User login
Oropouche Virus
The pediatrician’s first patient of the day was a 15-year-old boy complaining of fever, chills, and profound arthralgias. His exam, including a careful assessment of his joints, yielded no clues, and the pediatrician was ready to diagnose this as a routine viral illness. An additional bit of history provided by the patient’s mother prompted the pediatrician to pause and reconsider.
“A week ago, we returned from a visit to Cuba,” the mother reported. “Could this be Oropouche virus infection?”

Oropouche virus disease is an arboviral disease caused by the Oropouche virus (OROV). It is transmitted to humans through midge or mosquito bites. Although largely unknown to most United States clinicians until recently, this vector-borne virus is not new. The first human Oropouche virus infection was identified in Trinidad and Tobago in 1955 and since then, there have been intermittent outbreaks in the Amazon region. In recent months, though, the epidemiology of Oropouche virus infections has changed. Infections are being identified in new geographic areas, including Cuba. According to the Pan American Health Organization, 506 cases of Oropouche virus infection have been identified in Cuba since May 27, 2024.
Two deaths from Oropouche virus infection have been reported in previously healthy people. Evolving data suggests adverse outcomes associated with vertical transmission during pregnancy. One fetal death and child with congenital anomalies have been reported in Brazil. Additional fetal deaths, miscarriages, and congenital anomalies are under investigation.
Travel-associated cases have been reported in the United States. As of September 10, 2024, 52 Oropouche virus disease cases had been reported from five states in the United States. The Centers for Disease Control and Prevention confirmed that the first 31 of these cases were travelers returning from Cuba. The CDC issued a health advisory on August 16, 2024: Increased Oropouche Virus Activity and Associated Risk to Travelers.
The pediatrician quickly reviewed the signs and symptoms of Oropouche virus infection. Disease typically presents as an abrupt onset of fever, severe headache, chills, myalgia, and arthralgia 3 to 10 days after the bite of infected mosquito. Some patients develop a maculopapular rash that starts on the trunk and spreads to the extremities. Meningitis and encephalitis develop in less than 1 in 20 people. The symptoms of Oropouche virus infection overlap with those of other arboviruses such as dengue, chikungunya, and Zika viruses. The disease can also mimic malaria or rickettsial infection. Approximately 60% of people with Oropouche virus infection experience a recurrence of symptoms within days to weeks of the initial resolution of symptoms.
Testing for Oropouche virus infection is available through the CDC’s Arbovirus Diagnostic Laboratory. In people who are acutely ill, reverse transcription-polymerase chain reaction testing can be used to identify the virus in serum and cerebrospinal fluid. Serologic testing is also available for people who have been symptomatic for at least 6 days.
The pediatrician contacted his local health department to discuss the possibility of Oropouche virus infection. After reviewing the case definition, public health authorities recommended laboratory testing for Oropouche virus, dengue, and Zika virus.
Back in the exam room, the pediatrician provided anticipatory guidance to the patient and his mother. There are no antiviral medications to treat Oropouche virus infection, so the pediatrician recommended supportive care, including acetaminophen for fever and pain. He also advised avoiding aspirin or other nonsteroidal anti-inflammatory drugs (NSAIDs) until dengue could be ruled out to reduce the risk of bleeding. After confirming that no one else in the home was sick with similar symptoms, he counseled about prevention strategies.
To date, transmission of Oropouche virus in the United States has not been documented, but vectors potentially capable of transmitting the virus are present in some areas of the United States. When people who are infected with Oropouche are bitten, they can spread the virus through their blood to biting midges or mosquitoes. The insects can then spread the virus to other people. To reduce to potential for local transmission, people who are sick with suspected Oropouche virus infection are advised to avoid biting-midge and mosquito bites for the first week of their illness. Any person who has recently traveled to an area where Oropouche virus transmission is occurring should also avoid insect bites for 3 weeks after returning home to account for the potential incubation period of the virus. This includes wearing an EPA-registered insect repellent.
A suspect case is a patient who has been in an area with documented or suspected OROV circulation* within 2 weeks of initial symptom onset (as patients may experience recurrent symptoms) and the following:
- Abrupt onset of reported fever, headache, and one or more of the following: myalgia, arthralgia, photophobia, retro-orbital/eye pain, or signs and symptoms of neuroinvasive disease (eg, stiff neck, altered mental status, seizures, limb weakness, or cerebrospinal fluid pleocytosis).
- Tested negative for other possible diseases, in particular dengue.†
- Absence of a more likely clinical explanation.
*If concern exists for local transmission in a nonendemic area, consider if the patient shared an exposure location with a person with confirmed OROV infection, lives in an area where travel-related cases have been identified, or has known vector exposure (eg, mosquitoes or biting midges).
†If strong suspicion of OROV disease exists based on the patient’s clinical features and history of travel to an area with virus circulation, do not wait on negative testing before sending specimens to CDC.
Adapted from: Centers for Disease Control and Prevention. Response to Oropouche Virus Disease Cases in U.S. States and Territories in the Americas. Available at: https.//www.cdc.gov/oropouche/media/pdfs/2024/09/response-to-oropouche-virus-disease.pdf
Dr. Bryant is a pediatrician specializing in infectious diseases at the University of Louisville (Ky.) and Norton Children’s Hospital, also in Louisville. She is a member of the AAP’s Committee on Infectious Diseases and one of the lead authors of the AAP’s Recommendations for Prevention and Control of Influenza in Children, 2022-2023. The opinions expressed in this article are her own. Dr. Bryant discloses that she has served as an investigator on clinical trials funded by Pfizer, Enanta and Gilead. Email her at [email protected]. (Also [email protected])
The pediatrician’s first patient of the day was a 15-year-old boy complaining of fever, chills, and profound arthralgias. His exam, including a careful assessment of his joints, yielded no clues, and the pediatrician was ready to diagnose this as a routine viral illness. An additional bit of history provided by the patient’s mother prompted the pediatrician to pause and reconsider.
“A week ago, we returned from a visit to Cuba,” the mother reported. “Could this be Oropouche virus infection?”
Oropouche virus disease is an arboviral disease caused by the Oropouche virus (OROV). It is transmitted to humans through midge or mosquito bites. Although largely unknown to most United States clinicians until recently, this vector-borne virus is not new. The first human Oropouche virus infection was identified in Trinidad and Tobago in 1955 and since then, there have been intermittent outbreaks in the Amazon region. In recent months, though, the epidemiology of Oropouche virus infections has changed. Infections are being identified in new geographic areas, including Cuba. According to the Pan American Health Organization, 506 cases of Oropouche virus infection have been identified in Cuba since May 27, 2024.
Two deaths from Oropouche virus infection have been reported in previously healthy people. Evolving data suggests adverse outcomes associated with vertical transmission during pregnancy. One fetal death and child with congenital anomalies have been reported in Brazil. Additional fetal deaths, miscarriages, and congenital anomalies are under investigation.
Travel-associated cases have been reported in the United States. As of September 10, 2024, 52 Oropouche virus disease cases had been reported from five states in the United States. The Centers for Disease Control and Prevention confirmed that the first 31 of these cases were travelers returning from Cuba. The CDC issued a health advisory on August 16, 2024: Increased Oropouche Virus Activity and Associated Risk to Travelers.
The pediatrician quickly reviewed the signs and symptoms of Oropouche virus infection. Disease typically presents as an abrupt onset of fever, severe headache, chills, myalgia, and arthralgia 3 to 10 days after the bite of infected mosquito. Some patients develop a maculopapular rash that starts on the trunk and spreads to the extremities. Meningitis and encephalitis develop in less than 1 in 20 people. The symptoms of Oropouche virus infection overlap with those of other arboviruses such as dengue, chikungunya, and Zika viruses. The disease can also mimic malaria or rickettsial infection. Approximately 60% of people with Oropouche virus infection experience a recurrence of symptoms within days to weeks of the initial resolution of symptoms.
Testing for Oropouche virus infection is available through the CDC’s Arbovirus Diagnostic Laboratory. In people who are acutely ill, reverse transcription-polymerase chain reaction testing can be used to identify the virus in serum and cerebrospinal fluid. Serologic testing is also available for people who have been symptomatic for at least 6 days.
The pediatrician contacted his local health department to discuss the possibility of Oropouche virus infection. After reviewing the case definition, public health authorities recommended laboratory testing for Oropouche virus, dengue, and Zika virus.
Back in the exam room, the pediatrician provided anticipatory guidance to the patient and his mother. There are no antiviral medications to treat Oropouche virus infection, so the pediatrician recommended supportive care, including acetaminophen for fever and pain. He also advised avoiding aspirin or other nonsteroidal anti-inflammatory drugs (NSAIDs) until dengue could be ruled out to reduce the risk of bleeding. After confirming that no one else in the home was sick with similar symptoms, he counseled about prevention strategies.
To date, transmission of Oropouche virus in the United States has not been documented, but vectors potentially capable of transmitting the virus are present in some areas of the United States. When people who are infected with Oropouche are bitten, they can spread the virus through their blood to biting midges or mosquitoes. The insects can then spread the virus to other people. To reduce to potential for local transmission, people who are sick with suspected Oropouche virus infection are advised to avoid biting-midge and mosquito bites for the first week of their illness. Any person who has recently traveled to an area where Oropouche virus transmission is occurring should also avoid insect bites for 3 weeks after returning home to account for the potential incubation period of the virus. This includes wearing an EPA-registered insect repellent.
A suspect case is a patient who has been in an area with documented or suspected OROV circulation* within 2 weeks of initial symptom onset (as patients may experience recurrent symptoms) and the following:
- Abrupt onset of reported fever, headache, and one or more of the following: myalgia, arthralgia, photophobia, retro-orbital/eye pain, or signs and symptoms of neuroinvasive disease (eg, stiff neck, altered mental status, seizures, limb weakness, or cerebrospinal fluid pleocytosis).
- Tested negative for other possible diseases, in particular dengue.†
- Absence of a more likely clinical explanation.
*If concern exists for local transmission in a nonendemic area, consider if the patient shared an exposure location with a person with confirmed OROV infection, lives in an area where travel-related cases have been identified, or has known vector exposure (eg, mosquitoes or biting midges).
†If strong suspicion of OROV disease exists based on the patient’s clinical features and history of travel to an area with virus circulation, do not wait on negative testing before sending specimens to CDC.
Adapted from: Centers for Disease Control and Prevention. Response to Oropouche Virus Disease Cases in U.S. States and Territories in the Americas. Available at: https.//www.cdc.gov/oropouche/media/pdfs/2024/09/response-to-oropouche-virus-disease.pdf
Dr. Bryant is a pediatrician specializing in infectious diseases at the University of Louisville (Ky.) and Norton Children’s Hospital, also in Louisville. She is a member of the AAP’s Committee on Infectious Diseases and one of the lead authors of the AAP’s Recommendations for Prevention and Control of Influenza in Children, 2022-2023. The opinions expressed in this article are her own. Dr. Bryant discloses that she has served as an investigator on clinical trials funded by Pfizer, Enanta and Gilead. Email her at [email protected]. (Also [email protected])
The pediatrician’s first patient of the day was a 15-year-old boy complaining of fever, chills, and profound arthralgias. His exam, including a careful assessment of his joints, yielded no clues, and the pediatrician was ready to diagnose this as a routine viral illness. An additional bit of history provided by the patient’s mother prompted the pediatrician to pause and reconsider.
“A week ago, we returned from a visit to Cuba,” the mother reported. “Could this be Oropouche virus infection?”
Oropouche virus disease is an arboviral disease caused by the Oropouche virus (OROV). It is transmitted to humans through midge or mosquito bites. Although largely unknown to most United States clinicians until recently, this vector-borne virus is not new. The first human Oropouche virus infection was identified in Trinidad and Tobago in 1955 and since then, there have been intermittent outbreaks in the Amazon region. In recent months, though, the epidemiology of Oropouche virus infections has changed. Infections are being identified in new geographic areas, including Cuba. According to the Pan American Health Organization, 506 cases of Oropouche virus infection have been identified in Cuba since May 27, 2024.
Two deaths from Oropouche virus infection have been reported in previously healthy people. Evolving data suggests adverse outcomes associated with vertical transmission during pregnancy. One fetal death and child with congenital anomalies have been reported in Brazil. Additional fetal deaths, miscarriages, and congenital anomalies are under investigation.
Travel-associated cases have been reported in the United States. As of September 10, 2024, 52 Oropouche virus disease cases had been reported from five states in the United States. The Centers for Disease Control and Prevention confirmed that the first 31 of these cases were travelers returning from Cuba. The CDC issued a health advisory on August 16, 2024: Increased Oropouche Virus Activity and Associated Risk to Travelers.
The pediatrician quickly reviewed the signs and symptoms of Oropouche virus infection. Disease typically presents as an abrupt onset of fever, severe headache, chills, myalgia, and arthralgia 3 to 10 days after the bite of infected mosquito. Some patients develop a maculopapular rash that starts on the trunk and spreads to the extremities. Meningitis and encephalitis develop in less than 1 in 20 people. The symptoms of Oropouche virus infection overlap with those of other arboviruses such as dengue, chikungunya, and Zika viruses. The disease can also mimic malaria or rickettsial infection. Approximately 60% of people with Oropouche virus infection experience a recurrence of symptoms within days to weeks of the initial resolution of symptoms.
Testing for Oropouche virus infection is available through the CDC’s Arbovirus Diagnostic Laboratory. In people who are acutely ill, reverse transcription-polymerase chain reaction testing can be used to identify the virus in serum and cerebrospinal fluid. Serologic testing is also available for people who have been symptomatic for at least 6 days.
The pediatrician contacted his local health department to discuss the possibility of Oropouche virus infection. After reviewing the case definition, public health authorities recommended laboratory testing for Oropouche virus, dengue, and Zika virus.
Back in the exam room, the pediatrician provided anticipatory guidance to the patient and his mother. There are no antiviral medications to treat Oropouche virus infection, so the pediatrician recommended supportive care, including acetaminophen for fever and pain. He also advised avoiding aspirin or other nonsteroidal anti-inflammatory drugs (NSAIDs) until dengue could be ruled out to reduce the risk of bleeding. After confirming that no one else in the home was sick with similar symptoms, he counseled about prevention strategies.
To date, transmission of Oropouche virus in the United States has not been documented, but vectors potentially capable of transmitting the virus are present in some areas of the United States. When people who are infected with Oropouche are bitten, they can spread the virus through their blood to biting midges or mosquitoes. The insects can then spread the virus to other people. To reduce to potential for local transmission, people who are sick with suspected Oropouche virus infection are advised to avoid biting-midge and mosquito bites for the first week of their illness. Any person who has recently traveled to an area where Oropouche virus transmission is occurring should also avoid insect bites for 3 weeks after returning home to account for the potential incubation period of the virus. This includes wearing an EPA-registered insect repellent.
A suspect case is a patient who has been in an area with documented or suspected OROV circulation* within 2 weeks of initial symptom onset (as patients may experience recurrent symptoms) and the following:
- Abrupt onset of reported fever, headache, and one or more of the following: myalgia, arthralgia, photophobia, retro-orbital/eye pain, or signs and symptoms of neuroinvasive disease (eg, stiff neck, altered mental status, seizures, limb weakness, or cerebrospinal fluid pleocytosis).
- Tested negative for other possible diseases, in particular dengue.†
- Absence of a more likely clinical explanation.
*If concern exists for local transmission in a nonendemic area, consider if the patient shared an exposure location with a person with confirmed OROV infection, lives in an area where travel-related cases have been identified, or has known vector exposure (eg, mosquitoes or biting midges).
†If strong suspicion of OROV disease exists based on the patient’s clinical features and history of travel to an area with virus circulation, do not wait on negative testing before sending specimens to CDC.
Adapted from: Centers for Disease Control and Prevention. Response to Oropouche Virus Disease Cases in U.S. States and Territories in the Americas. Available at: https.//www.cdc.gov/oropouche/media/pdfs/2024/09/response-to-oropouche-virus-disease.pdf
Dr. Bryant is a pediatrician specializing in infectious diseases at the University of Louisville (Ky.) and Norton Children’s Hospital, also in Louisville. She is a member of the AAP’s Committee on Infectious Diseases and one of the lead authors of the AAP’s Recommendations for Prevention and Control of Influenza in Children, 2022-2023. The opinions expressed in this article are her own. Dr. Bryant discloses that she has served as an investigator on clinical trials funded by Pfizer, Enanta and Gilead. Email her at [email protected]. (Also [email protected])
No Matched Sibling Donor? Sickle Cell Experts Debate Next-Best Option
“If there is an indication for intervention, for a curative therapy, in the absence of a matched sibling donor, gene therapy is the first choice,” Jaap-Jan Boelens, MD, PhD, of Memorial Sloan Kettering Cancer Center, New York City, argued in a presentation at the annual meeting of the Society of Hematologic Oncology (SOHO) in Houston.
“In the registries, alternative transplant outcomes are pretty poor, although there is some encouraging data coming up. The time is not there yet when this is the [best] choice.”
But Adetola Kassim, MBBS, of Vanderbilt University Medical Center in Nashville, Tennessee, said patients with sickle cell disease (SCD) who don’t qualify for a matched sibling donor transplant can still have good transplant options. And the results can be impressive.
“Once you’re engrafted, and you don’t lose your graft, the effect in transplant is lifelong,” he said. When it comes to long-lasting effects, he added, “we’re not sure yet about gene therapy.”
As Dr. Kassim noted, SCD continues to take a huge toll.
