User login
Review of Radiologic Considerations in an Immunocompetent Patient With Primary Central Nervous System Lymphoma (FULL)
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
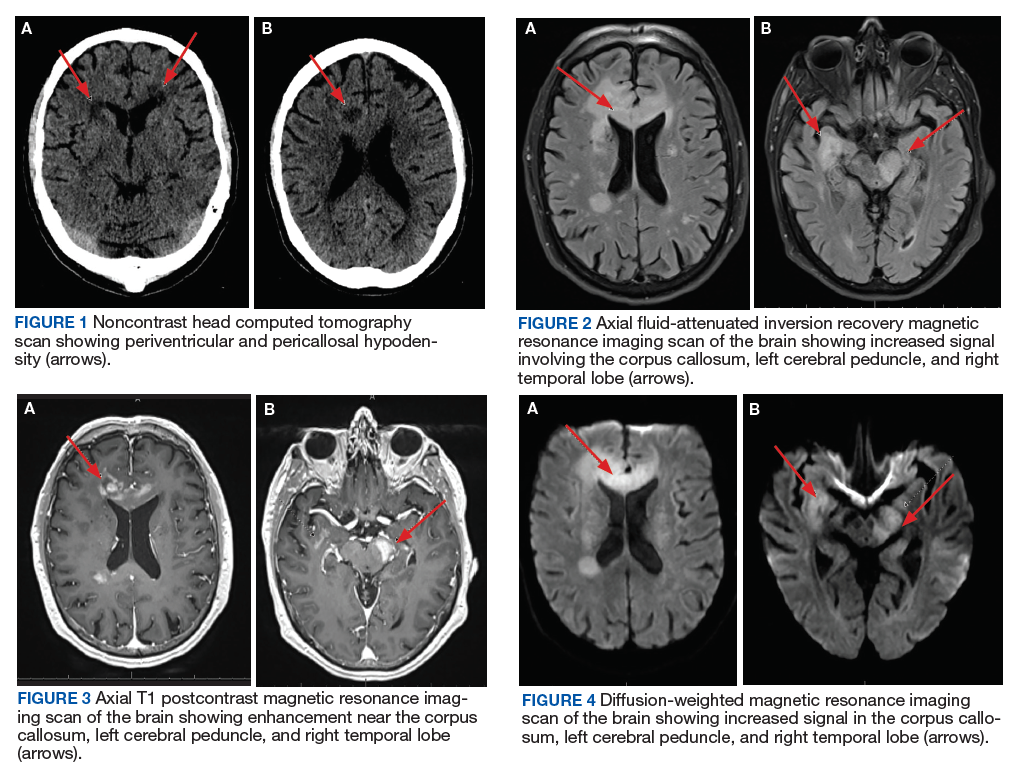
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
Which birth defects are associated with childhood cancer risk?

Accuracy of Endoscopic Ultrasound in Staging of Early Rectal Cancer (FULL)
Endoscopic ultrasound can be highly accurate for the staging of neoplasms in early rectal cancer.
Colorectal cancer is the second most common cause of cancer death in the US, with one-third of all colorectal cancers occurring within the rectum. Each year, an estimated 40000 Americans are diagnosed with rectal cancer (RC).1,2 The prognosis and treatment of RC depends on both T and N stage at the time of diagnosis.3-5 According to the most recent National Comprehensive Cancer Network guidelines from May 2019, patients with T1 to T2N0 tumors should undergo transanal or transabdominal surgery upfront, whereas patients with T3 to T4N0 or any TN1 to 2 should start with neoadjuvant therapy for better locoregional control, followed by surgery.6 Therefore, the appropriate management of RC requires adequate staging.
Endoscopic ultrasound (EUS), magnetic resonance imaging (MRI), and computed tomography (CT) are the imaging techniques currently used to stage RC. In a meta-analysis of 90 articles published between 1985 and 2002 that compared the 3 radiologic modalities, Bipat and colleagues found that MRI and EUS had a similar sensitivity of 94%, whereas the specificity of EUS (86%) was significantly higher than that of MRI (69%) for muscularis propria invasion.7 CT was performed only in a limited number of trials because CT was considered inadequate to assess early T stage. For perirectal tissue invasion, the sensitivity of EUS was statistically higher than that of CT and MRI imaging: 90% compared with 79% and 82%, respectively. The specificity estimates for EUS, CT, and MRI were comparable: 75%, 78%, and 76%, respectively. The respective sensitivity and specificity of the 3 imaging modalities to evaluate lymph nodes were also comparable: EUS, 67% and 78%; CT, 55% and 74%; and MRI, 66% and 76%.
The role of EUS in the diagnosis and treatment of RC has long been validated.1,2-5 A meta-analysis of 42 studies involving 5039 patients found EUS to be highly accurate for differentiating various T stages.8 However, EUS cannot assess iliac and mesenteric lymph nodes or posterior tumor extension beyond endopelvic fascia in advanced RC. Notable heterogeneity was found among the studies in the meta-analyses with regard to the type of equipment used for staging, as well as the criteria used to assess the depth of penetration and nodal status. The recent introduction of phased-array coils and the development of T2-weighted fast spin sequences have improved the resolution of MRI. The MERCURY trial showed that extension of tumor to within 1 mm of the circumferential margin on high-resolution MRI correctly predicted margin involvement at the time of surgery in 92% of the patients.9 In the retrospective study by Balyasnikova and colleagues, MRI was found to correctly identify partial submucosal invasion and suitability for local excision in 89% of the cases.10
Therefore, both EUS and MRI are useful, more so than CT, in assessment of the depth of tumor invasion, nodal staging, and predicting the circumferential resection margin. The use of EUS, however, does not preclude the use of MRI, or vice versa. Rather, the 2 modalities can complement each other in staging and proper patient selection for treatment.11
Despite data supporting the value of EUS in staging RC, its use is limited by a high degree of operator dependence and a substantial learning curve,12-17 which may explain the low EUS accuracy observed in some reports.7,13,15 Given the presence of recognized alternatives such as MRI, we decided to reevaluate EUS accuracy for the staging of RC outside high-volume specialized centers and prospective clinical trials.
Methods
A retrospective chart review was performed that included all consecutive patients undergoing rectal ultrasound from January 2011 to August 2015 at the US Department of Veterans Affairs Medical Center (VAMC) in Memphis, Tennessee. Sixty-five patients with short-stocked or sessile lesions < 15 cm from anal margin staged T2N0M0 or lower by endorectal ultrasound (ERUS) were included. The patients with neoplasms staged in excess of T2 or N0 were excluded from the study because treatment protocol dictates immediate neoadjuvant treatment, the administration of which would affect subsequent histopathology.
For the 37 patients included in the final analysis, ERUS results were compared with surgical pathology to ascertain accuracy. The resections were performed endoscopically or surgically with a goal of obtaining clear margins. The choice of procedure depended on size, shape, location, and depth of invasion. All patients underwent clinical and endoscopic surveillance with flexible sigmoidoscopy/EUS every 3 to 6 months for the first 2 years. We used 2 different gold standards for surveillance depending on the type of procedure performed to remove the lesion. A pathology report was the gold standard used for patients who underwent surgery. In patients who underwent endoscopic resection, we used the lack of recurrent disease, determined by normal endoscopic and endoscopic ultrasound examination, to signify complete endoscopic resection and therefore adequate staging as an early neoplasm.
Results
From January 2011 to August 2015, 65 rectal ultrasounds were performed. All EUS procedures were performed by 1 physician (C Ruben Tombazzi). All patients had previous endoscopic evaluation and tissue diagnoses. Twenty-eight patients were excluded: 18 had T3 or N1 disease, 2 had T2N0 but refused surgery, 2 had anal cancer, 3 patients with suspected cancer had benign nonneoplastic disease (2 radiation proctitis, 1 normal rectal wall), and 3 underwent EUS for benign tumors (1 ganglioneuroma and 2 lipomas).
Thirty-seven patients were included in the study, 3 of whom were staged as T2N0 and 34 as T1N0 or lower by EUS. All patients were men ranging in age from 43 to 73 years (mean, 59 years). All 37 patients underwent endoscopic or surgical resection of their early rectal neoplasm. The final pathologic evaluation of the specimens demonstrated 14 carcinoid tumors, 11 adenocarcinomas, 6 tubular adenomas with high-grade dysplasia, and 6 benign adenomas. The preoperative EUS staging was confirmed for all patients, with 100% sensitivity, specificity, and accuracy. None of the patients who underwent endoscopic or surgical transanal resection had recurrence, determined by normal endoscopic and endoscopic ultrasound appearance, during a mean of 32.6 months surveillance.
Discussion
EUS has long been a recognized method for T and N staging of RC.1,3-5,7,8 Our data confirm that, in experienced hands, EUS is highly accurate in the staging of early rectal cancers.
The impact of EUS on the management of RC was demonstrated in a Mayo Clinic prospective blinded study.1 In that cohort of 80 consecutive patients who had previously had a CT for staging, EUS altered patient management in about 30% of cases. The most common change precipatated by EUS was the indication for additional neoadjuvant treatment.
However, the results have not been as encouraging when ERUS is performed outside of strict research protocol. A multicenter, prospective, country-wide quality assurance study from > 300 German hospitals was designed to assess the diagnostic accuracy of EUS in RC.13 Of 29206 patients, 7096 underwent surgery, without neoadjuvant treatment, and were included in the final analysis. The correspondence of tumor invasion with histopathology was 64.7%, with understaging of 18% and overstaging of 17.3%.13 These numbers were better in hospitals with greater experience performing ERUS: 73% accuracy in the centers with a case load of > 30 cases per year compared with 63.2% accuracy for the centers with < 10 cases a year. Marusch and colleagues had previously demonstrated an EUS accuracy of 63.3% in a study of 1463 patients with RC in Germany.14 Another study based out of the UK had similar findings. Ashraf and colleagues performed a database analyses from 20 UK centers and identified 165 patients with RC who underwent ERUS and endoscopic microsurgery.15 Compared with histopathology, EUS had 57.1% sensitivity, 73% specificity, and 42.9% accuracy for T1 cancers; EUS accuracy was 50% for T2 and 58% for T3 tumors. The authors concluded that the general accuracy of EUS in determining stage was around 50%, the statistical equivalent of flipping a coin.
The low accuracy of EUS observed by German and British multicenter studies13-15 was attributed to the difference that may exist in clinical trials at specialized centers compared with wider use of EUS in a community setting. As seen by our data, the Memphis VAMC is not a high-volume center for the treatment of RC. However, all our EUS procedures were performed and interpreted by a single operator (C. Ruben Tombazzi) with 18 years of EUS experience. We cannot conclude that no patient was overstaged, as patients receiving a stage of T3N0 or T > N0 received neoadjuvant treatment and were not included. However, we can conclude that no patient was understaged. All patients deemed to be T1 to T2N0 included in our study received accurate staging. Our results are consistent with the high accuracy of EUS reported from other centers with experience in diagnosis and treatment of RC.1,3-5,17,18
Although EUS is accurate in differentiating T1 from T2 tumors, it cannot reliably differentiate T1 from T0 lesions. In one study, 57.6% of adenomas and 30.7% of carcinomas in situ were staged as T1 on EUS, while almost half of T1 cancers were interpreted as T0.17 This drawback is a well-known limitation of EUS; although, the misinterpretation does not affect treatment, as both T0 and T1 lesions can be treated successfully by local excision alone, which was the algorithm used for our patients. The choice of the specific procedure for local excision was left to the clinicians and included transanal endoscopic or surgical resections. At a mean follow-up of 32.6 months, none of the 37 patients who underwent endoscopic or surgical transanal resection had evidence of recurrent disease.
A limitation of EUS, or any other imaging modality, is differentiating tumor invasion from peritumoral inflammation. The inflammation can render images of tumor borders ill-defined and irregular, which hinders precise staging. However, the accurate identification of tumors with deep involvement of the submucosa (T1sm3) is of importance, because these tumors are more advanced than the superficial and intermediate T1 lesions (T1sm1 and T1sm2, respectively).
Patients with RC whose lesions are considered T1sm3 are at higher risk of harboring lymph node metastases.18 Nascimbeni and colleagues had shown that the invasion into the lower third of the submucosa (sm3) was an independent risk factor for lower cancer-free survival among patients with T1 RC.19
Unlike rectal adenocarcinomas, the prognosis for carcinoid tumors correlates not only with the depth of invasion but also with the size of the tumor. The other adverse prognostic features include poor differentiation, high mitosis index, and lymphovascular invasion.20
EUS had been shown to be highly accurate in determining the precise carcinoid tumor size, depth of invasion, and lymph node metastases.20,21 In a study of 66 resected rectal carcinoid tumors by Ishii and colleagues, 57 lesions had a diameter of ≤ 10 mm and 9 lesions had a diameter of > 10 mm.21 All of the 57 carcinoid tumors with a diameter of ≤ 10 mm were confined to the submucosa. In contrast, 5 of the 9 lesions > 10 mm invaded the muscularis propria, 6 had a lymphovascular invasion, 4 were lymph node metastases, and 1 was a liver metastasis.
In our series, 4 of the 14 carcinoid tumors were > 10 mm but none were > 20 mm. None of the carcinoids with a diameter ≤ 10 mm invaded the muscularis propria. Of the 4 carcinoids > 10 mm, 1 was T2N0 and 3 were T1N0. All carcinoid tumors in our series were low grade and with low proliferation indexes, and all were treated successfully by local excision.
Conclusion
We believe our study shows that EUS can be highly accurate in staging rectal lesions, specifically lesions that are T1-T2N0, be they adenocarcinoma or carcinoid. Although we could not assess overstaging for lesions that were staged > T2 or > N0, we w
1. Harewood GC, Wiersema MJ, Nelson H, et al. A prospective, blinded assessment of the impact of preoperative staging on the management of rectal cancer. Gastroenterology. 2002;123(1):24-32.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
3. Ahuja NK, Sauer BG, Wang AY, et al. Performance of endoscopic ultrasound in staging rectal adenocarcinoma appropriate for primary surgical resection. Clin Gastroenterol Hepatol. 2015;13:339-44.
4. Doornebosch PG, Bronkhorst PJ, Hop WC, Bode WA, Sing AK, de Graaf EJ. The role of endorectal ultrasound in therapeutic decision-making for local vs. transabdominal resection of rectal tumors. Dis Colon Rectum. 2008;51(1):38-42.
5. Santoro GA, Gizzi G, Pellegrini L, Battistella G, Di Falco G. The value of high-resolution three-dimensional endorectal ultrasonography in the management of submucosal invasive rectal tumors. Dis Colon Rectum. 2009;52(11):1837-1843.
6. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: rectal cancer, version 2.2019. https://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf. Published May 15, 2019. Accessed July 19, 2019.
7. Bipat S, Glas AS, Slors FJ, Zwinderman AH, Bossuyt PM, Stoker J. Rectal cancer: local staging and assessment of lymph node involvement with endoluminal US, CT, and MR imaging—a meta-analysis. Radiology. 2004;232(3):773-783.
8. Puli SR, Bechtold ML, Reddy JB, Choudhary A, Antillon MR, Brugge WR. How good is endoscopic ultrasound in differentiating various T stages of rectal cancer? Meta-analysis and systematic review. Ann Surg Oncol. 2009;16(2):254-265.
