User login
56-year-old woman • worsening pain in left upper arm • influenza vaccination in the arm a few days prior to pain onset • Dx?
THE CASE
A 56-year-old woman presented with a 3-day complaint of worsening left upper arm pain. She denied having any specific initiating factors but reported receiving an influenza vaccination in the arm a few days prior to the onset of pain. The patient did not have any associated numbness or tingling in the arm. She reported that the pain was worse with movement—especially abduction. The patient reported taking an over-the-counter nonsteroidal anti-inflammatory drug (NSAID) without much relief.
On physical examination, the patient had difficulty with active range of motion and had erythema, swelling, and tenderness to palpation along the subacromial space and the proximal deltoid. Further examination of the shoulder revealed a positive Neer Impingement Test and a positive Hawkins–Kennedy Test. (For more on these tests, visit “MSK Clinic: Evaluating shoulder pain using IPASS.”). The patient demonstrated full passive range of motion, but her pain was exacerbated with abduction.
THE DIAGNOSIS
In light of the soft-tissue findings and the absence of trauma, magnetic resonance imaging (MRI), rather than an x-ray, of the upper extremity was ordered. Imaging revealed subacromial subdeltoid bursal inflammation (FIGURE).

DISCUSSION
Shoulder injury related to vaccine administration (SIRVA) is the result of accidental injection of a vaccine into the tissue lying underneath the deltoid muscle or joint space, leading to a suspected immune-mediated inflammatory reaction.
A report from the National Vaccine Advisory Committee of the US Department of Health & Human Services showed an increase in the number of reported cases of SIRVA (59 reported cases in 2011-2014 and 202 cases reported in 2016).1 Additionally, in 2016 more than $29 million was awarded in compensation to patients with SIRVA.1,2 In a 2011 report, an Institute of Medicine committee found convincing evidence of a causal relationship between injection of vaccine, independent of the antigen involved, and deltoid bursitis, or frozen shoulder, characterized by shoulder pain and loss of motion.3
A review of 13 cases revealed that 50% of the patients reported pain immediately after the injection and 90% had developed pain within 24 hours.2 On physical exam, a limited range of motion and pain were the most common findings, while weakness and sensory changes were uncommon. In some cases, the pain lasted several years and 30% of the patients required surgery. Forty-six percent of the patients reported apprehension concerning the administration of the vaccine, specifically that the injection was administered “too high” into the deltoid.2
In the review of cases, routine x-rays of the shoulder did not provide beneficial diagnostic information; however, when an MRI was performed, it revealed fluid collections in the deep deltoid or overlying the rotator cuff tendons; bursitis; tendonitis; and rotator cuff tears.2
Continue to: Management of SIRVA
Management of SIRVA
Management of SIRVA is similar to that of other shoulder injuries. Treatment may include icing the shoulder, NSAIDs, intra-articular steroid injections, and physical therapy. If conservative management does not resolve the patient’s pain and improve function, then a consult with an orthopedic surgeon is recommended to determine if surgical intervention is required.
Another case report from Japan reported that a 45-year-old woman developed acute pain following a third injection of Cervarix, the prophylactic human papillomavirus-16/18 vaccine. An x-ray was ordered and was normal, but an MRI revealed acute subacromial bursitis. In an attempt to relieve the pain and improve her mobility, multiple cortisone injections were administered and physical therapy was performed. Despite the conservative treatment efforts, she continued to have pain and limited mobility in the shoulder 6 months following the onset of symptoms. As a result, the patient underwent arthroscopic synovectomy and subacromial decompression. One week following the surgery, the patient’s pain improved and at 1 year she had no pain and full range of motion.4
Prevention of SIRVA
By using appropriate techniques when administering intramuscular vaccinations, SIRVA can be prevented. The manufacturer recommended route of administration is based on studies showing maximum safety and immunogenicity, and should therefore be followed by the individual administering the vaccine.5 The Centers for Disease Control and Prevention recommends using a 22- to 25-gauge needle that is long enough to reach into the muscle and may range from ⅝" to 1½" depending on the patient’s weight.6 The vaccine should be injected at a 90° angle into the central and thickest portion of the deltoid muscle, about 2" below the acromion process and above the level of the axilla.5
Our patient’s outcome. The patient’s symptoms resolved within 10 days of receiving a steroid injection into the subacromial space. Although this case was the result of the influenza vaccine, any intramuscularly injected vaccine could lead to SIRVA.
THE TAKEAWAY
Inappropriate administration of routine intramuscularly injected vaccinations can lead to significant patient harm, including pain and disability. It is important for physicians to be aware of SIRVA and to be able to identify the signs and symptoms. Although an MRI of the shoulder is helpful in confirming the diagnosis, it is not necessary if the physician takes a thorough history and performs a comprehensive shoulder exam. Routine x-rays do not provide any beneficial clinical information.
CORRESPONDENCE
Bryan Farford, DO, Department of Family Medicine, Mayo Clinic, Davis Building, 4500 San Pablo Road South #358, Jacksonville, FL 32224; [email protected]
1. Nair N. Update on SIRVA National Vaccine Advisory Committee. U.S. Department of Health & Human Services. Health Resources and Services Administration (HRSA). www.hhs.gov/sites/default/files/Nair_Special%20Highlight_SIRVA%20remediated.pdf. Accessed January 14, 2020.
2. Atanasoff S, Ryan T, Lightfoot R, et al. Shoulder injury related to vaccine administration (SIRVA). Vaccine. 2010;28:8049-8052.
3. Institute of Medicine of the National Academies. Adverse Effects of Vaccines: Evidence and Causality. Washington DC: The National Academies Press; 2011.
4. Uchida S, Sakai A, Nakamura T. Subacromial bursitis following human papilloma virus vaccine misinjection. Vaccine. 2012;31:27-30.
5. Meissner HC. Shoulder injury related to vaccine administration reported more frequently. AAP News. September 1, 2017. www.aappublications.org/news/2017/09/01/IDSnapshot082917. Accessed January 14, 2020.
6. Immunization Action Coalition. How to administer intramuscular and subcutaneous vaccine injections to adults. https://www.immunize.org/catg.d/p2020a.pdf. Accessed January 14, 2020.
THE CASE
A 56-year-old woman presented with a 3-day complaint of worsening left upper arm pain. She denied having any specific initiating factors but reported receiving an influenza vaccination in the arm a few days prior to the onset of pain. The patient did not have any associated numbness or tingling in the arm. She reported that the pain was worse with movement—especially abduction. The patient reported taking an over-the-counter nonsteroidal anti-inflammatory drug (NSAID) without much relief.
On physical examination, the patient had difficulty with active range of motion and had erythema, swelling, and tenderness to palpation along the subacromial space and the proximal deltoid. Further examination of the shoulder revealed a positive Neer Impingement Test and a positive Hawkins–Kennedy Test. (For more on these tests, visit “MSK Clinic: Evaluating shoulder pain using IPASS.”). The patient demonstrated full passive range of motion, but her pain was exacerbated with abduction.
THE DIAGNOSIS
In light of the soft-tissue findings and the absence of trauma, magnetic resonance imaging (MRI), rather than an x-ray, of the upper extremity was ordered. Imaging revealed subacromial subdeltoid bursal inflammation (FIGURE).
DISCUSSION
Shoulder injury related to vaccine administration (SIRVA) is the result of accidental injection of a vaccine into the tissue lying underneath the deltoid muscle or joint space, leading to a suspected immune-mediated inflammatory reaction.
A report from the National Vaccine Advisory Committee of the US Department of Health & Human Services showed an increase in the number of reported cases of SIRVA (59 reported cases in 2011-2014 and 202 cases reported in 2016).1 Additionally, in 2016 more than $29 million was awarded in compensation to patients with SIRVA.1,2 In a 2011 report, an Institute of Medicine committee found convincing evidence of a causal relationship between injection of vaccine, independent of the antigen involved, and deltoid bursitis, or frozen shoulder, characterized by shoulder pain and loss of motion.3
A review of 13 cases revealed that 50% of the patients reported pain immediately after the injection and 90% had developed pain within 24 hours.2 On physical exam, a limited range of motion and pain were the most common findings, while weakness and sensory changes were uncommon. In some cases, the pain lasted several years and 30% of the patients required surgery. Forty-six percent of the patients reported apprehension concerning the administration of the vaccine, specifically that the injection was administered “too high” into the deltoid.2
In the review of cases, routine x-rays of the shoulder did not provide beneficial diagnostic information; however, when an MRI was performed, it revealed fluid collections in the deep deltoid or overlying the rotator cuff tendons; bursitis; tendonitis; and rotator cuff tears.2
Continue to: Management of SIRVA
Management of SIRVA
Management of SIRVA is similar to that of other shoulder injuries. Treatment may include icing the shoulder, NSAIDs, intra-articular steroid injections, and physical therapy. If conservative management does not resolve the patient’s pain and improve function, then a consult with an orthopedic surgeon is recommended to determine if surgical intervention is required.
Another case report from Japan reported that a 45-year-old woman developed acute pain following a third injection of Cervarix, the prophylactic human papillomavirus-16/18 vaccine. An x-ray was ordered and was normal, but an MRI revealed acute subacromial bursitis. In an attempt to relieve the pain and improve her mobility, multiple cortisone injections were administered and physical therapy was performed. Despite the conservative treatment efforts, she continued to have pain and limited mobility in the shoulder 6 months following the onset of symptoms. As a result, the patient underwent arthroscopic synovectomy and subacromial decompression. One week following the surgery, the patient’s pain improved and at 1 year she had no pain and full range of motion.4
Prevention of SIRVA
By using appropriate techniques when administering intramuscular vaccinations, SIRVA can be prevented. The manufacturer recommended route of administration is based on studies showing maximum safety and immunogenicity, and should therefore be followed by the individual administering the vaccine.5 The Centers for Disease Control and Prevention recommends using a 22- to 25-gauge needle that is long enough to reach into the muscle and may range from ⅝" to 1½" depending on the patient’s weight.6 The vaccine should be injected at a 90° angle into the central and thickest portion of the deltoid muscle, about 2" below the acromion process and above the level of the axilla.5
Our patient’s outcome. The patient’s symptoms resolved within 10 days of receiving a steroid injection into the subacromial space. Although this case was the result of the influenza vaccine, any intramuscularly injected vaccine could lead to SIRVA.
THE TAKEAWAY
Inappropriate administration of routine intramuscularly injected vaccinations can lead to significant patient harm, including pain and disability. It is important for physicians to be aware of SIRVA and to be able to identify the signs and symptoms. Although an MRI of the shoulder is helpful in confirming the diagnosis, it is not necessary if the physician takes a thorough history and performs a comprehensive shoulder exam. Routine x-rays do not provide any beneficial clinical information.
CORRESPONDENCE
Bryan Farford, DO, Department of Family Medicine, Mayo Clinic, Davis Building, 4500 San Pablo Road South #358, Jacksonville, FL 32224; [email protected]
THE CASE
A 56-year-old woman presented with a 3-day complaint of worsening left upper arm pain. She denied having any specific initiating factors but reported receiving an influenza vaccination in the arm a few days prior to the onset of pain. The patient did not have any associated numbness or tingling in the arm. She reported that the pain was worse with movement—especially abduction. The patient reported taking an over-the-counter nonsteroidal anti-inflammatory drug (NSAID) without much relief.
On physical examination, the patient had difficulty with active range of motion and had erythema, swelling, and tenderness to palpation along the subacromial space and the proximal deltoid. Further examination of the shoulder revealed a positive Neer Impingement Test and a positive Hawkins–Kennedy Test. (For more on these tests, visit “MSK Clinic: Evaluating shoulder pain using IPASS.”). The patient demonstrated full passive range of motion, but her pain was exacerbated with abduction.
THE DIAGNOSIS
In light of the soft-tissue findings and the absence of trauma, magnetic resonance imaging (MRI), rather than an x-ray, of the upper extremity was ordered. Imaging revealed subacromial subdeltoid bursal inflammation (FIGURE).
DISCUSSION
Shoulder injury related to vaccine administration (SIRVA) is the result of accidental injection of a vaccine into the tissue lying underneath the deltoid muscle or joint space, leading to a suspected immune-mediated inflammatory reaction.
A report from the National Vaccine Advisory Committee of the US Department of Health & Human Services showed an increase in the number of reported cases of SIRVA (59 reported cases in 2011-2014 and 202 cases reported in 2016).1 Additionally, in 2016 more than $29 million was awarded in compensation to patients with SIRVA.1,2 In a 2011 report, an Institute of Medicine committee found convincing evidence of a causal relationship between injection of vaccine, independent of the antigen involved, and deltoid bursitis, or frozen shoulder, characterized by shoulder pain and loss of motion.3
A review of 13 cases revealed that 50% of the patients reported pain immediately after the injection and 90% had developed pain within 24 hours.2 On physical exam, a limited range of motion and pain were the most common findings, while weakness and sensory changes were uncommon. In some cases, the pain lasted several years and 30% of the patients required surgery. Forty-six percent of the patients reported apprehension concerning the administration of the vaccine, specifically that the injection was administered “too high” into the deltoid.2
In the review of cases, routine x-rays of the shoulder did not provide beneficial diagnostic information; however, when an MRI was performed, it revealed fluid collections in the deep deltoid or overlying the rotator cuff tendons; bursitis; tendonitis; and rotator cuff tears.2
Continue to: Management of SIRVA
Management of SIRVA
Management of SIRVA is similar to that of other shoulder injuries. Treatment may include icing the shoulder, NSAIDs, intra-articular steroid injections, and physical therapy. If conservative management does not resolve the patient’s pain and improve function, then a consult with an orthopedic surgeon is recommended to determine if surgical intervention is required.
Another case report from Japan reported that a 45-year-old woman developed acute pain following a third injection of Cervarix, the prophylactic human papillomavirus-16/18 vaccine. An x-ray was ordered and was normal, but an MRI revealed acute subacromial bursitis. In an attempt to relieve the pain and improve her mobility, multiple cortisone injections were administered and physical therapy was performed. Despite the conservative treatment efforts, she continued to have pain and limited mobility in the shoulder 6 months following the onset of symptoms. As a result, the patient underwent arthroscopic synovectomy and subacromial decompression. One week following the surgery, the patient’s pain improved and at 1 year she had no pain and full range of motion.4
Prevention of SIRVA
By using appropriate techniques when administering intramuscular vaccinations, SIRVA can be prevented. The manufacturer recommended route of administration is based on studies showing maximum safety and immunogenicity, and should therefore be followed by the individual administering the vaccine.5 The Centers for Disease Control and Prevention recommends using a 22- to 25-gauge needle that is long enough to reach into the muscle and may range from ⅝" to 1½" depending on the patient’s weight.6 The vaccine should be injected at a 90° angle into the central and thickest portion of the deltoid muscle, about 2" below the acromion process and above the level of the axilla.5
Our patient’s outcome. The patient’s symptoms resolved within 10 days of receiving a steroid injection into the subacromial space. Although this case was the result of the influenza vaccine, any intramuscularly injected vaccine could lead to SIRVA.
THE TAKEAWAY
Inappropriate administration of routine intramuscularly injected vaccinations can lead to significant patient harm, including pain and disability. It is important for physicians to be aware of SIRVA and to be able to identify the signs and symptoms. Although an MRI of the shoulder is helpful in confirming the diagnosis, it is not necessary if the physician takes a thorough history and performs a comprehensive shoulder exam. Routine x-rays do not provide any beneficial clinical information.
CORRESPONDENCE
Bryan Farford, DO, Department of Family Medicine, Mayo Clinic, Davis Building, 4500 San Pablo Road South #358, Jacksonville, FL 32224; [email protected]
1. Nair N. Update on SIRVA National Vaccine Advisory Committee. U.S. Department of Health & Human Services. Health Resources and Services Administration (HRSA). www.hhs.gov/sites/default/files/Nair_Special%20Highlight_SIRVA%20remediated.pdf. Accessed January 14, 2020.
2. Atanasoff S, Ryan T, Lightfoot R, et al. Shoulder injury related to vaccine administration (SIRVA). Vaccine. 2010;28:8049-8052.
3. Institute of Medicine of the National Academies. Adverse Effects of Vaccines: Evidence and Causality. Washington DC: The National Academies Press; 2011.
4. Uchida S, Sakai A, Nakamura T. Subacromial bursitis following human papilloma virus vaccine misinjection. Vaccine. 2012;31:27-30.
5. Meissner HC. Shoulder injury related to vaccine administration reported more frequently. AAP News. September 1, 2017. www.aappublications.org/news/2017/09/01/IDSnapshot082917. Accessed January 14, 2020.
6. Immunization Action Coalition. How to administer intramuscular and subcutaneous vaccine injections to adults. https://www.immunize.org/catg.d/p2020a.pdf. Accessed January 14, 2020.
1. Nair N. Update on SIRVA National Vaccine Advisory Committee. U.S. Department of Health & Human Services. Health Resources and Services Administration (HRSA). www.hhs.gov/sites/default/files/Nair_Special%20Highlight_SIRVA%20remediated.pdf. Accessed January 14, 2020.
2. Atanasoff S, Ryan T, Lightfoot R, et al. Shoulder injury related to vaccine administration (SIRVA). Vaccine. 2010;28:8049-8052.
3. Institute of Medicine of the National Academies. Adverse Effects of Vaccines: Evidence and Causality. Washington DC: The National Academies Press; 2011.
4. Uchida S, Sakai A, Nakamura T. Subacromial bursitis following human papilloma virus vaccine misinjection. Vaccine. 2012;31:27-30.
5. Meissner HC. Shoulder injury related to vaccine administration reported more frequently. AAP News. September 1, 2017. www.aappublications.org/news/2017/09/01/IDSnapshot082917. Accessed January 14, 2020.
6. Immunization Action Coalition. How to administer intramuscular and subcutaneous vaccine injections to adults. https://www.immunize.org/catg.d/p2020a.pdf. Accessed January 14, 2020.

33-year-old man • flaccid paralysis in limbs • 30-lb weight loss • thyromegaly without nodules • Dx?
THE CASE
A 33-year-old Hispanic man with no significant past medical history presented to the emergency department with generalized flaccid paralysis in both arms and legs. Two days before, he had been working on a construction site in hot weather. The following day, he woke up with very little energy or strength to perform his daily activities, and he had pain in the inguinal area and both calves. He denied taking any medications or supplements.
The patient had complete muscle weakness and was unable to move his arms and legs. He reported dysphagia and an unintentional weight loss of 30 lb during the previous month.
On physical examination, the patient’s vital signs were within the normal range, and mild thyromegaly without nodules was present. Neurologic examination revealed decreased deep tendon reflexes with intact sensation. Muscle strength in his arms and legs was 0/5.
Initial laboratory test results included a potassium level of 2.2 mEq/L (normal range, 3.5–5 mEq/L) and normal acid-basic status that was confirmed by an arterial blood gas measurement. Serum magnesium was 1.6 mg/dL (normal range, 1.6–2.5 mg/dL); phosphorus, 1.9 mg/dL (normal range, 2.7–4.5 mg/dL); and random urinary potassium, 16 mEq/L (normal range, 25–125 mEq/L). An initial chest x-ray was normal, and an electrocardiogram showed a prolonged QT interval, flattening of the T wave, and a prominent U wave consistent with hypokalemia.
THE DIAGNOSIS
The initial clinical diagnosis was hypokalemic paralysis. The patient was treated with intravenous (IV) potassium chloride 40 mEq
Evaluation of the patient’s hypokalemia revealed the following: thyroid-stimulating hormone (TSH) level, < 0.01 microIU/mL (normal range, 0.27–4.2 microIU/mL); free T4 (thyroxine) level, 4.47 ng/dL (normal range, 0.08–1.70 ng/dL); total T3 (triiodothyronine) level, 17.5 ng/dL (normal range, 2.6–4.4 ng/dL).
The patient was diagnosed with hypokalemic periodic paralysis (HPP) secondary to thyrotoxicosis, also known as thyrotoxicosis periodic paralysis (TPP). His hyperthyroidism was treated with oral atenolol 25 mg/d and oral methimazole 10 mg tid.
Continue to: Within a few hours...
Within a few hours of this treatment, the patient experienced significant improvement in muscle strength and complete resolution of weakness in his arms and legs. Serial measurements of potassium levels normalized.
Further workup revealed that the patient’s thyroid-stimulating immunoglobulin (TSI) was 4.2 on the TSI index (normal, ≤ 1.3) and his thyroid peroxidase (TPO) antibody level was 133.4 IU/mL (normal, < 34 IU/mL). Ultrasonography showed decreased echogenicity of the thyroid gland, consistent with the acute phase of Hashimoto thyroiditis or Graves disease.
The patient was unaware that he had any thyroid disorder previously. He was a private-pay, undocumented immigrant and did not have a regular primary care physician. On discharge, he was referred to a local primary care physician as well as an endocrinologist. He was discharged on atenolol and methimazole.
DISCUSSION
A rare neuromuscular disorder known as periodic paralysis can be precipitated by a hypokalemic or hyperkalemic state; HPP is more common and can be either familial (a defect in the gene) or acquired (secondary to thyrotoxicosis; TPP).1,2 In both forms of periodic paralysis, patients present with hypokalemia and paralysis. Physicians need to look closely at thyroid lab test results so as not to miss the cause of the paralysis.
TPP is most commonly seen in Asian populations, and 95% of cases reported occur in males, despite the higher incidence of hyperthyroidism in females.3 TPP can be precipitated by emotional stress, steroid use, beta-adrenergic bronchodilators, heavy exercise, fasting, or high-carbohydrate meals.2-4 In our patient, heavy exercise and fasting likely were the triggers.
Continue to: The pathophysiology for the hypokalemia...
The pathophysiology for the hypokalemia in TPP is thought to involve the sodium/potassium–adenosine triphosphatase (Na+/K+–ATPase) pump. This pump activity is increased in skeletal muscle and platelets in patients with TPP vs patients with thyrotoxicosis alone.3,5
The role of Hashimoto thyrotoxicosis. Most acquired cases of TPP are mainly secondary to Graves disease with elevated levels of TSI and mildly elevated or normal levels of TPO. In this case, the patient was in the acute phase of Hashimoto thyrotoxicosis (“hashitoxicosis”) with elevated levels of TPO and only mildly elevated TSI.Imaging studies to support the diagnosis, such as a thyroid uptake scan or ultrasonography, are not necessary to determine the cause of thyrotoxicosis. In the absence of test results for TPO and TSI antibodies, however, a scan can be helpful.6,7
Treatment of TPP consists of early recognition and supportive management by correcting the potassium deficit; failure to do so could cause severe complications, such as respiratory failure and psychosis.8 Because of the risk for rebound hyperkalemia, serial potassium levels must be measured until a stable potassium level in the normal range is achieved.
Nonselective beta-blockers, such as propranolol (3 mg/kg) 4 times per day, have been reported to ameliorate the periodic paralysis and prevent rebound hyperkalemia.9 Finally, restoring a euthyroid state will prevent the patient from experiencing future attacks.
THE TAKEAWAY
Few medical conditions result in complete muscle paralysis in a matter of hours. Clinicians should consider the possibility of TPP in any patient who presents with acute onset of paralysis.
CORRESPONDENCE
Jorge Luis Chavez, MD; 8405 E. San Pedro Drive, Scottsdale, AZ 85258; [email protected].
1. Fontaine B. Periodic paralysis. Adv Genet. 2008;63:3-23.
2. Ober KP. Thyrotoxic periodic paralysis in the United States. Report of 7 cases and review of the literature. Medicine (Baltimore).1992;71:109-120.
3. Lin YF, Wu CC, Pei D, et al. Diagnosing thyrotoxic periodic paralysis in the ED. Am J Emerg Med. 2003;21:339-342.
4. Yu TS, Tseng CF, Chuang YY, et al. Potassium chloride supplementation alone may not improve hypokalemia in thyrotoxic hypokalemic periodic paralysis. J Emerg Med. 2007;32:263-265.
5. Chan A, Shinde R, Chow CC, et al. In vivo and in vitro sodium pump activity in subjects with thyrotoxic periodic paralysis. BMJ. 1991;303:1096-1099.
6. Harsch IA, Hahn EG, Strobel D. Hashitoxicosis—three cases and a review of the literature. Eur Endocrinol. 2008;4:70-72. 7. Pou Ucha JL. Imaging in hyperthyroidism. In: Díaz-Soto G, ed. Thyroid Disorders: Focus on Hyperthyroidism. InTechOpen; 2014. www.intechopen.com/books/thyroid-disorders-focus-on-hyperthyroidism/imaging-in-hyperthyroidism. Accessed January 14, 2020.
8. Abbasi B, Sharif Z, Sprabery LR. Hypokalemic thyrotoxic periodic paralysis with thyrotoxic psychosis and hypercapnic respiratory failure. Am J Med Sci. 2010;340:147-153.
9. Lin SH, Lin YF. Propranolol rapidly reverses paralysis, hypokalemia, and hypophosphatemia in thyrotoxic periodic paralysis. Am J Kidney Dis. 2001;37:620-623.
THE CASE
A 33-year-old Hispanic man with no significant past medical history presented to the emergency department with generalized flaccid paralysis in both arms and legs. Two days before, he had been working on a construction site in hot weather. The following day, he woke up with very little energy or strength to perform his daily activities, and he had pain in the inguinal area and both calves. He denied taking any medications or supplements.
The patient had complete muscle weakness and was unable to move his arms and legs. He reported dysphagia and an unintentional weight loss of 30 lb during the previous month.
On physical examination, the patient’s vital signs were within the normal range, and mild thyromegaly without nodules was present. Neurologic examination revealed decreased deep tendon reflexes with intact sensation. Muscle strength in his arms and legs was 0/5.
Initial laboratory test results included a potassium level of 2.2 mEq/L (normal range, 3.5–5 mEq/L) and normal acid-basic status that was confirmed by an arterial blood gas measurement. Serum magnesium was 1.6 mg/dL (normal range, 1.6–2.5 mg/dL); phosphorus, 1.9 mg/dL (normal range, 2.7–4.5 mg/dL); and random urinary potassium, 16 mEq/L (normal range, 25–125 mEq/L). An initial chest x-ray was normal, and an electrocardiogram showed a prolonged QT interval, flattening of the T wave, and a prominent U wave consistent with hypokalemia.
THE DIAGNOSIS
The initial clinical diagnosis was hypokalemic paralysis. The patient was treated with intravenous (IV) potassium chloride 40 mEq
Evaluation of the patient’s hypokalemia revealed the following: thyroid-stimulating hormone (TSH) level, < 0.01 microIU/mL (normal range, 0.27–4.2 microIU/mL); free T4 (thyroxine) level, 4.47 ng/dL (normal range, 0.08–1.70 ng/dL); total T3 (triiodothyronine) level, 17.5 ng/dL (normal range, 2.6–4.4 ng/dL).
The patient was diagnosed with hypokalemic periodic paralysis (HPP) secondary to thyrotoxicosis, also known as thyrotoxicosis periodic paralysis (TPP). His hyperthyroidism was treated with oral atenolol 25 mg/d and oral methimazole 10 mg tid.
Continue to: Within a few hours...
Within a few hours of this treatment, the patient experienced significant improvement in muscle strength and complete resolution of weakness in his arms and legs. Serial measurements of potassium levels normalized.
Further workup revealed that the patient’s thyroid-stimulating immunoglobulin (TSI) was 4.2 on the TSI index (normal, ≤ 1.3) and his thyroid peroxidase (TPO) antibody level was 133.4 IU/mL (normal, < 34 IU/mL). Ultrasonography showed decreased echogenicity of the thyroid gland, consistent with the acute phase of Hashimoto thyroiditis or Graves disease.
The patient was unaware that he had any thyroid disorder previously. He was a private-pay, undocumented immigrant and did not have a regular primary care physician. On discharge, he was referred to a local primary care physician as well as an endocrinologist. He was discharged on atenolol and methimazole.
DISCUSSION
A rare neuromuscular disorder known as periodic paralysis can be precipitated by a hypokalemic or hyperkalemic state; HPP is more common and can be either familial (a defect in the gene) or acquired (secondary to thyrotoxicosis; TPP).1,2 In both forms of periodic paralysis, patients present with hypokalemia and paralysis. Physicians need to look closely at thyroid lab test results so as not to miss the cause of the paralysis.
TPP is most commonly seen in Asian populations, and 95% of cases reported occur in males, despite the higher incidence of hyperthyroidism in females.3 TPP can be precipitated by emotional stress, steroid use, beta-adrenergic bronchodilators, heavy exercise, fasting, or high-carbohydrate meals.2-4 In our patient, heavy exercise and fasting likely were the triggers.
Continue to: The pathophysiology for the hypokalemia...
The pathophysiology for the hypokalemia in TPP is thought to involve the sodium/potassium–adenosine triphosphatase (Na+/K+–ATPase) pump. This pump activity is increased in skeletal muscle and platelets in patients with TPP vs patients with thyrotoxicosis alone.3,5
The role of Hashimoto thyrotoxicosis. Most acquired cases of TPP are mainly secondary to Graves disease with elevated levels of TSI and mildly elevated or normal levels of TPO. In this case, the patient was in the acute phase of Hashimoto thyrotoxicosis (“hashitoxicosis”) with elevated levels of TPO and only mildly elevated TSI.Imaging studies to support the diagnosis, such as a thyroid uptake scan or ultrasonography, are not necessary to determine the cause of thyrotoxicosis. In the absence of test results for TPO and TSI antibodies, however, a scan can be helpful.6,7
Treatment of TPP consists of early recognition and supportive management by correcting the potassium deficit; failure to do so could cause severe complications, such as respiratory failure and psychosis.8 Because of the risk for rebound hyperkalemia, serial potassium levels must be measured until a stable potassium level in the normal range is achieved.
Nonselective beta-blockers, such as propranolol (3 mg/kg) 4 times per day, have been reported to ameliorate the periodic paralysis and prevent rebound hyperkalemia.9 Finally, restoring a euthyroid state will prevent the patient from experiencing future attacks.
THE TAKEAWAY
Few medical conditions result in complete muscle paralysis in a matter of hours. Clinicians should consider the possibility of TPP in any patient who presents with acute onset of paralysis.
CORRESPONDENCE
Jorge Luis Chavez, MD; 8405 E. San Pedro Drive, Scottsdale, AZ 85258; [email protected].
THE CASE
A 33-year-old Hispanic man with no significant past medical history presented to the emergency department with generalized flaccid paralysis in both arms and legs. Two days before, he had been working on a construction site in hot weather. The following day, he woke up with very little energy or strength to perform his daily activities, and he had pain in the inguinal area and both calves. He denied taking any medications or supplements.
The patient had complete muscle weakness and was unable to move his arms and legs. He reported dysphagia and an unintentional weight loss of 30 lb during the previous month.
On physical examination, the patient’s vital signs were within the normal range, and mild thyromegaly without nodules was present. Neurologic examination revealed decreased deep tendon reflexes with intact sensation. Muscle strength in his arms and legs was 0/5.
Initial laboratory test results included a potassium level of 2.2 mEq/L (normal range, 3.5–5 mEq/L) and normal acid-basic status that was confirmed by an arterial blood gas measurement. Serum magnesium was 1.6 mg/dL (normal range, 1.6–2.5 mg/dL); phosphorus, 1.9 mg/dL (normal range, 2.7–4.5 mg/dL); and random urinary potassium, 16 mEq/L (normal range, 25–125 mEq/L). An initial chest x-ray was normal, and an electrocardiogram showed a prolonged QT interval, flattening of the T wave, and a prominent U wave consistent with hypokalemia.