“Median survival for patients with sickle cell anemia remains stuck in the fifth decade of life with no change in 25 years,” he said. Heart, lung, and kidney complications account for 50% of identifiable causes of death, followed by about 26% attributed to cardiovascular disease, he said. “The question here is about which therapy can impact the most debilitating complication in children, which is stroke, and improve survival in adults with progressive organ dysfunction.”
Dr. Boelens said there are “huge barriers” to stem cell transplant in SCD because only 15% of patients eligible for the treatment have a matched related donor, and only 10% have a matched related or unrelated donor.
“There’s also a lack of financial and psychosocial support in many of the families. There is also parental refusal because of the mortality risk, and there’s also physician refusal because hematologists aren’t always in the same hospitals as the transplant programs.”
Dr. Boelens highlighted a 2019 study of data from 2008-2017 that found outcomes in unmatched donor transplantations are “not great,” with higher risk for mortality and graft failure.
As an alternative, he said, two gene therapies, both gene “additions,” are now approved by the US Food and Drug Administration (FDA). They are exagamglogene autotemcel (exa-cel, Casgevy) and betibeglogene autotemcel (LentiGlobin, Zynteglo). There’s also a gene “correction” option in the works, but it’s not yet ready for prime time, he said.
In the two approved gene therapy treatments, stem cells are removed from the patient, modified/manufactured in an outside facility, and then engrafted.
The advantages of gene therapy include no need to find a donor or worry about graft resistance, and there’s no need for immunosuppression, he said. However, the process takes a long time, there’s limited long-term data, and there’s a risk for loss of fertility and other chemotherapy-related adverse effects.
For his part, Dr. Kassim noted how several groups are excluded from the strong outcomes in matched sibling donor stem-cell transplants: Children with strokes and no eligible donors, others without eligible donors, and adults with severe disease and organ dysfunction who are typically excluded.
“We need transplants with less toxicity and alternative donors,” he said. Another challenge: “How do we decrease graft failure without increasing transplant-related mortality?”
Researchers are exploring several strategies to adjust drug therapy during conditioning, Dr. Kassim said, and he led a promising phase II study that explored one approach. The results of that study were recently published in the journal Blood. Graft failures were very low in both adults and children, he said, and 2-year survival among 70 patients was 94.8%. The five deaths were related to infection.
The evidence about the various strategies shows that “virtually all SCD patients, except those with severe heart, lung, or kidney disease” can benefit from a curative transplant, Dr. Kassim said.
Dr. Boelens had no disclosures. Disclosures for Dr. Kassim were not provided, but he recently reported no disclosures in a report about transplants in SCD.
A version of this article appeared on Medscape.com.
“If there is an indication for intervention, for a curative therapy, in the absence of a matched sibling donor, gene therapy is the first choice,” Jaap-Jan Boelens, MD, PhD, of Memorial Sloan Kettering Cancer Center, New York City, argued in a presentation at the annual meeting of the Society of Hematologic Oncology (SOHO) in Houston.
“In the registries, alternative transplant outcomes are pretty poor, although there is some encouraging data coming up. The time is not there yet when this is the [best] choice.”
But Adetola Kassim, MBBS, of Vanderbilt University Medical Center in Nashville, Tennessee, said patients with sickle cell disease (SCD) who don’t qualify for a matched sibling donor transplant can still have good transplant options. And the results can be impressive.
“Once you’re engrafted, and you don’t lose your graft, the effect in transplant is lifelong,” he said. When it comes to long-lasting effects, he added, “we’re not sure yet about gene therapy.”
As Dr. Kassim noted, SCD continues to take a huge toll.
“Median survival for patients with sickle cell anemia remains stuck in the fifth decade of life with no change in 25 years,” he said. Heart, lung, and kidney complications account for 50% of identifiable causes of death, followed by about 26% attributed to cardiovascular disease, he said. “The question here is about which therapy can impact the most debilitating complication in children, which is stroke, and improve survival in adults with progressive organ dysfunction.”
Dr. Boelens said there are “huge barriers” to stem cell transplant in SCD because only 15% of patients eligible for the treatment have a matched related donor, and only 10% have a matched related or unrelated donor.
“There’s also a lack of financial and psychosocial support in many of the families. There is also parental refusal because of the mortality risk, and there’s also physician refusal because hematologists aren’t always in the same hospitals as the transplant programs.”
Dr. Boelens highlighted a 2019 study of data from 2008-2017 that found outcomes in unmatched donor transplantations are “not great,” with higher risk for mortality and graft failure.
As an alternative, he said, two gene therapies, both gene “additions,” are now approved by the US Food and Drug Administration (FDA). They are exagamglogene autotemcel (exa-cel, Casgevy) and betibeglogene autotemcel (LentiGlobin, Zynteglo). There’s also a gene “correction” option in the works, but it’s not yet ready for prime time, he said.
In the two approved gene therapy treatments, stem cells are removed from the patient, modified/manufactured in an outside facility, and then engrafted.
The advantages of gene therapy include no need to find a donor or worry about graft resistance, and there’s no need for immunosuppression, he said. However, the process takes a long time, there’s limited long-term data, and there’s a risk for loss of fertility and other chemotherapy-related adverse effects.
For his part, Dr. Kassim noted how several groups are excluded from the strong outcomes in matched sibling donor stem-cell transplants: Children with strokes and no eligible donors, others without eligible donors, and adults with severe disease and organ dysfunction who are typically excluded.
“We need transplants with less toxicity and alternative donors,” he said. Another challenge: “How do we decrease graft failure without increasing transplant-related mortality?”
Researchers are exploring several strategies to adjust drug therapy during conditioning, Dr. Kassim said, and he led a promising phase II study that explored one approach. The results of that study were recently published in the journal Blood. Graft failures were very low in both adults and children, he said, and 2-year survival among 70 patients was 94.8%. The five deaths were related to infection.
The evidence about the various strategies shows that “virtually all SCD patients, except those with severe heart, lung, or kidney disease” can benefit from a curative transplant, Dr. Kassim said.
Dr. Boelens had no disclosures. Disclosures for Dr. Kassim were not provided, but he recently reported no disclosures in a report about transplants in SCD.
A version of this article appeared on Medscape.com.
“If there is an indication for intervention, for a curative therapy, in the absence of a matched sibling donor, gene therapy is the first choice,” Jaap-Jan Boelens, MD, PhD, of Memorial Sloan Kettering Cancer Center, New York City, argued in a presentation at the annual meeting of the Society of Hematologic Oncology (SOHO) in Houston.
“In the registries, alternative transplant outcomes are pretty poor, although there is some encouraging data coming up. The time is not there yet when this is the [best] choice.”
But Adetola Kassim, MBBS, of Vanderbilt University Medical Center in Nashville, Tennessee, said patients with sickle cell disease (SCD) who don’t qualify for a matched sibling donor transplant can still have good transplant options. And the results can be impressive.
“Once you’re engrafted, and you don’t lose your graft, the effect in transplant is lifelong,” he said. When it comes to long-lasting effects, he added, “we’re not sure yet about gene therapy.”
As Dr. Kassim noted, SCD continues to take a huge toll.
“Median survival for patients with sickle cell anemia remains stuck in the fifth decade of life with no change in 25 years,” he said. Heart, lung, and kidney complications account for 50% of identifiable causes of death, followed by about 26% attributed to cardiovascular disease, he said. “The question here is about which therapy can impact the most debilitating complication in children, which is stroke, and improve survival in adults with progressive organ dysfunction.”
Dr. Boelens said there are “huge barriers” to stem cell transplant in SCD because only 15% of patients eligible for the treatment have a matched related donor, and only 10% have a matched related or unrelated donor.
“There’s also a lack of financial and psychosocial support in many of the families. There is also parental refusal because of the mortality risk, and there’s also physician refusal because hematologists aren’t always in the same hospitals as the transplant programs.”
Dr. Boelens highlighted a 2019 study of data from 2008-2017 that found outcomes in unmatched donor transplantations are “not great,” with higher risk for mortality and graft failure.
As an alternative, he said, two gene therapies, both gene “additions,” are now approved by the US Food and Drug Administration (FDA). They are exagamglogene autotemcel (exa-cel, Casgevy) and betibeglogene autotemcel (LentiGlobin, Zynteglo). There’s also a gene “correction” option in the works, but it’s not yet ready for prime time, he said.
In the two approved gene therapy treatments, stem cells are removed from the patient, modified/manufactured in an outside facility, and then engrafted.
The advantages of gene therapy include no need to find a donor or worry about graft resistance, and there’s no need for immunosuppression, he said. However, the process takes a long time, there’s limited long-term data, and there’s a risk for loss of fertility and other chemotherapy-related adverse effects.
For his part, Dr. Kassim noted how several groups are excluded from the strong outcomes in matched sibling donor stem-cell transplants: Children with strokes and no eligible donors, others without eligible donors, and adults with severe disease and organ dysfunction who are typically excluded.
“We need transplants with less toxicity and alternative donors,” he said. Another challenge: “How do we decrease graft failure without increasing transplant-related mortality?”
Researchers are exploring several strategies to adjust drug therapy during conditioning, Dr. Kassim said, and he led a promising phase II study that explored one approach. The results of that study were recently published in the journal Blood. Graft failures were very low in both adults and children, he said, and 2-year survival among 70 patients was 94.8%. The five deaths were related to infection.
The evidence about the various strategies shows that “virtually all SCD patients, except those with severe heart, lung, or kidney disease” can benefit from a curative transplant, Dr. Kassim said.
Dr. Boelens had no disclosures. Disclosures for Dr. Kassim were not provided, but he recently reported no disclosures in a report about transplants in SCD.
A version of this article appeared on Medscape.com.
FROM SOHO 2024
GI-Targeted Bitter Hop Extract Curbs Hunger, Food Cravings
TOPLINE:
METHODOLOGY:
- Researchers conducted a randomized, double-blind, crossover study with 30 normal-weight women ages 18-40 with BMIs of 18.5-25. The trial design was similar to that used previously to test the intervention among men.
- Participants engaged in a water-only fast for 24 hours (6 p.m. to 6 p.m.) on three occasions and were given an ad libitum meal to break each fast.
- Participants were randomly assigned to one of three treatments: placebo, a high-dose (500 mg) bitter hop–based appetite suppressant (Amarasate), or a low dose (250 mg) of the appetite suppressant.
- Treatment capsules of half the total dose were given twice daily (16 hours and 20 hours into the fast).
- Participants recorded their subjective feelings about appetite and food cravings using a visual analog scale at 30-minute intervals starting 16 hours into the fast.
TAKEAWAY:
- Both the high-dose and low-dose treatment groups experienced a significant reduction in appetite (ie, hunger, fullness, satisfaction, thoughts of food) and food cravings compared with the placebo group.
- Energy intake at the ad libitum meal was 14.3% lower in the high-dose group than the placebo group (P < .05). It was 8.1% lower in the low-dose group, but the difference was not statistically significant.
- In the high-dose group, two participants reported loose stools and one reported heartburn. In the low-dose group, one participant reported loose stools.
IN PRACTICE:
“These data suggest that appetite suppressant co-therapy may be useful in reducing hunger during fasting in women and show that gastrointestinal delivery of bitter compounds may also be an effective method of reducing cravings for food,” the authors wrote.
SOURCE:
The study, led by Edward Walker, PhD, of the New Zealand Institute for Plant and Food Research Limited, Auckland, New Zealand, was published online in Obesity Pillars.
LIMITATIONS:
The study has limitations that affect its application for weight management. First, the study included only normal-weight individuals, which limits extension of the results for weight loss treatments. Second, it involved acute fasting and does not assess any accommodation to the intervention that may occur over longer-term use. An additional limitation is the small number of participants.
DISCLOSURES:
The research was funded by Calocurb Limited. The article processing charge was funded by the New Zealand Institute for Plant and Food Research Limited, a New Zealand government–owned Crown Research Institute. The authors declare no competing financial interests. The New Zealand Institute for Plant and Food Research Limited has licensed a hop extract as a dietary supplement to Calocurb to commercialize and currently holds a minor shareholding in this company.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Researchers conducted a randomized, double-blind, crossover study with 30 normal-weight women ages 18-40 with BMIs of 18.5-25. The trial design was similar to that used previously to test the intervention among men.
- Participants engaged in a water-only fast for 24 hours (6 p.m. to 6 p.m.) on three occasions and were given an ad libitum meal to break each fast.
- Participants were randomly assigned to one of three treatments: placebo, a high-dose (500 mg) bitter hop–based appetite suppressant (Amarasate), or a low dose (250 mg) of the appetite suppressant.
- Treatment capsules of half the total dose were given twice daily (16 hours and 20 hours into the fast).
- Participants recorded their subjective feelings about appetite and food cravings using a visual analog scale at 30-minute intervals starting 16 hours into the fast.
TAKEAWAY:
- Both the high-dose and low-dose treatment groups experienced a significant reduction in appetite (ie, hunger, fullness, satisfaction, thoughts of food) and food cravings compared with the placebo group.
- Energy intake at the ad libitum meal was 14.3% lower in the high-dose group than the placebo group (P < .05). It was 8.1% lower in the low-dose group, but the difference was not statistically significant.
- In the high-dose group, two participants reported loose stools and one reported heartburn. In the low-dose group, one participant reported loose stools.
IN PRACTICE:
“These data suggest that appetite suppressant co-therapy may be useful in reducing hunger during fasting in women and show that gastrointestinal delivery of bitter compounds may also be an effective method of reducing cravings for food,” the authors wrote.
SOURCE:
The study, led by Edward Walker, PhD, of the New Zealand Institute for Plant and Food Research Limited, Auckland, New Zealand, was published online in Obesity Pillars.
LIMITATIONS:
The study has limitations that affect its application for weight management. First, the study included only normal-weight individuals, which limits extension of the results for weight loss treatments. Second, it involved acute fasting and does not assess any accommodation to the intervention that may occur over longer-term use. An additional limitation is the small number of participants.
DISCLOSURES:
The research was funded by Calocurb Limited. The article processing charge was funded by the New Zealand Institute for Plant and Food Research Limited, a New Zealand government–owned Crown Research Institute. The authors declare no competing financial interests. The New Zealand Institute for Plant and Food Research Limited has licensed a hop extract as a dietary supplement to Calocurb to commercialize and currently holds a minor shareholding in this company.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Researchers conducted a randomized, double-blind, crossover study with 30 normal-weight women ages 18-40 with BMIs of 18.5-25. The trial design was similar to that used previously to test the intervention among men.
- Participants engaged in a water-only fast for 24 hours (6 p.m. to 6 p.m.) on three occasions and were given an ad libitum meal to break each fast.
- Participants were randomly assigned to one of three treatments: placebo, a high-dose (500 mg) bitter hop–based appetite suppressant (Amarasate), or a low dose (250 mg) of the appetite suppressant.
- Treatment capsules of half the total dose were given twice daily (16 hours and 20 hours into the fast).
- Participants recorded their subjective feelings about appetite and food cravings using a visual analog scale at 30-minute intervals starting 16 hours into the fast.
TAKEAWAY:
- Both the high-dose and low-dose treatment groups experienced a significant reduction in appetite (ie, hunger, fullness, satisfaction, thoughts of food) and food cravings compared with the placebo group.
- Energy intake at the ad libitum meal was 14.3% lower in the high-dose group than the placebo group (P < .05). It was 8.1% lower in the low-dose group, but the difference was not statistically significant.
- In the high-dose group, two participants reported loose stools and one reported heartburn. In the low-dose group, one participant reported loose stools.
IN PRACTICE:
“These data suggest that appetite suppressant co-therapy may be useful in reducing hunger during fasting in women and show that gastrointestinal delivery of bitter compounds may also be an effective method of reducing cravings for food,” the authors wrote.
SOURCE:
The study, led by Edward Walker, PhD, of the New Zealand Institute for Plant and Food Research Limited, Auckland, New Zealand, was published online in Obesity Pillars.
LIMITATIONS:
The study has limitations that affect its application for weight management. First, the study included only normal-weight individuals, which limits extension of the results for weight loss treatments. Second, it involved acute fasting and does not assess any accommodation to the intervention that may occur over longer-term use. An additional limitation is the small number of participants.
DISCLOSURES:
The research was funded by Calocurb Limited. The article processing charge was funded by the New Zealand Institute for Plant and Food Research Limited, a New Zealand government–owned Crown Research Institute. The authors declare no competing financial interests. The New Zealand Institute for Plant and Food Research Limited has licensed a hop extract as a dietary supplement to Calocurb to commercialize and currently holds a minor shareholding in this company.
A version of this article first appeared on Medscape.com.
Stress Management
With the changing leaves and cooling temperatures, early autumn also brings the excitement of the new school year. While returning to sports, mastering new subjects, and spending time with old and new friends is exhilarating, this season can also be a time of intense stress.
For those high school students who are especially ambitious, the school year presents the challenge of a very high stakes performance, one whose success will be measured by admission to a prized college. Not only are there classes to study for, but schedules are packed with a maximum number of subjects, a maximum number of Advanced Placement courses and a maximum number of impressive extra-curricular activities. Varsity sports practice, SAT prep, Debate Club, volunteer hours, and on and on.
What is often missing is enough time for sleep, socializing, exploring new interests, and unwinding. When you hear your patients (or parents) describing the intense stress of their overloaded schedules compounded by a sense that “I have no choice,” you have an opportunity to complicate their thinking. Introduce the idea that there are smart approaches to performing your best under stress. Pushing themselves relentlessly will inevitably lead to burnout and exhaustion. This approach will help them learn to make wise choices and will better serve their healthy development.

