User login
Fear really does curdle blood, study suggests

Photo by Graham Colm
Horror movies may actually be “blood-curdling,” according to a study published in The BMJ.
The research showed that watching a scary movie was associated with an increase in the clotting protein factor VIII (FVIII).
Study subjects were more likely to experience an increase in FVIII while watching a horror film than an educational film.
So using the term “blood-curdling” to describe feeling extreme fear may be justified, researchers said.
The term dates back to medieval times and is based on the concept that fear would “run the blood cold” or “curdle” the blood, but the validity of this theory has never been studied.
So Banne Nemeth, MD, of Leiden University Medical Centre in The Netherlands, and his colleagues set out to assess whether acute fear can curdle blood.
Their study involved 24 healthy volunteers age 30 or younger. Fourteen subjects were assigned to watch a frightening movie followed by a non-threatening educational movie, and 10 were assigned to watch the movies in reverse order.
The movies were viewed more than a week apart at the same time of day and in a comfortable and relaxed environment. Both lasted approximately 90 minutes.
Before and after each movie (within 15 minutes), blood samples were taken and analyzed for markers of clotting activity.
After each movie, participants rated the fear they experienced using a visual analogue fear scale ranging from 0 (no fear at all) to 10 (worst fear imaginable). They also reported whether they had already seen the movie and completed a general questionnaire on lifestyle and favorite movie genre.
The horror movie was perceived to be more frightening than the educational movie, with a 5.4 mean difference in fear rating scores.
The difference in FVIII levels before and after watching the movies was higher for the horror movie than for the educational movie. The mean difference of differences was 11.1 IU/dL (95% confidence interval: 1.2 to 21.0 IU/dL).
FVIII levels increased in 12 (57%) subjects during the horror movie and in 3 (14%) during the educational movie. FVIII levels decreased in 18 (86%) participants during the educational movie and in 9 (43%) during the horror movie.
However, the researchers found no effect of either movie on thrombin-antithrombin complexes, D-dimer, or prothrombin fragments 1+2. They said this suggests that although acute fear may trigger coagulation, it does not lead to actual clot formation.
The team pointed out some study limitations but concluded that, in young and healthy adults, “watching blood-curdling movies is associated with an increase in blood coagulant factor VIII without actual thrombin formation.”
The researchers believe this phenomenon may confer an important evolutionary benefit by preparing the body for blood loss during life-threatening situations. ![]()
Photo by Graham Colm
Horror movies may actually be “blood-curdling,” according to a study published in The BMJ.
The research showed that watching a scary movie was associated with an increase in the clotting protein factor VIII (FVIII).
Study subjects were more likely to experience an increase in FVIII while watching a horror film than an educational film.
So using the term “blood-curdling” to describe feeling extreme fear may be justified, researchers said.
The term dates back to medieval times and is based on the concept that fear would “run the blood cold” or “curdle” the blood, but the validity of this theory has never been studied.
So Banne Nemeth, MD, of Leiden University Medical Centre in The Netherlands, and his colleagues set out to assess whether acute fear can curdle blood.
Their study involved 24 healthy volunteers age 30 or younger. Fourteen subjects were assigned to watch a frightening movie followed by a non-threatening educational movie, and 10 were assigned to watch the movies in reverse order.
The movies were viewed more than a week apart at the same time of day and in a comfortable and relaxed environment. Both lasted approximately 90 minutes.
Before and after each movie (within 15 minutes), blood samples were taken and analyzed for markers of clotting activity.
After each movie, participants rated the fear they experienced using a visual analogue fear scale ranging from 0 (no fear at all) to 10 (worst fear imaginable). They also reported whether they had already seen the movie and completed a general questionnaire on lifestyle and favorite movie genre.
The horror movie was perceived to be more frightening than the educational movie, with a 5.4 mean difference in fear rating scores.
The difference in FVIII levels before and after watching the movies was higher for the horror movie than for the educational movie. The mean difference of differences was 11.1 IU/dL (95% confidence interval: 1.2 to 21.0 IU/dL).
FVIII levels increased in 12 (57%) subjects during the horror movie and in 3 (14%) during the educational movie. FVIII levels decreased in 18 (86%) participants during the educational movie and in 9 (43%) during the horror movie.
However, the researchers found no effect of either movie on thrombin-antithrombin complexes, D-dimer, or prothrombin fragments 1+2. They said this suggests that although acute fear may trigger coagulation, it does not lead to actual clot formation.
The team pointed out some study limitations but concluded that, in young and healthy adults, “watching blood-curdling movies is associated with an increase in blood coagulant factor VIII without actual thrombin formation.”
The researchers believe this phenomenon may confer an important evolutionary benefit by preparing the body for blood loss during life-threatening situations. ![]()
Photo by Graham Colm
Horror movies may actually be “blood-curdling,” according to a study published in The BMJ.
The research showed that watching a scary movie was associated with an increase in the clotting protein factor VIII (FVIII).
Study subjects were more likely to experience an increase in FVIII while watching a horror film than an educational film.
So using the term “blood-curdling” to describe feeling extreme fear may be justified, researchers said.
The term dates back to medieval times and is based on the concept that fear would “run the blood cold” or “curdle” the blood, but the validity of this theory has never been studied.
So Banne Nemeth, MD, of Leiden University Medical Centre in The Netherlands, and his colleagues set out to assess whether acute fear can curdle blood.
Their study involved 24 healthy volunteers age 30 or younger. Fourteen subjects were assigned to watch a frightening movie followed by a non-threatening educational movie, and 10 were assigned to watch the movies in reverse order.
The movies were viewed more than a week apart at the same time of day and in a comfortable and relaxed environment. Both lasted approximately 90 minutes.
Before and after each movie (within 15 minutes), blood samples were taken and analyzed for markers of clotting activity.
After each movie, participants rated the fear they experienced using a visual analogue fear scale ranging from 0 (no fear at all) to 10 (worst fear imaginable). They also reported whether they had already seen the movie and completed a general questionnaire on lifestyle and favorite movie genre.
The horror movie was perceived to be more frightening than the educational movie, with a 5.4 mean difference in fear rating scores.
The difference in FVIII levels before and after watching the movies was higher for the horror movie than for the educational movie. The mean difference of differences was 11.1 IU/dL (95% confidence interval: 1.2 to 21.0 IU/dL).
FVIII levels increased in 12 (57%) subjects during the horror movie and in 3 (14%) during the educational movie. FVIII levels decreased in 18 (86%) participants during the educational movie and in 9 (43%) during the horror movie.
However, the researchers found no effect of either movie on thrombin-antithrombin complexes, D-dimer, or prothrombin fragments 1+2. They said this suggests that although acute fear may trigger coagulation, it does not lead to actual clot formation.
The team pointed out some study limitations but concluded that, in young and healthy adults, “watching blood-curdling movies is associated with an increase in blood coagulant factor VIII without actual thrombin formation.”
The researchers believe this phenomenon may confer an important evolutionary benefit by preparing the body for blood loss during life-threatening situations. ![]()
A potential target for IDH1-mutant cancers
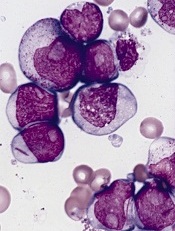
acute myeloid leukemia
Researchers say they have identified a potential therapeutic target for IDH1-mutant malignancies.
Preclinical experiments showed that tumors characterized by IDH1 mutations are “extremely vulnerable” to depletion of NAD+, a metabolic cofactor.
“Our finding . . . supports the proposal that medications that can decrease levels of this metabolite, which are in development, have the potential to specifically treat these cancers,” said Daniel Cahill, MD, PhD, of Massachusetts General Hospital in Boston.
Dr Cahill and his colleagues described their research in Cancer Cell.
IDH1 and IDH2 are known to play essential roles in cellular metabolism, including the processes by which cells convert glucose and other nutrients into ATP, providing the energy needed for cellular survival.
In 2008, researchers discovered that IDH1 mutations are involved in several cancers. The gene is mutated in around 20% of adult gliomas, in more than 10% of acute myeloid leukemias, and in a smaller percentage of other cancers.
One clear result of IDH1 mutation is a 100-fold elevation in levels of the metabolite 2-HG, which is known to mediate several properties that lead to tumor development. Drugs that decrease levels of 2-HG are currently under development.
Dr Cahill and his colleagues wanted to find additional ways of blocking the mutation’s effects, so they inhibited mutant IDH1 in tumor cells. They were surprised to find that this reduced 2-HG levels, but, in many cases, that reduction did not halt cell growth.
Detailed metabolic profiling of IDH1-mutant cells revealed that inhibiting the mutated enzyme greatly increased levels of NAD+, a cofactor that plays a role in several cellular energy processes.
Additional experiments showed that depletion of NAD+ induced the death of IDH1-mutant tumor cells and inhibited tumor growth in an animal model of glioma.
“Accumulation of excess 2-HG is known to drive several changes leading to tumor development, but our results indicate that simply depleting 2-HG levels was not sufficient to halt the growth of several types of later-stage IDH1-mutant tumors,” said Hiroaki Wakimoto, MD, PhD, of Massachusetts General Hospital.
“In addition, we found that mutant IDH1 reduces expression of an enzyme that maintains NAD+ levels, rendering IDH1-mutant tumor cells highly sensitive to direct NAD+ depletion. Several drugs that inhibit the synthesis of NAD+ are already in clinical trials, and these agents may prove useful for patients with IDH-mutant cancers. While we primarily focused on IDH1-mutant gliomas, we also found evidence that NAD+ inhibition could slow the growth of other types of cancer with this mutation.” ![]()
acute myeloid leukemia
Researchers say they have identified a potential therapeutic target for IDH1-mutant malignancies.
Preclinical experiments showed that tumors characterized by IDH1 mutations are “extremely vulnerable” to depletion of NAD+, a metabolic cofactor.
“Our finding . . . supports the proposal that medications that can decrease levels of this metabolite, which are in development, have the potential to specifically treat these cancers,” said Daniel Cahill, MD, PhD, of Massachusetts General Hospital in Boston.
Dr Cahill and his colleagues described their research in Cancer Cell.
IDH1 and IDH2 are known to play essential roles in cellular metabolism, including the processes by which cells convert glucose and other nutrients into ATP, providing the energy needed for cellular survival.
In 2008, researchers discovered that IDH1 mutations are involved in several cancers. The gene is mutated in around 20% of adult gliomas, in more than 10% of acute myeloid leukemias, and in a smaller percentage of other cancers.
One clear result of IDH1 mutation is a 100-fold elevation in levels of the metabolite 2-HG, which is known to mediate several properties that lead to tumor development. Drugs that decrease levels of 2-HG are currently under development.
Dr Cahill and his colleagues wanted to find additional ways of blocking the mutation’s effects, so they inhibited mutant IDH1 in tumor cells. They were surprised to find that this reduced 2-HG levels, but, in many cases, that reduction did not halt cell growth.
Detailed metabolic profiling of IDH1-mutant cells revealed that inhibiting the mutated enzyme greatly increased levels of NAD+, a cofactor that plays a role in several cellular energy processes.
Additional experiments showed that depletion of NAD+ induced the death of IDH1-mutant tumor cells and inhibited tumor growth in an animal model of glioma.
“Accumulation of excess 2-HG is known to drive several changes leading to tumor development, but our results indicate that simply depleting 2-HG levels was not sufficient to halt the growth of several types of later-stage IDH1-mutant tumors,” said Hiroaki Wakimoto, MD, PhD, of Massachusetts General Hospital.
“In addition, we found that mutant IDH1 reduces expression of an enzyme that maintains NAD+ levels, rendering IDH1-mutant tumor cells highly sensitive to direct NAD+ depletion. Several drugs that inhibit the synthesis of NAD+ are already in clinical trials, and these agents may prove useful for patients with IDH-mutant cancers. While we primarily focused on IDH1-mutant gliomas, we also found evidence that NAD+ inhibition could slow the growth of other types of cancer with this mutation.” ![]()
acute myeloid leukemia
Researchers say they have identified a potential therapeutic target for IDH1-mutant malignancies.
Preclinical experiments showed that tumors characterized by IDH1 mutations are “extremely vulnerable” to depletion of NAD+, a metabolic cofactor.
“Our finding . . . supports the proposal that medications that can decrease levels of this metabolite, which are in development, have the potential to specifically treat these cancers,” said Daniel Cahill, MD, PhD, of Massachusetts General Hospital in Boston.
Dr Cahill and his colleagues described their research in Cancer Cell.
IDH1 and IDH2 are known to play essential roles in cellular metabolism, including the processes by which cells convert glucose and other nutrients into ATP, providing the energy needed for cellular survival.
In 2008, researchers discovered that IDH1 mutations are involved in several cancers. The gene is mutated in around 20% of adult gliomas, in more than 10% of acute myeloid leukemias, and in a smaller percentage of other cancers.
One clear result of IDH1 mutation is a 100-fold elevation in levels of the metabolite 2-HG, which is known to mediate several properties that lead to tumor development. Drugs that decrease levels of 2-HG are currently under development.
Dr Cahill and his colleagues wanted to find additional ways of blocking the mutation’s effects, so they inhibited mutant IDH1 in tumor cells. They were surprised to find that this reduced 2-HG levels, but, in many cases, that reduction did not halt cell growth.
Detailed metabolic profiling of IDH1-mutant cells revealed that inhibiting the mutated enzyme greatly increased levels of NAD+, a cofactor that plays a role in several cellular energy processes.
Additional experiments showed that depletion of NAD+ induced the death of IDH1-mutant tumor cells and inhibited tumor growth in an animal model of glioma.
“Accumulation of excess 2-HG is known to drive several changes leading to tumor development, but our results indicate that simply depleting 2-HG levels was not sufficient to halt the growth of several types of later-stage IDH1-mutant tumors,” said Hiroaki Wakimoto, MD, PhD, of Massachusetts General Hospital.
“In addition, we found that mutant IDH1 reduces expression of an enzyme that maintains NAD+ levels, rendering IDH1-mutant tumor cells highly sensitive to direct NAD+ depletion. Several drugs that inhibit the synthesis of NAD+ are already in clinical trials, and these agents may prove useful for patients with IDH-mutant cancers. While we primarily focused on IDH1-mutant gliomas, we also found evidence that NAD+ inhibition could slow the growth of other types of cancer with this mutation.” ![]()
Antiplatelet agent shows promise in phase 1 trial

