User login
Academic hospitals offer better AML survival
ORLANDO – Patients with acute myeloid leukemia (AML) initially treated at an academic center lived significantly longer than those treated at nonacademic centers, a database analysis shows.
Median overall survival increased from 7 months at a nonacademic center to 12.6 months at an academic center (P less than .001).
One-year overall survival rates were also significantly better at 51% vs. 39% (P less than .001).

The difference remained significant even after controlling for important confounders including age, comorbidity burden, receipt of chemotherapy, transplant, and delay between diagnosis and treatment, Mr. Smith Giri reported at the annual meeting of the American Society of Hematology.
“From a policy perspective, it may be useful to know whether these results are due to higher volume of cases, more advanced technology, expanded role of specialists, or greater, round-the-clock availability of resident physicians,” said Mr. Giri of the University of Tennessee Health Science Center in Memphis.
Prior studies in cancer have suggested better overall survival among breast cancer patients treated at academic centers, but this is the first study looking at outcomes in AML, the most common acute leukemia in adults.
Using the National Cancer Database Participant User File, the investigators identified 7,823 patients with AML who received their initial therapy at the reporting facility from 1998 to 2011. The database collects information from more than 1,500 Commission on Cancer (CoC)–accredited facilities. Of the 7,823 patients, 4,681 (60%) were treated at an AC (academic/research program) and 3,142 at a non-AC (community cancer program/comprehensive community cancer program).
Patients treated at an AC were significantly younger than those treated at a non-AC (median 62 years vs. 67 years), tended to be of nonwhite race, less educated, have a lower income, and more comorbidities.
Receipt of chemotherapy (97.4% vs. 94.5%) and transplant (9% vs. 2.4%) were significantly higher at an AC than a non-AC (both P less than .001).
Kaplan Meier survival curves suggested disparate survival curves between the two groups, mainly within the first 5 years of follow-up (P less than .001), Mr. Giri said.
In multivariate model analysis, the non-AC group had significantly worse risk adjusted 30-day mortality than the AC group (odds ratio, 1.52; 95% confidence interval 1.33-1.74; P less than .001) and worse overall survival (hazard ratio, 1.13; 95% CI 1.07-1.19; P less than .001).
The study (Ab. 533) findings should be interpreted with caution because of its limitations, including the lack of information on AML risk type in the database, the fact that administrative datasets are prone to coding errors, and because the analysis did not adjust for hospital volume, which has been shown to affect survival, he said. Also, because there are more than 3,500 non–Coc approved hospitals, the sample may not be representative of overall U.S. hospitals.
During a discussion of the results, Mr. Giri acknowledged that patients treated at academic centers may have greater access to clinical trials and experimental agents. Future analyses should also distinguish patients with a diagnosis of acute promyelocytic leukemia, a distinct subset of AML.
ORLANDO – Patients with acute myeloid leukemia (AML) initially treated at an academic center lived significantly longer than those treated at nonacademic centers, a database analysis shows.
Median overall survival increased from 7 months at a nonacademic center to 12.6 months at an academic center (P less than .001).
One-year overall survival rates were also significantly better at 51% vs. 39% (P less than .001).
The difference remained significant even after controlling for important confounders including age, comorbidity burden, receipt of chemotherapy, transplant, and delay between diagnosis and treatment, Mr. Smith Giri reported at the annual meeting of the American Society of Hematology.
“From a policy perspective, it may be useful to know whether these results are due to higher volume of cases, more advanced technology, expanded role of specialists, or greater, round-the-clock availability of resident physicians,” said Mr. Giri of the University of Tennessee Health Science Center in Memphis.
Prior studies in cancer have suggested better overall survival among breast cancer patients treated at academic centers, but this is the first study looking at outcomes in AML, the most common acute leukemia in adults.
Using the National Cancer Database Participant User File, the investigators identified 7,823 patients with AML who received their initial therapy at the reporting facility from 1998 to 2011. The database collects information from more than 1,500 Commission on Cancer (CoC)–accredited facilities. Of the 7,823 patients, 4,681 (60%) were treated at an AC (academic/research program) and 3,142 at a non-AC (community cancer program/comprehensive community cancer program).
Patients treated at an AC were significantly younger than those treated at a non-AC (median 62 years vs. 67 years), tended to be of nonwhite race, less educated, have a lower income, and more comorbidities.
Receipt of chemotherapy (97.4% vs. 94.5%) and transplant (9% vs. 2.4%) were significantly higher at an AC than a non-AC (both P less than .001).
Kaplan Meier survival curves suggested disparate survival curves between the two groups, mainly within the first 5 years of follow-up (P less than .001), Mr. Giri said.
In multivariate model analysis, the non-AC group had significantly worse risk adjusted 30-day mortality than the AC group (odds ratio, 1.52; 95% confidence interval 1.33-1.74; P less than .001) and worse overall survival (hazard ratio, 1.13; 95% CI 1.07-1.19; P less than .001).
The study (Ab. 533) findings should be interpreted with caution because of its limitations, including the lack of information on AML risk type in the database, the fact that administrative datasets are prone to coding errors, and because the analysis did not adjust for hospital volume, which has been shown to affect survival, he said. Also, because there are more than 3,500 non–Coc approved hospitals, the sample may not be representative of overall U.S. hospitals.
During a discussion of the results, Mr. Giri acknowledged that patients treated at academic centers may have greater access to clinical trials and experimental agents. Future analyses should also distinguish patients with a diagnosis of acute promyelocytic leukemia, a distinct subset of AML.
ORLANDO – Patients with acute myeloid leukemia (AML) initially treated at an academic center lived significantly longer than those treated at nonacademic centers, a database analysis shows.
Median overall survival increased from 7 months at a nonacademic center to 12.6 months at an academic center (P less than .001).
One-year overall survival rates were also significantly better at 51% vs. 39% (P less than .001).
The difference remained significant even after controlling for important confounders including age, comorbidity burden, receipt of chemotherapy, transplant, and delay between diagnosis and treatment, Mr. Smith Giri reported at the annual meeting of the American Society of Hematology.
“From a policy perspective, it may be useful to know whether these results are due to higher volume of cases, more advanced technology, expanded role of specialists, or greater, round-the-clock availability of resident physicians,” said Mr. Giri of the University of Tennessee Health Science Center in Memphis.
Prior studies in cancer have suggested better overall survival among breast cancer patients treated at academic centers, but this is the first study looking at outcomes in AML, the most common acute leukemia in adults.
Using the National Cancer Database Participant User File, the investigators identified 7,823 patients with AML who received their initial therapy at the reporting facility from 1998 to 2011. The database collects information from more than 1,500 Commission on Cancer (CoC)–accredited facilities. Of the 7,823 patients, 4,681 (60%) were treated at an AC (academic/research program) and 3,142 at a non-AC (community cancer program/comprehensive community cancer program).
Patients treated at an AC were significantly younger than those treated at a non-AC (median 62 years vs. 67 years), tended to be of nonwhite race, less educated, have a lower income, and more comorbidities.
Receipt of chemotherapy (97.4% vs. 94.5%) and transplant (9% vs. 2.4%) were significantly higher at an AC than a non-AC (both P less than .001).
Kaplan Meier survival curves suggested disparate survival curves between the two groups, mainly within the first 5 years of follow-up (P less than .001), Mr. Giri said.
In multivariate model analysis, the non-AC group had significantly worse risk adjusted 30-day mortality than the AC group (odds ratio, 1.52; 95% confidence interval 1.33-1.74; P less than .001) and worse overall survival (hazard ratio, 1.13; 95% CI 1.07-1.19; P less than .001).
The study (Ab. 533) findings should be interpreted with caution because of its limitations, including the lack of information on AML risk type in the database, the fact that administrative datasets are prone to coding errors, and because the analysis did not adjust for hospital volume, which has been shown to affect survival, he said. Also, because there are more than 3,500 non–Coc approved hospitals, the sample may not be representative of overall U.S. hospitals.
During a discussion of the results, Mr. Giri acknowledged that patients treated at academic centers may have greater access to clinical trials and experimental agents. Future analyses should also distinguish patients with a diagnosis of acute promyelocytic leukemia, a distinct subset of AML.
AT ASH 2015
Key clinical point: Academic hospitals tend to have better short- and long-term mortality for patients with AML than nonacademic hospitals.
Major finding: Median overall survival was 12.6 months at an academic center vs. 7 months at a nonacademic center (P less than .001).
Data source: Retrospective analysis of 7,823 patients with AML.
Disclosures: The research was supported in part by a grant from the University of Nebraska Medical Center. The National Cancer Database is jointly sponsored by the American College of Surgeons and American Cancer Society. Mr. Giri reported having no relevant conflicts of interest.

AHA/ACC: Consensus recommendations for young athletes with congenital heart disease
Most children and young adult patients with congenital heart disease can and should engage in some form of physical activity and should avoid a sedentary lifestyle, according to a task force scientific statement from the American Heart Association and the American College of Cardiology (AHA/ACC).
This recommendation comes despite the fears of sudden cardiac death (SCD) in young athletes, which formed the initial impetus of the entire series of task force reports.
The recommended level of sports participation for patients with treated or untreated congenital heart defect, however, should consider the training and the competitive aspects of the sport itself and must be individualized to the patient. This means taking into account the patient’s current functional status, history of surgery, and the presence of implanted cardiac devices, according to the report by Dr. George F. Van Hare of Washington University, St. Louis, and his colleagues, which was published online in the Journal of the American College of Cardiology.

The report breaks down its specific recommendations based upon the various types of congenital heart defect (CHD). Full details and nuances of the recommendations and their specific levels of evidence for each individual condition and the many variants can be found in the online publication. Below is a brief and selected summary for some of the most common defects and some of those most pertinent to sudden cardiac death in young athletes.
Simple shunting lesions (atrial septal defect, ventricular septal defect, patent ductus arteriosus): Treated and untreated
In addressing the three most common subtypes of CHD – ventricular septal defect (VSD, 34%), atrial septal defect (ASD, 13%), and patent ductus arteriosus (PDA, 10%) – the committee found no data that children with these lesions are related to acknowledged episodes of sudden cardiac death (SCD). This applied whether the defects were closed or remained open. “With rare exceptions, patients with hemodynamically insignificant CHD such as VSD, ASD, and PDA may participate competitively in all sports,” it concluded. These recommendations fall under class I; level of evidence C for almost all of these patients, according to the writing committee.
Congenital coronary anomalies: Treated and untreated
Anomalies of coronary arteries are the second-most commonly identified structural causes of SCD in competitive athletes, accounting for about 17% of such deaths in the United States, according to the report. The vast majority of sudden deaths associated with coronary anomalies occur during or shortly after exercise. Despite being less commonly represented in patients, among athletes who have died suddenly, anomalous origin of the left main or left anterior descending coronary artery from the right sinus of Valsalva is far more prevalent. In addition, SCDs are most strongly associated with the pattern in which the anomalous left coronary artery passes between the aorta and main pulmonary artery. Recommended return to intense athletic activities is only to be permitted at least 3 months after surgery, and with a demonstration of the absence of ischemia on postoperative stress testing, with evidence levels depending on the type of anomaly. Of note, in contrast, the committee indicated that athletes with an anomalous origin of a right coronary artery from the left sinus of Valsalva should simply be evaluated by an exercise stress test, and for those without symptoms or a positive exercise stress test, permission to compete can be considered after adequate counseling (class IIa; level of evidence C).
Pulmonary valve stenosis: Treated and untreated
The committee determined that athletes with mild pulmonary stenosis (PS) and normal right ventricular (RV) function can participate in all competitive sports, although annual reevaluation also is recommended (class I; level of evidence B). In addition, athletes treated by operation or balloon valvuloplasty who have achieved adequate relief of PS (gradient less than 40 mm Hg by Doppler) can participate in all competitive sports (class I; level of evidence B). Other patients should be restricted to low-intensity sports, according to the committee.
Aortic valve stenosis: Treated and untreated
Children and adolescents with aortic stenosis (AS) are differentiated between those with mild, moderate, and severe AS by physical examination, ECG, and Doppler echocardiography. In all cases, regardless of the degree of stenosis, patients with a history of fatigue, light-headedness, dizziness, syncope, chest pain, or pallor on exercise deserve a full evaluation. Annual re-evaluation is required for all patients with AS because the disease can progress. Patients with severe AS are at risk of sudden death, particularly with exercise. The committee determined that athletes with mild AS can participate in all competitive sports (class I; level of evidence B), but that athletes with severe AS should be restricted from all competitive sports, with the possible exception of low-intensity sports (class III; level of evidence B).
Coarctation of the aorta: Treated and untreated
Before a decision is made regarding exercise participation, a detailed evaluation should be conducted, including a physical examination, ECG, chest radiograph, exercise testing, transthoracic echocardiographic evaluation of the aortic valve and aorta, and either magnetic resonance imaging or computed tomography angiography, according to the committee. The determination as to the level of sports participation permitted requires a complex assessment of these various test results and can range from full participation in the case of the least affected to restrictions to low-intensity sports in those more severely affected.
Cyanotic CHD, including tetralogy of Fallot
Full clinical assessment, including laboratory and exercise testing, should be considered before any physical activity because this population is at very high risk of sudden death, according to the committee. Recommendations are complex and depend on the level of repair and its success, but, in general, significant restrictions are recommended for all but the most effectively treated patients.
Transposition of the great arteries after atrial switch (Mustard or Senning operation)
This is a population highly at risk, according to the committee. They appear to have a unique response to exercise with reports that a high proportion of sudden death events occur during exertion. In addition, evidence of exercise-induced arrhythmias on routine clinical testing has not been shown to reliably predict exercise-induced SCD events. Although recommendations vary, including strong restrictions for many, at best the most successful of these patients should only be considered for low- to moderate-intensity competitive sports, according to the committee.
Other conditions assessed and evaluated by the committee included congenitally corrected TGA, TGA after the arterial switch, Fontan procedure, elevated pulmonary vascular resistence in CHD, ventricular dysfunction after CHD surgery, and Ebstein anomaly of the tricuspid valve.
In all cases, complete physical assessment of these patients is recommended, especially due to the often highly individualized nature of the patient’s presentation of these conditions and the variety and variability of interventions that may have been performed. Such differentials make recommendations regarding sports participation a complex calculus, which the committee attempts to provide, listing whatever evidence is available.
The majority of these patients, however, will not be considered for the highest levels of competitive sports participation. Although, in almost all cases, the need for physical activity as a contributor to patient health and well-being is stressed at whatever level of performance is possible.
