User login
Understanding Psychosis in a Veteran With a History of Combat and Multiple Sclerosis (FULL)
A patient with significant combat history and previous diagnoses of multiple sclerosis and unspecified schizophrenia spectrum and other psychotic disorder was admitted with acute psychosis inconsistent with expected clinical presentations.
Multiple sclerosis (MS) is an immune-mediated neurodegenerative disease that affects > 700,000 people in the US.1 The hallmarks of MS pathology are axonal or neuronal loss, demyelination, and astrocytic gliosis. Of these, axonal or neuronal loss is the main underlying mechanism of permanent clinical disability.
MS also has been associated with an increased prevalence of psychiatric illnesses, with mood disorders affecting up to 40% to 60% of the population, and psychosis being reported in 2% to 4% of patients.2 The link between MS and mood disorders, including bipolar disorder and depression, was documented as early as 1926,with mood disorders hypothesized to be manifestations of central nervous system (CNS) inflammation.3 More recently, inflammation-driven microglia have been hypothesized to impair hippocampal connectivity and activate glucocorticoid-insensitive inflammatory cells that then overstimulate the hypothalamic-pituitary-adrenal axis.4,5
Although the prevalence of psychosis in patients with MS is significantly rarer, averaging between 2% and 4%.6 A Canadian study by Patten and colleagues reviewed data from 2.45 million residents of Alberta and found that those who identified as having MS had a 2% to 3% prevalence of psychosis compared with 0.5% to 1% in the general population.7 The connection between psychosis and MS, similar to that between mood disorders and MS, has been described as a common regional demyelination process. Supporting this, MS manifesting as psychosis has been found to present with distinct magnetic resonance imaging (MRI) findings, such as diffuse periventricular lesions.8 Still, no conclusive criteria have been developed to distinguish MS presenting as psychosis from a primary psychiatric illness, such as schizophrenia.
In patients with combat history, it is possible that both neurodegenerative and psychotic symptoms can be explained by autoantibody formation in response to toxin exposure. When soldiers were deployed to Iraq and Afghanistan, they may have been exposed to multiple toxicities, including depleted uranium, dust and fumes, and numerous infectious diseases.9 Gulf War illness (GWI) or chronic multisymptom illness (CMI) encompass a cluster of symptoms, such as chronic pain, chronic fatigue, irritable bowel syndrome, dermatitis, and seizures, as well as mental health issues such as depression and anxiety experienced following exposure to these combat environments.10,11
In light of this diagnostic uncertainty, the authors detail a case of a patient with significant combat history previously diagnosed with MS and unspecified schizophrenia spectrum and other psychotic disorder (USS & OPD) presenting with acute psychosis.
Case Presentation
A 35-year-old male veteran, with a history of MS, USS & OPD, posttraumatic stress disorder, and traumatic brain injuries (TBIs) was admitted to the psychiatric unit after being found by the police lying in the middle of a busy intersection, internally preoccupied. On admission, he reported a week of auditory hallucinations from birds with whom he had been communicating telepathically, and a recurrent visual hallucination of a tall man in white and purple robes. He had discontinued his antipsychotic medication, aripiprazole 10 mg, a few weeks prior for unknown reasons. He was brought to the hospital by ambulance, where he presented with disorganized thinking, tangential thought process, and active auditory and visual hallucinations. The differential diagnoses included USS & OPD, schizophrenia, schizoaffective disorder and ruled out substance-induced psychotic disorder, and psychosis as a manifestation of MS.
The patient had 2 psychotic episodes prior to this presentation. He was hospitalized for his first psychotic break in 2015 at age 32, when he had tailed another car “to come back to reality” and ended up in a motor vehicle accident. During that admission, he reported weeks of thought broadcasting, conspiratorial delusions, and racing thoughts. Two years later, he was admitted to a psychiatric intensive care unit for his second episode of severe psychosis. After several trials of different antipsychotic medications, his most recent pharmacologic regimen was aripiprazole 10 mg once daily.
His medical history was complicated by 2 TBIs, in November 2014 and January 2015, with normal computed tomography (CT) scans. He was diagnosed with MS in December 2017, when he presented with intractable emesis, left facial numbness, right upper extremity ataxia, nystagmus, and imbalance. An MRI scan revealed multifocal bilateral hypodensities in his periventricular, subcortical, and brain stem white matter. Multiple areas of hyperintensity were visualized, including in the right periatrial region and left brachium pontis. More than 5 oligoclonal bands on lumbar puncture confirmed the diagnosis.
He was treated with IV methylprednisolone followed by a 2-week prednisone taper. Within 1 week, he returned to the psychiatric unit with worsening symptoms and received a second dose of IV steroids and plasma exchange treatment. In the following months, he completed a course of rituximab infusions and physical therapy for his dysarthria, gait abnormality, and vision impairment.
His social history was notable for multiple first-degree relatives with schizophrenia. He reported a history of sexual and verbal abuse and attempted suicide once at age 13 years by hanging himself with a bathrobe. He left home at age 18 years to serve in the Marine Corps (2001-2006). His service included deployment to Afghanistan, where he received a purple heart. Upon his return, he received BA and MS degrees. He married and had 2 daughters but became estranged from his wife. By his most recent admission, he was unemployed and living with his half-sister.
On the first day of this most recent psychiatric hospitalization, he was restarted on aripiprazole 10 mg daily, and a medicine consult was sought to evaluate the progression of his MS. No new onset neurologic symptoms were noted, but he had possible residual lower extremity hyperreflexia and tandem gait incoordination. The episodes of psychotic and neurologic symptoms appeared independent, given that his psychiatric history preceded the onset of his MS.
The patient reported no visual hallucinations starting day 2, and he no longer endorsed auditory hallucinations by day 3. However, he continued to appear internally preoccupied and was noticed to be pacing around the unit. On day 4 he presented with newly pressured speech and flights of ideas, while his affect remained euthymic and his sleep stayed consistent. In combination with his ongoing pacing, his newfound symptoms were hypothesized to be possibly akathisia, an adverse effect (AE) of aripiprazole. As such, on day 5 his dose was lowered to 5 mg daily. He continued to report no hallucinations and demonstrated progressively increased emotional range. A MRI scan was done on day 6 in case a new lesion could be identified, suggesting a primary MS flare-up; however, the scan identified no enhancing lesions, indicating no ongoing demyelination. After a neurology consult corroborated this conclusion, he was discharged in stable condition on day 7.
As is the case with the majority of patients with MS-induced psychosis, he continued to have relapsing psychiatric disease even after MS treatment had been started. Unfortunately, because this patient had stopped taking his atypical antipsychotic medication several weeks prior to his hospitalization, we cannot clarify whether his psychosis stems from a primary psychiatric vs MS process.
Discussion
Presently, treatment preferences for MS-related psychosis are divided between atypical antipsychotics and glucocorticoids. Some suggest that the treatment remains similar between MS-related psychosis and primary psychotic disorders in that atypical antipsychotics are the standard of care.12 A variety of atypical antipsychotics have been used successfully in case reports, including zipradisone, risperidone, olanzapine, quetiapine, and aripiprazole.13,14 First-generation antipsychotics and other psychotropic drugs that can precipitate extra-pyramidal AEs are not recommended given their potential additive effect to motor deficits associated with MS.12 Alternatively, several case reports have found that MS-related psychotic symptoms respond to glucocorticoids more effectively, while cautioning that glucocorticoids can precipitate psychosis and depression.15,16 One review article found that 90% of patients who received corticosteroids saw an improvement in their psychotic symptoms.2
Finally, it is possible that our patient’s neuropsychiatric symptoms can be explained by autoantibody formation in response to toxin exposure during his time in Afghanistan. In a pilot study of veterans with GWI, Abou-Donia and colleagues found 2-to-9 fold increase in autoantibody reactivity levels of the following neuronal and glial-specific proteins relative to healthy controls: neurofilament triplet proteins, tubulin, microtubule-associated tau proteins, microtubule-associated protein-2, myelin basic protein, myelin-associated glycoprotein, glial fibrillary acidic protein, and calcium-calmodulin kinase II.17,18 Many of these autoantibodies are longstanding explicit markers for neurodegenerative disorders, given that they target proteins and antigens that support axonal transport and myelination. Still Gulf War veteran status has yet to be explicitly linked to an increased risk of MS,19 making this hypothesis less likely for our patient. Future research should address the clinical and therapeutic implications of different autoantibody levels in combat veterans with psychosis.
Conclusion
For patients with MS, mood disorder and psychotic symptoms should warrant a MRI given the possibility of a psychiatric manifestation of MS relapse. Ultimately, our patient’s presentation was inconsistent with the expected clinical presentations of both a primary psychotic disorder and psychosis as a manifestation of MS. His late age at his first psychotic break is atypical for primary psychotic disease, and the lack of MRI imaging done at his initial psychotic episodes cannot exclude a primary MS diagnosis. Still, his lack of MRI findings at his most recent hospitalization, negative symptomatology, and strong history of schizophrenia make a primary psychotic disorder likely.
Following his future clinical course will be necessary to determine the etiology of his psychotic episodes. Future episodes of psychosis with neurologic symptoms would suggest a primary MS diagnosis and potential benefit of immunosuppressant treatment, whereas repeated psychotic breaks with minimal temporal lobe involvement or demyelination as seen on MRI would be suspicious for separate MS and psychotic disease processes. Further research on treatment regimens for patients experiencing psychosis as a manifestation of MS is still necessary.
1. Wallin MT, Culpepper WJ, Campbell JD, et al. The prevalence of MS in the United States: A population-based estimate using health claims data. Neurology. 2019;92(10):e1029-e1040.
2. Camara-Lemarroy CR, Ibarra-Yruegas BE, Rodriguez-Gutierrez R, Berrios-Morales I, Ionete C, Riskind P. The varieties of psychosis in multiple sclerosis: a systematic review of cases. Mult Scler Relat Disord. 2017;12:9-14.
3. Cottrel SS, Wilson SA. The affective symptomatology of disseminated sclerosis: a study of 100 cases. J Neurol Psychopathology. 1926;7(25):1-30.
4. Johansson V, Lundholm C, Hillert J, et al. Multiple sclerosis and psychiatric disorders: comorbidity and sibling risk in a nationwide Swedish cohort. Mult Scler. 2014;20(14):1881-1891.
5. Rossi S, Studer V, Motta C, et al. Neuroinflammation drives anxiety and depression in relapsing-remitting multiple sclerosis. Neurology. 2017;89(13):1338-1347.
6. Gilberthorpe TG, O’Connell KE, Carolan A, et al. The spectrum of psychosis in multiple sclerosis: a clinical case series. Neuropsychiatric disease and treatment. 2017;13:303.
7. Patten SB, Svenson LW, Metz LM. Psychotic disorders in MS: population-based evidence of an association. Neurology 2005;65(7):1123-1125.
8. Kosmidis MH, Giannakou M, Messinis L, Papathanasopoulos P. Psychotic features associated with multiple sclerosis. Int Rev Psychiatry. 2010; 22(1):55-66.
9. US Department of Veterans Affairs. Public health: military exposures. https://www.publichealth.va.gov/exposures/. Updated April 16, 2019. Accessed May 13, 2019.
10. DeBeer BB, Davidson D, Meyer EC, Kimbrel NA, Gulliver SB, Morissette SB. The association between toxic exposures and chronic multisymptom illness in veterans of the wars of Iraq and Afghanistan. J Occup Environ Med. 2017;59(1):54-60.
11. Kang HK, Li B, Mahan CM, Eisen SA, Engel CC. Health of US veterans of 1991 Gulf War: a follow-up survey in 10 years. J Occup Environ Med. 2009;51(4):401-410.
12. Murphy R, O’Donoghue S, Counihan T, et al. Neuropsychiatric syndromes of multiple sclerosis. J Neurol Neurosurg Psychiatry. 2017;88(8):697-708.
13. Davids E, Hartwig U, Gastpar, M. Antipsychotic treatment of psychosis associated with multiple sclerosis. Prog Neuro Psychopharmacol Biol Psychiatry. 2004;28(4):743-744.
14. Lo Fermo S, Barone R, Patti F, et al. Outcome of psychiatric symptoms presenting at onset of multiple sclerosis: a retrospective study. Mult Scler. 2010;16(6):742-748.
15. Enderami A, Fouladi R, Hosseini HS. First-episode psychosis as the initial presentation of multiple sclerosis: a case report. Int Medical Case Rep J. 2018;11:73-76.
16. Fragoso YD, Frota ER, Lopes JS, et al. Severe depression, suicide attempts, and ideation during the use of interferon beta by patients with multiple sclerosis. Clin Neuropharmacol. 2010;33(6):312-316.
17. Abou-Donia MB, Conboy LA, Kokkotou E, et al. Screening for novel central nervous system biomarkers in veterans with Gulf War Illness. Neurotoxicol Teratol. 2017;61:36-46.
18. Abou-Donia MB, Lieberman A, Curtis L. Neural autoantibodies in patients with neurological symptoms and histories of chemical/mold exposures. Toxicol Ind Health. 2018;34(1):44-53.
19. Wallin MT, Kurtzke JF, Culpepper WJ, et al. Multiple sclerosis in Gulf War era veterans. 2. Military deployment and risk of multiple sclerosis in the first Gulf War. Neuroepidemiology. 2014;42(4):226-234.
A patient with significant combat history and previous diagnoses of multiple sclerosis and unspecified schizophrenia spectrum and other psychotic disorder was admitted with acute psychosis inconsistent with expected clinical presentations.
A patient with significant combat history and previous diagnoses of multiple sclerosis and unspecified schizophrenia spectrum and other psychotic disorder was admitted with acute psychosis inconsistent with expected clinical presentations.
Multiple sclerosis (MS) is an immune-mediated neurodegenerative disease that affects > 700,000 people in the US.1 The hallmarks of MS pathology are axonal or neuronal loss, demyelination, and astrocytic gliosis. Of these, axonal or neuronal loss is the main underlying mechanism of permanent clinical disability.
MS also has been associated with an increased prevalence of psychiatric illnesses, with mood disorders affecting up to 40% to 60% of the population, and psychosis being reported in 2% to 4% of patients.2 The link between MS and mood disorders, including bipolar disorder and depression, was documented as early as 1926,with mood disorders hypothesized to be manifestations of central nervous system (CNS) inflammation.3 More recently, inflammation-driven microglia have been hypothesized to impair hippocampal connectivity and activate glucocorticoid-insensitive inflammatory cells that then overstimulate the hypothalamic-pituitary-adrenal axis.4,5
Although the prevalence of psychosis in patients with MS is significantly rarer, averaging between 2% and 4%.6 A Canadian study by Patten and colleagues reviewed data from 2.45 million residents of Alberta and found that those who identified as having MS had a 2% to 3% prevalence of psychosis compared with 0.5% to 1% in the general population.7 The connection between psychosis and MS, similar to that between mood disorders and MS, has been described as a common regional demyelination process. Supporting this, MS manifesting as psychosis has been found to present with distinct magnetic resonance imaging (MRI) findings, such as diffuse periventricular lesions.8 Still, no conclusive criteria have been developed to distinguish MS presenting as psychosis from a primary psychiatric illness, such as schizophrenia.
In patients with combat history, it is possible that both neurodegenerative and psychotic symptoms can be explained by autoantibody formation in response to toxin exposure. When soldiers were deployed to Iraq and Afghanistan, they may have been exposed to multiple toxicities, including depleted uranium, dust and fumes, and numerous infectious diseases.9 Gulf War illness (GWI) or chronic multisymptom illness (CMI) encompass a cluster of symptoms, such as chronic pain, chronic fatigue, irritable bowel syndrome, dermatitis, and seizures, as well as mental health issues such as depression and anxiety experienced following exposure to these combat environments.10,11
In light of this diagnostic uncertainty, the authors detail a case of a patient with significant combat history previously diagnosed with MS and unspecified schizophrenia spectrum and other psychotic disorder (USS & OPD) presenting with acute psychosis.
Case Presentation
A 35-year-old male veteran, with a history of MS, USS & OPD, posttraumatic stress disorder, and traumatic brain injuries (TBIs) was admitted to the psychiatric unit after being found by the police lying in the middle of a busy intersection, internally preoccupied. On admission, he reported a week of auditory hallucinations from birds with whom he had been communicating telepathically, and a recurrent visual hallucination of a tall man in white and purple robes. He had discontinued his antipsychotic medication, aripiprazole 10 mg, a few weeks prior for unknown reasons. He was brought to the hospital by ambulance, where he presented with disorganized thinking, tangential thought process, and active auditory and visual hallucinations. The differential diagnoses included USS & OPD, schizophrenia, schizoaffective disorder and ruled out substance-induced psychotic disorder, and psychosis as a manifestation of MS.
The patient had 2 psychotic episodes prior to this presentation. He was hospitalized for his first psychotic break in 2015 at age 32, when he had tailed another car “to come back to reality” and ended up in a motor vehicle accident. During that admission, he reported weeks of thought broadcasting, conspiratorial delusions, and racing thoughts. Two years later, he was admitted to a psychiatric intensive care unit for his second episode of severe psychosis. After several trials of different antipsychotic medications, his most recent pharmacologic regimen was aripiprazole 10 mg once daily.
His medical history was complicated by 2 TBIs, in November 2014 and January 2015, with normal computed tomography (CT) scans. He was diagnosed with MS in December 2017, when he presented with intractable emesis, left facial numbness, right upper extremity ataxia, nystagmus, and imbalance. An MRI scan revealed multifocal bilateral hypodensities in his periventricular, subcortical, and brain stem white matter. Multiple areas of hyperintensity were visualized, including in the right periatrial region and left brachium pontis. More than 5 oligoclonal bands on lumbar puncture confirmed the diagnosis.
He was treated with IV methylprednisolone followed by a 2-week prednisone taper. Within 1 week, he returned to the psychiatric unit with worsening symptoms and received a second dose of IV steroids and plasma exchange treatment. In the following months, he completed a course of rituximab infusions and physical therapy for his dysarthria, gait abnormality, and vision impairment.
His social history was notable for multiple first-degree relatives with schizophrenia. He reported a history of sexual and verbal abuse and attempted suicide once at age 13 years by hanging himself with a bathrobe. He left home at age 18 years to serve in the Marine Corps (2001-2006). His service included deployment to Afghanistan, where he received a purple heart. Upon his return, he received BA and MS degrees. He married and had 2 daughters but became estranged from his wife. By his most recent admission, he was unemployed and living with his half-sister.
On the first day of this most recent psychiatric hospitalization, he was restarted on aripiprazole 10 mg daily, and a medicine consult was sought to evaluate the progression of his MS. No new onset neurologic symptoms were noted, but he had possible residual lower extremity hyperreflexia and tandem gait incoordination. The episodes of psychotic and neurologic symptoms appeared independent, given that his psychiatric history preceded the onset of his MS.
The patient reported no visual hallucinations starting day 2, and he no longer endorsed auditory hallucinations by day 3. However, he continued to appear internally preoccupied and was noticed to be pacing around the unit. On day 4 he presented with newly pressured speech and flights of ideas, while his affect remained euthymic and his sleep stayed consistent. In combination with his ongoing pacing, his newfound symptoms were hypothesized to be possibly akathisia, an adverse effect (AE) of aripiprazole. As such, on day 5 his dose was lowered to 5 mg daily. He continued to report no hallucinations and demonstrated progressively increased emotional range. A MRI scan was done on day 6 in case a new lesion could be identified, suggesting a primary MS flare-up; however, the scan identified no enhancing lesions, indicating no ongoing demyelination. After a neurology consult corroborated this conclusion, he was discharged in stable condition on day 7.
As is the case with the majority of patients with MS-induced psychosis, he continued to have relapsing psychiatric disease even after MS treatment had been started. Unfortunately, because this patient had stopped taking his atypical antipsychotic medication several weeks prior to his hospitalization, we cannot clarify whether his psychosis stems from a primary psychiatric vs MS process.
Discussion
Presently, treatment preferences for MS-related psychosis are divided between atypical antipsychotics and glucocorticoids. Some suggest that the treatment remains similar between MS-related psychosis and primary psychotic disorders in that atypical antipsychotics are the standard of care.12 A variety of atypical antipsychotics have been used successfully in case reports, including zipradisone, risperidone, olanzapine, quetiapine, and aripiprazole.13,14 First-generation antipsychotics and other psychotropic drugs that can precipitate extra-pyramidal AEs are not recommended given their potential additive effect to motor deficits associated with MS.12 Alternatively, several case reports have found that MS-related psychotic symptoms respond to glucocorticoids more effectively, while cautioning that glucocorticoids can precipitate psychosis and depression.15,16 One review article found that 90% of patients who received corticosteroids saw an improvement in their psychotic symptoms.2
Finally, it is possible that our patient’s neuropsychiatric symptoms can be explained by autoantibody formation in response to toxin exposure during his time in Afghanistan. In a pilot study of veterans with GWI, Abou-Donia and colleagues found 2-to-9 fold increase in autoantibody reactivity levels of the following neuronal and glial-specific proteins relative to healthy controls: neurofilament triplet proteins, tubulin, microtubule-associated tau proteins, microtubule-associated protein-2, myelin basic protein, myelin-associated glycoprotein, glial fibrillary acidic protein, and calcium-calmodulin kinase II.17,18 Many of these autoantibodies are longstanding explicit markers for neurodegenerative disorders, given that they target proteins and antigens that support axonal transport and myelination. Still Gulf War veteran status has yet to be explicitly linked to an increased risk of MS,19 making this hypothesis less likely for our patient. Future research should address the clinical and therapeutic implications of different autoantibody levels in combat veterans with psychosis.
Conclusion
For patients with MS, mood disorder and psychotic symptoms should warrant a MRI given the possibility of a psychiatric manifestation of MS relapse. Ultimately, our patient’s presentation was inconsistent with the expected clinical presentations of both a primary psychotic disorder and psychosis as a manifestation of MS. His late age at his first psychotic break is atypical for primary psychotic disease, and the lack of MRI imaging done at his initial psychotic episodes cannot exclude a primary MS diagnosis. Still, his lack of MRI findings at his most recent hospitalization, negative symptomatology, and strong history of schizophrenia make a primary psychotic disorder likely.
Following his future clinical course will be necessary to determine the etiology of his psychotic episodes. Future episodes of psychosis with neurologic symptoms would suggest a primary MS diagnosis and potential benefit of immunosuppressant treatment, whereas repeated psychotic breaks with minimal temporal lobe involvement or demyelination as seen on MRI would be suspicious for separate MS and psychotic disease processes. Further research on treatment regimens for patients experiencing psychosis as a manifestation of MS is still necessary.
Multiple sclerosis (MS) is an immune-mediated neurodegenerative disease that affects > 700,000 people in the US.1 The hallmarks of MS pathology are axonal or neuronal loss, demyelination, and astrocytic gliosis. Of these, axonal or neuronal loss is the main underlying mechanism of permanent clinical disability.
MS also has been associated with an increased prevalence of psychiatric illnesses, with mood disorders affecting up to 40% to 60% of the population, and psychosis being reported in 2% to 4% of patients.2 The link between MS and mood disorders, including bipolar disorder and depression, was documented as early as 1926,with mood disorders hypothesized to be manifestations of central nervous system (CNS) inflammation.3 More recently, inflammation-driven microglia have been hypothesized to impair hippocampal connectivity and activate glucocorticoid-insensitive inflammatory cells that then overstimulate the hypothalamic-pituitary-adrenal axis.4,5
Although the prevalence of psychosis in patients with MS is significantly rarer, averaging between 2% and 4%.6 A Canadian study by Patten and colleagues reviewed data from 2.45 million residents of Alberta and found that those who identified as having MS had a 2% to 3% prevalence of psychosis compared with 0.5% to 1% in the general population.7 The connection between psychosis and MS, similar to that between mood disorders and MS, has been described as a common regional demyelination process. Supporting this, MS manifesting as psychosis has been found to present with distinct magnetic resonance imaging (MRI) findings, such as diffuse periventricular lesions.8 Still, no conclusive criteria have been developed to distinguish MS presenting as psychosis from a primary psychiatric illness, such as schizophrenia.
In patients with combat history, it is possible that both neurodegenerative and psychotic symptoms can be explained by autoantibody formation in response to toxin exposure. When soldiers were deployed to Iraq and Afghanistan, they may have been exposed to multiple toxicities, including depleted uranium, dust and fumes, and numerous infectious diseases.9 Gulf War illness (GWI) or chronic multisymptom illness (CMI) encompass a cluster of symptoms, such as chronic pain, chronic fatigue, irritable bowel syndrome, dermatitis, and seizures, as well as mental health issues such as depression and anxiety experienced following exposure to these combat environments.10,11
In light of this diagnostic uncertainty, the authors detail a case of a patient with significant combat history previously diagnosed with MS and unspecified schizophrenia spectrum and other psychotic disorder (USS & OPD) presenting with acute psychosis.
Case Presentation
A 35-year-old male veteran, with a history of MS, USS & OPD, posttraumatic stress disorder, and traumatic brain injuries (TBIs) was admitted to the psychiatric unit after being found by the police lying in the middle of a busy intersection, internally preoccupied. On admission, he reported a week of auditory hallucinations from birds with whom he had been communicating telepathically, and a recurrent visual hallucination of a tall man in white and purple robes. He had discontinued his antipsychotic medication, aripiprazole 10 mg, a few weeks prior for unknown reasons. He was brought to the hospital by ambulance, where he presented with disorganized thinking, tangential thought process, and active auditory and visual hallucinations. The differential diagnoses included USS & OPD, schizophrenia, schizoaffective disorder and ruled out substance-induced psychotic disorder, and psychosis as a manifestation of MS.
The patient had 2 psychotic episodes prior to this presentation. He was hospitalized for his first psychotic break in 2015 at age 32, when he had tailed another car “to come back to reality” and ended up in a motor vehicle accident. During that admission, he reported weeks of thought broadcasting, conspiratorial delusions, and racing thoughts. Two years later, he was admitted to a psychiatric intensive care unit for his second episode of severe psychosis. After several trials of different antipsychotic medications, his most recent pharmacologic regimen was aripiprazole 10 mg once daily.
His medical history was complicated by 2 TBIs, in November 2014 and January 2015, with normal computed tomography (CT) scans. He was diagnosed with MS in December 2017, when he presented with intractable emesis, left facial numbness, right upper extremity ataxia, nystagmus, and imbalance. An MRI scan revealed multifocal bilateral hypodensities in his periventricular, subcortical, and brain stem white matter. Multiple areas of hyperintensity were visualized, including in the right periatrial region and left brachium pontis. More than 5 oligoclonal bands on lumbar puncture confirmed the diagnosis.
He was treated with IV methylprednisolone followed by a 2-week prednisone taper. Within 1 week, he returned to the psychiatric unit with worsening symptoms and received a second dose of IV steroids and plasma exchange treatment. In the following months, he completed a course of rituximab infusions and physical therapy for his dysarthria, gait abnormality, and vision impairment.
His social history was notable for multiple first-degree relatives with schizophrenia. He reported a history of sexual and verbal abuse and attempted suicide once at age 13 years by hanging himself with a bathrobe. He left home at age 18 years to serve in the Marine Corps (2001-2006). His service included deployment to Afghanistan, where he received a purple heart. Upon his return, he received BA and MS degrees. He married and had 2 daughters but became estranged from his wife. By his most recent admission, he was unemployed and living with his half-sister.
On the first day of this most recent psychiatric hospitalization, he was restarted on aripiprazole 10 mg daily, and a medicine consult was sought to evaluate the progression of his MS. No new onset neurologic symptoms were noted, but he had possible residual lower extremity hyperreflexia and tandem gait incoordination. The episodes of psychotic and neurologic symptoms appeared independent, given that his psychiatric history preceded the onset of his MS.
The patient reported no visual hallucinations starting day 2, and he no longer endorsed auditory hallucinations by day 3. However, he continued to appear internally preoccupied and was noticed to be pacing around the unit. On day 4 he presented with newly pressured speech and flights of ideas, while his affect remained euthymic and his sleep stayed consistent. In combination with his ongoing pacing, his newfound symptoms were hypothesized to be possibly akathisia, an adverse effect (AE) of aripiprazole. As such, on day 5 his dose was lowered to 5 mg daily. He continued to report no hallucinations and demonstrated progressively increased emotional range. A MRI scan was done on day 6 in case a new lesion could be identified, suggesting a primary MS flare-up; however, the scan identified no enhancing lesions, indicating no ongoing demyelination. After a neurology consult corroborated this conclusion, he was discharged in stable condition on day 7.
As is the case with the majority of patients with MS-induced psychosis, he continued to have relapsing psychiatric disease even after MS treatment had been started. Unfortunately, because this patient had stopped taking his atypical antipsychotic medication several weeks prior to his hospitalization, we cannot clarify whether his psychosis stems from a primary psychiatric vs MS process.
Discussion
Presently, treatment preferences for MS-related psychosis are divided between atypical antipsychotics and glucocorticoids. Some suggest that the treatment remains similar between MS-related psychosis and primary psychotic disorders in that atypical antipsychotics are the standard of care.12 A variety of atypical antipsychotics have been used successfully in case reports, including zipradisone, risperidone, olanzapine, quetiapine, and aripiprazole.13,14 First-generation antipsychotics and other psychotropic drugs that can precipitate extra-pyramidal AEs are not recommended given their potential additive effect to motor deficits associated with MS.12 Alternatively, several case reports have found that MS-related psychotic symptoms respond to glucocorticoids more effectively, while cautioning that glucocorticoids can precipitate psychosis and depression.15,16 One review article found that 90% of patients who received corticosteroids saw an improvement in their psychotic symptoms.2
Finally, it is possible that our patient’s neuropsychiatric symptoms can be explained by autoantibody formation in response to toxin exposure during his time in Afghanistan. In a pilot study of veterans with GWI, Abou-Donia and colleagues found 2-to-9 fold increase in autoantibody reactivity levels of the following neuronal and glial-specific proteins relative to healthy controls: neurofilament triplet proteins, tubulin, microtubule-associated tau proteins, microtubule-associated protein-2, myelin basic protein, myelin-associated glycoprotein, glial fibrillary acidic protein, and calcium-calmodulin kinase II.17,18 Many of these autoantibodies are longstanding explicit markers for neurodegenerative disorders, given that they target proteins and antigens that support axonal transport and myelination. Still Gulf War veteran status has yet to be explicitly linked to an increased risk of MS,19 making this hypothesis less likely for our patient. Future research should address the clinical and therapeutic implications of different autoantibody levels in combat veterans with psychosis.
Conclusion
For patients with MS, mood disorder and psychotic symptoms should warrant a MRI given the possibility of a psychiatric manifestation of MS relapse. Ultimately, our patient’s presentation was inconsistent with the expected clinical presentations of both a primary psychotic disorder and psychosis as a manifestation of MS. His late age at his first psychotic break is atypical for primary psychotic disease, and the lack of MRI imaging done at his initial psychotic episodes cannot exclude a primary MS diagnosis. Still, his lack of MRI findings at his most recent hospitalization, negative symptomatology, and strong history of schizophrenia make a primary psychotic disorder likely.
Following his future clinical course will be necessary to determine the etiology of his psychotic episodes. Future episodes of psychosis with neurologic symptoms would suggest a primary MS diagnosis and potential benefit of immunosuppressant treatment, whereas repeated psychotic breaks with minimal temporal lobe involvement or demyelination as seen on MRI would be suspicious for separate MS and psychotic disease processes. Further research on treatment regimens for patients experiencing psychosis as a manifestation of MS is still necessary.
1. Wallin MT, Culpepper WJ, Campbell JD, et al. The prevalence of MS in the United States: A population-based estimate using health claims data. Neurology. 2019;92(10):e1029-e1040.
2. Camara-Lemarroy CR, Ibarra-Yruegas BE, Rodriguez-Gutierrez R, Berrios-Morales I, Ionete C, Riskind P. The varieties of psychosis in multiple sclerosis: a systematic review of cases. Mult Scler Relat Disord. 2017;12:9-14.
3. Cottrel SS, Wilson SA. The affective symptomatology of disseminated sclerosis: a study of 100 cases. J Neurol Psychopathology. 1926;7(25):1-30.
4. Johansson V, Lundholm C, Hillert J, et al. Multiple sclerosis and psychiatric disorders: comorbidity and sibling risk in a nationwide Swedish cohort. Mult Scler. 2014;20(14):1881-1891.
5. Rossi S, Studer V, Motta C, et al. Neuroinflammation drives anxiety and depression in relapsing-remitting multiple sclerosis. Neurology. 2017;89(13):1338-1347.
6. Gilberthorpe TG, O’Connell KE, Carolan A, et al. The spectrum of psychosis in multiple sclerosis: a clinical case series. Neuropsychiatric disease and treatment. 2017;13:303.
7. Patten SB, Svenson LW, Metz LM. Psychotic disorders in MS: population-based evidence of an association. Neurology 2005;65(7):1123-1125.
8. Kosmidis MH, Giannakou M, Messinis L, Papathanasopoulos P. Psychotic features associated with multiple sclerosis. Int Rev Psychiatry. 2010; 22(1):55-66.
9. US Department of Veterans Affairs. Public health: military exposures. https://www.publichealth.va.gov/exposures/. Updated April 16, 2019. Accessed May 13, 2019.
10. DeBeer BB, Davidson D, Meyer EC, Kimbrel NA, Gulliver SB, Morissette SB. The association between toxic exposures and chronic multisymptom illness in veterans of the wars of Iraq and Afghanistan. J Occup Environ Med. 2017;59(1):54-60.
11. Kang HK, Li B, Mahan CM, Eisen SA, Engel CC. Health of US veterans of 1991 Gulf War: a follow-up survey in 10 years. J Occup Environ Med. 2009;51(4):401-410.
12. Murphy R, O’Donoghue S, Counihan T, et al. Neuropsychiatric syndromes of multiple sclerosis. J Neurol Neurosurg Psychiatry. 2017;88(8):697-708.
13. Davids E, Hartwig U, Gastpar, M. Antipsychotic treatment of psychosis associated with multiple sclerosis. Prog Neuro Psychopharmacol Biol Psychiatry. 2004;28(4):743-744.
14. Lo Fermo S, Barone R, Patti F, et al. Outcome of psychiatric symptoms presenting at onset of multiple sclerosis: a retrospective study. Mult Scler. 2010;16(6):742-748.
15. Enderami A, Fouladi R, Hosseini HS. First-episode psychosis as the initial presentation of multiple sclerosis: a case report. Int Medical Case Rep J. 2018;11:73-76.
16. Fragoso YD, Frota ER, Lopes JS, et al. Severe depression, suicide attempts, and ideation during the use of interferon beta by patients with multiple sclerosis. Clin Neuropharmacol. 2010;33(6):312-316.
17. Abou-Donia MB, Conboy LA, Kokkotou E, et al. Screening for novel central nervous system biomarkers in veterans with Gulf War Illness. Neurotoxicol Teratol. 2017;61:36-46.
18. Abou-Donia MB, Lieberman A, Curtis L. Neural autoantibodies in patients with neurological symptoms and histories of chemical/mold exposures. Toxicol Ind Health. 2018;34(1):44-53.
19. Wallin MT, Kurtzke JF, Culpepper WJ, et al. Multiple sclerosis in Gulf War era veterans. 2. Military deployment and risk of multiple sclerosis in the first Gulf War. Neuroepidemiology. 2014;42(4):226-234.
1. Wallin MT, Culpepper WJ, Campbell JD, et al. The prevalence of MS in the United States: A population-based estimate using health claims data. Neurology. 2019;92(10):e1029-e1040.
2. Camara-Lemarroy CR, Ibarra-Yruegas BE, Rodriguez-Gutierrez R, Berrios-Morales I, Ionete C, Riskind P. The varieties of psychosis in multiple sclerosis: a systematic review of cases. Mult Scler Relat Disord. 2017;12:9-14.
3. Cottrel SS, Wilson SA. The affective symptomatology of disseminated sclerosis: a study of 100 cases. J Neurol Psychopathology. 1926;7(25):1-30.
4. Johansson V, Lundholm C, Hillert J, et al. Multiple sclerosis and psychiatric disorders: comorbidity and sibling risk in a nationwide Swedish cohort. Mult Scler. 2014;20(14):1881-1891.
5. Rossi S, Studer V, Motta C, et al. Neuroinflammation drives anxiety and depression in relapsing-remitting multiple sclerosis. Neurology. 2017;89(13):1338-1347.
6. Gilberthorpe TG, O’Connell KE, Carolan A, et al. The spectrum of psychosis in multiple sclerosis: a clinical case series. Neuropsychiatric disease and treatment. 2017;13:303.
7. Patten SB, Svenson LW, Metz LM. Psychotic disorders in MS: population-based evidence of an association. Neurology 2005;65(7):1123-1125.
8. Kosmidis MH, Giannakou M, Messinis L, Papathanasopoulos P. Psychotic features associated with multiple sclerosis. Int Rev Psychiatry. 2010; 22(1):55-66.
9. US Department of Veterans Affairs. Public health: military exposures. https://www.publichealth.va.gov/exposures/. Updated April 16, 2019. Accessed May 13, 2019.
10. DeBeer BB, Davidson D, Meyer EC, Kimbrel NA, Gulliver SB, Morissette SB. The association between toxic exposures and chronic multisymptom illness in veterans of the wars of Iraq and Afghanistan. J Occup Environ Med. 2017;59(1):54-60.
11. Kang HK, Li B, Mahan CM, Eisen SA, Engel CC. Health of US veterans of 1991 Gulf War: a follow-up survey in 10 years. J Occup Environ Med. 2009;51(4):401-410.
12. Murphy R, O’Donoghue S, Counihan T, et al. Neuropsychiatric syndromes of multiple sclerosis. J Neurol Neurosurg Psychiatry. 2017;88(8):697-708.
13. Davids E, Hartwig U, Gastpar, M. Antipsychotic treatment of psychosis associated with multiple sclerosis. Prog Neuro Psychopharmacol Biol Psychiatry. 2004;28(4):743-744.
14. Lo Fermo S, Barone R, Patti F, et al. Outcome of psychiatric symptoms presenting at onset of multiple sclerosis: a retrospective study. Mult Scler. 2010;16(6):742-748.
15. Enderami A, Fouladi R, Hosseini HS. First-episode psychosis as the initial presentation of multiple sclerosis: a case report. Int Medical Case Rep J. 2018;11:73-76.
16. Fragoso YD, Frota ER, Lopes JS, et al. Severe depression, suicide attempts, and ideation during the use of interferon beta by patients with multiple sclerosis. Clin Neuropharmacol. 2010;33(6):312-316.
17. Abou-Donia MB, Conboy LA, Kokkotou E, et al. Screening for novel central nervous system biomarkers in veterans with Gulf War Illness. Neurotoxicol Teratol. 2017;61:36-46.
18. Abou-Donia MB, Lieberman A, Curtis L. Neural autoantibodies in patients with neurological symptoms and histories of chemical/mold exposures. Toxicol Ind Health. 2018;34(1):44-53.
19. Wallin MT, Kurtzke JF, Culpepper WJ, et al. Multiple sclerosis in Gulf War era veterans. 2. Military deployment and risk of multiple sclerosis in the first Gulf War. Neuroepidemiology. 2014;42(4):226-234.
Hemolytic Uremic Syndrome With Severe Neurologic Complications in an Adult (FULL)
The case of a female presenting with Shiga toxin-producing Escherichia coli and hemolytic uremic syndrome highlights a severe neurologic complication that canbe associated with these conditions.
Hemolytic uremic syndrome (HUS) is a rare illness that can be acquired through the consumption of food products contaminated with strains of Shiga toxin-producing Escherichia coli (E coli; STEC).1 Between 6% and 15% of individuals infected with STEC develop HUS, with children affected more frequently than adults.2,3 This strain of E coli releases Shiga toxin into the systemic circulation, which causes a thrombotic microangiopathy resulting in the characteristic HUS triad of symptoms: acute renal insufficiency, thrombocytopenia, and hemolytic anemia.4-6
Although neurologic features are common in HUS, they have not been extensively studied, particularly in adults. We report a case of STEC 0157:H7 subtype HUS in an adult with severe neurologic complications. This case highlights the neurological sequelae in an adult with typical STEC-HUS. The use of treatment modalities, such as plasmapheresis and eculizumab, and their use in adult typical STEC-HUS also is explored.
Case
A 53-year-old white woman with no pertinent past medical history presented to the Bay Pines Veterans Affairs Healthcare System Emergency Department with a 2-day history of abdominal pain, vomiting, nausea, diarrhea, and bright bloody stools. She returned from a cruise to the Bahamas 3 days prior, where she ate local foods, including salads. She reported no fever, shortness of breath, chest pain, headache, and cognitive difficulties. She presented with a normal mental status and neurologic exam. Apart from leukocytosis and elevated glucose level, her laboratory results at initial presentation were normal, (Table). A stool sample showed occult blood with white blood cell counts (WBCs) but was negative for Clostridium difficile. She was started on ciprofloxacin 400 mg and metronidazole 500 mg on the day of admission.