9. MERCURY Study Group. Diagnostic accuracy of preoperative magnetic resonance imaging in predicting curative resection of rectal cancer: prospective observational study. BMJ. 2006;333(7572):779.
10. Balyasnikova S, Read J, Wotherspoon A, et al. Diagnostic accuracy of high-resolution MRI as a method to predict potentially safe endoscopic and surgical planes in patient with early rectal cancer. BMJ Open Gastroenterol. 2017;4(1):e000151.
11. Frasson M, Garcia-Granero E, Roda D, et al. Preoperative chemoradiation may not always be needed for patients with T3 and T2N+ rectal cancer. Cancer. 2011;117(14):3118-3125.
12. Rafaelsen SR, Sørensen T, Jakobsen A, Bisgaard C, Lindebjerg J. Transrectal ultrasonography and magnetic resonance imaging in the staging of rectal cancer. Effect of experience. Scand J Gastroenterol. 2008;43(4):440-446.
13. Marusch F, Ptok H, Sahm M, et al. Endorectal ultrasound in rectal carcinoma – do the literature results really correspond to the realities of routine clinical care? Endoscopy. 2011;43(5):425-431.
14. Marusch F, Koch A, Schmidt U, et al. Routine use of transrectal ultrasound in rectal carcinoma: results of a prospective multicenter study. Endoscopy. 2002;34(5):385-390.
15. Ashraf S, Hompes R, Slater A, et al; Association of Coloproctology of Great Britain and Ireland Transanal Endoscopic Microsurgery (TEM) Collaboration. A critical appraisal of endorectal ultrasound and transanal endoscopic microsurgery and decision-making in early rectal cancer. Colorectal Dis. 2012;14(7):821-826.
16. Harewood GC. Assessment of clinical impact of endoscopic ultrasound on rectal cancer. Am J Gastroenterol. 2004;99(4):623-627.
17. Zorcolo L, Fantola G, Cabras F, Marongiu L, D’Alia G, Casula G. Preoperative staging of patients with rectal tumors suitable for transanal endoscopic microsurgery (TEM): comparison of endorectal ultrasound and histopathologic findings. Surg Endosc. 2009;23(6):1384-1389.
18. Akasu T, Kondo H, Moriya Y, et al. Endoscopic ultrasonography and treatment of early stage rectal cancer. World J Surg. 2000;24(9):1061-1068.
19. Nascimbeni R, Nivatvongs S, Larson DR, Burgart LJ. Long-term survival after local excision for T1 carcinoma of the rectum. Dis Colon Rectum. 2004;47(11):1773-1779.
20. Park CH, Cheon JH, Kim JO, et al. Criteria for decision making after endoscopic resection of well-differentiated rectal carcinoids with regard to potential lymphatic spread. Endoscopy. 2011;43(9):790-795.
21. Ishii N, Horiki N, Itoh T, et al. Endoscopic submucosal dissection and preoperative assessment with endoscopic ultrasonography for the treatment of rectal carcinoid tumors. Surg Endosc. 2010;24(6):1413-1419.
Endoscopic ultrasound can be highly accurate for the staging of neoplasms in early rectal cancer.
Endoscopic ultrasound can be highly accurate for the staging of neoplasms in early rectal cancer.
Colorectal cancer is the second most common cause of cancer death in the US, with one-third of all colorectal cancers occurring within the rectum. Each year, an estimated 40000 Americans are diagnosed with rectal cancer (RC).1,2 The prognosis and treatment of RC depends on both T and N stage at the time of diagnosis.3-5 According to the most recent National Comprehensive Cancer Network guidelines from May 2019, patients with T1 to T2N0 tumors should undergo transanal or transabdominal surgery upfront, whereas patients with T3 to T4N0 or any TN1 to 2 should start with neoadjuvant therapy for better locoregional control, followed by surgery.6 Therefore, the appropriate management of RC requires adequate staging.
Endoscopic ultrasound (EUS), magnetic resonance imaging (MRI), and computed tomography (CT) are the imaging techniques currently used to stage RC. In a meta-analysis of 90 articles published between 1985 and 2002 that compared the 3 radiologic modalities, Bipat and colleagues found that MRI and EUS had a similar sensitivity of 94%, whereas the specificity of EUS (86%) was significantly higher than that of MRI (69%) for muscularis propria invasion.7 CT was performed only in a limited number of trials because CT was considered inadequate to assess early T stage. For perirectal tissue invasion, the sensitivity of EUS was statistically higher than that of CT and MRI imaging: 90% compared with 79% and 82%, respectively. The specificity estimates for EUS, CT, and MRI were comparable: 75%, 78%, and 76%, respectively. The respective sensitivity and specificity of the 3 imaging modalities to evaluate lymph nodes were also comparable: EUS, 67% and 78%; CT, 55% and 74%; and MRI, 66% and 76%.
The role of EUS in the diagnosis and treatment of RC has long been validated.1,2-5 A meta-analysis of 42 studies involving 5039 patients found EUS to be highly accurate for differentiating various T stages.8 However, EUS cannot assess iliac and mesenteric lymph nodes or posterior tumor extension beyond endopelvic fascia in advanced RC. Notable heterogeneity was found among the studies in the meta-analyses with regard to the type of equipment used for staging, as well as the criteria used to assess the depth of penetration and nodal status. The recent introduction of phased-array coils and the development of T2-weighted fast spin sequences have improved the resolution of MRI. The MERCURY trial showed that extension of tumor to within 1 mm of the circumferential margin on high-resolution MRI correctly predicted margin involvement at the time of surgery in 92% of the patients.9 In the retrospective study by Balyasnikova and colleagues, MRI was found to correctly identify partial submucosal invasion and suitability for local excision in 89% of the cases.10
Therefore, both EUS and MRI are useful, more so than CT, in assessment of the depth of tumor invasion, nodal staging, and predicting the circumferential resection margin. The use of EUS, however, does not preclude the use of MRI, or vice versa. Rather, the 2 modalities can complement each other in staging and proper patient selection for treatment.11
Despite data supporting the value of EUS in staging RC, its use is limited by a high degree of operator dependence and a substantial learning curve,12-17 which may explain the low EUS accuracy observed in some reports.7,13,15 Given the presence of recognized alternatives such as MRI, we decided to reevaluate EUS accuracy for the staging of RC outside high-volume specialized centers and prospective clinical trials.
Methods
A retrospective chart review was performed that included all consecutive patients undergoing rectal ultrasound from January 2011 to August 2015 at the US Department of Veterans Affairs Medical Center (VAMC) in Memphis, Tennessee. Sixty-five patients with short-stocked or sessile lesions < 15 cm from anal margin staged T2N0M0 or lower by endorectal ultrasound (ERUS) were included. The patients with neoplasms staged in excess of T2 or N0 were excluded from the study because treatment protocol dictates immediate neoadjuvant treatment, the administration of which would affect subsequent histopathology.
For the 37 patients included in the final analysis, ERUS results were compared with surgical pathology to ascertain accuracy. The resections were performed endoscopically or surgically with a goal of obtaining clear margins. The choice of procedure depended on size, shape, location, and depth of invasion. All patients underwent clinical and endoscopic surveillance with flexible sigmoidoscopy/EUS every 3 to 6 months for the first 2 years. We used 2 different gold standards for surveillance depending on the type of procedure performed to remove the lesion. A pathology report was the gold standard used for patients who underwent surgery. In patients who underwent endoscopic resection, we used the lack of recurrent disease, determined by normal endoscopic and endoscopic ultrasound examination, to signify complete endoscopic resection and therefore adequate staging as an early neoplasm.
Results
From January 2011 to August 2015, 65 rectal ultrasounds were performed. All EUS procedures were performed by 1 physician (C Ruben Tombazzi). All patients had previous endoscopic evaluation and tissue diagnoses. Twenty-eight patients were excluded: 18 had T3 or N1 disease, 2 had T2N0 but refused surgery, 2 had anal cancer, 3 patients with suspected cancer had benign nonneoplastic disease (2 radiation proctitis, 1 normal rectal wall), and 3 underwent EUS for benign tumors (1 ganglioneuroma and 2 lipomas).
Thirty-seven patients were included in the study, 3 of whom were staged as T2N0 and 34 as T1N0 or lower by EUS. All patients were men ranging in age from 43 to 73 years (mean, 59 years). All 37 patients underwent endoscopic or surgical resection of their early rectal neoplasm. The final pathologic evaluation of the specimens demonstrated 14 carcinoid tumors, 11 adenocarcinomas, 6 tubular adenomas with high-grade dysplasia, and 6 benign adenomas. The preoperative EUS staging was confirmed for all patients, with 100% sensitivity, specificity, and accuracy. None of the patients who underwent endoscopic or surgical transanal resection had recurrence, determined by normal endoscopic and endoscopic ultrasound appearance, during a mean of 32.6 months surveillance.
Discussion
EUS has long been a recognized method for T and N staging of RC.1,3-5,7,8 Our data confirm that, in experienced hands, EUS is highly accurate in the staging of early rectal cancers.
The impact of EUS on the management of RC was demonstrated in a Mayo Clinic prospective blinded study.1 In that cohort of 80 consecutive patients who had previously had a CT for staging, EUS altered patient management in about 30% of cases. The most common change precipatated by EUS was the indication for additional neoadjuvant treatment.
However, the results have not been as encouraging when ERUS is performed outside of strict research protocol. A multicenter, prospective, country-wide quality assurance study from > 300 German hospitals was designed to assess the diagnostic accuracy of EUS in RC.13 Of 29206 patients, 7096 underwent surgery, without neoadjuvant treatment, and were included in the final analysis. The correspondence of tumor invasion with histopathology was 64.7%, with understaging of 18% and overstaging of 17.3%.13 These numbers were better in hospitals with greater experience performing ERUS: 73% accuracy in the centers with a case load of > 30 cases per year compared with 63.2% accuracy for the centers with < 10 cases a year. Marusch and colleagues had previously demonstrated an EUS accuracy of 63.3% in a study of 1463 patients with RC in Germany.14 Another study based out of the UK had similar findings. Ashraf and colleagues performed a database analyses from 20 UK centers and identified 165 patients with RC who underwent ERUS and endoscopic microsurgery.15 Compared with histopathology, EUS had 57.1% sensitivity, 73% specificity, and 42.9% accuracy for T1 cancers; EUS accuracy was 50% for T2 and 58% for T3 tumors. The authors concluded that the general accuracy of EUS in determining stage was around 50%, the statistical equivalent of flipping a coin.
The low accuracy of EUS observed by German and British multicenter studies13-15 was attributed to the difference that may exist in clinical trials at specialized centers compared with wider use of EUS in a community setting. As seen by our data, the Memphis VAMC is not a high-volume center for the treatment of RC. However, all our EUS procedures were performed and interpreted by a single operator (C. Ruben Tombazzi) with 18 years of EUS experience. We cannot conclude that no patient was overstaged, as patients receiving a stage of T3N0 or T > N0 received neoadjuvant treatment and were not included. However, we can conclude that no patient was understaged. All patients deemed to be T1 to T2N0 included in our study received accurate staging. Our results are consistent with the high accuracy of EUS reported from other centers with experience in diagnosis and treatment of RC.1,3-5,17,18
Although EUS is accurate in differentiating T1 from T2 tumors, it cannot reliably differentiate T1 from T0 lesions. In one study, 57.6% of adenomas and 30.7% of carcinomas in situ were staged as T1 on EUS, while almost half of T1 cancers were interpreted as T0.17 This drawback is a well-known limitation of EUS; although, the misinterpretation does not affect treatment, as both T0 and T1 lesions can be treated successfully by local excision alone, which was the algorithm used for our patients. The choice of the specific procedure for local excision was left to the clinicians and included transanal endoscopic or surgical resections. At a mean follow-up of 32.6 months, none of the 37 patients who underwent endoscopic or surgical transanal resection had evidence of recurrent disease.
A limitation of EUS, or any other imaging modality, is differentiating tumor invasion from peritumoral inflammation. The inflammation can render images of tumor borders ill-defined and irregular, which hinders precise staging. However, the accurate identification of tumors with deep involvement of the submucosa (T1sm3) is of importance, because these tumors are more advanced than the superficial and intermediate T1 lesions (T1sm1 and T1sm2, respectively).
Patients with RC whose lesions are considered T1sm3 are at higher risk of harboring lymph node metastases.18 Nascimbeni and colleagues had shown that the invasion into the lower third of the submucosa (sm3) was an independent risk factor for lower cancer-free survival among patients with T1 RC.19
Unlike rectal adenocarcinomas, the prognosis for carcinoid tumors correlates not only with the depth of invasion but also with the size of the tumor. The other adverse prognostic features include poor differentiation, high mitosis index, and lymphovascular invasion.20
EUS had been shown to be highly accurate in determining the precise carcinoid tumor size, depth of invasion, and lymph node metastases.20,21 In a study of 66 resected rectal carcinoid tumors by Ishii and colleagues, 57 lesions had a diameter of ≤ 10 mm and 9 lesions had a diameter of > 10 mm.21 All of the 57 carcinoid tumors with a diameter of ≤ 10 mm were confined to the submucosa. In contrast, 5 of the 9 lesions > 10 mm invaded the muscularis propria, 6 had a lymphovascular invasion, 4 were lymph node metastases, and 1 was a liver metastasis.
In our series, 4 of the 14 carcinoid tumors were > 10 mm but none were > 20 mm. None of the carcinoids with a diameter ≤ 10 mm invaded the muscularis propria. Of the 4 carcinoids > 10 mm, 1 was T2N0 and 3 were T1N0. All carcinoid tumors in our series were low grade and with low proliferation indexes, and all were treated successfully by local excision.
Conclusion
We believe our study shows that EUS can be highly accurate in staging rectal lesions, specifically lesions that are T1-T2N0, be they adenocarcinoma or carcinoid. Although we could not assess overstaging for lesions that were staged > T2 or > N0, we w
Colorectal cancer is the second most common cause of cancer death in the US, with one-third of all colorectal cancers occurring within the rectum. Each year, an estimated 40000 Americans are diagnosed with rectal cancer (RC).1,2 The prognosis and treatment of RC depends on both T and N stage at the time of diagnosis.3-5 According to the most recent National Comprehensive Cancer Network guidelines from May 2019, patients with T1 to T2N0 tumors should undergo transanal or transabdominal surgery upfront, whereas patients with T3 to T4N0 or any TN1 to 2 should start with neoadjuvant therapy for better locoregional control, followed by surgery.6 Therefore, the appropriate management of RC requires adequate staging.
Endoscopic ultrasound (EUS), magnetic resonance imaging (MRI), and computed tomography (CT) are the imaging techniques currently used to stage RC. In a meta-analysis of 90 articles published between 1985 and 2002 that compared the 3 radiologic modalities, Bipat and colleagues found that MRI and EUS had a similar sensitivity of 94%, whereas the specificity of EUS (86%) was significantly higher than that of MRI (69%) for muscularis propria invasion.7 CT was performed only in a limited number of trials because CT was considered inadequate to assess early T stage. For perirectal tissue invasion, the sensitivity of EUS was statistically higher than that of CT and MRI imaging: 90% compared with 79% and 82%, respectively. The specificity estimates for EUS, CT, and MRI were comparable: 75%, 78%, and 76%, respectively. The respective sensitivity and specificity of the 3 imaging modalities to evaluate lymph nodes were also comparable: EUS, 67% and 78%; CT, 55% and 74%; and MRI, 66% and 76%.
