User login
Osteoporosis: What About Men?
› Order dual-energy x-ray absorptiometry of the spine and hip for men who are at increased risk for osteoporosis and candidates for pharmacotherapy. C
› Prescribe bisphosphonates for men with osteoporosis to reduce the risk of vertebral fractures. A
› Advise men who have, or are at risk for, osteoporosis to consume 1000 to 1200 mg of calcium and 600 to 800 IU of vitamin D daily. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
With older women in the United States about 4 times more likely than their male counterparts to develop osteoporosis,1,2 physicians often fail to screen for—or to treat—low bone mass in men. There are plenty of reasons why they should.
First and foremost: Osteoporosis is a leading cause of morbidity and mortality in the elderly.3 An estimated 8.8 million American men suffer from osteoporosis or osteopenia.3 And, although only about 20% of osteoporosis patients are male, men sustain between 30% and 40% of osteoporotic fractures.1,2 What’s more, hip fracture in men has a mortality rate of up to 37.5%—2 to 3 times higher than that of women with hip fracture.4,5
Clearly, then, it is crucial to be aware of the risks of osteoporosis faced by both men and women as they age. Here’s a look at what to consider, when to screen, and how to treat male patients who have, or are at risk for, osteoporosis.
Which men are at risk?
The incidence of fractures secondary to osteoporosis varies with race/ethnicity and geography. The highest rates worldwide occur in Scandinavia and among Caucasians in the United States; black, Asian, and Hispanic populations have the lowest rates.6,7 As with women, the risk of osteoporotic fracture in men increases with age. However, the peak incidence of fracture occurs about 10 years later in men than in women, starting at about age 70.8 Approximately 13% of white men older than 50 years will experience at least one osteoporotic fracture.9
There are 2 main types of osteoporosis: primary and secondary. Up to 40% of osteoporosis in men is primary,4 with bone loss due either to age (senile osteoporosis) or to an unknown cause (idiopathic osteoporosis).10 For men 70 years or older, osteoporosis is assumed to be age related. Idiopathic osteoporosis is diagnosed only in men younger than 70 who have no obvious secondary cause.10 There are numerous secondary causes, however, and most men with bone loss have at least one.4

Common secondary causes: Lifestyle, medical conditions, and meds
The most common causes of secondary osteoporosis in men are exposure to glucocorticoids, primary or secondary hypogonadism (low testosterone), diabetes, alcohol abuse, smoking, gastrointestinal (GI) disease, hypercalciuria, low body weight (body mass index <20 kg/m2), and immobility (TABLE 1).4,5,8,10
Chronic use of corticosteroids, often used to treat chronic obstructive pulmonary disease (COPD), asthma, and rheumatoid arthritis, directly affects the bone, decreasing skeletal muscle, increasing immobility, and reducing intestinal absorption of calcium as well as serum testosterone levels.10 Men with androgen deficiency (which may be due to androgen deprivation therapy to treat prostate cancer) or chronic use of opioids are also at increased risk.4,5,10-12
Diagnostic screening and criteria
The World Health Organization has established diagnostic criteria for osteoporosis using bone mineral density (BMD), reported as both T-scores and Z-scores as measured on dual-energy x-ray absorptiometry (DEXA) scan.13 The T-score represents the number of standard deviations above or below the mean BMD for young adults, matched for sex and race, but not age. It classifies individuals into 3 categories: normal; low (osteopenia), with a T-score between -1 and -2.5; and osteoporosis (T-score ≤-2.5).4,14 The Z-score indicates the number of standard deviations above or below the mean for age, as well as sex and race. A Z-score of ≤-2.0 is below the expected range, indicating an increased likelihood of a secondary form of osteoporosis.14
Which men to screen?
The US Preventive Services Task Force has concluded that evidence is insufficient to assess the balance of benefits and harms of screening for osteoporosis in men. It therefore makes no recommendation to screen men who don't have evidence of previous fractures or secondary causes of osteoporosis.15
Other organizations agree that there is insufficient evidence to recommend routine screening for men without known osteoporotic fractures or secondary causes for osteoporosis. There are, however, some guidelines that are useful in clinical practice.
The Endocrine Society, American College of Physicians (ACP), and National Osteoporosis Foundation (NOF) recommend screening men ages 70 years or older, and men ages 50 to 69 who have risk factors for fracture and/or a history of fracture sustained after age 50.5,16,17 (See “Did you know?”)1,2,4,5,9-12,16,17 Prior to screening, it is important to do a complete medical history and physical examination.
Screening considerations. The Endocrine Society, ACP, and NOF recommend a DEXA scan of the spine and hip for men who are at increased risk for osteoporosis and have no contraindications to drug therapy.5,16,17 In patients who have degenerative changes of the spine and hip that would likely obscure DEXA outcomes, a scan of the radius may provide a more accurate assessment of bone status. Men receiving androgen deprivation therapy for prostate cancer will have a greater decline of bone density in the radius than in the hip or spine and are therefore ideal candidates for DEXA of the forearm, as well.5,11 Keep in mind, however, that no studies have looked at how well, or whether, men with osteoporosis measured only in the radius respond to treatment.5
A DEXA scan is not always widely available, nor is it a perfect predictor of fracture risk. In addition, it is not always cost effective. For some patients, the use of a validated clinical predictive tool is preferable as an initial option.

The Male Osteoporosis Risk Estimation Score (MORES) uses age, weight, and history of COPD to identify men 60 years or older who are at risk for osteoporosis (TABLE 2).18 The score can be easily calculated during a clinical encounter and is beneficial for identifying men who should be referred for DEXA scan. A score of ≥6 has been found to yield an overall sensitivity of 0.93 (95% confidence interval [CI], 0.85-0.97) and a specificity of 0.59 (95% CI, 0.56-0.62), with a number needed to screen to prevent one additional hip fracture of 279.18
The Osteoporosis Self-assessment Tool (OST) (http://depts.washington.edu/osteoed/tools.php?type=ost) is a calculated value that uses age and weight to determine an individual’s risk for osteoporosis (risk score=weight [in kg] – age [in years]/5).16,19 Although there is not a defined value to determine a positive OST risk score, scores of -1 to 3 have been used in a variety of studies.16 In a study of 181 American men, the OST predicted osteoporosis with a sensitivity of 93% and a specificity of 66% when using a cutoff score of 3.20

Treating men at risk
Pharmacologic therapy is recommended for men at an increased risk for fracture. This includes men who have had a hip or vertebral fracture without major trauma, as well as those who have not had such a fracture but have a BMD of the spine, femoral neck, and/or total hip of ≤-2.5.5,17 This standard also applies to the radius when used as an alternative site.
The International Society for Clinical Densitometry and International Osteoporosis Foundation endorse the use of the Fracture Risk Assessment Tool (FRAX). Available at http://shef.ac.uk/FRAX/tool.aspx?country=9, FRAX is a computer-based calculator that uses risk factors and BMD of the femoral neck to estimate an individual’s 10-year fracture probability.21 Men who are 50 years or older, have a T-score between -1.0 and -2.5 in the spine, femoral neck, or total hip, and a 10-year risk of ≥20% of developing any fracture or ≥3% of developing a hip fracture based on FRAX, should be offered pharmacotherapy.5,17
Bisphosphonates are first-line therapy
Although oral bisphosphonates are first-line therapy for men who meet these criteria,4 pharmacotherapy should be individualized based on factors such as fracture history, severity of osteoporosis, comorbidities (eg, peptic ulcer disease, malignancy, renal disease, or malabsorption), and cost (TABLE 3).22,23