THE DIAGNOSIS
The initial clinical diagnosis was hypokalemic paralysis. The patient was treated with intravenous (IV) potassium chloride 40 mEq
Evaluation of the patient’s hypokalemia revealed the following: thyroid-stimulating hormone (TSH) level, < 0.01 microIU/mL (normal range, 0.27–4.2 microIU/mL); free T4 (thyroxine) level, 4.47 ng/dL (normal range, 0.08–1.70 ng/dL); total T3 (triiodothyronine) level, 17.5 ng/dL (normal range, 2.6–4.4 ng/dL).
The patient was diagnosed with hypokalemic periodic paralysis (HPP) secondary to thyrotoxicosis, also known as thyrotoxicosis periodic paralysis (TPP). His hyperthyroidism was treated with oral atenolol 25 mg/d and oral methimazole 10 mg tid.
Continue to: Within a few hours...
Within a few hours of this treatment, the patient experienced significant improvement in muscle strength and complete resolution of weakness in his arms and legs. Serial measurements of potassium levels normalized.
Further workup revealed that the patient’s thyroid-stimulating immunoglobulin (TSI) was 4.2 on the TSI index (normal, ≤ 1.3) and his thyroid peroxidase (TPO) antibody level was 133.4 IU/mL (normal, < 34 IU/mL). Ultrasonography showed decreased echogenicity of the thyroid gland, consistent with the acute phase of Hashimoto thyroiditis or Graves disease.
The patient was unaware that he had any thyroid disorder previously. He was a private-pay, undocumented immigrant and did not have a regular primary care physician. On discharge, he was referred to a local primary care physician as well as an endocrinologist. He was discharged on atenolol and methimazole.
DISCUSSION
A rare neuromuscular disorder known as periodic paralysis can be precipitated by a hypokalemic or hyperkalemic state; HPP is more common and can be either familial (a defect in the gene) or acquired (secondary to thyrotoxicosis; TPP).1,2 In both forms of periodic paralysis, patients present with hypokalemia and paralysis. Physicians need to look closely at thyroid lab test results so as not to miss the cause of the paralysis.
TPP is most commonly seen in Asian populations, and 95% of cases reported occur in males, despite the higher incidence of hyperthyroidism in females.3 TPP can be precipitated by emotional stress, steroid use, beta-adrenergic bronchodilators, heavy exercise, fasting, or high-carbohydrate meals.2-4 In our patient, heavy exercise and fasting likely were the triggers.
Continue to: The pathophysiology for the hypokalemia...
The pathophysiology for the hypokalemia in TPP is thought to involve the sodium/potassium–adenosine triphosphatase (Na+/K+–ATPase) pump. This pump activity is increased in skeletal muscle and platelets in patients with TPP vs patients with thyrotoxicosis alone.3,5
The role of Hashimoto thyrotoxicosis. Most acquired cases of TPP are mainly secondary to Graves disease with elevated levels of TSI and mildly elevated or normal levels of TPO. In this case, the patient was in the acute phase of Hashimoto thyrotoxicosis (“hashitoxicosis”) with elevated levels of TPO and only mildly elevated TSI.Imaging studies to support the diagnosis, such as a thyroid uptake scan or ultrasonography, are not necessary to determine the cause of thyrotoxicosis. In the absence of test results for TPO and TSI antibodies, however, a scan can be helpful.6,7
Treatment of TPP consists of early recognition and supportive management by correcting the potassium deficit; failure to do so could cause severe complications, such as respiratory failure and psychosis.8 Because of the risk for rebound hyperkalemia, serial potassium levels must be measured until a stable potassium level in the normal range is achieved.
Nonselective beta-blockers, such as propranolol (3 mg/kg) 4 times per day, have been reported to ameliorate the periodic paralysis and prevent rebound hyperkalemia.9 Finally, restoring a euthyroid state will prevent the patient from experiencing future attacks.
THE TAKEAWAY
Few medical conditions result in complete muscle paralysis in a matter of hours. Clinicians should consider the possibility of TPP in any patient who presents with acute onset of paralysis.
CORRESPONDENCE
Jorge Luis Chavez, MD; 8405 E. San Pedro Drive, Scottsdale, AZ 85258; [email protected].
1. Fontaine B. Periodic paralysis. Adv Genet. 2008;63:3-23.
2. Ober KP. Thyrotoxic periodic paralysis in the United States. Report of 7 cases and review of the literature. Medicine (Baltimore).1992;71:109-120.
3. Lin YF, Wu CC, Pei D, et al. Diagnosing thyrotoxic periodic paralysis in the ED. Am J Emerg Med. 2003;21:339-342.
4. Yu TS, Tseng CF, Chuang YY, et al. Potassium chloride supplementation alone may not improve hypokalemia in thyrotoxic hypokalemic periodic paralysis. J Emerg Med. 2007;32:263-265.
5. Chan A, Shinde R, Chow CC, et al. In vivo and in vitro sodium pump activity in subjects with thyrotoxic periodic paralysis. BMJ. 1991;303:1096-1099.
6. Harsch IA, Hahn EG, Strobel D. Hashitoxicosis—three cases and a review of the literature. Eur Endocrinol. 2008;4:70-72. 7. Pou Ucha JL. Imaging in hyperthyroidism. In: Díaz-Soto G, ed. Thyroid Disorders: Focus on Hyperthyroidism. InTechOpen; 2014. www.intechopen.com/books/thyroid-disorders-focus-on-hyperthyroidism/imaging-in-hyperthyroidism. Accessed January 14, 2020.
8. Abbasi B, Sharif Z, Sprabery LR. Hypokalemic thyrotoxic periodic paralysis with thyrotoxic psychosis and hypercapnic respiratory failure. Am J Med Sci. 2010;340:147-153.
9. Lin SH, Lin YF. Propranolol rapidly reverses paralysis, hypokalemia, and hypophosphatemia in thyrotoxic periodic paralysis. Am J Kidney Dis. 2001;37:620-623.
1. Fontaine B. Periodic paralysis. Adv Genet. 2008;63:3-23.
2. Ober KP. Thyrotoxic periodic paralysis in the United States. Report of 7 cases and review of the literature. Medicine (Baltimore).1992;71:109-120.
3. Lin YF, Wu CC, Pei D, et al. Diagnosing thyrotoxic periodic paralysis in the ED. Am J Emerg Med. 2003;21:339-342.
4. Yu TS, Tseng CF, Chuang YY, et al. Potassium chloride supplementation alone may not improve hypokalemia in thyrotoxic hypokalemic periodic paralysis. J Emerg Med. 2007;32:263-265.
5. Chan A, Shinde R, Chow CC, et al. In vivo and in vitro sodium pump activity in subjects with thyrotoxic periodic paralysis. BMJ. 1991;303:1096-1099.
6. Harsch IA, Hahn EG, Strobel D. Hashitoxicosis—three cases and a review of the literature. Eur Endocrinol. 2008;4:70-72. 7. Pou Ucha JL. Imaging in hyperthyroidism. In: Díaz-Soto G, ed. Thyroid Disorders: Focus on Hyperthyroidism. InTechOpen; 2014. www.intechopen.com/books/thyroid-disorders-focus-on-hyperthyroidism/imaging-in-hyperthyroidism. Accessed January 14, 2020.
8. Abbasi B, Sharif Z, Sprabery LR. Hypokalemic thyrotoxic periodic paralysis with thyrotoxic psychosis and hypercapnic respiratory failure. Am J Med Sci. 2010;340:147-153.
9. Lin SH, Lin YF. Propranolol rapidly reverses paralysis, hypokalemia, and hypophosphatemia in thyrotoxic periodic paralysis. Am J Kidney Dis. 2001;37:620-623.

Nonuremic Calciphylaxis Triggered by Rapid Weight Loss and Hypotension
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
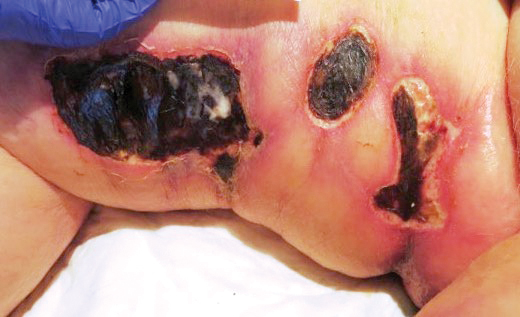
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).

Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
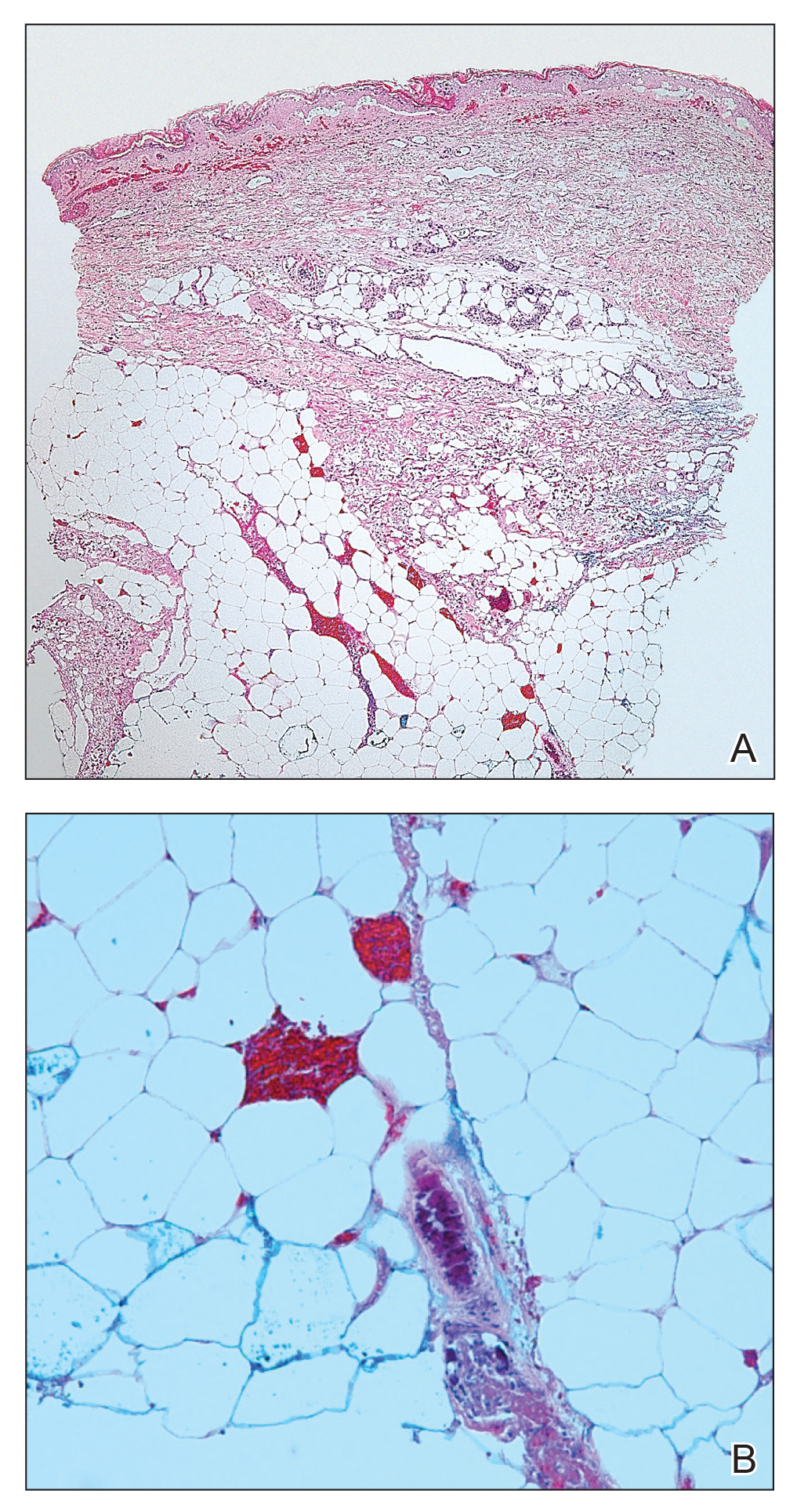
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).
Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).
Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
Practice Points
- Calciphylaxis is a potentially fatal disease caused by metastatic calcification of cutaneous small- and medium-sized blood vessels leading to ischemia and necrosis.
- Calciphylaxis most commonly is seen in patients with renal disease requiring dialysis, but it also may be triggered by nonuremic causes in patients with known risk factors for calciphylaxis.
- Risk factors for calciphylaxis include female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.
- The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature.
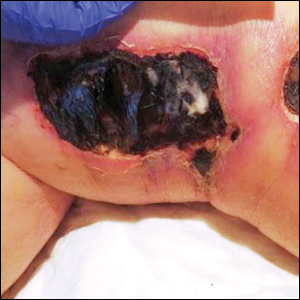
Scleromyxedema in a Patient With Thyroid Disease: An Atypical Case or a Case for Revised Criteria?
Scleromyxedema (SM) is a generalized papular and sclerodermoid form of lichen myxedematosus (LM), commonly referred to as papular mucinosis. It is a rare progressive disease of unknown etiology with systemic manifestations that cause serious morbidity and mortality. Diagnostic criteria were initially created by Montgomery and Underwood1 in 1953 and revised by Rongioletti and Rebora2 in 2001 as follows: (1) generalized papular and sclerodermoid eruption; (2) histologic triad of mucin deposition, fibroblast proliferation, and fibrosis; (3) monoclonal gammopathy; and (4) absence of thyroid disease. There are several reports of LM in association with hypothyroidism, most of which can be characterized as atypical.3-8 We present a case of SM in a patient with Hashimoto thyroiditis and propose that the presence of thyroid disease should not preclude the diagnosis of SM.
Case Report
A 44-year-old woman presented with a progressive eruption of thickened skin and papules spanning many months. The papules ranged from flesh colored to erythematous and covered more than 80% of the body surface area, most notably involving the face, neck, ears, arms, chest, abdomen, and thighs (Figures 1A and 2A). Review of systems was notable for pruritus, muscle pain but no weakness, dysphagia, and constipation. Her medical history included childhood atopic dermatitis and Hashimoto thyroiditis. Hypothyroidism was diagnosed with support of a thyroid ultrasound and thyroid peroxidase antibodies. It was treated with oral levothyroxine for 2 years prior to the skin eruption. Thyroid biopsy was not performed. Her thyroid-stimulating hormone levels notably fluctuated in the year prior to presentation despite close clinical and laboratory monitoring by an endocrinologist. Laboratory results are summarized in Table 1. Both skin and muscle9 biopsies were consistent with SM (Figure 3) and are summarized in Table 1.
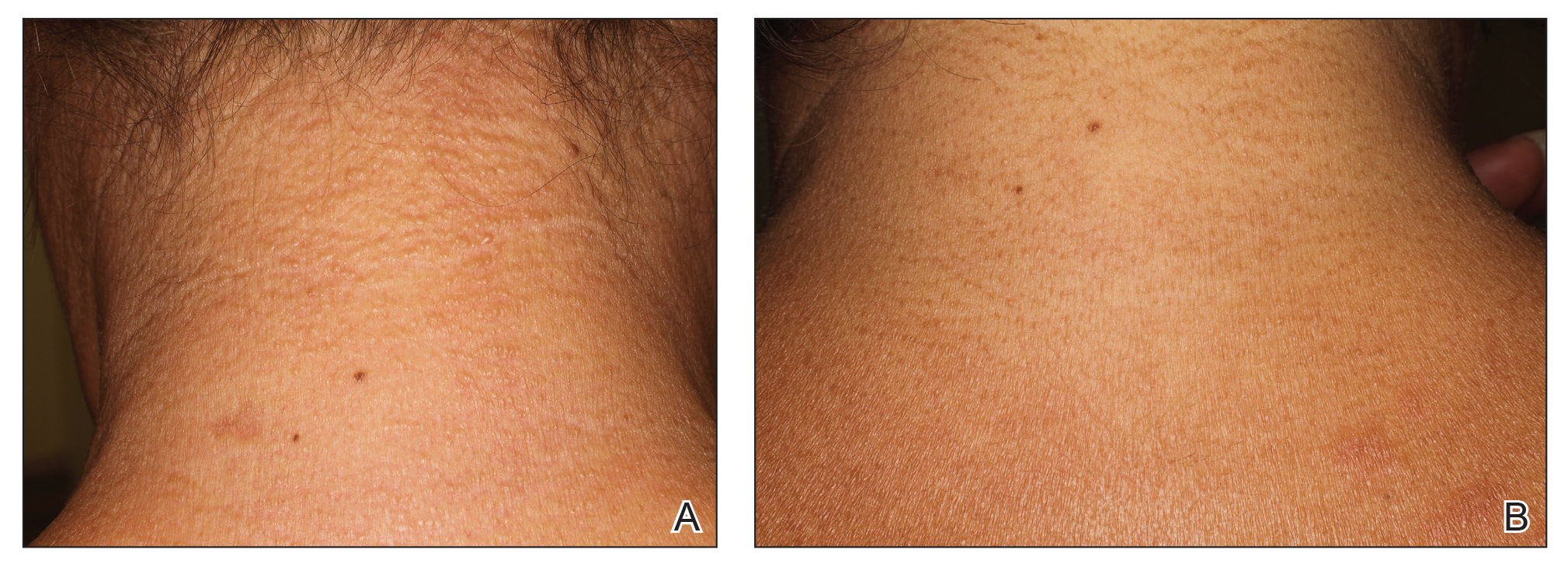

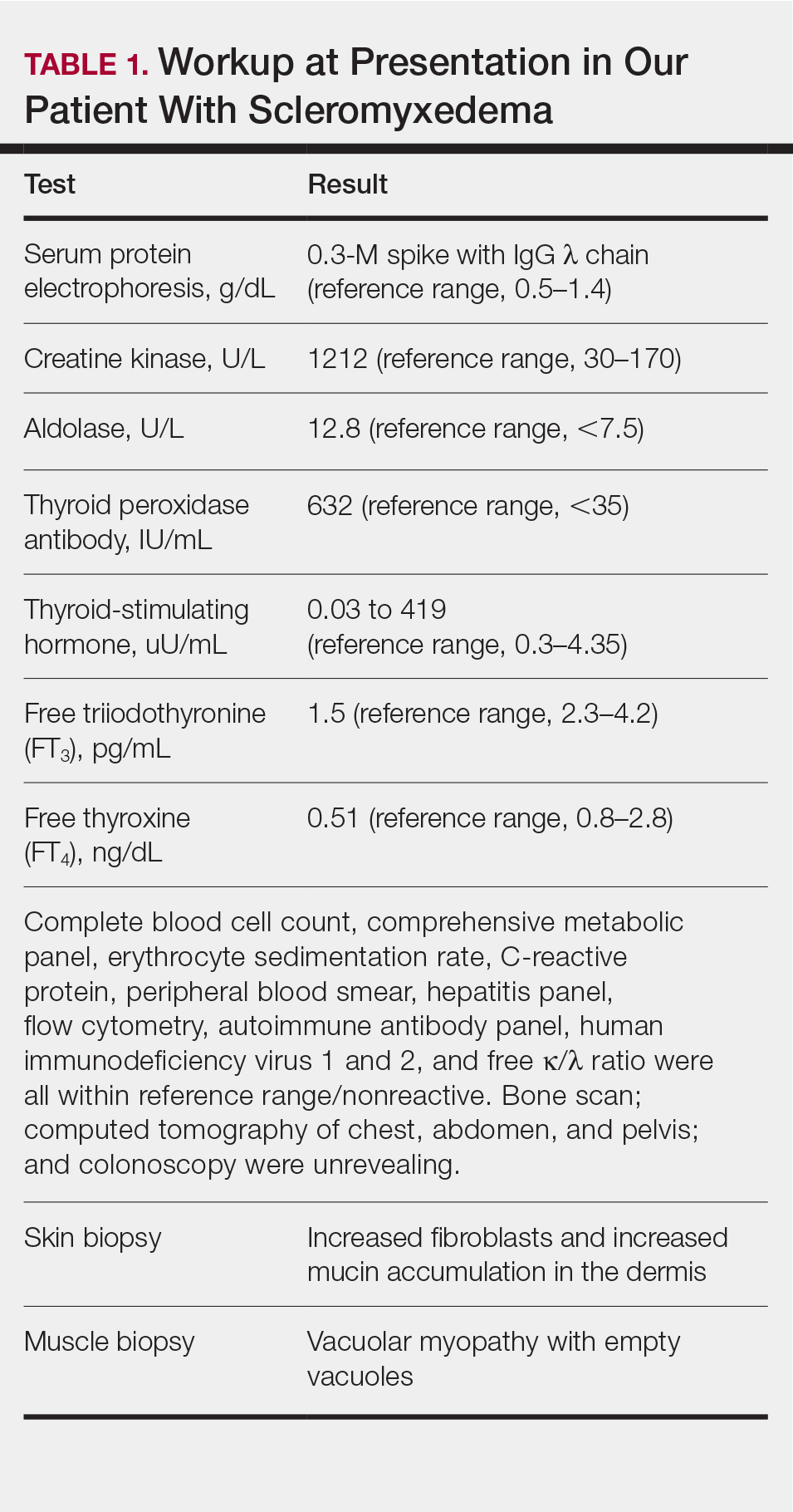
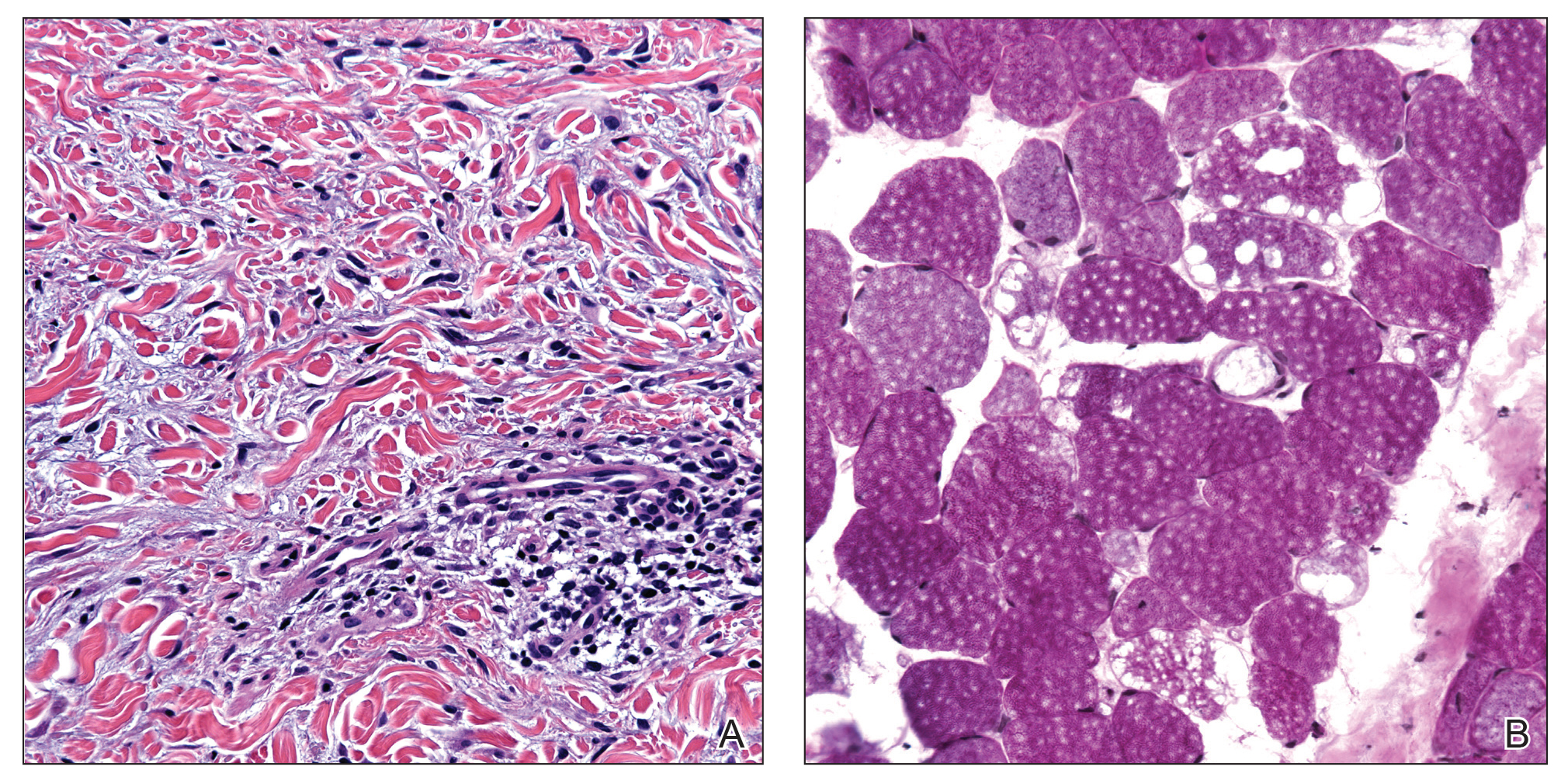
Shortly after presentation to our clinic the patient developed acute concerns of confusion and muscle weakness. She was admitted for further inpatient management due to concern for dermato-neuro syndrome, a rare but potentially fatal decline in neurological status that can progress to coma and death, rather than myxedema coma. On admission, a thyroid function test showed subclinical hypothyroidism with a thyroid-stimulating hormone level of 6.35 uU/mL (reference range, 0.3–4.35 uU/mL) and free thyroxine (FT4) level of 1.5 ng/dL (reference range, 0.8–2.8 ng/dL). While hospitalized she was started on intravenous levothyroxine, systemic steroids, and a course of intravenous immunoglobulin (IVIg) treatment consisting of 2 g/kg divided over 5 days. On this regimen, her mental status quickly returned to baseline and other symptoms improved, including the skin eruption (Figures 1B and 2B). She has been maintained on lenalidomide 25 mg/d for the first 3 weeks of each month as well as monthly IVIg infusions. Her thyroid levels have persistently fluctuated despite intramuscular levothyroxine dosing, but her skin has remained clear with continued SM-directed therapy.
Comment
Classification
Lichen myxedematosus is differentiated into localized and generalized forms. The former is limited to the skin and lacks monoclonal gammopathy. The latter, also known as SM, is associated with monoclonal gammopathy and systemic symptoms. Atypical LM is an umbrella term for intermediate cases.
Clinical Presentation
Skin manifestations of SM are described as 1- to 3-mm, firm, waxy, dome-shaped papules that commonly affect the hands, forearms, face, neck, trunk, and thighs. The surrounding skin may be reddish brown and edematous with evidence of skin thickening. Extracutaneous manifestations in SM are numerous and unpredictable. Any organ system can be involved, but gastrointestinal, rheumatologic, pulmonary, and cardiovascular complications are most common.10 A comprehensive multidisciplinary evaluation is necessary based on clinical symptoms and laboratory findings.
Management
Many treatments have been proposed for SM in case reports and case series. Prior treatments have had little success. Most recently, in one of the largest case series on SM, Rongioletti et al10 demonstrated IVIg to be a safe and effective treatment modality.
Differential Diagnosis
An important differential diagnosis is generalized myxedema, which is seen in long-standing hypothyroidism and may present with cutaneous mucinosis and systemic symptoms that resemble SM. Hypothyroid myxedema is associated with a widespread slowing of the body’s metabolic processes and deposition of mucin in various organs, including the skin, creating a generalized nonpitting edema. Classic clinical signs include macroglossia, periorbital puffiness, thick lips, and acral swelling. The skin tends to be cold, dry, and pale. Hair is characterized as being coarse, dry, and brittle with diffuse partial alopecia. Histologically, there is hyperkeratosis with follicular plugging and diffuse mucin and edema splaying between collagen fibers spanning the entire dermis.11 In contradistinction with SM, there is no fibroblast proliferation. The treatment is thyroid replacement therapy. Hyperthyroidism has distinct clinical and histologic changes. Clinically, there is moist and smooth skin with soft, fine, and sometimes alopecic hair. Graves disease, the most common cause of hyperthyroidism, is further characterized by Graves ophthalmopathy and pretibial myxedema, or pink to brown, raised, firm, indurated, asymmetric plaques most commonly affecting the shins. Histologically there is increased mucin in the lower to mid dermis without fibroblast proliferation. The epidermis can be hyperkeratotic, which will clinically correlate with verrucous lesions.12
Hypothyroid encephalopathy is a rare disorder that can cause a change in mental status. It is a steroid-responsive autoimmune process characterized by encephalopathy that is associated with cognitive impairment and psychiatric features. It is a diagnosis of exclusion and should be suspected in women with a history of autoimmune disease, especially antithyroid peroxidase antibodies, a negative infectious workup, and encephalitis with behavioral changes. Although typically highly responsive to systemic steroids, IVIg also has shown efficacy.13
Presence of Thyroid Disease
According to a PubMed search of articles indexed for MEDLINE using the terms scleromyxedema and lichen myxedematosus, there are 7 cases in the literature that potentially describe LM associated with hypothyroidism (Table 2).3-8 The majority of these cases lack monoclonal gammopathy; improved with thyroid replacement therapy; or had severely atypical clinical presentations, rendering them cases of atypical LM or atypical thyroid dermopathy.3-6 Macnab and Kenny7 presented a case of subclinical hypothyroidism with a generalized papular eruption, monoclonal gammopathy, and consistent histologic changes that responded to IVIg therapy. These findings are suggestive of SM, but limited to the current diagnostic criteria, the patient was diagnosed with atypical LM.7 Shenoy et al8 described 2 cases of LM with hypothyroidism. One patient had biopsy-proven SM that was responsive to IVIg as well as Hashimoto thyroiditis with delayed onset of monoclonal gammopathy. The second patient had a medical history of hypothyroidism and Hodgkin lymphoma with active rheumatoid arthritis and biopsy-proven LM that was responsive to systemic steroids.8
Current literature states that thyroid disorder precludes the diagnosis of SM. However, historic literature would suggest otherwise. Because of inconsistent reports and theories regarding the pathogenesis of various sclerodermoid and mucin deposition diseases, in 1953 Montgomery and Underwood1 sought to differentiate LM from scleroderma and generalized myxedema. They stressed clinical appearance and proposed diagnostic criteria for LM as generalized papular mucinosis in which “[n]o relation to disturbance of the thyroid or other endocrine glands is apparent,” whereas generalized myxedema was defined as a “[t]rue cutaneous myxedema, with diffuse edema and the usual commonly recognized changes” in patients with endocrine abnormalities.1 With this classification, the authors made a clear distinction between mucinosis caused by thyroid abnormalities and LM, which is not caused by a thyroid disorder. Since this original description was published, associations with monoclonal gammopathy and fibroblast proliferation have been made, ultimately culminating into the current 2001 criteria that incorporate the absence of thyroid disease.2

Conclusion
We believe our case is consistent with the classification initially proposed by Montgomery and Underwood1 and is strengthened with the more recent associations with monoclonal gammopathy and specific histopathologic findings. Although there is no definitive way to rule out myxedema coma or Hashimoto encephalopathy to describe our patient’s transient neurologic decline, her clinical symptoms, laboratory findings, and biopsy results all supported the diagnosis of SM. Furthermore, her response to SM-directed therapy, despite fluctuating thyroid function test results, also supported the diagnosis. In the setting of cutaneous mucinosis with conflicting findings for hypothyroid myxedema, LM should be ruled out. Given the features presented in this report and others, diagnostic criteria should allow for SM and thyroid dysfunction to be concurrent diagnoses. Most importantly, we believe it is essential to identify and diagnose SM in a timely manner to facilitate SM-directed therapy, namely IVIg, to potentially minimize the disease’s notable morbidity and mortality.
