User login
Metastatic angiosarcoma arising in a patient with long-standing treatment-refractory hemangioma
Angiosarcomas are malignant tumors of the vascular endothelium and are typically idiopathic. These tumors comprise 2% of all soft tissue sarcomas and have an estimated incidence of 2 per million.1,2 Known causes of angiosarcoma include genetic syndromes—such as von Hippel- Lindau, Chuvash polycythemia, Bannayan- Riley-Ruvalcaba, Cowden, and hamartomatous polyposis syndromes— chronic lymphedema, and exposure to radiation.3 Vinyl chloride, arsenicals, and thorotrast are known to increase the incidence of liver angiosarcoma.4
Malignant transformation of hemangioma is rare. We describe metastatic angiosarcoma in a patient with a large, longterm treatment-resistant subcutaneous hemangioma, illustrating such a possibility. We review similar cases and discuss the value of determining pathogenesis in such patients.
Case Presentation and Summary
A 55-year-old female with a long-standing childhood hemangioma of the left lower extremity was referred to Ochsner Medical Center for tissue diagnosis of new pulmonary nodules. Her medical history included a 7 pack-year smoking history; she had quit 3 years prior. Her family history included a sister who died from breast cancer. The patient initially had a progressive, intermittently bleeding tumor in the left foot at age 7. She was diagnosed with hemangioma in her twenties. At that point, her tumor began to involve the posterior calf and femur, causing deformity. She had multiple surgical resections but reportedly all pathology demonstrated benign hemangioma. She received radiation for pain, a routine treatment at the time, but developed a focus of progression in the heel. Above-knee amputation was considered but could not be performed when hemangioma was discovered in the hip area. She was lost to follow-up between 2001 and 2015. Lower extremity magnetic resonance imaging in 2015 was stable with imaging prior to 2001. A repeat biopsy in 2016 demonstrated hemangioma. The patient then received radiation to a wider field, including the femur, with minimal response. She completed a course of steroids as well. Bevacizumab was started in 2017 and improved foot deformity. She also briefly trialed pazopanib for 4 weeks in 2018 in an attempt to switch to oral medications. Despite partial response, she discontinued both agents in July 2018 because of toxicity and the burden of recurrent infusions.
Four months later, she presented with 2 months of intermittent hemoptysis and 18 months of metallic odors. Additionally, she lost 25 pounds in 3 months, which she attributed to a diet plan. At this visit, her left lower extremity exhibited multiple subcutaneous tumors and nodules.
Computed tomography (CT) with contrast demonstrated innumerable pulmonary nodules, the largest measuring 2.2 cm in the right lower lobe superior segment. Positron emission tomography (PET)/CT revealed 2 nodules with mild hypermetabolic activity; the largest nodule had a maximum standardized uptake value of 2.7. Bronchoalveolar lavage studies showed intra-alveolar hemorrhage with hemosiderin-laden macrophages. No malignancy, granuloma, or dysplasia was found in transbronchial needle aspirate of the largest nodule. The patient had no lymphadenopathy.
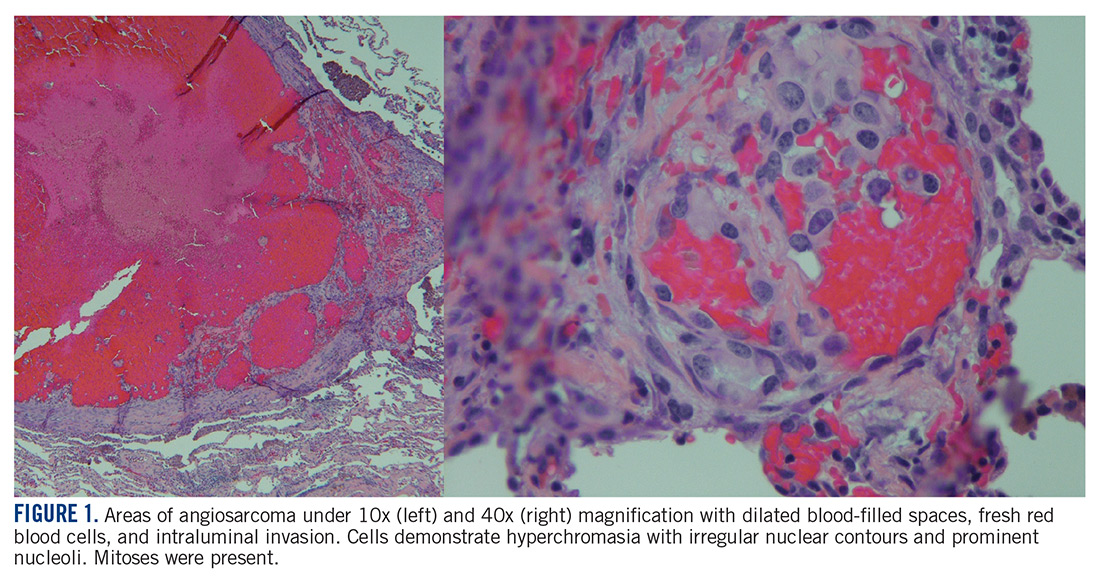
At this hospital, surgical resection by video-assisted thoracoscopic surgery confirmed multifocal malignant epithelioid neoplasm suspicious for angiosarcoma. Multiple areas showed proliferation of atypical epithelioid-to-spindle cells. There were prominent associated hemosiderin-laden macrophages, fresh red blood cells, and dilated blood-filled spaces. Cells demonstrated hyperchromasia with irregular nuclear contours, prominent nucleoli, and mitoses (FIGURE 1). Additionally, there were areas of focal organizing pneumonia. For atypical cells, staining was CD31-positive and CD34-negative. Staining was strongly positive for ERG. There was increased Ki-67 with retained INI expression and patchy weak reactivity for Fli-1.
Next-generation sequencing was performed. Specimen tumor content was 15%. Genomic findings included IDH1 p.R132C mutation, with variant allele frequency <10%. Testing was inconclusive for MSI and TMB mutations. PD-L1 assessment could not be performed. Unfortunately, the patient did not qualify for any clinical trials, as there were no matching alterations. This patient was lost to follow-up.
Discussion
Angiosarcoma accounts for 2% of soft tissue sarcomas.1 Cutaneous angiosarcomas most commonly occur in the face and scalp of the elderly, or in sites of chronic lymphedema. Angiosarcoma also develops following radiation therapy.5 For breast cancers and tumors of the head and neck, irradiation has <1% risk of inducing secondary malignancy, including angiosarcoma.6
This patient had a new diagnosis of angiosarcoma in the setting of long-standing benign hemangioma with history of radiation treatment. Thus, it is unclear whether this angiosarcoma was primary, radiation-induced, or secondary to transformation from the preexisting vascular tumor. Post-irradiation sarcoma carries a less favorable prognosis compared to de novo sarcoma; however, reports conflict on whether this holds for angiosarcoma subtypes.6 Determining etiology may benefit patients for prognostication and possibly inform future selection of treatment modalities.
The mutational signature in radiation- associated sarcomas differs from that of sporadic sarcomas. First, radiation- associated sarcomas demonstrate more frequent small deletions and balanced translocations. TP53 mutations are found in up to 1/3 of radiation-associated sarcomas and are more often due to small deletions than in sporadic sarcomas.7 High-level MYC amplification occurs in 54%-100% of secondary angiosarcomas, compared to 0-7% in sporadic angiosarcomas. Co-amplification of FLT4 occurs in 11%-25% of secondary angiosarcomas.8 Additionally, transcriptome analysis revealed differential expression of a 135-gene signature compared to non-radiation- induced sarcomas.7 Although this patient was not specifically analyzed for such alterations, such tests may differentiate post-irradiation angiosarcoma from sporadic etiologies.
In this patient, the R132C IDH1 mutation was identified and may be the first reported case in angiosarcoma. Typically, this mutation occurs in chondrosarcoma, myeloid neoplasms, gliomas, and cholangiocarcinomas. It is also found in spindle cell hemangiomas but not in other vascular tumors.9 The clinical significance of this mutation is uncertain at this time.
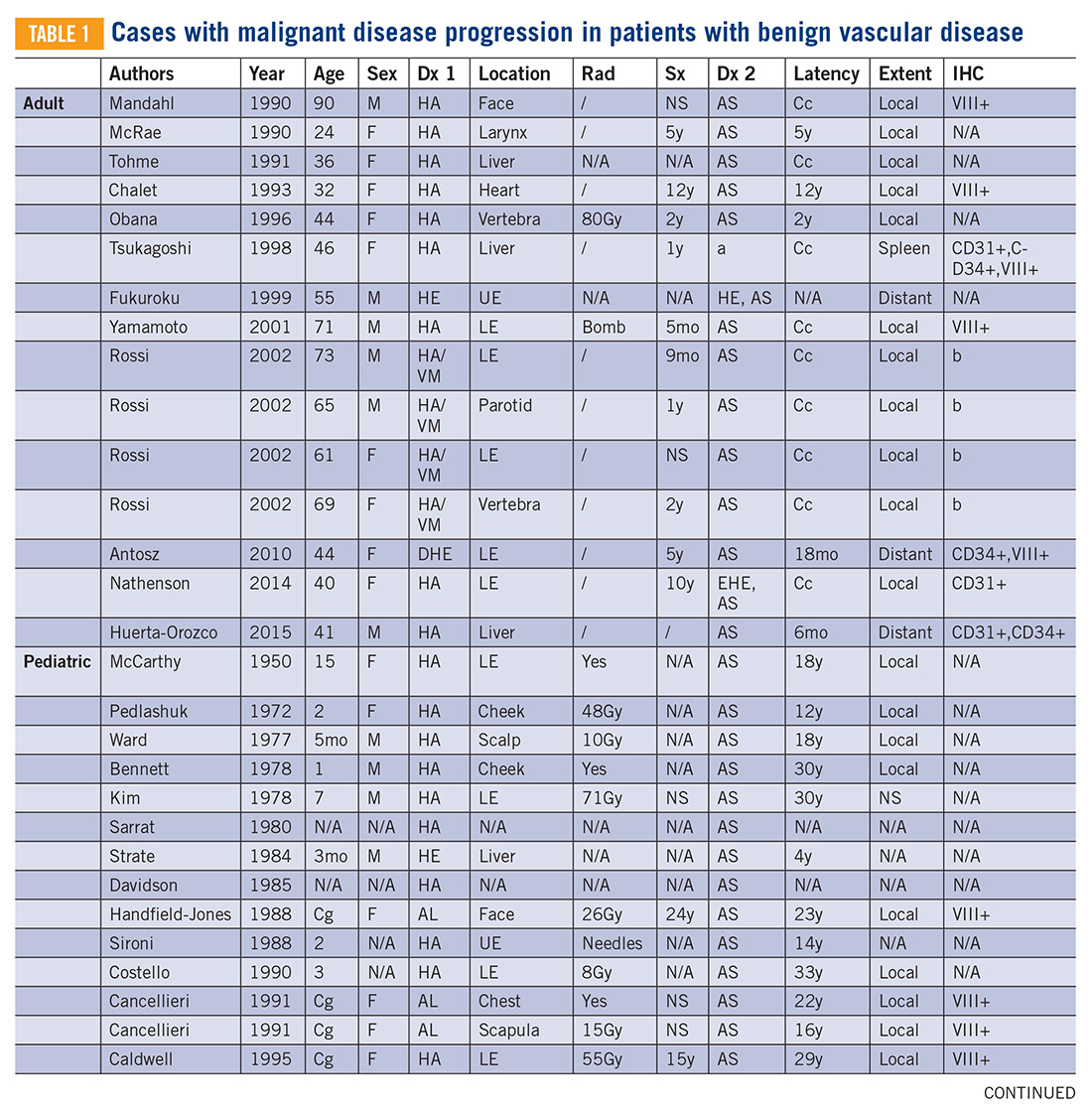
There are approximately 36 reported cases of malignant disease arising in patients with less aggressive vascular tumors (TABLE 1). Of these, 25 of 36 involve angiosarcoma arising in patients with hemangioma. Four cases of angiosarcoma were reported in patients with hemangioendothelioma, 1 case of hemangioendothelioma in a patient with hemangioma, 1 case of Dabska tumor in a patient with hemangioma, and 1 case of angiosarcoma in a patient with Dabska tumor. Fifteen cases involved initial disease with adult onset and 21 involved initial disease with pediatric onset, suggesting even distribution. Malignant disease mostly occurred in adulthood, in 26 out of 33 cases. Latency to malignancy ranged from concurrent discovery to 54 years. Mean latency, excluding cases with concurrent discovery, was shorter with adult-onset initial disease, at 4.2 years, compared to 16 years among patients with onset of initial disease in childhood. Longer latency in the pediatric-onset population correlated with longer latent periods for radiation-induced angiosarcoma following benign disease, which is reported to average 23 years.10 Thirteen of 19 cases with pediatric onset disease had a history of radiotherapy, while 2 of 13 cases with adult onset disease did. Sixteen cases involved tumor in the bone and soft tissue, as in this patient. Notably, 4 of these cases involved long-standing hemangioma for 10 years or more, as in this patient, suggesting a possible correlation between long-standing vascular tumors and malignant transformation. Angiosarcoma arising in non-irradiated patients suggests that malignant transformation and de novo transformation may compete with radiation-induced mutation in tumorigenesis. Further, 8 cases involved angiosarcoma growing within another vascular tumor, demonstrating the possibility of malignant transformation. Dehner and Ishak described a histological model for quantifying such a risk; a validated model may be particularly useful in patients with long-standing hemangioma.11
Etiology of tumorigenesis in cases of angiosarcoma arising in patients with a history of benign hemangioma may benefit prognostication and inform treatment selection in the future. Owing to long latent periods, radiation-associated angiosarcoma incidence may rise, as radiation therapy for benign hemangioma was recently routine. Future research may provide insight into disease progression and possibly predict the risk of angiosarcoma in patients with long-standing benign disease. TSJ
1. Tambe SA, Nayak CS. Metastatic angiosarcoma of lower extremity. Indian Dermatol Online J. 2018;9(3)177-181.
2. Cioffi A, Reichert S, Antonescu CR, Maki RG. Angiosarcomas and other sarcomas of endothelial origin. Hematol Oncol Clin North Am.2013;27(5):975-988.
3. Cohen SM, Storer RD, Criswell KA, et al. Hemangiosarcoma in rodents: mode-of-action evaluation and human relevance. Toxicol Sci. 2009;111(1):4-18.
4. Popper H, Thomas LB, Telles NC, Falk H, Selikoff IJ. Development of hepatic angiosarcoma in man induced by vinyl chloride, thorotrast, and arsenic. Comparison with cases of unknown etiology. Am J Pathol. 1978;92(2):349- 376.
5. Mark RJ, Bailet JW, Poen J, et al. Postirradiation sarcoma of the head and neck. Cancer. 1993;72(3):887-893.
6. Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20(4):1267-1274.
7. Mito JK, Mitra D, Doyle LA. Radiation-associated sarcomas: an update on clinical, histologic, and molecular features. Surg Pathol Clin. 2019;12(1):139-148.
8. Weidema ME, Versleijen-Jonkers YMH, Flucke UE, Desar IME, van der Graaf WTA. Targeting angiosarcomas of the soft tissues: A challenging effort in a heterogeneous and rare disease. Crit Rev Oncol Hematol. 2019;138:120-131.
9. Kurek KC, Pansuriya TC, van Ruler MAJH, et al. R132C IDH1 mutations are found in spindle cell hemangiomas and not in other vascular tumors or malformations. Am J Pathol. 2013;182(5):1494-1500.
10. Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
11. Dehner LP, Ishak KG. Vascular tumors of the liver in infants and children. A study of 30 cases and review of the literature. Arch Pathol. 1971;92(2):101-111.
Angiosarcomas are malignant tumors of the vascular endothelium and are typically idiopathic. These tumors comprise 2% of all soft tissue sarcomas and have an estimated incidence of 2 per million.1,2 Known causes of angiosarcoma include genetic syndromes—such as von Hippel- Lindau, Chuvash polycythemia, Bannayan- Riley-Ruvalcaba, Cowden, and hamartomatous polyposis syndromes— chronic lymphedema, and exposure to radiation.3 Vinyl chloride, arsenicals, and thorotrast are known to increase the incidence of liver angiosarcoma.4
Malignant transformation of hemangioma is rare. We describe metastatic angiosarcoma in a patient with a large, longterm treatment-resistant subcutaneous hemangioma, illustrating such a possibility. We review similar cases and discuss the value of determining pathogenesis in such patients.
Case Presentation and Summary
A 55-year-old female with a long-standing childhood hemangioma of the left lower extremity was referred to Ochsner Medical Center for tissue diagnosis of new pulmonary nodules. Her medical history included a 7 pack-year smoking history; she had quit 3 years prior. Her family history included a sister who died from breast cancer. The patient initially had a progressive, intermittently bleeding tumor in the left foot at age 7. She was diagnosed with hemangioma in her twenties. At that point, her tumor began to involve the posterior calf and femur, causing deformity. She had multiple surgical resections but reportedly all pathology demonstrated benign hemangioma. She received radiation for pain, a routine treatment at the time, but developed a focus of progression in the heel. Above-knee amputation was considered but could not be performed when hemangioma was discovered in the hip area. She was lost to follow-up between 2001 and 2015. Lower extremity magnetic resonance imaging in 2015 was stable with imaging prior to 2001. A repeat biopsy in 2016 demonstrated hemangioma. The patient then received radiation to a wider field, including the femur, with minimal response. She completed a course of steroids as well. Bevacizumab was started in 2017 and improved foot deformity. She also briefly trialed pazopanib for 4 weeks in 2018 in an attempt to switch to oral medications. Despite partial response, she discontinued both agents in July 2018 because of toxicity and the burden of recurrent infusions.
Four months later, she presented with 2 months of intermittent hemoptysis and 18 months of metallic odors. Additionally, she lost 25 pounds in 3 months, which she attributed to a diet plan. At this visit, her left lower extremity exhibited multiple subcutaneous tumors and nodules.
Computed tomography (CT) with contrast demonstrated innumerable pulmonary nodules, the largest measuring 2.2 cm in the right lower lobe superior segment. Positron emission tomography (PET)/CT revealed 2 nodules with mild hypermetabolic activity; the largest nodule had a maximum standardized uptake value of 2.7. Bronchoalveolar lavage studies showed intra-alveolar hemorrhage with hemosiderin-laden macrophages. No malignancy, granuloma, or dysplasia was found in transbronchial needle aspirate of the largest nodule. The patient had no lymphadenopathy.
At this hospital, surgical resection by video-assisted thoracoscopic surgery confirmed multifocal malignant epithelioid neoplasm suspicious for angiosarcoma. Multiple areas showed proliferation of atypical epithelioid-to-spindle cells. There were prominent associated hemosiderin-laden macrophages, fresh red blood cells, and dilated blood-filled spaces. Cells demonstrated hyperchromasia with irregular nuclear contours, prominent nucleoli, and mitoses (FIGURE 1). Additionally, there were areas of focal organizing pneumonia. For atypical cells, staining was CD31-positive and CD34-negative. Staining was strongly positive for ERG. There was increased Ki-67 with retained INI expression and patchy weak reactivity for Fli-1.
Next-generation sequencing was performed. Specimen tumor content was 15%. Genomic findings included IDH1 p.R132C mutation, with variant allele frequency <10%. Testing was inconclusive for MSI and TMB mutations. PD-L1 assessment could not be performed. Unfortunately, the patient did not qualify for any clinical trials, as there were no matching alterations. This patient was lost to follow-up.
Discussion
Angiosarcoma accounts for 2% of soft tissue sarcomas.1 Cutaneous angiosarcomas most commonly occur in the face and scalp of the elderly, or in sites of chronic lymphedema. Angiosarcoma also develops following radiation therapy.5 For breast cancers and tumors of the head and neck, irradiation has <1% risk of inducing secondary malignancy, including angiosarcoma.6
This patient had a new diagnosis of angiosarcoma in the setting of long-standing benign hemangioma with history of radiation treatment. Thus, it is unclear whether this angiosarcoma was primary, radiation-induced, or secondary to transformation from the preexisting vascular tumor. Post-irradiation sarcoma carries a less favorable prognosis compared to de novo sarcoma; however, reports conflict on whether this holds for angiosarcoma subtypes.6 Determining etiology may benefit patients for prognostication and possibly inform future selection of treatment modalities.
The mutational signature in radiation- associated sarcomas differs from that of sporadic sarcomas. First, radiation- associated sarcomas demonstrate more frequent small deletions and balanced translocations. TP53 mutations are found in up to 1/3 of radiation-associated sarcomas and are more often due to small deletions than in sporadic sarcomas.7 High-level MYC amplification occurs in 54%-100% of secondary angiosarcomas, compared to 0-7% in sporadic angiosarcomas. Co-amplification of FLT4 occurs in 11%-25% of secondary angiosarcomas.8 Additionally, transcriptome analysis revealed differential expression of a 135-gene signature compared to non-radiation- induced sarcomas.7 Although this patient was not specifically analyzed for such alterations, such tests may differentiate post-irradiation angiosarcoma from sporadic etiologies.
In this patient, the R132C IDH1 mutation was identified and may be the first reported case in angiosarcoma. Typically, this mutation occurs in chondrosarcoma, myeloid neoplasms, gliomas, and cholangiocarcinomas. It is also found in spindle cell hemangiomas but not in other vascular tumors.9 The clinical significance of this mutation is uncertain at this time.
There are approximately 36 reported cases of malignant disease arising in patients with less aggressive vascular tumors (TABLE 1). Of these, 25 of 36 involve angiosarcoma arising in patients with hemangioma. Four cases of angiosarcoma were reported in patients with hemangioendothelioma, 1 case of hemangioendothelioma in a patient with hemangioma, 1 case of Dabska tumor in a patient with hemangioma, and 1 case of angiosarcoma in a patient with Dabska tumor. Fifteen cases involved initial disease with adult onset and 21 involved initial disease with pediatric onset, suggesting even distribution. Malignant disease mostly occurred in adulthood, in 26 out of 33 cases. Latency to malignancy ranged from concurrent discovery to 54 years. Mean latency, excluding cases with concurrent discovery, was shorter with adult-onset initial disease, at 4.2 years, compared to 16 years among patients with onset of initial disease in childhood. Longer latency in the pediatric-onset population correlated with longer latent periods for radiation-induced angiosarcoma following benign disease, which is reported to average 23 years.10 Thirteen of 19 cases with pediatric onset disease had a history of radiotherapy, while 2 of 13 cases with adult onset disease did. Sixteen cases involved tumor in the bone and soft tissue, as in this patient. Notably, 4 of these cases involved long-standing hemangioma for 10 years or more, as in this patient, suggesting a possible correlation between long-standing vascular tumors and malignant transformation. Angiosarcoma arising in non-irradiated patients suggests that malignant transformation and de novo transformation may compete with radiation-induced mutation in tumorigenesis. Further, 8 cases involved angiosarcoma growing within another vascular tumor, demonstrating the possibility of malignant transformation. Dehner and Ishak described a histological model for quantifying such a risk; a validated model may be particularly useful in patients with long-standing hemangioma.11
Etiology of tumorigenesis in cases of angiosarcoma arising in patients with a history of benign hemangioma may benefit prognostication and inform treatment selection in the future. Owing to long latent periods, radiation-associated angiosarcoma incidence may rise, as radiation therapy for benign hemangioma was recently routine. Future research may provide insight into disease progression and possibly predict the risk of angiosarcoma in patients with long-standing benign disease. TSJ
Angiosarcomas are malignant tumors of the vascular endothelium and are typically idiopathic. These tumors comprise 2% of all soft tissue sarcomas and have an estimated incidence of 2 per million.1,2 Known causes of angiosarcoma include genetic syndromes—such as von Hippel- Lindau, Chuvash polycythemia, Bannayan- Riley-Ruvalcaba, Cowden, and hamartomatous polyposis syndromes— chronic lymphedema, and exposure to radiation.3 Vinyl chloride, arsenicals, and thorotrast are known to increase the incidence of liver angiosarcoma.4
Malignant transformation of hemangioma is rare. We describe metastatic angiosarcoma in a patient with a large, longterm treatment-resistant subcutaneous hemangioma, illustrating such a possibility. We review similar cases and discuss the value of determining pathogenesis in such patients.
Case Presentation and Summary
A 55-year-old female with a long-standing childhood hemangioma of the left lower extremity was referred to Ochsner Medical Center for tissue diagnosis of new pulmonary nodules. Her medical history included a 7 pack-year smoking history; she had quit 3 years prior. Her family history included a sister who died from breast cancer. The patient initially had a progressive, intermittently bleeding tumor in the left foot at age 7. She was diagnosed with hemangioma in her twenties. At that point, her tumor began to involve the posterior calf and femur, causing deformity. She had multiple surgical resections but reportedly all pathology demonstrated benign hemangioma. She received radiation for pain, a routine treatment at the time, but developed a focus of progression in the heel. Above-knee amputation was considered but could not be performed when hemangioma was discovered in the hip area. She was lost to follow-up between 2001 and 2015. Lower extremity magnetic resonance imaging in 2015 was stable with imaging prior to 2001. A repeat biopsy in 2016 demonstrated hemangioma. The patient then received radiation to a wider field, including the femur, with minimal response. She completed a course of steroids as well. Bevacizumab was started in 2017 and improved foot deformity. She also briefly trialed pazopanib for 4 weeks in 2018 in an attempt to switch to oral medications. Despite partial response, she discontinued both agents in July 2018 because of toxicity and the burden of recurrent infusions.
Four months later, she presented with 2 months of intermittent hemoptysis and 18 months of metallic odors. Additionally, she lost 25 pounds in 3 months, which she attributed to a diet plan. At this visit, her left lower extremity exhibited multiple subcutaneous tumors and nodules.
Computed tomography (CT) with contrast demonstrated innumerable pulmonary nodules, the largest measuring 2.2 cm in the right lower lobe superior segment. Positron emission tomography (PET)/CT revealed 2 nodules with mild hypermetabolic activity; the largest nodule had a maximum standardized uptake value of 2.7. Bronchoalveolar lavage studies showed intra-alveolar hemorrhage with hemosiderin-laden macrophages. No malignancy, granuloma, or dysplasia was found in transbronchial needle aspirate of the largest nodule. The patient had no lymphadenopathy.
At this hospital, surgical resection by video-assisted thoracoscopic surgery confirmed multifocal malignant epithelioid neoplasm suspicious for angiosarcoma. Multiple areas showed proliferation of atypical epithelioid-to-spindle cells. There were prominent associated hemosiderin-laden macrophages, fresh red blood cells, and dilated blood-filled spaces. Cells demonstrated hyperchromasia with irregular nuclear contours, prominent nucleoli, and mitoses (FIGURE 1). Additionally, there were areas of focal organizing pneumonia. For atypical cells, staining was CD31-positive and CD34-negative. Staining was strongly positive for ERG. There was increased Ki-67 with retained INI expression and patchy weak reactivity for Fli-1.
Next-generation sequencing was performed. Specimen tumor content was 15%. Genomic findings included IDH1 p.R132C mutation, with variant allele frequency <10%. Testing was inconclusive for MSI and TMB mutations. PD-L1 assessment could not be performed. Unfortunately, the patient did not qualify for any clinical trials, as there were no matching alterations. This patient was lost to follow-up.
Discussion
Angiosarcoma accounts for 2% of soft tissue sarcomas.1 Cutaneous angiosarcomas most commonly occur in the face and scalp of the elderly, or in sites of chronic lymphedema. Angiosarcoma also develops following radiation therapy.5 For breast cancers and tumors of the head and neck, irradiation has <1% risk of inducing secondary malignancy, including angiosarcoma.6
This patient had a new diagnosis of angiosarcoma in the setting of long-standing benign hemangioma with history of radiation treatment. Thus, it is unclear whether this angiosarcoma was primary, radiation-induced, or secondary to transformation from the preexisting vascular tumor. Post-irradiation sarcoma carries a less favorable prognosis compared to de novo sarcoma; however, reports conflict on whether this holds for angiosarcoma subtypes.6 Determining etiology may benefit patients for prognostication and possibly inform future selection of treatment modalities.
The mutational signature in radiation- associated sarcomas differs from that of sporadic sarcomas. First, radiation- associated sarcomas demonstrate more frequent small deletions and balanced translocations. TP53 mutations are found in up to 1/3 of radiation-associated sarcomas and are more often due to small deletions than in sporadic sarcomas.7 High-level MYC amplification occurs in 54%-100% of secondary angiosarcomas, compared to 0-7% in sporadic angiosarcomas. Co-amplification of FLT4 occurs in 11%-25% of secondary angiosarcomas.8 Additionally, transcriptome analysis revealed differential expression of a 135-gene signature compared to non-radiation- induced sarcomas.7 Although this patient was not specifically analyzed for such alterations, such tests may differentiate post-irradiation angiosarcoma from sporadic etiologies.
In this patient, the R132C IDH1 mutation was identified and may be the first reported case in angiosarcoma. Typically, this mutation occurs in chondrosarcoma, myeloid neoplasms, gliomas, and cholangiocarcinomas. It is also found in spindle cell hemangiomas but not in other vascular tumors.9 The clinical significance of this mutation is uncertain at this time.
