User login
Adolescent immunizations – Focus on HPV vaccine
The U.S. immunization program has been one of the country’s most successful initiatives and best investments. Prior to 2005, vaccines were targeted for administration to infants and young children. Adolescence was a period for catch-up immunizations. All that changed in 2005 when the first meningococcal conjugate vaccine (MCV) was recommended for administration to preteens at 11-12 years and college freshmen residing in dormitories by the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices (ACIP). Shortly thereafter in 2006, a new tetanus toxoid, diphtheria, and acellular pertussis vaccine (Tdap) was recommended, and in March 2007 the quadrivalent human papillomavirus vaccine (HPV4: types 6, 11, 16, and 18) was recommended for use in girls, starting at age 11-12 years, and young women up to 26 years of age. In 2009, a bivalent HPV vaccine (HPV2: types 16 and 18) was licensed, and in 2010, ACIP recommendations indicated that either HPV4 or HPV2 vaccine could be administered to girls and young women. In addition, the use of HPV4 vaccine in males was permitted. In 2011, ACIP recommended routine administration of HPV4 to boys and young adult males up to 21 years of age. Adolescents were the target population for these vaccines, and administration was recommended at the 11- to 12-year wellness visit. The primary role of the adolescent encounter was no longer to provide catch-up immunizations. A definitive adolescent immunization schedule had been established.
Why introduce the HPV vaccine so early?
HPV is the most common sexually transmitted infection in both men and women. Recent data suggest that approximately 79 million individuals are infected (Sex. Transm. Dis. 2013;40:187-93). Annually, about 14 million, mostly young adults are infected. Most sexually active individuals will acquire HPV. It is most common in teens and young adults, and intercourse is not required for transmission. It can be transmitted with any type of intimate sexual contact, and it has been isolated from virgins. The majority of these infections are asymptomatic and self- limited. However, persistent infection is associated with cervical and other types of anogenital cancer, and genital warts in both men and women. Complications of these infections may take years to manifest.
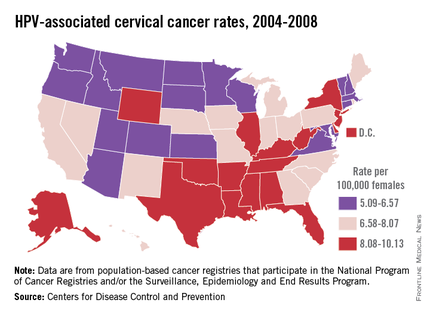
HPV is categorized by its epidemiologic association with cervical cancer. High-risk types cause cervical cancer, and HPV types 16 and 18 account for the majority of cervical cancers (66%) These two types are also associated with vaginal (55%), anal (79%), and oropharyngeal (62%) cancer (MMWR 2014 Jan. 31;63;69-72). It is estimated that each year there are 26,000 HPV-related cancers including 8,800 cases in men and 17,000 in women, 4,000 of whom will die of cervical cancer, according to the CDC. Low-risk types including HPV types 6 and 11 cause benign/low-grade cervical cell changes, recurrent papillomatosis, and 90% of the cases of genital warts.
Once a person is infected, HPV usually clears. If not, cervical intraepithelial neoplasia (CIN) may occur. The infection may still resolve spontaneously. If it persists, the degree of dysplasia can progress. Several years may pass before progression to invasive cancer. HPV vaccines are prophylactic like other vaccines. They cannot prevent disease progression and need to be administered before exposure to the viruses.
Compared with the introduction of other vaccines, such as Haemophilus influenzae type b and Prevnar7, some pediatric care providers may feel we may not have the benefit of realizing our efforts as immediately as in the past. However, encouraging vaccine effectiveness data in U.S. teens has been published. In one study, the investigators compared HPV prevalence data from the pre- and postvaccine era collected during the National Health and Nutrition Examination Survey. Among females aged 14-19 years, HPV prevalence (HPV-6, -11, -16, or -18 ) decreased from 11.5% in 2003-2006 to 5.1% in 2007-2010. That is a 56% reduction in vaccine type HPV prevalence. This decrease in prevalence occurred within 4 years of vaccine introduction and low vaccine uptake. (J. Infect. Dis. 2013;208:385-93). Studies conducted in Denmark, Australia, Germany, and New Zealand also have shown significant declines in HPV4 vaccine type infection prevalence.
Vaccination coverage
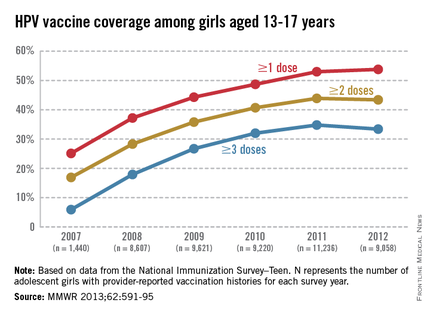
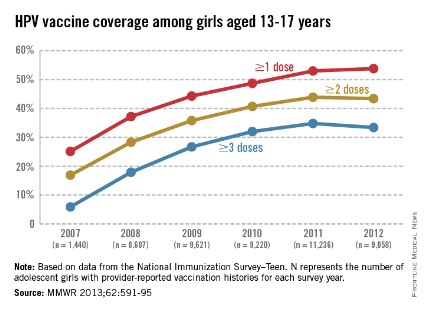
The CDC tracks vaccination coverage annually in the National Immunization Survey–Teen (NIS-Teen), with data obtained from the 50 states, the District of Columbia, the U.S. Virgin Islands, and six major urban areas (MMWR 2013;62:685-93). Vaccination coverage differed significantly although each vaccine is recommended to be routinely administered at the 11- to 12-year visit. Although an increase from 25% to 53% had been noted between 2007 and 2011, in 2012, coverage for receiving at least one dose of HPV among females was almost 54%, essentially unchanged since 2011. The number who had received the recommended three doses was also essentially unchanged from 2011 to 2012 (34.8% in 2011 and 33.4% in 2012). Receipt of a single dose of HPV in boys was 8.3% in 2011 and 20.8% in 2012, the first year after the vaccine was recommended. Completion of the series in boys was 6.3%, an increase from 1.3% in 2011.
In contrast, the 2012 coverage for Tdap increased to 85% and MCV4, to 74%. It has been suggested that the higher coverage of Tdap and MCV may be due to the 40 and 13 states, respectively, that require them for middle school entry.
The disparity in coverage between Tdap and other vaccines suggests there are numerous missed opportunities to vaccinate adolescents. Data revealed that missed opportunities for girls increased from 20.8% in 2007 to 84% in 2012. If all missed opportunities had been eliminated, HPV coverage for at least one dose could have reached 92.6%.Almost 25% of parents indicated that they had no plan to immunize their daughter. The top reasons parents stated for not immunizing their daughters included: not needed or necessary, 19.1%; not recommended by provider, 14.2%; safety concerns, 13.3%; lack of knowledge, 12.6%; and not sexually active, 10.1%. (MMWR 2013;62:591-5).
Vaccine safety also was addressed. All reported adverse events were consistent with prelicensure clinical trial data. Ninety two percent of all adverse events were nonserious and included syncope, dizziness, nausea, and fever. Reports peaked in 2008 and have declined each year thereafter.
Challenges for HPV prevention

Improving immunization coverage is critical. There are numerous strategies to increase coverage including reminder recall systems, standing orders, and educating parents, patients, health care providers, and office staff who interact with parents. Education should reemphasize why immunization is initiated at 11-12 years and that completion of the series is recommended by 13 years. School requirements have always led to an increase in vaccination coverage. Only the District of Columbia has one for HPV. In this case, eliminating missed opportunities is crucial. It is estimated that for every year coverage is delayed, an additional 4,400 women will develop cervical cancer. The reality is that the burden of HPV-related cancers will persist if coverage is not increased.
As Louis Pasteur once said, "When meditating over a disease, I never think of finding a remedy for it, but, instead a means of preventing it."
For additional resources to assist with discussions about HPV, click here.
Dr. Word is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. E-mail her at [email protected]. Scan this QR code or visit pediatricnews.com.
The U.S. immunization program has been one of the country’s most successful initiatives and best investments. Prior to 2005, vaccines were targeted for administration to infants and young children. Adolescence was a period for catch-up immunizations. All that changed in 2005 when the first meningococcal conjugate vaccine (MCV) was recommended for administration to preteens at 11-12 years and college freshmen residing in dormitories by the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices (ACIP). Shortly thereafter in 2006, a new tetanus toxoid, diphtheria, and acellular pertussis vaccine (Tdap) was recommended, and in March 2007 the quadrivalent human papillomavirus vaccine (HPV4: types 6, 11, 16, and 18) was recommended for use in girls, starting at age 11-12 years, and young women up to 26 years of age. In 2009, a bivalent HPV vaccine (HPV2: types 16 and 18) was licensed, and in 2010, ACIP recommendations indicated that either HPV4 or HPV2 vaccine could be administered to girls and young women. In addition, the use of HPV4 vaccine in males was permitted. In 2011, ACIP recommended routine administration of HPV4 to boys and young adult males up to 21 years of age. Adolescents were the target population for these vaccines, and administration was recommended at the 11- to 12-year wellness visit. The primary role of the adolescent encounter was no longer to provide catch-up immunizations. A definitive adolescent immunization schedule had been established.
Why introduce the HPV vaccine so early?
HPV is the most common sexually transmitted infection in both men and women. Recent data suggest that approximately 79 million individuals are infected (Sex. Transm. Dis. 2013;40:187-93). Annually, about 14 million, mostly young adults are infected. Most sexually active individuals will acquire HPV. It is most common in teens and young adults, and intercourse is not required for transmission. It can be transmitted with any type of intimate sexual contact, and it has been isolated from virgins. The majority of these infections are asymptomatic and self- limited. However, persistent infection is associated with cervical and other types of anogenital cancer, and genital warts in both men and women. Complications of these infections may take years to manifest.
HPV is categorized by its epidemiologic association with cervical cancer. High-risk types cause cervical cancer, and HPV types 16 and 18 account for the majority of cervical cancers (66%) These two types are also associated with vaginal (55%), anal (79%), and oropharyngeal (62%) cancer (MMWR 2014 Jan. 31;63;69-72). It is estimated that each year there are 26,000 HPV-related cancers including 8,800 cases in men and 17,000 in women, 4,000 of whom will die of cervical cancer, according to the CDC. Low-risk types including HPV types 6 and 11 cause benign/low-grade cervical cell changes, recurrent papillomatosis, and 90% of the cases of genital warts.
Once a person is infected, HPV usually clears. If not, cervical intraepithelial neoplasia (CIN) may occur. The infection may still resolve spontaneously. If it persists, the degree of dysplasia can progress. Several years may pass before progression to invasive cancer. HPV vaccines are prophylactic like other vaccines. They cannot prevent disease progression and need to be administered before exposure to the viruses.
Compared with the introduction of other vaccines, such as Haemophilus influenzae type b and Prevnar7, some pediatric care providers may feel we may not have the benefit of realizing our efforts as immediately as in the past. However, encouraging vaccine effectiveness data in U.S. teens has been published. In one study, the investigators compared HPV prevalence data from the pre- and postvaccine era collected during the National Health and Nutrition Examination Survey. Among females aged 14-19 years, HPV prevalence (HPV-6, -11, -16, or -18 ) decreased from 11.5% in 2003-2006 to 5.1% in 2007-2010. That is a 56% reduction in vaccine type HPV prevalence. This decrease in prevalence occurred within 4 years of vaccine introduction and low vaccine uptake. (J. Infect. Dis. 2013;208:385-93). Studies conducted in Denmark, Australia, Germany, and New Zealand also have shown significant declines in HPV4 vaccine type infection prevalence.
