User login
Some Readmission Risk Factors Not Captured by Medicare
Clinical question: Are there patient characteristics not currently measured by the Medicare readmission program that account for differences in hospital readmission rates?

Background: The Medicare Hospital Readmissions Reduction Program (HRRP) financially penalizes hospitals with higher than expected 30-day readmission rates. During 2014, more than 2,000 U.S. hospitals were fined $480 million for high readmission rates. HRRP accounts for differences in patient age, gender, discharge diagnosis, and diagnoses identified in Medicare claims over the previous 12 months; however, the impact of other factors is uncertain.
Study design: Survey data from the Health and Retirement Study, with linked Medicare claims.
Setting: Community-dwelling U.S. adults, older than 50 years.
Synopsis: Investigators analyzed more than 33,000 admissions from 2000 to 2012. They found 22 patient characteristics not included in the HRRP calculation that were statistically significantly predictive of hospital-wide, 30-day readmission and were more likely to be present among patients cared for in hospitals in the highest quintile of readmission rates. These characteristics reduced by 48% the differences in readmission rate between the highest- and lowest-performing quintiles. Examples include patient ethnicity, education level, personal as well as household income level, presence of prescription drug plan, Medicaid enrollment, cognitive status, and numerous others.
Bottom line: Patient characteristics account for much of the difference in readmission rates between high- and low-performing hospitals, suggesting that HRRP penalties reflect who hospitals treat as much as how well they treat them.
Citation: Barnett ML, Hsu J, McWilliams JM. Patient characteristics and differences in hospital readmission rates. JAMA Intern Med. 2015;175(11):1803-1812.
Clinical question: Are there patient characteristics not currently measured by the Medicare readmission program that account for differences in hospital readmission rates?
Background: The Medicare Hospital Readmissions Reduction Program (HRRP) financially penalizes hospitals with higher than expected 30-day readmission rates. During 2014, more than 2,000 U.S. hospitals were fined $480 million for high readmission rates. HRRP accounts for differences in patient age, gender, discharge diagnosis, and diagnoses identified in Medicare claims over the previous 12 months; however, the impact of other factors is uncertain.
Study design: Survey data from the Health and Retirement Study, with linked Medicare claims.
Setting: Community-dwelling U.S. adults, older than 50 years.
Synopsis: Investigators analyzed more than 33,000 admissions from 2000 to 2012. They found 22 patient characteristics not included in the HRRP calculation that were statistically significantly predictive of hospital-wide, 30-day readmission and were more likely to be present among patients cared for in hospitals in the highest quintile of readmission rates. These characteristics reduced by 48% the differences in readmission rate between the highest- and lowest-performing quintiles. Examples include patient ethnicity, education level, personal as well as household income level, presence of prescription drug plan, Medicaid enrollment, cognitive status, and numerous others.
Bottom line: Patient characteristics account for much of the difference in readmission rates between high- and low-performing hospitals, suggesting that HRRP penalties reflect who hospitals treat as much as how well they treat them.
Citation: Barnett ML, Hsu J, McWilliams JM. Patient characteristics and differences in hospital readmission rates. JAMA Intern Med. 2015;175(11):1803-1812.
Clinical question: Are there patient characteristics not currently measured by the Medicare readmission program that account for differences in hospital readmission rates?
Background: The Medicare Hospital Readmissions Reduction Program (HRRP) financially penalizes hospitals with higher than expected 30-day readmission rates. During 2014, more than 2,000 U.S. hospitals were fined $480 million for high readmission rates. HRRP accounts for differences in patient age, gender, discharge diagnosis, and diagnoses identified in Medicare claims over the previous 12 months; however, the impact of other factors is uncertain.
Study design: Survey data from the Health and Retirement Study, with linked Medicare claims.
Setting: Community-dwelling U.S. adults, older than 50 years.
Synopsis: Investigators analyzed more than 33,000 admissions from 2000 to 2012. They found 22 patient characteristics not included in the HRRP calculation that were statistically significantly predictive of hospital-wide, 30-day readmission and were more likely to be present among patients cared for in hospitals in the highest quintile of readmission rates. These characteristics reduced by 48% the differences in readmission rate between the highest- and lowest-performing quintiles. Examples include patient ethnicity, education level, personal as well as household income level, presence of prescription drug plan, Medicaid enrollment, cognitive status, and numerous others.
Bottom line: Patient characteristics account for much of the difference in readmission rates between high- and low-performing hospitals, suggesting that HRRP penalties reflect who hospitals treat as much as how well they treat them.
Citation: Barnett ML, Hsu J, McWilliams JM. Patient characteristics and differences in hospital readmission rates. JAMA Intern Med. 2015;175(11):1803-1812.

Review: Opioid prescriptions are the work of many physicians
A “broad swath” of Medicare providers wrote scripts for opioids in 2013, contradicting the idea that the overdose epidemic is mainly the work of “small groups of prolific prescribers and corrupt pill mills,” investigators wrote online in JAMA Internal Medicine.
“Contrary to the California workers’ compensation data showing a small subset of prescribers accounting for a disproportionately large percentage of opioid prescribing, Medicare opioid prescribing is distributed across many prescribers and is, if anything, less skewed than all drug prescribing,” said Dr. Jonathan H. Chen of the Veterans Affairs Palo Alto (Calif.) Health Care System, and his associates.
Their study included 808,020 prescribers and almost 1.2 billion Medicare Part D claims worth nearly $81 billion dollars. They focused on schedule II opioid prescriptions containing oxycodone, fentanyl, hydrocodone, morphine, methadone, hydromorphone, oxymorphone, meperidine, codeine, opium, or levorphanol (JAMA Intern Med. 2015 Dec 14. doi: 10.1001/jamainternmed.2015.6662).
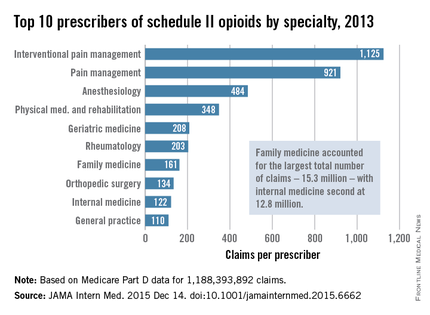
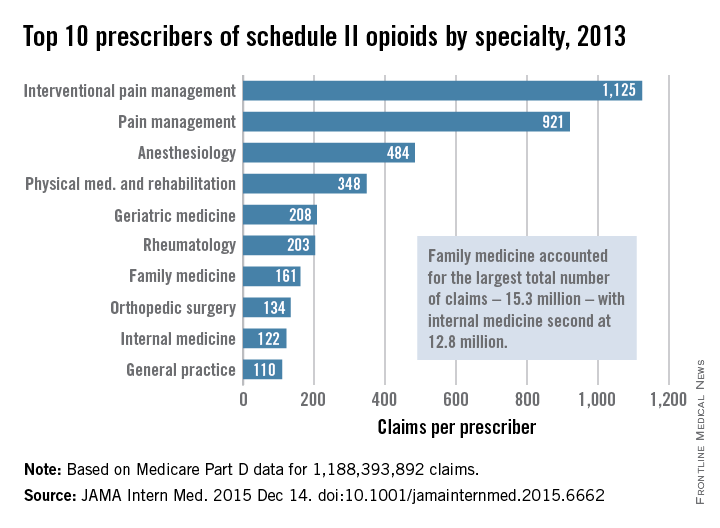
Not surprisingly, specialists in pain management, anesthesia, and physical medicine wrote the most prescriptions per provider. But family practitioners, internists, nurse practitioners, and physician assistants wrote 35,268,234 prescriptions – more than all other specialties combined. “The trends hold up across state lines, with negligible geographic variability,” the researchers said.
The findings contradict an analysis of California workers’ compensation data, in which 1% of prescribers accounted for a third of schedule II opioid prescriptions, and 10% of prescribers accounted for 80% of prescriptions, the investigators noted. Nonetheless, 10% of Medicare prescribers in Dr. Chen’s study accounted for 78% of the total cost of opioids, possibly because they were prescribing pricier formulations or higher doses.
Overall, the findings suggest that opioid prescribing is “widespread” and “relatively indifferent to individual physicians, specialty, or region” – and that efforts to stem the tide must be equally broad, the researchers concluded.
Their study was supported by the VA Office of Academic Affiliations, the VA Health Services Research and Development Service, the National Institute of General Medical Sciences, and the Peter F. McManus Charitable Trust. The researchers had no disclosures.
A “broad swath” of Medicare providers wrote scripts for opioids in 2013, contradicting the idea that the overdose epidemic is mainly the work of “small groups of prolific prescribers and corrupt pill mills,” investigators wrote online in JAMA Internal Medicine.
“Contrary to the California workers’ compensation data showing a small subset of prescribers accounting for a disproportionately large percentage of opioid prescribing, Medicare opioid prescribing is distributed across many prescribers and is, if anything, less skewed than all drug prescribing,” said Dr. Jonathan H. Chen of the Veterans Affairs Palo Alto (Calif.) Health Care System, and his associates.
Their study included 808,020 prescribers and almost 1.2 billion Medicare Part D claims worth nearly $81 billion dollars. They focused on schedule II opioid prescriptions containing oxycodone, fentanyl, hydrocodone, morphine, methadone, hydromorphone, oxymorphone, meperidine, codeine, opium, or levorphanol (JAMA Intern Med. 2015 Dec 14. doi: 10.1001/jamainternmed.2015.6662).
Not surprisingly, specialists in pain management, anesthesia, and physical medicine wrote the most prescriptions per provider. But family practitioners, internists, nurse practitioners, and physician assistants wrote 35,268,234 prescriptions – more than all other specialties combined. “The trends hold up across state lines, with negligible geographic variability,” the researchers said.
The findings contradict an analysis of California workers’ compensation data, in which 1% of prescribers accounted for a third of schedule II opioid prescriptions, and 10% of prescribers accounted for 80% of prescriptions, the investigators noted. Nonetheless, 10% of Medicare prescribers in Dr. Chen’s study accounted for 78% of the total cost of opioids, possibly because they were prescribing pricier formulations or higher doses.
Overall, the findings suggest that opioid prescribing is “widespread” and “relatively indifferent to individual physicians, specialty, or region” – and that efforts to stem the tide must be equally broad, the researchers concluded.
Their study was supported by the VA Office of Academic Affiliations, the VA Health Services Research and Development Service, the National Institute of General Medical Sciences, and the Peter F. McManus Charitable Trust. The researchers had no disclosures.
A “broad swath” of Medicare providers wrote scripts for opioids in 2013, contradicting the idea that the overdose epidemic is mainly the work of “small groups of prolific prescribers and corrupt pill mills,” investigators wrote online in JAMA Internal Medicine.
“Contrary to the California workers’ compensation data showing a small subset of prescribers accounting for a disproportionately large percentage of opioid prescribing, Medicare opioid prescribing is distributed across many prescribers and is, if anything, less skewed than all drug prescribing,” said Dr. Jonathan H. Chen of the Veterans Affairs Palo Alto (Calif.) Health Care System, and his associates.
Their study included 808,020 prescribers and almost 1.2 billion Medicare Part D claims worth nearly $81 billion dollars. They focused on schedule II opioid prescriptions containing oxycodone, fentanyl, hydrocodone, morphine, methadone, hydromorphone, oxymorphone, meperidine, codeine, opium, or levorphanol (JAMA Intern Med. 2015 Dec 14. doi: 10.1001/jamainternmed.2015.6662).
Not surprisingly, specialists in pain management, anesthesia, and physical medicine wrote the most prescriptions per provider. But family practitioners, internists, nurse practitioners, and physician assistants wrote 35,268,234 prescriptions – more than all other specialties combined. “The trends hold up across state lines, with negligible geographic variability,” the researchers said.
The findings contradict an analysis of California workers’ compensation data, in which 1% of prescribers accounted for a third of schedule II opioid prescriptions, and 10% of prescribers accounted for 80% of prescriptions, the investigators noted. Nonetheless, 10% of Medicare prescribers in Dr. Chen’s study accounted for 78% of the total cost of opioids, possibly because they were prescribing pricier formulations or higher doses.
Overall, the findings suggest that opioid prescribing is “widespread” and “relatively indifferent to individual physicians, specialty, or region” – and that efforts to stem the tide must be equally broad, the researchers concluded.
Their study was supported by the VA Office of Academic Affiliations, the VA Health Services Research and Development Service, the National Institute of General Medical Sciences, and the Peter F. McManus Charitable Trust. The researchers had no disclosures.
FROM JAMA INTERNAL MEDICINE
Key clinical point: Many different types of general practitioners and specialists often prescribe opioids to Medicare beneficiaries.
Major finding: Family practitioners, internists, nurse practitioners, and physician assistants wrote 35,268,234 prescriptions – more than all other specialties combined.
Data source: An analysis of nearly 1.2 billion Medicare part D claims from 2013.
Disclosures: The study was supported by the VA Office of Academic Affiliations, the VA Health Services Research and Development Service, the National Institute of General Medical Sciences, and the Peter F. McManus Charitable Trust. The researchers had no disclosures.