The role of EUS in the diagnosis and treatment of RC has long been validated.1,2-5 A meta-analysis of 42 studies involving 5039 patients found EUS to be highly accurate for differentiating various T stages.8 However, EUS cannot assess iliac and mesenteric lymph nodes or posterior tumor extension beyond endopelvic fascia in advanced RC. Notable heterogeneity was found among the studies in the meta-analyses with regard to the type of equipment used for staging, as well as the criteria used to assess the depth of penetration and nodal status. The recent introduction of phased-array coils and the development of T2-weighted fast spin sequences have improved the resolution of MRI. The MERCURY trial showed that extension of tumor to within 1 mm of the circumferential margin on high-resolution MRI correctly predicted margin involvement at the time of surgery in 92% of the patients.9 In the retrospective study by Balyasnikova and colleagues, MRI was found to correctly identify partial submucosal invasion and suitability for local excision in 89% of the cases.10
Therefore, both EUS and MRI are useful, more so than CT, in assessment of the depth of tumor invasion, nodal staging, and predicting the circumferential resection margin. The use of EUS, however, does not preclude the use of MRI, or vice versa. Rather, the 2 modalities can complement each other in staging and proper patient selection for treatment.11
Despite data supporting the value of EUS in staging RC, its use is limited by a high degree of operator dependence and a substantial learning curve,12-17 which may explain the low EUS accuracy observed in some reports.7,13,15 Given the presence of recognized alternatives such as MRI, we decided to reevaluate EUS accuracy for the staging of RC outside high-volume specialized centers and prospective clinical trials.
Methods
A retrospective chart review was performed that included all consecutive patients undergoing rectal ultrasound from January 2011 to August 2015 at the US Department of Veterans Affairs Medical Center (VAMC) in Memphis, Tennessee. Sixty-five patients with short-stocked or sessile lesions < 15 cm from anal margin staged T2N0M0 or lower by endorectal ultrasound (ERUS) were included. The patients with neoplasms staged in excess of T2 or N0 were excluded from the study because treatment protocol dictates immediate neoadjuvant treatment, the administration of which would affect subsequent histopathology.
For the 37 patients included in the final analysis, ERUS results were compared with surgical pathology to ascertain accuracy. The resections were performed endoscopically or surgically with a goal of obtaining clear margins. The choice of procedure depended on size, shape, location, and depth of invasion. All patients underwent clinical and endoscopic surveillance with flexible sigmoidoscopy/EUS every 3 to 6 months for the first 2 years. We used 2 different gold standards for surveillance depending on the type of procedure performed to remove the lesion. A pathology report was the gold standard used for patients who underwent surgery. In patients who underwent endoscopic resection, we used the lack of recurrent disease, determined by normal endoscopic and endoscopic ultrasound examination, to signify complete endoscopic resection and therefore adequate staging as an early neoplasm.
Results
From January 2011 to August 2015, 65 rectal ultrasounds were performed. All EUS procedures were performed by 1 physician (C Ruben Tombazzi). All patients had previous endoscopic evaluation and tissue diagnoses. Twenty-eight patients were excluded: 18 had T3 or N1 disease, 2 had T2N0 but refused surgery, 2 had anal cancer, 3 patients with suspected cancer had benign nonneoplastic disease (2 radiation proctitis, 1 normal rectal wall), and 3 underwent EUS for benign tumors (1 ganglioneuroma and 2 lipomas).
Thirty-seven patients were included in the study, 3 of whom were staged as T2N0 and 34 as T1N0 or lower by EUS. All patients were men ranging in age from 43 to 73 years (mean, 59 years). All 37 patients underwent endoscopic or surgical resection of their early rectal neoplasm. The final pathologic evaluation of the specimens demonstrated 14 carcinoid tumors, 11 adenocarcinomas, 6 tubular adenomas with high-grade dysplasia, and 6 benign adenomas. The preoperative EUS staging was confirmed for all patients, with 100% sensitivity, specificity, and accuracy. None of the patients who underwent endoscopic or surgical transanal resection had recurrence, determined by normal endoscopic and endoscopic ultrasound appearance, during a mean of 32.6 months surveillance.
Discussion
EUS has long been a recognized method for T and N staging of RC.1,3-5,7,8 Our data confirm that, in experienced hands, EUS is highly accurate in the staging of early rectal cancers.
The impact of EUS on the management of RC was demonstrated in a Mayo Clinic prospective blinded study.1 In that cohort of 80 consecutive patients who had previously had a CT for staging, EUS altered patient management in about 30% of cases. The most common change precipatated by EUS was the indication for additional neoadjuvant treatment.
However, the results have not been as encouraging when ERUS is performed outside of strict research protocol. A multicenter, prospective, country-wide quality assurance study from > 300 German hospitals was designed to assess the diagnostic accuracy of EUS in RC.13 Of 29206 patients, 7096 underwent surgery, without neoadjuvant treatment, and were included in the final analysis. The correspondence of tumor invasion with histopathology was 64.7%, with understaging of 18% and overstaging of 17.3%.13 These numbers were better in hospitals with greater experience performing ERUS: 73% accuracy in the centers with a case load of > 30 cases per year compared with 63.2% accuracy for the centers with < 10 cases a year. Marusch and colleagues had previously demonstrated an EUS accuracy of 63.3% in a study of 1463 patients with RC in Germany.14 Another study based out of the UK had similar findings. Ashraf and colleagues performed a database analyses from 20 UK centers and identified 165 patients with RC who underwent ERUS and endoscopic microsurgery.15 Compared with histopathology, EUS had 57.1% sensitivity, 73% specificity, and 42.9% accuracy for T1 cancers; EUS accuracy was 50% for T2 and 58% for T3 tumors. The authors concluded that the general accuracy of EUS in determining stage was around 50%, the statistical equivalent of flipping a coin.
The low accuracy of EUS observed by German and British multicenter studies13-15 was attributed to the difference that may exist in clinical trials at specialized centers compared with wider use of EUS in a community setting. As seen by our data, the Memphis VAMC is not a high-volume center for the treatment of RC. However, all our EUS procedures were performed and interpreted by a single operator (C. Ruben Tombazzi) with 18 years of EUS experience. We cannot conclude that no patient was overstaged, as patients receiving a stage of T3N0 or T > N0 received neoadjuvant treatment and were not included. However, we can conclude that no patient was understaged. All patients deemed to be T1 to T2N0 included in our study received accurate staging. Our results are consistent with the high accuracy of EUS reported from other centers with experience in diagnosis and treatment of RC.1,3-5,17,18
Although EUS is accurate in differentiating T1 from T2 tumors, it cannot reliably differentiate T1 from T0 lesions. In one study, 57.6% of adenomas and 30.7% of carcinomas in situ were staged as T1 on EUS, while almost half of T1 cancers were interpreted as T0.17 This drawback is a well-known limitation of EUS; although, the misinterpretation does not affect treatment, as both T0 and T1 lesions can be treated successfully by local excision alone, which was the algorithm used for our patients. The choice of the specific procedure for local excision was left to the clinicians and included transanal endoscopic or surgical resections. At a mean follow-up of 32.6 months, none of the 37 patients who underwent endoscopic or surgical transanal resection had evidence of recurrent disease.
A limitation of EUS, or any other imaging modality, is differentiating tumor invasion from peritumoral inflammation. The inflammation can render images of tumor borders ill-defined and irregular, which hinders precise staging. However, the accurate identification of tumors with deep involvement of the submucosa (T1sm3) is of importance, because these tumors are more advanced than the superficial and intermediate T1 lesions (T1sm1 and T1sm2, respectively).
Patients with RC whose lesions are considered T1sm3 are at higher risk of harboring lymph node metastases.18 Nascimbeni and colleagues had shown that the invasion into the lower third of the submucosa (sm3) was an independent risk factor for lower cancer-free survival among patients with T1 RC.19
Unlike rectal adenocarcinomas, the prognosis for carcinoid tumors correlates not only with the depth of invasion but also with the size of the tumor. The other adverse prognostic features include poor differentiation, high mitosis index, and lymphovascular invasion.20
EUS had been shown to be highly accurate in determining the precise carcinoid tumor size, depth of invasion, and lymph node metastases.20,21 In a study of 66 resected rectal carcinoid tumors by Ishii and colleagues, 57 lesions had a diameter of ≤ 10 mm and 9 lesions had a diameter of > 10 mm.21 All of the 57 carcinoid tumors with a diameter of ≤ 10 mm were confined to the submucosa. In contrast, 5 of the 9 lesions > 10 mm invaded the muscularis propria, 6 had a lymphovascular invasion, 4 were lymph node metastases, and 1 was a liver metastasis.
In our series, 4 of the 14 carcinoid tumors were > 10 mm but none were > 20 mm. None of the carcinoids with a diameter ≤ 10 mm invaded the muscularis propria. Of the 4 carcinoids > 10 mm, 1 was T2N0 and 3 were T1N0. All carcinoid tumors in our series were low grade and with low proliferation indexes, and all were treated successfully by local excision.
Conclusion
We believe our study shows that EUS can be highly accurate in staging rectal lesions, specifically lesions that are T1-T2N0, be they adenocarcinoma or carcinoid. Although we could not assess overstaging for lesions that were staged > T2 or > N0, we w
1. Harewood GC, Wiersema MJ, Nelson H, et al. A prospective, blinded assessment of the impact of preoperative staging on the management of rectal cancer. Gastroenterology. 2002;123(1):24-32.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
3. Ahuja NK, Sauer BG, Wang AY, et al. Performance of endoscopic ultrasound in staging rectal adenocarcinoma appropriate for primary surgical resection. Clin Gastroenterol Hepatol. 2015;13:339-44.
4. Doornebosch PG, Bronkhorst PJ, Hop WC, Bode WA, Sing AK, de Graaf EJ. The role of endorectal ultrasound in therapeutic decision-making for local vs. transabdominal resection of rectal tumors. Dis Colon Rectum. 2008;51(1):38-42.
5. Santoro GA, Gizzi G, Pellegrini L, Battistella G, Di Falco G. The value of high-resolution three-dimensional endorectal ultrasonography in the management of submucosal invasive rectal tumors. Dis Colon Rectum. 2009;52(11):1837-1843.
6. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: rectal cancer, version 2.2019. https://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf. Published May 15, 2019. Accessed July 19, 2019.
7. Bipat S, Glas AS, Slors FJ, Zwinderman AH, Bossuyt PM, Stoker J. Rectal cancer: local staging and assessment of lymph node involvement with endoluminal US, CT, and MR imaging—a meta-analysis. Radiology. 2004;232(3):773-783.
8. Puli SR, Bechtold ML, Reddy JB, Choudhary A, Antillon MR, Brugge WR. How good is endoscopic ultrasound in differentiating various T stages of rectal cancer? Meta-analysis and systematic review. Ann Surg Oncol. 2009;16(2):254-265.
9. MERCURY Study Group. Diagnostic accuracy of preoperative magnetic resonance imaging in predicting curative resection of rectal cancer: prospective observational study. BMJ. 2006;333(7572):779.
10. Balyasnikova S, Read J, Wotherspoon A, et al. Diagnostic accuracy of high-resolution MRI as a method to predict potentially safe endoscopic and surgical planes in patient with early rectal cancer. BMJ Open Gastroenterol. 2017;4(1):e000151.
11. Frasson M, Garcia-Granero E, Roda D, et al. Preoperative chemoradiation may not always be needed for patients with T3 and T2N+ rectal cancer. Cancer. 2011;117(14):3118-3125.
12. Rafaelsen SR, Sørensen T, Jakobsen A, Bisgaard C, Lindebjerg J. Transrectal ultrasonography and magnetic resonance imaging in the staging of rectal cancer. Effect of experience. Scand J Gastroenterol. 2008;43(4):440-446.
13. Marusch F, Ptok H, Sahm M, et al. Endorectal ultrasound in rectal carcinoma – do the literature results really correspond to the realities of routine clinical care? Endoscopy. 2011;43(5):425-431.
14. Marusch F, Koch A, Schmidt U, et al. Routine use of transrectal ultrasound in rectal carcinoma: results of a prospective multicenter study. Endoscopy. 2002;34(5):385-390.
15. Ashraf S, Hompes R, Slater A, et al; Association of Coloproctology of Great Britain and Ireland Transanal Endoscopic Microsurgery (TEM) Collaboration. A critical appraisal of endorectal ultrasound and transanal endoscopic microsurgery and decision-making in early rectal cancer. Colorectal Dis. 2012;14(7):821-826.
16. Harewood GC. Assessment of clinical impact of endoscopic ultrasound on rectal cancer. Am J Gastroenterol. 2004;99(4):623-627.
17. Zorcolo L, Fantola G, Cabras F, Marongiu L, D’Alia G, Casula G. Preoperative staging of patients with rectal tumors suitable for transanal endoscopic microsurgery (TEM): comparison of endorectal ultrasound and histopathologic findings. Surg Endosc. 2009;23(6):1384-1389.
18. Akasu T, Kondo H, Moriya Y, et al. Endoscopic ultrasonography and treatment of early stage rectal cancer. World J Surg. 2000;24(9):1061-1068.
19. Nascimbeni R, Nivatvongs S, Larson DR, Burgart LJ. Long-term survival after local excision for T1 carcinoma of the rectum. Dis Colon Rectum. 2004;47(11):1773-1779.
20. Park CH, Cheon JH, Kim JO, et al. Criteria for decision making after endoscopic resection of well-differentiated rectal carcinoids with regard to potential lymphatic spread. Endoscopy. 2011;43(9):790-795.
21. Ishii N, Horiki N, Itoh T, et al. Endoscopic submucosal dissection and preoperative assessment with endoscopic ultrasonography for the treatment of rectal carcinoid tumors. Surg Endosc. 2010;24(6):1413-1419.
1. Harewood GC, Wiersema MJ, Nelson H, et al. A prospective, blinded assessment of the impact of preoperative staging on the management of rectal cancer. Gastroenterology. 2002;123(1):24-32.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
3. Ahuja NK, Sauer BG, Wang AY, et al. Performance of endoscopic ultrasound in staging rectal adenocarcinoma appropriate for primary surgical resection. Clin Gastroenterol Hepatol. 2015;13:339-44.
4. Doornebosch PG, Bronkhorst PJ, Hop WC, Bode WA, Sing AK, de Graaf EJ. The role of endorectal ultrasound in therapeutic decision-making for local vs. transabdominal resection of rectal tumors. Dis Colon Rectum. 2008;51(1):38-42.
5. Santoro GA, Gizzi G, Pellegrini L, Battistella G, Di Falco G. The value of high-resolution three-dimensional endorectal ultrasonography in the management of submucosal invasive rectal tumors. Dis Colon Rectum. 2009;52(11):1837-1843.
6. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: rectal cancer, version 2.2019. https://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf. Published May 15, 2019. Accessed July 19, 2019.
7. Bipat S, Glas AS, Slors FJ, Zwinderman AH, Bossuyt PM, Stoker J. Rectal cancer: local staging and assessment of lymph node involvement with endoluminal US, CT, and MR imaging—a meta-analysis. Radiology. 2004;232(3):773-783.