Alendronate once weekly has been proven to increase BMD and to reduce the risk of fracture in men.24,25 A randomized, placebo-controlled trial of 241 men with osteoporosis found that alendronate increased BMD by 7.1% (±0.3) at the lumbar spine, 2.5% (±0.4) at the femoral neck, and 2% (±0.2) for the total body. Those in the placebo group had a 1.8% (±0.5) increase in BMD of the lumbar spine, with no significant change in femoral neck or total-body BMD—and a higher incidence of vertebral fractures (7.1% vs. 0.8% for those on alendronate; P=.02).24
Risedronate once daily has also been proven to increase BMD in the lumbar spine and hip, with a reduction in vertebral fractures.26 Another investigation—a 2-year, multicenter double-blind placebo-controlled study of 284 men with osteoporosis—found that risedronate given once a week increased BMD in the spine and hip, but did not reduce the incidence of either vertebral or nonvertebral fractures.27
Both alendronate and risedronate are effective for secondary causes of bone loss, such as corticosteroid use, androgen deprivation therapy/hypogonadism, and rheumatologic conditions.28 Oral bisphosphonates may cause GI irritation, however. Abdominal pain associated with alendronate use is between 1% and 7%, vs 2% to 12% for risedronate.23 Neither medication is recommended for use in patients with an estimated glomerular filtration rate <35 mL/min.23 There is no clearly established duration of therapy for men.
Zoledronic acid infusions, given intravenously (IV) once a year, are available for men who cannot tolerate oral bisphosphonates. In a multicenter double-blind, placebocontrolled trial, zoledronic acid was found to reduce the risk of vertebral fractures in men with primary or hypogonadism-associated osteoporosis by 67% (1.6% vertebral fractures in the treatment group after 24 months vs 4.9% with placebo).29 Given within 90 days of a hip fracture repair, zoledronic acid was associated with both a reduction in the rate of new fractures and an increased survival rate.30
Adverse effects of zoledronic acid include diffuse bone pain (3%-9%), fever (9%-22%) and flu-like symptoms (1%-11%). Osteonecrosis of the jaw has been reported in <1% of patients.23
Recombinant human parathyroid hormone stimulates bone growth
Teriparatide, administered subcutaneously (SC) once a day, directly stimulates bone formation. In a randomized placebo controlled trial of 437 men with a T-score of -2, teriparatide was found to increase BMD at the spine and femoral neck. Participants were randomized to receive teriparatide (20 or 40 mcg/d) or placebo. Those who received teriparatide had a doserelated increase in BMD from baseline at the spine (5.9% with 20 mcg and 9% with 40 mcg) and femoral neck (1.5% and 2.9%, respectively) compared with the placebo group.31 Teriparatide was shown to reduce vertebral fractures by 51% compared with placebo in a randomized study of 355 men with osteoporosis.32
Teriparatide is indicated for men with severe osteoporosis and those for whom bisphosphonate treatment has been unsuccessful. Its use is limited to 2 years due to a dose-dependent risk of osteosarcoma. Teriparatide is contraindicated in patients with skeletal metastasis and has been associated with transient hypercalcemia 4 to 6 hours after administration.23 Its use in combination with bisphosphonates is not recommended due to the lack of proven benefit, risk of adverse effects, and associated cost.5
Testosterone boosts bone density
Testosterone therapy is recommended for men with low levels of testosterone (<200 ng/dL), high risk for fracture, and contraindications to pharmacologic agents approved for the treatment of osteoporosis.5 Supplementation of testosterone to restore correct physiologic levels will decrease bone turnover and increase bone density.33 In a meta-analysis of 8 trials with a total of 365 participants, testosterone administered intramuscularly was found to increase lumbar BMD by 8% compared with placebo. The effect on fractures is not known.12
• Although US women are 4 times more likely than men to suffer from osteoporosis, men incur between 30% and 40% of osteoporotic fractures.
• Men who sustain hip fractures have a mortality rate of up to 37.5%—2 to 3 times that of women with hip fractures.
• Men treated with androgen deprivation therapy face an increased risk of osteoporosis.
• About 13% of white men older than 50 years will experience at least one osteoporotic fracture in their lifetime.
• The Endocrine Society, American College of Physicians, and National Osteoporosis Foundation recommend screening all men ages 70 years or older—and younger men with risk factors for fracture and/or a history of fracture after age 50—for osteoporosis.
Monoclonal antibody reduces fracture risk
Denosumab, a monoclonal antibody that prevents osteoclast formation leading to decreased bone resorption, is administered SC every 6 months.23 In a placebo-controlled trial of 242 men with low bone mass, denosumab increased BMD at the lumbar spine (5.7%), total hip (2.4%), femoral neck (2.1%), trochanter (3.1%), and one-third radius (0.6%) compared with placebo after one year.34 In men receiving androgen deprivation therapy for nonmetastatic prostate cancer, denosumab has been shown to increase BMD and reduce the incidence of vertebral fractures.35
Adverse effects include hypocalcemia, hypophosphatemia, fatigue, and back pain.23 No data exist on the ability of denosumab to reduce fracture risk in men without androgen deprivation.
Calcium and vitamin D for men at risk
Men who are at risk for or have osteoporosis should consume 1000 mg to 1200 mg of calcium per day. Ideally, this should come through dietary sources, but calcium supplementation may be added when diet is inadequate.5 The Institute of Medicine recommends a calcium intake of 1000 mg/d for men ages 51 to 70 years and 1200 mg/d for men ages 70 and older.36
Men with vitamin D levels below 30 ng/mL should receive vitamin D supplementation to attain blood 25(OH) D levels of at least 30 ng/mL.5 The Institute of Medicine recommends a daily intake of 600 international units (IU) of vitamin D for men ages 51 to 70 and 800 IU for men 70 and older.36 A recent Cochrane review on vitamin D and vitamin D analogues concluded that vitamin D alone was unlikely to prevent fractures in older people; when taken with calcium, however, it may have a preventive effect.37
Counseling and follow-up
Lifestyle modification is an important means of primary prevention for osteoporosis. Advise men at risk for osteoporosis to limit alcohol consumption to 2 drinks daily.4,5,8,10 Tell those who smoke that doing so increases their risk for osteoporotic fracture and refer them for smoking cessation counseling. Emphasize that weight-bearing exercise can improve BMD and should be done at least 3 days per week.4,5,8,10 It is important, too, to do a medication review to look for drug-drug interactions and to discuss fall prevention strategies, such as gait training and an environmental assessment and removal of fall hazards.
The evidence for monitoring treatment using BMD is not very strong.5,14 However, the Endocrine Society recommends that response to treatment be monitored using DEXA scans every one to 2 years, with reduced frequency once the BMD has stabilized.5 Any patient found to have a decrease in BMD after treatment is initiated should undergo further evaluation to determine the cause of the decline.
CORRESPONDENCE
Bryan Farford, DO, Mayo Clinic Division of Regional Medicine, 742 Marsh Landing Parkway, Jacksonville Beach, FL 32250; [email protected]
1. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
2. Bliuc D, Nguyen ND, Milch VE, et al. Mortality risk associated with low-trauma osteoporotic fracture and subsequent fracture in men and women. JAMA. 2009;301:513-521.
3. Gennari L, Bilezikian JP. Osteoporosis in men. Endocrinol Metab Clin North Am. 2007;36:399-419.
4. Ebeling PR. Clinical practice. Osteoporosis in men. N Engl J Med. 2008;358:1474-1482.
5. Watts NB, Adler RA, Bilezikian JP, et al; Endocrine Society. Osteoporosis in men: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:1802-1822.
6. Memon A, Pospula WM, Tantawy AY, et al. Incidence of hip fracture in Kuwait. Int J Epidemiol. 1998;27:860-865.
7. Maggi S, Kelsey JL, Litvak J, et al. Incidence of hip fractures in the elderly: a cross-national analysis. Osteoporos Int. 1991;1:232-241.
8. Rao SS, Budhwar N, Ashfaque A. Osteoporosis in men. Am Fam Physician. 2010;82:503-508.
9. Johnell O, Kanis J. Epidemiology of osteoporotic fractures. Osteoporos Int. 2005;16 (Suppl 2):S3-S7.
10. National Institutes of Health. NIH osteoporosis and related bone diseases national resource center. Osteoporosis in men. January 2012. National Institutes of Health Web site. Available at: http://www.niams.nih.gov/health_info/bone/osteoporosis/men.asp. Accessed April 22, 2015.
11. Bruder JM, Ma JZ, Basler JW, et al. Prevalence of osteopenia and osteoporosis by central and peripheral bone mineral density in men with prostate cancer during androgen-deprivation therapy. Urology. 2006;67:152-155.
12. Tracz MJ, Sideras K, Boloña ER, et al. Testosterone use in men and its effects on bone health. A systematic review and meta-analysis of randomized placebo-controlled trials. J Clin Endocrinol Metab. 2006;91:2011-2016.
13. World Health Organization. WHO scientific group on the assessment of osteoporosis at primary health care level. Summary meeting report. Geneva, Switzerland: World Health Organization. 2007. Available at: http://who.int/chp/topics/Osteoporosis.pdf. Accessed April 22, 2015.
14. The International Society for Clinical Densitometry. 2007 official positions & pediatric official positions of The International Society for Clinical Densitometry. The International Society for Clinical Densitometry Web site. Available at: http://www.iscd.org/wp-content/uploads/2012/10/ISCD2007OfficialPositions-Combined-AdultandPediatric.pdf. Accessed August 11, 2015.
15. U.S. Preventive Services Task Force. Screening for osteoporosis: U.S. preventive services task force recommendation statement. Ann Intern Med. 2011;154:356-364.
16. Qaseem A, Snow V, Shekelle P, et al; Clinical Efficacy Assessment Subcommittee of the American College of Physicians. Screening for osteoporosis in men: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;148:680-684.
17. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. National Osteoporosis Foundation Web site. Washington, DC: 2014. Available at: http://nof.org/files/nof/public/content/file/2791/upload/919.pdf. Accessed April 22, 2015.
18. Shepherd AJ, Cass AR, Carlson CA, et al. Development and internal validation of the male osteoporosis risk estimation score. Ann Fam Med. 2007;5:540-546.
19. Lynn HS, Woo J, Leung PC, et al; Osteoporotic Fractures in Men (MrOS) Study. An evaluation of osteoporosis screening tools for the osteoporotic fractures in men (MrOS) study. Osteoporos Int. 2008;19:1087-1092.
20. Adler RA, Tran MT, Petkov VI. Performance of the osteoporosis self-assessment screening tool for osteoporosis in American men. Mayo Clin Proc. 2003;78:723-727.
21. International Osteoporosis Foundation, The International Society for Clinical Densitometry. 2010 Official Positions on FRAX®. International Osteoporosis Foundation Web site. Available at: http://www.iofbonehealth.org/sites/default/files/PDFs/2010_Official_%20Positions_%20ISCD-IOF_%20FRAX.pdf. Accessed March 21, 2015.
22. Epocrates essentials. Epocrates Web site. Available at: www.epocrates.com. Accessed April 17, 2015.
23. American Pharmacist Association. Drug information handbook: a comprehensive resource for all clinicians and healthcare professionals. 21st ed. Alphen aan den Rijn, The Netherlands: Lexi-Comp, Inc. Wolters Kluwer; 2012-2013.
24. Orwoll E, Ettinger M, Weiss S, et al. Alendronate for the treatment of osteoporosis in men. N Engl J Med. 2000;343:604-610.
25. Ringe JD, Dorst A, Faber H, et al. Alendronate treatment of established primary osteoporosis in men: 3-year results of a prospective, comparative, two-arm study. Rheumatol Int. 2004;24:110-113.
26. Ringe JD, Faber H, Farahmand P, et al. Efficacy of risedronate in men with primary and secondary osteoporosis: results of a 1-year study. Rheumatol Int. 2006;26:427-431.
27. Boonen S, Orwoll ES, Wenderoth D, et al. Once-weekly risedronate in men with osteoporosis: results of a 2-year, placebocontrolled, double-blind, multicenter study. J Bone Miner Res. 2009;24:719-725.
28. Khosla S, Amin S, Orwoll E. Osteoporosis in men. Endocr Rev. 2008;29:441-464.
29. Boonen S, Reginster JY, Kaufman JM, et al. Fracture risk and zoledronic acid therapy in men with osteoporosis. N Engl J Med. 2012;367:1714-1723.
30. Lyles KW, Colón-Emeric CS, Magaziner JS, et al; HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
31. Orwoll ES, Scheele WH, Paul S, et al. The effect of teriparatide [human parathyroid hormone (1-34)] therapy on bone density in men with osteoporosis. J Bone Miner Res. 2003;18:9-17.
32. Kaufman JM, Orwoll E, Goemaere S, et al. Teriparatide effects on vertebral fractures and bone mineral density in men with osteoporosis: treatment and discontinuation of therapy. Osteoporos Int. 2005;16:510-516.
33. Snyder PJ, Peachey H, Hannoush P, et al. Effect of testosterone treatment on bone mineral density in men over 65 years of age. J Clin Endocrinol Metab. 1999;84:1966-1972.
34. Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab. 2012;97:3161-3169.
35. Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med. 2009;361:745-755.
36. Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Institute of Medicine. Dietary reference intakes for calcium and vitamin D. Institute of Medicine Web site. Available at: http://www.iom.edu/reports/2010/dietary-reference-intakes-for-calcium-and-vitamin-d.aspx. Accessed April 10, 2015.
37. Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227.
› Order dual-energy x-ray absorptiometry of the spine and hip for men who are at increased risk for osteoporosis and candidates for pharmacotherapy. C
› Prescribe bisphosphonates for men with osteoporosis to reduce the risk of vertebral fractures. A
› Advise men who have, or are at risk for, osteoporosis to consume 1000 to 1200 mg of calcium and 600 to 800 IU of vitamin D daily. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
With older women in the United States about 4 times more likely than their male counterparts to develop osteoporosis,1,2 physicians often fail to screen for—or to treat—low bone mass in men. There are plenty of reasons why they should.
First and foremost: Osteoporosis is a leading cause of morbidity and mortality in the elderly.3 An estimated 8.8 million American men suffer from osteoporosis or osteopenia.3 And, although only about 20% of osteoporosis patients are male, men sustain between 30% and 40% of osteoporotic fractures.1,2 What’s more, hip fracture in men has a mortality rate of up to 37.5%—2 to 3 times higher than that of women with hip fracture.4,5
Clearly, then, it is crucial to be aware of the risks of osteoporosis faced by both men and women as they age. Here’s a look at what to consider, when to screen, and how to treat male patients who have, or are at risk for, osteoporosis.
Which men are at risk?
The incidence of fractures secondary to osteoporosis varies with race/ethnicity and geography. The highest rates worldwide occur in Scandinavia and among Caucasians in the United States; black, Asian, and Hispanic populations have the lowest rates.6,7 As with women, the risk of osteoporotic fracture in men increases with age. However, the peak incidence of fracture occurs about 10 years later in men than in women, starting at about age 70.8 Approximately 13% of white men older than 50 years will experience at least one osteoporotic fracture.9
There are 2 main types of osteoporosis: primary and secondary. Up to 40% of osteoporosis in men is primary,4 with bone loss due either to age (senile osteoporosis) or to an unknown cause (idiopathic osteoporosis).10 For men 70 years or older, osteoporosis is assumed to be age related. Idiopathic osteoporosis is diagnosed only in men younger than 70 who have no obvious secondary cause.10 There are numerous secondary causes, however, and most men with bone loss have at least one.4
Common secondary causes: Lifestyle, medical conditions, and meds
The most common causes of secondary osteoporosis in men are exposure to glucocorticoids, primary or secondary hypogonadism (low testosterone), diabetes, alcohol abuse, smoking, gastrointestinal (GI) disease, hypercalciuria, low body weight (body mass index <20 kg/m2), and immobility (TABLE 1).4,5,8,10
Chronic use of corticosteroids, often used to treat chronic obstructive pulmonary disease (COPD), asthma, and rheumatoid arthritis, directly affects the bone, decreasing skeletal muscle, increasing immobility, and reducing intestinal absorption of calcium as well as serum testosterone levels.10 Men with androgen deficiency (which may be due to androgen deprivation therapy to treat prostate cancer) or chronic use of opioids are also at increased risk.4,5,10-12
Diagnostic screening and criteria
The World Health Organization has established diagnostic criteria for osteoporosis using bone mineral density (BMD), reported as both T-scores and Z-scores as measured on dual-energy x-ray absorptiometry (DEXA) scan.13 The T-score represents the number of standard deviations above or below the mean BMD for young adults, matched for sex and race, but not age. It classifies individuals into 3 categories: normal; low (osteopenia), with a T-score between -1 and -2.5; and osteoporosis (T-score ≤-2.5).4,14 The Z-score indicates the number of standard deviations above or below the mean for age, as well as sex and race. A Z-score of ≤-2.0 is below the expected range, indicating an increased likelihood of a secondary form of osteoporosis.14
Which men to screen?
The US Preventive Services Task Force has concluded that evidence is insufficient to assess the balance of benefits and harms of screening for osteoporosis in men. It therefore makes no recommendation to screen men who don't have evidence of previous fractures or secondary causes of osteoporosis.15
Other organizations agree that there is insufficient evidence to recommend routine screening for men without known osteoporotic fractures or secondary causes for osteoporosis. There are, however, some guidelines that are useful in clinical practice.
The Endocrine Society, American College of Physicians (ACP), and National Osteoporosis Foundation (NOF) recommend screening men ages 70 years or older, and men ages 50 to 69 who have risk factors for fracture and/or a history of fracture sustained after age 50.5,16,17 (See “Did you know?”)1,2,4,5,9-12,16,17 Prior to screening, it is important to do a complete medical history and physical examination.
Screening considerations. The Endocrine Society, ACP, and NOF recommend a DEXA scan of the spine and hip for men who are at increased risk for osteoporosis and have no contraindications to drug therapy.5,16,17 In patients who have degenerative changes of the spine and hip that would likely obscure DEXA outcomes, a scan of the radius may provide a more accurate assessment of bone status. Men receiving androgen deprivation therapy for prostate cancer will have a greater decline of bone density in the radius than in the hip or spine and are therefore ideal candidates for DEXA of the forearm, as well.5,11 Keep in mind, however, that no studies have looked at how well, or whether, men with osteoporosis measured only in the radius respond to treatment.5
A DEXA scan is not always widely available, nor is it a perfect predictor of fracture risk. In addition, it is not always cost effective. For some patients, the use of a validated clinical predictive tool is preferable as an initial option.
The Male Osteoporosis Risk Estimation Score (MORES) uses age, weight, and history of COPD to identify men 60 years or older who are at risk for osteoporosis (TABLE 2).18 The score can be easily calculated during a clinical encounter and is beneficial for identifying men who should be referred for DEXA scan. A score of ≥6 has been found to yield an overall sensitivity of 0.93 (95% confidence interval [CI], 0.85-0.97) and a specificity of 0.59 (95% CI, 0.56-0.62), with a number needed to screen to prevent one additional hip fracture of 279.18
The Osteoporosis Self-assessment Tool (OST) (http://depts.washington.edu/osteoed/tools.php?type=ost) is a calculated value that uses age and weight to determine an individual’s risk for osteoporosis (risk score=weight [in kg] – age [in years]/5).16,19 Although there is not a defined value to determine a positive OST risk score, scores of -1 to 3 have been used in a variety of studies.16 In a study of 181 American men, the OST predicted osteoporosis with a sensitivity of 93% and a specificity of 66% when using a cutoff score of 3.20
Treating men at risk
Pharmacologic therapy is recommended for men at an increased risk for fracture. This includes men who have had a hip or vertebral fracture without major trauma, as well as those who have not had such a fracture but have a BMD of the spine, femoral neck, and/or total hip of ≤-2.5.5,17 This standard also applies to the radius when used as an alternative site.
The International Society for Clinical Densitometry and International Osteoporosis Foundation endorse the use of the Fracture Risk Assessment Tool (FRAX). Available at http://shef.ac.uk/FRAX/tool.aspx?country=9, FRAX is a computer-based calculator that uses risk factors and BMD of the femoral neck to estimate an individual’s 10-year fracture probability.21 Men who are 50 years or older, have a T-score between -1.0 and -2.5 in the spine, femoral neck, or total hip, and a 10-year risk of ≥20% of developing any fracture or ≥3% of developing a hip fracture based on FRAX, should be offered pharmacotherapy.5,17
Bisphosphonates are first-line therapy
Although oral bisphosphonates are first-line therapy for men who meet these criteria,4 pharmacotherapy should be individualized based on factors such as fracture history, severity of osteoporosis, comorbidities (eg, peptic ulcer disease, malignancy, renal disease, or malabsorption), and cost (TABLE 3).22,23
Alendronate once weekly has been proven to increase BMD and to reduce the risk of fracture in men.24,25 A randomized, placebo-controlled trial of 241 men with osteoporosis found that alendronate increased BMD by 7.1% (±0.3) at the lumbar spine, 2.5% (±0.4) at the femoral neck, and 2% (±0.2) for the total body. Those in the placebo group had a 1.8% (±0.5) increase in BMD of the lumbar spine, with no significant change in femoral neck or total-body BMD—and a higher incidence of vertebral fractures (7.1% vs. 0.8% for those on alendronate; P=.02).24
Risedronate once daily has also been proven to increase BMD in the lumbar spine and hip, with a reduction in vertebral fractures.26 Another investigation—a 2-year, multicenter double-blind placebo-controlled study of 284 men with osteoporosis—found that risedronate given once a week increased BMD in the spine and hip, but did not reduce the incidence of either vertebral or nonvertebral fractures.27
Both alendronate and risedronate are effective for secondary causes of bone loss, such as corticosteroid use, androgen deprivation therapy/hypogonadism, and rheumatologic conditions.28 Oral bisphosphonates may cause GI irritation, however. Abdominal pain associated with alendronate use is between 1% and 7%, vs 2% to 12% for risedronate.23 Neither medication is recommended for use in patients with an estimated glomerular filtration rate <35 mL/min.23 There is no clearly established duration of therapy for men.
Zoledronic acid infusions, given intravenously (IV) once a year, are available for men who cannot tolerate oral bisphosphonates. In a multicenter double-blind, placebocontrolled trial, zoledronic acid was found to reduce the risk of vertebral fractures in men with primary or hypogonadism-associated osteoporosis by 67% (1.6% vertebral fractures in the treatment group after 24 months vs 4.9% with placebo).29 Given within 90 days of a hip fracture repair, zoledronic acid was associated with both a reduction in the rate of new fractures and an increased survival rate.30
Adverse effects of zoledronic acid include diffuse bone pain (3%-9%), fever (9%-22%) and flu-like symptoms (1%-11%). Osteonecrosis of the jaw has been reported in <1% of patients.23
Recombinant human parathyroid hormone stimulates bone growth
Teriparatide, administered subcutaneously (SC) once a day, directly stimulates bone formation. In a randomized placebo controlled trial of 437 men with a T-score of -2, teriparatide was found to increase BMD at the spine and femoral neck. Participants were randomized to receive teriparatide (20 or 40 mcg/d) or placebo. Those who received teriparatide had a doserelated increase in BMD from baseline at the spine (5.9% with 20 mcg and 9% with 40 mcg) and femoral neck (1.5% and 2.9%, respectively) compared with the placebo group.31 Teriparatide was shown to reduce vertebral fractures by 51% compared with placebo in a randomized study of 355 men with osteoporosis.32
Teriparatide is indicated for men with severe osteoporosis and those for whom bisphosphonate treatment has been unsuccessful. Its use is limited to 2 years due to a dose-dependent risk of osteosarcoma. Teriparatide is contraindicated in patients with skeletal metastasis and has been associated with transient hypercalcemia 4 to 6 hours after administration.23 Its use in combination with bisphosphonates is not recommended due to the lack of proven benefit, risk of adverse effects, and associated cost.5
Testosterone boosts bone density
Testosterone therapy is recommended for men with low levels of testosterone (<200 ng/dL), high risk for fracture, and contraindications to pharmacologic agents approved for the treatment of osteoporosis.5 Supplementation of testosterone to restore correct physiologic levels will decrease bone turnover and increase bone density.33 In a meta-analysis of 8 trials with a total of 365 participants, testosterone administered intramuscularly was found to increase lumbar BMD by 8% compared with placebo. The effect on fractures is not known.12
• Although US women are 4 times more likely than men to suffer from osteoporosis, men incur between 30% and 40% of osteoporotic fractures.
• Men who sustain hip fractures have a mortality rate of up to 37.5%—2 to 3 times that of women with hip fractures.
• Men treated with androgen deprivation therapy face an increased risk of osteoporosis.
• About 13% of white men older than 50 years will experience at least one osteoporotic fracture in their lifetime.
• The Endocrine Society, American College of Physicians, and National Osteoporosis Foundation recommend screening all men ages 70 years or older—and younger men with risk factors for fracture and/or a history of fracture after age 50—for osteoporosis.
Monoclonal antibody reduces fracture risk
Denosumab, a monoclonal antibody that prevents osteoclast formation leading to decreased bone resorption, is administered SC every 6 months.23 In a placebo-controlled trial of 242 men with low bone mass, denosumab increased BMD at the lumbar spine (5.7%), total hip (2.4%), femoral neck (2.1%), trochanter (3.1%), and one-third radius (0.6%) compared with placebo after one year.34 In men receiving androgen deprivation therapy for nonmetastatic prostate cancer, denosumab has been shown to increase BMD and reduce the incidence of vertebral fractures.35
Adverse effects include hypocalcemia, hypophosphatemia, fatigue, and back pain.23 No data exist on the ability of denosumab to reduce fracture risk in men without androgen deprivation.
Calcium and vitamin D for men at risk
Men who are at risk for or have osteoporosis should consume 1000 mg to 1200 mg of calcium per day. Ideally, this should come through dietary sources, but calcium supplementation may be added when diet is inadequate.5 The Institute of Medicine recommends a calcium intake of 1000 mg/d for men ages 51 to 70 years and 1200 mg/d for men ages 70 and older.36
Men with vitamin D levels below 30 ng/mL should receive vitamin D supplementation to attain blood 25(OH) D levels of at least 30 ng/mL.5 The Institute of Medicine recommends a daily intake of 600 international units (IU) of vitamin D for men ages 51 to 70 and 800 IU for men 70 and older.36 A recent Cochrane review on vitamin D and vitamin D analogues concluded that vitamin D alone was unlikely to prevent fractures in older people; when taken with calcium, however, it may have a preventive effect.37
Counseling and follow-up
Lifestyle modification is an important means of primary prevention for osteoporosis. Advise men at risk for osteoporosis to limit alcohol consumption to 2 drinks daily.4,5,8,10 Tell those who smoke that doing so increases their risk for osteoporotic fracture and refer them for smoking cessation counseling. Emphasize that weight-bearing exercise can improve BMD and should be done at least 3 days per week.4,5,8,10 It is important, too, to do a medication review to look for drug-drug interactions and to discuss fall prevention strategies, such as gait training and an environmental assessment and removal of fall hazards.
The evidence for monitoring treatment using BMD is not very strong.5,14 However, the Endocrine Society recommends that response to treatment be monitored using DEXA scans every one to 2 years, with reduced frequency once the BMD has stabilized.5 Any patient found to have a decrease in BMD after treatment is initiated should undergo further evaluation to determine the cause of the decline.
CORRESPONDENCE
Bryan Farford, DO, Mayo Clinic Division of Regional Medicine, 742 Marsh Landing Parkway, Jacksonville Beach, FL 32250; [email protected]
› Order dual-energy x-ray absorptiometry of the spine and hip for men who are at increased risk for osteoporosis and candidates for pharmacotherapy. C
› Prescribe bisphosphonates for men with osteoporosis to reduce the risk of vertebral fractures. A
› Advise men who have, or are at risk for, osteoporosis to consume 1000 to 1200 mg of calcium and 600 to 800 IU of vitamin D daily. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
With older women in the United States about 4 times more likely than their male counterparts to develop osteoporosis,1,2 physicians often fail to screen for—or to treat—low bone mass in men. There are plenty of reasons why they should.
First and foremost: Osteoporosis is a leading cause of morbidity and mortality in the elderly.3 An estimated 8.8 million American men suffer from osteoporosis or osteopenia.3 And, although only about 20% of osteoporosis patients are male, men sustain between 30% and 40% of osteoporotic fractures.1,2 What’s more, hip fracture in men has a mortality rate of up to 37.5%—2 to 3 times higher than that of women with hip fracture.4,5
Clearly, then, it is crucial to be aware of the risks of osteoporosis faced by both men and women as they age. Here’s a look at what to consider, when to screen, and how to treat male patients who have, or are at risk for, osteoporosis.
Which men are at risk?
The incidence of fractures secondary to osteoporosis varies with race/ethnicity and geography. The highest rates worldwide occur in Scandinavia and among Caucasians in the United States; black, Asian, and Hispanic populations have the lowest rates.6,7 As with women, the risk of osteoporotic fracture in men increases with age. However, the peak incidence of fracture occurs about 10 years later in men than in women, starting at about age 70.8 Approximately 13% of white men older than 50 years will experience at least one osteoporotic fracture.9
There are 2 main types of osteoporosis: primary and secondary. Up to 40% of osteoporosis in men is primary,4 with bone loss due either to age (senile osteoporosis) or to an unknown cause (idiopathic osteoporosis).10 For men 70 years or older, osteoporosis is assumed to be age related. Idiopathic osteoporosis is diagnosed only in men younger than 70 who have no obvious secondary cause.10 There are numerous secondary causes, however, and most men with bone loss have at least one.4
Common secondary causes: Lifestyle, medical conditions, and meds
The most common causes of secondary osteoporosis in men are exposure to glucocorticoids, primary or secondary hypogonadism (low testosterone), diabetes, alcohol abuse, smoking, gastrointestinal (GI) disease, hypercalciuria, low body weight (body mass index <20 kg/m2), and immobility (TABLE 1).4,5,8,10
Chronic use of corticosteroids, often used to treat chronic obstructive pulmonary disease (COPD), asthma, and rheumatoid arthritis, directly affects the bone, decreasing skeletal muscle, increasing immobility, and reducing intestinal absorption of calcium as well as serum testosterone levels.10 Men with androgen deficiency (which may be due to androgen deprivation therapy to treat prostate cancer) or chronic use of opioids are also at increased risk.4,5,10-12
Diagnostic screening and criteria
The World Health Organization has established diagnostic criteria for osteoporosis using bone mineral density (BMD), reported as both T-scores and Z-scores as measured on dual-energy x-ray absorptiometry (DEXA) scan.13 The T-score represents the number of standard deviations above or below the mean BMD for young adults, matched for sex and race, but not age. It classifies individuals into 3 categories: normal; low (osteopenia), with a T-score between -1 and -2.5; and osteoporosis (T-score ≤-2.5).4,14 The Z-score indicates the number of standard deviations above or below the mean for age, as well as sex and race. A Z-score of ≤-2.0 is below the expected range, indicating an increased likelihood of a secondary form of osteoporosis.14
Which men to screen?
The US Preventive Services Task Force has concluded that evidence is insufficient to assess the balance of benefits and harms of screening for osteoporosis in men. It therefore makes no recommendation to screen men who don't have evidence of previous fractures or secondary causes of osteoporosis.15
Other organizations agree that there is insufficient evidence to recommend routine screening for men without known osteoporotic fractures or secondary causes for osteoporosis. There are, however, some guidelines that are useful in clinical practice.
The Endocrine Society, American College of Physicians (ACP), and National Osteoporosis Foundation (NOF) recommend screening men ages 70 years or older, and men ages 50 to 69 who have risk factors for fracture and/or a history of fracture sustained after age 50.5,16,17 (See “Did you know?”)1,2,4,5,9-12,16,17 Prior to screening, it is important to do a complete medical history and physical examination.
Screening considerations. The Endocrine Society, ACP, and NOF recommend a DEXA scan of the spine and hip for men who are at increased risk for osteoporosis and have no contraindications to drug therapy.5,16,17 In patients who have degenerative changes of the spine and hip that would likely obscure DEXA outcomes, a scan of the radius may provide a more accurate assessment of bone status. Men receiving androgen deprivation therapy for prostate cancer will have a greater decline of bone density in the radius than in the hip or spine and are therefore ideal candidates for DEXA of the forearm, as well.5,11 Keep in mind, however, that no studies have looked at how well, or whether, men with osteoporosis measured only in the radius respond to treatment.5
A DEXA scan is not always widely available, nor is it a perfect predictor of fracture risk. In addition, it is not always cost effective. For some patients, the use of a validated clinical predictive tool is preferable as an initial option.
The Male Osteoporosis Risk Estimation Score (MORES) uses age, weight, and history of COPD to identify men 60 years or older who are at risk for osteoporosis (TABLE 2).18 The score can be easily calculated during a clinical encounter and is beneficial for identifying men who should be referred for DEXA scan. A score of ≥6 has been found to yield an overall sensitivity of 0.93 (95% confidence interval [CI], 0.85-0.97) and a specificity of 0.59 (95% CI, 0.56-0.62), with a number needed to screen to prevent one additional hip fracture of 279.18
The Osteoporosis Self-assessment Tool (OST) (http://depts.washington.edu/osteoed/tools.php?type=ost) is a calculated value that uses age and weight to determine an individual’s risk for osteoporosis (risk score=weight [in kg] – age [in years]/5).16,19 Although there is not a defined value to determine a positive OST risk score, scores of -1 to 3 have been used in a variety of studies.16 In a study of 181 American men, the OST predicted osteoporosis with a sensitivity of 93% and a specificity of 66% when using a cutoff score of 3.20
Treating men at risk
Pharmacologic therapy is recommended for men at an increased risk for fracture. This includes men who have had a hip or vertebral fracture without major trauma, as well as those who have not had such a fracture but have a BMD of the spine, femoral neck, and/or total hip of ≤-2.5.5,17 This standard also applies to the radius when used as an alternative site.
The International Society for Clinical Densitometry and International Osteoporosis Foundation endorse the use of the Fracture Risk Assessment Tool (FRAX). Available at http://shef.ac.uk/FRAX/tool.aspx?country=9, FRAX is a computer-based calculator that uses risk factors and BMD of the femoral neck to estimate an individual’s 10-year fracture probability.21 Men who are 50 years or older, have a T-score between -1.0 and -2.5 in the spine, femoral neck, or total hip, and a 10-year risk of ≥20% of developing any fracture or ≥3% of developing a hip fracture based on FRAX, should be offered pharmacotherapy.5,17
Bisphosphonates are first-line therapy
Although oral bisphosphonates are first-line therapy for men who meet these criteria,4 pharmacotherapy should be individualized based on factors such as fracture history, severity of osteoporosis, comorbidities (eg, peptic ulcer disease, malignancy, renal disease, or malabsorption), and cost (TABLE 3).22,23
Alendronate once weekly has been proven to increase BMD and to reduce the risk of fracture in men.24,25 A randomized, placebo-controlled trial of 241 men with osteoporosis found that alendronate increased BMD by 7.1% (±0.3) at the lumbar spine, 2.5% (±0.4) at the femoral neck, and 2% (±0.2) for the total body. Those in the placebo group had a 1.8% (±0.5) increase in BMD of the lumbar spine, with no significant change in femoral neck or total-body BMD—and a higher incidence of vertebral fractures (7.1% vs. 0.8% for those on alendronate; P=.02).24
Risedronate once daily has also been proven to increase BMD in the lumbar spine and hip, with a reduction in vertebral fractures.26 Another investigation—a 2-year, multicenter double-blind placebo-controlled study of 284 men with osteoporosis—found that risedronate given once a week increased BMD in the spine and hip, but did not reduce the incidence of either vertebral or nonvertebral fractures.27
Both alendronate and risedronate are effective for secondary causes of bone loss, such as corticosteroid use, androgen deprivation therapy/hypogonadism, and rheumatologic conditions.28 Oral bisphosphonates may cause GI irritation, however. Abdominal pain associated with alendronate use is between 1% and 7%, vs 2% to 12% for risedronate.23 Neither medication is recommended for use in patients with an estimated glomerular filtration rate <35 mL/min.23 There is no clearly established duration of therapy for men.
Zoledronic acid infusions, given intravenously (IV) once a year, are available for men who cannot tolerate oral bisphosphonates. In a multicenter double-blind, placebocontrolled trial, zoledronic acid was found to reduce the risk of vertebral fractures in men with primary or hypogonadism-associated osteoporosis by 67% (1.6% vertebral fractures in the treatment group after 24 months vs 4.9% with placebo).29 Given within 90 days of a hip fracture repair, zoledronic acid was associated with both a reduction in the rate of new fractures and an increased survival rate.30
Adverse effects of zoledronic acid include diffuse bone pain (3%-9%), fever (9%-22%) and flu-like symptoms (1%-11%). Osteonecrosis of the jaw has been reported in <1% of patients.23
Recombinant human parathyroid hormone stimulates bone growth
Teriparatide, administered subcutaneously (SC) once a day, directly stimulates bone formation. In a randomized placebo controlled trial of 437 men with a T-score of -2, teriparatide was found to increase BMD at the spine and femoral neck. Participants were randomized to receive teriparatide (20 or 40 mcg/d) or placebo. Those who received teriparatide had a doserelated increase in BMD from baseline at the spine (5.9% with 20 mcg and 9% with 40 mcg) and femoral neck (1.5% and 2.9%, respectively) compared with the placebo group.31 Teriparatide was shown to reduce vertebral fractures by 51% compared with placebo in a randomized study of 355 men with osteoporosis.32
Teriparatide is indicated for men with severe osteoporosis and those for whom bisphosphonate treatment has been unsuccessful. Its use is limited to 2 years due to a dose-dependent risk of osteosarcoma. Teriparatide is contraindicated in patients with skeletal metastasis and has been associated with transient hypercalcemia 4 to 6 hours after administration.23 Its use in combination with bisphosphonates is not recommended due to the lack of proven benefit, risk of adverse effects, and associated cost.5
Testosterone boosts bone density
Testosterone therapy is recommended for men with low levels of testosterone (<200 ng/dL), high risk for fracture, and contraindications to pharmacologic agents approved for the treatment of osteoporosis.5 Supplementation of testosterone to restore correct physiologic levels will decrease bone turnover and increase bone density.33 In a meta-analysis of 8 trials with a total of 365 participants, testosterone administered intramuscularly was found to increase lumbar BMD by 8% compared with placebo. The effect on fractures is not known.12
• Although US women are 4 times more likely than men to suffer from osteoporosis, men incur between 30% and 40% of osteoporotic fractures.
• Men who sustain hip fractures have a mortality rate of up to 37.5%—2 to 3 times that of women with hip fractures.
• Men treated with androgen deprivation therapy face an increased risk of osteoporosis.
• About 13% of white men older than 50 years will experience at least one osteoporotic fracture in their lifetime.
• The Endocrine Society, American College of Physicians, and National Osteoporosis Foundation recommend screening all men ages 70 years or older—and younger men with risk factors for fracture and/or a history of fracture after age 50—for osteoporosis.
Monoclonal antibody reduces fracture risk
Denosumab, a monoclonal antibody that prevents osteoclast formation leading to decreased bone resorption, is administered SC every 6 months.23 In a placebo-controlled trial of 242 men with low bone mass, denosumab increased BMD at the lumbar spine (5.7%), total hip (2.4%), femoral neck (2.1%), trochanter (3.1%), and one-third radius (0.6%) compared with placebo after one year.34 In men receiving androgen deprivation therapy for nonmetastatic prostate cancer, denosumab has been shown to increase BMD and reduce the incidence of vertebral fractures.35
Adverse effects include hypocalcemia, hypophosphatemia, fatigue, and back pain.23 No data exist on the ability of denosumab to reduce fracture risk in men without androgen deprivation.
Calcium and vitamin D for men at risk
Men who are at risk for or have osteoporosis should consume 1000 mg to 1200 mg of calcium per day. Ideally, this should come through dietary sources, but calcium supplementation may be added when diet is inadequate.5 The Institute of Medicine recommends a calcium intake of 1000 mg/d for men ages 51 to 70 years and 1200 mg/d for men ages 70 and older.36
Men with vitamin D levels below 30 ng/mL should receive vitamin D supplementation to attain blood 25(OH) D levels of at least 30 ng/mL.5 The Institute of Medicine recommends a daily intake of 600 international units (IU) of vitamin D for men ages 51 to 70 and 800 IU for men 70 and older.36 A recent Cochrane review on vitamin D and vitamin D analogues concluded that vitamin D alone was unlikely to prevent fractures in older people; when taken with calcium, however, it may have a preventive effect.37
Counseling and follow-up
Lifestyle modification is an important means of primary prevention for osteoporosis. Advise men at risk for osteoporosis to limit alcohol consumption to 2 drinks daily.4,5,8,10 Tell those who smoke that doing so increases their risk for osteoporotic fracture and refer them for smoking cessation counseling. Emphasize that weight-bearing exercise can improve BMD and should be done at least 3 days per week.4,5,8,10 It is important, too, to do a medication review to look for drug-drug interactions and to discuss fall prevention strategies, such as gait training and an environmental assessment and removal of fall hazards.
The evidence for monitoring treatment using BMD is not very strong.5,14 However, the Endocrine Society recommends that response to treatment be monitored using DEXA scans every one to 2 years, with reduced frequency once the BMD has stabilized.5 Any patient found to have a decrease in BMD after treatment is initiated should undergo further evaluation to determine the cause of the decline.
CORRESPONDENCE
Bryan Farford, DO, Mayo Clinic Division of Regional Medicine, 742 Marsh Landing Parkway, Jacksonville Beach, FL 32250; [email protected]
1. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
2. Bliuc D, Nguyen ND, Milch VE, et al. Mortality risk associated with low-trauma osteoporotic fracture and subsequent fracture in men and women. JAMA. 2009;301:513-521.
3. Gennari L, Bilezikian JP. Osteoporosis in men. Endocrinol Metab Clin North Am. 2007;36:399-419.
4. Ebeling PR. Clinical practice. Osteoporosis in men. N Engl J Med. 2008;358:1474-1482.
5. Watts NB, Adler RA, Bilezikian JP, et al; Endocrine Society. Osteoporosis in men: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:1802-1822.
6. Memon A, Pospula WM, Tantawy AY, et al. Incidence of hip fracture in Kuwait. Int J Epidemiol. 1998;27:860-865.
7. Maggi S, Kelsey JL, Litvak J, et al. Incidence of hip fractures in the elderly: a cross-national analysis. Osteoporos Int. 1991;1:232-241.
8. Rao SS, Budhwar N, Ashfaque A. Osteoporosis in men. Am Fam Physician. 2010;82:503-508.
9. Johnell O, Kanis J. Epidemiology of osteoporotic fractures. Osteoporos Int. 2005;16 (Suppl 2):S3-S7.
10. National Institutes of Health. NIH osteoporosis and related bone diseases national resource center. Osteoporosis in men. January 2012. National Institutes of Health Web site. Available at: http://www.niams.nih.gov/health_info/bone/osteoporosis/men.asp. Accessed April 22, 2015.
11. Bruder JM, Ma JZ, Basler JW, et al. Prevalence of osteopenia and osteoporosis by central and peripheral bone mineral density in men with prostate cancer during androgen-deprivation therapy. Urology. 2006;67:152-155.
12. Tracz MJ, Sideras K, Boloña ER, et al. Testosterone use in men and its effects on bone health. A systematic review and meta-analysis of randomized placebo-controlled trials. J Clin Endocrinol Metab. 2006;91:2011-2016.
13. World Health Organization. WHO scientific group on the assessment of osteoporosis at primary health care level. Summary meeting report. Geneva, Switzerland: World Health Organization. 2007. Available at: http://who.int/chp/topics/Osteoporosis.pdf. Accessed April 22, 2015.
14. The International Society for Clinical Densitometry. 2007 official positions & pediatric official positions of The International Society for Clinical Densitometry. The International Society for Clinical Densitometry Web site. Available at: http://www.iscd.org/wp-content/uploads/2012/10/ISCD2007OfficialPositions-Combined-AdultandPediatric.pdf. Accessed August 11, 2015.
15. U.S. Preventive Services Task Force. Screening for osteoporosis: U.S. preventive services task force recommendation statement. Ann Intern Med. 2011;154:356-364.
16. Qaseem A, Snow V, Shekelle P, et al; Clinical Efficacy Assessment Subcommittee of the American College of Physicians. Screening for osteoporosis in men: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;148:680-684.
17. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. National Osteoporosis Foundation Web site. Washington, DC: 2014. Available at: http://nof.org/files/nof/public/content/file/2791/upload/919.pdf. Accessed April 22, 2015.
18. Shepherd AJ, Cass AR, Carlson CA, et al. Development and internal validation of the male osteoporosis risk estimation score. Ann Fam Med. 2007;5:540-546.
19. Lynn HS, Woo J, Leung PC, et al; Osteoporotic Fractures in Men (MrOS) Study. An evaluation of osteoporosis screening tools for the osteoporotic fractures in men (MrOS) study. Osteoporos Int. 2008;19:1087-1092.
20. Adler RA, Tran MT, Petkov VI. Performance of the osteoporosis self-assessment screening tool for osteoporosis in American men. Mayo Clin Proc. 2003;78:723-727.
21. International Osteoporosis Foundation, The International Society for Clinical Densitometry. 2010 Official Positions on FRAX®. International Osteoporosis Foundation Web site. Available at: http://www.iofbonehealth.org/sites/default/files/PDFs/2010_Official_%20Positions_%20ISCD-IOF_%20FRAX.pdf. Accessed March 21, 2015.
22. Epocrates essentials. Epocrates Web site. Available at: www.epocrates.com. Accessed April 17, 2015.
23. American Pharmacist Association. Drug information handbook: a comprehensive resource for all clinicians and healthcare professionals. 21st ed. Alphen aan den Rijn, The Netherlands: Lexi-Comp, Inc. Wolters Kluwer; 2012-2013.
24. Orwoll E, Ettinger M, Weiss S, et al. Alendronate for the treatment of osteoporosis in men. N Engl J Med. 2000;343:604-610.
25. Ringe JD, Dorst A, Faber H, et al. Alendronate treatment of established primary osteoporosis in men: 3-year results of a prospective, comparative, two-arm study. Rheumatol Int. 2004;24:110-113.
26. Ringe JD, Faber H, Farahmand P, et al. Efficacy of risedronate in men with primary and secondary osteoporosis: results of a 1-year study. Rheumatol Int. 2006;26:427-431.
27. Boonen S, Orwoll ES, Wenderoth D, et al. Once-weekly risedronate in men with osteoporosis: results of a 2-year, placebocontrolled, double-blind, multicenter study. J Bone Miner Res. 2009;24:719-725.
28. Khosla S, Amin S, Orwoll E. Osteoporosis in men. Endocr Rev. 2008;29:441-464.
29. Boonen S, Reginster JY, Kaufman JM, et al. Fracture risk and zoledronic acid therapy in men with osteoporosis. N Engl J Med. 2012;367:1714-1723.
30. Lyles KW, Colón-Emeric CS, Magaziner JS, et al; HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
31. Orwoll ES, Scheele WH, Paul S, et al. The effect of teriparatide [human parathyroid hormone (1-34)] therapy on bone density in men with osteoporosis. J Bone Miner Res. 2003;18:9-17.
32. Kaufman JM, Orwoll E, Goemaere S, et al. Teriparatide effects on vertebral fractures and bone mineral density in men with osteoporosis: treatment and discontinuation of therapy. Osteoporos Int. 2005;16:510-516.
33. Snyder PJ, Peachey H, Hannoush P, et al. Effect of testosterone treatment on bone mineral density in men over 65 years of age. J Clin Endocrinol Metab. 1999;84:1966-1972.
34. Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab. 2012;97:3161-3169.
35. Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med. 2009;361:745-755.
36. Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Institute of Medicine. Dietary reference intakes for calcium and vitamin D. Institute of Medicine Web site. Available at: http://www.iom.edu/reports/2010/dietary-reference-intakes-for-calcium-and-vitamin-d.aspx. Accessed April 10, 2015.
37. Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227.
1. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
2. Bliuc D, Nguyen ND, Milch VE, et al. Mortality risk associated with low-trauma osteoporotic fracture and subsequent fracture in men and women. JAMA. 2009;301:513-521.
3. Gennari L, Bilezikian JP. Osteoporosis in men. Endocrinol Metab Clin North Am. 2007;36:399-419.
4. Ebeling PR. Clinical practice. Osteoporosis in men. N Engl J Med. 2008;358:1474-1482.
5. Watts NB, Adler RA, Bilezikian JP, et al; Endocrine Society. Osteoporosis in men: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:1802-1822.
6. Memon A, Pospula WM, Tantawy AY, et al. Incidence of hip fracture in Kuwait. Int J Epidemiol. 1998;27:860-865.
7. Maggi S, Kelsey JL, Litvak J, et al. Incidence of hip fractures in the elderly: a cross-national analysis. Osteoporos Int. 1991;1:232-241.
8. Rao SS, Budhwar N, Ashfaque A. Osteoporosis in men. Am Fam Physician. 2010;82:503-508.
9. Johnell O, Kanis J. Epidemiology of osteoporotic fractures. Osteoporos Int. 2005;16 (Suppl 2):S3-S7.
10. National Institutes of Health. NIH osteoporosis and related bone diseases national resource center. Osteoporosis in men. January 2012. National Institutes of Health Web site. Available at: http://www.niams.nih.gov/health_info/bone/osteoporosis/men.asp. Accessed April 22, 2015.
11. Bruder JM, Ma JZ, Basler JW, et al. Prevalence of osteopenia and osteoporosis by central and peripheral bone mineral density in men with prostate cancer during androgen-deprivation therapy. Urology. 2006;67:152-155.
12. Tracz MJ, Sideras K, Boloña ER, et al. Testosterone use in men and its effects on bone health. A systematic review and meta-analysis of randomized placebo-controlled trials. J Clin Endocrinol Metab. 2006;91:2011-2016.
13. World Health Organization. WHO scientific group on the assessment of osteoporosis at primary health care level. Summary meeting report. Geneva, Switzerland: World Health Organization. 2007. Available at: http://who.int/chp/topics/Osteoporosis.pdf. Accessed April 22, 2015.
14. The International Society for Clinical Densitometry. 2007 official positions & pediatric official positions of The International Society for Clinical Densitometry. The International Society for Clinical Densitometry Web site. Available at: http://www.iscd.org/wp-content/uploads/2012/10/ISCD2007OfficialPositions-Combined-AdultandPediatric.pdf. Accessed August 11, 2015.
15. U.S. Preventive Services Task Force. Screening for osteoporosis: U.S. preventive services task force recommendation statement. Ann Intern Med. 2011;154:356-364.
16. Qaseem A, Snow V, Shekelle P, et al; Clinical Efficacy Assessment Subcommittee of the American College of Physicians. Screening for osteoporosis in men: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;148:680-684.
17. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. National Osteoporosis Foundation Web site. Washington, DC: 2014. Available at: http://nof.org/files/nof/public/content/file/2791/upload/919.pdf. Accessed April 22, 2015.
18. Shepherd AJ, Cass AR, Carlson CA, et al. Development and internal validation of the male osteoporosis risk estimation score. Ann Fam Med. 2007;5:540-546.
19. Lynn HS, Woo J, Leung PC, et al; Osteoporotic Fractures in Men (MrOS) Study. An evaluation of osteoporosis screening tools for the osteoporotic fractures in men (MrOS) study. Osteoporos Int. 2008;19:1087-1092.
20. Adler RA, Tran MT, Petkov VI. Performance of the osteoporosis self-assessment screening tool for osteoporosis in American men. Mayo Clin Proc. 2003;78:723-727.
21. International Osteoporosis Foundation, The International Society for Clinical Densitometry. 2010 Official Positions on FRAX®. International Osteoporosis Foundation Web site. Available at: http://www.iofbonehealth.org/sites/default/files/PDFs/2010_Official_%20Positions_%20ISCD-IOF_%20FRAX.pdf. Accessed March 21, 2015.
22. Epocrates essentials. Epocrates Web site. Available at: www.epocrates.com. Accessed April 17, 2015.
23. American Pharmacist Association. Drug information handbook: a comprehensive resource for all clinicians and healthcare professionals. 21st ed. Alphen aan den Rijn, The Netherlands: Lexi-Comp, Inc. Wolters Kluwer; 2012-2013.
24. Orwoll E, Ettinger M, Weiss S, et al. Alendronate for the treatment of osteoporosis in men. N Engl J Med. 2000;343:604-610.
25. Ringe JD, Dorst A, Faber H, et al. Alendronate treatment of established primary osteoporosis in men: 3-year results of a prospective, comparative, two-arm study. Rheumatol Int. 2004;24:110-113.
26. Ringe JD, Faber H, Farahmand P, et al. Efficacy of risedronate in men with primary and secondary osteoporosis: results of a 1-year study. Rheumatol Int. 2006;26:427-431.
27. Boonen S, Orwoll ES, Wenderoth D, et al. Once-weekly risedronate in men with osteoporosis: results of a 2-year, placebocontrolled, double-blind, multicenter study. J Bone Miner Res. 2009;24:719-725.
28. Khosla S, Amin S, Orwoll E. Osteoporosis in men. Endocr Rev. 2008;29:441-464.
29. Boonen S, Reginster JY, Kaufman JM, et al. Fracture risk and zoledronic acid therapy in men with osteoporosis. N Engl J Med. 2012;367:1714-1723.
30. Lyles KW, Colón-Emeric CS, Magaziner JS, et al; HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
31. Orwoll ES, Scheele WH, Paul S, et al. The effect of teriparatide [human parathyroid hormone (1-34)] therapy on bone density in men with osteoporosis. J Bone Miner Res. 2003;18:9-17.
32. Kaufman JM, Orwoll E, Goemaere S, et al. Teriparatide effects on vertebral fractures and bone mineral density in men with osteoporosis: treatment and discontinuation of therapy. Osteoporos Int. 2005;16:510-516.
33. Snyder PJ, Peachey H, Hannoush P, et al. Effect of testosterone treatment on bone mineral density in men over 65 years of age. J Clin Endocrinol Metab. 1999;84:1966-1972.
34. Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab. 2012;97:3161-3169.
35. Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med. 2009;361:745-755.
36. Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Institute of Medicine. Dietary reference intakes for calcium and vitamin D. Institute of Medicine Web site. Available at: http://www.iom.edu/reports/2010/dietary-reference-intakes-for-calcium-and-vitamin-d.aspx. Accessed April 10, 2015.
37. Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227.