- Montgomery H, Underwood LJ. Lichen myxedematosus; differentiation from cutaneous myxedemas or mucoid states. J Invest Dermatol. 1953;20:213-236.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Archibald GC, Calvert HT. Hypothyroidsm and lichen myxedematosus. Arch Dermatol. 1977;113:684.
- Schaeffer D, Bruce S, Rosen T. Cutaneous mucinosis associated with thyroid dysfunction. Cutis. 1983;11:449-456.
- Martin-Ezquerra G, Sanchez-Regaña M, Massana-Gil J, et al. Papular mucinosis associated with subclinical hypothyroidism: improvement with thyroxine therapy. J Eur Acad Dermatol Venereol. 2006;20:1340-1341.
- Volpato MB, Jaime TJ, Proença MP, et al. Papular mucinosis associated with hypothyroidism. An Bras Dermatol. 2010;85:89-92.
- Macnab M, Kenny P. Successful intravenous immunoglobulin treatment of atypical lichen myxedematosus associated with hypothyroidism and central nervous system. involvement: case report and discussion of the literature. J Cutan Med Surg. 2013;17:69-73.
- Shenoy A, Steixner J, Beltrani V, et al. Discrete papular lichen myxedematosus and scleromyxedema with hypothyroidism: a report of two cases. Case Rep Dermatol. 2019;11:64-70.
- Helfrich DJ, Walker ER, Martinez AJ, et al. Scleromyxedema myopathy: case report and review of the literature. Arthritis Rheum. 1988;31:1437-1441.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Jackson EM, English JC 3rd. Diffuse cutaneous mucinoses. Dermatol Clin. 2002;20:493-501.
- Leonhardt JM, Heymann WR. Thyroid disease and the skin. Dermatol Clin. 2002;20:473-481.
- Zhou JY, Xu B, Lopes J, et al. Hashimoto encephalopathy: literature review. Acta Neurol Scand. 2017;135:285-290.
Scleromyxedema (SM) is a generalized papular and sclerodermoid form of lichen myxedematosus (LM), commonly referred to as papular mucinosis. It is a rare progressive disease of unknown etiology with systemic manifestations that cause serious morbidity and mortality. Diagnostic criteria were initially created by Montgomery and Underwood1 in 1953 and revised by Rongioletti and Rebora2 in 2001 as follows: (1) generalized papular and sclerodermoid eruption; (2) histologic triad of mucin deposition, fibroblast proliferation, and fibrosis; (3) monoclonal gammopathy; and (4) absence of thyroid disease. There are several reports of LM in association with hypothyroidism, most of which can be characterized as atypical.3-8 We present a case of SM in a patient with Hashimoto thyroiditis and propose that the presence of thyroid disease should not preclude the diagnosis of SM.
Case Report
A 44-year-old woman presented with a progressive eruption of thickened skin and papules spanning many months. The papules ranged from flesh colored to erythematous and covered more than 80% of the body surface area, most notably involving the face, neck, ears, arms, chest, abdomen, and thighs (Figures 1A and 2A). Review of systems was notable for pruritus, muscle pain but no weakness, dysphagia, and constipation. Her medical history included childhood atopic dermatitis and Hashimoto thyroiditis. Hypothyroidism was diagnosed with support of a thyroid ultrasound and thyroid peroxidase antibodies. It was treated with oral levothyroxine for 2 years prior to the skin eruption. Thyroid biopsy was not performed. Her thyroid-stimulating hormone levels notably fluctuated in the year prior to presentation despite close clinical and laboratory monitoring by an endocrinologist. Laboratory results are summarized in Table 1. Both skin and muscle9 biopsies were consistent with SM (Figure 3) and are summarized in Table 1.
Shortly after presentation to our clinic the patient developed acute concerns of confusion and muscle weakness. She was admitted for further inpatient management due to concern for dermato-neuro syndrome, a rare but potentially fatal decline in neurological status that can progress to coma and death, rather than myxedema coma. On admission, a thyroid function test showed subclinical hypothyroidism with a thyroid-stimulating hormone level of 6.35 uU/mL (reference range, 0.3–4.35 uU/mL) and free thyroxine (FT4) level of 1.5 ng/dL (reference range, 0.8–2.8 ng/dL). While hospitalized she was started on intravenous levothyroxine, systemic steroids, and a course of intravenous immunoglobulin (IVIg) treatment consisting of 2 g/kg divided over 5 days. On this regimen, her mental status quickly returned to baseline and other symptoms improved, including the skin eruption (Figures 1B and 2B). She has been maintained on lenalidomide 25 mg/d for the first 3 weeks of each month as well as monthly IVIg infusions. Her thyroid levels have persistently fluctuated despite intramuscular levothyroxine dosing, but her skin has remained clear with continued SM-directed therapy.
Comment
Classification
Lichen myxedematosus is differentiated into localized and generalized forms. The former is limited to the skin and lacks monoclonal gammopathy. The latter, also known as SM, is associated with monoclonal gammopathy and systemic symptoms. Atypical LM is an umbrella term for intermediate cases.
Clinical Presentation
Skin manifestations of SM are described as 1- to 3-mm, firm, waxy, dome-shaped papules that commonly affect the hands, forearms, face, neck, trunk, and thighs. The surrounding skin may be reddish brown and edematous with evidence of skin thickening. Extracutaneous manifestations in SM are numerous and unpredictable. Any organ system can be involved, but gastrointestinal, rheumatologic, pulmonary, and cardiovascular complications are most common.10 A comprehensive multidisciplinary evaluation is necessary based on clinical symptoms and laboratory findings.
Management
Many treatments have been proposed for SM in case reports and case series. Prior treatments have had little success. Most recently, in one of the largest case series on SM, Rongioletti et al10 demonstrated IVIg to be a safe and effective treatment modality.
Differential Diagnosis
An important differential diagnosis is generalized myxedema, which is seen in long-standing hypothyroidism and may present with cutaneous mucinosis and systemic symptoms that resemble SM. Hypothyroid myxedema is associated with a widespread slowing of the body’s metabolic processes and deposition of mucin in various organs, including the skin, creating a generalized nonpitting edema. Classic clinical signs include macroglossia, periorbital puffiness, thick lips, and acral swelling. The skin tends to be cold, dry, and pale. Hair is characterized as being coarse, dry, and brittle with diffuse partial alopecia. Histologically, there is hyperkeratosis with follicular plugging and diffuse mucin and edema splaying between collagen fibers spanning the entire dermis.11 In contradistinction with SM, there is no fibroblast proliferation. The treatment is thyroid replacement therapy. Hyperthyroidism has distinct clinical and histologic changes. Clinically, there is moist and smooth skin with soft, fine, and sometimes alopecic hair. Graves disease, the most common cause of hyperthyroidism, is further characterized by Graves ophthalmopathy and pretibial myxedema, or pink to brown, raised, firm, indurated, asymmetric plaques most commonly affecting the shins. Histologically there is increased mucin in the lower to mid dermis without fibroblast proliferation. The epidermis can be hyperkeratotic, which will clinically correlate with verrucous lesions.12
Hypothyroid encephalopathy is a rare disorder that can cause a change in mental status. It is a steroid-responsive autoimmune process characterized by encephalopathy that is associated with cognitive impairment and psychiatric features. It is a diagnosis of exclusion and should be suspected in women with a history of autoimmune disease, especially antithyroid peroxidase antibodies, a negative infectious workup, and encephalitis with behavioral changes. Although typically highly responsive to systemic steroids, IVIg also has shown efficacy.13
Presence of Thyroid Disease
According to a PubMed search of articles indexed for MEDLINE using the terms scleromyxedema and lichen myxedematosus, there are 7 cases in the literature that potentially describe LM associated with hypothyroidism (Table 2).3-8 The majority of these cases lack monoclonal gammopathy; improved with thyroid replacement therapy; or had severely atypical clinical presentations, rendering them cases of atypical LM or atypical thyroid dermopathy.3-6 Macnab and Kenny7 presented a case of subclinical hypothyroidism with a generalized papular eruption, monoclonal gammopathy, and consistent histologic changes that responded to IVIg therapy. These findings are suggestive of SM, but limited to the current diagnostic criteria, the patient was diagnosed with atypical LM.7 Shenoy et al8 described 2 cases of LM with hypothyroidism. One patient had biopsy-proven SM that was responsive to IVIg as well as Hashimoto thyroiditis with delayed onset of monoclonal gammopathy. The second patient had a medical history of hypothyroidism and Hodgkin lymphoma with active rheumatoid arthritis and biopsy-proven LM that was responsive to systemic steroids.8
Current literature states that thyroid disorder precludes the diagnosis of SM. However, historic literature would suggest otherwise. Because of inconsistent reports and theories regarding the pathogenesis of various sclerodermoid and mucin deposition diseases, in 1953 Montgomery and Underwood1 sought to differentiate LM from scleroderma and generalized myxedema. They stressed clinical appearance and proposed diagnostic criteria for LM as generalized papular mucinosis in which “[n]o relation to disturbance of the thyroid or other endocrine glands is apparent,” whereas generalized myxedema was defined as a “[t]rue cutaneous myxedema, with diffuse edema and the usual commonly recognized changes” in patients with endocrine abnormalities.1 With this classification, the authors made a clear distinction between mucinosis caused by thyroid abnormalities and LM, which is not caused by a thyroid disorder. Since this original description was published, associations with monoclonal gammopathy and fibroblast proliferation have been made, ultimately culminating into the current 2001 criteria that incorporate the absence of thyroid disease.2
Conclusion
We believe our case is consistent with the classification initially proposed by Montgomery and Underwood1 and is strengthened with the more recent associations with monoclonal gammopathy and specific histopathologic findings. Although there is no definitive way to rule out myxedema coma or Hashimoto encephalopathy to describe our patient’s transient neurologic decline, her clinical symptoms, laboratory findings, and biopsy results all supported the diagnosis of SM. Furthermore, her response to SM-directed therapy, despite fluctuating thyroid function test results, also supported the diagnosis. In the setting of cutaneous mucinosis with conflicting findings for hypothyroid myxedema, LM should be ruled out. Given the features presented in this report and others, diagnostic criteria should allow for SM and thyroid dysfunction to be concurrent diagnoses. Most importantly, we believe it is essential to identify and diagnose SM in a timely manner to facilitate SM-directed therapy, namely IVIg, to potentially minimize the disease’s notable morbidity and mortality.
Scleromyxedema (SM) is a generalized papular and sclerodermoid form of lichen myxedematosus (LM), commonly referred to as papular mucinosis. It is a rare progressive disease of unknown etiology with systemic manifestations that cause serious morbidity and mortality. Diagnostic criteria were initially created by Montgomery and Underwood1 in 1953 and revised by Rongioletti and Rebora2 in 2001 as follows: (1) generalized papular and sclerodermoid eruption; (2) histologic triad of mucin deposition, fibroblast proliferation, and fibrosis; (3) monoclonal gammopathy; and (4) absence of thyroid disease. There are several reports of LM in association with hypothyroidism, most of which can be characterized as atypical.3-8 We present a case of SM in a patient with Hashimoto thyroiditis and propose that the presence of thyroid disease should not preclude the diagnosis of SM.
Case Report
A 44-year-old woman presented with a progressive eruption of thickened skin and papules spanning many months. The papules ranged from flesh colored to erythematous and covered more than 80% of the body surface area, most notably involving the face, neck, ears, arms, chest, abdomen, and thighs (Figures 1A and 2A). Review of systems was notable for pruritus, muscle pain but no weakness, dysphagia, and constipation. Her medical history included childhood atopic dermatitis and Hashimoto thyroiditis. Hypothyroidism was diagnosed with support of a thyroid ultrasound and thyroid peroxidase antibodies. It was treated with oral levothyroxine for 2 years prior to the skin eruption. Thyroid biopsy was not performed. Her thyroid-stimulating hormone levels notably fluctuated in the year prior to presentation despite close clinical and laboratory monitoring by an endocrinologist. Laboratory results are summarized in Table 1. Both skin and muscle9 biopsies were consistent with SM (Figure 3) and are summarized in Table 1.
Shortly after presentation to our clinic the patient developed acute concerns of confusion and muscle weakness. She was admitted for further inpatient management due to concern for dermato-neuro syndrome, a rare but potentially fatal decline in neurological status that can progress to coma and death, rather than myxedema coma. On admission, a thyroid function test showed subclinical hypothyroidism with a thyroid-stimulating hormone level of 6.35 uU/mL (reference range, 0.3–4.35 uU/mL) and free thyroxine (FT4) level of 1.5 ng/dL (reference range, 0.8–2.8 ng/dL). While hospitalized she was started on intravenous levothyroxine, systemic steroids, and a course of intravenous immunoglobulin (IVIg) treatment consisting of 2 g/kg divided over 5 days. On this regimen, her mental status quickly returned to baseline and other symptoms improved, including the skin eruption (Figures 1B and 2B). She has been maintained on lenalidomide 25 mg/d for the first 3 weeks of each month as well as monthly IVIg infusions. Her thyroid levels have persistently fluctuated despite intramuscular levothyroxine dosing, but her skin has remained clear with continued SM-directed therapy.
Comment
Classification
Lichen myxedematosus is differentiated into localized and generalized forms. The former is limited to the skin and lacks monoclonal gammopathy. The latter, also known as SM, is associated with monoclonal gammopathy and systemic symptoms. Atypical LM is an umbrella term for intermediate cases.
Clinical Presentation
Skin manifestations of SM are described as 1- to 3-mm, firm, waxy, dome-shaped papules that commonly affect the hands, forearms, face, neck, trunk, and thighs. The surrounding skin may be reddish brown and edematous with evidence of skin thickening. Extracutaneous manifestations in SM are numerous and unpredictable. Any organ system can be involved, but gastrointestinal, rheumatologic, pulmonary, and cardiovascular complications are most common.10 A comprehensive multidisciplinary evaluation is necessary based on clinical symptoms and laboratory findings.
Management
Many treatments have been proposed for SM in case reports and case series. Prior treatments have had little success. Most recently, in one of the largest case series on SM, Rongioletti et al10 demonstrated IVIg to be a safe and effective treatment modality.
Differential Diagnosis
An important differential diagnosis is generalized myxedema, which is seen in long-standing hypothyroidism and may present with cutaneous mucinosis and systemic symptoms that resemble SM. Hypothyroid myxedema is associated with a widespread slowing of the body’s metabolic processes and deposition of mucin in various organs, including the skin, creating a generalized nonpitting edema. Classic clinical signs include macroglossia, periorbital puffiness, thick lips, and acral swelling. The skin tends to be cold, dry, and pale. Hair is characterized as being coarse, dry, and brittle with diffuse partial alopecia. Histologically, there is hyperkeratosis with follicular plugging and diffuse mucin and edema splaying between collagen fibers spanning the entire dermis.11 In contradistinction with SM, there is no fibroblast proliferation. The treatment is thyroid replacement therapy. Hyperthyroidism has distinct clinical and histologic changes. Clinically, there is moist and smooth skin with soft, fine, and sometimes alopecic hair. Graves disease, the most common cause of hyperthyroidism, is further characterized by Graves ophthalmopathy and pretibial myxedema, or pink to brown, raised, firm, indurated, asymmetric plaques most commonly affecting the shins. Histologically there is increased mucin in the lower to mid dermis without fibroblast proliferation. The epidermis can be hyperkeratotic, which will clinically correlate with verrucous lesions.12
Hypothyroid encephalopathy is a rare disorder that can cause a change in mental status. It is a steroid-responsive autoimmune process characterized by encephalopathy that is associated with cognitive impairment and psychiatric features. It is a diagnosis of exclusion and should be suspected in women with a history of autoimmune disease, especially antithyroid peroxidase antibodies, a negative infectious workup, and encephalitis with behavioral changes. Although typically highly responsive to systemic steroids, IVIg also has shown efficacy.13
Presence of Thyroid Disease
According to a PubMed search of articles indexed for MEDLINE using the terms scleromyxedema and lichen myxedematosus, there are 7 cases in the literature that potentially describe LM associated with hypothyroidism (Table 2).3-8 The majority of these cases lack monoclonal gammopathy; improved with thyroid replacement therapy; or had severely atypical clinical presentations, rendering them cases of atypical LM or atypical thyroid dermopathy.3-6 Macnab and Kenny7 presented a case of subclinical hypothyroidism with a generalized papular eruption, monoclonal gammopathy, and consistent histologic changes that responded to IVIg therapy. These findings are suggestive of SM, but limited to the current diagnostic criteria, the patient was diagnosed with atypical LM.7 Shenoy et al8 described 2 cases of LM with hypothyroidism. One patient had biopsy-proven SM that was responsive to IVIg as well as Hashimoto thyroiditis with delayed onset of monoclonal gammopathy. The second patient had a medical history of hypothyroidism and Hodgkin lymphoma with active rheumatoid arthritis and biopsy-proven LM that was responsive to systemic steroids.8
Current literature states that thyroid disorder precludes the diagnosis of SM. However, historic literature would suggest otherwise. Because of inconsistent reports and theories regarding the pathogenesis of various sclerodermoid and mucin deposition diseases, in 1953 Montgomery and Underwood1 sought to differentiate LM from scleroderma and generalized myxedema. They stressed clinical appearance and proposed diagnostic criteria for LM as generalized papular mucinosis in which “[n]o relation to disturbance of the thyroid or other endocrine glands is apparent,” whereas generalized myxedema was defined as a “[t]rue cutaneous myxedema, with diffuse edema and the usual commonly recognized changes” in patients with endocrine abnormalities.1 With this classification, the authors made a clear distinction between mucinosis caused by thyroid abnormalities and LM, which is not caused by a thyroid disorder. Since this original description was published, associations with monoclonal gammopathy and fibroblast proliferation have been made, ultimately culminating into the current 2001 criteria that incorporate the absence of thyroid disease.2
Conclusion
We believe our case is consistent with the classification initially proposed by Montgomery and Underwood1 and is strengthened with the more recent associations with monoclonal gammopathy and specific histopathologic findings. Although there is no definitive way to rule out myxedema coma or Hashimoto encephalopathy to describe our patient’s transient neurologic decline, her clinical symptoms, laboratory findings, and biopsy results all supported the diagnosis of SM. Furthermore, her response to SM-directed therapy, despite fluctuating thyroid function test results, also supported the diagnosis. In the setting of cutaneous mucinosis with conflicting findings for hypothyroid myxedema, LM should be ruled out. Given the features presented in this report and others, diagnostic criteria should allow for SM and thyroid dysfunction to be concurrent diagnoses. Most importantly, we believe it is essential to identify and diagnose SM in a timely manner to facilitate SM-directed therapy, namely IVIg, to potentially minimize the disease’s notable morbidity and mortality.
- Montgomery H, Underwood LJ. Lichen myxedematosus; differentiation from cutaneous myxedemas or mucoid states. J Invest Dermatol. 1953;20:213-236.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Archibald GC, Calvert HT. Hypothyroidsm and lichen myxedematosus. Arch Dermatol. 1977;113:684.
- Schaeffer D, Bruce S, Rosen T. Cutaneous mucinosis associated with thyroid dysfunction. Cutis. 1983;11:449-456.
- Martin-Ezquerra G, Sanchez-Regaña M, Massana-Gil J, et al. Papular mucinosis associated with subclinical hypothyroidism: improvement with thyroxine therapy. J Eur Acad Dermatol Venereol. 2006;20:1340-1341.
- Volpato MB, Jaime TJ, Proença MP, et al. Papular mucinosis associated with hypothyroidism. An Bras Dermatol. 2010;85:89-92.
- Macnab M, Kenny P. Successful intravenous immunoglobulin treatment of atypical lichen myxedematosus associated with hypothyroidism and central nervous system. involvement: case report and discussion of the literature. J Cutan Med Surg. 2013;17:69-73.
- Shenoy A, Steixner J, Beltrani V, et al. Discrete papular lichen myxedematosus and scleromyxedema with hypothyroidism: a report of two cases. Case Rep Dermatol. 2019;11:64-70.
- Helfrich DJ, Walker ER, Martinez AJ, et al. Scleromyxedema myopathy: case report and review of the literature. Arthritis Rheum. 1988;31:1437-1441.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Jackson EM, English JC 3rd. Diffuse cutaneous mucinoses. Dermatol Clin. 2002;20:493-501.
- Leonhardt JM, Heymann WR. Thyroid disease and the skin. Dermatol Clin. 2002;20:473-481.
- Zhou JY, Xu B, Lopes J, et al. Hashimoto encephalopathy: literature review. Acta Neurol Scand. 2017;135:285-290.
- Montgomery H, Underwood LJ. Lichen myxedematosus; differentiation from cutaneous myxedemas or mucoid states. J Invest Dermatol. 1953;20:213-236.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Archibald GC, Calvert HT. Hypothyroidsm and lichen myxedematosus. Arch Dermatol. 1977;113:684.
- Schaeffer D, Bruce S, Rosen T. Cutaneous mucinosis associated with thyroid dysfunction. Cutis. 1983;11:449-456.
- Martin-Ezquerra G, Sanchez-Regaña M, Massana-Gil J, et al. Papular mucinosis associated with subclinical hypothyroidism: improvement with thyroxine therapy. J Eur Acad Dermatol Venereol. 2006;20:1340-1341.
- Volpato MB, Jaime TJ, Proença MP, et al. Papular mucinosis associated with hypothyroidism. An Bras Dermatol. 2010;85:89-92.
- Macnab M, Kenny P. Successful intravenous immunoglobulin treatment of atypical lichen myxedematosus associated with hypothyroidism and central nervous system. involvement: case report and discussion of the literature. J Cutan Med Surg. 2013;17:69-73.
- Shenoy A, Steixner J, Beltrani V, et al. Discrete papular lichen myxedematosus and scleromyxedema with hypothyroidism: a report of two cases. Case Rep Dermatol. 2019;11:64-70.
- Helfrich DJ, Walker ER, Martinez AJ, et al. Scleromyxedema myopathy: case report and review of the literature. Arthritis Rheum. 1988;31:1437-1441.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Jackson EM, English JC 3rd. Diffuse cutaneous mucinoses. Dermatol Clin. 2002;20:493-501.
- Leonhardt JM, Heymann WR. Thyroid disease and the skin. Dermatol Clin. 2002;20:473-481.
- Zhou JY, Xu B, Lopes J, et al. Hashimoto encephalopathy: literature review. Acta Neurol Scand. 2017;135:285-290.
Practice Points
- Scleromyxedema (SM) is progressive disease of unknown etiology with unpredictable behavior.
- Systemic manifestations associated with SM can cause serious morbidity and mortality.
- Intravenous immunoglobulin is the most effective treatment modality in SM.
- The presence of thyroid disease should not preclude the diagnosis of SM.
Mystery Burns and Nocturnal Seizure Safety
Patients with seizures are placed at an increased risk for sustaining burn injuries, which may occur during common daily activities such as cooking, showering, and using heaters.1 Although patients are warned of the risks of injury at the time of their epilepsy diagnosis, patients still experience injuries that commonly occur during the seizure or the postictal phase. In a study of 134 patients with epilepsy, only 38% recalled being burned during a seizure, with approximately 9% being burned multiple times.2 Another study investigated the circumstances resulting in burns in this patient population and found that cooking on a stove was the most common cause, followed by hot water while showering and exposed room heaters.1 Another study found that the majority of burns in seizure patients were from spilled hot drinks.3
We report 2 patients who presented to the dermatology clinic with second-degree burns following nocturnal seizures. In both cases, the patients were sleeping next to exposed heaters, which led to burn injuries from seizures that occurred in the night.
Case Reports
Patient 1
A 30-year-old woman with a history of a seizure disorder presented with painful second-degree blistering burns along the left arm and flank (Figure 1). One day prior to presentation, she had woken up to find these lesions and visited the emergency department where she was prescribed silver sulfadiazine cream to prevent infection of the wound site and was referred to our dermatology clinic. Initially, the patient had difficulty pinpointing the source of the burn lesions and thought that it may have been due to sleeping with her cell phone, but she later realized that they were due to the space heater placed next to her bed. Because of the unclear etiology at the initial presentation, a skin biopsy of a lesion was taken while she was at the clinic.
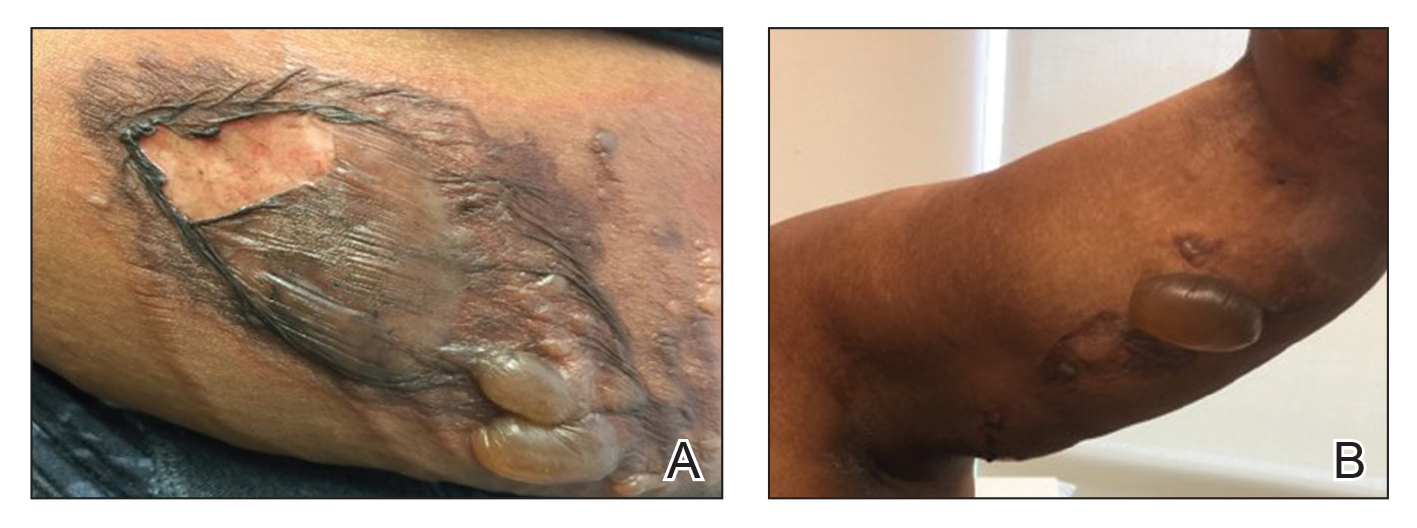
Biopsy of the lesions exhibited separation of the epidermal and dermal layers (Figure 2). Thermal damage was seen extending into the dermal layers with notable edema present. A few inflammatory cells, neutrophils, and monocytes were noted in the biopsy. The initial pathology results showed the epidermis was necrotic with edema, spongiform vesicles, and few neutrophils. The histologic findings aligned with the timeline of the injury occurring 2 days prior to the biopsy. She was treated supportively using mupirocin ointment to prevent secondary infection.
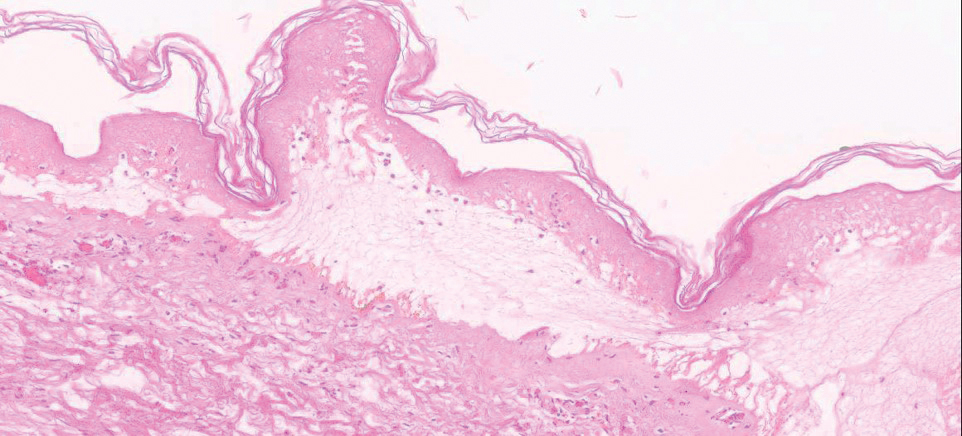
Case 2
A 27-year-old woman with a history of epilepsy presented to the dermatology clinic with painful blistering lesions along the right upper arm (Figure 3). She was found to have notable second-degree burns along the right arm. She reported placing her bed near a baseboard heater to stay warm overnight. She noticed the painful lesions after waking up next to the heater following a suspected seizure. She was treated supportively using mupirocin ointment to prevent secondary infection.
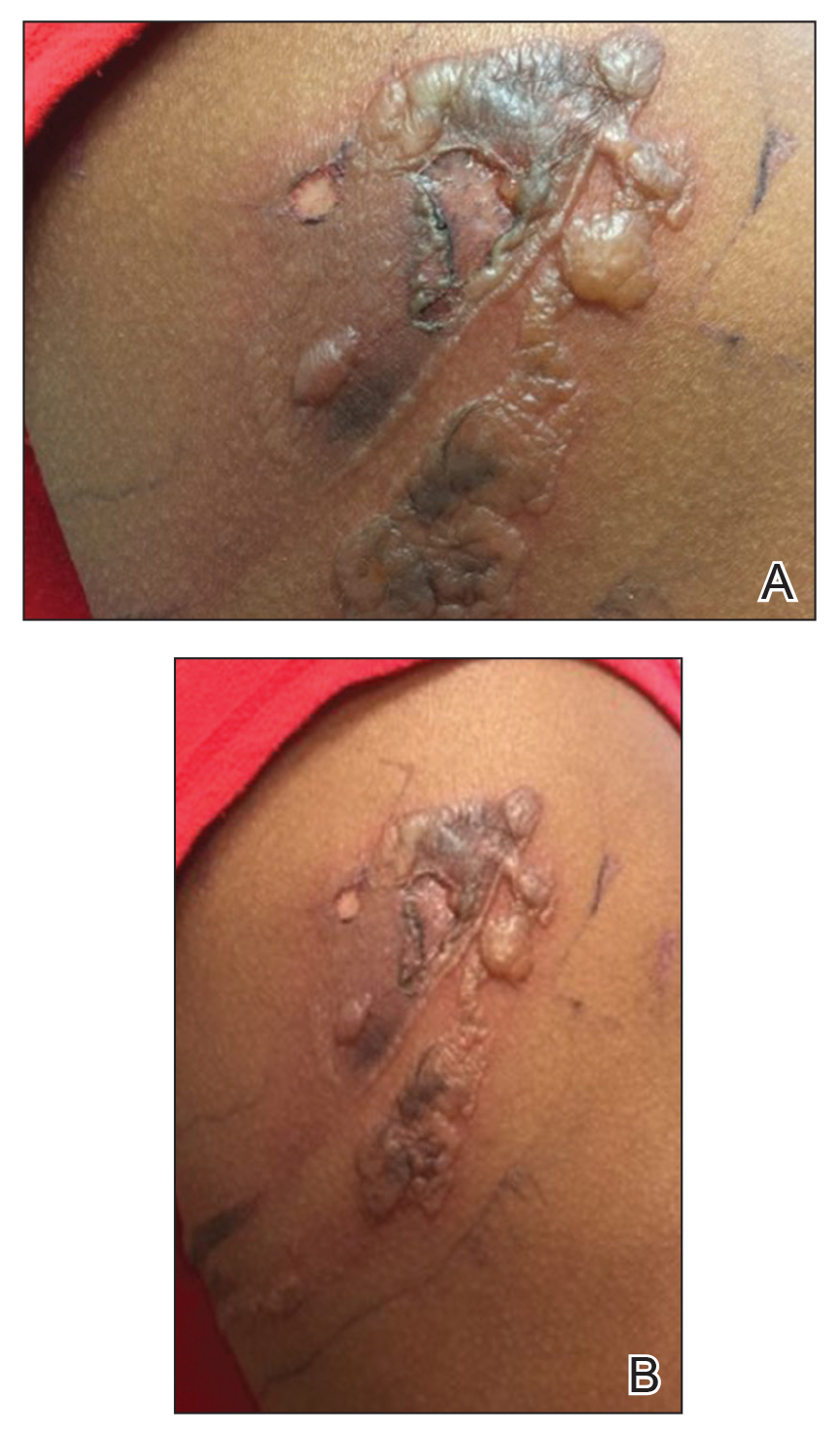
Comment
Classification of Burns and Damage
According to the World Health Organization, nonfatal burn injuries are a leading cause of morbidity and occur mainly in the home and workplace.4 There are many types of burns: radiation, electrical, chemical, friction, and thermal. The most common type of burns are thermal burns,4 which can be further subdivided into wet and dry. Both of our patients experienced dry thermal burns.