Start by acknowledging the stress of high-stakes performance. Telling your patients that they need to lower the temperature by not putting so much pressure on themselves is likely to be experienced as a lack of confidence in them and is unlikely to get any traction. Instead, ask your patients what matters to them the most: Is it admission to the college of their choice? Achieving a certain score or GPA? Is it their competitiveness and drive to win? There is no wrong answer, but it is helpful for them to be able to reflect on what matters to them. Are they hoping to impress someone else? Are they worried about their future financial health and convinced that getting into a certain college will secure their financial success? Do they think this matters more to their parents than to themselves? Or have they discovered an intense interest in theoretical physics and want to be able to study at Caltech? If their ambition is meaningfully connected to an authentic interest or to their emerging identity, their sense of purpose will be much deeper and able to sustain them.
Even with talent and a strong sense of purpose, performing well is very difficult and demanding. It is important to consider the cycle of performance as including preparation, performance itself, and effective rest and recovery, just as with athletic performance. Whether the performance is the SATs, an AP test, a debate or big game, there were probably hours of preparation for every hour of performance. Help them to consider the importance of this practice or preparation time, and how to use that time effectively. Are they able to work in environments where there are few distractions? Do they have the support or useful feedback they need? How are they able to know when it is time for a break or when they are ready? It can be helpful for them to appreciate whether preparation or performance is more challenging for them, as the former requires focus and patience, while the latter requires courage and tenacity. If they are aware of which is harder for them, they can be thoughtful about how to effectively handle those challenges.

What can be most valuable for your patients is hearing from their pediatricians that they need to have time protected for rest and recharging, and not only for preparation and performance. Any athlete knows that failing to do so will lead to exhaustion and injury, and performance inevitably suffers. Rest is unwinding and slowing down, and a restful activity will leave them feeling calm, relaxed, and ready for sleep. A recharging activity is one that leaves them feeling refreshed and energized. Some common restful activities are a hot bath or shower, a distracting activity such as watching a show or surfing the web, playing a simple video game or puzzle or listening to music. Some recharging activities are creative ones (making art or music), engaging in hobbies, reading, or talking with a good friend. A few activities — sleep, exercise, and mindfulness meditation, are powerful in that they pack both rest and recharge into the same activity. Your patients should be discovering and learning which activities they find restful or recharging. The college application process or preparing for a varsity tryout will both add stress and give them an opportunity to learn what rests and recharges them. They should aim to have a list of at least five effective strategies that they can turn to when it’s time to rest or to recharge. Help them turn their work ethic to building a deeper well of self-knowledge that will serve them when they face challenges in high school or when they are on their own in college. This time of stress can be a time of growth, too.
Of course, remind your patients that this is a critical time to focus on basic self-care: They need consistently adequate, restful sleep, good nutrition, and physical activity. They will benefit from regular time in nature and time spent with friends that nourish them. They can find ways to compound these activities: Go for a walk with a friend, eat dinner with family, play a relaxing game while enjoying music. Lastly, ask your patients what is the last new thing they tried. It is easy to become so focused on an ambitious project that there is no time for exploration and play. Play is important throughout life, but adolescents are actively discovering their interests, talents, tastes, and values. To do this they need to be trying things that are new and maybe less purpose-driven. I call this type of activity “senseless fun.” Splashing in the pool is senseless fun, swimming laps is purposeful exercise that my contribute to recharging, and competing in a swim meet is often more on the stressful side. As they discover new talents, deeply engaging interests, what relaxes and recharges them, they will be learning who they are. Regardless of the outcome of a test, a big game, or where they go to college, it is this emerging knowledge about themselves that will carry them into adulthood. The pediatrician’s goal: Encouraging aspiration, exploration, and self-awareness in the context of giving permission for rest, recharging, and senseless fun.
Dr. Swick is physician in chief at Ohana, Center for Child and Adolescent Behavioral Health, Community Hospital of the Monterey (Calif.) Peninsula. Dr. Jellinek is professor emeritus of psychiatry and pediatrics, Harvard Medical School, Boston. Email them at [email protected].
With the changing leaves and cooling temperatures, early autumn also brings the excitement of the new school year. While returning to sports, mastering new subjects, and spending time with old and new friends is exhilarating, this season can also be a time of intense stress.
For those high school students who are especially ambitious, the school year presents the challenge of a very high stakes performance, one whose success will be measured by admission to a prized college. Not only are there classes to study for, but schedules are packed with a maximum number of subjects, a maximum number of Advanced Placement courses and a maximum number of impressive extra-curricular activities. Varsity sports practice, SAT prep, Debate Club, volunteer hours, and on and on.
What is often missing is enough time for sleep, socializing, exploring new interests, and unwinding. When you hear your patients (or parents) describing the intense stress of their overloaded schedules compounded by a sense that “I have no choice,” you have an opportunity to complicate their thinking. Introduce the idea that there are smart approaches to performing your best under stress. Pushing themselves relentlessly will inevitably lead to burnout and exhaustion. This approach will help them learn to make wise choices and will better serve their healthy development.
Start by acknowledging the stress of high-stakes performance. Telling your patients that they need to lower the temperature by not putting so much pressure on themselves is likely to be experienced as a lack of confidence in them and is unlikely to get any traction. Instead, ask your patients what matters to them the most: Is it admission to the college of their choice? Achieving a certain score or GPA? Is it their competitiveness and drive to win? There is no wrong answer, but it is helpful for them to be able to reflect on what matters to them. Are they hoping to impress someone else? Are they worried about their future financial health and convinced that getting into a certain college will secure their financial success? Do they think this matters more to their parents than to themselves? Or have they discovered an intense interest in theoretical physics and want to be able to study at Caltech? If their ambition is meaningfully connected to an authentic interest or to their emerging identity, their sense of purpose will be much deeper and able to sustain them.
Even with talent and a strong sense of purpose, performing well is very difficult and demanding. It is important to consider the cycle of performance as including preparation, performance itself, and effective rest and recovery, just as with athletic performance. Whether the performance is the SATs, an AP test, a debate or big game, there were probably hours of preparation for every hour of performance. Help them to consider the importance of this practice or preparation time, and how to use that time effectively. Are they able to work in environments where there are few distractions? Do they have the support or useful feedback they need? How are they able to know when it is time for a break or when they are ready? It can be helpful for them to appreciate whether preparation or performance is more challenging for them, as the former requires focus and patience, while the latter requires courage and tenacity. If they are aware of which is harder for them, they can be thoughtful about how to effectively handle those challenges.
What can be most valuable for your patients is hearing from their pediatricians that they need to have time protected for rest and recharging, and not only for preparation and performance. Any athlete knows that failing to do so will lead to exhaustion and injury, and performance inevitably suffers. Rest is unwinding and slowing down, and a restful activity will leave them feeling calm, relaxed, and ready for sleep. A recharging activity is one that leaves them feeling refreshed and energized. Some common restful activities are a hot bath or shower, a distracting activity such as watching a show or surfing the web, playing a simple video game or puzzle or listening to music. Some recharging activities are creative ones (making art or music), engaging in hobbies, reading, or talking with a good friend. A few activities — sleep, exercise, and mindfulness meditation, are powerful in that they pack both rest and recharge into the same activity. Your patients should be discovering and learning which activities they find restful or recharging. The college application process or preparing for a varsity tryout will both add stress and give them an opportunity to learn what rests and recharges them. They should aim to have a list of at least five effective strategies that they can turn to when it’s time to rest or to recharge. Help them turn their work ethic to building a deeper well of self-knowledge that will serve them when they face challenges in high school or when they are on their own in college. This time of stress can be a time of growth, too.
Of course, remind your patients that this is a critical time to focus on basic self-care: They need consistently adequate, restful sleep, good nutrition, and physical activity. They will benefit from regular time in nature and time spent with friends that nourish them. They can find ways to compound these activities: Go for a walk with a friend, eat dinner with family, play a relaxing game while enjoying music. Lastly, ask your patients what is the last new thing they tried. It is easy to become so focused on an ambitious project that there is no time for exploration and play. Play is important throughout life, but adolescents are actively discovering their interests, talents, tastes, and values. To do this they need to be trying things that are new and maybe less purpose-driven. I call this type of activity “senseless fun.” Splashing in the pool is senseless fun, swimming laps is purposeful exercise that my contribute to recharging, and competing in a swim meet is often more on the stressful side. As they discover new talents, deeply engaging interests, what relaxes and recharges them, they will be learning who they are. Regardless of the outcome of a test, a big game, or where they go to college, it is this emerging knowledge about themselves that will carry them into adulthood. The pediatrician’s goal: Encouraging aspiration, exploration, and self-awareness in the context of giving permission for rest, recharging, and senseless fun.
Dr. Swick is physician in chief at Ohana, Center for Child and Adolescent Behavioral Health, Community Hospital of the Monterey (Calif.) Peninsula. Dr. Jellinek is professor emeritus of psychiatry and pediatrics, Harvard Medical School, Boston. Email them at [email protected].
With the changing leaves and cooling temperatures, early autumn also brings the excitement of the new school year. While returning to sports, mastering new subjects, and spending time with old and new friends is exhilarating, this season can also be a time of intense stress.
For those high school students who are especially ambitious, the school year presents the challenge of a very high stakes performance, one whose success will be measured by admission to a prized college. Not only are there classes to study for, but schedules are packed with a maximum number of subjects, a maximum number of Advanced Placement courses and a maximum number of impressive extra-curricular activities. Varsity sports practice, SAT prep, Debate Club, volunteer hours, and on and on.
What is often missing is enough time for sleep, socializing, exploring new interests, and unwinding. When you hear your patients (or parents) describing the intense stress of their overloaded schedules compounded by a sense that “I have no choice,” you have an opportunity to complicate their thinking. Introduce the idea that there are smart approaches to performing your best under stress. Pushing themselves relentlessly will inevitably lead to burnout and exhaustion. This approach will help them learn to make wise choices and will better serve their healthy development.
Start by acknowledging the stress of high-stakes performance. Telling your patients that they need to lower the temperature by not putting so much pressure on themselves is likely to be experienced as a lack of confidence in them and is unlikely to get any traction. Instead, ask your patients what matters to them the most: Is it admission to the college of their choice? Achieving a certain score or GPA? Is it their competitiveness and drive to win? There is no wrong answer, but it is helpful for them to be able to reflect on what matters to them. Are they hoping to impress someone else? Are they worried about their future financial health and convinced that getting into a certain college will secure their financial success? Do they think this matters more to their parents than to themselves? Or have they discovered an intense interest in theoretical physics and want to be able to study at Caltech? If their ambition is meaningfully connected to an authentic interest or to their emerging identity, their sense of purpose will be much deeper and able to sustain them.
Even with talent and a strong sense of purpose, performing well is very difficult and demanding. It is important to consider the cycle of performance as including preparation, performance itself, and effective rest and recovery, just as with athletic performance. Whether the performance is the SATs, an AP test, a debate or big game, there were probably hours of preparation for every hour of performance. Help them to consider the importance of this practice or preparation time, and how to use that time effectively. Are they able to work in environments where there are few distractions? Do they have the support or useful feedback they need? How are they able to know when it is time for a break or when they are ready? It can be helpful for them to appreciate whether preparation or performance is more challenging for them, as the former requires focus and patience, while the latter requires courage and tenacity. If they are aware of which is harder for them, they can be thoughtful about how to effectively handle those challenges.
What can be most valuable for your patients is hearing from their pediatricians that they need to have time protected for rest and recharging, and not only for preparation and performance. Any athlete knows that failing to do so will lead to exhaustion and injury, and performance inevitably suffers. Rest is unwinding and slowing down, and a restful activity will leave them feeling calm, relaxed, and ready for sleep. A recharging activity is one that leaves them feeling refreshed and energized. Some common restful activities are a hot bath or shower, a distracting activity such as watching a show or surfing the web, playing a simple video game or puzzle or listening to music. Some recharging activities are creative ones (making art or music), engaging in hobbies, reading, or talking with a good friend. A few activities — sleep, exercise, and mindfulness meditation, are powerful in that they pack both rest and recharge into the same activity. Your patients should be discovering and learning which activities they find restful or recharging. The college application process or preparing for a varsity tryout will both add stress and give them an opportunity to learn what rests and recharges them. They should aim to have a list of at least five effective strategies that they can turn to when it’s time to rest or to recharge. Help them turn their work ethic to building a deeper well of self-knowledge that will serve them when they face challenges in high school or when they are on their own in college. This time of stress can be a time of growth, too.
Of course, remind your patients that this is a critical time to focus on basic self-care: They need consistently adequate, restful sleep, good nutrition, and physical activity. They will benefit from regular time in nature and time spent with friends that nourish them. They can find ways to compound these activities: Go for a walk with a friend, eat dinner with family, play a relaxing game while enjoying music. Lastly, ask your patients what is the last new thing they tried. It is easy to become so focused on an ambitious project that there is no time for exploration and play. Play is important throughout life, but adolescents are actively discovering their interests, talents, tastes, and values. To do this they need to be trying things that are new and maybe less purpose-driven. I call this type of activity “senseless fun.” Splashing in the pool is senseless fun, swimming laps is purposeful exercise that my contribute to recharging, and competing in a swim meet is often more on the stressful side. As they discover new talents, deeply engaging interests, what relaxes and recharges them, they will be learning who they are. Regardless of the outcome of a test, a big game, or where they go to college, it is this emerging knowledge about themselves that will carry them into adulthood. The pediatrician’s goal: Encouraging aspiration, exploration, and self-awareness in the context of giving permission for rest, recharging, and senseless fun.
Dr. Swick is physician in chief at Ohana, Center for Child and Adolescent Behavioral Health, Community Hospital of the Monterey (Calif.) Peninsula. Dr. Jellinek is professor emeritus of psychiatry and pediatrics, Harvard Medical School, Boston. Email them at [email protected].
Is It Time for Universal Suicide Screening?
US suicide rates have reached alarming levels, with data from Centers for Disease Control and Prevention (CDC) showing a 37% increase from 2000 to 2022. Nearly 49,000 people died by suicide in 2022 alone, translating to one death every 11 minutes.
The increase in suicide rates has prompted calls for expansion of universal suicide screening, in which all individuals in medical or mental health care settings are screened for suicide risk, regardless of the purpose for their visit. But the psychiatric field is split on the issue, with some experts citing false positives and a lack of mental health care resources for those deemed at risk.
In 2022, when the US Preventative Services Task Force released its recommendations on suicide prevention, first in children and adolescents, and then in adults, the authors said there was insufficient evidence to support universal suicide screening.
Proponents of the practice pushed back on that finding, arguing that universal suicide screening could help identify those at high risk who might otherwise go undiagnosed, leading to earlier, potentially lifesaving, intervention.
So, what is the case for — and against — universal screening?
Sounding an Alert
The introduction of universal screening was driven by a confluence of factors that began with a 1999 report by then-US Surgeon General David Satcher, MD. This was followed by a report in 2016 from the Joint Commission on Detecting and Treating Suicidal Ideation that called for healthcare organizations to improve detection and treatment of suicidal ideation in all healthcare care settings.
Data from the alert showed that a significant number of people who died by suicide had a healthcare visit before their death. Half had seen a clinician a month before their death; nearly 30% had a medical visit just the week before — all with no detection of increased suicide risk.
It was that sort of finding that led Parkland Health and Hospital System in Dallas to become the first US hospital to implement universal suicide screening. Since the program launched in 2015, the system has screened more than 4.3 million patients in its emergency department, inpatient units, and 20 primary care clinics.
“Since the program began, we’ve completed between 40,000 to 50,000 screenings per month,” said Kimberly Roaten, PhD, associate chief quality and safety officer for behavioral health at Parkland Health.
Clinicians at Parkland use the five-item Ask Suicide-Screening Questions to assess suicidal intent, a commonly used tool that was originally developed for use in pediatric emergency rooms (ERs). The tool, which takes about 20 seconds to administer, has since been validated in both children and adults.
Based on a patient’s response, a clinical decision support system integrated into the electronic health record classifies suicide risk as none, moderate, or high.
Patients identified as moderate risk are offered a more in-depth assessment with a mental health clinician, though participation is not mandatory, said Dr. Roaten. Those at high risk receive a more thorough evaluation.
The proportion of ER patients at Parkland who screen positive for any suicidal intent has consistently remained at about 7%, and at 2% in the primary care clinics, she said.
To better understand what the program may have had on suicide prevention, Dr. Roaten is leading a National Institute of Mental Health–funded study to link a decade of mortality data from the state of Texas to patient data from Parkland Health. Investigators will analyze information about patients identified at risk for suicide, those patients’ characteristics, and who dies by suicide.
Universal Screening Expands
Other health systems have adopted universal suicide screening including the Indian Health Service and the US Veterans Health Administration. Universal suicide screening is also in place in a growing number of primary care practices and hospitals throughout the United States and will be mandatory for patients aged 12 years and older in all acute care hospitals in California beginning in 2025.
There is also a push for universal screening to be coordinated through local, state, and federal government, nonprofit, and private sectors. The National Action Alliance for Suicide Prevention is charged with advancing the White House’s 2024 National Strategy for Suicide Prevention, a 10-year plan to address gaps in suicide prevention in the United States.
Sarah Brummett, JD, director of the National Action Alliance for Suicide Prevention’s executive committee, said that universal suicide screening is part of the 2024 strategy. “We know there are barriers to universal screening, and so it’s important to recognize what they are so we can address them,” said Ms. Brummett.
Barriers may include adequate staffing, or a system in place to triage patients who screen positive.
At Parkland, cost and workload have been minimal, Dr. Roaten said. “We built a model that only dedicates our highest-value resources to the most at-risk patients.”
She also noted that relief may be on the horizon for health systems where cost is an obstacle to universal screening and subsequent intervention. “There are efforts at the federal level to increase funding for suicide assessment and crisis response,” she said.
