User login
Granulomatous Cheilitis: A Stiff Upper Lip
To the Editor:
A 51-year-old woman presented to her dermatologist with recurrent and progressive upper lip swelling of 2 years’ duration. Her condition was previously evaluated by several other physicians without a diagnosis or resolution of the symptoms. The swelling began on the right side of the upper lip and right cheek; however, over the course of 2 years, the swelling had progressed to involve the entire upper lip with complete sparing of the lower lip. She denied pain but reported numbness of the upper lip. The patient visited her dentist who ruled out periodontal infection as the cause of the swelling. Diphenhydramine provided no relief; however, the cheek swelling resolved after a course of antibiotics prescribed by an ear, nose, and throat physician.
She consulted her primary care physician and was subsequently referred to a neurologist and allergist who were unable to provide a definitive diagnosis or complete relief of the symptoms. She denied any history of hypersensitivity reactions, odontogenic infections, gastrointestinal concerns, or any other signs or symptoms of systemic granulomatous disease.
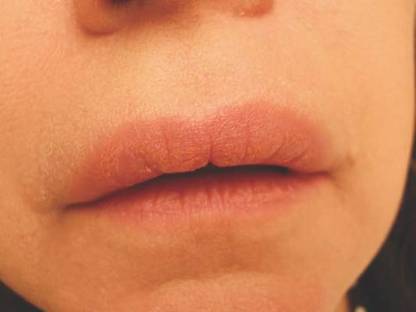
On physical examination, the upper lip was swollen symmetrically without evidence of ulceration, fissuring, or scaling (Figure 1). Palpation of the upper lip was notable for firm, nontender, nonpitting edema without nodularity. The oral mucosa did not appear swollen or erythematous. Examination did not reveal ulceration or a cobblestone appearance.
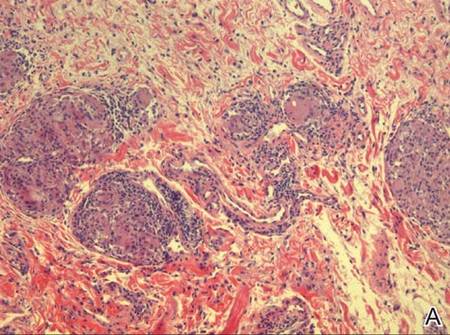
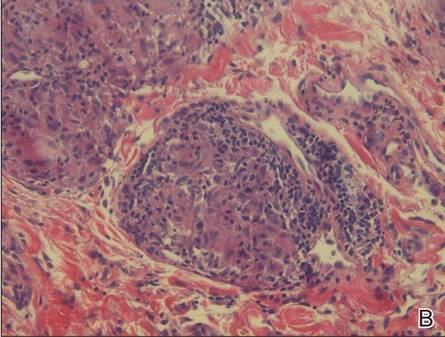
A full-thickness skin biopsy of the upper lip was performed. Histopathology revealed perivascular nonnecrotizing granulomas adjacent to ectatic vascular channels with associated lymphoplasmacytic infiltrate (Figure 2). Periodic acid–Schiff stain was negative for fungal hyphae, tissue Gram stain was negative for bacteria, Fite and acid-fast bacillus stains were both negative for acid-fast organisms, and polariscopy was negative for polarizable foreign material. In this clinical context, the morphologic findings were consistent with the diagnosis of granulomatous cheilitis (GC).
|
Figure 2. Upper lip biopsy showed dermal edema, vascular ectasia, perivascular nonnecrotizing granulomas, and perivascular lymphocyte predominant inflammatory infiltrate (A)(H&E, original magnification ×100). Higher magnification of granulomas with perivascular lymphoplasmacytic infiltrate (B)(H&E, original magnification ×200). |
Granulomatous cheilitis is a rare disorder of the lips and orofacial mucosa that was first described by Meischer1 in 1945 as persistent or recurrent orofacial swelling secondary to lymphatic obstruction by granulomatous proliferation. It often has been described as a monosymptomatic form of Melkersson-Rosenthal syndrome (MRS). In its entirety, MRS constitutes a triad of GC, facial nerve palsy, and lingua plicata (also known as fissured tongue).2,3 Although many authors agree that GC is associated with MRS, some believe that GC is a distinct entity because the majority of patients who present with GC subsequently do not develop MRS.4 Despite its relationship to MRS, the true incidence of GC largely is unknown. The onset of disease usually occurs in early adulthood but can present in middle-aged or older individuals.
The typical course of GC is relapsing and remitting, nontender and nonpitting swelling of the lips that eventually becomes permanent, leading to possible facial distortion and disability. Involvement of the upper lip is the most common, followed by (in order of decreasing frequency) the lower lip and cheeks.5 The swelling may be unilateral or bilateral and generally is not associated with ulceration, fissuring, or scaling; however, these complications have been reported in the terminal stages of the disease in which the macrocheilia has become permanent.
Despite the controversy over the etiology, pathophysiology, and classification of GC, it largely is accepted that when a patient presents clinically with a history of recurrent or persistent lip swelling, a full-thickness skin biopsy of the involved oral mucosa should be taken. Conditions that are considered in the differential diagnosis of orofacial granulomatosis are systemic granulomatous diseases that are known to have oral manifestations including Crohn disease, sarcoidosis, and mycobacterial infections. Given the many causes of orofacial and labial swelling, GC is a diagnosis of exclusion based on a thorough history and physical examination as well as appropriate diagnostic studies, with the cornerstone of the diagnosis resting on the histologic appearance of the lesion. Histologically, the diagnosis lies in the demonstration of granuloma formation, consisting of collections of epithelioid histiocytes and Langerhans giant cells. Once granuloma formation is documented, special stains are used to rule out other granulomatous diseases.
Intralesional steroids have been reported to provide the greatest improvement; however, in the majority of patients, multiple treatments are required.6,7 Allen et al8 suggested that the efficacy of intralesional therapy increases when preceded by local anesthesia of the lip, thus allowing larger doses of triamcinolone to be tolerated by the patient. Systemic corticosteroids also have been used with moderate success, but the side effects of long-term systemic corticosteroid therapy make this treatment option less appealing.9 Other agents with known anti-inflammatory properties also have been used that may offer better side-effect profiles when used for long-term suppressive therapy, including clofazimine, dapsone, sulfapyridine, danazol, hydroxychloroquine, and antibiotics such as doxycycline and metronidazole.10
In severe or recalcitrant cases, surgical intervention by way of a reduction cheiloplasty is considered by some to be an appropriate next step in therapy but is rarely needed. Postoperative intralesional steroid injections are necessary due to reported cases of worsening disease when injections are discontinued after cheiloplasty.11,12
Our patient was treated with 5 mg of intralesional triamcinolone acetonide with 10 separate injections of 0.5 cc each along the affected portions of the upper lip. She also was given doxycycline 100 mg once daily for 30 days. The patient reported complete resolution of the upper lip swelling 7 days after the initiation of therapy. At 1-month follow-up, she reported that the swelling had completely resolved. However, 1 day prior to the scheduled visit, shortly after finishing the course of doxycycline, she noted recurrent swelling. Due to the concomitant initial administration of both the steroid injections and doxycycline, it was unclear which treatment had provided relief. To avoid, or at least delay, the need for chronic intralesional steroid injections, another course of 40 mg doxycycline daily was prescribed. After 2 weeks, the patient reported that the swelling had markedly improved. The patient has maintained remission of the symptoms for approximately 6 months on daily suppressive therapy with 40 mg of doxycycline.
The recurrence of lip swelling after therapy, as in our patient, is typical of GC, and most cases require multiple follow-up visits and frequent alterations in therapy, which is often frustrating for both the patient and physician. However, awareness of this disease entity, its natural course, and the therapeutic options will allow physicians to more appropriately counsel and educate patients of this uncommon disease process.
1. Meischer G. Über essentielle granulomatöse makrocheilie (cheilitis granulomatosa). Dermatologica. 1945;91:57-85.
2. Melkersson E. Ett Fall av recidiverande facialispares i samband med angioneurotiskt ödem. Hygiea (Stockh). 1928;90:737-741.
3. Rosenthal C. Klinish-erbbiologischer beitrag zur konstitutionspathologie: gemeinsames auftreten von (rezidiverender familiärer) facialislähmung, angioneurotischem gesichtsödem und lingua plicata in arthritismus-familien. Z Ges Neurol Psychiat. 1931;131:475-501.
4. van der Waal RI, Schulten EA, van der Meij EH, et al. Cheilitis granulomatosa: overview of 13 patients with long-term follow up–results of management. Int J Dermatol. 2002;41:225-229.
5. Worsaae N, Christensen KC, Schiødt M, et al. Melkersson-Rosenthal syndrome and cheilitis granulomatosa. a clinical pathological study of thirty-three patients with special reference to their oral lesions. Oral Surg Oral Med Oral Pathol. 1982;54:404-413.
6. El-Hakim M, Chauvin P. Orofacial granulomatosis presenting as persistent lip swelling: review of 6 new cases. J Oral Maxillofac Surg. 2004;62:1114-1117.
7. Williams PM, Greenberg MS. Management of cheilitis granulomatosa. Oral Surg Oral Med Oral Pathol. 1991;72:436-439.
8. Allen CM, Camisa C, Hamzeh S, et al. Cheilitis granulomatosa: report of six cases and review of the literature. J Am Acad Dermatol. 1990;23(3, pt 1):444-450.
9. Banks T, Gada S. A comprehensive review of current treatments for granulomatous cheilitis. Br J Dermatol. 2012;166:934-937.
10. Sciubba JJ, Said-Al-Naief N. Orofacial granulomatosis: presentation, pathology and management of 13 cases. J Oral Pathol Med. 2003;32:576-585.
11. Glickman LT, Gruss JS, Birt BD, et al. The surgical management of Melkersson-Rosenthal syndrome. Plast Reconstr Surg. 1992;89:815-821.
12. Krutchkoff D, James R. Cheilitis granulomatosa. successful treatment with combined local triamcinolone injections and surgery. Arch Dermatol. 1978;114:1203-1206.
To the Editor:
A 51-year-old woman presented to her dermatologist with recurrent and progressive upper lip swelling of 2 years’ duration. Her condition was previously evaluated by several other physicians without a diagnosis or resolution of the symptoms. The swelling began on the right side of the upper lip and right cheek; however, over the course of 2 years, the swelling had progressed to involve the entire upper lip with complete sparing of the lower lip. She denied pain but reported numbness of the upper lip. The patient visited her dentist who ruled out periodontal infection as the cause of the swelling. Diphenhydramine provided no relief; however, the cheek swelling resolved after a course of antibiotics prescribed by an ear, nose, and throat physician.
She consulted her primary care physician and was subsequently referred to a neurologist and allergist who were unable to provide a definitive diagnosis or complete relief of the symptoms. She denied any history of hypersensitivity reactions, odontogenic infections, gastrointestinal concerns, or any other signs or symptoms of systemic granulomatous disease.
On physical examination, the upper lip was swollen symmetrically without evidence of ulceration, fissuring, or scaling (Figure 1). Palpation of the upper lip was notable for firm, nontender, nonpitting edema without nodularity. The oral mucosa did not appear swollen or erythematous. Examination did not reveal ulceration or a cobblestone appearance.
A full-thickness skin biopsy of the upper lip was performed. Histopathology revealed perivascular nonnecrotizing granulomas adjacent to ectatic vascular channels with associated lymphoplasmacytic infiltrate (Figure 2). Periodic acid–Schiff stain was negative for fungal hyphae, tissue Gram stain was negative for bacteria, Fite and acid-fast bacillus stains were both negative for acid-fast organisms, and polariscopy was negative for polarizable foreign material. In this clinical context, the morphologic findings were consistent with the diagnosis of granulomatous cheilitis (GC).
|
|
Figure 2. Upper lip biopsy showed dermal edema, vascular ectasia, perivascular nonnecrotizing granulomas, and perivascular lymphocyte predominant inflammatory infiltrate (A)(H&E, original magnification ×100). Higher magnification of granulomas with perivascular lymphoplasmacytic infiltrate (B)(H&E, original magnification ×200). |
Granulomatous cheilitis is a rare disorder of the lips and orofacial mucosa that was first described by Meischer1 in 1945 as persistent or recurrent orofacial swelling secondary to lymphatic obstruction by granulomatous proliferation. It often has been described as a monosymptomatic form of Melkersson-Rosenthal syndrome (MRS). In its entirety, MRS constitutes a triad of GC, facial nerve palsy, and lingua plicata (also known as fissured tongue).2,3 Although many authors agree that GC is associated with MRS, some believe that GC is a distinct entity because the majority of patients who present with GC subsequently do not develop MRS.4 Despite its relationship to MRS, the true incidence of GC largely is unknown. The onset of disease usually occurs in early adulthood but can present in middle-aged or older individuals.
The typical course of GC is relapsing and remitting, nontender and nonpitting swelling of the lips that eventually becomes permanent, leading to possible facial distortion and disability. Involvement of the upper lip is the most common, followed by (in order of decreasing frequency) the lower lip and cheeks.5 The swelling may be unilateral or bilateral and generally is not associated with ulceration, fissuring, or scaling; however, these complications have been reported in the terminal stages of the disease in which the macrocheilia has become permanent.
Despite the controversy over the etiology, pathophysiology, and classification of GC, it largely is accepted that when a patient presents clinically with a history of recurrent or persistent lip swelling, a full-thickness skin biopsy of the involved oral mucosa should be taken. Conditions that are considered in the differential diagnosis of orofacial granulomatosis are systemic granulomatous diseases that are known to have oral manifestations including Crohn disease, sarcoidosis, and mycobacterial infections. Given the many causes of orofacial and labial swelling, GC is a diagnosis of exclusion based on a thorough history and physical examination as well as appropriate diagnostic studies, with the cornerstone of the diagnosis resting on the histologic appearance of the lesion. Histologically, the diagnosis lies in the demonstration of granuloma formation, consisting of collections of epithelioid histiocytes and Langerhans giant cells. Once granuloma formation is documented, special stains are used to rule out other granulomatous diseases.
Intralesional steroids have been reported to provide the greatest improvement; however, in the majority of patients, multiple treatments are required.6,7 Allen et al8 suggested that the efficacy of intralesional therapy increases when preceded by local anesthesia of the lip, thus allowing larger doses of triamcinolone to be tolerated by the patient. Systemic corticosteroids also have been used with moderate success, but the side effects of long-term systemic corticosteroid therapy make this treatment option less appealing.9 Other agents with known anti-inflammatory properties also have been used that may offer better side-effect profiles when used for long-term suppressive therapy, including clofazimine, dapsone, sulfapyridine, danazol, hydroxychloroquine, and antibiotics such as doxycycline and metronidazole.10
In severe or recalcitrant cases, surgical intervention by way of a reduction cheiloplasty is considered by some to be an appropriate next step in therapy but is rarely needed. Postoperative intralesional steroid injections are necessary due to reported cases of worsening disease when injections are discontinued after cheiloplasty.11,12
Our patient was treated with 5 mg of intralesional triamcinolone acetonide with 10 separate injections of 0.5 cc each along the affected portions of the upper lip. She also was given doxycycline 100 mg once daily for 30 days. The patient reported complete resolution of the upper lip swelling 7 days after the initiation of therapy. At 1-month follow-up, she reported that the swelling had completely resolved. However, 1 day prior to the scheduled visit, shortly after finishing the course of doxycycline, she noted recurrent swelling. Due to the concomitant initial administration of both the steroid injections and doxycycline, it was unclear which treatment had provided relief. To avoid, or at least delay, the need for chronic intralesional steroid injections, another course of 40 mg doxycycline daily was prescribed. After 2 weeks, the patient reported that the swelling had markedly improved. The patient has maintained remission of the symptoms for approximately 6 months on daily suppressive therapy with 40 mg of doxycycline.
The recurrence of lip swelling after therapy, as in our patient, is typical of GC, and most cases require multiple follow-up visits and frequent alterations in therapy, which is often frustrating for both the patient and physician. However, awareness of this disease entity, its natural course, and the therapeutic options will allow physicians to more appropriately counsel and educate patients of this uncommon disease process.
To the Editor:
A 51-year-old woman presented to her dermatologist with recurrent and progressive upper lip swelling of 2 years’ duration. Her condition was previously evaluated by several other physicians without a diagnosis or resolution of the symptoms. The swelling began on the right side of the upper lip and right cheek; however, over the course of 2 years, the swelling had progressed to involve the entire upper lip with complete sparing of the lower lip. She denied pain but reported numbness of the upper lip. The patient visited her dentist who ruled out periodontal infection as the cause of the swelling. Diphenhydramine provided no relief; however, the cheek swelling resolved after a course of antibiotics prescribed by an ear, nose, and throat physician.
She consulted her primary care physician and was subsequently referred to a neurologist and allergist who were unable to provide a definitive diagnosis or complete relief of the symptoms. She denied any history of hypersensitivity reactions, odontogenic infections, gastrointestinal concerns, or any other signs or symptoms of systemic granulomatous disease.
On physical examination, the upper lip was swollen symmetrically without evidence of ulceration, fissuring, or scaling (Figure 1). Palpation of the upper lip was notable for firm, nontender, nonpitting edema without nodularity. The oral mucosa did not appear swollen or erythematous. Examination did not reveal ulceration or a cobblestone appearance.
A full-thickness skin biopsy of the upper lip was performed. Histopathology revealed perivascular nonnecrotizing granulomas adjacent to ectatic vascular channels with associated lymphoplasmacytic infiltrate (Figure 2). Periodic acid–Schiff stain was negative for fungal hyphae, tissue Gram stain was negative for bacteria, Fite and acid-fast bacillus stains were both negative for acid-fast organisms, and polariscopy was negative for polarizable foreign material. In this clinical context, the morphologic findings were consistent with the diagnosis of granulomatous cheilitis (GC).
|
|
Figure 2. Upper lip biopsy showed dermal edema, vascular ectasia, perivascular nonnecrotizing granulomas, and perivascular lymphocyte predominant inflammatory infiltrate (A)(H&E, original magnification ×100). Higher magnification of granulomas with perivascular lymphoplasmacytic infiltrate (B)(H&E, original magnification ×200). |
Granulomatous cheilitis is a rare disorder of the lips and orofacial mucosa that was first described by Meischer1 in 1945 as persistent or recurrent orofacial swelling secondary to lymphatic obstruction by granulomatous proliferation. It often has been described as a monosymptomatic form of Melkersson-Rosenthal syndrome (MRS). In its entirety, MRS constitutes a triad of GC, facial nerve palsy, and lingua plicata (also known as fissured tongue).2,3 Although many authors agree that GC is associated with MRS, some believe that GC is a distinct entity because the majority of patients who present with GC subsequently do not develop MRS.4 Despite its relationship to MRS, the true incidence of GC largely is unknown. The onset of disease usually occurs in early adulthood but can present in middle-aged or older individuals.
The typical course of GC is relapsing and remitting, nontender and nonpitting swelling of the lips that eventually becomes permanent, leading to possible facial distortion and disability. Involvement of the upper lip is the most common, followed by (in order of decreasing frequency) the lower lip and cheeks.5 The swelling may be unilateral or bilateral and generally is not associated with ulceration, fissuring, or scaling; however, these complications have been reported in the terminal stages of the disease in which the macrocheilia has become permanent.
Despite the controversy over the etiology, pathophysiology, and classification of GC, it largely is accepted that when a patient presents clinically with a history of recurrent or persistent lip swelling, a full-thickness skin biopsy of the involved oral mucosa should be taken. Conditions that are considered in the differential diagnosis of orofacial granulomatosis are systemic granulomatous diseases that are known to have oral manifestations including Crohn disease, sarcoidosis, and mycobacterial infections. Given the many causes of orofacial and labial swelling, GC is a diagnosis of exclusion based on a thorough history and physical examination as well as appropriate diagnostic studies, with the cornerstone of the diagnosis resting on the histologic appearance of the lesion. Histologically, the diagnosis lies in the demonstration of granuloma formation, consisting of collections of epithelioid histiocytes and Langerhans giant cells. Once granuloma formation is documented, special stains are used to rule out other granulomatous diseases.
Intralesional steroids have been reported to provide the greatest improvement; however, in the majority of patients, multiple treatments are required.6,7 Allen et al8 suggested that the efficacy of intralesional therapy increases when preceded by local anesthesia of the lip, thus allowing larger doses of triamcinolone to be tolerated by the patient. Systemic corticosteroids also have been used with moderate success, but the side effects of long-term systemic corticosteroid therapy make this treatment option less appealing.9 Other agents with known anti-inflammatory properties also have been used that may offer better side-effect profiles when used for long-term suppressive therapy, including clofazimine, dapsone, sulfapyridine, danazol, hydroxychloroquine, and antibiotics such as doxycycline and metronidazole.10
In severe or recalcitrant cases, surgical intervention by way of a reduction cheiloplasty is considered by some to be an appropriate next step in therapy but is rarely needed. Postoperative intralesional steroid injections are necessary due to reported cases of worsening disease when injections are discontinued after cheiloplasty.11,12
Our patient was treated with 5 mg of intralesional triamcinolone acetonide with 10 separate injections of 0.5 cc each along the affected portions of the upper lip. She also was given doxycycline 100 mg once daily for 30 days. The patient reported complete resolution of the upper lip swelling 7 days after the initiation of therapy. At 1-month follow-up, she reported that the swelling had completely resolved. However, 1 day prior to the scheduled visit, shortly after finishing the course of doxycycline, she noted recurrent swelling. Due to the concomitant initial administration of both the steroid injections and doxycycline, it was unclear which treatment had provided relief. To avoid, or at least delay, the need for chronic intralesional steroid injections, another course of 40 mg doxycycline daily was prescribed. After 2 weeks, the patient reported that the swelling had markedly improved. The patient has maintained remission of the symptoms for approximately 6 months on daily suppressive therapy with 40 mg of doxycycline.
The recurrence of lip swelling after therapy, as in our patient, is typical of GC, and most cases require multiple follow-up visits and frequent alterations in therapy, which is often frustrating for both the patient and physician. However, awareness of this disease entity, its natural course, and the therapeutic options will allow physicians to more appropriately counsel and educate patients of this uncommon disease process.
1. Meischer G. Über essentielle granulomatöse makrocheilie (cheilitis granulomatosa). Dermatologica. 1945;91:57-85.
2. Melkersson E. Ett Fall av recidiverande facialispares i samband med angioneurotiskt ödem. Hygiea (Stockh). 1928;90:737-741.
3. Rosenthal C. Klinish-erbbiologischer beitrag zur konstitutionspathologie: gemeinsames auftreten von (rezidiverender familiärer) facialislähmung, angioneurotischem gesichtsödem und lingua plicata in arthritismus-familien. Z Ges Neurol Psychiat. 1931;131:475-501.
4. van der Waal RI, Schulten EA, van der Meij EH, et al. Cheilitis granulomatosa: overview of 13 patients with long-term follow up–results of management. Int J Dermatol. 2002;41:225-229.
5. Worsaae N, Christensen KC, Schiødt M, et al. Melkersson-Rosenthal syndrome and cheilitis granulomatosa. a clinical pathological study of thirty-three patients with special reference to their oral lesions. Oral Surg Oral Med Oral Pathol. 1982;54:404-413.
6. El-Hakim M, Chauvin P. Orofacial granulomatosis presenting as persistent lip swelling: review of 6 new cases. J Oral Maxillofac Surg. 2004;62:1114-1117.
7. Williams PM, Greenberg MS. Management of cheilitis granulomatosa. Oral Surg Oral Med Oral Pathol. 1991;72:436-439.
8. Allen CM, Camisa C, Hamzeh S, et al. Cheilitis granulomatosa: report of six cases and review of the literature. J Am Acad Dermatol. 1990;23(3, pt 1):444-450.
9. Banks T, Gada S. A comprehensive review of current treatments for granulomatous cheilitis. Br J Dermatol. 2012;166:934-937.
10. Sciubba JJ, Said-Al-Naief N. Orofacial granulomatosis: presentation, pathology and management of 13 cases. J Oral Pathol Med. 2003;32:576-585.
11. Glickman LT, Gruss JS, Birt BD, et al. The surgical management of Melkersson-Rosenthal syndrome. Plast Reconstr Surg. 1992;89:815-821.
12. Krutchkoff D, James R. Cheilitis granulomatosa. successful treatment with combined local triamcinolone injections and surgery. Arch Dermatol. 1978;114:1203-1206.
1. Meischer G. Über essentielle granulomatöse makrocheilie (cheilitis granulomatosa). Dermatologica. 1945;91:57-85.
2. Melkersson E. Ett Fall av recidiverande facialispares i samband med angioneurotiskt ödem. Hygiea (Stockh). 1928;90:737-741.
3. Rosenthal C. Klinish-erbbiologischer beitrag zur konstitutionspathologie: gemeinsames auftreten von (rezidiverender familiärer) facialislähmung, angioneurotischem gesichtsödem und lingua plicata in arthritismus-familien. Z Ges Neurol Psychiat. 1931;131:475-501.
4. van der Waal RI, Schulten EA, van der Meij EH, et al. Cheilitis granulomatosa: overview of 13 patients with long-term follow up–results of management. Int J Dermatol. 2002;41:225-229.
5. Worsaae N, Christensen KC, Schiødt M, et al. Melkersson-Rosenthal syndrome and cheilitis granulomatosa. a clinical pathological study of thirty-three patients with special reference to their oral lesions. Oral Surg Oral Med Oral Pathol. 1982;54:404-413.
6. El-Hakim M, Chauvin P. Orofacial granulomatosis presenting as persistent lip swelling: review of 6 new cases. J Oral Maxillofac Surg. 2004;62:1114-1117.
7. Williams PM, Greenberg MS. Management of cheilitis granulomatosa. Oral Surg Oral Med Oral Pathol. 1991;72:436-439.
8. Allen CM, Camisa C, Hamzeh S, et al. Cheilitis granulomatosa: report of six cases and review of the literature. J Am Acad Dermatol. 1990;23(3, pt 1):444-450.
9. Banks T, Gada S. A comprehensive review of current treatments for granulomatous cheilitis. Br J Dermatol. 2012;166:934-937.
10. Sciubba JJ, Said-Al-Naief N. Orofacial granulomatosis: presentation, pathology and management of 13 cases. J Oral Pathol Med. 2003;32:576-585.
11. Glickman LT, Gruss JS, Birt BD, et al. The surgical management of Melkersson-Rosenthal syndrome. Plast Reconstr Surg. 1992;89:815-821.
12. Krutchkoff D, James R. Cheilitis granulomatosa. successful treatment with combined local triamcinolone injections and surgery. Arch Dermatol. 1978;114:1203-1206.
Painful Skin Lesions on the Hands Following Black Henna Application
The Diagnosis: Allergic Contact Dermatitis to Para-phenylenediamine
To darken the color of henna and increase penetrance and staining, para-phenylenediamine (PPD) is added.1 Allergic contact dermatitis is the most common type of hypersensitivity to PPD.2 A retrospective study that examined severe adverse events from applying henna dyes in children found that angioedema of mucosal tissues was the most common severe adverse event; others included renal failure and shock.3
Black henna is associated with multiple cultural practices. For example, Indian weddings contain a henna decoration ceremony for the bride based on the belief that the longer the henna lasts, the longer the marriage lasts. Black henna is favored for this practice, as it lasts longer than red henna.

Henna (Lawsonia inermis) is a plant that contains the molecule lawsone (naphthoquinone). Lawsone has an intense affinity for keratin; as a result, lawsone is frequently added to temporary body tattoos and hair dyes to create a relatively permanent change in skin or hair color.4 Henna is mixed with hennotannic acid to release the lawsone from the plant. Lawsone and hennotannic acid rarely cause allergic reactions.1,5-7 Once applied to skin, henna takes a few hours to dry, and the resulting color is orange to red.8 Often, PPD is added to henna paste to create a black color, to speed up the drying process, and to increase its longevity.
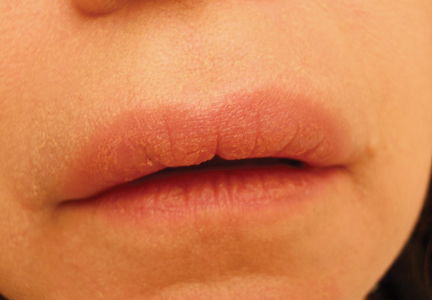
Para-phenylenediamine has been repeatedly reported to cause allergic contact dermatitis. We describe a case of allergic contact dermatitis secondary to PPD in black henna. Our patient is a clear example that PPD is the allergen in black henna given that there was no reaction to the natural red henna tattoo that was applied at the same time to the palmar surfaces of the hands (Figure). Aside from the bullous reaction to black henna dye described here, other reported presentations include erythema multiforme–like and exudative erythema reactions.9,10
Contact dermatitis lesions from black henna dye can be treated with topical corticosteroids. Patients may develop residual postinflammatory hyperpigmentation or hypopigmentation, leukoderma, keloids, or scars.1,11,12
- Onder M, Atahan CA, Oztas P, et al. Temporary henna tattoo reactions in children. Int J Dermatol. 2001;40:577-579.
- Marcoux D, Couture-Trudel PM, Rboulet-Delmas G, et al. Sensitization to paraphenylenediame from a streetside temporary tattoo. Pediatr Dermatol. 2002;19:498-502.
- Hashim S, Hamza Y, Yahia B, et al. Poisoning from henna dye and para-phenylenediamine mixtures in children in Khartoum. Ann Trop Paediatr. 1992;12:3-6.
- Hijji Y, Barare B, Zhang Y. Lawsone (2- hydroxy-1, 4-naphthoquinone) as a sensitive cyanide and acetate sensor. Sensors and Actuators B: Chemical. 2012;169:106-112.
- Neri I, Guareschi E, Savoia F, et al. Childhood allergic contact dermatitis from henna tattoo. Pediatr Dermatol. 2002;19:503-505.
- Evans CC, Fleming JD. Allergic contact dermatitis from a henna tattoo. N Engl J Med. 2008;359:627.
- Belhadjali H, Akkari H, Youssef M, et al. Bullous allergic contact dermatitis to pure henna in a 3-year-old girl. Pediatr Dermatol. 2011;28:580-581.
- Najem N, Bagher Zadeh V. Allergic contact dermatitis to black henna. Acta Dermatovenerol Alp Pannonica Adriat. 2011;20:87-88.
- Sidwell RU, Francis ND, Basarab T, et al. Vesicular erythema multiforme-like reaction to para-phenylenediamine in a henna tattoo. Pediatr Dermatol. 2008;25:201-204.
- Jovanovic DL, Slavkovic-Jovanovic MR. Allergic contact dermatitis from temporary henna tattoo. J Dermatol. 2009;36:63-65.
- Valsecchi R, Leghissa P, Di Landro A, et al. Persistent leukoderma after henna tattoo. Contact Dermatitis. 2007;56:108-109.
- Gunasti S, Aksungur VL. Severe inflammatory and keloidal, allergic reaction due to para-phenylenediamine in temporary tattoos. Indian J Dermatol Venereol Leprol. 2010;76:165-167.
The Diagnosis: Allergic Contact Dermatitis to Para-phenylenediamine
To darken the color of henna and increase penetrance and staining, para-phenylenediamine (PPD) is added.1 Allergic contact dermatitis is the most common type of hypersensitivity to PPD.2 A retrospective study that examined severe adverse events from applying henna dyes in children found that angioedema of mucosal tissues was the most common severe adverse event; others included renal failure and shock.3
Black henna is associated with multiple cultural practices. For example, Indian weddings contain a henna decoration ceremony for the bride based on the belief that the longer the henna lasts, the longer the marriage lasts. Black henna is favored for this practice, as it lasts longer than red henna.
Henna (Lawsonia inermis) is a plant that contains the molecule lawsone (naphthoquinone). Lawsone has an intense affinity for keratin; as a result, lawsone is frequently added to temporary body tattoos and hair dyes to create a relatively permanent change in skin or hair color.4 Henna is mixed with hennotannic acid to release the lawsone from the plant. Lawsone and hennotannic acid rarely cause allergic reactions.1,5-7 Once applied to skin, henna takes a few hours to dry, and the resulting color is orange to red.8 Often, PPD is added to henna paste to create a black color, to speed up the drying process, and to increase its longevity.
Para-phenylenediamine has been repeatedly reported to cause allergic contact dermatitis. We describe a case of allergic contact dermatitis secondary to PPD in black henna. Our patient is a clear example that PPD is the allergen in black henna given that there was no reaction to the natural red henna tattoo that was applied at the same time to the palmar surfaces of the hands (Figure). Aside from the bullous reaction to black henna dye described here, other reported presentations include erythema multiforme–like and exudative erythema reactions.9,10
Contact dermatitis lesions from black henna dye can be treated with topical corticosteroids. Patients may develop residual postinflammatory hyperpigmentation or hypopigmentation, leukoderma, keloids, or scars.1,11,12
The Diagnosis: Allergic Contact Dermatitis to Para-phenylenediamine
To darken the color of henna and increase penetrance and staining, para-phenylenediamine (PPD) is added.1 Allergic contact dermatitis is the most common type of hypersensitivity to PPD.2 A retrospective study that examined severe adverse events from applying henna dyes in children found that angioedema of mucosal tissues was the most common severe adverse event; others included renal failure and shock.3
Black henna is associated with multiple cultural practices. For example, Indian weddings contain a henna decoration ceremony for the bride based on the belief that the longer the henna lasts, the longer the marriage lasts. Black henna is favored for this practice, as it lasts longer than red henna.