Based on the skin tissue layers involved in the thermal damage, burn wounds are further divided into first-degree burns, superficial second-degree burns, deep second-degree burns, and third-degree burns.5 These classifications each have characteristic gross features. Based on these criteria, our patients both presented with blistering and ruptured bullae and no eschar formation, which is classified as second-degree superficial burns.
Following thermal insult to the skin, 3 zones are formed. The central zone consists of irreparable damage referred to as the zone of coagulation. The zone of stasis lies between the completely damaged central region and the outermost regions of the burn lesion, and it receives slightly less blood flow. This area can fully recover after complete perfusion is returned early in the healing process. The outermost zone of hyperemia can fully recover and is an area marked by intense vasodilation from inflammatory reactions.5
Wound Healing
During the healing process, metabolic activity is remarkably increased, which leads to formation of
Burns in Patients With Seizure Disorders
Burns pose a serious risk to patients with seizure disorders that often is underappreciated by patients and health care providers. Although many burns are first-degree burns, up to 10% of burns require medical attention.1 In the initial phase following a thermal insult, the skin’s microflora is killed off, but within a week the sterile skin can become infected.5 The most common microbial invasions seen in blistering wounds are due to Pseudomonas aeruginosa and Staphylococcus aureus.8 With larger burns associated with immunocompromising factors such as diabetes mellitus or older age, patients are at an increased risk for becoming septic. Prior to the period of infection, the damage caused by the heat leads to vasodilation of the microvasculature surrounding the injured area. In addition, release of cytokines leads to migration of inflammatory cells. With the vasodilation of vasculature, proteinaceous fluids from the intravascular space can collect between the dead epidermal and dermal layers to form blisters.5 In larger burns, the fluid shifts will lead to severe oncotic pressure decreases intravascularly and can lead to hypotensive shock.6 When burns have a more severe global effect, aggressive resuscitation and vasopressors are required to maintain perfusion of vital organs.
Both of our patients experienced painful lesions, but they were fortunate to have factors of youth, superficial damage, and low total body surface area burns for a smaller risk for infection, fluid loss, and severely disfiguring scars.8 Because the duration of the postictal phase can vary, there is potential for more severe burns that can leave a lifelong reminder of the event. Depending on the skin type and the depth of the thermal insult, evidence of injury may last many years in the form of hypertrophic scars, contractures, and changes in skin pigmentation.5 At distances 30 cm or less from the standard blow-dryer, it takes 2 minutes to cause cell death.9 In comparison to a heat source that is meant to provide warmth to a room, there is a notable difference in potential for severe burns with the standard heater vs the standard blow-dryer.
Along with the physical pain, the visual reminders of the injurious event can have notable psychological effects. Scars can decrease self-esteem and lead to depression, anxiety, body image problems, and sexuality issues.10
Given the immense risks associated with burn injuries and the many unfortunate outcomes, emphasis should be placed on patient education regarding safety precautions with seizure disorders. In one study, it was found that only 5% of patients recall receiving a warning about the risk for burn injuries with seizures.2 It is important for patients and physicians to develop a written comprehensive safety plan that addresses the risks for daily activities during the day and night. Although patients may not remember being told about the risks, a written safety plan likely will increase patient awareness and reduce avoidable injuries. In addition to written safety plans, prior recommendations for reducing burn injuries in seizure patients include the use of fire and heater guards as well as flame-retardant clothing and blankets.11
- Spitz MC, Towbin JA, Shantz D, et al. Risk factors for burns as a consequence of seizures in persons with epilepsy. Epilepsia. 1994;35:764-767.
- Hampton KK, Peatfield RC, Pullar T, et al. Burns because of epilepsy. Br Med J (Clin Res Ed). 1988;296:1659-1660.
- Kinton L, Duncan JS. Frequency, causes, and consequences of burns in patients with epilepsy. J Neurol Neurosurg Psychiatry. 1998;65:404-405.
- World Health Organization. Burns. http://www.who.int/news-room/fact-sheets/detail/burns. Published March 6, 2018. Accessed December 13, 2019.
- Tiwari VK. Burn wound: how it differs from other wounds? Indian J Plast Surg. 2012;45:364-373.
- Nielson CB, Duethman NC, Howard JM, et al. Burns: pathophysiology of systemic complications and current management. J Burn Care Res. 2017;38:E469-E481.
- Travers JB, Murphy RC, Johnson CA, et al. Identification and pharmacological characterization of platelet-activating factor and related 1-palmitoyl species found in human inflammatory blistering diseases. Prostaglandins Other Lipid Mediat. 1998;5:305-324.
- Church D, Elsayed S, Reid O, et al. Burn wound infections. Clin Microbiol Rev. 2006;19:403-434.
- Aslam A, Khoo CT. No sense; no sensibility—a tale of two adult hair-drier burns. Burns. 1997;23:454-457.
- Van Loey NE, Van Son MJ. Psychopathology and psychological problems in patients with burn scars: epidemiology and management. Am J Clin Dermatol. 2003;4:245-272.
- Josty IC, Narayanan V, Dickson WA. Burns in patients with epilepsy: changes in epidemiology and implications for burn treatment and prevention. Epilepsia. 2000;41:453-456.
Patients with seizures are placed at an increased risk for sustaining burn injuries, which may occur during common daily activities such as cooking, showering, and using heaters.1 Although patients are warned of the risks of injury at the time of their epilepsy diagnosis, patients still experience injuries that commonly occur during the seizure or the postictal phase. In a study of 134 patients with epilepsy, only 38% recalled being burned during a seizure, with approximately 9% being burned multiple times.2 Another study investigated the circumstances resulting in burns in this patient population and found that cooking on a stove was the most common cause, followed by hot water while showering and exposed room heaters.1 Another study found that the majority of burns in seizure patients were from spilled hot drinks.3
We report 2 patients who presented to the dermatology clinic with second-degree burns following nocturnal seizures. In both cases, the patients were sleeping next to exposed heaters, which led to burn injuries from seizures that occurred in the night.
Case Reports
Patient 1
A 30-year-old woman with a history of a seizure disorder presented with painful second-degree blistering burns along the left arm and flank (Figure 1). One day prior to presentation, she had woken up to find these lesions and visited the emergency department where she was prescribed silver sulfadiazine cream to prevent infection of the wound site and was referred to our dermatology clinic. Initially, the patient had difficulty pinpointing the source of the burn lesions and thought that it may have been due to sleeping with her cell phone, but she later realized that they were due to the space heater placed next to her bed. Because of the unclear etiology at the initial presentation, a skin biopsy of a lesion was taken while she was at the clinic.
Biopsy of the lesions exhibited separation of the epidermal and dermal layers (Figure 2). Thermal damage was seen extending into the dermal layers with notable edema present. A few inflammatory cells, neutrophils, and monocytes were noted in the biopsy. The initial pathology results showed the epidermis was necrotic with edema, spongiform vesicles, and few neutrophils. The histologic findings aligned with the timeline of the injury occurring 2 days prior to the biopsy. She was treated supportively using mupirocin ointment to prevent secondary infection.
Case 2
A 27-year-old woman with a history of epilepsy presented to the dermatology clinic with painful blistering lesions along the right upper arm (Figure 3). She was found to have notable second-degree burns along the right arm. She reported placing her bed near a baseboard heater to stay warm overnight. She noticed the painful lesions after waking up next to the heater following a suspected seizure. She was treated supportively using mupirocin ointment to prevent secondary infection.
Comment
Classification of Burns and Damage
According to the World Health Organization, nonfatal burn injuries are a leading cause of morbidity and occur mainly in the home and workplace.4 There are many types of burns: radiation, electrical, chemical, friction, and thermal. The most common type of burns are thermal burns,4 which can be further subdivided into wet and dry. Both of our patients experienced dry thermal burns.
Based on the skin tissue layers involved in the thermal damage, burn wounds are further divided into first-degree burns, superficial second-degree burns, deep second-degree burns, and third-degree burns.5 These classifications each have characteristic gross features. Based on these criteria, our patients both presented with blistering and ruptured bullae and no eschar formation, which is classified as second-degree superficial burns.
Following thermal insult to the skin, 3 zones are formed. The central zone consists of irreparable damage referred to as the zone of coagulation. The zone of stasis lies between the completely damaged central region and the outermost regions of the burn lesion, and it receives slightly less blood flow. This area can fully recover after complete perfusion is returned early in the healing process. The outermost zone of hyperemia can fully recover and is an area marked by intense vasodilation from inflammatory reactions.5
Wound Healing
During the healing process, metabolic activity is remarkably increased, which leads to formation of
Burns in Patients With Seizure Disorders
Burns pose a serious risk to patients with seizure disorders that often is underappreciated by patients and health care providers. Although many burns are first-degree burns, up to 10% of burns require medical attention.1 In the initial phase following a thermal insult, the skin’s microflora is killed off, but within a week the sterile skin can become infected.5 The most common microbial invasions seen in blistering wounds are due to Pseudomonas aeruginosa and Staphylococcus aureus.8 With larger burns associated with immunocompromising factors such as diabetes mellitus or older age, patients are at an increased risk for becoming septic. Prior to the period of infection, the damage caused by the heat leads to vasodilation of the microvasculature surrounding the injured area. In addition, release of cytokines leads to migration of inflammatory cells. With the vasodilation of vasculature, proteinaceous fluids from the intravascular space can collect between the dead epidermal and dermal layers to form blisters.5 In larger burns, the fluid shifts will lead to severe oncotic pressure decreases intravascularly and can lead to hypotensive shock.6 When burns have a more severe global effect, aggressive resuscitation and vasopressors are required to maintain perfusion of vital organs.
Both of our patients experienced painful lesions, but they were fortunate to have factors of youth, superficial damage, and low total body surface area burns for a smaller risk for infection, fluid loss, and severely disfiguring scars.8 Because the duration of the postictal phase can vary, there is potential for more severe burns that can leave a lifelong reminder of the event. Depending on the skin type and the depth of the thermal insult, evidence of injury may last many years in the form of hypertrophic scars, contractures, and changes in skin pigmentation.5 At distances 30 cm or less from the standard blow-dryer, it takes 2 minutes to cause cell death.9 In comparison to a heat source that is meant to provide warmth to a room, there is a notable difference in potential for severe burns with the standard heater vs the standard blow-dryer.
Along with the physical pain, the visual reminders of the injurious event can have notable psychological effects. Scars can decrease self-esteem and lead to depression, anxiety, body image problems, and sexuality issues.10
Given the immense risks associated with burn injuries and the many unfortunate outcomes, emphasis should be placed on patient education regarding safety precautions with seizure disorders. In one study, it was found that only 5% of patients recall receiving a warning about the risk for burn injuries with seizures.2 It is important for patients and physicians to develop a written comprehensive safety plan that addresses the risks for daily activities during the day and night. Although patients may not remember being told about the risks, a written safety plan likely will increase patient awareness and reduce avoidable injuries. In addition to written safety plans, prior recommendations for reducing burn injuries in seizure patients include the use of fire and heater guards as well as flame-retardant clothing and blankets.11
Patients with seizures are placed at an increased risk for sustaining burn injuries, which may occur during common daily activities such as cooking, showering, and using heaters.1 Although patients are warned of the risks of injury at the time of their epilepsy diagnosis, patients still experience injuries that commonly occur during the seizure or the postictal phase. In a study of 134 patients with epilepsy, only 38% recalled being burned during a seizure, with approximately 9% being burned multiple times.2 Another study investigated the circumstances resulting in burns in this patient population and found that cooking on a stove was the most common cause, followed by hot water while showering and exposed room heaters.1 Another study found that the majority of burns in seizure patients were from spilled hot drinks.3
We report 2 patients who presented to the dermatology clinic with second-degree burns following nocturnal seizures. In both cases, the patients were sleeping next to exposed heaters, which led to burn injuries from seizures that occurred in the night.
Case Reports
Patient 1
A 30-year-old woman with a history of a seizure disorder presented with painful second-degree blistering burns along the left arm and flank (Figure 1). One day prior to presentation, she had woken up to find these lesions and visited the emergency department where she was prescribed silver sulfadiazine cream to prevent infection of the wound site and was referred to our dermatology clinic. Initially, the patient had difficulty pinpointing the source of the burn lesions and thought that it may have been due to sleeping with her cell phone, but she later realized that they were due to the space heater placed next to her bed. Because of the unclear etiology at the initial presentation, a skin biopsy of a lesion was taken while she was at the clinic.
Biopsy of the lesions exhibited separation of the epidermal and dermal layers (Figure 2). Thermal damage was seen extending into the dermal layers with notable edema present. A few inflammatory cells, neutrophils, and monocytes were noted in the biopsy. The initial pathology results showed the epidermis was necrotic with edema, spongiform vesicles, and few neutrophils. The histologic findings aligned with the timeline of the injury occurring 2 days prior to the biopsy. She was treated supportively using mupirocin ointment to prevent secondary infection.
Case 2
A 27-year-old woman with a history of epilepsy presented to the dermatology clinic with painful blistering lesions along the right upper arm (Figure 3). She was found to have notable second-degree burns along the right arm. She reported placing her bed near a baseboard heater to stay warm overnight. She noticed the painful lesions after waking up next to the heater following a suspected seizure. She was treated supportively using mupirocin ointment to prevent secondary infection.
Comment
Classification of Burns and Damage
According to the World Health Organization, nonfatal burn injuries are a leading cause of morbidity and occur mainly in the home and workplace.4 There are many types of burns: radiation, electrical, chemical, friction, and thermal. The most common type of burns are thermal burns,4 which can be further subdivided into wet and dry. Both of our patients experienced dry thermal burns.
Based on the skin tissue layers involved in the thermal damage, burn wounds are further divided into first-degree burns, superficial second-degree burns, deep second-degree burns, and third-degree burns.5 These classifications each have characteristic gross features. Based on these criteria, our patients both presented with blistering and ruptured bullae and no eschar formation, which is classified as second-degree superficial burns.
Following thermal insult to the skin, 3 zones are formed. The central zone consists of irreparable damage referred to as the zone of coagulation. The zone of stasis lies between the completely damaged central region and the outermost regions of the burn lesion, and it receives slightly less blood flow. This area can fully recover after complete perfusion is returned early in the healing process. The outermost zone of hyperemia can fully recover and is an area marked by intense vasodilation from inflammatory reactions.5
Wound Healing
During the healing process, metabolic activity is remarkably increased, which leads to formation of
Burns in Patients With Seizure Disorders
Burns pose a serious risk to patients with seizure disorders that often is underappreciated by patients and health care providers. Although many burns are first-degree burns, up to 10% of burns require medical attention.1 In the initial phase following a thermal insult, the skin’s microflora is killed off, but within a week the sterile skin can become infected.5 The most common microbial invasions seen in blistering wounds are due to Pseudomonas aeruginosa and Staphylococcus aureus.8 With larger burns associated with immunocompromising factors such as diabetes mellitus or older age, patients are at an increased risk for becoming septic. Prior to the period of infection, the damage caused by the heat leads to vasodilation of the microvasculature surrounding the injured area. In addition, release of cytokines leads to migration of inflammatory cells. With the vasodilation of vasculature, proteinaceous fluids from the intravascular space can collect between the dead epidermal and dermal layers to form blisters.5 In larger burns, the fluid shifts will lead to severe oncotic pressure decreases intravascularly and can lead to hypotensive shock.6 When burns have a more severe global effect, aggressive resuscitation and vasopressors are required to maintain perfusion of vital organs.
Both of our patients experienced painful lesions, but they were fortunate to have factors of youth, superficial damage, and low total body surface area burns for a smaller risk for infection, fluid loss, and severely disfiguring scars.8 Because the duration of the postictal phase can vary, there is potential for more severe burns that can leave a lifelong reminder of the event. Depending on the skin type and the depth of the thermal insult, evidence of injury may last many years in the form of hypertrophic scars, contractures, and changes in skin pigmentation.5 At distances 30 cm or less from the standard blow-dryer, it takes 2 minutes to cause cell death.9 In comparison to a heat source that is meant to provide warmth to a room, there is a notable difference in potential for severe burns with the standard heater vs the standard blow-dryer.
Along with the physical pain, the visual reminders of the injurious event can have notable psychological effects. Scars can decrease self-esteem and lead to depression, anxiety, body image problems, and sexuality issues.10
Given the immense risks associated with burn injuries and the many unfortunate outcomes, emphasis should be placed on patient education regarding safety precautions with seizure disorders. In one study, it was found that only 5% of patients recall receiving a warning about the risk for burn injuries with seizures.2 It is important for patients and physicians to develop a written comprehensive safety plan that addresses the risks for daily activities during the day and night. Although patients may not remember being told about the risks, a written safety plan likely will increase patient awareness and reduce avoidable injuries. In addition to written safety plans, prior recommendations for reducing burn injuries in seizure patients include the use of fire and heater guards as well as flame-retardant clothing and blankets.11
- Spitz MC, Towbin JA, Shantz D, et al. Risk factors for burns as a consequence of seizures in persons with epilepsy. Epilepsia. 1994;35:764-767.
- Hampton KK, Peatfield RC, Pullar T, et al. Burns because of epilepsy. Br Med J (Clin Res Ed). 1988;296:1659-1660.
- Kinton L, Duncan JS. Frequency, causes, and consequences of burns in patients with epilepsy. J Neurol Neurosurg Psychiatry. 1998;65:404-405.
- World Health Organization. Burns. http://www.who.int/news-room/fact-sheets/detail/burns. Published March 6, 2018. Accessed December 13, 2019.
- Tiwari VK. Burn wound: how it differs from other wounds? Indian J Plast Surg. 2012;45:364-373.
- Nielson CB, Duethman NC, Howard JM, et al. Burns: pathophysiology of systemic complications and current management. J Burn Care Res. 2017;38:E469-E481.
- Travers JB, Murphy RC, Johnson CA, et al. Identification and pharmacological characterization of platelet-activating factor and related 1-palmitoyl species found in human inflammatory blistering diseases. Prostaglandins Other Lipid Mediat. 1998;5:305-324.
- Church D, Elsayed S, Reid O, et al. Burn wound infections. Clin Microbiol Rev. 2006;19:403-434.
- Aslam A, Khoo CT. No sense; no sensibility—a tale of two adult hair-drier burns. Burns. 1997;23:454-457.
- Van Loey NE, Van Son MJ. Psychopathology and psychological problems in patients with burn scars: epidemiology and management. Am J Clin Dermatol. 2003;4:245-272.
- Josty IC, Narayanan V, Dickson WA. Burns in patients with epilepsy: changes in epidemiology and implications for burn treatment and prevention. Epilepsia. 2000;41:453-456.
- Spitz MC, Towbin JA, Shantz D, et al. Risk factors for burns as a consequence of seizures in persons with epilepsy. Epilepsia. 1994;35:764-767.
- Hampton KK, Peatfield RC, Pullar T, et al. Burns because of epilepsy. Br Med J (Clin Res Ed). 1988;296:1659-1660.
- Kinton L, Duncan JS. Frequency, causes, and consequences of burns in patients with epilepsy. J Neurol Neurosurg Psychiatry. 1998;65:404-405.
- World Health Organization. Burns. http://www.who.int/news-room/fact-sheets/detail/burns. Published March 6, 2018. Accessed December 13, 2019.
- Tiwari VK. Burn wound: how it differs from other wounds? Indian J Plast Surg. 2012;45:364-373.
- Nielson CB, Duethman NC, Howard JM, et al. Burns: pathophysiology of systemic complications and current management. J Burn Care Res. 2017;38:E469-E481.
- Travers JB, Murphy RC, Johnson CA, et al. Identification and pharmacological characterization of platelet-activating factor and related 1-palmitoyl species found in human inflammatory blistering diseases. Prostaglandins Other Lipid Mediat. 1998;5:305-324.
- Church D, Elsayed S, Reid O, et al. Burn wound infections. Clin Microbiol Rev. 2006;19:403-434.
- Aslam A, Khoo CT. No sense; no sensibility—a tale of two adult hair-drier burns. Burns. 1997;23:454-457.
- Van Loey NE, Van Son MJ. Psychopathology and psychological problems in patients with burn scars: epidemiology and management. Am J Clin Dermatol. 2003;4:245-272.
- Josty IC, Narayanan V, Dickson WA. Burns in patients with epilepsy: changes in epidemiology and implications for burn treatment and prevention. Epilepsia. 2000;41:453-456.
Practice Points
- Burns and scars from burns can lead to both life-threatening consequences and lifelong psychological effects.
- Many epileptic patients who present with thermal burn injuries do not remember getting burned.
- Clinicians should be aware of all the potential dangers that patients with epilepsy may encounter both during the day and night.
Incidentally Discovered Ochronosis Explaining Decades of Chronic Pain
Alkaptonuria is a rare autosomal recessive disorder uniquely known for causing black, or darkened, urine when left standing due to the renal excretion of excess homogentisic acid (HGA). When this disorder goes undiagnosed, as demonstrated in this case, patients experience its many complications without a unifying explanation. The disorder has 3 clinical stages that occur in a predictable order: clinical silence, clinical ochronosis, and ochronotic arthropathy. These stages lead to multiple musculoskeletal, cardiovascular (CV), and renal complications that can be mitigated with management focused on decreasing homogentisic acid buildup, alleviating symptoms, and close monitoring for these complications.
Case Presentation
A 61-year-old African American male with a medical history of multiple traumatic fractures, right Achilles tendon injury, early-onset multijoint osteoarthritis, chronic low back pain, and recurrent nephrolithiasis presented to the emergency department with sudden onset of sharp left ankle pain while moving furniture. His physical exam revealed a positive Thompson test—lack of foot plantar flexion with calf squeeze—and a subsequent magnetic resonance image (MRI) showed evidence of an acute Achilles tendon rupture.
Given these findings the patient was treated with nonsteroidal anti-inflammatory drugs (NSAIDs) and rest to allow for resolution of swelling and inflammation, followed by elective surgery a month later to repair the ruptured tendon. An operative report following his surgery described “black ends to the area where the Achilles was ruptured…and tendinopathy of the flexor hallucis longus with blackening of the flexor.”
A more in-depth patient history revealed that he underwent multiple invasive and noninvasive interventions for his chronic low back and joint pain with medical management of a prior right Achilles tendon injury. His medical history also included multiple nonspecific diagnoses, such as premature atherosclerosis (diagnosed in his third decade), severe lumbar degenerative disc disease, several tendonopathies and cartilage injuries (Figure 1), pseudogout (following calcium pyrophosphate dehydrate crystals found from a left knee aspirate), and chronic pain syndrome. Along this diagnostic journey, he had several health care providers (HCPs) in rheumatology, orthopedic surgery, pain management, and podiatry who offered a range of symptom management options, including physical therapy, NSAIDs, opioid agonists, tricyclic antidepressants, gabapentin, colchicine, and epidural steroid injections, all of which provided little or no relief of his pain. The patient reported that he told a HCP, “I’ll just live with [the pain].”

At the postsurgery follow-up, the patient reported that he had noticed dark urine and dark spots on his ears in the past. He also recounted that chronic joint pain was common in his family, with both his mother and brother receiving bilateral total knee replacements. Taking into consideration the surgical report and this new history, a urine assessment for HGA was ordered and yielded a diagnosis of alkaptonuria.
He later suffered an acute myocardial infarction leading to an incidental discovery of severe aortic stenosis on echocardiography, requiring coronary stent placements and transcatheter aortic valve replacement, respectively. He reported that with CV interventions and joint replacement surgeries, including bilateral knees and hips, his symptoms and quality of life began to significantly improve. However, he continued to have diffuse chronic joint pain unimproved with any single agent or intervention.
Discussion
Alkaptonuria is a rare autosomal recessive disorder, with a prevalence of about 1 in 100,000 to 250,000, which results from an enzyme error in an essential amino acid metabolism pathway (Figure 2).1 This inheritable gene mutation leads to ineffective homogentisate 1,2-dioxygenase (HGD), an enzyme required to break down HGA—which is a product of phenylalanine and tyrosine metabolism.2 As these patients engage in normal dietary protein intake, which includes essential amino acid phenylalanine, they develop clinically evident manifestations of the buildup and deposition of HGA.
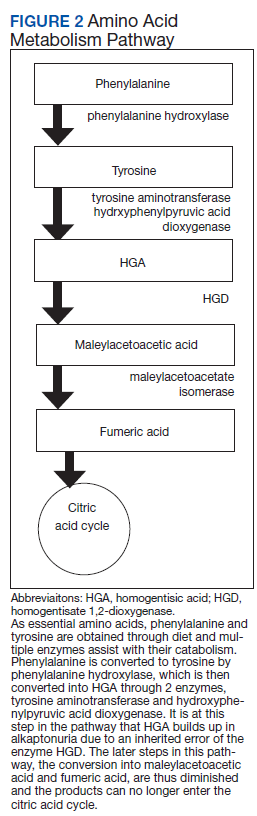
The rarity of alkaptonuria combined with the gradual buildup of HGA makes it difficult to diagnose. A common diagnostic technique is the visualization of discolored cartilage during surgical procedures, especially when discoloration in urine or skin is not immediately evident. A few case reports have noted surgical diagnosis of black or darkening tissue, known as ochronosis, following tendon rupture—a common complication of this disorder.3-5 Additional intervention-related case reports linked to the discovery of ochronosis include aortic valve replacement, lumbar discectomy, and bronchoscopy.6-9 Cases like these illustrate the complex, disabling, and unclear nature of this disorder when not diagnosed early in life.
The patient in this case communicated via e-mail about his tendon repair surgery. “Something very interesting was found during the surgery,” the patient explained. “I was diagnosed with the disease called ochronosis. I don’t know much about this disease but I am beginning to know why some of the things are happening to me and why I am always in constant pain.” This was the first recognized clue toward a diagnosis of alkaptonuria.
Pathophysiology
The pathophysiology of alkaptonuria is based on the extensive deposition of HGA throughout the body. Its progression is based on 3 clinical stages: clinical silence, clinical ochronosis, and ochronotic arthropathy.1 In the first stage the disorder is asymptomatic but includes its most notable feature—the gradual darkening of urine when exposed to air through oxidation of the renally excreted HGA. A similar process occurs in the blood through formed HGA-melanin compounds, which cause discoloration in cartilage.1 This internal metabolic disruption accounts for the disorder’s eventual second stage, clinical ochronosis, usually with an onset in the second or third decade. Prominent features noted on physical examination primarily include discoloration of ear pinnae and eye sclera but can involve the nose, gums, teeth, and hands. The third, final, and symptomatic stage, ochronotic arthropathy, occurs by the patient’s fourth to fifth decade and presents as joint pain, usually starting with the vertebrae and larger joints like hips, knees, and shoulders, that can appear as advanced early osteoarthritis on imaging.
Treatment
This clinical manifestation of alkaptonuria requires that HCPs manage patients with 3 strategies: decrease HGA buildup, alleviate symptoms, and monitor for disorder complications. Decreasing HGA buildup is a difficult aspect of management given the natural physiology of protein intake and metabolism. Three approaches to limit HGA buildup incorporate decreasing protein intake, inhibiting enzyme production of HGA, and increasing HGA excretion. Phenylalanine is an essential amino acid—meaning its levels are dependent on dietary protein intake. Patients should be advised to adhere to a low protein diet, especially phenylalanine and tyrosine, to lessen HGA concentrations.
Manipulating the metabolic pathway of phenylalanine with medication is a second option. An example of this is nitisinone, a US Food and Drug Administration-approved medication for treatment of tyrosinemia. It acts by inhibiting hydroxyphenylpyruvic acid dioxygenase, one of the enzymes that converts tyrosine into HGA, to prevent the buildup of damaging tyrosine byproducts. At low doses it has been effective in decreasing HGA concentrations in alkaptonuria and tyrosinemia.10,11 Due to this mechanism of action, nitisinone directly causes increased tyrosine levels. Therefore, tyrosine toxicity, usually not predicted by tyrosine levels, has been associated with eye-related adverse effects (AEs), including keratopathy, diminished visual acuity, and corneal tissue damage.1,2,10 Incidence of these AEs have not been clearly documented, but routine monitoring should include patient education on ocular symptoms and slit-lamp examinations.12
In addition, case reports have shown that high-dose ascorbic acid (vitamin C) promotes HGA, tyrosine, and phenylalanine excretion in urine, which may slow the progression of alkaptonuria, but clinical effect has not been proven.13 Additionally, high vitamin C intake is considered a risk factor for nephrolithiasis, which must be balanced with the increased risk of stone formation from HGA excretion.14 These dietary and medical options can be considered, especially in the setting of severe symptoms or complications, but the risks must be discussed with patients.
A second and commonly utilized strategy for caring for these patients is symptom management. As demonstrated through this case report, there is no clear medication that adequately addresses the pain caused by HGA deposition. Patients should be referred to a pain specialist to allow for single provider prescribing of pain medications. This patient found most relief and least AEs with tramadol but eventually self-discontinued due to diminishing pain relief. Given the eventual involvement of large joints, these patients will often require further symptom management with joint replacement surgery, usually much earlier than patients who undergo these surgeries for age-related osteoarthritis. The imperative aspect of symptom management is to engage patients in shared decision making with clear expectation setting.