Vaccination coverage
The CDC tracks vaccination coverage annually in the National Immunization Survey–Teen (NIS-Teen), with data obtained from the 50 states, the District of Columbia, the U.S. Virgin Islands, and six major urban areas (MMWR 2013;62:685-93). Vaccination coverage differed significantly although each vaccine is recommended to be routinely administered at the 11- to 12-year visit. Although an increase from 25% to 53% had been noted between 2007 and 2011, in 2012, coverage for receiving at least one dose of HPV among females was almost 54%, essentially unchanged since 2011. The number who had received the recommended three doses was also essentially unchanged from 2011 to 2012 (34.8% in 2011 and 33.4% in 2012). Receipt of a single dose of HPV in boys was 8.3% in 2011 and 20.8% in 2012, the first year after the vaccine was recommended. Completion of the series in boys was 6.3%, an increase from 1.3% in 2011.
In contrast, the 2012 coverage for Tdap increased to 85% and MCV4, to 74%. It has been suggested that the higher coverage of Tdap and MCV may be due to the 40 and 13 states, respectively, that require them for middle school entry.
The disparity in coverage between Tdap and other vaccines suggests there are numerous missed opportunities to vaccinate adolescents. Data revealed that missed opportunities for girls increased from 20.8% in 2007 to 84% in 2012. If all missed opportunities had been eliminated, HPV coverage for at least one dose could have reached 92.6%.Almost 25% of parents indicated that they had no plan to immunize their daughter. The top reasons parents stated for not immunizing their daughters included: not needed or necessary, 19.1%; not recommended by provider, 14.2%; safety concerns, 13.3%; lack of knowledge, 12.6%; and not sexually active, 10.1%. (MMWR 2013;62:591-5).
Vaccine safety also was addressed. All reported adverse events were consistent with prelicensure clinical trial data. Ninety two percent of all adverse events were nonserious and included syncope, dizziness, nausea, and fever. Reports peaked in 2008 and have declined each year thereafter.
Challenges for HPV prevention
Improving immunization coverage is critical. There are numerous strategies to increase coverage including reminder recall systems, standing orders, and educating parents, patients, health care providers, and office staff who interact with parents. Education should reemphasize why immunization is initiated at 11-12 years and that completion of the series is recommended by 13 years. School requirements have always led to an increase in vaccination coverage. Only the District of Columbia has one for HPV. In this case, eliminating missed opportunities is crucial. It is estimated that for every year coverage is delayed, an additional 4,400 women will develop cervical cancer. The reality is that the burden of HPV-related cancers will persist if coverage is not increased.
As Louis Pasteur once said, "When meditating over a disease, I never think of finding a remedy for it, but, instead a means of preventing it."
For additional resources to assist with discussions about HPV, click here.
Dr. Word is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. E-mail her at [email protected]. Scan this QR code or visit pediatricnews.com.
The U.S. immunization program has been one of the country’s most successful initiatives and best investments. Prior to 2005, vaccines were targeted for administration to infants and young children. Adolescence was a period for catch-up immunizations. All that changed in 2005 when the first meningococcal conjugate vaccine (MCV) was recommended for administration to preteens at 11-12 years and college freshmen residing in dormitories by the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices (ACIP). Shortly thereafter in 2006, a new tetanus toxoid, diphtheria, and acellular pertussis vaccine (Tdap) was recommended, and in March 2007 the quadrivalent human papillomavirus vaccine (HPV4: types 6, 11, 16, and 18) was recommended for use in girls, starting at age 11-12 years, and young women up to 26 years of age. In 2009, a bivalent HPV vaccine (HPV2: types 16 and 18) was licensed, and in 2010, ACIP recommendations indicated that either HPV4 or HPV2 vaccine could be administered to girls and young women. In addition, the use of HPV4 vaccine in males was permitted. In 2011, ACIP recommended routine administration of HPV4 to boys and young adult males up to 21 years of age. Adolescents were the target population for these vaccines, and administration was recommended at the 11- to 12-year wellness visit. The primary role of the adolescent encounter was no longer to provide catch-up immunizations. A definitive adolescent immunization schedule had been established.
Why introduce the HPV vaccine so early?
HPV is the most common sexually transmitted infection in both men and women. Recent data suggest that approximately 79 million individuals are infected (Sex. Transm. Dis. 2013;40:187-93). Annually, about 14 million, mostly young adults are infected. Most sexually active individuals will acquire HPV. It is most common in teens and young adults, and intercourse is not required for transmission. It can be transmitted with any type of intimate sexual contact, and it has been isolated from virgins. The majority of these infections are asymptomatic and self- limited. However, persistent infection is associated with cervical and other types of anogenital cancer, and genital warts in both men and women. Complications of these infections may take years to manifest.
HPV is categorized by its epidemiologic association with cervical cancer. High-risk types cause cervical cancer, and HPV types 16 and 18 account for the majority of cervical cancers (66%) These two types are also associated with vaginal (55%), anal (79%), and oropharyngeal (62%) cancer (MMWR 2014 Jan. 31;63;69-72). It is estimated that each year there are 26,000 HPV-related cancers including 8,800 cases in men and 17,000 in women, 4,000 of whom will die of cervical cancer, according to the CDC. Low-risk types including HPV types 6 and 11 cause benign/low-grade cervical cell changes, recurrent papillomatosis, and 90% of the cases of genital warts.
Once a person is infected, HPV usually clears. If not, cervical intraepithelial neoplasia (CIN) may occur. The infection may still resolve spontaneously. If it persists, the degree of dysplasia can progress. Several years may pass before progression to invasive cancer. HPV vaccines are prophylactic like other vaccines. They cannot prevent disease progression and need to be administered before exposure to the viruses.
Compared with the introduction of other vaccines, such as Haemophilus influenzae type b and Prevnar7, some pediatric care providers may feel we may not have the benefit of realizing our efforts as immediately as in the past. However, encouraging vaccine effectiveness data in U.S. teens has been published. In one study, the investigators compared HPV prevalence data from the pre- and postvaccine era collected during the National Health and Nutrition Examination Survey. Among females aged 14-19 years, HPV prevalence (HPV-6, -11, -16, or -18 ) decreased from 11.5% in 2003-2006 to 5.1% in 2007-2010. That is a 56% reduction in vaccine type HPV prevalence. This decrease in prevalence occurred within 4 years of vaccine introduction and low vaccine uptake. (J. Infect. Dis. 2013;208:385-93). Studies conducted in Denmark, Australia, Germany, and New Zealand also have shown significant declines in HPV4 vaccine type infection prevalence.
Vaccination coverage
The CDC tracks vaccination coverage annually in the National Immunization Survey–Teen (NIS-Teen), with data obtained from the 50 states, the District of Columbia, the U.S. Virgin Islands, and six major urban areas (MMWR 2013;62:685-93). Vaccination coverage differed significantly although each vaccine is recommended to be routinely administered at the 11- to 12-year visit. Although an increase from 25% to 53% had been noted between 2007 and 2011, in 2012, coverage for receiving at least one dose of HPV among females was almost 54%, essentially unchanged since 2011. The number who had received the recommended three doses was also essentially unchanged from 2011 to 2012 (34.8% in 2011 and 33.4% in 2012). Receipt of a single dose of HPV in boys was 8.3% in 2011 and 20.8% in 2012, the first year after the vaccine was recommended. Completion of the series in boys was 6.3%, an increase from 1.3% in 2011.
In contrast, the 2012 coverage for Tdap increased to 85% and MCV4, to 74%. It has been suggested that the higher coverage of Tdap and MCV may be due to the 40 and 13 states, respectively, that require them for middle school entry.
The disparity in coverage between Tdap and other vaccines suggests there are numerous missed opportunities to vaccinate adolescents. Data revealed that missed opportunities for girls increased from 20.8% in 2007 to 84% in 2012. If all missed opportunities had been eliminated, HPV coverage for at least one dose could have reached 92.6%.Almost 25% of parents indicated that they had no plan to immunize their daughter. The top reasons parents stated for not immunizing their daughters included: not needed or necessary, 19.1%; not recommended by provider, 14.2%; safety concerns, 13.3%; lack of knowledge, 12.6%; and not sexually active, 10.1%. (MMWR 2013;62:591-5).
Vaccine safety also was addressed. All reported adverse events were consistent with prelicensure clinical trial data. Ninety two percent of all adverse events were nonserious and included syncope, dizziness, nausea, and fever. Reports peaked in 2008 and have declined each year thereafter.
Challenges for HPV prevention
Improving immunization coverage is critical. There are numerous strategies to increase coverage including reminder recall systems, standing orders, and educating parents, patients, health care providers, and office staff who interact with parents. Education should reemphasize why immunization is initiated at 11-12 years and that completion of the series is recommended by 13 years. School requirements have always led to an increase in vaccination coverage. Only the District of Columbia has one for HPV. In this case, eliminating missed opportunities is crucial. It is estimated that for every year coverage is delayed, an additional 4,400 women will develop cervical cancer. The reality is that the burden of HPV-related cancers will persist if coverage is not increased.
As Louis Pasteur once said, "When meditating over a disease, I never think of finding a remedy for it, but, instead a means of preventing it."
For additional resources to assist with discussions about HPV, click here.
Dr. Word is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. E-mail her at [email protected]. Scan this QR code or visit pediatricnews.com.
Defect causes bone marrow failure, group finds
Credit: Daniel E. Sabath
Researchers say they’ve discovered a distinct bone-marrow-failure syndrome and the genetic defect that causes it.
In its natural form, the gene ERCC6L2 plays a role in DNA repair and mitochondrial function.
But investigators found evidence to suggest that mutations in ERCC6L2, and the subsequent DNA damage, were the underlying cause of tri-lineage bone marrow failure in a pair of patients with neurological dysfunction.
“New DNA sequencing technology has enabled us to identify and define a new gene defect which causes a particular type of bone marrow failure,” said Inderjeet Dokal, MD, of Queen Mary University of London in the UK.
“Clinicians treating patients with bone marrow failure should now include analysis for this gene in their investigation.”
Dr Dokal and his colleagues described this research in The American Journal of Human Genetics.
The team performed exome sequencing in 3 patients with genetically uncharacterized, tri-lineage bone marrow failure.
The patients came from consanguineous families (their parents were first-degree cousins), they had developmental delays characterized by learning disabilities, and 2 of the patients had microcephaly.
The sequencing did not uncover variations in any of the known genes associated with bone marrow failure. And the researchers could not find any obvious disease-causing variants in 1 of the patients.
However, the other 2 patients shared homozygous truncating mutations in ERCC6L2—c.1963C>T (p.Arg655*) and c.1236_1239delAACA (p.Thr413Cysfs*2). The c.1963C>T variant had already been identified, but, to the researchers’ knowledge, the other variant had not.
Additional experiments suggested that these mutations affect the subcellular localization and stability of ERCC6L2.
The investigators then speculated that ERCC6L2 plays a role in the DNA-damage response. To test that theory, they mimicked the truncating mutations by knocking down ERCC6L2 expression in human A549 cells.
This significantly reduced cell viability when the cells were exposed to the DNA-damaging agents mitomycin C and irofulven.
To further confirm their theory, the researchers looked at another marker of DNA damage. Previous research had suggested that Snf2 protein complexes are involved in the recruitment of γH2AX, a phosphorylated form of histone 2A, to sites of DNA damage.
So the team performed immunostaining with a γH2AX-specific antibody. And they found that ERCC6L2-knockdown cells displayed H2AX phosphorylation, an effect that increased upon genotoxic stress (treatment with irofulven).
These results indicate that ERCC6L2 plays a role in the DNA-damage-response pathway, and knockdown of this gene sensitizes cells to genotoxic agents.
Additional experiments showed that ERCC6L2 translocated to the mitochondria and the nucleus in response to DNA damage. And ERCC6L2 knockdown induced intracellular reactive oxygen species (ROS).
But introducing the ROS scavenger N-acetyl cysteine diminished the cytotoxicity induced by irofulven, and it halted ERCCGL2 traffic to the mitochondria and nucleus.
The investigators said these results point to a distinct bone-marrow-failure syndrome resulting from mutations in ERCC6L2.