Status Report From the American Acne & Rosacea Society on Medical Management of Acne in Adult Women, Part 3: Oral Therapies
Selection of oral agents for treatment of AV in adult women is dependent on multiple factors including the patient’s age, medication history, child-bearing potential, clinical presentation, and treatment preference following a discussion of the anticipated benefits versus potential risks.1,2 In patients with the mixed inflammatory and comedonal clinical pattern of AV, oral antibiotics can be used concurrently with topical therapies when moderate to severe inflammatory lesions are noted.3,4 However, many adult women who had AV as teenagers have already utilized oral antibiotic therapies in the past and often are interested in alternative options, express concerns regarding antibiotic resistance, report a history of antibiotic-associated yeast infections or other side effects, and/or encounter issues related to drug-drug interactions.3,5-8 Oral hormonal therapies such as combination oral contraceptives (COCs) or spironolactone often are utilized to treat adult women with AV, sometimes in combination with each other or other agents. Combination oral contraceptives appear to be especially effective in the management of the U-shaped clinical pattern or predominantly inflammatory, late-onset AV.1,5,9,10 Potential warnings, contraindications, adverse effects, and drug-drug interactions are important to keep in mind when considering the use of oral hormonal therapies.8-10 Oral isotretinoin, which should be prescribed with strict adherence to the iPLEDGE™ program (https://www.ipledgeprogram.com/), remains a viable option for cases of severe nodular AV and selected cases of refractory inflammatory AV, especially when scarring and/or marked psychosocial distress are noted.1,2,5,11 Although it is recognized that adult women with AV typically present with either a mixed inflammatory and comedonal or U-shaped clinical pattern predominantly involving the lower face and anterolateral neck, the available data do not adequately differentiate the relative responsiveness of these clinical patterns to specific therapeutic agents.
Combination Oral Contraceptives
Combination oral contraceptives are commonly used to treat AV in adult women, including those without and those with measurable androgen excess (eg, polycystic ovary syndrome [PCOS]). Combination oral contraceptives contain ethinyl estradiol and a progestational agent (eg, progestin); the latter varies in terms of its nonselective receptor interactions and the relative magnitude or absence of androgenic effects.10,12,13 Although some COCs are approved by the US Food and Drug Administration (FDA) for AV, there is little data available to determine the comparative efficacy among these and other COCs.10,14 When choosing a COC for treatment of AV, it is best to select an agent whose effectiveness is supported by evidence from clinical studies.10,15
Mechanisms of Action
The reported mechanisms of action for COCs include inhibition of ovarian androgen production and ovulation through gonadotropin suppression; upregulated synthesis of sex hormone–binding globulin, which decreases free testosterone levels through receptor binding; and inhibition of 5α-reductase (by some progestins), which reduces conversion of testosterone to dihydrotestosterone, the active derivative that induces androgenic effects at peripheral target tissues.10,13,16,17
Therapeutic Benefits
Use of COCs to treat AV in adult women who do not have measurable androgen excess is most rational in patients who also desire a method of contraception. Multiple monotherapy studies have demonstrated the efficacy of COCs in the treatment of AV on the face and trunk.4,10,12,15,17,18 It may take a minimum of 3 monthly cycles of use before acne lesion counts begin to appreciably decrease.12,15,19-21 Initiating COC therapy during menstruation ensures the absence of pregnancy. Combination oral contraceptives may be used with other topical and oral therapies for AV.2,3,9,10 Potential ancillary benefits of COCs include normalization of the menstrual cycle; reduced premenstrual dysphoric disorder symptoms; and reduced risk of endometrial cancer (approximately 50%), ovarian cancer (approximately 40%), and colorectal cancer.22-24
Risks and Contraindications
It is important to consider the potential risks associated with the use of COCs, especially in women with AV who are not seeking a method of contraception. Side effects of COCs can include nausea, breast tenderness, breakthrough bleeding, and weight gain.25,26 Potential adverse associations of COCs are described in the Table. The major potential vascular associations include venous thromboembolism, myocardial infarction, and cerebrovascular accident, all of which are influenced by concurrent factors such as a history of smoking, age (≥35 years), and hypertension.27-32 It is recommended that blood pressure be measured before initiating COC therapy as part of the general examination.33

The potential increase in breast cancer risk appears to be low, while the cervical cancer risk is reported to increase relative to the duration of use.34-37 This latter observation may be due to the greater likelihood of unprotected sex in women using a COC and exposure to multiple sexual partners in some cases, which may increase the likelihood of oncogenic human papillomavirus infection of the cervix. If a dermatologist elects to prescribe a COC to treat AV, it has been suggested that the patient also consult with her general practitioner or gynecologist to undergo pelvic and breast examinations and a Papanicolaou test.33 The recommendation for initial screening for cervical cancer is within 3 years of initiation of sexual intercourse or by 21 years of age, whichever is first.33,38,39
Combination oral contraceptives are not ideal for all adult women with AV. Absolute contraindications are pregnancy and history of thromboembolic, cardiac, or hepatic disease; in women aged 35 years and older who smoke, relative contraindications include hypertension, diabetes, migraines, breastfeeding, and current breast or liver cancer.33 In adult women with AV who have relative contra-indications but are likely to benefit from the use of a COC when other options are limited or not viable, consultation with a gynecologist is prudent. Other than rifamycin antibiotics (eg, rifampin) and griseofulvin, there is no definitive evidence that oral antibiotics (eg, tetracycline) or oral antifungal agents reduce the contraceptive efficacy of COCs, although cautions remain in print within some approved package inserts.8
Spironolactone
Available since 1957, spironolactone is an oral aldos-terone antagonist and potassium-sparing diuretic used to treat hypertension and congestive heart failure.9 Recognition of its antiandrogenic effects led to its use in dermatology to treat certain dermatologic disorders in women (eg, hirsutism, alopecia, AV).1,4,5,9,10 Spironolactone is not approved for AV by the FDA; therefore, available data from multiple independent studies and retrospective analyses that have been collectively reviewed support its efficacy when used as both monotherapy or in combination with other agents in adult women with AV, especially those with a U-shaped pattern and/or late-onset AV.9,40-43
Mechanism of Action
Spironolactone inhibits sebaceous gland activity through peripheral androgen receptor blockade, inhibition of 5α-reductase, decrease in androgen production, and increase in sex hormone–binding globulin.9,10,40
Therapeutic Benefits
Good to excellent improvement of AV in women, many of whom are postadolescent, has ranged from 66% to 100% in published reports9,40-43; however, inclusion and exclusion criteria, dosing regimens, and concomitant therapies were not usually controlled. Spironolactone has been used to treat AV in adult women as monotherapy or in combination with topical agents, oral antibiotics, and COCs.9,40-42 Additionally, dose-ranging studies have not been completed with spironolactone for AV.9,40 The suggested dose range is 50 mg to 200 mg daily; however, it usually is best to start at 50 mg daily and increase to 100 mg daily if clinical response is not adequate after 2 to 3 months. The gastrointestinal (GI) absorption of spironolactone is increased when ingested with a high-fat meal.9,10
Once effective control of AV is achieved, it is optimal to use the lowest dose needed to continue reasonable suppression of new AV lesions. There is no defined end point for spironolactone use in AV, with or without concurrent PCOS, as many adult women usually continue treatment with low-dose therapy because they experience marked flaring shortly after the drug is stopped.9
Risks and Contraindications
Side effects associated with spironolactone are dose related and include increased diuresis, migraines, menstrual irregularities, breast tenderness, gynecomastia, fatigue, and dizziness.9,10,40-44 Side effects (particularly menstrual irregularities and breast tenderness) are more common at doses higher than 100 mg daily, especially when used as monotherapy without concurrent use of a COC.9,40
Spironolactone-associated hyperkalemia is most clinically relevant in patients on higher doses (eg, 100–200 mg daily), in those with renal impairment and/or congestive heart failure, and when used concurrently with certain other medications. In any patient on spironolactone, the risk of clinically relevant hyperkalemia may be increased by coingestion of potassium supplements, potassium-based salt substitutes, potassium-sparing diuretics (eg, amiloride, triamterene); aldosterone antagonists and angiotensin-converting enzyme inhibitors (eg, lisinopril, benazepril); angiotensin II receptor blockers (eg, losartan, valsartan); and tri-methoprim (with or without sulfamethoxazole).8,9,40,45 Spironolactone may also increase serum levels of lithium or digoxin.9,40,45,46 For management of AV, it is best that spironolactone be avoided in patients taking any of these medications.9
In healthy adult women with AV who are not on medications or supplements that interact adversely with spironolactone, there is no definitive recommendation regarding monitoring of serum potassium levels during treatment with spironolactone, and it has been suggested that monitoring serum potassium levels in this subgroup is not necessary.47 However, each clinician is advised to choose whether or not they wish to obtain baseline and/or periodic serum potassium levels when prescribing spironolactone for AV based on their degree of comfort and the patient’s history. Baseline and periodic blood testing to evaluate serum electrolytes and renal function are reasonable, especially as adult women with AV are usually treated with spironolactone over a prolonged period of time.9
The FDA black box warning for spironolactone states that it is tumorigenic in chronic toxicity studies in rats and refers to exposures 25- to 100-fold higher than those administered to humans.9,48 Although continued vigilance is warranted, evaluation of large populations of women treated with spironolactone do not suggest an association with increased risk of breast cancer.49,50
Spironolactone is a category C drug and thus should be avoided during pregnancy, primarily due to animal data suggesting risks of hypospadias and feminization in male fetuses.9 Importantly, there is an absence of reports linking exposure during pregnancy with congenital defects in humans, including in 2 known cases of high-dose exposures for maternal Bartter syndrome.9
The active metabolite, canrenone, is known to be present in breast milk at 0.2% of the maternal daily dose, but breastfeeding is generally believed to be safe with spironolactone based on evidence to date.9
Oral Antibiotics
Oral antibiotic therapy may be used in combination with a topical regimen to treat AV in adult women, keeping in mind some important caveats.1-7 For instance, monotherapy with oral antibiotics should be avoided, and concomitant use of benzoyl peroxide is suggested to reduce emergence of antibiotic-resistant Propionibacterium acnes strains.3,4 A therapeutic exit plan also is suggested when prescribing oral antibiotics to limit treatment to 3 to 4 months, if possible, to help mitigate the emergence of antibiotic-resistant bacteria (eg, staphylococci and streptococci).3-5,51
Tetracyclines, especially doxycycline and minocycline, are the most commonly prescribed agents. Doxycycline use warrants patient education on measures to limit the risks of esophageal and GI side effects and phototoxicity; enteric-coated and small tablet formulations have been shown to reduce GI side effects, especially when administered with food.3,52-55 In addition to vestibular side effects and hyperpigmentation, minocycline may be associated with rare but potentially severe adverse reactions such as drug hypersensitivity syndrome, autoimmune hepatitis, and lupus-like syndrome, which are reported more commonly in women.5,52,54 Vestibular side effects have been shown to decrease with use of extended-release tablets with weight-based dosing.53
Oral Isotretinoin
Oral isotretinoin is well established as highly effective for treatment of severe, recalcitrant AV, including nodular acne on the face and trunk.4,56 Currently available oral isotretinoins are branded generic formulations based on the pharmacokinetic profile of the original brand (Accutane [Roche Pharmaceuticals]) and with the use of Lidose Technology (Absorica [Cipher Pharmaceuticals]), which substantially increases GI absorption of isotretinoin in the absence of ingestion with a high-calorie, high-fat meal.57 The short- and long-term efficacy, dosing regimens, safety considerations, and serious teratogenic risks for oral isotretinoin are well published.4,56-58 Importantly, oral isotretinoin must be prescribed with strict adherence to the federally mandated iPLEDGE risk management program.
Low-dose oral isotretinoin therapy (<0.5 mg/kg–1 mg/kg daily) administered over several months longer than conventional regimens (ie, 16–20 weeks) has been suggested with demonstrated efficacy.57 However, this approach is not optimal due to the lack of established sustained clearance of AV after discontinuation of therapy and the greater potential for exposure to isotretinoin during pregnancy. Recurrences of AV do occur after completion of isotretinoin therapy, especially if cumulative systemic exposure to the drug during the initial course of treatment was inadequate.56,57
Oral isotretinoin has been shown to be effective in AV in adult women with or without PCOS with 0.5 mg/kg to 1 mg/kg daily and a total cumulative exposure of 120 mg/kg to 150 mg/kg.59 In one study, the presence of PCOS and greater number of nodules at baseline were predictive of a higher risk of relapse during the second year posttreatment.59
Conclusion
All oral therapies that are used to treat AV in adult women warrant individual consideration of possible benefits versus risks. Careful attention to possible side effects, patient-related risk factors, and potential drug-drug interactions is important. End points of therapy are not well established, with the exception of oral isotretinoin therapy. Clinicians must use their judgment in each case along with obtaining feedback from patients regarding the selection of therapy after a discussion of the available options.
- Holzmann R, Shakery K. Postadolescent acne in females. Skin Pharmacol Physiol. 2014;27(suppl 1):3-8.
- Villasenor J, Berson DS, Kroshinsky D. Treatment guidelines in adult women. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:198-207.
- Del Rosso JQ, Kim G. Optimizing use of oral antibiotics in acne vulgaris. Dermatol Clin. 2009;27:33-42.
- Gollnick H, Cunliffe W, Berson D, et al. Management of acne: report from a Global Alliance to Improve Outcomes in Acne. J Am Acad Dermatol. 2003;49(suppl 1):S1-S37.
- Fisk WA, Lev-Tov HA, Sivamani RK. Epidemiology and management of acne in adult women. Curr Derm Rep. 2014;3:29-39.
- Del Rosso JQ, Leyden JJ. Status report on antibiotic resistance: implications for the dermatologist. Dermatol Clin. 2007;25:127-132.
- Bowe WP, Leyden JJ. Clinical implications of antibiotic resistance: risk of systemic infection from Staphylococcus and Streptococcus. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:125-133.
- Del Rosso JQ. Oral antibiotic drug interactions of clinical significance to dermatologists. Dermatol Clin. 2009;27:91-94.
- Kim GK, Del Rosso JQ. Oral spironolactone in post-teenage female patients with acne vulgaris: practical considerations for the clinician based on current data and clinical experience. J Clin Aesthet Dermatol. 2012;5:37-50.
- Keri J, Berson DS, Thiboutot DM. Hormonal treatment of acne in women. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:146-155.