Given the progressive nature of alkaptonuria, providers must monitor and address complications that are a result of this disorder. HGA becomes pathologic by binding to and weakening collagen fibers.5 This gradual buildup leads to degenerative changes in weight-bearing lower vertebrae and large joints that can become severe. Due to HGA’s interaction with collagen fibers, tendon involvement leading to inflammation, calcification, and rupture can result as patients enter the third stage, ochronotic arthropathy, of the disorder (Figure 3).15 Many of these arthropathies will require medical and surgical management and can be urgent in situations like tendon ruptures and meniscal tears. Understanding the pathophysiology of tendinopathies in patients with alkaptonuria also can aid orthopedic surgeons during the postoperative period where patients may be at risk for poor healing.5
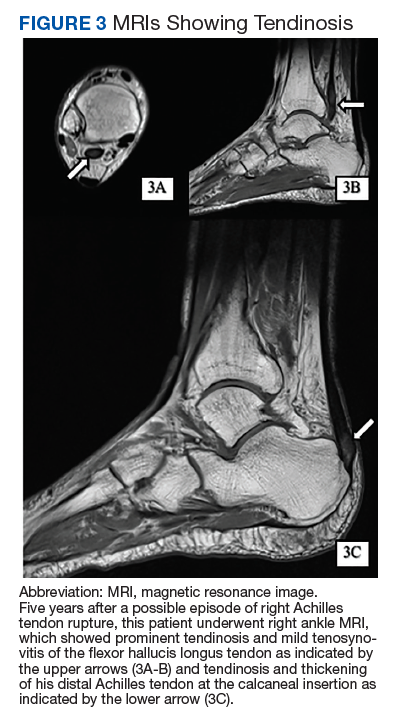
A second area of complications includes CV involvement. This patient was diagnosed with premature atherosclerosis and underwent cardiac interventions, including coronary stent placement and valve replacements at age 63 years. This early cardiac involvement was likely due in part to the deposition of HGA and collagen injury in CV tissue leading to damage of the endocardium, aortic intima, heart valves, and coronary arteries.1 HCPs should monitor for these manifestations with regular visits, chest computed tomography, and echocardiographic studies.2
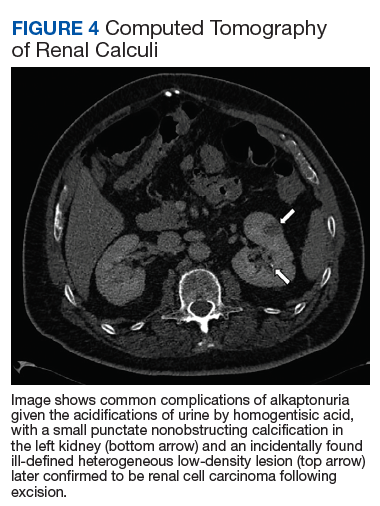
The most classic aspect of this rare disorder is urine darkening due to the renal excretion of HGA and comprises the third area of complications. This process leads to chronically acidic urine—every urinalysis in this patient’s chart displayed the lowest pH measurable—and an increased risk for calcification and precipitation of solutes within the kidney and urinary tract (Figure 4). Both X-ray and ultrasound imaging should be used to identify kidney and prostate stones in the setting of abdominal or genitourinary pain or infection. Patients with diminished renal function may manifest a more severe and rapidly progressing form of alkaptonuria that exacerbates symptoms and complications, but direct damage to the kidneys by HGA is not evident.
Conclusion
Alkaptonuria is a rare autosomal recessive metabolic disorder that has a progressively debilitating pathophysiologic course spanning decades of a patient’s life. Its low prevalence and gradually progressive nature make it a difficult diagnosis to make without clinical suspicion. In patients with early-onset degenerative joint disease, tendinopathy, and cartilage or skin discoloration, congenital metabolic disorders like alkaptonuria should be considered.
As this case shows, suspicion and diagnosis can occur during surgical intervention in which tendon discoloration is directly visualized, especially in patients without prominent skin or cartilage discoloration. Once the diagnosis is made through elevated levels of urine HGA, there are 3 management strategies, including decreasing homogentisic acid buildup, providing symptom management, and monitoring for arthropathic, CV, and genitourinary complications.
1. Aquaron R. Alkaptonuria: a very rare metabolic disorder. Indian J Biochem Biophys. 2013;50(5):339-344.
2. Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111-2121.
3. Alajoulin OA, Alsbou MS, Ja’afreh SO, Kalbouneh HM. Spontaneous Achilles tendon rupture in alkaptonuria. Saudi Med J. 2015;36(12):1486-1489.
4. Manoj Kumar RV, Rajasekaran S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br. 2003;85(6):883-886.
5. Tanoglu O, Arican G, Ozmeric A, Alemdaroglu KB, Caydere M. Calcaneal avulsion of an ochronotic Achilles tendon: a case report. J Foot Ankle Surg. 2018;57(1):179-183.
6. Schuuring MJ, Delemarre B, Keyhan-Falsafi AM, van der Bilt IA. Mending a darkened heart: alkaptonuria discovered during aortic valve replacement. Circulation. 2016;133(12):e444-445.
7. Hiroyoshi J, Saito A, Panthee N, et al. Aortic valve replacement for aortic stenosis caused by alkaptonuria. Ann Thorac Surg. 2013;95(3):1076-1079.
8. Parambil JG, Daniels CE, Zehr KJ, Utz JP. Alkaptonuria diagnosed by flexible bronchoscopy. Chest. 2005;128(5):3678-3680.
9. Farzannia A, Shokouhi G, Hadidchi S. Alkaptonuria and lumbar disc herniation. Report of three cases. J Neurosurg. 2003;98(suppl 1):87-89.
10. Introne WJ, Perry MB, Troendle J, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307-314.
11. Gissen P, Preece MA, Willshaw HA, McKiernan PJ. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. J Inherit Metab Dis. 2003;26(1):13-16.
12. Khedr M, Judd S, Briggs MC, et al. Asymptomatic corneal keratopathy secondary to hypertyrosinaemia following low dose nitisinone and a literature review of tyrosine keratopathy in alkaptonuria. JIMD Rep. 2018;40:31-37.
13. Wolff JA, Barshop B, Nyhan WL, et al. Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res. 1989;26(2):140-144.
14. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232.
15. Abate M, Salini V, Andia I. Tendons involvement in congenital metabolic disorders. Adv Exp Med Biol. 2016;920:117-122.
Alkaptonuria is a rare autosomal recessive disorder uniquely known for causing black, or darkened, urine when left standing due to the renal excretion of excess homogentisic acid (HGA). When this disorder goes undiagnosed, as demonstrated in this case, patients experience its many complications without a unifying explanation. The disorder has 3 clinical stages that occur in a predictable order: clinical silence, clinical ochronosis, and ochronotic arthropathy. These stages lead to multiple musculoskeletal, cardiovascular (CV), and renal complications that can be mitigated with management focused on decreasing homogentisic acid buildup, alleviating symptoms, and close monitoring for these complications.
Case Presentation
A 61-year-old African American male with a medical history of multiple traumatic fractures, right Achilles tendon injury, early-onset multijoint osteoarthritis, chronic low back pain, and recurrent nephrolithiasis presented to the emergency department with sudden onset of sharp left ankle pain while moving furniture. His physical exam revealed a positive Thompson test—lack of foot plantar flexion with calf squeeze—and a subsequent magnetic resonance image (MRI) showed evidence of an acute Achilles tendon rupture.
Given these findings the patient was treated with nonsteroidal anti-inflammatory drugs (NSAIDs) and rest to allow for resolution of swelling and inflammation, followed by elective surgery a month later to repair the ruptured tendon. An operative report following his surgery described “black ends to the area where the Achilles was ruptured…and tendinopathy of the flexor hallucis longus with blackening of the flexor.”
A more in-depth patient history revealed that he underwent multiple invasive and noninvasive interventions for his chronic low back and joint pain with medical management of a prior right Achilles tendon injury. His medical history also included multiple nonspecific diagnoses, such as premature atherosclerosis (diagnosed in his third decade), severe lumbar degenerative disc disease, several tendonopathies and cartilage injuries (Figure 1), pseudogout (following calcium pyrophosphate dehydrate crystals found from a left knee aspirate), and chronic pain syndrome. Along this diagnostic journey, he had several health care providers (HCPs) in rheumatology, orthopedic surgery, pain management, and podiatry who offered a range of symptom management options, including physical therapy, NSAIDs, opioid agonists, tricyclic antidepressants, gabapentin, colchicine, and epidural steroid injections, all of which provided little or no relief of his pain. The patient reported that he told a HCP, “I’ll just live with [the pain].”
At the postsurgery follow-up, the patient reported that he had noticed dark urine and dark spots on his ears in the past. He also recounted that chronic joint pain was common in his family, with both his mother and brother receiving bilateral total knee replacements. Taking into consideration the surgical report and this new history, a urine assessment for HGA was ordered and yielded a diagnosis of alkaptonuria.
He later suffered an acute myocardial infarction leading to an incidental discovery of severe aortic stenosis on echocardiography, requiring coronary stent placements and transcatheter aortic valve replacement, respectively. He reported that with CV interventions and joint replacement surgeries, including bilateral knees and hips, his symptoms and quality of life began to significantly improve. However, he continued to have diffuse chronic joint pain unimproved with any single agent or intervention.
Discussion
Alkaptonuria is a rare autosomal recessive disorder, with a prevalence of about 1 in 100,000 to 250,000, which results from an enzyme error in an essential amino acid metabolism pathway (Figure 2).1 This inheritable gene mutation leads to ineffective homogentisate 1,2-dioxygenase (HGD), an enzyme required to break down HGA—which is a product of phenylalanine and tyrosine metabolism.2 As these patients engage in normal dietary protein intake, which includes essential amino acid phenylalanine, they develop clinically evident manifestations of the buildup and deposition of HGA.
The rarity of alkaptonuria combined with the gradual buildup of HGA makes it difficult to diagnose. A common diagnostic technique is the visualization of discolored cartilage during surgical procedures, especially when discoloration in urine or skin is not immediately evident. A few case reports have noted surgical diagnosis of black or darkening tissue, known as ochronosis, following tendon rupture—a common complication of this disorder.3-5 Additional intervention-related case reports linked to the discovery of ochronosis include aortic valve replacement, lumbar discectomy, and bronchoscopy.6-9 Cases like these illustrate the complex, disabling, and unclear nature of this disorder when not diagnosed early in life.
The patient in this case communicated via e-mail about his tendon repair surgery. “Something very interesting was found during the surgery,” the patient explained. “I was diagnosed with the disease called ochronosis. I don’t know much about this disease but I am beginning to know why some of the things are happening to me and why I am always in constant pain.” This was the first recognized clue toward a diagnosis of alkaptonuria.
Pathophysiology
The pathophysiology of alkaptonuria is based on the extensive deposition of HGA throughout the body. Its progression is based on 3 clinical stages: clinical silence, clinical ochronosis, and ochronotic arthropathy.1 In the first stage the disorder is asymptomatic but includes its most notable feature—the gradual darkening of urine when exposed to air through oxidation of the renally excreted HGA. A similar process occurs in the blood through formed HGA-melanin compounds, which cause discoloration in cartilage.1 This internal metabolic disruption accounts for the disorder’s eventual second stage, clinical ochronosis, usually with an onset in the second or third decade. Prominent features noted on physical examination primarily include discoloration of ear pinnae and eye sclera but can involve the nose, gums, teeth, and hands. The third, final, and symptomatic stage, ochronotic arthropathy, occurs by the patient’s fourth to fifth decade and presents as joint pain, usually starting with the vertebrae and larger joints like hips, knees, and shoulders, that can appear as advanced early osteoarthritis on imaging.
Treatment
This clinical manifestation of alkaptonuria requires that HCPs manage patients with 3 strategies: decrease HGA buildup, alleviate symptoms, and monitor for disorder complications. Decreasing HGA buildup is a difficult aspect of management given the natural physiology of protein intake and metabolism. Three approaches to limit HGA buildup incorporate decreasing protein intake, inhibiting enzyme production of HGA, and increasing HGA excretion. Phenylalanine is an essential amino acid—meaning its levels are dependent on dietary protein intake. Patients should be advised to adhere to a low protein diet, especially phenylalanine and tyrosine, to lessen HGA concentrations.
Manipulating the metabolic pathway of phenylalanine with medication is a second option. An example of this is nitisinone, a US Food and Drug Administration-approved medication for treatment of tyrosinemia. It acts by inhibiting hydroxyphenylpyruvic acid dioxygenase, one of the enzymes that converts tyrosine into HGA, to prevent the buildup of damaging tyrosine byproducts. At low doses it has been effective in decreasing HGA concentrations in alkaptonuria and tyrosinemia.10,11 Due to this mechanism of action, nitisinone directly causes increased tyrosine levels. Therefore, tyrosine toxicity, usually not predicted by tyrosine levels, has been associated with eye-related adverse effects (AEs), including keratopathy, diminished visual acuity, and corneal tissue damage.1,2,10 Incidence of these AEs have not been clearly documented, but routine monitoring should include patient education on ocular symptoms and slit-lamp examinations.12
In addition, case reports have shown that high-dose ascorbic acid (vitamin C) promotes HGA, tyrosine, and phenylalanine excretion in urine, which may slow the progression of alkaptonuria, but clinical effect has not been proven.13 Additionally, high vitamin C intake is considered a risk factor for nephrolithiasis, which must be balanced with the increased risk of stone formation from HGA excretion.14 These dietary and medical options can be considered, especially in the setting of severe symptoms or complications, but the risks must be discussed with patients.
A second and commonly utilized strategy for caring for these patients is symptom management. As demonstrated through this case report, there is no clear medication that adequately addresses the pain caused by HGA deposition. Patients should be referred to a pain specialist to allow for single provider prescribing of pain medications. This patient found most relief and least AEs with tramadol but eventually self-discontinued due to diminishing pain relief. Given the eventual involvement of large joints, these patients will often require further symptom management with joint replacement surgery, usually much earlier than patients who undergo these surgeries for age-related osteoarthritis. The imperative aspect of symptom management is to engage patients in shared decision making with clear expectation setting.
Given the progressive nature of alkaptonuria, providers must monitor and address complications that are a result of this disorder. HGA becomes pathologic by binding to and weakening collagen fibers.5 This gradual buildup leads to degenerative changes in weight-bearing lower vertebrae and large joints that can become severe. Due to HGA’s interaction with collagen fibers, tendon involvement leading to inflammation, calcification, and rupture can result as patients enter the third stage, ochronotic arthropathy, of the disorder (Figure 3).15 Many of these arthropathies will require medical and surgical management and can be urgent in situations like tendon ruptures and meniscal tears. Understanding the pathophysiology of tendinopathies in patients with alkaptonuria also can aid orthopedic surgeons during the postoperative period where patients may be at risk for poor healing.5
A second area of complications includes CV involvement. This patient was diagnosed with premature atherosclerosis and underwent cardiac interventions, including coronary stent placement and valve replacements at age 63 years. This early cardiac involvement was likely due in part to the deposition of HGA and collagen injury in CV tissue leading to damage of the endocardium, aortic intima, heart valves, and coronary arteries.1 HCPs should monitor for these manifestations with regular visits, chest computed tomography, and echocardiographic studies.2
The most classic aspect of this rare disorder is urine darkening due to the renal excretion of HGA and comprises the third area of complications. This process leads to chronically acidic urine—every urinalysis in this patient’s chart displayed the lowest pH measurable—and an increased risk for calcification and precipitation of solutes within the kidney and urinary tract (Figure 4). Both X-ray and ultrasound imaging should be used to identify kidney and prostate stones in the setting of abdominal or genitourinary pain or infection. Patients with diminished renal function may manifest a more severe and rapidly progressing form of alkaptonuria that exacerbates symptoms and complications, but direct damage to the kidneys by HGA is not evident.
Conclusion
Alkaptonuria is a rare autosomal recessive metabolic disorder that has a progressively debilitating pathophysiologic course spanning decades of a patient’s life. Its low prevalence and gradually progressive nature make it a difficult diagnosis to make without clinical suspicion. In patients with early-onset degenerative joint disease, tendinopathy, and cartilage or skin discoloration, congenital metabolic disorders like alkaptonuria should be considered.
As this case shows, suspicion and diagnosis can occur during surgical intervention in which tendon discoloration is directly visualized, especially in patients without prominent skin or cartilage discoloration. Once the diagnosis is made through elevated levels of urine HGA, there are 3 management strategies, including decreasing homogentisic acid buildup, providing symptom management, and monitoring for arthropathic, CV, and genitourinary complications.
Alkaptonuria is a rare autosomal recessive disorder uniquely known for causing black, or darkened, urine when left standing due to the renal excretion of excess homogentisic acid (HGA). When this disorder goes undiagnosed, as demonstrated in this case, patients experience its many complications without a unifying explanation. The disorder has 3 clinical stages that occur in a predictable order: clinical silence, clinical ochronosis, and ochronotic arthropathy. These stages lead to multiple musculoskeletal, cardiovascular (CV), and renal complications that can be mitigated with management focused on decreasing homogentisic acid buildup, alleviating symptoms, and close monitoring for these complications.
Case Presentation
A 61-year-old African American male with a medical history of multiple traumatic fractures, right Achilles tendon injury, early-onset multijoint osteoarthritis, chronic low back pain, and recurrent nephrolithiasis presented to the emergency department with sudden onset of sharp left ankle pain while moving furniture. His physical exam revealed a positive Thompson test—lack of foot plantar flexion with calf squeeze—and a subsequent magnetic resonance image (MRI) showed evidence of an acute Achilles tendon rupture.
Given these findings the patient was treated with nonsteroidal anti-inflammatory drugs (NSAIDs) and rest to allow for resolution of swelling and inflammation, followed by elective surgery a month later to repair the ruptured tendon. An operative report following his surgery described “black ends to the area where the Achilles was ruptured…and tendinopathy of the flexor hallucis longus with blackening of the flexor.”
A more in-depth patient history revealed that he underwent multiple invasive and noninvasive interventions for his chronic low back and joint pain with medical management of a prior right Achilles tendon injury. His medical history also included multiple nonspecific diagnoses, such as premature atherosclerosis (diagnosed in his third decade), severe lumbar degenerative disc disease, several tendonopathies and cartilage injuries (Figure 1), pseudogout (following calcium pyrophosphate dehydrate crystals found from a left knee aspirate), and chronic pain syndrome. Along this diagnostic journey, he had several health care providers (HCPs) in rheumatology, orthopedic surgery, pain management, and podiatry who offered a range of symptom management options, including physical therapy, NSAIDs, opioid agonists, tricyclic antidepressants, gabapentin, colchicine, and epidural steroid injections, all of which provided little or no relief of his pain. The patient reported that he told a HCP, “I’ll just live with [the pain].”
At the postsurgery follow-up, the patient reported that he had noticed dark urine and dark spots on his ears in the past. He also recounted that chronic joint pain was common in his family, with both his mother and brother receiving bilateral total knee replacements. Taking into consideration the surgical report and this new history, a urine assessment for HGA was ordered and yielded a diagnosis of alkaptonuria.
He later suffered an acute myocardial infarction leading to an incidental discovery of severe aortic stenosis on echocardiography, requiring coronary stent placements and transcatheter aortic valve replacement, respectively. He reported that with CV interventions and joint replacement surgeries, including bilateral knees and hips, his symptoms and quality of life began to significantly improve. However, he continued to have diffuse chronic joint pain unimproved with any single agent or intervention.
Discussion
Alkaptonuria is a rare autosomal recessive disorder, with a prevalence of about 1 in 100,000 to 250,000, which results from an enzyme error in an essential amino acid metabolism pathway (Figure 2).1 This inheritable gene mutation leads to ineffective homogentisate 1,2-dioxygenase (HGD), an enzyme required to break down HGA—which is a product of phenylalanine and tyrosine metabolism.2 As these patients engage in normal dietary protein intake, which includes essential amino acid phenylalanine, they develop clinically evident manifestations of the buildup and deposition of HGA.
The rarity of alkaptonuria combined with the gradual buildup of HGA makes it difficult to diagnose. A common diagnostic technique is the visualization of discolored cartilage during surgical procedures, especially when discoloration in urine or skin is not immediately evident. A few case reports have noted surgical diagnosis of black or darkening tissue, known as ochronosis, following tendon rupture—a common complication of this disorder.3-5 Additional intervention-related case reports linked to the discovery of ochronosis include aortic valve replacement, lumbar discectomy, and bronchoscopy.6-9 Cases like these illustrate the complex, disabling, and unclear nature of this disorder when not diagnosed early in life.
The patient in this case communicated via e-mail about his tendon repair surgery. “Something very interesting was found during the surgery,” the patient explained. “I was diagnosed with the disease called ochronosis. I don’t know much about this disease but I am beginning to know why some of the things are happening to me and why I am always in constant pain.” This was the first recognized clue toward a diagnosis of alkaptonuria.
Pathophysiology
The pathophysiology of alkaptonuria is based on the extensive deposition of HGA throughout the body. Its progression is based on 3 clinical stages: clinical silence, clinical ochronosis, and ochronotic arthropathy.1 In the first stage the disorder is asymptomatic but includes its most notable feature—the gradual darkening of urine when exposed to air through oxidation of the renally excreted HGA. A similar process occurs in the blood through formed HGA-melanin compounds, which cause discoloration in cartilage.1 This internal metabolic disruption accounts for the disorder’s eventual second stage, clinical ochronosis, usually with an onset in the second or third decade. Prominent features noted on physical examination primarily include discoloration of ear pinnae and eye sclera but can involve the nose, gums, teeth, and hands. The third, final, and symptomatic stage, ochronotic arthropathy, occurs by the patient’s fourth to fifth decade and presents as joint pain, usually starting with the vertebrae and larger joints like hips, knees, and shoulders, that can appear as advanced early osteoarthritis on imaging.
Treatment
This clinical manifestation of alkaptonuria requires that HCPs manage patients with 3 strategies: decrease HGA buildup, alleviate symptoms, and monitor for disorder complications. Decreasing HGA buildup is a difficult aspect of management given the natural physiology of protein intake and metabolism. Three approaches to limit HGA buildup incorporate decreasing protein intake, inhibiting enzyme production of HGA, and increasing HGA excretion. Phenylalanine is an essential amino acid—meaning its levels are dependent on dietary protein intake. Patients should be advised to adhere to a low protein diet, especially phenylalanine and tyrosine, to lessen HGA concentrations.
Manipulating the metabolic pathway of phenylalanine with medication is a second option. An example of this is nitisinone, a US Food and Drug Administration-approved medication for treatment of tyrosinemia. It acts by inhibiting hydroxyphenylpyruvic acid dioxygenase, one of the enzymes that converts tyrosine into HGA, to prevent the buildup of damaging tyrosine byproducts. At low doses it has been effective in decreasing HGA concentrations in alkaptonuria and tyrosinemia.10,11 Due to this mechanism of action, nitisinone directly causes increased tyrosine levels. Therefore, tyrosine toxicity, usually not predicted by tyrosine levels, has been associated with eye-related adverse effects (AEs), including keratopathy, diminished visual acuity, and corneal tissue damage.1,2,10 Incidence of these AEs have not been clearly documented, but routine monitoring should include patient education on ocular symptoms and slit-lamp examinations.12
In addition, case reports have shown that high-dose ascorbic acid (vitamin C) promotes HGA, tyrosine, and phenylalanine excretion in urine, which may slow the progression of alkaptonuria, but clinical effect has not been proven.13 Additionally, high vitamin C intake is considered a risk factor for nephrolithiasis, which must be balanced with the increased risk of stone formation from HGA excretion.14 These dietary and medical options can be considered, especially in the setting of severe symptoms or complications, but the risks must be discussed with patients.
A second and commonly utilized strategy for caring for these patients is symptom management. As demonstrated through this case report, there is no clear medication that adequately addresses the pain caused by HGA deposition. Patients should be referred to a pain specialist to allow for single provider prescribing of pain medications. This patient found most relief and least AEs with tramadol but eventually self-discontinued due to diminishing pain relief. Given the eventual involvement of large joints, these patients will often require further symptom management with joint replacement surgery, usually much earlier than patients who undergo these surgeries for age-related osteoarthritis. The imperative aspect of symptom management is to engage patients in shared decision making with clear expectation setting.
Given the progressive nature of alkaptonuria, providers must monitor and address complications that are a result of this disorder. HGA becomes pathologic by binding to and weakening collagen fibers.5 This gradual buildup leads to degenerative changes in weight-bearing lower vertebrae and large joints that can become severe. Due to HGA’s interaction with collagen fibers, tendon involvement leading to inflammation, calcification, and rupture can result as patients enter the third stage, ochronotic arthropathy, of the disorder (Figure 3).15 Many of these arthropathies will require medical and surgical management and can be urgent in situations like tendon ruptures and meniscal tears. Understanding the pathophysiology of tendinopathies in patients with alkaptonuria also can aid orthopedic surgeons during the postoperative period where patients may be at risk for poor healing.5
A second area of complications includes CV involvement. This patient was diagnosed with premature atherosclerosis and underwent cardiac interventions, including coronary stent placement and valve replacements at age 63 years. This early cardiac involvement was likely due in part to the deposition of HGA and collagen injury in CV tissue leading to damage of the endocardium, aortic intima, heart valves, and coronary arteries.1 HCPs should monitor for these manifestations with regular visits, chest computed tomography, and echocardiographic studies.2
The most classic aspect of this rare disorder is urine darkening due to the renal excretion of HGA and comprises the third area of complications. This process leads to chronically acidic urine—every urinalysis in this patient’s chart displayed the lowest pH measurable—and an increased risk for calcification and precipitation of solutes within the kidney and urinary tract (Figure 4). Both X-ray and ultrasound imaging should be used to identify kidney and prostate stones in the setting of abdominal or genitourinary pain or infection. Patients with diminished renal function may manifest a more severe and rapidly progressing form of alkaptonuria that exacerbates symptoms and complications, but direct damage to the kidneys by HGA is not evident.
Conclusion
Alkaptonuria is a rare autosomal recessive metabolic disorder that has a progressively debilitating pathophysiologic course spanning decades of a patient’s life. Its low prevalence and gradually progressive nature make it a difficult diagnosis to make without clinical suspicion. In patients with early-onset degenerative joint disease, tendinopathy, and cartilage or skin discoloration, congenital metabolic disorders like alkaptonuria should be considered.
As this case shows, suspicion and diagnosis can occur during surgical intervention in which tendon discoloration is directly visualized, especially in patients without prominent skin or cartilage discoloration. Once the diagnosis is made through elevated levels of urine HGA, there are 3 management strategies, including decreasing homogentisic acid buildup, providing symptom management, and monitoring for arthropathic, CV, and genitourinary complications.
1. Aquaron R. Alkaptonuria: a very rare metabolic disorder. Indian J Biochem Biophys. 2013;50(5):339-344.
2. Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111-2121.
3. Alajoulin OA, Alsbou MS, Ja’afreh SO, Kalbouneh HM. Spontaneous Achilles tendon rupture in alkaptonuria. Saudi Med J. 2015;36(12):1486-1489.
4. Manoj Kumar RV, Rajasekaran S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br. 2003;85(6):883-886.
5. Tanoglu O, Arican G, Ozmeric A, Alemdaroglu KB, Caydere M. Calcaneal avulsion of an ochronotic Achilles tendon: a case report. J Foot Ankle Surg. 2018;57(1):179-183.
6. Schuuring MJ, Delemarre B, Keyhan-Falsafi AM, van der Bilt IA. Mending a darkened heart: alkaptonuria discovered during aortic valve replacement. Circulation. 2016;133(12):e444-445.
7. Hiroyoshi J, Saito A, Panthee N, et al. Aortic valve replacement for aortic stenosis caused by alkaptonuria. Ann Thorac Surg. 2013;95(3):1076-1079.
8. Parambil JG, Daniels CE, Zehr KJ, Utz JP. Alkaptonuria diagnosed by flexible bronchoscopy. Chest. 2005;128(5):3678-3680.
9. Farzannia A, Shokouhi G, Hadidchi S. Alkaptonuria and lumbar disc herniation. Report of three cases. J Neurosurg. 2003;98(suppl 1):87-89.
10. Introne WJ, Perry MB, Troendle J, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307-314.
11. Gissen P, Preece MA, Willshaw HA, McKiernan PJ. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. J Inherit Metab Dis. 2003;26(1):13-16.
12. Khedr M, Judd S, Briggs MC, et al. Asymptomatic corneal keratopathy secondary to hypertyrosinaemia following low dose nitisinone and a literature review of tyrosine keratopathy in alkaptonuria. JIMD Rep. 2018;40:31-37.
13. Wolff JA, Barshop B, Nyhan WL, et al. Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res. 1989;26(2):140-144.
14. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232.
15. Abate M, Salini V, Andia I. Tendons involvement in congenital metabolic disorders. Adv Exp Med Biol. 2016;920:117-122.
1. Aquaron R. Alkaptonuria: a very rare metabolic disorder. Indian J Biochem Biophys. 2013;50(5):339-344.
2. Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111-2121.
3. Alajoulin OA, Alsbou MS, Ja’afreh SO, Kalbouneh HM. Spontaneous Achilles tendon rupture in alkaptonuria. Saudi Med J. 2015;36(12):1486-1489.
4. Manoj Kumar RV, Rajasekaran S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br. 2003;85(6):883-886.
5. Tanoglu O, Arican G, Ozmeric A, Alemdaroglu KB, Caydere M. Calcaneal avulsion of an ochronotic Achilles tendon: a case report. J Foot Ankle Surg. 2018;57(1):179-183.
6. Schuuring MJ, Delemarre B, Keyhan-Falsafi AM, van der Bilt IA. Mending a darkened heart: alkaptonuria discovered during aortic valve replacement. Circulation. 2016;133(12):e444-445.
7. Hiroyoshi J, Saito A, Panthee N, et al. Aortic valve replacement for aortic stenosis caused by alkaptonuria. Ann Thorac Surg. 2013;95(3):1076-1079.
8. Parambil JG, Daniels CE, Zehr KJ, Utz JP. Alkaptonuria diagnosed by flexible bronchoscopy. Chest. 2005;128(5):3678-3680.
9. Farzannia A, Shokouhi G, Hadidchi S. Alkaptonuria and lumbar disc herniation. Report of three cases. J Neurosurg. 2003;98(suppl 1):87-89.
10. Introne WJ, Perry MB, Troendle J, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307-314.
11. Gissen P, Preece MA, Willshaw HA, McKiernan PJ. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. J Inherit Metab Dis. 2003;26(1):13-16.
12. Khedr M, Judd S, Briggs MC, et al. Asymptomatic corneal keratopathy secondary to hypertyrosinaemia following low dose nitisinone and a literature review of tyrosine keratopathy in alkaptonuria. JIMD Rep. 2018;40:31-37.
13. Wolff JA, Barshop B, Nyhan WL, et al. Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res. 1989;26(2):140-144.
14. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232.
15. Abate M, Salini V, Andia I. Tendons involvement in congenital metabolic disorders. Adv Exp Med Biol. 2016;920:117-122.
A Veteran With a Solitary Pulmonary Nodule
Case Presentation. A 69-year-old veteran presented with an intermittent, waxing and waning cough. He had never smoked and had no family history of lung cancer. His primary care physician ordered a chest radiograph, which revealed a nodular opacity within the lingula concerning for a parenchymal nodule. Further characterization with a chest computed tomography (CT) demonstrated a 1.4-cm left upper lobe subpleural nodule with small satellite nodules (Figure 1). Given these imaging findings, the patient was referred to the pulmonary clinic.