8. Puli SR, Bechtold ML, Reddy JB, Choudhary A, Antillon MR, Brugge WR. How good is endoscopic ultrasound in differentiating various T stages of rectal cancer? Meta-analysis and systematic review. Ann Surg Oncol. 2009;16(2):254-265.
9. MERCURY Study Group. Diagnostic accuracy of preoperative magnetic resonance imaging in predicting curative resection of rectal cancer: prospective observational study. BMJ. 2006;333(7572):779.
10. Balyasnikova S, Read J, Wotherspoon A, et al. Diagnostic accuracy of high-resolution MRI as a method to predict potentially safe endoscopic and surgical planes in patient with early rectal cancer. BMJ Open Gastroenterol. 2017;4(1):e000151.
11. Frasson M, Garcia-Granero E, Roda D, et al. Preoperative chemoradiation may not always be needed for patients with T3 and T2N+ rectal cancer. Cancer. 2011;117(14):3118-3125.
12. Rafaelsen SR, Sørensen T, Jakobsen A, Bisgaard C, Lindebjerg J. Transrectal ultrasonography and magnetic resonance imaging in the staging of rectal cancer. Effect of experience. Scand J Gastroenterol. 2008;43(4):440-446.
13. Marusch F, Ptok H, Sahm M, et al. Endorectal ultrasound in rectal carcinoma – do the literature results really correspond to the realities of routine clinical care? Endoscopy. 2011;43(5):425-431.
14. Marusch F, Koch A, Schmidt U, et al. Routine use of transrectal ultrasound in rectal carcinoma: results of a prospective multicenter study. Endoscopy. 2002;34(5):385-390.
15. Ashraf S, Hompes R, Slater A, et al; Association of Coloproctology of Great Britain and Ireland Transanal Endoscopic Microsurgery (TEM) Collaboration. A critical appraisal of endorectal ultrasound and transanal endoscopic microsurgery and decision-making in early rectal cancer. Colorectal Dis. 2012;14(7):821-826.
16. Harewood GC. Assessment of clinical impact of endoscopic ultrasound on rectal cancer. Am J Gastroenterol. 2004;99(4):623-627.
17. Zorcolo L, Fantola G, Cabras F, Marongiu L, D’Alia G, Casula G. Preoperative staging of patients with rectal tumors suitable for transanal endoscopic microsurgery (TEM): comparison of endorectal ultrasound and histopathologic findings. Surg Endosc. 2009;23(6):1384-1389.
18. Akasu T, Kondo H, Moriya Y, et al. Endoscopic ultrasonography and treatment of early stage rectal cancer. World J Surg. 2000;24(9):1061-1068.
19. Nascimbeni R, Nivatvongs S, Larson DR, Burgart LJ. Long-term survival after local excision for T1 carcinoma of the rectum. Dis Colon Rectum. 2004;47(11):1773-1779.
20. Park CH, Cheon JH, Kim JO, et al. Criteria for decision making after endoscopic resection of well-differentiated rectal carcinoids with regard to potential lymphatic spread. Endoscopy. 2011;43(9):790-795.
21. Ishii N, Horiki N, Itoh T, et al. Endoscopic submucosal dissection and preoperative assessment with endoscopic ultrasonography for the treatment of rectal carcinoid tumors. Surg Endosc. 2010;24(6):1413-1419.
T cells and IL-2 drive acute celiac symptoms
CD4+ T-cell reactivation and interleukin (IL)–2 release are responsible for acute gastrointestinal symptoms when patients with celiac disease are exposed to gluten, according to investigators.

Although T cells have been well studied in previous celiac disease research, clinical symptoms after acute gluten exposure have never been linked with specific cytokine changes, reported lead author Gautam Goel, PhD, of Massachusetts General Hospital in Boston, and colleagues.
“If treated [celiac disease] patients, i.e., those following a strict [gluten-free diet], are exposed to gluten-containing food, they typically suffer from gastrointestinal reactions occurring 1 to 2 hours after the gluten exposure,” the investigators wrote in Science Advances. “There is currently no explanation for the acute gluten-induced symptoms seen in treated [celiac disease] patients.”
The current study was prompted by two phase 1 trials involving the therapeutic vaccine Nexvax2, which uses peptide fragments of gluten proteins to desensitize celiac patients to gluten, the investigators explained. During those trials, intradermal injections of Nexvax2 above a certain dose threshold led to gastrointestinal symptoms within 2-5 hours, but not injection site reactions, which would have been indicative of a cutaneous response to recall antigen.
“Our observations from these phase 1 studies led us to hypothesize that cytokine release occurs following natural gluten exposure and could be used to implicate which arms of the immune system drive early symptoms.”
Of the 28 patients in the two trials, all underwent intradermal testing, while 19 also participated in an oral gluten challenge. Following intradermal injection of gluten peptides, patients exhibited gastrointestinal symptoms, along with coordinated elevations of least 15 plasma cytokines; most significantly IL-2, MCP-1, IL-8, IL-10, MIP-1beta, IP-10, and eotaxin. The first cytokines to respond to injection were IL-2 and IL-8, rising within 2 hours, prior to symptoms. At 4 hours, when symptoms were present, peak IL-2 elevations were most dramatic, with a 272-fold elevation, followed by IL-8 (11-fold) and IL-10 (1.2-fold).
“IL-2 is both the earliest and most sensitive marker for the coordinated cytokine release that was almost universal in HLA-DQ2.5 + [celiac disease] patients administered gluten peptides,” the investigators wrote.
Similar to intradermal testing, oral challenge with gluten caused IL-2, IL-8, and IL-10 to elevate within 2 hours, and peak within 4-6 hours. Again, IL-2 was most sensitive, with a 15-fold increase at 4 hours. This increase in IL-2 correlated with IL-8 and IL-10 elevations, although IL-2 increases were at least six times greater than the other two cytokines.
“Together, the serum cytokine profile following gluten ingestion is less prominent but qualitatively similar and over a corresponding time course to that after injecting gluten peptides, which is consistent with activated CD4+ T cells being the driver of cytokine release in both scenarios,” the investigators wrote.
Further testing showed that, after gluten challenge, plasma levels of IL-2, IL-8, and IL-10 negatively correlated with duodenal villous height-to-crypt depth ratios. In addition, high levels of IL-2 correlated with severe nausea and vomiting, adding to the evidence that celiac symptoms were linked with specific cytokine elevations.
“The link between immune activation and symptoms was further strengthened by showing that postdose symptoms and cytokine release were both lessened after three weekly doses and absent after 16 twice-weekly injections of gluten peptides,” the investigators wrote. “These findings are consistent with the difference in severity of symptoms after gluten ingestion compared to gluten peptide injection being related to potency of the antigen challenge and T-cell activation measured by circulating IL-2 concentration at 4 hours.”
Even though IL-2 elevations appeared to drive celiac symptoms, the source of IL-2 was initially unknown. “Activated T cells are the primary source of IL-2, but [dendritic cells] can also secrete IL-2 following ligation of specific pathogen recognition receptors; mast cells also secrete IL-2 following exposure to IL-33 or IL-9,” the investigators explained. Still, CD4+ T cells are known to be key players in celiac disease, and the timing and magnitude of IL-2 release made T cells the most likely candidates. To test this hypothesis, the investigators collected blood from patients 6 days after gluten food challenge and incubated these samples for 24 hours with gluten peptides. Results of this test suggested that gluten-specific CD4+ T cells were the most likely source of IL-2.
The connection between particular cytokines and gastrointestinal symptoms is now supported with evidence; however, the investigators pointed out that a relationship between cytokines and other symptoms of celiac disease remains to be seen. “Whether cytokines elevated in blood after injecting gluten peptides or ingesting gluten have any direct extraintestinal effects is unclear,” the investigators wrote. “Fatigue, headache, and ‘brain fog’ are the commonly reported extraintestinal symptoms in [celiac disease] patients. However, symptoms being focused on the upper gastrointestinal tract suggest that cytokines increased in blood have clinical and immunological effects that selectively affect the tissue from which they originate.”
“Future studies should test whether cytokine concentrations are substantially higher in gut mucosal tissue than in blood after gluten challenge or injection of gluten peptides and determine whether alterations in local cytokine levels are matched by immune and inflammatory cell infiltration,” the investigators wrote.
The study was supported by the University of Chicago Celiac Disease Center and the University of Oslo KG Jebsen Coeliac Disease Research Centre. The investigators reported additional relationships The investigators reported additional relationships with several government and nonprofit organizations. Multiple investigators are employees of ImmusanT, which is developing Nexvax2.
SOURCE: Goel G et al. Science Advances. 2019 Aug 7. doi: 10.1126/sciadv.aaw7756.
CD4+ T-cell reactivation and interleukin (IL)–2 release are responsible for acute gastrointestinal symptoms when patients with celiac disease are exposed to gluten, according to investigators.
Although T cells have been well studied in previous celiac disease research, clinical symptoms after acute gluten exposure have never been linked with specific cytokine changes, reported lead author Gautam Goel, PhD, of Massachusetts General Hospital in Boston, and colleagues.
“If treated [celiac disease] patients, i.e., those following a strict [gluten-free diet], are exposed to gluten-containing food, they typically suffer from gastrointestinal reactions occurring 1 to 2 hours after the gluten exposure,” the investigators wrote in Science Advances. “There is currently no explanation for the acute gluten-induced symptoms seen in treated [celiac disease] patients.”
The current study was prompted by two phase 1 trials involving the therapeutic vaccine Nexvax2, which uses peptide fragments of gluten proteins to desensitize celiac patients to gluten, the investigators explained. During those trials, intradermal injections of Nexvax2 above a certain dose threshold led to gastrointestinal symptoms within 2-5 hours, but not injection site reactions, which would have been indicative of a cutaneous response to recall antigen.
“Our observations from these phase 1 studies led us to hypothesize that cytokine release occurs following natural gluten exposure and could be used to implicate which arms of the immune system drive early symptoms.”
Of the 28 patients in the two trials, all underwent intradermal testing, while 19 also participated in an oral gluten challenge. Following intradermal injection of gluten peptides, patients exhibited gastrointestinal symptoms, along with coordinated elevations of least 15 plasma cytokines; most significantly IL-2, MCP-1, IL-8, IL-10, MIP-1beta, IP-10, and eotaxin. The first cytokines to respond to injection were IL-2 and IL-8, rising within 2 hours, prior to symptoms. At 4 hours, when symptoms were present, peak IL-2 elevations were most dramatic, with a 272-fold elevation, followed by IL-8 (11-fold) and IL-10 (1.2-fold).
“IL-2 is both the earliest and most sensitive marker for the coordinated cytokine release that was almost universal in HLA-DQ2.5 + [celiac disease] patients administered gluten peptides,” the investigators wrote.
Similar to intradermal testing, oral challenge with gluten caused IL-2, IL-8, and IL-10 to elevate within 2 hours, and peak within 4-6 hours. Again, IL-2 was most sensitive, with a 15-fold increase at 4 hours. This increase in IL-2 correlated with IL-8 and IL-10 elevations, although IL-2 increases were at least six times greater than the other two cytokines.
“Together, the serum cytokine profile following gluten ingestion is less prominent but qualitatively similar and over a corresponding time course to that after injecting gluten peptides, which is consistent with activated CD4+ T cells being the driver of cytokine release in both scenarios,” the investigators wrote.
Further testing showed that, after gluten challenge, plasma levels of IL-2, IL-8, and IL-10 negatively correlated with duodenal villous height-to-crypt depth ratios. In addition, high levels of IL-2 correlated with severe nausea and vomiting, adding to the evidence that celiac symptoms were linked with specific cytokine elevations.
“The link between immune activation and symptoms was further strengthened by showing that postdose symptoms and cytokine release were both lessened after three weekly doses and absent after 16 twice-weekly injections of gluten peptides,” the investigators wrote. “These findings are consistent with the difference in severity of symptoms after gluten ingestion compared to gluten peptide injection being related to potency of the antigen challenge and T-cell activation measured by circulating IL-2 concentration at 4 hours.”
Even though IL-2 elevations appeared to drive celiac symptoms, the source of IL-2 was initially unknown. “Activated T cells are the primary source of IL-2, but [dendritic cells] can also secrete IL-2 following ligation of specific pathogen recognition receptors; mast cells also secrete IL-2 following exposure to IL-33 or IL-9,” the investigators explained. Still, CD4+ T cells are known to be key players in celiac disease, and the timing and magnitude of IL-2 release made T cells the most likely candidates. To test this hypothesis, the investigators collected blood from patients 6 days after gluten food challenge and incubated these samples for 24 hours with gluten peptides. Results of this test suggested that gluten-specific CD4+ T cells were the most likely source of IL-2.
The connection between particular cytokines and gastrointestinal symptoms is now supported with evidence; however, the investigators pointed out that a relationship between cytokines and other symptoms of celiac disease remains to be seen. “Whether cytokines elevated in blood after injecting gluten peptides or ingesting gluten have any direct extraintestinal effects is unclear,” the investigators wrote. “Fatigue, headache, and ‘brain fog’ are the commonly reported extraintestinal symptoms in [celiac disease] patients. However, symptoms being focused on the upper gastrointestinal tract suggest that cytokines increased in blood have clinical and immunological effects that selectively affect the tissue from which they originate.”
“Future studies should test whether cytokine concentrations are substantially higher in gut mucosal tissue than in blood after gluten challenge or injection of gluten peptides and determine whether alterations in local cytokine levels are matched by immune and inflammatory cell infiltration,” the investigators wrote.
The study was supported by the University of Chicago Celiac Disease Center and the University of Oslo KG Jebsen Coeliac Disease Research Centre. The investigators reported additional relationships The investigators reported additional relationships with several government and nonprofit organizations. Multiple investigators are employees of ImmusanT, which is developing Nexvax2.
SOURCE: Goel G et al. Science Advances. 2019 Aug 7. doi: 10.1126/sciadv.aaw7756.
CD4+ T-cell reactivation and interleukin (IL)–2 release are responsible for acute gastrointestinal symptoms when patients with celiac disease are exposed to gluten, according to investigators.
Although T cells have been well studied in previous celiac disease research, clinical symptoms after acute gluten exposure have never been linked with specific cytokine changes, reported lead author Gautam Goel, PhD, of Massachusetts General Hospital in Boston, and colleagues.
“If treated [celiac disease] patients, i.e., those following a strict [gluten-free diet], are exposed to gluten-containing food, they typically suffer from gastrointestinal reactions occurring 1 to 2 hours after the gluten exposure,” the investigators wrote in Science Advances. “There is currently no explanation for the acute gluten-induced symptoms seen in treated [celiac disease] patients.”
The current study was prompted by two phase 1 trials involving the therapeutic vaccine Nexvax2, which uses peptide fragments of gluten proteins to desensitize celiac patients to gluten, the investigators explained. During those trials, intradermal injections of Nexvax2 above a certain dose threshold led to gastrointestinal symptoms within 2-5 hours, but not injection site reactions, which would have been indicative of a cutaneous response to recall antigen.
“Our observations from these phase 1 studies led us to hypothesize that cytokine release occurs following natural gluten exposure and could be used to implicate which arms of the immune system drive early symptoms.”
