User login
Coordination of Care in Breast Cancer Survivors: An Overview
Volume 9, Issue 6, November-December 2011, Pages 210-215
| doi:10.1016/j.suponc.2011.06.008 | How to Cite or Link Using DOI |
| Permissions & Reprints |
How We Do It

TO READ THE ENTIRE ARTICLE, CLICK ON THE ADJACENT LINK TO THE PDF FILE
Abstract
The number of breast cancer survivors in the United States is increasing. With longer survival, there has been an increase in the complexity and duration of posttreatment care. Multidisciplinary care teams are needed to participate across the broad spectrum of issues that breast cancer survivors face. In this setting, the need for well-established patterns of communication between care providers is increasingly apparent. We have created a multidisciplinary approach to the management of breast cancer survivors to improve communication and education between providers and patients. This approach could be extended to the care and management of survivors of other types of cancer.
Case
Vitae
Dr. Peairs is from the Johns Hopkins School of Medicine, Baltimore, Maryland. |
Dr. Wolff is from the Johns Hopkins School of Medicine, Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, Maryland. |
Dr. Olsenis from the Johns Hopkins School of Nursing, Baltimore, Maryland. |
Dr. Bantugis from the Johns Hopkins School of Medicine, Baltimore, Maryland. |
Dr. Shockney is from the Johns Hopkins School of Medicine, Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, Maryland. |
Dr. Kantsiper is from the Johns Hopkins School of Medicine, Baltimore, Maryland. |
Dr. Carrino-Tamasi is from the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, Maryland. |
Dr. Snyder is from the Johns Hopkins School of Medicine, Baltimore, Maryland. |
Volume 9, Issue 6, November-December 2011, Pages 210-215
Volume 9, Issue 6, November-December 2011, Pages 210-215
| doi:10.1016/j.suponc.2011.06.008 | How to Cite or Link Using DOI |
| Permissions & Reprints |
How We Do It
TO READ THE ENTIRE ARTICLE, CLICK ON THE ADJACENT LINK TO THE PDF FILE
Abstract
The number of breast cancer survivors in the United States is increasing. With longer survival, there has been an increase in the complexity and duration of posttreatment care. Multidisciplinary care teams are needed to participate across the broad spectrum of issues that breast cancer survivors face. In this setting, the need for well-established patterns of communication between care providers is increasingly apparent. We have created a multidisciplinary approach to the management of breast cancer survivors to improve communication and education between providers and patients. This approach could be extended to the care and management of survivors of other types of cancer.
Case
Vitae
Dr. Peairs is from the Johns Hopkins School of Medicine, Baltimore, Maryland. |
Dr. Wolff is from the Johns Hopkins School of Medicine, Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, Maryland. |
Dr. Olsenis from the Johns Hopkins School of Nursing, Baltimore, Maryland. |
Dr. Bantugis from the Johns Hopkins School of Medicine, Baltimore, Maryland. |
Dr. Shockney is from the Johns Hopkins School of Medicine, Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, Maryland. |
Dr. Kantsiper is from the Johns Hopkins School of Medicine, Baltimore, Maryland. |
Dr. Carrino-Tamasi is from the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, Maryland. |
Dr. Snyder is from the Johns Hopkins School of Medicine, Baltimore, Maryland. |
Volume 9, Issue 6, November-December 2011, Pages 210-215
Volume 9, Issue 6, November-December 2011, Pages 210-215
| doi:10.1016/j.suponc.2011.06.008 | How to Cite or Link Using DOI |
| Permissions & Reprints |
How We Do It
TO READ THE ENTIRE ARTICLE, CLICK ON THE ADJACENT LINK TO THE PDF FILE
Abstract
The number of breast cancer survivors in the United States is increasing. With longer survival, there has been an increase in the complexity and duration of posttreatment care. Multidisciplinary care teams are needed to participate across the broad spectrum of issues that breast cancer survivors face. In this setting, the need for well-established patterns of communication between care providers is increasingly apparent. We have created a multidisciplinary approach to the management of breast cancer survivors to improve communication and education between providers and patients. This approach could be extended to the care and management of survivors of other types of cancer.
Case
Vitae
Dr. Peairs is from the Johns Hopkins School of Medicine, Baltimore, Maryland. |
Dr. Wolff is from the Johns Hopkins School of Medicine, Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, Maryland. |
Dr. Olsenis from the Johns Hopkins School of Nursing, Baltimore, Maryland. |
Dr. Bantugis from the Johns Hopkins School of Medicine, Baltimore, Maryland. |
Dr. Shockney is from the Johns Hopkins School of Medicine, Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, Maryland. |
Dr. Kantsiper is from the Johns Hopkins School of Medicine, Baltimore, Maryland. |
Dr. Carrino-Tamasi is from the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, Maryland. |
Dr. Snyder is from the Johns Hopkins School of Medicine, Baltimore, Maryland. |
Volume 9, Issue 6, November-December 2011, Pages 210-215
UPDATED: Vytorin Gets FDA Panel Nod for Some CKD Patients
[UPDATED]SILVER SPRING, MD. – Vytorin should be approved for preventing major vascular events in patients who have chronic kidney disease but are not on dialysis, according to a unanimous vote by a Food and Drug Administration advisory panel.
The panel also voted 10-6, however, that the safety and effectiveness data did not support approval of the combination of 10 mg of ezetimibe with 20 mg of simvastatin for the same indication in patients with end-stage renal disease who are on dialysis.
Their votes at the Nov. 2 meeting were based on the results of the Study of Heart and Renal Protection (SHARP), which evaluated the effects of reducing LDL cholesterol on the risk of coronary vascular disease in patients with chronic kidney disease who are at an increased risk of cardiovascular morbidity and mortality. About two-thirds of the patients in the trial were not on dialysis at baseline, and in these patients, there was a 23% reduction in the primary end point – the risk of a major vascular event (nonfatal MI or cardiac death, stroke, or a revascularization procedure that excluded dialysis access-related procedures) – compared with those on placebo over a mean of 5 years (Lancet 2011;377:2181-92).
But in the patients who were on dialysis at baseline, the risk was reduced by about 6% over placebo. Panelists who did not support approval in this group cited the lower degree of effectiveness, and the fact that end stage renal disease patients on dialysis are very different from predialysis patients – and that patients with ESRD in the United States are different than those elsewhere. They noted that only 4% of the patients in SHARP were in the United States, and management and outcomes of patients with ESRD are different in the United States than in other parts of the world.

Dr. Lamont Weide, chief of diabetes and endocrinology, at the University of Missouri, Kansas City, backed approval for predialysis patients, but not for those on dialysis. Like several other panelists, he also considered the results of two previous large studies of about 4,000 CKD patients on dialysis, which found no significant beneficial effects of treatment with other statins on cardiovascular outcomes. While these were three different studies with different agents and different entry criteria, he said that the results go in the same direction as SHARP, in a large group of patients "without any clear indication of benefit." Those studies were the 4D study (N. Engl. J. Med. 2005;353:238-48) and the AURORA study (N. Engl J. Med. 2009;360:1395-407).
Also splitting her votes, Dr. Julia Lewis, professor of medicine in the department of nephrology, Vanderbilt University, Nashville, referred to those two trials and added that she considers her dialysis patients considerably different than her clinic patients who are not on dialysis. She commended the SHARP study and investigators for "providing what I think is going to be a wonderful addition to the care of CKD patients. It is going to change care of CKD patients and prevent the bad [cardiovascular] outcomes that affect them," she said.
In the study overall, the risk of major vascular events was reduced by 16% among those treated with the combination as compared to those on placebo, which was primarily driven by a reduction in the revascularization component. Cancers and all-cause mortality were similar in treated patients and those on placebo, and the panel agreed there were no new safety concerns at the dose studied. (About one-quarter of those enrolled died during the study.)
Merck, the manufacturer of ezetimibe (Zetia) and Vytorin, filed for approval of the claim that 10 mg of ezetimibe plus 20 mg of simvastatin (in the fixed-dose combination pill or taken separately) reduces the risk of major cardiovascular events in patients with chronic kidney disease on the basis of the SHARP results. SHARP was funded by Merck and Schering-Plough, but was independently conducted by the Oxford (England) University Clinical Trials Service Unit. Ezetimibe was used to make it possible to use a lower dose of simvastatin in the CKD patients, who are at a greater risk of myopathy and other adverse effects of statins.
Panelists emphasized that the 10 mg of ezetimibe with the 20-mg dose of simvastatin was the dose combination studied and was shown to be safe, and that doses should not be increased in this population of patients.

The two drugs are only approved for lipid-lowering indications: Ezetimibe, a selective inhibitor of the absorption of intestinal cholesterol and related phytosterol marketed as Zetia, was approved by the FDA in 2002; Vytorin, a combination of ezetimibe and the HMG-CoA reductase inhibitor simvastatin was approved in 2004; simvastatin was approved in 1991.
The mean age of the patients in the SHARP trial was 61 years; they did not have a history of MI or coronary revascularization.
The six panelists who voted in favor of approval for patient not yet on dialysis and those on dialysis included Dr. William Hiatt, professor of medicine, division of cardiology, at the University of Colorado, Denver, voted in favor of approval for both populations, based on the overall trial results. The differences between the two populations could be explained in the Vytorin label, and "the consistency between the vascular and atherosclerotic events led me to believe that the overall trial had integrity and the various components of the primary outcome were relatively consistent," he said. "To deny an indication to extend to patients who are on chronic hemodialysis ... would not respect the totality of the data," he added.
Dr. Lawrence Hunsicker, professor of medicine in the nephrology division, and emeritus medical director of organ transplantation, University of Iowa, Iowa City, also voted in favor of approval for both groups, but added that the FDA should ensure that advertising and detailing of the product should reflect that "the data are far more clear for patients not on dialysis."
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of disclosures, although in some cases, they are given a waiver-but not at this meeting.
[UPDATED]SILVER SPRING, MD. – Vytorin should be approved for preventing major vascular events in patients who have chronic kidney disease but are not on dialysis, according to a unanimous vote by a Food and Drug Administration advisory panel.
The panel also voted 10-6, however, that the safety and effectiveness data did not support approval of the combination of 10 mg of ezetimibe with 20 mg of simvastatin for the same indication in patients with end-stage renal disease who are on dialysis.
Their votes at the Nov. 2 meeting were based on the results of the Study of Heart and Renal Protection (SHARP), which evaluated the effects of reducing LDL cholesterol on the risk of coronary vascular disease in patients with chronic kidney disease who are at an increased risk of cardiovascular morbidity and mortality. About two-thirds of the patients in the trial were not on dialysis at baseline, and in these patients, there was a 23% reduction in the primary end point – the risk of a major vascular event (nonfatal MI or cardiac death, stroke, or a revascularization procedure that excluded dialysis access-related procedures) – compared with those on placebo over a mean of 5 years (Lancet 2011;377:2181-92).
But in the patients who were on dialysis at baseline, the risk was reduced by about 6% over placebo. Panelists who did not support approval in this group cited the lower degree of effectiveness, and the fact that end stage renal disease patients on dialysis are very different from predialysis patients – and that patients with ESRD in the United States are different than those elsewhere. They noted that only 4% of the patients in SHARP were in the United States, and management and outcomes of patients with ESRD are different in the United States than in other parts of the world.
Dr. Lamont Weide, chief of diabetes and endocrinology, at the University of Missouri, Kansas City, backed approval for predialysis patients, but not for those on dialysis. Like several other panelists, he also considered the results of two previous large studies of about 4,000 CKD patients on dialysis, which found no significant beneficial effects of treatment with other statins on cardiovascular outcomes. While these were three different studies with different agents and different entry criteria, he said that the results go in the same direction as SHARP, in a large group of patients "without any clear indication of benefit." Those studies were the 4D study (N. Engl. J. Med. 2005;353:238-48) and the AURORA study (N. Engl J. Med. 2009;360:1395-407).
Also splitting her votes, Dr. Julia Lewis, professor of medicine in the department of nephrology, Vanderbilt University, Nashville, referred to those two trials and added that she considers her dialysis patients considerably different than her clinic patients who are not on dialysis. She commended the SHARP study and investigators for "providing what I think is going to be a wonderful addition to the care of CKD patients. It is going to change care of CKD patients and prevent the bad [cardiovascular] outcomes that affect them," she said.
In the study overall, the risk of major vascular events was reduced by 16% among those treated with the combination as compared to those on placebo, which was primarily driven by a reduction in the revascularization component. Cancers and all-cause mortality were similar in treated patients and those on placebo, and the panel agreed there were no new safety concerns at the dose studied. (About one-quarter of those enrolled died during the study.)
Merck, the manufacturer of ezetimibe (Zetia) and Vytorin, filed for approval of the claim that 10 mg of ezetimibe plus 20 mg of simvastatin (in the fixed-dose combination pill or taken separately) reduces the risk of major cardiovascular events in patients with chronic kidney disease on the basis of the SHARP results. SHARP was funded by Merck and Schering-Plough, but was independently conducted by the Oxford (England) University Clinical Trials Service Unit. Ezetimibe was used to make it possible to use a lower dose of simvastatin in the CKD patients, who are at a greater risk of myopathy and other adverse effects of statins.
Panelists emphasized that the 10 mg of ezetimibe with the 20-mg dose of simvastatin was the dose combination studied and was shown to be safe, and that doses should not be increased in this population of patients.
The two drugs are only approved for lipid-lowering indications: Ezetimibe, a selective inhibitor of the absorption of intestinal cholesterol and related phytosterol marketed as Zetia, was approved by the FDA in 2002; Vytorin, a combination of ezetimibe and the HMG-CoA reductase inhibitor simvastatin was approved in 2004; simvastatin was approved in 1991.
The mean age of the patients in the SHARP trial was 61 years; they did not have a history of MI or coronary revascularization.
The six panelists who voted in favor of approval for patient not yet on dialysis and those on dialysis included Dr. William Hiatt, professor of medicine, division of cardiology, at the University of Colorado, Denver, voted in favor of approval for both populations, based on the overall trial results. The differences between the two populations could be explained in the Vytorin label, and "the consistency between the vascular and atherosclerotic events led me to believe that the overall trial had integrity and the various components of the primary outcome were relatively consistent," he said. "To deny an indication to extend to patients who are on chronic hemodialysis ... would not respect the totality of the data," he added.
Dr. Lawrence Hunsicker, professor of medicine in the nephrology division, and emeritus medical director of organ transplantation, University of Iowa, Iowa City, also voted in favor of approval for both groups, but added that the FDA should ensure that advertising and detailing of the product should reflect that "the data are far more clear for patients not on dialysis."
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of disclosures, although in some cases, they are given a waiver-but not at this meeting.
[UPDATED]SILVER SPRING, MD. – Vytorin should be approved for preventing major vascular events in patients who have chronic kidney disease but are not on dialysis, according to a unanimous vote by a Food and Drug Administration advisory panel.
The panel also voted 10-6, however, that the safety and effectiveness data did not support approval of the combination of 10 mg of ezetimibe with 20 mg of simvastatin for the same indication in patients with end-stage renal disease who are on dialysis.
Their votes at the Nov. 2 meeting were based on the results of the Study of Heart and Renal Protection (SHARP), which evaluated the effects of reducing LDL cholesterol on the risk of coronary vascular disease in patients with chronic kidney disease who are at an increased risk of cardiovascular morbidity and mortality. About two-thirds of the patients in the trial were not on dialysis at baseline, and in these patients, there was a 23% reduction in the primary end point – the risk of a major vascular event (nonfatal MI or cardiac death, stroke, or a revascularization procedure that excluded dialysis access-related procedures) – compared with those on placebo over a mean of 5 years (Lancet 2011;377:2181-92).
But in the patients who were on dialysis at baseline, the risk was reduced by about 6% over placebo. Panelists who did not support approval in this group cited the lower degree of effectiveness, and the fact that end stage renal disease patients on dialysis are very different from predialysis patients – and that patients with ESRD in the United States are different than those elsewhere. They noted that only 4% of the patients in SHARP were in the United States, and management and outcomes of patients with ESRD are different in the United States than in other parts of the world.
Dr. Lamont Weide, chief of diabetes and endocrinology, at the University of Missouri, Kansas City, backed approval for predialysis patients, but not for those on dialysis. Like several other panelists, he also considered the results of two previous large studies of about 4,000 CKD patients on dialysis, which found no significant beneficial effects of treatment with other statins on cardiovascular outcomes. While these were three different studies with different agents and different entry criteria, he said that the results go in the same direction as SHARP, in a large group of patients "without any clear indication of benefit." Those studies were the 4D study (N. Engl. J. Med. 2005;353:238-48) and the AURORA study (N. Engl J. Med. 2009;360:1395-407).
Also splitting her votes, Dr. Julia Lewis, professor of medicine in the department of nephrology, Vanderbilt University, Nashville, referred to those two trials and added that she considers her dialysis patients considerably different than her clinic patients who are not on dialysis. She commended the SHARP study and investigators for "providing what I think is going to be a wonderful addition to the care of CKD patients. It is going to change care of CKD patients and prevent the bad [cardiovascular] outcomes that affect them," she said.
In the study overall, the risk of major vascular events was reduced by 16% among those treated with the combination as compared to those on placebo, which was primarily driven by a reduction in the revascularization component. Cancers and all-cause mortality were similar in treated patients and those on placebo, and the panel agreed there were no new safety concerns at the dose studied. (About one-quarter of those enrolled died during the study.)
Merck, the manufacturer of ezetimibe (Zetia) and Vytorin, filed for approval of the claim that 10 mg of ezetimibe plus 20 mg of simvastatin (in the fixed-dose combination pill or taken separately) reduces the risk of major cardiovascular events in patients with chronic kidney disease on the basis of the SHARP results. SHARP was funded by Merck and Schering-Plough, but was independently conducted by the Oxford (England) University Clinical Trials Service Unit. Ezetimibe was used to make it possible to use a lower dose of simvastatin in the CKD patients, who are at a greater risk of myopathy and other adverse effects of statins.
Panelists emphasized that the 10 mg of ezetimibe with the 20-mg dose of simvastatin was the dose combination studied and was shown to be safe, and that doses should not be increased in this population of patients.
The two drugs are only approved for lipid-lowering indications: Ezetimibe, a selective inhibitor of the absorption of intestinal cholesterol and related phytosterol marketed as Zetia, was approved by the FDA in 2002; Vytorin, a combination of ezetimibe and the HMG-CoA reductase inhibitor simvastatin was approved in 2004; simvastatin was approved in 1991.
The mean age of the patients in the SHARP trial was 61 years; they did not have a history of MI or coronary revascularization.
The six panelists who voted in favor of approval for patient not yet on dialysis and those on dialysis included Dr. William Hiatt, professor of medicine, division of cardiology, at the University of Colorado, Denver, voted in favor of approval for both populations, based on the overall trial results. The differences between the two populations could be explained in the Vytorin label, and "the consistency between the vascular and atherosclerotic events led me to believe that the overall trial had integrity and the various components of the primary outcome were relatively consistent," he said. "To deny an indication to extend to patients who are on chronic hemodialysis ... would not respect the totality of the data," he added.
Dr. Lawrence Hunsicker, professor of medicine in the nephrology division, and emeritus medical director of organ transplantation, University of Iowa, Iowa City, also voted in favor of approval for both groups, but added that the FDA should ensure that advertising and detailing of the product should reflect that "the data are far more clear for patients not on dialysis."
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of disclosures, although in some cases, they are given a waiver-but not at this meeting.
FROM A MEETING OF THE FDA’S ENDOCRINOLOGIC AND METABOLIC DRUGS ADVISORY PANEL
Wasted Effort?
A research team reports in the Annals of Internal Medicine (2011;155:520-528) that of the 43 recent English-language studies of care-transition strategies they reviewed, none was associated with a reduced risk for 30-day rehospitalization.
The team, from Northwestern Feinberg School of Medicine in Chicago, defined 12 distinct activities done before, after, and during hospital discharge to reduce readmissions. The activities might be familiar to hospitalists who follow this subject, such as medication reconciliation, scheduling of follow-up appointments before discharge, placing follow-up phone calls, and the use of transitions coaches.
As with many such reviews, the Annals article leaves open the question of whether this negative finding reflects limitations in the research literature, "or does it reflect an absolute truth about care-transitions strategies?" says lead author Luke Hansen, MD, MHS. "So you have to make inferences. But we clearly don't have a strong research base."
The study is timely, as many HM groups are preparing for a Medicare policy to start in October 2012 that would penalize hospitals with higher-than-expected readmission rates.
"Hospitals have to change, but unfortunately they'll have to do it without a lot of evidence," Dr. Hansen says. "You probably will have to bundle several strategies together, and the more components you include, the more likely you are to achieve the needed cultural change."
A research team reports in the Annals of Internal Medicine (2011;155:520-528) that of the 43 recent English-language studies of care-transition strategies they reviewed, none was associated with a reduced risk for 30-day rehospitalization.
The team, from Northwestern Feinberg School of Medicine in Chicago, defined 12 distinct activities done before, after, and during hospital discharge to reduce readmissions. The activities might be familiar to hospitalists who follow this subject, such as medication reconciliation, scheduling of follow-up appointments before discharge, placing follow-up phone calls, and the use of transitions coaches.
As with many such reviews, the Annals article leaves open the question of whether this negative finding reflects limitations in the research literature, "or does it reflect an absolute truth about care-transitions strategies?" says lead author Luke Hansen, MD, MHS. "So you have to make inferences. But we clearly don't have a strong research base."
The study is timely, as many HM groups are preparing for a Medicare policy to start in October 2012 that would penalize hospitals with higher-than-expected readmission rates.
"Hospitals have to change, but unfortunately they'll have to do it without a lot of evidence," Dr. Hansen says. "You probably will have to bundle several strategies together, and the more components you include, the more likely you are to achieve the needed cultural change."
A research team reports in the Annals of Internal Medicine (2011;155:520-528) that of the 43 recent English-language studies of care-transition strategies they reviewed, none was associated with a reduced risk for 30-day rehospitalization.
The team, from Northwestern Feinberg School of Medicine in Chicago, defined 12 distinct activities done before, after, and during hospital discharge to reduce readmissions. The activities might be familiar to hospitalists who follow this subject, such as medication reconciliation, scheduling of follow-up appointments before discharge, placing follow-up phone calls, and the use of transitions coaches.
As with many such reviews, the Annals article leaves open the question of whether this negative finding reflects limitations in the research literature, "or does it reflect an absolute truth about care-transitions strategies?" says lead author Luke Hansen, MD, MHS. "So you have to make inferences. But we clearly don't have a strong research base."
The study is timely, as many HM groups are preparing for a Medicare policy to start in October 2012 that would penalize hospitals with higher-than-expected readmission rates.
"Hospitals have to change, but unfortunately they'll have to do it without a lot of evidence," Dr. Hansen says. "You probably will have to bundle several strategies together, and the more components you include, the more likely you are to achieve the needed cultural change."
In the Literature: Research You Need to Know
Clinical question: When do venous thromboembolism (VTE) events occur after cancer surgery?
Background: Cancer is a known risk factor for VTE. Prophylaxis for VTE after cancer surgery is commonly stopped at the time of hospital discharge despite evidence for extended-duration treatment.
Study design: Retrospective cohort.
Setting: Patients reported to the American College of Surgeons National Surgical Quality Improvement Program (ACS NSQIP) database.