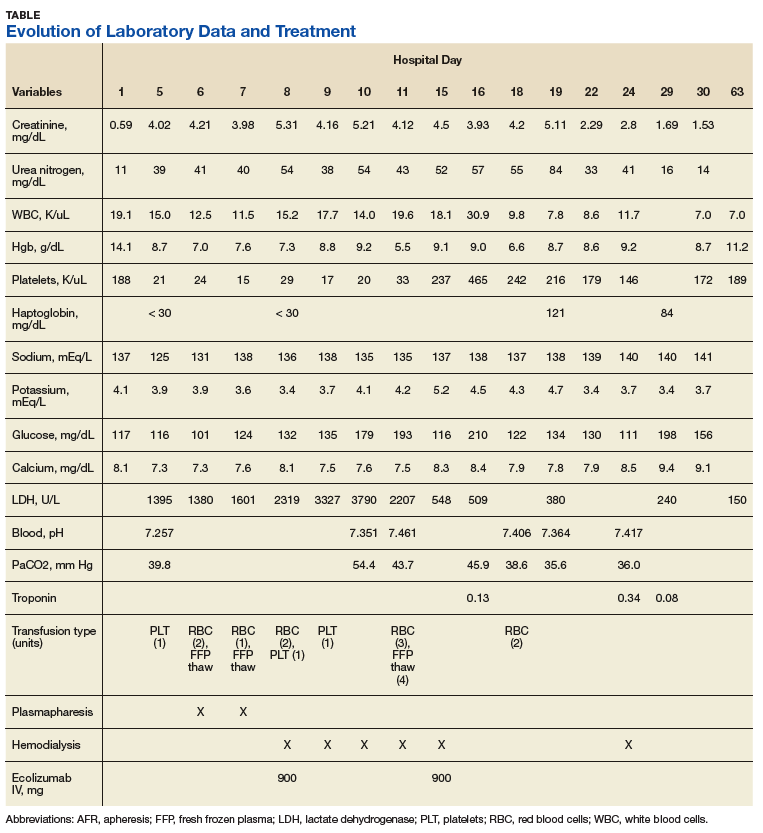
Hematuria was found on hospital day 2. On hospital day 4, the patient exhibited word finding difficulties. Blood studies revealed anemia, thrombocytopenia, leukocytosis, and increasing blood urea nitrogen (BUN) and creatinine. A computed tomography scan of the head was normal. Laboratory analysis showed schistocytes in the peripheral blood smear.
The patient’s cognitive functioning deteriorated on hospital day 5. She was not oriented to time or place. Her laboratory results showed complement level C3 at 70 mg/dL (ref: 83-193 mg/dL) complement C4 at 12 mg/dL (ref: 15-57mg/dL). The patient exhibited oliguria and hyponatremia, as well as both metabolic and respiratory acidosis; dialysis was then initiated. Results from the stool sample that was collected on hospital day 1 were received and tested positive for Shiga toxin.
At this point, the patient’s presentation of hemolytic anemia and thrombocytopenia in the setting of acute bloody diarrheal illness with known Shiga toxin, schistocytes on blood smear, and lack of pertinent medical history for other causes of this presentation made STEC-HUS the leading differential diagnosis. Plasmapheresis was ordered and performed on hospital day 6 and 7. Shiga toxin was no longer detected in the stool and antibiotics were stopped on hospital day 7.
The patient’s progressive deterioration in mental status continued on hospital day 8. She was not oriented to time or place, unable to perform simple calculations, and could not spell the word “hand” backwards. Physicians observed repetitive jerking motions of the upper extremities that were worse on the left side. An electroencephalogram (EEG) revealed right hemispheric sharp waves that were thought to be epileptiform (Figure 1). The patient began taking levetiracetam 1500 mg IV with 750 mg bid maintenance for seizure control. Plasmapheresis was discontinued due to her continued neurologic deterioration on this therapy. Consequently, eculizumab 900 mg IV was given along with the Neisseria meningitidis (N meningitidis) vaccine and a 19-day course of azithromycin 250 mg po as prophylaxis for encapsulated bacteria.
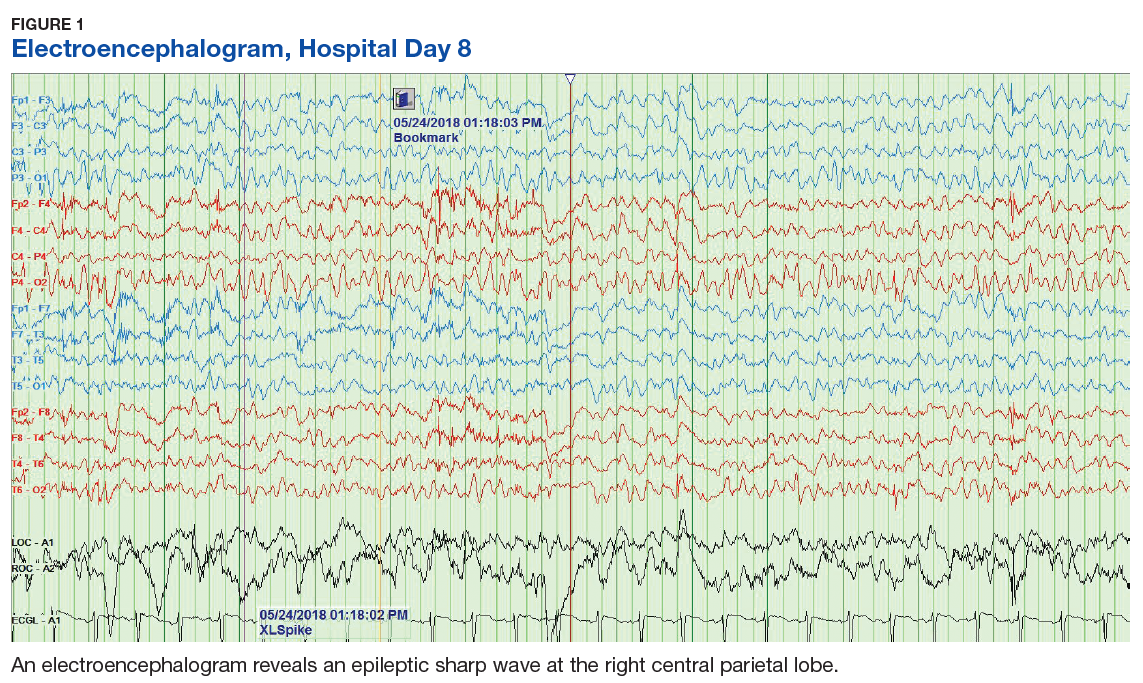
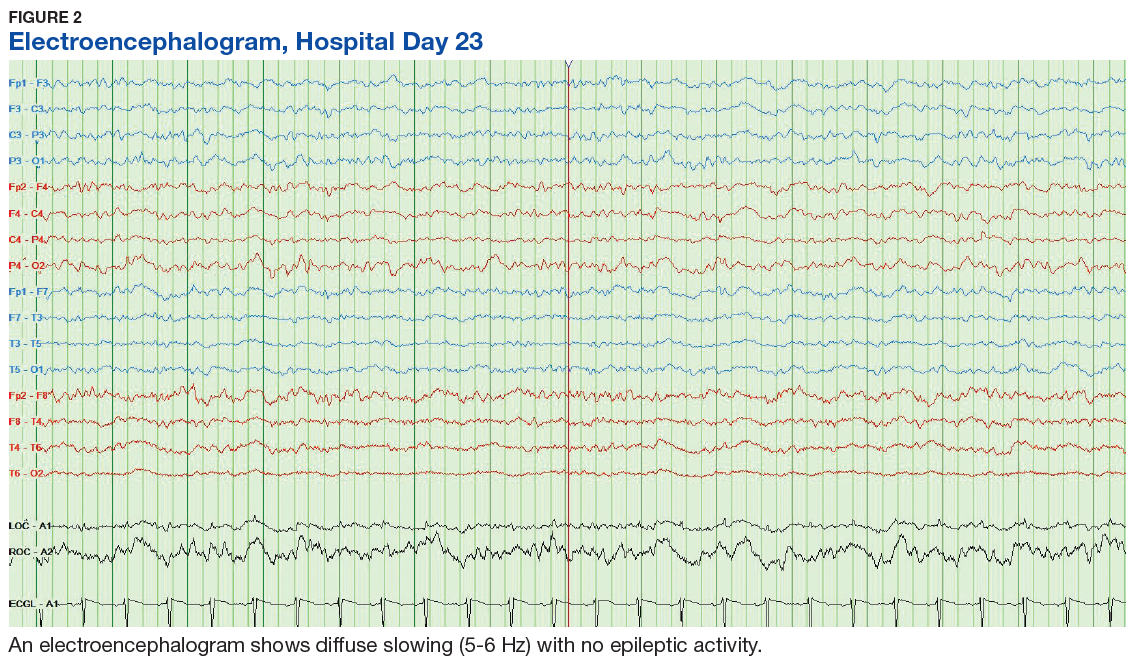
The patient continued to seize on hospital days 10 through 13. Oculocephalic maneuvers showed a tendency to keep her eyes deviated to the right. Her pupils continued to react to light. A repeat EEG showed diffuse slowing (5-6 Hz) with no epileptic activity seen (Figure 2). A second dose of eculizumab 900 mg IV was administered on hospital day 15. The patient experienced cardiac arrest on hospital day 16 and was successfully resuscitated. On hospital day 25 (10 days after receiving her second dose of eculizumab), the patient was able to speak and follow simple commands but exhibited difficulty concentrating and poor impulse control.
The patient was alert and oriented to person, place, time, and situation on hospital day 28 (6 days after the third and final dose of eculizumab). A neurologic exam was significant only for a slight intention tremor. She was continued on levetiracetam with a plan to be maintained on the medication for the next 6 months for seizure control. She was discharged on hospital day 30.
Twenty-eight days postdischarge (57 days postadmission), the patient showed marked recovery. She had returned to her previous employment as a business administrator on a part-time basis and exhibited no deficiencies in executive functioning or handling activities of daily living. Although she had been very active prior to this illness, she now experienced decreased physical and mental endurance; however, this gradually improved with physical therapy. She planned on returning to work on a full-time basis when she had regained her stamina. She also noticed difficulties in retaining short term memory since her discharge but believed that these symptoms were remitting. On examination her mental status and neurologic exam was significant for inability to continue serial 7s, left sided 4/5 muscle strength in quadriceps and thumb to 5th metacarpal adduction, bilateral 1+ reflexes in muscle groups tested (triceps, biceps, brachioradialis, patellar, and Achilles), loss of dull pinprick sensation bilaterally at web of hands, deficit in tandem gait while looking away, and slight intention tremor on finger to nose testing bilaterally (with left hand tremor more pronounced than right). Her complete blood count was normal. Her recovery continues to be monitored in an outpatient setting.
Discussion
HUS is characterized by 3 core clinical features: microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury.4 Schistocytes are seen on peripheral blood smear and occur due to the passage of red blood cells over the microvascular thrombi induced by the disease. HUS can be classified as typical, atypical, or occurring with a coexisting disease. Typical HUS is associated with STEC 0157:H7 subtype, a bacterium known to be acquired through contaminated food and via human-to-human transmission.6-8 In the case of typical STEC 0157:H7, the bacterium releases a verotoxin that damages the vascular endothelium, thereby leading to activation of the coagulation cascade and eventually the formation of thrombi.4 It has been hypothesized that the Shiga toxin also activates the alternative complement pathway directly, which could contribute to thrombosis.9 This would explain the findings of low complement levels in our patient. Atypical HUS is primarily attributable to mutations in the alternative complement pathway. Causes for the third type of HUS can include Streptococcus pneumoniae, HIV, drug toxicity, and alterations in the metabolism of cobalamin C.
Epidemiologically, 15.3% of children aged < 5 years develop typical HUS after exposure to STEC compared with 1.2% of adults aged 18 to 59 years. The median age of patients who developed HUS from STEC exposure was 4 years compared with 16 years for those who did not develop HUS.2
Neurologic manifestations increase mortality for HUS patients.10 These have been described in the pediatric population as alteration in consciousness (85%), seizures (71%), pyramidal syndrome (52%), and extrapyramidal syndrome with hypertonia (42%).11 Brain imaging in children has demonstrated hemorrhagic lesions involving the pons, basal ganglia, and occipital cortex.11 Blood flow to areas such as the cerebellum, brainstem, and orbitofrontal area can be compromised.10 Adult patients with HUS can present without lesions on cranial magnetic resonance imaging (MRI), but instead with transient symmetric vasogenic edema of the central brain stem.12 Unfortunately in this case, MRI was not performed because it was thought to provide limited aid in diagnosis and to avoid unnecessary testing for the acutely ill patient.
The underlying pathophysiology of neurologic manifestations in patients may be due to a metabolic disturbance, toxin-mediated damage of the vascular endothelium, or toxin-induced cytokine release resulting in death of neural cells and subsequent neuroinflammation. However, the most likely mechanism is parenchymal ischemic changes related to microangiopathy.11,13 Pediatric patients often experience seizures and altered mental status, and their EEGs display delta waves.13 This patient’s diffuse slowing on her second EEG and altered mental status suggests that the neuropathologic mechanisms for typical HUS in adults may be similar to those in children.
HUS Treatment
The treatment and management of adults with typical STEC-HUS is evolving. The patient was first suspected to have an infectious colitis and empiric antibiotics were initiated. Some studies suggest that antibiotic administration may worsen the course of HUS in children as it may lead to release and subsequent absorption of Shiga toxin in the intestine.9,14 However, there is little evidence to suggest harm or efficacy of administration in adults. It is unclear what role antibiotic administration played in the recovery time of HUS given the co-administration of other treatments such as eculizumab and plasmapheresis, but it does appear to have helped with the initial E coli infection.
Plasmapheresis was subsequently administered, due to its documented benefit in the treatment of HUS.15 However, it should be noted that even though plasmapheresis is currently used in patients with CNS involvement, it remains unproven with conflicting information on its efficacy.3,16 The mechanism of action is unclear, but it has been hypothesized that plasmapheresis prevents microangiopathy caused by microthrombi.3,16 For this reason, eculizumab is becoming the mainstay for treatment of STEC-HUS with neurologic complications given the lack of well researched alternative treatments. In this case study, the use of plasmapheresis did not result in clinical improvement, and was abandoned after 2 days of treatment.
Eculizumab is a humanized, recombinant monoclonal IgG antibody that is a terminal complement inhibitor of the alternative complement system at the final step to cleave C5.17 The Shiga toxin may directly activate the complement system via the alternative pathway, which can result in uncontrolled platelet and white blood cell activation and depletion, endothelial cell damage, and hemolysis. The galvanized complement system leads to a series of cascading events that contribute to organ damage and death.9 Eculizumab is FDA approved for use in atypical HUS.18 It also can be used off-label to treat typical-HUS in adults with neurologic complications.
Eculizumab interferes with the immune response against encapsulated bacteria because it inhibits the alternative complement pathway. Thus, vaccination against N meningitides is recommended 2 weeks prior to the administration of eculizumab. However, in situations where the risks of delaying eculizumab for 2 weeks are greater than the risk of developing an N meningitides infection, eculizumab may be given without delay.18 Given the rapid deterioration of our patient’s condition, the vaccine and eculizumab were given together with prophylactic azithromycin. Although penicillin is the standard for prophylaxis in this situation, the patient’s penicillin allergy led to the use of azithromycin 250 mg po once a day. Literature also suggests azithromycin reduces the carriage duration of E coli-induced colitis.19 As such, it is possible that some improvement in the patient’s condition could be attributed to the elimination of the pathogen and toxin.
Conclusion
Three doses of eculizumab were administered at weekly intervals, with the first dose on hospital day 8 and the final dose on hospital day 22. Prior to the first dose, the patient displayed significant decline in mental status with EEG findings of right hemisphere epileptogenic discharges. After her third dose, she was found to have a drastically improved mental status exam and a normal EEG. One week later, she was discharged home. At the time of her 1-month follow-up, she was independent in all activities of daily living and had returned to part-time work. Apart from subtle cognitive changes, the remainder of her neurologic exam was normal.
There is evidence that supports the efficacy of eculizumab in children with HUS with neurologic symptoms on dialysis.20 However, its use in adults is not well established.21 This patient required dialysis and had neurologic symptoms similar to pediatric patients described in the literature, and responded similarly to the eculizumab. The rationale for the use of eculizumab in STEC-HUS also is evidenced by in vitro demonstrations of complement activation in STEC-HUS.22-25 This case report adds to the literature supporting the use of eculizumab in adult patients with typical HUS with neurological complications. Further research is necessary to develop guidelines in the treatment of adult STEC-HUS with regards to neurologic complications.
Acknowledgments
The authors would like to thank Pete DiStaso, REEGT for his work on obtaining the electroencephalograms and Anthony Rinaldi, PsyD; Julie Cessnapalas, PsyD; and Syed Faizan Sagheer for proof-reading the article.
1. Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365(9464):1073-1086.
2. Gould LH, Demma L, Jones TF, et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin Infect Dis. 2009;49(10):1480-1485.
3. Boyce TG, Swerdlow DL, Griffin PM. Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med. 1995;333(6):364-368.
4. Rondeau E, Peraldi MN. Escherichia coli and the hemolytic-uremic syndrome. N Engl J Med. 1996;335(9):660-662.
5. Te Loo DM, van Hinsbergh VW, van den Heuvel LP, Monnens LA. Detection of verocytotoxin bound to circulating polymorphonuclear leukocytes of patients with hemolytic uremic syndrome. J Am Soc Nephrol. 2001;12(4):800-806.
6. Tran SL, Jenkins C, Livrelli V, Schüller S. Shiga toxin 2 translocation across intestinal epithelium is linked to virulence of Shiga toxin-producing Escherichia coli in humans. Microbiology. 2018;164(4):509-516.
7. Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847-2856.
8. Ferens WA, Hovde CJ. Escherichia coli O157:H7: animal reservoir and sources of human infection. Foodborne Pathog Dis. 2011;8(4):465-487.
9. Percheron L, Gramada R, Tellier S, et al. Eculizumab treatment in severe pediatric STEC-HUS: a multicenter retrospective study. Pediatr Nephrol. 2018;33(8):1385-1394.
10. Hosaka T, Nakamagoe K, Tamaoka A. Hemolytic uremic syndrome-associated encephalopathy successfully treated with corticosteroids. Intern Med. 2017;56(21):2937-2941.
11. Nathanson S, Kwon T, Elmaleh M, et al. Acute neurological involvement in diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2010;5(7):1218-1228.
12. Wengenroth M, Hoeltje J, Repenthin J, et al. Central nervous system involvement in adults with epidemic hemolytic uremic syndrome. AJNR Am J Neuroradiol. 2013;34(5):1016-1021, S1.
13. Eriksson KJ, Boyd SG, Tasker RC. Acute neurology and neurophysiology of haemolytic-uraemic syndrome. Arch Dis Child. 2001;84(5):434-435.
14. Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342(26):1930-1936.
15. Nguyen TC, Kiss JE, Goldman JR, Carcillo JA. The role of plasmapheresis in critical illness. Crit Care Clin. 2012;28(3):453-468, vii.
16. Loos S, Ahlenstiel T, Kranz B, et al. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: presentation and short-term outcome in children. Clin Infect Dis. 2012;55(6):753-759.
17. Hossain MA, Cheema A, Kalathil S, et al. Atypical hemolytic uremic syndrome: Laboratory characteristics, complement-amplifying conditions, renal biopsy, and genetic mutations. Saudi J Kidney Dis Transpl. 2018;29(2):276-283.
18. Soliris (eculizumab) [package insert]. Cheshire, CT: Alexion Pharmaceuticals, Inc; 2011.
19. Keenswijk W, Raes A, Vande Walle J. Is eculizumab efficacious in Shigatoxin-associated hemolytic uremic syndrome? A narrative review of current evidence. Eur J Pediatr. 2018;177(3):311-318.
20. Lapeyraque AL, Malina M, Fremeaux-Bacchi V, et al. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med. 2011;364(26):2561-2563.
21. Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, Ahlenstiel-Grunow T. Eculizumab in typical hemolytic uremic syndrome (HUS) with neurological involvement. Medicine (Baltimore). 2015;94(24):e1000.
22. Kim Y, Miller K, Michael AF. Breakdown products of C3 and factor B in hemolytic-uremic syndrome. J Lab Clin Med. 1977;89(4):845-850.
23. Monnens L, Molenaar J, Lambert PH, Proesmans W, van Munster P. The complement system in hemolytic-uremic syndrome in childhood. Clin Nephrol. 1980;13(4):168-171.
24. Thurman JM, Marians R, Emlen W, et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2009;4(12):1920-1924.
25. Ståhl AL, Sartz L, Karpman D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood. 2011;117(20):5503-5513.
The case of a female presenting with Shiga toxin-producing Escherichia coli and hemolytic uremic syndrome highlights a severe neurologic complication that canbe associated with these conditions.
The case of a female presenting with Shiga toxin-producing Escherichia coli and hemolytic uremic syndrome highlights a severe neurologic complication that canbe associated with these conditions.
Hemolytic uremic syndrome (HUS) is a rare illness that can be acquired through the consumption of food products contaminated with strains of Shiga toxin-producing Escherichia coli (E coli; STEC).1 Between 6% and 15% of individuals infected with STEC develop HUS, with children affected more frequently than adults.2,3 This strain of E coli releases Shiga toxin into the systemic circulation, which causes a thrombotic microangiopathy resulting in the characteristic HUS triad of symptoms: acute renal insufficiency, thrombocytopenia, and hemolytic anemia.4-6
Although neurologic features are common in HUS, they have not been extensively studied, particularly in adults. We report a case of STEC 0157:H7 subtype HUS in an adult with severe neurologic complications. This case highlights the neurological sequelae in an adult with typical STEC-HUS. The use of treatment modalities, such as plasmapheresis and eculizumab, and their use in adult typical STEC-HUS also is explored.
Case
A 53-year-old white woman with no pertinent past medical history presented to the Bay Pines Veterans Affairs Healthcare System Emergency Department with a 2-day history of abdominal pain, vomiting, nausea, diarrhea, and bright bloody stools. She returned from a cruise to the Bahamas 3 days prior, where she ate local foods, including salads. She reported no fever, shortness of breath, chest pain, headache, and cognitive difficulties. She presented with a normal mental status and neurologic exam. Apart from leukocytosis and elevated glucose level, her laboratory results at initial presentation were normal, (Table). A stool sample showed occult blood with white blood cell counts (WBCs) but was negative for Clostridium difficile. She was started on ciprofloxacin 400 mg and metronidazole 500 mg on the day of admission.
Hematuria was found on hospital day 2. On hospital day 4, the patient exhibited word finding difficulties. Blood studies revealed anemia, thrombocytopenia, leukocytosis, and increasing blood urea nitrogen (BUN) and creatinine. A computed tomography scan of the head was normal. Laboratory analysis showed schistocytes in the peripheral blood smear.
The patient’s cognitive functioning deteriorated on hospital day 5. She was not oriented to time or place. Her laboratory results showed complement level C3 at 70 mg/dL (ref: 83-193 mg/dL) complement C4 at 12 mg/dL (ref: 15-57mg/dL). The patient exhibited oliguria and hyponatremia, as well as both metabolic and respiratory acidosis; dialysis was then initiated. Results from the stool sample that was collected on hospital day 1 were received and tested positive for Shiga toxin.
At this point, the patient’s presentation of hemolytic anemia and thrombocytopenia in the setting of acute bloody diarrheal illness with known Shiga toxin, schistocytes on blood smear, and lack of pertinent medical history for other causes of this presentation made STEC-HUS the leading differential diagnosis. Plasmapheresis was ordered and performed on hospital day 6 and 7. Shiga toxin was no longer detected in the stool and antibiotics were stopped on hospital day 7.
The patient’s progressive deterioration in mental status continued on hospital day 8. She was not oriented to time or place, unable to perform simple calculations, and could not spell the word “hand” backwards. Physicians observed repetitive jerking motions of the upper extremities that were worse on the left side. An electroencephalogram (EEG) revealed right hemispheric sharp waves that were thought to be epileptiform (Figure 1). The patient began taking levetiracetam 1500 mg IV with 750 mg bid maintenance for seizure control. Plasmapheresis was discontinued due to her continued neurologic deterioration on this therapy. Consequently, eculizumab 900 mg IV was given along with the Neisseria meningitidis (N meningitidis) vaccine and a 19-day course of azithromycin 250 mg po as prophylaxis for encapsulated bacteria.
The patient continued to seize on hospital days 10 through 13. Oculocephalic maneuvers showed a tendency to keep her eyes deviated to the right. Her pupils continued to react to light. A repeat EEG showed diffuse slowing (5-6 Hz) with no epileptic activity seen (Figure 2). A second dose of eculizumab 900 mg IV was administered on hospital day 15. The patient experienced cardiac arrest on hospital day 16 and was successfully resuscitated. On hospital day 25 (10 days after receiving her second dose of eculizumab), the patient was able to speak and follow simple commands but exhibited difficulty concentrating and poor impulse control.
The patient was alert and oriented to person, place, time, and situation on hospital day 28 (6 days after the third and final dose of eculizumab). A neurologic exam was significant only for a slight intention tremor. She was continued on levetiracetam with a plan to be maintained on the medication for the next 6 months for seizure control. She was discharged on hospital day 30.
Twenty-eight days postdischarge (57 days postadmission), the patient showed marked recovery. She had returned to her previous employment as a business administrator on a part-time basis and exhibited no deficiencies in executive functioning or handling activities of daily living. Although she had been very active prior to this illness, she now experienced decreased physical and mental endurance; however, this gradually improved with physical therapy. She planned on returning to work on a full-time basis when she had regained her stamina. She also noticed difficulties in retaining short term memory since her discharge but believed that these symptoms were remitting. On examination her mental status and neurologic exam was significant for inability to continue serial 7s, left sided 4/5 muscle strength in quadriceps and thumb to 5th metacarpal adduction, bilateral 1+ reflexes in muscle groups tested (triceps, biceps, brachioradialis, patellar, and Achilles), loss of dull pinprick sensation bilaterally at web of hands, deficit in tandem gait while looking away, and slight intention tremor on finger to nose testing bilaterally (with left hand tremor more pronounced than right). Her complete blood count was normal. Her recovery continues to be monitored in an outpatient setting.
Discussion
HUS is characterized by 3 core clinical features: microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury.4 Schistocytes are seen on peripheral blood smear and occur due to the passage of red blood cells over the microvascular thrombi induced by the disease. HUS can be classified as typical, atypical, or occurring with a coexisting disease. Typical HUS is associated with STEC 0157:H7 subtype, a bacterium known to be acquired through contaminated food and via human-to-human transmission.6-8 In the case of typical STEC 0157:H7, the bacterium releases a verotoxin that damages the vascular endothelium, thereby leading to activation of the coagulation cascade and eventually the formation of thrombi.4 It has been hypothesized that the Shiga toxin also activates the alternative complement pathway directly, which could contribute to thrombosis.9 This would explain the findings of low complement levels in our patient. Atypical HUS is primarily attributable to mutations in the alternative complement pathway. Causes for the third type of HUS can include Streptococcus pneumoniae, HIV, drug toxicity, and alterations in the metabolism of cobalamin C.
Epidemiologically, 15.3% of children aged < 5 years develop typical HUS after exposure to STEC compared with 1.2% of adults aged 18 to 59 years. The median age of patients who developed HUS from STEC exposure was 4 years compared with 16 years for those who did not develop HUS.2
Neurologic manifestations increase mortality for HUS patients.10 These have been described in the pediatric population as alteration in consciousness (85%), seizures (71%), pyramidal syndrome (52%), and extrapyramidal syndrome with hypertonia (42%).11 Brain imaging in children has demonstrated hemorrhagic lesions involving the pons, basal ganglia, and occipital cortex.11 Blood flow to areas such as the cerebellum, brainstem, and orbitofrontal area can be compromised.10 Adult patients with HUS can present without lesions on cranial magnetic resonance imaging (MRI), but instead with transient symmetric vasogenic edema of the central brain stem.12 Unfortunately in this case, MRI was not performed because it was thought to provide limited aid in diagnosis and to avoid unnecessary testing for the acutely ill patient.
The underlying pathophysiology of neurologic manifestations in patients may be due to a metabolic disturbance, toxin-mediated damage of the vascular endothelium, or toxin-induced cytokine release resulting in death of neural cells and subsequent neuroinflammation. However, the most likely mechanism is parenchymal ischemic changes related to microangiopathy.11,13 Pediatric patients often experience seizures and altered mental status, and their EEGs display delta waves.13 This patient’s diffuse slowing on her second EEG and altered mental status suggests that the neuropathologic mechanisms for typical HUS in adults may be similar to those in children.
HUS Treatment
The treatment and management of adults with typical STEC-HUS is evolving. The patient was first suspected to have an infectious colitis and empiric antibiotics were initiated. Some studies suggest that antibiotic administration may worsen the course of HUS in children as it may lead to release and subsequent absorption of Shiga toxin in the intestine.9,14 However, there is little evidence to suggest harm or efficacy of administration in adults. It is unclear what role antibiotic administration played in the recovery time of HUS given the co-administration of other treatments such as eculizumab and plasmapheresis, but it does appear to have helped with the initial E coli infection.
Plasmapheresis was subsequently administered, due to its documented benefit in the treatment of HUS.15 However, it should be noted that even though plasmapheresis is currently used in patients with CNS involvement, it remains unproven with conflicting information on its efficacy.3,16 The mechanism of action is unclear, but it has been hypothesized that plasmapheresis prevents microangiopathy caused by microthrombi.3,16 For this reason, eculizumab is becoming the mainstay for treatment of STEC-HUS with neurologic complications given the lack of well researched alternative treatments. In this case study, the use of plasmapheresis did not result in clinical improvement, and was abandoned after 2 days of treatment.
Eculizumab is a humanized, recombinant monoclonal IgG antibody that is a terminal complement inhibitor of the alternative complement system at the final step to cleave C5.17 The Shiga toxin may directly activate the complement system via the alternative pathway, which can result in uncontrolled platelet and white blood cell activation and depletion, endothelial cell damage, and hemolysis. The galvanized complement system leads to a series of cascading events that contribute to organ damage and death.9 Eculizumab is FDA approved for use in atypical HUS.18 It also can be used off-label to treat typical-HUS in adults with neurologic complications.
Eculizumab interferes with the immune response against encapsulated bacteria because it inhibits the alternative complement pathway. Thus, vaccination against N meningitides is recommended 2 weeks prior to the administration of eculizumab. However, in situations where the risks of delaying eculizumab for 2 weeks are greater than the risk of developing an N meningitides infection, eculizumab may be given without delay.18 Given the rapid deterioration of our patient’s condition, the vaccine and eculizumab were given together with prophylactic azithromycin. Although penicillin is the standard for prophylaxis in this situation, the patient’s penicillin allergy led to the use of azithromycin 250 mg po once a day. Literature also suggests azithromycin reduces the carriage duration of E coli-induced colitis.19 As such, it is possible that some improvement in the patient’s condition could be attributed to the elimination of the pathogen and toxin.
Conclusion
Three doses of eculizumab were administered at weekly intervals, with the first dose on hospital day 8 and the final dose on hospital day 22. Prior to the first dose, the patient displayed significant decline in mental status with EEG findings of right hemisphere epileptogenic discharges. After her third dose, she was found to have a drastically improved mental status exam and a normal EEG. One week later, she was discharged home. At the time of her 1-month follow-up, she was independent in all activities of daily living and had returned to part-time work. Apart from subtle cognitive changes, the remainder of her neurologic exam was normal.
There is evidence that supports the efficacy of eculizumab in children with HUS with neurologic symptoms on dialysis.20 However, its use in adults is not well established.21 This patient required dialysis and had neurologic symptoms similar to pediatric patients described in the literature, and responded similarly to the eculizumab. The rationale for the use of eculizumab in STEC-HUS also is evidenced by in vitro demonstrations of complement activation in STEC-HUS.22-25 This case report adds to the literature supporting the use of eculizumab in adult patients with typical HUS with neurological complications. Further research is necessary to develop guidelines in the treatment of adult STEC-HUS with regards to neurologic complications.
Acknowledgments
The authors would like to thank Pete DiStaso, REEGT for his work on obtaining the electroencephalograms and Anthony Rinaldi, PsyD; Julie Cessnapalas, PsyD; and Syed Faizan Sagheer for proof-reading the article.
Hemolytic uremic syndrome (HUS) is a rare illness that can be acquired through the consumption of food products contaminated with strains of Shiga toxin-producing Escherichia coli (E coli; STEC).1 Between 6% and 15% of individuals infected with STEC develop HUS, with children affected more frequently than adults.2,3 This strain of E coli releases Shiga toxin into the systemic circulation, which causes a thrombotic microangiopathy resulting in the characteristic HUS triad of symptoms: acute renal insufficiency, thrombocytopenia, and hemolytic anemia.4-6
Although neurologic features are common in HUS, they have not been extensively studied, particularly in adults. We report a case of STEC 0157:H7 subtype HUS in an adult with severe neurologic complications. This case highlights the neurological sequelae in an adult with typical STEC-HUS. The use of treatment modalities, such as plasmapheresis and eculizumab, and their use in adult typical STEC-HUS also is explored.
Case
A 53-year-old white woman with no pertinent past medical history presented to the Bay Pines Veterans Affairs Healthcare System Emergency Department with a 2-day history of abdominal pain, vomiting, nausea, diarrhea, and bright bloody stools. She returned from a cruise to the Bahamas 3 days prior, where she ate local foods, including salads. She reported no fever, shortness of breath, chest pain, headache, and cognitive difficulties. She presented with a normal mental status and neurologic exam. Apart from leukocytosis and elevated glucose level, her laboratory results at initial presentation were normal, (Table). A stool sample showed occult blood with white blood cell counts (WBCs) but was negative for Clostridium difficile. She was started on ciprofloxacin 400 mg and metronidazole 500 mg on the day of admission.
Hematuria was found on hospital day 2. On hospital day 4, the patient exhibited word finding difficulties. Blood studies revealed anemia, thrombocytopenia, leukocytosis, and increasing blood urea nitrogen (BUN) and creatinine. A computed tomography scan of the head was normal. Laboratory analysis showed schistocytes in the peripheral blood smear.
The patient’s cognitive functioning deteriorated on hospital day 5. She was not oriented to time or place. Her laboratory results showed complement level C3 at 70 mg/dL (ref: 83-193 mg/dL) complement C4 at 12 mg/dL (ref: 15-57mg/dL). The patient exhibited oliguria and hyponatremia, as well as both metabolic and respiratory acidosis; dialysis was then initiated. Results from the stool sample that was collected on hospital day 1 were received and tested positive for Shiga toxin.
At this point, the patient’s presentation of hemolytic anemia and thrombocytopenia in the setting of acute bloody diarrheal illness with known Shiga toxin, schistocytes on blood smear, and lack of pertinent medical history for other causes of this presentation made STEC-HUS the leading differential diagnosis. Plasmapheresis was ordered and performed on hospital day 6 and 7. Shiga toxin was no longer detected in the stool and antibiotics were stopped on hospital day 7.
The patient’s progressive deterioration in mental status continued on hospital day 8. She was not oriented to time or place, unable to perform simple calculations, and could not spell the word “hand” backwards. Physicians observed repetitive jerking motions of the upper extremities that were worse on the left side. An electroencephalogram (EEG) revealed right hemispheric sharp waves that were thought to be epileptiform (Figure 1). The patient began taking levetiracetam 1500 mg IV with 750 mg bid maintenance for seizure control. Plasmapheresis was discontinued due to her continued neurologic deterioration on this therapy. Consequently, eculizumab 900 mg IV was given along with the Neisseria meningitidis (N meningitidis) vaccine and a 19-day course of azithromycin 250 mg po as prophylaxis for encapsulated bacteria.
The patient continued to seize on hospital days 10 through 13. Oculocephalic maneuvers showed a tendency to keep her eyes deviated to the right. Her pupils continued to react to light. A repeat EEG showed diffuse slowing (5-6 Hz) with no epileptic activity seen (Figure 2). A second dose of eculizumab 900 mg IV was administered on hospital day 15. The patient experienced cardiac arrest on hospital day 16 and was successfully resuscitated. On hospital day 25 (10 days after receiving her second dose of eculizumab), the patient was able to speak and follow simple commands but exhibited difficulty concentrating and poor impulse control.
The patient was alert and oriented to person, place, time, and situation on hospital day 28 (6 days after the third and final dose of eculizumab). A neurologic exam was significant only for a slight intention tremor. She was continued on levetiracetam with a plan to be maintained on the medication for the next 6 months for seizure control. She was discharged on hospital day 30.
Twenty-eight days postdischarge (57 days postadmission), the patient showed marked recovery. She had returned to her previous employment as a business administrator on a part-time basis and exhibited no deficiencies in executive functioning or handling activities of daily living. Although she had been very active prior to this illness, she now experienced decreased physical and mental endurance; however, this gradually improved with physical therapy. She planned on returning to work on a full-time basis when she had regained her stamina. She also noticed difficulties in retaining short term memory since her discharge but believed that these symptoms were remitting. On examination her mental status and neurologic exam was significant for inability to continue serial 7s, left sided 4/5 muscle strength in quadriceps and thumb to 5th metacarpal adduction, bilateral 1+ reflexes in muscle groups tested (triceps, biceps, brachioradialis, patellar, and Achilles), loss of dull pinprick sensation bilaterally at web of hands, deficit in tandem gait while looking away, and slight intention tremor on finger to nose testing bilaterally (with left hand tremor more pronounced than right). Her complete blood count was normal. Her recovery continues to be monitored in an outpatient setting.
Discussion
HUS is characterized by 3 core clinical features: microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury.4 Schistocytes are seen on peripheral blood smear and occur due to the passage of red blood cells over the microvascular thrombi induced by the disease. HUS can be classified as typical, atypical, or occurring with a coexisting disease. Typical HUS is associated with STEC 0157:H7 subtype, a bacterium known to be acquired through contaminated food and via human-to-human transmission.6-8 In the case of typical STEC 0157:H7, the bacterium releases a verotoxin that damages the vascular endothelium, thereby leading to activation of the coagulation cascade and eventually the formation of thrombi.4 It has been hypothesized that the Shiga toxin also activates the alternative complement pathway directly, which could contribute to thrombosis.9 This would explain the findings of low complement levels in our patient. Atypical HUS is primarily attributable to mutations in the alternative complement pathway. Causes for the third type of HUS can include Streptococcus pneumoniae, HIV, drug toxicity, and alterations in the metabolism of cobalamin C.
Epidemiologically, 15.3% of children aged < 5 years develop typical HUS after exposure to STEC compared with 1.2% of adults aged 18 to 59 years. The median age of patients who developed HUS from STEC exposure was 4 years compared with 16 years for those who did not develop HUS.2
Neurologic manifestations increase mortality for HUS patients.10 These have been described in the pediatric population as alteration in consciousness (85%), seizures (71%), pyramidal syndrome (52%), and extrapyramidal syndrome with hypertonia (42%).11 Brain imaging in children has demonstrated hemorrhagic lesions involving the pons, basal ganglia, and occipital cortex.11 Blood flow to areas such as the cerebellum, brainstem, and orbitofrontal area can be compromised.10 Adult patients with HUS can present without lesions on cranial magnetic resonance imaging (MRI), but instead with transient symmetric vasogenic edema of the central brain stem.12 Unfortunately in this case, MRI was not performed because it was thought to provide limited aid in diagnosis and to avoid unnecessary testing for the acutely ill patient.
The underlying pathophysiology of neurologic manifestations in patients may be due to a metabolic disturbance, toxin-mediated damage of the vascular endothelium, or toxin-induced cytokine release resulting in death of neural cells and subsequent neuroinflammation. However, the most likely mechanism is parenchymal ischemic changes related to microangiopathy.11,13 Pediatric patients often experience seizures and altered mental status, and their EEGs display delta waves.13 This patient’s diffuse slowing on her second EEG and altered mental status suggests that the neuropathologic mechanisms for typical HUS in adults may be similar to those in children.
HUS Treatment
The treatment and management of adults with typical STEC-HUS is evolving. The patient was first suspected to have an infectious colitis and empiric antibiotics were initiated. Some studies suggest that antibiotic administration may worsen the course of HUS in children as it may lead to release and subsequent absorption of Shiga toxin in the intestine.9,14 However, there is little evidence to suggest harm or efficacy of administration in adults. It is unclear what role antibiotic administration played in the recovery time of HUS given the co-administration of other treatments such as eculizumab and plasmapheresis, but it does appear to have helped with the initial E coli infection.
Plasmapheresis was subsequently administered, due to its documented benefit in the treatment of HUS.15 However, it should be noted that even though plasmapheresis is currently used in patients with CNS involvement, it remains unproven with conflicting information on its efficacy.3,16 The mechanism of action is unclear, but it has been hypothesized that plasmapheresis prevents microangiopathy caused by microthrombi.3,16 For this reason, eculizumab is becoming the mainstay for treatment of STEC-HUS with neurologic complications given the lack of well researched alternative treatments. In this case study, the use of plasmapheresis did not result in clinical improvement, and was abandoned after 2 days of treatment.
Eculizumab is a humanized, recombinant monoclonal IgG antibody that is a terminal complement inhibitor of the alternative complement system at the final step to cleave C5.17 The Shiga toxin may directly activate the complement system via the alternative pathway, which can result in uncontrolled platelet and white blood cell activation and depletion, endothelial cell damage, and hemolysis. The galvanized complement system leads to a series of cascading events that contribute to organ damage and death.9 Eculizumab is FDA approved for use in atypical HUS.18 It also can be used off-label to treat typical-HUS in adults with neurologic complications.