Image by Andre E.X. Brown
Results of a phase 1 study suggest the experimental agent PZ-128 provides rapid and reversible inhibition of platelet aggregation.
PZ-128 inhibited platelet aggregation in a cohort of patients who had vascular disease or multiple risk factors for coronary artery disease.
This effect was dose-dependent and diminished over time. The drug did not affect bleeding, coagulation, clinical chemistry, or ECG parameters.
Investigators reported these results in Arteriosclerosis, Thrombosis and Vascular Biology.
The team explained that PZ-128 is a protease-activated receptor-1 (PAR1)-based pepducin intended as an antiplatelet agent. Pepducins are membrane-tethered, cell-penetrating lipopeptides that target the cytoplasmic surface of their cognate receptor.
The current study is the first demonstration of pepducins’ potential benefits in humans, said study author Athan Kuliopulos, MD, PhD, of Tufts Medical Center in Boston, Massachusetts.
He and his colleagues tested PZ-128 in 31 patients, ages 43 to 74. The patients had vascular disease (22%)—coronary artery disease, previous myocardial infarction, etc.—or multiple coronary artery disease risk factors—dyslipidemia (81%), hypertension (69%), diabetes (34%), etc.
Many of the patients were taking other medications at baseline, including blood pressure medications (66%), aspirin (63%), lipid-lowering medications (56%), and diabetes medications (22%).
The patients received PZ-128 by 1- to 2-hour-long intravenous infusion at doses ranging from 0.01 mg/kg to 2 mg/kg. The investigators evaluated patients at baseline and 0.5, 1, 2, 6, and 24 hours after dosing, as well as 7 to 10 days after dosing.
PZ-128 had a dose-dependent inhibitory effect on platelet aggregation stimulated by the PAR1 agonist SFLLRN (8 µmol/L). At 30 minutes to 6 hours after dosing, the investigators observed 20% to 40% inhibition with PZ-128 at 0.3 mg/kg, 40% to 60% inhibition at 0.5 mg/kg, and 80% to 100% inhibition at 1 to 2 mg/kg.
Patients who were receiving aspirin in the 0.5 mg/kg and 1 mg/kg dose cohorts had 65% to 100% inhibition of final aggregation to SFLLRN at 30 minutes to 2 hours after dosing and 95% to 100% inhibition by 6 hours.
The impact of PZ-128 was reversible, with 50% recovery of aggregation to SFLLRN by 24 hours and near-complete recovery by 192 hours.
The investigators said PZ-128 did not have any significant effects on aggregation induced by AYPGKF, ADP, or collagen, which suggests the observed effects were specific to PAR1.
Likewise, the team said PZ-128 did not have any significant effects on bleeding, coagulation, clinical chemistry, or ECG parameters.
The investigators noted that there are currently no drugs on the market that target PAR1 and can be used during procedures when the risk of serious complications is high.
The PAR1 inhibitor vorapaxar is available for non-acute use in patients with a prior myocardial infarction or current peripheral artery disease. But the drug, whose effects build slowly and are long-lasting, was not approved for use during cardiac procedures due to a risk of excessive bleeding, Dr Kuliopulos said.
By contrast, PZ-128 appears able to block PAR1 fast enough to be used in an urgent procedure and for a time short enough to limit bleeding risk afterward, he said. Of course, more research is needed to confirm this.
To that end, the investigators are planning a phase 2 study of PZ-128 in up to 600 patients who have acute coronary syndromes or are undergoing percutaneous coronary intervention. ![]()
Image by Andre E.X. Brown
Results of a phase 1 study suggest the experimental agent PZ-128 provides rapid and reversible inhibition of platelet aggregation.
PZ-128 inhibited platelet aggregation in a cohort of patients who had vascular disease or multiple risk factors for coronary artery disease.
This effect was dose-dependent and diminished over time. The drug did not affect bleeding, coagulation, clinical chemistry, or ECG parameters.
Investigators reported these results in Arteriosclerosis, Thrombosis and Vascular Biology.
The team explained that PZ-128 is a protease-activated receptor-1 (PAR1)-based pepducin intended as an antiplatelet agent. Pepducins are membrane-tethered, cell-penetrating lipopeptides that target the cytoplasmic surface of their cognate receptor.
The current study is the first demonstration of pepducins’ potential benefits in humans, said study author Athan Kuliopulos, MD, PhD, of Tufts Medical Center in Boston, Massachusetts.
He and his colleagues tested PZ-128 in 31 patients, ages 43 to 74. The patients had vascular disease (22%)—coronary artery disease, previous myocardial infarction, etc.—or multiple coronary artery disease risk factors—dyslipidemia (81%), hypertension (69%), diabetes (34%), etc.
Many of the patients were taking other medications at baseline, including blood pressure medications (66%), aspirin (63%), lipid-lowering medications (56%), and diabetes medications (22%).
The patients received PZ-128 by 1- to 2-hour-long intravenous infusion at doses ranging from 0.01 mg/kg to 2 mg/kg. The investigators evaluated patients at baseline and 0.5, 1, 2, 6, and 24 hours after dosing, as well as 7 to 10 days after dosing.
PZ-128 had a dose-dependent inhibitory effect on platelet aggregation stimulated by the PAR1 agonist SFLLRN (8 µmol/L). At 30 minutes to 6 hours after dosing, the investigators observed 20% to 40% inhibition with PZ-128 at 0.3 mg/kg, 40% to 60% inhibition at 0.5 mg/kg, and 80% to 100% inhibition at 1 to 2 mg/kg.
Patients who were receiving aspirin in the 0.5 mg/kg and 1 mg/kg dose cohorts had 65% to 100% inhibition of final aggregation to SFLLRN at 30 minutes to 2 hours after dosing and 95% to 100% inhibition by 6 hours.
The impact of PZ-128 was reversible, with 50% recovery of aggregation to SFLLRN by 24 hours and near-complete recovery by 192 hours.
The investigators said PZ-128 did not have any significant effects on aggregation induced by AYPGKF, ADP, or collagen, which suggests the observed effects were specific to PAR1.
Likewise, the team said PZ-128 did not have any significant effects on bleeding, coagulation, clinical chemistry, or ECG parameters.
The investigators noted that there are currently no drugs on the market that target PAR1 and can be used during procedures when the risk of serious complications is high.
The PAR1 inhibitor vorapaxar is available for non-acute use in patients with a prior myocardial infarction or current peripheral artery disease. But the drug, whose effects build slowly and are long-lasting, was not approved for use during cardiac procedures due to a risk of excessive bleeding, Dr Kuliopulos said.
By contrast, PZ-128 appears able to block PAR1 fast enough to be used in an urgent procedure and for a time short enough to limit bleeding risk afterward, he said. Of course, more research is needed to confirm this.
To that end, the investigators are planning a phase 2 study of PZ-128 in up to 600 patients who have acute coronary syndromes or are undergoing percutaneous coronary intervention. ![]()
Image by Andre E.X. Brown
Results of a phase 1 study suggest the experimental agent PZ-128 provides rapid and reversible inhibition of platelet aggregation.
PZ-128 inhibited platelet aggregation in a cohort of patients who had vascular disease or multiple risk factors for coronary artery disease.
This effect was dose-dependent and diminished over time. The drug did not affect bleeding, coagulation, clinical chemistry, or ECG parameters.
Investigators reported these results in Arteriosclerosis, Thrombosis and Vascular Biology.
The team explained that PZ-128 is a protease-activated receptor-1 (PAR1)-based pepducin intended as an antiplatelet agent. Pepducins are membrane-tethered, cell-penetrating lipopeptides that target the cytoplasmic surface of their cognate receptor.
The current study is the first demonstration of pepducins’ potential benefits in humans, said study author Athan Kuliopulos, MD, PhD, of Tufts Medical Center in Boston, Massachusetts.
He and his colleagues tested PZ-128 in 31 patients, ages 43 to 74. The patients had vascular disease (22%)—coronary artery disease, previous myocardial infarction, etc.—or multiple coronary artery disease risk factors—dyslipidemia (81%), hypertension (69%), diabetes (34%), etc.
Many of the patients were taking other medications at baseline, including blood pressure medications (66%), aspirin (63%), lipid-lowering medications (56%), and diabetes medications (22%).
The patients received PZ-128 by 1- to 2-hour-long intravenous infusion at doses ranging from 0.01 mg/kg to 2 mg/kg. The investigators evaluated patients at baseline and 0.5, 1, 2, 6, and 24 hours after dosing, as well as 7 to 10 days after dosing.
PZ-128 had a dose-dependent inhibitory effect on platelet aggregation stimulated by the PAR1 agonist SFLLRN (8 µmol/L). At 30 minutes to 6 hours after dosing, the investigators observed 20% to 40% inhibition with PZ-128 at 0.3 mg/kg, 40% to 60% inhibition at 0.5 mg/kg, and 80% to 100% inhibition at 1 to 2 mg/kg.
Patients who were receiving aspirin in the 0.5 mg/kg and 1 mg/kg dose cohorts had 65% to 100% inhibition of final aggregation to SFLLRN at 30 minutes to 2 hours after dosing and 95% to 100% inhibition by 6 hours.
The impact of PZ-128 was reversible, with 50% recovery of aggregation to SFLLRN by 24 hours and near-complete recovery by 192 hours.
The investigators said PZ-128 did not have any significant effects on aggregation induced by AYPGKF, ADP, or collagen, which suggests the observed effects were specific to PAR1.
Likewise, the team said PZ-128 did not have any significant effects on bleeding, coagulation, clinical chemistry, or ECG parameters.
The investigators noted that there are currently no drugs on the market that target PAR1 and can be used during procedures when the risk of serious complications is high.
The PAR1 inhibitor vorapaxar is available for non-acute use in patients with a prior myocardial infarction or current peripheral artery disease. But the drug, whose effects build slowly and are long-lasting, was not approved for use during cardiac procedures due to a risk of excessive bleeding, Dr Kuliopulos said.
By contrast, PZ-128 appears able to block PAR1 fast enough to be used in an urgent procedure and for a time short enough to limit bleeding risk afterward, he said. Of course, more research is needed to confirm this.
To that end, the investigators are planning a phase 2 study of PZ-128 in up to 600 patients who have acute coronary syndromes or are undergoing percutaneous coronary intervention. ![]()
ASH: First shot across the bow for CAR T cells in multiple myeloma
ORLANDO – Chimeric antigen receptor (CAR) T cells targeting BCMA eradicated myeloma cells in a patient with a heavy burden of chemotherapy-resistant multiple myeloma.
The patient had received three prior myeloma therapies and relapsed with myeloma making up more than 90% of his bone marrow cells just 3 months after autologous transplant.