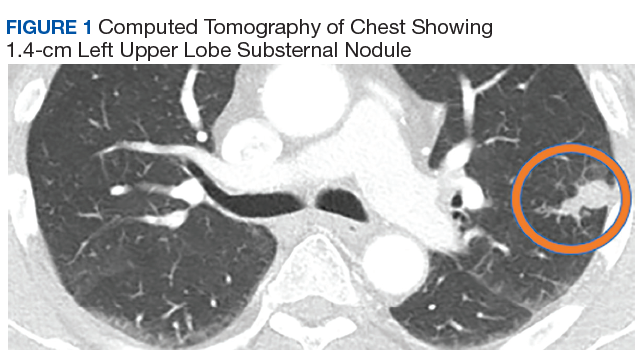
►Lauren Kearney, MD, Medical Resident, VA Boston Healthcare System (VABHS) and Boston Medical Center. What is the differential diagnosis of a solitary pulmonary nodule? What characteristics of the nodule do you consider to differentiate these diagnoses?
►Renda Wiener, MD, Pulmonary and Critical Care, VABHS, and Assistant Professor of Medicine, Boston University School of Medicine. Pulmonary nodules are well-defined lesions < 3 cm in diameter that are surrounded by lung parenchyma. Although cancer is a possibility (including primary lung cancers, metastatic cancers, or carcinoid tumors), most small nodules do not turn out to be malignant.1 Benign etiologies include infections, benign tumors, vascular malformations, and inflammatory conditions. Infectious causes of nodules are often granulomatous in nature, including fungi, Mycobacterium tuberculosis, and nontuberculous mycobacteria. Benign tumors are most commonly hamartomas, and these may be clearly distinguished based on imaging characteristics. Pulmonary arteriovenous malformations, hematomas, and infarcts may present as nodules as well. Inflammatory causes of nodules are important and relatively common, including granulomatosis with polyangiitis, rheumatoid arthritis, sarcoidosis, amyloidosis, and rounded atelectasis.
To distinguish benign from malignant etiologies, we look for several features of pulmonary nodules on imaging. Larger size, irregular borders, and upper lobe location all increase the likelihood of cancer, whereas solid attenuation and calcification make cancer less likely. One of the most reassuring findings that suggests a benign etiology is lack of growth over a period of surveillance; after 2 years without growth we typically consider a nodule benign.1 And of course, we also consider the patient’s symptoms and risk factors: weight loss, hemoptysis, a history of cigarette smoking or asbestos exposure, or family history of cancer all increase the likelihood of malignancy.
►Dr. Kearney. Given that the differential diagnosis is so broad, how do you think about the next step in evaluating a pulmonary nodule? How do you approach shared decision making with the patient?
►Dr. Wiener. The characteristics of the patient, the nodule, and the circumstances in which the nodule were discovered are all important to consider. Incidental pulmonary nodules are often found on chest imaging. The imaging characteristics of the nodule are important, as are the patient’s risk factors. A similarly appearing nodule can have very different implications if the patient is a never-smoker exposed to endemic fungi, or a long-time smoker enrolled in a lung cancer screening program. Consultation with a pulmonologist is often appropriate.
It’s important to note that we lack high-quality evidence on the optimal strategy to evaluate pulmonary nodules, and there is no single “right answer“ for all patients. For patients with a low risk of malignancy (< 5%-10%)—which comprises the majority of the incidental nodules discovered—we typically favor serial CT surveillance of the nodule over a period of a few years, whereas for patients at high risk of malignancy (> 65%), we favor early surgical resection if the patient is able to tolerate that. For patients with an intermediate risk of malignancy (~5%-65%), we might consider serial CT surveillance, positron emission tomography (PET) scan, or biopsy.1 The American College of Chest Physicians guidelines for pulmonary nodule evaluation recommend discussing with patients the different options and the trade-offs of these options in a shared decision-making process.1
►Dr. Kearney. The patient’s pulmonologist laid out options, including monitoring with serial CT scans, obtaining a PET scan, performing CT-guided needle biopsy, or referring for surgical excision. In this case, the patient elected to undergo CT-guided needle biopsy. Dr. Huang, can you discuss the pathology results?
►Qin Huang, MD, Pathology and Laboratory Medicine, VABHS, and Assistant Professor of Pathology, Harvard Medical School (HMS). The microscopic examination of the needle biopsy of the lung mass revealed rare clusters of atypical cells with crushed cells adjacent to an extensive area of necrosis with scarring. The atypical cells were suspicious for carcinoma. The Gomori methenamine silver (GMS) and periodic acid-Schiff (PAS) stains were negative for common bacterial and fungal microorganisms.
►Dr. Kearney. The tumor board, pulmonologist, and patient decide to move forward with video-assisted excisional biopsy with lymphadenectomy. Dr. Huang, can you interpret the pathology?
►Dr. Huang. Figure 2 showed an hemotoxylin and eosin (H&E)-stained lung resection tissue section with multiple caseating necrotic granulomas. No foreign bodies were identified. There was no evidence of malignancy. The GMS stain revealed a fungal microorganism oval with morphology typical of histoplasma capsulatum (Figure 3).
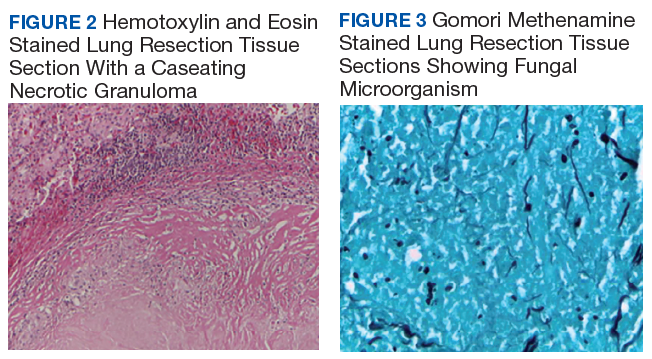
►Dr. Kearney. What are some of the different ways histoplasmosis can present? Which of these diagnoses fits this patient’s presentation?
►Judy Strymish, MD, Infectious Disease, VABHS, and Assistant Professor of Medicine, HMS. Most patients who inhale histoplasmosis spores develop asymptomatic or self-limited infection that is usually not detected. Patients at risk of symptomatic and clinically relevant disease include those who are immunocompromised, at extremes of ages, or exposed to larger inoculums. Acute pulmonary histoplasmosis can present with cough, shortness of breath, fever, chills, and less commonly, rheumatologic complaints such as erythema nodosum or erythema multiforme. Imaging often shows patchy infiltrates and enlarged mediastinal and hilar lymphadenopathy. Patients can go on to develop subacute or chronic pulmonary disease with focal opacities and mediastinal and hilar lymphadenopathy. Those with chronic disease can have cavitary lesions similar to patients with tuberculosis. Progressive disseminated histoplasmosis can develop in immunocompromised patients and disseminate through the reticuloendothelial system to other organs with the gastrointestinal tract, central nervous system, and adrenal glands.2
Pulmonary nodules are common incidental finding on chest imaging of patients who reside in histoplasmosis endemic regions, and they are often hard to differentiate from malignancies. There are 3 mediastinal manifestations: adenitis, granuloma, and fibrosis. Usually the syndromes are subclinical, but occasionally the nodes cause symptoms by impinging on other structures.2
This patient had a solitary pulmonary nodule with none of the associated features mentioned above. Pathology showed caseating granuloma and confirmed histoplasmosis.
►Dr. Kearney. Given the diagnosis of solitary histoplasmoma, how should this patient be managed?
►Dr. Strymish. The optimal therapy for histoplasmosis depends on the patient’s clinical syndrome. Most infections are self-limited and require no therapy. However, patients who are immunocompromised, exposed to large inoculum, and have progressive disease require antifungal treatment, usually with itraconazole for mild-to-moderate disease and a combination of azole therapy and amphotericin B with extensive disease. Patients with few solitary pulmonary nodules do not benefit from antifungal therapy as the nodule could represent quiescent disease that is unlikely to have clinical impact; in this case, the treatment would be higher risk than the nodule.3
►Dr. Kearney. While the discussion of the diagnosis is interesting, it is also important to acknowledge what the patient went through to arrive at this, an essentially benign diagnosis: 8 months, multiple imaging studies, and 2 invasive diagnostic procedures. Further, the patient had to grapple with the possibility of a diagnosis of cancer. Dr. Wiener, can you talk about the challenges in communicating with patients about pulmonary nodules when cancer is on the differential? What are some of the harms patients face and how can clinicians work to mitigate these harms?

►Dr. Wiener. My colleague Dr. Christopher Slatore of the Portland VA Medical Center and I studied communication about pulmonary nodules in a series of surveys and qualitative studies of patients with pulmonary nodules and the clinicians who take care of them. We found that there seems to be a disconnect between patients’ perceptions of pulmonary nodules and their clinicians, often due to inadequate communication about the nodule. Many clinicians indicated that they do not tell patients about the chance that a nodule may be cancer, because the clinicians know that cancer is unlikely (< 5% of incidentally detected pulmonary nodules turn out to be malignant), and they do not want to alarm patients unnecessarily. However, we found that patients almost immediately wondered about cancer when they learned about their pulmonary nodule, and without hearing explicitly from their clinician that cancer was unlikely, patients tended to overestimate the likelihood of a malignant nodule. Moreover, patients often were not told much about the evaluation plan for the nodule or the rationale for CT surveillance of small nodules instead of biopsy. This uncertainty about the risk of cancer and the plan for evaluating the nodule was difficult for some patients to live with; we found that about one-quarter of patients with a small pulmonary nodule experienced mild-moderate distress during the period of radiographic surveillance. Reassuringly, high-quality patient-clinician communication was associated with lower distress and higher adherence to pulmonary nodule evaluation.4
►Dr. Kearney. The patient was educated about his diagnosis of solitary histoplasmoma. Given that the patient was otherwise well appearing with no complicating factors, he was not treated with antifungal therapy. After an 8-month-long workup, the patient was relieved to receive a diagnosis that excluded cancer and did not require any further treatment. His case provides a good example of how to proceed in the workup of a solitary pulmonary nodule and on the importance of communication and shared decision making with our patients.
1. Gould MK, Donington J, Lynch WR, et al. Evaluation of individuals with pulmonary nodules: when is it lung cancer? Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(suppl 5):e93S-e120S.
2. Azar MM, Hage CA. Clinical perspectives in the diagnosis and management of histoplasmosis. Clin Chest Med. 2017;38(3):403-415.
3. Wheat LJ, Freifeld A, Kleiman MB, et al. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis. 2007;45(7):807-825.
4. Slatore CG, Wiener RS. Pulmonary nodules: a small problem for many, severe distress for some, and how to communicate about it. Chest. 2018;153(4):1004-1015.
Case Presentation. A 69-year-old veteran presented with an intermittent, waxing and waning cough. He had never smoked and had no family history of lung cancer. His primary care physician ordered a chest radiograph, which revealed a nodular opacity within the lingula concerning for a parenchymal nodule. Further characterization with a chest computed tomography (CT) demonstrated a 1.4-cm left upper lobe subpleural nodule with small satellite nodules (Figure 1). Given these imaging findings, the patient was referred to the pulmonary clinic.
►Lauren Kearney, MD, Medical Resident, VA Boston Healthcare System (VABHS) and Boston Medical Center. What is the differential diagnosis of a solitary pulmonary nodule? What characteristics of the nodule do you consider to differentiate these diagnoses?
►Renda Wiener, MD, Pulmonary and Critical Care, VABHS, and Assistant Professor of Medicine, Boston University School of Medicine. Pulmonary nodules are well-defined lesions < 3 cm in diameter that are surrounded by lung parenchyma. Although cancer is a possibility (including primary lung cancers, metastatic cancers, or carcinoid tumors), most small nodules do not turn out to be malignant.1 Benign etiologies include infections, benign tumors, vascular malformations, and inflammatory conditions. Infectious causes of nodules are often granulomatous in nature, including fungi, Mycobacterium tuberculosis, and nontuberculous mycobacteria. Benign tumors are most commonly hamartomas, and these may be clearly distinguished based on imaging characteristics. Pulmonary arteriovenous malformations, hematomas, and infarcts may present as nodules as well. Inflammatory causes of nodules are important and relatively common, including granulomatosis with polyangiitis, rheumatoid arthritis, sarcoidosis, amyloidosis, and rounded atelectasis.
To distinguish benign from malignant etiologies, we look for several features of pulmonary nodules on imaging. Larger size, irregular borders, and upper lobe location all increase the likelihood of cancer, whereas solid attenuation and calcification make cancer less likely. One of the most reassuring findings that suggests a benign etiology is lack of growth over a period of surveillance; after 2 years without growth we typically consider a nodule benign.1 And of course, we also consider the patient’s symptoms and risk factors: weight loss, hemoptysis, a history of cigarette smoking or asbestos exposure, or family history of cancer all increase the likelihood of malignancy.
►Dr. Kearney. Given that the differential diagnosis is so broad, how do you think about the next step in evaluating a pulmonary nodule? How do you approach shared decision making with the patient?
►Dr. Wiener. The characteristics of the patient, the nodule, and the circumstances in which the nodule were discovered are all important to consider. Incidental pulmonary nodules are often found on chest imaging. The imaging characteristics of the nodule are important, as are the patient’s risk factors. A similarly appearing nodule can have very different implications if the patient is a never-smoker exposed to endemic fungi, or a long-time smoker enrolled in a lung cancer screening program. Consultation with a pulmonologist is often appropriate.
It’s important to note that we lack high-quality evidence on the optimal strategy to evaluate pulmonary nodules, and there is no single “right answer“ for all patients. For patients with a low risk of malignancy (< 5%-10%)—which comprises the majority of the incidental nodules discovered—we typically favor serial CT surveillance of the nodule over a period of a few years, whereas for patients at high risk of malignancy (> 65%), we favor early surgical resection if the patient is able to tolerate that. For patients with an intermediate risk of malignancy (~5%-65%), we might consider serial CT surveillance, positron emission tomography (PET) scan, or biopsy.1 The American College of Chest Physicians guidelines for pulmonary nodule evaluation recommend discussing with patients the different options and the trade-offs of these options in a shared decision-making process.1
►Dr. Kearney. The patient’s pulmonologist laid out options, including monitoring with serial CT scans, obtaining a PET scan, performing CT-guided needle biopsy, or referring for surgical excision. In this case, the patient elected to undergo CT-guided needle biopsy. Dr. Huang, can you discuss the pathology results?
►Qin Huang, MD, Pathology and Laboratory Medicine, VABHS, and Assistant Professor of Pathology, Harvard Medical School (HMS). The microscopic examination of the needle biopsy of the lung mass revealed rare clusters of atypical cells with crushed cells adjacent to an extensive area of necrosis with scarring. The atypical cells were suspicious for carcinoma. The Gomori methenamine silver (GMS) and periodic acid-Schiff (PAS) stains were negative for common bacterial and fungal microorganisms.
►Dr. Kearney. The tumor board, pulmonologist, and patient decide to move forward with video-assisted excisional biopsy with lymphadenectomy. Dr. Huang, can you interpret the pathology?
►Dr. Huang. Figure 2 showed an hemotoxylin and eosin (H&E)-stained lung resection tissue section with multiple caseating necrotic granulomas. No foreign bodies were identified. There was no evidence of malignancy. The GMS stain revealed a fungal microorganism oval with morphology typical of histoplasma capsulatum (Figure 3).
►Dr. Kearney. What are some of the different ways histoplasmosis can present? Which of these diagnoses fits this patient’s presentation?
►Judy Strymish, MD, Infectious Disease, VABHS, and Assistant Professor of Medicine, HMS. Most patients who inhale histoplasmosis spores develop asymptomatic or self-limited infection that is usually not detected. Patients at risk of symptomatic and clinically relevant disease include those who are immunocompromised, at extremes of ages, or exposed to larger inoculums. Acute pulmonary histoplasmosis can present with cough, shortness of breath, fever, chills, and less commonly, rheumatologic complaints such as erythema nodosum or erythema multiforme. Imaging often shows patchy infiltrates and enlarged mediastinal and hilar lymphadenopathy. Patients can go on to develop subacute or chronic pulmonary disease with focal opacities and mediastinal and hilar lymphadenopathy. Those with chronic disease can have cavitary lesions similar to patients with tuberculosis. Progressive disseminated histoplasmosis can develop in immunocompromised patients and disseminate through the reticuloendothelial system to other organs with the gastrointestinal tract, central nervous system, and adrenal glands.2
Pulmonary nodules are common incidental finding on chest imaging of patients who reside in histoplasmosis endemic regions, and they are often hard to differentiate from malignancies. There are 3 mediastinal manifestations: adenitis, granuloma, and fibrosis. Usually the syndromes are subclinical, but occasionally the nodes cause symptoms by impinging on other structures.2
This patient had a solitary pulmonary nodule with none of the associated features mentioned above. Pathology showed caseating granuloma and confirmed histoplasmosis.
►Dr. Kearney. Given the diagnosis of solitary histoplasmoma, how should this patient be managed?
►Dr. Strymish. The optimal therapy for histoplasmosis depends on the patient’s clinical syndrome. Most infections are self-limited and require no therapy. However, patients who are immunocompromised, exposed to large inoculum, and have progressive disease require antifungal treatment, usually with itraconazole for mild-to-moderate disease and a combination of azole therapy and amphotericin B with extensive disease. Patients with few solitary pulmonary nodules do not benefit from antifungal therapy as the nodule could represent quiescent disease that is unlikely to have clinical impact; in this case, the treatment would be higher risk than the nodule.3
►Dr. Kearney. While the discussion of the diagnosis is interesting, it is also important to acknowledge what the patient went through to arrive at this, an essentially benign diagnosis: 8 months, multiple imaging studies, and 2 invasive diagnostic procedures. Further, the patient had to grapple with the possibility of a diagnosis of cancer. Dr. Wiener, can you talk about the challenges in communicating with patients about pulmonary nodules when cancer is on the differential? What are some of the harms patients face and how can clinicians work to mitigate these harms?
►Dr. Wiener. My colleague Dr. Christopher Slatore of the Portland VA Medical Center and I studied communication about pulmonary nodules in a series of surveys and qualitative studies of patients with pulmonary nodules and the clinicians who take care of them. We found that there seems to be a disconnect between patients’ perceptions of pulmonary nodules and their clinicians, often due to inadequate communication about the nodule. Many clinicians indicated that they do not tell patients about the chance that a nodule may be cancer, because the clinicians know that cancer is unlikely (< 5% of incidentally detected pulmonary nodules turn out to be malignant), and they do not want to alarm patients unnecessarily. However, we found that patients almost immediately wondered about cancer when they learned about their pulmonary nodule, and without hearing explicitly from their clinician that cancer was unlikely, patients tended to overestimate the likelihood of a malignant nodule. Moreover, patients often were not told much about the evaluation plan for the nodule or the rationale for CT surveillance of small nodules instead of biopsy. This uncertainty about the risk of cancer and the plan for evaluating the nodule was difficult for some patients to live with; we found that about one-quarter of patients with a small pulmonary nodule experienced mild-moderate distress during the period of radiographic surveillance. Reassuringly, high-quality patient-clinician communication was associated with lower distress and higher adherence to pulmonary nodule evaluation.4
►Dr. Kearney. The patient was educated about his diagnosis of solitary histoplasmoma. Given that the patient was otherwise well appearing with no complicating factors, he was not treated with antifungal therapy. After an 8-month-long workup, the patient was relieved to receive a diagnosis that excluded cancer and did not require any further treatment. His case provides a good example of how to proceed in the workup of a solitary pulmonary nodule and on the importance of communication and shared decision making with our patients.
Case Presentation. A 69-year-old veteran presented with an intermittent, waxing and waning cough. He had never smoked and had no family history of lung cancer. His primary care physician ordered a chest radiograph, which revealed a nodular opacity within the lingula concerning for a parenchymal nodule. Further characterization with a chest computed tomography (CT) demonstrated a 1.4-cm left upper lobe subpleural nodule with small satellite nodules (Figure 1). Given these imaging findings, the patient was referred to the pulmonary clinic.
►Lauren Kearney, MD, Medical Resident, VA Boston Healthcare System (VABHS) and Boston Medical Center. What is the differential diagnosis of a solitary pulmonary nodule? What characteristics of the nodule do you consider to differentiate these diagnoses?
►Renda Wiener, MD, Pulmonary and Critical Care, VABHS, and Assistant Professor of Medicine, Boston University School of Medicine. Pulmonary nodules are well-defined lesions < 3 cm in diameter that are surrounded by lung parenchyma. Although cancer is a possibility (including primary lung cancers, metastatic cancers, or carcinoid tumors), most small nodules do not turn out to be malignant.1 Benign etiologies include infections, benign tumors, vascular malformations, and inflammatory conditions. Infectious causes of nodules are often granulomatous in nature, including fungi, Mycobacterium tuberculosis, and nontuberculous mycobacteria. Benign tumors are most commonly hamartomas, and these may be clearly distinguished based on imaging characteristics. Pulmonary arteriovenous malformations, hematomas, and infarcts may present as nodules as well. Inflammatory causes of nodules are important and relatively common, including granulomatosis with polyangiitis, rheumatoid arthritis, sarcoidosis, amyloidosis, and rounded atelectasis.
To distinguish benign from malignant etiologies, we look for several features of pulmonary nodules on imaging. Larger size, irregular borders, and upper lobe location all increase the likelihood of cancer, whereas solid attenuation and calcification make cancer less likely. One of the most reassuring findings that suggests a benign etiology is lack of growth over a period of surveillance; after 2 years without growth we typically consider a nodule benign.1 And of course, we also consider the patient’s symptoms and risk factors: weight loss, hemoptysis, a history of cigarette smoking or asbestos exposure, or family history of cancer all increase the likelihood of malignancy.
►Dr. Kearney. Given that the differential diagnosis is so broad, how do you think about the next step in evaluating a pulmonary nodule? How do you approach shared decision making with the patient?
►Dr. Wiener. The characteristics of the patient, the nodule, and the circumstances in which the nodule were discovered are all important to consider. Incidental pulmonary nodules are often found on chest imaging. The imaging characteristics of the nodule are important, as are the patient’s risk factors. A similarly appearing nodule can have very different implications if the patient is a never-smoker exposed to endemic fungi, or a long-time smoker enrolled in a lung cancer screening program. Consultation with a pulmonologist is often appropriate.
It’s important to note that we lack high-quality evidence on the optimal strategy to evaluate pulmonary nodules, and there is no single “right answer“ for all patients. For patients with a low risk of malignancy (< 5%-10%)—which comprises the majority of the incidental nodules discovered—we typically favor serial CT surveillance of the nodule over a period of a few years, whereas for patients at high risk of malignancy (> 65%), we favor early surgical resection if the patient is able to tolerate that. For patients with an intermediate risk of malignancy (~5%-65%), we might consider serial CT surveillance, positron emission tomography (PET) scan, or biopsy.1 The American College of Chest Physicians guidelines for pulmonary nodule evaluation recommend discussing with patients the different options and the trade-offs of these options in a shared decision-making process.1
►Dr. Kearney. The patient’s pulmonologist laid out options, including monitoring with serial CT scans, obtaining a PET scan, performing CT-guided needle biopsy, or referring for surgical excision. In this case, the patient elected to undergo CT-guided needle biopsy. Dr. Huang, can you discuss the pathology results?
►Qin Huang, MD, Pathology and Laboratory Medicine, VABHS, and Assistant Professor of Pathology, Harvard Medical School (HMS). The microscopic examination of the needle biopsy of the lung mass revealed rare clusters of atypical cells with crushed cells adjacent to an extensive area of necrosis with scarring. The atypical cells were suspicious for carcinoma. The Gomori methenamine silver (GMS) and periodic acid-Schiff (PAS) stains were negative for common bacterial and fungal microorganisms.
►Dr. Kearney. The tumor board, pulmonologist, and patient decide to move forward with video-assisted excisional biopsy with lymphadenectomy. Dr. Huang, can you interpret the pathology?
►Dr. Huang. Figure 2 showed an hemotoxylin and eosin (H&E)-stained lung resection tissue section with multiple caseating necrotic granulomas. No foreign bodies were identified. There was no evidence of malignancy. The GMS stain revealed a fungal microorganism oval with morphology typical of histoplasma capsulatum (Figure 3).
►Dr. Kearney. What are some of the different ways histoplasmosis can present? Which of these diagnoses fits this patient’s presentation?
►Judy Strymish, MD, Infectious Disease, VABHS, and Assistant Professor of Medicine, HMS. Most patients who inhale histoplasmosis spores develop asymptomatic or self-limited infection that is usually not detected. Patients at risk of symptomatic and clinically relevant disease include those who are immunocompromised, at extremes of ages, or exposed to larger inoculums. Acute pulmonary histoplasmosis can present with cough, shortness of breath, fever, chills, and less commonly, rheumatologic complaints such as erythema nodosum or erythema multiforme. Imaging often shows patchy infiltrates and enlarged mediastinal and hilar lymphadenopathy. Patients can go on to develop subacute or chronic pulmonary disease with focal opacities and mediastinal and hilar lymphadenopathy. Those with chronic disease can have cavitary lesions similar to patients with tuberculosis. Progressive disseminated histoplasmosis can develop in immunocompromised patients and disseminate through the reticuloendothelial system to other organs with the gastrointestinal tract, central nervous system, and adrenal glands.2
Pulmonary nodules are common incidental finding on chest imaging of patients who reside in histoplasmosis endemic regions, and they are often hard to differentiate from malignancies. There are 3 mediastinal manifestations: adenitis, granuloma, and fibrosis. Usually the syndromes are subclinical, but occasionally the nodes cause symptoms by impinging on other structures.2
This patient had a solitary pulmonary nodule with none of the associated features mentioned above. Pathology showed caseating granuloma and confirmed histoplasmosis.
►Dr. Kearney. Given the diagnosis of solitary histoplasmoma, how should this patient be managed?
►Dr. Strymish. The optimal therapy for histoplasmosis depends on the patient’s clinical syndrome. Most infections are self-limited and require no therapy. However, patients who are immunocompromised, exposed to large inoculum, and have progressive disease require antifungal treatment, usually with itraconazole for mild-to-moderate disease and a combination of azole therapy and amphotericin B with extensive disease. Patients with few solitary pulmonary nodules do not benefit from antifungal therapy as the nodule could represent quiescent disease that is unlikely to have clinical impact; in this case, the treatment would be higher risk than the nodule.3
►Dr. Kearney. While the discussion of the diagnosis is interesting, it is also important to acknowledge what the patient went through to arrive at this, an essentially benign diagnosis: 8 months, multiple imaging studies, and 2 invasive diagnostic procedures. Further, the patient had to grapple with the possibility of a diagnosis of cancer. Dr. Wiener, can you talk about the challenges in communicating with patients about pulmonary nodules when cancer is on the differential? What are some of the harms patients face and how can clinicians work to mitigate these harms?
►Dr. Wiener. My colleague Dr. Christopher Slatore of the Portland VA Medical Center and I studied communication about pulmonary nodules in a series of surveys and qualitative studies of patients with pulmonary nodules and the clinicians who take care of them. We found that there seems to be a disconnect between patients’ perceptions of pulmonary nodules and their clinicians, often due to inadequate communication about the nodule. Many clinicians indicated that they do not tell patients about the chance that a nodule may be cancer, because the clinicians know that cancer is unlikely (< 5% of incidentally detected pulmonary nodules turn out to be malignant), and they do not want to alarm patients unnecessarily. However, we found that patients almost immediately wondered about cancer when they learned about their pulmonary nodule, and without hearing explicitly from their clinician that cancer was unlikely, patients tended to overestimate the likelihood of a malignant nodule. Moreover, patients often were not told much about the evaluation plan for the nodule or the rationale for CT surveillance of small nodules instead of biopsy. This uncertainty about the risk of cancer and the plan for evaluating the nodule was difficult for some patients to live with; we found that about one-quarter of patients with a small pulmonary nodule experienced mild-moderate distress during the period of radiographic surveillance. Reassuringly, high-quality patient-clinician communication was associated with lower distress and higher adherence to pulmonary nodule evaluation.4
►Dr. Kearney. The patient was educated about his diagnosis of solitary histoplasmoma. Given that the patient was otherwise well appearing with no complicating factors, he was not treated with antifungal therapy. After an 8-month-long workup, the patient was relieved to receive a diagnosis that excluded cancer and did not require any further treatment. His case provides a good example of how to proceed in the workup of a solitary pulmonary nodule and on the importance of communication and shared decision making with our patients.
1. Gould MK, Donington J, Lynch WR, et al. Evaluation of individuals with pulmonary nodules: when is it lung cancer? Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(suppl 5):e93S-e120S.
2. Azar MM, Hage CA. Clinical perspectives in the diagnosis and management of histoplasmosis. Clin Chest Med. 2017;38(3):403-415.
3. Wheat LJ, Freifeld A, Kleiman MB, et al. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis. 2007;45(7):807-825.
4. Slatore CG, Wiener RS. Pulmonary nodules: a small problem for many, severe distress for some, and how to communicate about it. Chest. 2018;153(4):1004-1015.
1. Gould MK, Donington J, Lynch WR, et al. Evaluation of individuals with pulmonary nodules: when is it lung cancer? Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(suppl 5):e93S-e120S.
2. Azar MM, Hage CA. Clinical perspectives in the diagnosis and management of histoplasmosis. Clin Chest Med. 2017;38(3):403-415.
3. Wheat LJ, Freifeld A, Kleiman MB, et al. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis. 2007;45(7):807-825.
4. Slatore CG, Wiener RS. Pulmonary nodules: a small problem for many, severe distress for some, and how to communicate about it. Chest. 2018;153(4):1004-1015.
Remote-Onset Alopecia Areata Attributed to Ipilimumab
Cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) is a key co-stimulatory receptor expressed on activated T cells that negatively regulates T-cell activation.1-3 It exerts its effects in part by the prevention of IL-2 transcription and inhibition of cell-cycle progression.4 Cytotoxic T-lymphocyte–associated antigen 4 also is expressed by a subset of CD25+CD4+ regulatory T cells (Tregs), where it plays a role in immune tolerance.5 Blockade has demonstrated antitumor activity as well as immune activation, and CTLA-4 dysregulation has been implicated in autoimmune diseases such as alopecia areata (AA).6
Ipilimumab is a fully humanized monoclonal antibody against CTLA-4 and one of a growing class of immune checkpoint inhibitor therapies for metastatic melanoma. Phase 2 and 3 clinical trials have shown an improved survival effect of ipilimumab in patients with advanced melanoma,7-10 with 3-year survival rates ranging from 20.8% to 46.5%.10,11 The US Food and Drug Administration approved ipilimumab in 2011 for treatment of unresectable or metastatic melanoma.12 The most common toxicities of ipilimumab are immune-related adverse effects (irAEs), which represent loss of tolerance to self-antigens.13 Immune-related adverse effects occur in 64.2% of patients,14 with severe or life-threatening irAEs in 17.8% of patients.14 Rates of irAEs appear dose dependent but consistent across increased doses.15 Cutaneous irAEs occur in more than 47% of patients16 and commonly manifest as pruritus with or without a diffuse morbilliform rash,10,17 though less common skin reactions, including vitiligo, vasculitis, and Stevens-Johnson syndrome/toxic epidermal necrolysis, have been documented.9,18
Generalized AA and its more widespread variant, alopecia universalis, have been reported as adverse effects of ipilimumab monotherapy in 2 prior cases in the English-language literature (Table).17,19 Alopecia areata also has been attributed to combination immune checkpoint inhibitor therapy.20,21 We report a case of AA attributable to ipilimumab monotherapy that was localized exclusively to the scalp and remote in onset following treatment.