There are approximately 36 reported cases of malignant disease arising in patients with less aggressive vascular tumors (TABLE 1). Of these, 25 of 36 involve angiosarcoma arising in patients with hemangioma. Four cases of angiosarcoma were reported in patients with hemangioendothelioma, 1 case of hemangioendothelioma in a patient with hemangioma, 1 case of Dabska tumor in a patient with hemangioma, and 1 case of angiosarcoma in a patient with Dabska tumor. Fifteen cases involved initial disease with adult onset and 21 involved initial disease with pediatric onset, suggesting even distribution. Malignant disease mostly occurred in adulthood, in 26 out of 33 cases. Latency to malignancy ranged from concurrent discovery to 54 years. Mean latency, excluding cases with concurrent discovery, was shorter with adult-onset initial disease, at 4.2 years, compared to 16 years among patients with onset of initial disease in childhood. Longer latency in the pediatric-onset population correlated with longer latent periods for radiation-induced angiosarcoma following benign disease, which is reported to average 23 years.10 Thirteen of 19 cases with pediatric onset disease had a history of radiotherapy, while 2 of 13 cases with adult onset disease did. Sixteen cases involved tumor in the bone and soft tissue, as in this patient. Notably, 4 of these cases involved long-standing hemangioma for 10 years or more, as in this patient, suggesting a possible correlation between long-standing vascular tumors and malignant transformation. Angiosarcoma arising in non-irradiated patients suggests that malignant transformation and de novo transformation may compete with radiation-induced mutation in tumorigenesis. Further, 8 cases involved angiosarcoma growing within another vascular tumor, demonstrating the possibility of malignant transformation. Dehner and Ishak described a histological model for quantifying such a risk; a validated model may be particularly useful in patients with long-standing hemangioma.11
Etiology of tumorigenesis in cases of angiosarcoma arising in patients with a history of benign hemangioma may benefit prognostication and inform treatment selection in the future. Owing to long latent periods, radiation-associated angiosarcoma incidence may rise, as radiation therapy for benign hemangioma was recently routine. Future research may provide insight into disease progression and possibly predict the risk of angiosarcoma in patients with long-standing benign disease. TSJ
1. Tambe SA, Nayak CS. Metastatic angiosarcoma of lower extremity. Indian Dermatol Online J. 2018;9(3)177-181.
2. Cioffi A, Reichert S, Antonescu CR, Maki RG. Angiosarcomas and other sarcomas of endothelial origin. Hematol Oncol Clin North Am.2013;27(5):975-988.
3. Cohen SM, Storer RD, Criswell KA, et al. Hemangiosarcoma in rodents: mode-of-action evaluation and human relevance. Toxicol Sci. 2009;111(1):4-18.
4. Popper H, Thomas LB, Telles NC, Falk H, Selikoff IJ. Development of hepatic angiosarcoma in man induced by vinyl chloride, thorotrast, and arsenic. Comparison with cases of unknown etiology. Am J Pathol. 1978;92(2):349- 376.
5. Mark RJ, Bailet JW, Poen J, et al. Postirradiation sarcoma of the head and neck. Cancer. 1993;72(3):887-893.
6. Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20(4):1267-1274.
7. Mito JK, Mitra D, Doyle LA. Radiation-associated sarcomas: an update on clinical, histologic, and molecular features. Surg Pathol Clin. 2019;12(1):139-148.
8. Weidema ME, Versleijen-Jonkers YMH, Flucke UE, Desar IME, van der Graaf WTA. Targeting angiosarcomas of the soft tissues: A challenging effort in a heterogeneous and rare disease. Crit Rev Oncol Hematol. 2019;138:120-131.
9. Kurek KC, Pansuriya TC, van Ruler MAJH, et al. R132C IDH1 mutations are found in spindle cell hemangiomas and not in other vascular tumors or malformations. Am J Pathol. 2013;182(5):1494-1500.
10. Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
11. Dehner LP, Ishak KG. Vascular tumors of the liver in infants and children. A study of 30 cases and review of the literature. Arch Pathol. 1971;92(2):101-111.
1. Tambe SA, Nayak CS. Metastatic angiosarcoma of lower extremity. Indian Dermatol Online J. 2018;9(3)177-181.
2. Cioffi A, Reichert S, Antonescu CR, Maki RG. Angiosarcomas and other sarcomas of endothelial origin. Hematol Oncol Clin North Am.2013;27(5):975-988.
3. Cohen SM, Storer RD, Criswell KA, et al. Hemangiosarcoma in rodents: mode-of-action evaluation and human relevance. Toxicol Sci. 2009;111(1):4-18.
4. Popper H, Thomas LB, Telles NC, Falk H, Selikoff IJ. Development of hepatic angiosarcoma in man induced by vinyl chloride, thorotrast, and arsenic. Comparison with cases of unknown etiology. Am J Pathol. 1978;92(2):349- 376.
5. Mark RJ, Bailet JW, Poen J, et al. Postirradiation sarcoma of the head and neck. Cancer. 1993;72(3):887-893.
6. Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20(4):1267-1274.
7. Mito JK, Mitra D, Doyle LA. Radiation-associated sarcomas: an update on clinical, histologic, and molecular features. Surg Pathol Clin. 2019;12(1):139-148.
8. Weidema ME, Versleijen-Jonkers YMH, Flucke UE, Desar IME, van der Graaf WTA. Targeting angiosarcomas of the soft tissues: A challenging effort in a heterogeneous and rare disease. Crit Rev Oncol Hematol. 2019;138:120-131.
9. Kurek KC, Pansuriya TC, van Ruler MAJH, et al. R132C IDH1 mutations are found in spindle cell hemangiomas and not in other vascular tumors or malformations. Am J Pathol. 2013;182(5):1494-1500.
10. Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
11. Dehner LP, Ishak KG. Vascular tumors of the liver in infants and children. A study of 30 cases and review of the literature. Arch Pathol. 1971;92(2):101-111.
Was this patient's transdermal Tx making her dog sick?
THE CASE
A 56-year-old postmenopausal woman with a history of anxiety, depression, alcohol abuse, fatigue, insomnia, and mental fogginess presented to the family medicine clinic with concerns about her companion animal because of symptoms possibly associated with the patient’s medication. Of note, the patient’s physical exam was unremarkable.
The patient noticed that her 5-year-old, 4.5-lb spayed female Chihuahua dog was exhibiting peculiar behaviors, including excessive licking of the abdomen, nipples, and vulvar areas and straining with urination. The dog’s symptoms had started 1 week after the patient began using estradiol transdermal spray (Evamist) for her menopause symptoms. The patient’s menopause symptoms included hot flushes, insomnia, and mental fogginess.
The patient had been applying the estradiol transdermal spray on her inner forearm twice daily, in the morning and at bedtime. She would let the applied medication dry for approximately 2 hours before allowing her arm to come in contact with other items. She worried that some of the hormone may have wiped off onto her couch, pillows, blankets, and other surfaces. In addition, she often cradled the dog in her arms, which allowed the canine’s back to come in contact with her inner forearms. To her knowledge, the dog did not lick or ingest the medication.
The patient had taken the dog to her veterinarian. On physical exam, the veterinarian noted that the dog had nipple and vulvar enlargement but no vaginal discharge, vaginal bleeding, skin changes, or urine abnormalities.
THE (PET’S) DIAGNOSIS, THE PATIENT’S Rx
The veterinarian diagnosed the Chihuahua with vaginal hyperplasia and vulvar enlargement secondary to hyperestrogenism. The animal’s symptoms were likely caused by exposure to the owner’s hormone replacement therapy (HRT) medication—the estradiol spray. The veterinarian advised the woman to return to her family physician to discuss her use of the topical estrogen.
The patient asked her physician (SS) to change her HRT formulation. She was given a prescription for an estradiol 0.05 mg/24-hour transdermal patch to be placed on her abdomen twice weekly. After 2 weeks of using the patch therapy, the patient’s menopausal symptoms were reported to be well controlled. In addition, the companion animal’s breast and vulvar changes resolved, as did the dog’s licking behavior.
DISCUSSION
Estrogen therapy, with or without progesterone, is the most effective treatment for postmenopausal vasomotor symptoms.1 Given the concerns raised in the Women’s Health Initiative (WHI) and other clinical trials regarding hormone therapy and cardiovascular and breast cancer findings, many clinicians look to alternative, nonoral dosage forms to improve the safety profile.
Continue to: Safety of nonroal estrogen therapy
Safety of nonoral estrogen therapy. Administration of nonoral estrogen is associated with avoidance of hepatic first-pass metabolism and a resulting lower impact on hepatic proteins. Thus, data indicate a potentially lower risk for venous thromboembolic events with transdermal estrogen compared to oral estrogen.1 Since the publication of the results of the WHI trials, prescribing patterns in the United States indicate a general decline in the proportion of oral hormones, while transdermal prescription volume has remained steady, and the use of vaginal formulations has increased.2
Topical estrogen formulations. Transdermal or topical delivery of estrogen can be achieved through various formulations, including patches, gels, and a spray. While patches are simple to use, some women display hypersensitivity to the adhesive. Use of gel and spray formulations avoids exposure to adhesives, but these pose a risk of transfer of hormonal ingredients that are not covered by a patch. This risk is amplified by the relative accessibility of the product-specific application sites, which include the arms or thighs. Each manufacturer recommends careful handwashing after handling the product, a specific drying time before the user covers the site with clothing, and avoidance of contact with the application site for a prescribed period of time, usually at least 1 to 2 hours.3-6
Our patient. This case illustrates the importance of discussing the risk of medication transfer to both humans and animals when prescribing individualized hormone therapy. While the Evamist prescribing information specifically addresses the risk of unintentional medication transfer to children, it does not discuss other contact risks.6 In the literature, there have been a limited number of reports on the adverse effects from transdermal or topical human medication transfer to pets. Notably, the American Pet Products Association estimates that in the United States, approximately 90 million dogs and 94 million cats are owned as a pet in 67% of households.7
THE TAKEAWAY
Use of HRT, including transdermal or topical estrogen formulations, is common. Given the large number of companion animals in the United States, physicians should consider that all members of a patient’s household—including pets—may be subject to unintentional secondary exposure to topical estrogen formulations and that they may experience adverse effects. This presents an opportunity for patient education, which can have a larger impact on all occupants of the home.
CORRESPONDENCE
Shannon Scott, DO, FACOFP, Clinical Associate Professor, Arizona College of Osteopathic Medicine, 19389 North 59th Avenue, Glendale, AZ 85308; [email protected].
1. The NAMS 2017 Hormone Therapy Position Statement Advisory Panel. The 2017 hormone therapy position statement of The North American Menopause Society. Menopause. 2017;24:728-753.
2. Steinkellner AR, Denison SE, Eldridge SL, et al. A decade of postmenopausal hormone therapy prescribing in the United States: long-term effects of the Women’s Health Initiative. Menopause. 2012;19:616-621.
3. Divigel [package insert]. Bridgewater, NJ: Vertical Pharmaceuticals, LLC; 2014.
4. Elestrin [package insert]. Somerset, NJ: Meda Pharmaceuticals; 2014.
5. Estrogel [package insert]. Herndon, VA: Ascend Therapeutics; 2018.
6. Evamist [package insert]. Minneapolis, MN: Perrigo; 2017.
7. American Pet Products Association. Pet Industry Market Size & Ownership Statistics. www.americanpetproducts.org/press_industrytrends.asp. Accessed November 1, 2019.
THE CASE
A 56-year-old postmenopausal woman with a history of anxiety, depression, alcohol abuse, fatigue, insomnia, and mental fogginess presented to the family medicine clinic with concerns about her companion animal because of symptoms possibly associated with the patient’s medication. Of note, the patient’s physical exam was unremarkable.
The patient noticed that her 5-year-old, 4.5-lb spayed female Chihuahua dog was exhibiting peculiar behaviors, including excessive licking of the abdomen, nipples, and vulvar areas and straining with urination. The dog’s symptoms had started 1 week after the patient began using estradiol transdermal spray (Evamist) for her menopause symptoms. The patient’s menopause symptoms included hot flushes, insomnia, and mental fogginess.
The patient had been applying the estradiol transdermal spray on her inner forearm twice daily, in the morning and at bedtime. She would let the applied medication dry for approximately 2 hours before allowing her arm to come in contact with other items. She worried that some of the hormone may have wiped off onto her couch, pillows, blankets, and other surfaces. In addition, she often cradled the dog in her arms, which allowed the canine’s back to come in contact with her inner forearms. To her knowledge, the dog did not lick or ingest the medication.
The patient had taken the dog to her veterinarian. On physical exam, the veterinarian noted that the dog had nipple and vulvar enlargement but no vaginal discharge, vaginal bleeding, skin changes, or urine abnormalities.
THE (PET’S) DIAGNOSIS, THE PATIENT’S Rx
The veterinarian diagnosed the Chihuahua with vaginal hyperplasia and vulvar enlargement secondary to hyperestrogenism. The animal’s symptoms were likely caused by exposure to the owner’s hormone replacement therapy (HRT) medication—the estradiol spray. The veterinarian advised the woman to return to her family physician to discuss her use of the topical estrogen.
The patient asked her physician (SS) to change her HRT formulation. She was given a prescription for an estradiol 0.05 mg/24-hour transdermal patch to be placed on her abdomen twice weekly. After 2 weeks of using the patch therapy, the patient’s menopausal symptoms were reported to be well controlled. In addition, the companion animal’s breast and vulvar changes resolved, as did the dog’s licking behavior.
DISCUSSION
Estrogen therapy, with or without progesterone, is the most effective treatment for postmenopausal vasomotor symptoms.1 Given the concerns raised in the Women’s Health Initiative (WHI) and other clinical trials regarding hormone therapy and cardiovascular and breast cancer findings, many clinicians look to alternative, nonoral dosage forms to improve the safety profile.
Continue to: Safety of nonroal estrogen therapy
Safety of nonoral estrogen therapy. Administration of nonoral estrogen is associated with avoidance of hepatic first-pass metabolism and a resulting lower impact on hepatic proteins. Thus, data indicate a potentially lower risk for venous thromboembolic events with transdermal estrogen compared to oral estrogen.1 Since the publication of the results of the WHI trials, prescribing patterns in the United States indicate a general decline in the proportion of oral hormones, while transdermal prescription volume has remained steady, and the use of vaginal formulations has increased.2
Topical estrogen formulations. Transdermal or topical delivery of estrogen can be achieved through various formulations, including patches, gels, and a spray. While patches are simple to use, some women display hypersensitivity to the adhesive. Use of gel and spray formulations avoids exposure to adhesives, but these pose a risk of transfer of hormonal ingredients that are not covered by a patch. This risk is amplified by the relative accessibility of the product-specific application sites, which include the arms or thighs. Each manufacturer recommends careful handwashing after handling the product, a specific drying time before the user covers the site with clothing, and avoidance of contact with the application site for a prescribed period of time, usually at least 1 to 2 hours.3-6
Our patient. This case illustrates the importance of discussing the risk of medication transfer to both humans and animals when prescribing individualized hormone therapy. While the Evamist prescribing information specifically addresses the risk of unintentional medication transfer to children, it does not discuss other contact risks.6 In the literature, there have been a limited number of reports on the adverse effects from transdermal or topical human medication transfer to pets. Notably, the American Pet Products Association estimates that in the United States, approximately 90 million dogs and 94 million cats are owned as a pet in 67% of households.7
THE TAKEAWAY
Use of HRT, including transdermal or topical estrogen formulations, is common. Given the large number of companion animals in the United States, physicians should consider that all members of a patient’s household—including pets—may be subject to unintentional secondary exposure to topical estrogen formulations and that they may experience adverse effects. This presents an opportunity for patient education, which can have a larger impact on all occupants of the home.
CORRESPONDENCE
Shannon Scott, DO, FACOFP, Clinical Associate Professor, Arizona College of Osteopathic Medicine, 19389 North 59th Avenue, Glendale, AZ 85308; [email protected].
THE CASE
A 56-year-old postmenopausal woman with a history of anxiety, depression, alcohol abuse, fatigue, insomnia, and mental fogginess presented to the family medicine clinic with concerns about her companion animal because of symptoms possibly associated with the patient’s medication. Of note, the patient’s physical exam was unremarkable.
The patient noticed that her 5-year-old, 4.5-lb spayed female Chihuahua dog was exhibiting peculiar behaviors, including excessive licking of the abdomen, nipples, and vulvar areas and straining with urination. The dog’s symptoms had started 1 week after the patient began using estradiol transdermal spray (Evamist) for her menopause symptoms. The patient’s menopause symptoms included hot flushes, insomnia, and mental fogginess.
The patient had been applying the estradiol transdermal spray on her inner forearm twice daily, in the morning and at bedtime. She would let the applied medication dry for approximately 2 hours before allowing her arm to come in contact with other items. She worried that some of the hormone may have wiped off onto her couch, pillows, blankets, and other surfaces. In addition, she often cradled the dog in her arms, which allowed the canine’s back to come in contact with her inner forearms. To her knowledge, the dog did not lick or ingest the medication.
The patient had taken the dog to her veterinarian. On physical exam, the veterinarian noted that the dog had nipple and vulvar enlargement but no vaginal discharge, vaginal bleeding, skin changes, or urine abnormalities.
THE (PET’S) DIAGNOSIS, THE PATIENT’S Rx
The veterinarian diagnosed the Chihuahua with vaginal hyperplasia and vulvar enlargement secondary to hyperestrogenism. The animal’s symptoms were likely caused by exposure to the owner’s hormone replacement therapy (HRT) medication—the estradiol spray. The veterinarian advised the woman to return to her family physician to discuss her use of the topical estrogen.
The patient asked her physician (SS) to change her HRT formulation. She was given a prescription for an estradiol 0.05 mg/24-hour transdermal patch to be placed on her abdomen twice weekly. After 2 weeks of using the patch therapy, the patient’s menopausal symptoms were reported to be well controlled. In addition, the companion animal’s breast and vulvar changes resolved, as did the dog’s licking behavior.
DISCUSSION
Estrogen therapy, with or without progesterone, is the most effective treatment for postmenopausal vasomotor symptoms.1 Given the concerns raised in the Women’s Health Initiative (WHI) and other clinical trials regarding hormone therapy and cardiovascular and breast cancer findings, many clinicians look to alternative, nonoral dosage forms to improve the safety profile.
Continue to: Safety of nonroal estrogen therapy
Safety of nonoral estrogen therapy. Administration of nonoral estrogen is associated with avoidance of hepatic first-pass metabolism and a resulting lower impact on hepatic proteins. Thus, data indicate a potentially lower risk for venous thromboembolic events with transdermal estrogen compared to oral estrogen.1 Since the publication of the results of the WHI trials, prescribing patterns in the United States indicate a general decline in the proportion of oral hormones, while transdermal prescription volume has remained steady, and the use of vaginal formulations has increased.2
Topical estrogen formulations. Transdermal or topical delivery of estrogen can be achieved through various formulations, including patches, gels, and a spray. While patches are simple to use, some women display hypersensitivity to the adhesive. Use of gel and spray formulations avoids exposure to adhesives, but these pose a risk of transfer of hormonal ingredients that are not covered by a patch. This risk is amplified by the relative accessibility of the product-specific application sites, which include the arms or thighs. Each manufacturer recommends careful handwashing after handling the product, a specific drying time before the user covers the site with clothing, and avoidance of contact with the application site for a prescribed period of time, usually at least 1 to 2 hours.3-6
Our patient. This case illustrates the importance of discussing the risk of medication transfer to both humans and animals when prescribing individualized hormone therapy. While the Evamist prescribing information specifically addresses the risk of unintentional medication transfer to children, it does not discuss other contact risks.6 In the literature, there have been a limited number of reports on the adverse effects from transdermal or topical human medication transfer to pets. Notably, the American Pet Products Association estimates that in the United States, approximately 90 million dogs and 94 million cats are owned as a pet in 67% of households.7
THE TAKEAWAY
Use of HRT, including transdermal or topical estrogen formulations, is common. Given the large number of companion animals in the United States, physicians should consider that all members of a patient’s household—including pets—may be subject to unintentional secondary exposure to topical estrogen formulations and that they may experience adverse effects. This presents an opportunity for patient education, which can have a larger impact on all occupants of the home.
CORRESPONDENCE
Shannon Scott, DO, FACOFP, Clinical Associate Professor, Arizona College of Osteopathic Medicine, 19389 North 59th Avenue, Glendale, AZ 85308; [email protected].
1. The NAMS 2017 Hormone Therapy Position Statement Advisory Panel. The 2017 hormone therapy position statement of The North American Menopause Society. Menopause. 2017;24:728-753.
2. Steinkellner AR, Denison SE, Eldridge SL, et al. A decade of postmenopausal hormone therapy prescribing in the United States: long-term effects of the Women’s Health Initiative. Menopause. 2012;19:616-621.
3. Divigel [package insert]. Bridgewater, NJ: Vertical Pharmaceuticals, LLC; 2014.
4. Elestrin [package insert]. Somerset, NJ: Meda Pharmaceuticals; 2014.
5. Estrogel [package insert]. Herndon, VA: Ascend Therapeutics; 2018.
6. Evamist [package insert]. Minneapolis, MN: Perrigo; 2017.
7. American Pet Products Association. Pet Industry Market Size & Ownership Statistics. www.americanpetproducts.org/press_industrytrends.asp. Accessed November 1, 2019.
1. The NAMS 2017 Hormone Therapy Position Statement Advisory Panel. The 2017 hormone therapy position statement of The North American Menopause Society. Menopause. 2017;24:728-753.
2. Steinkellner AR, Denison SE, Eldridge SL, et al. A decade of postmenopausal hormone therapy prescribing in the United States: long-term effects of the Women’s Health Initiative. Menopause. 2012;19:616-621.
3. Divigel [package insert]. Bridgewater, NJ: Vertical Pharmaceuticals, LLC; 2014.
4. Elestrin [package insert]. Somerset, NJ: Meda Pharmaceuticals; 2014.
5. Estrogel [package insert]. Herndon, VA: Ascend Therapeutics; 2018.
6. Evamist [package insert]. Minneapolis, MN: Perrigo; 2017.
7. American Pet Products Association. Pet Industry Market Size & Ownership Statistics. www.americanpetproducts.org/press_industrytrends.asp. Accessed November 1, 2019.

Pyoderma Gangrenosum Developing After Chest Tube Placement in a Patient With Chronic Lymphocytic Leukemia
Diagnosis of a neutrophilic dermatosis, such as pyoderma gangrenosum (PG), often is challenging at onset because it can be impossible to distinguish clinically and histopathologically from acute infection in an immunosuppressed patient, necessitating a detailed history as well as correlation pathology with microbial tissue cultures. The dermatologist’s ability to distinguish a neutrophilic dermatosis from active infection is of paramount importance because the decision to treat with surgical debridement, in addition to an antibiotic regimen, can have grave consequences in the misdiagnosed patient.
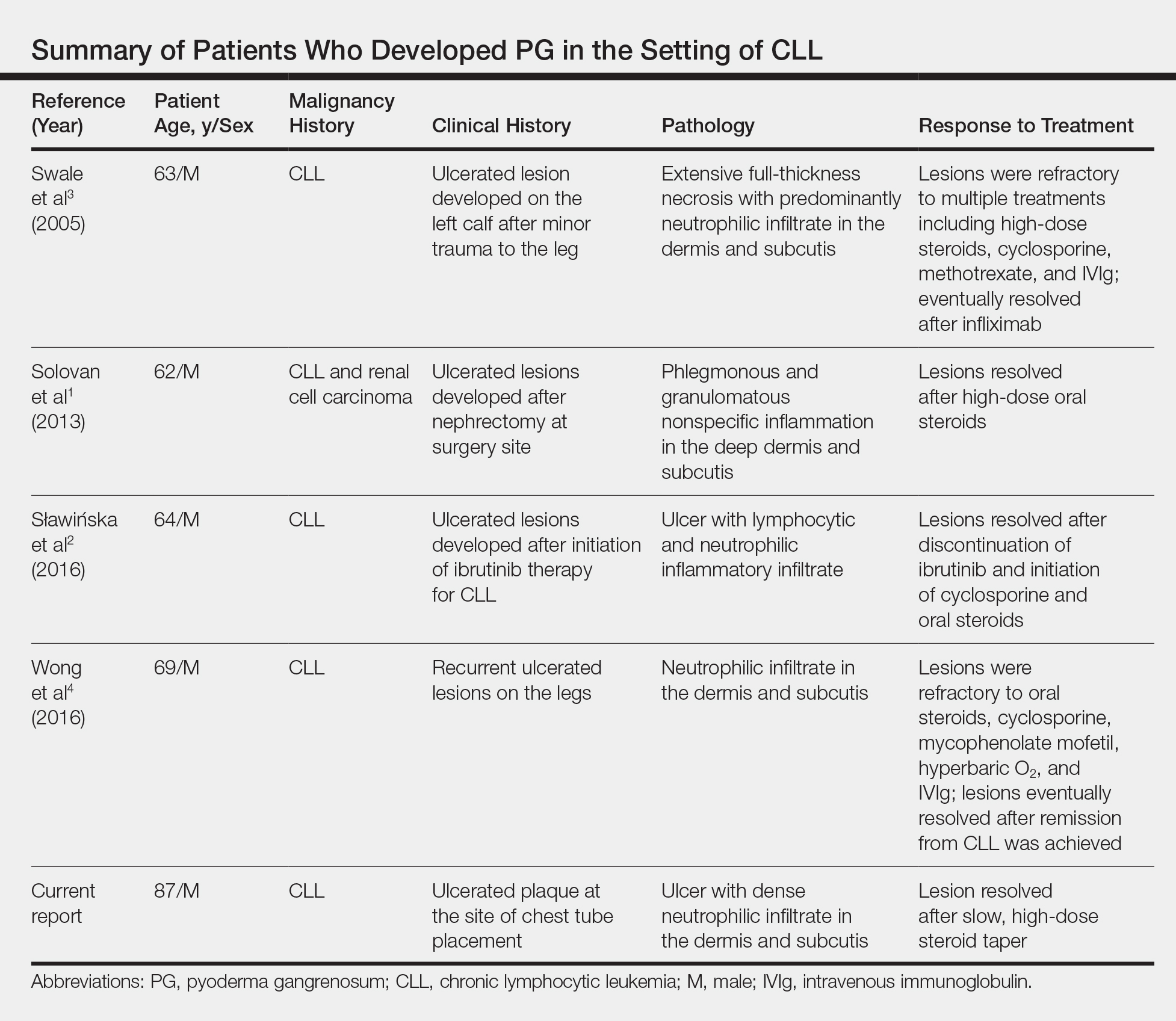
Pyoderma gangrenosum is a neutrophilic dermatosis histologically characterized by a pandermal neutrophilic infiltrate without evidence of an infectious cause or true vasculitis. It is classically associated with inflammatory bowel disease or an underlying hematologic malignancy. Pyoderma gangrenosum in the setting of chronic lymphocytic leukemia (CLL) is rare, with as few as 4 cases having been described in the literature and only 1 case of PG developing after a surgical procedure.1-4 We present a case of PG occurring at a chest tube site in a patient with CLL. We highlight the challenges and therapeutic importance of arriving at the correct diagnosis.
