User login
Severe Phymatous Rosacea of the Nose, Cheeks, and Chin Treated With Hydrosurgery
Phymatous rosacea is a rare and severe form of rosacea that manifests as disfiguring soft-tissue hypertrophy and hyperplasia as well as fibrosis of the sebaceous glands. 1 Treatments for phymatous rosacea include pharmacotherapeutic and surgical modalities; most cases are treated surgically. Surgical modalities vary, ranging from cryosurgery to conventional excision, and consensus guidelines for surgical management do not exist because data are largely limited to case reports and small case series. 2 The Versajet II Hydrosurgery System (Smith-Nephew) is a high-pressure, pulsatile lavage system that has been used for phymatous rosacea and then only for rosacea of the nose (rhinophyma). We present the case of a patient with phymatous rosacea of the nose, cheeks, and chin who was successfully treated with the Versajet II Hydrosurgery System beyond just the nose region.
Case Report
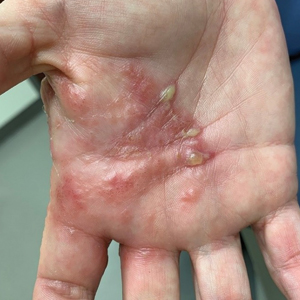
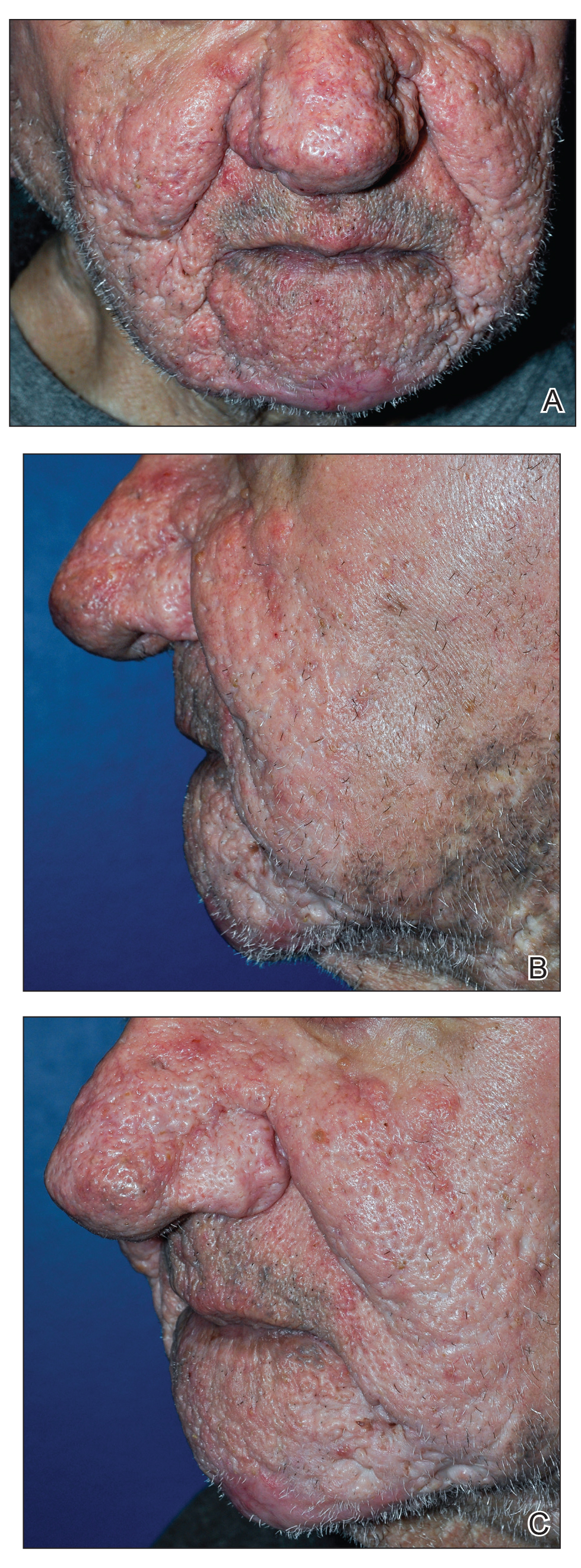
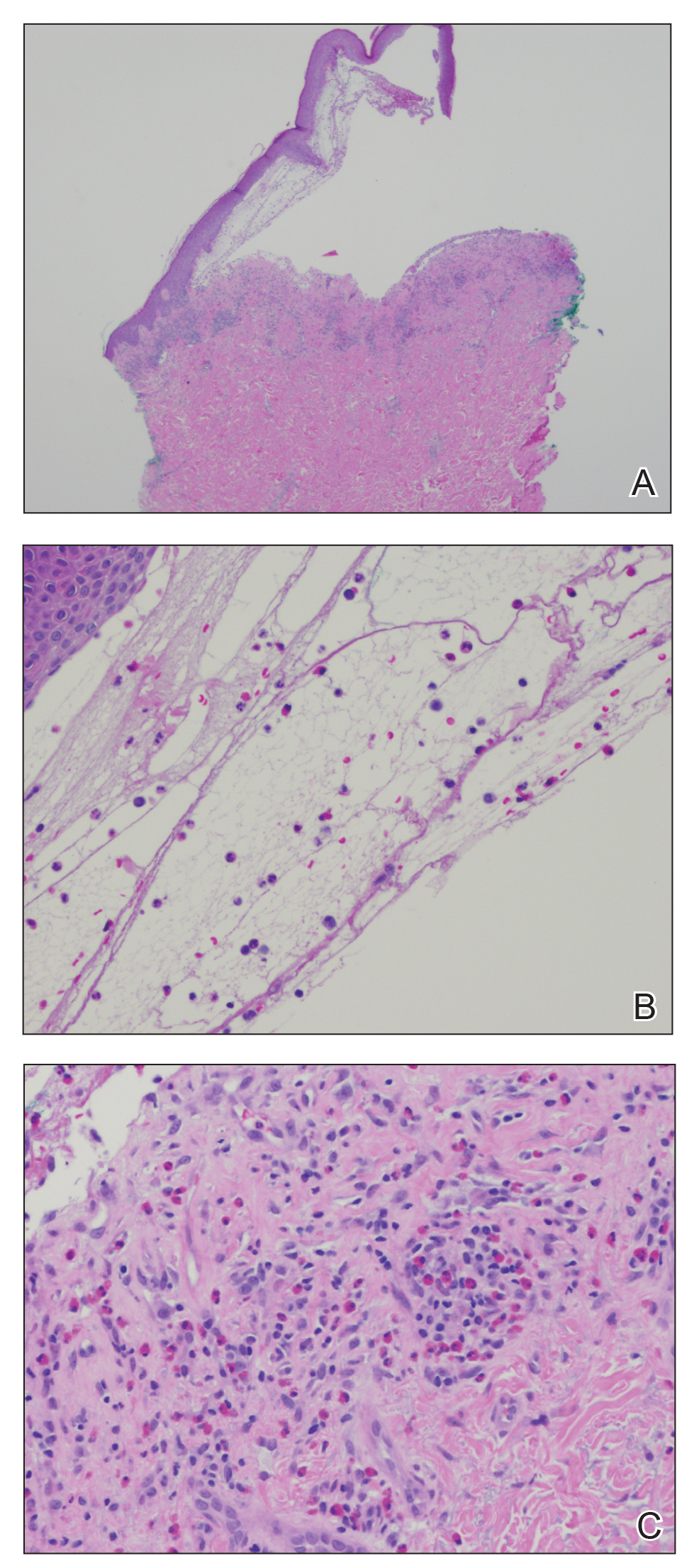
A 75-year-old man presented to the dermatology clinic for evaluation of severe phymatous rosacea of the nose, cheeks, and chin that had been present for several years. Examination revealed verruciform, thickened, erythematous skin of the nose, cheeks, and chin; marked blue-gray hyperpigmentation on the neck and hands; generalized facial redness; and cystic and depressed scars (Figure 1). The patient had been treated with topical metronidazole without response, and isotretinoin worsened the symptoms. He also was taking minocycline but stopped it at our request because of concern that the drug was causing the blue-gray hyperpigmentation. The patient was referred to plastic surgery and tangential excision was recommended. Fractional ablative laser therapy was considered but deferred because the patient wanted quicker results.

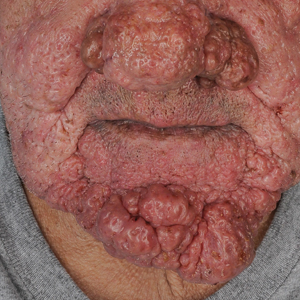
The patient received tangential excision of the phymatous areas of the chin, bilateral cheeks, and nose with the Versajet II Hydrosurgery System until a pleasing contour was noted. At 1-month follow-up, the patient had an excellent contour of the nose, cheeks, and chin (Figure 2).
Comment
Phymatous rosacea is a rare disfiguring disease that most commonly presents on the nose but also can affect the chin, cheeks, eyelids, ears, and forehead. Incidence is greater in individuals of Scottish descent and in men due to the influence of androgens. The etiology of the condition is unknown.1
Aside from clinical findings of hyperplastic and fibrotic sebaceous glands in conjunction with enlargement of the affected facial areas, histopathologic findings of phymatous rosacea vary but typically include hypertrophy of subcutaneous tissue, enlarged sebaceous ducts filled with keratin and sebum, atrophy of the dermis, and abnormal vascular development in the form of telangiectases.
Phymatous rosacea adversely affects patients’ physical, mental, and social well-being. Left untreated, it can cause nasal obstruction and recurrent bacterial infections. Furthermore, because of the potential extent of facial deformity, phymatous rosacea can be highly stigmatizing.3 Nonmelanoma skin cancers have been reported within phymatous skin, but evidence of an association between the 2 diseases remains inconclusive.4 Excised tissue from our patient was not submitted to pathology for analysis.
Given the far-reaching physical and psychological consequences of phymatous rosacea, treatment is critical but, regrettably, challenging. Although medical and surgical interventions exist, surgery is the most common practice. Oral isotretinoin may help, but many cases are recalcitrant, as was the disease in our patient. Therefore, procedural remedies often are sought, including scalpel excision, cryosurgery, argon laser, CO2 laser, dermabrasion, and electrocautery.2
Our patient underwent Versajet II Hydrosurgery System treatment of the phymatous rosacea on the nose, cheeks, and chin. Versajet is not yet commonly used to treat phymatous rosacea, likely due to the upfront cost of obtaining a new device, lack of physician familiarity, and few reports of its use for phymatous skin. A search of PubMed, EMBASE, and the Web of Science using the terms Rosacea AND (Versajet OR Hydrosurgery) yielded only 6 cases of rosacea treated by hydrosurgery; all were limited to rhinophyma and reported excellent cosmetic and functional results.5-10 Our case was unique in that hydrosurgery was used to treat phymatous rosacea beyond the nose.
Hydrosurgery has many advantages in the treatment of phymatous rosacea and other conditions in which surgical debridement is necessary, such as burns and wounds. A randomized clinical trial demonstrated that hydrosurgery is more cost-effective than conventional excision because of decreased operative time and intraoperative blood loss, fewer debridement procedures, and fewer postoperative complications.11
Rennekampff et al12 showed that Versajet debridement is superior to conventional surgery in contouring facial and acral sites and has a lower probability of infection. They proposed that by running a highly pressurized constant stream of saline across the device, Versajet clears blood and debris from the surgical site during excision.12 Hydrosurgical debridement also has been shown to reduce Staphylococcus aureus inoculate levels from in vitro–contaminated equine models significantly more than conventional debridement methods (P<.05).13
Versajet surgery appears to be well tolerated, with side effects comparable to those of classic surgical excision. A randomized controlled trial in burn patients in which treatment with Versajet was compared to traditional debridement found no significant difference in postoperative pain, healing time, and contracture rate.13
Overall, tangential excision of our patient’s phymatous rosacea using the Versajet II Hydrosurgery System yielded excellent contouring. However, due to the paucity of literature on the subject, it is difficult to discern the optimal treatment modality. Therefore, more research—ideally randomized trials—should be pursued to examine the comparative effectiveness of different interventions for phymatous rosacea.
- Curnier A, Choudhary S. Rhinophyma: dispelling the myths. Plast Reconstr Surg. 2004;114:351-354.
- Sadick H, Goepel B, Bersch C, et al. Rhinophyma: diagnosis and treatment options for a disfiguring tumor of the nose. Ann Plast Surg. 2008;61:114-120.
- Dirschka T, Micali G, Papadopoulos L, et al. Perceptions on the psychological impact of facial erythema associated with rosacea: results of international survey. Dermatol Ther (Heidelb). 2015;5:117-127.
- Lazzeri D, Colizzi L, Licata G, et al. Malignancies within rhinophyma: report of three new cases and review of the literature. Aesthetic Plast Surg. 2012;36:396-405.
- Dunne JA, Saleh DB, Rawlins JM. Management of rhinophyma with Versajet™ and ReCell®. Br J Oral Maxillofac Surg. 2013;51:e282-e284.
- Yildiz K, Kayan BR, Dulgeroglu T, et al. Treatment of rhinophyma with the Versajet™ Hydrosurgery System and autologous cell suspension (ReCELL®): a case report. J Cosmet Laser Ther. 2018;20:114-116.
- Nicolas J, Garmi R, Labbé D, et al. The role of Versajet in the surgical treatment of rhinophyma. case report. Ann Chir Plast Esthet. 2009;54:78-81.
- Novati FC, Franchi A, Roggio T, et al. Treatment of a double-giant rhinophyma with electrocautery and Versajet Hydrosurgery System. Ann Ital Chir. 2015;86. pii: S2239253X15023269.
- Taghizadeh R, Mackay SP, Gilbert PM. Treatment of rhinophyma with the Versajet Hydrosurgery System. J Plast Reconstr Aesthet Surg. 2008;61:330-333.
- Wong WL, Wong She R, Mathy JA. Rhinophyma treatment using Versajet Hydrosurgery. ANZ J Surg. 2017;87:E331-E332.
- Liu J, Ko JH, Secretov E, et al. Comparing the hydrosurgery system to conventional debridement techniques for the treatment of delayed healing wounds: a prospective, randomised clinical trial to investigate clinical efficacy and cost-effectiveness. Int Wound J. 2015;12:456-461.
- Rennekampff H-O, Schaller H-E, Wisser D, et al. Debridement of burn wounds with a water jet surgical tool. Burns. 2006;32:64-69.
- Skarlina EM, Wilmink JM, Fall N, et al. Effectiveness of conventional and hydrosurgical debridement methods in reducing Staphylococcus aureus inoculation of equine muscle in vitro. Equine Vet J. 2015;47:218-222.
Phymatous rosacea is a rare and severe form of rosacea that manifests as disfiguring soft-tissue hypertrophy and hyperplasia as well as fibrosis of the sebaceous glands. 1 Treatments for phymatous rosacea include pharmacotherapeutic and surgical modalities; most cases are treated surgically. Surgical modalities vary, ranging from cryosurgery to conventional excision, and consensus guidelines for surgical management do not exist because data are largely limited to case reports and small case series. 2 The Versajet II Hydrosurgery System (Smith-Nephew) is a high-pressure, pulsatile lavage system that has been used for phymatous rosacea and then only for rosacea of the nose (rhinophyma). We present the case of a patient with phymatous rosacea of the nose, cheeks, and chin who was successfully treated with the Versajet II Hydrosurgery System beyond just the nose region.
Case Report
A 75-year-old man presented to the dermatology clinic for evaluation of severe phymatous rosacea of the nose, cheeks, and chin that had been present for several years. Examination revealed verruciform, thickened, erythematous skin of the nose, cheeks, and chin; marked blue-gray hyperpigmentation on the neck and hands; generalized facial redness; and cystic and depressed scars (Figure 1). The patient had been treated with topical metronidazole without response, and isotretinoin worsened the symptoms. He also was taking minocycline but stopped it at our request because of concern that the drug was causing the blue-gray hyperpigmentation. The patient was referred to plastic surgery and tangential excision was recommended. Fractional ablative laser therapy was considered but deferred because the patient wanted quicker results.
The patient received tangential excision of the phymatous areas of the chin, bilateral cheeks, and nose with the Versajet II Hydrosurgery System until a pleasing contour was noted. At 1-month follow-up, the patient had an excellent contour of the nose, cheeks, and chin (Figure 2).
Comment
Phymatous rosacea is a rare disfiguring disease that most commonly presents on the nose but also can affect the chin, cheeks, eyelids, ears, and forehead. Incidence is greater in individuals of Scottish descent and in men due to the influence of androgens. The etiology of the condition is unknown.1
Aside from clinical findings of hyperplastic and fibrotic sebaceous glands in conjunction with enlargement of the affected facial areas, histopathologic findings of phymatous rosacea vary but typically include hypertrophy of subcutaneous tissue, enlarged sebaceous ducts filled with keratin and sebum, atrophy of the dermis, and abnormal vascular development in the form of telangiectases.
Phymatous rosacea adversely affects patients’ physical, mental, and social well-being. Left untreated, it can cause nasal obstruction and recurrent bacterial infections. Furthermore, because of the potential extent of facial deformity, phymatous rosacea can be highly stigmatizing.3 Nonmelanoma skin cancers have been reported within phymatous skin, but evidence of an association between the 2 diseases remains inconclusive.4 Excised tissue from our patient was not submitted to pathology for analysis.
Given the far-reaching physical and psychological consequences of phymatous rosacea, treatment is critical but, regrettably, challenging. Although medical and surgical interventions exist, surgery is the most common practice. Oral isotretinoin may help, but many cases are recalcitrant, as was the disease in our patient. Therefore, procedural remedies often are sought, including scalpel excision, cryosurgery, argon laser, CO2 laser, dermabrasion, and electrocautery.2
Our patient underwent Versajet II Hydrosurgery System treatment of the phymatous rosacea on the nose, cheeks, and chin. Versajet is not yet commonly used to treat phymatous rosacea, likely due to the upfront cost of obtaining a new device, lack of physician familiarity, and few reports of its use for phymatous skin. A search of PubMed, EMBASE, and the Web of Science using the terms Rosacea AND (Versajet OR Hydrosurgery) yielded only 6 cases of rosacea treated by hydrosurgery; all were limited to rhinophyma and reported excellent cosmetic and functional results.5-10 Our case was unique in that hydrosurgery was used to treat phymatous rosacea beyond the nose.
Hydrosurgery has many advantages in the treatment of phymatous rosacea and other conditions in which surgical debridement is necessary, such as burns and wounds. A randomized clinical trial demonstrated that hydrosurgery is more cost-effective than conventional excision because of decreased operative time and intraoperative blood loss, fewer debridement procedures, and fewer postoperative complications.11
Rennekampff et al12 showed that Versajet debridement is superior to conventional surgery in contouring facial and acral sites and has a lower probability of infection. They proposed that by running a highly pressurized constant stream of saline across the device, Versajet clears blood and debris from the surgical site during excision.12 Hydrosurgical debridement also has been shown to reduce Staphylococcus aureus inoculate levels from in vitro–contaminated equine models significantly more than conventional debridement methods (P<.05).13
Versajet surgery appears to be well tolerated, with side effects comparable to those of classic surgical excision. A randomized controlled trial in burn patients in which treatment with Versajet was compared to traditional debridement found no significant difference in postoperative pain, healing time, and contracture rate.13
Overall, tangential excision of our patient’s phymatous rosacea using the Versajet II Hydrosurgery System yielded excellent contouring. However, due to the paucity of literature on the subject, it is difficult to discern the optimal treatment modality. Therefore, more research—ideally randomized trials—should be pursued to examine the comparative effectiveness of different interventions for phymatous rosacea.
Phymatous rosacea is a rare and severe form of rosacea that manifests as disfiguring soft-tissue hypertrophy and hyperplasia as well as fibrosis of the sebaceous glands. 1 Treatments for phymatous rosacea include pharmacotherapeutic and surgical modalities; most cases are treated surgically. Surgical modalities vary, ranging from cryosurgery to conventional excision, and consensus guidelines for surgical management do not exist because data are largely limited to case reports and small case series. 2 The Versajet II Hydrosurgery System (Smith-Nephew) is a high-pressure, pulsatile lavage system that has been used for phymatous rosacea and then only for rosacea of the nose (rhinophyma). We present the case of a patient with phymatous rosacea of the nose, cheeks, and chin who was successfully treated with the Versajet II Hydrosurgery System beyond just the nose region.
Case Report
A 75-year-old man presented to the dermatology clinic for evaluation of severe phymatous rosacea of the nose, cheeks, and chin that had been present for several years. Examination revealed verruciform, thickened, erythematous skin of the nose, cheeks, and chin; marked blue-gray hyperpigmentation on the neck and hands; generalized facial redness; and cystic and depressed scars (Figure 1). The patient had been treated with topical metronidazole without response, and isotretinoin worsened the symptoms. He also was taking minocycline but stopped it at our request because of concern that the drug was causing the blue-gray hyperpigmentation. The patient was referred to plastic surgery and tangential excision was recommended. Fractional ablative laser therapy was considered but deferred because the patient wanted quicker results.
The patient received tangential excision of the phymatous areas of the chin, bilateral cheeks, and nose with the Versajet II Hydrosurgery System until a pleasing contour was noted. At 1-month follow-up, the patient had an excellent contour of the nose, cheeks, and chin (Figure 2).
Comment
Phymatous rosacea is a rare disfiguring disease that most commonly presents on the nose but also can affect the chin, cheeks, eyelids, ears, and forehead. Incidence is greater in individuals of Scottish descent and in men due to the influence of androgens. The etiology of the condition is unknown.1
Aside from clinical findings of hyperplastic and fibrotic sebaceous glands in conjunction with enlargement of the affected facial areas, histopathologic findings of phymatous rosacea vary but typically include hypertrophy of subcutaneous tissue, enlarged sebaceous ducts filled with keratin and sebum, atrophy of the dermis, and abnormal vascular development in the form of telangiectases.
Phymatous rosacea adversely affects patients’ physical, mental, and social well-being. Left untreated, it can cause nasal obstruction and recurrent bacterial infections. Furthermore, because of the potential extent of facial deformity, phymatous rosacea can be highly stigmatizing.3 Nonmelanoma skin cancers have been reported within phymatous skin, but evidence of an association between the 2 diseases remains inconclusive.4 Excised tissue from our patient was not submitted to pathology for analysis.
Given the far-reaching physical and psychological consequences of phymatous rosacea, treatment is critical but, regrettably, challenging. Although medical and surgical interventions exist, surgery is the most common practice. Oral isotretinoin may help, but many cases are recalcitrant, as was the disease in our patient. Therefore, procedural remedies often are sought, including scalpel excision, cryosurgery, argon laser, CO2 laser, dermabrasion, and electrocautery.2
Our patient underwent Versajet II Hydrosurgery System treatment of the phymatous rosacea on the nose, cheeks, and chin. Versajet is not yet commonly used to treat phymatous rosacea, likely due to the upfront cost of obtaining a new device, lack of physician familiarity, and few reports of its use for phymatous skin. A search of PubMed, EMBASE, and the Web of Science using the terms Rosacea AND (Versajet OR Hydrosurgery) yielded only 6 cases of rosacea treated by hydrosurgery; all were limited to rhinophyma and reported excellent cosmetic and functional results.5-10 Our case was unique in that hydrosurgery was used to treat phymatous rosacea beyond the nose.
Hydrosurgery has many advantages in the treatment of phymatous rosacea and other conditions in which surgical debridement is necessary, such as burns and wounds. A randomized clinical trial demonstrated that hydrosurgery is more cost-effective than conventional excision because of decreased operative time and intraoperative blood loss, fewer debridement procedures, and fewer postoperative complications.11
Rennekampff et al12 showed that Versajet debridement is superior to conventional surgery in contouring facial and acral sites and has a lower probability of infection. They proposed that by running a highly pressurized constant stream of saline across the device, Versajet clears blood and debris from the surgical site during excision.12 Hydrosurgical debridement also has been shown to reduce Staphylococcus aureus inoculate levels from in vitro–contaminated equine models significantly more than conventional debridement methods (P<.05).13
Versajet surgery appears to be well tolerated, with side effects comparable to those of classic surgical excision. A randomized controlled trial in burn patients in which treatment with Versajet was compared to traditional debridement found no significant difference in postoperative pain, healing time, and contracture rate.13
Overall, tangential excision of our patient’s phymatous rosacea using the Versajet II Hydrosurgery System yielded excellent contouring. However, due to the paucity of literature on the subject, it is difficult to discern the optimal treatment modality. Therefore, more research—ideally randomized trials—should be pursued to examine the comparative effectiveness of different interventions for phymatous rosacea.
- Curnier A, Choudhary S. Rhinophyma: dispelling the myths. Plast Reconstr Surg. 2004;114:351-354.
- Sadick H, Goepel B, Bersch C, et al. Rhinophyma: diagnosis and treatment options for a disfiguring tumor of the nose. Ann Plast Surg. 2008;61:114-120.
- Dirschka T, Micali G, Papadopoulos L, et al. Perceptions on the psychological impact of facial erythema associated with rosacea: results of international survey. Dermatol Ther (Heidelb). 2015;5:117-127.
- Lazzeri D, Colizzi L, Licata G, et al. Malignancies within rhinophyma: report of three new cases and review of the literature. Aesthetic Plast Surg. 2012;36:396-405.
- Dunne JA, Saleh DB, Rawlins JM. Management of rhinophyma with Versajet™ and ReCell®. Br J Oral Maxillofac Surg. 2013;51:e282-e284.
- Yildiz K, Kayan BR, Dulgeroglu T, et al. Treatment of rhinophyma with the Versajet™ Hydrosurgery System and autologous cell suspension (ReCELL®): a case report. J Cosmet Laser Ther. 2018;20:114-116.
- Nicolas J, Garmi R, Labbé D, et al. The role of Versajet in the surgical treatment of rhinophyma. case report. Ann Chir Plast Esthet. 2009;54:78-81.
- Novati FC, Franchi A, Roggio T, et al. Treatment of a double-giant rhinophyma with electrocautery and Versajet Hydrosurgery System. Ann Ital Chir. 2015;86. pii: S2239253X15023269.
- Taghizadeh R, Mackay SP, Gilbert PM. Treatment of rhinophyma with the Versajet Hydrosurgery System. J Plast Reconstr Aesthet Surg. 2008;61:330-333.
- Wong WL, Wong She R, Mathy JA. Rhinophyma treatment using Versajet Hydrosurgery. ANZ J Surg. 2017;87:E331-E332.
- Liu J, Ko JH, Secretov E, et al. Comparing the hydrosurgery system to conventional debridement techniques for the treatment of delayed healing wounds: a prospective, randomised clinical trial to investigate clinical efficacy and cost-effectiveness. Int Wound J. 2015;12:456-461.
- Rennekampff H-O, Schaller H-E, Wisser D, et al. Debridement of burn wounds with a water jet surgical tool. Burns. 2006;32:64-69.
- Skarlina EM, Wilmink JM, Fall N, et al. Effectiveness of conventional and hydrosurgical debridement methods in reducing Staphylococcus aureus inoculation of equine muscle in vitro. Equine Vet J. 2015;47:218-222.
- Curnier A, Choudhary S. Rhinophyma: dispelling the myths. Plast Reconstr Surg. 2004;114:351-354.
- Sadick H, Goepel B, Bersch C, et al. Rhinophyma: diagnosis and treatment options for a disfiguring tumor of the nose. Ann Plast Surg. 2008;61:114-120.
- Dirschka T, Micali G, Papadopoulos L, et al. Perceptions on the psychological impact of facial erythema associated with rosacea: results of international survey. Dermatol Ther (Heidelb). 2015;5:117-127.
- Lazzeri D, Colizzi L, Licata G, et al. Malignancies within rhinophyma: report of three new cases and review of the literature. Aesthetic Plast Surg. 2012;36:396-405.
- Dunne JA, Saleh DB, Rawlins JM. Management of rhinophyma with Versajet™ and ReCell®. Br J Oral Maxillofac Surg. 2013;51:e282-e284.
- Yildiz K, Kayan BR, Dulgeroglu T, et al. Treatment of rhinophyma with the Versajet™ Hydrosurgery System and autologous cell suspension (ReCELL®): a case report. J Cosmet Laser Ther. 2018;20:114-116.
- Nicolas J, Garmi R, Labbé D, et al. The role of Versajet in the surgical treatment of rhinophyma. case report. Ann Chir Plast Esthet. 2009;54:78-81.
- Novati FC, Franchi A, Roggio T, et al. Treatment of a double-giant rhinophyma with electrocautery and Versajet Hydrosurgery System. Ann Ital Chir. 2015;86. pii: S2239253X15023269.
- Taghizadeh R, Mackay SP, Gilbert PM. Treatment of rhinophyma with the Versajet Hydrosurgery System. J Plast Reconstr Aesthet Surg. 2008;61:330-333.
- Wong WL, Wong She R, Mathy JA. Rhinophyma treatment using Versajet Hydrosurgery. ANZ J Surg. 2017;87:E331-E332.
- Liu J, Ko JH, Secretov E, et al. Comparing the hydrosurgery system to conventional debridement techniques for the treatment of delayed healing wounds: a prospective, randomised clinical trial to investigate clinical efficacy and cost-effectiveness. Int Wound J. 2015;12:456-461.
- Rennekampff H-O, Schaller H-E, Wisser D, et al. Debridement of burn wounds with a water jet surgical tool. Burns. 2006;32:64-69.
- Skarlina EM, Wilmink JM, Fall N, et al. Effectiveness of conventional and hydrosurgical debridement methods in reducing Staphylococcus aureus inoculation of equine muscle in vitro. Equine Vet J. 2015;47:218-222.
Practice Points
- Phymatous rosacea is a rare disfiguring disease that most commonly affects men and can have considerable effects on a patient’s physical, mental, and social well-being.
- Treatment of phymatous rosacea usually is surgical; however, no consensus guidelines exist for best surgical management.
- The Versajet II Hydrosurgery System can be useful and effective for the treatment of phymatous rosacea, not only on the nose but elsewhere on the face.
Anti–PD1 Immune Checkpoint Inhibitor–Induced Bullous Pemphigoid in Metastatic Melanoma and Non–Small Cell Lung Cancer
Immune checkpoint inhibitors are used for a variety of advanced malignancies, including melanoma, non–small cell lung cancer, urothelial cancer, and renal cell carcinoma. Anti–programmed cell death 1 (PD1) targeted therapies, such as pembrolizumab and nivolumab, are improving patient survival. This class of immunotherapy is revolutionary but is associated with autoimmune adverse effects. A rare but increasingly reported adverse effect of anti-PD1 therapy is bullous pemphigoid (BP), an autoimmune blistering disease directed against
High clinical suspicion, early diagnosis, and proper management of immunotherapy-related BP are imperative for keeping patients on life-prolonging treatment. We present 3 cases of BP secondary to anti-PD1 immunotherapy in patients with melanoma or non–small cell lung cancer to highlight the diagnosis and treatment of BP as well as emphasize the importance of the dermatologist in the care of patients with immunotherapy-related skin disease.
Case Reports
Patient 1
A 72-year-old woman with metastatic BRAF-mutated melanoma from an unknown primary site presented with intensely pruritic papules on the back, chest, and extremities of 4 months’ duration. She described her symptoms as insidious in onset and refractory to clobetasol ointment, oral diphenhydramine, and over-the-counter anti-itch creams. The patient had been treated with oral dabrafenib 150 mg twice daily and trametinib 2 mg/d but was switched to pembrolizumab when the disease progressed. After 8 months, she had a complete radiologic response to pembrolizumab 2 mg/kg every 3 weeks, which was discontinued in favor of observation 3 months prior to presentation to dermatology.