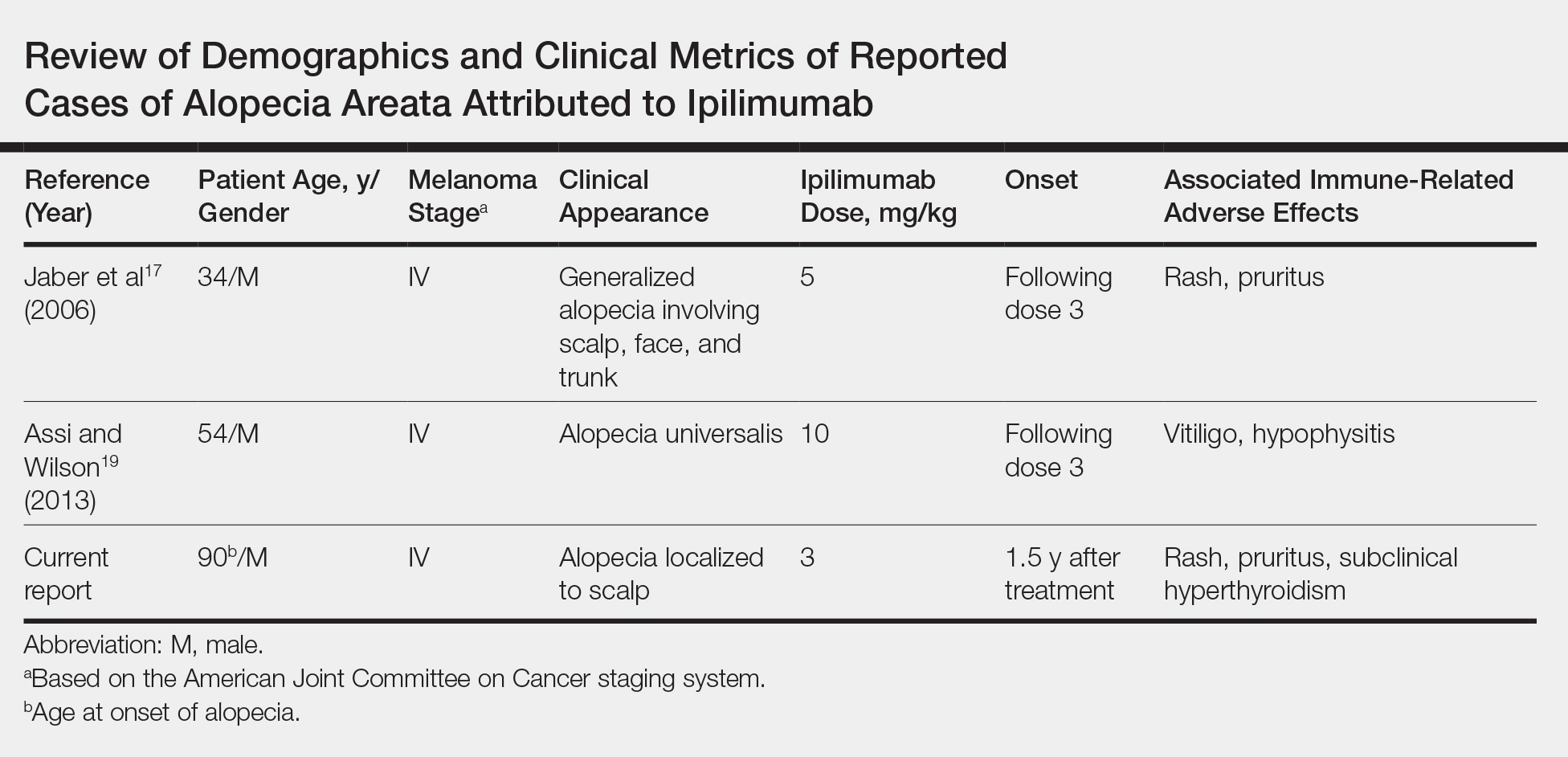
Case Report
An 88-year-old man with pT3bpN3 nodular melanoma of the back demonstrated multiple lung metastases by positron emission tomography–computed tomography. Lactate dehydrogenase was within reference range, and his Eastern Cooperative Oncology Group performance status was 0 (fully active). One month later, he was started on ipilimumab 3 mg/kg intravenous infusion every 3 weeks for a total of 4 doses. At approximately week 6, his course was complicated by mild fatigue, a faintly erythematous morbilliform rash, and mild pruritus, with laboratory evidence of subclinical hyperthyroidism. Follow-up positron emission tomography–computed tomography at the conclusion of treatment demonstrated complete regression of previously noted hypermetabolic foci. His symptoms and subclinical hyperthyroidism resolved several months later.
Seventeen months after completion of ipilimumab therapy (at age 90 years), the patient’s barber noted new-onset hair loss on the right occipital scalp. Physical examination demonstrated a well-circumscribed patch of nonscarring alopecia (approximately 6 cm) that was clinically consistent with AA (Figure). There were no associated symptoms or other involved areas of hair loss. He denied any personal or family history of AA. The patient’s melanoma has remained in remission to date.
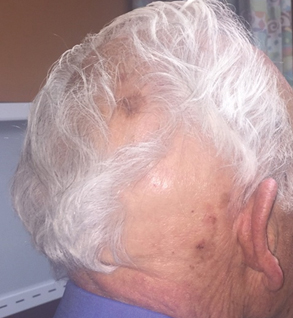
Comment
This case is unique in that AA was localized to a single circumscribed patch on the scalp and occurred nearly 1.5 years after treatment with ipilimumab, which may indicate a robust blockade of CTLA-4 given the remote development of autoimmunity in the setting of persistent remission of melanoma. Although the appearance of AA may be coincidental, onset at 90 years of age would be unusual. The mean age of onset of AA has been reported between 25.2 and 36.3 years,22,23 and its incidence in men older than 60 years is only 6.4 per 100,000 person-years.24
Although AA is a rare irAE of CTLA-4 blockade, the disease has been increasingly linked to CTLA-4 dysregulation in both animal models and humans.6,25,26 A genome-wide association study of 1054 patients with AA and 3278 controls implicated several genes controlling activation and proliferation of Tregs, including CTLA-4.27 More specifically, single-nucleotide polymorphisms of the CTLA-4 gene were found to be associated with AA in a study of 1196 unrelated patients and 1280 controls,28 and Megiorni et al
Given the role of CTLA-4 dysregulation in the pathogenesis of AA, the very low rates of AA in ipilimumab are somewhat surprising, which may represent a reporting bias. Alternatively, there may be sufficient Treg activity to prevent high rates of AA at a lower ipilimumab dose of 3 mg/kg but insufficient activity to prevent development of other irAEs. With US Food and Drug Administration approval of ipilimumab at a higher dose of 10 mg/kg for use as adjuvant therapy for stage III melanomas,12 less common cutaneous irAEs such as AA may be seen with increased frequency. Clinicians planning ipilimumab therapy should discuss this side effect and other potential irAEs with their patients before initiation of treatment.
- Brunet JF, Denizot F, Luciani MF, et al. A new member of the immunoglobulin superfamily--CTLA-4. Nature. 1987;328:267-270.
- Scalapino KJ, Daikh DI. CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev. 2008;223:143-155.
- Buchbinder E, Hodi FS. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J Clin Invest. 2015;125:3377-3383.
- Brunner MC, Chambers CA, Chan FK, et al. CTLA-4-mediated inhibition of early events of T cell proliferation. J Immunol. 1999;162:5813-5820.
- Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303-310.
- Carroll JM, McElwee KJ, E King L, et al. Gene array profiling and immunomodulation studies define a cell-mediated immune response underlying the pathogenesis of alopecia areata in a mouse model and humans. J Invest Dermatol. 2002;119:392-402.
- Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15:5591-5598.
- O’Day SJ, Maio M, Chiarion-Sileni V, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann Oncol. 2010;21:1712-1717.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
- Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015;16:522-530.
- Yervoy (ipilimumab)[package insert]. Princeton, NJ: Bristol-Myers Squibb; 2019.
- Weber J. Review: anti-CTLA-4 antibody ipilimumab: case studies of clinical response and immune-related adverse events. Oncologist. 2007;12:864-872.
- Ibrahim RA, Berman DM, DePril V, et al. Ipilimumab safety profile: summary of findings from completed trials in advanced melanoma [abstract]. J Clin Oncol. 2011;29(suppl):8583.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155-164.
- Kähler KC, Hauschild A. Treatment and side effect management of CTLA-4 antibody therapy in metastatic melanoma. J Dtsch Dermatol Ges. 2011;9:277-286.
- Jaber SH, Cowen EW, Haworth LR, et al. Skin reactions in a subset of patients with stage IV melanoma treated with anti-cytotoxic T-lymphocyte antigen 4 monoclonal antibody as a single agent. Arch Dermatol. 2006;142:166-172.
- Voskens CJ, Goldinger SM, Loquai C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 2013;8:E537545.
- Assi H, Wilson KS. Immune toxicities and long remission duration after ipilimumab therapy for metastatic melanoma: two illustrative cases. Curr Oncol. 2013;20:E165-E169.
- Zarbo A, Belum VR, Sibaud V, et al. Immune-related alopecia (areata and universalis) in cancer patients receiving immune checkpoint inhibitors. Br J Dermatol. 2017;176:1649-1652.
- Lakhmiri M, Cavelier-Balloy B, Lacoste C, et al. Nivolumab-induced alopecia areata: a reversible factor of good prognosis? JAAD Case Rep. 2018;4:761-765.
- Tan E, Tay YK, Goh CL, et al. The pattern and profile of alopecia areata in Singapore–a study of 219 Asians. Int J Dermatol. 2002;41:748-753.
- Goh C, Finkel M, Christos PJ, et al. Profile of 513 patients with alopecia areata: associations of disease subtypes with atopy, autoimmune disease and positive family history. J Eur Acad Dermatol Venereol. 2006;20:1055-1060.
- Mirzoyev SA, Schrum AG, Davis MD, et al. Lifetime incidence risk of alopecia areata estimated at 2.1% by Rochester Epidemiology Project, 1990-2009. J Invest Dermatol. 2014;134:1141-1142.
- Zöller M, McElwee KJ, Engel P, et al. Transient CD44 variant isoform expression and reduction in CD4(+)/CD25(+) regulatory T cells in C3H/HeJ mice with alopecia areata. J Invest Dermatol. 2002;118:983-992.
- Zöller M, McElwee KJ, Vitacolonna M, et al. The progressive state, in contrast to the stable or regressive state of alopecia areata, is reflected in peripheral blood mononuclear cells. Exp Dermatol. 2004;13:435-444.
- Petukhova L, Duvic M, Hordinsky M, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466:113-117.
- John KK, Brockschmidt FF, Redler S, et al. Genetic variants in CTLA4 are strongly associated with alopecia areata. J Invest Dermatol. 2011;131:1169-1172.
- Megiorni F, Mora B, Maxia C, et al. Cytotoxic T-lymphocyte antigen 4 (CTLA4) +49AG and CT60 gene polymorphisms in alopecia areata: a case-control association study in the Italian population. Arch Dermatol Res. 2013;305:665-670
Cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) is a key co-stimulatory receptor expressed on activated T cells that negatively regulates T-cell activation.1-3 It exerts its effects in part by the prevention of IL-2 transcription and inhibition of cell-cycle progression.4 Cytotoxic T-lymphocyte–associated antigen 4 also is expressed by a subset of CD25+CD4+ regulatory T cells (Tregs), where it plays a role in immune tolerance.5 Blockade has demonstrated antitumor activity as well as immune activation, and CTLA-4 dysregulation has been implicated in autoimmune diseases such as alopecia areata (AA).6
Ipilimumab is a fully humanized monoclonal antibody against CTLA-4 and one of a growing class of immune checkpoint inhibitor therapies for metastatic melanoma. Phase 2 and 3 clinical trials have shown an improved survival effect of ipilimumab in patients with advanced melanoma,7-10 with 3-year survival rates ranging from 20.8% to 46.5%.10,11 The US Food and Drug Administration approved ipilimumab in 2011 for treatment of unresectable or metastatic melanoma.12 The most common toxicities of ipilimumab are immune-related adverse effects (irAEs), which represent loss of tolerance to self-antigens.13 Immune-related adverse effects occur in 64.2% of patients,14 with severe or life-threatening irAEs in 17.8% of patients.14 Rates of irAEs appear dose dependent but consistent across increased doses.15 Cutaneous irAEs occur in more than 47% of patients16 and commonly manifest as pruritus with or without a diffuse morbilliform rash,10,17 though less common skin reactions, including vitiligo, vasculitis, and Stevens-Johnson syndrome/toxic epidermal necrolysis, have been documented.9,18
Generalized AA and its more widespread variant, alopecia universalis, have been reported as adverse effects of ipilimumab monotherapy in 2 prior cases in the English-language literature (Table).17,19 Alopecia areata also has been attributed to combination immune checkpoint inhibitor therapy.20,21 We report a case of AA attributable to ipilimumab monotherapy that was localized exclusively to the scalp and remote in onset following treatment.
Case Report
An 88-year-old man with pT3bpN3 nodular melanoma of the back demonstrated multiple lung metastases by positron emission tomography–computed tomography. Lactate dehydrogenase was within reference range, and his Eastern Cooperative Oncology Group performance status was 0 (fully active). One month later, he was started on ipilimumab 3 mg/kg intravenous infusion every 3 weeks for a total of 4 doses. At approximately week 6, his course was complicated by mild fatigue, a faintly erythematous morbilliform rash, and mild pruritus, with laboratory evidence of subclinical hyperthyroidism. Follow-up positron emission tomography–computed tomography at the conclusion of treatment demonstrated complete regression of previously noted hypermetabolic foci. His symptoms and subclinical hyperthyroidism resolved several months later.
Seventeen months after completion of ipilimumab therapy (at age 90 years), the patient’s barber noted new-onset hair loss on the right occipital scalp. Physical examination demonstrated a well-circumscribed patch of nonscarring alopecia (approximately 6 cm) that was clinically consistent with AA (Figure). There were no associated symptoms or other involved areas of hair loss. He denied any personal or family history of AA. The patient’s melanoma has remained in remission to date.
Comment
This case is unique in that AA was localized to a single circumscribed patch on the scalp and occurred nearly 1.5 years after treatment with ipilimumab, which may indicate a robust blockade of CTLA-4 given the remote development of autoimmunity in the setting of persistent remission of melanoma. Although the appearance of AA may be coincidental, onset at 90 years of age would be unusual. The mean age of onset of AA has been reported between 25.2 and 36.3 years,22,23 and its incidence in men older than 60 years is only 6.4 per 100,000 person-years.24
Although AA is a rare irAE of CTLA-4 blockade, the disease has been increasingly linked to CTLA-4 dysregulation in both animal models and humans.6,25,26 A genome-wide association study of 1054 patients with AA and 3278 controls implicated several genes controlling activation and proliferation of Tregs, including CTLA-4.27 More specifically, single-nucleotide polymorphisms of the CTLA-4 gene were found to be associated with AA in a study of 1196 unrelated patients and 1280 controls,28 and Megiorni et al
Given the role of CTLA-4 dysregulation in the pathogenesis of AA, the very low rates of AA in ipilimumab are somewhat surprising, which may represent a reporting bias. Alternatively, there may be sufficient Treg activity to prevent high rates of AA at a lower ipilimumab dose of 3 mg/kg but insufficient activity to prevent development of other irAEs. With US Food and Drug Administration approval of ipilimumab at a higher dose of 10 mg/kg for use as adjuvant therapy for stage III melanomas,12 less common cutaneous irAEs such as AA may be seen with increased frequency. Clinicians planning ipilimumab therapy should discuss this side effect and other potential irAEs with their patients before initiation of treatment.
Cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) is a key co-stimulatory receptor expressed on activated T cells that negatively regulates T-cell activation.1-3 It exerts its effects in part by the prevention of IL-2 transcription and inhibition of cell-cycle progression.4 Cytotoxic T-lymphocyte–associated antigen 4 also is expressed by a subset of CD25+CD4+ regulatory T cells (Tregs), where it plays a role in immune tolerance.5 Blockade has demonstrated antitumor activity as well as immune activation, and CTLA-4 dysregulation has been implicated in autoimmune diseases such as alopecia areata (AA).6
Ipilimumab is a fully humanized monoclonal antibody against CTLA-4 and one of a growing class of immune checkpoint inhibitor therapies for metastatic melanoma. Phase 2 and 3 clinical trials have shown an improved survival effect of ipilimumab in patients with advanced melanoma,7-10 with 3-year survival rates ranging from 20.8% to 46.5%.10,11 The US Food and Drug Administration approved ipilimumab in 2011 for treatment of unresectable or metastatic melanoma.12 The most common toxicities of ipilimumab are immune-related adverse effects (irAEs), which represent loss of tolerance to self-antigens.13 Immune-related adverse effects occur in 64.2% of patients,14 with severe or life-threatening irAEs in 17.8% of patients.14 Rates of irAEs appear dose dependent but consistent across increased doses.15 Cutaneous irAEs occur in more than 47% of patients16 and commonly manifest as pruritus with or without a diffuse morbilliform rash,10,17 though less common skin reactions, including vitiligo, vasculitis, and Stevens-Johnson syndrome/toxic epidermal necrolysis, have been documented.9,18
Generalized AA and its more widespread variant, alopecia universalis, have been reported as adverse effects of ipilimumab monotherapy in 2 prior cases in the English-language literature (Table).17,19 Alopecia areata also has been attributed to combination immune checkpoint inhibitor therapy.20,21 We report a case of AA attributable to ipilimumab monotherapy that was localized exclusively to the scalp and remote in onset following treatment.
Case Report
An 88-year-old man with pT3bpN3 nodular melanoma of the back demonstrated multiple lung metastases by positron emission tomography–computed tomography. Lactate dehydrogenase was within reference range, and his Eastern Cooperative Oncology Group performance status was 0 (fully active). One month later, he was started on ipilimumab 3 mg/kg intravenous infusion every 3 weeks for a total of 4 doses. At approximately week 6, his course was complicated by mild fatigue, a faintly erythematous morbilliform rash, and mild pruritus, with laboratory evidence of subclinical hyperthyroidism. Follow-up positron emission tomography–computed tomography at the conclusion of treatment demonstrated complete regression of previously noted hypermetabolic foci. His symptoms and subclinical hyperthyroidism resolved several months later.
Seventeen months after completion of ipilimumab therapy (at age 90 years), the patient’s barber noted new-onset hair loss on the right occipital scalp. Physical examination demonstrated a well-circumscribed patch of nonscarring alopecia (approximately 6 cm) that was clinically consistent with AA (Figure). There were no associated symptoms or other involved areas of hair loss. He denied any personal or family history of AA. The patient’s melanoma has remained in remission to date.
Comment
This case is unique in that AA was localized to a single circumscribed patch on the scalp and occurred nearly 1.5 years after treatment with ipilimumab, which may indicate a robust blockade of CTLA-4 given the remote development of autoimmunity in the setting of persistent remission of melanoma. Although the appearance of AA may be coincidental, onset at 90 years of age would be unusual. The mean age of onset of AA has been reported between 25.2 and 36.3 years,22,23 and its incidence in men older than 60 years is only 6.4 per 100,000 person-years.24
Although AA is a rare irAE of CTLA-4 blockade, the disease has been increasingly linked to CTLA-4 dysregulation in both animal models and humans.6,25,26 A genome-wide association study of 1054 patients with AA and 3278 controls implicated several genes controlling activation and proliferation of Tregs, including CTLA-4.27 More specifically, single-nucleotide polymorphisms of the CTLA-4 gene were found to be associated with AA in a study of 1196 unrelated patients and 1280 controls,28 and Megiorni et al
Given the role of CTLA-4 dysregulation in the pathogenesis of AA, the very low rates of AA in ipilimumab are somewhat surprising, which may represent a reporting bias. Alternatively, there may be sufficient Treg activity to prevent high rates of AA at a lower ipilimumab dose of 3 mg/kg but insufficient activity to prevent development of other irAEs. With US Food and Drug Administration approval of ipilimumab at a higher dose of 10 mg/kg for use as adjuvant therapy for stage III melanomas,12 less common cutaneous irAEs such as AA may be seen with increased frequency. Clinicians planning ipilimumab therapy should discuss this side effect and other potential irAEs with their patients before initiation of treatment.
- Brunet JF, Denizot F, Luciani MF, et al. A new member of the immunoglobulin superfamily--CTLA-4. Nature. 1987;328:267-270.
- Scalapino KJ, Daikh DI. CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev. 2008;223:143-155.
- Buchbinder E, Hodi FS. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J Clin Invest. 2015;125:3377-3383.
- Brunner MC, Chambers CA, Chan FK, et al. CTLA-4-mediated inhibition of early events of T cell proliferation. J Immunol. 1999;162:5813-5820.
- Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303-310.
- Carroll JM, McElwee KJ, E King L, et al. Gene array profiling and immunomodulation studies define a cell-mediated immune response underlying the pathogenesis of alopecia areata in a mouse model and humans. J Invest Dermatol. 2002;119:392-402.
- Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15:5591-5598.
- O’Day SJ, Maio M, Chiarion-Sileni V, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann Oncol. 2010;21:1712-1717.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
- Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015;16:522-530.
- Yervoy (ipilimumab)[package insert]. Princeton, NJ: Bristol-Myers Squibb; 2019.
- Weber J. Review: anti-CTLA-4 antibody ipilimumab: case studies of clinical response and immune-related adverse events. Oncologist. 2007;12:864-872.
- Ibrahim RA, Berman DM, DePril V, et al. Ipilimumab safety profile: summary of findings from completed trials in advanced melanoma [abstract]. J Clin Oncol. 2011;29(suppl):8583.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155-164.
- Kähler KC, Hauschild A. Treatment and side effect management of CTLA-4 antibody therapy in metastatic melanoma. J Dtsch Dermatol Ges. 2011;9:277-286.
- Jaber SH, Cowen EW, Haworth LR, et al. Skin reactions in a subset of patients with stage IV melanoma treated with anti-cytotoxic T-lymphocyte antigen 4 monoclonal antibody as a single agent. Arch Dermatol. 2006;142:166-172.
- Voskens CJ, Goldinger SM, Loquai C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 2013;8:E537545.
- Assi H, Wilson KS. Immune toxicities and long remission duration after ipilimumab therapy for metastatic melanoma: two illustrative cases. Curr Oncol. 2013;20:E165-E169.
- Zarbo A, Belum VR, Sibaud V, et al. Immune-related alopecia (areata and universalis) in cancer patients receiving immune checkpoint inhibitors. Br J Dermatol. 2017;176:1649-1652.
- Lakhmiri M, Cavelier-Balloy B, Lacoste C, et al. Nivolumab-induced alopecia areata: a reversible factor of good prognosis? JAAD Case Rep. 2018;4:761-765.
- Tan E, Tay YK, Goh CL, et al. The pattern and profile of alopecia areata in Singapore–a study of 219 Asians. Int J Dermatol. 2002;41:748-753.
- Goh C, Finkel M, Christos PJ, et al. Profile of 513 patients with alopecia areata: associations of disease subtypes with atopy, autoimmune disease and positive family history. J Eur Acad Dermatol Venereol. 2006;20:1055-1060.
- Mirzoyev SA, Schrum AG, Davis MD, et al. Lifetime incidence risk of alopecia areata estimated at 2.1% by Rochester Epidemiology Project, 1990-2009. J Invest Dermatol. 2014;134:1141-1142.
- Zöller M, McElwee KJ, Engel P, et al. Transient CD44 variant isoform expression and reduction in CD4(+)/CD25(+) regulatory T cells in C3H/HeJ mice with alopecia areata. J Invest Dermatol. 2002;118:983-992.
- Zöller M, McElwee KJ, Vitacolonna M, et al. The progressive state, in contrast to the stable or regressive state of alopecia areata, is reflected in peripheral blood mononuclear cells. Exp Dermatol. 2004;13:435-444.
- Petukhova L, Duvic M, Hordinsky M, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466:113-117.
- John KK, Brockschmidt FF, Redler S, et al. Genetic variants in CTLA4 are strongly associated with alopecia areata. J Invest Dermatol. 2011;131:1169-1172.
- Megiorni F, Mora B, Maxia C, et al. Cytotoxic T-lymphocyte antigen 4 (CTLA4) +49AG and CT60 gene polymorphisms in alopecia areata: a case-control association study in the Italian population. Arch Dermatol Res. 2013;305:665-670
- Brunet JF, Denizot F, Luciani MF, et al. A new member of the immunoglobulin superfamily--CTLA-4. Nature. 1987;328:267-270.
- Scalapino KJ, Daikh DI. CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev. 2008;223:143-155.
- Buchbinder E, Hodi FS. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J Clin Invest. 2015;125:3377-3383.
- Brunner MC, Chambers CA, Chan FK, et al. CTLA-4-mediated inhibition of early events of T cell proliferation. J Immunol. 1999;162:5813-5820.
- Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303-310.
- Carroll JM, McElwee KJ, E King L, et al. Gene array profiling and immunomodulation studies define a cell-mediated immune response underlying the pathogenesis of alopecia areata in a mouse model and humans. J Invest Dermatol. 2002;119:392-402.
- Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15:5591-5598.
- O’Day SJ, Maio M, Chiarion-Sileni V, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann Oncol. 2010;21:1712-1717.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
- Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015;16:522-530.
- Yervoy (ipilimumab)[package insert]. Princeton, NJ: Bristol-Myers Squibb; 2019.
- Weber J. Review: anti-CTLA-4 antibody ipilimumab: case studies of clinical response and immune-related adverse events. Oncologist. 2007;12:864-872.
- Ibrahim RA, Berman DM, DePril V, et al. Ipilimumab safety profile: summary of findings from completed trials in advanced melanoma [abstract]. J Clin Oncol. 2011;29(suppl):8583.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155-164.
- Kähler KC, Hauschild A. Treatment and side effect management of CTLA-4 antibody therapy in metastatic melanoma. J Dtsch Dermatol Ges. 2011;9:277-286.
- Jaber SH, Cowen EW, Haworth LR, et al. Skin reactions in a subset of patients with stage IV melanoma treated with anti-cytotoxic T-lymphocyte antigen 4 monoclonal antibody as a single agent. Arch Dermatol. 2006;142:166-172.
- Voskens CJ, Goldinger SM, Loquai C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 2013;8:E537545.
- Assi H, Wilson KS. Immune toxicities and long remission duration after ipilimumab therapy for metastatic melanoma: two illustrative cases. Curr Oncol. 2013;20:E165-E169.
- Zarbo A, Belum VR, Sibaud V, et al. Immune-related alopecia (areata and universalis) in cancer patients receiving immune checkpoint inhibitors. Br J Dermatol. 2017;176:1649-1652.
- Lakhmiri M, Cavelier-Balloy B, Lacoste C, et al. Nivolumab-induced alopecia areata: a reversible factor of good prognosis? JAAD Case Rep. 2018;4:761-765.
- Tan E, Tay YK, Goh CL, et al. The pattern and profile of alopecia areata in Singapore–a study of 219 Asians. Int J Dermatol. 2002;41:748-753.
- Goh C, Finkel M, Christos PJ, et al. Profile of 513 patients with alopecia areata: associations of disease subtypes with atopy, autoimmune disease and positive family history. J Eur Acad Dermatol Venereol. 2006;20:1055-1060.
- Mirzoyev SA, Schrum AG, Davis MD, et al. Lifetime incidence risk of alopecia areata estimated at 2.1% by Rochester Epidemiology Project, 1990-2009. J Invest Dermatol. 2014;134:1141-1142.
- Zöller M, McElwee KJ, Engel P, et al. Transient CD44 variant isoform expression and reduction in CD4(+)/CD25(+) regulatory T cells in C3H/HeJ mice with alopecia areata. J Invest Dermatol. 2002;118:983-992.
- Zöller M, McElwee KJ, Vitacolonna M, et al. The progressive state, in contrast to the stable or regressive state of alopecia areata, is reflected in peripheral blood mononuclear cells. Exp Dermatol. 2004;13:435-444.
- Petukhova L, Duvic M, Hordinsky M, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466:113-117.
- John KK, Brockschmidt FF, Redler S, et al. Genetic variants in CTLA4 are strongly associated with alopecia areata. J Invest Dermatol. 2011;131:1169-1172.
- Megiorni F, Mora B, Maxia C, et al. Cytotoxic T-lymphocyte antigen 4 (CTLA4) +49AG and CT60 gene polymorphisms in alopecia areata: a case-control association study in the Italian population. Arch Dermatol Res. 2013;305:665-670
Practice Points
- Cutaneous immune-related adverse effects (irAEs) are among the most common adverse effects of ipilimumab, a fully humanized monoclonal antibody directed against cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) used to treat advanced-stage melanoma.
- Alopecia areata is a rarely reported irAE, but its connection to CTLA-4 dysregulation may mean that clinicians see an increased incidence at higher ipilimumab doses.
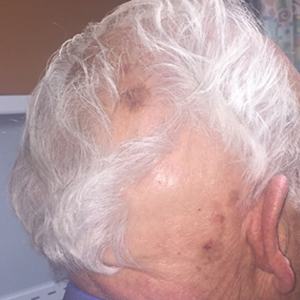
North American Blastomycosis in an Immunocompromised Patient
Blastomycosis is a systemic fungal infection that is endemic in the South Central, Midwest, and southeastern regions of the United States, as well as in provinces of Canada bordering the Great Lakes. After inhalation of Blastomyces dermatitidis spores, which are taken up by bronchopulmonary macrophages, there is an approximate 30- to 45-day incubation period. The initial response at the infected site is suppurative, which progresses to granuloma formation. Blastomyces dermatitidis most commonly infects the lungs, followed by the skin, bones, prostate, and central nervous system (CNS). Therapy for blastomycosis is determined by the severity of the clinical presentation and consideration of the toxicities of the antifungal agent.
We present the case of a 38-year-old man with a medical history of human immunodeficiency virus (HIV) infection and AIDS who reported a 3- to 4-week history of respiratory and cutaneous symptoms. Initial clinical impression favored secondary syphilis; however, after laboratory evaluation and lack of response to treatment for syphilis, further investigation revealed a diagnosis of widespread cutaneous North American blastomycosis.
Case Report
A 38-year-old man with a medical history of HIV infection and AIDS presented to the emergency department at a medical center in Minneapolis, Minnesota, with a cough; chest discomfort; and concomitant nonpainful, mildly pruritic papules and plaques of 3 to 4 weeks’ duration that initially appeared on the face and ears and spread to the trunk, arms, palms, legs, and feet. He had a nonpainful ulcer on the glans penis. Symptoms began while he was living in Atlanta, Georgia, before relocating to Minneapolis. A chest radiograph was negative.
The initial clinical impression favored secondary syphilis. Intramuscular penicillin G benzathine (2.4 million U) weekly for 3 weeks was initiated by the primary care team based on clinical suspicion alone without laboratory evidence of a positive rapid plasma reagin or VDRL test. Because laboratory evaluation and lack of response to treatment did not support syphilis, dermatology consultation was requested.
The patient had a history of crack cocaine abuse. He reported sexual activity with a single female partner while living in a halfway house in the Minneapolis–St. Paul area. Physical examination showed an age-appropriate man in no acute distress who was alert and oriented. He had well-demarcated papules and plaques on the forehead, ears, nose, cutaneous and mucosal lips, chest, back, arms, legs, palms, and soles. Many of the facial papules were pink, nonscaly, and concentrated around the nose and mouth; some were umbilicated (Figure 1). Trunk and extensor papules and plaques were well demarcated, oval, and scaly; some had erosions centrally and were excoriated. Palmar papules were round and had peripheral brown hyperpigmentation and central scale (Figure 2). A 1-cm, shallow, nontender, oval ulceration withraised borders was located on the glans penis under the foreskin (Figure 3).