Synopsis: The authors examined the records of 46,656 patients who underwent surgery for one of nine specified cancers. Overall VTE rate was 1.6% (1.0% deep venous thrombosis and 0.6% pulmonary embolism), with 33.4% of VTE events occurring after hospital discharge. VTE risk was highest after esophagogastric and hepatopancreaticobiliary surgery, followed by lung, rectum, ovary/uterus, colon, and prostate. Breast and thyroid/parathyroid surgeries had the lowest incidence of VTE. VTE was associated with increased 30-day mortality. Use of VTE prophylaxis during or after hospitalization was not recorded.
Bottom line: Elevated VTE risk persists following hospital discharge after cancer surgery and consideration should be given to extended-duration thromboprophylaxis. Optimal duration of prophylaxis and its risks and benefits remain poorly defined.
Citation: Merkow RP, Bilimoria KY, McCarter MD, et al. Post-discharge venous thromboembolism after cancer surgery: extending the case for extended prophylaxis. Ann Surg. 2011;254:131-137.
For more physician reviews of HM-related literature, visit our website.
Clinical question: When do venous thromboembolism (VTE) events occur after cancer surgery?
Background: Cancer is a known risk factor for VTE. Prophylaxis for VTE after cancer surgery is commonly stopped at the time of hospital discharge despite evidence for extended-duration treatment.
Study design: Retrospective cohort.
Setting: Patients reported to the American College of Surgeons National Surgical Quality Improvement Program (ACS NSQIP) database.
Synopsis: The authors examined the records of 46,656 patients who underwent surgery for one of nine specified cancers. Overall VTE rate was 1.6% (1.0% deep venous thrombosis and 0.6% pulmonary embolism), with 33.4% of VTE events occurring after hospital discharge. VTE risk was highest after esophagogastric and hepatopancreaticobiliary surgery, followed by lung, rectum, ovary/uterus, colon, and prostate. Breast and thyroid/parathyroid surgeries had the lowest incidence of VTE. VTE was associated with increased 30-day mortality. Use of VTE prophylaxis during or after hospitalization was not recorded.
Bottom line: Elevated VTE risk persists following hospital discharge after cancer surgery and consideration should be given to extended-duration thromboprophylaxis. Optimal duration of prophylaxis and its risks and benefits remain poorly defined.
Citation: Merkow RP, Bilimoria KY, McCarter MD, et al. Post-discharge venous thromboembolism after cancer surgery: extending the case for extended prophylaxis. Ann Surg. 2011;254:131-137.
For more physician reviews of HM-related literature, visit our website.
Clinical question: When do venous thromboembolism (VTE) events occur after cancer surgery?
Background: Cancer is a known risk factor for VTE. Prophylaxis for VTE after cancer surgery is commonly stopped at the time of hospital discharge despite evidence for extended-duration treatment.
Study design: Retrospective cohort.
Setting: Patients reported to the American College of Surgeons National Surgical Quality Improvement Program (ACS NSQIP) database.
Synopsis: The authors examined the records of 46,656 patients who underwent surgery for one of nine specified cancers. Overall VTE rate was 1.6% (1.0% deep venous thrombosis and 0.6% pulmonary embolism), with 33.4% of VTE events occurring after hospital discharge. VTE risk was highest after esophagogastric and hepatopancreaticobiliary surgery, followed by lung, rectum, ovary/uterus, colon, and prostate. Breast and thyroid/parathyroid surgeries had the lowest incidence of VTE. VTE was associated with increased 30-day mortality. Use of VTE prophylaxis during or after hospitalization was not recorded.
Bottom line: Elevated VTE risk persists following hospital discharge after cancer surgery and consideration should be given to extended-duration thromboprophylaxis. Optimal duration of prophylaxis and its risks and benefits remain poorly defined.
Citation: Merkow RP, Bilimoria KY, McCarter MD, et al. Post-discharge venous thromboembolism after cancer surgery: extending the case for extended prophylaxis. Ann Surg. 2011;254:131-137.
For more physician reviews of HM-related literature, visit our website.
Expectations Exceeded

When HM pioneers identified potential candidates to become editor-in-chief of a new peer-reviewed journal dedicated to their specialty, they found themselves working from a short list. The term “hospitalist” had been part of the American healthcare lexicon for less than a decade, and only a select few in the young field possessed the leadership, management experience, and research credibility to fill the role.
Mark Williams, MD, FACP, FHM, then a professor and director of the hospital medicine unit at Emory University School of Medicine in Atlanta, met the criteria. He also demonstrated two attributes that distinguished him from other finalists. First, he understood the Journal of Hospital Medicine’s mission, having led an SHM task force that created a development plan for the publication. More importantly, he had the personality to sell JHM as a valuable tool for researchers and frontline hospitalists long before the first issue rolled off the press, says Robert Wachter, MD, MHM, professor and associate chairman of the Department of Medicine at the University of California at San Francisco (UCSF), chief of the division of hospital medicine and chief of medical service at UCSF Medical Center, and one of the HM leaders who advocated for the journal’s launch.
“The early phase is particularly tricky in that you are trying to get an entire specialty interested in something that is conceptual,” Dr. Wachter says. “If you can’t, you don’t ever develop the momentum to build the thing you’re talking about. If you can, you get people excited and jazzed about it before it’s real, so when it becomes real, you have accomplished, talented people truly engaged. The latter was the experience with Mark.”
Dr. Williams did more than generate excitement. He assembled a diverse editorial team and developed a comprehensive content plan that, over the next six years, helped JHM evolve into a frequently cited, well-respected publication.
“It has exceeded my expectations,” Dr. Wachter says, “and my expectations were pretty high.”

Getting Off the Ground
By the turn of the century, HM achieved many of the milestones its leaders believed were necessary to solidify itself as a specialty—it had formed a society, published textbooks, and held regular conferences. The next step, they believed, was the launch of a peer-reviewed journal.
Discussions continued for a few years until proponents believed a sizable readership base existed, and that HM had enough established researchers and authors to sustain a journal. In March 2005, SHM appointed Dr. Williams, who had reviewed and written journal articles but never served as an editor, as JHM ’s editor-in-chief and scheduled a February 2006 launch.
“Off we went, with me not really having any clear idea what I was getting myself into,” says Dr. Williams, a former SHM president who now is a professor and chief of the Division of Hospital Medicine at Northwestern University’s Feinberg School of Medicine in Chicago.
He immediately immersed himself in other top-tier journals he hoped to emulate. He also began formulating strategies to tackle two significant challenges. The first—promoting the publication—meshed perfectly with his persona, says Vineet Arora, MD, FHM, assistant dean for scholarship and discovery at the University of Chicago’s Pritzker School of Medicine.
During HM05 in Chicago, Dr. Williams handed out business cards to annual meeting attendees and encouraged every presenter to submit his or her research to the journal.
“Everyone was so excited to meet him and to find out there was a home for their work,” Dr. Arora recalls. “Mark was really successful, right from the start, at building those bridges and making sure everybody felt part of the team.”
—Brian Harte, MD, SFHM, chief operating officer, Hillcrest Hospital, chairman of hospital medicine, Cleveland Clinic, JHM deputy editor
The second challenge—lining up content for the inaugural issue—proved easier than anticipated. Diane Meier, MD, FACP, an internationally recognized expert on palliative care, and C. Seth Landefeld, MD, FACP, chief of the Division of Geriatrics at UCSF, submitted review articles. Christine Cassel, MD, FACP, president and CEO of the American Board of Internal Medicine, wrote an editorial. Diane Payne, publications director for the Board of Regents for the University System of Georgia, submitted what remains Dr. Williams’ favorite JHM article, a patient commentary titled “Hospitals Foreign Soil for Those Who Don’t Work There.”
The first issue also included The Core Competencies in Hospital Medicine: A Framework for Curriculum Development, a blueprint created by SHM to help medical schools and post-graduate programs develop standardized curricula for teaching HM. The supplement remains the most-cited article in JHM history.
“We were told over and over the biggest problem we’d face would be getting enough content,” Dr. Williams says. “We were flooded with content from day one. That tells me we probably could have started this journal a year or two earlier, but this ensured our success.”
Success from the Start
JHM ’s success continued beyond the inaugural issue. Less than a year after the launch, it was selected for indexing and inclusion in MEDLINE, a U.S. National Library of Medicine bibliographic database that contains more than 18 million references to journal articles in medicine and other life sciences.
In summer 2009, it received a debut 3.163 Impact Factor, an industry metric that calculates average citations received by peer-reviewed journals. The score ranked JHM in the top 20% of its cohort, a stronger-than-expected showing for a journal in its fourth year of publication.
An increasing amount of original research helped JHM become a valuable educational tool, and nearly 10,000 journal articles have been downloaded since its inception, Dr. Williams says.
The journal’s clinical vignettes and articles that explain how political developments affect HM are especially beneficial, says James Neviackas, MD, a hospitalist at Decatur Memorial Hospital in Illinois. The format also serves hospitalists well.
“The articles are short and hard-hitting, so they enable me to get as much information as I can in as little time as possible,” Dr. Neviackas says.
Dr. Williams deserves credit for making the journal a viable and valuable publication, says Dana P. Edelson, MD, FHM, assistant professor at The University of Chicago’s Department of Medicine and a JHM assistant editor.
“To build it from nothing into a well-regarded academic journal in a matter of few years is pretty amazing,” Dr. Edelson says. “From a success standpoint, it’s truly remarkable.”
—Mark Williams, MD, SFHM, professor, chief, division of hospital medicine, Feinberg School of Medicine, Northwestern University, Chicago, former SHM president, JHM editor-in-chief, 2006-2011
Dr. Williams inspires members of his editorial staff “to bring their A game” by giving them considerable authority, valuing their opinions, and demonstrating a willingness to support their decisions, even when they risk angering authors whose articles are rejected, Dr. Edelson says.
“Mark has shown a combination of operational capabilities, organizational skills, and servant leadership that is really inspirational,” adds deputy editor Brian Harte, MD, SFHM, chief operating officer of Hillcrest Hospital in Ohio and chairman of hospital medicine at The Cleveland Clinic. “He sets the strategic vision and empowers his team to execute. He is always open, and he encourages ideas. He’s a facilitator, which is what a great leader is.”
Dr. Williams, in turn, credits the support of JHM ’s publisher, John Wiley & Sons Inc., which also publishes The Hospitalist, and his editorial team for the journal’s achievements. He also praises his team for ensuring his greatest fear—constant complaints from authors whose papers were rejected—never came to fruition.
“I thought I’d get nasty emails saying, ‘Why are you rejecting my article? Clearly you don’t understand what I’m doing,’” Dr. Williams says. “Invariably, I get emails along the lines of, ‘Thank you so much for carefully reviewing the article. I deeply appreciate the insightful comments from the reviewers.’
“That has been very rewarding,” he adds. “It demonstrates we have done a terrific job of candidly and fairly reviewing articles … and that the amount of effort we put into providing those reviews is recognized and welcomed and appreciated.”
The Transition
Dr. Williams will serve as the journal’s editor-in-chief through the end of the year. Andrew Auerbach, MD, MPH, SFHM, associate professor of medicine at UCSF and director of research for the Division of Hospital Medicine, will take over in January.
Dr. Auerbach, who will serve a five-year term, says Dr. Williams has “done a remarkable job” developing HM’s only peer-reviewed journal. “He raised the visibility of the journal inside the field of hospital medicine and outside,” Dr. Auerbach says. “He built a publication that is really aligned with what hospitalists are doing and what they want to do.”
Dr. Williams is helping Dr. Auerbach develop a strategic plan for the first 18 months of his term, but he looks forward to having more time to mentor junior faculty at Northwestern. He’ll leave the editor’s chair with two pieces of unfinished business: The economic downturn thwarted his effort to increase JHM ’s publishing frequency from nine to 12 times a year, a move he hopes is made next year; he also fell short of his goal to feature regular patient commentaries, such as Diane Payne’s editorial in the inaugural issue.
Although he takes pride in the journal’s cover design, which includes three photos that he says convey HM is about caring for people, he hopes patients’ voices are better represented in future issues.
“We’re all about taking care of patients. That’s our purpose,” he says. “Too often, health care providers get busy and they forget that. They don’t realize how difficult it is for patients to go through the struggles of obtaining healthcare and being in a hospital when they are incredibly sick.”
‘A Big Tent’
Despite the challenges associated with starting a journal from scratch, Dr. Williams says his six years at the helm went more smoothly than he could have imagined. The effort has paid off.
JHM’s Impact Factor, although down from its debut figure, rose to 1.951 last year from 1.496 in 2009, ranking it 40th out of 151 journals in its cohort.
The success, Dr. Wachter says, shows Dr. Williams was the right choice to lead JHM from birth through toddlerhood.
More importantly, Dr. Williams embraced the vision of HM leaders who believed the journal needed to be a big tent in order to succeed. “We wanted to try to somehow hit the sweet spot of being relevant and interesting to folks who practice hospital medicine in a wide array of circumstances,” Dr. Wachter says, “while also being a go-to place for researchers to submit their research. That was ambitious, and that could have failed in all sorts of directions. It could have been quite relevant to clinicians, but not rigorous enough for researchers. It could have been perfect for researchers, but the clinicians could have felt it wasn’t relative to their day-to-day life. I think the journal has done a masterful job negotiating that tight wire.”
Mark Leiser is a freelance writer based in New Jersey.
When HM pioneers identified potential candidates to become editor-in-chief of a new peer-reviewed journal dedicated to their specialty, they found themselves working from a short list. The term “hospitalist” had been part of the American healthcare lexicon for less than a decade, and only a select few in the young field possessed the leadership, management experience, and research credibility to fill the role.
Mark Williams, MD, FACP, FHM, then a professor and director of the hospital medicine unit at Emory University School of Medicine in Atlanta, met the criteria. He also demonstrated two attributes that distinguished him from other finalists. First, he understood the Journal of Hospital Medicine’s mission, having led an SHM task force that created a development plan for the publication. More importantly, he had the personality to sell JHM as a valuable tool for researchers and frontline hospitalists long before the first issue rolled off the press, says Robert Wachter, MD, MHM, professor and associate chairman of the Department of Medicine at the University of California at San Francisco (UCSF), chief of the division of hospital medicine and chief of medical service at UCSF Medical Center, and one of the HM leaders who advocated for the journal’s launch.
“The early phase is particularly tricky in that you are trying to get an entire specialty interested in something that is conceptual,” Dr. Wachter says. “If you can’t, you don’t ever develop the momentum to build the thing you’re talking about. If you can, you get people excited and jazzed about it before it’s real, so when it becomes real, you have accomplished, talented people truly engaged. The latter was the experience with Mark.”
Dr. Williams did more than generate excitement. He assembled a diverse editorial team and developed a comprehensive content plan that, over the next six years, helped JHM evolve into a frequently cited, well-respected publication.
“It has exceeded my expectations,” Dr. Wachter says, “and my expectations were pretty high.”
Getting Off the Ground
By the turn of the century, HM achieved many of the milestones its leaders believed were necessary to solidify itself as a specialty—it had formed a society, published textbooks, and held regular conferences. The next step, they believed, was the launch of a peer-reviewed journal.
Discussions continued for a few years until proponents believed a sizable readership base existed, and that HM had enough established researchers and authors to sustain a journal. In March 2005, SHM appointed Dr. Williams, who had reviewed and written journal articles but never served as an editor, as JHM ’s editor-in-chief and scheduled a February 2006 launch.
“Off we went, with me not really having any clear idea what I was getting myself into,” says Dr. Williams, a former SHM president who now is a professor and chief of the Division of Hospital Medicine at Northwestern University’s Feinberg School of Medicine in Chicago.
He immediately immersed himself in other top-tier journals he hoped to emulate. He also began formulating strategies to tackle two significant challenges. The first—promoting the publication—meshed perfectly with his persona, says Vineet Arora, MD, FHM, assistant dean for scholarship and discovery at the University of Chicago’s Pritzker School of Medicine.
During HM05 in Chicago, Dr. Williams handed out business cards to annual meeting attendees and encouraged every presenter to submit his or her research to the journal.
“Everyone was so excited to meet him and to find out there was a home for their work,” Dr. Arora recalls. “Mark was really successful, right from the start, at building those bridges and making sure everybody felt part of the team.”
—Brian Harte, MD, SFHM, chief operating officer, Hillcrest Hospital, chairman of hospital medicine, Cleveland Clinic, JHM deputy editor
The second challenge—lining up content for the inaugural issue—proved easier than anticipated. Diane Meier, MD, FACP, an internationally recognized expert on palliative care, and C. Seth Landefeld, MD, FACP, chief of the Division of Geriatrics at UCSF, submitted review articles. Christine Cassel, MD, FACP, president and CEO of the American Board of Internal Medicine, wrote an editorial. Diane Payne, publications director for the Board of Regents for the University System of Georgia, submitted what remains Dr. Williams’ favorite JHM article, a patient commentary titled “Hospitals Foreign Soil for Those Who Don’t Work There.”
The first issue also included The Core Competencies in Hospital Medicine: A Framework for Curriculum Development, a blueprint created by SHM to help medical schools and post-graduate programs develop standardized curricula for teaching HM. The supplement remains the most-cited article in JHM history.
“We were told over and over the biggest problem we’d face would be getting enough content,” Dr. Williams says. “We were flooded with content from day one. That tells me we probably could have started this journal a year or two earlier, but this ensured our success.”
Success from the Start
JHM ’s success continued beyond the inaugural issue. Less than a year after the launch, it was selected for indexing and inclusion in MEDLINE, a U.S. National Library of Medicine bibliographic database that contains more than 18 million references to journal articles in medicine and other life sciences.
In summer 2009, it received a debut 3.163 Impact Factor, an industry metric that calculates average citations received by peer-reviewed journals. The score ranked JHM in the top 20% of its cohort, a stronger-than-expected showing for a journal in its fourth year of publication.
An increasing amount of original research helped JHM become a valuable educational tool, and nearly 10,000 journal articles have been downloaded since its inception, Dr. Williams says.
The journal’s clinical vignettes and articles that explain how political developments affect HM are especially beneficial, says James Neviackas, MD, a hospitalist at Decatur Memorial Hospital in Illinois. The format also serves hospitalists well.
“The articles are short and hard-hitting, so they enable me to get as much information as I can in as little time as possible,” Dr. Neviackas says.
Dr. Williams deserves credit for making the journal a viable and valuable publication, says Dana P. Edelson, MD, FHM, assistant professor at The University of Chicago’s Department of Medicine and a JHM assistant editor.
“To build it from nothing into a well-regarded academic journal in a matter of few years is pretty amazing,” Dr. Edelson says. “From a success standpoint, it’s truly remarkable.”
—Mark Williams, MD, SFHM, professor, chief, division of hospital medicine, Feinberg School of Medicine, Northwestern University, Chicago, former SHM president, JHM editor-in-chief, 2006-2011
Dr. Williams inspires members of his editorial staff “to bring their A game” by giving them considerable authority, valuing their opinions, and demonstrating a willingness to support their decisions, even when they risk angering authors whose articles are rejected, Dr. Edelson says.
“Mark has shown a combination of operational capabilities, organizational skills, and servant leadership that is really inspirational,” adds deputy editor Brian Harte, MD, SFHM, chief operating officer of Hillcrest Hospital in Ohio and chairman of hospital medicine at The Cleveland Clinic. “He sets the strategic vision and empowers his team to execute. He is always open, and he encourages ideas. He’s a facilitator, which is what a great leader is.”
Dr. Williams, in turn, credits the support of JHM ’s publisher, John Wiley & Sons Inc., which also publishes The Hospitalist, and his editorial team for the journal’s achievements. He also praises his team for ensuring his greatest fear—constant complaints from authors whose papers were rejected—never came to fruition.
“I thought I’d get nasty emails saying, ‘Why are you rejecting my article? Clearly you don’t understand what I’m doing,’” Dr. Williams says. “Invariably, I get emails along the lines of, ‘Thank you so much for carefully reviewing the article. I deeply appreciate the insightful comments from the reviewers.’
“That has been very rewarding,” he adds. “It demonstrates we have done a terrific job of candidly and fairly reviewing articles … and that the amount of effort we put into providing those reviews is recognized and welcomed and appreciated.”
The Transition
Dr. Williams will serve as the journal’s editor-in-chief through the end of the year. Andrew Auerbach, MD, MPH, SFHM, associate professor of medicine at UCSF and director of research for the Division of Hospital Medicine, will take over in January.
Dr. Auerbach, who will serve a five-year term, says Dr. Williams has “done a remarkable job” developing HM’s only peer-reviewed journal. “He raised the visibility of the journal inside the field of hospital medicine and outside,” Dr. Auerbach says. “He built a publication that is really aligned with what hospitalists are doing and what they want to do.”
Dr. Williams is helping Dr. Auerbach develop a strategic plan for the first 18 months of his term, but he looks forward to having more time to mentor junior faculty at Northwestern. He’ll leave the editor’s chair with two pieces of unfinished business: The economic downturn thwarted his effort to increase JHM ’s publishing frequency from nine to 12 times a year, a move he hopes is made next year; he also fell short of his goal to feature regular patient commentaries, such as Diane Payne’s editorial in the inaugural issue.
Although he takes pride in the journal’s cover design, which includes three photos that he says convey HM is about caring for people, he hopes patients’ voices are better represented in future issues.
“We’re all about taking care of patients. That’s our purpose,” he says. “Too often, health care providers get busy and they forget that. They don’t realize how difficult it is for patients to go through the struggles of obtaining healthcare and being in a hospital when they are incredibly sick.”
‘A Big Tent’
Despite the challenges associated with starting a journal from scratch, Dr. Williams says his six years at the helm went more smoothly than he could have imagined. The effort has paid off.
JHM’s Impact Factor, although down from its debut figure, rose to 1.951 last year from 1.496 in 2009, ranking it 40th out of 151 journals in its cohort.
The success, Dr. Wachter says, shows Dr. Williams was the right choice to lead JHM from birth through toddlerhood.
More importantly, Dr. Williams embraced the vision of HM leaders who believed the journal needed to be a big tent in order to succeed. “We wanted to try to somehow hit the sweet spot of being relevant and interesting to folks who practice hospital medicine in a wide array of circumstances,” Dr. Wachter says, “while also being a go-to place for researchers to submit their research. That was ambitious, and that could have failed in all sorts of directions. It could have been quite relevant to clinicians, but not rigorous enough for researchers. It could have been perfect for researchers, but the clinicians could have felt it wasn’t relative to their day-to-day life. I think the journal has done a masterful job negotiating that tight wire.”
Mark Leiser is a freelance writer based in New Jersey.
When HM pioneers identified potential candidates to become editor-in-chief of a new peer-reviewed journal dedicated to their specialty, they found themselves working from a short list. The term “hospitalist” had been part of the American healthcare lexicon for less than a decade, and only a select few in the young field possessed the leadership, management experience, and research credibility to fill the role.
Mark Williams, MD, FACP, FHM, then a professor and director of the hospital medicine unit at Emory University School of Medicine in Atlanta, met the criteria. He also demonstrated two attributes that distinguished him from other finalists. First, he understood the Journal of Hospital Medicine’s mission, having led an SHM task force that created a development plan for the publication. More importantly, he had the personality to sell JHM as a valuable tool for researchers and frontline hospitalists long before the first issue rolled off the press, says Robert Wachter, MD, MHM, professor and associate chairman of the Department of Medicine at the University of California at San Francisco (UCSF), chief of the division of hospital medicine and chief of medical service at UCSF Medical Center, and one of the HM leaders who advocated for the journal’s launch.
