User login
Dog Walking Can Be Hazardous to Cutaneous Health
Studies have recommended dog walking as an activity designed to improve the overall health of older adults.1,2 Benefits purportedly associated with dog walking include lower body mass index, fewer chronic diseases, reduction in the number of physician visits, and decreased limitations of activities of daily living.2 The Arthritis Foundation even recommends dog walking to relieve arthritis symptoms.3 Of course, dogs also provide comfort in companionship, and dog walking can be an enjoyable way for a pet and owner to spend time together.
However, this seemingly benign activity poses a notable and perhaps grossly underrecognized risk for injury in older adults. The annual number of patients 65 years and older who presented to US emergency departments (EDs) for fractures directly associated with walking leashed dogs more than doubled from 2004 to 2017.4 Interestingly, this dramatic increase parallels a nationwide trend in dog ownership demographics. Between 2006 and 2016, the median age of dog owners in the United States rose from 46 to 49 years.5
These trends raise concern for more than just the health of older Americans’ bones. Intuitively, a dog- walking accident that results in a bone fracture will likely also lead to some degree of skin trauma. Older adults have thin fragile skin due to flattening of the dermoepidermal junction and disintegration or degeneration of dermal collagen and elastin.6 This loss of connective tissue as well as subcutaneous tissue in some body areas facilitates shearing injury; concurrently, weakened perivascular support increases the risk for vascular injury and bruising.7 Therefore, when an older person falls while walking a dog, trauma can easily damage delicate aged skin.
Older adults are particularly susceptible to falls, the leading cause of fatal and nonfatal injuries in this age group.8 There are multiple risk factors for falls, including polypharmacy, impaired balance and gait, visual impairments, and cognitive decline, among others.9
Also, many older adults with atrial fibrillation or venous thromboembolism take an anticoagulant drug to prevent stroke. The use of anticoagulants is associated with an increased risk for bleeding, ranging from minor cutaneous bleeding to fatal intracranial hemorrhage.10
A predisposition to falling and bleeding can be hazardous for a dog owner whose excited pet suddenly jumps, runs, or scratches. The use of a leash, mandatory in many urban jurisdictions, tethers the human to the dog, which expedites a fall associated with any sudden, forceful forward or lateral movement by the dog. The following case reports describe a variety of cutaneous injuries experienced by older adults while dog walking.
Case Reports
Patient 1
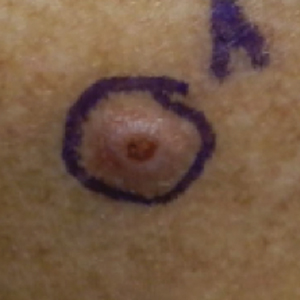
A 79-year-old woman was quietly walking her dog when the dog spotted a squirrel climbing a tree. The dog became excited, turned to the owner, and jumped on her, which caused the dog’s claws to dig into the owner’s fragile forearm skin, creating several superficial but painful abrasions and lacerations (Figure 1). These injuries healed well with conservative therapy including application of an occlusive ointment.
Patient 2
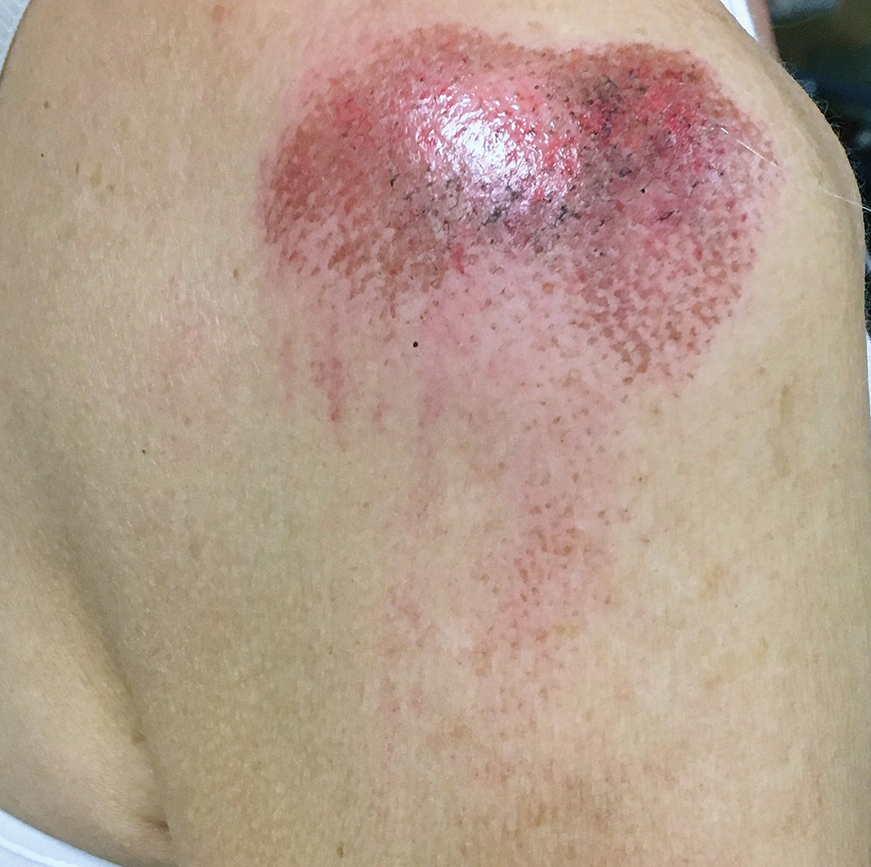
A 68-year-old woman was walking her dog when the dog saw a cat running across the street. The dog suddenly leaped toward the cat, causing the owner to fall forward as the animal’s momentum was transferred through the leash. The owner fell awkwardly on her side, leading to an extensive abrasion and contusion of the shoulder (Figure 2). The lesion healed well with conservative management, albeit with moderate postinflammatory hypochromia.
Patient 3
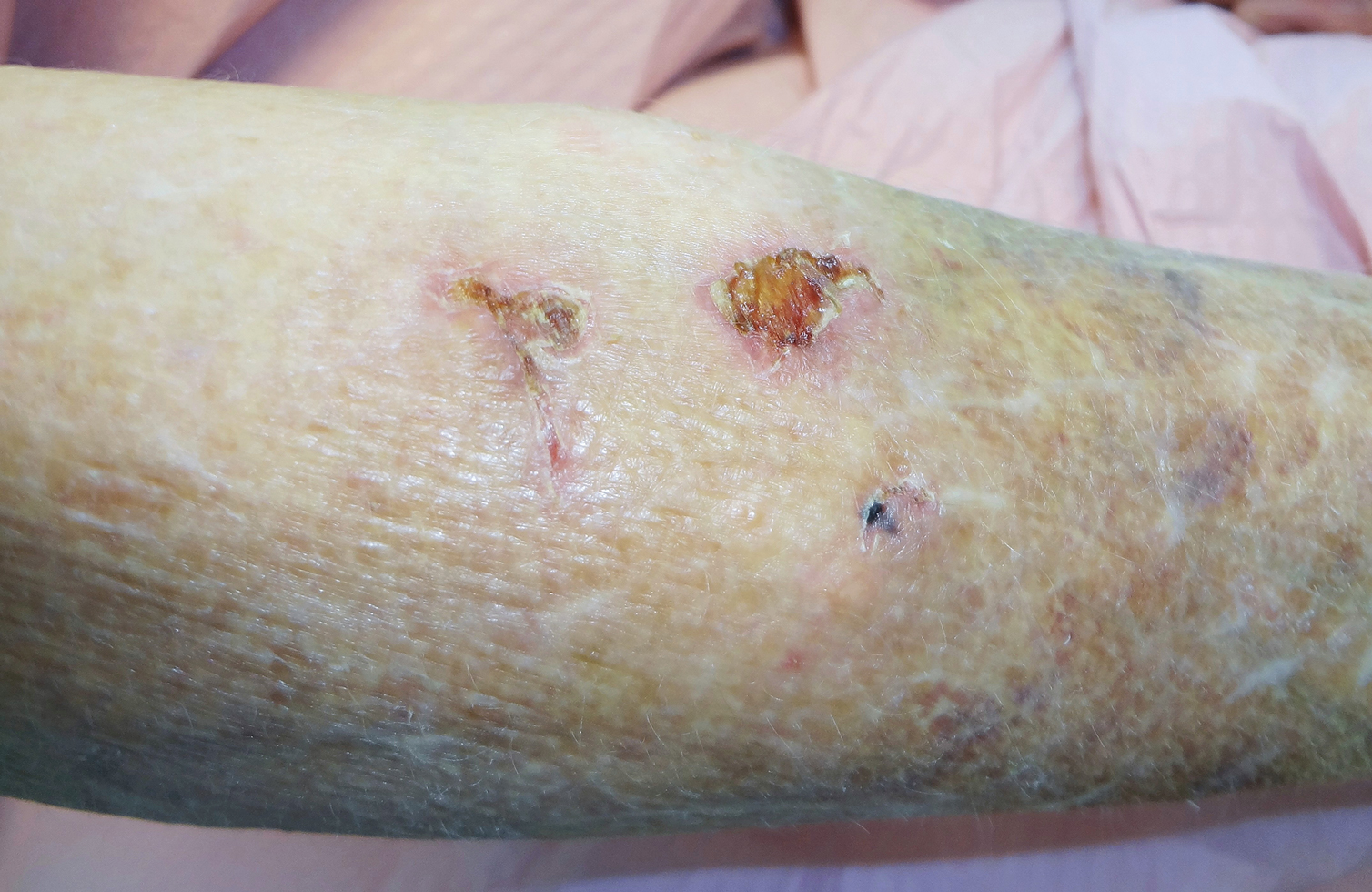
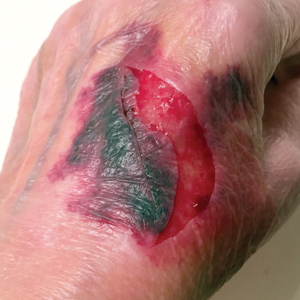
A 65-year-old woman was walking her dog and they heard a loud noise. The dog started to run forward—likely, startled. The owner did not fall, but the leash, which was wrapped around her hand, exerted enough force to avulse a 5×3-cm piece of skin from the dorsum of the hand (Figure 3). The painful abrasion and concomitant bruise eventually healed with conservative management but left a noticeable hemosiderin stain.
Patient 4
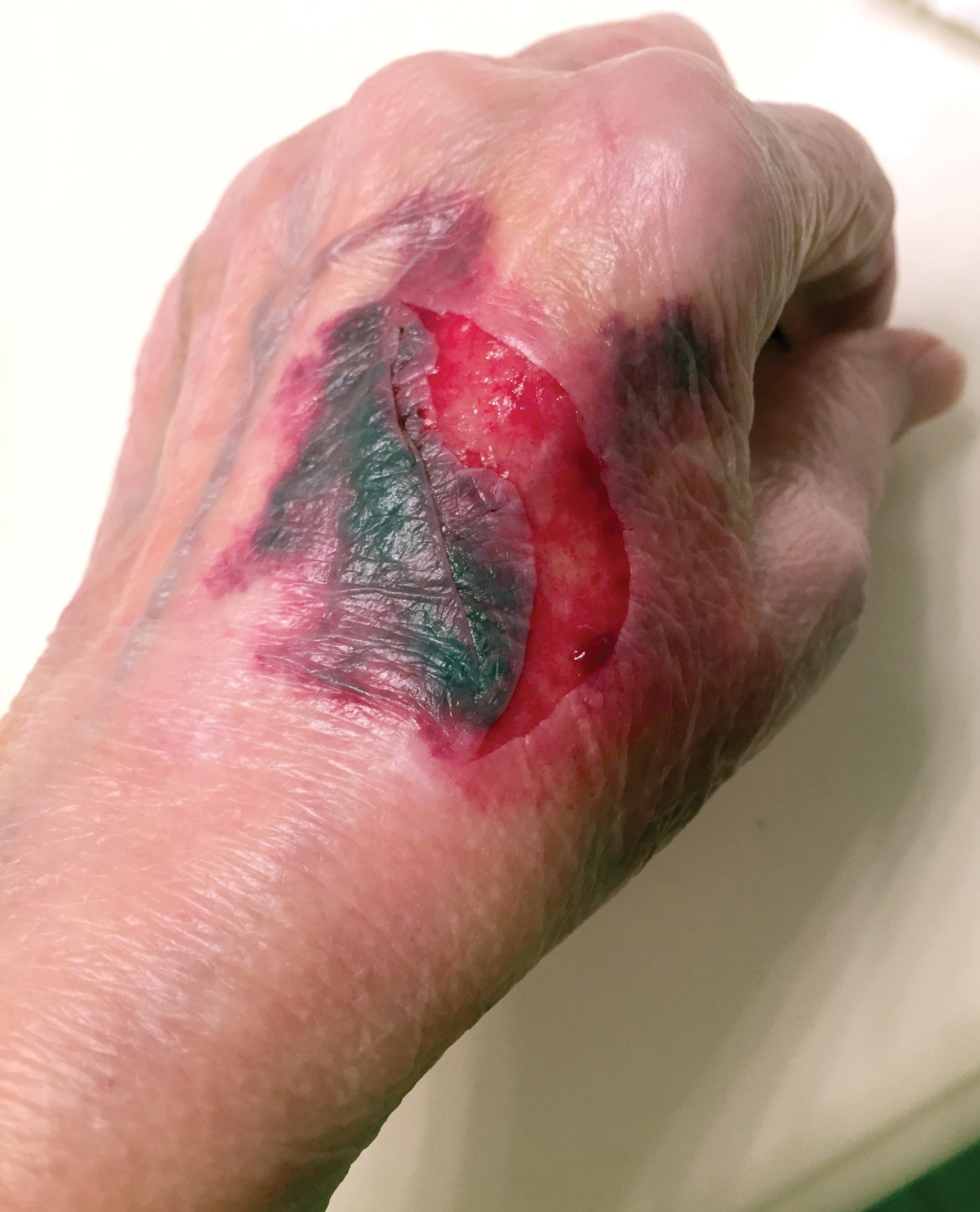
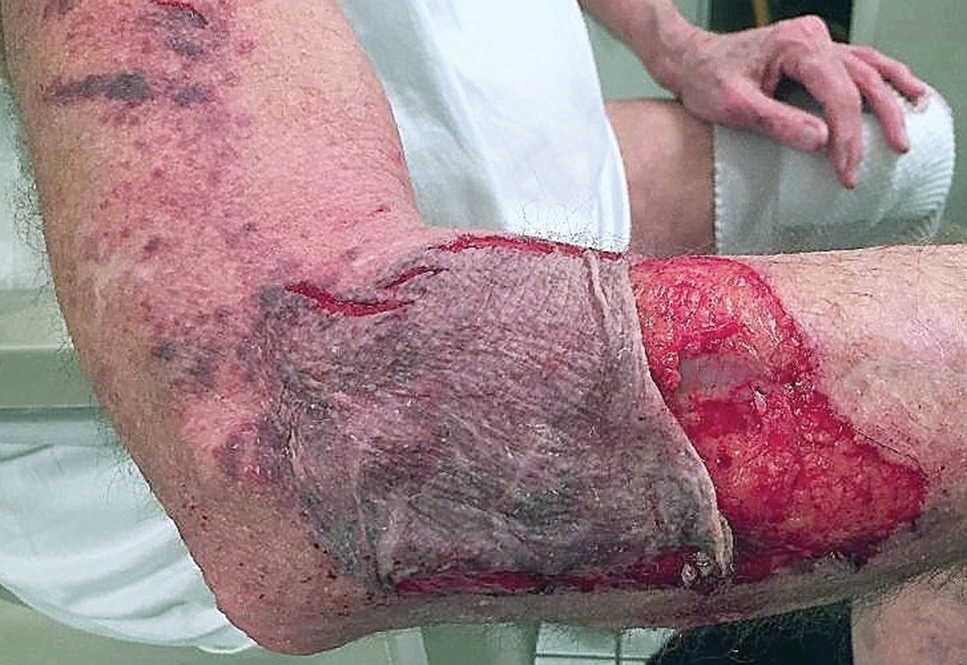
A 66-year-old man was walking a large Rottweiler when the dog lurched toward another dog that was being walked across the street. The owner, taken by surprise by this sudden motion, fell on the concrete sidewalk and was dragged several feet by the dog. This unexpected and off-balance fall caused multiple injuries, including bruises on the upper arm, a large avulsion of epidermal forearm skin (Figure 4), a gouge in the dermis down to fat, and a large abrasion of the contralateral knee. The patient received a tetanus booster and conservative therapy. The affected area healed with an atrophic hypopigmented scar.
Patient 5
An 82-year-old woman with known atrial fibrillation who was taking chronic anticoagulation medication was walking her dog. For no apparent reason, the dog sped up the pace. The woman lost her balance and fell face first onto the sidewalk. She did not lose consciousness but did develop a large bruise on the forehead with a tender fluctuant nodule in the center (Figure 5).
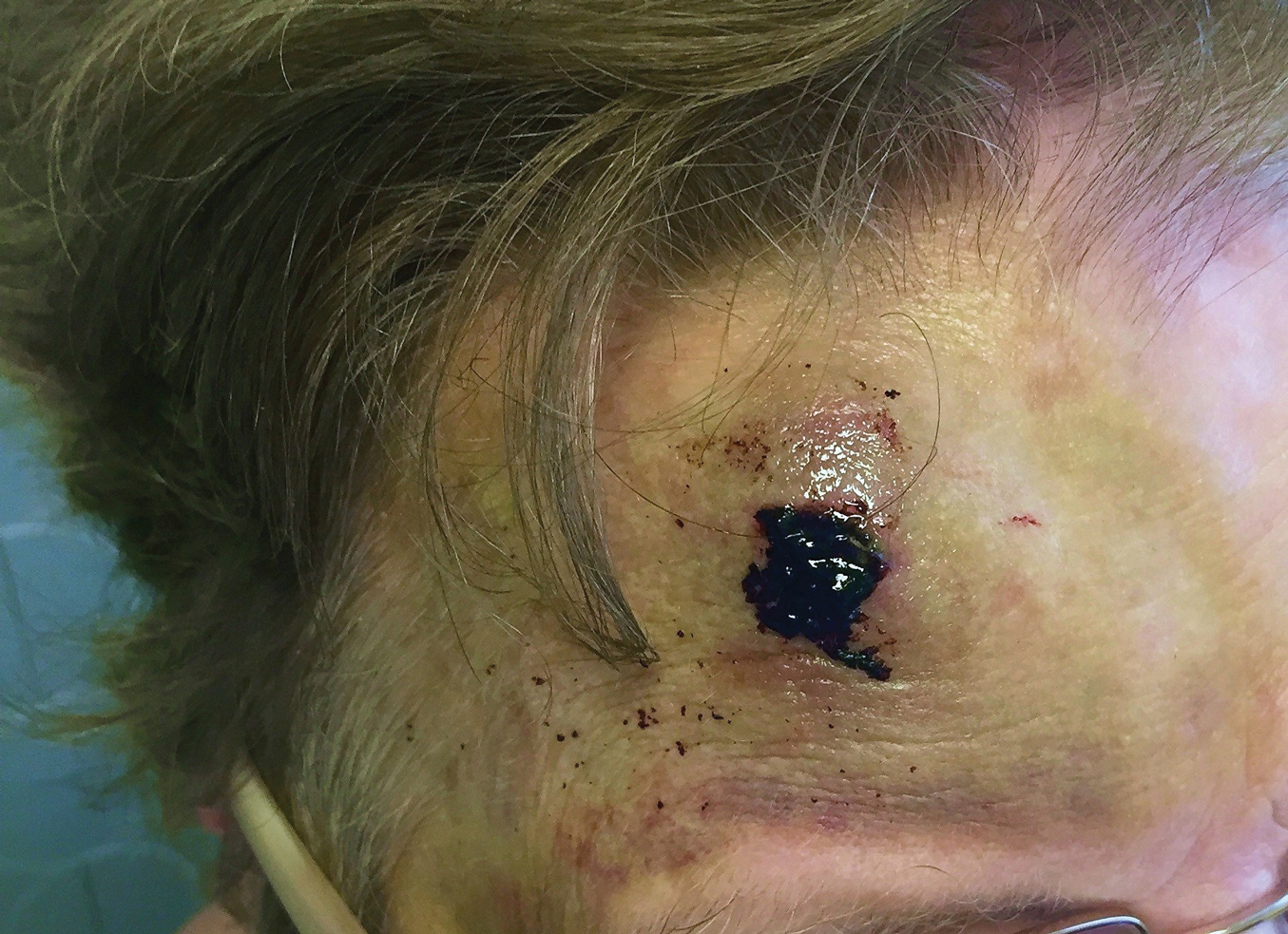
The patient presented the next day, requesting drainage of the forehead hematoma. However, a brief review of systems revealed a persistent severe headache and nausea with vomiting since the prior day. She was immediately transported to the nearest ED where complete neurologic workup revealed a moderate-sized subdural hematoma that was treated by trephination. Recovery was uneventful.
Comment
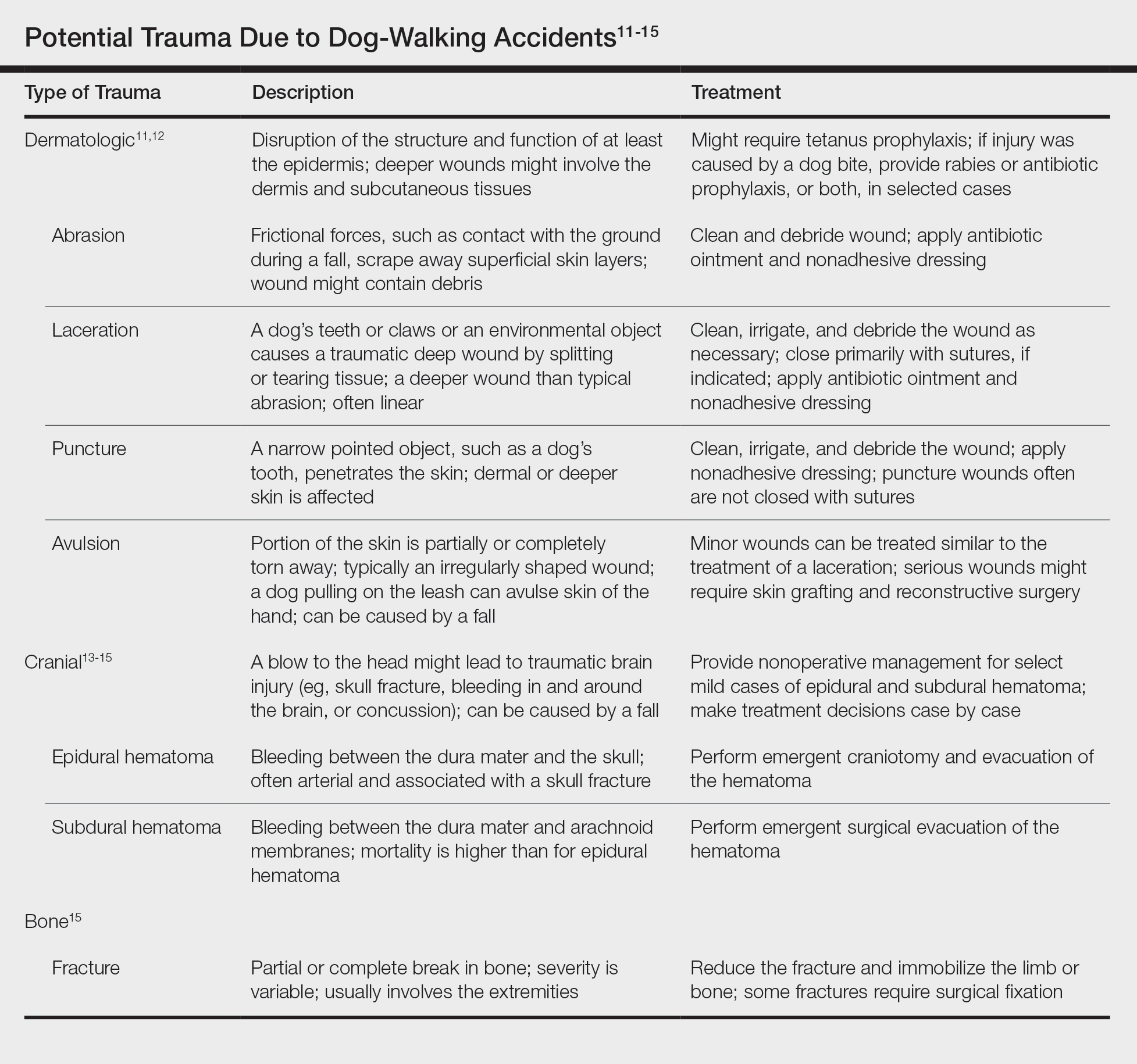
These 5 cases illustrate the notable skin (and neurologic) trauma that can occur due to a dog-walking accident (Table).11-15
Regrettably, obtaining an accurate national estimate of the annual incidence of cutaneous dog-walking injuries is difficult. Researchers who have described the rise in dog walking–associated bone fractures queried the US Consumer Product Safety Commission’s National Electronic Injury Surveillance System database for its numbers.4 This public database generates incidence estimates of activity- or product-related injuries based on data from a nationally representative sample of approximately 100 hospital EDs.16
We queried the same database for the diagnoses avulsion, abrasion or contusion, and laceration.17 These terms were searched in association with pet supplies, including leashes, and patients 65 years and older. This search yielded fewer than 800 total cases from 2008 to 2017, resulting in unreliable estimates for each year.
The National Electronic Injury Surveillance System database no doubt underestimates the true incidence of dog walking–related skin trauma; the great majority of patients with cutaneous injury, as illustrated here, likely never present to the ED, unlike patients with bone fracture. Moreover, data do not capture cases handled by providers outside the ED and self-treated injuries.
In the absence of accurate estimates of cutaneous morbidity related to dog-walking injury, the case reports here are clearly a cautionary tale. Physicians and older adults need to be cognizant of the hazards of this activity. Providers should discuss with older patients the potential risks of dog walking before recommending or condoning this exercise.
The presence of other comorbidities that could hamper a person’s ability to control a leashed dog warrants special consideration. Older prospective dog owners might consider adopting a small, easily manageable breed. These measures can help protect older adults’ fragile skin (and bones) from avoidable minor to potentially life-threatening trauma.
- Christian H, Bauman A, Epping JN, et al. Encouraging dog walking for health promotion and disease prevention. Am J Lifestyle Med. 2016;12:233-243.
- Curl AL, Bibbo J, Johnson RA. Dog walking, the human–animal bond and older adults’ physical health. Gerontologist. 2017;57:930-939.
- Dunkin MA. Walking strategies. Arthritis Foundation website. https://arthritis.org/health-wellness/healthy-living/physical-activity/walking/5-walking-strategies. Accessed March 16, 2020.
- Pirruccio K, Yoon YM, Ahn J. Fractures in elderly Americans associated with walking leashed dogs. JAMA Surg. 2019;154:458-459.
- Sprinkle D. Pet owner demographics get grayer, more golden. Petfood Industry website. https://www.petfoodindustry.com/articles/6315-pet-owner-demographics-get-grayer-more-golden?v=preview. Published March 10, 2017. Accessed March 16, 2020.
- Quan T, Fisher GJ. Role of age-associated alterations of the dermal extracellular matrix microenvironment in human skin aging: a mini-review. Gerontology. 2015;61:427-434.
- Aging & painful skin. Cleveland Clinic website. https://my.clevelandclinic.org/health/diseases/16725-aging--painful-skin. Accessed March 16, 2020.
- Bergen G, Stevens MR, Burns ER. Falls and fall injuries among adults aged ≥65 years—United States, 2014. MMWR Morb Mortal Wkly Rep. 2016;65:993-998.
- Ambrose AF, Paul G, Hausdorff JM. Risk factors for falls among older adults: a review of the literature. Maturitas. 2013;75:51-61.
- January CT, Wann LS, Alpert JS, et al; . 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014;64:E1-E76.
- Armstrong DG, Meyr AJ. Basic principles of wound management. UpToDate. https://www.uptodate.com/contents/basic-principles-of-wound-management. Accessed March 18, 2020.
- Trott AT. Wounds and Lacerations: Emergency Care and Closure. 4th ed. Philadelphia, PA: Saunders; 2012.
- Head injuries in adults: what is it? Harvard Health Publishing website. www.health.harvard.edu/a_to_z/head-injury-in-adults-a-to-z. Published October 2018. Accessed January 30, 2020.
- McBride W. Intracranial epidural hematoma in adults. UpToDate. https://www.uptodate.com/contents/intracranial-epidural-hematoma-in-adults. Updated July 23, 2018. Accessed March 18, 2020.
- McBride W. Subdural hematoma in adults: prognosis and management. UpToDate. https://www.uptodate.com/contents/subdural-hematoma-in-adults-prognosis-and-management. Updated July 11, 2019. Accessed March 18, 2020.
- Schroeder T, Ault K. The NEISS sample: design and implementation. Washington, DC: US Consumer Product Safety Commission, Division of Hazard and Injury Data Systems; June 2001. https://cpsc.gov/s3fs-public/pdfs/blk_media_2001d011-6b6.pdf. Accessed January 30, 2020.
- National Electronic Injury Surveillance System (NEISS). Bethesda, MD: US Consumer Product Safety Commission; 2018. https://www.cpsc.gov/Research--Statistics/NEISS-Injury-Data. Accessed March 16, 2020.
Studies have recommended dog walking as an activity designed to improve the overall health of older adults.1,2 Benefits purportedly associated with dog walking include lower body mass index, fewer chronic diseases, reduction in the number of physician visits, and decreased limitations of activities of daily living.2 The Arthritis Foundation even recommends dog walking to relieve arthritis symptoms.3 Of course, dogs also provide comfort in companionship, and dog walking can be an enjoyable way for a pet and owner to spend time together.
However, this seemingly benign activity poses a notable and perhaps grossly underrecognized risk for injury in older adults. The annual number of patients 65 years and older who presented to US emergency departments (EDs) for fractures directly associated with walking leashed dogs more than doubled from 2004 to 2017.4 Interestingly, this dramatic increase parallels a nationwide trend in dog ownership demographics. Between 2006 and 2016, the median age of dog owners in the United States rose from 46 to 49 years.5
These trends raise concern for more than just the health of older Americans’ bones. Intuitively, a dog- walking accident that results in a bone fracture will likely also lead to some degree of skin trauma. Older adults have thin fragile skin due to flattening of the dermoepidermal junction and disintegration or degeneration of dermal collagen and elastin.6 This loss of connective tissue as well as subcutaneous tissue in some body areas facilitates shearing injury; concurrently, weakened perivascular support increases the risk for vascular injury and bruising.7 Therefore, when an older person falls while walking a dog, trauma can easily damage delicate aged skin.
Older adults are particularly susceptible to falls, the leading cause of fatal and nonfatal injuries in this age group.8 There are multiple risk factors for falls, including polypharmacy, impaired balance and gait, visual impairments, and cognitive decline, among others.9
Also, many older adults with atrial fibrillation or venous thromboembolism take an anticoagulant drug to prevent stroke. The use of anticoagulants is associated with an increased risk for bleeding, ranging from minor cutaneous bleeding to fatal intracranial hemorrhage.10
A predisposition to falling and bleeding can be hazardous for a dog owner whose excited pet suddenly jumps, runs, or scratches. The use of a leash, mandatory in many urban jurisdictions, tethers the human to the dog, which expedites a fall associated with any sudden, forceful forward or lateral movement by the dog. The following case reports describe a variety of cutaneous injuries experienced by older adults while dog walking.
Case Reports
Patient 1
A 79-year-old woman was quietly walking her dog when the dog spotted a squirrel climbing a tree. The dog became excited, turned to the owner, and jumped on her, which caused the dog’s claws to dig into the owner’s fragile forearm skin, creating several superficial but painful abrasions and lacerations (Figure 1). These injuries healed well with conservative therapy including application of an occlusive ointment.