Eculizumab interferes with the immune response against encapsulated bacteria because it inhibits the alternative complement pathway. Thus, vaccination against N meningitides is recommended 2 weeks prior to the administration of eculizumab. However, in situations where the risks of delaying eculizumab for 2 weeks are greater than the risk of developing an N meningitides infection, eculizumab may be given without delay.18 Given the rapid deterioration of our patient’s condition, the vaccine and eculizumab were given together with prophylactic azithromycin. Although penicillin is the standard for prophylaxis in this situation, the patient’s penicillin allergy led to the use of azithromycin 250 mg po once a day. Literature also suggests azithromycin reduces the carriage duration of E coli-induced colitis.19 As such, it is possible that some improvement in the patient’s condition could be attributed to the elimination of the pathogen and toxin.
Conclusion
Three doses of eculizumab were administered at weekly intervals, with the first dose on hospital day 8 and the final dose on hospital day 22. Prior to the first dose, the patient displayed significant decline in mental status with EEG findings of right hemisphere epileptogenic discharges. After her third dose, she was found to have a drastically improved mental status exam and a normal EEG. One week later, she was discharged home. At the time of her 1-month follow-up, she was independent in all activities of daily living and had returned to part-time work. Apart from subtle cognitive changes, the remainder of her neurologic exam was normal.
There is evidence that supports the efficacy of eculizumab in children with HUS with neurologic symptoms on dialysis.20 However, its use in adults is not well established.21 This patient required dialysis and had neurologic symptoms similar to pediatric patients described in the literature, and responded similarly to the eculizumab. The rationale for the use of eculizumab in STEC-HUS also is evidenced by in vitro demonstrations of complement activation in STEC-HUS.22-25 This case report adds to the literature supporting the use of eculizumab in adult patients with typical HUS with neurological complications. Further research is necessary to develop guidelines in the treatment of adult STEC-HUS with regards to neurologic complications.
Acknowledgments
The authors would like to thank Pete DiStaso, REEGT for his work on obtaining the electroencephalograms and Anthony Rinaldi, PsyD; Julie Cessnapalas, PsyD; and Syed Faizan Sagheer for proof-reading the article.
1. Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365(9464):1073-1086.
2. Gould LH, Demma L, Jones TF, et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin Infect Dis. 2009;49(10):1480-1485.
3. Boyce TG, Swerdlow DL, Griffin PM. Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med. 1995;333(6):364-368.
4. Rondeau E, Peraldi MN. Escherichia coli and the hemolytic-uremic syndrome. N Engl J Med. 1996;335(9):660-662.
5. Te Loo DM, van Hinsbergh VW, van den Heuvel LP, Monnens LA. Detection of verocytotoxin bound to circulating polymorphonuclear leukocytes of patients with hemolytic uremic syndrome. J Am Soc Nephrol. 2001;12(4):800-806.
6. Tran SL, Jenkins C, Livrelli V, Schüller S. Shiga toxin 2 translocation across intestinal epithelium is linked to virulence of Shiga toxin-producing Escherichia coli in humans. Microbiology. 2018;164(4):509-516.
7. Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847-2856.
8. Ferens WA, Hovde CJ. Escherichia coli O157:H7: animal reservoir and sources of human infection. Foodborne Pathog Dis. 2011;8(4):465-487.
9. Percheron L, Gramada R, Tellier S, et al. Eculizumab treatment in severe pediatric STEC-HUS: a multicenter retrospective study. Pediatr Nephrol. 2018;33(8):1385-1394.
10. Hosaka T, Nakamagoe K, Tamaoka A. Hemolytic uremic syndrome-associated encephalopathy successfully treated with corticosteroids. Intern Med. 2017;56(21):2937-2941.
11. Nathanson S, Kwon T, Elmaleh M, et al. Acute neurological involvement in diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2010;5(7):1218-1228.
12. Wengenroth M, Hoeltje J, Repenthin J, et al. Central nervous system involvement in adults with epidemic hemolytic uremic syndrome. AJNR Am J Neuroradiol. 2013;34(5):1016-1021, S1.
13. Eriksson KJ, Boyd SG, Tasker RC. Acute neurology and neurophysiology of haemolytic-uraemic syndrome. Arch Dis Child. 2001;84(5):434-435.
14. Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342(26):1930-1936.
15. Nguyen TC, Kiss JE, Goldman JR, Carcillo JA. The role of plasmapheresis in critical illness. Crit Care Clin. 2012;28(3):453-468, vii.
16. Loos S, Ahlenstiel T, Kranz B, et al. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: presentation and short-term outcome in children. Clin Infect Dis. 2012;55(6):753-759.
17. Hossain MA, Cheema A, Kalathil S, et al. Atypical hemolytic uremic syndrome: Laboratory characteristics, complement-amplifying conditions, renal biopsy, and genetic mutations. Saudi J Kidney Dis Transpl. 2018;29(2):276-283.
18. Soliris (eculizumab) [package insert]. Cheshire, CT: Alexion Pharmaceuticals, Inc; 2011.
19. Keenswijk W, Raes A, Vande Walle J. Is eculizumab efficacious in Shigatoxin-associated hemolytic uremic syndrome? A narrative review of current evidence. Eur J Pediatr. 2018;177(3):311-318.
20. Lapeyraque AL, Malina M, Fremeaux-Bacchi V, et al. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med. 2011;364(26):2561-2563.
21. Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, Ahlenstiel-Grunow T. Eculizumab in typical hemolytic uremic syndrome (HUS) with neurological involvement. Medicine (Baltimore). 2015;94(24):e1000.
22. Kim Y, Miller K, Michael AF. Breakdown products of C3 and factor B in hemolytic-uremic syndrome. J Lab Clin Med. 1977;89(4):845-850.
23. Monnens L, Molenaar J, Lambert PH, Proesmans W, van Munster P. The complement system in hemolytic-uremic syndrome in childhood. Clin Nephrol. 1980;13(4):168-171.
24. Thurman JM, Marians R, Emlen W, et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2009;4(12):1920-1924.
25. Ståhl AL, Sartz L, Karpman D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood. 2011;117(20):5503-5513.
1. Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365(9464):1073-1086.
2. Gould LH, Demma L, Jones TF, et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin Infect Dis. 2009;49(10):1480-1485.
3. Boyce TG, Swerdlow DL, Griffin PM. Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med. 1995;333(6):364-368.
4. Rondeau E, Peraldi MN. Escherichia coli and the hemolytic-uremic syndrome. N Engl J Med. 1996;335(9):660-662.
5. Te Loo DM, van Hinsbergh VW, van den Heuvel LP, Monnens LA. Detection of verocytotoxin bound to circulating polymorphonuclear leukocytes of patients with hemolytic uremic syndrome. J Am Soc Nephrol. 2001;12(4):800-806.
6. Tran SL, Jenkins C, Livrelli V, Schüller S. Shiga toxin 2 translocation across intestinal epithelium is linked to virulence of Shiga toxin-producing Escherichia coli in humans. Microbiology. 2018;164(4):509-516.
7. Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847-2856.
8. Ferens WA, Hovde CJ. Escherichia coli O157:H7: animal reservoir and sources of human infection. Foodborne Pathog Dis. 2011;8(4):465-487.
9. Percheron L, Gramada R, Tellier S, et al. Eculizumab treatment in severe pediatric STEC-HUS: a multicenter retrospective study. Pediatr Nephrol. 2018;33(8):1385-1394.
10. Hosaka T, Nakamagoe K, Tamaoka A. Hemolytic uremic syndrome-associated encephalopathy successfully treated with corticosteroids. Intern Med. 2017;56(21):2937-2941.
11. Nathanson S, Kwon T, Elmaleh M, et al. Acute neurological involvement in diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2010;5(7):1218-1228.
12. Wengenroth M, Hoeltje J, Repenthin J, et al. Central nervous system involvement in adults with epidemic hemolytic uremic syndrome. AJNR Am J Neuroradiol. 2013;34(5):1016-1021, S1.
13. Eriksson KJ, Boyd SG, Tasker RC. Acute neurology and neurophysiology of haemolytic-uraemic syndrome. Arch Dis Child. 2001;84(5):434-435.
14. Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342(26):1930-1936.
15. Nguyen TC, Kiss JE, Goldman JR, Carcillo JA. The role of plasmapheresis in critical illness. Crit Care Clin. 2012;28(3):453-468, vii.
16. Loos S, Ahlenstiel T, Kranz B, et al. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: presentation and short-term outcome in children. Clin Infect Dis. 2012;55(6):753-759.
17. Hossain MA, Cheema A, Kalathil S, et al. Atypical hemolytic uremic syndrome: Laboratory characteristics, complement-amplifying conditions, renal biopsy, and genetic mutations. Saudi J Kidney Dis Transpl. 2018;29(2):276-283.
18. Soliris (eculizumab) [package insert]. Cheshire, CT: Alexion Pharmaceuticals, Inc; 2011.
19. Keenswijk W, Raes A, Vande Walle J. Is eculizumab efficacious in Shigatoxin-associated hemolytic uremic syndrome? A narrative review of current evidence. Eur J Pediatr. 2018;177(3):311-318.
20. Lapeyraque AL, Malina M, Fremeaux-Bacchi V, et al. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med. 2011;364(26):2561-2563.
21. Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, Ahlenstiel-Grunow T. Eculizumab in typical hemolytic uremic syndrome (HUS) with neurological involvement. Medicine (Baltimore). 2015;94(24):e1000.
22. Kim Y, Miller K, Michael AF. Breakdown products of C3 and factor B in hemolytic-uremic syndrome. J Lab Clin Med. 1977;89(4):845-850.
23. Monnens L, Molenaar J, Lambert PH, Proesmans W, van Munster P. The complement system in hemolytic-uremic syndrome in childhood. Clin Nephrol. 1980;13(4):168-171.
24. Thurman JM, Marians R, Emlen W, et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2009;4(12):1920-1924.
25. Ståhl AL, Sartz L, Karpman D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood. 2011;117(20):5503-5513.
Bacteroides Fragilis Vertebral Osteomyelitis and Discitis: “Back” to Susceptibility Testing
Acute pyogenic vertebral osteomyelitis is often due to hematogenous spread of aerobic bacteria.1-4 Conversely, only 0.5% of anaerobic bacteremias lead to osteomyelitis.5 Anaerobic osteomyelitis typically results from the contiguous spread of polymicrobial infections through breaks in the gut mucosal barrier and involves the vertebral bodies in only 2% to 5% of cases.5,6 Although Bacteroides fragilis (B fragilis) is the most common anaerobic pathogen cultivated from blood, accounting for about half of all anaerobic blood isolates, it seldom leads to osteomyelitis.1,2,7-11 We report an uncommon case of B fragilis bacteremia and vertebral osteomyelitis confounded by uncertainties in anaerobic identification and susceptibilities.
Case Presentation
A healthy-appearing male aged 55 years presented to the Naval Medical Center Portsmouth (NMCP) with subacute low back pain and fevers of 103 °F for > 3 weeks. While traveling 4 weeks prior, he completed a course of oseltamivir for influenza B infection; afterward, he was diagnosed with community-acquired pneumonia and treated with a dose of ceftriaxone and a 7-day course of doxycycline. The patient presented to the same facility a week later for low back pain and nonresolving respiratory symptoms, and his therapy was changed to azithromycin, cefuroxime, prednisone, and inhalers. Additionally, after being treated for influenza, he developed constipation and hematochezia for which he did not seek care. The hematochezia was similar to a previous episode from an anal fissure 1 year prior that resolved with stool softeners. When he was finally seen at NMCP after 3 weeks of worsening back pain and fevers, lumbosacral magnetic resonance imaging (MRI) demonstrated vertebral osteomyelitis and discitis at L4-L5 and admitted to the hospital (Figure 1).

After a fluoroscopy-guided biopsy of the L4 vertebral body on hospital day 1, the patient was started on cefepime and vancomycin. The biopsy sample was inoculated onto solid media (blood agar, chocolate agar, and MacConkey agar) and incubated at 36 °C for 24 hours in a 5% CO2 atmosphere, as well as onto Shaedler agar with vitamin K and chopped meat glucose broth and incubated at 36 °C for 48 hours under anaerobic conditions. Metronidazole was added and vancomycin discontinued after 2 anaerobic blood culture vials obtained on hospital day 1, incubated in a Becton Dickinson BACTEC FX automated system, which demonstrated Gram-negative bacilli after 48 hours. The blood culture isolates demonstrated a > 99% probability of being identified as ß-lactamase positive Prevotella loescheii using Thermo Fischer Scientific RapID ANA II biochemical testing. Nitrocefinase discs were used to detect ß-lactamase activity.
The biopsy demonstrated nongranulomatous focal areas of necrotic bone and neutrophilia in a hematopoietic background consistent with acute osteomyelitis (Figure 2); on hospital day 4, ß-lactamase positive B fragilis grew from the bone culture. Additionally, 1 anaerobic vial from a surveillance blood culture set that was obtained on hospital day 3 grew ß-lactamasepositive B fragilis using the same identification methods. With these results he was thought to have a polymicrobial infection (B fragilis and Prevotella loescheii [P loescheii]) from a suspected bowel source based on his hematochezia and history of anal fissure. No aerobic, Gram-negative enterobacteriaceae were isolated, but he had previously been on cefuroxime, which has potential activity against these organisms, for ≥ 2 weeks prior to hospitalization and cultures. He was discharged on moxifloxacin and metronidazole pending final culture results, including requested anaerobic susceptibility testing.
At 1-week follow-up, both aerobic and anaerobic vials from surveillance blood cultures remained negative for any microbes, so antibiotics were deescalated to moxifloxacin monotherapy. However, after 3 days the patient was readmitted for increasing C-reactive protein (CRP) levels and intractable back pain with worsening bilateral radiculopathy. A repeat MRI demonstrated interval disease progression with near obliteration of the L4-L5 disc space and hyperenhancement of the prevertebral soft tissues and adjacent psoas musculature without focal rim-enhancing fluid collection (Figure 3). After repeat L4 biopsy, metronidazole was restarted and ertapenem added for enterobacteriaceae coverage, given the known B fragilis and potential suppression from previous cephalosporin therapy; moxifloxacin was discontinued. L4 biopsy cultures showed no growth, and CRP levels trended down from 154.2 mg/L (start of first admission) to 42.4 mg/L (start of second admission) to 14.9 mg/L (day of discharge) (reference range, 5-9.9 mg/L). He was discharged on ertapenem and metronidazole. He completed a 6-week course without further complication.

During antibiotic therapy he had an unremarkable colonoscopy, CRP normalized to 2.6 mg/L (reference range, 0-4.9 mg/L), and he underwent successful L4-L5 transforaminal lumbar interbody fusion 2 weeks after finishing antibiotics.
We retroactively sent both P loescheii isolates and the 1 B fragilis isolate that grew from the surveillance blood culture to the Multidrug-resistant Organism Repository and Surveillance Network (MRSN) at the Walter Reed Army Institute of Research for identification confirmation and susceptibility analysis. Whole genome sequencing with single nucleotide polymorphism (SNP)-based analysis revealed all isolates were 100% identical and consistent with B fragilis and not P loescheii, based on clustering around other B fragilis sequences found in the National Center for Biotechnology Information (NCBI) Genbank database (Figure 4). All isolates carried the antibiotic resistance genes— cepA, sul(2), tetQ— encoding for possible resistance to cephalosporins, sulphonamides, and tetracyclines, respectively; as well as a point mutation in the gyrA gene (Ser82Phe). None of the isolates carried the nim gene, and screening for the 3 subtypes of B fragilis enterotoxin gene (bft-1, bft-2, bft-3) was negative. Eventual susceptibility testing at the Mayo Clinic several months after the conclusion of the case indicated that the B fragilis isolate was sensitive to piperacillin-tazobactam, ertapenem, clindamycin, and metronidazole; however, testing was not performed against moxifloxacin.
Discussion
In the era of growing antibiotic resistance patterns, antimicrobial stewardship programs recommend interventions to improve antimicrobial use through targeted narrow- spectrum antibiotics.12 The Clinical and Laboratory Standards Institute (CLSI) maintains guidelines on the major indications for anaerobic antimicrobial susceptibility testing (AST) to help direct narrow-targeted antimicrobial therapy. However, in a 2008 practice survey Goldstein and colleagues reported that less than half of US hospitals performed anaerobic AST, and only 21% of these facilities did it in-house, while the remainder sent out their isolates for testing.11-14 The CLSI major indications for AST include situations in which the selection of agents is important because of the (1) known resistance of a particular species; (2) confirmation of appropriate therapy for severe infections or for those that may require long-term therapy; (3) persistence of infection despite adequate treatment with an appropriate therapeutic regimen; and (4) difficulty in making empirical decisions based on precedent.14 Additionally, isolates from brain abscess, endocarditis, osteomyelitis, joint infection, infection of prosthetic devices or vascular grafts, bacteremia, and normally sterile body sites (unless contamination suspected) should be tested.14
Because of the lack of anaerobic AST, health care providers must base empiric treatment on reported sensitivities from the medical literature. Empiric selection of antimicrobials for anaerobic infections is made even more challenging by the increased rates of resistance reported in the literature, leading to recommendations to increase susceptibility testing to guide therapy.13,15,16 Empiric therapy of deep-seated anaerobic infections may lead to use of inactive agents or overly broad-spectrum antibiotics. Current antimicrobial stewardship initiatives recognize the importance of narrow-spectrum antibiotics to minimize risk of adverse events and selective pressure for antimicrobial resistance.
Although we attempted to confirm the identification of the anaerobic isolates via commercially available methods, it was not until we performed genetic testing that we were able verify the isolates as B fragilis. Furthermore, earlier susceptibility testing would have allowed for more narrow-targeted antimicrobial therapy and could have potentially prevented our patient’s readmission and use of ertapenem, despite its > 98% susceptibility rates against B fragilis.13,17
All of the B fragilis isolates carried the cepA gene, which is a cephalosporinase that encodes for resistance to cephalosporins and aminopenicillins but not to ß-lactam ß-lactamase inhibitor combinations.13 Although not a substitution for susceptibility analysis, genetic testing showed that all of the isolates carried a nonsynonymous mutation from serine to a phenylalanine at amino acid position 82 (S82F) in the gyrA gene. The S82F mutation has been implicated in fluoroquinolone resistance, via inhibition of substrate–target recognition and binding between fluoroquinolones and the target topoisomerase protein,18 and may potentially explain why our patient clinically worsened while on moxifloxacin monotherapy. Although moxifloxacin susceptibility was not performed, susceptibility rates remain highly variable, ranging from 50% to 70% for B fragilis.13,15,16
It is important to note that the metronidazole the patient received during his first hospital admission could have sterilized the vertebral body without completely eradicating the microbe; thus could explain his clinical worsening while on moxifloxacin monotherapy despite no growth from the repeat biopsy culture. Our rationale for initially continuing moxifloxacin was based on its excellent bioavailability and bone penetration properties. Additionally, of the fluoroquinolones it has the most reliable anaerobic activity and is the only one recommended as monotherapy for complicated intraabdominal infections.19 However, guidelines recommend avoiding its use in patients who have received a fluoroquinolone in the past 90 days or at institutions with high rates of resistance. At our institution Escherichia coli has a > 90% susceptibility rate to fluoroquinolones. Given this rate and our concern that the patient had a polymicrobial infection, we felt that moxifloxacin would provide appropriate anaerobic and aerobic coverage, especially since he had no previous fluoroquinolone exposure.
Additionally, none of the isolates carried the nim or bft toxin genes. Although the nim gene is associated with metronidazole resistance,its presence does not invariably result in resistant strains of B fragilis; in fact, metronidazole resistance is relatively uncommon, with the majority of B fragilis showing < 1% resistance, based on CLSI breakpoints (≥ 32 mg/L).13,20,21 However, one recent epidemiologic study on anaerobic wound isolates from Iraq and Afghanistan casualties found that 12% (2/17) of B fragilis isolates were resistant to metronidazole.15 Given the improvement of the patient’s symptoms while on metronidazole, it is likely that the B fragilis was susceptible. Nevertheless, susceptibility testing with minimum inhibitory concentrations is necessary to verify this result. Also, although enterotoxigenic strains of B fragilis have been associated with bloodstream infections, our patient’s isolates lacked the 3 subtypes of B fragilis enterotoxin gene.22
Conclusions
We report a case of B fragilis bacteremia and vertebral osteomyelitis complicated by challenges in anaerobic identification and sensitivities that led to brief use of a possibly inactive antimicrobial and the subsequent use of carbapenem therapy, which may have been avoided if susceptibility testing were more readily available. This case led to changes in our hospital’s processing of anaerobic isolates to include susceptibility testing on request.
Acknowledgments
We thank Keith Thompson, MD (staff pathologist, Naval Medical Center Portsmouth Virginia), for providing the pathology images from the initial vertebral biopsy, and Dr. Kate Hinkle (director, Multidrug-Resistant Organism Repository and Surveillance Network, Silver Spring, Maryland ) for providing the whole genome sequencing results from the B fragilis isolates.
1. Zimmerli W. Vertebral osteomyelitis. N Eng J Med. 2010;362(11):1022-1029.
2. Chazan B, Strahilevitz J, Millgram MA, Kaufmann S, Raz R. Bacteroides fragilis vertebral osteomyelitis secondary to anal dilatation. Spine (Phila PA 1976). 2001;26(16):E377-E378.
3. Kierzkowska M, Pedzisz P, Babiak I, et al. Orthopedic infections caused by obligatory anaerobic Gram-negative rods: report of two cases. Med Microbiol Immunol. 2017;206(5):363-366.
4. McHenry M, Easley K, Locker G. Vertebral osteomyelitis: long-term outcome for 253 patients from 7 Cleveland-area hospitals. Clin Infect Dis. 2002;34(10):1342-1350.
5. Raff MJ, Melo JC. Anaerobic osteomyelitis. Medicine (Baltimore).1978;57(1):83-103.
6. Lewis R, Sutter V, Finegold S. Bone infections involving anaerobic bacteria. Medicine (Baltimore). 1978;57(1):279-305.
7. Brook I. The role of anaerobic bacteria in bacteremia. Anaerobe. 2010;16(3):183-189.
8. Lassmann B, Gustafson DR, Wood CM, Rosenblatt JE. Reemergence of anaerobic bacteremia. Clin Infect Dis. 2007;44(7):895-900.
9. Lazarovitch T, Freimann S, Shapira G, Blank H. Decrease in anaerobe-related bacteraemias and increase in Bacteroides species isolation rate from 1998 to 2007: a retrospective study. Anaerobe. 2010;16(3):201-205.
10. Keukeleire S, Wybo I, Naessens A, et al. Anaerobic bacteraemia: a 10-year retrospective epidemiological survey. Anaerobe. 2016;39:54-59.
11. Goldstein EJC, Citron DM, Goldman PJ, Goldman RJ. National hospital survey of anaerobic culture and susceptibility methods: III. Anaerobe. 2008;14(2):68-72.
12. Barlam TF, Cosgrove SE, Abbo LM, et al. Implementing an antibiotic stewardship program: Guidelines by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America. Clin Infect Dis. 2016;62(10):e51-e77.
13. Schuetz AN. Antimicrobial resistance and susceptibility testing of anaerobic bacteria. Antimicr Resist. 2014;59(5):698-705.
14. Clinical and Laboratory Standards Institute. M11-A8: Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria; Approved Standard. 8th ed. Wayne, PA: Clinical and Laboratory Standards Institute; 2012.
15. White B, Mende K, Weintrob A, et al; Infectious Disease Clinical Research Program Trauma Infectious Disease Outcome Study Group. Epidemiology and antimicrobial susceptibilities of wound isolates of obligate anaerobes from combat casualties. Diagn Mircrobiol Infect Dis. 2016;84(2):144-150.
16. Hastey CJ, Boyd H, Schuetz AN, et al; Ad Hoc Working Group on Antimicrobial Susceptibility Testing of Anaerobic Bacteria of CLSI. Changes in the antibiotic susceptibility of anaerobic bacteria from 2007-2009 to 2010-2012 based on the CLSI methodology. Anaerobe. 2016;42:27-30.
17. Brook I, Wexler HM, Goldstein EJC. Antianaerobic antimicrobials: spectrum and susceptibility testing. Clin Microbiol Rev. 2013;26(3):526-546.
18. Pumbwe L, Wareham D, Aduse-Opoku J, Brazier JS, Wexler HM. Genetic analysis of mechanisms of multidrug resistance in a clinical isolate of Bacteroides fragilis. Clin Microbiol Infect. 2007;13(2):183-189.
19. Solomkin J, Mazuski J, Bradley J, et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin Infect Dis. 2010;50(2):133-164.
20. Breuil J, Dublanchet A, Truffaut N, Sebald M. Transferable 5-nitroimidazole resistance Bacteroides fragilis group. Plasmid. 1989;21(2):151-154.
21. Nagy E, Urbán E, Nord CE; ESCMID Study Group on Antimicrobial Resistance in Anaerobic Bacteria. Antimicrobial susceptibility of Bacteroides fragilis group isolates in Europe: 20 years of experience. Clin Microbiol Infect. 2011;17(3):371-379.
22. Avila-Campos M, Liu C, Song Y, Rowlinson M-C, Finegold SM. Determination of bft gene subtypes in Bacteroides fragilis clinical isolates. J Clin Microbiol. 2007;45(4):1336-1338.
Acute pyogenic vertebral osteomyelitis is often due to hematogenous spread of aerobic bacteria.1-4 Conversely, only 0.5% of anaerobic bacteremias lead to osteomyelitis.5 Anaerobic osteomyelitis typically results from the contiguous spread of polymicrobial infections through breaks in the gut mucosal barrier and involves the vertebral bodies in only 2% to 5% of cases.5,6 Although Bacteroides fragilis (B fragilis) is the most common anaerobic pathogen cultivated from blood, accounting for about half of all anaerobic blood isolates, it seldom leads to osteomyelitis.1,2,7-11 We report an uncommon case of B fragilis bacteremia and vertebral osteomyelitis confounded by uncertainties in anaerobic identification and susceptibilities.
Case Presentation
A healthy-appearing male aged 55 years presented to the Naval Medical Center Portsmouth (NMCP) with subacute low back pain and fevers of 103 °F for > 3 weeks. While traveling 4 weeks prior, he completed a course of oseltamivir for influenza B infection; afterward, he was diagnosed with community-acquired pneumonia and treated with a dose of ceftriaxone and a 7-day course of doxycycline. The patient presented to the same facility a week later for low back pain and nonresolving respiratory symptoms, and his therapy was changed to azithromycin, cefuroxime, prednisone, and inhalers. Additionally, after being treated for influenza, he developed constipation and hematochezia for which he did not seek care. The hematochezia was similar to a previous episode from an anal fissure 1 year prior that resolved with stool softeners. When he was finally seen at NMCP after 3 weeks of worsening back pain and fevers, lumbosacral magnetic resonance imaging (MRI) demonstrated vertebral osteomyelitis and discitis at L4-L5 and admitted to the hospital (Figure 1).
After a fluoroscopy-guided biopsy of the L4 vertebral body on hospital day 1, the patient was started on cefepime and vancomycin. The biopsy sample was inoculated onto solid media (blood agar, chocolate agar, and MacConkey agar) and incubated at 36 °C for 24 hours in a 5% CO2 atmosphere, as well as onto Shaedler agar with vitamin K and chopped meat glucose broth and incubated at 36 °C for 48 hours under anaerobic conditions. Metronidazole was added and vancomycin discontinued after 2 anaerobic blood culture vials obtained on hospital day 1, incubated in a Becton Dickinson BACTEC FX automated system, which demonstrated Gram-negative bacilli after 48 hours. The blood culture isolates demonstrated a > 99% probability of being identified as ß-lactamase positive Prevotella loescheii using Thermo Fischer Scientific RapID ANA II biochemical testing. Nitrocefinase discs were used to detect ß-lactamase activity.
The biopsy demonstrated nongranulomatous focal areas of necrotic bone and neutrophilia in a hematopoietic background consistent with acute osteomyelitis (Figure 2); on hospital day 4, ß-lactamase positive B fragilis grew from the bone culture. Additionally, 1 anaerobic vial from a surveillance blood culture set that was obtained on hospital day 3 grew ß-lactamasepositive B fragilis using the same identification methods. With these results he was thought to have a polymicrobial infection (B fragilis and Prevotella loescheii [P loescheii]) from a suspected bowel source based on his hematochezia and history of anal fissure. No aerobic, Gram-negative enterobacteriaceae were isolated, but he had previously been on cefuroxime, which has potential activity against these organisms, for ≥ 2 weeks prior to hospitalization and cultures. He was discharged on moxifloxacin and metronidazole pending final culture results, including requested anaerobic susceptibility testing.
At 1-week follow-up, both aerobic and anaerobic vials from surveillance blood cultures remained negative for any microbes, so antibiotics were deescalated to moxifloxacin monotherapy. However, after 3 days the patient was readmitted for increasing C-reactive protein (CRP) levels and intractable back pain with worsening bilateral radiculopathy. A repeat MRI demonstrated interval disease progression with near obliteration of the L4-L5 disc space and hyperenhancement of the prevertebral soft tissues and adjacent psoas musculature without focal rim-enhancing fluid collection (Figure 3). After repeat L4 biopsy, metronidazole was restarted and ertapenem added for enterobacteriaceae coverage, given the known B fragilis and potential suppression from previous cephalosporin therapy; moxifloxacin was discontinued. L4 biopsy cultures showed no growth, and CRP levels trended down from 154.2 mg/L (start of first admission) to 42.4 mg/L (start of second admission) to 14.9 mg/L (day of discharge) (reference range, 5-9.9 mg/L). He was discharged on ertapenem and metronidazole. He completed a 6-week course without further complication.
During antibiotic therapy he had an unremarkable colonoscopy, CRP normalized to 2.6 mg/L (reference range, 0-4.9 mg/L), and he underwent successful L4-L5 transforaminal lumbar interbody fusion 2 weeks after finishing antibiotics.
We retroactively sent both P loescheii isolates and the 1 B fragilis isolate that grew from the surveillance blood culture to the Multidrug-resistant Organism Repository and Surveillance Network (MRSN) at the Walter Reed Army Institute of Research for identification confirmation and susceptibility analysis. Whole genome sequencing with single nucleotide polymorphism (SNP)-based analysis revealed all isolates were 100% identical and consistent with B fragilis and not P loescheii, based on clustering around other B fragilis sequences found in the National Center for Biotechnology Information (NCBI) Genbank database (Figure 4). All isolates carried the antibiotic resistance genes— cepA, sul(2), tetQ— encoding for possible resistance to cephalosporins, sulphonamides, and tetracyclines, respectively; as well as a point mutation in the gyrA gene (Ser82Phe). None of the isolates carried the nim gene, and screening for the 3 subtypes of B fragilis enterotoxin gene (bft-1, bft-2, bft-3) was negative. Eventual susceptibility testing at the Mayo Clinic several months after the conclusion of the case indicated that the B fragilis isolate was sensitive to piperacillin-tazobactam, ertapenem, clindamycin, and metronidazole; however, testing was not performed against moxifloxacin.
Discussion
In the era of growing antibiotic resistance patterns, antimicrobial stewardship programs recommend interventions to improve antimicrobial use through targeted narrow- spectrum antibiotics.12 The Clinical and Laboratory Standards Institute (CLSI) maintains guidelines on the major indications for anaerobic antimicrobial susceptibility testing (AST) to help direct narrow-targeted antimicrobial therapy. However, in a 2008 practice survey Goldstein and colleagues reported that less than half of US hospitals performed anaerobic AST, and only 21% of these facilities did it in-house, while the remainder sent out their isolates for testing.11-14 The CLSI major indications for AST include situations in which the selection of agents is important because of the (1) known resistance of a particular species; (2) confirmation of appropriate therapy for severe infections or for those that may require long-term therapy; (3) persistence of infection despite adequate treatment with an appropriate therapeutic regimen; and (4) difficulty in making empirical decisions based on precedent.14 Additionally, isolates from brain abscess, endocarditis, osteomyelitis, joint infection, infection of prosthetic devices or vascular grafts, bacteremia, and normally sterile body sites (unless contamination suspected) should be tested.14
Because of the lack of anaerobic AST, health care providers must base empiric treatment on reported sensitivities from the medical literature. Empiric selection of antimicrobials for anaerobic infections is made even more challenging by the increased rates of resistance reported in the literature, leading to recommendations to increase susceptibility testing to guide therapy.13,15,16 Empiric therapy of deep-seated anaerobic infections may lead to use of inactive agents or overly broad-spectrum antibiotics. Current antimicrobial stewardship initiatives recognize the importance of narrow-spectrum antibiotics to minimize risk of adverse events and selective pressure for antimicrobial resistance.
Although we attempted to confirm the identification of the anaerobic isolates via commercially available methods, it was not until we performed genetic testing that we were able verify the isolates as B fragilis. Furthermore, earlier susceptibility testing would have allowed for more narrow-targeted antimicrobial therapy and could have potentially prevented our patient’s readmission and use of ertapenem, despite its > 98% susceptibility rates against B fragilis.13,17
All of the B fragilis isolates carried the cepA gene, which is a cephalosporinase that encodes for resistance to cephalosporins and aminopenicillins but not to ß-lactam ß-lactamase inhibitor combinations.13 Although not a substitution for susceptibility analysis, genetic testing showed that all of the isolates carried a nonsynonymous mutation from serine to a phenylalanine at amino acid position 82 (S82F) in the gyrA gene. The S82F mutation has been implicated in fluoroquinolone resistance, via inhibition of substrate–target recognition and binding between fluoroquinolones and the target topoisomerase protein,18 and may potentially explain why our patient clinically worsened while on moxifloxacin monotherapy. Although moxifloxacin susceptibility was not performed, susceptibility rates remain highly variable, ranging from 50% to 70% for B fragilis.13,15,16
It is important to note that the metronidazole the patient received during his first hospital admission could have sterilized the vertebral body without completely eradicating the microbe; thus could explain his clinical worsening while on moxifloxacin monotherapy despite no growth from the repeat biopsy culture. Our rationale for initially continuing moxifloxacin was based on its excellent bioavailability and bone penetration properties. Additionally, of the fluoroquinolones it has the most reliable anaerobic activity and is the only one recommended as monotherapy for complicated intraabdominal infections.19 However, guidelines recommend avoiding its use in patients who have received a fluoroquinolone in the past 90 days or at institutions with high rates of resistance. At our institution Escherichia coli has a > 90% susceptibility rate to fluoroquinolones. Given this rate and our concern that the patient had a polymicrobial infection, we felt that moxifloxacin would provide appropriate anaerobic and aerobic coverage, especially since he had no previous fluoroquinolone exposure.
Additionally, none of the isolates carried the nim or bft toxin genes. Although the nim gene is associated with metronidazole resistance,its presence does not invariably result in resistant strains of B fragilis; in fact, metronidazole resistance is relatively uncommon, with the majority of B fragilis showing < 1% resistance, based on CLSI breakpoints (≥ 32 mg/L).13,20,21 However, one recent epidemiologic study on anaerobic wound isolates from Iraq and Afghanistan casualties found that 12% (2/17) of B fragilis isolates were resistant to metronidazole.15 Given the improvement of the patient’s symptoms while on metronidazole, it is likely that the B fragilis was susceptible. Nevertheless, susceptibility testing with minimum inhibitory concentrations is necessary to verify this result. Also, although enterotoxigenic strains of B fragilis have been associated with bloodstream infections, our patient’s isolates lacked the 3 subtypes of B fragilis enterotoxin gene.22
Conclusions
We report a case of B fragilis bacteremia and vertebral osteomyelitis complicated by challenges in anaerobic identification and sensitivities that led to brief use of a possibly inactive antimicrobial and the subsequent use of carbapenem therapy, which may have been avoided if susceptibility testing were more readily available. This case led to changes in our hospital’s processing of anaerobic isolates to include susceptibility testing on request.
Acknowledgments
We thank Keith Thompson, MD (staff pathologist, Naval Medical Center Portsmouth Virginia), for providing the pathology images from the initial vertebral biopsy, and Dr. Kate Hinkle (director, Multidrug-Resistant Organism Repository and Surveillance Network, Silver Spring, Maryland ) for providing the whole genome sequencing results from the B fragilis isolates.
Acute pyogenic vertebral osteomyelitis is often due to hematogenous spread of aerobic bacteria.1-4 Conversely, only 0.5% of anaerobic bacteremias lead to osteomyelitis.5 Anaerobic osteomyelitis typically results from the contiguous spread of polymicrobial infections through breaks in the gut mucosal barrier and involves the vertebral bodies in only 2% to 5% of cases.5,6 Although Bacteroides fragilis (B fragilis) is the most common anaerobic pathogen cultivated from blood, accounting for about half of all anaerobic blood isolates, it seldom leads to osteomyelitis.1,2,7-11 We report an uncommon case of B fragilis bacteremia and vertebral osteomyelitis confounded by uncertainties in anaerobic identification and susceptibilities.
Case Presentation
A healthy-appearing male aged 55 years presented to the Naval Medical Center Portsmouth (NMCP) with subacute low back pain and fevers of 103 °F for > 3 weeks. While traveling 4 weeks prior, he completed a course of oseltamivir for influenza B infection; afterward, he was diagnosed with community-acquired pneumonia and treated with a dose of ceftriaxone and a 7-day course of doxycycline. The patient presented to the same facility a week later for low back pain and nonresolving respiratory symptoms, and his therapy was changed to azithromycin, cefuroxime, prednisone, and inhalers. Additionally, after being treated for influenza, he developed constipation and hematochezia for which he did not seek care. The hematochezia was similar to a previous episode from an anal fissure 1 year prior that resolved with stool softeners. When he was finally seen at NMCP after 3 weeks of worsening back pain and fevers, lumbosacral magnetic resonance imaging (MRI) demonstrated vertebral osteomyelitis and discitis at L4-L5 and admitted to the hospital (Figure 1).
After a fluoroscopy-guided biopsy of the L4 vertebral body on hospital day 1, the patient was started on cefepime and vancomycin. The biopsy sample was inoculated onto solid media (blood agar, chocolate agar, and MacConkey agar) and incubated at 36 °C for 24 hours in a 5% CO2 atmosphere, as well as onto Shaedler agar with vitamin K and chopped meat glucose broth and incubated at 36 °C for 48 hours under anaerobic conditions. Metronidazole was added and vancomycin discontinued after 2 anaerobic blood culture vials obtained on hospital day 1, incubated in a Becton Dickinson BACTEC FX automated system, which demonstrated Gram-negative bacilli after 48 hours. The blood culture isolates demonstrated a > 99% probability of being identified as ß-lactamase positive Prevotella loescheii using Thermo Fischer Scientific RapID ANA II biochemical testing. Nitrocefinase discs were used to detect ß-lactamase activity.
The biopsy demonstrated nongranulomatous focal areas of necrotic bone and neutrophilia in a hematopoietic background consistent with acute osteomyelitis (Figure 2); on hospital day 4, ß-lactamase positive B fragilis grew from the bone culture. Additionally, 1 anaerobic vial from a surveillance blood culture set that was obtained on hospital day 3 grew ß-lactamasepositive B fragilis using the same identification methods. With these results he was thought to have a polymicrobial infection (B fragilis and Prevotella loescheii [P loescheii]) from a suspected bowel source based on his hematochezia and history of anal fissure. No aerobic, Gram-negative enterobacteriaceae were isolated, but he had previously been on cefuroxime, which has potential activity against these organisms, for ≥ 2 weeks prior to hospitalization and cultures. He was discharged on moxifloxacin and metronidazole pending final culture results, including requested anaerobic susceptibility testing.
At 1-week follow-up, both aerobic and anaerobic vials from surveillance blood cultures remained negative for any microbes, so antibiotics were deescalated to moxifloxacin monotherapy. However, after 3 days the patient was readmitted for increasing C-reactive protein (CRP) levels and intractable back pain with worsening bilateral radiculopathy. A repeat MRI demonstrated interval disease progression with near obliteration of the L4-L5 disc space and hyperenhancement of the prevertebral soft tissues and adjacent psoas musculature without focal rim-enhancing fluid collection (Figure 3). After repeat L4 biopsy, metronidazole was restarted and ertapenem added for enterobacteriaceae coverage, given the known B fragilis and potential suppression from previous cephalosporin therapy; moxifloxacin was discontinued. L4 biopsy cultures showed no growth, and CRP levels trended down from 154.2 mg/L (start of first admission) to 42.4 mg/L (start of second admission) to 14.9 mg/L (day of discharge) (reference range, 5-9.9 mg/L). He was discharged on ertapenem and metronidazole. He completed a 6-week course without further complication.
During antibiotic therapy he had an unremarkable colonoscopy, CRP normalized to 2.6 mg/L (reference range, 0-4.9 mg/L), and he underwent successful L4-L5 transforaminal lumbar interbody fusion 2 weeks after finishing antibiotics.