“The early phase is particularly tricky in that you are trying to get an entire specialty interested in something that is conceptual,” Dr. Wachter says. “If you can’t, you don’t ever develop the momentum to build the thing you’re talking about. If you can, you get people excited and jazzed about it before it’s real, so when it becomes real, you have accomplished, talented people truly engaged. The latter was the experience with Mark.”
Dr. Williams did more than generate excitement. He assembled a diverse editorial team and developed a comprehensive content plan that, over the next six years, helped JHM evolve into a frequently cited, well-respected publication.
“It has exceeded my expectations,” Dr. Wachter says, “and my expectations were pretty high.”
Getting Off the Ground
By the turn of the century, HM achieved many of the milestones its leaders believed were necessary to solidify itself as a specialty—it had formed a society, published textbooks, and held regular conferences. The next step, they believed, was the launch of a peer-reviewed journal.
Discussions continued for a few years until proponents believed a sizable readership base existed, and that HM had enough established researchers and authors to sustain a journal. In March 2005, SHM appointed Dr. Williams, who had reviewed and written journal articles but never served as an editor, as JHM ’s editor-in-chief and scheduled a February 2006 launch.
“Off we went, with me not really having any clear idea what I was getting myself into,” says Dr. Williams, a former SHM president who now is a professor and chief of the Division of Hospital Medicine at Northwestern University’s Feinberg School of Medicine in Chicago.
He immediately immersed himself in other top-tier journals he hoped to emulate. He also began formulating strategies to tackle two significant challenges. The first—promoting the publication—meshed perfectly with his persona, says Vineet Arora, MD, FHM, assistant dean for scholarship and discovery at the University of Chicago’s Pritzker School of Medicine.
During HM05 in Chicago, Dr. Williams handed out business cards to annual meeting attendees and encouraged every presenter to submit his or her research to the journal.
“Everyone was so excited to meet him and to find out there was a home for their work,” Dr. Arora recalls. “Mark was really successful, right from the start, at building those bridges and making sure everybody felt part of the team.”
—Brian Harte, MD, SFHM, chief operating officer, Hillcrest Hospital, chairman of hospital medicine, Cleveland Clinic, JHM deputy editor
The second challenge—lining up content for the inaugural issue—proved easier than anticipated. Diane Meier, MD, FACP, an internationally recognized expert on palliative care, and C. Seth Landefeld, MD, FACP, chief of the Division of Geriatrics at UCSF, submitted review articles. Christine Cassel, MD, FACP, president and CEO of the American Board of Internal Medicine, wrote an editorial. Diane Payne, publications director for the Board of Regents for the University System of Georgia, submitted what remains Dr. Williams’ favorite JHM article, a patient commentary titled “Hospitals Foreign Soil for Those Who Don’t Work There.”
The first issue also included The Core Competencies in Hospital Medicine: A Framework for Curriculum Development, a blueprint created by SHM to help medical schools and post-graduate programs develop standardized curricula for teaching HM. The supplement remains the most-cited article in JHM history.
“We were told over and over the biggest problem we’d face would be getting enough content,” Dr. Williams says. “We were flooded with content from day one. That tells me we probably could have started this journal a year or two earlier, but this ensured our success.”
Success from the Start
JHM ’s success continued beyond the inaugural issue. Less than a year after the launch, it was selected for indexing and inclusion in MEDLINE, a U.S. National Library of Medicine bibliographic database that contains more than 18 million references to journal articles in medicine and other life sciences.
In summer 2009, it received a debut 3.163 Impact Factor, an industry metric that calculates average citations received by peer-reviewed journals. The score ranked JHM in the top 20% of its cohort, a stronger-than-expected showing for a journal in its fourth year of publication.
An increasing amount of original research helped JHM become a valuable educational tool, and nearly 10,000 journal articles have been downloaded since its inception, Dr. Williams says.
The journal’s clinical vignettes and articles that explain how political developments affect HM are especially beneficial, says James Neviackas, MD, a hospitalist at Decatur Memorial Hospital in Illinois. The format also serves hospitalists well.
“The articles are short and hard-hitting, so they enable me to get as much information as I can in as little time as possible,” Dr. Neviackas says.
Dr. Williams deserves credit for making the journal a viable and valuable publication, says Dana P. Edelson, MD, FHM, assistant professor at The University of Chicago’s Department of Medicine and a JHM assistant editor.
“To build it from nothing into a well-regarded academic journal in a matter of few years is pretty amazing,” Dr. Edelson says. “From a success standpoint, it’s truly remarkable.”
—Mark Williams, MD, SFHM, professor, chief, division of hospital medicine, Feinberg School of Medicine, Northwestern University, Chicago, former SHM president, JHM editor-in-chief, 2006-2011
Dr. Williams inspires members of his editorial staff “to bring their A game” by giving them considerable authority, valuing their opinions, and demonstrating a willingness to support their decisions, even when they risk angering authors whose articles are rejected, Dr. Edelson says.
“Mark has shown a combination of operational capabilities, organizational skills, and servant leadership that is really inspirational,” adds deputy editor Brian Harte, MD, SFHM, chief operating officer of Hillcrest Hospital in Ohio and chairman of hospital medicine at The Cleveland Clinic. “He sets the strategic vision and empowers his team to execute. He is always open, and he encourages ideas. He’s a facilitator, which is what a great leader is.”
Dr. Williams, in turn, credits the support of JHM ’s publisher, John Wiley & Sons Inc., which also publishes The Hospitalist, and his editorial team for the journal’s achievements. He also praises his team for ensuring his greatest fear—constant complaints from authors whose papers were rejected—never came to fruition.
“I thought I’d get nasty emails saying, ‘Why are you rejecting my article? Clearly you don’t understand what I’m doing,’” Dr. Williams says. “Invariably, I get emails along the lines of, ‘Thank you so much for carefully reviewing the article. I deeply appreciate the insightful comments from the reviewers.’
“That has been very rewarding,” he adds. “It demonstrates we have done a terrific job of candidly and fairly reviewing articles … and that the amount of effort we put into providing those reviews is recognized and welcomed and appreciated.”
The Transition
Dr. Williams will serve as the journal’s editor-in-chief through the end of the year. Andrew Auerbach, MD, MPH, SFHM, associate professor of medicine at UCSF and director of research for the Division of Hospital Medicine, will take over in January.
Dr. Auerbach, who will serve a five-year term, says Dr. Williams has “done a remarkable job” developing HM’s only peer-reviewed journal. “He raised the visibility of the journal inside the field of hospital medicine and outside,” Dr. Auerbach says. “He built a publication that is really aligned with what hospitalists are doing and what they want to do.”
Dr. Williams is helping Dr. Auerbach develop a strategic plan for the first 18 months of his term, but he looks forward to having more time to mentor junior faculty at Northwestern. He’ll leave the editor’s chair with two pieces of unfinished business: The economic downturn thwarted his effort to increase JHM ’s publishing frequency from nine to 12 times a year, a move he hopes is made next year; he also fell short of his goal to feature regular patient commentaries, such as Diane Payne’s editorial in the inaugural issue.
Although he takes pride in the journal’s cover design, which includes three photos that he says convey HM is about caring for people, he hopes patients’ voices are better represented in future issues.
“We’re all about taking care of patients. That’s our purpose,” he says. “Too often, health care providers get busy and they forget that. They don’t realize how difficult it is for patients to go through the struggles of obtaining healthcare and being in a hospital when they are incredibly sick.”
‘A Big Tent’
Despite the challenges associated with starting a journal from scratch, Dr. Williams says his six years at the helm went more smoothly than he could have imagined. The effort has paid off.
JHM’s Impact Factor, although down from its debut figure, rose to 1.951 last year from 1.496 in 2009, ranking it 40th out of 151 journals in its cohort.
The success, Dr. Wachter says, shows Dr. Williams was the right choice to lead JHM from birth through toddlerhood.
More importantly, Dr. Williams embraced the vision of HM leaders who believed the journal needed to be a big tent in order to succeed. “We wanted to try to somehow hit the sweet spot of being relevant and interesting to folks who practice hospital medicine in a wide array of circumstances,” Dr. Wachter says, “while also being a go-to place for researchers to submit their research. That was ambitious, and that could have failed in all sorts of directions. It could have been quite relevant to clinicians, but not rigorous enough for researchers. It could have been perfect for researchers, but the clinicians could have felt it wasn’t relative to their day-to-day life. I think the journal has done a masterful job negotiating that tight wire.”
Mark Leiser is a freelance writer based in New Jersey.
A New Direction

Andrew Auerbach, MD, MPH, SFHM, is an associate professor at one of the most highly regarded academic medical centers in the country, and he is a nationally respected researcher whose work has been published in prominent scientific publications.
His peers believe both roles prepared him well for his newest endeavor: editor-in-chief of the Journal of Hospital Medicine. His five-year term begins in January.
Dr. Auerbach’s affiliation with the University of California at San Francisco (UCSF), where he also serves as director of research for the division of hospital medicine and associate director of the General Medicine Research Fellowship, lends instant legitimacy to the journal, editorial team members say. His research has focused on evaluation of care-delivery models and methods for improving the measurement of quality of care.
That background, medical publishing experts believe, gives Dr. Auerbach a foundation in scientific accuracy and reporting transparency that is integral to a new editor’s effort to expand a journal’s reach and increase the quality of work it publishes.
Dr. Auerbach intends to do both, continuing JHM’s growth and solidifying it as the go-to resource for hospitalists. “It is an incredibly good platform for hospitalists to publish their work,” he adds. “I want people to look back at the journal in five years and say it’s even better than it is now.”
As Dr. Auerbach prepares to take over as editor-in-chief, he is focusing his efforts on how to best build on the success the journal has enjoyed since its 2006 launch.
“JHM’s core values are to reflect the field of hospital medicine broadly, to provide a venue where the field’s current scholarly work can be published, and to provide a point of reference for where future scholarly work might be directed and published,” he says. “JHM has been very successful at the first two. I think there is an opportunity to be somewhat more strategic in providing the reference point for future research directions, largely through the input of the editorial team and by paying close attention to JHM readers.”
The Right Choice
An eight-person committee embarked on a five-month search to identify the ideal candidate to replace JHM ’s founding editor-in-chief, Mark Williams, MD, FACP, FHM. The intense process reflected the search committee’s goal that the new editor strengthen the journal and the society it represents, says committee member Harold C. Sox, MD, MACP, a noted internist and author who served as editor of Annals of Internal Medicine from 2001 to 2009.
“A journal published by a professional organization is far and away the most visible manifestation of the organization and its values and ambitions,” Dr. Sox says. “The appointment was anything but pro forma. It had to be the right choice.”
Committee members wanted to select a physician who had a strong scientific background, the judgment required to be a leader, and a desire to better position the journal as a source for first-rate articles in the competitive landscape of medical publishing, Dr. Sox says. They also searched for a candidate who had considerable experience writing for publication.
Dr. Auerbach, who served as deputy editor of the Journal of General Internal Medicine from 2004 to 2007 and as a JHM reviewer, has been published in the New England Journal of Medicine, the Journal of the American Medical Association, Annals of Internal Medicine, and the Archives of Internal Medicine.
As editor of Annals, Dr. Sox shared responsibility for publishing two of Dr. Auerbach’s articles.
“I know something about how Dr. Auerbach thinks about science and about his own personal scientific standards—wanting to get it right, writing it in a way that people know exactly what happened, and interpreting the results in a way that will stand up over time, rather than trying to read too much into the results,” Dr. Sox says. “We also talked with some of the candidates about their strategy for trying to make the journal better, and I thought he showed himself to be very analytic and strategic in his thinking. I’m confident he’s going to move the journal forward.”
Dr. Auerbach believes his experience as a deputy editor and his mentoring role at UCSF, which requires him to provide constructive feedback on investigators’ papers, has prepared him for his new role.
“I have a good sense for how to approach the review process with the goal of getting a paper to the point where it can be accepted into a journal and, if not, have it leave the review process much better than it was when it arrived,” he says.
His research background also complements his new role, he says.
“Research is a way to take care of patients I never see and to disseminate things that are broadly applicable … that other people can look at and use to improve their healthcare delivery,” he says. “I see the journal as doing many of the same things.”
Strategies for Success
Dr. Auerbach, who has worked closely with Dr. Williams in recent months to ensure a smooth transition, says he does not intend to make substantial changes to the journal’s focus. But he is formulating strategies to make it more valuable and relevant to the hospitalists it serves.
Increasing the journal’s Impact Factor—currently 1.951, ranking it 40th out of 151 publications in its cohort—is one way to increase JHM ’s value, making it a destination for the highest-quality research, Dr. Auerbach says.
He also intends to solicit as many scholarly works as possible, employing “a marketing strategy and recruitment strategy all in one” to make sure JHM is on researchers’ short list of publications in which they consider submitting their work.

—Robert Wachter, MD, MHM, professor, associate chair, Department of Medicine, University of California at San Francisco, chief, division of hospital medicine, chief, medical service, UCSF Medical Center, former SHM president
He wants to take advantage of the growing number of hospitalists who are doing research and expand the journal’s reach beyond its traditional peer groups. “Are there intensive-care doctors, pulmonologists, cardiologists, or infectious-disease doctors who are doing work with hospitalists—or work that would align with hospital medicine—that could be published in JHM?” Dr. Auerbach asks, rhetorically.
He intends to empower members of his editorial team to serve as JHM ambassadors who promote the journal at professional meetings and encourage investigators who are pursuing interesting projects to submit their findings.
Dr. Auerbach expects to devise other content strategies in consultation with members of his editorial team. Certain aspects of content, such as case reports and scholarly reviews of conundrums in HM cases, could be enhanced, he says. Specific sections of the journal could be developed for effectiveness research or implementation research.
He also says he wants the journal to explore new content areas so that it appeals to readers who aren’t interested in randomized controlled trials.
He hopes to put the journal in the position to publish work supported by such new initiatives as the Patient-Centered Outcomes Research Institute and the Centers for Medicare & Medicaid Services’ Center for Medicare and Medicaid Innovation.
That might mean providing editorials or inviting papers that outline the importance of hospitalists’ involvement in those initiatives, then making sure the journal is opportunistic in reviewing and publishing high-quality work, he explains.
Andrew Auerbach, MD, MPH, SFHM

Age: 44
Academic rank: Associate professor of medicine, University of California at San Francisco’s Division of Hospital Medicine
Administrative titles at UCSF: Attending physician, Department of Medicine; director of research for division of hospital medicine; associate director of the General Medicine Research Fellowship; chair of the Clinical Content Governance Committee at UCSF Medical Center
Education: Bachelor’s degree in biochemistry from Bowdoin College, Brunswick, Maine (1988); medical degree from Dartmouth Medical School, Hanover, N.H. (1992); master’s of public health in clinical effectiveness from Harvard School of Public Health, Boston (1998)
Clinical training: Internship in internal medicine, Yale-New Haven Hospital, New Haven, Conn. (1992-1993); residency in internal medicine, Yale-New Haven Hospital, New Haven, Conn. (1993-1995); fellowship in general internal medicine, Beth Israel-Deaconess Medical Center, Boston (1996-1998)
Principal positions held: Attending physician at Moffitt-Long Hospitals in San Francisco (1998-present); clinical fellow at Beth Israel Deaconess Medical Center, Boston (1996-1998); emergency room physician at Milton Hospital, Milton, Mass. (1996-1998); physician and co-director of student health services at University of New Haven, New Haven, Conn. (1995-1996); hospitalist at Hospital of Saint Raphael, New Haven, Conn. (1995-1996); admitting officer at West Haven Veterans Administration Hospital, West Haven, Conn. (1994-1996)
Awards and honors: Western Society for Clinical Investigation, Investigator of the Year Award (2010); Orthopaedic Research Society Harris Award for Outstanding Research (2010); fellow, American College of Physicians (2009); James Rand Award for Outstanding Research Project (2008); Society of Hospital Medicine Young Investigator Award (2004)
Professional memberships: Association of Chiefs of General Internal Medicine, Society of Hospital Medicine, American Medical Association, American College of Physicians, Society of General Internal Medicine
Most Significant Co-Authored Publications
- Rothberg M, Maselli J, Pekow P, Lindenauer P, Auerbach AD. Cost-effectiveness of changes in care at U.S. hospitals from 2000-2004. In press, Health Affairs.
- Auerbach AD, Wachter RM, Cheng H, et al. Effects of a neurosurgery-hospitalist comanagement model on outcomes of patients with neurosurgical illnesses. In press, Arch Int Med.
- Auerbach AD, Hilton JF, Maselli JM, Pekow P, Rothberg M, Lindenauer PK. Follow the crowd or shop for the best: How volume and care quality influence outcomes of cardiac surgery. Annals Int Med. 2009;150(10):696-704.
- Auerbach AD, Landefeld CS, Shojania KS. The tension between needing to improve care and knowing how to do it. N Engl J Med. 2007;357(6):608-613.
- Lindenauer PK, Pekow P, Rothberg M, Benjamin E, Auerbach AD. Outcomes of patients treated by hospitalists, general internists, and family physicians. N Engl J Med. 2007;357(25):2589-2600.
Bold New Direction
The JHM team, which partners SHM leaders and the journal’s publisher, Wiley-Blackwell Inc., is revisiting the journal’s press relations and editorial strategy (Wiley-Blackwell also publishes The Hospitalist). Discussions also are under way about whether to increase the journal’s publishing frequency, as well as how the journal can increase its digital footprint and take advantage of social media outlets to increase the usability and visibility of its offerings, Dr. Auerbach says.
“One side effect of JHM ’s growth has been growing constraints on space to publish excellent work,” he says. “Our team will focus a great deal on strategies to increase JHM ’s ability to review and publish the many outstanding papers submitted each year.”
Dr. Williams characterizes Dr. Auerbach’s appointment as “a software upgrade to JHM 2.0.”
Dr. Auerbach’s connection to the top-ranked division of hospital medicine in the country provides instant credibility to the journal, and his diverse contacts should help his efforts to solicit an increasing volume of high-quality submissions, Dr. Williams says.
His appointment also can reinvigorate members of the editorial team, motivating them to step out of their comfort zone and embrace the challenge of adapting to the rapidly changing world of hospital medicine.
“He’ll bring renewed enthusiasm,” says Dr. Williams, a former SHM president who is a professor and chief of the Division of Hospital Medicine at Northwestern University’s Feinberg School of Medicine in Chicago. “I firmly believe that’s essential for any growing enterprise. You need an infusion of new energy and fresh thinking.”
With Dr. Auerbach at the helm, JHM is well-positioned to attract well-done, systematic reviews while appealing to authors who are writing about such relevant topics as change management, collaboration, models of care, and transitional care, says deputy editor Brian Harte, MD, SFHM, chief operating officer of Hillcrest Hospital in Mayfield Heights, Ohio, and chairman of hospital medicine at The Cleveland Clinic.
“I hope people say under Dr. Auerbach’s tenure we continued to innovate and do things as an editorial group that other journals hadn’t thought of doing, were not nimble enough to do, or were not creative enough to do,” Dr. Harte says. “And that it was an incubator of novel and innovative and, ultimately, very effective ideas that took the journal into strategic directions that other journals weren’t bold enough to go in.”
Dr. Auerbach is more than capable of steering JHM in those new directions, according to his mentor, Robert Wachter, MD, MHM, professor and associate chairman of the Department of Medicine at the University of California at San Francisco, and chief of the division of hospital medicine and chief of medical service at UCSF Medical Center, former SHM president, and author of the blog Wachter’s World (www.wachtersworld.com).
Dr. Auerbach is a broad thinker who is capable of recognizing what issues are important to his field before they become obvious to others, a trait that will help him to use the journal to help chart the course for HM, Dr. Wachter says.
He also has the perfect personality for the job. “He is the most doggedly persistent person I’ve ever met,” Dr. Wachter says. “I’ve seen him go through setbacks that would have caused lesser mortals to give up their ideas.…He’s like a prizefighter. He sits in the corner for a little bit, has someone dab the wounds, and then comes back out again for the next round and swings a little bit harder.
“I’ve learned never to underestimate him. It’s an amazing characteristic, and it’s one that is very useful doing something like running a journal.”
Mark Leiser is a freelance writer based in New Jersey.
Andrew Auerbach, MD, MPH, SFHM, is an associate professor at one of the most highly regarded academic medical centers in the country, and he is a nationally respected researcher whose work has been published in prominent scientific publications.
His peers believe both roles prepared him well for his newest endeavor: editor-in-chief of the Journal of Hospital Medicine. His five-year term begins in January.
Dr. Auerbach’s affiliation with the University of California at San Francisco (UCSF), where he also serves as director of research for the division of hospital medicine and associate director of the General Medicine Research Fellowship, lends instant legitimacy to the journal, editorial team members say. His research has focused on evaluation of care-delivery models and methods for improving the measurement of quality of care.
That background, medical publishing experts believe, gives Dr. Auerbach a foundation in scientific accuracy and reporting transparency that is integral to a new editor’s effort to expand a journal’s reach and increase the quality of work it publishes.
Dr. Auerbach intends to do both, continuing JHM’s growth and solidifying it as the go-to resource for hospitalists. “It is an incredibly good platform for hospitalists to publish their work,” he adds. “I want people to look back at the journal in five years and say it’s even better than it is now.”
As Dr. Auerbach prepares to take over as editor-in-chief, he is focusing his efforts on how to best build on the success the journal has enjoyed since its 2006 launch.
“JHM’s core values are to reflect the field of hospital medicine broadly, to provide a venue where the field’s current scholarly work can be published, and to provide a point of reference for where future scholarly work might be directed and published,” he says. “JHM has been very successful at the first two. I think there is an opportunity to be somewhat more strategic in providing the reference point for future research directions, largely through the input of the editorial team and by paying close attention to JHM readers.”
The Right Choice
An eight-person committee embarked on a five-month search to identify the ideal candidate to replace JHM ’s founding editor-in-chief, Mark Williams, MD, FACP, FHM. The intense process reflected the search committee’s goal that the new editor strengthen the journal and the society it represents, says committee member Harold C. Sox, MD, MACP, a noted internist and author who served as editor of Annals of Internal Medicine from 2001 to 2009.
“A journal published by a professional organization is far and away the most visible manifestation of the organization and its values and ambitions,” Dr. Sox says. “The appointment was anything but pro forma. It had to be the right choice.”
Committee members wanted to select a physician who had a strong scientific background, the judgment required to be a leader, and a desire to better position the journal as a source for first-rate articles in the competitive landscape of medical publishing, Dr. Sox says. They also searched for a candidate who had considerable experience writing for publication.
Dr. Auerbach, who served as deputy editor of the Journal of General Internal Medicine from 2004 to 2007 and as a JHM reviewer, has been published in the New England Journal of Medicine, the Journal of the American Medical Association, Annals of Internal Medicine, and the Archives of Internal Medicine.
As editor of Annals, Dr. Sox shared responsibility for publishing two of Dr. Auerbach’s articles.