Of the 28 patients in the two trials, all underwent intradermal testing, while 19 also participated in an oral gluten challenge. Following intradermal injection of gluten peptides, patients exhibited gastrointestinal symptoms, along with coordinated elevations of least 15 plasma cytokines; most significantly IL-2, MCP-1, IL-8, IL-10, MIP-1beta, IP-10, and eotaxin. The first cytokines to respond to injection were IL-2 and IL-8, rising within 2 hours, prior to symptoms. At 4 hours, when symptoms were present, peak IL-2 elevations were most dramatic, with a 272-fold elevation, followed by IL-8 (11-fold) and IL-10 (1.2-fold).
“IL-2 is both the earliest and most sensitive marker for the coordinated cytokine release that was almost universal in HLA-DQ2.5 + [celiac disease] patients administered gluten peptides,” the investigators wrote.
Similar to intradermal testing, oral challenge with gluten caused IL-2, IL-8, and IL-10 to elevate within 2 hours, and peak within 4-6 hours. Again, IL-2 was most sensitive, with a 15-fold increase at 4 hours. This increase in IL-2 correlated with IL-8 and IL-10 elevations, although IL-2 increases were at least six times greater than the other two cytokines.
“Together, the serum cytokine profile following gluten ingestion is less prominent but qualitatively similar and over a corresponding time course to that after injecting gluten peptides, which is consistent with activated CD4+ T cells being the driver of cytokine release in both scenarios,” the investigators wrote.
Further testing showed that, after gluten challenge, plasma levels of IL-2, IL-8, and IL-10 negatively correlated with duodenal villous height-to-crypt depth ratios. In addition, high levels of IL-2 correlated with severe nausea and vomiting, adding to the evidence that celiac symptoms were linked with specific cytokine elevations.
“The link between immune activation and symptoms was further strengthened by showing that postdose symptoms and cytokine release were both lessened after three weekly doses and absent after 16 twice-weekly injections of gluten peptides,” the investigators wrote. “These findings are consistent with the difference in severity of symptoms after gluten ingestion compared to gluten peptide injection being related to potency of the antigen challenge and T-cell activation measured by circulating IL-2 concentration at 4 hours.”
Even though IL-2 elevations appeared to drive celiac symptoms, the source of IL-2 was initially unknown. “Activated T cells are the primary source of IL-2, but [dendritic cells] can also secrete IL-2 following ligation of specific pathogen recognition receptors; mast cells also secrete IL-2 following exposure to IL-33 or IL-9,” the investigators explained. Still, CD4+ T cells are known to be key players in celiac disease, and the timing and magnitude of IL-2 release made T cells the most likely candidates. To test this hypothesis, the investigators collected blood from patients 6 days after gluten food challenge and incubated these samples for 24 hours with gluten peptides. Results of this test suggested that gluten-specific CD4+ T cells were the most likely source of IL-2.
The connection between particular cytokines and gastrointestinal symptoms is now supported with evidence; however, the investigators pointed out that a relationship between cytokines and other symptoms of celiac disease remains to be seen. “Whether cytokines elevated in blood after injecting gluten peptides or ingesting gluten have any direct extraintestinal effects is unclear,” the investigators wrote. “Fatigue, headache, and ‘brain fog’ are the commonly reported extraintestinal symptoms in [celiac disease] patients. However, symptoms being focused on the upper gastrointestinal tract suggest that cytokines increased in blood have clinical and immunological effects that selectively affect the tissue from which they originate.”
“Future studies should test whether cytokine concentrations are substantially higher in gut mucosal tissue than in blood after gluten challenge or injection of gluten peptides and determine whether alterations in local cytokine levels are matched by immune and inflammatory cell infiltration,” the investigators wrote.
The study was supported by the University of Chicago Celiac Disease Center and the University of Oslo KG Jebsen Coeliac Disease Research Centre. The investigators reported additional relationships The investigators reported additional relationships with several government and nonprofit organizations. Multiple investigators are employees of ImmusanT, which is developing Nexvax2.
SOURCE: Goel G et al. Science Advances. 2019 Aug 7. doi: 10.1126/sciadv.aaw7756.
FROM SCIENCE ADVANCES
Vitamin D supplementation may improve ulcerative colitis
Vitamin D supplementation may lead to significant improvements in ulcerative colitis (UC), based on a placebo-controlled trial involving 60 patients with active disease.

Those who achieved vitamin D levels greater than 40 ng/mL were most likely to benefit, reported lead author Rizwan Ahamed Z, MD, of the Postgraduate Institute of Medical Education and Research in Chandigarh, India, and colleagues. They noted that the findings contribute much-needed clinical data to a largely theoretical subject area.
“[T]he discovery of vitamin D receptors on lymphocytes, monocytes, and dendritic cells initiated various studies which have highlighted the role of vitamin D in regulating gut mucosal immunity and gut barrier,” the investigators wrote in Journal of Clinical Gastroenterology. “In experimental interleukin (IL)-10 knockout mice models, vitamin D deficiency was found to result in severe colitis, progressive wasting, and high mortality. However, vitamin D supplementation not only prevented but also ameliorated symptoms of colitis in the mice model.”
Human studies have revealed similar associations between vitamin D supplementation and inflammatory bowel disease, such as a study by Jørgensen and colleagues that found a lower risk of relapse in Crohn’s disease, and another by Sharifi and colleagues that showed injectable vitamin D could reduce erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) in patients with UC. Still, the investigators suggested that more clinical data are needed, particularly for outcomes after vitamin D therapy. In addition to providing such data, the present trial was also the first of its kind to test oral nano vitamin D3, which may have better bioavailability than conventional supplements.
The investigators initially recruited 110 patients with active UC who had an ulcerative colitis disease activity index (UCDAI) of at least 3. After screening, 50 patients were excluded because they had vitamin D levels greater than 40 ng/mL, were already taking a vitamin D supplement, had severe UC requiring hospitalization, or exhibited severe systemic illness. The remaining 60 patients were randomized in a 1:1 ratio to receive either 60,000 IU nano vitamin D3 once daily for 8 days, or placebo. Disease parameters, which were measured at baseline and then again at 4 weeks, included UCDAI, ESR, CRP, and fecal calprotectin. The primary outcome was response, defined as a UCDAI reduction of at least 3 points. Secondary outcome measures included stool frequency, stool consistency, and remission (UCDAI less than 3); in addition, the investigators evaluated histologic, endoscopic, fecal, and serum inflammatory markers.
The majority of patients in the study were men (60%), with a mean age of 36 years. Most patients had moderate UC (73.3%), while smaller proportions had severe (18%) or mild (8%) disease. All patients were taking a 5-aminosalicylic acid oral compound and some (16.6%) were also taking azathioprine. At baseline, the mean vitamin D level was 14 ng/mL. Most patients (70%) were diagnosed with vitamin D deficiency, based on measurements below 20 ng/mL. The remaining patients were diagnosed with insufficiency (13%; 20-30 ng/mL) or suboptimal levels (17%; 30-40 ng/mL).
From baseline to 4-week follow-up, median vitamin D level in the supplement group increased from 15.4 to 40.83 ng/mL, compared with a much smaller increase in the placebo group, from 13.45 to 18.85 ng/mL. Compared with the placebo group, significantly more patients given nano vitamin D3 achieved a UCDAI 3-point reduction (53% vs 13%; P = .001); this translated to a Pearson correlation coefficient (rho) of –0.713, between vitamin D level and UCDAI. Similar, albeit less strong, inverse relationships were detected between vitamin D level and CRP (rho = −0.603) and calprotectin (rho = −0.368).
Benefits observed in the supplement group also extended to stool frequency, stool consistency, and histologic measures. Those who achieved a vitamin D level greater than 40 ng/mL were 4 times more likely to have a UCDAI 3-point reduction than those who did not meet the same criteria (80% vs 20%; P = .038). Independent predictors of response included baseline histologic activity (odds ratio, 1.92), and to a greater extent, vitamin D supplementation (OR, 9.17). No patients achieved remission, which the investigators attributed to the relatively short study duration.
Minor, self-limiting side effects occurred in 13.3% and 10% of patients given the vitamin D supplement and placebo, respectively.
“[T]he present study showed significant improvement in all inflammatory parameters of the disease including clinical, endoscopic, histopathologic, and serum and fecal markers of inflammation, all of which paralleled each other in showing [the benefit of] oral nano vitamin D supplementation,” the investigators concluded. They advised that larger, longer-term studies are needed before the findings can be generalized to all patients with active UC.
The investigators disclosed no external funding or conflicts of interest.
SOURCE: Ahamed R et al. J Clin Gastroenterol. 2019 Jul 24. doi: 10.1097/MCG.0000000000001233.
Vitamin D supplementation may lead to significant improvements in ulcerative colitis (UC), based on a placebo-controlled trial involving 60 patients with active disease.
Those who achieved vitamin D levels greater than 40 ng/mL were most likely to benefit, reported lead author Rizwan Ahamed Z, MD, of the Postgraduate Institute of Medical Education and Research in Chandigarh, India, and colleagues. They noted that the findings contribute much-needed clinical data to a largely theoretical subject area.
“[T]he discovery of vitamin D receptors on lymphocytes, monocytes, and dendritic cells initiated various studies which have highlighted the role of vitamin D in regulating gut mucosal immunity and gut barrier,” the investigators wrote in Journal of Clinical Gastroenterology. “In experimental interleukin (IL)-10 knockout mice models, vitamin D deficiency was found to result in severe colitis, progressive wasting, and high mortality. However, vitamin D supplementation not only prevented but also ameliorated symptoms of colitis in the mice model.”
Human studies have revealed similar associations between vitamin D supplementation and inflammatory bowel disease, such as a study by Jørgensen and colleagues that found a lower risk of relapse in Crohn’s disease, and another by Sharifi and colleagues that showed injectable vitamin D could reduce erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) in patients with UC. Still, the investigators suggested that more clinical data are needed, particularly for outcomes after vitamin D therapy. In addition to providing such data, the present trial was also the first of its kind to test oral nano vitamin D3, which may have better bioavailability than conventional supplements.
The investigators initially recruited 110 patients with active UC who had an ulcerative colitis disease activity index (UCDAI) of at least 3. After screening, 50 patients were excluded because they had vitamin D levels greater than 40 ng/mL, were already taking a vitamin D supplement, had severe UC requiring hospitalization, or exhibited severe systemic illness. The remaining 60 patients were randomized in a 1:1 ratio to receive either 60,000 IU nano vitamin D3 once daily for 8 days, or placebo. Disease parameters, which were measured at baseline and then again at 4 weeks, included UCDAI, ESR, CRP, and fecal calprotectin. The primary outcome was response, defined as a UCDAI reduction of at least 3 points. Secondary outcome measures included stool frequency, stool consistency, and remission (UCDAI less than 3); in addition, the investigators evaluated histologic, endoscopic, fecal, and serum inflammatory markers.
The majority of patients in the study were men (60%), with a mean age of 36 years. Most patients had moderate UC (73.3%), while smaller proportions had severe (18%) or mild (8%) disease. All patients were taking a 5-aminosalicylic acid oral compound and some (16.6%) were also taking azathioprine. At baseline, the mean vitamin D level was 14 ng/mL. Most patients (70%) were diagnosed with vitamin D deficiency, based on measurements below 20 ng/mL. The remaining patients were diagnosed with insufficiency (13%; 20-30 ng/mL) or suboptimal levels (17%; 30-40 ng/mL).
From baseline to 4-week follow-up, median vitamin D level in the supplement group increased from 15.4 to 40.83 ng/mL, compared with a much smaller increase in the placebo group, from 13.45 to 18.85 ng/mL. Compared with the placebo group, significantly more patients given nano vitamin D3 achieved a UCDAI 3-point reduction (53% vs 13%; P = .001); this translated to a Pearson correlation coefficient (rho) of –0.713, between vitamin D level and UCDAI. Similar, albeit less strong, inverse relationships were detected between vitamin D level and CRP (rho = −0.603) and calprotectin (rho = −0.368).
Benefits observed in the supplement group also extended to stool frequency, stool consistency, and histologic measures. Those who achieved a vitamin D level greater than 40 ng/mL were 4 times more likely to have a UCDAI 3-point reduction than those who did not meet the same criteria (80% vs 20%; P = .038). Independent predictors of response included baseline histologic activity (odds ratio, 1.92), and to a greater extent, vitamin D supplementation (OR, 9.17). No patients achieved remission, which the investigators attributed to the relatively short study duration.
Minor, self-limiting side effects occurred in 13.3% and 10% of patients given the vitamin D supplement and placebo, respectively.
“[T]he present study showed significant improvement in all inflammatory parameters of the disease including clinical, endoscopic, histopathologic, and serum and fecal markers of inflammation, all of which paralleled each other in showing [the benefit of] oral nano vitamin D supplementation,” the investigators concluded. They advised that larger, longer-term studies are needed before the findings can be generalized to all patients with active UC.
The investigators disclosed no external funding or conflicts of interest.
SOURCE: Ahamed R et al. J Clin Gastroenterol. 2019 Jul 24. doi: 10.1097/MCG.0000000000001233.
Vitamin D supplementation may lead to significant improvements in ulcerative colitis (UC), based on a placebo-controlled trial involving 60 patients with active disease.
Those who achieved vitamin D levels greater than 40 ng/mL were most likely to benefit, reported lead author Rizwan Ahamed Z, MD, of the Postgraduate Institute of Medical Education and Research in Chandigarh, India, and colleagues. They noted that the findings contribute much-needed clinical data to a largely theoretical subject area.
“[T]he discovery of vitamin D receptors on lymphocytes, monocytes, and dendritic cells initiated various studies which have highlighted the role of vitamin D in regulating gut mucosal immunity and gut barrier,” the investigators wrote in Journal of Clinical Gastroenterology. “In experimental interleukin (IL)-10 knockout mice models, vitamin D deficiency was found to result in severe colitis, progressive wasting, and high mortality. However, vitamin D supplementation not only prevented but also ameliorated symptoms of colitis in the mice model.”
Human studies have revealed similar associations between vitamin D supplementation and inflammatory bowel disease, such as a study by Jørgensen and colleagues that found a lower risk of relapse in Crohn’s disease, and another by Sharifi and colleagues that showed injectable vitamin D could reduce erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) in patients with UC. Still, the investigators suggested that more clinical data are needed, particularly for outcomes after vitamin D therapy. In addition to providing such data, the present trial was also the first of its kind to test oral nano vitamin D3, which may have better bioavailability than conventional supplements.
The investigators initially recruited 110 patients with active UC who had an ulcerative colitis disease activity index (UCDAI) of at least 3. After screening, 50 patients were excluded because they had vitamin D levels greater than 40 ng/mL, were already taking a vitamin D supplement, had severe UC requiring hospitalization, or exhibited severe systemic illness. The remaining 60 patients were randomized in a 1:1 ratio to receive either 60,000 IU nano vitamin D3 once daily for 8 days, or placebo. Disease parameters, which were measured at baseline and then again at 4 weeks, included UCDAI, ESR, CRP, and fecal calprotectin. The primary outcome was response, defined as a UCDAI reduction of at least 3 points. Secondary outcome measures included stool frequency, stool consistency, and remission (UCDAI less than 3); in addition, the investigators evaluated histologic, endoscopic, fecal, and serum inflammatory markers.