We retroactively sent both P loescheii isolates and the 1 B fragilis isolate that grew from the surveillance blood culture to the Multidrug-resistant Organism Repository and Surveillance Network (MRSN) at the Walter Reed Army Institute of Research for identification confirmation and susceptibility analysis. Whole genome sequencing with single nucleotide polymorphism (SNP)-based analysis revealed all isolates were 100% identical and consistent with B fragilis and not P loescheii, based on clustering around other B fragilis sequences found in the National Center for Biotechnology Information (NCBI) Genbank database (Figure 4). All isolates carried the antibiotic resistance genes— cepA, sul(2), tetQ— encoding for possible resistance to cephalosporins, sulphonamides, and tetracyclines, respectively; as well as a point mutation in the gyrA gene (Ser82Phe). None of the isolates carried the nim gene, and screening for the 3 subtypes of B fragilis enterotoxin gene (bft-1, bft-2, bft-3) was negative. Eventual susceptibility testing at the Mayo Clinic several months after the conclusion of the case indicated that the B fragilis isolate was sensitive to piperacillin-tazobactam, ertapenem, clindamycin, and metronidazole; however, testing was not performed against moxifloxacin.
Discussion
In the era of growing antibiotic resistance patterns, antimicrobial stewardship programs recommend interventions to improve antimicrobial use through targeted narrow- spectrum antibiotics.12 The Clinical and Laboratory Standards Institute (CLSI) maintains guidelines on the major indications for anaerobic antimicrobial susceptibility testing (AST) to help direct narrow-targeted antimicrobial therapy. However, in a 2008 practice survey Goldstein and colleagues reported that less than half of US hospitals performed anaerobic AST, and only 21% of these facilities did it in-house, while the remainder sent out their isolates for testing.11-14 The CLSI major indications for AST include situations in which the selection of agents is important because of the (1) known resistance of a particular species; (2) confirmation of appropriate therapy for severe infections or for those that may require long-term therapy; (3) persistence of infection despite adequate treatment with an appropriate therapeutic regimen; and (4) difficulty in making empirical decisions based on precedent.14 Additionally, isolates from brain abscess, endocarditis, osteomyelitis, joint infection, infection of prosthetic devices or vascular grafts, bacteremia, and normally sterile body sites (unless contamination suspected) should be tested.14
Because of the lack of anaerobic AST, health care providers must base empiric treatment on reported sensitivities from the medical literature. Empiric selection of antimicrobials for anaerobic infections is made even more challenging by the increased rates of resistance reported in the literature, leading to recommendations to increase susceptibility testing to guide therapy.13,15,16 Empiric therapy of deep-seated anaerobic infections may lead to use of inactive agents or overly broad-spectrum antibiotics. Current antimicrobial stewardship initiatives recognize the importance of narrow-spectrum antibiotics to minimize risk of adverse events and selective pressure for antimicrobial resistance.
Although we attempted to confirm the identification of the anaerobic isolates via commercially available methods, it was not until we performed genetic testing that we were able verify the isolates as B fragilis. Furthermore, earlier susceptibility testing would have allowed for more narrow-targeted antimicrobial therapy and could have potentially prevented our patient’s readmission and use of ertapenem, despite its > 98% susceptibility rates against B fragilis.13,17
All of the B fragilis isolates carried the cepA gene, which is a cephalosporinase that encodes for resistance to cephalosporins and aminopenicillins but not to ß-lactam ß-lactamase inhibitor combinations.13 Although not a substitution for susceptibility analysis, genetic testing showed that all of the isolates carried a nonsynonymous mutation from serine to a phenylalanine at amino acid position 82 (S82F) in the gyrA gene. The S82F mutation has been implicated in fluoroquinolone resistance, via inhibition of substrate–target recognition and binding between fluoroquinolones and the target topoisomerase protein,18 and may potentially explain why our patient clinically worsened while on moxifloxacin monotherapy. Although moxifloxacin susceptibility was not performed, susceptibility rates remain highly variable, ranging from 50% to 70% for B fragilis.13,15,16
It is important to note that the metronidazole the patient received during his first hospital admission could have sterilized the vertebral body without completely eradicating the microbe; thus could explain his clinical worsening while on moxifloxacin monotherapy despite no growth from the repeat biopsy culture. Our rationale for initially continuing moxifloxacin was based on its excellent bioavailability and bone penetration properties. Additionally, of the fluoroquinolones it has the most reliable anaerobic activity and is the only one recommended as monotherapy for complicated intraabdominal infections.19 However, guidelines recommend avoiding its use in patients who have received a fluoroquinolone in the past 90 days or at institutions with high rates of resistance. At our institution Escherichia coli has a > 90% susceptibility rate to fluoroquinolones. Given this rate and our concern that the patient had a polymicrobial infection, we felt that moxifloxacin would provide appropriate anaerobic and aerobic coverage, especially since he had no previous fluoroquinolone exposure.
Additionally, none of the isolates carried the nim or bft toxin genes. Although the nim gene is associated with metronidazole resistance,its presence does not invariably result in resistant strains of B fragilis; in fact, metronidazole resistance is relatively uncommon, with the majority of B fragilis showing < 1% resistance, based on CLSI breakpoints (≥ 32 mg/L).13,20,21 However, one recent epidemiologic study on anaerobic wound isolates from Iraq and Afghanistan casualties found that 12% (2/17) of B fragilis isolates were resistant to metronidazole.15 Given the improvement of the patient’s symptoms while on metronidazole, it is likely that the B fragilis was susceptible. Nevertheless, susceptibility testing with minimum inhibitory concentrations is necessary to verify this result. Also, although enterotoxigenic strains of B fragilis have been associated with bloodstream infections, our patient’s isolates lacked the 3 subtypes of B fragilis enterotoxin gene.22
Conclusions
We report a case of B fragilis bacteremia and vertebral osteomyelitis complicated by challenges in anaerobic identification and sensitivities that led to brief use of a possibly inactive antimicrobial and the subsequent use of carbapenem therapy, which may have been avoided if susceptibility testing were more readily available. This case led to changes in our hospital’s processing of anaerobic isolates to include susceptibility testing on request.
Acknowledgments
We thank Keith Thompson, MD (staff pathologist, Naval Medical Center Portsmouth Virginia), for providing the pathology images from the initial vertebral biopsy, and Dr. Kate Hinkle (director, Multidrug-Resistant Organism Repository and Surveillance Network, Silver Spring, Maryland ) for providing the whole genome sequencing results from the B fragilis isolates.
1. Zimmerli W. Vertebral osteomyelitis. N Eng J Med. 2010;362(11):1022-1029.
2. Chazan B, Strahilevitz J, Millgram MA, Kaufmann S, Raz R. Bacteroides fragilis vertebral osteomyelitis secondary to anal dilatation. Spine (Phila PA 1976). 2001;26(16):E377-E378.
3. Kierzkowska M, Pedzisz P, Babiak I, et al. Orthopedic infections caused by obligatory anaerobic Gram-negative rods: report of two cases. Med Microbiol Immunol. 2017;206(5):363-366.
4. McHenry M, Easley K, Locker G. Vertebral osteomyelitis: long-term outcome for 253 patients from 7 Cleveland-area hospitals. Clin Infect Dis. 2002;34(10):1342-1350.
5. Raff MJ, Melo JC. Anaerobic osteomyelitis. Medicine (Baltimore).1978;57(1):83-103.
6. Lewis R, Sutter V, Finegold S. Bone infections involving anaerobic bacteria. Medicine (Baltimore). 1978;57(1):279-305.
7. Brook I. The role of anaerobic bacteria in bacteremia. Anaerobe. 2010;16(3):183-189.
8. Lassmann B, Gustafson DR, Wood CM, Rosenblatt JE. Reemergence of anaerobic bacteremia. Clin Infect Dis. 2007;44(7):895-900.
9. Lazarovitch T, Freimann S, Shapira G, Blank H. Decrease in anaerobe-related bacteraemias and increase in Bacteroides species isolation rate from 1998 to 2007: a retrospective study. Anaerobe. 2010;16(3):201-205.
10. Keukeleire S, Wybo I, Naessens A, et al. Anaerobic bacteraemia: a 10-year retrospective epidemiological survey. Anaerobe. 2016;39:54-59.
11. Goldstein EJC, Citron DM, Goldman PJ, Goldman RJ. National hospital survey of anaerobic culture and susceptibility methods: III. Anaerobe. 2008;14(2):68-72.
12. Barlam TF, Cosgrove SE, Abbo LM, et al. Implementing an antibiotic stewardship program: Guidelines by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America. Clin Infect Dis. 2016;62(10):e51-e77.
13. Schuetz AN. Antimicrobial resistance and susceptibility testing of anaerobic bacteria. Antimicr Resist. 2014;59(5):698-705.
14. Clinical and Laboratory Standards Institute. M11-A8: Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria; Approved Standard. 8th ed. Wayne, PA: Clinical and Laboratory Standards Institute; 2012.
15. White B, Mende K, Weintrob A, et al; Infectious Disease Clinical Research Program Trauma Infectious Disease Outcome Study Group. Epidemiology and antimicrobial susceptibilities of wound isolates of obligate anaerobes from combat casualties. Diagn Mircrobiol Infect Dis. 2016;84(2):144-150.
16. Hastey CJ, Boyd H, Schuetz AN, et al; Ad Hoc Working Group on Antimicrobial Susceptibility Testing of Anaerobic Bacteria of CLSI. Changes in the antibiotic susceptibility of anaerobic bacteria from 2007-2009 to 2010-2012 based on the CLSI methodology. Anaerobe. 2016;42:27-30.
17. Brook I, Wexler HM, Goldstein EJC. Antianaerobic antimicrobials: spectrum and susceptibility testing. Clin Microbiol Rev. 2013;26(3):526-546.
18. Pumbwe L, Wareham D, Aduse-Opoku J, Brazier JS, Wexler HM. Genetic analysis of mechanisms of multidrug resistance in a clinical isolate of Bacteroides fragilis. Clin Microbiol Infect. 2007;13(2):183-189.
19. Solomkin J, Mazuski J, Bradley J, et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin Infect Dis. 2010;50(2):133-164.
20. Breuil J, Dublanchet A, Truffaut N, Sebald M. Transferable 5-nitroimidazole resistance Bacteroides fragilis group. Plasmid. 1989;21(2):151-154.
21. Nagy E, Urbán E, Nord CE; ESCMID Study Group on Antimicrobial Resistance in Anaerobic Bacteria. Antimicrobial susceptibility of Bacteroides fragilis group isolates in Europe: 20 years of experience. Clin Microbiol Infect. 2011;17(3):371-379.
22. Avila-Campos M, Liu C, Song Y, Rowlinson M-C, Finegold SM. Determination of bft gene subtypes in Bacteroides fragilis clinical isolates. J Clin Microbiol. 2007;45(4):1336-1338.
1. Zimmerli W. Vertebral osteomyelitis. N Eng J Med. 2010;362(11):1022-1029.
2. Chazan B, Strahilevitz J, Millgram MA, Kaufmann S, Raz R. Bacteroides fragilis vertebral osteomyelitis secondary to anal dilatation. Spine (Phila PA 1976). 2001;26(16):E377-E378.
3. Kierzkowska M, Pedzisz P, Babiak I, et al. Orthopedic infections caused by obligatory anaerobic Gram-negative rods: report of two cases. Med Microbiol Immunol. 2017;206(5):363-366.
4. McHenry M, Easley K, Locker G. Vertebral osteomyelitis: long-term outcome for 253 patients from 7 Cleveland-area hospitals. Clin Infect Dis. 2002;34(10):1342-1350.
5. Raff MJ, Melo JC. Anaerobic osteomyelitis. Medicine (Baltimore).1978;57(1):83-103.
6. Lewis R, Sutter V, Finegold S. Bone infections involving anaerobic bacteria. Medicine (Baltimore). 1978;57(1):279-305.
7. Brook I. The role of anaerobic bacteria in bacteremia. Anaerobe. 2010;16(3):183-189.
8. Lassmann B, Gustafson DR, Wood CM, Rosenblatt JE. Reemergence of anaerobic bacteremia. Clin Infect Dis. 2007;44(7):895-900.
9. Lazarovitch T, Freimann S, Shapira G, Blank H. Decrease in anaerobe-related bacteraemias and increase in Bacteroides species isolation rate from 1998 to 2007: a retrospective study. Anaerobe. 2010;16(3):201-205.
10. Keukeleire S, Wybo I, Naessens A, et al. Anaerobic bacteraemia: a 10-year retrospective epidemiological survey. Anaerobe. 2016;39:54-59.
11. Goldstein EJC, Citron DM, Goldman PJ, Goldman RJ. National hospital survey of anaerobic culture and susceptibility methods: III. Anaerobe. 2008;14(2):68-72.
12. Barlam TF, Cosgrove SE, Abbo LM, et al. Implementing an antibiotic stewardship program: Guidelines by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America. Clin Infect Dis. 2016;62(10):e51-e77.
13. Schuetz AN. Antimicrobial resistance and susceptibility testing of anaerobic bacteria. Antimicr Resist. 2014;59(5):698-705.
14. Clinical and Laboratory Standards Institute. M11-A8: Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria; Approved Standard. 8th ed. Wayne, PA: Clinical and Laboratory Standards Institute; 2012.
15. White B, Mende K, Weintrob A, et al; Infectious Disease Clinical Research Program Trauma Infectious Disease Outcome Study Group. Epidemiology and antimicrobial susceptibilities of wound isolates of obligate anaerobes from combat casualties. Diagn Mircrobiol Infect Dis. 2016;84(2):144-150.
16. Hastey CJ, Boyd H, Schuetz AN, et al; Ad Hoc Working Group on Antimicrobial Susceptibility Testing of Anaerobic Bacteria of CLSI. Changes in the antibiotic susceptibility of anaerobic bacteria from 2007-2009 to 2010-2012 based on the CLSI methodology. Anaerobe. 2016;42:27-30.
17. Brook I, Wexler HM, Goldstein EJC. Antianaerobic antimicrobials: spectrum and susceptibility testing. Clin Microbiol Rev. 2013;26(3):526-546.
18. Pumbwe L, Wareham D, Aduse-Opoku J, Brazier JS, Wexler HM. Genetic analysis of mechanisms of multidrug resistance in a clinical isolate of Bacteroides fragilis. Clin Microbiol Infect. 2007;13(2):183-189.
19. Solomkin J, Mazuski J, Bradley J, et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin Infect Dis. 2010;50(2):133-164.
20. Breuil J, Dublanchet A, Truffaut N, Sebald M. Transferable 5-nitroimidazole resistance Bacteroides fragilis group. Plasmid. 1989;21(2):151-154.
21. Nagy E, Urbán E, Nord CE; ESCMID Study Group on Antimicrobial Resistance in Anaerobic Bacteria. Antimicrobial susceptibility of Bacteroides fragilis group isolates in Europe: 20 years of experience. Clin Microbiol Infect. 2011;17(3):371-379.
22. Avila-Campos M, Liu C, Song Y, Rowlinson M-C, Finegold SM. Determination of bft gene subtypes in Bacteroides fragilis clinical isolates. J Clin Microbiol. 2007;45(4):1336-1338.
Postpartum IUD placement • breastfeeding • difficulty maintaining milk supply • Dx?
THE CASE
A 28-year-old G1P1 initially presented to the family medicine clinic 4 weeks postpartum to discuss possibilities for contraception. She had received her prenatal care through a midwife and had had a successful home delivery. She was exclusively breastfeeding her infant daughter but wanted to ensure adequate spacing between her pregnancies.
During the discussion of possible options, the patient revealed that she had previously had an intrauterine device (IUD) placed and expressed interest in using this method again. A levonorgestrel-releasing IUD (Mirena) was placed at 6 weeks postpartum, after a negative pregnancy test was obtained.
The patient returned to the clinic about 6 months later with complaints of increased difficulty maintaining her milk supply.
THE DIAGNOSIS
The patient had taken a home pregnancy test, which was positive—a finding confirmed in clinic via a urine pregnancy test.
Gestational age. Since the patient had an IUD in place and had been exclusively breastfeeding, gestational age was difficult to determine. A quantitative human chorionic gonadotropin (hCG) test showed an hCG level of 12,469 U/L, consistent with a 4-to-8-week pregnancy. An ultrasound performed the next day showed a single intrauterine pregnancy at 21 weeks.
IUD location. There was also the question of the location of the IUD and whether it would interfere with the patient’s ability to maintain the pregnancy. On ultrasound, the IUD was noted within the cervix and myometrium. After discussion of the risks, the patient chose to leave it in place.
DISCUSSION
IUDs are among the most effective forms of contraception; levonorgestrel-releasing IUDs are more effective than copper IUDs.1 The rates of failure in the first year of use are 0.8% and 0.2% for copper and levonorgestrel-releasing IUDs, respectively.1
Continue to: The Lactational Amenorrhea Method
The Lactational Amenorrhea Method (LAM), which is defined as providing infant nutrition exclusively through breastmilk during the first 6 months postpartum, also provides protection against pregnancy. LAM has a failure rate of 0% to 1.5%.2
It is not surprising that this patient thought she was adequately protected against pregnancy. That said, no contraceptive method is foolproof (as this case demonstrates).
Risks to the pregnancy. When pregnancy does occur with an IUD in place, the patient should be informed of the possible risks to the pregnancy. These include complications such as spontaneous abortion, chorioamnionitis, and preterm delivery.3 Risk is further increased if the IUD is malpositioned (as this one was), meaning that any part of the IUD is located in the lower uterine segment, myometrium, or endocervical canal.4,5
Removal of the IUD is generally recommended if the device and its strings can be located, although removal does not completely mitigate risk. In a study done in Egypt, 46 of 52 IUDs were removed successfully, with 2 spontaneous abortions as a result.6 Of note, the IUDs extracted in this study were Lippes loop and copper models, not levonorgestrel-releasing IUDs such as our patient had. There is a single case report7 of a patient who had a Mirena inserted very early in a pregnancy; the IUD had to be left in place due to the risk for miscarriage, but she was able to carry the infant to term and did not experience any adverse effects.
Our patient
The patient delivered a male infant vaginally at term without issue. However, the IUD was not expelled during this process. Ultrasound showed that it was embedded in the posterior myometrium with a hypoechoic tract. The patient was referred to Gynecology, and the IUD was successfully removed.
Continue to: THE TAKEAWAY
THE TAKEAWAY
Even the most reliable method of contraception can fail—so pregnancy should always be in the differential diagnosis for a sexually active woman. Location of IUD placement is important; it must be in the right place to be effective. The tenets of LAM must be followed precisely in order for breastfeeding to provide protection against pregnancy. Patients can successfully carry a pregnancy to term with an IUD, as this patient did, but it places them at higher risk for ectopic pregnancy, premature rupture of membranes, and infection.
The author thanks Jenny Walters, lactation consultant, for her assistance in the preparation of the manuscript.
CORRESPONDENCE
Hannah Maxfield, MD, 3901 Rainbow Boulevard, MS 4010, Kansas City, KS 66160; [email protected]
1. Heinemann K, Reed S, Moehner S, et al. Comparative contraceptive effectiveness of levonorgestrel-releasing and copper intrauterine devices: the European Active Surveillance Study for Intrauterine Devices. Contraception. 2015;91:280-283.
2. Labbok MH. Postpartum sexuality and the Lactational Amenorrhea Method for contraception. Clin Obstet Gynecol. 2015;58:915-927.
3. Ganer H, Levy A, Ohel I, et al. Pregnancy outcome in women with an intrauterine contraceptive device. Am J Obstet Gynecol. 2009;201:381.e1-e5.
4. Moschos E, Twickler D. Intrauterine devices in early pregnancy: findings on ultrasound and clinical outcomes. Am J Obstet Gynecol. 2011;204:427.e1-e6.
5. Ozgu-Erdinc AS, Tasdemir UG, Uygur D, et al. Outcome of intrauterine pregnancies with intrauterine device in place and effects of device location on prognosis. Contraception. 2014;89:426-430.
6. Assaf A, Gohar M, Saad S, et al. Removal of intrauterine devices with missing tails during early pregnancy. Contraception. 1992;45:541-546.
7. Gardyszewska A, Czajkowski K. Application of levonorgestrel-releasing intrauterine system in early pregnancy: a case report [article in Polish]. Ginekol Pol. 2012;83:950-952.
THE CASE
A 28-year-old G1P1 initially presented to the family medicine clinic 4 weeks postpartum to discuss possibilities for contraception. She had received her prenatal care through a midwife and had had a successful home delivery. She was exclusively breastfeeding her infant daughter but wanted to ensure adequate spacing between her pregnancies.
During the discussion of possible options, the patient revealed that she had previously had an intrauterine device (IUD) placed and expressed interest in using this method again. A levonorgestrel-releasing IUD (Mirena) was placed at 6 weeks postpartum, after a negative pregnancy test was obtained.
The patient returned to the clinic about 6 months later with complaints of increased difficulty maintaining her milk supply.
THE DIAGNOSIS
The patient had taken a home pregnancy test, which was positive—a finding confirmed in clinic via a urine pregnancy test.
Gestational age. Since the patient had an IUD in place and had been exclusively breastfeeding, gestational age was difficult to determine. A quantitative human chorionic gonadotropin (hCG) test showed an hCG level of 12,469 U/L, consistent with a 4-to-8-week pregnancy. An ultrasound performed the next day showed a single intrauterine pregnancy at 21 weeks.
IUD location. There was also the question of the location of the IUD and whether it would interfere with the patient’s ability to maintain the pregnancy. On ultrasound, the IUD was noted within the cervix and myometrium. After discussion of the risks, the patient chose to leave it in place.
DISCUSSION
IUDs are among the most effective forms of contraception; levonorgestrel-releasing IUDs are more effective than copper IUDs.1 The rates of failure in the first year of use are 0.8% and 0.2% for copper and levonorgestrel-releasing IUDs, respectively.1
Continue to: The Lactational Amenorrhea Method
The Lactational Amenorrhea Method (LAM), which is defined as providing infant nutrition exclusively through breastmilk during the first 6 months postpartum, also provides protection against pregnancy. LAM has a failure rate of 0% to 1.5%.2
It is not surprising that this patient thought she was adequately protected against pregnancy. That said, no contraceptive method is foolproof (as this case demonstrates).
Risks to the pregnancy. When pregnancy does occur with an IUD in place, the patient should be informed of the possible risks to the pregnancy. These include complications such as spontaneous abortion, chorioamnionitis, and preterm delivery.3 Risk is further increased if the IUD is malpositioned (as this one was), meaning that any part of the IUD is located in the lower uterine segment, myometrium, or endocervical canal.4,5
Removal of the IUD is generally recommended if the device and its strings can be located, although removal does not completely mitigate risk. In a study done in Egypt, 46 of 52 IUDs were removed successfully, with 2 spontaneous abortions as a result.6 Of note, the IUDs extracted in this study were Lippes loop and copper models, not levonorgestrel-releasing IUDs such as our patient had. There is a single case report7 of a patient who had a Mirena inserted very early in a pregnancy; the IUD had to be left in place due to the risk for miscarriage, but she was able to carry the infant to term and did not experience any adverse effects.
Our patient
The patient delivered a male infant vaginally at term without issue. However, the IUD was not expelled during this process. Ultrasound showed that it was embedded in the posterior myometrium with a hypoechoic tract. The patient was referred to Gynecology, and the IUD was successfully removed.
Continue to: THE TAKEAWAY
THE TAKEAWAY
Even the most reliable method of contraception can fail—so pregnancy should always be in the differential diagnosis for a sexually active woman. Location of IUD placement is important; it must be in the right place to be effective. The tenets of LAM must be followed precisely in order for breastfeeding to provide protection against pregnancy. Patients can successfully carry a pregnancy to term with an IUD, as this patient did, but it places them at higher risk for ectopic pregnancy, premature rupture of membranes, and infection.
The author thanks Jenny Walters, lactation consultant, for her assistance in the preparation of the manuscript.
CORRESPONDENCE
Hannah Maxfield, MD, 3901 Rainbow Boulevard, MS 4010, Kansas City, KS 66160; [email protected]
THE CASE
A 28-year-old G1P1 initially presented to the family medicine clinic 4 weeks postpartum to discuss possibilities for contraception. She had received her prenatal care through a midwife and had had a successful home delivery. She was exclusively breastfeeding her infant daughter but wanted to ensure adequate spacing between her pregnancies.
During the discussion of possible options, the patient revealed that she had previously had an intrauterine device (IUD) placed and expressed interest in using this method again. A levonorgestrel-releasing IUD (Mirena) was placed at 6 weeks postpartum, after a negative pregnancy test was obtained.
The patient returned to the clinic about 6 months later with complaints of increased difficulty maintaining her milk supply.
THE DIAGNOSIS
The patient had taken a home pregnancy test, which was positive—a finding confirmed in clinic via a urine pregnancy test.
Gestational age. Since the patient had an IUD in place and had been exclusively breastfeeding, gestational age was difficult to determine. A quantitative human chorionic gonadotropin (hCG) test showed an hCG level of 12,469 U/L, consistent with a 4-to-8-week pregnancy. An ultrasound performed the next day showed a single intrauterine pregnancy at 21 weeks.
IUD location. There was also the question of the location of the IUD and whether it would interfere with the patient’s ability to maintain the pregnancy. On ultrasound, the IUD was noted within the cervix and myometrium. After discussion of the risks, the patient chose to leave it in place.
DISCUSSION
IUDs are among the most effective forms of contraception; levonorgestrel-releasing IUDs are more effective than copper IUDs.1 The rates of failure in the first year of use are 0.8% and 0.2% for copper and levonorgestrel-releasing IUDs, respectively.1
Continue to: The Lactational Amenorrhea Method
The Lactational Amenorrhea Method (LAM), which is defined as providing infant nutrition exclusively through breastmilk during the first 6 months postpartum, also provides protection against pregnancy. LAM has a failure rate of 0% to 1.5%.2
It is not surprising that this patient thought she was adequately protected against pregnancy. That said, no contraceptive method is foolproof (as this case demonstrates).
Risks to the pregnancy. When pregnancy does occur with an IUD in place, the patient should be informed of the possible risks to the pregnancy. These include complications such as spontaneous abortion, chorioamnionitis, and preterm delivery.3 Risk is further increased if the IUD is malpositioned (as this one was), meaning that any part of the IUD is located in the lower uterine segment, myometrium, or endocervical canal.4,5
Removal of the IUD is generally recommended if the device and its strings can be located, although removal does not completely mitigate risk. In a study done in Egypt, 46 of 52 IUDs were removed successfully, with 2 spontaneous abortions as a result.6 Of note, the IUDs extracted in this study were Lippes loop and copper models, not levonorgestrel-releasing IUDs such as our patient had. There is a single case report7 of a patient who had a Mirena inserted very early in a pregnancy; the IUD had to be left in place due to the risk for miscarriage, but she was able to carry the infant to term and did not experience any adverse effects.
Our patient
The patient delivered a male infant vaginally at term without issue. However, the IUD was not expelled during this process. Ultrasound showed that it was embedded in the posterior myometrium with a hypoechoic tract. The patient was referred to Gynecology, and the IUD was successfully removed.
Continue to: THE TAKEAWAY
THE TAKEAWAY
Even the most reliable method of contraception can fail—so pregnancy should always be in the differential diagnosis for a sexually active woman. Location of IUD placement is important; it must be in the right place to be effective. The tenets of LAM must be followed precisely in order for breastfeeding to provide protection against pregnancy. Patients can successfully carry a pregnancy to term with an IUD, as this patient did, but it places them at higher risk for ectopic pregnancy, premature rupture of membranes, and infection.
The author thanks Jenny Walters, lactation consultant, for her assistance in the preparation of the manuscript.
CORRESPONDENCE
Hannah Maxfield, MD, 3901 Rainbow Boulevard, MS 4010, Kansas City, KS 66160; [email protected]
1. Heinemann K, Reed S, Moehner S, et al. Comparative contraceptive effectiveness of levonorgestrel-releasing and copper intrauterine devices: the European Active Surveillance Study for Intrauterine Devices. Contraception. 2015;91:280-283.
2. Labbok MH. Postpartum sexuality and the Lactational Amenorrhea Method for contraception. Clin Obstet Gynecol. 2015;58:915-927.
3. Ganer H, Levy A, Ohel I, et al. Pregnancy outcome in women with an intrauterine contraceptive device. Am J Obstet Gynecol. 2009;201:381.e1-e5.
4. Moschos E, Twickler D. Intrauterine devices in early pregnancy: findings on ultrasound and clinical outcomes. Am J Obstet Gynecol. 2011;204:427.e1-e6.
5. Ozgu-Erdinc AS, Tasdemir UG, Uygur D, et al. Outcome of intrauterine pregnancies with intrauterine device in place and effects of device location on prognosis. Contraception. 2014;89:426-430.
6. Assaf A, Gohar M, Saad S, et al. Removal of intrauterine devices with missing tails during early pregnancy. Contraception. 1992;45:541-546.
7. Gardyszewska A, Czajkowski K. Application of levonorgestrel-releasing intrauterine system in early pregnancy: a case report [article in Polish]. Ginekol Pol. 2012;83:950-952.
1. Heinemann K, Reed S, Moehner S, et al. Comparative contraceptive effectiveness of levonorgestrel-releasing and copper intrauterine devices: the European Active Surveillance Study for Intrauterine Devices. Contraception. 2015;91:280-283.
2. Labbok MH. Postpartum sexuality and the Lactational Amenorrhea Method for contraception. Clin Obstet Gynecol. 2015;58:915-927.
3. Ganer H, Levy A, Ohel I, et al. Pregnancy outcome in women with an intrauterine contraceptive device. Am J Obstet Gynecol. 2009;201:381.e1-e5.
4. Moschos E, Twickler D. Intrauterine devices in early pregnancy: findings on ultrasound and clinical outcomes. Am J Obstet Gynecol. 2011;204:427.e1-e6.
5. Ozgu-Erdinc AS, Tasdemir UG, Uygur D, et al. Outcome of intrauterine pregnancies with intrauterine device in place and effects of device location on prognosis. Contraception. 2014;89:426-430.
6. Assaf A, Gohar M, Saad S, et al. Removal of intrauterine devices with missing tails during early pregnancy. Contraception. 1992;45:541-546.
7. Gardyszewska A, Czajkowski K. Application of levonorgestrel-releasing intrauterine system in early pregnancy: a case report [article in Polish]. Ginekol Pol. 2012;83:950-952.

Testicular Swelling as an Initial Presentation of a Patient With Metastatic Gastric Cancer (FULL)
Gastric cancer is one of the most common cancers and the second most common cause of cancer-related death worldwide.1 Patients with an early-stage gastric cancer are often asymptomatic, and about 30% to 40% of patients present with distant metastases.2 The most common sites of metastasis are the liver, peritoneum, and the lymph nodes. In more advanced stages, gastric cancers spread to the lungs, brain, bones, soft tissues, and other sites. Krukenberg tumors are classic but rare occurrences of gastric cancer metastasis to ovaries in females. In men, it is rare for gastric cancer to metastasize to the testes. Only a few cases of testicular metastasis from gastric cancer have been reported in the literature.3-5
Primary testicular neoplasms typically present with unilateral testicular swelling. In rare instances, testicular swelling can be an initial presentation of a metastatic cancer. In this article, we report a man who initially presented with right testicular swelling and was eventually diagnosed with a secondary testicular cancer from an aggressive metastatic gastric adenocarcinoma.
Case Presentation
A 43-year-old male presented to his primary care physician with a 6-month history of right testicular swelling and unintentional weight loss of 33 pounds. An ultrasound of the scrotum and the testes showed a 4.5-cm hypoechoic and predominantly cystic mass in the right testis. Serum levels of β-human chorionic gonadotropin, α-feto protein and lactate dehydrogenase were within normal limits.
A primary testicular neoplasm was suspected. The patient underwent a right-sided inguinal orchiectomy. The pathology of the resected testis revealed an intestinal type adenocarcinoma with mucinous and signet-ring cell features (Figure). 
A staging Fluorine-18 fluorodeoxyglucose positron emission tomography scan revealed extensive hypermetabolic peritoneal nodules consistent with peritoneal carcinomatosis. The patient started palliative chemotherapy with 5-fluorouracil and oxaliplatin but rapidly declined with progressive abdominal symptoms after completion of 2 cycles. He discontinued chemotherapy and eventually opted for supportive care focusing on comfort.
Discussion
Testicular neoplasm is the most common solid tumor in men aged 20 to 34 years in the US.6 More than 90% of testicular neoplasms are germ-cell tumors and originate from the testes.6 Secondary neoplasms of the testes from solid tumors are rare. In an autopsy series, secondary neoplasms from solid tumors represented 4.6% of all testicular neoplasms.7 The reported frequencies of primary sites of origin vary based on the report. In a relatively large series, the most common solid tumors of origin were the prostate (35%) and lungs (20%).8 Among hematologic malignancies, lymphomas may originate from or secondarily involve the testes. Primary testicular lymphomas account for approximately 5% of testicular neoplasms and commonly occur in men aged > 60 years.9 Testicular involvement of acute myeloid or lymphoid leukemia may happen at diagnosis, but relapses of acute leukemia in a testicular site are more common and well recognized.10,11
Our report shows a rare occurrence of a testicular swelling as an initial presentation of an aggressive metastatic gastric adenocarcinoma. When a patient presents with a testicular swelling, a testicular neoplasm is suspected based on the clinical examination, serum tumor markers, and ultrasonographic examination of the testes. Inguinal orchiectomy is the initial standard of care, and pathologic examination of a resected testis renders the confirmatory diagnosis. The overwhelming majority of these patients have primary testicular neoplasms, predominantly testicular germ-cell tumors. A small group may have atypical and rare tumors requiring additional workup to establish a diagnosis.
Metastatic spread of a solid tumor to a testicular site is rare. A testis is somewhat protected by the distinct anatomy that is offered by the blood-testis barrier. The tunica vaginalis, an external fibrous covering sheath of the testis, separates the testis from the peritoneal cavity and provides additional protection. Nevertheless, possible routes of secondary metastatic spread may include hematogenous and lymphovascular spread, cavitary dissemination and peritoneal seeding. Among gastric cancers, diffuse gastric cancer subtype is commonly associated with signet-ring cells. These cells lack adhesion and invade as single cells or small groups, leading to scattered tumor cells and a higher probability of seeding.
Our patient exhibited extensive peritoneal carcinomatosis, so peritoneal seeding likely played a dominant role in disseminating the cancer to the testis. It should be noted that possible primary sites of signet-ring cell metastatic adenocarcinomas are broad and may include the stomach and other GI and pancreaticobiliary sites, as well as the lung, bladder, breast, and gynecologic tract. Immunohistochemistry is often of limited value in establishing the primary site. Diagnosis is best established on clinical grounds, as was done in our patient.
When signet-ring cells are encountered in an orchiectomy specimen and a primary extratesticular site of origin cannot be identified, it is important to consider rare variants of primary testicular tumors in the differential diagnosis. A number of primary testicular tumors have signetring morphology. These include primary signetring cell stromal tumor of the testis (PSRST),12 seminoma with signet-ring cell predominance,13 and a primary signet-ring cell germ-cell carcinoma of the testes (PSRGCT).14
Seminoma and PSRST variants generally are differentiated from adenocarcinoma by a lack of mucin production and the absence of keratin expression. PSRGCT is considered a
malignant somatic-type transformation of a testicular germ-cell tumor and germ-cell clonality is established by abnormal 12p chromosome through fluorescent in situ hybridization testing.13 PSRST is a benign tumor with excellent prognosis. Seminoma variants with signet-ring cell predominance and PSRGCT are managed as germ-cell tumors and are also considered to have good outcome. In contrast, testicular involvements from metastatic adenocarcinomas, including gastric cancers, are managed with multidisciplinary approach, including systemic chemotherapy. Despite this, patients have a poor outcome with a median survival of about 1 year.2
Conclusion
Advanced gastric adenocarcinoma can metastasize to the testes, and patients may present with a testicular swelling as an initial presentation. It is important to differentiate secondary testicular cancers from rare variants of primary testicular tumors because the prognosis and management vary substantially.
1. Global Burden of Disease Cancer Collaboration, Fitzmaurice C, Dicker D, et al. The global burden of cancer 2013. JAMA Oncol. 2015;1(4):505-527.
2. Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388(10060):2654-2664.
3. Qazi HA, Manikandan R, Foster CS, Fordham MV. Testicular metastasis from gastric carcinoma. Urology. 2006;68(4):890.e7-e8.
4. Civelek B, Aksoy S, Kos T, et al. Isolated testicular metastasis of gastric cancer. J Gastrointest Canc. 2012;43(suppl 1):S64-S66.
5. Li B, Cai H, Kang ZC, et al. Testicular metastasis from gastric carcinoma: a case report. World J of Gastroenterol. 2015;21(21):6764-6768.
6. National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: testicular cancer. https://seer.cancer.gov/statfacts/html/testis.html. Updated September 10, 2018. Accessed April 10, 2019.
7. Dutt N, Bates AW, Baithun SI. Secondary neoplasms of the male genital tract with different patterns of involvement in adults and children. Histopathology. 2000;37(4):323-331.
8. Haupt HM, Mann RB, Trump DL, Abeloff MD. Metastatic carcinoma involving the testis. Clinical and pathologic distinction from primary testicular neoplasms. Cancer.1984;54 (4):709-714.
9. Cheah CY, Wirth A, Seymour JF. Primary testicular lymphoma. Blood. 2014;123(4):486-493.
10. Kawamoto K, Miyoshi H, Yoshida N, Takizawa J, Sone H, Ohshima K. Clinicopathological, cytogenetic, and prognostic analysis of 131 myeloid sarcoma patients. Am J Surg Pathol. 2016;40(11):1473-1483.
11. Kulkarni KP, Marwaha RK, Trehan A, Bansal D. Testicular relapse in childhood acute lymphoblastic leukemia: the challenges and lessons. Indian J Cancer. 2010;47(2):134-138.
12. Kuo CY, Wen MC, Wang J, Jan YJ. Signet-ring stromal tumor of the testis: a case report and literature review. Hum Pathol. 2009;40(4):584-587.
13. Ulbright TM, Young RH. Seminoma with conspicuous signet ring cells: a rare, previously uncharacterized morphologic variant. Am J Surg Pathol. 2008;32(8):1175-1181.
14. Williamson SR, Kum JB, Shah SR, et al. Signet ring cell carcinoma of the testis: clinicopathologic and molecular evidence for germ cell tumor origin—a case report. Am J
Surg Pathol. 2012;36(2):311-315.
Gastric cancer is one of the most common cancers and the second most common cause of cancer-related death worldwide.1 Patients with an early-stage gastric cancer are often asymptomatic, and about 30% to 40% of patients present with distant metastases.2 The most common sites of metastasis are the liver, peritoneum, and the lymph nodes. In more advanced stages, gastric cancers spread to the lungs, brain, bones, soft tissues, and other sites. Krukenberg tumors are classic but rare occurrences of gastric cancer metastasis to ovaries in females. In men, it is rare for gastric cancer to metastasize to the testes. Only a few cases of testicular metastasis from gastric cancer have been reported in the literature.3-5
Primary testicular neoplasms typically present with unilateral testicular swelling. In rare instances, testicular swelling can be an initial presentation of a metastatic cancer. In this article, we report a man who initially presented with right testicular swelling and was eventually diagnosed with a secondary testicular cancer from an aggressive metastatic gastric adenocarcinoma.
Case Presentation
A 43-year-old male presented to his primary care physician with a 6-month history of right testicular swelling and unintentional weight loss of 33 pounds. An ultrasound of the scrotum and the testes showed a 4.5-cm hypoechoic and predominantly cystic mass in the right testis. Serum levels of β-human chorionic gonadotropin, α-feto protein and lactate dehydrogenase were within normal limits.
A primary testicular neoplasm was suspected. The patient underwent a right-sided inguinal orchiectomy. The pathology of the resected testis revealed an intestinal type adenocarcinoma with mucinous and signet-ring cell features (Figure).
A staging Fluorine-18 fluorodeoxyglucose positron emission tomography scan revealed extensive hypermetabolic peritoneal nodules consistent with peritoneal carcinomatosis. The patient started palliative chemotherapy with 5-fluorouracil and oxaliplatin but rapidly declined with progressive abdominal symptoms after completion of 2 cycles. He discontinued chemotherapy and eventually opted for supportive care focusing on comfort.
Discussion
Testicular neoplasm is the most common solid tumor in men aged 20 to 34 years in the US.6 More than 90% of testicular neoplasms are germ-cell tumors and originate from the testes.6 Secondary neoplasms of the testes from solid tumors are rare. In an autopsy series, secondary neoplasms from solid tumors represented 4.6% of all testicular neoplasms.7 The reported frequencies of primary sites of origin vary based on the report. In a relatively large series, the most common solid tumors of origin were the prostate (35%) and lungs (20%).8 Among hematologic malignancies, lymphomas may originate from or secondarily involve the testes. Primary testicular lymphomas account for approximately 5% of testicular neoplasms and commonly occur in men aged > 60 years.9 Testicular involvement of acute myeloid or lymphoid leukemia may happen at diagnosis, but relapses of acute leukemia in a testicular site are more common and well recognized.10,11
Our report shows a rare occurrence of a testicular swelling as an initial presentation of an aggressive metastatic gastric adenocarcinoma. When a patient presents with a testicular swelling, a testicular neoplasm is suspected based on the clinical examination, serum tumor markers, and ultrasonographic examination of the testes. Inguinal orchiectomy is the initial standard of care, and pathologic examination of a resected testis renders the confirmatory diagnosis. The overwhelming majority of these patients have primary testicular neoplasms, predominantly testicular germ-cell tumors. A small group may have atypical and rare tumors requiring additional workup to establish a diagnosis.