- American Academy of Dermatology. Position statement on isotretinoin. AAD Web site. https://www.aad.org /Forms/Policies/Uploads/PS/PS-Isotretinoin.pdf. Updated November 13, 2010. Accessed October 28, 2015.
- Arowojolu AO, Gallo MF, Lopez LM, et al. Combined oral contraceptive pills for treatment of acne. Cochrane Database Syst Rev. June 2012;7:CD004425.
- Sitruk-Ware R. Pharmacology of different progestogens: the special case of drospirenone. Climacteric. 2005;8 (suppl 3):4-12.
- Arowojolu AO, Gallo MF, Lopez LM, et al. Combined oral contraceptive pills for the treatment of acne. Cochrane Database Syst Rev. July 2012;7:CD004425.
- Thiboutot D, Archer DF, Lemay A, et al. A randomized, controlled trial of a low-dose contraceptive containing 20 microg of ethinyl estradiol and 100 microg of levonogestrel for acne treatment. Fertil Steril. 2001;76:461-468.
- Koulianos GT. Treatment of acne with oral contraceptives: criteria for pill selection. Cutis. 2000;66:281-286.
- Rabe T, Kowald A, Ortmann J, et al. Inhibition of skin 5-alpha reductase by oral contraceptive progestins in vitro. Gynecol Endocrinol. 2000;14:223-230.
- Palli MB, Reyes-Habito CM, Lima XT, et al. A single-center, randomized double-blind, parallel-group study to examine the safety and efficacy of 3mg drospirenone/0.02mg ethinyl estradiol compared with placebo in the treatment of moderate truncal acne vulgaris. J Drugs Dermatol. 2013;12:633-637.
- Koltun W, Maloney JM, Marr J, et al. Treatment of moderate acne vulgaris using a combined oral contraceptive containing ethinylestradiol 20 μg plus drospirenone 3 mg administered in a 24/4 regimen: a pooled analysis. Eur J Obstet Gynecol Reprod Biol. 2011;155:171-175.
- Maloney JM, Dietze P, Watson D, et al. A randomized controlled trial of a low-dose combined oral contraceptive containing 3 mg drospirenone plus 20 μg ethinylestradiol in the treatment of acne vulgaris: lesion counts, investigator ratings and subject self-assessment. J Drugs Dermatol. 2009;8:837-844.
- Lucky AW, Koltun W, Thiboutot D, et al. A combined oral contraceptive containing 3-mg drospirenone/20-μg ethinyl estradiol in the treatment of acne vulgaris: a randomized, double-blind, placebo-controlled study evaluating lesion counts and participant self-assessment. Cutis. 2008;82:143-150.
- Burkman R, Schlesselman JJ, Zieman M. Safety concerns and health benefits associated with oral contraception. Am J Obstet Gynecol. 2004;190(suppl 4):S5-S22.
- Maguire K, Westhoff C. The state of hormonal contraception today: established and emerging noncontraceptive health benefits. Am J Obstet Gynecol. 2011;205 (suppl 4):S4-S8.
- Weiss NS, Sayvetz TA. Incidence of endometrial cancer in relation to the use of oral contraceptives. N Engl J Med. 1980;302:551-554.
- Tyler KH, Zirwas MJ. Contraception and the dermatologist. J Am Acad Dermatol. 2013;68:1022-1029.
- Gallo MF, Lopez LM, Grimes DA, et al. Combination contraceptives: effects on weight. Cochrane Database Syst Rev. 2008;4:CD003987.
- de Bastos M, Stegeman BH, Rosendaal FR, et al. Combined oral contraceptives: venous thrombosis. Cochrane Database Syst Rev. 2014;3:CD010813.
- Raymond EG, Burke AE, Espey E. Combined hormonal contraceptives and venous thromboembolism: putting the risks into perspective. Obstet Gynecol. 2012;119:1039-1044.
- Jick SS, Hernandez RK. Risk of non-fatal venous thromboembolism in women using oral contraceptives containing drospirenone compared with women using oral contraceptives containing levonorgestrel: case-control study using United States claims data. BMJ. 2011;342:d2151.
- US Food and Drug Administration Office of Surveillance and Epidemiology. Combined hormonal contraceptives (CHCs) and the risk of cardiovascular disease endpoints. US Food and Drug Administration Web site. http://www.fda.gov/downloads/Drugs /Drug Safety/UCM277384.pdf. Accessed October 28, 2015.
- The American College of Obstetricians and Gynecologists Committee on Gynecologic Practice. Risk of venous thromboembolism among users of drospirenone-containing oral contraceptive pills. Obstet Gynecol. 2012;120:1239-1242.
- World Health Organization. Cardiovascular Disease and Steroid Hormone Contraception: Report of a WHO Scientific Group. Geneva, Switzerland: World Health Organization; 1998. Technical Report Series 877.
- Frangos JE, Alavian CN, Kimball AB. Acne and oral contraceptives: update on women’s health screening guidelines. J Am Acad Dermatol. 2008;58:781-786.
- Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormonal contraceptives: collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Lancet. 1996;347:1713-1727.
- Gierisch JM, Coeytaux RR, Urrutia RP, et al. Oral contraceptive use and risk of breast, cervical, colorectal, and endometrial cancers: a systematic review. Cancer Epidemiol Biomarkers Prev. 2013;22:1931-1943.
- International Collaboration of Epidemiological Studies of Cervical Cancer. Cervical cancer and hormonal contraceptives: collaborative reanalysis of individual data for 16 573 women with cervical cancer and 35 509 women without cervical cancer from 24 epidemiological studies. Lancet. 2007;370:1609-1621.
- Agostino H, Di Meglio G. Low-dose oral contraceptives in adolescents: how low can you go? J Pediatr Adolesc Gynecol. 2010;23:195-201.
- Buzney E, Sheu J, Buzney C, et al. Polycystic ovary syndrome: a review for dermatologists: part II. Treatment. J Am Acad Dermatol. 2014;71:859.e1-859.e15.
- Stewart FH, Harper CC, Ellertson CE, et al. Clinical breast and pelvic examination requirements for hormonal contraception: current practice vs evidence. JAMA. 2001;285:2232-2239.
- Sawaya ME, Somani N. Antiandrogens and androgen inhibitors. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelpha, PA: Saunders; 2013:361-374.
- Muhlemann MF, Carter GD, Cream JJ, et al. Oral spironolactone: an effective treatment for acne vulgaris in women. Br J Dermatol. 1986;115:227-232.
- Shaw JC. Low-dose adjunctive spironolactone in the treatment of acne in women: a retrospective analysis of 85 consecutively treated patients. J Am Acad Dermatol. 2000;43:498-502.
- Sato K, Matsumoto D, Iizuka F, et al. Anti-androgenic therapy using oral spironolactone for acne vulgaris in Asians. Aesth Plast Surg. 2006;30:689-694.
- Shaw JC, White LE. Long-term safety of spironolactone in acne: results of an 8-year follow-up study. J Cutan Med Surg. 2002;6:541-545.
- Stockley I. Antihypertensive drug interactions. In: Stockley I, ed. Drug Interactions. 5th ed. London, United Kingdom: Pharmaceutical Press; 1999:335-347.
- Antoniou T, Gomes T, Mamdani MM, et al. Trimethoprim-sulfamethoxazole induced hyperkalaemia in elderly patients receiving spironolactone: nested case-control study. BMJ. 2011;343:d5228.
- Plovanich M, Weng QY, Mostaghimi A. Low usefulness of potassium monitoring among healthy young women taking spironolactone for acne. JAMA Dermatol. 2015;151:941-944.
- Aldactone [package insert]. New York, NY: Pfizer Inc; 2008.
- Biggar RJ, Andersen EW, Wohlfahrt J, et al. Spironolactone use and the risk of breast and gynecologic cancers. Cancer Epidemiol. 2013;37:870-875.
- Mackenzie IS, Macdonald TM, Thompson A, et al. Spironolactone and risk of incident breast cancer in women older than 55 years: retrospective, matched cohort study. BMJ. 2012;345:e4447.
- Dreno B, Thiboutot D, Gollnick H, et al. Antibiotic stewardship in dermatology: limiting antibiotic use in acne. Eur J Dermatol. 2014;24:330-334.
- Kim S, Michaels BD, Kim GK, et al. Systemic antibacterial agents. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelpha, PA: Saunders; 2013:61-97.
- Leyden JJ, Del Rosso JQ. Oral antibiotic therapy for acne vulgaris: pharmacokinetic and pharmacodynamics perspectives. J Clin Aesthet Dermatol. 2011;4:40-47.
- Del Rosso JQ. Oral antibiotics. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:113-124.
- Del Rosso JQ. Oral doxycycline in the management of acne vulgaris: current perspectives on clinical use and recent findings with a new double-scored small tablet formulation. J Clin Aesthet Dermatol. 2015;8:19-26.
- Osofsky MG, Strauss JS. Isotretinoin. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:134-145.
- Leyden JJ, Del Rosso JQ, Baum EW. The use of isotretinoin in the treatment of acne vulgaris: clinical considerations and future directions. J Clin Aesthet Dermatol. 2014;7(suppl 2):S3-S21.
- Patton TJ, Ferris LK. Systemic retinoids. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelpha, PA: Saunders; 2013:252-268.
- Cakir GA, Erdogan FG, Gurler A. Isotretinoin treatment in nodulocystic acne with and without polycystic ovary syndrome: efficacy and determinants of relapse. Int J Dermatol. 2013;52:371-376.
Selection of oral agents for treatment of AV in adult women is dependent on multiple factors including the patient’s age, medication history, child-bearing potential, clinical presentation, and treatment preference following a discussion of the anticipated benefits versus potential risks.1,2 In patients with the mixed inflammatory and comedonal clinical pattern of AV, oral antibiotics can be used concurrently with topical therapies when moderate to severe inflammatory lesions are noted.3,4 However, many adult women who had AV as teenagers have already utilized oral antibiotic therapies in the past and often are interested in alternative options, express concerns regarding antibiotic resistance, report a history of antibiotic-associated yeast infections or other side effects, and/or encounter issues related to drug-drug interactions.3,5-8 Oral hormonal therapies such as combination oral contraceptives (COCs) or spironolactone often are utilized to treat adult women with AV, sometimes in combination with each other or other agents. Combination oral contraceptives appear to be especially effective in the management of the U-shaped clinical pattern or predominantly inflammatory, late-onset AV.1,5,9,10 Potential warnings, contraindications, adverse effects, and drug-drug interactions are important to keep in mind when considering the use of oral hormonal therapies.8-10 Oral isotretinoin, which should be prescribed with strict adherence to the iPLEDGE™ program (https://www.ipledgeprogram.com/), remains a viable option for cases of severe nodular AV and selected cases of refractory inflammatory AV, especially when scarring and/or marked psychosocial distress are noted.1,2,5,11 Although it is recognized that adult women with AV typically present with either a mixed inflammatory and comedonal or U-shaped clinical pattern predominantly involving the lower face and anterolateral neck, the available data do not adequately differentiate the relative responsiveness of these clinical patterns to specific therapeutic agents.