“I know something about how Dr. Auerbach thinks about science and about his own personal scientific standards—wanting to get it right, writing it in a way that people know exactly what happened, and interpreting the results in a way that will stand up over time, rather than trying to read too much into the results,” Dr. Sox says. “We also talked with some of the candidates about their strategy for trying to make the journal better, and I thought he showed himself to be very analytic and strategic in his thinking. I’m confident he’s going to move the journal forward.”
Dr. Auerbach believes his experience as a deputy editor and his mentoring role at UCSF, which requires him to provide constructive feedback on investigators’ papers, has prepared him for his new role.
“I have a good sense for how to approach the review process with the goal of getting a paper to the point where it can be accepted into a journal and, if not, have it leave the review process much better than it was when it arrived,” he says.
His research background also complements his new role, he says.
“Research is a way to take care of patients I never see and to disseminate things that are broadly applicable … that other people can look at and use to improve their healthcare delivery,” he says. “I see the journal as doing many of the same things.”
Strategies for Success
Dr. Auerbach, who has worked closely with Dr. Williams in recent months to ensure a smooth transition, says he does not intend to make substantial changes to the journal’s focus. But he is formulating strategies to make it more valuable and relevant to the hospitalists it serves.
Increasing the journal’s Impact Factor—currently 1.951, ranking it 40th out of 151 publications in its cohort—is one way to increase JHM ’s value, making it a destination for the highest-quality research, Dr. Auerbach says.
He also intends to solicit as many scholarly works as possible, employing “a marketing strategy and recruitment strategy all in one” to make sure JHM is on researchers’ short list of publications in which they consider submitting their work.
—Robert Wachter, MD, MHM, professor, associate chair, Department of Medicine, University of California at San Francisco, chief, division of hospital medicine, chief, medical service, UCSF Medical Center, former SHM president
He wants to take advantage of the growing number of hospitalists who are doing research and expand the journal’s reach beyond its traditional peer groups. “Are there intensive-care doctors, pulmonologists, cardiologists, or infectious-disease doctors who are doing work with hospitalists—or work that would align with hospital medicine—that could be published in JHM?” Dr. Auerbach asks, rhetorically.
He intends to empower members of his editorial team to serve as JHM ambassadors who promote the journal at professional meetings and encourage investigators who are pursuing interesting projects to submit their findings.
Dr. Auerbach expects to devise other content strategies in consultation with members of his editorial team. Certain aspects of content, such as case reports and scholarly reviews of conundrums in HM cases, could be enhanced, he says. Specific sections of the journal could be developed for effectiveness research or implementation research.
He also says he wants the journal to explore new content areas so that it appeals to readers who aren’t interested in randomized controlled trials.
He hopes to put the journal in the position to publish work supported by such new initiatives as the Patient-Centered Outcomes Research Institute and the Centers for Medicare & Medicaid Services’ Center for Medicare and Medicaid Innovation.
That might mean providing editorials or inviting papers that outline the importance of hospitalists’ involvement in those initiatives, then making sure the journal is opportunistic in reviewing and publishing high-quality work, he explains.
Andrew Auerbach, MD, MPH, SFHM
Age: 44
Academic rank: Associate professor of medicine, University of California at San Francisco’s Division of Hospital Medicine
Administrative titles at UCSF: Attending physician, Department of Medicine; director of research for division of hospital medicine; associate director of the General Medicine Research Fellowship; chair of the Clinical Content Governance Committee at UCSF Medical Center
Education: Bachelor’s degree in biochemistry from Bowdoin College, Brunswick, Maine (1988); medical degree from Dartmouth Medical School, Hanover, N.H. (1992); master’s of public health in clinical effectiveness from Harvard School of Public Health, Boston (1998)
Clinical training: Internship in internal medicine, Yale-New Haven Hospital, New Haven, Conn. (1992-1993); residency in internal medicine, Yale-New Haven Hospital, New Haven, Conn. (1993-1995); fellowship in general internal medicine, Beth Israel-Deaconess Medical Center, Boston (1996-1998)
Principal positions held: Attending physician at Moffitt-Long Hospitals in San Francisco (1998-present); clinical fellow at Beth Israel Deaconess Medical Center, Boston (1996-1998); emergency room physician at Milton Hospital, Milton, Mass. (1996-1998); physician and co-director of student health services at University of New Haven, New Haven, Conn. (1995-1996); hospitalist at Hospital of Saint Raphael, New Haven, Conn. (1995-1996); admitting officer at West Haven Veterans Administration Hospital, West Haven, Conn. (1994-1996)
Awards and honors: Western Society for Clinical Investigation, Investigator of the Year Award (2010); Orthopaedic Research Society Harris Award for Outstanding Research (2010); fellow, American College of Physicians (2009); James Rand Award for Outstanding Research Project (2008); Society of Hospital Medicine Young Investigator Award (2004)
Professional memberships: Association of Chiefs of General Internal Medicine, Society of Hospital Medicine, American Medical Association, American College of Physicians, Society of General Internal Medicine
Most Significant Co-Authored Publications
- Rothberg M, Maselli J, Pekow P, Lindenauer P, Auerbach AD. Cost-effectiveness of changes in care at U.S. hospitals from 2000-2004. In press, Health Affairs.
- Auerbach AD, Wachter RM, Cheng H, et al. Effects of a neurosurgery-hospitalist comanagement model on outcomes of patients with neurosurgical illnesses. In press, Arch Int Med.
- Auerbach AD, Hilton JF, Maselli JM, Pekow P, Rothberg M, Lindenauer PK. Follow the crowd or shop for the best: How volume and care quality influence outcomes of cardiac surgery. Annals Int Med. 2009;150(10):696-704.
- Auerbach AD, Landefeld CS, Shojania KS. The tension between needing to improve care and knowing how to do it. N Engl J Med. 2007;357(6):608-613.
- Lindenauer PK, Pekow P, Rothberg M, Benjamin E, Auerbach AD. Outcomes of patients treated by hospitalists, general internists, and family physicians. N Engl J Med. 2007;357(25):2589-2600.
Bold New Direction
The JHM team, which partners SHM leaders and the journal’s publisher, Wiley-Blackwell Inc., is revisiting the journal’s press relations and editorial strategy (Wiley-Blackwell also publishes The Hospitalist). Discussions also are under way about whether to increase the journal’s publishing frequency, as well as how the journal can increase its digital footprint and take advantage of social media outlets to increase the usability and visibility of its offerings, Dr. Auerbach says.
“One side effect of JHM ’s growth has been growing constraints on space to publish excellent work,” he says. “Our team will focus a great deal on strategies to increase JHM ’s ability to review and publish the many outstanding papers submitted each year.”
Dr. Williams characterizes Dr. Auerbach’s appointment as “a software upgrade to JHM 2.0.”
Dr. Auerbach’s connection to the top-ranked division of hospital medicine in the country provides instant credibility to the journal, and his diverse contacts should help his efforts to solicit an increasing volume of high-quality submissions, Dr. Williams says.
His appointment also can reinvigorate members of the editorial team, motivating them to step out of their comfort zone and embrace the challenge of adapting to the rapidly changing world of hospital medicine.
“He’ll bring renewed enthusiasm,” says Dr. Williams, a former SHM president who is a professor and chief of the Division of Hospital Medicine at Northwestern University’s Feinberg School of Medicine in Chicago. “I firmly believe that’s essential for any growing enterprise. You need an infusion of new energy and fresh thinking.”
With Dr. Auerbach at the helm, JHM is well-positioned to attract well-done, systematic reviews while appealing to authors who are writing about such relevant topics as change management, collaboration, models of care, and transitional care, says deputy editor Brian Harte, MD, SFHM, chief operating officer of Hillcrest Hospital in Mayfield Heights, Ohio, and chairman of hospital medicine at The Cleveland Clinic.
“I hope people say under Dr. Auerbach’s tenure we continued to innovate and do things as an editorial group that other journals hadn’t thought of doing, were not nimble enough to do, or were not creative enough to do,” Dr. Harte says. “And that it was an incubator of novel and innovative and, ultimately, very effective ideas that took the journal into strategic directions that other journals weren’t bold enough to go in.”
Dr. Auerbach is more than capable of steering JHM in those new directions, according to his mentor, Robert Wachter, MD, MHM, professor and associate chairman of the Department of Medicine at the University of California at San Francisco, and chief of the division of hospital medicine and chief of medical service at UCSF Medical Center, former SHM president, and author of the blog Wachter’s World (www.wachtersworld.com).
Dr. Auerbach is a broad thinker who is capable of recognizing what issues are important to his field before they become obvious to others, a trait that will help him to use the journal to help chart the course for HM, Dr. Wachter says.
He also has the perfect personality for the job. “He is the most doggedly persistent person I’ve ever met,” Dr. Wachter says. “I’ve seen him go through setbacks that would have caused lesser mortals to give up their ideas.…He’s like a prizefighter. He sits in the corner for a little bit, has someone dab the wounds, and then comes back out again for the next round and swings a little bit harder.
“I’ve learned never to underestimate him. It’s an amazing characteristic, and it’s one that is very useful doing something like running a journal.”
Mark Leiser is a freelance writer based in New Jersey.
Andrew Auerbach, MD, MPH, SFHM, is an associate professor at one of the most highly regarded academic medical centers in the country, and he is a nationally respected researcher whose work has been published in prominent scientific publications.
His peers believe both roles prepared him well for his newest endeavor: editor-in-chief of the Journal of Hospital Medicine. His five-year term begins in January.
Dr. Auerbach’s affiliation with the University of California at San Francisco (UCSF), where he also serves as director of research for the division of hospital medicine and associate director of the General Medicine Research Fellowship, lends instant legitimacy to the journal, editorial team members say. His research has focused on evaluation of care-delivery models and methods for improving the measurement of quality of care.
That background, medical publishing experts believe, gives Dr. Auerbach a foundation in scientific accuracy and reporting transparency that is integral to a new editor’s effort to expand a journal’s reach and increase the quality of work it publishes.
Dr. Auerbach intends to do both, continuing JHM’s growth and solidifying it as the go-to resource for hospitalists. “It is an incredibly good platform for hospitalists to publish their work,” he adds. “I want people to look back at the journal in five years and say it’s even better than it is now.”
As Dr. Auerbach prepares to take over as editor-in-chief, he is focusing his efforts on how to best build on the success the journal has enjoyed since its 2006 launch.
“JHM’s core values are to reflect the field of hospital medicine broadly, to provide a venue where the field’s current scholarly work can be published, and to provide a point of reference for where future scholarly work might be directed and published,” he says. “JHM has been very successful at the first two. I think there is an opportunity to be somewhat more strategic in providing the reference point for future research directions, largely through the input of the editorial team and by paying close attention to JHM readers.”
The Right Choice
An eight-person committee embarked on a five-month search to identify the ideal candidate to replace JHM ’s founding editor-in-chief, Mark Williams, MD, FACP, FHM. The intense process reflected the search committee’s goal that the new editor strengthen the journal and the society it represents, says committee member Harold C. Sox, MD, MACP, a noted internist and author who served as editor of Annals of Internal Medicine from 2001 to 2009.
“A journal published by a professional organization is far and away the most visible manifestation of the organization and its values and ambitions,” Dr. Sox says. “The appointment was anything but pro forma. It had to be the right choice.”
Committee members wanted to select a physician who had a strong scientific background, the judgment required to be a leader, and a desire to better position the journal as a source for first-rate articles in the competitive landscape of medical publishing, Dr. Sox says. They also searched for a candidate who had considerable experience writing for publication.
Dr. Auerbach, who served as deputy editor of the Journal of General Internal Medicine from 2004 to 2007 and as a JHM reviewer, has been published in the New England Journal of Medicine, the Journal of the American Medical Association, Annals of Internal Medicine, and the Archives of Internal Medicine.
As editor of Annals, Dr. Sox shared responsibility for publishing two of Dr. Auerbach’s articles.
“I know something about how Dr. Auerbach thinks about science and about his own personal scientific standards—wanting to get it right, writing it in a way that people know exactly what happened, and interpreting the results in a way that will stand up over time, rather than trying to read too much into the results,” Dr. Sox says. “We also talked with some of the candidates about their strategy for trying to make the journal better, and I thought he showed himself to be very analytic and strategic in his thinking. I’m confident he’s going to move the journal forward.”
Dr. Auerbach believes his experience as a deputy editor and his mentoring role at UCSF, which requires him to provide constructive feedback on investigators’ papers, has prepared him for his new role.
“I have a good sense for how to approach the review process with the goal of getting a paper to the point where it can be accepted into a journal and, if not, have it leave the review process much better than it was when it arrived,” he says.
His research background also complements his new role, he says.
“Research is a way to take care of patients I never see and to disseminate things that are broadly applicable … that other people can look at and use to improve their healthcare delivery,” he says. “I see the journal as doing many of the same things.”
Strategies for Success
Dr. Auerbach, who has worked closely with Dr. Williams in recent months to ensure a smooth transition, says he does not intend to make substantial changes to the journal’s focus. But he is formulating strategies to make it more valuable and relevant to the hospitalists it serves.
Increasing the journal’s Impact Factor—currently 1.951, ranking it 40th out of 151 publications in its cohort—is one way to increase JHM ’s value, making it a destination for the highest-quality research, Dr. Auerbach says.
He also intends to solicit as many scholarly works as possible, employing “a marketing strategy and recruitment strategy all in one” to make sure JHM is on researchers’ short list of publications in which they consider submitting their work.
—Robert Wachter, MD, MHM, professor, associate chair, Department of Medicine, University of California at San Francisco, chief, division of hospital medicine, chief, medical service, UCSF Medical Center, former SHM president
He wants to take advantage of the growing number of hospitalists who are doing research and expand the journal’s reach beyond its traditional peer groups. “Are there intensive-care doctors, pulmonologists, cardiologists, or infectious-disease doctors who are doing work with hospitalists—or work that would align with hospital medicine—that could be published in JHM?” Dr. Auerbach asks, rhetorically.
He intends to empower members of his editorial team to serve as JHM ambassadors who promote the journal at professional meetings and encourage investigators who are pursuing interesting projects to submit their findings.
Dr. Auerbach expects to devise other content strategies in consultation with members of his editorial team. Certain aspects of content, such as case reports and scholarly reviews of conundrums in HM cases, could be enhanced, he says. Specific sections of the journal could be developed for effectiveness research or implementation research.
He also says he wants the journal to explore new content areas so that it appeals to readers who aren’t interested in randomized controlled trials.
He hopes to put the journal in the position to publish work supported by such new initiatives as the Patient-Centered Outcomes Research Institute and the Centers for Medicare & Medicaid Services’ Center for Medicare and Medicaid Innovation.
That might mean providing editorials or inviting papers that outline the importance of hospitalists’ involvement in those initiatives, then making sure the journal is opportunistic in reviewing and publishing high-quality work, he explains.
Andrew Auerbach, MD, MPH, SFHM
Age: 44
Academic rank: Associate professor of medicine, University of California at San Francisco’s Division of Hospital Medicine
Administrative titles at UCSF: Attending physician, Department of Medicine; director of research for division of hospital medicine; associate director of the General Medicine Research Fellowship; chair of the Clinical Content Governance Committee at UCSF Medical Center
Education: Bachelor’s degree in biochemistry from Bowdoin College, Brunswick, Maine (1988); medical degree from Dartmouth Medical School, Hanover, N.H. (1992); master’s of public health in clinical effectiveness from Harvard School of Public Health, Boston (1998)
Clinical training: Internship in internal medicine, Yale-New Haven Hospital, New Haven, Conn. (1992-1993); residency in internal medicine, Yale-New Haven Hospital, New Haven, Conn. (1993-1995); fellowship in general internal medicine, Beth Israel-Deaconess Medical Center, Boston (1996-1998)
Principal positions held: Attending physician at Moffitt-Long Hospitals in San Francisco (1998-present); clinical fellow at Beth Israel Deaconess Medical Center, Boston (1996-1998); emergency room physician at Milton Hospital, Milton, Mass. (1996-1998); physician and co-director of student health services at University of New Haven, New Haven, Conn. (1995-1996); hospitalist at Hospital of Saint Raphael, New Haven, Conn. (1995-1996); admitting officer at West Haven Veterans Administration Hospital, West Haven, Conn. (1994-1996)
Awards and honors: Western Society for Clinical Investigation, Investigator of the Year Award (2010); Orthopaedic Research Society Harris Award for Outstanding Research (2010); fellow, American College of Physicians (2009); James Rand Award for Outstanding Research Project (2008); Society of Hospital Medicine Young Investigator Award (2004)
Professional memberships: Association of Chiefs of General Internal Medicine, Society of Hospital Medicine, American Medical Association, American College of Physicians, Society of General Internal Medicine
Most Significant Co-Authored Publications
- Rothberg M, Maselli J, Pekow P, Lindenauer P, Auerbach AD. Cost-effectiveness of changes in care at U.S. hospitals from 2000-2004. In press, Health Affairs.
- Auerbach AD, Wachter RM, Cheng H, et al. Effects of a neurosurgery-hospitalist comanagement model on outcomes of patients with neurosurgical illnesses. In press, Arch Int Med.
- Auerbach AD, Hilton JF, Maselli JM, Pekow P, Rothberg M, Lindenauer PK. Follow the crowd or shop for the best: How volume and care quality influence outcomes of cardiac surgery. Annals Int Med. 2009;150(10):696-704.
- Auerbach AD, Landefeld CS, Shojania KS. The tension between needing to improve care and knowing how to do it. N Engl J Med. 2007;357(6):608-613.
- Lindenauer PK, Pekow P, Rothberg M, Benjamin E, Auerbach AD. Outcomes of patients treated by hospitalists, general internists, and family physicians. N Engl J Med. 2007;357(25):2589-2600.
Bold New Direction
The JHM team, which partners SHM leaders and the journal’s publisher, Wiley-Blackwell Inc., is revisiting the journal’s press relations and editorial strategy (Wiley-Blackwell also publishes The Hospitalist). Discussions also are under way about whether to increase the journal’s publishing frequency, as well as how the journal can increase its digital footprint and take advantage of social media outlets to increase the usability and visibility of its offerings, Dr. Auerbach says.
“One side effect of JHM ’s growth has been growing constraints on space to publish excellent work,” he says. “Our team will focus a great deal on strategies to increase JHM ’s ability to review and publish the many outstanding papers submitted each year.”
Dr. Williams characterizes Dr. Auerbach’s appointment as “a software upgrade to JHM 2.0.”
Dr. Auerbach’s connection to the top-ranked division of hospital medicine in the country provides instant credibility to the journal, and his diverse contacts should help his efforts to solicit an increasing volume of high-quality submissions, Dr. Williams says.
His appointment also can reinvigorate members of the editorial team, motivating them to step out of their comfort zone and embrace the challenge of adapting to the rapidly changing world of hospital medicine.
“He’ll bring renewed enthusiasm,” says Dr. Williams, a former SHM president who is a professor and chief of the Division of Hospital Medicine at Northwestern University’s Feinberg School of Medicine in Chicago. “I firmly believe that’s essential for any growing enterprise. You need an infusion of new energy and fresh thinking.”
With Dr. Auerbach at the helm, JHM is well-positioned to attract well-done, systematic reviews while appealing to authors who are writing about such relevant topics as change management, collaboration, models of care, and transitional care, says deputy editor Brian Harte, MD, SFHM, chief operating officer of Hillcrest Hospital in Mayfield Heights, Ohio, and chairman of hospital medicine at The Cleveland Clinic.
“I hope people say under Dr. Auerbach’s tenure we continued to innovate and do things as an editorial group that other journals hadn’t thought of doing, were not nimble enough to do, or were not creative enough to do,” Dr. Harte says. “And that it was an incubator of novel and innovative and, ultimately, very effective ideas that took the journal into strategic directions that other journals weren’t bold enough to go in.”
Dr. Auerbach is more than capable of steering JHM in those new directions, according to his mentor, Robert Wachter, MD, MHM, professor and associate chairman of the Department of Medicine at the University of California at San Francisco, and chief of the division of hospital medicine and chief of medical service at UCSF Medical Center, former SHM president, and author of the blog Wachter’s World (www.wachtersworld.com).
Dr. Auerbach is a broad thinker who is capable of recognizing what issues are important to his field before they become obvious to others, a trait that will help him to use the journal to help chart the course for HM, Dr. Wachter says.
He also has the perfect personality for the job. “He is the most doggedly persistent person I’ve ever met,” Dr. Wachter says. “I’ve seen him go through setbacks that would have caused lesser mortals to give up their ideas.…He’s like a prizefighter. He sits in the corner for a little bit, has someone dab the wounds, and then comes back out again for the next round and swings a little bit harder.
“I’ve learned never to underestimate him. It’s an amazing characteristic, and it’s one that is very useful doing something like running a journal.”
Mark Leiser is a freelance writer based in New Jersey.
ONLINE EXCLUSIVE: Physician leaders discuss JHM's new editor-in-chief
Click here to listen to Dr. Wachter.
Click here to listen to Dr. Sox.
Click here to listen to Dr. Wachter.
Click here to listen to Dr. Sox.
Click here to listen to Dr. Wachter.
Click here to listen to Dr. Sox.
What Is the Best Approach for the Evaluation and Management of Endocrine Incidentalomas?
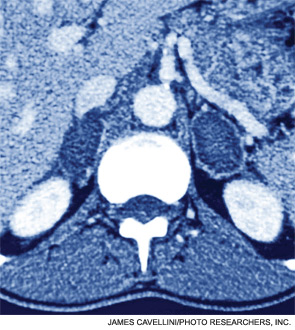
Case
A 54-year-old man with a history of hypertension treated with hydrocholorothiazide and Type 2 diabetes mellitus is admitted with abdominal pain and found to have an incidental 2.1-cm left adrenal mass on CT scan of the abdomen. He denies symptoms of headache, palpitations, weight gain, or muscle weakness. His exam is significant for mildly elevated blood pressure. What is the best approach for evaluation and management of this incidental finding?
Overview
Incidentalomas are mass lesions that are inadvertently discovered during radiolographic diagnostic testing or treatment for other clinical conditions that are unrelated to the incidental mass. In recent decades, improvements in radiographic diagnostic techniques and sensitivity have led to increasing discovery of incidental lesions that are often in the absence of clinical signs or symptoms.1 Three commonly discovered lesions by hospitalists are pituitary, thyroid, and adrenal incidentalomas.2 The concerns associated with these findings relate to the potential for dysfunctional hormone secretion or malignancy.
Patients found with pituitary incidentalomas can be susceptible to several types of adverse outcomes: hormonal hypersecretion, hypopituitarism, neurologic morbidity due to tumor size, and malignancy in rare cases. Thyroid incidentalomas are impalpable nodules discovered in the setting of ultrasound or cross-sectional neck scans, such as positron emission tomography (PET) scans. Discovery of a thyroid incidentaloma raises concern for thyroid malignancy.3 The increased use of abdominal ultrasound, CT scans, and MRI has fueled the growing incidence of adrenal incidentalomas (AIs).
The discovery of an endocrine incidentaloma in the inpatient setting warrants a systematic approach that includes both diagnostic and potentially therapeutic management. A hospitalist should consider an approach that includes (see Table 1):
- Characterization of the incidentaloma, including clinical signs and symptoms, size, hormonal function, and malignant potential;
- Immediate management, including medical versus surgical treatment; and
- Post-discharge management, including monitoring.