The report ”Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 4: congenital heart disease: a scientific statement from the American Heart Association and American College of Cardiology,” was prepared by Dr. Van Hare and his colleagues on behalf of the American Heart Association Electrocardiography and Arrhythmias Committee of the Council on Clinical Cardiology, Council on Cardiovascular Disease in the Young, Council on Cardiovascular and Stroke Nursing, Council on Functional Genomics and Translational Biology, and the American College of Cardiology (doi: 10.1016/j.jacc.2015.09.032).
This report is one of the assessments and recommendations of 15 task forces on eligibility and disqualification recommendations for young athletes, nine of which are disease or multidisease related. The other six task forces focus on a variety of relevant topics and issues regarding the risks of young athletes on the field, including screening, the use of automated external defibrillators on the field, the use of dietary supplements and performance-enhancing drugs, sudden death, and the medical-legal perspectives involved.
All 15 task force reports were simultaneously published online in the Journal of the American College of Cardiology and the journal Circulation.
Dr. Van Hare and all but one member of the writing group had no disclosures. One member disclosed consultant/advisory committee associations with a variety of medical device companies.
For many busy clinicians, societal guidelines, task force recommendations, expert consensus statements, and similar authoritative tomes are resources that are scarcely ever read carefully. This is likely not a reflection of the inherent value of such documents, but rather related to the observation that updated guidelines generally reflect, at most, a small change from predecessor versions. (It also should be mentioned that many such contributions are fairly heavy going for even the most determined reader.)
Occasionally, however, a new guideline may signal a dramatic shift in practice, and the recently published AHA/ACC Scientific Statement on Eligibility and Disqualification Recommendations for Competitive Athletes with Cardiovascular Abnormalities (Congenital Heart Disease) contains such a change.

|
Dr. Robert Jaquiss |
In particular, the new recommendation suggests that athletes with anomalous aortic origin of the right coronary from the left coronary sinus, who have neither symptoms nor a positive stress test, may be allowed to participate in competitive athletics without undergoing surgical repair. As before, those with anomalous left coronary should not be allowed to participate until after surgical treatment.
Prior guidelines suggested that all patients, both anomalous left from right sinus and right from left sinus, be restricted prior to surgery. Because anomalous right coronary is five to six times more common than anomalous left coronary and because it is certainly much less ominous, the previous “one size fits all” approach almost certainly resulted in overtreatment, unnecessary restriction of participation, or both. Furthermore, because anomalous aortic of a coronary artery is so common, occurring in 0.1%-0.2% of the population (300,000 to 600,000 people in the United States), many thousands of competitive athletes will be impacted by the changed guidelines.
Most cardiologists, surgeons, and, most especially, patients will welcome the updated recommendations. Nonetheless, it must be emphasized that anomalous coronary arteries, even anomalous right coronary arteries, may indicate an increased risk of sudden death and that a complete assessment, including stress testing when feasible, and thorough discussion with expert clinicians is still absolutely necessary for such patients and their families.
Dr. Robert Jaquiss of Duke University, Durham, N.C., is the congenital heart section associate medical editor for Thoracic Surgery News.
For many busy clinicians, societal guidelines, task force recommendations, expert consensus statements, and similar authoritative tomes are resources that are scarcely ever read carefully. This is likely not a reflection of the inherent value of such documents, but rather related to the observation that updated guidelines generally reflect, at most, a small change from predecessor versions. (It also should be mentioned that many such contributions are fairly heavy going for even the most determined reader.)
Occasionally, however, a new guideline may signal a dramatic shift in practice, and the recently published AHA/ACC Scientific Statement on Eligibility and Disqualification Recommendations for Competitive Athletes with Cardiovascular Abnormalities (Congenital Heart Disease) contains such a change.
|
|
Dr. Robert Jaquiss |
In particular, the new recommendation suggests that athletes with anomalous aortic origin of the right coronary from the left coronary sinus, who have neither symptoms nor a positive stress test, may be allowed to participate in competitive athletics without undergoing surgical repair. As before, those with anomalous left coronary should not be allowed to participate until after surgical treatment.
Prior guidelines suggested that all patients, both anomalous left from right sinus and right from left sinus, be restricted prior to surgery. Because anomalous right coronary is five to six times more common than anomalous left coronary and because it is certainly much less ominous, the previous “one size fits all” approach almost certainly resulted in overtreatment, unnecessary restriction of participation, or both. Furthermore, because anomalous aortic of a coronary artery is so common, occurring in 0.1%-0.2% of the population (300,000 to 600,000 people in the United States), many thousands of competitive athletes will be impacted by the changed guidelines.
Most cardiologists, surgeons, and, most especially, patients will welcome the updated recommendations. Nonetheless, it must be emphasized that anomalous coronary arteries, even anomalous right coronary arteries, may indicate an increased risk of sudden death and that a complete assessment, including stress testing when feasible, and thorough discussion with expert clinicians is still absolutely necessary for such patients and their families.
Dr. Robert Jaquiss of Duke University, Durham, N.C., is the congenital heart section associate medical editor for Thoracic Surgery News.
For many busy clinicians, societal guidelines, task force recommendations, expert consensus statements, and similar authoritative tomes are resources that are scarcely ever read carefully. This is likely not a reflection of the inherent value of such documents, but rather related to the observation that updated guidelines generally reflect, at most, a small change from predecessor versions. (It also should be mentioned that many such contributions are fairly heavy going for even the most determined reader.)
Occasionally, however, a new guideline may signal a dramatic shift in practice, and the recently published AHA/ACC Scientific Statement on Eligibility and Disqualification Recommendations for Competitive Athletes with Cardiovascular Abnormalities (Congenital Heart Disease) contains such a change.
|
|
Dr. Robert Jaquiss |
In particular, the new recommendation suggests that athletes with anomalous aortic origin of the right coronary from the left coronary sinus, who have neither symptoms nor a positive stress test, may be allowed to participate in competitive athletics without undergoing surgical repair. As before, those with anomalous left coronary should not be allowed to participate until after surgical treatment.
Prior guidelines suggested that all patients, both anomalous left from right sinus and right from left sinus, be restricted prior to surgery. Because anomalous right coronary is five to six times more common than anomalous left coronary and because it is certainly much less ominous, the previous “one size fits all” approach almost certainly resulted in overtreatment, unnecessary restriction of participation, or both. Furthermore, because anomalous aortic of a coronary artery is so common, occurring in 0.1%-0.2% of the population (300,000 to 600,000 people in the United States), many thousands of competitive athletes will be impacted by the changed guidelines.
Most cardiologists, surgeons, and, most especially, patients will welcome the updated recommendations. Nonetheless, it must be emphasized that anomalous coronary arteries, even anomalous right coronary arteries, may indicate an increased risk of sudden death and that a complete assessment, including stress testing when feasible, and thorough discussion with expert clinicians is still absolutely necessary for such patients and their families.
Dr. Robert Jaquiss of Duke University, Durham, N.C., is the congenital heart section associate medical editor for Thoracic Surgery News.
Most children and young adult patients with congenital heart disease can and should engage in some form of physical activity and should avoid a sedentary lifestyle, according to a task force scientific statement from the American Heart Association and the American College of Cardiology (AHA/ACC).
This recommendation comes despite the fears of sudden cardiac death (SCD) in young athletes, which formed the initial impetus of the entire series of task force reports.
The recommended level of sports participation for patients with treated or untreated congenital heart defect, however, should consider the training and the competitive aspects of the sport itself and must be individualized to the patient. This means taking into account the patient’s current functional status, history of surgery, and the presence of implanted cardiac devices, according to the report by Dr. George F. Van Hare of Washington University, St. Louis, and his colleagues, which was published online in the Journal of the American College of Cardiology.
The report breaks down its specific recommendations based upon the various types of congenital heart defect (CHD). Full details and nuances of the recommendations and their specific levels of evidence for each individual condition and the many variants can be found in the online publication. Below is a brief and selected summary for some of the most common defects and some of those most pertinent to sudden cardiac death in young athletes.
Simple shunting lesions (atrial septal defect, ventricular septal defect, patent ductus arteriosus): Treated and untreated
In addressing the three most common subtypes of CHD – ventricular septal defect (VSD, 34%), atrial septal defect (ASD, 13%), and patent ductus arteriosus (PDA, 10%) – the committee found no data that children with these lesions are related to acknowledged episodes of sudden cardiac death (SCD). This applied whether the defects were closed or remained open. “With rare exceptions, patients with hemodynamically insignificant CHD such as VSD, ASD, and PDA may participate competitively in all sports,” it concluded. These recommendations fall under class I; level of evidence C for almost all of these patients, according to the writing committee.
Congenital coronary anomalies: Treated and untreated
Anomalies of coronary arteries are the second-most commonly identified structural causes of SCD in competitive athletes, accounting for about 17% of such deaths in the United States, according to the report. The vast majority of sudden deaths associated with coronary anomalies occur during or shortly after exercise. Despite being less commonly represented in patients, among athletes who have died suddenly, anomalous origin of the left main or left anterior descending coronary artery from the right sinus of Valsalva is far more prevalent. In addition, SCDs are most strongly associated with the pattern in which the anomalous left coronary artery passes between the aorta and main pulmonary artery. Recommended return to intense athletic activities is only to be permitted at least 3 months after surgery, and with a demonstration of the absence of ischemia on postoperative stress testing, with evidence levels depending on the type of anomaly. Of note, in contrast, the committee indicated that athletes with an anomalous origin of a right coronary artery from the left sinus of Valsalva should simply be evaluated by an exercise stress test, and for those without symptoms or a positive exercise stress test, permission to compete can be considered after adequate counseling (class IIa; level of evidence C).
Pulmonary valve stenosis: Treated and untreated
The committee determined that athletes with mild pulmonary stenosis (PS) and normal right ventricular (RV) function can participate in all competitive sports, although annual reevaluation also is recommended (class I; level of evidence B). In addition, athletes treated by operation or balloon valvuloplasty who have achieved adequate relief of PS (gradient less than 40 mm Hg by Doppler) can participate in all competitive sports (class I; level of evidence B). Other patients should be restricted to low-intensity sports, according to the committee.
Aortic valve stenosis: Treated and untreated
Children and adolescents with aortic stenosis (AS) are differentiated between those with mild, moderate, and severe AS by physical examination, ECG, and Doppler echocardiography. In all cases, regardless of the degree of stenosis, patients with a history of fatigue, light-headedness, dizziness, syncope, chest pain, or pallor on exercise deserve a full evaluation. Annual re-evaluation is required for all patients with AS because the disease can progress. Patients with severe AS are at risk of sudden death, particularly with exercise. The committee determined that athletes with mild AS can participate in all competitive sports (class I; level of evidence B), but that athletes with severe AS should be restricted from all competitive sports, with the possible exception of low-intensity sports (class III; level of evidence B).
Coarctation of the aorta: Treated and untreated
Before a decision is made regarding exercise participation, a detailed evaluation should be conducted, including a physical examination, ECG, chest radiograph, exercise testing, transthoracic echocardiographic evaluation of the aortic valve and aorta, and either magnetic resonance imaging or computed tomography angiography, according to the committee. The determination as to the level of sports participation permitted requires a complex assessment of these various test results and can range from full participation in the case of the least affected to restrictions to low-intensity sports in those more severely affected.
Cyanotic CHD, including tetralogy of Fallot
Full clinical assessment, including laboratory and exercise testing, should be considered before any physical activity because this population is at very high risk of sudden death, according to the committee. Recommendations are complex and depend on the level of repair and its success, but, in general, significant restrictions are recommended for all but the most effectively treated patients.
Transposition of the great arteries after atrial switch (Mustard or Senning operation)
This is a population highly at risk, according to the committee. They appear to have a unique response to exercise with reports that a high proportion of sudden death events occur during exertion. In addition, evidence of exercise-induced arrhythmias on routine clinical testing has not been shown to reliably predict exercise-induced SCD events. Although recommendations vary, including strong restrictions for many, at best the most successful of these patients should only be considered for low- to moderate-intensity competitive sports, according to the committee.
Other conditions assessed and evaluated by the committee included congenitally corrected TGA, TGA after the arterial switch, Fontan procedure, elevated pulmonary vascular resistence in CHD, ventricular dysfunction after CHD surgery, and Ebstein anomaly of the tricuspid valve.
In all cases, complete physical assessment of these patients is recommended, especially due to the often highly individualized nature of the patient’s presentation of these conditions and the variety and variability of interventions that may have been performed. Such differentials make recommendations regarding sports participation a complex calculus, which the committee attempts to provide, listing whatever evidence is available.
The majority of these patients, however, will not be considered for the highest levels of competitive sports participation. Although, in almost all cases, the need for physical activity as a contributor to patient health and well-being is stressed at whatever level of performance is possible.
The report ”Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 4: congenital heart disease: a scientific statement from the American Heart Association and American College of Cardiology,” was prepared by Dr. Van Hare and his colleagues on behalf of the American Heart Association Electrocardiography and Arrhythmias Committee of the Council on Clinical Cardiology, Council on Cardiovascular Disease in the Young, Council on Cardiovascular and Stroke Nursing, Council on Functional Genomics and Translational Biology, and the American College of Cardiology (doi: 10.1016/j.jacc.2015.09.032).
This report is one of the assessments and recommendations of 15 task forces on eligibility and disqualification recommendations for young athletes, nine of which are disease or multidisease related. The other six task forces focus on a variety of relevant topics and issues regarding the risks of young athletes on the field, including screening, the use of automated external defibrillators on the field, the use of dietary supplements and performance-enhancing drugs, sudden death, and the medical-legal perspectives involved.
All 15 task force reports were simultaneously published online in the Journal of the American College of Cardiology and the journal Circulation.
Dr. Van Hare and all but one member of the writing group had no disclosures. One member disclosed consultant/advisory committee associations with a variety of medical device companies.
Most children and young adult patients with congenital heart disease can and should engage in some form of physical activity and should avoid a sedentary lifestyle, according to a task force scientific statement from the American Heart Association and the American College of Cardiology (AHA/ACC).
This recommendation comes despite the fears of sudden cardiac death (SCD) in young athletes, which formed the initial impetus of the entire series of task force reports.
The recommended level of sports participation for patients with treated or untreated congenital heart defect, however, should consider the training and the competitive aspects of the sport itself and must be individualized to the patient. This means taking into account the patient’s current functional status, history of surgery, and the presence of implanted cardiac devices, according to the report by Dr. George F. Van Hare of Washington University, St. Louis, and his colleagues, which was published online in the Journal of the American College of Cardiology.