Metastatic spread of a solid tumor to a testicular site is rare. A testis is somewhat protected by the distinct anatomy that is offered by the blood-testis barrier. The tunica vaginalis, an external fibrous covering sheath of the testis, separates the testis from the peritoneal cavity and provides additional protection. Nevertheless, possible routes of secondary metastatic spread may include hematogenous and lymphovascular spread, cavitary dissemination and peritoneal seeding. Among gastric cancers, diffuse gastric cancer subtype is commonly associated with signet-ring cells. These cells lack adhesion and invade as single cells or small groups, leading to scattered tumor cells and a higher probability of seeding.
Our patient exhibited extensive peritoneal carcinomatosis, so peritoneal seeding likely played a dominant role in disseminating the cancer to the testis. It should be noted that possible primary sites of signet-ring cell metastatic adenocarcinomas are broad and may include the stomach and other GI and pancreaticobiliary sites, as well as the lung, bladder, breast, and gynecologic tract. Immunohistochemistry is often of limited value in establishing the primary site. Diagnosis is best established on clinical grounds, as was done in our patient.
When signet-ring cells are encountered in an orchiectomy specimen and a primary extratesticular site of origin cannot be identified, it is important to consider rare variants of primary testicular tumors in the differential diagnosis. A number of primary testicular tumors have signetring morphology. These include primary signetring cell stromal tumor of the testis (PSRST),12 seminoma with signet-ring cell predominance,13 and a primary signet-ring cell germ-cell carcinoma of the testes (PSRGCT).14
Seminoma and PSRST variants generally are differentiated from adenocarcinoma by a lack of mucin production and the absence of keratin expression. PSRGCT is considered a
malignant somatic-type transformation of a testicular germ-cell tumor and germ-cell clonality is established by abnormal 12p chromosome through fluorescent in situ hybridization testing.13 PSRST is a benign tumor with excellent prognosis. Seminoma variants with signet-ring cell predominance and PSRGCT are managed as germ-cell tumors and are also considered to have good outcome. In contrast, testicular involvements from metastatic adenocarcinomas, including gastric cancers, are managed with multidisciplinary approach, including systemic chemotherapy. Despite this, patients have a poor outcome with a median survival of about 1 year.2
Conclusion
Advanced gastric adenocarcinoma can metastasize to the testes, and patients may present with a testicular swelling as an initial presentation. It is important to differentiate secondary testicular cancers from rare variants of primary testicular tumors because the prognosis and management vary substantially.
Gastric cancer is one of the most common cancers and the second most common cause of cancer-related death worldwide.1 Patients with an early-stage gastric cancer are often asymptomatic, and about 30% to 40% of patients present with distant metastases.2 The most common sites of metastasis are the liver, peritoneum, and the lymph nodes. In more advanced stages, gastric cancers spread to the lungs, brain, bones, soft tissues, and other sites. Krukenberg tumors are classic but rare occurrences of gastric cancer metastasis to ovaries in females. In men, it is rare for gastric cancer to metastasize to the testes. Only a few cases of testicular metastasis from gastric cancer have been reported in the literature.3-5
Primary testicular neoplasms typically present with unilateral testicular swelling. In rare instances, testicular swelling can be an initial presentation of a metastatic cancer. In this article, we report a man who initially presented with right testicular swelling and was eventually diagnosed with a secondary testicular cancer from an aggressive metastatic gastric adenocarcinoma.
Case Presentation
A 43-year-old male presented to his primary care physician with a 6-month history of right testicular swelling and unintentional weight loss of 33 pounds. An ultrasound of the scrotum and the testes showed a 4.5-cm hypoechoic and predominantly cystic mass in the right testis. Serum levels of β-human chorionic gonadotropin, α-feto protein and lactate dehydrogenase were within normal limits.
A primary testicular neoplasm was suspected. The patient underwent a right-sided inguinal orchiectomy. The pathology of the resected testis revealed an intestinal type adenocarcinoma with mucinous and signet-ring cell features (Figure).
A staging Fluorine-18 fluorodeoxyglucose positron emission tomography scan revealed extensive hypermetabolic peritoneal nodules consistent with peritoneal carcinomatosis. The patient started palliative chemotherapy with 5-fluorouracil and oxaliplatin but rapidly declined with progressive abdominal symptoms after completion of 2 cycles. He discontinued chemotherapy and eventually opted for supportive care focusing on comfort.
Discussion
Testicular neoplasm is the most common solid tumor in men aged 20 to 34 years in the US.6 More than 90% of testicular neoplasms are germ-cell tumors and originate from the testes.6 Secondary neoplasms of the testes from solid tumors are rare. In an autopsy series, secondary neoplasms from solid tumors represented 4.6% of all testicular neoplasms.7 The reported frequencies of primary sites of origin vary based on the report. In a relatively large series, the most common solid tumors of origin were the prostate (35%) and lungs (20%).8 Among hematologic malignancies, lymphomas may originate from or secondarily involve the testes. Primary testicular lymphomas account for approximately 5% of testicular neoplasms and commonly occur in men aged > 60 years.9 Testicular involvement of acute myeloid or lymphoid leukemia may happen at diagnosis, but relapses of acute leukemia in a testicular site are more common and well recognized.10,11
Our report shows a rare occurrence of a testicular swelling as an initial presentation of an aggressive metastatic gastric adenocarcinoma. When a patient presents with a testicular swelling, a testicular neoplasm is suspected based on the clinical examination, serum tumor markers, and ultrasonographic examination of the testes. Inguinal orchiectomy is the initial standard of care, and pathologic examination of a resected testis renders the confirmatory diagnosis. The overwhelming majority of these patients have primary testicular neoplasms, predominantly testicular germ-cell tumors. A small group may have atypical and rare tumors requiring additional workup to establish a diagnosis.
Metastatic spread of a solid tumor to a testicular site is rare. A testis is somewhat protected by the distinct anatomy that is offered by the blood-testis barrier. The tunica vaginalis, an external fibrous covering sheath of the testis, separates the testis from the peritoneal cavity and provides additional protection. Nevertheless, possible routes of secondary metastatic spread may include hematogenous and lymphovascular spread, cavitary dissemination and peritoneal seeding. Among gastric cancers, diffuse gastric cancer subtype is commonly associated with signet-ring cells. These cells lack adhesion and invade as single cells or small groups, leading to scattered tumor cells and a higher probability of seeding.
Our patient exhibited extensive peritoneal carcinomatosis, so peritoneal seeding likely played a dominant role in disseminating the cancer to the testis. It should be noted that possible primary sites of signet-ring cell metastatic adenocarcinomas are broad and may include the stomach and other GI and pancreaticobiliary sites, as well as the lung, bladder, breast, and gynecologic tract. Immunohistochemistry is often of limited value in establishing the primary site. Diagnosis is best established on clinical grounds, as was done in our patient.
When signet-ring cells are encountered in an orchiectomy specimen and a primary extratesticular site of origin cannot be identified, it is important to consider rare variants of primary testicular tumors in the differential diagnosis. A number of primary testicular tumors have signetring morphology. These include primary signetring cell stromal tumor of the testis (PSRST),12 seminoma with signet-ring cell predominance,13 and a primary signet-ring cell germ-cell carcinoma of the testes (PSRGCT).14
Seminoma and PSRST variants generally are differentiated from adenocarcinoma by a lack of mucin production and the absence of keratin expression. PSRGCT is considered a
malignant somatic-type transformation of a testicular germ-cell tumor and germ-cell clonality is established by abnormal 12p chromosome through fluorescent in situ hybridization testing.13 PSRST is a benign tumor with excellent prognosis. Seminoma variants with signet-ring cell predominance and PSRGCT are managed as germ-cell tumors and are also considered to have good outcome. In contrast, testicular involvements from metastatic adenocarcinomas, including gastric cancers, are managed with multidisciplinary approach, including systemic chemotherapy. Despite this, patients have a poor outcome with a median survival of about 1 year.2
Conclusion
Advanced gastric adenocarcinoma can metastasize to the testes, and patients may present with a testicular swelling as an initial presentation. It is important to differentiate secondary testicular cancers from rare variants of primary testicular tumors because the prognosis and management vary substantially.
1. Global Burden of Disease Cancer Collaboration, Fitzmaurice C, Dicker D, et al. The global burden of cancer 2013. JAMA Oncol. 2015;1(4):505-527.
2. Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388(10060):2654-2664.
3. Qazi HA, Manikandan R, Foster CS, Fordham MV. Testicular metastasis from gastric carcinoma. Urology. 2006;68(4):890.e7-e8.
4. Civelek B, Aksoy S, Kos T, et al. Isolated testicular metastasis of gastric cancer. J Gastrointest Canc. 2012;43(suppl 1):S64-S66.
5. Li B, Cai H, Kang ZC, et al. Testicular metastasis from gastric carcinoma: a case report. World J of Gastroenterol. 2015;21(21):6764-6768.
6. National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: testicular cancer. https://seer.cancer.gov/statfacts/html/testis.html. Updated September 10, 2018. Accessed April 10, 2019.
7. Dutt N, Bates AW, Baithun SI. Secondary neoplasms of the male genital tract with different patterns of involvement in adults and children. Histopathology. 2000;37(4):323-331.
8. Haupt HM, Mann RB, Trump DL, Abeloff MD. Metastatic carcinoma involving the testis. Clinical and pathologic distinction from primary testicular neoplasms. Cancer.1984;54 (4):709-714.
9. Cheah CY, Wirth A, Seymour JF. Primary testicular lymphoma. Blood. 2014;123(4):486-493.
10. Kawamoto K, Miyoshi H, Yoshida N, Takizawa J, Sone H, Ohshima K. Clinicopathological, cytogenetic, and prognostic analysis of 131 myeloid sarcoma patients. Am J Surg Pathol. 2016;40(11):1473-1483.
11. Kulkarni KP, Marwaha RK, Trehan A, Bansal D. Testicular relapse in childhood acute lymphoblastic leukemia: the challenges and lessons. Indian J Cancer. 2010;47(2):134-138.
12. Kuo CY, Wen MC, Wang J, Jan YJ. Signet-ring stromal tumor of the testis: a case report and literature review. Hum Pathol. 2009;40(4):584-587.
13. Ulbright TM, Young RH. Seminoma with conspicuous signet ring cells: a rare, previously uncharacterized morphologic variant. Am J Surg Pathol. 2008;32(8):1175-1181.
14. Williamson SR, Kum JB, Shah SR, et al. Signet ring cell carcinoma of the testis: clinicopathologic and molecular evidence for germ cell tumor origin—a case report. Am J
Surg Pathol. 2012;36(2):311-315.
1. Global Burden of Disease Cancer Collaboration, Fitzmaurice C, Dicker D, et al. The global burden of cancer 2013. JAMA Oncol. 2015;1(4):505-527.
2. Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388(10060):2654-2664.
3. Qazi HA, Manikandan R, Foster CS, Fordham MV. Testicular metastasis from gastric carcinoma. Urology. 2006;68(4):890.e7-e8.
4. Civelek B, Aksoy S, Kos T, et al. Isolated testicular metastasis of gastric cancer. J Gastrointest Canc. 2012;43(suppl 1):S64-S66.
5. Li B, Cai H, Kang ZC, et al. Testicular metastasis from gastric carcinoma: a case report. World J of Gastroenterol. 2015;21(21):6764-6768.
6. National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: testicular cancer. https://seer.cancer.gov/statfacts/html/testis.html. Updated September 10, 2018. Accessed April 10, 2019.
7. Dutt N, Bates AW, Baithun SI. Secondary neoplasms of the male genital tract with different patterns of involvement in adults and children. Histopathology. 2000;37(4):323-331.
8. Haupt HM, Mann RB, Trump DL, Abeloff MD. Metastatic carcinoma involving the testis. Clinical and pathologic distinction from primary testicular neoplasms. Cancer.1984;54 (4):709-714.
9. Cheah CY, Wirth A, Seymour JF. Primary testicular lymphoma. Blood. 2014;123(4):486-493.
10. Kawamoto K, Miyoshi H, Yoshida N, Takizawa J, Sone H, Ohshima K. Clinicopathological, cytogenetic, and prognostic analysis of 131 myeloid sarcoma patients. Am J Surg Pathol. 2016;40(11):1473-1483.
11. Kulkarni KP, Marwaha RK, Trehan A, Bansal D. Testicular relapse in childhood acute lymphoblastic leukemia: the challenges and lessons. Indian J Cancer. 2010;47(2):134-138.
12. Kuo CY, Wen MC, Wang J, Jan YJ. Signet-ring stromal tumor of the testis: a case report and literature review. Hum Pathol. 2009;40(4):584-587.
13. Ulbright TM, Young RH. Seminoma with conspicuous signet ring cells: a rare, previously uncharacterized morphologic variant. Am J Surg Pathol. 2008;32(8):1175-1181.
14. Williamson SR, Kum JB, Shah SR, et al. Signet ring cell carcinoma of the testis: clinicopathologic and molecular evidence for germ cell tumor origin—a case report. Am J
Surg Pathol. 2012;36(2):311-315.
Paranoid delusions • ideas of reference • sleep problems • Dx?
THE CASE
A 58-year-old married Asian woman with no apparent psychiatric history presented to the emergency department (ED) in an acute state with ideas of reference, paranoid delusions, and multiple, vague somatic symptoms.
Based on information in the patient’s medical record, there had been suspicion of an underlying psychiatric disorder 6 years earlier. At that time, the patient had presented to her primary care provider (PCP) with vague somatic complaints, including diffuse body pain, dry cough, chills, weakness, facial numbness, and concerns about infections. A physical examination and work-up did not reveal the source of her complaints. Unfortunately, the patient’s complaints increased in number and severity over time.
Her medical records also indicated that she had been assessed for depression severity using the Patient Health Questionnaire-9 (PHQ-9), with scores of 0 (4 years earlier) and 3 (3 years earlier). The scores suggested that she was not suffering from depression.
During this time, the patient also saw a psychiatrist; however, it was unclear whether her symptoms met the criteria for delusional disorder or schizophrenia because she did not exhibit negative symptoms or sensory hallucinations. In addition, the patient was extremely high-functioning in the community—she participated in dance classes and other social events—and had the equivalent of a medical degree from another country. Based on chart review, when she went to the psychiatrist 3 years prior to her current presentation, there were no antipsychotics prescribed.
In the weeks leading up to her current presentation, the patient reported that she was struggling with sleep, sometimes spending days in bed and other times needing unspecified medication obtained overseas to help her sleep. Her husband reported that she had become increasingly withdrawn and stopped attending her dance classes and social events.
The patient believed the government was trying to poison her via radiation and that unknown people were trying to harm her via an online messaging application. Immediately prior to her arrival in the ED, the police were called to pull her away from oncoming traffic because she ran into the road to find the assassins that were stalking her.
During this recent visit to the ED, the patient presented with labile affect, rapid speech, and anxious and angry mood. She complained about darkened spots on her arm (inflicted through radiation by the media), vaginal bleeding, paralysis below the waist (although she was pacing around), and unspecific pain around her belly. Physical examination revealed no obvious signs of head trauma, intact extraocular movements, no coughing or wheezing, regular heart rate and rhythm, a nontender abdomen to palpation, and normal bowel sounds. No focal neurological deficits were appreciated. She had no rashes, bruises, or skin abrasions on her abdomen or upper extremities.
Continue to: The patient tried to...
The patient tried to leave the ED, saying that her third eye could see the radiation. She required medication and 4-point restraints.
Her initial laboratory work-up for heavy metals, Lyme disease, human immunodeficiency virus (HIV), syphilis, delirium, and drug use were all negative. She also underwent head imaging studies that were also found to be negative. Her mental status exam was notable for a tangential thought process, preservation of delusions with loose associations, labile mood, and dysphoric affect. The patient demonstrated limited insight and judgment, although she was fully oriented to person, place, and time, which suggested against delirium at the time of evaluation.
THE DIAGNOSIS
Based on the patient’s current presentation and in light of her medical history, the health care team arrived at a
DISCUSSION
Schizophrenia is a severe, lifelong mental disorder characterized by at least 2 symptoms of delusions, hallucinations, disorganized speech, disorganized or catatonic behavior, or negative symptoms for at least 6 months, with significant social, occupational, and functional deterioration. Current models attribute the neurodevelopmental deregulation of the brain in patients with schizophrenia to dopaminergic hyperactivity and hypofunction of the glutamatergic neurotransmitter system, explaining why its onset is usually in adolescence or young adulthood.1,2 However, 23% of patients present with symptoms after age 40, with 7% of patients being diagnosed between the ages of 51 and 60.3
Late-onset vs early-onset schizophrenia. LOS is often a missed diagnosis because the clinical presentation is different from early-onset schizophrenia (EOS). Although the prodromal symptoms of EOS and LOS are similar and include marked isolation that subsequently progresses to suspiciousness and ideas of reference,4 patients with EOS often also have prodromal negative symptoms. These prodromal negative symptoms associated with EOS may include loss of motivation, social passivity, and disorganized behavior. These symptoms are hypothesized to be caused by dopaminergic dysregulation in the anterior cingulate cortex. EOS is characterized by the patient experiencing more negative symptoms than LOS, which is characterized by the patient experiencing more positive symptoms.
Continue to: Patients with late-onset schizophrenia...
Patients with LOS typically do not exhibit negative symptoms because remodeling and myelination of neuronal circuitry matures by late adulthood, and thus becomes more resistant to impairment of motivational processes in the anterior cingulate gyrus.4,5,6
LOS is characterized by paranoid personality with predominantly positive symptoms, likely due to disruptions in
Other features of LOS include a high female:male ratio and symptomatic improvement with antipsychotics.7,10 Studies show that the LOS ratio of women:men can range from 2.2:1 to 22.5:1, which could be explained by the effect of dopaminergic-modulating estrogen from different sex-specific aging brain patterns.8,11,12 Finally, patients with LOS are less likely to seek care for sensory deficits than their age-equivalent counterparts.8,10 Fortunately, many of the characteristics of LOS predict good prognosis: Patients are usually female, display positive symptoms, have acute onset of symptoms, and are married with social support.10
Diagnosing LOS
LOS can be challenging to diagnose because of its atypical presentation compared with EOS, relative rarity in the population, and its propensity to be confused with progressive Alzheimer disease/dementia, delusional disorder, and major depressive disorder with psychotic features.3,6 Patients with no prior psychiatric history often do not have ready access to psychiatrists and depend on PCPs and other clinicians to identify mental health issues. A careful history, including familial involvement, utilization of the Montreal Cognitive Assessment (MoCA) test, and evaluation of environmental factors, are crucial to arriving at the proper diagnosis.
Continue to: Differential diagnosis
Differential diagnosis. When psychosis appears later in life, it is important to consider a broad differential (TABLE13-18), which includes the following:
Alzheimer disease. LOS can be easily differentiated from psychosis associated with Alzheimer disease or dementia through findings from neuropsychologic assessments and brain imaging. The initial first-line assessment for Alzheimer disease includes determining time course of daily living impairment and memory with follow-up brain imaging. Magnetic resonance imaging of patients with Alzheimer disease shows clear atrophy of the medial temporal lobes and general brain atrophy.19 Other than hypoperfusion in the frontal and temporal area, brain imaging of patients with LOS will not reveal any pathology.1

Delusional disorder and LOS are often more challenging to differentiate because symptoms can overlap, and many of the negative symptoms that would otherwise help clinicians diagnose schizophrenia in a younger population are absent in LOS. The milder symptoms of LOS may also lead clinicians to favor a diagnosis of delusional disorder. However, the following differences can help physicians differentiate between LOS and delusional disorder. Delusional disorder20-22:
- often will include paranoid beliefs, but these beliefs will not be bizarre, and the patient’s daily functioning will not be impaired, whereas patients with schizophrenia would have an increase in isolation and impairment in functioning that tends to be distinct from baseline.
- is more rare than schizophrenia. Delusional disorder has a prevalence of 0.05% to 0.1% compared to 1% for schizophrenia.
Major depressive disorder (MDD) with psychotic features. Major depressive disorder with psychotic features is an important differential to consider in this setting because the treatment intervention can be considerably different. Among patients who have MDD with psychotic features, a significant mood component is present, and treatment typically focuses on optimizing a selective serotonin reuptake inhibitor (SSRI); depending on severity, electroconvulsive therapy (ECT) also may be warranted.19
Continue to: For patients with LOS...
For patients with LOS, optimizing an antipsychotic medication is the typical course of treatment, and ECT would likely have less of an impact than it does with MDD with psychotic features.
Other. Finally, in an acute setting, other differential diagnoses for mental status changes (depending on clinical findings) might include:
- drug/medication use
- delirium
- nutrient deficiencies
- acute head trauma
- chronic subdural hematoma
- syphilis
- Lyme disease
- HIV encephalitis
- heavy metal toxicity.
Treatment involves antipsychotics—especially certain ones
Antipsychotic medications are utilized for the treatment of patients with LOS. A Cochrane review concluded that there are no trial-based evidence guidelines for the treatment of patients with LOS, and that physicians should continue with their current practice and use clinical judgment and prescribing patterns to guide their selection of antipsychotic medications.22,23 Pearlson et al24 found that 76% of patients with schizophrenia achieved at least partial remission and 48% achieved full remission with antipsychotic treatment.
The preferred treatment for patients with schizophrenia is low doses of newer antipsychotics (atypical or second-generation antipsychotics [SGAs]) because they are less likely to cause extrapyramidal symptoms/adverse effects than first-generation antipsychotics. Examples of SGAs include aripiprazole, risperidone, olanzapine, quetiapine, and ziprasidone.
Effective treatment for LOS includes antipsychotics at a quarter to one-half of the usual therapeutic doses. In patients with very late-onset schizophrenia, doses should be started at a tenth of therapeutic dose.1,23 Physicians should titrate up carefully, as needed.
Continue to: As with any significant mental illness...
As with any significant mental illness, to improve clinical outcomes, family support may help patients’ medication adherence and ensure they attend scheduled medical appointments.
Our patient was eventually stabilized on long-acting injectable risperidone, 25 mg, with improvement in symptoms. Unfortunately, she was not convinced that her symptoms were psychiatric in nature and did not continue with her medications as an outpatient.
The patient’s nonadherence to her medication regimen led to 2 more hospitalizations with similar presentations over the following 2 years. On her most recent discharge, she was stabilized on oral olanzapine, 10 mg every night at bedtime, with close outpatient follow-up and family education.
THE TAKEAWAY
The prodromal phase of patients with LOS is similar to patients with EOS and includes withdrawal and isolation from others, making it difficult for physicians to evaluate and treat patients. Patients with LOS predominantly experience positive symptoms that may include delusions and hallucinations. Brain imaging studies can help rule out progressive dementia diseases. A neuropsychological evaluation can assess the patient’s functional level and types of delusions, which helps to differentiate LOS from other late-age psychoses. Treatment with SGAs make for a good prognosis; however, this requires patients to be adherent to treatment.
CORRESPONDENCE
Sandy Chan, MD, Department of Internal Medicine, UMass Memorial Medical Center, 55N Lake Avenue, Worcester, MA 01605; [email protected]
1. Howard R, Rabins P, Seeman M, et al. Late-onset schizophrenia and very-late-onset schizophrenia-like psychosis: an international crisis. Am J Psychiatry 2000;157:172-178.
2. Pickard B. Progress in defining the biological causes of schizophrenia. Expert Rev Mol Med. 2011;13:e25.
3. Jeste D, Symonds L, Harris M, et al. Nondementia nonpraecox dementia praecox? Am J Geriatr Psychiatry. 1997;5:302-317.
4. Gourzis P, Katrivanou A, Beratis S. Symptomatology of the initial prodromal phase of schizophrenia. Schizophr Bull. 2002;28:415-429.
5. Dolan R, Fletcher P, Frith C, et al. Dopaminergic modulation of impaired cognitive activation in the anterior cingulate cortex in schizophrenia. Nature. 1995;378:180-182.
6. Skokou M, Katrivanou A, Andriopoulos I, et al. Active and prodromal phase symptomatology of young-onset and late-onset paranoid schizophrenia. Rev Psiquiatr Salud Ment. 2012;5:150-159.
7. Riecher-Rossler A, Loffler W, Munk-Jorgensen P. What do we really know about late-onset schizophrenia? Eur Arch Psychiatry Clin Neurosci. 1997;247:195-208.
8. Lubman D, Castle D. Late-onset schizophrenia: make the right diagnosis when psychosis emerges after age 60. Current Psychiatry. 2002;1:35-44.
9. Howard R, Castle D, Wessely S, et al. A comparative study of 470 cases of early-onset and late-onset schizophrenia. British Journal of Psychiatry. 1993;163:352-357.
10. Harris M, Jeste D. Late-onset schizophrenia: an overview. Schizophr Bull. 1988;14:39-55.
11. Castle D, Murray R. The epidemiology of late-onset schizophrenia. Schizophr Bull. 1993;19:691-700.
12. Lindamer L, Lohr J, Harris M, Jeste D. Gender, estrogen, and schizophrenia. Psychopharmacol Bull. 1997;33:221-228.
13. Gaudiano BA, Dalrymple KL, Zimmerman M. Prevalence and clinical characteristics of psychotic versus non-psychotic major depression in a general psychiatric outpatient clinic. Depress Anxiety. 2009;26:54-64.
14. Saha S, Chant D, Welham J, et al. A systematic review of the prevalence of schizophrenia. PLoS Med. 2005;2:e141.
15. Gao S, Hendrie H, Hall K. The relationships between age, sex, and the incidence of dementia and Alzheimer Disease. JAMA Psychiatry. 1998;55:809-815.
16. Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer Disease. Nature Reviews Neurology. 2011;7:137-152
17. Winokur G. Delusional Disorder (Paranoia). Comprehensive Psychiatry. 1977;18:511-521.
18. Scheltens P, Leys D, Huglo D, et al. Atrophy of medial temporal lobes on MRI in “probable” Alzheimer’s disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol, Neurosurg Psychiatry. 1992;55:967-972.
19. Copeland J, Dewey M, Scott A, et al. Schizophrenia and delusional disorder in older age: community prevalence, incidence, comorbidity, and outcome. Schizophr Bull. 1998;24:153-161.
20. Kendler K. Demography of paranoid psychosis (delusional disorder): a review and comparison with schizophrenia and affective illness. Arch Gen Psychiatry 1982;39:890-902.
21. McGrath J, Saha S, Chant D, et al. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol Rev. 2008;30:67.
22. Essali A, Ali G. Antipsychotic drug treatment for elderly people with late-onset schizophrenia. Cochrane Database Syst Rev. 2012;(2):CD004162.
23. Sweet R, Pollock B. New atypical antipsychotics- experience and utility in the elderly. Drugs Aging. 1998;12:115-127.
24. Pearlson G, Kreger L, Rabins P, et al. A chart review study of late-onset and early-onset schizophrenia. Am J Psychiatry.1989;146:1568-1574.
THE CASE
A 58-year-old married Asian woman with no apparent psychiatric history presented to the emergency department (ED) in an acute state with ideas of reference, paranoid delusions, and multiple, vague somatic symptoms.
Based on information in the patient’s medical record, there had been suspicion of an underlying psychiatric disorder 6 years earlier. At that time, the patient had presented to her primary care provider (PCP) with vague somatic complaints, including diffuse body pain, dry cough, chills, weakness, facial numbness, and concerns about infections. A physical examination and work-up did not reveal the source of her complaints. Unfortunately, the patient’s complaints increased in number and severity over time.
Her medical records also indicated that she had been assessed for depression severity using the Patient Health Questionnaire-9 (PHQ-9), with scores of 0 (4 years earlier) and 3 (3 years earlier). The scores suggested that she was not suffering from depression.
During this time, the patient also saw a psychiatrist; however, it was unclear whether her symptoms met the criteria for delusional disorder or schizophrenia because she did not exhibit negative symptoms or sensory hallucinations. In addition, the patient was extremely high-functioning in the community—she participated in dance classes and other social events—and had the equivalent of a medical degree from another country. Based on chart review, when she went to the psychiatrist 3 years prior to her current presentation, there were no antipsychotics prescribed.
In the weeks leading up to her current presentation, the patient reported that she was struggling with sleep, sometimes spending days in bed and other times needing unspecified medication obtained overseas to help her sleep. Her husband reported that she had become increasingly withdrawn and stopped attending her dance classes and social events.
The patient believed the government was trying to poison her via radiation and that unknown people were trying to harm her via an online messaging application. Immediately prior to her arrival in the ED, the police were called to pull her away from oncoming traffic because she ran into the road to find the assassins that were stalking her.
During this recent visit to the ED, the patient presented with labile affect, rapid speech, and anxious and angry mood. She complained about darkened spots on her arm (inflicted through radiation by the media), vaginal bleeding, paralysis below the waist (although she was pacing around), and unspecific pain around her belly. Physical examination revealed no obvious signs of head trauma, intact extraocular movements, no coughing or wheezing, regular heart rate and rhythm, a nontender abdomen to palpation, and normal bowel sounds. No focal neurological deficits were appreciated. She had no rashes, bruises, or skin abrasions on her abdomen or upper extremities.
Continue to: The patient tried to...
The patient tried to leave the ED, saying that her third eye could see the radiation. She required medication and 4-point restraints.
Her initial laboratory work-up for heavy metals, Lyme disease, human immunodeficiency virus (HIV), syphilis, delirium, and drug use were all negative. She also underwent head imaging studies that were also found to be negative. Her mental status exam was notable for a tangential thought process, preservation of delusions with loose associations, labile mood, and dysphoric affect. The patient demonstrated limited insight and judgment, although she was fully oriented to person, place, and time, which suggested against delirium at the time of evaluation.
THE DIAGNOSIS
Based on the patient’s current presentation and in light of her medical history, the health care team arrived at a
DISCUSSION
Schizophrenia is a severe, lifelong mental disorder characterized by at least 2 symptoms of delusions, hallucinations, disorganized speech, disorganized or catatonic behavior, or negative symptoms for at least 6 months, with significant social, occupational, and functional deterioration. Current models attribute the neurodevelopmental deregulation of the brain in patients with schizophrenia to dopaminergic hyperactivity and hypofunction of the glutamatergic neurotransmitter system, explaining why its onset is usually in adolescence or young adulthood.1,2 However, 23% of patients present with symptoms after age 40, with 7% of patients being diagnosed between the ages of 51 and 60.3
Late-onset vs early-onset schizophrenia. LOS is often a missed diagnosis because the clinical presentation is different from early-onset schizophrenia (EOS). Although the prodromal symptoms of EOS and LOS are similar and include marked isolation that subsequently progresses to suspiciousness and ideas of reference,4 patients with EOS often also have prodromal negative symptoms. These prodromal negative symptoms associated with EOS may include loss of motivation, social passivity, and disorganized behavior. These symptoms are hypothesized to be caused by dopaminergic dysregulation in the anterior cingulate cortex. EOS is characterized by the patient experiencing more negative symptoms than LOS, which is characterized by the patient experiencing more positive symptoms.
Continue to: Patients with late-onset schizophrenia...
Patients with LOS typically do not exhibit negative symptoms because remodeling and myelination of neuronal circuitry matures by late adulthood, and thus becomes more resistant to impairment of motivational processes in the anterior cingulate gyrus.4,5,6
LOS is characterized by paranoid personality with predominantly positive symptoms, likely due to disruptions in
Other features of LOS include a high female:male ratio and symptomatic improvement with antipsychotics.7,10 Studies show that the LOS ratio of women:men can range from 2.2:1 to 22.5:1, which could be explained by the effect of dopaminergic-modulating estrogen from different sex-specific aging brain patterns.8,11,12 Finally, patients with LOS are less likely to seek care for sensory deficits than their age-equivalent counterparts.8,10 Fortunately, many of the characteristics of LOS predict good prognosis: Patients are usually female, display positive symptoms, have acute onset of symptoms, and are married with social support.10
Diagnosing LOS
LOS can be challenging to diagnose because of its atypical presentation compared with EOS, relative rarity in the population, and its propensity to be confused with progressive Alzheimer disease/dementia, delusional disorder, and major depressive disorder with psychotic features.3,6 Patients with no prior psychiatric history often do not have ready access to psychiatrists and depend on PCPs and other clinicians to identify mental health issues. A careful history, including familial involvement, utilization of the Montreal Cognitive Assessment (MoCA) test, and evaluation of environmental factors, are crucial to arriving at the proper diagnosis.
Continue to: Differential diagnosis
Differential diagnosis. When psychosis appears later in life, it is important to consider a broad differential (TABLE13-18), which includes the following:
Alzheimer disease. LOS can be easily differentiated from psychosis associated with Alzheimer disease or dementia through findings from neuropsychologic assessments and brain imaging. The initial first-line assessment for Alzheimer disease includes determining time course of daily living impairment and memory with follow-up brain imaging. Magnetic resonance imaging of patients with Alzheimer disease shows clear atrophy of the medial temporal lobes and general brain atrophy.19 Other than hypoperfusion in the frontal and temporal area, brain imaging of patients with LOS will not reveal any pathology.1
Delusional disorder and LOS are often more challenging to differentiate because symptoms can overlap, and many of the negative symptoms that would otherwise help clinicians diagnose schizophrenia in a younger population are absent in LOS. The milder symptoms of LOS may also lead clinicians to favor a diagnosis of delusional disorder. However, the following differences can help physicians differentiate between LOS and delusional disorder. Delusional disorder20-22:
- often will include paranoid beliefs, but these beliefs will not be bizarre, and the patient’s daily functioning will not be impaired, whereas patients with schizophrenia would have an increase in isolation and impairment in functioning that tends to be distinct from baseline.
- is more rare than schizophrenia. Delusional disorder has a prevalence of 0.05% to 0.1% compared to 1% for schizophrenia.
Major depressive disorder (MDD) with psychotic features. Major depressive disorder with psychotic features is an important differential to consider in this setting because the treatment intervention can be considerably different. Among patients who have MDD with psychotic features, a significant mood component is present, and treatment typically focuses on optimizing a selective serotonin reuptake inhibitor (SSRI); depending on severity, electroconvulsive therapy (ECT) also may be warranted.19
Continue to: For patients with LOS...
For patients with LOS, optimizing an antipsychotic medication is the typical course of treatment, and ECT would likely have less of an impact than it does with MDD with psychotic features.
Other. Finally, in an acute setting, other differential diagnoses for mental status changes (depending on clinical findings) might include:
- drug/medication use
- delirium
- nutrient deficiencies
- acute head trauma
- chronic subdural hematoma
- syphilis
- Lyme disease
- HIV encephalitis
- heavy metal toxicity.
Treatment involves antipsychotics—especially certain ones
Antipsychotic medications are utilized for the treatment of patients with LOS. A Cochrane review concluded that there are no trial-based evidence guidelines for the treatment of patients with LOS, and that physicians should continue with their current practice and use clinical judgment and prescribing patterns to guide their selection of antipsychotic medications.22,23 Pearlson et al24 found that 76% of patients with schizophrenia achieved at least partial remission and 48% achieved full remission with antipsychotic treatment.
The preferred treatment for patients with schizophrenia is low doses of newer antipsychotics (atypical or second-generation antipsychotics [SGAs]) because they are less likely to cause extrapyramidal symptoms/adverse effects than first-generation antipsychotics. Examples of SGAs include aripiprazole, risperidone, olanzapine, quetiapine, and ziprasidone.
Effective treatment for LOS includes antipsychotics at a quarter to one-half of the usual therapeutic doses. In patients with very late-onset schizophrenia, doses should be started at a tenth of therapeutic dose.1,23 Physicians should titrate up carefully, as needed.
Continue to: As with any significant mental illness...
As with any significant mental illness, to improve clinical outcomes, family support may help patients’ medication adherence and ensure they attend scheduled medical appointments.
Our patient was eventually stabilized on long-acting injectable risperidone, 25 mg, with improvement in symptoms. Unfortunately, she was not convinced that her symptoms were psychiatric in nature and did not continue with her medications as an outpatient.
The patient’s nonadherence to her medication regimen led to 2 more hospitalizations with similar presentations over the following 2 years. On her most recent discharge, she was stabilized on oral olanzapine, 10 mg every night at bedtime, with close outpatient follow-up and family education.
THE TAKEAWAY
The prodromal phase of patients with LOS is similar to patients with EOS and includes withdrawal and isolation from others, making it difficult for physicians to evaluate and treat patients. Patients with LOS predominantly experience positive symptoms that may include delusions and hallucinations. Brain imaging studies can help rule out progressive dementia diseases. A neuropsychological evaluation can assess the patient’s functional level and types of delusions, which helps to differentiate LOS from other late-age psychoses. Treatment with SGAs make for a good prognosis; however, this requires patients to be adherent to treatment.
CORRESPONDENCE
Sandy Chan, MD, Department of Internal Medicine, UMass Memorial Medical Center, 55N Lake Avenue, Worcester, MA 01605; [email protected]
THE CASE
A 58-year-old married Asian woman with no apparent psychiatric history presented to the emergency department (ED) in an acute state with ideas of reference, paranoid delusions, and multiple, vague somatic symptoms.
Based on information in the patient’s medical record, there had been suspicion of an underlying psychiatric disorder 6 years earlier. At that time, the patient had presented to her primary care provider (PCP) with vague somatic complaints, including diffuse body pain, dry cough, chills, weakness, facial numbness, and concerns about infections. A physical examination and work-up did not reveal the source of her complaints. Unfortunately, the patient’s complaints increased in number and severity over time.
Her medical records also indicated that she had been assessed for depression severity using the Patient Health Questionnaire-9 (PHQ-9), with scores of 0 (4 years earlier) and 3 (3 years earlier). The scores suggested that she was not suffering from depression.
During this time, the patient also saw a psychiatrist; however, it was unclear whether her symptoms met the criteria for delusional disorder or schizophrenia because she did not exhibit negative symptoms or sensory hallucinations. In addition, the patient was extremely high-functioning in the community—she participated in dance classes and other social events—and had the equivalent of a medical degree from another country. Based on chart review, when she went to the psychiatrist 3 years prior to her current presentation, there were no antipsychotics prescribed.
In the weeks leading up to her current presentation, the patient reported that she was struggling with sleep, sometimes spending days in bed and other times needing unspecified medication obtained overseas to help her sleep. Her husband reported that she had become increasingly withdrawn and stopped attending her dance classes and social events.
The patient believed the government was trying to poison her via radiation and that unknown people were trying to harm her via an online messaging application. Immediately prior to her arrival in the ED, the police were called to pull her away from oncoming traffic because she ran into the road to find the assassins that were stalking her.
During this recent visit to the ED, the patient presented with labile affect, rapid speech, and anxious and angry mood. She complained about darkened spots on her arm (inflicted through radiation by the media), vaginal bleeding, paralysis below the waist (although she was pacing around), and unspecific pain around her belly. Physical examination revealed no obvious signs of head trauma, intact extraocular movements, no coughing or wheezing, regular heart rate and rhythm, a nontender abdomen to palpation, and normal bowel sounds. No focal neurological deficits were appreciated. She had no rashes, bruises, or skin abrasions on her abdomen or upper extremities.
Continue to: The patient tried to...
The patient tried to leave the ED, saying that her third eye could see the radiation. She required medication and 4-point restraints.
Her initial laboratory work-up for heavy metals, Lyme disease, human immunodeficiency virus (HIV), syphilis, delirium, and drug use were all negative. She also underwent head imaging studies that were also found to be negative. Her mental status exam was notable for a tangential thought process, preservation of delusions with loose associations, labile mood, and dysphoric affect. The patient demonstrated limited insight and judgment, although she was fully oriented to person, place, and time, which suggested against delirium at the time of evaluation.
THE DIAGNOSIS
Based on the patient’s current presentation and in light of her medical history, the health care team arrived at a
DISCUSSION
Schizophrenia is a severe, lifelong mental disorder characterized by at least 2 symptoms of delusions, hallucinations, disorganized speech, disorganized or catatonic behavior, or negative symptoms for at least 6 months, with significant social, occupational, and functional deterioration. Current models attribute the neurodevelopmental deregulation of the brain in patients with schizophrenia to dopaminergic hyperactivity and hypofunction of the glutamatergic neurotransmitter system, explaining why its onset is usually in adolescence or young adulthood.1,2 However, 23% of patients present with symptoms after age 40, with 7% of patients being diagnosed between the ages of 51 and 60.3
Late-onset vs early-onset schizophrenia. LOS is often a missed diagnosis because the clinical presentation is different from early-onset schizophrenia (EOS). Although the prodromal symptoms of EOS and LOS are similar and include marked isolation that subsequently progresses to suspiciousness and ideas of reference,4 patients with EOS often also have prodromal negative symptoms. These prodromal negative symptoms associated with EOS may include loss of motivation, social passivity, and disorganized behavior. These symptoms are hypothesized to be caused by dopaminergic dysregulation in the anterior cingulate cortex. EOS is characterized by the patient experiencing more negative symptoms than LOS, which is characterized by the patient experiencing more positive symptoms.
Continue to: Patients with late-onset schizophrenia...