Combination Oral Contraceptives
Combination oral contraceptives are commonly used to treat AV in adult women, including those without and those with measurable androgen excess (eg, polycystic ovary syndrome [PCOS]). Combination oral contraceptives contain ethinyl estradiol and a progestational agent (eg, progestin); the latter varies in terms of its nonselective receptor interactions and the relative magnitude or absence of androgenic effects.10,12,13 Although some COCs are approved by the US Food and Drug Administration (FDA) for AV, there is little data available to determine the comparative efficacy among these and other COCs.10,14 When choosing a COC for treatment of AV, it is best to select an agent whose effectiveness is supported by evidence from clinical studies.10,15
Mechanisms of Action
The reported mechanisms of action for COCs include inhibition of ovarian androgen production and ovulation through gonadotropin suppression; upregulated synthesis of sex hormone–binding globulin, which decreases free testosterone levels through receptor binding; and inhibition of 5α-reductase (by some progestins), which reduces conversion of testosterone to dihydrotestosterone, the active derivative that induces androgenic effects at peripheral target tissues.10,13,16,17
Therapeutic Benefits
Use of COCs to treat AV in adult women who do not have measurable androgen excess is most rational in patients who also desire a method of contraception. Multiple monotherapy studies have demonstrated the efficacy of COCs in the treatment of AV on the face and trunk.4,10,12,15,17,18 It may take a minimum of 3 monthly cycles of use before acne lesion counts begin to appreciably decrease.12,15,19-21 Initiating COC therapy during menstruation ensures the absence of pregnancy. Combination oral contraceptives may be used with other topical and oral therapies for AV.2,3,9,10 Potential ancillary benefits of COCs include normalization of the menstrual cycle; reduced premenstrual dysphoric disorder symptoms; and reduced risk of endometrial cancer (approximately 50%), ovarian cancer (approximately 40%), and colorectal cancer.22-24
Risks and Contraindications
It is important to consider the potential risks associated with the use of COCs, especially in women with AV who are not seeking a method of contraception. Side effects of COCs can include nausea, breast tenderness, breakthrough bleeding, and weight gain.25,26 Potential adverse associations of COCs are described in the Table. The major potential vascular associations include venous thromboembolism, myocardial infarction, and cerebrovascular accident, all of which are influenced by concurrent factors such as a history of smoking, age (≥35 years), and hypertension.27-32 It is recommended that blood pressure be measured before initiating COC therapy as part of the general examination.33
The potential increase in breast cancer risk appears to be low, while the cervical cancer risk is reported to increase relative to the duration of use.34-37 This latter observation may be due to the greater likelihood of unprotected sex in women using a COC and exposure to multiple sexual partners in some cases, which may increase the likelihood of oncogenic human papillomavirus infection of the cervix. If a dermatologist elects to prescribe a COC to treat AV, it has been suggested that the patient also consult with her general practitioner or gynecologist to undergo pelvic and breast examinations and a Papanicolaou test.33 The recommendation for initial screening for cervical cancer is within 3 years of initiation of sexual intercourse or by 21 years of age, whichever is first.33,38,39
Combination oral contraceptives are not ideal for all adult women with AV. Absolute contraindications are pregnancy and history of thromboembolic, cardiac, or hepatic disease; in women aged 35 years and older who smoke, relative contraindications include hypertension, diabetes, migraines, breastfeeding, and current breast or liver cancer.33 In adult women with AV who have relative contra-indications but are likely to benefit from the use of a COC when other options are limited or not viable, consultation with a gynecologist is prudent. Other than rifamycin antibiotics (eg, rifampin) and griseofulvin, there is no definitive evidence that oral antibiotics (eg, tetracycline) or oral antifungal agents reduce the contraceptive efficacy of COCs, although cautions remain in print within some approved package inserts.8
Spironolactone
Available since 1957, spironolactone is an oral aldos-terone antagonist and potassium-sparing diuretic used to treat hypertension and congestive heart failure.9 Recognition of its antiandrogenic effects led to its use in dermatology to treat certain dermatologic disorders in women (eg, hirsutism, alopecia, AV).1,4,5,9,10 Spironolactone is not approved for AV by the FDA; therefore, available data from multiple independent studies and retrospective analyses that have been collectively reviewed support its efficacy when used as both monotherapy or in combination with other agents in adult women with AV, especially those with a U-shaped pattern and/or late-onset AV.9,40-43
Mechanism of Action
Spironolactone inhibits sebaceous gland activity through peripheral androgen receptor blockade, inhibition of 5α-reductase, decrease in androgen production, and increase in sex hormone–binding globulin.9,10,40
Therapeutic Benefits
Good to excellent improvement of AV in women, many of whom are postadolescent, has ranged from 66% to 100% in published reports9,40-43; however, inclusion and exclusion criteria, dosing regimens, and concomitant therapies were not usually controlled. Spironolactone has been used to treat AV in adult women as monotherapy or in combination with topical agents, oral antibiotics, and COCs.9,40-42 Additionally, dose-ranging studies have not been completed with spironolactone for AV.9,40 The suggested dose range is 50 mg to 200 mg daily; however, it usually is best to start at 50 mg daily and increase to 100 mg daily if clinical response is not adequate after 2 to 3 months. The gastrointestinal (GI) absorption of spironolactone is increased when ingested with a high-fat meal.9,10
Once effective control of AV is achieved, it is optimal to use the lowest dose needed to continue reasonable suppression of new AV lesions. There is no defined end point for spironolactone use in AV, with or without concurrent PCOS, as many adult women usually continue treatment with low-dose therapy because they experience marked flaring shortly after the drug is stopped.9
Risks and Contraindications
Side effects associated with spironolactone are dose related and include increased diuresis, migraines, menstrual irregularities, breast tenderness, gynecomastia, fatigue, and dizziness.9,10,40-44 Side effects (particularly menstrual irregularities and breast tenderness) are more common at doses higher than 100 mg daily, especially when used as monotherapy without concurrent use of a COC.9,40
Spironolactone-associated hyperkalemia is most clinically relevant in patients on higher doses (eg, 100–200 mg daily), in those with renal impairment and/or congestive heart failure, and when used concurrently with certain other medications. In any patient on spironolactone, the risk of clinically relevant hyperkalemia may be increased by coingestion of potassium supplements, potassium-based salt substitutes, potassium-sparing diuretics (eg, amiloride, triamterene); aldosterone antagonists and angiotensin-converting enzyme inhibitors (eg, lisinopril, benazepril); angiotensin II receptor blockers (eg, losartan, valsartan); and tri-methoprim (with or without sulfamethoxazole).8,9,40,45 Spironolactone may also increase serum levels of lithium or digoxin.9,40,45,46 For management of AV, it is best that spironolactone be avoided in patients taking any of these medications.9
In healthy adult women with AV who are not on medications or supplements that interact adversely with spironolactone, there is no definitive recommendation regarding monitoring of serum potassium levels during treatment with spironolactone, and it has been suggested that monitoring serum potassium levels in this subgroup is not necessary.47 However, each clinician is advised to choose whether or not they wish to obtain baseline and/or periodic serum potassium levels when prescribing spironolactone for AV based on their degree of comfort and the patient’s history. Baseline and periodic blood testing to evaluate serum electrolytes and renal function are reasonable, especially as adult women with AV are usually treated with spironolactone over a prolonged period of time.9
The FDA black box warning for spironolactone states that it is tumorigenic in chronic toxicity studies in rats and refers to exposures 25- to 100-fold higher than those administered to humans.9,48 Although continued vigilance is warranted, evaluation of large populations of women treated with spironolactone do not suggest an association with increased risk of breast cancer.49,50
Spironolactone is a category C drug and thus should be avoided during pregnancy, primarily due to animal data suggesting risks of hypospadias and feminization in male fetuses.9 Importantly, there is an absence of reports linking exposure during pregnancy with congenital defects in humans, including in 2 known cases of high-dose exposures for maternal Bartter syndrome.9
The active metabolite, canrenone, is known to be present in breast milk at 0.2% of the maternal daily dose, but breastfeeding is generally believed to be safe with spironolactone based on evidence to date.9
Oral Antibiotics
Oral antibiotic therapy may be used in combination with a topical regimen to treat AV in adult women, keeping in mind some important caveats.1-7 For instance, monotherapy with oral antibiotics should be avoided, and concomitant use of benzoyl peroxide is suggested to reduce emergence of antibiotic-resistant Propionibacterium acnes strains.3,4 A therapeutic exit plan also is suggested when prescribing oral antibiotics to limit treatment to 3 to 4 months, if possible, to help mitigate the emergence of antibiotic-resistant bacteria (eg, staphylococci and streptococci).3-5,51
Tetracyclines, especially doxycycline and minocycline, are the most commonly prescribed agents. Doxycycline use warrants patient education on measures to limit the risks of esophageal and GI side effects and phototoxicity; enteric-coated and small tablet formulations have been shown to reduce GI side effects, especially when administered with food.3,52-55 In addition to vestibular side effects and hyperpigmentation, minocycline may be associated with rare but potentially severe adverse reactions such as drug hypersensitivity syndrome, autoimmune hepatitis, and lupus-like syndrome, which are reported more commonly in women.5,52,54 Vestibular side effects have been shown to decrease with use of extended-release tablets with weight-based dosing.53
Oral Isotretinoin
Oral isotretinoin is well established as highly effective for treatment of severe, recalcitrant AV, including nodular acne on the face and trunk.4,56 Currently available oral isotretinoins are branded generic formulations based on the pharmacokinetic profile of the original brand (Accutane [Roche Pharmaceuticals]) and with the use of Lidose Technology (Absorica [Cipher Pharmaceuticals]), which substantially increases GI absorption of isotretinoin in the absence of ingestion with a high-calorie, high-fat meal.57 The short- and long-term efficacy, dosing regimens, safety considerations, and serious teratogenic risks for oral isotretinoin are well published.4,56-58 Importantly, oral isotretinoin must be prescribed with strict adherence to the federally mandated iPLEDGE risk management program.
Low-dose oral isotretinoin therapy (<0.5 mg/kg–1 mg/kg daily) administered over several months longer than conventional regimens (ie, 16–20 weeks) has been suggested with demonstrated efficacy.57 However, this approach is not optimal due to the lack of established sustained clearance of AV after discontinuation of therapy and the greater potential for exposure to isotretinoin during pregnancy. Recurrences of AV do occur after completion of isotretinoin therapy, especially if cumulative systemic exposure to the drug during the initial course of treatment was inadequate.56,57
Oral isotretinoin has been shown to be effective in AV in adult women with or without PCOS with 0.5 mg/kg to 1 mg/kg daily and a total cumulative exposure of 120 mg/kg to 150 mg/kg.59 In one study, the presence of PCOS and greater number of nodules at baseline were predictive of a higher risk of relapse during the second year posttreatment.59
Conclusion
All oral therapies that are used to treat AV in adult women warrant individual consideration of possible benefits versus risks. Careful attention to possible side effects, patient-related risk factors, and potential drug-drug interactions is important. End points of therapy are not well established, with the exception of oral isotretinoin therapy. Clinicians must use their judgment in each case along with obtaining feedback from patients regarding the selection of therapy after a discussion of the available options.
Selection of oral agents for treatment of AV in adult women is dependent on multiple factors including the patient’s age, medication history, child-bearing potential, clinical presentation, and treatment preference following a discussion of the anticipated benefits versus potential risks.1,2 In patients with the mixed inflammatory and comedonal clinical pattern of AV, oral antibiotics can be used concurrently with topical therapies when moderate to severe inflammatory lesions are noted.3,4 However, many adult women who had AV as teenagers have already utilized oral antibiotic therapies in the past and often are interested in alternative options, express concerns regarding antibiotic resistance, report a history of antibiotic-associated yeast infections or other side effects, and/or encounter issues related to drug-drug interactions.3,5-8 Oral hormonal therapies such as combination oral contraceptives (COCs) or spironolactone often are utilized to treat adult women with AV, sometimes in combination with each other or other agents. Combination oral contraceptives appear to be especially effective in the management of the U-shaped clinical pattern or predominantly inflammatory, late-onset AV.1,5,9,10 Potential warnings, contraindications, adverse effects, and drug-drug interactions are important to keep in mind when considering the use of oral hormonal therapies.8-10 Oral isotretinoin, which should be prescribed with strict adherence to the iPLEDGE™ program (https://www.ipledgeprogram.com/), remains a viable option for cases of severe nodular AV and selected cases of refractory inflammatory AV, especially when scarring and/or marked psychosocial distress are noted.1,2,5,11 Although it is recognized that adult women with AV typically present with either a mixed inflammatory and comedonal or U-shaped clinical pattern predominantly involving the lower face and anterolateral neck, the available data do not adequately differentiate the relative responsiveness of these clinical patterns to specific therapeutic agents.