Patient 2
A 68-year-old woman was walking her dog when the dog saw a cat running across the street. The dog suddenly leaped toward the cat, causing the owner to fall forward as the animal’s momentum was transferred through the leash. The owner fell awkwardly on her side, leading to an extensive abrasion and contusion of the shoulder (Figure 2). The lesion healed well with conservative management, albeit with moderate postinflammatory hypochromia.
Patient 3
A 65-year-old woman was walking her dog and they heard a loud noise. The dog started to run forward—likely, startled. The owner did not fall, but the leash, which was wrapped around her hand, exerted enough force to avulse a 5×3-cm piece of skin from the dorsum of the hand (Figure 3). The painful abrasion and concomitant bruise eventually healed with conservative management but left a noticeable hemosiderin stain.
Patient 4
A 66-year-old man was walking a large Rottweiler when the dog lurched toward another dog that was being walked across the street. The owner, taken by surprise by this sudden motion, fell on the concrete sidewalk and was dragged several feet by the dog. This unexpected and off-balance fall caused multiple injuries, including bruises on the upper arm, a large avulsion of epidermal forearm skin (Figure 4), a gouge in the dermis down to fat, and a large abrasion of the contralateral knee. The patient received a tetanus booster and conservative therapy. The affected area healed with an atrophic hypopigmented scar.
Patient 5
An 82-year-old woman with known atrial fibrillation who was taking chronic anticoagulation medication was walking her dog. For no apparent reason, the dog sped up the pace. The woman lost her balance and fell face first onto the sidewalk. She did not lose consciousness but did develop a large bruise on the forehead with a tender fluctuant nodule in the center (Figure 5).
The patient presented the next day, requesting drainage of the forehead hematoma. However, a brief review of systems revealed a persistent severe headache and nausea with vomiting since the prior day. She was immediately transported to the nearest ED where complete neurologic workup revealed a moderate-sized subdural hematoma that was treated by trephination. Recovery was uneventful.
Comment
These 5 cases illustrate the notable skin (and neurologic) trauma that can occur due to a dog-walking accident (Table).11-15
Regrettably, obtaining an accurate national estimate of the annual incidence of cutaneous dog-walking injuries is difficult. Researchers who have described the rise in dog walking–associated bone fractures queried the US Consumer Product Safety Commission’s National Electronic Injury Surveillance System database for its numbers.4 This public database generates incidence estimates of activity- or product-related injuries based on data from a nationally representative sample of approximately 100 hospital EDs.16
We queried the same database for the diagnoses avulsion, abrasion or contusion, and laceration.17 These terms were searched in association with pet supplies, including leashes, and patients 65 years and older. This search yielded fewer than 800 total cases from 2008 to 2017, resulting in unreliable estimates for each year.
The National Electronic Injury Surveillance System database no doubt underestimates the true incidence of dog walking–related skin trauma; the great majority of patients with cutaneous injury, as illustrated here, likely never present to the ED, unlike patients with bone fracture. Moreover, data do not capture cases handled by providers outside the ED and self-treated injuries.
In the absence of accurate estimates of cutaneous morbidity related to dog-walking injury, the case reports here are clearly a cautionary tale. Physicians and older adults need to be cognizant of the hazards of this activity. Providers should discuss with older patients the potential risks of dog walking before recommending or condoning this exercise.
The presence of other comorbidities that could hamper a person’s ability to control a leashed dog warrants special consideration. Older prospective dog owners might consider adopting a small, easily manageable breed. These measures can help protect older adults’ fragile skin (and bones) from avoidable minor to potentially life-threatening trauma.
Studies have recommended dog walking as an activity designed to improve the overall health of older adults.1,2 Benefits purportedly associated with dog walking include lower body mass index, fewer chronic diseases, reduction in the number of physician visits, and decreased limitations of activities of daily living.2 The Arthritis Foundation even recommends dog walking to relieve arthritis symptoms.3 Of course, dogs also provide comfort in companionship, and dog walking can be an enjoyable way for a pet and owner to spend time together.
However, this seemingly benign activity poses a notable and perhaps grossly underrecognized risk for injury in older adults. The annual number of patients 65 years and older who presented to US emergency departments (EDs) for fractures directly associated with walking leashed dogs more than doubled from 2004 to 2017.4 Interestingly, this dramatic increase parallels a nationwide trend in dog ownership demographics. Between 2006 and 2016, the median age of dog owners in the United States rose from 46 to 49 years.5
These trends raise concern for more than just the health of older Americans’ bones. Intuitively, a dog- walking accident that results in a bone fracture will likely also lead to some degree of skin trauma. Older adults have thin fragile skin due to flattening of the dermoepidermal junction and disintegration or degeneration of dermal collagen and elastin.6 This loss of connective tissue as well as subcutaneous tissue in some body areas facilitates shearing injury; concurrently, weakened perivascular support increases the risk for vascular injury and bruising.7 Therefore, when an older person falls while walking a dog, trauma can easily damage delicate aged skin.
Older adults are particularly susceptible to falls, the leading cause of fatal and nonfatal injuries in this age group.8 There are multiple risk factors for falls, including polypharmacy, impaired balance and gait, visual impairments, and cognitive decline, among others.9
Also, many older adults with atrial fibrillation or venous thromboembolism take an anticoagulant drug to prevent stroke. The use of anticoagulants is associated with an increased risk for bleeding, ranging from minor cutaneous bleeding to fatal intracranial hemorrhage.10
A predisposition to falling and bleeding can be hazardous for a dog owner whose excited pet suddenly jumps, runs, or scratches. The use of a leash, mandatory in many urban jurisdictions, tethers the human to the dog, which expedites a fall associated with any sudden, forceful forward or lateral movement by the dog. The following case reports describe a variety of cutaneous injuries experienced by older adults while dog walking.
Case Reports
Patient 1
A 79-year-old woman was quietly walking her dog when the dog spotted a squirrel climbing a tree. The dog became excited, turned to the owner, and jumped on her, which caused the dog’s claws to dig into the owner’s fragile forearm skin, creating several superficial but painful abrasions and lacerations (Figure 1). These injuries healed well with conservative therapy including application of an occlusive ointment.
Patient 2
A 68-year-old woman was walking her dog when the dog saw a cat running across the street. The dog suddenly leaped toward the cat, causing the owner to fall forward as the animal’s momentum was transferred through the leash. The owner fell awkwardly on her side, leading to an extensive abrasion and contusion of the shoulder (Figure 2). The lesion healed well with conservative management, albeit with moderate postinflammatory hypochromia.
Patient 3
A 65-year-old woman was walking her dog and they heard a loud noise. The dog started to run forward—likely, startled. The owner did not fall, but the leash, which was wrapped around her hand, exerted enough force to avulse a 5×3-cm piece of skin from the dorsum of the hand (Figure 3). The painful abrasion and concomitant bruise eventually healed with conservative management but left a noticeable hemosiderin stain.
Patient 4
A 66-year-old man was walking a large Rottweiler when the dog lurched toward another dog that was being walked across the street. The owner, taken by surprise by this sudden motion, fell on the concrete sidewalk and was dragged several feet by the dog. This unexpected and off-balance fall caused multiple injuries, including bruises on the upper arm, a large avulsion of epidermal forearm skin (Figure 4), a gouge in the dermis down to fat, and a large abrasion of the contralateral knee. The patient received a tetanus booster and conservative therapy. The affected area healed with an atrophic hypopigmented scar.
Patient 5
An 82-year-old woman with known atrial fibrillation who was taking chronic anticoagulation medication was walking her dog. For no apparent reason, the dog sped up the pace. The woman lost her balance and fell face first onto the sidewalk. She did not lose consciousness but did develop a large bruise on the forehead with a tender fluctuant nodule in the center (Figure 5).
The patient presented the next day, requesting drainage of the forehead hematoma. However, a brief review of systems revealed a persistent severe headache and nausea with vomiting since the prior day. She was immediately transported to the nearest ED where complete neurologic workup revealed a moderate-sized subdural hematoma that was treated by trephination. Recovery was uneventful.
Comment
These 5 cases illustrate the notable skin (and neurologic) trauma that can occur due to a dog-walking accident (Table).11-15
Regrettably, obtaining an accurate national estimate of the annual incidence of cutaneous dog-walking injuries is difficult. Researchers who have described the rise in dog walking–associated bone fractures queried the US Consumer Product Safety Commission’s National Electronic Injury Surveillance System database for its numbers.4 This public database generates incidence estimates of activity- or product-related injuries based on data from a nationally representative sample of approximately 100 hospital EDs.16
We queried the same database for the diagnoses avulsion, abrasion or contusion, and laceration.17 These terms were searched in association with pet supplies, including leashes, and patients 65 years and older. This search yielded fewer than 800 total cases from 2008 to 2017, resulting in unreliable estimates for each year.
The National Electronic Injury Surveillance System database no doubt underestimates the true incidence of dog walking–related skin trauma; the great majority of patients with cutaneous injury, as illustrated here, likely never present to the ED, unlike patients with bone fracture. Moreover, data do not capture cases handled by providers outside the ED and self-treated injuries.
In the absence of accurate estimates of cutaneous morbidity related to dog-walking injury, the case reports here are clearly a cautionary tale. Physicians and older adults need to be cognizant of the hazards of this activity. Providers should discuss with older patients the potential risks of dog walking before recommending or condoning this exercise.
The presence of other comorbidities that could hamper a person’s ability to control a leashed dog warrants special consideration. Older prospective dog owners might consider adopting a small, easily manageable breed. These measures can help protect older adults’ fragile skin (and bones) from avoidable minor to potentially life-threatening trauma.
- Christian H, Bauman A, Epping JN, et al. Encouraging dog walking for health promotion and disease prevention. Am J Lifestyle Med. 2016;12:233-243.
- Curl AL, Bibbo J, Johnson RA. Dog walking, the human–animal bond and older adults’ physical health. Gerontologist. 2017;57:930-939.
- Dunkin MA. Walking strategies. Arthritis Foundation website. https://arthritis.org/health-wellness/healthy-living/physical-activity/walking/5-walking-strategies. Accessed March 16, 2020.
- Pirruccio K, Yoon YM, Ahn J. Fractures in elderly Americans associated with walking leashed dogs. JAMA Surg. 2019;154:458-459.
- Sprinkle D. Pet owner demographics get grayer, more golden. Petfood Industry website. https://www.petfoodindustry.com/articles/6315-pet-owner-demographics-get-grayer-more-golden?v=preview. Published March 10, 2017. Accessed March 16, 2020.
- Quan T, Fisher GJ. Role of age-associated alterations of the dermal extracellular matrix microenvironment in human skin aging: a mini-review. Gerontology. 2015;61:427-434.
- Aging & painful skin. Cleveland Clinic website. https://my.clevelandclinic.org/health/diseases/16725-aging--painful-skin. Accessed March 16, 2020.
- Bergen G, Stevens MR, Burns ER. Falls and fall injuries among adults aged ≥65 years—United States, 2014. MMWR Morb Mortal Wkly Rep. 2016;65:993-998.
- Ambrose AF, Paul G, Hausdorff JM. Risk factors for falls among older adults: a review of the literature. Maturitas. 2013;75:51-61.
- January CT, Wann LS, Alpert JS, et al; . 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014;64:E1-E76.
- Armstrong DG, Meyr AJ. Basic principles of wound management. UpToDate. https://www.uptodate.com/contents/basic-principles-of-wound-management. Accessed March 18, 2020.
- Trott AT. Wounds and Lacerations: Emergency Care and Closure. 4th ed. Philadelphia, PA: Saunders; 2012.
- Head injuries in adults: what is it? Harvard Health Publishing website. www.health.harvard.edu/a_to_z/head-injury-in-adults-a-to-z. Published October 2018. Accessed January 30, 2020.
- McBride W. Intracranial epidural hematoma in adults. UpToDate. https://www.uptodate.com/contents/intracranial-epidural-hematoma-in-adults. Updated July 23, 2018. Accessed March 18, 2020.
- McBride W. Subdural hematoma in adults: prognosis and management. UpToDate. https://www.uptodate.com/contents/subdural-hematoma-in-adults-prognosis-and-management. Updated July 11, 2019. Accessed March 18, 2020.
- Schroeder T, Ault K. The NEISS sample: design and implementation. Washington, DC: US Consumer Product Safety Commission, Division of Hazard and Injury Data Systems; June 2001. https://cpsc.gov/s3fs-public/pdfs/blk_media_2001d011-6b6.pdf. Accessed January 30, 2020.
- National Electronic Injury Surveillance System (NEISS). Bethesda, MD: US Consumer Product Safety Commission; 2018. https://www.cpsc.gov/Research--Statistics/NEISS-Injury-Data. Accessed March 16, 2020.
- Christian H, Bauman A, Epping JN, et al. Encouraging dog walking for health promotion and disease prevention. Am J Lifestyle Med. 2016;12:233-243.
- Curl AL, Bibbo J, Johnson RA. Dog walking, the human–animal bond and older adults’ physical health. Gerontologist. 2017;57:930-939.
- Dunkin MA. Walking strategies. Arthritis Foundation website. https://arthritis.org/health-wellness/healthy-living/physical-activity/walking/5-walking-strategies. Accessed March 16, 2020.
- Pirruccio K, Yoon YM, Ahn J. Fractures in elderly Americans associated with walking leashed dogs. JAMA Surg. 2019;154:458-459.
- Sprinkle D. Pet owner demographics get grayer, more golden. Petfood Industry website. https://www.petfoodindustry.com/articles/6315-pet-owner-demographics-get-grayer-more-golden?v=preview. Published March 10, 2017. Accessed March 16, 2020.
- Quan T, Fisher GJ. Role of age-associated alterations of the dermal extracellular matrix microenvironment in human skin aging: a mini-review. Gerontology. 2015;61:427-434.
- Aging & painful skin. Cleveland Clinic website. https://my.clevelandclinic.org/health/diseases/16725-aging--painful-skin. Accessed March 16, 2020.
- Bergen G, Stevens MR, Burns ER. Falls and fall injuries among adults aged ≥65 years—United States, 2014. MMWR Morb Mortal Wkly Rep. 2016;65:993-998.
- Ambrose AF, Paul G, Hausdorff JM. Risk factors for falls among older adults: a review of the literature. Maturitas. 2013;75:51-61.
- January CT, Wann LS, Alpert JS, et al; . 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014;64:E1-E76.
- Armstrong DG, Meyr AJ. Basic principles of wound management. UpToDate. https://www.uptodate.com/contents/basic-principles-of-wound-management. Accessed March 18, 2020.
- Trott AT. Wounds and Lacerations: Emergency Care and Closure. 4th ed. Philadelphia, PA: Saunders; 2012.
- Head injuries in adults: what is it? Harvard Health Publishing website. www.health.harvard.edu/a_to_z/head-injury-in-adults-a-to-z. Published October 2018. Accessed January 30, 2020.
- McBride W. Intracranial epidural hematoma in adults. UpToDate. https://www.uptodate.com/contents/intracranial-epidural-hematoma-in-adults. Updated July 23, 2018. Accessed March 18, 2020.
- McBride W. Subdural hematoma in adults: prognosis and management. UpToDate. https://www.uptodate.com/contents/subdural-hematoma-in-adults-prognosis-and-management. Updated July 11, 2019. Accessed March 18, 2020.
- Schroeder T, Ault K. The NEISS sample: design and implementation. Washington, DC: US Consumer Product Safety Commission, Division of Hazard and Injury Data Systems; June 2001. https://cpsc.gov/s3fs-public/pdfs/blk_media_2001d011-6b6.pdf. Accessed January 30, 2020.
- National Electronic Injury Surveillance System (NEISS). Bethesda, MD: US Consumer Product Safety Commission; 2018. https://www.cpsc.gov/Research--Statistics/NEISS-Injury-Data. Accessed March 16, 2020.
Practice Points
- Dog walking is a good source of exercise but can lead to serious skin/soft tissue injury.
- When evaluating cutaneous trauma related to dog walking, remember to consider the possibility of an underlying bone fracture.
- Cutaneous trauma may overlay serious internal injury, such as epidural or subdural hematoma.
Sharp lower back pain • left-side paraspinal tenderness • anterior thigh sensory loss • Dx?
THE CASE
A 64-year-old woman with a history of late-onset type 1 diabetes mellitus, Hashimoto thyroiditis, and scoliosis presented to the sports medicine clinic with acute-onset, sharp, nonradiating right lower back pain that began when she bent forward to apply lotion. At presentation, she denied fever, chills, numbness, tingling, aggravation of pain with movement, weakness, and incontinence. Her neuromuscular examination was unremarkable except for left-side paraspinal tenderness. She was prescribed cyclobenzaprine for symptomatic relief.
Two days later, she was seen for worsening pain. Her physical exam was unchanged. She was prescribed tramadol and advised to start physical therapy gradually. As the day progressed, however, she developed anterior thigh sensory loss, which gradually extended distally.
The following day, she was brought to the emergency department with severe left-side weakness without urinary incontinence. Her mental status and cranial nerve exams were normal. On examination, strength of the iliopsoas and quadriceps was 1/5 bilaterally, and of the peroneal tendon and gastrocnemius, 3/5 bilaterally. Reflexes of triceps, biceps, knee, and Achilles tendon were symmetric and 3+ with bilateral clonus of the ankle. The Babinski sign was positive bilaterally. The patient had diminished pain sensation bilaterally, extending down from the T11 dermatome (left more than right side) with diminished vibration sensation at the left ankle. Her perianal sensation, bilateral temperature sensation, and cerebellar examination were normal.
Magnetic resonance imaging (MRI) without contrast of the lumbar spine demonstrated ischemia findings corresponding to T12-L1. Degenerative changes from L1-S1 were noted, with multiple osteophytes impinging on the neural foramina without cord compression.
THE DIAGNOSIS
The initial presentation was consistent with mechanical low back pain with signs of anterior spinal artery infarction and medial lemniscus pathway involvement 48 hours after initial presentation. Spinal cord infarction occurs more commonly in women and in the young than does cerebral infarction,1 with better reemployment rates.1,2 Similar to other strokes, long-term prognosis is primarily determined by the initial severity of motor impairment, which is linked to long-term immobility and need for bladder catheterization.3
Neurogenic pain developing years after spinal cord infarction is most often observed in anterior spinal artery infarction4 without functional limitations.
Initial treatment. Our patient was started on aspirin 325 mg/d and clopidogrel 75 mg/d. Her mean arterial blood pressure was maintained above 80 mm Hg. Computed tomography angiography of the abdomen and pelvis was negative for aortic dissection. Lumbar puncture for cerebrospinal fluid analysis was unremarkable. Results of antineutrophil cytoplasmic antibody testing, antinuclear antibody testing, a hepatitis panel, and an antiphospholipid panel were all negative. The patient was started on IV steroids with a plan for gradual tapering. The neurosurgical team agreed with medical management.
Continue to: DISCUSSION
DISCUSSION
Possible etiologies for acute spinal cord infarction include spinal cord ischemia from compression of the vessels, fibrocartilaginous embolism, and arterial thrombosis or atherosclerosis, especially in patients with diabetes.5
The majority (86%) of spinal strokes are due to spontaneous occlusion of the vessels with no identifiable cause; much less frequently (9% of cases), hemorrhage is the causative factor.1 A retrospective study demonstrated that 10 of 27 patients with spinal stroke had an anterior spinal infarct. Of those 10 patients, 6 reported a mechanical triggering movement (similar to this case), indicating potential compression of the radicular arteries due to said movement.4
Fibrocartilaginous embolism (FCE) is worth considering as a possible cause, because it accounts for 5.5% of all cases of acute spinal cord infarction.3 FCE is thought to arise after a precipitating event such as minor trauma, heavy lifting, physical exertion, or Valsalva maneuver causing embolization of the fragments of nucleus pulposus to the arterial system. In a case series of 8 patients, 2 had possible FCE with precipitating events occurring within the prior 24 hours. This was also demonstrated in another case series6 in which 7 of 9 patients had precipitating events.
Although FCE can only definitively be diagnosed postmortem, the researchers6 proposed clinical criteria for its diagnosis in living patients, based on 40 postmortem and 11 suspected antemortem cases of FCE. These criteria include a rapid evolution of symptoms consistent with vascular etiology, with or without preceding minor trauma or Valsalva maneuver; MRI changes consistent with ischemic myelopathy, with or without evidence of disc herniation; and no more than 2 vascular risk factors.
Our patient had no trauma (although there was a triggering movement), no signs of disc herniation, and 2 risk factors (> 60 years and diabetes mellitus). Also, a neurologically symptom-free interval between the painful movement and the onset of neurologic manifestations in our case parallels the clinical picture of FCE.
Continue to: The role of factor V Leiden (FVL) mutation
The role of factor V Leiden (FVL) mutation in arterial thrombosis is questionable. Previous reports demonstrate a risk for venous thrombosis 7 to 10 times higher with heterozygous FVL mutation and 100 times higher with homozygous mutation, with a less established role in arterial thrombosis.7 A retrospective Turkish study compared the incidence of FVL mutation in patients with arterial thrombosis vs healthy subjects; incidence was significantly higher in female patients than female controls (37.5% vs. 2%).7 A meta-analysis of published studies showed an association between arterial ischemic events and FVL mutation to be modest, with an odds ratio of 1.21 (95% CI, 0.99-1.49).8
In contrast, a 3.4-year longitudinal health study of patients ages 65 and older found no significant difference in the occurrence of myocardial infarction, transient ischemic attack, stroke, or angina for more than 5000 patients with heterozygous FVL mutation compared to fewer than 500 controls.9 The case patient’s clinical course did not fit a thrombotic clinical picture.
Evaluating for “red flags” is crucial in any case of low back pain to exclude serious pathologies. Red flag symptoms include signs of myelopathy, signs of infection, history of trauma with focal tenderness to palpation, and steroid or anticoagulant use (to rule out medication adverse effects).10 Our patient lacked these classical signs, but she did have subjective pain out of proportion to the clinical exam findings.
Of note: The above red flags for low back pain are all based on expert opinion,11 and the positive predictive value of a red flag is always low because of the low prevalence of serious spinal pathologies.12
Striking a proper balance. This case emphasizes the necessity to keep uncommon causes—such as nontraumatic spinal stroke, which has a prevalence of about 5% to 8% of all acute myelopathies—in the differential diagnosis.3
Continue to: We recommend watchful...
We recommend watchful waiting coupled with communication with the patient regarding monitoring for changes in symptoms over time.11 Any changes in symptoms concerning for underlying spinal cord injury indicate necessity for transfer to a tertiary care center (if possible), along with immediate evaluation with imaging—including computed tomography angiography of the abdomen to rule out aortic dissection (1%-2% of all spinal cord infarcts), followed by a specialist consultation based on the findings.3
Our patient
Our patient was discharged to rehabilitation on hospital Day 5, after progressive return of lower extremity strength. At the 2-month follow-up visit, she demonstrated grade 4+ strength throughout her lower extremities bilaterally. Weakness was predominant at the hip flexors and ankle dorsiflexors, which was consistent with her status at discharge. She had burning pain in the distribution of the L1 dermatome that responded to ibuprofen.
Hypercoagulability work-up was positive for heterozygous FVL mutation without any previous history of venous thromboembolic disease. She was continued on aspirin 325 mg/d, as per American College of Chest Physicians antithrombotic guidelines.13
One year later, our patient underwent a follow-up MRI of the thoracic spine, which showed an “owl’s eye” hyperintensity in the anterior cord (FIGURE), a sign that’s often seen in bilateral spinal cord infarction

THE TAKEAWAY
Spinal stroke is rare, but a missed diagnosis and lack of treatment can result in long-term morbidity. Therefore, it is prudent to consider this diagnosis in the differential—especially when the patient’s subjective back pain is out of proportion to the clinical examination findings.
CORRESPONDENCE
Srikanth Nithyanandam, MBBS, MS, University of Kentucky Family and Community Medicine, 2195 Harrodsburg Road, Suite 125, Lexington, KY 40504-3504; [email protected].
1. Romi F, Naess H. Spinal cord infarction in clinical neurology: a review of characteristics and long-term prognosis in comparison to cerebral infarction. Eur Neurol. 2016;76:95-98.
2. Hanson SR, Romi F, Rekand T, et al. Long-term outcome after spinal cord infarctions. Acta Neurol Scand. 2015;131:253-257.
3. Rigney L, Cappelen-Smith C, Sebire D, et al. Nontraumatic spinal cord ischaemic syndrome. J Clin Neurosci. 2015;22:1544-1549.
4. Novy J, Carruzzo A, Maeder P, Bogousslavsky J. Spinal cord ischemia: clinical and imaging patterns, pathogenesis, and outcomes in 27 patients. Arch Neurol. 2006;63:1113-1120.
5. Goldstein LB, Adams R, Alberts MJ, et al; American Heart Association; American Stroke Association Stroke Council. Primary prevention of ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council: cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2006;113:e873-e923.
6. Mateen FJ, Monrad PA, Hunderfund AN, et al. Clinically suspected fibrocartilaginous embolism: clinical characteristics, treatments, and outcomes. Eur J Neurol. 2011;18:218-225.
7. Ozmen F, Ozmen MM, Ozalp N, et al. The prevalence of factor V (G1691A), MTHFR (C677T) and PT (G20210A) gene mutations in arterial thrombosis. Ulus Travma Acil Cerrahi Derg. 2009;15:113-119.
8. Kim RJ, Becker RC. Association between factor V Leiden, prothrombin G20210A, and methylenetetrahydrofolate reductase C677T mutations and events of the arterial circulatory system: a meta-analysis of published studies. Am Heart J. 2003;146:948-957.
9. Cushman M, Rosendaal FR, Psaty BM, et al. Factor V Leiden is not a risk factor for arterial vascular disease in the elderly: results from the Cardiovascular Health Study. Thromb Haemost. 1998;79:912-915.
10. Strudwick K, McPhee M, Bell A, et al. Review article: best practice management of low back pain in the emergency department (part 1 of the musculoskeletal injuries rapid review series). Emerg Med Australas. 2018;30:18-35.
11. Cook CE, George SZ, Reiman MP. Red flag screening for low back pain: nothing to see here, move along: a narrative review. Br J Sports Med. 2018;52:493-496.
12. Grunau GL, Darlow B, Flynn T, et al. Red flags or red herrings? Redefining the role of red flags in low back pain to reduce overimaging. Br J Sports Med. 2018;52:488-489.
13. Lansberg MG, O’Donnell MJ, Khatri P, et al. Antithrombotic and thrombolytic therapy for ischemic stroke: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 suppl):e601S-e636S.
14. Pikija S, Mutzenbach JS, Kunz AB, et al. Delayed hospital presentation and neuroimaging in non-surgical spinal cord infarction. Front Neurol. 2017;8:143.
THE CASE
A 64-year-old woman with a history of late-onset type 1 diabetes mellitus, Hashimoto thyroiditis, and scoliosis presented to the sports medicine clinic with acute-onset, sharp, nonradiating right lower back pain that began when she bent forward to apply lotion. At presentation, she denied fever, chills, numbness, tingling, aggravation of pain with movement, weakness, and incontinence. Her neuromuscular examination was unremarkable except for left-side paraspinal tenderness. She was prescribed cyclobenzaprine for symptomatic relief.
Two days later, she was seen for worsening pain. Her physical exam was unchanged. She was prescribed tramadol and advised to start physical therapy gradually. As the day progressed, however, she developed anterior thigh sensory loss, which gradually extended distally.
The following day, she was brought to the emergency department with severe left-side weakness without urinary incontinence. Her mental status and cranial nerve exams were normal. On examination, strength of the iliopsoas and quadriceps was 1/5 bilaterally, and of the peroneal tendon and gastrocnemius, 3/5 bilaterally. Reflexes of triceps, biceps, knee, and Achilles tendon were symmetric and 3+ with bilateral clonus of the ankle. The Babinski sign was positive bilaterally. The patient had diminished pain sensation bilaterally, extending down from the T11 dermatome (left more than right side) with diminished vibration sensation at the left ankle. Her perianal sensation, bilateral temperature sensation, and cerebellar examination were normal.
Magnetic resonance imaging (MRI) without contrast of the lumbar spine demonstrated ischemia findings corresponding to T12-L1. Degenerative changes from L1-S1 were noted, with multiple osteophytes impinging on the neural foramina without cord compression.
THE DIAGNOSIS
The initial presentation was consistent with mechanical low back pain with signs of anterior spinal artery infarction and medial lemniscus pathway involvement 48 hours after initial presentation. Spinal cord infarction occurs more commonly in women and in the young than does cerebral infarction,1 with better reemployment rates.1,2 Similar to other strokes, long-term prognosis is primarily determined by the initial severity of motor impairment, which is linked to long-term immobility and need for bladder catheterization.3
Neurogenic pain developing years after spinal cord infarction is most often observed in anterior spinal artery infarction4 without functional limitations.
Initial treatment. Our patient was started on aspirin 325 mg/d and clopidogrel 75 mg/d. Her mean arterial blood pressure was maintained above 80 mm Hg. Computed tomography angiography of the abdomen and pelvis was negative for aortic dissection. Lumbar puncture for cerebrospinal fluid analysis was unremarkable. Results of antineutrophil cytoplasmic antibody testing, antinuclear antibody testing, a hepatitis panel, and an antiphospholipid panel were all negative. The patient was started on IV steroids with a plan for gradual tapering. The neurosurgical team agreed with medical management.
Continue to: DISCUSSION
DISCUSSION
Possible etiologies for acute spinal cord infarction include spinal cord ischemia from compression of the vessels, fibrocartilaginous embolism, and arterial thrombosis or atherosclerosis, especially in patients with diabetes.5
The majority (86%) of spinal strokes are due to spontaneous occlusion of the vessels with no identifiable cause; much less frequently (9% of cases), hemorrhage is the causative factor.1 A retrospective study demonstrated that 10 of 27 patients with spinal stroke had an anterior spinal infarct. Of those 10 patients, 6 reported a mechanical triggering movement (similar to this case), indicating potential compression of the radicular arteries due to said movement.4
Fibrocartilaginous embolism (FCE) is worth considering as a possible cause, because it accounts for 5.5% of all cases of acute spinal cord infarction.3 FCE is thought to arise after a precipitating event such as minor trauma, heavy lifting, physical exertion, or Valsalva maneuver causing embolization of the fragments of nucleus pulposus to the arterial system. In a case series of 8 patients, 2 had possible FCE with precipitating events occurring within the prior 24 hours. This was also demonstrated in another case series6 in which 7 of 9 patients had precipitating events.
Although FCE can only definitively be diagnosed postmortem, the researchers6 proposed clinical criteria for its diagnosis in living patients, based on 40 postmortem and 11 suspected antemortem cases of FCE. These criteria include a rapid evolution of symptoms consistent with vascular etiology, with or without preceding minor trauma or Valsalva maneuver; MRI changes consistent with ischemic myelopathy, with or without evidence of disc herniation; and no more than 2 vascular risk factors.
Our patient had no trauma (although there was a triggering movement), no signs of disc herniation, and 2 risk factors (> 60 years and diabetes mellitus). Also, a neurologically symptom-free interval between the painful movement and the onset of neurologic manifestations in our case parallels the clinical picture of FCE.
Continue to: The role of factor V Leiden (FVL) mutation
The role of factor V Leiden (FVL) mutation in arterial thrombosis is questionable. Previous reports demonstrate a risk for venous thrombosis 7 to 10 times higher with heterozygous FVL mutation and 100 times higher with homozygous mutation, with a less established role in arterial thrombosis.7 A retrospective Turkish study compared the incidence of FVL mutation in patients with arterial thrombosis vs healthy subjects; incidence was significantly higher in female patients than female controls (37.5% vs. 2%).7 A meta-analysis of published studies showed an association between arterial ischemic events and FVL mutation to be modest, with an odds ratio of 1.21 (95% CI, 0.99-1.49).8
In contrast, a 3.4-year longitudinal health study of patients ages 65 and older found no significant difference in the occurrence of myocardial infarction, transient ischemic attack, stroke, or angina for more than 5000 patients with heterozygous FVL mutation compared to fewer than 500 controls.9 The case patient’s clinical course did not fit a thrombotic clinical picture.
