socioeconomic status, patients with limited literacy, and the homeless.2,4 Federal regulations require that Medicare Part D plans target beneficiaries who meet specific criteria for MTM programs. Under these rules, plans must target beneficiaries with ≥ 3 chronic diseases and ≥ 8 chronic medications, although plans also may include patients with fewer medications and diseases.3 Although the Wheel Model of Pharmaceutical Care is postulated to be an accurate model for the ideal care of all patients, initial implementation should be targeted toward populations who are likely to benefit the most from intervention. For these reasons, elderly Native American patients who have ≥ 2 chronic diseases and who take ≥ 5 chronic medications were targeted for initial enrollment in MOSAIC at GIMC.
Overview
In MOSAIC, pharmacists act as the hub of the pharmaceutical care wheel. Pharmacists work to ensure optimization of the patient’s comprehensive, integrated care plan—the rim of the wheel. As a part of this optimization process, MOSAIC pharmacists facilitate synchronization of the patient’s prescriptions to a monthly or quarterly target fill date. The patient’s current medication therapy is organized, and pharmacists track which medications are due to be filled instead of depending on the patient to request each prescription refill. This process effectively changes pharmacy from a requested service to a provided service.
Pharmacists also monitor the air in the tire to promote adherence. This is accomplished by providing efficient monthly or quarterly telephone or in-person consultations, which helps the patient better understand his or her comprehensive, integrated care plan. MOSAIC eliminates the possibility of nonadherence due to running out of refills. Specialized packaging, such as pill boxes or blister packs, can also improve adherence for certain patients.
MOSAIC ensures that pharmacists stay connected with the spokes, which represent a patient’s numerous prescribers, and close communication loops. Pharmacists can make prescribers aware of potential gaps or overlaps in treatment and assist them in the optimization and development of the patient’s comprehensive, integrated care plan. Pharmacists also make sure that the patient’s medication profile is current and accurate in the electronic health record (EHR). Any pertinent information discovered during MOSAIC encounters, such as abnormal laboratory results or changes in medications or disease, is documented in an EHR note. The patient’s prescribers are made aware of this information by tagging them as additional signers to the note in the EHR.
Keeping patients—the tires—healthy will ensure smooth operation of the vehicle and have a positive impact on public health. MOSAIC is expected to not only improve individual patient outcomes, but also decrease health care costs for patients and society due to nonadherence, suboptimal regimens, stockpiled home medications, and preventable hospital admissions.
Traditionally, pharmacy has been a requested service: A patient requests each of their prescriptions to be refilled, and the pharmacy fills the prescription. Ideally, pharmacy must become a provided service, with pharmacists keeping track of when a patient’s medications are due to be filled and actively looking for medication therapy optimization opportunities. This is accomplished by synchronizing the patient’s medications to the same monthly or quarterly fill date; screening for any potentially inappropriate medications, including high-risk medications in elderly patients, duplications, and omissions; verifying any medication changes with the patient each fill; and then providing all needed medications to the patient at a scheduled time.
To facilitate this process, custom software was developed for MOSAIC. In addition, a collaborative practice agreement (CPA) was drafted that allowed MOSAIC pharmacists to make certain medication therapy optimizations on behalf of the patient’s primary care provider. As part of this CPA, pharmacists also may order and act on certain laboratory tests, which helps to monitor disease progression, ensure safe medication use, and meet Government Performance and Results Act (GPRA) measures. As a novel model of pharmaceutical care, the effects of this approach are not yet known; however, research suggests that increased communication among HCPs and patient-centered approaches to care are beneficial to patient outcomes, adherence, and public health.1,5
Investigated Outcomes
As patients continue to enroll in MOSAIC, the effectiveness of the clinic will be evaluated. Specifically, quality of life, patient and HCP satisfaction with the program, adherence metrics, hospitalization rates, and all-cause mortality will be assessed for patients enrolled in MOSAIC as well as similar patients who are not enrolled in MOSAIC. Also, pharmacists will log all recommended medication therapy interventions so that the optimization component of MOSAIC may be quantified. GPRA measures and the financial implications of the interventions made by MOSAIC will also be evaluated.
Discussion
There are a number of factors, such as MTM services and interprofessional care teams, that research has shown to independently improve patient outcomes, adherence, or public health. By synthesizing these factors, a completely new approach—the Wheel Model of Pharmaceutical Care—was developed. This model presents a radical departure from traditional, requested-service practices and posits pharmacy as a provided service instead. Although the ideas of MTM and interprofessional care teams are not new, there has never been a practical way to truly integrate community pharmacists into the patient care team or to ensure adequate communication among all of the patient’s HCPs. The Wheel Model of Pharmaceutical Care includes public health as one of its core components and provides a framework for pharmacies to meaningfully impact health outcomes for patients.
The Wheel Model of Pharmaceutical Care was designed to minimize the likelihood of nonadherence. Despite this, patients might willfully choose to be nonadherent, forget to take their medications, or neglect to pick up their medications. Additionally, in health care systems where patients must pay for their medications, prescription drug costs might be a barrier to adherence.
When nonadherence is suspected, the Wheel Model of Pharmaceutical Care directs pharmacists in MOSAIC to take action. First, the underlying cause of the nonadherence must be determined. For example, if a patient is nonadherent because of an adverse drug reaction, a therapy change may be indicated. If a patient is nonadherent due to apathy toward their health or therapy, the patient may benefit from education about their condition and treatment options; thus, the patient can make shared, informed decisions and feel more actively involved with his or her health. If a patients is nonadherent due to forgetfulness, adherence packaging dispense methods should be considered as an alternative to traditional vials. Depending on the services offered by a given pharmacy, adherence packaging options may include blister packs, pill boxes, or strips prepared by robotic dispensing systems. The use of medication reminders, whether in the form of a smartphone application or a simple alarm clock, should be discussed with the patient. If the patient does not pick up their medications on time, a pharmacist can contact the patient to determine why the medications were not picked up and to assess any nonadherence. In this case, mail order pharmacy services, if available, should be offered to patients as a more convenient option.
The medication regimen optimization component of MOSAIC helps reduce the workload of primary care providers and allows pharmacists to act autonomously based on clinical judgment, within the scope of the CPA. This can prevent delays in care caused by no refills remaining on a prescription. The laboratory monitoring component allows pharmacists to track diseases and take action if necessary, which should have a favorable impact on GPRA measures. Medication optimizations can reduce wasted resources by identifying cost-saving formulary alternatives, potentially inappropriate medications, and suboptimal doses.
Since many Indian Health Service beneficiaries do not have private insurance and therefore do not generate third-party reimbursements for services and care provided by GIMC, keeping patients healthy and out of the hospital is a top priority. As more patients are enrolled in MOSAIC, the program is expected to have a favorable impact on pharmacy workload and workflow as well. Prescriptions are anticipated and filled in advance, which decreases the amount of patients calling and presenting to the pharmacy for same-day refill requests. Scheduling when MOSAIC patients’ medications are to be filled and dispensed creates a predictable workload that allows the pharmacy staff to be managed more efficiently.
Conclusion
Adherence is the responsibility of the patient, but the Wheel Model of Pharmaceutical Care aims to provide pharmacists with a framework to monitor and encourage adherence in their patients. By taking this patient-centered approach, MOSAIC is expected to improve outcomes and decrease hospitalizations for high-risk patients who simply need a little extra help with their medications.
1. Bosworth HB, Granger BB, Mendys P, et al. Medication adherence: a call for action. Am Heart J. 2011;162(3):412-424.
2. Vlasnik JJ, Aliotta SL, DeLor B. Medication adherence: factors influencing compliance with prescribed medication plans. Case Manager. 2005;16(2):47-51.
3. Drug utilization management, quality assurance, and medication therapy management programs (MTMPs). Fed Regist. 2012;77(71):2207-22175. To be codified at 42 CFR § 423.153.
4. Thiruchselvam T, Naglie G, Moineddin R, et al. Risk factors for medication nonadherence in older adults with cognitive impairment who live alone. Int J Geriatr Psychiatry. 2012;27(12):1275-1282.
5. Liddy C, Blazkho V, Mill K. Challenges of self-management when living with multiple chronic conditions: systematic review of the qualitative literature. Can Fam Physician. 2014;60(12):1123-1133.
Nonadherence is a significant problem that has a negative impact on both patients and public health. Patients with multiple diseases often have complicated medication regimens, which can be difficult for them to manage. Unfortunately, nonadherence in these high-risk patients can have drastic consequences, including disease progression, hospitalization, and death, resulting in billions of dollars in unnecessary costs nationwide.1,2 The Wheel Model of Pharmaceutical Care (Figure) is a novel care model developed at the Gallup Indian Medical Center (GIMC) in New Mexico to address these problems by positioning pharmacy as a proactive service. The Wheel Model of Pharmaceutical Care was designed to improve adherence and patient outcomes and to encourage communication among the patient, pharmacists, prescribers, and other health care team members.
Pharmacists are central to managing patients’ medication therapies and coordinating communication among the health care providers (HCPs).1,3 Medication therapy management (MTM), a required component of Medicare Part D plans, helps ensure appropriate drug use and reduce the risk of adverse events.3 Since pharmacists receive prescriptions from all of the patient’s HCPs, patients may see pharmacists more often than they see any other HCP. GIMC is currently piloting a new clinic, the Medication Optimization, Synchronization, and Adherence Improvement Clinic (MOSAIC), that was created to implement the Wheel Model of Pharmaceutical Care. MOSAIC aims to provide proactive pharmacy services and continuous MTM to high-risk patients and will enable the effectiveness of this new pharmaceutical care model to be assessed.
Methods
Studies have identified certain populations who are at an increased risk for nonadherence: the elderly, patients with complex or extensive medication regimens, patients with multiple chronic medical conditions, substance misusers, certain ethnicities, patients of lower socioeconomic status, patients with limited literacy, and the homeless.2,4 Federal regulations require that Medicare Part D plans target beneficiaries who meet specific criteria for MTM programs. Under these rules, plans must target beneficiaries with ≥ 3 chronic diseases and ≥ 8 chronic medications, although plans also may include patients with fewer medications and diseases.3 Although the Wheel Model of Pharmaceutical Care is postulated to be an accurate model for the ideal care of all patients, initial implementation should be targeted toward populations who are likely to benefit the most from intervention. For these reasons, elderly Native American patients who have ≥ 2 chronic diseases and who take ≥ 5 chronic medications were targeted for initial enrollment in MOSAIC at GIMC.
Overview
In MOSAIC, pharmacists act as the hub of the pharmaceutical care wheel. Pharmacists work to ensure optimization of the patient’s comprehensive, integrated care plan—the rim of the wheel. As a part of this optimization process, MOSAIC pharmacists facilitate synchronization of the patient’s prescriptions to a monthly or quarterly target fill date. The patient’s current medication therapy is organized, and pharmacists track which medications are due to be filled instead of depending on the patient to request each prescription refill. This process effectively changes pharmacy from a requested service to a provided service.
Pharmacists also monitor the air in the tire to promote adherence. This is accomplished by providing efficient monthly or quarterly telephone or in-person consultations, which helps the patient better understand his or her comprehensive, integrated care plan. MOSAIC eliminates the possibility of nonadherence due to running out of refills. Specialized packaging, such as pill boxes or blister packs, can also improve adherence for certain patients.
MOSAIC ensures that pharmacists stay connected with the spokes, which represent a patient’s numerous prescribers, and close communication loops. Pharmacists can make prescribers aware of potential gaps or overlaps in treatment and assist them in the optimization and development of the patient’s comprehensive, integrated care plan. Pharmacists also make sure that the patient’s medication profile is current and accurate in the electronic health record (EHR). Any pertinent information discovered during MOSAIC encounters, such as abnormal laboratory results or changes in medications or disease, is documented in an EHR note. The patient’s prescribers are made aware of this information by tagging them as additional signers to the note in the EHR.
Keeping patients—the tires—healthy will ensure smooth operation of the vehicle and have a positive impact on public health. MOSAIC is expected to not only improve individual patient outcomes, but also decrease health care costs for patients and society due to nonadherence, suboptimal regimens, stockpiled home medications, and preventable hospital admissions.
Traditionally, pharmacy has been a requested service: A patient requests each of their prescriptions to be refilled, and the pharmacy fills the prescription. Ideally, pharmacy must become a provided service, with pharmacists keeping track of when a patient’s medications are due to be filled and actively looking for medication therapy optimization opportunities. This is accomplished by synchronizing the patient’s medications to the same monthly or quarterly fill date; screening for any potentially inappropriate medications, including high-risk medications in elderly patients, duplications, and omissions; verifying any medication changes with the patient each fill; and then providing all needed medications to the patient at a scheduled time.
To facilitate this process, custom software was developed for MOSAIC. In addition, a collaborative practice agreement (CPA) was drafted that allowed MOSAIC pharmacists to make certain medication therapy optimizations on behalf of the patient’s primary care provider. As part of this CPA, pharmacists also may order and act on certain laboratory tests, which helps to monitor disease progression, ensure safe medication use, and meet Government Performance and Results Act (GPRA) measures. As a novel model of pharmaceutical care, the effects of this approach are not yet known; however, research suggests that increased communication among HCPs and patient-centered approaches to care are beneficial to patient outcomes, adherence, and public health.1,5
Investigated Outcomes
As patients continue to enroll in MOSAIC, the effectiveness of the clinic will be evaluated. Specifically, quality of life, patient and HCP satisfaction with the program, adherence metrics, hospitalization rates, and all-cause mortality will be assessed for patients enrolled in MOSAIC as well as similar patients who are not enrolled in MOSAIC. Also, pharmacists will log all recommended medication therapy interventions so that the optimization component of MOSAIC may be quantified. GPRA measures and the financial implications of the interventions made by MOSAIC will also be evaluated.
Discussion
There are a number of factors, such as MTM services and interprofessional care teams, that research has shown to independently improve patient outcomes, adherence, or public health. By synthesizing these factors, a completely new approach—the Wheel Model of Pharmaceutical Care—was developed. This model presents a radical departure from traditional, requested-service practices and posits pharmacy as a provided service instead. Although the ideas of MTM and interprofessional care teams are not new, there has never been a practical way to truly integrate community pharmacists into the patient care team or to ensure adequate communication among all of the patient’s HCPs. The Wheel Model of Pharmaceutical Care includes public health as one of its core components and provides a framework for pharmacies to meaningfully impact health outcomes for patients.
The Wheel Model of Pharmaceutical Care was designed to minimize the likelihood of nonadherence. Despite this, patients might willfully choose to be nonadherent, forget to take their medications, or neglect to pick up their medications. Additionally, in health care systems where patients must pay for their medications, prescription drug costs might be a barrier to adherence.
When nonadherence is suspected, the Wheel Model of Pharmaceutical Care directs pharmacists in MOSAIC to take action. First, the underlying cause of the nonadherence must be determined. For example, if a patient is nonadherent because of an adverse drug reaction, a therapy change may be indicated. If a patient is nonadherent due to apathy toward their health or therapy, the patient may benefit from education about their condition and treatment options; thus, the patient can make shared, informed decisions and feel more actively involved with his or her health. If a patients is nonadherent due to forgetfulness, adherence packaging dispense methods should be considered as an alternative to traditional vials. Depending on the services offered by a given pharmacy, adherence packaging options may include blister packs, pill boxes, or strips prepared by robotic dispensing systems. The use of medication reminders, whether in the form of a smartphone application or a simple alarm clock, should be discussed with the patient. If the patient does not pick up their medications on time, a pharmacist can contact the patient to determine why the medications were not picked up and to assess any nonadherence. In this case, mail order pharmacy services, if available, should be offered to patients as a more convenient option.
The medication regimen optimization component of MOSAIC helps reduce the workload of primary care providers and allows pharmacists to act autonomously based on clinical judgment, within the scope of the CPA. This can prevent delays in care caused by no refills remaining on a prescription. The laboratory monitoring component allows pharmacists to track diseases and take action if necessary, which should have a favorable impact on GPRA measures. Medication optimizations can reduce wasted resources by identifying cost-saving formulary alternatives, potentially inappropriate medications, and suboptimal doses.
Since many Indian Health Service beneficiaries do not have private insurance and therefore do not generate third-party reimbursements for services and care provided by GIMC, keeping patients healthy and out of the hospital is a top priority. As more patients are enrolled in MOSAIC, the program is expected to have a favorable impact on pharmacy workload and workflow as well. Prescriptions are anticipated and filled in advance, which decreases the amount of patients calling and presenting to the pharmacy for same-day refill requests. Scheduling when MOSAIC patients’ medications are to be filled and dispensed creates a predictable workload that allows the pharmacy staff to be managed more efficiently.
Conclusion
Adherence is the responsibility of the patient, but the Wheel Model of Pharmaceutical Care aims to provide pharmacists with a framework to monitor and encourage adherence in their patients. By taking this patient-centered approach, MOSAIC is expected to improve outcomes and decrease hospitalizations for high-risk patients who simply need a little extra help with their medications.
Nonadherence is a significant problem that has a negative impact on both patients and public health. Patients with multiple diseases often have complicated medication regimens, which can be difficult for them to manage. Unfortunately, nonadherence in these high-risk patients can have drastic consequences, including disease progression, hospitalization, and death, resulting in billions of dollars in unnecessary costs nationwide.1,2 The Wheel Model of Pharmaceutical Care (Figure) is a novel care model developed at the Gallup Indian Medical Center (GIMC) in New Mexico to address these problems by positioning pharmacy as a proactive service. The Wheel Model of Pharmaceutical Care was designed to improve adherence and patient outcomes and to encourage communication among the patient, pharmacists, prescribers, and other health care team members.
Pharmacists are central to managing patients’ medication therapies and coordinating communication among the health care providers (HCPs).1,3 Medication therapy management (MTM), a required component of Medicare Part D plans, helps ensure appropriate drug use and reduce the risk of adverse events.3 Since pharmacists receive prescriptions from all of the patient’s HCPs, patients may see pharmacists more often than they see any other HCP. GIMC is currently piloting a new clinic, the Medication Optimization, Synchronization, and Adherence Improvement Clinic (MOSAIC), that was created to implement the Wheel Model of Pharmaceutical Care. MOSAIC aims to provide proactive pharmacy services and continuous MTM to high-risk patients and will enable the effectiveness of this new pharmaceutical care model to be assessed.
Methods
Studies have identified certain populations who are at an increased risk for nonadherence: the elderly, patients with complex or extensive medication regimens, patients with multiple chronic medical conditions, substance misusers, certain ethnicities, patients of lower socioeconomic status, patients with limited literacy, and the homeless.2,4 Federal regulations require that Medicare Part D plans target beneficiaries who meet specific criteria for MTM programs. Under these rules, plans must target beneficiaries with ≥ 3 chronic diseases and ≥ 8 chronic medications, although plans also may include patients with fewer medications and diseases.3 Although the Wheel Model of Pharmaceutical Care is postulated to be an accurate model for the ideal care of all patients, initial implementation should be targeted toward populations who are likely to benefit the most from intervention. For these reasons, elderly Native American patients who have ≥ 2 chronic diseases and who take ≥ 5 chronic medications were targeted for initial enrollment in MOSAIC at GIMC.
Overview
In MOSAIC, pharmacists act as the hub of the pharmaceutical care wheel. Pharmacists work to ensure optimization of the patient’s comprehensive, integrated care plan—the rim of the wheel. As a part of this optimization process, MOSAIC pharmacists facilitate synchronization of the patient’s prescriptions to a monthly or quarterly target fill date. The patient’s current medication therapy is organized, and pharmacists track which medications are due to be filled instead of depending on the patient to request each prescription refill. This process effectively changes pharmacy from a requested service to a provided service.
Pharmacists also monitor the air in the tire to promote adherence. This is accomplished by providing efficient monthly or quarterly telephone or in-person consultations, which helps the patient better understand his or her comprehensive, integrated care plan. MOSAIC eliminates the possibility of nonadherence due to running out of refills. Specialized packaging, such as pill boxes or blister packs, can also improve adherence for certain patients.
MOSAIC ensures that pharmacists stay connected with the spokes, which represent a patient’s numerous prescribers, and close communication loops. Pharmacists can make prescribers aware of potential gaps or overlaps in treatment and assist them in the optimization and development of the patient’s comprehensive, integrated care plan. Pharmacists also make sure that the patient’s medication profile is current and accurate in the electronic health record (EHR). Any pertinent information discovered during MOSAIC encounters, such as abnormal laboratory results or changes in medications or disease, is documented in an EHR note. The patient’s prescribers are made aware of this information by tagging them as additional signers to the note in the EHR.
Keeping patients—the tires—healthy will ensure smooth operation of the vehicle and have a positive impact on public health. MOSAIC is expected to not only improve individual patient outcomes, but also decrease health care costs for patients and society due to nonadherence, suboptimal regimens, stockpiled home medications, and preventable hospital admissions.
Traditionally, pharmacy has been a requested service: A patient requests each of their prescriptions to be refilled, and the pharmacy fills the prescription. Ideally, pharmacy must become a provided service, with pharmacists keeping track of when a patient’s medications are due to be filled and actively looking for medication therapy optimization opportunities. This is accomplished by synchronizing the patient’s medications to the same monthly or quarterly fill date; screening for any potentially inappropriate medications, including high-risk medications in elderly patients, duplications, and omissions; verifying any medication changes with the patient each fill; and then providing all needed medications to the patient at a scheduled time.
To facilitate this process, custom software was developed for MOSAIC. In addition, a collaborative practice agreement (CPA) was drafted that allowed MOSAIC pharmacists to make certain medication therapy optimizations on behalf of the patient’s primary care provider. As part of this CPA, pharmacists also may order and act on certain laboratory tests, which helps to monitor disease progression, ensure safe medication use, and meet Government Performance and Results Act (GPRA) measures. As a novel model of pharmaceutical care, the effects of this approach are not yet known; however, research suggests that increased communication among HCPs and patient-centered approaches to care are beneficial to patient outcomes, adherence, and public health.1,5
Investigated Outcomes
As patients continue to enroll in MOSAIC, the effectiveness of the clinic will be evaluated. Specifically, quality of life, patient and HCP satisfaction with the program, adherence metrics, hospitalization rates, and all-cause mortality will be assessed for patients enrolled in MOSAIC as well as similar patients who are not enrolled in MOSAIC. Also, pharmacists will log all recommended medication therapy interventions so that the optimization component of MOSAIC may be quantified. GPRA measures and the financial implications of the interventions made by MOSAIC will also be evaluated.
Discussion
There are a number of factors, such as MTM services and interprofessional care teams, that research has shown to independently improve patient outcomes, adherence, or public health. By synthesizing these factors, a completely new approach—the Wheel Model of Pharmaceutical Care—was developed. This model presents a radical departure from traditional, requested-service practices and posits pharmacy as a provided service instead. Although the ideas of MTM and interprofessional care teams are not new, there has never been a practical way to truly integrate community pharmacists into the patient care team or to ensure adequate communication among all of the patient’s HCPs. The Wheel Model of Pharmaceutical Care includes public health as one of its core components and provides a framework for pharmacies to meaningfully impact health outcomes for patients.
The Wheel Model of Pharmaceutical Care was designed to minimize the likelihood of nonadherence. Despite this, patients might willfully choose to be nonadherent, forget to take their medications, or neglect to pick up their medications. Additionally, in health care systems where patients must pay for their medications, prescription drug costs might be a barrier to adherence.
When nonadherence is suspected, the Wheel Model of Pharmaceutical Care directs pharmacists in MOSAIC to take action. First, the underlying cause of the nonadherence must be determined. For example, if a patient is nonadherent because of an adverse drug reaction, a therapy change may be indicated. If a patient is nonadherent due to apathy toward their health or therapy, the patient may benefit from education about their condition and treatment options; thus, the patient can make shared, informed decisions and feel more actively involved with his or her health. If a patients is nonadherent due to forgetfulness, adherence packaging dispense methods should be considered as an alternative to traditional vials. Depending on the services offered by a given pharmacy, adherence packaging options may include blister packs, pill boxes, or strips prepared by robotic dispensing systems. The use of medication reminders, whether in the form of a smartphone application or a simple alarm clock, should be discussed with the patient. If the patient does not pick up their medications on time, a pharmacist can contact the patient to determine why the medications were not picked up and to assess any nonadherence. In this case, mail order pharmacy services, if available, should be offered to patients as a more convenient option.
The medication regimen optimization component of MOSAIC helps reduce the workload of primary care providers and allows pharmacists to act autonomously based on clinical judgment, within the scope of the CPA. This can prevent delays in care caused by no refills remaining on a prescription. The laboratory monitoring component allows pharmacists to track diseases and take action if necessary, which should have a favorable impact on GPRA measures. Medication optimizations can reduce wasted resources by identifying cost-saving formulary alternatives, potentially inappropriate medications, and suboptimal doses.
Since many Indian Health Service beneficiaries do not have private insurance and therefore do not generate third-party reimbursements for services and care provided by GIMC, keeping patients healthy and out of the hospital is a top priority. As more patients are enrolled in MOSAIC, the program is expected to have a favorable impact on pharmacy workload and workflow as well. Prescriptions are anticipated and filled in advance, which decreases the amount of patients calling and presenting to the pharmacy for same-day refill requests. Scheduling when MOSAIC patients’ medications are to be filled and dispensed creates a predictable workload that allows the pharmacy staff to be managed more efficiently.
Conclusion
Adherence is the responsibility of the patient, but the Wheel Model of Pharmaceutical Care aims to provide pharmacists with a framework to monitor and encourage adherence in their patients. By taking this patient-centered approach, MOSAIC is expected to improve outcomes and decrease hospitalizations for high-risk patients who simply need a little extra help with their medications.
1. Bosworth HB, Granger BB, Mendys P, et al. Medication adherence: a call for action. Am Heart J. 2011;162(3):412-424.
2. Vlasnik JJ, Aliotta SL, DeLor B. Medication adherence: factors influencing compliance with prescribed medication plans. Case Manager. 2005;16(2):47-51.
3. Drug utilization management, quality assurance, and medication therapy management programs (MTMPs). Fed Regist. 2012;77(71):2207-22175. To be codified at 42 CFR § 423.153.
4. Thiruchselvam T, Naglie G, Moineddin R, et al. Risk factors for medication nonadherence in older adults with cognitive impairment who live alone. Int J Geriatr Psychiatry. 2012;27(12):1275-1282.
5. Liddy C, Blazkho V, Mill K. Challenges of self-management when living with multiple chronic conditions: systematic review of the qualitative literature. Can Fam Physician. 2014;60(12):1123-1133.
1. Bosworth HB, Granger BB, Mendys P, et al. Medication adherence: a call for action. Am Heart J. 2011;162(3):412-424.
2. Vlasnik JJ, Aliotta SL, DeLor B. Medication adherence: factors influencing compliance with prescribed medication plans. Case Manager. 2005;16(2):47-51.
3. Drug utilization management, quality assurance, and medication therapy management programs (MTMPs). Fed Regist. 2012;77(71):2207-22175. To be codified at 42 CFR § 423.153.
4. Thiruchselvam T, Naglie G, Moineddin R, et al. Risk factors for medication nonadherence in older adults with cognitive impairment who live alone. Int J Geriatr Psychiatry. 2012;27(12):1275-1282.
5. Liddy C, Blazkho V, Mill K. Challenges of self-management when living with multiple chronic conditions: systematic review of the qualitative literature. Can Fam Physician. 2014;60(12):1123-1133.
Sacroiliac Joint Dysfunction in Patients With Low Back Pain
Patients experiencing sacroiliac joint (SIJ) dysfunction might show symptoms that overlap with those seen in lumbar spine pathology. This article reviews diagnostic tools that assist practitioners to discern the true pain generator in patients with low back pain (LBP) and therapeutic approaches when the cause is SIJ dysfunction.
Prevalence
Most of the US population will experience LBP at some point in their lives. A 2002 National Health Interview survey found that more than one-quarter (26.4%) of 31 044 respondents had complained of LBP in the previous 3 months.1 About 74 million individuals in the US experienced LBP in the past 3 months.1 A full 10% of the US population is expected to suffer from chronic LBP, and it is estimated that 2.3% of all visits to physicians are related to LBP.1
The etiology of LBP often is unclear even after thorough clinical and radiographic evaluation because of the myriad possible mechanisms. Degenerative disc disease, facet arthropathy, ligamentous hypertrophy, muscle spasm, hip arthropathy, and SIJ dysfunction are potential pain generators and exact clinical and radiographic correlation is not always possible. Compounding this difficulty is the lack of specificity with current diagnostic techniques. For example, many patients will have disc desiccation or herniation without any LBP or radicular symptoms on radiographic studies, such as X-rays, computed tomography (CT), and magnetic resonance imaging (MRI). As such, providers of patients with diffuse radiographic abnormalities often have to identify a specific pain generator, which might not have any role in the patient’s pain.
Other tests, such as electromyographic studies, positron emission tomography (PET) scans, discography, and epidural steroid injections, can help pinpoint a specific pain generator. These tests might help determine whether the patient has a surgically treatable condition and could help predict whether a patient’s symptoms will respond to surgery.
However, the standard spine surgery workup often fails to identify an obvious pain generator in many individuals. The significant number of patients that fall into this category has prompted spine surgeons to consider other potential etiologies for LBP, and SIJ dysfunction has become a rapidly developing field of research.
Sacroiliac Joint Dysfunction
The SIJ is a bilateral, C-shaped synovial joint surrounded by a fibrous capsule and affixes the sacrum to the ilia. Several sacral ligaments and pelvic muscles support the SIJ. The L5 nerve ventral ramus and lumbosacral trunk pass anteriorly and the S1 nerve ventral ramus passes inferiorly to the joint capsule. The SIJ is innervated by the dorsal rami of L4-S3 nerve roots, transmitting nociception and temperature. Mechanisms of injury to the SIJ could arise from intra- and extra-articular etiologies, including capsular disruption, ligamentous tension, muscular inflammation, shearing, fractures, arthritis, and infection.2 Patients could develop SIJ pain spontaneously or after a traumatic event or repetitive shear.3 Risk factors for developing SIJ dysfunction include a history of lumbar fusion, scoliosis, leg length discrepancies, sustained athletic activity, pregnancy, seronegative HLA-B27 spondyloarthropathies, or gait abnormalities. Inflammation of the SIJ and surrounding structures secondary to an environmental insult in susceptible individuals is a common theme among these etiologies.2
Pain from the SIJ is localized to an area of approximately 3 cm × 10 cm that is inferior to the ipsilateral posterior superior iliac spine.4 Referred pain maps from SIJ dysfunction extend in the L5-S1 nerve distributions, commonly seen in the buttocks, groin, posterior thigh, and lower leg with radicular symptoms. However, this pain distribution demonstrates extensive variability among patients and bears strong similarities to discogenic or facet joint sources of LBP.5-7 Direct communication has been shown between the SIJ and adjacent neural structures, namely the L5 nerve, sacral foramina, and the lumbosacral plexus. These direct pathways could explain an inflammatory mechanism for lower extremity symptoms seen in SIJ dysfunction.8
The prevalence of SIJ dysfunction among patients with LBP is estimated to be 15% to 30%, an extraordinary number given the total number of patients presenting with LBP every year.9 These patients might represent a significant segment of patients with an unrevealing standard spine evaluation. Despite the large number of patients who experience SIJ dysfunction, there is disagreement about optimal methods for diagnosis and treatment.
Diagnosis
The International Association for the Study of Pain has proposed criteria for evaluating patients who have suspected SIJ dysfunction: Pain must be in the SIJ area, should be reproducible by performing specific provocative maneuvers, and must be relieved by injection of local anesthetic into the SIJ.10 These criteria provide a sound foundation, but in clinical practice, patients often defy categorization.
The presence of pain in the area inferior to the posterior superior iliac spine and lateral to the gluteal fold with pain referral patterns in the L5-S1 nerve distributions is highly sensitive for identifying patients with SIJ dysfunction. Furthermore, pain arising from the SIJ will not be above the level of the L5 nerve sensory distribution. However, this diagnostic finding alone is not specific and might represent other etiologies known to produce similar pain, such as intervertebral discs and facet joints. Patients with SIJ dysfunction often describe their pain as sciatica-like, recurrent, and triggered with bending or twisting motions. It is worsened with any activity loading the SIJ, such as walking, climbing stairs, standing, or sitting upright. SIJ pain might be accompanied by dyspareunia and changes in bladder function because of the nerves involved.11
The use of provocative maneuvers for testing SIJ dysfunction is controversial because of the high rate of false positives and the inability to distinguish whether the SIJ or an adjacent structure is affected. However, the diagnostic utility of specific stress tests has been studied, and clusters of tests are recommended if a health care provider (HCP) suspects SIJ dysfunction. A diagnostic algorithm should first focus on using the distraction test and the thigh thrust test. Distraction is done by applying vertically oriented pressure to the anterior superior iliac spine while aiming posteriorly, therefore distracting the SIJ. During the thigh thrust test the examiner fixates the patient’s sacrum against the table with the left hand and applies a vertical force through the line of the femur aiming posteriorly, producing a posterior shearing force at the SIJ. Studies show that the thigh thrust test is the most sensitive, and the distraction test is the most specific. If both tests are positive, there is reasonable evidence to suggest SIJ dysfunction as the source of LBP.
If there are not 2 positive results, the addition of the compression test, followed by the sacral thrust test also can point to the diagnosis. The compression test is performed with vertical downward force applied to the iliac crest with the patient lying on each side, compressing the SIJ by transverse pressure across the pelvis. The sacral thrust test is performed with vertical force applied to the midline posterior sacrum at its apex directed anteriorly with the patient lying prone, producing a shearing force at the SIJs. The Gaenslen test uses a torsion force by applying a superior and posterior force to the right knee and posteriorly directed force to the left knee. Omitting the Gaenslen test has not been shown to compromise diagnostic efficacy of the other tests and can be safely excluded.12
A HCP can rule out SIJ dysfunction if these provocation tests are negative. However, the diagnostic predictive value of these tests is subject to variability among HCPs, and their reliability is increased when used in clusters.9,13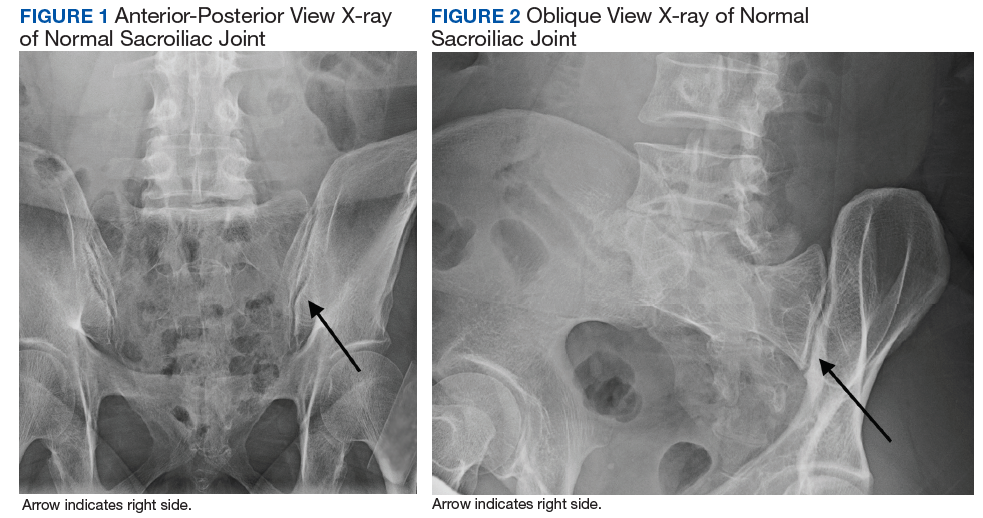
Imaging for the SIJ should begin with anterior/posterior, oblique, and lateral view plain X-rays of the pelvis (Figures 1 and 2), which will rule out other pathologies by identifying other sources of LBP, such as spondylolisthesis or hip osteoarthritis. HCPs should obtain lumbar and pelvis CT images to identify inflammatory or degenerative changes within the SIJ. CT images provide the high resolution that is needed to identify pathologies, such as fractures and tumors within the pelvic ring that could cause similar pain. MRI does not reliably depict a dysfunctional ligamentous apparatus within the SIJ; however, it can help identify inflammatory sacroiliitis, such as is seen in the spondyloarthropathies.11,14 Recent studies show combined single photon emission tomography and CT (SPECT-CT) might be the most promising imaging modality to reveal mechanical failure of load transfer with increased scintigraphic uptake in the posterior and superior SIJ ligamentous attachments. The joint loses its characteristic “dumbbell” shape in affected patients with about 50% higher uptake than unaffected joints. These findings were evident in patients who experienced pelvic trauma or during the peripartum period.15,16