Henna (Lawsonia inermis) is a plant that contains the molecule lawsone (naphthoquinone). Lawsone has an intense affinity for keratin; as a result, lawsone is frequently added to temporary body tattoos and hair dyes to create a relatively permanent change in skin or hair color.4 Henna is mixed with hennotannic acid to release the lawsone from the plant. Lawsone and hennotannic acid rarely cause allergic reactions.1,5-7 Once applied to skin, henna takes a few hours to dry, and the resulting color is orange to red.8 Often, PPD is added to henna paste to create a black color, to speed up the drying process, and to increase its longevity.
Para-phenylenediamine has been repeatedly reported to cause allergic contact dermatitis. We describe a case of allergic contact dermatitis secondary to PPD in black henna. Our patient is a clear example that PPD is the allergen in black henna given that there was no reaction to the natural red henna tattoo that was applied at the same time to the palmar surfaces of the hands (Figure). Aside from the bullous reaction to black henna dye described here, other reported presentations include erythema multiforme–like and exudative erythema reactions.9,10
Contact dermatitis lesions from black henna dye can be treated with topical corticosteroids. Patients may develop residual postinflammatory hyperpigmentation or hypopigmentation, leukoderma, keloids, or scars.1,11,12
- Onder M, Atahan CA, Oztas P, et al. Temporary henna tattoo reactions in children. Int J Dermatol. 2001;40:577-579.
- Marcoux D, Couture-Trudel PM, Rboulet-Delmas G, et al. Sensitization to paraphenylenediame from a streetside temporary tattoo. Pediatr Dermatol. 2002;19:498-502.
- Hashim S, Hamza Y, Yahia B, et al. Poisoning from henna dye and para-phenylenediamine mixtures in children in Khartoum. Ann Trop Paediatr. 1992;12:3-6.
- Hijji Y, Barare B, Zhang Y. Lawsone (2- hydroxy-1, 4-naphthoquinone) as a sensitive cyanide and acetate sensor. Sensors and Actuators B: Chemical. 2012;169:106-112.
- Neri I, Guareschi E, Savoia F, et al. Childhood allergic contact dermatitis from henna tattoo. Pediatr Dermatol. 2002;19:503-505.
- Evans CC, Fleming JD. Allergic contact dermatitis from a henna tattoo. N Engl J Med. 2008;359:627.
- Belhadjali H, Akkari H, Youssef M, et al. Bullous allergic contact dermatitis to pure henna in a 3-year-old girl. Pediatr Dermatol. 2011;28:580-581.
- Najem N, Bagher Zadeh V. Allergic contact dermatitis to black henna. Acta Dermatovenerol Alp Pannonica Adriat. 2011;20:87-88.
- Sidwell RU, Francis ND, Basarab T, et al. Vesicular erythema multiforme-like reaction to para-phenylenediamine in a henna tattoo. Pediatr Dermatol. 2008;25:201-204.
- Jovanovic DL, Slavkovic-Jovanovic MR. Allergic contact dermatitis from temporary henna tattoo. J Dermatol. 2009;36:63-65.
- Valsecchi R, Leghissa P, Di Landro A, et al. Persistent leukoderma after henna tattoo. Contact Dermatitis. 2007;56:108-109.
- Gunasti S, Aksungur VL. Severe inflammatory and keloidal, allergic reaction due to para-phenylenediamine in temporary tattoos. Indian J Dermatol Venereol Leprol. 2010;76:165-167.
- Onder M, Atahan CA, Oztas P, et al. Temporary henna tattoo reactions in children. Int J Dermatol. 2001;40:577-579.
- Marcoux D, Couture-Trudel PM, Rboulet-Delmas G, et al. Sensitization to paraphenylenediame from a streetside temporary tattoo. Pediatr Dermatol. 2002;19:498-502.
- Hashim S, Hamza Y, Yahia B, et al. Poisoning from henna dye and para-phenylenediamine mixtures in children in Khartoum. Ann Trop Paediatr. 1992;12:3-6.
- Hijji Y, Barare B, Zhang Y. Lawsone (2- hydroxy-1, 4-naphthoquinone) as a sensitive cyanide and acetate sensor. Sensors and Actuators B: Chemical. 2012;169:106-112.
- Neri I, Guareschi E, Savoia F, et al. Childhood allergic contact dermatitis from henna tattoo. Pediatr Dermatol. 2002;19:503-505.
- Evans CC, Fleming JD. Allergic contact dermatitis from a henna tattoo. N Engl J Med. 2008;359:627.
- Belhadjali H, Akkari H, Youssef M, et al. Bullous allergic contact dermatitis to pure henna in a 3-year-old girl. Pediatr Dermatol. 2011;28:580-581.
- Najem N, Bagher Zadeh V. Allergic contact dermatitis to black henna. Acta Dermatovenerol Alp Pannonica Adriat. 2011;20:87-88.
- Sidwell RU, Francis ND, Basarab T, et al. Vesicular erythema multiforme-like reaction to para-phenylenediamine in a henna tattoo. Pediatr Dermatol. 2008;25:201-204.
- Jovanovic DL, Slavkovic-Jovanovic MR. Allergic contact dermatitis from temporary henna tattoo. J Dermatol. 2009;36:63-65.
- Valsecchi R, Leghissa P, Di Landro A, et al. Persistent leukoderma after henna tattoo. Contact Dermatitis. 2007;56:108-109.
- Gunasti S, Aksungur VL. Severe inflammatory and keloidal, allergic reaction due to para-phenylenediamine in temporary tattoos. Indian J Dermatol Venereol Leprol. 2010;76:165-167.

A 14-year-old adolescent girl presented with painful skin lesions on the dorsal aspect of the hands of 10 days’ duration. She reported having received red henna tattoo on the palmar surface of the hands and black henna tattoo on the dorsal surface of the hands 1 day prior to development of the lesions. Within 1 day of receiving the tattoo, she developed pruritus, blisters, and pain on the dorsal aspect of the hands. The palms were unaffected. Physical examination revealed erythematous, brown to black bullae and crusts that followed the contours of the henna design on the dorsal aspect of the hands. There were orange and brown henna designs on the patient’s palms, but no erythema, bullae, or induration was noted.

Is the smartphone recording while the patient is under anesthesia?
CASE: Physician defames sedated patient
Our case takes us to the Commonwealth of Virginia. A male patient preparing to undergo a colonoscopy was concerned that, because of grogginess brought on by anesthesia, he might misunderstand postprocedure instructions or advice. He, therefore, turned his cell phone’s record function “on” and put it with his clothes. His clothes were put in a plastic bag, which ended up under the table with him in the operating room.
Following the procedure, as his wife drove him home, the patient replayed the instructions on the cell phone and realized that it had recorded the entire procedure. It quickly became apparent that the medical personnel had engaged in a series of inappropriate and insulting comments at the patient’s expense.
The anesthesiologist, talking to the now-unconscious patient, said, “after five minutes of talking to you in pre-op, I wanted to punch you in the face.” The patient had reported he was taking medication for a mild penile rash. The anesthesiologist warned an assistant not to touch it or “you might get syphilis on your arm or something,” but then noted, “it’s probably tuberculosis of the penis, so you’ll be all right.” There was further mocking of the patient, including a question of whether he was homosexual.
The anesthesiologist and gastroenterologist wanted to avoid talking to the patient after the procedure, and the gastroenterologist instructed an assistant to lie to the patient and convince the patient that the gastroenterologist had already spoken to him following the colonoscopy but, “you just don’t remember it.” In addition, the anesthesiologist announced that she was going to mark “hemorrhoids” on the patient’s chart, which she knew was a false diagnosis.
The patient, who is identified only by initials, is an attorney.1 Of course, the smartphone was “good documentation” of what came out of what the health care team said.
The lawsuit
The patient (now plaintiff) claimed that he was verbally brutalized and suffered anxiety, embarrassment, and loss of sleep for several months.
On the first day of trial, the gastroenterologist was dismissed from the case. The trial went on against the anesthesiologist and the anesthesia practice.
What’s the verdict?
The patient was awarded $500,000, as follows:
- $100,000 for defamation, ($50,000 each for the syphilis and tuberculosis comments),
- $200,000 for medical malpractice
- $200,000 in punitive damages (including $50,000 the jury found that the anesthesia practice should pay).
Caveat. The above facts about this case come from the plaintiff’s complaint1 and various professional commentaries and news sources.2–5 Such sources are not always reliable, so they may not describe accurately all of the relevant events and statements.
Neither of the authors of this column attended the trial or heard the testimony presented. For the purposes of discussing the issues below, however, we treat as true the facts stated above. In addition, some of the legal claims in this case are uncertain. It is entirely possible that an appeal will be made and accepted, and some or all of the damages could be reduced by the trial court or an appellate court. The jury award, therefore, is not necessarily the last word.
Medicolegal takeaways from this case
This case raises a number of professional, ethical, and legal issues. Most fundamentally, the health care team is always expected to prioritize the patient’s best interest. Respect for the patient is an essential element of that.
Behaviors such as those reported about these physicians are “absolutely not to engage in any time,” stated President of the American Society of Anesthesiologists John Absentein, MD.6 A former president of the Academy of Anesthesiology, Kathryn McGoldrick, MD, added some common sense advice that such discussions are “not only offensive but frankly stupid.” As she notes, “we can never be certain that our patients are asleep and wouldn’t have recall.”7
The actions of the physicians also may violate ethical obligations. The very first principle of medical ethics is that the physician shall provide care “with compassion and respect for human dignity and rights.”8
The legal claims included defamation, infliction of emotional distress, privacy (related to medical records), and malpractice. We will take a very brief look at each of those causes of action and then say a word about punitive damages (which the jury awarded in this case).
It is important to remember that state law, rather than federal, is providing the legal principles on which these claims were decided. Federal law might provide some relevant principles in such cases (for example, the First Amendment freedom of speech limits the state defamation rules), but that is the exception. State law is the rule.
Patient−physician recordings and the law
State laws differ regarding when it is legal to record in-person conversations. When everyone in the conversations knows about the recording, it is permissible and can be used in a court of law. In most states it is legal to record when only one party to the conversation has agreed to it, even though others in the conversation are not aware of it (which was the situation in the case discussed here).
In theory, physicians (by contract with patients) might try to limit patients’ rights to record medical services. But that practice would be difficult to implement or enforce in many circumstances. The reality is that audio and video recording devices are so ubiquitous that it is not sensible to avoid all recording of patient contact.
Physicians also might consider the potential such recordings have in some circumstances to improve communication with patients. Permitting the patient to record the patient−physician exchange, for instance, allows the patient the ability to review the advice after having left the office. This could be beneficial from a patient care perspective.
Defamation—award of damages
At its core, defamation is publishing (that is, telling someone other than the plaintiff) something untrue that may be harmful to another person. Generally the harm is reputational and the plaintiff may be affected by loss of business, mental suffering, or loss of esteem in the community.9
Defamation claims are not typical in health care cases. However, these claims are not rare: instances of health care professionals defaming other health care professionals, patients giving negative “reviews,” or health care professionals releasing false information to employers certainly do exist.
In this case, in addition to saying that the patient had syphilis and tuberculosis (both untrue), the physicians said he was a “wimp.” One interesting concept of defamation law that has developed over the centuries is “negligence per se.” This means a falsehood has been published about someone and the falsehood is likely to cause serious reputational harm. Claims that someone has a contagious disease traditionally have been considered negligence per se. Syphilis and tuberculosis fall in that category. On the other hand, saying someone needs to “man up” is usually a matter of opinion, so defamation for such comments is unlikely without special circumstances.
From the anesthesiologist’s perspective, the question is whether anyone who heard the publication really believed that the patient had either of the diseases. A joke that nobody believes to be based on fact generally is not defamatory because it has not harmed the plaintiff.10 It is apparent that the jury felt the patient had been defamed, however, given the $100,000 award for defamation.
In the United States there is special sensitivity to defamation awards because they may implicate the First Amendment’s protection of free speech. That being the case, this award may be particularly open to review by the judge and appellate courts.
Emotional distress—no award of damages
There are 2 kinds of “emotional distress” claimed in this case:
- intentional infliction
- negligent infliction.
Intentional infliction usually requires outrageous conduct by the defendant who acts intentionally or recklessly to inflict severe mental pain on the plaintiff.11 In this case, the element of “intentional” or “reckless” is interesting. While the conduct was outrageous, it is doubtful that there was any way the anesthesiologist could have imagined that these outrageous statements would have been transmitted to the patient/plaintiff.
As for negligent infliction of emotional distress, most states have been wary of opening a Pandora’s Box of litigation. Therefore, they generally require significant physical manifestations of great stress to allow recovery.12 It appears that the jury did not find the elements of either intentional or negligent infliction of emotional distress in this case.
As a side-note, this kind of emotional distress is viewed by the law as different from emotional distress that is incidental to a physical injury (pain and suffering). All states recognize that form of emotional distress.
Privacy—no award of damages
The privacy of medical records has, of course, become a major concern in the last few years. Both federal and state law provides significant penalties for the unauthorized release of medical information. However, in this case, it does not appear that medical information was improp- erly revealed.13
The patient’s complaint suggested that the anesthesiologist’s discussion during the colonoscopy of the medication for the penile rash was unnecessary for health care purposes.1 Therefore, it claims, the discussion violated the state health records privacy law. At the same time there was no indication in the public reports that this caused any harm to the patient.
Medical malpractice—award of damages
Malpractice usually involves professional practice that is unacceptable to the profession itself. It most commonly is negligence, or carelessness, that causes injury to the patient. The gross disregard for professional medical standards here was certainly negligence.14 The plaintiff claimed that discussing the medication for the penile rash and falsification of the medical records constituted malpractice.1
Presumably the jury award for medical malpractice means the jury found that the misconduct of the medical staff caused the emotional harm that the plaintiff experienced (described as embarrassment, loss of sleep, mental anguish, and anxiety), and that those injuries warranted a $200,000 award.
Punitive damage—award of damages
The jury also awarded $200,000 in “punitive” or “exemplary” damages. These are unusual damages, given not so much to compensate the victim but rather as a deterrent for the future. Generally the defendant’s conduct must have been egregious and completely unacceptable.15 Those elements were apparent to the jury from the facts of this case.
What about loss of practice privileges?
It is not unlikely that one or more of the medical professionals might, beyond civil liability, be subject to licensure discipline by the Virginia board. In addition, there are other secondary consequences of this lawsuit. The employment of those involved may be interrupted. (The anesthesiologist is said to have moved to another state, for example.) Hospital privileges also may be affected, as may insurance rates. The results of this award likely will have to be reported to the National Practitioner Data Bank.
As physicians, what’s our takeaway?
Conduct unbecoming a physician remains front and center with a recent essay published in the internal medicine literature.16 The anonymous author attests to witnessing a male gynecologist making sexual comments regarding the patient at the time of vaginal surgery preparation and an obstetrician singing and dancing to a Mexican song while treating his Hispanic patient for postpartum bleeding.
The unusual case of the anesthesiologist that we address was made even more unusual by the fact that it was recorded. Recordings, however, are likely to become ever more common. The advice of everyone’s grandmother is well taken: “Always act as though what you do will be published on the front page of the newspaper.” The ubiquitous presence of video and audio cameras and untold other devices means that someone may well be watching.
Aside from the risk of getting caught, respect for patients and clients is the very foundation of respect and professional care. It is distressing that the anesthesiologist was so disrespectful of a patient. It is equally disappointing that nobody put a stop to it.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- D.B. v Safe Sedation, Complaint, Civil Action 2014-05265, Circuit Court of Fairfax County.
- Abbott R. Unconscious patient says doctors mocked him. Courthouse News Service. http://www.courthousenews.com/2014/04/22/67225.htm. Updated April 22, 2014. Accessed August 19, 2015.
- Jackman T. Anesthesiologist trashes sedated patient—and it ends up costing her. Washington Post. June 23, 2015. http://www.washingtonpost.com/local/anesthesiologist-trashes-sedated-patient-jury-orders-her-to-pay-500000/2015/06/23/cae05c00-18f3-11e5-ab92-c75ae6ab94b5_story.html. Accessed August 19, 2015.
- Waibel E. Patient says Bethesda practitioners mocked him during the colonoscopy. GazetteNet. May 13, 2014. http://www.gazette.net/article/20140513/NEWS/140519703/1070/patient-says-bethesda-practitioners-mocked-him-during-colonoscopy&template=gazette. Accessed July 15, 2015.
- Vieth P. Fairfax County Circuit Court: Doctors allegedly mocked their unconscious patient. Virginia Lawyers Weekly. May 1, 2014. http://valawyersweekly.com/2014/05/01/doctors-allegedly-mocked-their-unconscious-patient. Accessed August 19, 2015.
- Welch A. Patient sues anesthesiologist who mocked him while sedated. CBS News. http://www.cbsnews.com/news/patient-sues-anesthesiloigst-who mocked-him-while sedated. Accessed July 15, 2015.
- Leins C. Anesthesiologist derides subdued patient, loses lawsuit. US News. June 24, 2015. http://www.usnews.com/news/articles/2015/06/24/anesthesiologist-derides-subdued-patient-loses-lawsuit. Accessed August 19, 2015.
- Principles of medical ethics. American Medical Association Web site. http://www.ama-assn.org/ama/pub/physician-resources/medical-ethics/code-medical-ethics/principles-medical-ethics.page. Accessed August 19, 2015.
- Instruction nos. 24 and 25. Virginia Defamation Lawyer Web site. http://www.virginiadefamationlawyer.com/Instr%2024%20and%2025.pdf. Accessed August 19, 2015.
- Berlik LE. The Virginia model jury instructions for defamation lead to bad verdicts. The Virginia Defamation Law Blog. June 27, 2015. http://www.virginiadefamationlawyer.com/2015/06/the-virginia-model-jury-instructions-for-defamation-lead-to-bad-verdicts.html#more. Accessed August 19, 2015.
- Russo v White, 241 Va 23 (1991).
- Hughes v Moore, 214 Va 27 (1973).
- Law on patient health records/privacy. Virginia Department of Health Professions Web site.
- http://webcache.googleusercontent.com/search?q=cache:asc1xQmBefoJ:https://www.dhp.virginia.gov/dhp_laws/Law_Patient%2520Health%2520Records.doc+&cd=2&hl=en&ct=clnk&gl=us. Accessed August 19, 2015.
- Virginia Medical Malpractice Act. Va Code Ann. 8.01-230, 8.01-243(A).
- Ford CR. Pleading and understanding punitive-damages claims in Virginia. Litigation News. 2008;8(10):1-11.
Joseph S. Sanfilippo, MD, MBA, and Steven R. Smith, JD
In this quarterly column, these medical and legal experts and educators present a case-based discussion and provide clear teaching points and takeaways for your practice.

Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology & Infertility, at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the OBG Management Board of Editors.

Mr. Smith is Professor of Law and Dean Emeritus at California Western School of Law, San Diego, California.
The authors report no financial relationships relevant to this article.
Joseph S. Sanfilippo, MD, MBA, and Steven R. Smith, JD
In this quarterly column, these medical and legal experts and educators present a case-based discussion and provide clear teaching points and takeaways for your practice.
Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology & Infertility, at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the OBG Management Board of Editors.
Mr. Smith is Professor of Law and Dean Emeritus at California Western School of Law, San Diego, California.
The authors report no financial relationships relevant to this article.
Joseph S. Sanfilippo, MD, MBA, and Steven R. Smith, JD
In this quarterly column, these medical and legal experts and educators present a case-based discussion and provide clear teaching points and takeaways for your practice.
Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology & Infertility, at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the OBG Management Board of Editors.
Mr. Smith is Professor of Law and Dean Emeritus at California Western School of Law, San Diego, California.
The authors report no financial relationships relevant to this article.
CASE: Physician defames sedated patient
Our case takes us to the Commonwealth of Virginia. A male patient preparing to undergo a colonoscopy was concerned that, because of grogginess brought on by anesthesia, he might misunderstand postprocedure instructions or advice. He, therefore, turned his cell phone’s record function “on” and put it with his clothes. His clothes were put in a plastic bag, which ended up under the table with him in the operating room.
Following the procedure, as his wife drove him home, the patient replayed the instructions on the cell phone and realized that it had recorded the entire procedure. It quickly became apparent that the medical personnel had engaged in a series of inappropriate and insulting comments at the patient’s expense.
The anesthesiologist, talking to the now-unconscious patient, said, “after five minutes of talking to you in pre-op, I wanted to punch you in the face.” The patient had reported he was taking medication for a mild penile rash. The anesthesiologist warned an assistant not to touch it or “you might get syphilis on your arm or something,” but then noted, “it’s probably tuberculosis of the penis, so you’ll be all right.” There was further mocking of the patient, including a question of whether he was homosexual.
The anesthesiologist and gastroenterologist wanted to avoid talking to the patient after the procedure, and the gastroenterologist instructed an assistant to lie to the patient and convince the patient that the gastroenterologist had already spoken to him following the colonoscopy but, “you just don’t remember it.” In addition, the anesthesiologist announced that she was going to mark “hemorrhoids” on the patient’s chart, which she knew was a false diagnosis.
The patient, who is identified only by initials, is an attorney.1 Of course, the smartphone was “good documentation” of what came out of what the health care team said.
The lawsuit
The patient (now plaintiff) claimed that he was verbally brutalized and suffered anxiety, embarrassment, and loss of sleep for several months.
On the first day of trial, the gastroenterologist was dismissed from the case. The trial went on against the anesthesiologist and the anesthesia practice.
What’s the verdict?
The patient was awarded $500,000, as follows:
- $100,000 for defamation, ($50,000 each for the syphilis and tuberculosis comments),
- $200,000 for medical malpractice
- $200,000 in punitive damages (including $50,000 the jury found that the anesthesia practice should pay).
Caveat. The above facts about this case come from the plaintiff’s complaint1 and various professional commentaries and news sources.2–5 Such sources are not always reliable, so they may not describe accurately all of the relevant events and statements.
Neither of the authors of this column attended the trial or heard the testimony presented. For the purposes of discussing the issues below, however, we treat as true the facts stated above. In addition, some of the legal claims in this case are uncertain. It is entirely possible that an appeal will be made and accepted, and some or all of the damages could be reduced by the trial court or an appellate court. The jury award, therefore, is not necessarily the last word.
Medicolegal takeaways from this case
This case raises a number of professional, ethical, and legal issues. Most fundamentally, the health care team is always expected to prioritize the patient’s best interest. Respect for the patient is an essential element of that.
Behaviors such as those reported about these physicians are “absolutely not to engage in any time,” stated President of the American Society of Anesthesiologists John Absentein, MD.6 A former president of the Academy of Anesthesiology, Kathryn McGoldrick, MD, added some common sense advice that such discussions are “not only offensive but frankly stupid.” As she notes, “we can never be certain that our patients are asleep and wouldn’t have recall.”7
The actions of the physicians also may violate ethical obligations. The very first principle of medical ethics is that the physician shall provide care “with compassion and respect for human dignity and rights.”8
The legal claims included defamation, infliction of emotional distress, privacy (related to medical records), and malpractice. We will take a very brief look at each of those causes of action and then say a word about punitive damages (which the jury awarded in this case).
It is important to remember that state law, rather than federal, is providing the legal principles on which these claims were decided. Federal law might provide some relevant principles in such cases (for example, the First Amendment freedom of speech limits the state defamation rules), but that is the exception. State law is the rule.
Patient−physician recordings and the law
State laws differ regarding when it is legal to record in-person conversations. When everyone in the conversations knows about the recording, it is permissible and can be used in a court of law. In most states it is legal to record when only one party to the conversation has agreed to it, even though others in the conversation are not aware of it (which was the situation in the case discussed here).
In theory, physicians (by contract with patients) might try to limit patients’ rights to record medical services. But that practice would be difficult to implement or enforce in many circumstances. The reality is that audio and video recording devices are so ubiquitous that it is not sensible to avoid all recording of patient contact.
Physicians also might consider the potential such recordings have in some circumstances to improve communication with patients. Permitting the patient to record the patient−physician exchange, for instance, allows the patient the ability to review the advice after having left the office. This could be beneficial from a patient care perspective.
Defamation—award of damages
At its core, defamation is publishing (that is, telling someone other than the plaintiff) something untrue that may be harmful to another person. Generally the harm is reputational and the plaintiff may be affected by loss of business, mental suffering, or loss of esteem in the community.9
Defamation claims are not typical in health care cases. However, these claims are not rare: instances of health care professionals defaming other health care professionals, patients giving negative “reviews,” or health care professionals releasing false information to employers certainly do exist.
In this case, in addition to saying that the patient had syphilis and tuberculosis (both untrue), the physicians said he was a “wimp.” One interesting concept of defamation law that has developed over the centuries is “negligence per se.” This means a falsehood has been published about someone and the falsehood is likely to cause serious reputational harm. Claims that someone has a contagious disease traditionally have been considered negligence per se. Syphilis and tuberculosis fall in that category. On the other hand, saying someone needs to “man up” is usually a matter of opinion, so defamation for such comments is unlikely without special circumstances.
From the anesthesiologist’s perspective, the question is whether anyone who heard the publication really believed that the patient had either of the diseases. A joke that nobody believes to be based on fact generally is not defamatory because it has not harmed the plaintiff.10 It is apparent that the jury felt the patient had been defamed, however, given the $100,000 award for defamation.
In the United States there is special sensitivity to defamation awards because they may implicate the First Amendment’s protection of free speech. That being the case, this award may be particularly open to review by the judge and appellate courts.
Emotional distress—no award of damages
There are 2 kinds of “emotional distress” claimed in this case:
- intentional infliction
- negligent infliction.
Intentional infliction usually requires outrageous conduct by the defendant who acts intentionally or recklessly to inflict severe mental pain on the plaintiff.11 In this case, the element of “intentional” or “reckless” is interesting. While the conduct was outrageous, it is doubtful that there was any way the anesthesiologist could have imagined that these outrageous statements would have been transmitted to the patient/plaintiff.
As for negligent infliction of emotional distress, most states have been wary of opening a Pandora’s Box of litigation. Therefore, they generally require significant physical manifestations of great stress to allow recovery.12 It appears that the jury did not find the elements of either intentional or negligent infliction of emotional distress in this case.
As a side-note, this kind of emotional distress is viewed by the law as different from emotional distress that is incidental to a physical injury (pain and suffering). All states recognize that form of emotional distress.
Privacy—no award of damages
The privacy of medical records has, of course, become a major concern in the last few years. Both federal and state law provides significant penalties for the unauthorized release of medical information. However, in this case, it does not appear that medical information was improp- erly revealed.13
The patient’s complaint suggested that the anesthesiologist’s discussion during the colonoscopy of the medication for the penile rash was unnecessary for health care purposes.1 Therefore, it claims, the discussion violated the state health records privacy law. At the same time there was no indication in the public reports that this caused any harm to the patient.
Medical malpractice—award of damages
Malpractice usually involves professional practice that is unacceptable to the profession itself. It most commonly is negligence, or carelessness, that causes injury to the patient. The gross disregard for professional medical standards here was certainly negligence.14 The plaintiff claimed that discussing the medication for the penile rash and falsification of the medical records constituted malpractice.1
Presumably the jury award for medical malpractice means the jury found that the misconduct of the medical staff caused the emotional harm that the plaintiff experienced (described as embarrassment, loss of sleep, mental anguish, and anxiety), and that those injuries warranted a $200,000 award.
Punitive damage—award of damages
The jury also awarded $200,000 in “punitive” or “exemplary” damages. These are unusual damages, given not so much to compensate the victim but rather as a deterrent for the future. Generally the defendant’s conduct must have been egregious and completely unacceptable.15 Those elements were apparent to the jury from the facts of this case.
What about loss of practice privileges?
It is not unlikely that one or more of the medical professionals might, beyond civil liability, be subject to licensure discipline by the Virginia board. In addition, there are other secondary consequences of this lawsuit. The employment of those involved may be interrupted. (The anesthesiologist is said to have moved to another state, for example.) Hospital privileges also may be affected, as may insurance rates. The results of this award likely will have to be reported to the National Practitioner Data Bank.
As physicians, what’s our takeaway?
Conduct unbecoming a physician remains front and center with a recent essay published in the internal medicine literature.16 The anonymous author attests to witnessing a male gynecologist making sexual comments regarding the patient at the time of vaginal surgery preparation and an obstetrician singing and dancing to a Mexican song while treating his Hispanic patient for postpartum bleeding.
The unusual case of the anesthesiologist that we address was made even more unusual by the fact that it was recorded. Recordings, however, are likely to become ever more common. The advice of everyone’s grandmother is well taken: “Always act as though what you do will be published on the front page of the newspaper.” The ubiquitous presence of video and audio cameras and untold other devices means that someone may well be watching.
Aside from the risk of getting caught, respect for patients and clients is the very foundation of respect and professional care. It is distressing that the anesthesiologist was so disrespectful of a patient. It is equally disappointing that nobody put a stop to it.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
CASE: Physician defames sedated patient
Our case takes us to the Commonwealth of Virginia. A male patient preparing to undergo a colonoscopy was concerned that, because of grogginess brought on by anesthesia, he might misunderstand postprocedure instructions or advice. He, therefore, turned his cell phone’s record function “on” and put it with his clothes. His clothes were put in a plastic bag, which ended up under the table with him in the operating room.
Following the procedure, as his wife drove him home, the patient replayed the instructions on the cell phone and realized that it had recorded the entire procedure. It quickly became apparent that the medical personnel had engaged in a series of inappropriate and insulting comments at the patient’s expense.
The anesthesiologist, talking to the now-unconscious patient, said, “after five minutes of talking to you in pre-op, I wanted to punch you in the face.” The patient had reported he was taking medication for a mild penile rash. The anesthesiologist warned an assistant not to touch it or “you might get syphilis on your arm or something,” but then noted, “it’s probably tuberculosis of the penis, so you’ll be all right.” There was further mocking of the patient, including a question of whether he was homosexual.
The anesthesiologist and gastroenterologist wanted to avoid talking to the patient after the procedure, and the gastroenterologist instructed an assistant to lie to the patient and convince the patient that the gastroenterologist had already spoken to him following the colonoscopy but, “you just don’t remember it.” In addition, the anesthesiologist announced that she was going to mark “hemorrhoids” on the patient’s chart, which she knew was a false diagnosis.
The patient, who is identified only by initials, is an attorney.1 Of course, the smartphone was “good documentation” of what came out of what the health care team said.
The lawsuit
The patient (now plaintiff) claimed that he was verbally brutalized and suffered anxiety, embarrassment, and loss of sleep for several months.
On the first day of trial, the gastroenterologist was dismissed from the case. The trial went on against the anesthesiologist and the anesthesia practice.
What’s the verdict?
The patient was awarded $500,000, as follows:
- $100,000 for defamation, ($50,000 each for the syphilis and tuberculosis comments),
- $200,000 for medical malpractice
- $200,000 in punitive damages (including $50,000 the jury found that the anesthesia practice should pay).
Caveat. The above facts about this case come from the plaintiff’s complaint1 and various professional commentaries and news sources.2–5 Such sources are not always reliable, so they may not describe accurately all of the relevant events and statements.
Neither of the authors of this column attended the trial or heard the testimony presented. For the purposes of discussing the issues below, however, we treat as true the facts stated above. In addition, some of the legal claims in this case are uncertain. It is entirely possible that an appeal will be made and accepted, and some or all of the damages could be reduced by the trial court or an appellate court. The jury award, therefore, is not necessarily the last word.
Medicolegal takeaways from this case
This case raises a number of professional, ethical, and legal issues. Most fundamentally, the health care team is always expected to prioritize the patient’s best interest. Respect for the patient is an essential element of that.
Behaviors such as those reported about these physicians are “absolutely not to engage in any time,” stated President of the American Society of Anesthesiologists John Absentein, MD.6 A former president of the Academy of Anesthesiology, Kathryn McGoldrick, MD, added some common sense advice that such discussions are “not only offensive but frankly stupid.” As she notes, “we can never be certain that our patients are asleep and wouldn’t have recall.”7
The actions of the physicians also may violate ethical obligations. The very first principle of medical ethics is that the physician shall provide care “with compassion and respect for human dignity and rights.”8
The legal claims included defamation, infliction of emotional distress, privacy (related to medical records), and malpractice. We will take a very brief look at each of those causes of action and then say a word about punitive damages (which the jury awarded in this case).
It is important to remember that state law, rather than federal, is providing the legal principles on which these claims were decided. Federal law might provide some relevant principles in such cases (for example, the First Amendment freedom of speech limits the state defamation rules), but that is the exception. State law is the rule.
Patient−physician recordings and the law
State laws differ regarding when it is legal to record in-person conversations. When everyone in the conversations knows about the recording, it is permissible and can be used in a court of law. In most states it is legal to record when only one party to the conversation has agreed to it, even though others in the conversation are not aware of it (which was the situation in the case discussed here).
In theory, physicians (by contract with patients) might try to limit patients’ rights to record medical services. But that practice would be difficult to implement or enforce in many circumstances. The reality is that audio and video recording devices are so ubiquitous that it is not sensible to avoid all recording of patient contact.