“This is a promising finding which we hope, one day, could lead to finding an effective treatment for this type of gene defect,” Dr Dokal said. “Now [that] we know this research technique works, we plan to carry out further studies to shed more light on the genetic basis of many other cases of bone marrow failure.” ![]()
Credit: Daniel E. Sabath
Researchers say they’ve discovered a distinct bone-marrow-failure syndrome and the genetic defect that causes it.
In its natural form, the gene ERCC6L2 plays a role in DNA repair and mitochondrial function.
But investigators found evidence to suggest that mutations in ERCC6L2, and the subsequent DNA damage, were the underlying cause of tri-lineage bone marrow failure in a pair of patients with neurological dysfunction.
“New DNA sequencing technology has enabled us to identify and define a new gene defect which causes a particular type of bone marrow failure,” said Inderjeet Dokal, MD, of Queen Mary University of London in the UK.
“Clinicians treating patients with bone marrow failure should now include analysis for this gene in their investigation.”
Dr Dokal and his colleagues described this research in The American Journal of Human Genetics.
The team performed exome sequencing in 3 patients with genetically uncharacterized, tri-lineage bone marrow failure.
The patients came from consanguineous families (their parents were first-degree cousins), they had developmental delays characterized by learning disabilities, and 2 of the patients had microcephaly.
The sequencing did not uncover variations in any of the known genes associated with bone marrow failure. And the researchers could not find any obvious disease-causing variants in 1 of the patients.
However, the other 2 patients shared homozygous truncating mutations in ERCC6L2—c.1963C>T (p.Arg655*) and c.1236_1239delAACA (p.Thr413Cysfs*2). The c.1963C>T variant had already been identified, but, to the researchers’ knowledge, the other variant had not.
Additional experiments suggested that these mutations affect the subcellular localization and stability of ERCC6L2.
The investigators then speculated that ERCC6L2 plays a role in the DNA-damage response. To test that theory, they mimicked the truncating mutations by knocking down ERCC6L2 expression in human A549 cells.
This significantly reduced cell viability when the cells were exposed to the DNA-damaging agents mitomycin C and irofulven.
To further confirm their theory, the researchers looked at another marker of DNA damage. Previous research had suggested that Snf2 protein complexes are involved in the recruitment of γH2AX, a phosphorylated form of histone 2A, to sites of DNA damage.
So the team performed immunostaining with a γH2AX-specific antibody. And they found that ERCC6L2-knockdown cells displayed H2AX phosphorylation, an effect that increased upon genotoxic stress (treatment with irofulven).
These results indicate that ERCC6L2 plays a role in the DNA-damage-response pathway, and knockdown of this gene sensitizes cells to genotoxic agents.
Additional experiments showed that ERCC6L2 translocated to the mitochondria and the nucleus in response to DNA damage. And ERCC6L2 knockdown induced intracellular reactive oxygen species (ROS).
But introducing the ROS scavenger N-acetyl cysteine diminished the cytotoxicity induced by irofulven, and it halted ERCCGL2 traffic to the mitochondria and nucleus.
The investigators said these results point to a distinct bone-marrow-failure syndrome resulting from mutations in ERCC6L2.
“This is a promising finding which we hope, one day, could lead to finding an effective treatment for this type of gene defect,” Dr Dokal said. “Now [that] we know this research technique works, we plan to carry out further studies to shed more light on the genetic basis of many other cases of bone marrow failure.” ![]()
Credit: Daniel E. Sabath
Researchers say they’ve discovered a distinct bone-marrow-failure syndrome and the genetic defect that causes it.
In its natural form, the gene ERCC6L2 plays a role in DNA repair and mitochondrial function.
But investigators found evidence to suggest that mutations in ERCC6L2, and the subsequent DNA damage, were the underlying cause of tri-lineage bone marrow failure in a pair of patients with neurological dysfunction.
“New DNA sequencing technology has enabled us to identify and define a new gene defect which causes a particular type of bone marrow failure,” said Inderjeet Dokal, MD, of Queen Mary University of London in the UK.
“Clinicians treating patients with bone marrow failure should now include analysis for this gene in their investigation.”
Dr Dokal and his colleagues described this research in The American Journal of Human Genetics.
The team performed exome sequencing in 3 patients with genetically uncharacterized, tri-lineage bone marrow failure.
The patients came from consanguineous families (their parents were first-degree cousins), they had developmental delays characterized by learning disabilities, and 2 of the patients had microcephaly.
The sequencing did not uncover variations in any of the known genes associated with bone marrow failure. And the researchers could not find any obvious disease-causing variants in 1 of the patients.
However, the other 2 patients shared homozygous truncating mutations in ERCC6L2—c.1963C>T (p.Arg655*) and c.1236_1239delAACA (p.Thr413Cysfs*2). The c.1963C>T variant had already been identified, but, to the researchers’ knowledge, the other variant had not.
Additional experiments suggested that these mutations affect the subcellular localization and stability of ERCC6L2.
The investigators then speculated that ERCC6L2 plays a role in the DNA-damage response. To test that theory, they mimicked the truncating mutations by knocking down ERCC6L2 expression in human A549 cells.
This significantly reduced cell viability when the cells were exposed to the DNA-damaging agents mitomycin C and irofulven.
To further confirm their theory, the researchers looked at another marker of DNA damage. Previous research had suggested that Snf2 protein complexes are involved in the recruitment of γH2AX, a phosphorylated form of histone 2A, to sites of DNA damage.
So the team performed immunostaining with a γH2AX-specific antibody. And they found that ERCC6L2-knockdown cells displayed H2AX phosphorylation, an effect that increased upon genotoxic stress (treatment with irofulven).
These results indicate that ERCC6L2 plays a role in the DNA-damage-response pathway, and knockdown of this gene sensitizes cells to genotoxic agents.
Additional experiments showed that ERCC6L2 translocated to the mitochondria and the nucleus in response to DNA damage. And ERCC6L2 knockdown induced intracellular reactive oxygen species (ROS).
But introducing the ROS scavenger N-acetyl cysteine diminished the cytotoxicity induced by irofulven, and it halted ERCCGL2 traffic to the mitochondria and nucleus.
The investigators said these results point to a distinct bone-marrow-failure syndrome resulting from mutations in ERCC6L2.
“This is a promising finding which we hope, one day, could lead to finding an effective treatment for this type of gene defect,” Dr Dokal said. “Now [that] we know this research technique works, we plan to carry out further studies to shed more light on the genetic basis of many other cases of bone marrow failure.” ![]()
Allo-SCT can be effective in advanced SS, MF
SAN FRANCISCO—A single-center study suggests that transplant can induce remissions and improve survival in certain patients with advanced cutaneous T-cell lymphomas.
Allogeneic stem cell transplant (SCT) proved particularly effective in patients with Sézary syndrome (SS).
It also conferred benefits to mycosis fungoides (MF) patients with large-cell transformation (LCT), but patients with SS and LCT did not fare as well.
Madeleine Duvic, MD, of the MD Anderson Cancer Center in Houston, presented these results at the 6th Annual T-cell Lymphoma Forum. The data were updated from a previously published report (Duvic et al, JCO 2010).
Patient characteristics
Dr Duvic and her colleagues evaluated 48 patients who had biopsy-proven MF or SS. They underwent SCT at MD Anderson between July 2001 and September 2013.
The patients were in good health but had advanced disease. They had received a median of 6 prior therapies (range, 2-11).
The median age was 51.5 years (range, 19-72 years), and 54% of patients were female. Sixty-nine percent were Caucasian, 23% were African American, and 8% were Hispanic.
Fourteen patients had SS only, 16 had MF with LCT, 9 had SS and LCT, 5 had stage IVA or IIB disease (4 nodal, 1 tumor), and 4 had folliculotropic MF.
Transplant and other treatment
Patients had to have a 9/10 or 10/10 HLA-matched donor (related or unrelated). Most of the stem cells were collected via apheresis, but bone marrow aspiration was used for patients receiving mismatched transplants.
Forty-three of the patients underwent tumor and skin debulking with total skin electron beam (TBSEB) radiation (35 Gy) 1 or 2 months prior to SCT.
Most patients received a conditioning regimen of fludarabine and melphalan, but a few received fludarabine and cyclophosphamide. Patients received tacrolimus and methotrexate as graft-vs-host disease (GVHD) prophylaxis, as well as extracorporeal photopheresis if they developed GVHD.
All of the SS patients received vancomycin, fluconazole, and valacyclovir to ward off infections.
Response and GVHD
The overall complete response rate was 58% (28/48). Eight percent of patients did not engraft—3 MF patients with LCT and 1 SS patient.
“The response rate was much higher in Sézary patients [than in the rest of the cohort],” Dr Duvic said. “The worst prognosis was for patients who had both Sézary and large-cell transformation, who relapsed early and were generally refractory to prior therapies.”
Complete responses occurred in 79% of SS patients, 56% of MF patients with LCT, 44% of patients with SS and LCT, 40% of patients with stage IVA/IIB disease, and 50% of those with folliculotropic MF.
Among patients who received TBSEB, 58% (25/43) achieved a complete response. Of the 5 patients who did not receive TBSEB, 3 had a complete response (60%).
Sixty percent of patients developed GVHD (29/48). Eighteen patients had acute skin GVHD, 9 had acute gastrointestinal GVHD, 13 had chronic skin GVHD, and 6 had chronic gastrointestinal GVHD.
Relapse and survival
Overall, the relapse rate was 33% (16/48). Twenty-one percent of SS patients relapsed, as did 25% of MF patients with LCT and 56% of patients with SS and LCT.
The mortality rate was 44% (21/48). Patients died of relapsed MF, sepsis, infection, second malignancy, and other causes.
The overall survival (OS) was 10.2 years from diagnosis and 5.7 years from SCT. The progression-free survival (PFS) was 6 years from diagnosis and 1.8 years from SCT.
“We also looked at whether large-cell transformation had an effect on survival and therapy,” Dr Duvic said. “Large-cell transformation in MF has been reported to have a more aggressive course and a shorter overall survival than untransformed MF.”
“In our cohort of patients, we found an overall survival of 4.79 years in patients with large-cell transformation, which is a little bit higher than [survival rates in] the literature.”
Among MF patients with LCT, OS was 84% at 1 year from SCT and 38% at both 5 years and 10 years. PFS was 55% at 1 year, 16% at 5 years, and 0% at 10 years.
In comparison, among SS patients without LCT, OS was 88% at 1 year from SCT and 70% at both 5 years and 10 years. PFS was 63% at 1 year and 49% at 5 years and 10 years. ![]()
SAN FRANCISCO—A single-center study suggests that transplant can induce remissions and improve survival in certain patients with advanced cutaneous T-cell lymphomas.
Allogeneic stem cell transplant (SCT) proved particularly effective in patients with Sézary syndrome (SS).
It also conferred benefits to mycosis fungoides (MF) patients with large-cell transformation (LCT), but patients with SS and LCT did not fare as well.
Madeleine Duvic, MD, of the MD Anderson Cancer Center in Houston, presented these results at the 6th Annual T-cell Lymphoma Forum. The data were updated from a previously published report (Duvic et al, JCO 2010).
Patient characteristics
Dr Duvic and her colleagues evaluated 48 patients who had biopsy-proven MF or SS. They underwent SCT at MD Anderson between July 2001 and September 2013.
The patients were in good health but had advanced disease. They had received a median of 6 prior therapies (range, 2-11).
The median age was 51.5 years (range, 19-72 years), and 54% of patients were female. Sixty-nine percent were Caucasian, 23% were African American, and 8% were Hispanic.
Fourteen patients had SS only, 16 had MF with LCT, 9 had SS and LCT, 5 had stage IVA or IIB disease (4 nodal, 1 tumor), and 4 had folliculotropic MF.
Transplant and other treatment
Patients had to have a 9/10 or 10/10 HLA-matched donor (related or unrelated). Most of the stem cells were collected via apheresis, but bone marrow aspiration was used for patients receiving mismatched transplants.
Forty-three of the patients underwent tumor and skin debulking with total skin electron beam (TBSEB) radiation (35 Gy) 1 or 2 months prior to SCT.