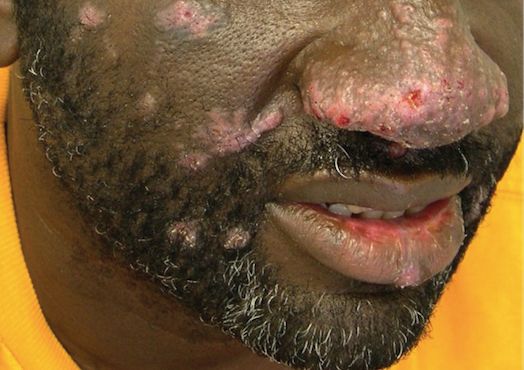
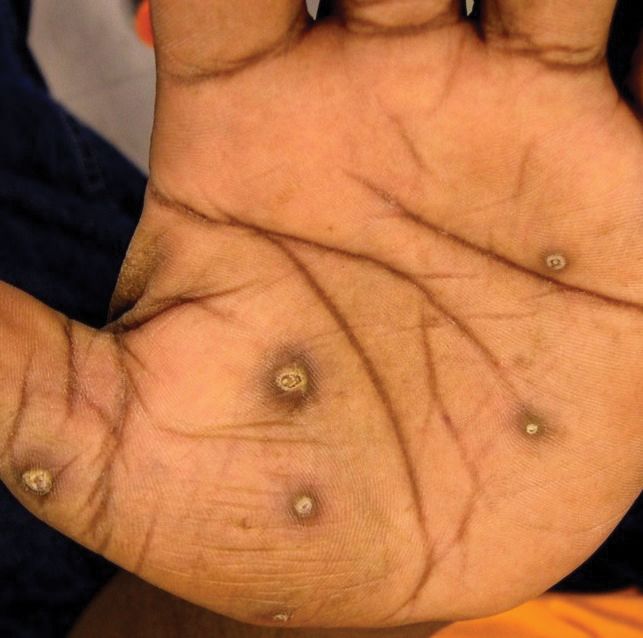
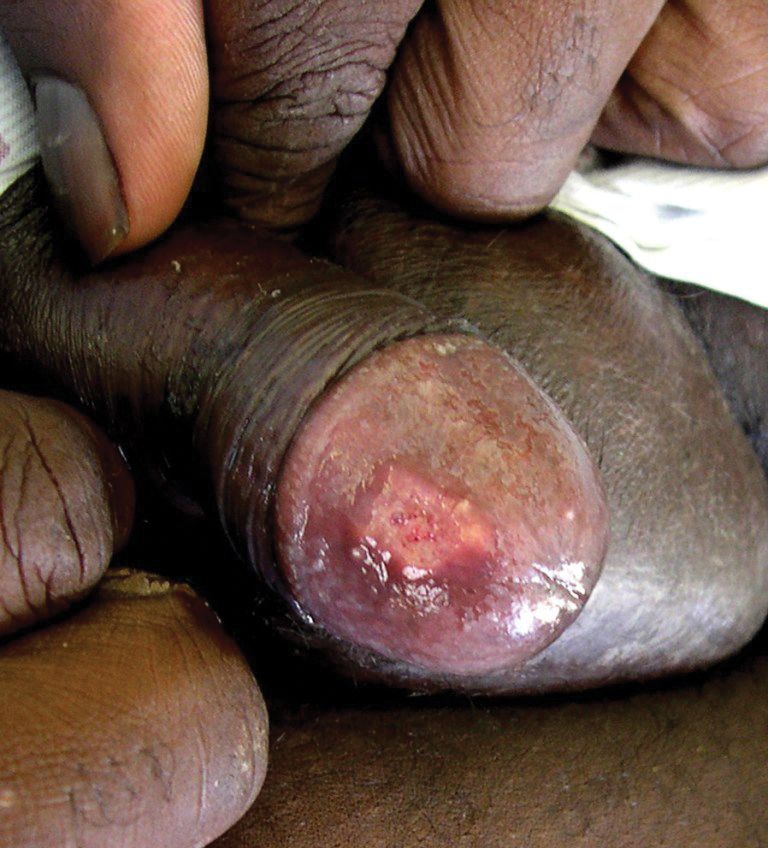
A rapid plasma reagin test was nonreactive; a fluorescent treponemal antibody absorption test was negative. Chest radiograph, magnetic resonance imaging, and electroencephalogram were normal. In addition, spinal fluid drawn from a tap was negative on India ink and Gram stain preparations and was negative for cryptococcal antigen. In addition, spinal fluid was negative for fungal and bacterial growth, as were blood cultures.
Abnormal tests included a positive enzyme-linked immunosorbent assay and Western blot test for HIV, with an absolute CD4 count of 6 cells/mL and a viral load more than 100,000 copies/mL. Urine histoplasmosis antigen was markedly elevated. A potassium hydroxide preparation was performed on the skin of the right forearm, revealing broad-based budding yeast, later confirmed on skin and sputum cultures to be B dermatitidis.
Punch biopsy from the upper back revealed a mixed acute and granulomatous infiltrate with numerous yeast forms (Figure 4A) that were highlighted by Grocott-Gomori methenamine-silver (Figure 4B) and periodic acid–Schiff (Figure 4C) stains.
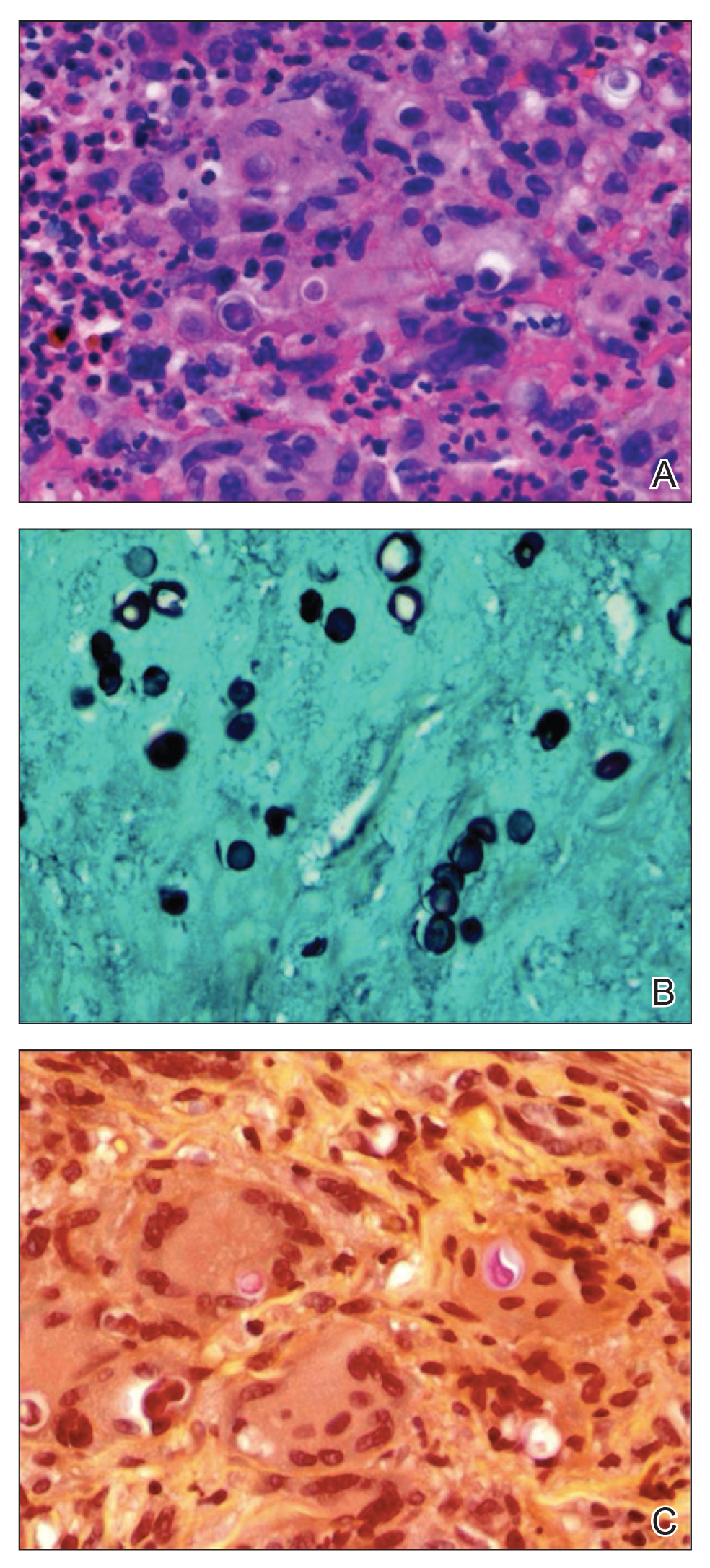
The patient was treated with intravenous amphotericin with improvement in skin lesions. A healing ointment and occlusive dressing were used on eroded skin lesions. The patient was discharged on oral itraconazole 200 mg twice daily for 6 months (for blastomycosis); oral sulfamethoxazole-trimethoprim 15 mg/kg/d every 8 hours for 21 days (for Pneumocystis carinii pneumonia prophylaxis); oral azithromycin 500 mg daily (for Mycobacterium avium-intracellulare prophylaxis); oral levetiracetam 500 mg every 12 hours (as an antiseizure agent); albuterol 90 µg per actuation; and healing ointment. He continues his chemical dependency program and is being followed by the neurology seizure clinic as well as the outpatient HIV infectious disease clinic for planned reinitiation of highly active antiretroviral therapy.
Comment
Diagnosis
Our patient had an interesting and dramatic presentation of widespread cutaneous North American blastomycosis that was initially considered to be secondary syphilis because of involvement of the palms and soles and the presence of the painless penile ulcer. In addition, the initial skin biopsy finding was considered morphologically consistent with Cryptococcus neoformans based on positive Grocott-Gomori methenamine-silver and periodic acid–Schiff stains and an equivocal mucicarmine stain. However, the potassium hydroxide preparation of skin and positive urine histoplasmosis antigen strongly suggested blastomycosis, which was confirmed by culture of B dermatitidis. The urine histoplasmosis antigen can cross-react with B dermatitidis and other mycoses (eg, Paracoccidioides brasiliensis and Penicillium marneffei); however, because the treatment of either of these mycoses is similar, the value of the test remains high.1
Skin tests and serologic markers are useful epidemiologic tools but are of inadequate sensitivity and specificity to be diagnostic for B dermatitidis. Diagnosis depends on direct examination of tissue or isolation of the fungus in culture.2
Source of Infection
The probable occult source of cutaneous infection was the lungs, given the natural history of disseminated blastomycosis; the history of cough and chest discomfort; the widespread nature of skin lesions; and the ultimate growth of rare yeast forms in sputum. Cutaneous infection generally is from disseminated disease and rarely from direct inoculation.
Unlike many other systemic dimorphic mycoses, blastomycosis usually occurs in healthy hosts and is frequently associated with point-source outbreak. Immunosuppressed patients typically develop infection following exposure to the organism, but reactivation also can occur. Blastomycosis is uncommon among HIV-infected individuals and is not recognized as an AIDS-defining illness.
In a review from Canada of 133 patients with blastomycosis, nearly half had an underlying medical condition but not one typically associated with marked immunosuppression.3 Only 2 of 133 patients had HIV infection. Overall mortality was 6.3%, and the average duration of symptoms before diagnosis was less in those who died vs those who survived the disease.3 In the setting of AIDS or other marked immunosuppression, disease usually is more severe, with multiple-system involvement, including the CNS, and can progress rapidly to death.2
Treatment
Therapy for blastomycosis is determined by the severity of the clinical presentation and consideration of the toxicities of the antifungal agent. There are no randomized, blinded trials comparing antifungal agents, and data on the treatment of blastomycosis in patients infected with HIV are limited. Amphotericin B 3 mg/kg every 24 hours is recommended in life-threatening systemic disease and CNS disease as well as in patients with immune suppression, including AIDS.4 In a retrospective study of 326 patients with blastomycosis, those receiving amphotericin B had a cure rate of 86.5% with a relapse rate of 3.9%; patients receiving ketoconazole had a cure rate of 81.7% with a relapse rate of 14%.4 Although data are limited, chronic suppressive therapy generally is recommended in patients with HIV who have been treated for blastomycosis. Fluconazole, itraconazole, and ketoconazole are all used as chronic suppressive therapy; however, given the higher relapse rate observed with ketoconazole, itraconazole is preferred. Because neither ketoconazole nor itraconazole penetrates the blood-brain barrier, these drugs are not recommended in cases of CNS involvement. Patients with CNS disease or intolerance to itraconazole should be treated with fluconazole for chronic suppression.3
- Wheat J, Wheat H, Connolly P, et al. Cross-reactivity in Histoplasma capsulatum variety capsulatum antigen assays of urine samples from patients with endemic mycoses. Clin Infect Dis. 1997;24:1169-1171.
- Pappas PG, Pottage JC, Powderly WG, et al. Blastomycosis in patients with the acquired immunodeficiency syndrome. Ann Intern Med. 1992;116:847-853.
- Crampton TL, Light RB, Berg GM, et al. Epidemiology and clinical spectrum of blastomycosis diagnosed at Manitoba hospitals. Clin Infect Dis. 2002;34:1310-1316. Cited by: Aberg JA. Blastomycosis and HIV. HIV In Site Knowledge Base Chapter. http://hivinsite.ucsf.edu/InSite?page=kb-05-02-09#SIX. Published April 2003. Updated January 2006. Accessed December 16, 2019.
- Chapman SW, Bradsher RW Jr, Campbell GD Jr, et al. Practice guidelines for the management of patients with blastomycosis. Infectious Diseases Society of America. Clin Infect Dis. 2000;30:679-683.
Blastomycosis is a systemic fungal infection that is endemic in the South Central, Midwest, and southeastern regions of the United States, as well as in provinces of Canada bordering the Great Lakes. After inhalation of Blastomyces dermatitidis spores, which are taken up by bronchopulmonary macrophages, there is an approximate 30- to 45-day incubation period. The initial response at the infected site is suppurative, which progresses to granuloma formation. Blastomyces dermatitidis most commonly infects the lungs, followed by the skin, bones, prostate, and central nervous system (CNS). Therapy for blastomycosis is determined by the severity of the clinical presentation and consideration of the toxicities of the antifungal agent.
We present the case of a 38-year-old man with a medical history of human immunodeficiency virus (HIV) infection and AIDS who reported a 3- to 4-week history of respiratory and cutaneous symptoms. Initial clinical impression favored secondary syphilis; however, after laboratory evaluation and lack of response to treatment for syphilis, further investigation revealed a diagnosis of widespread cutaneous North American blastomycosis.
Case Report
A 38-year-old man with a medical history of HIV infection and AIDS presented to the emergency department at a medical center in Minneapolis, Minnesota, with a cough; chest discomfort; and concomitant nonpainful, mildly pruritic papules and plaques of 3 to 4 weeks’ duration that initially appeared on the face and ears and spread to the trunk, arms, palms, legs, and feet. He had a nonpainful ulcer on the glans penis. Symptoms began while he was living in Atlanta, Georgia, before relocating to Minneapolis. A chest radiograph was negative.
The initial clinical impression favored secondary syphilis. Intramuscular penicillin G benzathine (2.4 million U) weekly for 3 weeks was initiated by the primary care team based on clinical suspicion alone without laboratory evidence of a positive rapid plasma reagin or VDRL test. Because laboratory evaluation and lack of response to treatment did not support syphilis, dermatology consultation was requested.
The patient had a history of crack cocaine abuse. He reported sexual activity with a single female partner while living in a halfway house in the Minneapolis–St. Paul area. Physical examination showed an age-appropriate man in no acute distress who was alert and oriented. He had well-demarcated papules and plaques on the forehead, ears, nose, cutaneous and mucosal lips, chest, back, arms, legs, palms, and soles. Many of the facial papules were pink, nonscaly, and concentrated around the nose and mouth; some were umbilicated (Figure 1). Trunk and extensor papules and plaques were well demarcated, oval, and scaly; some had erosions centrally and were excoriated. Palmar papules were round and had peripheral brown hyperpigmentation and central scale (Figure 2). A 1-cm, shallow, nontender, oval ulceration withraised borders was located on the glans penis under the foreskin (Figure 3).
A rapid plasma reagin test was nonreactive; a fluorescent treponemal antibody absorption test was negative. Chest radiograph, magnetic resonance imaging, and electroencephalogram were normal. In addition, spinal fluid drawn from a tap was negative on India ink and Gram stain preparations and was negative for cryptococcal antigen. In addition, spinal fluid was negative for fungal and bacterial growth, as were blood cultures.
Abnormal tests included a positive enzyme-linked immunosorbent assay and Western blot test for HIV, with an absolute CD4 count of 6 cells/mL and a viral load more than 100,000 copies/mL. Urine histoplasmosis antigen was markedly elevated. A potassium hydroxide preparation was performed on the skin of the right forearm, revealing broad-based budding yeast, later confirmed on skin and sputum cultures to be B dermatitidis.
Punch biopsy from the upper back revealed a mixed acute and granulomatous infiltrate with numerous yeast forms (Figure 4A) that were highlighted by Grocott-Gomori methenamine-silver (Figure 4B) and periodic acid–Schiff (Figure 4C) stains.
The patient was treated with intravenous amphotericin with improvement in skin lesions. A healing ointment and occlusive dressing were used on eroded skin lesions. The patient was discharged on oral itraconazole 200 mg twice daily for 6 months (for blastomycosis); oral sulfamethoxazole-trimethoprim 15 mg/kg/d every 8 hours for 21 days (for Pneumocystis carinii pneumonia prophylaxis); oral azithromycin 500 mg daily (for Mycobacterium avium-intracellulare prophylaxis); oral levetiracetam 500 mg every 12 hours (as an antiseizure agent); albuterol 90 µg per actuation; and healing ointment. He continues his chemical dependency program and is being followed by the neurology seizure clinic as well as the outpatient HIV infectious disease clinic for planned reinitiation of highly active antiretroviral therapy.
Comment
Diagnosis
Our patient had an interesting and dramatic presentation of widespread cutaneous North American blastomycosis that was initially considered to be secondary syphilis because of involvement of the palms and soles and the presence of the painless penile ulcer. In addition, the initial skin biopsy finding was considered morphologically consistent with Cryptococcus neoformans based on positive Grocott-Gomori methenamine-silver and periodic acid–Schiff stains and an equivocal mucicarmine stain. However, the potassium hydroxide preparation of skin and positive urine histoplasmosis antigen strongly suggested blastomycosis, which was confirmed by culture of B dermatitidis. The urine histoplasmosis antigen can cross-react with B dermatitidis and other mycoses (eg, Paracoccidioides brasiliensis and Penicillium marneffei); however, because the treatment of either of these mycoses is similar, the value of the test remains high.1
Skin tests and serologic markers are useful epidemiologic tools but are of inadequate sensitivity and specificity to be diagnostic for B dermatitidis. Diagnosis depends on direct examination of tissue or isolation of the fungus in culture.2
Source of Infection
The probable occult source of cutaneous infection was the lungs, given the natural history of disseminated blastomycosis; the history of cough and chest discomfort; the widespread nature of skin lesions; and the ultimate growth of rare yeast forms in sputum. Cutaneous infection generally is from disseminated disease and rarely from direct inoculation.
Unlike many other systemic dimorphic mycoses, blastomycosis usually occurs in healthy hosts and is frequently associated with point-source outbreak. Immunosuppressed patients typically develop infection following exposure to the organism, but reactivation also can occur. Blastomycosis is uncommon among HIV-infected individuals and is not recognized as an AIDS-defining illness.
In a review from Canada of 133 patients with blastomycosis, nearly half had an underlying medical condition but not one typically associated with marked immunosuppression.3 Only 2 of 133 patients had HIV infection. Overall mortality was 6.3%, and the average duration of symptoms before diagnosis was less in those who died vs those who survived the disease.3 In the setting of AIDS or other marked immunosuppression, disease usually is more severe, with multiple-system involvement, including the CNS, and can progress rapidly to death.2
Treatment
Therapy for blastomycosis is determined by the severity of the clinical presentation and consideration of the toxicities of the antifungal agent. There are no randomized, blinded trials comparing antifungal agents, and data on the treatment of blastomycosis in patients infected with HIV are limited. Amphotericin B 3 mg/kg every 24 hours is recommended in life-threatening systemic disease and CNS disease as well as in patients with immune suppression, including AIDS.4 In a retrospective study of 326 patients with blastomycosis, those receiving amphotericin B had a cure rate of 86.5% with a relapse rate of 3.9%; patients receiving ketoconazole had a cure rate of 81.7% with a relapse rate of 14%.4 Although data are limited, chronic suppressive therapy generally is recommended in patients with HIV who have been treated for blastomycosis. Fluconazole, itraconazole, and ketoconazole are all used as chronic suppressive therapy; however, given the higher relapse rate observed with ketoconazole, itraconazole is preferred. Because neither ketoconazole nor itraconazole penetrates the blood-brain barrier, these drugs are not recommended in cases of CNS involvement. Patients with CNS disease or intolerance to itraconazole should be treated with fluconazole for chronic suppression.3
Blastomycosis is a systemic fungal infection that is endemic in the South Central, Midwest, and southeastern regions of the United States, as well as in provinces of Canada bordering the Great Lakes. After inhalation of Blastomyces dermatitidis spores, which are taken up by bronchopulmonary macrophages, there is an approximate 30- to 45-day incubation period. The initial response at the infected site is suppurative, which progresses to granuloma formation. Blastomyces dermatitidis most commonly infects the lungs, followed by the skin, bones, prostate, and central nervous system (CNS). Therapy for blastomycosis is determined by the severity of the clinical presentation and consideration of the toxicities of the antifungal agent.
We present the case of a 38-year-old man with a medical history of human immunodeficiency virus (HIV) infection and AIDS who reported a 3- to 4-week history of respiratory and cutaneous symptoms. Initial clinical impression favored secondary syphilis; however, after laboratory evaluation and lack of response to treatment for syphilis, further investigation revealed a diagnosis of widespread cutaneous North American blastomycosis.
Case Report
A 38-year-old man with a medical history of HIV infection and AIDS presented to the emergency department at a medical center in Minneapolis, Minnesota, with a cough; chest discomfort; and concomitant nonpainful, mildly pruritic papules and plaques of 3 to 4 weeks’ duration that initially appeared on the face and ears and spread to the trunk, arms, palms, legs, and feet. He had a nonpainful ulcer on the glans penis. Symptoms began while he was living in Atlanta, Georgia, before relocating to Minneapolis. A chest radiograph was negative.
The initial clinical impression favored secondary syphilis. Intramuscular penicillin G benzathine (2.4 million U) weekly for 3 weeks was initiated by the primary care team based on clinical suspicion alone without laboratory evidence of a positive rapid plasma reagin or VDRL test. Because laboratory evaluation and lack of response to treatment did not support syphilis, dermatology consultation was requested.
The patient had a history of crack cocaine abuse. He reported sexual activity with a single female partner while living in a halfway house in the Minneapolis–St. Paul area. Physical examination showed an age-appropriate man in no acute distress who was alert and oriented. He had well-demarcated papules and plaques on the forehead, ears, nose, cutaneous and mucosal lips, chest, back, arms, legs, palms, and soles. Many of the facial papules were pink, nonscaly, and concentrated around the nose and mouth; some were umbilicated (Figure 1). Trunk and extensor papules and plaques were well demarcated, oval, and scaly; some had erosions centrally and were excoriated. Palmar papules were round and had peripheral brown hyperpigmentation and central scale (Figure 2). A 1-cm, shallow, nontender, oval ulceration withraised borders was located on the glans penis under the foreskin (Figure 3).
A rapid plasma reagin test was nonreactive; a fluorescent treponemal antibody absorption test was negative. Chest radiograph, magnetic resonance imaging, and electroencephalogram were normal. In addition, spinal fluid drawn from a tap was negative on India ink and Gram stain preparations and was negative for cryptococcal antigen. In addition, spinal fluid was negative for fungal and bacterial growth, as were blood cultures.
Abnormal tests included a positive enzyme-linked immunosorbent assay and Western blot test for HIV, with an absolute CD4 count of 6 cells/mL and a viral load more than 100,000 copies/mL. Urine histoplasmosis antigen was markedly elevated. A potassium hydroxide preparation was performed on the skin of the right forearm, revealing broad-based budding yeast, later confirmed on skin and sputum cultures to be B dermatitidis.
Punch biopsy from the upper back revealed a mixed acute and granulomatous infiltrate with numerous yeast forms (Figure 4A) that were highlighted by Grocott-Gomori methenamine-silver (Figure 4B) and periodic acid–Schiff (Figure 4C) stains.
The patient was treated with intravenous amphotericin with improvement in skin lesions. A healing ointment and occlusive dressing were used on eroded skin lesions. The patient was discharged on oral itraconazole 200 mg twice daily for 6 months (for blastomycosis); oral sulfamethoxazole-trimethoprim 15 mg/kg/d every 8 hours for 21 days (for Pneumocystis carinii pneumonia prophylaxis); oral azithromycin 500 mg daily (for Mycobacterium avium-intracellulare prophylaxis); oral levetiracetam 500 mg every 12 hours (as an antiseizure agent); albuterol 90 µg per actuation; and healing ointment. He continues his chemical dependency program and is being followed by the neurology seizure clinic as well as the outpatient HIV infectious disease clinic for planned reinitiation of highly active antiretroviral therapy.
Comment
Diagnosis
Our patient had an interesting and dramatic presentation of widespread cutaneous North American blastomycosis that was initially considered to be secondary syphilis because of involvement of the palms and soles and the presence of the painless penile ulcer. In addition, the initial skin biopsy finding was considered morphologically consistent with Cryptococcus neoformans based on positive Grocott-Gomori methenamine-silver and periodic acid–Schiff stains and an equivocal mucicarmine stain. However, the potassium hydroxide preparation of skin and positive urine histoplasmosis antigen strongly suggested blastomycosis, which was confirmed by culture of B dermatitidis. The urine histoplasmosis antigen can cross-react with B dermatitidis and other mycoses (eg, Paracoccidioides brasiliensis and Penicillium marneffei); however, because the treatment of either of these mycoses is similar, the value of the test remains high.1
Skin tests and serologic markers are useful epidemiologic tools but are of inadequate sensitivity and specificity to be diagnostic for B dermatitidis. Diagnosis depends on direct examination of tissue or isolation of the fungus in culture.2
Source of Infection
The probable occult source of cutaneous infection was the lungs, given the natural history of disseminated blastomycosis; the history of cough and chest discomfort; the widespread nature of skin lesions; and the ultimate growth of rare yeast forms in sputum. Cutaneous infection generally is from disseminated disease and rarely from direct inoculation.
Unlike many other systemic dimorphic mycoses, blastomycosis usually occurs in healthy hosts and is frequently associated with point-source outbreak. Immunosuppressed patients typically develop infection following exposure to the organism, but reactivation also can occur. Blastomycosis is uncommon among HIV-infected individuals and is not recognized as an AIDS-defining illness.
In a review from Canada of 133 patients with blastomycosis, nearly half had an underlying medical condition but not one typically associated with marked immunosuppression.3 Only 2 of 133 patients had HIV infection. Overall mortality was 6.3%, and the average duration of symptoms before diagnosis was less in those who died vs those who survived the disease.3 In the setting of AIDS or other marked immunosuppression, disease usually is more severe, with multiple-system involvement, including the CNS, and can progress rapidly to death.2
Treatment
Therapy for blastomycosis is determined by the severity of the clinical presentation and consideration of the toxicities of the antifungal agent. There are no randomized, blinded trials comparing antifungal agents, and data on the treatment of blastomycosis in patients infected with HIV are limited. Amphotericin B 3 mg/kg every 24 hours is recommended in life-threatening systemic disease and CNS disease as well as in patients with immune suppression, including AIDS.4 In a retrospective study of 326 patients with blastomycosis, those receiving amphotericin B had a cure rate of 86.5% with a relapse rate of 3.9%; patients receiving ketoconazole had a cure rate of 81.7% with a relapse rate of 14%.4 Although data are limited, chronic suppressive therapy generally is recommended in patients with HIV who have been treated for blastomycosis. Fluconazole, itraconazole, and ketoconazole are all used as chronic suppressive therapy; however, given the higher relapse rate observed with ketoconazole, itraconazole is preferred. Because neither ketoconazole nor itraconazole penetrates the blood-brain barrier, these drugs are not recommended in cases of CNS involvement. Patients with CNS disease or intolerance to itraconazole should be treated with fluconazole for chronic suppression.3
- Wheat J, Wheat H, Connolly P, et al. Cross-reactivity in Histoplasma capsulatum variety capsulatum antigen assays of urine samples from patients with endemic mycoses. Clin Infect Dis. 1997;24:1169-1171.
- Pappas PG, Pottage JC, Powderly WG, et al. Blastomycosis in patients with the acquired immunodeficiency syndrome. Ann Intern Med. 1992;116:847-853.
- Crampton TL, Light RB, Berg GM, et al. Epidemiology and clinical spectrum of blastomycosis diagnosed at Manitoba hospitals. Clin Infect Dis. 2002;34:1310-1316. Cited by: Aberg JA. Blastomycosis and HIV. HIV In Site Knowledge Base Chapter. http://hivinsite.ucsf.edu/InSite?page=kb-05-02-09#SIX. Published April 2003. Updated January 2006. Accessed December 16, 2019.
- Chapman SW, Bradsher RW Jr, Campbell GD Jr, et al. Practice guidelines for the management of patients with blastomycosis. Infectious Diseases Society of America. Clin Infect Dis. 2000;30:679-683.
- Wheat J, Wheat H, Connolly P, et al. Cross-reactivity in Histoplasma capsulatum variety capsulatum antigen assays of urine samples from patients with endemic mycoses. Clin Infect Dis. 1997;24:1169-1171.
- Pappas PG, Pottage JC, Powderly WG, et al. Blastomycosis in patients with the acquired immunodeficiency syndrome. Ann Intern Med. 1992;116:847-853.
- Crampton TL, Light RB, Berg GM, et al. Epidemiology and clinical spectrum of blastomycosis diagnosed at Manitoba hospitals. Clin Infect Dis. 2002;34:1310-1316. Cited by: Aberg JA. Blastomycosis and HIV. HIV In Site Knowledge Base Chapter. http://hivinsite.ucsf.edu/InSite?page=kb-05-02-09#SIX. Published April 2003. Updated January 2006. Accessed December 16, 2019.
- Chapman SW, Bradsher RW Jr, Campbell GD Jr, et al. Practice guidelines for the management of patients with blastomycosis. Infectious Diseases Society of America. Clin Infect Dis. 2000;30:679-683.
Practice Points
- Blastomycosis generally produces a pulmonary form of the disease and, to a lesser extent, extrapulmonary forms, such as cutaneous, osteoarticular, and genitourinary.
- Blastomycosis can be diagnosed by culture, direct visualization of the yeast in affected tissue, antigen testing, or a combination of these methods.
- After inhalation of Blastomyces dermatitidis spores, which are taken up by bronchopulmonary macrophages, there is an approximate 30- to 45-day incubation period.