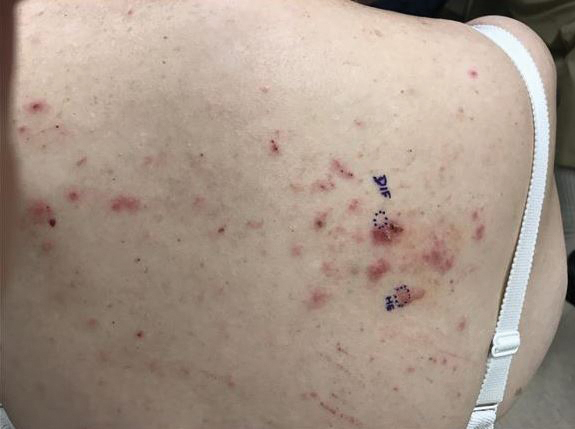
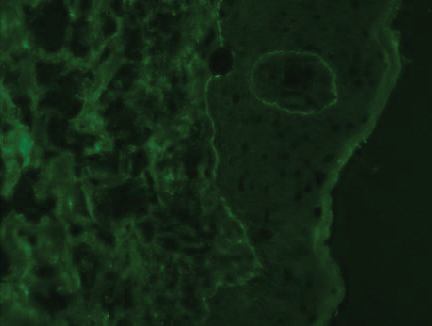
At the current presentation, physical examination revealed innumerable erythematous, excoriated, 2- to 4-mm, red papules diffusely scattered on the upper back, chest, abdomen, and thighs, with one 8×4-mm vesicle on the right side of the upper back (Figure 1). Discrete areas of depigmented macules, consistent with vitiligo, coalesced into patches on the legs, thighs, arms, and back. The patient was started on a 3-week oral prednisone taper for symptom relief. A hematoxylin and eosin (H&E)–stained punch biopsy of the back revealed a subepidermal split with eosinophils and a dense eosinophilic infiltrate in the dermis (Figure 2). Direct immunofluorescence (DIF) studies from a specimen adjacent to the biopsy collected for H&E staining showed linear deposition of IgA, IgG, and C3 along the dermoepidermal junction (Figure 3). Histologic findings were consistent with BP.
The patient was started on doxycycline 100 mg twice daily and clobetasol ointment 0.05% once daily to supplement the prednisone taper. At 3-week follow-up, she reported pruritus and a few erythematous macules but no new bullae. At 12 weeks, some papules persisted; however, the patient was averse to using systemic agents and decided that symptoms were adequately controlled with clobetasol ointment and oral doxycycline.
Because the patient currently remains in clinical and radiologic remission, anti-PD1 immune checkpoint inhibitors have not been restarted but remain an option for the future if disease recurs
Patient 2
An 82-year-old man with a history of stage IIC desmoplastic melanoma presented to dermatology with an intensely pruritic eruption on the legs, arms, waist, upper torso, and scalp of 3 weeks’ duration. Clobetasol ointment had provided minimal relief.
Six months prior to presenting to dermatology, the patient underwent immunotherapy with 4 cycles of ipilimumab 200 mg intravenous (IV) and nivolumab 240 mg IV every 2 weeks, receiving ipilimumab during the first cycle only because of a lack of availability at the pharmacy. He then received nivolumab 240 mg IV every 2 weeks as maintenance therapy. After the second dose of nivolumab maintenance therapy, however, he developed generalized bullae and pruritus. Dermatology was consulted during an oncology appointment, and his oncologist decided to hold nivolumab.
Physical examination revealed generalized tense and eroded bullae covering more than 50% of the body surface area and affecting the scalp, arms, legs, torso, and buttocks. Two punch biopsies were obtained. Hematoxylin and eosin staining revealed a subepidermal split with predominantly eosinophils and scattered neutrophils. Direct immunofluorescence studies showed linear deposition of IgG, IgA, and C3 along the dermoepidermal junction, consistent with BP.
The patient’s BP was difficult to control, requiring several hospital admissions for wound care, high-dose systemic steroids, and initiation of mycophenolate mofetil. After 4 months of waxing and waning symptoms, the BP was controlled with mycophenolate mofetil 1500 mg/d; clobetasol ointment 0.05%; and diphenhydramine for pruritus. Due to the prolonged recovery and severity of BP, the patient’s oncologist deemed that he was not a candidate for future immunotherapy.
Patient 3
A 68-year-old man with PD1-negative, metastatic, well-differentiated squamous cell carcinoma of the lung presented to dermatology with a pruritic rash of 3 weeks’ duration. He had been receiving nivolumab for 2 years after disease progressed on prior chemotherapies and experienced several grade 1 or grade 2 nivolumab-induced autoimmune reactions including thyroiditis, dermatitis, and nephritis, for which he was taking prednisone 5 mg/d for suppression.
Physical examination revealed psoriasiform pink plaques on the arms, chest, and legs. The differential diagnosis at the time favored psoriasiform dermatitis over lichenoid dermatitis. A punch biopsy revealed psoriasiform dermatitis. The patient was prescribed fluocinonide ointment 0.05% daily. His plaques improved with topical steroids.
The patient returned approximately 1 month later with a report of a new blistering rash on the legs. Physical examination revealed interval improvement of the psoriasiform plaques on the scalp, torso, and extremities, but tense bullae were seen on the thighs, with surrounding superficial erosions at sites of recent bullae. Punch biopsies of the skin for H&E staining and DIF showed BP.
Prednisone was increased to 50 mg/d for a 3-week taper. Doxycycline 100 mg twice daily was started. The patient’s skin disease continued to be difficult to control with therapy; nivolumab was held by his oncologist.
Comment
Immunotherapy with immune checkpoint blockade represents a successful application of immune recognition to treat metastatic cancers, including melanoma, non–small cell lung cancer, urothelial cancer, and renal cell carcinoma.
Anti-PD1 targeted therapies improve survival in solid and hematologic malignancies, with a response rate as high as 40% in melanoma.2 Although these medications can prolong survival, many are associated with loss of self-tolerance and severe autoimmunelike events that can limit therapy.3 An exception is PD1-induced vitiligo, which patient 1 developed and has been associated with a better response to therapy.4
Anti-PD1–induced BP is a newly reported adverse effect. In its early stages, BP can be difficult to differentiate from eczematous or urticarial dermatitis.5-8 Discontinuation of immunotherapy has been reported in more than 70% of patients who develop BP.1 There are reports of successful treatment of BP with a course of a PD1 inhibitor,9 but 2 of our patients had severe BP that led to discontinuation of immunotherapy.
Consider Prescreening
Given that development of BP often leads to cessation of therapy, identifying patients at risk prior to starting an immune checkpoint inhibitor might have clinical utility. Biopsy with DIF is the gold standard for diagnosis, but serologic testing can be a useful adjunct because enzyme-linked immunosorbent assay for BP antigen 1 and BP antigen 2 has a reported sensitivity and specificity of 87% and 98%, respectively.10 Serologic testing prior to starting therapy with an immune checkpoint inhibitor can provide a baseline for patients. A rise in titer, in conjunction with onset of a rash, might aid in earlier diagnosis, particularly because urticarial BP can be difficult to diagnose clinically.
Further study on the utility vs cost-benefit of these screening modalities is warranted. Their predictive utility might be limited, however, and positive serologic test results might have unanticipated consequences, such as hesitation in treating patients, thus leading to a delay in therapy or access to these medications.
Conclusion
The expanding use of immune checkpoint inhibitors is increasing survival in patients with metastatic melanoma and other malignancies. Adverse effects are part of the continuum of immune system stimulation, with overstimulation resulting in dermatitis; thyroiditis; pneumonitis; and less commonly hypophysitis, vitiligo, and colitis.
Rarely, immune checkpoint inhibition induces BP. Development of BP leads to discontinuation of therapy in more than half of reported cases due to lack of adequate treatment for this skin disease and its impact on quality of life. Therefore, quick diagnosis of BP in patients on immunotherapy and successful management techniques can prevent discontinuation of these lifesaving cancer therapies. For that reason, dermatologists play an important role in the management of patients on immune checkpoint inhibitors for cancer.
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669.
- Márquez-Rodas, I, Cerezuela P, Soria A, et al. Immune checkpoint inhibitors: therapeutic advances in melanoma. Ann Transl Med. 2015;3:267.
- Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors a review. JAMA Oncol. 2016;2:1346-1353.
- Hua C, Boussemart L, Mateus C, et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol. 2016;152:45-51.
- Hwang SJE, Carlos G, Chou S, et al. Bullous pemphigoid, an autoantibody-mediated disease, is a novel immune-related adverse event in patients treated with anti-programmed cell death 1 antibodies. Melanoma Res. 2016;26:413-416.
- Damsky W, Kole L, Tomayko MM. Development of bullous pemphigoid during nivolumab therapy. JAAD Case Rep. 2016;2:442-444.
- Garje R, Chau JJ, Chung J, et al. Acute flare of bullous pemphigus with pembrolizumab used for treatment of metastatic urothelial cancer. J Immunother. 2018;41:42-44.
- Ito M, Hoashi T, Endo Y, et al. Atypical pemphigus developed in a patient with urothelial carcinoma treated with nivolumab. J Dermatol. 2019;46:e90-e92.
- Chen W-S, Tetzlaff MT, Diwan H, et al. Suprabasal acantholytic dermatologic toxicities associated checkpoint inhibitor therapy: a spectrum of immune reactions from paraneoplastic pemphigus-like to Grover-like lesions. J Cutan Pathol. 2018;45:764-773.
- Muglia C, Bronsnick T, Kirkorian AY, et al. Questioning the specificity and sensitivity of ELISA for bullous pemphigoid diagnosis. Cutis. 2017;99:E27-E30.
Immune checkpoint inhibitors are used for a variety of advanced malignancies, including melanoma, non–small cell lung cancer, urothelial cancer, and renal cell carcinoma. Anti–programmed cell death 1 (PD1) targeted therapies, such as pembrolizumab and nivolumab, are improving patient survival. This class of immunotherapy is revolutionary but is associated with autoimmune adverse effects. A rare but increasingly reported adverse effect of anti-PD1 therapy is bullous pemphigoid (BP), an autoimmune blistering disease directed against
High clinical suspicion, early diagnosis, and proper management of immunotherapy-related BP are imperative for keeping patients on life-prolonging treatment. We present 3 cases of BP secondary to anti-PD1 immunotherapy in patients with melanoma or non–small cell lung cancer to highlight the diagnosis and treatment of BP as well as emphasize the importance of the dermatologist in the care of patients with immunotherapy-related skin disease.
Case Reports
Patient 1
A 72-year-old woman with metastatic BRAF-mutated melanoma from an unknown primary site presented with intensely pruritic papules on the back, chest, and extremities of 4 months’ duration. She described her symptoms as insidious in onset and refractory to clobetasol ointment, oral diphenhydramine, and over-the-counter anti-itch creams. The patient had been treated with oral dabrafenib 150 mg twice daily and trametinib 2 mg/d but was switched to pembrolizumab when the disease progressed. After 8 months, she had a complete radiologic response to pembrolizumab 2 mg/kg every 3 weeks, which was discontinued in favor of observation 3 months prior to presentation to dermatology.
At the current presentation, physical examination revealed innumerable erythematous, excoriated, 2- to 4-mm, red papules diffusely scattered on the upper back, chest, abdomen, and thighs, with one 8×4-mm vesicle on the right side of the upper back (Figure 1). Discrete areas of depigmented macules, consistent with vitiligo, coalesced into patches on the legs, thighs, arms, and back. The patient was started on a 3-week oral prednisone taper for symptom relief. A hematoxylin and eosin (H&E)–stained punch biopsy of the back revealed a subepidermal split with eosinophils and a dense eosinophilic infiltrate in the dermis (Figure 2). Direct immunofluorescence (DIF) studies from a specimen adjacent to the biopsy collected for H&E staining showed linear deposition of IgA, IgG, and C3 along the dermoepidermal junction (Figure 3). Histologic findings were consistent with BP.
The patient was started on doxycycline 100 mg twice daily and clobetasol ointment 0.05% once daily to supplement the prednisone taper. At 3-week follow-up, she reported pruritus and a few erythematous macules but no new bullae. At 12 weeks, some papules persisted; however, the patient was averse to using systemic agents and decided that symptoms were adequately controlled with clobetasol ointment and oral doxycycline.
Because the patient currently remains in clinical and radiologic remission, anti-PD1 immune checkpoint inhibitors have not been restarted but remain an option for the future if disease recurs
Patient 2
An 82-year-old man with a history of stage IIC desmoplastic melanoma presented to dermatology with an intensely pruritic eruption on the legs, arms, waist, upper torso, and scalp of 3 weeks’ duration. Clobetasol ointment had provided minimal relief.
Six months prior to presenting to dermatology, the patient underwent immunotherapy with 4 cycles of ipilimumab 200 mg intravenous (IV) and nivolumab 240 mg IV every 2 weeks, receiving ipilimumab during the first cycle only because of a lack of availability at the pharmacy. He then received nivolumab 240 mg IV every 2 weeks as maintenance therapy. After the second dose of nivolumab maintenance therapy, however, he developed generalized bullae and pruritus. Dermatology was consulted during an oncology appointment, and his oncologist decided to hold nivolumab.
Physical examination revealed generalized tense and eroded bullae covering more than 50% of the body surface area and affecting the scalp, arms, legs, torso, and buttocks. Two punch biopsies were obtained. Hematoxylin and eosin staining revealed a subepidermal split with predominantly eosinophils and scattered neutrophils. Direct immunofluorescence studies showed linear deposition of IgG, IgA, and C3 along the dermoepidermal junction, consistent with BP.
The patient’s BP was difficult to control, requiring several hospital admissions for wound care, high-dose systemic steroids, and initiation of mycophenolate mofetil. After 4 months of waxing and waning symptoms, the BP was controlled with mycophenolate mofetil 1500 mg/d; clobetasol ointment 0.05%; and diphenhydramine for pruritus. Due to the prolonged recovery and severity of BP, the patient’s oncologist deemed that he was not a candidate for future immunotherapy.
Patient 3
A 68-year-old man with PD1-negative, metastatic, well-differentiated squamous cell carcinoma of the lung presented to dermatology with a pruritic rash of 3 weeks’ duration. He had been receiving nivolumab for 2 years after disease progressed on prior chemotherapies and experienced several grade 1 or grade 2 nivolumab-induced autoimmune reactions including thyroiditis, dermatitis, and nephritis, for which he was taking prednisone 5 mg/d for suppression.
Physical examination revealed psoriasiform pink plaques on the arms, chest, and legs. The differential diagnosis at the time favored psoriasiform dermatitis over lichenoid dermatitis. A punch biopsy revealed psoriasiform dermatitis. The patient was prescribed fluocinonide ointment 0.05% daily. His plaques improved with topical steroids.
The patient returned approximately 1 month later with a report of a new blistering rash on the legs. Physical examination revealed interval improvement of the psoriasiform plaques on the scalp, torso, and extremities, but tense bullae were seen on the thighs, with surrounding superficial erosions at sites of recent bullae. Punch biopsies of the skin for H&E staining and DIF showed BP.
Prednisone was increased to 50 mg/d for a 3-week taper. Doxycycline 100 mg twice daily was started. The patient’s skin disease continued to be difficult to control with therapy; nivolumab was held by his oncologist.
Comment
Immunotherapy with immune checkpoint blockade represents a successful application of immune recognition to treat metastatic cancers, including melanoma, non–small cell lung cancer, urothelial cancer, and renal cell carcinoma.
Anti-PD1 targeted therapies improve survival in solid and hematologic malignancies, with a response rate as high as 40% in melanoma.2 Although these medications can prolong survival, many are associated with loss of self-tolerance and severe autoimmunelike events that can limit therapy.3 An exception is PD1-induced vitiligo, which patient 1 developed and has been associated with a better response to therapy.4
Anti-PD1–induced BP is a newly reported adverse effect. In its early stages, BP can be difficult to differentiate from eczematous or urticarial dermatitis.5-8 Discontinuation of immunotherapy has been reported in more than 70% of patients who develop BP.1 There are reports of successful treatment of BP with a course of a PD1 inhibitor,9 but 2 of our patients had severe BP that led to discontinuation of immunotherapy.
Consider Prescreening
Given that development of BP often leads to cessation of therapy, identifying patients at risk prior to starting an immune checkpoint inhibitor might have clinical utility. Biopsy with DIF is the gold standard for diagnosis, but serologic testing can be a useful adjunct because enzyme-linked immunosorbent assay for BP antigen 1 and BP antigen 2 has a reported sensitivity and specificity of 87% and 98%, respectively.10 Serologic testing prior to starting therapy with an immune checkpoint inhibitor can provide a baseline for patients. A rise in titer, in conjunction with onset of a rash, might aid in earlier diagnosis, particularly because urticarial BP can be difficult to diagnose clinically.
Further study on the utility vs cost-benefit of these screening modalities is warranted. Their predictive utility might be limited, however, and positive serologic test results might have unanticipated consequences, such as hesitation in treating patients, thus leading to a delay in therapy or access to these medications.
Conclusion
The expanding use of immune checkpoint inhibitors is increasing survival in patients with metastatic melanoma and other malignancies. Adverse effects are part of the continuum of immune system stimulation, with overstimulation resulting in dermatitis; thyroiditis; pneumonitis; and less commonly hypophysitis, vitiligo, and colitis.
Rarely, immune checkpoint inhibition induces BP. Development of BP leads to discontinuation of therapy in more than half of reported cases due to lack of adequate treatment for this skin disease and its impact on quality of life. Therefore, quick diagnosis of BP in patients on immunotherapy and successful management techniques can prevent discontinuation of these lifesaving cancer therapies. For that reason, dermatologists play an important role in the management of patients on immune checkpoint inhibitors for cancer.
Immune checkpoint inhibitors are used for a variety of advanced malignancies, including melanoma, non–small cell lung cancer, urothelial cancer, and renal cell carcinoma. Anti–programmed cell death 1 (PD1) targeted therapies, such as pembrolizumab and nivolumab, are improving patient survival. This class of immunotherapy is revolutionary but is associated with autoimmune adverse effects. A rare but increasingly reported adverse effect of anti-PD1 therapy is bullous pemphigoid (BP), an autoimmune blistering disease directed against
High clinical suspicion, early diagnosis, and proper management of immunotherapy-related BP are imperative for keeping patients on life-prolonging treatment. We present 3 cases of BP secondary to anti-PD1 immunotherapy in patients with melanoma or non–small cell lung cancer to highlight the diagnosis and treatment of BP as well as emphasize the importance of the dermatologist in the care of patients with immunotherapy-related skin disease.
Case Reports
Patient 1
A 72-year-old woman with metastatic BRAF-mutated melanoma from an unknown primary site presented with intensely pruritic papules on the back, chest, and extremities of 4 months’ duration. She described her symptoms as insidious in onset and refractory to clobetasol ointment, oral diphenhydramine, and over-the-counter anti-itch creams. The patient had been treated with oral dabrafenib 150 mg twice daily and trametinib 2 mg/d but was switched to pembrolizumab when the disease progressed. After 8 months, she had a complete radiologic response to pembrolizumab 2 mg/kg every 3 weeks, which was discontinued in favor of observation 3 months prior to presentation to dermatology.
At the current presentation, physical examination revealed innumerable erythematous, excoriated, 2- to 4-mm, red papules diffusely scattered on the upper back, chest, abdomen, and thighs, with one 8×4-mm vesicle on the right side of the upper back (Figure 1). Discrete areas of depigmented macules, consistent with vitiligo, coalesced into patches on the legs, thighs, arms, and back. The patient was started on a 3-week oral prednisone taper for symptom relief. A hematoxylin and eosin (H&E)–stained punch biopsy of the back revealed a subepidermal split with eosinophils and a dense eosinophilic infiltrate in the dermis (Figure 2). Direct immunofluorescence (DIF) studies from a specimen adjacent to the biopsy collected for H&E staining showed linear deposition of IgA, IgG, and C3 along the dermoepidermal junction (Figure 3). Histologic findings were consistent with BP.
The patient was started on doxycycline 100 mg twice daily and clobetasol ointment 0.05% once daily to supplement the prednisone taper. At 3-week follow-up, she reported pruritus and a few erythematous macules but no new bullae. At 12 weeks, some papules persisted; however, the patient was averse to using systemic agents and decided that symptoms were adequately controlled with clobetasol ointment and oral doxycycline.
Because the patient currently remains in clinical and radiologic remission, anti-PD1 immune checkpoint inhibitors have not been restarted but remain an option for the future if disease recurs
Patient 2
An 82-year-old man with a history of stage IIC desmoplastic melanoma presented to dermatology with an intensely pruritic eruption on the legs, arms, waist, upper torso, and scalp of 3 weeks’ duration. Clobetasol ointment had provided minimal relief.
Six months prior to presenting to dermatology, the patient underwent immunotherapy with 4 cycles of ipilimumab 200 mg intravenous (IV) and nivolumab 240 mg IV every 2 weeks, receiving ipilimumab during the first cycle only because of a lack of availability at the pharmacy. He then received nivolumab 240 mg IV every 2 weeks as maintenance therapy. After the second dose of nivolumab maintenance therapy, however, he developed generalized bullae and pruritus. Dermatology was consulted during an oncology appointment, and his oncologist decided to hold nivolumab.
Physical examination revealed generalized tense and eroded bullae covering more than 50% of the body surface area and affecting the scalp, arms, legs, torso, and buttocks. Two punch biopsies were obtained. Hematoxylin and eosin staining revealed a subepidermal split with predominantly eosinophils and scattered neutrophils. Direct immunofluorescence studies showed linear deposition of IgG, IgA, and C3 along the dermoepidermal junction, consistent with BP.
The patient’s BP was difficult to control, requiring several hospital admissions for wound care, high-dose systemic steroids, and initiation of mycophenolate mofetil. After 4 months of waxing and waning symptoms, the BP was controlled with mycophenolate mofetil 1500 mg/d; clobetasol ointment 0.05%; and diphenhydramine for pruritus. Due to the prolonged recovery and severity of BP, the patient’s oncologist deemed that he was not a candidate for future immunotherapy.
Patient 3
A 68-year-old man with PD1-negative, metastatic, well-differentiated squamous cell carcinoma of the lung presented to dermatology with a pruritic rash of 3 weeks’ duration. He had been receiving nivolumab for 2 years after disease progressed on prior chemotherapies and experienced several grade 1 or grade 2 nivolumab-induced autoimmune reactions including thyroiditis, dermatitis, and nephritis, for which he was taking prednisone 5 mg/d for suppression.
Physical examination revealed psoriasiform pink plaques on the arms, chest, and legs. The differential diagnosis at the time favored psoriasiform dermatitis over lichenoid dermatitis. A punch biopsy revealed psoriasiform dermatitis. The patient was prescribed fluocinonide ointment 0.05% daily. His plaques improved with topical steroids.
The patient returned approximately 1 month later with a report of a new blistering rash on the legs. Physical examination revealed interval improvement of the psoriasiform plaques on the scalp, torso, and extremities, but tense bullae were seen on the thighs, with surrounding superficial erosions at sites of recent bullae. Punch biopsies of the skin for H&E staining and DIF showed BP.
Prednisone was increased to 50 mg/d for a 3-week taper. Doxycycline 100 mg twice daily was started. The patient’s skin disease continued to be difficult to control with therapy; nivolumab was held by his oncologist.
Comment
Immunotherapy with immune checkpoint blockade represents a successful application of immune recognition to treat metastatic cancers, including melanoma, non–small cell lung cancer, urothelial cancer, and renal cell carcinoma.
Anti-PD1 targeted therapies improve survival in solid and hematologic malignancies, with a response rate as high as 40% in melanoma.2 Although these medications can prolong survival, many are associated with loss of self-tolerance and severe autoimmunelike events that can limit therapy.3 An exception is PD1-induced vitiligo, which patient 1 developed and has been associated with a better response to therapy.4
Anti-PD1–induced BP is a newly reported adverse effect. In its early stages, BP can be difficult to differentiate from eczematous or urticarial dermatitis.5-8 Discontinuation of immunotherapy has been reported in more than 70% of patients who develop BP.1 There are reports of successful treatment of BP with a course of a PD1 inhibitor,9 but 2 of our patients had severe BP that led to discontinuation of immunotherapy.
Consider Prescreening
Given that development of BP often leads to cessation of therapy, identifying patients at risk prior to starting an immune checkpoint inhibitor might have clinical utility. Biopsy with DIF is the gold standard for diagnosis, but serologic testing can be a useful adjunct because enzyme-linked immunosorbent assay for BP antigen 1 and BP antigen 2 has a reported sensitivity and specificity of 87% and 98%, respectively.10 Serologic testing prior to starting therapy with an immune checkpoint inhibitor can provide a baseline for patients. A rise in titer, in conjunction with onset of a rash, might aid in earlier diagnosis, particularly because urticarial BP can be difficult to diagnose clinically.
Further study on the utility vs cost-benefit of these screening modalities is warranted. Their predictive utility might be limited, however, and positive serologic test results might have unanticipated consequences, such as hesitation in treating patients, thus leading to a delay in therapy or access to these medications.
Conclusion
The expanding use of immune checkpoint inhibitors is increasing survival in patients with metastatic melanoma and other malignancies. Adverse effects are part of the continuum of immune system stimulation, with overstimulation resulting in dermatitis; thyroiditis; pneumonitis; and less commonly hypophysitis, vitiligo, and colitis.
Rarely, immune checkpoint inhibition induces BP. Development of BP leads to discontinuation of therapy in more than half of reported cases due to lack of adequate treatment for this skin disease and its impact on quality of life. Therefore, quick diagnosis of BP in patients on immunotherapy and successful management techniques can prevent discontinuation of these lifesaving cancer therapies. For that reason, dermatologists play an important role in the management of patients on immune checkpoint inhibitors for cancer.
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669.
- Márquez-Rodas, I, Cerezuela P, Soria A, et al. Immune checkpoint inhibitors: therapeutic advances in melanoma. Ann Transl Med. 2015;3:267.
- Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors a review. JAMA Oncol. 2016;2:1346-1353.
- Hua C, Boussemart L, Mateus C, et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol. 2016;152:45-51.
- Hwang SJE, Carlos G, Chou S, et al. Bullous pemphigoid, an autoantibody-mediated disease, is a novel immune-related adverse event in patients treated with anti-programmed cell death 1 antibodies. Melanoma Res. 2016;26:413-416.
- Damsky W, Kole L, Tomayko MM. Development of bullous pemphigoid during nivolumab therapy. JAAD Case Rep. 2016;2:442-444.
- Garje R, Chau JJ, Chung J, et al. Acute flare of bullous pemphigus with pembrolizumab used for treatment of metastatic urothelial cancer. J Immunother. 2018;41:42-44.
- Ito M, Hoashi T, Endo Y, et al. Atypical pemphigus developed in a patient with urothelial carcinoma treated with nivolumab. J Dermatol. 2019;46:e90-e92.
- Chen W-S, Tetzlaff MT, Diwan H, et al. Suprabasal acantholytic dermatologic toxicities associated checkpoint inhibitor therapy: a spectrum of immune reactions from paraneoplastic pemphigus-like to Grover-like lesions. J Cutan Pathol. 2018;45:764-773.
- Muglia C, Bronsnick T, Kirkorian AY, et al. Questioning the specificity and sensitivity of ELISA for bullous pemphigoid diagnosis. Cutis. 2017;99:E27-E30.
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669.
- Márquez-Rodas, I, Cerezuela P, Soria A, et al. Immune checkpoint inhibitors: therapeutic advances in melanoma. Ann Transl Med. 2015;3:267.
- Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors a review. JAMA Oncol. 2016;2:1346-1353.
- Hua C, Boussemart L, Mateus C, et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol. 2016;152:45-51.
- Hwang SJE, Carlos G, Chou S, et al. Bullous pemphigoid, an autoantibody-mediated disease, is a novel immune-related adverse event in patients treated with anti-programmed cell death 1 antibodies. Melanoma Res. 2016;26:413-416.
- Damsky W, Kole L, Tomayko MM. Development of bullous pemphigoid during nivolumab therapy. JAAD Case Rep. 2016;2:442-444.
- Garje R, Chau JJ, Chung J, et al. Acute flare of bullous pemphigus with pembrolizumab used for treatment of metastatic urothelial cancer. J Immunother. 2018;41:42-44.
- Ito M, Hoashi T, Endo Y, et al. Atypical pemphigus developed in a patient with urothelial carcinoma treated with nivolumab. J Dermatol. 2019;46:e90-e92.
- Chen W-S, Tetzlaff MT, Diwan H, et al. Suprabasal acantholytic dermatologic toxicities associated checkpoint inhibitor therapy: a spectrum of immune reactions from paraneoplastic pemphigus-like to Grover-like lesions. J Cutan Pathol. 2018;45:764-773.
- Muglia C, Bronsnick T, Kirkorian AY, et al. Questioning the specificity and sensitivity of ELISA for bullous pemphigoid diagnosis. Cutis. 2017;99:E27-E30.
Practice Points
- Anti–programmed cell death 1 (PD1) targeted therapies improve survival in solid and hematologic malignancies but are associated with autoimmune side effects, with bullous pemphigoid (BP) being the newest reported.
- Bullous pemphigoid can develop months into immunotherapy treatment.
- Bullous pemphigoid should be on the differential diagnosis in a patient who is on an anti-PD1 immune checkpoint inhibitor and develops 1 or more of the following: pruritus, dermatitis, and vesicles.
- Early diagnosis of BP is essential for keeping patients on immunotherapy because its severity often results in temporary or permanent discontinuation of treatment.
Pulmonary Neuroendocrine Tumor Presenting as a Left Pleural Effusion
Neuroendocrine tumors (NETs) account for about 0.5% of all newly diagnosed malignancies.1 Pulmonary NETs are rare, accounting for 1 to 2% of all invasive lung malignancies and involve about 20 to 25% of primary lung malignancies. 2,3 Their prevalence has increased by an estimated 6% per year over the past 30 years.2 Nonetheless, the time of diagnosis is frequently delayed because of nonspecific symptoms that may imitate other pulmonary conditions.
In the normal pleural space, there is a steady state in which there is a roughly equal rate of fluid formation and absorption. Any disequilibrium may produce a pleural effusion. Pleural fluids can be transudates or exudates. Transudates result from imbalances in hydrostatic and oncotic pressures in the pleural space. Exudates result primarily from pleural and/or lung inflammation or from impaired lymphatic drainage of the pleural space. Clinical manifestations include cough, wheezing, recurrent pneumonia, hemoptysis and pleural effusions. We present a case of a man who developed a large left pleural effusion with a pathology report suggesting a pulmonary NET as the etiology. Being aware of this rare entity may help improve prognosis by making an earlier diagnosis and starting treatment sooner.
Case Presentation
A 90-year-old man with a medical history of arterial hypertension, hyperlipidemia, type 2 diabetes mellitus, coronary artery disease, and vascular dementia presented to the emergency department with hypoactivity, poor appetite, productive cough, and shortness of breath. The patient was a former smoker (unknown pack-years) who quit smoking cigarettes 7 years prior. Vital signs showed sinus tachycardia and peripheral oxygen saturation of 90% at room air. The initial physical examination was remarkable for decreased breath sounds and crackles at the left lung base. Laboratory findings showed leukocytosis with neutrophilia and chronic normocytic anemia. Chest computed tomography (CT) showed a large left-sided pleural effusion occupying most of the left hemithorax with adjacent atelectatic lung, enlarged pretracheal, subcarinal, and left perihilar lymph nodes (Figure 1).