Most patients received a conditioning regimen of fludarabine and melphalan, but a few received fludarabine and cyclophosphamide. Patients received tacrolimus and methotrexate as graft-vs-host disease (GVHD) prophylaxis, as well as extracorporeal photopheresis if they developed GVHD.
All of the SS patients received vancomycin, fluconazole, and valacyclovir to ward off infections.
Response and GVHD
The overall complete response rate was 58% (28/48). Eight percent of patients did not engraft—3 MF patients with LCT and 1 SS patient.
“The response rate was much higher in Sézary patients [than in the rest of the cohort],” Dr Duvic said. “The worst prognosis was for patients who had both Sézary and large-cell transformation, who relapsed early and were generally refractory to prior therapies.”
Complete responses occurred in 79% of SS patients, 56% of MF patients with LCT, 44% of patients with SS and LCT, 40% of patients with stage IVA/IIB disease, and 50% of those with folliculotropic MF.
Among patients who received TBSEB, 58% (25/43) achieved a complete response. Of the 5 patients who did not receive TBSEB, 3 had a complete response (60%).
Sixty percent of patients developed GVHD (29/48). Eighteen patients had acute skin GVHD, 9 had acute gastrointestinal GVHD, 13 had chronic skin GVHD, and 6 had chronic gastrointestinal GVHD.
Relapse and survival
Overall, the relapse rate was 33% (16/48). Twenty-one percent of SS patients relapsed, as did 25% of MF patients with LCT and 56% of patients with SS and LCT.
The mortality rate was 44% (21/48). Patients died of relapsed MF, sepsis, infection, second malignancy, and other causes.
The overall survival (OS) was 10.2 years from diagnosis and 5.7 years from SCT. The progression-free survival (PFS) was 6 years from diagnosis and 1.8 years from SCT.
“We also looked at whether large-cell transformation had an effect on survival and therapy,” Dr Duvic said. “Large-cell transformation in MF has been reported to have a more aggressive course and a shorter overall survival than untransformed MF.”
“In our cohort of patients, we found an overall survival of 4.79 years in patients with large-cell transformation, which is a little bit higher than [survival rates in] the literature.”
Among MF patients with LCT, OS was 84% at 1 year from SCT and 38% at both 5 years and 10 years. PFS was 55% at 1 year, 16% at 5 years, and 0% at 10 years.
In comparison, among SS patients without LCT, OS was 88% at 1 year from SCT and 70% at both 5 years and 10 years. PFS was 63% at 1 year and 49% at 5 years and 10 years. ![]()
SAN FRANCISCO—A single-center study suggests that transplant can induce remissions and improve survival in certain patients with advanced cutaneous T-cell lymphomas.
Allogeneic stem cell transplant (SCT) proved particularly effective in patients with Sézary syndrome (SS).
It also conferred benefits to mycosis fungoides (MF) patients with large-cell transformation (LCT), but patients with SS and LCT did not fare as well.
Madeleine Duvic, MD, of the MD Anderson Cancer Center in Houston, presented these results at the 6th Annual T-cell Lymphoma Forum. The data were updated from a previously published report (Duvic et al, JCO 2010).
Patient characteristics
Dr Duvic and her colleagues evaluated 48 patients who had biopsy-proven MF or SS. They underwent SCT at MD Anderson between July 2001 and September 2013.
The patients were in good health but had advanced disease. They had received a median of 6 prior therapies (range, 2-11).
The median age was 51.5 years (range, 19-72 years), and 54% of patients were female. Sixty-nine percent were Caucasian, 23% were African American, and 8% were Hispanic.
Fourteen patients had SS only, 16 had MF with LCT, 9 had SS and LCT, 5 had stage IVA or IIB disease (4 nodal, 1 tumor), and 4 had folliculotropic MF.
Transplant and other treatment
Patients had to have a 9/10 or 10/10 HLA-matched donor (related or unrelated). Most of the stem cells were collected via apheresis, but bone marrow aspiration was used for patients receiving mismatched transplants.
Forty-three of the patients underwent tumor and skin debulking with total skin electron beam (TBSEB) radiation (35 Gy) 1 or 2 months prior to SCT.
Most patients received a conditioning regimen of fludarabine and melphalan, but a few received fludarabine and cyclophosphamide. Patients received tacrolimus and methotrexate as graft-vs-host disease (GVHD) prophylaxis, as well as extracorporeal photopheresis if they developed GVHD.
All of the SS patients received vancomycin, fluconazole, and valacyclovir to ward off infections.
Response and GVHD
The overall complete response rate was 58% (28/48). Eight percent of patients did not engraft—3 MF patients with LCT and 1 SS patient.
“The response rate was much higher in Sézary patients [than in the rest of the cohort],” Dr Duvic said. “The worst prognosis was for patients who had both Sézary and large-cell transformation, who relapsed early and were generally refractory to prior therapies.”
Complete responses occurred in 79% of SS patients, 56% of MF patients with LCT, 44% of patients with SS and LCT, 40% of patients with stage IVA/IIB disease, and 50% of those with folliculotropic MF.
Among patients who received TBSEB, 58% (25/43) achieved a complete response. Of the 5 patients who did not receive TBSEB, 3 had a complete response (60%).
Sixty percent of patients developed GVHD (29/48). Eighteen patients had acute skin GVHD, 9 had acute gastrointestinal GVHD, 13 had chronic skin GVHD, and 6 had chronic gastrointestinal GVHD.
Relapse and survival
Overall, the relapse rate was 33% (16/48). Twenty-one percent of SS patients relapsed, as did 25% of MF patients with LCT and 56% of patients with SS and LCT.
The mortality rate was 44% (21/48). Patients died of relapsed MF, sepsis, infection, second malignancy, and other causes.
The overall survival (OS) was 10.2 years from diagnosis and 5.7 years from SCT. The progression-free survival (PFS) was 6 years from diagnosis and 1.8 years from SCT.
“We also looked at whether large-cell transformation had an effect on survival and therapy,” Dr Duvic said. “Large-cell transformation in MF has been reported to have a more aggressive course and a shorter overall survival than untransformed MF.”
“In our cohort of patients, we found an overall survival of 4.79 years in patients with large-cell transformation, which is a little bit higher than [survival rates in] the literature.”
Among MF patients with LCT, OS was 84% at 1 year from SCT and 38% at both 5 years and 10 years. PFS was 55% at 1 year, 16% at 5 years, and 0% at 10 years.
In comparison, among SS patients without LCT, OS was 88% at 1 year from SCT and 70% at both 5 years and 10 years. PFS was 63% at 1 year and 49% at 5 years and 10 years. ![]()
Climate change may increase malaria incidence
Credit: James Gathany
New research indicates that, toward the end of the century, climate change may make tropical highland regions suitable breeding grounds for malaria. And this could greatly increase the incidence of the disease.
Scientists compared the latest predictions for global warming with a range of statistical models commonly used to predict the spread of malaria.
And the models suggested that the changing climate will allow malaria to move into higher altitudes during warmer seasons and become permanently resident in larger areas.
This would mainly affect Africa but would also have an impact in Asia and South America.
In eastern Africa, the change could result in an additional 100 million people being exposed to malaria by the end of the 2080s, the researchers calculated.
However, they noted that the size of the impact is highly variable, as it can be affected by a number of factors.
“[W]e expect increased urbanization in these areas over the next 70 years,” said study author Cyril Caminade, PhD, of the University of Liverpool in the UK.
“Other developments, such as land use changes, population movements, and economic growth, will also have to be accounted for in future studies. What is clear is that diseases such as malaria are going to be moving, and this is a crucial element of how we prepare for the effects of climate change.”
The comparison of these models has not been carried out before, and by doing so, the researchers were able to find that malaria spreading to tropical highland areas was the one area in which the models agreed.
There was distinct variation in other parts of the world. Two of the models predicted that malaria would move northward, eventually spreading into Europe, Russia, and North America. Others showed that malaria would move north, but only as far as North Africa, where it was eliminated in the 20th century.
Credit: James Gathany
New research indicates that, toward the end of the century, climate change may make tropical highland regions suitable breeding grounds for malaria. And this could greatly increase the incidence of the disease.
Scientists compared the latest predictions for global warming with a range of statistical models commonly used to predict the spread of malaria.
And the models suggested that the changing climate will allow malaria to move into higher altitudes during warmer seasons and become permanently resident in larger areas.
This would mainly affect Africa but would also have an impact in Asia and South America.
In eastern Africa, the change could result in an additional 100 million people being exposed to malaria by the end of the 2080s, the researchers calculated.
However, they noted that the size of the impact is highly variable, as it can be affected by a number of factors.
“[W]e expect increased urbanization in these areas over the next 70 years,” said study author Cyril Caminade, PhD, of the University of Liverpool in the UK.
“Other developments, such as land use changes, population movements, and economic growth, will also have to be accounted for in future studies. What is clear is that diseases such as malaria are going to be moving, and this is a crucial element of how we prepare for the effects of climate change.”
The comparison of these models has not been carried out before, and by doing so, the researchers were able to find that malaria spreading to tropical highland areas was the one area in which the models agreed.
There was distinct variation in other parts of the world. Two of the models predicted that malaria would move northward, eventually spreading into Europe, Russia, and North America. Others showed that malaria would move north, but only as far as North Africa, where it was eliminated in the 20th century.
Credit: James Gathany
New research indicates that, toward the end of the century, climate change may make tropical highland regions suitable breeding grounds for malaria. And this could greatly increase the incidence of the disease.
Scientists compared the latest predictions for global warming with a range of statistical models commonly used to predict the spread of malaria.
And the models suggested that the changing climate will allow malaria to move into higher altitudes during warmer seasons and become permanently resident in larger areas.
This would mainly affect Africa but would also have an impact in Asia and South America.
In eastern Africa, the change could result in an additional 100 million people being exposed to malaria by the end of the 2080s, the researchers calculated.
However, they noted that the size of the impact is highly variable, as it can be affected by a number of factors.
“[W]e expect increased urbanization in these areas over the next 70 years,” said study author Cyril Caminade, PhD, of the University of Liverpool in the UK.
“Other developments, such as land use changes, population movements, and economic growth, will also have to be accounted for in future studies. What is clear is that diseases such as malaria are going to be moving, and this is a crucial element of how we prepare for the effects of climate change.”
The comparison of these models has not been carried out before, and by doing so, the researchers were able to find that malaria spreading to tropical highland areas was the one area in which the models agreed.
There was distinct variation in other parts of the world. Two of the models predicted that malaria would move northward, eventually spreading into Europe, Russia, and North America. Others showed that malaria would move north, but only as far as North Africa, where it was eliminated in the 20th century.
Method captures images of multiple cell components

(green), mitochondria (purple),
Golgi apparatus (red), and
peroxisomes (yellow) in a cell
Credit: Maier Avendano
A new microscopy method allows scientists to image many cellular components at once, according to a paper published in Nature Methods.
Such images could shed light on complex cellular pathways and might lead to new ways to diagnose disease, track its progress, or monitor treatment effectiveness at a cellular level, the researchers said.
They noted that today’s imaging methods typically spot, at most, 3 or 4 types of biomolecules simultaneously.
But to truly understand complex cellular functions, it’s important to be able to visualize most or all of the molecules at once, said study author Peng Yin, PhD, of the Wyss Institute at Harvard Medical School in Boston.
“If you can see only a few things at a time, you are missing the big picture,” Dr Yin said.
So he and his colleagues sought a way to take aerial views of cells that could show dozens of types of biomolecules. They decided to build upon a method called DNA-PAINT, which can create snapshots of up to 3 molecules at once by labeling them with different colored dyes.
The team modified DNA-PAINT to create a method called Exchange-PAINT. Exchange-PAINT relies on the fact that DNA strands with the correct sequence of nucleotides bind specifically to partner strands with complementary sequences.
The researchers label a biomolecule they want to visualize with a short DNA tag. Then, they add to the solution a partner strand carrying a fluorescent dye that lights up only when the 2 strands pair up.