Fluoroscopy-guided intra-articular injection of a local anesthetic (lidocaine) and/or a corticosteroid (triamcinolone) has the dual functionality of diagnosis and treatment (Figure 3). It often is considered the most reliable method to diagnose SIJ dysfunction and has the benefit of pain relief for up to 1 year. However, intra-articular injections lack diagnostic validity because the solution often extravasates to extracapsular structures. This confounds the source of the pain and makes it difficult to interpret these diagnostic injections. In addition, the injection might not reach the entire SIJ capsule and could result in a false-negative diagnosis.17,18 Periarticular injections have been shown to result in better pain relief in patients diagnosed with SIJ dysfunction than intra-articular injections. Periarticular injections also are easier to perform and could be a first-step option for these patients.19
Treatment
Nonoperative management of SIJ dysfunction includes exercise programs, physical therapy, manual manipulation therapy, sacroiliac belts, and periodic articular injections. Efficacy of these methods is variable, and analgesics often do not significantly benefit this type of pain. Another nonoperative approach is radiofrequency ablation (RFA) of the lumbar dorsal rami and lateral sacral branches, which can vary based on the number of rami treated as well as the technique used. About two-thirds of patients report pain relief after RFA.2 When successful, pain is relieved for 6 to 12 months, which is a temporary yet effective option for patients experiencing SIJ dysfunction.14,20
Fusion Surgery
Cadaver studies show that biomechanical stabilization of the SIJ leads to decreased range of motion in flexion/extension, lateral bending, and axial rotation. This results in a decreased need for periarticular muscular and ligamentous support, therefore facilitating load transfer across the SIJ.21,22 Patients undergoing minimally invasive surgery report better pain relief compared with those receiving open surgery at 12 months postoperatively.23 The 2 main SIJ fusion approaches used are the lateral transarticular and the dorsal approaches. In the dorsal approach, the SIJ is distracted and allograft dowels or titanium cages with graft are inserted into the joint space posteriorly through the back. When approaching laterally, hollow screw implants filled with graft or triangular titanium implants are placed across the joint, accessing the SIJ through the iliac bones using imaging guidance. This lateral transiliac approach using porous titanium triangular rods currently is the most studied technique.24
A recent prospective, multicenter trial included 423 patients with SIJ dysfunction who were randomized to receive SIJ fusion with triangular titanium implants vs a control group who received nonoperative management. Patients in the SIJ fusion group showed substantially greater improvement in pain (81.4%) compared with that of the nonoperative group (26.1%) 6 months after surgery. Pain relief in the SIJ fusion group was maintained at > 80% at 1 and 2 year follow-up, while the nonoperative group’s pain relief decreased to < 10% at the follow-ups. Measures of quality of life and disability also improved for the SIJ fusion group compared with that of the nonoperative group. Patients who were crossed over from conservative management to SIJ fusion after 6 months demonstrated improvements that were similar to those in the SIJ fusion group by the end of the study. Only 3% of patients required surgical revision. The strongest predictor of pain relief after surgery was a diagnostic SIJ anesthetic block of 30 to 60 minutes, which resulted in > 75% pain reduction.21,25 Additional predictors of successful SIJ fusion include nonsmokers, nonopioid users, and older patients who have a longer time course of SIJ pain.26
Another study investigating the outcomes of SIJ fusion, RFA, and conservative management with a 6-year follow-up demonstrated similar results.27 This further confirms the durability of the surgical group’s outcome, which sustained significant improvement compared with RFA and conservative management group in pain relief, daily function, and opioid use.
HCPs should consider SIJ fusion for patients who have at least 6 months of unsuccessful nonoperative management, significant SIJ pain (> 5 in a 10-point scale), ≥ 3 positive provocation tests, and at least 50% pain relief (> 75% preferred) with diagnostic intra-articular anesthetic injection.14 It is reasonable for primary care providers to refer these patients to a neurosurgeon or orthopedic spine surgeon for possible fusion. Patients with earlier lumbar/lumbosacral spinal fusions and persistent LBP should be evaluated for potential SIJ dysfunction. SIJ dysfunction after lumbosacral fusion could be considered a form of distal pseudarthrosis resulting from increased motion at the joint. One study found its incidence correlated with the number of segments fused in the lumbar spine.28 Another study found that about one-third of patients with persistent LBP after lumbosacral fusion could be attributed to SIJ dysfunction.29
Case Presentation
A 27-year-old female army veteran presented with bilateral buttock pain, which she described as a dull, aching pain across her sacral region, 8 out of 10 in severity. The pain was in a L5-S1 pattern. The pain was bilateral, with the right side worse than the left, and worsened with lateral bending and load transferring. She reported no numbness, tingling, or weakness.
On physical examination, she had full strength in her lower extremities and intact sensation. She reported tenderness to palpation of the sacrum and SIJ. Her gait was normal. The patient had positive thigh thrust and distraction tests. Lumbar spine X-ray, CT, MRI, and electromyographic studies did not show any pathology. She described little or no relief with analgesics or physical therapy. Previous L4-L5 and L5-S1 facet anesthetic injections and transforaminal epidural steroid injections provided minimal pain relief immediately after the procedures. Bilateral SIJ anesthetic injections under fluoroscopic guidance decreased her pain severity from a 7 to 3 out of 10 for 2 to 3 months before returning to her baseline. Radiofrequency ablation of the right SIJ under fluoroscopy provided moderate relief for about 4 months.
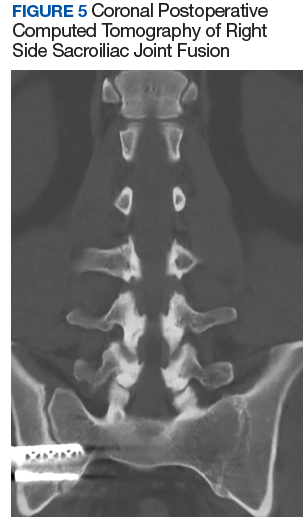
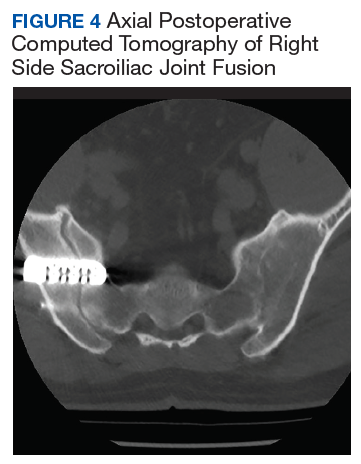
After exhausting nonoperative management for SIJ dysfunction without adequate pain control, the patient was referred to neurosurgery for surgical fusion. The patient was deemed an appropriate surgical candidate and underwent a right-sided SIJ fusion (Figures 4 and 5). At her 6-month and 1-year follow-up appointments, she had lasting pain relief, 2 out of 10.
Conclusion
SIJ dysfunction is widely overlooked because of the difficulty in distinguishing it from other similarly presenting syndromes. However, with a detailed history, appropriate physical maneuvers, imaging, and adequate response to intra-articular anesthetic, providers can reach an accurate diagnosis that will inform subsequent treatments. After failure of nonsurgical methods, patients with SIJ dysfunction should be considered for minimally invasive fusion techniques, which have proven to be a safe, effective, and viable treatment option.
1. Zaidi HA, Montoure AJ, Dickman CA. Surgical and clinical efficacy of sacroiliac joint fusion: a systematic review of the literature. J Neurosurg Spine. 2015;23(1):59-66.
2. Cohen SP. Sacroiliac joint pain: a comprehensive review of anatomy, diagnosis, and treatment. Anesth Analg. 2005;101(5):1440-1453.
3. Chou LH, Slipman CW, Bhagia SM, et al. Inciting events initiating injection‐proven sacroiliac joint syndrome. Pain Med. 2004;5(1):26-32.
4. Dreyfuss P, Dreyer SJ, Cole A, Mayo K. Sacroiliac joint pain. J Am Acad Orthop Surg. 2004;12(4):255-265.
5. Buijs E, Visser L, Groen G. Sciatica and the sacroiliac joint: a forgotten concept. Br J Anaesth. 2007;99(5):713-716.
6. Fortin JD, Dwyer AP, West S, Pier J. Sacroiliac joint: pain referral maps upon applying a new injection/arthrography technique. Part I: asymptomatic volunteers. Spine (Phila Pa 1976). 1994;19(13):1475-1482.
7. Schwarzer AC, Aprill CN, Bogduk N. The sacroiliac joint in chronic low back pain. Spine (Phila Pa 1976). 1995;20(1):31-37.
8. Fortin JD, Washington WJ, Falco FJ. Three pathways between the sacroiliac joint and neural structures. ANJR Am J Neuroradiol. 1999;20(8):1429-1434.
9. Szadek KM, van der Wurff P, van Tulder MW, Zuurmond WW, Perez RS. Diagnostic validity of criteria for sacroiliac joint pain: a systematic review. J Pain. 2009;10(4):354-368.
10. Merskey H, Bogduk N, eds. Classification of Chronic Pain: Descriptions of Chronic Pain Syndromes and Definitions of Pain Terms. 2nd ed. Seattle, WA: IASP Press; 1994.
11. Cusi MF. Paradigm for assessment and treatment of SIJ mechanical dysfunction. J Bodyw Mov Ther. 2010;14(2):152-161.
12. Laslett M, Aprill CN, McDonald B, Young SB. Diagnosis of sacroiliac joint pain: validity of individual provocation tests and composites of tests. Man Ther. 2005;10(3):207-218.
13. Laslett M. Evidence-based diagnosis and treatment of the painful sacroiliac joint. J Man Manip Ther. 2008;16(3):142-152.
14. Polly DW Jr. The sacroiliac joint. Neurosurg Clin N Am. 2017;28(3):301-312.
15. Cusi M, Van Der Wall H, Saunders J, Fogelman I. Metabolic disturbances identified by SPECT-CT in patients with a clinical diagnosis of sacroiliac joint incompetence. Eur Spine J. 2013;22(7):1674-1682.
16. Tofuku K, Koga H, Komiya S. The diagnostic value of single-photon emission computed tomography/computed tomography for severe sacroiliac joint dysfunction. Eur Spine J. 2015;24(4):859-863.
17. Kennedy DJ, Engel A, Kreiner DS, Nampiaparampil D, Duszynski B, MacVicar J. Fluoroscopically guided diagnostic and therapeutic intra‐articular sacroiliac joint injections: a systematic review. Pain Med. 2015;16(8):1500-1518.
18. Schneider BJ, Huynh L, Levin J, Rinkaekan P, Kordi R, Kennedy DJ. Does immediate pain relief after an injection into the sacroiliac joint with anesthetic and corticosteroid predict subsequent pain relief? Pain Med. 2018;19(2):244-251.
19. Murakami E, Tanaka Y, Aizawa T, Ishizuka M, Kokubun S. Effect of periarticular and intraarticular lidocaine injections for sacroiliac joint pain: prospective comparative study. J Orthop Sci. 2007;12(3):274-280.
20. Cohen SP, Hurley RW, Buckenmaier CC 3rd, Kurihara C, Morlando B, Dragovich A. Randomized placebo-controlled study evaluating lateral branch radiofrequency denervation for sacroiliac joint pain. Anesthesiology. 2008;109(2):279-288.
21. Polly DW, Cher DJ, Wine KD, et al; INSITE Study Group. Randomized controlled trial of minimally invasive sacroiliac joint fusion using triangular titanium implants vs nonsurgical management for sacroiliac joint dysfunction: 12-month outcomes. Neurosurgery. 2015;77(5):674-690.
22. Soriano-Baron H, Lindsey DP, Rodriguez-Martinez N, et al. The effect of implant placement on sacroiliac joint range of motion: posterior versus transarticular. Spine. 2015;40(9):E525-E530.
23. Smith AG, Capobianco R, Cher D, et al. Open versus minimally invasive sacroiliac joint fusion: a multi-center comparison of perioperative measures and clinical outcomes. Ann Surg Innov Res. 2013;7(1):14.
24. Rashbaum RF, Ohnmeiss DD, Lindley EM, Kitchel SH, Patel VV. Sacroiliac joint pain and its treatment. Clin Spine Surg. 2016;29(2):42-48.
25. Polly DW, Swofford J, Whang PG, et al. Two-year outcomes from a randomized controlled trial of minimally invasive sacroiliac joint fusion vs. non-surgical management for sacroiliac joint dysfunction. Int J Spine Surg. 2016;10:28.
26. Dengler J, Duhon B, Whang P, et al. Predictors of outcome in conservative and minimally invasive surgical management of pain originating from the sacroiliac joint: a pooled analysis. Spine (Phila Pa 1976). 2017;42(21):1664-1673.
27. Vanaclocha V, Herrera JM, Sáiz-Sapena N, Rivera-Paz M, Verdú-López F. Minimally invasive sacroiliac joint fusion, radiofrequency denervation, and conservative management for sacroiliac joint pain: 6-year comparative case series. Neurosurgery. 2018;82(1):48-55.
28. Unoki E, Abe E, Murai H, Kobayashi T, Abe T. Fusion of multiple segments can increase the incidence of sacroiliac joint pain after lumbar or lumbosacral fusion. Spine (Phila Pa 1976). 2016;41(12):999-1005.
29. Katz V, Schofferman J, Reynolds J. The sacroiliac joint: a potential cause of pain after lumbar fusion to the sacrum. J Spinal Disord Tech. 2003;16(1):96-99.
Patients experiencing sacroiliac joint (SIJ) dysfunction might show symptoms that overlap with those seen in lumbar spine pathology. This article reviews diagnostic tools that assist practitioners to discern the true pain generator in patients with low back pain (LBP) and therapeutic approaches when the cause is SIJ dysfunction.
Prevalence
Most of the US population will experience LBP at some point in their lives. A 2002 National Health Interview survey found that more than one-quarter (26.4%) of 31 044 respondents had complained of LBP in the previous 3 months.1 About 74 million individuals in the US experienced LBP in the past 3 months.1 A full 10% of the US population is expected to suffer from chronic LBP, and it is estimated that 2.3% of all visits to physicians are related to LBP.1
The etiology of LBP often is unclear even after thorough clinical and radiographic evaluation because of the myriad possible mechanisms. Degenerative disc disease, facet arthropathy, ligamentous hypertrophy, muscle spasm, hip arthropathy, and SIJ dysfunction are potential pain generators and exact clinical and radiographic correlation is not always possible. Compounding this difficulty is the lack of specificity with current diagnostic techniques. For example, many patients will have disc desiccation or herniation without any LBP or radicular symptoms on radiographic studies, such as X-rays, computed tomography (CT), and magnetic resonance imaging (MRI). As such, providers of patients with diffuse radiographic abnormalities often have to identify a specific pain generator, which might not have any role in the patient’s pain.
Other tests, such as electromyographic studies, positron emission tomography (PET) scans, discography, and epidural steroid injections, can help pinpoint a specific pain generator. These tests might help determine whether the patient has a surgically treatable condition and could help predict whether a patient’s symptoms will respond to surgery.
However, the standard spine surgery workup often fails to identify an obvious pain generator in many individuals. The significant number of patients that fall into this category has prompted spine surgeons to consider other potential etiologies for LBP, and SIJ dysfunction has become a rapidly developing field of research.
Sacroiliac Joint Dysfunction
The SIJ is a bilateral, C-shaped synovial joint surrounded by a fibrous capsule and affixes the sacrum to the ilia. Several sacral ligaments and pelvic muscles support the SIJ. The L5 nerve ventral ramus and lumbosacral trunk pass anteriorly and the S1 nerve ventral ramus passes inferiorly to the joint capsule. The SIJ is innervated by the dorsal rami of L4-S3 nerve roots, transmitting nociception and temperature. Mechanisms of injury to the SIJ could arise from intra- and extra-articular etiologies, including capsular disruption, ligamentous tension, muscular inflammation, shearing, fractures, arthritis, and infection.2 Patients could develop SIJ pain spontaneously or after a traumatic event or repetitive shear.3 Risk factors for developing SIJ dysfunction include a history of lumbar fusion, scoliosis, leg length discrepancies, sustained athletic activity, pregnancy, seronegative HLA-B27 spondyloarthropathies, or gait abnormalities. Inflammation of the SIJ and surrounding structures secondary to an environmental insult in susceptible individuals is a common theme among these etiologies.2
Pain from the SIJ is localized to an area of approximately 3 cm × 10 cm that is inferior to the ipsilateral posterior superior iliac spine.4 Referred pain maps from SIJ dysfunction extend in the L5-S1 nerve distributions, commonly seen in the buttocks, groin, posterior thigh, and lower leg with radicular symptoms. However, this pain distribution demonstrates extensive variability among patients and bears strong similarities to discogenic or facet joint sources of LBP.5-7 Direct communication has been shown between the SIJ and adjacent neural structures, namely the L5 nerve, sacral foramina, and the lumbosacral plexus. These direct pathways could explain an inflammatory mechanism for lower extremity symptoms seen in SIJ dysfunction.8
The prevalence of SIJ dysfunction among patients with LBP is estimated to be 15% to 30%, an extraordinary number given the total number of patients presenting with LBP every year.9 These patients might represent a significant segment of patients with an unrevealing standard spine evaluation. Despite the large number of patients who experience SIJ dysfunction, there is disagreement about optimal methods for diagnosis and treatment.
Diagnosis
The International Association for the Study of Pain has proposed criteria for evaluating patients who have suspected SIJ dysfunction: Pain must be in the SIJ area, should be reproducible by performing specific provocative maneuvers, and must be relieved by injection of local anesthetic into the SIJ.10 These criteria provide a sound foundation, but in clinical practice, patients often defy categorization.
The presence of pain in the area inferior to the posterior superior iliac spine and lateral to the gluteal fold with pain referral patterns in the L5-S1 nerve distributions is highly sensitive for identifying patients with SIJ dysfunction. Furthermore, pain arising from the SIJ will not be above the level of the L5 nerve sensory distribution. However, this diagnostic finding alone is not specific and might represent other etiologies known to produce similar pain, such as intervertebral discs and facet joints. Patients with SIJ dysfunction often describe their pain as sciatica-like, recurrent, and triggered with bending or twisting motions. It is worsened with any activity loading the SIJ, such as walking, climbing stairs, standing, or sitting upright. SIJ pain might be accompanied by dyspareunia and changes in bladder function because of the nerves involved.11
The use of provocative maneuvers for testing SIJ dysfunction is controversial because of the high rate of false positives and the inability to distinguish whether the SIJ or an adjacent structure is affected. However, the diagnostic utility of specific stress tests has been studied, and clusters of tests are recommended if a health care provider (HCP) suspects SIJ dysfunction. A diagnostic algorithm should first focus on using the distraction test and the thigh thrust test. Distraction is done by applying vertically oriented pressure to the anterior superior iliac spine while aiming posteriorly, therefore distracting the SIJ. During the thigh thrust test the examiner fixates the patient’s sacrum against the table with the left hand and applies a vertical force through the line of the femur aiming posteriorly, producing a posterior shearing force at the SIJ. Studies show that the thigh thrust test is the most sensitive, and the distraction test is the most specific. If both tests are positive, there is reasonable evidence to suggest SIJ dysfunction as the source of LBP.
If there are not 2 positive results, the addition of the compression test, followed by the sacral thrust test also can point to the diagnosis. The compression test is performed with vertical downward force applied to the iliac crest with the patient lying on each side, compressing the SIJ by transverse pressure across the pelvis. The sacral thrust test is performed with vertical force applied to the midline posterior sacrum at its apex directed anteriorly with the patient lying prone, producing a shearing force at the SIJs. The Gaenslen test uses a torsion force by applying a superior and posterior force to the right knee and posteriorly directed force to the left knee. Omitting the Gaenslen test has not been shown to compromise diagnostic efficacy of the other tests and can be safely excluded.12
A HCP can rule out SIJ dysfunction if these provocation tests are negative. However, the diagnostic predictive value of these tests is subject to variability among HCPs, and their reliability is increased when used in clusters.9,13
Imaging for the SIJ should begin with anterior/posterior, oblique, and lateral view plain X-rays of the pelvis (Figures 1 and 2), which will rule out other pathologies by identifying other sources of LBP, such as spondylolisthesis or hip osteoarthritis. HCPs should obtain lumbar and pelvis CT images to identify inflammatory or degenerative changes within the SIJ. CT images provide the high resolution that is needed to identify pathologies, such as fractures and tumors within the pelvic ring that could cause similar pain. MRI does not reliably depict a dysfunctional ligamentous apparatus within the SIJ; however, it can help identify inflammatory sacroiliitis, such as is seen in the spondyloarthropathies.11,14 Recent studies show combined single photon emission tomography and CT (SPECT-CT) might be the most promising imaging modality to reveal mechanical failure of load transfer with increased scintigraphic uptake in the posterior and superior SIJ ligamentous attachments. The joint loses its characteristic “dumbbell” shape in affected patients with about 50% higher uptake than unaffected joints. These findings were evident in patients who experienced pelvic trauma or during the peripartum period.15,16
Fluoroscopy-guided intra-articular injection of a local anesthetic (lidocaine) and/or a corticosteroid (triamcinolone) has the dual functionality of diagnosis and treatment (Figure 3). It often is considered the most reliable method to diagnose SIJ dysfunction and has the benefit of pain relief for up to 1 year. However, intra-articular injections lack diagnostic validity because the solution often extravasates to extracapsular structures. This confounds the source of the pain and makes it difficult to interpret these diagnostic injections. In addition, the injection might not reach the entire SIJ capsule and could result in a false-negative diagnosis.17,18 Periarticular injections have been shown to result in better pain relief in patients diagnosed with SIJ dysfunction than intra-articular injections. Periarticular injections also are easier to perform and could be a first-step option for these patients.19
Treatment
Nonoperative management of SIJ dysfunction includes exercise programs, physical therapy, manual manipulation therapy, sacroiliac belts, and periodic articular injections. Efficacy of these methods is variable, and analgesics often do not significantly benefit this type of pain. Another nonoperative approach is radiofrequency ablation (RFA) of the lumbar dorsal rami and lateral sacral branches, which can vary based on the number of rami treated as well as the technique used. About two-thirds of patients report pain relief after RFA.2 When successful, pain is relieved for 6 to 12 months, which is a temporary yet effective option for patients experiencing SIJ dysfunction.14,20
Fusion Surgery
Cadaver studies show that biomechanical stabilization of the SIJ leads to decreased range of motion in flexion/extension, lateral bending, and axial rotation. This results in a decreased need for periarticular muscular and ligamentous support, therefore facilitating load transfer across the SIJ.21,22 Patients undergoing minimally invasive surgery report better pain relief compared with those receiving open surgery at 12 months postoperatively.23 The 2 main SIJ fusion approaches used are the lateral transarticular and the dorsal approaches. In the dorsal approach, the SIJ is distracted and allograft dowels or titanium cages with graft are inserted into the joint space posteriorly through the back. When approaching laterally, hollow screw implants filled with graft or triangular titanium implants are placed across the joint, accessing the SIJ through the iliac bones using imaging guidance. This lateral transiliac approach using porous titanium triangular rods currently is the most studied technique.24
A recent prospective, multicenter trial included 423 patients with SIJ dysfunction who were randomized to receive SIJ fusion with triangular titanium implants vs a control group who received nonoperative management. Patients in the SIJ fusion group showed substantially greater improvement in pain (81.4%) compared with that of the nonoperative group (26.1%) 6 months after surgery. Pain relief in the SIJ fusion group was maintained at > 80% at 1 and 2 year follow-up, while the nonoperative group’s pain relief decreased to < 10% at the follow-ups. Measures of quality of life and disability also improved for the SIJ fusion group compared with that of the nonoperative group. Patients who were crossed over from conservative management to SIJ fusion after 6 months demonstrated improvements that were similar to those in the SIJ fusion group by the end of the study. Only 3% of patients required surgical revision. The strongest predictor of pain relief after surgery was a diagnostic SIJ anesthetic block of 30 to 60 minutes, which resulted in > 75% pain reduction.21,25 Additional predictors of successful SIJ fusion include nonsmokers, nonopioid users, and older patients who have a longer time course of SIJ pain.26
Another study investigating the outcomes of SIJ fusion, RFA, and conservative management with a 6-year follow-up demonstrated similar results.27 This further confirms the durability of the surgical group’s outcome, which sustained significant improvement compared with RFA and conservative management group in pain relief, daily function, and opioid use.
HCPs should consider SIJ fusion for patients who have at least 6 months of unsuccessful nonoperative management, significant SIJ pain (> 5 in a 10-point scale), ≥ 3 positive provocation tests, and at least 50% pain relief (> 75% preferred) with diagnostic intra-articular anesthetic injection.14 It is reasonable for primary care providers to refer these patients to a neurosurgeon or orthopedic spine surgeon for possible fusion. Patients with earlier lumbar/lumbosacral spinal fusions and persistent LBP should be evaluated for potential SIJ dysfunction. SIJ dysfunction after lumbosacral fusion could be considered a form of distal pseudarthrosis resulting from increased motion at the joint. One study found its incidence correlated with the number of segments fused in the lumbar spine.28 Another study found that about one-third of patients with persistent LBP after lumbosacral fusion could be attributed to SIJ dysfunction.29
Case Presentation
A 27-year-old female army veteran presented with bilateral buttock pain, which she described as a dull, aching pain across her sacral region, 8 out of 10 in severity. The pain was in a L5-S1 pattern. The pain was bilateral, with the right side worse than the left, and worsened with lateral bending and load transferring. She reported no numbness, tingling, or weakness.
On physical examination, she had full strength in her lower extremities and intact sensation. She reported tenderness to palpation of the sacrum and SIJ. Her gait was normal. The patient had positive thigh thrust and distraction tests. Lumbar spine X-ray, CT, MRI, and electromyographic studies did not show any pathology. She described little or no relief with analgesics or physical therapy. Previous L4-L5 and L5-S1 facet anesthetic injections and transforaminal epidural steroid injections provided minimal pain relief immediately after the procedures. Bilateral SIJ anesthetic injections under fluoroscopic guidance decreased her pain severity from a 7 to 3 out of 10 for 2 to 3 months before returning to her baseline. Radiofrequency ablation of the right SIJ under fluoroscopy provided moderate relief for about 4 months.
After exhausting nonoperative management for SIJ dysfunction without adequate pain control, the patient was referred to neurosurgery for surgical fusion. The patient was deemed an appropriate surgical candidate and underwent a right-sided SIJ fusion (Figures 4 and 5). At her 6-month and 1-year follow-up appointments, she had lasting pain relief, 2 out of 10.
Conclusion
SIJ dysfunction is widely overlooked because of the difficulty in distinguishing it from other similarly presenting syndromes. However, with a detailed history, appropriate physical maneuvers, imaging, and adequate response to intra-articular anesthetic, providers can reach an accurate diagnosis that will inform subsequent treatments. After failure of nonsurgical methods, patients with SIJ dysfunction should be considered for minimally invasive fusion techniques, which have proven to be a safe, effective, and viable treatment option.
Patients experiencing sacroiliac joint (SIJ) dysfunction might show symptoms that overlap with those seen in lumbar spine pathology. This article reviews diagnostic tools that assist practitioners to discern the true pain generator in patients with low back pain (LBP) and therapeutic approaches when the cause is SIJ dysfunction.
Prevalence
Most of the US population will experience LBP at some point in their lives. A 2002 National Health Interview survey found that more than one-quarter (26.4%) of 31 044 respondents had complained of LBP in the previous 3 months.1 About 74 million individuals in the US experienced LBP in the past 3 months.1 A full 10% of the US population is expected to suffer from chronic LBP, and it is estimated that 2.3% of all visits to physicians are related to LBP.1
The etiology of LBP often is unclear even after thorough clinical and radiographic evaluation because of the myriad possible mechanisms. Degenerative disc disease, facet arthropathy, ligamentous hypertrophy, muscle spasm, hip arthropathy, and SIJ dysfunction are potential pain generators and exact clinical and radiographic correlation is not always possible. Compounding this difficulty is the lack of specificity with current diagnostic techniques. For example, many patients will have disc desiccation or herniation without any LBP or radicular symptoms on radiographic studies, such as X-rays, computed tomography (CT), and magnetic resonance imaging (MRI). As such, providers of patients with diffuse radiographic abnormalities often have to identify a specific pain generator, which might not have any role in the patient’s pain.
Other tests, such as electromyographic studies, positron emission tomography (PET) scans, discography, and epidural steroid injections, can help pinpoint a specific pain generator. These tests might help determine whether the patient has a surgically treatable condition and could help predict whether a patient’s symptoms will respond to surgery.
However, the standard spine surgery workup often fails to identify an obvious pain generator in many individuals. The significant number of patients that fall into this category has prompted spine surgeons to consider other potential etiologies for LBP, and SIJ dysfunction has become a rapidly developing field of research.
Sacroiliac Joint Dysfunction
The SIJ is a bilateral, C-shaped synovial joint surrounded by a fibrous capsule and affixes the sacrum to the ilia. Several sacral ligaments and pelvic muscles support the SIJ. The L5 nerve ventral ramus and lumbosacral trunk pass anteriorly and the S1 nerve ventral ramus passes inferiorly to the joint capsule. The SIJ is innervated by the dorsal rami of L4-S3 nerve roots, transmitting nociception and temperature. Mechanisms of injury to the SIJ could arise from intra- and extra-articular etiologies, including capsular disruption, ligamentous tension, muscular inflammation, shearing, fractures, arthritis, and infection.2 Patients could develop SIJ pain spontaneously or after a traumatic event or repetitive shear.3 Risk factors for developing SIJ dysfunction include a history of lumbar fusion, scoliosis, leg length discrepancies, sustained athletic activity, pregnancy, seronegative HLA-B27 spondyloarthropathies, or gait abnormalities. Inflammation of the SIJ and surrounding structures secondary to an environmental insult in susceptible individuals is a common theme among these etiologies.2
Pain from the SIJ is localized to an area of approximately 3 cm × 10 cm that is inferior to the ipsilateral posterior superior iliac spine.4 Referred pain maps from SIJ dysfunction extend in the L5-S1 nerve distributions, commonly seen in the buttocks, groin, posterior thigh, and lower leg with radicular symptoms. However, this pain distribution demonstrates extensive variability among patients and bears strong similarities to discogenic or facet joint sources of LBP.5-7 Direct communication has been shown between the SIJ and adjacent neural structures, namely the L5 nerve, sacral foramina, and the lumbosacral plexus. These direct pathways could explain an inflammatory mechanism for lower extremity symptoms seen in SIJ dysfunction.8
The prevalence of SIJ dysfunction among patients with LBP is estimated to be 15% to 30%, an extraordinary number given the total number of patients presenting with LBP every year.9 These patients might represent a significant segment of patients with an unrevealing standard spine evaluation. Despite the large number of patients who experience SIJ dysfunction, there is disagreement about optimal methods for diagnosis and treatment.
Diagnosis
The International Association for the Study of Pain has proposed criteria for evaluating patients who have suspected SIJ dysfunction: Pain must be in the SIJ area, should be reproducible by performing specific provocative maneuvers, and must be relieved by injection of local anesthetic into the SIJ.10 These criteria provide a sound foundation, but in clinical practice, patients often defy categorization.
The presence of pain in the area inferior to the posterior superior iliac spine and lateral to the gluteal fold with pain referral patterns in the L5-S1 nerve distributions is highly sensitive for identifying patients with SIJ dysfunction. Furthermore, pain arising from the SIJ will not be above the level of the L5 nerve sensory distribution. However, this diagnostic finding alone is not specific and might represent other etiologies known to produce similar pain, such as intervertebral discs and facet joints. Patients with SIJ dysfunction often describe their pain as sciatica-like, recurrent, and triggered with bending or twisting motions. It is worsened with any activity loading the SIJ, such as walking, climbing stairs, standing, or sitting upright. SIJ pain might be accompanied by dyspareunia and changes in bladder function because of the nerves involved.11
The use of provocative maneuvers for testing SIJ dysfunction is controversial because of the high rate of false positives and the inability to distinguish whether the SIJ or an adjacent structure is affected. However, the diagnostic utility of specific stress tests has been studied, and clusters of tests are recommended if a health care provider (HCP) suspects SIJ dysfunction. A diagnostic algorithm should first focus on using the distraction test and the thigh thrust test. Distraction is done by applying vertically oriented pressure to the anterior superior iliac spine while aiming posteriorly, therefore distracting the SIJ. During the thigh thrust test the examiner fixates the patient’s sacrum against the table with the left hand and applies a vertical force through the line of the femur aiming posteriorly, producing a posterior shearing force at the SIJ. Studies show that the thigh thrust test is the most sensitive, and the distraction test is the most specific. If both tests are positive, there is reasonable evidence to suggest SIJ dysfunction as the source of LBP.
If there are not 2 positive results, the addition of the compression test, followed by the sacral thrust test also can point to the diagnosis. The compression test is performed with vertical downward force applied to the iliac crest with the patient lying on each side, compressing the SIJ by transverse pressure across the pelvis. The sacral thrust test is performed with vertical force applied to the midline posterior sacrum at its apex directed anteriorly with the patient lying prone, producing a shearing force at the SIJs. The Gaenslen test uses a torsion force by applying a superior and posterior force to the right knee and posteriorly directed force to the left knee. Omitting the Gaenslen test has not been shown to compromise diagnostic efficacy of the other tests and can be safely excluded.12
A HCP can rule out SIJ dysfunction if these provocation tests are negative. However, the diagnostic predictive value of these tests is subject to variability among HCPs, and their reliability is increased when used in clusters.9,13
Imaging for the SIJ should begin with anterior/posterior, oblique, and lateral view plain X-rays of the pelvis (Figures 1 and 2), which will rule out other pathologies by identifying other sources of LBP, such as spondylolisthesis or hip osteoarthritis. HCPs should obtain lumbar and pelvis CT images to identify inflammatory or degenerative changes within the SIJ. CT images provide the high resolution that is needed to identify pathologies, such as fractures and tumors within the pelvic ring that could cause similar pain. MRI does not reliably depict a dysfunctional ligamentous apparatus within the SIJ; however, it can help identify inflammatory sacroiliitis, such as is seen in the spondyloarthropathies.11,14 Recent studies show combined single photon emission tomography and CT (SPECT-CT) might be the most promising imaging modality to reveal mechanical failure of load transfer with increased scintigraphic uptake in the posterior and superior SIJ ligamentous attachments. The joint loses its characteristic “dumbbell” shape in affected patients with about 50% higher uptake than unaffected joints. These findings were evident in patients who experienced pelvic trauma or during the peripartum period.15,16
Fluoroscopy-guided intra-articular injection of a local anesthetic (lidocaine) and/or a corticosteroid (triamcinolone) has the dual functionality of diagnosis and treatment (Figure 3). It often is considered the most reliable method to diagnose SIJ dysfunction and has the benefit of pain relief for up to 1 year. However, intra-articular injections lack diagnostic validity because the solution often extravasates to extracapsular structures. This confounds the source of the pain and makes it difficult to interpret these diagnostic injections. In addition, the injection might not reach the entire SIJ capsule and could result in a false-negative diagnosis.17,18 Periarticular injections have been shown to result in better pain relief in patients diagnosed with SIJ dysfunction than intra-articular injections. Periarticular injections also are easier to perform and could be a first-step option for these patients.19
Treatment
Nonoperative management of SIJ dysfunction includes exercise programs, physical therapy, manual manipulation therapy, sacroiliac belts, and periodic articular injections. Efficacy of these methods is variable, and analgesics often do not significantly benefit this type of pain. Another nonoperative approach is radiofrequency ablation (RFA) of the lumbar dorsal rami and lateral sacral branches, which can vary based on the number of rami treated as well as the technique used. About two-thirds of patients report pain relief after RFA.2 When successful, pain is relieved for 6 to 12 months, which is a temporary yet effective option for patients experiencing SIJ dysfunction.14,20
Fusion Surgery
Cadaver studies show that biomechanical stabilization of the SIJ leads to decreased range of motion in flexion/extension, lateral bending, and axial rotation. This results in a decreased need for periarticular muscular and ligamentous support, therefore facilitating load transfer across the SIJ.21,22 Patients undergoing minimally invasive surgery report better pain relief compared with those receiving open surgery at 12 months postoperatively.23 The 2 main SIJ fusion approaches used are the lateral transarticular and the dorsal approaches. In the dorsal approach, the SIJ is distracted and allograft dowels or titanium cages with graft are inserted into the joint space posteriorly through the back. When approaching laterally, hollow screw implants filled with graft or triangular titanium implants are placed across the joint, accessing the SIJ through the iliac bones using imaging guidance. This lateral transiliac approach using porous titanium triangular rods currently is the most studied technique.24
A recent prospective, multicenter trial included 423 patients with SIJ dysfunction who were randomized to receive SIJ fusion with triangular titanium implants vs a control group who received nonoperative management. Patients in the SIJ fusion group showed substantially greater improvement in pain (81.4%) compared with that of the nonoperative group (26.1%) 6 months after surgery. Pain relief in the SIJ fusion group was maintained at > 80% at 1 and 2 year follow-up, while the nonoperative group’s pain relief decreased to < 10% at the follow-ups. Measures of quality of life and disability also improved for the SIJ fusion group compared with that of the nonoperative group. Patients who were crossed over from conservative management to SIJ fusion after 6 months demonstrated improvements that were similar to those in the SIJ fusion group by the end of the study. Only 3% of patients required surgical revision. The strongest predictor of pain relief after surgery was a diagnostic SIJ anesthetic block of 30 to 60 minutes, which resulted in > 75% pain reduction.21,25 Additional predictors of successful SIJ fusion include nonsmokers, nonopioid users, and older patients who have a longer time course of SIJ pain.26
Another study investigating the outcomes of SIJ fusion, RFA, and conservative management with a 6-year follow-up demonstrated similar results.27 This further confirms the durability of the surgical group’s outcome, which sustained significant improvement compared with RFA and conservative management group in pain relief, daily function, and opioid use.
HCPs should consider SIJ fusion for patients who have at least 6 months of unsuccessful nonoperative management, significant SIJ pain (> 5 in a 10-point scale), ≥ 3 positive provocation tests, and at least 50% pain relief (> 75% preferred) with diagnostic intra-articular anesthetic injection.14 It is reasonable for primary care providers to refer these patients to a neurosurgeon or orthopedic spine surgeon for possible fusion. Patients with earlier lumbar/lumbosacral spinal fusions and persistent LBP should be evaluated for potential SIJ dysfunction. SIJ dysfunction after lumbosacral fusion could be considered a form of distal pseudarthrosis resulting from increased motion at the joint. One study found its incidence correlated with the number of segments fused in the lumbar spine.28 Another study found that about one-third of patients with persistent LBP after lumbosacral fusion could be attributed to SIJ dysfunction.29
Case Presentation
A 27-year-old female army veteran presented with bilateral buttock pain, which she described as a dull, aching pain across her sacral region, 8 out of 10 in severity. The pain was in a L5-S1 pattern. The pain was bilateral, with the right side worse than the left, and worsened with lateral bending and load transferring. She reported no numbness, tingling, or weakness.
On physical examination, she had full strength in her lower extremities and intact sensation. She reported tenderness to palpation of the sacrum and SIJ. Her gait was normal. The patient had positive thigh thrust and distraction tests. Lumbar spine X-ray, CT, MRI, and electromyographic studies did not show any pathology. She described little or no relief with analgesics or physical therapy. Previous L4-L5 and L5-S1 facet anesthetic injections and transforaminal epidural steroid injections provided minimal pain relief immediately after the procedures. Bilateral SIJ anesthetic injections under fluoroscopic guidance decreased her pain severity from a 7 to 3 out of 10 for 2 to 3 months before returning to her baseline. Radiofrequency ablation of the right SIJ under fluoroscopy provided moderate relief for about 4 months.
After exhausting nonoperative management for SIJ dysfunction without adequate pain control, the patient was referred to neurosurgery for surgical fusion. The patient was deemed an appropriate surgical candidate and underwent a right-sided SIJ fusion (Figures 4 and 5). At her 6-month and 1-year follow-up appointments, she had lasting pain relief, 2 out of 10.
Conclusion
SIJ dysfunction is widely overlooked because of the difficulty in distinguishing it from other similarly presenting syndromes. However, with a detailed history, appropriate physical maneuvers, imaging, and adequate response to intra-articular anesthetic, providers can reach an accurate diagnosis that will inform subsequent treatments. After failure of nonsurgical methods, patients with SIJ dysfunction should be considered for minimally invasive fusion techniques, which have proven to be a safe, effective, and viable treatment option.
1. Zaidi HA, Montoure AJ, Dickman CA. Surgical and clinical efficacy of sacroiliac joint fusion: a systematic review of the literature. J Neurosurg Spine. 2015;23(1):59-66.
2. Cohen SP. Sacroiliac joint pain: a comprehensive review of anatomy, diagnosis, and treatment. Anesth Analg. 2005;101(5):1440-1453.
3. Chou LH, Slipman CW, Bhagia SM, et al. Inciting events initiating injection‐proven sacroiliac joint syndrome. Pain Med. 2004;5(1):26-32.
4. Dreyfuss P, Dreyer SJ, Cole A, Mayo K. Sacroiliac joint pain. J Am Acad Orthop Surg. 2004;12(4):255-265.
5. Buijs E, Visser L, Groen G. Sciatica and the sacroiliac joint: a forgotten concept. Br J Anaesth. 2007;99(5):713-716.
6. Fortin JD, Dwyer AP, West S, Pier J. Sacroiliac joint: pain referral maps upon applying a new injection/arthrography technique. Part I: asymptomatic volunteers. Spine (Phila Pa 1976). 1994;19(13):1475-1482.
7. Schwarzer AC, Aprill CN, Bogduk N. The sacroiliac joint in chronic low back pain. Spine (Phila Pa 1976). 1995;20(1):31-37.
8. Fortin JD, Washington WJ, Falco FJ. Three pathways between the sacroiliac joint and neural structures. ANJR Am J Neuroradiol. 1999;20(8):1429-1434.
9. Szadek KM, van der Wurff P, van Tulder MW, Zuurmond WW, Perez RS. Diagnostic validity of criteria for sacroiliac joint pain: a systematic review. J Pain. 2009;10(4):354-368.
10. Merskey H, Bogduk N, eds. Classification of Chronic Pain: Descriptions of Chronic Pain Syndromes and Definitions of Pain Terms. 2nd ed. Seattle, WA: IASP Press; 1994.
11. Cusi MF. Paradigm for assessment and treatment of SIJ mechanical dysfunction. J Bodyw Mov Ther. 2010;14(2):152-161.
12. Laslett M, Aprill CN, McDonald B, Young SB. Diagnosis of sacroiliac joint pain: validity of individual provocation tests and composites of tests. Man Ther. 2005;10(3):207-218.
13. Laslett M. Evidence-based diagnosis and treatment of the painful sacroiliac joint. J Man Manip Ther. 2008;16(3):142-152.
14. Polly DW Jr. The sacroiliac joint. Neurosurg Clin N Am. 2017;28(3):301-312.
15. Cusi M, Van Der Wall H, Saunders J, Fogelman I. Metabolic disturbances identified by SPECT-CT in patients with a clinical diagnosis of sacroiliac joint incompetence. Eur Spine J. 2013;22(7):1674-1682.
16. Tofuku K, Koga H, Komiya S. The diagnostic value of single-photon emission computed tomography/computed tomography for severe sacroiliac joint dysfunction. Eur Spine J. 2015;24(4):859-863.
17. Kennedy DJ, Engel A, Kreiner DS, Nampiaparampil D, Duszynski B, MacVicar J. Fluoroscopically guided diagnostic and therapeutic intra‐articular sacroiliac joint injections: a systematic review. Pain Med. 2015;16(8):1500-1518.
18. Schneider BJ, Huynh L, Levin J, Rinkaekan P, Kordi R, Kennedy DJ. Does immediate pain relief after an injection into the sacroiliac joint with anesthetic and corticosteroid predict subsequent pain relief? Pain Med. 2018;19(2):244-251.
19. Murakami E, Tanaka Y, Aizawa T, Ishizuka M, Kokubun S. Effect of periarticular and intraarticular lidocaine injections for sacroiliac joint pain: prospective comparative study. J Orthop Sci. 2007;12(3):274-280.
20. Cohen SP, Hurley RW, Buckenmaier CC 3rd, Kurihara C, Morlando B, Dragovich A. Randomized placebo-controlled study evaluating lateral branch radiofrequency denervation for sacroiliac joint pain. Anesthesiology. 2008;109(2):279-288.
21. Polly DW, Cher DJ, Wine KD, et al; INSITE Study Group. Randomized controlled trial of minimally invasive sacroiliac joint fusion using triangular titanium implants vs nonsurgical management for sacroiliac joint dysfunction: 12-month outcomes. Neurosurgery. 2015;77(5):674-690.
22. Soriano-Baron H, Lindsey DP, Rodriguez-Martinez N, et al. The effect of implant placement on sacroiliac joint range of motion: posterior versus transarticular. Spine. 2015;40(9):E525-E530.
23. Smith AG, Capobianco R, Cher D, et al. Open versus minimally invasive sacroiliac joint fusion: a multi-center comparison of perioperative measures and clinical outcomes. Ann Surg Innov Res. 2013;7(1):14.
24. Rashbaum RF, Ohnmeiss DD, Lindley EM, Kitchel SH, Patel VV. Sacroiliac joint pain and its treatment. Clin Spine Surg. 2016;29(2):42-48.
25. Polly DW, Swofford J, Whang PG, et al. Two-year outcomes from a randomized controlled trial of minimally invasive sacroiliac joint fusion vs. non-surgical management for sacroiliac joint dysfunction. Int J Spine Surg. 2016;10:28.
26. Dengler J, Duhon B, Whang P, et al. Predictors of outcome in conservative and minimally invasive surgical management of pain originating from the sacroiliac joint: a pooled analysis. Spine (Phila Pa 1976). 2017;42(21):1664-1673.
27. Vanaclocha V, Herrera JM, Sáiz-Sapena N, Rivera-Paz M, Verdú-López F. Minimally invasive sacroiliac joint fusion, radiofrequency denervation, and conservative management for sacroiliac joint pain: 6-year comparative case series. Neurosurgery. 2018;82(1):48-55.
28. Unoki E, Abe E, Murai H, Kobayashi T, Abe T. Fusion of multiple segments can increase the incidence of sacroiliac joint pain after lumbar or lumbosacral fusion. Spine (Phila Pa 1976). 2016;41(12):999-1005.
29. Katz V, Schofferman J, Reynolds J. The sacroiliac joint: a potential cause of pain after lumbar fusion to the sacrum. J Spinal Disord Tech. 2003;16(1):96-99.
1. Zaidi HA, Montoure AJ, Dickman CA. Surgical and clinical efficacy of sacroiliac joint fusion: a systematic review of the literature. J Neurosurg Spine. 2015;23(1):59-66.
2. Cohen SP. Sacroiliac joint pain: a comprehensive review of anatomy, diagnosis, and treatment. Anesth Analg. 2005;101(5):1440-1453.
3. Chou LH, Slipman CW, Bhagia SM, et al. Inciting events initiating injection‐proven sacroiliac joint syndrome. Pain Med. 2004;5(1):26-32.
4. Dreyfuss P, Dreyer SJ, Cole A, Mayo K. Sacroiliac joint pain. J Am Acad Orthop Surg. 2004;12(4):255-265.
5. Buijs E, Visser L, Groen G. Sciatica and the sacroiliac joint: a forgotten concept. Br J Anaesth. 2007;99(5):713-716.
6. Fortin JD, Dwyer AP, West S, Pier J. Sacroiliac joint: pain referral maps upon applying a new injection/arthrography technique. Part I: asymptomatic volunteers. Spine (Phila Pa 1976). 1994;19(13):1475-1482.
7. Schwarzer AC, Aprill CN, Bogduk N. The sacroiliac joint in chronic low back pain. Spine (Phila Pa 1976). 1995;20(1):31-37.
8. Fortin JD, Washington WJ, Falco FJ. Three pathways between the sacroiliac joint and neural structures. ANJR Am J Neuroradiol. 1999;20(8):1429-1434.
9. Szadek KM, van der Wurff P, van Tulder MW, Zuurmond WW, Perez RS. Diagnostic validity of criteria for sacroiliac joint pain: a systematic review. J Pain. 2009;10(4):354-368.
10. Merskey H, Bogduk N, eds. Classification of Chronic Pain: Descriptions of Chronic Pain Syndromes and Definitions of Pain Terms. 2nd ed. Seattle, WA: IASP Press; 1994.
11. Cusi MF. Paradigm for assessment and treatment of SIJ mechanical dysfunction. J Bodyw Mov Ther. 2010;14(2):152-161.
12. Laslett M, Aprill CN, McDonald B, Young SB. Diagnosis of sacroiliac joint pain: validity of individual provocation tests and composites of tests. Man Ther. 2005;10(3):207-218.
13. Laslett M. Evidence-based diagnosis and treatment of the painful sacroiliac joint. J Man Manip Ther. 2008;16(3):142-152.
14. Polly DW Jr. The sacroiliac joint. Neurosurg Clin N Am. 2017;28(3):301-312.
15. Cusi M, Van Der Wall H, Saunders J, Fogelman I. Metabolic disturbances identified by SPECT-CT in patients with a clinical diagnosis of sacroiliac joint incompetence. Eur Spine J. 2013;22(7):1674-1682.
16. Tofuku K, Koga H, Komiya S. The diagnostic value of single-photon emission computed tomography/computed tomography for severe sacroiliac joint dysfunction. Eur Spine J. 2015;24(4):859-863.
17. Kennedy DJ, Engel A, Kreiner DS, Nampiaparampil D, Duszynski B, MacVicar J. Fluoroscopically guided diagnostic and therapeutic intra‐articular sacroiliac joint injections: a systematic review. Pain Med. 2015;16(8):1500-1518.
18. Schneider BJ, Huynh L, Levin J, Rinkaekan P, Kordi R, Kennedy DJ. Does immediate pain relief after an injection into the sacroiliac joint with anesthetic and corticosteroid predict subsequent pain relief? Pain Med. 2018;19(2):244-251.
19. Murakami E, Tanaka Y, Aizawa T, Ishizuka M, Kokubun S. Effect of periarticular and intraarticular lidocaine injections for sacroiliac joint pain: prospective comparative study. J Orthop Sci. 2007;12(3):274-280.
20. Cohen SP, Hurley RW, Buckenmaier CC 3rd, Kurihara C, Morlando B, Dragovich A. Randomized placebo-controlled study evaluating lateral branch radiofrequency denervation for sacroiliac joint pain. Anesthesiology. 2008;109(2):279-288.
21. Polly DW, Cher DJ, Wine KD, et al; INSITE Study Group. Randomized controlled trial of minimally invasive sacroiliac joint fusion using triangular titanium implants vs nonsurgical management for sacroiliac joint dysfunction: 12-month outcomes. Neurosurgery. 2015;77(5):674-690.
22. Soriano-Baron H, Lindsey DP, Rodriguez-Martinez N, et al. The effect of implant placement on sacroiliac joint range of motion: posterior versus transarticular. Spine. 2015;40(9):E525-E530.
23. Smith AG, Capobianco R, Cher D, et al. Open versus minimally invasive sacroiliac joint fusion: a multi-center comparison of perioperative measures and clinical outcomes. Ann Surg Innov Res. 2013;7(1):14.
24. Rashbaum RF, Ohnmeiss DD, Lindley EM, Kitchel SH, Patel VV. Sacroiliac joint pain and its treatment. Clin Spine Surg. 2016;29(2):42-48.
25. Polly DW, Swofford J, Whang PG, et al. Two-year outcomes from a randomized controlled trial of minimally invasive sacroiliac joint fusion vs. non-surgical management for sacroiliac joint dysfunction. Int J Spine Surg. 2016;10:28.
26. Dengler J, Duhon B, Whang P, et al. Predictors of outcome in conservative and minimally invasive surgical management of pain originating from the sacroiliac joint: a pooled analysis. Spine (Phila Pa 1976). 2017;42(21):1664-1673.
27. Vanaclocha V, Herrera JM, Sáiz-Sapena N, Rivera-Paz M, Verdú-López F. Minimally invasive sacroiliac joint fusion, radiofrequency denervation, and conservative management for sacroiliac joint pain: 6-year comparative case series. Neurosurgery. 2018;82(1):48-55.
28. Unoki E, Abe E, Murai H, Kobayashi T, Abe T. Fusion of multiple segments can increase the incidence of sacroiliac joint pain after lumbar or lumbosacral fusion. Spine (Phila Pa 1976). 2016;41(12):999-1005.
29. Katz V, Schofferman J, Reynolds J. The sacroiliac joint: a potential cause of pain after lumbar fusion to the sacrum. J Spinal Disord Tech. 2003;16(1):96-99.
HCV-infected people who inject drugs also have substantial alcohol use
Curing hepatitis C virus (HCV) infection without addressing the high rate of alcohol use disorder in many patients may undermine the benefits of treatment to long-term liver health, according to the results of a large cohort study.