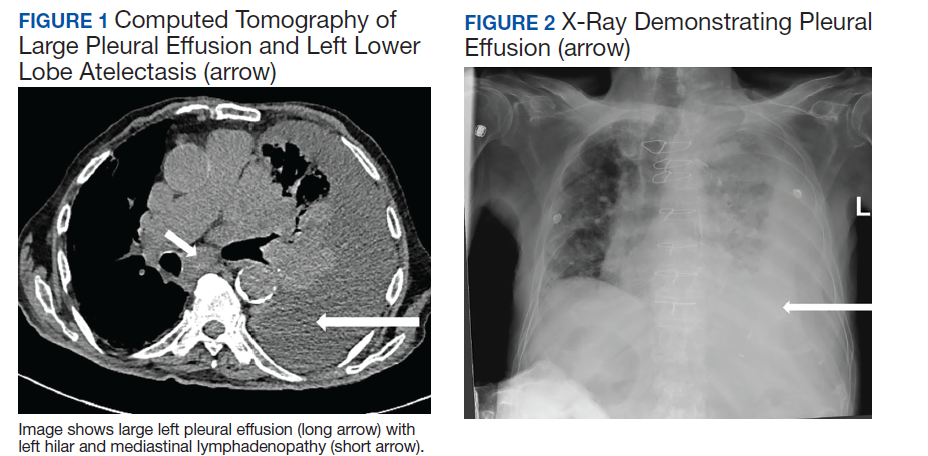
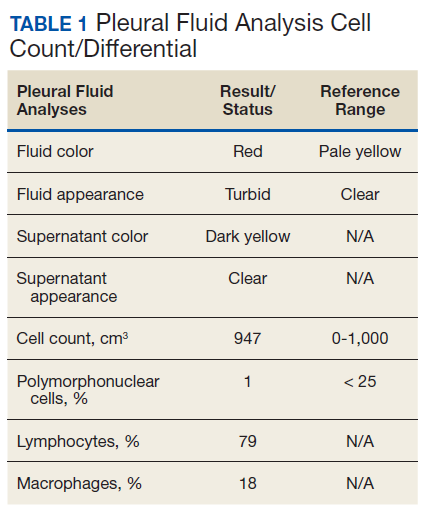
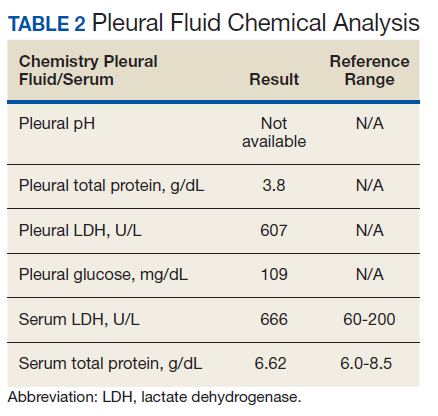
The patient was admitted to the internal medicine ward with the diagnosis of left pneumonic process and started on IV levofloxacin. However, despite 7 days of antibiotic therapy, the patient’s respiratory symptoms worsened. This clinical deterioration prompted pulmonary service consultation. Chest radiography demonstrated an enlarging left pleural effusion (Figure 2). A thoracentesis drained 1.2 L of serosanguineous pleural fluid. Pleural fluid analysis showed a cell count of 947/cm3 with 79% of lymphocytes, total protein 3.8 g/dL, lactic dehydrogenase (LDH) level 607 U/L, and glucose level 109 mg/dL. Serum total protein was 6.62 g/dL, LDH 666 U/L and glucose 92 mg/dL (Tables 1 and 2). Alanine transaminase (ALT) and aspartate aminotransferase (AST) were 11 U/L and 21 U/L, respectively. Using Light criteria, the pleural:serum protein ratio was 0.57, the pleural:serum LDH ratio was 0.91, and the pleural LDH was more than two-thirds of the serum LDH. These calculations were consistent with an exudative effusion. An infectious disease workup, including blood and pleural fluid cultures, was negative.
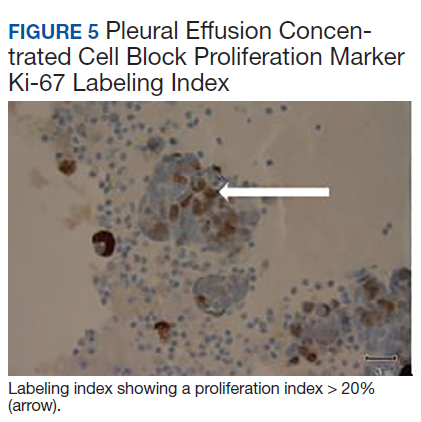
The pleural fluid concentrated cell block hematoxylin and eosin (H&E) staining showed chromatin, prominent nucleoli, and nuclear molding, which was compatible with high-grade lung NET (Figure 3). The cell block immunohistochemistry (IHC) was positive for synaptophysin, chromogranin A, and neuron specific enolase (NSE) also consistent with a high-grade pulmonary NET (Figure 4). The proliferation marker protein Ki-67 labeling index (LI) showed a proliferation index > 20% (Figure 5). The patient did not have decision-making capacity given vascular dementia. Multiple attempts to contact the next of kin or family members were unsuccessful. Risks vs benefits were evaluated, and given the patient’s advanced age and multiple comorbidities, a conservative management approach under palliative care was chosen. For this reason, further genomic studies were not done.
Discussion
NETs are a group of neoplasms that differ in site, amount of cell propagation, and clinical manifestations.4 These tumors are rare with an estimated incidence of 25 to 50 per 100,000.4 The most commonly affected organ systems are the gastroenteropancreatic and the bronchopulmonary tracts, accounting for 60% and 25% of the tumors, respectively.4 The incidence is increasing over the past years in part because of novel diagnostic techniques.

The average age of diagnosis is between the fourth and sixth decades, affecting more women than men.5 Smoking has been identified as a possible culprit for the development of these neoplasms; nonetheless, the association is still not clear.4 For example, poorly differentiated pulmonary NETs have a strong association with smoking but not well-differentiated pulmonary NETs.2
Patients typically present with cough, wheezing, hemoptysis, and recurrent pneumonias, which are in part a consequence of obstruction caused by the mass.2 Sometimes, obstruction may yield persistent pleural effusions. Hemoptysis may be seen secondary to the vascularity of pulmonary NETs.
The diagnosis is often delayed because patients are frequently treated for infection before being diagnosed with the malignancy, such as in our case. Radiologic image findings include round opacities, central masses, and atelectasis. Pulmonary NETs are frequently found incidentally as solitary lung nodules. The CT scan is the most common diagnostic modality and can provide information about the borders of the tumor, the location and surrounding structures, including the presence of atelectasis.5 Pulmonary NETs are usually centrally located in an accessible region for lung biopsy. In cases where the mass is not easily reachable, thoracentesis may provide the only available specimen.
The 2015 World Health Organization classification has identified 4 histologic types of pulmonary NETs, namely, typical carcinoid (TC), atypical carcinoid (AC), large cell neuroendocrine carcinoma (LCNEC) and small cell lung carcinoma (SCLC).6 The low-grade pulmonary NET, the typical carcinoid, is slow growing and has lower rates of metastasis. The intermediate-grade NET, the atypical carcinoid, is more aggressive. The highgrade NETs, the LCNEC and the SCLC, are aggressive and spread quickly to other places.6 Consequently, LCNEC and SCLC have higher mortalities with a 5-year survival, ranging from 13 to 57% and 5%, respectively.7
Tumors may be histomorphologically classified by H&E staining. The main characteristics that differentiate the low- and high-grade NETs are the presence of necrosis and the mitotic rate. Both categories form neuropeptides and have dense granular cores when seen with an electron microscopy.6 The TC and AC have welldefined, organized histologic patterns, no necrosis, and scarce mitosis. On the other hand, the LCNEC and SCLC are poorly differentiated tumors with necrosis, atypia, and mitosis.6 LCNEC can be separated from SCLC and other tumors by IHC staining, whereas SCLC is primarily distinguished by morphology.
If the biopsy sample size is small, then IHC morphology and markers are helpful for subclassification.8 IHC is used to discern between neuroendocrine (NE) vs non-NE. The evaluation of pleural fluid includes preparation of cell blocks. Cell block staining is deemed better for IHC because it mimics a small biopsy that enables superior stains.9 The need for a pleural biopsy in cases where the cytology is negative depends on treatment aims, the kind of tumor, and the presence of metastasis.10 In almost 80% of cases, pleural biopsy and cytology are the only specimens obtained for analysis.Therefore, identification of these markers is practical for diagnosis.10 For this reason, pleural effusion samples are appropriate options to lung biopsy for molecular studies.10
Ki-67 LI in samples has the highest specificity and sensitivity for low-tointermediate- grade vs high-grade tumors. It is being used for guiding clinical and treatment decisions.6 In SCLC, the Ki-67 LI is not necessary for diagnosis but will be about 80%.11 The tumor cells will show epithelial characteristics with positive cytokeratin AE1/AE3 and monoclonal antibody CAM5.2 and neuroendocrine markers, including NCAM/CD56, chromogranin A, and synaptophysin.11
Thyroid transcription factor-1 (TTF- 1) is positive in most cases. In LCNEC, the Ki-67 LI is between 40% and 80%. NCAM/ CD56, chromogranin A, and synaptophysin are present in 92 to 100%, 80 to 85%, and 50 to 60%, respectively.11 TTF-1 is identified in half of the tumors. All these tumors express pancytokeratin (AE1/AE3), cytokeratin 7 or low-molecular-weight cytokeratin. Likewise, the carcinoids will show markers, such as chromogranin A, synaptophysin, CD56, and epithelial markers like pancytokeratin.11 However, the high-molecular-weight cytokeratin and TTF-1 are negative. Furthermore, NSE is considered a good tumor marker in the diagnosis and prognosis of SCLC. NSE also has been reported in NSCLC. The level of NSE correlates with tumor burden, number of metastatic sites, and response to treatment. 12 A potentially useful marker is the insulinoma-associated protein 1, which is a nuclear determinant of NE differentiation that stains all types of pulmonary NETs irrespective of the histology but does not stain adenocarcinoma or squamous cell carcinoma (SCC).6
Recently, genomic studies have identified gene alterations that have become standard of care for diagnosis and targeted therapies.8 For example, epidermal growth factor receptor (EGFR) and echinoderm microtubule- associated proteinlike 4, and anaplastic lymphoma kinase (EML4-ALK) mutations have been found in about 25% of lung adenocarcinomas. 8 Other abnormalities in LKB1/STK11, NF1, CDKN2A, SMARCA4 and KEAP1, KRAS, MET, ROS1, and RET have also been identified.8 On the other hand, SCC rarely have derangements in EGFR and EML4-ALK, but do show changes in RTKs, DDR2M, FGGRs, among others.8 In TC and AC, observed molecular alterations include MEN1 mutations, mTOR, and SSTRs pathway activation, and GC/ CEACAM1 and CD44/OTP expression.13 LCNEC and SCLC have shown TP53 and RB1 mutations and CDX2/VIL1/BAI3 expression. DLL3 expression and MET mutations may be present in SCLC.13 Last, chromatin remodeling gene mutations have been identified in all these lung NET types.13
Furthermore, neuropeptides and neuroamines may be measured in the blood and urine.14 Pulmonary NETs may be functional and secrete these substances, leading to systemic symptoms based on the released molecules.15 However, pulmonary NETs produce less serotonin than gastrointestinal NETs; therefore, carcinoid syndrome is less frequent in pulmonary NETs.16 Liver metastasis is often present when it occurs.5 Other possible clinical features include Cushing syndrome and acromegaly depending on the secreted hormones.5
In a recent metanalysis, serum LDH has been found to have a prognostic role in Ewing sarcoma, urologic cancers, malignant mesothelioma, among others.17 It demonstrated that a higher LDH concentration is associated with worse survival in patients with lung cancer.17 Serum LDH is an enzyme that catalyzes the reaction between lactic acid and pyruvic acid that typically takes place in anaerobic conditions.17 LDH levels are elevated in malignancies because tumors have an anaerobic environment. Elevated LDH levels correlate with the anaerobic metabolism in the tumor. Other studies also have noted that patients with high metastatic score have higher LDH levels.17 Therefore, LDH may reflect tumor extension.
In addition, other techniques, such as somatostatin- receptor imaging are specifically beneficial in tumors that express the somatostatin receptor.16 For this reason, this type of study is typically indicated in patients with known metastasis, not in patients with low-grade tumors. Abdominal CT scans are done because the liver is a common site for metastasis.
Our case report demonstrates how biomarkers help diagnose these potentially aggressive and life-threatening tumors that may present as a common condition such as a pleural effusion. Using a less invasive and quicker approach with thoracentesis rather than with lung biopsies is a diagnostic tool in this entity. IHC in cell blocks is a reasonable diagnostic method especially in patients in whom performing a lung biopsy is difficult.
Conclusions
The presence of a symptomatic and recurrent unilateral pleural effusion must urge physicians to consider thoracentesis with mindful use of biomarkers not only for therapeutic purposes, but also for diagnosis of a variety of etiologies, both benign and malignant.
1. Oronsky B, Ma PC, Morgensztern D, Carter CA. Nothing but NET: a review of neuroendocrine tumors and carcinomas. Neoplasia. 2017;19(12):991-1002. doi: 10.1016/j.neo.2017.09.002
2. Hendifar AE, Marchevsky AM, Tuli R. Neuroendocrine tumors of the lung: current challenges and advances in the diagnosis and management of well-differentiated disease. J Thorac Oncol. 2017;12(3):425-436. doi: 10.1016/j.jtho.2016.11.2222
3. Fisseler-Eckhoff A, Demes M. Neuroendocrine tumors of the lung. Cancers (Basel). 2012;4(3):777-798. doi: 10.3390/cancers4030777
4. Mandegaran R, David S, Screaton N. Cardiothoracic manifestations of neuroendocrine tumours. Br J Radiol. 2016;89(1060). doi: 10.1259/bjr.20150787
5. Caplin ME, Baudin E, Ferolla P, et al; ENETS consensus conference participants. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol. 2015;26(8):1604-1620. doi: 10.1093/annonc/mdv041
6. Pelosi G, Sonzogni A, Harari S, et al. Classification of pulmonary neuroendocrine tumors: new insights. Transl Lung Cancer Res. 2017;6(5):513-529. doi: 10.21037/tlcr.2017.09.04
7. Rossi G, Bertero L, Marchiò C, Papotti M. Molecular alterations of neuroendocrine tumours of the lung. Histopathology. 2018;72(1):142-152. doi: 10.1111/his.13394.
8. Osmani L, Askin F, Gabrielson E, Li QK. Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): moving from targeted therapy to immunotherapy. Semin Cancer Biol. 2018;52(pt 1):103-109. doi: 10.1016/j.semcancer.2017.11.019
9. Kaur G, Nijhawan R, Gupta N, Singh N, Rajwanshi A. Pleural fluid cytology samples in cases of suspected lung cancer: an experience from a tertiary care centre. Diagn Cytopathol. 2017;45(3):195-201.
10. Porcel JM. Biomarkers in the diagnosis of pleural diseases: a 2018 update. Ther Adv Respir Dis. 2018;12. doi: 10.1177/1753466618808660
11. Kim JY, Hong SM, Ro JY. Recent updates on grading and classification of neuroendocrine tumors. Ann Diagn Pathol. 2017;29:11-16. doi: 10.1016/j.anndiagpath.2017.04.005
12. Isgrò MA, Bottoni P, Scatena R. Neuron-specific enolase as a biomarker: biochemical and clinical aspects. Adv Exp Med Biol. 2015;867:125-143. doi: 10.1007/978-94-017-7215-0_9
13. Rossi G, Bertero L, Marchiò C, Papotti M. Molecular alterations of neuroendocrine tumours of the lung. Histopathology. 2018;72(1):142-152. doi: 10.1111/his.13394
14. Eriksson B, Oberg K, Stridsberg M. Tumor markers in neuroendocrine tumors. Digestion. 2000;62(suppl 1):33-38.
15. Melosky B. Low grade neuroendocrine tumors of the lung. Front Oncol. 2017;7:119. doi: 10.3389/fonc.2017.00119
16. Gustafsson BI, Kidd M, Chan A, Malfertheiner MV, Modlin IM. Bronchopulmonary neuroendocrine tumors. Cancer. 2001;113(1):5-21. https://doi.org/10.1002/cncr.23542
17. Deng T, Zhang J, Meng Y, Zhou Y, Li W. Higher pretreatment lactate dehydrogenase concentration predicts worse overall survival in patients with lung cancer. Medicine (Baltimore). 2018;97(38):e12524
Neuroendocrine tumors (NETs) account for about 0.5% of all newly diagnosed malignancies.1 Pulmonary NETs are rare, accounting for 1 to 2% of all invasive lung malignancies and involve about 20 to 25% of primary lung malignancies. 2,3 Their prevalence has increased by an estimated 6% per year over the past 30 years.2 Nonetheless, the time of diagnosis is frequently delayed because of nonspecific symptoms that may imitate other pulmonary conditions.
In the normal pleural space, there is a steady state in which there is a roughly equal rate of fluid formation and absorption. Any disequilibrium may produce a pleural effusion. Pleural fluids can be transudates or exudates. Transudates result from imbalances in hydrostatic and oncotic pressures in the pleural space. Exudates result primarily from pleural and/or lung inflammation or from impaired lymphatic drainage of the pleural space. Clinical manifestations include cough, wheezing, recurrent pneumonia, hemoptysis and pleural effusions. We present a case of a man who developed a large left pleural effusion with a pathology report suggesting a pulmonary NET as the etiology. Being aware of this rare entity may help improve prognosis by making an earlier diagnosis and starting treatment sooner.
Case Presentation
A 90-year-old man with a medical history of arterial hypertension, hyperlipidemia, type 2 diabetes mellitus, coronary artery disease, and vascular dementia presented to the emergency department with hypoactivity, poor appetite, productive cough, and shortness of breath. The patient was a former smoker (unknown pack-years) who quit smoking cigarettes 7 years prior. Vital signs showed sinus tachycardia and peripheral oxygen saturation of 90% at room air. The initial physical examination was remarkable for decreased breath sounds and crackles at the left lung base. Laboratory findings showed leukocytosis with neutrophilia and chronic normocytic anemia. Chest computed tomography (CT) showed a large left-sided pleural effusion occupying most of the left hemithorax with adjacent atelectatic lung, enlarged pretracheal, subcarinal, and left perihilar lymph nodes (Figure 1).
The patient was admitted to the internal medicine ward with the diagnosis of left pneumonic process and started on IV levofloxacin. However, despite 7 days of antibiotic therapy, the patient’s respiratory symptoms worsened. This clinical deterioration prompted pulmonary service consultation. Chest radiography demonstrated an enlarging left pleural effusion (Figure 2). A thoracentesis drained 1.2 L of serosanguineous pleural fluid. Pleural fluid analysis showed a cell count of 947/cm3 with 79% of lymphocytes, total protein 3.8 g/dL, lactic dehydrogenase (LDH) level 607 U/L, and glucose level 109 mg/dL. Serum total protein was 6.62 g/dL, LDH 666 U/L and glucose 92 mg/dL (Tables 1 and 2). Alanine transaminase (ALT) and aspartate aminotransferase (AST) were 11 U/L and 21 U/L, respectively. Using Light criteria, the pleural:serum protein ratio was 0.57, the pleural:serum LDH ratio was 0.91, and the pleural LDH was more than two-thirds of the serum LDH. These calculations were consistent with an exudative effusion. An infectious disease workup, including blood and pleural fluid cultures, was negative.
The pleural fluid concentrated cell block hematoxylin and eosin (H&E) staining showed chromatin, prominent nucleoli, and nuclear molding, which was compatible with high-grade lung NET (Figure 3). The cell block immunohistochemistry (IHC) was positive for synaptophysin, chromogranin A, and neuron specific enolase (NSE) also consistent with a high-grade pulmonary NET (Figure 4). The proliferation marker protein Ki-67 labeling index (LI) showed a proliferation index > 20% (Figure 5). The patient did not have decision-making capacity given vascular dementia. Multiple attempts to contact the next of kin or family members were unsuccessful. Risks vs benefits were evaluated, and given the patient’s advanced age and multiple comorbidities, a conservative management approach under palliative care was chosen. For this reason, further genomic studies were not done.
Discussion
NETs are a group of neoplasms that differ in site, amount of cell propagation, and clinical manifestations.4 These tumors are rare with an estimated incidence of 25 to 50 per 100,000.4 The most commonly affected organ systems are the gastroenteropancreatic and the bronchopulmonary tracts, accounting for 60% and 25% of the tumors, respectively.4 The incidence is increasing over the past years in part because of novel diagnostic techniques.
The average age of diagnosis is between the fourth and sixth decades, affecting more women than men.5 Smoking has been identified as a possible culprit for the development of these neoplasms; nonetheless, the association is still not clear.4 For example, poorly differentiated pulmonary NETs have a strong association with smoking but not well-differentiated pulmonary NETs.2
Patients typically present with cough, wheezing, hemoptysis, and recurrent pneumonias, which are in part a consequence of obstruction caused by the mass.2 Sometimes, obstruction may yield persistent pleural effusions. Hemoptysis may be seen secondary to the vascularity of pulmonary NETs.
The diagnosis is often delayed because patients are frequently treated for infection before being diagnosed with the malignancy, such as in our case. Radiologic image findings include round opacities, central masses, and atelectasis. Pulmonary NETs are frequently found incidentally as solitary lung nodules. The CT scan is the most common diagnostic modality and can provide information about the borders of the tumor, the location and surrounding structures, including the presence of atelectasis.5 Pulmonary NETs are usually centrally located in an accessible region for lung biopsy. In cases where the mass is not easily reachable, thoracentesis may provide the only available specimen.
The 2015 World Health Organization classification has identified 4 histologic types of pulmonary NETs, namely, typical carcinoid (TC), atypical carcinoid (AC), large cell neuroendocrine carcinoma (LCNEC) and small cell lung carcinoma (SCLC).6 The low-grade pulmonary NET, the typical carcinoid, is slow growing and has lower rates of metastasis. The intermediate-grade NET, the atypical carcinoid, is more aggressive. The highgrade NETs, the LCNEC and the SCLC, are aggressive and spread quickly to other places.6 Consequently, LCNEC and SCLC have higher mortalities with a 5-year survival, ranging from 13 to 57% and 5%, respectively.7
Tumors may be histomorphologically classified by H&E staining. The main characteristics that differentiate the low- and high-grade NETs are the presence of necrosis and the mitotic rate. Both categories form neuropeptides and have dense granular cores when seen with an electron microscopy.6 The TC and AC have welldefined, organized histologic patterns, no necrosis, and scarce mitosis. On the other hand, the LCNEC and SCLC are poorly differentiated tumors with necrosis, atypia, and mitosis.6 LCNEC can be separated from SCLC and other tumors by IHC staining, whereas SCLC is primarily distinguished by morphology.
If the biopsy sample size is small, then IHC morphology and markers are helpful for subclassification.8 IHC is used to discern between neuroendocrine (NE) vs non-NE. The evaluation of pleural fluid includes preparation of cell blocks. Cell block staining is deemed better for IHC because it mimics a small biopsy that enables superior stains.9 The need for a pleural biopsy in cases where the cytology is negative depends on treatment aims, the kind of tumor, and the presence of metastasis.10 In almost 80% of cases, pleural biopsy and cytology are the only specimens obtained for analysis.Therefore, identification of these markers is practical for diagnosis.10 For this reason, pleural effusion samples are appropriate options to lung biopsy for molecular studies.10
Ki-67 LI in samples has the highest specificity and sensitivity for low-tointermediate- grade vs high-grade tumors. It is being used for guiding clinical and treatment decisions.6 In SCLC, the Ki-67 LI is not necessary for diagnosis but will be about 80%.11 The tumor cells will show epithelial characteristics with positive cytokeratin AE1/AE3 and monoclonal antibody CAM5.2 and neuroendocrine markers, including NCAM/CD56, chromogranin A, and synaptophysin.11
Thyroid transcription factor-1 (TTF- 1) is positive in most cases. In LCNEC, the Ki-67 LI is between 40% and 80%. NCAM/ CD56, chromogranin A, and synaptophysin are present in 92 to 100%, 80 to 85%, and 50 to 60%, respectively.11 TTF-1 is identified in half of the tumors. All these tumors express pancytokeratin (AE1/AE3), cytokeratin 7 or low-molecular-weight cytokeratin. Likewise, the carcinoids will show markers, such as chromogranin A, synaptophysin, CD56, and epithelial markers like pancytokeratin.11 However, the high-molecular-weight cytokeratin and TTF-1 are negative. Furthermore, NSE is considered a good tumor marker in the diagnosis and prognosis of SCLC. NSE also has been reported in NSCLC. The level of NSE correlates with tumor burden, number of metastatic sites, and response to treatment. 12 A potentially useful marker is the insulinoma-associated protein 1, which is a nuclear determinant of NE differentiation that stains all types of pulmonary NETs irrespective of the histology but does not stain adenocarcinoma or squamous cell carcinoma (SCC).6
Recently, genomic studies have identified gene alterations that have become standard of care for diagnosis and targeted therapies.8 For example, epidermal growth factor receptor (EGFR) and echinoderm microtubule- associated proteinlike 4, and anaplastic lymphoma kinase (EML4-ALK) mutations have been found in about 25% of lung adenocarcinomas. 8 Other abnormalities in LKB1/STK11, NF1, CDKN2A, SMARCA4 and KEAP1, KRAS, MET, ROS1, and RET have also been identified.8 On the other hand, SCC rarely have derangements in EGFR and EML4-ALK, but do show changes in RTKs, DDR2M, FGGRs, among others.8 In TC and AC, observed molecular alterations include MEN1 mutations, mTOR, and SSTRs pathway activation, and GC/ CEACAM1 and CD44/OTP expression.13 LCNEC and SCLC have shown TP53 and RB1 mutations and CDX2/VIL1/BAI3 expression. DLL3 expression and MET mutations may be present in SCLC.13 Last, chromatin remodeling gene mutations have been identified in all these lung NET types.13
Furthermore, neuropeptides and neuroamines may be measured in the blood and urine.14 Pulmonary NETs may be functional and secrete these substances, leading to systemic symptoms based on the released molecules.15 However, pulmonary NETs produce less serotonin than gastrointestinal NETs; therefore, carcinoid syndrome is less frequent in pulmonary NETs.16 Liver metastasis is often present when it occurs.5 Other possible clinical features include Cushing syndrome and acromegaly depending on the secreted hormones.5
In a recent metanalysis, serum LDH has been found to have a prognostic role in Ewing sarcoma, urologic cancers, malignant mesothelioma, among others.17 It demonstrated that a higher LDH concentration is associated with worse survival in patients with lung cancer.17 Serum LDH is an enzyme that catalyzes the reaction between lactic acid and pyruvic acid that typically takes place in anaerobic conditions.17 LDH levels are elevated in malignancies because tumors have an anaerobic environment. Elevated LDH levels correlate with the anaerobic metabolism in the tumor. Other studies also have noted that patients with high metastatic score have higher LDH levels.17 Therefore, LDH may reflect tumor extension.
In addition, other techniques, such as somatostatin- receptor imaging are specifically beneficial in tumors that express the somatostatin receptor.16 For this reason, this type of study is typically indicated in patients with known metastasis, not in patients with low-grade tumors. Abdominal CT scans are done because the liver is a common site for metastasis.
Our case report demonstrates how biomarkers help diagnose these potentially aggressive and life-threatening tumors that may present as a common condition such as a pleural effusion. Using a less invasive and quicker approach with thoracentesis rather than with lung biopsies is a diagnostic tool in this entity. IHC in cell blocks is a reasonable diagnostic method especially in patients in whom performing a lung biopsy is difficult.
Conclusions
The presence of a symptomatic and recurrent unilateral pleural effusion must urge physicians to consider thoracentesis with mindful use of biomarkers not only for therapeutic purposes, but also for diagnosis of a variety of etiologies, both benign and malignant.
Neuroendocrine tumors (NETs) account for about 0.5% of all newly diagnosed malignancies.1 Pulmonary NETs are rare, accounting for 1 to 2% of all invasive lung malignancies and involve about 20 to 25% of primary lung malignancies. 2,3 Their prevalence has increased by an estimated 6% per year over the past 30 years.2 Nonetheless, the time of diagnosis is frequently delayed because of nonspecific symptoms that may imitate other pulmonary conditions.
In the normal pleural space, there is a steady state in which there is a roughly equal rate of fluid formation and absorption. Any disequilibrium may produce a pleural effusion. Pleural fluids can be transudates or exudates. Transudates result from imbalances in hydrostatic and oncotic pressures in the pleural space. Exudates result primarily from pleural and/or lung inflammation or from impaired lymphatic drainage of the pleural space. Clinical manifestations include cough, wheezing, recurrent pneumonia, hemoptysis and pleural effusions. We present a case of a man who developed a large left pleural effusion with a pathology report suggesting a pulmonary NET as the etiology. Being aware of this rare entity may help improve prognosis by making an earlier diagnosis and starting treatment sooner.
Case Presentation
A 90-year-old man with a medical history of arterial hypertension, hyperlipidemia, type 2 diabetes mellitus, coronary artery disease, and vascular dementia presented to the emergency department with hypoactivity, poor appetite, productive cough, and shortness of breath. The patient was a former smoker (unknown pack-years) who quit smoking cigarettes 7 years prior. Vital signs showed sinus tachycardia and peripheral oxygen saturation of 90% at room air. The initial physical examination was remarkable for decreased breath sounds and crackles at the left lung base. Laboratory findings showed leukocytosis with neutrophilia and chronic normocytic anemia. Chest computed tomography (CT) showed a large left-sided pleural effusion occupying most of the left hemithorax with adjacent atelectatic lung, enlarged pretracheal, subcarinal, and left perihilar lymph nodes (Figure 1).
The patient was admitted to the internal medicine ward with the diagnosis of left pneumonic process and started on IV levofloxacin. However, despite 7 days of antibiotic therapy, the patient’s respiratory symptoms worsened. This clinical deterioration prompted pulmonary service consultation. Chest radiography demonstrated an enlarging left pleural effusion (Figure 2). A thoracentesis drained 1.2 L of serosanguineous pleural fluid. Pleural fluid analysis showed a cell count of 947/cm3 with 79% of lymphocytes, total protein 3.8 g/dL, lactic dehydrogenase (LDH) level 607 U/L, and glucose level 109 mg/dL. Serum total protein was 6.62 g/dL, LDH 666 U/L and glucose 92 mg/dL (Tables 1 and 2). Alanine transaminase (ALT) and aspartate aminotransferase (AST) were 11 U/L and 21 U/L, respectively. Using Light criteria, the pleural:serum protein ratio was 0.57, the pleural:serum LDH ratio was 0.91, and the pleural LDH was more than two-thirds of the serum LDH. These calculations were consistent with an exudative effusion. An infectious disease workup, including blood and pleural fluid cultures, was negative.
The pleural fluid concentrated cell block hematoxylin and eosin (H&E) staining showed chromatin, prominent nucleoli, and nuclear molding, which was compatible with high-grade lung NET (Figure 3). The cell block immunohistochemistry (IHC) was positive for synaptophysin, chromogranin A, and neuron specific enolase (NSE) also consistent with a high-grade pulmonary NET (Figure 4). The proliferation marker protein Ki-67 labeling index (LI) showed a proliferation index > 20% (Figure 5). The patient did not have decision-making capacity given vascular dementia. Multiple attempts to contact the next of kin or family members were unsuccessful. Risks vs benefits were evaluated, and given the patient’s advanced age and multiple comorbidities, a conservative management approach under palliative care was chosen. For this reason, further genomic studies were not done.
Discussion
NETs are a group of neoplasms that differ in site, amount of cell propagation, and clinical manifestations.4 These tumors are rare with an estimated incidence of 25 to 50 per 100,000.4 The most commonly affected organ systems are the gastroenteropancreatic and the bronchopulmonary tracts, accounting for 60% and 25% of the tumors, respectively.4 The incidence is increasing over the past years in part because of novel diagnostic techniques.
The average age of diagnosis is between the fourth and sixth decades, affecting more women than men.5 Smoking has been identified as a possible culprit for the development of these neoplasms; nonetheless, the association is still not clear.4 For example, poorly differentiated pulmonary NETs have a strong association with smoking but not well-differentiated pulmonary NETs.2
Patients typically present with cough, wheezing, hemoptysis, and recurrent pneumonias, which are in part a consequence of obstruction caused by the mass.2 Sometimes, obstruction may yield persistent pleural effusions. Hemoptysis may be seen secondary to the vascularity of pulmonary NETs.
The diagnosis is often delayed because patients are frequently treated for infection before being diagnosed with the malignancy, such as in our case. Radiologic image findings include round opacities, central masses, and atelectasis. Pulmonary NETs are frequently found incidentally as solitary lung nodules. The CT scan is the most common diagnostic modality and can provide information about the borders of the tumor, the location and surrounding structures, including the presence of atelectasis.5 Pulmonary NETs are usually centrally located in an accessible region for lung biopsy. In cases where the mass is not easily reachable, thoracentesis may provide the only available specimen.