CD138-positive plasma cells were completely absent, however, one month after infusion of CAR T cells directed against the B-cell maturation antigen (BCMA), and remain undetectable 14 weeks after infusion.
“We have demonstrated for the first time that CAR T cells can have powerful activity against measurable multiple myeloma,” Dr. James N. Kochenderfer of the National Cancer Institute in Bethesda, Md., said at the annual meeting of the American Society of Hematology.
A second patient, who had five prior lines of myeloma therapy and myeloma in 80% of his bone marrow cells, also experienced a dramatic reduction in myeloma, resulting so far in a partial response. The patient has returned to full-time work and myeloma cells continue to decrease 6 weeks after infusion.
The two ongoing responses, however, were associated with the most severe clinical signs of cytokine release syndrome in the late-breaking abstract study (LBA-1), Dr. Kochenderfer said.
The complete responder experienced cytokine release toxicities including fever, tachycardia, hypotension, elevated liver enzymes, and elevated creatinine kinase that resolved. The patient was also platelet transfusion-dependent for 9 weeks after infusion and a baseline absolute neutrophil count of less than 500 microliters remained at that level for 40 days after infusion. All symptoms resolved within two weeks, he said.
The ongoing partial responder experienced fever, tachycardia, hypotension, acute kidney injury, dyspnea, delirium, and prolonged thrombocytopenia and all completely resolved.
Both patients had much higher serum levels of interleukin-6 than the other patients, he noted.
Both patients also received the highest dose level of anti-BCMA CAR T cells. Going forward, that dose will now only be administered to patients with less than 50% bone marrow plasma cells, Dr. Kochenderfer said.
Other responses among the 12 patients treated thus far include 1 transient very good partial response of 8-week duration, 1 transient partial response lasting 2 weeks, and 8 responses of stable disease.
While three new drugs (elotuzumab, daratumumab, ixazomib) were approved for the treatment of multiple myeloma in the month of November alone, this is early days for CAR T-cell therapy in multiple myeloma.
“Almost all of the CAR T-cell work has been in B-cell malignancies, acute lymphocytic leukemia, and chronic lymphocytic leukemia, so is this the right antigen, we don’t know? Is this the right construct, we don’t know? But there’s clearly activity and that is very exciting,” session comoderator Dr. David P. Steensma of the Dana-Farber Cancer Institute in Boston, said in an interview.
Dana-Farber is conducting a CAR T cell trial in myeloma using a different target and NKG2D ligands. Other centers are also using CAR T cells with different targets. “So maybe other targets would have different results and a better safety profile,” he added.
BCMA is an appropriate target for CAR T cell therapy for multiple reasons, Dr. Kochenderfer said. Multiple myeloma is still an incurable disease and BCMA, a member of the tumor necrosis factor superfamily, is uniformly expressed in 60% to 70% of cases.
Preclinical studies by the team also show that BCMA is not detectable in normal human tissues, but is selectively expressed only in bone marrow, lymphoid organs, and organs known to have plasma cells in their lamina propria and by plasma cells and a small fraction of B cells.
The investigators genetically modified autologous T cells to express an anti-BCMA CAR and ligated it into a replication-incompetent gamma retrovirus. The T cells were stimulated with the anti-CD3 monoclonal antibody OKT3 before transduction, with the entire culture process taking 9 days from start to finish. T cells expressing this CAR recognize BCMA with great specificity, Dr. Kochenderfer observed.
The 12 patients received 300 mg/m2 of cyclophosphamide and 30 mg/m2 of fludarabine (Fludara) daily for 3 days before a single infusion of anti-BCMA CAR at dose levels of 0.3 x 106 to 9 x 106 T cells/kg. All patients had at least 3 prior therapies and normal organ function. Five had amyloid light chain only, 4 had immunoglobulin gamma disease, and 4 patients, including the complete responder, had immunoglobulin alpha disease.
It’s unclear why only a few patients responded to the CAR T cells, but dose level was likely a big factor and there is a lot of patient variability with T-cell therapies, Dr. Kochenderfer said at a press briefing.
“I wish we knew exactly why some patients respond and some don’t,” he added. “The CAR T cells have shown remarkable activity against really previously refractory diseases like acute lymphocytic leukemia,” noted Dr. George Daley of Harvard Medical School, Boston, in an interview at the meeting. “There’s been a lot of excitement that maybe we can extend the principles that work in leukemia to other types of diseases.”
The study is still ongoing and accruing patients, but a multicenter trial is also being initiated in myeloma with Bluebird Bio using “a closely related, but slightly different CAR,” Dr. Kochenderfer said.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ORLANDO – Chimeric antigen receptor (CAR) T cells targeting BCMA eradicated myeloma cells in a patient with a heavy burden of chemotherapy-resistant multiple myeloma.
The patient had received three prior myeloma therapies and relapsed with myeloma making up more than 90% of his bone marrow cells just 3 months after autologous transplant.
CD138-positive plasma cells were completely absent, however, one month after infusion of CAR T cells directed against the B-cell maturation antigen (BCMA), and remain undetectable 14 weeks after infusion.
“We have demonstrated for the first time that CAR T cells can have powerful activity against measurable multiple myeloma,” Dr. James N. Kochenderfer of the National Cancer Institute in Bethesda, Md., said at the annual meeting of the American Society of Hematology.
A second patient, who had five prior lines of myeloma therapy and myeloma in 80% of his bone marrow cells, also experienced a dramatic reduction in myeloma, resulting so far in a partial response. The patient has returned to full-time work and myeloma cells continue to decrease 6 weeks after infusion.
The two ongoing responses, however, were associated with the most severe clinical signs of cytokine release syndrome in the late-breaking abstract study (LBA-1), Dr. Kochenderfer said.
The complete responder experienced cytokine release toxicities including fever, tachycardia, hypotension, elevated liver enzymes, and elevated creatinine kinase that resolved. The patient was also platelet transfusion-dependent for 9 weeks after infusion and a baseline absolute neutrophil count of less than 500 microliters remained at that level for 40 days after infusion. All symptoms resolved within two weeks, he said.
The ongoing partial responder experienced fever, tachycardia, hypotension, acute kidney injury, dyspnea, delirium, and prolonged thrombocytopenia and all completely resolved.
Both patients had much higher serum levels of interleukin-6 than the other patients, he noted.
Both patients also received the highest dose level of anti-BCMA CAR T cells. Going forward, that dose will now only be administered to patients with less than 50% bone marrow plasma cells, Dr. Kochenderfer said.
Other responses among the 12 patients treated thus far include 1 transient very good partial response of 8-week duration, 1 transient partial response lasting 2 weeks, and 8 responses of stable disease.
While three new drugs (elotuzumab, daratumumab, ixazomib) were approved for the treatment of multiple myeloma in the month of November alone, this is early days for CAR T-cell therapy in multiple myeloma.
“Almost all of the CAR T-cell work has been in B-cell malignancies, acute lymphocytic leukemia, and chronic lymphocytic leukemia, so is this the right antigen, we don’t know? Is this the right construct, we don’t know? But there’s clearly activity and that is very exciting,” session comoderator Dr. David P. Steensma of the Dana-Farber Cancer Institute in Boston, said in an interview.
Dana-Farber is conducting a CAR T cell trial in myeloma using a different target and NKG2D ligands. Other centers are also using CAR T cells with different targets. “So maybe other targets would have different results and a better safety profile,” he added.
BCMA is an appropriate target for CAR T cell therapy for multiple reasons, Dr. Kochenderfer said. Multiple myeloma is still an incurable disease and BCMA, a member of the tumor necrosis factor superfamily, is uniformly expressed in 60% to 70% of cases.
Preclinical studies by the team also show that BCMA is not detectable in normal human tissues, but is selectively expressed only in bone marrow, lymphoid organs, and organs known to have plasma cells in their lamina propria and by plasma cells and a small fraction of B cells.
The investigators genetically modified autologous T cells to express an anti-BCMA CAR and ligated it into a replication-incompetent gamma retrovirus. The T cells were stimulated with the anti-CD3 monoclonal antibody OKT3 before transduction, with the entire culture process taking 9 days from start to finish. T cells expressing this CAR recognize BCMA with great specificity, Dr. Kochenderfer observed.
The 12 patients received 300 mg/m2 of cyclophosphamide and 30 mg/m2 of fludarabine (Fludara) daily for 3 days before a single infusion of anti-BCMA CAR at dose levels of 0.3 x 106 to 9 x 106 T cells/kg. All patients had at least 3 prior therapies and normal organ function. Five had amyloid light chain only, 4 had immunoglobulin gamma disease, and 4 patients, including the complete responder, had immunoglobulin alpha disease.
It’s unclear why only a few patients responded to the CAR T cells, but dose level was likely a big factor and there is a lot of patient variability with T-cell therapies, Dr. Kochenderfer said at a press briefing.
“I wish we knew exactly why some patients respond and some don’t,” he added. “The CAR T cells have shown remarkable activity against really previously refractory diseases like acute lymphocytic leukemia,” noted Dr. George Daley of Harvard Medical School, Boston, in an interview at the meeting. “There’s been a lot of excitement that maybe we can extend the principles that work in leukemia to other types of diseases.”
The study is still ongoing and accruing patients, but a multicenter trial is also being initiated in myeloma with Bluebird Bio using “a closely related, but slightly different CAR,” Dr. Kochenderfer said.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ORLANDO – Chimeric antigen receptor (CAR) T cells targeting BCMA eradicated myeloma cells in a patient with a heavy burden of chemotherapy-resistant multiple myeloma.
The patient had received three prior myeloma therapies and relapsed with myeloma making up more than 90% of his bone marrow cells just 3 months after autologous transplant.
CD138-positive plasma cells were completely absent, however, one month after infusion of CAR T cells directed against the B-cell maturation antigen (BCMA), and remain undetectable 14 weeks after infusion.
“We have demonstrated for the first time that CAR T cells can have powerful activity against measurable multiple myeloma,” Dr. James N. Kochenderfer of the National Cancer Institute in Bethesda, Md., said at the annual meeting of the American Society of Hematology.
A second patient, who had five prior lines of myeloma therapy and myeloma in 80% of his bone marrow cells, also experienced a dramatic reduction in myeloma, resulting so far in a partial response. The patient has returned to full-time work and myeloma cells continue to decrease 6 weeks after infusion.
The two ongoing responses, however, were associated with the most severe clinical signs of cytokine release syndrome in the late-breaking abstract study (LBA-1), Dr. Kochenderfer said.
The complete responder experienced cytokine release toxicities including fever, tachycardia, hypotension, elevated liver enzymes, and elevated creatinine kinase that resolved. The patient was also platelet transfusion-dependent for 9 weeks after infusion and a baseline absolute neutrophil count of less than 500 microliters remained at that level for 40 days after infusion. All symptoms resolved within two weeks, he said.
The ongoing partial responder experienced fever, tachycardia, hypotension, acute kidney injury, dyspnea, delirium, and prolonged thrombocytopenia and all completely resolved.
Both patients had much higher serum levels of interleukin-6 than the other patients, he noted.
Both patients also received the highest dose level of anti-BCMA CAR T cells. Going forward, that dose will now only be administered to patients with less than 50% bone marrow plasma cells, Dr. Kochenderfer said.
Other responses among the 12 patients treated thus far include 1 transient very good partial response of 8-week duration, 1 transient partial response lasting 2 weeks, and 8 responses of stable disease.
While three new drugs (elotuzumab, daratumumab, ixazomib) were approved for the treatment of multiple myeloma in the month of November alone, this is early days for CAR T-cell therapy in multiple myeloma.
“Almost all of the CAR T-cell work has been in B-cell malignancies, acute lymphocytic leukemia, and chronic lymphocytic leukemia, so is this the right antigen, we don’t know? Is this the right construct, we don’t know? But there’s clearly activity and that is very exciting,” session comoderator Dr. David P. Steensma of the Dana-Farber Cancer Institute in Boston, said in an interview.
Dana-Farber is conducting a CAR T cell trial in myeloma using a different target and NKG2D ligands. Other centers are also using CAR T cells with different targets. “So maybe other targets would have different results and a better safety profile,” he added.
BCMA is an appropriate target for CAR T cell therapy for multiple reasons, Dr. Kochenderfer said. Multiple myeloma is still an incurable disease and BCMA, a member of the tumor necrosis factor superfamily, is uniformly expressed in 60% to 70% of cases.
Preclinical studies by the team also show that BCMA is not detectable in normal human tissues, but is selectively expressed only in bone marrow, lymphoid organs, and organs known to have plasma cells in their lamina propria and by plasma cells and a small fraction of B cells.
The investigators genetically modified autologous T cells to express an anti-BCMA CAR and ligated it into a replication-incompetent gamma retrovirus. The T cells were stimulated with the anti-CD3 monoclonal antibody OKT3 before transduction, with the entire culture process taking 9 days from start to finish. T cells expressing this CAR recognize BCMA with great specificity, Dr. Kochenderfer observed.
The 12 patients received 300 mg/m2 of cyclophosphamide and 30 mg/m2 of fludarabine (Fludara) daily for 3 days before a single infusion of anti-BCMA CAR at dose levels of 0.3 x 106 to 9 x 106 T cells/kg. All patients had at least 3 prior therapies and normal organ function. Five had amyloid light chain only, 4 had immunoglobulin gamma disease, and 4 patients, including the complete responder, had immunoglobulin alpha disease.
It’s unclear why only a few patients responded to the CAR T cells, but dose level was likely a big factor and there is a lot of patient variability with T-cell therapies, Dr. Kochenderfer said at a press briefing.
“I wish we knew exactly why some patients respond and some don’t,” he added. “The CAR T cells have shown remarkable activity against really previously refractory diseases like acute lymphocytic leukemia,” noted Dr. George Daley of Harvard Medical School, Boston, in an interview at the meeting. “There’s been a lot of excitement that maybe we can extend the principles that work in leukemia to other types of diseases.”
The study is still ongoing and accruing patients, but a multicenter trial is also being initiated in myeloma with Bluebird Bio using “a closely related, but slightly different CAR,” Dr. Kochenderfer said.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ASH 2015
Key clinical point: The first clinical trial of a CAR targeting BCMA demonstrated strong antimyeloma activity in patients with at least 3 prior lines of myeloma therapy.
Major finding: Myeloma in plasma cells was reduced from 90% to 0% in one patient.
Data source: Phase I study in 12 patients with at least 3 prior lines of multiple myeloma therapy.
Disclosures: Dr. Kochenderfer reported consultancy with Bluebird Bio and off-label use of cyclophosphamide and fludarabine.

Anti-BCL2, CD20 combo safe, effective in untreated CLL
ORLANDO – An experimental combination of obinutuzumab and venetoclax appears safe as frontline therapy for patients with active, untreated chronic lymphocytic leukemia and comorbidities.
In the safety run-in portion of a phase 3, open-label trial comparing the combination of obinutuzumab (Gazyva) and the investigational Bcl-2 inhibitor venetoclax with obinutuzumab and its usual partner chlorambucil in 12 patients, only two of seven patients classified as being at high risk for the tumor lysis syndrome (TLS) developed laboratory-defined TLS, and no patients had clinical TLS.
The combination did not meet any of the pre-specified stopping criteria, and early data hinted at efficacy for the combination, said Dr. Kirsten Fischer, from the Center for Integrated Oncology at University Hospital Cologne in Germany.