Pushback on Universal Screening
Universal suicide screening has its detractors, including critics who say expansion is unlikely to reduce suicide rates.
“The issue with suicidal ideation is that it is very dynamic. Suicidal ideation changes very quickly — sometimes within hours,” said Craig Bryan, PsyD, professor of psychiatry and behavioral health at Ohio State University in Columbus, Ohio.
Universal screening can also lead to false positives, where a patient who screens positive for suicidal ideation has no actual intention of attempting suicide, potentially creating unnecessary concern and burden on health care resources, Dr. Bryan noted.
“What do you do with everyone who screens positive?” Dr. Bryan said. “I’ve spoken with leaders of many health systems in the United States, and there is pushback against universal screening because they don’t have enough mental health resources to handle all of the referrals.”
Suicide screening also doesn’t predict who will die by suicide, Dr. Bryan added. It only identifies those willing to disclose suicidal thoughts. There is a significant number of people without mental illness who may never seek medical care, so “the warning signs we’re teaching people to recognize — depression, anxiety, and substance abuse — might not be evident in these individuals,” he said.
“Life sideswipes them suddenly, and they go from 0 to 60 ... and they may have access to a highly lethal method [of suicide] which weaponizes that moment of despair,” said Dr. Bryan. No amount of screening could possibly predict those types of suicides, he added.
Paul Nestadt, MD, associate professor of psychiatry and behavioral sciences at Johns Hopkins School of Medicine, agrees with Dr. Bryan and noted there isn’t a strong correlation between suicidal ideation and death by suicide.
“Suicidal thoughts are very common, but suicide is a rare event,” he said.
He cited a study that showed that two thirds of individuals who died by suicide had denied experiencing suicidal thoughts when asked, and half of them died within 2 days of this denial. Other research suggests that as many as 98% of people who express suicidal ideation do not die by suicide, Dr. Nestadt said.
A Public Health Issue
If universal screening is not the answer to predicting and preventing suicide, what is? One way would be to approach suicide as a public health issue, Dr. Nestadt said.
“How did we reduce the rate of motor vehicle deaths? We didn’t test each driver’s reaction time behind the wheel,” he said. “Instead, we passed seatbelt and airbag legislation, implemented federal speed limits, and as a result, the number of motor vehicle fatalities decreased.”
Dr. Nestadt is an advocate for stronger gun safety legislation, which has proven effective in reducing suicide rates. A study published this year showed that states with child access prevention laws, negligent storage laws, and mandatory waiting periods for gun purchases reported fewer suicide deaths than those without that legislation.
Other measures might be applied in cases of extreme individual suicide risk, including extreme risk protection orders, also known as “red flag” laws, he added. This type of legislation provides a pathway for law enforcement to temporarily remove firearms from individuals who pose a risk to themselves or others.
“These have been shown to be very effective in saving lives,” Dr. Nestadt said.
Dr. Nestadt and others are also using machine learning models to predict suicide risk. Those identified as high-risk may be flagged on their electronic medical record. Ideally, when the algorithm becomes more accurate at predicting suicide, anyone treating this patient can then decide if action is needed, said Dr. Nestadt.
In his work with suicidal military personnel, Dr. Bryan and his colleagues established a brief form of cognitive behavioral therapy (BCBT) to help participants challenge cognitive distortions and build coping strategies to deal with feel with intense feelings of distress. Data show that BCBT reduced suicide attempts among active-duty soldiers by 60% compared with standard mental health treatment. It has since been shown to work in civilians as well.
Dr. Bryan is also researching fluctuations in the wish to live versus the wish to die relative to one another and mapping the trajectory of risk states along the way.
The goal is that these and other suicide prevention strategies currently under study by his team and others will help stem the rise in suicide deaths.
“Overall, we need to train mental health providers to implement suicide prevention therapies and establish suicide risk programs,” Dr. Bryan said. “But until we build one of these suicide prevention interventions to scale, we’re putting the cart before the horse.”
Dr. Roaten, Ms. Brummett, Dr. Bryan, and Dr. Nestadt reported no relevant disclosures.
A version of this article appeared on Medscape.com.
US suicide rates have reached alarming levels, with data from Centers for Disease Control and Prevention (CDC) showing a 37% increase from 2000 to 2022. Nearly 49,000 people died by suicide in 2022 alone, translating to one death every 11 minutes.
The increase in suicide rates has prompted calls for expansion of universal suicide screening, in which all individuals in medical or mental health care settings are screened for suicide risk, regardless of the purpose for their visit. But the psychiatric field is split on the issue, with some experts citing false positives and a lack of mental health care resources for those deemed at risk.
In 2022, when the US Preventative Services Task Force released its recommendations on suicide prevention, first in children and adolescents, and then in adults, the authors said there was insufficient evidence to support universal suicide screening.
Proponents of the practice pushed back on that finding, arguing that universal suicide screening could help identify those at high risk who might otherwise go undiagnosed, leading to earlier, potentially lifesaving, intervention.
So, what is the case for — and against — universal screening?
Sounding an Alert
The introduction of universal screening was driven by a confluence of factors that began with a 1999 report by then-US Surgeon General David Satcher, MD. This was followed by a report in 2016 from the Joint Commission on Detecting and Treating Suicidal Ideation that called for healthcare organizations to improve detection and treatment of suicidal ideation in all healthcare care settings.
Data from the alert showed that a significant number of people who died by suicide had a healthcare visit before their death. Half had seen a clinician a month before their death; nearly 30% had a medical visit just the week before — all with no detection of increased suicide risk.
It was that sort of finding that led Parkland Health and Hospital System in Dallas to become the first US hospital to implement universal suicide screening. Since the program launched in 2015, the system has screened more than 4.3 million patients in its emergency department, inpatient units, and 20 primary care clinics.
“Since the program began, we’ve completed between 40,000 to 50,000 screenings per month,” said Kimberly Roaten, PhD, associate chief quality and safety officer for behavioral health at Parkland Health.
Clinicians at Parkland use the five-item Ask Suicide-Screening Questions to assess suicidal intent, a commonly used tool that was originally developed for use in pediatric emergency rooms (ERs). The tool, which takes about 20 seconds to administer, has since been validated in both children and adults.
Based on a patient’s response, a clinical decision support system integrated into the electronic health record classifies suicide risk as none, moderate, or high.
Patients identified as moderate risk are offered a more in-depth assessment with a mental health clinician, though participation is not mandatory, said Dr. Roaten. Those at high risk receive a more thorough evaluation.
The proportion of ER patients at Parkland who screen positive for any suicidal intent has consistently remained at about 7%, and at 2% in the primary care clinics, she said.
To better understand what the program may have had on suicide prevention, Dr. Roaten is leading a National Institute of Mental Health–funded study to link a decade of mortality data from the state of Texas to patient data from Parkland Health. Investigators will analyze information about patients identified at risk for suicide, those patients’ characteristics, and who dies by suicide.
Universal Screening Expands
Other health systems have adopted universal suicide screening including the Indian Health Service and the US Veterans Health Administration. Universal suicide screening is also in place in a growing number of primary care practices and hospitals throughout the United States and will be mandatory for patients aged 12 years and older in all acute care hospitals in California beginning in 2025.
There is also a push for universal screening to be coordinated through local, state, and federal government, nonprofit, and private sectors. The National Action Alliance for Suicide Prevention is charged with advancing the White House’s 2024 National Strategy for Suicide Prevention, a 10-year plan to address gaps in suicide prevention in the United States.
Sarah Brummett, JD, director of the National Action Alliance for Suicide Prevention’s executive committee, said that universal suicide screening is part of the 2024 strategy. “We know there are barriers to universal screening, and so it’s important to recognize what they are so we can address them,” said Ms. Brummett.
Barriers may include adequate staffing, or a system in place to triage patients who screen positive.
At Parkland, cost and workload have been minimal, Dr. Roaten said. “We built a model that only dedicates our highest-value resources to the most at-risk patients.”
She also noted that relief may be on the horizon for health systems where cost is an obstacle to universal screening and subsequent intervention. “There are efforts at the federal level to increase funding for suicide assessment and crisis response,” she said.
Pushback on Universal Screening
Universal suicide screening has its detractors, including critics who say expansion is unlikely to reduce suicide rates.
“The issue with suicidal ideation is that it is very dynamic. Suicidal ideation changes very quickly — sometimes within hours,” said Craig Bryan, PsyD, professor of psychiatry and behavioral health at Ohio State University in Columbus, Ohio.
Universal screening can also lead to false positives, where a patient who screens positive for suicidal ideation has no actual intention of attempting suicide, potentially creating unnecessary concern and burden on health care resources, Dr. Bryan noted.
“What do you do with everyone who screens positive?” Dr. Bryan said. “I’ve spoken with leaders of many health systems in the United States, and there is pushback against universal screening because they don’t have enough mental health resources to handle all of the referrals.”
Suicide screening also doesn’t predict who will die by suicide, Dr. Bryan added. It only identifies those willing to disclose suicidal thoughts. There is a significant number of people without mental illness who may never seek medical care, so “the warning signs we’re teaching people to recognize — depression, anxiety, and substance abuse — might not be evident in these individuals,” he said.
“Life sideswipes them suddenly, and they go from 0 to 60 ... and they may have access to a highly lethal method [of suicide] which weaponizes that moment of despair,” said Dr. Bryan. No amount of screening could possibly predict those types of suicides, he added.
Paul Nestadt, MD, associate professor of psychiatry and behavioral sciences at Johns Hopkins School of Medicine, agrees with Dr. Bryan and noted there isn’t a strong correlation between suicidal ideation and death by suicide.
“Suicidal thoughts are very common, but suicide is a rare event,” he said.
He cited a study that showed that two thirds of individuals who died by suicide had denied experiencing suicidal thoughts when asked, and half of them died within 2 days of this denial. Other research suggests that as many as 98% of people who express suicidal ideation do not die by suicide, Dr. Nestadt said.
A Public Health Issue
If universal screening is not the answer to predicting and preventing suicide, what is? One way would be to approach suicide as a public health issue, Dr. Nestadt said.
“How did we reduce the rate of motor vehicle deaths? We didn’t test each driver’s reaction time behind the wheel,” he said. “Instead, we passed seatbelt and airbag legislation, implemented federal speed limits, and as a result, the number of motor vehicle fatalities decreased.”
Dr. Nestadt is an advocate for stronger gun safety legislation, which has proven effective in reducing suicide rates. A study published this year showed that states with child access prevention laws, negligent storage laws, and mandatory waiting periods for gun purchases reported fewer suicide deaths than those without that legislation.
Other measures might be applied in cases of extreme individual suicide risk, including extreme risk protection orders, also known as “red flag” laws, he added. This type of legislation provides a pathway for law enforcement to temporarily remove firearms from individuals who pose a risk to themselves or others.
“These have been shown to be very effective in saving lives,” Dr. Nestadt said.
Dr. Nestadt and others are also using machine learning models to predict suicide risk. Those identified as high-risk may be flagged on their electronic medical record. Ideally, when the algorithm becomes more accurate at predicting suicide, anyone treating this patient can then decide if action is needed, said Dr. Nestadt.
In his work with suicidal military personnel, Dr. Bryan and his colleagues established a brief form of cognitive behavioral therapy (BCBT) to help participants challenge cognitive distortions and build coping strategies to deal with feel with intense feelings of distress. Data show that BCBT reduced suicide attempts among active-duty soldiers by 60% compared with standard mental health treatment. It has since been shown to work in civilians as well.
Dr. Bryan is also researching fluctuations in the wish to live versus the wish to die relative to one another and mapping the trajectory of risk states along the way.
The goal is that these and other suicide prevention strategies currently under study by his team and others will help stem the rise in suicide deaths.
“Overall, we need to train mental health providers to implement suicide prevention therapies and establish suicide risk programs,” Dr. Bryan said. “But until we build one of these suicide prevention interventions to scale, we’re putting the cart before the horse.”
Dr. Roaten, Ms. Brummett, Dr. Bryan, and Dr. Nestadt reported no relevant disclosures.
A version of this article appeared on Medscape.com.
US suicide rates have reached alarming levels, with data from Centers for Disease Control and Prevention (CDC) showing a 37% increase from 2000 to 2022. Nearly 49,000 people died by suicide in 2022 alone, translating to one death every 11 minutes.
The increase in suicide rates has prompted calls for expansion of universal suicide screening, in which all individuals in medical or mental health care settings are screened for suicide risk, regardless of the purpose for their visit. But the psychiatric field is split on the issue, with some experts citing false positives and a lack of mental health care resources for those deemed at risk.
In 2022, when the US Preventative Services Task Force released its recommendations on suicide prevention, first in children and adolescents, and then in adults, the authors said there was insufficient evidence to support universal suicide screening.
Proponents of the practice pushed back on that finding, arguing that universal suicide screening could help identify those at high risk who might otherwise go undiagnosed, leading to earlier, potentially lifesaving, intervention.
So, what is the case for — and against — universal screening?
Sounding an Alert
The introduction of universal screening was driven by a confluence of factors that began with a 1999 report by then-US Surgeon General David Satcher, MD. This was followed by a report in 2016 from the Joint Commission on Detecting and Treating Suicidal Ideation that called for healthcare organizations to improve detection and treatment of suicidal ideation in all healthcare care settings.
Data from the alert showed that a significant number of people who died by suicide had a healthcare visit before their death. Half had seen a clinician a month before their death; nearly 30% had a medical visit just the week before — all with no detection of increased suicide risk.
It was that sort of finding that led Parkland Health and Hospital System in Dallas to become the first US hospital to implement universal suicide screening. Since the program launched in 2015, the system has screened more than 4.3 million patients in its emergency department, inpatient units, and 20 primary care clinics.
“Since the program began, we’ve completed between 40,000 to 50,000 screenings per month,” said Kimberly Roaten, PhD, associate chief quality and safety officer for behavioral health at Parkland Health.
Clinicians at Parkland use the five-item Ask Suicide-Screening Questions to assess suicidal intent, a commonly used tool that was originally developed for use in pediatric emergency rooms (ERs). The tool, which takes about 20 seconds to administer, has since been validated in both children and adults.
Based on a patient’s response, a clinical decision support system integrated into the electronic health record classifies suicide risk as none, moderate, or high.
Patients identified as moderate risk are offered a more in-depth assessment with a mental health clinician, though participation is not mandatory, said Dr. Roaten. Those at high risk receive a more thorough evaluation.
The proportion of ER patients at Parkland who screen positive for any suicidal intent has consistently remained at about 7%, and at 2% in the primary care clinics, she said.
To better understand what the program may have had on suicide prevention, Dr. Roaten is leading a National Institute of Mental Health–funded study to link a decade of mortality data from the state of Texas to patient data from Parkland Health. Investigators will analyze information about patients identified at risk for suicide, those patients’ characteristics, and who dies by suicide.
Universal Screening Expands
Other health systems have adopted universal suicide screening including the Indian Health Service and the US Veterans Health Administration. Universal suicide screening is also in place in a growing number of primary care practices and hospitals throughout the United States and will be mandatory for patients aged 12 years and older in all acute care hospitals in California beginning in 2025.
There is also a push for universal screening to be coordinated through local, state, and federal government, nonprofit, and private sectors. The National Action Alliance for Suicide Prevention is charged with advancing the White House’s 2024 National Strategy for Suicide Prevention, a 10-year plan to address gaps in suicide prevention in the United States.
Sarah Brummett, JD, director of the National Action Alliance for Suicide Prevention’s executive committee, said that universal suicide screening is part of the 2024 strategy. “We know there are barriers to universal screening, and so it’s important to recognize what they are so we can address them,” said Ms. Brummett.
Barriers may include adequate staffing, or a system in place to triage patients who screen positive.
At Parkland, cost and workload have been minimal, Dr. Roaten said. “We built a model that only dedicates our highest-value resources to the most at-risk patients.”
She also noted that relief may be on the horizon for health systems where cost is an obstacle to universal screening and subsequent intervention. “There are efforts at the federal level to increase funding for suicide assessment and crisis response,” she said.
Pushback on Universal Screening
Universal suicide screening has its detractors, including critics who say expansion is unlikely to reduce suicide rates.
“The issue with suicidal ideation is that it is very dynamic. Suicidal ideation changes very quickly — sometimes within hours,” said Craig Bryan, PsyD, professor of psychiatry and behavioral health at Ohio State University in Columbus, Ohio.
Universal screening can also lead to false positives, where a patient who screens positive for suicidal ideation has no actual intention of attempting suicide, potentially creating unnecessary concern and burden on health care resources, Dr. Bryan noted.
“What do you do with everyone who screens positive?” Dr. Bryan said. “I’ve spoken with leaders of many health systems in the United States, and there is pushback against universal screening because they don’t have enough mental health resources to handle all of the referrals.”
Suicide screening also doesn’t predict who will die by suicide, Dr. Bryan added. It only identifies those willing to disclose suicidal thoughts. There is a significant number of people without mental illness who may never seek medical care, so “the warning signs we’re teaching people to recognize — depression, anxiety, and substance abuse — might not be evident in these individuals,” he said.
“Life sideswipes them suddenly, and they go from 0 to 60 ... and they may have access to a highly lethal method [of suicide] which weaponizes that moment of despair,” said Dr. Bryan. No amount of screening could possibly predict those types of suicides, he added.
Paul Nestadt, MD, associate professor of psychiatry and behavioral sciences at Johns Hopkins School of Medicine, agrees with Dr. Bryan and noted there isn’t a strong correlation between suicidal ideation and death by suicide.
“Suicidal thoughts are very common, but suicide is a rare event,” he said.