Physicians also might consider the potential such recordings have in some circumstances to improve communication with patients. Permitting the patient to record the patient−physician exchange, for instance, allows the patient the ability to review the advice after having left the office. This could be beneficial from a patient care perspective.
Defamation—award of damages
At its core, defamation is publishing (that is, telling someone other than the plaintiff) something untrue that may be harmful to another person. Generally the harm is reputational and the plaintiff may be affected by loss of business, mental suffering, or loss of esteem in the community.9
Defamation claims are not typical in health care cases. However, these claims are not rare: instances of health care professionals defaming other health care professionals, patients giving negative “reviews,” or health care professionals releasing false information to employers certainly do exist.
In this case, in addition to saying that the patient had syphilis and tuberculosis (both untrue), the physicians said he was a “wimp.” One interesting concept of defamation law that has developed over the centuries is “negligence per se.” This means a falsehood has been published about someone and the falsehood is likely to cause serious reputational harm. Claims that someone has a contagious disease traditionally have been considered negligence per se. Syphilis and tuberculosis fall in that category. On the other hand, saying someone needs to “man up” is usually a matter of opinion, so defamation for such comments is unlikely without special circumstances.
From the anesthesiologist’s perspective, the question is whether anyone who heard the publication really believed that the patient had either of the diseases. A joke that nobody believes to be based on fact generally is not defamatory because it has not harmed the plaintiff.10 It is apparent that the jury felt the patient had been defamed, however, given the $100,000 award for defamation.
In the United States there is special sensitivity to defamation awards because they may implicate the First Amendment’s protection of free speech. That being the case, this award may be particularly open to review by the judge and appellate courts.
Emotional distress—no award of damages
There are 2 kinds of “emotional distress” claimed in this case:
- intentional infliction
- negligent infliction.
Intentional infliction usually requires outrageous conduct by the defendant who acts intentionally or recklessly to inflict severe mental pain on the plaintiff.11 In this case, the element of “intentional” or “reckless” is interesting. While the conduct was outrageous, it is doubtful that there was any way the anesthesiologist could have imagined that these outrageous statements would have been transmitted to the patient/plaintiff.
As for negligent infliction of emotional distress, most states have been wary of opening a Pandora’s Box of litigation. Therefore, they generally require significant physical manifestations of great stress to allow recovery.12 It appears that the jury did not find the elements of either intentional or negligent infliction of emotional distress in this case.
As a side-note, this kind of emotional distress is viewed by the law as different from emotional distress that is incidental to a physical injury (pain and suffering). All states recognize that form of emotional distress.
Privacy—no award of damages
The privacy of medical records has, of course, become a major concern in the last few years. Both federal and state law provides significant penalties for the unauthorized release of medical information. However, in this case, it does not appear that medical information was improp- erly revealed.13
The patient’s complaint suggested that the anesthesiologist’s discussion during the colonoscopy of the medication for the penile rash was unnecessary for health care purposes.1 Therefore, it claims, the discussion violated the state health records privacy law. At the same time there was no indication in the public reports that this caused any harm to the patient.
Medical malpractice—award of damages
Malpractice usually involves professional practice that is unacceptable to the profession itself. It most commonly is negligence, or carelessness, that causes injury to the patient. The gross disregard for professional medical standards here was certainly negligence.14 The plaintiff claimed that discussing the medication for the penile rash and falsification of the medical records constituted malpractice.1
Presumably the jury award for medical malpractice means the jury found that the misconduct of the medical staff caused the emotional harm that the plaintiff experienced (described as embarrassment, loss of sleep, mental anguish, and anxiety), and that those injuries warranted a $200,000 award.
Punitive damage—award of damages
The jury also awarded $200,000 in “punitive” or “exemplary” damages. These are unusual damages, given not so much to compensate the victim but rather as a deterrent for the future. Generally the defendant’s conduct must have been egregious and completely unacceptable.15 Those elements were apparent to the jury from the facts of this case.
What about loss of practice privileges?
It is not unlikely that one or more of the medical professionals might, beyond civil liability, be subject to licensure discipline by the Virginia board. In addition, there are other secondary consequences of this lawsuit. The employment of those involved may be interrupted. (The anesthesiologist is said to have moved to another state, for example.) Hospital privileges also may be affected, as may insurance rates. The results of this award likely will have to be reported to the National Practitioner Data Bank.
As physicians, what’s our takeaway?
Conduct unbecoming a physician remains front and center with a recent essay published in the internal medicine literature.16 The anonymous author attests to witnessing a male gynecologist making sexual comments regarding the patient at the time of vaginal surgery preparation and an obstetrician singing and dancing to a Mexican song while treating his Hispanic patient for postpartum bleeding.
The unusual case of the anesthesiologist that we address was made even more unusual by the fact that it was recorded. Recordings, however, are likely to become ever more common. The advice of everyone’s grandmother is well taken: “Always act as though what you do will be published on the front page of the newspaper.” The ubiquitous presence of video and audio cameras and untold other devices means that someone may well be watching.
Aside from the risk of getting caught, respect for patients and clients is the very foundation of respect and professional care. It is distressing that the anesthesiologist was so disrespectful of a patient. It is equally disappointing that nobody put a stop to it.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- D.B. v Safe Sedation, Complaint, Civil Action 2014-05265, Circuit Court of Fairfax County.
- Abbott R. Unconscious patient says doctors mocked him. Courthouse News Service. http://www.courthousenews.com/2014/04/22/67225.htm. Updated April 22, 2014. Accessed August 19, 2015.
- Jackman T. Anesthesiologist trashes sedated patient—and it ends up costing her. Washington Post. June 23, 2015. http://www.washingtonpost.com/local/anesthesiologist-trashes-sedated-patient-jury-orders-her-to-pay-500000/2015/06/23/cae05c00-18f3-11e5-ab92-c75ae6ab94b5_story.html. Accessed August 19, 2015.
- Waibel E. Patient says Bethesda practitioners mocked him during the colonoscopy. GazetteNet. May 13, 2014. http://www.gazette.net/article/20140513/NEWS/140519703/1070/patient-says-bethesda-practitioners-mocked-him-during-colonoscopy&template=gazette. Accessed July 15, 2015.
- Vieth P. Fairfax County Circuit Court: Doctors allegedly mocked their unconscious patient. Virginia Lawyers Weekly. May 1, 2014. http://valawyersweekly.com/2014/05/01/doctors-allegedly-mocked-their-unconscious-patient. Accessed August 19, 2015.
- Welch A. Patient sues anesthesiologist who mocked him while sedated. CBS News. http://www.cbsnews.com/news/patient-sues-anesthesiloigst-who mocked-him-while sedated. Accessed July 15, 2015.
- Leins C. Anesthesiologist derides subdued patient, loses lawsuit. US News. June 24, 2015. http://www.usnews.com/news/articles/2015/06/24/anesthesiologist-derides-subdued-patient-loses-lawsuit. Accessed August 19, 2015.
- Principles of medical ethics. American Medical Association Web site. http://www.ama-assn.org/ama/pub/physician-resources/medical-ethics/code-medical-ethics/principles-medical-ethics.page. Accessed August 19, 2015.
- Instruction nos. 24 and 25. Virginia Defamation Lawyer Web site. http://www.virginiadefamationlawyer.com/Instr%2024%20and%2025.pdf. Accessed August 19, 2015.
- Berlik LE. The Virginia model jury instructions for defamation lead to bad verdicts. The Virginia Defamation Law Blog. June 27, 2015. http://www.virginiadefamationlawyer.com/2015/06/the-virginia-model-jury-instructions-for-defamation-lead-to-bad-verdicts.html#more. Accessed August 19, 2015.
- Russo v White, 241 Va 23 (1991).
- Hughes v Moore, 214 Va 27 (1973).
- Law on patient health records/privacy. Virginia Department of Health Professions Web site.
- http://webcache.googleusercontent.com/search?q=cache:asc1xQmBefoJ:https://www.dhp.virginia.gov/dhp_laws/Law_Patient%2520Health%2520Records.doc+&cd=2&hl=en&ct=clnk&gl=us. Accessed August 19, 2015.
- Virginia Medical Malpractice Act. Va Code Ann. 8.01-230, 8.01-243(A).
- Ford CR. Pleading and understanding punitive-damages claims in Virginia. Litigation News. 2008;8(10):1-11.
- D.B. v Safe Sedation, Complaint, Civil Action 2014-05265, Circuit Court of Fairfax County.
- Abbott R. Unconscious patient says doctors mocked him. Courthouse News Service. http://www.courthousenews.com/2014/04/22/67225.htm. Updated April 22, 2014. Accessed August 19, 2015.
- Jackman T. Anesthesiologist trashes sedated patient—and it ends up costing her. Washington Post. June 23, 2015. http://www.washingtonpost.com/local/anesthesiologist-trashes-sedated-patient-jury-orders-her-to-pay-500000/2015/06/23/cae05c00-18f3-11e5-ab92-c75ae6ab94b5_story.html. Accessed August 19, 2015.
- Waibel E. Patient says Bethesda practitioners mocked him during the colonoscopy. GazetteNet. May 13, 2014. http://www.gazette.net/article/20140513/NEWS/140519703/1070/patient-says-bethesda-practitioners-mocked-him-during-colonoscopy&template=gazette. Accessed July 15, 2015.
- Vieth P. Fairfax County Circuit Court: Doctors allegedly mocked their unconscious patient. Virginia Lawyers Weekly. May 1, 2014. http://valawyersweekly.com/2014/05/01/doctors-allegedly-mocked-their-unconscious-patient. Accessed August 19, 2015.
- Welch A. Patient sues anesthesiologist who mocked him while sedated. CBS News. http://www.cbsnews.com/news/patient-sues-anesthesiloigst-who mocked-him-while sedated. Accessed July 15, 2015.
- Leins C. Anesthesiologist derides subdued patient, loses lawsuit. US News. June 24, 2015. http://www.usnews.com/news/articles/2015/06/24/anesthesiologist-derides-subdued-patient-loses-lawsuit. Accessed August 19, 2015.
- Principles of medical ethics. American Medical Association Web site. http://www.ama-assn.org/ama/pub/physician-resources/medical-ethics/code-medical-ethics/principles-medical-ethics.page. Accessed August 19, 2015.
- Instruction nos. 24 and 25. Virginia Defamation Lawyer Web site. http://www.virginiadefamationlawyer.com/Instr%2024%20and%2025.pdf. Accessed August 19, 2015.
- Berlik LE. The Virginia model jury instructions for defamation lead to bad verdicts. The Virginia Defamation Law Blog. June 27, 2015. http://www.virginiadefamationlawyer.com/2015/06/the-virginia-model-jury-instructions-for-defamation-lead-to-bad-verdicts.html#more. Accessed August 19, 2015.
- Russo v White, 241 Va 23 (1991).
- Hughes v Moore, 214 Va 27 (1973).
- Law on patient health records/privacy. Virginia Department of Health Professions Web site.
- http://webcache.googleusercontent.com/search?q=cache:asc1xQmBefoJ:https://www.dhp.virginia.gov/dhp_laws/Law_Patient%2520Health%2520Records.doc+&cd=2&hl=en&ct=clnk&gl=us. Accessed August 19, 2015.
- Virginia Medical Malpractice Act. Va Code Ann. 8.01-230, 8.01-243(A).
- Ford CR. Pleading and understanding punitive-damages claims in Virginia. Litigation News. 2008;8(10):1-11.
In this Article
- The patient’s claims in this case
- Recordings and the law

Ankle pain in a young woman with Gaucher disease
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
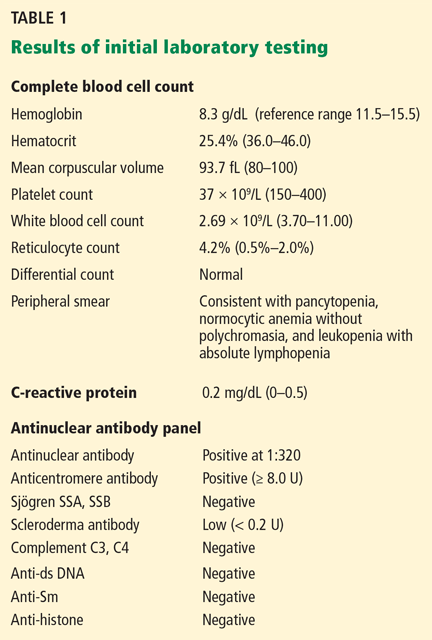
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
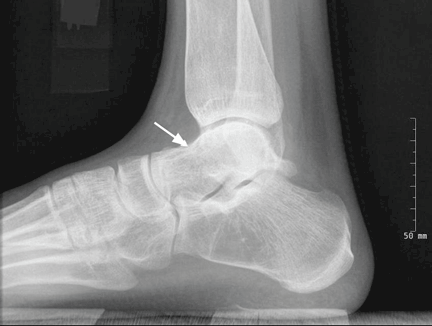
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
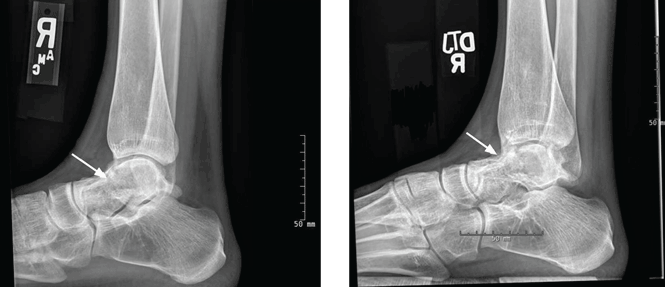
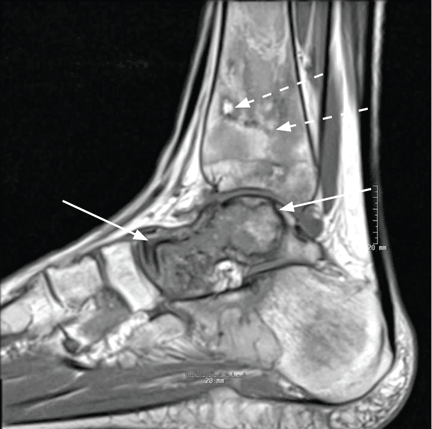
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
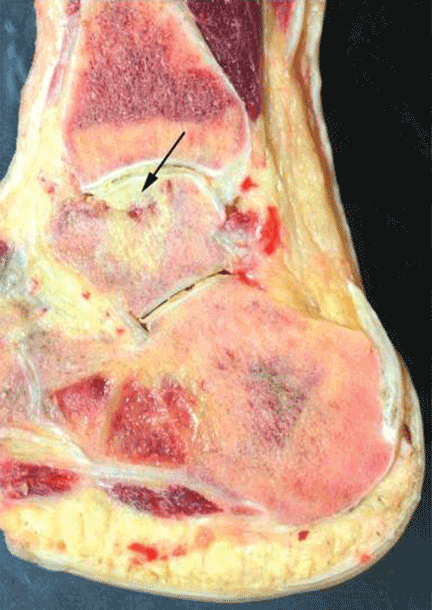
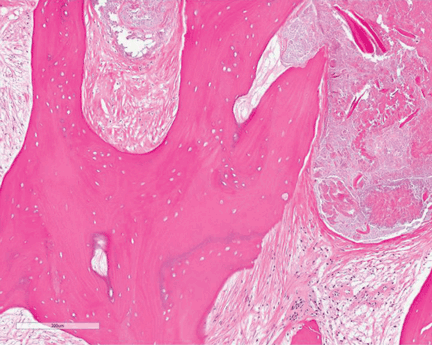
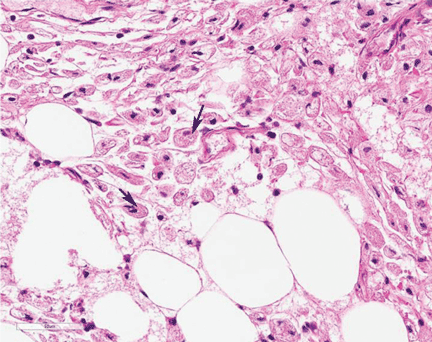
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
A 20-year-old woman with Gaucher disease presents with pain in her right ankle and in her back. She has had the ankle pain for the past 12 months and the back pain for the past 2 years. She describes the ankle pain as stabbing and moderately severe. It is constant, present both at rest and during physical activity, but aggravated by walking and twisting movements. She has noticed grinding and clicking sounds as she moves her ankle. The ankle pain has worsened over the past several months.
She says her back pain is similar to her ankle pain but less severe. She also reports generalized mild aches and bone pain. No other joints are involved. She has no history of fever, chills, or trauma.
A COMPLICATED MEDICAL HISTORY
Her Gaucher disease was diagnosed at age 4 when she presented with failure to thrive and with thrombocytopenia and splenomegaly. She and was found to have an N370S/IVS2+1 mutation of the GBA gene. She underwent removal of 90% of her spleen at the time of diagnosis and was on enzyme replacement therapy with imiglucerase until 3 years ago, when the treatment was stopped because the drug had become unavailable (because of a temporary closure of the manufacturing facility), and because she had developed neutralizing antibodies to it. Despite a dosage as high as 120 U/kg every 2 weeks (the recommended range is 2.5 U/kg three times a week up to 60 U/kg every 2 weeks), her anemia and thrombocytopenia worsened to the point that she became dependent on transfusion of red blood cells and platelets. She has also taken glucocorticoids at various times in the past as a premedication before enzyme replacement therapy.
About 3 years ago, she developed dryness of the skin, pruritus, shiny skin, hardening of the skin, and decreased oral aperture, which was diagnosed as scleroderma.
During the past 5 years, she has had multiple episodes of pale coloration of her skin on exposure to cold, suggestive of Raynaud phenomenon. And for the past 5 months, she has noticed a burning sensation in her throat and retrosternal pain, suggestive of gastroesophageal reflux disease.
She is a college student, with no history of smoking or use of alcohol or recreational drugs. She is sexually active, with no history of sexually transmitted disease, and she uses condoms and oral contraceptives for contraception.
Her father and mother are both carriers of Gaucher disease. She is not of Ashkenazi Jewish descent.
FINDINGS ON PHYSICAL EXAMINATION
On physical examination, her temperature, blood pressure, pulse, and respiratory rate are within normal limits. She has extensive tattooing on her upper chest to hide scarring from previous cannulation ports. The right ankle joint is moderately swollen but shows no other signs of inflammation; its range of motion is limited by severe pain. She has tenderness of the spinous processes and paraspinal area, in addition to multiple tender points in the thoracolumbar area. Palpation of the right hip reveals tenderness of the groin and trochanteric bursa.
No lymphadenopathy, hepatomegaly, splenomegaly, or abdominal masses are noted. Neurologic examination is essentially nonfocal.
Her current medications include omeprazole, ergocalciferol, calcium carbonate, gabapentin, citalopram, and celecoxib. She also takes a multivitamin daily.
1. Which is the most likely underlying cause of her ankle pain?
- Rheumatoid arthritis
- Gaucher disease
- Septic arthritis
- Avascular necrosis secondary to steroid use
Rheumatoid arthritis varies in its presentation. It is usually insidious in onset, migratory, and intermittent, with polyarticular or even monoarticular involvement, and it presents with pain, stiffness, and swelling of the joint.1 Most often affected are the metacarpophalangeal, proximal interphalangeal, wrist, and metatarsophalangeal joints. Involvement of large joints of the upper and lower limbs is also common.2 This is not the most likely cause of this patient’s symptoms, based on the history and the current presentation.
Gaucher disease is a lipidosis caused by accumulation of cellular glycolipids, especially glucocerebrosides, due to deficiency of the enzyme beta-glucosidase. Clinical manifestations include hepatomegaly, splenomegaly, and bone marrow disease presenting as anemia, thrombocytopenia, or skeletal disease.3 Skeletal involvement in Gaucher disease includes bone pain, bone infarcts, and lytic lesions.
Whether splenectomy predisposes the patient to bone manifestations is controversial. Some believe that splenectomy decreases the total body reservoir for the storage of glycolipids and predisposes to their deposition in bone, which in turn results in cortical thinning, impaired remodeling, and decreased intraosseous blood flow, leading to osteonecrosis and fractures.4 This is more common in patients with type 1 Gaucher disease who have undergone splenectomy. (Types 2 and 3 are much rarer, occurring mainly in children; central nervous system involvement is a key feature. A discussion of these types is beyond the focus of this paper.) However, some studies suggest that the increase in bone manifestations after splenectomy may be simply because of severe disease.5 It should be noted that, since the advent of enzyme replacement therapy for Gaucher disease, splenectomy is now rarely performed.6
Anemia is also considered an independent risk factor for the development of avascular necrosis in type 1 Gaucher disease.7 Osteonecrosis due to Gaucher disease is relatively common in the femur, tibia, and humerus and uncommon in the ankle joints.8
Septic arthritis is unlikely in this patient in the absence of fever or signs of inflammation of the joint. Her long-standing history of ankle pain would also be unusual for infection, but a superimposed infectious process should always be suspected in an arthritic joint.
Avascular necrosis secondary to steroid use. Glucocorticoids are notorious for their adverse effects on bone. They induce osteocyte apoptosis and a decrease in bone remodeling, potentially predisposing to osteonecrosis.9 There is a high incidence of osteoporosis, osteonecrosis, and fracture risk with glucocorticoid therapy, and the incidence is dose-dependent. Discontinuation of the drug only partially restores fracture risk to baseline levels.10,11
A meta-analysis of cohort studies with a total sample size of about 42,000 reported an increased risk of fracture at all ages with the use of glucocorticoids.12 Because the minimum dosage and duration of therapy to prevent glucocorticoid-induced osteoporosis are not known, the only recommendation is to keep the dosage as low as possible.13
Glucocorticoid therapy is the most common cause of nontraumatic avascular necrosis. The risk of osteonecrosis in patients on long-term glucocorticoid therapy may be as high as 40%.14 The risk is increased with prolonged treatment and with high doses, but it can also occur with short-term exposure to high doses. The increased risk has been shown to persist for as long as 2 years after the drugs are discontinued.15 Glucocorticoid-induced bone disease commonly affects the hip and vertebrae.
At this stage of the workup, we cannot completely rule out glucocorticoid use as the cause. However, after considering this patient’s presentation and the key features of the other diagnoses, her ankle pain and back pain are more likely caused by her preexisting Gaucher disease.
CONTINUED EVALUATION
Initial laboratory tests (Table 1) reveal severe anemia and thrombocytopenia. Bone marrow biopsy of the iliac crest done as part of the workup for these conditions shows extensive bone marrow space replacement by histiocytic infiltrate, consistent with Gaucher disease. No other marrow process is observed.
Radiography of the ankle (Figure 1) shows a subtle lucency in the talar dome with minimal subarticular collapse seen on the lateral view, suggestive of avascular necrosis and diffuse osteopenia. Joint spaces are maintained.
Magnetic resonance imaging (MRI) of the ankle shows numerous bone infarcts with an approximately 15-mm region of mild articular surface collapse in the central and lateral aspect of the talar dome.
MRI of the back shows extensive abnormal bone marrow signal intensity throughout the spine, compatible with a marrow replacement process. Patchy nonexpansile T2/stir hyperintensity with serpiginous enhancement within the T9, T11, T12, L2, and L3 vertebral bodies as well as throughout the entire sacrum is consistent with bone infarct.
2. Based on the results of radiographic studies, which is most likely the immediate cause of her ankle pain?
- Talar avascular necrosis secondary to rheumatoid arthritis
- Talar avascular necrosis secondary to Gaucher disease
- Trauma-induced fracture of the talus
- Plantar fasciitis
Of the bones of the feet, the talus is unique. It is the second largest of the tarsal bones and does not have muscular or tendinous attachments. Sixty percent of the talus bone is covered by articular cartilage,16 so only a limited area is available for penetration of blood vessels. Also, small nutrient vessels and variations of intraosseous anastomoses with a lack of collateral circulation predispose the talus to osteonecrosis when the vascular supply is compromised.16
Radiographic evidence of avascular necrosis is the presence of bone that is more radiopaque than normal bone; this is necrotic bone surrounded by osteopenic bone. Avascular necrosis causes hyperemia and resorption of bone. The resorption does not take place in necrotic bone because of the lack of a vascular supply, and so it appears radiopaque, whereas the bone surrounding the necrotic bone becomes osteopenic and radiolucent.
The sclerotic rim of a bone infarct is also enhanced by an attempted healing process in which new bone forms on the surface of necrotic trabeculae, a process known as “creeping substitution.” This gives a typical sclerotic picture of the talus.
MRI is the most sensitive technique for detecting osteonecrosis. A characteristic radiographic pattern is seen with osteonecrosis of the talus starting with talar dome opacity, followed by deformity and, in severe cases, articular collapse and bone fragmentation.17
The radiograph in our patient’s case is not consistent with features of rheumatoid arthritis or traumatic fracture of the talus. In plantar fasciitis, radiographs are used to rule out other pathologies of the foot, and the only finding may be a bone spur seen at the site of pain. The bone spur is not the cause of pain in plantar fasciitis but may be a result of the plantar fasciitis itself.
Therefore, avascular necrosis secondary to Gaucher disease is most likely the immediate cause of her ankle pain.
THE COURSE OF TREATMENT
The patient is started on enzyme replacement therapy with taliglucerase alfa (see discussion of enzyme replacement below). For the ankle pain, conservative management is prescribed, with application of a splint and a boot.
After 4 months of conservative management, radiography (Figure 2) and magnetic resonance imaging (Figure 3) show progressive deterioration of the talus body, and her ankle pain has worsened. A 6-week trial of an ankle brace also proves futile. Her pain continues to worsen and is not controllable with high doses of pain medication. She requests below-the-knee amputation.
Given the complexity of this patient’s medical condition, fusion of the ankle and hindfoot—which in some patients is preferable to amputation—is not considered because of her extensive bone involvement and ongoing thrombocytopenia, which would impede healing after the procedure. Below-the-knee amputation is performed without complications.
Study of the specimen after amputation reveals talar bone necrosis and bone marrow infiltration by foamy macrophages, consistent with Gaucher disease (Figures 4–6).
GAUCHER DISEASE
Pharmacologic treatments, effective only for type 1 Gaucher disease, target hepatosplenomegaly, cytopenia, and bone manifestations. Two approaches are enzyme replacement therapy—ie, to replace the defective enzyme—and substrate reduction therapy—ie, to reduce the production and thus the accumulation of glucocerebroside. Enzyme replacement is the first choice of therapy; substrate reduction is reserved for patients unable to tolerate enzyme replacement therapy.
Enzyme replacement
Current drugs for enzyme replacement therapy are imiglucerase, taliglucerase alfa, and velaglucerase alfa. The drugs are given by intravenous infusion over 1 to 2 hours in an outpatient clinic or office every 2 weeks.
These drugs are extremely expensive. Currently, the estimated cost of therapy for 1 year would be $432,978 for imiglucerase, $324,870 for taliglucerase alfa, and $368,550 for velaglucerase alfa. (The estimated costs are for 1 year of treatment for a 70-kg patient at 60 U/kg every 2 weeks.)18 Taliglucerase alfa is less expensive than the other two because it is plant-derived and thus can be more readily produced on a large scale.19
Substrate reduction
Current drugs for substrate reduction therapy are eliglustat and miglustat. They are given orally. Eliglustat is the first oral drug approved as a first-line treatment for Gaucher disease.20 Miglustat is approved only for mild to moderate disease when enzyme replacement fails or is not tolerated.
Patients can develop antibodies to any of the enzyme replacement drugs. It is not known whether this antibody response differs among the three drugs.21
Avascular necrosis of bone can occur in many clinical settings especially after a fracture, particularly of the head of the femur, which leads to interruption of blood supply to the area. Patients with sickle cell disease, those on corticosteroids or bisphosphonates (the latter causing osteonecrosis of the jaw), and those who have pancreatitis or human immunodeficiency virus infection are more prone to this bone complication.
In Gaucher disease, osteonecrosis is associated with splenectomy and severe disease and tends to occur at a younger age than in patients with other diagnoses.8 The plasma chitotriosidase activity and pulmonary and activation-regulated chemokines (PARC/CCL18), which are 10 to 40 times higher than normal in symptomatic patients with Gaucher disease, can be used as a biomarker of disease activity.8 Only plasma chitotriosidase is clinically available and used on a routine basis.
Bone involvement is seen in approximately 75% of the patients with type 1 Gaucher disease,22 and osteonecrosis is a severe form of bone involvement. Monitoring of patients for bone involvement is recommended. Enzyme replacement therapy for Gaucher disease needs to be started even if visceral disease is absent if the patient has evidence of bone involvement in the form of avascular necrosis.7 Prospective studies have shown that enzyme replacement therapy reduces the incidence of osteonecrosis.23
FOLLOW-UP MANAGEMENT OF OUR PATIENT
Avascular necrosis in Gaucher disease more typically involves the hips and shoulders. In the case of our patient, the talus was the most affected bone. Other contributing factors may have been the use of steroids as a premedication (often unnecessary) for her enzyme replacement therapy, as well as the coexistent scleroderma.24
The decision to switch from imiglucerase, to which she developed antibodies, to taliglucerase was made in the hope that the antibodies would not cross-react. After she started taliglucerase, her complete blood count values improved steadily. She did not require transfusions for more than 1 year. Her platelet count rose to 90 × 109/L, and her hemoglobin to 12 g/dL.
A multidisciplinary approach with regular monitoring and appropriate initiation of therapy is necessary to prevent disastrous complications in patients with Gaucher disease.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet 2001; 358:903–911.
- Fleming A, Crown JM, Corbett M. Early rheumatoid disease. I. Onset. Ann Rheum Dis 1976; 35:357–360.
- Grabowski GA, Andria G, Baldellou A, et al. Pediatric non-neuronopathic Gaucher disease: presentation, diagnosis, and assessment. Consensus statements. Eur J Pediatr 2004; 163:58–66.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999; May (362):201–207.
- Lee RE. The pathology of Gaucher disease. Prog Clin Biol Res 1982; 95:177–217.
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis 2008; 31:319–336.
- Khan A, Hangartner T, Weinreb NJ, Taylor JS, Mistry PK. Risk factors for fractures and avascular osteonecrosis in type 1 Gaucher disease: a study from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. J Bone Miner Res 2012; 27:1839–1848.
- Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore) 2011; 90:52–60.
- Weinstein RS. Glucocorticoid-induced osteonecrosis. Endocrine 2012; 41:183–190.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johnell O, Oden A, et al. The risk and burden of vertebral fractures in Sweden. Osteoporos Int 2004; 15:20–26.
- Seguro LP, Rosario C, Shoenfeld Y. Long-term complications of past glucocorticoid use. Autoimmun Rev 2013; 12:629–632.
- Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am 2012; 41:595–611.
- Cooper C, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int 2010; 21:569–577.
- Mulfinger GL, Trueta J. The blood supply of the talus. J Bone Joint Surg Br 1970; 52:160–167.
- Pearce DH, Mongiardi CN, Fornasier VL, Daniels TR. Avascular necrosis of the talus: a pictoral essay. Radiographics 2005; 25:399–410.
- In brief: Taliglucerase (Elelyso) for Gaucher disease. Med Lett Drugs Ther 2012 Jul 9; 54(1394):56.
- Hollak CE. An evidence-based review of the potential benefits of taliglucerase alfa in the treatment of patients with Gaucher disease. Core Evid 2012; 7:15–20.
- Poole RM. Eliglustat: first global approval. Drugs 2014; 74:1829–1836.
- Bennett LL, Mohan D. Gaucher disease and its treatment options. Ann Pharmacother 2013; 47:1182–1193.
- Germain DP. Gaucher’s disease: a paradigm for interventional genetics. Clin Genet 2004; 65:77–86.
- Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008; 73:430–440.
- Rennie C, Britton J, Prouse P. Bilateral avascular necrosis of the lunate in a patient with severe Raynaud’s phenomenon and scleroderma. J Clin Rheumatol 1999; 5:165–168.
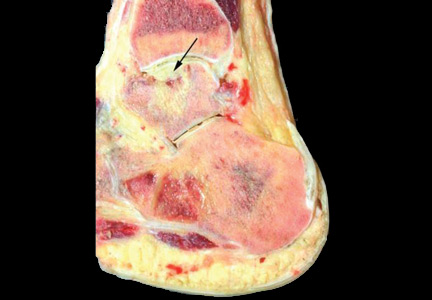
EMR notes should communicate and educate
To the Editor: Dr. Venkat1 was spot on when he identified the need for electronic medical records to communicate and educate, rather than document. Short and actionable notes are best. But with the focus on billing and compliance, annotated, informative assessments are actually discouraged. Our billing and coding department performs periodic chart audits and considers the note “out of compliance” if there is a difference between the list of free text assessments and the International Classification of Diseases, Ninth Revision (ICD-9) codes chosen. Therefore, many physicians just use the billing codes as their assessment and skip the free text assessment section of a SOAP (subjective-objective-assessment-plan) note, which means the notes convey even less of what the physician is thinking. A classic example is the note of a patient whom I knew had pernio, yet the assessment blandly reported “circulatory disorder.” The plan likewise is often reduced to the imported structured text of the tests and medications ordered rather than a rich discussion of the differential diagnosis and medical reasoning.