“As with previous reports, our data confirm rapid and profound reduction in lymphocyte counts after the first dose obinutuzumab in all 12 [evaluable] patients,” she said at the American Society of Hematology annual meeting.
In the CLL 11 trial, investigators in the German CLL group previously showed that the combination of the anti-CD20 antibody obinutuzumab and chlorambucil resulted in improved overall survival compared to chlorambucil alone in patients with previously untreated CLL and coexisting medical conditions. The combination is approved in the United States for adults with treatment-naïve CLL.
Venetoclax has been shown to have good efficacy against relapsed/refractory CLL both as monotherapy and in combination with rituximab, prompting the investigators to explore it in combination with the rituximab follow-on drug obinutuzumab.
In the safety run-in phase of the CLL 14 trial, the investigators enrolled 13 adults (median age 75, range 59-88 years) with newly diagnosed, active confirmed CLL and coexisting medical conditions as determined either by a score greater 6 on the cumulative illness rating scale (CIRS) or by estimated creatinine clearance less than 70 mL/min.
The patients were treated with 6 cycles of obinutuzumab and venetoclax followed by 6 additional cycles of venetoclax. Obinutuzumab was administered intravenously 100 mg on day 1 and 900 mg on day 2, with the option to deliver 1,000 mg on day 1 instead, then 1,000 mg on days 8 and 15 of cycle 1, and 1,000 mg on day 1 for cycles 2-6.
The dose of venetoclax was titrated upward gradually, with doses of 20 mg, 50 mg, 100 mg, 200 mg, and up to 400 mg administered starting on day 22 of cycle 1.
Planned stopping criteria were one treatment-related death or a grade 4 adverse event related to clinical TLS despite prophylaxis as specified by the protocol.
At the time of data cutoff (October 2015), 12 patients had been on treatment for at least 4 weeks and had reached the maximum venetoclax dose. Two patients had reached 11 cycles, three had reached 10 cycles, and seven had reached 8 cycles.
Grade 1 or 2 adverse events in all 13 enrolled patients included infusion-related reactions in 8; infections in 6; diarrhea, hyperkalemia, and constipation in 5; nausea, dizziness and cough in 4; and fatigue, headache and pruritus in 3.
Grade 3 or 4 adverse events were neutropenia in five patients; infusion related reactions; syncope, thrombocytopenia and laboratory-defined TLS in two patients; and bradycardia, hyperglycemia, influenza, leucopenia, pyrexia, respiratory tract infection, and elevated transaminases in one patient each.
As noted, all 12 evaluable patients had rapid drops in absolute lymphocyte counts, and all but one had complete resolution of lymphadenopathy after three cycles, with the improvement maintained after six cycles. The remaining patient had a decrease to near normal after both three and six cycles.
Of the 12 patients, 11 had a partial response after three cycles, and the remaining patient had stable disease, for an overall response rate of 92%. The overall response rate after six cycles was 100%, with all patients having a partial response.
The data were sufficiently good to justify continuing with the randomized phase, which began in August 2015, Dr. Fischer noted.
The study is sponsored by Hoffman-La Roche and AbbVie. Dr. Fischer disclosed receiving travel grants from Hoffman-La Roche.
ORLANDO – An experimental combination of obinutuzumab and venetoclax appears safe as frontline therapy for patients with active, untreated chronic lymphocytic leukemia and comorbidities.
In the safety run-in portion of a phase 3, open-label trial comparing the combination of obinutuzumab (Gazyva) and the investigational Bcl-2 inhibitor venetoclax with obinutuzumab and its usual partner chlorambucil in 12 patients, only two of seven patients classified as being at high risk for the tumor lysis syndrome (TLS) developed laboratory-defined TLS, and no patients had clinical TLS.
The combination did not meet any of the pre-specified stopping criteria, and early data hinted at efficacy for the combination, said Dr. Kirsten Fischer, from the Center for Integrated Oncology at University Hospital Cologne in Germany.
“As with previous reports, our data confirm rapid and profound reduction in lymphocyte counts after the first dose obinutuzumab in all 12 [evaluable] patients,” she said at the American Society of Hematology annual meeting.
In the CLL 11 trial, investigators in the German CLL group previously showed that the combination of the anti-CD20 antibody obinutuzumab and chlorambucil resulted in improved overall survival compared to chlorambucil alone in patients with previously untreated CLL and coexisting medical conditions. The combination is approved in the United States for adults with treatment-naïve CLL.
Venetoclax has been shown to have good efficacy against relapsed/refractory CLL both as monotherapy and in combination with rituximab, prompting the investigators to explore it in combination with the rituximab follow-on drug obinutuzumab.
In the safety run-in phase of the CLL 14 trial, the investigators enrolled 13 adults (median age 75, range 59-88 years) with newly diagnosed, active confirmed CLL and coexisting medical conditions as determined either by a score greater 6 on the cumulative illness rating scale (CIRS) or by estimated creatinine clearance less than 70 mL/min.
The patients were treated with 6 cycles of obinutuzumab and venetoclax followed by 6 additional cycles of venetoclax. Obinutuzumab was administered intravenously 100 mg on day 1 and 900 mg on day 2, with the option to deliver 1,000 mg on day 1 instead, then 1,000 mg on days 8 and 15 of cycle 1, and 1,000 mg on day 1 for cycles 2-6.
The dose of venetoclax was titrated upward gradually, with doses of 20 mg, 50 mg, 100 mg, 200 mg, and up to 400 mg administered starting on day 22 of cycle 1.
Planned stopping criteria were one treatment-related death or a grade 4 adverse event related to clinical TLS despite prophylaxis as specified by the protocol.
At the time of data cutoff (October 2015), 12 patients had been on treatment for at least 4 weeks and had reached the maximum venetoclax dose. Two patients had reached 11 cycles, three had reached 10 cycles, and seven had reached 8 cycles.
Grade 1 or 2 adverse events in all 13 enrolled patients included infusion-related reactions in 8; infections in 6; diarrhea, hyperkalemia, and constipation in 5; nausea, dizziness and cough in 4; and fatigue, headache and pruritus in 3.
Grade 3 or 4 adverse events were neutropenia in five patients; infusion related reactions; syncope, thrombocytopenia and laboratory-defined TLS in two patients; and bradycardia, hyperglycemia, influenza, leucopenia, pyrexia, respiratory tract infection, and elevated transaminases in one patient each.
As noted, all 12 evaluable patients had rapid drops in absolute lymphocyte counts, and all but one had complete resolution of lymphadenopathy after three cycles, with the improvement maintained after six cycles. The remaining patient had a decrease to near normal after both three and six cycles.
Of the 12 patients, 11 had a partial response after three cycles, and the remaining patient had stable disease, for an overall response rate of 92%. The overall response rate after six cycles was 100%, with all patients having a partial response.
The data were sufficiently good to justify continuing with the randomized phase, which began in August 2015, Dr. Fischer noted.
The study is sponsored by Hoffman-La Roche and AbbVie. Dr. Fischer disclosed receiving travel grants from Hoffman-La Roche.
ORLANDO – An experimental combination of obinutuzumab and venetoclax appears safe as frontline therapy for patients with active, untreated chronic lymphocytic leukemia and comorbidities.
In the safety run-in portion of a phase 3, open-label trial comparing the combination of obinutuzumab (Gazyva) and the investigational Bcl-2 inhibitor venetoclax with obinutuzumab and its usual partner chlorambucil in 12 patients, only two of seven patients classified as being at high risk for the tumor lysis syndrome (TLS) developed laboratory-defined TLS, and no patients had clinical TLS.
The combination did not meet any of the pre-specified stopping criteria, and early data hinted at efficacy for the combination, said Dr. Kirsten Fischer, from the Center for Integrated Oncology at University Hospital Cologne in Germany.
“As with previous reports, our data confirm rapid and profound reduction in lymphocyte counts after the first dose obinutuzumab in all 12 [evaluable] patients,” she said at the American Society of Hematology annual meeting.
In the CLL 11 trial, investigators in the German CLL group previously showed that the combination of the anti-CD20 antibody obinutuzumab and chlorambucil resulted in improved overall survival compared to chlorambucil alone in patients with previously untreated CLL and coexisting medical conditions. The combination is approved in the United States for adults with treatment-naïve CLL.
Venetoclax has been shown to have good efficacy against relapsed/refractory CLL both as monotherapy and in combination with rituximab, prompting the investigators to explore it in combination with the rituximab follow-on drug obinutuzumab.
In the safety run-in phase of the CLL 14 trial, the investigators enrolled 13 adults (median age 75, range 59-88 years) with newly diagnosed, active confirmed CLL and coexisting medical conditions as determined either by a score greater 6 on the cumulative illness rating scale (CIRS) or by estimated creatinine clearance less than 70 mL/min.
The patients were treated with 6 cycles of obinutuzumab and venetoclax followed by 6 additional cycles of venetoclax. Obinutuzumab was administered intravenously 100 mg on day 1 and 900 mg on day 2, with the option to deliver 1,000 mg on day 1 instead, then 1,000 mg on days 8 and 15 of cycle 1, and 1,000 mg on day 1 for cycles 2-6.
The dose of venetoclax was titrated upward gradually, with doses of 20 mg, 50 mg, 100 mg, 200 mg, and up to 400 mg administered starting on day 22 of cycle 1.
Planned stopping criteria were one treatment-related death or a grade 4 adverse event related to clinical TLS despite prophylaxis as specified by the protocol.
At the time of data cutoff (October 2015), 12 patients had been on treatment for at least 4 weeks and had reached the maximum venetoclax dose. Two patients had reached 11 cycles, three had reached 10 cycles, and seven had reached 8 cycles.
Grade 1 or 2 adverse events in all 13 enrolled patients included infusion-related reactions in 8; infections in 6; diarrhea, hyperkalemia, and constipation in 5; nausea, dizziness and cough in 4; and fatigue, headache and pruritus in 3.
Grade 3 or 4 adverse events were neutropenia in five patients; infusion related reactions; syncope, thrombocytopenia and laboratory-defined TLS in two patients; and bradycardia, hyperglycemia, influenza, leucopenia, pyrexia, respiratory tract infection, and elevated transaminases in one patient each.
As noted, all 12 evaluable patients had rapid drops in absolute lymphocyte counts, and all but one had complete resolution of lymphadenopathy after three cycles, with the improvement maintained after six cycles. The remaining patient had a decrease to near normal after both three and six cycles.
Of the 12 patients, 11 had a partial response after three cycles, and the remaining patient had stable disease, for an overall response rate of 92%. The overall response rate after six cycles was 100%, with all patients having a partial response.
The data were sufficiently good to justify continuing with the randomized phase, which began in August 2015, Dr. Fischer noted.
The study is sponsored by Hoffman-La Roche and AbbVie. Dr. Fischer disclosed receiving travel grants from Hoffman-La Roche.
AT ASH 2015
Key clinical point: Patients with CLL and comorbidities were able to tolerate an experimental regimen of obinutuzumab and venetoclax.
Major finding: Two of 12 evaluable patients had evidence of laboratory-defined but not clinical tumor lysis syndrome.
Data source: Safety run-in phase of a randomized phase 3 trial with 13 patients with chronic lymphocytic leukemia and comorbidities.
Disclosures: The study is sponsored by Hoffman-La Roche and AbbVie. Dr. Fischer disclosed receiving travel grants from Hoffman-La Roche.

Bystander CPR rising in children with cardiac arrest
ORLANDO – Bystander CPR was provided in 49% of U.S. cases of pediatric out-of-hospital cardiac arrest during 2013-2014, a major improvement over the 35% rate in a prior study 15 years ago, Dr. Maryam Y. Naim reported at the American Heart Association scientific sessions.
She presented an analysis of 2,176 out-of-hospital cardiac arrests (OHCA) in patients up to age 18 years who were included in the Cardiac Arrest Registry to Enhance Survival (CARES), the nation’s largest OHCA registry. Patients with traumatic OHCA and those whose bystander CPR (BCPR) was provided by a health care professional weren’t included.