The 2015 World Health Organization classification has identified 4 histologic types of pulmonary NETs, namely, typical carcinoid (TC), atypical carcinoid (AC), large cell neuroendocrine carcinoma (LCNEC) and small cell lung carcinoma (SCLC).6 The low-grade pulmonary NET, the typical carcinoid, is slow growing and has lower rates of metastasis. The intermediate-grade NET, the atypical carcinoid, is more aggressive. The highgrade NETs, the LCNEC and the SCLC, are aggressive and spread quickly to other places.6 Consequently, LCNEC and SCLC have higher mortalities with a 5-year survival, ranging from 13 to 57% and 5%, respectively.7
Tumors may be histomorphologically classified by H&E staining. The main characteristics that differentiate the low- and high-grade NETs are the presence of necrosis and the mitotic rate. Both categories form neuropeptides and have dense granular cores when seen with an electron microscopy.6 The TC and AC have welldefined, organized histologic patterns, no necrosis, and scarce mitosis. On the other hand, the LCNEC and SCLC are poorly differentiated tumors with necrosis, atypia, and mitosis.6 LCNEC can be separated from SCLC and other tumors by IHC staining, whereas SCLC is primarily distinguished by morphology.
If the biopsy sample size is small, then IHC morphology and markers are helpful for subclassification.8 IHC is used to discern between neuroendocrine (NE) vs non-NE. The evaluation of pleural fluid includes preparation of cell blocks. Cell block staining is deemed better for IHC because it mimics a small biopsy that enables superior stains.9 The need for a pleural biopsy in cases where the cytology is negative depends on treatment aims, the kind of tumor, and the presence of metastasis.10 In almost 80% of cases, pleural biopsy and cytology are the only specimens obtained for analysis.Therefore, identification of these markers is practical for diagnosis.10 For this reason, pleural effusion samples are appropriate options to lung biopsy for molecular studies.10
Ki-67 LI in samples has the highest specificity and sensitivity for low-tointermediate- grade vs high-grade tumors. It is being used for guiding clinical and treatment decisions.6 In SCLC, the Ki-67 LI is not necessary for diagnosis but will be about 80%.11 The tumor cells will show epithelial characteristics with positive cytokeratin AE1/AE3 and monoclonal antibody CAM5.2 and neuroendocrine markers, including NCAM/CD56, chromogranin A, and synaptophysin.11
Thyroid transcription factor-1 (TTF- 1) is positive in most cases. In LCNEC, the Ki-67 LI is between 40% and 80%. NCAM/ CD56, chromogranin A, and synaptophysin are present in 92 to 100%, 80 to 85%, and 50 to 60%, respectively.11 TTF-1 is identified in half of the tumors. All these tumors express pancytokeratin (AE1/AE3), cytokeratin 7 or low-molecular-weight cytokeratin. Likewise, the carcinoids will show markers, such as chromogranin A, synaptophysin, CD56, and epithelial markers like pancytokeratin.11 However, the high-molecular-weight cytokeratin and TTF-1 are negative. Furthermore, NSE is considered a good tumor marker in the diagnosis and prognosis of SCLC. NSE also has been reported in NSCLC. The level of NSE correlates with tumor burden, number of metastatic sites, and response to treatment. 12 A potentially useful marker is the insulinoma-associated protein 1, which is a nuclear determinant of NE differentiation that stains all types of pulmonary NETs irrespective of the histology but does not stain adenocarcinoma or squamous cell carcinoma (SCC).6
Recently, genomic studies have identified gene alterations that have become standard of care for diagnosis and targeted therapies.8 For example, epidermal growth factor receptor (EGFR) and echinoderm microtubule- associated proteinlike 4, and anaplastic lymphoma kinase (EML4-ALK) mutations have been found in about 25% of lung adenocarcinomas. 8 Other abnormalities in LKB1/STK11, NF1, CDKN2A, SMARCA4 and KEAP1, KRAS, MET, ROS1, and RET have also been identified.8 On the other hand, SCC rarely have derangements in EGFR and EML4-ALK, but do show changes in RTKs, DDR2M, FGGRs, among others.8 In TC and AC, observed molecular alterations include MEN1 mutations, mTOR, and SSTRs pathway activation, and GC/ CEACAM1 and CD44/OTP expression.13 LCNEC and SCLC have shown TP53 and RB1 mutations and CDX2/VIL1/BAI3 expression. DLL3 expression and MET mutations may be present in SCLC.13 Last, chromatin remodeling gene mutations have been identified in all these lung NET types.13
Furthermore, neuropeptides and neuroamines may be measured in the blood and urine.14 Pulmonary NETs may be functional and secrete these substances, leading to systemic symptoms based on the released molecules.15 However, pulmonary NETs produce less serotonin than gastrointestinal NETs; therefore, carcinoid syndrome is less frequent in pulmonary NETs.16 Liver metastasis is often present when it occurs.5 Other possible clinical features include Cushing syndrome and acromegaly depending on the secreted hormones.5
In a recent metanalysis, serum LDH has been found to have a prognostic role in Ewing sarcoma, urologic cancers, malignant mesothelioma, among others.17 It demonstrated that a higher LDH concentration is associated with worse survival in patients with lung cancer.17 Serum LDH is an enzyme that catalyzes the reaction between lactic acid and pyruvic acid that typically takes place in anaerobic conditions.17 LDH levels are elevated in malignancies because tumors have an anaerobic environment. Elevated LDH levels correlate with the anaerobic metabolism in the tumor. Other studies also have noted that patients with high metastatic score have higher LDH levels.17 Therefore, LDH may reflect tumor extension.
In addition, other techniques, such as somatostatin- receptor imaging are specifically beneficial in tumors that express the somatostatin receptor.16 For this reason, this type of study is typically indicated in patients with known metastasis, not in patients with low-grade tumors. Abdominal CT scans are done because the liver is a common site for metastasis.
Our case report demonstrates how biomarkers help diagnose these potentially aggressive and life-threatening tumors that may present as a common condition such as a pleural effusion. Using a less invasive and quicker approach with thoracentesis rather than with lung biopsies is a diagnostic tool in this entity. IHC in cell blocks is a reasonable diagnostic method especially in patients in whom performing a lung biopsy is difficult.
Conclusions
The presence of a symptomatic and recurrent unilateral pleural effusion must urge physicians to consider thoracentesis with mindful use of biomarkers not only for therapeutic purposes, but also for diagnosis of a variety of etiologies, both benign and malignant.
1. Oronsky B, Ma PC, Morgensztern D, Carter CA. Nothing but NET: a review of neuroendocrine tumors and carcinomas. Neoplasia. 2017;19(12):991-1002. doi: 10.1016/j.neo.2017.09.002
2. Hendifar AE, Marchevsky AM, Tuli R. Neuroendocrine tumors of the lung: current challenges and advances in the diagnosis and management of well-differentiated disease. J Thorac Oncol. 2017;12(3):425-436. doi: 10.1016/j.jtho.2016.11.2222
3. Fisseler-Eckhoff A, Demes M. Neuroendocrine tumors of the lung. Cancers (Basel). 2012;4(3):777-798. doi: 10.3390/cancers4030777
4. Mandegaran R, David S, Screaton N. Cardiothoracic manifestations of neuroendocrine tumours. Br J Radiol. 2016;89(1060). doi: 10.1259/bjr.20150787
5. Caplin ME, Baudin E, Ferolla P, et al; ENETS consensus conference participants. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol. 2015;26(8):1604-1620. doi: 10.1093/annonc/mdv041
6. Pelosi G, Sonzogni A, Harari S, et al. Classification of pulmonary neuroendocrine tumors: new insights. Transl Lung Cancer Res. 2017;6(5):513-529. doi: 10.21037/tlcr.2017.09.04
7. Rossi G, Bertero L, Marchiò C, Papotti M. Molecular alterations of neuroendocrine tumours of the lung. Histopathology. 2018;72(1):142-152. doi: 10.1111/his.13394.
8. Osmani L, Askin F, Gabrielson E, Li QK. Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): moving from targeted therapy to immunotherapy. Semin Cancer Biol. 2018;52(pt 1):103-109. doi: 10.1016/j.semcancer.2017.11.019
9. Kaur G, Nijhawan R, Gupta N, Singh N, Rajwanshi A. Pleural fluid cytology samples in cases of suspected lung cancer: an experience from a tertiary care centre. Diagn Cytopathol. 2017;45(3):195-201.
10. Porcel JM. Biomarkers in the diagnosis of pleural diseases: a 2018 update. Ther Adv Respir Dis. 2018;12. doi: 10.1177/1753466618808660
11. Kim JY, Hong SM, Ro JY. Recent updates on grading and classification of neuroendocrine tumors. Ann Diagn Pathol. 2017;29:11-16. doi: 10.1016/j.anndiagpath.2017.04.005
12. Isgrò MA, Bottoni P, Scatena R. Neuron-specific enolase as a biomarker: biochemical and clinical aspects. Adv Exp Med Biol. 2015;867:125-143. doi: 10.1007/978-94-017-7215-0_9
13. Rossi G, Bertero L, Marchiò C, Papotti M. Molecular alterations of neuroendocrine tumours of the lung. Histopathology. 2018;72(1):142-152. doi: 10.1111/his.13394
14. Eriksson B, Oberg K, Stridsberg M. Tumor markers in neuroendocrine tumors. Digestion. 2000;62(suppl 1):33-38.
15. Melosky B. Low grade neuroendocrine tumors of the lung. Front Oncol. 2017;7:119. doi: 10.3389/fonc.2017.00119
16. Gustafsson BI, Kidd M, Chan A, Malfertheiner MV, Modlin IM. Bronchopulmonary neuroendocrine tumors. Cancer. 2001;113(1):5-21. https://doi.org/10.1002/cncr.23542
17. Deng T, Zhang J, Meng Y, Zhou Y, Li W. Higher pretreatment lactate dehydrogenase concentration predicts worse overall survival in patients with lung cancer. Medicine (Baltimore). 2018;97(38):e12524
1. Oronsky B, Ma PC, Morgensztern D, Carter CA. Nothing but NET: a review of neuroendocrine tumors and carcinomas. Neoplasia. 2017;19(12):991-1002. doi: 10.1016/j.neo.2017.09.002
2. Hendifar AE, Marchevsky AM, Tuli R. Neuroendocrine tumors of the lung: current challenges and advances in the diagnosis and management of well-differentiated disease. J Thorac Oncol. 2017;12(3):425-436. doi: 10.1016/j.jtho.2016.11.2222
3. Fisseler-Eckhoff A, Demes M. Neuroendocrine tumors of the lung. Cancers (Basel). 2012;4(3):777-798. doi: 10.3390/cancers4030777
4. Mandegaran R, David S, Screaton N. Cardiothoracic manifestations of neuroendocrine tumours. Br J Radiol. 2016;89(1060). doi: 10.1259/bjr.20150787
5. Caplin ME, Baudin E, Ferolla P, et al; ENETS consensus conference participants. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol. 2015;26(8):1604-1620. doi: 10.1093/annonc/mdv041
6. Pelosi G, Sonzogni A, Harari S, et al. Classification of pulmonary neuroendocrine tumors: new insights. Transl Lung Cancer Res. 2017;6(5):513-529. doi: 10.21037/tlcr.2017.09.04
7. Rossi G, Bertero L, Marchiò C, Papotti M. Molecular alterations of neuroendocrine tumours of the lung. Histopathology. 2018;72(1):142-152. doi: 10.1111/his.13394.
8. Osmani L, Askin F, Gabrielson E, Li QK. Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): moving from targeted therapy to immunotherapy. Semin Cancer Biol. 2018;52(pt 1):103-109. doi: 10.1016/j.semcancer.2017.11.019
9. Kaur G, Nijhawan R, Gupta N, Singh N, Rajwanshi A. Pleural fluid cytology samples in cases of suspected lung cancer: an experience from a tertiary care centre. Diagn Cytopathol. 2017;45(3):195-201.
10. Porcel JM. Biomarkers in the diagnosis of pleural diseases: a 2018 update. Ther Adv Respir Dis. 2018;12. doi: 10.1177/1753466618808660
11. Kim JY, Hong SM, Ro JY. Recent updates on grading and classification of neuroendocrine tumors. Ann Diagn Pathol. 2017;29:11-16. doi: 10.1016/j.anndiagpath.2017.04.005
12. Isgrò MA, Bottoni P, Scatena R. Neuron-specific enolase as a biomarker: biochemical and clinical aspects. Adv Exp Med Biol. 2015;867:125-143. doi: 10.1007/978-94-017-7215-0_9
13. Rossi G, Bertero L, Marchiò C, Papotti M. Molecular alterations of neuroendocrine tumours of the lung. Histopathology. 2018;72(1):142-152. doi: 10.1111/his.13394
14. Eriksson B, Oberg K, Stridsberg M. Tumor markers in neuroendocrine tumors. Digestion. 2000;62(suppl 1):33-38.
15. Melosky B. Low grade neuroendocrine tumors of the lung. Front Oncol. 2017;7:119. doi: 10.3389/fonc.2017.00119
16. Gustafsson BI, Kidd M, Chan A, Malfertheiner MV, Modlin IM. Bronchopulmonary neuroendocrine tumors. Cancer. 2001;113(1):5-21. https://doi.org/10.1002/cncr.23542
17. Deng T, Zhang J, Meng Y, Zhou Y, Li W. Higher pretreatment lactate dehydrogenase concentration predicts worse overall survival in patients with lung cancer. Medicine (Baltimore). 2018;97(38):e12524
Difluoroethane Inhalant Abuse, Skeletal Fluorosis, and Withdrawal
Difluoroethane (DFE) is an easily acquired and inexpensive volatile substance that can be inhaled recreationally. 1 It is found in common household items, including compressed air dusters, refrigerants, and propellants. DFE is a central nervous system (CNS) depressant associated with a brief sensation of euphoria when inhaled.2 Prolonged or excessive use is associated with toxicity, and abrupt cessation can induce withdrawal.3-5 We present a case of DFE abuse associated with skeletal fluorosis and withdrawal psychosis.
Case Presentation
A 39-year-old man with a 6-month history of inhaling 20 to 25 cans of DFE per day presented to the emergency department after abruptly stopping use 6 days prior. He described irritability, agitation, auditory hallucinations, and delusions of “demons trying to harm him.”
On presentation, the patient was afebrile with a mild sinus tachycardia. He was calm and cooperative but reported delusions and auditory hallucinations. He denied suicidal or homicidal ideation. His physical examination was remarkable for bony deformities of his hands (Figure 1).
The initial workup included a complete blood count; basic metabolic panel; liver function tests; urine toxicology; and testing for hepatitis B/C and HIV; all unremarkable. Psychiatry and poison control were consulted, and he was admitted.
After 72 hours, the patient's irritability, agitation, and sinus tachycardia resolved; however, his psychosis and hallucinations persisted. He was started on olanzapine and transferred to inpatient psychiatry. Additional laboratory tests revealed a serum fluoride of 0.35 mg/L (normal, 1-47 ug/L), C-telopeptide of 2,663 pg/mL (normal, 70-780 pg/mL), and hand X-rays showing diffuse bilateral periosteal reaction in the phalanges and distal ulnas (Figure 2).6
Discussion
DFE acts as a CNS depressant via glutamate and γ-aminobutyric acid receptors, causing a brief euphoria when inhaled.2 Acute toxicity can cause nausea, vomiting, abdominal pain, and altered mental status. Severe complications include loss of consciousness, mucosal frostbite, angioedema, cardiac arrhythmias, and skeletal fluorosis.2,7
Skeletal fluorosis is a rare ramification of excessive or prolonged DFE inhalation. DFE is metabolized into a fluorinated compound that accumulates and leaches calcium from bone, altering its structure. This can manifest as bony deformities with diffuse periosteal reaction and elevated serum fluoride levels. Furthermore, the elevated C-telopeptide level seen in this case may suggest increased bone turnover.
Approximately 50% of patients report withdrawal symptoms, but the timing, duration, and associated symptoms are not well understood.3 Withdrawal can include tremors, diaphoresis, nausea, vomiting, depression, anxiety, irritability, psychosis, and hallucinations. Symptoms typically start within 24 to 48 hours of cessation and last for 3 to 7 days.5 Psychotic symptoms often abate quickly; however, anxiety and insomnia can persist for weeks.5 There are no formal treatment guidelines, but poison control suggests observation and as-needed benzodiazepines. Although this patient’s irritability and agitation resolved, his psychosis and hallucinations persisted, raising concern for an underlying psychiatric diagnosis and prompting transfer to inpatient psychiatry.
Conslusion
Health care providers should recognize the symptoms of DFE toxicity, its complications, and withdrawal. Collaborating with psychiatry and poison control is beneficial in providing guidelines for supportive care.
1. Arroyo JP, Johnson DC, Lewis JB, et al. Treatment of acute intoxication from inhaled 1,2-difluoroethane. Ann Intern Med. 2018;169(11):820‐822. doi:10.7326/L18-0186
2. National Library of Medicine, PubChem. Hazardous Substance Data Bank (HSDB) 1,1-Difluoroethane. https:// pubchem.ncbi.nlm.nih.gov/source/hsdb/5205. Updated October 25, 2016. Accessed May 20, 2020.
3. Perron BE, Glass JE, Ahmedani BK, Vaughn MG, Roberts DE, Wu LT. The prevalence and clinical significance of inhalant withdrawal symptoms among a national sample. Subst Abuse Rehabil. 2011;2011(2):69‐76. doi:10.2147/SAR.S14937
4. Perron BE, Howard MO, Vaughn MG, Jarman CN. Inhalant withdrawal as a clinically significant feature of inhalant dependence disorder. Med Hypotheses. 2009;73(6):935‐937. doi:10.1016/j.mehy.2009.06.036
5. Addiction Center. Inhalant withdrawal and detox. https://www.addictioncenter.com/drugs/inhalants /withdrawal-detox. Accessed May 18, 2020.
6. Torra M, Rodamilans M, Corbella J. Serum and urine ionic fluoride: normal range in a nonexposed population. Biol Trace Elem Res. 1998;63(1):67‐71. doi:10.1007/BF02785278 7. Cohen E, Hsu RY, Evangelista P, Aaron R, Rubin LE. Rapid-onset diffuse skeletal fluorosis from inhalant abuse: a case report. JBJS Case Connect. 2014;4(4):e108. doi:10.2106/JBJS.CC.N.00085
Difluoroethane (DFE) is an easily acquired and inexpensive volatile substance that can be inhaled recreationally. 1 It is found in common household items, including compressed air dusters, refrigerants, and propellants. DFE is a central nervous system (CNS) depressant associated with a brief sensation of euphoria when inhaled.2 Prolonged or excessive use is associated with toxicity, and abrupt cessation can induce withdrawal.3-5 We present a case of DFE abuse associated with skeletal fluorosis and withdrawal psychosis.
Case Presentation
A 39-year-old man with a 6-month history of inhaling 20 to 25 cans of DFE per day presented to the emergency department after abruptly stopping use 6 days prior. He described irritability, agitation, auditory hallucinations, and delusions of “demons trying to harm him.”
On presentation, the patient was afebrile with a mild sinus tachycardia. He was calm and cooperative but reported delusions and auditory hallucinations. He denied suicidal or homicidal ideation. His physical examination was remarkable for bony deformities of his hands (Figure 1).
The initial workup included a complete blood count; basic metabolic panel; liver function tests; urine toxicology; and testing for hepatitis B/C and HIV; all unremarkable. Psychiatry and poison control were consulted, and he was admitted.
After 72 hours, the patient's irritability, agitation, and sinus tachycardia resolved; however, his psychosis and hallucinations persisted. He was started on olanzapine and transferred to inpatient psychiatry. Additional laboratory tests revealed a serum fluoride of 0.35 mg/L (normal, 1-47 ug/L), C-telopeptide of 2,663 pg/mL (normal, 70-780 pg/mL), and hand X-rays showing diffuse bilateral periosteal reaction in the phalanges and distal ulnas (Figure 2).6
Discussion
DFE acts as a CNS depressant via glutamate and γ-aminobutyric acid receptors, causing a brief euphoria when inhaled.2 Acute toxicity can cause nausea, vomiting, abdominal pain, and altered mental status. Severe complications include loss of consciousness, mucosal frostbite, angioedema, cardiac arrhythmias, and skeletal fluorosis.2,7
Skeletal fluorosis is a rare ramification of excessive or prolonged DFE inhalation. DFE is metabolized into a fluorinated compound that accumulates and leaches calcium from bone, altering its structure. This can manifest as bony deformities with diffuse periosteal reaction and elevated serum fluoride levels. Furthermore, the elevated C-telopeptide level seen in this case may suggest increased bone turnover.
Approximately 50% of patients report withdrawal symptoms, but the timing, duration, and associated symptoms are not well understood.3 Withdrawal can include tremors, diaphoresis, nausea, vomiting, depression, anxiety, irritability, psychosis, and hallucinations. Symptoms typically start within 24 to 48 hours of cessation and last for 3 to 7 days.5 Psychotic symptoms often abate quickly; however, anxiety and insomnia can persist for weeks.5 There are no formal treatment guidelines, but poison control suggests observation and as-needed benzodiazepines. Although this patient’s irritability and agitation resolved, his psychosis and hallucinations persisted, raising concern for an underlying psychiatric diagnosis and prompting transfer to inpatient psychiatry.
Conslusion
Health care providers should recognize the symptoms of DFE toxicity, its complications, and withdrawal. Collaborating with psychiatry and poison control is beneficial in providing guidelines for supportive care.
Difluoroethane (DFE) is an easily acquired and inexpensive volatile substance that can be inhaled recreationally. 1 It is found in common household items, including compressed air dusters, refrigerants, and propellants. DFE is a central nervous system (CNS) depressant associated with a brief sensation of euphoria when inhaled.2 Prolonged or excessive use is associated with toxicity, and abrupt cessation can induce withdrawal.3-5 We present a case of DFE abuse associated with skeletal fluorosis and withdrawal psychosis.
Case Presentation
A 39-year-old man with a 6-month history of inhaling 20 to 25 cans of DFE per day presented to the emergency department after abruptly stopping use 6 days prior. He described irritability, agitation, auditory hallucinations, and delusions of “demons trying to harm him.”
On presentation, the patient was afebrile with a mild sinus tachycardia. He was calm and cooperative but reported delusions and auditory hallucinations. He denied suicidal or homicidal ideation. His physical examination was remarkable for bony deformities of his hands (Figure 1).
The initial workup included a complete blood count; basic metabolic panel; liver function tests; urine toxicology; and testing for hepatitis B/C and HIV; all unremarkable. Psychiatry and poison control were consulted, and he was admitted.
After 72 hours, the patient's irritability, agitation, and sinus tachycardia resolved; however, his psychosis and hallucinations persisted. He was started on olanzapine and transferred to inpatient psychiatry. Additional laboratory tests revealed a serum fluoride of 0.35 mg/L (normal, 1-47 ug/L), C-telopeptide of 2,663 pg/mL (normal, 70-780 pg/mL), and hand X-rays showing diffuse bilateral periosteal reaction in the phalanges and distal ulnas (Figure 2).6
Discussion
DFE acts as a CNS depressant via glutamate and γ-aminobutyric acid receptors, causing a brief euphoria when inhaled.2 Acute toxicity can cause nausea, vomiting, abdominal pain, and altered mental status. Severe complications include loss of consciousness, mucosal frostbite, angioedema, cardiac arrhythmias, and skeletal fluorosis.2,7
Skeletal fluorosis is a rare ramification of excessive or prolonged DFE inhalation. DFE is metabolized into a fluorinated compound that accumulates and leaches calcium from bone, altering its structure. This can manifest as bony deformities with diffuse periosteal reaction and elevated serum fluoride levels. Furthermore, the elevated C-telopeptide level seen in this case may suggest increased bone turnover.
Approximately 50% of patients report withdrawal symptoms, but the timing, duration, and associated symptoms are not well understood.3 Withdrawal can include tremors, diaphoresis, nausea, vomiting, depression, anxiety, irritability, psychosis, and hallucinations. Symptoms typically start within 24 to 48 hours of cessation and last for 3 to 7 days.5 Psychotic symptoms often abate quickly; however, anxiety and insomnia can persist for weeks.5 There are no formal treatment guidelines, but poison control suggests observation and as-needed benzodiazepines. Although this patient’s irritability and agitation resolved, his psychosis and hallucinations persisted, raising concern for an underlying psychiatric diagnosis and prompting transfer to inpatient psychiatry.
Conslusion
Health care providers should recognize the symptoms of DFE toxicity, its complications, and withdrawal. Collaborating with psychiatry and poison control is beneficial in providing guidelines for supportive care.
1. Arroyo JP, Johnson DC, Lewis JB, et al. Treatment of acute intoxication from inhaled 1,2-difluoroethane. Ann Intern Med. 2018;169(11):820‐822. doi:10.7326/L18-0186
2. National Library of Medicine, PubChem. Hazardous Substance Data Bank (HSDB) 1,1-Difluoroethane. https:// pubchem.ncbi.nlm.nih.gov/source/hsdb/5205. Updated October 25, 2016. Accessed May 20, 2020.
3. Perron BE, Glass JE, Ahmedani BK, Vaughn MG, Roberts DE, Wu LT. The prevalence and clinical significance of inhalant withdrawal symptoms among a national sample. Subst Abuse Rehabil. 2011;2011(2):69‐76. doi:10.2147/SAR.S14937
4. Perron BE, Howard MO, Vaughn MG, Jarman CN. Inhalant withdrawal as a clinically significant feature of inhalant dependence disorder. Med Hypotheses. 2009;73(6):935‐937. doi:10.1016/j.mehy.2009.06.036
5. Addiction Center. Inhalant withdrawal and detox. https://www.addictioncenter.com/drugs/inhalants /withdrawal-detox. Accessed May 18, 2020.
6. Torra M, Rodamilans M, Corbella J. Serum and urine ionic fluoride: normal range in a nonexposed population. Biol Trace Elem Res. 1998;63(1):67‐71. doi:10.1007/BF02785278 7. Cohen E, Hsu RY, Evangelista P, Aaron R, Rubin LE. Rapid-onset diffuse skeletal fluorosis from inhalant abuse: a case report. JBJS Case Connect. 2014;4(4):e108. doi:10.2106/JBJS.CC.N.00085
1. Arroyo JP, Johnson DC, Lewis JB, et al. Treatment of acute intoxication from inhaled 1,2-difluoroethane. Ann Intern Med. 2018;169(11):820‐822. doi:10.7326/L18-0186
2. National Library of Medicine, PubChem. Hazardous Substance Data Bank (HSDB) 1,1-Difluoroethane. https:// pubchem.ncbi.nlm.nih.gov/source/hsdb/5205. Updated October 25, 2016. Accessed May 20, 2020.
3. Perron BE, Glass JE, Ahmedani BK, Vaughn MG, Roberts DE, Wu LT. The prevalence and clinical significance of inhalant withdrawal symptoms among a national sample. Subst Abuse Rehabil. 2011;2011(2):69‐76. doi:10.2147/SAR.S14937
4. Perron BE, Howard MO, Vaughn MG, Jarman CN. Inhalant withdrawal as a clinically significant feature of inhalant dependence disorder. Med Hypotheses. 2009;73(6):935‐937. doi:10.1016/j.mehy.2009.06.036
5. Addiction Center. Inhalant withdrawal and detox. https://www.addictioncenter.com/drugs/inhalants /withdrawal-detox. Accessed May 18, 2020.
6. Torra M, Rodamilans M, Corbella J. Serum and urine ionic fluoride: normal range in a nonexposed population. Biol Trace Elem Res. 1998;63(1):67‐71. doi:10.1007/BF02785278 7. Cohen E, Hsu RY, Evangelista P, Aaron R, Rubin LE. Rapid-onset diffuse skeletal fluorosis from inhalant abuse: a case report. JBJS Case Connect. 2014;4(4):e108. doi:10.2106/JBJS.CC.N.00085
24-year-old man • prednisone therapy for nephrotic syndrome • diffuse maculopapular rash • pruritis
THE CASE
A 24-year-old man with no past medical history was referred to a nephrologist for a 5-month history of leg swelling and weight gain. His only medication was furosemide 40 mg/d, prescribed by his primary care physician. His physical examination was unremarkable except for lower extremity and scrotal edema.
Laboratory values included a creatinine of 0.8 mg/dL (reference range, 0.6 to 1.2 mg/dL); hemoglobin concentration, 14.4 g/dL (reference range, 14 to 18 g/dL); albumin, 1.9 g/dL (reference range, 3.5 to 5.5 g/dL); and glucose, 80 mg/dL (reference range, 74 to 106 mg/dL). Electrolyte levels were normal. Urinalysis revealed 3+ blood and 4+ protein on dipstick, as well as the presence of granular and lipid casts on microscopic exam. A 24-hour urine collection contained 10.5 g of protein. Antinuclear antibody titers, complement levels, hepatitis serologies, and antineutrophil cytoplasmic antibody titers were all normal.
A renal biopsy revealed idiopathic focal segmental glomerulosclerosis. The patient was started on oral prednisone 40 mg twice daily.
Two days later, he developed a diffuse pruritic maculopapular rash. He stopped taking the prednisone, and the rash resolved over the next 3 to 5 days. He was then instructed to restart the prednisone for his nephrotic syndrome. When he developed a new but similar rash, the prednisone was discontinued. The rash again resolved.
THE DIAGNOSIS
Since the patient had already been taking furosemide for 6 weeks without an adverse reaction, it was presumed that the prednisone tablet was causing his rash. It would be unusual for prednisone itself to cause a drug eruption, so an additive or coloring agent in the tablet was thought to be responsible for the reaction.
We noted that the patient had been taking a 20-mg orange tablet of prednisone. So we opted to “tweak” the prescription and prescribe the same daily dose but in the form of 10-mg white tablets. The patient tolerated this new regimen without any adverse effects and completed a full 9 months of prednisone therapy without any recurrence of skin lesions. His glomerular disease went into remission.
DISCUSSION
Excipients are inert substances that are added to a food or drug to provide the desired consistency, appearance, or form. They are also used as a preservative for substance stabilization.
Continue to: There are many reports in the literature...
There are many reports in the literature of adverse reactions to excipients.1-3 These include skin rashes induced by the coloring agent in the capsule shell of rifampicin2 and a rash that developed from a coloring agent in oral iron.3 Other reports have noted dyes in foods and even toothpaste as triggers.4,5
Hypersensitivity. Although a specific reaction to prednisone was considered unlikely in this case, type IV delayed hypersensitivity reactions to corticosteroids have been reported. The most common type of corticosteroid-related allergy is contact dermatitis associated with topical corticosteroid use.6 Many cases of delayed maculopapular reactions are thought to be T-cell–mediated type IV reactions.6
Type I immediate hypersensitivity reactions to corticosteroids are also well documented. In a literature review of 120 immediate hypersensitivity reactions to corticosteroids, anaphylactic symptoms were more commonly reported than urticaria or angioedema.7 Intravenous exposure was most frequently associated with reactions, followed by the intra-articular and oral routes of administration.7
Causative agents. The same literature review identified methylprednisolone as the most common steroid to cause a reaction; dexamethasone and prednisone were the least frequently associated with reactions.7 Pharmacologically inactive ingredients were implicated in 28% of the corticosteroid hypersensitivity reactions.7
Additives suspected to be triggers include succinate and phosphate esters, carboxymethylcellulose, polyethylene glycol, and lactose. Interestingly, there have been reports of acute allergic reactions to methylprednisolone sodium succinate 40 mg/mL intravenous preparation in children with milk allergy, due to lactose contaminated with milk protein.8,9
Continue to: Yellow dye was to blame
Yellow dye was to blame. In our case, the 20-mg tablet that the patient had been taking contained the coloring agent FD&C yellow #6, an azo dye also known as sunset yellow or E-110 in Europe. Several reports have described adverse reactions to this coloring agent.1,3 There were other additives in the 20-mg tablet, but a comparison revealed that the 10-mg tablet contained identical substances—but no dye. Thus, it was most likely that the coloring agent was the cause of the patient’s probable type IV exanthematous drug reaction.
Our patient
The patient was instructed to avoid all medications and food containing FD&C yellow #6. No formal allergy testing or re-challenge was performed, since the patient did well under the care of his nephrologist.
THE TAKEAWAY
It’s important to recognize that adverse drug reactions can occur from any medication—not only from the drug itself, but also from excipients contained within. This case reminds us that when a patient complains of an adverse effect to a medication, dyes and inactive ingredients need to be considered as possible inciting agents.