Osteoporosis: What about men?
› Order dual-energy x-ray absorptiometry of the spine and hip for men who are at increased risk for osteoporosis and candidates for pharmacotherapy. C
› Prescribe bisphosphonates for men with osteoporosis to reduce the risk of vertebral fractures. A
› Advise men who have, or are at risk for, osteoporosis to consume 1000 to 1200 mg of calcium and 600 to 800 IU of vitamin D daily. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
With older women in the United States about 4 times more likely than their male counterparts to develop osteoporosis,1,2 physicians often fail to screen for—or to treat—low bone mass in men. There are plenty of reasons why they should.
First and foremost: Osteoporosis is a leading cause of morbidity and mortality in the elderly.3 An estimated 8.8 million American men suffer from osteoporosis or osteopenia.3 And, although only about 20% of osteoporosis patients are male, men sustain between 30% and 40% of osteoporotic fractures.1,2 What’s more, hip fracture in men has a mortality rate of up to 37.5%—2 to 3 times higher than that of women with hip fracture.4,5
Clearly, then, it is crucial to be aware of the risks of osteoporosis faced by both men and women as they age. Here’s a look at what to consider, when to screen, and how to treat male patients who have, or are at risk for, osteoporosis.
Which men are at risk?
The incidence of fractures secondary to osteoporosis varies with race/ethnicity and geography. The highest rates worldwide occur in Scandinavia and among Caucasians in the United States; black, Asian, and Hispanic populations have the lowest rates.6,7 As with women, the risk of osteoporotic fracture in men increases with age. However, the peak incidence of fracture occurs about 10 years later in men than in women, starting at about age 70.8 Approximately 13% of white men older than 50 years will experience at least one osteoporotic fracture.9
There are 2 main types of osteoporosis: primary and secondary. Up to 40% of osteoporosis in men is primary,4 with bone loss due either to age (senile osteoporosis) or to an unknown cause (idiopathic osteoporosis).10 For men 70 years or older, osteoporosis is assumed to be age related. Idiopathic osteoporosis is diagnosed only in men younger than 70 who have no obvious secondary cause.10 There are numerous secondary causes, however, and most men with bone loss have at least one.4

Common secondary causes: Lifestyle, medical conditions, and meds
The most common causes of secondary osteoporosis in men are exposure to glucocorticoids, primary or secondary hypogonadism (low testosterone), diabetes, alcohol abuse, smoking, gastrointestinal (GI) disease, hypercalciuria, low body weight (body mass index <20 kg/m2), and immobility (TABLE 1).4,5,8,10
Chronic use of corticosteroids, often used to treat chronic obstructive pulmonary disease (COPD), asthma, and rheumatoid arthritis, directly affects the bone, decreasing skeletal muscle, increasing immobility, and reducing intestinal absorption of calcium as well as serum testosterone levels.10 Men with androgen deficiency (which may be due to androgen deprivation therapy to treat prostate cancer) or chronic use of opioids are also at increased risk.4,5,10-12
Diagnostic screening and criteria
The World Health Organization has established diagnostic criteria for osteoporosis using bone mineral density (BMD), reported as both T-scores and Z-scores as measured on dual-energy x-ray absorptiometry (DEXA) scan.13 The T-score represents the number of standard deviations above or below the mean BMD for young adults, matched for sex and race, but not age. It classifies individuals into 3 categories: normal; low (osteopenia), with a T-score between -1 and -2.5; and osteoporosis (T-score ≤-2.5).4,14 The Z-score indicates the number of standard deviations above or below the mean for age, as well as sex and race. A Z-score of ≤-2.0 is below the expected range, indicating an increased likelihood of a secondary form of osteoporosis.14
Which men to screen?
The US Preventive Services Task Force has concluded that evidence is insufficient to assess the balance of benefits and harms of screening for osteoporosis in men. It therefore makes no recommendation to screen men who don't have evidence of previous fractures or secondary causes of osteoporosis.15
Other organizations agree that there is insufficient evidence to recommend routine screening for men without known osteoporotic fractures or secondary causes for osteoporosis. There are, however, some guidelines that are useful in clinical practice.
The Endocrine Society, American College of Physicians (ACP), and National Osteoporosis Foundation (NOF) recommend screening men ages 70 years or older, and men ages 50 to 69 who have risk factors for fracture and/or a history of fracture sustained after age 50.5,16,17 (See “Did you know?”)1,2,4,5,9-12,16,17 Prior to screening, it is important to do a complete medical history and physical examination.
Screening considerations. The Endocrine Society, ACP, and NOF recommend a DEXA scan of the spine and hip for men who are at increased risk for osteoporosis and have no contraindications to drug therapy.5,16,17 In patients who have degenerative changes of the spine and hip that would likely obscure DEXA outcomes, a scan of the radius may provide a more accurate assessment of bone status. Men receiving androgen deprivation therapy for prostate cancer will have a greater decline of bone density in the radius than in the hip or spine and are therefore ideal candidates for DEXA of the forearm, as well.5,11 Keep in mind, however, that no studies have looked at how well, or whether, men with osteoporosis measured only in the radius respond to treatment.5
A DEXA scan is not always widely available, nor is it a perfect predictor of fracture risk. In addition, it is not always cost effective. For some patients, the use of a validated clinical predictive tool is preferable as an initial option.

The Male Osteoporosis Risk Estimation Score (MORES) uses age, weight, and history of COPD to identify men 60 years or older who are at risk for osteoporosis (TABLE 2).18 The score can be easily calculated during a clinical encounter and is beneficial for identifying men who should be referred for DEXA scan. A score of ≥6 has been found to yield an overall sensitivity of 0.93 (95% confidence interval [CI], 0.85-0.97) and a specificity of 0.59 (95% CI, 0.56-0.62), with a number needed to screen to prevent one additional hip fracture of 279.18
The Osteoporosis Self-assessment Tool (OST) (http://depts.washington.edu/osteoed/tools.php?type=ost) is a calculated value that uses age and weight to determine an individual’s risk for osteoporosis (risk score=weight [in kg] – age [in years]/5).16,19 Although there is not a defined value to determine a positive OST risk score, scores of -1 to 3 have been used in a variety of studies.16 In a study of 181 American men, the OST predicted osteoporosis with a sensitivity of 93% and a specificity of 66% when using a cutoff score of 3.20

Treating men at risk
Pharmacologic therapy is recommended for men at an increased risk for fracture. This includes men who have had a hip or vertebral fracture without major trauma, as well as those who have not had such a fracture but have a BMD of the spine, femoral neck, and/or total hip of ≤-2.5.5,17 This standard also applies to the radius when used as an alternative site.
The International Society for Clinical Densitometry and International Osteoporosis Foundation endorse the use of the Fracture Risk Assessment Tool (FRAX). Available at http://shef.ac.uk/FRAX/tool.aspx?country=9, FRAX is a computer-based calculator that uses risk factors and BMD of the femoral neck to estimate an individual’s 10-year fracture probability.21 Men who are 50 years or older, have a T-score between -1.0 and -2.5 in the spine, femoral neck, or total hip, and a 10-year risk of ≥20% of developing any fracture or ≥3% of developing a hip fracture based on FRAX, should be offered pharmacotherapy.5,17
Bisphosphonates are first-line therapy
Although oral bisphosphonates are first-line therapy for men who meet these criteria,4 pharmacotherapy should be individualized based on factors such as fracture history, severity of osteoporosis, comorbidities (eg, peptic ulcer disease, malignancy, renal disease, or malabsorption), and cost (TABLE 3).22,23