Patients with LOS typically do not exhibit negative symptoms because remodeling and myelination of neuronal circuitry matures by late adulthood, and thus becomes more resistant to impairment of motivational processes in the anterior cingulate gyrus.4,5,6
LOS is characterized by paranoid personality with predominantly positive symptoms, likely due to disruptions in
Other features of LOS include a high female:male ratio and symptomatic improvement with antipsychotics.7,10 Studies show that the LOS ratio of women:men can range from 2.2:1 to 22.5:1, which could be explained by the effect of dopaminergic-modulating estrogen from different sex-specific aging brain patterns.8,11,12 Finally, patients with LOS are less likely to seek care for sensory deficits than their age-equivalent counterparts.8,10 Fortunately, many of the characteristics of LOS predict good prognosis: Patients are usually female, display positive symptoms, have acute onset of symptoms, and are married with social support.10
Diagnosing LOS
LOS can be challenging to diagnose because of its atypical presentation compared with EOS, relative rarity in the population, and its propensity to be confused with progressive Alzheimer disease/dementia, delusional disorder, and major depressive disorder with psychotic features.3,6 Patients with no prior psychiatric history often do not have ready access to psychiatrists and depend on PCPs and other clinicians to identify mental health issues. A careful history, including familial involvement, utilization of the Montreal Cognitive Assessment (MoCA) test, and evaluation of environmental factors, are crucial to arriving at the proper diagnosis.
Continue to: Differential diagnosis
Differential diagnosis. When psychosis appears later in life, it is important to consider a broad differential (TABLE13-18), which includes the following:
Alzheimer disease. LOS can be easily differentiated from psychosis associated with Alzheimer disease or dementia through findings from neuropsychologic assessments and brain imaging. The initial first-line assessment for Alzheimer disease includes determining time course of daily living impairment and memory with follow-up brain imaging. Magnetic resonance imaging of patients with Alzheimer disease shows clear atrophy of the medial temporal lobes and general brain atrophy.19 Other than hypoperfusion in the frontal and temporal area, brain imaging of patients with LOS will not reveal any pathology.1
Delusional disorder and LOS are often more challenging to differentiate because symptoms can overlap, and many of the negative symptoms that would otherwise help clinicians diagnose schizophrenia in a younger population are absent in LOS. The milder symptoms of LOS may also lead clinicians to favor a diagnosis of delusional disorder. However, the following differences can help physicians differentiate between LOS and delusional disorder. Delusional disorder20-22:
- often will include paranoid beliefs, but these beliefs will not be bizarre, and the patient’s daily functioning will not be impaired, whereas patients with schizophrenia would have an increase in isolation and impairment in functioning that tends to be distinct from baseline.
- is more rare than schizophrenia. Delusional disorder has a prevalence of 0.05% to 0.1% compared to 1% for schizophrenia.
Major depressive disorder (MDD) with psychotic features. Major depressive disorder with psychotic features is an important differential to consider in this setting because the treatment intervention can be considerably different. Among patients who have MDD with psychotic features, a significant mood component is present, and treatment typically focuses on optimizing a selective serotonin reuptake inhibitor (SSRI); depending on severity, electroconvulsive therapy (ECT) also may be warranted.19
Continue to: For patients with LOS...
For patients with LOS, optimizing an antipsychotic medication is the typical course of treatment, and ECT would likely have less of an impact than it does with MDD with psychotic features.
Other. Finally, in an acute setting, other differential diagnoses for mental status changes (depending on clinical findings) might include:
- drug/medication use
- delirium
- nutrient deficiencies
- acute head trauma
- chronic subdural hematoma
- syphilis
- Lyme disease
- HIV encephalitis
- heavy metal toxicity.
Treatment involves antipsychotics—especially certain ones
Antipsychotic medications are utilized for the treatment of patients with LOS. A Cochrane review concluded that there are no trial-based evidence guidelines for the treatment of patients with LOS, and that physicians should continue with their current practice and use clinical judgment and prescribing patterns to guide their selection of antipsychotic medications.22,23 Pearlson et al24 found that 76% of patients with schizophrenia achieved at least partial remission and 48% achieved full remission with antipsychotic treatment.
The preferred treatment for patients with schizophrenia is low doses of newer antipsychotics (atypical or second-generation antipsychotics [SGAs]) because they are less likely to cause extrapyramidal symptoms/adverse effects than first-generation antipsychotics. Examples of SGAs include aripiprazole, risperidone, olanzapine, quetiapine, and ziprasidone.
Effective treatment for LOS includes antipsychotics at a quarter to one-half of the usual therapeutic doses. In patients with very late-onset schizophrenia, doses should be started at a tenth of therapeutic dose.1,23 Physicians should titrate up carefully, as needed.
Continue to: As with any significant mental illness...
As with any significant mental illness, to improve clinical outcomes, family support may help patients’ medication adherence and ensure they attend scheduled medical appointments.
Our patient was eventually stabilized on long-acting injectable risperidone, 25 mg, with improvement in symptoms. Unfortunately, she was not convinced that her symptoms were psychiatric in nature and did not continue with her medications as an outpatient.
The patient’s nonadherence to her medication regimen led to 2 more hospitalizations with similar presentations over the following 2 years. On her most recent discharge, she was stabilized on oral olanzapine, 10 mg every night at bedtime, with close outpatient follow-up and family education.
THE TAKEAWAY
The prodromal phase of patients with LOS is similar to patients with EOS and includes withdrawal and isolation from others, making it difficult for physicians to evaluate and treat patients. Patients with LOS predominantly experience positive symptoms that may include delusions and hallucinations. Brain imaging studies can help rule out progressive dementia diseases. A neuropsychological evaluation can assess the patient’s functional level and types of delusions, which helps to differentiate LOS from other late-age psychoses. Treatment with SGAs make for a good prognosis; however, this requires patients to be adherent to treatment.
CORRESPONDENCE
Sandy Chan, MD, Department of Internal Medicine, UMass Memorial Medical Center, 55N Lake Avenue, Worcester, MA 01605; [email protected]
1. Howard R, Rabins P, Seeman M, et al. Late-onset schizophrenia and very-late-onset schizophrenia-like psychosis: an international crisis. Am J Psychiatry 2000;157:172-178.
2. Pickard B. Progress in defining the biological causes of schizophrenia. Expert Rev Mol Med. 2011;13:e25.
3. Jeste D, Symonds L, Harris M, et al. Nondementia nonpraecox dementia praecox? Am J Geriatr Psychiatry. 1997;5:302-317.
4. Gourzis P, Katrivanou A, Beratis S. Symptomatology of the initial prodromal phase of schizophrenia. Schizophr Bull. 2002;28:415-429.
5. Dolan R, Fletcher P, Frith C, et al. Dopaminergic modulation of impaired cognitive activation in the anterior cingulate cortex in schizophrenia. Nature. 1995;378:180-182.
6. Skokou M, Katrivanou A, Andriopoulos I, et al. Active and prodromal phase symptomatology of young-onset and late-onset paranoid schizophrenia. Rev Psiquiatr Salud Ment. 2012;5:150-159.
7. Riecher-Rossler A, Loffler W, Munk-Jorgensen P. What do we really know about late-onset schizophrenia? Eur Arch Psychiatry Clin Neurosci. 1997;247:195-208.
8. Lubman D, Castle D. Late-onset schizophrenia: make the right diagnosis when psychosis emerges after age 60. Current Psychiatry. 2002;1:35-44.
9. Howard R, Castle D, Wessely S, et al. A comparative study of 470 cases of early-onset and late-onset schizophrenia. British Journal of Psychiatry. 1993;163:352-357.
10. Harris M, Jeste D. Late-onset schizophrenia: an overview. Schizophr Bull. 1988;14:39-55.
11. Castle D, Murray R. The epidemiology of late-onset schizophrenia. Schizophr Bull. 1993;19:691-700.
12. Lindamer L, Lohr J, Harris M, Jeste D. Gender, estrogen, and schizophrenia. Psychopharmacol Bull. 1997;33:221-228.
13. Gaudiano BA, Dalrymple KL, Zimmerman M. Prevalence and clinical characteristics of psychotic versus non-psychotic major depression in a general psychiatric outpatient clinic. Depress Anxiety. 2009;26:54-64.
14. Saha S, Chant D, Welham J, et al. A systematic review of the prevalence of schizophrenia. PLoS Med. 2005;2:e141.
15. Gao S, Hendrie H, Hall K. The relationships between age, sex, and the incidence of dementia and Alzheimer Disease. JAMA Psychiatry. 1998;55:809-815.
16. Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer Disease. Nature Reviews Neurology. 2011;7:137-152
17. Winokur G. Delusional Disorder (Paranoia). Comprehensive Psychiatry. 1977;18:511-521.
18. Scheltens P, Leys D, Huglo D, et al. Atrophy of medial temporal lobes on MRI in “probable” Alzheimer’s disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol, Neurosurg Psychiatry. 1992;55:967-972.
19. Copeland J, Dewey M, Scott A, et al. Schizophrenia and delusional disorder in older age: community prevalence, incidence, comorbidity, and outcome. Schizophr Bull. 1998;24:153-161.
20. Kendler K. Demography of paranoid psychosis (delusional disorder): a review and comparison with schizophrenia and affective illness. Arch Gen Psychiatry 1982;39:890-902.
21. McGrath J, Saha S, Chant D, et al. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol Rev. 2008;30:67.
22. Essali A, Ali G. Antipsychotic drug treatment for elderly people with late-onset schizophrenia. Cochrane Database Syst Rev. 2012;(2):CD004162.
23. Sweet R, Pollock B. New atypical antipsychotics- experience and utility in the elderly. Drugs Aging. 1998;12:115-127.
24. Pearlson G, Kreger L, Rabins P, et al. A chart review study of late-onset and early-onset schizophrenia. Am J Psychiatry.1989;146:1568-1574.
1. Howard R, Rabins P, Seeman M, et al. Late-onset schizophrenia and very-late-onset schizophrenia-like psychosis: an international crisis. Am J Psychiatry 2000;157:172-178.
2. Pickard B. Progress in defining the biological causes of schizophrenia. Expert Rev Mol Med. 2011;13:e25.
3. Jeste D, Symonds L, Harris M, et al. Nondementia nonpraecox dementia praecox? Am J Geriatr Psychiatry. 1997;5:302-317.
4. Gourzis P, Katrivanou A, Beratis S. Symptomatology of the initial prodromal phase of schizophrenia. Schizophr Bull. 2002;28:415-429.
5. Dolan R, Fletcher P, Frith C, et al. Dopaminergic modulation of impaired cognitive activation in the anterior cingulate cortex in schizophrenia. Nature. 1995;378:180-182.
6. Skokou M, Katrivanou A, Andriopoulos I, et al. Active and prodromal phase symptomatology of young-onset and late-onset paranoid schizophrenia. Rev Psiquiatr Salud Ment. 2012;5:150-159.
7. Riecher-Rossler A, Loffler W, Munk-Jorgensen P. What do we really know about late-onset schizophrenia? Eur Arch Psychiatry Clin Neurosci. 1997;247:195-208.
8. Lubman D, Castle D. Late-onset schizophrenia: make the right diagnosis when psychosis emerges after age 60. Current Psychiatry. 2002;1:35-44.
9. Howard R, Castle D, Wessely S, et al. A comparative study of 470 cases of early-onset and late-onset schizophrenia. British Journal of Psychiatry. 1993;163:352-357.
10. Harris M, Jeste D. Late-onset schizophrenia: an overview. Schizophr Bull. 1988;14:39-55.
11. Castle D, Murray R. The epidemiology of late-onset schizophrenia. Schizophr Bull. 1993;19:691-700.
12. Lindamer L, Lohr J, Harris M, Jeste D. Gender, estrogen, and schizophrenia. Psychopharmacol Bull. 1997;33:221-228.
13. Gaudiano BA, Dalrymple KL, Zimmerman M. Prevalence and clinical characteristics of psychotic versus non-psychotic major depression in a general psychiatric outpatient clinic. Depress Anxiety. 2009;26:54-64.
14. Saha S, Chant D, Welham J, et al. A systematic review of the prevalence of schizophrenia. PLoS Med. 2005;2:e141.
15. Gao S, Hendrie H, Hall K. The relationships between age, sex, and the incidence of dementia and Alzheimer Disease. JAMA Psychiatry. 1998;55:809-815.
16. Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer Disease. Nature Reviews Neurology. 2011;7:137-152
17. Winokur G. Delusional Disorder (Paranoia). Comprehensive Psychiatry. 1977;18:511-521.
18. Scheltens P, Leys D, Huglo D, et al. Atrophy of medial temporal lobes on MRI in “probable” Alzheimer’s disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol, Neurosurg Psychiatry. 1992;55:967-972.
19. Copeland J, Dewey M, Scott A, et al. Schizophrenia and delusional disorder in older age: community prevalence, incidence, comorbidity, and outcome. Schizophr Bull. 1998;24:153-161.
20. Kendler K. Demography of paranoid psychosis (delusional disorder): a review and comparison with schizophrenia and affective illness. Arch Gen Psychiatry 1982;39:890-902.
21. McGrath J, Saha S, Chant D, et al. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol Rev. 2008;30:67.
22. Essali A, Ali G. Antipsychotic drug treatment for elderly people with late-onset schizophrenia. Cochrane Database Syst Rev. 2012;(2):CD004162.
23. Sweet R, Pollock B. New atypical antipsychotics- experience and utility in the elderly. Drugs Aging. 1998;12:115-127.
24. Pearlson G, Kreger L, Rabins P, et al. A chart review study of late-onset and early-onset schizophrenia. Am J Psychiatry.1989;146:1568-1574.

A Veteran Presenting With Altered Mental Status and Clonus
►Zachary Reese, MD, Chief Medical Resident, VABHS and Beth Israel Deaconess Medical Center (BIDMC):Dr. Weller, the differential diagnosis for altered mental status is quite broad. How does the presence of clonus change or focus your approach to altered mental status?
►Jason Weller, MD, Instructor of Neurology, Boston Medical Center (BMC) and VABHS:The presence of clonus does not significantly narrow the differential. It does, however, suggest a central component to the patient’s altered mental status. Specifically, it implies that the underlying process, whether systemic or neurologic, interferes with central nervous system (CNS) control of the neuromuscular system.1 The differential is still quite broad and includes metabolic derangements (eg, uremia, electrolyte disturbances, hypercarbia, and thyroid dysfunction), medication toxicity from olanzapine or duloxetine, and vascular processes (eg, CNS vasculitis). Infectious etiologies, both within the CNS and systemically, can cause encephalopathy, as can autoimmune processes, such as immune-mediated encephalitis. Finally, primary neurologic conditions such as myoclonic epilepsy can be considered. Given the patient’s medical history, serotonin syndrome must be considered.
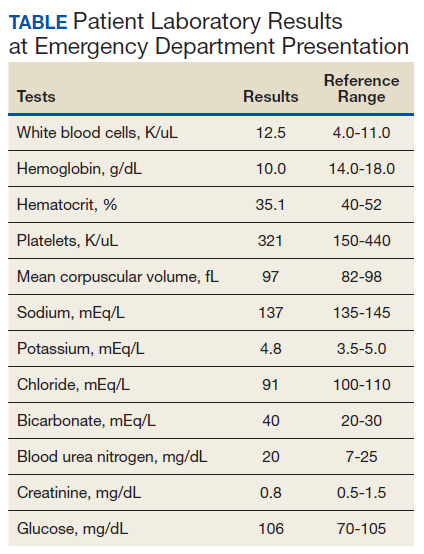
►Dr. Reese: Given the concern for serotonin syndrome, the admitting medical team discontinued the patient’s duloxetine. Dr. Weller, what is the pathophysiology of serotonin syndrome, and how is it diagnosed?
►Dr. Weller: Serotonin is ubiquitous throughout the body and brain. Serotonin syndrome is caused by excess endogenous or exogenous serotonin, and this is usually caused by a variety of medications. The symptoms range from tachycardia, agitation, and diaphoresis to sustained clonus, hyperthermia, and shock.2,3 The extent of serotonin syndrome is typically thought to reflect the degree of serotonergic activity.4
Serotonin syndrome is a clinical diagnosis. While there are no tests that can confirm the diagnosis, the Hunter criteria can be used to assist with making the diagnosis.5 Per the Hunter criteria, a patient can be diagnosed with serotonin syndrome if they have taken a serotonergic agent and have at least 1 of the following: spontaneous clonus, inducible or ocular clonus with agitation or diaphoresis, tremor and hyperreflexia, or hypertonia with fever and clonus. This patient had taken duloxetine and had inducible clonus and diaphoresis, thus suggesting a diagnosis of serotonin syndrome.
►Dr. Reese: Aside from selective serotonin reuptake inhibitors (SSRIs), are there other medications that we typically prescribe that can cause serotonin syndrome?
►Dr. Weller: In addition to SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs), other commonly prescribed medications that can cause serotonin syndrome are 5-HT3 antagonists (eg, ondansetron), 5-HT agonists (eg, triptans), and opioids (eg, fentanyl and tramadol). There are also case reports of atypical antipsychotics (eg, olanzapine) causing serotonin syndrome because of their antagonism of the 5-HT2 and 5-HT3 receptors.2 Additionally, linezolid is commonly overlooked as a cause of serotonin syndrome given its action as a monoamine oxidase inhibitor.4 In this patient, it would be prudent to discontinue olanzapine and duloxetine.
►Dr. Reese: Duloxetine, olanzapine, and buprenorphine/naloxone were discontinuedgiven concern for serotonin syndrome. Although there are not strong data that buprenorphine/ naloxone can cause serotonin syndrome, the team discontinued the medication in case it might be contributing to the patient’s encephalopathy, while closely monitoring the patient for withdrawal. There was a rapid improvement in the patient’s symptoms over the 24 hours after discontinuation of the 3 medications.
As part of the initial workup, the patient received a computed tomography (CT) scan of his chest to follow up pulmonary nodules identified 16 months prior. The CT scan showed interval growth of the pulmonary nodules in the right lower lobe to 2 cm with extension into the major fissure, which was concerning for malignancy. Plans were made for an outpatient positron emission tomography (PET) scan after hospital discharge.
Dr. Schlechter and Dr. Rangachari, what factors can help us determine whether or not further workup of a malignancy should occur before discharge or can be deferred to the outpatient setting?
►Benjamin Schlechter, MD, Instructor in Medicine, BIDMC; and Deepa Rangachari, MD, Assistant Professor of Medicine, BIDMC: Key considerations in this domain include rapidity of growth and any threat to critical end-organ function (ie, brain, heart, lungs, kidney, and liver). If the malignancy is bulky and/or rapidly progressing to the point that the patient has significant symptoms burden and/or end-organ dysfunction, then initiating the evaluation as an inpatient may be necessary. For suspected intrathoracic malignancies, considering whether this may be a high-grade process (ie, small cell lung cancer) is often a vital branch point. Key considerations in this regard are the following: Is it a bulky central tumor? Is there evidence of widespread metastatic disease, an obstructing mass, and/or tumor lysis? One final and critical aspect to consider is whether there are any patient- specific barriers to timely and reliable outpatient follow-up. If there is no evidence of rapid progression, bulky disease with threatened end-organ involvement, and/or issues with timely and reliable follow-up, then outpatient evaluation is often the best approach to ensure a comprehensive and well-coordinated effort on the patient’s behalf.
►Dr. Reese: Buprenorphine/naloxone was restarted without return of the symptoms. The patient was discharged home with an outpatient PET scan scheduled the following week. Unfortunately, the patient was unable to keep this appointment. Three weeks after hospital discharge, the patient presented again to the emergency department with gradually worsening altered mental status, confusion, visual hallucinations, and myoclonic jerking of the arms and legs. Medication adherence was confirmed by the patient’s wife, resulting in a low concern for serotonin syndrome. Physical examination revealed confusion, dysarthria, diffuse, arrhythmic, myoclonic jerking in all extremities, asterixis in the upper extremities, and hyperreflexia.
A CT scan of the brain did not reveal an intracranial process. A spot electroencephalograph (EEG) and magnetic resonance image (MRI) of the brain were obtained. Dr. Weller, what is the utility of spot EEG vs 24-hour EEG? When might we choose one over the other?
►Dr. Weller: If a patient is persistently altered, then a spot EEG would be sufficient to capture a seizure if that is what is causing the patient’s altered mental status. However, if the patient’s mental status is waxing and waning, then that may warrant a 24-hour EEG because the patient may need to be monitored for longer periods to capture an event that is causing intermittent alterations in mental status.6 Additionally, patients who are acutely ill may require long-term monitoring for the purpose of treatment and outcome management.
►Dr. Reese: The spot EEG showed nearly continuous generalized slowing indicative of a diffuse encephalopathy. The MRI of the brain showed scattered, nonspecific periventricular T2 hyperintense foci, suggestive of advanced chronic microvascular ischemic changes.
A PET CT was obtained and revealed mildly fluorodeoxyglucose (FDG)-avid, enlarging nodules within the right lower lobe, which was suspicious for malignancy. There were no other areas of FDG avidity on the PET scan. Valproic acid was initiated for treatment of myoclonus with transition to clonazepam when no improvement was seen. After starting clonazepam, the patient’s condition stabilized.
Dr. Weller, given the additional history, how has your differential diagnosis changed?
►Dr. Weller: Given the patient’s laboratory findings, we can be quite sure that there is not a contributing metabolic process. The findings suggestive of metastatic cancer, along with the profound neurologic changes, are most concerning for a paraneoplastic syndrome. I would suggest biopsy and consideration of a lumbar puncture. One can also send serum markers, including a paraneoplastic antibody panel.
►Dr. Reese: Biopsy of the mass in his right lower lobe revealed squamous cell lung cancer. Dr. Schlechter and Dr. Rangachari, do you have a framework for the different forms of lung cancer?
►Dr. Schlechter/Dr. Rangachari: The 2 broad categories of lung cancer are small cell and non-small cell (NSCLC). Small cell lung cancer has a tight association with tobacco exposure and is often clinically defined by rapid, bulky progression (ie, weeks to months).7,8 NSCLCs are also commonly seen in those with tobacco exposure, though not always. The main subgroups in this category are adenocarcinoma and squamous cell carcinoma. These cancers often evolve at a slower pace (ie, months to years).8 While small cell lung cancers are highgrade tumors and exquisitely sensitive to chemotherapy and radiation, NSCLCs tend to be less responsive to such therapies. The staging evaluation for either entity is the same and consists of defining localized vs metastatic disease.
►Dr. Reese: Because this patient had an MRI and PET scan that were both negative for metastatic disease, can we assume that this patient had stage I NSCLC?
►Dr. Schlechter/Dr. Rangachari: Not necessarily. While PET and MRI brain are exceptionally helpful in detecting distant metastases, they may over- or underestimate intrathoracic lymph node involvement by as much as 20%.9 As such, dedicated lymph node staging—either via bronchoscopy (endobronchial ultrasound) or surgically (mediastinoscopy) is indicated as lymph node involvement can significantly alter the stage, prognosis, and optimal therapeutic approach.10,11
►Dr. Reese: After this diagnosis was made, the teams caring for this patient attributed his altered mental status to a paraneoplastic syndrome. What is a paraneoplastic syndrome, and how does a paraneoplastic syndrome from malignancy present? Does its presence worsen a patient’s prognosis?

►Dr. Schlechter/Dr. Rangachari: A paraneoplastic syndrome is defined by an immunologic response to the cancer that ends up erroneously targeting self-antigens. Paraneoplastic syndromes are associated with a broad array of clinical findings—from endocrinopathy to encephalopathy—and certain neoplasms are more commonly associated with these syndromes than others (eg, small cell lung cancer and thymoma). Further, severity and onset of a paraneoplastic syndrome does not correlate with the burden of visible disease—and the syndrome may predate the cancer diagnosis by months to years.11 While treatment of the cancer affords the best hope of resolving the paraneoplastic syndrome, the cancer and the paraneoplastic process may have a discordant trajectory, with the paraneoplastic syndrome persisting even after the cancer is maximally treated. Although one might assume that paraneoplastic syndromes portend worse outcomes, in some cases, a presentation with the paraneoplastic syndrome may afford sooner detection of an otherwise occult/asymptomatic malignancy.
►Dr. Reese: The following week, the serum paraneoplastic antibody panel that tested for anti-Yo antibody, anti-Ri antibody,and anti-Hu antibody came back negative. Dr. Weller, what does this mean? Since we have yet to obtain a lumbar puncture, might his symptoms still be caused by a paraneoplastic syndrome?
►Dr. Weller: The negative serum test just means that he does not have antibodies to those 3 antibodies. There are now over 30 different paraneoplastic antibodies that have been discovered, and there are always more that are being discovered. So this negative test result does not exclude a paraneoplastic syndrome in the appropriate clinical context.12 Furthermore, the sensitivity and specificity for certain antibodies are different based upon source fluid, and cerebrospinal fluid testing would provide more diagnostic clarity. A negative test for paraneoplastic syndrome, by itself, would similarly not exclude a paraneoplastic syndrome. Often, empiric treatment is the best diagnostic option for paraneoplastic and autoimmune encephalopathies.
►Dr. Reese: The following week, the patient was discharged to rehabilitation with clonazepam for his symptoms and a scheduled follow-up. Given the patient’s frailty and medical comorbidities, thoracic surgery recommended consultation with radiation oncology. Dr. Schlechter and Dr. Rangachari, when do we decide to use radiation vs chemotherapy for someone with lung cancer?
►Dr. Schlechter/Dr. Rangachari: Patients with early stage, nonmetastatic NSCLC may not always be candidates for surgical resection on the basis of pulmonary function, other medical comorbidities (as in this case), anatomic considerations, and/or patient preference. In these cases, if there is lung-limited disease without lymph node involvement (ie, stage I/II NSCLC) and the patient is not felt to be an operative candidate, then alternatives to surgery include either radiation or ablation.13,14 As we care for an aging and comorbid population, evolving evidence suggests that well-selected patients with early stage disease undergoing these nonoperative approaches have roughly equivalent outcomes to those undergoing conventional surgical resection.13 In such cases, multidisciplinary consultation with a team having dedicated expertise in these various operative and nonoperative modalities is essential.
►Dr. Reese: The patient followed up with radiation oncology for consideration of radiation treatment, but his simulation CT scan showed some ground-glass opacity that were concerning for inflammation vs infection. The patient’s case was discussed at the multidisciplinary tumor board, and it was determined to treat him with antibiotics for a possible pneumonia before proceeding with radiation therapy. After he completed antibiotic treatment, he underwent 10 fractions of radiation treatment, which he tolerated well.
1. Kojovic M, Cordivari C, Bhatia K. Myoclonic disorders: a practical approach for diagnosis and treatment. Ther Adv Neurol Disord. 2011;4(1):47-62.
2. Volpi-Abadie J, Kaye AM, Kaye AD. Serotonin syndrome. Ochsner J. 2013;13(4):533-540.
3. Arora B, Kannikeswaran N. The serotonin syndrome-the need for physician’s awareness. Int J Emerg Med. 2010;3(4):373-377.
4. Boyer EW, Shannon M. The serotonin syndrome [published correction appears in N Engl J Med. 2007;356(23):2437 and N Engl J Med. 2009;361(17):1714]. N Engl J Med.
2005;352(11):1112-1120.
5. Dunkley EJC, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM.
2003;96(9):635-642.
6. Nordli DR Jr. Usefulness of video-EEG monitoring. Epilepsia. 2006;47(suppl 1):26-30.
7. Ettinger DS, Aisner J. Changing face of small-cell lung cancer: real and artifact. J Clin Oncol. 2006;24(28):4526-4527.
8. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10(9):1243-1260.
9. Cerfolio RJ, Bryant AS, Ojha B, Eloubeidi M. Improving the inaccuracies of clinical staging of patients with NSCLC: a prospective trial. Ann Thorac Surg. 2005;80(4):1207-1214.
10. El-Osta H, Jani P, Mansour A, Rascoe P, Jafri S. Endobronchial ultrasound for nodal staging of patients with non-smallcell lung cancer with radiologically normal mediastinum. A meta-analysis. Ann Am Thorac Soc. 2018;15(7):864-874.
11. Darnell RB, Posner JB. Paraneoplastic syndromes involving the nervous system. N Engl J Med. 2003;349(16):1543-1554.
12. McKeon A. Autoimmune Encephalopathies and Dementias. Continuum (Minneap Minn). 2016;22(2 Dementia): 538-558.
13. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Nonsmall cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584-594.
14. Ettinger DS, Aisner DL, Wood DE, et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 5.2018. J Natl Compr Canc Netw. 2018;16(7):807-821.
►Zachary Reese, MD, Chief Medical Resident, VABHS and Beth Israel Deaconess Medical Center (BIDMC):Dr. Weller, the differential diagnosis for altered mental status is quite broad. How does the presence of clonus change or focus your approach to altered mental status?
►Jason Weller, MD, Instructor of Neurology, Boston Medical Center (BMC) and VABHS:The presence of clonus does not significantly narrow the differential. It does, however, suggest a central component to the patient’s altered mental status. Specifically, it implies that the underlying process, whether systemic or neurologic, interferes with central nervous system (CNS) control of the neuromuscular system.1 The differential is still quite broad and includes metabolic derangements (eg, uremia, electrolyte disturbances, hypercarbia, and thyroid dysfunction), medication toxicity from olanzapine or duloxetine, and vascular processes (eg, CNS vasculitis). Infectious etiologies, both within the CNS and systemically, can cause encephalopathy, as can autoimmune processes, such as immune-mediated encephalitis. Finally, primary neurologic conditions such as myoclonic epilepsy can be considered. Given the patient’s medical history, serotonin syndrome must be considered.
►Dr. Reese: Given the concern for serotonin syndrome, the admitting medical team discontinued the patient’s duloxetine. Dr. Weller, what is the pathophysiology of serotonin syndrome, and how is it diagnosed?
►Dr. Weller: Serotonin is ubiquitous throughout the body and brain. Serotonin syndrome is caused by excess endogenous or exogenous serotonin, and this is usually caused by a variety of medications. The symptoms range from tachycardia, agitation, and diaphoresis to sustained clonus, hyperthermia, and shock.2,3 The extent of serotonin syndrome is typically thought to reflect the degree of serotonergic activity.4
Serotonin syndrome is a clinical diagnosis. While there are no tests that can confirm the diagnosis, the Hunter criteria can be used to assist with making the diagnosis.5 Per the Hunter criteria, a patient can be diagnosed with serotonin syndrome if they have taken a serotonergic agent and have at least 1 of the following: spontaneous clonus, inducible or ocular clonus with agitation or diaphoresis, tremor and hyperreflexia, or hypertonia with fever and clonus. This patient had taken duloxetine and had inducible clonus and diaphoresis, thus suggesting a diagnosis of serotonin syndrome.
►Dr. Reese: Aside from selective serotonin reuptake inhibitors (SSRIs), are there other medications that we typically prescribe that can cause serotonin syndrome?
►Dr. Weller: In addition to SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs), other commonly prescribed medications that can cause serotonin syndrome are 5-HT3 antagonists (eg, ondansetron), 5-HT agonists (eg, triptans), and opioids (eg, fentanyl and tramadol). There are also case reports of atypical antipsychotics (eg, olanzapine) causing serotonin syndrome because of their antagonism of the 5-HT2 and 5-HT3 receptors.2 Additionally, linezolid is commonly overlooked as a cause of serotonin syndrome given its action as a monoamine oxidase inhibitor.4 In this patient, it would be prudent to discontinue olanzapine and duloxetine.
►Dr. Reese: Duloxetine, olanzapine, and buprenorphine/naloxone were discontinuedgiven concern for serotonin syndrome. Although there are not strong data that buprenorphine/ naloxone can cause serotonin syndrome, the team discontinued the medication in case it might be contributing to the patient’s encephalopathy, while closely monitoring the patient for withdrawal. There was a rapid improvement in the patient’s symptoms over the 24 hours after discontinuation of the 3 medications.
As part of the initial workup, the patient received a computed tomography (CT) scan of his chest to follow up pulmonary nodules identified 16 months prior. The CT scan showed interval growth of the pulmonary nodules in the right lower lobe to 2 cm with extension into the major fissure, which was concerning for malignancy. Plans were made for an outpatient positron emission tomography (PET) scan after hospital discharge.
Dr. Schlechter and Dr. Rangachari, what factors can help us determine whether or not further workup of a malignancy should occur before discharge or can be deferred to the outpatient setting?
►Benjamin Schlechter, MD, Instructor in Medicine, BIDMC; and Deepa Rangachari, MD, Assistant Professor of Medicine, BIDMC: Key considerations in this domain include rapidity of growth and any threat to critical end-organ function (ie, brain, heart, lungs, kidney, and liver). If the malignancy is bulky and/or rapidly progressing to the point that the patient has significant symptoms burden and/or end-organ dysfunction, then initiating the evaluation as an inpatient may be necessary. For suspected intrathoracic malignancies, considering whether this may be a high-grade process (ie, small cell lung cancer) is often a vital branch point. Key considerations in this regard are the following: Is it a bulky central tumor? Is there evidence of widespread metastatic disease, an obstructing mass, and/or tumor lysis? One final and critical aspect to consider is whether there are any patient- specific barriers to timely and reliable outpatient follow-up. If there is no evidence of rapid progression, bulky disease with threatened end-organ involvement, and/or issues with timely and reliable follow-up, then outpatient evaluation is often the best approach to ensure a comprehensive and well-coordinated effort on the patient’s behalf.
►Dr. Reese: Buprenorphine/naloxone was restarted without return of the symptoms. The patient was discharged home with an outpatient PET scan scheduled the following week. Unfortunately, the patient was unable to keep this appointment. Three weeks after hospital discharge, the patient presented again to the emergency department with gradually worsening altered mental status, confusion, visual hallucinations, and myoclonic jerking of the arms and legs. Medication adherence was confirmed by the patient’s wife, resulting in a low concern for serotonin syndrome. Physical examination revealed confusion, dysarthria, diffuse, arrhythmic, myoclonic jerking in all extremities, asterixis in the upper extremities, and hyperreflexia.
A CT scan of the brain did not reveal an intracranial process. A spot electroencephalograph (EEG) and magnetic resonance image (MRI) of the brain were obtained. Dr. Weller, what is the utility of spot EEG vs 24-hour EEG? When might we choose one over the other?
►Dr. Weller: If a patient is persistently altered, then a spot EEG would be sufficient to capture a seizure if that is what is causing the patient’s altered mental status. However, if the patient’s mental status is waxing and waning, then that may warrant a 24-hour EEG because the patient may need to be monitored for longer periods to capture an event that is causing intermittent alterations in mental status.6 Additionally, patients who are acutely ill may require long-term monitoring for the purpose of treatment and outcome management.
►Dr. Reese: The spot EEG showed nearly continuous generalized slowing indicative of a diffuse encephalopathy. The MRI of the brain showed scattered, nonspecific periventricular T2 hyperintense foci, suggestive of advanced chronic microvascular ischemic changes.
A PET CT was obtained and revealed mildly fluorodeoxyglucose (FDG)-avid, enlarging nodules within the right lower lobe, which was suspicious for malignancy. There were no other areas of FDG avidity on the PET scan. Valproic acid was initiated for treatment of myoclonus with transition to clonazepam when no improvement was seen. After starting clonazepam, the patient’s condition stabilized.
Dr. Weller, given the additional history, how has your differential diagnosis changed?
►Dr. Weller: Given the patient’s laboratory findings, we can be quite sure that there is not a contributing metabolic process. The findings suggestive of metastatic cancer, along with the profound neurologic changes, are most concerning for a paraneoplastic syndrome. I would suggest biopsy and consideration of a lumbar puncture. One can also send serum markers, including a paraneoplastic antibody panel.
►Dr. Reese: Biopsy of the mass in his right lower lobe revealed squamous cell lung cancer. Dr. Schlechter and Dr. Rangachari, do you have a framework for the different forms of lung cancer?
►Dr. Schlechter/Dr. Rangachari: The 2 broad categories of lung cancer are small cell and non-small cell (NSCLC). Small cell lung cancer has a tight association with tobacco exposure and is often clinically defined by rapid, bulky progression (ie, weeks to months).7,8 NSCLCs are also commonly seen in those with tobacco exposure, though not always. The main subgroups in this category are adenocarcinoma and squamous cell carcinoma. These cancers often evolve at a slower pace (ie, months to years).8 While small cell lung cancers are highgrade tumors and exquisitely sensitive to chemotherapy and radiation, NSCLCs tend to be less responsive to such therapies. The staging evaluation for either entity is the same and consists of defining localized vs metastatic disease.
►Dr. Reese: Because this patient had an MRI and PET scan that were both negative for metastatic disease, can we assume that this patient had stage I NSCLC?
►Dr. Schlechter/Dr. Rangachari: Not necessarily. While PET and MRI brain are exceptionally helpful in detecting distant metastases, they may over- or underestimate intrathoracic lymph node involvement by as much as 20%.9 As such, dedicated lymph node staging—either via bronchoscopy (endobronchial ultrasound) or surgically (mediastinoscopy) is indicated as lymph node involvement can significantly alter the stage, prognosis, and optimal therapeutic approach.10,11
►Dr. Reese: After this diagnosis was made, the teams caring for this patient attributed his altered mental status to a paraneoplastic syndrome. What is a paraneoplastic syndrome, and how does a paraneoplastic syndrome from malignancy present? Does its presence worsen a patient’s prognosis?
►Dr. Schlechter/Dr. Rangachari: A paraneoplastic syndrome is defined by an immunologic response to the cancer that ends up erroneously targeting self-antigens. Paraneoplastic syndromes are associated with a broad array of clinical findings—from endocrinopathy to encephalopathy—and certain neoplasms are more commonly associated with these syndromes than others (eg, small cell lung cancer and thymoma). Further, severity and onset of a paraneoplastic syndrome does not correlate with the burden of visible disease—and the syndrome may predate the cancer diagnosis by months to years.11 While treatment of the cancer affords the best hope of resolving the paraneoplastic syndrome, the cancer and the paraneoplastic process may have a discordant trajectory, with the paraneoplastic syndrome persisting even after the cancer is maximally treated. Although one might assume that paraneoplastic syndromes portend worse outcomes, in some cases, a presentation with the paraneoplastic syndrome may afford sooner detection of an otherwise occult/asymptomatic malignancy.
►Dr. Reese: The following week, the serum paraneoplastic antibody panel that tested for anti-Yo antibody, anti-Ri antibody,and anti-Hu antibody came back negative. Dr. Weller, what does this mean? Since we have yet to obtain a lumbar puncture, might his symptoms still be caused by a paraneoplastic syndrome?
►Dr. Weller: The negative serum test just means that he does not have antibodies to those 3 antibodies. There are now over 30 different paraneoplastic antibodies that have been discovered, and there are always more that are being discovered. So this negative test result does not exclude a paraneoplastic syndrome in the appropriate clinical context.12 Furthermore, the sensitivity and specificity for certain antibodies are different based upon source fluid, and cerebrospinal fluid testing would provide more diagnostic clarity. A negative test for paraneoplastic syndrome, by itself, would similarly not exclude a paraneoplastic syndrome. Often, empiric treatment is the best diagnostic option for paraneoplastic and autoimmune encephalopathies.
►Dr. Reese: The following week, the patient was discharged to rehabilitation with clonazepam for his symptoms and a scheduled follow-up. Given the patient’s frailty and medical comorbidities, thoracic surgery recommended consultation with radiation oncology. Dr. Schlechter and Dr. Rangachari, when do we decide to use radiation vs chemotherapy for someone with lung cancer?
►Dr. Schlechter/Dr. Rangachari: Patients with early stage, nonmetastatic NSCLC may not always be candidates for surgical resection on the basis of pulmonary function, other medical comorbidities (as in this case), anatomic considerations, and/or patient preference. In these cases, if there is lung-limited disease without lymph node involvement (ie, stage I/II NSCLC) and the patient is not felt to be an operative candidate, then alternatives to surgery include either radiation or ablation.13,14 As we care for an aging and comorbid population, evolving evidence suggests that well-selected patients with early stage disease undergoing these nonoperative approaches have roughly equivalent outcomes to those undergoing conventional surgical resection.13 In such cases, multidisciplinary consultation with a team having dedicated expertise in these various operative and nonoperative modalities is essential.
►Dr. Reese: The patient followed up with radiation oncology for consideration of radiation treatment, but his simulation CT scan showed some ground-glass opacity that were concerning for inflammation vs infection. The patient’s case was discussed at the multidisciplinary tumor board, and it was determined to treat him with antibiotics for a possible pneumonia before proceeding with radiation therapy. After he completed antibiotic treatment, he underwent 10 fractions of radiation treatment, which he tolerated well.
►Zachary Reese, MD, Chief Medical Resident, VABHS and Beth Israel Deaconess Medical Center (BIDMC):Dr. Weller, the differential diagnosis for altered mental status is quite broad. How does the presence of clonus change or focus your approach to altered mental status?