CORRESPONDENCE
Neil E. Soifer, MD, Lakeside Nephrology, 2277 West Howard, Chicago, IL 60645; [email protected]
1. Swerlick RA, Campbell CF. Medication dyes as a source of drug allergy. J Drugs Dermatol. 2013;12:99-102.
2. Calişkaner Z, Oztürk S, Karaayvaz M. Not all adverse drug reactions originate from active component: coloring agent-induced skin eruption in a patient treated with rifampicin. Allergy. 2003;58:1077-1079.
3. Rogkakou A, Guerra L, Scordamaglia A, et al. Severe skin reaction to excipients of an oral iron treatment. Allergy. 2007;62:334-335.
4. Zaknun D, Schroecksnadel S, Kurz K, et al. Potential role of antioxidant food supplements, preservatives and colorants in the pathogenesis of allergy and asthma. Int Arch Allergy Immunol. 2012;157:113-124.
5. Barbaud A. Place of excipients in systemic drug allergy. Immunol Allergy Clin N Am. 2014;34:671-679.
6. Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. Drug allergy: an updated practice parameter. Ann Allergy Asthma Immunol. 2010;105:259-273.
7. Patel A, Bahna S. Immediate hypersensitivity reactions to corticosteroids. Ann Allergy Asthma Immunol. 2015;115:178-182.
8. Eda A, Sugai K, Shioya H, et al. Acute allergic reaction due to milk proteins contaminating lactose added to corticosteroid for injection. Allergol Int. 2009;58:137-139.
9. Levy Y, Segal N, Nahum A, et al. Hypersensitivity to methylprednisolone sodium succinate in children with milk allergy. J Allergy Clin Immunol Pract. 2014;2:471-474.
THE CASE
A 24-year-old man with no past medical history was referred to a nephrologist for a 5-month history of leg swelling and weight gain. His only medication was furosemide 40 mg/d, prescribed by his primary care physician. His physical examination was unremarkable except for lower extremity and scrotal edema.
Laboratory values included a creatinine of 0.8 mg/dL (reference range, 0.6 to 1.2 mg/dL); hemoglobin concentration, 14.4 g/dL (reference range, 14 to 18 g/dL); albumin, 1.9 g/dL (reference range, 3.5 to 5.5 g/dL); and glucose, 80 mg/dL (reference range, 74 to 106 mg/dL). Electrolyte levels were normal. Urinalysis revealed 3+ blood and 4+ protein on dipstick, as well as the presence of granular and lipid casts on microscopic exam. A 24-hour urine collection contained 10.5 g of protein. Antinuclear antibody titers, complement levels, hepatitis serologies, and antineutrophil cytoplasmic antibody titers were all normal.
A renal biopsy revealed idiopathic focal segmental glomerulosclerosis. The patient was started on oral prednisone 40 mg twice daily.
Two days later, he developed a diffuse pruritic maculopapular rash. He stopped taking the prednisone, and the rash resolved over the next 3 to 5 days. He was then instructed to restart the prednisone for his nephrotic syndrome. When he developed a new but similar rash, the prednisone was discontinued. The rash again resolved.
THE DIAGNOSIS
Since the patient had already been taking furosemide for 6 weeks without an adverse reaction, it was presumed that the prednisone tablet was causing his rash. It would be unusual for prednisone itself to cause a drug eruption, so an additive or coloring agent in the tablet was thought to be responsible for the reaction.
We noted that the patient had been taking a 20-mg orange tablet of prednisone. So we opted to “tweak” the prescription and prescribe the same daily dose but in the form of 10-mg white tablets. The patient tolerated this new regimen without any adverse effects and completed a full 9 months of prednisone therapy without any recurrence of skin lesions. His glomerular disease went into remission.
DISCUSSION
Excipients are inert substances that are added to a food or drug to provide the desired consistency, appearance, or form. They are also used as a preservative for substance stabilization.
Continue to: There are many reports in the literature...
There are many reports in the literature of adverse reactions to excipients.1-3 These include skin rashes induced by the coloring agent in the capsule shell of rifampicin2 and a rash that developed from a coloring agent in oral iron.3 Other reports have noted dyes in foods and even toothpaste as triggers.4,5
Hypersensitivity. Although a specific reaction to prednisone was considered unlikely in this case, type IV delayed hypersensitivity reactions to corticosteroids have been reported. The most common type of corticosteroid-related allergy is contact dermatitis associated with topical corticosteroid use.6 Many cases of delayed maculopapular reactions are thought to be T-cell–mediated type IV reactions.6
Type I immediate hypersensitivity reactions to corticosteroids are also well documented. In a literature review of 120 immediate hypersensitivity reactions to corticosteroids, anaphylactic symptoms were more commonly reported than urticaria or angioedema.7 Intravenous exposure was most frequently associated with reactions, followed by the intra-articular and oral routes of administration.7
Causative agents. The same literature review identified methylprednisolone as the most common steroid to cause a reaction; dexamethasone and prednisone were the least frequently associated with reactions.7 Pharmacologically inactive ingredients were implicated in 28% of the corticosteroid hypersensitivity reactions.7
Additives suspected to be triggers include succinate and phosphate esters, carboxymethylcellulose, polyethylene glycol, and lactose. Interestingly, there have been reports of acute allergic reactions to methylprednisolone sodium succinate 40 mg/mL intravenous preparation in children with milk allergy, due to lactose contaminated with milk protein.8,9
Continue to: Yellow dye was to blame
Yellow dye was to blame. In our case, the 20-mg tablet that the patient had been taking contained the coloring agent FD&C yellow #6, an azo dye also known as sunset yellow or E-110 in Europe. Several reports have described adverse reactions to this coloring agent.1,3 There were other additives in the 20-mg tablet, but a comparison revealed that the 10-mg tablet contained identical substances—but no dye. Thus, it was most likely that the coloring agent was the cause of the patient’s probable type IV exanthematous drug reaction.
Our patient
The patient was instructed to avoid all medications and food containing FD&C yellow #6. No formal allergy testing or re-challenge was performed, since the patient did well under the care of his nephrologist.
THE TAKEAWAY
It’s important to recognize that adverse drug reactions can occur from any medication—not only from the drug itself, but also from excipients contained within. This case reminds us that when a patient complains of an adverse effect to a medication, dyes and inactive ingredients need to be considered as possible inciting agents.
CORRESPONDENCE
Neil E. Soifer, MD, Lakeside Nephrology, 2277 West Howard, Chicago, IL 60645; [email protected]
THE CASE
A 24-year-old man with no past medical history was referred to a nephrologist for a 5-month history of leg swelling and weight gain. His only medication was furosemide 40 mg/d, prescribed by his primary care physician. His physical examination was unremarkable except for lower extremity and scrotal edema.
Laboratory values included a creatinine of 0.8 mg/dL (reference range, 0.6 to 1.2 mg/dL); hemoglobin concentration, 14.4 g/dL (reference range, 14 to 18 g/dL); albumin, 1.9 g/dL (reference range, 3.5 to 5.5 g/dL); and glucose, 80 mg/dL (reference range, 74 to 106 mg/dL). Electrolyte levels were normal. Urinalysis revealed 3+ blood and 4+ protein on dipstick, as well as the presence of granular and lipid casts on microscopic exam. A 24-hour urine collection contained 10.5 g of protein. Antinuclear antibody titers, complement levels, hepatitis serologies, and antineutrophil cytoplasmic antibody titers were all normal.
A renal biopsy revealed idiopathic focal segmental glomerulosclerosis. The patient was started on oral prednisone 40 mg twice daily.
Two days later, he developed a diffuse pruritic maculopapular rash. He stopped taking the prednisone, and the rash resolved over the next 3 to 5 days. He was then instructed to restart the prednisone for his nephrotic syndrome. When he developed a new but similar rash, the prednisone was discontinued. The rash again resolved.
THE DIAGNOSIS
Since the patient had already been taking furosemide for 6 weeks without an adverse reaction, it was presumed that the prednisone tablet was causing his rash. It would be unusual for prednisone itself to cause a drug eruption, so an additive or coloring agent in the tablet was thought to be responsible for the reaction.
We noted that the patient had been taking a 20-mg orange tablet of prednisone. So we opted to “tweak” the prescription and prescribe the same daily dose but in the form of 10-mg white tablets. The patient tolerated this new regimen without any adverse effects and completed a full 9 months of prednisone therapy without any recurrence of skin lesions. His glomerular disease went into remission.
DISCUSSION
Excipients are inert substances that are added to a food or drug to provide the desired consistency, appearance, or form. They are also used as a preservative for substance stabilization.
Continue to: There are many reports in the literature...
There are many reports in the literature of adverse reactions to excipients.1-3 These include skin rashes induced by the coloring agent in the capsule shell of rifampicin2 and a rash that developed from a coloring agent in oral iron.3 Other reports have noted dyes in foods and even toothpaste as triggers.4,5
Hypersensitivity. Although a specific reaction to prednisone was considered unlikely in this case, type IV delayed hypersensitivity reactions to corticosteroids have been reported. The most common type of corticosteroid-related allergy is contact dermatitis associated with topical corticosteroid use.6 Many cases of delayed maculopapular reactions are thought to be T-cell–mediated type IV reactions.6
Type I immediate hypersensitivity reactions to corticosteroids are also well documented. In a literature review of 120 immediate hypersensitivity reactions to corticosteroids, anaphylactic symptoms were more commonly reported than urticaria or angioedema.7 Intravenous exposure was most frequently associated with reactions, followed by the intra-articular and oral routes of administration.7
Causative agents. The same literature review identified methylprednisolone as the most common steroid to cause a reaction; dexamethasone and prednisone were the least frequently associated with reactions.7 Pharmacologically inactive ingredients were implicated in 28% of the corticosteroid hypersensitivity reactions.7
Additives suspected to be triggers include succinate and phosphate esters, carboxymethylcellulose, polyethylene glycol, and lactose. Interestingly, there have been reports of acute allergic reactions to methylprednisolone sodium succinate 40 mg/mL intravenous preparation in children with milk allergy, due to lactose contaminated with milk protein.8,9
Continue to: Yellow dye was to blame
Yellow dye was to blame. In our case, the 20-mg tablet that the patient had been taking contained the coloring agent FD&C yellow #6, an azo dye also known as sunset yellow or E-110 in Europe. Several reports have described adverse reactions to this coloring agent.1,3 There were other additives in the 20-mg tablet, but a comparison revealed that the 10-mg tablet contained identical substances—but no dye. Thus, it was most likely that the coloring agent was the cause of the patient’s probable type IV exanthematous drug reaction.
Our patient
The patient was instructed to avoid all medications and food containing FD&C yellow #6. No formal allergy testing or re-challenge was performed, since the patient did well under the care of his nephrologist.
THE TAKEAWAY
It’s important to recognize that adverse drug reactions can occur from any medication—not only from the drug itself, but also from excipients contained within. This case reminds us that when a patient complains of an adverse effect to a medication, dyes and inactive ingredients need to be considered as possible inciting agents.
CORRESPONDENCE
Neil E. Soifer, MD, Lakeside Nephrology, 2277 West Howard, Chicago, IL 60645; [email protected]
1. Swerlick RA, Campbell CF. Medication dyes as a source of drug allergy. J Drugs Dermatol. 2013;12:99-102.
2. Calişkaner Z, Oztürk S, Karaayvaz M. Not all adverse drug reactions originate from active component: coloring agent-induced skin eruption in a patient treated with rifampicin. Allergy. 2003;58:1077-1079.
3. Rogkakou A, Guerra L, Scordamaglia A, et al. Severe skin reaction to excipients of an oral iron treatment. Allergy. 2007;62:334-335.
4. Zaknun D, Schroecksnadel S, Kurz K, et al. Potential role of antioxidant food supplements, preservatives and colorants in the pathogenesis of allergy and asthma. Int Arch Allergy Immunol. 2012;157:113-124.
5. Barbaud A. Place of excipients in systemic drug allergy. Immunol Allergy Clin N Am. 2014;34:671-679.
6. Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. Drug allergy: an updated practice parameter. Ann Allergy Asthma Immunol. 2010;105:259-273.
7. Patel A, Bahna S. Immediate hypersensitivity reactions to corticosteroids. Ann Allergy Asthma Immunol. 2015;115:178-182.
8. Eda A, Sugai K, Shioya H, et al. Acute allergic reaction due to milk proteins contaminating lactose added to corticosteroid for injection. Allergol Int. 2009;58:137-139.
9. Levy Y, Segal N, Nahum A, et al. Hypersensitivity to methylprednisolone sodium succinate in children with milk allergy. J Allergy Clin Immunol Pract. 2014;2:471-474.
1. Swerlick RA, Campbell CF. Medication dyes as a source of drug allergy. J Drugs Dermatol. 2013;12:99-102.
2. Calişkaner Z, Oztürk S, Karaayvaz M. Not all adverse drug reactions originate from active component: coloring agent-induced skin eruption in a patient treated with rifampicin. Allergy. 2003;58:1077-1079.
3. Rogkakou A, Guerra L, Scordamaglia A, et al. Severe skin reaction to excipients of an oral iron treatment. Allergy. 2007;62:334-335.
4. Zaknun D, Schroecksnadel S, Kurz K, et al. Potential role of antioxidant food supplements, preservatives and colorants in the pathogenesis of allergy and asthma. Int Arch Allergy Immunol. 2012;157:113-124.
5. Barbaud A. Place of excipients in systemic drug allergy. Immunol Allergy Clin N Am. 2014;34:671-679.
6. Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. Drug allergy: an updated practice parameter. Ann Allergy Asthma Immunol. 2010;105:259-273.
7. Patel A, Bahna S. Immediate hypersensitivity reactions to corticosteroids. Ann Allergy Asthma Immunol. 2015;115:178-182.
8. Eda A, Sugai K, Shioya H, et al. Acute allergic reaction due to milk proteins contaminating lactose added to corticosteroid for injection. Allergol Int. 2009;58:137-139.
9. Levy Y, Segal N, Nahum A, et al. Hypersensitivity to methylprednisolone sodium succinate in children with milk allergy. J Allergy Clin Immunol Pract. 2014;2:471-474.

Hemiballismus in Patients With Poorly Controlled Type 2 Diabetes Mellitus
Hemiballismus is an acquired hyperkinetic movement disorder characterized by unilateral, involuntary, often large-amplitude limb movements. Ballistic movements are now considered to be on the choreiform spectrum.1 Movements usually involve both the arm and leg, and in half of cases, facial movements such as tongue clucking and grimacing are seen.2,3 Presentations of hemiballismus vary in severity from intermittent to nearly continuous movements, which, in some cases, may lead to exhaustion, injury, or disability. Some patients are unable to ambulate or feed themselves with the affected limb.
Background
The 2 most common causes of hemichorea-hemiballismus are stroke and hyperglycemia, with an incidence of 4% and unknown incidence, respectively.1,3,4 Other causes include HIV, traumatic brain injury, encephalitis, vasculitis, mass effect, multiple sclerosis, and adverse drug reactions. 4-7 Acute or subacute hemiballismus is classically attributed to a lesion in subthalamic nucleus (STN), but this is true only in a minority of cases. Hemiballismus can be caused by any abnormality in various subnuclei of the basal ganglia, including the classic location in the STN, striatum, and globus pallidus.4 Evidence shows the lesions typically involve a functional network connected to the posterolateral putamen.8
Although not commonly recognized, hyperglycemia in patients with type 2 diabetes mellitus (T2DM) is the second most common cause of hemichoreahemiballismus. 3 Over the past 90 years, numerous case reports have described patients with DM with acute and subacute onset of hemiballistic and hemichoreiform movements while in a hyperglycemic state or after its resolution. Reported cases have been limited to small numbers of patients with only a few larger-scale reviews of more than 20 patients.7,9 Most reported cases involve geriatric patients and more commonly, females of Eastern Asian descent with an average age of onset of 71 years.4,10 Patients typically present with glucose levels from 500 to 1,000 mg/dL and hemoglobin A1c (HbA1c) levels almost double the normal values. Interestingly, neuroimaging findings in these patients have consistently shown hyperintense signal in the contralateral basal ganglia on T1-weighted magnetic resonance images (MRIs). Noncontrast computed tomography (CT) shows well-defined unilateral increased density in the contralateral basal ganglia without mass effect.1,9,11
This report aims to illustrate and enhance the understanding of hemiballismus associated with hyperglycemia. One patient presented to the US Department of Veterans Affairs (VA) Bay Pines VA Healthcare System (BPVAHCS) in Florida, which motivated us to search for other similar cases. We reviewed the charts of 2 other patients who presented to BPVAHCS over the past 10 years. The first case presented with severe hyperglycemia and abnormal movements that were not clearly diagnosed as hemiballismus. MRI findings were characteristic and assisted in making the diagnosis. The second case was misdiagnosed as hemiballismus secondary to ischemic stroke. The third case was initially diagnosed as conversion disorder until movements worsened and the correct diagnosis of hyperglycemia-induced hemichorea hemiballismus was confirmed by the pathognomonic neuroimaging findings.
Case Presentations
Case 1
A 65-year-old male with a history of uncontrolled T2DM presented with repetitive twitching and kicking movements that involved his left upper and lower extremities for 3 weeks. The patient reported that he did not take his medications or follow the recommended diabetes diet. His HbA1c on admission was 12.2% with a serum glucose of 254 mg/dL. The MRI showed a hyperintense T1 signal within the right basal ganglia including the right caudate with sparing of the internal capsule (Figure 1). There was no associated mass effect or restricted diffusion. It was compatible with a diagnosis of hyperglycemia- induced hemichorea-hemiballismus. The patient was advised to resume taking glipizide 10 mg daily, metformin 1,000 mg by mouth twice daily, and to begin 10 units of 70/30 insulin aspart 15 minutes before meals twice daily, and to follow a low carbohydrate diet, with reduce dietary intake of sugar. At his 1-month follow-up visit, the patient reported an improvement in his involuntary movements. At the 5-month follow-up, the patient’s HbA1c level was 10.4% and his hyperkinetic movements had completely resolved.
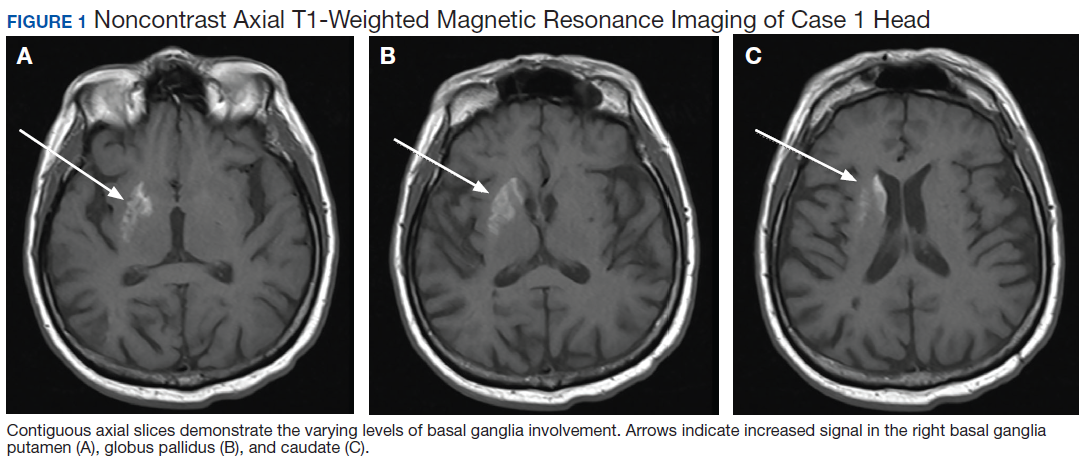
Case 2
of T2DM, hypertension, and hyperlipidemia was admitted due to increased jerky movements in the left upper extremity. On admission, his vital signs were within normal limits and his physical examination demonstrated choreoathetoid movements with ballistic components of his left upper extremity. His laboratory results showed a glucose level of 528 mg/dL with a HbA1c of 16.3%. An initial CT obtained in the emergency department (ED) demonstrated a well-defined hyperdensity in the striatal (caudate and lentiform nucleus) region (Figure 2). There was no associated edema/mass effect that would be typical for an intracranial hemorrhage.
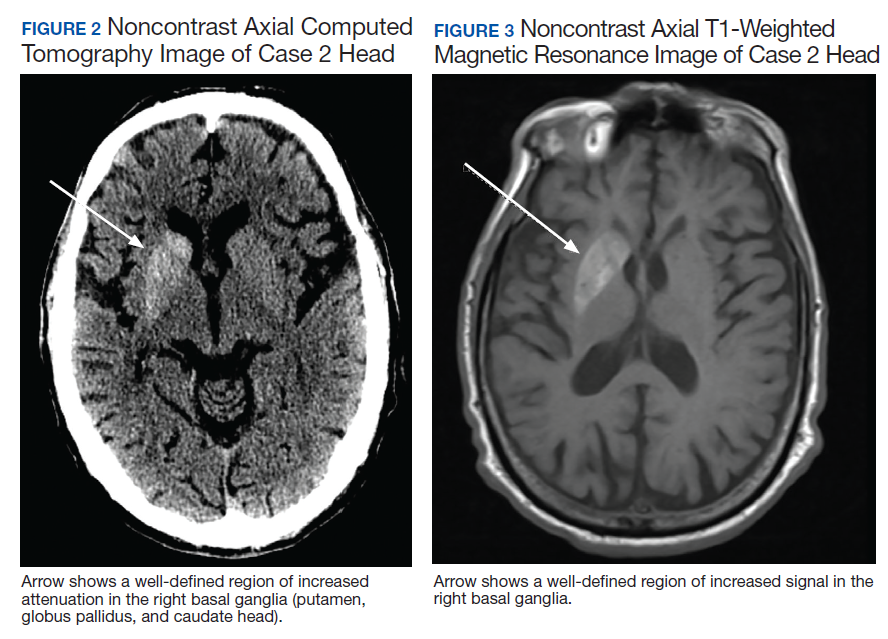
An MRI obtained 1 week later showed hyperintense TI signal corresponding to the basal ganglia (Figure 3). In addition, there was a questionable lacunar infarct in the right internal capsule. Due to lack of awareness regarding hyperglycemic associated basal ganglia changes, the patient’s movement disorder was presumed to be ischemic in etiology. The patient was prescribed oral amantadine 100 mg 3 times daily for the hemiballismus in conjunction with treatment of his T2DM. The only follow-up occurred 5 weeks later, which showed no improvement of uncontrollable movements. Imaging at that time (not available) indicated the persistence of the abnormal signal in the right basal ganglia. This patient died later that year without further follow-up.
Case 3
A 78-year-old white male with a history of syncope, transient ischemic attacks (TIAs), and poorly controlled T2DM presented with a 1-month history of progressively worsening involuntary, left-sided movements that began in his left shoulder and advanced to involve his arm, hand, and leg, and the left side of his face with grimacing and clucking of his tongue. Three weeks earlier, the patient had been discharged from the ED with a diagnosis of conversion disorder particularly because he experienced decreased movements when given a dose of Vitamin D. It was overlooked that administration of haloperidol had occurred a few hours before, and because the sounds made by his tongue were not felt to be consistent with a known movement disorder. A MRI of the brain was read as normal.
The patient returned 3 weeks later (the original presentation) due to his inability to perform activities of daily living because of his worsening involuntary movements. On admission, his HbA1c was 11.1% and his glucose was 167 mg/dL. On chart review, it was revealed that the patient’s HbA1c had been > 9% for the past 3 years with an increase from 10.1% to 11.1% in the 3 months preceding the onset of his symptoms.
On admission a MRI showed a unilateral right-sided T1 hyperintensity in the basal ganglia, no acute ischemia (Figure 4). In retrospect, subtle increased T1 signal can be seen on the earlier MRI (Figure 5). In view of the patient’s left-sided symptoms, DM, and MRI findings, a diagnosis of hyperglycemia-induced hemichorea- hemiballismus was made as the etiology of the patient’s symptoms.
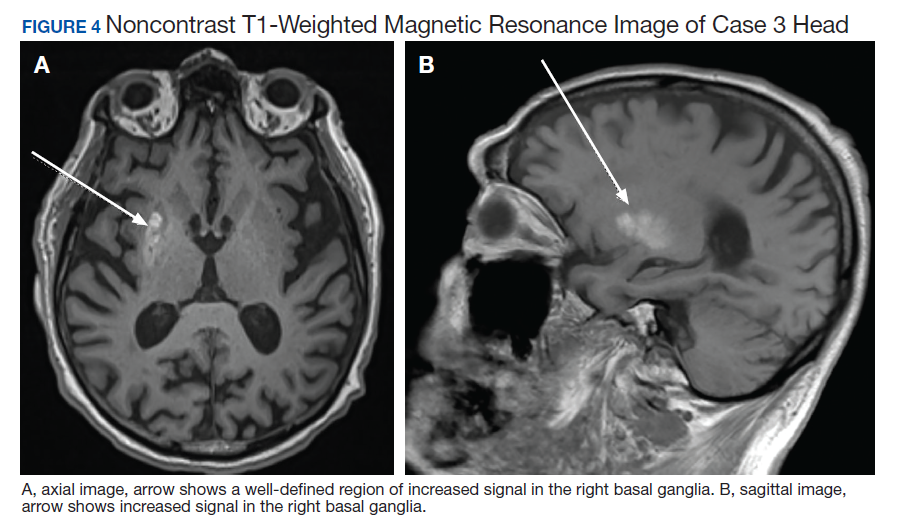
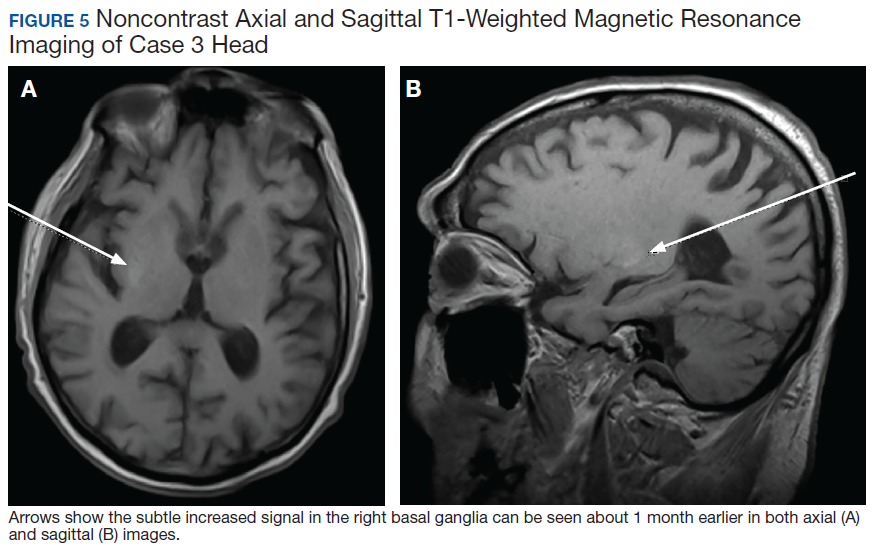
The patient was prescribed numerous medications to control his hyperkinesia including (and in combination): benztropine, gabapentin, baclofen, diphenhydramine, benzodiazepines, risperidone, olanzapine, and valproic acid, which did not control his movements. Ultimately, his hyperglycemic hemiballismus improved with tight glycemic control and oral tetrabenazine 12.5 mg twice daily. This patient underwent a protracted course of treatment with 17 days of inpatient medical admission, 3 weeks inpatient rehabilitation, and subsequent transfer to an assisted living facility.
Discussion
The 3 cases presented in this report contribute to the evidence that severe persistent hyperglycemia can result in movement disorders that mimic those seen after basal ganglia strokes. As with Case 2, past literature describes many cases of acute hyperglycemic episodes with glucose ranging from 500 to 1,000 mg/mL presenting with hemiballismus.1,3 However, there are many cases that describe hemiballismus occurring after glycemic correction, persisting despite glycemic correction, and presenting without an acute hyperglycemic episode, but in the setting of elevated HbA1c, as in Case 3.12,13 Notably, all 3 cases in this series had marked elevation in their HbA1c levels, which suggests that a more chronic hyperglycemic state or multiple shorter periods of hyperglycemia may be necessary to produce the described hyperkinetic movements.
Case reports describe the pathognomonic T1 hyperintensity of the basal ganglia that is identified in all 3 cases presented here. While the exact etiology remains unclear, the to metabolic derangements caused by hyperviscosity of the blood in the small end arteries feeding the basal ganglia.3,11 These abnormalities in turn interrupt the signaling cascade with abnormal firing rates or firing patterns, leading to reduced inhibition of the motor thalamus and ultimately present as hemiballismus.1,3,7 While most cases presented with unilateral hyperkinesis and associated contralateral basal ganglia abnormalities, there are reports of both unilateral and bilateral movements associated with bilateral basal ganglia hyperintensities on imaging. 9 The predilection for unilateral brain lesions may be explained by the varying degree of small vessel disease in different areas of the brain leading to perfusion deficits worsened by hyper viscosity. Further research into this is required to elucidate the exact pathophysiologic mechanism.
The course of disease for patients ranges from resolution within hours of tight glycemic control to persistent movements for > 3 months with a gradual improvement in severity.12,13 Treatments center on the importance of tight glycemic control to protect against the protracted course described in Case 3. Swift recognition of this rare condition is critical because improved glycemic control decreases the severity and duration of this disease. The significant disability associated with Case 3 highlights the need for prompt recognition and early, aggressive glycemic management to prevent the progression of hemiballismus. In addition to glycemic control, various CNS medications such as typical and atypical antipsychotics and tetrabenazine are firstline therapy with chemodenervation and surgical lesioning in cases unresponsive to medication therapy.
When unrecognized, hyperglycemic hemiballismus is associated with significant morbidity and mortality. The patients presented in this report were subject to either delayed diagnosis or misdiagnosis as stroke or psychiatric disorder. The rarity of the disorder, lack of evidence delineating pathogenesis and causality, low level of awareness, and varying presentations of patients all contribute to the challenge of recognizing, diagnosing, and treating hemiballismus due to hyperglycemia. This challenge can subsequently result in deteriorating symptoms, prolonged hospital stays, and unnecessary health care costs.
Conclusion
While hemiballismus due to severe persistent hyperglycemia is rare, the goal of this report is to highlight its occurrence in patients with T2DM. Further research can help develop a standardized, effective treatment strategy for these patients. Currently, lowering and maintaining appropriate glucose and HbA1c levels is the most effective treatment approach. Potential areas of research include alternative medical and surgical treatment interventions for patients while glycemic control is being achieved or for those who fail to benefit from glycemic control alone. Some success has been demonstrated with the use of antidopaminergic medications such as atypical antipsychotics and tetrabenazine and these medications should be considered when tight, sustained glycemic control alone is not successful in treating this disorder in the acute stages. Hopefully, with increasing awareness and recognition of hemiballismus related to hyperglycemia, more large-scale clinical trials can be conducted that will result in an effective treatment strategy for this devastating disorder.