Case Report
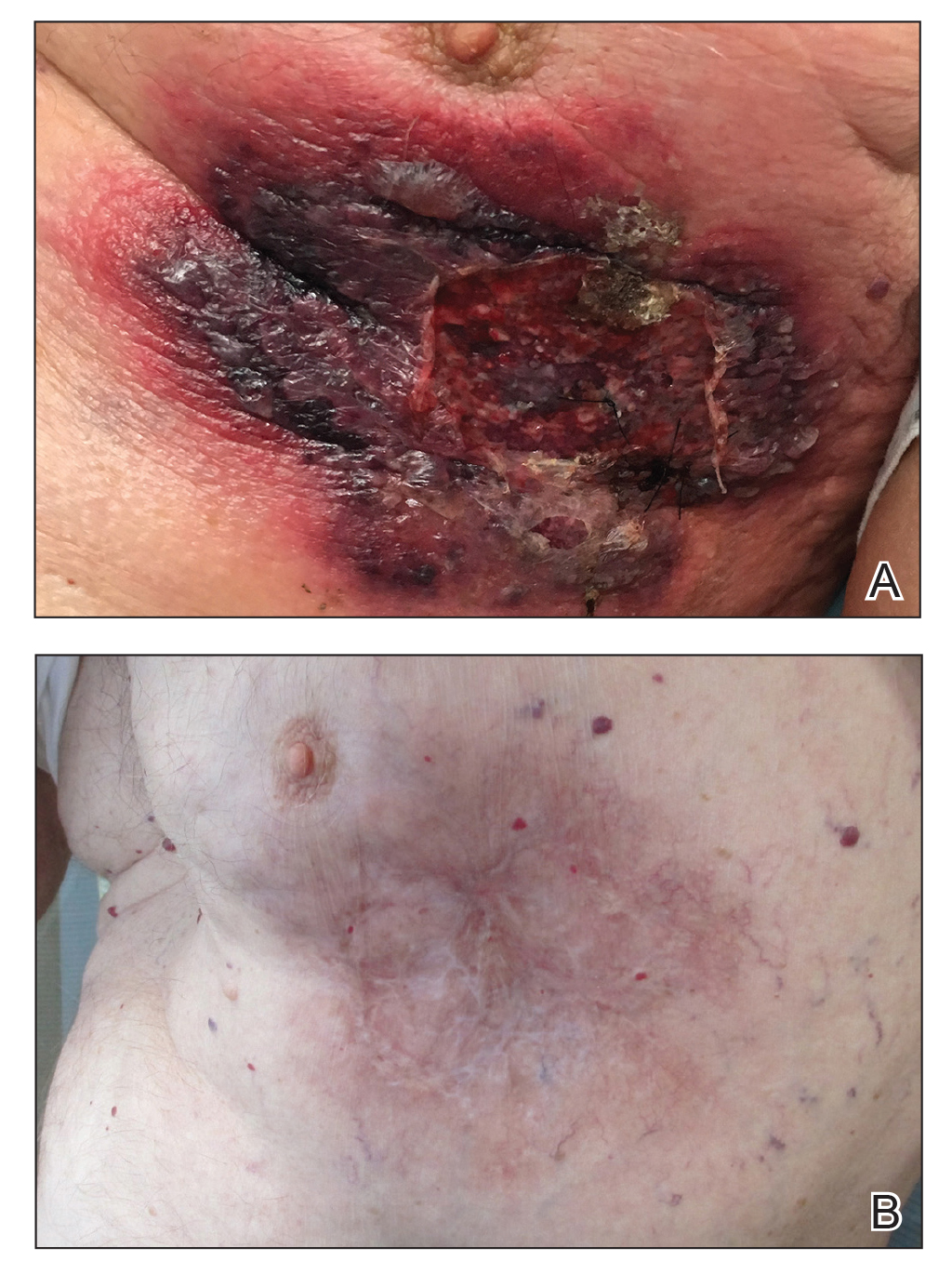
An 87-year-old man with a history of refractory CLL was admitted to the hospital with pneumonia and pleural effusion requiring chest tube placement (left). His most recent therapeutic regimen for CLL was rituximab and bendamustine, which was administered 9 days prior to admission. After removal of the chest tube, an erythematous plaque with central necrosis surrounding the chest tube site developed (Figure 1A). During this time period, the patient had documented intermittent fevers, leukopenia, and neutropenia. Serial blood cultures yielded no growth. Because the patient was on broad-spectrum antibiotic coverage, dermatology was consulted for possible angioinvasive fungal infection.
Physical examination revealed an indurated, erythematous-violaceous, targetoid, well-defined, ulcerated plaque with central necrosis on the left side of the chest. Notably, we observed an isolated bulla with an erythematous base within the right antecubital fossa at the site of intravenous placement, suggesting pathergy.
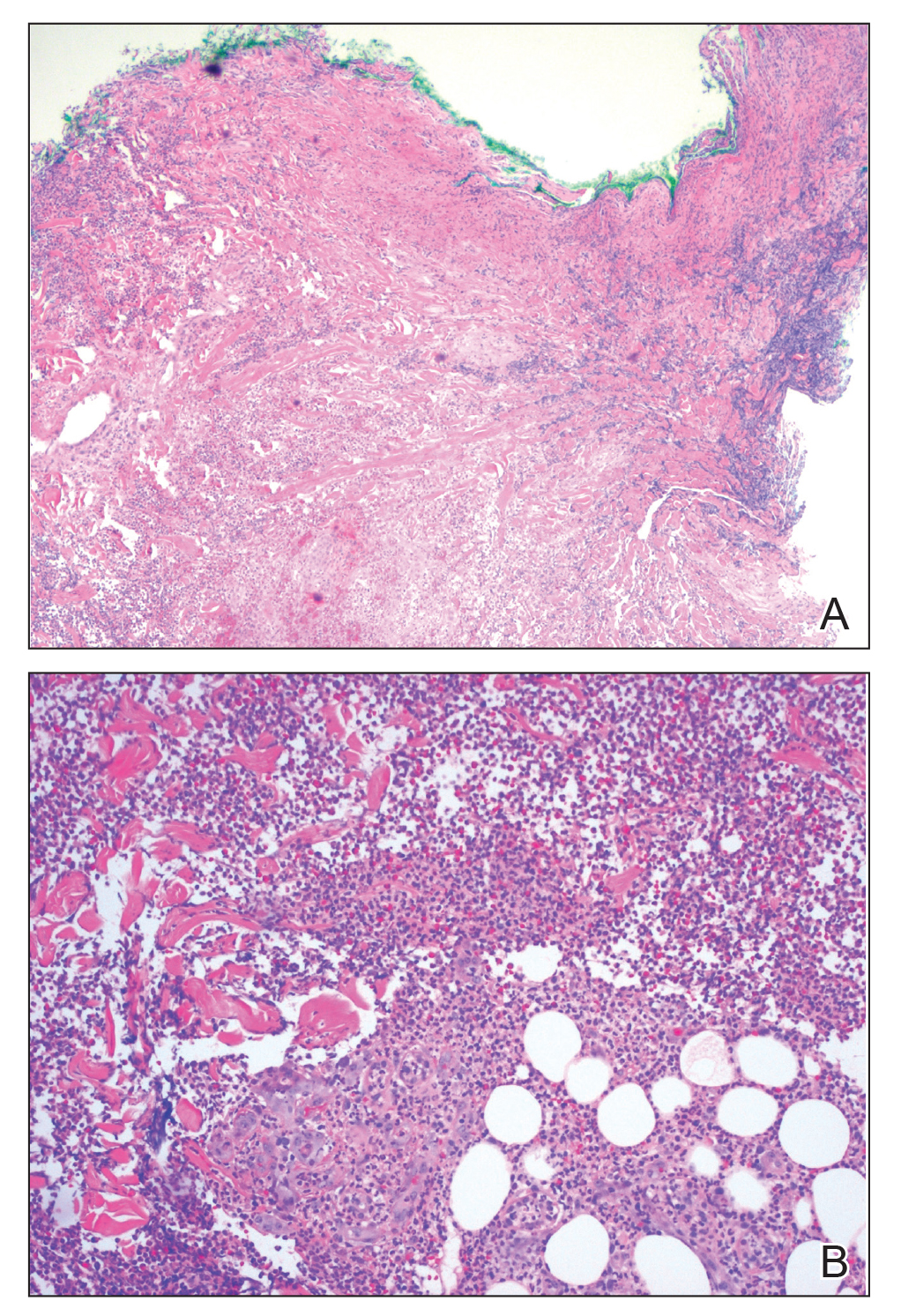
Multiple punch biopsies revealed an ulcer with an underlying dense neutrophilic infiltrate within the dermis and subcutaneous tissues (Figure 2). Grocott-Gomori methenamine-silver, periodic acid–Schiff, and acid-fast bacillus stains were all negative for organisms. Tissue cultures for bacterial, fungal, and acid-fast bacilli revealed no growth. Due to the rapidly expanding nature of the plaque and the possibility of infection despite negative microbial stains and cultures, the patient was scheduled for surgical debridement by the surgical team.
Opportunely, after thoughtful consideration of the clinical history, histopathology, and negative tissue cultures, we made a diagnosis of PG, a condition that would have been further exacerbated by debridement and unimproved with antibiotics. Based on our recommendation, the patient received immunosuppressive treatment with prednisone 60 mg/d and triamcinolone ointment 0.1%. He experienced immediate clinical improvement, allowing him to be discharged to the care of dermatology as an outpatient. He continued to receive a monthly rituximab infusion. We intentionally tapered the patient’s prednisone dosage slowly over 4 months and photodocumented steady improvement with eventual resolution of the PG (Figure 1B).
Comment
Pathogenesis of PG
Pyoderma gangrenosum lies in the spectrum of neutrophilic dermatoses, which are characterized histologically by a pandermal neutrophilic infiltrate without evidence of an infectious cause or true vasculitis. Clinically, PG typically presents as a steadily expanding ulceration with an undermined or slightly raised border, and often is associated with the pathergy phenomenon. Historically, PG is classically linked to inflammatory bowel disease; however, association with underlying malignancy, including acute myelogenous leukemia, chronic myelogenous leukemia, myeloma, and myeloid metaplasia, also has been described.5
Pathogenesis of CLL
Chronic lymphocytic leukemia represents the most prevalent form of leukemia in US adults, with the second highest annual incidence.6 Cutaneous findings are seen in 25% of patients with CLL, varying from leukemia cutis to secondary findings such as vasculitis, purpura, generalized pruritus, exfoliative erythroderma, paraneoplastic pemphigus, infections, and rarely neutrophilic dermatoses.7 According to a PubMed search of articles indexed for MEDLINE using the term pyoderma gangrenosum in CLL, only 4 cases of PG occurring in the setting of CLL exist in the literature, with 1 case demonstrating development after a surgical procedure, making ours the second such case (Table).1-4
Diagnosis
Making the diagnosis of a neutrophilic dermatosis such as PG or Sweet syndrome (SS) in the hospital setting is not only difficult but also imperative, considering that the counterdiagnosis more often is an infectious process. The distinction between individual neutrophilic dermatoses is less crucial at the onset because the initial treatment is the same.
Sweet syndrome is classically the most challenging entity within the spectrum to differentiate from PG. However, our case outlines several key distinguishing features:
• The lesion in classic PG is a rapidly expanding ulceration with undermined borders, whereas SS is less commonly associated with ulceration and instead classically presents with multiple edematous papules that progress to juicy plaques.8
• The pathergy phenomenon has been reported in SS, though it is more commonly associated with PG.9
• In reported cases of SS that were related to cutaneous trauma, lesions developed outside the area of trauma and there was documented leukocytosis and neutrophilia.10-14
• Although leukocytosis is part of the minor diagnostic criteria for SS, it is not required for the diagnosis of PG. Considering that our patient had ulcerated lesions, lesions only at the site of trauma, and leukopenia with intermittent neutropenia, the diagnosis was consistent with PG.
The primary value of early recognition and diagnosis of PG lies in the physician’s ability to distinguish PG from an infectious process, which can be challenging in an immunosuppressed patient with an underlying hematologic malignancy.
Conclusion
This case report represents our experience in arriving at the correct diagnosis of PG in a febrile neutrophilic patient with CLL. In the case of PG in a complicated patient, it is critical to initiate appropriate treatment and avoid inappropriate therapies. Aggressive surgical debridement could have resulted in a fatal outcome for our patient, highlighting the need for dermatologists to raise physician awareness of this challenging disease.
Acknowledgments
The authors acknowledge the contributions of Sarah Shalin, MD, PhD; Nikhil Meena, MD; and Aditya Chada, MD (all from Little Rock, Arkansas), for excellent patient care.
- Solovan C, Smiszek R, Wickenhauser C, et al. Postoperative pyoderma gangrenosum in association with renal cell carcinoma and chronic lymphocytic leukemia. Infect Dis Ther. 2013;2:75-80.
- Sławińska M, Barańska-Rybak W, Sobjanek M, et al. Ibrutinib-induced pyoderma gangrenosum. Pol Arch Med Wewn. 2016;126:710-711.
- Swale VJ, Saha M, Kapur N, et al. Pyoderma gangrenosum outside the context of inflammatory bowel disease treated successfully with infliximab. Clin Exp Dermatol. 2005;30:134-136.
- Wong SM, McComish J, Douglass J, et al. Rare skin manifestations successfully treated with primary B-cell chronic lymphocytic leukemia treatment. J Cutan Pathol. 2016;43:552-555.
- Jockenhöfer F, Herberger K, Schaller J, et al. Tricenter analysis of cofactors and comorbidity in patients with pyoderma gangrenosum. J Dtsch Dermatol Ges. 2016;14:1023-1030.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30. 7.
- Robak E, Robak T. Skin lesions in chronic lymphocytic leukemia. Leuk Lymphoma. 2007;48:855-865.
- Beasley JM, Sluzevich JC. A recurrent vesiculobullous eruption on the head, trunk, and extremities. Bullous Sweet’s syndrome. Int J Dermatol. 2016;55:149-150.
- Awan F, Hamadani M, Devine S. Paraneoplastic Sweet’s syndrome and the pathergy phenomenon. Ann Hematol. 2007;86:613-614.
- de Moya MA, Wong JT, Kroshinsky D, et al. Case records of the Massachusetts General Hospital. Case 28-2012. A 30-year-old woman with shock and abdominal-wall necrosis after cesarean section. N Engl J Med. 2012;367:1046-1057.
- Minocha R, Sebaratnam DF, Choi JY. Sweet’s syndrome following surgery: cutaneous trauma as a possible aetiological co-factor in neutrophilic dermatoses. Australas J Dermatol. 2015;56:
e74-e76. - Phua YS, Al-Ani SA, She RB, et al. Sweet’s syndrome triggered by scalding: a case study and review of the literature. Burns. 2010;36:e49-e52.
- Schwarz RE, Quinn MA, Molina A. Acute postoperative dermatosis at the site of the electrocautery pad: sweet diagnosis of a burning issue. Surg Today. 2000;30:207-209.
- Tan AW, Tan HH, Lim PL. Bullous Sweet’s syndrome following influenza vaccination in a HIV-infected patient. Int J Dermatol. 2006;45:1254-1255.
Diagnosis of a neutrophilic dermatosis, such as pyoderma gangrenosum (PG), often is challenging at onset because it can be impossible to distinguish clinically and histopathologically from acute infection in an immunosuppressed patient, necessitating a detailed history as well as correlation pathology with microbial tissue cultures. The dermatologist’s ability to distinguish a neutrophilic dermatosis from active infection is of paramount importance because the decision to treat with surgical debridement, in addition to an antibiotic regimen, can have grave consequences in the misdiagnosed patient.
Pyoderma gangrenosum is a neutrophilic dermatosis histologically characterized by a pandermal neutrophilic infiltrate without evidence of an infectious cause or true vasculitis. It is classically associated with inflammatory bowel disease or an underlying hematologic malignancy. Pyoderma gangrenosum in the setting of chronic lymphocytic leukemia (CLL) is rare, with as few as 4 cases having been described in the literature and only 1 case of PG developing after a surgical procedure.1-4 We present a case of PG occurring at a chest tube site in a patient with CLL. We highlight the challenges and therapeutic importance of arriving at the correct diagnosis.
Case Report
An 87-year-old man with a history of refractory CLL was admitted to the hospital with pneumonia and pleural effusion requiring chest tube placement (left). His most recent therapeutic regimen for CLL was rituximab and bendamustine, which was administered 9 days prior to admission. After removal of the chest tube, an erythematous plaque with central necrosis surrounding the chest tube site developed (Figure 1A). During this time period, the patient had documented intermittent fevers, leukopenia, and neutropenia. Serial blood cultures yielded no growth. Because the patient was on broad-spectrum antibiotic coverage, dermatology was consulted for possible angioinvasive fungal infection.
Physical examination revealed an indurated, erythematous-violaceous, targetoid, well-defined, ulcerated plaque with central necrosis on the left side of the chest. Notably, we observed an isolated bulla with an erythematous base within the right antecubital fossa at the site of intravenous placement, suggesting pathergy.
Multiple punch biopsies revealed an ulcer with an underlying dense neutrophilic infiltrate within the dermis and subcutaneous tissues (Figure 2). Grocott-Gomori methenamine-silver, periodic acid–Schiff, and acid-fast bacillus stains were all negative for organisms. Tissue cultures for bacterial, fungal, and acid-fast bacilli revealed no growth. Due to the rapidly expanding nature of the plaque and the possibility of infection despite negative microbial stains and cultures, the patient was scheduled for surgical debridement by the surgical team.
Opportunely, after thoughtful consideration of the clinical history, histopathology, and negative tissue cultures, we made a diagnosis of PG, a condition that would have been further exacerbated by debridement and unimproved with antibiotics. Based on our recommendation, the patient received immunosuppressive treatment with prednisone 60 mg/d and triamcinolone ointment 0.1%. He experienced immediate clinical improvement, allowing him to be discharged to the care of dermatology as an outpatient. He continued to receive a monthly rituximab infusion. We intentionally tapered the patient’s prednisone dosage slowly over 4 months and photodocumented steady improvement with eventual resolution of the PG (Figure 1B).
Comment
Pathogenesis of PG
Pyoderma gangrenosum lies in the spectrum of neutrophilic dermatoses, which are characterized histologically by a pandermal neutrophilic infiltrate without evidence of an infectious cause or true vasculitis. Clinically, PG typically presents as a steadily expanding ulceration with an undermined or slightly raised border, and often is associated with the pathergy phenomenon. Historically, PG is classically linked to inflammatory bowel disease; however, association with underlying malignancy, including acute myelogenous leukemia, chronic myelogenous leukemia, myeloma, and myeloid metaplasia, also has been described.5
Pathogenesis of CLL
Chronic lymphocytic leukemia represents the most prevalent form of leukemia in US adults, with the second highest annual incidence.6 Cutaneous findings are seen in 25% of patients with CLL, varying from leukemia cutis to secondary findings such as vasculitis, purpura, generalized pruritus, exfoliative erythroderma, paraneoplastic pemphigus, infections, and rarely neutrophilic dermatoses.7 According to a PubMed search of articles indexed for MEDLINE using the term pyoderma gangrenosum in CLL, only 4 cases of PG occurring in the setting of CLL exist in the literature, with 1 case demonstrating development after a surgical procedure, making ours the second such case (Table).1-4
Diagnosis
Making the diagnosis of a neutrophilic dermatosis such as PG or Sweet syndrome (SS) in the hospital setting is not only difficult but also imperative, considering that the counterdiagnosis more often is an infectious process. The distinction between individual neutrophilic dermatoses is less crucial at the onset because the initial treatment is the same.
Sweet syndrome is classically the most challenging entity within the spectrum to differentiate from PG. However, our case outlines several key distinguishing features:
• The lesion in classic PG is a rapidly expanding ulceration with undermined borders, whereas SS is less commonly associated with ulceration and instead classically presents with multiple edematous papules that progress to juicy plaques.8
• The pathergy phenomenon has been reported in SS, though it is more commonly associated with PG.9
• In reported cases of SS that were related to cutaneous trauma, lesions developed outside the area of trauma and there was documented leukocytosis and neutrophilia.10-14
• Although leukocytosis is part of the minor diagnostic criteria for SS, it is not required for the diagnosis of PG. Considering that our patient had ulcerated lesions, lesions only at the site of trauma, and leukopenia with intermittent neutropenia, the diagnosis was consistent with PG.
The primary value of early recognition and diagnosis of PG lies in the physician’s ability to distinguish PG from an infectious process, which can be challenging in an immunosuppressed patient with an underlying hematologic malignancy.
Conclusion
This case report represents our experience in arriving at the correct diagnosis of PG in a febrile neutrophilic patient with CLL. In the case of PG in a complicated patient, it is critical to initiate appropriate treatment and avoid inappropriate therapies. Aggressive surgical debridement could have resulted in a fatal outcome for our patient, highlighting the need for dermatologists to raise physician awareness of this challenging disease.
Acknowledgments
The authors acknowledge the contributions of Sarah Shalin, MD, PhD; Nikhil Meena, MD; and Aditya Chada, MD (all from Little Rock, Arkansas), for excellent patient care.
Diagnosis of a neutrophilic dermatosis, such as pyoderma gangrenosum (PG), often is challenging at onset because it can be impossible to distinguish clinically and histopathologically from acute infection in an immunosuppressed patient, necessitating a detailed history as well as correlation pathology with microbial tissue cultures. The dermatologist’s ability to distinguish a neutrophilic dermatosis from active infection is of paramount importance because the decision to treat with surgical debridement, in addition to an antibiotic regimen, can have grave consequences in the misdiagnosed patient.
Pyoderma gangrenosum is a neutrophilic dermatosis histologically characterized by a pandermal neutrophilic infiltrate without evidence of an infectious cause or true vasculitis. It is classically associated with inflammatory bowel disease or an underlying hematologic malignancy. Pyoderma gangrenosum in the setting of chronic lymphocytic leukemia (CLL) is rare, with as few as 4 cases having been described in the literature and only 1 case of PG developing after a surgical procedure.1-4 We present a case of PG occurring at a chest tube site in a patient with CLL. We highlight the challenges and therapeutic importance of arriving at the correct diagnosis.
Case Report
An 87-year-old man with a history of refractory CLL was admitted to the hospital with pneumonia and pleural effusion requiring chest tube placement (left). His most recent therapeutic regimen for CLL was rituximab and bendamustine, which was administered 9 days prior to admission. After removal of the chest tube, an erythematous plaque with central necrosis surrounding the chest tube site developed (Figure 1A). During this time period, the patient had documented intermittent fevers, leukopenia, and neutropenia. Serial blood cultures yielded no growth. Because the patient was on broad-spectrum antibiotic coverage, dermatology was consulted for possible angioinvasive fungal infection.
Physical examination revealed an indurated, erythematous-violaceous, targetoid, well-defined, ulcerated plaque with central necrosis on the left side of the chest. Notably, we observed an isolated bulla with an erythematous base within the right antecubital fossa at the site of intravenous placement, suggesting pathergy.
Multiple punch biopsies revealed an ulcer with an underlying dense neutrophilic infiltrate within the dermis and subcutaneous tissues (Figure 2). Grocott-Gomori methenamine-silver, periodic acid–Schiff, and acid-fast bacillus stains were all negative for organisms. Tissue cultures for bacterial, fungal, and acid-fast bacilli revealed no growth. Due to the rapidly expanding nature of the plaque and the possibility of infection despite negative microbial stains and cultures, the patient was scheduled for surgical debridement by the surgical team.
Opportunely, after thoughtful consideration of the clinical history, histopathology, and negative tissue cultures, we made a diagnosis of PG, a condition that would have been further exacerbated by debridement and unimproved with antibiotics. Based on our recommendation, the patient received immunosuppressive treatment with prednisone 60 mg/d and triamcinolone ointment 0.1%. He experienced immediate clinical improvement, allowing him to be discharged to the care of dermatology as an outpatient. He continued to receive a monthly rituximab infusion. We intentionally tapered the patient’s prednisone dosage slowly over 4 months and photodocumented steady improvement with eventual resolution of the PG (Figure 1B).
Comment
Pathogenesis of PG
Pyoderma gangrenosum lies in the spectrum of neutrophilic dermatoses, which are characterized histologically by a pandermal neutrophilic infiltrate without evidence of an infectious cause or true vasculitis. Clinically, PG typically presents as a steadily expanding ulceration with an undermined or slightly raised border, and often is associated with the pathergy phenomenon. Historically, PG is classically linked to inflammatory bowel disease; however, association with underlying malignancy, including acute myelogenous leukemia, chronic myelogenous leukemia, myeloma, and myeloid metaplasia, also has been described.5
Pathogenesis of CLL
Chronic lymphocytic leukemia represents the most prevalent form of leukemia in US adults, with the second highest annual incidence.6 Cutaneous findings are seen in 25% of patients with CLL, varying from leukemia cutis to secondary findings such as vasculitis, purpura, generalized pruritus, exfoliative erythroderma, paraneoplastic pemphigus, infections, and rarely neutrophilic dermatoses.7 According to a PubMed search of articles indexed for MEDLINE using the term pyoderma gangrenosum in CLL, only 4 cases of PG occurring in the setting of CLL exist in the literature, with 1 case demonstrating development after a surgical procedure, making ours the second such case (Table).1-4
Diagnosis
Making the diagnosis of a neutrophilic dermatosis such as PG or Sweet syndrome (SS) in the hospital setting is not only difficult but also imperative, considering that the counterdiagnosis more often is an infectious process. The distinction between individual neutrophilic dermatoses is less crucial at the onset because the initial treatment is the same.
Sweet syndrome is classically the most challenging entity within the spectrum to differentiate from PG. However, our case outlines several key distinguishing features:
• The lesion in classic PG is a rapidly expanding ulceration with undermined borders, whereas SS is less commonly associated with ulceration and instead classically presents with multiple edematous papules that progress to juicy plaques.8
• The pathergy phenomenon has been reported in SS, though it is more commonly associated with PG.9
• In reported cases of SS that were related to cutaneous trauma, lesions developed outside the area of trauma and there was documented leukocytosis and neutrophilia.10-14
• Although leukocytosis is part of the minor diagnostic criteria for SS, it is not required for the diagnosis of PG. Considering that our patient had ulcerated lesions, lesions only at the site of trauma, and leukopenia with intermittent neutropenia, the diagnosis was consistent with PG.
The primary value of early recognition and diagnosis of PG lies in the physician’s ability to distinguish PG from an infectious process, which can be challenging in an immunosuppressed patient with an underlying hematologic malignancy.
Conclusion
This case report represents our experience in arriving at the correct diagnosis of PG in a febrile neutrophilic patient with CLL. In the case of PG in a complicated patient, it is critical to initiate appropriate treatment and avoid inappropriate therapies. Aggressive surgical debridement could have resulted in a fatal outcome for our patient, highlighting the need for dermatologists to raise physician awareness of this challenging disease.
Acknowledgments
The authors acknowledge the contributions of Sarah Shalin, MD, PhD; Nikhil Meena, MD; and Aditya Chada, MD (all from Little Rock, Arkansas), for excellent patient care.
- Solovan C, Smiszek R, Wickenhauser C, et al. Postoperative pyoderma gangrenosum in association with renal cell carcinoma and chronic lymphocytic leukemia. Infect Dis Ther. 2013;2:75-80.
- Sławińska M, Barańska-Rybak W, Sobjanek M, et al. Ibrutinib-induced pyoderma gangrenosum. Pol Arch Med Wewn. 2016;126:710-711.
- Swale VJ, Saha M, Kapur N, et al. Pyoderma gangrenosum outside the context of inflammatory bowel disease treated successfully with infliximab. Clin Exp Dermatol. 2005;30:134-136.
- Wong SM, McComish J, Douglass J, et al. Rare skin manifestations successfully treated with primary B-cell chronic lymphocytic leukemia treatment. J Cutan Pathol. 2016;43:552-555.
- Jockenhöfer F, Herberger K, Schaller J, et al. Tricenter analysis of cofactors and comorbidity in patients with pyoderma gangrenosum. J Dtsch Dermatol Ges. 2016;14:1023-1030.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30. 7.
- Robak E, Robak T. Skin lesions in chronic lymphocytic leukemia. Leuk Lymphoma. 2007;48:855-865.
- Beasley JM, Sluzevich JC. A recurrent vesiculobullous eruption on the head, trunk, and extremities. Bullous Sweet’s syndrome. Int J Dermatol. 2016;55:149-150.
- Awan F, Hamadani M, Devine S. Paraneoplastic Sweet’s syndrome and the pathergy phenomenon. Ann Hematol. 2007;86:613-614.
- de Moya MA, Wong JT, Kroshinsky D, et al. Case records of the Massachusetts General Hospital. Case 28-2012. A 30-year-old woman with shock and abdominal-wall necrosis after cesarean section. N Engl J Med. 2012;367:1046-1057.
- Minocha R, Sebaratnam DF, Choi JY. Sweet’s syndrome following surgery: cutaneous trauma as a possible aetiological co-factor in neutrophilic dermatoses. Australas J Dermatol. 2015;56:
e74-e76. - Phua YS, Al-Ani SA, She RB, et al. Sweet’s syndrome triggered by scalding: a case study and review of the literature. Burns. 2010;36:e49-e52.
- Schwarz RE, Quinn MA, Molina A. Acute postoperative dermatosis at the site of the electrocautery pad: sweet diagnosis of a burning issue. Surg Today. 2000;30:207-209.
- Tan AW, Tan HH, Lim PL. Bullous Sweet’s syndrome following influenza vaccination in a HIV-infected patient. Int J Dermatol. 2006;45:1254-1255.
- Solovan C, Smiszek R, Wickenhauser C, et al. Postoperative pyoderma gangrenosum in association with renal cell carcinoma and chronic lymphocytic leukemia. Infect Dis Ther. 2013;2:75-80.
- Sławińska M, Barańska-Rybak W, Sobjanek M, et al. Ibrutinib-induced pyoderma gangrenosum. Pol Arch Med Wewn. 2016;126:710-711.
- Swale VJ, Saha M, Kapur N, et al. Pyoderma gangrenosum outside the context of inflammatory bowel disease treated successfully with infliximab. Clin Exp Dermatol. 2005;30:134-136.
- Wong SM, McComish J, Douglass J, et al. Rare skin manifestations successfully treated with primary B-cell chronic lymphocytic leukemia treatment. J Cutan Pathol. 2016;43:552-555.
- Jockenhöfer F, Herberger K, Schaller J, et al. Tricenter analysis of cofactors and comorbidity in patients with pyoderma gangrenosum. J Dtsch Dermatol Ges. 2016;14:1023-1030.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30. 7.
- Robak E, Robak T. Skin lesions in chronic lymphocytic leukemia. Leuk Lymphoma. 2007;48:855-865.