Because excess alcohol use is known to accelerate liver disease progression, researchers Risha Irvin, MD, and her colleagues from Johns Hopkins University, Baltimore, examined the prevalence of alcohol use in HCV-infected people who inject drugs (PWID). Their study examined the prevalence and associated correlates of alcohol use (Addictive Behaviors 2019;96:56-61).
They followed a large cohort of 1,623 HCV-antibody positive PWID from 2005 to 2013 from the AIDS Linked to the Intravenous Experience (ALIVE) study. They characterized alcohol use with the Alcohol Use Disorders Identification Test (AUDIT-C) questionnaire. Multivariable logistic regression with generalized estimated equations was used to examine sociodemographic, clinical, and substance use correlates of alcohol use.
At baseline, the median age was 47 years, 67% were men, 81% were black, and 34% were HIV positive. The majority (60%) reported injection drug use in the prior 6 months, while 46% reported noninjection cocaine or heroin, 31% reported street-acquired prescription drugs, and 22% reported marijuana use in the same time period. According to the AUDIT-C results, 41% of the patients reported no alcohol use, 21% reported moderate alcohol use, and 38% reported heavy alcohol use at their baseline visit.
The factors that were significantly associated with heavy alcohol use included male sex, black race, income of $5,000 or less, a Center for Epidemiologic Studies Depression Scale (range 0-60) score of 23 or greater, being homeless, being incarcerated, marijuana use, use of street-acquired prescription drugs, noninjection cocaine/heroin, injection drug use, and cigarette smoking. In a model that included the composite summary variable for substance use intensity, one drug type (adjusted odds ratio, 1.92), two drug types (AOR, 2.93), and three drug types (AOR, 3.65) were significantly associated with heavy alcohol use.
“While clinicians are undoubtedly concerned about any level of alcohol use in the setting of HCV infection due to the acceleration of liver fibrosis, there is particular concern for individuals with heavy alcohol use and their increased risk for cirrhosis and liver failure even after HCV cure. Without intervention, alcohol use will persist after HCV is cured with the potential to undermine the benefit of HCV cure. Therefore, our data point to the need to invest in and develop programs that effectively address alcohol use and co-occurring substance use in this population of PWID with HCV,” the researchers concluded.
The study was supported by the U.S. National Institute on Drug Abuse, the National Institute of Allergy and Infectious Diseases, and the National Institute on Alcohol Abuse and Alcoholism. The authors declared that they had no conflicts.
SOURCE: Irvin R et al. Addictive Behaviors. 2019;96:56-61.
Curing hepatitis C virus (HCV) infection without addressing the high rate of alcohol use disorder in many patients may undermine the benefits of treatment to long-term liver health, according to the results of a large cohort study.
Because excess alcohol use is known to accelerate liver disease progression, researchers Risha Irvin, MD, and her colleagues from Johns Hopkins University, Baltimore, examined the prevalence of alcohol use in HCV-infected people who inject drugs (PWID). Their study examined the prevalence and associated correlates of alcohol use (Addictive Behaviors 2019;96:56-61).
They followed a large cohort of 1,623 HCV-antibody positive PWID from 2005 to 2013 from the AIDS Linked to the Intravenous Experience (ALIVE) study. They characterized alcohol use with the Alcohol Use Disorders Identification Test (AUDIT-C) questionnaire. Multivariable logistic regression with generalized estimated equations was used to examine sociodemographic, clinical, and substance use correlates of alcohol use.
At baseline, the median age was 47 years, 67% were men, 81% were black, and 34% were HIV positive. The majority (60%) reported injection drug use in the prior 6 months, while 46% reported noninjection cocaine or heroin, 31% reported street-acquired prescription drugs, and 22% reported marijuana use in the same time period. According to the AUDIT-C results, 41% of the patients reported no alcohol use, 21% reported moderate alcohol use, and 38% reported heavy alcohol use at their baseline visit.
The factors that were significantly associated with heavy alcohol use included male sex, black race, income of $5,000 or less, a Center for Epidemiologic Studies Depression Scale (range 0-60) score of 23 or greater, being homeless, being incarcerated, marijuana use, use of street-acquired prescription drugs, noninjection cocaine/heroin, injection drug use, and cigarette smoking. In a model that included the composite summary variable for substance use intensity, one drug type (adjusted odds ratio, 1.92), two drug types (AOR, 2.93), and three drug types (AOR, 3.65) were significantly associated with heavy alcohol use.
“While clinicians are undoubtedly concerned about any level of alcohol use in the setting of HCV infection due to the acceleration of liver fibrosis, there is particular concern for individuals with heavy alcohol use and their increased risk for cirrhosis and liver failure even after HCV cure. Without intervention, alcohol use will persist after HCV is cured with the potential to undermine the benefit of HCV cure. Therefore, our data point to the need to invest in and develop programs that effectively address alcohol use and co-occurring substance use in this population of PWID with HCV,” the researchers concluded.
The study was supported by the U.S. National Institute on Drug Abuse, the National Institute of Allergy and Infectious Diseases, and the National Institute on Alcohol Abuse and Alcoholism. The authors declared that they had no conflicts.
SOURCE: Irvin R et al. Addictive Behaviors. 2019;96:56-61.
Curing hepatitis C virus (HCV) infection without addressing the high rate of alcohol use disorder in many patients may undermine the benefits of treatment to long-term liver health, according to the results of a large cohort study.
Because excess alcohol use is known to accelerate liver disease progression, researchers Risha Irvin, MD, and her colleagues from Johns Hopkins University, Baltimore, examined the prevalence of alcohol use in HCV-infected people who inject drugs (PWID). Their study examined the prevalence and associated correlates of alcohol use (Addictive Behaviors 2019;96:56-61).
They followed a large cohort of 1,623 HCV-antibody positive PWID from 2005 to 2013 from the AIDS Linked to the Intravenous Experience (ALIVE) study. They characterized alcohol use with the Alcohol Use Disorders Identification Test (AUDIT-C) questionnaire. Multivariable logistic regression with generalized estimated equations was used to examine sociodemographic, clinical, and substance use correlates of alcohol use.
At baseline, the median age was 47 years, 67% were men, 81% were black, and 34% were HIV positive. The majority (60%) reported injection drug use in the prior 6 months, while 46% reported noninjection cocaine or heroin, 31% reported street-acquired prescription drugs, and 22% reported marijuana use in the same time period. According to the AUDIT-C results, 41% of the patients reported no alcohol use, 21% reported moderate alcohol use, and 38% reported heavy alcohol use at their baseline visit.
The factors that were significantly associated with heavy alcohol use included male sex, black race, income of $5,000 or less, a Center for Epidemiologic Studies Depression Scale (range 0-60) score of 23 or greater, being homeless, being incarcerated, marijuana use, use of street-acquired prescription drugs, noninjection cocaine/heroin, injection drug use, and cigarette smoking. In a model that included the composite summary variable for substance use intensity, one drug type (adjusted odds ratio, 1.92), two drug types (AOR, 2.93), and three drug types (AOR, 3.65) were significantly associated with heavy alcohol use.
“While clinicians are undoubtedly concerned about any level of alcohol use in the setting of HCV infection due to the acceleration of liver fibrosis, there is particular concern for individuals with heavy alcohol use and their increased risk for cirrhosis and liver failure even after HCV cure. Without intervention, alcohol use will persist after HCV is cured with the potential to undermine the benefit of HCV cure. Therefore, our data point to the need to invest in and develop programs that effectively address alcohol use and co-occurring substance use in this population of PWID with HCV,” the researchers concluded.
The study was supported by the U.S. National Institute on Drug Abuse, the National Institute of Allergy and Infectious Diseases, and the National Institute on Alcohol Abuse and Alcoholism. The authors declared that they had no conflicts.
SOURCE: Irvin R et al. Addictive Behaviors. 2019;96:56-61.
FROM ADDICTIVE BEHAVIORS
Quality of Care for Veterans With In-Hospital Stroke
Stroke is a leading cause of death and long-term disability in the US.1 Quality improvement efforts for acute stroke care delivery have successfully led to increased rates of thrombolytic utilization.2 Increasing attention is now being paid to additional quality metrics for stroke care, including hospital management and initiation of appropriate secondary stroke prevention measures at discharge. Many organizations, including the Veterans Health Administration (VHA), use these measures to monitor health care quality and certify centers that are committed to excellence in stroke care.3-6 It is anticipated that collection, evaluation, and feedback from these data may lead to improvements in outcomes after stroke.7
Patients who experience onset of stroke symptoms while already admitted to a hospital may be uniquely suited for quality improvement strategies. In-hospital strokes (IHS) are not uncommon and have been associated with higher stroke severity and increased mortality compared with patients with stroke symptoms prior to arriving at the emergency department (ED).8-10 A potential reason for the higher observed mortality is that patients with IHS may have poorer access to acute stroke resources, such as stroke teams and neuroimaging, as well as increased rates of medical comorbidities.9,11,12 Furthermore, stroke management protocols are typically created based on ED resources, which may not be equivalent to resources available on inpatient settings.
Although many studies have examined clinical characteristics of patients with IHS, few studies have looked at the quality of stroke care for IHS. Information on stroke quality data is even more limited in VHA hospitals due to the small number of admitted patients with stroke.13 VHA released a directive on Acute Stroke Treatment (Directive 2011-03) in 2011 with a recent update in 2018, which aimed to implement quality improvement strategies for stroke care in VHA hospitals.14 Although focusing primarily on acute stroke care in the ED, this directive has led to increased awareness of areas for improvement, particularly among larger VHA hospitals. Prior to this directive, although national stroke guidelines were well-defined, more variability likely existed in stroke protocols and the manner in which stroke care was delivered across care settings. As efforts to measure and improve stroke care evolve, it is important to ensure that strategies used in ED settings also are implemented for patients already admitted to the hospital. This study seeks to define the quality of care in VHA hospitals between patients having an in-hospital ischemic stroke compared with those presenting to the ED.
Methods
As a secondary analysis, we examined stroke care quality data from an 11-site VHA stroke quality improvement study.15 Sites participating in this study were high stroke volume VHA hospitals from various geographic regions of the US. This study collected data on ICD-9 discharge diagnosis-defined ischemic stroke admissions between January 2009 and June 2012. Patient charts were reviewed by a group of central, trained abstractors who collected information on patient demographics, clinical history, and stroke characteristics. Stroke severity was defined using the National Institutes of Health Stroke Scale (NIHSS), assessed by standardized retrospective review of admission physical examination documentation.16 A multidisciplinary team defined 11 stroke quality indicators (QIs; the 8 Joint Commission indictors and 3 additional indicators: smoking cessation and dysphagia screening, and NIHSS assessment), and the chart abstractors’ data were used to evaluate eligibility and passing rates for each QI.
For our analysis, patients were stratified into 2 categories: patients admitted to the hospital for another diagnosis who developed an IHS, and patients presenting with stroke to the ED. We excluded patients transferred from other facilities. We then compared the demographic and clinical features of the 2 groups as well as eligibility and passing rates for each of the 11 QIs. Patients were recorded as eligible if they did not have any clinical contraindication to receiving the assessment or intervention measured by the quality metric. Passing rates were defined by the presence of clear documentation in the patient record that the quality metric was met or fulfilled. Comparisons were made using nonparametric Mann-Whitney U tests and chi-square tests. All tests were performed at α .05 level.
Results
A total of 1823 patients were included in this analysis: 35 IHS and 1788 ED strokes. The 2 groups did not differ with respect to age, race, or sex (Table 1). Patients with IHS had higher stroke severity (mean NIHSS 11.3 vs 5.1, P <.01) and longer length of stay than did ED patients with stroke (mean 12.8 vs 7.3 days, P < .01). Patients with IHS also were less likely to be discharged home when compared with ED patients with stroke (34.3% vs 63.8%, P < .01).
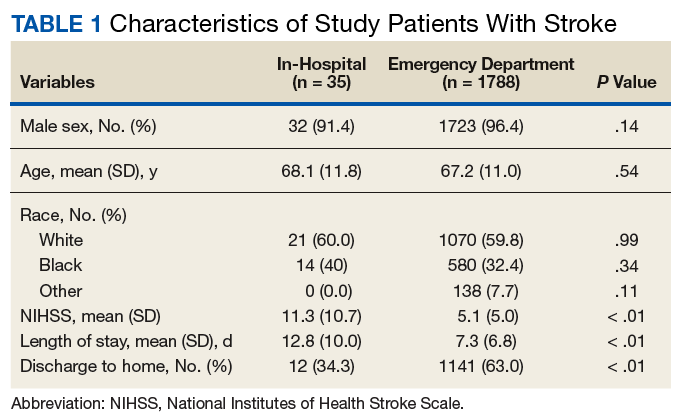
Table 2 summarizes our findings on eligibility and passing rates for the 11 QIs. For acute care metrics, we found that stroke severity documentation rates did not differ but were low for each patient group (51% vs 48%, P = .07). Patients with IHS were more likely to be eligible for IV tissue plasminogen activator (tPA; P < .01) although utilization rates did not differ. Only 2% of ED patients met eligibility criteria to receive tPA (36 of 1788), and among these patients only 16 actually received the drug. By comparison, 5 of 6 of eligible patients with IHS received tPA. Rates of dysphagia screening also were low for both groups, and patients with IHS were less likely to receive this screen prior to initiation of oral intake than were ED patients with stroke (27% vs 50%, P = .01).
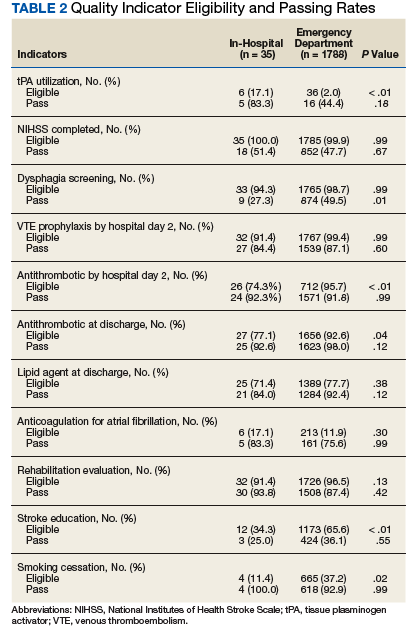
Beyond the acute period, we found that patients with IHS were less likely than were ED patients with stroke to be eligible to receive antithrombotic therapy by 2 days after their initial stroke evaluation (74% vs 96%, P < .01), although treatment rates were similar between the 2 groups (P = .99). In patients with documented atrial fibrillation, initiation of anticoagulation therapy also did not differ (P = .99). The 2 groups were similar with respect to initiation of venous thromboembolism (VTE) prophylaxis (P = .596) and evaluation for rehabilitation needs (P = .42). Although rates of smoking cessation counseling and stroke education prior to discharge did not differ, overall rates of stroke education were very low for both groups (25% vs 36%, P = .55).
Similar to initiation of antithrombotic therapy in the hospital, we found lower rates of eligibility to receive antithrombotic therapy on discharge in the IHS group when compared with the ED group (77% vs 93%, P = .04). However, actual treatment initiation rates did not differ (P = .12). Use of lipid-lowering agents was similar for the 2 groups (P = .12).
Discussion
Our study found that veterans who develop an IHS received similar quality of care as did those presenting to the ED with stroke symptoms for many QIs, although there were some notable differences. We were pleased to find that overall rates of secondary stroke prevention initiation (antithrombotic and statin therapy), VTE prophylaxis, rehabilitation evaluations, and smoking cessation counseling were high for both groups, in keeping with evidence-based guidelines.17 This likely reflected the fact that these metrics typically involve care outside of the acute period and are less likely to be influenced by the location of initial stroke evaluation. Furthermore, efforts to improve smoking cessation and VTE prophylaxis are not exclusive to stroke care and have been the target of several nonstroke quality projects in the VHA. Many aspects of acute stroke care did differ, and present opportunities for quality improvement in the future.
In our sample, patients with IHS had higher IV thrombolytic eligibility, which has not typically been reported in other samples.10,11,18 In these studies, hospitalized patients have been reported to more often have contraindications to tPA, such as recent surgery or lack of stroke symptom recognition due to delirium or medication effects. Interestingly, patients presenting to VHA EDs had extremely low rates of tPA eligibility (2%), which is lower than many reported estimates of tPA eligibility outside of the VHA.19,20 This may be due to multiple influences, such as geographic barriers, patient perceptions about stroke symptoms, access to emergency medical services (EMS), EMS routing patterns, and social/cultural factors. Although not statistically significant due to small sample size, tPA use also was twice as high in the IHS group.
Given that a significant proportion of patients with IHS in the VHA system may be eligible for acute thrombolysis, our findings highlight the need for acute stroke protocols to ensure that patients with IHS receive the same rapid stroke assessment and access to thrombolytics as do patients evaluated in the ED. Further investigation is needed to determine whether there are unique features of patients with IHS in VHA hospitals, which may make them more eligible for IV thrombolysis.
Dysphagia is associated with increased risks for aspiration pneumonia in stroke patients.21 We found that patients with IHS were less likely to receive dysphagia screening compared with that of stroke patients admitted through the ED. This finding is consistent with the fact that care for patients with IHS is less frequently guided by specific stroke care protocols and algorithms that are more often used in EDs.8,11 Although attention to swallowing function may lead to improved outcomes in stroke, this can be easily overlooked in patients with IHS.22 However, low dysphagia screening also was found in patients admitted through the ED, suggesting that low screening rates cannot be solely explained by differences in where the initial stroke evaluation is occurring. These findings suggest a need for novel approaches to dysphagia screening in VHA stroke patients that can be universally implemented throughout the hospital.
Finally, we also found very low rates of stroke education prior to discharge for both groups. Given the risk of stroke recurrence and the overall poor level of public knowledge about stroke, providing patients with stroke with formal oral and written information on stroke is a critical component of secondary prevention.23,24 Educational tools, including those that are veteran specific, are now available for use in VHA hospitals and should be incorporated into quality improvement strategies for stroke care in VHA hospitals.
In 2012, the VHA Acute Stroke Treatment Directive was published in an effort to improve stroke care systemwide. Several of the metrics examined in this study are addressed in this directive. The data presented in this study is one of the only samples of stroke quality metrics within the VHA that largely predates the directive and can serve as a baseline comparator for future work examining stroke care after release of the directive. At present, although continuous internal reviews of quality data are ongoing, longitudinal description of stroke care quality since publication of the directive will help to inform future efforts to improve stroke care for veterans.
Limitations
Despite the strength of being a multicenter sampling of stroke care in high volume VHA hospitals, our study had several limitations. The IHS sample size was small, which limited our ability to evaluate differences between the groups, to evaluate generalizability, and account for estimation error.13 It is possible that differences existed between the groups that could not be observed in this sample due to small size (type II error) or that patient-specific characteristics not captured by these data could influence these metrics. Assessments of eligibility and passing were based on retrospective chart review and post hoc coding. Our sample assessed only patients who presented to larger VHA hospitals with higher stroke volumes, thus these findings may not be generalizable to smaller VHA hospitals with less systematized stroke care. This sample did not describe the specialty care services that were received by each patient, which may have influenced their stroke care. Finally, this study is an analysis of use of QIs in stroke care and did not examine how these indicators affect outcomes.
Conclusion
Despite reassuring findings for several inpatient ischemic stroke quality metrics, we found several differences in stroke care between patients with IHS compared with those presenting to the ED, emphasizing the need for standardized approaches to stroke care regardless of care setting. Although patients with IHS may be more likely to be eligible for tPA, these patients received dysphagia screening and less often than did ED patients with stroke. Ongoing quality initiatives should continue to place emphasis on improving all quality metrics (particularly dysphagia screening, stroke severity documentation, and stroke education) for patients with stroke at VHA hospitals across all care settings. Future work will be needed to examine how specific patient characteristics and revisions to stroke protocols may affect stroke quality metrics and outcomes between patients with IHS and those presenting to the ED.
Acknowledgments
The authors would like to thank Danielle Sager for her contributions to this project.
1. Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation. 2014;129:e28-e292.
2. Schwamm LH, Ali SF, Reeves MJ, et al. Temporal trends in patient characteristics and treatment with intravenous thrombolysis among acute ischemic stroke patients at Get With the Guidelines—Stroke hospitals. Circ Cardiovasc Qual Outcomes. 2013;6(5):543-549.
3. Reeves MJ, Parker C, Fonarow GC, Smith EE, Schwamm LH. Development of stroke performance measures: definitions, methods, and current measures. Stroke. 2010;41(7):1573-1578.
4. The Joint Commission. Certificate of distinction for primary stroke centers. https://www.jointcommission.org/certificate_of_distinction_for_primary_stroke_centers_/.Published April 30, 2012. Accessed July 9, 2019.
5. US Department of Veterans Affairs. Center highlight: acute ischemic stroke care for veterans. https://www.queri.research.va.gov/center_highlights/stroke.cfm. Updated February 20, 2014. Accessed July 16, 2019.
6. Chumbler NR, Jia H, Phipps MS, et al. Does inpatient quality of care differ by age among US veterans with ischemic stroke? J Stroke Cerebrovasc Dis. 2012;21(8):844-851.
7. Katzan IL, Spertus J, Bettger JP, et al; American Heart Association Stroke Council; Council on Quality of Care and Outcomes Research; Council on Cardiovascular and Stroke Nursing; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Risk adjustment of ischemic stroke outcomes for comparing hospital performance: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45(3):918-944.
8. Cumbler E, Wald H, Bhatt DL, et al. Quality of care and outcomes for in-hospital ischemic stroke: findings from the National Get With the Guidelines—Stroke. Stroke. 2014;45(1):231-238.
9. Blacker DJ. In-hospital stroke. Lancet Neurol. 2003;2(12):741-746.
10. Farooq MU, Reeves MJ, Gargano J, Wehner S, Hickenbottom S, Majid A; Paul Coverdell National Acute Stroke Registry Michigan Prototype Investigators. In-hospital stroke in a statewide stroke registry. Cerebrovascular Dis. 2008;25(1-2):12-20.
11. Bhalla A, Smeeton N, Rudd AG, Heuschmann P, Wolfe CD. A comparison of characteristics and resource use between in-hospital and admitted patients with stroke. J Stroke Cerebrovasc Dis. 2010;19:(5)357-363.
12. Garcia-Santibanez R, Liang J, Walker A, Matos-Diaz I, Kahkeshani K, Boniece I. Comparison of stroke codes in the emergency room and inpatient setting. J Stroke Cerebrovasc Dis. 2015;24(8):1948-1950.
13. Arling G, Reeves M, Ross J, et al. Estimating and reporting on the quality of inpatient stroke care by Veterans Health Administration medical centers. Circ Cardiovasc Qual Outcomes. 2012;5(1):44-51.
14. US Department of Veterans Affairs. Treatment of Acute Ischemic Stroke (AIS). VHA Directive 2011-038. https://www.hsrd.research.va.gov/news/feature/stroke.cfm. Updated January 20, 2014. Accessed July 17, 2019.
15. Williams LS, Daggett V, Slaven J, et al. Abstract 18: Does quality improvement training add to audit and feedback for inpatient stroke care processes? [International Stroke Conference abstract 18] Stroke. 2014;45(suppl 1):A18.
16. Williams LS, Yilmaz EY, Lopez-Yunez AM. Retrospective assessment of initial stroke severity with the NIH Stroke Scale. Stroke. 2000;31(4):858-862.
17. Jauch EC, Saver JL, Adams HP Jr, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44(3):870-947.
18. Park HJ, Cho HJ, Kim YD, et al. Comparison of the characteristics for in-hospital and out-of-hospital ischaemic strokes. Eur J Neurol. 2009;16(5):582-588.
19. Messé SR, Fonarow GC, Smith EE, et al. Use of tissue-type plasminogen activator before and after publication of the European Cooperative Acute Stroke Study III in Get With the Guidelines-Stroke. Circ Cardiovasc Qual Outcomes. 2012;5(3):321-326.
20. Allen NB, Kaltenbach L, Goldstein LB, et al. Regional variation in recommended treatments for ischemic stroke and TIA: Get With the Guidelines—Stroke 2003-2010. Stroke. 2012;43(7):1858-1864.
21. Martino R, Foley N, Bhogal S, Diamant N, Speechley M, Teasell R. Dysphagia after stroke: incidence, diagnosis, and pulmonary complications. Stroke. 2005;36(12):2756-2763.
22. Bravata DM, Wells CK, Lo AC, et al. Processes of care associated with acute stroke outcomes. Arch Intern Med. 2010;170(9):804-810.
23. Mosley I, Nicol M, Donnan G, Patrick I, Dewey H. Stroke symptoms and the decision to call for an ambulance. Stroke; a journal of cerebral circulation. 2007;38(2):361-366.
24. Jurkowski JM, Maniccia DM, Dennison BA, Samuels SJ, Spicer DA. Awareness of necessity to call 9-1-1 for stroke symptoms, upstate New York. Prev Chronic Dis. 2008;5(2):A41.
Stroke is a leading cause of death and long-term disability in the US.1 Quality improvement efforts for acute stroke care delivery have successfully led to increased rates of thrombolytic utilization.2 Increasing attention is now being paid to additional quality metrics for stroke care, including hospital management and initiation of appropriate secondary stroke prevention measures at discharge. Many organizations, including the Veterans Health Administration (VHA), use these measures to monitor health care quality and certify centers that are committed to excellence in stroke care.3-6 It is anticipated that collection, evaluation, and feedback from these data may lead to improvements in outcomes after stroke.7
Patients who experience onset of stroke symptoms while already admitted to a hospital may be uniquely suited for quality improvement strategies. In-hospital strokes (IHS) are not uncommon and have been associated with higher stroke severity and increased mortality compared with patients with stroke symptoms prior to arriving at the emergency department (ED).8-10 A potential reason for the higher observed mortality is that patients with IHS may have poorer access to acute stroke resources, such as stroke teams and neuroimaging, as well as increased rates of medical comorbidities.9,11,12 Furthermore, stroke management protocols are typically created based on ED resources, which may not be equivalent to resources available on inpatient settings.
Although many studies have examined clinical characteristics of patients with IHS, few studies have looked at the quality of stroke care for IHS. Information on stroke quality data is even more limited in VHA hospitals due to the small number of admitted patients with stroke.13 VHA released a directive on Acute Stroke Treatment (Directive 2011-03) in 2011 with a recent update in 2018, which aimed to implement quality improvement strategies for stroke care in VHA hospitals.14 Although focusing primarily on acute stroke care in the ED, this directive has led to increased awareness of areas for improvement, particularly among larger VHA hospitals. Prior to this directive, although national stroke guidelines were well-defined, more variability likely existed in stroke protocols and the manner in which stroke care was delivered across care settings. As efforts to measure and improve stroke care evolve, it is important to ensure that strategies used in ED settings also are implemented for patients already admitted to the hospital. This study seeks to define the quality of care in VHA hospitals between patients having an in-hospital ischemic stroke compared with those presenting to the ED.
Methods
As a secondary analysis, we examined stroke care quality data from an 11-site VHA stroke quality improvement study.15 Sites participating in this study were high stroke volume VHA hospitals from various geographic regions of the US. This study collected data on ICD-9 discharge diagnosis-defined ischemic stroke admissions between January 2009 and June 2012. Patient charts were reviewed by a group of central, trained abstractors who collected information on patient demographics, clinical history, and stroke characteristics. Stroke severity was defined using the National Institutes of Health Stroke Scale (NIHSS), assessed by standardized retrospective review of admission physical examination documentation.16 A multidisciplinary team defined 11 stroke quality indicators (QIs; the 8 Joint Commission indictors and 3 additional indicators: smoking cessation and dysphagia screening, and NIHSS assessment), and the chart abstractors’ data were used to evaluate eligibility and passing rates for each QI.
For our analysis, patients were stratified into 2 categories: patients admitted to the hospital for another diagnosis who developed an IHS, and patients presenting with stroke to the ED. We excluded patients transferred from other facilities. We then compared the demographic and clinical features of the 2 groups as well as eligibility and passing rates for each of the 11 QIs. Patients were recorded as eligible if they did not have any clinical contraindication to receiving the assessment or intervention measured by the quality metric. Passing rates were defined by the presence of clear documentation in the patient record that the quality metric was met or fulfilled. Comparisons were made using nonparametric Mann-Whitney U tests and chi-square tests. All tests were performed at α .05 level.
Results
A total of 1823 patients were included in this analysis: 35 IHS and 1788 ED strokes. The 2 groups did not differ with respect to age, race, or sex (Table 1). Patients with IHS had higher stroke severity (mean NIHSS 11.3 vs 5.1, P <.01) and longer length of stay than did ED patients with stroke (mean 12.8 vs 7.3 days, P < .01). Patients with IHS also were less likely to be discharged home when compared with ED patients with stroke (34.3% vs 63.8%, P < .01).
Table 2 summarizes our findings on eligibility and passing rates for the 11 QIs. For acute care metrics, we found that stroke severity documentation rates did not differ but were low for each patient group (51% vs 48%, P = .07). Patients with IHS were more likely to be eligible for IV tissue plasminogen activator (tPA; P < .01) although utilization rates did not differ. Only 2% of ED patients met eligibility criteria to receive tPA (36 of 1788), and among these patients only 16 actually received the drug. By comparison, 5 of 6 of eligible patients with IHS received tPA. Rates of dysphagia screening also were low for both groups, and patients with IHS were less likely to receive this screen prior to initiation of oral intake than were ED patients with stroke (27% vs 50%, P = .01).
Beyond the acute period, we found that patients with IHS were less likely than were ED patients with stroke to be eligible to receive antithrombotic therapy by 2 days after their initial stroke evaluation (74% vs 96%, P < .01), although treatment rates were similar between the 2 groups (P = .99). In patients with documented atrial fibrillation, initiation of anticoagulation therapy also did not differ (P = .99). The 2 groups were similar with respect to initiation of venous thromboembolism (VTE) prophylaxis (P = .596) and evaluation for rehabilitation needs (P = .42). Although rates of smoking cessation counseling and stroke education prior to discharge did not differ, overall rates of stroke education were very low for both groups (25% vs 36%, P = .55).
Similar to initiation of antithrombotic therapy in the hospital, we found lower rates of eligibility to receive antithrombotic therapy on discharge in the IHS group when compared with the ED group (77% vs 93%, P = .04). However, actual treatment initiation rates did not differ (P = .12). Use of lipid-lowering agents was similar for the 2 groups (P = .12).
Discussion
Our study found that veterans who develop an IHS received similar quality of care as did those presenting to the ED with stroke symptoms for many QIs, although there were some notable differences. We were pleased to find that overall rates of secondary stroke prevention initiation (antithrombotic and statin therapy), VTE prophylaxis, rehabilitation evaluations, and smoking cessation counseling were high for both groups, in keeping with evidence-based guidelines.17 This likely reflected the fact that these metrics typically involve care outside of the acute period and are less likely to be influenced by the location of initial stroke evaluation. Furthermore, efforts to improve smoking cessation and VTE prophylaxis are not exclusive to stroke care and have been the target of several nonstroke quality projects in the VHA. Many aspects of acute stroke care did differ, and present opportunities for quality improvement in the future.
In our sample, patients with IHS had higher IV thrombolytic eligibility, which has not typically been reported in other samples.10,11,18 In these studies, hospitalized patients have been reported to more often have contraindications to tPA, such as recent surgery or lack of stroke symptom recognition due to delirium or medication effects. Interestingly, patients presenting to VHA EDs had extremely low rates of tPA eligibility (2%), which is lower than many reported estimates of tPA eligibility outside of the VHA.19,20 This may be due to multiple influences, such as geographic barriers, patient perceptions about stroke symptoms, access to emergency medical services (EMS), EMS routing patterns, and social/cultural factors. Although not statistically significant due to small sample size, tPA use also was twice as high in the IHS group.
Given that a significant proportion of patients with IHS in the VHA system may be eligible for acute thrombolysis, our findings highlight the need for acute stroke protocols to ensure that patients with IHS receive the same rapid stroke assessment and access to thrombolytics as do patients evaluated in the ED. Further investigation is needed to determine whether there are unique features of patients with IHS in VHA hospitals, which may make them more eligible for IV thrombolysis.
Dysphagia is associated with increased risks for aspiration pneumonia in stroke patients.21 We found that patients with IHS were less likely to receive dysphagia screening compared with that of stroke patients admitted through the ED. This finding is consistent with the fact that care for patients with IHS is less frequently guided by specific stroke care protocols and algorithms that are more often used in EDs.8,11 Although attention to swallowing function may lead to improved outcomes in stroke, this can be easily overlooked in patients with IHS.22 However, low dysphagia screening also was found in patients admitted through the ED, suggesting that low screening rates cannot be solely explained by differences in where the initial stroke evaluation is occurring. These findings suggest a need for novel approaches to dysphagia screening in VHA stroke patients that can be universally implemented throughout the hospital.
Finally, we also found very low rates of stroke education prior to discharge for both groups. Given the risk of stroke recurrence and the overall poor level of public knowledge about stroke, providing patients with stroke with formal oral and written information on stroke is a critical component of secondary prevention.23,24 Educational tools, including those that are veteran specific, are now available for use in VHA hospitals and should be incorporated into quality improvement strategies for stroke care in VHA hospitals.
In 2012, the VHA Acute Stroke Treatment Directive was published in an effort to improve stroke care systemwide. Several of the metrics examined in this study are addressed in this directive. The data presented in this study is one of the only samples of stroke quality metrics within the VHA that largely predates the directive and can serve as a baseline comparator for future work examining stroke care after release of the directive. At present, although continuous internal reviews of quality data are ongoing, longitudinal description of stroke care quality since publication of the directive will help to inform future efforts to improve stroke care for veterans.
Limitations
Despite the strength of being a multicenter sampling of stroke care in high volume VHA hospitals, our study had several limitations. The IHS sample size was small, which limited our ability to evaluate differences between the groups, to evaluate generalizability, and account for estimation error.13 It is possible that differences existed between the groups that could not be observed in this sample due to small size (type II error) or that patient-specific characteristics not captured by these data could influence these metrics. Assessments of eligibility and passing were based on retrospective chart review and post hoc coding. Our sample assessed only patients who presented to larger VHA hospitals with higher stroke volumes, thus these findings may not be generalizable to smaller VHA hospitals with less systematized stroke care. This sample did not describe the specialty care services that were received by each patient, which may have influenced their stroke care. Finally, this study is an analysis of use of QIs in stroke care and did not examine how these indicators affect outcomes.
Conclusion
Despite reassuring findings for several inpatient ischemic stroke quality metrics, we found several differences in stroke care between patients with IHS compared with those presenting to the ED, emphasizing the need for standardized approaches to stroke care regardless of care setting. Although patients with IHS may be more likely to be eligible for tPA, these patients received dysphagia screening and less often than did ED patients with stroke. Ongoing quality initiatives should continue to place emphasis on improving all quality metrics (particularly dysphagia screening, stroke severity documentation, and stroke education) for patients with stroke at VHA hospitals across all care settings. Future work will be needed to examine how specific patient characteristics and revisions to stroke protocols may affect stroke quality metrics and outcomes between patients with IHS and those presenting to the ED.
Acknowledgments
The authors would like to thank Danielle Sager for her contributions to this project.
Stroke is a leading cause of death and long-term disability in the US.1 Quality improvement efforts for acute stroke care delivery have successfully led to increased rates of thrombolytic utilization.2 Increasing attention is now being paid to additional quality metrics for stroke care, including hospital management and initiation of appropriate secondary stroke prevention measures at discharge. Many organizations, including the Veterans Health Administration (VHA), use these measures to monitor health care quality and certify centers that are committed to excellence in stroke care.3-6 It is anticipated that collection, evaluation, and feedback from these data may lead to improvements in outcomes after stroke.7
Patients who experience onset of stroke symptoms while already admitted to a hospital may be uniquely suited for quality improvement strategies. In-hospital strokes (IHS) are not uncommon and have been associated with higher stroke severity and increased mortality compared with patients with stroke symptoms prior to arriving at the emergency department (ED).8-10 A potential reason for the higher observed mortality is that patients with IHS may have poorer access to acute stroke resources, such as stroke teams and neuroimaging, as well as increased rates of medical comorbidities.9,11,12 Furthermore, stroke management protocols are typically created based on ED resources, which may not be equivalent to resources available on inpatient settings.
Although many studies have examined clinical characteristics of patients with IHS, few studies have looked at the quality of stroke care for IHS. Information on stroke quality data is even more limited in VHA hospitals due to the small number of admitted patients with stroke.13 VHA released a directive on Acute Stroke Treatment (Directive 2011-03) in 2011 with a recent update in 2018, which aimed to implement quality improvement strategies for stroke care in VHA hospitals.14 Although focusing primarily on acute stroke care in the ED, this directive has led to increased awareness of areas for improvement, particularly among larger VHA hospitals. Prior to this directive, although national stroke guidelines were well-defined, more variability likely existed in stroke protocols and the manner in which stroke care was delivered across care settings. As efforts to measure and improve stroke care evolve, it is important to ensure that strategies used in ED settings also are implemented for patients already admitted to the hospital. This study seeks to define the quality of care in VHA hospitals between patients having an in-hospital ischemic stroke compared with those presenting to the ED.
Methods
As a secondary analysis, we examined stroke care quality data from an 11-site VHA stroke quality improvement study.15 Sites participating in this study were high stroke volume VHA hospitals from various geographic regions of the US. This study collected data on ICD-9 discharge diagnosis-defined ischemic stroke admissions between January 2009 and June 2012. Patient charts were reviewed by a group of central, trained abstractors who collected information on patient demographics, clinical history, and stroke characteristics. Stroke severity was defined using the National Institutes of Health Stroke Scale (NIHSS), assessed by standardized retrospective review of admission physical examination documentation.16 A multidisciplinary team defined 11 stroke quality indicators (QIs; the 8 Joint Commission indictors and 3 additional indicators: smoking cessation and dysphagia screening, and NIHSS assessment), and the chart abstractors’ data were used to evaluate eligibility and passing rates for each QI.
For our analysis, patients were stratified into 2 categories: patients admitted to the hospital for another diagnosis who developed an IHS, and patients presenting with stroke to the ED. We excluded patients transferred from other facilities. We then compared the demographic and clinical features of the 2 groups as well as eligibility and passing rates for each of the 11 QIs. Patients were recorded as eligible if they did not have any clinical contraindication to receiving the assessment or intervention measured by the quality metric. Passing rates were defined by the presence of clear documentation in the patient record that the quality metric was met or fulfilled. Comparisons were made using nonparametric Mann-Whitney U tests and chi-square tests. All tests were performed at α .05 level.
Results
A total of 1823 patients were included in this analysis: 35 IHS and 1788 ED strokes. The 2 groups did not differ with respect to age, race, or sex (Table 1). Patients with IHS had higher stroke severity (mean NIHSS 11.3 vs 5.1, P <.01) and longer length of stay than did ED patients with stroke (mean 12.8 vs 7.3 days, P < .01). Patients with IHS also were less likely to be discharged home when compared with ED patients with stroke (34.3% vs 63.8%, P < .01).
Table 2 summarizes our findings on eligibility and passing rates for the 11 QIs. For acute care metrics, we found that stroke severity documentation rates did not differ but were low for each patient group (51% vs 48%, P = .07). Patients with IHS were more likely to be eligible for IV tissue plasminogen activator (tPA; P < .01) although utilization rates did not differ. Only 2% of ED patients met eligibility criteria to receive tPA (36 of 1788), and among these patients only 16 actually received the drug. By comparison, 5 of 6 of eligible patients with IHS received tPA. Rates of dysphagia screening also were low for both groups, and patients with IHS were less likely to receive this screen prior to initiation of oral intake than were ED patients with stroke (27% vs 50%, P = .01).
Beyond the acute period, we found that patients with IHS were less likely than were ED patients with stroke to be eligible to receive antithrombotic therapy by 2 days after their initial stroke evaluation (74% vs 96%, P < .01), although treatment rates were similar between the 2 groups (P = .99). In patients with documented atrial fibrillation, initiation of anticoagulation therapy also did not differ (P = .99). The 2 groups were similar with respect to initiation of venous thromboembolism (VTE) prophylaxis (P = .596) and evaluation for rehabilitation needs (P = .42). Although rates of smoking cessation counseling and stroke education prior to discharge did not differ, overall rates of stroke education were very low for both groups (25% vs 36%, P = .55).
Similar to initiation of antithrombotic therapy in the hospital, we found lower rates of eligibility to receive antithrombotic therapy on discharge in the IHS group when compared with the ED group (77% vs 93%, P = .04). However, actual treatment initiation rates did not differ (P = .12). Use of lipid-lowering agents was similar for the 2 groups (P = .12).
Discussion
Our study found that veterans who develop an IHS received similar quality of care as did those presenting to the ED with stroke symptoms for many QIs, although there were some notable differences. We were pleased to find that overall rates of secondary stroke prevention initiation (antithrombotic and statin therapy), VTE prophylaxis, rehabilitation evaluations, and smoking cessation counseling were high for both groups, in keeping with evidence-based guidelines.17 This likely reflected the fact that these metrics typically involve care outside of the acute period and are less likely to be influenced by the location of initial stroke evaluation. Furthermore, efforts to improve smoking cessation and VTE prophylaxis are not exclusive to stroke care and have been the target of several nonstroke quality projects in the VHA. Many aspects of acute stroke care did differ, and present opportunities for quality improvement in the future.
In our sample, patients with IHS had higher IV thrombolytic eligibility, which has not typically been reported in other samples.10,11,18 In these studies, hospitalized patients have been reported to more often have contraindications to tPA, such as recent surgery or lack of stroke symptom recognition due to delirium or medication effects. Interestingly, patients presenting to VHA EDs had extremely low rates of tPA eligibility (2%), which is lower than many reported estimates of tPA eligibility outside of the VHA.19,20 This may be due to multiple influences, such as geographic barriers, patient perceptions about stroke symptoms, access to emergency medical services (EMS), EMS routing patterns, and social/cultural factors. Although not statistically significant due to small sample size, tPA use also was twice as high in the IHS group.
Given that a significant proportion of patients with IHS in the VHA system may be eligible for acute thrombolysis, our findings highlight the need for acute stroke protocols to ensure that patients with IHS receive the same rapid stroke assessment and access to thrombolytics as do patients evaluated in the ED. Further investigation is needed to determine whether there are unique features of patients with IHS in VHA hospitals, which may make them more eligible for IV thrombolysis.
Dysphagia is associated with increased risks for aspiration pneumonia in stroke patients.21 We found that patients with IHS were less likely to receive dysphagia screening compared with that of stroke patients admitted through the ED. This finding is consistent with the fact that care for patients with IHS is less frequently guided by specific stroke care protocols and algorithms that are more often used in EDs.8,11 Although attention to swallowing function may lead to improved outcomes in stroke, this can be easily overlooked in patients with IHS.22 However, low dysphagia screening also was found in patients admitted through the ED, suggesting that low screening rates cannot be solely explained by differences in where the initial stroke evaluation is occurring. These findings suggest a need for novel approaches to dysphagia screening in VHA stroke patients that can be universally implemented throughout the hospital.
Finally, we also found very low rates of stroke education prior to discharge for both groups. Given the risk of stroke recurrence and the overall poor level of public knowledge about stroke, providing patients with stroke with formal oral and written information on stroke is a critical component of secondary prevention.23,24 Educational tools, including those that are veteran specific, are now available for use in VHA hospitals and should be incorporated into quality improvement strategies for stroke care in VHA hospitals.
In 2012, the VHA Acute Stroke Treatment Directive was published in an effort to improve stroke care systemwide. Several of the metrics examined in this study are addressed in this directive. The data presented in this study is one of the only samples of stroke quality metrics within the VHA that largely predates the directive and can serve as a baseline comparator for future work examining stroke care after release of the directive. At present, although continuous internal reviews of quality data are ongoing, longitudinal description of stroke care quality since publication of the directive will help to inform future efforts to improve stroke care for veterans.
Limitations
Despite the strength of being a multicenter sampling of stroke care in high volume VHA hospitals, our study had several limitations. The IHS sample size was small, which limited our ability to evaluate differences between the groups, to evaluate generalizability, and account for estimation error.13 It is possible that differences existed between the groups that could not be observed in this sample due to small size (type II error) or that patient-specific characteristics not captured by these data could influence these metrics. Assessments of eligibility and passing were based on retrospective chart review and post hoc coding. Our sample assessed only patients who presented to larger VHA hospitals with higher stroke volumes, thus these findings may not be generalizable to smaller VHA hospitals with less systematized stroke care. This sample did not describe the specialty care services that were received by each patient, which may have influenced their stroke care. Finally, this study is an analysis of use of QIs in stroke care and did not examine how these indicators affect outcomes.
Conclusion
Despite reassuring findings for several inpatient ischemic stroke quality metrics, we found several differences in stroke care between patients with IHS compared with those presenting to the ED, emphasizing the need for standardized approaches to stroke care regardless of care setting. Although patients with IHS may be more likely to be eligible for tPA, these patients received dysphagia screening and less often than did ED patients with stroke. Ongoing quality initiatives should continue to place emphasis on improving all quality metrics (particularly dysphagia screening, stroke severity documentation, and stroke education) for patients with stroke at VHA hospitals across all care settings. Future work will be needed to examine how specific patient characteristics and revisions to stroke protocols may affect stroke quality metrics and outcomes between patients with IHS and those presenting to the ED.
Acknowledgments
The authors would like to thank Danielle Sager for her contributions to this project.
1. Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation. 2014;129:e28-e292.
2. Schwamm LH, Ali SF, Reeves MJ, et al. Temporal trends in patient characteristics and treatment with intravenous thrombolysis among acute ischemic stroke patients at Get With the Guidelines—Stroke hospitals. Circ Cardiovasc Qual Outcomes. 2013;6(5):543-549.
3. Reeves MJ, Parker C, Fonarow GC, Smith EE, Schwamm LH. Development of stroke performance measures: definitions, methods, and current measures. Stroke. 2010;41(7):1573-1578.
4. The Joint Commission. Certificate of distinction for primary stroke centers. https://www.jointcommission.org/certificate_of_distinction_for_primary_stroke_centers_/.Published April 30, 2012. Accessed July 9, 2019.
5. US Department of Veterans Affairs. Center highlight: acute ischemic stroke care for veterans. https://www.queri.research.va.gov/center_highlights/stroke.cfm. Updated February 20, 2014. Accessed July 16, 2019.
6. Chumbler NR, Jia H, Phipps MS, et al. Does inpatient quality of care differ by age among US veterans with ischemic stroke? J Stroke Cerebrovasc Dis. 2012;21(8):844-851.
7. Katzan IL, Spertus J, Bettger JP, et al; American Heart Association Stroke Council; Council on Quality of Care and Outcomes Research; Council on Cardiovascular and Stroke Nursing; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Risk adjustment of ischemic stroke outcomes for comparing hospital performance: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45(3):918-944.
8. Cumbler E, Wald H, Bhatt DL, et al. Quality of care and outcomes for in-hospital ischemic stroke: findings from the National Get With the Guidelines—Stroke. Stroke. 2014;45(1):231-238.
9. Blacker DJ. In-hospital stroke. Lancet Neurol. 2003;2(12):741-746.
10. Farooq MU, Reeves MJ, Gargano J, Wehner S, Hickenbottom S, Majid A; Paul Coverdell National Acute Stroke Registry Michigan Prototype Investigators. In-hospital stroke in a statewide stroke registry. Cerebrovascular Dis. 2008;25(1-2):12-20.
11. Bhalla A, Smeeton N, Rudd AG, Heuschmann P, Wolfe CD. A comparison of characteristics and resource use between in-hospital and admitted patients with stroke. J Stroke Cerebrovasc Dis. 2010;19:(5)357-363.
12. Garcia-Santibanez R, Liang J, Walker A, Matos-Diaz I, Kahkeshani K, Boniece I. Comparison of stroke codes in the emergency room and inpatient setting. J Stroke Cerebrovasc Dis. 2015;24(8):1948-1950.
13. Arling G, Reeves M, Ross J, et al. Estimating and reporting on the quality of inpatient stroke care by Veterans Health Administration medical centers. Circ Cardiovasc Qual Outcomes. 2012;5(1):44-51.
14. US Department of Veterans Affairs. Treatment of Acute Ischemic Stroke (AIS). VHA Directive 2011-038. https://www.hsrd.research.va.gov/news/feature/stroke.cfm. Updated January 20, 2014. Accessed July 17, 2019.
15. Williams LS, Daggett V, Slaven J, et al. Abstract 18: Does quality improvement training add to audit and feedback for inpatient stroke care processes? [International Stroke Conference abstract 18] Stroke. 2014;45(suppl 1):A18.
16. Williams LS, Yilmaz EY, Lopez-Yunez AM. Retrospective assessment of initial stroke severity with the NIH Stroke Scale. Stroke. 2000;31(4):858-862.
17. Jauch EC, Saver JL, Adams HP Jr, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44(3):870-947.
18. Park HJ, Cho HJ, Kim YD, et al. Comparison of the characteristics for in-hospital and out-of-hospital ischaemic strokes. Eur J Neurol. 2009;16(5):582-588.
19. Messé SR, Fonarow GC, Smith EE, et al. Use of tissue-type plasminogen activator before and after publication of the European Cooperative Acute Stroke Study III in Get With the Guidelines-Stroke. Circ Cardiovasc Qual Outcomes. 2012;5(3):321-326.
20. Allen NB, Kaltenbach L, Goldstein LB, et al. Regional variation in recommended treatments for ischemic stroke and TIA: Get With the Guidelines—Stroke 2003-2010. Stroke. 2012;43(7):1858-1864.
21. Martino R, Foley N, Bhogal S, Diamant N, Speechley M, Teasell R. Dysphagia after stroke: incidence, diagnosis, and pulmonary complications. Stroke. 2005;36(12):2756-2763.
22. Bravata DM, Wells CK, Lo AC, et al. Processes of care associated with acute stroke outcomes. Arch Intern Med. 2010;170(9):804-810.
23. Mosley I, Nicol M, Donnan G, Patrick I, Dewey H. Stroke symptoms and the decision to call for an ambulance. Stroke; a journal of cerebral circulation. 2007;38(2):361-366.
24. Jurkowski JM, Maniccia DM, Dennison BA, Samuels SJ, Spicer DA. Awareness of necessity to call 9-1-1 for stroke symptoms, upstate New York. Prev Chronic Dis. 2008;5(2):A41.
1. Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation. 2014;129:e28-e292.
2. Schwamm LH, Ali SF, Reeves MJ, et al. Temporal trends in patient characteristics and treatment with intravenous thrombolysis among acute ischemic stroke patients at Get With the Guidelines—Stroke hospitals. Circ Cardiovasc Qual Outcomes. 2013;6(5):543-549.
3. Reeves MJ, Parker C, Fonarow GC, Smith EE, Schwamm LH. Development of stroke performance measures: definitions, methods, and current measures. Stroke. 2010;41(7):1573-1578.
4. The Joint Commission. Certificate of distinction for primary stroke centers. https://www.jointcommission.org/certificate_of_distinction_for_primary_stroke_centers_/.Published April 30, 2012. Accessed July 9, 2019.
5. US Department of Veterans Affairs. Center highlight: acute ischemic stroke care for veterans. https://www.queri.research.va.gov/center_highlights/stroke.cfm. Updated February 20, 2014. Accessed July 16, 2019.
6. Chumbler NR, Jia H, Phipps MS, et al. Does inpatient quality of care differ by age among US veterans with ischemic stroke? J Stroke Cerebrovasc Dis. 2012;21(8):844-851.
7. Katzan IL, Spertus J, Bettger JP, et al; American Heart Association Stroke Council; Council on Quality of Care and Outcomes Research; Council on Cardiovascular and Stroke Nursing; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Risk adjustment of ischemic stroke outcomes for comparing hospital performance: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45(3):918-944.
8. Cumbler E, Wald H, Bhatt DL, et al. Quality of care and outcomes for in-hospital ischemic stroke: findings from the National Get With the Guidelines—Stroke. Stroke. 2014;45(1):231-238.
9. Blacker DJ. In-hospital stroke. Lancet Neurol. 2003;2(12):741-746.
10. Farooq MU, Reeves MJ, Gargano J, Wehner S, Hickenbottom S, Majid A; Paul Coverdell National Acute Stroke Registry Michigan Prototype Investigators. In-hospital stroke in a statewide stroke registry. Cerebrovascular Dis. 2008;25(1-2):12-20.
11. Bhalla A, Smeeton N, Rudd AG, Heuschmann P, Wolfe CD. A comparison of characteristics and resource use between in-hospital and admitted patients with stroke. J Stroke Cerebrovasc Dis. 2010;19:(5)357-363.
12. Garcia-Santibanez R, Liang J, Walker A, Matos-Diaz I, Kahkeshani K, Boniece I. Comparison of stroke codes in the emergency room and inpatient setting. J Stroke Cerebrovasc Dis. 2015;24(8):1948-1950.
13. Arling G, Reeves M, Ross J, et al. Estimating and reporting on the quality of inpatient stroke care by Veterans Health Administration medical centers. Circ Cardiovasc Qual Outcomes. 2012;5(1):44-51.
14. US Department of Veterans Affairs. Treatment of Acute Ischemic Stroke (AIS). VHA Directive 2011-038. https://www.hsrd.research.va.gov/news/feature/stroke.cfm. Updated January 20, 2014. Accessed July 17, 2019.
15. Williams LS, Daggett V, Slaven J, et al. Abstract 18: Does quality improvement training add to audit and feedback for inpatient stroke care processes? [International Stroke Conference abstract 18] Stroke. 2014;45(suppl 1):A18.
16. Williams LS, Yilmaz EY, Lopez-Yunez AM. Retrospective assessment of initial stroke severity with the NIH Stroke Scale. Stroke. 2000;31(4):858-862.
17. Jauch EC, Saver JL, Adams HP Jr, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44(3):870-947.
18. Park HJ, Cho HJ, Kim YD, et al. Comparison of the characteristics for in-hospital and out-of-hospital ischaemic strokes. Eur J Neurol. 2009;16(5):582-588.
19. Messé SR, Fonarow GC, Smith EE, et al. Use of tissue-type plasminogen activator before and after publication of the European Cooperative Acute Stroke Study III in Get With the Guidelines-Stroke. Circ Cardiovasc Qual Outcomes. 2012;5(3):321-326.
20. Allen NB, Kaltenbach L, Goldstein LB, et al. Regional variation in recommended treatments for ischemic stroke and TIA: Get With the Guidelines—Stroke 2003-2010. Stroke. 2012;43(7):1858-1864.
21. Martino R, Foley N, Bhogal S, Diamant N, Speechley M, Teasell R. Dysphagia after stroke: incidence, diagnosis, and pulmonary complications. Stroke. 2005;36(12):2756-2763.
22. Bravata DM, Wells CK, Lo AC, et al. Processes of care associated with acute stroke outcomes. Arch Intern Med. 2010;170(9):804-810.
23. Mosley I, Nicol M, Donnan G, Patrick I, Dewey H. Stroke symptoms and the decision to call for an ambulance. Stroke; a journal of cerebral circulation. 2007;38(2):361-366.
24. Jurkowski JM, Maniccia DM, Dennison BA, Samuels SJ, Spicer DA. Awareness of necessity to call 9-1-1 for stroke symptoms, upstate New York. Prev Chronic Dis. 2008;5(2):A41.
Using Optical Coherence Tomography in the Management of Postoperative Wound Leaks After Cataract Surgery
The term cataract is derived from the Latin word “catarractes,” which means “waterfall,” as the foamy white opacity of an advanced cataract can be likened to a tempestuous cascade. Cataract is the leading cause of preventable blindness worldwide.1,2 It is no surprise, therefore, that cataract surgery is the most frequently performed ophthalmic surgical procedure worldwide. Cataract surgeries may reach 30 million annual cases by 2020.3 Given the large number of surgeries being performed, postsurgical complications are not uncommon.
Early postoperative complications from lens exchange (cataract) surgery include increased intraocular pressure (IOP), corneal edema, and corneal wound leakage.4 Corneal wound leakage is not uncommon; one study showed that, in 100 cases, almost one-third of incisions leaked.5 A 2014 prospective study of 500 postcataract surgery eyes revealed that 48.8% had fluid egress.6 Early detection is important so that efforts to restore corneal integrity can immediately be implemented. If not caught early, patients are at risk for developing a cascade of sequelae, including endophthalmitis.
The majority of corneal wound leaks postphacoemulsification are self-limiting and self-sealing. Moderate wound leaks require treatment, as in the following case. Strategies to detect, image, and treat wound leaks are covered in this discussion.
Case Presentation
A 69-year-old male veteran presented with no complaints for a 1-day postoperative visit following right eye phacoemulsification cataract extraction. His best corrected visual acuity in the right eye was 20/40, and his pinhole visual acuity was 20/25+2. On slit-lamp examination, the temporally located main incision appeared well-adhered and was found to be Seidel negative; however, the inferior paracentesis wound was found to be Seidel positive, demonstrating a slow leak. Intraocular pressure (IOP) measured with tonopen was 9 mm Hg.
A bandage soft contact lens was placed on the eye. The patient was instructed not to rub or place any pressure on the eye and to avoid bending and heavy lifting. He was also instructed to continue his postoperative medications (prednisolone 1% every 2 hours and polymyxin B sulfate 4 times daily) in his right eye. A follow-up appointment was scheduled for the next day.