1. Hawley JS, Weiner WJ. Hemiballismus: current concepts and review. Parkinsonism Relat Disord. 2012;18(2):125‐129. doi:10.1016/j.parkreldis.2011.08.015
2. Gasca-Salas C, Lang AE. Paroxysmal Hemiballism/ Hemichorea Resulting from Transient Ischemic Attacks. Mov Disord Clin Pract. 2015;3(3):303‐305. doi:10.1002/mdc3.12268
3. Garcia-Grimshaw MA, Jimenez-Ruiz A, Ornelas-Velazquez A, Luna-Armenta A, Gutierrez-Manjarrez FA. New-onset diabetes presenting as monoballism secondary to a mixed hyperglycemic crisis. Cureus. 2018;10(6):e2882. doi:10.7759/cureus.2882
4. Postuma RB, Lang AE. Hemiballism: revisiting a classic disorder. Lancet Neurol. 2003;2(11):661‐668. doi:10.1016/s1474-4422(03)00554-4
5. Gallo BV, Shulman LM, Weiner WJ, Petito CK, Berger JR. HIV encephalitis presenting with severe generalized chorea. Neurology. 1996;46(4):1163‐1165. doi:10.1212/wnl.46.4.1163
6. Provenzale JM, Glass JP. Hemiballismus: CT and MR findings. J Comput Assist Tomogr. 1995;19(4):537‐540.
7. Hodde M, Rowe KE, Surapaneni K, Terrigno P, Brighenti A, Altschuler EL. Management of severe hemiballismus: treatment challenges in the acute inpatient rehabilitation setting: a case presentation. PMR. 2017;9(7):732‐735. doi:10.1016/j.pmrj.2016.10.023
8. Laganiere S, Boes AD, Fox MD. Network localization of hemichorea-hemiballismus. Neurology. 2016;86(23):2187‐2195. doi:10.1212/WNL.0000000000002741
9. Cosentino C, Torres L, Nuñez Y, Suarez R, Velez M, Flores M. Hemichorea/hemiballism associated with hyperglycemia: report of 20 cases. Tremor Other Hyperkinet Mov (NY). 2016;6:402. doi:10.7916/D8DN454P
10. Oh SH, Lee KY, Im JH, Lee MS. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a metaanalysis of 53 cases including four present cases. J Neurol Sci. 2002;200(1-2):57‐62. doi:10.1016/s0022-510x(02)00133-8
11. Carrion DM, Carrion AF. Non-ketotic hyperglycaemia hemichorea-hemiballismus and acute ischaemic stroke. BMJ Case Rep. 2013;2013:bcr2012008359. doi:10.1136/bcr-2012-008359
12. Cho HS, Hong CT, Chan L. Hemichorea after hyperglycemia correction: a case report and a short review of hyperglycemia-related hemichorea at the euglycemic state. Medicine (Baltimore). 2018;97(10):e0076. doi:10.1097/MD.0000000000010076
13. Lin YC, Lin YC. Prolonged hemiballism after the remission of non-ketotic hyperosmolar syndrome. BMJ Case Rep. 2012;2012:bcr0120125627. doi:10.1136/bcr.01.2012.5627
Hemiballismus is an acquired hyperkinetic movement disorder characterized by unilateral, involuntary, often large-amplitude limb movements. Ballistic movements are now considered to be on the choreiform spectrum.1 Movements usually involve both the arm and leg, and in half of cases, facial movements such as tongue clucking and grimacing are seen.2,3 Presentations of hemiballismus vary in severity from intermittent to nearly continuous movements, which, in some cases, may lead to exhaustion, injury, or disability. Some patients are unable to ambulate or feed themselves with the affected limb.
Background
The 2 most common causes of hemichorea-hemiballismus are stroke and hyperglycemia, with an incidence of 4% and unknown incidence, respectively.1,3,4 Other causes include HIV, traumatic brain injury, encephalitis, vasculitis, mass effect, multiple sclerosis, and adverse drug reactions. 4-7 Acute or subacute hemiballismus is classically attributed to a lesion in subthalamic nucleus (STN), but this is true only in a minority of cases. Hemiballismus can be caused by any abnormality in various subnuclei of the basal ganglia, including the classic location in the STN, striatum, and globus pallidus.4 Evidence shows the lesions typically involve a functional network connected to the posterolateral putamen.8
Although not commonly recognized, hyperglycemia in patients with type 2 diabetes mellitus (T2DM) is the second most common cause of hemichoreahemiballismus. 3 Over the past 90 years, numerous case reports have described patients with DM with acute and subacute onset of hemiballistic and hemichoreiform movements while in a hyperglycemic state or after its resolution. Reported cases have been limited to small numbers of patients with only a few larger-scale reviews of more than 20 patients.7,9 Most reported cases involve geriatric patients and more commonly, females of Eastern Asian descent with an average age of onset of 71 years.4,10 Patients typically present with glucose levels from 500 to 1,000 mg/dL and hemoglobin A1c (HbA1c) levels almost double the normal values. Interestingly, neuroimaging findings in these patients have consistently shown hyperintense signal in the contralateral basal ganglia on T1-weighted magnetic resonance images (MRIs). Noncontrast computed tomography (CT) shows well-defined unilateral increased density in the contralateral basal ganglia without mass effect.1,9,11
This report aims to illustrate and enhance the understanding of hemiballismus associated with hyperglycemia. One patient presented to the US Department of Veterans Affairs (VA) Bay Pines VA Healthcare System (BPVAHCS) in Florida, which motivated us to search for other similar cases. We reviewed the charts of 2 other patients who presented to BPVAHCS over the past 10 years. The first case presented with severe hyperglycemia and abnormal movements that were not clearly diagnosed as hemiballismus. MRI findings were characteristic and assisted in making the diagnosis. The second case was misdiagnosed as hemiballismus secondary to ischemic stroke. The third case was initially diagnosed as conversion disorder until movements worsened and the correct diagnosis of hyperglycemia-induced hemichorea hemiballismus was confirmed by the pathognomonic neuroimaging findings.
Case Presentations
Case 1
A 65-year-old male with a history of uncontrolled T2DM presented with repetitive twitching and kicking movements that involved his left upper and lower extremities for 3 weeks. The patient reported that he did not take his medications or follow the recommended diabetes diet. His HbA1c on admission was 12.2% with a serum glucose of 254 mg/dL. The MRI showed a hyperintense T1 signal within the right basal ganglia including the right caudate with sparing of the internal capsule (Figure 1). There was no associated mass effect or restricted diffusion. It was compatible with a diagnosis of hyperglycemia- induced hemichorea-hemiballismus. The patient was advised to resume taking glipizide 10 mg daily, metformin 1,000 mg by mouth twice daily, and to begin 10 units of 70/30 insulin aspart 15 minutes before meals twice daily, and to follow a low carbohydrate diet, with reduce dietary intake of sugar. At his 1-month follow-up visit, the patient reported an improvement in his involuntary movements. At the 5-month follow-up, the patient’s HbA1c level was 10.4% and his hyperkinetic movements had completely resolved.
Case 2
of T2DM, hypertension, and hyperlipidemia was admitted due to increased jerky movements in the left upper extremity. On admission, his vital signs were within normal limits and his physical examination demonstrated choreoathetoid movements with ballistic components of his left upper extremity. His laboratory results showed a glucose level of 528 mg/dL with a HbA1c of 16.3%. An initial CT obtained in the emergency department (ED) demonstrated a well-defined hyperdensity in the striatal (caudate and lentiform nucleus) region (Figure 2). There was no associated edema/mass effect that would be typical for an intracranial hemorrhage.
An MRI obtained 1 week later showed hyperintense TI signal corresponding to the basal ganglia (Figure 3). In addition, there was a questionable lacunar infarct in the right internal capsule. Due to lack of awareness regarding hyperglycemic associated basal ganglia changes, the patient’s movement disorder was presumed to be ischemic in etiology. The patient was prescribed oral amantadine 100 mg 3 times daily for the hemiballismus in conjunction with treatment of his T2DM. The only follow-up occurred 5 weeks later, which showed no improvement of uncontrollable movements. Imaging at that time (not available) indicated the persistence of the abnormal signal in the right basal ganglia. This patient died later that year without further follow-up.
Case 3
A 78-year-old white male with a history of syncope, transient ischemic attacks (TIAs), and poorly controlled T2DM presented with a 1-month history of progressively worsening involuntary, left-sided movements that began in his left shoulder and advanced to involve his arm, hand, and leg, and the left side of his face with grimacing and clucking of his tongue. Three weeks earlier, the patient had been discharged from the ED with a diagnosis of conversion disorder particularly because he experienced decreased movements when given a dose of Vitamin D. It was overlooked that administration of haloperidol had occurred a few hours before, and because the sounds made by his tongue were not felt to be consistent with a known movement disorder. A MRI of the brain was read as normal.
The patient returned 3 weeks later (the original presentation) due to his inability to perform activities of daily living because of his worsening involuntary movements. On admission, his HbA1c was 11.1% and his glucose was 167 mg/dL. On chart review, it was revealed that the patient’s HbA1c had been > 9% for the past 3 years with an increase from 10.1% to 11.1% in the 3 months preceding the onset of his symptoms.
On admission a MRI showed a unilateral right-sided T1 hyperintensity in the basal ganglia, no acute ischemia (Figure 4). In retrospect, subtle increased T1 signal can be seen on the earlier MRI (Figure 5). In view of the patient’s left-sided symptoms, DM, and MRI findings, a diagnosis of hyperglycemia-induced hemichorea- hemiballismus was made as the etiology of the patient’s symptoms.
The patient was prescribed numerous medications to control his hyperkinesia including (and in combination): benztropine, gabapentin, baclofen, diphenhydramine, benzodiazepines, risperidone, olanzapine, and valproic acid, which did not control his movements. Ultimately, his hyperglycemic hemiballismus improved with tight glycemic control and oral tetrabenazine 12.5 mg twice daily. This patient underwent a protracted course of treatment with 17 days of inpatient medical admission, 3 weeks inpatient rehabilitation, and subsequent transfer to an assisted living facility.
Discussion
The 3 cases presented in this report contribute to the evidence that severe persistent hyperglycemia can result in movement disorders that mimic those seen after basal ganglia strokes. As with Case 2, past literature describes many cases of acute hyperglycemic episodes with glucose ranging from 500 to 1,000 mg/mL presenting with hemiballismus.1,3 However, there are many cases that describe hemiballismus occurring after glycemic correction, persisting despite glycemic correction, and presenting without an acute hyperglycemic episode, but in the setting of elevated HbA1c, as in Case 3.12,13 Notably, all 3 cases in this series had marked elevation in their HbA1c levels, which suggests that a more chronic hyperglycemic state or multiple shorter periods of hyperglycemia may be necessary to produce the described hyperkinetic movements.
Case reports describe the pathognomonic T1 hyperintensity of the basal ganglia that is identified in all 3 cases presented here. While the exact etiology remains unclear, the to metabolic derangements caused by hyperviscosity of the blood in the small end arteries feeding the basal ganglia.3,11 These abnormalities in turn interrupt the signaling cascade with abnormal firing rates or firing patterns, leading to reduced inhibition of the motor thalamus and ultimately present as hemiballismus.1,3,7 While most cases presented with unilateral hyperkinesis and associated contralateral basal ganglia abnormalities, there are reports of both unilateral and bilateral movements associated with bilateral basal ganglia hyperintensities on imaging. 9 The predilection for unilateral brain lesions may be explained by the varying degree of small vessel disease in different areas of the brain leading to perfusion deficits worsened by hyper viscosity. Further research into this is required to elucidate the exact pathophysiologic mechanism.
The course of disease for patients ranges from resolution within hours of tight glycemic control to persistent movements for > 3 months with a gradual improvement in severity.12,13 Treatments center on the importance of tight glycemic control to protect against the protracted course described in Case 3. Swift recognition of this rare condition is critical because improved glycemic control decreases the severity and duration of this disease. The significant disability associated with Case 3 highlights the need for prompt recognition and early, aggressive glycemic management to prevent the progression of hemiballismus. In addition to glycemic control, various CNS medications such as typical and atypical antipsychotics and tetrabenazine are firstline therapy with chemodenervation and surgical lesioning in cases unresponsive to medication therapy.
When unrecognized, hyperglycemic hemiballismus is associated with significant morbidity and mortality. The patients presented in this report were subject to either delayed diagnosis or misdiagnosis as stroke or psychiatric disorder. The rarity of the disorder, lack of evidence delineating pathogenesis and causality, low level of awareness, and varying presentations of patients all contribute to the challenge of recognizing, diagnosing, and treating hemiballismus due to hyperglycemia. This challenge can subsequently result in deteriorating symptoms, prolonged hospital stays, and unnecessary health care costs.
Conclusion
While hemiballismus due to severe persistent hyperglycemia is rare, the goal of this report is to highlight its occurrence in patients with T2DM. Further research can help develop a standardized, effective treatment strategy for these patients. Currently, lowering and maintaining appropriate glucose and HbA1c levels is the most effective treatment approach. Potential areas of research include alternative medical and surgical treatment interventions for patients while glycemic control is being achieved or for those who fail to benefit from glycemic control alone. Some success has been demonstrated with the use of antidopaminergic medications such as atypical antipsychotics and tetrabenazine and these medications should be considered when tight, sustained glycemic control alone is not successful in treating this disorder in the acute stages. Hopefully, with increasing awareness and recognition of hemiballismus related to hyperglycemia, more large-scale clinical trials can be conducted that will result in an effective treatment strategy for this devastating disorder.
Hemiballismus is an acquired hyperkinetic movement disorder characterized by unilateral, involuntary, often large-amplitude limb movements. Ballistic movements are now considered to be on the choreiform spectrum.1 Movements usually involve both the arm and leg, and in half of cases, facial movements such as tongue clucking and grimacing are seen.2,3 Presentations of hemiballismus vary in severity from intermittent to nearly continuous movements, which, in some cases, may lead to exhaustion, injury, or disability. Some patients are unable to ambulate or feed themselves with the affected limb.
Background
The 2 most common causes of hemichorea-hemiballismus are stroke and hyperglycemia, with an incidence of 4% and unknown incidence, respectively.1,3,4 Other causes include HIV, traumatic brain injury, encephalitis, vasculitis, mass effect, multiple sclerosis, and adverse drug reactions. 4-7 Acute or subacute hemiballismus is classically attributed to a lesion in subthalamic nucleus (STN), but this is true only in a minority of cases. Hemiballismus can be caused by any abnormality in various subnuclei of the basal ganglia, including the classic location in the STN, striatum, and globus pallidus.4 Evidence shows the lesions typically involve a functional network connected to the posterolateral putamen.8
Although not commonly recognized, hyperglycemia in patients with type 2 diabetes mellitus (T2DM) is the second most common cause of hemichoreahemiballismus. 3 Over the past 90 years, numerous case reports have described patients with DM with acute and subacute onset of hemiballistic and hemichoreiform movements while in a hyperglycemic state or after its resolution. Reported cases have been limited to small numbers of patients with only a few larger-scale reviews of more than 20 patients.7,9 Most reported cases involve geriatric patients and more commonly, females of Eastern Asian descent with an average age of onset of 71 years.4,10 Patients typically present with glucose levels from 500 to 1,000 mg/dL and hemoglobin A1c (HbA1c) levels almost double the normal values. Interestingly, neuroimaging findings in these patients have consistently shown hyperintense signal in the contralateral basal ganglia on T1-weighted magnetic resonance images (MRIs). Noncontrast computed tomography (CT) shows well-defined unilateral increased density in the contralateral basal ganglia without mass effect.1,9,11
This report aims to illustrate and enhance the understanding of hemiballismus associated with hyperglycemia. One patient presented to the US Department of Veterans Affairs (VA) Bay Pines VA Healthcare System (BPVAHCS) in Florida, which motivated us to search for other similar cases. We reviewed the charts of 2 other patients who presented to BPVAHCS over the past 10 years. The first case presented with severe hyperglycemia and abnormal movements that were not clearly diagnosed as hemiballismus. MRI findings were characteristic and assisted in making the diagnosis. The second case was misdiagnosed as hemiballismus secondary to ischemic stroke. The third case was initially diagnosed as conversion disorder until movements worsened and the correct diagnosis of hyperglycemia-induced hemichorea hemiballismus was confirmed by the pathognomonic neuroimaging findings.
Case Presentations
Case 1
A 65-year-old male with a history of uncontrolled T2DM presented with repetitive twitching and kicking movements that involved his left upper and lower extremities for 3 weeks. The patient reported that he did not take his medications or follow the recommended diabetes diet. His HbA1c on admission was 12.2% with a serum glucose of 254 mg/dL. The MRI showed a hyperintense T1 signal within the right basal ganglia including the right caudate with sparing of the internal capsule (Figure 1). There was no associated mass effect or restricted diffusion. It was compatible with a diagnosis of hyperglycemia- induced hemichorea-hemiballismus. The patient was advised to resume taking glipizide 10 mg daily, metformin 1,000 mg by mouth twice daily, and to begin 10 units of 70/30 insulin aspart 15 minutes before meals twice daily, and to follow a low carbohydrate diet, with reduce dietary intake of sugar. At his 1-month follow-up visit, the patient reported an improvement in his involuntary movements. At the 5-month follow-up, the patient’s HbA1c level was 10.4% and his hyperkinetic movements had completely resolved.
Case 2
of T2DM, hypertension, and hyperlipidemia was admitted due to increased jerky movements in the left upper extremity. On admission, his vital signs were within normal limits and his physical examination demonstrated choreoathetoid movements with ballistic components of his left upper extremity. His laboratory results showed a glucose level of 528 mg/dL with a HbA1c of 16.3%. An initial CT obtained in the emergency department (ED) demonstrated a well-defined hyperdensity in the striatal (caudate and lentiform nucleus) region (Figure 2). There was no associated edema/mass effect that would be typical for an intracranial hemorrhage.
An MRI obtained 1 week later showed hyperintense TI signal corresponding to the basal ganglia (Figure 3). In addition, there was a questionable lacunar infarct in the right internal capsule. Due to lack of awareness regarding hyperglycemic associated basal ganglia changes, the patient’s movement disorder was presumed to be ischemic in etiology. The patient was prescribed oral amantadine 100 mg 3 times daily for the hemiballismus in conjunction with treatment of his T2DM. The only follow-up occurred 5 weeks later, which showed no improvement of uncontrollable movements. Imaging at that time (not available) indicated the persistence of the abnormal signal in the right basal ganglia. This patient died later that year without further follow-up.
Case 3
A 78-year-old white male with a history of syncope, transient ischemic attacks (TIAs), and poorly controlled T2DM presented with a 1-month history of progressively worsening involuntary, left-sided movements that began in his left shoulder and advanced to involve his arm, hand, and leg, and the left side of his face with grimacing and clucking of his tongue. Three weeks earlier, the patient had been discharged from the ED with a diagnosis of conversion disorder particularly because he experienced decreased movements when given a dose of Vitamin D. It was overlooked that administration of haloperidol had occurred a few hours before, and because the sounds made by his tongue were not felt to be consistent with a known movement disorder. A MRI of the brain was read as normal.
The patient returned 3 weeks later (the original presentation) due to his inability to perform activities of daily living because of his worsening involuntary movements. On admission, his HbA1c was 11.1% and his glucose was 167 mg/dL. On chart review, it was revealed that the patient’s HbA1c had been > 9% for the past 3 years with an increase from 10.1% to 11.1% in the 3 months preceding the onset of his symptoms.
On admission a MRI showed a unilateral right-sided T1 hyperintensity in the basal ganglia, no acute ischemia (Figure 4). In retrospect, subtle increased T1 signal can be seen on the earlier MRI (Figure 5). In view of the patient’s left-sided symptoms, DM, and MRI findings, a diagnosis of hyperglycemia-induced hemichorea- hemiballismus was made as the etiology of the patient’s symptoms.
The patient was prescribed numerous medications to control his hyperkinesia including (and in combination): benztropine, gabapentin, baclofen, diphenhydramine, benzodiazepines, risperidone, olanzapine, and valproic acid, which did not control his movements. Ultimately, his hyperglycemic hemiballismus improved with tight glycemic control and oral tetrabenazine 12.5 mg twice daily. This patient underwent a protracted course of treatment with 17 days of inpatient medical admission, 3 weeks inpatient rehabilitation, and subsequent transfer to an assisted living facility.
Discussion
The 3 cases presented in this report contribute to the evidence that severe persistent hyperglycemia can result in movement disorders that mimic those seen after basal ganglia strokes. As with Case 2, past literature describes many cases of acute hyperglycemic episodes with glucose ranging from 500 to 1,000 mg/mL presenting with hemiballismus.1,3 However, there are many cases that describe hemiballismus occurring after glycemic correction, persisting despite glycemic correction, and presenting without an acute hyperglycemic episode, but in the setting of elevated HbA1c, as in Case 3.12,13 Notably, all 3 cases in this series had marked elevation in their HbA1c levels, which suggests that a more chronic hyperglycemic state or multiple shorter periods of hyperglycemia may be necessary to produce the described hyperkinetic movements.
Case reports describe the pathognomonic T1 hyperintensity of the basal ganglia that is identified in all 3 cases presented here. While the exact etiology remains unclear, the to metabolic derangements caused by hyperviscosity of the blood in the small end arteries feeding the basal ganglia.3,11 These abnormalities in turn interrupt the signaling cascade with abnormal firing rates or firing patterns, leading to reduced inhibition of the motor thalamus and ultimately present as hemiballismus.1,3,7 While most cases presented with unilateral hyperkinesis and associated contralateral basal ganglia abnormalities, there are reports of both unilateral and bilateral movements associated with bilateral basal ganglia hyperintensities on imaging. 9 The predilection for unilateral brain lesions may be explained by the varying degree of small vessel disease in different areas of the brain leading to perfusion deficits worsened by hyper viscosity. Further research into this is required to elucidate the exact pathophysiologic mechanism.
The course of disease for patients ranges from resolution within hours of tight glycemic control to persistent movements for > 3 months with a gradual improvement in severity.12,13 Treatments center on the importance of tight glycemic control to protect against the protracted course described in Case 3. Swift recognition of this rare condition is critical because improved glycemic control decreases the severity and duration of this disease. The significant disability associated with Case 3 highlights the need for prompt recognition and early, aggressive glycemic management to prevent the progression of hemiballismus. In addition to glycemic control, various CNS medications such as typical and atypical antipsychotics and tetrabenazine are firstline therapy with chemodenervation and surgical lesioning in cases unresponsive to medication therapy.
When unrecognized, hyperglycemic hemiballismus is associated with significant morbidity and mortality. The patients presented in this report were subject to either delayed diagnosis or misdiagnosis as stroke or psychiatric disorder. The rarity of the disorder, lack of evidence delineating pathogenesis and causality, low level of awareness, and varying presentations of patients all contribute to the challenge of recognizing, diagnosing, and treating hemiballismus due to hyperglycemia. This challenge can subsequently result in deteriorating symptoms, prolonged hospital stays, and unnecessary health care costs.
Conclusion
While hemiballismus due to severe persistent hyperglycemia is rare, the goal of this report is to highlight its occurrence in patients with T2DM. Further research can help develop a standardized, effective treatment strategy for these patients. Currently, lowering and maintaining appropriate glucose and HbA1c levels is the most effective treatment approach. Potential areas of research include alternative medical and surgical treatment interventions for patients while glycemic control is being achieved or for those who fail to benefit from glycemic control alone. Some success has been demonstrated with the use of antidopaminergic medications such as atypical antipsychotics and tetrabenazine and these medications should be considered when tight, sustained glycemic control alone is not successful in treating this disorder in the acute stages. Hopefully, with increasing awareness and recognition of hemiballismus related to hyperglycemia, more large-scale clinical trials can be conducted that will result in an effective treatment strategy for this devastating disorder.
1. Hawley JS, Weiner WJ. Hemiballismus: current concepts and review. Parkinsonism Relat Disord. 2012;18(2):125‐129. doi:10.1016/j.parkreldis.2011.08.015
2. Gasca-Salas C, Lang AE. Paroxysmal Hemiballism/ Hemichorea Resulting from Transient Ischemic Attacks. Mov Disord Clin Pract. 2015;3(3):303‐305. doi:10.1002/mdc3.12268
3. Garcia-Grimshaw MA, Jimenez-Ruiz A, Ornelas-Velazquez A, Luna-Armenta A, Gutierrez-Manjarrez FA. New-onset diabetes presenting as monoballism secondary to a mixed hyperglycemic crisis. Cureus. 2018;10(6):e2882. doi:10.7759/cureus.2882
4. Postuma RB, Lang AE. Hemiballism: revisiting a classic disorder. Lancet Neurol. 2003;2(11):661‐668. doi:10.1016/s1474-4422(03)00554-4
5. Gallo BV, Shulman LM, Weiner WJ, Petito CK, Berger JR. HIV encephalitis presenting with severe generalized chorea. Neurology. 1996;46(4):1163‐1165. doi:10.1212/wnl.46.4.1163
6. Provenzale JM, Glass JP. Hemiballismus: CT and MR findings. J Comput Assist Tomogr. 1995;19(4):537‐540.
7. Hodde M, Rowe KE, Surapaneni K, Terrigno P, Brighenti A, Altschuler EL. Management of severe hemiballismus: treatment challenges in the acute inpatient rehabilitation setting: a case presentation. PMR. 2017;9(7):732‐735. doi:10.1016/j.pmrj.2016.10.023
8. Laganiere S, Boes AD, Fox MD. Network localization of hemichorea-hemiballismus. Neurology. 2016;86(23):2187‐2195. doi:10.1212/WNL.0000000000002741
9. Cosentino C, Torres L, Nuñez Y, Suarez R, Velez M, Flores M. Hemichorea/hemiballism associated with hyperglycemia: report of 20 cases. Tremor Other Hyperkinet Mov (NY). 2016;6:402. doi:10.7916/D8DN454P
10. Oh SH, Lee KY, Im JH, Lee MS. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a metaanalysis of 53 cases including four present cases. J Neurol Sci. 2002;200(1-2):57‐62. doi:10.1016/s0022-510x(02)00133-8
11. Carrion DM, Carrion AF. Non-ketotic hyperglycaemia hemichorea-hemiballismus and acute ischaemic stroke. BMJ Case Rep. 2013;2013:bcr2012008359. doi:10.1136/bcr-2012-008359
12. Cho HS, Hong CT, Chan L. Hemichorea after hyperglycemia correction: a case report and a short review of hyperglycemia-related hemichorea at the euglycemic state. Medicine (Baltimore). 2018;97(10):e0076. doi:10.1097/MD.0000000000010076
13. Lin YC, Lin YC. Prolonged hemiballism after the remission of non-ketotic hyperosmolar syndrome. BMJ Case Rep. 2012;2012:bcr0120125627. doi:10.1136/bcr.01.2012.5627
1. Hawley JS, Weiner WJ. Hemiballismus: current concepts and review. Parkinsonism Relat Disord. 2012;18(2):125‐129. doi:10.1016/j.parkreldis.2011.08.015
2. Gasca-Salas C, Lang AE. Paroxysmal Hemiballism/ Hemichorea Resulting from Transient Ischemic Attacks. Mov Disord Clin Pract. 2015;3(3):303‐305. doi:10.1002/mdc3.12268
3. Garcia-Grimshaw MA, Jimenez-Ruiz A, Ornelas-Velazquez A, Luna-Armenta A, Gutierrez-Manjarrez FA. New-onset diabetes presenting as monoballism secondary to a mixed hyperglycemic crisis. Cureus. 2018;10(6):e2882. doi:10.7759/cureus.2882
4. Postuma RB, Lang AE. Hemiballism: revisiting a classic disorder. Lancet Neurol. 2003;2(11):661‐668. doi:10.1016/s1474-4422(03)00554-4
5. Gallo BV, Shulman LM, Weiner WJ, Petito CK, Berger JR. HIV encephalitis presenting with severe generalized chorea. Neurology. 1996;46(4):1163‐1165. doi:10.1212/wnl.46.4.1163
6. Provenzale JM, Glass JP. Hemiballismus: CT and MR findings. J Comput Assist Tomogr. 1995;19(4):537‐540.
7. Hodde M, Rowe KE, Surapaneni K, Terrigno P, Brighenti A, Altschuler EL. Management of severe hemiballismus: treatment challenges in the acute inpatient rehabilitation setting: a case presentation. PMR. 2017;9(7):732‐735. doi:10.1016/j.pmrj.2016.10.023
8. Laganiere S, Boes AD, Fox MD. Network localization of hemichorea-hemiballismus. Neurology. 2016;86(23):2187‐2195. doi:10.1212/WNL.0000000000002741
9. Cosentino C, Torres L, Nuñez Y, Suarez R, Velez M, Flores M. Hemichorea/hemiballism associated with hyperglycemia: report of 20 cases. Tremor Other Hyperkinet Mov (NY). 2016;6:402. doi:10.7916/D8DN454P
10. Oh SH, Lee KY, Im JH, Lee MS. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a metaanalysis of 53 cases including four present cases. J Neurol Sci. 2002;200(1-2):57‐62. doi:10.1016/s0022-510x(02)00133-8
11. Carrion DM, Carrion AF. Non-ketotic hyperglycaemia hemichorea-hemiballismus and acute ischaemic stroke. BMJ Case Rep. 2013;2013:bcr2012008359. doi:10.1136/bcr-2012-008359
12. Cho HS, Hong CT, Chan L. Hemichorea after hyperglycemia correction: a case report and a short review of hyperglycemia-related hemichorea at the euglycemic state. Medicine (Baltimore). 2018;97(10):e0076. doi:10.1097/MD.0000000000010076
13. Lin YC, Lin YC. Prolonged hemiballism after the remission of non-ketotic hyperosmolar syndrome. BMJ Case Rep. 2012;2012:bcr0120125627. doi:10.1136/bcr.01.2012.5627
36-year-old man • persistent dry cough • frequent sinus congestion • hemoptysis
THE CASE
A 36-year-old nonsmoking white man presented with an episodic 3-month history of dry cough and nasal allergy symptoms. He reported a past history of sinus allergies but no history of asthma. His illness began with a flu-like syndrome, and he had been treated with antibiotics (amoxicillin and azithromycin) and oral steroids (methylprednisolone) by 2 other physicians for “viral syndrome” and “bronchitis.”
The patient reported some tactile fever initially but none thereafter. Symptoms included episodic wheezing but no overt shortness of breath. In addition to the persistent dry cough, he complained of frequent sinus congestion, post-nasal drip, and sneezing. He became concerned when he noticed a fleck of blood in his phlegm.
Physical exam was unimpressive, except for nasal congestion. His breath sounds were clear. Chest x-ray showed a benign-appearing granuloma in the right lower lobe (no previous films available for comparison). Peak-flow measurements taken in the office were persistently low (58%-70%) but improved with steroids and inhaled albuterol.
Over the following 7 weeks, the patient experienced waxing and waning symptoms. At his follow-up visit, he appeared well; chest auscultation revealed normal breath sounds. He was treated with an additional round of antibiotics (levofloxacin), oral steroids, nasal steroids, and inhaled albuterol.
At 13 weeks from his initial presentation, he developed frank hemoptysis and was diagnosed with a right lower-lobe pneumonia in the emergency department. While hospitalized, his clinical status deteriorated, requiring chest tube placement for a large pleural effusion.
Shortly thereafter, he underwent right middle and lower lobectomies and decortication. Multiple organisms were cultured from the pleural fluid. Tuberculosis testing and acid-fast bacilli stains were negative. No malignant cells were identified. Pathologic examination of the resected lung tissue confirmed the chest x-ray finding of a benign calcified granuloma. Additional testing, including a thin barium esophagram, was performed.
THE DIAGNOSIS
Results of the esophagram revealed a congenital bronchoesophageal fistula (C-BEF) between the patient’s esophagus and right mainstem bronchus, located 15 cm distal to his trachea.
Continue to: DISCUSSION
DISCUSSION
Fistulous connections between the esophagus and bronchi are rare but may arise in the setting of malignancy, trauma, inflammation, or congenital malformation.1 While the precise etiology of C-BEF remains unknown, it is believed to be a consequence of failed tracheoesophageal separation during the early stages of embryonic development.
Prevalence and epidemiology. C-BEF has been reported to occur in 1 in 3000 to 4000 live births, often with concomitant esophageal atresia.2 Infants with esophageal atresia demonstrate clinically significant respiratory symptoms and failure to thrive. However, C-BEF without esophageal atresia may be asymptomatic for years to decades.
Age at diagnosis ranges from 9 days to 83 years.3 Several explanations exist for the prolonged asymptomatic phase of this disease: (1) presence of a membrane overlying the fistula during childhood that subsequently ruptures; (2) presence of a proximal fold of esophageal mucosa overlapping the orifice; (3) antigravitational or upward extension of the fistulous tract from the esophagus; and (4) spasm of the smooth muscle of the fistula.4
Four subtypes. Type I fistulas are associated with a wide-necked congenital diverticulum of the esophagus, which may become inflamed and allow perforation into the nearby lung. Type II fistulas (most common) consist of a short tract running directly from the esophagus to a nearby lobar or segmental bronchus. Type III fistulas involve a communication between the esophagus and a cystic structure within the lung parenchyma. Type IV fistulas run from the esophagus into a sequestered pulmonary segment.1 Our patient had a type II fistula.
Is there a nonspecific cough? The most common signs and symptoms of C-BEF are nonspecific cough, cough after ingestion of fluids or meals, and hemoptysis.5,6 Symptoms may persist for decades prior to diagnosis, and the indolent course of C-BEF may lead to fatal complications such as recalcitrant pneumonia, bronchiectasis, and abscess formation.