He cited a study that showed that two thirds of individuals who died by suicide had denied experiencing suicidal thoughts when asked, and half of them died within 2 days of this denial. Other research suggests that as many as 98% of people who express suicidal ideation do not die by suicide, Dr. Nestadt said.
A Public Health Issue
If universal screening is not the answer to predicting and preventing suicide, what is? One way would be to approach suicide as a public health issue, Dr. Nestadt said.
“How did we reduce the rate of motor vehicle deaths? We didn’t test each driver’s reaction time behind the wheel,” he said. “Instead, we passed seatbelt and airbag legislation, implemented federal speed limits, and as a result, the number of motor vehicle fatalities decreased.”
Dr. Nestadt is an advocate for stronger gun safety legislation, which has proven effective in reducing suicide rates. A study published this year showed that states with child access prevention laws, negligent storage laws, and mandatory waiting periods for gun purchases reported fewer suicide deaths than those without that legislation.
Other measures might be applied in cases of extreme individual suicide risk, including extreme risk protection orders, also known as “red flag” laws, he added. This type of legislation provides a pathway for law enforcement to temporarily remove firearms from individuals who pose a risk to themselves or others.
“These have been shown to be very effective in saving lives,” Dr. Nestadt said.
Dr. Nestadt and others are also using machine learning models to predict suicide risk. Those identified as high-risk may be flagged on their electronic medical record. Ideally, when the algorithm becomes more accurate at predicting suicide, anyone treating this patient can then decide if action is needed, said Dr. Nestadt.
In his work with suicidal military personnel, Dr. Bryan and his colleagues established a brief form of cognitive behavioral therapy (BCBT) to help participants challenge cognitive distortions and build coping strategies to deal with feel with intense feelings of distress. Data show that BCBT reduced suicide attempts among active-duty soldiers by 60% compared with standard mental health treatment. It has since been shown to work in civilians as well.
Dr. Bryan is also researching fluctuations in the wish to live versus the wish to die relative to one another and mapping the trajectory of risk states along the way.
The goal is that these and other suicide prevention strategies currently under study by his team and others will help stem the rise in suicide deaths.
“Overall, we need to train mental health providers to implement suicide prevention therapies and establish suicide risk programs,” Dr. Bryan said. “But until we build one of these suicide prevention interventions to scale, we’re putting the cart before the horse.”
Dr. Roaten, Ms. Brummett, Dr. Bryan, and Dr. Nestadt reported no relevant disclosures.
A version of this article appeared on Medscape.com.
Moving Beyond Traditional Methods for Treatment of Acne Keloidalis Nuchae
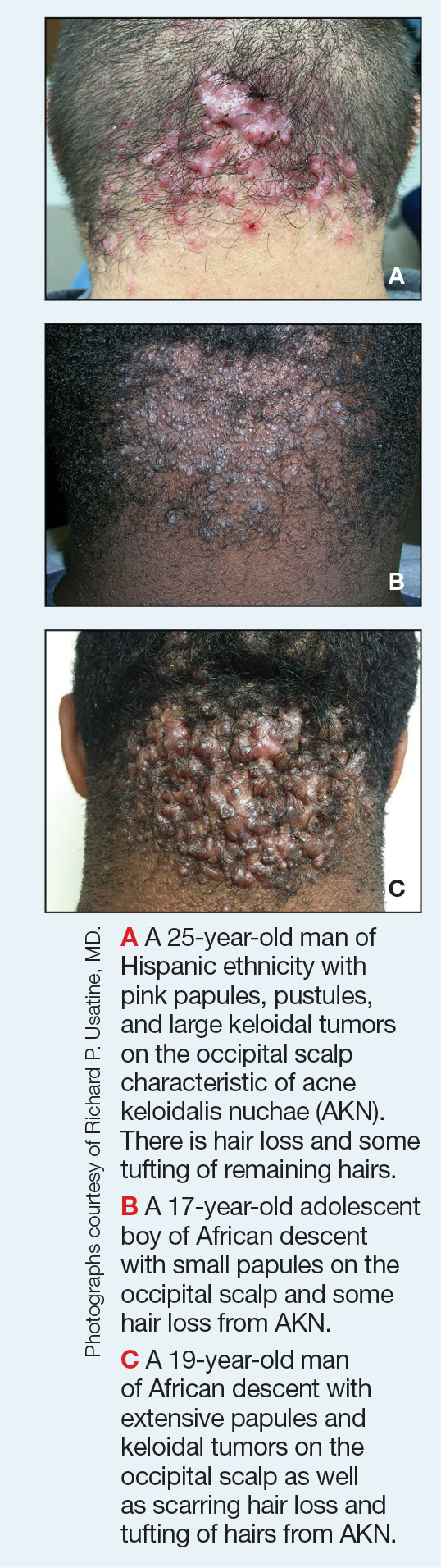
Acne keloidalis nuchae (AKN) is a chronic inflammatory condition commonly affecting the occipital scalp and posterior neck. It causes discrete or extensive fibrosing papules that may coalesce to form pronounced tumorlike masses1,2 with scarring alopecia (Figure, A–C).3 Pustules, hair tufts, secondary bacterial infections, abscesses, and sinus tracts also may occur.1 The pathogenesis of AKN has been characterized as varying stages of follicular inflammation at the infundibular and isthmus levels followed by fibrotic occlusion of the follicular lumen.4 Pruritus, pain, bleeding, oozing, and a feeling of scalp tightness may occur.1,5
Umar et al6 performed a retrospective review of 108 men with AKN—58% of African descent, 37% Hispanic, 3% Asian, and 2% Middle Eastern—and proposed a 3-tier classification system for AKN. Tier 1 focused on the distribution and sagittal spread of AKN lesions between the clinical demarcation lines of the occipital notch and posterior hairline. Tier 2 focused on the type of lesions present—discrete papules or nodules, coalescing/abutting lesions, plaques (raised, atrophic, or indurated), or dome-shaped tumoral masses. Tier 3 focused on the presence or absence of co-existing dissecting cellulitis or folliculitis decalvans.6
Epidemiology
Acne keloidalis nuchae primarily manifests in adolescent and adult men of African or Afro-Caribbean descent.7 Among African American men, the prevalence of AKN ranges from 0.5% to 13.6%.8 Similar ranges have been reported among Nigerian, South African, and West African men.1 Acne keloidalis nuchae also affects Asian and Hispanic men but rarely is seen in non-Hispanic White men or in women of any ethnicity.9,10 The male to female ratio is 20:1.1,11 Hair texture, hairstyling practices such as closely shaved or faded haircuts, and genetics likely contribute to development of AKN. Sports and occupations that require the use of headgear or a tight collar may increase the risk for AKN.12
Key clinical features in people with darker skin tones
- The lesions of AKN range in color from pink to dark brown or black. Postinflammatory hyperpigmentation or hyperchromia may be present around AKN lesions.
- Chronicity of AKN may lead to extended use of high-potency topical or intralesional corticosteroids, which causes transient or long-lasting hypopigmentation, especially in those with darker skin tones.
Worth noting
- Acne keloidalis nuchae can be disfiguring, which negatively impacts quality of life and self-esteem.12
- Some occupations (eg, military, police) have hair policies that may not be favorable to those with or at risk for AKN.
- Patients with AKN are 2 to 3 times more likely to present with metabolic syndrome, hypertension, type 2 diabetes mellitus, or obesity.13
Treatment
There are no treatments approved by the US Food and Drug Administration specifically for AKN. Treatment approaches are based on the pathophysiology, secondary impacts on the skin, and disease severity. Growing out the hair may prevent worsening and/or decrease the risk for new lesions.6
- Options include but are not limited to topical and systemic therapies (eg, topical corticosteroids, oral or topical antibiotics, isotretinoin, topical retinoids, imiquimod, pimecrolimus), light devices (eg, phototherapy, laser), ablative therapies (eg, laser, cryotherapy, radiotherapy), and surgery (eg, excision, follicular unit excision), often in combination.6,14,15
- Intralesional triamcinolone injections are considered standard of care. Adotama et al found that injecting triamcinolone into the deep dermis in the area of flat or papular AKN yielded better control of inflammation and decreased appearance of lesions compared with injecting individual lesions.16
- For extensive AKN lesions that do not respond to less-invasive therapies, consider surgical techniques,6,17 such as follicular unit excision18 and more extensive surgical excisions building on approaches from pioneers Drs. John Kenney and Harold Pierce.19 An innovative surgical approach for removal of large AKNs is the bat excision technique—wound shape resembles a bat in a spread-eagled position—with secondary intention healing with or without debridement and/or tension sutures. The resulting linear scar acts as a new posterior hair line.20
Health disparity highlights
Access to a dermatologic or plastic surgeon with expertise in the surgical treatment of large AKNs may be challenging but is needed to reduce risk for recurrence and adverse events.
Close-cropped haircuts on the occipital scalp, which are particularly popular among men of African descent, increase the risk for AKN.5 Although this grooming style may be a personal preference, other hairstyles commonly worn by those with tightly coiled hair may be deemed “unprofessional” in society or the workplace, which leads to hairstyling practices that may increase the risk for AKN.21
Acne keloidalis nuchae remains an understudied entity that adversely affects patients with skin of color.
- Ogunbiyi A. Acne keloidalis nuchae: prevalence, impact, and management challenges. Clin Cosmet Investig Dermatol. 2016;9:483-489. doi:10.2147/CCID.S99225
- Al Aboud DM, Badri T. Acne keloidalis nuchae. In: StatPearls [Internet]. Updated July 31, 2023. Accessed August 2, 2024. https://www.ncbi.nlm.nih.gov/books/NBK459135/
- Sperling LC, Homoky C, Pratt L, et al. Acne keloidalis is a form of primary scarring alopecia. Arch Dermatol. 2000;136:479-484.
- Herzberg AJ, Dinehart SM, Kerns BJ, et al. Acne keloidalis: transverse microscopy, immunohistochemistry, and electron microscopy. Am J Dermatopathol. 1990;12:109-121. doi:10.1097/00000372-199004000-00001
- Saka B, Akakpo A-S, Téclessou JN, et al. Risk factors associated with acne keloidalis nuchae in black subjects: a case-control study. Ann Dermatol Venereol. 2020;147:350-354. doi:10.1016/j.annder.2020.01.007
- Umar S, Lee DJ, Lullo JJ. A retrospective cohort study and clinical classification system of acne keloidalis nuchae. J Clin Aesthet Dermatol. 2021;14:E61-E67.
- Reja M, Silverberg NB. Acne keloidalis nuchae. In: Silverberg NB, Durán-McKinster C, Tay YK, eds. Pediatric Skin of Color. Springer; 2015:141-145. doi:10.1007/978-1-4614-6654-3_16
- Knable AL Jr, Hanke CW, Gonin R. Prevalence of acne keloidalis nuchae in football players. J Am Acad Dermatol. 1997;37:570-574. doi:10.1016/s0190-9622(97)70173-7
- Umar S, Ton D, Carter MJ, et al. Unveiling a shared precursor condition for acne keloidalis nuchae and primary cicatricial alopecias. Clin Cosmet Investig Dermatol. 2023;16:2315-2327. doi:10.2147/CCID.S422310
- Na K, Oh SH, Kim SK. Acne keloidalis nuchae in Asian: a single institutional experience. PLoS One. 2017;12:e0189790. doi:10.1371/journal.pone.0189790
- Ogunbiyi A, George A. Acne keloidalis in females: case report and review of literature. J Natl Med Assoc. 2005;97:736-738.
- Alexis A, Heath CR, Halder RM. Folliculitis keloidalis nuchae and pseudofolliculitis barbae: are prevention and effective treatment within reach? Dermatol Clin. 2014;32:183-191. doi:10.1016/j.det.2013.12.001
- Kridin K, Solomon A, Tzur-Bitan D, et al. Acne keloidalis nuchae and the metabolic syndrome: a population-based study. Am J Clin Dermatol. 2020;21:733-739. doi:10.1007/s40257-020-00541-z
- Smart K, Rodriguez I, Worswick S. Comorbidities and treatment options for acne keloidalis nuchae. Dermatol Ther. Published online May 25, 2024. doi:10.1155/2024/8336926
- Callender VD, Young CM, Haverstock CL, et al. An open label study of clobetasol propionate 0.05% and betamethasone valerate 0.12% foams in the treatment of mild to moderate acne keloidalis. Cutis. 2005;75:317-321.
- Adotama P, Grullon K, Ali S, et al. How we do it: our method for triamcinolone injections of acne keloidalis nuchae. Dermatol Surg. 2023;49:713-714. doi:10.1097/DSS.0000000000003803
- Beckett N, Lawson C, Cohen G. Electrosurgical excision of acne keloidalis nuchae with secondary intention healing. J Clin Aesthet Dermatol. 2011;4:36-39.
- Esmat SM, Abdel Hay RM, Abu Zeid OM, et al. The efficacy of laser-assisted hair removal in the treatment of acne keloidalis nuchae; a pilot study. Eur J Dermatol. 2012;22:645-650. doi:10.1684/ejd.2012.1830
- Dillard AD, Quarles FN. African-American pioneers in dermatology. In: Taylor SC, Kelly AP, Lim HW, et al, eds. Dermatology for Skin of Color. 2nd ed. McGraw-Hill Education; 2016:717-730.
- Umar S, David CV, Castillo JR, et al. Innovative surgical approaches and selection criteria of large acne keloidalis nuchae lesions. Plast Reconstr Surg Glob Open. 2019;7:E2215. doi:10.1097/GOX.0000000000002215
- Lee MS, Nambudiri VE. The CROWN act and dermatology: taking a stand against race-based hair discrimination. J Am Acad Dermatol. 2021;84:1181-1182. doi:10.1016/j.jaad.2020.11.065
Acne keloidalis nuchae (AKN) is a chronic inflammatory condition commonly affecting the occipital scalp and posterior neck. It causes discrete or extensive fibrosing papules that may coalesce to form pronounced tumorlike masses1,2 with scarring alopecia (Figure, A–C).3 Pustules, hair tufts, secondary bacterial infections, abscesses, and sinus tracts also may occur.1 The pathogenesis of AKN has been characterized as varying stages of follicular inflammation at the infundibular and isthmus levels followed by fibrotic occlusion of the follicular lumen.4 Pruritus, pain, bleeding, oozing, and a feeling of scalp tightness may occur.1,5
Umar et al6 performed a retrospective review of 108 men with AKN—58% of African descent, 37% Hispanic, 3% Asian, and 2% Middle Eastern—and proposed a 3-tier classification system for AKN. Tier 1 focused on the distribution and sagittal spread of AKN lesions between the clinical demarcation lines of the occipital notch and posterior hairline. Tier 2 focused on the type of lesions present—discrete papules or nodules, coalescing/abutting lesions, plaques (raised, atrophic, or indurated), or dome-shaped tumoral masses. Tier 3 focused on the presence or absence of co-existing dissecting cellulitis or folliculitis decalvans.6
Epidemiology
Acne keloidalis nuchae primarily manifests in adolescent and adult men of African or Afro-Caribbean descent.7 Among African American men, the prevalence of AKN ranges from 0.5% to 13.6%.8 Similar ranges have been reported among Nigerian, South African, and West African men.1 Acne keloidalis nuchae also affects Asian and Hispanic men but rarely is seen in non-Hispanic White men or in women of any ethnicity.9,10 The male to female ratio is 20:1.1,11 Hair texture, hairstyling practices such as closely shaved or faded haircuts, and genetics likely contribute to development of AKN. Sports and occupations that require the use of headgear or a tight collar may increase the risk for AKN.12
Key clinical features in people with darker skin tones
- The lesions of AKN range in color from pink to dark brown or black. Postinflammatory hyperpigmentation or hyperchromia may be present around AKN lesions.
- Chronicity of AKN may lead to extended use of high-potency topical or intralesional corticosteroids, which causes transient or long-lasting hypopigmentation, especially in those with darker skin tones.
Worth noting
- Acne keloidalis nuchae can be disfiguring, which negatively impacts quality of life and self-esteem.12
- Some occupations (eg, military, police) have hair policies that may not be favorable to those with or at risk for AKN.
- Patients with AKN are 2 to 3 times more likely to present with metabolic syndrome, hypertension, type 2 diabetes mellitus, or obesity.13
Treatment
There are no treatments approved by the US Food and Drug Administration specifically for AKN. Treatment approaches are based on the pathophysiology, secondary impacts on the skin, and disease severity. Growing out the hair may prevent worsening and/or decrease the risk for new lesions.6
- Options include but are not limited to topical and systemic therapies (eg, topical corticosteroids, oral or topical antibiotics, isotretinoin, topical retinoids, imiquimod, pimecrolimus), light devices (eg, phototherapy, laser), ablative therapies (eg, laser, cryotherapy, radiotherapy), and surgery (eg, excision, follicular unit excision), often in combination.6,14,15
- Intralesional triamcinolone injections are considered standard of care. Adotama et al found that injecting triamcinolone into the deep dermis in the area of flat or papular AKN yielded better control of inflammation and decreased appearance of lesions compared with injecting individual lesions.16
- For extensive AKN lesions that do not respond to less-invasive therapies, consider surgical techniques,6,17 such as follicular unit excision18 and more extensive surgical excisions building on approaches from pioneers Drs. John Kenney and Harold Pierce.19 An innovative surgical approach for removal of large AKNs is the bat excision technique—wound shape resembles a bat in a spread-eagled position—with secondary intention healing with or without debridement and/or tension sutures. The resulting linear scar acts as a new posterior hair line.20
Health disparity highlights
Access to a dermatologic or plastic surgeon with expertise in the surgical treatment of large AKNs may be challenging but is needed to reduce risk for recurrence and adverse events.