Combination Oral Contraceptives
Combination oral contraceptives are commonly used to treat AV in adult women, including those without and those with measurable androgen excess (eg, polycystic ovary syndrome [PCOS]). Combination oral contraceptives contain ethinyl estradiol and a progestational agent (eg, progestin); the latter varies in terms of its nonselective receptor interactions and the relative magnitude or absence of androgenic effects.10,12,13 Although some COCs are approved by the US Food and Drug Administration (FDA) for AV, there is little data available to determine the comparative efficacy among these and other COCs.10,14 When choosing a COC for treatment of AV, it is best to select an agent whose effectiveness is supported by evidence from clinical studies.10,15
Mechanisms of Action
The reported mechanisms of action for COCs include inhibition of ovarian androgen production and ovulation through gonadotropin suppression; upregulated synthesis of sex hormone–binding globulin, which decreases free testosterone levels through receptor binding; and inhibition of 5α-reductase (by some progestins), which reduces conversion of testosterone to dihydrotestosterone, the active derivative that induces androgenic effects at peripheral target tissues.10,13,16,17
Therapeutic Benefits
Use of COCs to treat AV in adult women who do not have measurable androgen excess is most rational in patients who also desire a method of contraception. Multiple monotherapy studies have demonstrated the efficacy of COCs in the treatment of AV on the face and trunk.4,10,12,15,17,18 It may take a minimum of 3 monthly cycles of use before acne lesion counts begin to appreciably decrease.12,15,19-21 Initiating COC therapy during menstruation ensures the absence of pregnancy. Combination oral contraceptives may be used with other topical and oral therapies for AV.2,3,9,10 Potential ancillary benefits of COCs include normalization of the menstrual cycle; reduced premenstrual dysphoric disorder symptoms; and reduced risk of endometrial cancer (approximately 50%), ovarian cancer (approximately 40%), and colorectal cancer.22-24
Risks and Contraindications
It is important to consider the potential risks associated with the use of COCs, especially in women with AV who are not seeking a method of contraception. Side effects of COCs can include nausea, breast tenderness, breakthrough bleeding, and weight gain.25,26 Potential adverse associations of COCs are described in the Table. The major potential vascular associations include venous thromboembolism, myocardial infarction, and cerebrovascular accident, all of which are influenced by concurrent factors such as a history of smoking, age (≥35 years), and hypertension.27-32 It is recommended that blood pressure be measured before initiating COC therapy as part of the general examination.33
The potential increase in breast cancer risk appears to be low, while the cervical cancer risk is reported to increase relative to the duration of use.34-37 This latter observation may be due to the greater likelihood of unprotected sex in women using a COC and exposure to multiple sexual partners in some cases, which may increase the likelihood of oncogenic human papillomavirus infection of the cervix. If a dermatologist elects to prescribe a COC to treat AV, it has been suggested that the patient also consult with her general practitioner or gynecologist to undergo pelvic and breast examinations and a Papanicolaou test.33 The recommendation for initial screening for cervical cancer is within 3 years of initiation of sexual intercourse or by 21 years of age, whichever is first.33,38,39
Combination oral contraceptives are not ideal for all adult women with AV. Absolute contraindications are pregnancy and history of thromboembolic, cardiac, or hepatic disease; in women aged 35 years and older who smoke, relative contraindications include hypertension, diabetes, migraines, breastfeeding, and current breast or liver cancer.33 In adult women with AV who have relative contra-indications but are likely to benefit from the use of a COC when other options are limited or not viable, consultation with a gynecologist is prudent. Other than rifamycin antibiotics (eg, rifampin) and griseofulvin, there is no definitive evidence that oral antibiotics (eg, tetracycline) or oral antifungal agents reduce the contraceptive efficacy of COCs, although cautions remain in print within some approved package inserts.8
Spironolactone
Available since 1957, spironolactone is an oral aldos-terone antagonist and potassium-sparing diuretic used to treat hypertension and congestive heart failure.9 Recognition of its antiandrogenic effects led to its use in dermatology to treat certain dermatologic disorders in women (eg, hirsutism, alopecia, AV).1,4,5,9,10 Spironolactone is not approved for AV by the FDA; therefore, available data from multiple independent studies and retrospective analyses that have been collectively reviewed support its efficacy when used as both monotherapy or in combination with other agents in adult women with AV, especially those with a U-shaped pattern and/or late-onset AV.9,40-43
Mechanism of Action
Spironolactone inhibits sebaceous gland activity through peripheral androgen receptor blockade, inhibition of 5α-reductase, decrease in androgen production, and increase in sex hormone–binding globulin.9,10,40
Therapeutic Benefits
Good to excellent improvement of AV in women, many of whom are postadolescent, has ranged from 66% to 100% in published reports9,40-43; however, inclusion and exclusion criteria, dosing regimens, and concomitant therapies were not usually controlled. Spironolactone has been used to treat AV in adult women as monotherapy or in combination with topical agents, oral antibiotics, and COCs.9,40-42 Additionally, dose-ranging studies have not been completed with spironolactone for AV.9,40 The suggested dose range is 50 mg to 200 mg daily; however, it usually is best to start at 50 mg daily and increase to 100 mg daily if clinical response is not adequate after 2 to 3 months. The gastrointestinal (GI) absorption of spironolactone is increased when ingested with a high-fat meal.9,10
Once effective control of AV is achieved, it is optimal to use the lowest dose needed to continue reasonable suppression of new AV lesions. There is no defined end point for spironolactone use in AV, with or without concurrent PCOS, as many adult women usually continue treatment with low-dose therapy because they experience marked flaring shortly after the drug is stopped.9
Risks and Contraindications
Side effects associated with spironolactone are dose related and include increased diuresis, migraines, menstrual irregularities, breast tenderness, gynecomastia, fatigue, and dizziness.9,10,40-44 Side effects (particularly menstrual irregularities and breast tenderness) are more common at doses higher than 100 mg daily, especially when used as monotherapy without concurrent use of a COC.9,40
Spironolactone-associated hyperkalemia is most clinically relevant in patients on higher doses (eg, 100–200 mg daily), in those with renal impairment and/or congestive heart failure, and when used concurrently with certain other medications. In any patient on spironolactone, the risk of clinically relevant hyperkalemia may be increased by coingestion of potassium supplements, potassium-based salt substitutes, potassium-sparing diuretics (eg, amiloride, triamterene); aldosterone antagonists and angiotensin-converting enzyme inhibitors (eg, lisinopril, benazepril); angiotensin II receptor blockers (eg, losartan, valsartan); and tri-methoprim (with or without sulfamethoxazole).8,9,40,45 Spironolactone may also increase serum levels of lithium or digoxin.9,40,45,46 For management of AV, it is best that spironolactone be avoided in patients taking any of these medications.9
In healthy adult women with AV who are not on medications or supplements that interact adversely with spironolactone, there is no definitive recommendation regarding monitoring of serum potassium levels during treatment with spironolactone, and it has been suggested that monitoring serum potassium levels in this subgroup is not necessary.47 However, each clinician is advised to choose whether or not they wish to obtain baseline and/or periodic serum potassium levels when prescribing spironolactone for AV based on their degree of comfort and the patient’s history. Baseline and periodic blood testing to evaluate serum electrolytes and renal function are reasonable, especially as adult women with AV are usually treated with spironolactone over a prolonged period of time.9
The FDA black box warning for spironolactone states that it is tumorigenic in chronic toxicity studies in rats and refers to exposures 25- to 100-fold higher than those administered to humans.9,48 Although continued vigilance is warranted, evaluation of large populations of women treated with spironolactone do not suggest an association with increased risk of breast cancer.49,50
Spironolactone is a category C drug and thus should be avoided during pregnancy, primarily due to animal data suggesting risks of hypospadias and feminization in male fetuses.9 Importantly, there is an absence of reports linking exposure during pregnancy with congenital defects in humans, including in 2 known cases of high-dose exposures for maternal Bartter syndrome.9
The active metabolite, canrenone, is known to be present in breast milk at 0.2% of the maternal daily dose, but breastfeeding is generally believed to be safe with spironolactone based on evidence to date.9
Oral Antibiotics
Oral antibiotic therapy may be used in combination with a topical regimen to treat AV in adult women, keeping in mind some important caveats.1-7 For instance, monotherapy with oral antibiotics should be avoided, and concomitant use of benzoyl peroxide is suggested to reduce emergence of antibiotic-resistant Propionibacterium acnes strains.3,4 A therapeutic exit plan also is suggested when prescribing oral antibiotics to limit treatment to 3 to 4 months, if possible, to help mitigate the emergence of antibiotic-resistant bacteria (eg, staphylococci and streptococci).3-5,51
Tetracyclines, especially doxycycline and minocycline, are the most commonly prescribed agents. Doxycycline use warrants patient education on measures to limit the risks of esophageal and GI side effects and phototoxicity; enteric-coated and small tablet formulations have been shown to reduce GI side effects, especially when administered with food.3,52-55 In addition to vestibular side effects and hyperpigmentation, minocycline may be associated with rare but potentially severe adverse reactions such as drug hypersensitivity syndrome, autoimmune hepatitis, and lupus-like syndrome, which are reported more commonly in women.5,52,54 Vestibular side effects have been shown to decrease with use of extended-release tablets with weight-based dosing.53
Oral Isotretinoin
Oral isotretinoin is well established as highly effective for treatment of severe, recalcitrant AV, including nodular acne on the face and trunk.4,56 Currently available oral isotretinoins are branded generic formulations based on the pharmacokinetic profile of the original brand (Accutane [Roche Pharmaceuticals]) and with the use of Lidose Technology (Absorica [Cipher Pharmaceuticals]), which substantially increases GI absorption of isotretinoin in the absence of ingestion with a high-calorie, high-fat meal.57 The short- and long-term efficacy, dosing regimens, safety considerations, and serious teratogenic risks for oral isotretinoin are well published.4,56-58 Importantly, oral isotretinoin must be prescribed with strict adherence to the federally mandated iPLEDGE risk management program.
Low-dose oral isotretinoin therapy (<0.5 mg/kg–1 mg/kg daily) administered over several months longer than conventional regimens (ie, 16–20 weeks) has been suggested with demonstrated efficacy.57 However, this approach is not optimal due to the lack of established sustained clearance of AV after discontinuation of therapy and the greater potential for exposure to isotretinoin during pregnancy. Recurrences of AV do occur after completion of isotretinoin therapy, especially if cumulative systemic exposure to the drug during the initial course of treatment was inadequate.56,57
Oral isotretinoin has been shown to be effective in AV in adult women with or without PCOS with 0.5 mg/kg to 1 mg/kg daily and a total cumulative exposure of 120 mg/kg to 150 mg/kg.59 In one study, the presence of PCOS and greater number of nodules at baseline were predictive of a higher risk of relapse during the second year posttreatment.59
Conclusion
All oral therapies that are used to treat AV in adult women warrant individual consideration of possible benefits versus risks. Careful attention to possible side effects, patient-related risk factors, and potential drug-drug interactions is important. End points of therapy are not well established, with the exception of oral isotretinoin therapy. Clinicians must use their judgment in each case along with obtaining feedback from patients regarding the selection of therapy after a discussion of the available options.
- Holzmann R, Shakery K. Postadolescent acne in females. Skin Pharmacol Physiol. 2014;27(suppl 1):3-8.
- Villasenor J, Berson DS, Kroshinsky D. Treatment guidelines in adult women. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:198-207.
- Del Rosso JQ, Kim G. Optimizing use of oral antibiotics in acne vulgaris. Dermatol Clin. 2009;27:33-42.
- Gollnick H, Cunliffe W, Berson D, et al. Management of acne: report from a Global Alliance to Improve Outcomes in Acne. J Am Acad Dermatol. 2003;49(suppl 1):S1-S37.
- Fisk WA, Lev-Tov HA, Sivamani RK. Epidemiology and management of acne in adult women. Curr Derm Rep. 2014;3:29-39.
- Del Rosso JQ, Leyden JJ. Status report on antibiotic resistance: implications for the dermatologist. Dermatol Clin. 2007;25:127-132.
- Bowe WP, Leyden JJ. Clinical implications of antibiotic resistance: risk of systemic infection from Staphylococcus and Streptococcus. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:125-133.
- Del Rosso JQ. Oral antibiotic drug interactions of clinical significance to dermatologists. Dermatol Clin. 2009;27:91-94.
- Kim GK, Del Rosso JQ. Oral spironolactone in post-teenage female patients with acne vulgaris: practical considerations for the clinician based on current data and clinical experience. J Clin Aesthet Dermatol. 2012;5:37-50.
- Keri J, Berson DS, Thiboutot DM. Hormonal treatment of acne in women. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:146-155.
- American Academy of Dermatology. Position statement on isotretinoin. AAD Web site. https://www.aad.org /Forms/Policies/Uploads/PS/PS-Isotretinoin.pdf. Updated November 13, 2010. Accessed October 28, 2015.
- Arowojolu AO, Gallo MF, Lopez LM, et al. Combined oral contraceptive pills for treatment of acne. Cochrane Database Syst Rev. June 2012;7:CD004425.
- Sitruk-Ware R. Pharmacology of different progestogens: the special case of drospirenone. Climacteric. 2005;8 (suppl 3):4-12.
- Arowojolu AO, Gallo MF, Lopez LM, et al. Combined oral contraceptive pills for the treatment of acne. Cochrane Database Syst Rev. July 2012;7:CD004425.
- Thiboutot D, Archer DF, Lemay A, et al. A randomized, controlled trial of a low-dose contraceptive containing 20 microg of ethinyl estradiol and 100 microg of levonogestrel for acne treatment. Fertil Steril. 2001;76:461-468.
- Koulianos GT. Treatment of acne with oral contraceptives: criteria for pill selection. Cutis. 2000;66:281-286.
- Rabe T, Kowald A, Ortmann J, et al. Inhibition of skin 5-alpha reductase by oral contraceptive progestins in vitro. Gynecol Endocrinol. 2000;14:223-230.
- Palli MB, Reyes-Habito CM, Lima XT, et al. A single-center, randomized double-blind, parallel-group study to examine the safety and efficacy of 3mg drospirenone/0.02mg ethinyl estradiol compared with placebo in the treatment of moderate truncal acne vulgaris. J Drugs Dermatol. 2013;12:633-637.
- Koltun W, Maloney JM, Marr J, et al. Treatment of moderate acne vulgaris using a combined oral contraceptive containing ethinylestradiol 20 μg plus drospirenone 3 mg administered in a 24/4 regimen: a pooled analysis. Eur J Obstet Gynecol Reprod Biol. 2011;155:171-175.
- Maloney JM, Dietze P, Watson D, et al. A randomized controlled trial of a low-dose combined oral contraceptive containing 3 mg drospirenone plus 20 μg ethinylestradiol in the treatment of acne vulgaris: lesion counts, investigator ratings and subject self-assessment. J Drugs Dermatol. 2009;8:837-844.
- Lucky AW, Koltun W, Thiboutot D, et al. A combined oral contraceptive containing 3-mg drospirenone/20-μg ethinyl estradiol in the treatment of acne vulgaris: a randomized, double-blind, placebo-controlled study evaluating lesion counts and participant self-assessment. Cutis. 2008;82:143-150.
- Burkman R, Schlesselman JJ, Zieman M. Safety concerns and health benefits associated with oral contraception. Am J Obstet Gynecol. 2004;190(suppl 4):S5-S22.
- Maguire K, Westhoff C. The state of hormonal contraception today: established and emerging noncontraceptive health benefits. Am J Obstet Gynecol. 2011;205 (suppl 4):S4-S8.
- Weiss NS, Sayvetz TA. Incidence of endometrial cancer in relation to the use of oral contraceptives. N Engl J Med. 1980;302:551-554.
- Tyler KH, Zirwas MJ. Contraception and the dermatologist. J Am Acad Dermatol. 2013;68:1022-1029.
- Gallo MF, Lopez LM, Grimes DA, et al. Combination contraceptives: effects on weight. Cochrane Database Syst Rev. 2008;4:CD003987.
- de Bastos M, Stegeman BH, Rosendaal FR, et al. Combined oral contraceptives: venous thrombosis. Cochrane Database Syst Rev. 2014;3:CD010813.
- Raymond EG, Burke AE, Espey E. Combined hormonal contraceptives and venous thromboembolism: putting the risks into perspective. Obstet Gynecol. 2012;119:1039-1044.
- Jick SS, Hernandez RK. Risk of non-fatal venous thromboembolism in women using oral contraceptives containing drospirenone compared with women using oral contraceptives containing levonorgestrel: case-control study using United States claims data. BMJ. 2011;342:d2151.
- US Food and Drug Administration Office of Surveillance and Epidemiology. Combined hormonal contraceptives (CHCs) and the risk of cardiovascular disease endpoints. US Food and Drug Administration Web site. http://www.fda.gov/downloads/Drugs /Drug Safety/UCM277384.pdf. Accessed October 28, 2015.
- The American College of Obstetricians and Gynecologists Committee on Gynecologic Practice. Risk of venous thromboembolism among users of drospirenone-containing oral contraceptive pills. Obstet Gynecol. 2012;120:1239-1242.
- World Health Organization. Cardiovascular Disease and Steroid Hormone Contraception: Report of a WHO Scientific Group. Geneva, Switzerland: World Health Organization; 1998. Technical Report Series 877.
- Frangos JE, Alavian CN, Kimball AB. Acne and oral contraceptives: update on women’s health screening guidelines. J Am Acad Dermatol. 2008;58:781-786.
- Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormonal contraceptives: collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Lancet. 1996;347:1713-1727.