Continue to: One test bests others for diagnosis
One test bests others for diagnosis. Plain chest x-ray may indicate enlarged lymph nodes or surrounding airspace disease but will not be able to identify a C-BEF. Computed tomography (CT) of the chest may detect the presence of a C-BEF but does not rule it out. Barium esophagram is the most sensitive test for BEF. Esophagoscopy and bronchoscopy may be helpful once the BEF has been identified, but neither has demonstrated reliability as a first-line test. In this particular case, the C-BEF was not seen on chest CT but was later
To confirm the congenital nature of the fistula, histopathology should be examined. C-BEFs will have a mucosal layer and definitive muscularis layer within the fistulous tract.3,5
Treatment. The preferred method of treatment for C-BEF is thoracotomy with resection of the fistula and insertion of a pleural or muscular flap graft to close the defects in the bronchus and esophagus.7 Alternatively, obliteration of the esophageal defect can be performed using biological glue or silver nitrate. Prognosis after surgical repair is excellent.
Our patient
Two weeks after hospital discharge, the patient was re-admitted for hydropneumothorax and underwent additional surgeries. Unfortunately, he died in the ICU due to a tension pneumothorax while intubated.
THE TAKEAWAY
C-BEF is a rare, insidious condition that may remain asymptomatic into adulthood. After common causes are ruled out, patients with adult-onset nonspecific cough, episodes of coughing after eating/drinking, and hemoptysis should be evaluated for BEF. The most useful diagnostic investigation is barium esophagram. Once C-BEF is identified, prompt surgical management is warranted. Because C-BEF persisting into adulthood is so rare, recommendations regarding diagnosis and treatment are based on expert opinion.
CORRESPONDENCE
Rade N. Pejic, MD, MMM, Department of Family Medicine, Tulane University School of Medicine, 1430 Tulane Avenue, Mailbox #8033, New Orleans, LA 70112; [email protected].
1. Braimbridge MV, Keith HI. Oesophago-bronchial fistula in the adult. Thorax. 1965;20:226-233.
2. Taira N, Kawasaki H, Atsumi E, et al. A rare case of congenital bronchoesophageal fistula in an adult. Int J Surg Case Rep. 2017;36:182-184.
3. Risher WH, Arensman RM, Ochsner JL. Congenital bronchoesophageal fistula. Ann Thorac Surg. 1990;49:500-505.
4. Paul M, John S, Ashton R. Recurrent pneumonia in a 51-year-old woman due to congenital bronchoesophageal fistula. Respir Care. 2011;56:1203-1205.
5. Rämö OJ, Salo JA, Mattila SP. Congenital bronchoesophageal fistula in the adult. Ann Thorac Surg. 1995;59:887-889.
6. Zhang B-S, Zhou N-K, Yu C-H. Congenital bronchoesophageal fistula in adults. World J Gastroenterol. 2011;17:1358-1361.
7. Su L, Wei X-Q, Zhi X-Y, et al. Congenital bronchoesophageal fistula in an adult: a case report. World J Gastroenterol. 2007;13:3776–3777.
THE CASE
A 36-year-old nonsmoking white man presented with an episodic 3-month history of dry cough and nasal allergy symptoms. He reported a past history of sinus allergies but no history of asthma. His illness began with a flu-like syndrome, and he had been treated with antibiotics (amoxicillin and azithromycin) and oral steroids (methylprednisolone) by 2 other physicians for “viral syndrome” and “bronchitis.”
The patient reported some tactile fever initially but none thereafter. Symptoms included episodic wheezing but no overt shortness of breath. In addition to the persistent dry cough, he complained of frequent sinus congestion, post-nasal drip, and sneezing. He became concerned when he noticed a fleck of blood in his phlegm.
Physical exam was unimpressive, except for nasal congestion. His breath sounds were clear. Chest x-ray showed a benign-appearing granuloma in the right lower lobe (no previous films available for comparison). Peak-flow measurements taken in the office were persistently low (58%-70%) but improved with steroids and inhaled albuterol.
Over the following 7 weeks, the patient experienced waxing and waning symptoms. At his follow-up visit, he appeared well; chest auscultation revealed normal breath sounds. He was treated with an additional round of antibiotics (levofloxacin), oral steroids, nasal steroids, and inhaled albuterol.
At 13 weeks from his initial presentation, he developed frank hemoptysis and was diagnosed with a right lower-lobe pneumonia in the emergency department. While hospitalized, his clinical status deteriorated, requiring chest tube placement for a large pleural effusion.
Shortly thereafter, he underwent right middle and lower lobectomies and decortication. Multiple organisms were cultured from the pleural fluid. Tuberculosis testing and acid-fast bacilli stains were negative. No malignant cells were identified. Pathologic examination of the resected lung tissue confirmed the chest x-ray finding of a benign calcified granuloma. Additional testing, including a thin barium esophagram, was performed.
THE DIAGNOSIS
Results of the esophagram revealed a congenital bronchoesophageal fistula (C-BEF) between the patient’s esophagus and right mainstem bronchus, located 15 cm distal to his trachea.
Continue to: DISCUSSION
DISCUSSION
Fistulous connections between the esophagus and bronchi are rare but may arise in the setting of malignancy, trauma, inflammation, or congenital malformation.1 While the precise etiology of C-BEF remains unknown, it is believed to be a consequence of failed tracheoesophageal separation during the early stages of embryonic development.
Prevalence and epidemiology. C-BEF has been reported to occur in 1 in 3000 to 4000 live births, often with concomitant esophageal atresia.2 Infants with esophageal atresia demonstrate clinically significant respiratory symptoms and failure to thrive. However, C-BEF without esophageal atresia may be asymptomatic for years to decades.
Age at diagnosis ranges from 9 days to 83 years.3 Several explanations exist for the prolonged asymptomatic phase of this disease: (1) presence of a membrane overlying the fistula during childhood that subsequently ruptures; (2) presence of a proximal fold of esophageal mucosa overlapping the orifice; (3) antigravitational or upward extension of the fistulous tract from the esophagus; and (4) spasm of the smooth muscle of the fistula.4
Four subtypes. Type I fistulas are associated with a wide-necked congenital diverticulum of the esophagus, which may become inflamed and allow perforation into the nearby lung. Type II fistulas (most common) consist of a short tract running directly from the esophagus to a nearby lobar or segmental bronchus. Type III fistulas involve a communication between the esophagus and a cystic structure within the lung parenchyma. Type IV fistulas run from the esophagus into a sequestered pulmonary segment.1 Our patient had a type II fistula.
Is there a nonspecific cough? The most common signs and symptoms of C-BEF are nonspecific cough, cough after ingestion of fluids or meals, and hemoptysis.5,6 Symptoms may persist for decades prior to diagnosis, and the indolent course of C-BEF may lead to fatal complications such as recalcitrant pneumonia, bronchiectasis, and abscess formation.
Continue to: One test bests others for diagnosis
One test bests others for diagnosis. Plain chest x-ray may indicate enlarged lymph nodes or surrounding airspace disease but will not be able to identify a C-BEF. Computed tomography (CT) of the chest may detect the presence of a C-BEF but does not rule it out. Barium esophagram is the most sensitive test for BEF. Esophagoscopy and bronchoscopy may be helpful once the BEF has been identified, but neither has demonstrated reliability as a first-line test. In this particular case, the C-BEF was not seen on chest CT but was later
To confirm the congenital nature of the fistula, histopathology should be examined. C-BEFs will have a mucosal layer and definitive muscularis layer within the fistulous tract.3,5
Treatment. The preferred method of treatment for C-BEF is thoracotomy with resection of the fistula and insertion of a pleural or muscular flap graft to close the defects in the bronchus and esophagus.7 Alternatively, obliteration of the esophageal defect can be performed using biological glue or silver nitrate. Prognosis after surgical repair is excellent.
Our patient
Two weeks after hospital discharge, the patient was re-admitted for hydropneumothorax and underwent additional surgeries. Unfortunately, he died in the ICU due to a tension pneumothorax while intubated.
THE TAKEAWAY
C-BEF is a rare, insidious condition that may remain asymptomatic into adulthood. After common causes are ruled out, patients with adult-onset nonspecific cough, episodes of coughing after eating/drinking, and hemoptysis should be evaluated for BEF. The most useful diagnostic investigation is barium esophagram. Once C-BEF is identified, prompt surgical management is warranted. Because C-BEF persisting into adulthood is so rare, recommendations regarding diagnosis and treatment are based on expert opinion.
CORRESPONDENCE
Rade N. Pejic, MD, MMM, Department of Family Medicine, Tulane University School of Medicine, 1430 Tulane Avenue, Mailbox #8033, New Orleans, LA 70112; [email protected].
THE CASE
A 36-year-old nonsmoking white man presented with an episodic 3-month history of dry cough and nasal allergy symptoms. He reported a past history of sinus allergies but no history of asthma. His illness began with a flu-like syndrome, and he had been treated with antibiotics (amoxicillin and azithromycin) and oral steroids (methylprednisolone) by 2 other physicians for “viral syndrome” and “bronchitis.”
The patient reported some tactile fever initially but none thereafter. Symptoms included episodic wheezing but no overt shortness of breath. In addition to the persistent dry cough, he complained of frequent sinus congestion, post-nasal drip, and sneezing. He became concerned when he noticed a fleck of blood in his phlegm.
Physical exam was unimpressive, except for nasal congestion. His breath sounds were clear. Chest x-ray showed a benign-appearing granuloma in the right lower lobe (no previous films available for comparison). Peak-flow measurements taken in the office were persistently low (58%-70%) but improved with steroids and inhaled albuterol.
Over the following 7 weeks, the patient experienced waxing and waning symptoms. At his follow-up visit, he appeared well; chest auscultation revealed normal breath sounds. He was treated with an additional round of antibiotics (levofloxacin), oral steroids, nasal steroids, and inhaled albuterol.
At 13 weeks from his initial presentation, he developed frank hemoptysis and was diagnosed with a right lower-lobe pneumonia in the emergency department. While hospitalized, his clinical status deteriorated, requiring chest tube placement for a large pleural effusion.
Shortly thereafter, he underwent right middle and lower lobectomies and decortication. Multiple organisms were cultured from the pleural fluid. Tuberculosis testing and acid-fast bacilli stains were negative. No malignant cells were identified. Pathologic examination of the resected lung tissue confirmed the chest x-ray finding of a benign calcified granuloma. Additional testing, including a thin barium esophagram, was performed.
THE DIAGNOSIS
Results of the esophagram revealed a congenital bronchoesophageal fistula (C-BEF) between the patient’s esophagus and right mainstem bronchus, located 15 cm distal to his trachea.
Continue to: DISCUSSION
DISCUSSION
Fistulous connections between the esophagus and bronchi are rare but may arise in the setting of malignancy, trauma, inflammation, or congenital malformation.1 While the precise etiology of C-BEF remains unknown, it is believed to be a consequence of failed tracheoesophageal separation during the early stages of embryonic development.
Prevalence and epidemiology. C-BEF has been reported to occur in 1 in 3000 to 4000 live births, often with concomitant esophageal atresia.2 Infants with esophageal atresia demonstrate clinically significant respiratory symptoms and failure to thrive. However, C-BEF without esophageal atresia may be asymptomatic for years to decades.
Age at diagnosis ranges from 9 days to 83 years.3 Several explanations exist for the prolonged asymptomatic phase of this disease: (1) presence of a membrane overlying the fistula during childhood that subsequently ruptures; (2) presence of a proximal fold of esophageal mucosa overlapping the orifice; (3) antigravitational or upward extension of the fistulous tract from the esophagus; and (4) spasm of the smooth muscle of the fistula.4
Four subtypes. Type I fistulas are associated with a wide-necked congenital diverticulum of the esophagus, which may become inflamed and allow perforation into the nearby lung. Type II fistulas (most common) consist of a short tract running directly from the esophagus to a nearby lobar or segmental bronchus. Type III fistulas involve a communication between the esophagus and a cystic structure within the lung parenchyma. Type IV fistulas run from the esophagus into a sequestered pulmonary segment.1 Our patient had a type II fistula.
Is there a nonspecific cough? The most common signs and symptoms of C-BEF are nonspecific cough, cough after ingestion of fluids or meals, and hemoptysis.5,6 Symptoms may persist for decades prior to diagnosis, and the indolent course of C-BEF may lead to fatal complications such as recalcitrant pneumonia, bronchiectasis, and abscess formation.
Continue to: One test bests others for diagnosis
One test bests others for diagnosis. Plain chest x-ray may indicate enlarged lymph nodes or surrounding airspace disease but will not be able to identify a C-BEF. Computed tomography (CT) of the chest may detect the presence of a C-BEF but does not rule it out. Barium esophagram is the most sensitive test for BEF. Esophagoscopy and bronchoscopy may be helpful once the BEF has been identified, but neither has demonstrated reliability as a first-line test. In this particular case, the C-BEF was not seen on chest CT but was later
To confirm the congenital nature of the fistula, histopathology should be examined. C-BEFs will have a mucosal layer and definitive muscularis layer within the fistulous tract.3,5
Treatment. The preferred method of treatment for C-BEF is thoracotomy with resection of the fistula and insertion of a pleural or muscular flap graft to close the defects in the bronchus and esophagus.7 Alternatively, obliteration of the esophageal defect can be performed using biological glue or silver nitrate. Prognosis after surgical repair is excellent.
Our patient
Two weeks after hospital discharge, the patient was re-admitted for hydropneumothorax and underwent additional surgeries. Unfortunately, he died in the ICU due to a tension pneumothorax while intubated.
THE TAKEAWAY
C-BEF is a rare, insidious condition that may remain asymptomatic into adulthood. After common causes are ruled out, patients with adult-onset nonspecific cough, episodes of coughing after eating/drinking, and hemoptysis should be evaluated for BEF. The most useful diagnostic investigation is barium esophagram. Once C-BEF is identified, prompt surgical management is warranted. Because C-BEF persisting into adulthood is so rare, recommendations regarding diagnosis and treatment are based on expert opinion.
CORRESPONDENCE
Rade N. Pejic, MD, MMM, Department of Family Medicine, Tulane University School of Medicine, 1430 Tulane Avenue, Mailbox #8033, New Orleans, LA 70112; [email protected].
1. Braimbridge MV, Keith HI. Oesophago-bronchial fistula in the adult. Thorax. 1965;20:226-233.
2. Taira N, Kawasaki H, Atsumi E, et al. A rare case of congenital bronchoesophageal fistula in an adult. Int J Surg Case Rep. 2017;36:182-184.
3. Risher WH, Arensman RM, Ochsner JL. Congenital bronchoesophageal fistula. Ann Thorac Surg. 1990;49:500-505.
4. Paul M, John S, Ashton R. Recurrent pneumonia in a 51-year-old woman due to congenital bronchoesophageal fistula. Respir Care. 2011;56:1203-1205.
5. Rämö OJ, Salo JA, Mattila SP. Congenital bronchoesophageal fistula in the adult. Ann Thorac Surg. 1995;59:887-889.
6. Zhang B-S, Zhou N-K, Yu C-H. Congenital bronchoesophageal fistula in adults. World J Gastroenterol. 2011;17:1358-1361.
7. Su L, Wei X-Q, Zhi X-Y, et al. Congenital bronchoesophageal fistula in an adult: a case report. World J Gastroenterol. 2007;13:3776–3777.
1. Braimbridge MV, Keith HI. Oesophago-bronchial fistula in the adult. Thorax. 1965;20:226-233.
2. Taira N, Kawasaki H, Atsumi E, et al. A rare case of congenital bronchoesophageal fistula in an adult. Int J Surg Case Rep. 2017;36:182-184.
3. Risher WH, Arensman RM, Ochsner JL. Congenital bronchoesophageal fistula. Ann Thorac Surg. 1990;49:500-505.
4. Paul M, John S, Ashton R. Recurrent pneumonia in a 51-year-old woman due to congenital bronchoesophageal fistula. Respir Care. 2011;56:1203-1205.
5. Rämö OJ, Salo JA, Mattila SP. Congenital bronchoesophageal fistula in the adult. Ann Thorac Surg. 1995;59:887-889.
6. Zhang B-S, Zhou N-K, Yu C-H. Congenital bronchoesophageal fistula in adults. World J Gastroenterol. 2011;17:1358-1361.
7. Su L, Wei X-Q, Zhi X-Y, et al. Congenital bronchoesophageal fistula in an adult: a case report. World J Gastroenterol. 2007;13:3776–3777.

Brilliant Green Staining of the Fingernails
Case Report
A 92-year-old Eastern European woman presented to our nail clinic with a history of onychodystrophy and arthralgia of the digits of several months’ duration. Her dermatologic history was notable for irritant hand dermatitis. A prior nail plate clipping with histopathologic examination was negative for fungal elements. Physical examination revealed onychorrhexis of all fingernails as well as onycholysis and subungual hyperkeratosis of the right fourth fingernail. Blue-green staining was incidentally noted on the right second and third fingernails and nail folds (Figure 1). Contact dermoscopy using ultrasound gel revealed translucent areas with sparse pigment, though denser areas had a fine branching pattern (Figure 2). When questioned, the patient reported use of “zelyonka,” a brilliant green solution, to self-treat the nails. Histopathology on repeat nail clippings showed parakeratosis and serum, which was most consistent with her known history of irritant hand dermatitis. Radiographs of the hands revealed osteoarthritis that was most prominent at the distal interphalangeal joints.
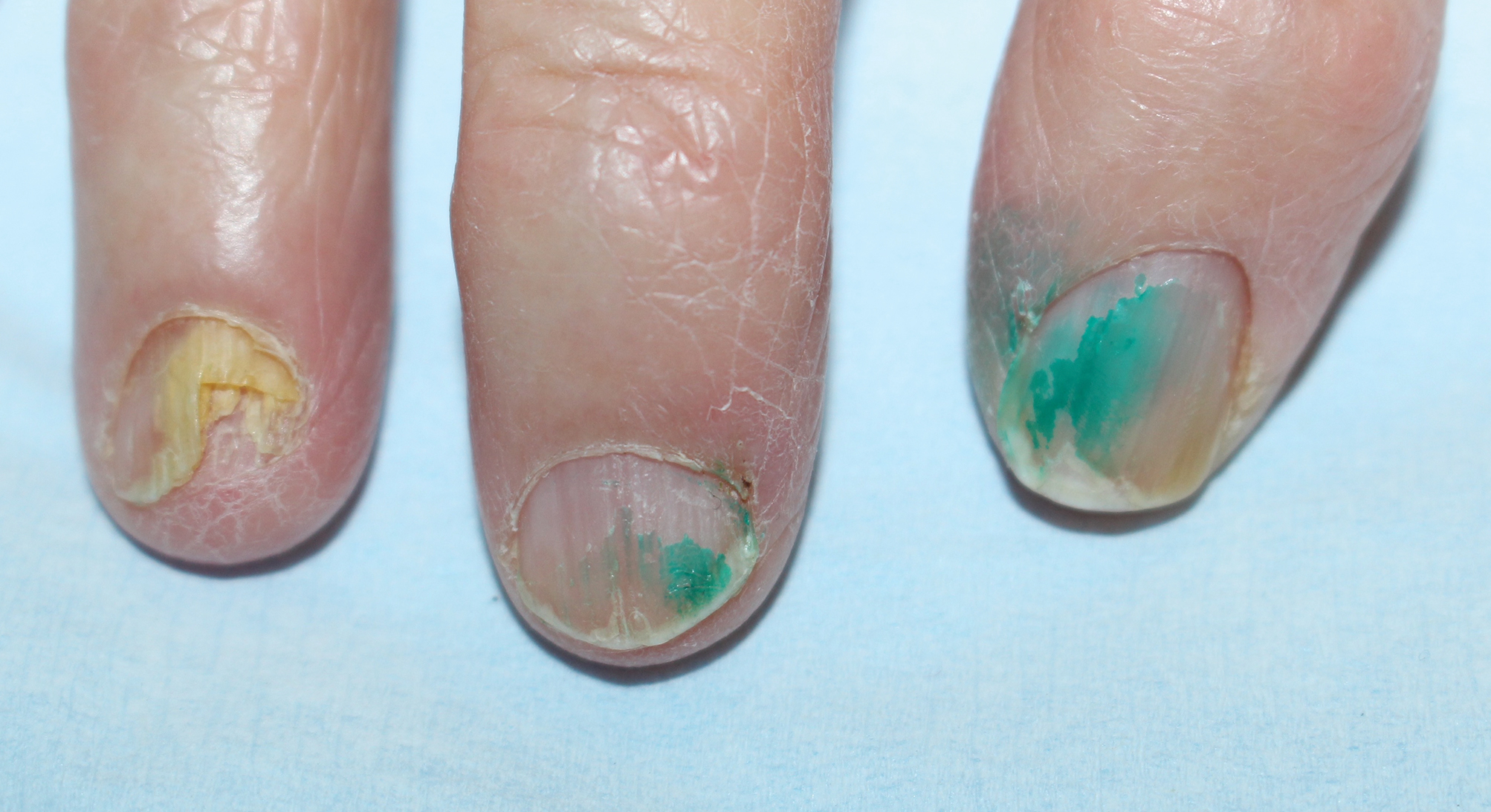
Comment
Brilliant green is a triphenylmethane dye commonly used in Eastern Europe and other regions for the treatment of superficial skin infections and onychomycosis.1 Its use as an antiseptic and wound healing agent has been investigated in the scientific literature since at least the early 20th century.2 Brilliant green typically is applied in a 0.1% to 2% ethanol solution.1 The dye has bactericidal activity against gram-positive organisms, particularly staphylococci and streptococci.2,3 It has been used for the treatment of fungal skin and nail infections since at least the early 20th century, with anecdotal success.4 Although there have been no studies investigating use of brilliant green alone for the treatment of onychomycosis, it is sometimes used in combination with conventional oral agents for this purpose.5 Because of its availability, safety, ease of use, and low cost, brilliant green has been promoted as an antiseptic in resource-poor settings.3 The revival of brilliant green and other antiseptic dyes in these settings has been suggested as an alternative to oral antibiotic agents, to which resistance is rising, and as a potential cancer therapy.6,7 Although brilliant green’s mechanism of action in treating skin infections is unclear, it has been shown to form covalent adducts with thioredoxin reductase 2, a protein conserved from bacteria to humans with an essential function for cellular activity.7
Early case studies suggested that brilliant green was beneficial in treating wounds2; however, this indication is controversial. In a guinea pig study, brilliant green was shown to inhibit wound healing and the formation of granulation tissue.8 It also should be noted that when used topically, brilliant green may cause skin sensitization, necrotic skin reactions, and permanent staining of clothing. It has no known anti-inflammatory properties and also may cause skin irritation.8 Brilliant green may cause blindness if it comes in contact with the eyes.1
Brilliant green has other potential dermatologic indications. For example, a combination of brilliant green and gentian violet, a related dye, has demonstrated efficacy in the treatment of cutaneous hemangiomas in mouse models by blocking expression of angiopoietin-2.7
Dermatologists should be familiar with brilliant green and its common uses as well as adverse effects. Brilliant green is commercially available for a low cost ($5 to $20) in specialty pharmacies or online (eg, Amazon). It is sold alone or in combination with gentian violet and proflavine hemisulfate, and a prescription is not required. Due to its low cost and accessibility, patients may use brilliant green to self-treat dermatologic conditions. Green nails due to staining with brilliant green dye must be distinguished from other etiologies causing green nail discoloration, such as infection with Pseudomonas aeruginosa or Aspergillus, bullous disorders, jaundice, “old” hematomas, nail polish, and other exogenous pigments.
- Balabanova M, Popova L, Tchipeva R. Dyes in dermatology. Clin Dermatol. 2003;21:2-6.
- Browning CH, Gulbransen R, Kennaway EL, et al. Flavine and brilliant green, powerful antiseptics with low toxicity to the tissues: their use in the treatment of infected wounds. Br Med J. 1917;1:73-78.
- Bakker P, Doorne H, Gooskens V, et al. Activity of gentian violet and brilliant green against some microorganisms associated with skin infections. Int J Dermatol. 1992;31:210-213.
- Montgomery RM, Casper EA. Cutaneous manifestations of the fungi causing dermatophytosis and onychomycosis and their treatment. J Am Med Assoc. 1945;128:77-83.
- Tchernev G, Cardoso JC, Ali MM, et al. Primary onychomycosis with granulomatous Tinea faciei. Braz J Infect Dis. 2010;14:546-547.
- Berrios RL, Arbiser JL. Effectiveness of gentian violet and similar products commonly used to treat pyodermas. Dermatol Clin. 2011;29:69-73.
- Maley AM, Arbiser JL. Gentian violet: a 19th century drug re-emerges in the 21st century. Exp Dermatol. 2013;22:775-80.
- Niedner R, Schöpf E. Inhibition of wound healing by antiseptics. Br J Dermatol. 1986;115:41-44
Case Report
A 92-year-old Eastern European woman presented to our nail clinic with a history of onychodystrophy and arthralgia of the digits of several months’ duration. Her dermatologic history was notable for irritant hand dermatitis. A prior nail plate clipping with histopathologic examination was negative for fungal elements. Physical examination revealed onychorrhexis of all fingernails as well as onycholysis and subungual hyperkeratosis of the right fourth fingernail. Blue-green staining was incidentally noted on the right second and third fingernails and nail folds (Figure 1). Contact dermoscopy using ultrasound gel revealed translucent areas with sparse pigment, though denser areas had a fine branching pattern (Figure 2). When questioned, the patient reported use of “zelyonka,” a brilliant green solution, to self-treat the nails. Histopathology on repeat nail clippings showed parakeratosis and serum, which was most consistent with her known history of irritant hand dermatitis. Radiographs of the hands revealed osteoarthritis that was most prominent at the distal interphalangeal joints.
Comment
Brilliant green is a triphenylmethane dye commonly used in Eastern Europe and other regions for the treatment of superficial skin infections and onychomycosis.1 Its use as an antiseptic and wound healing agent has been investigated in the scientific literature since at least the early 20th century.2 Brilliant green typically is applied in a 0.1% to 2% ethanol solution.1 The dye has bactericidal activity against gram-positive organisms, particularly staphylococci and streptococci.2,3 It has been used for the treatment of fungal skin and nail infections since at least the early 20th century, with anecdotal success.4 Although there have been no studies investigating use of brilliant green alone for the treatment of onychomycosis, it is sometimes used in combination with conventional oral agents for this purpose.5 Because of its availability, safety, ease of use, and low cost, brilliant green has been promoted as an antiseptic in resource-poor settings.3 The revival of brilliant green and other antiseptic dyes in these settings has been suggested as an alternative to oral antibiotic agents, to which resistance is rising, and as a potential cancer therapy.6,7 Although brilliant green’s mechanism of action in treating skin infections is unclear, it has been shown to form covalent adducts with thioredoxin reductase 2, a protein conserved from bacteria to humans with an essential function for cellular activity.7
Early case studies suggested that brilliant green was beneficial in treating wounds2; however, this indication is controversial. In a guinea pig study, brilliant green was shown to inhibit wound healing and the formation of granulation tissue.8 It also should be noted that when used topically, brilliant green may cause skin sensitization, necrotic skin reactions, and permanent staining of clothing. It has no known anti-inflammatory properties and also may cause skin irritation.8 Brilliant green may cause blindness if it comes in contact with the eyes.1
Brilliant green has other potential dermatologic indications. For example, a combination of brilliant green and gentian violet, a related dye, has demonstrated efficacy in the treatment of cutaneous hemangiomas in mouse models by blocking expression of angiopoietin-2.7
Dermatologists should be familiar with brilliant green and its common uses as well as adverse effects. Brilliant green is commercially available for a low cost ($5 to $20) in specialty pharmacies or online (eg, Amazon). It is sold alone or in combination with gentian violet and proflavine hemisulfate, and a prescription is not required. Due to its low cost and accessibility, patients may use brilliant green to self-treat dermatologic conditions. Green nails due to staining with brilliant green dye must be distinguished from other etiologies causing green nail discoloration, such as infection with Pseudomonas aeruginosa or Aspergillus, bullous disorders, jaundice, “old” hematomas, nail polish, and other exogenous pigments.
Case Report
A 92-year-old Eastern European woman presented to our nail clinic with a history of onychodystrophy and arthralgia of the digits of several months’ duration. Her dermatologic history was notable for irritant hand dermatitis. A prior nail plate clipping with histopathologic examination was negative for fungal elements. Physical examination revealed onychorrhexis of all fingernails as well as onycholysis and subungual hyperkeratosis of the right fourth fingernail. Blue-green staining was incidentally noted on the right second and third fingernails and nail folds (Figure 1). Contact dermoscopy using ultrasound gel revealed translucent areas with sparse pigment, though denser areas had a fine branching pattern (Figure 2). When questioned, the patient reported use of “zelyonka,” a brilliant green solution, to self-treat the nails. Histopathology on repeat nail clippings showed parakeratosis and serum, which was most consistent with her known history of irritant hand dermatitis. Radiographs of the hands revealed osteoarthritis that was most prominent at the distal interphalangeal joints.
Comment
Brilliant green is a triphenylmethane dye commonly used in Eastern Europe and other regions for the treatment of superficial skin infections and onychomycosis.1 Its use as an antiseptic and wound healing agent has been investigated in the scientific literature since at least the early 20th century.2 Brilliant green typically is applied in a 0.1% to 2% ethanol solution.1 The dye has bactericidal activity against gram-positive organisms, particularly staphylococci and streptococci.2,3 It has been used for the treatment of fungal skin and nail infections since at least the early 20th century, with anecdotal success.4 Although there have been no studies investigating use of brilliant green alone for the treatment of onychomycosis, it is sometimes used in combination with conventional oral agents for this purpose.5 Because of its availability, safety, ease of use, and low cost, brilliant green has been promoted as an antiseptic in resource-poor settings.3 The revival of brilliant green and other antiseptic dyes in these settings has been suggested as an alternative to oral antibiotic agents, to which resistance is rising, and as a potential cancer therapy.6,7 Although brilliant green’s mechanism of action in treating skin infections is unclear, it has been shown to form covalent adducts with thioredoxin reductase 2, a protein conserved from bacteria to humans with an essential function for cellular activity.7
Early case studies suggested that brilliant green was beneficial in treating wounds2; however, this indication is controversial. In a guinea pig study, brilliant green was shown to inhibit wound healing and the formation of granulation tissue.8 It also should be noted that when used topically, brilliant green may cause skin sensitization, necrotic skin reactions, and permanent staining of clothing. It has no known anti-inflammatory properties and also may cause skin irritation.8 Brilliant green may cause blindness if it comes in contact with the eyes.1
Brilliant green has other potential dermatologic indications. For example, a combination of brilliant green and gentian violet, a related dye, has demonstrated efficacy in the treatment of cutaneous hemangiomas in mouse models by blocking expression of angiopoietin-2.7
Dermatologists should be familiar with brilliant green and its common uses as well as adverse effects. Brilliant green is commercially available for a low cost ($5 to $20) in specialty pharmacies or online (eg, Amazon). It is sold alone or in combination with gentian violet and proflavine hemisulfate, and a prescription is not required. Due to its low cost and accessibility, patients may use brilliant green to self-treat dermatologic conditions. Green nails due to staining with brilliant green dye must be distinguished from other etiologies causing green nail discoloration, such as infection with Pseudomonas aeruginosa or Aspergillus, bullous disorders, jaundice, “old” hematomas, nail polish, and other exogenous pigments.
- Balabanova M, Popova L, Tchipeva R. Dyes in dermatology. Clin Dermatol. 2003;21:2-6.
- Browning CH, Gulbransen R, Kennaway EL, et al. Flavine and brilliant green, powerful antiseptics with low toxicity to the tissues: their use in the treatment of infected wounds. Br Med J. 1917;1:73-78.
- Bakker P, Doorne H, Gooskens V, et al. Activity of gentian violet and brilliant green against some microorganisms associated with skin infections. Int J Dermatol. 1992;31:210-213.
- Montgomery RM, Casper EA. Cutaneous manifestations of the fungi causing dermatophytosis and onychomycosis and their treatment. J Am Med Assoc. 1945;128:77-83.