Close-cropped haircuts on the occipital scalp, which are particularly popular among men of African descent, increase the risk for AKN.5 Although this grooming style may be a personal preference, other hairstyles commonly worn by those with tightly coiled hair may be deemed “unprofessional” in society or the workplace, which leads to hairstyling practices that may increase the risk for AKN.21
Acne keloidalis nuchae remains an understudied entity that adversely affects patients with skin of color.
Acne keloidalis nuchae (AKN) is a chronic inflammatory condition commonly affecting the occipital scalp and posterior neck. It causes discrete or extensive fibrosing papules that may coalesce to form pronounced tumorlike masses1,2 with scarring alopecia (Figure, A–C).3 Pustules, hair tufts, secondary bacterial infections, abscesses, and sinus tracts also may occur.1 The pathogenesis of AKN has been characterized as varying stages of follicular inflammation at the infundibular and isthmus levels followed by fibrotic occlusion of the follicular lumen.4 Pruritus, pain, bleeding, oozing, and a feeling of scalp tightness may occur.1,5
Umar et al6 performed a retrospective review of 108 men with AKN—58% of African descent, 37% Hispanic, 3% Asian, and 2% Middle Eastern—and proposed a 3-tier classification system for AKN. Tier 1 focused on the distribution and sagittal spread of AKN lesions between the clinical demarcation lines of the occipital notch and posterior hairline. Tier 2 focused on the type of lesions present—discrete papules or nodules, coalescing/abutting lesions, plaques (raised, atrophic, or indurated), or dome-shaped tumoral masses. Tier 3 focused on the presence or absence of co-existing dissecting cellulitis or folliculitis decalvans.6
Epidemiology
Acne keloidalis nuchae primarily manifests in adolescent and adult men of African or Afro-Caribbean descent.7 Among African American men, the prevalence of AKN ranges from 0.5% to 13.6%.8 Similar ranges have been reported among Nigerian, South African, and West African men.1 Acne keloidalis nuchae also affects Asian and Hispanic men but rarely is seen in non-Hispanic White men or in women of any ethnicity.9,10 The male to female ratio is 20:1.1,11 Hair texture, hairstyling practices such as closely shaved or faded haircuts, and genetics likely contribute to development of AKN. Sports and occupations that require the use of headgear or a tight collar may increase the risk for AKN.12
Key clinical features in people with darker skin tones
- The lesions of AKN range in color from pink to dark brown or black. Postinflammatory hyperpigmentation or hyperchromia may be present around AKN lesions.
- Chronicity of AKN may lead to extended use of high-potency topical or intralesional corticosteroids, which causes transient or long-lasting hypopigmentation, especially in those with darker skin tones.
Worth noting
- Acne keloidalis nuchae can be disfiguring, which negatively impacts quality of life and self-esteem.12
- Some occupations (eg, military, police) have hair policies that may not be favorable to those with or at risk for AKN.
- Patients with AKN are 2 to 3 times more likely to present with metabolic syndrome, hypertension, type 2 diabetes mellitus, or obesity.13
Treatment
There are no treatments approved by the US Food and Drug Administration specifically for AKN. Treatment approaches are based on the pathophysiology, secondary impacts on the skin, and disease severity. Growing out the hair may prevent worsening and/or decrease the risk for new lesions.6
- Options include but are not limited to topical and systemic therapies (eg, topical corticosteroids, oral or topical antibiotics, isotretinoin, topical retinoids, imiquimod, pimecrolimus), light devices (eg, phototherapy, laser), ablative therapies (eg, laser, cryotherapy, radiotherapy), and surgery (eg, excision, follicular unit excision), often in combination.6,14,15
- Intralesional triamcinolone injections are considered standard of care. Adotama et al found that injecting triamcinolone into the deep dermis in the area of flat or papular AKN yielded better control of inflammation and decreased appearance of lesions compared with injecting individual lesions.16
- For extensive AKN lesions that do not respond to less-invasive therapies, consider surgical techniques,6,17 such as follicular unit excision18 and more extensive surgical excisions building on approaches from pioneers Drs. John Kenney and Harold Pierce.19 An innovative surgical approach for removal of large AKNs is the bat excision technique—wound shape resembles a bat in a spread-eagled position—with secondary intention healing with or without debridement and/or tension sutures. The resulting linear scar acts as a new posterior hair line.20
Health disparity highlights
Access to a dermatologic or plastic surgeon with expertise in the surgical treatment of large AKNs may be challenging but is needed to reduce risk for recurrence and adverse events.
Close-cropped haircuts on the occipital scalp, which are particularly popular among men of African descent, increase the risk for AKN.5 Although this grooming style may be a personal preference, other hairstyles commonly worn by those with tightly coiled hair may be deemed “unprofessional” in society or the workplace, which leads to hairstyling practices that may increase the risk for AKN.21
Acne keloidalis nuchae remains an understudied entity that adversely affects patients with skin of color.
- Ogunbiyi A. Acne keloidalis nuchae: prevalence, impact, and management challenges. Clin Cosmet Investig Dermatol. 2016;9:483-489. doi:10.2147/CCID.S99225
- Al Aboud DM, Badri T. Acne keloidalis nuchae. In: StatPearls [Internet]. Updated July 31, 2023. Accessed August 2, 2024. https://www.ncbi.nlm.nih.gov/books/NBK459135/
- Sperling LC, Homoky C, Pratt L, et al. Acne keloidalis is a form of primary scarring alopecia. Arch Dermatol. 2000;136:479-484.
- Herzberg AJ, Dinehart SM, Kerns BJ, et al. Acne keloidalis: transverse microscopy, immunohistochemistry, and electron microscopy. Am J Dermatopathol. 1990;12:109-121. doi:10.1097/00000372-199004000-00001
- Saka B, Akakpo A-S, Téclessou JN, et al. Risk factors associated with acne keloidalis nuchae in black subjects: a case-control study. Ann Dermatol Venereol. 2020;147:350-354. doi:10.1016/j.annder.2020.01.007
- Umar S, Lee DJ, Lullo JJ. A retrospective cohort study and clinical classification system of acne keloidalis nuchae. J Clin Aesthet Dermatol. 2021;14:E61-E67.
- Reja M, Silverberg NB. Acne keloidalis nuchae. In: Silverberg NB, Durán-McKinster C, Tay YK, eds. Pediatric Skin of Color. Springer; 2015:141-145. doi:10.1007/978-1-4614-6654-3_16
- Knable AL Jr, Hanke CW, Gonin R. Prevalence of acne keloidalis nuchae in football players. J Am Acad Dermatol. 1997;37:570-574. doi:10.1016/s0190-9622(97)70173-7
- Umar S, Ton D, Carter MJ, et al. Unveiling a shared precursor condition for acne keloidalis nuchae and primary cicatricial alopecias. Clin Cosmet Investig Dermatol. 2023;16:2315-2327. doi:10.2147/CCID.S422310
- Na K, Oh SH, Kim SK. Acne keloidalis nuchae in Asian: a single institutional experience. PLoS One. 2017;12:e0189790. doi:10.1371/journal.pone.0189790
- Ogunbiyi A, George A. Acne keloidalis in females: case report and review of literature. J Natl Med Assoc. 2005;97:736-738.
- Alexis A, Heath CR, Halder RM. Folliculitis keloidalis nuchae and pseudofolliculitis barbae: are prevention and effective treatment within reach? Dermatol Clin. 2014;32:183-191. doi:10.1016/j.det.2013.12.001
- Kridin K, Solomon A, Tzur-Bitan D, et al. Acne keloidalis nuchae and the metabolic syndrome: a population-based study. Am J Clin Dermatol. 2020;21:733-739. doi:10.1007/s40257-020-00541-z
- Smart K, Rodriguez I, Worswick S. Comorbidities and treatment options for acne keloidalis nuchae. Dermatol Ther. Published online May 25, 2024. doi:10.1155/2024/8336926
- Callender VD, Young CM, Haverstock CL, et al. An open label study of clobetasol propionate 0.05% and betamethasone valerate 0.12% foams in the treatment of mild to moderate acne keloidalis. Cutis. 2005;75:317-321.
- Adotama P, Grullon K, Ali S, et al. How we do it: our method for triamcinolone injections of acne keloidalis nuchae. Dermatol Surg. 2023;49:713-714. doi:10.1097/DSS.0000000000003803
- Beckett N, Lawson C, Cohen G. Electrosurgical excision of acne keloidalis nuchae with secondary intention healing. J Clin Aesthet Dermatol. 2011;4:36-39.
- Esmat SM, Abdel Hay RM, Abu Zeid OM, et al. The efficacy of laser-assisted hair removal in the treatment of acne keloidalis nuchae; a pilot study. Eur J Dermatol. 2012;22:645-650. doi:10.1684/ejd.2012.1830
- Dillard AD, Quarles FN. African-American pioneers in dermatology. In: Taylor SC, Kelly AP, Lim HW, et al, eds. Dermatology for Skin of Color. 2nd ed. McGraw-Hill Education; 2016:717-730.
- Umar S, David CV, Castillo JR, et al. Innovative surgical approaches and selection criteria of large acne keloidalis nuchae lesions. Plast Reconstr Surg Glob Open. 2019;7:E2215. doi:10.1097/GOX.0000000000002215
- Lee MS, Nambudiri VE. The CROWN act and dermatology: taking a stand against race-based hair discrimination. J Am Acad Dermatol. 2021;84:1181-1182. doi:10.1016/j.jaad.2020.11.065
- Ogunbiyi A. Acne keloidalis nuchae: prevalence, impact, and management challenges. Clin Cosmet Investig Dermatol. 2016;9:483-489. doi:10.2147/CCID.S99225
- Al Aboud DM, Badri T. Acne keloidalis nuchae. In: StatPearls [Internet]. Updated July 31, 2023. Accessed August 2, 2024. https://www.ncbi.nlm.nih.gov/books/NBK459135/
- Sperling LC, Homoky C, Pratt L, et al. Acne keloidalis is a form of primary scarring alopecia. Arch Dermatol. 2000;136:479-484.
- Herzberg AJ, Dinehart SM, Kerns BJ, et al. Acne keloidalis: transverse microscopy, immunohistochemistry, and electron microscopy. Am J Dermatopathol. 1990;12:109-121. doi:10.1097/00000372-199004000-00001
- Saka B, Akakpo A-S, Téclessou JN, et al. Risk factors associated with acne keloidalis nuchae in black subjects: a case-control study. Ann Dermatol Venereol. 2020;147:350-354. doi:10.1016/j.annder.2020.01.007
- Umar S, Lee DJ, Lullo JJ. A retrospective cohort study and clinical classification system of acne keloidalis nuchae. J Clin Aesthet Dermatol. 2021;14:E61-E67.
- Reja M, Silverberg NB. Acne keloidalis nuchae. In: Silverberg NB, Durán-McKinster C, Tay YK, eds. Pediatric Skin of Color. Springer; 2015:141-145. doi:10.1007/978-1-4614-6654-3_16
- Knable AL Jr, Hanke CW, Gonin R. Prevalence of acne keloidalis nuchae in football players. J Am Acad Dermatol. 1997;37:570-574. doi:10.1016/s0190-9622(97)70173-7
- Umar S, Ton D, Carter MJ, et al. Unveiling a shared precursor condition for acne keloidalis nuchae and primary cicatricial alopecias. Clin Cosmet Investig Dermatol. 2023;16:2315-2327. doi:10.2147/CCID.S422310
- Na K, Oh SH, Kim SK. Acne keloidalis nuchae in Asian: a single institutional experience. PLoS One. 2017;12:e0189790. doi:10.1371/journal.pone.0189790
- Ogunbiyi A, George A. Acne keloidalis in females: case report and review of literature. J Natl Med Assoc. 2005;97:736-738.
- Alexis A, Heath CR, Halder RM. Folliculitis keloidalis nuchae and pseudofolliculitis barbae: are prevention and effective treatment within reach? Dermatol Clin. 2014;32:183-191. doi:10.1016/j.det.2013.12.001
- Kridin K, Solomon A, Tzur-Bitan D, et al. Acne keloidalis nuchae and the metabolic syndrome: a population-based study. Am J Clin Dermatol. 2020;21:733-739. doi:10.1007/s40257-020-00541-z
- Smart K, Rodriguez I, Worswick S. Comorbidities and treatment options for acne keloidalis nuchae. Dermatol Ther. Published online May 25, 2024. doi:10.1155/2024/8336926
- Callender VD, Young CM, Haverstock CL, et al. An open label study of clobetasol propionate 0.05% and betamethasone valerate 0.12% foams in the treatment of mild to moderate acne keloidalis. Cutis. 2005;75:317-321.
- Adotama P, Grullon K, Ali S, et al. How we do it: our method for triamcinolone injections of acne keloidalis nuchae. Dermatol Surg. 2023;49:713-714. doi:10.1097/DSS.0000000000003803
- Beckett N, Lawson C, Cohen G. Electrosurgical excision of acne keloidalis nuchae with secondary intention healing. J Clin Aesthet Dermatol. 2011;4:36-39.
- Esmat SM, Abdel Hay RM, Abu Zeid OM, et al. The efficacy of laser-assisted hair removal in the treatment of acne keloidalis nuchae; a pilot study. Eur J Dermatol. 2012;22:645-650. doi:10.1684/ejd.2012.1830
- Dillard AD, Quarles FN. African-American pioneers in dermatology. In: Taylor SC, Kelly AP, Lim HW, et al, eds. Dermatology for Skin of Color. 2nd ed. McGraw-Hill Education; 2016:717-730.
- Umar S, David CV, Castillo JR, et al. Innovative surgical approaches and selection criteria of large acne keloidalis nuchae lesions. Plast Reconstr Surg Glob Open. 2019;7:E2215. doi:10.1097/GOX.0000000000002215
- Lee MS, Nambudiri VE. The CROWN act and dermatology: taking a stand against race-based hair discrimination. J Am Acad Dermatol. 2021;84:1181-1182. doi:10.1016/j.jaad.2020.11.065
SGLT2 Inhibitor Reduces Risk for Neurodegenerative Diseases in T2D
MADRID — Patients with type 2 diabetes treated with sodium-glucose cotransporter 2 inhibitors (SGLT2is) show significant reductions in the risk of developing neurodegenerative disorders including Alzheimer’s disease, vascular dementia, and Parkinson’s disease, compared with those treated with other antidiabetic drugs, results from a large population-based cohort show.
“This was the largest nationwide population-based longitudinal cohort study to investigate the association between the use of SGLT2 inhibitors and the incidence of all-cause dementia and Parkinson’s disease,” said first author Hae Kyung Kim, MD, of the Department of Internal Medicine, Yonsei University College of Medicine, Seoul, South Korea, in presenting the findings at the annual meeting of the European Association for the Study of Diabetes.
Type 2 diabetes is known to increase the risk for neurodegenerative diseases such as dementia or Alzheimer’s disease, said Dr. Kim. Key factors attributed to the risk include shared pathophysiological mechanisms such as central nervous system insulin resistance and reduced cerebral glucose metabolism.
While research is lacking on the role of antidiabetic drugs in the treatment of neurodegenerative diseases, the researcher noted that “SGLT2 inhibitors, which have shown significant cardiorenal benefits and enhanced energy metabolism through ketogenesis, offer promise.”
To further investigate, Dr. Kim and her colleagues conducted the retrospective study, evaluating data on more than 1.3 million enrollees in Korea’s National Health Insurance Service Database who were aged 40 years or older, diagnosed with type 2 diabetes, and had initiated antidiabetic drugs between September 2014 and December 2019.
In the propensity score analysis, 358,862 patients were matched 1:1, in groups of 179,431 participants each, based on whether they were treated with SGLT2is or other oral antidiabetic drugs. Patients with a history of neurodegenerative disease, cancer, or use of glucagon-like peptide 1 receptor agonists were excluded.
The patients had a mean age of 57.8 years, 57.9% were men, and 6837 had incident dementia or Parkinson’s disease events reported.
With a mean follow-up of 2.88 years, after adjustment for key variables, those treated with SGLT2is had a 19% reduced risk of developing Alzheimer’s disease (adjusted hazard ratio [aHR], 0.81), a 31% reduced risk for vascular dementia (aHR, 0.69), and a 20% reduced risk for Parkinson’s disease (aHR, 0.80) compared with the non-SGLT2i group.
Furthermore, those receiving SGLT2i treatment had a 21% reduced risk for all-cause dementia (aHR, 0.79) and a 22% reduced risk for all-cause dementia and Parkinson’s disease compared with the oral antidiabetic drug group (aHR, 0.78) with a 6-month drug use lag period.
The association was observed regardless of SGLT2i exposure duration. Subgroup analyses indicated that the reductions in neurodegenerative disorders among those receiving SGLT2is were not associated with factors including age, sex, body mass index, blood pressure, glucose, lipid profiles, kidney function, health behaviors, comorbidities, diabetic complications, or other medication use.
Dr. Kim speculated that mechanisms underlying the reduced dementia risk could include SGLT2i effects of mitigating the common severe risk factors of type 2 diabetes and neurodegenerative diseases, including hypertension, heart failure, and chronic kidney disease, and improving hyperperfusion in the heart and cerebral vascular insufficiency.
Commenting on the study to this news organization, Erik H. Serné, MD, of the VU University Medical Centre, Amsterdam, the Netherlands, who comoderated the session, noted that “people with type 2 diabetes have a 50%-100% increased risk of developing dementia, particularly Alzheimer’s disease and vascular dementia.”
“The increasing prevalence of both conditions poses significant public health challenges, highlighting the need for effective prevention strategies and interventions.”
Currently, treatments for dementia are limited, with most primarily addressing symptoms and not the underlying cause of the neurodegenerative disease, he said.