Evaluating for “red flags” is crucial in any case of low back pain to exclude serious pathologies. Red flag symptoms include signs of myelopathy, signs of infection, history of trauma with focal tenderness to palpation, and steroid or anticoagulant use (to rule out medication adverse effects).10 Our patient lacked these classical signs, but she did have subjective pain out of proportion to the clinical exam findings.
Of note: The above red flags for low back pain are all based on expert opinion,11 and the positive predictive value of a red flag is always low because of the low prevalence of serious spinal pathologies.12
Striking a proper balance. This case emphasizes the necessity to keep uncommon causes—such as nontraumatic spinal stroke, which has a prevalence of about 5% to 8% of all acute myelopathies—in the differential diagnosis.3
Continue to: We recommend watchful...
We recommend watchful waiting coupled with communication with the patient regarding monitoring for changes in symptoms over time.11 Any changes in symptoms concerning for underlying spinal cord injury indicate necessity for transfer to a tertiary care center (if possible), along with immediate evaluation with imaging—including computed tomography angiography of the abdomen to rule out aortic dissection (1%-2% of all spinal cord infarcts), followed by a specialist consultation based on the findings.3
Our patient
Our patient was discharged to rehabilitation on hospital Day 5, after progressive return of lower extremity strength. At the 2-month follow-up visit, she demonstrated grade 4+ strength throughout her lower extremities bilaterally. Weakness was predominant at the hip flexors and ankle dorsiflexors, which was consistent with her status at discharge. She had burning pain in the distribution of the L1 dermatome that responded to ibuprofen.
Hypercoagulability work-up was positive for heterozygous FVL mutation without any previous history of venous thromboembolic disease. She was continued on aspirin 325 mg/d, as per American College of Chest Physicians antithrombotic guidelines.13
One year later, our patient underwent a follow-up MRI of the thoracic spine, which showed an “owl’s eye” hyperintensity in the anterior cord (FIGURE), a sign that’s often seen in bilateral spinal cord infarction
THE TAKEAWAY
Spinal stroke is rare, but a missed diagnosis and lack of treatment can result in long-term morbidity. Therefore, it is prudent to consider this diagnosis in the differential—especially when the patient’s subjective back pain is out of proportion to the clinical examination findings.
CORRESPONDENCE
Srikanth Nithyanandam, MBBS, MS, University of Kentucky Family and Community Medicine, 2195 Harrodsburg Road, Suite 125, Lexington, KY 40504-3504; [email protected].
THE CASE
A 64-year-old woman with a history of late-onset type 1 diabetes mellitus, Hashimoto thyroiditis, and scoliosis presented to the sports medicine clinic with acute-onset, sharp, nonradiating right lower back pain that began when she bent forward to apply lotion. At presentation, she denied fever, chills, numbness, tingling, aggravation of pain with movement, weakness, and incontinence. Her neuromuscular examination was unremarkable except for left-side paraspinal tenderness. She was prescribed cyclobenzaprine for symptomatic relief.
Two days later, she was seen for worsening pain. Her physical exam was unchanged. She was prescribed tramadol and advised to start physical therapy gradually. As the day progressed, however, she developed anterior thigh sensory loss, which gradually extended distally.
The following day, she was brought to the emergency department with severe left-side weakness without urinary incontinence. Her mental status and cranial nerve exams were normal. On examination, strength of the iliopsoas and quadriceps was 1/5 bilaterally, and of the peroneal tendon and gastrocnemius, 3/5 bilaterally. Reflexes of triceps, biceps, knee, and Achilles tendon were symmetric and 3+ with bilateral clonus of the ankle. The Babinski sign was positive bilaterally. The patient had diminished pain sensation bilaterally, extending down from the T11 dermatome (left more than right side) with diminished vibration sensation at the left ankle. Her perianal sensation, bilateral temperature sensation, and cerebellar examination were normal.
Magnetic resonance imaging (MRI) without contrast of the lumbar spine demonstrated ischemia findings corresponding to T12-L1. Degenerative changes from L1-S1 were noted, with multiple osteophytes impinging on the neural foramina without cord compression.
THE DIAGNOSIS
The initial presentation was consistent with mechanical low back pain with signs of anterior spinal artery infarction and medial lemniscus pathway involvement 48 hours after initial presentation. Spinal cord infarction occurs more commonly in women and in the young than does cerebral infarction,1 with better reemployment rates.1,2 Similar to other strokes, long-term prognosis is primarily determined by the initial severity of motor impairment, which is linked to long-term immobility and need for bladder catheterization.3
Neurogenic pain developing years after spinal cord infarction is most often observed in anterior spinal artery infarction4 without functional limitations.
Initial treatment. Our patient was started on aspirin 325 mg/d and clopidogrel 75 mg/d. Her mean arterial blood pressure was maintained above 80 mm Hg. Computed tomography angiography of the abdomen and pelvis was negative for aortic dissection. Lumbar puncture for cerebrospinal fluid analysis was unremarkable. Results of antineutrophil cytoplasmic antibody testing, antinuclear antibody testing, a hepatitis panel, and an antiphospholipid panel were all negative. The patient was started on IV steroids with a plan for gradual tapering. The neurosurgical team agreed with medical management.
Continue to: DISCUSSION
DISCUSSION
Possible etiologies for acute spinal cord infarction include spinal cord ischemia from compression of the vessels, fibrocartilaginous embolism, and arterial thrombosis or atherosclerosis, especially in patients with diabetes.5
The majority (86%) of spinal strokes are due to spontaneous occlusion of the vessels with no identifiable cause; much less frequently (9% of cases), hemorrhage is the causative factor.1 A retrospective study demonstrated that 10 of 27 patients with spinal stroke had an anterior spinal infarct. Of those 10 patients, 6 reported a mechanical triggering movement (similar to this case), indicating potential compression of the radicular arteries due to said movement.4
Fibrocartilaginous embolism (FCE) is worth considering as a possible cause, because it accounts for 5.5% of all cases of acute spinal cord infarction.3 FCE is thought to arise after a precipitating event such as minor trauma, heavy lifting, physical exertion, or Valsalva maneuver causing embolization of the fragments of nucleus pulposus to the arterial system. In a case series of 8 patients, 2 had possible FCE with precipitating events occurring within the prior 24 hours. This was also demonstrated in another case series6 in which 7 of 9 patients had precipitating events.
Although FCE can only definitively be diagnosed postmortem, the researchers6 proposed clinical criteria for its diagnosis in living patients, based on 40 postmortem and 11 suspected antemortem cases of FCE. These criteria include a rapid evolution of symptoms consistent with vascular etiology, with or without preceding minor trauma or Valsalva maneuver; MRI changes consistent with ischemic myelopathy, with or without evidence of disc herniation; and no more than 2 vascular risk factors.
Our patient had no trauma (although there was a triggering movement), no signs of disc herniation, and 2 risk factors (> 60 years and diabetes mellitus). Also, a neurologically symptom-free interval between the painful movement and the onset of neurologic manifestations in our case parallels the clinical picture of FCE.
Continue to: The role of factor V Leiden (FVL) mutation
The role of factor V Leiden (FVL) mutation in arterial thrombosis is questionable. Previous reports demonstrate a risk for venous thrombosis 7 to 10 times higher with heterozygous FVL mutation and 100 times higher with homozygous mutation, with a less established role in arterial thrombosis.7 A retrospective Turkish study compared the incidence of FVL mutation in patients with arterial thrombosis vs healthy subjects; incidence was significantly higher in female patients than female controls (37.5% vs. 2%).7 A meta-analysis of published studies showed an association between arterial ischemic events and FVL mutation to be modest, with an odds ratio of 1.21 (95% CI, 0.99-1.49).8
In contrast, a 3.4-year longitudinal health study of patients ages 65 and older found no significant difference in the occurrence of myocardial infarction, transient ischemic attack, stroke, or angina for more than 5000 patients with heterozygous FVL mutation compared to fewer than 500 controls.9 The case patient’s clinical course did not fit a thrombotic clinical picture.
Evaluating for “red flags” is crucial in any case of low back pain to exclude serious pathologies. Red flag symptoms include signs of myelopathy, signs of infection, history of trauma with focal tenderness to palpation, and steroid or anticoagulant use (to rule out medication adverse effects).10 Our patient lacked these classical signs, but she did have subjective pain out of proportion to the clinical exam findings.
Of note: The above red flags for low back pain are all based on expert opinion,11 and the positive predictive value of a red flag is always low because of the low prevalence of serious spinal pathologies.12
Striking a proper balance. This case emphasizes the necessity to keep uncommon causes—such as nontraumatic spinal stroke, which has a prevalence of about 5% to 8% of all acute myelopathies—in the differential diagnosis.3
Continue to: We recommend watchful...
We recommend watchful waiting coupled with communication with the patient regarding monitoring for changes in symptoms over time.11 Any changes in symptoms concerning for underlying spinal cord injury indicate necessity for transfer to a tertiary care center (if possible), along with immediate evaluation with imaging—including computed tomography angiography of the abdomen to rule out aortic dissection (1%-2% of all spinal cord infarcts), followed by a specialist consultation based on the findings.3
Our patient
Our patient was discharged to rehabilitation on hospital Day 5, after progressive return of lower extremity strength. At the 2-month follow-up visit, she demonstrated grade 4+ strength throughout her lower extremities bilaterally. Weakness was predominant at the hip flexors and ankle dorsiflexors, which was consistent with her status at discharge. She had burning pain in the distribution of the L1 dermatome that responded to ibuprofen.
Hypercoagulability work-up was positive for heterozygous FVL mutation without any previous history of venous thromboembolic disease. She was continued on aspirin 325 mg/d, as per American College of Chest Physicians antithrombotic guidelines.13
One year later, our patient underwent a follow-up MRI of the thoracic spine, which showed an “owl’s eye” hyperintensity in the anterior cord (FIGURE), a sign that’s often seen in bilateral spinal cord infarction
THE TAKEAWAY
Spinal stroke is rare, but a missed diagnosis and lack of treatment can result in long-term morbidity. Therefore, it is prudent to consider this diagnosis in the differential—especially when the patient’s subjective back pain is out of proportion to the clinical examination findings.
CORRESPONDENCE
Srikanth Nithyanandam, MBBS, MS, University of Kentucky Family and Community Medicine, 2195 Harrodsburg Road, Suite 125, Lexington, KY 40504-3504; [email protected].
1. Romi F, Naess H. Spinal cord infarction in clinical neurology: a review of characteristics and long-term prognosis in comparison to cerebral infarction. Eur Neurol. 2016;76:95-98.
2. Hanson SR, Romi F, Rekand T, et al. Long-term outcome after spinal cord infarctions. Acta Neurol Scand. 2015;131:253-257.
3. Rigney L, Cappelen-Smith C, Sebire D, et al. Nontraumatic spinal cord ischaemic syndrome. J Clin Neurosci. 2015;22:1544-1549.
4. Novy J, Carruzzo A, Maeder P, Bogousslavsky J. Spinal cord ischemia: clinical and imaging patterns, pathogenesis, and outcomes in 27 patients. Arch Neurol. 2006;63:1113-1120.
5. Goldstein LB, Adams R, Alberts MJ, et al; American Heart Association; American Stroke Association Stroke Council. Primary prevention of ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council: cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2006;113:e873-e923.
6. Mateen FJ, Monrad PA, Hunderfund AN, et al. Clinically suspected fibrocartilaginous embolism: clinical characteristics, treatments, and outcomes. Eur J Neurol. 2011;18:218-225.
7. Ozmen F, Ozmen MM, Ozalp N, et al. The prevalence of factor V (G1691A), MTHFR (C677T) and PT (G20210A) gene mutations in arterial thrombosis. Ulus Travma Acil Cerrahi Derg. 2009;15:113-119.
8. Kim RJ, Becker RC. Association between factor V Leiden, prothrombin G20210A, and methylenetetrahydrofolate reductase C677T mutations and events of the arterial circulatory system: a meta-analysis of published studies. Am Heart J. 2003;146:948-957.
9. Cushman M, Rosendaal FR, Psaty BM, et al. Factor V Leiden is not a risk factor for arterial vascular disease in the elderly: results from the Cardiovascular Health Study. Thromb Haemost. 1998;79:912-915.
10. Strudwick K, McPhee M, Bell A, et al. Review article: best practice management of low back pain in the emergency department (part 1 of the musculoskeletal injuries rapid review series). Emerg Med Australas. 2018;30:18-35.
11. Cook CE, George SZ, Reiman MP. Red flag screening for low back pain: nothing to see here, move along: a narrative review. Br J Sports Med. 2018;52:493-496.
12. Grunau GL, Darlow B, Flynn T, et al. Red flags or red herrings? Redefining the role of red flags in low back pain to reduce overimaging. Br J Sports Med. 2018;52:488-489.
13. Lansberg MG, O’Donnell MJ, Khatri P, et al. Antithrombotic and thrombolytic therapy for ischemic stroke: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 suppl):e601S-e636S.
14. Pikija S, Mutzenbach JS, Kunz AB, et al. Delayed hospital presentation and neuroimaging in non-surgical spinal cord infarction. Front Neurol. 2017;8:143.
1. Romi F, Naess H. Spinal cord infarction in clinical neurology: a review of characteristics and long-term prognosis in comparison to cerebral infarction. Eur Neurol. 2016;76:95-98.
2. Hanson SR, Romi F, Rekand T, et al. Long-term outcome after spinal cord infarctions. Acta Neurol Scand. 2015;131:253-257.
3. Rigney L, Cappelen-Smith C, Sebire D, et al. Nontraumatic spinal cord ischaemic syndrome. J Clin Neurosci. 2015;22:1544-1549.
4. Novy J, Carruzzo A, Maeder P, Bogousslavsky J. Spinal cord ischemia: clinical and imaging patterns, pathogenesis, and outcomes in 27 patients. Arch Neurol. 2006;63:1113-1120.
5. Goldstein LB, Adams R, Alberts MJ, et al; American Heart Association; American Stroke Association Stroke Council. Primary prevention of ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council: cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2006;113:e873-e923.
6. Mateen FJ, Monrad PA, Hunderfund AN, et al. Clinically suspected fibrocartilaginous embolism: clinical characteristics, treatments, and outcomes. Eur J Neurol. 2011;18:218-225.
7. Ozmen F, Ozmen MM, Ozalp N, et al. The prevalence of factor V (G1691A), MTHFR (C677T) and PT (G20210A) gene mutations in arterial thrombosis. Ulus Travma Acil Cerrahi Derg. 2009;15:113-119.
8. Kim RJ, Becker RC. Association between factor V Leiden, prothrombin G20210A, and methylenetetrahydrofolate reductase C677T mutations and events of the arterial circulatory system: a meta-analysis of published studies. Am Heart J. 2003;146:948-957.
9. Cushman M, Rosendaal FR, Psaty BM, et al. Factor V Leiden is not a risk factor for arterial vascular disease in the elderly: results from the Cardiovascular Health Study. Thromb Haemost. 1998;79:912-915.
10. Strudwick K, McPhee M, Bell A, et al. Review article: best practice management of low back pain in the emergency department (part 1 of the musculoskeletal injuries rapid review series). Emerg Med Australas. 2018;30:18-35.
11. Cook CE, George SZ, Reiman MP. Red flag screening for low back pain: nothing to see here, move along: a narrative review. Br J Sports Med. 2018;52:493-496.
12. Grunau GL, Darlow B, Flynn T, et al. Red flags or red herrings? Redefining the role of red flags in low back pain to reduce overimaging. Br J Sports Med. 2018;52:488-489.
13. Lansberg MG, O’Donnell MJ, Khatri P, et al. Antithrombotic and thrombolytic therapy for ischemic stroke: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 suppl):e601S-e636S.
14. Pikija S, Mutzenbach JS, Kunz AB, et al. Delayed hospital presentation and neuroimaging in non-surgical spinal cord infarction. Front Neurol. 2017;8:143.

A Nervous Recipient of a “Tongue Lashing”
Self-injurious behaviors are common and can be either volitional or unintentional. Often people who perform these behaviors receive “tongue lashings” from family, friends, and loved ones. We recently treated a patient whose lesion in the oral cavity was thought to be caused by some form of self-injury, though the prognosis clearly depended on the true culprit. It is important for clinicians to identify the cause of the injury when encountering patients with oral cavity lesions.
Case Presentation
A 40-year-old white male with a medical history of bipolar disorder, posttraumatic stress disorder, polysubstance abuse, and recently diagnosed temporomandibular joint (TMJ) syndrome was seen in outpatient primary care for a bleeding lesion in his mouth for the past 3 weeks. The lesion was under the surface of his right tongue. He first noted the lesion after he had burned himself tasting some homemade rice pudding while under the influence of marijuana. The next day, an impression was taken of his mouth by a dental assistant who was fitting him for an oral appliance for his TMJ syndrome; according to his history, she did not perform a visual inspection of his mouth nor could he recall his last dental examination. He had neither lost weight nor experienced dysphagia. He was not taking any prescribed medications, had an 8 pack-year history of smoking cigarettes, and had smoked crack cocaine intermittently for several years. The also patient had chewed one-half tin per day of chewing tobacco for 5 years, though he had quit 7 years before presentation. He was consuming 6 alcoholic drinks daily and had no history of chewing betel nuts.
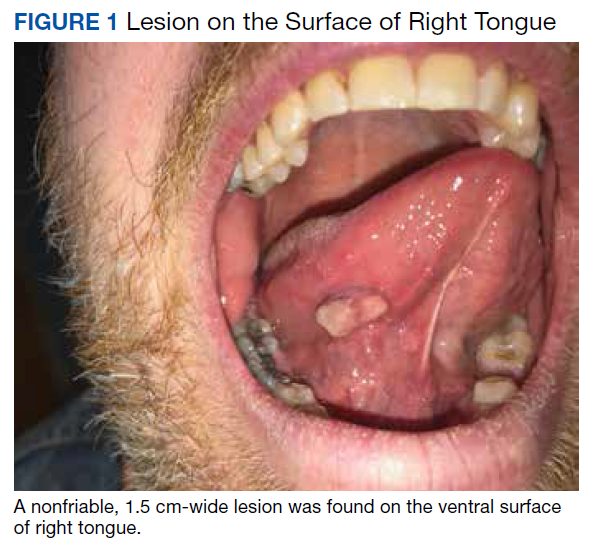
On physical examination, the patient seemed extremely anxious, but his vital signs were unremarkable. The nasal dorsum was straight, and the nares were widely patent. There were no suspicious cutaneous lesions noted of the face, head, trunk, or extremities. The salivary glands were soft and showed no lesions or masses within the parotid or submandibular glands bilaterally. There was no obvious obstruction of Stenson or Wharton ducts bilaterally. He had normal lips and oral competence. The dentition was noted to be fair.
A nonfriable, 1.5 cm-wide lesion was found on the ventral surface of the right tongue (Figure 1). The tongue was mobile. The mouth floor was soft and without evidence of masses or lesions. The tonsils, tonsillar pillars, palate, and base of tongue did not show any concerning lesions or masses. The neck revealed a nonenlarged thyroid and no lymphadenopathy. The remainder of the examination was unremarkable.
Diagnosis
Given his risk factors of alcohol use disorder and a history of both inhaled and chewing tobacco, oral squamous cell carcinoma (SCC) was considered. The differential diagnosis also included pyogenic granuloma, mucocele, sublingual fibroma, and metastasis to the oral soft tissue. Due to its implications with respect to morbidity and mortality, we thought it necessary to rule out SCC of the oral cavity. SCC comprises more than 90% of oral malignancies, and tobacco-related products, alcohol, and human papilloma virus are well-established risk factors.1
Pyogenic granuloma, also known as eruptive hemangioma and lobular capillary hemangioma, is a relatively common benign lesion of the skin and mucosal surfaces that often presents as a solitary, rapidly enlarging papule or nodule that is extremely friable.2 Interestingly, pyogenic granuloma is a misnomer, since it is neither infectious in origin nor granulomatous when visualized under the microscope and is thought to arise from an exuberant tissue response to localized irritation or trauma. An individual lesion can range in size from a few millimeters to a few centimeters and generally reaches its maximum size within a matter of weeks; they often arise at sites of minor trauma.3 While the pathogenesis of pyogenic granuloma has not been clearly established, it seems to be related to an imbalance of angiogenesis secondary to overexpression of vascular endothelial growth factor and basic fibroblast growth factor.4 While they can occur at any age, pyogenic granulomas are frequently seen in pediatric patients and during pregnancy.
A fibroma, also known as an irritation fibroma, is one of the more common fibrous tumorlike growths and is often caused by trauma or irritation. It usually presents as a smooth-surfaced, painless solid lesion, though it can be nodular and histopathologically shows collagen and connective tissue.5 While fibromas can occur anywhere in the oral cavity, they commonly arise on the buccal mucosa along the plane of occlusion between the maxillary and mandibular teeth.
Mucoceles are the most common benign lesions in the mouth and are commonly found on the lower lip and are mucus-filled cavities, arising from the accumulation of mucus from trauma or lip-biting and alteration of minor salivary glands.6 Our patient’s rapid evolution and history of trauma were consistent with a mucocele. Although the lower lip is the most common site of involvement, mucoceles also occur on the tongue, cheek, palate, and mouth floor.Metastases to the oral cavity are rare and comprise only 1% of all oral cavity malignancies.7 Although most commonly seen in the jaw, nearly one-third of oral cavity metastases are in the soft tissue.8 They generally occur late in the course of disease, and the time between appearance and death is usually short.8 Our patient’s lack of known primary malignancy and lack of weight loss rendered this diagnosis unlikely.
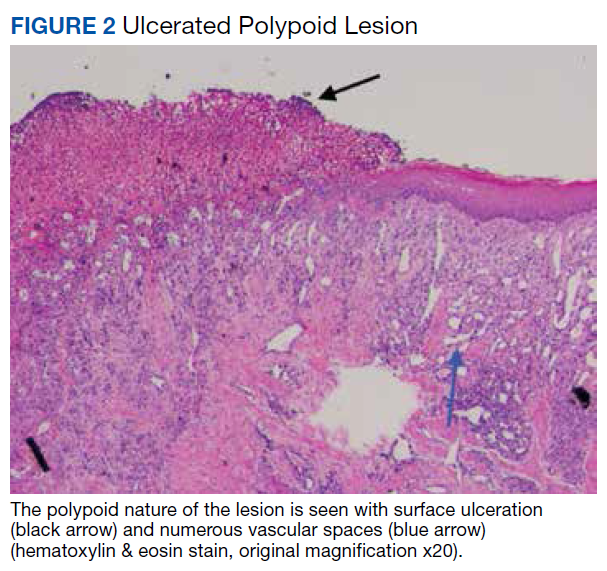
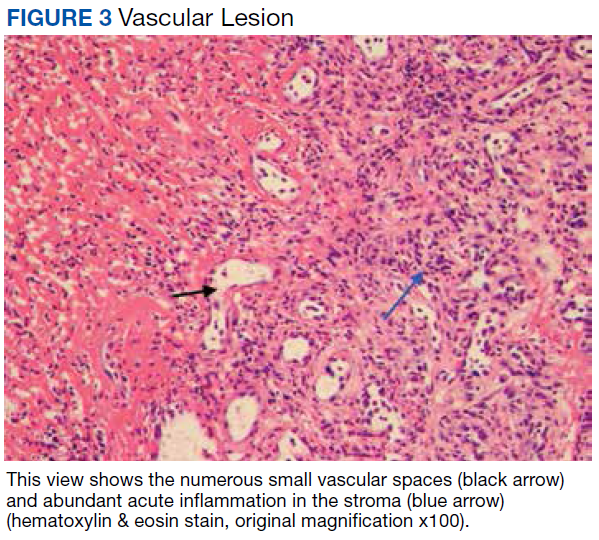
Other possibilities include peripheral giant cell granuloma, a reactive hyperplastic lesion of the oral cavity originating from the periosteum or periodontal membrane following local irritation or chronic trauma,9 and peripheral ossifying fibroma, a reactive soft tissue growth usually seen on the interdental papilla.10
Surgical excision was performed and revealed reactive epidermal hyperplasia, ulceration, granulation tissue formation, and marked inflammation with reactive changes. There was no evidence of malignancy and was interpreted as consistent with pyogenic granuloma (Figures 2 and 3) likely due to the trauma from the thermal burn or poor dentition.
Management
The patient was relieved to be informed of the diagnosis of an unusual presentation of pyogenic granuloma with no evidence of cancer. Current treatment strategies for pyogenic granuloma include surgical excision, shave excision with cautery, cryotherapy, sclerotherapy, carbon dioxide or pulsed dye laser, as well as expectant management. However, recurrence after initial treatment can occur, with lower recurrence rates occurring with surgical excision.11
Although we wouldn’t state that we gave the patient a “tongue-lashing,” we strongly advised him that he return to his dentist and abstain from tobacco products, alcohol, illicit drugs, and taste-testing scalding food directly from the pot.
1. Khot KP, Deshmane S, Choudhari S. Human papilloma virus in oral squamous cell carcinoma-the enigma unraveled. Clin J Dent Res. 2016;19(1):17-23.
2. Bolognia JL, Jorizzo JL, Rapini RP, eds. Neoplasms of the skin. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. St. Louis, MO: Mosby; 2007:1627-1901.
3. Tatusov M, Reddy S, Federman DG. Pyogenic granuloma: yet another motorcycle peril. Postgrad Med. 2012;124(6):124-126.
4. Yuan K, Jin YT, Lin MT. The detection and comparison of angiogenesis-associated factors in pyogenic granuloma by immunohistochemistry. J Periodontol. 2000;71(5):701-709.
5. Krishnan V, Shunmugavelu K. A clinical challenging situation of intra oral fibroma mimicking pyogenic granuloma. J Pan African Med. 2015;22(1):263.
6. Nallasivam KU, Sudha BR. Oral mucocele: review of literature and a case report. J Pharm Bioallied Sci. 2015;7(suppl 2):S731-S733.
7. Zachariades N. Neoplasms metastatic to the mouth, jaws, and surrounding tissues. J Craniomaxillofac Surg. 1989;17(6):283-290.
8. Irani S. Metastasis to the oral soft tissues: a review of 412 cases. J Int Soc Prev Community Dent. 2016;6(5):393-401.
9. Shadman N, Ebrahimi SF, Jafari S, Eslami M. Peripheral giant cell granuloma: a review of 123 cases. Dent Res J (Isfahan). 2009;6(1):47-50.
10. Poonacha KS, Shigli AL, Shirol D. Peripheral ossifying fibroma: a clinical report. Contemp Clin Dent. 2010;1(1):54-56.
11. Gilmore A, Kelsberg G, Safranek G. Clinical inquiries. What’s the best treatment for pyogenic granuloma? J Fam Pract. 2010;59(1):40-42.
Self-injurious behaviors are common and can be either volitional or unintentional. Often people who perform these behaviors receive “tongue lashings” from family, friends, and loved ones. We recently treated a patient whose lesion in the oral cavity was thought to be caused by some form of self-injury, though the prognosis clearly depended on the true culprit. It is important for clinicians to identify the cause of the injury when encountering patients with oral cavity lesions.
Case Presentation
A 40-year-old white male with a medical history of bipolar disorder, posttraumatic stress disorder, polysubstance abuse, and recently diagnosed temporomandibular joint (TMJ) syndrome was seen in outpatient primary care for a bleeding lesion in his mouth for the past 3 weeks. The lesion was under the surface of his right tongue. He first noted the lesion after he had burned himself tasting some homemade rice pudding while under the influence of marijuana. The next day, an impression was taken of his mouth by a dental assistant who was fitting him for an oral appliance for his TMJ syndrome; according to his history, she did not perform a visual inspection of his mouth nor could he recall his last dental examination. He had neither lost weight nor experienced dysphagia. He was not taking any prescribed medications, had an 8 pack-year history of smoking cigarettes, and had smoked crack cocaine intermittently for several years. The also patient had chewed one-half tin per day of chewing tobacco for 5 years, though he had quit 7 years before presentation. He was consuming 6 alcoholic drinks daily and had no history of chewing betel nuts.
On physical examination, the patient seemed extremely anxious, but his vital signs were unremarkable. The nasal dorsum was straight, and the nares were widely patent. There were no suspicious cutaneous lesions noted of the face, head, trunk, or extremities. The salivary glands were soft and showed no lesions or masses within the parotid or submandibular glands bilaterally. There was no obvious obstruction of Stenson or Wharton ducts bilaterally. He had normal lips and oral competence. The dentition was noted to be fair.
A nonfriable, 1.5 cm-wide lesion was found on the ventral surface of the right tongue (Figure 1). The tongue was mobile. The mouth floor was soft and without evidence of masses or lesions. The tonsils, tonsillar pillars, palate, and base of tongue did not show any concerning lesions or masses. The neck revealed a nonenlarged thyroid and no lymphadenopathy. The remainder of the examination was unremarkable.
Diagnosis
Given his risk factors of alcohol use disorder and a history of both inhaled and chewing tobacco, oral squamous cell carcinoma (SCC) was considered. The differential diagnosis also included pyogenic granuloma, mucocele, sublingual fibroma, and metastasis to the oral soft tissue. Due to its implications with respect to morbidity and mortality, we thought it necessary to rule out SCC of the oral cavity. SCC comprises more than 90% of oral malignancies, and tobacco-related products, alcohol, and human papilloma virus are well-established risk factors.1
Pyogenic granuloma, also known as eruptive hemangioma and lobular capillary hemangioma, is a relatively common benign lesion of the skin and mucosal surfaces that often presents as a solitary, rapidly enlarging papule or nodule that is extremely friable.2 Interestingly, pyogenic granuloma is a misnomer, since it is neither infectious in origin nor granulomatous when visualized under the microscope and is thought to arise from an exuberant tissue response to localized irritation or trauma. An individual lesion can range in size from a few millimeters to a few centimeters and generally reaches its maximum size within a matter of weeks; they often arise at sites of minor trauma.3 While the pathogenesis of pyogenic granuloma has not been clearly established, it seems to be related to an imbalance of angiogenesis secondary to overexpression of vascular endothelial growth factor and basic fibroblast growth factor.4 While they can occur at any age, pyogenic granulomas are frequently seen in pediatric patients and during pregnancy.
A fibroma, also known as an irritation fibroma, is one of the more common fibrous tumorlike growths and is often caused by trauma or irritation. It usually presents as a smooth-surfaced, painless solid lesion, though it can be nodular and histopathologically shows collagen and connective tissue.5 While fibromas can occur anywhere in the oral cavity, they commonly arise on the buccal mucosa along the plane of occlusion between the maxillary and mandibular teeth.
Mucoceles are the most common benign lesions in the mouth and are commonly found on the lower lip and are mucus-filled cavities, arising from the accumulation of mucus from trauma or lip-biting and alteration of minor salivary glands.6 Our patient’s rapid evolution and history of trauma were consistent with a mucocele. Although the lower lip is the most common site of involvement, mucoceles also occur on the tongue, cheek, palate, and mouth floor.Metastases to the oral cavity are rare and comprise only 1% of all oral cavity malignancies.7 Although most commonly seen in the jaw, nearly one-third of oral cavity metastases are in the soft tissue.8 They generally occur late in the course of disease, and the time between appearance and death is usually short.8 Our patient’s lack of known primary malignancy and lack of weight loss rendered this diagnosis unlikely.