- Beasley JM, Sluzevich JC. A recurrent vesiculobullous eruption on the head, trunk, and extremities. Bullous Sweet’s syndrome. Int J Dermatol. 2016;55:149-150.
- Awan F, Hamadani M, Devine S. Paraneoplastic Sweet’s syndrome and the pathergy phenomenon. Ann Hematol. 2007;86:613-614.
- de Moya MA, Wong JT, Kroshinsky D, et al. Case records of the Massachusetts General Hospital. Case 28-2012. A 30-year-old woman with shock and abdominal-wall necrosis after cesarean section. N Engl J Med. 2012;367:1046-1057.
- Minocha R, Sebaratnam DF, Choi JY. Sweet’s syndrome following surgery: cutaneous trauma as a possible aetiological co-factor in neutrophilic dermatoses. Australas J Dermatol. 2015;56:
e74-e76. - Phua YS, Al-Ani SA, She RB, et al. Sweet’s syndrome triggered by scalding: a case study and review of the literature. Burns. 2010;36:e49-e52.
- Schwarz RE, Quinn MA, Molina A. Acute postoperative dermatosis at the site of the electrocautery pad: sweet diagnosis of a burning issue. Surg Today. 2000;30:207-209.
- Tan AW, Tan HH, Lim PL. Bullous Sweet’s syndrome following influenza vaccination in a HIV-infected patient. Int J Dermatol. 2006;45:1254-1255.
Practice Points
- The primary value of early recognition and diagnosis of pyoderma gangrenosum (PG) lies in the physician’s ability to distinguish PG from an infectious process.
- Surgical debridement would further exacerbate PG, making proper diagnosis of a neutrophilic dermatosis of paramount importance to avoid treatments that could have grave consequences in the misdiagnosed patient.
- Cutaneous findings are seen in one-quarter of patients with chronic lymphocytic leukemia.
- Pyoderma gangrenosum is commonly associated with inflammatory bowel disease but also can be seen in many hematologic malignancies. Physicians should be aware of this association to ensure these patients are diagnosed properly.
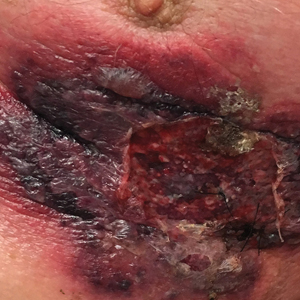
Recurrent Angiotensin-Converting Enzyme Inhibitor-Induced Angioedema Refractory to Fresh Frozen Plasma
Angioedema induced by angiotensin-converting enzyme inhibitors (ACEIs) is present in from 0.1% to 0.7% of treated patients and more often involves the head, neck, face, lips, tongue, and larynx.1 ACEI-induced angioedema results from inhibition of angiotensin-converting enzyme (ACE), which results in reduced degradation and resultant accumulation of bradykinin, a potent inflammatory mediator.2
The treatment of choice is discontinuing all ACEIs; however, the patient may be at increased risk of a subsequent angioedema attack for many weeks.3 Antihistamines (H1 and H2 receptor blockade), epinephrine, and glucocorticoids are effective in allergic/histaminergic angioedema but are usually ineffective for hereditary angioedema or ACEI angioedema and are not recommended for acute therapy.4 Kallikrein-bradykinin pathway targeted therapies are now approved by the Food and Drug Administration (FDA) for hereditary angioedema attacks and have been studied for ACEI-induced angioedema. Ecallantide and icatibant inhibit conversion of precursors to bradykinin. Multiple randomized trials of ecallantide have not shown any advantage over traditional therapies.5 On the other hand, icatibant has shown resolution of angioedema in several case reports and in a randomized trial.6 Icatibant for ACEI-induced angioedema continues to be off-label because the data are conflicting.
Case Presentation
A 67-year-old man presented with a medical history of arterial hypertension (diagnosed 17 years previously), hypercholesterolemia, type 2 diabetes mellitus, alcohol dependence, and obesity. His outpatient medications included simvastatin, aripiprazole, losartan/hydrochlorothiazide, and amlodipine. He was voluntarily admitted for inpatient detoxification. After evaluation by the internist, medication reconciliation was done, and the therapy was adjusted according to medication availability. He reported having no drug allergies, and the losartan was changed for lisinopril. About 24 hours after the first dose of lisinopril, the patient developed swelling of the lips. Antihistamine and IV steroids were administered, and the ACEI was discontinued. His baseline vital signs were temperature 98° F, heart rate 83 beats per minute, respiratory rate 19 breaths per minute, blood pressure 150/94, and oxygen saturation 98% by pulse oximeter.
During the night shift the patient’s symptoms worsened, developing difficulty swallowing and shortness of breath. He was transferred to the medicine intensive care unit (MICU), intubated, and placed on mechanical ventilation to protect his airway. Laryngoscopic examination was notable for edematous tongue, uvula, and larynx. Also, the patient had mild stridor. His laboratory test results showed normal levels of complement, tryptase, and C1 esterase. On the fourth day after admission to MICU (Figure 1), the patient extubated himself. At that time, he did not present stridor or respiratory distress and remained at the MICU for 24 hours for close monitoring.
Thirty-six hours after self-extubation the patient developed stridor and shortness of breath at the general medicine ward. In view of his clinical presentation of recurrent ACEI-induced angioedema, the Anesthesiology Service was consulted. Direct visualization of the airways showed edema of the epiglottis and vocal cords, requiring nasotracheal intubation. Two units of fresh frozen plasma (FFP) were administered. Complete resolution of angioedema took at least 72 hours even after the administration of FFP. As part of the ventilator-associated pneumonia prevention bundle, the patient continued with daily spontaneous breathing trials. On the fourth day, he was he was extubated after a cuff-leak test was positive and his rapid shallow breathing index was adequate.
The cuff-leak test is usually done to predict postextubation stridor. It consists of deflating the endotracheal tube cuff to verify if gas can pass around the tube. Absence of cuff leak is suggestive of airway edema, a risk factor for postextubation stridor and failure of extubation. For example, if the patient has an endotracheal tube that is too large in relation to the patient’s airway, the leak test can result in a false negative. In this case, fiber optic visualization of the airway can confirm the endotracheal tube occluding all the airway even with the cuff deflated and without evidence of swelling of the vocal cords. The rapid shallow breathing index is a ratio of respiratory rate over tidal volume in liters and is used to predict successful extubation. Values < 105 have a high sensitivity for successful extubation.
The patient remained under observation for 24 hours in the MICU and then was transferred to the general medicine ward. Unfortunately, 36 hours after, the patient had a new episode of angioedema requiring endotracheal intubation and placement on mechanical ventilation. This was his third episode of angioedema; he had a difficult airway classified as a Cormack-Lehane grade 3, requiring intubation with fiber-optic laryngoscope. In view of the recurrent events, a tracheostomy was done several days later. Figure 2 shows posttracheostomy X-ray with adequate position of the tracheostomy tube.
The patient was transferred to the Respiratory Care Unit and weaned off mechanical ventilation. He completed an intensive physical rehabilitation program and was discharged home. On discharge, he was followed by the Otorhinolaryngology Service and was decannulated about 5 months after. After tracheostomy decannulation, he developed asymptomatic stridor. A neck computer tomography scan revealed soft tissue thickening at the anterior and lateral aspects of the proximal tracheal likely representing granulation tissue/scarring. The findings were consistent with proximal tracheal stenosis sequelae of tracheostomy and intubation. In Figure 3, the upper portion of the curve represents the expiratory limb of the forced vital capacity and the lower portion represents inspiration. The flow-volume loop graph showed flattening of the inspiratory limb. There was a plateau in the inspiratory limb, suggestive of limitation of inspiratory flow as seen in variable extrathoracic lesions, such as glotticstricture, tumors, and vocal cord paralysis.7 The findings on the flow-volume loop were consistent with the subglottic stenosis identified by laryngoscopic examination. The patient was reluctant to undergo further interventions.
Discussion
The standard therapy for ACEI-inducedangioedema continues to be airway management and discontinuation of medication. However, life-threatening progression of symptoms have led to the use of off-label therapies, including FFP and bradykinin receptor antagonists, such as icatibant, which has been approved by the FDA for the treatment of hereditary angioedema. Icatibant is expensive and most hospitals do not have access to it. When considering the bradykinin pathway for therapy, FFP is commonly used. The cases described in the literature that have reported success with the use of FFP have used up to 2 units. There is no reported benefit of its use beyond 2 units. The initial randomized trials of icatibant for ACEI angioedema showed decreased time of resolution of angioedema.6 However, repeated trials showed conflicting results. At Veterans Affairs Caribbean Healthcare System, this medication was not available, and we decided to use FFP to improve the patient’s symptoms.
The administration of 2 units of FFP has been documented on case reports as a method to decrease the time of resolution of angioedema and the risk of recurrence. The mechanism of action thought to be involved includes the degradation of bradykinin by the enzyme ACE into inactive peptides and by supplying C1 inhibitor.8 No randomized clinical trial has investigated the use of FFP for the treatment of ACEI-induced angioedema. However, a retrospective cohort study report compared patients who presented with acute (nonhereditary) angioedema and airway compromise and received FFP with patients who were not treated with FFP.9 The study suggested a shorter ICU stay in the group treated with FFP, but the findings did not present statistical outcomes.
Nevertheless, our patient had recurrent ACEI-induced angioedema refractory to FFP. In addition to ACE or kininase II, FFP contains high-molecular weight-kininogen and kallikrein, the substrates that form bradykinin, which explained the mechanism of worsening angioedema.10 No randomized trials have investigated the use of FFP for the treatment of bradykinin-induced angioedema nor the appropriate dose.
Conclusion
In view of the emerging case reports of the effectiveness of FFP, this case of refractory angioedema raises concern for its true effectiveness and other possible factors involved in the mechanism of recurrence. Probably it would be unwise to conduct randomized studies in clinical situations such as the ones outlined. A collection of case series where FFP administration was done may be a more reasonable source of conclusions to be analyzed by a panel of experts.
1. Sánchez-Borges M, González-Aveledo LA. Angiotensin-converting enzyme inhibitors and angioedema. Allergy Asthma Immunol Res. 2010;2(3):195-198.
2. Kaplan AP. Angioedema. World Allergy Organ J. 2008;1(6):103-113.
3. Moellman JJ, Bernstein JA, Lindsell C, et al; American College of Allergy, Asthma & Immunology (ACAAI); Society for Academic Emergency Medicine (SAEM). A consensus parameter for the evaluation and management of angioedema in the emergency department. Acad Emerg Med. 2014;21(4):469-484.
4. LoVerde D, Files DC, Krishnaswamy G. Angioedema. Crit Care Med. 2017;45(4):725-735.
5. van den Elzen M, Go MFLC, Knulst AC, Blankestijn MA, van Os-Medendorp H, Otten HG. Efficacy of treatment of non-hereditary angioedema. Clinic Rev Allerg Immunol. 2018;54(3):412-431.
6. Bas M, Greve J, Stelter S, et al. A randomized trial of icatibant in ace-inhibitor–induced angioedema. N Engl J Med. 2015;372(5):418-425.
7. Diaz J, Casal J, Rodriguez W. Flow-volume loops: clinical correlation. PR Health Sci J. 2008;27(2):181-182.
8. Stewart M, McGlone R. Fresh frozen plasma in the treatment of ACE inhibitor-induced angioedema. BMJ Case Rep. 2012;2012:pii:bcr2012006849.
9. Saeb A, Hagglund KH, Cigolle CT. Using fresh frozen plasma for acute airway angioedema to prevent intubation in the emergency department: a retrospective cohort study. Emerg Med Int. 2016;2016:6091510.
10. Brown T, Gonzalez J, Monteleone C. Angiotensin-converting enzyme inhibitor-induced angioedema: a review of the literature. J Clin Hypertens (Greenwich). 2017;19(12):1377-1382.
Angioedema induced by angiotensin-converting enzyme inhibitors (ACEIs) is present in from 0.1% to 0.7% of treated patients and more often involves the head, neck, face, lips, tongue, and larynx.1 ACEI-induced angioedema results from inhibition of angiotensin-converting enzyme (ACE), which results in reduced degradation and resultant accumulation of bradykinin, a potent inflammatory mediator.2
The treatment of choice is discontinuing all ACEIs; however, the patient may be at increased risk of a subsequent angioedema attack for many weeks.3 Antihistamines (H1 and H2 receptor blockade), epinephrine, and glucocorticoids are effective in allergic/histaminergic angioedema but are usually ineffective for hereditary angioedema or ACEI angioedema and are not recommended for acute therapy.4 Kallikrein-bradykinin pathway targeted therapies are now approved by the Food and Drug Administration (FDA) for hereditary angioedema attacks and have been studied for ACEI-induced angioedema. Ecallantide and icatibant inhibit conversion of precursors to bradykinin. Multiple randomized trials of ecallantide have not shown any advantage over traditional therapies.5 On the other hand, icatibant has shown resolution of angioedema in several case reports and in a randomized trial.6 Icatibant for ACEI-induced angioedema continues to be off-label because the data are conflicting.
Case Presentation
A 67-year-old man presented with a medical history of arterial hypertension (diagnosed 17 years previously), hypercholesterolemia, type 2 diabetes mellitus, alcohol dependence, and obesity. His outpatient medications included simvastatin, aripiprazole, losartan/hydrochlorothiazide, and amlodipine. He was voluntarily admitted for inpatient detoxification. After evaluation by the internist, medication reconciliation was done, and the therapy was adjusted according to medication availability. He reported having no drug allergies, and the losartan was changed for lisinopril. About 24 hours after the first dose of lisinopril, the patient developed swelling of the lips. Antihistamine and IV steroids were administered, and the ACEI was discontinued. His baseline vital signs were temperature 98° F, heart rate 83 beats per minute, respiratory rate 19 breaths per minute, blood pressure 150/94, and oxygen saturation 98% by pulse oximeter.
During the night shift the patient’s symptoms worsened, developing difficulty swallowing and shortness of breath. He was transferred to the medicine intensive care unit (MICU), intubated, and placed on mechanical ventilation to protect his airway. Laryngoscopic examination was notable for edematous tongue, uvula, and larynx. Also, the patient had mild stridor. His laboratory test results showed normal levels of complement, tryptase, and C1 esterase. On the fourth day after admission to MICU (Figure 1), the patient extubated himself. At that time, he did not present stridor or respiratory distress and remained at the MICU for 24 hours for close monitoring.
Thirty-six hours after self-extubation the patient developed stridor and shortness of breath at the general medicine ward. In view of his clinical presentation of recurrent ACEI-induced angioedema, the Anesthesiology Service was consulted. Direct visualization of the airways showed edema of the epiglottis and vocal cords, requiring nasotracheal intubation. Two units of fresh frozen plasma (FFP) were administered. Complete resolution of angioedema took at least 72 hours even after the administration of FFP. As part of the ventilator-associated pneumonia prevention bundle, the patient continued with daily spontaneous breathing trials. On the fourth day, he was he was extubated after a cuff-leak test was positive and his rapid shallow breathing index was adequate.
The cuff-leak test is usually done to predict postextubation stridor. It consists of deflating the endotracheal tube cuff to verify if gas can pass around the tube. Absence of cuff leak is suggestive of airway edema, a risk factor for postextubation stridor and failure of extubation. For example, if the patient has an endotracheal tube that is too large in relation to the patient’s airway, the leak test can result in a false negative. In this case, fiber optic visualization of the airway can confirm the endotracheal tube occluding all the airway even with the cuff deflated and without evidence of swelling of the vocal cords. The rapid shallow breathing index is a ratio of respiratory rate over tidal volume in liters and is used to predict successful extubation. Values < 105 have a high sensitivity for successful extubation.
The patient remained under observation for 24 hours in the MICU and then was transferred to the general medicine ward. Unfortunately, 36 hours after, the patient had a new episode of angioedema requiring endotracheal intubation and placement on mechanical ventilation. This was his third episode of angioedema; he had a difficult airway classified as a Cormack-Lehane grade 3, requiring intubation with fiber-optic laryngoscope. In view of the recurrent events, a tracheostomy was done several days later. Figure 2 shows posttracheostomy X-ray with adequate position of the tracheostomy tube.
The patient was transferred to the Respiratory Care Unit and weaned off mechanical ventilation. He completed an intensive physical rehabilitation program and was discharged home. On discharge, he was followed by the Otorhinolaryngology Service and was decannulated about 5 months after. After tracheostomy decannulation, he developed asymptomatic stridor. A neck computer tomography scan revealed soft tissue thickening at the anterior and lateral aspects of the proximal tracheal likely representing granulation tissue/scarring. The findings were consistent with proximal tracheal stenosis sequelae of tracheostomy and intubation. In Figure 3, the upper portion of the curve represents the expiratory limb of the forced vital capacity and the lower portion represents inspiration. The flow-volume loop graph showed flattening of the inspiratory limb. There was a plateau in the inspiratory limb, suggestive of limitation of inspiratory flow as seen in variable extrathoracic lesions, such as glotticstricture, tumors, and vocal cord paralysis.7 The findings on the flow-volume loop were consistent with the subglottic stenosis identified by laryngoscopic examination. The patient was reluctant to undergo further interventions.
Discussion
The standard therapy for ACEI-inducedangioedema continues to be airway management and discontinuation of medication. However, life-threatening progression of symptoms have led to the use of off-label therapies, including FFP and bradykinin receptor antagonists, such as icatibant, which has been approved by the FDA for the treatment of hereditary angioedema. Icatibant is expensive and most hospitals do not have access to it. When considering the bradykinin pathway for therapy, FFP is commonly used. The cases described in the literature that have reported success with the use of FFP have used up to 2 units. There is no reported benefit of its use beyond 2 units. The initial randomized trials of icatibant for ACEI angioedema showed decreased time of resolution of angioedema.6 However, repeated trials showed conflicting results. At Veterans Affairs Caribbean Healthcare System, this medication was not available, and we decided to use FFP to improve the patient’s symptoms.
The administration of 2 units of FFP has been documented on case reports as a method to decrease the time of resolution of angioedema and the risk of recurrence. The mechanism of action thought to be involved includes the degradation of bradykinin by the enzyme ACE into inactive peptides and by supplying C1 inhibitor.8 No randomized clinical trial has investigated the use of FFP for the treatment of ACEI-induced angioedema. However, a retrospective cohort study report compared patients who presented with acute (nonhereditary) angioedema and airway compromise and received FFP with patients who were not treated with FFP.9 The study suggested a shorter ICU stay in the group treated with FFP, but the findings did not present statistical outcomes.
Nevertheless, our patient had recurrent ACEI-induced angioedema refractory to FFP. In addition to ACE or kininase II, FFP contains high-molecular weight-kininogen and kallikrein, the substrates that form bradykinin, which explained the mechanism of worsening angioedema.10 No randomized trials have investigated the use of FFP for the treatment of bradykinin-induced angioedema nor the appropriate dose.
Conclusion
In view of the emerging case reports of the effectiveness of FFP, this case of refractory angioedema raises concern for its true effectiveness and other possible factors involved in the mechanism of recurrence. Probably it would be unwise to conduct randomized studies in clinical situations such as the ones outlined. A collection of case series where FFP administration was done may be a more reasonable source of conclusions to be analyzed by a panel of experts.
Angioedema induced by angiotensin-converting enzyme inhibitors (ACEIs) is present in from 0.1% to 0.7% of treated patients and more often involves the head, neck, face, lips, tongue, and larynx.1 ACEI-induced angioedema results from inhibition of angiotensin-converting enzyme (ACE), which results in reduced degradation and resultant accumulation of bradykinin, a potent inflammatory mediator.2
The treatment of choice is discontinuing all ACEIs; however, the patient may be at increased risk of a subsequent angioedema attack for many weeks.3 Antihistamines (H1 and H2 receptor blockade), epinephrine, and glucocorticoids are effective in allergic/histaminergic angioedema but are usually ineffective for hereditary angioedema or ACEI angioedema and are not recommended for acute therapy.4 Kallikrein-bradykinin pathway targeted therapies are now approved by the Food and Drug Administration (FDA) for hereditary angioedema attacks and have been studied for ACEI-induced angioedema. Ecallantide and icatibant inhibit conversion of precursors to bradykinin. Multiple randomized trials of ecallantide have not shown any advantage over traditional therapies.5 On the other hand, icatibant has shown resolution of angioedema in several case reports and in a randomized trial.6 Icatibant for ACEI-induced angioedema continues to be off-label because the data are conflicting.
Case Presentation
A 67-year-old man presented with a medical history of arterial hypertension (diagnosed 17 years previously), hypercholesterolemia, type 2 diabetes mellitus, alcohol dependence, and obesity. His outpatient medications included simvastatin, aripiprazole, losartan/hydrochlorothiazide, and amlodipine. He was voluntarily admitted for inpatient detoxification. After evaluation by the internist, medication reconciliation was done, and the therapy was adjusted according to medication availability. He reported having no drug allergies, and the losartan was changed for lisinopril. About 24 hours after the first dose of lisinopril, the patient developed swelling of the lips. Antihistamine and IV steroids were administered, and the ACEI was discontinued. His baseline vital signs were temperature 98° F, heart rate 83 beats per minute, respiratory rate 19 breaths per minute, blood pressure 150/94, and oxygen saturation 98% by pulse oximeter.
During the night shift the patient’s symptoms worsened, developing difficulty swallowing and shortness of breath. He was transferred to the medicine intensive care unit (MICU), intubated, and placed on mechanical ventilation to protect his airway. Laryngoscopic examination was notable for edematous tongue, uvula, and larynx. Also, the patient had mild stridor. His laboratory test results showed normal levels of complement, tryptase, and C1 esterase. On the fourth day after admission to MICU (Figure 1), the patient extubated himself. At that time, he did not present stridor or respiratory distress and remained at the MICU for 24 hours for close monitoring.
Thirty-six hours after self-extubation the patient developed stridor and shortness of breath at the general medicine ward. In view of his clinical presentation of recurrent ACEI-induced angioedema, the Anesthesiology Service was consulted. Direct visualization of the airways showed edema of the epiglottis and vocal cords, requiring nasotracheal intubation. Two units of fresh frozen plasma (FFP) were administered. Complete resolution of angioedema took at least 72 hours even after the administration of FFP. As part of the ventilator-associated pneumonia prevention bundle, the patient continued with daily spontaneous breathing trials. On the fourth day, he was he was extubated after a cuff-leak test was positive and his rapid shallow breathing index was adequate.
The cuff-leak test is usually done to predict postextubation stridor. It consists of deflating the endotracheal tube cuff to verify if gas can pass around the tube. Absence of cuff leak is suggestive of airway edema, a risk factor for postextubation stridor and failure of extubation. For example, if the patient has an endotracheal tube that is too large in relation to the patient’s airway, the leak test can result in a false negative. In this case, fiber optic visualization of the airway can confirm the endotracheal tube occluding all the airway even with the cuff deflated and without evidence of swelling of the vocal cords. The rapid shallow breathing index is a ratio of respiratory rate over tidal volume in liters and is used to predict successful extubation. Values < 105 have a high sensitivity for successful extubation.
The patient remained under observation for 24 hours in the MICU and then was transferred to the general medicine ward. Unfortunately, 36 hours after, the patient had a new episode of angioedema requiring endotracheal intubation and placement on mechanical ventilation. This was his third episode of angioedema; he had a difficult airway classified as a Cormack-Lehane grade 3, requiring intubation with fiber-optic laryngoscope. In view of the recurrent events, a tracheostomy was done several days later. Figure 2 shows posttracheostomy X-ray with adequate position of the tracheostomy tube.
The patient was transferred to the Respiratory Care Unit and weaned off mechanical ventilation. He completed an intensive physical rehabilitation program and was discharged home. On discharge, he was followed by the Otorhinolaryngology Service and was decannulated about 5 months after. After tracheostomy decannulation, he developed asymptomatic stridor. A neck computer tomography scan revealed soft tissue thickening at the anterior and lateral aspects of the proximal tracheal likely representing granulation tissue/scarring. The findings were consistent with proximal tracheal stenosis sequelae of tracheostomy and intubation. In Figure 3, the upper portion of the curve represents the expiratory limb of the forced vital capacity and the lower portion represents inspiration. The flow-volume loop graph showed flattening of the inspiratory limb. There was a plateau in the inspiratory limb, suggestive of limitation of inspiratory flow as seen in variable extrathoracic lesions, such as glotticstricture, tumors, and vocal cord paralysis.7 The findings on the flow-volume loop were consistent with the subglottic stenosis identified by laryngoscopic examination. The patient was reluctant to undergo further interventions.
Discussion
The standard therapy for ACEI-inducedangioedema continues to be airway management and discontinuation of medication. However, life-threatening progression of symptoms have led to the use of off-label therapies, including FFP and bradykinin receptor antagonists, such as icatibant, which has been approved by the FDA for the treatment of hereditary angioedema. Icatibant is expensive and most hospitals do not have access to it. When considering the bradykinin pathway for therapy, FFP is commonly used. The cases described in the literature that have reported success with the use of FFP have used up to 2 units. There is no reported benefit of its use beyond 2 units. The initial randomized trials of icatibant for ACEI angioedema showed decreased time of resolution of angioedema.6 However, repeated trials showed conflicting results. At Veterans Affairs Caribbean Healthcare System, this medication was not available, and we decided to use FFP to improve the patient’s symptoms.
The administration of 2 units of FFP has been documented on case reports as a method to decrease the time of resolution of angioedema and the risk of recurrence. The mechanism of action thought to be involved includes the degradation of bradykinin by the enzyme ACE into inactive peptides and by supplying C1 inhibitor.8 No randomized clinical trial has investigated the use of FFP for the treatment of ACEI-induced angioedema. However, a retrospective cohort study report compared patients who presented with acute (nonhereditary) angioedema and airway compromise and received FFP with patients who were not treated with FFP.9 The study suggested a shorter ICU stay in the group treated with FFP, but the findings did not present statistical outcomes.
Nevertheless, our patient had recurrent ACEI-induced angioedema refractory to FFP. In addition to ACE or kininase II, FFP contains high-molecular weight-kininogen and kallikrein, the substrates that form bradykinin, which explained the mechanism of worsening angioedema.10 No randomized trials have investigated the use of FFP for the treatment of bradykinin-induced angioedema nor the appropriate dose.
Conclusion
In view of the emerging case reports of the effectiveness of FFP, this case of refractory angioedema raises concern for its true effectiveness and other possible factors involved in the mechanism of recurrence. Probably it would be unwise to conduct randomized studies in clinical situations such as the ones outlined. A collection of case series where FFP administration was done may be a more reasonable source of conclusions to be analyzed by a panel of experts.
1. Sánchez-Borges M, González-Aveledo LA. Angiotensin-converting enzyme inhibitors and angioedema. Allergy Asthma Immunol Res. 2010;2(3):195-198.
2. Kaplan AP. Angioedema. World Allergy Organ J. 2008;1(6):103-113.
3. Moellman JJ, Bernstein JA, Lindsell C, et al; American College of Allergy, Asthma & Immunology (ACAAI); Society for Academic Emergency Medicine (SAEM). A consensus parameter for the evaluation and management of angioedema in the emergency department. Acad Emerg Med. 2014;21(4):469-484.
4. LoVerde D, Files DC, Krishnaswamy G. Angioedema. Crit Care Med. 2017;45(4):725-735.
5. van den Elzen M, Go MFLC, Knulst AC, Blankestijn MA, van Os-Medendorp H, Otten HG. Efficacy of treatment of non-hereditary angioedema. Clinic Rev Allerg Immunol. 2018;54(3):412-431.
6. Bas M, Greve J, Stelter S, et al. A randomized trial of icatibant in ace-inhibitor–induced angioedema. N Engl J Med. 2015;372(5):418-425.
7. Diaz J, Casal J, Rodriguez W. Flow-volume loops: clinical correlation. PR Health Sci J. 2008;27(2):181-182.
8. Stewart M, McGlone R. Fresh frozen plasma in the treatment of ACE inhibitor-induced angioedema. BMJ Case Rep. 2012;2012:pii:bcr2012006849.
9. Saeb A, Hagglund KH, Cigolle CT. Using fresh frozen plasma for acute airway angioedema to prevent intubation in the emergency department: a retrospective cohort study. Emerg Med Int. 2016;2016:6091510.
10. Brown T, Gonzalez J, Monteleone C. Angiotensin-converting enzyme inhibitor-induced angioedema: a review of the literature. J Clin Hypertens (Greenwich). 2017;19(12):1377-1382.
1. Sánchez-Borges M, González-Aveledo LA. Angiotensin-converting enzyme inhibitors and angioedema. Allergy Asthma Immunol Res. 2010;2(3):195-198.
2. Kaplan AP. Angioedema. World Allergy Organ J. 2008;1(6):103-113.
3. Moellman JJ, Bernstein JA, Lindsell C, et al; American College of Allergy, Asthma & Immunology (ACAAI); Society for Academic Emergency Medicine (SAEM). A consensus parameter for the evaluation and management of angioedema in the emergency department. Acad Emerg Med. 2014;21(4):469-484.