The patient presented for his postoperative day-2 visit with a best corrected visual acuity in the right eye of 20/20. He reported no visual problems, no eye pain, and mentioned that he had had a comfortable night sleep. A slit-lamp examination revealed trace diffuse injection in the operative eye, predominantly central Descemet membrane folds, 1+ stromal edema, and a Seidel negative main incision wound. However, the inferior paracentesis wound showed a moderate leak (Seidel positive), and the anterior chamber showed a 1+ cell and flare. Goldmann tonometry revealed an IOP of 5 mm Hg, indicating hypotony.

Anterior segment cube 512 x 128 optical coherence tomography (OCT) was obtained with the bandage contact lens (Figures 1 and 2), and then repeated with the bandage contact lens removed (Figures 3 and 4). OCT imaging confirmed epithelial and endothelial gaping, loss of coaptation, and a localized detachment of the Descemet membrane. The veteran was referred to his surgeon that same day, and 2 limbal vicryl sutures were placed. The patient was instructed to continue prednisolone 1% 4 times daily and polymyxin B sulfate every 2 hours; erythromycin ointment 3 times daily was added to his regimen.
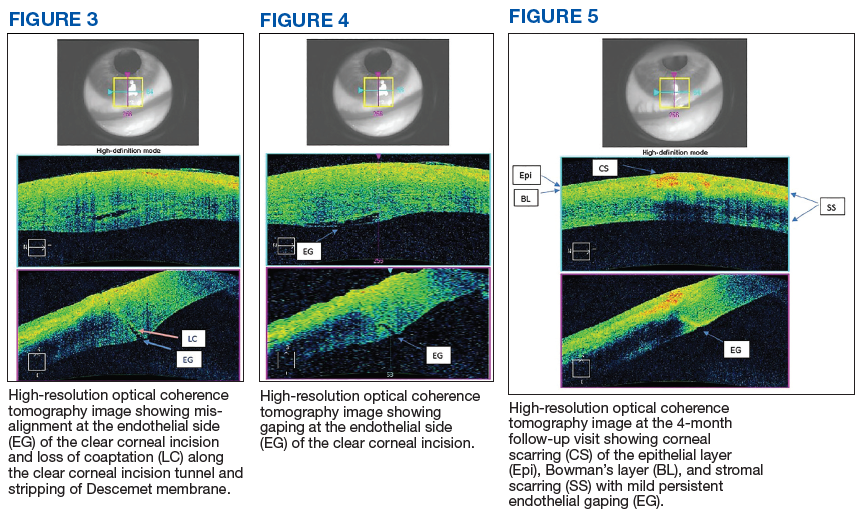
He was scheduled for a follow-up examination 1 week later. At that visit, the wound was no longer leaking and IOP had risen to a preoperative value of 17 mm Hg. The corneal sutures were removed at the 1-month postoperative examination and a follow-up was scheduled for 4 months later. An anterior segment OCT was obtained (Figure 5).
Discussion
In July 1967, Charles Kelman, MD, suggested using a dental ultrasonic tool, normally employed to clean teeth, to fragment the nucleus of the crystalline lens. Dr. Kelman’s first operation using phacoemulsification on a human eye took 3 hours.7 As the procedure for cataract removal has been refined, complication rates and surgical times have vastly improved.
Phacoemulsification is the most commonly performed outpatient surgery in the US; about 3 million cases are performed annually. Due to the high volume of cases, adverse events (AEs) are not uncommon. The incidence of complications following phacoemulsification is < 5%; the frequency of severe complications has been estimated at < 0.7%.8 Severe complications include endophthalmitis, suprachoroidal hemorrhage, and/or retinal detachment.9 Studies have shown a decline in rates of sight-threatening AEs from 1994 to 2006.9 A retrospective study of 45,082 veterans from 2005 to 2007 identified that a preoperative disease burden such as diabetes mellitus, chronic pulmonary disease, age-related macular degeneration, and diabetes with ophthalmic manifestations, was positively associated with a greater risk of cataract surgical complications.10
Complications
The level of a surgeon’s proficiency with phacoemulsification is directly correlated to the number of operations performed; there is a lower complication rate among more experienced surgeons, including those who work in high-volume settings.11,12 One study identified that the AE rate within 14 days of surgery was 0.8% for surgeons performing 50 to 250 cataract surgeries per year, but only 0.1% for those performing > 1000 cataract surgeries annually.12
Potential postoperative lens exchange complications include increased IOP, corneal wound leakage, corneal edema, bullous keratopathy, cystoid macular edema, retinal detachment, and endophthalmitis (Table 1). A corneal wound leak can provide a potential ingress for bacteria, putting the patient at risk for endophthalmitis, perhaps the most devastating complication following cataract surgery.

Endophthalmitis
Endophthalmitis has been reported to occur in .001% to .327% of patients during postoperative care.5,13-17 Early detection is important to maintain corneal integrity and prevent a cascade of detrimental ocular sequalae including the potential for endophthalmitis. According to Zaida and colleagues, endophthalmitis occurred in fewer than 1 of 1000 consecutive cases.14 A leaking clear corneal incision wound on the first day postoperatively has been associated with a 44-fold increased risk of endophthalmitis.13
Causes of endophthalmitis
In a retrospective case-controlled series of 57 patients with postcataract endophthalmitis, implantation of an intraocular lens with a resultant wound abnormality was thought to be the causative factor in 5%.17 Another source of endophthalmitis can be the intraocular lens (IOL), which may act as a vector for bacteria. By placing the IOL against the conjunctiva or exposing it to the theater air during surgery, bacteria can be introduced prior to implantation.17 Immunosuppressive treatment is the only patient antecedent factor that can be considered a predictor for endopthalmitis.17

The internal corneal seal is IOP dependent, and postoperative ocular hypotony may cause a seemingly watertight wound to leak. Taban and colleagues used anterior segment OCT to image numerous self-sealing incisions. They found that the corneal incision wound more tightly seals at higher IOPs. Additionally, more perpendicular (larger angle) incisions seal better at a lower IOP while less perpendicular (smaller angle) incisions seal better at a higher IOP (Figure 6).18
Incision Placement
Studies have shown that the main incision site is more clinically competent than is the side port incision site, as in our case study.19 Side-port incisions have a 1- or 2-plane architectural profile in contrast to the 3-plane profile typical of a main incision.19 Recent advances including the conversion to clear-corneal incisions of diminishing size, techniques used for wound construction, phacoemulsification machine design, and small-incision IOLs, should further reduce the prevalence and complications of wound compromise.20
Seidel Testing
Seidel testing is the most common method to evaluate corneal wound integrity and identify leaks. A drop of topical anesthetic is instilled in the eye and then a fluorescein strip (not fluorescein sodium and benoxinate hydrochloride ophthalmic solution, which may become less sterile since it has a multiuse container) is applied to the superior conjunctiva. The clinician then looks for evidence of fluid egress using the cobalt blue filter. The patient is instructed to blink once. Fluid egress appears as a black stream as the fluorescein dye becomes diluted by aqueous humor escaping the nonintact wound and the appearance of bright green dye surrounds the leak site. The term Seidel positive indicates a leak. An estimate should be made of the rate and volume of fluid exiting the wound.
Gonioscopy
Gonioscopy can be used to evaluate the postsurgical incision, more specifically for identification and management of internal incision wound gape. On gonioscopy, internal wound gape appears as an elongated oval opening resembling a fish mouth. If internal incision wound gape is identified gonioscopically before surgery is complete, the leak can be managed intraoperatively. The surgeon can irrigate along the length of the incision to remove cortical fragments or viscoelastic that may cause internal wound gaping. If unsuccessful, rapidly deepening the anterior chamber with balanced salt solution through the paracentesis incision may be employed. These methods may improve wound stability, reduce risk of postoperative hyphema, lower the incidence of endophthalmitis, and lessen the likelihood of late against-the-rule drift.21
Anterior Segment Optical Coherence Tomography
Instances when Seidel testing was negative despite actual wound gaping have been described.22,23 Anterior segment OCT is useful to evaluate incision architecture. A 2007 United Kingdom study investigated the corneal architecture in the immediate postoperative period following phacoemulsification using anterior segment OCT. This study showed the benefits of identifying architectural features such as epithelial gaping, endothelial gaping, stripping of Descemet membrane, and loss of coaptation. These features were found to be more common at low IOP and could represent a significant risk factor for endophthalmitis.24 Another study published by Behrens and colleagues indicated that a localized detachment of Descemet membrane may be more common than observed with slit-lamp (Figure 7). Corneal gaping, especially if along the entire length of the surgical wound, may lead to inadvertent bacterial access into the anterior chamber.25

Anterior segment OCT imaging was first described by Izatt and colleagues in 1994.26 Unlike posterior segment OCT, anterior segment OCT requires a greater depth of field and higher energy levels as images are commonly distorted by refraction at boundaries where the refractive index changes. Longer infrared wavelengths improve the penetration through tissues that scatter light, such as the sclera and limbus, which allows visualization, for example, of the iridocorneal angle.27,28

Two main scan patterns are used for anterior segment OCT: 512 x 128 cube scan (4-mm width x 4-mm length) and 5-line raster (3-mm length) with adjustable rotation and spacing. A recent software update allows measurement of corneal thickness, visualization of anterior chamber angle structures along with topographic analysis, anterior and posterior elevation maps of the cornea, and reliable pachymetric maps.29,30 The anterior segment cube acquires a series of 128 horizontal scan lines each composed of 512 A-scans. These high-definition scans acquire vertical and horizontal directions composed of 1024 A-scans each. This cube may be used to measure corneal thickness and visualize corneal architecture, creating a 3-D image of the data (Figure 8). The anterior segment 5-line raster scans through 5 parallel lines of equal length to view high-resolution images of the anterior chamber angle and cornea. Each line, fixed at 3-mm in length, is composed of 4096 A-scans.31 Anterior segment cube OCT allows identification of subtle variations in incision architecture at different locations across the width of the OCT image.
Bandage Soft Contact Lens
Upon reviewing the anterior segment OCT images of our patient with the bandage contact lens in place, it was evident that the adherent ocular bandage was protecting the incision. A tighter fitting bandage contact lens is ideal and adheres firmly to any area of epithelial damage and epithelial gaping to help seal the incision, protecting the wound and improving structural integrity. The bandage contact lens is gradually replaced by new cells via re-epithelialization; thus, it behaves as an adjunct to natural wound healing. A bandage contact lens also improves patient comfort.
It is hypothesized that a bandage contact lens improves the structural integrity of the incision site and helps prevent leaking, hypotony, and minor wound leaks. One study revealed a statistically significant lower IOP in nonbandage contact lens patients by an average of 6 mm Hg (mean [SD] 13.4 mm Hg [5.3]; range, 5 - 23 mm Hg) vs patients with a bandage contact lens (mean [SD] 19.4 mm Hg [5.9]; range, 11 - 29 mm Hg) in the immediate postoperative period.32 The authors suggested that the bandage contact lens may prevent microleaks, resulting in a higher IOP.
Aqueous Suppressants
Aqueous suppressants are a great option when IOP is abnormally elevated by decreasing the IOP and allowing the cornea to heal and self-seal.Effective aqueous suppressants are β blockers and carbonic anhydrase inhibitors.
After phacoemulsification ocular hypotony (< 6 mm Hg) occurs most commonly due to wound leakage or excessive intraocular inflammation. However, with the presence of corneal wound leakage and ocular hypotony, aqueous suppressants are not the best option.
Further Management of Wound Leaks
Management of a postoperative wound leak will vary based on severity. The majority of mild leaks are self-sealing. Anterior segment OCT helps the clinician to identify microleaks in an otherwise Seidel negative eye. If wound leakage is moderate with a formed anterior chamber, the use of a bandage contact lens is a good option, as can be the prescription of aqueous suppressants, depending on IOP.33
If the anterior chamber is flat, iris prolapse is apparent, or extremely low IOP exists, the patient needs to be referred to the surgeon. Current standard of care directs the surgeon to use sutures to further manage corneal wound leak. However, several studies have recognized the increased risk of suture-related complications, such as induced astigmatism, corneal opacities, incomplete wound closure, and corneal neovascularization.6,34-38 Other wound closure options include polyethylene glycol-based products, corneal welding, cyanoacrylate, or fibrin (Table 2).39 Traditionally nylon sutures have been used for clear corneal incision wound closure. However, tissue adhesives are gaining popularity as a substitute for sutures in wound closure.40