Overall, the rate of neurologically favorable survival in pediatric recipients of BCPR was 11%, compared with 7% when BCPR wasn’t provided. But the results were far more impressive in the 14% of cardiac arrests that occurred outside the home, where the rate of neurologically favorable survival in BCPR recipients was 34%, more than twice the 15% figure for nonrecipients, according to Dr. Naim, a pediatrician and cardiac intensivist at Children’s Hospital of Philadelphia and the University of Pennsylvania.
Infants accounted for 47% of all pediatric OHCA, and in these youngest patients BCPR was of no benefit.
“The most common etiology of cardiac arrest in infants is sudden infant death syndrome. These are children who are found unresponsive in their cribs, and sometimes they’ve been dead a long time. We need to find something different for this population: perhaps developing a monitor to signal when an infant stops breathing or the heart rate goes down,” she said.
The fact that the BCPR rate in pediatric OHCA has climbed to 49% speaks well for public health efforts to improve education and awareness. Of those who received BCPR during 2013 and 2014, half got compression-only CPR, suggesting increasing adherence to the 2010 AHA guidelines for CPR and emergency cardiovascular care, which emphasized compression-only CPR as a viable alternative to conventional CPR, Dr. Naim added.
Her study highlighted a racial disparity in the application of BCPR in children and adolescents: Sixty percent of white youths with OHCA received BCPR, compared with 42% of blacks and 48% of Hispanics.
“About 70% of all bystander CPR was provided by a family member at home. So there’s really an opportunity there, especially in minority communities, to further increase education and awareness about bystander CPR, teaching family members to do it and also how to call 911 to start the chain of response,” she said.
Dr. Naim reported having no financial conflicts regarding her study.
ORLANDO – Bystander CPR was provided in 49% of U.S. cases of pediatric out-of-hospital cardiac arrest during 2013-2014, a major improvement over the 35% rate in a prior study 15 years ago, Dr. Maryam Y. Naim reported at the American Heart Association scientific sessions.
She presented an analysis of 2,176 out-of-hospital cardiac arrests (OHCA) in patients up to age 18 years who were included in the Cardiac Arrest Registry to Enhance Survival (CARES), the nation’s largest OHCA registry. Patients with traumatic OHCA and those whose bystander CPR (BCPR) was provided by a health care professional weren’t included.
Overall, the rate of neurologically favorable survival in pediatric recipients of BCPR was 11%, compared with 7% when BCPR wasn’t provided. But the results were far more impressive in the 14% of cardiac arrests that occurred outside the home, where the rate of neurologically favorable survival in BCPR recipients was 34%, more than twice the 15% figure for nonrecipients, according to Dr. Naim, a pediatrician and cardiac intensivist at Children’s Hospital of Philadelphia and the University of Pennsylvania.
Infants accounted for 47% of all pediatric OHCA, and in these youngest patients BCPR was of no benefit.
“The most common etiology of cardiac arrest in infants is sudden infant death syndrome. These are children who are found unresponsive in their cribs, and sometimes they’ve been dead a long time. We need to find something different for this population: perhaps developing a monitor to signal when an infant stops breathing or the heart rate goes down,” she said.
The fact that the BCPR rate in pediatric OHCA has climbed to 49% speaks well for public health efforts to improve education and awareness. Of those who received BCPR during 2013 and 2014, half got compression-only CPR, suggesting increasing adherence to the 2010 AHA guidelines for CPR and emergency cardiovascular care, which emphasized compression-only CPR as a viable alternative to conventional CPR, Dr. Naim added.
Her study highlighted a racial disparity in the application of BCPR in children and adolescents: Sixty percent of white youths with OHCA received BCPR, compared with 42% of blacks and 48% of Hispanics.
“About 70% of all bystander CPR was provided by a family member at home. So there’s really an opportunity there, especially in minority communities, to further increase education and awareness about bystander CPR, teaching family members to do it and also how to call 911 to start the chain of response,” she said.
Dr. Naim reported having no financial conflicts regarding her study.
ORLANDO – Bystander CPR was provided in 49% of U.S. cases of pediatric out-of-hospital cardiac arrest during 2013-2014, a major improvement over the 35% rate in a prior study 15 years ago, Dr. Maryam Y. Naim reported at the American Heart Association scientific sessions.
She presented an analysis of 2,176 out-of-hospital cardiac arrests (OHCA) in patients up to age 18 years who were included in the Cardiac Arrest Registry to Enhance Survival (CARES), the nation’s largest OHCA registry. Patients with traumatic OHCA and those whose bystander CPR (BCPR) was provided by a health care professional weren’t included.
Overall, the rate of neurologically favorable survival in pediatric recipients of BCPR was 11%, compared with 7% when BCPR wasn’t provided. But the results were far more impressive in the 14% of cardiac arrests that occurred outside the home, where the rate of neurologically favorable survival in BCPR recipients was 34%, more than twice the 15% figure for nonrecipients, according to Dr. Naim, a pediatrician and cardiac intensivist at Children’s Hospital of Philadelphia and the University of Pennsylvania.
Infants accounted for 47% of all pediatric OHCA, and in these youngest patients BCPR was of no benefit.
“The most common etiology of cardiac arrest in infants is sudden infant death syndrome. These are children who are found unresponsive in their cribs, and sometimes they’ve been dead a long time. We need to find something different for this population: perhaps developing a monitor to signal when an infant stops breathing or the heart rate goes down,” she said.
The fact that the BCPR rate in pediatric OHCA has climbed to 49% speaks well for public health efforts to improve education and awareness. Of those who received BCPR during 2013 and 2014, half got compression-only CPR, suggesting increasing adherence to the 2010 AHA guidelines for CPR and emergency cardiovascular care, which emphasized compression-only CPR as a viable alternative to conventional CPR, Dr. Naim added.
Her study highlighted a racial disparity in the application of BCPR in children and adolescents: Sixty percent of white youths with OHCA received BCPR, compared with 42% of blacks and 48% of Hispanics.
“About 70% of all bystander CPR was provided by a family member at home. So there’s really an opportunity there, especially in minority communities, to further increase education and awareness about bystander CPR, teaching family members to do it and also how to call 911 to start the chain of response,” she said.
Dr. Naim reported having no financial conflicts regarding her study.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: Nearly half of all children and adolescents with out-of-hospital cardiac arrest in 2013-2014 got bystander CPR.
Major finding: The rate of neurologically favorable survival in pediatric patients with out-of-hospital cardiac arrest who receive bystander CPR is 34%, compared with 15% in those who don’t get the intervention.
Data source: An analysis of 2,176 out-of-hospital cardiac arrests in the Cardiac Arrest Registry to Enhance Survival during 2013 and 2014.
Disclosures: The presenter reported having no financial conflicts regarding her study.

Does cannabis cause psychosis? A brief review of the evidence
As more and more states consider legalizing marijuana for recreational use, the widely held belief that cannabis is associated with few serious health consequences has been challenged by many medical and substance use professionals. One potential risk that has been discussed is the possibility that cannabis use increases the risk for psychotic symptoms that may be long lasting and develop into schizophrenia. The data, however, have not been completely consistent and often are methodologically flawed, leading proponents of legalization to downplay this possible risk. This debate has even made its way to prominent science journals such as Nature where scholars have presented opposing views (Nature. 2015 Sep 24;525[7570]:S14and Nature. 2015 Nov 19;527[7578]:305).
These divergent opinions can lead to some confusion and hesitancy on the part of pediatricians who may be asked to offer an opinion about the dangers of cannabis use to individual patients and families during this time of public debate. Thus, this column will attempt to offer a brief overview and synthesis of the evidence that cannabis plays a causal role in the progression of psychotic disorders.

A recent review of the subject examined 10 epidemiological studies that have now been performed on the association between cannabis and psychotic disorders. Overall, a nearly 50% increased risk of psychosis was found among cannabis users, compared to nonusers (Biol Psychiatry. 2015 Aug 12. pii: S0006-3223[15]00647-2). This association rises among heavier cannabis users (Lancet. 2007 Jul 28;370[9584]:319-28). Because all of these longitudinal studies were observational in nature, however, proving causation in the face of association has remained challenging. Many of these studies have attempted to control for baseline psychotic symptoms to address the “reverse causation hypothesis,” which posits that early psychotic symptoms leads to cannabis use rather than the other way around. It is also worth pointing out that the inevitable limitations and potential biases of these studies could potentially lead to both overestimation and underestimation of the actual risk.
Putting all of this together, the authors concluded that “there is a strong body of epidemiologic evidence to support the view that regular or heavy cannabis use increases the risk of developing psychotic disorders that persist beyond the direct effects of exogenous cannabinoids.” In making this conclusion, despite the inherent uncertainties of interpreting observational studies, the authors describe a number of lines of evidence that support the likelihood of a causal connection. These include the following:

• The well-known fact that acute intoxication of cannabis can produce transient psychotic symptoms.
• The replicated finding that there is a dose-dependent response between amount of cannabis use and psychosis.
• An increased risk of psychosis among cannabis users who carry specific risk genes (Biol Psychiatry. 2012 Nov 15;72[10]:811-6).
• Increasing evidence that the more potent marijuana that is available now may be associated with additional risk.
• The finding that the link between cannabis and psychosis is not equal for all age groups, but may be stronger for adolescents.
One line of argument against a causal role of cannabis in the development of psychotic disorders is that the rate of schizophrenia has remained relatively flat over the years that cannabis use has increased. Countering that assertion, however, Large and colleagues pointed out that some studies do show increasing rates of schizophrenia (Nature. 2015 Nov 19;527[7578]:305). Further, it is somewhat precarious to conclude that a possible risk factor is not consequential when it moves in a different direction than a multifactorial disorder such as schizophrenia. Lead toxicity, for example, is an accepted risk factor for attention-deficit/hyperactivity disorder (ADHD), yet exposure has been decreasing while rates of ADHD climb.
Overall, the data appear to be strengthening that cannabis does play a causal role in the development of psychosis and psychotic disorders. This risk is combined with data showing links between cannabis use and decreased IQ, academic underachievement, car accidents, and use of other types of drugs (Addiction. 2015 Jan;110[1]:19-35). These dangers need to be articulated in discussions about the wisdom of legalizing cannabis at the state and federal level.
Dr. Rettew is associate professor of psychiatry and pediatrics at the University of Vermont, Burlington. He said he has no relevant financial disclosures. Follow him on Twitter @pedipsych. E-mail him at [email protected].
As more and more states consider legalizing marijuana for recreational use, the widely held belief that cannabis is associated with few serious health consequences has been challenged by many medical and substance use professionals. One potential risk that has been discussed is the possibility that cannabis use increases the risk for psychotic symptoms that may be long lasting and develop into schizophrenia. The data, however, have not been completely consistent and often are methodologically flawed, leading proponents of legalization to downplay this possible risk. This debate has even made its way to prominent science journals such as Nature where scholars have presented opposing views (Nature. 2015 Sep 24;525[7570]:S14and Nature. 2015 Nov 19;527[7578]:305).
These divergent opinions can lead to some confusion and hesitancy on the part of pediatricians who may be asked to offer an opinion about the dangers of cannabis use to individual patients and families during this time of public debate. Thus, this column will attempt to offer a brief overview and synthesis of the evidence that cannabis plays a causal role in the progression of psychotic disorders.
A recent review of the subject examined 10 epidemiological studies that have now been performed on the association between cannabis and psychotic disorders. Overall, a nearly 50% increased risk of psychosis was found among cannabis users, compared to nonusers (Biol Psychiatry. 2015 Aug 12. pii: S0006-3223[15]00647-2). This association rises among heavier cannabis users (Lancet. 2007 Jul 28;370[9584]:319-28). Because all of these longitudinal studies were observational in nature, however, proving causation in the face of association has remained challenging. Many of these studies have attempted to control for baseline psychotic symptoms to address the “reverse causation hypothesis,” which posits that early psychotic symptoms leads to cannabis use rather than the other way around. It is also worth pointing out that the inevitable limitations and potential biases of these studies could potentially lead to both overestimation and underestimation of the actual risk.
Putting all of this together, the authors concluded that “there is a strong body of epidemiologic evidence to support the view that regular or heavy cannabis use increases the risk of developing psychotic disorders that persist beyond the direct effects of exogenous cannabinoids.” In making this conclusion, despite the inherent uncertainties of interpreting observational studies, the authors describe a number of lines of evidence that support the likelihood of a causal connection. These include the following:
• The well-known fact that acute intoxication of cannabis can produce transient psychotic symptoms.
• The replicated finding that there is a dose-dependent response between amount of cannabis use and psychosis.
• An increased risk of psychosis among cannabis users who carry specific risk genes (Biol Psychiatry. 2012 Nov 15;72[10]:811-6).
• Increasing evidence that the more potent marijuana that is available now may be associated with additional risk.
• The finding that the link between cannabis and psychosis is not equal for all age groups, but may be stronger for adolescents.
One line of argument against a causal role of cannabis in the development of psychotic disorders is that the rate of schizophrenia has remained relatively flat over the years that cannabis use has increased. Countering that assertion, however, Large and colleagues pointed out that some studies do show increasing rates of schizophrenia (Nature. 2015 Nov 19;527[7578]:305). Further, it is somewhat precarious to conclude that a possible risk factor is not consequential when it moves in a different direction than a multifactorial disorder such as schizophrenia. Lead toxicity, for example, is an accepted risk factor for attention-deficit/hyperactivity disorder (ADHD), yet exposure has been decreasing while rates of ADHD climb.
Overall, the data appear to be strengthening that cannabis does play a causal role in the development of psychosis and psychotic disorders. This risk is combined with data showing links between cannabis use and decreased IQ, academic underachievement, car accidents, and use of other types of drugs (Addiction. 2015 Jan;110[1]:19-35). These dangers need to be articulated in discussions about the wisdom of legalizing cannabis at the state and federal level.
Dr. Rettew is associate professor of psychiatry and pediatrics at the University of Vermont, Burlington. He said he has no relevant financial disclosures. Follow him on Twitter @pedipsych. E-mail him at [email protected].
As more and more states consider legalizing marijuana for recreational use, the widely held belief that cannabis is associated with few serious health consequences has been challenged by many medical and substance use professionals. One potential risk that has been discussed is the possibility that cannabis use increases the risk for psychotic symptoms that may be long lasting and develop into schizophrenia. The data, however, have not been completely consistent and often are methodologically flawed, leading proponents of legalization to downplay this possible risk. This debate has even made its way to prominent science journals such as Nature where scholars have presented opposing views (Nature. 2015 Sep 24;525[7570]:S14and Nature. 2015 Nov 19;527[7578]:305).
These divergent opinions can lead to some confusion and hesitancy on the part of pediatricians who may be asked to offer an opinion about the dangers of cannabis use to individual patients and families during this time of public debate. Thus, this column will attempt to offer a brief overview and synthesis of the evidence that cannabis plays a causal role in the progression of psychotic disorders.
A recent review of the subject examined 10 epidemiological studies that have now been performed on the association between cannabis and psychotic disorders. Overall, a nearly 50% increased risk of psychosis was found among cannabis users, compared to nonusers (Biol Psychiatry. 2015 Aug 12. pii: S0006-3223[15]00647-2). This association rises among heavier cannabis users (Lancet. 2007 Jul 28;370[9584]:319-28). Because all of these longitudinal studies were observational in nature, however, proving causation in the face of association has remained challenging. Many of these studies have attempted to control for baseline psychotic symptoms to address the “reverse causation hypothesis,” which posits that early psychotic symptoms leads to cannabis use rather than the other way around. It is also worth pointing out that the inevitable limitations and potential biases of these studies could potentially lead to both overestimation and underestimation of the actual risk.
Putting all of this together, the authors concluded that “there is a strong body of epidemiologic evidence to support the view that regular or heavy cannabis use increases the risk of developing psychotic disorders that persist beyond the direct effects of exogenous cannabinoids.” In making this conclusion, despite the inherent uncertainties of interpreting observational studies, the authors describe a number of lines of evidence that support the likelihood of a causal connection. These include the following:
• The well-known fact that acute intoxication of cannabis can produce transient psychotic symptoms.
• The replicated finding that there is a dose-dependent response between amount of cannabis use and psychosis.
• An increased risk of psychosis among cannabis users who carry specific risk genes (Biol Psychiatry. 2012 Nov 15;72[10]:811-6).
• Increasing evidence that the more potent marijuana that is available now may be associated with additional risk.
• The finding that the link between cannabis and psychosis is not equal for all age groups, but may be stronger for adolescents.
One line of argument against a causal role of cannabis in the development of psychotic disorders is that the rate of schizophrenia has remained relatively flat over the years that cannabis use has increased. Countering that assertion, however, Large and colleagues pointed out that some studies do show increasing rates of schizophrenia (Nature. 2015 Nov 19;527[7578]:305). Further, it is somewhat precarious to conclude that a possible risk factor is not consequential when it moves in a different direction than a multifactorial disorder such as schizophrenia. Lead toxicity, for example, is an accepted risk factor for attention-deficit/hyperactivity disorder (ADHD), yet exposure has been decreasing while rates of ADHD climb.
Overall, the data appear to be strengthening that cannabis does play a causal role in the development of psychosis and psychotic disorders. This risk is combined with data showing links between cannabis use and decreased IQ, academic underachievement, car accidents, and use of other types of drugs (Addiction. 2015 Jan;110[1]:19-35). These dangers need to be articulated in discussions about the wisdom of legalizing cannabis at the state and federal level.
Dr. Rettew is associate professor of psychiatry and pediatrics at the University of Vermont, Burlington. He said he has no relevant financial disclosures. Follow him on Twitter @pedipsych. E-mail him at [email protected].