Review of the Data
Pituitary incidentalomas. The prevalence of pituitary incidentalomas found by CT ranges from 3.7% to 20%, while the prevalence found by MRI approximates 10%. Autopsy studies have revealed a prevalence ranging from 1.5% to 26.7% for adenomas less than 10 mm, considered to be microadenomas. Broad categories of etiologies should be considered: pituitary adenoma, nonpituitary tumors, vascular lesions, infiltrative disorders, and others (see Table 2). The majority of pituitary adenomas secrete prolactin (30% to 40%) or are nonsecreting (30% to 40%). Adenomas secreting adrenocorticotropin hormone (ACTH, 2% to 10%), growth hormone (GH, 2% to 10%), thyroid-stimulating hormone (TSH, <1%), follicle-stimulating hormone (FSH), and luteinizing hormone (LH) are much less common.2 Significant morbidity and premature mortality are associated with hyperprolactinemia, acromegaly (growth hormone excess), Cushing’s syndrome, and hyperthyroidism. Additionally, up to 41% of patients with macroadenomas were found to have varying degrees of hypopituitarism due to compression of the hypothalamus, the hypothalamic-pituitary stalk, or the pituitary itself.4
Recently, the Endocrine Society released consensus recommendations to guide the evaluation and treatment of pituitary incidentalomas, which are included in the approach outlined below.5 A detailed history and physical examination should be obtained with specific inquiry as to signs and symptoms of hormonal excess and mass effect from the tumor. Examples of symptoms of hormone excess can include:
- Prolactin: menstrual irregularity, anovulation, infertility, decreased libido, impotence, osteoporosis;
- Growth hormone: high frequency of colonic polyps and colon cancer (chronic excess);
- TSH: thyrotoxicosis, atrial fibrillation; and
- ACTH: hypertension, osteoporosis, accelerated vascular disease.
Symptoms related to the mass effect of the tumor include visual field defects and hypopituitarism related to the deficient hormone, including:
- FSH/LH: oligomenorrhea, decreased libido, infertility;
- TSH: hypothyroidism (weight gain, constipation, cold intolerance);
- ACTH: adrenal insufficiency (hypotension, hypoglycemia, weight loss); and
- ADH: polyuria, polydypsia.
The size and location of the pituitary lesion must be assessed. Lesions greater than 10 mm are considered macroademonas, and their size will affect their management. If the lesion was initially identified by CT scan, an MRI is recommended to better evaluate it.5 If the MRI locates the incidentaloma abutting the optic nerve or chiasm, then the patient should undergo a formal visual field examination.
Indications for an inpatient surgical referral for treatment include: a lesion larger than 2 cm, evidence of mass effect such as visual field defects, neurologic compromise, opthalmoplegia, hypopituitarism, a tumor abutting the optic nerve or chiasm, pituitary apoplexy, and hypersecretion of hormones other than prolactin. Patients with prolactinomas warrant an inpatient endo-crinology consult and may need medical management with a dopamine agonist. Hormone replacement therapy can also be provided for patients with hypopituitarism.2,5
For patients who do not meet the criteria for inpatient surgical therapy, follow-up management must be arranged at the time of discharge. Clinical, laboratory assessment, and an MRI should be scheduled six months after the initial finding of the incidentaloma with the patient’s PCP or with an endocrinologist.5
Thyroid incidentalomas. The prevalence of thyroid nodules based on ultrasound studies ranges from 19% to 46%, with autopsy studies estimating an incidence of approximately 50%.2,6 Incidence of thyroid nodules also increases with age, as almost 60% of people over the age of 60 harbor a thyroid incidentaloma. The rate of malignancy in the general population has ranged between 8% and 24%; however, in the last decade, the rates have increased by 2.4 times as more sophisticated ultrasound techniques and liberal use of fine-needle aspiration (FNA) biopsies have detected subclinical disease.7,8
Etiologies for incidental thyroid nodules can be divided into benign and malignant causes. Benign etiologies include thyroid cyst (simple or complex), multinodular goiter, and Hashimoto’s thryoiditis, while malignant causes include papillary, medullary, follicular, Hurthle cell, and anaplastic carcinomas, thyroid lymphomas, and rare instances of metastatic cancers.2,3
Targeted history and physical examination helps to characterize the thyroid incidentaloma. Historical features, such as palpitations, weight loss, anxiety, new onset atrial fibrillation, or menstrual irregularities, coupled with tachycardia, tremors, proximal muscle weakness, and a palpable nodule aid in the diagnosis of hyperthyroidism. Findings such as a family history of thyroid cancer, symptoms of hoarseness or dysphagia, rapid growth of the nodule, environmental or history of head or neck irradiation along with physical findings of a hard, fixed nodule, or cervical lymphadenopathy increase the suspicion for malignancy.2,7
The functionality of the nodule can be assessed by checking TSH, free T3, and free T4 levels. Suppression of TSH (< 0.1 mU/L) with elevated levels of free T3 and T4 indicates nodule production of excess thyroid hormone and warrants thyroid scintography. Thyroid scintography will identify the nodule as “hot” (hyperfunctioning) or “cold” (nonfunctioning).2
Regardless of the radiographic modality that initially identified the thyroid incidentaloma, a dedicated thyroid high-resolution ultrasound should be ordered to assess the size, multiplicity (single or multinodular), location, and character (solid, cystic, or mixed).7
Recommendations for proceeding to FNA to evaluate for malignancy differ among subspecialty societies. Generally, nodules larger than 1 cm or nodules smaller than 1 cm with risk factors for malignancy should be referred for FNA.2,7
If diagnostic workup identifies a patient with hyperthyroidism due to an autonomously functional nodule or a nodule that may be at high risk for malignancy, it is appropriate to involve an endocrinologist and possibly a surgical subspecialist prior to discharge. Management of hyperthyroidism can include starting antithyroid agents (methimazole or propylthiouracil), radioactive iodine ablation, or referral for surgery.
Preparation for discharge of the patient whose incidentaloma is nonfunctional or does not appear to be malignant should include appointments to recheck thyroid hormone levels, including TSH as well as a thyroid ultrasound within one year of the initial discovery.
Adrenal incidentaloma. The prevalence of AIs found by CT of the abdomen ranges from 0.4% to 4%, while autopsy studies have found a prevalence of 1.4% to 9% with increasing prevalence with age.2,9,10 The majority of AIs are benign and nonfunctioning adenomas, in the absence of known malignancy. Other differential diagnoses include Cushing’s syndrome, pheochromocytoma, adrenocortical adenoma, aldosteronoma, and metastatic lesions.
Because functioning adrenal incidentalomas may be clinically silent, any patient found with an AI must undergo biochemical workup as part of their evaluation to assess for pheochromocytoma, Cushing’s syndrome, and if he or she has a history of hypertension or hyperaldosteronism (Conn’s syndrome). Table 3 outlines the approach for characterizing adrenal incidentalomas.2,11,12 An important point is that imaging studies are not useful in distinguishing a functioning versus nonfunctioning tumor but rather can help to discriminate malignant lesions.11
Inpatient surgical consult for resection is indicated if the patient is found to have pheochromocytoma, clinically apparent functioning adrenocortical adenoma, or a tumor size greater than 4 cm. Consultation with an endocrinologist is also recommended if biochemical tests are positive. If the diagnostic workup leads to suspicion for infection or metastatic disease, the patient should be referred for FNA.2,12
For patients whose lesions do not require surgical resection, repeat CT scan of the abdomen is recommended six months from the initial finding. Hospitalists should also arrange for the patient to repeat biochemical testing, including an overnight dexamethasone test.12,13
Back to the Case
The patient underwent biochemical testing and was found to have normal levels of plasma-free metanephrines, a plasma aldosterone, plasma renin activity ratio less than 20, and a serum cortisol level of 7 mg/dL after his overnight dexamethasone suppression test. The 24-hour urine collection for free cortisol revealed elevated levels of cortisol in the urine, and the ACTH level was low.
Endocrinology and endocrine surgery teams were consulted, and recommended surgical resection. After surgical resection of his tumor, the patient was started on glucocorticoid replacement and was discharged with a follow-up appointment with endocrinology.
Bottom Line
An inpatient approach to endocrine incidentalomas should include characterization of the clinical signs and symptoms, size, function, and malignant potential of the lesion. Based on this, inpatient surgical or medical management can be determined. Post-discharge management should include arrangements for surveillance testing and follow-up with appropriate subspecialists.
Dr. Tad-y is assistant professor of medicine and a hospitalist at the University of Colorado Denver.
References
- Aron DC, Howlett TA. Pituitary incidentalomas. Endocrinol Metab Clin North Am. 2000;29:205-221.
- Shirodkar M, Jabbour SA. Endocrine incidentalomas. Int J Clin Pract. 2008;62:1423-1431.
- Burguera B, Gharib H. Thyroid incidentalomas. Prevalence, diagnosis, significance, and management. Endocrinol Metab Clin North Am. 2000;29:187-203.
- Molitch ME. Nonfunctioning pituitary tumors and pituitary incidentalomas. Endocrinol Metab Clin North Am. 2008;37:151-171, xi.
- Freda PU, Beckers AM, Katznelson L, et al. Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:894-904.
- Gough J, Scott-Coombes D, Fausto Palazzo F. Thyroid incidentaloma: an evidence-based assessment of management strategy. World J Surg. 2008;32:1264-1268.
- Iyer NG, Shaha AR, Silver CE, et al. Thyroid incidentalomas: to treat or not to treat. Eur Arch Otorhinolaryngol. 2010;267:1019-1026.
- Jin J, Wilhelm SM, McHenry CR. Incidental thyroid nodule: patterns of diagnosis and rate of malignancy. Am J Surg. 2009;197:320-324.
- Davenport C, Liew L, Doherty B, et al. The prevalence of adrenal incidentaloma in routine clinical practice. Endocrine. 2011;40:80-83.
- Zeiger MA, Siegelman SS, Hamrahian AH. Medical and surgical evaluation and treatment of adrenal incidentalomas. J Clin Endocrinol Metab. 2011;96: 2004-2015.
- Zeiger MA, Thompson GB, Duh QY, et al. American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas: executive summary of recommendations. Endocr Pract. 2009;15:450-453.
- NIH state-of-the-science statement on management of the clinically inapparent adrenal mass (“incidentaloma”). NIH Consens State Sci Statements. 2002;19:1-25.
- Young WF. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med. 2007;356:601-610.
- Chidiac RM, Aron DC. Incidentalomas. A disease of modern technology. Endocrinol Metab Clin North Am. 1997;26:233-253.
Case
A 54-year-old man with a history of hypertension treated with hydrocholorothiazide and Type 2 diabetes mellitus is admitted with abdominal pain and found to have an incidental 2.1-cm left adrenal mass on CT scan of the abdomen. He denies symptoms of headache, palpitations, weight gain, or muscle weakness. His exam is significant for mildly elevated blood pressure. What is the best approach for evaluation and management of this incidental finding?
Overview
Incidentalomas are mass lesions that are inadvertently discovered during radiolographic diagnostic testing or treatment for other clinical conditions that are unrelated to the incidental mass. In recent decades, improvements in radiographic diagnostic techniques and sensitivity have led to increasing discovery of incidental lesions that are often in the absence of clinical signs or symptoms.1 Three commonly discovered lesions by hospitalists are pituitary, thyroid, and adrenal incidentalomas.2 The concerns associated with these findings relate to the potential for dysfunctional hormone secretion or malignancy.
Patients found with pituitary incidentalomas can be susceptible to several types of adverse outcomes: hormonal hypersecretion, hypopituitarism, neurologic morbidity due to tumor size, and malignancy in rare cases. Thyroid incidentalomas are impalpable nodules discovered in the setting of ultrasound or cross-sectional neck scans, such as positron emission tomography (PET) scans. Discovery of a thyroid incidentaloma raises concern for thyroid malignancy.3 The increased use of abdominal ultrasound, CT scans, and MRI has fueled the growing incidence of adrenal incidentalomas (AIs).
The discovery of an endocrine incidentaloma in the inpatient setting warrants a systematic approach that includes both diagnostic and potentially therapeutic management. A hospitalist should consider an approach that includes (see Table 1):
- Characterization of the incidentaloma, including clinical signs and symptoms, size, hormonal function, and malignant potential;
- Immediate management, including medical versus surgical treatment; and
- Post-discharge management, including monitoring.
Review of the Data
Pituitary incidentalomas. The prevalence of pituitary incidentalomas found by CT ranges from 3.7% to 20%, while the prevalence found by MRI approximates 10%. Autopsy studies have revealed a prevalence ranging from 1.5% to 26.7% for adenomas less than 10 mm, considered to be microadenomas. Broad categories of etiologies should be considered: pituitary adenoma, nonpituitary tumors, vascular lesions, infiltrative disorders, and others (see Table 2). The majority of pituitary adenomas secrete prolactin (30% to 40%) or are nonsecreting (30% to 40%). Adenomas secreting adrenocorticotropin hormone (ACTH, 2% to 10%), growth hormone (GH, 2% to 10%), thyroid-stimulating hormone (TSH, <1%), follicle-stimulating hormone (FSH), and luteinizing hormone (LH) are much less common.2 Significant morbidity and premature mortality are associated with hyperprolactinemia, acromegaly (growth hormone excess), Cushing’s syndrome, and hyperthyroidism. Additionally, up to 41% of patients with macroadenomas were found to have varying degrees of hypopituitarism due to compression of the hypothalamus, the hypothalamic-pituitary stalk, or the pituitary itself.4
Recently, the Endocrine Society released consensus recommendations to guide the evaluation and treatment of pituitary incidentalomas, which are included in the approach outlined below.5 A detailed history and physical examination should be obtained with specific inquiry as to signs and symptoms of hormonal excess and mass effect from the tumor. Examples of symptoms of hormone excess can include:
- Prolactin: menstrual irregularity, anovulation, infertility, decreased libido, impotence, osteoporosis;
- Growth hormone: high frequency of colonic polyps and colon cancer (chronic excess);
- TSH: thyrotoxicosis, atrial fibrillation; and
- ACTH: hypertension, osteoporosis, accelerated vascular disease.
Symptoms related to the mass effect of the tumor include visual field defects and hypopituitarism related to the deficient hormone, including:
- FSH/LH: oligomenorrhea, decreased libido, infertility;
- TSH: hypothyroidism (weight gain, constipation, cold intolerance);
- ACTH: adrenal insufficiency (hypotension, hypoglycemia, weight loss); and
- ADH: polyuria, polydypsia.
The size and location of the pituitary lesion must be assessed. Lesions greater than 10 mm are considered macroademonas, and their size will affect their management. If the lesion was initially identified by CT scan, an MRI is recommended to better evaluate it.5 If the MRI locates the incidentaloma abutting the optic nerve or chiasm, then the patient should undergo a formal visual field examination.
Indications for an inpatient surgical referral for treatment include: a lesion larger than 2 cm, evidence of mass effect such as visual field defects, neurologic compromise, opthalmoplegia, hypopituitarism, a tumor abutting the optic nerve or chiasm, pituitary apoplexy, and hypersecretion of hormones other than prolactin. Patients with prolactinomas warrant an inpatient endo-crinology consult and may need medical management with a dopamine agonist. Hormone replacement therapy can also be provided for patients with hypopituitarism.2,5
For patients who do not meet the criteria for inpatient surgical therapy, follow-up management must be arranged at the time of discharge. Clinical, laboratory assessment, and an MRI should be scheduled six months after the initial finding of the incidentaloma with the patient’s PCP or with an endocrinologist.5
Thyroid incidentalomas. The prevalence of thyroid nodules based on ultrasound studies ranges from 19% to 46%, with autopsy studies estimating an incidence of approximately 50%.2,6 Incidence of thyroid nodules also increases with age, as almost 60% of people over the age of 60 harbor a thyroid incidentaloma. The rate of malignancy in the general population has ranged between 8% and 24%; however, in the last decade, the rates have increased by 2.4 times as more sophisticated ultrasound techniques and liberal use of fine-needle aspiration (FNA) biopsies have detected subclinical disease.7,8
Etiologies for incidental thyroid nodules can be divided into benign and malignant causes. Benign etiologies include thyroid cyst (simple or complex), multinodular goiter, and Hashimoto’s thryoiditis, while malignant causes include papillary, medullary, follicular, Hurthle cell, and anaplastic carcinomas, thyroid lymphomas, and rare instances of metastatic cancers.2,3
Targeted history and physical examination helps to characterize the thyroid incidentaloma. Historical features, such as palpitations, weight loss, anxiety, new onset atrial fibrillation, or menstrual irregularities, coupled with tachycardia, tremors, proximal muscle weakness, and a palpable nodule aid in the diagnosis of hyperthyroidism. Findings such as a family history of thyroid cancer, symptoms of hoarseness or dysphagia, rapid growth of the nodule, environmental or history of head or neck irradiation along with physical findings of a hard, fixed nodule, or cervical lymphadenopathy increase the suspicion for malignancy.2,7
The functionality of the nodule can be assessed by checking TSH, free T3, and free T4 levels. Suppression of TSH (< 0.1 mU/L) with elevated levels of free T3 and T4 indicates nodule production of excess thyroid hormone and warrants thyroid scintography. Thyroid scintography will identify the nodule as “hot” (hyperfunctioning) or “cold” (nonfunctioning).2
Regardless of the radiographic modality that initially identified the thyroid incidentaloma, a dedicated thyroid high-resolution ultrasound should be ordered to assess the size, multiplicity (single or multinodular), location, and character (solid, cystic, or mixed).7
Recommendations for proceeding to FNA to evaluate for malignancy differ among subspecialty societies. Generally, nodules larger than 1 cm or nodules smaller than 1 cm with risk factors for malignancy should be referred for FNA.2,7
If diagnostic workup identifies a patient with hyperthyroidism due to an autonomously functional nodule or a nodule that may be at high risk for malignancy, it is appropriate to involve an endocrinologist and possibly a surgical subspecialist prior to discharge. Management of hyperthyroidism can include starting antithyroid agents (methimazole or propylthiouracil), radioactive iodine ablation, or referral for surgery.
Preparation for discharge of the patient whose incidentaloma is nonfunctional or does not appear to be malignant should include appointments to recheck thyroid hormone levels, including TSH as well as a thyroid ultrasound within one year of the initial discovery.
Adrenal incidentaloma. The prevalence of AIs found by CT of the abdomen ranges from 0.4% to 4%, while autopsy studies have found a prevalence of 1.4% to 9% with increasing prevalence with age.2,9,10 The majority of AIs are benign and nonfunctioning adenomas, in the absence of known malignancy. Other differential diagnoses include Cushing’s syndrome, pheochromocytoma, adrenocortical adenoma, aldosteronoma, and metastatic lesions.
Because functioning adrenal incidentalomas may be clinically silent, any patient found with an AI must undergo biochemical workup as part of their evaluation to assess for pheochromocytoma, Cushing’s syndrome, and if he or she has a history of hypertension or hyperaldosteronism (Conn’s syndrome). Table 3 outlines the approach for characterizing adrenal incidentalomas.2,11,12 An important point is that imaging studies are not useful in distinguishing a functioning versus nonfunctioning tumor but rather can help to discriminate malignant lesions.11
Inpatient surgical consult for resection is indicated if the patient is found to have pheochromocytoma, clinically apparent functioning adrenocortical adenoma, or a tumor size greater than 4 cm. Consultation with an endocrinologist is also recommended if biochemical tests are positive. If the diagnostic workup leads to suspicion for infection or metastatic disease, the patient should be referred for FNA.2,12
For patients whose lesions do not require surgical resection, repeat CT scan of the abdomen is recommended six months from the initial finding. Hospitalists should also arrange for the patient to repeat biochemical testing, including an overnight dexamethasone test.12,13
Back to the Case
The patient underwent biochemical testing and was found to have normal levels of plasma-free metanephrines, a plasma aldosterone, plasma renin activity ratio less than 20, and a serum cortisol level of 7 mg/dL after his overnight dexamethasone suppression test. The 24-hour urine collection for free cortisol revealed elevated levels of cortisol in the urine, and the ACTH level was low.
Endocrinology and endocrine surgery teams were consulted, and recommended surgical resection. After surgical resection of his tumor, the patient was started on glucocorticoid replacement and was discharged with a follow-up appointment with endocrinology.
Bottom Line
An inpatient approach to endocrine incidentalomas should include characterization of the clinical signs and symptoms, size, function, and malignant potential of the lesion. Based on this, inpatient surgical or medical management can be determined. Post-discharge management should include arrangements for surveillance testing and follow-up with appropriate subspecialists.
Dr. Tad-y is assistant professor of medicine and a hospitalist at the University of Colorado Denver.
References
- Aron DC, Howlett TA. Pituitary incidentalomas. Endocrinol Metab Clin North Am. 2000;29:205-221.
- Shirodkar M, Jabbour SA. Endocrine incidentalomas. Int J Clin Pract. 2008;62:1423-1431.
- Burguera B, Gharib H. Thyroid incidentalomas. Prevalence, diagnosis, significance, and management. Endocrinol Metab Clin North Am. 2000;29:187-203.
- Molitch ME. Nonfunctioning pituitary tumors and pituitary incidentalomas. Endocrinol Metab Clin North Am. 2008;37:151-171, xi.
- Freda PU, Beckers AM, Katznelson L, et al. Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:894-904.
- Gough J, Scott-Coombes D, Fausto Palazzo F. Thyroid incidentaloma: an evidence-based assessment of management strategy. World J Surg. 2008;32:1264-1268.
- Iyer NG, Shaha AR, Silver CE, et al. Thyroid incidentalomas: to treat or not to treat. Eur Arch Otorhinolaryngol. 2010;267:1019-1026.
- Jin J, Wilhelm SM, McHenry CR. Incidental thyroid nodule: patterns of diagnosis and rate of malignancy. Am J Surg. 2009;197:320-324.
- Davenport C, Liew L, Doherty B, et al. The prevalence of adrenal incidentaloma in routine clinical practice. Endocrine. 2011;40:80-83.
- Zeiger MA, Siegelman SS, Hamrahian AH. Medical and surgical evaluation and treatment of adrenal incidentalomas. J Clin Endocrinol Metab. 2011;96: 2004-2015.
- Zeiger MA, Thompson GB, Duh QY, et al. American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas: executive summary of recommendations. Endocr Pract. 2009;15:450-453.
- NIH state-of-the-science statement on management of the clinically inapparent adrenal mass (“incidentaloma”). NIH Consens State Sci Statements. 2002;19:1-25.
- Young WF. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med. 2007;356:601-610.
- Chidiac RM, Aron DC. Incidentalomas. A disease of modern technology. Endocrinol Metab Clin North Am. 1997;26:233-253.
Case
A 54-year-old man with a history of hypertension treated with hydrocholorothiazide and Type 2 diabetes mellitus is admitted with abdominal pain and found to have an incidental 2.1-cm left adrenal mass on CT scan of the abdomen. He denies symptoms of headache, palpitations, weight gain, or muscle weakness. His exam is significant for mildly elevated blood pressure. What is the best approach for evaluation and management of this incidental finding?
Overview
Incidentalomas are mass lesions that are inadvertently discovered during radiolographic diagnostic testing or treatment for other clinical conditions that are unrelated to the incidental mass. In recent decades, improvements in radiographic diagnostic techniques and sensitivity have led to increasing discovery of incidental lesions that are often in the absence of clinical signs or symptoms.1 Three commonly discovered lesions by hospitalists are pituitary, thyroid, and adrenal incidentalomas.2 The concerns associated with these findings relate to the potential for dysfunctional hormone secretion or malignancy.