Imagine the notes we might write if their primary purpose was communication to ourselves and the others involved in our patients’ care. Imagine if the notes made us more knowledgeable about the uniqueness of this particular patient and also contributed to a continuous learning environment. More meaning, less filler. The notes would be shorter and sweeter, as Dr. Venkat suggested.
- Venkat KK. Short and sweet: writing better consult notes in the era of the electronic medical record. Cleve Clin J Med 2015; 82:13–17.
To the Editor: Dr. Venkat1 was spot on when he identified the need for electronic medical records to communicate and educate, rather than document. Short and actionable notes are best. But with the focus on billing and compliance, annotated, informative assessments are actually discouraged. Our billing and coding department performs periodic chart audits and considers the note “out of compliance” if there is a difference between the list of free text assessments and the International Classification of Diseases, Ninth Revision (ICD-9) codes chosen. Therefore, many physicians just use the billing codes as their assessment and skip the free text assessment section of a SOAP (subjective-objective-assessment-plan) note, which means the notes convey even less of what the physician is thinking. A classic example is the note of a patient whom I knew had pernio, yet the assessment blandly reported “circulatory disorder.” The plan likewise is often reduced to the imported structured text of the tests and medications ordered rather than a rich discussion of the differential diagnosis and medical reasoning.
Imagine the notes we might write if their primary purpose was communication to ourselves and the others involved in our patients’ care. Imagine if the notes made us more knowledgeable about the uniqueness of this particular patient and also contributed to a continuous learning environment. More meaning, less filler. The notes would be shorter and sweeter, as Dr. Venkat suggested.
To the Editor: Dr. Venkat1 was spot on when he identified the need for electronic medical records to communicate and educate, rather than document. Short and actionable notes are best. But with the focus on billing and compliance, annotated, informative assessments are actually discouraged. Our billing and coding department performs periodic chart audits and considers the note “out of compliance” if there is a difference between the list of free text assessments and the International Classification of Diseases, Ninth Revision (ICD-9) codes chosen. Therefore, many physicians just use the billing codes as their assessment and skip the free text assessment section of a SOAP (subjective-objective-assessment-plan) note, which means the notes convey even less of what the physician is thinking. A classic example is the note of a patient whom I knew had pernio, yet the assessment blandly reported “circulatory disorder.” The plan likewise is often reduced to the imported structured text of the tests and medications ordered rather than a rich discussion of the differential diagnosis and medical reasoning.
Imagine the notes we might write if their primary purpose was communication to ourselves and the others involved in our patients’ care. Imagine if the notes made us more knowledgeable about the uniqueness of this particular patient and also contributed to a continuous learning environment. More meaning, less filler. The notes would be shorter and sweeter, as Dr. Venkat suggested.
- Venkat KK. Short and sweet: writing better consult notes in the era of the electronic medical record. Cleve Clin J Med 2015; 82:13–17.
- Venkat KK. Short and sweet: writing better consult notes in the era of the electronic medical record. Cleve Clin J Med 2015; 82:13–17.
Sleep apnea ABCs
To the Editor: We read with interest the paper by Dr. Reena Mehra,“Sleep apnea ABCs: Airway, breathing, circulation.”1 It was very consistent and informative. However, we feel that some considerations on the pathogenesis warrant more discussion.
The pathophysiologic heterogeneity of sympathetic nervous system activity enhancement is complex and involves both intermittent hypoxia and arousal. We agree with Dr. Mehra about the importance of intermittent hypoxia in sympathetic activation, and we would like to point out the importance of effects of arousal from sleep on autonomic outflow. In some patients with obstructive sleep apnea (OSA) in whom respiratory events are not followed by oxygen desaturation, sympathetic activation cannot be explained by intermittent hypoxia. Arousal has been reported to be associated with an acute increase in sympathetic activity in the absence of hypercapnia or hypoxia.2 Cortical arousals from sleep have been historically assumed to be important in restoring airflow at the end of OSA breathing events.3 Furthermore, arousals often precede upper-airway opening in patients with OSA.4
In Figure 1 of Dr. Mehra’s paper, all the respiratory events were associated with microarousals. According to the conventional definition, cortical arousal is an abrupt shift in the electroencephalogram lasting more than 3 seconds. In Figure 1, the beginning of arousals must be scored a few seconds before breathing recovery, just at the beginning of electroencephalogram acceleration. The second respiratory event was scored as obstructive apnea, or the apnea started out as central apnea, where all respiratory channels are flat and then the chest and abdominal belts start moving, making it look like typical mixed apnea.
In the title of the paper, the “A” of ABCs referred to airway and, more specifically, to the collapse of the upper airway in sleep, which is the cause of OSA. We think that the “A” can be attributed to arousal, which is specific to sleep and contributes to the pathogenesis of OSA.
- Mehra R. Sleep apnea ABCs: airway, breathing, circulation. Cleve Clin J Med 2014; 81:479–489.
- O’Driscoll DM, Meadows GE, Corfield DR, Simonds AK, Morrell MJ. Cardiovascular response to arousal from sleep under controlled conditions of central and peripheral chemoreceptor stimulation in humans. J Appl Physiol 2004; 96:865–870.
- Eckert DJ, Younes MK. Arousal from sleep: implications for obstructive sleep apnea pathogenesis and treatment. J Appl Physiol 2014; 116:302–313.
- Younes M. Role of arousals in the pathogenesis of obstructive sleep apnea. Am J Respir Crit Care Med 2004; 69:623–633.
To the Editor: We read with interest the paper by Dr. Reena Mehra,“Sleep apnea ABCs: Airway, breathing, circulation.”1 It was very consistent and informative. However, we feel that some considerations on the pathogenesis warrant more discussion.
The pathophysiologic heterogeneity of sympathetic nervous system activity enhancement is complex and involves both intermittent hypoxia and arousal. We agree with Dr. Mehra about the importance of intermittent hypoxia in sympathetic activation, and we would like to point out the importance of effects of arousal from sleep on autonomic outflow. In some patients with obstructive sleep apnea (OSA) in whom respiratory events are not followed by oxygen desaturation, sympathetic activation cannot be explained by intermittent hypoxia. Arousal has been reported to be associated with an acute increase in sympathetic activity in the absence of hypercapnia or hypoxia.2 Cortical arousals from sleep have been historically assumed to be important in restoring airflow at the end of OSA breathing events.3 Furthermore, arousals often precede upper-airway opening in patients with OSA.4
In Figure 1 of Dr. Mehra’s paper, all the respiratory events were associated with microarousals. According to the conventional definition, cortical arousal is an abrupt shift in the electroencephalogram lasting more than 3 seconds. In Figure 1, the beginning of arousals must be scored a few seconds before breathing recovery, just at the beginning of electroencephalogram acceleration. The second respiratory event was scored as obstructive apnea, or the apnea started out as central apnea, where all respiratory channels are flat and then the chest and abdominal belts start moving, making it look like typical mixed apnea.
In the title of the paper, the “A” of ABCs referred to airway and, more specifically, to the collapse of the upper airway in sleep, which is the cause of OSA. We think that the “A” can be attributed to arousal, which is specific to sleep and contributes to the pathogenesis of OSA.
To the Editor: We read with interest the paper by Dr. Reena Mehra,“Sleep apnea ABCs: Airway, breathing, circulation.”1 It was very consistent and informative. However, we feel that some considerations on the pathogenesis warrant more discussion.
The pathophysiologic heterogeneity of sympathetic nervous system activity enhancement is complex and involves both intermittent hypoxia and arousal. We agree with Dr. Mehra about the importance of intermittent hypoxia in sympathetic activation, and we would like to point out the importance of effects of arousal from sleep on autonomic outflow. In some patients with obstructive sleep apnea (OSA) in whom respiratory events are not followed by oxygen desaturation, sympathetic activation cannot be explained by intermittent hypoxia. Arousal has been reported to be associated with an acute increase in sympathetic activity in the absence of hypercapnia or hypoxia.2 Cortical arousals from sleep have been historically assumed to be important in restoring airflow at the end of OSA breathing events.3 Furthermore, arousals often precede upper-airway opening in patients with OSA.4
In Figure 1 of Dr. Mehra’s paper, all the respiratory events were associated with microarousals. According to the conventional definition, cortical arousal is an abrupt shift in the electroencephalogram lasting more than 3 seconds. In Figure 1, the beginning of arousals must be scored a few seconds before breathing recovery, just at the beginning of electroencephalogram acceleration. The second respiratory event was scored as obstructive apnea, or the apnea started out as central apnea, where all respiratory channels are flat and then the chest and abdominal belts start moving, making it look like typical mixed apnea.
In the title of the paper, the “A” of ABCs referred to airway and, more specifically, to the collapse of the upper airway in sleep, which is the cause of OSA. We think that the “A” can be attributed to arousal, which is specific to sleep and contributes to the pathogenesis of OSA.
- Mehra R. Sleep apnea ABCs: airway, breathing, circulation. Cleve Clin J Med 2014; 81:479–489.
- O’Driscoll DM, Meadows GE, Corfield DR, Simonds AK, Morrell MJ. Cardiovascular response to arousal from sleep under controlled conditions of central and peripheral chemoreceptor stimulation in humans. J Appl Physiol 2004; 96:865–870.
- Eckert DJ, Younes MK. Arousal from sleep: implications for obstructive sleep apnea pathogenesis and treatment. J Appl Physiol 2014; 116:302–313.
- Younes M. Role of arousals in the pathogenesis of obstructive sleep apnea. Am J Respir Crit Care Med 2004; 69:623–633.
- Mehra R. Sleep apnea ABCs: airway, breathing, circulation. Cleve Clin J Med 2014; 81:479–489.
- O’Driscoll DM, Meadows GE, Corfield DR, Simonds AK, Morrell MJ. Cardiovascular response to arousal from sleep under controlled conditions of central and peripheral chemoreceptor stimulation in humans. J Appl Physiol 2004; 96:865–870.
- Eckert DJ, Younes MK. Arousal from sleep: implications for obstructive sleep apnea pathogenesis and treatment. J Appl Physiol 2014; 116:302–313.
- Younes M. Role of arousals in the pathogenesis of obstructive sleep apnea. Am J Respir Crit Care Med 2004; 69:623–633.
In reply: Sleep apnea ABCs
In Reply: We thank Dr. Abouda for underscoring the role of arousals in the pathophysiology of obstructive sleep apnea (OSA). Although the focus of the referenced article was to provide a general overview of the epidemiology, diagnostic testing, and cardiovascular ramifications of untreated OSA and not a detailed summary of the underlying pathophysiology, we welcome the comments from Dr. Abouda to highlight the importance of cortical or microarousals in OSA.
Whether cortical arousal during sleep is bad or good is controversial. During the development of the American Academy of Sleep Medicine respiratory event guidelines, the assignment of detriment or benefit to the arousal when considering defining and scoring of a hypopnea event was a topic of much discussion.1,2 Supporters of including arousal in the hypopnea definition cite data that sleep fragmentation without attendant hypoxia is associated with symptoms such as excessive daytime somnolence, which is recognized to be effectively addressed with OSA treatment.3,4 Moreover, experimental data indicate that arousals lead to activation of the sympathetic nervous system.5 On the other hand, those who question the inclusion of cortical arousal in the hypopnea definition cite large-scale epidemiologic studies that have failed to find a significantly increased cardiovascular risk in relation to increasing arousal index, as well as the enhanced potential to introduce measurement variability.1
The effects of cortical arousals as a purported source of sympathetic activation may operate in concert with hypoxic influences, the latter resulting in sustained increases in blood pressure in both animal models and human studies.6,7 Gottlieb et al8 examined the effect of supplemental oxygen vs continuous positive airway pressure (CPAP) on 24-hour mean arterial pressure in a multicenter randomized controlled trial. Although CPAP reduced blood pressure, as expected, the somewhat unanticipated finding that supplemental oxygen did not suggests that other factors such as hypercapnia and cortical arousals with attendant sympathetic activation may represent potential culprits. Along these lines, in patients with OSA and increased loop gain, benefit in response to sedative hypnotics has been shown to reduce ventilatory instability through an increase in arousal threshold.9 A genetic predisposition may influence the intensity of cortical arousals and accompanying cardiovascular influences that appear to be consistent within individuals but that are heterogeneous within populations.10
Few studies have identified increased cortical arousals as a cardiovascular risk factor. In the Cleveland Family Study, an elevated arousal index was associated with hypertension, but respiratory event-specific arousals was not specifically examined.11 Not only have large-scale epidemiologic studies failed to identify an association between arousal index and cardiovascular outcomes, existing data appear to support the contrary. For example, the extent of incident white matter disease identified on brain magnetic resonance imaging was inversely related to the arousal index in a subset of participants of the Sleep Heart Health Study, a large population-based study focused on sleep and cardiovascular outcomes.12 Furthermore, elevated arousal indices in women were associated with reduced incidence of stroke in the Sleep Heart Health Study.13 These data suggest that arousals may represent beneficial, protective biomarkers reflecting truncation of respiratory events translating into reduced duration of hypoxic exposure and decreased work of breathing.
Needed is further investigation dedicated to understanding the impact of cortical arousals on health outcomes in population-based studies and elucidating the mechanistic role of cortical arousals in the autonomic nervous system physiology in various subtypes of sleep-disordered breathing (eg, obstructive vs central sleep apnea) as well as periodic limb movements.
As the upper Airway is central to the pathophysiology of OSA leading to compromise in Breathing and Circulatory or Cardiovascular ramifications, we think it logical that the “A” in ABCs should stand for “airway.” Hopefully, future research will allow us to better understand the associated benefit vs detriment of cortical arousals as they pertain to subgroup susceptibilities and enhance our ability to tailor a personalized medicine approach to the treatment of sleep disorders.
- Berry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med 2012; 8:597–619.
- Ruehland WR, Rochford PD, O’Donoghue FJ, Pierce RJ, Singh P, Thornton AT. The new AASM criteria for scoring hypopneas: impact on the apnea hypopnea index. Sleep 2009; 32:150-157.
- Guilleminault C, Stoohs R, Clerk A, Cetel M, Maistros P. A cause of excessive daytime sleepiness. The upper airway resistance syndrome. Chest 1993; 104:781–787.
- Bonnet MH, Doghramji K, Roehrs T, et al. The scoring of arousal in sleep: reliability, validity, and alternatives. J Clin Sleep Med 2007; 3:133–145.
- Loredo JS, Ziegler MG, Ancoli-Israel S, Clausen JL, Dimsdale JE. Relationship of arousals from sleep to sympathetic nervous system activity and BP in obstructive sleep apnea. Chest J 1999; 116:655–659.
- Fletcher EC, Lesske J, Culman J, Miller CC, Unger T. Sympathetic denervation blocks blood pressure elevation in episodic hypoxia. Hypertension 1992; 20:612–619.
- Tamisier R, Pépin JL, Rémy J, et al. 14 nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur Respir J 2011; 37:119–128.
- Gottlieb DJ, Punjabi NM, Mehra R, et al. CPAP versus oxygen in obstructive sleep apnea. N Engl J Med 2014; 370:2276–2285.
- Eckert DJ, Owens RL, Kehlmann GB, et al. Eszopiclone increases the respiratory arousal threshold and lowers the apnoea/hypopnoea index in obstructive sleep apnoea patients with a low arousal threshold. Clin Sci Lond Engl 1979. 2011; 120:505–514.
- Azarbarzin A, Ostrowski M, Hanly P, Younes M. Relationship between arousal intensity and heart rate response to arousal. Sleep 2014; 37:645–653.
- Sulit L, Storfer-Isser A, Kirchner HL, Redline S. Differences in polysomnography predictors for hypertension and impaired glucose tolerance. Sleep 2006; 29:777–783.
- Ding J, Nieto FJ, Beauchamp NJ, et al. Sleep-disordered breathing and white matter disease in the brainstem in older adults. Sleep 2004; 27:474–479.
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the Sleep Heart Health Study. Am J Respir Crit Care Med 2010; 182:269–277.
In Reply: We thank Dr. Abouda for underscoring the role of arousals in the pathophysiology of obstructive sleep apnea (OSA). Although the focus of the referenced article was to provide a general overview of the epidemiology, diagnostic testing, and cardiovascular ramifications of untreated OSA and not a detailed summary of the underlying pathophysiology, we welcome the comments from Dr. Abouda to highlight the importance of cortical or microarousals in OSA.
Whether cortical arousal during sleep is bad or good is controversial. During the development of the American Academy of Sleep Medicine respiratory event guidelines, the assignment of detriment or benefit to the arousal when considering defining and scoring of a hypopnea event was a topic of much discussion.1,2 Supporters of including arousal in the hypopnea definition cite data that sleep fragmentation without attendant hypoxia is associated with symptoms such as excessive daytime somnolence, which is recognized to be effectively addressed with OSA treatment.3,4 Moreover, experimental data indicate that arousals lead to activation of the sympathetic nervous system.5 On the other hand, those who question the inclusion of cortical arousal in the hypopnea definition cite large-scale epidemiologic studies that have failed to find a significantly increased cardiovascular risk in relation to increasing arousal index, as well as the enhanced potential to introduce measurement variability.1
The effects of cortical arousals as a purported source of sympathetic activation may operate in concert with hypoxic influences, the latter resulting in sustained increases in blood pressure in both animal models and human studies.6,7 Gottlieb et al8 examined the effect of supplemental oxygen vs continuous positive airway pressure (CPAP) on 24-hour mean arterial pressure in a multicenter randomized controlled trial. Although CPAP reduced blood pressure, as expected, the somewhat unanticipated finding that supplemental oxygen did not suggests that other factors such as hypercapnia and cortical arousals with attendant sympathetic activation may represent potential culprits. Along these lines, in patients with OSA and increased loop gain, benefit in response to sedative hypnotics has been shown to reduce ventilatory instability through an increase in arousal threshold.9 A genetic predisposition may influence the intensity of cortical arousals and accompanying cardiovascular influences that appear to be consistent within individuals but that are heterogeneous within populations.10
Few studies have identified increased cortical arousals as a cardiovascular risk factor. In the Cleveland Family Study, an elevated arousal index was associated with hypertension, but respiratory event-specific arousals was not specifically examined.11 Not only have large-scale epidemiologic studies failed to identify an association between arousal index and cardiovascular outcomes, existing data appear to support the contrary. For example, the extent of incident white matter disease identified on brain magnetic resonance imaging was inversely related to the arousal index in a subset of participants of the Sleep Heart Health Study, a large population-based study focused on sleep and cardiovascular outcomes.12 Furthermore, elevated arousal indices in women were associated with reduced incidence of stroke in the Sleep Heart Health Study.13 These data suggest that arousals may represent beneficial, protective biomarkers reflecting truncation of respiratory events translating into reduced duration of hypoxic exposure and decreased work of breathing.
Needed is further investigation dedicated to understanding the impact of cortical arousals on health outcomes in population-based studies and elucidating the mechanistic role of cortical arousals in the autonomic nervous system physiology in various subtypes of sleep-disordered breathing (eg, obstructive vs central sleep apnea) as well as periodic limb movements.
As the upper Airway is central to the pathophysiology of OSA leading to compromise in Breathing and Circulatory or Cardiovascular ramifications, we think it logical that the “A” in ABCs should stand for “airway.” Hopefully, future research will allow us to better understand the associated benefit vs detriment of cortical arousals as they pertain to subgroup susceptibilities and enhance our ability to tailor a personalized medicine approach to the treatment of sleep disorders.
In Reply: We thank Dr. Abouda for underscoring the role of arousals in the pathophysiology of obstructive sleep apnea (OSA). Although the focus of the referenced article was to provide a general overview of the epidemiology, diagnostic testing, and cardiovascular ramifications of untreated OSA and not a detailed summary of the underlying pathophysiology, we welcome the comments from Dr. Abouda to highlight the importance of cortical or microarousals in OSA.
Whether cortical arousal during sleep is bad or good is controversial. During the development of the American Academy of Sleep Medicine respiratory event guidelines, the assignment of detriment or benefit to the arousal when considering defining and scoring of a hypopnea event was a topic of much discussion.1,2 Supporters of including arousal in the hypopnea definition cite data that sleep fragmentation without attendant hypoxia is associated with symptoms such as excessive daytime somnolence, which is recognized to be effectively addressed with OSA treatment.3,4 Moreover, experimental data indicate that arousals lead to activation of the sympathetic nervous system.5 On the other hand, those who question the inclusion of cortical arousal in the hypopnea definition cite large-scale epidemiologic studies that have failed to find a significantly increased cardiovascular risk in relation to increasing arousal index, as well as the enhanced potential to introduce measurement variability.1
The effects of cortical arousals as a purported source of sympathetic activation may operate in concert with hypoxic influences, the latter resulting in sustained increases in blood pressure in both animal models and human studies.6,7 Gottlieb et al8 examined the effect of supplemental oxygen vs continuous positive airway pressure (CPAP) on 24-hour mean arterial pressure in a multicenter randomized controlled trial. Although CPAP reduced blood pressure, as expected, the somewhat unanticipated finding that supplemental oxygen did not suggests that other factors such as hypercapnia and cortical arousals with attendant sympathetic activation may represent potential culprits. Along these lines, in patients with OSA and increased loop gain, benefit in response to sedative hypnotics has been shown to reduce ventilatory instability through an increase in arousal threshold.9 A genetic predisposition may influence the intensity of cortical arousals and accompanying cardiovascular influences that appear to be consistent within individuals but that are heterogeneous within populations.10
Few studies have identified increased cortical arousals as a cardiovascular risk factor. In the Cleveland Family Study, an elevated arousal index was associated with hypertension, but respiratory event-specific arousals was not specifically examined.11 Not only have large-scale epidemiologic studies failed to identify an association between arousal index and cardiovascular outcomes, existing data appear to support the contrary. For example, the extent of incident white matter disease identified on brain magnetic resonance imaging was inversely related to the arousal index in a subset of participants of the Sleep Heart Health Study, a large population-based study focused on sleep and cardiovascular outcomes.12 Furthermore, elevated arousal indices in women were associated with reduced incidence of stroke in the Sleep Heart Health Study.13 These data suggest that arousals may represent beneficial, protective biomarkers reflecting truncation of respiratory events translating into reduced duration of hypoxic exposure and decreased work of breathing.
Needed is further investigation dedicated to understanding the impact of cortical arousals on health outcomes in population-based studies and elucidating the mechanistic role of cortical arousals in the autonomic nervous system physiology in various subtypes of sleep-disordered breathing (eg, obstructive vs central sleep apnea) as well as periodic limb movements.
As the upper Airway is central to the pathophysiology of OSA leading to compromise in Breathing and Circulatory or Cardiovascular ramifications, we think it logical that the “A” in ABCs should stand for “airway.” Hopefully, future research will allow us to better understand the associated benefit vs detriment of cortical arousals as they pertain to subgroup susceptibilities and enhance our ability to tailor a personalized medicine approach to the treatment of sleep disorders.
- Berry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med 2012; 8:597–619.
- Ruehland WR, Rochford PD, O’Donoghue FJ, Pierce RJ, Singh P, Thornton AT. The new AASM criteria for scoring hypopneas: impact on the apnea hypopnea index. Sleep 2009; 32:150-157.
- Guilleminault C, Stoohs R, Clerk A, Cetel M, Maistros P. A cause of excessive daytime sleepiness. The upper airway resistance syndrome. Chest 1993; 104:781–787.
- Bonnet MH, Doghramji K, Roehrs T, et al. The scoring of arousal in sleep: reliability, validity, and alternatives. J Clin Sleep Med 2007; 3:133–145.
- Loredo JS, Ziegler MG, Ancoli-Israel S, Clausen JL, Dimsdale JE. Relationship of arousals from sleep to sympathetic nervous system activity and BP in obstructive sleep apnea. Chest J 1999; 116:655–659.
- Fletcher EC, Lesske J, Culman J, Miller CC, Unger T. Sympathetic denervation blocks blood pressure elevation in episodic hypoxia. Hypertension 1992; 20:612–619.
- Tamisier R, Pépin JL, Rémy J, et al. 14 nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur Respir J 2011; 37:119–128.
- Gottlieb DJ, Punjabi NM, Mehra R, et al. CPAP versus oxygen in obstructive sleep apnea. N Engl J Med 2014; 370:2276–2285.
- Eckert DJ, Owens RL, Kehlmann GB, et al. Eszopiclone increases the respiratory arousal threshold and lowers the apnoea/hypopnoea index in obstructive sleep apnoea patients with a low arousal threshold. Clin Sci Lond Engl 1979. 2011; 120:505–514.
- Azarbarzin A, Ostrowski M, Hanly P, Younes M. Relationship between arousal intensity and heart rate response to arousal. Sleep 2014; 37:645–653.
- Sulit L, Storfer-Isser A, Kirchner HL, Redline S. Differences in polysomnography predictors for hypertension and impaired glucose tolerance. Sleep 2006; 29:777–783.
- Ding J, Nieto FJ, Beauchamp NJ, et al. Sleep-disordered breathing and white matter disease in the brainstem in older adults. Sleep 2004; 27:474–479.
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the Sleep Heart Health Study. Am J Respir Crit Care Med 2010; 182:269–277.
- Berry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med 2012; 8:597–619.
- Ruehland WR, Rochford PD, O’Donoghue FJ, Pierce RJ, Singh P, Thornton AT. The new AASM criteria for scoring hypopneas: impact on the apnea hypopnea index. Sleep 2009; 32:150-157.
- Guilleminault C, Stoohs R, Clerk A, Cetel M, Maistros P. A cause of excessive daytime sleepiness. The upper airway resistance syndrome. Chest 1993; 104:781–787.
- Bonnet MH, Doghramji K, Roehrs T, et al. The scoring of arousal in sleep: reliability, validity, and alternatives. J Clin Sleep Med 2007; 3:133–145.
- Loredo JS, Ziegler MG, Ancoli-Israel S, Clausen JL, Dimsdale JE. Relationship of arousals from sleep to sympathetic nervous system activity and BP in obstructive sleep apnea. Chest J 1999; 116:655–659.
- Fletcher EC, Lesske J, Culman J, Miller CC, Unger T. Sympathetic denervation blocks blood pressure elevation in episodic hypoxia. Hypertension 1992; 20:612–619.
- Tamisier R, Pépin JL, Rémy J, et al. 14 nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur Respir J 2011; 37:119–128.
- Gottlieb DJ, Punjabi NM, Mehra R, et al. CPAP versus oxygen in obstructive sleep apnea. N Engl J Med 2014; 370:2276–2285.
- Eckert DJ, Owens RL, Kehlmann GB, et al. Eszopiclone increases the respiratory arousal threshold and lowers the apnoea/hypopnoea index in obstructive sleep apnoea patients with a low arousal threshold. Clin Sci Lond Engl 1979. 2011; 120:505–514.
- Azarbarzin A, Ostrowski M, Hanly P, Younes M. Relationship between arousal intensity and heart rate response to arousal. Sleep 2014; 37:645–653.
- Sulit L, Storfer-Isser A, Kirchner HL, Redline S. Differences in polysomnography predictors for hypertension and impaired glucose tolerance. Sleep 2006; 29:777–783.
- Ding J, Nieto FJ, Beauchamp NJ, et al. Sleep-disordered breathing and white matter disease in the brainstem in older adults. Sleep 2004; 27:474–479.
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the Sleep Heart Health Study. Am J Respir Crit Care Med 2010; 182:269–277.
Lactic acidosis: Clinical implications and management strategies
Physicians are paying more attention to serum lactate levels in hospitalized patients than in the past, especially with the advent of point-of-care testing. Elevated lactate levels are associated with tissue hypoxia and hypoperfusion but can also be found in a number of other conditions. Therefore, confusion can arise as to how to interpret elevated levels and subsequently manage these patients in a variety of settings.
In this review, we discuss the mechanisms underlying lactic acidosis, its prognostic implications, and its use as a therapeutic target in treating patients in septic shock and other serious disorders.
LACTATE IS A PRODUCT OF ANAEROBIC RESPIRATION
Lactate, or lactic acid, is produced from pyruvate as an end product of glycolysis under anaerobic conditions (Figure 1). It is produced in most tissues in the body, but primarily in skeletal muscle, brain, intestine, and red blood cells. During times of stress, lactate is also produced in the lungs, white blood cells, and splanchnic organs.
Most lactate in the blood is cleared by the liver, where it is the substrate for gluconeogenesis, and a small amount is cleared by the kidneys.1,2 The entire pathway by which lactate is produced and converted back to glucose is called the Cori cycle.
NORMAL LEVELS ARE LESS THAN ABOUT 2.0 MMOL/L
In this review, we will present lactate levels in the SI units of mmol/L (1 mmol/L = 9 mg/dL).
Basal lactate production is approximately 0.8 mmol/kg body weight/hour. The average normal arterial blood lactate level is approximately 0.620 mmol/L and the venous level is slightly higher at 0.997 mmol/L,3 but overall, arterial and venous lactate levels correlate well.
Normal lactate levels are less than 2 mmol/L,4 intermediate levels range from 2 to less than 4 mmol/L, and high levels are 4 mmol/L or higher.5
To minimize variations in measurement, blood samples should be drawn without a tourniquet into tubes containing fluoride, placed on ice, and processed quickly (ideally within 15 minutes).
INCREASED PRODUCTION, DECREASED CLEARANCE, OR BOTH
An elevated lactate level can be the result of increased production, decreased clearance, or both (as in liver dysfunction).
Type A lactic acidosis—due to hypoperfusion and hypoxia—occurs when there is a mismatch between oxygen delivery and consumption, with resultant anaerobic glycolysis.

The guidelines from the Surviving Sepsis Campaign6 emphasize using lactate levels to diagnose patients with sepsis-induced hypoperfusion. However, hyperlactatemia can indicate inadequate oxygen delivery due to any type of shock (Table 1).
Type B lactic acidosis—not due to hypoperfusion—occurs in a variety of conditions (Table 1), including liver disease, malignancy, use of certain medications (eg, metformin, epinephrine), total parenteral nutrition, human immunodeficiency virus infection, thiamine deficiency, mitochondrial myopathies, and congenital lactic acidosis.1–3,7 Yet other causes include trauma, excessive exercise, diabetic ketoacidosis, ethanol intoxication, dysfunction of the enzyme pyruvate dehydrogenase, and increased muscle degradation leading to increased production of pyruvate. In these latter scenarios, glucose metabolism exceeds the oxidation capacity of the mitochondria, and the rise in pyruvate concentration drives lactate production.8,9 Mitochondrial dysfunction and subsequent deficits in cellular oxygen use can also result in persistently high lactate levels.10
In some situations, patients with mildly elevated lactic acid levels in type B lactic acidosis can be monitored to ensure stability, rather than be treated aggressively.
HIGHER LEVELS AND LOWER CLEARANCE PREDICT DEATH
The higher the lactate level and the slower the rate of normalization (lactate clearance), the higher the risk of death.
Lactate levels and mortality rate
Shapiro et al11 showed that increases in lactate level are associated with proportional increases in the mortality rate. Mikkelsen et al12 showed that intermediate levels (2.0–3.9 mmol/L) and high levels (≥ 4 mmol/L) of serum lactate are associated with increased risk of death independent of organ failure and shock. Patients with mildly elevated and intermediate lactate levels and sepsis have higher rates of in-hospital and 30-day mortality, which correlate with the baseline lactate level.13
In a post hoc analysis of a randomized controlled trial, patients with septic shock who presented to the emergency department with hypotension and a lactate level higher than 2 mmol/L had a significantly higher in-hospital mortality rate than those who presented with hypotension and a lactate level of 2 mmol/L or less (26% vs 9%, P < .0001).14 These data suggest that elevated lactate levels may have a significant prognostic role, independent of blood pressure.
Slower clearance
The prognostic implications of lactate clearance (reductions in lactate levels over time, as opposed to a single value in time), have also been evaluated.
Lactate clearance of at least 10% at 6 hours after presentation has been associated with a lower mortality rate than nonclearance (19% vs 60%) in patients with sepsis or septic shock with elevated levels.15–17 Similar findings have been reported in a general intensive care unit population,18 as well as a surgical intensive care population.sup>19
Puskarich et al20 have also shown that lactate normalization to less than 2 mmol/L during early sepsis resuscitation is the strongest predictor of survival (odds ratio [OR] 5.2), followed by lactate clearance of 50% (OR 4.0) within the first 6 hours of presentation. Not only is lactate clearance associated with improved outcomes, but a faster rate of clearance after initial presentation is also beneficial.15,16,18
Lactate clearance over a longer period (> 6 hours) has not been studied in patients with septic shock. However, in the general intensive care unit population, therapy guided by lactate clearance for the first 8 hours after presentation has shown a reduction in mortality rate.18 There are no data available on outcomes of lactate-directed therapy beyond 8 hours, but lactate concentration and lactate clearance at 24 hours correlate with the 28-day mortality rate.21
Cryptic shock
Cryptic shock describes a state in a subgroup of patients who have elevated lactate levels and global tissue hypoxia despite being normotensive or even hypertensive. These patients have a higher mortality rate independent of blood pressure. Jansen et al18 found that patients with a lactate level higher than 4 mmol/L and preserved blood pressure had a mortality rate of 15%, while those without shock or hyperlactatemia had a mortality rate of 2.5%. In addition, patients with an elevated lactate level in the absence of hypotension have mortality rates similar to those in patients with high lactate levels and hypotension refractory to fluid boluses, suggesting the presence of tissue hypoxia even in these normotensive patients.6
HOW TO APPROACH AN ELEVATED LACTATE LEVEL
An elevated lactate level should prompt an evaluation for causes of decreased oxygen delivery, due either to a systemic low-flow state (as a result of decreased cardiac output) or severe anemia, or to regionally decreased perfusion, (eg, limb or mesenteric ischemia). If tissue hypoxia is ruled out after an exhaustive workup, consideration should be given to causes of hyperlactatemia without concomitant tissue hypoxia (type B acidosis).