Cyanoacrylate
Numerous studies have been published on the efficacy of cyanoacrylate as a substitute for sutures, specifically in clear corneal incisions. AEs of cyanoacrylate include a transient foreign-body sensation and diffuse or focal bulbar conjunctival hyperemia.41,42 Shigemitsu and Majima found that fibrin and cyanoacrylate glue had tensile strength similar to sutures when used in cataract surgery.39 Polyethylene glycol-based products, also used in artificial tears and contact lens materials, may also help seal wound leaks. Another agent is ReSure (Ocular Therapeutix, Bedford, MA), an FDA-approved synthetic, polyethylene glycol hydrogel sealant that is 90% water after polymerization. ReSure has been shown to be safe and effective in sealing cataract surgical clear corneal incisions.6,43 ReSure takes about 20 seconds to prepare, and placement is aided by the use of a blue dye that dissipates within hours. This hydrogel will gradually slough off in the tears once the tissue has fully regenerated; there is no need to remove the sealant.44
Rossi and colleagues evaluated the efficacy of corneal welding to close wounds after cataract surgery. The technique involves laser-assisted closure of the corneal wound(s) by a diode laser that welds the stroma.45 Corneal welding takes seconds to achieve good closure without significant astigmatism or inflammation; however very careful application of the light absorbing dyes is required as they are toxic if allowed to enter the anterior chamber.45-47
Conclusion
Optometrists may be called to manage patients during both the preoperative and postoperative phases of cataract surgical care. Those who participate in postoperative care should carefully evaluate for the presence of wound leak or wound gape as a potential complication. The OCT may be employed to evaluate patients suspected of having these leaks or gapes. Proficiency in the interpretation of OCT results and more traditional evaluation methods allows for successful detection of wound leaks or gapes. The timely diagnosis and treatment of postoperative wound leaks allow for the best possible outcomes for cataract surgery patients.
1. Thylefors B, Négrel AD, Pararajasegaram R, Dadzie KY. Global data on blindness. Bull World Health Organ. 1995;73(1):115-121.
2. Flaxman SR, Bourne RRA, Resnikoff S, et al; Vision Loss Expert Group of the Global Burden of Disease Study. Global causes of blindness and distance vision impairment 1990-2020: a systematic review and meta-analysis. Lancet Glob Health. 2017;5(12):e1221-e1224.
3. Congdon N, Vingerling JR, Klein BE, et al; Eye Diseases Prevalence Research Group. Prevalence of cataract and pseudophakia/aphakia among adults in the United States. Arch Ophthalmol. 2004;122(4):487-494.
4. Kurt E, Mayalı H. Early post-operative complications in cataract surgery. In: Zaidi FH, ed. Cataract Surgery. IntechOpen; 2013. https://www.intechopen.com/books/cataract-surgery/post-operative-infections-associated-with-cataract-surgery. Accessed July 15, 2019.
5. Chee SP. Clear corneal incision leakage after phacoemulsification--detection using povidone iodine 5%. Int Ophthalmol. 2005;26(4-5):175-179.
6. Masket S, Hovanesian JA, Levenson J, et al. Hydrogel sealant versus sutures to prevent fluid egress after cataract surgery. J Cataract Refract Surg. 2014;40(12):2057-2066.
7. Kelman CD. Phaco-emulsification and aspiration: a new technique of cataract removal. A preliminary report. Am J Ophthalmol. 1967;64(1):23-35.
8. Powe NR, Schein OD, Gieser SC, et al. Synthesis of the literature on visual acuity and complications following cataract extraction with intraocular lens implantation. Cataract Patient Outcome Research Team [published correction appears in Arch Ophthalmol. 1994;112(7):889]. Arch Ophthalmol. 1994;112(2):239-252.
9. Stein JD, Grossman DS, Mundy KM, Sugar A, Sloan FA. Severe adverse events after cataract surgery among medicare beneficiaries. Ophthalmology. 2011;118(9):1716-1723.
10. Greenberg PB, Tseng VL, Wu WC, et al. Prevalence and predictors of ocular complications associated with cataract surgery in United States veterans. Ophthalmology. 2011;118(3):507-514.
11. Mangan MS, Atalay E, Anci C, Tuncer I, Bilqec MD. Comparison of different types of complications in the phacoemulsification surgery learning curve according to number of operations performed. Turk J Ophthalmol. 2016;46(1):7-10.
12. Bell CM, Hatch WV, Cernat G, Urbach DR. Surgeon volumes and selected patient outcomes in cataract surgery: a population-based analysis. Ophthalmology. 2007;114(3):405-410.
13. Wallin T, Parker J, Jin Y, Kefalopoulos G, Olson RJ. Cohort study of 27 cases of endophthalmitis at a single institution. J Cataract Refract Surg. 2005;31(4):735-741.
14. Zaidi FH, Corbett MC, Burton BJ, Bloom PA. Raising the benchmark for the 21st century--the 1000 cataract operations audit and survey: outcomes, consultant-supervised training and sourcing NHS choice. Br J Ophthalmol. 2007;91(6):731-736.
15. Nichamin LD, Chang DF, Johnson SH, et al; American Society of Cataract and Refractive Surgery Cataract Clinical Committee. ASCRS white paper: what is the association between clear corneal cataract incisions and postoperative endophthalmitis? J Cataract Refract Surg. 2006;32(9):1556-1559.
16. Packer M, Chang DF, Dewey SH, et al; ASCRS Cataract Clinical Committee. Prevention, diagnosis, and management of acute postoperative bacterial endophthalmitis. J Cataract Refract Surg. 2011;37(9):1699-1714.
17. Montan PG, Koranyi G, Setterquist HE, Stridh A, Philipson BT, Wiklund K. Endophthalmitis after cataract surgery: risk factors relating to technique and events of the operation and patient history: a retrospective case-control study. Ophthalmology. 1998;105(12):2171-2177.
18. Taban M, Rao B, Reznik J, Zhang J, Chen Z, McDonnell PJ. Dynamic morphology of sutureless cataract wounds—effect of incision angle and location. Surv Ophthalmol. 2004;49(suppl 2):S62-S72.
19. Chee SP, Ti SE, Lim L, Chan AS, Jap A. Anterior segment optical coherence tomography evaluation of the integrity of clear corneal incisions: a comparison between 2.2-mm and 2.65-mm main incisions. Am J Ophthalmol. 2010;149(5):768-776.e1.
20. Koch DD, Nacke RE, Wang L, Novak KD. Issues in wound management. In: Steinert R, ed. Cataract Surgery. 3rd ed. New York: Elsevier; 2009:581-588.
21. Gimbel HV, Sun R, DeBroff GM. Recognition and management of internal wound gape. J Cataract Refract Surg. 1995;21(2):121-124.
22. May WN, Castro-Combs J, Quinto GG, Kashiwabuchi R, Gower EW, Behrens A. Standardized Seidel test to evaluate different sutureless cataract incision configurations. J Cataract Refract Surg. 2010;36(6):1011-1017.
23. Kashiwabuchi FK, Khan YA, Rodrigues MW Jr, Wang J, McDonnell PJ, Daoud YJ. Seidel and India ink tests assessment of different clear cornea side-port incision configurations. Graefes Arch Clin Exp Ophthalmol. 2013;251(8):1961-1965.
24. Calladine D, Packard R. Clear corneal incision architecture in the immediate postoperative period evaluated using optical coherence tomography. J Cataract Refract Surg. 2007;33(8):1429-1435.
25. Behrens WJ, Stark KA, Pratzer, McDonnell PJ. Dynamics of small-incision clear cornea wounds after phacoemulsification surgery using optical coherence tomography in the early postoperative period. J Refractive Surgery. 2008;24(1):46-49.
26. Izatt JA, Hee MR, Swanson EA, et al. Micrometer-scale resolution imaging of the anterior eye in vivo with optical coherence tomography. Arch Ophthalmol. 1994;112(12):1584-1589.
27. Hurmeric V, Yoo SH, Mutlu FM. Optical coherence tomography in cornea and refractive surgery. Expert Rev Ophthalmol. 2012;7(3):241-250.
28. Schuman JS, Puliafito CA, Fujimoto JG, Duker JS. Optical Coherence Tomography of Ocular Diseases. 3rd ed. Thorofare, NJ: Slack Inc; 2013.
29. Salim S. The role of anterior segment optical coherence tomography in glaucoma. J Ophthalmol. 2012;2012:476801.
30. Kharousi NA, Wali UK, Azeem S. Current applications of optical coherence tomography in ophthalmology. In: Kawasaki M, ed. Optical Coherence Tomography. IntechOpen; 2013. https://www.intechopen.com/books/optical-coherence-tomography. Accessed July 31, 2019.
31. Rodrigues EB, Johanson M, Penha FM. Anterior segment tomography with the cirrus optical coherence tomography. J Ophthalmol. 2012;2012:806989.
32. Calladine D, Ward M, Packard R. Adherent ocular bandage for clear corneal incisions used in cataract surgery. J Cataract Refract Surg. 2010;36(11):1839-1848.
33. Haldar K, Saraff R. Closure technique for leaking wound resulting from thermal injury during phacoemulsification. J Cataract Refract Surg. 2014;40(9):1412-1414.
34. Zoghby JT, Cohen KL. Phacoemulsification-related corneal incision contracture. https://www.aao.org/eyenet/article/phacoemulsification-related-corneal-incision-contr. Published December 2012. Accessed June 16, 2019.
35. Bhatia SS. Ocular surface sealants and adhesives. Ocul Surf. 2006;4(3):146-154.
36. May WN, Castro-Combs J, Kashiwabuchi RT, et al. Bacterial-sized particle inflow through sutured clear corneal incisions in a laboratory human model. J Cataract Refract Surg. 2011;37(6):1140-1146.
37. Meskin SW, Ritterband DC, Shapiro DE, et al. Liquid bandage (2-octyl cyanoacrylate) as a temporary wound barrier in clear corneal cataract surgery. Ophthalmology. 2005;112(11):2015-2021.
38. Heaven CJ, Davison CR, Cockcroft PM. Bacterial contamination of nylon corneal sutures. Eye (Lond). 1995;9(pt 1):116-118.
39. Shigemitsu T, Majima Y. The utilization of a biological adhesive for wound treatment: comparison of suture, self-sealing sutureless and cyanoacrylate closure in the tensile strength test. Int Ophthalmol. 1996-1997;20:323-328.
40. Uy HS, Kenyon KR. Surgical outcomes after application of a liquid adhesive ocular bandage to clear corneal incisions during cataract surgery. J Cataract Refract Surg. 2013;39(11):1668-1674.
41. Meskin SW, Ritterband DC, Shapiro DE, et al. Liquid bandage (2-octyl cyanoacrylate) as a temporary wound barrier in clear corneal cataract surgery. Ophthalmology. 2005;112(11):2015-2021.
42. Tong AY, Gupta PK, Kim T. Wound closure and tissue adhesives in clear corneal incision cataract surgery. Curr Opin Ophthalmol. 2018;29(1):14-18.
43. US Food and Drug Administration. Summary of Safety and Effectiveness Data. Ophthalmic sealant: ReSure Sealant. https://www.accessdata.fda.gov/cdrh_docs/pdf13/P130004b.pdf. Published September 13, 2013. Accessed July 9, 2019.
44. About ReSure sealant. https://www.resuresealant.com/overview. Accessed July 31, 2019.
45. Menabuoni L, Pini R, Rossi F, Lenzetti I, Yoo SH, Parel JM. Laser-assisted corneal welding in cataract surgery: retrospective study. J Cataract Refract Surg. 2007;33(9):1608-1612.
46. Rasier R, Ozeren M, Artunay O, et al. Corneal tissue welding with infrared laser irradiation after clear corneal incision. Cornea. 2010;29(9):985-990.
47. Rossi F, Matteini P, Ratto F, Menabuoni L, Lenzetti I, Pini R. Laser tissue welding in ophthalmic surgery. J Biophotonics. 2008;1(4):331-342.
48. Taban M, Behrens A, Newcomb RL, et al. Acute endophthalmitis following cataract surgery: a systematic review of the literature. Arch Ophthalmol. 2005;123(5):613-620.
49. Taylor DM, Atlas BF, Romanchuk KG, Stern AL. Pseudophakic bullous keratopathy. Ophthalmology. 1983;90(1):19-24.
50. Lobo CL, Faria PM, Soares MA, Bernardes RC, Cunha-Vaz JG. Macular alterations after small-incision cataract surgery. J Cataract Refract Surg. 2004;30(4):752-760.
51. Flach AJ. The incidence, pathogenesis and treatment of cystoid macular edema following cataract surgery. Trans Am Ophthalmol Soc. 1998;96:557-634.
52. Wright PL, Wilkinson CP, Balyeat HD, Popham J, Reinke M. Angiographic cystoid macular edema after posterior chamber lens implantation. Arch Ophthalmol. 1988;106(6):740-744.
53. Kim SJ, Belair ML, Bressler NM, et al. A method of reporting macular edema after cataract surgery using optical coherence tomography. Retina. 2008;28(6):870-876.
54. Alio JL, Ruiz-Moreno JM, Shabayek MH, Lugo FL, Abd El Rahman AM. The risk of retinal detachment in high myopia after small incision coaxial phacoemulsification. Am J Ophthalmol. 2007;144(1):93-98.
55. Bhagwandien AC, Cheng YY, Wolfs RC, van Meurs JC, Luyten GP. Relationship between retinal detachment and biometry in 4262 cataractous eyes. Ophthalmology. 2006;113(4):643-649.
56. Boberg-Ans G, Henning V, Villumsen J, la Cour M. Longterm incidence of rhegmatogenous retinal detachment and survival in a defined population undergoing standardized phacoemulsification surgery. Acta Ophthalmol Scand. 2006;84(5):613-618.
57. Jakobsson G, Montan P, Zetterberg M, Stenevi U, Behndig A, Lundström M. Capsule complication during cataract surgery: retinal detachment after cataract surgery with capsule complication: Swedish Capsule Rupture Study Group report 4. J Cataract Refract Surg. 2009;35(10):1699-1705.
58. Neuhann IM, Neuhann TF, Heimann H, Schmickler S, Gerl RH, Foerster MH. Retinal detachment after phacoemulsification in high myopia: analysis of 2356 cases. J Cataract Refract Surg. 2008;34(10):1644-1657.
59. Russell M, Gaskin B, Russell D, Polkinghorne PJ. Pseudophakic retinal detachment after phacoemulsification cataract surgery: ten-year retrospective review. J Cataract Refract Surg. 2006;32(3):442-445.
60. Apple DJ, Solomon KD, Tetz MR, et al. Posterior capsule opacification. Surv Ophthalmol. 1992;37(2):73-116.
61. Wu S, Tong N, Pan L, et al. Retrospective analyses of potential risk factors for posterior capsule opacification after cataract surgery. J Ophthalmol. 2018;2018:9089285.
62. Clark A, Morlet N, Ng JQ, Preen DB, Semmens JB. Whole population trends in complications of cataract surgery over 22 years in Western Australia. Ophthalmology. 2011;118(6):1055-1061.
63. Adhikari S, Shrestha UD. Pediatric cataract surgery with hydrophilic acrylic intraocular lens implantation in Nepalese Children. Clin Ophthalmol. 2017;12:7-11.
64. Lee BJ, Smith SD, Jeng BH. Suture-related corneal infections after clear corneal cataract surgery. J Cataract Refract Surg. 2009;35(5):939-942.
65. May WN, Castro-Combs J, Kashiwabuchi RT, et al. Sutured clear corneal incision: wound apposition and permeability to bacterial-sized particles. Cornea. 2013;32(3):319-325.
66. Hillier RJ, Ajit RR, Kelly SP. Suture-related complications after cataract surgery: a patient safety issue. J Cataract Refract Surg. 2009;35(11):2035-2036.
67. Hovanesian JA, Karageozian VH. Watertight cataract incision closure using fibrin tissue adhesive. J Cataract Refract Surg. 2007;33(8):1461-1463.
The term cataract is derived from the Latin word “catarractes,” which means “waterfall,” as the foamy white opacity of an advanced cataract can be likened to a tempestuous cascade. Cataract is the leading cause of preventable blindness worldwide.1,2 It is no surprise, therefore, that cataract surgery is the most frequently performed ophthalmic surgical procedure worldwide. Cataract surgeries may reach 30 million annual cases by 2020.3 Given the large number of surgeries being performed, postsurgical complications are not uncommon.
Early postoperative complications from lens exchange (cataract) surgery include increased intraocular pressure (IOP), corneal edema, and corneal wound leakage.4 Corneal wound leakage is not uncommon; one study showed that, in 100 cases, almost one-third of incisions leaked.5 A 2014 prospective study of 500 postcataract surgery eyes revealed that 48.8% had fluid egress.6 Early detection is important so that efforts to restore corneal integrity can immediately be implemented. If not caught early, patients are at risk for developing a cascade of sequelae, including endophthalmitis.
The majority of corneal wound leaks postphacoemulsification are self-limiting and self-sealing. Moderate wound leaks require treatment, as in the following case. Strategies to detect, image, and treat wound leaks are covered in this discussion.
Case Presentation
A 69-year-old male veteran presented with no complaints for a 1-day postoperative visit following right eye phacoemulsification cataract extraction. His best corrected visual acuity in the right eye was 20/40, and his pinhole visual acuity was 20/25+2. On slit-lamp examination, the temporally located main incision appeared well-adhered and was found to be Seidel negative; however, the inferior paracentesis wound was found to be Seidel positive, demonstrating a slow leak. Intraocular pressure (IOP) measured with tonopen was 9 mm Hg.
A bandage soft contact lens was placed on the eye. The patient was instructed not to rub or place any pressure on the eye and to avoid bending and heavy lifting. He was also instructed to continue his postoperative medications (prednisolone 1% every 2 hours and polymyxin B sulfate 4 times daily) in his right eye. A follow-up appointment was scheduled for the next day.
The patient presented for his postoperative day-2 visit with a best corrected visual acuity in the right eye of 20/20. He reported no visual problems, no eye pain, and mentioned that he had had a comfortable night sleep. A slit-lamp examination revealed trace diffuse injection in the operative eye, predominantly central Descemet membrane folds, 1+ stromal edema, and a Seidel negative main incision wound. However, the inferior paracentesis wound showed a moderate leak (Seidel positive), and the anterior chamber showed a 1+ cell and flare. Goldmann tonometry revealed an IOP of 5 mm Hg, indicating hypotony.
Anterior segment cube 512 x 128 optical coherence tomography (OCT) was obtained with the bandage contact lens (Figures 1 and 2), and then repeated with the bandage contact lens removed (Figures 3 and 4). OCT imaging confirmed epithelial and endothelial gaping, loss of coaptation, and a localized detachment of the Descemet membrane. The veteran was referred to his surgeon that same day, and 2 limbal vicryl sutures were placed. The patient was instructed to continue prednisolone 1% 4 times daily and polymyxin B sulfate every 2 hours; erythromycin ointment 3 times daily was added to his regimen.
He was scheduled for a follow-up examination 1 week later. At that visit, the wound was no longer leaking and IOP had risen to a preoperative value of 17 mm Hg. The corneal sutures were removed at the 1-month postoperative examination and a follow-up was scheduled for 4 months later. An anterior segment OCT was obtained (Figure 5).
Discussion
In July 1967, Charles Kelman, MD, suggested using a dental ultrasonic tool, normally employed to clean teeth, to fragment the nucleus of the crystalline lens. Dr. Kelman’s first operation using phacoemulsification on a human eye took 3 hours.7 As the procedure for cataract removal has been refined, complication rates and surgical times have vastly improved.
Phacoemulsification is the most commonly performed outpatient surgery in the US; about 3 million cases are performed annually. Due to the high volume of cases, adverse events (AEs) are not uncommon. The incidence of complications following phacoemulsification is < 5%; the frequency of severe complications has been estimated at < 0.7%.8 Severe complications include endophthalmitis, suprachoroidal hemorrhage, and/or retinal detachment.9 Studies have shown a decline in rates of sight-threatening AEs from 1994 to 2006.9 A retrospective study of 45,082 veterans from 2005 to 2007 identified that a preoperative disease burden such as diabetes mellitus, chronic pulmonary disease, age-related macular degeneration, and diabetes with ophthalmic manifestations, was positively associated with a greater risk of cataract surgical complications.10
Complications
The level of a surgeon’s proficiency with phacoemulsification is directly correlated to the number of operations performed; there is a lower complication rate among more experienced surgeons, including those who work in high-volume settings.11,12 One study identified that the AE rate within 14 days of surgery was 0.8% for surgeons performing 50 to 250 cataract surgeries per year, but only 0.1% for those performing > 1000 cataract surgeries annually.12
Potential postoperative lens exchange complications include increased IOP, corneal wound leakage, corneal edema, bullous keratopathy, cystoid macular edema, retinal detachment, and endophthalmitis (Table 1). A corneal wound leak can provide a potential ingress for bacteria, putting the patient at risk for endophthalmitis, perhaps the most devastating complication following cataract surgery.
Endophthalmitis
Endophthalmitis has been reported to occur in .001% to .327% of patients during postoperative care.5,13-17 Early detection is important to maintain corneal integrity and prevent a cascade of detrimental ocular sequalae including the potential for endophthalmitis. According to Zaida and colleagues, endophthalmitis occurred in fewer than 1 of 1000 consecutive cases.14 A leaking clear corneal incision wound on the first day postoperatively has been associated with a 44-fold increased risk of endophthalmitis.13
Causes of endophthalmitis
In a retrospective case-controlled series of 57 patients with postcataract endophthalmitis, implantation of an intraocular lens with a resultant wound abnormality was thought to be the causative factor in 5%.17 Another source of endophthalmitis can be the intraocular lens (IOL), which may act as a vector for bacteria. By placing the IOL against the conjunctiva or exposing it to the theater air during surgery, bacteria can be introduced prior to implantation.17 Immunosuppressive treatment is the only patient antecedent factor that can be considered a predictor for endopthalmitis.17
The internal corneal seal is IOP dependent, and postoperative ocular hypotony may cause a seemingly watertight wound to leak. Taban and colleagues used anterior segment OCT to image numerous self-sealing incisions. They found that the corneal incision wound more tightly seals at higher IOPs. Additionally, more perpendicular (larger angle) incisions seal better at a lower IOP while less perpendicular (smaller angle) incisions seal better at a higher IOP (Figure 6).18
Incision Placement
Studies have shown that the main incision site is more clinically competent than is the side port incision site, as in our case study.19 Side-port incisions have a 1- or 2-plane architectural profile in contrast to the 3-plane profile typical of a main incision.19 Recent advances including the conversion to clear-corneal incisions of diminishing size, techniques used for wound construction, phacoemulsification machine design, and small-incision IOLs, should further reduce the prevalence and complications of wound compromise.20
Seidel Testing
Seidel testing is the most common method to evaluate corneal wound integrity and identify leaks. A drop of topical anesthetic is instilled in the eye and then a fluorescein strip (not fluorescein sodium and benoxinate hydrochloride ophthalmic solution, which may become less sterile since it has a multiuse container) is applied to the superior conjunctiva. The clinician then looks for evidence of fluid egress using the cobalt blue filter. The patient is instructed to blink once. Fluid egress appears as a black stream as the fluorescein dye becomes diluted by aqueous humor escaping the nonintact wound and the appearance of bright green dye surrounds the leak site. The term Seidel positive indicates a leak. An estimate should be made of the rate and volume of fluid exiting the wound.
Gonioscopy
Gonioscopy can be used to evaluate the postsurgical incision, more specifically for identification and management of internal incision wound gape. On gonioscopy, internal wound gape appears as an elongated oval opening resembling a fish mouth. If internal incision wound gape is identified gonioscopically before surgery is complete, the leak can be managed intraoperatively. The surgeon can irrigate along the length of the incision to remove cortical fragments or viscoelastic that may cause internal wound gaping. If unsuccessful, rapidly deepening the anterior chamber with balanced salt solution through the paracentesis incision may be employed. These methods may improve wound stability, reduce risk of postoperative hyphema, lower the incidence of endophthalmitis, and lessen the likelihood of late against-the-rule drift.21
Anterior Segment Optical Coherence Tomography
Instances when Seidel testing was negative despite actual wound gaping have been described.22,23 Anterior segment OCT is useful to evaluate incision architecture. A 2007 United Kingdom study investigated the corneal architecture in the immediate postoperative period following phacoemulsification using anterior segment OCT. This study showed the benefits of identifying architectural features such as epithelial gaping, endothelial gaping, stripping of Descemet membrane, and loss of coaptation. These features were found to be more common at low IOP and could represent a significant risk factor for endophthalmitis.24 Another study published by Behrens and colleagues indicated that a localized detachment of Descemet membrane may be more common than observed with slit-lamp (Figure 7). Corneal gaping, especially if along the entire length of the surgical wound, may lead to inadvertent bacterial access into the anterior chamber.25
Anterior segment OCT imaging was first described by Izatt and colleagues in 1994.26 Unlike posterior segment OCT, anterior segment OCT requires a greater depth of field and higher energy levels as images are commonly distorted by refraction at boundaries where the refractive index changes. Longer infrared wavelengths improve the penetration through tissues that scatter light, such as the sclera and limbus, which allows visualization, for example, of the iridocorneal angle.27,28
Two main scan patterns are used for anterior segment OCT: 512 x 128 cube scan (4-mm width x 4-mm length) and 5-line raster (3-mm length) with adjustable rotation and spacing. A recent software update allows measurement of corneal thickness, visualization of anterior chamber angle structures along with topographic analysis, anterior and posterior elevation maps of the cornea, and reliable pachymetric maps.29,30 The anterior segment cube acquires a series of 128 horizontal scan lines each composed of 512 A-scans. These high-definition scans acquire vertical and horizontal directions composed of 1024 A-scans each. This cube may be used to measure corneal thickness and visualize corneal architecture, creating a 3-D image of the data (Figure 8). The anterior segment 5-line raster scans through 5 parallel lines of equal length to view high-resolution images of the anterior chamber angle and cornea. Each line, fixed at 3-mm in length, is composed of 4096 A-scans.31 Anterior segment cube OCT allows identification of subtle variations in incision architecture at different locations across the width of the OCT image.
Bandage Soft Contact Lens
Upon reviewing the anterior segment OCT images of our patient with the bandage contact lens in place, it was evident that the adherent ocular bandage was protecting the incision. A tighter fitting bandage contact lens is ideal and adheres firmly to any area of epithelial damage and epithelial gaping to help seal the incision, protecting the wound and improving structural integrity. The bandage contact lens is gradually replaced by new cells via re-epithelialization; thus, it behaves as an adjunct to natural wound healing. A bandage contact lens also improves patient comfort.
It is hypothesized that a bandage contact lens improves the structural integrity of the incision site and helps prevent leaking, hypotony, and minor wound leaks. One study revealed a statistically significant lower IOP in nonbandage contact lens patients by an average of 6 mm Hg (mean [SD] 13.4 mm Hg [5.3]; range, 5 - 23 mm Hg) vs patients with a bandage contact lens (mean [SD] 19.4 mm Hg [5.9]; range, 11 - 29 mm Hg) in the immediate postoperative period.32 The authors suggested that the bandage contact lens may prevent microleaks, resulting in a higher IOP.
Aqueous Suppressants
Aqueous suppressants are a great option when IOP is abnormally elevated by decreasing the IOP and allowing the cornea to heal and self-seal.Effective aqueous suppressants are β blockers and carbonic anhydrase inhibitors.
After phacoemulsification ocular hypotony (< 6 mm Hg) occurs most commonly due to wound leakage or excessive intraocular inflammation. However, with the presence of corneal wound leakage and ocular hypotony, aqueous suppressants are not the best option.
Further Management of Wound Leaks
Management of a postoperative wound leak will vary based on severity. The majority of mild leaks are self-sealing. Anterior segment OCT helps the clinician to identify microleaks in an otherwise Seidel negative eye. If wound leakage is moderate with a formed anterior chamber, the use of a bandage contact lens is a good option, as can be the prescription of aqueous suppressants, depending on IOP.33
If the anterior chamber is flat, iris prolapse is apparent, or extremely low IOP exists, the patient needs to be referred to the surgeon. Current standard of care directs the surgeon to use sutures to further manage corneal wound leak. However, several studies have recognized the increased risk of suture-related complications, such as induced astigmatism, corneal opacities, incomplete wound closure, and corneal neovascularization.6,34-38 Other wound closure options include polyethylene glycol-based products, corneal welding, cyanoacrylate, or fibrin (Table 2).39 Traditionally nylon sutures have been used for clear corneal incision wound closure. However, tissue adhesives are gaining popularity as a substitute for sutures in wound closure.40
Cyanoacrylate
Numerous studies have been published on the efficacy of cyanoacrylate as a substitute for sutures, specifically in clear corneal incisions. AEs of cyanoacrylate include a transient foreign-body sensation and diffuse or focal bulbar conjunctival hyperemia.41,42 Shigemitsu and Majima found that fibrin and cyanoacrylate glue had tensile strength similar to sutures when used in cataract surgery.39 Polyethylene glycol-based products, also used in artificial tears and contact lens materials, may also help seal wound leaks. Another agent is ReSure (Ocular Therapeutix, Bedford, MA), an FDA-approved synthetic, polyethylene glycol hydrogel sealant that is 90% water after polymerization. ReSure has been shown to be safe and effective in sealing cataract surgical clear corneal incisions.6,43 ReSure takes about 20 seconds to prepare, and placement is aided by the use of a blue dye that dissipates within hours. This hydrogel will gradually slough off in the tears once the tissue has fully regenerated; there is no need to remove the sealant.44
Rossi and colleagues evaluated the efficacy of corneal welding to close wounds after cataract surgery. The technique involves laser-assisted closure of the corneal wound(s) by a diode laser that welds the stroma.45 Corneal welding takes seconds to achieve good closure without significant astigmatism or inflammation; however very careful application of the light absorbing dyes is required as they are toxic if allowed to enter the anterior chamber.45-47
Conclusion
Optometrists may be called to manage patients during both the preoperative and postoperative phases of cataract surgical care. Those who participate in postoperative care should carefully evaluate for the presence of wound leak or wound gape as a potential complication. The OCT may be employed to evaluate patients suspected of having these leaks or gapes. Proficiency in the interpretation of OCT results and more traditional evaluation methods allows for successful detection of wound leaks or gapes. The timely diagnosis and treatment of postoperative wound leaks allow for the best possible outcomes for cataract surgery patients.
The term cataract is derived from the Latin word “catarractes,” which means “waterfall,” as the foamy white opacity of an advanced cataract can be likened to a tempestuous cascade. Cataract is the leading cause of preventable blindness worldwide.1,2 It is no surprise, therefore, that cataract surgery is the most frequently performed ophthalmic surgical procedure worldwide. Cataract surgeries may reach 30 million annual cases by 2020.3 Given the large number of surgeries being performed, postsurgical complications are not uncommon.
Early postoperative complications from lens exchange (cataract) surgery include increased intraocular pressure (IOP), corneal edema, and corneal wound leakage.4 Corneal wound leakage is not uncommon; one study showed that, in 100 cases, almost one-third of incisions leaked.5 A 2014 prospective study of 500 postcataract surgery eyes revealed that 48.8% had fluid egress.6 Early detection is important so that efforts to restore corneal integrity can immediately be implemented. If not caught early, patients are at risk for developing a cascade of sequelae, including endophthalmitis.
The majority of corneal wound leaks postphacoemulsification are self-limiting and self-sealing. Moderate wound leaks require treatment, as in the following case. Strategies to detect, image, and treat wound leaks are covered in this discussion.
Case Presentation
A 69-year-old male veteran presented with no complaints for a 1-day postoperative visit following right eye phacoemulsification cataract extraction. His best corrected visual acuity in the right eye was 20/40, and his pinhole visual acuity was 20/25+2. On slit-lamp examination, the temporally located main incision appeared well-adhered and was found to be Seidel negative; however, the inferior paracentesis wound was found to be Seidel positive, demonstrating a slow leak. Intraocular pressure (IOP) measured with tonopen was 9 mm Hg.
A bandage soft contact lens was placed on the eye. The patient was instructed not to rub or place any pressure on the eye and to avoid bending and heavy lifting. He was also instructed to continue his postoperative medications (prednisolone 1% every 2 hours and polymyxin B sulfate 4 times daily) in his right eye. A follow-up appointment was scheduled for the next day.
The patient presented for his postoperative day-2 visit with a best corrected visual acuity in the right eye of 20/20. He reported no visual problems, no eye pain, and mentioned that he had had a comfortable night sleep. A slit-lamp examination revealed trace diffuse injection in the operative eye, predominantly central Descemet membrane folds, 1+ stromal edema, and a Seidel negative main incision wound. However, the inferior paracentesis wound showed a moderate leak (Seidel positive), and the anterior chamber showed a 1+ cell and flare. Goldmann tonometry revealed an IOP of 5 mm Hg, indicating hypotony.
Anterior segment cube 512 x 128 optical coherence tomography (OCT) was obtained with the bandage contact lens (Figures 1 and 2), and then repeated with the bandage contact lens removed (Figures 3 and 4). OCT imaging confirmed epithelial and endothelial gaping, loss of coaptation, and a localized detachment of the Descemet membrane. The veteran was referred to his surgeon that same day, and 2 limbal vicryl sutures were placed. The patient was instructed to continue prednisolone 1% 4 times daily and polymyxin B sulfate every 2 hours; erythromycin ointment 3 times daily was added to his regimen.
He was scheduled for a follow-up examination 1 week later. At that visit, the wound was no longer leaking and IOP had risen to a preoperative value of 17 mm Hg. The corneal sutures were removed at the 1-month postoperative examination and a follow-up was scheduled for 4 months later. An anterior segment OCT was obtained (Figure 5).
Discussion
In July 1967, Charles Kelman, MD, suggested using a dental ultrasonic tool, normally employed to clean teeth, to fragment the nucleus of the crystalline lens. Dr. Kelman’s first operation using phacoemulsification on a human eye took 3 hours.7 As the procedure for cataract removal has been refined, complication rates and surgical times have vastly improved.
Phacoemulsification is the most commonly performed outpatient surgery in the US; about 3 million cases are performed annually. Due to the high volume of cases, adverse events (AEs) are not uncommon. The incidence of complications following phacoemulsification is < 5%; the frequency of severe complications has been estimated at < 0.7%.8 Severe complications include endophthalmitis, suprachoroidal hemorrhage, and/or retinal detachment.9 Studies have shown a decline in rates of sight-threatening AEs from 1994 to 2006.9 A retrospective study of 45,082 veterans from 2005 to 2007 identified that a preoperative disease burden such as diabetes mellitus, chronic pulmonary disease, age-related macular degeneration, and diabetes with ophthalmic manifestations, was positively associated with a greater risk of cataract surgical complications.10
Complications
The level of a surgeon’s proficiency with phacoemulsification is directly correlated to the number of operations performed; there is a lower complication rate among more experienced surgeons, including those who work in high-volume settings.11,12 One study identified that the AE rate within 14 days of surgery was 0.8% for surgeons performing 50 to 250 cataract surgeries per year, but only 0.1% for those performing > 1000 cataract surgeries annually.12
Potential postoperative lens exchange complications include increased IOP, corneal wound leakage, corneal edema, bullous keratopathy, cystoid macular edema, retinal detachment, and endophthalmitis (Table 1). A corneal wound leak can provide a potential ingress for bacteria, putting the patient at risk for endophthalmitis, perhaps the most devastating complication following cataract surgery.
Endophthalmitis
Endophthalmitis has been reported to occur in .001% to .327% of patients during postoperative care.5,13-17 Early detection is important to maintain corneal integrity and prevent a cascade of detrimental ocular sequalae including the potential for endophthalmitis. According to Zaida and colleagues, endophthalmitis occurred in fewer than 1 of 1000 consecutive cases.14 A leaking clear corneal incision wound on the first day postoperatively has been associated with a 44-fold increased risk of endophthalmitis.13
Causes of endophthalmitis
In a retrospective case-controlled series of 57 patients with postcataract endophthalmitis, implantation of an intraocular lens with a resultant wound abnormality was thought to be the causative factor in 5%.17 Another source of endophthalmitis can be the intraocular lens (IOL), which may act as a vector for bacteria. By placing the IOL against the conjunctiva or exposing it to the theater air during surgery, bacteria can be introduced prior to implantation.17 Immunosuppressive treatment is the only patient antecedent factor that can be considered a predictor for endopthalmitis.17
The internal corneal seal is IOP dependent, and postoperative ocular hypotony may cause a seemingly watertight wound to leak. Taban and colleagues used anterior segment OCT to image numerous self-sealing incisions. They found that the corneal incision wound more tightly seals at higher IOPs. Additionally, more perpendicular (larger angle) incisions seal better at a lower IOP while less perpendicular (smaller angle) incisions seal better at a higher IOP (Figure 6).18
Incision Placement
Studies have shown that the main incision site is more clinically competent than is the side port incision site, as in our case study.19 Side-port incisions have a 1- or 2-plane architectural profile in contrast to the 3-plane profile typical of a main incision.19 Recent advances including the conversion to clear-corneal incisions of diminishing size, techniques used for wound construction, phacoemulsification machine design, and small-incision IOLs, should further reduce the prevalence and complications of wound compromise.20
Seidel Testing
Seidel testing is the most common method to evaluate corneal wound integrity and identify leaks. A drop of topical anesthetic is instilled in the eye and then a fluorescein strip (not fluorescein sodium and benoxinate hydrochloride ophthalmic solution, which may become less sterile since it has a multiuse container) is applied to the superior conjunctiva. The clinician then looks for evidence of fluid egress using the cobalt blue filter. The patient is instructed to blink once. Fluid egress appears as a black stream as the fluorescein dye becomes diluted by aqueous humor escaping the nonintact wound and the appearance of bright green dye surrounds the leak site. The term Seidel positive indicates a leak. An estimate should be made of the rate and volume of fluid exiting the wound.
Gonioscopy
Gonioscopy can be used to evaluate the postsurgical incision, more specifically for identification and management of internal incision wound gape. On gonioscopy, internal wound gape appears as an elongated oval opening resembling a fish mouth. If internal incision wound gape is identified gonioscopically before surgery is complete, the leak can be managed intraoperatively. The surgeon can irrigate along the length of the incision to remove cortical fragments or viscoelastic that may cause internal wound gaping. If unsuccessful, rapidly deepening the anterior chamber with balanced salt solution through the paracentesis incision may be employed. These methods may improve wound stability, reduce risk of postoperative hyphema, lower the incidence of endophthalmitis, and lessen the likelihood of late against-the-rule drift.21
Anterior Segment Optical Coherence Tomography
Instances when Seidel testing was negative despite actual wound gaping have been described.22,23 Anterior segment OCT is useful to evaluate incision architecture. A 2007 United Kingdom study investigated the corneal architecture in the immediate postoperative period following phacoemulsification using anterior segment OCT. This study showed the benefits of identifying architectural features such as epithelial gaping, endothelial gaping, stripping of Descemet membrane, and loss of coaptation. These features were found to be more common at low IOP and could represent a significant risk factor for endophthalmitis.24 Another study published by Behrens and colleagues indicated that a localized detachment of Descemet membrane may be more common than observed with slit-lamp (Figure 7). Corneal gaping, especially if along the entire length of the surgical wound, may lead to inadvertent bacterial access into the anterior chamber.25
Anterior segment OCT imaging was first described by Izatt and colleagues in 1994.26 Unlike posterior segment OCT, anterior segment OCT requires a greater depth of field and higher energy levels as images are commonly distorted by refraction at boundaries where the refractive index changes. Longer infrared wavelengths improve the penetration through tissues that scatter light, such as the sclera and limbus, which allows visualization, for example, of the iridocorneal angle.27,28
Two main scan patterns are used for anterior segment OCT: 512 x 128 cube scan (4-mm width x 4-mm length) and 5-line raster (3-mm length) with adjustable rotation and spacing. A recent software update allows measurement of corneal thickness, visualization of anterior chamber angle structures along with topographic analysis, anterior and posterior elevation maps of the cornea, and reliable pachymetric maps.29,30 The anterior segment cube acquires a series of 128 horizontal scan lines each composed of 512 A-scans. These high-definition scans acquire vertical and horizontal directions composed of 1024 A-scans each. This cube may be used to measure corneal thickness and visualize corneal architecture, creating a 3-D image of the data (Figure 8). The anterior segment 5-line raster scans through 5 parallel lines of equal length to view high-resolution images of the anterior chamber angle and cornea. Each line, fixed at 3-mm in length, is composed of 4096 A-scans.31 Anterior segment cube OCT allows identification of subtle variations in incision architecture at different locations across the width of the OCT image.
Bandage Soft Contact Lens
Upon reviewing the anterior segment OCT images of our patient with the bandage contact lens in place, it was evident that the adherent ocular bandage was protecting the incision. A tighter fitting bandage contact lens is ideal and adheres firmly to any area of epithelial damage and epithelial gaping to help seal the incision, protecting the wound and improving structural integrity. The bandage contact lens is gradually replaced by new cells via re-epithelialization; thus, it behaves as an adjunct to natural wound healing. A bandage contact lens also improves patient comfort.
It is hypothesized that a bandage contact lens improves the structural integrity of the incision site and helps prevent leaking, hypotony, and minor wound leaks. One study revealed a statistically significant lower IOP in nonbandage contact lens patients by an average of 6 mm Hg (mean [SD] 13.4 mm Hg [5.3]; range, 5 - 23 mm Hg) vs patients with a bandage contact lens (mean [SD] 19.4 mm Hg [5.9]; range, 11 - 29 mm Hg) in the immediate postoperative period.32 The authors suggested that the bandage contact lens may prevent microleaks, resulting in a higher IOP.
Aqueous Suppressants
Aqueous suppressants are a great option when IOP is abnormally elevated by decreasing the IOP and allowing the cornea to heal and self-seal.Effective aqueous suppressants are β blockers and carbonic anhydrase inhibitors.
After phacoemulsification ocular hypotony (< 6 mm Hg) occurs most commonly due to wound leakage or excessive intraocular inflammation. However, with the presence of corneal wound leakage and ocular hypotony, aqueous suppressants are not the best option.
Further Management of Wound Leaks
Management of a postoperative wound leak will vary based on severity. The majority of mild leaks are self-sealing. Anterior segment OCT helps the clinician to identify microleaks in an otherwise Seidel negative eye. If wound leakage is moderate with a formed anterior chamber, the use of a bandage contact lens is a good option, as can be the prescription of aqueous suppressants, depending on IOP.33
If the anterior chamber is flat, iris prolapse is apparent, or extremely low IOP exists, the patient needs to be referred to the surgeon. Current standard of care directs the surgeon to use sutures to further manage corneal wound leak. However, several studies have recognized the increased risk of suture-related complications, such as induced astigmatism, corneal opacities, incomplete wound closure, and corneal neovascularization.6,34-38 Other wound closure options include polyethylene glycol-based products, corneal welding, cyanoacrylate, or fibrin (Table 2).39 Traditionally nylon sutures have been used for clear corneal incision wound closure. However, tissue adhesives are gaining popularity as a substitute for sutures in wound closure.40
Cyanoacrylate
Numerous studies have been published on the efficacy of cyanoacrylate as a substitute for sutures, specifically in clear corneal incisions. AEs of cyanoacrylate include a transient foreign-body sensation and diffuse or focal bulbar conjunctival hyperemia.41,42 Shigemitsu and Majima found that fibrin and cyanoacrylate glue had tensile strength similar to sutures when used in cataract surgery.39 Polyethylene glycol-based products, also used in artificial tears and contact lens materials, may also help seal wound leaks. Another agent is ReSure (Ocular Therapeutix, Bedford, MA), an FDA-approved synthetic, polyethylene glycol hydrogel sealant that is 90% water after polymerization. ReSure has been shown to be safe and effective in sealing cataract surgical clear corneal incisions.6,43 ReSure takes about 20 seconds to prepare, and placement is aided by the use of a blue dye that dissipates within hours. This hydrogel will gradually slough off in the tears once the tissue has fully regenerated; there is no need to remove the sealant.44
Rossi and colleagues evaluated the efficacy of corneal welding to close wounds after cataract surgery. The technique involves laser-assisted closure of the corneal wound(s) by a diode laser that welds the stroma.45 Corneal welding takes seconds to achieve good closure without significant astigmatism or inflammation; however very careful application of the light absorbing dyes is required as they are toxic if allowed to enter the anterior chamber.45-47
Conclusion
Optometrists may be called to manage patients during both the preoperative and postoperative phases of cataract surgical care. Those who participate in postoperative care should carefully evaluate for the presence of wound leak or wound gape as a potential complication. The OCT may be employed to evaluate patients suspected of having these leaks or gapes. Proficiency in the interpretation of OCT results and more traditional evaluation methods allows for successful detection of wound leaks or gapes. The timely diagnosis and treatment of postoperative wound leaks allow for the best possible outcomes for cataract surgery patients.
1. Thylefors B, Négrel AD, Pararajasegaram R, Dadzie KY. Global data on blindness. Bull World Health Organ. 1995;73(1):115-121.
2. Flaxman SR, Bourne RRA, Resnikoff S, et al; Vision Loss Expert Group of the Global Burden of Disease Study. Global causes of blindness and distance vision impairment 1990-2020: a systematic review and meta-analysis. Lancet Glob Health. 2017;5(12):e1221-e1224.
3. Congdon N, Vingerling JR, Klein BE, et al; Eye Diseases Prevalence Research Group. Prevalence of cataract and pseudophakia/aphakia among adults in the United States. Arch Ophthalmol. 2004;122(4):487-494.
4. Kurt E, Mayalı H. Early post-operative complications in cataract surgery. In: Zaidi FH, ed. Cataract Surgery. IntechOpen; 2013. https://www.intechopen.com/books/cataract-surgery/post-operative-infections-associated-with-cataract-surgery. Accessed July 15, 2019.
5. Chee SP. Clear corneal incision leakage after phacoemulsification--detection using povidone iodine 5%. Int Ophthalmol. 2005;26(4-5):175-179.
6. Masket S, Hovanesian JA, Levenson J, et al. Hydrogel sealant versus sutures to prevent fluid egress after cataract surgery. J Cataract Refract Surg. 2014;40(12):2057-2066.
7. Kelman CD. Phaco-emulsification and aspiration: a new technique of cataract removal. A preliminary report. Am J Ophthalmol. 1967;64(1):23-35.
8. Powe NR, Schein OD, Gieser SC, et al. Synthesis of the literature on visual acuity and complications following cataract extraction with intraocular lens implantation. Cataract Patient Outcome Research Team [published correction appears in Arch Ophthalmol. 1994;112(7):889]. Arch Ophthalmol. 1994;112(2):239-252.
9. Stein JD, Grossman DS, Mundy KM, Sugar A, Sloan FA. Severe adverse events after cataract surgery among medicare beneficiaries. Ophthalmology. 2011;118(9):1716-1723.
10. Greenberg PB, Tseng VL, Wu WC, et al. Prevalence and predictors of ocular complications associated with cataract surgery in United States veterans. Ophthalmology. 2011;118(3):507-514.
11. Mangan MS, Atalay E, Anci C, Tuncer I, Bilqec MD. Comparison of different types of complications in the phacoemulsification surgery learning curve according to number of operations performed. Turk J Ophthalmol. 2016;46(1):7-10.
12. Bell CM, Hatch WV, Cernat G, Urbach DR. Surgeon volumes and selected patient outcomes in cataract surgery: a population-based analysis. Ophthalmology. 2007;114(3):405-410.
13. Wallin T, Parker J, Jin Y, Kefalopoulos G, Olson RJ. Cohort study of 27 cases of endophthalmitis at a single institution. J Cataract Refract Surg. 2005;31(4):735-741.
14. Zaidi FH, Corbett MC, Burton BJ, Bloom PA. Raising the benchmark for the 21st century--the 1000 cataract operations audit and survey: outcomes, consultant-supervised training and sourcing NHS choice. Br J Ophthalmol. 2007;91(6):731-736.
15. Nichamin LD, Chang DF, Johnson SH, et al; American Society of Cataract and Refractive Surgery Cataract Clinical Committee. ASCRS white paper: what is the association between clear corneal cataract incisions and postoperative endophthalmitis? J Cataract Refract Surg. 2006;32(9):1556-1559.
16. Packer M, Chang DF, Dewey SH, et al; ASCRS Cataract Clinical Committee. Prevention, diagnosis, and management of acute postoperative bacterial endophthalmitis. J Cataract Refract Surg. 2011;37(9):1699-1714.
17. Montan PG, Koranyi G, Setterquist HE, Stridh A, Philipson BT, Wiklund K. Endophthalmitis after cataract surgery: risk factors relating to technique and events of the operation and patient history: a retrospective case-control study. Ophthalmology. 1998;105(12):2171-2177.
18. Taban M, Rao B, Reznik J, Zhang J, Chen Z, McDonnell PJ. Dynamic morphology of sutureless cataract wounds—effect of incision angle and location. Surv Ophthalmol. 2004;49(suppl 2):S62-S72.
19. Chee SP, Ti SE, Lim L, Chan AS, Jap A. Anterior segment optical coherence tomography evaluation of the integrity of clear corneal incisions: a comparison between 2.2-mm and 2.65-mm main incisions. Am J Ophthalmol. 2010;149(5):768-776.e1.
20. Koch DD, Nacke RE, Wang L, Novak KD. Issues in wound management. In: Steinert R, ed. Cataract Surgery. 3rd ed. New York: Elsevier; 2009:581-588.
21. Gimbel HV, Sun R, DeBroff GM. Recognition and management of internal wound gape. J Cataract Refract Surg. 1995;21(2):121-124.
22. May WN, Castro-Combs J, Quinto GG, Kashiwabuchi R, Gower EW, Behrens A. Standardized Seidel test to evaluate different sutureless cataract incision configurations. J Cataract Refract Surg. 2010;36(6):1011-1017.
23. Kashiwabuchi FK, Khan YA, Rodrigues MW Jr, Wang J, McDonnell PJ, Daoud YJ. Seidel and India ink tests assessment of different clear cornea side-port incision configurations. Graefes Arch Clin Exp Ophthalmol. 2013;251(8):1961-1965.
24. Calladine D, Packard R. Clear corneal incision architecture in the immediate postoperative period evaluated using optical coherence tomography. J Cataract Refract Surg. 2007;33(8):1429-1435.
25. Behrens WJ, Stark KA, Pratzer, McDonnell PJ. Dynamics of small-incision clear cornea wounds after phacoemulsification surgery using optical coherence tomography in the early postoperative period. J Refractive Surgery. 2008;24(1):46-49.
26. Izatt JA, Hee MR, Swanson EA, et al. Micrometer-scale resolution imaging of the anterior eye in vivo with optical coherence tomography. Arch Ophthalmol. 1994;112(12):1584-1589.
27. Hurmeric V, Yoo SH, Mutlu FM. Optical coherence tomography in cornea and refractive surgery. Expert Rev Ophthalmol. 2012;7(3):241-250.
28. Schuman JS, Puliafito CA, Fujimoto JG, Duker JS. Optical Coherence Tomography of Ocular Diseases. 3rd ed. Thorofare, NJ: Slack Inc; 2013.
29. Salim S. The role of anterior segment optical coherence tomography in glaucoma. J Ophthalmol. 2012;2012:476801.
30. Kharousi NA, Wali UK, Azeem S. Current applications of optical coherence tomography in ophthalmology. In: Kawasaki M, ed. Optical Coherence Tomography. IntechOpen; 2013. https://www.intechopen.com/books/optical-coherence-tomography. Accessed July 31, 2019.
31. Rodrigues EB, Johanson M, Penha FM. Anterior segment tomography with the cirrus optical coherence tomography. J Ophthalmol. 2012;2012:806989.
32. Calladine D, Ward M, Packard R. Adherent ocular bandage for clear corneal incisions used in cataract surgery. J Cataract Refract Surg. 2010;36(11):1839-1848.
33. Haldar K, Saraff R. Closure technique for leaking wound resulting from thermal injury during phacoemulsification. J Cataract Refract Surg. 2014;40(9):1412-1414.
34. Zoghby JT, Cohen KL. Phacoemulsification-related corneal incision contracture. https://www.aao.org/eyenet/article/phacoemulsification-related-corneal-incision-contr. Published December 2012. Accessed June 16, 2019.
35. Bhatia SS. Ocular surface sealants and adhesives. Ocul Surf. 2006;4(3):146-154.
36. May WN, Castro-Combs J, Kashiwabuchi RT, et al. Bacterial-sized particle inflow through sutured clear corneal incisions in a laboratory human model. J Cataract Refract Surg. 2011;37(6):1140-1146.
37. Meskin SW, Ritterband DC, Shapiro DE, et al. Liquid bandage (2-octyl cyanoacrylate) as a temporary wound barrier in clear corneal cataract surgery. Ophthalmology. 2005;112(11):2015-2021.
38. Heaven CJ, Davison CR, Cockcroft PM. Bacterial contamination of nylon corneal sutures. Eye (Lond). 1995;9(pt 1):116-118.
39. Shigemitsu T, Majima Y. The utilization of a biological adhesive for wound treatment: comparison of suture, self-sealing sutureless and cyanoacrylate closure in the tensile strength test. Int Ophthalmol. 1996-1997;20:323-328.
40. Uy HS, Kenyon KR. Surgical outcomes after application of a liquid adhesive ocular bandage to clear corneal incisions during cataract surgery. J Cataract Refract Surg. 2013;39(11):1668-1674.
41. Meskin SW, Ritterband DC, Shapiro DE, et al. Liquid bandage (2-octyl cyanoacrylate) as a temporary wound barrier in clear corneal cataract surgery. Ophthalmology. 2005;112(11):2015-2021.
42. Tong AY, Gupta PK, Kim T. Wound closure and tissue adhesives in clear corneal incision cataract surgery. Curr Opin Ophthalmol. 2018;29(1):14-18.
43. US Food and Drug Administration. Summary of Safety and Effectiveness Data. Ophthalmic sealant: ReSure Sealant. https://www.accessdata.fda.gov/cdrh_docs/pdf13/P130004b.pdf. Published September 13, 2013. Accessed July 9, 2019.
44. About ReSure sealant. https://www.resuresealant.com/overview. Accessed July 31, 2019.
45. Menabuoni L, Pini R, Rossi F, Lenzetti I, Yoo SH, Parel JM. Laser-assisted corneal welding in cataract surgery: retrospective study. J Cataract Refract Surg. 2007;33(9):1608-1612.
46. Rasier R, Ozeren M, Artunay O, et al. Corneal tissue welding with infrared laser irradiation after clear corneal incision. Cornea. 2010;29(9):985-990.
47. Rossi F, Matteini P, Ratto F, Menabuoni L, Lenzetti I, Pini R. Laser tissue welding in ophthalmic surgery. J Biophotonics. 2008;1(4):331-342.
48. Taban M, Behrens A, Newcomb RL, et al. Acute endophthalmitis following cataract surgery: a systematic review of the literature. Arch Ophthalmol. 2005;123(5):613-620.
49. Taylor DM, Atlas BF, Romanchuk KG, Stern AL. Pseudophakic bullous keratopathy. Ophthalmology. 1983;90(1):19-24.
50. Lobo CL, Faria PM, Soares MA, Bernardes RC, Cunha-Vaz JG. Macular alterations after small-incision cataract surgery. J Cataract Refract Surg. 2004;30(4):752-760.
51. Flach AJ. The incidence, pathogenesis and treatment of cystoid macular edema following cataract surgery. Trans Am Ophthalmol Soc. 1998;96:557-634.
52. Wright PL, Wilkinson CP, Balyeat HD, Popham J, Reinke M. Angiographic cystoid macular edema after posterior chamber lens implantation. Arch Ophthalmol. 1988;106(6):740-744.
53. Kim SJ, Belair ML, Bressler NM, et al. A method of reporting macular edema after cataract surgery using optical coherence tomography. Retina. 2008;28(6):870-876.
54. Alio JL, Ruiz-Moreno JM, Shabayek MH, Lugo FL, Abd El Rahman AM. The risk of retinal detachment in high myopia after small incision coaxial phacoemulsification. Am J Ophthalmol. 2007;144(1):93-98.
55. Bhagwandien AC, Cheng YY, Wolfs RC, van Meurs JC, Luyten GP. Relationship between retinal detachment and biometry in 4262 cataractous eyes. Ophthalmology. 2006;113(4):643-649.
56. Boberg-Ans G, Henning V, Villumsen J, la Cour M. Longterm incidence of rhegmatogenous retinal detachment and survival in a defined population undergoing standardized phacoemulsification surgery. Acta Ophthalmol Scand. 2006;84(5):613-618.
57. Jakobsson G, Montan P, Zetterberg M, Stenevi U, Behndig A, Lundström M. Capsule complication during cataract surgery: retinal detachment after cataract surgery with capsule complication: Swedish Capsule Rupture Study Group report 4. J Cataract Refract Surg. 2009;35(10):1699-1705.
58. Neuhann IM, Neuhann TF, Heimann H, Schmickler S, Gerl RH, Foerster MH. Retinal detachment after phacoemulsification in high myopia: analysis of 2356 cases. J Cataract Refract Surg. 2008;34(10):1644-1657.
59. Russell M, Gaskin B, Russell D, Polkinghorne PJ. Pseudophakic retinal detachment after phacoemulsification cataract surgery: ten-year retrospective review. J Cataract Refract Surg. 2006;32(3):442-445.
60. Apple DJ, Solomon KD, Tetz MR, et al. Posterior capsule opacification. Surv Ophthalmol. 1992;37(2):73-116.
61. Wu S, Tong N, Pan L, et al. Retrospective analyses of potential risk factors for posterior capsule opacification after cataract surgery. J Ophthalmol. 2018;2018:9089285.
62. Clark A, Morlet N, Ng JQ, Preen DB, Semmens JB. Whole population trends in complications of cataract surgery over 22 years in Western Australia. Ophthalmology. 2011;118(6):1055-1061.
63. Adhikari S, Shrestha UD. Pediatric cataract surgery with hydrophilic acrylic intraocular lens implantation in Nepalese Children. Clin Ophthalmol. 2017;12:7-11.
64. Lee BJ, Smith SD, Jeng BH. Suture-related corneal infections after clear corneal cataract surgery. J Cataract Refract Surg. 2009;35(5):939-942.
65. May WN, Castro-Combs J, Kashiwabuchi RT, et al. Sutured clear corneal incision: wound apposition and permeability to bacterial-sized particles. Cornea. 2013;32(3):319-325.
66. Hillier RJ, Ajit RR, Kelly SP. Suture-related complications after cataract surgery: a patient safety issue. J Cataract Refract Surg. 2009;35(11):2035-2036.
67. Hovanesian JA, Karageozian VH. Watertight cataract incision closure using fibrin tissue adhesive. J Cataract Refract Surg. 2007;33(8):1461-1463.
1. Thylefors B, Négrel AD, Pararajasegaram R, Dadzie KY. Global data on blindness. Bull World Health Organ. 1995;73(1):115-121.
2. Flaxman SR, Bourne RRA, Resnikoff S, et al; Vision Loss Expert Group of the Global Burden of Disease Study. Global causes of blindness and distance vision impairment 1990-2020: a systematic review and meta-analysis. Lancet Glob Health. 2017;5(12):e1221-e1224.
3. Congdon N, Vingerling JR, Klein BE, et al; Eye Diseases Prevalence Research Group. Prevalence of cataract and pseudophakia/aphakia among adults in the United States. Arch Ophthalmol. 2004;122(4):487-494.
4. Kurt E, Mayalı H. Early post-operative complications in cataract surgery. In: Zaidi FH, ed. Cataract Surgery. IntechOpen; 2013. https://www.intechopen.com/books/cataract-surgery/post-operative-infections-associated-with-cataract-surgery. Accessed July 15, 2019.
5. Chee SP. Clear corneal incision leakage after phacoemulsification--detection using povidone iodine 5%. Int Ophthalmol. 2005;26(4-5):175-179.
6. Masket S, Hovanesian JA, Levenson J, et al. Hydrogel sealant versus sutures to prevent fluid egress after cataract surgery. J Cataract Refract Surg. 2014;40(12):2057-2066.
7. Kelman CD. Phaco-emulsification and aspiration: a new technique of cataract removal. A preliminary report. Am J Ophthalmol. 1967;64(1):23-35.
8. Powe NR, Schein OD, Gieser SC, et al. Synthesis of the literature on visual acuity and complications following cataract extraction with intraocular lens implantation. Cataract Patient Outcome Research Team [published correction appears in Arch Ophthalmol. 1994;112(7):889]. Arch Ophthalmol. 1994;112(2):239-252.
9. Stein JD, Grossman DS, Mundy KM, Sugar A, Sloan FA. Severe adverse events after cataract surgery among medicare beneficiaries. Ophthalmology. 2011;118(9):1716-1723.
10. Greenberg PB, Tseng VL, Wu WC, et al. Prevalence and predictors of ocular complications associated with cataract surgery in United States veterans. Ophthalmology. 2011;118(3):507-514.
11. Mangan MS, Atalay E, Anci C, Tuncer I, Bilqec MD. Comparison of different types of complications in the phacoemulsification surgery learning curve according to number of operations performed. Turk J Ophthalmol. 2016;46(1):7-10.
12. Bell CM, Hatch WV, Cernat G, Urbach DR. Surgeon volumes and selected patient outcomes in cataract surgery: a population-based analysis. Ophthalmology. 2007;114(3):405-410.
13. Wallin T, Parker J, Jin Y, Kefalopoulos G, Olson RJ. Cohort study of 27 cases of endophthalmitis at a single institution. J Cataract Refract Surg. 2005;31(4):735-741.
14. Zaidi FH, Corbett MC, Burton BJ, Bloom PA. Raising the benchmark for the 21st century--the 1000 cataract operations audit and survey: outcomes, consultant-supervised training and sourcing NHS choice. Br J Ophthalmol. 2007;91(6):731-736.
15. Nichamin LD, Chang DF, Johnson SH, et al; American Society of Cataract and Refractive Surgery Cataract Clinical Committee. ASCRS white paper: what is the association between clear corneal cataract incisions and postoperative endophthalmitis? J Cataract Refract Surg. 2006;32(9):1556-1559.
16. Packer M, Chang DF, Dewey SH, et al; ASCRS Cataract Clinical Committee. Prevention, diagnosis, and management of acute postoperative bacterial endophthalmitis. J Cataract Refract Surg. 2011;37(9):1699-1714.
17. Montan PG, Koranyi G, Setterquist HE, Stridh A, Philipson BT, Wiklund K. Endophthalmitis after cataract surgery: risk factors relating to technique and events of the operation and patient history: a retrospective case-control study. Ophthalmology. 1998;105(12):2171-2177.
18. Taban M, Rao B, Reznik J, Zhang J, Chen Z, McDonnell PJ. Dynamic morphology of sutureless cataract wounds—effect of incision angle and location. Surv Ophthalmol. 2004;49(suppl 2):S62-S72.
19. Chee SP, Ti SE, Lim L, Chan AS, Jap A. Anterior segment optical coherence tomography evaluation of the integrity of clear corneal incisions: a comparison between 2.2-mm and 2.65-mm main incisions. Am J Ophthalmol. 2010;149(5):768-776.e1.
20. Koch DD, Nacke RE, Wang L, Novak KD. Issues in wound management. In: Steinert R, ed. Cataract Surgery. 3rd ed. New York: Elsevier; 2009:581-588.
21. Gimbel HV, Sun R, DeBroff GM. Recognition and management of internal wound gape. J Cataract Refract Surg. 1995;21(2):121-124.
22. May WN, Castro-Combs J, Quinto GG, Kashiwabuchi R, Gower EW, Behrens A. Standardized Seidel test to evaluate different sutureless cataract incision configurations. J Cataract Refract Surg. 2010;36(6):1011-1017.
23. Kashiwabuchi FK, Khan YA, Rodrigues MW Jr, Wang J, McDonnell PJ, Daoud YJ. Seidel and India ink tests assessment of different clear cornea side-port incision configurations. Graefes Arch Clin Exp Ophthalmol. 2013;251(8):1961-1965.
24. Calladine D, Packard R. Clear corneal incision architecture in the immediate postoperative period evaluated using optical coherence tomography. J Cataract Refract Surg. 2007;33(8):1429-1435.
25. Behrens WJ, Stark KA, Pratzer, McDonnell PJ. Dynamics of small-incision clear cornea wounds after phacoemulsification surgery using optical coherence tomography in the early postoperative period. J Refractive Surgery. 2008;24(1):46-49.
26. Izatt JA, Hee MR, Swanson EA, et al. Micrometer-scale resolution imaging of the anterior eye in vivo with optical coherence tomography. Arch Ophthalmol. 1994;112(12):1584-1589.
27. Hurmeric V, Yoo SH, Mutlu FM. Optical coherence tomography in cornea and refractive surgery. Expert Rev Ophthalmol. 2012;7(3):241-250.
28. Schuman JS, Puliafito CA, Fujimoto JG, Duker JS. Optical Coherence Tomography of Ocular Diseases. 3rd ed. Thorofare, NJ: Slack Inc; 2013.
29. Salim S. The role of anterior segment optical coherence tomography in glaucoma. J Ophthalmol. 2012;2012:476801.
30. Kharousi NA, Wali UK, Azeem S. Current applications of optical coherence tomography in ophthalmology. In: Kawasaki M, ed. Optical Coherence Tomography. IntechOpen; 2013. https://www.intechopen.com/books/optical-coherence-tomography. Accessed July 31, 2019.
31. Rodrigues EB, Johanson M, Penha FM. Anterior segment tomography with the cirrus optical coherence tomography. J Ophthalmol. 2012;2012:806989.
32. Calladine D, Ward M, Packard R. Adherent ocular bandage for clear corneal incisions used in cataract surgery. J Cataract Refract Surg. 2010;36(11):1839-1848.
33. Haldar K, Saraff R. Closure technique for leaking wound resulting from thermal injury during phacoemulsification. J Cataract Refract Surg. 2014;40(9):1412-1414.
34. Zoghby JT, Cohen KL. Phacoemulsification-related corneal incision contracture. https://www.aao.org/eyenet/article/phacoemulsification-related-corneal-incision-contr. Published December 2012. Accessed June 16, 2019.
35. Bhatia SS. Ocular surface sealants and adhesives. Ocul Surf. 2006;4(3):146-154.
36. May WN, Castro-Combs J, Kashiwabuchi RT, et al. Bacterial-sized particle inflow through sutured clear corneal incisions in a laboratory human model. J Cataract Refract Surg. 2011;37(6):1140-1146.
37. Meskin SW, Ritterband DC, Shapiro DE, et al. Liquid bandage (2-octyl cyanoacrylate) as a temporary wound barrier in clear corneal cataract surgery. Ophthalmology. 2005;112(11):2015-2021.
38. Heaven CJ, Davison CR, Cockcroft PM. Bacterial contamination of nylon corneal sutures. Eye (Lond). 1995;9(pt 1):116-118.
39. Shigemitsu T, Majima Y. The utilization of a biological adhesive for wound treatment: comparison of suture, self-sealing sutureless and cyanoacrylate closure in the tensile strength test. Int Ophthalmol. 1996-1997;20:323-328.
40. Uy HS, Kenyon KR. Surgical outcomes after application of a liquid adhesive ocular bandage to clear corneal incisions during cataract surgery. J Cataract Refract Surg. 2013;39(11):1668-1674.
41. Meskin SW, Ritterband DC, Shapiro DE, et al. Liquid bandage (2-octyl cyanoacrylate) as a temporary wound barrier in clear corneal cataract surgery. Ophthalmology. 2005;112(11):2015-2021.
42. Tong AY, Gupta PK, Kim T. Wound closure and tissue adhesives in clear corneal incision cataract surgery. Curr Opin Ophthalmol. 2018;29(1):14-18.
43. US Food and Drug Administration. Summary of Safety and Effectiveness Data. Ophthalmic sealant: ReSure Sealant. https://www.accessdata.fda.gov/cdrh_docs/pdf13/P130004b.pdf. Published September 13, 2013. Accessed July 9, 2019.
44. About ReSure sealant. https://www.resuresealant.com/overview. Accessed July 31, 2019.
45. Menabuoni L, Pini R, Rossi F, Lenzetti I, Yoo SH, Parel JM. Laser-assisted corneal welding in cataract surgery: retrospective study. J Cataract Refract Surg. 2007;33(9):1608-1612.
46. Rasier R, Ozeren M, Artunay O, et al. Corneal tissue welding with infrared laser irradiation after clear corneal incision. Cornea. 2010;29(9):985-990.
47. Rossi F, Matteini P, Ratto F, Menabuoni L, Lenzetti I, Pini R. Laser tissue welding in ophthalmic surgery. J Biophotonics. 2008;1(4):331-342.
48. Taban M, Behrens A, Newcomb RL, et al. Acute endophthalmitis following cataract surgery: a systematic review of the literature. Arch Ophthalmol. 2005;123(5):613-620.
49. Taylor DM, Atlas BF, Romanchuk KG, Stern AL. Pseudophakic bullous keratopathy. Ophthalmology. 1983;90(1):19-24.
50. Lobo CL, Faria PM, Soares MA, Bernardes RC, Cunha-Vaz JG. Macular alterations after small-incision cataract surgery. J Cataract Refract Surg. 2004;30(4):752-760.
51. Flach AJ. The incidence, pathogenesis and treatment of cystoid macular edema following cataract surgery. Trans Am Ophthalmol Soc. 1998;96:557-634.
52. Wright PL, Wilkinson CP, Balyeat HD, Popham J, Reinke M. Angiographic cystoid macular edema after posterior chamber lens implantation. Arch Ophthalmol. 1988;106(6):740-744.
53. Kim SJ, Belair ML, Bressler NM, et al. A method of reporting macular edema after cataract surgery using optical coherence tomography. Retina. 2008;28(6):870-876.
54. Alio JL, Ruiz-Moreno JM, Shabayek MH, Lugo FL, Abd El Rahman AM. The risk of retinal detachment in high myopia after small incision coaxial phacoemulsification. Am J Ophthalmol. 2007;144(1):93-98.
55. Bhagwandien AC, Cheng YY, Wolfs RC, van Meurs JC, Luyten GP. Relationship between retinal detachment and biometry in 4262 cataractous eyes. Ophthalmology. 2006;113(4):643-649.
56. Boberg-Ans G, Henning V, Villumsen J, la Cour M. Longterm incidence of rhegmatogenous retinal detachment and survival in a defined population undergoing standardized phacoemulsification surgery. Acta Ophthalmol Scand. 2006;84(5):613-618.
57. Jakobsson G, Montan P, Zetterberg M, Stenevi U, Behndig A, Lundström M. Capsule complication during cataract surgery: retinal detachment after cataract surgery with capsule complication: Swedish Capsule Rupture Study Group report 4. J Cataract Refract Surg. 2009;35(10):1699-1705.
58. Neuhann IM, Neuhann TF, Heimann H, Schmickler S, Gerl RH, Foerster MH. Retinal detachment after phacoemulsification in high myopia: analysis of 2356 cases. J Cataract Refract Surg. 2008;34(10):1644-1657.
59. Russell M, Gaskin B, Russell D, Polkinghorne PJ. Pseudophakic retinal detachment after phacoemulsification cataract surgery: ten-year retrospective review. J Cataract Refract Surg. 2006;32(3):442-445.
60. Apple DJ, Solomon KD, Tetz MR, et al. Posterior capsule opacification. Surv Ophthalmol. 1992;37(2):73-116.
61. Wu S, Tong N, Pan L, et al. Retrospective analyses of potential risk factors for posterior capsule opacification after cataract surgery. J Ophthalmol. 2018;2018:9089285.
62. Clark A, Morlet N, Ng JQ, Preen DB, Semmens JB. Whole population trends in complications of cataract surgery over 22 years in Western Australia. Ophthalmology. 2011;118(6):1055-1061.
63. Adhikari S, Shrestha UD. Pediatric cataract surgery with hydrophilic acrylic intraocular lens implantation in Nepalese Children. Clin Ophthalmol. 2017;12:7-11.
64. Lee BJ, Smith SD, Jeng BH. Suture-related corneal infections after clear corneal cataract surgery. J Cataract Refract Surg. 2009;35(5):939-942.
65. May WN, Castro-Combs J, Kashiwabuchi RT, et al. Sutured clear corneal incision: wound apposition and permeability to bacterial-sized particles. Cornea. 2013;32(3):319-325.
66. Hillier RJ, Ajit RR, Kelly SP. Suture-related complications after cataract surgery: a patient safety issue. J Cataract Refract Surg. 2009;35(11):2035-2036.
67. Hovanesian JA, Karageozian VH. Watertight cataract incision closure using fibrin tissue adhesive. J Cataract Refract Surg. 2007;33(8):1461-1463.
USPSTF reaffirms recommendation against pancreatic cancer screening in asymptomatic adults
The U.S. Preventive Services Task Force has issued a recommendation statement reaffirming its 2004 guideline, which recommended against screening for pancreatic cancer in asymptomatic adults, according to task force member Douglas K. Owens, MD, of the Veterans Affairs Palo Alto (Calif.) Health Care System and associates.
Pancreatic cancer is uncommon, with an age-adjusted annual incidence of 12.9 cases per 100,000 person-years; however, pancreatic cancer is the third most common cause of cancer death because mortality is high. The mortality rate is 11.0 deaths per 100,000 person-years, and an estimated 45,750 people will die from the disease in 2019.
In 2004, the USPSTF issued a D recommendation for pancreatic cancer screening in asymptomatic adults without a family history of pancreatic cancer or a genetic disorder that increases the risk of cancer. For the 2019 update, the task force conducted a systematic review of 13 studies that assessed the benefits and harms of screening for pancreatic cancer, the diagnostic accuracy of screening tests for pancreatic cancer, and the benefits and harms of treating screen-detected or asymptomatic pancreatic cancer.
According to the USPSTF, the studies included in the review found no evidence that screening for pancreatic cancer or treatment of screen-detected pancreatic cancer improves morbidity or mortality, found adequate evidence that the magnitude of the benefits of screening for pancreatic cancer in asymptomatic adults can be bounded as no greater than small, and found adequate evidence that the magnitude of the harms of screening for pancreatic cancer and treatment of screen-detected pancreatic cancer can be bounded as at least moderate.
Because no new evidence was found supporting pancreatic cancer screening in asymptomatic adults, “the USPSTF reaffirms its previous conclusion that the potential benefits of screening for pancreatic cancer in asymptomatic adults do not outweigh the potential harms,” the task force members noted.
The task force authors reported no disclosures related to the recommendation statement.
SOURCE: Owens DK et al. JAMA. 2019 Aug 6. doi: 10.1001/jama.2019.10232.
The U.S. Preventive Services Task Force has issued a recommendation statement reaffirming its 2004 guideline, which recommended against screening for pancreatic cancer in asymptomatic adults, according to task force member Douglas K. Owens, MD, of the Veterans Affairs Palo Alto (Calif.) Health Care System and associates.
Pancreatic cancer is uncommon, with an age-adjusted annual incidence of 12.9 cases per 100,000 person-years; however, pancreatic cancer is the third most common cause of cancer death because mortality is high. The mortality rate is 11.0 deaths per 100,000 person-years, and an estimated 45,750 people will die from the disease in 2019.
In 2004, the USPSTF issued a D recommendation for pancreatic cancer screening in asymptomatic adults without a family history of pancreatic cancer or a genetic disorder that increases the risk of cancer. For the 2019 update, the task force conducted a systematic review of 13 studies that assessed the benefits and harms of screening for pancreatic cancer, the diagnostic accuracy of screening tests for pancreatic cancer, and the benefits and harms of treating screen-detected or asymptomatic pancreatic cancer.
According to the USPSTF, the studies included in the review found no evidence that screening for pancreatic cancer or treatment of screen-detected pancreatic cancer improves morbidity or mortality, found adequate evidence that the magnitude of the benefits of screening for pancreatic cancer in asymptomatic adults can be bounded as no greater than small, and found adequate evidence that the magnitude of the harms of screening for pancreatic cancer and treatment of screen-detected pancreatic cancer can be bounded as at least moderate.
Because no new evidence was found supporting pancreatic cancer screening in asymptomatic adults, “the USPSTF reaffirms its previous conclusion that the potential benefits of screening for pancreatic cancer in asymptomatic adults do not outweigh the potential harms,” the task force members noted.
The task force authors reported no disclosures related to the recommendation statement.
SOURCE: Owens DK et al. JAMA. 2019 Aug 6. doi: 10.1001/jama.2019.10232.
The U.S. Preventive Services Task Force has issued a recommendation statement reaffirming its 2004 guideline, which recommended against screening for pancreatic cancer in asymptomatic adults, according to task force member Douglas K. Owens, MD, of the Veterans Affairs Palo Alto (Calif.) Health Care System and associates.
Pancreatic cancer is uncommon, with an age-adjusted annual incidence of 12.9 cases per 100,000 person-years; however, pancreatic cancer is the third most common cause of cancer death because mortality is high. The mortality rate is 11.0 deaths per 100,000 person-years, and an estimated 45,750 people will die from the disease in 2019.
In 2004, the USPSTF issued a D recommendation for pancreatic cancer screening in asymptomatic adults without a family history of pancreatic cancer or a genetic disorder that increases the risk of cancer. For the 2019 update, the task force conducted a systematic review of 13 studies that assessed the benefits and harms of screening for pancreatic cancer, the diagnostic accuracy of screening tests for pancreatic cancer, and the benefits and harms of treating screen-detected or asymptomatic pancreatic cancer.
According to the USPSTF, the studies included in the review found no evidence that screening for pancreatic cancer or treatment of screen-detected pancreatic cancer improves morbidity or mortality, found adequate evidence that the magnitude of the benefits of screening for pancreatic cancer in asymptomatic adults can be bounded as no greater than small, and found adequate evidence that the magnitude of the harms of screening for pancreatic cancer and treatment of screen-detected pancreatic cancer can be bounded as at least moderate.
Because no new evidence was found supporting pancreatic cancer screening in asymptomatic adults, “the USPSTF reaffirms its previous conclusion that the potential benefits of screening for pancreatic cancer in asymptomatic adults do not outweigh the potential harms,” the task force members noted.
The task force authors reported no disclosures related to the recommendation statement.
SOURCE: Owens DK et al. JAMA. 2019 Aug 6. doi: 10.1001/jama.2019.10232.
FROM JAMA
Early pregnancy loss and abortion: Medical management is safe, effective
NASHVILLE, TENN. – Medical management of abortion and early pregnancy loss is best achieved with both mifepristone and misoprostol, according to Sarah W. Prager, MD.