4. LoVerde D, Files DC, Krishnaswamy G. Angioedema. Crit Care Med. 2017;45(4):725-735.
5. van den Elzen M, Go MFLC, Knulst AC, Blankestijn MA, van Os-Medendorp H, Otten HG. Efficacy of treatment of non-hereditary angioedema. Clinic Rev Allerg Immunol. 2018;54(3):412-431.
6. Bas M, Greve J, Stelter S, et al. A randomized trial of icatibant in ace-inhibitor–induced angioedema. N Engl J Med. 2015;372(5):418-425.
7. Diaz J, Casal J, Rodriguez W. Flow-volume loops: clinical correlation. PR Health Sci J. 2008;27(2):181-182.
8. Stewart M, McGlone R. Fresh frozen plasma in the treatment of ACE inhibitor-induced angioedema. BMJ Case Rep. 2012;2012:pii:bcr2012006849.
9. Saeb A, Hagglund KH, Cigolle CT. Using fresh frozen plasma for acute airway angioedema to prevent intubation in the emergency department: a retrospective cohort study. Emerg Med Int. 2016;2016:6091510.
10. Brown T, Gonzalez J, Monteleone C. Angiotensin-converting enzyme inhibitor-induced angioedema: a review of the literature. J Clin Hypertens (Greenwich). 2017;19(12):1377-1382.
Application of Hand Therapy Extensor Tendon Protocol to Toe Extensor Tendon Rehabilitation
Plastic and orthopedic surgeons worked closely with therapists in military hospitals to rehabilitate soldiers afflicted with upper extremity trauma during World War II. Together, they developed treatment protocols. In 1975, the American Society for Hand Therapists (ASHT) was created during the American Society for Surgery of the Hand meeting. The ASHT application process required case studies, patient logs, and clinical hours, so membership was equivalent to competency. In May 1991, the first hand certification examination took place and designated the first group of certified hand therapists (CHT).1
In the US Department of Veterans Affairs collaboration takes place between different services and communication is facilitated using the electronic heath record. The case presented here is an example of several services (emergency medicine, plastic/hand surgery, and occupational therapy) working together to develop a treatment plan for a condition that often goes undiagnosed or untreated. This article describes an innovative application of hand extensor tendon therapy clinical decision making to rehabilitate foot extensor tendons when the plastic surgery service was called on to work outside its usual comfort zone of the hand and upper extremity. The hand therapist applied hand extensor tendon rehabilitation principles to recover toe extensor lacerations.
Certified hand therapists (CHTs) are key to a successful hand surgery practice. The Plastic Surgery Service at the Malcom Randall VA Medical Center in Gainesville, Florida, relies heavily on the CHTs to optimize patient outcomes. The hand surgery clinic and hand therapy clinics are in the same hospital building, allowing for easy face-to-face communication. Hand therapy students are able to observe cases in the operating room. Immediately after surgery, follow-up consults are scheduled to coordinate postoperative care between the services.
Case Presentation
The next day, the patient was examined in the plastic surgery clinic and found to have a completely lacerated extensor digitorum brevis to the second toe and a completely lacerated extensor digitorum longus to the third toe. These were located proximal to the metatarsal phalangeal joints. Surgery was scheduled for the following week.
In surgery, the tendons were sharply debrided and repaired using a 3.0 Ethibond suture placed in a modified Kessler technique followed by a horizontal mattress for a total of a 4-core repair. This was reinforced with a No. 6 Prolene to the paratendon. The surgery was performed under IV sedation and an ankle block, using 17 minutes of tourniquet time.
On postoperative day 1, the patient was seen in plastic surgery and occupational therapy clinic. The hand therapist modified the hand extensor tendon repair protocol since there was no known protocol for repairs of the foot and toe extensor tendon. The patient was placed in an ankle foot orthosis with a toe extension device created by heating and molding a low-temperature thermoplastic sheet (Figure 2). The toes were boosted into slight hyper extension. This was done to reduce tension across the extensor tendon repair site. All of the toes were held in about 20°of extension, as the extensor digitorum longus (EDL) has a common origin, to aide in adherence of wearing and for comfort. No standing or weight bearing was permitted for 3 weeks.
A wheelchair was issued in lieu of crutches to inhibit the work of toe extension with gait swing-through. Otherwise, the patient would generate tension on the extensor tendon in order for the toes to clear the ground. It was postulated that it would be difficult to turn off the toe extensors while using crutches. Maximal laxity was desired because edema and early scar formation could increase tension on the repair, resulting in rupture if the patient tried to fire the muscle belly even while in passive extension.
The patient kept his appointments and progressed steadily. He started passive toe extension and relaxation once per day for 30 repetitions at 1 week to aide in tendon glide. He started place and hold techniques in toe extension at 3 weeks. This progressed to active extension 50% effort plus active flexion at 4 weeks after surgery, then 75% extension effort plus toe towel crunches at 5 weeks. Toe crunches are toe flexion exercises with a washcloth on the floor with active bending of the toes with light resistance similar to picking up a marble with the toes. He was found to have a third toe extensor lag at that time that was correctible. The patient was actively able to flex and extend the toe independently. The early extension lag was felt to be secondary to edema and scar formation, which, over time are anticipated to resolve and contract and effectively shorten the tendon. Tendon gliding, and scar massage were reviewed. The patient’s last therapy session occurred 7 weeks after surgery, and he was cleared for full activity at 12 weeks. There was no further follow-up as he was planning on back surgery 2 weeks later.
Discussion
The North Florida/South Georgia Veterans Health System is fortunate to have 4 CHTs on staff. CHTs take a 200 question 4 hour certifying exam after being licensed for a minimum of 3 years as a physical or occupational therapist and completing 4,000 hours of direct upper extremity patient experience. Pass rates from 2008 to 2018 ranged from 52% to 68%.3 These clinicians are key to the success of our hand surgery service, utilizing their education and skills on our elective and trauma cases. The hand therapy service applied their knowledge of hand extensor rehabilitation protocols to rehabilitate the patient’s toe extensor in the absence of clear guidelines.
Hand extensor tendon rehabilitation protocols are based on the location of the repair on the hand or forearm. Nine extensor zones are named, distal to proximal, from the distal interphalangeal joints to the proximal forearm (Figure 3). In his review of extensor hallucis longus (EHL) repairs, Al-Qattan described 6 foot-extensor tendon zones, distal to proximal, from the first toe at the insertion of the big toe extensor to the distal leg proximal to the extensor retinaculum (Figure 4).4 Zone 3 is over the metatarsophalangeal joint; zone 5 is under the extensor retinaculum. The extensor tendon repairs described in this report were in dorsal foot zone 4 (proximal to the metatarsophalangeal joint and over the metatarsals), which would be most comparable to hand extensor zone 6 (proximal to the metacarpal phalangeal joint and over the metacarpals).

The EDL originates on the lateral condyle of the tibia and anterior surface of the fibula and the interosseous membrane, passes under the extensor retinaculum, and divides into 4 separate tendons. The 4 tendons split into 3 slips; the central one inserts on the middle phalanx, and the lateral ones insert onto the distal phalanx of the 4 lateral toes, which allows for toe extension.5 The EDL common origin for the muscle belly that serves 4 tendon slips has clinical significance because rehabilitation for one digit will affect the others. Knowledge of the anatomical structures guides the clinical decision making whether it is in the hand or foot. The EDL works synergistically with the extensor digitorum brevis (EDBr) to dorsiflex (extend) the toe phalanges. The EDB originates at the supralateral surface of the calcaneus, lateral talocalcaneal ligament and cruciate crural ligament and inserts at the lateral side of the EDL of second, third, and fourth toes at the level of the metatarsophalangeal joint.6
Repair of lacerated extensor tendons in the foot is the recommended treatment. Chronic extensor lag of the phalanges can result in a claw toe deformity, difficulty controlling the toes when putting on shoes or socks, and catching of the toe on fabric or insoles.7 The extensor tendons are close to the deep and superficial peroneal nerves and to the dorsalis pedis artery, none of which were involved in this case report.
There are case reports and series of EHL repairs that all involves at least 3 weeks of immobilization.4,8,9 The EHL dorsiflexes the big toe. Al-Qattan’s series involved placing K wires across the interphalangeal joint of the big toe and across the metatarsophalangeal joint, which were removed at 6 weeks, in addition to 3.0 polypropylene tendon mattress sutures. All patients in this series healed without tendon rupture or infection. Our PubMed search did not reveal any specific protocol for the EDL or EDB tendons, which are anatomically most comparable to the extensor digitorum communis (EDC) tendons in the hand. The EDC originates at the lateral epicondyle of the humerus, also divides into 4 separate tendons and is responsible for extending the 4 ulnar sided fingers at the metacarpophalangeal joint.10
Tendon repair protocols are a balance between preventing tendon rupture by too aggressive therapy and with preventing tendon adhesions from prolonged immobilization. Orthotic fabrication plays a key early role with blocking possible forces creating unacceptable strain or tension across the surgical repair site. Traditionally, extensor tendon repairs in the hand were immobilized for at least 3 weeks to prevent rupture. This is still the preferred protocol for the patient unwilling or unable to follow instructions. The downside to this method is extension lags, extrinsic tightness, and adhesions that prevent flexion, which can require prolonged therapy or tenolysis surgery to correct.11-13
Early passive motion (EPM) was promoted in the 1980s when studies found better functional outcomes and fewer adhesions. This involved either a dynamic extension splint that relied on elastic bands (Louisville protocol) to keep tension off the repair or the Duran protocol that relied on a static splint and the patient doing the passive exercises with his other uninjured hand. Critics of the EPM protocol point to the costs of the splints and demands of postoperative hand therapy.11
Early active motion (EAM) is the most recent development in hand tendon rehabilitation and starts within days of surgery. Studies have found an earlier regain of total active motion in patients who are mobilized earlier.12 EAM protocols can be divided into controlled active motion (CAM) and relative motion extension splinting (RMES). CAM splints are forearm based and cross more joints. Relative motion splinting is the least restrictive, which makes it less likely that the patient will remove it. Patient friendly splints are ideal because tendon ruptures are often secondary to nonadherence.13 The yoke splint is an example of a RMES, which places the repaired digit in slightly greater extension at the metacarpal phalangeal joint than the other digits (Figure 5), allowing use of the uninjured digits.
The toe extensors do not have the juncturae tendinum connecting the individual EDL tendons to each other, as found between the EDC tendons in the hand. These connective bands can mask a single extensor tendon laceration in the hand when the patient is still able to extend the digit to neutral in the event of a more proximal dorsal hand laceration. A case can be made for closing the skin only in lesser toe extensor injuries in poor surgical candidates because the extensor lag would not be appreciated functionally when wearing shoes. There would be less functional impact when letting a toe extensor go untreated compared with that of a hand extensor. Routine activities such as typing or getting the fingers into a tight pocket could be challenging if hand extensors were untreated. The rehabilitation for toe extensors is more inconvenient when a patient is nonweight bearing, compared with wearing a hand yoke splint.
Conclusion
The case described used an early passive motion protocol without the dynamic splint to rehabilitate the third toe EDL and second toe EDB. This was felt to be the most patient and therapist friendly option, given the previously unchartered territory. The foot orthosis was in stock at the adjacent physical therapy clinic, and the toe booster was created in the hand therapy clinic with readily available supplies. Ideally, one would like to return structures to their anatomic site and control the healing process in the event of a traumatic injury to prevent nonanatomic healing between structures and painful scar adhesions in an area with little subcutaneous tissue. This patient’s tendon repair was still intact at 7 weeks and on his way to recovery, demonstrating good scar management techniques. The risks and benefits to lesser toe tendon repair and recovery would have to be weighed on an individual basis.
Acknowledgments
This project is the result of work supported with resources and use of facilities at the Malcom Randall VA Medical Center in Gainesville, Florida.
1. Hand Therapy Certification Commission. History of HTCC. https://www.htcc.org/consumer-information/about-htcc/history-of-htcc. Accessed November 8, 2019.
2. Coady-Fariborzian L, McGreane A. Comparison of hand emergency triage before and after specialty templates (2007 vs 2012). Hand (N Y). 2015;10(2):215-220.
3. Hand Therapy Certification Commission. Passing rates for the CHT exam. https://www.htcc.org/certify/exam-results/passing-rates. Accessed November 8, 2019.
4. Al-Qattan MM. Surgical treatment and results in 17 cases of open lacerations of the extensor hallucis longus tendon. J Plast Reconstr Aesthet Surg. 2007;60(4):360-367.
5. Wheeless CR. Wheeless’ textbook of orthopaedics: extensor digitorum longus. http://www.wheelessonline.com/ortho/extensor_digitorum_longus. Updated December 8, 2011. Accessed November 8, 2019.
6. Wheeless CR. Wheeless’ textbook of orthopaedics: extensor digitorum brevis. http://www.wheelessonline.com/ortho/extensor_digitorum_brevis. Updated March 4, 2018. Accessed November 8, 2019.
7. Coughlin M, Schon L. Disorders of tendons. https://musculoskeletalkey.com/disorders-of-tendons-2/#s0035. Published August 27, 2016. Accessed November 8, 2019.
8. Bronner S, Ojofeitimi S, Rose D. Repair and rehabilitation of extensor hallucis longus and brevis tendon lacerations in a professional dancer. J Orthop Sports Phys Ther. 2008;38(6):362-370.
9. Wong JC, Daniel JN, Raikin SM. Repair of acute extensor hallucis longus tendon injuries: a retrospective review. Foot Ankle Spec. 2014;7(1):45-51.
10. Wheeless CR. Wheeless’ textbook of orthopaedics: extensor digitorum communis. http://www.wheelessonline.com/ortho/extensor_digitorum_communis. Updated March 4, 2018. Accessed November 8, 2019.
11. Hall B, Lee H, Page R, Rosenwax L, Lee AH. Comparing three postoperative treatment protocols for extensor tendon repair in zones V and VI of the hand. Am J Occup Ther. 2010;64(5):682-688.
12. Wong AL, Wilson M, Girnary S, Nojoomi M, Acharya S, Paul SM. The optimal orthosis and motion protocol for extensor tendon injury in zones IV-VIII: a systematic review. J Hand Ther. 2017;30(4):447-456.
13. Collocott SJ, Kelly E, Ellis RF. Optimal early active mobilisation protocol after extensor tendon repairs in zones V and VI: a systematic review of literature. Hand Ther. 2018;23(1):3-18.
Plastic and orthopedic surgeons worked closely with therapists in military hospitals to rehabilitate soldiers afflicted with upper extremity trauma during World War II. Together, they developed treatment protocols. In 1975, the American Society for Hand Therapists (ASHT) was created during the American Society for Surgery of the Hand meeting. The ASHT application process required case studies, patient logs, and clinical hours, so membership was equivalent to competency. In May 1991, the first hand certification examination took place and designated the first group of certified hand therapists (CHT).1
In the US Department of Veterans Affairs collaboration takes place between different services and communication is facilitated using the electronic heath record. The case presented here is an example of several services (emergency medicine, plastic/hand surgery, and occupational therapy) working together to develop a treatment plan for a condition that often goes undiagnosed or untreated. This article describes an innovative application of hand extensor tendon therapy clinical decision making to rehabilitate foot extensor tendons when the plastic surgery service was called on to work outside its usual comfort zone of the hand and upper extremity. The hand therapist applied hand extensor tendon rehabilitation principles to recover toe extensor lacerations.
Certified hand therapists (CHTs) are key to a successful hand surgery practice. The Plastic Surgery Service at the Malcom Randall VA Medical Center in Gainesville, Florida, relies heavily on the CHTs to optimize patient outcomes. The hand surgery clinic and hand therapy clinics are in the same hospital building, allowing for easy face-to-face communication. Hand therapy students are able to observe cases in the operating room. Immediately after surgery, follow-up consults are scheduled to coordinate postoperative care between the services.
Case Presentation
The next day, the patient was examined in the plastic surgery clinic and found to have a completely lacerated extensor digitorum brevis to the second toe and a completely lacerated extensor digitorum longus to the third toe. These were located proximal to the metatarsal phalangeal joints. Surgery was scheduled for the following week.
In surgery, the tendons were sharply debrided and repaired using a 3.0 Ethibond suture placed in a modified Kessler technique followed by a horizontal mattress for a total of a 4-core repair. This was reinforced with a No. 6 Prolene to the paratendon. The surgery was performed under IV sedation and an ankle block, using 17 minutes of tourniquet time.
On postoperative day 1, the patient was seen in plastic surgery and occupational therapy clinic. The hand therapist modified the hand extensor tendon repair protocol since there was no known protocol for repairs of the foot and toe extensor tendon. The patient was placed in an ankle foot orthosis with a toe extension device created by heating and molding a low-temperature thermoplastic sheet (Figure 2). The toes were boosted into slight hyper extension. This was done to reduce tension across the extensor tendon repair site. All of the toes were held in about 20°of extension, as the extensor digitorum longus (EDL) has a common origin, to aide in adherence of wearing and for comfort. No standing or weight bearing was permitted for 3 weeks.
A wheelchair was issued in lieu of crutches to inhibit the work of toe extension with gait swing-through. Otherwise, the patient would generate tension on the extensor tendon in order for the toes to clear the ground. It was postulated that it would be difficult to turn off the toe extensors while using crutches. Maximal laxity was desired because edema and early scar formation could increase tension on the repair, resulting in rupture if the patient tried to fire the muscle belly even while in passive extension.
The patient kept his appointments and progressed steadily. He started passive toe extension and relaxation once per day for 30 repetitions at 1 week to aide in tendon glide. He started place and hold techniques in toe extension at 3 weeks. This progressed to active extension 50% effort plus active flexion at 4 weeks after surgery, then 75% extension effort plus toe towel crunches at 5 weeks. Toe crunches are toe flexion exercises with a washcloth on the floor with active bending of the toes with light resistance similar to picking up a marble with the toes. He was found to have a third toe extensor lag at that time that was correctible. The patient was actively able to flex and extend the toe independently. The early extension lag was felt to be secondary to edema and scar formation, which, over time are anticipated to resolve and contract and effectively shorten the tendon. Tendon gliding, and scar massage were reviewed. The patient’s last therapy session occurred 7 weeks after surgery, and he was cleared for full activity at 12 weeks. There was no further follow-up as he was planning on back surgery 2 weeks later.
Discussion
The North Florida/South Georgia Veterans Health System is fortunate to have 4 CHTs on staff. CHTs take a 200 question 4 hour certifying exam after being licensed for a minimum of 3 years as a physical or occupational therapist and completing 4,000 hours of direct upper extremity patient experience. Pass rates from 2008 to 2018 ranged from 52% to 68%.3 These clinicians are key to the success of our hand surgery service, utilizing their education and skills on our elective and trauma cases. The hand therapy service applied their knowledge of hand extensor rehabilitation protocols to rehabilitate the patient’s toe extensor in the absence of clear guidelines.
Hand extensor tendon rehabilitation protocols are based on the location of the repair on the hand or forearm. Nine extensor zones are named, distal to proximal, from the distal interphalangeal joints to the proximal forearm (Figure 3). In his review of extensor hallucis longus (EHL) repairs, Al-Qattan described 6 foot-extensor tendon zones, distal to proximal, from the first toe at the insertion of the big toe extensor to the distal leg proximal to the extensor retinaculum (Figure 4).4 Zone 3 is over the metatarsophalangeal joint; zone 5 is under the extensor retinaculum. The extensor tendon repairs described in this report were in dorsal foot zone 4 (proximal to the metatarsophalangeal joint and over the metatarsals), which would be most comparable to hand extensor zone 6 (proximal to the metacarpal phalangeal joint and over the metacarpals).
The EDL originates on the lateral condyle of the tibia and anterior surface of the fibula and the interosseous membrane, passes under the extensor retinaculum, and divides into 4 separate tendons. The 4 tendons split into 3 slips; the central one inserts on the middle phalanx, and the lateral ones insert onto the distal phalanx of the 4 lateral toes, which allows for toe extension.5 The EDL common origin for the muscle belly that serves 4 tendon slips has clinical significance because rehabilitation for one digit will affect the others. Knowledge of the anatomical structures guides the clinical decision making whether it is in the hand or foot. The EDL works synergistically with the extensor digitorum brevis (EDBr) to dorsiflex (extend) the toe phalanges. The EDB originates at the supralateral surface of the calcaneus, lateral talocalcaneal ligament and cruciate crural ligament and inserts at the lateral side of the EDL of second, third, and fourth toes at the level of the metatarsophalangeal joint.6
Repair of lacerated extensor tendons in the foot is the recommended treatment. Chronic extensor lag of the phalanges can result in a claw toe deformity, difficulty controlling the toes when putting on shoes or socks, and catching of the toe on fabric or insoles.7 The extensor tendons are close to the deep and superficial peroneal nerves and to the dorsalis pedis artery, none of which were involved in this case report.
There are case reports and series of EHL repairs that all involves at least 3 weeks of immobilization.4,8,9 The EHL dorsiflexes the big toe. Al-Qattan’s series involved placing K wires across the interphalangeal joint of the big toe and across the metatarsophalangeal joint, which were removed at 6 weeks, in addition to 3.0 polypropylene tendon mattress sutures. All patients in this series healed without tendon rupture or infection. Our PubMed search did not reveal any specific protocol for the EDL or EDB tendons, which are anatomically most comparable to the extensor digitorum communis (EDC) tendons in the hand. The EDC originates at the lateral epicondyle of the humerus, also divides into 4 separate tendons and is responsible for extending the 4 ulnar sided fingers at the metacarpophalangeal joint.10
Tendon repair protocols are a balance between preventing tendon rupture by too aggressive therapy and with preventing tendon adhesions from prolonged immobilization. Orthotic fabrication plays a key early role with blocking possible forces creating unacceptable strain or tension across the surgical repair site. Traditionally, extensor tendon repairs in the hand were immobilized for at least 3 weeks to prevent rupture. This is still the preferred protocol for the patient unwilling or unable to follow instructions. The downside to this method is extension lags, extrinsic tightness, and adhesions that prevent flexion, which can require prolonged therapy or tenolysis surgery to correct.11-13
Early passive motion (EPM) was promoted in the 1980s when studies found better functional outcomes and fewer adhesions. This involved either a dynamic extension splint that relied on elastic bands (Louisville protocol) to keep tension off the repair or the Duran protocol that relied on a static splint and the patient doing the passive exercises with his other uninjured hand. Critics of the EPM protocol point to the costs of the splints and demands of postoperative hand therapy.11
Early active motion (EAM) is the most recent development in hand tendon rehabilitation and starts within days of surgery. Studies have found an earlier regain of total active motion in patients who are mobilized earlier.12 EAM protocols can be divided into controlled active motion (CAM) and relative motion extension splinting (RMES). CAM splints are forearm based and cross more joints. Relative motion splinting is the least restrictive, which makes it less likely that the patient will remove it. Patient friendly splints are ideal because tendon ruptures are often secondary to nonadherence.13 The yoke splint is an example of a RMES, which places the repaired digit in slightly greater extension at the metacarpal phalangeal joint than the other digits (Figure 5), allowing use of the uninjured digits.
The toe extensors do not have the juncturae tendinum connecting the individual EDL tendons to each other, as found between the EDC tendons in the hand. These connective bands can mask a single extensor tendon laceration in the hand when the patient is still able to extend the digit to neutral in the event of a more proximal dorsal hand laceration. A case can be made for closing the skin only in lesser toe extensor injuries in poor surgical candidates because the extensor lag would not be appreciated functionally when wearing shoes. There would be less functional impact when letting a toe extensor go untreated compared with that of a hand extensor. Routine activities such as typing or getting the fingers into a tight pocket could be challenging if hand extensors were untreated. The rehabilitation for toe extensors is more inconvenient when a patient is nonweight bearing, compared with wearing a hand yoke splint.
Conclusion
The case described used an early passive motion protocol without the dynamic splint to rehabilitate the third toe EDL and second toe EDB. This was felt to be the most patient and therapist friendly option, given the previously unchartered territory. The foot orthosis was in stock at the adjacent physical therapy clinic, and the toe booster was created in the hand therapy clinic with readily available supplies. Ideally, one would like to return structures to their anatomic site and control the healing process in the event of a traumatic injury to prevent nonanatomic healing between structures and painful scar adhesions in an area with little subcutaneous tissue. This patient’s tendon repair was still intact at 7 weeks and on his way to recovery, demonstrating good scar management techniques. The risks and benefits to lesser toe tendon repair and recovery would have to be weighed on an individual basis.
Acknowledgments
This project is the result of work supported with resources and use of facilities at the Malcom Randall VA Medical Center in Gainesville, Florida.
Plastic and orthopedic surgeons worked closely with therapists in military hospitals to rehabilitate soldiers afflicted with upper extremity trauma during World War II. Together, they developed treatment protocols. In 1975, the American Society for Hand Therapists (ASHT) was created during the American Society for Surgery of the Hand meeting. The ASHT application process required case studies, patient logs, and clinical hours, so membership was equivalent to competency. In May 1991, the first hand certification examination took place and designated the first group of certified hand therapists (CHT).1
In the US Department of Veterans Affairs collaboration takes place between different services and communication is facilitated using the electronic heath record. The case presented here is an example of several services (emergency medicine, plastic/hand surgery, and occupational therapy) working together to develop a treatment plan for a condition that often goes undiagnosed or untreated. This article describes an innovative application of hand extensor tendon therapy clinical decision making to rehabilitate foot extensor tendons when the plastic surgery service was called on to work outside its usual comfort zone of the hand and upper extremity. The hand therapist applied hand extensor tendon rehabilitation principles to recover toe extensor lacerations.
Certified hand therapists (CHTs) are key to a successful hand surgery practice. The Plastic Surgery Service at the Malcom Randall VA Medical Center in Gainesville, Florida, relies heavily on the CHTs to optimize patient outcomes. The hand surgery clinic and hand therapy clinics are in the same hospital building, allowing for easy face-to-face communication. Hand therapy students are able to observe cases in the operating room. Immediately after surgery, follow-up consults are scheduled to coordinate postoperative care between the services.
Case Presentation
The next day, the patient was examined in the plastic surgery clinic and found to have a completely lacerated extensor digitorum brevis to the second toe and a completely lacerated extensor digitorum longus to the third toe. These were located proximal to the metatarsal phalangeal joints. Surgery was scheduled for the following week.
In surgery, the tendons were sharply debrided and repaired using a 3.0 Ethibond suture placed in a modified Kessler technique followed by a horizontal mattress for a total of a 4-core repair. This was reinforced with a No. 6 Prolene to the paratendon. The surgery was performed under IV sedation and an ankle block, using 17 minutes of tourniquet time.
On postoperative day 1, the patient was seen in plastic surgery and occupational therapy clinic. The hand therapist modified the hand extensor tendon repair protocol since there was no known protocol for repairs of the foot and toe extensor tendon. The patient was placed in an ankle foot orthosis with a toe extension device created by heating and molding a low-temperature thermoplastic sheet (Figure 2). The toes were boosted into slight hyper extension. This was done to reduce tension across the extensor tendon repair site. All of the toes were held in about 20°of extension, as the extensor digitorum longus (EDL) has a common origin, to aide in adherence of wearing and for comfort. No standing or weight bearing was permitted for 3 weeks.
A wheelchair was issued in lieu of crutches to inhibit the work of toe extension with gait swing-through. Otherwise, the patient would generate tension on the extensor tendon in order for the toes to clear the ground. It was postulated that it would be difficult to turn off the toe extensors while using crutches. Maximal laxity was desired because edema and early scar formation could increase tension on the repair, resulting in rupture if the patient tried to fire the muscle belly even while in passive extension.
The patient kept his appointments and progressed steadily. He started passive toe extension and relaxation once per day for 30 repetitions at 1 week to aide in tendon glide. He started place and hold techniques in toe extension at 3 weeks. This progressed to active extension 50% effort plus active flexion at 4 weeks after surgery, then 75% extension effort plus toe towel crunches at 5 weeks. Toe crunches are toe flexion exercises with a washcloth on the floor with active bending of the toes with light resistance similar to picking up a marble with the toes. He was found to have a third toe extensor lag at that time that was correctible. The patient was actively able to flex and extend the toe independently. The early extension lag was felt to be secondary to edema and scar formation, which, over time are anticipated to resolve and contract and effectively shorten the tendon. Tendon gliding, and scar massage were reviewed. The patient’s last therapy session occurred 7 weeks after surgery, and he was cleared for full activity at 12 weeks. There was no further follow-up as he was planning on back surgery 2 weeks later.
Discussion
The North Florida/South Georgia Veterans Health System is fortunate to have 4 CHTs on staff. CHTs take a 200 question 4 hour certifying exam after being licensed for a minimum of 3 years as a physical or occupational therapist and completing 4,000 hours of direct upper extremity patient experience. Pass rates from 2008 to 2018 ranged from 52% to 68%.3 These clinicians are key to the success of our hand surgery service, utilizing their education and skills on our elective and trauma cases. The hand therapy service applied their knowledge of hand extensor rehabilitation protocols to rehabilitate the patient’s toe extensor in the absence of clear guidelines.