Treatment differs depending on the underlying mechanism of the lactate elevation; nevertheless, treatment is mostly related to optimizing oxygen delivery by giving fluids, packed red blood cells, and vasopressors or inotropic agents, or both (Figure 2). The specific treatment differs based on the shock state, but there are similarities that can guide the clinician.
FLUID SUPPORT
Giving fluids, with a goal of improving cardiac output, remains a cornerstone of initial therapy for most shock states.22,23
How much fluid?
Fluids should be given until the patient is no longer preload-dependent, although there is much debate about which assessment strategy should be used to determine if cardiac output will improve with more fluid (ie, fluid-responsiveness).24 In many cases, fluid resuscitation alone may be enough to restore hemodynamic stability, improve tissue perfusion, and reduce elevated lactate concentrations.25
The decision to give more fluids should not be made lightly, though, as a more positive fluid balance early in the course of septic shock and over 4 days has been associated with a higher mortality rate.26 Additionally, pushing fluids in patients with cardiogenic shock due to impaired left ventricular systolic function may lead to or worsen pulmonary edema. Therefore, the indiscriminate use of fluids should be avoided.
Which fluids?
Despite years of research, controversy persists about whether crystalloids or colloids are better for resuscitation. Randomized trials in heterogeneous intensive care unit patients have not detected differences in 28-day mortality rates between those allocated to crystalloids or 4% albumin27 and those allocated to crystalloids or hydroxyethyl starch.28
Hydroxyethyl starch may not be best. In a study of patients with severe sepsis, those randomized to receive hydroxyethyl starch had a higher 90-day mortality rate than patients randomized to crystalloids (51% vs 43%, P = .03).29 A sequential prospective before-and-after study did not detect a difference in the time to normalization (< 2.2 mmol/L) of lactate (P = .68) or cessation of vasopressors (P = .11) in patients with severe sepsis who received fluid resuscitation with crystalloids, gelatin, or hydroxyethyl starch. More patients who received hydroxyethyl starch in these studies developed acute kidney injury than those receiving crystalloids.28–30
Taken together, these data strongly suggest hydroxyethyl starch should not be used for fluid resuscitation in the intensive care unit.
Normal saline or albumin? Although some data suggest that albumin may be preferable to 0.9% sodium chloride in patients with severe sepsis,31,32 these analyses should be viewed as hypothesis-generating. There do not seem to be differences between fluid types in terms of subsequent serum lactate concentrations or achievement of lactate clearance goals.28–30 Until further studies are completed, both albumin and crystalloids are reasonable for resuscitation.
Caironi et al33 performed an open-label study comparing albumin replacement (with a goal serum albumin concentration of 3 g/dL) plus a crystalloid solution vs a crystalloid solution alone in patients with severe sepsis or septic shock. They detected no difference between the albumin and crystalloid groups in mortality rates at 28 days (31.8% vs 32.0%, P = .94) or 90 days (41.1% vs 43.6%, P = .29). However, patients in the albumin group had a shorter time to cessation of vasoactive agents (median 3 vs 4 days, P = .007) and lower cardiovascular Sequential Organ Failure Assessment subscores (median 1.20 vs 1.42, P = .03), and more frequently achieved a mean arterial pressure of at least 65 mm Hg within 6 hours of randomization (86.0% vs 82.5%, P = .04).
Although serum lactate levels were lower in the albumin group at baseline (1.7 mmol/L vs 1.8 mmol/L, P = .05), inspection of the data appears to show a similar daily lactate clearance rate between groups over the first 7 study days (although these data were not analyzed by the authors). Achievement of a lactate level lower than 2 mmol/L on the first day of therapy was not significantly different between groups (73.4% vs 72.5%, P = .11).33
In a post hoc subgroup analysis, patients with septic shock at baseline randomized to albumin had a lower 90-day mortality rate than patients randomized to crystalloid solutions (RR 0.87, 95% CI 0.77–0.99). There was no difference in the 90-day mortality rate in patients without septic shock (RR 1.13, 95% CI 0.92–1.39, P = .03 for heterogeneity).33
These data suggest that albumin replacement may not improve outcomes in patients with severe sepsis, but may have advantages in terms of hemodynamic variables (and potentially mortality) in patients with septic shock. The role of albumin replacement in patients with septic shock warrants further study.
VASOPRESSORS
Vasopressors, inotropes, or both should be given to patients who have signs of hypoperfusion (including elevated lactate levels) despite preload optimization or ongoing fluid administration. The most appropriate drug depends on the goal: vasopressors are used to increase systemic vascular resistance, while inotropes are used to improve cardiac output and oxygen delivery.
Blood pressure target
The Surviving Sepsis Campaign guidelines recommend a mean arterial blood pressure target of at least 65 mm Hg during initial resuscitation and when vasopressors are applied for patients with septic shock.22 This recommendation is based on small studies that did not show differences in serum lactate levels or regional blood flow when the mean arterial pressure was elevated above 65 mm Hg with norepinephrine.34,35 However, the campaign guidelines note that the mean arterial pressure goal must be individualized in order to achieve optimal perfusion.
A large, open-label trial36 detected no difference in 28-day mortality rates in patients with septic shock between those allocated to a mean arterial pressure goal of 80 to 85 mm Hg or 65 to 70 mm Hg (36.6% vs 34.0%, P = .57). Although lactate levels did not differ between groups, the incidence of new-onset atrial fibrillation was higher in the higher-target group (6.7% vs 2.8%, P = .02). Fewer patients with chronic hypertension needed renal replacement therapy in the higher pressure group, further emphasizing the need to individualize the mean arterial pressure goal for patients in shock.36
Which vasopressor agent?
Dopamine and norepinephrine have traditionally been the preferred initial vasopressors for patients with shock. Until recently there were few data to guide selection between the two, but this is changing.
In a 2010 study of 1,679 patients with shock requiring vasopressors, there was no difference in the 28-day mortality rate between patients randomized to dopamine or norepinephrine (53% vs 49%, P = .10).37 Patients allocated to dopamine, though, had a higher incidence of arrhythmias (24% vs 12%, P < .001) and more frequently required open-label norepinephrine (26% vs 20%, P < .001). Although lactate levels and the time to achievement of a mean arterial pressure of 65 mm Hg were similar between groups, patients allocated to norepinephrine had more vasopressor-free days through day 28.
An a priori-planned subgroup analysis evaluated the influence of the type of shock on patient outcome. Patients with cardiogenic shock randomized to dopamine had a higher mortality rate than those randomized to norepinephrine (P = .03). However, the overall effect of treatment did not differ among the shock subgroups (interaction P = .87), suggesting that the reported differences in mortality according to subgroup may be spurious.
In a 2012 meta-analysis of patients with septic shock, dopamine use was associated with a higher mortality rate than norepinephrine use.38
In light of these data, norepinephrine should be preferred over dopamine as the initial vasopressor in most types of shock.
Epinephrine does not offer an outcome advantage over norepinephrine and may be associated with a higher incidence of adverse events.39–42 Indeed, in a study of patients with septic shock, lactate concentrations on the first day after randomization were significantly higher in patients allocated to epinephrine than in patients allocated to norepinephrine plus dobutamine.39 Similar effects on lactate concentrations with epinephrine were seen in patients with various types of shock40 and in those with cardiogenic shock.42
These differences in lactate concentrations may be directly attributable to epinephrine. Epinephrine can increase lactate concentrations through glycolysis and pyruvate dehydrogenase activation by stimulation of sodium-potassium ATPase activity via beta-2 adrenergic receptors in skeletal muscles,43 as well as decrease splanchnic perfusion.42,44,45 These effects may preclude using lactate clearance as a resuscitation goal in patients receiving epinephrine. Epinephrine is likely best reserved for patients with refractory shock,22 particularly those in whom cardiac output is known to be low.
Phenylephrine, essentially a pure vasoconstrictor, should be avoided in low cardiac output states and is best reserved for patients who develop a tachyarrhythmia on norepinephrine.22
Vasopressin, also a pure vasoconstrictor that should be avoided in low cardiac output states, has been best studied in patients with vasodilatory shock. Although controversy exists on the mortality benefits of vasopressin in vasodilatory shock, it is a relatively safe drug with consistent norepinephrine-sparing effects when added to existing norepinephrine therapy.46,47 In patients with less severe septic shock, including those with low lactate concentrations, adding vasopressin to norepinephrine instead of continuing norepinephrine alone may confer a mortality advantage.48
OTHER MEASURES TO OPTIMIZE OXYGEN DELIVERY
In circulatory shock from any cause, tissue oxygen demand exceeds oxygen delivery. Once arterial oxygenation and hemoglobin levels (by packed red blood cell transfusion) have been optimized, cardiac output is the critical determinant of oxygen delivery. Cardiac output may be augmented by ensuring adequate preload (by fluid resuscitation) or by giving inotropes or vasodilators.
The optimal cardiac output is difficult to define, and the exact marker for determining when cardiac output should be augmented is unclear. A strategy of increasing cardiac output to predefined “supranormal” levels was not associated with a lower mortality rate.49 Therefore, the decision to augment cardiac output must be individualized and will likely vary in the same patient over time.23
A reasonable approach to determining when augmentation of cardiac output is necessary was proposed in a study by Rivers et al.50 In that study, in patients randomized to early goal-directed therapy, inotropes were recommended when the central venous oxygenation saturation (Scvo2) was below 70% despite adequate fluid resuscitation (central venous pressure ≥ 8 mm Hg) and hematocrits were higher than 30%.
When an inotrope is indicated to improve cardiac output, dobutamine is usually the preferred agent. Dobutamine has a shorter half-life (allowing for easier titration) and causes less hypotension (assuming preload has been optimized) than phosphodiesterase type III inhibitors such as milrinone.
Mechanical support devices, such as intra-aortic balloon counterpulsation, and vasodilators can also be used to improve tissue perfusion in selected patients with low cardiac output syndromes.
USING LACTATE LEVELS TO GUIDE THERAPY
Lactate levels above 4.0 mmol/L
Lactate may be a useful marker for determining whether organ dysfunction is present and, hence, what course of therapy should be given, especially in sepsis. A serum lactate level higher than 4.0 mmol/L has been used as the trigger to start aggressive resuscitation in patients with sepsis.50,51
Traditionally, as delineated by Rivers et al50 in their landmark study of early goal-directed therapy, this entailed placing an arterial line and a central line for hemodynamic monitoring, with specific interventions directed at increasing the central venous pressure, mean arterial pressure, and central venous oxygen saturation.50 However, a recent study in a similar population of patients with sepsis with elevated lactate found no significant advantage of protocol-based resuscitation over care provided according to physician judgment, and no significant benefit in central venous catheterization and hemodynamic monitoring in all patients.51
Lactate clearance: 10% or above at 8 hours?
Regardless of the approach chosen, decreasing lactate levels can be interpreted as an adequate response to the interventions provided. As a matter of fact, several groups of investigators have also demonstrated the merits of lactate clearance alone as a prognostic indicator in patients requiring hemodynamic support.
McNelis et al52 retrospectively evaluated 95 postsurgical patients who required hemodynamic monitoring.52,53 The authors found that the slower the lactate clearance, the higher the mortality rate.
Given the prognostic implications of lactate clearance, investigators have evaluated whether lactate clearance could be used as a surrogate resuscitation goal for optimizing oxygen delivery. Using lactate clearance may have significant practical advantages over using central venous oxygen saturation, since it does not require a central venous catheter or continuous oximetric monitoring.
In a study comparing these two resuscitation end points, patients were randomized to a goal of either central venous oxygen saturation of 70% or more or lactate clearance of 10% or more within the first 6 hours after presentation as a marker of oxygen delivery.53 Mortality rates were similar with either strategy. Of note, only 10% of the patients actually required therapies to improve their oxygen delivery. Furthermore, there were no differences in the treatments given (including fluids, vasopressors, inotropes, packed red blood cells) throughout the treatment period.
These findings provide several insights. First, few patients admitted to the emergency department with severe sepsis and treated with an initial quantitative resuscitation protocol require additional therapy for augmenting oxygen delivery. Second, lactate clearance, in a setting where initial resuscitation with fluids and vasopressors restores adequate oxygen delivery for the majority of patients, is likely as good a target for resuscitation as central venous oxygen saturation.
This study, however, does not address the question of whether lactate clearance is useful as an additional marker of oxygen delivery (in conjunction with central venous oxygen saturation). Indeed, caution should be taken to target central venous oxygen saturation goals alone, as patients with septic shock presenting with venous hyperoxia (central venous oxygen saturation > 89%) have been shown to have a higher mortality rate than patients with normoxia (central venous oxygen saturation 71%–89%).54
This was further demonstrated by Arnold et al in a study of patients presenting to the emergency department with severe sepsis.15 In this study, significant discordance between central venous oxygen saturation and lactate clearance was seen, where 79% of patients with less than 10% lactate clearance had concomitant central venous oxygen saturation of 70% or greater.
Jansen et al18 evaluated the role of targeting lactate clearance in conjunction with central venous oxygen saturation monitoring. In this study, critically ill patients with elevated lactate and inadequate lactate clearance were randomized to usual care or to resuscitation to adequate lactate clearance (20% or more). The therapies to optimize oxygen delivery were given according to the central venous oxygen saturation. Overall, after adjustment for predefined risk factors, the in-hospital mortality rate was lower in the lactate clearance group. This may signify that patients with sepsis and central venous oxygen saturation of 70% or more may continue to have poor lactate clearance, warranting further treatment.
Taken together, serum lactate may be helpful for prognostication, determination of course of therapy, and quantification for tissue hypoperfusion for targeted therapies. Figure 2 presents our approach to an elevated lactate level. As performed in the study by Jansen et al,18 it seems reasonable to measure lactate levels every 2 hours for the first 8 hours of resuscitation in patients with type A lactic acidosis. These levels should be interpreted in the context of lactate clearance (at least 10%, but preferably 20%) and normalization, and should be treated with an approach similar to the one outlined in Figure 2.
TREATING TYPE B LACTIC ACIDOSIS (NORMAL PERFUSION AND OXYGENATION)
Treating type B lactic acidosis is quite different because the goal is not to correct mismatches in oxygen consumption and delivery. Since most cases are due to underlying conditions such as malignancy or medications, treatment should be centered around eliminating the cause (eg, treat the malignancy, discontinue the offending medication). The main reason for treatment is to alleviate the harmful effects of acidosis. For example, acidosis can result in a negative inotropic effect.
Sodium bicarbonate, dichloroacetate, carbicarb, and tromethamine have all been studied in the management of type B lactic acidosis, with little success.55,56
Renal replacement therapy has had some success in drug-induced lactic acidosis.57,58
l-carnitine has had promising results in treating patients with human immunodeficiency virus infection, since these patients are carnitine-deficient and carnitine plays an important role in mitochondrial function.59
Thiamine and biotin deficiencies can occur in patients receiving total parenteral nutrition without vitamins and in patients who drink alcohol heavily and can cause lactic acidosis. These nutrients should be supplemented accordingly.
Treatment of mitochondrial disorders includes antioxidants (coenzyme Q10, vitamin C, vitamin E) and amino acids (l-arginine).60
- Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc 2013; 88:1127–1140.
- Fuller BM, Dellinger RP. Lactate as a hemodynamic marker in the critically ill. Curr Opin Crit Care 2012; 18:267–272.
- Fall PJ, Szerlip HM. Lactic acidosis: from sour milk to septic shock. J Intensive Care Med 2005; 20:255–271.
- Kruse O, Grunnet N, Barfod C. Blood lactate as a predictor for in-hospital mortality in patients admitted acutely to hospital: a systematic review. Scand J Trauma Resusc Emerg Med 2011;19:74.
- Howell MD, Donnino M, Clardy P, Talmor D, Shapiro NI. Occult hypoperfusion and mortality in patients with suspected infection. Intensive Care Med 2007; 33:1892–1899.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Outcomes of patients undergoing early sepsis resuscitation for cryptic shock compared with overt shock. Resuscitation 2011; 82:1289–1293.
- Bakker J, Nijsten MW, Jansen TC. Clinical use of lactate monitoring in critically ill patients. Ann Intensive Care 2013; 3:12.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Shapiro NI, Howell MD, Talmor D, et al. Serum lactate as a predictor of mortality in emergency department patients with infection. Ann Emerg Med 2005; 45:524–528.
- Mikkelsen ME, Miltiades AN, Gaieski DF, et al. Serum lactate is associated with mortality in severe sepsis independent of organ failure and shock. Crit Care Med 2009; 37:1670–1677.
- Liu V, Morehouse JW, Soule J, Whippy A, Escobar GJ. Fluid volume, lactate values, and mortality in sepsis patients with intermediate lactate values. Ann Am Thorac Soc 2013; 10:466–473.
- Sterling SA, Puskarich MA, Shapiro NI, et al; Emergency Medicine Shock Research Network (EMShockNET). Characteristics and outcomes of patients with vasoplegic versus tissue dysoxic septic shock. Shock 2013; 40:11–14.
- Arnold RC, Shapiro NI, Jones AE, et al; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Jones AE. Lactate clearance for assessing response to resuscitation in severe sepsis. Acad Emerg Med 2013; 20:844–847.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Jansen TC, van Bommel J, Schoonderbeek FJ, et al; LACTATE study group. Early lactate-guided therapy in intensive care unit patients: a multicenter, open-label, randomized controlled trial. Am J Respir Crit Care Med 2010; 182:752–761.
- Husain FA, Martin MJ, Mullenix PS, Steele SR, Elliott DC. Serum lactate and base deficit as predictors of mortality and morbidity. Am J Surg 2003; 185:485–491.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Whole blood lactate kinetics in patients undergoing quantitative resuscitation for severe sepsis and septic shock. Chest 2013; 143:1548–1553.
- Marty P, Roquilly A, Vallee F, et al. Lactate clearance for death prediction in severe sepsis or septic shock patients during the first 24 hours in intensive care unit: an observational study. Ann Intensive Care 2013; 3:3.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Vincent JL, De Backer D. Circulatory shock. N Engl J Med 2013; 369:1726–1734.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Vincent JL, Dufaye P, Berré J, Leeman M, Degaute JP, Kahn RJ. Serial lactate determinations during circulatory shock. Crit Care Med 1983; 11:449–451.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R; SAFE Study Investigators. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Myburgh JA, Finfer S, Bellomo R, et al; CHEST Investigators; Australian and New Zealand Intensive Care Society Clinical Trials Group. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Perner A, Haase N, Guttormsen AB, et al; 6S Trial Group; Scandinavian Critical Care Trials Group. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Bayer O, Reinhart K, Kohl M, et al. Effects of fluid resuscitation with synthetic colloids or crystalloids alone on shock reversal, fluid balance, and patient outcomes in patients with severe sepsis: a prospective sequential analysis. Crit Care Med 2012; 40:2543–2551.
- Delaney AP, Dan A, McCaffrey J, Finfer S. The role of albumin as a resuscitation fluid for patients with sepsis: a systematic review and meta-analysis. Crit Care Med 2011; 39:386–391.
- SAFE Study Investigators; Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Caironi P, Tognoni G, Masson S, et al; ALBIOS Study Investigators. Albumin replacement in patients with severe sepsis or septic shock. N Engl J Med 2014; 370:1412–1421.
- Bourgoin A, Leone M, Delmas A, Garnier F, Albanèse J, Martin C. Increasing mean arterial pressure in patients with septic shock: effects on oxygen variables and renal function. Crit Care Med 2005; 33:780–786.
- LeDoux D, Astiz ME, Carpati CM, Rackow EC. Effects of perfusion pressure on tissue perfusion in septic shock. Crit Care Med 2000; 28:2729–2732.
- Asfar P, Meziani F, Hamel JF, et al; SEPSISPAM Investigators. High versus low blood-pressure target in patients with septic shock. N Engl J Med 2014; 370:1583–1593.
- De Backer D, Biston P, Devriendt J, et al; SOAP II Investigators. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- De Backer D, Aldecoa C, Njimi H, Vincent JL. Dopamine versus norepinephrine in the treatment of septic shock: a meta-analysis. Crit Care Med 2012; 40:725–730.
- Annane D, Vignon P, Renault A, et al: CATS Study Group. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Myburgh JA, Higgins A, Jovanovska A, Lipman J, Ramakrishnan N, Santamaria J; CAT Study investigators. A comparison of epinephrine and norepinephrine in critically ill patients. Intensive Care Med 2008; 34:2226–2234.
- Schmittinger CA, Torgersen C, Luckner G, Schröder DC, Lorenz I, Dünser MW. Adverse cardiac events during catecholamine vasopressor therapy: a prospective observational study. Intensive Care Med 2012; 38:950–958.
- Levy B, Perez P, Perny J, Thivilier C, Gerard A. Comparison of norepinephrine-dobutamine to epinephrine for hemodynamics, lactate metabolism, and organ function variables in cardiogenic shock. A prospective, randomized pilot study. Crit Care Med 2011; 39:450–455.
- Watt MJ, Howlett KF, Febbraio MA, Spriet LL, Hargreaves M. Adrenaline increases skeletal muscle glycogenolysis, pyruvate dehydrogenase activation and carbohydrate oxidation during moderate exercise in humans. J Physiol 2001; 534:269–278.
- De Backer D, Creteur J, Silva E, Vincent JL. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? Crit Care Med 2003; 31:1659–1667.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Polito A, Parisini E, Ricci Z, Picardo S, Annane D. Vasopressin for treatment of vasodilatory shock: an ESICM systematic review and meta-analysis. Intensive Care Med 2012; 38:9–19.
- Serpa Neto A, Nassar APJ, Cardoso SO, et al. Vasopressin and terlipressin in adult vasodilatory shock: a systematic review and meta-analysis of nine randomized controlled trials. Crit Care 2012; 16:R154.
- Russell JA, Walley KR, Singer J, et al; VASST Investigators. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Gattinoni L, Brazzi L, Pelosi P, et al; for the SvO2 Collaborative Group. A trial of goal-oriented hemodynamic therapy in critically ill patients. N Engl J Med 1995; 333:1025–1032.
- Rivers E, Nguyen B, Havstad S, et al; Early Goal-Directed Therapy Collaborative Group. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- ProCESS Investigators; Yealy DM, Kellum JA, Huang DT, et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med 2014; 370:1683–1693.
- McNelis J, Marini CP, Jurkiewicz A, et al. Prolonged lactate clearance is associated with increased mortality in the surgical intensive care unit. Am J Surg 2001; 182:481–485.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Pope JV, Jones AE, Gaieski DF, Arnold RC, Trzeciak S, Shapiro NI; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of central venous oxygen saturation (ScvO2) as a predictor of mortality in patients with sepsis. Ann Emerg Med 2010; 55:40–46.e1
- Kraut JA, Kurtz I. Use of base in the treatment of severe acidemic states. Am J Kidney Dis 2001; 38:703–727.
- Levraut J, Grimaud D. Treatment of metabolic acidosis. Curr Opin Crit Care 2003; 9:260–265.
- Orija AA, Jenks CL. Nucleoside analog reverse transcriptase inhibitor induced lactic acidosis treated with continuous renal replacement in the medical intensive care unit. Crit Care & Shock 2012; 15:9–11.
- Friesecke S, Abel P, Kraft M, Gerner A, Runge S. Combined renal replacement therapy for severe metformin-induced lactic acidosis. Nephrol Dial Transplant 2006; 21:2038–2039.
- Claessens YE, Cariou A, Monchi M, et al. Detecting life-threatening lactic acidosis related to nucleoside-analog treatment of human immunodeficiency virus-infected patients, and treatment with l-carnitine. Crit Care Med 2003; 31:1042–1047.
- Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R; Medicine Society TM. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol 2009; 11:414–430.
Physicians are paying more attention to serum lactate levels in hospitalized patients than in the past, especially with the advent of point-of-care testing. Elevated lactate levels are associated with tissue hypoxia and hypoperfusion but can also be found in a number of other conditions. Therefore, confusion can arise as to how to interpret elevated levels and subsequently manage these patients in a variety of settings.
In this review, we discuss the mechanisms underlying lactic acidosis, its prognostic implications, and its use as a therapeutic target in treating patients in septic shock and other serious disorders.
LACTATE IS A PRODUCT OF ANAEROBIC RESPIRATION
Lactate, or lactic acid, is produced from pyruvate as an end product of glycolysis under anaerobic conditions (Figure 1). It is produced in most tissues in the body, but primarily in skeletal muscle, brain, intestine, and red blood cells. During times of stress, lactate is also produced in the lungs, white blood cells, and splanchnic organs.
Most lactate in the blood is cleared by the liver, where it is the substrate for gluconeogenesis, and a small amount is cleared by the kidneys.1,2 The entire pathway by which lactate is produced and converted back to glucose is called the Cori cycle.
NORMAL LEVELS ARE LESS THAN ABOUT 2.0 MMOL/L
In this review, we will present lactate levels in the SI units of mmol/L (1 mmol/L = 9 mg/dL).
Basal lactate production is approximately 0.8 mmol/kg body weight/hour. The average normal arterial blood lactate level is approximately 0.620 mmol/L and the venous level is slightly higher at 0.997 mmol/L,3 but overall, arterial and venous lactate levels correlate well.
Normal lactate levels are less than 2 mmol/L,4 intermediate levels range from 2 to less than 4 mmol/L, and high levels are 4 mmol/L or higher.5
To minimize variations in measurement, blood samples should be drawn without a tourniquet into tubes containing fluoride, placed on ice, and processed quickly (ideally within 15 minutes).
INCREASED PRODUCTION, DECREASED CLEARANCE, OR BOTH
An elevated lactate level can be the result of increased production, decreased clearance, or both (as in liver dysfunction).
Type A lactic acidosis—due to hypoperfusion and hypoxia—occurs when there is a mismatch between oxygen delivery and consumption, with resultant anaerobic glycolysis.
The guidelines from the Surviving Sepsis Campaign6 emphasize using lactate levels to diagnose patients with sepsis-induced hypoperfusion. However, hyperlactatemia can indicate inadequate oxygen delivery due to any type of shock (Table 1).
Type B lactic acidosis—not due to hypoperfusion—occurs in a variety of conditions (Table 1), including liver disease, malignancy, use of certain medications (eg, metformin, epinephrine), total parenteral nutrition, human immunodeficiency virus infection, thiamine deficiency, mitochondrial myopathies, and congenital lactic acidosis.1–3,7 Yet other causes include trauma, excessive exercise, diabetic ketoacidosis, ethanol intoxication, dysfunction of the enzyme pyruvate dehydrogenase, and increased muscle degradation leading to increased production of pyruvate. In these latter scenarios, glucose metabolism exceeds the oxidation capacity of the mitochondria, and the rise in pyruvate concentration drives lactate production.8,9 Mitochondrial dysfunction and subsequent deficits in cellular oxygen use can also result in persistently high lactate levels.10
In some situations, patients with mildly elevated lactic acid levels in type B lactic acidosis can be monitored to ensure stability, rather than be treated aggressively.
HIGHER LEVELS AND LOWER CLEARANCE PREDICT DEATH
The higher the lactate level and the slower the rate of normalization (lactate clearance), the higher the risk of death.
Lactate levels and mortality rate
Shapiro et al11 showed that increases in lactate level are associated with proportional increases in the mortality rate. Mikkelsen et al12 showed that intermediate levels (2.0–3.9 mmol/L) and high levels (≥ 4 mmol/L) of serum lactate are associated with increased risk of death independent of organ failure and shock. Patients with mildly elevated and intermediate lactate levels and sepsis have higher rates of in-hospital and 30-day mortality, which correlate with the baseline lactate level.13
In a post hoc analysis of a randomized controlled trial, patients with septic shock who presented to the emergency department with hypotension and a lactate level higher than 2 mmol/L had a significantly higher in-hospital mortality rate than those who presented with hypotension and a lactate level of 2 mmol/L or less (26% vs 9%, P < .0001).14 These data suggest that elevated lactate levels may have a significant prognostic role, independent of blood pressure.
Slower clearance
The prognostic implications of lactate clearance (reductions in lactate levels over time, as opposed to a single value in time), have also been evaluated.
Lactate clearance of at least 10% at 6 hours after presentation has been associated with a lower mortality rate than nonclearance (19% vs 60%) in patients with sepsis or septic shock with elevated levels.15–17 Similar findings have been reported in a general intensive care unit population,18 as well as a surgical intensive care population.sup>19
Puskarich et al20 have also shown that lactate normalization to less than 2 mmol/L during early sepsis resuscitation is the strongest predictor of survival (odds ratio [OR] 5.2), followed by lactate clearance of 50% (OR 4.0) within the first 6 hours of presentation. Not only is lactate clearance associated with improved outcomes, but a faster rate of clearance after initial presentation is also beneficial.15,16,18
Lactate clearance over a longer period (> 6 hours) has not been studied in patients with septic shock. However, in the general intensive care unit population, therapy guided by lactate clearance for the first 8 hours after presentation has shown a reduction in mortality rate.18 There are no data available on outcomes of lactate-directed therapy beyond 8 hours, but lactate concentration and lactate clearance at 24 hours correlate with the 28-day mortality rate.21
Cryptic shock
Cryptic shock describes a state in a subgroup of patients who have elevated lactate levels and global tissue hypoxia despite being normotensive or even hypertensive. These patients have a higher mortality rate independent of blood pressure. Jansen et al18 found that patients with a lactate level higher than 4 mmol/L and preserved blood pressure had a mortality rate of 15%, while those without shock or hyperlactatemia had a mortality rate of 2.5%. In addition, patients with an elevated lactate level in the absence of hypotension have mortality rates similar to those in patients with high lactate levels and hypotension refractory to fluid boluses, suggesting the presence of tissue hypoxia even in these normotensive patients.6
HOW TO APPROACH AN ELEVATED LACTATE LEVEL
An elevated lactate level should prompt an evaluation for causes of decreased oxygen delivery, due either to a systemic low-flow state (as a result of decreased cardiac output) or severe anemia, or to regionally decreased perfusion, (eg, limb or mesenteric ischemia). If tissue hypoxia is ruled out after an exhaustive workup, consideration should be given to causes of hyperlactatemia without concomitant tissue hypoxia (type B acidosis).
Treatment differs depending on the underlying mechanism of the lactate elevation; nevertheless, treatment is mostly related to optimizing oxygen delivery by giving fluids, packed red blood cells, and vasopressors or inotropic agents, or both (Figure 2). The specific treatment differs based on the shock state, but there are similarities that can guide the clinician.
FLUID SUPPORT
Giving fluids, with a goal of improving cardiac output, remains a cornerstone of initial therapy for most shock states.22,23
How much fluid?
Fluids should be given until the patient is no longer preload-dependent, although there is much debate about which assessment strategy should be used to determine if cardiac output will improve with more fluid (ie, fluid-responsiveness).24 In many cases, fluid resuscitation alone may be enough to restore hemodynamic stability, improve tissue perfusion, and reduce elevated lactate concentrations.25
The decision to give more fluids should not be made lightly, though, as a more positive fluid balance early in the course of septic shock and over 4 days has been associated with a higher mortality rate.26 Additionally, pushing fluids in patients with cardiogenic shock due to impaired left ventricular systolic function may lead to or worsen pulmonary edema. Therefore, the indiscriminate use of fluids should be avoided.
Which fluids?
Despite years of research, controversy persists about whether crystalloids or colloids are better for resuscitation. Randomized trials in heterogeneous intensive care unit patients have not detected differences in 28-day mortality rates between those allocated to crystalloids or 4% albumin27 and those allocated to crystalloids or hydroxyethyl starch.28
Hydroxyethyl starch may not be best. In a study of patients with severe sepsis, those randomized to receive hydroxyethyl starch had a higher 90-day mortality rate than patients randomized to crystalloids (51% vs 43%, P = .03).29 A sequential prospective before-and-after study did not detect a difference in the time to normalization (< 2.2 mmol/L) of lactate (P = .68) or cessation of vasopressors (P = .11) in patients with severe sepsis who received fluid resuscitation with crystalloids, gelatin, or hydroxyethyl starch. More patients who received hydroxyethyl starch in these studies developed acute kidney injury than those receiving crystalloids.28–30
Taken together, these data strongly suggest hydroxyethyl starch should not be used for fluid resuscitation in the intensive care unit.
Normal saline or albumin? Although some data suggest that albumin may be preferable to 0.9% sodium chloride in patients with severe sepsis,31,32 these analyses should be viewed as hypothesis-generating. There do not seem to be differences between fluid types in terms of subsequent serum lactate concentrations or achievement of lactate clearance goals.28–30 Until further studies are completed, both albumin and crystalloids are reasonable for resuscitation.