The report breaks down its specific recommendations based upon the various types of congenital heart defect (CHD). Full details and nuances of the recommendations and their specific levels of evidence for each individual condition and the many variants can be found in the online publication. Below is a brief and selected summary for some of the most common defects and some of those most pertinent to sudden cardiac death in young athletes.
Simple shunting lesions (atrial septal defect, ventricular septal defect, patent ductus arteriosus): Treated and untreated
In addressing the three most common subtypes of CHD – ventricular septal defect (VSD, 34%), atrial septal defect (ASD, 13%), and patent ductus arteriosus (PDA, 10%) – the committee found no data that children with these lesions are related to acknowledged episodes of sudden cardiac death (SCD). This applied whether the defects were closed or remained open. “With rare exceptions, patients with hemodynamically insignificant CHD such as VSD, ASD, and PDA may participate competitively in all sports,” it concluded. These recommendations fall under class I; level of evidence C for almost all of these patients, according to the writing committee.
Congenital coronary anomalies: Treated and untreated
Anomalies of coronary arteries are the second-most commonly identified structural causes of SCD in competitive athletes, accounting for about 17% of such deaths in the United States, according to the report. The vast majority of sudden deaths associated with coronary anomalies occur during or shortly after exercise. Despite being less commonly represented in patients, among athletes who have died suddenly, anomalous origin of the left main or left anterior descending coronary artery from the right sinus of Valsalva is far more prevalent. In addition, SCDs are most strongly associated with the pattern in which the anomalous left coronary artery passes between the aorta and main pulmonary artery. Recommended return to intense athletic activities is only to be permitted at least 3 months after surgery, and with a demonstration of the absence of ischemia on postoperative stress testing, with evidence levels depending on the type of anomaly. Of note, in contrast, the committee indicated that athletes with an anomalous origin of a right coronary artery from the left sinus of Valsalva should simply be evaluated by an exercise stress test, and for those without symptoms or a positive exercise stress test, permission to compete can be considered after adequate counseling (class IIa; level of evidence C).
Pulmonary valve stenosis: Treated and untreated
The committee determined that athletes with mild pulmonary stenosis (PS) and normal right ventricular (RV) function can participate in all competitive sports, although annual reevaluation also is recommended (class I; level of evidence B). In addition, athletes treated by operation or balloon valvuloplasty who have achieved adequate relief of PS (gradient less than 40 mm Hg by Doppler) can participate in all competitive sports (class I; level of evidence B). Other patients should be restricted to low-intensity sports, according to the committee.
Aortic valve stenosis: Treated and untreated
Children and adolescents with aortic stenosis (AS) are differentiated between those with mild, moderate, and severe AS by physical examination, ECG, and Doppler echocardiography. In all cases, regardless of the degree of stenosis, patients with a history of fatigue, light-headedness, dizziness, syncope, chest pain, or pallor on exercise deserve a full evaluation. Annual re-evaluation is required for all patients with AS because the disease can progress. Patients with severe AS are at risk of sudden death, particularly with exercise. The committee determined that athletes with mild AS can participate in all competitive sports (class I; level of evidence B), but that athletes with severe AS should be restricted from all competitive sports, with the possible exception of low-intensity sports (class III; level of evidence B).
Coarctation of the aorta: Treated and untreated
Before a decision is made regarding exercise participation, a detailed evaluation should be conducted, including a physical examination, ECG, chest radiograph, exercise testing, transthoracic echocardiographic evaluation of the aortic valve and aorta, and either magnetic resonance imaging or computed tomography angiography, according to the committee. The determination as to the level of sports participation permitted requires a complex assessment of these various test results and can range from full participation in the case of the least affected to restrictions to low-intensity sports in those more severely affected.
Cyanotic CHD, including tetralogy of Fallot
Full clinical assessment, including laboratory and exercise testing, should be considered before any physical activity because this population is at very high risk of sudden death, according to the committee. Recommendations are complex and depend on the level of repair and its success, but, in general, significant restrictions are recommended for all but the most effectively treated patients.
Transposition of the great arteries after atrial switch (Mustard or Senning operation)
This is a population highly at risk, according to the committee. They appear to have a unique response to exercise with reports that a high proportion of sudden death events occur during exertion. In addition, evidence of exercise-induced arrhythmias on routine clinical testing has not been shown to reliably predict exercise-induced SCD events. Although recommendations vary, including strong restrictions for many, at best the most successful of these patients should only be considered for low- to moderate-intensity competitive sports, according to the committee.
Other conditions assessed and evaluated by the committee included congenitally corrected TGA, TGA after the arterial switch, Fontan procedure, elevated pulmonary vascular resistence in CHD, ventricular dysfunction after CHD surgery, and Ebstein anomaly of the tricuspid valve.
In all cases, complete physical assessment of these patients is recommended, especially due to the often highly individualized nature of the patient’s presentation of these conditions and the variety and variability of interventions that may have been performed. Such differentials make recommendations regarding sports participation a complex calculus, which the committee attempts to provide, listing whatever evidence is available.
The majority of these patients, however, will not be considered for the highest levels of competitive sports participation. Although, in almost all cases, the need for physical activity as a contributor to patient health and well-being is stressed at whatever level of performance is possible.
The report ”Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 4: congenital heart disease: a scientific statement from the American Heart Association and American College of Cardiology,” was prepared by Dr. Van Hare and his colleagues on behalf of the American Heart Association Electrocardiography and Arrhythmias Committee of the Council on Clinical Cardiology, Council on Cardiovascular Disease in the Young, Council on Cardiovascular and Stroke Nursing, Council on Functional Genomics and Translational Biology, and the American College of Cardiology (doi: 10.1016/j.jacc.2015.09.032).
This report is one of the assessments and recommendations of 15 task forces on eligibility and disqualification recommendations for young athletes, nine of which are disease or multidisease related. The other six task forces focus on a variety of relevant topics and issues regarding the risks of young athletes on the field, including screening, the use of automated external defibrillators on the field, the use of dietary supplements and performance-enhancing drugs, sudden death, and the medical-legal perspectives involved.
All 15 task force reports were simultaneously published online in the Journal of the American College of Cardiology and the journal Circulation.
Dr. Van Hare and all but one member of the writing group had no disclosures. One member disclosed consultant/advisory committee associations with a variety of medical device companies.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
Key clinical point: Recommendations for sports participation should consider the activity itself and take into account the patient’s functional status, history of surgery, and implanted devices.
Major finding: Congenital heart disease is the most common form of serious birth defect (8 per 1,000 live births) and, with dramatic improvements in survival, the issue of youth and young-adult participation in competitive sports must be addressed.
Data source: The AHA/ACC expert consensus recommendations were developed using the experience of the writing-group members and the available scientific evidence in the literature.
Disclosures: The review was sponsored by the AHA and the ACC. Dr. Van Hare and all but one member of the writing group had no disclosures. One member disclosed consultant/advisory committee associations with a variety of medical device companies.

Biomarkers beat DSM categories for capturing nuances in psychosis
Three biotypes surpass traditional diagnostic categories when it comes to identifying subgroups of psychosis, a study showed.
“Classification and treatment of brain diseases subsumed by psychiatry rely on clinical phenomenology, despite the call for alternatives,” wrote Brett A. Clementz, Ph.D., of the University of Georgia, Athens, and his coinvestigators. “There is overlap in susceptibility genes and phenotypes across bipolar disorder with psychosis and schizophrenia, and considerable similarity between different psychotic disorders on symptoms, illness course, cognition, psychophysiology, and neurobiology [while] drug treatments for these conditions overlap extensively” (Am J Psychiatry. 2015. doi: 10.1176/appi.ajp.2015.14091200).
The researchers recruited 711 people from Bipolar-Schizophrenia Network on Intermediate Phenotypes (B-SNIP) consortium sites (probands). All subjects had diagnoses of schizophrenia, schizoaffective disorder, or bipolar disorder with psychosis, and underwent interviews and laboratory data collection at the time of enrollment. In addition, 883 first-degree relatives of the 711 initial enrollees also were clinically evaluated, along with a cohort of 278 people deemed “demographically comparable [and] healthy” by investigators.
Biotypes for all individuals enrolled in the study were determined through laboratory tasks designed to “assess brain function at the neurocogntive/perceptual level.” These tasks consisted of the Brief Assessment of Cognition in Schizophrenia (BACS), pro- and antisaccade tasks, stop signal tasks, auditory paired stimuli and oddball evoked brain responses, and MRI acquisition and voxel-based morphometry.
The Structured Clinical Interview for DSM-IV and the Structured Interview for DSM-IV Personality Disorders were used for interviewing enrollees. Data compiled from these tests underwent multivariate taxometric analyses to compare biomarker variance across the three cohorts, in order to determine what, if any, heterogeneity exists in psychosis biotypes.
According to the results, diagnoses made with the clinical DSM guidelines yielded a single-severity continuum showing schizophrenia to be the most severe, followed by schizoaffective disorder and bipolar psychosis. However, biotypes showed significant variation, with investigators noting that “the three biotypes had distinctive patterns of abnormality across biomarkers that were neither entirely nor efficiently captured by a severity continuum.”
Larger separations were seen in biotype cohorts than in the DSM, specifically among probands. Among probands, group separation from healthy subjects was –2.58, –1.94, and –0.35 for biotype 1, 2, and 3 respectively for the BACS. Separation was –0.99, –0.78, and –0.05 for the stop signal task, and 3.32, 1.90, and 1.19 for the antisaccade errors. On the other hand, DSM diagnostics revealed group differences of –1.01, –1.51, and –1.83 for bipolar disorder psychosis, schizoaffective disorders, and schizophrenia, respectively, for the BACS test. Separation was –0.41, –0.61, and –0.55 for the stop signal task, and 1.36, 1.66, and 2.45 for antisaccade errors.
“Each biotype included all DSM psychosis categories, but probands diagnosed with schizophrenia were more numerous in biotype 1 (although 20% had bipolar disorder with psychosis), and probands diagnosed with bipolar disorder with psychosis were more numerous in biotype 3 (although 32% had schizophrenia), respectively,” the investigators noted.
The authors added that “when considered across proband and relative data, the biotype subgroups were superior to DSM diagnostic classes in between-group separations on external validating measures, illustrating the former scheme’s superiority for capturing neurobiological distinctiveness.”
Investigators noted that their approach did not use social functioning, brain structure, and characteristics of biological relatives in the creation of biotypes, which could have led to stronger results. Also, trial participants were mostly already on medication, classified as chronically psychotic, and tested at least once previously. The trial also had no replication sample for this sample population.
The study was supported by grants from the National Institute of Mental Health. Dr. Clementz did not report any relevant financial disclosures. Dr. Matcheri S. Keshavan reported receiving a grant from Sunovion and serving as a consultant to Forum Pharmaceuticals. Dr. Carol A. Tamminga also reported potential conflicts.
Three biotypes surpass traditional diagnostic categories when it comes to identifying subgroups of psychosis, a study showed.
“Classification and treatment of brain diseases subsumed by psychiatry rely on clinical phenomenology, despite the call for alternatives,” wrote Brett A. Clementz, Ph.D., of the University of Georgia, Athens, and his coinvestigators. “There is overlap in susceptibility genes and phenotypes across bipolar disorder with psychosis and schizophrenia, and considerable similarity between different psychotic disorders on symptoms, illness course, cognition, psychophysiology, and neurobiology [while] drug treatments for these conditions overlap extensively” (Am J Psychiatry. 2015. doi: 10.1176/appi.ajp.2015.14091200).
The researchers recruited 711 people from Bipolar-Schizophrenia Network on Intermediate Phenotypes (B-SNIP) consortium sites (probands). All subjects had diagnoses of schizophrenia, schizoaffective disorder, or bipolar disorder with psychosis, and underwent interviews and laboratory data collection at the time of enrollment. In addition, 883 first-degree relatives of the 711 initial enrollees also were clinically evaluated, along with a cohort of 278 people deemed “demographically comparable [and] healthy” by investigators.
Biotypes for all individuals enrolled in the study were determined through laboratory tasks designed to “assess brain function at the neurocogntive/perceptual level.” These tasks consisted of the Brief Assessment of Cognition in Schizophrenia (BACS), pro- and antisaccade tasks, stop signal tasks, auditory paired stimuli and oddball evoked brain responses, and MRI acquisition and voxel-based morphometry.
The Structured Clinical Interview for DSM-IV and the Structured Interview for DSM-IV Personality Disorders were used for interviewing enrollees. Data compiled from these tests underwent multivariate taxometric analyses to compare biomarker variance across the three cohorts, in order to determine what, if any, heterogeneity exists in psychosis biotypes.
According to the results, diagnoses made with the clinical DSM guidelines yielded a single-severity continuum showing schizophrenia to be the most severe, followed by schizoaffective disorder and bipolar psychosis. However, biotypes showed significant variation, with investigators noting that “the three biotypes had distinctive patterns of abnormality across biomarkers that were neither entirely nor efficiently captured by a severity continuum.”
Larger separations were seen in biotype cohorts than in the DSM, specifically among probands. Among probands, group separation from healthy subjects was –2.58, –1.94, and –0.35 for biotype 1, 2, and 3 respectively for the BACS. Separation was –0.99, –0.78, and –0.05 for the stop signal task, and 3.32, 1.90, and 1.19 for the antisaccade errors. On the other hand, DSM diagnostics revealed group differences of –1.01, –1.51, and –1.83 for bipolar disorder psychosis, schizoaffective disorders, and schizophrenia, respectively, for the BACS test. Separation was –0.41, –0.61, and –0.55 for the stop signal task, and 1.36, 1.66, and 2.45 for antisaccade errors.
“Each biotype included all DSM psychosis categories, but probands diagnosed with schizophrenia were more numerous in biotype 1 (although 20% had bipolar disorder with psychosis), and probands diagnosed with bipolar disorder with psychosis were more numerous in biotype 3 (although 32% had schizophrenia), respectively,” the investigators noted.
The authors added that “when considered across proband and relative data, the biotype subgroups were superior to DSM diagnostic classes in between-group separations on external validating measures, illustrating the former scheme’s superiority for capturing neurobiological distinctiveness.”
Investigators noted that their approach did not use social functioning, brain structure, and characteristics of biological relatives in the creation of biotypes, which could have led to stronger results. Also, trial participants were mostly already on medication, classified as chronically psychotic, and tested at least once previously. The trial also had no replication sample for this sample population.