Hand extensor tendon rehabilitation protocols are based on the location of the repair on the hand or forearm. Nine extensor zones are named, distal to proximal, from the distal interphalangeal joints to the proximal forearm (Figure 3). In his review of extensor hallucis longus (EHL) repairs, Al-Qattan described 6 foot-extensor tendon zones, distal to proximal, from the first toe at the insertion of the big toe extensor to the distal leg proximal to the extensor retinaculum (Figure 4).4 Zone 3 is over the metatarsophalangeal joint; zone 5 is under the extensor retinaculum. The extensor tendon repairs described in this report were in dorsal foot zone 4 (proximal to the metatarsophalangeal joint and over the metatarsals), which would be most comparable to hand extensor zone 6 (proximal to the metacarpal phalangeal joint and over the metacarpals).
The EDL originates on the lateral condyle of the tibia and anterior surface of the fibula and the interosseous membrane, passes under the extensor retinaculum, and divides into 4 separate tendons. The 4 tendons split into 3 slips; the central one inserts on the middle phalanx, and the lateral ones insert onto the distal phalanx of the 4 lateral toes, which allows for toe extension.5 The EDL common origin for the muscle belly that serves 4 tendon slips has clinical significance because rehabilitation for one digit will affect the others. Knowledge of the anatomical structures guides the clinical decision making whether it is in the hand or foot. The EDL works synergistically with the extensor digitorum brevis (EDBr) to dorsiflex (extend) the toe phalanges. The EDB originates at the supralateral surface of the calcaneus, lateral talocalcaneal ligament and cruciate crural ligament and inserts at the lateral side of the EDL of second, third, and fourth toes at the level of the metatarsophalangeal joint.6
Repair of lacerated extensor tendons in the foot is the recommended treatment. Chronic extensor lag of the phalanges can result in a claw toe deformity, difficulty controlling the toes when putting on shoes or socks, and catching of the toe on fabric or insoles.7 The extensor tendons are close to the deep and superficial peroneal nerves and to the dorsalis pedis artery, none of which were involved in this case report.
There are case reports and series of EHL repairs that all involves at least 3 weeks of immobilization.4,8,9 The EHL dorsiflexes the big toe. Al-Qattan’s series involved placing K wires across the interphalangeal joint of the big toe and across the metatarsophalangeal joint, which were removed at 6 weeks, in addition to 3.0 polypropylene tendon mattress sutures. All patients in this series healed without tendon rupture or infection. Our PubMed search did not reveal any specific protocol for the EDL or EDB tendons, which are anatomically most comparable to the extensor digitorum communis (EDC) tendons in the hand. The EDC originates at the lateral epicondyle of the humerus, also divides into 4 separate tendons and is responsible for extending the 4 ulnar sided fingers at the metacarpophalangeal joint.10
Tendon repair protocols are a balance between preventing tendon rupture by too aggressive therapy and with preventing tendon adhesions from prolonged immobilization. Orthotic fabrication plays a key early role with blocking possible forces creating unacceptable strain or tension across the surgical repair site. Traditionally, extensor tendon repairs in the hand were immobilized for at least 3 weeks to prevent rupture. This is still the preferred protocol for the patient unwilling or unable to follow instructions. The downside to this method is extension lags, extrinsic tightness, and adhesions that prevent flexion, which can require prolonged therapy or tenolysis surgery to correct.11-13
Early passive motion (EPM) was promoted in the 1980s when studies found better functional outcomes and fewer adhesions. This involved either a dynamic extension splint that relied on elastic bands (Louisville protocol) to keep tension off the repair or the Duran protocol that relied on a static splint and the patient doing the passive exercises with his other uninjured hand. Critics of the EPM protocol point to the costs of the splints and demands of postoperative hand therapy.11
Early active motion (EAM) is the most recent development in hand tendon rehabilitation and starts within days of surgery. Studies have found an earlier regain of total active motion in patients who are mobilized earlier.12 EAM protocols can be divided into controlled active motion (CAM) and relative motion extension splinting (RMES). CAM splints are forearm based and cross more joints. Relative motion splinting is the least restrictive, which makes it less likely that the patient will remove it. Patient friendly splints are ideal because tendon ruptures are often secondary to nonadherence.13 The yoke splint is an example of a RMES, which places the repaired digit in slightly greater extension at the metacarpal phalangeal joint than the other digits (Figure 5), allowing use of the uninjured digits.
The toe extensors do not have the juncturae tendinum connecting the individual EDL tendons to each other, as found between the EDC tendons in the hand. These connective bands can mask a single extensor tendon laceration in the hand when the patient is still able to extend the digit to neutral in the event of a more proximal dorsal hand laceration. A case can be made for closing the skin only in lesser toe extensor injuries in poor surgical candidates because the extensor lag would not be appreciated functionally when wearing shoes. There would be less functional impact when letting a toe extensor go untreated compared with that of a hand extensor. Routine activities such as typing or getting the fingers into a tight pocket could be challenging if hand extensors were untreated. The rehabilitation for toe extensors is more inconvenient when a patient is nonweight bearing, compared with wearing a hand yoke splint.
Conclusion
The case described used an early passive motion protocol without the dynamic splint to rehabilitate the third toe EDL and second toe EDB. This was felt to be the most patient and therapist friendly option, given the previously unchartered territory. The foot orthosis was in stock at the adjacent physical therapy clinic, and the toe booster was created in the hand therapy clinic with readily available supplies. Ideally, one would like to return structures to their anatomic site and control the healing process in the event of a traumatic injury to prevent nonanatomic healing between structures and painful scar adhesions in an area with little subcutaneous tissue. This patient’s tendon repair was still intact at 7 weeks and on his way to recovery, demonstrating good scar management techniques. The risks and benefits to lesser toe tendon repair and recovery would have to be weighed on an individual basis.
Acknowledgments
This project is the result of work supported with resources and use of facilities at the Malcom Randall VA Medical Center in Gainesville, Florida.
1. Hand Therapy Certification Commission. History of HTCC. https://www.htcc.org/consumer-information/about-htcc/history-of-htcc. Accessed November 8, 2019.
2. Coady-Fariborzian L, McGreane A. Comparison of hand emergency triage before and after specialty templates (2007 vs 2012). Hand (N Y). 2015;10(2):215-220.
3. Hand Therapy Certification Commission. Passing rates for the CHT exam. https://www.htcc.org/certify/exam-results/passing-rates. Accessed November 8, 2019.
4. Al-Qattan MM. Surgical treatment and results in 17 cases of open lacerations of the extensor hallucis longus tendon. J Plast Reconstr Aesthet Surg. 2007;60(4):360-367.
5. Wheeless CR. Wheeless’ textbook of orthopaedics: extensor digitorum longus. http://www.wheelessonline.com/ortho/extensor_digitorum_longus. Updated December 8, 2011. Accessed November 8, 2019.
6. Wheeless CR. Wheeless’ textbook of orthopaedics: extensor digitorum brevis. http://www.wheelessonline.com/ortho/extensor_digitorum_brevis. Updated March 4, 2018. Accessed November 8, 2019.
7. Coughlin M, Schon L. Disorders of tendons. https://musculoskeletalkey.com/disorders-of-tendons-2/#s0035. Published August 27, 2016. Accessed November 8, 2019.
8. Bronner S, Ojofeitimi S, Rose D. Repair and rehabilitation of extensor hallucis longus and brevis tendon lacerations in a professional dancer. J Orthop Sports Phys Ther. 2008;38(6):362-370.
9. Wong JC, Daniel JN, Raikin SM. Repair of acute extensor hallucis longus tendon injuries: a retrospective review. Foot Ankle Spec. 2014;7(1):45-51.
10. Wheeless CR. Wheeless’ textbook of orthopaedics: extensor digitorum communis. http://www.wheelessonline.com/ortho/extensor_digitorum_communis. Updated March 4, 2018. Accessed November 8, 2019.
11. Hall B, Lee H, Page R, Rosenwax L, Lee AH. Comparing three postoperative treatment protocols for extensor tendon repair in zones V and VI of the hand. Am J Occup Ther. 2010;64(5):682-688.
12. Wong AL, Wilson M, Girnary S, Nojoomi M, Acharya S, Paul SM. The optimal orthosis and motion protocol for extensor tendon injury in zones IV-VIII: a systematic review. J Hand Ther. 2017;30(4):447-456.
13. Collocott SJ, Kelly E, Ellis RF. Optimal early active mobilisation protocol after extensor tendon repairs in zones V and VI: a systematic review of literature. Hand Ther. 2018;23(1):3-18.
1. Hand Therapy Certification Commission. History of HTCC. https://www.htcc.org/consumer-information/about-htcc/history-of-htcc. Accessed November 8, 2019.
2. Coady-Fariborzian L, McGreane A. Comparison of hand emergency triage before and after specialty templates (2007 vs 2012). Hand (N Y). 2015;10(2):215-220.
3. Hand Therapy Certification Commission. Passing rates for the CHT exam. https://www.htcc.org/certify/exam-results/passing-rates. Accessed November 8, 2019.
4. Al-Qattan MM. Surgical treatment and results in 17 cases of open lacerations of the extensor hallucis longus tendon. J Plast Reconstr Aesthet Surg. 2007;60(4):360-367.
5. Wheeless CR. Wheeless’ textbook of orthopaedics: extensor digitorum longus. http://www.wheelessonline.com/ortho/extensor_digitorum_longus. Updated December 8, 2011. Accessed November 8, 2019.
6. Wheeless CR. Wheeless’ textbook of orthopaedics: extensor digitorum brevis. http://www.wheelessonline.com/ortho/extensor_digitorum_brevis. Updated March 4, 2018. Accessed November 8, 2019.
7. Coughlin M, Schon L. Disorders of tendons. https://musculoskeletalkey.com/disorders-of-tendons-2/#s0035. Published August 27, 2016. Accessed November 8, 2019.
8. Bronner S, Ojofeitimi S, Rose D. Repair and rehabilitation of extensor hallucis longus and brevis tendon lacerations in a professional dancer. J Orthop Sports Phys Ther. 2008;38(6):362-370.
9. Wong JC, Daniel JN, Raikin SM. Repair of acute extensor hallucis longus tendon injuries: a retrospective review. Foot Ankle Spec. 2014;7(1):45-51.
10. Wheeless CR. Wheeless’ textbook of orthopaedics: extensor digitorum communis. http://www.wheelessonline.com/ortho/extensor_digitorum_communis. Updated March 4, 2018. Accessed November 8, 2019.
11. Hall B, Lee H, Page R, Rosenwax L, Lee AH. Comparing three postoperative treatment protocols for extensor tendon repair in zones V and VI of the hand. Am J Occup Ther. 2010;64(5):682-688.
12. Wong AL, Wilson M, Girnary S, Nojoomi M, Acharya S, Paul SM. The optimal orthosis and motion protocol for extensor tendon injury in zones IV-VIII: a systematic review. J Hand Ther. 2017;30(4):447-456.
13. Collocott SJ, Kelly E, Ellis RF. Optimal early active mobilisation protocol after extensor tendon repairs in zones V and VI: a systematic review of literature. Hand Ther. 2018;23(1):3-18.
Chondrodermatitis Nodularis Helicis in an Adolescent Boy: Not Just for Old Men
Chondrodermatitis nodularis helicis (CNH) is a chronic painful or crusted, 4- to 6-mm, solitary nodule, primarily on the upper part of the ear (most commonly on the right side). The presence of pain, which increases the likelihood that a person will seek treatment, clinically distinguishes CNH from other cutaneous tumors in the differential diagnosis that produce painless ulceration.
It is roughly 5 times more prevalent in males (72.9%),1 with an average age of onset of 65 years.2 However, CNH has been reported in females3 and rarely in individuals younger than 20 years. According to a PubMed search of articles indexed for MEDLINE and a Google Scholar search using the terms chrondodermatitis nodularis helices child, only 6 cases of CNH have been reported in the pediatric population.4-8 The youngest reported case was a 9-month-old infant.8 Including the present case, males and females in the pediatric population are equally affected; 4 patients had an underlying dermatomyositis,7 rheumatoid nodule,8 or systemic disease, including systemic lupus erythematosus and Beckwith-Wiedemann syndrome.5,9 Chronic intermittent pressure from headwear was the etiologic agent in the remaining cases.4 Recognizing that CNH can occur in young patients and can be associated with underlying autoimmune disease helps direct management and avoid overly invasive treatment.
Case Report
A 17-year-old adolescent boy presented with a painful ulcerated papule on the right upper helix of 3 months’ duration (Figure 1). The patient habitually slept on the right side, pressed a cell phone to that ear, and wore a tight-fitting visor while lifeguarding, which, along with solar damage, all may have contributed to the disease process. He was otherwise in good health, without a history of underlying systemic disease. Given the patient’s extensive occupational sun exposure, biopsy of the lesion was taken under the impression of CNH vs squamous cell carcinoma or basal cell carcinoma.

Histopathologic analysis revealed a central area of ulceration with edematous degenerated dermal collagen and overlying inflammatory crust, characteristic of CNH (Figure 2A). Biopsy in this patient demonstrated classic histopathologic findings of CNH, including a central area of epidermal ulceration capped by an inflammatory crust and an underlying edematous degenerated dermal collagen (Figure 2B).
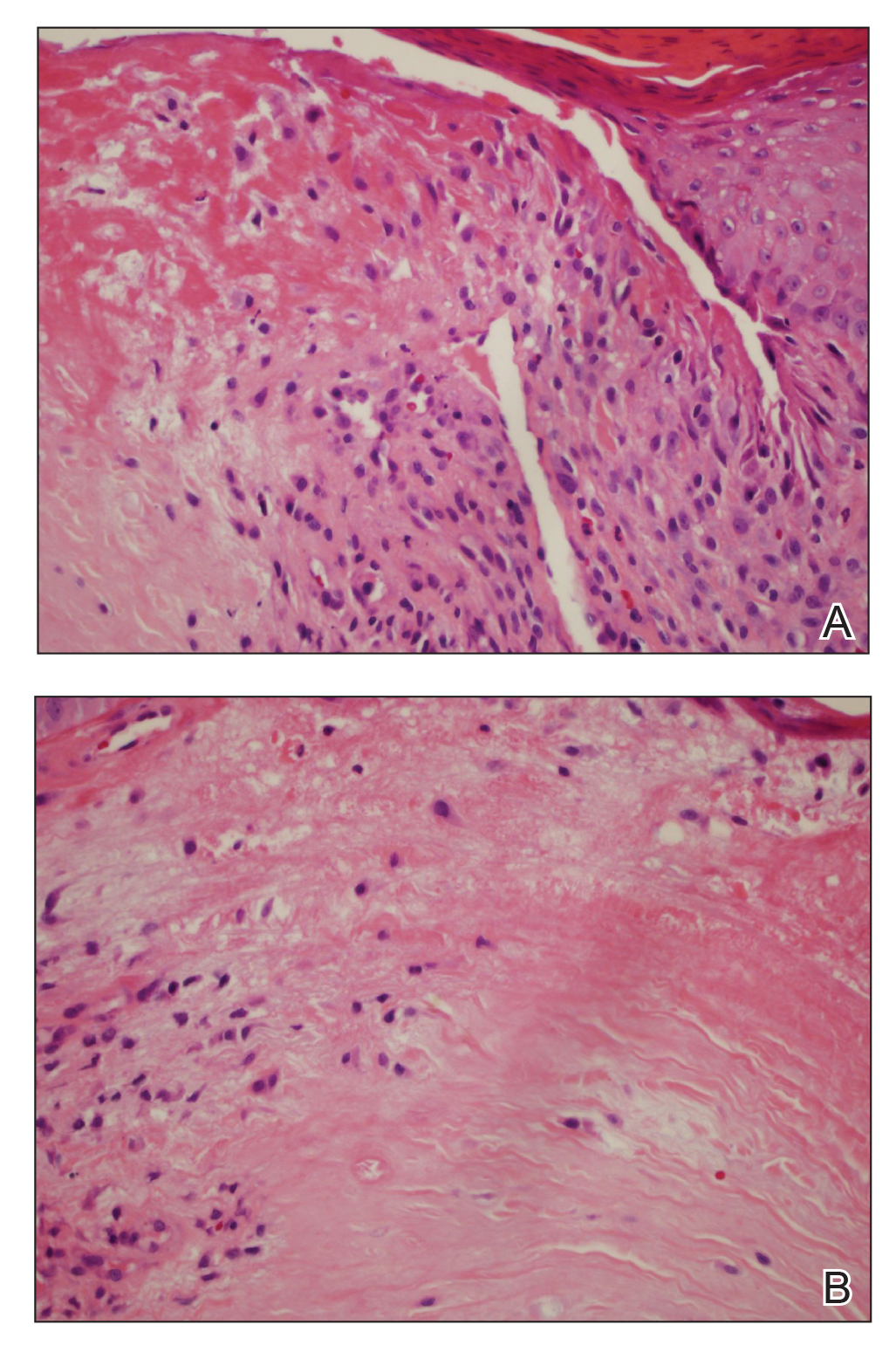
Following biopsy, the patient was advised of this diagnosis and recommended to avoid applying pressure to the area with cell phones or hats or when sleeping to prevent recurrence. At 3-month follow-up, no residual lesion remained.
Comment
Pathogenesis
The exact cause of CNH is unknown but is probably the result of prolonged and excessive pressure on the ear that leads to ischemic injury to cartilage and skin. The external location of CNH, lack of bony support, and exquisitely thin padding or insulation in the form of subcutaneous tissue make the small dermal blood vessels supplying the outer ear vulnerable to compression. Dermal inflammation; edema; and necrosis from trauma, cold, or actinic damage also can help initiate CNH. This disruption of blood perfusion to the external ear also inhibits the ear’s ability to heal. A cycle of pressure from objects such as a pillow or cell phone, followed by inadequate healing, leads to secondary perichondritis and remodeling of perichondrial arterioles, which is demonstrated histologically by the presence of perichondrial fibrous thickening, mild chronic inflammation, collagen degeneration, hyalinization, and rarely necrosis or calcification. Healed lesions often show dermal fibrosis overlying perichondrium.
Repeated pressure can lead to vascular changes, but underlying vascular disease also can predispose a person to CNH at a younger age. A striking case of bilateral CNH was reported in an 8-year-old girl with a known history of dermatomyositis.7 Furthermore, in 24 patients with CNH (mean age, 43 years), Magro et al9 observed an association between CNH and collagen vascular disease, scleroderma, hypertension, thyroid disease, and heart disease, with a higher incidence of any of these medical problems in younger patients. Therefore, screening all patients presenting with CNH, particularly those younger than their fourth decade, for underlying vasculopathy and an autoimmune connective tissue disorder is advised.9
Other findings of CNH reported in the literature include loss of elastic fibers in the central area of degenerated dermal collagen and nerve hyperplasia, which might account for pain.6 Many of the biopsies in cases of CNH reported in the literature also demonstrate perichondrial fibrous thickening, mild chronic inflammation, and degenerative changes in collagen, including hyalinization and rarely necrosis and calcification. Skin at the periphery of the lesion usually contains granulation tissue, with a mild to moderate inflammatory infiltrate and dilated vessels extending beyond the lesion.2
Genetics might play a role in the disorder, which is suggested by the occurrence of CNH in monozygotic twins10 and in an otherwise healthy 16-year-old adolescent girl with CNH of the right ear who screened negative for underlying connective tissue disease—serologic tests included antinuclear antibody, anti-Sm, anti-SCL-70, anti-Ro, anti-La, and rheumatoid factor—but who had a family history of a maternal grandmother with CNH, also on the right side.6
In the present case, there was no family history or signs and symptoms of underlying systemic disease at the time of diagnosis. The social history revealed excessive occupational sun exposure, habitually wearing a tight visor, and frequent cell phone use, all of which might have contributed to CNH.
Management
Medical management is geared toward relieving pressure at the site of the lesion, which was accomplished by use of an off-loading, ring-shaped, foam pillow at night in a 9-month-old girl with CNH, in which the smaller of her 2 left-sided lesions completely resolved by 6-month follow-up.8 However, it often is difficult to achieve adequate relief of pressure because of the patient’s preference for holding a cell phone to a particular ear or unconscious sleeping habits that perpetuate lesions. There are many creative physical interventions to offload aggravating pressure from the area during sleep. A prosthesis can be fashioned by cutting a hole from the center of a bath sponge and securing it with a headband,11 or a crescentic or rectangular piece of self-adhering foam sponge can be applied to the non–hair-bearing postauricular scalp during sleep.12 Topical antibiotics might relieve pain caused by secondary infection.
Surgical intervention, with or without placement of a full-thickness skin graft, is the mainstay of therapy. Excision was performed in 3 previously reported pediatric cases, with no recurrence reported at 6- to 24-month follow-up. Other treatments employed to varying effect include topical and intralesional steroids, collagen injection, cryotherapy, nitroglycerin paste 2% twice daily,13 and electrodesiccation and curettage.14 In adults, if full resolution is desired, multiple surgeries might be required to remove underlying protuberant cartilage; however, this strategy is not without risk of complication, including formation of adjacent cartilaginous nodules that can become site(s) of CNH recurrence due to a change in pressure points.
Conclusion
Although uncommon, CNH can present on the ears of young patients. A causal link between underlying vasculopathy and CNH has yet to be determined, but the association discovered by Magro et al9 merits obtaining a more detailed rheumatologic history and examination, followed by serologic testing (if indicated). Once the diagnosis of CNH is determined, patient education is paramount to prevent recurrence. Increased awareness of habits that inflict persistent repetitive trauma or pressure to the site—from sleeping patterns to cell phone use—will help to extinguish the behavior and therefore the lesion.
- Rex J, Rivera M, Bielsa I, et al. Narrow elliptical skin excision and cartilage shaving for treatment of chondrodermatitis nodularis. Dermatol Surg. 2006;32:400-404.
- Wettlé C, Keller F, Will F, et al. Chondrodermatitis nodularis chronical helicis: a descriptive study of 99 patients [in French]. Ann Dermatol Venereol. 2013;140:687-692.
- Oelzner S, Elsner P. Bilateral chondrodermatitis nodularis chronica helicis on the free border of the helix in a woman. J Am Acad Dermatol. 2003;49:720-722.
- Grigoryants V, Qureshi H, Patterson J, et al. Pediatric chondrodermatitis nodularis helicis. J Craniofac Surg. 2007;18:228-231.
- Fix WC, Cornejo C, Duffy KA, et al. Pediatric chondrodermatitis nodularis helicis (CNH) in a child with Beckwith-Wiedemann syndrome (BWS). Pediatr Dermatol. 2019;36:388-390.
- Rogers NE, Farris PK, Wang AR. Juvenile chondrodermatitis nodularis helicis: case report and literature review. Pediatr Dermatol. 2003;20:488-490.
- Sasaki T, Nishizawa H, Sugita Y. Chondrodermatitis nodularis helicis in childhood dermatomyositis. Br J Dermatol. 1999;141:363-365.
- Tsai TH, Lin YC, Chen HC. Infantile chondrodermatitis nodularis. Pediatr Dermatol. 2007;24:337-339.
- Magro CM, Frambach GE, Crowson AN. Chondrodermatitis nodularis helicis as a marker of internal disease associated with microvascular injury. J Cutan Pathol. 2005;32:329-333.
- Chan HP, Neuhaus IM, Maibach HI. Chondrodermatitis nodularis chronica helicis in monozygotic twins. Clin Exp Dermatol. 2009;34:358-359.
- Moncrieff M, Sassoon EM. Effective treatment of chondrodermatitis nodularis chronica helicis using a conservative approach. Br J Dermatol. 2004;150:892-894.
- Travelute CR. Self-adhering foam: a simple method for pressure relief during sleep in patients with chondrodermatitis nodularis helicis. Dermatol Surg. 2013;39:317-319.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;65:531-536.
- Kromann N, Høyer H, Reymann F. Chondrodermatitis nodularis chronica helicis treated with curettage and electrocauterization: follow-up of a 15-year material. Acta Derm Venereol. 1983;63:85-87.
Chondrodermatitis nodularis helicis (CNH) is a chronic painful or crusted, 4- to 6-mm, solitary nodule, primarily on the upper part of the ear (most commonly on the right side). The presence of pain, which increases the likelihood that a person will seek treatment, clinically distinguishes CNH from other cutaneous tumors in the differential diagnosis that produce painless ulceration.
It is roughly 5 times more prevalent in males (72.9%),1 with an average age of onset of 65 years.2 However, CNH has been reported in females3 and rarely in individuals younger than 20 years. According to a PubMed search of articles indexed for MEDLINE and a Google Scholar search using the terms chrondodermatitis nodularis helices child, only 6 cases of CNH have been reported in the pediatric population.4-8 The youngest reported case was a 9-month-old infant.8 Including the present case, males and females in the pediatric population are equally affected; 4 patients had an underlying dermatomyositis,7 rheumatoid nodule,8 or systemic disease, including systemic lupus erythematosus and Beckwith-Wiedemann syndrome.5,9 Chronic intermittent pressure from headwear was the etiologic agent in the remaining cases.4 Recognizing that CNH can occur in young patients and can be associated with underlying autoimmune disease helps direct management and avoid overly invasive treatment.
Case Report
A 17-year-old adolescent boy presented with a painful ulcerated papule on the right upper helix of 3 months’ duration (Figure 1). The patient habitually slept on the right side, pressed a cell phone to that ear, and wore a tight-fitting visor while lifeguarding, which, along with solar damage, all may have contributed to the disease process. He was otherwise in good health, without a history of underlying systemic disease. Given the patient’s extensive occupational sun exposure, biopsy of the lesion was taken under the impression of CNH vs squamous cell carcinoma or basal cell carcinoma.
Histopathologic analysis revealed a central area of ulceration with edematous degenerated dermal collagen and overlying inflammatory crust, characteristic of CNH (Figure 2A). Biopsy in this patient demonstrated classic histopathologic findings of CNH, including a central area of epidermal ulceration capped by an inflammatory crust and an underlying edematous degenerated dermal collagen (Figure 2B).
Following biopsy, the patient was advised of this diagnosis and recommended to avoid applying pressure to the area with cell phones or hats or when sleeping to prevent recurrence. At 3-month follow-up, no residual lesion remained.
Comment
Pathogenesis
The exact cause of CNH is unknown but is probably the result of prolonged and excessive pressure on the ear that leads to ischemic injury to cartilage and skin. The external location of CNH, lack of bony support, and exquisitely thin padding or insulation in the form of subcutaneous tissue make the small dermal blood vessels supplying the outer ear vulnerable to compression. Dermal inflammation; edema; and necrosis from trauma, cold, or actinic damage also can help initiate CNH. This disruption of blood perfusion to the external ear also inhibits the ear’s ability to heal. A cycle of pressure from objects such as a pillow or cell phone, followed by inadequate healing, leads to secondary perichondritis and remodeling of perichondrial arterioles, which is demonstrated histologically by the presence of perichondrial fibrous thickening, mild chronic inflammation, collagen degeneration, hyalinization, and rarely necrosis or calcification. Healed lesions often show dermal fibrosis overlying perichondrium.
Repeated pressure can lead to vascular changes, but underlying vascular disease also can predispose a person to CNH at a younger age. A striking case of bilateral CNH was reported in an 8-year-old girl with a known history of dermatomyositis.7 Furthermore, in 24 patients with CNH (mean age, 43 years), Magro et al9 observed an association between CNH and collagen vascular disease, scleroderma, hypertension, thyroid disease, and heart disease, with a higher incidence of any of these medical problems in younger patients. Therefore, screening all patients presenting with CNH, particularly those younger than their fourth decade, for underlying vasculopathy and an autoimmune connective tissue disorder is advised.9
Other findings of CNH reported in the literature include loss of elastic fibers in the central area of degenerated dermal collagen and nerve hyperplasia, which might account for pain.6 Many of the biopsies in cases of CNH reported in the literature also demonstrate perichondrial fibrous thickening, mild chronic inflammation, and degenerative changes in collagen, including hyalinization and rarely necrosis and calcification. Skin at the periphery of the lesion usually contains granulation tissue, with a mild to moderate inflammatory infiltrate and dilated vessels extending beyond the lesion.2
Genetics might play a role in the disorder, which is suggested by the occurrence of CNH in monozygotic twins10 and in an otherwise healthy 16-year-old adolescent girl with CNH of the right ear who screened negative for underlying connective tissue disease—serologic tests included antinuclear antibody, anti-Sm, anti-SCL-70, anti-Ro, anti-La, and rheumatoid factor—but who had a family history of a maternal grandmother with CNH, also on the right side.6
In the present case, there was no family history or signs and symptoms of underlying systemic disease at the time of diagnosis. The social history revealed excessive occupational sun exposure, habitually wearing a tight visor, and frequent cell phone use, all of which might have contributed to CNH.
Management
Medical management is geared toward relieving pressure at the site of the lesion, which was accomplished by use of an off-loading, ring-shaped, foam pillow at night in a 9-month-old girl with CNH, in which the smaller of her 2 left-sided lesions completely resolved by 6-month follow-up.8 However, it often is difficult to achieve adequate relief of pressure because of the patient’s preference for holding a cell phone to a particular ear or unconscious sleeping habits that perpetuate lesions. There are many creative physical interventions to offload aggravating pressure from the area during sleep. A prosthesis can be fashioned by cutting a hole from the center of a bath sponge and securing it with a headband,11 or a crescentic or rectangular piece of self-adhering foam sponge can be applied to the non–hair-bearing postauricular scalp during sleep.12 Topical antibiotics might relieve pain caused by secondary infection.