A Look at Speakers, Educational Tracks Planned for Hospital Medicine 2016

Get ready for hospital medicine’s main event—Hospital Medicine 2016 (HM16). SHM remains at the forefront of healthcare, leading the charge to provide the best care for hospitalized patients. We invite you to join us on the sunny shores of San Diego from March 6-9, to learn about the latest advances in hospital medicine and connect with over 3,000 hospital-based professionals.
On hand this year will be some world-class faculty, who will examine today’s issues and challenge everyone to be a part of the solution. HM16’s renowned speakers are leaders in the field. We proudly welcome:
Karen DeSalvo, MD, MPH, MSc

Acting Assistant Secretary for Health, U.S. Department of Health and Human Services
Dr. DeSalvo has made a tremendous impact on quality in healthcare through direct patient care, medical education, policy and administrative roles, research, and public service. She has received many honors, including recognition as a “Woman of Excellence in Health Care” by the Louisiana Legislative Women’s Caucus. Join her for her featured address, “Public Health 3.0, the Role of the Hospitalist and Hospital.”
Robert M. Wachter, MD, MHM

Professor and interim chair, department of medicine, University of California San Francisco, author of “The Digital Doctor: Hope, Hype and Harm at the Dawn of Medicine’s Computer Age,” and of the HM-focused blog, “Wachter’s World."
Dr. Wachter, national thought leader in healthcare quality improvement, in his always entertaining style, will address important and interesting work going on in medical leadership, teamwork, artificial intelligence, and physician evaluation. Join him for his closing keynote address: “Hospital Medicine Turns 20: Celebrating the Past While Charting a New Course.”
In addition to these much anticipated keynotes, be sure to check out new tracks for HM16, including:
• Co-Management/Perioperative Medicine: This hospitalist core competency increases in complexity, yet many physicians were not even taught the basics in residency. This new track explores the perioperative and consultative medicine questions that challenge hospitalists on a daily basis.
• Health IT for Hospitalists: Technology has changed the practice of medicine. The new Health IT for Hospitalists track focuses on topics to help frontline clinicians perform better by using technology, while helping those with leadership roles in Health IT obtain tools to make them more effective. HM2016 will mark the beginning of an annual update on the best mobile apps in the healthcare sector.
• Post-Acute Care: This track targets two audiences: 1) mainstream hospitalists, who increasingly are being asked to assume responsibility for the full episode of care, including post-acute care services after discharge; 2) hospitalists in programs who have assumed responsibility for post-acute care services, either in skilled nursing facilities, independent rehabilitation facilities, or long-term acute care hospitals. This track will provide information on how hospitalists can build and operate a successful post-acute care practice.
These are sessions you won’t want to miss! If you haven’t registered, now is the time. SHM members save $325 on HM16 registration, and if you’re new to SHM, you can receive one free year of membership after registering.
Get the scoop on all things HM16, and register now, at www.hospitalmedicine2016.org.
See you in San Diego! TH
Brett Radler is SHM’s communications coordinator.
Get ready for hospital medicine’s main event—Hospital Medicine 2016 (HM16). SHM remains at the forefront of healthcare, leading the charge to provide the best care for hospitalized patients. We invite you to join us on the sunny shores of San Diego from March 6-9, to learn about the latest advances in hospital medicine and connect with over 3,000 hospital-based professionals.
On hand this year will be some world-class faculty, who will examine today’s issues and challenge everyone to be a part of the solution. HM16’s renowned speakers are leaders in the field. We proudly welcome:
Karen DeSalvo, MD, MPH, MSc
Acting Assistant Secretary for Health, U.S. Department of Health and Human Services
Dr. DeSalvo has made a tremendous impact on quality in healthcare through direct patient care, medical education, policy and administrative roles, research, and public service. She has received many honors, including recognition as a “Woman of Excellence in Health Care” by the Louisiana Legislative Women’s Caucus. Join her for her featured address, “Public Health 3.0, the Role of the Hospitalist and Hospital.”
Robert M. Wachter, MD, MHM
Professor and interim chair, department of medicine, University of California San Francisco, author of “The Digital Doctor: Hope, Hype and Harm at the Dawn of Medicine’s Computer Age,” and of the HM-focused blog, “Wachter’s World."
Dr. Wachter, national thought leader in healthcare quality improvement, in his always entertaining style, will address important and interesting work going on in medical leadership, teamwork, artificial intelligence, and physician evaluation. Join him for his closing keynote address: “Hospital Medicine Turns 20: Celebrating the Past While Charting a New Course.”
In addition to these much anticipated keynotes, be sure to check out new tracks for HM16, including:
• Co-Management/Perioperative Medicine: This hospitalist core competency increases in complexity, yet many physicians were not even taught the basics in residency. This new track explores the perioperative and consultative medicine questions that challenge hospitalists on a daily basis.
• Health IT for Hospitalists: Technology has changed the practice of medicine. The new Health IT for Hospitalists track focuses on topics to help frontline clinicians perform better by using technology, while helping those with leadership roles in Health IT obtain tools to make them more effective. HM2016 will mark the beginning of an annual update on the best mobile apps in the healthcare sector.
• Post-Acute Care: This track targets two audiences: 1) mainstream hospitalists, who increasingly are being asked to assume responsibility for the full episode of care, including post-acute care services after discharge; 2) hospitalists in programs who have assumed responsibility for post-acute care services, either in skilled nursing facilities, independent rehabilitation facilities, or long-term acute care hospitals. This track will provide information on how hospitalists can build and operate a successful post-acute care practice.
These are sessions you won’t want to miss! If you haven’t registered, now is the time. SHM members save $325 on HM16 registration, and if you’re new to SHM, you can receive one free year of membership after registering.
Get the scoop on all things HM16, and register now, at www.hospitalmedicine2016.org.
See you in San Diego! TH
Brett Radler is SHM’s communications coordinator.
Get ready for hospital medicine’s main event—Hospital Medicine 2016 (HM16). SHM remains at the forefront of healthcare, leading the charge to provide the best care for hospitalized patients. We invite you to join us on the sunny shores of San Diego from March 6-9, to learn about the latest advances in hospital medicine and connect with over 3,000 hospital-based professionals.
On hand this year will be some world-class faculty, who will examine today’s issues and challenge everyone to be a part of the solution. HM16’s renowned speakers are leaders in the field. We proudly welcome:
Karen DeSalvo, MD, MPH, MSc
Acting Assistant Secretary for Health, U.S. Department of Health and Human Services
Dr. DeSalvo has made a tremendous impact on quality in healthcare through direct patient care, medical education, policy and administrative roles, research, and public service. She has received many honors, including recognition as a “Woman of Excellence in Health Care” by the Louisiana Legislative Women’s Caucus. Join her for her featured address, “Public Health 3.0, the Role of the Hospitalist and Hospital.”
Robert M. Wachter, MD, MHM
Professor and interim chair, department of medicine, University of California San Francisco, author of “The Digital Doctor: Hope, Hype and Harm at the Dawn of Medicine’s Computer Age,” and of the HM-focused blog, “Wachter’s World."
Dr. Wachter, national thought leader in healthcare quality improvement, in his always entertaining style, will address important and interesting work going on in medical leadership, teamwork, artificial intelligence, and physician evaluation. Join him for his closing keynote address: “Hospital Medicine Turns 20: Celebrating the Past While Charting a New Course.”
In addition to these much anticipated keynotes, be sure to check out new tracks for HM16, including:
• Co-Management/Perioperative Medicine: This hospitalist core competency increases in complexity, yet many physicians were not even taught the basics in residency. This new track explores the perioperative and consultative medicine questions that challenge hospitalists on a daily basis.
• Health IT for Hospitalists: Technology has changed the practice of medicine. The new Health IT for Hospitalists track focuses on topics to help frontline clinicians perform better by using technology, while helping those with leadership roles in Health IT obtain tools to make them more effective. HM2016 will mark the beginning of an annual update on the best mobile apps in the healthcare sector.
• Post-Acute Care: This track targets two audiences: 1) mainstream hospitalists, who increasingly are being asked to assume responsibility for the full episode of care, including post-acute care services after discharge; 2) hospitalists in programs who have assumed responsibility for post-acute care services, either in skilled nursing facilities, independent rehabilitation facilities, or long-term acute care hospitals. This track will provide information on how hospitalists can build and operate a successful post-acute care practice.
These are sessions you won’t want to miss! If you haven’t registered, now is the time. SHM members save $325 on HM16 registration, and if you’re new to SHM, you can receive one free year of membership after registering.
Get the scoop on all things HM16, and register now, at www.hospitalmedicine2016.org.
See you in San Diego! TH
Brett Radler is SHM’s communications coordinator.