- Gierisch JM, Coeytaux RR, Urrutia RP, et al. Oral contraceptive use and risk of breast, cervical, colorectal, and endometrial cancers: a systematic review. Cancer Epidemiol Biomarkers Prev. 2013;22:1931-1943.
- International Collaboration of Epidemiological Studies of Cervical Cancer. Cervical cancer and hormonal contraceptives: collaborative reanalysis of individual data for 16 573 women with cervical cancer and 35 509 women without cervical cancer from 24 epidemiological studies. Lancet. 2007;370:1609-1621.
- Agostino H, Di Meglio G. Low-dose oral contraceptives in adolescents: how low can you go? J Pediatr Adolesc Gynecol. 2010;23:195-201.
- Buzney E, Sheu J, Buzney C, et al. Polycystic ovary syndrome: a review for dermatologists: part II. Treatment. J Am Acad Dermatol. 2014;71:859.e1-859.e15.
- Stewart FH, Harper CC, Ellertson CE, et al. Clinical breast and pelvic examination requirements for hormonal contraception: current practice vs evidence. JAMA. 2001;285:2232-2239.
- Sawaya ME, Somani N. Antiandrogens and androgen inhibitors. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelpha, PA: Saunders; 2013:361-374.
- Muhlemann MF, Carter GD, Cream JJ, et al. Oral spironolactone: an effective treatment for acne vulgaris in women. Br J Dermatol. 1986;115:227-232.
- Shaw JC. Low-dose adjunctive spironolactone in the treatment of acne in women: a retrospective analysis of 85 consecutively treated patients. J Am Acad Dermatol. 2000;43:498-502.
- Sato K, Matsumoto D, Iizuka F, et al. Anti-androgenic therapy using oral spironolactone for acne vulgaris in Asians. Aesth Plast Surg. 2006;30:689-694.
- Shaw JC, White LE. Long-term safety of spironolactone in acne: results of an 8-year follow-up study. J Cutan Med Surg. 2002;6:541-545.
- Stockley I. Antihypertensive drug interactions. In: Stockley I, ed. Drug Interactions. 5th ed. London, United Kingdom: Pharmaceutical Press; 1999:335-347.
- Antoniou T, Gomes T, Mamdani MM, et al. Trimethoprim-sulfamethoxazole induced hyperkalaemia in elderly patients receiving spironolactone: nested case-control study. BMJ. 2011;343:d5228.
- Plovanich M, Weng QY, Mostaghimi A. Low usefulness of potassium monitoring among healthy young women taking spironolactone for acne. JAMA Dermatol. 2015;151:941-944.
- Aldactone [package insert]. New York, NY: Pfizer Inc; 2008.
- Biggar RJ, Andersen EW, Wohlfahrt J, et al. Spironolactone use and the risk of breast and gynecologic cancers. Cancer Epidemiol. 2013;37:870-875.
- Mackenzie IS, Macdonald TM, Thompson A, et al. Spironolactone and risk of incident breast cancer in women older than 55 years: retrospective, matched cohort study. BMJ. 2012;345:e4447.
- Dreno B, Thiboutot D, Gollnick H, et al. Antibiotic stewardship in dermatology: limiting antibiotic use in acne. Eur J Dermatol. 2014;24:330-334.
- Kim S, Michaels BD, Kim GK, et al. Systemic antibacterial agents. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelpha, PA: Saunders; 2013:61-97.
- Leyden JJ, Del Rosso JQ. Oral antibiotic therapy for acne vulgaris: pharmacokinetic and pharmacodynamics perspectives. J Clin Aesthet Dermatol. 2011;4:40-47.
- Del Rosso JQ. Oral antibiotics. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:113-124.
- Del Rosso JQ. Oral doxycycline in the management of acne vulgaris: current perspectives on clinical use and recent findings with a new double-scored small tablet formulation. J Clin Aesthet Dermatol. 2015;8:19-26.
- Osofsky MG, Strauss JS. Isotretinoin. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:134-145.
- Leyden JJ, Del Rosso JQ, Baum EW. The use of isotretinoin in the treatment of acne vulgaris: clinical considerations and future directions. J Clin Aesthet Dermatol. 2014;7(suppl 2):S3-S21.
- Patton TJ, Ferris LK. Systemic retinoids. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelpha, PA: Saunders; 2013:252-268.
- Cakir GA, Erdogan FG, Gurler A. Isotretinoin treatment in nodulocystic acne with and without polycystic ovary syndrome: efficacy and determinants of relapse. Int J Dermatol. 2013;52:371-376.
- Holzmann R, Shakery K. Postadolescent acne in females. Skin Pharmacol Physiol. 2014;27(suppl 1):3-8.
- Villasenor J, Berson DS, Kroshinsky D. Treatment guidelines in adult women. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:198-207.
- Del Rosso JQ, Kim G. Optimizing use of oral antibiotics in acne vulgaris. Dermatol Clin. 2009;27:33-42.
- Gollnick H, Cunliffe W, Berson D, et al. Management of acne: report from a Global Alliance to Improve Outcomes in Acne. J Am Acad Dermatol. 2003;49(suppl 1):S1-S37.
- Fisk WA, Lev-Tov HA, Sivamani RK. Epidemiology and management of acne in adult women. Curr Derm Rep. 2014;3:29-39.
- Del Rosso JQ, Leyden JJ. Status report on antibiotic resistance: implications for the dermatologist. Dermatol Clin. 2007;25:127-132.
- Bowe WP, Leyden JJ. Clinical implications of antibiotic resistance: risk of systemic infection from Staphylococcus and Streptococcus. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:125-133.
- Del Rosso JQ. Oral antibiotic drug interactions of clinical significance to dermatologists. Dermatol Clin. 2009;27:91-94.
- Kim GK, Del Rosso JQ. Oral spironolactone in post-teenage female patients with acne vulgaris: practical considerations for the clinician based on current data and clinical experience. J Clin Aesthet Dermatol. 2012;5:37-50.
- Keri J, Berson DS, Thiboutot DM. Hormonal treatment of acne in women. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:146-155.
- American Academy of Dermatology. Position statement on isotretinoin. AAD Web site. https://www.aad.org /Forms/Policies/Uploads/PS/PS-Isotretinoin.pdf. Updated November 13, 2010. Accessed October 28, 2015.
- Arowojolu AO, Gallo MF, Lopez LM, et al. Combined oral contraceptive pills for treatment of acne. Cochrane Database Syst Rev. June 2012;7:CD004425.
- Sitruk-Ware R. Pharmacology of different progestogens: the special case of drospirenone. Climacteric. 2005;8 (suppl 3):4-12.
- Arowojolu AO, Gallo MF, Lopez LM, et al. Combined oral contraceptive pills for the treatment of acne. Cochrane Database Syst Rev. July 2012;7:CD004425.
- Thiboutot D, Archer DF, Lemay A, et al. A randomized, controlled trial of a low-dose contraceptive containing 20 microg of ethinyl estradiol and 100 microg of levonogestrel for acne treatment. Fertil Steril. 2001;76:461-468.
- Koulianos GT. Treatment of acne with oral contraceptives: criteria for pill selection. Cutis. 2000;66:281-286.
- Rabe T, Kowald A, Ortmann J, et al. Inhibition of skin 5-alpha reductase by oral contraceptive progestins in vitro. Gynecol Endocrinol. 2000;14:223-230.
- Palli MB, Reyes-Habito CM, Lima XT, et al. A single-center, randomized double-blind, parallel-group study to examine the safety and efficacy of 3mg drospirenone/0.02mg ethinyl estradiol compared with placebo in the treatment of moderate truncal acne vulgaris. J Drugs Dermatol. 2013;12:633-637.
- Koltun W, Maloney JM, Marr J, et al. Treatment of moderate acne vulgaris using a combined oral contraceptive containing ethinylestradiol 20 μg plus drospirenone 3 mg administered in a 24/4 regimen: a pooled analysis. Eur J Obstet Gynecol Reprod Biol. 2011;155:171-175.
- Maloney JM, Dietze P, Watson D, et al. A randomized controlled trial of a low-dose combined oral contraceptive containing 3 mg drospirenone plus 20 μg ethinylestradiol in the treatment of acne vulgaris: lesion counts, investigator ratings and subject self-assessment. J Drugs Dermatol. 2009;8:837-844.
- Lucky AW, Koltun W, Thiboutot D, et al. A combined oral contraceptive containing 3-mg drospirenone/20-μg ethinyl estradiol in the treatment of acne vulgaris: a randomized, double-blind, placebo-controlled study evaluating lesion counts and participant self-assessment. Cutis. 2008;82:143-150.
- Burkman R, Schlesselman JJ, Zieman M. Safety concerns and health benefits associated with oral contraception. Am J Obstet Gynecol. 2004;190(suppl 4):S5-S22.
- Maguire K, Westhoff C. The state of hormonal contraception today: established and emerging noncontraceptive health benefits. Am J Obstet Gynecol. 2011;205 (suppl 4):S4-S8.
- Weiss NS, Sayvetz TA. Incidence of endometrial cancer in relation to the use of oral contraceptives. N Engl J Med. 1980;302:551-554.
- Tyler KH, Zirwas MJ. Contraception and the dermatologist. J Am Acad Dermatol. 2013;68:1022-1029.
- Gallo MF, Lopez LM, Grimes DA, et al. Combination contraceptives: effects on weight. Cochrane Database Syst Rev. 2008;4:CD003987.
- de Bastos M, Stegeman BH, Rosendaal FR, et al. Combined oral contraceptives: venous thrombosis. Cochrane Database Syst Rev. 2014;3:CD010813.
- Raymond EG, Burke AE, Espey E. Combined hormonal contraceptives and venous thromboembolism: putting the risks into perspective. Obstet Gynecol. 2012;119:1039-1044.
- Jick SS, Hernandez RK. Risk of non-fatal venous thromboembolism in women using oral contraceptives containing drospirenone compared with women using oral contraceptives containing levonorgestrel: case-control study using United States claims data. BMJ. 2011;342:d2151.
- US Food and Drug Administration Office of Surveillance and Epidemiology. Combined hormonal contraceptives (CHCs) and the risk of cardiovascular disease endpoints. US Food and Drug Administration Web site. http://www.fda.gov/downloads/Drugs /Drug Safety/UCM277384.pdf. Accessed October 28, 2015.
- The American College of Obstetricians and Gynecologists Committee on Gynecologic Practice. Risk of venous thromboembolism among users of drospirenone-containing oral contraceptive pills. Obstet Gynecol. 2012;120:1239-1242.
- World Health Organization. Cardiovascular Disease and Steroid Hormone Contraception: Report of a WHO Scientific Group. Geneva, Switzerland: World Health Organization; 1998. Technical Report Series 877.
- Frangos JE, Alavian CN, Kimball AB. Acne and oral contraceptives: update on women’s health screening guidelines. J Am Acad Dermatol. 2008;58:781-786.
- Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormonal contraceptives: collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Lancet. 1996;347:1713-1727.
- Gierisch JM, Coeytaux RR, Urrutia RP, et al. Oral contraceptive use and risk of breast, cervical, colorectal, and endometrial cancers: a systematic review. Cancer Epidemiol Biomarkers Prev. 2013;22:1931-1943.
- International Collaboration of Epidemiological Studies of Cervical Cancer. Cervical cancer and hormonal contraceptives: collaborative reanalysis of individual data for 16 573 women with cervical cancer and 35 509 women without cervical cancer from 24 epidemiological studies. Lancet. 2007;370:1609-1621.
- Agostino H, Di Meglio G. Low-dose oral contraceptives in adolescents: how low can you go? J Pediatr Adolesc Gynecol. 2010;23:195-201.
- Buzney E, Sheu J, Buzney C, et al. Polycystic ovary syndrome: a review for dermatologists: part II. Treatment. J Am Acad Dermatol. 2014;71:859.e1-859.e15.
- Stewart FH, Harper CC, Ellertson CE, et al. Clinical breast and pelvic examination requirements for hormonal contraception: current practice vs evidence. JAMA. 2001;285:2232-2239.
- Sawaya ME, Somani N. Antiandrogens and androgen inhibitors. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelpha, PA: Saunders; 2013:361-374.
- Muhlemann MF, Carter GD, Cream JJ, et al. Oral spironolactone: an effective treatment for acne vulgaris in women. Br J Dermatol. 1986;115:227-232.
- Shaw JC. Low-dose adjunctive spironolactone in the treatment of acne in women: a retrospective analysis of 85 consecutively treated patients. J Am Acad Dermatol. 2000;43:498-502.
- Sato K, Matsumoto D, Iizuka F, et al. Anti-androgenic therapy using oral spironolactone for acne vulgaris in Asians. Aesth Plast Surg. 2006;30:689-694.
- Shaw JC, White LE. Long-term safety of spironolactone in acne: results of an 8-year follow-up study. J Cutan Med Surg. 2002;6:541-545.
- Stockley I. Antihypertensive drug interactions. In: Stockley I, ed. Drug Interactions. 5th ed. London, United Kingdom: Pharmaceutical Press; 1999:335-347.
- Antoniou T, Gomes T, Mamdani MM, et al. Trimethoprim-sulfamethoxazole induced hyperkalaemia in elderly patients receiving spironolactone: nested case-control study. BMJ. 2011;343:d5228.
- Plovanich M, Weng QY, Mostaghimi A. Low usefulness of potassium monitoring among healthy young women taking spironolactone for acne. JAMA Dermatol. 2015;151:941-944.
- Aldactone [package insert]. New York, NY: Pfizer Inc; 2008.
- Biggar RJ, Andersen EW, Wohlfahrt J, et al. Spironolactone use and the risk of breast and gynecologic cancers. Cancer Epidemiol. 2013;37:870-875.