He noted that, in addition to the effects mentioned by Dr. Kim, SGLT2is are also speculated to provide potential neuroprotective effects through improved glycemic control and insulin sensitivity, reduced inflammation and oxidative stress, enhanced mitochondrial function and energy metabolism, and reduced beta-amyloid and tau pathology.
“These mechanisms collectively may reduce the risk of cognitive decline, particularly in diabetic patients, and warrant further investigation in clinical trials to solidify the neuroprotective role of SGLT2 inhibitors,” said Dr. Serné.
In addition to their benefits in type 2 diabetes, SGLT2is “now offer hope in the prevention of dementia, a disease that has very limited therapeutic options thus far. The current data [presented by Dr. Kim] seem to corroborate this,” he added.
Dr. Kim and Dr. Serné had no disclosures to report.
A version of this article first appeared on Medscape.com.
MADRID — Patients with type 2 diabetes treated with sodium-glucose cotransporter 2 inhibitors (SGLT2is) show significant reductions in the risk of developing neurodegenerative disorders including Alzheimer’s disease, vascular dementia, and Parkinson’s disease, compared with those treated with other antidiabetic drugs, results from a large population-based cohort show.
“This was the largest nationwide population-based longitudinal cohort study to investigate the association between the use of SGLT2 inhibitors and the incidence of all-cause dementia and Parkinson’s disease,” said first author Hae Kyung Kim, MD, of the Department of Internal Medicine, Yonsei University College of Medicine, Seoul, South Korea, in presenting the findings at the annual meeting of the European Association for the Study of Diabetes.
Type 2 diabetes is known to increase the risk for neurodegenerative diseases such as dementia or Alzheimer’s disease, said Dr. Kim. Key factors attributed to the risk include shared pathophysiological mechanisms such as central nervous system insulin resistance and reduced cerebral glucose metabolism.
While research is lacking on the role of antidiabetic drugs in the treatment of neurodegenerative diseases, the researcher noted that “SGLT2 inhibitors, which have shown significant cardiorenal benefits and enhanced energy metabolism through ketogenesis, offer promise.”
To further investigate, Dr. Kim and her colleagues conducted the retrospective study, evaluating data on more than 1.3 million enrollees in Korea’s National Health Insurance Service Database who were aged 40 years or older, diagnosed with type 2 diabetes, and had initiated antidiabetic drugs between September 2014 and December 2019.
In the propensity score analysis, 358,862 patients were matched 1:1, in groups of 179,431 participants each, based on whether they were treated with SGLT2is or other oral antidiabetic drugs. Patients with a history of neurodegenerative disease, cancer, or use of glucagon-like peptide 1 receptor agonists were excluded.
The patients had a mean age of 57.8 years, 57.9% were men, and 6837 had incident dementia or Parkinson’s disease events reported.
With a mean follow-up of 2.88 years, after adjustment for key variables, those treated with SGLT2is had a 19% reduced risk of developing Alzheimer’s disease (adjusted hazard ratio [aHR], 0.81), a 31% reduced risk for vascular dementia (aHR, 0.69), and a 20% reduced risk for Parkinson’s disease (aHR, 0.80) compared with the non-SGLT2i group.
Furthermore, those receiving SGLT2i treatment had a 21% reduced risk for all-cause dementia (aHR, 0.79) and a 22% reduced risk for all-cause dementia and Parkinson’s disease compared with the oral antidiabetic drug group (aHR, 0.78) with a 6-month drug use lag period.
The association was observed regardless of SGLT2i exposure duration. Subgroup analyses indicated that the reductions in neurodegenerative disorders among those receiving SGLT2is were not associated with factors including age, sex, body mass index, blood pressure, glucose, lipid profiles, kidney function, health behaviors, comorbidities, diabetic complications, or other medication use.
Dr. Kim speculated that mechanisms underlying the reduced dementia risk could include SGLT2i effects of mitigating the common severe risk factors of type 2 diabetes and neurodegenerative diseases, including hypertension, heart failure, and chronic kidney disease, and improving hyperperfusion in the heart and cerebral vascular insufficiency.
Commenting on the study to this news organization, Erik H. Serné, MD, of the VU University Medical Centre, Amsterdam, the Netherlands, who comoderated the session, noted that “people with type 2 diabetes have a 50%-100% increased risk of developing dementia, particularly Alzheimer’s disease and vascular dementia.”
“The increasing prevalence of both conditions poses significant public health challenges, highlighting the need for effective prevention strategies and interventions.”
Currently, treatments for dementia are limited, with most primarily addressing symptoms and not the underlying cause of the neurodegenerative disease, he said.
He noted that, in addition to the effects mentioned by Dr. Kim, SGLT2is are also speculated to provide potential neuroprotective effects through improved glycemic control and insulin sensitivity, reduced inflammation and oxidative stress, enhanced mitochondrial function and energy metabolism, and reduced beta-amyloid and tau pathology.
“These mechanisms collectively may reduce the risk of cognitive decline, particularly in diabetic patients, and warrant further investigation in clinical trials to solidify the neuroprotective role of SGLT2 inhibitors,” said Dr. Serné.
In addition to their benefits in type 2 diabetes, SGLT2is “now offer hope in the prevention of dementia, a disease that has very limited therapeutic options thus far. The current data [presented by Dr. Kim] seem to corroborate this,” he added.
Dr. Kim and Dr. Serné had no disclosures to report.
A version of this article first appeared on Medscape.com.
MADRID — Patients with type 2 diabetes treated with sodium-glucose cotransporter 2 inhibitors (SGLT2is) show significant reductions in the risk of developing neurodegenerative disorders including Alzheimer’s disease, vascular dementia, and Parkinson’s disease, compared with those treated with other antidiabetic drugs, results from a large population-based cohort show.
“This was the largest nationwide population-based longitudinal cohort study to investigate the association between the use of SGLT2 inhibitors and the incidence of all-cause dementia and Parkinson’s disease,” said first author Hae Kyung Kim, MD, of the Department of Internal Medicine, Yonsei University College of Medicine, Seoul, South Korea, in presenting the findings at the annual meeting of the European Association for the Study of Diabetes.
Type 2 diabetes is known to increase the risk for neurodegenerative diseases such as dementia or Alzheimer’s disease, said Dr. Kim. Key factors attributed to the risk include shared pathophysiological mechanisms such as central nervous system insulin resistance and reduced cerebral glucose metabolism.
While research is lacking on the role of antidiabetic drugs in the treatment of neurodegenerative diseases, the researcher noted that “SGLT2 inhibitors, which have shown significant cardiorenal benefits and enhanced energy metabolism through ketogenesis, offer promise.”
To further investigate, Dr. Kim and her colleagues conducted the retrospective study, evaluating data on more than 1.3 million enrollees in Korea’s National Health Insurance Service Database who were aged 40 years or older, diagnosed with type 2 diabetes, and had initiated antidiabetic drugs between September 2014 and December 2019.
In the propensity score analysis, 358,862 patients were matched 1:1, in groups of 179,431 participants each, based on whether they were treated with SGLT2is or other oral antidiabetic drugs. Patients with a history of neurodegenerative disease, cancer, or use of glucagon-like peptide 1 receptor agonists were excluded.
The patients had a mean age of 57.8 years, 57.9% were men, and 6837 had incident dementia or Parkinson’s disease events reported.
With a mean follow-up of 2.88 years, after adjustment for key variables, those treated with SGLT2is had a 19% reduced risk of developing Alzheimer’s disease (adjusted hazard ratio [aHR], 0.81), a 31% reduced risk for vascular dementia (aHR, 0.69), and a 20% reduced risk for Parkinson’s disease (aHR, 0.80) compared with the non-SGLT2i group.
Furthermore, those receiving SGLT2i treatment had a 21% reduced risk for all-cause dementia (aHR, 0.79) and a 22% reduced risk for all-cause dementia and Parkinson’s disease compared with the oral antidiabetic drug group (aHR, 0.78) with a 6-month drug use lag period.
The association was observed regardless of SGLT2i exposure duration. Subgroup analyses indicated that the reductions in neurodegenerative disorders among those receiving SGLT2is were not associated with factors including age, sex, body mass index, blood pressure, glucose, lipid profiles, kidney function, health behaviors, comorbidities, diabetic complications, or other medication use.
Dr. Kim speculated that mechanisms underlying the reduced dementia risk could include SGLT2i effects of mitigating the common severe risk factors of type 2 diabetes and neurodegenerative diseases, including hypertension, heart failure, and chronic kidney disease, and improving hyperperfusion in the heart and cerebral vascular insufficiency.
Commenting on the study to this news organization, Erik H. Serné, MD, of the VU University Medical Centre, Amsterdam, the Netherlands, who comoderated the session, noted that “people with type 2 diabetes have a 50%-100% increased risk of developing dementia, particularly Alzheimer’s disease and vascular dementia.”
“The increasing prevalence of both conditions poses significant public health challenges, highlighting the need for effective prevention strategies and interventions.”
Currently, treatments for dementia are limited, with most primarily addressing symptoms and not the underlying cause of the neurodegenerative disease, he said.
He noted that, in addition to the effects mentioned by Dr. Kim, SGLT2is are also speculated to provide potential neuroprotective effects through improved glycemic control and insulin sensitivity, reduced inflammation and oxidative stress, enhanced mitochondrial function and energy metabolism, and reduced beta-amyloid and tau pathology.
“These mechanisms collectively may reduce the risk of cognitive decline, particularly in diabetic patients, and warrant further investigation in clinical trials to solidify the neuroprotective role of SGLT2 inhibitors,” said Dr. Serné.
In addition to their benefits in type 2 diabetes, SGLT2is “now offer hope in the prevention of dementia, a disease that has very limited therapeutic options thus far. The current data [presented by Dr. Kim] seem to corroborate this,” he added.
Dr. Kim and Dr. Serné had no disclosures to report.
A version of this article first appeared on Medscape.com.
FROM EASD 2024
NSAIDs Offer No Relief for Pain From IUD Placement
Research on pain management during placement of intrauterine devices (IUD) is lacking, but most studies so far indicate that nonsteroidal anti-inflammatory drugs (NSAIDs) are not effective, according to a poster presented at Pain Week 2024 in Las Vegas.
Roughly 79% of the 14 studies included in the systematic review found NSAIDs — one of the most common drugs clinicians advise patients to take before placement — did not diminish discomfort.
“We’re challenging the current practice of using just NSAIDs as a first-line of treatment,” said Kevin Rowland, PhD, professor and chair of biomedical sciences at Tilman J. Fertitta Family College of Medicine in Houston, who helped conduct the meta-analysis. “We need additional measures.”
Some studies found the drugs offered virtually no improvement for patients, while the biggest drop in pain shown in one study was about 40%. The range of pain levels women reported while using NSAIDs was between 1.8 and 7.3 on the visual analog scale (VAS), with an average score of 4.25.
The review included 10 types of NSAIDs and dosages administered to patients before the procedure. One intramuscular NSAID was included while the remaining were oral. All studies were peer-reviewed, used the VAS pain scale, and were not limited to any specific population.
The findings highlight a longstanding but unresolved problem in reproductive health: An overall lack of effective pain management strategies for gynecologic procedures.
“We went into this having a pretty good idea of what we were going to find because [the lack of NSAID efficacy] has been shown before, it’s been talked about before, and we’re just not listening as a medical community,” said Isabella D. Martingano, an MD candidate at Tilman J. Fertitta Family College of Medicine, who led the review.
The research also points to a lack of robust studies on pain during IUD placement, said Emma Lakey, a coauthor and medical student at Tilman J. Fertitta Family College of Medicine.
“We were only able to review 14 studies, which was enough to go off of, but considering we were looking for trials about pain control for a procedure that helps prevent pregnancy, that’s just not enough research,” Ms. Lakey said.
Discomfort associated with IUD placement ranges from mild to severe, can last for over a week, and includes cramping, bleeding, lightheadedness, nausea, and fainting. Some research suggests that providers may underestimate the level of pain the procedures cause.
“Unfortunately, the pain associated with IUD insertion and removal has been underplayed for a long time and many practitioners in the field likely haven’t counseled patients fully on what the procedure will feel like,” said Jennifer Chin, MD, an ob.gyn. and assistant professor of obstetrics and gynecology at the University of Washington in Seattle.
NSAIDs are not mentioned in the recently expanded guidelines on IUD placement from the US Centers for Disease Control and Prevention (CDC). The CDC recommends lidocaine paracervical blocks, gels, sprays, and creams, plus counseling women about pain ahead of the procedures.
IUDs are one of the most effective forms of birth control, with a failure rate below 1%.
Yet hearing about painful placement keeps many women from seeking out an IUD or replacing an existing device, Dr. Rowland said. The review adds to the body of evidence that current strategies are not working and that more research is needed, he said.
According to Dr. Chin, making IUDs more accessible means taking a more personalized approach to pain management while understanding that what may be a painless procedure for one patient may be excruciating for another.
Dr. Chin offers a range of options for her patients, including NSAIDs, lorazepam for anxiety, paracervical blocks, lidocaine jelly and spray, intravenous sedation, and general anesthesia. She also talks to her patients through the procedure and provides guided imagery and meditation.
“We should always make sure we’re prioritizing the patients and providing evidence-based, compassionate, and individualized care,” said Dr. Chin. “Each patient comes to us in a particular context and with a specific set of experiences and history that will make a difference in how we’re best able to take care of them.”
The authors reported no disclosures and no sources of funding. Dr. Chin reported no disclosures.
A version of this article first appeared on Medscape.com.
Research on pain management during placement of intrauterine devices (IUD) is lacking, but most studies so far indicate that nonsteroidal anti-inflammatory drugs (NSAIDs) are not effective, according to a poster presented at Pain Week 2024 in Las Vegas.
Roughly 79% of the 14 studies included in the systematic review found NSAIDs — one of the most common drugs clinicians advise patients to take before placement — did not diminish discomfort.
“We’re challenging the current practice of using just NSAIDs as a first-line of treatment,” said Kevin Rowland, PhD, professor and chair of biomedical sciences at Tilman J. Fertitta Family College of Medicine in Houston, who helped conduct the meta-analysis. “We need additional measures.”
Some studies found the drugs offered virtually no improvement for patients, while the biggest drop in pain shown in one study was about 40%. The range of pain levels women reported while using NSAIDs was between 1.8 and 7.3 on the visual analog scale (VAS), with an average score of 4.25.
The review included 10 types of NSAIDs and dosages administered to patients before the procedure. One intramuscular NSAID was included while the remaining were oral. All studies were peer-reviewed, used the VAS pain scale, and were not limited to any specific population.
The findings highlight a longstanding but unresolved problem in reproductive health: An overall lack of effective pain management strategies for gynecologic procedures.
“We went into this having a pretty good idea of what we were going to find because [the lack of NSAID efficacy] has been shown before, it’s been talked about before, and we’re just not listening as a medical community,” said Isabella D. Martingano, an MD candidate at Tilman J. Fertitta Family College of Medicine, who led the review.
The research also points to a lack of robust studies on pain during IUD placement, said Emma Lakey, a coauthor and medical student at Tilman J. Fertitta Family College of Medicine.
“We were only able to review 14 studies, which was enough to go off of, but considering we were looking for trials about pain control for a procedure that helps prevent pregnancy, that’s just not enough research,” Ms. Lakey said.
Discomfort associated with IUD placement ranges from mild to severe, can last for over a week, and includes cramping, bleeding, lightheadedness, nausea, and fainting. Some research suggests that providers may underestimate the level of pain the procedures cause.
“Unfortunately, the pain associated with IUD insertion and removal has been underplayed for a long time and many practitioners in the field likely haven’t counseled patients fully on what the procedure will feel like,” said Jennifer Chin, MD, an ob.gyn. and assistant professor of obstetrics and gynecology at the University of Washington in Seattle.
NSAIDs are not mentioned in the recently expanded guidelines on IUD placement from the US Centers for Disease Control and Prevention (CDC). The CDC recommends lidocaine paracervical blocks, gels, sprays, and creams, plus counseling women about pain ahead of the procedures.
IUDs are one of the most effective forms of birth control, with a failure rate below 1%.
Yet hearing about painful placement keeps many women from seeking out an IUD or replacing an existing device, Dr. Rowland said. The review adds to the body of evidence that current strategies are not working and that more research is needed, he said.
According to Dr. Chin, making IUDs more accessible means taking a more personalized approach to pain management while understanding that what may be a painless procedure for one patient may be excruciating for another.
Dr. Chin offers a range of options for her patients, including NSAIDs, lorazepam for anxiety, paracervical blocks, lidocaine jelly and spray, intravenous sedation, and general anesthesia. She also talks to her patients through the procedure and provides guided imagery and meditation.
“We should always make sure we’re prioritizing the patients and providing evidence-based, compassionate, and individualized care,” said Dr. Chin. “Each patient comes to us in a particular context and with a specific set of experiences and history that will make a difference in how we’re best able to take care of them.”
The authors reported no disclosures and no sources of funding. Dr. Chin reported no disclosures.
A version of this article first appeared on Medscape.com.
Research on pain management during placement of intrauterine devices (IUD) is lacking, but most studies so far indicate that nonsteroidal anti-inflammatory drugs (NSAIDs) are not effective, according to a poster presented at Pain Week 2024 in Las Vegas.
Roughly 79% of the 14 studies included in the systematic review found NSAIDs — one of the most common drugs clinicians advise patients to take before placement — did not diminish discomfort.
“We’re challenging the current practice of using just NSAIDs as a first-line of treatment,” said Kevin Rowland, PhD, professor and chair of biomedical sciences at Tilman J. Fertitta Family College of Medicine in Houston, who helped conduct the meta-analysis. “We need additional measures.”