Other possibilities include peripheral giant cell granuloma, a reactive hyperplastic lesion of the oral cavity originating from the periosteum or periodontal membrane following local irritation or chronic trauma,9 and peripheral ossifying fibroma, a reactive soft tissue growth usually seen on the interdental papilla.10
Surgical excision was performed and revealed reactive epidermal hyperplasia, ulceration, granulation tissue formation, and marked inflammation with reactive changes. There was no evidence of malignancy and was interpreted as consistent with pyogenic granuloma (Figures 2 and 3) likely due to the trauma from the thermal burn or poor dentition.
Management
The patient was relieved to be informed of the diagnosis of an unusual presentation of pyogenic granuloma with no evidence of cancer. Current treatment strategies for pyogenic granuloma include surgical excision, shave excision with cautery, cryotherapy, sclerotherapy, carbon dioxide or pulsed dye laser, as well as expectant management. However, recurrence after initial treatment can occur, with lower recurrence rates occurring with surgical excision.11
Although we wouldn’t state that we gave the patient a “tongue-lashing,” we strongly advised him that he return to his dentist and abstain from tobacco products, alcohol, illicit drugs, and taste-testing scalding food directly from the pot.
Self-injurious behaviors are common and can be either volitional or unintentional. Often people who perform these behaviors receive “tongue lashings” from family, friends, and loved ones. We recently treated a patient whose lesion in the oral cavity was thought to be caused by some form of self-injury, though the prognosis clearly depended on the true culprit. It is important for clinicians to identify the cause of the injury when encountering patients with oral cavity lesions.
Case Presentation
A 40-year-old white male with a medical history of bipolar disorder, posttraumatic stress disorder, polysubstance abuse, and recently diagnosed temporomandibular joint (TMJ) syndrome was seen in outpatient primary care for a bleeding lesion in his mouth for the past 3 weeks. The lesion was under the surface of his right tongue. He first noted the lesion after he had burned himself tasting some homemade rice pudding while under the influence of marijuana. The next day, an impression was taken of his mouth by a dental assistant who was fitting him for an oral appliance for his TMJ syndrome; according to his history, she did not perform a visual inspection of his mouth nor could he recall his last dental examination. He had neither lost weight nor experienced dysphagia. He was not taking any prescribed medications, had an 8 pack-year history of smoking cigarettes, and had smoked crack cocaine intermittently for several years. The also patient had chewed one-half tin per day of chewing tobacco for 5 years, though he had quit 7 years before presentation. He was consuming 6 alcoholic drinks daily and had no history of chewing betel nuts.
On physical examination, the patient seemed extremely anxious, but his vital signs were unremarkable. The nasal dorsum was straight, and the nares were widely patent. There were no suspicious cutaneous lesions noted of the face, head, trunk, or extremities. The salivary glands were soft and showed no lesions or masses within the parotid or submandibular glands bilaterally. There was no obvious obstruction of Stenson or Wharton ducts bilaterally. He had normal lips and oral competence. The dentition was noted to be fair.
A nonfriable, 1.5 cm-wide lesion was found on the ventral surface of the right tongue (Figure 1). The tongue was mobile. The mouth floor was soft and without evidence of masses or lesions. The tonsils, tonsillar pillars, palate, and base of tongue did not show any concerning lesions or masses. The neck revealed a nonenlarged thyroid and no lymphadenopathy. The remainder of the examination was unremarkable.
Diagnosis
Given his risk factors of alcohol use disorder and a history of both inhaled and chewing tobacco, oral squamous cell carcinoma (SCC) was considered. The differential diagnosis also included pyogenic granuloma, mucocele, sublingual fibroma, and metastasis to the oral soft tissue. Due to its implications with respect to morbidity and mortality, we thought it necessary to rule out SCC of the oral cavity. SCC comprises more than 90% of oral malignancies, and tobacco-related products, alcohol, and human papilloma virus are well-established risk factors.1
Pyogenic granuloma, also known as eruptive hemangioma and lobular capillary hemangioma, is a relatively common benign lesion of the skin and mucosal surfaces that often presents as a solitary, rapidly enlarging papule or nodule that is extremely friable.2 Interestingly, pyogenic granuloma is a misnomer, since it is neither infectious in origin nor granulomatous when visualized under the microscope and is thought to arise from an exuberant tissue response to localized irritation or trauma. An individual lesion can range in size from a few millimeters to a few centimeters and generally reaches its maximum size within a matter of weeks; they often arise at sites of minor trauma.3 While the pathogenesis of pyogenic granuloma has not been clearly established, it seems to be related to an imbalance of angiogenesis secondary to overexpression of vascular endothelial growth factor and basic fibroblast growth factor.4 While they can occur at any age, pyogenic granulomas are frequently seen in pediatric patients and during pregnancy.
A fibroma, also known as an irritation fibroma, is one of the more common fibrous tumorlike growths and is often caused by trauma or irritation. It usually presents as a smooth-surfaced, painless solid lesion, though it can be nodular and histopathologically shows collagen and connective tissue.5 While fibromas can occur anywhere in the oral cavity, they commonly arise on the buccal mucosa along the plane of occlusion between the maxillary and mandibular teeth.
Mucoceles are the most common benign lesions in the mouth and are commonly found on the lower lip and are mucus-filled cavities, arising from the accumulation of mucus from trauma or lip-biting and alteration of minor salivary glands.6 Our patient’s rapid evolution and history of trauma were consistent with a mucocele. Although the lower lip is the most common site of involvement, mucoceles also occur on the tongue, cheek, palate, and mouth floor.Metastases to the oral cavity are rare and comprise only 1% of all oral cavity malignancies.7 Although most commonly seen in the jaw, nearly one-third of oral cavity metastases are in the soft tissue.8 They generally occur late in the course of disease, and the time between appearance and death is usually short.8 Our patient’s lack of known primary malignancy and lack of weight loss rendered this diagnosis unlikely.
Other possibilities include peripheral giant cell granuloma, a reactive hyperplastic lesion of the oral cavity originating from the periosteum or periodontal membrane following local irritation or chronic trauma,9 and peripheral ossifying fibroma, a reactive soft tissue growth usually seen on the interdental papilla.10
Surgical excision was performed and revealed reactive epidermal hyperplasia, ulceration, granulation tissue formation, and marked inflammation with reactive changes. There was no evidence of malignancy and was interpreted as consistent with pyogenic granuloma (Figures 2 and 3) likely due to the trauma from the thermal burn or poor dentition.
Management
The patient was relieved to be informed of the diagnosis of an unusual presentation of pyogenic granuloma with no evidence of cancer. Current treatment strategies for pyogenic granuloma include surgical excision, shave excision with cautery, cryotherapy, sclerotherapy, carbon dioxide or pulsed dye laser, as well as expectant management. However, recurrence after initial treatment can occur, with lower recurrence rates occurring with surgical excision.11
Although we wouldn’t state that we gave the patient a “tongue-lashing,” we strongly advised him that he return to his dentist and abstain from tobacco products, alcohol, illicit drugs, and taste-testing scalding food directly from the pot.
1. Khot KP, Deshmane S, Choudhari S. Human papilloma virus in oral squamous cell carcinoma-the enigma unraveled. Clin J Dent Res. 2016;19(1):17-23.
2. Bolognia JL, Jorizzo JL, Rapini RP, eds. Neoplasms of the skin. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. St. Louis, MO: Mosby; 2007:1627-1901.
3. Tatusov M, Reddy S, Federman DG. Pyogenic granuloma: yet another motorcycle peril. Postgrad Med. 2012;124(6):124-126.
4. Yuan K, Jin YT, Lin MT. The detection and comparison of angiogenesis-associated factors in pyogenic granuloma by immunohistochemistry. J Periodontol. 2000;71(5):701-709.
5. Krishnan V, Shunmugavelu K. A clinical challenging situation of intra oral fibroma mimicking pyogenic granuloma. J Pan African Med. 2015;22(1):263.
6. Nallasivam KU, Sudha BR. Oral mucocele: review of literature and a case report. J Pharm Bioallied Sci. 2015;7(suppl 2):S731-S733.
7. Zachariades N. Neoplasms metastatic to the mouth, jaws, and surrounding tissues. J Craniomaxillofac Surg. 1989;17(6):283-290.
8. Irani S. Metastasis to the oral soft tissues: a review of 412 cases. J Int Soc Prev Community Dent. 2016;6(5):393-401.
9. Shadman N, Ebrahimi SF, Jafari S, Eslami M. Peripheral giant cell granuloma: a review of 123 cases. Dent Res J (Isfahan). 2009;6(1):47-50.
10. Poonacha KS, Shigli AL, Shirol D. Peripheral ossifying fibroma: a clinical report. Contemp Clin Dent. 2010;1(1):54-56.
11. Gilmore A, Kelsberg G, Safranek G. Clinical inquiries. What’s the best treatment for pyogenic granuloma? J Fam Pract. 2010;59(1):40-42.
1. Khot KP, Deshmane S, Choudhari S. Human papilloma virus in oral squamous cell carcinoma-the enigma unraveled. Clin J Dent Res. 2016;19(1):17-23.
2. Bolognia JL, Jorizzo JL, Rapini RP, eds. Neoplasms of the skin. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. St. Louis, MO: Mosby; 2007:1627-1901.
3. Tatusov M, Reddy S, Federman DG. Pyogenic granuloma: yet another motorcycle peril. Postgrad Med. 2012;124(6):124-126.
4. Yuan K, Jin YT, Lin MT. The detection and comparison of angiogenesis-associated factors in pyogenic granuloma by immunohistochemistry. J Periodontol. 2000;71(5):701-709.
5. Krishnan V, Shunmugavelu K. A clinical challenging situation of intra oral fibroma mimicking pyogenic granuloma. J Pan African Med. 2015;22(1):263.
6. Nallasivam KU, Sudha BR. Oral mucocele: review of literature and a case report. J Pharm Bioallied Sci. 2015;7(suppl 2):S731-S733.
7. Zachariades N. Neoplasms metastatic to the mouth, jaws, and surrounding tissues. J Craniomaxillofac Surg. 1989;17(6):283-290.
8. Irani S. Metastasis to the oral soft tissues: a review of 412 cases. J Int Soc Prev Community Dent. 2016;6(5):393-401.
9. Shadman N, Ebrahimi SF, Jafari S, Eslami M. Peripheral giant cell granuloma: a review of 123 cases. Dent Res J (Isfahan). 2009;6(1):47-50.
10. Poonacha KS, Shigli AL, Shirol D. Peripheral ossifying fibroma: a clinical report. Contemp Clin Dent. 2010;1(1):54-56.
11. Gilmore A, Kelsberg G, Safranek G. Clinical inquiries. What’s the best treatment for pyogenic granuloma? J Fam Pract. 2010;59(1):40-42.
Small Bowel Obstruction in a Surgically Naïve Abdomen
A 53-year-old male veteran with a history of heavy tobacco and alcohol use presented with abdominal pain, emesis, and no bowel movements for 2 days. He had no history of surgical procedures, malignancies, diverticulitis, inflammatory bowel disease, traveling abroad, parasitic infections, tuberculosis exposure, or hospital admissions for abdominal pain. He reported experiencing no flushing, diarrhea, or cardiac symptoms. His medical history included hypertension, depression, and osteoarthritis. His vital signs were within normal limits.
A physical examination revealed a distended abdomen with mild tenderness. He had no inguinal or ventral hernias. He also had no abnormal skin lesions. A rectal examination did not reveal any masses or blood. His laboratory values were normal. X-ray and computed tomography (CT) scan revealed dilated loops of proximal small bowel, mild wall thickening in a segment of the midileum, and narrowing of the distal small bowel suggestive of a partial small bowel obstruction (Figure 1). A 1-cm nonspecific omental nodule also was seen on the CT scan, but no enlarged lymph nodes or mesenteric calcifications were seen. There was no thickening of the terminal ileum.
The patient underwent an exploratory laparotomy, which revealed no adhesions. In the midileum there was an area of thickened bowel with some nodularity associated with the thickness, but no discrete mass. In the mesentery there were multiple hard, white, calcified nodules, with the majority clustered near the thickened ileal segment. There also was a 1-cm hard, peritoneal mass on the anterior abdominal wall. The segment of thickened ileum, the adjacent mesentery, and the peritoneal nodule were resected.
Pathologic examination of the resected tissue showed immunohistochemical stains that were positive for CD79a, CD10, and BCL-2 and negative for CD23, CD5, and CD3. Nineteen mesenteric lymph nodes were negative for malignancy. The postoperative staging positron emission tomography (PET) scan did not reveal any fluorodeoxyglucose avid masses anywhere else, and bone marrow biopsy showed no infiltration.
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
Based on the pathologic examination of the resected tissue and immunohistochemical stains, this patient was diagnosed with malignant non-Hodgkin B-cell lymphoma, follicular type, grade 1. PET scan and bone marrow biopsy revealed no other lesions, making this a primary lymphoma of the small intestine. The resected tissue showed negative margins and negative lymph nodes, indicating the full extent of the patient’s tumor was removed. He then underwent nasogastric tube decompression and IV fluid resuscitation. Two days later, he had a large bowel movement, and his abdominal pain resolved. He was provided the treatment options of observation only, radiation therapy, or rituximab treatment. Based on the high risk of enteritis following radiation therapy, the patient elected for observation only, with a repeat scan in 6 months. He also was counseled on alcohol and tobacco cessation. At the 6-month oncology follow-up, the patient showed no evidence of disease recurrence.
Discussion
Small bowel obstruction accounts for about 350,000 hospitalizations annually in the US.1 The incidence is equal in men and women and can present at any age.2,3 Patients typically present nonspecifically, with intermittent, colicky abdominal pain, nausea, vomiting, and constipation.2 A physical examination may reveal abdominal distention, rigidity, and hypoactive or absent bowel sounds.1 The 2 most common etiologies of small bowel obstruction are adhesions from prior abdominal surgery (65%) and incarcerated inguinal hernias (10%).1 However, in a patient presenting with a small bowel obstruction in a surgically naïve abdomen with no hernias, a more detailed history covering current malignancies, past hospital admissions for abdominal pains, pelvic inflammatory disease, diverticulitis, inflammatory bowel disease, and risks for parasite infection must be taken. The differential should include intraluminal causes, including small bowel malignancy, which accounts for 5% of small bowel obstructions,1 as well as extraluminal causes, including adhesions from diverticulitis, Meckel diverticulum, Ladd bands, and undiagnosed prior appendicitis.
To provide a tissue diagnosis and definitive treatment, surgical exploration was needed for this patient. Exploratory laparotomy revealed an area of thickened ileum and calcified nodules in its mesentery. Pathologic examination of the resected tissue revealed large lymphoid nodules in a follicular pattern with coarse chromatin (Figure 2). Taken together with the immunohistochemical stains, this was consistent with malignant B-cell non-Hodgkin lymphoma, follicular type, grade 1.
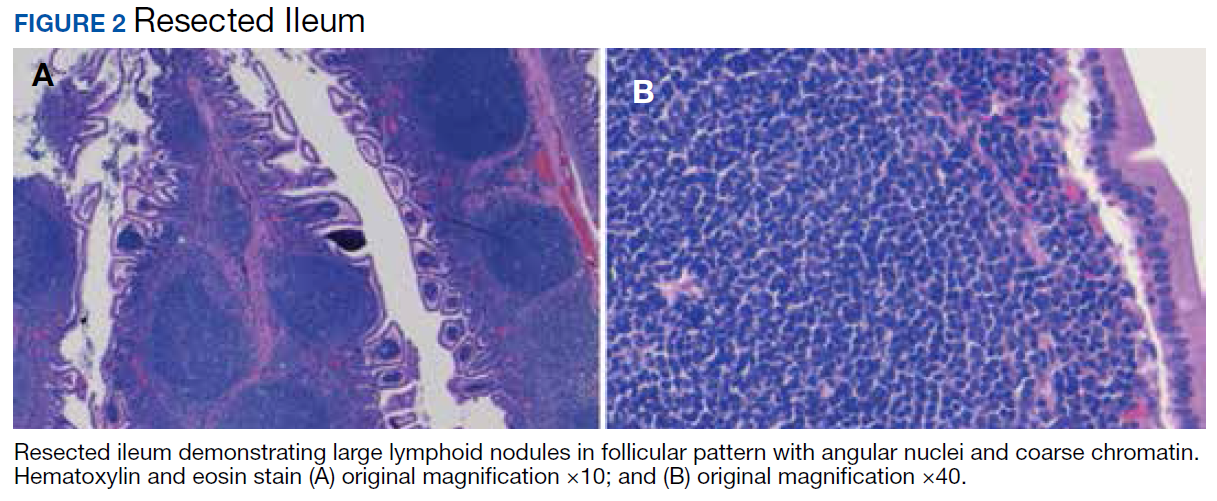
Small bowel malignancy accounts for > 5% of all gastrointestinal tumors.4 Of these, small bowel neuroendocrine tumors are the most common, followed by adenocarcinomas, lymphomas, and stromal tumors.4 Primary follicular lymphoma (PFL) is a B-cell non-Hodgkin lymphoma, and comprises between 3.8% and 11% of gastrointestinal lymphomas, commonly in the duodenum and terminal ileum.5
PFL typically occurs in middle-aged females and can be difficult to diagnose, as most patients are asymptomatic or present with unspecified abdominal pain. Many are diagnosed incidentally when endoscopy biopsies are performed for other reasons.4,5 Histologically, PFL is composed of a mixed population of small (centrocytes) and large (centroblasts) lymphoid cells, with higher proportions of centroblasts corresponding to a higher grade lymphoma.6 The classic immunophenotype of PFL shows coexpression of CD79a (or CD20), CD10, and BCL-2; however, in rare cases, low-grade PFL may stain negative for BCL-2 and have diminished staining for CD10 in interfollicular areas.7
PFL generally carries a favorable prognosis. Most patients achieving complete disease regression or stable disease following treatment and a low recurrence rate. Treatment can include surgical resection, radiation, rituximab therapy, chemotherapy, or observation.8 Patient also should be counseled in alcohol and tobacco cessation to reduce recurrence risk.
Other small bowel malignancies may present as small bowel obstructions as well. Neuroendocrine tumors and adenocarcinomas are both more common than small bowel lymphomas and can present as small bowel obstruction. However, neuroendocrine tumors are derived from serotonin-expressing enterochromaffin cells of the midgut and often present with classic carcinoid syndrome symptoms, including diarrhea, flushing, and right heart fibrosis, which the patient lacked.9 Immunohistology of small bowel adenocarcinoma often shows expression of MUC1 or MUC5AC with tumor markers CEA and CA 19-9.10
Primary intestinal melanoma, another small bowel malignancy, is extremely rare. More commonly, the etiology of intestinal melanoma is cutaneous melanoma that metastasizes to the gastrointestinal tract.11 This patient had no skin lesions to suggest metastatic melanoma. With intestinal melanoma, immunohistochemical evaluation may show S-100, the most sensitive marker for melanoma, or HMB-45, MART-1/Melan-A, tyrosinase, and MITF.12
Conclusion
This case is notable because it highlights the importance of examining the cause of small bowel obstruction in a surgically naïve abdomen, as exploration led to the discovery and curative treatment of a primary intestinal malignancy. It also underscores the nonspecific presentation that PFLs of the small intestine can have and the importance of understanding the different histopathology and immunohistochemical profiles of small bowel malignancies.
1. Rami Reddy SR, Cappell MS. A systematic review of the clinical presentation, diagnosis, and treatment of small bowel obstruction. Curr Gastroenterol Rep. 2017;19(6):28.
2. Smith DA, Nehring SM. Bowel obstruction. https://www.ncbi.nlm.nih.gov/books/NBK441975. Updated November 12, 2019. Accessed February 6, 2020.
3. Popoola D, Lou MA, Mansour AY, Sims EH. Small bowel obstruction: review of nine years of experience. J Natl Med Assoc. 1984;76(11):1089-1094.
4. Bilimoria KY, Bentrem DJ, Wayne JD, Ko CY, Bennett CL, Talamonti MS. Small bowel cancer in the United States: changes in epidemiology, treatment, and survival over the last 20 years. Ann Surg. 2009;249(1):63-71.
5. Freedman AS. Clinical presentation and diagnosis of primary gastrointestinal lymphomas. https://www.uptodate.com/contents/clinical-presentation-and-diagnosis-of-primary-gastrointestinal-lymphomas. Updated March 26, 2019. Accessed February 6, 2020.
6. Moy BT, Wilmot J, Ballesteros E, Forouhar F, Vaziri H. Primary follicular lymphoma of the gastrointestinal tract: casereport and review. J Gastrointest Cancer. 2016;47(3):255-263.
7. Choi SM, Betz BL, Perry AM. Follicular lymphoma diagnostic caveats and updates. Arch Pathol Lab Med. 2018;142(11):1330-1340.
8. Schmatz AI, Streubel B, Kretschmer-Chott E, et al. Primary follicular lymphoma of the duodenum is a distinct mucosal/submucosal variant of follicular lymphoma: a retrospective study of 63 cases. J Clin Oncol. 2011;29(11):1445-1451.
9. Grin A, Streutker CJ. Neuroendocrine tumors of the luminal gastrointestinal tract. Arch Pathol Lab Med. 2015;139(6):750-756.
10. Chang H-K, Yu E, Kim J, et al; Korean Small Intestinal Cancer Study Group. Adenocarcinoma of the small intestine: a multi-institutional study of 197 surgically resected cases. Hum Pathol. 2010;41(8):1087-1096.
11. Lens M, Bataille V, Krivokapic Z. Melanoma of the small intestine. Lancet Oncol. 2009;10(5):516-521.
12. Ohsie SJ, Sarantopoulos GP, Cochran AJ, Binder SW. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 2008;35(5):433-444.
A 53-year-old male veteran with a history of heavy tobacco and alcohol use presented with abdominal pain, emesis, and no bowel movements for 2 days. He had no history of surgical procedures, malignancies, diverticulitis, inflammatory bowel disease, traveling abroad, parasitic infections, tuberculosis exposure, or hospital admissions for abdominal pain. He reported experiencing no flushing, diarrhea, or cardiac symptoms. His medical history included hypertension, depression, and osteoarthritis. His vital signs were within normal limits.
A physical examination revealed a distended abdomen with mild tenderness. He had no inguinal or ventral hernias. He also had no abnormal skin lesions. A rectal examination did not reveal any masses or blood. His laboratory values were normal. X-ray and computed tomography (CT) scan revealed dilated loops of proximal small bowel, mild wall thickening in a segment of the midileum, and narrowing of the distal small bowel suggestive of a partial small bowel obstruction (Figure 1). A 1-cm nonspecific omental nodule also was seen on the CT scan, but no enlarged lymph nodes or mesenteric calcifications were seen. There was no thickening of the terminal ileum.
The patient underwent an exploratory laparotomy, which revealed no adhesions. In the midileum there was an area of thickened bowel with some nodularity associated with the thickness, but no discrete mass. In the mesentery there were multiple hard, white, calcified nodules, with the majority clustered near the thickened ileal segment. There also was a 1-cm hard, peritoneal mass on the anterior abdominal wall. The segment of thickened ileum, the adjacent mesentery, and the peritoneal nodule were resected.
Pathologic examination of the resected tissue showed immunohistochemical stains that were positive for CD79a, CD10, and BCL-2 and negative for CD23, CD5, and CD3. Nineteen mesenteric lymph nodes were negative for malignancy. The postoperative staging positron emission tomography (PET) scan did not reveal any fluorodeoxyglucose avid masses anywhere else, and bone marrow biopsy showed no infiltration.
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
Based on the pathologic examination of the resected tissue and immunohistochemical stains, this patient was diagnosed with malignant non-Hodgkin B-cell lymphoma, follicular type, grade 1. PET scan and bone marrow biopsy revealed no other lesions, making this a primary lymphoma of the small intestine. The resected tissue showed negative margins and negative lymph nodes, indicating the full extent of the patient’s tumor was removed. He then underwent nasogastric tube decompression and IV fluid resuscitation. Two days later, he had a large bowel movement, and his abdominal pain resolved. He was provided the treatment options of observation only, radiation therapy, or rituximab treatment. Based on the high risk of enteritis following radiation therapy, the patient elected for observation only, with a repeat scan in 6 months. He also was counseled on alcohol and tobacco cessation. At the 6-month oncology follow-up, the patient showed no evidence of disease recurrence.
Discussion
Small bowel obstruction accounts for about 350,000 hospitalizations annually in the US.1 The incidence is equal in men and women and can present at any age.2,3 Patients typically present nonspecifically, with intermittent, colicky abdominal pain, nausea, vomiting, and constipation.2 A physical examination may reveal abdominal distention, rigidity, and hypoactive or absent bowel sounds.1 The 2 most common etiologies of small bowel obstruction are adhesions from prior abdominal surgery (65%) and incarcerated inguinal hernias (10%).1 However, in a patient presenting with a small bowel obstruction in a surgically naïve abdomen with no hernias, a more detailed history covering current malignancies, past hospital admissions for abdominal pains, pelvic inflammatory disease, diverticulitis, inflammatory bowel disease, and risks for parasite infection must be taken. The differential should include intraluminal causes, including small bowel malignancy, which accounts for 5% of small bowel obstructions,1 as well as extraluminal causes, including adhesions from diverticulitis, Meckel diverticulum, Ladd bands, and undiagnosed prior appendicitis.
To provide a tissue diagnosis and definitive treatment, surgical exploration was needed for this patient. Exploratory laparotomy revealed an area of thickened ileum and calcified nodules in its mesentery. Pathologic examination of the resected tissue revealed large lymphoid nodules in a follicular pattern with coarse chromatin (Figure 2). Taken together with the immunohistochemical stains, this was consistent with malignant B-cell non-Hodgkin lymphoma, follicular type, grade 1.
Small bowel malignancy accounts for > 5% of all gastrointestinal tumors.4 Of these, small bowel neuroendocrine tumors are the most common, followed by adenocarcinomas, lymphomas, and stromal tumors.4 Primary follicular lymphoma (PFL) is a B-cell non-Hodgkin lymphoma, and comprises between 3.8% and 11% of gastrointestinal lymphomas, commonly in the duodenum and terminal ileum.5
PFL typically occurs in middle-aged females and can be difficult to diagnose, as most patients are asymptomatic or present with unspecified abdominal pain. Many are diagnosed incidentally when endoscopy biopsies are performed for other reasons.4,5 Histologically, PFL is composed of a mixed population of small (centrocytes) and large (centroblasts) lymphoid cells, with higher proportions of centroblasts corresponding to a higher grade lymphoma.6 The classic immunophenotype of PFL shows coexpression of CD79a (or CD20), CD10, and BCL-2; however, in rare cases, low-grade PFL may stain negative for BCL-2 and have diminished staining for CD10 in interfollicular areas.7
PFL generally carries a favorable prognosis. Most patients achieving complete disease regression or stable disease following treatment and a low recurrence rate. Treatment can include surgical resection, radiation, rituximab therapy, chemotherapy, or observation.8 Patient also should be counseled in alcohol and tobacco cessation to reduce recurrence risk.
Other small bowel malignancies may present as small bowel obstructions as well. Neuroendocrine tumors and adenocarcinomas are both more common than small bowel lymphomas and can present as small bowel obstruction. However, neuroendocrine tumors are derived from serotonin-expressing enterochromaffin cells of the midgut and often present with classic carcinoid syndrome symptoms, including diarrhea, flushing, and right heart fibrosis, which the patient lacked.9 Immunohistology of small bowel adenocarcinoma often shows expression of MUC1 or MUC5AC with tumor markers CEA and CA 19-9.10
Primary intestinal melanoma, another small bowel malignancy, is extremely rare. More commonly, the etiology of intestinal melanoma is cutaneous melanoma that metastasizes to the gastrointestinal tract.11 This patient had no skin lesions to suggest metastatic melanoma. With intestinal melanoma, immunohistochemical evaluation may show S-100, the most sensitive marker for melanoma, or HMB-45, MART-1/Melan-A, tyrosinase, and MITF.12
Conclusion
This case is notable because it highlights the importance of examining the cause of small bowel obstruction in a surgically naïve abdomen, as exploration led to the discovery and curative treatment of a primary intestinal malignancy. It also underscores the nonspecific presentation that PFLs of the small intestine can have and the importance of understanding the different histopathology and immunohistochemical profiles of small bowel malignancies.
A 53-year-old male veteran with a history of heavy tobacco and alcohol use presented with abdominal pain, emesis, and no bowel movements for 2 days. He had no history of surgical procedures, malignancies, diverticulitis, inflammatory bowel disease, traveling abroad, parasitic infections, tuberculosis exposure, or hospital admissions for abdominal pain. He reported experiencing no flushing, diarrhea, or cardiac symptoms. His medical history included hypertension, depression, and osteoarthritis. His vital signs were within normal limits.
A physical examination revealed a distended abdomen with mild tenderness. He had no inguinal or ventral hernias. He also had no abnormal skin lesions. A rectal examination did not reveal any masses or blood. His laboratory values were normal. X-ray and computed tomography (CT) scan revealed dilated loops of proximal small bowel, mild wall thickening in a segment of the midileum, and narrowing of the distal small bowel suggestive of a partial small bowel obstruction (Figure 1). A 1-cm nonspecific omental nodule also was seen on the CT scan, but no enlarged lymph nodes or mesenteric calcifications were seen. There was no thickening of the terminal ileum.
The patient underwent an exploratory laparotomy, which revealed no adhesions. In the midileum there was an area of thickened bowel with some nodularity associated with the thickness, but no discrete mass. In the mesentery there were multiple hard, white, calcified nodules, with the majority clustered near the thickened ileal segment. There also was a 1-cm hard, peritoneal mass on the anterior abdominal wall. The segment of thickened ileum, the adjacent mesentery, and the peritoneal nodule were resected.
Pathologic examination of the resected tissue showed immunohistochemical stains that were positive for CD79a, CD10, and BCL-2 and negative for CD23, CD5, and CD3. Nineteen mesenteric lymph nodes were negative for malignancy. The postoperative staging positron emission tomography (PET) scan did not reveal any fluorodeoxyglucose avid masses anywhere else, and bone marrow biopsy showed no infiltration.
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
Based on the pathologic examination of the resected tissue and immunohistochemical stains, this patient was diagnosed with malignant non-Hodgkin B-cell lymphoma, follicular type, grade 1. PET scan and bone marrow biopsy revealed no other lesions, making this a primary lymphoma of the small intestine. The resected tissue showed negative margins and negative lymph nodes, indicating the full extent of the patient’s tumor was removed. He then underwent nasogastric tube decompression and IV fluid resuscitation. Two days later, he had a large bowel movement, and his abdominal pain resolved. He was provided the treatment options of observation only, radiation therapy, or rituximab treatment. Based on the high risk of enteritis following radiation therapy, the patient elected for observation only, with a repeat scan in 6 months. He also was counseled on alcohol and tobacco cessation. At the 6-month oncology follow-up, the patient showed no evidence of disease recurrence.
Discussion
Small bowel obstruction accounts for about 350,000 hospitalizations annually in the US.1 The incidence is equal in men and women and can present at any age.2,3 Patients typically present nonspecifically, with intermittent, colicky abdominal pain, nausea, vomiting, and constipation.2 A physical examination may reveal abdominal distention, rigidity, and hypoactive or absent bowel sounds.1 The 2 most common etiologies of small bowel obstruction are adhesions from prior abdominal surgery (65%) and incarcerated inguinal hernias (10%).1 However, in a patient presenting with a small bowel obstruction in a surgically naïve abdomen with no hernias, a more detailed history covering current malignancies, past hospital admissions for abdominal pains, pelvic inflammatory disease, diverticulitis, inflammatory bowel disease, and risks for parasite infection must be taken. The differential should include intraluminal causes, including small bowel malignancy, which accounts for 5% of small bowel obstructions,1 as well as extraluminal causes, including adhesions from diverticulitis, Meckel diverticulum, Ladd bands, and undiagnosed prior appendicitis.