When that partner strand binds the tagged biomolecule, it lights up, then lets go, causing the biomolecule to “blink” at a precise rate the researchers can control. The team uses this blinking to obtain ultra-sharp images.
They repeat the process to visualize a second target, a third, and so on. Then, they overlay the resulting images to create a composite image in which each biomolecule is assigned a different color.
This allows them to create false-color images that simultaneously show many types of biomolecules—far more than they could simultaneously visualize by labeling them with different colored dyes. And these false-color images allow them to spot enough biomolecules at once to capture the entire scene.
To test Exchange-PAINT, the researchers created 10 unique pieces of folded DNA that resembled the numerals 0 through 9. These numerals could be resolved with less than 10 nanometers resolution, or 1/20th of the diffraction limit.
The team was able to use Exchange-PAINT to capture clear images of the 10 different DNA origami structures in one image. They also used the method to capture images of fixed human cells, with each color tagging a different cellular component—microtubules, mitochondria, Golgi apparatus, or peroxisomes.
Dr Yin expects that, with further development, this method could be used to visualize dozens of cellular components at once. ![]()
(green), mitochondria (purple),
Golgi apparatus (red), and
peroxisomes (yellow) in a cell
Credit: Maier Avendano
A new microscopy method allows scientists to image many cellular components at once, according to a paper published in Nature Methods.
Such images could shed light on complex cellular pathways and might lead to new ways to diagnose disease, track its progress, or monitor treatment effectiveness at a cellular level, the researchers said.
They noted that today’s imaging methods typically spot, at most, 3 or 4 types of biomolecules simultaneously.
But to truly understand complex cellular functions, it’s important to be able to visualize most or all of the molecules at once, said study author Peng Yin, PhD, of the Wyss Institute at Harvard Medical School in Boston.
“If you can see only a few things at a time, you are missing the big picture,” Dr Yin said.
So he and his colleagues sought a way to take aerial views of cells that could show dozens of types of biomolecules. They decided to build upon a method called DNA-PAINT, which can create snapshots of up to 3 molecules at once by labeling them with different colored dyes.
The team modified DNA-PAINT to create a method called Exchange-PAINT. Exchange-PAINT relies on the fact that DNA strands with the correct sequence of nucleotides bind specifically to partner strands with complementary sequences.
The researchers label a biomolecule they want to visualize with a short DNA tag. Then, they add to the solution a partner strand carrying a fluorescent dye that lights up only when the 2 strands pair up.
When that partner strand binds the tagged biomolecule, it lights up, then lets go, causing the biomolecule to “blink” at a precise rate the researchers can control. The team uses this blinking to obtain ultra-sharp images.
They repeat the process to visualize a second target, a third, and so on. Then, they overlay the resulting images to create a composite image in which each biomolecule is assigned a different color.
This allows them to create false-color images that simultaneously show many types of biomolecules—far more than they could simultaneously visualize by labeling them with different colored dyes. And these false-color images allow them to spot enough biomolecules at once to capture the entire scene.
To test Exchange-PAINT, the researchers created 10 unique pieces of folded DNA that resembled the numerals 0 through 9. These numerals could be resolved with less than 10 nanometers resolution, or 1/20th of the diffraction limit.
The team was able to use Exchange-PAINT to capture clear images of the 10 different DNA origami structures in one image. They also used the method to capture images of fixed human cells, with each color tagging a different cellular component—microtubules, mitochondria, Golgi apparatus, or peroxisomes.
Dr Yin expects that, with further development, this method could be used to visualize dozens of cellular components at once. ![]()
(green), mitochondria (purple),
Golgi apparatus (red), and
peroxisomes (yellow) in a cell
Credit: Maier Avendano
A new microscopy method allows scientists to image many cellular components at once, according to a paper published in Nature Methods.
Such images could shed light on complex cellular pathways and might lead to new ways to diagnose disease, track its progress, or monitor treatment effectiveness at a cellular level, the researchers said.
They noted that today’s imaging methods typically spot, at most, 3 or 4 types of biomolecules simultaneously.
But to truly understand complex cellular functions, it’s important to be able to visualize most or all of the molecules at once, said study author Peng Yin, PhD, of the Wyss Institute at Harvard Medical School in Boston.
“If you can see only a few things at a time, you are missing the big picture,” Dr Yin said.
So he and his colleagues sought a way to take aerial views of cells that could show dozens of types of biomolecules. They decided to build upon a method called DNA-PAINT, which can create snapshots of up to 3 molecules at once by labeling them with different colored dyes.
The team modified DNA-PAINT to create a method called Exchange-PAINT. Exchange-PAINT relies on the fact that DNA strands with the correct sequence of nucleotides bind specifically to partner strands with complementary sequences.
The researchers label a biomolecule they want to visualize with a short DNA tag. Then, they add to the solution a partner strand carrying a fluorescent dye that lights up only when the 2 strands pair up.
When that partner strand binds the tagged biomolecule, it lights up, then lets go, causing the biomolecule to “blink” at a precise rate the researchers can control. The team uses this blinking to obtain ultra-sharp images.
They repeat the process to visualize a second target, a third, and so on. Then, they overlay the resulting images to create a composite image in which each biomolecule is assigned a different color.
This allows them to create false-color images that simultaneously show many types of biomolecules—far more than they could simultaneously visualize by labeling them with different colored dyes. And these false-color images allow them to spot enough biomolecules at once to capture the entire scene.
To test Exchange-PAINT, the researchers created 10 unique pieces of folded DNA that resembled the numerals 0 through 9. These numerals could be resolved with less than 10 nanometers resolution, or 1/20th of the diffraction limit.
The team was able to use Exchange-PAINT to capture clear images of the 10 different DNA origami structures in one image. They also used the method to capture images of fixed human cells, with each color tagging a different cellular component—microtubules, mitochondria, Golgi apparatus, or peroxisomes.
Dr Yin expects that, with further development, this method could be used to visualize dozens of cellular components at once. ![]()
New stroke guidelines focus on women’s risks
Newly released guidelines provide the first evidence-based recommendations for preventing stroke in women.
The document addresses the issues that uniquely increase stroke risk in women – pregnancy, hormonal therapy, contraception, and migraine – along with factors like atrial fibrillation and obesity, Dr. Cheryl Bushnell and her colleagues wrote in the February issue of Stroke.
"If you are a woman, you share many of the same risk factors for stroke with men, but your risk is also influenced by hormones, reproductive health, pregnancy, childbirth, and other sex-related factors," Dr. Bushnell noted in a press statement.
The document – created by the American Heart Association and American Stroke Association – is the first to look at these gender-specific issues, wrote Dr. Bushnell, director of the Stroke Center at Wake Forest Baptist Medical Center in Winston-Salem, N.C. (Stroke 2014 [doi:10.1161/01.str.0000442009.06663.48]).
It provides graded evidence for preventive strategies in a number of risk categories. Evidence was obtained by examining dozens of studies numbering hundreds of thousands of women. But despite the extant literature, Dr. Bushnell and her colleagues said more research needs to be conducted.

"There is a need for recognition of women’s unique, sex-specific stroke risk factors, and a risk score that includes these factors would thereby identify women at risk," they wrote. "Similarly, it is important to improve stroke awareness and provide more rigorous education to women at younger ages, including childbearing ages."
The guidelines are aimed at primary care providers, who have the biggest interface with women at a prevention level – and intended to help them forge an active partnership with patients.
"More importantly," the authors wrote, "this guideline may empower women and their families to understand their own risk and how they can minimize the chances of having a stroke."
Pregnancy
For recommendations on pregnancy outcomes and stroke related to preeclampsia, the guidelines drew on evidence from 17 studies.
For women with chronic primary or secondary hypertension, or with a history of pregnancy-related hypertension, Level A evidence supports using low-dose aspirin during the second and third trimester. Level A evidence also supports calcium supplementation to prevent preeclampsia in women with low dietary intake.
There was also a Level A recommendation to treat severe hypertension during pregnancy with safe antihypertensives (methyldopa, labetalol, and nifedipine). Level B evidence supported treating moderate hypertension. The use of atenolol, angiotensin receptor blockers, and direct renin inhibitors is contraindicated because of teratogenicity.
Because preeclampsia increases lifelong stroke risk, the guidelines also recommended evaluating these women within 1 year of giving birth, and, based on their individual and family risk factors, possibly treating them for cardiovascular risk factors.
Oral contraceptives
Four studies comprising about 800,000 women examined the risk of stroke in women using hormonal birth control.
Level A evidence did not support routine screening for prothrombotic mutations before starting oral contraception. But there was Level B evidence that oral contraceptives may be harmful in women who had risk factors, including cigarette use and prior thromboembolic events.
Menopause-related hormone therapy
Seven studies – including the Women’s Health Initiative – examined the links between stroke and hormone therapy in about 37,000 women. Two recommendations supported by Level A evidence were made.
Hormone therapy should not be used for either primary or secondary stroke prevention in postmenopausal women.
Selective estrogen receptor modulators (raloxifene, tamoxifen, and tibolone) should not be used for primary prevention of stroke.
Migraine with aura
There is scant literature examining the link between migraine with aura and stroke, although what does exist suggests that the risk may be doubled overall. The addition of another factor, like pregnancy or preeclampsia, dramatically increases the risk. But because these data are low in number, the recommendations are the same as they are for men.
Level B evidence supports smoking cessation in women with migraine and aura. Level C evidence suggests that treatments that reduce the frequency of migraine may also reduce the risk of stroke.
Obesity and metabolic syndrome
A healthy lifestyle of eating whole foods, exercise, and abstaining from tobacco has been shown to lower stroke incidence in both women and men. But subgroup analyses hint that men derive the most benefit. Women-only studies of these interventions have posted mixed results about their ability to reduce stroke in women.
The authors said much more research is necessary to target interventions that are especially beneficial for women. Until then, Level B evidence supports maintaining a lifestyle of exercise, healthy eating, no tobacco use, and moderate alcohol intake (a drink a day or less) for women who aren’t pregnant.
Atrial fibrillation
Overall, similar numbers of women and men have atrial fibrillation. But the condition becomes more common with age, and women have a longer life expectancy than do men. Therefore, the authors noted, atrial fibrillation will become more common as the population of elderly women increases.
They recommend that primary care physicians actively screen women for atrial fibrillation once they reach age 75 years. The screening method, supported by Level B evidence, should be pulse followed by an electrocardiogram.
For women aged 65 years and younger who have atrial fibrillation but no other risk factors, there is no evidence supporting oral anticoagulation. Level B evidence does support antiplatelet therapy.
Dr. Bushnell had no financial disclosures. One of the 16 coauthors reported relationships with several pharmaceutical companies
On Twitter @alz_gal
Newly released guidelines provide the first evidence-based recommendations for preventing stroke in women.
The document addresses the issues that uniquely increase stroke risk in women – pregnancy, hormonal therapy, contraception, and migraine – along with factors like atrial fibrillation and obesity, Dr. Cheryl Bushnell and her colleagues wrote in the February issue of Stroke.
"If you are a woman, you share many of the same risk factors for stroke with men, but your risk is also influenced by hormones, reproductive health, pregnancy, childbirth, and other sex-related factors," Dr. Bushnell noted in a press statement.
The document – created by the American Heart Association and American Stroke Association – is the first to look at these gender-specific issues, wrote Dr. Bushnell, director of the Stroke Center at Wake Forest Baptist Medical Center in Winston-Salem, N.C. (Stroke 2014 [doi:10.1161/01.str.0000442009.06663.48]).
It provides graded evidence for preventive strategies in a number of risk categories. Evidence was obtained by examining dozens of studies numbering hundreds of thousands of women. But despite the extant literature, Dr. Bushnell and her colleagues said more research needs to be conducted.
"There is a need for recognition of women’s unique, sex-specific stroke risk factors, and a risk score that includes these factors would thereby identify women at risk," they wrote. "Similarly, it is important to improve stroke awareness and provide more rigorous education to women at younger ages, including childbearing ages."
The guidelines are aimed at primary care providers, who have the biggest interface with women at a prevention level – and intended to help them forge an active partnership with patients.