The study was supported by grants from the National Institute of Mental Health. Dr. Clementz did not report any relevant financial disclosures. Dr. Matcheri S. Keshavan reported receiving a grant from Sunovion and serving as a consultant to Forum Pharmaceuticals. Dr. Carol A. Tamminga also reported potential conflicts.
Three biotypes surpass traditional diagnostic categories when it comes to identifying subgroups of psychosis, a study showed.
“Classification and treatment of brain diseases subsumed by psychiatry rely on clinical phenomenology, despite the call for alternatives,” wrote Brett A. Clementz, Ph.D., of the University of Georgia, Athens, and his coinvestigators. “There is overlap in susceptibility genes and phenotypes across bipolar disorder with psychosis and schizophrenia, and considerable similarity between different psychotic disorders on symptoms, illness course, cognition, psychophysiology, and neurobiology [while] drug treatments for these conditions overlap extensively” (Am J Psychiatry. 2015. doi: 10.1176/appi.ajp.2015.14091200).
The researchers recruited 711 people from Bipolar-Schizophrenia Network on Intermediate Phenotypes (B-SNIP) consortium sites (probands). All subjects had diagnoses of schizophrenia, schizoaffective disorder, or bipolar disorder with psychosis, and underwent interviews and laboratory data collection at the time of enrollment. In addition, 883 first-degree relatives of the 711 initial enrollees also were clinically evaluated, along with a cohort of 278 people deemed “demographically comparable [and] healthy” by investigators.
Biotypes for all individuals enrolled in the study were determined through laboratory tasks designed to “assess brain function at the neurocogntive/perceptual level.” These tasks consisted of the Brief Assessment of Cognition in Schizophrenia (BACS), pro- and antisaccade tasks, stop signal tasks, auditory paired stimuli and oddball evoked brain responses, and MRI acquisition and voxel-based morphometry.
The Structured Clinical Interview for DSM-IV and the Structured Interview for DSM-IV Personality Disorders were used for interviewing enrollees. Data compiled from these tests underwent multivariate taxometric analyses to compare biomarker variance across the three cohorts, in order to determine what, if any, heterogeneity exists in psychosis biotypes.
According to the results, diagnoses made with the clinical DSM guidelines yielded a single-severity continuum showing schizophrenia to be the most severe, followed by schizoaffective disorder and bipolar psychosis. However, biotypes showed significant variation, with investigators noting that “the three biotypes had distinctive patterns of abnormality across biomarkers that were neither entirely nor efficiently captured by a severity continuum.”
Larger separations were seen in biotype cohorts than in the DSM, specifically among probands. Among probands, group separation from healthy subjects was –2.58, –1.94, and –0.35 for biotype 1, 2, and 3 respectively for the BACS. Separation was –0.99, –0.78, and –0.05 for the stop signal task, and 3.32, 1.90, and 1.19 for the antisaccade errors. On the other hand, DSM diagnostics revealed group differences of –1.01, –1.51, and –1.83 for bipolar disorder psychosis, schizoaffective disorders, and schizophrenia, respectively, for the BACS test. Separation was –0.41, –0.61, and –0.55 for the stop signal task, and 1.36, 1.66, and 2.45 for antisaccade errors.
“Each biotype included all DSM psychosis categories, but probands diagnosed with schizophrenia were more numerous in biotype 1 (although 20% had bipolar disorder with psychosis), and probands diagnosed with bipolar disorder with psychosis were more numerous in biotype 3 (although 32% had schizophrenia), respectively,” the investigators noted.
The authors added that “when considered across proband and relative data, the biotype subgroups were superior to DSM diagnostic classes in between-group separations on external validating measures, illustrating the former scheme’s superiority for capturing neurobiological distinctiveness.”
Investigators noted that their approach did not use social functioning, brain structure, and characteristics of biological relatives in the creation of biotypes, which could have led to stronger results. Also, trial participants were mostly already on medication, classified as chronically psychotic, and tested at least once previously. The trial also had no replication sample for this sample population.
The study was supported by grants from the National Institute of Mental Health. Dr. Clementz did not report any relevant financial disclosures. Dr. Matcheri S. Keshavan reported receiving a grant from Sunovion and serving as a consultant to Forum Pharmaceuticals. Dr. Carol A. Tamminga also reported potential conflicts.
FROM THE AMERICAN JOURNAL OF PSYCHIATRY
Key clinical point: Brain scans capture gray matter volume differences among people with schizophrenia, schizoaffective disorder, and bipolar disorder that are missed by DSM diagnoses.
Major finding: Individuals with schizophrenia, schizoaffective disorder, and bipolar disorder with psychosis were compared with first-degree relatives and “demographically comparable healthy subjects” for biomarker variance; three psychosis variants were identified that were neurobiologically distinct and did not conform to accepted diagnostic boundaries.
Data source: A prospective cohort study of 711 individuals with schizophrenia, schizoaffective disorder, and bipolar disorder with psychosis, along with 883 first-degree relatives and 278 “demographically comparable healthy subjects.”
Disclosures: The study was supported by grants from the National Institute of Mental Health. Dr. Clementz did not report any relevant financial disclosures. Dr. Matcheri S. Keshavan reported receiving a grant from Sunovion and serving as a consultant to Forum Pharmaceuticals. Dr. Carol A. Tamminga also reported potential conflicts.
High NLR predicts poor survival in trauma patients
CHICAGO – A high neutrophil to lymphocyte ratio on days 2 and 5 of surgical ICU hospitalization independently predicts increased mortality in critically ill trauma patients, an award-winning study showed.
The risk of death was two times higher for patients with a neutrophil to lymphocyte ratio (NLR) of at least 10.45 on day 2 (adjusted hazard ratio, 2.07; 95% confidence interval, 1.38-3.13; P = .001) and 5.7 times higher for those with an NLR of at least 7.91 on day 5 (adjusted HR, 5.79; 95% CI, 2.93-11.44; P less than .001) in a multivariate analysis, after adjustment for age 65 years or older, male sex, a systolic blood pressure of 90 mm Hg or less, a Glasgow Coma Scale (GCS) score of 8 or less, an Injury Severity Score (ISS) of at least 25, and operation on admission.

“The neutrophil to lymphocyte ratio is easily accessible, the calculation is simple, it adds no additional costs, and virtually all critically ill patients will have these labs,” study author Dr. Evren Dilektasli said at the American College of Surgeons annual clinical congress.
The simple calculation has been shown to be useful in the diagnosis of appendicitis and to be associated with overall survivalin metastatic colorectal cancer, but its association with mortality in trauma patients is not known, he said.
The retrospective cohort comprised 1,356 trauma patients, at least 16 years old, admitted to the Los Angeles County–University of Southern California Medical Center surgical ICU between January 2013 and January 2014. The median NLR was calculated for each day of the surgical ICU stay. At baseline, 16% of patients had an ISS of at least 25, 16.5% had a GCS of 8 or less, 4.5% had a systolic BP of 90 mm Hg or less, 74.3% were male, 23.7% were aged 65 or older, and 86% had a blunt injury. The most common operations on admission were laparotomy (39.6%) and craniectomy/craniotomy (20.7%).
In receiver operating characteristic (ROC) analysis for the first 10 days of hospitalization, the area under the curve (AUC) values for predicting mortality were between 0.55 on day 1 and a high of 0.79 on day 5, Dr. Dilektasli reported.
Starting from day 2 to day 10, the AUCs were statistically significant for predicting mortality.
The NLRs on day 2 (AUC, 0.73; P less than .001) and day 5 (AUC, 0.79; P less than .001) were selected in order to adjust for the clinical probability of early and late complications, he said.
Subsequent ROC curve analysis revealed an NLR cutoff of 10.45 on day 2 (AUC, 0.73; sensitivity, 73.2%; specificity, 61.8%) and a cutoff of 7.91 on day 5 (AUC, 0.79; sensitivity, 82.8%; specificity, 65.2%).
A high NLR on day 2 (at least 10.45) versus a low NLR (less than 10.45) was associated with significantly more ventilator days (5 days vs. 3 days), a longer surgical ICU length of stay (5 days vs. 3 days), a longer hospital stay (11 days vs. 8 days), and greater mortality (18.3% vs. 4.8%; all P values less than .001), reported Dr. Dilektasli, who was a research fellow at USC at the time of the study and has returned to Turkey to continue his training.
On day 5, a high NLR (at least 7.91) versus a low NLR (less than 7.91) was associated with significantly more ventilator days (7 days vs. 4 days; P less than .001), a longer surgical ICU stay (9 days vs. 5 days; P less than .001), and increased mortality (20.4% vs. 2.8%; P less than .001), but not a longer hospital stay (17 days vs. 14 days; P = .119).
In Kaplan-Meier analysis, a significant difference was observed between the high and low NLR groups on day 2 (log rank P less than .001) and day 5 (log rank P less than .001), he said.
“NLR may be a promising tool for assessing the risk of in-hospital mortality,” Dr. Dilektasli concluded. “Prospective external validation is warranted in a larger heterogeneous trauma population.”
During a discussion of the study, it was noted that the ROC curves were impressive, but that other biologic markers known to be associated with poor survival such as C-reactive protein level and class II major hepatitis C expression should have been included in the analysis.
When asked whether any patients with a low NLR on day 2 went on to have a high NLR on day 5, Dr. Dilektasli said there were such patients and that they also had an increased risk of death.
The findings of the study, which earned an excellence in research award, are only an “observation” at this point and are not being used in clinical practice, he added.
The authors reported having no relevant financial conflicts of interest.
CHICAGO – A high neutrophil to lymphocyte ratio on days 2 and 5 of surgical ICU hospitalization independently predicts increased mortality in critically ill trauma patients, an award-winning study showed.
The risk of death was two times higher for patients with a neutrophil to lymphocyte ratio (NLR) of at least 10.45 on day 2 (adjusted hazard ratio, 2.07; 95% confidence interval, 1.38-3.13; P = .001) and 5.7 times higher for those with an NLR of at least 7.91 on day 5 (adjusted HR, 5.79; 95% CI, 2.93-11.44; P less than .001) in a multivariate analysis, after adjustment for age 65 years or older, male sex, a systolic blood pressure of 90 mm Hg or less, a Glasgow Coma Scale (GCS) score of 8 or less, an Injury Severity Score (ISS) of at least 25, and operation on admission.
“The neutrophil to lymphocyte ratio is easily accessible, the calculation is simple, it adds no additional costs, and virtually all critically ill patients will have these labs,” study author Dr. Evren Dilektasli said at the American College of Surgeons annual clinical congress.
The simple calculation has been shown to be useful in the diagnosis of appendicitis and to be associated with overall survivalin metastatic colorectal cancer, but its association with mortality in trauma patients is not known, he said.
The retrospective cohort comprised 1,356 trauma patients, at least 16 years old, admitted to the Los Angeles County–University of Southern California Medical Center surgical ICU between January 2013 and January 2014. The median NLR was calculated for each day of the surgical ICU stay. At baseline, 16% of patients had an ISS of at least 25, 16.5% had a GCS of 8 or less, 4.5% had a systolic BP of 90 mm Hg or less, 74.3% were male, 23.7% were aged 65 or older, and 86% had a blunt injury. The most common operations on admission were laparotomy (39.6%) and craniectomy/craniotomy (20.7%).
In receiver operating characteristic (ROC) analysis for the first 10 days of hospitalization, the area under the curve (AUC) values for predicting mortality were between 0.55 on day 1 and a high of 0.79 on day 5, Dr. Dilektasli reported.
Starting from day 2 to day 10, the AUCs were statistically significant for predicting mortality.
The NLRs on day 2 (AUC, 0.73; P less than .001) and day 5 (AUC, 0.79; P less than .001) were selected in order to adjust for the clinical probability of early and late complications, he said.
Subsequent ROC curve analysis revealed an NLR cutoff of 10.45 on day 2 (AUC, 0.73; sensitivity, 73.2%; specificity, 61.8%) and a cutoff of 7.91 on day 5 (AUC, 0.79; sensitivity, 82.8%; specificity, 65.2%).
A high NLR on day 2 (at least 10.45) versus a low NLR (less than 10.45) was associated with significantly more ventilator days (5 days vs. 3 days), a longer surgical ICU length of stay (5 days vs. 3 days), a longer hospital stay (11 days vs. 8 days), and greater mortality (18.3% vs. 4.8%; all P values less than .001), reported Dr. Dilektasli, who was a research fellow at USC at the time of the study and has returned to Turkey to continue his training.
On day 5, a high NLR (at least 7.91) versus a low NLR (less than 7.91) was associated with significantly more ventilator days (7 days vs. 4 days; P less than .001), a longer surgical ICU stay (9 days vs. 5 days; P less than .001), and increased mortality (20.4% vs. 2.8%; P less than .001), but not a longer hospital stay (17 days vs. 14 days; P = .119).
In Kaplan-Meier analysis, a significant difference was observed between the high and low NLR groups on day 2 (log rank P less than .001) and day 5 (log rank P less than .001), he said.
“NLR may be a promising tool for assessing the risk of in-hospital mortality,” Dr. Dilektasli concluded. “Prospective external validation is warranted in a larger heterogeneous trauma population.”
During a discussion of the study, it was noted that the ROC curves were impressive, but that other biologic markers known to be associated with poor survival such as C-reactive protein level and class II major hepatitis C expression should have been included in the analysis.
When asked whether any patients with a low NLR on day 2 went on to have a high NLR on day 5, Dr. Dilektasli said there were such patients and that they also had an increased risk of death.
The findings of the study, which earned an excellence in research award, are only an “observation” at this point and are not being used in clinical practice, he added.
The authors reported having no relevant financial conflicts of interest.
CHICAGO – A high neutrophil to lymphocyte ratio on days 2 and 5 of surgical ICU hospitalization independently predicts increased mortality in critically ill trauma patients, an award-winning study showed.
The risk of death was two times higher for patients with a neutrophil to lymphocyte ratio (NLR) of at least 10.45 on day 2 (adjusted hazard ratio, 2.07; 95% confidence interval, 1.38-3.13; P = .001) and 5.7 times higher for those with an NLR of at least 7.91 on day 5 (adjusted HR, 5.79; 95% CI, 2.93-11.44; P less than .001) in a multivariate analysis, after adjustment for age 65 years or older, male sex, a systolic blood pressure of 90 mm Hg or less, a Glasgow Coma Scale (GCS) score of 8 or less, an Injury Severity Score (ISS) of at least 25, and operation on admission.
“The neutrophil to lymphocyte ratio is easily accessible, the calculation is simple, it adds no additional costs, and virtually all critically ill patients will have these labs,” study author Dr. Evren Dilektasli said at the American College of Surgeons annual clinical congress.