Some studies found the drugs offered virtually no improvement for patients, while the biggest drop in pain shown in one study was about 40%. The range of pain levels women reported while using NSAIDs was between 1.8 and 7.3 on the visual analog scale (VAS), with an average score of 4.25.
The review included 10 types of NSAIDs and dosages administered to patients before the procedure. One intramuscular NSAID was included while the remaining were oral. All studies were peer-reviewed, used the VAS pain scale, and were not limited to any specific population.
The findings highlight a longstanding but unresolved problem in reproductive health: An overall lack of effective pain management strategies for gynecologic procedures.
“We went into this having a pretty good idea of what we were going to find because [the lack of NSAID efficacy] has been shown before, it’s been talked about before, and we’re just not listening as a medical community,” said Isabella D. Martingano, an MD candidate at Tilman J. Fertitta Family College of Medicine, who led the review.
The research also points to a lack of robust studies on pain during IUD placement, said Emma Lakey, a coauthor and medical student at Tilman J. Fertitta Family College of Medicine.
“We were only able to review 14 studies, which was enough to go off of, but considering we were looking for trials about pain control for a procedure that helps prevent pregnancy, that’s just not enough research,” Ms. Lakey said.
Discomfort associated with IUD placement ranges from mild to severe, can last for over a week, and includes cramping, bleeding, lightheadedness, nausea, and fainting. Some research suggests that providers may underestimate the level of pain the procedures cause.
“Unfortunately, the pain associated with IUD insertion and removal has been underplayed for a long time and many practitioners in the field likely haven’t counseled patients fully on what the procedure will feel like,” said Jennifer Chin, MD, an ob.gyn. and assistant professor of obstetrics and gynecology at the University of Washington in Seattle.
NSAIDs are not mentioned in the recently expanded guidelines on IUD placement from the US Centers for Disease Control and Prevention (CDC). The CDC recommends lidocaine paracervical blocks, gels, sprays, and creams, plus counseling women about pain ahead of the procedures.
IUDs are one of the most effective forms of birth control, with a failure rate below 1%.
Yet hearing about painful placement keeps many women from seeking out an IUD or replacing an existing device, Dr. Rowland said. The review adds to the body of evidence that current strategies are not working and that more research is needed, he said.
According to Dr. Chin, making IUDs more accessible means taking a more personalized approach to pain management while understanding that what may be a painless procedure for one patient may be excruciating for another.
Dr. Chin offers a range of options for her patients, including NSAIDs, lorazepam for anxiety, paracervical blocks, lidocaine jelly and spray, intravenous sedation, and general anesthesia. She also talks to her patients through the procedure and provides guided imagery and meditation.
“We should always make sure we’re prioritizing the patients and providing evidence-based, compassionate, and individualized care,” said Dr. Chin. “Each patient comes to us in a particular context and with a specific set of experiences and history that will make a difference in how we’re best able to take care of them.”
The authors reported no disclosures and no sources of funding. Dr. Chin reported no disclosures.
A version of this article first appeared on Medscape.com.
Long-Term Exposure to Road Traffic Noise and Air Pollution Linked to Infertility
TOPLINE:
Long-term exposure to particulate matter < 2.5 μm in diameter (PM2.5) is linked to a higher risk for infertility in men. Exposure to road traffic noise is associated with a higher risk for infertility in women aged > 35 years and possibly in men aged > 37 years.
METHODOLOGY:
- This nationwide prospective cohort study evaluated the association between long-term exposure to road traffic noise and PM2.5 and infertility in 526,056 men (mean age, 33.6 years) and 377,850 women (mean age, 32.7 years) who were cohabiting or married, had fewer than two children, and lived in Denmark between 2000 and 2017.
- Residential exposure to road traffic noise (most exposed facade of the home) and PM2.5 was estimated using validated models and linked to data from national registers.
- Diagnoses of infertility were identified in men and women from the Danish National Patient Register over a mean follow-up of 4.3 years and 4.2 years, respectively.
TAKEAWAY:
- Each 2.9 µg/m3 increase in the 5-year average exposure to PM2.5 was associated with a 24% increase in the risk for infertility in men aged 30-45 years (adjusted hazard ratio [aHR], 1.24).
- No significant association was found between exposure to PM2.5 and infertility in women.
- Each 10.2 dB increase in the 5-year average exposure to road traffic noise was associated with a 14% increase in infertility (aHR, 1.14; 95% CI, 1.10-1.18) in women aged 35-45 years.
- Exposure to noise was associated with a reduced risk for infertility in men aged 30.0-36.9 years (aHR, 0.93; 95% CI, 0.91-0.96) and an increased risk in those aged 37-45 years (aHR, 1.06; 95% CI, 1.02-1.11).
IN PRACTICE:
“As many Western countries are facing declining birth rates and increasing maternal age at the birth of a first child, knowledge on environmental pollutants affecting fertility is crucial,” the authors of the study wrote. “It suggests that political implementation of air pollution and noise mitigations may be important tools for improving birth rates in the Western world,” they added.
SOURCE:
The study, led by Mette Sorensen, of the Danish Cancer Institute in Copenhagen, Denmark, was published online in The BMJ.
LIMITATIONS:
The study’s reliance on register data meant information on lifestyle factors such as alcohol use, body mass index, and smoking was unavailable. The lack of data on exposure to noise and PM2.5 at work and during leisure activities may affect the size and statistical precision of risk estimates.
DISCLOSURES:
The study did not receive any external funding. The authors declared no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
TOPLINE:
Long-term exposure to particulate matter < 2.5 μm in diameter (PM2.5) is linked to a higher risk for infertility in men. Exposure to road traffic noise is associated with a higher risk for infertility in women aged > 35 years and possibly in men aged > 37 years.
METHODOLOGY:
- This nationwide prospective cohort study evaluated the association between long-term exposure to road traffic noise and PM2.5 and infertility in 526,056 men (mean age, 33.6 years) and 377,850 women (mean age, 32.7 years) who were cohabiting or married, had fewer than two children, and lived in Denmark between 2000 and 2017.
- Residential exposure to road traffic noise (most exposed facade of the home) and PM2.5 was estimated using validated models and linked to data from national registers.
- Diagnoses of infertility were identified in men and women from the Danish National Patient Register over a mean follow-up of 4.3 years and 4.2 years, respectively.
TAKEAWAY:
- Each 2.9 µg/m3 increase in the 5-year average exposure to PM2.5 was associated with a 24% increase in the risk for infertility in men aged 30-45 years (adjusted hazard ratio [aHR], 1.24).
- No significant association was found between exposure to PM2.5 and infertility in women.
- Each 10.2 dB increase in the 5-year average exposure to road traffic noise was associated with a 14% increase in infertility (aHR, 1.14; 95% CI, 1.10-1.18) in women aged 35-45 years.
- Exposure to noise was associated with a reduced risk for infertility in men aged 30.0-36.9 years (aHR, 0.93; 95% CI, 0.91-0.96) and an increased risk in those aged 37-45 years (aHR, 1.06; 95% CI, 1.02-1.11).
IN PRACTICE:
“As many Western countries are facing declining birth rates and increasing maternal age at the birth of a first child, knowledge on environmental pollutants affecting fertility is crucial,” the authors of the study wrote. “It suggests that political implementation of air pollution and noise mitigations may be important tools for improving birth rates in the Western world,” they added.
SOURCE:
The study, led by Mette Sorensen, of the Danish Cancer Institute in Copenhagen, Denmark, was published online in The BMJ.
LIMITATIONS:
The study’s reliance on register data meant information on lifestyle factors such as alcohol use, body mass index, and smoking was unavailable. The lack of data on exposure to noise and PM2.5 at work and during leisure activities may affect the size and statistical precision of risk estimates.
DISCLOSURES:
The study did not receive any external funding. The authors declared no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
TOPLINE:
Long-term exposure to particulate matter < 2.5 μm in diameter (PM2.5) is linked to a higher risk for infertility in men. Exposure to road traffic noise is associated with a higher risk for infertility in women aged > 35 years and possibly in men aged > 37 years.
METHODOLOGY:
- This nationwide prospective cohort study evaluated the association between long-term exposure to road traffic noise and PM2.5 and infertility in 526,056 men (mean age, 33.6 years) and 377,850 women (mean age, 32.7 years) who were cohabiting or married, had fewer than two children, and lived in Denmark between 2000 and 2017.
- Residential exposure to road traffic noise (most exposed facade of the home) and PM2.5 was estimated using validated models and linked to data from national registers.
- Diagnoses of infertility were identified in men and women from the Danish National Patient Register over a mean follow-up of 4.3 years and 4.2 years, respectively.
TAKEAWAY:
- Each 2.9 µg/m3 increase in the 5-year average exposure to PM2.5 was associated with a 24% increase in the risk for infertility in men aged 30-45 years (adjusted hazard ratio [aHR], 1.24).
- No significant association was found between exposure to PM2.5 and infertility in women.
- Each 10.2 dB increase in the 5-year average exposure to road traffic noise was associated with a 14% increase in infertility (aHR, 1.14; 95% CI, 1.10-1.18) in women aged 35-45 years.
- Exposure to noise was associated with a reduced risk for infertility in men aged 30.0-36.9 years (aHR, 0.93; 95% CI, 0.91-0.96) and an increased risk in those aged 37-45 years (aHR, 1.06; 95% CI, 1.02-1.11).
IN PRACTICE:
“As many Western countries are facing declining birth rates and increasing maternal age at the birth of a first child, knowledge on environmental pollutants affecting fertility is crucial,” the authors of the study wrote. “It suggests that political implementation of air pollution and noise mitigations may be important tools for improving birth rates in the Western world,” they added.
SOURCE:
The study, led by Mette Sorensen, of the Danish Cancer Institute in Copenhagen, Denmark, was published online in The BMJ.
LIMITATIONS:
The study’s reliance on register data meant information on lifestyle factors such as alcohol use, body mass index, and smoking was unavailable. The lack of data on exposure to noise and PM2.5 at work and during leisure activities may affect the size and statistical precision of risk estimates.
DISCLOSURES:
The study did not receive any external funding. The authors declared no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
Laughter: Comic Relief for Dry Eyes?
TOPLINE:
Laughter exercise practiced four times a day was noninferior to 0.1% sodium hyaluronic acid in alleviating the symptoms of dry eye disease and improved tear film stability and the function of the meibomian gland, researchers found.
METHODOLOGY:
- Researchers aimed to assess the effectiveness and safety of laughter exercise in patients with symptomatic dry eye disease by conducting a two-arm clinical trial at the largest ophthalmic center in southern China.
- They included 299 patients aged 18-45 years (74% women) with symptomatic dry eye disease who were randomly assigned to receive either laughter exercise or eye drops with 0.1% sodium hyaluronic acid four times daily for 8 weeks.
- All the participants were required to have an ocular surface disease index score, a measure of symptoms related to dry eye disease, between 18 and 80 points and a fluorescein tear film breakup time of no more than 8 seconds.
- Participants in the laughter exercise group watched an instructional video and were requested to repeat the phrases “Hee hee hee, hah hah hah, cheese cheese cheese, cheek cheek cheek, hah hah hah hah hah hah” 30 times per 5-minute session.
- The primary outcome was the mean change in the ocular surface disease index score from baseline to 8 weeks.
TAKEAWAY:
- At 8 weeks, the ocular surface disease index score reduced by 10.50 points (95% CI, −13.1 to −7.82) in the laughter exercise group and by 8.83 points (95% CI, −11.7 to −6.02) in the group prescribed eye drops.
- At 12 weeks, patients in the laughter exercise group showed a significantly greater reduction in the ocular surface disease index score than those in the group prescribed eye drops (mean between-group difference, −4.08 points; P = .024).
- Laughter exercise also led to a more significant improvement in the noninvasive tear breakup time than the use of eye drops (mean between-group difference, 2.30 sec; P < .001).
- No adverse events were reported in either of the groups during the study period.
IN PRACTICE:
“As a safe, environmentally friendly, and low-cost intervention, laughter exercise could serve as a first-line, home-based treatment for people with symptomatic dry eye disease and limited corneal staining,” the authors of the study reported.
SOURCE:
This study was led by Jing Li from the State Key Laboratory of Ophthalmology at the Zhongshan Ophthalmic Center in Sun Yat-sen University in Guangzhou, China, and was published online on September 11, 2024, in The BMJ.
LIMITATIONS:
The study lacked a double-blinded design. The laughter exercise required a greater time investment than the application of eye drops, which may affect adherence in the long run.
DISCLOSURES:
This study was supported by grants from the National Natural Science Foundation of China and High-level Hospital Construction Project. The authors declared receiving support from the funding agencies.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
Laughter exercise practiced four times a day was noninferior to 0.1% sodium hyaluronic acid in alleviating the symptoms of dry eye disease and improved tear film stability and the function of the meibomian gland, researchers found.
METHODOLOGY:
- Researchers aimed to assess the effectiveness and safety of laughter exercise in patients with symptomatic dry eye disease by conducting a two-arm clinical trial at the largest ophthalmic center in southern China.
- They included 299 patients aged 18-45 years (74% women) with symptomatic dry eye disease who were randomly assigned to receive either laughter exercise or eye drops with 0.1% sodium hyaluronic acid four times daily for 8 weeks.
- All the participants were required to have an ocular surface disease index score, a measure of symptoms related to dry eye disease, between 18 and 80 points and a fluorescein tear film breakup time of no more than 8 seconds.
- Participants in the laughter exercise group watched an instructional video and were requested to repeat the phrases “Hee hee hee, hah hah hah, cheese cheese cheese, cheek cheek cheek, hah hah hah hah hah hah” 30 times per 5-minute session.
- The primary outcome was the mean change in the ocular surface disease index score from baseline to 8 weeks.
TAKEAWAY:
- At 8 weeks, the ocular surface disease index score reduced by 10.50 points (95% CI, −13.1 to −7.82) in the laughter exercise group and by 8.83 points (95% CI, −11.7 to −6.02) in the group prescribed eye drops.
- At 12 weeks, patients in the laughter exercise group showed a significantly greater reduction in the ocular surface disease index score than those in the group prescribed eye drops (mean between-group difference, −4.08 points; P = .024).
- Laughter exercise also led to a more significant improvement in the noninvasive tear breakup time than the use of eye drops (mean between-group difference, 2.30 sec; P < .001).
- No adverse events were reported in either of the groups during the study period.
IN PRACTICE:
“As a safe, environmentally friendly, and low-cost intervention, laughter exercise could serve as a first-line, home-based treatment for people with symptomatic dry eye disease and limited corneal staining,” the authors of the study reported.
SOURCE:
This study was led by Jing Li from the State Key Laboratory of Ophthalmology at the Zhongshan Ophthalmic Center in Sun Yat-sen University in Guangzhou, China, and was published online on September 11, 2024, in The BMJ.
LIMITATIONS:
The study lacked a double-blinded design. The laughter exercise required a greater time investment than the application of eye drops, which may affect adherence in the long run.
DISCLOSURES:
This study was supported by grants from the National Natural Science Foundation of China and High-level Hospital Construction Project. The authors declared receiving support from the funding agencies.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
Laughter exercise practiced four times a day was noninferior to 0.1% sodium hyaluronic acid in alleviating the symptoms of dry eye disease and improved tear film stability and the function of the meibomian gland, researchers found.
METHODOLOGY:
- Researchers aimed to assess the effectiveness and safety of laughter exercise in patients with symptomatic dry eye disease by conducting a two-arm clinical trial at the largest ophthalmic center in southern China.
- They included 299 patients aged 18-45 years (74% women) with symptomatic dry eye disease who were randomly assigned to receive either laughter exercise or eye drops with 0.1% sodium hyaluronic acid four times daily for 8 weeks.
- All the participants were required to have an ocular surface disease index score, a measure of symptoms related to dry eye disease, between 18 and 80 points and a fluorescein tear film breakup time of no more than 8 seconds.
- Participants in the laughter exercise group watched an instructional video and were requested to repeat the phrases “Hee hee hee, hah hah hah, cheese cheese cheese, cheek cheek cheek, hah hah hah hah hah hah” 30 times per 5-minute session.
- The primary outcome was the mean change in the ocular surface disease index score from baseline to 8 weeks.
TAKEAWAY:
- At 8 weeks, the ocular surface disease index score reduced by 10.50 points (95% CI, −13.1 to −7.82) in the laughter exercise group and by 8.83 points (95% CI, −11.7 to −6.02) in the group prescribed eye drops.
- At 12 weeks, patients in the laughter exercise group showed a significantly greater reduction in the ocular surface disease index score than those in the group prescribed eye drops (mean between-group difference, −4.08 points; P = .024).
- Laughter exercise also led to a more significant improvement in the noninvasive tear breakup time than the use of eye drops (mean between-group difference, 2.30 sec; P < .001).
- No adverse events were reported in either of the groups during the study period.
IN PRACTICE:
“As a safe, environmentally friendly, and low-cost intervention, laughter exercise could serve as a first-line, home-based treatment for people with symptomatic dry eye disease and limited corneal staining,” the authors of the study reported.
SOURCE:
This study was led by Jing Li from the State Key Laboratory of Ophthalmology at the Zhongshan Ophthalmic Center in Sun Yat-sen University in Guangzhou, China, and was published online on September 11, 2024, in The BMJ.
LIMITATIONS:
The study lacked a double-blinded design. The laughter exercise required a greater time investment than the application of eye drops, which may affect adherence in the long run.
DISCLOSURES:
This study was supported by grants from the National Natural Science Foundation of China and High-level Hospital Construction Project. The authors declared receiving support from the funding agencies.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.