To provide a tissue diagnosis and definitive treatment, surgical exploration was needed for this patient. Exploratory laparotomy revealed an area of thickened ileum and calcified nodules in its mesentery. Pathologic examination of the resected tissue revealed large lymphoid nodules in a follicular pattern with coarse chromatin (Figure 2). Taken together with the immunohistochemical stains, this was consistent with malignant B-cell non-Hodgkin lymphoma, follicular type, grade 1.
Small bowel malignancy accounts for > 5% of all gastrointestinal tumors.4 Of these, small bowel neuroendocrine tumors are the most common, followed by adenocarcinomas, lymphomas, and stromal tumors.4 Primary follicular lymphoma (PFL) is a B-cell non-Hodgkin lymphoma, and comprises between 3.8% and 11% of gastrointestinal lymphomas, commonly in the duodenum and terminal ileum.5
PFL typically occurs in middle-aged females and can be difficult to diagnose, as most patients are asymptomatic or present with unspecified abdominal pain. Many are diagnosed incidentally when endoscopy biopsies are performed for other reasons.4,5 Histologically, PFL is composed of a mixed population of small (centrocytes) and large (centroblasts) lymphoid cells, with higher proportions of centroblasts corresponding to a higher grade lymphoma.6 The classic immunophenotype of PFL shows coexpression of CD79a (or CD20), CD10, and BCL-2; however, in rare cases, low-grade PFL may stain negative for BCL-2 and have diminished staining for CD10 in interfollicular areas.7
PFL generally carries a favorable prognosis. Most patients achieving complete disease regression or stable disease following treatment and a low recurrence rate. Treatment can include surgical resection, radiation, rituximab therapy, chemotherapy, or observation.8 Patient also should be counseled in alcohol and tobacco cessation to reduce recurrence risk.
Other small bowel malignancies may present as small bowel obstructions as well. Neuroendocrine tumors and adenocarcinomas are both more common than small bowel lymphomas and can present as small bowel obstruction. However, neuroendocrine tumors are derived from serotonin-expressing enterochromaffin cells of the midgut and often present with classic carcinoid syndrome symptoms, including diarrhea, flushing, and right heart fibrosis, which the patient lacked.9 Immunohistology of small bowel adenocarcinoma often shows expression of MUC1 or MUC5AC with tumor markers CEA and CA 19-9.10
Primary intestinal melanoma, another small bowel malignancy, is extremely rare. More commonly, the etiology of intestinal melanoma is cutaneous melanoma that metastasizes to the gastrointestinal tract.11 This patient had no skin lesions to suggest metastatic melanoma. With intestinal melanoma, immunohistochemical evaluation may show S-100, the most sensitive marker for melanoma, or HMB-45, MART-1/Melan-A, tyrosinase, and MITF.12
Conclusion
This case is notable because it highlights the importance of examining the cause of small bowel obstruction in a surgically naïve abdomen, as exploration led to the discovery and curative treatment of a primary intestinal malignancy. It also underscores the nonspecific presentation that PFLs of the small intestine can have and the importance of understanding the different histopathology and immunohistochemical profiles of small bowel malignancies.
1. Rami Reddy SR, Cappell MS. A systematic review of the clinical presentation, diagnosis, and treatment of small bowel obstruction. Curr Gastroenterol Rep. 2017;19(6):28.
2. Smith DA, Nehring SM. Bowel obstruction. https://www.ncbi.nlm.nih.gov/books/NBK441975. Updated November 12, 2019. Accessed February 6, 2020.
3. Popoola D, Lou MA, Mansour AY, Sims EH. Small bowel obstruction: review of nine years of experience. J Natl Med Assoc. 1984;76(11):1089-1094.
4. Bilimoria KY, Bentrem DJ, Wayne JD, Ko CY, Bennett CL, Talamonti MS. Small bowel cancer in the United States: changes in epidemiology, treatment, and survival over the last 20 years. Ann Surg. 2009;249(1):63-71.
5. Freedman AS. Clinical presentation and diagnosis of primary gastrointestinal lymphomas. https://www.uptodate.com/contents/clinical-presentation-and-diagnosis-of-primary-gastrointestinal-lymphomas. Updated March 26, 2019. Accessed February 6, 2020.
6. Moy BT, Wilmot J, Ballesteros E, Forouhar F, Vaziri H. Primary follicular lymphoma of the gastrointestinal tract: casereport and review. J Gastrointest Cancer. 2016;47(3):255-263.
7. Choi SM, Betz BL, Perry AM. Follicular lymphoma diagnostic caveats and updates. Arch Pathol Lab Med. 2018;142(11):1330-1340.
8. Schmatz AI, Streubel B, Kretschmer-Chott E, et al. Primary follicular lymphoma of the duodenum is a distinct mucosal/submucosal variant of follicular lymphoma: a retrospective study of 63 cases. J Clin Oncol. 2011;29(11):1445-1451.
9. Grin A, Streutker CJ. Neuroendocrine tumors of the luminal gastrointestinal tract. Arch Pathol Lab Med. 2015;139(6):750-756.
10. Chang H-K, Yu E, Kim J, et al; Korean Small Intestinal Cancer Study Group. Adenocarcinoma of the small intestine: a multi-institutional study of 197 surgically resected cases. Hum Pathol. 2010;41(8):1087-1096.
11. Lens M, Bataille V, Krivokapic Z. Melanoma of the small intestine. Lancet Oncol. 2009;10(5):516-521.
12. Ohsie SJ, Sarantopoulos GP, Cochran AJ, Binder SW. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 2008;35(5):433-444.
1. Rami Reddy SR, Cappell MS. A systematic review of the clinical presentation, diagnosis, and treatment of small bowel obstruction. Curr Gastroenterol Rep. 2017;19(6):28.
2. Smith DA, Nehring SM. Bowel obstruction. https://www.ncbi.nlm.nih.gov/books/NBK441975. Updated November 12, 2019. Accessed February 6, 2020.
3. Popoola D, Lou MA, Mansour AY, Sims EH. Small bowel obstruction: review of nine years of experience. J Natl Med Assoc. 1984;76(11):1089-1094.
4. Bilimoria KY, Bentrem DJ, Wayne JD, Ko CY, Bennett CL, Talamonti MS. Small bowel cancer in the United States: changes in epidemiology, treatment, and survival over the last 20 years. Ann Surg. 2009;249(1):63-71.
5. Freedman AS. Clinical presentation and diagnosis of primary gastrointestinal lymphomas. https://www.uptodate.com/contents/clinical-presentation-and-diagnosis-of-primary-gastrointestinal-lymphomas. Updated March 26, 2019. Accessed February 6, 2020.
6. Moy BT, Wilmot J, Ballesteros E, Forouhar F, Vaziri H. Primary follicular lymphoma of the gastrointestinal tract: casereport and review. J Gastrointest Cancer. 2016;47(3):255-263.
7. Choi SM, Betz BL, Perry AM. Follicular lymphoma diagnostic caveats and updates. Arch Pathol Lab Med. 2018;142(11):1330-1340.
8. Schmatz AI, Streubel B, Kretschmer-Chott E, et al. Primary follicular lymphoma of the duodenum is a distinct mucosal/submucosal variant of follicular lymphoma: a retrospective study of 63 cases. J Clin Oncol. 2011;29(11):1445-1451.
9. Grin A, Streutker CJ. Neuroendocrine tumors of the luminal gastrointestinal tract. Arch Pathol Lab Med. 2015;139(6):750-756.
10. Chang H-K, Yu E, Kim J, et al; Korean Small Intestinal Cancer Study Group. Adenocarcinoma of the small intestine: a multi-institutional study of 197 surgically resected cases. Hum Pathol. 2010;41(8):1087-1096.
11. Lens M, Bataille V, Krivokapic Z. Melanoma of the small intestine. Lancet Oncol. 2009;10(5):516-521.
12. Ohsie SJ, Sarantopoulos GP, Cochran AJ, Binder SW. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 2008;35(5):433-444.
Concurrent Beau Lines, Onychomadesis, and Retronychia Following Scurvy
Beau lines are palpable transverse depressions on the dorsal aspect of the nail plate that result from a temporary slowing of nail plate production by the proximal nail matrix. Onychomadesis is a separation of the proximal nail plate from the distal nail plate leading to shedding of the nail. It occurs due to a complete growth arrest in the nail matrix and is thought to be on a continuum with Beau lines. The etiologies of these 2 conditions overlap and include trauma, inflammatory diseases, systemic illnesses, hereditary conditions, and infections.1-5 In almost all cases of both conditions, normal nail plate production ensues upon identification and removal of the inciting agent or recuperation from the causal illness.3,4,6 Beau lines will move distally as the nail grows out and can be clipped. In onychomadesis, the affected nails will be shed with time. Resolution of these nail defects can be estimated from average nail growth rates (1 mm/mo for fingernails and 2–3 mm/mo for toenails).7
Retronychia is defined as a proximal ingrowing of the nail plate into the ventral surface of the proximal nail fold.4,6 It is thought to occur via vertical progression of the nail plate into the proximal nail fold, repetitive nail matrix trauma, or shearing forces, resulting in inflammation that leads to nail plate stacking.8,9 Although conservative treatment using topical corticosteroids may be attempted, proximal nail plate avulsion typically is required for treatment.10
Braswell et al1 suggested a unifying hypothesis for a common pathophysiologic basis for these 3 conditions; that is, nail matrix injury results in slowing and/or cessation of nail plate production, followed by recommencement of nail plate production by the nail matrix after removal of the insult. We report a case of a patient presenting with concurrent Beau lines, onychomadesis, and retronychia following scurvy, thus supporting the hypothesis that these 3 nail conditions lie on a continuum.
Case Report
A 41-year-old woman with a history of thyroiditis, gastroesophageal reflux disease, endometriosis, osteoarthritis, gastric ulcer, pancreatitis, fatty liver, and polycystic ovarian syndrome presented with lines on the toenails and no growth of the right second toenail of several months’ duration. She denied any pain or prior trauma to the nails, participation in sports activities, or wearing tight or high-heeled shoes. She had presented 6 months prior for evaluation of perifollicular erythema on the posterior thighs, legs, and abdomen, as well as gingival bleeding.11 At that time, one of the authors (S.R.L.) found that she was vitamin C deficient, and a diagnosis of scurvy was made. The rash and gingival bleeding resolved with vitamin C supplementation.11
At the current presentation, physical examination revealed transverse grooves involving several fingernails but most evident on the left thumbnail (Figure, A). The grooves did not span the entire breadth of the nail, which was consistent with Beau lines. Several toenails had parallel transverse grooves spanning the entire width of the nail plate such that the proximal nail plate was discontinuous with the distal nail plate, which was consistent with onychomadesis (Figure, B). The right second toenail was yellow and thickened with layered nail plates, indicative of retronychia (Figure, B). Histopathology of a nail plate clipping from the right second toenail was negative for fungal hyphae, and a radiograph was negative for bony changes or exostosis.
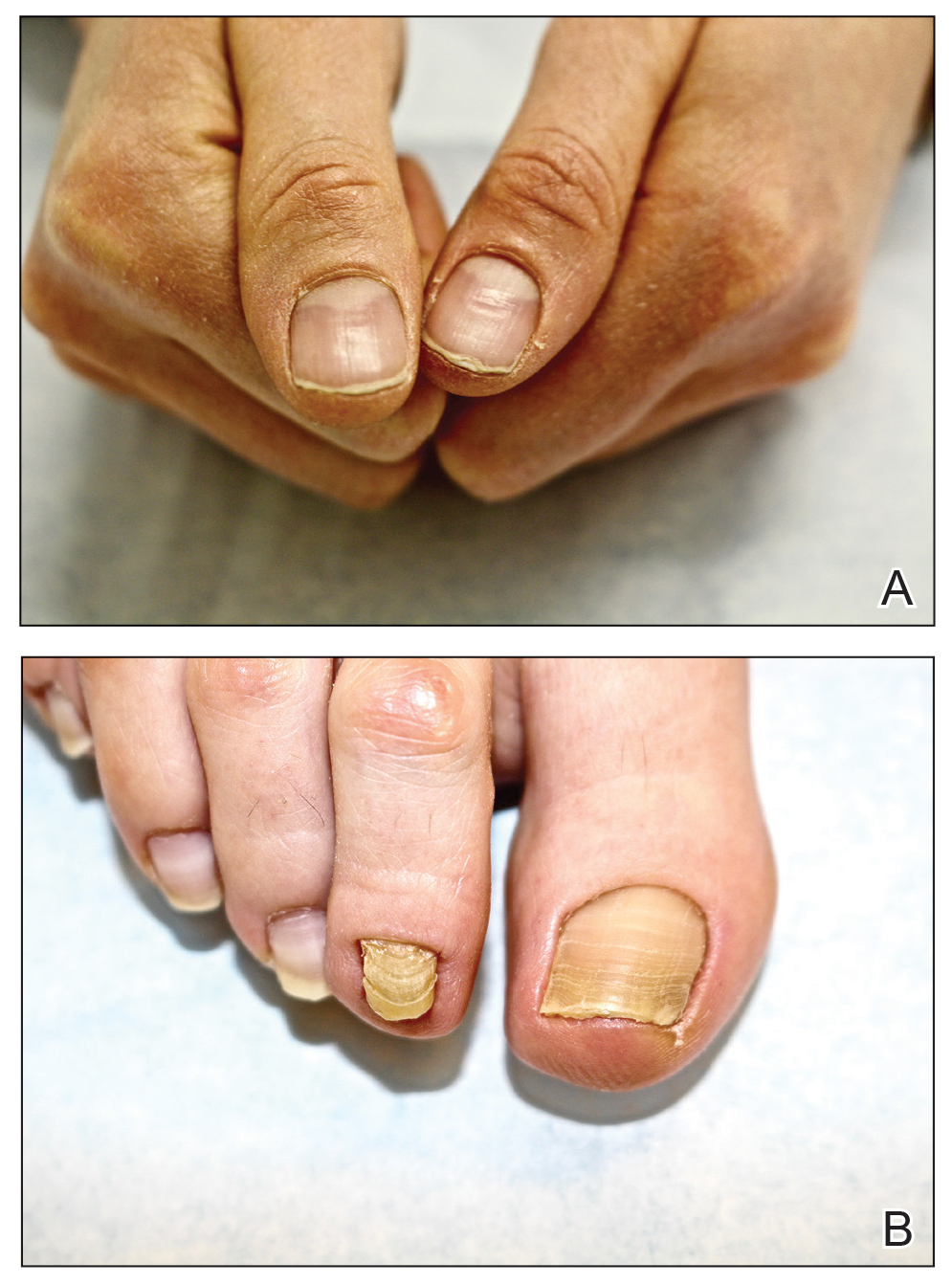
Comment
The nail matrix is responsible for nail plate production, and the newly formed nail plate then moves outward over the nail bed. It is hypothesized that the pathophysiologic basis for Beau lines, onychomadesis, and retronychia lies on a continuum such that all 3 conditions are caused by an insult to the nail matrix that results in slowing and/or halting of nail plate growth. Beau lines result from slowing or disruption in cell growth from the nail matrix, whereas onychomadesis is associated with a complete halt in nail plate production.1,3 In retronychia, the new nail growing from the matrix pushes the old one upward, interrupting the longitudinal growth of the nail and leading to nail plate stacking.10
Our patient presented with concurrent Beau lines, onychomadesis, and retronychia. Although Beau lines and onychomadesis have been reported together in some instances,12-14 retronychia is not commonly reported with either of these conditions. The exact incidence of each condition has not been studied, but Beau lines are relatively common, onychomadesis is less common, and retronychia is seen infrequently; therefore, the concurrent presentation of these 3 conditions in the same patient is exceedingly rare. Thus, it was most likely that one etiology accounted for all 3 nail findings.
Because the patient had been diagnosed with scurvy 6 months prior to presentation, we hypothesized that the associated vitamin C deficiency caused a systemic insult to the nail matrix, which resulted in cessation of nail growth. The mechanism of nail matrix arrest in the setting of systemic disease is thought to be due to inhibition of cellular proliferation or a change in the quality of the newly manufactured nail plate, which becomes thinner and more dystrophic.15 Vitamin C (ascorbic acid) deficiency causes scurvy, which is characterized by cutaneous signs such as perifollicular hemorrhage and purpura, corkscrew hairs, bruising, gingivitis, arthralgia, and impaired wound healing.16 These clinical manifestations are due to impaired collagen synthesis and disordered connective tissue. Ascorbic acid also is involved in fatty acid transport, neurotransmitter synthesis, prostaglandin metabolism, and nitric oxide synthesis.17 Ascorbic acid has not been studied for its role in nail plate synthesis18; however, given the role that ascorbic acid plays in a myriad of biologic processes, the deficiency associated with scurvy likely had a considerable systemic effect in our patient that halted nail plate synthesis and resulted in the concurrent presentation of Beau lines, onychomadesis, and retronychia.
- Braswell MA, Daniel CR III, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Lipner SR. Onychomadesis following a fish pedicure. JAMA Dermatol. 2018;154:1091-1092.
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Lawry M, Daniel CR III. Nails in systemic disease. In: Scher RK, Daniel CR III, eds. Nails: Diagnosis, Therapy, Surgery. 3rd ed. Oxford, England: Elsevier Saunders; 2005:147-176.
- Lipner SR, Scher RK. Evaluation of nail lines: color and shape hold clues. Cleve Clin J Med. 2016;83:385.
- Rich P. Nail signs and symptoms. In: Scher RK, Daniel CR III, eds. Nails: Diagnosis, Therapy, Surgery. 3rd ed. Oxford, England: Elsevier Saunders; 2005:1-6.
- Lipner SR, Scher RK. Nail growth evaluation and factors affecting nail growth. In: Humbert P, Fanian F, Maibach H, et al, eds. Agache’s Measuring the Skin. Cham, Switzerland: Springer; 2017:1-15.
- de Berker DA, Richert B, Duhard E, et al. Retronychia: proximal ingrowing of the nail plate. J Am Acad Dermatol. 2008;58:978-983.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Piraccini BM, Richert B, de Berker DA, et al. Retronychia in children, adolescents, and young adults: a case series. J Am Acad Dermatol. 2014;70:388-390.
- Lipner S. A classic case of scurvy. Lancet. 2018;392:431.
- Jacobsen L, Zimmerman S, Lohr J. Nail findings in hand-foot-and-mouth disease. Pediatr Infect Dis J. 2015;34:449-450.
- Damevska K, Gocev G, Pollozhani N, et al. Onychomadesis following cutaneous vasculitis. Acta Dermatovenerol Croat. 2017;25:77-79.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand‐foot‐mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Weismann K. J.H.S Beau and his descriptions of transverse depressions on nails. Br J Dermatol. 1977;97:571-572.
- Abdullah M, Jamil RT, Attia FN. Vitamin C (ascorbic acid). Treasure Island, FL: StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK499877/. Updated October 21, 2019. Accessed February 24, 2020.
- Pazirandeh S, Burns DL. Overview of water-soluble vitamins. UpToDate. https://www.uptodate.com/contents/overview-of-water-soluble-vitamins. Updated January 29, 2020. Accessed February 24, 2020.
- Scheinfeld N, Dahdah MJ, Scher RK. Vitamins and minerals: their role in nail health and disease. J Drugs Dermatol. 2007;6:782-787.
Beau lines are palpable transverse depressions on the dorsal aspect of the nail plate that result from a temporary slowing of nail plate production by the proximal nail matrix. Onychomadesis is a separation of the proximal nail plate from the distal nail plate leading to shedding of the nail. It occurs due to a complete growth arrest in the nail matrix and is thought to be on a continuum with Beau lines. The etiologies of these 2 conditions overlap and include trauma, inflammatory diseases, systemic illnesses, hereditary conditions, and infections.1-5 In almost all cases of both conditions, normal nail plate production ensues upon identification and removal of the inciting agent or recuperation from the causal illness.3,4,6 Beau lines will move distally as the nail grows out and can be clipped. In onychomadesis, the affected nails will be shed with time. Resolution of these nail defects can be estimated from average nail growth rates (1 mm/mo for fingernails and 2–3 mm/mo for toenails).7
Retronychia is defined as a proximal ingrowing of the nail plate into the ventral surface of the proximal nail fold.4,6 It is thought to occur via vertical progression of the nail plate into the proximal nail fold, repetitive nail matrix trauma, or shearing forces, resulting in inflammation that leads to nail plate stacking.8,9 Although conservative treatment using topical corticosteroids may be attempted, proximal nail plate avulsion typically is required for treatment.10
Braswell et al1 suggested a unifying hypothesis for a common pathophysiologic basis for these 3 conditions; that is, nail matrix injury results in slowing and/or cessation of nail plate production, followed by recommencement of nail plate production by the nail matrix after removal of the insult. We report a case of a patient presenting with concurrent Beau lines, onychomadesis, and retronychia following scurvy, thus supporting the hypothesis that these 3 nail conditions lie on a continuum.
Case Report
A 41-year-old woman with a history of thyroiditis, gastroesophageal reflux disease, endometriosis, osteoarthritis, gastric ulcer, pancreatitis, fatty liver, and polycystic ovarian syndrome presented with lines on the toenails and no growth of the right second toenail of several months’ duration. She denied any pain or prior trauma to the nails, participation in sports activities, or wearing tight or high-heeled shoes. She had presented 6 months prior for evaluation of perifollicular erythema on the posterior thighs, legs, and abdomen, as well as gingival bleeding.11 At that time, one of the authors (S.R.L.) found that she was vitamin C deficient, and a diagnosis of scurvy was made. The rash and gingival bleeding resolved with vitamin C supplementation.11
At the current presentation, physical examination revealed transverse grooves involving several fingernails but most evident on the left thumbnail (Figure, A). The grooves did not span the entire breadth of the nail, which was consistent with Beau lines. Several toenails had parallel transverse grooves spanning the entire width of the nail plate such that the proximal nail plate was discontinuous with the distal nail plate, which was consistent with onychomadesis (Figure, B). The right second toenail was yellow and thickened with layered nail plates, indicative of retronychia (Figure, B). Histopathology of a nail plate clipping from the right second toenail was negative for fungal hyphae, and a radiograph was negative for bony changes or exostosis.
Comment
The nail matrix is responsible for nail plate production, and the newly formed nail plate then moves outward over the nail bed. It is hypothesized that the pathophysiologic basis for Beau lines, onychomadesis, and retronychia lies on a continuum such that all 3 conditions are caused by an insult to the nail matrix that results in slowing and/or halting of nail plate growth. Beau lines result from slowing or disruption in cell growth from the nail matrix, whereas onychomadesis is associated with a complete halt in nail plate production.1,3 In retronychia, the new nail growing from the matrix pushes the old one upward, interrupting the longitudinal growth of the nail and leading to nail plate stacking.10
Our patient presented with concurrent Beau lines, onychomadesis, and retronychia. Although Beau lines and onychomadesis have been reported together in some instances,12-14 retronychia is not commonly reported with either of these conditions. The exact incidence of each condition has not been studied, but Beau lines are relatively common, onychomadesis is less common, and retronychia is seen infrequently; therefore, the concurrent presentation of these 3 conditions in the same patient is exceedingly rare. Thus, it was most likely that one etiology accounted for all 3 nail findings.
Because the patient had been diagnosed with scurvy 6 months prior to presentation, we hypothesized that the associated vitamin C deficiency caused a systemic insult to the nail matrix, which resulted in cessation of nail growth. The mechanism of nail matrix arrest in the setting of systemic disease is thought to be due to inhibition of cellular proliferation or a change in the quality of the newly manufactured nail plate, which becomes thinner and more dystrophic.15 Vitamin C (ascorbic acid) deficiency causes scurvy, which is characterized by cutaneous signs such as perifollicular hemorrhage and purpura, corkscrew hairs, bruising, gingivitis, arthralgia, and impaired wound healing.16 These clinical manifestations are due to impaired collagen synthesis and disordered connective tissue. Ascorbic acid also is involved in fatty acid transport, neurotransmitter synthesis, prostaglandin metabolism, and nitric oxide synthesis.17 Ascorbic acid has not been studied for its role in nail plate synthesis18; however, given the role that ascorbic acid plays in a myriad of biologic processes, the deficiency associated with scurvy likely had a considerable systemic effect in our patient that halted nail plate synthesis and resulted in the concurrent presentation of Beau lines, onychomadesis, and retronychia.
Beau lines are palpable transverse depressions on the dorsal aspect of the nail plate that result from a temporary slowing of nail plate production by the proximal nail matrix. Onychomadesis is a separation of the proximal nail plate from the distal nail plate leading to shedding of the nail. It occurs due to a complete growth arrest in the nail matrix and is thought to be on a continuum with Beau lines. The etiologies of these 2 conditions overlap and include trauma, inflammatory diseases, systemic illnesses, hereditary conditions, and infections.1-5 In almost all cases of both conditions, normal nail plate production ensues upon identification and removal of the inciting agent or recuperation from the causal illness.3,4,6 Beau lines will move distally as the nail grows out and can be clipped. In onychomadesis, the affected nails will be shed with time. Resolution of these nail defects can be estimated from average nail growth rates (1 mm/mo for fingernails and 2–3 mm/mo for toenails).7
Retronychia is defined as a proximal ingrowing of the nail plate into the ventral surface of the proximal nail fold.4,6 It is thought to occur via vertical progression of the nail plate into the proximal nail fold, repetitive nail matrix trauma, or shearing forces, resulting in inflammation that leads to nail plate stacking.8,9 Although conservative treatment using topical corticosteroids may be attempted, proximal nail plate avulsion typically is required for treatment.10
Braswell et al1 suggested a unifying hypothesis for a common pathophysiologic basis for these 3 conditions; that is, nail matrix injury results in slowing and/or cessation of nail plate production, followed by recommencement of nail plate production by the nail matrix after removal of the insult. We report a case of a patient presenting with concurrent Beau lines, onychomadesis, and retronychia following scurvy, thus supporting the hypothesis that these 3 nail conditions lie on a continuum.
Case Report
A 41-year-old woman with a history of thyroiditis, gastroesophageal reflux disease, endometriosis, osteoarthritis, gastric ulcer, pancreatitis, fatty liver, and polycystic ovarian syndrome presented with lines on the toenails and no growth of the right second toenail of several months’ duration. She denied any pain or prior trauma to the nails, participation in sports activities, or wearing tight or high-heeled shoes. She had presented 6 months prior for evaluation of perifollicular erythema on the posterior thighs, legs, and abdomen, as well as gingival bleeding.11 At that time, one of the authors (S.R.L.) found that she was vitamin C deficient, and a diagnosis of scurvy was made. The rash and gingival bleeding resolved with vitamin C supplementation.11
At the current presentation, physical examination revealed transverse grooves involving several fingernails but most evident on the left thumbnail (Figure, A). The grooves did not span the entire breadth of the nail, which was consistent with Beau lines. Several toenails had parallel transverse grooves spanning the entire width of the nail plate such that the proximal nail plate was discontinuous with the distal nail plate, which was consistent with onychomadesis (Figure, B). The right second toenail was yellow and thickened with layered nail plates, indicative of retronychia (Figure, B). Histopathology of a nail plate clipping from the right second toenail was negative for fungal hyphae, and a radiograph was negative for bony changes or exostosis.
Comment
The nail matrix is responsible for nail plate production, and the newly formed nail plate then moves outward over the nail bed. It is hypothesized that the pathophysiologic basis for Beau lines, onychomadesis, and retronychia lies on a continuum such that all 3 conditions are caused by an insult to the nail matrix that results in slowing and/or halting of nail plate growth. Beau lines result from slowing or disruption in cell growth from the nail matrix, whereas onychomadesis is associated with a complete halt in nail plate production.1,3 In retronychia, the new nail growing from the matrix pushes the old one upward, interrupting the longitudinal growth of the nail and leading to nail plate stacking.10
Our patient presented with concurrent Beau lines, onychomadesis, and retronychia. Although Beau lines and onychomadesis have been reported together in some instances,12-14 retronychia is not commonly reported with either of these conditions. The exact incidence of each condition has not been studied, but Beau lines are relatively common, onychomadesis is less common, and retronychia is seen infrequently; therefore, the concurrent presentation of these 3 conditions in the same patient is exceedingly rare. Thus, it was most likely that one etiology accounted for all 3 nail findings.
Because the patient had been diagnosed with scurvy 6 months prior to presentation, we hypothesized that the associated vitamin C deficiency caused a systemic insult to the nail matrix, which resulted in cessation of nail growth. The mechanism of nail matrix arrest in the setting of systemic disease is thought to be due to inhibition of cellular proliferation or a change in the quality of the newly manufactured nail plate, which becomes thinner and more dystrophic.15 Vitamin C (ascorbic acid) deficiency causes scurvy, which is characterized by cutaneous signs such as perifollicular hemorrhage and purpura, corkscrew hairs, bruising, gingivitis, arthralgia, and impaired wound healing.16 These clinical manifestations are due to impaired collagen synthesis and disordered connective tissue. Ascorbic acid also is involved in fatty acid transport, neurotransmitter synthesis, prostaglandin metabolism, and nitric oxide synthesis.17 Ascorbic acid has not been studied for its role in nail plate synthesis18; however, given the role that ascorbic acid plays in a myriad of biologic processes, the deficiency associated with scurvy likely had a considerable systemic effect in our patient that halted nail plate synthesis and resulted in the concurrent presentation of Beau lines, onychomadesis, and retronychia.
- Braswell MA, Daniel CR III, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Lipner SR. Onychomadesis following a fish pedicure. JAMA Dermatol. 2018;154:1091-1092.
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Lawry M, Daniel CR III. Nails in systemic disease. In: Scher RK, Daniel CR III, eds. Nails: Diagnosis, Therapy, Surgery. 3rd ed. Oxford, England: Elsevier Saunders; 2005:147-176.
- Lipner SR, Scher RK. Evaluation of nail lines: color and shape hold clues. Cleve Clin J Med. 2016;83:385.
- Rich P. Nail signs and symptoms. In: Scher RK, Daniel CR III, eds. Nails: Diagnosis, Therapy, Surgery. 3rd ed. Oxford, England: Elsevier Saunders; 2005:1-6.
- Lipner SR, Scher RK. Nail growth evaluation and factors affecting nail growth. In: Humbert P, Fanian F, Maibach H, et al, eds. Agache’s Measuring the Skin. Cham, Switzerland: Springer; 2017:1-15.
- de Berker DA, Richert B, Duhard E, et al. Retronychia: proximal ingrowing of the nail plate. J Am Acad Dermatol. 2008;58:978-983.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Piraccini BM, Richert B, de Berker DA, et al. Retronychia in children, adolescents, and young adults: a case series. J Am Acad Dermatol. 2014;70:388-390.
- Lipner S. A classic case of scurvy. Lancet. 2018;392:431.
- Jacobsen L, Zimmerman S, Lohr J. Nail findings in hand-foot-and-mouth disease. Pediatr Infect Dis J. 2015;34:449-450.
- Damevska K, Gocev G, Pollozhani N, et al. Onychomadesis following cutaneous vasculitis. Acta Dermatovenerol Croat. 2017;25:77-79.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand‐foot‐mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Weismann K. J.H.S Beau and his descriptions of transverse depressions on nails. Br J Dermatol. 1977;97:571-572.
- Abdullah M, Jamil RT, Attia FN. Vitamin C (ascorbic acid). Treasure Island, FL: StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK499877/. Updated October 21, 2019. Accessed February 24, 2020.
- Pazirandeh S, Burns DL. Overview of water-soluble vitamins. UpToDate. https://www.uptodate.com/contents/overview-of-water-soluble-vitamins. Updated January 29, 2020. Accessed February 24, 2020.
- Scheinfeld N, Dahdah MJ, Scher RK. Vitamins and minerals: their role in nail health and disease. J Drugs Dermatol. 2007;6:782-787.
- Braswell MA, Daniel CR III, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Lipner SR. Onychomadesis following a fish pedicure. JAMA Dermatol. 2018;154:1091-1092.