The simple calculation has been shown to be useful in the diagnosis of appendicitis and to be associated with overall survivalin metastatic colorectal cancer, but its association with mortality in trauma patients is not known, he said.
The retrospective cohort comprised 1,356 trauma patients, at least 16 years old, admitted to the Los Angeles County–University of Southern California Medical Center surgical ICU between January 2013 and January 2014. The median NLR was calculated for each day of the surgical ICU stay. At baseline, 16% of patients had an ISS of at least 25, 16.5% had a GCS of 8 or less, 4.5% had a systolic BP of 90 mm Hg or less, 74.3% were male, 23.7% were aged 65 or older, and 86% had a blunt injury. The most common operations on admission were laparotomy (39.6%) and craniectomy/craniotomy (20.7%).
In receiver operating characteristic (ROC) analysis for the first 10 days of hospitalization, the area under the curve (AUC) values for predicting mortality were between 0.55 on day 1 and a high of 0.79 on day 5, Dr. Dilektasli reported.
Starting from day 2 to day 10, the AUCs were statistically significant for predicting mortality.
The NLRs on day 2 (AUC, 0.73; P less than .001) and day 5 (AUC, 0.79; P less than .001) were selected in order to adjust for the clinical probability of early and late complications, he said.
Subsequent ROC curve analysis revealed an NLR cutoff of 10.45 on day 2 (AUC, 0.73; sensitivity, 73.2%; specificity, 61.8%) and a cutoff of 7.91 on day 5 (AUC, 0.79; sensitivity, 82.8%; specificity, 65.2%).
A high NLR on day 2 (at least 10.45) versus a low NLR (less than 10.45) was associated with significantly more ventilator days (5 days vs. 3 days), a longer surgical ICU length of stay (5 days vs. 3 days), a longer hospital stay (11 days vs. 8 days), and greater mortality (18.3% vs. 4.8%; all P values less than .001), reported Dr. Dilektasli, who was a research fellow at USC at the time of the study and has returned to Turkey to continue his training.
On day 5, a high NLR (at least 7.91) versus a low NLR (less than 7.91) was associated with significantly more ventilator days (7 days vs. 4 days; P less than .001), a longer surgical ICU stay (9 days vs. 5 days; P less than .001), and increased mortality (20.4% vs. 2.8%; P less than .001), but not a longer hospital stay (17 days vs. 14 days; P = .119).
In Kaplan-Meier analysis, a significant difference was observed between the high and low NLR groups on day 2 (log rank P less than .001) and day 5 (log rank P less than .001), he said.
“NLR may be a promising tool for assessing the risk of in-hospital mortality,” Dr. Dilektasli concluded. “Prospective external validation is warranted in a larger heterogeneous trauma population.”
During a discussion of the study, it was noted that the ROC curves were impressive, but that other biologic markers known to be associated with poor survival such as C-reactive protein level and class II major hepatitis C expression should have been included in the analysis.
When asked whether any patients with a low NLR on day 2 went on to have a high NLR on day 5, Dr. Dilektasli said there were such patients and that they also had an increased risk of death.
The findings of the study, which earned an excellence in research award, are only an “observation” at this point and are not being used in clinical practice, he added.
The authors reported having no relevant financial conflicts of interest.
AT THE ACS CLINICAL CONGRESS
Key clinical point: A high neutrophil to lymphocyte ratio on day 2 and day 5 of surgical ICU admission may be a useful predictor of poor survival in trauma patients.
Major finding: The adjusted hazard ratios for mortality were 2.07 with a neutrophil to lymphocyte ratio of at least 10.45 on day 2 (95% CI, 1.38-3.13; P = .001) and 5.79 with an NLR of at least 7.91 on day 5 (95% CI, 2.93-11.44; P less than .001).
Data source: A retrospective study involving 1,356 trauma patients.
Disclosures: The authors reported having no relevant financial conflicts of interest.

Residents’ Forum: Searching for a Thoracic Job in 2016
In the thick of a job search for this coming July, I fondly remember my first experience at the 47th Annual STS meeting in San Diego in 2011. The presidential address was delivered by Dr. Mathisen and focused nearly an entire hour on encouraging and mentoring young trainees to pursue a career in cardiothoracic surgery. Citing concerns about job availability and security, declining compensation, and increasing scrutiny on outcome measures, the president attempted to individually address these.
Most vividly I remember the emphasis on baby boomers getting older, median CT surgeon age approaching the retirement point, and the overall increased need for young, motivated, and highly skilled junior attendings. Until this year, the number of STS and CTSnet job posts was abysmally low. This led many new graduates to consider advanced fellowships or alternative avenues. Half a decade after Dr. Mathisen’s prediction at STS, the proverbial flood gates have opened.

This past year, I’ve seen at least a dozen quality academic positions looking for new graduates in thoracic surgery. Additionally, at least another dozen hybrid practice or private/community practice groups are looking to hire junior attendings. What is also interesting is the direction that many practices are headed: thoracic service lines, where volume is accrued from surrounding community practices and brought to centralized, multidisciplinary tertiary care institutions.
In many models, mentorship has been built in with involved senior partners, especially now that expertise in advanced techniques in bronchoscopy (EBUS, navigational bronchoscopy), endoscopy (RFA, EMR, and other ablative strategies) and minimally invasive surgery (VATS, MIS esophagectomy, robotic) are commonly expected by patients. These skills are initially learned in training and, with the right support structure, are perfected early in practice.
With more precise earlier stage screening and detection of esophageal and lung malignancies and the general stability or increase in the year-to-year incidence of these cancers, I expect the job market will continue to accommodate a growing need for thoracic surgeons in the United States. This should greatly help recruitment of the best young medical talent into what is truly an exciting and blossoming field of surgery.
Reference
Mathisen DJ. “It’s Not the Destination, It’s the Journey: Lessons Learned”. 47th Annual STS Presidential Address. San Diego, CA. January 2011. www.sts.org/news/mathisen-delivers-presidential-address.
Dr. Shersher is at Rush University Medical Center, Chicago.
In the thick of a job search for this coming July, I fondly remember my first experience at the 47th Annual STS meeting in San Diego in 2011. The presidential address was delivered by Dr. Mathisen and focused nearly an entire hour on encouraging and mentoring young trainees to pursue a career in cardiothoracic surgery. Citing concerns about job availability and security, declining compensation, and increasing scrutiny on outcome measures, the president attempted to individually address these.
Most vividly I remember the emphasis on baby boomers getting older, median CT surgeon age approaching the retirement point, and the overall increased need for young, motivated, and highly skilled junior attendings. Until this year, the number of STS and CTSnet job posts was abysmally low. This led many new graduates to consider advanced fellowships or alternative avenues. Half a decade after Dr. Mathisen’s prediction at STS, the proverbial flood gates have opened.
This past year, I’ve seen at least a dozen quality academic positions looking for new graduates in thoracic surgery. Additionally, at least another dozen hybrid practice or private/community practice groups are looking to hire junior attendings. What is also interesting is the direction that many practices are headed: thoracic service lines, where volume is accrued from surrounding community practices and brought to centralized, multidisciplinary tertiary care institutions.
In many models, mentorship has been built in with involved senior partners, especially now that expertise in advanced techniques in bronchoscopy (EBUS, navigational bronchoscopy), endoscopy (RFA, EMR, and other ablative strategies) and minimally invasive surgery (VATS, MIS esophagectomy, robotic) are commonly expected by patients. These skills are initially learned in training and, with the right support structure, are perfected early in practice.
With more precise earlier stage screening and detection of esophageal and lung malignancies and the general stability or increase in the year-to-year incidence of these cancers, I expect the job market will continue to accommodate a growing need for thoracic surgeons in the United States. This should greatly help recruitment of the best young medical talent into what is truly an exciting and blossoming field of surgery.
Reference
Mathisen DJ. “It’s Not the Destination, It’s the Journey: Lessons Learned”. 47th Annual STS Presidential Address. San Diego, CA. January 2011. www.sts.org/news/mathisen-delivers-presidential-address.
Dr. Shersher is at Rush University Medical Center, Chicago.
In the thick of a job search for this coming July, I fondly remember my first experience at the 47th Annual STS meeting in San Diego in 2011. The presidential address was delivered by Dr. Mathisen and focused nearly an entire hour on encouraging and mentoring young trainees to pursue a career in cardiothoracic surgery. Citing concerns about job availability and security, declining compensation, and increasing scrutiny on outcome measures, the president attempted to individually address these.
Most vividly I remember the emphasis on baby boomers getting older, median CT surgeon age approaching the retirement point, and the overall increased need for young, motivated, and highly skilled junior attendings. Until this year, the number of STS and CTSnet job posts was abysmally low. This led many new graduates to consider advanced fellowships or alternative avenues. Half a decade after Dr. Mathisen’s prediction at STS, the proverbial flood gates have opened.
This past year, I’ve seen at least a dozen quality academic positions looking for new graduates in thoracic surgery. Additionally, at least another dozen hybrid practice or private/community practice groups are looking to hire junior attendings. What is also interesting is the direction that many practices are headed: thoracic service lines, where volume is accrued from surrounding community practices and brought to centralized, multidisciplinary tertiary care institutions.
In many models, mentorship has been built in with involved senior partners, especially now that expertise in advanced techniques in bronchoscopy (EBUS, navigational bronchoscopy), endoscopy (RFA, EMR, and other ablative strategies) and minimally invasive surgery (VATS, MIS esophagectomy, robotic) are commonly expected by patients. These skills are initially learned in training and, with the right support structure, are perfected early in practice.
With more precise earlier stage screening and detection of esophageal and lung malignancies and the general stability or increase in the year-to-year incidence of these cancers, I expect the job market will continue to accommodate a growing need for thoracic surgeons in the United States. This should greatly help recruitment of the best young medical talent into what is truly an exciting and blossoming field of surgery.
Reference
Mathisen DJ. “It’s Not the Destination, It’s the Journey: Lessons Learned”. 47th Annual STS Presidential Address. San Diego, CA. January 2011. www.sts.org/news/mathisen-delivers-presidential-address.
Dr. Shersher is at Rush University Medical Center, Chicago.

Erratum (Cutis. 2015;96:297, 326)
The article “What Is Your Diagnosis? Tinea Corporis” (Cutis. 2015;96:297, 326) contained an error in the author affiliations. The text should have stated:
Ms. Gyurjyan is from the University of Virginia School of Medicine, Charlottesville. Dr. Wilson is from the Department of Dermatology, University of Virginia Health System, Charlottesville.
The staff of Cutis® makes every possible effort to ensure accuracy in its articles and apologizes for the mistake.
The article “What Is Your Diagnosis? Tinea Corporis” (Cutis. 2015;96:297, 326) contained an error in the author affiliations. The text should have stated:
Ms. Gyurjyan is from the University of Virginia School of Medicine, Charlottesville. Dr. Wilson is from the Department of Dermatology, University of Virginia Health System, Charlottesville.
The staff of Cutis® makes every possible effort to ensure accuracy in its articles and apologizes for the mistake.
The article “What Is Your Diagnosis? Tinea Corporis” (Cutis. 2015;96:297, 326) contained an error in the author affiliations. The text should have stated:
Ms. Gyurjyan is from the University of Virginia School of Medicine, Charlottesville. Dr. Wilson is from the Department of Dermatology, University of Virginia Health System, Charlottesville.
The staff of Cutis® makes every possible effort to ensure accuracy in its articles and apologizes for the mistake.
Cutaneous Leishmaniasis
Cutaneous leishmaniasis is a parasitic infection caused by intracellular organisms found in tropical climates. Old World leishmaniasis is endemic to Asia, Africa, and parts of Europe, while New World leishmaniasis is native to Central and South Americas.1 Depending upon a host’s immune status and the specific Leishmania species, clinical presentations vary in appearance and severity, ranging from self-limited, localized cutaneous disease to potentially fatal visceral and mucocutaneous involvement. Most cutaneous manifestations of leishmaniasis begin as distinct, painless papules that may progress to nodules or become ulcerated over time.1 Histologically, leishmaniasis is diagnosed by the identification of intracellular organisms that characteristically align along the peripheral rim inside the vacuole of a histiocyte.2 This unique finding is called the “marquee sign” due to its resemblance to light bulbs arranged around a dressing room mirror (Figure 1).2Leishmania amastigotes (also known as Leishman-Donovan bodies) have kinetoplasts that are helpful in diagnosis but also may be difficult to detect.2 Along with the Leishmania parasites, there typically is a mixed inflammatory infiltrate of plasma cells, lymphocytes, histiocytes, and neutrophils (Figure 2).1,2 There also may be varying degrees of pseudoepitheliomatous hyperplasia and overlying epidermal ulceration.1
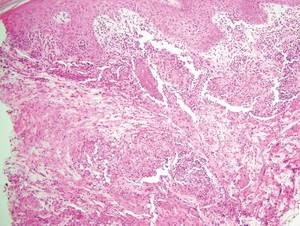

Cutaneous botryomycosis can present clinically as a number of various primary lesions, including papules, nodules, or ulcers that may resemble leishmaniasis.3 Botryomycosis represents a specific histologic collection of bacterial granules, most commonly caused by Staphylococcus aureus.3 The dermal granulomatous infiltrate seen in botryomycosis often is similar to that seen in chronic leishmaniasis; however, one histologic feature unique to botryomycosis is the presence of characteristic basophilic staphylococcal grains that are arranged in clusters resembling bunches of grapes (the term botryo means “bunch of grapes” in Greek).3 A thin, eosinophilic rim consisting of antibodies, bacterial debris, and complement proteins and glycoproteins may encircle the basophilic grains but does not need to be present for diagnosis (Figure 3).3
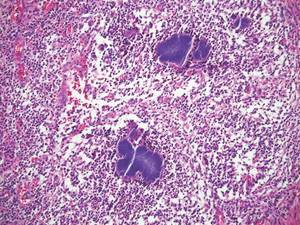
Lepromatous leprosy presents as a symmetric, widespread eruption of macules, patches, plaques, or papules that are most prominent in acral areas.4 Perivascular infiltration of lymphocytes and histiocytes is characteristic of lepromatous leprosy.2 Mycobacteria bacilli also are seen within histiocytic vacuoles, similarly to leishmaniasis; however, collections of these bacilli congregate within the center of a foamy histiocyte to form a distinctive histologic finding known as a globus. These individual histiocytes containing central globi are called Virchow cells (Figure 4).2 However, lepromatous leprosy can be distinguished from leishmaniasis histologically by carefully observing the intracellular location of the infectious organism. Mycobacteria bacilli are located in the center of a histiocyte vacuole whereas Leishmania parasites demonstrate a peripheral alignment along a histiocyte vacuole. If any uncertainty remains between a diagnosis of leishmaniasis and lepromatous leprosy, positive Fite staining for mycobacteria easily differentiates between the 2 conditions.2,4
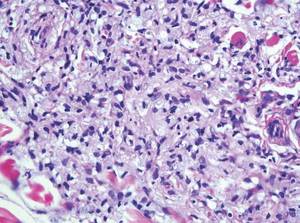
Cutaneous lobomycosis, a rare fungal infection transmitted by dolphins, manifests clinically as an asymptomatic nodule that is similar in appearance to a keloid. Histologic similarities to leishmaniasis include pseudoepitheliomatous hyperplasia and dermal granulomatous inflammation.4 The most distinguishing characteristic of lobomycosis is the presence of round, thick-walled, white organisms connected in a “string of beads” or chainlike configuration (Figure 5).2 Unlike leishmaniasis, lobomycosis fungal organisms would stain positive on periodic acid–Schiff staining.4

Cutaneous protothecosis is a rare clinical entity that presents as an isolated nodule or plaque or bursitis.4 It occurs following minor trauma and inoculation with Prototheca organisms, a genus of algae found in contaminated water.2,4 In its morula form, Prototheca adopts a characteristic arrangement within histiocytes that strikingly resembles a soccer ball (Figure 6).2 Conversely, nonmorulating forms of protothecosis can also be seen; these exhibit a central basophilic, dotlike structure within the histiocytes surrounded by a white halo.2 Definitive diagnosis of protothecosis can only be made upon successful culture of the algae.5
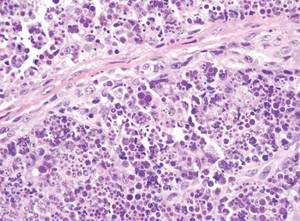
- Kevric I, Cappel MA, Keeling JH. New World and Old World leishmania infections: a practical review. Dermatol Clin. 2015;33:579-593.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 2nd ed. London, England: Elsevier Saunders; 2013.