Patients found with pituitary incidentalomas can be susceptible to several types of adverse outcomes: hormonal hypersecretion, hypopituitarism, neurologic morbidity due to tumor size, and malignancy in rare cases. Thyroid incidentalomas are impalpable nodules discovered in the setting of ultrasound or cross-sectional neck scans, such as positron emission tomography (PET) scans. Discovery of a thyroid incidentaloma raises concern for thyroid malignancy.3 The increased use of abdominal ultrasound, CT scans, and MRI has fueled the growing incidence of adrenal incidentalomas (AIs).
The discovery of an endocrine incidentaloma in the inpatient setting warrants a systematic approach that includes both diagnostic and potentially therapeutic management. A hospitalist should consider an approach that includes (see Table 1):
- Characterization of the incidentaloma, including clinical signs and symptoms, size, hormonal function, and malignant potential;
- Immediate management, including medical versus surgical treatment; and
- Post-discharge management, including monitoring.
Review of the Data
Pituitary incidentalomas. The prevalence of pituitary incidentalomas found by CT ranges from 3.7% to 20%, while the prevalence found by MRI approximates 10%. Autopsy studies have revealed a prevalence ranging from 1.5% to 26.7% for adenomas less than 10 mm, considered to be microadenomas. Broad categories of etiologies should be considered: pituitary adenoma, nonpituitary tumors, vascular lesions, infiltrative disorders, and others (see Table 2). The majority of pituitary adenomas secrete prolactin (30% to 40%) or are nonsecreting (30% to 40%). Adenomas secreting adrenocorticotropin hormone (ACTH, 2% to 10%), growth hormone (GH, 2% to 10%), thyroid-stimulating hormone (TSH, <1%), follicle-stimulating hormone (FSH), and luteinizing hormone (LH) are much less common.2 Significant morbidity and premature mortality are associated with hyperprolactinemia, acromegaly (growth hormone excess), Cushing’s syndrome, and hyperthyroidism. Additionally, up to 41% of patients with macroadenomas were found to have varying degrees of hypopituitarism due to compression of the hypothalamus, the hypothalamic-pituitary stalk, or the pituitary itself.4
Recently, the Endocrine Society released consensus recommendations to guide the evaluation and treatment of pituitary incidentalomas, which are included in the approach outlined below.5 A detailed history and physical examination should be obtained with specific inquiry as to signs and symptoms of hormonal excess and mass effect from the tumor. Examples of symptoms of hormone excess can include:
- Prolactin: menstrual irregularity, anovulation, infertility, decreased libido, impotence, osteoporosis;
- Growth hormone: high frequency of colonic polyps and colon cancer (chronic excess);
- TSH: thyrotoxicosis, atrial fibrillation; and
- ACTH: hypertension, osteoporosis, accelerated vascular disease.
Symptoms related to the mass effect of the tumor include visual field defects and hypopituitarism related to the deficient hormone, including:
- FSH/LH: oligomenorrhea, decreased libido, infertility;
- TSH: hypothyroidism (weight gain, constipation, cold intolerance);
- ACTH: adrenal insufficiency (hypotension, hypoglycemia, weight loss); and
- ADH: polyuria, polydypsia.
The size and location of the pituitary lesion must be assessed. Lesions greater than 10 mm are considered macroademonas, and their size will affect their management. If the lesion was initially identified by CT scan, an MRI is recommended to better evaluate it.5 If the MRI locates the incidentaloma abutting the optic nerve or chiasm, then the patient should undergo a formal visual field examination.
Indications for an inpatient surgical referral for treatment include: a lesion larger than 2 cm, evidence of mass effect such as visual field defects, neurologic compromise, opthalmoplegia, hypopituitarism, a tumor abutting the optic nerve or chiasm, pituitary apoplexy, and hypersecretion of hormones other than prolactin. Patients with prolactinomas warrant an inpatient endo-crinology consult and may need medical management with a dopamine agonist. Hormone replacement therapy can also be provided for patients with hypopituitarism.2,5
For patients who do not meet the criteria for inpatient surgical therapy, follow-up management must be arranged at the time of discharge. Clinical, laboratory assessment, and an MRI should be scheduled six months after the initial finding of the incidentaloma with the patient’s PCP or with an endocrinologist.5
Thyroid incidentalomas. The prevalence of thyroid nodules based on ultrasound studies ranges from 19% to 46%, with autopsy studies estimating an incidence of approximately 50%.2,6 Incidence of thyroid nodules also increases with age, as almost 60% of people over the age of 60 harbor a thyroid incidentaloma. The rate of malignancy in the general population has ranged between 8% and 24%; however, in the last decade, the rates have increased by 2.4 times as more sophisticated ultrasound techniques and liberal use of fine-needle aspiration (FNA) biopsies have detected subclinical disease.7,8
Etiologies for incidental thyroid nodules can be divided into benign and malignant causes. Benign etiologies include thyroid cyst (simple or complex), multinodular goiter, and Hashimoto’s thryoiditis, while malignant causes include papillary, medullary, follicular, Hurthle cell, and anaplastic carcinomas, thyroid lymphomas, and rare instances of metastatic cancers.2,3
Targeted history and physical examination helps to characterize the thyroid incidentaloma. Historical features, such as palpitations, weight loss, anxiety, new onset atrial fibrillation, or menstrual irregularities, coupled with tachycardia, tremors, proximal muscle weakness, and a palpable nodule aid in the diagnosis of hyperthyroidism. Findings such as a family history of thyroid cancer, symptoms of hoarseness or dysphagia, rapid growth of the nodule, environmental or history of head or neck irradiation along with physical findings of a hard, fixed nodule, or cervical lymphadenopathy increase the suspicion for malignancy.2,7
The functionality of the nodule can be assessed by checking TSH, free T3, and free T4 levels. Suppression of TSH (< 0.1 mU/L) with elevated levels of free T3 and T4 indicates nodule production of excess thyroid hormone and warrants thyroid scintography. Thyroid scintography will identify the nodule as “hot” (hyperfunctioning) or “cold” (nonfunctioning).2
Regardless of the radiographic modality that initially identified the thyroid incidentaloma, a dedicated thyroid high-resolution ultrasound should be ordered to assess the size, multiplicity (single or multinodular), location, and character (solid, cystic, or mixed).7
Recommendations for proceeding to FNA to evaluate for malignancy differ among subspecialty societies. Generally, nodules larger than 1 cm or nodules smaller than 1 cm with risk factors for malignancy should be referred for FNA.2,7
If diagnostic workup identifies a patient with hyperthyroidism due to an autonomously functional nodule or a nodule that may be at high risk for malignancy, it is appropriate to involve an endocrinologist and possibly a surgical subspecialist prior to discharge. Management of hyperthyroidism can include starting antithyroid agents (methimazole or propylthiouracil), radioactive iodine ablation, or referral for surgery.
Preparation for discharge of the patient whose incidentaloma is nonfunctional or does not appear to be malignant should include appointments to recheck thyroid hormone levels, including TSH as well as a thyroid ultrasound within one year of the initial discovery.
Adrenal incidentaloma. The prevalence of AIs found by CT of the abdomen ranges from 0.4% to 4%, while autopsy studies have found a prevalence of 1.4% to 9% with increasing prevalence with age.2,9,10 The majority of AIs are benign and nonfunctioning adenomas, in the absence of known malignancy. Other differential diagnoses include Cushing’s syndrome, pheochromocytoma, adrenocortical adenoma, aldosteronoma, and metastatic lesions.
Because functioning adrenal incidentalomas may be clinically silent, any patient found with an AI must undergo biochemical workup as part of their evaluation to assess for pheochromocytoma, Cushing’s syndrome, and if he or she has a history of hypertension or hyperaldosteronism (Conn’s syndrome). Table 3 outlines the approach for characterizing adrenal incidentalomas.2,11,12 An important point is that imaging studies are not useful in distinguishing a functioning versus nonfunctioning tumor but rather can help to discriminate malignant lesions.11
Inpatient surgical consult for resection is indicated if the patient is found to have pheochromocytoma, clinically apparent functioning adrenocortical adenoma, or a tumor size greater than 4 cm. Consultation with an endocrinologist is also recommended if biochemical tests are positive. If the diagnostic workup leads to suspicion for infection or metastatic disease, the patient should be referred for FNA.2,12
For patients whose lesions do not require surgical resection, repeat CT scan of the abdomen is recommended six months from the initial finding. Hospitalists should also arrange for the patient to repeat biochemical testing, including an overnight dexamethasone test.12,13
Back to the Case
The patient underwent biochemical testing and was found to have normal levels of plasma-free metanephrines, a plasma aldosterone, plasma renin activity ratio less than 20, and a serum cortisol level of 7 mg/dL after his overnight dexamethasone suppression test. The 24-hour urine collection for free cortisol revealed elevated levels of cortisol in the urine, and the ACTH level was low.
Endocrinology and endocrine surgery teams were consulted, and recommended surgical resection. After surgical resection of his tumor, the patient was started on glucocorticoid replacement and was discharged with a follow-up appointment with endocrinology.
Bottom Line
An inpatient approach to endocrine incidentalomas should include characterization of the clinical signs and symptoms, size, function, and malignant potential of the lesion. Based on this, inpatient surgical or medical management can be determined. Post-discharge management should include arrangements for surveillance testing and follow-up with appropriate subspecialists.
Dr. Tad-y is assistant professor of medicine and a hospitalist at the University of Colorado Denver.
References
- Aron DC, Howlett TA. Pituitary incidentalomas. Endocrinol Metab Clin North Am. 2000;29:205-221.
- Shirodkar M, Jabbour SA. Endocrine incidentalomas. Int J Clin Pract. 2008;62:1423-1431.
- Burguera B, Gharib H. Thyroid incidentalomas. Prevalence, diagnosis, significance, and management. Endocrinol Metab Clin North Am. 2000;29:187-203.
- Molitch ME. Nonfunctioning pituitary tumors and pituitary incidentalomas. Endocrinol Metab Clin North Am. 2008;37:151-171, xi.
- Freda PU, Beckers AM, Katznelson L, et al. Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:894-904.
- Gough J, Scott-Coombes D, Fausto Palazzo F. Thyroid incidentaloma: an evidence-based assessment of management strategy. World J Surg. 2008;32:1264-1268.
- Iyer NG, Shaha AR, Silver CE, et al. Thyroid incidentalomas: to treat or not to treat. Eur Arch Otorhinolaryngol. 2010;267:1019-1026.
- Jin J, Wilhelm SM, McHenry CR. Incidental thyroid nodule: patterns of diagnosis and rate of malignancy. Am J Surg. 2009;197:320-324.
- Davenport C, Liew L, Doherty B, et al. The prevalence of adrenal incidentaloma in routine clinical practice. Endocrine. 2011;40:80-83.
- Zeiger MA, Siegelman SS, Hamrahian AH. Medical and surgical evaluation and treatment of adrenal incidentalomas. J Clin Endocrinol Metab. 2011;96: 2004-2015.
- Zeiger MA, Thompson GB, Duh QY, et al. American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas: executive summary of recommendations. Endocr Pract. 2009;15:450-453.
- NIH state-of-the-science statement on management of the clinically inapparent adrenal mass (“incidentaloma”). NIH Consens State Sci Statements. 2002;19:1-25.
- Young WF. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med. 2007;356:601-610.
- Chidiac RM, Aron DC. Incidentalomas. A disease of modern technology. Endocrinol Metab Clin North Am. 1997;26:233-253.
Does Hospital Medicine Reinforce the Pillars of Career Satisfaction?
Gregory Misky, MD, describes it as a “deer in the headlights” moment. About four years ago, Dr. Misky, assistant professor of medicine at the University of Colorado Denver, and Mark Reid, MD, assistant professor at Denver Health Medical Center, were trying to figure out what being an academic hospitalist was all about. What were the expectations of them, and how could they combine their clinical duties with scholarly work, especially given the significant lack of mentorship?
The duo wondered if other young hospitalists were feeling the same uncertainty about their chosen career, and whether there were any variables that might help predict success or burnout among their fellow doctors.
They haven’t been alone. Regardless of the practice model and location, physicians within the fastest-spreading medical specialty in the U.S. have noted both the promise and unsettled nature of HM. “We are still a relatively young profession, and I think over the past five to 10 years, we’ve been seeing the growing pains of the profession,” says Tosha Wetterneck, MD, MS, FACP, associate professor of medicine at the University of Wisconsin School of Medicine and Public Health in Madison.
In response to mounting concerns over multiple career-satisfaction-related issues, SHM assembled a Career Satisfaction Task Force that produced a detailed white paper at the end of 2006 (available from the “White Papers” tab under the “Publications” heading at www.hospitalmedicine.org).
One tangible outcome of the paper was the establishment of “Four Pillars of Career Satisfaction” for hospitalists:
- Reward and recognition;
- Workload and schedule;
- Autonomy and control; and
- Community and environment.
The paper included definitions for each pillar, and assembled scorecards, action steps, tools, and recommendations for both HM leaders and individual hospitalists to help shore up perceived weak spots.
So how strong are those pillars in practice? If hospitalists are the future of healthcare, as SHM and other medical groups assert, what do current studies suggest about the prospects of HM solidifying into a satisfying and sustainable career choice?
The Evidence
One outgrowth of Dr. Misky and Dr. Reid’s frustration was a study in which they and their collaborators emailed a 61-question survey to hospitalists at 20 academic medical centers. Among the results, the researchers found that 75% of respondents reported either “high” or “somewhat high” satisfaction with their current job. At the same time, though, 67% felt “high” or “somewhat high” stress levels at work, and nearly 1 in 4 (24%) reported some degree of burnout, based on their own definition of the word.1
As one of the first hospitalists in his group, Dr. Misky recalls the stress he felt over whether the hospital, division, and department would all buy into the idea of an academic hospitalist, and what his role would be. “I think we spent a lot of our early years trying to carve out our niche and proving ourselves and trying to balance the clinical needs that people had for us with other expectations of being an academic,” he says. Dr. Misky likens the experience to the adrenaline rush of mountain-biking straight down a hill. The feeling that too many things are going on at once, though, might also partially explain the apparent dichotomy of high overall satisfaction but a worrisome degree of burnout.
The profession hasn’t been around long enough for good longitudinal studies, and surveys have worded questions on satisfaction and burnout in different ways, complicating attempts at direct comparisons over time. A 2001 study, for example, reported that 12.9% of community and academic hospitalists were burned out, with another 25% at risk, but the survey was limited to dues-paying members of the National Association of Inpatient Physicians, the precursor to SHM.2
Nor has it been easy to compare hospitalist satisfaction and burnout levels to those of other specialists. “We haven’t really defined what a sustained, long-term career in hospital medicine is going to be,” Dr. Wetterneck says. “And in that way, it’s hard to say, ‘Compared to other professions, are we happier or not?’”

One of her recent studies, however, generally agrees with the handful of surveys addressing satisfaction and burnout among hospitalists. Overall, 63% of respondents reported high satisfaction with their job, while 69% were highly satisfied with their specialty. Roughly 30%, however, also reported feeling symptom of job burnout.3
Kelki Hinami, MD, MS, assistant of professor of medicine at Northwestern University Feinberg School of Medicine in Chicago and a coauthor of the study, says one take-home message is that hospitalists do fairly well in finding jobs that match their individual needs. “To further illustrate this, we found that hospitalists working in various practice models have different ideas about what is most important to their job,” he says.
Autonomy, for example, is considered most important by more local group hospitalists than by those of any other model, while recognition by leaders and having a variety of tasks at work are particularly important to academic hospitalists. Unlike other hospitalists, however, fewer academics consider pay to be the most important job characteristic.
A third study, led by John Yoon, MD, assistant professor in the section of hospital medicine at the University of Chicago, has examined career satisfaction, burnout, and morale among primary-care physicians (PCPs) and hospitalists. So far, the results he reported at HM11 largely agree with the other recent surveys: Combined, 85% of hospitalists report being either somewhat or very satisfied with their overall career. Conversely, 24% of hospitalists regretted choosing medicine as a career and 38% say they would have chosen a different medical specialty if they had to do it over again.4
Dr. Yoon says his data, compiled from two survey samples of about 1,000 generalists each, have revealed few differences between hospitalists and PCPs. “I thought hospitalists would be more satisfied than primary-care physicians, given the declining satisfaction rates of PCPs that we know about, and that students and trainees are less likely to go into primary care,” he says. Even burnout rates are similar, however; Dr. Yoon says he’s noticed a trend toward hospitalists reporting less burnout than PCPs, but the difference is not yet statistically significant.
Choice of a New Generation?
HM’s attractiveness to medical residents offers other clues about its ability to provide a sustainable and satisfying career choice. Salary, part of the “reward and recognition” pillar, has long been one perceived weakness. Anecdotally, however, Dr. Yoon says many general medicine residents see HM as a better financial option than primary care. “Some of the residents I work with, when I asked them, ‘Will you be a primary-care physician or a hospitalist?’ a lot of them say, ‘Probably hospitalist,’” he says. “And generally the reason is because they have to pay off their debt.”
It’s true that hospitalists’ salaries lag behind that of most of other specialists. Nevertheless, researchers like Colin West, MD, PhD, associate professor of medicine and biostatistics at the Mayo Clinic in Rochester, Minn., say many medical residents are prioritizing financial considerations as relatively low on the scale of general preferences.

—John Yoon, MD, assistant professor, section of hospital medicine, University of Chicago
Dr. West, an associate program director for the internal-medicine residency program at Mayo, sees a generational sea change in the career considerations deemed most important. Based on a career decision survey filled out by nearly 15,000 internal-medical residents, he found that roughly 70% of respondents said time with family was of “high” or “very high” importance to their career decisions.5 The category, which relates to SHM’s “workload and schedule” pillar, beat out eight others as the most important factor overall, while global financial considerations scored relatively low.
Residents who placed high value on time with family were more likely to choose careers in more predictable, outpatient-based specialties, such as endocrinology or rheumatology. HM also fared well in this category. Dr. West says the results suggest that residents considering a hospitalist career are attracted to the specialty’s flexibility and predictability of the largely shift-based scheduling.
William Cors, MD, chief medical quality officer at Pocono Health System in East Stroudsburg, Pa., says more physicians are looking for job security, predictable shifts, and a better work-life balance. As HM matures and demonstrates that it can address those needs, Dr. Cors sees it becoming more attractive for medical students and residents.
In practice, though, other research suggests a career in HM doesn’t always meet expectations. Dr. Wetterneck and Dr. Hinami, for example, highlighted both compensation and work-life balance as points of concern in their study: For both factors, only about 30% of hospitalists were optimally satisfied.
Separately, Dr. Misky and his colleagues reported that roughly half of academic hospitalists were satisfied with the ability to control their schedule, and with their amount of personal and family time. Those who were unsatisfied with either of these categories, the survey found, were at higher risk for burnout. Similarly, Dr. Yoon found that physicians who reported having no control over their work hours or their call schedule, part of SHM’s “autonomy and control” pillar, were more likely to report burnout.
So why is HM stumbling on perceived selling points like family friendliness and autonomy? Dr. Wetterneck believes too many unfilled jobs and rapid turnover could be putting more pressure on existing hospitalists and interfering with their ability to balance home and work life. “There’s a huge need for hospitalists everywhere,” she says, and reliance on them has been especially acute at academic centers and large community hospitals contending with the recently imposed limits on residents’ work hours.
The Hospitalist: A People Person
Another shift may be occurring in the types of relationships necessary for a satisfying work environment, a big part of the “community and environment” pillar. Although Dr. Yoon says long-term connections with students and trainees have added meaning to his job, he is mourning the absence of other bonds. “One loss I’m starting to feel keenly as an academic hospitalist, having spent my early training years as a primary-care doc, really is the loss of having long-term relationships with patients,” he says. “My clinical encounters with patients these days as a hospitalist are very intense, but also very brief.”
Dr. Yoon has pondered whether the HM field can rearrange practice settings to promote more satisfying relationships. Such a change, he says, might occur through innovative models that aid coordination with medical homes, or provide more chronic care for high-risk patients. “In my view, the trajectory of hospital medicine is pretty wide open for creativity and new models of care,” he says. “I think it will be partly driven by the need to want to have more meaningful interactions with patients.”
Those relationships need not be long-term, however. One recent study found high satisfaction among hospitalists and laborists working within the fast-growing OBGYN hospitalist field.6
Dr. Hinami says collaborative care that involves close working relationships with specialists and other care providers might help propel the hospitalist movement forward. In his survey with Dr. Wetterneck, hospitalists ranked relationships with staff and colleagues among the most satisfying of any of the domains; hospitalists also indicated high levels of satisfaction with their patient relationships. “Clearly, relationships are critical to overall job satisfaction, and hospitalists, I think, are doing a fairly good job at maintaining those relationships,” Dr. Hinami says.

—Keiki Hinami, MD, assistant professor of medicine, Northwestern University Feinberg School of Medicine, Chicago
A 2002 survey-based study reinforces the importance of such bonds. Job burnout and intent to remain in the hospitalist career, its authors concluded, were more highly influenced by “favorable social relations” involving colleagues, coworkers, and patients than by such factors as reduced autonomy and the use of financial incentives.7
The focus on maintaining multiple relationships fits well with the collaborative approach to care that many hospitalists say they value highly. One big satisfier for hospitalists, Dr. Cors says, will be “a sense that they’re really part of a healthcare team and not just punching the clock and doing their shifts.”
The Verdict
Despite the difficulty in discerning long-term trends, studies suggest that overall satisfaction with the specialty of hospital medicine remains high, a promising sign for the maturing field. Career hospitalists also seem adept at relationships with peers and other providers, a skill that will serve them well as collaborative-care models gain steam.
Nonetheless, surveys also suggest a worrisome rate of burnout and less-than-optimal satisfaction with elements that should be the strong suits of HM, such as work-life balance and autonomy. Academics are searching for their own clinical-research balance. And Dr. West says the jury’s still out on the future pitfalls that might get in the way of a sustainable career path for older practitioners, such as overnight shifts.
Hospitalist-led efforts, however, may be starting to pay dividends. At the University of California at San Francisco, a faculty development program for first-year hospitalists has included a coaching relationship with a senior faculty member, a teaching course, newly established divisional grand rounds, and a framework for meeting scholarly expectations. Upon its implementation, the program has led to higher job satisfaction, skill-set comfort, and academic production among participants.8
Given the expanding range of HM duties and practice models, hospitals, division chiefs, and team leaders cannot rely on a single recipe for happy and productive hospitalists. “I don’t know if there is a cookbook; I think it’s highly variable depending on your institution and the needs of the academic facility where you are,” Dr. Misky says.
SHM’s 2006 white paper stated that the best career satisfaction strategy is to find a job that fits an individual’s preferences and attitudes. “People who are unhappy with their job don’t tend to stay in it, and from what we know about hospital medicine right now, you can find pretty much any type of job anywhere you want, so the job market is very open,” Dr. Wetterneck says.
Ensuring the right fit for doctors within HM, though, will require institutional support. “It’s going to be up to hospitals and hospitalist programs to create jobs that are sustainable that people like,” she says, “so that hospitalists will stay long in their job and in the profession.”
Bryn Nelson is a freelance medical writer based in Seattle.