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Lawry M, Daniel CR III. Nails in systemic disease. In: Scher RK, Daniel CR III, eds. Nails: Diagnosis, Therapy, Surgery. 3rd ed. Oxford, England: Elsevier Saunders; 2005:147-176.
- Lipner SR, Scher RK. Evaluation of nail lines: color and shape hold clues. Cleve Clin J Med. 2016;83:385.
- Rich P. Nail signs and symptoms. In: Scher RK, Daniel CR III, eds. Nails: Diagnosis, Therapy, Surgery. 3rd ed. Oxford, England: Elsevier Saunders; 2005:1-6.
- Lipner SR, Scher RK. Nail growth evaluation and factors affecting nail growth. In: Humbert P, Fanian F, Maibach H, et al, eds. Agache’s Measuring the Skin. Cham, Switzerland: Springer; 2017:1-15.
- de Berker DA, Richert B, Duhard E, et al. Retronychia: proximal ingrowing of the nail plate. J Am Acad Dermatol. 2008;58:978-983.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Piraccini BM, Richert B, de Berker DA, et al. Retronychia in children, adolescents, and young adults: a case series. J Am Acad Dermatol. 2014;70:388-390.
- Lipner S. A classic case of scurvy. Lancet. 2018;392:431.
- Jacobsen L, Zimmerman S, Lohr J. Nail findings in hand-foot-and-mouth disease. Pediatr Infect Dis J. 2015;34:449-450.
- Damevska K, Gocev G, Pollozhani N, et al. Onychomadesis following cutaneous vasculitis. Acta Dermatovenerol Croat. 2017;25:77-79.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand‐foot‐mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Weismann K. J.H.S Beau and his descriptions of transverse depressions on nails. Br J Dermatol. 1977;97:571-572.
- Abdullah M, Jamil RT, Attia FN. Vitamin C (ascorbic acid). Treasure Island, FL: StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK499877/. Updated October 21, 2019. Accessed February 24, 2020.
- Pazirandeh S, Burns DL. Overview of water-soluble vitamins. UpToDate. https://www.uptodate.com/contents/overview-of-water-soluble-vitamins. Updated January 29, 2020. Accessed February 24, 2020.
- Scheinfeld N, Dahdah MJ, Scher RK. Vitamins and minerals: their role in nail health and disease. J Drugs Dermatol. 2007;6:782-787.
Practice Points
- Beau lines, onychomadesis, and retronychia are nail conditions with distinct clinical findings.
- Beau lines and onychomadesis may be seen concurrently following trauma, inflammatory diseases, systemic illnesses, hereditary conditions, and infections.
- Retronychia shares a common pathophysiology with Beau lines and onychomadesis, and all reflect slowing or cessation of nail plate production.
Granulomatous Reaction After Cholla Cactus Spine Injury
Skin injuries caused by spines of various species of cactus are common in the southwestern United States and Mexico and have been described worldwide.1 Effects of injury vary depending on localization, surface extension, and skin conditions (eg, preexisting erosions, ulcerations, sunburns).
Case Report
A 22-year-old woman presented to the outpatient department with extremely painful, erythematous papules on the second, third, and fourth fingers of the left hand, as well as diffuse swelling of the entire metacarpophalangeal and interphalangeal joints (Figure 1). She reported accidentally falling on a cholla cactus (genus Cylindropuntia) 2 weeks earlier while walking on a cholla cactus trail during a vacation in California. She reported that the symptoms had worsened over the last week. Class 3 corticosteroid ointments did not provide benefit. The patient had no comorbidities and was allergic to penicillin.

Radiographs of the left hand excluded concomitant fracture. Digital dermoscopy showed multiple white homogeneous areas with a central pustule (Figure 2A). Frequency-domain optical coherence tomography (OCT) displayed round hyperrefractive structures in the dermis suggestive of granulomas, as well as a small needlelike hyperrefractive structure, a foreign body (Figure 2B).
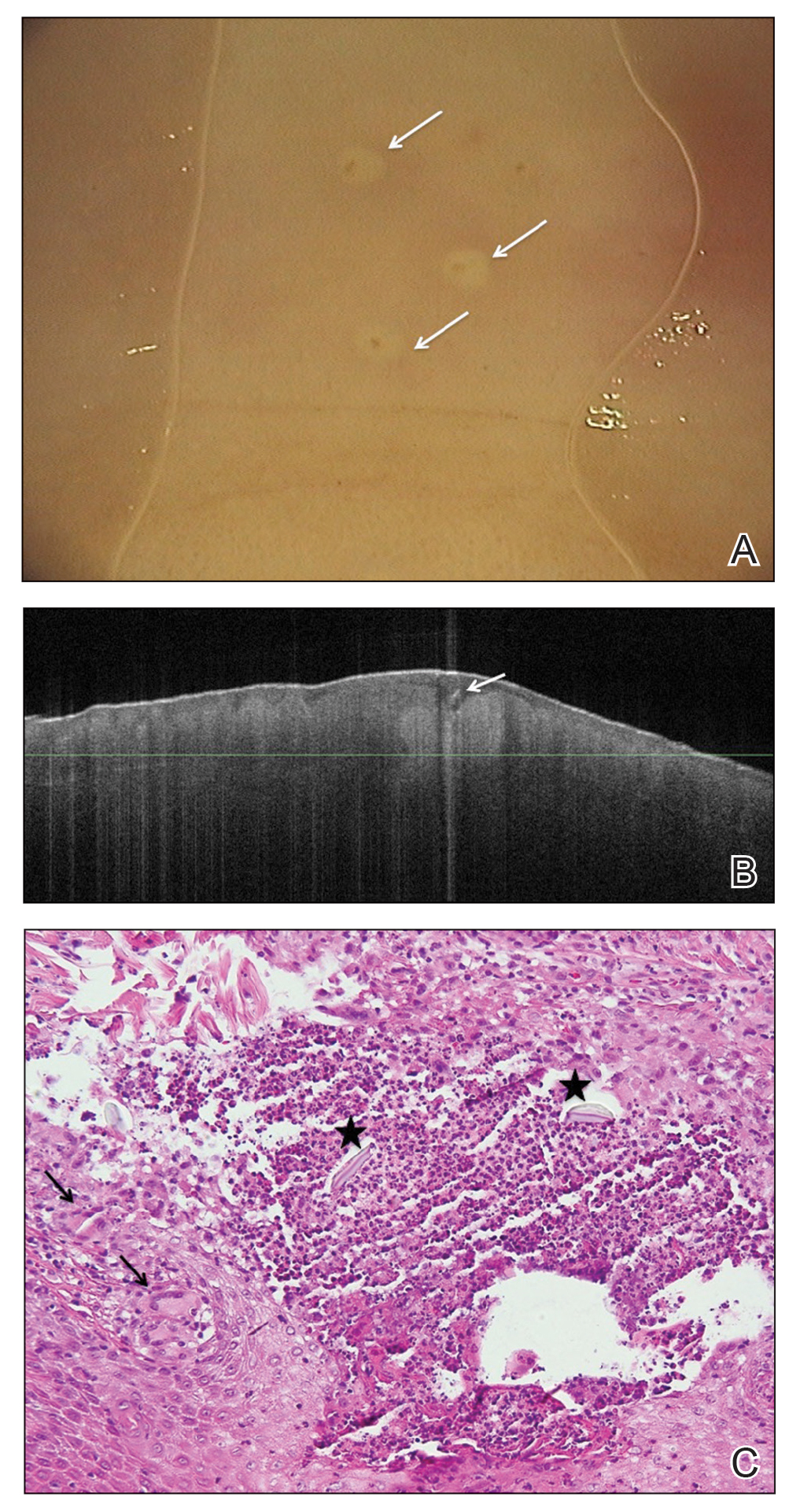
The few visible spines were immediately removed with tweezers; the patient remained symptom free for approximately 2 weeks. Subsequently, extreme pain developed in the left hand; the clinical presentation and pain did not respond to empiric intravenous antibiotic therapy with weight-calculated clarithromycin (500 mg twice daily), systemic analgesia with nonsteroidal anti-inflammatory drugs, and local therapy with antiseptics and class 3 corticosteroid ointment. Four days later, all 27 papules were excised with 3- and 4-mm punch biopsies using digital nerve blocks. Histology showed classic foreign body granulomas with hematoxylin and eosin stain (Figure 2C).
One week later, pain, erythema, and swelling had disappeared; no additional lesions had developed (Figure 3). Follow-up OCT showed no foreign bodies. At 4-week follow-up, the inflammatory component had disappeared, and no granulomas were evident. Six months later, the lesions healed with minimal scarring that could later be treated with fractional laser therapy (Figure 4).
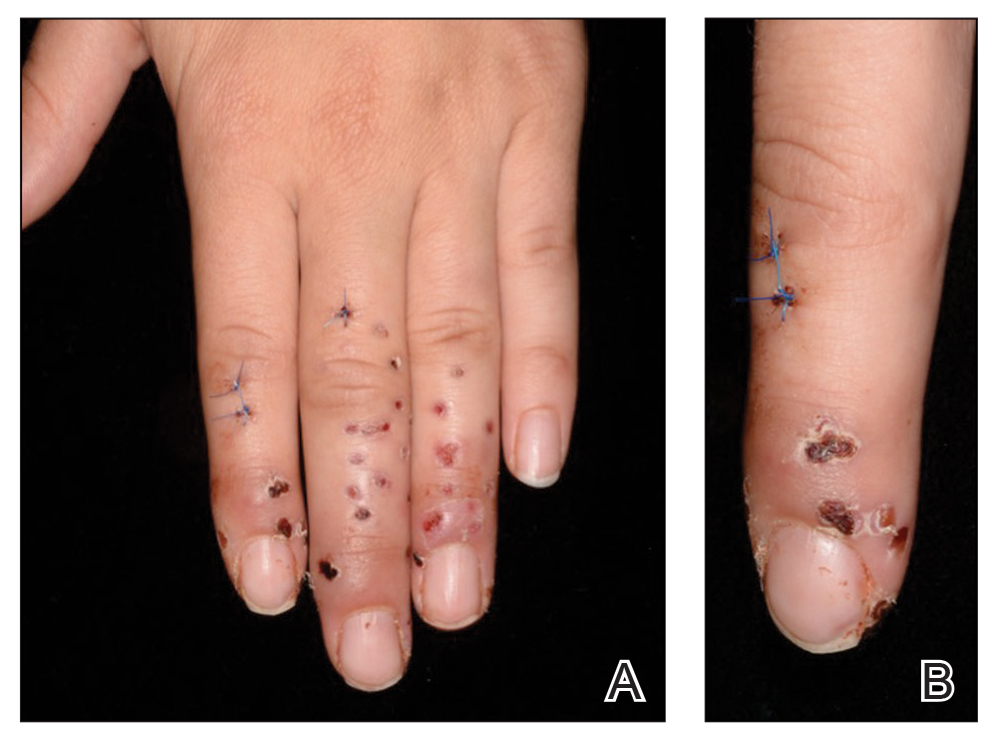
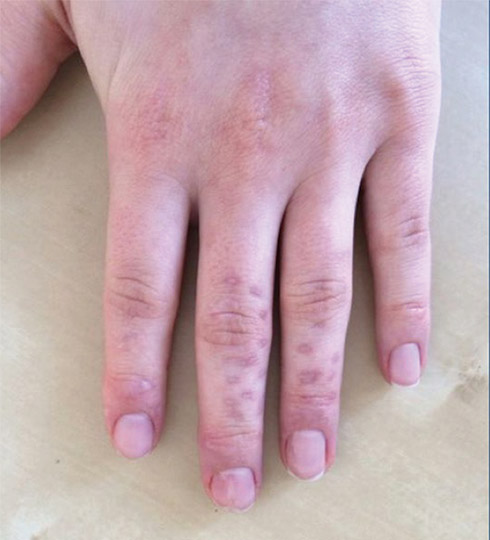
Comment
Pathogenesis and Presentation
Cactus spines are included in the possible causes of foreign body granulomas of the skin (eTable).2,3 However, granulomatous inflammation after cactus spine injury rarely has been described in the medical literature. In the first known case report in 1955, Winer and Zeilenga4 described a woman who developed multiple hand granulomas that were partially removed by curettage, while the spines underwent slow spontaneous expulsion.
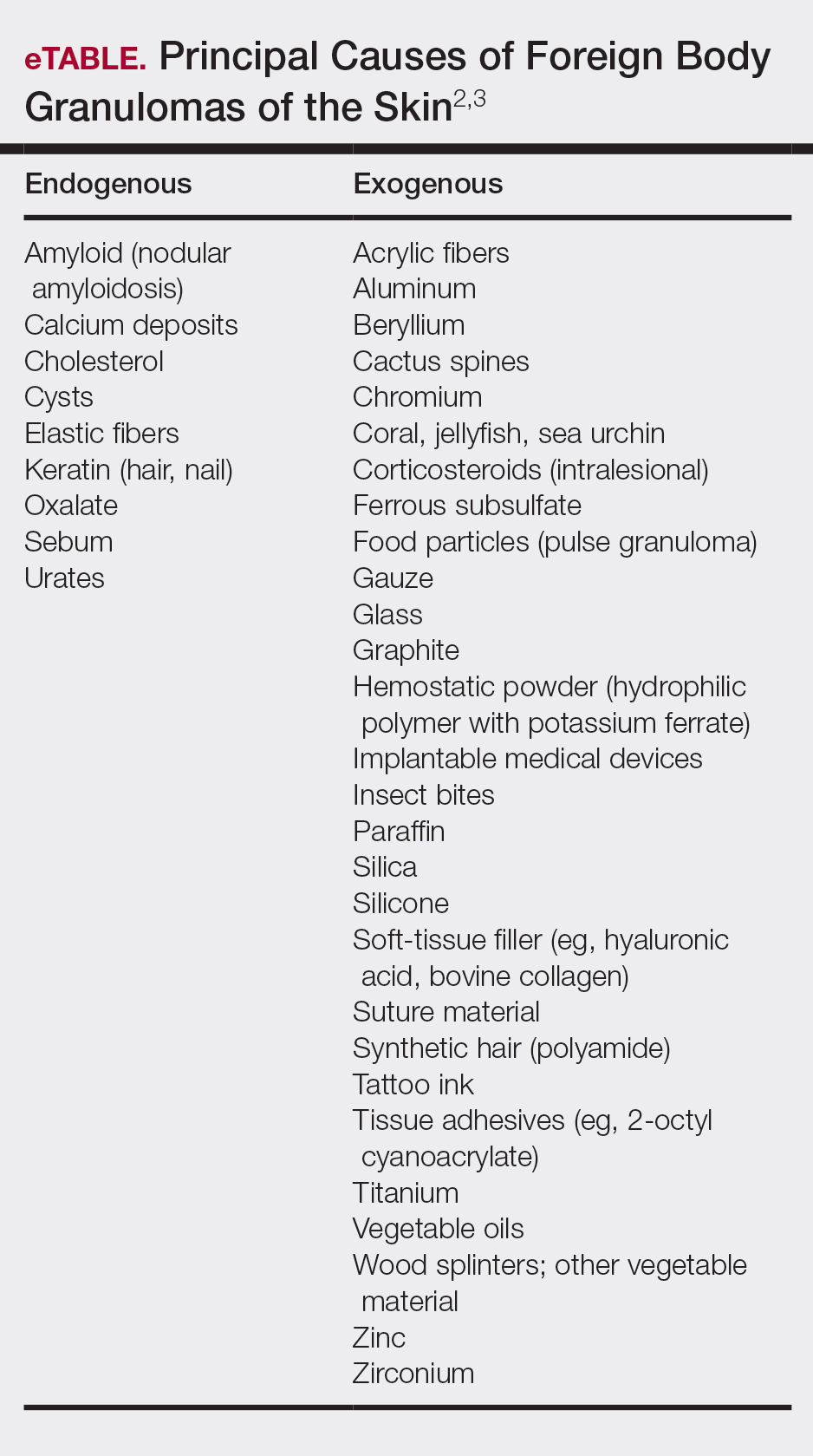
In 1971, Schreiber et al5 hypothesized a type 2 allergic response to cactus spines based on the variability of reactions in different cases. Doctoroff et al6 proposed an unroofing technique based on the removal of spines under microscopy, which brought faster (2–4 months) healing. Madkan et al7 reported that complete response is possible only with punch excision of the largest lesions.
The cholla (Cylindropuntia) cactus has been described as the species most commonly implicated in granulomatous reactions to cactus spines.8,9 Two principal pathogenic mechanisms have been described—foreign body granuloma and allergic reaction to cactus antigens—because not every patient develops granulomatous lesions.
Sequelae
Complications of injury from cactus spines are common, especially when spines are not completely removed, including local inflammation, superinfection, necrosis, allergic reactions, granulomas, scarring, and chronic pain. Rare consequences of cactus injury include bacterial infection with Staphylococcus aureus; Enterobacter species; atypical mycobacteria, including Mycobacterium marinum; Nocardia species; and Clostridium tetani, as well as deep fungal infection, especially in immunocompromised patients.10 In our case, bacterial culture and polymerase chain reaction testing for mycobacteria were negative.
Diagnosis
Cactus spine injuries usually are easy to diagnose based on the clear-cut anamnesis and clinical picture; however, it might be interesting to assess the presence of foreign body granulomas without biopsy. Optical coherence tomography is a noninvasive optical imaging technique based on low-coherence interferometry that uses a low-intensity, 1310-nm infrared laser. Widespread in ophthalmology, OCT has gained importance in dermatologic diagnostics, especially for nonmelanoma skin cancer.11 Moreover, it has demonstrated its usefulness in various dermatologic fields, including granulomatous lesions.12 Further methods include reflectance confocal microscopy, based on a near-infrared laser, and 7.5-MHz ultrasonography. In our experience, however, 7.5-MHz ultrasonography has been ineffective in detecting cactus spines in the current patient as well as others. Preoperative and postoperative monitoring with dermoscopy and OCT helped us evaluate the nature, size, and location of spines and lesions and effective healing.
Treatment
Management strategies are still debated and include watchful waiting, corticosteroid ointment, partial removal of spines, and unroofing.1,2,4-10,13-18 We treated our patient with an innovative radical surgical approach using punch excision for granulomas that developed after cholla cactus spine injury. Our approach resulted in rapid relief of pain and reduced complications, a good aesthetic result, and no recurrence.
- Lindsey D, Lindsey WE. Cactus spine injuries. Am J Emerg Med. 1988;6:362-369.
Molina-Ruiz AM, Requena L. Foreign body granulomas. Dermatol Clin. 2015;33:497-523. - Patterson JW. Weedon’s Skin Pathology. 4th ed. New York, NY: Elsevier; 2016.
- Winer LH, Zeilenga RH. Cactus granulomas of the skin; report of a case. AMA Arch Derm. 1955;72:566-569.
- Schreiber MM, Shapiro SI, Berry CZ. Cactus granulomas of the skin. an allergic phenomenon. Arch Dermatol. 1971;104:374-379.
- Doctoroff A, Vidimos AT, Taylor JS. Cactus skin injuries. Cutis. 2000;65:290-292.
- Madkan VK, Abraham T, Lesher JL Jr. Cactus spine granuloma. Cutis. 2007;79:208-210.
- Spoerke DG, Spoerke SE. Granuloma formation induced by spines of the cactus, Opuntia acanthocarpa. Vet Hum Toxicol. 1991;33:342-344.
- Suzuki H, Baba S. Cactus granuloma of the skin. J Dermatol. 1993;20:424-427.
- Burrell SR, Ostlie DJ, Saubolle M, et al. Apophysomyces elegans infection associated with cactus spine injury in an immunocompetent pediatric patient. Pediatr Infect Dis J. 1998;17:663-664.
- von BraunmT. Optical coherence tomography. Hautarzt. 2015;66:499-503.
- Banzhaf C, Jemec GB. Imaging granulomatous lesions with optical coherence tomography. Case Rep Dermatol. 2012;4:14-18.
- Putnam MH. Simple cactus spine removal. J Pediatr. 1981;98:333.
- Snyder RA, Schwartz RA. Cactus bristle implantation. Report of an unusual case initially seen with rows of yellow hairs. Arch Dermatol. 1983;119:152-154.
- Schunk JE, Corneli HM. Cactus spine removal. J Pediatr. 1987;110:667.
- Gutierrez Ortega MC, Martin Moreno L, Arias Palomo D, et al. Facial granuloma caused by cactus bristles. Med Cutan Ibero Lat Am. 1990;18:197-200.
- Dieter RA Jr, Whitehouse LR, Gulliver R. Cactus spine wounds: a case report and short review of the literature. Wounds. 2017;29:E18-E21.
- O’Neill PJ, Sinha M, McArthur RA, et al. Penetrating cactus spine injury to the mediastinum of a child. J Pediatr Surg. 2008;43:E33-E35.
Skin injuries caused by spines of various species of cactus are common in the southwestern United States and Mexico and have been described worldwide.1 Effects of injury vary depending on localization, surface extension, and skin conditions (eg, preexisting erosions, ulcerations, sunburns).
Case Report
A 22-year-old woman presented to the outpatient department with extremely painful, erythematous papules on the second, third, and fourth fingers of the left hand, as well as diffuse swelling of the entire metacarpophalangeal and interphalangeal joints (Figure 1). She reported accidentally falling on a cholla cactus (genus Cylindropuntia) 2 weeks earlier while walking on a cholla cactus trail during a vacation in California. She reported that the symptoms had worsened over the last week. Class 3 corticosteroid ointments did not provide benefit. The patient had no comorbidities and was allergic to penicillin.
Radiographs of the left hand excluded concomitant fracture. Digital dermoscopy showed multiple white homogeneous areas with a central pustule (Figure 2A). Frequency-domain optical coherence tomography (OCT) displayed round hyperrefractive structures in the dermis suggestive of granulomas, as well as a small needlelike hyperrefractive structure, a foreign body (Figure 2B).
The few visible spines were immediately removed with tweezers; the patient remained symptom free for approximately 2 weeks. Subsequently, extreme pain developed in the left hand; the clinical presentation and pain did not respond to empiric intravenous antibiotic therapy with weight-calculated clarithromycin (500 mg twice daily), systemic analgesia with nonsteroidal anti-inflammatory drugs, and local therapy with antiseptics and class 3 corticosteroid ointment. Four days later, all 27 papules were excised with 3- and 4-mm punch biopsies using digital nerve blocks. Histology showed classic foreign body granulomas with hematoxylin and eosin stain (Figure 2C).
One week later, pain, erythema, and swelling had disappeared; no additional lesions had developed (Figure 3). Follow-up OCT showed no foreign bodies. At 4-week follow-up, the inflammatory component had disappeared, and no granulomas were evident. Six months later, the lesions healed with minimal scarring that could later be treated with fractional laser therapy (Figure 4).
Comment
Pathogenesis and Presentation
Cactus spines are included in the possible causes of foreign body granulomas of the skin (eTable).2,3 However, granulomatous inflammation after cactus spine injury rarely has been described in the medical literature. In the first known case report in 1955, Winer and Zeilenga4 described a woman who developed multiple hand granulomas that were partially removed by curettage, while the spines underwent slow spontaneous expulsion.
In 1971, Schreiber et al5 hypothesized a type 2 allergic response to cactus spines based on the variability of reactions in different cases. Doctoroff et al6 proposed an unroofing technique based on the removal of spines under microscopy, which brought faster (2–4 months) healing. Madkan et al7 reported that complete response is possible only with punch excision of the largest lesions.
The cholla (Cylindropuntia) cactus has been described as the species most commonly implicated in granulomatous reactions to cactus spines.8,9 Two principal pathogenic mechanisms have been described—foreign body granuloma and allergic reaction to cactus antigens—because not every patient develops granulomatous lesions.
Sequelae
Complications of injury from cactus spines are common, especially when spines are not completely removed, including local inflammation, superinfection, necrosis, allergic reactions, granulomas, scarring, and chronic pain. Rare consequences of cactus injury include bacterial infection with Staphylococcus aureus; Enterobacter species; atypical mycobacteria, including Mycobacterium marinum; Nocardia species; and Clostridium tetani, as well as deep fungal infection, especially in immunocompromised patients.10 In our case, bacterial culture and polymerase chain reaction testing for mycobacteria were negative.
Diagnosis
Cactus spine injuries usually are easy to diagnose based on the clear-cut anamnesis and clinical picture; however, it might be interesting to assess the presence of foreign body granulomas without biopsy. Optical coherence tomography is a noninvasive optical imaging technique based on low-coherence interferometry that uses a low-intensity, 1310-nm infrared laser. Widespread in ophthalmology, OCT has gained importance in dermatologic diagnostics, especially for nonmelanoma skin cancer.11 Moreover, it has demonstrated its usefulness in various dermatologic fields, including granulomatous lesions.12 Further methods include reflectance confocal microscopy, based on a near-infrared laser, and 7.5-MHz ultrasonography. In our experience, however, 7.5-MHz ultrasonography has been ineffective in detecting cactus spines in the current patient as well as others. Preoperative and postoperative monitoring with dermoscopy and OCT helped us evaluate the nature, size, and location of spines and lesions and effective healing.
Treatment
Management strategies are still debated and include watchful waiting, corticosteroid ointment, partial removal of spines, and unroofing.1,2,4-10,13-18 We treated our patient with an innovative radical surgical approach using punch excision for granulomas that developed after cholla cactus spine injury. Our approach resulted in rapid relief of pain and reduced complications, a good aesthetic result, and no recurrence.
Skin injuries caused by spines of various species of cactus are common in the southwestern United States and Mexico and have been described worldwide.1 Effects of injury vary depending on localization, surface extension, and skin conditions (eg, preexisting erosions, ulcerations, sunburns).
Case Report
A 22-year-old woman presented to the outpatient department with extremely painful, erythematous papules on the second, third, and fourth fingers of the left hand, as well as diffuse swelling of the entire metacarpophalangeal and interphalangeal joints (Figure 1). She reported accidentally falling on a cholla cactus (genus Cylindropuntia) 2 weeks earlier while walking on a cholla cactus trail during a vacation in California. She reported that the symptoms had worsened over the last week. Class 3 corticosteroid ointments did not provide benefit. The patient had no comorbidities and was allergic to penicillin.
Radiographs of the left hand excluded concomitant fracture. Digital dermoscopy showed multiple white homogeneous areas with a central pustule (Figure 2A). Frequency-domain optical coherence tomography (OCT) displayed round hyperrefractive structures in the dermis suggestive of granulomas, as well as a small needlelike hyperrefractive structure, a foreign body (Figure 2B).
The few visible spines were immediately removed with tweezers; the patient remained symptom free for approximately 2 weeks. Subsequently, extreme pain developed in the left hand; the clinical presentation and pain did not respond to empiric intravenous antibiotic therapy with weight-calculated clarithromycin (500 mg twice daily), systemic analgesia with nonsteroidal anti-inflammatory drugs, and local therapy with antiseptics and class 3 corticosteroid ointment. Four days later, all 27 papules were excised with 3- and 4-mm punch biopsies using digital nerve blocks. Histology showed classic foreign body granulomas with hematoxylin and eosin stain (Figure 2C).
One week later, pain, erythema, and swelling had disappeared; no additional lesions had developed (Figure 3). Follow-up OCT showed no foreign bodies. At 4-week follow-up, the inflammatory component had disappeared, and no granulomas were evident. Six months later, the lesions healed with minimal scarring that could later be treated with fractional laser therapy (Figure 4).
Comment
Pathogenesis and Presentation
Cactus spines are included in the possible causes of foreign body granulomas of the skin (eTable).2,3 However, granulomatous inflammation after cactus spine injury rarely has been described in the medical literature. In the first known case report in 1955, Winer and Zeilenga4 described a woman who developed multiple hand granulomas that were partially removed by curettage, while the spines underwent slow spontaneous expulsion.
In 1971, Schreiber et al5 hypothesized a type 2 allergic response to cactus spines based on the variability of reactions in different cases. Doctoroff et al6 proposed an unroofing technique based on the removal of spines under microscopy, which brought faster (2–4 months) healing. Madkan et al7 reported that complete response is possible only with punch excision of the largest lesions.
The cholla (Cylindropuntia) cactus has been described as the species most commonly implicated in granulomatous reactions to cactus spines.8,9 Two principal pathogenic mechanisms have been described—foreign body granuloma and allergic reaction to cactus antigens—because not every patient develops granulomatous lesions.
Sequelae
Complications of injury from cactus spines are common, especially when spines are not completely removed, including local inflammation, superinfection, necrosis, allergic reactions, granulomas, scarring, and chronic pain. Rare consequences of cactus injury include bacterial infection with Staphylococcus aureus; Enterobacter species; atypical mycobacteria, including Mycobacterium marinum; Nocardia species; and Clostridium tetani, as well as deep fungal infection, especially in immunocompromised patients.10 In our case, bacterial culture and polymerase chain reaction testing for mycobacteria were negative.
Diagnosis
Cactus spine injuries usually are easy to diagnose based on the clear-cut anamnesis and clinical picture; however, it might be interesting to assess the presence of foreign body granulomas without biopsy. Optical coherence tomography is a noninvasive optical imaging technique based on low-coherence interferometry that uses a low-intensity, 1310-nm infrared laser. Widespread in ophthalmology, OCT has gained importance in dermatologic diagnostics, especially for nonmelanoma skin cancer.11 Moreover, it has demonstrated its usefulness in various dermatologic fields, including granulomatous lesions.12 Further methods include reflectance confocal microscopy, based on a near-infrared laser, and 7.5-MHz ultrasonography. In our experience, however, 7.5-MHz ultrasonography has been ineffective in detecting cactus spines in the current patient as well as others. Preoperative and postoperative monitoring with dermoscopy and OCT helped us evaluate the nature, size, and location of spines and lesions and effective healing.
Treatment
Management strategies are still debated and include watchful waiting, corticosteroid ointment, partial removal of spines, and unroofing.1,2,4-10,13-18 We treated our patient with an innovative radical surgical approach using punch excision for granulomas that developed after cholla cactus spine injury. Our approach resulted in rapid relief of pain and reduced complications, a good aesthetic result, and no recurrence.
- Lindsey D, Lindsey WE. Cactus spine injuries. Am J Emerg Med. 1988;6:362-369.
Molina-Ruiz AM, Requena L. Foreign body granulomas. Dermatol Clin. 2015;33:497-523. - Patterson JW. Weedon’s Skin Pathology. 4th ed. New York, NY: Elsevier; 2016.
- Winer LH, Zeilenga RH. Cactus granulomas of the skin; report of a case. AMA Arch Derm. 1955;72:566-569.
- Schreiber MM, Shapiro SI, Berry CZ. Cactus granulomas of the skin. an allergic phenomenon. Arch Dermatol. 1971;104:374-379.
- Doctoroff A, Vidimos AT, Taylor JS. Cactus skin injuries. Cutis. 2000;65:290-292.
- Madkan VK, Abraham T, Lesher JL Jr. Cactus spine granuloma. Cutis. 2007;79:208-210.
- Spoerke DG, Spoerke SE. Granuloma formation induced by spines of the cactus, Opuntia acanthocarpa. Vet Hum Toxicol. 1991;33:342-344.
- Suzuki H, Baba S. Cactus granuloma of the skin. J Dermatol. 1993;20:424-427.
- Burrell SR, Ostlie DJ, Saubolle M, et al. Apophysomyces elegans infection associated with cactus spine injury in an immunocompetent pediatric patient. Pediatr Infect Dis J. 1998;17:663-664.
- von BraunmT. Optical coherence tomography. Hautarzt. 2015;66:499-503.
- Banzhaf C, Jemec GB. Imaging granulomatous lesions with optical coherence tomography. Case Rep Dermatol. 2012;4:14-18.
- Putnam MH. Simple cactus spine removal. J Pediatr. 1981;98:333.
- Snyder RA, Schwartz RA. Cactus bristle implantation. Report of an unusual case initially seen with rows of yellow hairs. Arch Dermatol. 1983;119:152-154.