- De Vries HJ, Van Noesel CJ, Hoekzema R, et al. Botryomycosis in an HIV-positive subject. J Eur Acad Dermatol Venereol. 2003;17:87-90.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Health Sciences UK; 2012.
- Hillesheim PB, Bahrami S. Cutaneous protothecosis. Arch Pathol Lab Med. 2011;135:941-944.
Cutaneous leishmaniasis is a parasitic infection caused by intracellular organisms found in tropical climates. Old World leishmaniasis is endemic to Asia, Africa, and parts of Europe, while New World leishmaniasis is native to Central and South Americas.1 Depending upon a host’s immune status and the specific Leishmania species, clinical presentations vary in appearance and severity, ranging from self-limited, localized cutaneous disease to potentially fatal visceral and mucocutaneous involvement. Most cutaneous manifestations of leishmaniasis begin as distinct, painless papules that may progress to nodules or become ulcerated over time.1 Histologically, leishmaniasis is diagnosed by the identification of intracellular organisms that characteristically align along the peripheral rim inside the vacuole of a histiocyte.2 This unique finding is called the “marquee sign” due to its resemblance to light bulbs arranged around a dressing room mirror (Figure 1).2Leishmania amastigotes (also known as Leishman-Donovan bodies) have kinetoplasts that are helpful in diagnosis but also may be difficult to detect.2 Along with the Leishmania parasites, there typically is a mixed inflammatory infiltrate of plasma cells, lymphocytes, histiocytes, and neutrophils (Figure 2).1,2 There also may be varying degrees of pseudoepitheliomatous hyperplasia and overlying epidermal ulceration.1
Cutaneous botryomycosis can present clinically as a number of various primary lesions, including papules, nodules, or ulcers that may resemble leishmaniasis.3 Botryomycosis represents a specific histologic collection of bacterial granules, most commonly caused by Staphylococcus aureus.3 The dermal granulomatous infiltrate seen in botryomycosis often is similar to that seen in chronic leishmaniasis; however, one histologic feature unique to botryomycosis is the presence of characteristic basophilic staphylococcal grains that are arranged in clusters resembling bunches of grapes (the term botryo means “bunch of grapes” in Greek).3 A thin, eosinophilic rim consisting of antibodies, bacterial debris, and complement proteins and glycoproteins may encircle the basophilic grains but does not need to be present for diagnosis (Figure 3).3
Lepromatous leprosy presents as a symmetric, widespread eruption of macules, patches, plaques, or papules that are most prominent in acral areas.4 Perivascular infiltration of lymphocytes and histiocytes is characteristic of lepromatous leprosy.2 Mycobacteria bacilli also are seen within histiocytic vacuoles, similarly to leishmaniasis; however, collections of these bacilli congregate within the center of a foamy histiocyte to form a distinctive histologic finding known as a globus. These individual histiocytes containing central globi are called Virchow cells (Figure 4).2 However, lepromatous leprosy can be distinguished from leishmaniasis histologically by carefully observing the intracellular location of the infectious organism. Mycobacteria bacilli are located in the center of a histiocyte vacuole whereas Leishmania parasites demonstrate a peripheral alignment along a histiocyte vacuole. If any uncertainty remains between a diagnosis of leishmaniasis and lepromatous leprosy, positive Fite staining for mycobacteria easily differentiates between the 2 conditions.2,4
Cutaneous lobomycosis, a rare fungal infection transmitted by dolphins, manifests clinically as an asymptomatic nodule that is similar in appearance to a keloid. Histologic similarities to leishmaniasis include pseudoepitheliomatous hyperplasia and dermal granulomatous inflammation.4 The most distinguishing characteristic of lobomycosis is the presence of round, thick-walled, white organisms connected in a “string of beads” or chainlike configuration (Figure 5).2 Unlike leishmaniasis, lobomycosis fungal organisms would stain positive on periodic acid–Schiff staining.4
Cutaneous protothecosis is a rare clinical entity that presents as an isolated nodule or plaque or bursitis.4 It occurs following minor trauma and inoculation with Prototheca organisms, a genus of algae found in contaminated water.2,4 In its morula form, Prototheca adopts a characteristic arrangement within histiocytes that strikingly resembles a soccer ball (Figure 6).2 Conversely, nonmorulating forms of protothecosis can also be seen; these exhibit a central basophilic, dotlike structure within the histiocytes surrounded by a white halo.2 Definitive diagnosis of protothecosis can only be made upon successful culture of the algae.5
Cutaneous leishmaniasis is a parasitic infection caused by intracellular organisms found in tropical climates. Old World leishmaniasis is endemic to Asia, Africa, and parts of Europe, while New World leishmaniasis is native to Central and South Americas.1 Depending upon a host’s immune status and the specific Leishmania species, clinical presentations vary in appearance and severity, ranging from self-limited, localized cutaneous disease to potentially fatal visceral and mucocutaneous involvement. Most cutaneous manifestations of leishmaniasis begin as distinct, painless papules that may progress to nodules or become ulcerated over time.1 Histologically, leishmaniasis is diagnosed by the identification of intracellular organisms that characteristically align along the peripheral rim inside the vacuole of a histiocyte.2 This unique finding is called the “marquee sign” due to its resemblance to light bulbs arranged around a dressing room mirror (Figure 1).2Leishmania amastigotes (also known as Leishman-Donovan bodies) have kinetoplasts that are helpful in diagnosis but also may be difficult to detect.2 Along with the Leishmania parasites, there typically is a mixed inflammatory infiltrate of plasma cells, lymphocytes, histiocytes, and neutrophils (Figure 2).1,2 There also may be varying degrees of pseudoepitheliomatous hyperplasia and overlying epidermal ulceration.1
Cutaneous botryomycosis can present clinically as a number of various primary lesions, including papules, nodules, or ulcers that may resemble leishmaniasis.3 Botryomycosis represents a specific histologic collection of bacterial granules, most commonly caused by Staphylococcus aureus.3 The dermal granulomatous infiltrate seen in botryomycosis often is similar to that seen in chronic leishmaniasis; however, one histologic feature unique to botryomycosis is the presence of characteristic basophilic staphylococcal grains that are arranged in clusters resembling bunches of grapes (the term botryo means “bunch of grapes” in Greek).3 A thin, eosinophilic rim consisting of antibodies, bacterial debris, and complement proteins and glycoproteins may encircle the basophilic grains but does not need to be present for diagnosis (Figure 3).3
Lepromatous leprosy presents as a symmetric, widespread eruption of macules, patches, plaques, or papules that are most prominent in acral areas.4 Perivascular infiltration of lymphocytes and histiocytes is characteristic of lepromatous leprosy.2 Mycobacteria bacilli also are seen within histiocytic vacuoles, similarly to leishmaniasis; however, collections of these bacilli congregate within the center of a foamy histiocyte to form a distinctive histologic finding known as a globus. These individual histiocytes containing central globi are called Virchow cells (Figure 4).2 However, lepromatous leprosy can be distinguished from leishmaniasis histologically by carefully observing the intracellular location of the infectious organism. Mycobacteria bacilli are located in the center of a histiocyte vacuole whereas Leishmania parasites demonstrate a peripheral alignment along a histiocyte vacuole. If any uncertainty remains between a diagnosis of leishmaniasis and lepromatous leprosy, positive Fite staining for mycobacteria easily differentiates between the 2 conditions.2,4
Cutaneous lobomycosis, a rare fungal infection transmitted by dolphins, manifests clinically as an asymptomatic nodule that is similar in appearance to a keloid. Histologic similarities to leishmaniasis include pseudoepitheliomatous hyperplasia and dermal granulomatous inflammation.4 The most distinguishing characteristic of lobomycosis is the presence of round, thick-walled, white organisms connected in a “string of beads” or chainlike configuration (Figure 5).2 Unlike leishmaniasis, lobomycosis fungal organisms would stain positive on periodic acid–Schiff staining.4
Cutaneous protothecosis is a rare clinical entity that presents as an isolated nodule or plaque or bursitis.4 It occurs following minor trauma and inoculation with Prototheca organisms, a genus of algae found in contaminated water.2,4 In its morula form, Prototheca adopts a characteristic arrangement within histiocytes that strikingly resembles a soccer ball (Figure 6).2 Conversely, nonmorulating forms of protothecosis can also be seen; these exhibit a central basophilic, dotlike structure within the histiocytes surrounded by a white halo.2 Definitive diagnosis of protothecosis can only be made upon successful culture of the algae.5
- Kevric I, Cappel MA, Keeling JH. New World and Old World leishmania infections: a practical review. Dermatol Clin. 2015;33:579-593.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 2nd ed. London, England: Elsevier Saunders; 2013.
- De Vries HJ, Van Noesel CJ, Hoekzema R, et al. Botryomycosis in an HIV-positive subject. J Eur Acad Dermatol Venereol. 2003;17:87-90.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Health Sciences UK; 2012.
- Hillesheim PB, Bahrami S. Cutaneous protothecosis. Arch Pathol Lab Med. 2011;135:941-944.
- Kevric I, Cappel MA, Keeling JH. New World and Old World leishmania infections: a practical review. Dermatol Clin. 2015;33:579-593.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 2nd ed. London, England: Elsevier Saunders; 2013.
- De Vries HJ, Van Noesel CJ, Hoekzema R, et al. Botryomycosis in an HIV-positive subject. J Eur Acad Dermatol Venereol. 2003;17:87-90.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Health Sciences UK; 2012.
- Hillesheim PB, Bahrami S. Cutaneous protothecosis. Arch Pathol Lab Med. 2011;135:941-944.
Expert shares ‘recipe’ for kidney stone disease
SAN DIEGO – The prevalence of kidney stone disease appears to be rising in the United States.
According to an analysis of responses from the 2007-2012 National Health and Nutrition Examination Survey (NHANES), 8.8% of people in the United States have kidney stone disease (Eur Urol. 2012 Jul;62[1]:160-5), up from a prevalence of 5.2% observed in the 1988-1994 NHANES.

“There’s also been a marked increase in emergency room visits for kidney stones: 91% between 1994 and 2006,” Dr. Anna L. Zisman said at the meeting sponsored by the American Society of Nephrology.
“Unfortunately, it doesn’t only affect adults. There has been an increased incidence in ER visits for kids as well.” Though good national data on the incidence of kidney stone disease in children are lacking, one study conducted in South Carolina found that the incidence of ER visits in children rose from 8 per 100,000 in 1996 to more than 18 per 100,000 in 2007 (J Pediatr. 2010 Jul;157[1]:132-7).
What’s driving these increases? Dr. Zisman, a nephrologist at the University of Chicago, discussed a “recipe” for how to create a kidney stone, with heredity as the first step.
“Pick your parents well,” she said. “The familial clustering index is higher for nephrolithiasis than for diabetes and hypertension. A family history of stone disease is present in 16%-37% of stone formers, compared with 4%-12% of healthy controls. And the heritability estimates – how much of a given disease or trait can be attributable to genetic predisposition – is somewhere between 46% and 63%.”
According to Mayo Clinic researchers, heritable traits for kidney stone disease based on 24-hour urine measurements, adjusted for diet, include calcium, magnesium, pH, and citrate (Clin J Am Soc Nephrol. 2014 May;9[5]:943-50).
“Hypercalciuria is the most well-established risk factor for stone disease,” Dr. Zisman said. “Up to 50% of subjects with stones have a history of hypercalciuria, and 43% of first-degree relatives of hypercalciuric patients have hypercalciuria.”
Race and gender are two factors people can’t control in their risk for kidney stone disease. NHANES data suggest that non-Hispanic whites are at highest risk for stone formation, compared with Hispanics and non-Hispanic blacks. However, among whites and all of the racial categories, males have a higher risk than females.
Step two in the recipe for stone formation is timing: Age matters.
According to an analysis of 49,976 men who participated in the Health Professionals Follow-Up Study, the highest risk of stone formation was in male patients in their 40s (J Am Soc Nephrol. 2004 Dec;15[12]:3225-32). By the time white males reach their 70s, the prevalence is almost 19%, while the prevalence for white women in their 70s is 9.4% (Eur Urol. 2012 Jul;62[1]:160-5).
Step three in the recipe is location. According to Dr. Zisman, the prevalence of kidney stone disease across the world is quite varied. “That likely has to do with both genetic predispositions as well as environmental factors,” she noted. “For example, Iran, which has a pretty warm climate, has a prevalence of 5.7%, Greece 15.2%, whereas Argentina only 4%. In the United States, data suggest that the highest incidence of stone disease is in the south. It is suspected that this is due to more men working outside in manual labor in the heat, but that’s just a hypothesis.”