Venetoclax produces deep responses in ultra-high-risk CLL

Photo courtesy of ASH
ORLANDO, FL—The pivotal phase 2 study of venetoclax monotherapy in patients with relapsed/refractory 17p-deleted chronic lymphocytic leukemia (CLL) has achieved unprecedented deep responses, according to investigators.
More than 10% of patients had a complete response (CR), complete response with incomplete blood count recovery (CRi), or near partial response (nPR), as confirmed by an independent review committee (IRC).
And more than 20% of responders became negative for minimal residual disease (MRD).
Venetoclax is an orally bioavailable, selective BCL-2 inhibitor that directly induces apoptosis in CLL cells independent of p53.
The US Food and Drug Administration granted venetoclax breakthrough therapy designation for relapsed/refractory CLL earlier this year.
“Patients with a 17p deletion in CLL have very poor prognosis,” said Stephan Stilgenbauer, MD, of University of Ulm in Germany, “and limited treatment options.”
The median progression-free survival (PFS) with frontline chemoimmunotherapy in this population is less than 12 months.
The first-in-human study of venetoclax, which was recently published in NEJM, showed a 79% overall response rate (ORR) in relapsed/refractory CLL patients.
Dr Stilgenbauer presented the pivotal phase 2 results at the 2015 ASH Annual Meeting as LBA-6.
Study overview
The primary objective of the trial was ORR by independent review committee. The secondary endpoints were CR/PR rates, time to first response, duration of response, PFS, overall survival (OS), and safety.
Investigators also included the exploratory endpoint of MRD as determined by flow cytometry with a sensitivity of less than 10-4.
Patients had to have an ECOG score of 2 or less, an absolute neutrophil count of 1000/μL or greater, a platelet count of 40,000/mm3 or higher, and a hemoglobin count of at least 8 g/dL. They also had to have a creatinine clearance of 50 mL/min or more.
“With regard to performance status, blood counts, and creatinine clearance,” Dr Stilgenbauer said, “inclusion criteria were relatively liberal, allowing patients with comorbidity on the trial.”
Patients were excluded if they had prior allogeneic stem cell transplantation, Richter’s transformation, uncontrolled autoimmune cytopenia, other malignancy, or major organ dysfunction.
Trial design
Patients received an oral dose of venetoclax once daily continuously until disease progression or discontinuation for another reason.
Because tumor lysis syndrome (TLS) was a concern, investigators devised a step-wise weekly ramp-up with risk-based prophylaxis to mitigate TLS.
Patients started on a dose of 20 mg on day 1. If they did not experience any electrolyte abnormalities, they received a 50 mg daily dose for the rest of the first week, escalating to 100 mg, 200 mg, and to the target dose of 400 mg daily on subsequent weeks. Patients continued on 400 mg daily for the remainder of the study.
The investigators assessed response using iwCLL 2008 criteria with monthly physical exams and blood counts, CT scans to confirm clinical response at week 36, and a bone marrow biopsy to confirm CR.
Patient population and disposition
Investigators enrolled 107 patients with a median age of 67 (range, 37–85). Seventy (65%) were male.
Patients had a median of 2 prior therapies (range, 1–10): 54 (50%) had prior bendamustine and 38 (70%) were refractory to it; 78 (73%) had prior fludarabine and 34 (44%) were refractory to it; and 90 (84%) had a prior CD20 monoclonal antibody.
About half (52%) were ECOG grade 1, 53% had 1 or more nodes 5 cm or larger, and 51% had absolute lymphocyte (ALC) levels 25 x 109/L or higher.
Eighty-two percent of patients were in the medium and high TLS risk categories, slightly less than half were Rai stage III or IV, and 81% were IGHV unmutated.
As of the data lock on April 30, 2015, patients remained a median of 12.1 months on study (range, 0.03–21.5). Seventy are still active on venetoclax, and 37 discontinued the treatment.
Eleven patients discontinued due to Richter’s transformation, 11 due to CLL progression, and 9 due to adverse events. Three patients proceeded to stem cell transplant, 2 withdrew consent, and 1 was noncompliant.
Eighteen patients died, 14 due to disease progression.
Response
Eighty-five patients responded, for an ORR of 79.4% by IRC. Eight patients (7.5%) achieved a CR or CRi, 3 (2.8%) had an nPR, and 74 (69.2%) had a PR. Twenty-two patients (20.6%) had no response.
Twenty-five of 48 patients had no evidence of CLL in their bone marrow by immunohistochemistry, and 18 of 45 patients assessed were MRD-negative in the peripheral blood.
Reduction in lymphocytosis “was quite a universal phenomenon across this trial,” Dr Stilgenbauer said. Only 4 patients of 87 with baseline lymphocytosis failed to reduce their lymphocyte count to below 4 x 109/L, the usual threshold for a CR. And the median time to normalization was 22 days (range, 2–122).
Eighty-nine of 96 patients had 50% or more reduction in their nodal size in a median of 2.7 months (range, 0.7–8.4).
The median time to first response was 0.8 months (range, 0.1–8.1), and the median time to CR/CRi was 8.2 months (range, 3.0–16.3).
“And this number still appears to evolve over the duration of the trial,” Dr Stilgenbauer said.
The median duration of response has not yet been reached. But investigators estimated that of the 85 responders, 84.7% would maintain their response at 12 months, 100% of patients in the CR/CRi and nPR groups would maintain their response, and 94.4% of patients who were MRD-negative would maintain their response.
The median PFS and OS have not been reached. The PFS estimate for 12 months was 72.0%, and the OS estimate was 86.7%.
Adverse events
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%).
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%).
Dr Stilgenbauer pointed out that 22.4% of patients had neutropenia at baseline. Neutropenia was managed with dose interruption or reduction, G-CSF, and/or antibiotics.
Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher.
“The types of infections were the usual expected ones,” Dr Stilgenbauer said.
Laboratory TLS occurred in 5 patients exclusively during the ramp-up period. Two required a dose interruption of 1 day each. There were no clinical TLS events.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
The investigators concluded that venetoclax offers a favorable risk-benefit profile. The risk of TLS can be effectively mitigated with no clinical TLS, and the incidence of neutropenia and infection are similar to frontline chemoimmunotherapy.
“Venetoclax may provide an attractive treatment option for 17p-deleted CLL as monotherapy or as a component of novel combination strategies,” Dr Stilgenbauer said.
AbbVie and Genentech, collaborators in the development of venetoclax, provided financial support for the study design, study conduct, analysis, data interpretation, writing, and review. ![]()
Photo courtesy of ASH
ORLANDO, FL—The pivotal phase 2 study of venetoclax monotherapy in patients with relapsed/refractory 17p-deleted chronic lymphocytic leukemia (CLL) has achieved unprecedented deep responses, according to investigators.
More than 10% of patients had a complete response (CR), complete response with incomplete blood count recovery (CRi), or near partial response (nPR), as confirmed by an independent review committee (IRC).
And more than 20% of responders became negative for minimal residual disease (MRD).
Venetoclax is an orally bioavailable, selective BCL-2 inhibitor that directly induces apoptosis in CLL cells independent of p53.
The US Food and Drug Administration granted venetoclax breakthrough therapy designation for relapsed/refractory CLL earlier this year.
“Patients with a 17p deletion in CLL have very poor prognosis,” said Stephan Stilgenbauer, MD, of University of Ulm in Germany, “and limited treatment options.”
The median progression-free survival (PFS) with frontline chemoimmunotherapy in this population is less than 12 months.
The first-in-human study of venetoclax, which was recently published in NEJM, showed a 79% overall response rate (ORR) in relapsed/refractory CLL patients.
Dr Stilgenbauer presented the pivotal phase 2 results at the 2015 ASH Annual Meeting as LBA-6.
Study overview
The primary objective of the trial was ORR by independent review committee. The secondary endpoints were CR/PR rates, time to first response, duration of response, PFS, overall survival (OS), and safety.
Investigators also included the exploratory endpoint of MRD as determined by flow cytometry with a sensitivity of less than 10-4.
Patients had to have an ECOG score of 2 or less, an absolute neutrophil count of 1000/μL or greater, a platelet count of 40,000/mm3 or higher, and a hemoglobin count of at least 8 g/dL. They also had to have a creatinine clearance of 50 mL/min or more.
“With regard to performance status, blood counts, and creatinine clearance,” Dr Stilgenbauer said, “inclusion criteria were relatively liberal, allowing patients with comorbidity on the trial.”
Patients were excluded if they had prior allogeneic stem cell transplantation, Richter’s transformation, uncontrolled autoimmune cytopenia, other malignancy, or major organ dysfunction.
Trial design
Patients received an oral dose of venetoclax once daily continuously until disease progression or discontinuation for another reason.
Because tumor lysis syndrome (TLS) was a concern, investigators devised a step-wise weekly ramp-up with risk-based prophylaxis to mitigate TLS.
Patients started on a dose of 20 mg on day 1. If they did not experience any electrolyte abnormalities, they received a 50 mg daily dose for the rest of the first week, escalating to 100 mg, 200 mg, and to the target dose of 400 mg daily on subsequent weeks. Patients continued on 400 mg daily for the remainder of the study.
The investigators assessed response using iwCLL 2008 criteria with monthly physical exams and blood counts, CT scans to confirm clinical response at week 36, and a bone marrow biopsy to confirm CR.
Patient population and disposition
Investigators enrolled 107 patients with a median age of 67 (range, 37–85). Seventy (65%) were male.
Patients had a median of 2 prior therapies (range, 1–10): 54 (50%) had prior bendamustine and 38 (70%) were refractory to it; 78 (73%) had prior fludarabine and 34 (44%) were refractory to it; and 90 (84%) had a prior CD20 monoclonal antibody.
About half (52%) were ECOG grade 1, 53% had 1 or more nodes 5 cm or larger, and 51% had absolute lymphocyte (ALC) levels 25 x 109/L or higher.
Eighty-two percent of patients were in the medium and high TLS risk categories, slightly less than half were Rai stage III or IV, and 81% were IGHV unmutated.
As of the data lock on April 30, 2015, patients remained a median of 12.1 months on study (range, 0.03–21.5). Seventy are still active on venetoclax, and 37 discontinued the treatment.
Eleven patients discontinued due to Richter’s transformation, 11 due to CLL progression, and 9 due to adverse events. Three patients proceeded to stem cell transplant, 2 withdrew consent, and 1 was noncompliant.
Eighteen patients died, 14 due to disease progression.
Response
Eighty-five patients responded, for an ORR of 79.4% by IRC. Eight patients (7.5%) achieved a CR or CRi, 3 (2.8%) had an nPR, and 74 (69.2%) had a PR. Twenty-two patients (20.6%) had no response.
Twenty-five of 48 patients had no evidence of CLL in their bone marrow by immunohistochemistry, and 18 of 45 patients assessed were MRD-negative in the peripheral blood.
Reduction in lymphocytosis “was quite a universal phenomenon across this trial,” Dr Stilgenbauer said. Only 4 patients of 87 with baseline lymphocytosis failed to reduce their lymphocyte count to below 4 x 109/L, the usual threshold for a CR. And the median time to normalization was 22 days (range, 2–122).
Eighty-nine of 96 patients had 50% or more reduction in their nodal size in a median of 2.7 months (range, 0.7–8.4).
The median time to first response was 0.8 months (range, 0.1–8.1), and the median time to CR/CRi was 8.2 months (range, 3.0–16.3).
“And this number still appears to evolve over the duration of the trial,” Dr Stilgenbauer said.
The median duration of response has not yet been reached. But investigators estimated that of the 85 responders, 84.7% would maintain their response at 12 months, 100% of patients in the CR/CRi and nPR groups would maintain their response, and 94.4% of patients who were MRD-negative would maintain their response.
The median PFS and OS have not been reached. The PFS estimate for 12 months was 72.0%, and the OS estimate was 86.7%.
Adverse events
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%).
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%).
Dr Stilgenbauer pointed out that 22.4% of patients had neutropenia at baseline. Neutropenia was managed with dose interruption or reduction, G-CSF, and/or antibiotics.
Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher.
“The types of infections were the usual expected ones,” Dr Stilgenbauer said.
Laboratory TLS occurred in 5 patients exclusively during the ramp-up period. Two required a dose interruption of 1 day each. There were no clinical TLS events.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
The investigators concluded that venetoclax offers a favorable risk-benefit profile. The risk of TLS can be effectively mitigated with no clinical TLS, and the incidence of neutropenia and infection are similar to frontline chemoimmunotherapy.
“Venetoclax may provide an attractive treatment option for 17p-deleted CLL as monotherapy or as a component of novel combination strategies,” Dr Stilgenbauer said.
AbbVie and Genentech, collaborators in the development of venetoclax, provided financial support for the study design, study conduct, analysis, data interpretation, writing, and review. ![]()
Photo courtesy of ASH
ORLANDO, FL—The pivotal phase 2 study of venetoclax monotherapy in patients with relapsed/refractory 17p-deleted chronic lymphocytic leukemia (CLL) has achieved unprecedented deep responses, according to investigators.
More than 10% of patients had a complete response (CR), complete response with incomplete blood count recovery (CRi), or near partial response (nPR), as confirmed by an independent review committee (IRC).
And more than 20% of responders became negative for minimal residual disease (MRD).
Venetoclax is an orally bioavailable, selective BCL-2 inhibitor that directly induces apoptosis in CLL cells independent of p53.
The US Food and Drug Administration granted venetoclax breakthrough therapy designation for relapsed/refractory CLL earlier this year.
“Patients with a 17p deletion in CLL have very poor prognosis,” said Stephan Stilgenbauer, MD, of University of Ulm in Germany, “and limited treatment options.”
The median progression-free survival (PFS) with frontline chemoimmunotherapy in this population is less than 12 months.
The first-in-human study of venetoclax, which was recently published in NEJM, showed a 79% overall response rate (ORR) in relapsed/refractory CLL patients.
Dr Stilgenbauer presented the pivotal phase 2 results at the 2015 ASH Annual Meeting as LBA-6.