Surgical intervention, with or without placement of a full-thickness skin graft, is the mainstay of therapy. Excision was performed in 3 previously reported pediatric cases, with no recurrence reported at 6- to 24-month follow-up. Other treatments employed to varying effect include topical and intralesional steroids, collagen injection, cryotherapy, nitroglycerin paste 2% twice daily,13 and electrodesiccation and curettage.14 In adults, if full resolution is desired, multiple surgeries might be required to remove underlying protuberant cartilage; however, this strategy is not without risk of complication, including formation of adjacent cartilaginous nodules that can become site(s) of CNH recurrence due to a change in pressure points.
Conclusion
Although uncommon, CNH can present on the ears of young patients. A causal link between underlying vasculopathy and CNH has yet to be determined, but the association discovered by Magro et al9 merits obtaining a more detailed rheumatologic history and examination, followed by serologic testing (if indicated). Once the diagnosis of CNH is determined, patient education is paramount to prevent recurrence. Increased awareness of habits that inflict persistent repetitive trauma or pressure to the site—from sleeping patterns to cell phone use—will help to extinguish the behavior and therefore the lesion.
Chondrodermatitis nodularis helicis (CNH) is a chronic painful or crusted, 4- to 6-mm, solitary nodule, primarily on the upper part of the ear (most commonly on the right side). The presence of pain, which increases the likelihood that a person will seek treatment, clinically distinguishes CNH from other cutaneous tumors in the differential diagnosis that produce painless ulceration.
It is roughly 5 times more prevalent in males (72.9%),1 with an average age of onset of 65 years.2 However, CNH has been reported in females3 and rarely in individuals younger than 20 years. According to a PubMed search of articles indexed for MEDLINE and a Google Scholar search using the terms chrondodermatitis nodularis helices child, only 6 cases of CNH have been reported in the pediatric population.4-8 The youngest reported case was a 9-month-old infant.8 Including the present case, males and females in the pediatric population are equally affected; 4 patients had an underlying dermatomyositis,7 rheumatoid nodule,8 or systemic disease, including systemic lupus erythematosus and Beckwith-Wiedemann syndrome.5,9 Chronic intermittent pressure from headwear was the etiologic agent in the remaining cases.4 Recognizing that CNH can occur in young patients and can be associated with underlying autoimmune disease helps direct management and avoid overly invasive treatment.
Case Report
A 17-year-old adolescent boy presented with a painful ulcerated papule on the right upper helix of 3 months’ duration (Figure 1). The patient habitually slept on the right side, pressed a cell phone to that ear, and wore a tight-fitting visor while lifeguarding, which, along with solar damage, all may have contributed to the disease process. He was otherwise in good health, without a history of underlying systemic disease. Given the patient’s extensive occupational sun exposure, biopsy of the lesion was taken under the impression of CNH vs squamous cell carcinoma or basal cell carcinoma.
Histopathologic analysis revealed a central area of ulceration with edematous degenerated dermal collagen and overlying inflammatory crust, characteristic of CNH (Figure 2A). Biopsy in this patient demonstrated classic histopathologic findings of CNH, including a central area of epidermal ulceration capped by an inflammatory crust and an underlying edematous degenerated dermal collagen (Figure 2B).
Following biopsy, the patient was advised of this diagnosis and recommended to avoid applying pressure to the area with cell phones or hats or when sleeping to prevent recurrence. At 3-month follow-up, no residual lesion remained.
Comment
Pathogenesis
The exact cause of CNH is unknown but is probably the result of prolonged and excessive pressure on the ear that leads to ischemic injury to cartilage and skin. The external location of CNH, lack of bony support, and exquisitely thin padding or insulation in the form of subcutaneous tissue make the small dermal blood vessels supplying the outer ear vulnerable to compression. Dermal inflammation; edema; and necrosis from trauma, cold, or actinic damage also can help initiate CNH. This disruption of blood perfusion to the external ear also inhibits the ear’s ability to heal. A cycle of pressure from objects such as a pillow or cell phone, followed by inadequate healing, leads to secondary perichondritis and remodeling of perichondrial arterioles, which is demonstrated histologically by the presence of perichondrial fibrous thickening, mild chronic inflammation, collagen degeneration, hyalinization, and rarely necrosis or calcification. Healed lesions often show dermal fibrosis overlying perichondrium.
Repeated pressure can lead to vascular changes, but underlying vascular disease also can predispose a person to CNH at a younger age. A striking case of bilateral CNH was reported in an 8-year-old girl with a known history of dermatomyositis.7 Furthermore, in 24 patients with CNH (mean age, 43 years), Magro et al9 observed an association between CNH and collagen vascular disease, scleroderma, hypertension, thyroid disease, and heart disease, with a higher incidence of any of these medical problems in younger patients. Therefore, screening all patients presenting with CNH, particularly those younger than their fourth decade, for underlying vasculopathy and an autoimmune connective tissue disorder is advised.9
Other findings of CNH reported in the literature include loss of elastic fibers in the central area of degenerated dermal collagen and nerve hyperplasia, which might account for pain.6 Many of the biopsies in cases of CNH reported in the literature also demonstrate perichondrial fibrous thickening, mild chronic inflammation, and degenerative changes in collagen, including hyalinization and rarely necrosis and calcification. Skin at the periphery of the lesion usually contains granulation tissue, with a mild to moderate inflammatory infiltrate and dilated vessels extending beyond the lesion.2
Genetics might play a role in the disorder, which is suggested by the occurrence of CNH in monozygotic twins10 and in an otherwise healthy 16-year-old adolescent girl with CNH of the right ear who screened negative for underlying connective tissue disease—serologic tests included antinuclear antibody, anti-Sm, anti-SCL-70, anti-Ro, anti-La, and rheumatoid factor—but who had a family history of a maternal grandmother with CNH, also on the right side.6
In the present case, there was no family history or signs and symptoms of underlying systemic disease at the time of diagnosis. The social history revealed excessive occupational sun exposure, habitually wearing a tight visor, and frequent cell phone use, all of which might have contributed to CNH.
Management
Medical management is geared toward relieving pressure at the site of the lesion, which was accomplished by use of an off-loading, ring-shaped, foam pillow at night in a 9-month-old girl with CNH, in which the smaller of her 2 left-sided lesions completely resolved by 6-month follow-up.8 However, it often is difficult to achieve adequate relief of pressure because of the patient’s preference for holding a cell phone to a particular ear or unconscious sleeping habits that perpetuate lesions. There are many creative physical interventions to offload aggravating pressure from the area during sleep. A prosthesis can be fashioned by cutting a hole from the center of a bath sponge and securing it with a headband,11 or a crescentic or rectangular piece of self-adhering foam sponge can be applied to the non–hair-bearing postauricular scalp during sleep.12 Topical antibiotics might relieve pain caused by secondary infection.
Surgical intervention, with or without placement of a full-thickness skin graft, is the mainstay of therapy. Excision was performed in 3 previously reported pediatric cases, with no recurrence reported at 6- to 24-month follow-up. Other treatments employed to varying effect include topical and intralesional steroids, collagen injection, cryotherapy, nitroglycerin paste 2% twice daily,13 and electrodesiccation and curettage.14 In adults, if full resolution is desired, multiple surgeries might be required to remove underlying protuberant cartilage; however, this strategy is not without risk of complication, including formation of adjacent cartilaginous nodules that can become site(s) of CNH recurrence due to a change in pressure points.
Conclusion
Although uncommon, CNH can present on the ears of young patients. A causal link between underlying vasculopathy and CNH has yet to be determined, but the association discovered by Magro et al9 merits obtaining a more detailed rheumatologic history and examination, followed by serologic testing (if indicated). Once the diagnosis of CNH is determined, patient education is paramount to prevent recurrence. Increased awareness of habits that inflict persistent repetitive trauma or pressure to the site—from sleeping patterns to cell phone use—will help to extinguish the behavior and therefore the lesion.
- Rex J, Rivera M, Bielsa I, et al. Narrow elliptical skin excision and cartilage shaving for treatment of chondrodermatitis nodularis. Dermatol Surg. 2006;32:400-404.
- Wettlé C, Keller F, Will F, et al. Chondrodermatitis nodularis chronical helicis: a descriptive study of 99 patients [in French]. Ann Dermatol Venereol. 2013;140:687-692.
- Oelzner S, Elsner P. Bilateral chondrodermatitis nodularis chronica helicis on the free border of the helix in a woman. J Am Acad Dermatol. 2003;49:720-722.
- Grigoryants V, Qureshi H, Patterson J, et al. Pediatric chondrodermatitis nodularis helicis. J Craniofac Surg. 2007;18:228-231.
- Fix WC, Cornejo C, Duffy KA, et al. Pediatric chondrodermatitis nodularis helicis (CNH) in a child with Beckwith-Wiedemann syndrome (BWS). Pediatr Dermatol. 2019;36:388-390.
- Rogers NE, Farris PK, Wang AR. Juvenile chondrodermatitis nodularis helicis: case report and literature review. Pediatr Dermatol. 2003;20:488-490.
- Sasaki T, Nishizawa H, Sugita Y. Chondrodermatitis nodularis helicis in childhood dermatomyositis. Br J Dermatol. 1999;141:363-365.
- Tsai TH, Lin YC, Chen HC. Infantile chondrodermatitis nodularis. Pediatr Dermatol. 2007;24:337-339.
- Magro CM, Frambach GE, Crowson AN. Chondrodermatitis nodularis helicis as a marker of internal disease associated with microvascular injury. J Cutan Pathol. 2005;32:329-333.
- Chan HP, Neuhaus IM, Maibach HI. Chondrodermatitis nodularis chronica helicis in monozygotic twins. Clin Exp Dermatol. 2009;34:358-359.
- Moncrieff M, Sassoon EM. Effective treatment of chondrodermatitis nodularis chronica helicis using a conservative approach. Br J Dermatol. 2004;150:892-894.
- Travelute CR. Self-adhering foam: a simple method for pressure relief during sleep in patients with chondrodermatitis nodularis helicis. Dermatol Surg. 2013;39:317-319.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;65:531-536.
- Kromann N, Høyer H, Reymann F. Chondrodermatitis nodularis chronica helicis treated with curettage and electrocauterization: follow-up of a 15-year material. Acta Derm Venereol. 1983;63:85-87.
- Rex J, Rivera M, Bielsa I, et al. Narrow elliptical skin excision and cartilage shaving for treatment of chondrodermatitis nodularis. Dermatol Surg. 2006;32:400-404.
- Wettlé C, Keller F, Will F, et al. Chondrodermatitis nodularis chronical helicis: a descriptive study of 99 patients [in French]. Ann Dermatol Venereol. 2013;140:687-692.
- Oelzner S, Elsner P. Bilateral chondrodermatitis nodularis chronica helicis on the free border of the helix in a woman. J Am Acad Dermatol. 2003;49:720-722.
- Grigoryants V, Qureshi H, Patterson J, et al. Pediatric chondrodermatitis nodularis helicis. J Craniofac Surg. 2007;18:228-231.
- Fix WC, Cornejo C, Duffy KA, et al. Pediatric chondrodermatitis nodularis helicis (CNH) in a child with Beckwith-Wiedemann syndrome (BWS). Pediatr Dermatol. 2019;36:388-390.
- Rogers NE, Farris PK, Wang AR. Juvenile chondrodermatitis nodularis helicis: case report and literature review. Pediatr Dermatol. 2003;20:488-490.
- Sasaki T, Nishizawa H, Sugita Y. Chondrodermatitis nodularis helicis in childhood dermatomyositis. Br J Dermatol. 1999;141:363-365.
- Tsai TH, Lin YC, Chen HC. Infantile chondrodermatitis nodularis. Pediatr Dermatol. 2007;24:337-339.
- Magro CM, Frambach GE, Crowson AN. Chondrodermatitis nodularis helicis as a marker of internal disease associated with microvascular injury. J Cutan Pathol. 2005;32:329-333.
- Chan HP, Neuhaus IM, Maibach HI. Chondrodermatitis nodularis chronica helicis in monozygotic twins. Clin Exp Dermatol. 2009;34:358-359.
- Moncrieff M, Sassoon EM. Effective treatment of chondrodermatitis nodularis chronica helicis using a conservative approach. Br J Dermatol. 2004;150:892-894.
- Travelute CR. Self-adhering foam: a simple method for pressure relief during sleep in patients with chondrodermatitis nodularis helicis. Dermatol Surg. 2013;39:317-319.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;65:531-536.
- Kromann N, Høyer H, Reymann F. Chondrodermatitis nodularis chronica helicis treated with curettage and electrocauterization: follow-up of a 15-year material. Acta Derm Venereol. 1983;63:85-87.
Practice Points
- Chondrodermatitis nodularis helicis should be in the differential for nodular lesions on the ears of adolescents, as societal shifts in behavior have altered the epidemiology of this condition such that it is no longer exclusive to the geriatric population.
- Make sure to get a thorough history of potential pressure triggers when evaluating nodules on the ears of adolescents.
45-year-old woman • fever and chills • diffuse abdominal pain • shortness of breath • Dx?
THE CASE
A 45-year-old white woman presented to our emergency department (ED) with a 3-day history of fever, chills, diffuse abdominal pain, severe headache, and shortness of breath.
The patient’s medical and surgical history was notable for acromegaly secondary to pituitary microadenoma, pituitary resection, and complete thyroidectomy 4 years earlier. Her medications included lanreotide, levothyroxine, gabapentin, alprazolam, and zolpidem. She had no history of cardiac disease, diabetes mellitus, immunodeficiency, or injection drug use. Three months prior to presenting to the ED, she underwent an outpatient gynecologic procedure for insertion of a levonorgestrel-releasing intrauterine device (IUD) for menorrhagia.
In the ED, the patient had a fever (101.5°F) and an elevated white blood cell count of 13,600/mm3 (reference range, 4,000–10,000/mm3). Cardiac auscultation revealed a regular heart rate and rhythm, with normal S1 and S2 sounds without murmur. Electrocardiogram documented normal sinus rhythm with no abnormalities. The physical examination revealed a diffusely tender lower abdomen without rebound or guarding. A pelvic examination was not conducted, and there was no collection of a vaginal swab sample to test for gonorrhea, chlamydia, or group B Streptococcus (GBS). Further workups for infection, including urinalysis, lumbar puncture, and chest x-ray, all yielded normal results.
Shortly after she was discharged from the ED, the patient was called to return to the hospital after blood cultures grew GBS; she was admitted for treatment.
THE DIAGNOSIS
A diagnosis of sepsis secondary to GBS bacteremia was made. However, the source of the GBS bacteremia and the patient’s abdominal symptoms remained unclear. Further workup included computed tomography (CT) of the abdomen, pelvis, and head, and magnetic resonance imaging of the brain; all imaging revealed no acute findings. Blood work (chem-7 panel, complete blood count, human immunodeficiency virus testing) was unremarkable except for an elevated level of C-reactive protein of 90 mg/L (reference range, 0–10 mg/L).
Radiography confirmed that the IUD was in the correct intrauterine position. However, transesophageal echocardiography (TEE) showed vegetations on the mitral and aortic valves, with preserved cardiac function. A diagnosis of GBS endocarditis was made, and infectious disease specialists were consulted. Because the patient had an anaphylactic allergy to penicillin, she was treated with intravenous vancomycin for 4 weeks. One month later, she had the IUD removed because of persistent abdominal pain.
DISCUSSION
Although the source of GBS bacteremia and endocarditis in our patient remained nondefinitive, the recent insertion of the IUD continued to be the suspected source and leading diagnosis.
Continue to: Other sources of GBS bacteremia...
Other sources of GBS bacteremia were unlikely based on the examination and imaging results. The patient’s abdominal exam was benign, and no intra-abdominal abscess was detected on CT. Although Streptococcus viridans, S bovis, and enterococcus are far more common pathogens for infective endocarditis,1 there was no evidence of dental caries, gastrointestinal pathology, or urinary tract infection to suggest misidentification of bacteria.
Theoretically, GBS bacteremia after a gynecologic procedure is possible since GBS frequently colonizes the vagina.2 However, most reports document transient rather than persistent bacteremia and/or endocarditis.3,4
IUD insertion as a cause of bacteremia. The medical literature offers scant evidence of endocarditis or severe GBS bacteremia related to IUD insertion. Of 124 gynecology-related reports of infective endocarditis between 1946 and 1986, only 3 were associated with IUDs.5 All 3 women had underlying cardiac disease, and 2 of the 3 had identifiable pelvic infections.5
Among 12 case reports of endocarditis related to gynecologic procedures from 1985 to 2003, therapeutic abortion was the most common antecedent event, and no cases were related to IUD insertion.2 Compared with cases reported before 1985, in these cases most patients (64%) did not have underlying valvular disease, and most had a subacute course with low mortality but high morbidity (8 of 11 patients had clinically significant emboli).2 The study authors also mentioned a case of endocarditis following a Pap smear test, suggesting that minimally invasive procedures may result in infective endocarditis.2
THE TAKEAWAY
Our patient presented with fever, fatigue, and abdominal pain in the setting of recent IUD insertion. She was found to have GBS bacteremia with endocarditis based on TEE and positive blood culture growth. Her clinical situation was suspicious for a gynecologic source of bacteremia.
Continue to: There is no definitive way...
There is no definitive way to confirm that IUD insertion 3 months prior caused the GBS bacteremia. However, this case illustrates that it is important to consider a usually benign gynecologic procedure as the source of clinically significant persistent bacteremia.
Evidence is insufficient to recommend prophylactic antibiotic use prior to a gynecologic procedure, and it is not recommended by current practice guidelines of the American College of Obstetricians and Gynecologists or the European Society of Cardiology.6,7
This patient case raises our suspicion for IUD-related bacteremia as an adverse reaction in healthy women with recent IUD insertion who present with fever and diffuse abdominal pain without apparent signs of a pelvic infection. Prompt antibiotic treatment is necessary to prevent significant morbidity and mortality.
CORRESPONDENCE
Lauren Cowen, MD, 777 South Clinton Avenue, Rochester, NY 14620; [email protected]
1. Baddour LM, Wilson WR, Bayer AS, et al; American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and Stroke Council. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation. 2015;132:1435-1486.
2. Crespo A, Retter AS, Lorber B. Group B streptococcal endocarditis in obstetric and gynecologic practice. Infect Dis Obstet Gynecol. 2003;11:109-115.
3. Murray S, Hickey JB, Houang E. Significant bacteremia associated with replacement of intrauterine contraceptive device. Am J Obstet Gynecol. 1987;156:698-700.
4. Everett ED, Reller LB, Droegemueller W, et al. Absence of bacteremia after insertion or removal of intrauterine devices. Obstet Gynecol. 1976;47:207-209.
5. Seaworth BJ, Durack DT. Infective endocarditis in obstetric and gynecologic practice. Am J Obstet Gynecol. 1986;154:180-188.
6. ACOG Committee on Practice Bulletins–Gynecology. Practice bulletin no. 186: Long-acting reversible contraception: implants and intrauterine devices. Obstet Gynecol. 2017;130:e251-e269.
7. Habib G, Lancellotti P, Antunes MJ, et al; ESC Scientific Document Group. 2015 ESC guidelines for the management of infective endocarditis: the Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Eur Heart J. 2015;36:3075-3128.
THE CASE
A 45-year-old white woman presented to our emergency department (ED) with a 3-day history of fever, chills, diffuse abdominal pain, severe headache, and shortness of breath.
The patient’s medical and surgical history was notable for acromegaly secondary to pituitary microadenoma, pituitary resection, and complete thyroidectomy 4 years earlier. Her medications included lanreotide, levothyroxine, gabapentin, alprazolam, and zolpidem. She had no history of cardiac disease, diabetes mellitus, immunodeficiency, or injection drug use. Three months prior to presenting to the ED, she underwent an outpatient gynecologic procedure for insertion of a levonorgestrel-releasing intrauterine device (IUD) for menorrhagia.
In the ED, the patient had a fever (101.5°F) and an elevated white blood cell count of 13,600/mm3 (reference range, 4,000–10,000/mm3). Cardiac auscultation revealed a regular heart rate and rhythm, with normal S1 and S2 sounds without murmur. Electrocardiogram documented normal sinus rhythm with no abnormalities. The physical examination revealed a diffusely tender lower abdomen without rebound or guarding. A pelvic examination was not conducted, and there was no collection of a vaginal swab sample to test for gonorrhea, chlamydia, or group B Streptococcus (GBS). Further workups for infection, including urinalysis, lumbar puncture, and chest x-ray, all yielded normal results.
Shortly after she was discharged from the ED, the patient was called to return to the hospital after blood cultures grew GBS; she was admitted for treatment.
THE DIAGNOSIS
A diagnosis of sepsis secondary to GBS bacteremia was made. However, the source of the GBS bacteremia and the patient’s abdominal symptoms remained unclear. Further workup included computed tomography (CT) of the abdomen, pelvis, and head, and magnetic resonance imaging of the brain; all imaging revealed no acute findings. Blood work (chem-7 panel, complete blood count, human immunodeficiency virus testing) was unremarkable except for an elevated level of C-reactive protein of 90 mg/L (reference range, 0–10 mg/L).
Radiography confirmed that the IUD was in the correct intrauterine position. However, transesophageal echocardiography (TEE) showed vegetations on the mitral and aortic valves, with preserved cardiac function. A diagnosis of GBS endocarditis was made, and infectious disease specialists were consulted. Because the patient had an anaphylactic allergy to penicillin, she was treated with intravenous vancomycin for 4 weeks. One month later, she had the IUD removed because of persistent abdominal pain.
DISCUSSION
Although the source of GBS bacteremia and endocarditis in our patient remained nondefinitive, the recent insertion of the IUD continued to be the suspected source and leading diagnosis.
Continue to: Other sources of GBS bacteremia...
Other sources of GBS bacteremia were unlikely based on the examination and imaging results. The patient’s abdominal exam was benign, and no intra-abdominal abscess was detected on CT. Although Streptococcus viridans, S bovis, and enterococcus are far more common pathogens for infective endocarditis,1 there was no evidence of dental caries, gastrointestinal pathology, or urinary tract infection to suggest misidentification of bacteria.
Theoretically, GBS bacteremia after a gynecologic procedure is possible since GBS frequently colonizes the vagina.2 However, most reports document transient rather than persistent bacteremia and/or endocarditis.3,4
IUD insertion as a cause of bacteremia. The medical literature offers scant evidence of endocarditis or severe GBS bacteremia related to IUD insertion. Of 124 gynecology-related reports of infective endocarditis between 1946 and 1986, only 3 were associated with IUDs.5 All 3 women had underlying cardiac disease, and 2 of the 3 had identifiable pelvic infections.5
Among 12 case reports of endocarditis related to gynecologic procedures from 1985 to 2003, therapeutic abortion was the most common antecedent event, and no cases were related to IUD insertion.2 Compared with cases reported before 1985, in these cases most patients (64%) did not have underlying valvular disease, and most had a subacute course with low mortality but high morbidity (8 of 11 patients had clinically significant emboli).2 The study authors also mentioned a case of endocarditis following a Pap smear test, suggesting that minimally invasive procedures may result in infective endocarditis.2
THE TAKEAWAY
Our patient presented with fever, fatigue, and abdominal pain in the setting of recent IUD insertion. She was found to have GBS bacteremia with endocarditis based on TEE and positive blood culture growth. Her clinical situation was suspicious for a gynecologic source of bacteremia.
Continue to: There is no definitive way...
There is no definitive way to confirm that IUD insertion 3 months prior caused the GBS bacteremia. However, this case illustrates that it is important to consider a usually benign gynecologic procedure as the source of clinically significant persistent bacteremia.
Evidence is insufficient to recommend prophylactic antibiotic use prior to a gynecologic procedure, and it is not recommended by current practice guidelines of the American College of Obstetricians and Gynecologists or the European Society of Cardiology.6,7
This patient case raises our suspicion for IUD-related bacteremia as an adverse reaction in healthy women with recent IUD insertion who present with fever and diffuse abdominal pain without apparent signs of a pelvic infection. Prompt antibiotic treatment is necessary to prevent significant morbidity and mortality.
CORRESPONDENCE
Lauren Cowen, MD, 777 South Clinton Avenue, Rochester, NY 14620; [email protected]
THE CASE
A 45-year-old white woman presented to our emergency department (ED) with a 3-day history of fever, chills, diffuse abdominal pain, severe headache, and shortness of breath.
The patient’s medical and surgical history was notable for acromegaly secondary to pituitary microadenoma, pituitary resection, and complete thyroidectomy 4 years earlier. Her medications included lanreotide, levothyroxine, gabapentin, alprazolam, and zolpidem. She had no history of cardiac disease, diabetes mellitus, immunodeficiency, or injection drug use. Three months prior to presenting to the ED, she underwent an outpatient gynecologic procedure for insertion of a levonorgestrel-releasing intrauterine device (IUD) for menorrhagia.
In the ED, the patient had a fever (101.5°F) and an elevated white blood cell count of 13,600/mm3 (reference range, 4,000–10,000/mm3). Cardiac auscultation revealed a regular heart rate and rhythm, with normal S1 and S2 sounds without murmur. Electrocardiogram documented normal sinus rhythm with no abnormalities. The physical examination revealed a diffusely tender lower abdomen without rebound or guarding. A pelvic examination was not conducted, and there was no collection of a vaginal swab sample to test for gonorrhea, chlamydia, or group B Streptococcus (GBS). Further workups for infection, including urinalysis, lumbar puncture, and chest x-ray, all yielded normal results.
Shortly after she was discharged from the ED, the patient was called to return to the hospital after blood cultures grew GBS; she was admitted for treatment.
THE DIAGNOSIS
A diagnosis of sepsis secondary to GBS bacteremia was made. However, the source of the GBS bacteremia and the patient’s abdominal symptoms remained unclear. Further workup included computed tomography (CT) of the abdomen, pelvis, and head, and magnetic resonance imaging of the brain; all imaging revealed no acute findings. Blood work (chem-7 panel, complete blood count, human immunodeficiency virus testing) was unremarkable except for an elevated level of C-reactive protein of 90 mg/L (reference range, 0–10 mg/L).
Radiography confirmed that the IUD was in the correct intrauterine position. However, transesophageal echocardiography (TEE) showed vegetations on the mitral and aortic valves, with preserved cardiac function. A diagnosis of GBS endocarditis was made, and infectious disease specialists were consulted. Because the patient had an anaphylactic allergy to penicillin, she was treated with intravenous vancomycin for 4 weeks. One month later, she had the IUD removed because of persistent abdominal pain.
DISCUSSION
Although the source of GBS bacteremia and endocarditis in our patient remained nondefinitive, the recent insertion of the IUD continued to be the suspected source and leading diagnosis.
Continue to: Other sources of GBS bacteremia...
Other sources of GBS bacteremia were unlikely based on the examination and imaging results. The patient’s abdominal exam was benign, and no intra-abdominal abscess was detected on CT. Although Streptococcus viridans, S bovis, and enterococcus are far more common pathogens for infective endocarditis,1 there was no evidence of dental caries, gastrointestinal pathology, or urinary tract infection to suggest misidentification of bacteria.
Theoretically, GBS bacteremia after a gynecologic procedure is possible since GBS frequently colonizes the vagina.2 However, most reports document transient rather than persistent bacteremia and/or endocarditis.3,4
IUD insertion as a cause of bacteremia. The medical literature offers scant evidence of endocarditis or severe GBS bacteremia related to IUD insertion. Of 124 gynecology-related reports of infective endocarditis between 1946 and 1986, only 3 were associated with IUDs.5 All 3 women had underlying cardiac disease, and 2 of the 3 had identifiable pelvic infections.5
Among 12 case reports of endocarditis related to gynecologic procedures from 1985 to 2003, therapeutic abortion was the most common antecedent event, and no cases were related to IUD insertion.2 Compared with cases reported before 1985, in these cases most patients (64%) did not have underlying valvular disease, and most had a subacute course with low mortality but high morbidity (8 of 11 patients had clinically significant emboli).2 The study authors also mentioned a case of endocarditis following a Pap smear test, suggesting that minimally invasive procedures may result in infective endocarditis.2
THE TAKEAWAY
Our patient presented with fever, fatigue, and abdominal pain in the setting of recent IUD insertion. She was found to have GBS bacteremia with endocarditis based on TEE and positive blood culture growth. Her clinical situation was suspicious for a gynecologic source of bacteremia.
Continue to: There is no definitive way...
There is no definitive way to confirm that IUD insertion 3 months prior caused the GBS bacteremia. However, this case illustrates that it is important to consider a usually benign gynecologic procedure as the source of clinically significant persistent bacteremia.