"More importantly," the authors wrote, "this guideline may empower women and their families to understand their own risk and how they can minimize the chances of having a stroke."
Pregnancy
For recommendations on pregnancy outcomes and stroke related to preeclampsia, the guidelines drew on evidence from 17 studies.
For women with chronic primary or secondary hypertension, or with a history of pregnancy-related hypertension, Level A evidence supports using low-dose aspirin during the second and third trimester. Level A evidence also supports calcium supplementation to prevent preeclampsia in women with low dietary intake.
There was also a Level A recommendation to treat severe hypertension during pregnancy with safe antihypertensives (methyldopa, labetalol, and nifedipine). Level B evidence supported treating moderate hypertension. The use of atenolol, angiotensin receptor blockers, and direct renin inhibitors is contraindicated because of teratogenicity.
Because preeclampsia increases lifelong stroke risk, the guidelines also recommended evaluating these women within 1 year of giving birth, and, based on their individual and family risk factors, possibly treating them for cardiovascular risk factors.
Oral contraceptives
Four studies comprising about 800,000 women examined the risk of stroke in women using hormonal birth control.
Level A evidence did not support routine screening for prothrombotic mutations before starting oral contraception. But there was Level B evidence that oral contraceptives may be harmful in women who had risk factors, including cigarette use and prior thromboembolic events.
Menopause-related hormone therapy
Seven studies – including the Women’s Health Initiative – examined the links between stroke and hormone therapy in about 37,000 women. Two recommendations supported by Level A evidence were made.
Hormone therapy should not be used for either primary or secondary stroke prevention in postmenopausal women.
Selective estrogen receptor modulators (raloxifene, tamoxifen, and tibolone) should not be used for primary prevention of stroke.
Migraine with aura
There is scant literature examining the link between migraine with aura and stroke, although what does exist suggests that the risk may be doubled overall. The addition of another factor, like pregnancy or preeclampsia, dramatically increases the risk. But because these data are low in number, the recommendations are the same as they are for men.
Level B evidence supports smoking cessation in women with migraine and aura. Level C evidence suggests that treatments that reduce the frequency of migraine may also reduce the risk of stroke.
Obesity and metabolic syndrome
A healthy lifestyle of eating whole foods, exercise, and abstaining from tobacco has been shown to lower stroke incidence in both women and men. But subgroup analyses hint that men derive the most benefit. Women-only studies of these interventions have posted mixed results about their ability to reduce stroke in women.
The authors said much more research is necessary to target interventions that are especially beneficial for women. Until then, Level B evidence supports maintaining a lifestyle of exercise, healthy eating, no tobacco use, and moderate alcohol intake (a drink a day or less) for women who aren’t pregnant.
Atrial fibrillation
Overall, similar numbers of women and men have atrial fibrillation. But the condition becomes more common with age, and women have a longer life expectancy than do men. Therefore, the authors noted, atrial fibrillation will become more common as the population of elderly women increases.
They recommend that primary care physicians actively screen women for atrial fibrillation once they reach age 75 years. The screening method, supported by Level B evidence, should be pulse followed by an electrocardiogram.
For women aged 65 years and younger who have atrial fibrillation but no other risk factors, there is no evidence supporting oral anticoagulation. Level B evidence does support antiplatelet therapy.
Dr. Bushnell had no financial disclosures. One of the 16 coauthors reported relationships with several pharmaceutical companies
On Twitter @alz_gal
Newly released guidelines provide the first evidence-based recommendations for preventing stroke in women.
The document addresses the issues that uniquely increase stroke risk in women – pregnancy, hormonal therapy, contraception, and migraine – along with factors like atrial fibrillation and obesity, Dr. Cheryl Bushnell and her colleagues wrote in the February issue of Stroke.
"If you are a woman, you share many of the same risk factors for stroke with men, but your risk is also influenced by hormones, reproductive health, pregnancy, childbirth, and other sex-related factors," Dr. Bushnell noted in a press statement.
The document – created by the American Heart Association and American Stroke Association – is the first to look at these gender-specific issues, wrote Dr. Bushnell, director of the Stroke Center at Wake Forest Baptist Medical Center in Winston-Salem, N.C. (Stroke 2014 [doi:10.1161/01.str.0000442009.06663.48]).
It provides graded evidence for preventive strategies in a number of risk categories. Evidence was obtained by examining dozens of studies numbering hundreds of thousands of women. But despite the extant literature, Dr. Bushnell and her colleagues said more research needs to be conducted.
"There is a need for recognition of women’s unique, sex-specific stroke risk factors, and a risk score that includes these factors would thereby identify women at risk," they wrote. "Similarly, it is important to improve stroke awareness and provide more rigorous education to women at younger ages, including childbearing ages."
The guidelines are aimed at primary care providers, who have the biggest interface with women at a prevention level – and intended to help them forge an active partnership with patients.
"More importantly," the authors wrote, "this guideline may empower women and their families to understand their own risk and how they can minimize the chances of having a stroke."
Pregnancy
For recommendations on pregnancy outcomes and stroke related to preeclampsia, the guidelines drew on evidence from 17 studies.
For women with chronic primary or secondary hypertension, or with a history of pregnancy-related hypertension, Level A evidence supports using low-dose aspirin during the second and third trimester. Level A evidence also supports calcium supplementation to prevent preeclampsia in women with low dietary intake.
There was also a Level A recommendation to treat severe hypertension during pregnancy with safe antihypertensives (methyldopa, labetalol, and nifedipine). Level B evidence supported treating moderate hypertension. The use of atenolol, angiotensin receptor blockers, and direct renin inhibitors is contraindicated because of teratogenicity.
Because preeclampsia increases lifelong stroke risk, the guidelines also recommended evaluating these women within 1 year of giving birth, and, based on their individual and family risk factors, possibly treating them for cardiovascular risk factors.
Oral contraceptives
Four studies comprising about 800,000 women examined the risk of stroke in women using hormonal birth control.
Level A evidence did not support routine screening for prothrombotic mutations before starting oral contraception. But there was Level B evidence that oral contraceptives may be harmful in women who had risk factors, including cigarette use and prior thromboembolic events.
Menopause-related hormone therapy
Seven studies – including the Women’s Health Initiative – examined the links between stroke and hormone therapy in about 37,000 women. Two recommendations supported by Level A evidence were made.
Hormone therapy should not be used for either primary or secondary stroke prevention in postmenopausal women.
Selective estrogen receptor modulators (raloxifene, tamoxifen, and tibolone) should not be used for primary prevention of stroke.
Migraine with aura
There is scant literature examining the link between migraine with aura and stroke, although what does exist suggests that the risk may be doubled overall. The addition of another factor, like pregnancy or preeclampsia, dramatically increases the risk. But because these data are low in number, the recommendations are the same as they are for men.
Level B evidence supports smoking cessation in women with migraine and aura. Level C evidence suggests that treatments that reduce the frequency of migraine may also reduce the risk of stroke.
Obesity and metabolic syndrome
A healthy lifestyle of eating whole foods, exercise, and abstaining from tobacco has been shown to lower stroke incidence in both women and men. But subgroup analyses hint that men derive the most benefit. Women-only studies of these interventions have posted mixed results about their ability to reduce stroke in women.
The authors said much more research is necessary to target interventions that are especially beneficial for women. Until then, Level B evidence supports maintaining a lifestyle of exercise, healthy eating, no tobacco use, and moderate alcohol intake (a drink a day or less) for women who aren’t pregnant.
Atrial fibrillation
Overall, similar numbers of women and men have atrial fibrillation. But the condition becomes more common with age, and women have a longer life expectancy than do men. Therefore, the authors noted, atrial fibrillation will become more common as the population of elderly women increases.
They recommend that primary care physicians actively screen women for atrial fibrillation once they reach age 75 years. The screening method, supported by Level B evidence, should be pulse followed by an electrocardiogram.
For women aged 65 years and younger who have atrial fibrillation but no other risk factors, there is no evidence supporting oral anticoagulation. Level B evidence does support antiplatelet therapy.
Dr. Bushnell had no financial disclosures. One of the 16 coauthors reported relationships with several pharmaceutical companies
On Twitter @alz_gal
FROM STROKE

Combo may overcome drug resistance in ALL

Credit: Linda Bartlett
Adding the alkylating agent cyclophosphamide to treatment with a monoclonal antibody (mAb) can overcome drug resistance in mice with acute lymphoblastic leukemia (ALL), researchers have reported in Cell.
mAbs such as rituximab and alemtuzumab are designed to bind to proteins found on the surfaces of tumor cells.
Once the mAbs flag the tumor cells, macrophages destroy them. But the drugs have little effect on tumor cells that hide out in the bone marrow.
Experiments in mice with B-cell ALL revealed that cyclophosphamide stimulates the immune response in bone marrow, eliminating the reservoir of cancer cells that can produce new tumors after treatment with a mAb.
Finding hidden ALL cells
Michael Hemann, PhD, of MIT’s Koch Institute for Integrative Cancer Research in Cambridge, Massachusetts, and his colleagues began this research by administering alemtuzumab to the mice.
The drug successfully cleared most ALL cells, but some remained hidden in the bone marrow, which has been identified as a site of drug resistance in many cancers.
The researchers found that, within the bone marrow, alemtuzumab successfully binds to ALL cells. But macrophages do not attack the cells due to the presence of lipid compounds called prostaglandins, which repress macrophage activity.
Scientists believe the bone marrow naturally produces prostaglandins to help protect the immune cells maturing there. Tumor cells that reach the bone marrow can exploit this protective environment to aid their own survival.
The finding is an important contribution to scientists’ understanding of how mAbs act against ALL, according to Ravi Majeti, MD, PhD, of Stanford University in California, who was not involved in this research.
“There clearly has been a lack of understanding about why antibody therapies have been relatively unsuccessful as monotherapies,” Dr Majeti said.
Tricking the immune system
Dr Hemann and his colleagues then tested a variety of anticancer drugs in combination with alemtuzumab. And they discovered that cyclophosphamide can “rewire” the bone marrow microenvironment to make it much more receptive to macrophages, allowing them to destroy the tumor cells hiding there.
“After you treat with cyclophosphamide, you get this flux of macrophages into the bone marrow, and these macrophages are now active and very capable of consuming the targeted tumor cells,” Dr Hemann said.
“Essentially, we are tricking the immune system to suddenly recognize an entity that it wouldn’t typically recognize and aggressively go after antibody-bound tumor cells.”
Following treatment with this combination, the mice survived and remained free of ALL for the duration of the study, which was about 18 months.
However, the researchers found that timing of drug delivery was critical. Alemtuzumab and cyclophosphamide must be administered together so that cyclophosphamide can create the right type of environment for macrophages to become activated in the bone marrow.
The team also obtained good results by combining cyclophosphamide with rituximab.
They now plan to test cyclophosphamide with other mAbs and begin testing the alemtuzumab-cyclophosphamide combination in patients. ![]()
Credit: Linda Bartlett
Adding the alkylating agent cyclophosphamide to treatment with a monoclonal antibody (mAb) can overcome drug resistance in mice with acute lymphoblastic leukemia (ALL), researchers have reported in Cell.
mAbs such as rituximab and alemtuzumab are designed to bind to proteins found on the surfaces of tumor cells.
Once the mAbs flag the tumor cells, macrophages destroy them. But the drugs have little effect on tumor cells that hide out in the bone marrow.
Experiments in mice with B-cell ALL revealed that cyclophosphamide stimulates the immune response in bone marrow, eliminating the reservoir of cancer cells that can produce new tumors after treatment with a mAb.
Finding hidden ALL cells
Michael Hemann, PhD, of MIT’s Koch Institute for Integrative Cancer Research in Cambridge, Massachusetts, and his colleagues began this research by administering alemtuzumab to the mice.
The drug successfully cleared most ALL cells, but some remained hidden in the bone marrow, which has been identified as a site of drug resistance in many cancers.