- Schunk JE, Corneli HM. Cactus spine removal. J Pediatr. 1987;110:667.
- Gutierrez Ortega MC, Martin Moreno L, Arias Palomo D, et al. Facial granuloma caused by cactus bristles. Med Cutan Ibero Lat Am. 1990;18:197-200.
- Dieter RA Jr, Whitehouse LR, Gulliver R. Cactus spine wounds: a case report and short review of the literature. Wounds. 2017;29:E18-E21.
- O’Neill PJ, Sinha M, McArthur RA, et al. Penetrating cactus spine injury to the mediastinum of a child. J Pediatr Surg. 2008;43:E33-E35.
- Lindsey D, Lindsey WE. Cactus spine injuries. Am J Emerg Med. 1988;6:362-369.
Molina-Ruiz AM, Requena L. Foreign body granulomas. Dermatol Clin. 2015;33:497-523. - Patterson JW. Weedon’s Skin Pathology. 4th ed. New York, NY: Elsevier; 2016.
- Winer LH, Zeilenga RH. Cactus granulomas of the skin; report of a case. AMA Arch Derm. 1955;72:566-569.
- Schreiber MM, Shapiro SI, Berry CZ. Cactus granulomas of the skin. an allergic phenomenon. Arch Dermatol. 1971;104:374-379.
- Doctoroff A, Vidimos AT, Taylor JS. Cactus skin injuries. Cutis. 2000;65:290-292.
- Madkan VK, Abraham T, Lesher JL Jr. Cactus spine granuloma. Cutis. 2007;79:208-210.
- Spoerke DG, Spoerke SE. Granuloma formation induced by spines of the cactus, Opuntia acanthocarpa. Vet Hum Toxicol. 1991;33:342-344.
- Suzuki H, Baba S. Cactus granuloma of the skin. J Dermatol. 1993;20:424-427.
- Burrell SR, Ostlie DJ, Saubolle M, et al. Apophysomyces elegans infection associated with cactus spine injury in an immunocompetent pediatric patient. Pediatr Infect Dis J. 1998;17:663-664.
- von BraunmT. Optical coherence tomography. Hautarzt. 2015;66:499-503.
- Banzhaf C, Jemec GB. Imaging granulomatous lesions with optical coherence tomography. Case Rep Dermatol. 2012;4:14-18.
- Putnam MH. Simple cactus spine removal. J Pediatr. 1981;98:333.
- Snyder RA, Schwartz RA. Cactus bristle implantation. Report of an unusual case initially seen with rows of yellow hairs. Arch Dermatol. 1983;119:152-154.
- Schunk JE, Corneli HM. Cactus spine removal. J Pediatr. 1987;110:667.
- Gutierrez Ortega MC, Martin Moreno L, Arias Palomo D, et al. Facial granuloma caused by cactus bristles. Med Cutan Ibero Lat Am. 1990;18:197-200.
- Dieter RA Jr, Whitehouse LR, Gulliver R. Cactus spine wounds: a case report and short review of the literature. Wounds. 2017;29:E18-E21.
- O’Neill PJ, Sinha M, McArthur RA, et al. Penetrating cactus spine injury to the mediastinum of a child. J Pediatr Surg. 2008;43:E33-E35.
Practice Points
- Cactus spine injuries are an important source of morbidity in sports and leisure.
- Even after removal of cactus spines, painful granulomas can develop and persist for a long period of time. Patient education on early treatment can prevent further complications.
- Immediate and complete removal of spines as well as avoidance of bacterial superinfections should be given priority in cactus spine injuries. In case of granulomas, a surgical approach can result in rapid relief of symptoms.

5-year-old boy • behavioral issues • elevated ALT and AST levels • Dx?
THE CASE
A 5-year-old boy was brought into his primary care clinic by his mother, who expressed concern about her son’s increasing impulsiveness, aggression, and difficulty staying on task at preschool and at home. The child’s medical history was unremarkable, and he was taking no medications. The family history was negative for hepatic or metabolic disease and positive for attention deficit-hyperactivity disorder (ADHD; father).
The child’s growth was normal. His physical exam was remarkable for a liver edge 1 cm below his costal margin. No Kayser-Fleischer rings were present.
Screening included a complete metabolic panel. Notable results included an alanine aminotransferase (ALT) level of 208 U/dL (normal range, < 30 U/dL), an aspartate transaminase (AST) level of 125 U/dL (normal range, 10-34 U/dL), and an alkaline phosphatase (ALP) of 470 U/dL (normal range, 93-309 U/dL). Subsequent repeat laboratory testing confirmed these elevations (ALT, 248 U/dL; AST, 137 U/dL; ALP, 462 U/dL). Ceruloplasmin levels were low (11 mg/dL; normal range, 18-35 mg/dL), and 24-hour urinary copper was not obtainable. Prothrombin/partial thromboplastin time, ammonia, lactate, total and direct bilirubin, and gamma-glutamyltransferase levels were normal.
Further evaluation included abdominal ultrasound and brain magnetic resonance imaging, both of which yielded normal results. Testing for Epstein-Barr virus; cytomegalovirus; hepatitis A, B, and C titers; and antinuclear, anti-smooth muscle, and anti–liver-kidney microsomal antibodies was negative.
THE DIAGNOSIS
The patient’s low ceruloplasmin prompted referral to Pediatric Gastroenterology for consultation and liver biopsy due to concern for Wilson disease. Biopsy results were consistent with, and quantitative liver copper confirmatory for, this diagnosis (FIGURE).

Genetic testing for mutations in the ATP7B gene was performed on the patient, his mother, and his siblings (his father was unavailable). The patient, his mother, and his sister were all positive for His1069Gln mutation; only the patient was positive for a 3990_3993 del mutation (his half-brother was negative for both mutations). The presence of 2 different mutant alleles for the ATP7B gene, one on each chromosome—the common substitution mutation, His1069Gln, in exon 14 and a 3990_3993 del TTAT mutation in exon 19—qualified the patient as a compound heterozygote.
The 3990_3993 del TTAT mutation—which to our knowledge has not been previously reported—produced a translational frame shift and premature stop codon. As others have pointed out, frame shift and missense mutations produce a more severe phenotype.1
Continue to: Further testing was prompted...
Further testing was prompted by a report suggesting that codon 129 mutations of the human prion gene (HPG) influence Wilson disease.2 Compared with patients who are heterozygous (M129V) or homozygous (V129V) for valine, those who are homozygous for methionine (M129M) have delayed symptom onset.2 Our patient was heterozygous (M129V). It is interesting to speculate that HPG heterozygosity, combined with a mutation causing a stop codon, predisposed our patient to more rapid accumulation of copper and earlier age of onset.
DISCUSSION
Wilson disease is an inherited disorder of copper metabolism.3 An inherent difficulty in its recognition, diagnosis, and management is its rarity: global prevalence is estimated as 1/30,000, although this varies by geographic location.1 In contrast, ADHD has a prevalence of 7.2%,4 making it 2400 times more prevalent than Wilson disease. Furthermore, abnormal liver function tests are common in children; the differential diagnosis includes etiologies such as infection (both viral and nonviral), immune-mediated inflammatory disease, drug toxicity (iatrogenic or medication-induced), anatomic abnormalities, and nonalcoholic fatty liver disease.5
Wilson disease is remarkable, however, for being easily treatable if detected and devastating if not. Although liver abnormalities often improve with treatment, delayed diagnosis and management significantly impact neurologic recovery: An 18-month delay results in 38% of patients continuing to have major neurologic disabilities.6 Untreated, Wilson disease may be fatal within 5 years of development of neurologic symptoms.7 Thus, it has been suggested that evaluation for Wilson disease be considered in any child older than 1 year who presents with unexplained liver disease, including asymptomatic elevations of serum transaminases.8
Mutations in ATP7B on chromosome 13 are responsible for the pathology of Wilson disease9; more than 250 mutations have been identified, including substitutions, deletions, and missense mutations.10 Affected patients may be compound heterozygotes11 and/or may possess new mutations, as seen in our patient.
Although copper absorption is normal, impaired excretion causes toxic accumulation in affected organs. ATP7B’s product, ATPase 2, regulates copper excretion, as well as copper binding to apoceruloplasmin to form the carrier protein ceruloplasmin. An ATP7B abnormality would prevent the latter—making ceruloplasmin a useful screening biomarker and a reliable marker for Wilson disease by age 1 year.8
Continue to: Hepatic and neurocognitive effects
Hepatic and neurocognitive effects. Excess copper in hepatocytes causes oxidative damage and release of copper into the circulation, with accumulation in susceptible organs (eg, brain, kidneys). Hepatocyte apoptosis is accelerated by copper’s negative effect on inhibitor of apoptosis protein.12,13 Renal tubular damage leads to Fanconi syndrome,14 in which substances such as glucose, phosphates, and potassium are excreted in urine rather than absorbed into the bloodstream by the kidneys. Excess copper deposition in the Descemet membrane may lead to Kayser-Fleisher ring formation.15 In the brain, copper deposition may occur in the lenticular nuclei,3 as well as in the thalamus, subthalamus, brainstem, and frontal cortex—resulting in extrapyramidal, cerebral, and mild cerebellar symptoms.6
Cognitive impairment, which may be subtle, includes increased impulsivity, impaired judgment, apathy, poor decision making, decreased attention, increased lability, slowed thinking, and memory loss.6 Behavioral manifestations include changes in school or work performance and outbursts mimicking ADHD12,16,17 as well as paranoia, depression, and bizarre behaviors.16,18 Neuropsychiatric abnormalities include personality changes, pseudoparkinsonism, dyskinesia/dysarthria, and ataxia/tremor. Younger patients with psychiatric symptoms may be labelled with depression, anxiety, obsessive-compulsive disorder, bipolar disorder, or antisocial disorder.6,16,18
Hepatic disease manifestations range from asymptomatic elevations in AST/ALT to acute hepatitis, mimicking infectious processes. Cirrhosis is the end result of untreated Wilson disease, with liver transplantation required if end-stage liver disease results. Rarely, patients present in fulminant hepatic failure, with death occurring if emergent liver transplantation is not performed.6,8,10
Of note, before age 10, > 80% of patients with Wilson disease present with hepatic symptoms; those ages 10 to 18 often manifest psychiatric changes.17 Kayser-Fleisher rings are common in patients with neurologic manifestations but less so in those who have hepatic presentations or are presymptomatic.6,15
Effective disease-mitigating treatment is indicated and available for both symptomatic and asymptomatic individuals and includes the copper chelators D-penicillamine (starting dose, 150-300 mg/d with a gradual weekly increase to 20 mg/kg/d) and trientine hydrochloride (a heavy metal chelating compound; starting dose, 20 mg/kg/d to a maximum of 1000 mg/d in young adults). Adverse effects of D-penicillamine include cutaneous eruptions, neutropenia, thrombocytopenia, proteinuria, and a lupus-like syndrome; therefore, trientine is increasingly being used as first-line therapy.8
Continue to: For asymptomatic...
For asymptomatic patients who have had effective chelation therapy and proven de-coppering, zinc salts are a useful follow-on therapy. Zinc’s proposed mechanism of action is induction of metallothionein in enterocytes, which promotes copper trapping and eventual excretion into the lumen. Importantly, treatment for Wilson disease is lifelong and monitoring of compliance is essential.8
Our 5-year-old patient was started on oral trientine at 20 mg/kg/d and a low copper diet. In response to this initial treatment, the patient’s liver function tests (LFTs) normalized, and he was switched to 25 mg tid of a zinc chelate, with continuation of the low copper diet. His LFTs have remained normal, although his urine copper levels are still elevated. He continues to be monitored periodically with LFTs and measurement of urine copper levels. He is also being treated for ADHD, as his presenting behavioral abnormalities suggestive of ADHD have not resolved.
THE TAKEAWAY
Although children presenting with symptoms consistent with ADHD often have ADHD, as was true in this case, it is important to consider other diagnoses. Unexplained elevations of liver function test values in children older than 1 year should prompt screening for Wilson disease.5,8 Additionally, other family members should be evaluated; if they have the disease, treatment should be started by age 2 years, even if the patient is asymptomatic.
In our patient’s case, routine screening saved the day. The complete metabolic panel revealed elevated ALT and AST levels, prompting further evaluation. Without this testing, his diagnosis likely would have been delayed, leading to progressive liver and central nervous system disease. With early identification and treatment, it is possible to stop the progression of Wilson disease.
CORRESPONDENCE
Jeffrey Taylor, MD, MS, Evangelical Community Hospital, Department of Pediatrics, 1 Hospital Drive, Lewisburg PA, 17837; [email protected].
1. Wu F, Wang J, Pu C, et al. Wilson’s disease: a comprehensive review of the molecular mechanisms. Int J Mol Sci. 2015;16:6419-6431.
2. Merle U, Stremmel W, Gessner R. Influence of homozygosity for methionine at codon 129 of the human prion gene on the onset of neurological and hepatic symptoms in Wilson disease. Arch Neurol. 2006;63:982-985.
3. Compston A. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver, by S. A. Kinnier Wilson, (From the National Hospital, and the Laboratory of the National Hospital, Queen Square, London) Brain 1912: 34; 295-509. Brain. 2009;132(pt 8):1997-2001.
4. Thomas R, Sanders S, Doust J, et al. Prevalence of attention-deficit/hyperactivity disorder: a systematic review and meta-analysis. Pediatrics. 2015;135:e994-e1001.
5. Kang K. Abnormality on liver function test. Pediatr Gastroenterol Hepatol Nutr. 2013;16:225-232.
6. Lorincz M. Neurologic Wilson’s disease. Ann NY Acad Sci. 2010;1184:173-187.
7. Dening TR, Berrios GE, Walshe JM. Wilson’s disease and epilepsy. Brain. 1988;111(pt 5):1139-1155.
8. Socha P, Janczyk W, Dhawan A, et al. Wilson’s disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutrit. 2018;66:334-344.
9. Bull PC, Thomas GR, Rommens JM, et al. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327-337.
10. Ala A, Schilsky ML. Wilson disease: pathophysiology, diagnosis, treatment and screening. Clin Liver Dis. 2004;8:787-805, viii.
11. Thomas GR, Forbes JR, Roberts EA, et al. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet. 1995;9:210-217.
12. Pfeiffer RF. Wilson’s disease. Semin Neurol. 2007;27:123-132.
13. Das SK, Ray K. Wilson’s disease: an update. Nat Clin Pract Neurol. 2006;2:482-493.
14. Morgan HG, Stewart WK, Lowe KG, et al. Wilson’s disease and the Fanconi syndrome. Q J Med. 1962;31:361-384.
15. Wiebers DO, Hollenhorst RW, Goldstein NP. The ophthalmologic manifestations of Wilson’s disease. Mayo Clin Proc. 1977;52:409-416.
16. Jackson GH, Meyer A, Lippmann S. Wilson’s disease: psychiatric manifestations may be the clinical presentation. Postgrad Med. 1994;95:135-138.
17. O’Conner JA, Sokol RJ. Copper metabolism and copper storage disorders. In: Suchy FJ, Sokol RJ, Balistreri WF, eds. Liver Disease in Children. 3rd ed. New York, NY: Cambridge University Press; 2007:626-660.
18. Dening TR, Berrios GE. Wilson’s disease: a longitudinal study of psychiatric symptoms. Biol Psychiatry. 1990;28:255-265.
THE CASE
A 5-year-old boy was brought into his primary care clinic by his mother, who expressed concern about her son’s increasing impulsiveness, aggression, and difficulty staying on task at preschool and at home. The child’s medical history was unremarkable, and he was taking no medications. The family history was negative for hepatic or metabolic disease and positive for attention deficit-hyperactivity disorder (ADHD; father).
The child’s growth was normal. His physical exam was remarkable for a liver edge 1 cm below his costal margin. No Kayser-Fleischer rings were present.
Screening included a complete metabolic panel. Notable results included an alanine aminotransferase (ALT) level of 208 U/dL (normal range, < 30 U/dL), an aspartate transaminase (AST) level of 125 U/dL (normal range, 10-34 U/dL), and an alkaline phosphatase (ALP) of 470 U/dL (normal range, 93-309 U/dL). Subsequent repeat laboratory testing confirmed these elevations (ALT, 248 U/dL; AST, 137 U/dL; ALP, 462 U/dL). Ceruloplasmin levels were low (11 mg/dL; normal range, 18-35 mg/dL), and 24-hour urinary copper was not obtainable. Prothrombin/partial thromboplastin time, ammonia, lactate, total and direct bilirubin, and gamma-glutamyltransferase levels were normal.
Further evaluation included abdominal ultrasound and brain magnetic resonance imaging, both of which yielded normal results. Testing for Epstein-Barr virus; cytomegalovirus; hepatitis A, B, and C titers; and antinuclear, anti-smooth muscle, and anti–liver-kidney microsomal antibodies was negative.
THE DIAGNOSIS
The patient’s low ceruloplasmin prompted referral to Pediatric Gastroenterology for consultation and liver biopsy due to concern for Wilson disease. Biopsy results were consistent with, and quantitative liver copper confirmatory for, this diagnosis (FIGURE).
Genetic testing for mutations in the ATP7B gene was performed on the patient, his mother, and his siblings (his father was unavailable). The patient, his mother, and his sister were all positive for His1069Gln mutation; only the patient was positive for a 3990_3993 del mutation (his half-brother was negative for both mutations). The presence of 2 different mutant alleles for the ATP7B gene, one on each chromosome—the common substitution mutation, His1069Gln, in exon 14 and a 3990_3993 del TTAT mutation in exon 19—qualified the patient as a compound heterozygote.
The 3990_3993 del TTAT mutation—which to our knowledge has not been previously reported—produced a translational frame shift and premature stop codon. As others have pointed out, frame shift and missense mutations produce a more severe phenotype.1
Continue to: Further testing was prompted...
Further testing was prompted by a report suggesting that codon 129 mutations of the human prion gene (HPG) influence Wilson disease.2 Compared with patients who are heterozygous (M129V) or homozygous (V129V) for valine, those who are homozygous for methionine (M129M) have delayed symptom onset.2 Our patient was heterozygous (M129V). It is interesting to speculate that HPG heterozygosity, combined with a mutation causing a stop codon, predisposed our patient to more rapid accumulation of copper and earlier age of onset.
DISCUSSION
Wilson disease is an inherited disorder of copper metabolism.3 An inherent difficulty in its recognition, diagnosis, and management is its rarity: global prevalence is estimated as 1/30,000, although this varies by geographic location.1 In contrast, ADHD has a prevalence of 7.2%,4 making it 2400 times more prevalent than Wilson disease. Furthermore, abnormal liver function tests are common in children; the differential diagnosis includes etiologies such as infection (both viral and nonviral), immune-mediated inflammatory disease, drug toxicity (iatrogenic or medication-induced), anatomic abnormalities, and nonalcoholic fatty liver disease.5
Wilson disease is remarkable, however, for being easily treatable if detected and devastating if not. Although liver abnormalities often improve with treatment, delayed diagnosis and management significantly impact neurologic recovery: An 18-month delay results in 38% of patients continuing to have major neurologic disabilities.6 Untreated, Wilson disease may be fatal within 5 years of development of neurologic symptoms.7 Thus, it has been suggested that evaluation for Wilson disease be considered in any child older than 1 year who presents with unexplained liver disease, including asymptomatic elevations of serum transaminases.8
Mutations in ATP7B on chromosome 13 are responsible for the pathology of Wilson disease9; more than 250 mutations have been identified, including substitutions, deletions, and missense mutations.10 Affected patients may be compound heterozygotes11 and/or may possess new mutations, as seen in our patient.
Although copper absorption is normal, impaired excretion causes toxic accumulation in affected organs. ATP7B’s product, ATPase 2, regulates copper excretion, as well as copper binding to apoceruloplasmin to form the carrier protein ceruloplasmin. An ATP7B abnormality would prevent the latter—making ceruloplasmin a useful screening biomarker and a reliable marker for Wilson disease by age 1 year.8
Continue to: Hepatic and neurocognitive effects
Hepatic and neurocognitive effects. Excess copper in hepatocytes causes oxidative damage and release of copper into the circulation, with accumulation in susceptible organs (eg, brain, kidneys). Hepatocyte apoptosis is accelerated by copper’s negative effect on inhibitor of apoptosis protein.12,13 Renal tubular damage leads to Fanconi syndrome,14 in which substances such as glucose, phosphates, and potassium are excreted in urine rather than absorbed into the bloodstream by the kidneys. Excess copper deposition in the Descemet membrane may lead to Kayser-Fleisher ring formation.15 In the brain, copper deposition may occur in the lenticular nuclei,3 as well as in the thalamus, subthalamus, brainstem, and frontal cortex—resulting in extrapyramidal, cerebral, and mild cerebellar symptoms.6
Cognitive impairment, which may be subtle, includes increased impulsivity, impaired judgment, apathy, poor decision making, decreased attention, increased lability, slowed thinking, and memory loss.6 Behavioral manifestations include changes in school or work performance and outbursts mimicking ADHD12,16,17 as well as paranoia, depression, and bizarre behaviors.16,18 Neuropsychiatric abnormalities include personality changes, pseudoparkinsonism, dyskinesia/dysarthria, and ataxia/tremor. Younger patients with psychiatric symptoms may be labelled with depression, anxiety, obsessive-compulsive disorder, bipolar disorder, or antisocial disorder.6,16,18
Hepatic disease manifestations range from asymptomatic elevations in AST/ALT to acute hepatitis, mimicking infectious processes. Cirrhosis is the end result of untreated Wilson disease, with liver transplantation required if end-stage liver disease results. Rarely, patients present in fulminant hepatic failure, with death occurring if emergent liver transplantation is not performed.6,8,10
Of note, before age 10, > 80% of patients with Wilson disease present with hepatic symptoms; those ages 10 to 18 often manifest psychiatric changes.17 Kayser-Fleisher rings are common in patients with neurologic manifestations but less so in those who have hepatic presentations or are presymptomatic.6,15
Effective disease-mitigating treatment is indicated and available for both symptomatic and asymptomatic individuals and includes the copper chelators D-penicillamine (starting dose, 150-300 mg/d with a gradual weekly increase to 20 mg/kg/d) and trientine hydrochloride (a heavy metal chelating compound; starting dose, 20 mg/kg/d to a maximum of 1000 mg/d in young adults). Adverse effects of D-penicillamine include cutaneous eruptions, neutropenia, thrombocytopenia, proteinuria, and a lupus-like syndrome; therefore, trientine is increasingly being used as first-line therapy.8
Continue to: For asymptomatic...
For asymptomatic patients who have had effective chelation therapy and proven de-coppering, zinc salts are a useful follow-on therapy. Zinc’s proposed mechanism of action is induction of metallothionein in enterocytes, which promotes copper trapping and eventual excretion into the lumen. Importantly, treatment for Wilson disease is lifelong and monitoring of compliance is essential.8
Our 5-year-old patient was started on oral trientine at 20 mg/kg/d and a low copper diet. In response to this initial treatment, the patient’s liver function tests (LFTs) normalized, and he was switched to 25 mg tid of a zinc chelate, with continuation of the low copper diet. His LFTs have remained normal, although his urine copper levels are still elevated. He continues to be monitored periodically with LFTs and measurement of urine copper levels. He is also being treated for ADHD, as his presenting behavioral abnormalities suggestive of ADHD have not resolved.
THE TAKEAWAY
Although children presenting with symptoms consistent with ADHD often have ADHD, as was true in this case, it is important to consider other diagnoses. Unexplained elevations of liver function test values in children older than 1 year should prompt screening for Wilson disease.5,8 Additionally, other family members should be evaluated; if they have the disease, treatment should be started by age 2 years, even if the patient is asymptomatic.
In our patient’s case, routine screening saved the day. The complete metabolic panel revealed elevated ALT and AST levels, prompting further evaluation. Without this testing, his diagnosis likely would have been delayed, leading to progressive liver and central nervous system disease. With early identification and treatment, it is possible to stop the progression of Wilson disease.
CORRESPONDENCE
Jeffrey Taylor, MD, MS, Evangelical Community Hospital, Department of Pediatrics, 1 Hospital Drive, Lewisburg PA, 17837; [email protected].
THE CASE
A 5-year-old boy was brought into his primary care clinic by his mother, who expressed concern about her son’s increasing impulsiveness, aggression, and difficulty staying on task at preschool and at home. The child’s medical history was unremarkable, and he was taking no medications. The family history was negative for hepatic or metabolic disease and positive for attention deficit-hyperactivity disorder (ADHD; father).
The child’s growth was normal. His physical exam was remarkable for a liver edge 1 cm below his costal margin. No Kayser-Fleischer rings were present.
Screening included a complete metabolic panel. Notable results included an alanine aminotransferase (ALT) level of 208 U/dL (normal range, < 30 U/dL), an aspartate transaminase (AST) level of 125 U/dL (normal range, 10-34 U/dL), and an alkaline phosphatase (ALP) of 470 U/dL (normal range, 93-309 U/dL). Subsequent repeat laboratory testing confirmed these elevations (ALT, 248 U/dL; AST, 137 U/dL; ALP, 462 U/dL). Ceruloplasmin levels were low (11 mg/dL; normal range, 18-35 mg/dL), and 24-hour urinary copper was not obtainable. Prothrombin/partial thromboplastin time, ammonia, lactate, total and direct bilirubin, and gamma-glutamyltransferase levels were normal.
Further evaluation included abdominal ultrasound and brain magnetic resonance imaging, both of which yielded normal results. Testing for Epstein-Barr virus; cytomegalovirus; hepatitis A, B, and C titers; and antinuclear, anti-smooth muscle, and anti–liver-kidney microsomal antibodies was negative.
THE DIAGNOSIS
The patient’s low ceruloplasmin prompted referral to Pediatric Gastroenterology for consultation and liver biopsy due to concern for Wilson disease. Biopsy results were consistent with, and quantitative liver copper confirmatory for, this diagnosis (FIGURE).
Genetic testing for mutations in the ATP7B gene was performed on the patient, his mother, and his siblings (his father was unavailable). The patient, his mother, and his sister were all positive for His1069Gln mutation; only the patient was positive for a 3990_3993 del mutation (his half-brother was negative for both mutations). The presence of 2 different mutant alleles for the ATP7B gene, one on each chromosome—the common substitution mutation, His1069Gln, in exon 14 and a 3990_3993 del TTAT mutation in exon 19—qualified the patient as a compound heterozygote.
The 3990_3993 del TTAT mutation—which to our knowledge has not been previously reported—produced a translational frame shift and premature stop codon. As others have pointed out, frame shift and missense mutations produce a more severe phenotype.1
Continue to: Further testing was prompted...
Further testing was prompted by a report suggesting that codon 129 mutations of the human prion gene (HPG) influence Wilson disease.2 Compared with patients who are heterozygous (M129V) or homozygous (V129V) for valine, those who are homozygous for methionine (M129M) have delayed symptom onset.2 Our patient was heterozygous (M129V). It is interesting to speculate that HPG heterozygosity, combined with a mutation causing a stop codon, predisposed our patient to more rapid accumulation of copper and earlier age of onset.
DISCUSSION
Wilson disease is an inherited disorder of copper metabolism.3 An inherent difficulty in its recognition, diagnosis, and management is its rarity: global prevalence is estimated as 1/30,000, although this varies by geographic location.1 In contrast, ADHD has a prevalence of 7.2%,4 making it 2400 times more prevalent than Wilson disease. Furthermore, abnormal liver function tests are common in children; the differential diagnosis includes etiologies such as infection (both viral and nonviral), immune-mediated inflammatory disease, drug toxicity (iatrogenic or medication-induced), anatomic abnormalities, and nonalcoholic fatty liver disease.5
Wilson disease is remarkable, however, for being easily treatable if detected and devastating if not. Although liver abnormalities often improve with treatment, delayed diagnosis and management significantly impact neurologic recovery: An 18-month delay results in 38% of patients continuing to have major neurologic disabilities.6 Untreated, Wilson disease may be fatal within 5 years of development of neurologic symptoms.7 Thus, it has been suggested that evaluation for Wilson disease be considered in any child older than 1 year who presents with unexplained liver disease, including asymptomatic elevations of serum transaminases.8
Mutations in ATP7B on chromosome 13 are responsible for the pathology of Wilson disease9; more than 250 mutations have been identified, including substitutions, deletions, and missense mutations.10 Affected patients may be compound heterozygotes11 and/or may possess new mutations, as seen in our patient.
Although copper absorption is normal, impaired excretion causes toxic accumulation in affected organs. ATP7B’s product, ATPase 2, regulates copper excretion, as well as copper binding to apoceruloplasmin to form the carrier protein ceruloplasmin. An ATP7B abnormality would prevent the latter—making ceruloplasmin a useful screening biomarker and a reliable marker for Wilson disease by age 1 year.8
Continue to: Hepatic and neurocognitive effects
Hepatic and neurocognitive effects. Excess copper in hepatocytes causes oxidative damage and release of copper into the circulation, with accumulation in susceptible organs (eg, brain, kidneys). Hepatocyte apoptosis is accelerated by copper’s negative effect on inhibitor of apoptosis protein.12,13 Renal tubular damage leads to Fanconi syndrome,14 in which substances such as glucose, phosphates, and potassium are excreted in urine rather than absorbed into the bloodstream by the kidneys. Excess copper deposition in the Descemet membrane may lead to Kayser-Fleisher ring formation.15 In the brain, copper deposition may occur in the lenticular nuclei,3 as well as in the thalamus, subthalamus, brainstem, and frontal cortex—resulting in extrapyramidal, cerebral, and mild cerebellar symptoms.6
Cognitive impairment, which may be subtle, includes increased impulsivity, impaired judgment, apathy, poor decision making, decreased attention, increased lability, slowed thinking, and memory loss.6 Behavioral manifestations include changes in school or work performance and outbursts mimicking ADHD12,16,17 as well as paranoia, depression, and bizarre behaviors.16,18 Neuropsychiatric abnormalities include personality changes, pseudoparkinsonism, dyskinesia/dysarthria, and ataxia/tremor. Younger patients with psychiatric symptoms may be labelled with depression, anxiety, obsessive-compulsive disorder, bipolar disorder, or antisocial disorder.6,16,18
Hepatic disease manifestations range from asymptomatic elevations in AST/ALT to acute hepatitis, mimicking infectious processes. Cirrhosis is the end result of untreated Wilson disease, with liver transplantation required if end-stage liver disease results. Rarely, patients present in fulminant hepatic failure, with death occurring if emergent liver transplantation is not performed.6,8,10
Of note, before age 10, > 80% of patients with Wilson disease present with hepatic symptoms; those ages 10 to 18 often manifest psychiatric changes.17 Kayser-Fleisher rings are common in patients with neurologic manifestations but less so in those who have hepatic presentations or are presymptomatic.6,15
Effective disease-mitigating treatment is indicated and available for both symptomatic and asymptomatic individuals and includes the copper chelators D-penicillamine (starting dose, 150-300 mg/d with a gradual weekly increase to 20 mg/kg/d) and trientine hydrochloride (a heavy metal chelating compound; starting dose, 20 mg/kg/d to a maximum of 1000 mg/d in young adults). Adverse effects of D-penicillamine include cutaneous eruptions, neutropenia, thrombocytopenia, proteinuria, and a lupus-like syndrome; therefore, trientine is increasingly being used as first-line therapy.8
Continue to: For asymptomatic...
For asymptomatic patients who have had effective chelation therapy and proven de-coppering, zinc salts are a useful follow-on therapy. Zinc’s proposed mechanism of action is induction of metallothionein in enterocytes, which promotes copper trapping and eventual excretion into the lumen. Importantly, treatment for Wilson disease is lifelong and monitoring of compliance is essential.8
Our 5-year-old patient was started on oral trientine at 20 mg/kg/d and a low copper diet. In response to this initial treatment, the patient’s liver function tests (LFTs) normalized, and he was switched to 25 mg tid of a zinc chelate, with continuation of the low copper diet. His LFTs have remained normal, although his urine copper levels are still elevated. He continues to be monitored periodically with LFTs and measurement of urine copper levels. He is also being treated for ADHD, as his presenting behavioral abnormalities suggestive of ADHD have not resolved.