Evidence is insufficient to recommend prophylactic antibiotic use prior to a gynecologic procedure, and it is not recommended by current practice guidelines of the American College of Obstetricians and Gynecologists or the European Society of Cardiology.6,7
This patient case raises our suspicion for IUD-related bacteremia as an adverse reaction in healthy women with recent IUD insertion who present with fever and diffuse abdominal pain without apparent signs of a pelvic infection. Prompt antibiotic treatment is necessary to prevent significant morbidity and mortality.
CORRESPONDENCE
Lauren Cowen, MD, 777 South Clinton Avenue, Rochester, NY 14620; [email protected]
1. Baddour LM, Wilson WR, Bayer AS, et al; American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and Stroke Council. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation. 2015;132:1435-1486.
2. Crespo A, Retter AS, Lorber B. Group B streptococcal endocarditis in obstetric and gynecologic practice. Infect Dis Obstet Gynecol. 2003;11:109-115.
3. Murray S, Hickey JB, Houang E. Significant bacteremia associated with replacement of intrauterine contraceptive device. Am J Obstet Gynecol. 1987;156:698-700.
4. Everett ED, Reller LB, Droegemueller W, et al. Absence of bacteremia after insertion or removal of intrauterine devices. Obstet Gynecol. 1976;47:207-209.
5. Seaworth BJ, Durack DT. Infective endocarditis in obstetric and gynecologic practice. Am J Obstet Gynecol. 1986;154:180-188.
6. ACOG Committee on Practice Bulletins–Gynecology. Practice bulletin no. 186: Long-acting reversible contraception: implants and intrauterine devices. Obstet Gynecol. 2017;130:e251-e269.
7. Habib G, Lancellotti P, Antunes MJ, et al; ESC Scientific Document Group. 2015 ESC guidelines for the management of infective endocarditis: the Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Eur Heart J. 2015;36:3075-3128.
1. Baddour LM, Wilson WR, Bayer AS, et al; American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and Stroke Council. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation. 2015;132:1435-1486.
2. Crespo A, Retter AS, Lorber B. Group B streptococcal endocarditis in obstetric and gynecologic practice. Infect Dis Obstet Gynecol. 2003;11:109-115.
3. Murray S, Hickey JB, Houang E. Significant bacteremia associated with replacement of intrauterine contraceptive device. Am J Obstet Gynecol. 1987;156:698-700.
4. Everett ED, Reller LB, Droegemueller W, et al. Absence of bacteremia after insertion or removal of intrauterine devices. Obstet Gynecol. 1976;47:207-209.
5. Seaworth BJ, Durack DT. Infective endocarditis in obstetric and gynecologic practice. Am J Obstet Gynecol. 1986;154:180-188.
6. ACOG Committee on Practice Bulletins–Gynecology. Practice bulletin no. 186: Long-acting reversible contraception: implants and intrauterine devices. Obstet Gynecol. 2017;130:e251-e269.
7. Habib G, Lancellotti P, Antunes MJ, et al; ESC Scientific Document Group. 2015 ESC guidelines for the management of infective endocarditis: the Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Eur Heart J. 2015;36:3075-3128.

Worsening nausea, vomiting, and dizziness • 20-pound weight loss in 2 months • mild hearing loss • reoccurring episodes of falls • Dx?
THE CASE
A 26-year-old Hispanic/African American woman presented to our clinic with a 2-month history of nausea and vomiting, along with dizziness. The nausea and vomiting persistently worsened, and she was only able to tolerate apples and berries. During this 2-month period, she lost 20 pounds and her symptoms progressed to include pruritus, ataxia, and mild hearing loss, with reoccurring episodes of falls.
THE DIAGNOSIS
On examination, she was found to be bradycardic with a heart rate of 47 beats/min, right- axis deviation, and inverted T waves in leads I, II, and augmented vector left. Her family history included the death of an aunt who was in her early 30s due to an unknown heart condition.
Echocardiogram identified mild mitral valve regurgitation with an ejection fraction of 55% to 60% (reference range: 55%-70%). Cardiology determined that her bradycardia was not the source of her symptoms. A neurologic exam identified 3+ hyperreflexia (indicating the reflex was increased), tandem gait instability, and left oculomotor dysfunction.
Brain magnetic resonance imaging (MRI) identified bilateral parietal white matter lesions where a demyelinating process could not be excluded (FIGURE 1A). The patient’s symptoms of nausea and vomiting continued, and she only tolerated peanuts and liquids. An MRI of the spine was negative.

Laboratory testing revealed that the patient was negative for human immunodeficiency virus (HIV), syphilis, Lyme disease, and lupus. Her thyroid-stimulating hormone level was 1.7 mIU/L (reference range: 0.4-4.2 mIU/L), and her vitamin B12 level was 504 pg/mL (reference range: 160-950 pg/mL).
The patient’s lumbar puncture was negative for oligoclonal bands. The IgG synthesis rate/index cerebrospinal fluid (CSF) was –3.9, ruling out multiple sclerosis. Her CSF culture was negative, with a glucose level of 42 mg/dL (reference range: 70-110 mg/dL), colorless appearance, 1 white blood cell, and spinal albumin of 12.2 mg/dL (reference range: 8-42 mg/dL). The visual evoked potential was negative. The aquaporin-4 (AQP4) antibody was positive at 3.4 U/mL, and the myelin oligodendrocyte glycoprotein (MOG) antibody was positive.
Gastroenterology concluded a normal gastric accommodation and unremarkable computed tomography (CT) enterography. Moderate erosions were identified in the stomach with an erythematous gastropathy. The patient was placed on a proton pump inhibitor.
Continue to: Following the examination...
Following the examination and laboratory testing, the patient was admitted under our family medicine service for neuromyelitis optica (NMO) affecting the area postrema. NMO, also known as Devic’s disease, is an autoimmune disorder that affects the spinal cord and optic nerves. Autoantibodies against AQP4 are created in the periphery and are directed against astrocytes in the central nervous system. These antibodies bind to the foot processes of astrocytes, inducing complement-mediated cell damage and granulocyte infiltration.1-5
Intravenous methylprednisolone was initiated at 250 mg every 6 hours for 3 days. A repeat brain MRI demonstrated nonspecific multiple scattered foci of hyperintense signal involving the subcortical supratentorial white matter without abnormal enhancement, most likely representing nonactive demyelinating plaques (FIGURES 1B and 1C).
Dx is revisited. Our patient was referred to an NMO clinic for evaluation. After further testing (including a repeat MRI based on the neurologist’s specifications, anti-aquaporin antibody testing, and MOG-antibody testing) and case discussion, it was determined that the patient had MOG-antibody disease. This disease, along with NMO, comprise a spectrum of disorders referred to as neuromyelitis optica spectrum disorder (NMOSD).
The patient was subsequently prescribed a rituximab infusion, 500 mg/50 mL, to treat the current attack. One infusion was to be completed weekly for 2 weeks with plans to repeat treatment every 6 months to prevent flares of NMO. During the first dose, the patient had a reaction to the treatment, which caused pruritus and chest tightness. She was able to complete the infusion after being treated with diphenhydramine.
Tx continued. In order to complete the second of 2 infusions of rituximab, the patient was pretreated with oral methylprednisolone the night before the infusion, along with diphenhydramine and acetaminophen on the day of treatment. Fortunately, the patient tolerated the infusion well with no adverse effects or reactions.
Continue to: DISCUSSION
DISCUSSION
Within the NMO spectrum, the MOG antibody is positive in up to 42% of AQP4-seronegative cases.6 MOG is a minor myelin component that is expressed exclusively in the central nervous system on the surface of myelin and oligodendrocyte processes. The role of this glycoprotein is not well understood but is hypothesized to function as a cell surface receptor or cell adhesion molecule.7
Among a cohort of 252 patients from the United Kingdom who tested positive for the MOG-IgG1 antibody, optic neuritis was seen in 55%, while 18% experienced transverse myelitis, and 15% had a history of area postrema syndrome. A brain MRI identified lesions in all areas of the brain including the brain stem, cerebellum, and cerebral hemispheres.8
Risk factors for NMOSD include female gender, Asian and African ethnicities, Epstein Barr virus seropositivity, and tobacco abuse.
Differential diagnosis. Many diseases or conditions that are inflammatory, autoimmune, infectious, or neoplastic can involve the central nervous system and mimic the clinical and radiologic phenotypes of NMOSD-AQP4. They include lupus, SjÖgren’s syndrome, multiple sclerosis, sarcoidosis, acute disseminated encephalomyelitis, HIV, and vitamin B12 deficiency.
Treatment. The standard treatment is intravenous methylprednisolone, 1 g/d for 3 to 5 days followed by a steroid taper. Therapeutic plasma exchange is recommended for refractory cases and in patients with spinal cord demyelination.9-11 Rituximab is the first-line therapy for attack prevention12-15 in NMOSD broadly and may be effective in MOG antibody disease, as well. In an open-label study of patients with NMOSD treated with rituximab, 64% were relapse free at follow-up, which ranged from 12 to 67 months.13 In a long-term study of patients treated with rituximab, 87% maintained a reduced relapse rate and 93% had improvement or stability over a 5-year follow-up.14
Continue to: Our patient
Our patient. After her diagnosis of NMOSD/MOG-antibody disease, our patient’s symptoms progressed to include vertigo, vestibular ataxia, pruritus, left foot drop, lower extremity numbness, and decreased hearing. After the second rituximab infusion her symptoms continued, but over time stabilized and have not worsened. She currently receives gabapentin 300 mg every 8 hours, as needed, for extremity numbness (which has been working well) along with sertraline 100 mg/d for depression.
Subsequent office visits have showed no further weight loss. Based on the current response to the rituximab, her prognosis is undetermined by Neurology as they continue to monitor for progression.
THE TAKEAWAY
Vestibular ataxia, foot drop, pruritus, vertigo, decreased hearing, numbness, and oculomotor dysfunction in the presence of nausea and vomiting should raise suspicion for NMOSD. The presence of AQP4 antibodies along with demyelinating central nervous system lesions, is highly indicative of NMO. The presence of MOG antibodies may indicate NMOSD/MOG-antibody disease. The initial treatment of NMOSD is intravenous methylprednisolone, which can be followed by treatment with rituximab to achieve remission.
CORRESPONDENCE
Daniel Murphy, MD, FAAFP, Department of Family and Community Medicine, Texas Tech University Health Science Center El Paso, 9849 Kenworthy Street, El Paso, Texas 79924; [email protected]
1. Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69:2221-2231.
2. Ratelade J, Zhang H, Saadoun S, et al. Neuromyelitis optica IgG and natural killer cells Produce NMO lesions in mice without myelin loss. Acta Neuropathol. 2012;123:861-872.
3. Saadoun S, Waters P, Bell BA, et al. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349-361.
4. Takahashi T, Fujihara K, Nakashima I, et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titer. Brain. 2007;130:1235-1243.
5. Jarius S, Aboul-Enein F, Waters P, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain. 2008;131:3072-3080.
6. Hamid SHM, Whittam D, Mutch K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are Mog-IgG positive? A cross sectional study of 132 patients. J Neurol. 2017; 264:2088-2094.
7. Peschl P, Bradi M, Hoftberger R, et al. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. 2017;8:529.
8. Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140:3128-3138.
9. Sellner J, Boggild M, Clanet M, et al. EFNS Guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17:1019-1032.
10. Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79:206-216.
11. Watanabe S, Nakashima I, Misu T, et al. Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult Scler. 2007;13:128-132.
12. Collongues N, Brassat D, Maillart E, et al. Efficacy of rituximab in refractory neuromyelitis optica. Mult Scler. 2016;22:955-959.
13. Collongues N, de Seze J. An update on the evidence for the efficacy and safety of rituximab in the management of neuromyelitis optica. Ther Adv Neurol Disord. 2016;9:180-188.
14. Kim SH, Huh SY, Lee SJ, et al. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013;70:1110-1117.
15. Kim SH, Kim W, Li XF, et al. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol. 2011;68:1412-1420.
THE CASE
A 26-year-old Hispanic/African American woman presented to our clinic with a 2-month history of nausea and vomiting, along with dizziness. The nausea and vomiting persistently worsened, and she was only able to tolerate apples and berries. During this 2-month period, she lost 20 pounds and her symptoms progressed to include pruritus, ataxia, and mild hearing loss, with reoccurring episodes of falls.
THE DIAGNOSIS
On examination, she was found to be bradycardic with a heart rate of 47 beats/min, right- axis deviation, and inverted T waves in leads I, II, and augmented vector left. Her family history included the death of an aunt who was in her early 30s due to an unknown heart condition.
Echocardiogram identified mild mitral valve regurgitation with an ejection fraction of 55% to 60% (reference range: 55%-70%). Cardiology determined that her bradycardia was not the source of her symptoms. A neurologic exam identified 3+ hyperreflexia (indicating the reflex was increased), tandem gait instability, and left oculomotor dysfunction.
Brain magnetic resonance imaging (MRI) identified bilateral parietal white matter lesions where a demyelinating process could not be excluded (FIGURE 1A). The patient’s symptoms of nausea and vomiting continued, and she only tolerated peanuts and liquids. An MRI of the spine was negative.
Laboratory testing revealed that the patient was negative for human immunodeficiency virus (HIV), syphilis, Lyme disease, and lupus. Her thyroid-stimulating hormone level was 1.7 mIU/L (reference range: 0.4-4.2 mIU/L), and her vitamin B12 level was 504 pg/mL (reference range: 160-950 pg/mL).
The patient’s lumbar puncture was negative for oligoclonal bands. The IgG synthesis rate/index cerebrospinal fluid (CSF) was –3.9, ruling out multiple sclerosis. Her CSF culture was negative, with a glucose level of 42 mg/dL (reference range: 70-110 mg/dL), colorless appearance, 1 white blood cell, and spinal albumin of 12.2 mg/dL (reference range: 8-42 mg/dL). The visual evoked potential was negative. The aquaporin-4 (AQP4) antibody was positive at 3.4 U/mL, and the myelin oligodendrocyte glycoprotein (MOG) antibody was positive.
Gastroenterology concluded a normal gastric accommodation and unremarkable computed tomography (CT) enterography. Moderate erosions were identified in the stomach with an erythematous gastropathy. The patient was placed on a proton pump inhibitor.
Continue to: Following the examination...
Following the examination and laboratory testing, the patient was admitted under our family medicine service for neuromyelitis optica (NMO) affecting the area postrema. NMO, also known as Devic’s disease, is an autoimmune disorder that affects the spinal cord and optic nerves. Autoantibodies against AQP4 are created in the periphery and are directed against astrocytes in the central nervous system. These antibodies bind to the foot processes of astrocytes, inducing complement-mediated cell damage and granulocyte infiltration.1-5
Intravenous methylprednisolone was initiated at 250 mg every 6 hours for 3 days. A repeat brain MRI demonstrated nonspecific multiple scattered foci of hyperintense signal involving the subcortical supratentorial white matter without abnormal enhancement, most likely representing nonactive demyelinating plaques (FIGURES 1B and 1C).
Dx is revisited. Our patient was referred to an NMO clinic for evaluation. After further testing (including a repeat MRI based on the neurologist’s specifications, anti-aquaporin antibody testing, and MOG-antibody testing) and case discussion, it was determined that the patient had MOG-antibody disease. This disease, along with NMO, comprise a spectrum of disorders referred to as neuromyelitis optica spectrum disorder (NMOSD).
The patient was subsequently prescribed a rituximab infusion, 500 mg/50 mL, to treat the current attack. One infusion was to be completed weekly for 2 weeks with plans to repeat treatment every 6 months to prevent flares of NMO. During the first dose, the patient had a reaction to the treatment, which caused pruritus and chest tightness. She was able to complete the infusion after being treated with diphenhydramine.
Tx continued. In order to complete the second of 2 infusions of rituximab, the patient was pretreated with oral methylprednisolone the night before the infusion, along with diphenhydramine and acetaminophen on the day of treatment. Fortunately, the patient tolerated the infusion well with no adverse effects or reactions.
Continue to: DISCUSSION
DISCUSSION
Within the NMO spectrum, the MOG antibody is positive in up to 42% of AQP4-seronegative cases.6 MOG is a minor myelin component that is expressed exclusively in the central nervous system on the surface of myelin and oligodendrocyte processes. The role of this glycoprotein is not well understood but is hypothesized to function as a cell surface receptor or cell adhesion molecule.7
Among a cohort of 252 patients from the United Kingdom who tested positive for the MOG-IgG1 antibody, optic neuritis was seen in 55%, while 18% experienced transverse myelitis, and 15% had a history of area postrema syndrome. A brain MRI identified lesions in all areas of the brain including the brain stem, cerebellum, and cerebral hemispheres.8
Risk factors for NMOSD include female gender, Asian and African ethnicities, Epstein Barr virus seropositivity, and tobacco abuse.
Differential diagnosis. Many diseases or conditions that are inflammatory, autoimmune, infectious, or neoplastic can involve the central nervous system and mimic the clinical and radiologic phenotypes of NMOSD-AQP4. They include lupus, SjÖgren’s syndrome, multiple sclerosis, sarcoidosis, acute disseminated encephalomyelitis, HIV, and vitamin B12 deficiency.
Treatment. The standard treatment is intravenous methylprednisolone, 1 g/d for 3 to 5 days followed by a steroid taper. Therapeutic plasma exchange is recommended for refractory cases and in patients with spinal cord demyelination.9-11 Rituximab is the first-line therapy for attack prevention12-15 in NMOSD broadly and may be effective in MOG antibody disease, as well. In an open-label study of patients with NMOSD treated with rituximab, 64% were relapse free at follow-up, which ranged from 12 to 67 months.13 In a long-term study of patients treated with rituximab, 87% maintained a reduced relapse rate and 93% had improvement or stability over a 5-year follow-up.14
Continue to: Our patient
Our patient. After her diagnosis of NMOSD/MOG-antibody disease, our patient’s symptoms progressed to include vertigo, vestibular ataxia, pruritus, left foot drop, lower extremity numbness, and decreased hearing. After the second rituximab infusion her symptoms continued, but over time stabilized and have not worsened. She currently receives gabapentin 300 mg every 8 hours, as needed, for extremity numbness (which has been working well) along with sertraline 100 mg/d for depression.
Subsequent office visits have showed no further weight loss. Based on the current response to the rituximab, her prognosis is undetermined by Neurology as they continue to monitor for progression.
THE TAKEAWAY
Vestibular ataxia, foot drop, pruritus, vertigo, decreased hearing, numbness, and oculomotor dysfunction in the presence of nausea and vomiting should raise suspicion for NMOSD. The presence of AQP4 antibodies along with demyelinating central nervous system lesions, is highly indicative of NMO. The presence of MOG antibodies may indicate NMOSD/MOG-antibody disease. The initial treatment of NMOSD is intravenous methylprednisolone, which can be followed by treatment with rituximab to achieve remission.
CORRESPONDENCE
Daniel Murphy, MD, FAAFP, Department of Family and Community Medicine, Texas Tech University Health Science Center El Paso, 9849 Kenworthy Street, El Paso, Texas 79924; [email protected]
THE CASE
A 26-year-old Hispanic/African American woman presented to our clinic with a 2-month history of nausea and vomiting, along with dizziness. The nausea and vomiting persistently worsened, and she was only able to tolerate apples and berries. During this 2-month period, she lost 20 pounds and her symptoms progressed to include pruritus, ataxia, and mild hearing loss, with reoccurring episodes of falls.
THE DIAGNOSIS
On examination, she was found to be bradycardic with a heart rate of 47 beats/min, right- axis deviation, and inverted T waves in leads I, II, and augmented vector left. Her family history included the death of an aunt who was in her early 30s due to an unknown heart condition.
Echocardiogram identified mild mitral valve regurgitation with an ejection fraction of 55% to 60% (reference range: 55%-70%). Cardiology determined that her bradycardia was not the source of her symptoms. A neurologic exam identified 3+ hyperreflexia (indicating the reflex was increased), tandem gait instability, and left oculomotor dysfunction.
Brain magnetic resonance imaging (MRI) identified bilateral parietal white matter lesions where a demyelinating process could not be excluded (FIGURE 1A). The patient’s symptoms of nausea and vomiting continued, and she only tolerated peanuts and liquids. An MRI of the spine was negative.
Laboratory testing revealed that the patient was negative for human immunodeficiency virus (HIV), syphilis, Lyme disease, and lupus. Her thyroid-stimulating hormone level was 1.7 mIU/L (reference range: 0.4-4.2 mIU/L), and her vitamin B12 level was 504 pg/mL (reference range: 160-950 pg/mL).
The patient’s lumbar puncture was negative for oligoclonal bands. The IgG synthesis rate/index cerebrospinal fluid (CSF) was –3.9, ruling out multiple sclerosis. Her CSF culture was negative, with a glucose level of 42 mg/dL (reference range: 70-110 mg/dL), colorless appearance, 1 white blood cell, and spinal albumin of 12.2 mg/dL (reference range: 8-42 mg/dL). The visual evoked potential was negative. The aquaporin-4 (AQP4) antibody was positive at 3.4 U/mL, and the myelin oligodendrocyte glycoprotein (MOG) antibody was positive.
Gastroenterology concluded a normal gastric accommodation and unremarkable computed tomography (CT) enterography. Moderate erosions were identified in the stomach with an erythematous gastropathy. The patient was placed on a proton pump inhibitor.
Continue to: Following the examination...
Following the examination and laboratory testing, the patient was admitted under our family medicine service for neuromyelitis optica (NMO) affecting the area postrema. NMO, also known as Devic’s disease, is an autoimmune disorder that affects the spinal cord and optic nerves. Autoantibodies against AQP4 are created in the periphery and are directed against astrocytes in the central nervous system. These antibodies bind to the foot processes of astrocytes, inducing complement-mediated cell damage and granulocyte infiltration.1-5
Intravenous methylprednisolone was initiated at 250 mg every 6 hours for 3 days. A repeat brain MRI demonstrated nonspecific multiple scattered foci of hyperintense signal involving the subcortical supratentorial white matter without abnormal enhancement, most likely representing nonactive demyelinating plaques (FIGURES 1B and 1C).
Dx is revisited. Our patient was referred to an NMO clinic for evaluation. After further testing (including a repeat MRI based on the neurologist’s specifications, anti-aquaporin antibody testing, and MOG-antibody testing) and case discussion, it was determined that the patient had MOG-antibody disease. This disease, along with NMO, comprise a spectrum of disorders referred to as neuromyelitis optica spectrum disorder (NMOSD).
The patient was subsequently prescribed a rituximab infusion, 500 mg/50 mL, to treat the current attack. One infusion was to be completed weekly for 2 weeks with plans to repeat treatment every 6 months to prevent flares of NMO. During the first dose, the patient had a reaction to the treatment, which caused pruritus and chest tightness. She was able to complete the infusion after being treated with diphenhydramine.
Tx continued. In order to complete the second of 2 infusions of rituximab, the patient was pretreated with oral methylprednisolone the night before the infusion, along with diphenhydramine and acetaminophen on the day of treatment. Fortunately, the patient tolerated the infusion well with no adverse effects or reactions.
Continue to: DISCUSSION
DISCUSSION
Within the NMO spectrum, the MOG antibody is positive in up to 42% of AQP4-seronegative cases.6 MOG is a minor myelin component that is expressed exclusively in the central nervous system on the surface of myelin and oligodendrocyte processes. The role of this glycoprotein is not well understood but is hypothesized to function as a cell surface receptor or cell adhesion molecule.7
Among a cohort of 252 patients from the United Kingdom who tested positive for the MOG-IgG1 antibody, optic neuritis was seen in 55%, while 18% experienced transverse myelitis, and 15% had a history of area postrema syndrome. A brain MRI identified lesions in all areas of the brain including the brain stem, cerebellum, and cerebral hemispheres.8
Risk factors for NMOSD include female gender, Asian and African ethnicities, Epstein Barr virus seropositivity, and tobacco abuse.
Differential diagnosis. Many diseases or conditions that are inflammatory, autoimmune, infectious, or neoplastic can involve the central nervous system and mimic the clinical and radiologic phenotypes of NMOSD-AQP4. They include lupus, SjÖgren’s syndrome, multiple sclerosis, sarcoidosis, acute disseminated encephalomyelitis, HIV, and vitamin B12 deficiency.
Treatment. The standard treatment is intravenous methylprednisolone, 1 g/d for 3 to 5 days followed by a steroid taper. Therapeutic plasma exchange is recommended for refractory cases and in patients with spinal cord demyelination.9-11 Rituximab is the first-line therapy for attack prevention12-15 in NMOSD broadly and may be effective in MOG antibody disease, as well. In an open-label study of patients with NMOSD treated with rituximab, 64% were relapse free at follow-up, which ranged from 12 to 67 months.13 In a long-term study of patients treated with rituximab, 87% maintained a reduced relapse rate and 93% had improvement or stability over a 5-year follow-up.14
Continue to: Our patient
Our patient. After her diagnosis of NMOSD/MOG-antibody disease, our patient’s symptoms progressed to include vertigo, vestibular ataxia, pruritus, left foot drop, lower extremity numbness, and decreased hearing. After the second rituximab infusion her symptoms continued, but over time stabilized and have not worsened. She currently receives gabapentin 300 mg every 8 hours, as needed, for extremity numbness (which has been working well) along with sertraline 100 mg/d for depression.
Subsequent office visits have showed no further weight loss. Based on the current response to the rituximab, her prognosis is undetermined by Neurology as they continue to monitor for progression.
THE TAKEAWAY
Vestibular ataxia, foot drop, pruritus, vertigo, decreased hearing, numbness, and oculomotor dysfunction in the presence of nausea and vomiting should raise suspicion for NMOSD. The presence of AQP4 antibodies along with demyelinating central nervous system lesions, is highly indicative of NMO. The presence of MOG antibodies may indicate NMOSD/MOG-antibody disease. The initial treatment of NMOSD is intravenous methylprednisolone, which can be followed by treatment with rituximab to achieve remission.
CORRESPONDENCE
Daniel Murphy, MD, FAAFP, Department of Family and Community Medicine, Texas Tech University Health Science Center El Paso, 9849 Kenworthy Street, El Paso, Texas 79924; [email protected]
1. Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69:2221-2231.
2. Ratelade J, Zhang H, Saadoun S, et al. Neuromyelitis optica IgG and natural killer cells Produce NMO lesions in mice without myelin loss. Acta Neuropathol. 2012;123:861-872.
3. Saadoun S, Waters P, Bell BA, et al. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349-361.
4. Takahashi T, Fujihara K, Nakashima I, et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titer. Brain. 2007;130:1235-1243.
5. Jarius S, Aboul-Enein F, Waters P, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain. 2008;131:3072-3080.
6. Hamid SHM, Whittam D, Mutch K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are Mog-IgG positive? A cross sectional study of 132 patients. J Neurol. 2017; 264:2088-2094.
7. Peschl P, Bradi M, Hoftberger R, et al. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. 2017;8:529.
8. Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140:3128-3138.
9. Sellner J, Boggild M, Clanet M, et al. EFNS Guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17:1019-1032.
10. Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79:206-216.
11. Watanabe S, Nakashima I, Misu T, et al. Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult Scler. 2007;13:128-132.
12. Collongues N, Brassat D, Maillart E, et al. Efficacy of rituximab in refractory neuromyelitis optica. Mult Scler. 2016;22:955-959.
13. Collongues N, de Seze J. An update on the evidence for the efficacy and safety of rituximab in the management of neuromyelitis optica. Ther Adv Neurol Disord. 2016;9:180-188.
14. Kim SH, Huh SY, Lee SJ, et al. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013;70:1110-1117.
15. Kim SH, Kim W, Li XF, et al. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol. 2011;68:1412-1420.
1. Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69:2221-2231.
2. Ratelade J, Zhang H, Saadoun S, et al. Neuromyelitis optica IgG and natural killer cells Produce NMO lesions in mice without myelin loss. Acta Neuropathol. 2012;123:861-872.
3. Saadoun S, Waters P, Bell BA, et al. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349-361.
4. Takahashi T, Fujihara K, Nakashima I, et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titer. Brain. 2007;130:1235-1243.
5. Jarius S, Aboul-Enein F, Waters P, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain. 2008;131:3072-3080.
6. Hamid SHM, Whittam D, Mutch K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are Mog-IgG positive? A cross sectional study of 132 patients. J Neurol. 2017; 264:2088-2094.
7. Peschl P, Bradi M, Hoftberger R, et al. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. 2017;8:529.
8. Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140:3128-3138.
9. Sellner J, Boggild M, Clanet M, et al. EFNS Guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17:1019-1032.
10. Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79:206-216.
11. Watanabe S, Nakashima I, Misu T, et al. Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult Scler. 2007;13:128-132.
12. Collongues N, Brassat D, Maillart E, et al. Efficacy of rituximab in refractory neuromyelitis optica. Mult Scler. 2016;22:955-959.
13. Collongues N, de Seze J. An update on the evidence for the efficacy and safety of rituximab in the management of neuromyelitis optica. Ther Adv Neurol Disord. 2016;9:180-188.
14. Kim SH, Huh SY, Lee SJ, et al. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013;70:1110-1117.
15. Kim SH, Kim W, Li XF, et al. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol. 2011;68:1412-1420.