The majority of patients in the study were men (60%), with a mean age of 36 years. Most patients had moderate UC (73.3%), while smaller proportions had severe (18%) or mild (8%) disease. All patients were taking a 5-aminosalicylic acid oral compound and some (16.6%) were also taking azathioprine. At baseline, the mean vitamin D level was 14 ng/mL. Most patients (70%) were diagnosed with vitamin D deficiency, based on measurements below 20 ng/mL. The remaining patients were diagnosed with insufficiency (13%; 20-30 ng/mL) or suboptimal levels (17%; 30-40 ng/mL).
From baseline to 4-week follow-up, median vitamin D level in the supplement group increased from 15.4 to 40.83 ng/mL, compared with a much smaller increase in the placebo group, from 13.45 to 18.85 ng/mL. Compared with the placebo group, significantly more patients given nano vitamin D3 achieved a UCDAI 3-point reduction (53% vs 13%; P = .001); this translated to a Pearson correlation coefficient (rho) of –0.713, between vitamin D level and UCDAI. Similar, albeit less strong, inverse relationships were detected between vitamin D level and CRP (rho = −0.603) and calprotectin (rho = −0.368).
Benefits observed in the supplement group also extended to stool frequency, stool consistency, and histologic measures. Those who achieved a vitamin D level greater than 40 ng/mL were 4 times more likely to have a UCDAI 3-point reduction than those who did not meet the same criteria (80% vs 20%; P = .038). Independent predictors of response included baseline histologic activity (odds ratio, 1.92), and to a greater extent, vitamin D supplementation (OR, 9.17). No patients achieved remission, which the investigators attributed to the relatively short study duration.
Minor, self-limiting side effects occurred in 13.3% and 10% of patients given the vitamin D supplement and placebo, respectively.
“[T]he present study showed significant improvement in all inflammatory parameters of the disease including clinical, endoscopic, histopathologic, and serum and fecal markers of inflammation, all of which paralleled each other in showing [the benefit of] oral nano vitamin D supplementation,” the investigators concluded. They advised that larger, longer-term studies are needed before the findings can be generalized to all patients with active UC.
The investigators disclosed no external funding or conflicts of interest.
SOURCE: Ahamed R et al. J Clin Gastroenterol. 2019 Jul 24. doi: 10.1097/MCG.0000000000001233.
FROM THE JOURNAL OF CLINICAL GASTROENTEROLOGY

Hospital slashes S. aureus vancomycin resistance
LJUBLJANA, SLOVENIA – , Johannes Huebner, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
He presented a retrospective analysis of S. aureus isolates obtained from 540 patients at the Dr. von Hauner Children’s Hospital, Munich, from 2002 to 2017. All were either newly identified methicillin-resistant S. aureus (MRSA) or specimens from bacteremic children with invasive MRSA or methicillin-sensitive S. aureus (MSSA). The strains were tested for vancomycin resistance and minimum inhibitory concentration (MIC). The results from the 200 isolates obtained from 2002 to 2009 were then compared to the 340 specimens from 2010 to 2017, when antibiotic stewardship programs rose to the fore at the pediatric hospital.
All samples proved to be vancomycin sensitive. The further good news was there was absolutely no evidence of the worrisome vancomycin MIC creep that has been described at some centers. On the contrary, the MIC was significantly lower in the later samples, at 0.99 mcg/mL, compared with 1.11 mcg/mL in the earlier period. Moreover, the prevalence of heterogeneous glycopeptide-intermediate S. aureus (hGISA) – a phenotype that has been associated with increased rates of treatment failure – improved from 25% in the earlier period to 6% during the later period, reported Dr. Huebner, head of the division of pediatric infectious diseases at the children’s hospital, part of the University of Munich.
Vancomycin MICs weren’t significantly different between the MRSA and MSSA samples.
Based upon this favorable institutional experience, vancomycin remains the first-line treatment for suspected severe gram-positive cocci infections as well as proven infections involving MRSA at Dr. von Hauner Children’s Hospital.
These vancomycin MIC and hGISA data underscore the importance of periodically monitoring local S. aureus antimicrobial susceptibilities, which, as in this case, can differ from the broader global trends. The vancomycin MIC creep issue hadn’t been studied previously in German hospitals, according to Dr. Huebner.
He and his coworkers have published details of the elements of pediatric antibiotic stewardship programs they have found to be most effective (Infection. 2017 Aug;45[4]:493-504) as well as a systematic review of studies on the favorable economic impact of such programs (J Hosp Infect. 2019 Aug;102[4]:369-376).
Dr. Huebner reported having no financial conflicts regarding his study, which was conducted free of commercial support.
LJUBLJANA, SLOVENIA – , Johannes Huebner, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
He presented a retrospective analysis of S. aureus isolates obtained from 540 patients at the Dr. von Hauner Children’s Hospital, Munich, from 2002 to 2017. All were either newly identified methicillin-resistant S. aureus (MRSA) or specimens from bacteremic children with invasive MRSA or methicillin-sensitive S. aureus (MSSA). The strains were tested for vancomycin resistance and minimum inhibitory concentration (MIC). The results from the 200 isolates obtained from 2002 to 2009 were then compared to the 340 specimens from 2010 to 2017, when antibiotic stewardship programs rose to the fore at the pediatric hospital.
All samples proved to be vancomycin sensitive. The further good news was there was absolutely no evidence of the worrisome vancomycin MIC creep that has been described at some centers. On the contrary, the MIC was significantly lower in the later samples, at 0.99 mcg/mL, compared with 1.11 mcg/mL in the earlier period. Moreover, the prevalence of heterogeneous glycopeptide-intermediate S. aureus (hGISA) – a phenotype that has been associated with increased rates of treatment failure – improved from 25% in the earlier period to 6% during the later period, reported Dr. Huebner, head of the division of pediatric infectious diseases at the children’s hospital, part of the University of Munich.
Vancomycin MICs weren’t significantly different between the MRSA and MSSA samples.
Based upon this favorable institutional experience, vancomycin remains the first-line treatment for suspected severe gram-positive cocci infections as well as proven infections involving MRSA at Dr. von Hauner Children’s Hospital.
These vancomycin MIC and hGISA data underscore the importance of periodically monitoring local S. aureus antimicrobial susceptibilities, which, as in this case, can differ from the broader global trends. The vancomycin MIC creep issue hadn’t been studied previously in German hospitals, according to Dr. Huebner.
He and his coworkers have published details of the elements of pediatric antibiotic stewardship programs they have found to be most effective (Infection. 2017 Aug;45[4]:493-504) as well as a systematic review of studies on the favorable economic impact of such programs (J Hosp Infect. 2019 Aug;102[4]:369-376).
Dr. Huebner reported having no financial conflicts regarding his study, which was conducted free of commercial support.
LJUBLJANA, SLOVENIA – , Johannes Huebner, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
He presented a retrospective analysis of S. aureus isolates obtained from 540 patients at the Dr. von Hauner Children’s Hospital, Munich, from 2002 to 2017. All were either newly identified methicillin-resistant S. aureus (MRSA) or specimens from bacteremic children with invasive MRSA or methicillin-sensitive S. aureus (MSSA). The strains were tested for vancomycin resistance and minimum inhibitory concentration (MIC). The results from the 200 isolates obtained from 2002 to 2009 were then compared to the 340 specimens from 2010 to 2017, when antibiotic stewardship programs rose to the fore at the pediatric hospital.
All samples proved to be vancomycin sensitive. The further good news was there was absolutely no evidence of the worrisome vancomycin MIC creep that has been described at some centers. On the contrary, the MIC was significantly lower in the later samples, at 0.99 mcg/mL, compared with 1.11 mcg/mL in the earlier period. Moreover, the prevalence of heterogeneous glycopeptide-intermediate S. aureus (hGISA) – a phenotype that has been associated with increased rates of treatment failure – improved from 25% in the earlier period to 6% during the later period, reported Dr. Huebner, head of the division of pediatric infectious diseases at the children’s hospital, part of the University of Munich.
Vancomycin MICs weren’t significantly different between the MRSA and MSSA samples.
Based upon this favorable institutional experience, vancomycin remains the first-line treatment for suspected severe gram-positive cocci infections as well as proven infections involving MRSA at Dr. von Hauner Children’s Hospital.
These vancomycin MIC and hGISA data underscore the importance of periodically monitoring local S. aureus antimicrobial susceptibilities, which, as in this case, can differ from the broader global trends. The vancomycin MIC creep issue hadn’t been studied previously in German hospitals, according to Dr. Huebner.
He and his coworkers have published details of the elements of pediatric antibiotic stewardship programs they have found to be most effective (Infection. 2017 Aug;45[4]:493-504) as well as a systematic review of studies on the favorable economic impact of such programs (J Hosp Infect. 2019 Aug;102[4]:369-376).
Dr. Huebner reported having no financial conflicts regarding his study, which was conducted free of commercial support.
REPORTING FROM ESPID 2019
Key clinical point: Staphylococcus aureus vancomycin MIC creep is reversible through dedicated antimicrobial stewardship.
Major finding: The prevalence of hGISA in MRSA and MSSA specimens improved from 25% during 2002-2009 to 6% during 2010-2017 at one German tertiary children’s hospital.
Study details: This was a retrospective single-center analysis of vancomycin resistance trends over time in 540 S. aureus specimens gathered in 2002-2017.
Disclosures: The presenter reported having no financial conflicts regarding this study, which was conducted free of commercial support.
Urine albumin testing is crucial for patients at risk for CKD, but drop the 24-hour urine test
SAN DIEGO – Whatever you do, don’t order a 24-hour urine test. Do encourage “pork holidays.” And choose between an ACE inhibitor and an ARB – don’t give them both to a single patient, according to Kim Zuber, PA-C, MS, a nephrology physician assistant from St. Petersburg, Fla., in a presentation about kidney disease, hypertension, and diabetes at the Metabolic & Endocrine Disease Summit, sponsored by Global Academy for Medical Education.

Ms. Zuber, who is the executive director of the American Academy of Nephrology PAs and the outreach chair of the National Kidney Foundation, outlined some approaches for the diagnosis, management, and treatment of chronic kidney disease (CKD) with comorbid hypertension and diabetes.
- Use the right urine test. Ms. Zuber said, although it’s often not performed. In fact, research suggests that most Medicare patients with diabetes, hypertension, or both do not have this test, she said. Order a urine albumin-to-creatinine ratio (UACR) test at least once a year in at-risk patients, she recommended, and more frequently if they show signs of abnormal values. But be aware, she said, some labs might refer to the test as microalbuminuria instead of UACR, and be prepared to calculate the UACR yourself if your institution provides only albumin and creatinine levels. Also watch out for mix-ups regarding UACR measurements. Nephrotic-range proteinuria starts at 3 g/dL or 3,000 mg/dL, she said, and residents often confuse those two sets of units. “Many have gotten in trouble with that,” she said.
- Don’t go near a 24-hour urine test. Thinking about ordering a 24-hour urine test that requires a patients to collect all their urine for a day? Think again. “We’ve been telling you almost 20 years not to do this,” Ms. Zuber said. These tests “are unreliable, and they don’t work.”
- Don’t focus on tight blood pressure control. Studies provide little insight into the ideal blood pressure readings for patients with diabetes and CKD, according to Ms. Zuber, but some findings suggest that tight control can be harmful to the kidneys. She urges her patients to treat hypertension in part by embracing lifestyle change. “I tell them that if you improve your lifestyle, you can give up one of your drugs. When they average 15 drugs a day, that becomes popular.” Physical activity, the DASH diet, salt restriction, moderate alcohol consumption, weight loss, stress reduction, and smoking cessation can all lower blood pressure, she added.
- Talk up the “pork holiday.” For patients with hypertension, “sodium restriction is huge,” Ms. Zuber said, especially among black patients. She urges her patients to take “pork holidays”, that is, eat pork only four times a year, on holidays such as the Fourth of July. She also urges them to prepare food in ways that begin with B, as in bake, boil, and barbecue. “You’ll notice that ‘fry’ doesn’t start with a B.”
- Try an ACE inhibitor or an ARB, but not both. In patients with hypertension plus diabetes and/or CKD, Ms. Zuber suggests using an angiotensin-converting enzyme inhibitor or angiotensin receptor blockers, but not both. One or the other can improve albuminuria, she said, but together they can boost risk of CKD, hyperkalemia, and hypotension.
Consider factors such as formularies and personal experience when trying to decide which drug to use, she said. If a patient still has hypertension, consider a diuretic and then move to a calcium channel or beta blocker. However, she cautioned, although beta blockers, they can cause erectile dysfunction.
Global Academy for Medical Education and this news organization are owned by the same parent company. Ms. Zuber reported no disclosures.
SAN DIEGO – Whatever you do, don’t order a 24-hour urine test. Do encourage “pork holidays.” And choose between an ACE inhibitor and an ARB – don’t give them both to a single patient, according to Kim Zuber, PA-C, MS, a nephrology physician assistant from St. Petersburg, Fla., in a presentation about kidney disease, hypertension, and diabetes at the Metabolic & Endocrine Disease Summit, sponsored by Global Academy for Medical Education.
Ms. Zuber, who is the executive director of the American Academy of Nephrology PAs and the outreach chair of the National Kidney Foundation, outlined some approaches for the diagnosis, management, and treatment of chronic kidney disease (CKD) with comorbid hypertension and diabetes.
- Use the right urine test. Ms. Zuber said, although it’s often not performed. In fact, research suggests that most Medicare patients with diabetes, hypertension, or both do not have this test, she said. Order a urine albumin-to-creatinine ratio (UACR) test at least once a year in at-risk patients, she recommended, and more frequently if they show signs of abnormal values. But be aware, she said, some labs might refer to the test as microalbuminuria instead of UACR, and be prepared to calculate the UACR yourself if your institution provides only albumin and creatinine levels. Also watch out for mix-ups regarding UACR measurements. Nephrotic-range proteinuria starts at 3 g/dL or 3,000 mg/dL, she said, and residents often confuse those two sets of units. “Many have gotten in trouble with that,” she said.
- Don’t go near a 24-hour urine test. Thinking about ordering a 24-hour urine test that requires a patients to collect all their urine for a day? Think again. “We’ve been telling you almost 20 years not to do this,” Ms. Zuber said. These tests “are unreliable, and they don’t work.”
- Don’t focus on tight blood pressure control. Studies provide little insight into the ideal blood pressure readings for patients with diabetes and CKD, according to Ms. Zuber, but some findings suggest that tight control can be harmful to the kidneys. She urges her patients to treat hypertension in part by embracing lifestyle change. “I tell them that if you improve your lifestyle, you can give up one of your drugs. When they average 15 drugs a day, that becomes popular.” Physical activity, the DASH diet, salt restriction, moderate alcohol consumption, weight loss, stress reduction, and smoking cessation can all lower blood pressure, she added.
- Talk up the “pork holiday.” For patients with hypertension, “sodium restriction is huge,” Ms. Zuber said, especially among black patients. She urges her patients to take “pork holidays”, that is, eat pork only four times a year, on holidays such as the Fourth of July. She also urges them to prepare food in ways that begin with B, as in bake, boil, and barbecue. “You’ll notice that ‘fry’ doesn’t start with a B.”
- Try an ACE inhibitor or an ARB, but not both. In patients with hypertension plus diabetes and/or CKD, Ms. Zuber suggests using an angiotensin-converting enzyme inhibitor or angiotensin receptor blockers, but not both. One or the other can improve albuminuria, she said, but together they can boost risk of CKD, hyperkalemia, and hypotension.