THE TAKEAWAY
Although children presenting with symptoms consistent with ADHD often have ADHD, as was true in this case, it is important to consider other diagnoses. Unexplained elevations of liver function test values in children older than 1 year should prompt screening for Wilson disease.5,8 Additionally, other family members should be evaluated; if they have the disease, treatment should be started by age 2 years, even if the patient is asymptomatic.
In our patient’s case, routine screening saved the day. The complete metabolic panel revealed elevated ALT and AST levels, prompting further evaluation. Without this testing, his diagnosis likely would have been delayed, leading to progressive liver and central nervous system disease. With early identification and treatment, it is possible to stop the progression of Wilson disease.
CORRESPONDENCE
Jeffrey Taylor, MD, MS, Evangelical Community Hospital, Department of Pediatrics, 1 Hospital Drive, Lewisburg PA, 17837; [email protected].
1. Wu F, Wang J, Pu C, et al. Wilson’s disease: a comprehensive review of the molecular mechanisms. Int J Mol Sci. 2015;16:6419-6431.
2. Merle U, Stremmel W, Gessner R. Influence of homozygosity for methionine at codon 129 of the human prion gene on the onset of neurological and hepatic symptoms in Wilson disease. Arch Neurol. 2006;63:982-985.
3. Compston A. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver, by S. A. Kinnier Wilson, (From the National Hospital, and the Laboratory of the National Hospital, Queen Square, London) Brain 1912: 34; 295-509. Brain. 2009;132(pt 8):1997-2001.
4. Thomas R, Sanders S, Doust J, et al. Prevalence of attention-deficit/hyperactivity disorder: a systematic review and meta-analysis. Pediatrics. 2015;135:e994-e1001.
5. Kang K. Abnormality on liver function test. Pediatr Gastroenterol Hepatol Nutr. 2013;16:225-232.
6. Lorincz M. Neurologic Wilson’s disease. Ann NY Acad Sci. 2010;1184:173-187.
7. Dening TR, Berrios GE, Walshe JM. Wilson’s disease and epilepsy. Brain. 1988;111(pt 5):1139-1155.
8. Socha P, Janczyk W, Dhawan A, et al. Wilson’s disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutrit. 2018;66:334-344.
9. Bull PC, Thomas GR, Rommens JM, et al. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327-337.
10. Ala A, Schilsky ML. Wilson disease: pathophysiology, diagnosis, treatment and screening. Clin Liver Dis. 2004;8:787-805, viii.
11. Thomas GR, Forbes JR, Roberts EA, et al. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet. 1995;9:210-217.
12. Pfeiffer RF. Wilson’s disease. Semin Neurol. 2007;27:123-132.
13. Das SK, Ray K. Wilson’s disease: an update. Nat Clin Pract Neurol. 2006;2:482-493.
14. Morgan HG, Stewart WK, Lowe KG, et al. Wilson’s disease and the Fanconi syndrome. Q J Med. 1962;31:361-384.
15. Wiebers DO, Hollenhorst RW, Goldstein NP. The ophthalmologic manifestations of Wilson’s disease. Mayo Clin Proc. 1977;52:409-416.
16. Jackson GH, Meyer A, Lippmann S. Wilson’s disease: psychiatric manifestations may be the clinical presentation. Postgrad Med. 1994;95:135-138.
17. O’Conner JA, Sokol RJ. Copper metabolism and copper storage disorders. In: Suchy FJ, Sokol RJ, Balistreri WF, eds. Liver Disease in Children. 3rd ed. New York, NY: Cambridge University Press; 2007:626-660.
18. Dening TR, Berrios GE. Wilson’s disease: a longitudinal study of psychiatric symptoms. Biol Psychiatry. 1990;28:255-265.
1. Wu F, Wang J, Pu C, et al. Wilson’s disease: a comprehensive review of the molecular mechanisms. Int J Mol Sci. 2015;16:6419-6431.
2. Merle U, Stremmel W, Gessner R. Influence of homozygosity for methionine at codon 129 of the human prion gene on the onset of neurological and hepatic symptoms in Wilson disease. Arch Neurol. 2006;63:982-985.
3. Compston A. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver, by S. A. Kinnier Wilson, (From the National Hospital, and the Laboratory of the National Hospital, Queen Square, London) Brain 1912: 34; 295-509. Brain. 2009;132(pt 8):1997-2001.
4. Thomas R, Sanders S, Doust J, et al. Prevalence of attention-deficit/hyperactivity disorder: a systematic review and meta-analysis. Pediatrics. 2015;135:e994-e1001.
5. Kang K. Abnormality on liver function test. Pediatr Gastroenterol Hepatol Nutr. 2013;16:225-232.
6. Lorincz M. Neurologic Wilson’s disease. Ann NY Acad Sci. 2010;1184:173-187.
7. Dening TR, Berrios GE, Walshe JM. Wilson’s disease and epilepsy. Brain. 1988;111(pt 5):1139-1155.
8. Socha P, Janczyk W, Dhawan A, et al. Wilson’s disease in children: a position paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutrit. 2018;66:334-344.
9. Bull PC, Thomas GR, Rommens JM, et al. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327-337.
10. Ala A, Schilsky ML. Wilson disease: pathophysiology, diagnosis, treatment and screening. Clin Liver Dis. 2004;8:787-805, viii.
11. Thomas GR, Forbes JR, Roberts EA, et al. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet. 1995;9:210-217.
12. Pfeiffer RF. Wilson’s disease. Semin Neurol. 2007;27:123-132.
13. Das SK, Ray K. Wilson’s disease: an update. Nat Clin Pract Neurol. 2006;2:482-493.
14. Morgan HG, Stewart WK, Lowe KG, et al. Wilson’s disease and the Fanconi syndrome. Q J Med. 1962;31:361-384.
15. Wiebers DO, Hollenhorst RW, Goldstein NP. The ophthalmologic manifestations of Wilson’s disease. Mayo Clin Proc. 1977;52:409-416.
16. Jackson GH, Meyer A, Lippmann S. Wilson’s disease: psychiatric manifestations may be the clinical presentation. Postgrad Med. 1994;95:135-138.
17. O’Conner JA, Sokol RJ. Copper metabolism and copper storage disorders. In: Suchy FJ, Sokol RJ, Balistreri WF, eds. Liver Disease in Children. 3rd ed. New York, NY: Cambridge University Press; 2007:626-660.
18. Dening TR, Berrios GE. Wilson’s disease: a longitudinal study of psychiatric symptoms. Biol Psychiatry. 1990;28:255-265.

Metastatic Melanoma Mimicking Eruptive Keratoacanthomas
To the Editor:
Melanoma is the third most common skin cancer. It is estimated that 18% of melanoma patients will develop skin metastases, with skin being the first site of involvement in 56% of cases.1 Of all cancers, it is estimated that 5% will develop skin metastases. It is the presenting sign in nearly 1% of visceral cancers.2 Melanoma and nonmelanoma metastases can have sundry presentations. We present a case of metastatic melanoma with multiple keratoacanthoma (KA)–like skin lesions in a patient with a known history of nonmelanoma skin cancer (NMSC) as well as melanoma.
A 76-year-old man with a history of pT2aNXMX melanoma on the left upper back presented for a routine 3-month follow-up and reported several new asymptomatic bumps on the chest, back, and right upper extremity within the last 2 weeks. The melanoma was removed via wide local excision 2 years prior at an outside facility with a Breslow depth of 1.05 mm and a negative sentinel lymph node biopsy. The mitotic rate or ulceration status was unknown. He also had a history of several NMSCs, as well as a medical history of coronary artery disease, myocardial infarction, and ventricular tachycardia with cardiac defibrillator placement. Physical examination revealed 5 pink, volcano-shaped nodules with central keratotic plugs on the upper back (Figure 1), chest, and right upper extremity, in addition to 1 pink pearly nodule on the right side of the chest. The history and appearance of the lesions were suspicious for eruptive KAs. There was no evidence of cancer recurrence at the prior melanoma and NMSC sites.
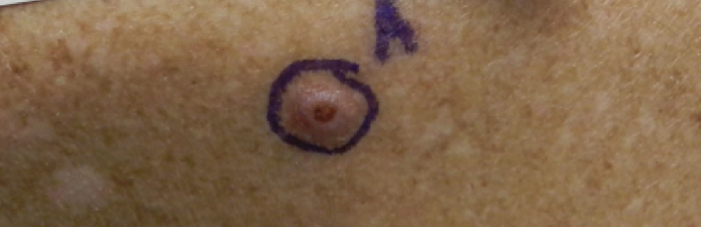
A deep shave skin biopsy was performed at all 6 sites. Histopathology showed a diffuse dermal infiltrate of elongated nests of melanocytes and nonnested melanocytes. Marked cytologic atypia and ulceration were present. Minimal connection to the overlying epidermis and a lack of junctional nests was noted. Immunohistochemical studies revealed scattered positivity for Melan-A and negative staining for AE1, AE3, cytokeratin 5, and cytokeratin 6 at all 6 sites (Figure 2). A subsequent metastatic workup showed widespread metastatic disease in the liver, bone, lung, and inferior vena cava. Computed tomography of the head was unremarkable. Magnetic resonance imaging of the brain was not performed due to the cardiac defibrillator. The patient’s lactate dehydrogenase level showed a mild increase compared to 2 months prior to the metastatic melanoma diagnosis (144 U/L vs 207 U/L [reference range, 100–200 U/L]).
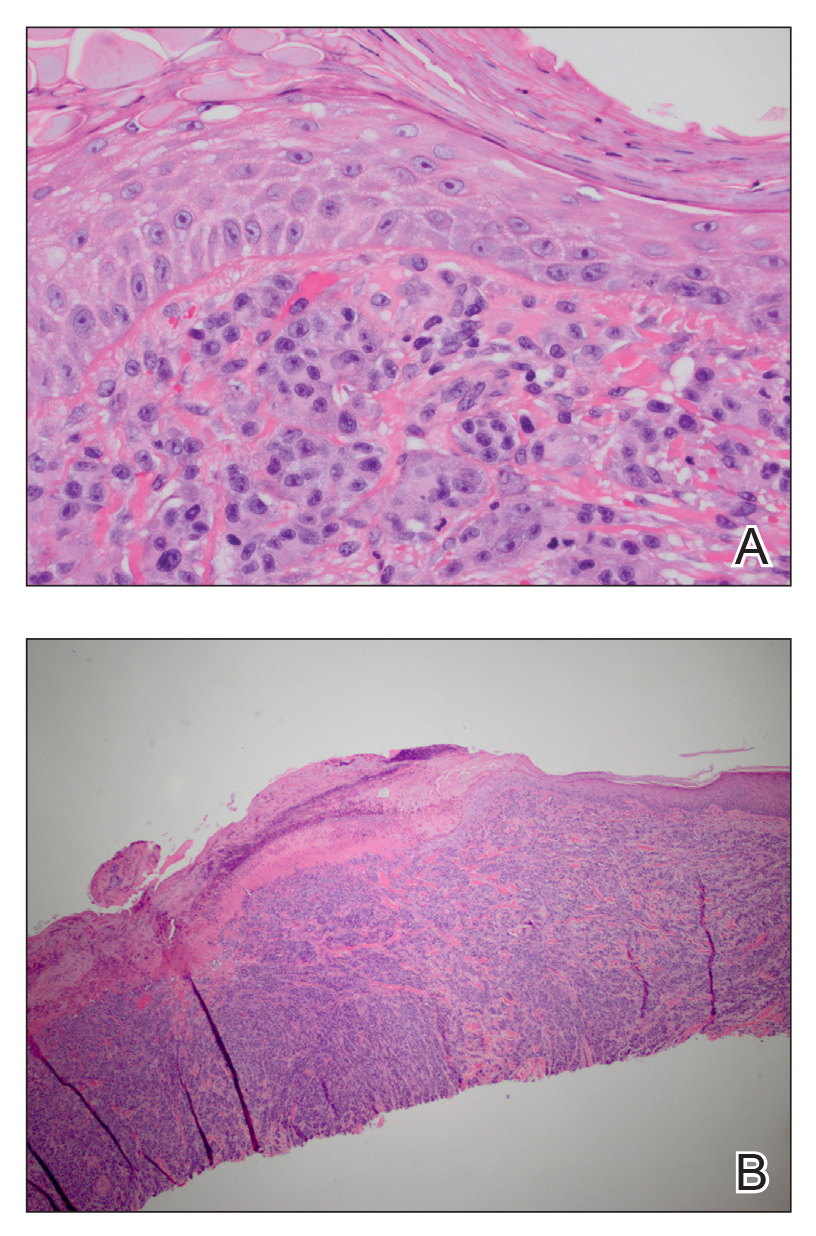
The patient had no systemic symptoms at follow-up 5 weeks later. He was already evaluated by an oncologist and received his first dose of ipilimumab. He was BRAF-mutation negative. He had developed 2 new skin metastases. Five of 6 initially biopsied metastases returned and were growing; they were tender and friable with intermittent bleeding. He was subsequently referred to surgical oncology for excision of symptomatic nodules as palliative care.
Although melanoma is well known to metastasize years and even decades later, KA-like lesions have not been reported as manifestations of metastatic melanoma.4,5 Our patient likely had a primary amelanotic melanoma, as the medical records from the outside facility stated that basal cell carcinoma was ruled out via biopsy. The amelanotic nature of the primary melanoma may have influenced the amelanotic appearance of the metastases. Our patient had no signs of immunosuppression that could have contributed to the sudden skin metastases.
Depending on the subtype of cutaneous metastases (eg, satellitosis, in-transit disease, distant cutaneous metastases), the location prevalence of the primary melanoma varies. In a study of 4865 melanoma patients who were diagnosed and followed prospectively over a 30-year period, skin metastases were mostly locoregional and presentation on the leg and foot were disproportionate.1 In contrast, the trunk was overrepresented for distant metastases. Distant metastases also were more associated with concurrent metastases to the viscera.1 Accordingly, a patient’s prognosis and management will differ depending on the subtype of cutaneous metastases.
Eruptive or multiple KAs classically have been associated with the Grzybowski variant, the Ferguson-Smith familial variant, and Muir-Torre syndrome. It was reported as a paraneoplastic syndrome associated with colon cancer, ovarian cancer, and once with myelodysplastic syndrome.3 Keratoacanthomas are being classified as well-differentiated squamous cell carcinomas and have metastatic potential. A biopsy is recommended to diagnose KAs as opposed to historically being monitored for resolution. A skin biopsy is the standard of care in management of KAs.
In addition to being associated with Muir-Torre syndrome and classified as a paraneoplastic syndrome,3 eruptive KAs can occur following skin resurfacing for actinic damage, fractional photothermolysis, cryotherapy, Jessner peels, and trichloroacetic acid peels.6 A couple other uncommon settings include a case report of an arc welder with job-associated radiation and multiple reports of tattoo-induced KAs.7,8 There is the new increasingly common association of squamous cell carcinomas with BRAF inhibitors, such as vemurafenib, for metastatic melanoma.9
In a 2012 review article on cutaneous metastases, Riahi and Cohen10 found 8 cases of cutaneous metastases presenting as KA-like lesions; none were metastatic melanoma. All were solitary lesions, not multiple lesions, as in our patient. The sources were lung (3 cases), breast, esophagus, chondrosarcoma, bronchial, and mesothelioma. The most common location was the upper lip. Additionally, similar to our patient, they behaved clinically as KAs with rapid growth and keratotic plugs and were asymptomatic.10
Metastatic melanoma may mimic many other cutaneous processes that may make the diagnosis more difficult. Our case indicates that cutaneous metastases may mimic KAs. Although multiple KA-like lesions can spontaneously occur, a paraneoplastic syndrome and other underlying etiologies should be considered.
- Savoia P, Fava P, Nardò T, et al. Skin metastases of malignant melanoma: a clinical and prognostic study. Melanoma Res. 2009;19:321-326.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma. J Am Acad Dermatol. 1990;22:19-26.
- Behzad M, Michl C, Pfützner W. Multiple eruptive keratoacanthomas associated with myelodysplastic syndrome. J Dtsch Dermatol Ges. 2012;10:359-360.
- Cheung WL, Patel RR, Leonard A, et al. Amelanotic melanoma: a detailed morphologic analysis with clinicopathologic correlation of 75 cases. J Cutan Pathol. 2012;39:33-39.
- Ferrari A, Piccolo D, Fargnoli MC, et al. Cutaneous amelanotic melanoma metastasis and dermatofibromas showing a dotted vascular pattern. Acta Dermato Venereologica. 2004;84:164-165.
- Mohr B, Fernandez MP, Krejci-Manwaring J. Eruptive keratoacanthoma after Jessner’s and trichloroacetic acid peel for actinic keratosis. Dermatol Surg. 2013;39:331-333.
- Wolfe CM, Green WH, Cognetta AB, et al. Multiple squamous cell carcinomas and eruptive keratoacanthomas in an arc welder. Dermatol Surg. 2013;39:328-330.
- Kluger N, Phan A, Debarbieux S, et al. Skin cancers arising in tattoos: coincidental or not? Dermatology. 2008;217:219-221.
- Mays R, Curry J, Kim K, et al. Eruptive squamous cell carcinomas after vemurafenib therapy. J Cutan Med Surg. 2013;17:419-422.
- Riahi RR, Cohen PR. Clinical manifestations of cutaneous metastases: a review with special emphasis on cutaneous metastases mimicking keratoacanthoma. Am J Clin Dermatol. 2012;13:103-112.
To the Editor:
Melanoma is the third most common skin cancer. It is estimated that 18% of melanoma patients will develop skin metastases, with skin being the first site of involvement in 56% of cases.1 Of all cancers, it is estimated that 5% will develop skin metastases. It is the presenting sign in nearly 1% of visceral cancers.2 Melanoma and nonmelanoma metastases can have sundry presentations. We present a case of metastatic melanoma with multiple keratoacanthoma (KA)–like skin lesions in a patient with a known history of nonmelanoma skin cancer (NMSC) as well as melanoma.
A 76-year-old man with a history of pT2aNXMX melanoma on the left upper back presented for a routine 3-month follow-up and reported several new asymptomatic bumps on the chest, back, and right upper extremity within the last 2 weeks. The melanoma was removed via wide local excision 2 years prior at an outside facility with a Breslow depth of 1.05 mm and a negative sentinel lymph node biopsy. The mitotic rate or ulceration status was unknown. He also had a history of several NMSCs, as well as a medical history of coronary artery disease, myocardial infarction, and ventricular tachycardia with cardiac defibrillator placement. Physical examination revealed 5 pink, volcano-shaped nodules with central keratotic plugs on the upper back (Figure 1), chest, and right upper extremity, in addition to 1 pink pearly nodule on the right side of the chest. The history and appearance of the lesions were suspicious for eruptive KAs. There was no evidence of cancer recurrence at the prior melanoma and NMSC sites.
A deep shave skin biopsy was performed at all 6 sites. Histopathology showed a diffuse dermal infiltrate of elongated nests of melanocytes and nonnested melanocytes. Marked cytologic atypia and ulceration were present. Minimal connection to the overlying epidermis and a lack of junctional nests was noted. Immunohistochemical studies revealed scattered positivity for Melan-A and negative staining for AE1, AE3, cytokeratin 5, and cytokeratin 6 at all 6 sites (Figure 2). A subsequent metastatic workup showed widespread metastatic disease in the liver, bone, lung, and inferior vena cava. Computed tomography of the head was unremarkable. Magnetic resonance imaging of the brain was not performed due to the cardiac defibrillator. The patient’s lactate dehydrogenase level showed a mild increase compared to 2 months prior to the metastatic melanoma diagnosis (144 U/L vs 207 U/L [reference range, 100–200 U/L]).
The patient had no systemic symptoms at follow-up 5 weeks later. He was already evaluated by an oncologist and received his first dose of ipilimumab. He was BRAF-mutation negative. He had developed 2 new skin metastases. Five of 6 initially biopsied metastases returned and were growing; they were tender and friable with intermittent bleeding. He was subsequently referred to surgical oncology for excision of symptomatic nodules as palliative care.
Although melanoma is well known to metastasize years and even decades later, KA-like lesions have not been reported as manifestations of metastatic melanoma.4,5 Our patient likely had a primary amelanotic melanoma, as the medical records from the outside facility stated that basal cell carcinoma was ruled out via biopsy. The amelanotic nature of the primary melanoma may have influenced the amelanotic appearance of the metastases. Our patient had no signs of immunosuppression that could have contributed to the sudden skin metastases.
Depending on the subtype of cutaneous metastases (eg, satellitosis, in-transit disease, distant cutaneous metastases), the location prevalence of the primary melanoma varies. In a study of 4865 melanoma patients who were diagnosed and followed prospectively over a 30-year period, skin metastases were mostly locoregional and presentation on the leg and foot were disproportionate.1 In contrast, the trunk was overrepresented for distant metastases. Distant metastases also were more associated with concurrent metastases to the viscera.1 Accordingly, a patient’s prognosis and management will differ depending on the subtype of cutaneous metastases.
Eruptive or multiple KAs classically have been associated with the Grzybowski variant, the Ferguson-Smith familial variant, and Muir-Torre syndrome. It was reported as a paraneoplastic syndrome associated with colon cancer, ovarian cancer, and once with myelodysplastic syndrome.3 Keratoacanthomas are being classified as well-differentiated squamous cell carcinomas and have metastatic potential. A biopsy is recommended to diagnose KAs as opposed to historically being monitored for resolution. A skin biopsy is the standard of care in management of KAs.
In addition to being associated with Muir-Torre syndrome and classified as a paraneoplastic syndrome,3 eruptive KAs can occur following skin resurfacing for actinic damage, fractional photothermolysis, cryotherapy, Jessner peels, and trichloroacetic acid peels.6 A couple other uncommon settings include a case report of an arc welder with job-associated radiation and multiple reports of tattoo-induced KAs.7,8 There is the new increasingly common association of squamous cell carcinomas with BRAF inhibitors, such as vemurafenib, for metastatic melanoma.9
In a 2012 review article on cutaneous metastases, Riahi and Cohen10 found 8 cases of cutaneous metastases presenting as KA-like lesions; none were metastatic melanoma. All were solitary lesions, not multiple lesions, as in our patient. The sources were lung (3 cases), breast, esophagus, chondrosarcoma, bronchial, and mesothelioma. The most common location was the upper lip. Additionally, similar to our patient, they behaved clinically as KAs with rapid growth and keratotic plugs and were asymptomatic.10
Metastatic melanoma may mimic many other cutaneous processes that may make the diagnosis more difficult. Our case indicates that cutaneous metastases may mimic KAs. Although multiple KA-like lesions can spontaneously occur, a paraneoplastic syndrome and other underlying etiologies should be considered.
To the Editor:
Melanoma is the third most common skin cancer. It is estimated that 18% of melanoma patients will develop skin metastases, with skin being the first site of involvement in 56% of cases.1 Of all cancers, it is estimated that 5% will develop skin metastases. It is the presenting sign in nearly 1% of visceral cancers.2 Melanoma and nonmelanoma metastases can have sundry presentations. We present a case of metastatic melanoma with multiple keratoacanthoma (KA)–like skin lesions in a patient with a known history of nonmelanoma skin cancer (NMSC) as well as melanoma.
A 76-year-old man with a history of pT2aNXMX melanoma on the left upper back presented for a routine 3-month follow-up and reported several new asymptomatic bumps on the chest, back, and right upper extremity within the last 2 weeks. The melanoma was removed via wide local excision 2 years prior at an outside facility with a Breslow depth of 1.05 mm and a negative sentinel lymph node biopsy. The mitotic rate or ulceration status was unknown. He also had a history of several NMSCs, as well as a medical history of coronary artery disease, myocardial infarction, and ventricular tachycardia with cardiac defibrillator placement. Physical examination revealed 5 pink, volcano-shaped nodules with central keratotic plugs on the upper back (Figure 1), chest, and right upper extremity, in addition to 1 pink pearly nodule on the right side of the chest. The history and appearance of the lesions were suspicious for eruptive KAs. There was no evidence of cancer recurrence at the prior melanoma and NMSC sites.
A deep shave skin biopsy was performed at all 6 sites. Histopathology showed a diffuse dermal infiltrate of elongated nests of melanocytes and nonnested melanocytes. Marked cytologic atypia and ulceration were present. Minimal connection to the overlying epidermis and a lack of junctional nests was noted. Immunohistochemical studies revealed scattered positivity for Melan-A and negative staining for AE1, AE3, cytokeratin 5, and cytokeratin 6 at all 6 sites (Figure 2). A subsequent metastatic workup showed widespread metastatic disease in the liver, bone, lung, and inferior vena cava. Computed tomography of the head was unremarkable. Magnetic resonance imaging of the brain was not performed due to the cardiac defibrillator. The patient’s lactate dehydrogenase level showed a mild increase compared to 2 months prior to the metastatic melanoma diagnosis (144 U/L vs 207 U/L [reference range, 100–200 U/L]).
The patient had no systemic symptoms at follow-up 5 weeks later. He was already evaluated by an oncologist and received his first dose of ipilimumab. He was BRAF-mutation negative. He had developed 2 new skin metastases. Five of 6 initially biopsied metastases returned and were growing; they were tender and friable with intermittent bleeding. He was subsequently referred to surgical oncology for excision of symptomatic nodules as palliative care.
Although melanoma is well known to metastasize years and even decades later, KA-like lesions have not been reported as manifestations of metastatic melanoma.4,5 Our patient likely had a primary amelanotic melanoma, as the medical records from the outside facility stated that basal cell carcinoma was ruled out via biopsy. The amelanotic nature of the primary melanoma may have influenced the amelanotic appearance of the metastases. Our patient had no signs of immunosuppression that could have contributed to the sudden skin metastases.
Depending on the subtype of cutaneous metastases (eg, satellitosis, in-transit disease, distant cutaneous metastases), the location prevalence of the primary melanoma varies. In a study of 4865 melanoma patients who were diagnosed and followed prospectively over a 30-year period, skin metastases were mostly locoregional and presentation on the leg and foot were disproportionate.1 In contrast, the trunk was overrepresented for distant metastases. Distant metastases also were more associated with concurrent metastases to the viscera.1 Accordingly, a patient’s prognosis and management will differ depending on the subtype of cutaneous metastases.
Eruptive or multiple KAs classically have been associated with the Grzybowski variant, the Ferguson-Smith familial variant, and Muir-Torre syndrome. It was reported as a paraneoplastic syndrome associated with colon cancer, ovarian cancer, and once with myelodysplastic syndrome.3 Keratoacanthomas are being classified as well-differentiated squamous cell carcinomas and have metastatic potential. A biopsy is recommended to diagnose KAs as opposed to historically being monitored for resolution. A skin biopsy is the standard of care in management of KAs.
In addition to being associated with Muir-Torre syndrome and classified as a paraneoplastic syndrome,3 eruptive KAs can occur following skin resurfacing for actinic damage, fractional photothermolysis, cryotherapy, Jessner peels, and trichloroacetic acid peels.6 A couple other uncommon settings include a case report of an arc welder with job-associated radiation and multiple reports of tattoo-induced KAs.7,8 There is the new increasingly common association of squamous cell carcinomas with BRAF inhibitors, such as vemurafenib, for metastatic melanoma.9
In a 2012 review article on cutaneous metastases, Riahi and Cohen10 found 8 cases of cutaneous metastases presenting as KA-like lesions; none were metastatic melanoma. All were solitary lesions, not multiple lesions, as in our patient. The sources were lung (3 cases), breast, esophagus, chondrosarcoma, bronchial, and mesothelioma. The most common location was the upper lip. Additionally, similar to our patient, they behaved clinically as KAs with rapid growth and keratotic plugs and were asymptomatic.10
Metastatic melanoma may mimic many other cutaneous processes that may make the diagnosis more difficult. Our case indicates that cutaneous metastases may mimic KAs. Although multiple KA-like lesions can spontaneously occur, a paraneoplastic syndrome and other underlying etiologies should be considered.
- Savoia P, Fava P, Nardò T, et al. Skin metastases of malignant melanoma: a clinical and prognostic study. Melanoma Res. 2009;19:321-326.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma. J Am Acad Dermatol. 1990;22:19-26.
- Behzad M, Michl C, Pfützner W. Multiple eruptive keratoacanthomas associated with myelodysplastic syndrome. J Dtsch Dermatol Ges. 2012;10:359-360.
- Cheung WL, Patel RR, Leonard A, et al. Amelanotic melanoma: a detailed morphologic analysis with clinicopathologic correlation of 75 cases. J Cutan Pathol. 2012;39:33-39.
- Ferrari A, Piccolo D, Fargnoli MC, et al. Cutaneous amelanotic melanoma metastasis and dermatofibromas showing a dotted vascular pattern. Acta Dermato Venereologica. 2004;84:164-165.
- Mohr B, Fernandez MP, Krejci-Manwaring J. Eruptive keratoacanthoma after Jessner’s and trichloroacetic acid peel for actinic keratosis. Dermatol Surg. 2013;39:331-333.
- Wolfe CM, Green WH, Cognetta AB, et al. Multiple squamous cell carcinomas and eruptive keratoacanthomas in an arc welder. Dermatol Surg. 2013;39:328-330.
- Kluger N, Phan A, Debarbieux S, et al. Skin cancers arising in tattoos: coincidental or not? Dermatology. 2008;217:219-221.
- Mays R, Curry J, Kim K, et al. Eruptive squamous cell carcinomas after vemurafenib therapy. J Cutan Med Surg. 2013;17:419-422.
- Riahi RR, Cohen PR. Clinical manifestations of cutaneous metastases: a review with special emphasis on cutaneous metastases mimicking keratoacanthoma. Am J Clin Dermatol. 2012;13:103-112.
- Savoia P, Fava P, Nardò T, et al. Skin metastases of malignant melanoma: a clinical and prognostic study. Melanoma Res. 2009;19:321-326.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma. J Am Acad Dermatol. 1990;22:19-26.
- Behzad M, Michl C, Pfützner W. Multiple eruptive keratoacanthomas associated with myelodysplastic syndrome. J Dtsch Dermatol Ges. 2012;10:359-360.
- Cheung WL, Patel RR, Leonard A, et al. Amelanotic melanoma: a detailed morphologic analysis with clinicopathologic correlation of 75 cases. J Cutan Pathol. 2012;39:33-39.
- Ferrari A, Piccolo D, Fargnoli MC, et al. Cutaneous amelanotic melanoma metastasis and dermatofibromas showing a dotted vascular pattern. Acta Dermato Venereologica. 2004;84:164-165.
- Mohr B, Fernandez MP, Krejci-Manwaring J. Eruptive keratoacanthoma after Jessner’s and trichloroacetic acid peel for actinic keratosis. Dermatol Surg. 2013;39:331-333.
- Wolfe CM, Green WH, Cognetta AB, et al. Multiple squamous cell carcinomas and eruptive keratoacanthomas in an arc welder. Dermatol Surg. 2013;39:328-330.
- Kluger N, Phan A, Debarbieux S, et al. Skin cancers arising in tattoos: coincidental or not? Dermatology. 2008;217:219-221.
- Mays R, Curry J, Kim K, et al. Eruptive squamous cell carcinomas after vemurafenib therapy. J Cutan Med Surg. 2013;17:419-422.
- Riahi RR, Cohen PR. Clinical manifestations of cutaneous metastases: a review with special emphasis on cutaneous metastases mimicking keratoacanthoma. Am J Clin Dermatol. 2012;13:103-112.
Practice Points
- Cutaneous metastatic melanoma can have variable clinical presentations.
- Patients with a history of melanoma should be monitored closely with a low threshold for biopsy of new skin lesions.