Caironi et al33 performed an open-label study comparing albumin replacement (with a goal serum albumin concentration of 3 g/dL) plus a crystalloid solution vs a crystalloid solution alone in patients with severe sepsis or septic shock. They detected no difference between the albumin and crystalloid groups in mortality rates at 28 days (31.8% vs 32.0%, P = .94) or 90 days (41.1% vs 43.6%, P = .29). However, patients in the albumin group had a shorter time to cessation of vasoactive agents (median 3 vs 4 days, P = .007) and lower cardiovascular Sequential Organ Failure Assessment subscores (median 1.20 vs 1.42, P = .03), and more frequently achieved a mean arterial pressure of at least 65 mm Hg within 6 hours of randomization (86.0% vs 82.5%, P = .04).
Although serum lactate levels were lower in the albumin group at baseline (1.7 mmol/L vs 1.8 mmol/L, P = .05), inspection of the data appears to show a similar daily lactate clearance rate between groups over the first 7 study days (although these data were not analyzed by the authors). Achievement of a lactate level lower than 2 mmol/L on the first day of therapy was not significantly different between groups (73.4% vs 72.5%, P = .11).33
In a post hoc subgroup analysis, patients with septic shock at baseline randomized to albumin had a lower 90-day mortality rate than patients randomized to crystalloid solutions (RR 0.87, 95% CI 0.77–0.99). There was no difference in the 90-day mortality rate in patients without septic shock (RR 1.13, 95% CI 0.92–1.39, P = .03 for heterogeneity).33
These data suggest that albumin replacement may not improve outcomes in patients with severe sepsis, but may have advantages in terms of hemodynamic variables (and potentially mortality) in patients with septic shock. The role of albumin replacement in patients with septic shock warrants further study.
VASOPRESSORS
Vasopressors, inotropes, or both should be given to patients who have signs of hypoperfusion (including elevated lactate levels) despite preload optimization or ongoing fluid administration. The most appropriate drug depends on the goal: vasopressors are used to increase systemic vascular resistance, while inotropes are used to improve cardiac output and oxygen delivery.
Blood pressure target
The Surviving Sepsis Campaign guidelines recommend a mean arterial blood pressure target of at least 65 mm Hg during initial resuscitation and when vasopressors are applied for patients with septic shock.22 This recommendation is based on small studies that did not show differences in serum lactate levels or regional blood flow when the mean arterial pressure was elevated above 65 mm Hg with norepinephrine.34,35 However, the campaign guidelines note that the mean arterial pressure goal must be individualized in order to achieve optimal perfusion.
A large, open-label trial36 detected no difference in 28-day mortality rates in patients with septic shock between those allocated to a mean arterial pressure goal of 80 to 85 mm Hg or 65 to 70 mm Hg (36.6% vs 34.0%, P = .57). Although lactate levels did not differ between groups, the incidence of new-onset atrial fibrillation was higher in the higher-target group (6.7% vs 2.8%, P = .02). Fewer patients with chronic hypertension needed renal replacement therapy in the higher pressure group, further emphasizing the need to individualize the mean arterial pressure goal for patients in shock.36
Which vasopressor agent?
Dopamine and norepinephrine have traditionally been the preferred initial vasopressors for patients with shock. Until recently there were few data to guide selection between the two, but this is changing.
In a 2010 study of 1,679 patients with shock requiring vasopressors, there was no difference in the 28-day mortality rate between patients randomized to dopamine or norepinephrine (53% vs 49%, P = .10).37 Patients allocated to dopamine, though, had a higher incidence of arrhythmias (24% vs 12%, P < .001) and more frequently required open-label norepinephrine (26% vs 20%, P < .001). Although lactate levels and the time to achievement of a mean arterial pressure of 65 mm Hg were similar between groups, patients allocated to norepinephrine had more vasopressor-free days through day 28.
An a priori-planned subgroup analysis evaluated the influence of the type of shock on patient outcome. Patients with cardiogenic shock randomized to dopamine had a higher mortality rate than those randomized to norepinephrine (P = .03). However, the overall effect of treatment did not differ among the shock subgroups (interaction P = .87), suggesting that the reported differences in mortality according to subgroup may be spurious.
In a 2012 meta-analysis of patients with septic shock, dopamine use was associated with a higher mortality rate than norepinephrine use.38
In light of these data, norepinephrine should be preferred over dopamine as the initial vasopressor in most types of shock.
Epinephrine does not offer an outcome advantage over norepinephrine and may be associated with a higher incidence of adverse events.39–42 Indeed, in a study of patients with septic shock, lactate concentrations on the first day after randomization were significantly higher in patients allocated to epinephrine than in patients allocated to norepinephrine plus dobutamine.39 Similar effects on lactate concentrations with epinephrine were seen in patients with various types of shock40 and in those with cardiogenic shock.42
These differences in lactate concentrations may be directly attributable to epinephrine. Epinephrine can increase lactate concentrations through glycolysis and pyruvate dehydrogenase activation by stimulation of sodium-potassium ATPase activity via beta-2 adrenergic receptors in skeletal muscles,43 as well as decrease splanchnic perfusion.42,44,45 These effects may preclude using lactate clearance as a resuscitation goal in patients receiving epinephrine. Epinephrine is likely best reserved for patients with refractory shock,22 particularly those in whom cardiac output is known to be low.
Phenylephrine, essentially a pure vasoconstrictor, should be avoided in low cardiac output states and is best reserved for patients who develop a tachyarrhythmia on norepinephrine.22
Vasopressin, also a pure vasoconstrictor that should be avoided in low cardiac output states, has been best studied in patients with vasodilatory shock. Although controversy exists on the mortality benefits of vasopressin in vasodilatory shock, it is a relatively safe drug with consistent norepinephrine-sparing effects when added to existing norepinephrine therapy.46,47 In patients with less severe septic shock, including those with low lactate concentrations, adding vasopressin to norepinephrine instead of continuing norepinephrine alone may confer a mortality advantage.48
OTHER MEASURES TO OPTIMIZE OXYGEN DELIVERY
In circulatory shock from any cause, tissue oxygen demand exceeds oxygen delivery. Once arterial oxygenation and hemoglobin levels (by packed red blood cell transfusion) have been optimized, cardiac output is the critical determinant of oxygen delivery. Cardiac output may be augmented by ensuring adequate preload (by fluid resuscitation) or by giving inotropes or vasodilators.
The optimal cardiac output is difficult to define, and the exact marker for determining when cardiac output should be augmented is unclear. A strategy of increasing cardiac output to predefined “supranormal” levels was not associated with a lower mortality rate.49 Therefore, the decision to augment cardiac output must be individualized and will likely vary in the same patient over time.23
A reasonable approach to determining when augmentation of cardiac output is necessary was proposed in a study by Rivers et al.50 In that study, in patients randomized to early goal-directed therapy, inotropes were recommended when the central venous oxygenation saturation (Scvo2) was below 70% despite adequate fluid resuscitation (central venous pressure ≥ 8 mm Hg) and hematocrits were higher than 30%.
When an inotrope is indicated to improve cardiac output, dobutamine is usually the preferred agent. Dobutamine has a shorter half-life (allowing for easier titration) and causes less hypotension (assuming preload has been optimized) than phosphodiesterase type III inhibitors such as milrinone.
Mechanical support devices, such as intra-aortic balloon counterpulsation, and vasodilators can also be used to improve tissue perfusion in selected patients with low cardiac output syndromes.
USING LACTATE LEVELS TO GUIDE THERAPY
Lactate levels above 4.0 mmol/L
Lactate may be a useful marker for determining whether organ dysfunction is present and, hence, what course of therapy should be given, especially in sepsis. A serum lactate level higher than 4.0 mmol/L has been used as the trigger to start aggressive resuscitation in patients with sepsis.50,51
Traditionally, as delineated by Rivers et al50 in their landmark study of early goal-directed therapy, this entailed placing an arterial line and a central line for hemodynamic monitoring, with specific interventions directed at increasing the central venous pressure, mean arterial pressure, and central venous oxygen saturation.50 However, a recent study in a similar population of patients with sepsis with elevated lactate found no significant advantage of protocol-based resuscitation over care provided according to physician judgment, and no significant benefit in central venous catheterization and hemodynamic monitoring in all patients.51
Lactate clearance: 10% or above at 8 hours?
Regardless of the approach chosen, decreasing lactate levels can be interpreted as an adequate response to the interventions provided. As a matter of fact, several groups of investigators have also demonstrated the merits of lactate clearance alone as a prognostic indicator in patients requiring hemodynamic support.
McNelis et al52 retrospectively evaluated 95 postsurgical patients who required hemodynamic monitoring.52,53 The authors found that the slower the lactate clearance, the higher the mortality rate.
Given the prognostic implications of lactate clearance, investigators have evaluated whether lactate clearance could be used as a surrogate resuscitation goal for optimizing oxygen delivery. Using lactate clearance may have significant practical advantages over using central venous oxygen saturation, since it does not require a central venous catheter or continuous oximetric monitoring.
In a study comparing these two resuscitation end points, patients were randomized to a goal of either central venous oxygen saturation of 70% or more or lactate clearance of 10% or more within the first 6 hours after presentation as a marker of oxygen delivery.53 Mortality rates were similar with either strategy. Of note, only 10% of the patients actually required therapies to improve their oxygen delivery. Furthermore, there were no differences in the treatments given (including fluids, vasopressors, inotropes, packed red blood cells) throughout the treatment period.
These findings provide several insights. First, few patients admitted to the emergency department with severe sepsis and treated with an initial quantitative resuscitation protocol require additional therapy for augmenting oxygen delivery. Second, lactate clearance, in a setting where initial resuscitation with fluids and vasopressors restores adequate oxygen delivery for the majority of patients, is likely as good a target for resuscitation as central venous oxygen saturation.
This study, however, does not address the question of whether lactate clearance is useful as an additional marker of oxygen delivery (in conjunction with central venous oxygen saturation). Indeed, caution should be taken to target central venous oxygen saturation goals alone, as patients with septic shock presenting with venous hyperoxia (central venous oxygen saturation > 89%) have been shown to have a higher mortality rate than patients with normoxia (central venous oxygen saturation 71%–89%).54
This was further demonstrated by Arnold et al in a study of patients presenting to the emergency department with severe sepsis.15 In this study, significant discordance between central venous oxygen saturation and lactate clearance was seen, where 79% of patients with less than 10% lactate clearance had concomitant central venous oxygen saturation of 70% or greater.
Jansen et al18 evaluated the role of targeting lactate clearance in conjunction with central venous oxygen saturation monitoring. In this study, critically ill patients with elevated lactate and inadequate lactate clearance were randomized to usual care or to resuscitation to adequate lactate clearance (20% or more). The therapies to optimize oxygen delivery were given according to the central venous oxygen saturation. Overall, after adjustment for predefined risk factors, the in-hospital mortality rate was lower in the lactate clearance group. This may signify that patients with sepsis and central venous oxygen saturation of 70% or more may continue to have poor lactate clearance, warranting further treatment.
Taken together, serum lactate may be helpful for prognostication, determination of course of therapy, and quantification for tissue hypoperfusion for targeted therapies. Figure 2 presents our approach to an elevated lactate level. As performed in the study by Jansen et al,18 it seems reasonable to measure lactate levels every 2 hours for the first 8 hours of resuscitation in patients with type A lactic acidosis. These levels should be interpreted in the context of lactate clearance (at least 10%, but preferably 20%) and normalization, and should be treated with an approach similar to the one outlined in Figure 2.
TREATING TYPE B LACTIC ACIDOSIS (NORMAL PERFUSION AND OXYGENATION)
Treating type B lactic acidosis is quite different because the goal is not to correct mismatches in oxygen consumption and delivery. Since most cases are due to underlying conditions such as malignancy or medications, treatment should be centered around eliminating the cause (eg, treat the malignancy, discontinue the offending medication). The main reason for treatment is to alleviate the harmful effects of acidosis. For example, acidosis can result in a negative inotropic effect.
Sodium bicarbonate, dichloroacetate, carbicarb, and tromethamine have all been studied in the management of type B lactic acidosis, with little success.55,56
Renal replacement therapy has had some success in drug-induced lactic acidosis.57,58
l-carnitine has had promising results in treating patients with human immunodeficiency virus infection, since these patients are carnitine-deficient and carnitine plays an important role in mitochondrial function.59
Thiamine and biotin deficiencies can occur in patients receiving total parenteral nutrition without vitamins and in patients who drink alcohol heavily and can cause lactic acidosis. These nutrients should be supplemented accordingly.
Treatment of mitochondrial disorders includes antioxidants (coenzyme Q10, vitamin C, vitamin E) and amino acids (l-arginine).60
Physicians are paying more attention to serum lactate levels in hospitalized patients than in the past, especially with the advent of point-of-care testing. Elevated lactate levels are associated with tissue hypoxia and hypoperfusion but can also be found in a number of other conditions. Therefore, confusion can arise as to how to interpret elevated levels and subsequently manage these patients in a variety of settings.
In this review, we discuss the mechanisms underlying lactic acidosis, its prognostic implications, and its use as a therapeutic target in treating patients in septic shock and other serious disorders.
LACTATE IS A PRODUCT OF ANAEROBIC RESPIRATION
Lactate, or lactic acid, is produced from pyruvate as an end product of glycolysis under anaerobic conditions (Figure 1). It is produced in most tissues in the body, but primarily in skeletal muscle, brain, intestine, and red blood cells. During times of stress, lactate is also produced in the lungs, white blood cells, and splanchnic organs.
Most lactate in the blood is cleared by the liver, where it is the substrate for gluconeogenesis, and a small amount is cleared by the kidneys.1,2 The entire pathway by which lactate is produced and converted back to glucose is called the Cori cycle.
NORMAL LEVELS ARE LESS THAN ABOUT 2.0 MMOL/L
In this review, we will present lactate levels in the SI units of mmol/L (1 mmol/L = 9 mg/dL).
Basal lactate production is approximately 0.8 mmol/kg body weight/hour. The average normal arterial blood lactate level is approximately 0.620 mmol/L and the venous level is slightly higher at 0.997 mmol/L,3 but overall, arterial and venous lactate levels correlate well.
Normal lactate levels are less than 2 mmol/L,4 intermediate levels range from 2 to less than 4 mmol/L, and high levels are 4 mmol/L or higher.5
To minimize variations in measurement, blood samples should be drawn without a tourniquet into tubes containing fluoride, placed on ice, and processed quickly (ideally within 15 minutes).
INCREASED PRODUCTION, DECREASED CLEARANCE, OR BOTH
An elevated lactate level can be the result of increased production, decreased clearance, or both (as in liver dysfunction).
Type A lactic acidosis—due to hypoperfusion and hypoxia—occurs when there is a mismatch between oxygen delivery and consumption, with resultant anaerobic glycolysis.
The guidelines from the Surviving Sepsis Campaign6 emphasize using lactate levels to diagnose patients with sepsis-induced hypoperfusion. However, hyperlactatemia can indicate inadequate oxygen delivery due to any type of shock (Table 1).
Type B lactic acidosis—not due to hypoperfusion—occurs in a variety of conditions (Table 1), including liver disease, malignancy, use of certain medications (eg, metformin, epinephrine), total parenteral nutrition, human immunodeficiency virus infection, thiamine deficiency, mitochondrial myopathies, and congenital lactic acidosis.1–3,7 Yet other causes include trauma, excessive exercise, diabetic ketoacidosis, ethanol intoxication, dysfunction of the enzyme pyruvate dehydrogenase, and increased muscle degradation leading to increased production of pyruvate. In these latter scenarios, glucose metabolism exceeds the oxidation capacity of the mitochondria, and the rise in pyruvate concentration drives lactate production.8,9 Mitochondrial dysfunction and subsequent deficits in cellular oxygen use can also result in persistently high lactate levels.10
In some situations, patients with mildly elevated lactic acid levels in type B lactic acidosis can be monitored to ensure stability, rather than be treated aggressively.
HIGHER LEVELS AND LOWER CLEARANCE PREDICT DEATH
The higher the lactate level and the slower the rate of normalization (lactate clearance), the higher the risk of death.
Lactate levels and mortality rate
Shapiro et al11 showed that increases in lactate level are associated with proportional increases in the mortality rate. Mikkelsen et al12 showed that intermediate levels (2.0–3.9 mmol/L) and high levels (≥ 4 mmol/L) of serum lactate are associated with increased risk of death independent of organ failure and shock. Patients with mildly elevated and intermediate lactate levels and sepsis have higher rates of in-hospital and 30-day mortality, which correlate with the baseline lactate level.13
In a post hoc analysis of a randomized controlled trial, patients with septic shock who presented to the emergency department with hypotension and a lactate level higher than 2 mmol/L had a significantly higher in-hospital mortality rate than those who presented with hypotension and a lactate level of 2 mmol/L or less (26% vs 9%, P < .0001).14 These data suggest that elevated lactate levels may have a significant prognostic role, independent of blood pressure.
Slower clearance
The prognostic implications of lactate clearance (reductions in lactate levels over time, as opposed to a single value in time), have also been evaluated.
Lactate clearance of at least 10% at 6 hours after presentation has been associated with a lower mortality rate than nonclearance (19% vs 60%) in patients with sepsis or septic shock with elevated levels.15–17 Similar findings have been reported in a general intensive care unit population,18 as well as a surgical intensive care population.sup>19
Puskarich et al20 have also shown that lactate normalization to less than 2 mmol/L during early sepsis resuscitation is the strongest predictor of survival (odds ratio [OR] 5.2), followed by lactate clearance of 50% (OR 4.0) within the first 6 hours of presentation. Not only is lactate clearance associated with improved outcomes, but a faster rate of clearance after initial presentation is also beneficial.15,16,18
Lactate clearance over a longer period (> 6 hours) has not been studied in patients with septic shock. However, in the general intensive care unit population, therapy guided by lactate clearance for the first 8 hours after presentation has shown a reduction in mortality rate.18 There are no data available on outcomes of lactate-directed therapy beyond 8 hours, but lactate concentration and lactate clearance at 24 hours correlate with the 28-day mortality rate.21
Cryptic shock
Cryptic shock describes a state in a subgroup of patients who have elevated lactate levels and global tissue hypoxia despite being normotensive or even hypertensive. These patients have a higher mortality rate independent of blood pressure. Jansen et al18 found that patients with a lactate level higher than 4 mmol/L and preserved blood pressure had a mortality rate of 15%, while those without shock or hyperlactatemia had a mortality rate of 2.5%. In addition, patients with an elevated lactate level in the absence of hypotension have mortality rates similar to those in patients with high lactate levels and hypotension refractory to fluid boluses, suggesting the presence of tissue hypoxia even in these normotensive patients.6
HOW TO APPROACH AN ELEVATED LACTATE LEVEL
An elevated lactate level should prompt an evaluation for causes of decreased oxygen delivery, due either to a systemic low-flow state (as a result of decreased cardiac output) or severe anemia, or to regionally decreased perfusion, (eg, limb or mesenteric ischemia). If tissue hypoxia is ruled out after an exhaustive workup, consideration should be given to causes of hyperlactatemia without concomitant tissue hypoxia (type B acidosis).
Treatment differs depending on the underlying mechanism of the lactate elevation; nevertheless, treatment is mostly related to optimizing oxygen delivery by giving fluids, packed red blood cells, and vasopressors or inotropic agents, or both (Figure 2). The specific treatment differs based on the shock state, but there are similarities that can guide the clinician.
FLUID SUPPORT
Giving fluids, with a goal of improving cardiac output, remains a cornerstone of initial therapy for most shock states.22,23
How much fluid?
Fluids should be given until the patient is no longer preload-dependent, although there is much debate about which assessment strategy should be used to determine if cardiac output will improve with more fluid (ie, fluid-responsiveness).24 In many cases, fluid resuscitation alone may be enough to restore hemodynamic stability, improve tissue perfusion, and reduce elevated lactate concentrations.25
The decision to give more fluids should not be made lightly, though, as a more positive fluid balance early in the course of septic shock and over 4 days has been associated with a higher mortality rate.26 Additionally, pushing fluids in patients with cardiogenic shock due to impaired left ventricular systolic function may lead to or worsen pulmonary edema. Therefore, the indiscriminate use of fluids should be avoided.
Which fluids?
Despite years of research, controversy persists about whether crystalloids or colloids are better for resuscitation. Randomized trials in heterogeneous intensive care unit patients have not detected differences in 28-day mortality rates between those allocated to crystalloids or 4% albumin27 and those allocated to crystalloids or hydroxyethyl starch.28
Hydroxyethyl starch may not be best. In a study of patients with severe sepsis, those randomized to receive hydroxyethyl starch had a higher 90-day mortality rate than patients randomized to crystalloids (51% vs 43%, P = .03).29 A sequential prospective before-and-after study did not detect a difference in the time to normalization (< 2.2 mmol/L) of lactate (P = .68) or cessation of vasopressors (P = .11) in patients with severe sepsis who received fluid resuscitation with crystalloids, gelatin, or hydroxyethyl starch. More patients who received hydroxyethyl starch in these studies developed acute kidney injury than those receiving crystalloids.28–30
Taken together, these data strongly suggest hydroxyethyl starch should not be used for fluid resuscitation in the intensive care unit.
Normal saline or albumin? Although some data suggest that albumin may be preferable to 0.9% sodium chloride in patients with severe sepsis,31,32 these analyses should be viewed as hypothesis-generating. There do not seem to be differences between fluid types in terms of subsequent serum lactate concentrations or achievement of lactate clearance goals.28–30 Until further studies are completed, both albumin and crystalloids are reasonable for resuscitation.
Caironi et al33 performed an open-label study comparing albumin replacement (with a goal serum albumin concentration of 3 g/dL) plus a crystalloid solution vs a crystalloid solution alone in patients with severe sepsis or septic shock. They detected no difference between the albumin and crystalloid groups in mortality rates at 28 days (31.8% vs 32.0%, P = .94) or 90 days (41.1% vs 43.6%, P = .29). However, patients in the albumin group had a shorter time to cessation of vasoactive agents (median 3 vs 4 days, P = .007) and lower cardiovascular Sequential Organ Failure Assessment subscores (median 1.20 vs 1.42, P = .03), and more frequently achieved a mean arterial pressure of at least 65 mm Hg within 6 hours of randomization (86.0% vs 82.5%, P = .04).
Although serum lactate levels were lower in the albumin group at baseline (1.7 mmol/L vs 1.8 mmol/L, P = .05), inspection of the data appears to show a similar daily lactate clearance rate between groups over the first 7 study days (although these data were not analyzed by the authors). Achievement of a lactate level lower than 2 mmol/L on the first day of therapy was not significantly different between groups (73.4% vs 72.5%, P = .11).33
In a post hoc subgroup analysis, patients with septic shock at baseline randomized to albumin had a lower 90-day mortality rate than patients randomized to crystalloid solutions (RR 0.87, 95% CI 0.77–0.99). There was no difference in the 90-day mortality rate in patients without septic shock (RR 1.13, 95% CI 0.92–1.39, P = .03 for heterogeneity).33
These data suggest that albumin replacement may not improve outcomes in patients with severe sepsis, but may have advantages in terms of hemodynamic variables (and potentially mortality) in patients with septic shock. The role of albumin replacement in patients with septic shock warrants further study.
VASOPRESSORS
Vasopressors, inotropes, or both should be given to patients who have signs of hypoperfusion (including elevated lactate levels) despite preload optimization or ongoing fluid administration. The most appropriate drug depends on the goal: vasopressors are used to increase systemic vascular resistance, while inotropes are used to improve cardiac output and oxygen delivery.
Blood pressure target
The Surviving Sepsis Campaign guidelines recommend a mean arterial blood pressure target of at least 65 mm Hg during initial resuscitation and when vasopressors are applied for patients with septic shock.22 This recommendation is based on small studies that did not show differences in serum lactate levels or regional blood flow when the mean arterial pressure was elevated above 65 mm Hg with norepinephrine.34,35 However, the campaign guidelines note that the mean arterial pressure goal must be individualized in order to achieve optimal perfusion.
A large, open-label trial36 detected no difference in 28-day mortality rates in patients with septic shock between those allocated to a mean arterial pressure goal of 80 to 85 mm Hg or 65 to 70 mm Hg (36.6% vs 34.0%, P = .57). Although lactate levels did not differ between groups, the incidence of new-onset atrial fibrillation was higher in the higher-target group (6.7% vs 2.8%, P = .02). Fewer patients with chronic hypertension needed renal replacement therapy in the higher pressure group, further emphasizing the need to individualize the mean arterial pressure goal for patients in shock.36
Which vasopressor agent?
Dopamine and norepinephrine have traditionally been the preferred initial vasopressors for patients with shock. Until recently there were few data to guide selection between the two, but this is changing.
In a 2010 study of 1,679 patients with shock requiring vasopressors, there was no difference in the 28-day mortality rate between patients randomized to dopamine or norepinephrine (53% vs 49%, P = .10).37 Patients allocated to dopamine, though, had a higher incidence of arrhythmias (24% vs 12%, P < .001) and more frequently required open-label norepinephrine (26% vs 20%, P < .001). Although lactate levels and the time to achievement of a mean arterial pressure of 65 mm Hg were similar between groups, patients allocated to norepinephrine had more vasopressor-free days through day 28.
An a priori-planned subgroup analysis evaluated the influence of the type of shock on patient outcome. Patients with cardiogenic shock randomized to dopamine had a higher mortality rate than those randomized to norepinephrine (P = .03). However, the overall effect of treatment did not differ among the shock subgroups (interaction P = .87), suggesting that the reported differences in mortality according to subgroup may be spurious.
In a 2012 meta-analysis of patients with septic shock, dopamine use was associated with a higher mortality rate than norepinephrine use.38
In light of these data, norepinephrine should be preferred over dopamine as the initial vasopressor in most types of shock.
Epinephrine does not offer an outcome advantage over norepinephrine and may be associated with a higher incidence of adverse events.39–42 Indeed, in a study of patients with septic shock, lactate concentrations on the first day after randomization were significantly higher in patients allocated to epinephrine than in patients allocated to norepinephrine plus dobutamine.39 Similar effects on lactate concentrations with epinephrine were seen in patients with various types of shock40 and in those with cardiogenic shock.42
These differences in lactate concentrations may be directly attributable to epinephrine. Epinephrine can increase lactate concentrations through glycolysis and pyruvate dehydrogenase activation by stimulation of sodium-potassium ATPase activity via beta-2 adrenergic receptors in skeletal muscles,43 as well as decrease splanchnic perfusion.42,44,45 These effects may preclude using lactate clearance as a resuscitation goal in patients receiving epinephrine. Epinephrine is likely best reserved for patients with refractory shock,22 particularly those in whom cardiac output is known to be low.
Phenylephrine, essentially a pure vasoconstrictor, should be avoided in low cardiac output states and is best reserved for patients who develop a tachyarrhythmia on norepinephrine.22
Vasopressin, also a pure vasoconstrictor that should be avoided in low cardiac output states, has been best studied in patients with vasodilatory shock. Although controversy exists on the mortality benefits of vasopressin in vasodilatory shock, it is a relatively safe drug with consistent norepinephrine-sparing effects when added to existing norepinephrine therapy.46,47 In patients with less severe septic shock, including those with low lactate concentrations, adding vasopressin to norepinephrine instead of continuing norepinephrine alone may confer a mortality advantage.48
OTHER MEASURES TO OPTIMIZE OXYGEN DELIVERY
In circulatory shock from any cause, tissue oxygen demand exceeds oxygen delivery. Once arterial oxygenation and hemoglobin levels (by packed red blood cell transfusion) have been optimized, cardiac output is the critical determinant of oxygen delivery. Cardiac output may be augmented by ensuring adequate preload (by fluid resuscitation) or by giving inotropes or vasodilators.
The optimal cardiac output is difficult to define, and the exact marker for determining when cardiac output should be augmented is unclear. A strategy of increasing cardiac output to predefined “supranormal” levels was not associated with a lower mortality rate.49 Therefore, the decision to augment cardiac output must be individualized and will likely vary in the same patient over time.23
A reasonable approach to determining when augmentation of cardiac output is necessary was proposed in a study by Rivers et al.50 In that study, in patients randomized to early goal-directed therapy, inotropes were recommended when the central venous oxygenation saturation (Scvo2) was below 70% despite adequate fluid resuscitation (central venous pressure ≥ 8 mm Hg) and hematocrits were higher than 30%.
When an inotrope is indicated to improve cardiac output, dobutamine is usually the preferred agent. Dobutamine has a shorter half-life (allowing for easier titration) and causes less hypotension (assuming preload has been optimized) than phosphodiesterase type III inhibitors such as milrinone.
Mechanical support devices, such as intra-aortic balloon counterpulsation, and vasodilators can also be used to improve tissue perfusion in selected patients with low cardiac output syndromes.
USING LACTATE LEVELS TO GUIDE THERAPY
Lactate levels above 4.0 mmol/L
Lactate may be a useful marker for determining whether organ dysfunction is present and, hence, what course of therapy should be given, especially in sepsis. A serum lactate level higher than 4.0 mmol/L has been used as the trigger to start aggressive resuscitation in patients with sepsis.50,51
Traditionally, as delineated by Rivers et al50 in their landmark study of early goal-directed therapy, this entailed placing an arterial line and a central line for hemodynamic monitoring, with specific interventions directed at increasing the central venous pressure, mean arterial pressure, and central venous oxygen saturation.50 However, a recent study in a similar population of patients with sepsis with elevated lactate found no significant advantage of protocol-based resuscitation over care provided according to physician judgment, and no significant benefit in central venous catheterization and hemodynamic monitoring in all patients.51
Lactate clearance: 10% or above at 8 hours?
Regardless of the approach chosen, decreasing lactate levels can be interpreted as an adequate response to the interventions provided. As a matter of fact, several groups of investigators have also demonstrated the merits of lactate clearance alone as a prognostic indicator in patients requiring hemodynamic support.
McNelis et al52 retrospectively evaluated 95 postsurgical patients who required hemodynamic monitoring.52,53 The authors found that the slower the lactate clearance, the higher the mortality rate.
Given the prognostic implications of lactate clearance, investigators have evaluated whether lactate clearance could be used as a surrogate resuscitation goal for optimizing oxygen delivery. Using lactate clearance may have significant practical advantages over using central venous oxygen saturation, since it does not require a central venous catheter or continuous oximetric monitoring.
In a study comparing these two resuscitation end points, patients were randomized to a goal of either central venous oxygen saturation of 70% or more or lactate clearance of 10% or more within the first 6 hours after presentation as a marker of oxygen delivery.53 Mortality rates were similar with either strategy. Of note, only 10% of the patients actually required therapies to improve their oxygen delivery. Furthermore, there were no differences in the treatments given (including fluids, vasopressors, inotropes, packed red blood cells) throughout the treatment period.
These findings provide several insights. First, few patients admitted to the emergency department with severe sepsis and treated with an initial quantitative resuscitation protocol require additional therapy for augmenting oxygen delivery. Second, lactate clearance, in a setting where initial resuscitation with fluids and vasopressors restores adequate oxygen delivery for the majority of patients, is likely as good a target for resuscitation as central venous oxygen saturation.
This study, however, does not address the question of whether lactate clearance is useful as an additional marker of oxygen delivery (in conjunction with central venous oxygen saturation). Indeed, caution should be taken to target central venous oxygen saturation goals alone, as patients with septic shock presenting with venous hyperoxia (central venous oxygen saturation > 89%) have been shown to have a higher mortality rate than patients with normoxia (central venous oxygen saturation 71%–89%).54
This was further demonstrated by Arnold et al in a study of patients presenting to the emergency department with severe sepsis.15 In this study, significant discordance between central venous oxygen saturation and lactate clearance was seen, where 79% of patients with less than 10% lactate clearance had concomitant central venous oxygen saturation of 70% or greater.
Jansen et al18 evaluated the role of targeting lactate clearance in conjunction with central venous oxygen saturation monitoring. In this study, critically ill patients with elevated lactate and inadequate lactate clearance were randomized to usual care or to resuscitation to adequate lactate clearance (20% or more). The therapies to optimize oxygen delivery were given according to the central venous oxygen saturation. Overall, after adjustment for predefined risk factors, the in-hospital mortality rate was lower in the lactate clearance group. This may signify that patients with sepsis and central venous oxygen saturation of 70% or more may continue to have poor lactate clearance, warranting further treatment.
Taken together, serum lactate may be helpful for prognostication, determination of course of therapy, and quantification for tissue hypoperfusion for targeted therapies. Figure 2 presents our approach to an elevated lactate level. As performed in the study by Jansen et al,18 it seems reasonable to measure lactate levels every 2 hours for the first 8 hours of resuscitation in patients with type A lactic acidosis. These levels should be interpreted in the context of lactate clearance (at least 10%, but preferably 20%) and normalization, and should be treated with an approach similar to the one outlined in Figure 2.
TREATING TYPE B LACTIC ACIDOSIS (NORMAL PERFUSION AND OXYGENATION)
Treating type B lactic acidosis is quite different because the goal is not to correct mismatches in oxygen consumption and delivery. Since most cases are due to underlying conditions such as malignancy or medications, treatment should be centered around eliminating the cause (eg, treat the malignancy, discontinue the offending medication). The main reason for treatment is to alleviate the harmful effects of acidosis. For example, acidosis can result in a negative inotropic effect.