Step four in the recipe involves the role of certain nutrients. For example, a higher daily calcium intake is associated with a lower risk of kidney stone formation. “The theory is that with higher dietary calcium, your urine oxalate tends to drop,” she said. Increased intake of magnesium, protein, potassium, and fluid are also associated with decreased stone formation.
On the other hand, a higher daily vitamin C intake is associated with an increased risk of stone formation. Specifically, a daily intake of more than 1,000 mg confers a 41% increased risk, compared with a lower intake. “The theory there is that vitamin C intake, once absorbed, results in a higher urine oxalate,” Dr. Zisman said.
Current epidemiology literature draws no clear association between a high-sodium diet and the development of kidney stones. From a clinical standpoint, however, “I think everyone would recommend a low-sodium diet because of the physiologic mechanisms leading to higher urine calcium,” she said.
Higher body mass index and increased waist circumference also impact the risk of developing kidney stones, especially among women. “The higher [they are], the greater the risk,” Dr. Zisman said. “We know that as weight goes up, urinary pH drops. Another potential reason is that as body weight goes up, urinary oxalate goes up as well.”
Step five in the recipe for stone formation is occupation. A study from Israel found that lifeguards in that country faced about a 20-fold increased risk of kidney stones, compared with that of the general population (Adv Exp Med Biol. 1980;128:467-72). Meanwhile, a study of glass factory workers in Italy found that the prevalence of kidney stones was 8.5% among those exposed to blast furnace sites, compared with 2.4% among those who worked in ambient temperatures (P = .03) (J Urol 1993 Dec;150[6]:1757-60). A similar finding was observed in a more recent study of steel factory workers in Brazil (Urology 2005;65[5]:858-61).
Variations to the “recipe” for kidney stones include certain genetic diseases such as primary hyperoxaluria and Dent disease; anatomic variations such as horseshoe kidney and ileal conduits; coexisting disease such as inflammatory bowel disease and primary hyperparathyroidism; and effects from medication such as acetazolamide/topiramate and prednisone.
“Mix up genetic predisposition, environmental factors, and dietary/lifestyle factors and add the magic ingredient,” Dr. Zisman concluded. “There is something that is affecting some people and not others. We don’t know what that is, and we clearly need more research.”
Dr. Zisman reported having no financial disclosures.
SAN DIEGO – The prevalence of kidney stone disease appears to be rising in the United States.
According to an analysis of responses from the 2007-2012 National Health and Nutrition Examination Survey (NHANES), 8.8% of people in the United States have kidney stone disease (Eur Urol. 2012 Jul;62[1]:160-5), up from a prevalence of 5.2% observed in the 1988-1994 NHANES.
“There’s also been a marked increase in emergency room visits for kidney stones: 91% between 1994 and 2006,” Dr. Anna L. Zisman said at the meeting sponsored by the American Society of Nephrology.
“Unfortunately, it doesn’t only affect adults. There has been an increased incidence in ER visits for kids as well.” Though good national data on the incidence of kidney stone disease in children are lacking, one study conducted in South Carolina found that the incidence of ER visits in children rose from 8 per 100,000 in 1996 to more than 18 per 100,000 in 2007 (J Pediatr. 2010 Jul;157[1]:132-7).
What’s driving these increases? Dr. Zisman, a nephrologist at the University of Chicago, discussed a “recipe” for how to create a kidney stone, with heredity as the first step.
“Pick your parents well,” she said. “The familial clustering index is higher for nephrolithiasis than for diabetes and hypertension. A family history of stone disease is present in 16%-37% of stone formers, compared with 4%-12% of healthy controls. And the heritability estimates – how much of a given disease or trait can be attributable to genetic predisposition – is somewhere between 46% and 63%.”
According to Mayo Clinic researchers, heritable traits for kidney stone disease based on 24-hour urine measurements, adjusted for diet, include calcium, magnesium, pH, and citrate (Clin J Am Soc Nephrol. 2014 May;9[5]:943-50).
“Hypercalciuria is the most well-established risk factor for stone disease,” Dr. Zisman said. “Up to 50% of subjects with stones have a history of hypercalciuria, and 43% of first-degree relatives of hypercalciuric patients have hypercalciuria.”
Race and gender are two factors people can’t control in their risk for kidney stone disease. NHANES data suggest that non-Hispanic whites are at highest risk for stone formation, compared with Hispanics and non-Hispanic blacks. However, among whites and all of the racial categories, males have a higher risk than females.
Step two in the recipe for stone formation is timing: Age matters.
According to an analysis of 49,976 men who participated in the Health Professionals Follow-Up Study, the highest risk of stone formation was in male patients in their 40s (J Am Soc Nephrol. 2004 Dec;15[12]:3225-32). By the time white males reach their 70s, the prevalence is almost 19%, while the prevalence for white women in their 70s is 9.4% (Eur Urol. 2012 Jul;62[1]:160-5).
Step three in the recipe is location. According to Dr. Zisman, the prevalence of kidney stone disease across the world is quite varied. “That likely has to do with both genetic predispositions as well as environmental factors,” she noted. “For example, Iran, which has a pretty warm climate, has a prevalence of 5.7%, Greece 15.2%, whereas Argentina only 4%. In the United States, data suggest that the highest incidence of stone disease is in the south. It is suspected that this is due to more men working outside in manual labor in the heat, but that’s just a hypothesis.”
Step four in the recipe involves the role of certain nutrients. For example, a higher daily calcium intake is associated with a lower risk of kidney stone formation. “The theory is that with higher dietary calcium, your urine oxalate tends to drop,” she said. Increased intake of magnesium, protein, potassium, and fluid are also associated with decreased stone formation.
On the other hand, a higher daily vitamin C intake is associated with an increased risk of stone formation. Specifically, a daily intake of more than 1,000 mg confers a 41% increased risk, compared with a lower intake. “The theory there is that vitamin C intake, once absorbed, results in a higher urine oxalate,” Dr. Zisman said.
Current epidemiology literature draws no clear association between a high-sodium diet and the development of kidney stones. From a clinical standpoint, however, “I think everyone would recommend a low-sodium diet because of the physiologic mechanisms leading to higher urine calcium,” she said.
Higher body mass index and increased waist circumference also impact the risk of developing kidney stones, especially among women. “The higher [they are], the greater the risk,” Dr. Zisman said. “We know that as weight goes up, urinary pH drops. Another potential reason is that as body weight goes up, urinary oxalate goes up as well.”
Step five in the recipe for stone formation is occupation. A study from Israel found that lifeguards in that country faced about a 20-fold increased risk of kidney stones, compared with that of the general population (Adv Exp Med Biol. 1980;128:467-72). Meanwhile, a study of glass factory workers in Italy found that the prevalence of kidney stones was 8.5% among those exposed to blast furnace sites, compared with 2.4% among those who worked in ambient temperatures (P = .03) (J Urol 1993 Dec;150[6]:1757-60). A similar finding was observed in a more recent study of steel factory workers in Brazil (Urology 2005;65[5]:858-61).
Variations to the “recipe” for kidney stones include certain genetic diseases such as primary hyperoxaluria and Dent disease; anatomic variations such as horseshoe kidney and ileal conduits; coexisting disease such as inflammatory bowel disease and primary hyperparathyroidism; and effects from medication such as acetazolamide/topiramate and prednisone.
“Mix up genetic predisposition, environmental factors, and dietary/lifestyle factors and add the magic ingredient,” Dr. Zisman concluded. “There is something that is affecting some people and not others. We don’t know what that is, and we clearly need more research.”
Dr. Zisman reported having no financial disclosures.
SAN DIEGO – The prevalence of kidney stone disease appears to be rising in the United States.
According to an analysis of responses from the 2007-2012 National Health and Nutrition Examination Survey (NHANES), 8.8% of people in the United States have kidney stone disease (Eur Urol. 2012 Jul;62[1]:160-5), up from a prevalence of 5.2% observed in the 1988-1994 NHANES.
“There’s also been a marked increase in emergency room visits for kidney stones: 91% between 1994 and 2006,” Dr. Anna L. Zisman said at the meeting sponsored by the American Society of Nephrology.
“Unfortunately, it doesn’t only affect adults. There has been an increased incidence in ER visits for kids as well.” Though good national data on the incidence of kidney stone disease in children are lacking, one study conducted in South Carolina found that the incidence of ER visits in children rose from 8 per 100,000 in 1996 to more than 18 per 100,000 in 2007 (J Pediatr. 2010 Jul;157[1]:132-7).
What’s driving these increases? Dr. Zisman, a nephrologist at the University of Chicago, discussed a “recipe” for how to create a kidney stone, with heredity as the first step.
“Pick your parents well,” she said. “The familial clustering index is higher for nephrolithiasis than for diabetes and hypertension. A family history of stone disease is present in 16%-37% of stone formers, compared with 4%-12% of healthy controls. And the heritability estimates – how much of a given disease or trait can be attributable to genetic predisposition – is somewhere between 46% and 63%.”
According to Mayo Clinic researchers, heritable traits for kidney stone disease based on 24-hour urine measurements, adjusted for diet, include calcium, magnesium, pH, and citrate (Clin J Am Soc Nephrol. 2014 May;9[5]:943-50).
“Hypercalciuria is the most well-established risk factor for stone disease,” Dr. Zisman said. “Up to 50% of subjects with stones have a history of hypercalciuria, and 43% of first-degree relatives of hypercalciuric patients have hypercalciuria.”
Race and gender are two factors people can’t control in their risk for kidney stone disease. NHANES data suggest that non-Hispanic whites are at highest risk for stone formation, compared with Hispanics and non-Hispanic blacks. However, among whites and all of the racial categories, males have a higher risk than females.
Step two in the recipe for stone formation is timing: Age matters.
According to an analysis of 49,976 men who participated in the Health Professionals Follow-Up Study, the highest risk of stone formation was in male patients in their 40s (J Am Soc Nephrol. 2004 Dec;15[12]:3225-32). By the time white males reach their 70s, the prevalence is almost 19%, while the prevalence for white women in their 70s is 9.4% (Eur Urol. 2012 Jul;62[1]:160-5).
Step three in the recipe is location. According to Dr. Zisman, the prevalence of kidney stone disease across the world is quite varied. “That likely has to do with both genetic predispositions as well as environmental factors,” she noted. “For example, Iran, which has a pretty warm climate, has a prevalence of 5.7%, Greece 15.2%, whereas Argentina only 4%. In the United States, data suggest that the highest incidence of stone disease is in the south. It is suspected that this is due to more men working outside in manual labor in the heat, but that’s just a hypothesis.”
Step four in the recipe involves the role of certain nutrients. For example, a higher daily calcium intake is associated with a lower risk of kidney stone formation. “The theory is that with higher dietary calcium, your urine oxalate tends to drop,” she said. Increased intake of magnesium, protein, potassium, and fluid are also associated with decreased stone formation.
On the other hand, a higher daily vitamin C intake is associated with an increased risk of stone formation. Specifically, a daily intake of more than 1,000 mg confers a 41% increased risk, compared with a lower intake. “The theory there is that vitamin C intake, once absorbed, results in a higher urine oxalate,” Dr. Zisman said.
Current epidemiology literature draws no clear association between a high-sodium diet and the development of kidney stones. From a clinical standpoint, however, “I think everyone would recommend a low-sodium diet because of the physiologic mechanisms leading to higher urine calcium,” she said.
Higher body mass index and increased waist circumference also impact the risk of developing kidney stones, especially among women. “The higher [they are], the greater the risk,” Dr. Zisman said. “We know that as weight goes up, urinary pH drops. Another potential reason is that as body weight goes up, urinary oxalate goes up as well.”
Step five in the recipe for stone formation is occupation. A study from Israel found that lifeguards in that country faced about a 20-fold increased risk of kidney stones, compared with that of the general population (Adv Exp Med Biol. 1980;128:467-72). Meanwhile, a study of glass factory workers in Italy found that the prevalence of kidney stones was 8.5% among those exposed to blast furnace sites, compared with 2.4% among those who worked in ambient temperatures (P = .03) (J Urol 1993 Dec;150[6]:1757-60). A similar finding was observed in a more recent study of steel factory workers in Brazil (Urology 2005;65[5]:858-61).
Variations to the “recipe” for kidney stones include certain genetic diseases such as primary hyperoxaluria and Dent disease; anatomic variations such as horseshoe kidney and ileal conduits; coexisting disease such as inflammatory bowel disease and primary hyperparathyroidism; and effects from medication such as acetazolamide/topiramate and prednisone.
“Mix up genetic predisposition, environmental factors, and dietary/lifestyle factors and add the magic ingredient,” Dr. Zisman concluded. “There is something that is affecting some people and not others. We don’t know what that is, and we clearly need more research.”
Dr. Zisman reported having no financial disclosures.
EXPERT ANALYSIS AT KIDNEY WEEK 2015

Decision making worse in suicide attempters with unipolar and bipolar disorders
There is a link between poor decision making and suicidal tendencies in patients with unipolar depressive disorder and bipolar disorder, according to Dr. Stéphane Richard-Devantoy and his associates.
In the first part of the study, suicide attempters with unipolar and bipolar disorders took the Iowa Gambling Task (IGT), as did a control group of non–suicide attempters with unipolar and bipolar disorders. IGT scores were significantly worse in the suicide-attempting group, compared with the control group, with most of the difference coming from patients with unipolar disorder. Women who attempted suicide also had significantly worse IGT scores than did male suicide attempters.
In a subsequent meta-analysis of 10 studies and 1,148 participants, impaired decision making in suicide attempters with unipolar and bipolar disorders, compared with nonattempters with unipolar and bipolar disorders was confirmed with a moderate effect size. The effect size was moderate between unipolar suicide attempters and a healthy control group, and large between bipolar suicide attempters and a healthy control group.
“Examination of sex differences in brain functioning appears to be necessary in [the] suicide field. Indeed, females consistently attempt suicide more than males,whereas males consistently die by suicide more frequently than females. In spite of the important role gender plays in the expression of suicidal behavior, the perspective of future tailor-made therapeutic modalities in suicide attempters is needed,” the investigators noted.
Find the full study in the Journal of Affective Disorders (doi: 10.1016/j.jad.2015.10.001).
There is a link between poor decision making and suicidal tendencies in patients with unipolar depressive disorder and bipolar disorder, according to Dr. Stéphane Richard-Devantoy and his associates.