Alendronate once weekly has been proven to increase BMD and to reduce the risk of fracture in men.24,25 A randomized, placebo-controlled trial of 241 men with osteoporosis found that alendronate increased BMD by 7.1% (±0.3) at the lumbar spine, 2.5% (±0.4) at the femoral neck, and 2% (±0.2) for the total body. Those in the placebo group had a 1.8% (±0.5) increase in BMD of the lumbar spine, with no significant change in femoral neck or total-body BMD—and a higher incidence of vertebral fractures (7.1% vs. 0.8% for those on alendronate; P=.02).24
Risedronate once daily has also been proven to increase BMD in the lumbar spine and hip, with a reduction in vertebral fractures.26 Another investigation—a 2-year, multicenter double-blind placebo-controlled study of 284 men with osteoporosis—found that risedronate given once a week increased BMD in the spine and hip, but did not reduce the incidence of either vertebral or nonvertebral fractures.27
Both alendronate and risedronate are effective for secondary causes of bone loss, such as corticosteroid use, androgen deprivation therapy/hypogonadism, and rheumatologic conditions.28 Oral bisphosphonates may cause GI irritation, however. Abdominal pain associated with alendronate use is between 1% and 7%, vs 2% to 12% for risedronate.23 Neither medication is recommended for use in patients with an estimated glomerular filtration rate <35 mL/min.23 There is no clearly established duration of therapy for men.
Zoledronic acid infusions, given intravenously (IV) once a year, are available for men who cannot tolerate oral bisphosphonates. In a multicenter double-blind, placebocontrolled trial, zoledronic acid was found to reduce the risk of vertebral fractures in men with primary or hypogonadism-associated osteoporosis by 67% (1.6% vertebral fractures in the treatment group after 24 months vs 4.9% with placebo).29 Given within 90 days of a hip fracture repair, zoledronic acid was associated with both a reduction in the rate of new fractures and an increased survival rate.30
Adverse effects of zoledronic acid include diffuse bone pain (3%-9%), fever (9%-22%) and flu-like symptoms (1%-11%). Osteonecrosis of the jaw has been reported in <1% of patients.23
Recombinant human parathyroid hormone stimulates bone growth
Teriparatide, administered subcutaneously (SC) once a day, directly stimulates bone formation. In a randomized placebo controlled trial of 437 men with a T-score of -2, teriparatide was found to increase BMD at the spine and femoral neck. Participants were randomized to receive teriparatide (20 or 40 mcg/d) or placebo. Those who received teriparatide had a doserelated increase in BMD from baseline at the spine (5.9% with 20 mcg and 9% with 40 mcg) and femoral neck (1.5% and 2.9%, respectively) compared with the placebo group.31 Teriparatide was shown to reduce vertebral fractures by 51% compared with placebo in a randomized study of 355 men with osteoporosis.32
Teriparatide is indicated for men with severe osteoporosis and those for whom bisphosphonate treatment has been unsuccessful. Its use is limited to 2 years due to a dose-dependent risk of osteosarcoma. Teriparatide is contraindicated in patients with skeletal metastasis and has been associated with transient hypercalcemia 4 to 6 hours after administration.23 Its use in combination with bisphosphonates is not recommended due to the lack of proven benefit, risk of adverse effects, and associated cost.5
Testosterone boosts bone density
Testosterone therapy is recommended for men with low levels of testosterone (<200 ng/dL), high risk for fracture, and contraindications to pharmacologic agents approved for the treatment of osteoporosis.5 Supplementation of testosterone to restore correct physiologic levels will decrease bone turnover and increase bone density.33 In a meta-analysis of 8 trials with a total of 365 participants, testosterone administered intramuscularly was found to increase lumbar BMD by 8% compared with placebo. The effect on fractures is not known.12
• Although US women are 4 times more likely than men to suffer from osteoporosis, men incur between 30% and 40% of osteoporotic fractures.
• Men who sustain hip fractures have a mortality rate of up to 37.5%—2 to 3 times that of women with hip fractures.
• Men treated with androgen deprivation therapy face an increased risk of osteoporosis.
• About 13% of white men older than 50 years will experience at least one osteoporotic fracture in their lifetime.
• The Endocrine Society, American College of Physicians, and National Osteoporosis Foundation recommend screening all men ages 70 years or older—and younger men with risk factors for fracture and/or a history of fracture after age 50—for osteoporosis.
Monoclonal antibody reduces fracture risk
Denosumab, a monoclonal antibody that prevents osteoclast formation leading to decreased bone resorption, is administered SC every 6 months.23 In a placebo-controlled trial of 242 men with low bone mass, denosumab increased BMD at the lumbar spine (5.7%), total hip (2.4%), femoral neck (2.1%), trochanter (3.1%), and one-third radius (0.6%) compared with placebo after one year.34 In men receiving androgen deprivation therapy for nonmetastatic prostate cancer, denosumab has been shown to increase BMD and reduce the incidence of vertebral fractures.35
Adverse effects include hypocalcemia, hypophosphatemia, fatigue, and back pain.23 No data exist on the ability of denosumab to reduce fracture risk in men without androgen deprivation.
Calcium and vitamin D for men at risk
Men who are at risk for or have osteoporosis should consume 1000 mg to 1200 mg of calcium per day. Ideally, this should come through dietary sources, but calcium supplementation may be added when diet is inadequate.5 The Institute of Medicine recommends a calcium intake of 1000 mg/d for men ages 51 to 70 years and 1200 mg/d for men ages 70 and older.36
Men with vitamin D levels below 30 ng/mL should receive vitamin D supplementation to attain blood 25(OH) D levels of at least 30 ng/mL.5 The Institute of Medicine recommends a daily intake of 600 international units (IU) of vitamin D for men ages 51 to 70 and 800 IU for men 70 and older.36 A recent Cochrane review on vitamin D and vitamin D analogues concluded that vitamin D alone was unlikely to prevent fractures in older people; when taken with calcium, however, it may have a preventive effect.37
Counseling and follow-up
Lifestyle modification is an important means of primary prevention for osteoporosis. Advise men at risk for osteoporosis to limit alcohol consumption to 2 drinks daily.4,5,8,10 Tell those who smoke that doing so increases their risk for osteoporotic fracture and refer them for smoking cessation counseling. Emphasize that weight-bearing exercise can improve BMD and should be done at least 3 days per week.4,5,8,10 It is important, too, to do a medication review to look for drug-drug interactions and to discuss fall prevention strategies, such as gait training and an environmental assessment and removal of fall hazards.
The evidence for monitoring treatment using BMD is not very strong.5,14 However, the Endocrine Society recommends that response to treatment be monitored using DEXA scans every one to 2 years, with reduced frequency once the BMD has stabilized.5 Any patient found to have a decrease in BMD after treatment is initiated should undergo further evaluation to determine the cause of the decline.
CORRESPONDENCE
Bryan Farford, DO, Mayo Clinic Division of Regional Medicine, 742 Marsh Landing Parkway, Jacksonville Beach, FL 32250; [email protected]
1. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
2. Bliuc D, Nguyen ND, Milch VE, et al. Mortality risk associated with low-trauma osteoporotic fracture and subsequent fracture in men and women. JAMA. 2009;301:513-521.
3. Gennari L, Bilezikian JP. Osteoporosis in men. Endocrinol Metab Clin North Am. 2007;36:399-419.
4. Ebeling PR. Clinical practice. Osteoporosis in men. N Engl J Med. 2008;358:1474-1482.
5. Watts NB, Adler RA, Bilezikian JP, et al; Endocrine Society. Osteoporosis in men: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:1802-1822.
6. Memon A, Pospula WM, Tantawy AY, et al. Incidence of hip fracture in Kuwait. Int J Epidemiol. 1998;27:860-865.
7. Maggi S, Kelsey JL, Litvak J, et al. Incidence of hip fractures in the elderly: a cross-national analysis. Osteoporos Int. 1991;1:232-241.
8. Rao SS, Budhwar N, Ashfaque A. Osteoporosis in men. Am Fam Physician. 2010;82:503-508.
9. Johnell O, Kanis J. Epidemiology of osteoporotic fractures. Osteoporos Int. 2005;16 (Suppl 2):S3-S7.
10. National Institutes of Health. NIH osteoporosis and related bone diseases national resource center. Osteoporosis in men. January 2012. National Institutes of Health Web site. Available at: http://www.niams.nih.gov/health_info/bone/osteoporosis/men.asp. Accessed April 22, 2015.
11. Bruder JM, Ma JZ, Basler JW, et al. Prevalence of osteopenia and osteoporosis by central and peripheral bone mineral density in men with prostate cancer during androgen-deprivation therapy. Urology. 2006;67:152-155.
12. Tracz MJ, Sideras K, Boloña ER, et al. Testosterone use in men and its effects on bone health. A systematic review and meta-analysis of randomized placebo-controlled trials. J Clin Endocrinol Metab. 2006;91:2011-2016.
13. World Health Organization. WHO scientific group on the assessment of osteoporosis at primary health care level. Summary meeting report. Geneva, Switzerland: World Health Organization. 2007. Available at: http://who.int/chp/topics/Osteoporosis.pdf. Accessed April 22, 2015.
14. The International Society for Clinical Densitometry. 2007 official positions & pediatric official positions of The International Society for Clinical Densitometry. The International Society for Clinical Densitometry Web site. Available at: http://www.iscd.org/wp-content/uploads/2012/10/ISCD2007OfficialPositions-Combined-AdultandPediatric.pdf. Accessed August 11, 2015.
15. U.S. Preventive Services Task Force. Screening for osteoporosis: U.S. preventive services task force recommendation statement. Ann Intern Med. 2011;154:356-364.
16. Qaseem A, Snow V, Shekelle P, et al; Clinical Efficacy Assessment Subcommittee of the American College of Physicians. Screening for osteoporosis in men: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;148:680-684.
17. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. National Osteoporosis Foundation Web site. Washington, DC: 2014. Available at: http://nof.org/files/nof/public/content/file/2791/upload/919.pdf. Accessed April 22, 2015.
18. Shepherd AJ, Cass AR, Carlson CA, et al. Development and internal validation of the male osteoporosis risk estimation score. Ann Fam Med. 2007;5:540-546.
19. Lynn HS, Woo J, Leung PC, et al; Osteoporotic Fractures in Men (MrOS) Study. An evaluation of osteoporosis screening tools for the osteoporotic fractures in men (MrOS) study. Osteoporos Int. 2008;19:1087-1092.
20. Adler RA, Tran MT, Petkov VI. Performance of the osteoporosis self-assessment screening tool for osteoporosis in American men. Mayo Clin Proc. 2003;78:723-727.
21. International Osteoporosis Foundation, The International Society for Clinical Densitometry. 2010 Official Positions on FRAX®. International Osteoporosis Foundation Web site. Available at: http://www.iofbonehealth.org/sites/default/files/PDFs/2010_Official_%20Positions_%20ISCD-IOF_%20FRAX.pdf. Accessed March 21, 2015.
22. Epocrates essentials. Epocrates Web site. Available at: www.epocrates.com. Accessed April 17, 2015.
23. American Pharmacist Association. Drug information handbook: a comprehensive resource for all clinicians and healthcare professionals. 21st ed. Alphen aan den Rijn, The Netherlands: Lexi-Comp, Inc. Wolters Kluwer; 2012-2013.
24. Orwoll E, Ettinger M, Weiss S, et al. Alendronate for the treatment of osteoporosis in men. N Engl J Med. 2000;343:604-610.
25. Ringe JD, Dorst A, Faber H, et al. Alendronate treatment of established primary osteoporosis in men: 3-year results of a prospective, comparative, two-arm study. Rheumatol Int. 2004;24:110-113.
26. Ringe JD, Faber H, Farahmand P, et al. Efficacy of risedronate in men with primary and secondary osteoporosis: results of a 1-year study. Rheumatol Int. 2006;26:427-431.
27. Boonen S, Orwoll ES, Wenderoth D, et al. Once-weekly risedronate in men with osteoporosis: results of a 2-year, placebocontrolled, double-blind, multicenter study. J Bone Miner Res. 2009;24:719-725.
28. Khosla S, Amin S, Orwoll E. Osteoporosis in men. Endocr Rev. 2008;29:441-464.
29. Boonen S, Reginster JY, Kaufman JM, et al. Fracture risk and zoledronic acid therapy in men with osteoporosis. N Engl J Med. 2012;367:1714-1723.
30. Lyles KW, Colón-Emeric CS, Magaziner JS, et al; HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
31. Orwoll ES, Scheele WH, Paul S, et al. The effect of teriparatide [human parathyroid hormone (1-34)] therapy on bone density in men with osteoporosis. J Bone Miner Res. 2003;18:9-17.
32. Kaufman JM, Orwoll E, Goemaere S, et al. Teriparatide effects on vertebral fractures and bone mineral density in men with osteoporosis: treatment and discontinuation of therapy. Osteoporos Int. 2005;16:510-516.
33. Snyder PJ, Peachey H, Hannoush P, et al. Effect of testosterone treatment on bone mineral density in men over 65 years of age. J Clin Endocrinol Metab. 1999;84:1966-1972.
34. Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab. 2012;97:3161-3169.
35. Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med. 2009;361:745-755.
36. Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Institute of Medicine. Dietary reference intakes for calcium and vitamin D. Institute of Medicine Web site. Available at: http://www.iom.edu/reports/2010/dietary-reference-intakes-for-calcium-and-vitamin-d.aspx. Accessed April 10, 2015.
37. Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227.
› Order dual-energy x-ray absorptiometry of the spine and hip for men who are at increased risk for osteoporosis and candidates for pharmacotherapy. C
› Prescribe bisphosphonates for men with osteoporosis to reduce the risk of vertebral fractures. A
› Advise men who have, or are at risk for, osteoporosis to consume 1000 to 1200 mg of calcium and 600 to 800 IU of vitamin D daily. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
With older women in the United States about 4 times more likely than their male counterparts to develop osteoporosis,1,2 physicians often fail to screen for—or to treat—low bone mass in men. There are plenty of reasons why they should.
First and foremost: Osteoporosis is a leading cause of morbidity and mortality in the elderly.3 An estimated 8.8 million American men suffer from osteoporosis or osteopenia.3 And, although only about 20% of osteoporosis patients are male, men sustain between 30% and 40% of osteoporotic fractures.1,2 What’s more, hip fracture in men has a mortality rate of up to 37.5%—2 to 3 times higher than that of women with hip fracture.4,5
Clearly, then, it is crucial to be aware of the risks of osteoporosis faced by both men and women as they age. Here’s a look at what to consider, when to screen, and how to treat male patients who have, or are at risk for, osteoporosis.
Which men are at risk?
The incidence of fractures secondary to osteoporosis varies with race/ethnicity and geography. The highest rates worldwide occur in Scandinavia and among Caucasians in the United States; black, Asian, and Hispanic populations have the lowest rates.6,7 As with women, the risk of osteoporotic fracture in men increases with age. However, the peak incidence of fracture occurs about 10 years later in men than in women, starting at about age 70.8 Approximately 13% of white men older than 50 years will experience at least one osteoporotic fracture.9
There are 2 main types of osteoporosis: primary and secondary. Up to 40% of osteoporosis in men is primary,4 with bone loss due either to age (senile osteoporosis) or to an unknown cause (idiopathic osteoporosis).10 For men 70 years or older, osteoporosis is assumed to be age related. Idiopathic osteoporosis is diagnosed only in men younger than 70 who have no obvious secondary cause.10 There are numerous secondary causes, however, and most men with bone loss have at least one.4
Common secondary causes: Lifestyle, medical conditions, and meds
The most common causes of secondary osteoporosis in men are exposure to glucocorticoids, primary or secondary hypogonadism (low testosterone), diabetes, alcohol abuse, smoking, gastrointestinal (GI) disease, hypercalciuria, low body weight (body mass index <20 kg/m2), and immobility (TABLE 1).4,5,8,10
Chronic use of corticosteroids, often used to treat chronic obstructive pulmonary disease (COPD), asthma, and rheumatoid arthritis, directly affects the bone, decreasing skeletal muscle, increasing immobility, and reducing intestinal absorption of calcium as well as serum testosterone levels.10 Men with androgen deficiency (which may be due to androgen deprivation therapy to treat prostate cancer) or chronic use of opioids are also at increased risk.4,5,10-12
Diagnostic screening and criteria
The World Health Organization has established diagnostic criteria for osteoporosis using bone mineral density (BMD), reported as both T-scores and Z-scores as measured on dual-energy x-ray absorptiometry (DEXA) scan.13 The T-score represents the number of standard deviations above or below the mean BMD for young adults, matched for sex and race, but not age. It classifies individuals into 3 categories: normal; low (osteopenia), with a T-score between -1 and -2.5; and osteoporosis (T-score ≤-2.5).4,14 The Z-score indicates the number of standard deviations above or below the mean for age, as well as sex and race. A Z-score of ≤-2.0 is below the expected range, indicating an increased likelihood of a secondary form of osteoporosis.14
Which men to screen?
The US Preventive Services Task Force has concluded that evidence is insufficient to assess the balance of benefits and harms of screening for osteoporosis in men. It therefore makes no recommendation to screen men who don't have evidence of previous fractures or secondary causes of osteoporosis.15
Other organizations agree that there is insufficient evidence to recommend routine screening for men without known osteoporotic fractures or secondary causes for osteoporosis. There are, however, some guidelines that are useful in clinical practice.
The Endocrine Society, American College of Physicians (ACP), and National Osteoporosis Foundation (NOF) recommend screening men ages 70 years or older, and men ages 50 to 69 who have risk factors for fracture and/or a history of fracture sustained after age 50.5,16,17 (See “Did you know?”)1,2,4,5,9-12,16,17 Prior to screening, it is important to do a complete medical history and physical examination.
Screening considerations. The Endocrine Society, ACP, and NOF recommend a DEXA scan of the spine and hip for men who are at increased risk for osteoporosis and have no contraindications to drug therapy.5,16,17 In patients who have degenerative changes of the spine and hip that would likely obscure DEXA outcomes, a scan of the radius may provide a more accurate assessment of bone status. Men receiving androgen deprivation therapy for prostate cancer will have a greater decline of bone density in the radius than in the hip or spine and are therefore ideal candidates for DEXA of the forearm, as well.5,11 Keep in mind, however, that no studies have looked at how well, or whether, men with osteoporosis measured only in the radius respond to treatment.5
A DEXA scan is not always widely available, nor is it a perfect predictor of fracture risk. In addition, it is not always cost effective. For some patients, the use of a validated clinical predictive tool is preferable as an initial option.
The Male Osteoporosis Risk Estimation Score (MORES) uses age, weight, and history of COPD to identify men 60 years or older who are at risk for osteoporosis (TABLE 2).18 The score can be easily calculated during a clinical encounter and is beneficial for identifying men who should be referred for DEXA scan. A score of ≥6 has been found to yield an overall sensitivity of 0.93 (95% confidence interval [CI], 0.85-0.97) and a specificity of 0.59 (95% CI, 0.56-0.62), with a number needed to screen to prevent one additional hip fracture of 279.18
The Osteoporosis Self-assessment Tool (OST) (http://depts.washington.edu/osteoed/tools.php?type=ost) is a calculated value that uses age and weight to determine an individual’s risk for osteoporosis (risk score=weight [in kg] – age [in years]/5).16,19 Although there is not a defined value to determine a positive OST risk score, scores of -1 to 3 have been used in a variety of studies.16 In a study of 181 American men, the OST predicted osteoporosis with a sensitivity of 93% and a specificity of 66% when using a cutoff score of 3.20
Treating men at risk
Pharmacologic therapy is recommended for men at an increased risk for fracture. This includes men who have had a hip or vertebral fracture without major trauma, as well as those who have not had such a fracture but have a BMD of the spine, femoral neck, and/or total hip of ≤-2.5.5,17 This standard also applies to the radius when used as an alternative site.
The International Society for Clinical Densitometry and International Osteoporosis Foundation endorse the use of the Fracture Risk Assessment Tool (FRAX). Available at http://shef.ac.uk/FRAX/tool.aspx?country=9, FRAX is a computer-based calculator that uses risk factors and BMD of the femoral neck to estimate an individual’s 10-year fracture probability.21 Men who are 50 years or older, have a T-score between -1.0 and -2.5 in the spine, femoral neck, or total hip, and a 10-year risk of ≥20% of developing any fracture or ≥3% of developing a hip fracture based on FRAX, should be offered pharmacotherapy.5,17
Bisphosphonates are first-line therapy
Although oral bisphosphonates are first-line therapy for men who meet these criteria,4 pharmacotherapy should be individualized based on factors such as fracture history, severity of osteoporosis, comorbidities (eg, peptic ulcer disease, malignancy, renal disease, or malabsorption), and cost (TABLE 3).22,23
Alendronate once weekly has been proven to increase BMD and to reduce the risk of fracture in men.24,25 A randomized, placebo-controlled trial of 241 men with osteoporosis found that alendronate increased BMD by 7.1% (±0.3) at the lumbar spine, 2.5% (±0.4) at the femoral neck, and 2% (±0.2) for the total body. Those in the placebo group had a 1.8% (±0.5) increase in BMD of the lumbar spine, with no significant change in femoral neck or total-body BMD—and a higher incidence of vertebral fractures (7.1% vs. 0.8% for those on alendronate; P=.02).24
Risedronate once daily has also been proven to increase BMD in the lumbar spine and hip, with a reduction in vertebral fractures.26 Another investigation—a 2-year, multicenter double-blind placebo-controlled study of 284 men with osteoporosis—found that risedronate given once a week increased BMD in the spine and hip, but did not reduce the incidence of either vertebral or nonvertebral fractures.27
Both alendronate and risedronate are effective for secondary causes of bone loss, such as corticosteroid use, androgen deprivation therapy/hypogonadism, and rheumatologic conditions.28 Oral bisphosphonates may cause GI irritation, however. Abdominal pain associated with alendronate use is between 1% and 7%, vs 2% to 12% for risedronate.23 Neither medication is recommended for use in patients with an estimated glomerular filtration rate <35 mL/min.23 There is no clearly established duration of therapy for men.
Zoledronic acid infusions, given intravenously (IV) once a year, are available for men who cannot tolerate oral bisphosphonates. In a multicenter double-blind, placebocontrolled trial, zoledronic acid was found to reduce the risk of vertebral fractures in men with primary or hypogonadism-associated osteoporosis by 67% (1.6% vertebral fractures in the treatment group after 24 months vs 4.9% with placebo).29 Given within 90 days of a hip fracture repair, zoledronic acid was associated with both a reduction in the rate of new fractures and an increased survival rate.30
Adverse effects of zoledronic acid include diffuse bone pain (3%-9%), fever (9%-22%) and flu-like symptoms (1%-11%). Osteonecrosis of the jaw has been reported in <1% of patients.23
Recombinant human parathyroid hormone stimulates bone growth
Teriparatide, administered subcutaneously (SC) once a day, directly stimulates bone formation. In a randomized placebo controlled trial of 437 men with a T-score of -2, teriparatide was found to increase BMD at the spine and femoral neck. Participants were randomized to receive teriparatide (20 or 40 mcg/d) or placebo. Those who received teriparatide had a doserelated increase in BMD from baseline at the spine (5.9% with 20 mcg and 9% with 40 mcg) and femoral neck (1.5% and 2.9%, respectively) compared with the placebo group.31 Teriparatide was shown to reduce vertebral fractures by 51% compared with placebo in a randomized study of 355 men with osteoporosis.32
Teriparatide is indicated for men with severe osteoporosis and those for whom bisphosphonate treatment has been unsuccessful. Its use is limited to 2 years due to a dose-dependent risk of osteosarcoma. Teriparatide is contraindicated in patients with skeletal metastasis and has been associated with transient hypercalcemia 4 to 6 hours after administration.23 Its use in combination with bisphosphonates is not recommended due to the lack of proven benefit, risk of adverse effects, and associated cost.5
Testosterone boosts bone density
Testosterone therapy is recommended for men with low levels of testosterone (<200 ng/dL), high risk for fracture, and contraindications to pharmacologic agents approved for the treatment of osteoporosis.5 Supplementation of testosterone to restore correct physiologic levels will decrease bone turnover and increase bone density.33 In a meta-analysis of 8 trials with a total of 365 participants, testosterone administered intramuscularly was found to increase lumbar BMD by 8% compared with placebo. The effect on fractures is not known.12
• Although US women are 4 times more likely than men to suffer from osteoporosis, men incur between 30% and 40% of osteoporotic fractures.
• Men who sustain hip fractures have a mortality rate of up to 37.5%—2 to 3 times that of women with hip fractures.
• Men treated with androgen deprivation therapy face an increased risk of osteoporosis.
• About 13% of white men older than 50 years will experience at least one osteoporotic fracture in their lifetime.
• The Endocrine Society, American College of Physicians, and National Osteoporosis Foundation recommend screening all men ages 70 years or older—and younger men with risk factors for fracture and/or a history of fracture after age 50—for osteoporosis.
Monoclonal antibody reduces fracture risk
Denosumab, a monoclonal antibody that prevents osteoclast formation leading to decreased bone resorption, is administered SC every 6 months.23 In a placebo-controlled trial of 242 men with low bone mass, denosumab increased BMD at the lumbar spine (5.7%), total hip (2.4%), femoral neck (2.1%), trochanter (3.1%), and one-third radius (0.6%) compared with placebo after one year.34 In men receiving androgen deprivation therapy for nonmetastatic prostate cancer, denosumab has been shown to increase BMD and reduce the incidence of vertebral fractures.35
Adverse effects include hypocalcemia, hypophosphatemia, fatigue, and back pain.23 No data exist on the ability of denosumab to reduce fracture risk in men without androgen deprivation.
Calcium and vitamin D for men at risk
Men who are at risk for or have osteoporosis should consume 1000 mg to 1200 mg of calcium per day. Ideally, this should come through dietary sources, but calcium supplementation may be added when diet is inadequate.5 The Institute of Medicine recommends a calcium intake of 1000 mg/d for men ages 51 to 70 years and 1200 mg/d for men ages 70 and older.36
Men with vitamin D levels below 30 ng/mL should receive vitamin D supplementation to attain blood 25(OH) D levels of at least 30 ng/mL.5 The Institute of Medicine recommends a daily intake of 600 international units (IU) of vitamin D for men ages 51 to 70 and 800 IU for men 70 and older.36 A recent Cochrane review on vitamin D and vitamin D analogues concluded that vitamin D alone was unlikely to prevent fractures in older people; when taken with calcium, however, it may have a preventive effect.37
Counseling and follow-up
Lifestyle modification is an important means of primary prevention for osteoporosis. Advise men at risk for osteoporosis to limit alcohol consumption to 2 drinks daily.4,5,8,10 Tell those who smoke that doing so increases their risk for osteoporotic fracture and refer them for smoking cessation counseling. Emphasize that weight-bearing exercise can improve BMD and should be done at least 3 days per week.4,5,8,10 It is important, too, to do a medication review to look for drug-drug interactions and to discuss fall prevention strategies, such as gait training and an environmental assessment and removal of fall hazards.
The evidence for monitoring treatment using BMD is not very strong.5,14 However, the Endocrine Society recommends that response to treatment be monitored using DEXA scans every one to 2 years, with reduced frequency once the BMD has stabilized.5 Any patient found to have a decrease in BMD after treatment is initiated should undergo further evaluation to determine the cause of the decline.
CORRESPONDENCE
Bryan Farford, DO, Mayo Clinic Division of Regional Medicine, 742 Marsh Landing Parkway, Jacksonville Beach, FL 32250; [email protected]
› Order dual-energy x-ray absorptiometry of the spine and hip for men who are at increased risk for osteoporosis and candidates for pharmacotherapy. C
› Prescribe bisphosphonates for men with osteoporosis to reduce the risk of vertebral fractures. A
› Advise men who have, or are at risk for, osteoporosis to consume 1000 to 1200 mg of calcium and 600 to 800 IU of vitamin D daily. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
With older women in the United States about 4 times more likely than their male counterparts to develop osteoporosis,1,2 physicians often fail to screen for—or to treat—low bone mass in men. There are plenty of reasons why they should.
First and foremost: Osteoporosis is a leading cause of morbidity and mortality in the elderly.3 An estimated 8.8 million American men suffer from osteoporosis or osteopenia.3 And, although only about 20% of osteoporosis patients are male, men sustain between 30% and 40% of osteoporotic fractures.1,2 What’s more, hip fracture in men has a mortality rate of up to 37.5%—2 to 3 times higher than that of women with hip fracture.4,5
Clearly, then, it is crucial to be aware of the risks of osteoporosis faced by both men and women as they age. Here’s a look at what to consider, when to screen, and how to treat male patients who have, or are at risk for, osteoporosis.
Which men are at risk?
The incidence of fractures secondary to osteoporosis varies with race/ethnicity and geography. The highest rates worldwide occur in Scandinavia and among Caucasians in the United States; black, Asian, and Hispanic populations have the lowest rates.6,7 As with women, the risk of osteoporotic fracture in men increases with age. However, the peak incidence of fracture occurs about 10 years later in men than in women, starting at about age 70.8 Approximately 13% of white men older than 50 years will experience at least one osteoporotic fracture.9
There are 2 main types of osteoporosis: primary and secondary. Up to 40% of osteoporosis in men is primary,4 with bone loss due either to age (senile osteoporosis) or to an unknown cause (idiopathic osteoporosis).10 For men 70 years or older, osteoporosis is assumed to be age related. Idiopathic osteoporosis is diagnosed only in men younger than 70 who have no obvious secondary cause.10 There are numerous secondary causes, however, and most men with bone loss have at least one.4
Common secondary causes: Lifestyle, medical conditions, and meds
The most common causes of secondary osteoporosis in men are exposure to glucocorticoids, primary or secondary hypogonadism (low testosterone), diabetes, alcohol abuse, smoking, gastrointestinal (GI) disease, hypercalciuria, low body weight (body mass index <20 kg/m2), and immobility (TABLE 1).4,5,8,10
Chronic use of corticosteroids, often used to treat chronic obstructive pulmonary disease (COPD), asthma, and rheumatoid arthritis, directly affects the bone, decreasing skeletal muscle, increasing immobility, and reducing intestinal absorption of calcium as well as serum testosterone levels.10 Men with androgen deficiency (which may be due to androgen deprivation therapy to treat prostate cancer) or chronic use of opioids are also at increased risk.4,5,10-12
Diagnostic screening and criteria
The World Health Organization has established diagnostic criteria for osteoporosis using bone mineral density (BMD), reported as both T-scores and Z-scores as measured on dual-energy x-ray absorptiometry (DEXA) scan.13 The T-score represents the number of standard deviations above or below the mean BMD for young adults, matched for sex and race, but not age. It classifies individuals into 3 categories: normal; low (osteopenia), with a T-score between -1 and -2.5; and osteoporosis (T-score ≤-2.5).4,14 The Z-score indicates the number of standard deviations above or below the mean for age, as well as sex and race. A Z-score of ≤-2.0 is below the expected range, indicating an increased likelihood of a secondary form of osteoporosis.14
Which men to screen?
The US Preventive Services Task Force has concluded that evidence is insufficient to assess the balance of benefits and harms of screening for osteoporosis in men. It therefore makes no recommendation to screen men who don't have evidence of previous fractures or secondary causes of osteoporosis.15
Other organizations agree that there is insufficient evidence to recommend routine screening for men without known osteoporotic fractures or secondary causes for osteoporosis. There are, however, some guidelines that are useful in clinical practice.
The Endocrine Society, American College of Physicians (ACP), and National Osteoporosis Foundation (NOF) recommend screening men ages 70 years or older, and men ages 50 to 69 who have risk factors for fracture and/or a history of fracture sustained after age 50.5,16,17 (See “Did you know?”)1,2,4,5,9-12,16,17 Prior to screening, it is important to do a complete medical history and physical examination.
Screening considerations. The Endocrine Society, ACP, and NOF recommend a DEXA scan of the spine and hip for men who are at increased risk for osteoporosis and have no contraindications to drug therapy.5,16,17 In patients who have degenerative changes of the spine and hip that would likely obscure DEXA outcomes, a scan of the radius may provide a more accurate assessment of bone status. Men receiving androgen deprivation therapy for prostate cancer will have a greater decline of bone density in the radius than in the hip or spine and are therefore ideal candidates for DEXA of the forearm, as well.5,11 Keep in mind, however, that no studies have looked at how well, or whether, men with osteoporosis measured only in the radius respond to treatment.5
A DEXA scan is not always widely available, nor is it a perfect predictor of fracture risk. In addition, it is not always cost effective. For some patients, the use of a validated clinical predictive tool is preferable as an initial option.
The Male Osteoporosis Risk Estimation Score (MORES) uses age, weight, and history of COPD to identify men 60 years or older who are at risk for osteoporosis (TABLE 2).18 The score can be easily calculated during a clinical encounter and is beneficial for identifying men who should be referred for DEXA scan. A score of ≥6 has been found to yield an overall sensitivity of 0.93 (95% confidence interval [CI], 0.85-0.97) and a specificity of 0.59 (95% CI, 0.56-0.62), with a number needed to screen to prevent one additional hip fracture of 279.18
The Osteoporosis Self-assessment Tool (OST) (http://depts.washington.edu/osteoed/tools.php?type=ost) is a calculated value that uses age and weight to determine an individual’s risk for osteoporosis (risk score=weight [in kg] – age [in years]/5).16,19 Although there is not a defined value to determine a positive OST risk score, scores of -1 to 3 have been used in a variety of studies.16 In a study of 181 American men, the OST predicted osteoporosis with a sensitivity of 93% and a specificity of 66% when using a cutoff score of 3.20
Treating men at risk
Pharmacologic therapy is recommended for men at an increased risk for fracture. This includes men who have had a hip or vertebral fracture without major trauma, as well as those who have not had such a fracture but have a BMD of the spine, femoral neck, and/or total hip of ≤-2.5.5,17 This standard also applies to the radius when used as an alternative site.
The International Society for Clinical Densitometry and International Osteoporosis Foundation endorse the use of the Fracture Risk Assessment Tool (FRAX). Available at http://shef.ac.uk/FRAX/tool.aspx?country=9, FRAX is a computer-based calculator that uses risk factors and BMD of the femoral neck to estimate an individual’s 10-year fracture probability.21 Men who are 50 years or older, have a T-score between -1.0 and -2.5 in the spine, femoral neck, or total hip, and a 10-year risk of ≥20% of developing any fracture or ≥3% of developing a hip fracture based on FRAX, should be offered pharmacotherapy.5,17
Bisphosphonates are first-line therapy
Although oral bisphosphonates are first-line therapy for men who meet these criteria,4 pharmacotherapy should be individualized based on factors such as fracture history, severity of osteoporosis, comorbidities (eg, peptic ulcer disease, malignancy, renal disease, or malabsorption), and cost (TABLE 3).22,23
Alendronate once weekly has been proven to increase BMD and to reduce the risk of fracture in men.24,25 A randomized, placebo-controlled trial of 241 men with osteoporosis found that alendronate increased BMD by 7.1% (±0.3) at the lumbar spine, 2.5% (±0.4) at the femoral neck, and 2% (±0.2) for the total body. Those in the placebo group had a 1.8% (±0.5) increase in BMD of the lumbar spine, with no significant change in femoral neck or total-body BMD—and a higher incidence of vertebral fractures (7.1% vs. 0.8% for those on alendronate; P=.02).24
Risedronate once daily has also been proven to increase BMD in the lumbar spine and hip, with a reduction in vertebral fractures.26 Another investigation—a 2-year, multicenter double-blind placebo-controlled study of 284 men with osteoporosis—found that risedronate given once a week increased BMD in the spine and hip, but did not reduce the incidence of either vertebral or nonvertebral fractures.27
Both alendronate and risedronate are effective for secondary causes of bone loss, such as corticosteroid use, androgen deprivation therapy/hypogonadism, and rheumatologic conditions.28 Oral bisphosphonates may cause GI irritation, however. Abdominal pain associated with alendronate use is between 1% and 7%, vs 2% to 12% for risedronate.23 Neither medication is recommended for use in patients with an estimated glomerular filtration rate <35 mL/min.23 There is no clearly established duration of therapy for men.
Zoledronic acid infusions, given intravenously (IV) once a year, are available for men who cannot tolerate oral bisphosphonates. In a multicenter double-blind, placebocontrolled trial, zoledronic acid was found to reduce the risk of vertebral fractures in men with primary or hypogonadism-associated osteoporosis by 67% (1.6% vertebral fractures in the treatment group after 24 months vs 4.9% with placebo).29 Given within 90 days of a hip fracture repair, zoledronic acid was associated with both a reduction in the rate of new fractures and an increased survival rate.30
Adverse effects of zoledronic acid include diffuse bone pain (3%-9%), fever (9%-22%) and flu-like symptoms (1%-11%). Osteonecrosis of the jaw has been reported in <1% of patients.23
Recombinant human parathyroid hormone stimulates bone growth
Teriparatide, administered subcutaneously (SC) once a day, directly stimulates bone formation. In a randomized placebo controlled trial of 437 men with a T-score of -2, teriparatide was found to increase BMD at the spine and femoral neck. Participants were randomized to receive teriparatide (20 or 40 mcg/d) or placebo. Those who received teriparatide had a doserelated increase in BMD from baseline at the spine (5.9% with 20 mcg and 9% with 40 mcg) and femoral neck (1.5% and 2.9%, respectively) compared with the placebo group.31 Teriparatide was shown to reduce vertebral fractures by 51% compared with placebo in a randomized study of 355 men with osteoporosis.32
Teriparatide is indicated for men with severe osteoporosis and those for whom bisphosphonate treatment has been unsuccessful. Its use is limited to 2 years due to a dose-dependent risk of osteosarcoma. Teriparatide is contraindicated in patients with skeletal metastasis and has been associated with transient hypercalcemia 4 to 6 hours after administration.23 Its use in combination with bisphosphonates is not recommended due to the lack of proven benefit, risk of adverse effects, and associated cost.5
Testosterone boosts bone density
Testosterone therapy is recommended for men with low levels of testosterone (<200 ng/dL), high risk for fracture, and contraindications to pharmacologic agents approved for the treatment of osteoporosis.5 Supplementation of testosterone to restore correct physiologic levels will decrease bone turnover and increase bone density.33 In a meta-analysis of 8 trials with a total of 365 participants, testosterone administered intramuscularly was found to increase lumbar BMD by 8% compared with placebo. The effect on fractures is not known.12
• Although US women are 4 times more likely than men to suffer from osteoporosis, men incur between 30% and 40% of osteoporotic fractures.
• Men who sustain hip fractures have a mortality rate of up to 37.5%—2 to 3 times that of women with hip fractures.
• Men treated with androgen deprivation therapy face an increased risk of osteoporosis.
• About 13% of white men older than 50 years will experience at least one osteoporotic fracture in their lifetime.
• The Endocrine Society, American College of Physicians, and National Osteoporosis Foundation recommend screening all men ages 70 years or older—and younger men with risk factors for fracture and/or a history of fracture after age 50—for osteoporosis.
Monoclonal antibody reduces fracture risk
Denosumab, a monoclonal antibody that prevents osteoclast formation leading to decreased bone resorption, is administered SC every 6 months.23 In a placebo-controlled trial of 242 men with low bone mass, denosumab increased BMD at the lumbar spine (5.7%), total hip (2.4%), femoral neck (2.1%), trochanter (3.1%), and one-third radius (0.6%) compared with placebo after one year.34 In men receiving androgen deprivation therapy for nonmetastatic prostate cancer, denosumab has been shown to increase BMD and reduce the incidence of vertebral fractures.35
Adverse effects include hypocalcemia, hypophosphatemia, fatigue, and back pain.23 No data exist on the ability of denosumab to reduce fracture risk in men without androgen deprivation.
Calcium and vitamin D for men at risk
Men who are at risk for or have osteoporosis should consume 1000 mg to 1200 mg of calcium per day. Ideally, this should come through dietary sources, but calcium supplementation may be added when diet is inadequate.5 The Institute of Medicine recommends a calcium intake of 1000 mg/d for men ages 51 to 70 years and 1200 mg/d for men ages 70 and older.36
Men with vitamin D levels below 30 ng/mL should receive vitamin D supplementation to attain blood 25(OH) D levels of at least 30 ng/mL.5 The Institute of Medicine recommends a daily intake of 600 international units (IU) of vitamin D for men ages 51 to 70 and 800 IU for men 70 and older.36 A recent Cochrane review on vitamin D and vitamin D analogues concluded that vitamin D alone was unlikely to prevent fractures in older people; when taken with calcium, however, it may have a preventive effect.37
Counseling and follow-up
Lifestyle modification is an important means of primary prevention for osteoporosis. Advise men at risk for osteoporosis to limit alcohol consumption to 2 drinks daily.4,5,8,10 Tell those who smoke that doing so increases their risk for osteoporotic fracture and refer them for smoking cessation counseling. Emphasize that weight-bearing exercise can improve BMD and should be done at least 3 days per week.4,5,8,10 It is important, too, to do a medication review to look for drug-drug interactions and to discuss fall prevention strategies, such as gait training and an environmental assessment and removal of fall hazards.
The evidence for monitoring treatment using BMD is not very strong.5,14 However, the Endocrine Society recommends that response to treatment be monitored using DEXA scans every one to 2 years, with reduced frequency once the BMD has stabilized.5 Any patient found to have a decrease in BMD after treatment is initiated should undergo further evaluation to determine the cause of the decline.
CORRESPONDENCE
Bryan Farford, DO, Mayo Clinic Division of Regional Medicine, 742 Marsh Landing Parkway, Jacksonville Beach, FL 32250; [email protected]
1. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
2. Bliuc D, Nguyen ND, Milch VE, et al. Mortality risk associated with low-trauma osteoporotic fracture and subsequent fracture in men and women. JAMA. 2009;301:513-521.
3. Gennari L, Bilezikian JP. Osteoporosis in men. Endocrinol Metab Clin North Am. 2007;36:399-419.
4. Ebeling PR. Clinical practice. Osteoporosis in men. N Engl J Med. 2008;358:1474-1482.
5. Watts NB, Adler RA, Bilezikian JP, et al; Endocrine Society. Osteoporosis in men: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:1802-1822.
6. Memon A, Pospula WM, Tantawy AY, et al. Incidence of hip fracture in Kuwait. Int J Epidemiol. 1998;27:860-865.
7. Maggi S, Kelsey JL, Litvak J, et al. Incidence of hip fractures in the elderly: a cross-national analysis. Osteoporos Int. 1991;1:232-241.
8. Rao SS, Budhwar N, Ashfaque A. Osteoporosis in men. Am Fam Physician. 2010;82:503-508.
9. Johnell O, Kanis J. Epidemiology of osteoporotic fractures. Osteoporos Int. 2005;16 (Suppl 2):S3-S7.
10. National Institutes of Health. NIH osteoporosis and related bone diseases national resource center. Osteoporosis in men. January 2012. National Institutes of Health Web site. Available at: http://www.niams.nih.gov/health_info/bone/osteoporosis/men.asp. Accessed April 22, 2015.
11. Bruder JM, Ma JZ, Basler JW, et al. Prevalence of osteopenia and osteoporosis by central and peripheral bone mineral density in men with prostate cancer during androgen-deprivation therapy. Urology. 2006;67:152-155.
12. Tracz MJ, Sideras K, Boloña ER, et al. Testosterone use in men and its effects on bone health. A systematic review and meta-analysis of randomized placebo-controlled trials. J Clin Endocrinol Metab. 2006;91:2011-2016.
13. World Health Organization. WHO scientific group on the assessment of osteoporosis at primary health care level. Summary meeting report. Geneva, Switzerland: World Health Organization. 2007. Available at: http://who.int/chp/topics/Osteoporosis.pdf. Accessed April 22, 2015.
14. The International Society for Clinical Densitometry. 2007 official positions & pediatric official positions of The International Society for Clinical Densitometry. The International Society for Clinical Densitometry Web site. Available at: http://www.iscd.org/wp-content/uploads/2012/10/ISCD2007OfficialPositions-Combined-AdultandPediatric.pdf. Accessed August 11, 2015.
15. U.S. Preventive Services Task Force. Screening for osteoporosis: U.S. preventive services task force recommendation statement. Ann Intern Med. 2011;154:356-364.
16. Qaseem A, Snow V, Shekelle P, et al; Clinical Efficacy Assessment Subcommittee of the American College of Physicians. Screening for osteoporosis in men: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;148:680-684.
17. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. National Osteoporosis Foundation Web site. Washington, DC: 2014. Available at: http://nof.org/files/nof/public/content/file/2791/upload/919.pdf. Accessed April 22, 2015.
18. Shepherd AJ, Cass AR, Carlson CA, et al. Development and internal validation of the male osteoporosis risk estimation score. Ann Fam Med. 2007;5:540-546.
19. Lynn HS, Woo J, Leung PC, et al; Osteoporotic Fractures in Men (MrOS) Study. An evaluation of osteoporosis screening tools for the osteoporotic fractures in men (MrOS) study. Osteoporos Int. 2008;19:1087-1092.
20. Adler RA, Tran MT, Petkov VI. Performance of the osteoporosis self-assessment screening tool for osteoporosis in American men. Mayo Clin Proc. 2003;78:723-727.
21. International Osteoporosis Foundation, The International Society for Clinical Densitometry. 2010 Official Positions on FRAX®. International Osteoporosis Foundation Web site. Available at: http://www.iofbonehealth.org/sites/default/files/PDFs/2010_Official_%20Positions_%20ISCD-IOF_%20FRAX.pdf. Accessed March 21, 2015.
22. Epocrates essentials. Epocrates Web site. Available at: www.epocrates.com. Accessed April 17, 2015.
23. American Pharmacist Association. Drug information handbook: a comprehensive resource for all clinicians and healthcare professionals. 21st ed. Alphen aan den Rijn, The Netherlands: Lexi-Comp, Inc. Wolters Kluwer; 2012-2013.
24. Orwoll E, Ettinger M, Weiss S, et al. Alendronate for the treatment of osteoporosis in men. N Engl J Med. 2000;343:604-610.
25. Ringe JD, Dorst A, Faber H, et al. Alendronate treatment of established primary osteoporosis in men: 3-year results of a prospective, comparative, two-arm study. Rheumatol Int. 2004;24:110-113.
26. Ringe JD, Faber H, Farahmand P, et al. Efficacy of risedronate in men with primary and secondary osteoporosis: results of a 1-year study. Rheumatol Int. 2006;26:427-431.
27. Boonen S, Orwoll ES, Wenderoth D, et al. Once-weekly risedronate in men with osteoporosis: results of a 2-year, placebocontrolled, double-blind, multicenter study. J Bone Miner Res. 2009;24:719-725.
28. Khosla S, Amin S, Orwoll E. Osteoporosis in men. Endocr Rev. 2008;29:441-464.
29. Boonen S, Reginster JY, Kaufman JM, et al. Fracture risk and zoledronic acid therapy in men with osteoporosis. N Engl J Med. 2012;367:1714-1723.
30. Lyles KW, Colón-Emeric CS, Magaziner JS, et al; HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
31. Orwoll ES, Scheele WH, Paul S, et al. The effect of teriparatide [human parathyroid hormone (1-34)] therapy on bone density in men with osteoporosis. J Bone Miner Res. 2003;18:9-17.
32. Kaufman JM, Orwoll E, Goemaere S, et al. Teriparatide effects on vertebral fractures and bone mineral density in men with osteoporosis: treatment and discontinuation of therapy. Osteoporos Int. 2005;16:510-516.
33. Snyder PJ, Peachey H, Hannoush P, et al. Effect of testosterone treatment on bone mineral density in men over 65 years of age. J Clin Endocrinol Metab. 1999;84:1966-1972.
34. Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab. 2012;97:3161-3169.
35. Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med. 2009;361:745-755.
36. Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Institute of Medicine. Dietary reference intakes for calcium and vitamin D. Institute of Medicine Web site. Available at: http://www.iom.edu/reports/2010/dietary-reference-intakes-for-calcium-and-vitamin-d.aspx. Accessed April 10, 2015.
37. Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227.
1. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465-475.
2. Bliuc D, Nguyen ND, Milch VE, et al. Mortality risk associated with low-trauma osteoporotic fracture and subsequent fracture in men and women. JAMA. 2009;301:513-521.
3. Gennari L, Bilezikian JP. Osteoporosis in men. Endocrinol Metab Clin North Am. 2007;36:399-419.
4. Ebeling PR. Clinical practice. Osteoporosis in men. N Engl J Med. 2008;358:1474-1482.
5. Watts NB, Adler RA, Bilezikian JP, et al; Endocrine Society. Osteoporosis in men: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:1802-1822.
6. Memon A, Pospula WM, Tantawy AY, et al. Incidence of hip fracture in Kuwait. Int J Epidemiol. 1998;27:860-865.
7. Maggi S, Kelsey JL, Litvak J, et al. Incidence of hip fractures in the elderly: a cross-national analysis. Osteoporos Int. 1991;1:232-241.
8. Rao SS, Budhwar N, Ashfaque A. Osteoporosis in men. Am Fam Physician. 2010;82:503-508.
9. Johnell O, Kanis J. Epidemiology of osteoporotic fractures. Osteoporos Int. 2005;16 (Suppl 2):S3-S7.
10. National Institutes of Health. NIH osteoporosis and related bone diseases national resource center. Osteoporosis in men. January 2012. National Institutes of Health Web site. Available at: http://www.niams.nih.gov/health_info/bone/osteoporosis/men.asp. Accessed April 22, 2015.
11. Bruder JM, Ma JZ, Basler JW, et al. Prevalence of osteopenia and osteoporosis by central and peripheral bone mineral density in men with prostate cancer during androgen-deprivation therapy. Urology. 2006;67:152-155.
12. Tracz MJ, Sideras K, Boloña ER, et al. Testosterone use in men and its effects on bone health. A systematic review and meta-analysis of randomized placebo-controlled trials. J Clin Endocrinol Metab. 2006;91:2011-2016.
13. World Health Organization. WHO scientific group on the assessment of osteoporosis at primary health care level. Summary meeting report. Geneva, Switzerland: World Health Organization. 2007. Available at: http://who.int/chp/topics/Osteoporosis.pdf. Accessed April 22, 2015.
14. The International Society for Clinical Densitometry. 2007 official positions & pediatric official positions of The International Society for Clinical Densitometry. The International Society for Clinical Densitometry Web site. Available at: http://www.iscd.org/wp-content/uploads/2012/10/ISCD2007OfficialPositions-Combined-AdultandPediatric.pdf. Accessed August 11, 2015.
15. U.S. Preventive Services Task Force. Screening for osteoporosis: U.S. preventive services task force recommendation statement. Ann Intern Med. 2011;154:356-364.
16. Qaseem A, Snow V, Shekelle P, et al; Clinical Efficacy Assessment Subcommittee of the American College of Physicians. Screening for osteoporosis in men: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;148:680-684.
17. National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. National Osteoporosis Foundation Web site. Washington, DC: 2014. Available at: http://nof.org/files/nof/public/content/file/2791/upload/919.pdf. Accessed April 22, 2015.
18. Shepherd AJ, Cass AR, Carlson CA, et al. Development and internal validation of the male osteoporosis risk estimation score. Ann Fam Med. 2007;5:540-546.
19. Lynn HS, Woo J, Leung PC, et al; Osteoporotic Fractures in Men (MrOS) Study. An evaluation of osteoporosis screening tools for the osteoporotic fractures in men (MrOS) study. Osteoporos Int. 2008;19:1087-1092.
20. Adler RA, Tran MT, Petkov VI. Performance of the osteoporosis self-assessment screening tool for osteoporosis in American men. Mayo Clin Proc. 2003;78:723-727.
21. International Osteoporosis Foundation, The International Society for Clinical Densitometry. 2010 Official Positions on FRAX®. International Osteoporosis Foundation Web site. Available at: http://www.iofbonehealth.org/sites/default/files/PDFs/2010_Official_%20Positions_%20ISCD-IOF_%20FRAX.pdf. Accessed March 21, 2015.
22. Epocrates essentials. Epocrates Web site. Available at: www.epocrates.com. Accessed April 17, 2015.
23. American Pharmacist Association. Drug information handbook: a comprehensive resource for all clinicians and healthcare professionals. 21st ed. Alphen aan den Rijn, The Netherlands: Lexi-Comp, Inc. Wolters Kluwer; 2012-2013.
24. Orwoll E, Ettinger M, Weiss S, et al. Alendronate for the treatment of osteoporosis in men. N Engl J Med. 2000;343:604-610.
25. Ringe JD, Dorst A, Faber H, et al. Alendronate treatment of established primary osteoporosis in men: 3-year results of a prospective, comparative, two-arm study. Rheumatol Int. 2004;24:110-113.
26. Ringe JD, Faber H, Farahmand P, et al. Efficacy of risedronate in men with primary and secondary osteoporosis: results of a 1-year study. Rheumatol Int. 2006;26:427-431.
27. Boonen S, Orwoll ES, Wenderoth D, et al. Once-weekly risedronate in men with osteoporosis: results of a 2-year, placebocontrolled, double-blind, multicenter study. J Bone Miner Res. 2009;24:719-725.
28. Khosla S, Amin S, Orwoll E. Osteoporosis in men. Endocr Rev. 2008;29:441-464.
29. Boonen S, Reginster JY, Kaufman JM, et al. Fracture risk and zoledronic acid therapy in men with osteoporosis. N Engl J Med. 2012;367:1714-1723.
30. Lyles KW, Colón-Emeric CS, Magaziner JS, et al; HORIZON Recurrent Fracture Trial. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357:1799-1809.
31. Orwoll ES, Scheele WH, Paul S, et al. The effect of teriparatide [human parathyroid hormone (1-34)] therapy on bone density in men with osteoporosis. J Bone Miner Res. 2003;18:9-17.
32. Kaufman JM, Orwoll E, Goemaere S, et al. Teriparatide effects on vertebral fractures and bone mineral density in men with osteoporosis: treatment and discontinuation of therapy. Osteoporos Int. 2005;16:510-516.
33. Snyder PJ, Peachey H, Hannoush P, et al. Effect of testosterone treatment on bone mineral density in men over 65 years of age. J Clin Endocrinol Metab. 1999;84:1966-1972.
34. Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab. 2012;97:3161-3169.
35. Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med. 2009;361:745-755.
36. Committee to Review Dietary Reference Intakes for Vitamin D and Calcium; Institute of Medicine. Dietary reference intakes for calcium and vitamin D. Institute of Medicine Web site. Available at: http://www.iom.edu/reports/2010/dietary-reference-intakes-for-calcium-and-vitamin-d.aspx. Accessed April 10, 2015.
37. Avenell A, Mak JC, O’Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227.