More Mentorship in Hospital Medicine? It’s Academic

Within the 2011 State of Hospital Medicine report, one statistic in particular points to the youth of the medical specialty: Just over 10% of surveyed hospitalists had reached the rank of associate professor or higher.
How might the potential lack of mentorship within this immature field affect the ability of hospitalists to successfully navigate academia? So asked Gregory Misky, MD, assistant professor of medicine at the University of Colorado Denver, and his colleagues in a survey-based study. The results agree with other recent assessments that mentors are in short supply. “Academic hospital medicine groups have an acute need for mentoring and career development programs,” one study concludes.
The research of Dr. Misky and his collaborators found that only 42% of academic hospitalists could identify a mentor, while only 31% reported that they were mentoring another academic hospitalist.1 Based on sheer numbers and experience, the pool of mentors may significantly expand as the field matures. But Dr. Misky also urges some flexibility, noting that his own mentor is a non-hospitalist.
In his own research, Colin West, MD, PhD, associate professor of medicine and biostatistics at the Mayo Clinic in Rochester, Minn., found that residents considering a career in HM placed less emphasis on the specialty or subspecialty of their mentor.5 Why? Very likely, he says, there just weren’t enough hospitalist mentors around to get a sense of what the career was all about.
Dr. West hopes the numbers suggest otherwise in the near future. “You want to recruit bright people into your specialty, but at the same time, you also want to recruit the right people,” he says. “And that means that you need to be able to expose people to a full breadth of what a decision to pursue a certain specialty really means.”
References
- Glasheen JJ, Misky GJ, Reid MB, Harrison RA, Sharpe B, Auerbach A. Career satisfaction and burnout in academic hospital medicine. Arch Intern Med. 2011;171(8) 782-785.
- Hoff TH, Whitcomb WF, Williams K, Nelson JR, Cheesman RA. Characteristics and work experiences of hospitalists in the United States. Arch Intern Med. 2001;161(6):851-858.
- Hinami K, Whelan CT, Wolosin RJ, Miller JA, Wetterneck TB. Worklife and satisfaction of hospitalists: toward flourishing careers [published online ahead of print July 20, 2011]. J Gen Intern Med. doi:10.1007/s116060-011-1780-z.
- Yoon J, Miller A, Rasinski K, Curlin F. Burnout, sense of calling, and career resilience among hospitalists and primary care physicians: a national survey. J Hosp Med. 2011;6(4):S90-S91.
- West CP, Drefahl MM, Popkave C, Kolars JC. Internal medicine resident self-report of factors associated with career decisions. J Gen Intern Med. 2009;24(8):946-949.
- Funk C, Anderson BL, Schulkin J, Weinstein L. Survey of obstetric and gynecologic hospitalists and laborists. Am J Obstet Gynecol. 2010;203(2):177.e1-177.e4.
- Hoff T, Whitcomb WF, Nelson JR. Thriving and surviving in a new medical career: the case of hospitalist physicians. J Health Soc Behav. 2002;43(1):72-91.
- Sehgal NL, Sharpe BA, Auerbach AA, Wachter RM. Investing in the future: Building an academic hospitalist faculty development program. J Hosp Med. 2011;6(3):161-166.
Gregory Misky, MD, describes it as a “deer in the headlights” moment. About four years ago, Dr. Misky, assistant professor of medicine at the University of Colorado Denver, and Mark Reid, MD, assistant professor at Denver Health Medical Center, were trying to figure out what being an academic hospitalist was all about. What were the expectations of them, and how could they combine their clinical duties with scholarly work, especially given the significant lack of mentorship?
The duo wondered if other young hospitalists were feeling the same uncertainty about their chosen career, and whether there were any variables that might help predict success or burnout among their fellow doctors.
They haven’t been alone. Regardless of the practice model and location, physicians within the fastest-spreading medical specialty in the U.S. have noted both the promise and unsettled nature of HM. “We are still a relatively young profession, and I think over the past five to 10 years, we’ve been seeing the growing pains of the profession,” says Tosha Wetterneck, MD, MS, FACP, associate professor of medicine at the University of Wisconsin School of Medicine and Public Health in Madison.
In response to mounting concerns over multiple career-satisfaction-related issues, SHM assembled a Career Satisfaction Task Force that produced a detailed white paper at the end of 2006 (available from the “White Papers” tab under the “Publications” heading at www.hospitalmedicine.org).
One tangible outcome of the paper was the establishment of “Four Pillars of Career Satisfaction” for hospitalists:
- Reward and recognition;
- Workload and schedule;
- Autonomy and control; and
- Community and environment.
The paper included definitions for each pillar, and assembled scorecards, action steps, tools, and recommendations for both HM leaders and individual hospitalists to help shore up perceived weak spots.
So how strong are those pillars in practice? If hospitalists are the future of healthcare, as SHM and other medical groups assert, what do current studies suggest about the prospects of HM solidifying into a satisfying and sustainable career choice?
The Evidence
One outgrowth of Dr. Misky and Dr. Reid’s frustration was a study in which they and their collaborators emailed a 61-question survey to hospitalists at 20 academic medical centers. Among the results, the researchers found that 75% of respondents reported either “high” or “somewhat high” satisfaction with their current job. At the same time, though, 67% felt “high” or “somewhat high” stress levels at work, and nearly 1 in 4 (24%) reported some degree of burnout, based on their own definition of the word.1
As one of the first hospitalists in his group, Dr. Misky recalls the stress he felt over whether the hospital, division, and department would all buy into the idea of an academic hospitalist, and what his role would be. “I think we spent a lot of our early years trying to carve out our niche and proving ourselves and trying to balance the clinical needs that people had for us with other expectations of being an academic,” he says. Dr. Misky likens the experience to the adrenaline rush of mountain-biking straight down a hill. The feeling that too many things are going on at once, though, might also partially explain the apparent dichotomy of high overall satisfaction but a worrisome degree of burnout.
The profession hasn’t been around long enough for good longitudinal studies, and surveys have worded questions on satisfaction and burnout in different ways, complicating attempts at direct comparisons over time. A 2001 study, for example, reported that 12.9% of community and academic hospitalists were burned out, with another 25% at risk, but the survey was limited to dues-paying members of the National Association of Inpatient Physicians, the precursor to SHM.2
Nor has it been easy to compare hospitalist satisfaction and burnout levels to those of other specialists. “We haven’t really defined what a sustained, long-term career in hospital medicine is going to be,” Dr. Wetterneck says. “And in that way, it’s hard to say, ‘Compared to other professions, are we happier or not?’”
One of her recent studies, however, generally agrees with the handful of surveys addressing satisfaction and burnout among hospitalists. Overall, 63% of respondents reported high satisfaction with their job, while 69% were highly satisfied with their specialty. Roughly 30%, however, also reported feeling symptom of job burnout.3
Kelki Hinami, MD, MS, assistant of professor of medicine at Northwestern University Feinberg School of Medicine in Chicago and a coauthor of the study, says one take-home message is that hospitalists do fairly well in finding jobs that match their individual needs. “To further illustrate this, we found that hospitalists working in various practice models have different ideas about what is most important to their job,” he says.
Autonomy, for example, is considered most important by more local group hospitalists than by those of any other model, while recognition by leaders and having a variety of tasks at work are particularly important to academic hospitalists. Unlike other hospitalists, however, fewer academics consider pay to be the most important job characteristic.
A third study, led by John Yoon, MD, assistant professor in the section of hospital medicine at the University of Chicago, has examined career satisfaction, burnout, and morale among primary-care physicians (PCPs) and hospitalists. So far, the results he reported at HM11 largely agree with the other recent surveys: Combined, 85% of hospitalists report being either somewhat or very satisfied with their overall career. Conversely, 24% of hospitalists regretted choosing medicine as a career and 38% say they would have chosen a different medical specialty if they had to do it over again.4
Dr. Yoon says his data, compiled from two survey samples of about 1,000 generalists each, have revealed few differences between hospitalists and PCPs. “I thought hospitalists would be more satisfied than primary-care physicians, given the declining satisfaction rates of PCPs that we know about, and that students and trainees are less likely to go into primary care,” he says. Even burnout rates are similar, however; Dr. Yoon says he’s noticed a trend toward hospitalists reporting less burnout than PCPs, but the difference is not yet statistically significant.
Choice of a New Generation?
HM’s attractiveness to medical residents offers other clues about its ability to provide a sustainable and satisfying career choice. Salary, part of the “reward and recognition” pillar, has long been one perceived weakness. Anecdotally, however, Dr. Yoon says many general medicine residents see HM as a better financial option than primary care. “Some of the residents I work with, when I asked them, ‘Will you be a primary-care physician or a hospitalist?’ a lot of them say, ‘Probably hospitalist,’” he says. “And generally the reason is because they have to pay off their debt.”
It’s true that hospitalists’ salaries lag behind that of most of other specialists. Nevertheless, researchers like Colin West, MD, PhD, associate professor of medicine and biostatistics at the Mayo Clinic in Rochester, Minn., say many medical residents are prioritizing financial considerations as relatively low on the scale of general preferences.
—John Yoon, MD, assistant professor, section of hospital medicine, University of Chicago
Dr. West, an associate program director for the internal-medicine residency program at Mayo, sees a generational sea change in the career considerations deemed most important. Based on a career decision survey filled out by nearly 15,000 internal-medical residents, he found that roughly 70% of respondents said time with family was of “high” or “very high” importance to their career decisions.5 The category, which relates to SHM’s “workload and schedule” pillar, beat out eight others as the most important factor overall, while global financial considerations scored relatively low.
Residents who placed high value on time with family were more likely to choose careers in more predictable, outpatient-based specialties, such as endocrinology or rheumatology. HM also fared well in this category. Dr. West says the results suggest that residents considering a hospitalist career are attracted to the specialty’s flexibility and predictability of the largely shift-based scheduling.
William Cors, MD, chief medical quality officer at Pocono Health System in East Stroudsburg, Pa., says more physicians are looking for job security, predictable shifts, and a better work-life balance. As HM matures and demonstrates that it can address those needs, Dr. Cors sees it becoming more attractive for medical students and residents.
In practice, though, other research suggests a career in HM doesn’t always meet expectations. Dr. Wetterneck and Dr. Hinami, for example, highlighted both compensation and work-life balance as points of concern in their study: For both factors, only about 30% of hospitalists were optimally satisfied.
Separately, Dr. Misky and his colleagues reported that roughly half of academic hospitalists were satisfied with the ability to control their schedule, and with their amount of personal and family time. Those who were unsatisfied with either of these categories, the survey found, were at higher risk for burnout. Similarly, Dr. Yoon found that physicians who reported having no control over their work hours or their call schedule, part of SHM’s “autonomy and control” pillar, were more likely to report burnout.
So why is HM stumbling on perceived selling points like family friendliness and autonomy? Dr. Wetterneck believes too many unfilled jobs and rapid turnover could be putting more pressure on existing hospitalists and interfering with their ability to balance home and work life. “There’s a huge need for hospitalists everywhere,” she says, and reliance on them has been especially acute at academic centers and large community hospitals contending with the recently imposed limits on residents’ work hours.
The Hospitalist: A People Person
Another shift may be occurring in the types of relationships necessary for a satisfying work environment, a big part of the “community and environment” pillar. Although Dr. Yoon says long-term connections with students and trainees have added meaning to his job, he is mourning the absence of other bonds. “One loss I’m starting to feel keenly as an academic hospitalist, having spent my early training years as a primary-care doc, really is the loss of having long-term relationships with patients,” he says. “My clinical encounters with patients these days as a hospitalist are very intense, but also very brief.”
Dr. Yoon has pondered whether the HM field can rearrange practice settings to promote more satisfying relationships. Such a change, he says, might occur through innovative models that aid coordination with medical homes, or provide more chronic care for high-risk patients. “In my view, the trajectory of hospital medicine is pretty wide open for creativity and new models of care,” he says. “I think it will be partly driven by the need to want to have more meaningful interactions with patients.”
Those relationships need not be long-term, however. One recent study found high satisfaction among hospitalists and laborists working within the fast-growing OBGYN hospitalist field.6
Dr. Hinami says collaborative care that involves close working relationships with specialists and other care providers might help propel the hospitalist movement forward. In his survey with Dr. Wetterneck, hospitalists ranked relationships with staff and colleagues among the most satisfying of any of the domains; hospitalists also indicated high levels of satisfaction with their patient relationships. “Clearly, relationships are critical to overall job satisfaction, and hospitalists, I think, are doing a fairly good job at maintaining those relationships,” Dr. Hinami says.
—Keiki Hinami, MD, assistant professor of medicine, Northwestern University Feinberg School of Medicine, Chicago
A 2002 survey-based study reinforces the importance of such bonds. Job burnout and intent to remain in the hospitalist career, its authors concluded, were more highly influenced by “favorable social relations” involving colleagues, coworkers, and patients than by such factors as reduced autonomy and the use of financial incentives.7
The focus on maintaining multiple relationships fits well with the collaborative approach to care that many hospitalists say they value highly. One big satisfier for hospitalists, Dr. Cors says, will be “a sense that they’re really part of a healthcare team and not just punching the clock and doing their shifts.”
The Verdict
Despite the difficulty in discerning long-term trends, studies suggest that overall satisfaction with the specialty of hospital medicine remains high, a promising sign for the maturing field. Career hospitalists also seem adept at relationships with peers and other providers, a skill that will serve them well as collaborative-care models gain steam.
Nonetheless, surveys also suggest a worrisome rate of burnout and less-than-optimal satisfaction with elements that should be the strong suits of HM, such as work-life balance and autonomy. Academics are searching for their own clinical-research balance. And Dr. West says the jury’s still out on the future pitfalls that might get in the way of a sustainable career path for older practitioners, such as overnight shifts.
Hospitalist-led efforts, however, may be starting to pay dividends. At the University of California at San Francisco, a faculty development program for first-year hospitalists has included a coaching relationship with a senior faculty member, a teaching course, newly established divisional grand rounds, and a framework for meeting scholarly expectations. Upon its implementation, the program has led to higher job satisfaction, skill-set comfort, and academic production among participants.8
Given the expanding range of HM duties and practice models, hospitals, division chiefs, and team leaders cannot rely on a single recipe for happy and productive hospitalists. “I don’t know if there is a cookbook; I think it’s highly variable depending on your institution and the needs of the academic facility where you are,” Dr. Misky says.
SHM’s 2006 white paper stated that the best career satisfaction strategy is to find a job that fits an individual’s preferences and attitudes. “People who are unhappy with their job don’t tend to stay in it, and from what we know about hospital medicine right now, you can find pretty much any type of job anywhere you want, so the job market is very open,” Dr. Wetterneck says.
Ensuring the right fit for doctors within HM, though, will require institutional support. “It’s going to be up to hospitals and hospitalist programs to create jobs that are sustainable that people like,” she says, “so that hospitalists will stay long in their job and in the profession.”
Bryn Nelson is a freelance medical writer based in Seattle.
More Mentorship in Hospital Medicine? It’s Academic
Within the 2011 State of Hospital Medicine report, one statistic in particular points to the youth of the medical specialty: Just over 10% of surveyed hospitalists had reached the rank of associate professor or higher.
How might the potential lack of mentorship within this immature field affect the ability of hospitalists to successfully navigate academia? So asked Gregory Misky, MD, assistant professor of medicine at the University of Colorado Denver, and his colleagues in a survey-based study. The results agree with other recent assessments that mentors are in short supply. “Academic hospital medicine groups have an acute need for mentoring and career development programs,” one study concludes.
The research of Dr. Misky and his collaborators found that only 42% of academic hospitalists could identify a mentor, while only 31% reported that they were mentoring another academic hospitalist.1 Based on sheer numbers and experience, the pool of mentors may significantly expand as the field matures. But Dr. Misky also urges some flexibility, noting that his own mentor is a non-hospitalist.
In his own research, Colin West, MD, PhD, associate professor of medicine and biostatistics at the Mayo Clinic in Rochester, Minn., found that residents considering a career in HM placed less emphasis on the specialty or subspecialty of their mentor.5 Why? Very likely, he says, there just weren’t enough hospitalist mentors around to get a sense of what the career was all about.
Dr. West hopes the numbers suggest otherwise in the near future. “You want to recruit bright people into your specialty, but at the same time, you also want to recruit the right people,” he says. “And that means that you need to be able to expose people to a full breadth of what a decision to pursue a certain specialty really means.”
References
- Glasheen JJ, Misky GJ, Reid MB, Harrison RA, Sharpe B, Auerbach A. Career satisfaction and burnout in academic hospital medicine. Arch Intern Med. 2011;171(8) 782-785.
- Hoff TH, Whitcomb WF, Williams K, Nelson JR, Cheesman RA. Characteristics and work experiences of hospitalists in the United States. Arch Intern Med. 2001;161(6):851-858.
- Hinami K, Whelan CT, Wolosin RJ, Miller JA, Wetterneck TB. Worklife and satisfaction of hospitalists: toward flourishing careers [published online ahead of print July 20, 2011]. J Gen Intern Med. doi:10.1007/s116060-011-1780-z.
- Yoon J, Miller A, Rasinski K, Curlin F. Burnout, sense of calling, and career resilience among hospitalists and primary care physicians: a national survey. J Hosp Med. 2011;6(4):S90-S91.
- West CP, Drefahl MM, Popkave C, Kolars JC. Internal medicine resident self-report of factors associated with career decisions. J Gen Intern Med. 2009;24(8):946-949.
- Funk C, Anderson BL, Schulkin J, Weinstein L. Survey of obstetric and gynecologic hospitalists and laborists. Am J Obstet Gynecol. 2010;203(2):177.e1-177.e4.
- Hoff T, Whitcomb WF, Nelson JR. Thriving and surviving in a new medical career: the case of hospitalist physicians. J Health Soc Behav. 2002;43(1):72-91.
- Sehgal NL, Sharpe BA, Auerbach AA, Wachter RM. Investing in the future: Building an academic hospitalist faculty development program. J Hosp Med. 2011;6(3):161-166.
Gregory Misky, MD, describes it as a “deer in the headlights” moment. About four years ago, Dr. Misky, assistant professor of medicine at the University of Colorado Denver, and Mark Reid, MD, assistant professor at Denver Health Medical Center, were trying to figure out what being an academic hospitalist was all about. What were the expectations of them, and how could they combine their clinical duties with scholarly work, especially given the significant lack of mentorship?
The duo wondered if other young hospitalists were feeling the same uncertainty about their chosen career, and whether there were any variables that might help predict success or burnout among their fellow doctors.
They haven’t been alone. Regardless of the practice model and location, physicians within the fastest-spreading medical specialty in the U.S. have noted both the promise and unsettled nature of HM. “We are still a relatively young profession, and I think over the past five to 10 years, we’ve been seeing the growing pains of the profession,” says Tosha Wetterneck, MD, MS, FACP, associate professor of medicine at the University of Wisconsin School of Medicine and Public Health in Madison.
In response to mounting concerns over multiple career-satisfaction-related issues, SHM assembled a Career Satisfaction Task Force that produced a detailed white paper at the end of 2006 (available from the “White Papers” tab under the “Publications” heading at www.hospitalmedicine.org).
One tangible outcome of the paper was the establishment of “Four Pillars of Career Satisfaction” for hospitalists:
- Reward and recognition;
- Workload and schedule;
- Autonomy and control; and
- Community and environment.
The paper included definitions for each pillar, and assembled scorecards, action steps, tools, and recommendations for both HM leaders and individual hospitalists to help shore up perceived weak spots.
So how strong are those pillars in practice? If hospitalists are the future of healthcare, as SHM and other medical groups assert, what do current studies suggest about the prospects of HM solidifying into a satisfying and sustainable career choice?
The Evidence
One outgrowth of Dr. Misky and Dr. Reid’s frustration was a study in which they and their collaborators emailed a 61-question survey to hospitalists at 20 academic medical centers. Among the results, the researchers found that 75% of respondents reported either “high” or “somewhat high” satisfaction with their current job. At the same time, though, 67% felt “high” or “somewhat high” stress levels at work, and nearly 1 in 4 (24%) reported some degree of burnout, based on their own definition of the word.1
As one of the first hospitalists in his group, Dr. Misky recalls the stress he felt over whether the hospital, division, and department would all buy into the idea of an academic hospitalist, and what his role would be. “I think we spent a lot of our early years trying to carve out our niche and proving ourselves and trying to balance the clinical needs that people had for us with other expectations of being an academic,” he says. Dr. Misky likens the experience to the adrenaline rush of mountain-biking straight down a hill. The feeling that too many things are going on at once, though, might also partially explain the apparent dichotomy of high overall satisfaction but a worrisome degree of burnout.
The profession hasn’t been around long enough for good longitudinal studies, and surveys have worded questions on satisfaction and burnout in different ways, complicating attempts at direct comparisons over time. A 2001 study, for example, reported that 12.9% of community and academic hospitalists were burned out, with another 25% at risk, but the survey was limited to dues-paying members of the National Association of Inpatient Physicians, the precursor to SHM.2
Nor has it been easy to compare hospitalist satisfaction and burnout levels to those of other specialists. “We haven’t really defined what a sustained, long-term career in hospital medicine is going to be,” Dr. Wetterneck says. “And in that way, it’s hard to say, ‘Compared to other professions, are we happier or not?’”
One of her recent studies, however, generally agrees with the handful of surveys addressing satisfaction and burnout among hospitalists. Overall, 63% of respondents reported high satisfaction with their job, while 69% were highly satisfied with their specialty. Roughly 30%, however, also reported feeling symptom of job burnout.3
Kelki Hinami, MD, MS, assistant of professor of medicine at Northwestern University Feinberg School of Medicine in Chicago and a coauthor of the study, says one take-home message is that hospitalists do fairly well in finding jobs that match their individual needs. “To further illustrate this, we found that hospitalists working in various practice models have different ideas about what is most important to their job,” he says.
Autonomy, for example, is considered most important by more local group hospitalists than by those of any other model, while recognition by leaders and having a variety of tasks at work are particularly important to academic hospitalists. Unlike other hospitalists, however, fewer academics consider pay to be the most important job characteristic.
A third study, led by John Yoon, MD, assistant professor in the section of hospital medicine at the University of Chicago, has examined career satisfaction, burnout, and morale among primary-care physicians (PCPs) and hospitalists. So far, the results he reported at HM11 largely agree with the other recent surveys: Combined, 85% of hospitalists report being either somewhat or very satisfied with their overall career. Conversely, 24% of hospitalists regretted choosing medicine as a career and 38% say they would have chosen a different medical specialty if they had to do it over again.4
Dr. Yoon says his data, compiled from two survey samples of about 1,000 generalists each, have revealed few differences between hospitalists and PCPs. “I thought hospitalists would be more satisfied than primary-care physicians, given the declining satisfaction rates of PCPs that we know about, and that students and trainees are less likely to go into primary care,” he says. Even burnout rates are similar, however; Dr. Yoon says he’s noticed a trend toward hospitalists reporting less burnout than PCPs, but the difference is not yet statistically significant.
Choice of a New Generation?
HM’s attractiveness to medical residents offers other clues about its ability to provide a sustainable and satisfying career choice. Salary, part of the “reward and recognition” pillar, has long been one perceived weakness. Anecdotally, however, Dr. Yoon says many general medicine residents see HM as a better financial option than primary care. “Some of the residents I work with, when I asked them, ‘Will you be a primary-care physician or a hospitalist?’ a lot of them say, ‘Probably hospitalist,’” he says. “And generally the reason is because they have to pay off their debt.”