The researchers found that, within the bone marrow, alemtuzumab successfully binds to ALL cells. But macrophages do not attack the cells due to the presence of lipid compounds called prostaglandins, which repress macrophage activity.
Scientists believe the bone marrow naturally produces prostaglandins to help protect the immune cells maturing there. Tumor cells that reach the bone marrow can exploit this protective environment to aid their own survival.
The finding is an important contribution to scientists’ understanding of how mAbs act against ALL, according to Ravi Majeti, MD, PhD, of Stanford University in California, who was not involved in this research.
“There clearly has been a lack of understanding about why antibody therapies have been relatively unsuccessful as monotherapies,” Dr Majeti said.
Tricking the immune system
Dr Hemann and his colleagues then tested a variety of anticancer drugs in combination with alemtuzumab. And they discovered that cyclophosphamide can “rewire” the bone marrow microenvironment to make it much more receptive to macrophages, allowing them to destroy the tumor cells hiding there.
“After you treat with cyclophosphamide, you get this flux of macrophages into the bone marrow, and these macrophages are now active and very capable of consuming the targeted tumor cells,” Dr Hemann said.
“Essentially, we are tricking the immune system to suddenly recognize an entity that it wouldn’t typically recognize and aggressively go after antibody-bound tumor cells.”
Following treatment with this combination, the mice survived and remained free of ALL for the duration of the study, which was about 18 months.
However, the researchers found that timing of drug delivery was critical. Alemtuzumab and cyclophosphamide must be administered together so that cyclophosphamide can create the right type of environment for macrophages to become activated in the bone marrow.
The team also obtained good results by combining cyclophosphamide with rituximab.
They now plan to test cyclophosphamide with other mAbs and begin testing the alemtuzumab-cyclophosphamide combination in patients. ![]()
Credit: Linda Bartlett
Adding the alkylating agent cyclophosphamide to treatment with a monoclonal antibody (mAb) can overcome drug resistance in mice with acute lymphoblastic leukemia (ALL), researchers have reported in Cell.
mAbs such as rituximab and alemtuzumab are designed to bind to proteins found on the surfaces of tumor cells.
Once the mAbs flag the tumor cells, macrophages destroy them. But the drugs have little effect on tumor cells that hide out in the bone marrow.
Experiments in mice with B-cell ALL revealed that cyclophosphamide stimulates the immune response in bone marrow, eliminating the reservoir of cancer cells that can produce new tumors after treatment with a mAb.
Finding hidden ALL cells
Michael Hemann, PhD, of MIT’s Koch Institute for Integrative Cancer Research in Cambridge, Massachusetts, and his colleagues began this research by administering alemtuzumab to the mice.
The drug successfully cleared most ALL cells, but some remained hidden in the bone marrow, which has been identified as a site of drug resistance in many cancers.
The researchers found that, within the bone marrow, alemtuzumab successfully binds to ALL cells. But macrophages do not attack the cells due to the presence of lipid compounds called prostaglandins, which repress macrophage activity.
Scientists believe the bone marrow naturally produces prostaglandins to help protect the immune cells maturing there. Tumor cells that reach the bone marrow can exploit this protective environment to aid their own survival.
The finding is an important contribution to scientists’ understanding of how mAbs act against ALL, according to Ravi Majeti, MD, PhD, of Stanford University in California, who was not involved in this research.
“There clearly has been a lack of understanding about why antibody therapies have been relatively unsuccessful as monotherapies,” Dr Majeti said.
Tricking the immune system
Dr Hemann and his colleagues then tested a variety of anticancer drugs in combination with alemtuzumab. And they discovered that cyclophosphamide can “rewire” the bone marrow microenvironment to make it much more receptive to macrophages, allowing them to destroy the tumor cells hiding there.
“After you treat with cyclophosphamide, you get this flux of macrophages into the bone marrow, and these macrophages are now active and very capable of consuming the targeted tumor cells,” Dr Hemann said.
“Essentially, we are tricking the immune system to suddenly recognize an entity that it wouldn’t typically recognize and aggressively go after antibody-bound tumor cells.”
Following treatment with this combination, the mice survived and remained free of ALL for the duration of the study, which was about 18 months.
However, the researchers found that timing of drug delivery was critical. Alemtuzumab and cyclophosphamide must be administered together so that cyclophosphamide can create the right type of environment for macrophages to become activated in the bone marrow.
The team also obtained good results by combining cyclophosphamide with rituximab.
They now plan to test cyclophosphamide with other mAbs and begin testing the alemtuzumab-cyclophosphamide combination in patients. ![]()
Experts offer guidance for preventing drug shortages

with chemotherapy drugs
Credit: Bill Branson
A group of healthcare experts has created a “blueprint” for managing and preventing drug shortages.
They devised 6 recommendations for preempting shortages of pediatric oncology drugs, but the suggestions can be applied to other drugs as well.
Some of the recommendations would represent new norms for healthcare practice, such as sharing scarce drugs among healthcare institutions and not giving preferential access to patients participating in clinical trials.
The suggestions appear in a consensus statement published in Pediatrics.
“Although our recommendations were developed with pediatric oncology in mind, and serve as a blueprint for preventing children with cancer from lacking access to essential life-saving medications, we believe that they apply more broadly across medicine to include pediatrics and adult medicine in general,” said statement author Yoram Unguru, MD, of the Johns Hopkins Berman Institute of Bioethics in Baltimore, Maryland.
He and his coauthors developed the following recommendations:
- Support national polices—and develop new measures—to prevent drug shortages
- Use drug supplies efficiently to reduce the likelihood of shortages
- Give equal priority to evidence-based uses of drugs, whether they occur within or outside clinical trials
- Develop an improved clearinghouse for sharing drug shortage information
- Explore the sharing of drug supplies among institutions
- Develop strategies to engage stakeholders in the management of drug shortages.
For each recommendation, the authors included potential barriers to its implementation. A centralized information source of drug supply information, for instance, comes with the risk that such information will encourage hoarding of existing supplies. This includes “gray market” suppliers that sell scarce drugs for inflated prices.
The authors also pointed to the marketplace economy as an obstacle to implementing their recommendations and preventing drug shortages. Drug manufacturers do not like to disclose manufacturing problems that lead to shortages, nor are competitive healthcare institutions accustomed to cooperating to share resources.
Nonetheless, the authors called for an exploration of “ways to facilitate inter-institutional and inter-state transfer of drugs, especially during shortages,” as well as the ethical obligation to patients inside vs outside a healthcare institution when there is a drug shortage.
“The reasons for drug shortages are complex, but we must not lose sight of the fact that, without access to these life-saving drugs, children and adults with cancer will almost certainly die,” Dr Unguru said. “It is untenable for this situation to continue any longer. We have a clear moral obligation to act to address this critical issue.” ![]()
with chemotherapy drugs
Credit: Bill Branson
A group of healthcare experts has created a “blueprint” for managing and preventing drug shortages.
They devised 6 recommendations for preempting shortages of pediatric oncology drugs, but the suggestions can be applied to other drugs as well.
Some of the recommendations would represent new norms for healthcare practice, such as sharing scarce drugs among healthcare institutions and not giving preferential access to patients participating in clinical trials.
The suggestions appear in a consensus statement published in Pediatrics.
“Although our recommendations were developed with pediatric oncology in mind, and serve as a blueprint for preventing children with cancer from lacking access to essential life-saving medications, we believe that they apply more broadly across medicine to include pediatrics and adult medicine in general,” said statement author Yoram Unguru, MD, of the Johns Hopkins Berman Institute of Bioethics in Baltimore, Maryland.
He and his coauthors developed the following recommendations:
- Support national polices—and develop new measures—to prevent drug shortages
- Use drug supplies efficiently to reduce the likelihood of shortages
- Give equal priority to evidence-based uses of drugs, whether they occur within or outside clinical trials
- Develop an improved clearinghouse for sharing drug shortage information
- Explore the sharing of drug supplies among institutions
- Develop strategies to engage stakeholders in the management of drug shortages.
For each recommendation, the authors included potential barriers to its implementation. A centralized information source of drug supply information, for instance, comes with the risk that such information will encourage hoarding of existing supplies. This includes “gray market” suppliers that sell scarce drugs for inflated prices.
The authors also pointed to the marketplace economy as an obstacle to implementing their recommendations and preventing drug shortages. Drug manufacturers do not like to disclose manufacturing problems that lead to shortages, nor are competitive healthcare institutions accustomed to cooperating to share resources.
Nonetheless, the authors called for an exploration of “ways to facilitate inter-institutional and inter-state transfer of drugs, especially during shortages,” as well as the ethical obligation to patients inside vs outside a healthcare institution when there is a drug shortage.
“The reasons for drug shortages are complex, but we must not lose sight of the fact that, without access to these life-saving drugs, children and adults with cancer will almost certainly die,” Dr Unguru said. “It is untenable for this situation to continue any longer. We have a clear moral obligation to act to address this critical issue.” ![]()
with chemotherapy drugs
Credit: Bill Branson
A group of healthcare experts has created a “blueprint” for managing and preventing drug shortages.
They devised 6 recommendations for preempting shortages of pediatric oncology drugs, but the suggestions can be applied to other drugs as well.
Some of the recommendations would represent new norms for healthcare practice, such as sharing scarce drugs among healthcare institutions and not giving preferential access to patients participating in clinical trials.
The suggestions appear in a consensus statement published in Pediatrics.
“Although our recommendations were developed with pediatric oncology in mind, and serve as a blueprint for preventing children with cancer from lacking access to essential life-saving medications, we believe that they apply more broadly across medicine to include pediatrics and adult medicine in general,” said statement author Yoram Unguru, MD, of the Johns Hopkins Berman Institute of Bioethics in Baltimore, Maryland.
He and his coauthors developed the following recommendations:
- Support national polices—and develop new measures—to prevent drug shortages
- Use drug supplies efficiently to reduce the likelihood of shortages
- Give equal priority to evidence-based uses of drugs, whether they occur within or outside clinical trials
- Develop an improved clearinghouse for sharing drug shortage information
- Explore the sharing of drug supplies among institutions
- Develop strategies to engage stakeholders in the management of drug shortages.
For each recommendation, the authors included potential barriers to its implementation. A centralized information source of drug supply information, for instance, comes with the risk that such information will encourage hoarding of existing supplies. This includes “gray market” suppliers that sell scarce drugs for inflated prices.
The authors also pointed to the marketplace economy as an obstacle to implementing their recommendations and preventing drug shortages. Drug manufacturers do not like to disclose manufacturing problems that lead to shortages, nor are competitive healthcare institutions accustomed to cooperating to share resources.
Nonetheless, the authors called for an exploration of “ways to facilitate inter-institutional and inter-state transfer of drugs, especially during shortages,” as well as the ethical obligation to patients inside vs outside a healthcare institution when there is a drug shortage.
“The reasons for drug shortages are complex, but we must not lose sight of the fact that, without access to these life-saving drugs, children and adults with cancer will almost certainly die,” Dr Unguru said. “It is untenable for this situation to continue any longer. We have a clear moral obligation to act to address this critical issue.” ![]()
Antibody prevents thrombosis in rabbits

oxygenator membranes
Credit: Kjell Hultenby
A newly developed antibody can prevent thrombosis without increasing the risk of bleeding in rabbits, according to research published in Science Translational Medicine.
The antibody, called 3F7, works by blocking a protein that’s active in the coagulation system factor XII (FXII).
Scientists have long known that humans deficient in FXII do not bleed excessively.
And in 2005, Thomas Renné, MD, PhD, of the Karolinska Institutet in Stockholm, Sweden, and his colleagues discovered that mice lacking FXII could have neither a stroke nor a pulmonary embolism, even though they had normal bleeding patterns.
“Since then, our goal has been to find an effective way to block FXII,” Dr Renné said. “Now, we have developed an antibody that blocks FXII in human blood, mice, and rabbits. This provides protection against thrombosis without increasing the risk of bleeding.”
The researchers tested 3F7 in rabbits during extracorporeal membrane oxygenation (ECMO) treatment. ECMO is an advanced heart-lung machine used in life-threatening conditions, but contact with the plastic tubing causes the blood to clot.