“A” Is for “Airway” (and “Accountability”)
A 3-month-old boy was diagnosed with severe respiratory syncytial virus infection at a North Carolina hospital in 2009. The infant was intubated, and a transfer to another hospital that had a pediatric ICU was ordered. The second hospital’s emergency transport service facilitated the transfer.
The ambulance was staffed by an EMT-paramedic and a transport registered nurse, both certified in Pediatric Advanced Life Support (PALS). During transport, the infant was intubated and medically paralyzed with vecuronium. About 10 minutes prior to arrival, the infant’s condition worsened, and he experienced cardiac arrest. Chest compressions and heart medications were provided.
Upon arrival at the second hospital, the infant was resuscitated. An emergency department physician, Dr S., ordered reintubation, and the infant again experienced cardiac arrest. For the next 10 minutes, Dr S. ordered chest compressions and heart medication; he eventually reintubated the infant, at which point cardiac arrest ceased with a spontaneous heartbeat.
After transfer to the pediatric ICU, the infant was diagnosed with permanent hypoxic ischemic brain injury caused by the cardiac episodes and low oxygen intake. At the time of trial, the child could not walk, talk, or hear very well and was fed via feeding tube. His vision and cognitive function were also impaired.
The plaintiff claimed that the intubation tube should have been adjusted or removed and re-inserted in the ambulance, but that did not happen. At trial, they called as an adverse witness the EMT-paramedic who had been in the ambulance with the plaintiff.
Continue for the outcome >>
OUTCOME
During the plaintiff’s presentation of evidence, the hospital agreed to a settlement of $13 million. The trial against Dr S. continued but ended in a mistrial. The plaintiffs were expected to re-try the claims against him.
COMMENT
This is a bad-airway case. Many medical malpractice cases are bad-airways cases: They are easy to bring and easy for jurors to understand, since hypoxic/anoxic injury is evident and plainly correlated with the airway missteps. These cases are also easy to prove, since the plaintiff can always hire an expert to testify that a reasonably prudent clinician would have been able to properly secure and monitor the airway—and the patient “would be standing here today,” unscathed.
Of course, securing an airway is not a “given” and can be challenging. Factor in variables such as anatomy, age, body habitus, intoxication, combativeness, medical comorbidities, and a full stomach, and the risk swells. If your employment requires that you manage airways, you are practicing in a high-legal-risk environment.
Make sure your skills are up to par. Practice often. Have a plan, a backup plan, and rescue backup plan. Have working suction ready. Check your equipment regularly. Run practice codes—particularly if your practice does not manage cardiorespiratory emergencies often. Does your staff know how to open the crash cart? Are the meds expired? Is the oxygen cylinder empty? Are roles clearly defined? As clinicians, we sit through our share of useless meetings (discussing things like who left what in the break room fridge). We should find time to drill on cardiorespiratory emergencies, because there is no time to “reacquaint oneself” on the fly.
In the case report, we are told that the patient was monitored by O2 saturation (SaO2) and end tidal CO2 (ETCO2). The plaintiff alleges that the transport team (EMT-paramedic and RN) failed to address tube placement and rather focused their efforts on chest compressions and medications. As we know, most cases of pediatric cardiac arrest are not primarily cardiac but instead follow primary progressive respiratory failure. Despite the emphasis on “airway, breathing, circulation” covered by PALS, the airway appears to have been missed and tube placement unquestioned after the child began to decompensate. Tube placement wasn’t reconsidered until 10 minutes after arrival at the second hospital, when the patient was reintubated and ventilatory status improved.
Airway cases—this one included—are “high damages” cases. A high damages case is one in which the injury is clear and expenses and costs to care for the patient are clear and immense. Such cases can be difficult to defend, because the enormity of the plaintiff’s plight (the limitations, years of required rehabilitation, hospitalization, PT, OT, nutritional care, etc) can overwhelm the jury, who may infer negligence based on the plaintiff’s condition. While jurors are required to consider damages only after negligence has been proven, lay jurors are human and many find it difficult to parse liability from damages. This is particularly true in airway cases involving a young, now-debilitated patient and an expert witness claiming an error that was preventable.
In this case, the plaintiff’s attorney made an unusual move, by calling as a “hostile witness” the paramedic who treated the 3-month-old boy. The paramedic would generally be called by the defense and later cross-examined by the plaintiff. Instead, the plaintiff chose to examine him first. Under evidence rules in most states (including North Carolina,1 where this case was heard), an adverse (or hostile) witness can be called by the opposing party.
Why is this important? Because during direct examination, you cannot lead the witness; during cross-examination, you can. Leading questions (if done correctly) generally produce the most effective and damaging moments during a trial. In this case, the plaintiffs were able to immediately examine one of the defendant’s principal actors using leading questions. Shortly thereafter, when the plaintiff was directly examining his own paramedic expert witnesses, the defense relented and settled for $13 million, with further recovery against the emergency physician still available.
Interestingly, the attorney suing the medical personnel in this case was none other than John Edwards. Yes, that John Edwards—the former vice presidential candidate famous for his $500 haircuts, campaign finance tribulations, and ethical lapses. He originally became famous (and rich) suing clinicians. After his political fall from grace, he is back in business suing clinicians—and for him, business is good.
IN SUM
This case was unfortunate. Always use care in securing the airway, particularly during patient movement. Once it is established, monitor the airway using SaO2, ETCO2, and keen observation. Airways are not in the “set it and forget it” camp. An airway must be maintained, safeguarded, and protected. Be prepared to act quickly should the airway become dislodged, migrate, or otherwise fail. —DML
REFERENCE
1. North Carolina Rules of Evidence Rule 611 (1983, c. 701, s. 1.).
A 3-month-old boy was diagnosed with severe respiratory syncytial virus infection at a North Carolina hospital in 2009. The infant was intubated, and a transfer to another hospital that had a pediatric ICU was ordered. The second hospital’s emergency transport service facilitated the transfer.
The ambulance was staffed by an EMT-paramedic and a transport registered nurse, both certified in Pediatric Advanced Life Support (PALS). During transport, the infant was intubated and medically paralyzed with vecuronium. About 10 minutes prior to arrival, the infant’s condition worsened, and he experienced cardiac arrest. Chest compressions and heart medications were provided.
Upon arrival at the second hospital, the infant was resuscitated. An emergency department physician, Dr S., ordered reintubation, and the infant again experienced cardiac arrest. For the next 10 minutes, Dr S. ordered chest compressions and heart medication; he eventually reintubated the infant, at which point cardiac arrest ceased with a spontaneous heartbeat.
After transfer to the pediatric ICU, the infant was diagnosed with permanent hypoxic ischemic brain injury caused by the cardiac episodes and low oxygen intake. At the time of trial, the child could not walk, talk, or hear very well and was fed via feeding tube. His vision and cognitive function were also impaired.
The plaintiff claimed that the intubation tube should have been adjusted or removed and re-inserted in the ambulance, but that did not happen. At trial, they called as an adverse witness the EMT-paramedic who had been in the ambulance with the plaintiff.
Continue for the outcome >>
OUTCOME
During the plaintiff’s presentation of evidence, the hospital agreed to a settlement of $13 million. The trial against Dr S. continued but ended in a mistrial. The plaintiffs were expected to re-try the claims against him.
COMMENT
This is a bad-airway case. Many medical malpractice cases are bad-airways cases: They are easy to bring and easy for jurors to understand, since hypoxic/anoxic injury is evident and plainly correlated with the airway missteps. These cases are also easy to prove, since the plaintiff can always hire an expert to testify that a reasonably prudent clinician would have been able to properly secure and monitor the airway—and the patient “would be standing here today,” unscathed.
Of course, securing an airway is not a “given” and can be challenging. Factor in variables such as anatomy, age, body habitus, intoxication, combativeness, medical comorbidities, and a full stomach, and the risk swells. If your employment requires that you manage airways, you are practicing in a high-legal-risk environment.
Make sure your skills are up to par. Practice often. Have a plan, a backup plan, and rescue backup plan. Have working suction ready. Check your equipment regularly. Run practice codes—particularly if your practice does not manage cardiorespiratory emergencies often. Does your staff know how to open the crash cart? Are the meds expired? Is the oxygen cylinder empty? Are roles clearly defined? As clinicians, we sit through our share of useless meetings (discussing things like who left what in the break room fridge). We should find time to drill on cardiorespiratory emergencies, because there is no time to “reacquaint oneself” on the fly.
In the case report, we are told that the patient was monitored by O2 saturation (SaO2) and end tidal CO2 (ETCO2). The plaintiff alleges that the transport team (EMT-paramedic and RN) failed to address tube placement and rather focused their efforts on chest compressions and medications. As we know, most cases of pediatric cardiac arrest are not primarily cardiac but instead follow primary progressive respiratory failure. Despite the emphasis on “airway, breathing, circulation” covered by PALS, the airway appears to have been missed and tube placement unquestioned after the child began to decompensate. Tube placement wasn’t reconsidered until 10 minutes after arrival at the second hospital, when the patient was reintubated and ventilatory status improved.
Airway cases—this one included—are “high damages” cases. A high damages case is one in which the injury is clear and expenses and costs to care for the patient are clear and immense. Such cases can be difficult to defend, because the enormity of the plaintiff’s plight (the limitations, years of required rehabilitation, hospitalization, PT, OT, nutritional care, etc) can overwhelm the jury, who may infer negligence based on the plaintiff’s condition. While jurors are required to consider damages only after negligence has been proven, lay jurors are human and many find it difficult to parse liability from damages. This is particularly true in airway cases involving a young, now-debilitated patient and an expert witness claiming an error that was preventable.
In this case, the plaintiff’s attorney made an unusual move, by calling as a “hostile witness” the paramedic who treated the 3-month-old boy. The paramedic would generally be called by the defense and later cross-examined by the plaintiff. Instead, the plaintiff chose to examine him first. Under evidence rules in most states (including North Carolina,1 where this case was heard), an adverse (or hostile) witness can be called by the opposing party.
Why is this important? Because during direct examination, you cannot lead the witness; during cross-examination, you can. Leading questions (if done correctly) generally produce the most effective and damaging moments during a trial. In this case, the plaintiffs were able to immediately examine one of the defendant’s principal actors using leading questions. Shortly thereafter, when the plaintiff was directly examining his own paramedic expert witnesses, the defense relented and settled for $13 million, with further recovery against the emergency physician still available.
Interestingly, the attorney suing the medical personnel in this case was none other than John Edwards. Yes, that John Edwards—the former vice presidential candidate famous for his $500 haircuts, campaign finance tribulations, and ethical lapses. He originally became famous (and rich) suing clinicians. After his political fall from grace, he is back in business suing clinicians—and for him, business is good.
IN SUM
This case was unfortunate. Always use care in securing the airway, particularly during patient movement. Once it is established, monitor the airway using SaO2, ETCO2, and keen observation. Airways are not in the “set it and forget it” camp. An airway must be maintained, safeguarded, and protected. Be prepared to act quickly should the airway become dislodged, migrate, or otherwise fail. —DML
REFERENCE
1. North Carolina Rules of Evidence Rule 611 (1983, c. 701, s. 1.).
A 3-month-old boy was diagnosed with severe respiratory syncytial virus infection at a North Carolina hospital in 2009. The infant was intubated, and a transfer to another hospital that had a pediatric ICU was ordered. The second hospital’s emergency transport service facilitated the transfer.
The ambulance was staffed by an EMT-paramedic and a transport registered nurse, both certified in Pediatric Advanced Life Support (PALS). During transport, the infant was intubated and medically paralyzed with vecuronium. About 10 minutes prior to arrival, the infant’s condition worsened, and he experienced cardiac arrest. Chest compressions and heart medications were provided.
Upon arrival at the second hospital, the infant was resuscitated. An emergency department physician, Dr S., ordered reintubation, and the infant again experienced cardiac arrest. For the next 10 minutes, Dr S. ordered chest compressions and heart medication; he eventually reintubated the infant, at which point cardiac arrest ceased with a spontaneous heartbeat.
After transfer to the pediatric ICU, the infant was diagnosed with permanent hypoxic ischemic brain injury caused by the cardiac episodes and low oxygen intake. At the time of trial, the child could not walk, talk, or hear very well and was fed via feeding tube. His vision and cognitive function were also impaired.
The plaintiff claimed that the intubation tube should have been adjusted or removed and re-inserted in the ambulance, but that did not happen. At trial, they called as an adverse witness the EMT-paramedic who had been in the ambulance with the plaintiff.
Continue for the outcome >>
OUTCOME
During the plaintiff’s presentation of evidence, the hospital agreed to a settlement of $13 million. The trial against Dr S. continued but ended in a mistrial. The plaintiffs were expected to re-try the claims against him.
COMMENT
This is a bad-airway case. Many medical malpractice cases are bad-airways cases: They are easy to bring and easy for jurors to understand, since hypoxic/anoxic injury is evident and plainly correlated with the airway missteps. These cases are also easy to prove, since the plaintiff can always hire an expert to testify that a reasonably prudent clinician would have been able to properly secure and monitor the airway—and the patient “would be standing here today,” unscathed.
Of course, securing an airway is not a “given” and can be challenging. Factor in variables such as anatomy, age, body habitus, intoxication, combativeness, medical comorbidities, and a full stomach, and the risk swells. If your employment requires that you manage airways, you are practicing in a high-legal-risk environment.
Make sure your skills are up to par. Practice often. Have a plan, a backup plan, and rescue backup plan. Have working suction ready. Check your equipment regularly. Run practice codes—particularly if your practice does not manage cardiorespiratory emergencies often. Does your staff know how to open the crash cart? Are the meds expired? Is the oxygen cylinder empty? Are roles clearly defined? As clinicians, we sit through our share of useless meetings (discussing things like who left what in the break room fridge). We should find time to drill on cardiorespiratory emergencies, because there is no time to “reacquaint oneself” on the fly.
In the case report, we are told that the patient was monitored by O2 saturation (SaO2) and end tidal CO2 (ETCO2). The plaintiff alleges that the transport team (EMT-paramedic and RN) failed to address tube placement and rather focused their efforts on chest compressions and medications. As we know, most cases of pediatric cardiac arrest are not primarily cardiac but instead follow primary progressive respiratory failure. Despite the emphasis on “airway, breathing, circulation” covered by PALS, the airway appears to have been missed and tube placement unquestioned after the child began to decompensate. Tube placement wasn’t reconsidered until 10 minutes after arrival at the second hospital, when the patient was reintubated and ventilatory status improved.
Airway cases—this one included—are “high damages” cases. A high damages case is one in which the injury is clear and expenses and costs to care for the patient are clear and immense. Such cases can be difficult to defend, because the enormity of the plaintiff’s plight (the limitations, years of required rehabilitation, hospitalization, PT, OT, nutritional care, etc) can overwhelm the jury, who may infer negligence based on the plaintiff’s condition. While jurors are required to consider damages only after negligence has been proven, lay jurors are human and many find it difficult to parse liability from damages. This is particularly true in airway cases involving a young, now-debilitated patient and an expert witness claiming an error that was preventable.
In this case, the plaintiff’s attorney made an unusual move, by calling as a “hostile witness” the paramedic who treated the 3-month-old boy. The paramedic would generally be called by the defense and later cross-examined by the plaintiff. Instead, the plaintiff chose to examine him first. Under evidence rules in most states (including North Carolina,1 where this case was heard), an adverse (or hostile) witness can be called by the opposing party.
Why is this important? Because during direct examination, you cannot lead the witness; during cross-examination, you can. Leading questions (if done correctly) generally produce the most effective and damaging moments during a trial. In this case, the plaintiffs were able to immediately examine one of the defendant’s principal actors using leading questions. Shortly thereafter, when the plaintiff was directly examining his own paramedic expert witnesses, the defense relented and settled for $13 million, with further recovery against the emergency physician still available.
Interestingly, the attorney suing the medical personnel in this case was none other than John Edwards. Yes, that John Edwards—the former vice presidential candidate famous for his $500 haircuts, campaign finance tribulations, and ethical lapses. He originally became famous (and rich) suing clinicians. After his political fall from grace, he is back in business suing clinicians—and for him, business is good.
IN SUM
This case was unfortunate. Always use care in securing the airway, particularly during patient movement. Once it is established, monitor the airway using SaO2, ETCO2, and keen observation. Airways are not in the “set it and forget it” camp. An airway must be maintained, safeguarded, and protected. Be prepared to act quickly should the airway become dislodged, migrate, or otherwise fail. —DML
REFERENCE
1. North Carolina Rules of Evidence Rule 611 (1983, c. 701, s. 1.).
VIDEO: LAA ablation safely treats long-standing, persistent Afib
LONDON – Routine catheter ablation that electrically isolates the left atrial appendage safely boosted the success rate of treatment for long-standing, persistent atrial fibrillation in a randomized trial with 173 patients.
This finding from the first prospective randomized trial to test adding routine left atrial appendage (LAA) electrical isolation to the established catheter-ablation protocol of pulmonary-vein isolation should encourage electrophysiologists to make LAA isolation a more standard part of their approach to treating long-standing, persistent atrial fibrillation (Afib), Dr. Jagmeet P. Singh commented in an interview at the annual congress of the European Society of Cardiology.
“A lot of us have, in the past, been hesitant to ablate the LAA” out of concern that it could render the LAA inert and more likely to become a source of blood clots, noted Dr. Singh, professor of medicine at Harvard Medical School and director of the cardiac resynchronization therapy program at Massachusetts General Hospital in Boston. “This study result provides, for the first time in a randomized fashion, direction on this area of ablation.” Based on the results, Dr. Singh said that in his practice now he would “look for LAA activity” when assessing an Afib patient in the electrophysiology laboratory, “and if the LAA was active I would ablate it,” he said.
The BELIEF (Effect of Empirical Left Atrial Appendage Isolation on Long-Term Procedure Outcome in Patients With Persistent or Long-Standing Persistent Atrial Fibrillation Undergoing Catheter Ablation) trial enrolled 173 patients with long-standing persistent Afib that was refractory to treatment with antiarrhythmic drugs at two U.S. centers and randomized them to receive either conventional pulmonary vein isolation alone, or pulmonary vein isolation and additional point ablations to also produce LAA isolation. The study’s primary endpoint was freedom from Afib episodes at 12 months after treatment.
At 12 months after treatment, freedom from Afib recurrence occurred in 48 of the 85 patients (56%) assigned to LAA isolation and in 25 of the 88 patients (25%) treated with pulmonary vein isolation only, a statistically significant difference, reported Dr. Luigi Di Biasi at the congress. In an analysis that adjusted for patient age, sex, and left atrial diameter the addition of LAA ablation linked with a statistically significant 55% reduction in Afib recurrence, said Dr. Di Biasi, director of the arrhythmia service at Montefiore Medical Center in New York.
Adding LAA isolation to the standard ablation procedure did not result in additional complications, said Dr. Di Biasi, although it did increase procedure time by about 15 minutes. The patients who underwent LAA isolation had no strokes during 2 years of follow-up, and no statistically significant change in the incidence of Afib-related hospitalizations or hospitalizations for heart failure, compared with control patients. One pericardial effusion occurred in each of the study arms during follow-up, and there were no deaths during follow-up in either group. LAA isolation resulted in impaired LAA function in about half of the patients who had the isolation procedure, detected by transesophageal echocardiography after the procedure, but the clinical outcomes indicated that this did not appear to affect patients’ stroke risk.
Dr. Singh has been a consultant to Boston Scientific, St. Jude, Medtronic, Sorin, and Biotronik. Dr. Di Biasi has been a consultant to Biosense Webster, Stereotaxis, and St. Jude, and a speaker for Biotronik, Medtronic, Boston Scientific, and Epi EP.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
LONDON – Routine catheter ablation that electrically isolates the left atrial appendage safely boosted the success rate of treatment for long-standing, persistent atrial fibrillation in a randomized trial with 173 patients.
This finding from the first prospective randomized trial to test adding routine left atrial appendage (LAA) electrical isolation to the established catheter-ablation protocol of pulmonary-vein isolation should encourage electrophysiologists to make LAA isolation a more standard part of their approach to treating long-standing, persistent atrial fibrillation (Afib), Dr. Jagmeet P. Singh commented in an interview at the annual congress of the European Society of Cardiology.
“A lot of us have, in the past, been hesitant to ablate the LAA” out of concern that it could render the LAA inert and more likely to become a source of blood clots, noted Dr. Singh, professor of medicine at Harvard Medical School and director of the cardiac resynchronization therapy program at Massachusetts General Hospital in Boston. “This study result provides, for the first time in a randomized fashion, direction on this area of ablation.” Based on the results, Dr. Singh said that in his practice now he would “look for LAA activity” when assessing an Afib patient in the electrophysiology laboratory, “and if the LAA was active I would ablate it,” he said.
The BELIEF (Effect of Empirical Left Atrial Appendage Isolation on Long-Term Procedure Outcome in Patients With Persistent or Long-Standing Persistent Atrial Fibrillation Undergoing Catheter Ablation) trial enrolled 173 patients with long-standing persistent Afib that was refractory to treatment with antiarrhythmic drugs at two U.S. centers and randomized them to receive either conventional pulmonary vein isolation alone, or pulmonary vein isolation and additional point ablations to also produce LAA isolation. The study’s primary endpoint was freedom from Afib episodes at 12 months after treatment.
At 12 months after treatment, freedom from Afib recurrence occurred in 48 of the 85 patients (56%) assigned to LAA isolation and in 25 of the 88 patients (25%) treated with pulmonary vein isolation only, a statistically significant difference, reported Dr. Luigi Di Biasi at the congress. In an analysis that adjusted for patient age, sex, and left atrial diameter the addition of LAA ablation linked with a statistically significant 55% reduction in Afib recurrence, said Dr. Di Biasi, director of the arrhythmia service at Montefiore Medical Center in New York.
Adding LAA isolation to the standard ablation procedure did not result in additional complications, said Dr. Di Biasi, although it did increase procedure time by about 15 minutes. The patients who underwent LAA isolation had no strokes during 2 years of follow-up, and no statistically significant change in the incidence of Afib-related hospitalizations or hospitalizations for heart failure, compared with control patients. One pericardial effusion occurred in each of the study arms during follow-up, and there were no deaths during follow-up in either group. LAA isolation resulted in impaired LAA function in about half of the patients who had the isolation procedure, detected by transesophageal echocardiography after the procedure, but the clinical outcomes indicated that this did not appear to affect patients’ stroke risk.
Dr. Singh has been a consultant to Boston Scientific, St. Jude, Medtronic, Sorin, and Biotronik. Dr. Di Biasi has been a consultant to Biosense Webster, Stereotaxis, and St. Jude, and a speaker for Biotronik, Medtronic, Boston Scientific, and Epi EP.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
LONDON – Routine catheter ablation that electrically isolates the left atrial appendage safely boosted the success rate of treatment for long-standing, persistent atrial fibrillation in a randomized trial with 173 patients.
This finding from the first prospective randomized trial to test adding routine left atrial appendage (LAA) electrical isolation to the established catheter-ablation protocol of pulmonary-vein isolation should encourage electrophysiologists to make LAA isolation a more standard part of their approach to treating long-standing, persistent atrial fibrillation (Afib), Dr. Jagmeet P. Singh commented in an interview at the annual congress of the European Society of Cardiology.
“A lot of us have, in the past, been hesitant to ablate the LAA” out of concern that it could render the LAA inert and more likely to become a source of blood clots, noted Dr. Singh, professor of medicine at Harvard Medical School and director of the cardiac resynchronization therapy program at Massachusetts General Hospital in Boston. “This study result provides, for the first time in a randomized fashion, direction on this area of ablation.” Based on the results, Dr. Singh said that in his practice now he would “look for LAA activity” when assessing an Afib patient in the electrophysiology laboratory, “and if the LAA was active I would ablate it,” he said.
The BELIEF (Effect of Empirical Left Atrial Appendage Isolation on Long-Term Procedure Outcome in Patients With Persistent or Long-Standing Persistent Atrial Fibrillation Undergoing Catheter Ablation) trial enrolled 173 patients with long-standing persistent Afib that was refractory to treatment with antiarrhythmic drugs at two U.S. centers and randomized them to receive either conventional pulmonary vein isolation alone, or pulmonary vein isolation and additional point ablations to also produce LAA isolation. The study’s primary endpoint was freedom from Afib episodes at 12 months after treatment.
At 12 months after treatment, freedom from Afib recurrence occurred in 48 of the 85 patients (56%) assigned to LAA isolation and in 25 of the 88 patients (25%) treated with pulmonary vein isolation only, a statistically significant difference, reported Dr. Luigi Di Biasi at the congress. In an analysis that adjusted for patient age, sex, and left atrial diameter the addition of LAA ablation linked with a statistically significant 55% reduction in Afib recurrence, said Dr. Di Biasi, director of the arrhythmia service at Montefiore Medical Center in New York.
Adding LAA isolation to the standard ablation procedure did not result in additional complications, said Dr. Di Biasi, although it did increase procedure time by about 15 minutes. The patients who underwent LAA isolation had no strokes during 2 years of follow-up, and no statistically significant change in the incidence of Afib-related hospitalizations or hospitalizations for heart failure, compared with control patients. One pericardial effusion occurred in each of the study arms during follow-up, and there were no deaths during follow-up in either group. LAA isolation resulted in impaired LAA function in about half of the patients who had the isolation procedure, detected by transesophageal echocardiography after the procedure, but the clinical outcomes indicated that this did not appear to affect patients’ stroke risk.
Dr. Singh has been a consultant to Boston Scientific, St. Jude, Medtronic, Sorin, and Biotronik. Dr. Di Biasi has been a consultant to Biosense Webster, Stereotaxis, and St. Jude, and a speaker for Biotronik, Medtronic, Boston Scientific, and Epi EP.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
AT THE ESC CONGRESS 2015