First-trimester procedures account for about 90% of elective abortions, with about two-thirds of those occurring before 8 weeks of gestation and 80% occurring in the first 10 weeks – and therefore considered eligible for medical management, Dr. Prager, director of the Family Planning Division and Family Planning Fellowship at the University of Washington, Seattle, said at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists.
“We estimate that it’s approximately 31% of all abortions that are done using medication, but it’s about 45% of those eligible by gestational age,” she noted.
The alternative is uterine aspiration, and in the absence of a clear contraindication, patient preference should determine management choice, she said.
The same is true for early pregnancy loss (spontaneous abortion), which is the most common complication of early pregnancy, occurring in about 20% of clinically recognized pregnancies.
“That means that there are about 1 million spontaneous abortions happening annually in the United States, and about 80% of those are in the first trimester,” Dr. Prager said.
Expectant management is an additional option for managing early pregnancy loss, she noted.
Candidates
Medical management is appropriate for patients who are undergoing elective abortion at up to about 70 days of gestation or with pregnancy loss in the first trimester.
“They should have stable vital signs, no evidence of infection, no allergies to the medications being used, no serious social or medical problems,” Dr. Prager said, explaining that “a shared decision making process” is important for patients with extreme anxiety or homelessness/lack of stable housing, for example, in order to make sure that medical management is a good option.
“While she definitely gets to have the final say, unless there is a real medical contraindication, it definitely should be part of that decision making,” Dr. Prager said, adding that adequate counseling and acceptance by the patient of the risks and side effects also are imperative.
Protocol
The most effective evidence-based treatment protocol for elective abortion through day 70 of gestation includes a 200-mg oral dose of mifepristone, followed 24-72 hours later with at-home buccal or vaginal administration of an 800-mcg dose of misoprostol, with follow up within 1-2 weeks, Dr. Prager said, citing a 2010 Cochrane review.
The Food and Drug Administration–approved protocol, which was updated in April 2016, adheres closely to those findings, except that it calls for misoprostol within 48 hours of mifepristone dosing. Optional repeat dosing of misoprostol is allowed, as well, she noted.
Buccal or vaginal administration of misoprostol is preferable to oral and sublingual administration because while the latter approaches provide more rapid onset, the former approaches provide significantly better sustained action over a 5-hour period of time.
“And by not having that big peak at the beginning, it actually decreases the side effects that women experience with the misoprostol medication,” she said.
Misoprostol can also be given alone for early pregnancy loss management – also at a dose of 800 mcg buccally or vaginally – with repeat dosing at 12-24 hours for incomplete abortion. However, new data suggest that, before about 63 days of gestation, giving two doses 3 hours apart is slightly more effective. That approach can also be repeated if necessary, Dr. Prager said.
Pain management is an important part of treatment, as both miscarriage and medication abortion can range from uncomfortable to extremely painful, depending on the patient, her prior obstetric experience, and her life experiences.
“I recommend talking to all your patients about pain management. For most people, just using some type of NSAID is probably going to be sufficient,” she said, noting that some women will require a narcotic.
Antiemetic medication may also be necessary, as some women will experience nausea and vomiting.
Complications and intervention
Major complications are rare with medical management of first-trimester abortion and early pregnancy loss, but can include ongoing pregnancy, which is infrequent but possible; incomplete abortion, which is easily managed; and allergic reactions, which are “extremely rare,” Dr. Prager said.
Hemorrhage can occur, but isn’t common and usually is at a level that doesn’t require blood transfusion. “But it does require somebody to come in, potentially needing uterine aspiration or sometimes just a second dose of misoprostol,” she said.
Serious infections are “extraordinarily uncommon,” with an actual risk of infectious death of 0.5 per 100,000, and therefore antibiotic prophylaxis is not recommended.
“This is not to say that there can’t be serious infectious problems with medication abortion, and actually also with spontaneous abortion ... but it’s extremely rare,” Dr. Prager said, adding that “there are also consequences to giving everybody antibiotics if they are not necessary. I, personally, am way more afraid of antibiotic resistance these days than I am about preventing an infection from an medication abortion.”
Intervention is necessary in certain situations, including when the gestational sac remains and when the patient continues to have clinical symptoms or has developed clinical symptoms, she said.
“Does she now show signs of infection? Is she bleeding very heavily or [is she] extremely uncomfortable with cramping? Those are all really great reasons to intervene,” she said.
Sometimes patients just prefer to switch to an alternative method of management, particularly in cases of early pregnancy loss when medical management has “not been successful after some period of time,” Dr. Prager added.
Outcomes
Studies have shown that the success rates with a single dose of 400-800 mcg of misoprostol range from 25% to 88%, and with repeat dosing for incomplete abortion at 24 hours, the success rate improves to between 80% and 88%. The success rate with placebo is 16%-60%; this indicates that “some miscarriages just happen expectantly,” Dr. Prager explained.
“We already knew that ... and that’s why expectant management is an option with early pregnancy loss,” she said, adding that expectant management works about 50% of the time – “if you wait long enough.”
However, success rates with medical management depend on the type of miscarriage; the rate is close to 100% with incomplete abortion, but for other types, such as anembryonic pregnancy or fetal demise, it is slightly less effective at about 87%, Dr. Prager noted.
When mifepristone and misoprostol are both used, success rates for early pregnancy loss range from 52% to 84% in observational trials and using nonstandard doses, and between 90% and 93% with standard dosing.
Other recent data, which led to a 2016 “reaffirmation” of an ACOG practice bulletin on medical management of first-trimester abortion, show an 83% success rate with the combination therapy in anembryonic pregnancies, and a 25% reduction in the need for further intervention (N Engl J Med. 2018;378:2161-70).
“So it really was significantly more effective to be using that addition of the mifepristone,” she said. “My take-home message about this is that, if mifepristone is something that you have easily available to you at your clinical site, absolutely use it, because it creates better outcomes for your patients. However, if it’s not available to you ... it is still perfectly reasonable for patients to choose medication management of their early pregnancy loss and use misoprostol only.
“It is effective enough, and that is just part of your informed consent.”
Postabortion care
Postmiscarriage care is important and involves several components, Dr. Prager said.
- RhoGAM treatment. The use of RhoGAM to prevent Rh immunization has been routine, but data increasingly suggest this is not necessary, and in some countries it is not given at all, particularly at 8 or fewer weeks of gestation and sometimes even during the whole first trimester for early pregnancy loss. “That is not common practice yet in the United States; I’m not recommending at this time that everybody change their practice ... but I will say that there are some really interesting studies going on right now in the United States that are looking specifically at this, and I think we may, within the next 10 years or so, change this practice of giving RhoGAM at all gestational ages,” she said.
- Counseling about bleeding. Light to moderate bleeding after abortion is common for about 2 weeks after abortion, with normal menses returning between 4 and 8 weeks, and typically around 6 weeks. “I usually ask patients to come back and see me if they have not had what seems to be a normal period to them 8 weeks following their completed process,” Dr. Prager said.
- Counseling about human chorionic gonadotropin levels. It is also helpful to inform patients that human chorionic gonadotropin may remain present for about 2-4 weeks after completed abortion, resulting in a positive pregnancy test during that time. A positive test at 4 weeks may still be normal, but warrants evaluation to determine why the patient is testing positive.
- Counseling about conception timing. Data do not support delaying repeat pregnancy after abortion. Studies show no difference in the ability to conceive or in pregnancy outcomes among women who conceive without delay after early pregnancy loss and in those who wait at least 3 months. “So what I now tell women is ‘when you’re emotionally ready to start trying to get pregnant again, it’s perfectly medically acceptable to do so. There’s no biologic reason why you have to wait,’ ” she said.
- Contraception initiation. Contraception, including IUDs, can be initiated right away after elective or spontaneous abortion. However, for IUD insertion after medical abortion, it is important to first use ultrasound to confirm complete abortion, Dr. Prager said.
- Grief counseling. This may be appropriate in cases of early pregnancy loss and for elective abortions. “Both groups of people may need some counseling, may be experiencing grief around this process – and they may not be,” she said. “I think we just need to be sensitive about asking our patients what their needs might be around this.”
Future directions
The future of medical management for first trimester abortion may involve “demedicalization,” Dr. Prager said.
“There are many papers coming out now about clinic versus home use of mifepristone,” she said, explaining that home use would require removing the FDA’s Risk Evaluation and Mitigation Strategy restriction that requires that the drug be dispensed in a clinic by a physician or physician extender.
Studies are also looking at prescriptions, pharmacist provision of mifepristone, and mailing of medications to women in rural areas.
Another area of research beyond these “really creative ways of using these medications” is whether medical management is effective beyond 10 weeks. A study that will soon be published is looking at mifepristone and two doses of misoprostol at 11 weeks, she noted.
“I think from pregnancy diagnosis through at least week 10 – soon we will see potentially week 11 – medical abortion techniques are safe, they’re effective, and they’re extremely well accepted by patients,” she said. “Also ... a diverse group of clinicians can be trained to offer medical abortion and provide back-up so that access can be improved.”
Dr. Prager reported having no financial disclosures.
NASHVILLE, TENN. – Medical management of abortion and early pregnancy loss is best achieved with both mifepristone and misoprostol, according to Sarah W. Prager, MD.
First-trimester procedures account for about 90% of elective abortions, with about two-thirds of those occurring before 8 weeks of gestation and 80% occurring in the first 10 weeks – and therefore considered eligible for medical management, Dr. Prager, director of the Family Planning Division and Family Planning Fellowship at the University of Washington, Seattle, said at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists.
“We estimate that it’s approximately 31% of all abortions that are done using medication, but it’s about 45% of those eligible by gestational age,” she noted.
The alternative is uterine aspiration, and in the absence of a clear contraindication, patient preference should determine management choice, she said.
The same is true for early pregnancy loss (spontaneous abortion), which is the most common complication of early pregnancy, occurring in about 20% of clinically recognized pregnancies.
“That means that there are about 1 million spontaneous abortions happening annually in the United States, and about 80% of those are in the first trimester,” Dr. Prager said.
Expectant management is an additional option for managing early pregnancy loss, she noted.
Candidates
Medical management is appropriate for patients who are undergoing elective abortion at up to about 70 days of gestation or with pregnancy loss in the first trimester.
“They should have stable vital signs, no evidence of infection, no allergies to the medications being used, no serious social or medical problems,” Dr. Prager said, explaining that “a shared decision making process” is important for patients with extreme anxiety or homelessness/lack of stable housing, for example, in order to make sure that medical management is a good option.
“While she definitely gets to have the final say, unless there is a real medical contraindication, it definitely should be part of that decision making,” Dr. Prager said, adding that adequate counseling and acceptance by the patient of the risks and side effects also are imperative.
Protocol
The most effective evidence-based treatment protocol for elective abortion through day 70 of gestation includes a 200-mg oral dose of mifepristone, followed 24-72 hours later with at-home buccal or vaginal administration of an 800-mcg dose of misoprostol, with follow up within 1-2 weeks, Dr. Prager said, citing a 2010 Cochrane review.
The Food and Drug Administration–approved protocol, which was updated in April 2016, adheres closely to those findings, except that it calls for misoprostol within 48 hours of mifepristone dosing. Optional repeat dosing of misoprostol is allowed, as well, she noted.
Buccal or vaginal administration of misoprostol is preferable to oral and sublingual administration because while the latter approaches provide more rapid onset, the former approaches provide significantly better sustained action over a 5-hour period of time.
“And by not having that big peak at the beginning, it actually decreases the side effects that women experience with the misoprostol medication,” she said.
Misoprostol can also be given alone for early pregnancy loss management – also at a dose of 800 mcg buccally or vaginally – with repeat dosing at 12-24 hours for incomplete abortion. However, new data suggest that, before about 63 days of gestation, giving two doses 3 hours apart is slightly more effective. That approach can also be repeated if necessary, Dr. Prager said.
Pain management is an important part of treatment, as both miscarriage and medication abortion can range from uncomfortable to extremely painful, depending on the patient, her prior obstetric experience, and her life experiences.
“I recommend talking to all your patients about pain management. For most people, just using some type of NSAID is probably going to be sufficient,” she said, noting that some women will require a narcotic.
Antiemetic medication may also be necessary, as some women will experience nausea and vomiting.
Complications and intervention
Major complications are rare with medical management of first-trimester abortion and early pregnancy loss, but can include ongoing pregnancy, which is infrequent but possible; incomplete abortion, which is easily managed; and allergic reactions, which are “extremely rare,” Dr. Prager said.
Hemorrhage can occur, but isn’t common and usually is at a level that doesn’t require blood transfusion. “But it does require somebody to come in, potentially needing uterine aspiration or sometimes just a second dose of misoprostol,” she said.
Serious infections are “extraordinarily uncommon,” with an actual risk of infectious death of 0.5 per 100,000, and therefore antibiotic prophylaxis is not recommended.
“This is not to say that there can’t be serious infectious problems with medication abortion, and actually also with spontaneous abortion ... but it’s extremely rare,” Dr. Prager said, adding that “there are also consequences to giving everybody antibiotics if they are not necessary. I, personally, am way more afraid of antibiotic resistance these days than I am about preventing an infection from an medication abortion.”
Intervention is necessary in certain situations, including when the gestational sac remains and when the patient continues to have clinical symptoms or has developed clinical symptoms, she said.
“Does she now show signs of infection? Is she bleeding very heavily or [is she] extremely uncomfortable with cramping? Those are all really great reasons to intervene,” she said.
Sometimes patients just prefer to switch to an alternative method of management, particularly in cases of early pregnancy loss when medical management has “not been successful after some period of time,” Dr. Prager added.
Outcomes
Studies have shown that the success rates with a single dose of 400-800 mcg of misoprostol range from 25% to 88%, and with repeat dosing for incomplete abortion at 24 hours, the success rate improves to between 80% and 88%. The success rate with placebo is 16%-60%; this indicates that “some miscarriages just happen expectantly,” Dr. Prager explained.
“We already knew that ... and that’s why expectant management is an option with early pregnancy loss,” she said, adding that expectant management works about 50% of the time – “if you wait long enough.”
However, success rates with medical management depend on the type of miscarriage; the rate is close to 100% with incomplete abortion, but for other types, such as anembryonic pregnancy or fetal demise, it is slightly less effective at about 87%, Dr. Prager noted.
When mifepristone and misoprostol are both used, success rates for early pregnancy loss range from 52% to 84% in observational trials and using nonstandard doses, and between 90% and 93% with standard dosing.
Other recent data, which led to a 2016 “reaffirmation” of an ACOG practice bulletin on medical management of first-trimester abortion, show an 83% success rate with the combination therapy in anembryonic pregnancies, and a 25% reduction in the need for further intervention (N Engl J Med. 2018;378:2161-70).
“So it really was significantly more effective to be using that addition of the mifepristone,” she said. “My take-home message about this is that, if mifepristone is something that you have easily available to you at your clinical site, absolutely use it, because it creates better outcomes for your patients. However, if it’s not available to you ... it is still perfectly reasonable for patients to choose medication management of their early pregnancy loss and use misoprostol only.
“It is effective enough, and that is just part of your informed consent.”
Postabortion care
Postmiscarriage care is important and involves several components, Dr. Prager said.
- RhoGAM treatment. The use of RhoGAM to prevent Rh immunization has been routine, but data increasingly suggest this is not necessary, and in some countries it is not given at all, particularly at 8 or fewer weeks of gestation and sometimes even during the whole first trimester for early pregnancy loss. “That is not common practice yet in the United States; I’m not recommending at this time that everybody change their practice ... but I will say that there are some really interesting studies going on right now in the United States that are looking specifically at this, and I think we may, within the next 10 years or so, change this practice of giving RhoGAM at all gestational ages,” she said.
- Counseling about bleeding. Light to moderate bleeding after abortion is common for about 2 weeks after abortion, with normal menses returning between 4 and 8 weeks, and typically around 6 weeks. “I usually ask patients to come back and see me if they have not had what seems to be a normal period to them 8 weeks following their completed process,” Dr. Prager said.
- Counseling about human chorionic gonadotropin levels. It is also helpful to inform patients that human chorionic gonadotropin may remain present for about 2-4 weeks after completed abortion, resulting in a positive pregnancy test during that time. A positive test at 4 weeks may still be normal, but warrants evaluation to determine why the patient is testing positive.
- Counseling about conception timing. Data do not support delaying repeat pregnancy after abortion. Studies show no difference in the ability to conceive or in pregnancy outcomes among women who conceive without delay after early pregnancy loss and in those who wait at least 3 months. “So what I now tell women is ‘when you’re emotionally ready to start trying to get pregnant again, it’s perfectly medically acceptable to do so. There’s no biologic reason why you have to wait,’ ” she said.
- Contraception initiation. Contraception, including IUDs, can be initiated right away after elective or spontaneous abortion. However, for IUD insertion after medical abortion, it is important to first use ultrasound to confirm complete abortion, Dr. Prager said.
- Grief counseling. This may be appropriate in cases of early pregnancy loss and for elective abortions. “Both groups of people may need some counseling, may be experiencing grief around this process – and they may not be,” she said. “I think we just need to be sensitive about asking our patients what their needs might be around this.”
Future directions
The future of medical management for first trimester abortion may involve “demedicalization,” Dr. Prager said.
“There are many papers coming out now about clinic versus home use of mifepristone,” she said, explaining that home use would require removing the FDA’s Risk Evaluation and Mitigation Strategy restriction that requires that the drug be dispensed in a clinic by a physician or physician extender.
Studies are also looking at prescriptions, pharmacist provision of mifepristone, and mailing of medications to women in rural areas.
Another area of research beyond these “really creative ways of using these medications” is whether medical management is effective beyond 10 weeks. A study that will soon be published is looking at mifepristone and two doses of misoprostol at 11 weeks, she noted.
“I think from pregnancy diagnosis through at least week 10 – soon we will see potentially week 11 – medical abortion techniques are safe, they’re effective, and they’re extremely well accepted by patients,” she said. “Also ... a diverse group of clinicians can be trained to offer medical abortion and provide back-up so that access can be improved.”
Dr. Prager reported having no financial disclosures.
NASHVILLE, TENN. – Medical management of abortion and early pregnancy loss is best achieved with both mifepristone and misoprostol, according to Sarah W. Prager, MD.
First-trimester procedures account for about 90% of elective abortions, with about two-thirds of those occurring before 8 weeks of gestation and 80% occurring in the first 10 weeks – and therefore considered eligible for medical management, Dr. Prager, director of the Family Planning Division and Family Planning Fellowship at the University of Washington, Seattle, said at the annual clinical and scientific meeting of the American College of Obstetricians and Gynecologists.
“We estimate that it’s approximately 31% of all abortions that are done using medication, but it’s about 45% of those eligible by gestational age,” she noted.
The alternative is uterine aspiration, and in the absence of a clear contraindication, patient preference should determine management choice, she said.
The same is true for early pregnancy loss (spontaneous abortion), which is the most common complication of early pregnancy, occurring in about 20% of clinically recognized pregnancies.
“That means that there are about 1 million spontaneous abortions happening annually in the United States, and about 80% of those are in the first trimester,” Dr. Prager said.
Expectant management is an additional option for managing early pregnancy loss, she noted.
Candidates
Medical management is appropriate for patients who are undergoing elective abortion at up to about 70 days of gestation or with pregnancy loss in the first trimester.
“They should have stable vital signs, no evidence of infection, no allergies to the medications being used, no serious social or medical problems,” Dr. Prager said, explaining that “a shared decision making process” is important for patients with extreme anxiety or homelessness/lack of stable housing, for example, in order to make sure that medical management is a good option.
“While she definitely gets to have the final say, unless there is a real medical contraindication, it definitely should be part of that decision making,” Dr. Prager said, adding that adequate counseling and acceptance by the patient of the risks and side effects also are imperative.
Protocol
The most effective evidence-based treatment protocol for elective abortion through day 70 of gestation includes a 200-mg oral dose of mifepristone, followed 24-72 hours later with at-home buccal or vaginal administration of an 800-mcg dose of misoprostol, with follow up within 1-2 weeks, Dr. Prager said, citing a 2010 Cochrane review.
The Food and Drug Administration–approved protocol, which was updated in April 2016, adheres closely to those findings, except that it calls for misoprostol within 48 hours of mifepristone dosing. Optional repeat dosing of misoprostol is allowed, as well, she noted.
Buccal or vaginal administration of misoprostol is preferable to oral and sublingual administration because while the latter approaches provide more rapid onset, the former approaches provide significantly better sustained action over a 5-hour period of time.
“And by not having that big peak at the beginning, it actually decreases the side effects that women experience with the misoprostol medication,” she said.
Misoprostol can also be given alone for early pregnancy loss management – also at a dose of 800 mcg buccally or vaginally – with repeat dosing at 12-24 hours for incomplete abortion. However, new data suggest that, before about 63 days of gestation, giving two doses 3 hours apart is slightly more effective. That approach can also be repeated if necessary, Dr. Prager said.
Pain management is an important part of treatment, as both miscarriage and medication abortion can range from uncomfortable to extremely painful, depending on the patient, her prior obstetric experience, and her life experiences.
“I recommend talking to all your patients about pain management. For most people, just using some type of NSAID is probably going to be sufficient,” she said, noting that some women will require a narcotic.
Antiemetic medication may also be necessary, as some women will experience nausea and vomiting.
Complications and intervention
Major complications are rare with medical management of first-trimester abortion and early pregnancy loss, but can include ongoing pregnancy, which is infrequent but possible; incomplete abortion, which is easily managed; and allergic reactions, which are “extremely rare,” Dr. Prager said.
Hemorrhage can occur, but isn’t common and usually is at a level that doesn’t require blood transfusion. “But it does require somebody to come in, potentially needing uterine aspiration or sometimes just a second dose of misoprostol,” she said.
Serious infections are “extraordinarily uncommon,” with an actual risk of infectious death of 0.5 per 100,000, and therefore antibiotic prophylaxis is not recommended.
“This is not to say that there can’t be serious infectious problems with medication abortion, and actually also with spontaneous abortion ... but it’s extremely rare,” Dr. Prager said, adding that “there are also consequences to giving everybody antibiotics if they are not necessary. I, personally, am way more afraid of antibiotic resistance these days than I am about preventing an infection from an medication abortion.”
Intervention is necessary in certain situations, including when the gestational sac remains and when the patient continues to have clinical symptoms or has developed clinical symptoms, she said.
“Does she now show signs of infection? Is she bleeding very heavily or [is she] extremely uncomfortable with cramping? Those are all really great reasons to intervene,” she said.
Sometimes patients just prefer to switch to an alternative method of management, particularly in cases of early pregnancy loss when medical management has “not been successful after some period of time,” Dr. Prager added.
Outcomes
Studies have shown that the success rates with a single dose of 400-800 mcg of misoprostol range from 25% to 88%, and with repeat dosing for incomplete abortion at 24 hours, the success rate improves to between 80% and 88%. The success rate with placebo is 16%-60%; this indicates that “some miscarriages just happen expectantly,” Dr. Prager explained.
“We already knew that ... and that’s why expectant management is an option with early pregnancy loss,” she said, adding that expectant management works about 50% of the time – “if you wait long enough.”
However, success rates with medical management depend on the type of miscarriage; the rate is close to 100% with incomplete abortion, but for other types, such as anembryonic pregnancy or fetal demise, it is slightly less effective at about 87%, Dr. Prager noted.
When mifepristone and misoprostol are both used, success rates for early pregnancy loss range from 52% to 84% in observational trials and using nonstandard doses, and between 90% and 93% with standard dosing.
Other recent data, which led to a 2016 “reaffirmation” of an ACOG practice bulletin on medical management of first-trimester abortion, show an 83% success rate with the combination therapy in anembryonic pregnancies, and a 25% reduction in the need for further intervention (N Engl J Med. 2018;378:2161-70).
“So it really was significantly more effective to be using that addition of the mifepristone,” she said. “My take-home message about this is that, if mifepristone is something that you have easily available to you at your clinical site, absolutely use it, because it creates better outcomes for your patients. However, if it’s not available to you ... it is still perfectly reasonable for patients to choose medication management of their early pregnancy loss and use misoprostol only.
“It is effective enough, and that is just part of your informed consent.”
Postabortion care
Postmiscarriage care is important and involves several components, Dr. Prager said.
- RhoGAM treatment. The use of RhoGAM to prevent Rh immunization has been routine, but data increasingly suggest this is not necessary, and in some countries it is not given at all, particularly at 8 or fewer weeks of gestation and sometimes even during the whole first trimester for early pregnancy loss. “That is not common practice yet in the United States; I’m not recommending at this time that everybody change their practice ... but I will say that there are some really interesting studies going on right now in the United States that are looking specifically at this, and I think we may, within the next 10 years or so, change this practice of giving RhoGAM at all gestational ages,” she said.
- Counseling about bleeding. Light to moderate bleeding after abortion is common for about 2 weeks after abortion, with normal menses returning between 4 and 8 weeks, and typically around 6 weeks. “I usually ask patients to come back and see me if they have not had what seems to be a normal period to them 8 weeks following their completed process,” Dr. Prager said.
- Counseling about human chorionic gonadotropin levels. It is also helpful to inform patients that human chorionic gonadotropin may remain present for about 2-4 weeks after completed abortion, resulting in a positive pregnancy test during that time. A positive test at 4 weeks may still be normal, but warrants evaluation to determine why the patient is testing positive.
- Counseling about conception timing. Data do not support delaying repeat pregnancy after abortion. Studies show no difference in the ability to conceive or in pregnancy outcomes among women who conceive without delay after early pregnancy loss and in those who wait at least 3 months. “So what I now tell women is ‘when you’re emotionally ready to start trying to get pregnant again, it’s perfectly medically acceptable to do so. There’s no biologic reason why you have to wait,’ ” she said.
- Contraception initiation. Contraception, including IUDs, can be initiated right away after elective or spontaneous abortion. However, for IUD insertion after medical abortion, it is important to first use ultrasound to confirm complete abortion, Dr. Prager said.
- Grief counseling. This may be appropriate in cases of early pregnancy loss and for elective abortions. “Both groups of people may need some counseling, may be experiencing grief around this process – and they may not be,” she said. “I think we just need to be sensitive about asking our patients what their needs might be around this.”
Future directions
The future of medical management for first trimester abortion may involve “demedicalization,” Dr. Prager said.
“There are many papers coming out now about clinic versus home use of mifepristone,” she said, explaining that home use would require removing the FDA’s Risk Evaluation and Mitigation Strategy restriction that requires that the drug be dispensed in a clinic by a physician or physician extender.
Studies are also looking at prescriptions, pharmacist provision of mifepristone, and mailing of medications to women in rural areas.
Another area of research beyond these “really creative ways of using these medications” is whether medical management is effective beyond 10 weeks. A study that will soon be published is looking at mifepristone and two doses of misoprostol at 11 weeks, she noted.
“I think from pregnancy diagnosis through at least week 10 – soon we will see potentially week 11 – medical abortion techniques are safe, they’re effective, and they’re extremely well accepted by patients,” she said. “Also ... a diverse group of clinicians can be trained to offer medical abortion and provide back-up so that access can be improved.”
Dr. Prager reported having no financial disclosures.
EXPERT ANALYSIS FROM ACOG 2019
Dr. Carl Bell always asked the hard questions
Carl C. Bell, MD, started his career by asking the hard questions that no one dared to ask. His curiosity, courage, and compassion for all communities would lead him to impact the world in ways that few psychiatrists could ever imagine.

His accomplishments were many and far-reaching, dating back over 4 decades of service and research. He was a prolific author and researcher, having written more than 400 books, chapters, and articles. His research covered a lot of ground, but four critical areas of focus were childhood trauma, violence prevention, criminal justice reform, and most recently, fetal alcohol spectrum disorders.
The word “visionary” is frequently overused. But when we apply it to Dr. Carl Bell, the word does not do him justice. If you take a closer look at all four of those areas, you can see a common thread: What are the elements of a society that can tear communities apart?
When I look at Dr. Bell’s research, I see a man with a dedicated vision to addressing each of those elements in systematic way, and a determination to bring the results of that research into his Southside Chicago community in numerous ways, including by serving as president and CEO of the Community Mental Health Council, and as director of the Institute for Juvenile Research at the University of Illinois at Chicago.
Dr. Bell was a leader for his patients as well as for black psychiatrists. He was a founding member of Black Psychiatrists of America and served as a mentor in some way to the vast majority of black psychiatrists currently practicing in the country. As a black male psychiatrist, I saw Dr. Bell as a source of inspiration in my career and the standard by which I measured myself. I’m not talking about awards or accomplishments, as Dr. Bell has countless accolades, including most recently, being presented this year with the American Psychiatric Association’s Adolph Meyer Award for Lifetime Achievement in Psychiatric Research and the National Medical Association’s Scroll of Merit. I am referring to Dr. Bell’s willingness to walk away from something he thought was wrong.
For every accolade he won or prestigious committee he served on, I would wager that he declined or stepped way from just as many. Dr. Bell’s character and vision for psychiatry in general, and the mental health of African American communities specifically, would not allow him to pay lip service to agendas that were self-serving, and did not push the field and communities forward.
During his service as an editorial advisory board member for Clinical Psychiatry News, Dr. Bell could always be relied upon to offer an insightful perspective to any discussion, ranging from violence prevention to the social determinants of health and their role in fetal alcohol spectrum disorders.
What I will remember most about Dr. Bell is his strong character. My guess is that he was never shy about stating the truth to a patient or a president of the United States. He possessed the intellect to back up any of his views while also having the humility of a dedicated community psychiatrist who worked for no other reason than to serve his patients.
When I first met Dr. Bell, he was giving a grand rounds at the George Washington University department of psychiatry. He was wearing a hat during the lecture and a belt with the Superman logo. I thought to myself, “Whoa, this is a different type of guy,” then I sat and listened to the talk, and was utterly astounded by his intellect, humor, honesty, and passion for his patients. I had never heard a psychiatrist speak with a combination of such command and approachability, and again, I thought to myself, “Whoa, this is a different type of guy.”
Little did I know that Dr. Bell and I would end up serving together on the editorial board of CPN. I loved seeing Dr. Bell, and catching up and gleaning from his wisdom. I was very humbled by how generous Dr. Bell was with his time and the extent to which he would make himself available as a mentor. During a CPN board meeting, we were trying to come up with a mantra that would capture the mission of the new MDedge Psychiatry website. Dr. Bell (in a hat, of course) let everyone else talk, and then, in a calm voice, said: “We ask the hard questions.” As editor in chief of MDedge Psychiatry, I knew this had to be the mantra. It was aspirational, gave us an identity, and held our feet to the fire – to always ask hard questions in the service of patients and readers. I looked forward to discussing the evolution of the site, seeing him at meetings and conferences, brainstorming the implications of new advances in the field, and simply walking down the street and laughing.
Those events will never occur again, because Carl left us on Thursday, Aug 1. Carl has left us with a legacy of work that we are still coming to appreciate. He left us with a mandate to pursue the truth and make an impact in our communities. He taught black psychiatrists what it meant to stand up unapologetically for your community and society. As I reflect on the scope of his life, I have one last hard question for Carl: “Why did you have to leave us so soon?”
Dr. Norris is assistant professor of psychiatry and behavioral sciences at George Washington University, Washington. He also serves as assistant dean of student affairs at the university, and medical director of psychiatric and behavioral sciences at GWU Hospital. Dr. Norris also is host of the MDedge Psychcast.
Carl C. Bell, MD, started his career by asking the hard questions that no one dared to ask. His curiosity, courage, and compassion for all communities would lead him to impact the world in ways that few psychiatrists could ever imagine.
His accomplishments were many and far-reaching, dating back over 4 decades of service and research. He was a prolific author and researcher, having written more than 400 books, chapters, and articles. His research covered a lot of ground, but four critical areas of focus were childhood trauma, violence prevention, criminal justice reform, and most recently, fetal alcohol spectrum disorders.
The word “visionary” is frequently overused. But when we apply it to Dr. Carl Bell, the word does not do him justice. If you take a closer look at all four of those areas, you can see a common thread: What are the elements of a society that can tear communities apart?
When I look at Dr. Bell’s research, I see a man with a dedicated vision to addressing each of those elements in systematic way, and a determination to bring the results of that research into his Southside Chicago community in numerous ways, including by serving as president and CEO of the Community Mental Health Council, and as director of the Institute for Juvenile Research at the University of Illinois at Chicago.
Dr. Bell was a leader for his patients as well as for black psychiatrists. He was a founding member of Black Psychiatrists of America and served as a mentor in some way to the vast majority of black psychiatrists currently practicing in the country. As a black male psychiatrist, I saw Dr. Bell as a source of inspiration in my career and the standard by which I measured myself. I’m not talking about awards or accomplishments, as Dr. Bell has countless accolades, including most recently, being presented this year with the American Psychiatric Association’s Adolph Meyer Award for Lifetime Achievement in Psychiatric Research and the National Medical Association’s Scroll of Merit. I am referring to Dr. Bell’s willingness to walk away from something he thought was wrong.
For every accolade he won or prestigious committee he served on, I would wager that he declined or stepped way from just as many. Dr. Bell’s character and vision for psychiatry in general, and the mental health of African American communities specifically, would not allow him to pay lip service to agendas that were self-serving, and did not push the field and communities forward.
During his service as an editorial advisory board member for Clinical Psychiatry News, Dr. Bell could always be relied upon to offer an insightful perspective to any discussion, ranging from violence prevention to the social determinants of health and their role in fetal alcohol spectrum disorders.
What I will remember most about Dr. Bell is his strong character. My guess is that he was never shy about stating the truth to a patient or a president of the United States. He possessed the intellect to back up any of his views while also having the humility of a dedicated community psychiatrist who worked for no other reason than to serve his patients.
When I first met Dr. Bell, he was giving a grand rounds at the George Washington University department of psychiatry. He was wearing a hat during the lecture and a belt with the Superman logo. I thought to myself, “Whoa, this is a different type of guy,” then I sat and listened to the talk, and was utterly astounded by his intellect, humor, honesty, and passion for his patients. I had never heard a psychiatrist speak with a combination of such command and approachability, and again, I thought to myself, “Whoa, this is a different type of guy.”
Little did I know that Dr. Bell and I would end up serving together on the editorial board of CPN. I loved seeing Dr. Bell, and catching up and gleaning from his wisdom. I was very humbled by how generous Dr. Bell was with his time and the extent to which he would make himself available as a mentor. During a CPN board meeting, we were trying to come up with a mantra that would capture the mission of the new MDedge Psychiatry website. Dr. Bell (in a hat, of course) let everyone else talk, and then, in a calm voice, said: “We ask the hard questions.” As editor in chief of MDedge Psychiatry, I knew this had to be the mantra. It was aspirational, gave us an identity, and held our feet to the fire – to always ask hard questions in the service of patients and readers. I looked forward to discussing the evolution of the site, seeing him at meetings and conferences, brainstorming the implications of new advances in the field, and simply walking down the street and laughing.
Those events will never occur again, because Carl left us on Thursday, Aug 1. Carl has left us with a legacy of work that we are still coming to appreciate. He left us with a mandate to pursue the truth and make an impact in our communities. He taught black psychiatrists what it meant to stand up unapologetically for your community and society. As I reflect on the scope of his life, I have one last hard question for Carl: “Why did you have to leave us so soon?”
Dr. Norris is assistant professor of psychiatry and behavioral sciences at George Washington University, Washington. He also serves as assistant dean of student affairs at the university, and medical director of psychiatric and behavioral sciences at GWU Hospital. Dr. Norris also is host of the MDedge Psychcast.
Carl C. Bell, MD, started his career by asking the hard questions that no one dared to ask. His curiosity, courage, and compassion for all communities would lead him to impact the world in ways that few psychiatrists could ever imagine.
His accomplishments were many and far-reaching, dating back over 4 decades of service and research. He was a prolific author and researcher, having written more than 400 books, chapters, and articles. His research covered a lot of ground, but four critical areas of focus were childhood trauma, violence prevention, criminal justice reform, and most recently, fetal alcohol spectrum disorders.
The word “visionary” is frequently overused. But when we apply it to Dr. Carl Bell, the word does not do him justice. If you take a closer look at all four of those areas, you can see a common thread: What are the elements of a society that can tear communities apart?
When I look at Dr. Bell’s research, I see a man with a dedicated vision to addressing each of those elements in systematic way, and a determination to bring the results of that research into his Southside Chicago community in numerous ways, including by serving as president and CEO of the Community Mental Health Council, and as director of the Institute for Juvenile Research at the University of Illinois at Chicago.
Dr. Bell was a leader for his patients as well as for black psychiatrists. He was a founding member of Black Psychiatrists of America and served as a mentor in some way to the vast majority of black psychiatrists currently practicing in the country. As a black male psychiatrist, I saw Dr. Bell as a source of inspiration in my career and the standard by which I measured myself. I’m not talking about awards or accomplishments, as Dr. Bell has countless accolades, including most recently, being presented this year with the American Psychiatric Association’s Adolph Meyer Award for Lifetime Achievement in Psychiatric Research and the National Medical Association’s Scroll of Merit. I am referring to Dr. Bell’s willingness to walk away from something he thought was wrong.
For every accolade he won or prestigious committee he served on, I would wager that he declined or stepped way from just as many. Dr. Bell’s character and vision for psychiatry in general, and the mental health of African American communities specifically, would not allow him to pay lip service to agendas that were self-serving, and did not push the field and communities forward.
During his service as an editorial advisory board member for Clinical Psychiatry News, Dr. Bell could always be relied upon to offer an insightful perspective to any discussion, ranging from violence prevention to the social determinants of health and their role in fetal alcohol spectrum disorders.
What I will remember most about Dr. Bell is his strong character. My guess is that he was never shy about stating the truth to a patient or a president of the United States. He possessed the intellect to back up any of his views while also having the humility of a dedicated community psychiatrist who worked for no other reason than to serve his patients.
When I first met Dr. Bell, he was giving a grand rounds at the George Washington University department of psychiatry. He was wearing a hat during the lecture and a belt with the Superman logo. I thought to myself, “Whoa, this is a different type of guy,” then I sat and listened to the talk, and was utterly astounded by his intellect, humor, honesty, and passion for his patients. I had never heard a psychiatrist speak with a combination of such command and approachability, and again, I thought to myself, “Whoa, this is a different type of guy.”
Little did I know that Dr. Bell and I would end up serving together on the editorial board of CPN. I loved seeing Dr. Bell, and catching up and gleaning from his wisdom. I was very humbled by how generous Dr. Bell was with his time and the extent to which he would make himself available as a mentor. During a CPN board meeting, we were trying to come up with a mantra that would capture the mission of the new MDedge Psychiatry website. Dr. Bell (in a hat, of course) let everyone else talk, and then, in a calm voice, said: “We ask the hard questions.” As editor in chief of MDedge Psychiatry, I knew this had to be the mantra. It was aspirational, gave us an identity, and held our feet to the fire – to always ask hard questions in the service of patients and readers. I looked forward to discussing the evolution of the site, seeing him at meetings and conferences, brainstorming the implications of new advances in the field, and simply walking down the street and laughing.
Those events will never occur again, because Carl left us on Thursday, Aug 1. Carl has left us with a legacy of work that we are still coming to appreciate. He left us with a mandate to pursue the truth and make an impact in our communities. He taught black psychiatrists what it meant to stand up unapologetically for your community and society. As I reflect on the scope of his life, I have one last hard question for Carl: “Why did you have to leave us so soon?”
Dr. Norris is assistant professor of psychiatry and behavioral sciences at George Washington University, Washington. He also serves as assistant dean of student affairs at the university, and medical director of psychiatric and behavioral sciences at GWU Hospital. Dr. Norris also is host of the MDedge Psychcast.
2019 Update on female sexual dysfunction
Hypoactive sexual desire disorder (HSDD) is the most prevalent sexual health problem in women of all ages, with population-based studies showing that about 36% to 39% of women report low sexual desire, and 8% to 10% meet the diagnostic criteria of low sexual desire and associated distress.1,2 An expanded definition of HSDD may include3:
- lack of motivation for sexual activity (reduced or absent spontaneous desire or responsive desire to erotic cues and stimulation; inability to maintain desire or interest through sexual activity)
- loss of desire to initiate or participate in sexual activity (including avoiding situations that could lead to sexual activity) combined with significant personal distress (frustration, loss, sadness, worry) (FIGURE).4
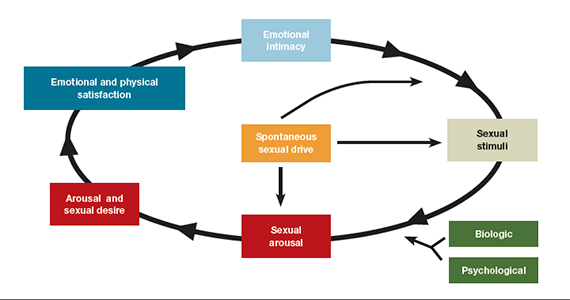
Despite the high prevalence of HSDD, patients often are uncomfortable and reluctant to voice concerns about low sexual desire to their ObGyn. Further, clinicians may feel ill equipped to diagnose and treat patients with HSDD. ObGyns, however, are well positioned to initiate a general discussion about sexual concerns with patients and use screening tools, such as the Decreased Sexual Desire Screener (DSDS), to facilitate a discussion and clarify a diagnosis of generalized acquired HSDD (TABLES 1 and 2).5 Helpful guidance on HSDD is available from the American College of Obstetricians and Gynecologists and the International Society for the Study of Women’s Sexual Health.6-8
Importantly, clinicians have a new treatment option they can offer to patients with HSDD. Bremelanotide was approved by the US Food and Drug Administration (FDA) on June 21, 2019, to treat acquired, generalized HSDD in premenopausal women. Up until this approval, flibanserin (approved in 2015) was the only drug FDA approved for the treatment of HSDD.
Assessing and treating HSDD today can be likened to managing depression 30 years ago, before selective serotonin receptor inhibitors were available. ObGyns would refer patients with depression to other health care providers, or not even ask patients about depressive symptoms because we had so little to offer. Once safe and effective antidepressants became available, knowing we could provide pharmacologic options made inquiring about depressive symptoms and the use of screening tools more readily incorporated into standard clinical practice. Depression is now recognized as a medical condition with biologic underpinnings, just like HSDD, and treatment options are available for both disorders.
For this Update, I had the opportunity to discuss the clinical trial experience with bremelanotide for HSDD with Dr. Sheryl Kingsberg, including efficacy and safety, dosage and administration, contraindications, and adverse events. She also details an ideal patient for treatment with bremelanotide, and we review pertinent aspects of flibanserin for comparative purposes.

Bremelanotide: A new therapeutic option
According to the product labeling for bremelanotide, the drug is indicated for the treatment of premenopausal women with acquired, generalized HSDD (low sexual desire that causes marked distress or interpersonal difficulty).9 This means that the HSDD developed in a woman who previously did not have problems with sexual desire, and that it occurred regardless of the type of stimulation, situation, or partner. In addition, the HSDD should not result from a coexisting medical or psychiatric condition, problems with the relationship, or the effects of a medication or drug substance.
Flibanserin also is indicated for the treatment of premenopausal women with HSDD. While both bremelanotide and flibanserin have indications only for premenopausal women, 2 studies of flibanserin in postmenopausal women have been published.10,11 Results from these studies in naturally menopausal women suggest that flibanserin may be efficacious in this population, with improvement in sexual desire, reduced distress associated with low desire, and improvement in the number of satisfying sexual events (SSEs).
No trials of bremelanotide in postmenopausal women have been published, but since this drug acts on central nervous system receptors, as does flibanserin, it may have similar effectiveness in postmenopausal women as well.

Continue to: Clinical trials show bremelanotide improves desire, reduces distress...
Clinical trials show bremelanotide improves desire, reduces distress
Two phase 3 clinical trials, dubbed the Reconnect studies, demonstrated that, compared with placebo, bremelanotide was associated with statistically significant improvements in sexual desire and levels of distress regarding sexual desire.
The 2 identical, randomized, placebo-controlled multicenter trials included 1,247 premenopausal women with HSDD of at least 6 months' duration.9,12 Bremelanotide 1.75 mg (or placebo) was self-administered subcutaneously with an autoinjector on an as-desired basis. The 24-week double-blind treatment period was followed by a 52-week open-label extension study.
The co-primary efficacy end points were the change from baseline to end-of-study (week 24 of the double-blind treatment period) in the 1) Female Sexual Function Index (FSFI) desire domain score and 2) feeling bothered by low sexual desire as measured by Question 13 on the Female Sexual Distress Scale (FSDS). An increase in the FSFI desire domain score over time denotes improvement in sexual desire, while a decrease in the FSDS Question 13 score over time indicates improvement in the level of distress associated with low sexual desire.
In the 2 clinical studies, the mean change from baseline (SD) in the FSFI desire domain score, which ranged from 1.2 to 6.0 at study outset (higher scores indicate greater desire), was:
- study 1: 0.5 (1.1) in the bremelanotide-treated women and 0.2 (1.0) in the placebo-treated women (P = .0002)
- study 2: 0.6 (1.0) in the bremelanotide group versus 0.2 (0.9) in the placebo group (P<.0001).
For FSDS Question 13, for which the score range was 0 to 4 (higher scores indicate greater bother), the mean change from baseline score was:
- study 1: -0.7 (1.2) in the bremelanotide-treated group compared with -0.4 (1.1) in the placebo-treated group (P<.0001)
- study 2: -0.7 (1.1) in the bremelanotide group and -0.4 (1.1) in the placebo group (P = .0053).
It should be noted that, in the past, SSEs were used as a primary end point in clinical studies. However, we have shifted from SSEs to desire and distress as an end point because SSEs have little to do with desire. Women worry about and are distressed by the fact that they no longer have sexual appetite. They no longer "want to want" even though their body will be responsive and they can have an orgasm. That is exemplified by the woman in our case scenario (see box, page 18), who very much wants the experience of being able to anticipate with pleasure the idea of having an enjoyable connection with her partner.
Continue to: Physiologic target: The melanocortin receptor...
Physiologic target: The melanocortin receptor
Bremelanotide's theorized mechanism of action is that it works to rebalance neurotransmitters that are implicated in causing HSDD, acting as an agonist on the melanocortin receptor to promote dopamine release and allow women to perceive sexual cues as rewarding. They can then respond to those cues the way they used to and therefore experience desire. Flibanserin has affinity for serotonin (5-hydroxytryptamine [5-HT]) receptors, with agonist and antagonist activity, as well as moderate antagonist activity on some dopamine receptors.
The bottom line is that we now have treatments to address the underlying biologic aspect of HSDD, which is a biopsychosocial disorder. Again, this has parallels to depression and its biologic mechanism, for which we have effective treatments.
Dosing is an as-needed injection
Unlike the daily nighttime oral dose required with flibanserin, bremelanotide is a 1.75-mg dose administered as a subcutaneous injection (in either the thigh or the abdomen) with a pen-like autoinjector, on an as-needed basis. It should be administered at least 45 minutes before anticipated sexual activity. That is a benefit for many women who do not want to take a daily pill when they know that their "desire to desire" may be once per week or once every other week.
Regarding the drug delivery mode, nobody dropped out of the bremelanotide clinical trials because of having to take an injection with an autoinjector, which employs a very thin needle and is virtually painless. A small number of bremelanotide-treated women, about 13%, had injection site reactions (compared with 8% in the placebo group), which is common with subcutaneous injection. Even in the phase 2 clinical trial, in which a syringe was used to administer the drug, no participants discontinued the study because of the injection mode.
There are no clear pharmacokinetic data on how long bremelanotide's effects last, but it may be anywhere from 8 to 16 hours. Patients should not take more than 1 dose within 24 hours--but since the effect may last up to 16 hours that should not be a problem--and use of more than 8 doses per month is not recommended.
While bremelanotide improves desire, certainly better than placebo, there is also some peripheral improvement in arousal, although women in the trials had only HSDD. We do not know whether bremelanotide would treat arousal disorder, but it will help women with or without arousal difficulties associated with their HSDD, as shown in a subgroup analysis in the trials.13
Counsel patients on treatment potentialities
Clinicians should be aware of several precautions with bremelanotide use.
Blood pressure increases. After each dose of bremelanotide, transient increases in blood pressure (6 mm Hg in systolic and 3 mm Hg in diastolic blood pressure) and reductions in heart rate (up to 5 beats per minute) occur; these measurements return to baseline usually within 12 hours postdose.9 When you think about whether having sexual desire will increase blood pressure, this may be physiologic. It is similar to walking up a flight of stairs.
The drug is not recommended, however, for use in patients at high risk for cardiovascular disease, and it is contraindicated in women with uncontrolled hypertension or known cardiovascular disease. Blood pressure should be well controlled before bremelanotide is initiated--use of antihypertensive agents is not contraindicated with bremelanotide as the drugs do not interact.
Clinicians are not required to participate in a Risk Evaluation and Mitigation Strategy (REMS) program to prescribe bremelanotide as they are with flibanserin (because of the increased risk of severe hypotension and syncope due to flibanserin's interaction with alcohol).
Drug interactions. Bremelanotide is a melanocortin receptor agonist--a unique compound. Antidepressants, other psychoactive medications, and oral contraceptives are not contraindicated with bremelanotide as there are no known interactions. Alcohol use also is not a contraindication or caution, in contrast to flibanserin. (In April, the FDA issued a labeling change order for flibanserin, specifying that alcohol does not have to be avoided completely when taking flibanserin, but that women should discontinue drinking alcohol at least 2 hours before taking the drug at bedtime, or skip the flibanserin dose that evening.14) Bremelanotide may slow gastric emptying, though, so when a patient is taking oral drugs that require threshold concentrations for efficacy, such as antibiotics, they should avoid bremelanotide. In addition, some drugs, such as indomethacin, may have a delayed onset of action with concomitant bremelanotide use.9
Importantly, patients should avoid using bremelanotide if they are taking an oral naltrexone product for treatment of alcohol or opioid addiction, because bremelanotide may decrease systemic exposure of oral naltrexone. That would potentially lead to naltrexone treatment failure and its consequences.9
Skin pigmentation changes. Hyperpigmentation occurred with bremelanotide use on the face, gingiva, and breasts, as reported in the clinical trials, in 1% of treated patients who received up to 8 doses per month, compared with no such occurrences in placebo-treated patients. In addition, 38% of patients who received bremelanotide daily for 8 days developed focal hyperpigmentation. It was not confirmed in all patients whether the hyperpigmentation resolved. Women with dark skin were more likely to develop hyperpigmentation.9
Common adverse reactions. The most common adverse reactions with bremelanotide treatment are nausea, flushing, injection site reactions, and headache, with most events being mild to moderate in intensity. In the clinical trials, 40% of the bremelanotide-treated women experienced nausea (compared with 1% of placebo-treated women), with most occurrences being mild; for most participants nausea improved with the second dose. Women had nausea that either went away or was intermittent, or it was mild enough that the drug benefits outweighed the tolerability costs--of women who experienced nausea, 92% continued in the trial, and 8% dropped out because of nausea.9
The following scenario describes the experience of HSDD in one of Dr. Kingsberg's patients.
CASE Woman avoids sex because of low desire; marriage is suffering
A 40-year-old woman, Sandra, who has been married for 19 years and has fraternal twins aged 8, presented to the behavioral medicine clinic with distressing symptoms of low sexual desire. For several years into the marriage the patient experienced excellent sex drive. After 6 to 7 years, she noticed that her desire had declined and that she was starting to avoid sex. She was irritated when her husband initiated sex, and she would make excuses as to why it was not the right time.
Her husband felt hurt, frustrated, and rejected. The couple was close to divorce because he was angry and resentful. Sandra recognized there was a problem but did not know how to fix it. She could not understand why her interest had waned since she still loved her husband and considered him objectively very attractive.
Sandra came to see Dr. Kingsberg at the behavioral medicine clinic. Using the 5-item validated diagnostic tool called the Decreased Sexual Desire Screener, Dr. Kingsberg diagnosed hypoactive sexual desire disorder (HSDD), a term Sandra had never heard of and did not know was a condition. The patient was relieved to know that she was one of several million women affected by HSDD and that the problem was not just that she was a "bad wife" or that she had some kind of psychological block. She emphasized how much she loved her husband and how she wanted desperately to "want to want desire," as she recalled feeling previously.
Sandra was treated with counseling and psychotherapy in which we addressed the relationship issues, the avoidance of sex, the comfort with being sexual, and the recognition that responsive desire can be helpful (as she was able to have arousal and orgasm and have a satisfying sexual event). The issue was that she had no motivation to seek out sex and had no interest in experiencing that pleasure. In subsequent couple's therapy, the husband recognized that his wife was not intentionally rejecting him, but that she had a real medical condition.
Although Sandra's relationship was now more stable and she and her husband were both working toward finding a solution to Sandra's loss of desire, she was still very distressed by her lack of desire. Sandra tried flibanserin for 3 months but unfortunately did not respond. Sandra heard about the recent approval of bremelanotide and is looking forward to the drug being available so that she can try it.
Final considerations
Asking patients about sexual function and using sexual function screening tools can help clinicians identify patients with the decreased sexual desire and associated distress characteristic of HSDD. ObGyns are the appropriate clinicians to treat these women and soon will have 2 pharmacologic options--bremelanotide (anticipated to be available in Fall 2019) and flibanserin--to offer patients with this biopsychosocial disorder that can adversely impact well-being and quality of life. Clinicians should individualize treatment, which may include psychotherapeutic counseling, and counsel patients on appropriate drug use and potential adverse effects.
AMAG Pharmaceuticals, Inc. has announced that they will have a copay assistance program for bremelanotide, where the first prescription of four autoinjectors will be a $0 copay, followed by a $99 copay or less for refills.15
- Shifren JL, Monz BU, Russo PA, et al. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol. 2008;112:970-978.
- West SL, D'Aloisio AA, Agans RP, et al. Prevalence of low sexual desire and hypoactive sexual desire disorder in a nationally representative sample of US women. Arch Intern Med. 2008;168:1441-1449.
- Parish SJ, Goldstein AT, Goldstein SW, et al. Toward a more evidence-based nosology and nomenclature for female sexual dysfunctions: part II. J Sex Med. 2016;13:1888-1906.
- Basson R. Using a different model for female sexual response to address women's problematic low sexual desire. J Sex Marital Ther. 2001;27:395-403.
- Clayton AH, Goldfischer ER, Goldstein I, et al. Validation of the Decreased Sexual Desire Screener (DSDS): a brief diagnostic instrument for generalized acquired female hypoactive sexual desire disorder (HSDD). J Sex Med. 2009;6:730-738.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins-Gynecology. ACOG practice bulletin no. 213: Female sexual dysfunction. Obstet Gynecol. 2019;134:e1-e18.
- Goldstein I, Kim NN, Clayton AH, et al. Hypoactive sexual desire disorder: International Society for the Study of Women's Sexual Health (ISSWSH) expert consensus panel review. Mayo Clin Proc. 2017;92:114-128.
- Clayton AH, Goldstein I, Kim NN, et al. The International Society for the Study of Women's Sexual Health process of care for management of hypoactive sexual desire disorder in women. Mayo Clin Proc. 2018;93:467-487.
- Vyleesi [package insert]. Waltham, MA: AMAG Pharmaceuticals; 2019.
- Simon JA, Kingsberg SA, Shumel B, et al. Efficacy and safety of flibanserin in postmenopausal women with hypoactive sexual desire disorder: results of the SNOWDROP trial. Menopause. 2014;21;633-640.
- Portman DJ, Brown L, Yuan, et al. Flibanserin in postmenopausal women with hypoactive sexual desire disorder: results of the PLUMERIA study. J Sex Med. 2017;14:834-842.
- Kingsberg SA, Clayton AH, Portman D, et al. Bremelanotide for the treatment of hypoactive sexual desire disorder: two randomized phase 3 trials. Obstet Gynecol. Forthcoming.
- Clayton AH, Lucas J, Jordon R, et al. Efficacy of the Investigational drug bremelanotide in the Reconnect studies. Poster presented at: 30th ECNP Congress of Applied and Translational Neuroscience; September 2-5, 2017, Paris, France.
- US Food and Drug Administration. FDA orders important safety labeling changes for Addyi [press release]. April 11, 2019. https://www.fda.gov/news-events/press-announcements/fda-orders-important-safety-labeling-changes-addyi. Accessed July 17, 2019.
- Vyleesi website (https://vyleesipro.com). Accessed August 5, 2019.