The incidence of thrombosis in rabbits on ECMO receiving 3F7 was as low as in rabbits receiving heparin. There were significantly fewer fibrin clots in the oxygenators of 3F7-treated rabbits and heparin-treated rabbits than in saline-treated controls—4 ± 3%, 8 ± 6%, and 100 ± 19%, respectively.
However, bleeding was more severe in the heparin-treated rabbits than in 3F7-treated rabbits and controls. Incision-provoked skin bleeding time was more than 600 seconds in the heparin group, 160 ± 40 s in the 3F7 group, and 130 ± 15 s in controls.
Cuticle bleeding time was more than 600 s in the heparin group, 165 ± 40 s in the 3F7 group, and 120 ± 30 s in controls. The mean blood loss from cuticle wounds was 5.1 ± 1.1 mL/10 min, 0.3 ± 0.1 mL/10 min, and 0.2 ± 0.05 mL/10 min, respectively.
“Blocking FXII appears to be an effective strategy against thrombus formation, and we have shown this in experiments on rabbits in a clinically relevant context,” Dr Renné said in conclusion.
“We plan to test the antibody in a phase 1 study. It is possible that the antibody also blocks inflammation mediated by FXII, an interesting area for future studies.” ![]()
oxygenator membranes
Credit: Kjell Hultenby
A newly developed antibody can prevent thrombosis without increasing the risk of bleeding in rabbits, according to research published in Science Translational Medicine.
The antibody, called 3F7, works by blocking a protein that’s active in the coagulation system factor XII (FXII).
Scientists have long known that humans deficient in FXII do not bleed excessively.
And in 2005, Thomas Renné, MD, PhD, of the Karolinska Institutet in Stockholm, Sweden, and his colleagues discovered that mice lacking FXII could have neither a stroke nor a pulmonary embolism, even though they had normal bleeding patterns.
“Since then, our goal has been to find an effective way to block FXII,” Dr Renné said. “Now, we have developed an antibody that blocks FXII in human blood, mice, and rabbits. This provides protection against thrombosis without increasing the risk of bleeding.”
The researchers tested 3F7 in rabbits during extracorporeal membrane oxygenation (ECMO) treatment. ECMO is an advanced heart-lung machine used in life-threatening conditions, but contact with the plastic tubing causes the blood to clot.
The incidence of thrombosis in rabbits on ECMO receiving 3F7 was as low as in rabbits receiving heparin. There were significantly fewer fibrin clots in the oxygenators of 3F7-treated rabbits and heparin-treated rabbits than in saline-treated controls—4 ± 3%, 8 ± 6%, and 100 ± 19%, respectively.
However, bleeding was more severe in the heparin-treated rabbits than in 3F7-treated rabbits and controls. Incision-provoked skin bleeding time was more than 600 seconds in the heparin group, 160 ± 40 s in the 3F7 group, and 130 ± 15 s in controls.
Cuticle bleeding time was more than 600 s in the heparin group, 165 ± 40 s in the 3F7 group, and 120 ± 30 s in controls. The mean blood loss from cuticle wounds was 5.1 ± 1.1 mL/10 min, 0.3 ± 0.1 mL/10 min, and 0.2 ± 0.05 mL/10 min, respectively.
“Blocking FXII appears to be an effective strategy against thrombus formation, and we have shown this in experiments on rabbits in a clinically relevant context,” Dr Renné said in conclusion.
“We plan to test the antibody in a phase 1 study. It is possible that the antibody also blocks inflammation mediated by FXII, an interesting area for future studies.” ![]()
oxygenator membranes
Credit: Kjell Hultenby
A newly developed antibody can prevent thrombosis without increasing the risk of bleeding in rabbits, according to research published in Science Translational Medicine.
The antibody, called 3F7, works by blocking a protein that’s active in the coagulation system factor XII (FXII).
Scientists have long known that humans deficient in FXII do not bleed excessively.
And in 2005, Thomas Renné, MD, PhD, of the Karolinska Institutet in Stockholm, Sweden, and his colleagues discovered that mice lacking FXII could have neither a stroke nor a pulmonary embolism, even though they had normal bleeding patterns.
“Since then, our goal has been to find an effective way to block FXII,” Dr Renné said. “Now, we have developed an antibody that blocks FXII in human blood, mice, and rabbits. This provides protection against thrombosis without increasing the risk of bleeding.”
The researchers tested 3F7 in rabbits during extracorporeal membrane oxygenation (ECMO) treatment. ECMO is an advanced heart-lung machine used in life-threatening conditions, but contact with the plastic tubing causes the blood to clot.
The incidence of thrombosis in rabbits on ECMO receiving 3F7 was as low as in rabbits receiving heparin. There were significantly fewer fibrin clots in the oxygenators of 3F7-treated rabbits and heparin-treated rabbits than in saline-treated controls—4 ± 3%, 8 ± 6%, and 100 ± 19%, respectively.
However, bleeding was more severe in the heparin-treated rabbits than in 3F7-treated rabbits and controls. Incision-provoked skin bleeding time was more than 600 seconds in the heparin group, 160 ± 40 s in the 3F7 group, and 130 ± 15 s in controls.
Cuticle bleeding time was more than 600 s in the heparin group, 165 ± 40 s in the 3F7 group, and 120 ± 30 s in controls. The mean blood loss from cuticle wounds was 5.1 ± 1.1 mL/10 min, 0.3 ± 0.1 mL/10 min, and 0.2 ± 0.05 mL/10 min, respectively.
“Blocking FXII appears to be an effective strategy against thrombus formation, and we have shown this in experiments on rabbits in a clinically relevant context,” Dr Renné said in conclusion.
“We plan to test the antibody in a phase 1 study. It is possible that the antibody also blocks inflammation mediated by FXII, an interesting area for future studies.”
Minnesota-based Hospital Readmissions Reduction Campaign Earns Prestigious Award
How's this for a quality-improvement success story? In 2011, three Minnesota-based institutions—the Institute for Clinical Systems Improvement (ICSI), the Minnesota Hospital Association, and Stratis Health—launched the Reducing Avoidable Readmissions Effectively (RARE) campaign to reduce avoidable hospital readmissions across the state. So far, the campaign's 82 participating hospitals and 100 community partners have prevented 6,211 readmissions between Jan. 1, 2011 and June 30, 2013.
The efforts were recognized last month when RARE was named a recipient of the 2013 John M. Eisenberg Patient Safety and Quality Award for Innovation in Patient Safety and Quality.
Launched in 2002 by National Quality Forum and Joint Commission, the award honors John M. Eisenberg, MD, MBA, a former administrator of the Agency for Healthcare Research and Quality and an advocate for patient safety and healthcare quality. SHM won the John M. Eisenberg Innovation in Patient Safety and Quality award in 2011 for its mentored-implementation program.
According to ICSI project manager Kathy Cummins, RN, MA, hospitals involved in RARE aren't given specific instructions on how to reduce readmissions, but are encouraged to focus their efforts on these areas:
- Comprehensive discharge planning;
- Medication management;
- Patient and family engagement;
- Transition-care support; and
- Transition communications.
While the campaign provides guidance and technical support, each hospital comes up with its own strategies for achieving these goals. Ms. Cummins, for example, describes how one hospital that was tasked with reducing readmissions without adding staff had paramedics use their downtime to visit recently discharged patients. The paramedics now check in to see if patients are exhibiting warning signs of illness and make sure they’re taking their prescribed medications.
SHM board member Howard Epstein, MD, FHM, ICSI's chief health systems officer, says the RARE campaign targets issues hospitalists have long struggled with.
"Hospitalists don't want to see their patients readmitted to the hospital," Dr. Epstein says. "It doesn't look good on their part, and it's not the best thing for their patients. The [RARE] campaign galvanized the system to support what hospitalists have been demanding for many years."
Stephanie C. Mackiewicz is a freelance author in California.
Visit our website for more information about the RARE campaign.
How's this for a quality-improvement success story? In 2011, three Minnesota-based institutions—the Institute for Clinical Systems Improvement (ICSI), the Minnesota Hospital Association, and Stratis Health—launched the Reducing Avoidable Readmissions Effectively (RARE) campaign to reduce avoidable hospital readmissions across the state. So far, the campaign's 82 participating hospitals and 100 community partners have prevented 6,211 readmissions between Jan. 1, 2011 and June 30, 2013.
The efforts were recognized last month when RARE was named a recipient of the 2013 John M. Eisenberg Patient Safety and Quality Award for Innovation in Patient Safety and Quality.
Launched in 2002 by National Quality Forum and Joint Commission, the award honors John M. Eisenberg, MD, MBA, a former administrator of the Agency for Healthcare Research and Quality and an advocate for patient safety and healthcare quality. SHM won the John M. Eisenberg Innovation in Patient Safety and Quality award in 2011 for its mentored-implementation program.
According to ICSI project manager Kathy Cummins, RN, MA, hospitals involved in RARE aren't given specific instructions on how to reduce readmissions, but are encouraged to focus their efforts on these areas:
- Comprehensive discharge planning;
- Medication management;
- Patient and family engagement;
- Transition-care support; and
- Transition communications.
While the campaign provides guidance and technical support, each hospital comes up with its own strategies for achieving these goals. Ms. Cummins, for example, describes how one hospital that was tasked with reducing readmissions without adding staff had paramedics use their downtime to visit recently discharged patients. The paramedics now check in to see if patients are exhibiting warning signs of illness and make sure they’re taking their prescribed medications.
SHM board member Howard Epstein, MD, FHM, ICSI's chief health systems officer, says the RARE campaign targets issues hospitalists have long struggled with.
"Hospitalists don't want to see their patients readmitted to the hospital," Dr. Epstein says. "It doesn't look good on their part, and it's not the best thing for their patients. The [RARE] campaign galvanized the system to support what hospitalists have been demanding for many years."
Stephanie C. Mackiewicz is a freelance author in California.
Visit our website for more information about the RARE campaign.
How's this for a quality-improvement success story? In 2011, three Minnesota-based institutions—the Institute for Clinical Systems Improvement (ICSI), the Minnesota Hospital Association, and Stratis Health—launched the Reducing Avoidable Readmissions Effectively (RARE) campaign to reduce avoidable hospital readmissions across the state. So far, the campaign's 82 participating hospitals and 100 community partners have prevented 6,211 readmissions between Jan. 1, 2011 and June 30, 2013.
The efforts were recognized last month when RARE was named a recipient of the 2013 John M. Eisenberg Patient Safety and Quality Award for Innovation in Patient Safety and Quality.
Launched in 2002 by National Quality Forum and Joint Commission, the award honors John M. Eisenberg, MD, MBA, a former administrator of the Agency for Healthcare Research and Quality and an advocate for patient safety and healthcare quality. SHM won the John M. Eisenberg Innovation in Patient Safety and Quality award in 2011 for its mentored-implementation program.
According to ICSI project manager Kathy Cummins, RN, MA, hospitals involved in RARE aren't given specific instructions on how to reduce readmissions, but are encouraged to focus their efforts on these areas:
- Comprehensive discharge planning;
- Medication management;
- Patient and family engagement;
- Transition-care support; and
- Transition communications.
While the campaign provides guidance and technical support, each hospital comes up with its own strategies for achieving these goals. Ms. Cummins, for example, describes how one hospital that was tasked with reducing readmissions without adding staff had paramedics use their downtime to visit recently discharged patients. The paramedics now check in to see if patients are exhibiting warning signs of illness and make sure they’re taking their prescribed medications.
SHM board member Howard Epstein, MD, FHM, ICSI's chief health systems officer, says the RARE campaign targets issues hospitalists have long struggled with.
"Hospitalists don't want to see their patients readmitted to the hospital," Dr. Epstein says. "It doesn't look good on their part, and it's not the best thing for their patients. The [RARE] campaign galvanized the system to support what hospitalists have been demanding for many years."
Stephanie C. Mackiewicz is a freelance author in California.
Visit our website for more information about the RARE campaign.