Management of Locally Advanced Rectal Adenocarcinoma
Colorectal cancers are among the most common cancers worldwide, and there is a high mortality rate for advanced-stage disease. Approximately 132,000 new cases of colorectal cancer will be diagnosed in the United States in 2015, and approximately 40,000 of these cases will be primary rectal cancers. The incidence and mortality rates have been steadily declining over the past two decades, largely through advances in screening and improvements in treatment. However, rectal cancer remains a significant cause of morbidity and mortality in the United States and worldwide.
To read the full article in PDF:
Colorectal cancers are among the most common cancers worldwide, and there is a high mortality rate for advanced-stage disease. Approximately 132,000 new cases of colorectal cancer will be diagnosed in the United States in 2015, and approximately 40,000 of these cases will be primary rectal cancers. The incidence and mortality rates have been steadily declining over the past two decades, largely through advances in screening and improvements in treatment. However, rectal cancer remains a significant cause of morbidity and mortality in the United States and worldwide.
To read the full article in PDF:
Colorectal cancers are among the most common cancers worldwide, and there is a high mortality rate for advanced-stage disease. Approximately 132,000 new cases of colorectal cancer will be diagnosed in the United States in 2015, and approximately 40,000 of these cases will be primary rectal cancers. The incidence and mortality rates have been steadily declining over the past two decades, largely through advances in screening and improvements in treatment. However, rectal cancer remains a significant cause of morbidity and mortality in the United States and worldwide.
To read the full article in PDF:
Liar, liar ...
Okay, I admit that from time to time I have embellished the anecdotes that I include in these letters. Sometimes, I feel I need to make sure that you are paying attention. But this time, I am relating this story in its true, unvarnished state.
Two mature women with whom I am acquainted (No, one was not my wife!) had just finished their habitual Saturday morning walk through a wooded upper middle class neighborhood here in town. It was nine o’clock in the morning and the sun was shining. Suddenly, a mangy-looking fox trotted out of the woods and down the road toward them. Aware that from time to time local raccoons, skunks, and foxes have tested positive for rabies, these women began to run and flagged down the first car they saw, and without a word hopped in the back seat.
The surprised occupants of the vehicle were two mature men. You might call them strangers, but here in Maine, we don’t have any strangers. We have tourists. If a fellow Mainer doesn’t know you, he probably knows two people with whom you are acquainted.
As the women began to breathlessly explain their actions, one of the women felt a searing pain in her right thigh and assumed she had torn a muscle as she sprinted away from the fox. Within a few hundred yards, the car began to fill with smoke. Believing that the vehicle was on fire, all four occupants tumbled out into the street like four carnival clowns.
It quickly became clear that the cause of the smoke and the searing pain was that the woman’s pants were on fire. Throwing all caution and modesty to the wind, she quickly shed her pants in the middle of the road and in full view of these men, with whom it turns out she does share several acquaintances.
The source of the fire was the woman’s cell phone. The resulting injury was a palm-size, painful, deep, second-degree burn of her anterior thigh. In a quick Internet search, you will discover several very similar stories – minus the fox and the strangers. Some of the victims were children.
It turns out some cell phones have a tendency to spontaneously explode and/or catch fire. There seems to be no common factor in the events, although some of the ultrathin and flexible cell phones may be more prone to conflagration. However, the victim in our scenario has a storied past with cell phones. She has dropped them in the toilet at least once (history is a little unclear here on the exact number). On another occasion, she placed one in the sink of a public restroom, we can assume to prevent a second or third toilet submersion. As she approached the sink to retrieve it, the clever water-saving faucet – sensing her presence – turned itself on. But in the fox and fire incident, she denies any previous submersions or unusual events with this particular phone. A lawyer is now involved.
So while you and I as pediatricians may be concerned about the relationship between cell phones and health of our patients primarily because cell phones can be a dangerous distraction for young drivers, cyclists, and pedestrians, I share this anecdote to make you aware of another of their health hazards. You also may want to reconsider where you carry your cell phone.
I am not worried myself. I have a little flip phone for which I pay $100 for 500 minutes of usage a year, way more than I need or use. It couldn’t be considered a smartphone as its only noteworthy skill is taking pictures of the inside of my pants pocket. I suspect that its battery must be so small and impotent that even if it decides to self-immolate, I doubt I will notice. However, I do worry about scraggly-looking foxes meandering through my neighborhood.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “Coping with a Picky Eater.” Email him at [email protected].
Okay, I admit that from time to time I have embellished the anecdotes that I include in these letters. Sometimes, I feel I need to make sure that you are paying attention. But this time, I am relating this story in its true, unvarnished state.
Two mature women with whom I am acquainted (No, one was not my wife!) had just finished their habitual Saturday morning walk through a wooded upper middle class neighborhood here in town. It was nine o’clock in the morning and the sun was shining. Suddenly, a mangy-looking fox trotted out of the woods and down the road toward them. Aware that from time to time local raccoons, skunks, and foxes have tested positive for rabies, these women began to run and flagged down the first car they saw, and without a word hopped in the back seat.
The surprised occupants of the vehicle were two mature men. You might call them strangers, but here in Maine, we don’t have any strangers. We have tourists. If a fellow Mainer doesn’t know you, he probably knows two people with whom you are acquainted.
As the women began to breathlessly explain their actions, one of the women felt a searing pain in her right thigh and assumed she had torn a muscle as she sprinted away from the fox. Within a few hundred yards, the car began to fill with smoke. Believing that the vehicle was on fire, all four occupants tumbled out into the street like four carnival clowns.
It quickly became clear that the cause of the smoke and the searing pain was that the woman’s pants were on fire. Throwing all caution and modesty to the wind, she quickly shed her pants in the middle of the road and in full view of these men, with whom it turns out she does share several acquaintances.
The source of the fire was the woman’s cell phone. The resulting injury was a palm-size, painful, deep, second-degree burn of her anterior thigh. In a quick Internet search, you will discover several very similar stories – minus the fox and the strangers. Some of the victims were children.
It turns out some cell phones have a tendency to spontaneously explode and/or catch fire. There seems to be no common factor in the events, although some of the ultrathin and flexible cell phones may be more prone to conflagration. However, the victim in our scenario has a storied past with cell phones. She has dropped them in the toilet at least once (history is a little unclear here on the exact number). On another occasion, she placed one in the sink of a public restroom, we can assume to prevent a second or third toilet submersion. As she approached the sink to retrieve it, the clever water-saving faucet – sensing her presence – turned itself on. But in the fox and fire incident, she denies any previous submersions or unusual events with this particular phone. A lawyer is now involved.
So while you and I as pediatricians may be concerned about the relationship between cell phones and health of our patients primarily because cell phones can be a dangerous distraction for young drivers, cyclists, and pedestrians, I share this anecdote to make you aware of another of their health hazards. You also may want to reconsider where you carry your cell phone.
I am not worried myself. I have a little flip phone for which I pay $100 for 500 minutes of usage a year, way more than I need or use. It couldn’t be considered a smartphone as its only noteworthy skill is taking pictures of the inside of my pants pocket. I suspect that its battery must be so small and impotent that even if it decides to self-immolate, I doubt I will notice. However, I do worry about scraggly-looking foxes meandering through my neighborhood.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “Coping with a Picky Eater.” Email him at [email protected].
Okay, I admit that from time to time I have embellished the anecdotes that I include in these letters. Sometimes, I feel I need to make sure that you are paying attention. But this time, I am relating this story in its true, unvarnished state.
Two mature women with whom I am acquainted (No, one was not my wife!) had just finished their habitual Saturday morning walk through a wooded upper middle class neighborhood here in town. It was nine o’clock in the morning and the sun was shining. Suddenly, a mangy-looking fox trotted out of the woods and down the road toward them. Aware that from time to time local raccoons, skunks, and foxes have tested positive for rabies, these women began to run and flagged down the first car they saw, and without a word hopped in the back seat.
The surprised occupants of the vehicle were two mature men. You might call them strangers, but here in Maine, we don’t have any strangers. We have tourists. If a fellow Mainer doesn’t know you, he probably knows two people with whom you are acquainted.
As the women began to breathlessly explain their actions, one of the women felt a searing pain in her right thigh and assumed she had torn a muscle as she sprinted away from the fox. Within a few hundred yards, the car began to fill with smoke. Believing that the vehicle was on fire, all four occupants tumbled out into the street like four carnival clowns.
It quickly became clear that the cause of the smoke and the searing pain was that the woman’s pants were on fire. Throwing all caution and modesty to the wind, she quickly shed her pants in the middle of the road and in full view of these men, with whom it turns out she does share several acquaintances.
The source of the fire was the woman’s cell phone. The resulting injury was a palm-size, painful, deep, second-degree burn of her anterior thigh. In a quick Internet search, you will discover several very similar stories – minus the fox and the strangers. Some of the victims were children.
It turns out some cell phones have a tendency to spontaneously explode and/or catch fire. There seems to be no common factor in the events, although some of the ultrathin and flexible cell phones may be more prone to conflagration. However, the victim in our scenario has a storied past with cell phones. She has dropped them in the toilet at least once (history is a little unclear here on the exact number). On another occasion, she placed one in the sink of a public restroom, we can assume to prevent a second or third toilet submersion. As she approached the sink to retrieve it, the clever water-saving faucet – sensing her presence – turned itself on. But in the fox and fire incident, she denies any previous submersions or unusual events with this particular phone. A lawyer is now involved.
So while you and I as pediatricians may be concerned about the relationship between cell phones and health of our patients primarily because cell phones can be a dangerous distraction for young drivers, cyclists, and pedestrians, I share this anecdote to make you aware of another of their health hazards. You also may want to reconsider where you carry your cell phone.
I am not worried myself. I have a little flip phone for which I pay $100 for 500 minutes of usage a year, way more than I need or use. It couldn’t be considered a smartphone as its only noteworthy skill is taking pictures of the inside of my pants pocket. I suspect that its battery must be so small and impotent that even if it decides to self-immolate, I doubt I will notice. However, I do worry about scraggly-looking foxes meandering through my neighborhood.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “Coping with a Picky Eater.” Email him at [email protected].