►Jason Weller, MD, Instructor of Neurology, Boston Medical Center (BMC) and VABHS:The presence of clonus does not significantly narrow the differential. It does, however, suggest a central component to the patient’s altered mental status. Specifically, it implies that the underlying process, whether systemic or neurologic, interferes with central nervous system (CNS) control of the neuromuscular system.1 The differential is still quite broad and includes metabolic derangements (eg, uremia, electrolyte disturbances, hypercarbia, and thyroid dysfunction), medication toxicity from olanzapine or duloxetine, and vascular processes (eg, CNS vasculitis). Infectious etiologies, both within the CNS and systemically, can cause encephalopathy, as can autoimmune processes, such as immune-mediated encephalitis. Finally, primary neurologic conditions such as myoclonic epilepsy can be considered. Given the patient’s medical history, serotonin syndrome must be considered.
►Dr. Reese: Given the concern for serotonin syndrome, the admitting medical team discontinued the patient’s duloxetine. Dr. Weller, what is the pathophysiology of serotonin syndrome, and how is it diagnosed?
►Dr. Weller: Serotonin is ubiquitous throughout the body and brain. Serotonin syndrome is caused by excess endogenous or exogenous serotonin, and this is usually caused by a variety of medications. The symptoms range from tachycardia, agitation, and diaphoresis to sustained clonus, hyperthermia, and shock.2,3 The extent of serotonin syndrome is typically thought to reflect the degree of serotonergic activity.4
Serotonin syndrome is a clinical diagnosis. While there are no tests that can confirm the diagnosis, the Hunter criteria can be used to assist with making the diagnosis.5 Per the Hunter criteria, a patient can be diagnosed with serotonin syndrome if they have taken a serotonergic agent and have at least 1 of the following: spontaneous clonus, inducible or ocular clonus with agitation or diaphoresis, tremor and hyperreflexia, or hypertonia with fever and clonus. This patient had taken duloxetine and had inducible clonus and diaphoresis, thus suggesting a diagnosis of serotonin syndrome.
►Dr. Reese: Aside from selective serotonin reuptake inhibitors (SSRIs), are there other medications that we typically prescribe that can cause serotonin syndrome?
►Dr. Weller: In addition to SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs), other commonly prescribed medications that can cause serotonin syndrome are 5-HT3 antagonists (eg, ondansetron), 5-HT agonists (eg, triptans), and opioids (eg, fentanyl and tramadol). There are also case reports of atypical antipsychotics (eg, olanzapine) causing serotonin syndrome because of their antagonism of the 5-HT2 and 5-HT3 receptors.2 Additionally, linezolid is commonly overlooked as a cause of serotonin syndrome given its action as a monoamine oxidase inhibitor.4 In this patient, it would be prudent to discontinue olanzapine and duloxetine.
►Dr. Reese: Duloxetine, olanzapine, and buprenorphine/naloxone were discontinuedgiven concern for serotonin syndrome. Although there are not strong data that buprenorphine/ naloxone can cause serotonin syndrome, the team discontinued the medication in case it might be contributing to the patient’s encephalopathy, while closely monitoring the patient for withdrawal. There was a rapid improvement in the patient’s symptoms over the 24 hours after discontinuation of the 3 medications.
As part of the initial workup, the patient received a computed tomography (CT) scan of his chest to follow up pulmonary nodules identified 16 months prior. The CT scan showed interval growth of the pulmonary nodules in the right lower lobe to 2 cm with extension into the major fissure, which was concerning for malignancy. Plans were made for an outpatient positron emission tomography (PET) scan after hospital discharge.
Dr. Schlechter and Dr. Rangachari, what factors can help us determine whether or not further workup of a malignancy should occur before discharge or can be deferred to the outpatient setting?
►Benjamin Schlechter, MD, Instructor in Medicine, BIDMC; and Deepa Rangachari, MD, Assistant Professor of Medicine, BIDMC: Key considerations in this domain include rapidity of growth and any threat to critical end-organ function (ie, brain, heart, lungs, kidney, and liver). If the malignancy is bulky and/or rapidly progressing to the point that the patient has significant symptoms burden and/or end-organ dysfunction, then initiating the evaluation as an inpatient may be necessary. For suspected intrathoracic malignancies, considering whether this may be a high-grade process (ie, small cell lung cancer) is often a vital branch point. Key considerations in this regard are the following: Is it a bulky central tumor? Is there evidence of widespread metastatic disease, an obstructing mass, and/or tumor lysis? One final and critical aspect to consider is whether there are any patient- specific barriers to timely and reliable outpatient follow-up. If there is no evidence of rapid progression, bulky disease with threatened end-organ involvement, and/or issues with timely and reliable follow-up, then outpatient evaluation is often the best approach to ensure a comprehensive and well-coordinated effort on the patient’s behalf.
►Dr. Reese: Buprenorphine/naloxone was restarted without return of the symptoms. The patient was discharged home with an outpatient PET scan scheduled the following week. Unfortunately, the patient was unable to keep this appointment. Three weeks after hospital discharge, the patient presented again to the emergency department with gradually worsening altered mental status, confusion, visual hallucinations, and myoclonic jerking of the arms and legs. Medication adherence was confirmed by the patient’s wife, resulting in a low concern for serotonin syndrome. Physical examination revealed confusion, dysarthria, diffuse, arrhythmic, myoclonic jerking in all extremities, asterixis in the upper extremities, and hyperreflexia.
A CT scan of the brain did not reveal an intracranial process. A spot electroencephalograph (EEG) and magnetic resonance image (MRI) of the brain were obtained. Dr. Weller, what is the utility of spot EEG vs 24-hour EEG? When might we choose one over the other?
►Dr. Weller: If a patient is persistently altered, then a spot EEG would be sufficient to capture a seizure if that is what is causing the patient’s altered mental status. However, if the patient’s mental status is waxing and waning, then that may warrant a 24-hour EEG because the patient may need to be monitored for longer periods to capture an event that is causing intermittent alterations in mental status.6 Additionally, patients who are acutely ill may require long-term monitoring for the purpose of treatment and outcome management.
►Dr. Reese: The spot EEG showed nearly continuous generalized slowing indicative of a diffuse encephalopathy. The MRI of the brain showed scattered, nonspecific periventricular T2 hyperintense foci, suggestive of advanced chronic microvascular ischemic changes.
A PET CT was obtained and revealed mildly fluorodeoxyglucose (FDG)-avid, enlarging nodules within the right lower lobe, which was suspicious for malignancy. There were no other areas of FDG avidity on the PET scan. Valproic acid was initiated for treatment of myoclonus with transition to clonazepam when no improvement was seen. After starting clonazepam, the patient’s condition stabilized.
Dr. Weller, given the additional history, how has your differential diagnosis changed?
►Dr. Weller: Given the patient’s laboratory findings, we can be quite sure that there is not a contributing metabolic process. The findings suggestive of metastatic cancer, along with the profound neurologic changes, are most concerning for a paraneoplastic syndrome. I would suggest biopsy and consideration of a lumbar puncture. One can also send serum markers, including a paraneoplastic antibody panel.
►Dr. Reese: Biopsy of the mass in his right lower lobe revealed squamous cell lung cancer. Dr. Schlechter and Dr. Rangachari, do you have a framework for the different forms of lung cancer?
►Dr. Schlechter/Dr. Rangachari: The 2 broad categories of lung cancer are small cell and non-small cell (NSCLC). Small cell lung cancer has a tight association with tobacco exposure and is often clinically defined by rapid, bulky progression (ie, weeks to months).7,8 NSCLCs are also commonly seen in those with tobacco exposure, though not always. The main subgroups in this category are adenocarcinoma and squamous cell carcinoma. These cancers often evolve at a slower pace (ie, months to years).8 While small cell lung cancers are highgrade tumors and exquisitely sensitive to chemotherapy and radiation, NSCLCs tend to be less responsive to such therapies. The staging evaluation for either entity is the same and consists of defining localized vs metastatic disease.
►Dr. Reese: Because this patient had an MRI and PET scan that were both negative for metastatic disease, can we assume that this patient had stage I NSCLC?
►Dr. Schlechter/Dr. Rangachari: Not necessarily. While PET and MRI brain are exceptionally helpful in detecting distant metastases, they may over- or underestimate intrathoracic lymph node involvement by as much as 20%.9 As such, dedicated lymph node staging—either via bronchoscopy (endobronchial ultrasound) or surgically (mediastinoscopy) is indicated as lymph node involvement can significantly alter the stage, prognosis, and optimal therapeutic approach.10,11
►Dr. Reese: After this diagnosis was made, the teams caring for this patient attributed his altered mental status to a paraneoplastic syndrome. What is a paraneoplastic syndrome, and how does a paraneoplastic syndrome from malignancy present? Does its presence worsen a patient’s prognosis?
►Dr. Schlechter/Dr. Rangachari: A paraneoplastic syndrome is defined by an immunologic response to the cancer that ends up erroneously targeting self-antigens. Paraneoplastic syndromes are associated with a broad array of clinical findings—from endocrinopathy to encephalopathy—and certain neoplasms are more commonly associated with these syndromes than others (eg, small cell lung cancer and thymoma). Further, severity and onset of a paraneoplastic syndrome does not correlate with the burden of visible disease—and the syndrome may predate the cancer diagnosis by months to years.11 While treatment of the cancer affords the best hope of resolving the paraneoplastic syndrome, the cancer and the paraneoplastic process may have a discordant trajectory, with the paraneoplastic syndrome persisting even after the cancer is maximally treated. Although one might assume that paraneoplastic syndromes portend worse outcomes, in some cases, a presentation with the paraneoplastic syndrome may afford sooner detection of an otherwise occult/asymptomatic malignancy.
►Dr. Reese: The following week, the serum paraneoplastic antibody panel that tested for anti-Yo antibody, anti-Ri antibody,and anti-Hu antibody came back negative. Dr. Weller, what does this mean? Since we have yet to obtain a lumbar puncture, might his symptoms still be caused by a paraneoplastic syndrome?
►Dr. Weller: The negative serum test just means that he does not have antibodies to those 3 antibodies. There are now over 30 different paraneoplastic antibodies that have been discovered, and there are always more that are being discovered. So this negative test result does not exclude a paraneoplastic syndrome in the appropriate clinical context.12 Furthermore, the sensitivity and specificity for certain antibodies are different based upon source fluid, and cerebrospinal fluid testing would provide more diagnostic clarity. A negative test for paraneoplastic syndrome, by itself, would similarly not exclude a paraneoplastic syndrome. Often, empiric treatment is the best diagnostic option for paraneoplastic and autoimmune encephalopathies.
►Dr. Reese: The following week, the patient was discharged to rehabilitation with clonazepam for his symptoms and a scheduled follow-up. Given the patient’s frailty and medical comorbidities, thoracic surgery recommended consultation with radiation oncology. Dr. Schlechter and Dr. Rangachari, when do we decide to use radiation vs chemotherapy for someone with lung cancer?
►Dr. Schlechter/Dr. Rangachari: Patients with early stage, nonmetastatic NSCLC may not always be candidates for surgical resection on the basis of pulmonary function, other medical comorbidities (as in this case), anatomic considerations, and/or patient preference. In these cases, if there is lung-limited disease without lymph node involvement (ie, stage I/II NSCLC) and the patient is not felt to be an operative candidate, then alternatives to surgery include either radiation or ablation.13,14 As we care for an aging and comorbid population, evolving evidence suggests that well-selected patients with early stage disease undergoing these nonoperative approaches have roughly equivalent outcomes to those undergoing conventional surgical resection.13 In such cases, multidisciplinary consultation with a team having dedicated expertise in these various operative and nonoperative modalities is essential.
►Dr. Reese: The patient followed up with radiation oncology for consideration of radiation treatment, but his simulation CT scan showed some ground-glass opacity that were concerning for inflammation vs infection. The patient’s case was discussed at the multidisciplinary tumor board, and it was determined to treat him with antibiotics for a possible pneumonia before proceeding with radiation therapy. After he completed antibiotic treatment, he underwent 10 fractions of radiation treatment, which he tolerated well.
1. Kojovic M, Cordivari C, Bhatia K. Myoclonic disorders: a practical approach for diagnosis and treatment. Ther Adv Neurol Disord. 2011;4(1):47-62.
2. Volpi-Abadie J, Kaye AM, Kaye AD. Serotonin syndrome. Ochsner J. 2013;13(4):533-540.
3. Arora B, Kannikeswaran N. The serotonin syndrome-the need for physician’s awareness. Int J Emerg Med. 2010;3(4):373-377.
4. Boyer EW, Shannon M. The serotonin syndrome [published correction appears in N Engl J Med. 2007;356(23):2437 and N Engl J Med. 2009;361(17):1714]. N Engl J Med.
2005;352(11):1112-1120.
5. Dunkley EJC, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM.
2003;96(9):635-642.
6. Nordli DR Jr. Usefulness of video-EEG monitoring. Epilepsia. 2006;47(suppl 1):26-30.
7. Ettinger DS, Aisner J. Changing face of small-cell lung cancer: real and artifact. J Clin Oncol. 2006;24(28):4526-4527.
8. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10(9):1243-1260.
9. Cerfolio RJ, Bryant AS, Ojha B, Eloubeidi M. Improving the inaccuracies of clinical staging of patients with NSCLC: a prospective trial. Ann Thorac Surg. 2005;80(4):1207-1214.
10. El-Osta H, Jani P, Mansour A, Rascoe P, Jafri S. Endobronchial ultrasound for nodal staging of patients with non-smallcell lung cancer with radiologically normal mediastinum. A meta-analysis. Ann Am Thorac Soc. 2018;15(7):864-874.
11. Darnell RB, Posner JB. Paraneoplastic syndromes involving the nervous system. N Engl J Med. 2003;349(16):1543-1554.
12. McKeon A. Autoimmune Encephalopathies and Dementias. Continuum (Minneap Minn). 2016;22(2 Dementia): 538-558.
13. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Nonsmall cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584-594.
14. Ettinger DS, Aisner DL, Wood DE, et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 5.2018. J Natl Compr Canc Netw. 2018;16(7):807-821.
1. Kojovic M, Cordivari C, Bhatia K. Myoclonic disorders: a practical approach for diagnosis and treatment. Ther Adv Neurol Disord. 2011;4(1):47-62.
2. Volpi-Abadie J, Kaye AM, Kaye AD. Serotonin syndrome. Ochsner J. 2013;13(4):533-540.
3. Arora B, Kannikeswaran N. The serotonin syndrome-the need for physician’s awareness. Int J Emerg Med. 2010;3(4):373-377.
4. Boyer EW, Shannon M. The serotonin syndrome [published correction appears in N Engl J Med. 2007;356(23):2437 and N Engl J Med. 2009;361(17):1714]. N Engl J Med.
2005;352(11):1112-1120.
5. Dunkley EJC, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM.
2003;96(9):635-642.
6. Nordli DR Jr. Usefulness of video-EEG monitoring. Epilepsia. 2006;47(suppl 1):26-30.
7. Ettinger DS, Aisner J. Changing face of small-cell lung cancer: real and artifact. J Clin Oncol. 2006;24(28):4526-4527.
8. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10(9):1243-1260.
9. Cerfolio RJ, Bryant AS, Ojha B, Eloubeidi M. Improving the inaccuracies of clinical staging of patients with NSCLC: a prospective trial. Ann Thorac Surg. 2005;80(4):1207-1214.
10. El-Osta H, Jani P, Mansour A, Rascoe P, Jafri S. Endobronchial ultrasound for nodal staging of patients with non-smallcell lung cancer with radiologically normal mediastinum. A meta-analysis. Ann Am Thorac Soc. 2018;15(7):864-874.
11. Darnell RB, Posner JB. Paraneoplastic syndromes involving the nervous system. N Engl J Med. 2003;349(16):1543-1554.
12. McKeon A. Autoimmune Encephalopathies and Dementias. Continuum (Minneap Minn). 2016;22(2 Dementia): 538-558.
13. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Nonsmall cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584-594.
14. Ettinger DS, Aisner DL, Wood DE, et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 5.2018. J Natl Compr Canc Netw. 2018;16(7):807-821.
Cervical Pannus Without Rheumatoid Arthritis or Trauma
Although usually seen in patients with rheumatoid arthritis, cervical pannus also can develop in patients who have had spine surgery.
Cervical pannus is a disease that could easily develop in an active-duty soldier or veteran. The disease has been associated with trauma and rheumatoid arthritis, or can be idiopathic. For years, cervical pannus has been closely tied to rheumatoid arthritis; however, a study published in 2019 showed that only 28% of patients with cervical pannus had an associated diagnosis of rheumatoid arthritis.1 In the same study, 18% of patients had undergone some type of prior cervical spine surgery as the next most common cause. The condition also can occur years after an injury.
Background
In the US, 42,000 veterans are living with spinal cord disease, and thousands of these veterans have surgery every year.2 Service men and women and veterans are at risk for cervical pannus as they age especially if they have a history of rheumatoid arthritis, cervical spine surgery, trauma, and numerous other causes. It is critical for health care providers who treat this population to understand cervical pannus, how to recognize it, and how to identify patients at risk. A cervical pannus can be life threatening if not detected and treated properly.
There is no clear definition for cervical pannus. Some researchers think of it as the chronically inflamed synovial membrane in patients with rheumatoid arthritis (RA); others consider it as a specialized synovial membrane derived from vascular soft tissue structures at or near the bone synovial membrane.3 The pathogenesis for developing a pannus is not well understood, and little is known when a pannus begins or its initial location. A pannus formation can occur in any synovial joint in the body, such as wrists, metacarpophalangeal joint, proximal interphalangeal joint, and cervical joints.
A cervical pannus can cause serious complications. It can lead to a cervical subluxation in up to 4% of patients with RA, or it also can occur spontaneously in some patients without RA especially those with trauma or cancer.4
There are 2 suggested mechanisms by which the synovial membrane proliferates. It was originally believed that T cells from the chronic inflamed joint lead to the pannus formation by initiating an autoimmune reaction through the production of different cytokines against arthritogenic agents.3-5 These cytokines increase inflammation by recruiting neutrophils and activating various kinds of macrophages that might lead to increased osteoclast activity.6 Osteoclastic activity can damage bone and allow the synovium to penetrate the bone, forming the pannus.
Another proposed mechanism is that the synovial cells hyperpolarize and hypertrophy automatically without T-cell help by expressing oncogenes and their proteins.3 In either case, angiogenesis follows this proliferation and increases the influx of inflammatory cells into the joints, which can lead to more destruction.7 This increase in blood supply to the synovial membrane is important in the growth of the pannus and can have a damaging effect to cartilage, bone, and joints.4,7
Cervical pannus can progress in patients with prolonged use of corticosteroids.8 Because a pannus can put pressure on any segment of the cervical spine and the cranio-cervical junction leading to cervical instability, patients with this condition may present with a variety of clinical symptoms.9 The most frequently reported clinical features include neck pain, easy fatigability, difficulty walking, abnormal gait, increased clumsiness, and numbness and tingling in the arms. Patients also may complain of neck stiffness and decreased neck motion.10Cervical pannus is most frequently seen in patients with RA. However, patients without a RA diagnosis and incidental atlantoaxial pannus on cervical spine magnetic resonance imaging (MRI) are unlikely to have previously undiagnosed RA.11
Case Presentation
A 70-year-old white woman presented to the neurology clinic at Gretna Medical Center in Virginia in December 2016 with constant headache and imbalance that started in September 2016. She characterized the pain as predominately pressure (6 on a 10-point pain scale) with occasional shooting pains. The pain started at the left occipital lobe and radiated toward the left temporal lobe and left eye. The patient also stated that it was very difficult to lay her head down on a pillow to sleep and that she had to use a recliner in order to sleep over the past 3 months. She reported that the headache felt slightly worse if she had a lot of repetitive head and neck movements during the day. There was no photophobia, phonophobia, nausea, vomiting, facial paresthesias, lacrimation, nasal congestion, confusion, or impaired speech.
The patient’s lack of balance, which resulted in an unsteady gait, had started 1 month before and had increased significantly in the past 2 to 3 weeks. She stated that the unsteady gait was associated with numbness in her right upper and lower extremities, although more intense in the right lower extremity. Aside from the headaches, paresthesia, and unsteady gait, the patient reported no other major symptoms. She did not smoke tobacco or drink alcohol. Her family history revealed that her brothers had heart disease.
The patient’s vital signs at physical examination included heart rate, 83 beats per minute; blood pressure, 159/75 mm hg; temporal temperature, 97.9 °F; and respiratory rate, 20 breaths per minute. The patient’s gait was unsteady, needing stabilization by holding on to her husband’s arm, slightly favoring right lower extremity. Finger-to-nose test, rapid alternating movements, heel-knee-shin testing were all normal. The Romberg sign was positive. The patient could rise on toes and heels with slight balance disturbance. Deep tendon reflexes and reflexes in the upper and lower extremities was symmetric 2+ bilaterally. Musculoskeletal examination revealed strength and tone in all major muscle groups and demonstrated symmetrical movements with no fasciculation noted. A rheumatologic evaluation showed no abnormalities, including inspection of hands, feet, major joints, and other range of motion, besides her neck. The rest of the physical, cognitive, and neurologic examination findings were otherwise unremarkable. A routine rheumatologic laboratory evaluation was negative.


A head computed tomography ordered before coming to the clinic showed normal results. An MRI of the head was obtained to evaluate for ischemic cause or structural abnormality (Figures 1 and 2). Given the patient’s presentation and the pattern seen on the MRI results, it was determined that large pannus posterior to the dens, severely narrowing the spinal canal, was most likely the diagnosis. A second opinion confirmed the diagnosis, and a second MRI revealed stabilization with no signs of enhancement.
The patient was advised to meet with a neurosurgeon to remove the pannus. The patient agreed on occiput to C2 posterior instrument arthrodesis as well as decompression. A plain film radiograph showed C2-occipital repair after surgery (Figure 3). The patient recovered in the neurosurgical intensive care unit, and the rest of the recovery was uncomplicated. She showed some improvement in her headaches and unsteady gait. A postoperative pathologic evaluation of tissue was not available. She was referred to a rheumatologist to rule out an autoimmune disease as the cause for this pannus, but no autoimmune disease was found.
Discussion
Cervical pannus is relatively uncommon in those without RA. However, there are multiple reasons that a patient could develop a cervical pannus. Cervical pannus in RA and cervical pannus without RA may mimic each other clinically, but medical management is distinctly different. Consequently, a rheumatology consult is necessary to ensure that there is no undiagnosed autoimmune disorder. Our patient did not have RA, and a neurosurgery intervention was needed to manage her headaches and unsteady gait. Although we could not isolate a cause of this patient’s cervical pannus development, we believed that nonintervention would adversely affect this patient.
The course of pannus progression can be fatal especially if left untreated.12 MRI can detect a pannus and may be helpful for planning surgery.13 Surgical resection has been the treatment of choice for patients with neurologic symptoms.14 However, some cases have reported resolution of pannus associated with RA and other forms of chronic atlantoaxial instability only after posterior stabilization.14In order to manage pannus, cervical spine examination for the diagnosis of cervical involvement is encouraged to prevent morbidity and mortality.13 There are new data that demonstrated the potential of using retinoid X receptor agonists, such as bexarotene, as a treatment against the development and progression of pannus.14
Conclusions
We present a patient with cervical pannus disease without RA whose diagnosis was based on the pathognomonic pattern seen on MRI. She showed a clinically significant recovery with an occiput to C2 posterior instrument arthrodesis as well as decompression. She showed marked improvements in her headaches and unsteady gait. This case report highlights the importance of realizing cervical pannus as a disease found in patients without RA. It serves as an alert to clinicians for timely detection, diagnosis, and initiation of treatment to prevent mortality and long-term neurologic sequelae of cervical pannus.
Although further studies of early diagnosis and treatment for cervical pannus are warranted, we propose that including pannus in a differential diagnosis for patients with no RA could be lifesaving.
1. Zvaifler NJ, Firestein GS. Pannus and pannocytes. Alternative models of joint destruction in rheumatoid arthritis. Arthritis Rheum. 1994;37(6):783-789.
2. Henderson DR. Vertical atlanto-axial subluxation in rheumatoid arthritis. Rheumatol Rehabil. 1975;14(1):31-38.
3. Skapenko A, Leipe J, Lipsky PE, Schulze-Koops H. The role of the T cell in autoimmune inflammation. Arthritis Res Ther. 2005;7(suppl 2):S4-S14.
4. Wang R, Zhang L, Zhang X, et al. Regulation of activation-induced receptor activator of NF-kappaB ligand (RANKL) expression in T cells. Eur J Immunol. 2002;32(4):1090-1098.
5. Koch AE. Angiogenesis as a target in rheumatoid arthritis. Ann Rheum Dis. 2003;62(suppl 2):ii60-ii67.
6. Reiter MF, Boden SD. Inflammatory disorders of the cervical spine. Spine (Phila Pa 1976). 1998;23(24):2755-2766.
7. Alaya Z, Lataoui S, Amri D, Zaghouani H, Bouajina E. Atlantoaxial instability: an exceptional complication of ankylosing spondylitis. Egypt Rheumatol. 2018;40(2):141-143.
8. Walter KD, Tassone JC. Atlantoaxial instability. In: Micheli LJ, ed. Encyclopedia of Sports Medicine. Thousand Oaks, CA: SAGE Reference; 2011:122-124.
9. Joyce AA, Williams JN, Shi J, Mandell JC, Isaac Z, Ermann J. Atlanto-axial pannus in patients with and without rheumatoid arthritis. J Rheumatol. 2019;46(11):1431-1437.
10. Neva MH, Myllykangas-Luosujärvi R, Kautiainen H, Kauppi M. Mortality associated with cervical spine disorders: a population-based study of 1666 patients with rheumatoid arthritis who died in Finland in 1989. Rheumatology (Oxford). 2001;40(2):123-127.
11. Mallory GW, Halasz SR, Clarke MJ. Advances in the treatment of cervical rheumatoid: less surgery and less morbidity. World J Orthop. 2014;5(3):292-303.
12. Lagares A, Arrese I, Pascual B, Gòmez PA, Ramos A, Lobato RD. Pannus resolution after occipitocervical fusion in a non-rheumatoid atlanto-axial instability. Eur Spine J. 2006;15(3):366-369.
13. Chung J, Bak KH, Yi H-J, Chun HJ, Ryu JI, Han M-H. Upper cervical subluxation and cervicomedullary junction compression in patients with rheumatoid arthritis. J Korean Neurosurg Soc. 2019;62(6):661-670.
14. Li Y, Xing Q, Wei Y, et al. Activation of RXR by bexarotene inhibits inflammatory conditions in human rheumatoid arthritis fibroblast‑like synoviocytes. Int J Mol Med. 2019;44(5):1963-1970.
Although usually seen in patients with rheumatoid arthritis, cervical pannus also can develop in patients who have had spine surgery.
Although usually seen in patients with rheumatoid arthritis, cervical pannus also can develop in patients who have had spine surgery.
Cervical pannus is a disease that could easily develop in an active-duty soldier or veteran. The disease has been associated with trauma and rheumatoid arthritis, or can be idiopathic. For years, cervical pannus has been closely tied to rheumatoid arthritis; however, a study published in 2019 showed that only 28% of patients with cervical pannus had an associated diagnosis of rheumatoid arthritis.1 In the same study, 18% of patients had undergone some type of prior cervical spine surgery as the next most common cause. The condition also can occur years after an injury.
Background
In the US, 42,000 veterans are living with spinal cord disease, and thousands of these veterans have surgery every year.2 Service men and women and veterans are at risk for cervical pannus as they age especially if they have a history of rheumatoid arthritis, cervical spine surgery, trauma, and numerous other causes. It is critical for health care providers who treat this population to understand cervical pannus, how to recognize it, and how to identify patients at risk. A cervical pannus can be life threatening if not detected and treated properly.
There is no clear definition for cervical pannus. Some researchers think of it as the chronically inflamed synovial membrane in patients with rheumatoid arthritis (RA); others consider it as a specialized synovial membrane derived from vascular soft tissue structures at or near the bone synovial membrane.3 The pathogenesis for developing a pannus is not well understood, and little is known when a pannus begins or its initial location. A pannus formation can occur in any synovial joint in the body, such as wrists, metacarpophalangeal joint, proximal interphalangeal joint, and cervical joints.
A cervical pannus can cause serious complications. It can lead to a cervical subluxation in up to 4% of patients with RA, or it also can occur spontaneously in some patients without RA especially those with trauma or cancer.4
There are 2 suggested mechanisms by which the synovial membrane proliferates. It was originally believed that T cells from the chronic inflamed joint lead to the pannus formation by initiating an autoimmune reaction through the production of different cytokines against arthritogenic agents.3-5 These cytokines increase inflammation by recruiting neutrophils and activating various kinds of macrophages that might lead to increased osteoclast activity.6 Osteoclastic activity can damage bone and allow the synovium to penetrate the bone, forming the pannus.
Another proposed mechanism is that the synovial cells hyperpolarize and hypertrophy automatically without T-cell help by expressing oncogenes and their proteins.3 In either case, angiogenesis follows this proliferation and increases the influx of inflammatory cells into the joints, which can lead to more destruction.7 This increase in blood supply to the synovial membrane is important in the growth of the pannus and can have a damaging effect to cartilage, bone, and joints.4,7
Cervical pannus can progress in patients with prolonged use of corticosteroids.8 Because a pannus can put pressure on any segment of the cervical spine and the cranio-cervical junction leading to cervical instability, patients with this condition may present with a variety of clinical symptoms.9 The most frequently reported clinical features include neck pain, easy fatigability, difficulty walking, abnormal gait, increased clumsiness, and numbness and tingling in the arms. Patients also may complain of neck stiffness and decreased neck motion.10Cervical pannus is most frequently seen in patients with RA. However, patients without a RA diagnosis and incidental atlantoaxial pannus on cervical spine magnetic resonance imaging (MRI) are unlikely to have previously undiagnosed RA.11
Case Presentation
A 70-year-old white woman presented to the neurology clinic at Gretna Medical Center in Virginia in December 2016 with constant headache and imbalance that started in September 2016. She characterized the pain as predominately pressure (6 on a 10-point pain scale) with occasional shooting pains. The pain started at the left occipital lobe and radiated toward the left temporal lobe and left eye. The patient also stated that it was very difficult to lay her head down on a pillow to sleep and that she had to use a recliner in order to sleep over the past 3 months. She reported that the headache felt slightly worse if she had a lot of repetitive head and neck movements during the day. There was no photophobia, phonophobia, nausea, vomiting, facial paresthesias, lacrimation, nasal congestion, confusion, or impaired speech.
The patient’s lack of balance, which resulted in an unsteady gait, had started 1 month before and had increased significantly in the past 2 to 3 weeks. She stated that the unsteady gait was associated with numbness in her right upper and lower extremities, although more intense in the right lower extremity. Aside from the headaches, paresthesia, and unsteady gait, the patient reported no other major symptoms. She did not smoke tobacco or drink alcohol. Her family history revealed that her brothers had heart disease.
The patient’s vital signs at physical examination included heart rate, 83 beats per minute; blood pressure, 159/75 mm hg; temporal temperature, 97.9 °F; and respiratory rate, 20 breaths per minute. The patient’s gait was unsteady, needing stabilization by holding on to her husband’s arm, slightly favoring right lower extremity. Finger-to-nose test, rapid alternating movements, heel-knee-shin testing were all normal. The Romberg sign was positive. The patient could rise on toes and heels with slight balance disturbance. Deep tendon reflexes and reflexes in the upper and lower extremities was symmetric 2+ bilaterally. Musculoskeletal examination revealed strength and tone in all major muscle groups and demonstrated symmetrical movements with no fasciculation noted. A rheumatologic evaluation showed no abnormalities, including inspection of hands, feet, major joints, and other range of motion, besides her neck. The rest of the physical, cognitive, and neurologic examination findings were otherwise unremarkable. A routine rheumatologic laboratory evaluation was negative.
A head computed tomography ordered before coming to the clinic showed normal results. An MRI of the head was obtained to evaluate for ischemic cause or structural abnormality (Figures 1 and 2). Given the patient’s presentation and the pattern seen on the MRI results, it was determined that large pannus posterior to the dens, severely narrowing the spinal canal, was most likely the diagnosis. A second opinion confirmed the diagnosis, and a second MRI revealed stabilization with no signs of enhancement.
The patient was advised to meet with a neurosurgeon to remove the pannus. The patient agreed on occiput to C2 posterior instrument arthrodesis as well as decompression. A plain film radiograph showed C2-occipital repair after surgery (Figure 3). The patient recovered in the neurosurgical intensive care unit, and the rest of the recovery was uncomplicated. She showed some improvement in her headaches and unsteady gait. A postoperative pathologic evaluation of tissue was not available. She was referred to a rheumatologist to rule out an autoimmune disease as the cause for this pannus, but no autoimmune disease was found.
Discussion
Cervical pannus is relatively uncommon in those without RA. However, there are multiple reasons that a patient could develop a cervical pannus. Cervical pannus in RA and cervical pannus without RA may mimic each other clinically, but medical management is distinctly different. Consequently, a rheumatology consult is necessary to ensure that there is no undiagnosed autoimmune disorder. Our patient did not have RA, and a neurosurgery intervention was needed to manage her headaches and unsteady gait. Although we could not isolate a cause of this patient’s cervical pannus development, we believed that nonintervention would adversely affect this patient.
The course of pannus progression can be fatal especially if left untreated.12 MRI can detect a pannus and may be helpful for planning surgery.13 Surgical resection has been the treatment of choice for patients with neurologic symptoms.14 However, some cases have reported resolution of pannus associated with RA and other forms of chronic atlantoaxial instability only after posterior stabilization.14In order to manage pannus, cervical spine examination for the diagnosis of cervical involvement is encouraged to prevent morbidity and mortality.13 There are new data that demonstrated the potential of using retinoid X receptor agonists, such as bexarotene, as a treatment against the development and progression of pannus.14
Conclusions
We present a patient with cervical pannus disease without RA whose diagnosis was based on the pathognomonic pattern seen on MRI. She showed a clinically significant recovery with an occiput to C2 posterior instrument arthrodesis as well as decompression. She showed marked improvements in her headaches and unsteady gait. This case report highlights the importance of realizing cervical pannus as a disease found in patients without RA. It serves as an alert to clinicians for timely detection, diagnosis, and initiation of treatment to prevent mortality and long-term neurologic sequelae of cervical pannus.
Although further studies of early diagnosis and treatment for cervical pannus are warranted, we propose that including pannus in a differential diagnosis for patients with no RA could be lifesaving.
Cervical pannus is a disease that could easily develop in an active-duty soldier or veteran. The disease has been associated with trauma and rheumatoid arthritis, or can be idiopathic. For years, cervical pannus has been closely tied to rheumatoid arthritis; however, a study published in 2019 showed that only 28% of patients with cervical pannus had an associated diagnosis of rheumatoid arthritis.1 In the same study, 18% of patients had undergone some type of prior cervical spine surgery as the next most common cause. The condition also can occur years after an injury.
Background
In the US, 42,000 veterans are living with spinal cord disease, and thousands of these veterans have surgery every year.2 Service men and women and veterans are at risk for cervical pannus as they age especially if they have a history of rheumatoid arthritis, cervical spine surgery, trauma, and numerous other causes. It is critical for health care providers who treat this population to understand cervical pannus, how to recognize it, and how to identify patients at risk. A cervical pannus can be life threatening if not detected and treated properly.
There is no clear definition for cervical pannus. Some researchers think of it as the chronically inflamed synovial membrane in patients with rheumatoid arthritis (RA); others consider it as a specialized synovial membrane derived from vascular soft tissue structures at or near the bone synovial membrane.3 The pathogenesis for developing a pannus is not well understood, and little is known when a pannus begins or its initial location. A pannus formation can occur in any synovial joint in the body, such as wrists, metacarpophalangeal joint, proximal interphalangeal joint, and cervical joints.
A cervical pannus can cause serious complications. It can lead to a cervical subluxation in up to 4% of patients with RA, or it also can occur spontaneously in some patients without RA especially those with trauma or cancer.4
There are 2 suggested mechanisms by which the synovial membrane proliferates. It was originally believed that T cells from the chronic inflamed joint lead to the pannus formation by initiating an autoimmune reaction through the production of different cytokines against arthritogenic agents.3-5 These cytokines increase inflammation by recruiting neutrophils and activating various kinds of macrophages that might lead to increased osteoclast activity.6 Osteoclastic activity can damage bone and allow the synovium to penetrate the bone, forming the pannus.
Another proposed mechanism is that the synovial cells hyperpolarize and hypertrophy automatically without T-cell help by expressing oncogenes and their proteins.3 In either case, angiogenesis follows this proliferation and increases the influx of inflammatory cells into the joints, which can lead to more destruction.7 This increase in blood supply to the synovial membrane is important in the growth of the pannus and can have a damaging effect to cartilage, bone, and joints.4,7
Cervical pannus can progress in patients with prolonged use of corticosteroids.8 Because a pannus can put pressure on any segment of the cervical spine and the cranio-cervical junction leading to cervical instability, patients with this condition may present with a variety of clinical symptoms.9 The most frequently reported clinical features include neck pain, easy fatigability, difficulty walking, abnormal gait, increased clumsiness, and numbness and tingling in the arms. Patients also may complain of neck stiffness and decreased neck motion.10Cervical pannus is most frequently seen in patients with RA. However, patients without a RA diagnosis and incidental atlantoaxial pannus on cervical spine magnetic resonance imaging (MRI) are unlikely to have previously undiagnosed RA.11
Case Presentation
A 70-year-old white woman presented to the neurology clinic at Gretna Medical Center in Virginia in December 2016 with constant headache and imbalance that started in September 2016. She characterized the pain as predominately pressure (6 on a 10-point pain scale) with occasional shooting pains. The pain started at the left occipital lobe and radiated toward the left temporal lobe and left eye. The patient also stated that it was very difficult to lay her head down on a pillow to sleep and that she had to use a recliner in order to sleep over the past 3 months. She reported that the headache felt slightly worse if she had a lot of repetitive head and neck movements during the day. There was no photophobia, phonophobia, nausea, vomiting, facial paresthesias, lacrimation, nasal congestion, confusion, or impaired speech.
The patient’s lack of balance, which resulted in an unsteady gait, had started 1 month before and had increased significantly in the past 2 to 3 weeks. She stated that the unsteady gait was associated with numbness in her right upper and lower extremities, although more intense in the right lower extremity. Aside from the headaches, paresthesia, and unsteady gait, the patient reported no other major symptoms. She did not smoke tobacco or drink alcohol. Her family history revealed that her brothers had heart disease.
The patient’s vital signs at physical examination included heart rate, 83 beats per minute; blood pressure, 159/75 mm hg; temporal temperature, 97.9 °F; and respiratory rate, 20 breaths per minute. The patient’s gait was unsteady, needing stabilization by holding on to her husband’s arm, slightly favoring right lower extremity. Finger-to-nose test, rapid alternating movements, heel-knee-shin testing were all normal. The Romberg sign was positive. The patient could rise on toes and heels with slight balance disturbance. Deep tendon reflexes and reflexes in the upper and lower extremities was symmetric 2+ bilaterally. Musculoskeletal examination revealed strength and tone in all major muscle groups and demonstrated symmetrical movements with no fasciculation noted. A rheumatologic evaluation showed no abnormalities, including inspection of hands, feet, major joints, and other range of motion, besides her neck. The rest of the physical, cognitive, and neurologic examination findings were otherwise unremarkable. A routine rheumatologic laboratory evaluation was negative.
A head computed tomography ordered before coming to the clinic showed normal results. An MRI of the head was obtained to evaluate for ischemic cause or structural abnormality (Figures 1 and 2). Given the patient’s presentation and the pattern seen on the MRI results, it was determined that large pannus posterior to the dens, severely narrowing the spinal canal, was most likely the diagnosis. A second opinion confirmed the diagnosis, and a second MRI revealed stabilization with no signs of enhancement.
The patient was advised to meet with a neurosurgeon to remove the pannus. The patient agreed on occiput to C2 posterior instrument arthrodesis as well as decompression. A plain film radiograph showed C2-occipital repair after surgery (Figure 3). The patient recovered in the neurosurgical intensive care unit, and the rest of the recovery was uncomplicated. She showed some improvement in her headaches and unsteady gait. A postoperative pathologic evaluation of tissue was not available. She was referred to a rheumatologist to rule out an autoimmune disease as the cause for this pannus, but no autoimmune disease was found.
Discussion
Cervical pannus is relatively uncommon in those without RA. However, there are multiple reasons that a patient could develop a cervical pannus. Cervical pannus in RA and cervical pannus without RA may mimic each other clinically, but medical management is distinctly different. Consequently, a rheumatology consult is necessary to ensure that there is no undiagnosed autoimmune disorder. Our patient did not have RA, and a neurosurgery intervention was needed to manage her headaches and unsteady gait. Although we could not isolate a cause of this patient’s cervical pannus development, we believed that nonintervention would adversely affect this patient.
The course of pannus progression can be fatal especially if left untreated.12 MRI can detect a pannus and may be helpful for planning surgery.13 Surgical resection has been the treatment of choice for patients with neurologic symptoms.14 However, some cases have reported resolution of pannus associated with RA and other forms of chronic atlantoaxial instability only after posterior stabilization.14In order to manage pannus, cervical spine examination for the diagnosis of cervical involvement is encouraged to prevent morbidity and mortality.13 There are new data that demonstrated the potential of using retinoid X receptor agonists, such as bexarotene, as a treatment against the development and progression of pannus.14
Conclusions
We present a patient with cervical pannus disease without RA whose diagnosis was based on the pathognomonic pattern seen on MRI. She showed a clinically significant recovery with an occiput to C2 posterior instrument arthrodesis as well as decompression. She showed marked improvements in her headaches and unsteady gait. This case report highlights the importance of realizing cervical pannus as a disease found in patients without RA. It serves as an alert to clinicians for timely detection, diagnosis, and initiation of treatment to prevent mortality and long-term neurologic sequelae of cervical pannus.