Consider factors such as formularies and personal experience when trying to decide which drug to use, she said. If a patient still has hypertension, consider a diuretic and then move to a calcium channel or beta blocker. However, she cautioned, although beta blockers, they can cause erectile dysfunction.
Global Academy for Medical Education and this news organization are owned by the same parent company. Ms. Zuber reported no disclosures.
SAN DIEGO – Whatever you do, don’t order a 24-hour urine test. Do encourage “pork holidays.” And choose between an ACE inhibitor and an ARB – don’t give them both to a single patient, according to Kim Zuber, PA-C, MS, a nephrology physician assistant from St. Petersburg, Fla., in a presentation about kidney disease, hypertension, and diabetes at the Metabolic & Endocrine Disease Summit, sponsored by Global Academy for Medical Education.
Ms. Zuber, who is the executive director of the American Academy of Nephrology PAs and the outreach chair of the National Kidney Foundation, outlined some approaches for the diagnosis, management, and treatment of chronic kidney disease (CKD) with comorbid hypertension and diabetes.
- Use the right urine test. Ms. Zuber said, although it’s often not performed. In fact, research suggests that most Medicare patients with diabetes, hypertension, or both do not have this test, she said. Order a urine albumin-to-creatinine ratio (UACR) test at least once a year in at-risk patients, she recommended, and more frequently if they show signs of abnormal values. But be aware, she said, some labs might refer to the test as microalbuminuria instead of UACR, and be prepared to calculate the UACR yourself if your institution provides only albumin and creatinine levels. Also watch out for mix-ups regarding UACR measurements. Nephrotic-range proteinuria starts at 3 g/dL or 3,000 mg/dL, she said, and residents often confuse those two sets of units. “Many have gotten in trouble with that,” she said.
- Don’t go near a 24-hour urine test. Thinking about ordering a 24-hour urine test that requires a patients to collect all their urine for a day? Think again. “We’ve been telling you almost 20 years not to do this,” Ms. Zuber said. These tests “are unreliable, and they don’t work.”
- Don’t focus on tight blood pressure control. Studies provide little insight into the ideal blood pressure readings for patients with diabetes and CKD, according to Ms. Zuber, but some findings suggest that tight control can be harmful to the kidneys. She urges her patients to treat hypertension in part by embracing lifestyle change. “I tell them that if you improve your lifestyle, you can give up one of your drugs. When they average 15 drugs a day, that becomes popular.” Physical activity, the DASH diet, salt restriction, moderate alcohol consumption, weight loss, stress reduction, and smoking cessation can all lower blood pressure, she added.
- Talk up the “pork holiday.” For patients with hypertension, “sodium restriction is huge,” Ms. Zuber said, especially among black patients. She urges her patients to take “pork holidays”, that is, eat pork only four times a year, on holidays such as the Fourth of July. She also urges them to prepare food in ways that begin with B, as in bake, boil, and barbecue. “You’ll notice that ‘fry’ doesn’t start with a B.”
- Try an ACE inhibitor or an ARB, but not both. In patients with hypertension plus diabetes and/or CKD, Ms. Zuber suggests using an angiotensin-converting enzyme inhibitor or angiotensin receptor blockers, but not both. One or the other can improve albuminuria, she said, but together they can boost risk of CKD, hyperkalemia, and hypotension.
Consider factors such as formularies and personal experience when trying to decide which drug to use, she said. If a patient still has hypertension, consider a diuretic and then move to a calcium channel or beta blocker. However, she cautioned, although beta blockers, they can cause erectile dysfunction.
Global Academy for Medical Education and this news organization are owned by the same parent company. Ms. Zuber reported no disclosures.
EXPERT ANALYSIS FROM MEDS 2019
Low-dose radiation therapy looks effective in hard-to-treat MCL
Low-dose radiation therapy – with or without concurrent chemotherapy – appears promising as a treatment for patients with relapsed or refractory mantle cell lymphoma (MCL) or at least a bridge to subsequent therapy, according to findings published in Blood Advances.

Matthew S. Ning, MD, of the department of radiation oncology at the University of Texas MD Anderson Cancer Center, Houston, and colleagues, said this is the first study to evaluate low-dose radiation therapy (LDRT) with chemotherapy as a treatment modality outside of palliative care for relapsed, multiple refractory MCL patients.
“Our findings indicate that LDRT imparts excellent [local control], minimal toxicity, and favorable outcomes in this setting,” the researchers said.
The study included 19 patients with a total of 98 sites of relapsed, refractory MCL who were treated from 2014 to 2018. The median follow-up was 51.3 months from initial diagnosis and 15.4 months from initial treatment with low-dose radiation therapy, given at a dose of 4 Gy.
These were hard-to-treat patients who had received multiple prior therapies since diagnosis, including carfilzomib, ibrutinib, bortezomib, anthracycline, and rituximab. In total, 8 of the patients had previously undergone autologous stem cell transplant and 11 were refractory to ibrutinib by the time of initial radiation therapy.
Median age of the patients was 69 years; 15 patients had classical histology and 4 had blastoid variant. Among the 98 tumor sites treated, the median tumor size was 2.8 cm.
In all, 14 patients received initial LDRT that was concurrent with chemotherapy. The remaining 5 patients had stopped chemotherapy prior to starting LDRT.
LDRT was given in 1-2 daily fractions via 3-dimensional conformal radiation therapy or electron beam.
Of the 98 tumor sites treated, complete response was achieved for 79 sites (81%) and the median time to complete response was 2.7 months after the start of LDRT. The researchers removed one patient who was an outlier with 27 tumor sites treated, and that dropped the complete response rate down to 76%. The overall response rate, which include an additional five sites with partial response, was 86%.
The researchers found links between complete response and soft tissue site versus non–soft tissue site (hazard ratio, 1.80; 1.12-2.90, P = .02). However, there were no associations between response and chemo-refractory status, ibrutinib-refractory status, prior chemotherapy courts, receipt of concurrent chemotherapy, tumor size, number of fractions, lesions treated per course, or blastoid variant.
The overall survival at 1 year after LDRT initiation was 90% and the 1-year progression-free survival was 55%. All five patients who died were refractory to ibrutinib.
The researchers reported finding no radiation therapy–related toxicities, even when patients received concurrent chemotherapy.
The use of LDRT has the potential to bridge refractory patients to subsequent therapies or to provide treatment breaks as patients recover from toxicities, the researchers said. However, they called for additional studies to confirm that this approach improves progression-free survival over chemotherapy alone.
The study was supported in part by a grant from the National Cancer Institute. The researchers reported having no competing financial interests.
SOURCE: Ning MS et al. Blood Adv. 2019. Jul 9;3(13):2035-9.
Low-dose radiation therapy – with or without concurrent chemotherapy – appears promising as a treatment for patients with relapsed or refractory mantle cell lymphoma (MCL) or at least a bridge to subsequent therapy, according to findings published in Blood Advances.
Matthew S. Ning, MD, of the department of radiation oncology at the University of Texas MD Anderson Cancer Center, Houston, and colleagues, said this is the first study to evaluate low-dose radiation therapy (LDRT) with chemotherapy as a treatment modality outside of palliative care for relapsed, multiple refractory MCL patients.
“Our findings indicate that LDRT imparts excellent [local control], minimal toxicity, and favorable outcomes in this setting,” the researchers said.
The study included 19 patients with a total of 98 sites of relapsed, refractory MCL who were treated from 2014 to 2018. The median follow-up was 51.3 months from initial diagnosis and 15.4 months from initial treatment with low-dose radiation therapy, given at a dose of 4 Gy.
These were hard-to-treat patients who had received multiple prior therapies since diagnosis, including carfilzomib, ibrutinib, bortezomib, anthracycline, and rituximab. In total, 8 of the patients had previously undergone autologous stem cell transplant and 11 were refractory to ibrutinib by the time of initial radiation therapy.
Median age of the patients was 69 years; 15 patients had classical histology and 4 had blastoid variant. Among the 98 tumor sites treated, the median tumor size was 2.8 cm.
In all, 14 patients received initial LDRT that was concurrent with chemotherapy. The remaining 5 patients had stopped chemotherapy prior to starting LDRT.
LDRT was given in 1-2 daily fractions via 3-dimensional conformal radiation therapy or electron beam.
Of the 98 tumor sites treated, complete response was achieved for 79 sites (81%) and the median time to complete response was 2.7 months after the start of LDRT. The researchers removed one patient who was an outlier with 27 tumor sites treated, and that dropped the complete response rate down to 76%. The overall response rate, which include an additional five sites with partial response, was 86%.
The researchers found links between complete response and soft tissue site versus non–soft tissue site (hazard ratio, 1.80; 1.12-2.90, P = .02). However, there were no associations between response and chemo-refractory status, ibrutinib-refractory status, prior chemotherapy courts, receipt of concurrent chemotherapy, tumor size, number of fractions, lesions treated per course, or blastoid variant.
The overall survival at 1 year after LDRT initiation was 90% and the 1-year progression-free survival was 55%. All five patients who died were refractory to ibrutinib.
The researchers reported finding no radiation therapy–related toxicities, even when patients received concurrent chemotherapy.
The use of LDRT has the potential to bridge refractory patients to subsequent therapies or to provide treatment breaks as patients recover from toxicities, the researchers said. However, they called for additional studies to confirm that this approach improves progression-free survival over chemotherapy alone.
The study was supported in part by a grant from the National Cancer Institute. The researchers reported having no competing financial interests.
SOURCE: Ning MS et al. Blood Adv. 2019. Jul 9;3(13):2035-9.
Low-dose radiation therapy – with or without concurrent chemotherapy – appears promising as a treatment for patients with relapsed or refractory mantle cell lymphoma (MCL) or at least a bridge to subsequent therapy, according to findings published in Blood Advances.
Matthew S. Ning, MD, of the department of radiation oncology at the University of Texas MD Anderson Cancer Center, Houston, and colleagues, said this is the first study to evaluate low-dose radiation therapy (LDRT) with chemotherapy as a treatment modality outside of palliative care for relapsed, multiple refractory MCL patients.
“Our findings indicate that LDRT imparts excellent [local control], minimal toxicity, and favorable outcomes in this setting,” the researchers said.
The study included 19 patients with a total of 98 sites of relapsed, refractory MCL who were treated from 2014 to 2018. The median follow-up was 51.3 months from initial diagnosis and 15.4 months from initial treatment with low-dose radiation therapy, given at a dose of 4 Gy.
These were hard-to-treat patients who had received multiple prior therapies since diagnosis, including carfilzomib, ibrutinib, bortezomib, anthracycline, and rituximab. In total, 8 of the patients had previously undergone autologous stem cell transplant and 11 were refractory to ibrutinib by the time of initial radiation therapy.
Median age of the patients was 69 years; 15 patients had classical histology and 4 had blastoid variant. Among the 98 tumor sites treated, the median tumor size was 2.8 cm.
In all, 14 patients received initial LDRT that was concurrent with chemotherapy. The remaining 5 patients had stopped chemotherapy prior to starting LDRT.
LDRT was given in 1-2 daily fractions via 3-dimensional conformal radiation therapy or electron beam.
Of the 98 tumor sites treated, complete response was achieved for 79 sites (81%) and the median time to complete response was 2.7 months after the start of LDRT. The researchers removed one patient who was an outlier with 27 tumor sites treated, and that dropped the complete response rate down to 76%. The overall response rate, which include an additional five sites with partial response, was 86%.
The researchers found links between complete response and soft tissue site versus non–soft tissue site (hazard ratio, 1.80; 1.12-2.90, P = .02). However, there were no associations between response and chemo-refractory status, ibrutinib-refractory status, prior chemotherapy courts, receipt of concurrent chemotherapy, tumor size, number of fractions, lesions treated per course, or blastoid variant.
The overall survival at 1 year after LDRT initiation was 90% and the 1-year progression-free survival was 55%. All five patients who died were refractory to ibrutinib.
The researchers reported finding no radiation therapy–related toxicities, even when patients received concurrent chemotherapy.
The use of LDRT has the potential to bridge refractory patients to subsequent therapies or to provide treatment breaks as patients recover from toxicities, the researchers said. However, they called for additional studies to confirm that this approach improves progression-free survival over chemotherapy alone.
The study was supported in part by a grant from the National Cancer Institute. The researchers reported having no competing financial interests.
SOURCE: Ning MS et al. Blood Adv. 2019. Jul 9;3(13):2035-9.
FROM BLOOD ADVANCES
Key clinical point:
Major finding: The overall survival was 90% at 1 year following the initiation of low-dose radiation therapy (4 Gy).
Study details: A study of 19 patients with relapsed, refractory mantle cell lymphoma who received low-dose radiation at doses of 4 Gy at 98 sites of disease.
Disclosures: The study was supported in part by a grant from the National Cancer Institute. The researchers reported having no competing financial interests.
Source: Ning MS et al. Blood Adv. 2019. Jul 9;3(13):2035-9.
Bevacizumab or pemetrexed, but not both, efficacious for NSCLC maintenance
Single-agent therapy with either bevacizumab or pemetrexed is efficacious as maintenance therapy for patients with advanced nonsquamous non–small cell lung cancer (NSCLC), but the combination of the two agents offers no survival benefit and is more toxic, results of the randomized ECOG-ACRIN 5508 study show.

For patients with no disease progression after four cycles of induction chemotherapy who were assigned to one of three maintenance therapy strategies, there were no differences in overall survival between those randomized to monotherapy with pemetrexed or bevacizumab or to a combination of the two agents, although progression-free survival (PFS) was better with the combination, reported Suresh S. Ramalingam, MD, from the Winship Cancer Institute of Emory University, Atlanta, and colleagues.
The incidence of grade 3 or greater adverse events was also significantly higher with the combination, compared with bevacizumab monotherapy.
Even in the age of immune checkpoint inhibitor therapy in the front line, “[i]t is clear that maintenance therapy will remain an integral part of the treatment approach to advanced nonsquamous NSCLC. The results of ECOG-ACRIN 5508 support the use of either pemetrexed or bevacizumab as a single agent in this setting,” the investigators wrote in the Journal of Clinical Oncology.
Although the combination of bevacizumab and pemetrexed was associated with a significant improvement in PFS in a randomized trial, the three maintenance strategies have never before been directly compared, Dr. Ramalingam and associates noted.
In ECOG-ACRIN 5508, 1,516 patients with advanced nonsquamous NSCLC who had not received prior systemic therapy were given standard induction chemotherapy, consisting of carboplatin to an area under the curve of 6, paclitaxel 200 mg/m2, and bevacizumab 15 mg/kg for up to four cycles.
Patients without progression after four cycles (874) were randomly assigned to maintenance therapy with bevacizumab at 15 mg/kg , pemetrexed 500 mg/m2, or a combination of the two agents.
For the primary endpoint of overall survival, with bevacizumab serving as the control group for comparison, the investigators found that, at a median follow-up of 50.6 months, median survival was 15.9 months with pemetrexed, compared with 14.4 months with bevacizumab, a difference that was not statistically significant. Median survival with the combination was 16.4 months and was also not significantly different from bevacizumab.
Median PFS was 4.2 months and 5.1 months for the pemetrexed and bevacizumab groups, respectively, with no significant difference. In contrast, median was 7.5 months with the combination, which was significantly better than controls, with a hazard ratio of 0.67 (P less than .001).