Nipple Raynaud’s can freeze out breastfeeding desire
NEW YORK – The “perfect storm” of pregnancy and lactation can throw breastfeeding moms into a painful deep freeze.
Almost 25% of women with lactation pain may actually be experiencing symptoms of Raynaud’s, Dr. Honor Fullerton Stone said at the American Academy of Dermatology summer meeting.

“Breastfeeding mothers are in a perfect storm,” of physical developments that predispose them to this vasoconstrictive phenomenon, said Dr. Fullerton Stone, who practices dermatology in Menlo Park, Ca. “Estrogen increases the alpha-adrenergic receptors on smooth muscle. Nerve irritation from constant breastfeeding upregulates those receptors. And emotional stress – crying baby, anyone? – increases epinephrine, which contributes further to vasoconstriction.”
She conducted a review of 88 of her own patients with nursing-related breast pain. Of these, about a quarter ended up with a diagnosis of nipple vasoconstriction. None of the women had a history of Raynaud’s disease.
Pain during a nursing session is the presenting complaint, but breastfeeding pain is incredibly nonspecific as a symptom. It’s the quality of the pain, Dr Stone said, that should ring a bell.
“There’s let-down pain, which occurs at latch and then comes on again later, with refill. There’s pain from candidiasis, which is dramatic at latch, like a radiating heat, but goes away rapidly after a few days of antifungals,” she noted. But with Raynaud’s, “the pain is persistent. It’s throbbing, which makes sense since it’s vascular. And it’s constant,” lasting through every nursing session, which she said is the kind of experience that makes mothers stop breastfeeding.
Since pain is such a pervasive symptom in breastfeeding complaints, women with vasoconstriction of the nipple are often misdiagnosed. They can go for months trying to improve latch technique or receiving antifungal therapy with no improvement, she said.
Dr. Stone considers a diagnosis of Raynaud’s if two of the following criteria are met:
• Color change of nipple, cold sensitivity, or color change of acral surfaces with cold exposure.
• Chronic deep breast pain for 4 or more weeks.
• Failure of oral antifungals and or antibiotics.
Treatment is both supportive and systemic and avoiding cold is key. She recalled a young mother who arrived in her office bundled up in a down parka on a warm California spring day. “I don’t know why, but this really seems to help,” she said.

“I suggest taking two hot showers a day and all the better if they can be right before nursing. Hot pads and compresses are not going to help. You need to warm up the entire body to calm this vascular reactivity.”
Women should also avoid consuming anything that can cause vasoconstriction, including caffeine and tobacco, she advised.
Nifedipine is a very effective medication, and is safe for nursing infants, Dr. Stone said. The sustained-released 30 mg/day dose is typically recommended, but she has changed her thinking on this a bit.
Her internal study showed that, although 83% responded very well to the drug, about a third of them also had a vasodilation-related side effect.
“For this reason, I usually now start with the 10 mg nonsustained form, and warn about things like headache, dizziness, postural hypotension, and fainting,” she said. Typically, patients adjust well to the drug’s effects and then the dose can be individually titrated.
Dr. Fullerton Stone had no relevant financial disclosures.
On Twitter @Alz_Gal
NEW YORK – The “perfect storm” of pregnancy and lactation can throw breastfeeding moms into a painful deep freeze.
Almost 25% of women with lactation pain may actually be experiencing symptoms of Raynaud’s, Dr. Honor Fullerton Stone said at the American Academy of Dermatology summer meeting.
“Breastfeeding mothers are in a perfect storm,” of physical developments that predispose them to this vasoconstrictive phenomenon, said Dr. Fullerton Stone, who practices dermatology in Menlo Park, Ca. “Estrogen increases the alpha-adrenergic receptors on smooth muscle. Nerve irritation from constant breastfeeding upregulates those receptors. And emotional stress – crying baby, anyone? – increases epinephrine, which contributes further to vasoconstriction.”
She conducted a review of 88 of her own patients with nursing-related breast pain. Of these, about a quarter ended up with a diagnosis of nipple vasoconstriction. None of the women had a history of Raynaud’s disease.
Pain during a nursing session is the presenting complaint, but breastfeeding pain is incredibly nonspecific as a symptom. It’s the quality of the pain, Dr Stone said, that should ring a bell.
“There’s let-down pain, which occurs at latch and then comes on again later, with refill. There’s pain from candidiasis, which is dramatic at latch, like a radiating heat, but goes away rapidly after a few days of antifungals,” she noted. But with Raynaud’s, “the pain is persistent. It’s throbbing, which makes sense since it’s vascular. And it’s constant,” lasting through every nursing session, which she said is the kind of experience that makes mothers stop breastfeeding.
Since pain is such a pervasive symptom in breastfeeding complaints, women with vasoconstriction of the nipple are often misdiagnosed. They can go for months trying to improve latch technique or receiving antifungal therapy with no improvement, she said.
Dr. Stone considers a diagnosis of Raynaud’s if two of the following criteria are met:
• Color change of nipple, cold sensitivity, or color change of acral surfaces with cold exposure.
• Chronic deep breast pain for 4 or more weeks.
• Failure of oral antifungals and or antibiotics.
Treatment is both supportive and systemic and avoiding cold is key. She recalled a young mother who arrived in her office bundled up in a down parka on a warm California spring day. “I don’t know why, but this really seems to help,” she said.
“I suggest taking two hot showers a day and all the better if they can be right before nursing. Hot pads and compresses are not going to help. You need to warm up the entire body to calm this vascular reactivity.”
Women should also avoid consuming anything that can cause vasoconstriction, including caffeine and tobacco, she advised.
Nifedipine is a very effective medication, and is safe for nursing infants, Dr. Stone said. The sustained-released 30 mg/day dose is typically recommended, but she has changed her thinking on this a bit.
Her internal study showed that, although 83% responded very well to the drug, about a third of them also had a vasodilation-related side effect.
“For this reason, I usually now start with the 10 mg nonsustained form, and warn about things like headache, dizziness, postural hypotension, and fainting,” she said. Typically, patients adjust well to the drug’s effects and then the dose can be individually titrated.
Dr. Fullerton Stone had no relevant financial disclosures.
On Twitter @Alz_Gal
NEW YORK – The “perfect storm” of pregnancy and lactation can throw breastfeeding moms into a painful deep freeze.
Almost 25% of women with lactation pain may actually be experiencing symptoms of Raynaud’s, Dr. Honor Fullerton Stone said at the American Academy of Dermatology summer meeting.
“Breastfeeding mothers are in a perfect storm,” of physical developments that predispose them to this vasoconstrictive phenomenon, said Dr. Fullerton Stone, who practices dermatology in Menlo Park, Ca. “Estrogen increases the alpha-adrenergic receptors on smooth muscle. Nerve irritation from constant breastfeeding upregulates those receptors. And emotional stress – crying baby, anyone? – increases epinephrine, which contributes further to vasoconstriction.”
She conducted a review of 88 of her own patients with nursing-related breast pain. Of these, about a quarter ended up with a diagnosis of nipple vasoconstriction. None of the women had a history of Raynaud’s disease.
Pain during a nursing session is the presenting complaint, but breastfeeding pain is incredibly nonspecific as a symptom. It’s the quality of the pain, Dr Stone said, that should ring a bell.
“There’s let-down pain, which occurs at latch and then comes on again later, with refill. There’s pain from candidiasis, which is dramatic at latch, like a radiating heat, but goes away rapidly after a few days of antifungals,” she noted. But with Raynaud’s, “the pain is persistent. It’s throbbing, which makes sense since it’s vascular. And it’s constant,” lasting through every nursing session, which she said is the kind of experience that makes mothers stop breastfeeding.
Since pain is such a pervasive symptom in breastfeeding complaints, women with vasoconstriction of the nipple are often misdiagnosed. They can go for months trying to improve latch technique or receiving antifungal therapy with no improvement, she said.
Dr. Stone considers a diagnosis of Raynaud’s if two of the following criteria are met:
• Color change of nipple, cold sensitivity, or color change of acral surfaces with cold exposure.
• Chronic deep breast pain for 4 or more weeks.
• Failure of oral antifungals and or antibiotics.
Treatment is both supportive and systemic and avoiding cold is key. She recalled a young mother who arrived in her office bundled up in a down parka on a warm California spring day. “I don’t know why, but this really seems to help,” she said.
“I suggest taking two hot showers a day and all the better if they can be right before nursing. Hot pads and compresses are not going to help. You need to warm up the entire body to calm this vascular reactivity.”
Women should also avoid consuming anything that can cause vasoconstriction, including caffeine and tobacco, she advised.
Nifedipine is a very effective medication, and is safe for nursing infants, Dr. Stone said. The sustained-released 30 mg/day dose is typically recommended, but she has changed her thinking on this a bit.
Her internal study showed that, although 83% responded very well to the drug, about a third of them also had a vasodilation-related side effect.
“For this reason, I usually now start with the 10 mg nonsustained form, and warn about things like headache, dizziness, postural hypotension, and fainting,” she said. Typically, patients adjust well to the drug’s effects and then the dose can be individually titrated.
Dr. Fullerton Stone had no relevant financial disclosures.
On Twitter @Alz_Gal
AT THE AAD SUMMER ACADEMY 2015

Problematic Medications: Antibiotics in Renal Patients
Q) At a lecture I recently attended, the speaker said sulfamethoxazole/trimethoprim is a potentially dangerous medication. I use it all the time. Is there any data to support her comments? Where did she get her information?
Sulfamethoxazole/trimethoprim (SMX/TMP) is a combination of two antibiotics, each of which has the potential to interact with other substances.
It is well documented that sulfamethoxazole can inhibit the metabolism of cytochrome P450 2C9 substrates. Frequently prescribed medications that also use the cytochrome substrate include warfarin and oral antihypoglycemic agents.
Trimethoprim’s distinct properties also lead to drug interactions. Trimethoprim inhibits sodium uptake by the appropriate channels in the distal tubule of the kidney, preventing reabsorption and altering the electrical balance of the tubular cells. As a result, the amount of potassium excreted into the urine is reduced, yielding an accumulation of serum potassium.1
High serum potassium retention can manifest as hyperkalemia in patients with chronic kidney disease (CKD). Use of potassium-sparing drugs by patients with comorbidities, including CKD, can increase risk for hyperkalemia; concurrent use of these drugs with ACE inhibitors or angiotensin II receptor blockers (ARBs) compounds the risk.2 The first reports of hyperkalemia with trimethoprim use occurred in HIV patients treated with large doses for Pneumocystis carinii infection.3
In a population-based case-control study, the results of which were published in the British Medical Journal, Fralick and colleagues analyzed data on older patients (age 66 or older) who were taking either ACE inhibitors or ARBs in combination with an antibiotic.4 They found a significantly increased risk for sudden death within seven days of prescription of SMX/TMP, compared to amoxicillin; a secondary analysis also revealed an increased risk for sudden death within 14 days with SMX/TMP. The researchers speculated that this excess risk, which translated to 3 sudden deaths in 1,000 patients taking SMX/TMP versus 1 sudden death in 1,000 patients taking amoxicillin, “reflects unrecognized arrhythmic death due to hyperkalemia.”
Since more than 250 million prescriptions for ACE inhibitors/ARBs and 20 million prescriptions for SMX/TMP are written each year, there will be instances of overlap. The prudent clinician would prescribe a different antibiotic or, if avoidance is not possible, use the lowest effective dose and duration of SMX/TMP. Close monitoring of serum potassium levels is warranted in patients with comorbidities, especially CKD, who are taking ACE inhibitors or ARBs—and of course, in our geriatric population. —DLC
Debra L. Coplon, DNP, DCC
City of Memphis Wellness Clinic, Tennessee
REFERENCES
1. Velazquez H, Perazella MA, Wright FS, Ellison DH. Renal mechanism of trimethoprim-induced hyperkalemia. Ann Intern Med. 1993;119:296-301.
2. Horn JR, Hansten PD. Trimethoprim and potassium-sparing drugs: a risk for hyperkalemia. www.pharmacytimes.com/publications/issue/2011/February2011/DrugInteractions-0211. Accessed August 24, 2015.
3. Medina I, Mills J, Leoung G, et al. Oral therapy for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome: a controlled trial of trimethoprim-sulfamethoxazole versus trimethoprim-dapsone. N Engl J Med. 1990;323:776-782.
4. Fralick M, Macdonald EM, Gomes T, et al. Co-trimoxazole and sudden death in patients receiving inhibitors of renin-angiotensin system: population based study. BMJ. 2014;349:g6196.
5. Gilbert SJ, Weiner DE, Gipson DS, et al. National Kidney Foundation’s Primer on Kidney Diseases. Philadelphia, PA: Elsevier; 2014.
6. Muriithi AK, Leung N, Valeri AM, et al. Biopsy-proven acute interstitial nephritis, 1993-2011: a case series. Am J Kidney Dis. 2014;64(4):558-566.
7. Blank ML, Parkin L, Paul C, Herbison P. A nationwide nested case-control study indicates an increased risk of acute interstitial nephritis with proton pump inhibitor use. Kidney Int. 2014;86(4):837-844.
8. Klepser DG, Collier DS, Cochran GL. Proton pump inhibitors and acute kidney injury: a nested case-control study. BMC Nephrology. 2013;14:150.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Debra L. Coplon, DNP, DCC, who practices at City of Memphis Wellness Clinic in Tennessee, and Cynthia A. Smith, DNP, APRN, FNP-BC, who practices with Renal Consultants PLLC in South Charleston, West Virginia.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Debra L. Coplon, DNP, DCC, who practices at City of Memphis Wellness Clinic in Tennessee, and Cynthia A. Smith, DNP, APRN, FNP-BC, who practices with Renal Consultants PLLC in South Charleston, West Virginia.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Debra L. Coplon, DNP, DCC, who practices at City of Memphis Wellness Clinic in Tennessee, and Cynthia A. Smith, DNP, APRN, FNP-BC, who practices with Renal Consultants PLLC in South Charleston, West Virginia.
Q) At a lecture I recently attended, the speaker said sulfamethoxazole/trimethoprim is a potentially dangerous medication. I use it all the time. Is there any data to support her comments? Where did she get her information?
Sulfamethoxazole/trimethoprim (SMX/TMP) is a combination of two antibiotics, each of which has the potential to interact with other substances.
It is well documented that sulfamethoxazole can inhibit the metabolism of cytochrome P450 2C9 substrates. Frequently prescribed medications that also use the cytochrome substrate include warfarin and oral antihypoglycemic agents.
Trimethoprim’s distinct properties also lead to drug interactions. Trimethoprim inhibits sodium uptake by the appropriate channels in the distal tubule of the kidney, preventing reabsorption and altering the electrical balance of the tubular cells. As a result, the amount of potassium excreted into the urine is reduced, yielding an accumulation of serum potassium.1
High serum potassium retention can manifest as hyperkalemia in patients with chronic kidney disease (CKD). Use of potassium-sparing drugs by patients with comorbidities, including CKD, can increase risk for hyperkalemia; concurrent use of these drugs with ACE inhibitors or angiotensin II receptor blockers (ARBs) compounds the risk.2 The first reports of hyperkalemia with trimethoprim use occurred in HIV patients treated with large doses for Pneumocystis carinii infection.3
In a population-based case-control study, the results of which were published in the British Medical Journal, Fralick and colleagues analyzed data on older patients (age 66 or older) who were taking either ACE inhibitors or ARBs in combination with an antibiotic.4 They found a significantly increased risk for sudden death within seven days of prescription of SMX/TMP, compared to amoxicillin; a secondary analysis also revealed an increased risk for sudden death within 14 days with SMX/TMP. The researchers speculated that this excess risk, which translated to 3 sudden deaths in 1,000 patients taking SMX/TMP versus 1 sudden death in 1,000 patients taking amoxicillin, “reflects unrecognized arrhythmic death due to hyperkalemia.”
Since more than 250 million prescriptions for ACE inhibitors/ARBs and 20 million prescriptions for SMX/TMP are written each year, there will be instances of overlap. The prudent clinician would prescribe a different antibiotic or, if avoidance is not possible, use the lowest effective dose and duration of SMX/TMP. Close monitoring of serum potassium levels is warranted in patients with comorbidities, especially CKD, who are taking ACE inhibitors or ARBs—and of course, in our geriatric population. —DLC
Debra L. Coplon, DNP, DCC
City of Memphis Wellness Clinic, Tennessee
REFERENCES
1. Velazquez H, Perazella MA, Wright FS, Ellison DH. Renal mechanism of trimethoprim-induced hyperkalemia. Ann Intern Med. 1993;119:296-301.
2. Horn JR, Hansten PD. Trimethoprim and potassium-sparing drugs: a risk for hyperkalemia. www.pharmacytimes.com/publications/issue/2011/February2011/DrugInteractions-0211. Accessed August 24, 2015.
3. Medina I, Mills J, Leoung G, et al. Oral therapy for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome: a controlled trial of trimethoprim-sulfamethoxazole versus trimethoprim-dapsone. N Engl J Med. 1990;323:776-782.
4. Fralick M, Macdonald EM, Gomes T, et al. Co-trimoxazole and sudden death in patients receiving inhibitors of renin-angiotensin system: population based study. BMJ. 2014;349:g6196.
5. Gilbert SJ, Weiner DE, Gipson DS, et al. National Kidney Foundation’s Primer on Kidney Diseases. Philadelphia, PA: Elsevier; 2014.
6. Muriithi AK, Leung N, Valeri AM, et al. Biopsy-proven acute interstitial nephritis, 1993-2011: a case series. Am J Kidney Dis. 2014;64(4):558-566.
7. Blank ML, Parkin L, Paul C, Herbison P. A nationwide nested case-control study indicates an increased risk of acute interstitial nephritis with proton pump inhibitor use. Kidney Int. 2014;86(4):837-844.
8. Klepser DG, Collier DS, Cochran GL. Proton pump inhibitors and acute kidney injury: a nested case-control study. BMC Nephrology. 2013;14:150.
Q) At a lecture I recently attended, the speaker said sulfamethoxazole/trimethoprim is a potentially dangerous medication. I use it all the time. Is there any data to support her comments? Where did she get her information?
Sulfamethoxazole/trimethoprim (SMX/TMP) is a combination of two antibiotics, each of which has the potential to interact with other substances.
It is well documented that sulfamethoxazole can inhibit the metabolism of cytochrome P450 2C9 substrates. Frequently prescribed medications that also use the cytochrome substrate include warfarin and oral antihypoglycemic agents.
Trimethoprim’s distinct properties also lead to drug interactions. Trimethoprim inhibits sodium uptake by the appropriate channels in the distal tubule of the kidney, preventing reabsorption and altering the electrical balance of the tubular cells. As a result, the amount of potassium excreted into the urine is reduced, yielding an accumulation of serum potassium.1
High serum potassium retention can manifest as hyperkalemia in patients with chronic kidney disease (CKD). Use of potassium-sparing drugs by patients with comorbidities, including CKD, can increase risk for hyperkalemia; concurrent use of these drugs with ACE inhibitors or angiotensin II receptor blockers (ARBs) compounds the risk.2 The first reports of hyperkalemia with trimethoprim use occurred in HIV patients treated with large doses for Pneumocystis carinii infection.3
In a population-based case-control study, the results of which were published in the British Medical Journal, Fralick and colleagues analyzed data on older patients (age 66 or older) who were taking either ACE inhibitors or ARBs in combination with an antibiotic.4 They found a significantly increased risk for sudden death within seven days of prescription of SMX/TMP, compared to amoxicillin; a secondary analysis also revealed an increased risk for sudden death within 14 days with SMX/TMP. The researchers speculated that this excess risk, which translated to 3 sudden deaths in 1,000 patients taking SMX/TMP versus 1 sudden death in 1,000 patients taking amoxicillin, “reflects unrecognized arrhythmic death due to hyperkalemia.”
Since more than 250 million prescriptions for ACE inhibitors/ARBs and 20 million prescriptions for SMX/TMP are written each year, there will be instances of overlap. The prudent clinician would prescribe a different antibiotic or, if avoidance is not possible, use the lowest effective dose and duration of SMX/TMP. Close monitoring of serum potassium levels is warranted in patients with comorbidities, especially CKD, who are taking ACE inhibitors or ARBs—and of course, in our geriatric population. —DLC
Debra L. Coplon, DNP, DCC
City of Memphis Wellness Clinic, Tennessee
REFERENCES
1. Velazquez H, Perazella MA, Wright FS, Ellison DH. Renal mechanism of trimethoprim-induced hyperkalemia. Ann Intern Med. 1993;119:296-301.
2. Horn JR, Hansten PD. Trimethoprim and potassium-sparing drugs: a risk for hyperkalemia. www.pharmacytimes.com/publications/issue/2011/February2011/DrugInteractions-0211. Accessed August 24, 2015.
3. Medina I, Mills J, Leoung G, et al. Oral therapy for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome: a controlled trial of trimethoprim-sulfamethoxazole versus trimethoprim-dapsone. N Engl J Med. 1990;323:776-782.
4. Fralick M, Macdonald EM, Gomes T, et al. Co-trimoxazole and sudden death in patients receiving inhibitors of renin-angiotensin system: population based study. BMJ. 2014;349:g6196.
5. Gilbert SJ, Weiner DE, Gipson DS, et al. National Kidney Foundation’s Primer on Kidney Diseases. Philadelphia, PA: Elsevier; 2014.
6. Muriithi AK, Leung N, Valeri AM, et al. Biopsy-proven acute interstitial nephritis, 1993-2011: a case series. Am J Kidney Dis. 2014;64(4):558-566.
7. Blank ML, Parkin L, Paul C, Herbison P. A nationwide nested case-control study indicates an increased risk of acute interstitial nephritis with proton pump inhibitor use. Kidney Int. 2014;86(4):837-844.
8. Klepser DG, Collier DS, Cochran GL. Proton pump inhibitors and acute kidney injury: a nested case-control study. BMC Nephrology. 2013;14:150.
Protecting pregnant women, infants from infections
Infectious disease morbidity and mortality continue to disproportionately impact pregnant women and young infants.
In California, the incidence of pertussis approximates 100 cases per 100,000 in infants less than 5 months of age; a rate threefold greater than any other age group. Seven of nine (77%) deaths in 2013/2014 occurred in infants less than 3 months of age (California Department of Public Health Pertussis Report, Aug. 3, 2015).