Study overview
The primary objective of the trial was ORR by independent review committee. The secondary endpoints were CR/PR rates, time to first response, duration of response, PFS, overall survival (OS), and safety.
Investigators also included the exploratory endpoint of MRD as determined by flow cytometry with a sensitivity of less than 10-4.
Patients had to have an ECOG score of 2 or less, an absolute neutrophil count of 1000/μL or greater, a platelet count of 40,000/mm3 or higher, and a hemoglobin count of at least 8 g/dL. They also had to have a creatinine clearance of 50 mL/min or more.
“With regard to performance status, blood counts, and creatinine clearance,” Dr Stilgenbauer said, “inclusion criteria were relatively liberal, allowing patients with comorbidity on the trial.”
Patients were excluded if they had prior allogeneic stem cell transplantation, Richter’s transformation, uncontrolled autoimmune cytopenia, other malignancy, or major organ dysfunction.
Trial design
Patients received an oral dose of venetoclax once daily continuously until disease progression or discontinuation for another reason.
Because tumor lysis syndrome (TLS) was a concern, investigators devised a step-wise weekly ramp-up with risk-based prophylaxis to mitigate TLS.
Patients started on a dose of 20 mg on day 1. If they did not experience any electrolyte abnormalities, they received a 50 mg daily dose for the rest of the first week, escalating to 100 mg, 200 mg, and to the target dose of 400 mg daily on subsequent weeks. Patients continued on 400 mg daily for the remainder of the study.
The investigators assessed response using iwCLL 2008 criteria with monthly physical exams and blood counts, CT scans to confirm clinical response at week 36, and a bone marrow biopsy to confirm CR.
Patient population and disposition
Investigators enrolled 107 patients with a median age of 67 (range, 37–85). Seventy (65%) were male.
Patients had a median of 2 prior therapies (range, 1–10): 54 (50%) had prior bendamustine and 38 (70%) were refractory to it; 78 (73%) had prior fludarabine and 34 (44%) were refractory to it; and 90 (84%) had a prior CD20 monoclonal antibody.
About half (52%) were ECOG grade 1, 53% had 1 or more nodes 5 cm or larger, and 51% had absolute lymphocyte (ALC) levels 25 x 109/L or higher.
Eighty-two percent of patients were in the medium and high TLS risk categories, slightly less than half were Rai stage III or IV, and 81% were IGHV unmutated.
As of the data lock on April 30, 2015, patients remained a median of 12.1 months on study (range, 0.03–21.5). Seventy are still active on venetoclax, and 37 discontinued the treatment.
Eleven patients discontinued due to Richter’s transformation, 11 due to CLL progression, and 9 due to adverse events. Three patients proceeded to stem cell transplant, 2 withdrew consent, and 1 was noncompliant.
Eighteen patients died, 14 due to disease progression.
Response
Eighty-five patients responded, for an ORR of 79.4% by IRC. Eight patients (7.5%) achieved a CR or CRi, 3 (2.8%) had an nPR, and 74 (69.2%) had a PR. Twenty-two patients (20.6%) had no response.
Twenty-five of 48 patients had no evidence of CLL in their bone marrow by immunohistochemistry, and 18 of 45 patients assessed were MRD-negative in the peripheral blood.
Reduction in lymphocytosis “was quite a universal phenomenon across this trial,” Dr Stilgenbauer said. Only 4 patients of 87 with baseline lymphocytosis failed to reduce their lymphocyte count to below 4 x 109/L, the usual threshold for a CR. And the median time to normalization was 22 days (range, 2–122).
Eighty-nine of 96 patients had 50% or more reduction in their nodal size in a median of 2.7 months (range, 0.7–8.4).
The median time to first response was 0.8 months (range, 0.1–8.1), and the median time to CR/CRi was 8.2 months (range, 3.0–16.3).
“And this number still appears to evolve over the duration of the trial,” Dr Stilgenbauer said.
The median duration of response has not yet been reached. But investigators estimated that of the 85 responders, 84.7% would maintain their response at 12 months, 100% of patients in the CR/CRi and nPR groups would maintain their response, and 94.4% of patients who were MRD-negative would maintain their response.
The median PFS and OS have not been reached. The PFS estimate for 12 months was 72.0%, and the OS estimate was 86.7%.
Adverse events
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%).
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%).
Dr Stilgenbauer pointed out that 22.4% of patients had neutropenia at baseline. Neutropenia was managed with dose interruption or reduction, G-CSF, and/or antibiotics.
Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher.
“The types of infections were the usual expected ones,” Dr Stilgenbauer said.
Laboratory TLS occurred in 5 patients exclusively during the ramp-up period. Two required a dose interruption of 1 day each. There were no clinical TLS events.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
The investigators concluded that venetoclax offers a favorable risk-benefit profile. The risk of TLS can be effectively mitigated with no clinical TLS, and the incidence of neutropenia and infection are similar to frontline chemoimmunotherapy.
“Venetoclax may provide an attractive treatment option for 17p-deleted CLL as monotherapy or as a component of novel combination strategies,” Dr Stilgenbauer said.
AbbVie and Genentech, collaborators in the development of venetoclax, provided financial support for the study design, study conduct, analysis, data interpretation, writing, and review. ![]()
CAR T cells persist for 3 years in young ALL patients
Photo courtesy of Penn Medicine
ORLANDO, FL—CTL019, a CD19 chimeric antigen receptor (CAR) T-cell therapy, can persist for 3 years or longer in children and young adults with relapsed/refractory acute lymphoblastic leukemia (ALL), according to the latest results of a pilot study.
This suggests CTL019 can offer long-term disease control without subsequent therapy, such as stem cell transplant, said study author Stephan Grupp, MD, PhD, of the University of Pennsylvania in Philadelphia.
Dr Grupp presented these results at the 2015 ASH Annual Meeting (abstract 681*).
Previously, Dr Grupp and his colleagues reported that CTL019 led to durable antitumor activity, including sustained complete responses (CRs) in adults and children with ALL (ASH 2012, NEJM 2013, ASH 2013, NEJM 2014, ASH 2014).
At this year’s ASH meeting, he reported on outcomes and longer follow-up of the first 59 patients with relapsed/refractory ALL treated in a pilot trial. The patients had a median age of 11 years.
The median follow-up was 12 months. Fifty-five patients (93%) achieved a CR at 1 month. Six patients went on to receive a transplant, and 1 patient went on to receive donor lymphocyte infusion.
The relapse-free survival was 76% at 6 months and 55% at 12 months. Overall survival was 79% at 12 months.
“There were no relapses past 1 year,” Dr Grupp said, noting that 18 patients beyond 1 year are in remission, and 13 patients have not received further therapy.
“We are able to get patients into remission,” he said, adding that response is similar at high and low disease burden.
“We see massive proliferation of CAR T cells. Prolonged CTL019 persistence is detected by flow cytometry. About 70% of patients maintain CAR T cells.”
Resistance was seen in 7% of patients due to failure of T cells to proliferate. One-third of recurrences occurred in those with CD19-positive relapse and two-thirds with CD19-negative relapse.
Dr Grupp noted that CTL019 has an impact on central nervous system (CNS) disease.
“We are able to control CNS disease,” he said. “We found no CNS relapses in patients who were treated, and 98% of them still have CTL019 in the cerebral spinal fluid.”
B-cell aplasia persists beyond 3.5 years in all responding patients. This is managed by immunoglobulin replacement.
“Patients in remission and with B-cell aplasia at 1 year are still alive at 2 to 3 years,” Dr Grupp said. “We do not know how long this will last and if B cells will recover.”
Severe cytokine release syndrome (CRS), observed in 88% of patients, is the principal toxicity. This is controlled with anti-IL6 therapy. Patients who are less likely to have CRS are those with a lower disease burden.
“IL-6 is a major player but does not predict CRS; it only correlates strongly with it,” Dr Grupp said.
Some toxicity is also associated with macrophage activation syndrome and neurotoxicity.
CTL019 was invented at The University of Pennsylvania but has been licensed to Novartis. Most of the researchers involved in this study reported research funding and/or consultancy payments from Novartis, and 1 researcher is employed by the company. ![]()
*Data in the abstract differ from the presentation.
Photo courtesy of Penn Medicine
ORLANDO, FL—CTL019, a CD19 chimeric antigen receptor (CAR) T-cell therapy, can persist for 3 years or longer in children and young adults with relapsed/refractory acute lymphoblastic leukemia (ALL), according to the latest results of a pilot study.
This suggests CTL019 can offer long-term disease control without subsequent therapy, such as stem cell transplant, said study author Stephan Grupp, MD, PhD, of the University of Pennsylvania in Philadelphia.
Dr Grupp presented these results at the 2015 ASH Annual Meeting (abstract 681*).
Previously, Dr Grupp and his colleagues reported that CTL019 led to durable antitumor activity, including sustained complete responses (CRs) in adults and children with ALL (ASH 2012, NEJM 2013, ASH 2013, NEJM 2014, ASH 2014).
At this year’s ASH meeting, he reported on outcomes and longer follow-up of the first 59 patients with relapsed/refractory ALL treated in a pilot trial. The patients had a median age of 11 years.
The median follow-up was 12 months. Fifty-five patients (93%) achieved a CR at 1 month. Six patients went on to receive a transplant, and 1 patient went on to receive donor lymphocyte infusion.
The relapse-free survival was 76% at 6 months and 55% at 12 months. Overall survival was 79% at 12 months.
“There were no relapses past 1 year,” Dr Grupp said, noting that 18 patients beyond 1 year are in remission, and 13 patients have not received further therapy.
“We are able to get patients into remission,” he said, adding that response is similar at high and low disease burden.
“We see massive proliferation of CAR T cells. Prolonged CTL019 persistence is detected by flow cytometry. About 70% of patients maintain CAR T cells.”
Resistance was seen in 7% of patients due to failure of T cells to proliferate. One-third of recurrences occurred in those with CD19-positive relapse and two-thirds with CD19-negative relapse.
Dr Grupp noted that CTL019 has an impact on central nervous system (CNS) disease.
“We are able to control CNS disease,” he said. “We found no CNS relapses in patients who were treated, and 98% of them still have CTL019 in the cerebral spinal fluid.”
B-cell aplasia persists beyond 3.5 years in all responding patients. This is managed by immunoglobulin replacement.
“Patients in remission and with B-cell aplasia at 1 year are still alive at 2 to 3 years,” Dr Grupp said. “We do not know how long this will last and if B cells will recover.”
Severe cytokine release syndrome (CRS), observed in 88% of patients, is the principal toxicity. This is controlled with anti-IL6 therapy. Patients who are less likely to have CRS are those with a lower disease burden.
“IL-6 is a major player but does not predict CRS; it only correlates strongly with it,” Dr Grupp said.
Some toxicity is also associated with macrophage activation syndrome and neurotoxicity.
CTL019 was invented at The University of Pennsylvania but has been licensed to Novartis. Most of the researchers involved in this study reported research funding and/or consultancy payments from Novartis, and 1 researcher is employed by the company. ![]()
*Data in the abstract differ from the presentation.
Photo courtesy of Penn Medicine
ORLANDO, FL—CTL019, a CD19 chimeric antigen receptor (CAR) T-cell therapy, can persist for 3 years or longer in children and young adults with relapsed/refractory acute lymphoblastic leukemia (ALL), according to the latest results of a pilot study.
This suggests CTL019 can offer long-term disease control without subsequent therapy, such as stem cell transplant, said study author Stephan Grupp, MD, PhD, of the University of Pennsylvania in Philadelphia.
Dr Grupp presented these results at the 2015 ASH Annual Meeting (abstract 681*).
Previously, Dr Grupp and his colleagues reported that CTL019 led to durable antitumor activity, including sustained complete responses (CRs) in adults and children with ALL (ASH 2012, NEJM 2013, ASH 2013, NEJM 2014, ASH 2014).
At this year’s ASH meeting, he reported on outcomes and longer follow-up of the first 59 patients with relapsed/refractory ALL treated in a pilot trial. The patients had a median age of 11 years.
The median follow-up was 12 months. Fifty-five patients (93%) achieved a CR at 1 month. Six patients went on to receive a transplant, and 1 patient went on to receive donor lymphocyte infusion.
The relapse-free survival was 76% at 6 months and 55% at 12 months. Overall survival was 79% at 12 months.
“There were no relapses past 1 year,” Dr Grupp said, noting that 18 patients beyond 1 year are in remission, and 13 patients have not received further therapy.
“We are able to get patients into remission,” he said, adding that response is similar at high and low disease burden.
“We see massive proliferation of CAR T cells. Prolonged CTL019 persistence is detected by flow cytometry. About 70% of patients maintain CAR T cells.”
Resistance was seen in 7% of patients due to failure of T cells to proliferate. One-third of recurrences occurred in those with CD19-positive relapse and two-thirds with CD19-negative relapse.
Dr Grupp noted that CTL019 has an impact on central nervous system (CNS) disease.
“We are able to control CNS disease,” he said. “We found no CNS relapses in patients who were treated, and 98% of them still have CTL019 in the cerebral spinal fluid.”
B-cell aplasia persists beyond 3.5 years in all responding patients. This is managed by immunoglobulin replacement.
“Patients in remission and with B-cell aplasia at 1 year are still alive at 2 to 3 years,” Dr Grupp said. “We do not know how long this will last and if B cells will recover.”
Severe cytokine release syndrome (CRS), observed in 88% of patients, is the principal toxicity. This is controlled with anti-IL6 therapy. Patients who are less likely to have CRS are those with a lower disease burden.
“IL-6 is a major player but does not predict CRS; it only correlates strongly with it,” Dr Grupp said.
Some toxicity is also associated with macrophage activation syndrome and neurotoxicity.
CTL019 was invented at The University of Pennsylvania but has been licensed to Novartis. Most of the researchers involved in this study reported research funding and/or consultancy payments from Novartis, and 1 researcher is employed by the company. ![]()
*Data in the abstract differ from the presentation.