Sodium bicarbonate, dichloroacetate, carbicarb, and tromethamine have all been studied in the management of type B lactic acidosis, with little success.55,56
Renal replacement therapy has had some success in drug-induced lactic acidosis.57,58
l-carnitine has had promising results in treating patients with human immunodeficiency virus infection, since these patients are carnitine-deficient and carnitine plays an important role in mitochondrial function.59
Thiamine and biotin deficiencies can occur in patients receiving total parenteral nutrition without vitamins and in patients who drink alcohol heavily and can cause lactic acidosis. These nutrients should be supplemented accordingly.
Treatment of mitochondrial disorders includes antioxidants (coenzyme Q10, vitamin C, vitamin E) and amino acids (l-arginine).60
- Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc 2013; 88:1127–1140.
- Fuller BM, Dellinger RP. Lactate as a hemodynamic marker in the critically ill. Curr Opin Crit Care 2012; 18:267–272.
- Fall PJ, Szerlip HM. Lactic acidosis: from sour milk to septic shock. J Intensive Care Med 2005; 20:255–271.
- Kruse O, Grunnet N, Barfod C. Blood lactate as a predictor for in-hospital mortality in patients admitted acutely to hospital: a systematic review. Scand J Trauma Resusc Emerg Med 2011;19:74.
- Howell MD, Donnino M, Clardy P, Talmor D, Shapiro NI. Occult hypoperfusion and mortality in patients with suspected infection. Intensive Care Med 2007; 33:1892–1899.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Outcomes of patients undergoing early sepsis resuscitation for cryptic shock compared with overt shock. Resuscitation 2011; 82:1289–1293.
- Bakker J, Nijsten MW, Jansen TC. Clinical use of lactate monitoring in critically ill patients. Ann Intensive Care 2013; 3:12.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Shapiro NI, Howell MD, Talmor D, et al. Serum lactate as a predictor of mortality in emergency department patients with infection. Ann Emerg Med 2005; 45:524–528.
- Mikkelsen ME, Miltiades AN, Gaieski DF, et al. Serum lactate is associated with mortality in severe sepsis independent of organ failure and shock. Crit Care Med 2009; 37:1670–1677.
- Liu V, Morehouse JW, Soule J, Whippy A, Escobar GJ. Fluid volume, lactate values, and mortality in sepsis patients with intermediate lactate values. Ann Am Thorac Soc 2013; 10:466–473.
- Sterling SA, Puskarich MA, Shapiro NI, et al; Emergency Medicine Shock Research Network (EMShockNET). Characteristics and outcomes of patients with vasoplegic versus tissue dysoxic septic shock. Shock 2013; 40:11–14.
- Arnold RC, Shapiro NI, Jones AE, et al; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Jones AE. Lactate clearance for assessing response to resuscitation in severe sepsis. Acad Emerg Med 2013; 20:844–847.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Jansen TC, van Bommel J, Schoonderbeek FJ, et al; LACTATE study group. Early lactate-guided therapy in intensive care unit patients: a multicenter, open-label, randomized controlled trial. Am J Respir Crit Care Med 2010; 182:752–761.
- Husain FA, Martin MJ, Mullenix PS, Steele SR, Elliott DC. Serum lactate and base deficit as predictors of mortality and morbidity. Am J Surg 2003; 185:485–491.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Whole blood lactate kinetics in patients undergoing quantitative resuscitation for severe sepsis and septic shock. Chest 2013; 143:1548–1553.
- Marty P, Roquilly A, Vallee F, et al. Lactate clearance for death prediction in severe sepsis or septic shock patients during the first 24 hours in intensive care unit: an observational study. Ann Intensive Care 2013; 3:3.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Vincent JL, De Backer D. Circulatory shock. N Engl J Med 2013; 369:1726–1734.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Vincent JL, Dufaye P, Berré J, Leeman M, Degaute JP, Kahn RJ. Serial lactate determinations during circulatory shock. Crit Care Med 1983; 11:449–451.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R; SAFE Study Investigators. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Myburgh JA, Finfer S, Bellomo R, et al; CHEST Investigators; Australian and New Zealand Intensive Care Society Clinical Trials Group. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Perner A, Haase N, Guttormsen AB, et al; 6S Trial Group; Scandinavian Critical Care Trials Group. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Bayer O, Reinhart K, Kohl M, et al. Effects of fluid resuscitation with synthetic colloids or crystalloids alone on shock reversal, fluid balance, and patient outcomes in patients with severe sepsis: a prospective sequential analysis. Crit Care Med 2012; 40:2543–2551.
- Delaney AP, Dan A, McCaffrey J, Finfer S. The role of albumin as a resuscitation fluid for patients with sepsis: a systematic review and meta-analysis. Crit Care Med 2011; 39:386–391.
- SAFE Study Investigators; Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Caironi P, Tognoni G, Masson S, et al; ALBIOS Study Investigators. Albumin replacement in patients with severe sepsis or septic shock. N Engl J Med 2014; 370:1412–1421.
- Bourgoin A, Leone M, Delmas A, Garnier F, Albanèse J, Martin C. Increasing mean arterial pressure in patients with septic shock: effects on oxygen variables and renal function. Crit Care Med 2005; 33:780–786.
- LeDoux D, Astiz ME, Carpati CM, Rackow EC. Effects of perfusion pressure on tissue perfusion in septic shock. Crit Care Med 2000; 28:2729–2732.
- Asfar P, Meziani F, Hamel JF, et al; SEPSISPAM Investigators. High versus low blood-pressure target in patients with septic shock. N Engl J Med 2014; 370:1583–1593.
- De Backer D, Biston P, Devriendt J, et al; SOAP II Investigators. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- De Backer D, Aldecoa C, Njimi H, Vincent JL. Dopamine versus norepinephrine in the treatment of septic shock: a meta-analysis. Crit Care Med 2012; 40:725–730.
- Annane D, Vignon P, Renault A, et al: CATS Study Group. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Myburgh JA, Higgins A, Jovanovska A, Lipman J, Ramakrishnan N, Santamaria J; CAT Study investigators. A comparison of epinephrine and norepinephrine in critically ill patients. Intensive Care Med 2008; 34:2226–2234.
- Schmittinger CA, Torgersen C, Luckner G, Schröder DC, Lorenz I, Dünser MW. Adverse cardiac events during catecholamine vasopressor therapy: a prospective observational study. Intensive Care Med 2012; 38:950–958.
- Levy B, Perez P, Perny J, Thivilier C, Gerard A. Comparison of norepinephrine-dobutamine to epinephrine for hemodynamics, lactate metabolism, and organ function variables in cardiogenic shock. A prospective, randomized pilot study. Crit Care Med 2011; 39:450–455.
- Watt MJ, Howlett KF, Febbraio MA, Spriet LL, Hargreaves M. Adrenaline increases skeletal muscle glycogenolysis, pyruvate dehydrogenase activation and carbohydrate oxidation during moderate exercise in humans. J Physiol 2001; 534:269–278.
- De Backer D, Creteur J, Silva E, Vincent JL. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? Crit Care Med 2003; 31:1659–1667.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Polito A, Parisini E, Ricci Z, Picardo S, Annane D. Vasopressin for treatment of vasodilatory shock: an ESICM systematic review and meta-analysis. Intensive Care Med 2012; 38:9–19.
- Serpa Neto A, Nassar APJ, Cardoso SO, et al. Vasopressin and terlipressin in adult vasodilatory shock: a systematic review and meta-analysis of nine randomized controlled trials. Crit Care 2012; 16:R154.
- Russell JA, Walley KR, Singer J, et al; VASST Investigators. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Gattinoni L, Brazzi L, Pelosi P, et al; for the SvO2 Collaborative Group. A trial of goal-oriented hemodynamic therapy in critically ill patients. N Engl J Med 1995; 333:1025–1032.
- Rivers E, Nguyen B, Havstad S, et al; Early Goal-Directed Therapy Collaborative Group. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- ProCESS Investigators; Yealy DM, Kellum JA, Huang DT, et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med 2014; 370:1683–1693.
- McNelis J, Marini CP, Jurkiewicz A, et al. Prolonged lactate clearance is associated with increased mortality in the surgical intensive care unit. Am J Surg 2001; 182:481–485.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Pope JV, Jones AE, Gaieski DF, Arnold RC, Trzeciak S, Shapiro NI; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of central venous oxygen saturation (ScvO2) as a predictor of mortality in patients with sepsis. Ann Emerg Med 2010; 55:40–46.e1
- Kraut JA, Kurtz I. Use of base in the treatment of severe acidemic states. Am J Kidney Dis 2001; 38:703–727.
- Levraut J, Grimaud D. Treatment of metabolic acidosis. Curr Opin Crit Care 2003; 9:260–265.
- Orija AA, Jenks CL. Nucleoside analog reverse transcriptase inhibitor induced lactic acidosis treated with continuous renal replacement in the medical intensive care unit. Crit Care & Shock 2012; 15:9–11.
- Friesecke S, Abel P, Kraft M, Gerner A, Runge S. Combined renal replacement therapy for severe metformin-induced lactic acidosis. Nephrol Dial Transplant 2006; 21:2038–2039.
- Claessens YE, Cariou A, Monchi M, et al. Detecting life-threatening lactic acidosis related to nucleoside-analog treatment of human immunodeficiency virus-infected patients, and treatment with l-carnitine. Crit Care Med 2003; 31:1042–1047.
- Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R; Medicine Society TM. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol 2009; 11:414–430.
- Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc 2013; 88:1127–1140.
- Fuller BM, Dellinger RP. Lactate as a hemodynamic marker in the critically ill. Curr Opin Crit Care 2012; 18:267–272.
- Fall PJ, Szerlip HM. Lactic acidosis: from sour milk to septic shock. J Intensive Care Med 2005; 20:255–271.
- Kruse O, Grunnet N, Barfod C. Blood lactate as a predictor for in-hospital mortality in patients admitted acutely to hospital: a systematic review. Scand J Trauma Resusc Emerg Med 2011;19:74.
- Howell MD, Donnino M, Clardy P, Talmor D, Shapiro NI. Occult hypoperfusion and mortality in patients with suspected infection. Intensive Care Med 2007; 33:1892–1899.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Outcomes of patients undergoing early sepsis resuscitation for cryptic shock compared with overt shock. Resuscitation 2011; 82:1289–1293.
- Bakker J, Nijsten MW, Jansen TC. Clinical use of lactate monitoring in critically ill patients. Ann Intensive Care 2013; 3:12.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Shapiro NI, Howell MD, Talmor D, et al. Serum lactate as a predictor of mortality in emergency department patients with infection. Ann Emerg Med 2005; 45:524–528.
- Mikkelsen ME, Miltiades AN, Gaieski DF, et al. Serum lactate is associated with mortality in severe sepsis independent of organ failure and shock. Crit Care Med 2009; 37:1670–1677.
- Liu V, Morehouse JW, Soule J, Whippy A, Escobar GJ. Fluid volume, lactate values, and mortality in sepsis patients with intermediate lactate values. Ann Am Thorac Soc 2013; 10:466–473.
- Sterling SA, Puskarich MA, Shapiro NI, et al; Emergency Medicine Shock Research Network (EMShockNET). Characteristics and outcomes of patients with vasoplegic versus tissue dysoxic septic shock. Shock 2013; 40:11–14.
- Arnold RC, Shapiro NI, Jones AE, et al; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Jones AE. Lactate clearance for assessing response to resuscitation in severe sepsis. Acad Emerg Med 2013; 20:844–847.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Jansen TC, van Bommel J, Schoonderbeek FJ, et al; LACTATE study group. Early lactate-guided therapy in intensive care unit patients: a multicenter, open-label, randomized controlled trial. Am J Respir Crit Care Med 2010; 182:752–761.
- Husain FA, Martin MJ, Mullenix PS, Steele SR, Elliott DC. Serum lactate and base deficit as predictors of mortality and morbidity. Am J Surg 2003; 185:485–491.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Whole blood lactate kinetics in patients undergoing quantitative resuscitation for severe sepsis and septic shock. Chest 2013; 143:1548–1553.
- Marty P, Roquilly A, Vallee F, et al. Lactate clearance for death prediction in severe sepsis or septic shock patients during the first 24 hours in intensive care unit: an observational study. Ann Intensive Care 2013; 3:3.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Vincent JL, De Backer D. Circulatory shock. N Engl J Med 2013; 369:1726–1734.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Vincent JL, Dufaye P, Berré J, Leeman M, Degaute JP, Kahn RJ. Serial lactate determinations during circulatory shock. Crit Care Med 1983; 11:449–451.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R; SAFE Study Investigators. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Myburgh JA, Finfer S, Bellomo R, et al; CHEST Investigators; Australian and New Zealand Intensive Care Society Clinical Trials Group. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Perner A, Haase N, Guttormsen AB, et al; 6S Trial Group; Scandinavian Critical Care Trials Group. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Bayer O, Reinhart K, Kohl M, et al. Effects of fluid resuscitation with synthetic colloids or crystalloids alone on shock reversal, fluid balance, and patient outcomes in patients with severe sepsis: a prospective sequential analysis. Crit Care Med 2012; 40:2543–2551.
- Delaney AP, Dan A, McCaffrey J, Finfer S. The role of albumin as a resuscitation fluid for patients with sepsis: a systematic review and meta-analysis. Crit Care Med 2011; 39:386–391.
- SAFE Study Investigators; Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Caironi P, Tognoni G, Masson S, et al; ALBIOS Study Investigators. Albumin replacement in patients with severe sepsis or septic shock. N Engl J Med 2014; 370:1412–1421.
- Bourgoin A, Leone M, Delmas A, Garnier F, Albanèse J, Martin C. Increasing mean arterial pressure in patients with septic shock: effects on oxygen variables and renal function. Crit Care Med 2005; 33:780–786.
- LeDoux D, Astiz ME, Carpati CM, Rackow EC. Effects of perfusion pressure on tissue perfusion in septic shock. Crit Care Med 2000; 28:2729–2732.
- Asfar P, Meziani F, Hamel JF, et al; SEPSISPAM Investigators. High versus low blood-pressure target in patients with septic shock. N Engl J Med 2014; 370:1583–1593.
- De Backer D, Biston P, Devriendt J, et al; SOAP II Investigators. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- De Backer D, Aldecoa C, Njimi H, Vincent JL. Dopamine versus norepinephrine in the treatment of septic shock: a meta-analysis. Crit Care Med 2012; 40:725–730.
- Annane D, Vignon P, Renault A, et al: CATS Study Group. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Myburgh JA, Higgins A, Jovanovska A, Lipman J, Ramakrishnan N, Santamaria J; CAT Study investigators. A comparison of epinephrine and norepinephrine in critically ill patients. Intensive Care Med 2008; 34:2226–2234.
- Schmittinger CA, Torgersen C, Luckner G, Schröder DC, Lorenz I, Dünser MW. Adverse cardiac events during catecholamine vasopressor therapy: a prospective observational study. Intensive Care Med 2012; 38:950–958.
- Levy B, Perez P, Perny J, Thivilier C, Gerard A. Comparison of norepinephrine-dobutamine to epinephrine for hemodynamics, lactate metabolism, and organ function variables in cardiogenic shock. A prospective, randomized pilot study. Crit Care Med 2011; 39:450–455.
- Watt MJ, Howlett KF, Febbraio MA, Spriet LL, Hargreaves M. Adrenaline increases skeletal muscle glycogenolysis, pyruvate dehydrogenase activation and carbohydrate oxidation during moderate exercise in humans. J Physiol 2001; 534:269–278.
- De Backer D, Creteur J, Silva E, Vincent JL. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? Crit Care Med 2003; 31:1659–1667.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Polito A, Parisini E, Ricci Z, Picardo S, Annane D. Vasopressin for treatment of vasodilatory shock: an ESICM systematic review and meta-analysis. Intensive Care Med 2012; 38:9–19.
- Serpa Neto A, Nassar APJ, Cardoso SO, et al. Vasopressin and terlipressin in adult vasodilatory shock: a systematic review and meta-analysis of nine randomized controlled trials. Crit Care 2012; 16:R154.
- Russell JA, Walley KR, Singer J, et al; VASST Investigators. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Gattinoni L, Brazzi L, Pelosi P, et al; for the SvO2 Collaborative Group. A trial of goal-oriented hemodynamic therapy in critically ill patients. N Engl J Med 1995; 333:1025–1032.
- Rivers E, Nguyen B, Havstad S, et al; Early Goal-Directed Therapy Collaborative Group. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- ProCESS Investigators; Yealy DM, Kellum JA, Huang DT, et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med 2014; 370:1683–1693.
- McNelis J, Marini CP, Jurkiewicz A, et al. Prolonged lactate clearance is associated with increased mortality in the surgical intensive care unit. Am J Surg 2001; 182:481–485.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Pope JV, Jones AE, Gaieski DF, Arnold RC, Trzeciak S, Shapiro NI; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of central venous oxygen saturation (ScvO2) as a predictor of mortality in patients with sepsis. Ann Emerg Med 2010; 55:40–46.e1
- Kraut JA, Kurtz I. Use of base in the treatment of severe acidemic states. Am J Kidney Dis 2001; 38:703–727.
- Levraut J, Grimaud D. Treatment of metabolic acidosis. Curr Opin Crit Care 2003; 9:260–265.
- Orija AA, Jenks CL. Nucleoside analog reverse transcriptase inhibitor induced lactic acidosis treated with continuous renal replacement in the medical intensive care unit. Crit Care & Shock 2012; 15:9–11.
- Friesecke S, Abel P, Kraft M, Gerner A, Runge S. Combined renal replacement therapy for severe metformin-induced lactic acidosis. Nephrol Dial Transplant 2006; 21:2038–2039.
- Claessens YE, Cariou A, Monchi M, et al. Detecting life-threatening lactic acidosis related to nucleoside-analog treatment of human immunodeficiency virus-infected patients, and treatment with l-carnitine. Crit Care Med 2003; 31:1042–1047.
- Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R; Medicine Society TM. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol 2009; 11:414–430.
KEY POINTS
- Serum lactate levels can become elevated by a variety of underlying processes, categorized as increased production in conditions of hypoperfusion and hypoxia (type A lactic acidosis), or as increased production or decreased clearance not due to hypoperfusion and hypoxia (type B).
- The higher the lactate level and the slower the rate of normalization (lactate clearance), the higher the risk of death.
- Treatments differ depending on the underlying mechanism of the lactate elevation. Thus, identifying the reason for hyperlactatemia and differentiating between type A and B lactic acidosis are of the utmost importance.
- Treatment of type A lactic acidosis aims to improve perfusion and match oxygen consumption with oxygen delivery by giving fluids, packed red blood cells, and vasopressors or inotropic agents, or both.
- Treatment of type B involves more specific management, such as discontinuing offending medications or supplementing key cofactors for anaerobic metabolism.
Middle East respiratory syndrome: SARS redux?
Middle East respiratory syndrome (MERS) is a potentially lethal illness caused by the Middle East respiratory syndrome coronavirus (MERS-CoV). The virus was first reported in 2012, when it was isolated from the sputum of a previously healthy man in Saudi Arabia who presented with acute pneumonia and subsequent renal failure with a fatal outcome.1 Retrospective studies subsequently identified an earlier outbreak that year involving 13 patients in Jordan, and since then cases have been reported in 25 countries across the Arabian Peninsula and in Asia, Europe, Africa, and the United States, with over 1,000 confirmed cases and 450 related deaths.2,3
At the time of this writing, two cases of MERS have been reported in the United States, both in May 2014. Both reported cases involved patients who had traveled from Saudi Arabia, and which did not result in secondary cases.4 Beginning in May 2015, the Republic of Korea had experienced the largest known outbreak of MERS outside the Arabian Peninsula, with over 100 cases.5
THE VIRUS
MERS-CoV is classified as a coronavirus, which is a family of single-stranded RNA viruses. In 2003, a previously unknown coronavirus (SARS-CoV) caused a global outbreak of pneumonia that resulted in approximately 800 deaths.6 The MERS-CoV virus attaches to dipeptidyl peptidase 4 to enter cells, and this receptor is believed to be critical for pathogenesis, as infection does not occur in its absence.7
The source and mode of transmission to humans is not completely defined. Early reports suggested that MERS-CoV originated in bats, as RNA sequences related to MERS-CoV have been found in several bat species, but the virus itself has not been isolated from bats.8 Camels have been found to have a high rate of anti-MERS-CoV antibodies and to have the virus in nose swabs, and evidence for camel-to-human transmission has been presented.9–11 However, the precise role of camels and other animals as reservoirs or vectors of infection is still under investigation.
The incubation period from exposure to the development of clinical disease is estimated at 5 to 14 days.
For MERS-CoV, the basic reproduction ratio (R0), which measures the average number of secondary cases from each infected person, is estimated12 to be less than 0.7. In diseases in which the R0 is less than 1.0, infections occur in isolated clusters as limited chains of transmission, and thus the sustained transmission of MERS-CoV resulting in a large epidemic is thought to be unlikely. As a comparison, the median R0 value for seasonal influenza is estimated13 at 1.28. “Superspreading” may result in limited outbreaks of secondary cases; however, the continued epidemic spread of infection is thought to be unlikely.14 Nevertheless, viral adaptation with increased transmissibility remains a concern and a potential threat.
CLINICAL PRESENTATION
MERS most commonly presents as a respiratory illness, although asymptomatic infection occurs. The percentage of patients who experience asymptomatic infection is unknown. A recent survey of 255 patients with laboratory-confirmed MERS-CoV found that 64 (25.1%) were reported as asymptomatic at time of specimen collection. However, when 33 (52%) of those patients were interviewed, 26 (79%) reported at least one symptom that was consistent with a viral respiratory illness.15
For symptomatic patients, the initial complaints are nonspecific, beginning with fever, cough, sore throat, chills, and myalgia. Patients experiencing severe infection progress to dyspnea and pneumonia, with requirements for ventilatory support, vasopressors, and renal replacement therapy.16 Gastrointestinal symptoms such as vomiting and diarrhea have been reported in about one-third of patients.17
In a study of 47 patients with MERS-CoV, most of whom had underlying medical illnesses, 42 (89%) required intensive care and 34 (72%) required mechanical ventilation.17 The case-fatality rate in this study was 60%, but other studies have reported rates closer to 30%.15
Laboratory findings in patients with MERS-CoV infection usually include leukopenia and thrombocytopenia. Severely ill patients may have evidence of acute kidney injury.
Radiographic findings of MERS are those of viral pneumonitis and acute respiratory distress syndrome. Computed tomographic findings include ground-glass opacities, with peripheral lower-lobe preference.18
DIAGNOSIS
As MERS is a respiratory illness, sampling of respiratory secretions provides the highest yield for diagnosis. A study of 112 patients with MERS-CoV reported that polymerase chain reaction (PCR) testing of tracheal aspirates and bronchoalveolar lavage samples yielded significantly higher MERS-CoV loads than nasopharyngeal swab samples and sputum samples.19 However, upper respiratory tract testing is less invasive, and a positive nasopharyngeal swab result may obviate the need for further testing.
The US Centers for Disease Control and Prevention (CDC) recommends collecting multiple specimens from different sites at different times after the onset of symptoms in order to increase the diagnostic yield. Specifically, it recommends testing a lower respiratory specimen (eg, sputum, bronchoalveolar lavage fluid, tracheal aspirate), a nasopharyngeal and oropharyngeal swab, and serum, using the CDC MERS-CoV rRT-PCR assay. In addition, for patients whose symptoms began more than 14 days earlier, the CDC also recommends testing a serum specimen with the CDC MERS-CoV serologic assay. As these guidelines are updated frequently, clinicians are advised to check the CDC website for the most up-to-date information (www.cdc.gov/coronavirus/mers/guidelines-clinical-specimens.html).20 The identification of MERS-CoV by virus isolation in cell culture is not recommended and, if pursued, must be performed in a biosafety level 3 facility. (Level 3 is the second-highest level of biosafety. The highest, level 4, is reserved for extremely dangerous agents such as Ebola virus).20
Given the nonspecific clinical presentation of MERS-CoV, clinicians may consider testing for other respiratory pathogens. A recent review of 54 travelers to California from MERS-CoV-affected areas found that while none tested positive for MERS-CoV, 32 (62%) of 52 travelers had other respiratory viruses.21 When testing for alternative pathogens, clinicians should order molecular or antigen-based detection methods.
TREATMENT
Unfortunately, treatment for MERS is primarily supportive.
Ribavirin and interferon alfa-2b demonstrated activity in an animal model, but the regimen was ineffective when given a median of 19 (range 10–22) days after admission in 5 critically ill patients who subsequently died.22 A retrospective analysis comparing 20 patients with severe MERS-CoV who received ribavirin and interferon alfa-2a with 24 patients who did not reported that while survival was improved at 14 days, the mortality rates were similar at 28 days.23
A systematic review of treatments used for severe acute respiratory syndrome (SARS) reported that most studies investigating steroid use were inconclusive and some showed possible harm, suggesting that systemic steroids should be avoided in coronavirus infections.24
PREVENTION
Healthcare-associated outbreaks of MERS are well described, and thus recognition of potential cases and prompt institution of appropriate infection control measures are critical.15,25

Healthcare providers should ask patients about recent travel history and ascertain if they meet the CDC criteria for a “patient under investigation” (PUI), ie, if they have both clinical features and an epidemiologic risk of MERS (Table 1). However, these recommendations for identification will assuredly change as the outbreak matures, and healthcare providers should refer to the CDC website for the most up-to-date information.
Once a PUI is identified, standard, contact, and airborne precautions are advised. These measures include performing hand hygiene and donning personal protective equipment, including gloves, gowns, eye protection, and respiratory protection (ie, a respirator) that is at least as protective as a fit-tested National Institute for Occupational Safety and Health-certified N95 filtering face-piece respirator. In addition, a patient with possible MERS should be placed in an airborne infection isolation room.
Traveler’s advice
The CDC does not currently recommend that Americans change their travel plans because of MERS. Clinicians performing pretravel evaluations should advise patients of current information on MERS. Patients at risk for MERS who develop a respiratory illness within 14 days of return should seek medical attention and inform healthcare providers of their travel history.
SUMMARY
Recent experience with SARS, Ebola virus disease, and now MERS-CoV highlights the impact of global air travel as a vector for the rapid worldwide dissemination of communicable diseases. Healthcare providers should elicit a travel history in all patients presenting with a febrile illness, as an infection acquired in one continent may not become manifest until the patient presents in another.
The scope of the current MERS-CoV outbreak is still evolving, with concerns that viral evolution could result in a SARS-like outbreak, as experienced almost a decade ago.
Healthcare providers are advised to screen patients at risk for MERS-CoV for respiratory symptoms, and to institute appropriate infection control measures. Through recognition and isolation, healthcare providers are at the front line in limiting the spread of this potentially lethal virus.
- Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012; 367:1814–1820.
- Al-Abdallat MM, Payne DC, Alqasrawi S, et al. Hospital-associated outbreak of Middle East respiratory syndrome coronavirus: a serologic, epidemiologic, and clinical description. Clin Infect Dis 2014; 59:1225–1233.
- World Health Organization. Frequently asked questions on Middle East respiratory syndrome coronavirus (MERS-CoV). www.who.int/csr/disease/coronavirus_infections/faq/en/. Accessed July 29, 2015.
- Bialek SR, Allen D, Alvarado-Ramy F, et al; Centers for Disease Control and Prevention (CDC). First confirmed cases of Middle East respiratory syndrome coronavirus (MERS-CoV) infection in the United States, updated information on the epidemiology of MERS-CoV infection, and guidance for the public, clinicians, and public health authorities—May 2014. MMWR Morb Mortal Wkly Rep 2014; 63:431–436.
- World Health Organization. Middle East respiratory syndrome coronavirus (MERS-CoV) – Republic of Korea. www.who.int/csr/don/12-june-2015-mers-korea/en/. Accessed July 29, 2015.
- Peiris JSM, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nat Med 2004; 10:S88–S97.
- van Doremalen N, Miazqowicz KL, Milne-Price S, et al. Host species restriction of Middle East respiratory syndrome coronavirus through its receptor, dipeptidyl peptidase 4. J Virol 2014; 88:9220–9232.
- Zumla A, Hui DS, Perlman S. Middle East respiratory syndrome. Lancet 2015; S0140-6736(15)60454-604548 (Epub ahead of print).
- Meyer B, Muller MA, Corman WM, et al. Antibodies against MERS coronavirus in dromedary camels, United Arab Emirates, 2003 and 2013. Emerg Infect Dis 2014; 20:552–559.
- Haagmans BL, Al Dhahiry SH, Reusken CB, et al. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect Dis 2014; 14:140–145.
- Azhar EI, El-Kafrawy SA, Farraj SA, et al. Evidence for camel-to-human transmission of MERS coronavirus. N Engl J Med 2014; 370:2499–2505.
- Chowell G, Blumberg S, Simonsen L, Miller MA, Viboud C. Synthesizing data and models for the spread of MERS-CoV, 2013: key role of index cases and hospital transmission. Epidemics 2014; 9:40–51.
- Biggerstaff M, Chauchemez S, Reed C, Gambhir M, Finelli L. Estimates of the reproduction number for seasonal, pandemic, and zoonotic influenza: a systematic review of the literature. BMC Infect Dis 2014: 14:480.
- Kucharski AJ, Althaus CL. The role of superspreading in Middle East respiratory syndrome coronavirus (MERS-CoV) transmission. Euro Surveill 2015; 20.
- Oboho I, Tomczyk S, Al-Asmari A, et al. 2014 MERS-CoV outbreak in Jeddah—a link to health care facilities. N Engl J Med 2015; 372:846–854.
- Arabi YM, Arifi AA, Balkhy HH, et al. Clinical course and outcomes of critically ill patients with Middle East respiratory syndrome coronavirus infection. Ann Intern Med 2014; 160:389–397.
- Assiri A, Al-Tawfig JA, Al-Rabeeah AA, et al. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: a descriptive study. Lancet Infect Dis 2013; 13:752–761.
- Das KM, Lee EY, Enani MA, et al. CT correlation with outcomes in 15 patients with acute Middle East respiratory syndrome coronavirus. AJR Am J Roentgenol 2015; 204:736–742.
- Memish ZA, Al-Tawfiq JA, Makhdoom HQ, et al. Respiratory tract samples, viral load, and genome fraction yield in patients with Middle East respiratory syndrome. J Infect Dis 2014; 210:1590–1594.
- Centers for Disease Control and Prevention. Middle East respiratory syndrome (MERS). Interim guidelines for collecting, handling, and testing clinical specimens from patients under investigation (PUIs) for Middle East respiratory syndrome coronavirus (MERS-CoV)—version 2.1. www.cdc.gov/coronavirus/mers/guidelines-clinical-specimens.html. Accessed July 29, 2015.
- Shakhkarami M, Yen C, Glaser CA, Xia D, Watt J, Wadford DA. Laboratory testing for Middle East respiratory syndrome coronavirus, California, USA, 2013–2014. Emerg Infect Dis 2015; 21: E-pub ahead of print. wwwnc.cdc.gov/eid/article/21/9/15-0476_article. Accessed July 29, 2015.
- Al-Tawfiq JA, Momattin H, Dib J, Memish ZA. Ribavirin and interferon therapy in patients infected with the Middle East respiratory syndrome coronavirus: an observational study. Int J Infect Dis 2014; 20:42–46.
- Omrani AS, Saad MM, Baig K, et al. Ribavirin and interferon alfa-2a for severe Middle East respiratory syndrome coronavirus infection: a retrospective cohort study. Lancet Infect Dis 2014; 14:1090–1095.
- Stockman LJ, Bellamy R, Garner, P. SARS: systematic review of treatment effects. PLoS Med 2006; 3:e343.
- Assiri A, McGeer A, Perl TM, et al; KSA MERS-CoV Investigation Team. Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med 2013; 369:407–416.
Middle East respiratory syndrome (MERS) is a potentially lethal illness caused by the Middle East respiratory syndrome coronavirus (MERS-CoV). The virus was first reported in 2012, when it was isolated from the sputum of a previously healthy man in Saudi Arabia who presented with acute pneumonia and subsequent renal failure with a fatal outcome.1 Retrospective studies subsequently identified an earlier outbreak that year involving 13 patients in Jordan, and since then cases have been reported in 25 countries across the Arabian Peninsula and in Asia, Europe, Africa, and the United States, with over 1,000 confirmed cases and 450 related deaths.2,3
At the time of this writing, two cases of MERS have been reported in the United States, both in May 2014. Both reported cases involved patients who had traveled from Saudi Arabia, and which did not result in secondary cases.4 Beginning in May 2015, the Republic of Korea had experienced the largest known outbreak of MERS outside the Arabian Peninsula, with over 100 cases.5
THE VIRUS
MERS-CoV is classified as a coronavirus, which is a family of single-stranded RNA viruses. In 2003, a previously unknown coronavirus (SARS-CoV) caused a global outbreak of pneumonia that resulted in approximately 800 deaths.6 The MERS-CoV virus attaches to dipeptidyl peptidase 4 to enter cells, and this receptor is believed to be critical for pathogenesis, as infection does not occur in its absence.7
The source and mode of transmission to humans is not completely defined. Early reports suggested that MERS-CoV originated in bats, as RNA sequences related to MERS-CoV have been found in several bat species, but the virus itself has not been isolated from bats.8 Camels have been found to have a high rate of anti-MERS-CoV antibodies and to have the virus in nose swabs, and evidence for camel-to-human transmission has been presented.9–11 However, the precise role of camels and other animals as reservoirs or vectors of infection is still under investigation.