Influenza severity and mortality is increased in pregnant women, and there is a greater risk of fetal morbidity and wastage. In the 2009 H1N1 pandemic, there was a 20% case fatality rate in women sick enough to be admitted to the ICU. The incidence of low birth weight also was increased among pregnant women delivering while hospitalized for influenza-related illness. These examples highlight the burden of vaccine-preventable disease in two vulnerable populations, pregnant women and infants too young to be protected by vaccines mandated by the U.S.immunization program.
The American College of Obstetricians and Gynecologists, the American Academy of Pediatrics, the Centers for Disease Control and Prevention, and many other national and state organizations endorse immunization of pregnant women to improve women’s and infants’ outcomes. Recent studies demonstrate that infants born to women vaccinated with influenza are 45%-48% less likely to be hospitalized for culture-proven influenza.
Benowitz et al. reported a 91.5% effectiveness for maternal influenza vaccination for prevention of hospitalization of infants caused by influenza in the first 6 months of life. The presumed mechanisms of protection are both the transplacental transfer of protective antibody as well as indirect protection from disease prevention in the mother (Clin Infect Dis. 2010 Dec 15;51(12):1355-61). The recommendation is that inactivated influenza vaccine can be given at any time during pregnancy; however, live attenuated influenza vaccine (LAIV; FluMist) is contraindicated, as are all live-virus vaccines. In contrast, Tdap is recommended for use either during pregnancy or post partum.
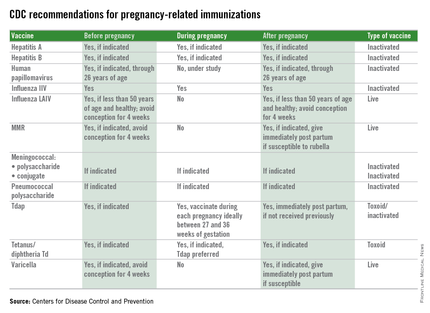
However, Healy et al. (Pediatr Infect Dis J. 2015;34(1):22-60) failed to demonstrate a benefit to postpartum immunization and cocooning for reducing pertussis illness in infants 6 months of age or younger. The likely explanation for this failure is revealed in a recent study in infant baboons where immunization with Tdap failed to decrease colonization or transmission of Bordetella pertussis, compared with natural disease or whole-cell pertussis. Thus, even though protective against disease, Tdap failure to prevent transmission within the community still occurs. The current Advisory Committee on Immunization Practices recommendation, immunization between 27 and 36 weeks, is designed to ensure high antibody concentrations in both mother and newborn at the time of birth and bridge the time period until infant immunization can elicit protective antibody.
The benefits achieved with maternal immunization must be weighed against potential for adverse events. There is no evidence of risk to either mother or infant from inactivated vaccines administered during pregnancy. Still, the recommendations for influenza and Tdap vaccine incorporate the high likelihood of exposure, the risk of morbidity or mortality from the infectious agent, and the likelihood of harm. During the H1N1 epidemic, a cohort study by Chambers et al. of H1N1 vaccine in exposed and unexposed pregnant women concluded that there was no increase in risk for major congenital defects, spontaneous abortion, or small for gestational age (Vaccine. 2013 Oct 17;31(44):5026-32). There was a signal for increase in prematurity, but the difference between H1N1-vaccinated and unvaccinated pregnancies was 3 days. In addition, a review of 11 studies, including one of 10,428 pregnant women, concluded there were no harmful maternal or fetal effects.
Additionally, no adverse risks have been identified in women who were inadvertently vaccinated during pregnancy with live-attenuated rubella, influenza, and yellow fever vaccines. Tetanus vaccination has been administered safely to several millions of pregnant women without documented serious adverse outcomes. Ongoing postmarketing surveillance continues as an important tool for identification of potential adverse effects.
One potential limitation is the blunting of infant immune responses to vaccination due to high serum antibody concentrations at the time of primary immunizations. Some studies have found lower antibody concentrations prior to booster vaccinations at 1 year of age. However, as morbidity and mortality is greater in the first months of life for many infectious diseases, this may be an acceptable trade off if high morbidity and mortality can be reduced in the first months of life.
Immunization during pregnancy represents only one aspect of prevention of vaccine preventable diseases. Preconception, prenatal, and postpartum visits with health care professionals represents an opportune time to discuss the benefits of immunization and their contribution to a healthy pregnancy outcome. Inactivated vaccines are safe for administration during pregnancy, live virus vaccines, despite being attenuated, are a theoretical risk if spread to the fetus occurs and therefore are contraindicated and should be administered during preconception counseling if indicated. The table below outlines vaccines that can be administered before, during, and after pregnancy.
Although once considered potentially contraindicated in pregnant women, evidence now supports specific vaccines as both safe for a pregnant woman and her fetus and effective for preventing serious disease in both. Universal immunization with influenza vaccine and Tdap, as recommended by multiple national professional medical organizations, will improve the outcome of pregnancy by prevention of morbidity and mortality from common community pathogens.
Dr. Pelton is chief of pediatric infectious disease and coordinator of the maternal-child HIV program at Boston Medical Center. E-mail him at [email protected].
Infectious disease morbidity and mortality continue to disproportionately impact pregnant women and young infants.
In California, the incidence of pertussis approximates 100 cases per 100,000 in infants less than 5 months of age; a rate threefold greater than any other age group. Seven of nine (77%) deaths in 2013/2014 occurred in infants less than 3 months of age (California Department of Public Health Pertussis Report, Aug. 3, 2015).
Influenza severity and mortality is increased in pregnant women, and there is a greater risk of fetal morbidity and wastage. In the 2009 H1N1 pandemic, there was a 20% case fatality rate in women sick enough to be admitted to the ICU. The incidence of low birth weight also was increased among pregnant women delivering while hospitalized for influenza-related illness. These examples highlight the burden of vaccine-preventable disease in two vulnerable populations, pregnant women and infants too young to be protected by vaccines mandated by the U.S.immunization program.
The American College of Obstetricians and Gynecologists, the American Academy of Pediatrics, the Centers for Disease Control and Prevention, and many other national and state organizations endorse immunization of pregnant women to improve women’s and infants’ outcomes. Recent studies demonstrate that infants born to women vaccinated with influenza are 45%-48% less likely to be hospitalized for culture-proven influenza.
Benowitz et al. reported a 91.5% effectiveness for maternal influenza vaccination for prevention of hospitalization of infants caused by influenza in the first 6 months of life. The presumed mechanisms of protection are both the transplacental transfer of protective antibody as well as indirect protection from disease prevention in the mother (Clin Infect Dis. 2010 Dec 15;51(12):1355-61). The recommendation is that inactivated influenza vaccine can be given at any time during pregnancy; however, live attenuated influenza vaccine (LAIV; FluMist) is contraindicated, as are all live-virus vaccines. In contrast, Tdap is recommended for use either during pregnancy or post partum.
However, Healy et al. (Pediatr Infect Dis J. 2015;34(1):22-60) failed to demonstrate a benefit to postpartum immunization and cocooning for reducing pertussis illness in infants 6 months of age or younger. The likely explanation for this failure is revealed in a recent study in infant baboons where immunization with Tdap failed to decrease colonization or transmission of Bordetella pertussis, compared with natural disease or whole-cell pertussis. Thus, even though protective against disease, Tdap failure to prevent transmission within the community still occurs. The current Advisory Committee on Immunization Practices recommendation, immunization between 27 and 36 weeks, is designed to ensure high antibody concentrations in both mother and newborn at the time of birth and bridge the time period until infant immunization can elicit protective antibody.
The benefits achieved with maternal immunization must be weighed against potential for adverse events. There is no evidence of risk to either mother or infant from inactivated vaccines administered during pregnancy. Still, the recommendations for influenza and Tdap vaccine incorporate the high likelihood of exposure, the risk of morbidity or mortality from the infectious agent, and the likelihood of harm. During the H1N1 epidemic, a cohort study by Chambers et al. of H1N1 vaccine in exposed and unexposed pregnant women concluded that there was no increase in risk for major congenital defects, spontaneous abortion, or small for gestational age (Vaccine. 2013 Oct 17;31(44):5026-32). There was a signal for increase in prematurity, but the difference between H1N1-vaccinated and unvaccinated pregnancies was 3 days. In addition, a review of 11 studies, including one of 10,428 pregnant women, concluded there were no harmful maternal or fetal effects.
Additionally, no adverse risks have been identified in women who were inadvertently vaccinated during pregnancy with live-attenuated rubella, influenza, and yellow fever vaccines. Tetanus vaccination has been administered safely to several millions of pregnant women without documented serious adverse outcomes. Ongoing postmarketing surveillance continues as an important tool for identification of potential adverse effects.
One potential limitation is the blunting of infant immune responses to vaccination due to high serum antibody concentrations at the time of primary immunizations. Some studies have found lower antibody concentrations prior to booster vaccinations at 1 year of age. However, as morbidity and mortality is greater in the first months of life for many infectious diseases, this may be an acceptable trade off if high morbidity and mortality can be reduced in the first months of life.
Immunization during pregnancy represents only one aspect of prevention of vaccine preventable diseases. Preconception, prenatal, and postpartum visits with health care professionals represents an opportune time to discuss the benefits of immunization and their contribution to a healthy pregnancy outcome. Inactivated vaccines are safe for administration during pregnancy, live virus vaccines, despite being attenuated, are a theoretical risk if spread to the fetus occurs and therefore are contraindicated and should be administered during preconception counseling if indicated. The table below outlines vaccines that can be administered before, during, and after pregnancy.
Although once considered potentially contraindicated in pregnant women, evidence now supports specific vaccines as both safe for a pregnant woman and her fetus and effective for preventing serious disease in both. Universal immunization with influenza vaccine and Tdap, as recommended by multiple national professional medical organizations, will improve the outcome of pregnancy by prevention of morbidity and mortality from common community pathogens.
Dr. Pelton is chief of pediatric infectious disease and coordinator of the maternal-child HIV program at Boston Medical Center. E-mail him at [email protected].
Infectious disease morbidity and mortality continue to disproportionately impact pregnant women and young infants.
In California, the incidence of pertussis approximates 100 cases per 100,000 in infants less than 5 months of age; a rate threefold greater than any other age group. Seven of nine (77%) deaths in 2013/2014 occurred in infants less than 3 months of age (California Department of Public Health Pertussis Report, Aug. 3, 2015).
Influenza severity and mortality is increased in pregnant women, and there is a greater risk of fetal morbidity and wastage. In the 2009 H1N1 pandemic, there was a 20% case fatality rate in women sick enough to be admitted to the ICU. The incidence of low birth weight also was increased among pregnant women delivering while hospitalized for influenza-related illness. These examples highlight the burden of vaccine-preventable disease in two vulnerable populations, pregnant women and infants too young to be protected by vaccines mandated by the U.S.immunization program.
The American College of Obstetricians and Gynecologists, the American Academy of Pediatrics, the Centers for Disease Control and Prevention, and many other national and state organizations endorse immunization of pregnant women to improve women’s and infants’ outcomes. Recent studies demonstrate that infants born to women vaccinated with influenza are 45%-48% less likely to be hospitalized for culture-proven influenza.
Benowitz et al. reported a 91.5% effectiveness for maternal influenza vaccination for prevention of hospitalization of infants caused by influenza in the first 6 months of life. The presumed mechanisms of protection are both the transplacental transfer of protective antibody as well as indirect protection from disease prevention in the mother (Clin Infect Dis. 2010 Dec 15;51(12):1355-61). The recommendation is that inactivated influenza vaccine can be given at any time during pregnancy; however, live attenuated influenza vaccine (LAIV; FluMist) is contraindicated, as are all live-virus vaccines. In contrast, Tdap is recommended for use either during pregnancy or post partum.
However, Healy et al. (Pediatr Infect Dis J. 2015;34(1):22-60) failed to demonstrate a benefit to postpartum immunization and cocooning for reducing pertussis illness in infants 6 months of age or younger. The likely explanation for this failure is revealed in a recent study in infant baboons where immunization with Tdap failed to decrease colonization or transmission of Bordetella pertussis, compared with natural disease or whole-cell pertussis. Thus, even though protective against disease, Tdap failure to prevent transmission within the community still occurs. The current Advisory Committee on Immunization Practices recommendation, immunization between 27 and 36 weeks, is designed to ensure high antibody concentrations in both mother and newborn at the time of birth and bridge the time period until infant immunization can elicit protective antibody.
The benefits achieved with maternal immunization must be weighed against potential for adverse events. There is no evidence of risk to either mother or infant from inactivated vaccines administered during pregnancy. Still, the recommendations for influenza and Tdap vaccine incorporate the high likelihood of exposure, the risk of morbidity or mortality from the infectious agent, and the likelihood of harm. During the H1N1 epidemic, a cohort study by Chambers et al. of H1N1 vaccine in exposed and unexposed pregnant women concluded that there was no increase in risk for major congenital defects, spontaneous abortion, or small for gestational age (Vaccine. 2013 Oct 17;31(44):5026-32). There was a signal for increase in prematurity, but the difference between H1N1-vaccinated and unvaccinated pregnancies was 3 days. In addition, a review of 11 studies, including one of 10,428 pregnant women, concluded there were no harmful maternal or fetal effects.
Additionally, no adverse risks have been identified in women who were inadvertently vaccinated during pregnancy with live-attenuated rubella, influenza, and yellow fever vaccines. Tetanus vaccination has been administered safely to several millions of pregnant women without documented serious adverse outcomes. Ongoing postmarketing surveillance continues as an important tool for identification of potential adverse effects.
One potential limitation is the blunting of infant immune responses to vaccination due to high serum antibody concentrations at the time of primary immunizations. Some studies have found lower antibody concentrations prior to booster vaccinations at 1 year of age. However, as morbidity and mortality is greater in the first months of life for many infectious diseases, this may be an acceptable trade off if high morbidity and mortality can be reduced in the first months of life.
Immunization during pregnancy represents only one aspect of prevention of vaccine preventable diseases. Preconception, prenatal, and postpartum visits with health care professionals represents an opportune time to discuss the benefits of immunization and their contribution to a healthy pregnancy outcome. Inactivated vaccines are safe for administration during pregnancy, live virus vaccines, despite being attenuated, are a theoretical risk if spread to the fetus occurs and therefore are contraindicated and should be administered during preconception counseling if indicated. The table below outlines vaccines that can be administered before, during, and after pregnancy.
Although once considered potentially contraindicated in pregnant women, evidence now supports specific vaccines as both safe for a pregnant woman and her fetus and effective for preventing serious disease in both. Universal immunization with influenza vaccine and Tdap, as recommended by multiple national professional medical organizations, will improve the outcome of pregnancy by prevention of morbidity and mortality from common community pathogens.
Dr. Pelton is chief of pediatric infectious disease and coordinator of the maternal-child HIV program at Boston Medical Center. E-mail him at [email protected].

A new, improved treatment approach for lymphoma?
Researchers believe they have discovered how therapy targeting CD47 harnesses the immune system to fight lymphoma and other cancers.
Conducting experiments in immune-competent mice, the team found that anti-CD47 therapy drives T-cell mediated elimination of lymphoma, colon cancer, and breast cancer.
The group’s research also revealed how the timing of chemotherapy administration affects anti-CD47 therapy.
Yang-Xin Fu, MD, PhD, of the University of Chicago in Illinois, and his colleagues described this research in Nature Medicine.
Previous research had shown that many cancer cells have CD47 on their surface. The protein instructs circulating macrophages not to devour the cells, but anti-CD47 therapy can negate this effect. This research relied on human tumors transplanted in immunocompromised mice.
With the current study, Dr Fu and his colleagues transplanted tumors from mice into genetically identical hosts with intact immune systems.
The team’s experiments revealed that anti-CD47-mediated tumor rejection requires both innate and adaptive immune responses. And the bulk of the therapeutic effect from CD47 blockade relies not on macrophages but on dendritic cells.
Dendritic cells proved more potent than macrophages at priming CD8+ T cells. Dendritic cells also caused type-1 interferon to boost adaptive immunity and activated the STING pathway, which was “absolutely essential for the antitumor effect of anti-CD47 therapy.”
The researchers also found evidence to suggest that chemotherapy should be administered before, rather than after, anti-CD47 therapy.
The team tested the anti-CD47 monoclonal antibody (mAb) MIAP301 in combination with clinically equivalent doses of cyclophosphamide or paclitaxel in mouse models of lymphoma (established A20 tumors).
When the chemotherapy was administered after the mAb, tumor regression was no faster than when the mAb was given alone.
In fact, the chemotherapy appeared to hinder antitumor memory responses generated by the mAb. When the researchers removed all tumors and rechallenged the mice with A20 cells, all of the mice that had received the mAb alone rejected the tumor rechallenge.
But mice that had received the mAb followed by chemotherapy were susceptible to tumor outgrowth—50% of cyclophosphamide-treated mice and 80% of paclitaxel-treated mice.
When chemotherapy was given before the mAb, however, it conferred benefits. A single dose of either chemotherapy drug synergized with the mAb to fight lymphoma.
And the treatment preserved the host memory response against relapsing tumors. All of the cyclophosphamide-treated mice and 80% of the paclitaxel-treated mice were resistant to tumor rechallenge.
The researchers said this suggests the order of treatment administration could have a major impact on primary and memory immune responses to tumors and alter outcomes of anti-CD47 therapy.
“Our results point to a new and more personalized strategy to modulate the tumor microenvironment,” Dr Fu said. “We think our approach, along with further investigation of scheduling and dosing, could improve survival and quality of life for patients battling advanced cancer.” ![]()
Researchers believe they have discovered how therapy targeting CD47 harnesses the immune system to fight lymphoma and other cancers.
Conducting experiments in immune-competent mice, the team found that anti-CD47 therapy drives T-cell mediated elimination of lymphoma, colon cancer, and breast cancer.
The group’s research also revealed how the timing of chemotherapy administration affects anti-CD47 therapy.
Yang-Xin Fu, MD, PhD, of the University of Chicago in Illinois, and his colleagues described this research in Nature Medicine.
Previous research had shown that many cancer cells have CD47 on their surface. The protein instructs circulating macrophages not to devour the cells, but anti-CD47 therapy can negate this effect. This research relied on human tumors transplanted in immunocompromised mice.
With the current study, Dr Fu and his colleagues transplanted tumors from mice into genetically identical hosts with intact immune systems.
The team’s experiments revealed that anti-CD47-mediated tumor rejection requires both innate and adaptive immune responses. And the bulk of the therapeutic effect from CD47 blockade relies not on macrophages but on dendritic cells.
Dendritic cells proved more potent than macrophages at priming CD8+ T cells. Dendritic cells also caused type-1 interferon to boost adaptive immunity and activated the STING pathway, which was “absolutely essential for the antitumor effect of anti-CD47 therapy.”
The researchers also found evidence to suggest that chemotherapy should be administered before, rather than after, anti-CD47 therapy.
The team tested the anti-CD47 monoclonal antibody (mAb) MIAP301 in combination with clinically equivalent doses of cyclophosphamide or paclitaxel in mouse models of lymphoma (established A20 tumors).
When the chemotherapy was administered after the mAb, tumor regression was no faster than when the mAb was given alone.
In fact, the chemotherapy appeared to hinder antitumor memory responses generated by the mAb. When the researchers removed all tumors and rechallenged the mice with A20 cells, all of the mice that had received the mAb alone rejected the tumor rechallenge.
But mice that had received the mAb followed by chemotherapy were susceptible to tumor outgrowth—50% of cyclophosphamide-treated mice and 80% of paclitaxel-treated mice.
When chemotherapy was given before the mAb, however, it conferred benefits. A single dose of either chemotherapy drug synergized with the mAb to fight lymphoma.
And the treatment preserved the host memory response against relapsing tumors. All of the cyclophosphamide-treated mice and 80% of the paclitaxel-treated mice were resistant to tumor rechallenge.
The researchers said this suggests the order of treatment administration could have a major impact on primary and memory immune responses to tumors and alter outcomes of anti-CD47 therapy.
“Our results point to a new and more personalized strategy to modulate the tumor microenvironment,” Dr Fu said. “We think our approach, along with further investigation of scheduling and dosing, could improve survival and quality of life for patients battling advanced cancer.” ![]()
Researchers believe they have discovered how therapy targeting CD47 harnesses the immune system to fight lymphoma and other cancers.
Conducting experiments in immune-competent mice, the team found that anti-CD47 therapy drives T-cell mediated elimination of lymphoma, colon cancer, and breast cancer.
The group’s research also revealed how the timing of chemotherapy administration affects anti-CD47 therapy.
Yang-Xin Fu, MD, PhD, of the University of Chicago in Illinois, and his colleagues described this research in Nature Medicine.
Previous research had shown that many cancer cells have CD47 on their surface. The protein instructs circulating macrophages not to devour the cells, but anti-CD47 therapy can negate this effect. This research relied on human tumors transplanted in immunocompromised mice.
With the current study, Dr Fu and his colleagues transplanted tumors from mice into genetically identical hosts with intact immune systems.
The team’s experiments revealed that anti-CD47-mediated tumor rejection requires both innate and adaptive immune responses. And the bulk of the therapeutic effect from CD47 blockade relies not on macrophages but on dendritic cells.
Dendritic cells proved more potent than macrophages at priming CD8+ T cells. Dendritic cells also caused type-1 interferon to boost adaptive immunity and activated the STING pathway, which was “absolutely essential for the antitumor effect of anti-CD47 therapy.”
The researchers also found evidence to suggest that chemotherapy should be administered before, rather than after, anti-CD47 therapy.
The team tested the anti-CD47 monoclonal antibody (mAb) MIAP301 in combination with clinically equivalent doses of cyclophosphamide or paclitaxel in mouse models of lymphoma (established A20 tumors).
When the chemotherapy was administered after the mAb, tumor regression was no faster than when the mAb was given alone.
In fact, the chemotherapy appeared to hinder antitumor memory responses generated by the mAb. When the researchers removed all tumors and rechallenged the mice with A20 cells, all of the mice that had received the mAb alone rejected the tumor rechallenge.
But mice that had received the mAb followed by chemotherapy were susceptible to tumor outgrowth—50% of cyclophosphamide-treated mice and 80% of paclitaxel-treated mice.
When chemotherapy was given before the mAb, however, it conferred benefits. A single dose of either chemotherapy drug synergized with the mAb to fight lymphoma.
And the treatment preserved the host memory response against relapsing tumors. All of the cyclophosphamide-treated mice and 80% of the paclitaxel-treated mice were resistant to tumor rechallenge.
The researchers said this suggests the order of treatment administration could have a major impact on primary and memory immune responses to tumors and alter outcomes of anti-CD47 therapy.
“Our results point to a new and more personalized strategy to modulate the tumor microenvironment,” Dr Fu said. “We think our approach, along with further investigation of scheduling and dosing, could improve survival and quality of life for patients battling advanced cancer.” ![]()