Barbara S. Levy, MD
Dr. Levy is Vice President for Health Policy at the American College of Obstetricians and Gynecologists, Washington, DC.

Sheryl A. Kingsberg, PhD
Dr. Kingsberg is Chief, Division of Behavioral Medicine, Department of Obstetrics and Gynecology, University Hospitals Cleveland Medical Center, and Professor, Departments of Reproductive Biology and Psychiatry, Case Western Reserve University School of Medicine, Cleveland, Ohio.
Dr. Levy reports no financial relationships relevant to this article. Dr. Kingsberg reports that she receives grant or research support from Endoceutics and Palatin Technologies; is a consultant to AMAG, Daré, Duchesnay, Emotional Brain, Endoceutics, IVIX, Lupin, Palatin Technologies, Pfizer, Shionogi, Sprout, SST, and TherapeuticsMD; and is a speaker for TherapeuticsMD.
Barbara S. Levy, MD
Dr. Levy is Vice President for Health Policy at the American College of Obstetricians and Gynecologists, Washington, DC.
Sheryl A. Kingsberg, PhD
Dr. Kingsberg is Chief, Division of Behavioral Medicine, Department of Obstetrics and Gynecology, University Hospitals Cleveland Medical Center, and Professor, Departments of Reproductive Biology and Psychiatry, Case Western Reserve University School of Medicine, Cleveland, Ohio.
Dr. Levy reports no financial relationships relevant to this article. Dr. Kingsberg reports that she receives grant or research support from Endoceutics and Palatin Technologies; is a consultant to AMAG, Daré, Duchesnay, Emotional Brain, Endoceutics, IVIX, Lupin, Palatin Technologies, Pfizer, Shionogi, Sprout, SST, and TherapeuticsMD; and is a speaker for TherapeuticsMD.
Barbara S. Levy, MD
Dr. Levy is Vice President for Health Policy at the American College of Obstetricians and Gynecologists, Washington, DC.
Sheryl A. Kingsberg, PhD
Dr. Kingsberg is Chief, Division of Behavioral Medicine, Department of Obstetrics and Gynecology, University Hospitals Cleveland Medical Center, and Professor, Departments of Reproductive Biology and Psychiatry, Case Western Reserve University School of Medicine, Cleveland, Ohio.
Dr. Levy reports no financial relationships relevant to this article. Dr. Kingsberg reports that she receives grant or research support from Endoceutics and Palatin Technologies; is a consultant to AMAG, Daré, Duchesnay, Emotional Brain, Endoceutics, IVIX, Lupin, Palatin Technologies, Pfizer, Shionogi, Sprout, SST, and TherapeuticsMD; and is a speaker for TherapeuticsMD.
Hypoactive sexual desire disorder (HSDD) is the most prevalent sexual health problem in women of all ages, with population-based studies showing that about 36% to 39% of women report low sexual desire, and 8% to 10% meet the diagnostic criteria of low sexual desire and associated distress.1,2 An expanded definition of HSDD may include3:
- lack of motivation for sexual activity (reduced or absent spontaneous desire or responsive desire to erotic cues and stimulation; inability to maintain desire or interest through sexual activity)
- loss of desire to initiate or participate in sexual activity (including avoiding situations that could lead to sexual activity) combined with significant personal distress (frustration, loss, sadness, worry) (FIGURE).4
Despite the high prevalence of HSDD, patients often are uncomfortable and reluctant to voice concerns about low sexual desire to their ObGyn. Further, clinicians may feel ill equipped to diagnose and treat patients with HSDD. ObGyns, however, are well positioned to initiate a general discussion about sexual concerns with patients and use screening tools, such as the Decreased Sexual Desire Screener (DSDS), to facilitate a discussion and clarify a diagnosis of generalized acquired HSDD (TABLES 1 and 2).5 Helpful guidance on HSDD is available from the American College of Obstetricians and Gynecologists and the International Society for the Study of Women’s Sexual Health.6-8
Importantly, clinicians have a new treatment option they can offer to patients with HSDD. Bremelanotide was approved by the US Food and Drug Administration (FDA) on June 21, 2019, to treat acquired, generalized HSDD in premenopausal women. Up until this approval, flibanserin (approved in 2015) was the only drug FDA approved for the treatment of HSDD.
Assessing and treating HSDD today can be likened to managing depression 30 years ago, before selective serotonin receptor inhibitors were available. ObGyns would refer patients with depression to other health care providers, or not even ask patients about depressive symptoms because we had so little to offer. Once safe and effective antidepressants became available, knowing we could provide pharmacologic options made inquiring about depressive symptoms and the use of screening tools more readily incorporated into standard clinical practice. Depression is now recognized as a medical condition with biologic underpinnings, just like HSDD, and treatment options are available for both disorders.
For this Update, I had the opportunity to discuss the clinical trial experience with bremelanotide for HSDD with Dr. Sheryl Kingsberg, including efficacy and safety, dosage and administration, contraindications, and adverse events. She also details an ideal patient for treatment with bremelanotide, and we review pertinent aspects of flibanserin for comparative purposes.
Bremelanotide: A new therapeutic option
According to the product labeling for bremelanotide, the drug is indicated for the treatment of premenopausal women with acquired, generalized HSDD (low sexual desire that causes marked distress or interpersonal difficulty).9 This means that the HSDD developed in a woman who previously did not have problems with sexual desire, and that it occurred regardless of the type of stimulation, situation, or partner. In addition, the HSDD should not result from a coexisting medical or psychiatric condition, problems with the relationship, or the effects of a medication or drug substance.
Flibanserin also is indicated for the treatment of premenopausal women with HSDD. While both bremelanotide and flibanserin have indications only for premenopausal women, 2 studies of flibanserin in postmenopausal women have been published.10,11 Results from these studies in naturally menopausal women suggest that flibanserin may be efficacious in this population, with improvement in sexual desire, reduced distress associated with low desire, and improvement in the number of satisfying sexual events (SSEs).
No trials of bremelanotide in postmenopausal women have been published, but since this drug acts on central nervous system receptors, as does flibanserin, it may have similar effectiveness in postmenopausal women as well.
Continue to: Clinical trials show bremelanotide improves desire, reduces distress...
Clinical trials show bremelanotide improves desire, reduces distress
Two phase 3 clinical trials, dubbed the Reconnect studies, demonstrated that, compared with placebo, bremelanotide was associated with statistically significant improvements in sexual desire and levels of distress regarding sexual desire.
The 2 identical, randomized, placebo-controlled multicenter trials included 1,247 premenopausal women with HSDD of at least 6 months' duration.9,12 Bremelanotide 1.75 mg (or placebo) was self-administered subcutaneously with an autoinjector on an as-desired basis. The 24-week double-blind treatment period was followed by a 52-week open-label extension study.
The co-primary efficacy end points were the change from baseline to end-of-study (week 24 of the double-blind treatment period) in the 1) Female Sexual Function Index (FSFI) desire domain score and 2) feeling bothered by low sexual desire as measured by Question 13 on the Female Sexual Distress Scale (FSDS). An increase in the FSFI desire domain score over time denotes improvement in sexual desire, while a decrease in the FSDS Question 13 score over time indicates improvement in the level of distress associated with low sexual desire.
In the 2 clinical studies, the mean change from baseline (SD) in the FSFI desire domain score, which ranged from 1.2 to 6.0 at study outset (higher scores indicate greater desire), was:
- study 1: 0.5 (1.1) in the bremelanotide-treated women and 0.2 (1.0) in the placebo-treated women (P = .0002)
- study 2: 0.6 (1.0) in the bremelanotide group versus 0.2 (0.9) in the placebo group (P<.0001).
For FSDS Question 13, for which the score range was 0 to 4 (higher scores indicate greater bother), the mean change from baseline score was:
- study 1: -0.7 (1.2) in the bremelanotide-treated group compared with -0.4 (1.1) in the placebo-treated group (P<.0001)
- study 2: -0.7 (1.1) in the bremelanotide group and -0.4 (1.1) in the placebo group (P = .0053).
It should be noted that, in the past, SSEs were used as a primary end point in clinical studies. However, we have shifted from SSEs to desire and distress as an end point because SSEs have little to do with desire. Women worry about and are distressed by the fact that they no longer have sexual appetite. They no longer "want to want" even though their body will be responsive and they can have an orgasm. That is exemplified by the woman in our case scenario (see box, page 18), who very much wants the experience of being able to anticipate with pleasure the idea of having an enjoyable connection with her partner.
Continue to: Physiologic target: The melanocortin receptor...
Physiologic target: The melanocortin receptor
Bremelanotide's theorized mechanism of action is that it works to rebalance neurotransmitters that are implicated in causing HSDD, acting as an agonist on the melanocortin receptor to promote dopamine release and allow women to perceive sexual cues as rewarding. They can then respond to those cues the way they used to and therefore experience desire. Flibanserin has affinity for serotonin (5-hydroxytryptamine [5-HT]) receptors, with agonist and antagonist activity, as well as moderate antagonist activity on some dopamine receptors.
The bottom line is that we now have treatments to address the underlying biologic aspect of HSDD, which is a biopsychosocial disorder. Again, this has parallels to depression and its biologic mechanism, for which we have effective treatments.
Dosing is an as-needed injection
Unlike the daily nighttime oral dose required with flibanserin, bremelanotide is a 1.75-mg dose administered as a subcutaneous injection (in either the thigh or the abdomen) with a pen-like autoinjector, on an as-needed basis. It should be administered at least 45 minutes before anticipated sexual activity. That is a benefit for many women who do not want to take a daily pill when they know that their "desire to desire" may be once per week or once every other week.
Regarding the drug delivery mode, nobody dropped out of the bremelanotide clinical trials because of having to take an injection with an autoinjector, which employs a very thin needle and is virtually painless. A small number of bremelanotide-treated women, about 13%, had injection site reactions (compared with 8% in the placebo group), which is common with subcutaneous injection. Even in the phase 2 clinical trial, in which a syringe was used to administer the drug, no participants discontinued the study because of the injection mode.
There are no clear pharmacokinetic data on how long bremelanotide's effects last, but it may be anywhere from 8 to 16 hours. Patients should not take more than 1 dose within 24 hours--but since the effect may last up to 16 hours that should not be a problem--and use of more than 8 doses per month is not recommended.
While bremelanotide improves desire, certainly better than placebo, there is also some peripheral improvement in arousal, although women in the trials had only HSDD. We do not know whether bremelanotide would treat arousal disorder, but it will help women with or without arousal difficulties associated with their HSDD, as shown in a subgroup analysis in the trials.13
Counsel patients on treatment potentialities
Clinicians should be aware of several precautions with bremelanotide use.
Blood pressure increases. After each dose of bremelanotide, transient increases in blood pressure (6 mm Hg in systolic and 3 mm Hg in diastolic blood pressure) and reductions in heart rate (up to 5 beats per minute) occur; these measurements return to baseline usually within 12 hours postdose.9 When you think about whether having sexual desire will increase blood pressure, this may be physiologic. It is similar to walking up a flight of stairs.
The drug is not recommended, however, for use in patients at high risk for cardiovascular disease, and it is contraindicated in women with uncontrolled hypertension or known cardiovascular disease. Blood pressure should be well controlled before bremelanotide is initiated--use of antihypertensive agents is not contraindicated with bremelanotide as the drugs do not interact.
Clinicians are not required to participate in a Risk Evaluation and Mitigation Strategy (REMS) program to prescribe bremelanotide as they are with flibanserin (because of the increased risk of severe hypotension and syncope due to flibanserin's interaction with alcohol).
Drug interactions. Bremelanotide is a melanocortin receptor agonist--a unique compound. Antidepressants, other psychoactive medications, and oral contraceptives are not contraindicated with bremelanotide as there are no known interactions. Alcohol use also is not a contraindication or caution, in contrast to flibanserin. (In April, the FDA issued a labeling change order for flibanserin, specifying that alcohol does not have to be avoided completely when taking flibanserin, but that women should discontinue drinking alcohol at least 2 hours before taking the drug at bedtime, or skip the flibanserin dose that evening.14) Bremelanotide may slow gastric emptying, though, so when a patient is taking oral drugs that require threshold concentrations for efficacy, such as antibiotics, they should avoid bremelanotide. In addition, some drugs, such as indomethacin, may have a delayed onset of action with concomitant bremelanotide use.9
Importantly, patients should avoid using bremelanotide if they are taking an oral naltrexone product for treatment of alcohol or opioid addiction, because bremelanotide may decrease systemic exposure of oral naltrexone. That would potentially lead to naltrexone treatment failure and its consequences.9
Skin pigmentation changes. Hyperpigmentation occurred with bremelanotide use on the face, gingiva, and breasts, as reported in the clinical trials, in 1% of treated patients who received up to 8 doses per month, compared with no such occurrences in placebo-treated patients. In addition, 38% of patients who received bremelanotide daily for 8 days developed focal hyperpigmentation. It was not confirmed in all patients whether the hyperpigmentation resolved. Women with dark skin were more likely to develop hyperpigmentation.9
Common adverse reactions. The most common adverse reactions with bremelanotide treatment are nausea, flushing, injection site reactions, and headache, with most events being mild to moderate in intensity. In the clinical trials, 40% of the bremelanotide-treated women experienced nausea (compared with 1% of placebo-treated women), with most occurrences being mild; for most participants nausea improved with the second dose. Women had nausea that either went away or was intermittent, or it was mild enough that the drug benefits outweighed the tolerability costs--of women who experienced nausea, 92% continued in the trial, and 8% dropped out because of nausea.9
The following scenario describes the experience of HSDD in one of Dr. Kingsberg's patients.
CASE Woman avoids sex because of low desire; marriage is suffering
A 40-year-old woman, Sandra, who has been married for 19 years and has fraternal twins aged 8, presented to the behavioral medicine clinic with distressing symptoms of low sexual desire. For several years into the marriage the patient experienced excellent sex drive. After 6 to 7 years, she noticed that her desire had declined and that she was starting to avoid sex. She was irritated when her husband initiated sex, and she would make excuses as to why it was not the right time.
Her husband felt hurt, frustrated, and rejected. The couple was close to divorce because he was angry and resentful. Sandra recognized there was a problem but did not know how to fix it. She could not understand why her interest had waned since she still loved her husband and considered him objectively very attractive.
Sandra came to see Dr. Kingsberg at the behavioral medicine clinic. Using the 5-item validated diagnostic tool called the Decreased Sexual Desire Screener, Dr. Kingsberg diagnosed hypoactive sexual desire disorder (HSDD), a term Sandra had never heard of and did not know was a condition. The patient was relieved to know that she was one of several million women affected by HSDD and that the problem was not just that she was a "bad wife" or that she had some kind of psychological block. She emphasized how much she loved her husband and how she wanted desperately to "want to want desire," as she recalled feeling previously.
Sandra was treated with counseling and psychotherapy in which we addressed the relationship issues, the avoidance of sex, the comfort with being sexual, and the recognition that responsive desire can be helpful (as she was able to have arousal and orgasm and have a satisfying sexual event). The issue was that she had no motivation to seek out sex and had no interest in experiencing that pleasure. In subsequent couple's therapy, the husband recognized that his wife was not intentionally rejecting him, but that she had a real medical condition.
Although Sandra's relationship was now more stable and she and her husband were both working toward finding a solution to Sandra's loss of desire, she was still very distressed by her lack of desire. Sandra tried flibanserin for 3 months but unfortunately did not respond. Sandra heard about the recent approval of bremelanotide and is looking forward to the drug being available so that she can try it.
Final considerations
Asking patients about sexual function and using sexual function screening tools can help clinicians identify patients with the decreased sexual desire and associated distress characteristic of HSDD. ObGyns are the appropriate clinicians to treat these women and soon will have 2 pharmacologic options--bremelanotide (anticipated to be available in Fall 2019) and flibanserin--to offer patients with this biopsychosocial disorder that can adversely impact well-being and quality of life. Clinicians should individualize treatment, which may include psychotherapeutic counseling, and counsel patients on appropriate drug use and potential adverse effects.
AMAG Pharmaceuticals, Inc. has announced that they will have a copay assistance program for bremelanotide, where the first prescription of four autoinjectors will be a $0 copay, followed by a $99 copay or less for refills.15
Hypoactive sexual desire disorder (HSDD) is the most prevalent sexual health problem in women of all ages, with population-based studies showing that about 36% to 39% of women report low sexual desire, and 8% to 10% meet the diagnostic criteria of low sexual desire and associated distress.1,2 An expanded definition of HSDD may include3:
- lack of motivation for sexual activity (reduced or absent spontaneous desire or responsive desire to erotic cues and stimulation; inability to maintain desire or interest through sexual activity)
- loss of desire to initiate or participate in sexual activity (including avoiding situations that could lead to sexual activity) combined with significant personal distress (frustration, loss, sadness, worry) (FIGURE).4
Despite the high prevalence of HSDD, patients often are uncomfortable and reluctant to voice concerns about low sexual desire to their ObGyn. Further, clinicians may feel ill equipped to diagnose and treat patients with HSDD. ObGyns, however, are well positioned to initiate a general discussion about sexual concerns with patients and use screening tools, such as the Decreased Sexual Desire Screener (DSDS), to facilitate a discussion and clarify a diagnosis of generalized acquired HSDD (TABLES 1 and 2).5 Helpful guidance on HSDD is available from the American College of Obstetricians and Gynecologists and the International Society for the Study of Women’s Sexual Health.6-8
Importantly, clinicians have a new treatment option they can offer to patients with HSDD. Bremelanotide was approved by the US Food and Drug Administration (FDA) on June 21, 2019, to treat acquired, generalized HSDD in premenopausal women. Up until this approval, flibanserin (approved in 2015) was the only drug FDA approved for the treatment of HSDD.
Assessing and treating HSDD today can be likened to managing depression 30 years ago, before selective serotonin receptor inhibitors were available. ObGyns would refer patients with depression to other health care providers, or not even ask patients about depressive symptoms because we had so little to offer. Once safe and effective antidepressants became available, knowing we could provide pharmacologic options made inquiring about depressive symptoms and the use of screening tools more readily incorporated into standard clinical practice. Depression is now recognized as a medical condition with biologic underpinnings, just like HSDD, and treatment options are available for both disorders.
For this Update, I had the opportunity to discuss the clinical trial experience with bremelanotide for HSDD with Dr. Sheryl Kingsberg, including efficacy and safety, dosage and administration, contraindications, and adverse events. She also details an ideal patient for treatment with bremelanotide, and we review pertinent aspects of flibanserin for comparative purposes.
Bremelanotide: A new therapeutic option
According to the product labeling for bremelanotide, the drug is indicated for the treatment of premenopausal women with acquired, generalized HSDD (low sexual desire that causes marked distress or interpersonal difficulty).9 This means that the HSDD developed in a woman who previously did not have problems with sexual desire, and that it occurred regardless of the type of stimulation, situation, or partner. In addition, the HSDD should not result from a coexisting medical or psychiatric condition, problems with the relationship, or the effects of a medication or drug substance.
Flibanserin also is indicated for the treatment of premenopausal women with HSDD. While both bremelanotide and flibanserin have indications only for premenopausal women, 2 studies of flibanserin in postmenopausal women have been published.10,11 Results from these studies in naturally menopausal women suggest that flibanserin may be efficacious in this population, with improvement in sexual desire, reduced distress associated with low desire, and improvement in the number of satisfying sexual events (SSEs).
No trials of bremelanotide in postmenopausal women have been published, but since this drug acts on central nervous system receptors, as does flibanserin, it may have similar effectiveness in postmenopausal women as well.
Continue to: Clinical trials show bremelanotide improves desire, reduces distress...
Clinical trials show bremelanotide improves desire, reduces distress
Two phase 3 clinical trials, dubbed the Reconnect studies, demonstrated that, compared with placebo, bremelanotide was associated with statistically significant improvements in sexual desire and levels of distress regarding sexual desire.
The 2 identical, randomized, placebo-controlled multicenter trials included 1,247 premenopausal women with HSDD of at least 6 months' duration.9,12 Bremelanotide 1.75 mg (or placebo) was self-administered subcutaneously with an autoinjector on an as-desired basis. The 24-week double-blind treatment period was followed by a 52-week open-label extension study.
The co-primary efficacy end points were the change from baseline to end-of-study (week 24 of the double-blind treatment period) in the 1) Female Sexual Function Index (FSFI) desire domain score and 2) feeling bothered by low sexual desire as measured by Question 13 on the Female Sexual Distress Scale (FSDS). An increase in the FSFI desire domain score over time denotes improvement in sexual desire, while a decrease in the FSDS Question 13 score over time indicates improvement in the level of distress associated with low sexual desire.
In the 2 clinical studies, the mean change from baseline (SD) in the FSFI desire domain score, which ranged from 1.2 to 6.0 at study outset (higher scores indicate greater desire), was:
- study 1: 0.5 (1.1) in the bremelanotide-treated women and 0.2 (1.0) in the placebo-treated women (P = .0002)
- study 2: 0.6 (1.0) in the bremelanotide group versus 0.2 (0.9) in the placebo group (P<.0001).
For FSDS Question 13, for which the score range was 0 to 4 (higher scores indicate greater bother), the mean change from baseline score was:
- study 1: -0.7 (1.2) in the bremelanotide-treated group compared with -0.4 (1.1) in the placebo-treated group (P<.0001)
- study 2: -0.7 (1.1) in the bremelanotide group and -0.4 (1.1) in the placebo group (P = .0053).
It should be noted that, in the past, SSEs were used as a primary end point in clinical studies. However, we have shifted from SSEs to desire and distress as an end point because SSEs have little to do with desire. Women worry about and are distressed by the fact that they no longer have sexual appetite. They no longer "want to want" even though their body will be responsive and they can have an orgasm. That is exemplified by the woman in our case scenario (see box, page 18), who very much wants the experience of being able to anticipate with pleasure the idea of having an enjoyable connection with her partner.
Continue to: Physiologic target: The melanocortin receptor...
Physiologic target: The melanocortin receptor
Bremelanotide's theorized mechanism of action is that it works to rebalance neurotransmitters that are implicated in causing HSDD, acting as an agonist on the melanocortin receptor to promote dopamine release and allow women to perceive sexual cues as rewarding. They can then respond to those cues the way they used to and therefore experience desire. Flibanserin has affinity for serotonin (5-hydroxytryptamine [5-HT]) receptors, with agonist and antagonist activity, as well as moderate antagonist activity on some dopamine receptors.
The bottom line is that we now have treatments to address the underlying biologic aspect of HSDD, which is a biopsychosocial disorder. Again, this has parallels to depression and its biologic mechanism, for which we have effective treatments.
Dosing is an as-needed injection
Unlike the daily nighttime oral dose required with flibanserin, bremelanotide is a 1.75-mg dose administered as a subcutaneous injection (in either the thigh or the abdomen) with a pen-like autoinjector, on an as-needed basis. It should be administered at least 45 minutes before anticipated sexual activity. That is a benefit for many women who do not want to take a daily pill when they know that their "desire to desire" may be once per week or once every other week.
Regarding the drug delivery mode, nobody dropped out of the bremelanotide clinical trials because of having to take an injection with an autoinjector, which employs a very thin needle and is virtually painless. A small number of bremelanotide-treated women, about 13%, had injection site reactions (compared with 8% in the placebo group), which is common with subcutaneous injection. Even in the phase 2 clinical trial, in which a syringe was used to administer the drug, no participants discontinued the study because of the injection mode.
There are no clear pharmacokinetic data on how long bremelanotide's effects last, but it may be anywhere from 8 to 16 hours. Patients should not take more than 1 dose within 24 hours--but since the effect may last up to 16 hours that should not be a problem--and use of more than 8 doses per month is not recommended.
While bremelanotide improves desire, certainly better than placebo, there is also some peripheral improvement in arousal, although women in the trials had only HSDD. We do not know whether bremelanotide would treat arousal disorder, but it will help women with or without arousal difficulties associated with their HSDD, as shown in a subgroup analysis in the trials.13
Counsel patients on treatment potentialities
Clinicians should be aware of several precautions with bremelanotide use.
Blood pressure increases. After each dose of bremelanotide, transient increases in blood pressure (6 mm Hg in systolic and 3 mm Hg in diastolic blood pressure) and reductions in heart rate (up to 5 beats per minute) occur; these measurements return to baseline usually within 12 hours postdose.9 When you think about whether having sexual desire will increase blood pressure, this may be physiologic. It is similar to walking up a flight of stairs.
The drug is not recommended, however, for use in patients at high risk for cardiovascular disease, and it is contraindicated in women with uncontrolled hypertension or known cardiovascular disease. Blood pressure should be well controlled before bremelanotide is initiated--use of antihypertensive agents is not contraindicated with bremelanotide as the drugs do not interact.
Clinicians are not required to participate in a Risk Evaluation and Mitigation Strategy (REMS) program to prescribe bremelanotide as they are with flibanserin (because of the increased risk of severe hypotension and syncope due to flibanserin's interaction with alcohol).
Drug interactions. Bremelanotide is a melanocortin receptor agonist--a unique compound. Antidepressants, other psychoactive medications, and oral contraceptives are not contraindicated with bremelanotide as there are no known interactions. Alcohol use also is not a contraindication or caution, in contrast to flibanserin. (In April, the FDA issued a labeling change order for flibanserin, specifying that alcohol does not have to be avoided completely when taking flibanserin, but that women should discontinue drinking alcohol at least 2 hours before taking the drug at bedtime, or skip the flibanserin dose that evening.14) Bremelanotide may slow gastric emptying, though, so when a patient is taking oral drugs that require threshold concentrations for efficacy, such as antibiotics, they should avoid bremelanotide. In addition, some drugs, such as indomethacin, may have a delayed onset of action with concomitant bremelanotide use.9
Importantly, patients should avoid using bremelanotide if they are taking an oral naltrexone product for treatment of alcohol or opioid addiction, because bremelanotide may decrease systemic exposure of oral naltrexone. That would potentially lead to naltrexone treatment failure and its consequences.9
Skin pigmentation changes. Hyperpigmentation occurred with bremelanotide use on the face, gingiva, and breasts, as reported in the clinical trials, in 1% of treated patients who received up to 8 doses per month, compared with no such occurrences in placebo-treated patients. In addition, 38% of patients who received bremelanotide daily for 8 days developed focal hyperpigmentation. It was not confirmed in all patients whether the hyperpigmentation resolved. Women with dark skin were more likely to develop hyperpigmentation.9
Common adverse reactions. The most common adverse reactions with bremelanotide treatment are nausea, flushing, injection site reactions, and headache, with most events being mild to moderate in intensity. In the clinical trials, 40% of the bremelanotide-treated women experienced nausea (compared with 1% of placebo-treated women), with most occurrences being mild; for most participants nausea improved with the second dose. Women had nausea that either went away or was intermittent, or it was mild enough that the drug benefits outweighed the tolerability costs--of women who experienced nausea, 92% continued in the trial, and 8% dropped out because of nausea.9
The following scenario describes the experience of HSDD in one of Dr. Kingsberg's patients.
CASE Woman avoids sex because of low desire; marriage is suffering
A 40-year-old woman, Sandra, who has been married for 19 years and has fraternal twins aged 8, presented to the behavioral medicine clinic with distressing symptoms of low sexual desire. For several years into the marriage the patient experienced excellent sex drive. After 6 to 7 years, she noticed that her desire had declined and that she was starting to avoid sex. She was irritated when her husband initiated sex, and she would make excuses as to why it was not the right time.
Her husband felt hurt, frustrated, and rejected. The couple was close to divorce because he was angry and resentful. Sandra recognized there was a problem but did not know how to fix it. She could not understand why her interest had waned since she still loved her husband and considered him objectively very attractive.
Sandra came to see Dr. Kingsberg at the behavioral medicine clinic. Using the 5-item validated diagnostic tool called the Decreased Sexual Desire Screener, Dr. Kingsberg diagnosed hypoactive sexual desire disorder (HSDD), a term Sandra had never heard of and did not know was a condition. The patient was relieved to know that she was one of several million women affected by HSDD and that the problem was not just that she was a "bad wife" or that she had some kind of psychological block. She emphasized how much she loved her husband and how she wanted desperately to "want to want desire," as she recalled feeling previously.
Sandra was treated with counseling and psychotherapy in which we addressed the relationship issues, the avoidance of sex, the comfort with being sexual, and the recognition that responsive desire can be helpful (as she was able to have arousal and orgasm and have a satisfying sexual event). The issue was that she had no motivation to seek out sex and had no interest in experiencing that pleasure. In subsequent couple's therapy, the husband recognized that his wife was not intentionally rejecting him, but that she had a real medical condition.
Although Sandra's relationship was now more stable and she and her husband were both working toward finding a solution to Sandra's loss of desire, she was still very distressed by her lack of desire. Sandra tried flibanserin for 3 months but unfortunately did not respond. Sandra heard about the recent approval of bremelanotide and is looking forward to the drug being available so that she can try it.
Final considerations
Asking patients about sexual function and using sexual function screening tools can help clinicians identify patients with the decreased sexual desire and associated distress characteristic of HSDD. ObGyns are the appropriate clinicians to treat these women and soon will have 2 pharmacologic options--bremelanotide (anticipated to be available in Fall 2019) and flibanserin--to offer patients with this biopsychosocial disorder that can adversely impact well-being and quality of life. Clinicians should individualize treatment, which may include psychotherapeutic counseling, and counsel patients on appropriate drug use and potential adverse effects.
AMAG Pharmaceuticals, Inc. has announced that they will have a copay assistance program for bremelanotide, where the first prescription of four autoinjectors will be a $0 copay, followed by a $99 copay or less for refills.15
- Shifren JL, Monz BU, Russo PA, et al. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol. 2008;112:970-978.
- West SL, D'Aloisio AA, Agans RP, et al. Prevalence of low sexual desire and hypoactive sexual desire disorder in a nationally representative sample of US women. Arch Intern Med. 2008;168:1441-1449.
- Parish SJ, Goldstein AT, Goldstein SW, et al. Toward a more evidence-based nosology and nomenclature for female sexual dysfunctions: part II. J Sex Med. 2016;13:1888-1906.
- Basson R. Using a different model for female sexual response to address women's problematic low sexual desire. J Sex Marital Ther. 2001;27:395-403.
- Clayton AH, Goldfischer ER, Goldstein I, et al. Validation of the Decreased Sexual Desire Screener (DSDS): a brief diagnostic instrument for generalized acquired female hypoactive sexual desire disorder (HSDD). J Sex Med. 2009;6:730-738.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins-Gynecology. ACOG practice bulletin no. 213: Female sexual dysfunction. Obstet Gynecol. 2019;134:e1-e18.
- Goldstein I, Kim NN, Clayton AH, et al. Hypoactive sexual desire disorder: International Society for the Study of Women's Sexual Health (ISSWSH) expert consensus panel review. Mayo Clin Proc. 2017;92:114-128.
- Clayton AH, Goldstein I, Kim NN, et al. The International Society for the Study of Women's Sexual Health process of care for management of hypoactive sexual desire disorder in women. Mayo Clin Proc. 2018;93:467-487.
- Vyleesi [package insert]. Waltham, MA: AMAG Pharmaceuticals; 2019.
- Simon JA, Kingsberg SA, Shumel B, et al. Efficacy and safety of flibanserin in postmenopausal women with hypoactive sexual desire disorder: results of the SNOWDROP trial. Menopause. 2014;21;633-640.
- Portman DJ, Brown L, Yuan, et al. Flibanserin in postmenopausal women with hypoactive sexual desire disorder: results of the PLUMERIA study. J Sex Med. 2017;14:834-842.
- Kingsberg SA, Clayton AH, Portman D, et al. Bremelanotide for the treatment of hypoactive sexual desire disorder: two randomized phase 3 trials. Obstet Gynecol. Forthcoming.
- Clayton AH, Lucas J, Jordon R, et al. Efficacy of the Investigational drug bremelanotide in the Reconnect studies. Poster presented at: 30th ECNP Congress of Applied and Translational Neuroscience; September 2-5, 2017, Paris, France.
- US Food and Drug Administration. FDA orders important safety labeling changes for Addyi [press release]. April 11, 2019. https://www.fda.gov/news-events/press-announcements/fda-orders-important-safety-labeling-changes-addyi. Accessed July 17, 2019.
- Vyleesi website (https://vyleesipro.com). Accessed August 5, 2019.
- Shifren JL, Monz BU, Russo PA, et al. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol. 2008;112:970-978.
- West SL, D'Aloisio AA, Agans RP, et al. Prevalence of low sexual desire and hypoactive sexual desire disorder in a nationally representative sample of US women. Arch Intern Med. 2008;168:1441-1449.
- Parish SJ, Goldstein AT, Goldstein SW, et al. Toward a more evidence-based nosology and nomenclature for female sexual dysfunctions: part II. J Sex Med. 2016;13:1888-1906.
- Basson R. Using a different model for female sexual response to address women's problematic low sexual desire. J Sex Marital Ther. 2001;27:395-403.
- Clayton AH, Goldfischer ER, Goldstein I, et al. Validation of the Decreased Sexual Desire Screener (DSDS): a brief diagnostic instrument for generalized acquired female hypoactive sexual desire disorder (HSDD). J Sex Med. 2009;6:730-738.
- American College of Obstetricians and Gynecologists Committee on Practice Bulletins-Gynecology. ACOG practice bulletin no. 213: Female sexual dysfunction. Obstet Gynecol. 2019;134:e1-e18.
- Goldstein I, Kim NN, Clayton AH, et al. Hypoactive sexual desire disorder: International Society for the Study of Women's Sexual Health (ISSWSH) expert consensus panel review. Mayo Clin Proc. 2017;92:114-128.
- Clayton AH, Goldstein I, Kim NN, et al. The International Society for the Study of Women's Sexual Health process of care for management of hypoactive sexual desire disorder in women. Mayo Clin Proc. 2018;93:467-487.
- Vyleesi [package insert]. Waltham, MA: AMAG Pharmaceuticals; 2019.
- Simon JA, Kingsberg SA, Shumel B, et al. Efficacy and safety of flibanserin in postmenopausal women with hypoactive sexual desire disorder: results of the SNOWDROP trial. Menopause. 2014;21;633-640.
- Portman DJ, Brown L, Yuan, et al. Flibanserin in postmenopausal women with hypoactive sexual desire disorder: results of the PLUMERIA study. J Sex Med. 2017;14:834-842.
- Kingsberg SA, Clayton AH, Portman D, et al. Bremelanotide for the treatment of hypoactive sexual desire disorder: two randomized phase 3 trials. Obstet Gynecol. Forthcoming.
- Clayton AH, Lucas J, Jordon R, et al. Efficacy of the Investigational drug bremelanotide in the Reconnect studies. Poster presented at: 30th ECNP Congress of Applied and Translational Neuroscience; September 2-5, 2017, Paris, France.
- US Food and Drug Administration. FDA orders important safety labeling changes for Addyi [press release]. April 11, 2019. https://www.fda.gov/news-events/press-announcements/fda-orders-important-safety-labeling-changes-addyi. Accessed July 17, 2019.
- Vyleesi website (https://vyleesipro.com). Accessed August 5, 2019.
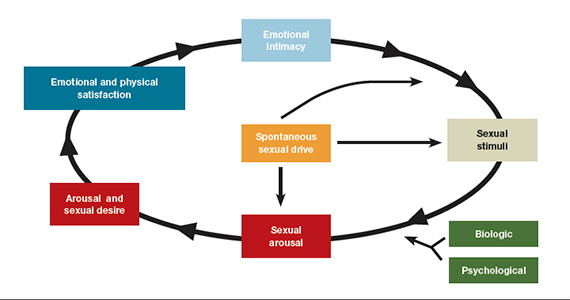
Morcellation use in gynecologic surgery: Current clinical recommendations and cautions
Morcellation of gynecologic surgical specimens became controversial after concerns arose about the potential for inadvertent spread of malignant cells throughout the abdomen and pelvis during tissue morcellation of suspected benign disease. In 2014, the US Food and Drug Administration (FDA) issued a warningagainst the use of laparoscopic power morcellation specifically for myomectomy or hysterectomy in the treatment of leiomyomas (fibroids) because of the risk of spreading undiagnosed malignancy throughout the abdomen and pelvis.1 This warning was issued after a high-profile case occurred in Boston in which an occult uterine sarcoma was morcellated during a supracervical robot-assisted hysterectomy for suspected benign fibroids.
Recently, the American College of Obstetricians and Gynecologists (ACOG) published a committee opinion with updated recommendations for practice detailing the risks associated with morcellation and suggestions for patient counseling regarding morcellation.2
In this review, we summarize the techniques and risks of morcellation, the epidemiology of undiagnosed uterine malignancies, practice changes noted at our institution, and clinical recommendations moving forward. A case scenario illustrates keys steps in preoperative evaluation and counseling.
Morcellation uses—and risks
Morcellation is the surgical process of dividing a large tissue specimen into smaller pieces to facilitate their removal through the small incisions made in minimally invasive surgery. Morcellation may be performed with a power instrument or manually.
In power morcellation, an electromechanical instrument is used to cut or shave the specimen; in manual morcellation, the surgeon uses a knife to carve the specimen. Power morcellation is performed through a laparoscopic incision, while the manual technique is performed through a minilaparotomy or vaginally after hysterectomy (TABLE). Unlike uncontained morcellation, contained morcellation involves the use of a laparoscopic bag to hold the specimen and therefore prevent tissue dissemination in the abdomen and pelvis.
Morcellation has greatly expanded our ability to perform minimally invasive surgery—for example, in patients with specimens that cannot be extracted en bloc through the vagina after hysterectomy or, in the case of myomectomy or supracervical hysterectomy without a colpotomy, through small laparoscopic ports. Minimally invasive surgery improves patient care, as it is associated with lower rates of infection, blood loss, venous thromboembolism, wound and bowel complications, postoperative pain, and shorter overall recovery time and hospital stay versus traditional open surgery.3,4 Furthermore, laparoscopic hysterectomy has a 3-fold lower risk of mortality compared with open hysterectomy.4 For these reasons, ACOG recommends choosing a minimally invasive approach for all benign hysterectomies whenever feasible.3
With abundant data supporting the use of a minimally invasive approach, laparoscopic morcellation allowed procedures involving larger tissue specimens to be accomplished without the addition of a minilaparotomy for tissue extraction. However, disseminating potentially malignant tissue throughout the abdomen and pelvis during the morcellation process remains a risk. While tissue spread can occur with either power or manual morcellation, the case that drew media attention to the controversy used power morcellation, and thus intense scrutiny focused on this technique. Morcellation has additional risks, including direct injury to surrounding organs, disruption of the pathologic specimen, and distribution of benign tissue throughout the abdomen and pelvis, such as fibroid, endometriosis, and adenomyosis implants.5-7