- Tchernev G, Cardoso JC, Ali MM, et al. Primary onychomycosis with granulomatous Tinea faciei. Braz J Infect Dis. 2010;14:546-547.
- Berrios RL, Arbiser JL. Effectiveness of gentian violet and similar products commonly used to treat pyodermas. Dermatol Clin. 2011;29:69-73.
- Maley AM, Arbiser JL. Gentian violet: a 19th century drug re-emerges in the 21st century. Exp Dermatol. 2013;22:775-80.
- Niedner R, Schöpf E. Inhibition of wound healing by antiseptics. Br J Dermatol. 1986;115:41-44
- Balabanova M, Popova L, Tchipeva R. Dyes in dermatology. Clin Dermatol. 2003;21:2-6.
- Browning CH, Gulbransen R, Kennaway EL, et al. Flavine and brilliant green, powerful antiseptics with low toxicity to the tissues: their use in the treatment of infected wounds. Br Med J. 1917;1:73-78.
- Bakker P, Doorne H, Gooskens V, et al. Activity of gentian violet and brilliant green against some microorganisms associated with skin infections. Int J Dermatol. 1992;31:210-213.
- Montgomery RM, Casper EA. Cutaneous manifestations of the fungi causing dermatophytosis and onychomycosis and their treatment. J Am Med Assoc. 1945;128:77-83.
- Tchernev G, Cardoso JC, Ali MM, et al. Primary onychomycosis with granulomatous Tinea faciei. Braz J Infect Dis. 2010;14:546-547.
- Berrios RL, Arbiser JL. Effectiveness of gentian violet and similar products commonly used to treat pyodermas. Dermatol Clin. 2011;29:69-73.
- Maley AM, Arbiser JL. Gentian violet: a 19th century drug re-emerges in the 21st century. Exp Dermatol. 2013;22:775-80.
- Niedner R, Schöpf E. Inhibition of wound healing by antiseptics. Br J Dermatol. 1986;115:41-44
Practice Points
- Chloronychia, or green nail syndrome, is due to Pseudomonas aeruginosaPalatino LT Std infection and is a common etiology of green nail discoloration. Green nail discoloration also may be secondary to use of the antiseptic dye brilliant green.
- Brilliant green is bactericidal but has no known antifungal or anti-inflammatory activity; it should be considered in the differential diagnosis of green nail discoloration and also may cause blindness with eye contact.

Bullous Eruption Caused by an Exotic Hedgehog Purchased as a Household Pet
Case Report
A 37-year-old woman presented to the dermatology clinic with an itchy rash involving the right hand. The rash had been present for 10 days but had become increasingly pruritic and vesicular over the last 5 days. She denied new exposures or other household members with similar symptoms. The patient reported that she had purchased a 4-toed, white-bellied African pygmy hedgehog (Atelerix albiventris) approximately 4 months prior. Upon questioning, she stated that she handled the hedgehog a couple of times a week and always washed her hands with soap and water immediately after. The patient’s medical and personal history were otherwise unremarkable.
Review of systems, including fevers, chills, and night sweats, was negative. Clinical examination revealed erythema with overlying vesicles and pustules on the right radial palm, radial dorsal hand, and interdigital web space of the first and second digit (Figure 1). The eruption was actively discharging serous exudate. No other lesions were present.
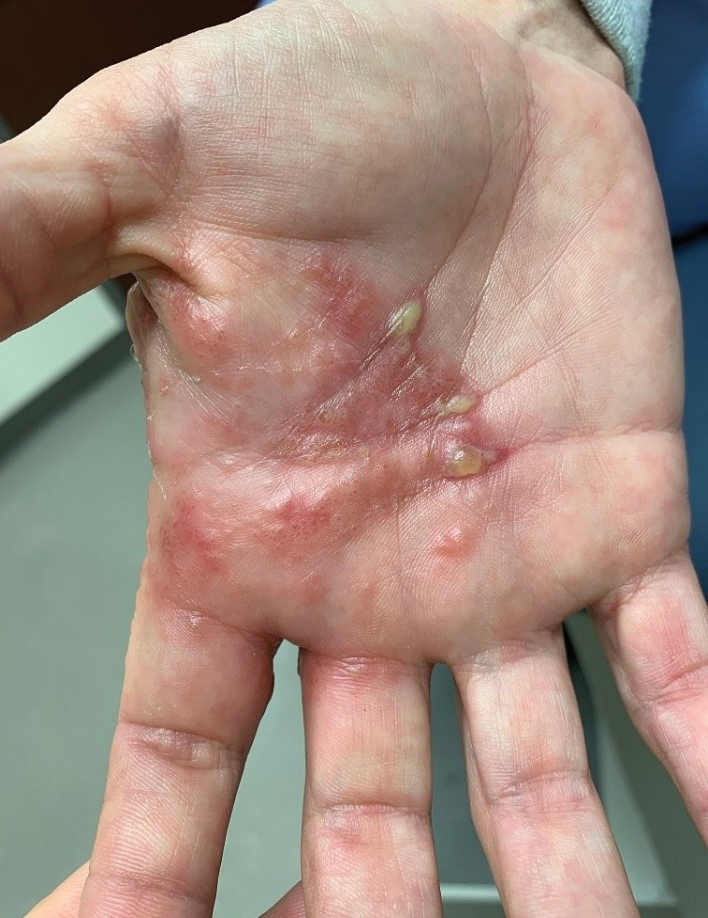
Unspecified acute contact dermatitis was the preliminary diagnosis based on clinical presentation and history. Other entities considered before making the diagnosis included psoriasis, eczema, and an infectious cause. Specimens were taken for bacterial and fungal cultures as well as a specimen for herpes simplex virus by polymerase chain reaction. Due to the intense pruritus and vesicular nature of the rash, the patient was treatedwith a 2-week, 60-40-20 prednisone taper and clobetasol propionate ointment 0.05% twice daily.
At 1-week follow-up, the eruption had improved, but the patient was still experiencing mild pruritus. Physical examination of the affected areas showed erythematous, violaceous, annular patches with slight scale at the periphery; all bullous lesions had resolved (Figure 2). Bacterial culture and herpes simplex virus by polymerase chain reaction were negative.
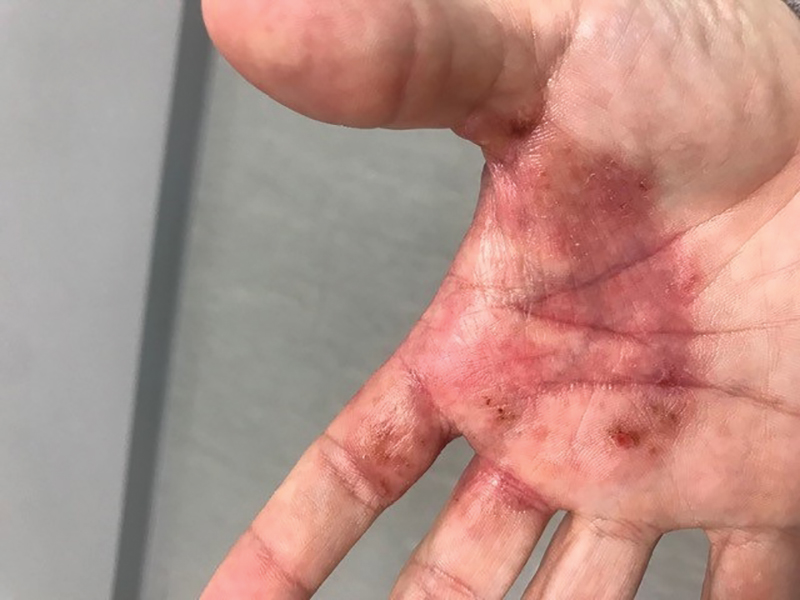
Two weeks after initial consultation, the fungal culture returned positive and showed growth of Trichophyton mentagrophytes. The patient was contacted and returned for re-evaluation. Physical examination showed decreased erythema and no bullous lesions; however, there was increased fine scale throughout the affected area on the right palm and first and second interdigital spaces (Figure 3). She reported mild pruritus. A confirmatory potassium hydroxide (KOH) preparation was positive for fungal hyphae. The patient was subsequently diagnosed with bullous tinea secondary to domestic hedgehog exposure that was now presenting as tinea manuum incognita. After 2 weeks of appropriate systemic and topical antifungal therapy, the patient’s skin eruption markedly improved (Figure 4).
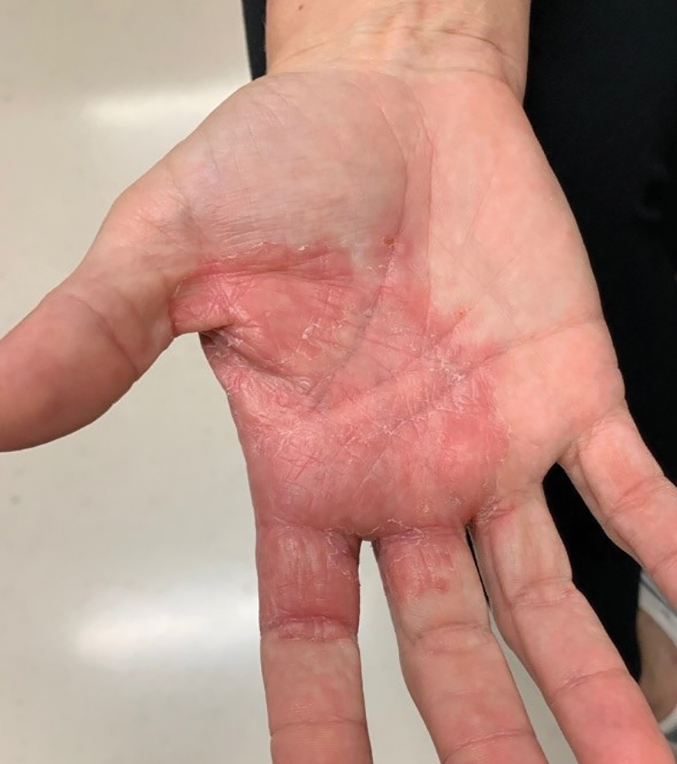
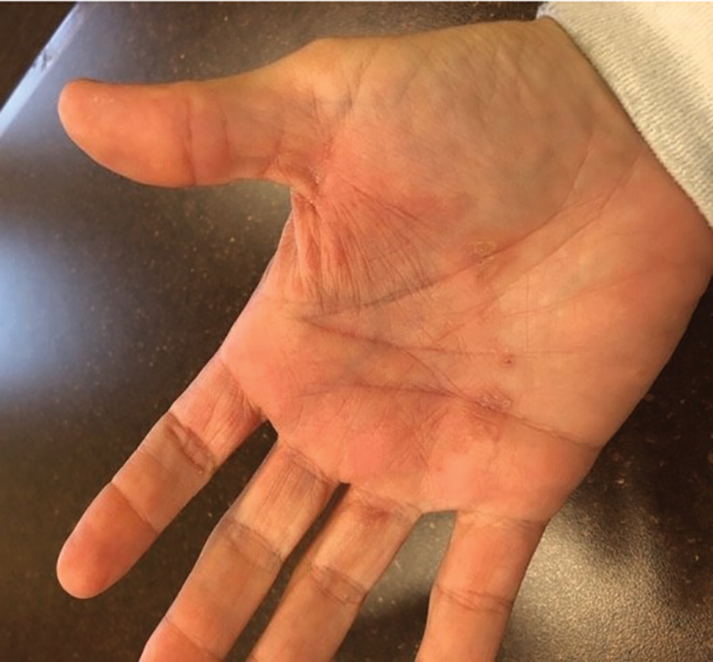
Comment
Tinea manuum is a dermatophytic epidermal infection of the hand. The most common causative organisms are Trichophyton rubrum, T mentagrophytes, and Epidermophyton floccosum. Infection can be acquired from contact with an infected person or animal, fomites, soil, or autoinoculation. Tinea manuum often is associated with tinea pedis. The hand that is used to excoriate the pruritic feet becomes infected, resulting in the classic two feet–one hand syndrome, which this patient did not have.1
Dermatophytes colonize keratin-containing tissues—skin, hair, and nails—utilizing the keratin for nutrients, and they do not invade living tissue in immunocompetent hosts. Dermatophytes cause clinical disease from an allergic host response to fungal antigens or their metabolic products.1 Tinea incognito results from the use of corticosteroids to treat a cutaneous fungal infection. The immunomodulatory effects of corticosteroids alter the appearance of the lesion. Hallmark signs and symptoms of a tinea infection, including scale, prominent border, erythema, and pruritus, can be reduced with corticosteroid use, giving the false impression that the lesion is resolving.2,3
The diagnosis of tinea manuum can be made clinically and often is supported with the findings of a KOH preparation. Scraping from an active scaling border generally provides the best results for obtaining fungal elements. For vesiculobullous lesions, the roof of a vesicle can provide an adequate specimen. Fungal culture and specific dermatophyte testing mediums can be used as confirmatory tests or allow for speciation, which help establish the diagnosis.1
Trichophyton mentagrophytes is a species complex—a group of closely related organisms that share morphologic appearance to the point that boundaries between them often are unclear. It can be identified by gross and microscopic morphology; however, variants of T mentagrophytes (eg, Trichophyton interdigitale, Trichophyton erinacei) require a confirmatory test or molecular analysis to be correctly identified.4-6 The laboratory used at our facility does not routinely attempt to identify the variant due to of lack of clinical significance.7,8
Anthropophilic fungi such as T rubrum, E floccosum, and T interdigitale generally do not cause a robust immunologic reaction. Infection usually is chronic in nature, though cases of pustular and vesicular tinea have been described.9,10Trichophyton erinacei and T mentagrophytes are zoophilic dermatophytes that cause an acute host response and are more likely to present with vesiculobullous lesions. Trichophyton erinacei is the most common fungal pathogen associated with A albiventris and has been isolated from its epidermal mites and quills,11,12 which likely facilitates interspecies transmission and compromises the cutaneous barrier of human hosts when the hedgehog is handled.
Atelerix albiventris is the most common domesticated hedgehog in the United States. These mild-mannered, nocturnal insectivores are unique, low-maintenance pets that have recently gained popularity. They are notable for their propensity to curl into a ball when frightened (Figure 5). The spines are not barbed and do not detach, as those of a porcupine do, but are still capable of piercing the skin. Atelerix albiventris is known to cause zoonotic dermatosis in humans and should be handled with gloves.13 Performing a KOH preparation early in the diagnostic workup can help initiate antifungal therapy, as results of fungal culture can take several weeks.

Conclusion
This case illustrates the importance of close follow-up of skin lesions that only partially respond to initial treatment and maintaining a high index of suspicion as exotic pets become popular.
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier; 2018:1329-1363.
- Habif T. Superficial fungal infections. In: Habif T. Clinical Dermatology. 6th ed. Philadelphia, PA: Elsevier; 2016:487-533.
- Lange M, Jasiel‐Walikowska E, Nowicki R, et al. Tinea incognito due to Trichophyton mentagrophytes. Mycoses. 2010;53:455-457.
- Pchelin IM, Azarov DV, Churina MA, et al. Species boundaries in the Trichophyton mentagrophytes/T. interdigitale species complex. Med Mycol. 2019;57:781-789.
- Makimura K, Mochizuki T, Hasegawa A, et al. Phylogenetic classification of Trichophyton mentagrophytes complex strains based on DNA sequences of nuclear ribosomal internal transcribed spacer 1 regions. J Clin Microbiol. 1998;36:2629-2633.
- de Hoog GS, Dukik K, Monod M, et al. Toward a novel multilocus phylogenetic taxonomy for the dermatophytes. Mycopathologia. 2017;182:5-31.
- Rudramurthy SM, Shankarnarayan SA, Dogra S, et al. Mutation in the squalene epoxidase gene of Trichophyton interdigitale and Trichophyton rubrum associated with allylamine resistance. Antimicrob Agents Chemother. 2018;62:e02522-17.
- Singh A, Masih A, Khurana A, et al. High terbinafine resistance in Trichophyton interdigitale isolates in Delhi, India harbouring mutations in the squalene epoxidase gene. Mycoses. 2018;61:477-484.
- Kawakami Y, Oyama N, Sakai E, et al. Childhood tinea incognito caused by Trichophyton mentagrophytes var. interdigitale mimicking pustular psoriasis. Pediatr Dermatol. 2011;28:738-739.
- Neri I, Piraccini BM, Guareschi E, et al. Bullous tinea pedis in two children. Mycoses. 2004;47:475-478.
- Abarca ML, Castellá G, Martorell J, et al. Trichophyton erinacei in pet hedgehogs in Spain: occurrence and revision of its taxonomic status. Med Mycol. 2016;55:164-172.
- Morris P, English MP. Transmission and course of Trichophyton erinacei infections in British hedgehogs. Sabouraudia. 1973;11:42-47.
- Riley PY, Chomel BB. Hedgehog zoonoses. Emerg Infect Dis. 2005;11:1-5.
Case Report
A 37-year-old woman presented to the dermatology clinic with an itchy rash involving the right hand. The rash had been present for 10 days but had become increasingly pruritic and vesicular over the last 5 days. She denied new exposures or other household members with similar symptoms. The patient reported that she had purchased a 4-toed, white-bellied African pygmy hedgehog (Atelerix albiventris) approximately 4 months prior. Upon questioning, she stated that she handled the hedgehog a couple of times a week and always washed her hands with soap and water immediately after. The patient’s medical and personal history were otherwise unremarkable.
Review of systems, including fevers, chills, and night sweats, was negative. Clinical examination revealed erythema with overlying vesicles and pustules on the right radial palm, radial dorsal hand, and interdigital web space of the first and second digit (Figure 1). The eruption was actively discharging serous exudate. No other lesions were present.
Unspecified acute contact dermatitis was the preliminary diagnosis based on clinical presentation and history. Other entities considered before making the diagnosis included psoriasis, eczema, and an infectious cause. Specimens were taken for bacterial and fungal cultures as well as a specimen for herpes simplex virus by polymerase chain reaction. Due to the intense pruritus and vesicular nature of the rash, the patient was treatedwith a 2-week, 60-40-20 prednisone taper and clobetasol propionate ointment 0.05% twice daily.
At 1-week follow-up, the eruption had improved, but the patient was still experiencing mild pruritus. Physical examination of the affected areas showed erythematous, violaceous, annular patches with slight scale at the periphery; all bullous lesions had resolved (Figure 2). Bacterial culture and herpes simplex virus by polymerase chain reaction were negative.
Two weeks after initial consultation, the fungal culture returned positive and showed growth of Trichophyton mentagrophytes. The patient was contacted and returned for re-evaluation. Physical examination showed decreased erythema and no bullous lesions; however, there was increased fine scale throughout the affected area on the right palm and first and second interdigital spaces (Figure 3). She reported mild pruritus. A confirmatory potassium hydroxide (KOH) preparation was positive for fungal hyphae. The patient was subsequently diagnosed with bullous tinea secondary to domestic hedgehog exposure that was now presenting as tinea manuum incognita. After 2 weeks of appropriate systemic and topical antifungal therapy, the patient’s skin eruption markedly improved (Figure 4).
Comment
Tinea manuum is a dermatophytic epidermal infection of the hand. The most common causative organisms are Trichophyton rubrum, T mentagrophytes, and Epidermophyton floccosum. Infection can be acquired from contact with an infected person or animal, fomites, soil, or autoinoculation. Tinea manuum often is associated with tinea pedis. The hand that is used to excoriate the pruritic feet becomes infected, resulting in the classic two feet–one hand syndrome, which this patient did not have.1
Dermatophytes colonize keratin-containing tissues—skin, hair, and nails—utilizing the keratin for nutrients, and they do not invade living tissue in immunocompetent hosts. Dermatophytes cause clinical disease from an allergic host response to fungal antigens or their metabolic products.1 Tinea incognito results from the use of corticosteroids to treat a cutaneous fungal infection. The immunomodulatory effects of corticosteroids alter the appearance of the lesion. Hallmark signs and symptoms of a tinea infection, including scale, prominent border, erythema, and pruritus, can be reduced with corticosteroid use, giving the false impression that the lesion is resolving.2,3
The diagnosis of tinea manuum can be made clinically and often is supported with the findings of a KOH preparation. Scraping from an active scaling border generally provides the best results for obtaining fungal elements. For vesiculobullous lesions, the roof of a vesicle can provide an adequate specimen. Fungal culture and specific dermatophyte testing mediums can be used as confirmatory tests or allow for speciation, which help establish the diagnosis.1
Trichophyton mentagrophytes is a species complex—a group of closely related organisms that share morphologic appearance to the point that boundaries between them often are unclear. It can be identified by gross and microscopic morphology; however, variants of T mentagrophytes (eg, Trichophyton interdigitale, Trichophyton erinacei) require a confirmatory test or molecular analysis to be correctly identified.4-6 The laboratory used at our facility does not routinely attempt to identify the variant due to of lack of clinical significance.7,8
Anthropophilic fungi such as T rubrum, E floccosum, and T interdigitale generally do not cause a robust immunologic reaction. Infection usually is chronic in nature, though cases of pustular and vesicular tinea have been described.9,10Trichophyton erinacei and T mentagrophytes are zoophilic dermatophytes that cause an acute host response and are more likely to present with vesiculobullous lesions. Trichophyton erinacei is the most common fungal pathogen associated with A albiventris and has been isolated from its epidermal mites and quills,11,12 which likely facilitates interspecies transmission and compromises the cutaneous barrier of human hosts when the hedgehog is handled.
Atelerix albiventris is the most common domesticated hedgehog in the United States. These mild-mannered, nocturnal insectivores are unique, low-maintenance pets that have recently gained popularity. They are notable for their propensity to curl into a ball when frightened (Figure 5). The spines are not barbed and do not detach, as those of a porcupine do, but are still capable of piercing the skin. Atelerix albiventris is known to cause zoonotic dermatosis in humans and should be handled with gloves.13 Performing a KOH preparation early in the diagnostic workup can help initiate antifungal therapy, as results of fungal culture can take several weeks.
Conclusion
This case illustrates the importance of close follow-up of skin lesions that only partially respond to initial treatment and maintaining a high index of suspicion as exotic pets become popular.
Case Report
A 37-year-old woman presented to the dermatology clinic with an itchy rash involving the right hand. The rash had been present for 10 days but had become increasingly pruritic and vesicular over the last 5 days. She denied new exposures or other household members with similar symptoms. The patient reported that she had purchased a 4-toed, white-bellied African pygmy hedgehog (Atelerix albiventris) approximately 4 months prior. Upon questioning, she stated that she handled the hedgehog a couple of times a week and always washed her hands with soap and water immediately after. The patient’s medical and personal history were otherwise unremarkable.
Review of systems, including fevers, chills, and night sweats, was negative. Clinical examination revealed erythema with overlying vesicles and pustules on the right radial palm, radial dorsal hand, and interdigital web space of the first and second digit (Figure 1). The eruption was actively discharging serous exudate. No other lesions were present.
Unspecified acute contact dermatitis was the preliminary diagnosis based on clinical presentation and history. Other entities considered before making the diagnosis included psoriasis, eczema, and an infectious cause. Specimens were taken for bacterial and fungal cultures as well as a specimen for herpes simplex virus by polymerase chain reaction. Due to the intense pruritus and vesicular nature of the rash, the patient was treatedwith a 2-week, 60-40-20 prednisone taper and clobetasol propionate ointment 0.05% twice daily.
At 1-week follow-up, the eruption had improved, but the patient was still experiencing mild pruritus. Physical examination of the affected areas showed erythematous, violaceous, annular patches with slight scale at the periphery; all bullous lesions had resolved (Figure 2). Bacterial culture and herpes simplex virus by polymerase chain reaction were negative.
Two weeks after initial consultation, the fungal culture returned positive and showed growth of Trichophyton mentagrophytes. The patient was contacted and returned for re-evaluation. Physical examination showed decreased erythema and no bullous lesions; however, there was increased fine scale throughout the affected area on the right palm and first and second interdigital spaces (Figure 3). She reported mild pruritus. A confirmatory potassium hydroxide (KOH) preparation was positive for fungal hyphae. The patient was subsequently diagnosed with bullous tinea secondary to domestic hedgehog exposure that was now presenting as tinea manuum incognita. After 2 weeks of appropriate systemic and topical antifungal therapy, the patient’s skin eruption markedly improved (Figure 4).
Comment
Tinea manuum is a dermatophytic epidermal infection of the hand. The most common causative organisms are Trichophyton rubrum, T mentagrophytes, and Epidermophyton floccosum. Infection can be acquired from contact with an infected person or animal, fomites, soil, or autoinoculation. Tinea manuum often is associated with tinea pedis. The hand that is used to excoriate the pruritic feet becomes infected, resulting in the classic two feet–one hand syndrome, which this patient did not have.1
Dermatophytes colonize keratin-containing tissues—skin, hair, and nails—utilizing the keratin for nutrients, and they do not invade living tissue in immunocompetent hosts. Dermatophytes cause clinical disease from an allergic host response to fungal antigens or their metabolic products.1 Tinea incognito results from the use of corticosteroids to treat a cutaneous fungal infection. The immunomodulatory effects of corticosteroids alter the appearance of the lesion. Hallmark signs and symptoms of a tinea infection, including scale, prominent border, erythema, and pruritus, can be reduced with corticosteroid use, giving the false impression that the lesion is resolving.2,3
The diagnosis of tinea manuum can be made clinically and often is supported with the findings of a KOH preparation. Scraping from an active scaling border generally provides the best results for obtaining fungal elements. For vesiculobullous lesions, the roof of a vesicle can provide an adequate specimen. Fungal culture and specific dermatophyte testing mediums can be used as confirmatory tests or allow for speciation, which help establish the diagnosis.1
Trichophyton mentagrophytes is a species complex—a group of closely related organisms that share morphologic appearance to the point that boundaries between them often are unclear. It can be identified by gross and microscopic morphology; however, variants of T mentagrophytes (eg, Trichophyton interdigitale, Trichophyton erinacei) require a confirmatory test or molecular analysis to be correctly identified.4-6 The laboratory used at our facility does not routinely attempt to identify the variant due to of lack of clinical significance.7,8
Anthropophilic fungi such as T rubrum, E floccosum, and T interdigitale generally do not cause a robust immunologic reaction. Infection usually is chronic in nature, though cases of pustular and vesicular tinea have been described.9,10Trichophyton erinacei and T mentagrophytes are zoophilic dermatophytes that cause an acute host response and are more likely to present with vesiculobullous lesions. Trichophyton erinacei is the most common fungal pathogen associated with A albiventris and has been isolated from its epidermal mites and quills,11,12 which likely facilitates interspecies transmission and compromises the cutaneous barrier of human hosts when the hedgehog is handled.
Atelerix albiventris is the most common domesticated hedgehog in the United States. These mild-mannered, nocturnal insectivores are unique, low-maintenance pets that have recently gained popularity. They are notable for their propensity to curl into a ball when frightened (Figure 5). The spines are not barbed and do not detach, as those of a porcupine do, but are still capable of piercing the skin. Atelerix albiventris is known to cause zoonotic dermatosis in humans and should be handled with gloves.13 Performing a KOH preparation early in the diagnostic workup can help initiate antifungal therapy, as results of fungal culture can take several weeks.
Conclusion
This case illustrates the importance of close follow-up of skin lesions that only partially respond to initial treatment and maintaining a high index of suspicion as exotic pets become popular.
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier; 2018:1329-1363.
- Habif T. Superficial fungal infections. In: Habif T. Clinical Dermatology. 6th ed. Philadelphia, PA: Elsevier; 2016:487-533.
- Lange M, Jasiel‐Walikowska E, Nowicki R, et al. Tinea incognito due to Trichophyton mentagrophytes. Mycoses. 2010;53:455-457.
- Pchelin IM, Azarov DV, Churina MA, et al. Species boundaries in the Trichophyton mentagrophytes/T. interdigitale species complex. Med Mycol. 2019;57:781-789.
- Makimura K, Mochizuki T, Hasegawa A, et al. Phylogenetic classification of Trichophyton mentagrophytes complex strains based on DNA sequences of nuclear ribosomal internal transcribed spacer 1 regions. J Clin Microbiol. 1998;36:2629-2633.
- de Hoog GS, Dukik K, Monod M, et al. Toward a novel multilocus phylogenetic taxonomy for the dermatophytes. Mycopathologia. 2017;182:5-31.
- Rudramurthy SM, Shankarnarayan SA, Dogra S, et al. Mutation in the squalene epoxidase gene of Trichophyton interdigitale and Trichophyton rubrum associated with allylamine resistance. Antimicrob Agents Chemother. 2018;62:e02522-17.
- Singh A, Masih A, Khurana A, et al. High terbinafine resistance in Trichophyton interdigitale isolates in Delhi, India harbouring mutations in the squalene epoxidase gene. Mycoses. 2018;61:477-484.
- Kawakami Y, Oyama N, Sakai E, et al. Childhood tinea incognito caused by Trichophyton mentagrophytes var. interdigitale mimicking pustular psoriasis. Pediatr Dermatol. 2011;28:738-739.
- Neri I, Piraccini BM, Guareschi E, et al. Bullous tinea pedis in two children. Mycoses. 2004;47:475-478.
- Abarca ML, Castellá G, Martorell J, et al. Trichophyton erinacei in pet hedgehogs in Spain: occurrence and revision of its taxonomic status. Med Mycol. 2016;55:164-172.
- Morris P, English MP. Transmission and course of Trichophyton erinacei infections in British hedgehogs. Sabouraudia. 1973;11:42-47.
- Riley PY, Chomel BB. Hedgehog zoonoses. Emerg Infect Dis. 2005;11:1-5.
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier; 2018:1329-1363.
- Habif T. Superficial fungal infections. In: Habif T. Clinical Dermatology. 6th ed. Philadelphia, PA: Elsevier; 2016:487-533.
- Lange M, Jasiel‐Walikowska E, Nowicki R, et al. Tinea incognito due to Trichophyton mentagrophytes. Mycoses. 2010;53:455-457.
- Pchelin IM, Azarov DV, Churina MA, et al. Species boundaries in the Trichophyton mentagrophytes/T. interdigitale species complex. Med Mycol. 2019;57:781-789.
- Makimura K, Mochizuki T, Hasegawa A, et al. Phylogenetic classification of Trichophyton mentagrophytes complex strains based on DNA sequences of nuclear ribosomal internal transcribed spacer 1 regions. J Clin Microbiol. 1998;36:2629-2633.
- de Hoog GS, Dukik K, Monod M, et al. Toward a novel multilocus phylogenetic taxonomy for the dermatophytes. Mycopathologia. 2017;182:5-31.
- Rudramurthy SM, Shankarnarayan SA, Dogra S, et al. Mutation in the squalene epoxidase gene of Trichophyton interdigitale and Trichophyton rubrum associated with allylamine resistance. Antimicrob Agents Chemother. 2018;62:e02522-17.
- Singh A, Masih A, Khurana A, et al. High terbinafine resistance in Trichophyton interdigitale isolates in Delhi, India harbouring mutations in the squalene epoxidase gene. Mycoses. 2018;61:477-484.
- Kawakami Y, Oyama N, Sakai E, et al. Childhood tinea incognito caused by Trichophyton mentagrophytes var. interdigitale mimicking pustular psoriasis. Pediatr Dermatol. 2011;28:738-739.
- Neri I, Piraccini BM, Guareschi E, et al. Bullous tinea pedis in two children. Mycoses. 2004;47:475-478.
- Abarca ML, Castellá G, Martorell J, et al. Trichophyton erinacei in pet hedgehogs in Spain: occurrence and revision of its taxonomic status. Med Mycol. 2016;55:164-172.
- Morris P, English MP. Transmission and course of Trichophyton erinacei infections in British hedgehogs. Sabouraudia. 1973;11:42-47.
- Riley PY, Chomel BB. Hedgehog zoonoses. Emerg Infect Dis. 2005;11:1-5.
Practice Points
- Bullous tinea may present with little or no scale, which can lead to confusion with acute contact dermatitis.
- The recent popularity of exotic pets may increase the incidence of fungal zoonotic dermatitis.
- Prompt recognition of tinea incognito is essential when treating lesions with corticosteroids.
- Skin lesions not responding appropriately to therapy warrant reassessment and further evaluation.