The incubation period from exposure to the development of clinical disease is estimated at 5 to 14 days.
For MERS-CoV, the basic reproduction ratio (R0), which measures the average number of secondary cases from each infected person, is estimated12 to be less than 0.7. In diseases in which the R0 is less than 1.0, infections occur in isolated clusters as limited chains of transmission, and thus the sustained transmission of MERS-CoV resulting in a large epidemic is thought to be unlikely. As a comparison, the median R0 value for seasonal influenza is estimated13 at 1.28. “Superspreading” may result in limited outbreaks of secondary cases; however, the continued epidemic spread of infection is thought to be unlikely.14 Nevertheless, viral adaptation with increased transmissibility remains a concern and a potential threat.
CLINICAL PRESENTATION
MERS most commonly presents as a respiratory illness, although asymptomatic infection occurs. The percentage of patients who experience asymptomatic infection is unknown. A recent survey of 255 patients with laboratory-confirmed MERS-CoV found that 64 (25.1%) were reported as asymptomatic at time of specimen collection. However, when 33 (52%) of those patients were interviewed, 26 (79%) reported at least one symptom that was consistent with a viral respiratory illness.15
For symptomatic patients, the initial complaints are nonspecific, beginning with fever, cough, sore throat, chills, and myalgia. Patients experiencing severe infection progress to dyspnea and pneumonia, with requirements for ventilatory support, vasopressors, and renal replacement therapy.16 Gastrointestinal symptoms such as vomiting and diarrhea have been reported in about one-third of patients.17
In a study of 47 patients with MERS-CoV, most of whom had underlying medical illnesses, 42 (89%) required intensive care and 34 (72%) required mechanical ventilation.17 The case-fatality rate in this study was 60%, but other studies have reported rates closer to 30%.15
Laboratory findings in patients with MERS-CoV infection usually include leukopenia and thrombocytopenia. Severely ill patients may have evidence of acute kidney injury.
Radiographic findings of MERS are those of viral pneumonitis and acute respiratory distress syndrome. Computed tomographic findings include ground-glass opacities, with peripheral lower-lobe preference.18
DIAGNOSIS
As MERS is a respiratory illness, sampling of respiratory secretions provides the highest yield for diagnosis. A study of 112 patients with MERS-CoV reported that polymerase chain reaction (PCR) testing of tracheal aspirates and bronchoalveolar lavage samples yielded significantly higher MERS-CoV loads than nasopharyngeal swab samples and sputum samples.19 However, upper respiratory tract testing is less invasive, and a positive nasopharyngeal swab result may obviate the need for further testing.
The US Centers for Disease Control and Prevention (CDC) recommends collecting multiple specimens from different sites at different times after the onset of symptoms in order to increase the diagnostic yield. Specifically, it recommends testing a lower respiratory specimen (eg, sputum, bronchoalveolar lavage fluid, tracheal aspirate), a nasopharyngeal and oropharyngeal swab, and serum, using the CDC MERS-CoV rRT-PCR assay. In addition, for patients whose symptoms began more than 14 days earlier, the CDC also recommends testing a serum specimen with the CDC MERS-CoV serologic assay. As these guidelines are updated frequently, clinicians are advised to check the CDC website for the most up-to-date information (www.cdc.gov/coronavirus/mers/guidelines-clinical-specimens.html).20 The identification of MERS-CoV by virus isolation in cell culture is not recommended and, if pursued, must be performed in a biosafety level 3 facility. (Level 3 is the second-highest level of biosafety. The highest, level 4, is reserved for extremely dangerous agents such as Ebola virus).20
Given the nonspecific clinical presentation of MERS-CoV, clinicians may consider testing for other respiratory pathogens. A recent review of 54 travelers to California from MERS-CoV-affected areas found that while none tested positive for MERS-CoV, 32 (62%) of 52 travelers had other respiratory viruses.21 When testing for alternative pathogens, clinicians should order molecular or antigen-based detection methods.
TREATMENT
Unfortunately, treatment for MERS is primarily supportive.
Ribavirin and interferon alfa-2b demonstrated activity in an animal model, but the regimen was ineffective when given a median of 19 (range 10–22) days after admission in 5 critically ill patients who subsequently died.22 A retrospective analysis comparing 20 patients with severe MERS-CoV who received ribavirin and interferon alfa-2a with 24 patients who did not reported that while survival was improved at 14 days, the mortality rates were similar at 28 days.23
A systematic review of treatments used for severe acute respiratory syndrome (SARS) reported that most studies investigating steroid use were inconclusive and some showed possible harm, suggesting that systemic steroids should be avoided in coronavirus infections.24
PREVENTION
Healthcare-associated outbreaks of MERS are well described, and thus recognition of potential cases and prompt institution of appropriate infection control measures are critical.15,25
Healthcare providers should ask patients about recent travel history and ascertain if they meet the CDC criteria for a “patient under investigation” (PUI), ie, if they have both clinical features and an epidemiologic risk of MERS (Table 1). However, these recommendations for identification will assuredly change as the outbreak matures, and healthcare providers should refer to the CDC website for the most up-to-date information.
Once a PUI is identified, standard, contact, and airborne precautions are advised. These measures include performing hand hygiene and donning personal protective equipment, including gloves, gowns, eye protection, and respiratory protection (ie, a respirator) that is at least as protective as a fit-tested National Institute for Occupational Safety and Health-certified N95 filtering face-piece respirator. In addition, a patient with possible MERS should be placed in an airborne infection isolation room.
Traveler’s advice
The CDC does not currently recommend that Americans change their travel plans because of MERS. Clinicians performing pretravel evaluations should advise patients of current information on MERS. Patients at risk for MERS who develop a respiratory illness within 14 days of return should seek medical attention and inform healthcare providers of their travel history.
SUMMARY
Recent experience with SARS, Ebola virus disease, and now MERS-CoV highlights the impact of global air travel as a vector for the rapid worldwide dissemination of communicable diseases. Healthcare providers should elicit a travel history in all patients presenting with a febrile illness, as an infection acquired in one continent may not become manifest until the patient presents in another.
The scope of the current MERS-CoV outbreak is still evolving, with concerns that viral evolution could result in a SARS-like outbreak, as experienced almost a decade ago.
Healthcare providers are advised to screen patients at risk for MERS-CoV for respiratory symptoms, and to institute appropriate infection control measures. Through recognition and isolation, healthcare providers are at the front line in limiting the spread of this potentially lethal virus.
Middle East respiratory syndrome (MERS) is a potentially lethal illness caused by the Middle East respiratory syndrome coronavirus (MERS-CoV). The virus was first reported in 2012, when it was isolated from the sputum of a previously healthy man in Saudi Arabia who presented with acute pneumonia and subsequent renal failure with a fatal outcome.1 Retrospective studies subsequently identified an earlier outbreak that year involving 13 patients in Jordan, and since then cases have been reported in 25 countries across the Arabian Peninsula and in Asia, Europe, Africa, and the United States, with over 1,000 confirmed cases and 450 related deaths.2,3
At the time of this writing, two cases of MERS have been reported in the United States, both in May 2014. Both reported cases involved patients who had traveled from Saudi Arabia, and which did not result in secondary cases.4 Beginning in May 2015, the Republic of Korea had experienced the largest known outbreak of MERS outside the Arabian Peninsula, with over 100 cases.5
THE VIRUS
MERS-CoV is classified as a coronavirus, which is a family of single-stranded RNA viruses. In 2003, a previously unknown coronavirus (SARS-CoV) caused a global outbreak of pneumonia that resulted in approximately 800 deaths.6 The MERS-CoV virus attaches to dipeptidyl peptidase 4 to enter cells, and this receptor is believed to be critical for pathogenesis, as infection does not occur in its absence.7
The source and mode of transmission to humans is not completely defined. Early reports suggested that MERS-CoV originated in bats, as RNA sequences related to MERS-CoV have been found in several bat species, but the virus itself has not been isolated from bats.8 Camels have been found to have a high rate of anti-MERS-CoV antibodies and to have the virus in nose swabs, and evidence for camel-to-human transmission has been presented.9–11 However, the precise role of camels and other animals as reservoirs or vectors of infection is still under investigation.
The incubation period from exposure to the development of clinical disease is estimated at 5 to 14 days.
For MERS-CoV, the basic reproduction ratio (R0), which measures the average number of secondary cases from each infected person, is estimated12 to be less than 0.7. In diseases in which the R0 is less than 1.0, infections occur in isolated clusters as limited chains of transmission, and thus the sustained transmission of MERS-CoV resulting in a large epidemic is thought to be unlikely. As a comparison, the median R0 value for seasonal influenza is estimated13 at 1.28. “Superspreading” may result in limited outbreaks of secondary cases; however, the continued epidemic spread of infection is thought to be unlikely.14 Nevertheless, viral adaptation with increased transmissibility remains a concern and a potential threat.
CLINICAL PRESENTATION
MERS most commonly presents as a respiratory illness, although asymptomatic infection occurs. The percentage of patients who experience asymptomatic infection is unknown. A recent survey of 255 patients with laboratory-confirmed MERS-CoV found that 64 (25.1%) were reported as asymptomatic at time of specimen collection. However, when 33 (52%) of those patients were interviewed, 26 (79%) reported at least one symptom that was consistent with a viral respiratory illness.15
For symptomatic patients, the initial complaints are nonspecific, beginning with fever, cough, sore throat, chills, and myalgia. Patients experiencing severe infection progress to dyspnea and pneumonia, with requirements for ventilatory support, vasopressors, and renal replacement therapy.16 Gastrointestinal symptoms such as vomiting and diarrhea have been reported in about one-third of patients.17
In a study of 47 patients with MERS-CoV, most of whom had underlying medical illnesses, 42 (89%) required intensive care and 34 (72%) required mechanical ventilation.17 The case-fatality rate in this study was 60%, but other studies have reported rates closer to 30%.15
Laboratory findings in patients with MERS-CoV infection usually include leukopenia and thrombocytopenia. Severely ill patients may have evidence of acute kidney injury.
Radiographic findings of MERS are those of viral pneumonitis and acute respiratory distress syndrome. Computed tomographic findings include ground-glass opacities, with peripheral lower-lobe preference.18
DIAGNOSIS
As MERS is a respiratory illness, sampling of respiratory secretions provides the highest yield for diagnosis. A study of 112 patients with MERS-CoV reported that polymerase chain reaction (PCR) testing of tracheal aspirates and bronchoalveolar lavage samples yielded significantly higher MERS-CoV loads than nasopharyngeal swab samples and sputum samples.19 However, upper respiratory tract testing is less invasive, and a positive nasopharyngeal swab result may obviate the need for further testing.
The US Centers for Disease Control and Prevention (CDC) recommends collecting multiple specimens from different sites at different times after the onset of symptoms in order to increase the diagnostic yield. Specifically, it recommends testing a lower respiratory specimen (eg, sputum, bronchoalveolar lavage fluid, tracheal aspirate), a nasopharyngeal and oropharyngeal swab, and serum, using the CDC MERS-CoV rRT-PCR assay. In addition, for patients whose symptoms began more than 14 days earlier, the CDC also recommends testing a serum specimen with the CDC MERS-CoV serologic assay. As these guidelines are updated frequently, clinicians are advised to check the CDC website for the most up-to-date information (www.cdc.gov/coronavirus/mers/guidelines-clinical-specimens.html).20 The identification of MERS-CoV by virus isolation in cell culture is not recommended and, if pursued, must be performed in a biosafety level 3 facility. (Level 3 is the second-highest level of biosafety. The highest, level 4, is reserved for extremely dangerous agents such as Ebola virus).20
Given the nonspecific clinical presentation of MERS-CoV, clinicians may consider testing for other respiratory pathogens. A recent review of 54 travelers to California from MERS-CoV-affected areas found that while none tested positive for MERS-CoV, 32 (62%) of 52 travelers had other respiratory viruses.21 When testing for alternative pathogens, clinicians should order molecular or antigen-based detection methods.
TREATMENT
Unfortunately, treatment for MERS is primarily supportive.
Ribavirin and interferon alfa-2b demonstrated activity in an animal model, but the regimen was ineffective when given a median of 19 (range 10–22) days after admission in 5 critically ill patients who subsequently died.22 A retrospective analysis comparing 20 patients with severe MERS-CoV who received ribavirin and interferon alfa-2a with 24 patients who did not reported that while survival was improved at 14 days, the mortality rates were similar at 28 days.23
A systematic review of treatments used for severe acute respiratory syndrome (SARS) reported that most studies investigating steroid use were inconclusive and some showed possible harm, suggesting that systemic steroids should be avoided in coronavirus infections.24
PREVENTION
Healthcare-associated outbreaks of MERS are well described, and thus recognition of potential cases and prompt institution of appropriate infection control measures are critical.15,25
Healthcare providers should ask patients about recent travel history and ascertain if they meet the CDC criteria for a “patient under investigation” (PUI), ie, if they have both clinical features and an epidemiologic risk of MERS (Table 1). However, these recommendations for identification will assuredly change as the outbreak matures, and healthcare providers should refer to the CDC website for the most up-to-date information.
Once a PUI is identified, standard, contact, and airborne precautions are advised. These measures include performing hand hygiene and donning personal protective equipment, including gloves, gowns, eye protection, and respiratory protection (ie, a respirator) that is at least as protective as a fit-tested National Institute for Occupational Safety and Health-certified N95 filtering face-piece respirator. In addition, a patient with possible MERS should be placed in an airborne infection isolation room.
Traveler’s advice
The CDC does not currently recommend that Americans change their travel plans because of MERS. Clinicians performing pretravel evaluations should advise patients of current information on MERS. Patients at risk for MERS who develop a respiratory illness within 14 days of return should seek medical attention and inform healthcare providers of their travel history.
SUMMARY
Recent experience with SARS, Ebola virus disease, and now MERS-CoV highlights the impact of global air travel as a vector for the rapid worldwide dissemination of communicable diseases. Healthcare providers should elicit a travel history in all patients presenting with a febrile illness, as an infection acquired in one continent may not become manifest until the patient presents in another.
The scope of the current MERS-CoV outbreak is still evolving, with concerns that viral evolution could result in a SARS-like outbreak, as experienced almost a decade ago.
Healthcare providers are advised to screen patients at risk for MERS-CoV for respiratory symptoms, and to institute appropriate infection control measures. Through recognition and isolation, healthcare providers are at the front line in limiting the spread of this potentially lethal virus.
- Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012; 367:1814–1820.
- Al-Abdallat MM, Payne DC, Alqasrawi S, et al. Hospital-associated outbreak of Middle East respiratory syndrome coronavirus: a serologic, epidemiologic, and clinical description. Clin Infect Dis 2014; 59:1225–1233.
- World Health Organization. Frequently asked questions on Middle East respiratory syndrome coronavirus (MERS-CoV). www.who.int/csr/disease/coronavirus_infections/faq/en/. Accessed July 29, 2015.
- Bialek SR, Allen D, Alvarado-Ramy F, et al; Centers for Disease Control and Prevention (CDC). First confirmed cases of Middle East respiratory syndrome coronavirus (MERS-CoV) infection in the United States, updated information on the epidemiology of MERS-CoV infection, and guidance for the public, clinicians, and public health authorities—May 2014. MMWR Morb Mortal Wkly Rep 2014; 63:431–436.
- World Health Organization. Middle East respiratory syndrome coronavirus (MERS-CoV) – Republic of Korea. www.who.int/csr/don/12-june-2015-mers-korea/en/. Accessed July 29, 2015.
- Peiris JSM, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nat Med 2004; 10:S88–S97.
- van Doremalen N, Miazqowicz KL, Milne-Price S, et al. Host species restriction of Middle East respiratory syndrome coronavirus through its receptor, dipeptidyl peptidase 4. J Virol 2014; 88:9220–9232.
- Zumla A, Hui DS, Perlman S. Middle East respiratory syndrome. Lancet 2015; S0140-6736(15)60454-604548 (Epub ahead of print).
- Meyer B, Muller MA, Corman WM, et al. Antibodies against MERS coronavirus in dromedary camels, United Arab Emirates, 2003 and 2013. Emerg Infect Dis 2014; 20:552–559.
- Haagmans BL, Al Dhahiry SH, Reusken CB, et al. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect Dis 2014; 14:140–145.
- Azhar EI, El-Kafrawy SA, Farraj SA, et al. Evidence for camel-to-human transmission of MERS coronavirus. N Engl J Med 2014; 370:2499–2505.
- Chowell G, Blumberg S, Simonsen L, Miller MA, Viboud C. Synthesizing data and models for the spread of MERS-CoV, 2013: key role of index cases and hospital transmission. Epidemics 2014; 9:40–51.
- Biggerstaff M, Chauchemez S, Reed C, Gambhir M, Finelli L. Estimates of the reproduction number for seasonal, pandemic, and zoonotic influenza: a systematic review of the literature. BMC Infect Dis 2014: 14:480.
- Kucharski AJ, Althaus CL. The role of superspreading in Middle East respiratory syndrome coronavirus (MERS-CoV) transmission. Euro Surveill 2015; 20.
- Oboho I, Tomczyk S, Al-Asmari A, et al. 2014 MERS-CoV outbreak in Jeddah—a link to health care facilities. N Engl J Med 2015; 372:846–854.
- Arabi YM, Arifi AA, Balkhy HH, et al. Clinical course and outcomes of critically ill patients with Middle East respiratory syndrome coronavirus infection. Ann Intern Med 2014; 160:389–397.
- Assiri A, Al-Tawfig JA, Al-Rabeeah AA, et al. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: a descriptive study. Lancet Infect Dis 2013; 13:752–761.
- Das KM, Lee EY, Enani MA, et al. CT correlation with outcomes in 15 patients with acute Middle East respiratory syndrome coronavirus. AJR Am J Roentgenol 2015; 204:736–742.
- Memish ZA, Al-Tawfiq JA, Makhdoom HQ, et al. Respiratory tract samples, viral load, and genome fraction yield in patients with Middle East respiratory syndrome. J Infect Dis 2014; 210:1590–1594.
- Centers for Disease Control and Prevention. Middle East respiratory syndrome (MERS). Interim guidelines for collecting, handling, and testing clinical specimens from patients under investigation (PUIs) for Middle East respiratory syndrome coronavirus (MERS-CoV)—version 2.1. www.cdc.gov/coronavirus/mers/guidelines-clinical-specimens.html. Accessed July 29, 2015.
- Shakhkarami M, Yen C, Glaser CA, Xia D, Watt J, Wadford DA. Laboratory testing for Middle East respiratory syndrome coronavirus, California, USA, 2013–2014. Emerg Infect Dis 2015; 21: E-pub ahead of print. wwwnc.cdc.gov/eid/article/21/9/15-0476_article. Accessed July 29, 2015.
- Al-Tawfiq JA, Momattin H, Dib J, Memish ZA. Ribavirin and interferon therapy in patients infected with the Middle East respiratory syndrome coronavirus: an observational study. Int J Infect Dis 2014; 20:42–46.
- Omrani AS, Saad MM, Baig K, et al. Ribavirin and interferon alfa-2a for severe Middle East respiratory syndrome coronavirus infection: a retrospective cohort study. Lancet Infect Dis 2014; 14:1090–1095.
- Stockman LJ, Bellamy R, Garner, P. SARS: systematic review of treatment effects. PLoS Med 2006; 3:e343.
- Assiri A, McGeer A, Perl TM, et al; KSA MERS-CoV Investigation Team. Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med 2013; 369:407–416.
- Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012; 367:1814–1820.
- Al-Abdallat MM, Payne DC, Alqasrawi S, et al. Hospital-associated outbreak of Middle East respiratory syndrome coronavirus: a serologic, epidemiologic, and clinical description. Clin Infect Dis 2014; 59:1225–1233.
- World Health Organization. Frequently asked questions on Middle East respiratory syndrome coronavirus (MERS-CoV). www.who.int/csr/disease/coronavirus_infections/faq/en/. Accessed July 29, 2015.
- Bialek SR, Allen D, Alvarado-Ramy F, et al; Centers for Disease Control and Prevention (CDC). First confirmed cases of Middle East respiratory syndrome coronavirus (MERS-CoV) infection in the United States, updated information on the epidemiology of MERS-CoV infection, and guidance for the public, clinicians, and public health authorities—May 2014. MMWR Morb Mortal Wkly Rep 2014; 63:431–436.
- World Health Organization. Middle East respiratory syndrome coronavirus (MERS-CoV) – Republic of Korea. www.who.int/csr/don/12-june-2015-mers-korea/en/. Accessed July 29, 2015.
- Peiris JSM, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nat Med 2004; 10:S88–S97.
- van Doremalen N, Miazqowicz KL, Milne-Price S, et al. Host species restriction of Middle East respiratory syndrome coronavirus through its receptor, dipeptidyl peptidase 4. J Virol 2014; 88:9220–9232.
- Zumla A, Hui DS, Perlman S. Middle East respiratory syndrome. Lancet 2015; S0140-6736(15)60454-604548 (Epub ahead of print).
- Meyer B, Muller MA, Corman WM, et al. Antibodies against MERS coronavirus in dromedary camels, United Arab Emirates, 2003 and 2013. Emerg Infect Dis 2014; 20:552–559.
- Haagmans BL, Al Dhahiry SH, Reusken CB, et al. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect Dis 2014; 14:140–145.
- Azhar EI, El-Kafrawy SA, Farraj SA, et al. Evidence for camel-to-human transmission of MERS coronavirus. N Engl J Med 2014; 370:2499–2505.
- Chowell G, Blumberg S, Simonsen L, Miller MA, Viboud C. Synthesizing data and models for the spread of MERS-CoV, 2013: key role of index cases and hospital transmission. Epidemics 2014; 9:40–51.
- Biggerstaff M, Chauchemez S, Reed C, Gambhir M, Finelli L. Estimates of the reproduction number for seasonal, pandemic, and zoonotic influenza: a systematic review of the literature. BMC Infect Dis 2014: 14:480.
- Kucharski AJ, Althaus CL. The role of superspreading in Middle East respiratory syndrome coronavirus (MERS-CoV) transmission. Euro Surveill 2015; 20.
- Oboho I, Tomczyk S, Al-Asmari A, et al. 2014 MERS-CoV outbreak in Jeddah—a link to health care facilities. N Engl J Med 2015; 372:846–854.
- Arabi YM, Arifi AA, Balkhy HH, et al. Clinical course and outcomes of critically ill patients with Middle East respiratory syndrome coronavirus infection. Ann Intern Med 2014; 160:389–397.
- Assiri A, Al-Tawfig JA, Al-Rabeeah AA, et al. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: a descriptive study. Lancet Infect Dis 2013; 13:752–761.
- Das KM, Lee EY, Enani MA, et al. CT correlation with outcomes in 15 patients with acute Middle East respiratory syndrome coronavirus. AJR Am J Roentgenol 2015; 204:736–742.
- Memish ZA, Al-Tawfiq JA, Makhdoom HQ, et al. Respiratory tract samples, viral load, and genome fraction yield in patients with Middle East respiratory syndrome. J Infect Dis 2014; 210:1590–1594.
- Centers for Disease Control and Prevention. Middle East respiratory syndrome (MERS). Interim guidelines for collecting, handling, and testing clinical specimens from patients under investigation (PUIs) for Middle East respiratory syndrome coronavirus (MERS-CoV)—version 2.1. www.cdc.gov/coronavirus/mers/guidelines-clinical-specimens.html. Accessed July 29, 2015.
- Shakhkarami M, Yen C, Glaser CA, Xia D, Watt J, Wadford DA. Laboratory testing for Middle East respiratory syndrome coronavirus, California, USA, 2013–2014. Emerg Infect Dis 2015; 21: E-pub ahead of print. wwwnc.cdc.gov/eid/article/21/9/15-0476_article. Accessed July 29, 2015.
- Al-Tawfiq JA, Momattin H, Dib J, Memish ZA. Ribavirin and interferon therapy in patients infected with the Middle East respiratory syndrome coronavirus: an observational study. Int J Infect Dis 2014; 20:42–46.
- Omrani AS, Saad MM, Baig K, et al. Ribavirin and interferon alfa-2a for severe Middle East respiratory syndrome coronavirus infection: a retrospective cohort study. Lancet Infect Dis 2014; 14:1090–1095.
- Stockman LJ, Bellamy R, Garner, P. SARS: systematic review of treatment effects. PLoS Med 2006; 3:e343.
- Assiri A, McGeer A, Perl TM, et al; KSA MERS-CoV Investigation Team. Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med 2013; 369:407–416.
KEY POINTS
- In MERS, initial complaints are of fever, cough, chills and myalgia. In a subset of patients, usually those with underlying illnesses, the disease can progress to fulminant sepsis with respiratory and renal failure and death.
- Healthcare providers should regularly visit the US Centers for Disease Control and Prevention website for current information on countries experiencing a MERS outbreak, and for advice on how to identify a potentially infected patient.
- MERS-CoV has caused several healthcare-related outbreaks, so prompt identification and isolation of infected patients is critical to limiting the spread of infection. A “patient under identification” (ie, a person who has both clinical features and an epidemiologic risk) should be cared for under standard, contact, and airborne precautions.
Herpes zoster triplex
A 77-year-old man presented with a 4-day history of painful eruptions on the left chest, right lower groin, and left thigh. He had been taking oral prednisolone 16 mg daily for interstitial pneumonia for 5 years. Ten days earlier, he had started to feel a stinging pain in these areas, but without eruptions.
Physical examination showed several grouped erythematous vesicles in the T2 dermatome of the left chest, L1 dermatome of the right groin, and L2 dermatome of the left upper anterior thigh (Figure 1).
Based on the presentation and a Tzanck smear of the lesions, a diagnosis of preherpetic neuralgia with herpes zoster triplex was made. The patient received intravenous acyclovir 750 mg/day for 7 days and continued to take the prednisolone. The lesions improved within 1 month, leaving scarring but no postherpetic neuralgia.
PREHERPETIC NEURALGIA
Herpes zoster usually occurs unilaterally in a single dermatome, with dermatomal pain appearing before the rash.1 Preherpetic neuralgia may be misdiagnosed as myocardial infarction or renal colic, especially in a case of zoster sine herpete.
Making the diagnosis of preherpetic neuralgia in our patient was difficult because it occurred simultaneously in three dermatomes. At first, his symptoms were suspected of being a recurrence of his past illnesses, including aortic dissection, gallstones, and diverticulitis. Anti-varicella-zoster virus immunoglobulin (Ig) M antibody was not detected, and the IgG antibody titer did not increase.
It has been suggested that cellular immunity is more important than humoral immunity for the surveillance and control of reactivations of herpes viruses. Risk factors for reactivation are increasing age, cancer, acquired immunodeficiency syndrome, and immunosuppressive medications.2,3 In addition, varicella-zoster virus can cause atypical lesions, including recurrent chickenpox, single-dermatomal herpes zoster with scattered rash, and herpes zoster in multiple dermatomes.4
Clinical suspicion for herpes zoster is important in the differential diagnosis of acute pain of uncertain origin, even if it occurs in multiple dermatomes in an immunocompromised patient.
- James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin. Clinical Dermatology, 10th ed. Philadelphia, PA: WB Saunders; 2006.
- Vu AQ, Radonich MA, Heald PW. Herpes zoster in seven disparate dermatomes (zoster multiplex): report of a case and review of literature. J Am Acad Dermatol 1999; 40:868–869.
- Failla V, Jacques J, Castronovo C, Nikkels AF. Herpes zoster in patients treated with biologicals. Dermatology 2012; 224:251–256.
- Kennedy PG, Steiner I. A molecular and cellular model to explain the differences in reactivation from latency by herpes simplex and varicella-zoster viruses. Neuropathol Appl Neurobiol 1994; 20:368–374.
A 77-year-old man presented with a 4-day history of painful eruptions on the left chest, right lower groin, and left thigh. He had been taking oral prednisolone 16 mg daily for interstitial pneumonia for 5 years. Ten days earlier, he had started to feel a stinging pain in these areas, but without eruptions.
Physical examination showed several grouped erythematous vesicles in the T2 dermatome of the left chest, L1 dermatome of the right groin, and L2 dermatome of the left upper anterior thigh (Figure 1).
Based on the presentation and a Tzanck smear of the lesions, a diagnosis of preherpetic neuralgia with herpes zoster triplex was made. The patient received intravenous acyclovir 750 mg/day for 7 days and continued to take the prednisolone. The lesions improved within 1 month, leaving scarring but no postherpetic neuralgia.
PREHERPETIC NEURALGIA
Herpes zoster usually occurs unilaterally in a single dermatome, with dermatomal pain appearing before the rash.1 Preherpetic neuralgia may be misdiagnosed as myocardial infarction or renal colic, especially in a case of zoster sine herpete.
Making the diagnosis of preherpetic neuralgia in our patient was difficult because it occurred simultaneously in three dermatomes. At first, his symptoms were suspected of being a recurrence of his past illnesses, including aortic dissection, gallstones, and diverticulitis. Anti-varicella-zoster virus immunoglobulin (Ig) M antibody was not detected, and the IgG antibody titer did not increase.
It has been suggested that cellular immunity is more important than humoral immunity for the surveillance and control of reactivations of herpes viruses. Risk factors for reactivation are increasing age, cancer, acquired immunodeficiency syndrome, and immunosuppressive medications.2,3 In addition, varicella-zoster virus can cause atypical lesions, including recurrent chickenpox, single-dermatomal herpes zoster with scattered rash, and herpes zoster in multiple dermatomes.4
Clinical suspicion for herpes zoster is important in the differential diagnosis of acute pain of uncertain origin, even if it occurs in multiple dermatomes in an immunocompromised patient.
A 77-year-old man presented with a 4-day history of painful eruptions on the left chest, right lower groin, and left thigh. He had been taking oral prednisolone 16 mg daily for interstitial pneumonia for 5 years. Ten days earlier, he had started to feel a stinging pain in these areas, but without eruptions.
Physical examination showed several grouped erythematous vesicles in the T2 dermatome of the left chest, L1 dermatome of the right groin, and L2 dermatome of the left upper anterior thigh (Figure 1).
Based on the presentation and a Tzanck smear of the lesions, a diagnosis of preherpetic neuralgia with herpes zoster triplex was made. The patient received intravenous acyclovir 750 mg/day for 7 days and continued to take the prednisolone. The lesions improved within 1 month, leaving scarring but no postherpetic neuralgia.
PREHERPETIC NEURALGIA
Herpes zoster usually occurs unilaterally in a single dermatome, with dermatomal pain appearing before the rash.1 Preherpetic neuralgia may be misdiagnosed as myocardial infarction or renal colic, especially in a case of zoster sine herpete.
Making the diagnosis of preherpetic neuralgia in our patient was difficult because it occurred simultaneously in three dermatomes. At first, his symptoms were suspected of being a recurrence of his past illnesses, including aortic dissection, gallstones, and diverticulitis. Anti-varicella-zoster virus immunoglobulin (Ig) M antibody was not detected, and the IgG antibody titer did not increase.
It has been suggested that cellular immunity is more important than humoral immunity for the surveillance and control of reactivations of herpes viruses. Risk factors for reactivation are increasing age, cancer, acquired immunodeficiency syndrome, and immunosuppressive medications.2,3 In addition, varicella-zoster virus can cause atypical lesions, including recurrent chickenpox, single-dermatomal herpes zoster with scattered rash, and herpes zoster in multiple dermatomes.4
Clinical suspicion for herpes zoster is important in the differential diagnosis of acute pain of uncertain origin, even if it occurs in multiple dermatomes in an immunocompromised patient.
- James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin. Clinical Dermatology, 10th ed. Philadelphia, PA: WB Saunders; 2006.
- Vu AQ, Radonich MA, Heald PW. Herpes zoster in seven disparate dermatomes (zoster multiplex): report of a case and review of literature. J Am Acad Dermatol 1999; 40:868–869.
- Failla V, Jacques J, Castronovo C, Nikkels AF. Herpes zoster in patients treated with biologicals. Dermatology 2012; 224:251–256.
- Kennedy PG, Steiner I. A molecular and cellular model to explain the differences in reactivation from latency by herpes simplex and varicella-zoster viruses. Neuropathol Appl Neurobiol 1994; 20:368–374.
- James WD, Berger TG, Elston DM. Andrew’s Diseases of the Skin. Clinical Dermatology, 10th ed. Philadelphia, PA: WB Saunders; 2006.
- Vu AQ, Radonich MA, Heald PW. Herpes zoster in seven disparate dermatomes (zoster multiplex): report of a case and review of literature. J Am Acad Dermatol 1999; 40:868–869.
- Failla V, Jacques J, Castronovo C, Nikkels AF. Herpes zoster in patients treated with biologicals. Dermatology 2012; 224:251–256.
- Kennedy PG, Steiner I. A molecular and cellular model to explain the differences in reactivation from latency by herpes simplex and varicella-zoster viruses. Neuropathol Appl Neurobiol 1994; 20:368–374.