Patients received a median of six maintenance therapy cycles for each of the single agents, and a median of eight for the combinations. The incidence of grade 3-4 toxicity was 29% with bevacizumab, 37% with pemetrexed, and 51% with the combination. The combination was associated with a significantly greater incidence of toxicities, compared with bevacizumab (P less than .001).
The study was supported by grants from the National Cancer Institute. Dr. Ramalingam reported a consulting/advisory role for bevacizumab maker Genentech/Roche and others. Multiple coauthors reported similar disclosures.
SOURCE: Ramalingam SS et al. J Clin Oncol. 2019 Jul 30. doi: 10.1200/JCO.19.01006.
Single-agent therapy with either bevacizumab or pemetrexed is efficacious as maintenance therapy for patients with advanced nonsquamous non–small cell lung cancer (NSCLC), but the combination of the two agents offers no survival benefit and is more toxic, results of the randomized ECOG-ACRIN 5508 study show.
For patients with no disease progression after four cycles of induction chemotherapy who were assigned to one of three maintenance therapy strategies, there were no differences in overall survival between those randomized to monotherapy with pemetrexed or bevacizumab or to a combination of the two agents, although progression-free survival (PFS) was better with the combination, reported Suresh S. Ramalingam, MD, from the Winship Cancer Institute of Emory University, Atlanta, and colleagues.
The incidence of grade 3 or greater adverse events was also significantly higher with the combination, compared with bevacizumab monotherapy.
Even in the age of immune checkpoint inhibitor therapy in the front line, “[i]t is clear that maintenance therapy will remain an integral part of the treatment approach to advanced nonsquamous NSCLC. The results of ECOG-ACRIN 5508 support the use of either pemetrexed or bevacizumab as a single agent in this setting,” the investigators wrote in the Journal of Clinical Oncology.
Although the combination of bevacizumab and pemetrexed was associated with a significant improvement in PFS in a randomized trial, the three maintenance strategies have never before been directly compared, Dr. Ramalingam and associates noted.
In ECOG-ACRIN 5508, 1,516 patients with advanced nonsquamous NSCLC who had not received prior systemic therapy were given standard induction chemotherapy, consisting of carboplatin to an area under the curve of 6, paclitaxel 200 mg/m2, and bevacizumab 15 mg/kg for up to four cycles.
Patients without progression after four cycles (874) were randomly assigned to maintenance therapy with bevacizumab at 15 mg/kg , pemetrexed 500 mg/m2, or a combination of the two agents.
For the primary endpoint of overall survival, with bevacizumab serving as the control group for comparison, the investigators found that, at a median follow-up of 50.6 months, median survival was 15.9 months with pemetrexed, compared with 14.4 months with bevacizumab, a difference that was not statistically significant. Median survival with the combination was 16.4 months and was also not significantly different from bevacizumab.
Median PFS was 4.2 months and 5.1 months for the pemetrexed and bevacizumab groups, respectively, with no significant difference. In contrast, median was 7.5 months with the combination, which was significantly better than controls, with a hazard ratio of 0.67 (P less than .001).
Patients received a median of six maintenance therapy cycles for each of the single agents, and a median of eight for the combinations. The incidence of grade 3-4 toxicity was 29% with bevacizumab, 37% with pemetrexed, and 51% with the combination. The combination was associated with a significantly greater incidence of toxicities, compared with bevacizumab (P less than .001).
The study was supported by grants from the National Cancer Institute. Dr. Ramalingam reported a consulting/advisory role for bevacizumab maker Genentech/Roche and others. Multiple coauthors reported similar disclosures.
SOURCE: Ramalingam SS et al. J Clin Oncol. 2019 Jul 30. doi: 10.1200/JCO.19.01006.
Single-agent therapy with either bevacizumab or pemetrexed is efficacious as maintenance therapy for patients with advanced nonsquamous non–small cell lung cancer (NSCLC), but the combination of the two agents offers no survival benefit and is more toxic, results of the randomized ECOG-ACRIN 5508 study show.
For patients with no disease progression after four cycles of induction chemotherapy who were assigned to one of three maintenance therapy strategies, there were no differences in overall survival between those randomized to monotherapy with pemetrexed or bevacizumab or to a combination of the two agents, although progression-free survival (PFS) was better with the combination, reported Suresh S. Ramalingam, MD, from the Winship Cancer Institute of Emory University, Atlanta, and colleagues.
The incidence of grade 3 or greater adverse events was also significantly higher with the combination, compared with bevacizumab monotherapy.
Even in the age of immune checkpoint inhibitor therapy in the front line, “[i]t is clear that maintenance therapy will remain an integral part of the treatment approach to advanced nonsquamous NSCLC. The results of ECOG-ACRIN 5508 support the use of either pemetrexed or bevacizumab as a single agent in this setting,” the investigators wrote in the Journal of Clinical Oncology.
Although the combination of bevacizumab and pemetrexed was associated with a significant improvement in PFS in a randomized trial, the three maintenance strategies have never before been directly compared, Dr. Ramalingam and associates noted.
In ECOG-ACRIN 5508, 1,516 patients with advanced nonsquamous NSCLC who had not received prior systemic therapy were given standard induction chemotherapy, consisting of carboplatin to an area under the curve of 6, paclitaxel 200 mg/m2, and bevacizumab 15 mg/kg for up to four cycles.
Patients without progression after four cycles (874) were randomly assigned to maintenance therapy with bevacizumab at 15 mg/kg , pemetrexed 500 mg/m2, or a combination of the two agents.
For the primary endpoint of overall survival, with bevacizumab serving as the control group for comparison, the investigators found that, at a median follow-up of 50.6 months, median survival was 15.9 months with pemetrexed, compared with 14.4 months with bevacizumab, a difference that was not statistically significant. Median survival with the combination was 16.4 months and was also not significantly different from bevacizumab.
Median PFS was 4.2 months and 5.1 months for the pemetrexed and bevacizumab groups, respectively, with no significant difference. In contrast, median was 7.5 months with the combination, which was significantly better than controls, with a hazard ratio of 0.67 (P less than .001).
Patients received a median of six maintenance therapy cycles for each of the single agents, and a median of eight for the combinations. The incidence of grade 3-4 toxicity was 29% with bevacizumab, 37% with pemetrexed, and 51% with the combination. The combination was associated with a significantly greater incidence of toxicities, compared with bevacizumab (P less than .001).
The study was supported by grants from the National Cancer Institute. Dr. Ramalingam reported a consulting/advisory role for bevacizumab maker Genentech/Roche and others. Multiple coauthors reported similar disclosures.
SOURCE: Ramalingam SS et al. J Clin Oncol. 2019 Jul 30. doi: 10.1200/JCO.19.01006.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
NSAIDs a significant mediator of cardiovascular risk in osteoarthritis

Writing in Arthritis & Rheumatology, researchers reported the outcomes of a longitudinal, population-based cohort study of 7,743 individuals with osteoarthritis patients and 23,229 age- and sex-matched controls without osteoarthritis.
“The prevailing hypothesis in the OA to CVD relationship has been that OA patients frequently take NSAIDs to control their pain and inflammation and that this may lead to them developing CVD,” wrote Mohammad Atiquzzaman, a PhD student at the University of British Columbia, Vancouver, and his coauthors. However they commented that no studies had so far examined this directly in patients with osteoarthritis.
Overall, people with osteoarthritis had a significant 23% higher risk of cardiovascular disease, compared with controls, after adjustment for factors such body mass index, hypertension, diabetes, hyperlipidemia, and socioeconomic status. They also had a 42% higher risk of congestive heart failure, 17% higher risk of ischemic heart disease, and 14% higher risk of stroke.
NSAID use was five times more common among people with osteoarthritis, and NSAIDs alone were associated with a greater than fourfold higher risk of cardiovascular disease, after adjusting for osteoarthritis and other potential confounders.
When the authors performed modeling to break down the effect of osteoarthritis on CVD risk into the direct effect of osteoarthritis itself and the indirect effect mediated by NSAID use, they concluded that 41% of the total effect of osteoarthritis on cardiovascular risk was mediated by NSAIDs. The effect of NSAIDs was particularly pronounced for stroke, in which cases they estimated that the drugs contributed to 64% of the increased in risk, and in ischemic heart disease, in which they contributed to 56% of the increased risk.
Subgroup analysis suggested that conventional NSAIDs were responsible for around 29% of the total increased risk of cardiovascular disease, while selective COX-2 inhibitors, or coxibs, such as celecoxib, lumiracoxib, rofecoxib, and valdecoxib mediated around 21%. For ischemic heart disease, conventional NSAIDs explained around 45% of the increased risk, while selective coxibs explained around 32% of the risk. Similarly, with congestive heart failure and stroke, the proportion of risk mediated by NSAIDs was higher for conventional NSAIDs, compared with coxibs.
The authors noted that while a number of previous studies have found osteoarthritis is an independent risk factor for cardiovascular disease, theirs was the first study to specifically examine the role that NSAIDs play in that increased risk.
However, they noted that their information on NSAID use was gleaned from prescription claims data, which did not include information on over-the-counter NSAID use. Their analysis was also unable to include information on family history of cardiovascular disease, smoking, and physical activity, which are important cardiovascular disease risk factors. They did observe that the rates of obesity were higher among the osteoarthritis group when compared with controls (29% vs. 20%), and hypertension and COPD were also more common among individuals with osteoarthritis.
There was no outside funding for the study, and the authors had no conflicts of interest to declare.
SOURCE: Atiquzzaman M et al. Arthritis Rheumatol. 2019 Aug 6. doi: 10.1002/art.41027
Writing in Arthritis & Rheumatology, researchers reported the outcomes of a longitudinal, population-based cohort study of 7,743 individuals with osteoarthritis patients and 23,229 age- and sex-matched controls without osteoarthritis.
“The prevailing hypothesis in the OA to CVD relationship has been that OA patients frequently take NSAIDs to control their pain and inflammation and that this may lead to them developing CVD,” wrote Mohammad Atiquzzaman, a PhD student at the University of British Columbia, Vancouver, and his coauthors. However they commented that no studies had so far examined this directly in patients with osteoarthritis.
Overall, people with osteoarthritis had a significant 23% higher risk of cardiovascular disease, compared with controls, after adjustment for factors such body mass index, hypertension, diabetes, hyperlipidemia, and socioeconomic status. They also had a 42% higher risk of congestive heart failure, 17% higher risk of ischemic heart disease, and 14% higher risk of stroke.
NSAID use was five times more common among people with osteoarthritis, and NSAIDs alone were associated with a greater than fourfold higher risk of cardiovascular disease, after adjusting for osteoarthritis and other potential confounders.
When the authors performed modeling to break down the effect of osteoarthritis on CVD risk into the direct effect of osteoarthritis itself and the indirect effect mediated by NSAID use, they concluded that 41% of the total effect of osteoarthritis on cardiovascular risk was mediated by NSAIDs. The effect of NSAIDs was particularly pronounced for stroke, in which cases they estimated that the drugs contributed to 64% of the increased in risk, and in ischemic heart disease, in which they contributed to 56% of the increased risk.
Subgroup analysis suggested that conventional NSAIDs were responsible for around 29% of the total increased risk of cardiovascular disease, while selective COX-2 inhibitors, or coxibs, such as celecoxib, lumiracoxib, rofecoxib, and valdecoxib mediated around 21%. For ischemic heart disease, conventional NSAIDs explained around 45% of the increased risk, while selective coxibs explained around 32% of the risk. Similarly, with congestive heart failure and stroke, the proportion of risk mediated by NSAIDs was higher for conventional NSAIDs, compared with coxibs.
The authors noted that while a number of previous studies have found osteoarthritis is an independent risk factor for cardiovascular disease, theirs was the first study to specifically examine the role that NSAIDs play in that increased risk.
However, they noted that their information on NSAID use was gleaned from prescription claims data, which did not include information on over-the-counter NSAID use. Their analysis was also unable to include information on family history of cardiovascular disease, smoking, and physical activity, which are important cardiovascular disease risk factors. They did observe that the rates of obesity were higher among the osteoarthritis group when compared with controls (29% vs. 20%), and hypertension and COPD were also more common among individuals with osteoarthritis.
There was no outside funding for the study, and the authors had no conflicts of interest to declare.
SOURCE: Atiquzzaman M et al. Arthritis Rheumatol. 2019 Aug 6. doi: 10.1002/art.41027
Writing in Arthritis & Rheumatology, researchers reported the outcomes of a longitudinal, population-based cohort study of 7,743 individuals with osteoarthritis patients and 23,229 age- and sex-matched controls without osteoarthritis.
“The prevailing hypothesis in the OA to CVD relationship has been that OA patients frequently take NSAIDs to control their pain and inflammation and that this may lead to them developing CVD,” wrote Mohammad Atiquzzaman, a PhD student at the University of British Columbia, Vancouver, and his coauthors. However they commented that no studies had so far examined this directly in patients with osteoarthritis.
Overall, people with osteoarthritis had a significant 23% higher risk of cardiovascular disease, compared with controls, after adjustment for factors such body mass index, hypertension, diabetes, hyperlipidemia, and socioeconomic status. They also had a 42% higher risk of congestive heart failure, 17% higher risk of ischemic heart disease, and 14% higher risk of stroke.
NSAID use was five times more common among people with osteoarthritis, and NSAIDs alone were associated with a greater than fourfold higher risk of cardiovascular disease, after adjusting for osteoarthritis and other potential confounders.
When the authors performed modeling to break down the effect of osteoarthritis on CVD risk into the direct effect of osteoarthritis itself and the indirect effect mediated by NSAID use, they concluded that 41% of the total effect of osteoarthritis on cardiovascular risk was mediated by NSAIDs. The effect of NSAIDs was particularly pronounced for stroke, in which cases they estimated that the drugs contributed to 64% of the increased in risk, and in ischemic heart disease, in which they contributed to 56% of the increased risk.
Subgroup analysis suggested that conventional NSAIDs were responsible for around 29% of the total increased risk of cardiovascular disease, while selective COX-2 inhibitors, or coxibs, such as celecoxib, lumiracoxib, rofecoxib, and valdecoxib mediated around 21%. For ischemic heart disease, conventional NSAIDs explained around 45% of the increased risk, while selective coxibs explained around 32% of the risk. Similarly, with congestive heart failure and stroke, the proportion of risk mediated by NSAIDs was higher for conventional NSAIDs, compared with coxibs.
The authors noted that while a number of previous studies have found osteoarthritis is an independent risk factor for cardiovascular disease, theirs was the first study to specifically examine the role that NSAIDs play in that increased risk.
However, they noted that their information on NSAID use was gleaned from prescription claims data, which did not include information on over-the-counter NSAID use. Their analysis was also unable to include information on family history of cardiovascular disease, smoking, and physical activity, which are important cardiovascular disease risk factors. They did observe that the rates of obesity were higher among the osteoarthritis group when compared with controls (29% vs. 20%), and hypertension and COPD were also more common among individuals with osteoarthritis.
There was no outside funding for the study, and the authors had no conflicts of interest to declare.
SOURCE: Atiquzzaman M et al. Arthritis Rheumatol. 2019 Aug 6. doi: 10.1002/art.41027
FROM ARTHRITIS & RHEUMATOLOGY