Continue to: The challenge of leiomyosarcoma...
The challenge of leiomyosarcoma
The primary controversy surrounding morcellation of fibroid tissue specimens is the potential for undiagnosed malignancy, namely uterine leiomyosarcoma or endometrial stromal sarcoma. While other gynecologic malignancies, including cervical and endometrial cancers, are more common and potentially could be disseminated by morcellation, these cancers are more reliably diagnosed preoperatively with cervical and endometrial biopsies, and they do not tend to mimic benign diseases.
Epidemiology and risk factors. Uterine leiomyosarcoma is rare, with an estimated incidence of 0.36 per 100,000 woman-years.8 However, leiomyosarcoma can mimic the appearance and clinical course of benign fibroids, making preoperative diagnosis difficult. Risk factors for leiomyosarcoma include postmenopausal status, with a median age of 54 years at diagnosis, tamoxifen use longer than 5 years, black race, history of pelvic radiation, and certain hereditary cancer syndromes, such as Lynch syndrome.9-11 Because of these risk factors, preoperative evaluation is crucial to determine the most appropriate surgical method for removal of a large, fibroid uterus (see “Employ shared decision making”).
Estimated incidence at benign hysterectomy. The incidence of leiomyosarcoma diagnosed at the time of benign hysterectomy or myomectomy has been studied extensively since the FDA’s 2014 warning was released, with varying rates identified.11,12 The FDA’s analysis cited a risk of 1 in 498 for unsuspected leiomyosarcoma and 1 in 352 for uterine sarcoma.1 Notably, this analysis excluded studies of women undergoing surgery for presumed fibroids in which no leiomyosarcoma was found on pathology, likely inflating the quoted prevalence. The FDA and other entities subsequently performed further analyses, but a systematic literature review and meta-analysis by the Agency for Healthcare Research and Quality (AHRQ) in 2017 is probably the most accurate. That review included 160 studies and reported a prevalence of less than 1 in 10,000 to 1 in 770, lower than the FDA-cited rate.13
Prognosis. The overall prognosis for women with leiomyosarcoma is poor. Studies indicate a 5-year survival rate of only 55.4%, even in stage 1 disease that is apparently confined to the uterus.9 Although evidence is limited linking morcellation to increased recurrence of leiomyosarcoma, data from small, single-center, retrospective studies cite a worse prognosis, higher risk of recurrence, and shorter progression-free survival after sarcoma morcellation compared with patients who underwent en bloc resection.12,14 Of note, these studies evaluated patients who underwent uncontained morcellation of specimens with unsuspected leiomyosarcoma.
CASE Woman with enlarged, irregular uterus and heavy bleeding
A 40-year-old woman (G2P2) with a history of 2 uncomplicated vaginal deliveries presents for evaluation of heavy uterine bleeding. She has regular periods, every 28 days, and she bleeds for 7 days, saturating 6 pads per day. She is currently taking only oral iron therapy as recommended by her primary care physician. Over the last 1 to 2 years she has felt that her abdomen has been getting larger and that her pants do not fit as well. She is otherwise in excellent health, exercises regularly, and has a full-time job. She has not been sexually active in several months.
The patient’s vitals are within normal limits and her body mass index (BMI) is 35 kg/m2.Pelvic examination reveals that she has an enlarged, irregular uterus with the fundus at the level of the umbilicus. The exam is otherwise unremarkable. On further questioning, the patient does not desire future fertility.
What next steps would you include in this patient’s workup, including imaging studies or lab tests? What surgical options would you give her? How would your management differ if this patient were 70 years old (postmenopausal)?
Continue to: Perform a thorough preoperative evaluation to optimize outcomes...
Perform a thorough preoperative evaluation to optimize outcomes
Women like this case patient who present with symptoms that may lead to treatment with myomectomy or hysterectomy should undergo appropriate preoperative testing to evaluate for malignancy.
According to ACOG guidance, patients should undergo a preoperative endometrial biopsy if they15:
- are older than 45 years with abnormal uterine bleeding
- are younger than 45 years with unopposed estrogen exposure (including obesity or polycystic ovary syndrome)
- have persistent bleeding, or
- failed medical management.
Our case patient is younger than 45 but is obese (BMI, 35) and therefore is a candidate for endometrial biopsy. Additionally, all patients should have up-to-date cervical cancer screening. ACOG also recommends appropriate use of imaging with ultrasonography or magnetic resonance imaging (MRI), although imaging is not recommended solely to evaluate for malignancy, as it cannot rule out the diagnosis of many gynecologic malignancies, including leiomyosarcoma.2
Currently, no tests are available to completely exclude a preoperative diagnosis of leiomyosarcoma. While studies have evaluated the use of MRI combined with lactate dehydrogenase isoenzyme testing, the evidence is weak, and this method is not recommended. Sarcoma is detected by endometrial sampling only 30% to 60% of the time, but it should be performed if the patient meets criteria for sampling or if she has other risk factors for malignancy.16 There are no data to support biopsy of presumed benign fibroids prior to surgical intervention. Patients should be evaluated with a careful history and physical examination for other uterine sarcoma risk factors.
Employ shared decision making
Clinicians should use shared decision making with patients to facilitate decisions on morcellation use in gynecologic surgeries for suspected benign fibroids. Informed consent must be obtained after thorough discussion and counseling regarding the literature on morcellation.17 For all patients, including the case patient described, this discussion should include alternative treatment options, surgical approach with associated risks, the use of morcellation, the incidence of leiomyosarcoma with presumed benign fibroids, leiomyosarcoma prognosis, and the risk of disseminating benign or undiagnosed cancerous tissue throughout the abdomen and pelvis.
Some would argue that the risks of laparotomy outweigh the possible risks associated with morcellation during a minimally invasive myomectomy or hysterectomy. However, this risk analysis is not uniform across all patients, and it is likely that in older women, because they have an a priori increased risk of malignancy in general, including leiomyosarcoma, the risks of power morcellation may outweigh the risks of open surgery.18 Younger women have a much lower risk of leiomyosarcoma, and thus discussion and consideration of the patient’s age should be a part of counseling. If the case patient described was 70 years of age, power morcellation might not be recommended, but these decisions require an in-depth discussion with the patient to make an informed decision and ensure patient autonomy.
The contained morcellation approach
Many surgeons who perform minimally invasive procedures use contained morcellation. In this approach, specimens are placed in a containment bag and morcellated with either power instruments or manually to ensure no dissemination of tissue. Manual contained morcellation can be done through a minilaparotomy or the vagina, depending on the procedure performed, while power contained morcellation is performed through a 15-mm laparoscopic incision.
Continue to: Currently, one containment bag has been...
Currently, one containment bag has been FDA approved for use in laparoscopic contained power morcellation.19 Use of a containment bag increases operative time by approximately 20 minutes, due to the additional steps required to accomplish the procedure.20 Its use, however, suggests a decrease in the risk of possible disease spread and it is feasible with appropriate surgeon training.
One study demonstrated the safety and feasibility of power morcellation within an insufflated containment bag, and subsequent follow-up revealed negative intraperitoneal washings.21,22 In another study evaluating tissue dissemination with contained morcellation of tissue stained with dye, the authors noted actual spillage of tissue fragments in only one case.23 Although more information is needed to confirm prevention of tissue dissemination and the safety of contained tissue morcellation, these studies provide promising data supporting the use of tissue morcellation in appropriate cases in order to perform minimally invasive surgery with larger specimens.
CASE Next steps and treatment outcome
The patient has up-to-date and negative cervical cancer screening. The complete blood count is notable for a hemoglobin level of 11.0 g/dL (normal range, 12.1 to 15.1 g/dL). You perform an endometrial biopsy; results are negative for malignancy. You order pelvic ultrasonography to better characterize the location and size of the fibroids. It shows multiple leiomyomas throughout the myometrium, with the 2 largest fibroids (measuring 5 and 7 cm) located in the left anterior and right posterolateral aspects of the uterus, respectively. Several 3- to 4-cm fibroids appear to be disrupting the endometrial canal, and there is no evidence of an endometrial polyp. There do not appear to be any cervical or lower uterine segment fibroids, which may have further complicated the proposed surgery.
You discuss treatment options for abnormal uterine bleeding with the patient, including initiation of combined oral contraceptive pills, placement of a levonorgestrel-containing intrauterine device, endometrial ablation, uterine artery embolization, and hysterectomy. You discuss the risks and benefits of each approach, keeping in mind the fibroids that are disrupting the contour of the endometrial canal and causing her bulk symptoms.
The patient ultimately decides to undergo a hysterectomy and would like it to be performed with a minimally invasive procedure, if possible. Because of the size of her uterus, you discuss the use of contained power morcellation, including the risks and benefits. You have a thorough discussion about the risk of occult malignancy, although she is at lower risk because of her age, and she consents.
The patient undergoes an uncomplicated total laparoscopic hysterectomy with bilateral salpingectomy. The specimen is removed using contained power morcellation through the umbilical port site. She has an unremarkable immediate postoperative course and is discharged on postoperative Day 1.
You see the patient in the clinic 2 weeks later. She reports minimal pain or discomfort and has no other complaints. Her abdominal incisions are healing well. You review the final pathology report with her, which showed no evidence of malignancy.
Society guidance on clinical applications
In current clinical practice, many surgeons have converted to exclusively performing contained morcellation in appropriate patients with a low risk of uterine leiomyosarcoma. At our institution, uncontained morcellation has not been performed since the FDA’s 2014 warning.
ACOG and AAGL (formerly the American Association of Gynecologic Laparoscopists) recommend use of containment bags as a solution to continue minimally invasive surgery for large specimens without the risk of possible tissue dissemination, although more in-depth surgeon training is likely required for accurate technique.2,24 The Society of Gynecologic Oncology (SGO) states that power morcellation or any other techniques that divide the uterus in the abdomen are contraindicated in patients with documented or highly suspected malignancy.25
With the presented data of risks associated with uncontained morcellation and agreement of the ACOG, AAGL, and SGO professional societies, we recommend that all morcellation be performed in a contained fashion to prevent the dissemination of benign or undiagnosed malignant tissue throughout the abdomen and pelvis. Shared decision making and counseling on the risks, benefits, and alternatives are paramount for patients to make informed decisions about their medical care. Continued exploration of techniques and methods for safe tissue extraction is still needed to improve minimally invasive surgical options for all women.
1. US Food and Drug Administration. Updated: Laparoscopic uterine power morcellation in hysterectomy and myomectomy: FDA safety communication. November 24, 2014; updated April 7, 2016. https://wayback.archiveit.org/7993/20170404182209/https:/www.fda.gov /MedicalDevices/Safety/AlertsandNotices/ucm424443.htm. Accessed July 23, 2019.
2. American College of Obstetricians and Gynecologists Committee on Gynecologic Practice. ACOG committee opinion no. 770: Uterine morcellation for presumed leiomyomas. Obstet Gynecol. 2019;133:e238-e248.
3. American College of Obstetricians and Gynecologists Committee on Gynecologic Practice. ACOG committee opinion no. 701: Choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2017;129:1149-1150.
4. Wiser A, Holcroft CA, Tolandi T, et al. Abdominal versus laparoscopic hysterectomies for benign diseases: evaluation of morbidity and mortality among 465,798 cases. Gynecol Surg. 2013;10:117-122.
5. Winner B, Biest S. Uterine morcellation: fact and fiction surrounding the recent controversy. Mo Med. 2017;114:176-180.
6. Tulandi T, Leung A, Jan N. Nonmalignant sequelae of unconfined morcellation at laparoscopic hysterectomy or myomectomy. J Minim Invasive Gynecol. 2016;23:331-337.
7. Milad MP, Milad EA. Laparoscopic morcellator-related complications. J Minim Invasive Gynecol. 2014;21:486-491.
8. Toro JR, Travis LB, Wu HJ, et al. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the Surveillance, Epidemiology and End Results program, 1978-2001: an analysis of 26,758 cases. Int J Cancer. 2006;119:2922-2930.
9. Seagle BL, Sobecki-Rausch J, Strohl AE, et al. Prognosis and treatment of uterine leiomyosarcoma: a National Cancer Database study. Gynecol Oncol. 2017;145:61-70.
10. Ricci S, Stone RL, Fader AN. Uterine leiomyosarcoma: epidemiology, contemporary treatment strategies and the impact of uterine morcellation. Gynecol Oncol. 2017;145:208-216.
11. Leibsohn S, d’Ablaing G, Mishell DR Jr, et al. Leiomyosarcoma in a series of hysterectomies performed for presumed uterine leiomyomas. Am J Obstet Gynecol. 1990;162:968-974. Discussion 974-976.
12. Rowland M, Lesnock J, Edwards R, et al. Occult uterine cancer in patients undergoing laparoscopic hysterectomy with morcellation [abstract]. Gynecol Oncol. 2012;127:S29.
13. Hartmann KE, Fonnesbeck C, Surawicz T, et al. Management of uterine fibroids. Comparative effectiveness review no. 195. AHRQ Publication No. 17(18)-EHC028-EF. Rockville, MD: Agency for Healthcare Research and Quality; 2017. https://effectivehealthcare.ahrq.gov/topics/uterine-fibroids /research-2017. Accessed July 23, 2019.
14. Pritts EA, Parker WH, Brown J, et al. Outcome of occult uterine leiomyosarcoma after surgery for presumed uterine fibroids: a systematic review. J Minim Invasive Gynecol. 2015;22:26-33.
15. American College of Obstetricians and Gynecologists Committee on Practice Bulletins–Gynecology. Practice bulletin no. 128: Diagnosis of abnormal uterine bleeding in reproductive-aged women. Obstet Gynecol. 2012;120:197-206.
16. Bansal N, Herzog TJ, Burke W, et al. The utility of preoperative endometrial sampling for the detection of uterine sarcomas. Gynecol Oncol. 2008 Jul;110(1):43–48.
17. American College of Obstetricians and Gynecologists Committee on Ethics. ACOG committee opinion no. 439: Informed consent. Obstet Gynecol. 2009;114:401-408.
18. Wright JD, Cui RR, Wang A, et al. Economic and survival implications of use of electric power morcellation for hysterectomy for presumed benign gynecologic disease. J Natl Cancer Inst. 2015;107:djv251.
19. US Food and Drug Administration. FDA allows marketing of first-of-kind tissue containment system for use with certain laparoscopic power morcellators in select patients [press release]. April 7, 2016. https://www.fda.gov/NewsEvents /Newsroom/PressAnnouncements/ucm494650.htm. Accessed July 23, 2019.
20. Winner B, Porter A, Velloze S, et al. S. Uncontained compared with contained power morcellation in total laparoscopic hysterectomy. Obstet Gynecol. 2015 Oct;126(4):834–8.
21. Cohen SL, Einarsson JI, Wang KC, et al. Contained power morcellation within an insufflated isolation bag. Obstet Gynecol. 2014;124:491-497.
22. Cohen SL, Greenberg JA, Wang KC, et al. Risk of leakage and tissue dissemination with various contained tissue extraction (CTE) techniques: an in vitro pilot study. J Minim Invasive Gynecol. 2014;21:935-939.
23. Cohen SL, Morris SN, Brown DN, et al. Contained tissue extraction using power morcellation: prospective evaluation of leakage parameters. Am J Obstet Gynecol. 2016;214(2):257. e1-257.e6.
24. AAGL. AAGL practice report: morcellation during uterine tissue extraction. J Minim Invasive Gynecol. 2014;21:517-530.
25. Society of Gynecologic Oncology. Position statement: morcellation. 2013. https://www.sgo.org/newsroom /position-statements-2/morcellation/.Accessed July 23, 2019.

Dr. Putman is Chief Resident, Department of Obstetrics and Gynecology, Barnes-Jewish Hospital/Washington University School of Medicine in St. Louis, St. Louis, Missouri.

Dr. Zamorano is Fellow, Division of Gynecologic Oncology, Department of Obstetrics and Gynecology, Washington University School of Medicine in St. Louis.

Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Vice Chair of Gynecology in the Division of Gynecologic Oncology, Department of Obstetrics and Gynecology, Washington University School of Medicine in St. Louis and Alvin J. Siteman Cancer Center. He serves on the OBG MANAGEMENT Board of Editors.
Dr. Mutch reports that he receives grant or research support from the National Institutes of Health and the GOG Foundation and that he is a consultant and speaker for Clovis and AstraZeneca. Dr. Putman and Dr. Zamorano report no financial relationships relevant to this article.
Dr. Putman is Chief Resident, Department of Obstetrics and Gynecology, Barnes-Jewish Hospital/Washington University School of Medicine in St. Louis, St. Louis, Missouri.
Dr. Zamorano is Fellow, Division of Gynecologic Oncology, Department of Obstetrics and Gynecology, Washington University School of Medicine in St. Louis.
Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Vice Chair of Gynecology in the Division of Gynecologic Oncology, Department of Obstetrics and Gynecology, Washington University School of Medicine in St. Louis and Alvin J. Siteman Cancer Center. He serves on the OBG MANAGEMENT Board of Editors.
Dr. Mutch reports that he receives grant or research support from the National Institutes of Health and the GOG Foundation and that he is a consultant and speaker for Clovis and AstraZeneca. Dr. Putman and Dr. Zamorano report no financial relationships relevant to this article.
Dr. Putman is Chief Resident, Department of Obstetrics and Gynecology, Barnes-Jewish Hospital/Washington University School of Medicine in St. Louis, St. Louis, Missouri.
Dr. Zamorano is Fellow, Division of Gynecologic Oncology, Department of Obstetrics and Gynecology, Washington University School of Medicine in St. Louis.
Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Vice Chair of Gynecology in the Division of Gynecologic Oncology, Department of Obstetrics and Gynecology, Washington University School of Medicine in St. Louis and Alvin J. Siteman Cancer Center. He serves on the OBG MANAGEMENT Board of Editors.
Dr. Mutch reports that he receives grant or research support from the National Institutes of Health and the GOG Foundation and that he is a consultant and speaker for Clovis and AstraZeneca. Dr. Putman and Dr. Zamorano report no financial relationships relevant to this article.
Morcellation of gynecologic surgical specimens became controversial after concerns arose about the potential for inadvertent spread of malignant cells throughout the abdomen and pelvis during tissue morcellation of suspected benign disease. In 2014, the US Food and Drug Administration (FDA) issued a warningagainst the use of laparoscopic power morcellation specifically for myomectomy or hysterectomy in the treatment of leiomyomas (fibroids) because of the risk of spreading undiagnosed malignancy throughout the abdomen and pelvis.1 This warning was issued after a high-profile case occurred in Boston in which an occult uterine sarcoma was morcellated during a supracervical robot-assisted hysterectomy for suspected benign fibroids.
Recently, the American College of Obstetricians and Gynecologists (ACOG) published a committee opinion with updated recommendations for practice detailing the risks associated with morcellation and suggestions for patient counseling regarding morcellation.2
In this review, we summarize the techniques and risks of morcellation, the epidemiology of undiagnosed uterine malignancies, practice changes noted at our institution, and clinical recommendations moving forward. A case scenario illustrates keys steps in preoperative evaluation and counseling.
Morcellation uses—and risks
Morcellation is the surgical process of dividing a large tissue specimen into smaller pieces to facilitate their removal through the small incisions made in minimally invasive surgery. Morcellation may be performed with a power instrument or manually.
In power morcellation, an electromechanical instrument is used to cut or shave the specimen; in manual morcellation, the surgeon uses a knife to carve the specimen. Power morcellation is performed through a laparoscopic incision, while the manual technique is performed through a minilaparotomy or vaginally after hysterectomy (TABLE). Unlike uncontained morcellation, contained morcellation involves the use of a laparoscopic bag to hold the specimen and therefore prevent tissue dissemination in the abdomen and pelvis.
Morcellation has greatly expanded our ability to perform minimally invasive surgery—for example, in patients with specimens that cannot be extracted en bloc through the vagina after hysterectomy or, in the case of myomectomy or supracervical hysterectomy without a colpotomy, through small laparoscopic ports. Minimally invasive surgery improves patient care, as it is associated with lower rates of infection, blood loss, venous thromboembolism, wound and bowel complications, postoperative pain, and shorter overall recovery time and hospital stay versus traditional open surgery.3,4 Furthermore, laparoscopic hysterectomy has a 3-fold lower risk of mortality compared with open hysterectomy.4 For these reasons, ACOG recommends choosing a minimally invasive approach for all benign hysterectomies whenever feasible.3
With abundant data supporting the use of a minimally invasive approach, laparoscopic morcellation allowed procedures involving larger tissue specimens to be accomplished without the addition of a minilaparotomy for tissue extraction. However, disseminating potentially malignant tissue throughout the abdomen and pelvis during the morcellation process remains a risk. While tissue spread can occur with either power or manual morcellation, the case that drew media attention to the controversy used power morcellation, and thus intense scrutiny focused on this technique. Morcellation has additional risks, including direct injury to surrounding organs, disruption of the pathologic specimen, and distribution of benign tissue throughout the abdomen and pelvis, such as fibroid, endometriosis, and adenomyosis implants.5-7
Continue to: The challenge of leiomyosarcoma...
The challenge of leiomyosarcoma
The primary controversy surrounding morcellation of fibroid tissue specimens is the potential for undiagnosed malignancy, namely uterine leiomyosarcoma or endometrial stromal sarcoma. While other gynecologic malignancies, including cervical and endometrial cancers, are more common and potentially could be disseminated by morcellation, these cancers are more reliably diagnosed preoperatively with cervical and endometrial biopsies, and they do not tend to mimic benign diseases.
Epidemiology and risk factors. Uterine leiomyosarcoma is rare, with an estimated incidence of 0.36 per 100,000 woman-years.8 However, leiomyosarcoma can mimic the appearance and clinical course of benign fibroids, making preoperative diagnosis difficult. Risk factors for leiomyosarcoma include postmenopausal status, with a median age of 54 years at diagnosis, tamoxifen use longer than 5 years, black race, history of pelvic radiation, and certain hereditary cancer syndromes, such as Lynch syndrome.9-11 Because of these risk factors, preoperative evaluation is crucial to determine the most appropriate surgical method for removal of a large, fibroid uterus (see “Employ shared decision making”).
Estimated incidence at benign hysterectomy. The incidence of leiomyosarcoma diagnosed at the time of benign hysterectomy or myomectomy has been studied extensively since the FDA’s 2014 warning was released, with varying rates identified.11,12 The FDA’s analysis cited a risk of 1 in 498 for unsuspected leiomyosarcoma and 1 in 352 for uterine sarcoma.1 Notably, this analysis excluded studies of women undergoing surgery for presumed fibroids in which no leiomyosarcoma was found on pathology, likely inflating the quoted prevalence. The FDA and other entities subsequently performed further analyses, but a systematic literature review and meta-analysis by the Agency for Healthcare Research and Quality (AHRQ) in 2017 is probably the most accurate. That review included 160 studies and reported a prevalence of less than 1 in 10,000 to 1 in 770, lower than the FDA-cited rate.13
Prognosis. The overall prognosis for women with leiomyosarcoma is poor. Studies indicate a 5-year survival rate of only 55.4%, even in stage 1 disease that is apparently confined to the uterus.9 Although evidence is limited linking morcellation to increased recurrence of leiomyosarcoma, data from small, single-center, retrospective studies cite a worse prognosis, higher risk of recurrence, and shorter progression-free survival after sarcoma morcellation compared with patients who underwent en bloc resection.12,14 Of note, these studies evaluated patients who underwent uncontained morcellation of specimens with unsuspected leiomyosarcoma.
CASE Woman with enlarged, irregular uterus and heavy bleeding
A 40-year-old woman (G2P2) with a history of 2 uncomplicated vaginal deliveries presents for evaluation of heavy uterine bleeding. She has regular periods, every 28 days, and she bleeds for 7 days, saturating 6 pads per day. She is currently taking only oral iron therapy as recommended by her primary care physician. Over the last 1 to 2 years she has felt that her abdomen has been getting larger and that her pants do not fit as well. She is otherwise in excellent health, exercises regularly, and has a full-time job. She has not been sexually active in several months.
The patient’s vitals are within normal limits and her body mass index (BMI) is 35 kg/m2.Pelvic examination reveals that she has an enlarged, irregular uterus with the fundus at the level of the umbilicus. The exam is otherwise unremarkable. On further questioning, the patient does not desire future fertility.
What next steps would you include in this patient’s workup, including imaging studies or lab tests? What surgical options would you give her? How would your management differ if this patient were 70 years old (postmenopausal)?
Continue to: Perform a thorough preoperative evaluation to optimize outcomes...
Perform a thorough preoperative evaluation to optimize outcomes
Women like this case patient who present with symptoms that may lead to treatment with myomectomy or hysterectomy should undergo appropriate preoperative testing to evaluate for malignancy.
According to ACOG guidance, patients should undergo a preoperative endometrial biopsy if they15:
- are older than 45 years with abnormal uterine bleeding
- are younger than 45 years with unopposed estrogen exposure (including obesity or polycystic ovary syndrome)
- have persistent bleeding, or
- failed medical management.
Our case patient is younger than 45 but is obese (BMI, 35) and therefore is a candidate for endometrial biopsy. Additionally, all patients should have up-to-date cervical cancer screening. ACOG also recommends appropriate use of imaging with ultrasonography or magnetic resonance imaging (MRI), although imaging is not recommended solely to evaluate for malignancy, as it cannot rule out the diagnosis of many gynecologic malignancies, including leiomyosarcoma.2
Currently, no tests are available to completely exclude a preoperative diagnosis of leiomyosarcoma. While studies have evaluated the use of MRI combined with lactate dehydrogenase isoenzyme testing, the evidence is weak, and this method is not recommended. Sarcoma is detected by endometrial sampling only 30% to 60% of the time, but it should be performed if the patient meets criteria for sampling or if she has other risk factors for malignancy.16 There are no data to support biopsy of presumed benign fibroids prior to surgical intervention. Patients should be evaluated with a careful history and physical examination for other uterine sarcoma risk factors.
Employ shared decision making
Clinicians should use shared decision making with patients to facilitate decisions on morcellation use in gynecologic surgeries for suspected benign fibroids. Informed consent must be obtained after thorough discussion and counseling regarding the literature on morcellation.17 For all patients, including the case patient described, this discussion should include alternative treatment options, surgical approach with associated risks, the use of morcellation, the incidence of leiomyosarcoma with presumed benign fibroids, leiomyosarcoma prognosis, and the risk of disseminating benign or undiagnosed cancerous tissue throughout the abdomen and pelvis.
Some would argue that the risks of laparotomy outweigh the possible risks associated with morcellation during a minimally invasive myomectomy or hysterectomy. However, this risk analysis is not uniform across all patients, and it is likely that in older women, because they have an a priori increased risk of malignancy in general, including leiomyosarcoma, the risks of power morcellation may outweigh the risks of open surgery.18 Younger women have a much lower risk of leiomyosarcoma, and thus discussion and consideration of the patient’s age should be a part of counseling. If the case patient described was 70 years of age, power morcellation might not be recommended, but these decisions require an in-depth discussion with the patient to make an informed decision and ensure patient autonomy.
The contained morcellation approach
Many surgeons who perform minimally invasive procedures use contained morcellation. In this approach, specimens are placed in a containment bag and morcellated with either power instruments or manually to ensure no dissemination of tissue. Manual contained morcellation can be done through a minilaparotomy or the vagina, depending on the procedure performed, while power contained morcellation is performed through a 15-mm laparoscopic incision.
Continue to: Currently, one containment bag has been...
Currently, one containment bag has been FDA approved for use in laparoscopic contained power morcellation.19 Use of a containment bag increases operative time by approximately 20 minutes, due to the additional steps required to accomplish the procedure.20 Its use, however, suggests a decrease in the risk of possible disease spread and it is feasible with appropriate surgeon training.
One study demonstrated the safety and feasibility of power morcellation within an insufflated containment bag, and subsequent follow-up revealed negative intraperitoneal washings.21,22 In another study evaluating tissue dissemination with contained morcellation of tissue stained with dye, the authors noted actual spillage of tissue fragments in only one case.23 Although more information is needed to confirm prevention of tissue dissemination and the safety of contained tissue morcellation, these studies provide promising data supporting the use of tissue morcellation in appropriate cases in order to perform minimally invasive surgery with larger specimens.
CASE Next steps and treatment outcome
The patient has up-to-date and negative cervical cancer screening. The complete blood count is notable for a hemoglobin level of 11.0 g/dL (normal range, 12.1 to 15.1 g/dL). You perform an endometrial biopsy; results are negative for malignancy. You order pelvic ultrasonography to better characterize the location and size of the fibroids. It shows multiple leiomyomas throughout the myometrium, with the 2 largest fibroids (measuring 5 and 7 cm) located in the left anterior and right posterolateral aspects of the uterus, respectively. Several 3- to 4-cm fibroids appear to be disrupting the endometrial canal, and there is no evidence of an endometrial polyp. There do not appear to be any cervical or lower uterine segment fibroids, which may have further complicated the proposed surgery.
You discuss treatment options for abnormal uterine bleeding with the patient, including initiation of combined oral contraceptive pills, placement of a levonorgestrel-containing intrauterine device, endometrial ablation, uterine artery embolization, and hysterectomy. You discuss the risks and benefits of each approach, keeping in mind the fibroids that are disrupting the contour of the endometrial canal and causing her bulk symptoms.
The patient ultimately decides to undergo a hysterectomy and would like it to be performed with a minimally invasive procedure, if possible. Because of the size of her uterus, you discuss the use of contained power morcellation, including the risks and benefits. You have a thorough discussion about the risk of occult malignancy, although she is at lower risk because of her age, and she consents.
The patient undergoes an uncomplicated total laparoscopic hysterectomy with bilateral salpingectomy. The specimen is removed using contained power morcellation through the umbilical port site. She has an unremarkable immediate postoperative course and is discharged on postoperative Day 1.
You see the patient in the clinic 2 weeks later. She reports minimal pain or discomfort and has no other complaints. Her abdominal incisions are healing well. You review the final pathology report with her, which showed no evidence of malignancy.
Society guidance on clinical applications
In current clinical practice, many surgeons have converted to exclusively performing contained morcellation in appropriate patients with a low risk of uterine leiomyosarcoma. At our institution, uncontained morcellation has not been performed since the FDA’s 2014 warning.
ACOG and AAGL (formerly the American Association of Gynecologic Laparoscopists) recommend use of containment bags as a solution to continue minimally invasive surgery for large specimens without the risk of possible tissue dissemination, although more in-depth surgeon training is likely required for accurate technique.2,24 The Society of Gynecologic Oncology (SGO) states that power morcellation or any other techniques that divide the uterus in the abdomen are contraindicated in patients with documented or highly suspected malignancy.25
With the presented data of risks associated with uncontained morcellation and agreement of the ACOG, AAGL, and SGO professional societies, we recommend that all morcellation be performed in a contained fashion to prevent the dissemination of benign or undiagnosed malignant tissue throughout the abdomen and pelvis. Shared decision making and counseling on the risks, benefits, and alternatives are paramount for patients to make informed decisions about their medical care. Continued exploration of techniques and methods for safe tissue extraction is still needed to improve minimally invasive surgical options for all women.
Morcellation of gynecologic surgical specimens became controversial after concerns arose about the potential for inadvertent spread of malignant cells throughout the abdomen and pelvis during tissue morcellation of suspected benign disease. In 2014, the US Food and Drug Administration (FDA) issued a warningagainst the use of laparoscopic power morcellation specifically for myomectomy or hysterectomy in the treatment of leiomyomas (fibroids) because of the risk of spreading undiagnosed malignancy throughout the abdomen and pelvis.1 This warning was issued after a high-profile case occurred in Boston in which an occult uterine sarcoma was morcellated during a supracervical robot-assisted hysterectomy for suspected benign fibroids.
Recently, the American College of Obstetricians and Gynecologists (ACOG) published a committee opinion with updated recommendations for practice detailing the risks associated with morcellation and suggestions for patient counseling regarding morcellation.2
In this review, we summarize the techniques and risks of morcellation, the epidemiology of undiagnosed uterine malignancies, practice changes noted at our institution, and clinical recommendations moving forward. A case scenario illustrates keys steps in preoperative evaluation and counseling.
Morcellation uses—and risks
Morcellation is the surgical process of dividing a large tissue specimen into smaller pieces to facilitate their removal through the small incisions made in minimally invasive surgery. Morcellation may be performed with a power instrument or manually.
In power morcellation, an electromechanical instrument is used to cut or shave the specimen; in manual morcellation, the surgeon uses a knife to carve the specimen. Power morcellation is performed through a laparoscopic incision, while the manual technique is performed through a minilaparotomy or vaginally after hysterectomy (TABLE). Unlike uncontained morcellation, contained morcellation involves the use of a laparoscopic bag to hold the specimen and therefore prevent tissue dissemination in the abdomen and pelvis.
Morcellation has greatly expanded our ability to perform minimally invasive surgery—for example, in patients with specimens that cannot be extracted en bloc through the vagina after hysterectomy or, in the case of myomectomy or supracervical hysterectomy without a colpotomy, through small laparoscopic ports. Minimally invasive surgery improves patient care, as it is associated with lower rates of infection, blood loss, venous thromboembolism, wound and bowel complications, postoperative pain, and shorter overall recovery time and hospital stay versus traditional open surgery.3,4 Furthermore, laparoscopic hysterectomy has a 3-fold lower risk of mortality compared with open hysterectomy.4 For these reasons, ACOG recommends choosing a minimally invasive approach for all benign hysterectomies whenever feasible.3
With abundant data supporting the use of a minimally invasive approach, laparoscopic morcellation allowed procedures involving larger tissue specimens to be accomplished without the addition of a minilaparotomy for tissue extraction. However, disseminating potentially malignant tissue throughout the abdomen and pelvis during the morcellation process remains a risk. While tissue spread can occur with either power or manual morcellation, the case that drew media attention to the controversy used power morcellation, and thus intense scrutiny focused on this technique. Morcellation has additional risks, including direct injury to surrounding organs, disruption of the pathologic specimen, and distribution of benign tissue throughout the abdomen and pelvis, such as fibroid, endometriosis, and adenomyosis implants.5-7
Continue to: The challenge of leiomyosarcoma...
The challenge of leiomyosarcoma
The primary controversy surrounding morcellation of fibroid tissue specimens is the potential for undiagnosed malignancy, namely uterine leiomyosarcoma or endometrial stromal sarcoma. While other gynecologic malignancies, including cervical and endometrial cancers, are more common and potentially could be disseminated by morcellation, these cancers are more reliably diagnosed preoperatively with cervical and endometrial biopsies, and they do not tend to mimic benign diseases.
Epidemiology and risk factors. Uterine leiomyosarcoma is rare, with an estimated incidence of 0.36 per 100,000 woman-years.8 However, leiomyosarcoma can mimic the appearance and clinical course of benign fibroids, making preoperative diagnosis difficult. Risk factors for leiomyosarcoma include postmenopausal status, with a median age of 54 years at diagnosis, tamoxifen use longer than 5 years, black race, history of pelvic radiation, and certain hereditary cancer syndromes, such as Lynch syndrome.9-11 Because of these risk factors, preoperative evaluation is crucial to determine the most appropriate surgical method for removal of a large, fibroid uterus (see “Employ shared decision making”).
Estimated incidence at benign hysterectomy. The incidence of leiomyosarcoma diagnosed at the time of benign hysterectomy or myomectomy has been studied extensively since the FDA’s 2014 warning was released, with varying rates identified.11,12 The FDA’s analysis cited a risk of 1 in 498 for unsuspected leiomyosarcoma and 1 in 352 for uterine sarcoma.1 Notably, this analysis excluded studies of women undergoing surgery for presumed fibroids in which no leiomyosarcoma was found on pathology, likely inflating the quoted prevalence. The FDA and other entities subsequently performed further analyses, but a systematic literature review and meta-analysis by the Agency for Healthcare Research and Quality (AHRQ) in 2017 is probably the most accurate. That review included 160 studies and reported a prevalence of less than 1 in 10,000 to 1 in 770, lower than the FDA-cited rate.13
Prognosis. The overall prognosis for women with leiomyosarcoma is poor. Studies indicate a 5-year survival rate of only 55.4%, even in stage 1 disease that is apparently confined to the uterus.9 Although evidence is limited linking morcellation to increased recurrence of leiomyosarcoma, data from small, single-center, retrospective studies cite a worse prognosis, higher risk of recurrence, and shorter progression-free survival after sarcoma morcellation compared with patients who underwent en bloc resection.12,14 Of note, these studies evaluated patients who underwent uncontained morcellation of specimens with unsuspected leiomyosarcoma.
CASE Woman with enlarged, irregular uterus and heavy bleeding
A 40-year-old woman (G2P2) with a history of 2 uncomplicated vaginal deliveries presents for evaluation of heavy uterine bleeding. She has regular periods, every 28 days, and she bleeds for 7 days, saturating 6 pads per day. She is currently taking only oral iron therapy as recommended by her primary care physician. Over the last 1 to 2 years she has felt that her abdomen has been getting larger and that her pants do not fit as well. She is otherwise in excellent health, exercises regularly, and has a full-time job. She has not been sexually active in several months.
The patient’s vitals are within normal limits and her body mass index (BMI) is 35 kg/m2.Pelvic examination reveals that she has an enlarged, irregular uterus with the fundus at the level of the umbilicus. The exam is otherwise unremarkable. On further questioning, the patient does not desire future fertility.
What next steps would you include in this patient’s workup, including imaging studies or lab tests? What surgical options would you give her? How would your management differ if this patient were 70 years old (postmenopausal)?
Continue to: Perform a thorough preoperative evaluation to optimize outcomes...
Perform a thorough preoperative evaluation to optimize outcomes
Women like this case patient who present with symptoms that may lead to treatment with myomectomy or hysterectomy should undergo appropriate preoperative testing to evaluate for malignancy.
According to ACOG guidance, patients should undergo a preoperative endometrial biopsy if they15:
- are older than 45 years with abnormal uterine bleeding
- are younger than 45 years with unopposed estrogen exposure (including obesity or polycystic ovary syndrome)
- have persistent bleeding, or
- failed medical management.
Our case patient is younger than 45 but is obese (BMI, 35) and therefore is a candidate for endometrial biopsy. Additionally, all patients should have up-to-date cervical cancer screening. ACOG also recommends appropriate use of imaging with ultrasonography or magnetic resonance imaging (MRI), although imaging is not recommended solely to evaluate for malignancy, as it cannot rule out the diagnosis of many gynecologic malignancies, including leiomyosarcoma.2
Currently, no tests are available to completely exclude a preoperative diagnosis of leiomyosarcoma. While studies have evaluated the use of MRI combined with lactate dehydrogenase isoenzyme testing, the evidence is weak, and this method is not recommended. Sarcoma is detected by endometrial sampling only 30% to 60% of the time, but it should be performed if the patient meets criteria for sampling or if she has other risk factors for malignancy.16 There are no data to support biopsy of presumed benign fibroids prior to surgical intervention. Patients should be evaluated with a careful history and physical examination for other uterine sarcoma risk factors.
Employ shared decision making
Clinicians should use shared decision making with patients to facilitate decisions on morcellation use in gynecologic surgeries for suspected benign fibroids. Informed consent must be obtained after thorough discussion and counseling regarding the literature on morcellation.17 For all patients, including the case patient described, this discussion should include alternative treatment options, surgical approach with associated risks, the use of morcellation, the incidence of leiomyosarcoma with presumed benign fibroids, leiomyosarcoma prognosis, and the risk of disseminating benign or undiagnosed cancerous tissue throughout the abdomen and pelvis.
Some would argue that the risks of laparotomy outweigh the possible risks associated with morcellation during a minimally invasive myomectomy or hysterectomy. However, this risk analysis is not uniform across all patients, and it is likely that in older women, because they have an a priori increased risk of malignancy in general, including leiomyosarcoma, the risks of power morcellation may outweigh the risks of open surgery.18 Younger women have a much lower risk of leiomyosarcoma, and thus discussion and consideration of the patient’s age should be a part of counseling. If the case patient described was 70 years of age, power morcellation might not be recommended, but these decisions require an in-depth discussion with the patient to make an informed decision and ensure patient autonomy.
The contained morcellation approach
Many surgeons who perform minimally invasive procedures use contained morcellation. In this approach, specimens are placed in a containment bag and morcellated with either power instruments or manually to ensure no dissemination of tissue. Manual contained morcellation can be done through a minilaparotomy or the vagina, depending on the procedure performed, while power contained morcellation is performed through a 15-mm laparoscopic incision.
Continue to: Currently, one containment bag has been...
Currently, one containment bag has been FDA approved for use in laparoscopic contained power morcellation.19 Use of a containment bag increases operative time by approximately 20 minutes, due to the additional steps required to accomplish the procedure.20 Its use, however, suggests a decrease in the risk of possible disease spread and it is feasible with appropriate surgeon training.
One study demonstrated the safety and feasibility of power morcellation within an insufflated containment bag, and subsequent follow-up revealed negative intraperitoneal washings.21,22 In another study evaluating tissue dissemination with contained morcellation of tissue stained with dye, the authors noted actual spillage of tissue fragments in only one case.23 Although more information is needed to confirm prevention of tissue dissemination and the safety of contained tissue morcellation, these studies provide promising data supporting the use of tissue morcellation in appropriate cases in order to perform minimally invasive surgery with larger specimens.
CASE Next steps and treatment outcome
The patient has up-to-date and negative cervical cancer screening. The complete blood count is notable for a hemoglobin level of 11.0 g/dL (normal range, 12.1 to 15.1 g/dL). You perform an endometrial biopsy; results are negative for malignancy. You order pelvic ultrasonography to better characterize the location and size of the fibroids. It shows multiple leiomyomas throughout the myometrium, with the 2 largest fibroids (measuring 5 and 7 cm) located in the left anterior and right posterolateral aspects of the uterus, respectively. Several 3- to 4-cm fibroids appear to be disrupting the endometrial canal, and there is no evidence of an endometrial polyp. There do not appear to be any cervical or lower uterine segment fibroids, which may have further complicated the proposed surgery.
You discuss treatment options for abnormal uterine bleeding with the patient, including initiation of combined oral contraceptive pills, placement of a levonorgestrel-containing intrauterine device, endometrial ablation, uterine artery embolization, and hysterectomy. You discuss the risks and benefits of each approach, keeping in mind the fibroids that are disrupting the contour of the endometrial canal and causing her bulk symptoms.
The patient ultimately decides to undergo a hysterectomy and would like it to be performed with a minimally invasive procedure, if possible. Because of the size of her uterus, you discuss the use of contained power morcellation, including the risks and benefits. You have a thorough discussion about the risk of occult malignancy, although she is at lower risk because of her age, and she consents.
The patient undergoes an uncomplicated total laparoscopic hysterectomy with bilateral salpingectomy. The specimen is removed using contained power morcellation through the umbilical port site. She has an unremarkable immediate postoperative course and is discharged on postoperative Day 1.
You see the patient in the clinic 2 weeks later. She reports minimal pain or discomfort and has no other complaints. Her abdominal incisions are healing well. You review the final pathology report with her, which showed no evidence of malignancy.
Society guidance on clinical applications
In current clinical practice, many surgeons have converted to exclusively performing contained morcellation in appropriate patients with a low risk of uterine leiomyosarcoma. At our institution, uncontained morcellation has not been performed since the FDA’s 2014 warning.
ACOG and AAGL (formerly the American Association of Gynecologic Laparoscopists) recommend use of containment bags as a solution to continue minimally invasive surgery for large specimens without the risk of possible tissue dissemination, although more in-depth surgeon training is likely required for accurate technique.2,24 The Society of Gynecologic Oncology (SGO) states that power morcellation or any other techniques that divide the uterus in the abdomen are contraindicated in patients with documented or highly suspected malignancy.25
With the presented data of risks associated with uncontained morcellation and agreement of the ACOG, AAGL, and SGO professional societies, we recommend that all morcellation be performed in a contained fashion to prevent the dissemination of benign or undiagnosed malignant tissue throughout the abdomen and pelvis. Shared decision making and counseling on the risks, benefits, and alternatives are paramount for patients to make informed decisions about their medical care. Continued exploration of techniques and methods for safe tissue extraction is still needed to improve minimally invasive surgical options for all women.
1. US Food and Drug Administration. Updated: Laparoscopic uterine power morcellation in hysterectomy and myomectomy: FDA safety communication. November 24, 2014; updated April 7, 2016. https://wayback.archiveit.org/7993/20170404182209/https:/www.fda.gov /MedicalDevices/Safety/AlertsandNotices/ucm424443.htm. Accessed July 23, 2019.
2. American College of Obstetricians and Gynecologists Committee on Gynecologic Practice. ACOG committee opinion no. 770: Uterine morcellation for presumed leiomyomas. Obstet Gynecol. 2019;133:e238-e248.
3. American College of Obstetricians and Gynecologists Committee on Gynecologic Practice. ACOG committee opinion no. 701: Choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2017;129:1149-1150.
4. Wiser A, Holcroft CA, Tolandi T, et al. Abdominal versus laparoscopic hysterectomies for benign diseases: evaluation of morbidity and mortality among 465,798 cases. Gynecol Surg. 2013;10:117-122.
5. Winner B, Biest S. Uterine morcellation: fact and fiction surrounding the recent controversy. Mo Med. 2017;114:176-180.
6. Tulandi T, Leung A, Jan N. Nonmalignant sequelae of unconfined morcellation at laparoscopic hysterectomy or myomectomy. J Minim Invasive Gynecol. 2016;23:331-337.
7. Milad MP, Milad EA. Laparoscopic morcellator-related complications. J Minim Invasive Gynecol. 2014;21:486-491.
8. Toro JR, Travis LB, Wu HJ, et al. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the Surveillance, Epidemiology and End Results program, 1978-2001: an analysis of 26,758 cases. Int J Cancer. 2006;119:2922-2930.
9. Seagle BL, Sobecki-Rausch J, Strohl AE, et al. Prognosis and treatment of uterine leiomyosarcoma: a National Cancer Database study. Gynecol Oncol. 2017;145:61-70.
10. Ricci S, Stone RL, Fader AN. Uterine leiomyosarcoma: epidemiology, contemporary treatment strategies and the impact of uterine morcellation. Gynecol Oncol. 2017;145:208-216.
11. Leibsohn S, d’Ablaing G, Mishell DR Jr, et al. Leiomyosarcoma in a series of hysterectomies performed for presumed uterine leiomyomas. Am J Obstet Gynecol. 1990;162:968-974. Discussion 974-976.
12. Rowland M, Lesnock J, Edwards R, et al. Occult uterine cancer in patients undergoing laparoscopic hysterectomy with morcellation [abstract]. Gynecol Oncol. 2012;127:S29.
13. Hartmann KE, Fonnesbeck C, Surawicz T, et al. Management of uterine fibroids. Comparative effectiveness review no. 195. AHRQ Publication No. 17(18)-EHC028-EF. Rockville, MD: Agency for Healthcare Research and Quality; 2017. https://effectivehealthcare.ahrq.gov/topics/uterine-fibroids /research-2017. Accessed July 23, 2019.
14. Pritts EA, Parker WH, Brown J, et al. Outcome of occult uterine leiomyosarcoma after surgery for presumed uterine fibroids: a systematic review. J Minim Invasive Gynecol. 2015;22:26-33.
15. American College of Obstetricians and Gynecologists Committee on Practice Bulletins–Gynecology. Practice bulletin no. 128: Diagnosis of abnormal uterine bleeding in reproductive-aged women. Obstet Gynecol. 2012;120:197-206.
16. Bansal N, Herzog TJ, Burke W, et al. The utility of preoperative endometrial sampling for the detection of uterine sarcomas. Gynecol Oncol. 2008 Jul;110(1):43–48.
17. American College of Obstetricians and Gynecologists Committee on Ethics. ACOG committee opinion no. 439: Informed consent. Obstet Gynecol. 2009;114:401-408.
18. Wright JD, Cui RR, Wang A, et al. Economic and survival implications of use of electric power morcellation for hysterectomy for presumed benign gynecologic disease. J Natl Cancer Inst. 2015;107:djv251.
19. US Food and Drug Administration. FDA allows marketing of first-of-kind tissue containment system for use with certain laparoscopic power morcellators in select patients [press release]. April 7, 2016. https://www.fda.gov/NewsEvents /Newsroom/PressAnnouncements/ucm494650.htm. Accessed July 23, 2019.
20. Winner B, Porter A, Velloze S, et al. S. Uncontained compared with contained power morcellation in total laparoscopic hysterectomy. Obstet Gynecol. 2015 Oct;126(4):834–8.
21. Cohen SL, Einarsson JI, Wang KC, et al. Contained power morcellation within an insufflated isolation bag. Obstet Gynecol. 2014;124:491-497.
22. Cohen SL, Greenberg JA, Wang KC, et al. Risk of leakage and tissue dissemination with various contained tissue extraction (CTE) techniques: an in vitro pilot study. J Minim Invasive Gynecol. 2014;21:935-939.
23. Cohen SL, Morris SN, Brown DN, et al. Contained tissue extraction using power morcellation: prospective evaluation of leakage parameters. Am J Obstet Gynecol. 2016;214(2):257. e1-257.e6.
24. AAGL. AAGL practice report: morcellation during uterine tissue extraction. J Minim Invasive Gynecol. 2014;21:517-530.
25. Society of Gynecologic Oncology. Position statement: morcellation. 2013. https://www.sgo.org/newsroom /position-statements-2/morcellation/.Accessed July 23, 2019.
1. US Food and Drug Administration. Updated: Laparoscopic uterine power morcellation in hysterectomy and myomectomy: FDA safety communication. November 24, 2014; updated April 7, 2016. https://wayback.archiveit.org/7993/20170404182209/https:/www.fda.gov /MedicalDevices/Safety/AlertsandNotices/ucm424443.htm. Accessed July 23, 2019.
2. American College of Obstetricians and Gynecologists Committee on Gynecologic Practice. ACOG committee opinion no. 770: Uterine morcellation for presumed leiomyomas. Obstet Gynecol. 2019;133:e238-e248.
3. American College of Obstetricians and Gynecologists Committee on Gynecologic Practice. ACOG committee opinion no. 701: Choosing the route of hysterectomy for benign disease. Obstet Gynecol. 2017;129:1149-1150.
4. Wiser A, Holcroft CA, Tolandi T, et al. Abdominal versus laparoscopic hysterectomies for benign diseases: evaluation of morbidity and mortality among 465,798 cases. Gynecol Surg. 2013;10:117-122.
5. Winner B, Biest S. Uterine morcellation: fact and fiction surrounding the recent controversy. Mo Med. 2017;114:176-180.
6. Tulandi T, Leung A, Jan N. Nonmalignant sequelae of unconfined morcellation at laparoscopic hysterectomy or myomectomy. J Minim Invasive Gynecol. 2016;23:331-337.
7. Milad MP, Milad EA. Laparoscopic morcellator-related complications. J Minim Invasive Gynecol. 2014;21:486-491.
8. Toro JR, Travis LB, Wu HJ, et al. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the Surveillance, Epidemiology and End Results program, 1978-2001: an analysis of 26,758 cases. Int J Cancer. 2006;119:2922-2930.
9. Seagle BL, Sobecki-Rausch J, Strohl AE, et al. Prognosis and treatment of uterine leiomyosarcoma: a National Cancer Database study. Gynecol Oncol. 2017;145:61-70.
10. Ricci S, Stone RL, Fader AN. Uterine leiomyosarcoma: epidemiology, contemporary treatment strategies and the impact of uterine morcellation. Gynecol Oncol. 2017;145:208-216.
11. Leibsohn S, d’Ablaing G, Mishell DR Jr, et al. Leiomyosarcoma in a series of hysterectomies performed for presumed uterine leiomyomas. Am J Obstet Gynecol. 1990;162:968-974. Discussion 974-976.
12. Rowland M, Lesnock J, Edwards R, et al. Occult uterine cancer in patients undergoing laparoscopic hysterectomy with morcellation [abstract]. Gynecol Oncol. 2012;127:S29.
13. Hartmann KE, Fonnesbeck C, Surawicz T, et al. Management of uterine fibroids. Comparative effectiveness review no. 195. AHRQ Publication No. 17(18)-EHC028-EF. Rockville, MD: Agency for Healthcare Research and Quality; 2017. https://effectivehealthcare.ahrq.gov/topics/uterine-fibroids /research-2017. Accessed July 23, 2019.
14. Pritts EA, Parker WH, Brown J, et al. Outcome of occult uterine leiomyosarcoma after surgery for presumed uterine fibroids: a systematic review. J Minim Invasive Gynecol. 2015;22:26-33.
15. American College of Obstetricians and Gynecologists Committee on Practice Bulletins–Gynecology. Practice bulletin no. 128: Diagnosis of abnormal uterine bleeding in reproductive-aged women. Obstet Gynecol. 2012;120:197-206.
16. Bansal N, Herzog TJ, Burke W, et al. The utility of preoperative endometrial sampling for the detection of uterine sarcomas. Gynecol Oncol. 2008 Jul;110(1):43–48.
17. American College of Obstetricians and Gynecologists Committee on Ethics. ACOG committee opinion no. 439: Informed consent. Obstet Gynecol. 2009;114:401-408.
18. Wright JD, Cui RR, Wang A, et al. Economic and survival implications of use of electric power morcellation for hysterectomy for presumed benign gynecologic disease. J Natl Cancer Inst. 2015;107:djv251.
19. US Food and Drug Administration. FDA allows marketing of first-of-kind tissue containment system for use with certain laparoscopic power morcellators in select patients [press release]. April 7, 2016. https://www.fda.gov/NewsEvents /Newsroom/PressAnnouncements/ucm494650.htm. Accessed July 23, 2019.
20. Winner B, Porter A, Velloze S, et al. S. Uncontained compared with contained power morcellation in total laparoscopic hysterectomy. Obstet Gynecol. 2015 Oct;126(4):834–8.
21. Cohen SL, Einarsson JI, Wang KC, et al. Contained power morcellation within an insufflated isolation bag. Obstet Gynecol. 2014;124:491-497.
22. Cohen SL, Greenberg JA, Wang KC, et al. Risk of leakage and tissue dissemination with various contained tissue extraction (CTE) techniques: an in vitro pilot study. J Minim Invasive Gynecol. 2014;21:935-939.
23. Cohen SL, Morris SN, Brown DN, et al. Contained tissue extraction using power morcellation: prospective evaluation of leakage parameters. Am J Obstet Gynecol. 2016;214(2):257. e1-257.e6.
24. AAGL. AAGL practice report: morcellation during uterine tissue extraction. J Minim Invasive Gynecol. 2014;21:517-530.
25. Society of Gynecologic Oncology. Position statement: morcellation. 2013. https://www.sgo.org/newsroom /position-statements-2/morcellation/.Accessed July 23, 2019.