Although further studies of early diagnosis and treatment for cervical pannus are warranted, we propose that including pannus in a differential diagnosis for patients with no RA could be lifesaving.
1. Zvaifler NJ, Firestein GS. Pannus and pannocytes. Alternative models of joint destruction in rheumatoid arthritis. Arthritis Rheum. 1994;37(6):783-789.
2. Henderson DR. Vertical atlanto-axial subluxation in rheumatoid arthritis. Rheumatol Rehabil. 1975;14(1):31-38.
3. Skapenko A, Leipe J, Lipsky PE, Schulze-Koops H. The role of the T cell in autoimmune inflammation. Arthritis Res Ther. 2005;7(suppl 2):S4-S14.
4. Wang R, Zhang L, Zhang X, et al. Regulation of activation-induced receptor activator of NF-kappaB ligand (RANKL) expression in T cells. Eur J Immunol. 2002;32(4):1090-1098.
5. Koch AE. Angiogenesis as a target in rheumatoid arthritis. Ann Rheum Dis. 2003;62(suppl 2):ii60-ii67.
6. Reiter MF, Boden SD. Inflammatory disorders of the cervical spine. Spine (Phila Pa 1976). 1998;23(24):2755-2766.
7. Alaya Z, Lataoui S, Amri D, Zaghouani H, Bouajina E. Atlantoaxial instability: an exceptional complication of ankylosing spondylitis. Egypt Rheumatol. 2018;40(2):141-143.
8. Walter KD, Tassone JC. Atlantoaxial instability. In: Micheli LJ, ed. Encyclopedia of Sports Medicine. Thousand Oaks, CA: SAGE Reference; 2011:122-124.
9. Joyce AA, Williams JN, Shi J, Mandell JC, Isaac Z, Ermann J. Atlanto-axial pannus in patients with and without rheumatoid arthritis. J Rheumatol. 2019;46(11):1431-1437.
10. Neva MH, Myllykangas-Luosujärvi R, Kautiainen H, Kauppi M. Mortality associated with cervical spine disorders: a population-based study of 1666 patients with rheumatoid arthritis who died in Finland in 1989. Rheumatology (Oxford). 2001;40(2):123-127.
11. Mallory GW, Halasz SR, Clarke MJ. Advances in the treatment of cervical rheumatoid: less surgery and less morbidity. World J Orthop. 2014;5(3):292-303.
12. Lagares A, Arrese I, Pascual B, Gòmez PA, Ramos A, Lobato RD. Pannus resolution after occipitocervical fusion in a non-rheumatoid atlanto-axial instability. Eur Spine J. 2006;15(3):366-369.
13. Chung J, Bak KH, Yi H-J, Chun HJ, Ryu JI, Han M-H. Upper cervical subluxation and cervicomedullary junction compression in patients with rheumatoid arthritis. J Korean Neurosurg Soc. 2019;62(6):661-670.
14. Li Y, Xing Q, Wei Y, et al. Activation of RXR by bexarotene inhibits inflammatory conditions in human rheumatoid arthritis fibroblast‑like synoviocytes. Int J Mol Med. 2019;44(5):1963-1970.
1. Zvaifler NJ, Firestein GS. Pannus and pannocytes. Alternative models of joint destruction in rheumatoid arthritis. Arthritis Rheum. 1994;37(6):783-789.
2. Henderson DR. Vertical atlanto-axial subluxation in rheumatoid arthritis. Rheumatol Rehabil. 1975;14(1):31-38.
3. Skapenko A, Leipe J, Lipsky PE, Schulze-Koops H. The role of the T cell in autoimmune inflammation. Arthritis Res Ther. 2005;7(suppl 2):S4-S14.
4. Wang R, Zhang L, Zhang X, et al. Regulation of activation-induced receptor activator of NF-kappaB ligand (RANKL) expression in T cells. Eur J Immunol. 2002;32(4):1090-1098.
5. Koch AE. Angiogenesis as a target in rheumatoid arthritis. Ann Rheum Dis. 2003;62(suppl 2):ii60-ii67.
6. Reiter MF, Boden SD. Inflammatory disorders of the cervical spine. Spine (Phila Pa 1976). 1998;23(24):2755-2766.
7. Alaya Z, Lataoui S, Amri D, Zaghouani H, Bouajina E. Atlantoaxial instability: an exceptional complication of ankylosing spondylitis. Egypt Rheumatol. 2018;40(2):141-143.
8. Walter KD, Tassone JC. Atlantoaxial instability. In: Micheli LJ, ed. Encyclopedia of Sports Medicine. Thousand Oaks, CA: SAGE Reference; 2011:122-124.
9. Joyce AA, Williams JN, Shi J, Mandell JC, Isaac Z, Ermann J. Atlanto-axial pannus in patients with and without rheumatoid arthritis. J Rheumatol. 2019;46(11):1431-1437.
10. Neva MH, Myllykangas-Luosujärvi R, Kautiainen H, Kauppi M. Mortality associated with cervical spine disorders: a population-based study of 1666 patients with rheumatoid arthritis who died in Finland in 1989. Rheumatology (Oxford). 2001;40(2):123-127.
11. Mallory GW, Halasz SR, Clarke MJ. Advances in the treatment of cervical rheumatoid: less surgery and less morbidity. World J Orthop. 2014;5(3):292-303.
12. Lagares A, Arrese I, Pascual B, Gòmez PA, Ramos A, Lobato RD. Pannus resolution after occipitocervical fusion in a non-rheumatoid atlanto-axial instability. Eur Spine J. 2006;15(3):366-369.
13. Chung J, Bak KH, Yi H-J, Chun HJ, Ryu JI, Han M-H. Upper cervical subluxation and cervicomedullary junction compression in patients with rheumatoid arthritis. J Korean Neurosurg Soc. 2019;62(6):661-670.
14. Li Y, Xing Q, Wei Y, et al. Activation of RXR by bexarotene inhibits inflammatory conditions in human rheumatoid arthritis fibroblast‑like synoviocytes. Int J Mol Med. 2019;44(5):1963-1970.
Infected Bronchogenic Cyst With Left Atrial, Pulmonary Artery, and Esophageal Compression
Bronchogenic cyst is a rare foregut malformation that typically presents during the second decade of life that arises due to aberrant development from the tracheobronchial tree.1 Mediastinal bronchogenic cyst is the most common primary cystic lesion of the mediastinum, and bronchogenic cysts of the mediastinum represent 18% of all primary mediastinal malformations.2 Patients with mediastinal bronchogenic cysts may present with symptoms of cough, dyspnea, or wheezing if there is encroachment on surrounding structures.
Rarely, bronchogenic cysts can become infected. Definitive treatment of bronchogenic cysts is surgical excision; however, endobronchial ultrasound (EBUS)-guided drainage also can be employed. EBUS-guided drainage may be used when the cyst cannot be distinguished from solid mass on computed tomography (CT) images, to relieve symptomatic compression of surrounding structures, or to provide a histologic or microbial diagnosis in cases where surgical excision is not immediately available. We present the first-ever described case of bronchogenic cyst infected with Actinomyces, diagnosed by EBUS-guided drainage as well as a review of the literature regarding infected bronchogenic cysts and management of cysts affecting mediastinal structures.
Case Presentation
A 57-year-old African American male presented with a 4-day history of continuous, sharp, substernal chest pain accompanied by dyspnea. Additionally, the patient reported progressive dysphagia to solids. The posteroanterior view of a chest X-ray showed a widened mediastinum with splaying of the carina. A contrast-enhanced CT of the chest showed a large, middle mediastinal mass of heterogenous density measuring 7.3. × 7.0 × 6.0 cm with compression of the right pulmonary artery, left atria, superior vena cava and esophagus (Figure 1).
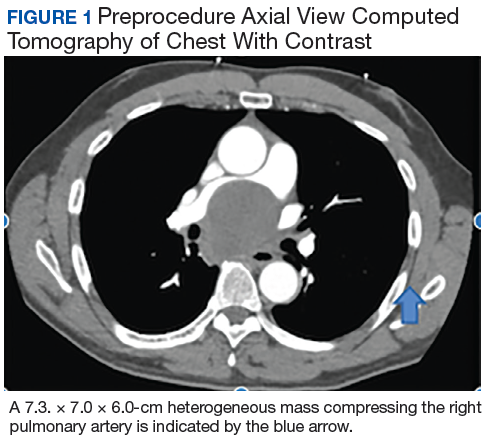
The mass demonstrated neither clear fluid-fluid level nor rounded structure with a distinct wall and uniform attenuation consistent with pure cystic structure and, in fact, was concerning for malignant process, such as lymphoma. Due to the malignancy concern and the findings of significant compression of surrounding mediastinal structures, the decision was made to proceed with bronchoscopy and EBUS-guided transbronchial needle aspiration (EBUS-TBNA) to assist in diagnosis and potentially provide symptomatic relief.
Under general anesthesia a P160 Olympus bronchoscope was advanced into the tracheobronchial tree; bronchoscopy with airway inspection revealed splayed carina with obtuse angle but was otherwise unremarkable. Next, an EBUS P160 fiber optic Olympus bronchoscope was advanced; ultrasound demonstrated a cystic structure. The EBUS-TBNA of cystic structure yielded 20 mL of brown, purulent fluid with decompression bringing pulmonary artery in ultrasound field (Figure 2). Rapid on-site cytology was performed with no preliminary findings of malignancy. The fluid was then sent for cytology and microbiologic evaluation.
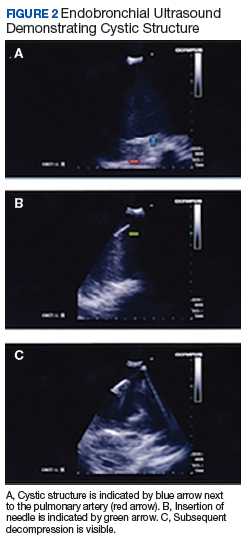
Following EBUS-guided aspiration, the patient reported significant improvement in chest pain, dyspnea, and dysphagia. A repeat chest CT demonstrated decrease in mass size to 5.9 × 5.5 × 4.6 cm with relief of the compression of the right pulmonary artery and decreased mass effect on the carina (Figure 3). Pathology ultimately demonstrated no evidence of malignancy but did demonstrate filamentous material with sulfur granules and anthracotic pigment suggestive of Actinomyces infection (Figure 4).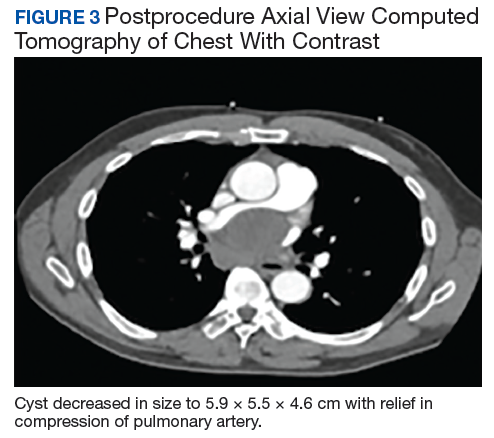
The patient was placed on amoxicillin/clavulanate 875 mg to 125 mg twice daily for 4 weeks based on antibiotic susceptibility testing to prevent progression to mediastinitis related to Actinomyces infection. The duration of therapy was extrapolated from treatment regimens described in case series of cervicofacial and abdominal Actinomyces infections.3 Thoracic surgery evaluation for definitive excision of cyst was recommended after the patient completed his course of antibiotics.
The patient underwent dental evaluation to identify the source of Actinomyces infection but there appeared to be no odontogenic source. The patient also had extensive skin survey with no findings of overt source of Actinomyces and CT abdomen/pelvis also identified no abscess that could be a potential source. He subsequently underwent thoracoscopic resection with pathology demonstrating a fibrous cyst wall lined with ciliated columnar epithelium consistent with diagnosis of bronchogenic cyst (Figure 5).
Discussion
Bronchogenic cysts can present at birth or later in life; patients may be asymptomatic for decades prior to discovery.4 Cysts located in the mediastinum can cause compression of the trachea and esophagus and cause cough, dyspnea, chest pain, and dysphagia.5 More life-threatening complications include infection, tracheal compression, malignant transformation, superior vena cava syndrome, or spontaneous rupture into the airway.6,7
Infection can occasionally occur, and various bacterial etiologies have been described. Hernandez-Solis and colleagues describe 12 cases of superinfected bronchogenic cysts with Staphylococcus aureus and Pseudomonas aeroginosa, the most commonly described organisms.8 Casal and colleagues describe a case of α-hemolytic Streptococci treated with amoxicillin.9 Liman and colleagues describe 2 cases of bronchogenic cyst infected with Mycobacterium and cite an additional case report by Lin and colleagues similarly infected by Mycobacterium.10,11 Only 1 case was identified to have direct bronchial communication as a potential source of introduction of infection into bronchogenic cyst. In other cases, potential sources of infection were not identified, though it was postulated that direct ventilation could be a potential source of inoculation.
Surgical resection of mediastinal bronchogenic cysts has traditionally been considered the definitive treatment of choice.12,13 However, bronchogenic cysts may sometimes be difficult to differentiate from soft tissue tumors by chest CT, especially in cases of cysts with nonserous fluid. In particular, cysts that are infected are likely to have increased density and high attenuation on imaging; therefore, surgical excision may be delayed until diagnosis is made.14 Due to low complication rates, EBUS is increasingly used in the diagnosis and therapeutic management of bronchogenic cysts as an alternative to surgery, particularly for those who are symptomatic.15,16 Ultrasound guidance can allow for complete aspiration of the cyst, causing complete collapse of the cystic space and can facilitate adhesion between the mucosal surfaces lining the cavity and reduce recurrence.17 Nonetheless, bronchogenic cysts that are found to be infected, recur, or have a malignant component should be resected for definitive treatment.18
The mass discovered on our patient’s imaging appeared to have heterogenous attenuation consistent with malignancy rather than homogenous attenuation surrounded by a clearly demarcated wall consistent with a cystic structure; therefore, EBUS-TBNA was initially pursued and yielded an expedited diagnosis of the first-ever described bronchogenic cyst with Actinomyces superinfection as well as dramatic symptomatic relief of compression of surrounding mediastinal structures, particularly of the right pulmonary artery. As this is a congenital malformation, the patient was likely asymptomatic until the cyst became infected, after which he likely experience cyst growth with subsequent encroachment of surrounding mediastinal structures. Additionally, identification of pathogen by TBNA allowed for treatment before surgical excision, possibly avoiding accidental spread of pathogen intraoperatively.
Conclusions
Our case adds to the literature on the use of EBUS-TBNA as a diagnostic and therapeutic modality for bronchogenic cyst. While cases of mediastinitis and pleural effusion following EBUS-guided aspiration of bronchogenic cysts have been reported, complications are extremely rare.19 EBUS is increasingly favored as a means of immediate diagnosis and treatment in cases where CT imaging may not overtly suggest cystic structure and in patients experiencing compression of critical mediastinal structures.
1. Weber T, Roth TC, Beshay M, Herrmann P, Stein R, Schmid RA. Video-assisted thoracoscopic surgery of mediastinal bronchogenic cysts in adults: a single-center experience. Ann Thorac Surg. 2004;78(3):987-991.
2. Martinod E, Pons F, Azorin J, et al. Thoracoscopic excision of mediastinal bronchogenic cysts: results in 20 cases. Ann Thorac Surg. 2000;69(5):1525-1528.
3. Könönen E, Wade WG. Actinomyces and related organisms in human infections. Clin Microbiol Rev. 2015;28(2):419-442.
4. Ribet ME, Copin MC, Gosselin BH. Bronchogenic cysts of the lung. Ann Thorac Surg. 1996;61(6):1636-1640.
5. Guillem P, Porte H, Marquette CH, Wurtz A. Progressive dysphonia and acute respiratory failure: revealing a bronchogenic cyst. Eur J Cardiothorac Surg. 1997;12(6):925-927.
6. McAdams HP, Kirejczyk WM, Rosado-de-Christenson ML, Matsumoto S. Bronchogenic cyst: imaging features with clinical and histopathologic correlation. Radiology. 2000;217(2):441-446.
7. Rammohan G, Berger HW, Lajam F, Buhain WJ. Superior vena cava syndrome caused by bronchogenic cyst. Chest. 1975;68(4):599-601.
8. Hernández-Solís A, Cruz-Ortiz H, Gutiérrez-Díaz Ceballos ME, Cicero-Sabido R. Quistes broncogénicos. Importancia de la infección en adultos. Estudio de 12 casos [Bronchogenic cysts. Importance of infection in adults. Study of 12 cases]. Cir Cir. 2015;83(2):112-116.
9. Casal RF, Jimenez CA, Mehran RJ, et al. Infected mediastinal bronchogenic cyst successfully treated by endobronchial ultrasound-guided fine-needle aspiration. Ann Thorac Surg. 2010;90(4):e52-e53.
10. Liman ST, Dogan Y, Topcu S, Karabulut N, Demirkan N, Keser Z. Mycobacterial infection of intraparenchymal bronchogenic cysts. Respir Med. 2006;100(11):2060-2062.
11. Lin SH, Lee LN, Chang YL, Lee YC, Ding LW, Hsueh PR. Infected bronchogenic cyst due to Mycobacterium avium in an immunocompetent patient. J Infect. 2005;51(3):e131-e133.
12. Gharagozloo F, Dausmann MJ, McReynolds SD, Sanderson DR, Helmers RA. Recurrent bronchogenic pseudocyst 24 years after incomplete excision. Report of a case. Chest. 1995;108(3):880-883.
13. Bolton JW, Shahian DM. Asymptomatic bronchogenic cysts: what is the best management? Ann Thorac Surg. 1992;53(6):1134-1137.
14. Sarper A, Ayten A, Golbasi I, Demircan A, Isin E. Bronchogenic cyst. Tex Heart Inst J. 2003;30(2):105-108.
15. Varela-Lema L, Fernández-Villar A, Ruano-Ravina A. Effectiveness and safety of endobronchial ultrasound-transbronchial needle aspiration: a systematic review. Eur Respir J. 2009;33(5):1156-1164.
16. Maturu VN, Dhooria S, Agarwal R. Efficacy and safety of transbronchial needle aspiration in diagnosis and treatment of mediastinal bronchogenic cysts: systematic review of case reports. J Bronchology Interv Pulmonol. 2015;22(3):195-203.
17. Galluccio G, Lucantoni G. Mediastinal bronchogenic cyst’s recurrence treated with EBUS-FNA with a long-term follow-up. Eur J Cardiothorac Surg. 2006;29(4):627-629.
18. Lee DH, Park CK, Kum DY, Kim JB, Hwang I. Clinical characteristics and management of intrathoracic bronchogenic cysts: a single center experience. Korean J Thorac Cardiovasc Surg. 2011;44(4):279-284.
19. Onuki T, Kuramochi M, Inagaki M. Mediastinitis of bronchogenic cyst caused by endobronchial ultrasound-guided transbronchial needle aspiration. Respirol Case Rep. 2014;2(2):73-75.
Bronchogenic cyst is a rare foregut malformation that typically presents during the second decade of life that arises due to aberrant development from the tracheobronchial tree.1 Mediastinal bronchogenic cyst is the most common primary cystic lesion of the mediastinum, and bronchogenic cysts of the mediastinum represent 18% of all primary mediastinal malformations.2 Patients with mediastinal bronchogenic cysts may present with symptoms of cough, dyspnea, or wheezing if there is encroachment on surrounding structures.
Rarely, bronchogenic cysts can become infected. Definitive treatment of bronchogenic cysts is surgical excision; however, endobronchial ultrasound (EBUS)-guided drainage also can be employed. EBUS-guided drainage may be used when the cyst cannot be distinguished from solid mass on computed tomography (CT) images, to relieve symptomatic compression of surrounding structures, or to provide a histologic or microbial diagnosis in cases where surgical excision is not immediately available. We present the first-ever described case of bronchogenic cyst infected with Actinomyces, diagnosed by EBUS-guided drainage as well as a review of the literature regarding infected bronchogenic cysts and management of cysts affecting mediastinal structures.
Case Presentation
A 57-year-old African American male presented with a 4-day history of continuous, sharp, substernal chest pain accompanied by dyspnea. Additionally, the patient reported progressive dysphagia to solids. The posteroanterior view of a chest X-ray showed a widened mediastinum with splaying of the carina. A contrast-enhanced CT of the chest showed a large, middle mediastinal mass of heterogenous density measuring 7.3. × 7.0 × 6.0 cm with compression of the right pulmonary artery, left atria, superior vena cava and esophagus (Figure 1).
The mass demonstrated neither clear fluid-fluid level nor rounded structure with a distinct wall and uniform attenuation consistent with pure cystic structure and, in fact, was concerning for malignant process, such as lymphoma. Due to the malignancy concern and the findings of significant compression of surrounding mediastinal structures, the decision was made to proceed with bronchoscopy and EBUS-guided transbronchial needle aspiration (EBUS-TBNA) to assist in diagnosis and potentially provide symptomatic relief.
Under general anesthesia a P160 Olympus bronchoscope was advanced into the tracheobronchial tree; bronchoscopy with airway inspection revealed splayed carina with obtuse angle but was otherwise unremarkable. Next, an EBUS P160 fiber optic Olympus bronchoscope was advanced; ultrasound demonstrated a cystic structure. The EBUS-TBNA of cystic structure yielded 20 mL of brown, purulent fluid with decompression bringing pulmonary artery in ultrasound field (Figure 2). Rapid on-site cytology was performed with no preliminary findings of malignancy. The fluid was then sent for cytology and microbiologic evaluation.
Following EBUS-guided aspiration, the patient reported significant improvement in chest pain, dyspnea, and dysphagia. A repeat chest CT demonstrated decrease in mass size to 5.9 × 5.5 × 4.6 cm with relief of the compression of the right pulmonary artery and decreased mass effect on the carina (Figure 3). Pathology ultimately demonstrated no evidence of malignancy but did demonstrate filamentous material with sulfur granules and anthracotic pigment suggestive of Actinomyces infection (Figure 4).
The patient was placed on amoxicillin/clavulanate 875 mg to 125 mg twice daily for 4 weeks based on antibiotic susceptibility testing to prevent progression to mediastinitis related to Actinomyces infection. The duration of therapy was extrapolated from treatment regimens described in case series of cervicofacial and abdominal Actinomyces infections.3 Thoracic surgery evaluation for definitive excision of cyst was recommended after the patient completed his course of antibiotics.
The patient underwent dental evaluation to identify the source of Actinomyces infection but there appeared to be no odontogenic source. The patient also had extensive skin survey with no findings of overt source of Actinomyces and CT abdomen/pelvis also identified no abscess that could be a potential source. He subsequently underwent thoracoscopic resection with pathology demonstrating a fibrous cyst wall lined with ciliated columnar epithelium consistent with diagnosis of bronchogenic cyst (Figure 5).
Discussion
Bronchogenic cysts can present at birth or later in life; patients may be asymptomatic for decades prior to discovery.4 Cysts located in the mediastinum can cause compression of the trachea and esophagus and cause cough, dyspnea, chest pain, and dysphagia.5 More life-threatening complications include infection, tracheal compression, malignant transformation, superior vena cava syndrome, or spontaneous rupture into the airway.6,7
Infection can occasionally occur, and various bacterial etiologies have been described. Hernandez-Solis and colleagues describe 12 cases of superinfected bronchogenic cysts with Staphylococcus aureus and Pseudomonas aeroginosa, the most commonly described organisms.8 Casal and colleagues describe a case of α-hemolytic Streptococci treated with amoxicillin.9 Liman and colleagues describe 2 cases of bronchogenic cyst infected with Mycobacterium and cite an additional case report by Lin and colleagues similarly infected by Mycobacterium.10,11 Only 1 case was identified to have direct bronchial communication as a potential source of introduction of infection into bronchogenic cyst. In other cases, potential sources of infection were not identified, though it was postulated that direct ventilation could be a potential source of inoculation.
Surgical resection of mediastinal bronchogenic cysts has traditionally been considered the definitive treatment of choice.12,13 However, bronchogenic cysts may sometimes be difficult to differentiate from soft tissue tumors by chest CT, especially in cases of cysts with nonserous fluid. In particular, cysts that are infected are likely to have increased density and high attenuation on imaging; therefore, surgical excision may be delayed until diagnosis is made.14 Due to low complication rates, EBUS is increasingly used in the diagnosis and therapeutic management of bronchogenic cysts as an alternative to surgery, particularly for those who are symptomatic.15,16 Ultrasound guidance can allow for complete aspiration of the cyst, causing complete collapse of the cystic space and can facilitate adhesion between the mucosal surfaces lining the cavity and reduce recurrence.17 Nonetheless, bronchogenic cysts that are found to be infected, recur, or have a malignant component should be resected for definitive treatment.18
The mass discovered on our patient’s imaging appeared to have heterogenous attenuation consistent with malignancy rather than homogenous attenuation surrounded by a clearly demarcated wall consistent with a cystic structure; therefore, EBUS-TBNA was initially pursued and yielded an expedited diagnosis of the first-ever described bronchogenic cyst with Actinomyces superinfection as well as dramatic symptomatic relief of compression of surrounding mediastinal structures, particularly of the right pulmonary artery. As this is a congenital malformation, the patient was likely asymptomatic until the cyst became infected, after which he likely experience cyst growth with subsequent encroachment of surrounding mediastinal structures. Additionally, identification of pathogen by TBNA allowed for treatment before surgical excision, possibly avoiding accidental spread of pathogen intraoperatively.
Conclusions
Our case adds to the literature on the use of EBUS-TBNA as a diagnostic and therapeutic modality for bronchogenic cyst. While cases of mediastinitis and pleural effusion following EBUS-guided aspiration of bronchogenic cysts have been reported, complications are extremely rare.19 EBUS is increasingly favored as a means of immediate diagnosis and treatment in cases where CT imaging may not overtly suggest cystic structure and in patients experiencing compression of critical mediastinal structures.
Bronchogenic cyst is a rare foregut malformation that typically presents during the second decade of life that arises due to aberrant development from the tracheobronchial tree.1 Mediastinal bronchogenic cyst is the most common primary cystic lesion of the mediastinum, and bronchogenic cysts of the mediastinum represent 18% of all primary mediastinal malformations.2 Patients with mediastinal bronchogenic cysts may present with symptoms of cough, dyspnea, or wheezing if there is encroachment on surrounding structures.
Rarely, bronchogenic cysts can become infected. Definitive treatment of bronchogenic cysts is surgical excision; however, endobronchial ultrasound (EBUS)-guided drainage also can be employed. EBUS-guided drainage may be used when the cyst cannot be distinguished from solid mass on computed tomography (CT) images, to relieve symptomatic compression of surrounding structures, or to provide a histologic or microbial diagnosis in cases where surgical excision is not immediately available. We present the first-ever described case of bronchogenic cyst infected with Actinomyces, diagnosed by EBUS-guided drainage as well as a review of the literature regarding infected bronchogenic cysts and management of cysts affecting mediastinal structures.
Case Presentation
A 57-year-old African American male presented with a 4-day history of continuous, sharp, substernal chest pain accompanied by dyspnea. Additionally, the patient reported progressive dysphagia to solids. The posteroanterior view of a chest X-ray showed a widened mediastinum with splaying of the carina. A contrast-enhanced CT of the chest showed a large, middle mediastinal mass of heterogenous density measuring 7.3. × 7.0 × 6.0 cm with compression of the right pulmonary artery, left atria, superior vena cava and esophagus (Figure 1).
The mass demonstrated neither clear fluid-fluid level nor rounded structure with a distinct wall and uniform attenuation consistent with pure cystic structure and, in fact, was concerning for malignant process, such as lymphoma. Due to the malignancy concern and the findings of significant compression of surrounding mediastinal structures, the decision was made to proceed with bronchoscopy and EBUS-guided transbronchial needle aspiration (EBUS-TBNA) to assist in diagnosis and potentially provide symptomatic relief.
Under general anesthesia a P160 Olympus bronchoscope was advanced into the tracheobronchial tree; bronchoscopy with airway inspection revealed splayed carina with obtuse angle but was otherwise unremarkable. Next, an EBUS P160 fiber optic Olympus bronchoscope was advanced; ultrasound demonstrated a cystic structure. The EBUS-TBNA of cystic structure yielded 20 mL of brown, purulent fluid with decompression bringing pulmonary artery in ultrasound field (Figure 2). Rapid on-site cytology was performed with no preliminary findings of malignancy. The fluid was then sent for cytology and microbiologic evaluation.
Following EBUS-guided aspiration, the patient reported significant improvement in chest pain, dyspnea, and dysphagia. A repeat chest CT demonstrated decrease in mass size to 5.9 × 5.5 × 4.6 cm with relief of the compression of the right pulmonary artery and decreased mass effect on the carina (Figure 3). Pathology ultimately demonstrated no evidence of malignancy but did demonstrate filamentous material with sulfur granules and anthracotic pigment suggestive of Actinomyces infection (Figure 4).
The patient was placed on amoxicillin/clavulanate 875 mg to 125 mg twice daily for 4 weeks based on antibiotic susceptibility testing to prevent progression to mediastinitis related to Actinomyces infection. The duration of therapy was extrapolated from treatment regimens described in case series of cervicofacial and abdominal Actinomyces infections.3 Thoracic surgery evaluation for definitive excision of cyst was recommended after the patient completed his course of antibiotics.
The patient underwent dental evaluation to identify the source of Actinomyces infection but there appeared to be no odontogenic source. The patient also had extensive skin survey with no findings of overt source of Actinomyces and CT abdomen/pelvis also identified no abscess that could be a potential source. He subsequently underwent thoracoscopic resection with pathology demonstrating a fibrous cyst wall lined with ciliated columnar epithelium consistent with diagnosis of bronchogenic cyst (Figure 5).
Discussion
Bronchogenic cysts can present at birth or later in life; patients may be asymptomatic for decades prior to discovery.4 Cysts located in the mediastinum can cause compression of the trachea and esophagus and cause cough, dyspnea, chest pain, and dysphagia.5 More life-threatening complications include infection, tracheal compression, malignant transformation, superior vena cava syndrome, or spontaneous rupture into the airway.6,7
Infection can occasionally occur, and various bacterial etiologies have been described. Hernandez-Solis and colleagues describe 12 cases of superinfected bronchogenic cysts with Staphylococcus aureus and Pseudomonas aeroginosa, the most commonly described organisms.8 Casal and colleagues describe a case of α-hemolytic Streptococci treated with amoxicillin.9 Liman and colleagues describe 2 cases of bronchogenic cyst infected with Mycobacterium and cite an additional case report by Lin and colleagues similarly infected by Mycobacterium.10,11 Only 1 case was identified to have direct bronchial communication as a potential source of introduction of infection into bronchogenic cyst. In other cases, potential sources of infection were not identified, though it was postulated that direct ventilation could be a potential source of inoculation.
Surgical resection of mediastinal bronchogenic cysts has traditionally been considered the definitive treatment of choice.12,13 However, bronchogenic cysts may sometimes be difficult to differentiate from soft tissue tumors by chest CT, especially in cases of cysts with nonserous fluid. In particular, cysts that are infected are likely to have increased density and high attenuation on imaging; therefore, surgical excision may be delayed until diagnosis is made.14 Due to low complication rates, EBUS is increasingly used in the diagnosis and therapeutic management of bronchogenic cysts as an alternative to surgery, particularly for those who are symptomatic.15,16 Ultrasound guidance can allow for complete aspiration of the cyst, causing complete collapse of the cystic space and can facilitate adhesion between the mucosal surfaces lining the cavity and reduce recurrence.17 Nonetheless, bronchogenic cysts that are found to be infected, recur, or have a malignant component should be resected for definitive treatment.18
The mass discovered on our patient’s imaging appeared to have heterogenous attenuation consistent with malignancy rather than homogenous attenuation surrounded by a clearly demarcated wall consistent with a cystic structure; therefore, EBUS-TBNA was initially pursued and yielded an expedited diagnosis of the first-ever described bronchogenic cyst with Actinomyces superinfection as well as dramatic symptomatic relief of compression of surrounding mediastinal structures, particularly of the right pulmonary artery. As this is a congenital malformation, the patient was likely asymptomatic until the cyst became infected, after which he likely experience cyst growth with subsequent encroachment of surrounding mediastinal structures. Additionally, identification of pathogen by TBNA allowed for treatment before surgical excision, possibly avoiding accidental spread of pathogen intraoperatively.
Conclusions
Our case adds to the literature on the use of EBUS-TBNA as a diagnostic and therapeutic modality for bronchogenic cyst. While cases of mediastinitis and pleural effusion following EBUS-guided aspiration of bronchogenic cysts have been reported, complications are extremely rare.19 EBUS is increasingly favored as a means of immediate diagnosis and treatment in cases where CT imaging may not overtly suggest cystic structure and in patients experiencing compression of critical mediastinal structures.
1. Weber T, Roth TC, Beshay M, Herrmann P, Stein R, Schmid RA. Video-assisted thoracoscopic surgery of mediastinal bronchogenic cysts in adults: a single-center experience. Ann Thorac Surg. 2004;78(3):987-991.
2. Martinod E, Pons F, Azorin J, et al. Thoracoscopic excision of mediastinal bronchogenic cysts: results in 20 cases. Ann Thorac Surg. 2000;69(5):1525-1528.
3. Könönen E, Wade WG. Actinomyces and related organisms in human infections. Clin Microbiol Rev. 2015;28(2):419-442.
4. Ribet ME, Copin MC, Gosselin BH. Bronchogenic cysts of the lung. Ann Thorac Surg. 1996;61(6):1636-1640.
5. Guillem P, Porte H, Marquette CH, Wurtz A. Progressive dysphonia and acute respiratory failure: revealing a bronchogenic cyst. Eur J Cardiothorac Surg. 1997;12(6):925-927.
6. McAdams HP, Kirejczyk WM, Rosado-de-Christenson ML, Matsumoto S. Bronchogenic cyst: imaging features with clinical and histopathologic correlation. Radiology. 2000;217(2):441-446.
7. Rammohan G, Berger HW, Lajam F, Buhain WJ. Superior vena cava syndrome caused by bronchogenic cyst. Chest. 1975;68(4):599-601.
8. Hernández-Solís A, Cruz-Ortiz H, Gutiérrez-Díaz Ceballos ME, Cicero-Sabido R. Quistes broncogénicos. Importancia de la infección en adultos. Estudio de 12 casos [Bronchogenic cysts. Importance of infection in adults. Study of 12 cases]. Cir Cir. 2015;83(2):112-116.
9. Casal RF, Jimenez CA, Mehran RJ, et al. Infected mediastinal bronchogenic cyst successfully treated by endobronchial ultrasound-guided fine-needle aspiration. Ann Thorac Surg. 2010;90(4):e52-e53.
10. Liman ST, Dogan Y, Topcu S, Karabulut N, Demirkan N, Keser Z. Mycobacterial infection of intraparenchymal bronchogenic cysts. Respir Med. 2006;100(11):2060-2062.
11. Lin SH, Lee LN, Chang YL, Lee YC, Ding LW, Hsueh PR. Infected bronchogenic cyst due to Mycobacterium avium in an immunocompetent patient. J Infect. 2005;51(3):e131-e133.
12. Gharagozloo F, Dausmann MJ, McReynolds SD, Sanderson DR, Helmers RA. Recurrent bronchogenic pseudocyst 24 years after incomplete excision. Report of a case. Chest. 1995;108(3):880-883.
13. Bolton JW, Shahian DM. Asymptomatic bronchogenic cysts: what is the best management? Ann Thorac Surg. 1992;53(6):1134-1137.
14. Sarper A, Ayten A, Golbasi I, Demircan A, Isin E. Bronchogenic cyst. Tex Heart Inst J. 2003;30(2):105-108.
15. Varela-Lema L, Fernández-Villar A, Ruano-Ravina A. Effectiveness and safety of endobronchial ultrasound-transbronchial needle aspiration: a systematic review. Eur Respir J. 2009;33(5):1156-1164.
16. Maturu VN, Dhooria S, Agarwal R. Efficacy and safety of transbronchial needle aspiration in diagnosis and treatment of mediastinal bronchogenic cysts: systematic review of case reports. J Bronchology Interv Pulmonol. 2015;22(3):195-203.
17. Galluccio G, Lucantoni G. Mediastinal bronchogenic cyst’s recurrence treated with EBUS-FNA with a long-term follow-up. Eur J Cardiothorac Surg. 2006;29(4):627-629.
18. Lee DH, Park CK, Kum DY, Kim JB, Hwang I. Clinical characteristics and management of intrathoracic bronchogenic cysts: a single center experience. Korean J Thorac Cardiovasc Surg. 2011;44(4):279-284.
19. Onuki T, Kuramochi M, Inagaki M. Mediastinitis of bronchogenic cyst caused by endobronchial ultrasound-guided transbronchial needle aspiration. Respirol Case Rep. 2014;2(2):73-75.
1. Weber T, Roth TC, Beshay M, Herrmann P, Stein R, Schmid RA. Video-assisted thoracoscopic surgery of mediastinal bronchogenic cysts in adults: a single-center experience. Ann Thorac Surg. 2004;78(3):987-991.
2. Martinod E, Pons F, Azorin J, et al. Thoracoscopic excision of mediastinal bronchogenic cysts: results in 20 cases. Ann Thorac Surg. 2000;69(5):1525-1528.
3. Könönen E, Wade WG. Actinomyces and related organisms in human infections. Clin Microbiol Rev. 2015;28(2):419-442.
4. Ribet ME, Copin MC, Gosselin BH. Bronchogenic cysts of the lung. Ann Thorac Surg. 1996;61(6):1636-1640.
5. Guillem P, Porte H, Marquette CH, Wurtz A. Progressive dysphonia and acute respiratory failure: revealing a bronchogenic cyst. Eur J Cardiothorac Surg. 1997;12(6):925-927.
6. McAdams HP, Kirejczyk WM, Rosado-de-Christenson ML, Matsumoto S. Bronchogenic cyst: imaging features with clinical and histopathologic correlation. Radiology. 2000;217(2):441-446.
7. Rammohan G, Berger HW, Lajam F, Buhain WJ. Superior vena cava syndrome caused by bronchogenic cyst. Chest. 1975;68(4):599-601.
8. Hernández-Solís A, Cruz-Ortiz H, Gutiérrez-Díaz Ceballos ME, Cicero-Sabido R. Quistes broncogénicos. Importancia de la infección en adultos. Estudio de 12 casos [Bronchogenic cysts. Importance of infection in adults. Study of 12 cases]. Cir Cir. 2015;83(2):112-116.
9. Casal RF, Jimenez CA, Mehran RJ, et al. Infected mediastinal bronchogenic cyst successfully treated by endobronchial ultrasound-guided fine-needle aspiration. Ann Thorac Surg. 2010;90(4):e52-e53.
10. Liman ST, Dogan Y, Topcu S, Karabulut N, Demirkan N, Keser Z. Mycobacterial infection of intraparenchymal bronchogenic cysts. Respir Med. 2006;100(11):2060-2062.
11. Lin SH, Lee LN, Chang YL, Lee YC, Ding LW, Hsueh PR. Infected bronchogenic cyst due to Mycobacterium avium in an immunocompetent patient. J Infect. 2005;51(3):e131-e133.
12. Gharagozloo F, Dausmann MJ, McReynolds SD, Sanderson DR, Helmers RA. Recurrent bronchogenic pseudocyst 24 years after incomplete excision. Report of a case. Chest. 1995;108(3):880-883.
13. Bolton JW, Shahian DM. Asymptomatic bronchogenic cysts: what is the best management? Ann Thorac Surg. 1992;53(6):1134-1137.
14. Sarper A, Ayten A, Golbasi I, Demircan A, Isin E. Bronchogenic cyst. Tex Heart Inst J. 2003;30(2):105-108.
15. Varela-Lema L, Fernández-Villar A, Ruano-Ravina A. Effectiveness and safety of endobronchial ultrasound-transbronchial needle aspiration: a systematic review. Eur Respir J. 2009;33(5):1156-1164.
16. Maturu VN, Dhooria S, Agarwal R. Efficacy and safety of transbronchial needle aspiration in diagnosis and treatment of mediastinal bronchogenic cysts: systematic review of case reports. J Bronchology Interv Pulmonol. 2015;22(3):195-203.
17. Galluccio G, Lucantoni G. Mediastinal bronchogenic cyst’s recurrence treated with EBUS-FNA with a long-term follow-up. Eur J Cardiothorac Surg. 2006;29(4):627-629.
18. Lee DH, Park CK, Kum DY, Kim JB, Hwang I. Clinical characteristics and management of intrathoracic bronchogenic cysts: a single center experience. Korean J Thorac Cardiovasc Surg. 2011;44(4):279-284.
19. Onuki T, Kuramochi M, Inagaki M. Mediastinitis of bronchogenic cyst caused by endobronchial ultrasound-guided transbronchial needle aspiration. Respirol Case Rep. 2014;2(2):73-75.