- Mackenzie IS, Macdonald TM, Thompson A, et al. Spironolactone and risk of incident breast cancer in women older than 55 years: retrospective, matched cohort study. BMJ. 2012;345:e4447.
- Dreno B, Thiboutot D, Gollnick H, et al. Antibiotic stewardship in dermatology: limiting antibiotic use in acne. Eur J Dermatol. 2014;24:330-334.
- Kim S, Michaels BD, Kim GK, et al. Systemic antibacterial agents. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelpha, PA: Saunders; 2013:61-97.
- Leyden JJ, Del Rosso JQ. Oral antibiotic therapy for acne vulgaris: pharmacokinetic and pharmacodynamics perspectives. J Clin Aesthet Dermatol. 2011;4:40-47.
- Del Rosso JQ. Oral antibiotics. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:113-124.
- Del Rosso JQ. Oral doxycycline in the management of acne vulgaris: current perspectives on clinical use and recent findings with a new double-scored small tablet formulation. J Clin Aesthet Dermatol. 2015;8:19-26.
- Osofsky MG, Strauss JS. Isotretinoin. In: Shalita AR, Del Rosso JQ, Webster GF, eds. Acne Vulgaris. London, United Kingdom: Informa Healthcare; 2011:134-145.
- Leyden JJ, Del Rosso JQ, Baum EW. The use of isotretinoin in the treatment of acne vulgaris: clinical considerations and future directions. J Clin Aesthet Dermatol. 2014;7(suppl 2):S3-S21.
- Patton TJ, Ferris LK. Systemic retinoids. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelpha, PA: Saunders; 2013:252-268.
- Cakir GA, Erdogan FG, Gurler A. Isotretinoin treatment in nodulocystic acne with and without polycystic ovary syndrome: efficacy and determinants of relapse. Int J Dermatol. 2013;52:371-376.
Practice Points
- Use of combination oral contraceptives to treat acne vulgaris (AV) in adult women who do not have measurable androgen excess is most rational in patients who also desire a method of contraception.
- Spironolactone is widely accepted as an oral agent that can be effective in treating adult women with AV and may be used in combination with other therapies.
- Monotherapy with oral antibiotics should be avoided in the treatment of adult women with AV, and concomitant use of benzoyl peroxide is suggested to reduce emergence of antibiotic-resistant Propionibacterium acnes strains.
- Oral isotretinoin use in adult women with AV warrants strict adherence to pregnancy prevention measures and requirements set forth by the federally mandated iPLEDGE™ risk management program.
Methylphenidate Linked to Sleep and Appetite Loss
LONDON - Researchers voiced concern on Nov. 25 about poor quality studies on the popular ADHD treatment methylphenidate, saying evidence of some benefits, but also of sleep problems and appetite loss, suggests the drug should be prescribed with caution.
Methylphenidate (brand names Ritalin, Concerta, Medikinet and Equasym) has been used to treat attention deficit hyperactivity disorder (ADHD) for more than 50 years.
The Cochrane Review researchers, who conducted a full assessment of studies on the benefits and harms of methylphenidate, said evidence on its use in children was poor.
"Our expectations of this treatment are probably greater than they should be," said Morris Zwi, a London-based consultant child and adolescent psychiatrist, who worked on the review.
"Whilst our review shows some evidence of benefit, we should bear in mind that this finding was based on very low-quality evidence. What we still need are large, well-conducted trials to clarify the risks versus the benefits."
Cochrane Reviews are conducted by international panels of independent researchers and considered as studies of the best available science on a topic.
Jonathan Green, a professor of child and adolescent psychiatry at Britain's Manchester University who was asked to comment on the Cochrane Review, said it would be "wrong to draw the conclusion... that methylphenidate is ineffective.
"In fact, clinical level evidence strongly supports the effectiveness of methylphenidate for many children with ADHD."
The Cochrane Review included data from 185 randomized controlled trials involving more than 12,000 children or adolescents. The studies were conducted mainly in the United States, Canada and Europe, and each one compared methylphenidate with either a placebo or no intervention.
In their review, the Cochrane researchers found that methylphenidate led to modest improvements in ADHD symptoms, general behavior, and quality of life, but that side-effects included a higher risk of sleep problems and loss of appetite.
The researchers added, however, that their confidence in the evidence was low since many of the trials were not conducted with sufficient rigor and results reporting was not complete.
"Clinicians prescribing methylphenidate must take account of the poor quality of the evidence, monitor treatment carefully, and weigh up the benefits and adverse effects," they said.
The analysis was published online Nov. 25 in the Cochrane Library.
LONDON - Researchers voiced concern on Nov. 25 about poor quality studies on the popular ADHD treatment methylphenidate, saying evidence of some benefits, but also of sleep problems and appetite loss, suggests the drug should be prescribed with caution.
Methylphenidate (brand names Ritalin, Concerta, Medikinet and Equasym) has been used to treat attention deficit hyperactivity disorder (ADHD) for more than 50 years.
The Cochrane Review researchers, who conducted a full assessment of studies on the benefits and harms of methylphenidate, said evidence on its use in children was poor.
"Our expectations of this treatment are probably greater than they should be," said Morris Zwi, a London-based consultant child and adolescent psychiatrist, who worked on the review.
"Whilst our review shows some evidence of benefit, we should bear in mind that this finding was based on very low-quality evidence. What we still need are large, well-conducted trials to clarify the risks versus the benefits."
Cochrane Reviews are conducted by international panels of independent researchers and considered as studies of the best available science on a topic.
Jonathan Green, a professor of child and adolescent psychiatry at Britain's Manchester University who was asked to comment on the Cochrane Review, said it would be "wrong to draw the conclusion... that methylphenidate is ineffective.
"In fact, clinical level evidence strongly supports the effectiveness of methylphenidate for many children with ADHD."
The Cochrane Review included data from 185 randomized controlled trials involving more than 12,000 children or adolescents. The studies were conducted mainly in the United States, Canada and Europe, and each one compared methylphenidate with either a placebo or no intervention.
In their review, the Cochrane researchers found that methylphenidate led to modest improvements in ADHD symptoms, general behavior, and quality of life, but that side-effects included a higher risk of sleep problems and loss of appetite.
The researchers added, however, that their confidence in the evidence was low since many of the trials were not conducted with sufficient rigor and results reporting was not complete.
"Clinicians prescribing methylphenidate must take account of the poor quality of the evidence, monitor treatment carefully, and weigh up the benefits and adverse effects," they said.
The analysis was published online Nov. 25 in the Cochrane Library.
LONDON - Researchers voiced concern on Nov. 25 about poor quality studies on the popular ADHD treatment methylphenidate, saying evidence of some benefits, but also of sleep problems and appetite loss, suggests the drug should be prescribed with caution.
Methylphenidate (brand names Ritalin, Concerta, Medikinet and Equasym) has been used to treat attention deficit hyperactivity disorder (ADHD) for more than 50 years.
The Cochrane Review researchers, who conducted a full assessment of studies on the benefits and harms of methylphenidate, said evidence on its use in children was poor.
"Our expectations of this treatment are probably greater than they should be," said Morris Zwi, a London-based consultant child and adolescent psychiatrist, who worked on the review.
"Whilst our review shows some evidence of benefit, we should bear in mind that this finding was based on very low-quality evidence. What we still need are large, well-conducted trials to clarify the risks versus the benefits."
Cochrane Reviews are conducted by international panels of independent researchers and considered as studies of the best available science on a topic.
Jonathan Green, a professor of child and adolescent psychiatry at Britain's Manchester University who was asked to comment on the Cochrane Review, said it would be "wrong to draw the conclusion... that methylphenidate is ineffective.
"In fact, clinical level evidence strongly supports the effectiveness of methylphenidate for many children with ADHD."
The Cochrane Review included data from 185 randomized controlled trials involving more than 12,000 children or adolescents. The studies were conducted mainly in the United States, Canada and Europe, and each one compared methylphenidate with either a placebo or no intervention.
In their review, the Cochrane researchers found that methylphenidate led to modest improvements in ADHD symptoms, general behavior, and quality of life, but that side-effects included a higher risk of sleep problems and loss of appetite.
The researchers added, however, that their confidence in the evidence was low since many of the trials were not conducted with sufficient rigor and results reporting was not complete.
"Clinicians prescribing methylphenidate must take account of the poor quality of the evidence, monitor treatment carefully, and weigh up the benefits and adverse effects," they said.
The analysis was published online Nov. 25 in the Cochrane Library.
Study characterizes intracerebral hemorrhage with new oral anticoagulants
Intracerebral hemorrhage related to non–vitamin-K antagonist oral anticoagulants carries a high mortality and frequently involves hematoma expansion, according to a report published online Dec. 14 in JAMA Neurology.
The characteristics and natural history of acute-phase non–vitamin-K antagonist oral anticoagulant (NOAC)-associated intracerebral hemorrhage (ICH) “are largely unknown,” and there are no prospective data concerning hematoma expansion or the effectiveness of prothrombin complex concentrate in limiting that expansion by reversing anticoagulation. Nevertheless, current recommendations suggest that clinicians consider administering prothrombin complex concentrate in this patient population, said Dr. Jan C. Purrucker of the department of neurology at Heidelberg (Germany) University and his associates (JAMA Neurol. 2015 Dec 14. doi: 10.1001/jamaneurol.2015.3682).
To characterize the clinical and radiologic course, management, and outcome of NOAC-associated intracerebral hemorrhage in routine clinical practice, Dr. Purrucker and his associates performed the ICH substudy of the Registry of Acute Stroke Under New Oral Anticoagulants (RASUNOA). This is a prospective registry involving 38 neurology departments with certified stroke units across Germany. For their substudy, the investigators focused on 61 adults with a mean age of 76 years (range, 46-97 years) who were taking NOACs (apixaban [Eliquis], dabigatran etexilate [Pradaxa], or rivaroxaban [Xarelto]) and had moderate to severe neurologic deficit and a median hematoma volume of 10.8 mL at presentation. Thirty-five of these patients (57%) were treated with prothrombin complex concentrate.
Mortality was high, at 16% (10 patients) during the acute inpatient stay and 28% (17 patients) at 3 months; 65% of the survivors had an unfavorable outcome. Substantial hematoma expansion – defined as a 33% or greater relative increase or 6 mL or greater absolute increase in ICH volume – was common, affecting 38% of patients. “This proportion was within the range reported for vitamin-K antagonist–associated intracerebral hemorrhage (36%-56%) and is higher, compared with that related to intracerebral hemorrhage in patients not receiving anticoagulation (12%-26%),” the investigators wrote.
Both larger hematoma volume at baseline (odds ratio, 2.37) and intraventricular extension at baseline (OR, 8.13) strongly correlated with adverse outcomes. In contrast, prothrombin complex concentrate failed to limit lesion expansion or avert adverse outcomes. This might be because patients given the treatment tended to have more severe initial neurologic deficits and more unfavorable hematoma location than did those who weren’t given prothrombin complex concentrate. In any case, “our study design, the limited sample size, and the potential for confounding by indication do not allow any [firm] conclusions regarding a potential association between prothrombin complex concentrate treatment and outcome,” they noted.
The RASUNOA registry was supported by the University Hospital Heidelberg. Dr. Purrucker reported receiving support from Pfizer unrelated to this study, and his associates reported ties to numerous industry sources.
It’s important to note that in the study by Dr. Purrucker and his colleagues, the median time from symptom onset to the first brain imaging was 14 hours and that fully 25% of patients presented for treatment more than 22 hours after noticing their initial symptoms.

|
Dr. Stephan A. Mayer |
In contrast, patients with spontaneous hypertensive intracerebral hemorrhage present much earlier, usually within 6 hours. This indicates that the bleeding in NOAC-associated hemorrhagic stroke often is gradual and prolonged, an “oozing” process rather than the explosive type of process seen in spontaneous hemorrhagic stroke.
It is almost certain that if this cohort had undergone imaging at 3 hours rather than at 14 hours after symptom onset, the frequency of hematoma expansion would have approached 100% rather than 38%.
Dr. Stephan A. Mayer is at Mount Sinai University, New York. He reported having no relevant financial disclosures. Dr. Mayer made these remarks in an editorial accompanying Dr. Purrucker’s report (JAMA Neurol. 2015 Dec 14. doi:10.1001/jamaneurol.2015.3884).
It’s important to note that in the study by Dr. Purrucker and his colleagues, the median time from symptom onset to the first brain imaging was 14 hours and that fully 25% of patients presented for treatment more than 22 hours after noticing their initial symptoms.
|
|
Dr. Stephan A. Mayer |
In contrast, patients with spontaneous hypertensive intracerebral hemorrhage present much earlier, usually within 6 hours. This indicates that the bleeding in NOAC-associated hemorrhagic stroke often is gradual and prolonged, an “oozing” process rather than the explosive type of process seen in spontaneous hemorrhagic stroke.
It is almost certain that if this cohort had undergone imaging at 3 hours rather than at 14 hours after symptom onset, the frequency of hematoma expansion would have approached 100% rather than 38%.
Dr. Stephan A. Mayer is at Mount Sinai University, New York. He reported having no relevant financial disclosures. Dr. Mayer made these remarks in an editorial accompanying Dr. Purrucker’s report (JAMA Neurol. 2015 Dec 14. doi:10.1001/jamaneurol.2015.3884).
It’s important to note that in the study by Dr. Purrucker and his colleagues, the median time from symptom onset to the first brain imaging was 14 hours and that fully 25% of patients presented for treatment more than 22 hours after noticing their initial symptoms.
|
|
Dr. Stephan A. Mayer |
In contrast, patients with spontaneous hypertensive intracerebral hemorrhage present much earlier, usually within 6 hours. This indicates that the bleeding in NOAC-associated hemorrhagic stroke often is gradual and prolonged, an “oozing” process rather than the explosive type of process seen in spontaneous hemorrhagic stroke.