It’s true that hospitalists’ salaries lag behind that of most of other specialists. Nevertheless, researchers like Colin West, MD, PhD, associate professor of medicine and biostatistics at the Mayo Clinic in Rochester, Minn., say many medical residents are prioritizing financial considerations as relatively low on the scale of general preferences.
—John Yoon, MD, assistant professor, section of hospital medicine, University of Chicago
Dr. West, an associate program director for the internal-medicine residency program at Mayo, sees a generational sea change in the career considerations deemed most important. Based on a career decision survey filled out by nearly 15,000 internal-medical residents, he found that roughly 70% of respondents said time with family was of “high” or “very high” importance to their career decisions.5 The category, which relates to SHM’s “workload and schedule” pillar, beat out eight others as the most important factor overall, while global financial considerations scored relatively low.
Residents who placed high value on time with family were more likely to choose careers in more predictable, outpatient-based specialties, such as endocrinology or rheumatology. HM also fared well in this category. Dr. West says the results suggest that residents considering a hospitalist career are attracted to the specialty’s flexibility and predictability of the largely shift-based scheduling.
William Cors, MD, chief medical quality officer at Pocono Health System in East Stroudsburg, Pa., says more physicians are looking for job security, predictable shifts, and a better work-life balance. As HM matures and demonstrates that it can address those needs, Dr. Cors sees it becoming more attractive for medical students and residents.
In practice, though, other research suggests a career in HM doesn’t always meet expectations. Dr. Wetterneck and Dr. Hinami, for example, highlighted both compensation and work-life balance as points of concern in their study: For both factors, only about 30% of hospitalists were optimally satisfied.
Separately, Dr. Misky and his colleagues reported that roughly half of academic hospitalists were satisfied with the ability to control their schedule, and with their amount of personal and family time. Those who were unsatisfied with either of these categories, the survey found, were at higher risk for burnout. Similarly, Dr. Yoon found that physicians who reported having no control over their work hours or their call schedule, part of SHM’s “autonomy and control” pillar, were more likely to report burnout.
So why is HM stumbling on perceived selling points like family friendliness and autonomy? Dr. Wetterneck believes too many unfilled jobs and rapid turnover could be putting more pressure on existing hospitalists and interfering with their ability to balance home and work life. “There’s a huge need for hospitalists everywhere,” she says, and reliance on them has been especially acute at academic centers and large community hospitals contending with the recently imposed limits on residents’ work hours.
The Hospitalist: A People Person
Another shift may be occurring in the types of relationships necessary for a satisfying work environment, a big part of the “community and environment” pillar. Although Dr. Yoon says long-term connections with students and trainees have added meaning to his job, he is mourning the absence of other bonds. “One loss I’m starting to feel keenly as an academic hospitalist, having spent my early training years as a primary-care doc, really is the loss of having long-term relationships with patients,” he says. “My clinical encounters with patients these days as a hospitalist are very intense, but also very brief.”
Dr. Yoon has pondered whether the HM field can rearrange practice settings to promote more satisfying relationships. Such a change, he says, might occur through innovative models that aid coordination with medical homes, or provide more chronic care for high-risk patients. “In my view, the trajectory of hospital medicine is pretty wide open for creativity and new models of care,” he says. “I think it will be partly driven by the need to want to have more meaningful interactions with patients.”
Those relationships need not be long-term, however. One recent study found high satisfaction among hospitalists and laborists working within the fast-growing OBGYN hospitalist field.6
Dr. Hinami says collaborative care that involves close working relationships with specialists and other care providers might help propel the hospitalist movement forward. In his survey with Dr. Wetterneck, hospitalists ranked relationships with staff and colleagues among the most satisfying of any of the domains; hospitalists also indicated high levels of satisfaction with their patient relationships. “Clearly, relationships are critical to overall job satisfaction, and hospitalists, I think, are doing a fairly good job at maintaining those relationships,” Dr. Hinami says.
—Keiki Hinami, MD, assistant professor of medicine, Northwestern University Feinberg School of Medicine, Chicago
A 2002 survey-based study reinforces the importance of such bonds. Job burnout and intent to remain in the hospitalist career, its authors concluded, were more highly influenced by “favorable social relations” involving colleagues, coworkers, and patients than by such factors as reduced autonomy and the use of financial incentives.7
The focus on maintaining multiple relationships fits well with the collaborative approach to care that many hospitalists say they value highly. One big satisfier for hospitalists, Dr. Cors says, will be “a sense that they’re really part of a healthcare team and not just punching the clock and doing their shifts.”
The Verdict
Despite the difficulty in discerning long-term trends, studies suggest that overall satisfaction with the specialty of hospital medicine remains high, a promising sign for the maturing field. Career hospitalists also seem adept at relationships with peers and other providers, a skill that will serve them well as collaborative-care models gain steam.
Nonetheless, surveys also suggest a worrisome rate of burnout and less-than-optimal satisfaction with elements that should be the strong suits of HM, such as work-life balance and autonomy. Academics are searching for their own clinical-research balance. And Dr. West says the jury’s still out on the future pitfalls that might get in the way of a sustainable career path for older practitioners, such as overnight shifts.
Hospitalist-led efforts, however, may be starting to pay dividends. At the University of California at San Francisco, a faculty development program for first-year hospitalists has included a coaching relationship with a senior faculty member, a teaching course, newly established divisional grand rounds, and a framework for meeting scholarly expectations. Upon its implementation, the program has led to higher job satisfaction, skill-set comfort, and academic production among participants.8
Given the expanding range of HM duties and practice models, hospitals, division chiefs, and team leaders cannot rely on a single recipe for happy and productive hospitalists. “I don’t know if there is a cookbook; I think it’s highly variable depending on your institution and the needs of the academic facility where you are,” Dr. Misky says.
SHM’s 2006 white paper stated that the best career satisfaction strategy is to find a job that fits an individual’s preferences and attitudes. “People who are unhappy with their job don’t tend to stay in it, and from what we know about hospital medicine right now, you can find pretty much any type of job anywhere you want, so the job market is very open,” Dr. Wetterneck says.
Ensuring the right fit for doctors within HM, though, will require institutional support. “It’s going to be up to hospitals and hospitalist programs to create jobs that are sustainable that people like,” she says, “so that hospitalists will stay long in their job and in the profession.”
Bryn Nelson is a freelance medical writer based in Seattle.
More Mentorship in Hospital Medicine? It’s Academic
Within the 2011 State of Hospital Medicine report, one statistic in particular points to the youth of the medical specialty: Just over 10% of surveyed hospitalists had reached the rank of associate professor or higher.
How might the potential lack of mentorship within this immature field affect the ability of hospitalists to successfully navigate academia? So asked Gregory Misky, MD, assistant professor of medicine at the University of Colorado Denver, and his colleagues in a survey-based study. The results agree with other recent assessments that mentors are in short supply. “Academic hospital medicine groups have an acute need for mentoring and career development programs,” one study concludes.
The research of Dr. Misky and his collaborators found that only 42% of academic hospitalists could identify a mentor, while only 31% reported that they were mentoring another academic hospitalist.1 Based on sheer numbers and experience, the pool of mentors may significantly expand as the field matures. But Dr. Misky also urges some flexibility, noting that his own mentor is a non-hospitalist.
In his own research, Colin West, MD, PhD, associate professor of medicine and biostatistics at the Mayo Clinic in Rochester, Minn., found that residents considering a career in HM placed less emphasis on the specialty or subspecialty of their mentor.5 Why? Very likely, he says, there just weren’t enough hospitalist mentors around to get a sense of what the career was all about.
Dr. West hopes the numbers suggest otherwise in the near future. “You want to recruit bright people into your specialty, but at the same time, you also want to recruit the right people,” he says. “And that means that you need to be able to expose people to a full breadth of what a decision to pursue a certain specialty really means.”
References
- Glasheen JJ, Misky GJ, Reid MB, Harrison RA, Sharpe B, Auerbach A. Career satisfaction and burnout in academic hospital medicine. Arch Intern Med. 2011;171(8) 782-785.
- Hoff TH, Whitcomb WF, Williams K, Nelson JR, Cheesman RA. Characteristics and work experiences of hospitalists in the United States. Arch Intern Med. 2001;161(6):851-858.
- Hinami K, Whelan CT, Wolosin RJ, Miller JA, Wetterneck TB. Worklife and satisfaction of hospitalists: toward flourishing careers [published online ahead of print July 20, 2011]. J Gen Intern Med. doi:10.1007/s116060-011-1780-z.
- Yoon J, Miller A, Rasinski K, Curlin F. Burnout, sense of calling, and career resilience among hospitalists and primary care physicians: a national survey. J Hosp Med. 2011;6(4):S90-S91.
- West CP, Drefahl MM, Popkave C, Kolars JC. Internal medicine resident self-report of factors associated with career decisions. J Gen Intern Med. 2009;24(8):946-949.
- Funk C, Anderson BL, Schulkin J, Weinstein L. Survey of obstetric and gynecologic hospitalists and laborists. Am J Obstet Gynecol. 2010;203(2):177.e1-177.e4.
- Hoff T, Whitcomb WF, Nelson JR. Thriving and surviving in a new medical career: the case of hospitalist physicians. J Health Soc Behav. 2002;43(1):72-91.
- Sehgal NL, Sharpe BA, Auerbach AA, Wachter RM. Investing in the future: Building an academic hospitalist faculty development program. J Hosp Med. 2011;6(3):161-166.
Good Citizenship

Hospital medicine is fortunate to have many very dedicated and professionally centered doctors who work enthusiastically to both provide excellent care to their patients and work to make their own practice and their hospital a better place. I am lucky to practice with many of them in our practice in Bellevue, Wash.
Yet a significant portion of hospitalists have chosen this work because they’re looking for relatively-low-commitment work. In essence, they see themselves as dating their practice rather than marrying it. Some of them might even say, “I thought I wanted a career. It turns out all I wanted was a paycheck.”
Most are skilled clinicians who find the energy to do a good job for the patients under their care but don’t have a mindset of owning their practice and investing time in making it perform better.
This gives rise to a dilemma: How can a practice turn these perfectly capable physicians into meaningfully engaged participants in the hospitalist practice itself and the hospital as a whole? What about a salary bonus based on good citizenship? Would that cause them to become more engaged and committed?
There is voluminous research and a whole row of books at your local Barnes & Noble that address these questions more completely that I can, so I’ll just share some real-world experience and insights from one book.
What Might a Citizenship Bonus Look Like?
There are a number of ways to consider designing a citizenship bonus. At a previous SHM practice-management course, Win Whitcomb, MD, MHM, presented one example from Mercy Medical Center in Springfield, Mass. (see Figure 1).
The following kinds of activities might be appropriate for a hospitalist to earn a citizenship bonus:
- Active participation on approved hospital committees (e.g. the pharmacy and therapeutic committees) and regular input from and feedback to the hospitalist group (e.g. via e-mail) about relevant activities of the committee;
- A project to improve clinical care (e.g. improved glycemic control, fall prevention, med reconciliation, discharge processes, readmission rates, ensuring follow-up of tests resulted after discharge, etc.);
- A project to improve business operations—for example, improve our billing/coding accuracy. Such a project could be to develop a new progress note template and collect data regarding its use and effectiveness;
- Work to improve communication and interaction with other hospital staff—for example, joint rounding with nurses, improve throughput, etc.; and
- Project(s) to increase the group’s social cohesion and engagement with hospital initiatives and goals.
Does a Citizenship Bonus Help or Hinder a Practice?
From the experience Mercy Hospital had with the citizenship bonus, Win concluded that many, but not all, hospitalists who don’t seem interested in quality improvement (QI) will become engaged if there is a reward/recognition structure. A relatively small dollar bonus is OK, as long as non-monetary rewards exist (e.g. improvement demonstrable, sense of teamwork, recognition). And hospitalists who were engaged prior to establishing the salary incentive are not likely to change their behavior, but their effort is now recognized—allowing for sustained engagement.
I’m sure many institutions would find a similar desirable outcome from putting into place a citizenship bonus. But it isn’t a guarantee. All performance bonus programs, whether based on “hard” outcomes like patient satisfaction scores or “soft” things like citizenship, are tricky to set up and operate effectively.
I have seen well-intentioned efforts to create a citizenship bonus lead to an increase in hospitalists working on projects outside of direct patient care, but at a cost of leading them to focus more intently on just how much they’re being paid for any work outside of direct patient care. It seems that the bonus might have ignited more frustration and concern about compensation, and any benefit to the practice might have been offset by harm to group culture. And if the bonus goes away, some doctors might be even less engaged than they were before it was turned on.
In “Drive: The Surprising Truth About What Motivates Us,” Daniel Pink makes a pretty convincing case that “the more prominent salary, perks, and benefits are in someone’s work life, the more they can inhibit creativity and unravel performance.” He makes the case that organizations are most demotivating “when they use rewards like money to motivate staff.”
“Effective organizations compensate people in amounts and ways that allow individuals to mostly forget about compensation and instead focus on the work itself,” Pink writes.
How do you allow individuals to forget about compensation? He says ensure internal and external fairness in compensation; pay more than average; and if you use performance metrics, make them wide-ranging, relevant, and hard to game.
So maybe financial compensation for citizenship, whether paid through a bonus, hourly, or some other separate salary element, isn’t such a good idea for a hospitalist practice (or any physician practice?). I don’t have a definitive answer, so you’ll have to decide this for yourself. But my hunch is that groups with a thriving culture might in some cases benefit from a well-designed citizenship bonus. That said, those groups also could be the ones less in need of it.
Groups that already have a weak or unhealthy culture, or are frustrated by what they see is inadequate compensation for clinical work, might find such a bonus leads to problems that offset its benefit.
Training in leadership, quality improvement, and other non-clinical areas that are critical for the success of a hospitalist practice is always worthwhile and might capture many of the benefits of a citizenship bonus without its drawbacks.
Dr. Nelson has been a practicing hospitalist since 1988 and is co-founder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is also course co-director and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.
Hospital medicine is fortunate to have many very dedicated and professionally centered doctors who work enthusiastically to both provide excellent care to their patients and work to make their own practice and their hospital a better place. I am lucky to practice with many of them in our practice in Bellevue, Wash.
Yet a significant portion of hospitalists have chosen this work because they’re looking for relatively-low-commitment work. In essence, they see themselves as dating their practice rather than marrying it. Some of them might even say, “I thought I wanted a career. It turns out all I wanted was a paycheck.”
Most are skilled clinicians who find the energy to do a good job for the patients under their care but don’t have a mindset of owning their practice and investing time in making it perform better.
This gives rise to a dilemma: How can a practice turn these perfectly capable physicians into meaningfully engaged participants in the hospitalist practice itself and the hospital as a whole? What about a salary bonus based on good citizenship? Would that cause them to become more engaged and committed?
There is voluminous research and a whole row of books at your local Barnes & Noble that address these questions more completely that I can, so I’ll just share some real-world experience and insights from one book.
What Might a Citizenship Bonus Look Like?
There are a number of ways to consider designing a citizenship bonus. At a previous SHM practice-management course, Win Whitcomb, MD, MHM, presented one example from Mercy Medical Center in Springfield, Mass. (see Figure 1).
The following kinds of activities might be appropriate for a hospitalist to earn a citizenship bonus:
- Active participation on approved hospital committees (e.g. the pharmacy and therapeutic committees) and regular input from and feedback to the hospitalist group (e.g. via e-mail) about relevant activities of the committee;
- A project to improve clinical care (e.g. improved glycemic control, fall prevention, med reconciliation, discharge processes, readmission rates, ensuring follow-up of tests resulted after discharge, etc.);
- A project to improve business operations—for example, improve our billing/coding accuracy. Such a project could be to develop a new progress note template and collect data regarding its use and effectiveness;
- Work to improve communication and interaction with other hospital staff—for example, joint rounding with nurses, improve throughput, etc.; and
- Project(s) to increase the group’s social cohesion and engagement with hospital initiatives and goals.
Does a Citizenship Bonus Help or Hinder a Practice?
From the experience Mercy Hospital had with the citizenship bonus, Win concluded that many, but not all, hospitalists who don’t seem interested in quality improvement (QI) will become engaged if there is a reward/recognition structure. A relatively small dollar bonus is OK, as long as non-monetary rewards exist (e.g. improvement demonstrable, sense of teamwork, recognition). And hospitalists who were engaged prior to establishing the salary incentive are not likely to change their behavior, but their effort is now recognized—allowing for sustained engagement.
I’m sure many institutions would find a similar desirable outcome from putting into place a citizenship bonus. But it isn’t a guarantee. All performance bonus programs, whether based on “hard” outcomes like patient satisfaction scores or “soft” things like citizenship, are tricky to set up and operate effectively.
I have seen well-intentioned efforts to create a citizenship bonus lead to an increase in hospitalists working on projects outside of direct patient care, but at a cost of leading them to focus more intently on just how much they’re being paid for any work outside of direct patient care. It seems that the bonus might have ignited more frustration and concern about compensation, and any benefit to the practice might have been offset by harm to group culture. And if the bonus goes away, some doctors might be even less engaged than they were before it was turned on.
In “Drive: The Surprising Truth About What Motivates Us,” Daniel Pink makes a pretty convincing case that “the more prominent salary, perks, and benefits are in someone’s work life, the more they can inhibit creativity and unravel performance.” He makes the case that organizations are most demotivating “when they use rewards like money to motivate staff.”
“Effective organizations compensate people in amounts and ways that allow individuals to mostly forget about compensation and instead focus on the work itself,” Pink writes.
How do you allow individuals to forget about compensation? He says ensure internal and external fairness in compensation; pay more than average; and if you use performance metrics, make them wide-ranging, relevant, and hard to game.
So maybe financial compensation for citizenship, whether paid through a bonus, hourly, or some other separate salary element, isn’t such a good idea for a hospitalist practice (or any physician practice?). I don’t have a definitive answer, so you’ll have to decide this for yourself. But my hunch is that groups with a thriving culture might in some cases benefit from a well-designed citizenship bonus. That said, those groups also could be the ones less in need of it.
Groups that already have a weak or unhealthy culture, or are frustrated by what they see is inadequate compensation for clinical work, might find such a bonus leads to problems that offset its benefit.
Training in leadership, quality improvement, and other non-clinical areas that are critical for the success of a hospitalist practice is always worthwhile and might capture many of the benefits of a citizenship bonus without its drawbacks.
Dr. Nelson has been a practicing hospitalist since 1988 and is co-founder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is also course co-director and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.
Hospital medicine is fortunate to have many very dedicated and professionally centered doctors who work enthusiastically to both provide excellent care to their patients and work to make their own practice and their hospital a better place. I am lucky to practice with many of them in our practice in Bellevue, Wash.
Yet a significant portion of hospitalists have chosen this work because they’re looking for relatively-low-commitment work. In essence, they see themselves as dating their practice rather than marrying it. Some of them might even say, “I thought I wanted a career. It turns out all I wanted was a paycheck.”
Most are skilled clinicians who find the energy to do a good job for the patients under their care but don’t have a mindset of owning their practice and investing time in making it perform better.
This gives rise to a dilemma: How can a practice turn these perfectly capable physicians into meaningfully engaged participants in the hospitalist practice itself and the hospital as a whole? What about a salary bonus based on good citizenship? Would that cause them to become more engaged and committed?
There is voluminous research and a whole row of books at your local Barnes & Noble that address these questions more completely that I can, so I’ll just share some real-world experience and insights from one book.
What Might a Citizenship Bonus Look Like?
There are a number of ways to consider designing a citizenship bonus. At a previous SHM practice-management course, Win Whitcomb, MD, MHM, presented one example from Mercy Medical Center in Springfield, Mass. (see Figure 1).
The following kinds of activities might be appropriate for a hospitalist to earn a citizenship bonus:
- Active participation on approved hospital committees (e.g. the pharmacy and therapeutic committees) and regular input from and feedback to the hospitalist group (e.g. via e-mail) about relevant activities of the committee;
- A project to improve clinical care (e.g. improved glycemic control, fall prevention, med reconciliation, discharge processes, readmission rates, ensuring follow-up of tests resulted after discharge, etc.);
- A project to improve business operations—for example, improve our billing/coding accuracy. Such a project could be to develop a new progress note template and collect data regarding its use and effectiveness;
- Work to improve communication and interaction with other hospital staff—for example, joint rounding with nurses, improve throughput, etc.; and
- Project(s) to increase the group’s social cohesion and engagement with hospital initiatives and goals.
Does a Citizenship Bonus Help or Hinder a Practice?
From the experience Mercy Hospital had with the citizenship bonus, Win concluded that many, but not all, hospitalists who don’t seem interested in quality improvement (QI) will become engaged if there is a reward/recognition structure. A relatively small dollar bonus is OK, as long as non-monetary rewards exist (e.g. improvement demonstrable, sense of teamwork, recognition). And hospitalists who were engaged prior to establishing the salary incentive are not likely to change their behavior, but their effort is now recognized—allowing for sustained engagement.
I’m sure many institutions would find a similar desirable outcome from putting into place a citizenship bonus. But it isn’t a guarantee. All performance bonus programs, whether based on “hard” outcomes like patient satisfaction scores or “soft” things like citizenship, are tricky to set up and operate effectively.
I have seen well-intentioned efforts to create a citizenship bonus lead to an increase in hospitalists working on projects outside of direct patient care, but at a cost of leading them to focus more intently on just how much they’re being paid for any work outside of direct patient care. It seems that the bonus might have ignited more frustration and concern about compensation, and any benefit to the practice might have been offset by harm to group culture. And if the bonus goes away, some doctors might be even less engaged than they were before it was turned on.
In “Drive: The Surprising Truth About What Motivates Us,” Daniel Pink makes a pretty convincing case that “the more prominent salary, perks, and benefits are in someone’s work life, the more they can inhibit creativity and unravel performance.” He makes the case that organizations are most demotivating “when they use rewards like money to motivate staff.”
“Effective organizations compensate people in amounts and ways that allow individuals to mostly forget about compensation and instead focus on the work itself,” Pink writes.
How do you allow individuals to forget about compensation? He says ensure internal and external fairness in compensation; pay more than average; and if you use performance metrics, make them wide-ranging, relevant, and hard to game.
So maybe financial compensation for citizenship, whether paid through a bonus, hourly, or some other separate salary element, isn’t such a good idea for a hospitalist practice (or any physician practice?). I don’t have a definitive answer, so you’ll have to decide this for yourself. But my hunch is that groups with a thriving culture might in some cases benefit from a well-designed citizenship bonus. That said, those groups also could be the ones less in need of it.
Groups that already have a weak or unhealthy culture, or are frustrated by what they see is inadequate compensation for clinical work, might find such a bonus leads to problems that offset its benefit.
Training in leadership, quality improvement, and other non-clinical areas that are critical for the success of a hospitalist practice is always worthwhile and might capture many of the benefits of a citizenship bonus without its drawbacks.
Dr. Nelson has been a practicing hospitalist since 1988 and is co-founder and past president of SHM. He is a principal in Nelson Flores Hospital Medicine Consultants, a national hospitalist practice management consulting firm (www.nelsonflores.com). He is also course co-director and faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official position of SHM.