In the first part of the study, suicide attempters with unipolar and bipolar disorders took the Iowa Gambling Task (IGT), as did a control group of non–suicide attempters with unipolar and bipolar disorders. IGT scores were significantly worse in the suicide-attempting group, compared with the control group, with most of the difference coming from patients with unipolar disorder. Women who attempted suicide also had significantly worse IGT scores than did male suicide attempters.
In a subsequent meta-analysis of 10 studies and 1,148 participants, impaired decision making in suicide attempters with unipolar and bipolar disorders, compared with nonattempters with unipolar and bipolar disorders was confirmed with a moderate effect size. The effect size was moderate between unipolar suicide attempters and a healthy control group, and large between bipolar suicide attempters and a healthy control group.
“Examination of sex differences in brain functioning appears to be necessary in [the] suicide field. Indeed, females consistently attempt suicide more than males,whereas males consistently die by suicide more frequently than females. In spite of the important role gender plays in the expression of suicidal behavior, the perspective of future tailor-made therapeutic modalities in suicide attempters is needed,” the investigators noted.
Find the full study in the Journal of Affective Disorders (doi: 10.1016/j.jad.2015.10.001).
There is a link between poor decision making and suicidal tendencies in patients with unipolar depressive disorder and bipolar disorder, according to Dr. Stéphane Richard-Devantoy and his associates.
In the first part of the study, suicide attempters with unipolar and bipolar disorders took the Iowa Gambling Task (IGT), as did a control group of non–suicide attempters with unipolar and bipolar disorders. IGT scores were significantly worse in the suicide-attempting group, compared with the control group, with most of the difference coming from patients with unipolar disorder. Women who attempted suicide also had significantly worse IGT scores than did male suicide attempters.
In a subsequent meta-analysis of 10 studies and 1,148 participants, impaired decision making in suicide attempters with unipolar and bipolar disorders, compared with nonattempters with unipolar and bipolar disorders was confirmed with a moderate effect size. The effect size was moderate between unipolar suicide attempters and a healthy control group, and large between bipolar suicide attempters and a healthy control group.
“Examination of sex differences in brain functioning appears to be necessary in [the] suicide field. Indeed, females consistently attempt suicide more than males,whereas males consistently die by suicide more frequently than females. In spite of the important role gender plays in the expression of suicidal behavior, the perspective of future tailor-made therapeutic modalities in suicide attempters is needed,” the investigators noted.
Find the full study in the Journal of Affective Disorders (doi: 10.1016/j.jad.2015.10.001).
FROM THE JOURNAL OF AFFECTIVE DISORDERS
Where Leading GOP Presidential Candidates Stand on Health Policies
As the long 2016 presidential election season draws on, Republican hopefuls strive to stand out among their fellow party candidates; however, many in the running remain tacit about specific policies on issues ranging from immigration to gun control and healthcare.
“Many of these candidates … do not feel like getting involved in an extensive policy discussion will influence whether they win Iowa and New Hampshire,” says Robert Blendon, ScD, professor of health policy and political analysis at the Harvard School of Public Health and Harvard Kennedy School of Government in Cambridge, Mass. “They see it as a distraction, because the people voting are not asking them.”
For physicians and others passionate about healthcare, “it’s very frustrating,” Dr. Blendon says. “People who are on the Republican side want a replacement [for the Affordable Care Act], but they are not driven—I have seen the surveys—to want to really know the details of that replacement.”
GOP candidates share many common ideas about the U.S. health system. Most say they want to allow people under age 26 to remain on their parents’ health plans and believe people with preexisting conditions should have access to coverage, generally through the creation of state-based, high-risk insurance pools. They believe expanded health savings accounts will give patients more skin in the game, and, across the board, they have vowed to “repeal and replace Obamacare.”
Listen to more of our interview with Robert Blendon, ScD
However, “with more than 10 candidates, there is going to be variation,” Dr. Blendon adds.
For instance, former Florida Governor Jeb Bush has proposed the Conservative Plan for 21st Century Health, which aims to “lower costs,” “promote innovation,” and “return power to states.”
Neurosurgeon Ben Carson originally suggested he would “abolish” Medicare and instead provide seniors with a $2,000-a-year federal subsidy to purchase private insurance. He has backtracked that idea and, in December 2015, issued a report highlighting the pillars of his health plan, which include creating “health empowerment accounts” and raising the Medicare age to 70.
New Jersey Governor Chris Christie’s plan suggests a priority for veterans, including the formation of a federal Secretary of Veterans Affairs, while Carly Fiorina says that “every healthcare provider “ought to publish its costs, its prices, its outcomes” so patients know what they are buying.
“As the field on the Republican side narrows, I think we will start to see more pressure on them to flesh those principles out a little bit more,” says Joshua Lenchus, DO, RPh, FACP, SFHM, a hospitalist at the University of Miami (Fla.) Jackson Memorial Hospital and a member of SHM’s Public Policy Committee.
Some GOP candidates, like Kentucky Senator and ophthalmologist Rand Paul, have proposed reforming medical malpractice. Some wish to make insurance portable from one job to the next, like former Arkansas Governor Mike Huckabee, or across state lines, as Ohio Governor John Kasich has proposed.
Some of these ideas, says hospitalist and SHM Public Policy Committee member Bradley Flansbaum, DO, MPH, MHM, “have been adequately dismembered, and they’re not going to carry weight.
“Buying insurance across state lines, fixing malpractice—that is not going to fix the healthcare system,” says Dr. Flansbaum, clinical professor of medicine at NYU School of Medicine in New York City.
Overall, a Republican-sponsored healthcare system will not guarantee the same level of comprehensive benefits patients have now under the ACA, Dr. Blendon says, and, in general, subsidies and tax credits will be less generous than they are today, in turn reducing federal expenditures.
Most Republican candidates are in favor of some version of free market healthcare, but Dr. Flansbaum points out that “there are so many imperfections in the market, everything from people having asymmetric information—a physician knows a lot more than a patient does—to opaque pricing,” he says. “It’s not exchanging goods like we are used to.”
Republicans are generally committed to “less federal government, less expenditures, more choices, and less expensive benefits,” in healthcare, but Dr. Blendon says the system “would not go back to 2009.”
For hospitalists interested in election-year or other healthcare policy issues, Dr. Flansbaum suggests getting involved in the SHM committee, visiting the advocacy section of the SHM website, and reaching out to local representatives and others who write and vote on laws.
“How do you affect change?” he asks. “It’s not sitting in the breakfast lounge at the hospital bellyaching to your colleagues.” TH
Editor's note: update Jan. 4, 2016.
Kelly April Tyrrell is a freelance writer in Madison, Wis.
As the long 2016 presidential election season draws on, Republican hopefuls strive to stand out among their fellow party candidates; however, many in the running remain tacit about specific policies on issues ranging from immigration to gun control and healthcare.
“Many of these candidates … do not feel like getting involved in an extensive policy discussion will influence whether they win Iowa and New Hampshire,” says Robert Blendon, ScD, professor of health policy and political analysis at the Harvard School of Public Health and Harvard Kennedy School of Government in Cambridge, Mass. “They see it as a distraction, because the people voting are not asking them.”
For physicians and others passionate about healthcare, “it’s very frustrating,” Dr. Blendon says. “People who are on the Republican side want a replacement [for the Affordable Care Act], but they are not driven—I have seen the surveys—to want to really know the details of that replacement.”
GOP candidates share many common ideas about the U.S. health system. Most say they want to allow people under age 26 to remain on their parents’ health plans and believe people with preexisting conditions should have access to coverage, generally through the creation of state-based, high-risk insurance pools. They believe expanded health savings accounts will give patients more skin in the game, and, across the board, they have vowed to “repeal and replace Obamacare.”
Listen to more of our interview with Robert Blendon, ScD
However, “with more than 10 candidates, there is going to be variation,” Dr. Blendon adds.
For instance, former Florida Governor Jeb Bush has proposed the Conservative Plan for 21st Century Health, which aims to “lower costs,” “promote innovation,” and “return power to states.”
Neurosurgeon Ben Carson originally suggested he would “abolish” Medicare and instead provide seniors with a $2,000-a-year federal subsidy to purchase private insurance. He has backtracked that idea and, in December 2015, issued a report highlighting the pillars of his health plan, which include creating “health empowerment accounts” and raising the Medicare age to 70.
New Jersey Governor Chris Christie’s plan suggests a priority for veterans, including the formation of a federal Secretary of Veterans Affairs, while Carly Fiorina says that “every healthcare provider “ought to publish its costs, its prices, its outcomes” so patients know what they are buying.
“As the field on the Republican side narrows, I think we will start to see more pressure on them to flesh those principles out a little bit more,” says Joshua Lenchus, DO, RPh, FACP, SFHM, a hospitalist at the University of Miami (Fla.) Jackson Memorial Hospital and a member of SHM’s Public Policy Committee.
Some GOP candidates, like Kentucky Senator and ophthalmologist Rand Paul, have proposed reforming medical malpractice. Some wish to make insurance portable from one job to the next, like former Arkansas Governor Mike Huckabee, or across state lines, as Ohio Governor John Kasich has proposed.
Some of these ideas, says hospitalist and SHM Public Policy Committee member Bradley Flansbaum, DO, MPH, MHM, “have been adequately dismembered, and they’re not going to carry weight.
“Buying insurance across state lines, fixing malpractice—that is not going to fix the healthcare system,” says Dr. Flansbaum, clinical professor of medicine at NYU School of Medicine in New York City.
Overall, a Republican-sponsored healthcare system will not guarantee the same level of comprehensive benefits patients have now under the ACA, Dr. Blendon says, and, in general, subsidies and tax credits will be less generous than they are today, in turn reducing federal expenditures.
Most Republican candidates are in favor of some version of free market healthcare, but Dr. Flansbaum points out that “there are so many imperfections in the market, everything from people having asymmetric information—a physician knows a lot more than a patient does—to opaque pricing,” he says. “It’s not exchanging goods like we are used to.”
Republicans are generally committed to “less federal government, less expenditures, more choices, and less expensive benefits,” in healthcare, but Dr. Blendon says the system “would not go back to 2009.”
For hospitalists interested in election-year or other healthcare policy issues, Dr. Flansbaum suggests getting involved in the SHM committee, visiting the advocacy section of the SHM website, and reaching out to local representatives and others who write and vote on laws.
“How do you affect change?” he asks. “It’s not sitting in the breakfast lounge at the hospital bellyaching to your colleagues.” TH
Editor's note: update Jan. 4, 2016.
Kelly April Tyrrell is a freelance writer in Madison, Wis.
As the long 2016 presidential election season draws on, Republican hopefuls strive to stand out among their fellow party candidates; however, many in the running remain tacit about specific policies on issues ranging from immigration to gun control and healthcare.
“Many of these candidates … do not feel like getting involved in an extensive policy discussion will influence whether they win Iowa and New Hampshire,” says Robert Blendon, ScD, professor of health policy and political analysis at the Harvard School of Public Health and Harvard Kennedy School of Government in Cambridge, Mass. “They see it as a distraction, because the people voting are not asking them.”
For physicians and others passionate about healthcare, “it’s very frustrating,” Dr. Blendon says. “People who are on the Republican side want a replacement [for the Affordable Care Act], but they are not driven—I have seen the surveys—to want to really know the details of that replacement.”
GOP candidates share many common ideas about the U.S. health system. Most say they want to allow people under age 26 to remain on their parents’ health plans and believe people with preexisting conditions should have access to coverage, generally through the creation of state-based, high-risk insurance pools. They believe expanded health savings accounts will give patients more skin in the game, and, across the board, they have vowed to “repeal and replace Obamacare.”
Listen to more of our interview with Robert Blendon, ScD
However, “with more than 10 candidates, there is going to be variation,” Dr. Blendon adds.
For instance, former Florida Governor Jeb Bush has proposed the Conservative Plan for 21st Century Health, which aims to “lower costs,” “promote innovation,” and “return power to states.”
Neurosurgeon Ben Carson originally suggested he would “abolish” Medicare and instead provide seniors with a $2,000-a-year federal subsidy to purchase private insurance. He has backtracked that idea and, in December 2015, issued a report highlighting the pillars of his health plan, which include creating “health empowerment accounts” and raising the Medicare age to 70.
New Jersey Governor Chris Christie’s plan suggests a priority for veterans, including the formation of a federal Secretary of Veterans Affairs, while Carly Fiorina says that “every healthcare provider “ought to publish its costs, its prices, its outcomes” so patients know what they are buying.
“As the field on the Republican side narrows, I think we will start to see more pressure on them to flesh those principles out a little bit more,” says Joshua Lenchus, DO, RPh, FACP, SFHM, a hospitalist at the University of Miami (Fla.) Jackson Memorial Hospital and a member of SHM’s Public Policy Committee.
Some GOP candidates, like Kentucky Senator and ophthalmologist Rand Paul, have proposed reforming medical malpractice. Some wish to make insurance portable from one job to the next, like former Arkansas Governor Mike Huckabee, or across state lines, as Ohio Governor John Kasich has proposed.
Some of these ideas, says hospitalist and SHM Public Policy Committee member Bradley Flansbaum, DO, MPH, MHM, “have been adequately dismembered, and they’re not going to carry weight.
“Buying insurance across state lines, fixing malpractice—that is not going to fix the healthcare system,” says Dr. Flansbaum, clinical professor of medicine at NYU School of Medicine in New York City.
Overall, a Republican-sponsored healthcare system will not guarantee the same level of comprehensive benefits patients have now under the ACA, Dr. Blendon says, and, in general, subsidies and tax credits will be less generous than they are today, in turn reducing federal expenditures.
Most Republican candidates are in favor of some version of free market healthcare, but Dr. Flansbaum points out that “there are so many imperfections in the market, everything from people having asymmetric information—a physician knows a lot more than a patient does—to opaque pricing,” he says. “It’s not exchanging goods like we are used to.”
Republicans are generally committed to “less federal government, less expenditures, more choices, and less expensive benefits,” in healthcare, but Dr. Blendon says the system “would not go back to 2009.”
For hospitalists interested in election-year or other healthcare policy issues, Dr. Flansbaum suggests getting involved in the SHM committee, visiting the advocacy section of the SHM website, and reaching out to local representatives and others who write and vote on laws.
“How do you affect change?” he asks. “It’s not sitting in the breakfast lounge at the hospital bellyaching to your colleagues.” TH
Editor's note: update Jan. 4, 2016.
Kelly April Tyrrell is a freelance writer in Madison, Wis.