It is almost certain that if this cohort had undergone imaging at 3 hours rather than at 14 hours after symptom onset, the frequency of hematoma expansion would have approached 100% rather than 38%.
Dr. Stephan A. Mayer is at Mount Sinai University, New York. He reported having no relevant financial disclosures. Dr. Mayer made these remarks in an editorial accompanying Dr. Purrucker’s report (JAMA Neurol. 2015 Dec 14. doi:10.1001/jamaneurol.2015.3884).
Intracerebral hemorrhage related to non–vitamin-K antagonist oral anticoagulants carries a high mortality and frequently involves hematoma expansion, according to a report published online Dec. 14 in JAMA Neurology.
The characteristics and natural history of acute-phase non–vitamin-K antagonist oral anticoagulant (NOAC)-associated intracerebral hemorrhage (ICH) “are largely unknown,” and there are no prospective data concerning hematoma expansion or the effectiveness of prothrombin complex concentrate in limiting that expansion by reversing anticoagulation. Nevertheless, current recommendations suggest that clinicians consider administering prothrombin complex concentrate in this patient population, said Dr. Jan C. Purrucker of the department of neurology at Heidelberg (Germany) University and his associates (JAMA Neurol. 2015 Dec 14. doi: 10.1001/jamaneurol.2015.3682).
To characterize the clinical and radiologic course, management, and outcome of NOAC-associated intracerebral hemorrhage in routine clinical practice, Dr. Purrucker and his associates performed the ICH substudy of the Registry of Acute Stroke Under New Oral Anticoagulants (RASUNOA). This is a prospective registry involving 38 neurology departments with certified stroke units across Germany. For their substudy, the investigators focused on 61 adults with a mean age of 76 years (range, 46-97 years) who were taking NOACs (apixaban [Eliquis], dabigatran etexilate [Pradaxa], or rivaroxaban [Xarelto]) and had moderate to severe neurologic deficit and a median hematoma volume of 10.8 mL at presentation. Thirty-five of these patients (57%) were treated with prothrombin complex concentrate.
Mortality was high, at 16% (10 patients) during the acute inpatient stay and 28% (17 patients) at 3 months; 65% of the survivors had an unfavorable outcome. Substantial hematoma expansion – defined as a 33% or greater relative increase or 6 mL or greater absolute increase in ICH volume – was common, affecting 38% of patients. “This proportion was within the range reported for vitamin-K antagonist–associated intracerebral hemorrhage (36%-56%) and is higher, compared with that related to intracerebral hemorrhage in patients not receiving anticoagulation (12%-26%),” the investigators wrote.
Both larger hematoma volume at baseline (odds ratio, 2.37) and intraventricular extension at baseline (OR, 8.13) strongly correlated with adverse outcomes. In contrast, prothrombin complex concentrate failed to limit lesion expansion or avert adverse outcomes. This might be because patients given the treatment tended to have more severe initial neurologic deficits and more unfavorable hematoma location than did those who weren’t given prothrombin complex concentrate. In any case, “our study design, the limited sample size, and the potential for confounding by indication do not allow any [firm] conclusions regarding a potential association between prothrombin complex concentrate treatment and outcome,” they noted.
The RASUNOA registry was supported by the University Hospital Heidelberg. Dr. Purrucker reported receiving support from Pfizer unrelated to this study, and his associates reported ties to numerous industry sources.
Intracerebral hemorrhage related to non–vitamin-K antagonist oral anticoagulants carries a high mortality and frequently involves hematoma expansion, according to a report published online Dec. 14 in JAMA Neurology.
The characteristics and natural history of acute-phase non–vitamin-K antagonist oral anticoagulant (NOAC)-associated intracerebral hemorrhage (ICH) “are largely unknown,” and there are no prospective data concerning hematoma expansion or the effectiveness of prothrombin complex concentrate in limiting that expansion by reversing anticoagulation. Nevertheless, current recommendations suggest that clinicians consider administering prothrombin complex concentrate in this patient population, said Dr. Jan C. Purrucker of the department of neurology at Heidelberg (Germany) University and his associates (JAMA Neurol. 2015 Dec 14. doi: 10.1001/jamaneurol.2015.3682).
To characterize the clinical and radiologic course, management, and outcome of NOAC-associated intracerebral hemorrhage in routine clinical practice, Dr. Purrucker and his associates performed the ICH substudy of the Registry of Acute Stroke Under New Oral Anticoagulants (RASUNOA). This is a prospective registry involving 38 neurology departments with certified stroke units across Germany. For their substudy, the investigators focused on 61 adults with a mean age of 76 years (range, 46-97 years) who were taking NOACs (apixaban [Eliquis], dabigatran etexilate [Pradaxa], or rivaroxaban [Xarelto]) and had moderate to severe neurologic deficit and a median hematoma volume of 10.8 mL at presentation. Thirty-five of these patients (57%) were treated with prothrombin complex concentrate.
Mortality was high, at 16% (10 patients) during the acute inpatient stay and 28% (17 patients) at 3 months; 65% of the survivors had an unfavorable outcome. Substantial hematoma expansion – defined as a 33% or greater relative increase or 6 mL or greater absolute increase in ICH volume – was common, affecting 38% of patients. “This proportion was within the range reported for vitamin-K antagonist–associated intracerebral hemorrhage (36%-56%) and is higher, compared with that related to intracerebral hemorrhage in patients not receiving anticoagulation (12%-26%),” the investigators wrote.
Both larger hematoma volume at baseline (odds ratio, 2.37) and intraventricular extension at baseline (OR, 8.13) strongly correlated with adverse outcomes. In contrast, prothrombin complex concentrate failed to limit lesion expansion or avert adverse outcomes. This might be because patients given the treatment tended to have more severe initial neurologic deficits and more unfavorable hematoma location than did those who weren’t given prothrombin complex concentrate. In any case, “our study design, the limited sample size, and the potential for confounding by indication do not allow any [firm] conclusions regarding a potential association between prothrombin complex concentrate treatment and outcome,” they noted.
The RASUNOA registry was supported by the University Hospital Heidelberg. Dr. Purrucker reported receiving support from Pfizer unrelated to this study, and his associates reported ties to numerous industry sources.
FROM JAMA NEUROLOGY
Key clinical point: Intracerebral hemorrhage related to new oral anticoagulants frequently involves hematoma expansion and doesn’t appear to respond to prothrombin complex concentrate.
Major finding: Mortality was 28%, 65% of survivors had unfavorable outcomes, and substantial hematoma expansion occurred in 38% of patients.
Data source: A prospective, multicenter, observational study involving 61 patients treated during a 3-year period in Germany.
Disclosures: The RASUNOA registry was supported by the University Hospital Heidelberg. Dr. Purrucker reported receiving support from Pfizer unrelated to this study, and his associates reported ties to numerous industry sources.

Make the Diagnosis - January 2016
Diagnosis: Urticaria pigmentosa
Urticaria pigmentosa (UP), also known as cutaneous mastocytosis, is characterized by the presence of pigmented macular and/or papular lesions associated with severe pruritis that can appear on any part of the body. With increased scratching and/or exposure to heat, the lesions become elevated. This phenomenon is known as Darier’s sign. The urticarial lesions can progress to become fluid-filled blisters. UP can rarely progress to systemic mastocytosis and present with systemic symptoms that are more common in adults, including headache, fatigue, abdominal pain, diarrhea, and tachycardia.
UP has been associated with increased inflammatory mast cells that abnormally collect in the skin. Mast cells specialize in producing histamine. In UP, the overproliferation of mast cells, secondary to point mutations in proto-oncogene c-kit binding to mast cell growth factor (MCGF), leads to an abundance of inflammatory chemicals, which produces the characteristic itching and presenting symptoms.
Although the pathophysiology of UP is known, the exact etiology is unclear. Certain medications that can cause mast-cell degranulation have been implicated, such as aspirin, NSAIDs, narcotics, alcohol, and anticholinergics. Children with allergies such as asthma are known to have an increased predisposition to UP, which typically presents in the first year of life and is self-limited by adolescence. Some cases sporadically appear, but others may be secondary to genetic inheritance as an autosomal dominant trait.
Diagnosis of UP is made by the presence of the characteristic skin lesions but can be confirmed by microscopic evaluation. A positive Darier’s sign on physical exam and lab testing for elevated histamine can aid in the diagnosis. UP is often mistaken for moles or insect bites on initial presentation; however, the persistence of the lesions for months to years is a distinguishing factor.
Given the self-limiting nature of UP, the treatment is symptomatic and supportive. Topical steroids and antihistamines can be useful to treat severe pruritis. In addition, PUVA has been shown to be an effective treatment for UP in adults.
This case and photo were submitted by Dr. Parteek Singla and Dr. Damon McClain of Naval Hospital Camp Lejeune, N.C.
Dr. Bilu Martin is in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit your case for possible publication, send an email to [email protected].
Diagnosis: Urticaria pigmentosa
Urticaria pigmentosa (UP), also known as cutaneous mastocytosis, is characterized by the presence of pigmented macular and/or papular lesions associated with severe pruritis that can appear on any part of the body. With increased scratching and/or exposure to heat, the lesions become elevated. This phenomenon is known as Darier’s sign. The urticarial lesions can progress to become fluid-filled blisters. UP can rarely progress to systemic mastocytosis and present with systemic symptoms that are more common in adults, including headache, fatigue, abdominal pain, diarrhea, and tachycardia.
UP has been associated with increased inflammatory mast cells that abnormally collect in the skin. Mast cells specialize in producing histamine. In UP, the overproliferation of mast cells, secondary to point mutations in proto-oncogene c-kit binding to mast cell growth factor (MCGF), leads to an abundance of inflammatory chemicals, which produces the characteristic itching and presenting symptoms.
Although the pathophysiology of UP is known, the exact etiology is unclear. Certain medications that can cause mast-cell degranulation have been implicated, such as aspirin, NSAIDs, narcotics, alcohol, and anticholinergics. Children with allergies such as asthma are known to have an increased predisposition to UP, which typically presents in the first year of life and is self-limited by adolescence. Some cases sporadically appear, but others may be secondary to genetic inheritance as an autosomal dominant trait.
Diagnosis of UP is made by the presence of the characteristic skin lesions but can be confirmed by microscopic evaluation. A positive Darier’s sign on physical exam and lab testing for elevated histamine can aid in the diagnosis. UP is often mistaken for moles or insect bites on initial presentation; however, the persistence of the lesions for months to years is a distinguishing factor.
Given the self-limiting nature of UP, the treatment is symptomatic and supportive. Topical steroids and antihistamines can be useful to treat severe pruritis. In addition, PUVA has been shown to be an effective treatment for UP in adults.
This case and photo were submitted by Dr. Parteek Singla and Dr. Damon McClain of Naval Hospital Camp Lejeune, N.C.
Dr. Bilu Martin is in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit your case for possible publication, send an email to [email protected].
Diagnosis: Urticaria pigmentosa
Urticaria pigmentosa (UP), also known as cutaneous mastocytosis, is characterized by the presence of pigmented macular and/or papular lesions associated with severe pruritis that can appear on any part of the body. With increased scratching and/or exposure to heat, the lesions become elevated. This phenomenon is known as Darier’s sign. The urticarial lesions can progress to become fluid-filled blisters. UP can rarely progress to systemic mastocytosis and present with systemic symptoms that are more common in adults, including headache, fatigue, abdominal pain, diarrhea, and tachycardia.
UP has been associated with increased inflammatory mast cells that abnormally collect in the skin. Mast cells specialize in producing histamine. In UP, the overproliferation of mast cells, secondary to point mutations in proto-oncogene c-kit binding to mast cell growth factor (MCGF), leads to an abundance of inflammatory chemicals, which produces the characteristic itching and presenting symptoms.
Although the pathophysiology of UP is known, the exact etiology is unclear. Certain medications that can cause mast-cell degranulation have been implicated, such as aspirin, NSAIDs, narcotics, alcohol, and anticholinergics. Children with allergies such as asthma are known to have an increased predisposition to UP, which typically presents in the first year of life and is self-limited by adolescence. Some cases sporadically appear, but others may be secondary to genetic inheritance as an autosomal dominant trait.
Diagnosis of UP is made by the presence of the characteristic skin lesions but can be confirmed by microscopic evaluation. A positive Darier’s sign on physical exam and lab testing for elevated histamine can aid in the diagnosis. UP is often mistaken for moles or insect bites on initial presentation; however, the persistence of the lesions for months to years is a distinguishing factor.
Given the self-limiting nature of UP, the treatment is symptomatic and supportive. Topical steroids and antihistamines can be useful to treat severe pruritis. In addition, PUVA has been shown to be an effective treatment for UP in adults.
This case and photo were submitted by Dr. Parteek Singla and Dr. Damon McClain of Naval Hospital Camp Lejeune, N.C.
Dr. Bilu Martin is in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit your case for possible publication, send an email to [email protected].
Otherwise healthy 11-month-old female twins presented with a mildly pruritic rash that had been present since birth. There was no history of prolonged nausea, diarrhea, or vomiting. There was no family history of mast cell diseases. The girls were not taking any medications. On exam, there were hyperpigmented macules, patches, and papules mostly on the trunk but also on the extremities. A Darier’s sign was noted when firmly scratching with a tongue depressor
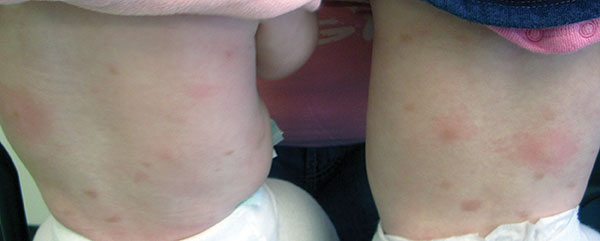
Differentiation of Latex Allergy From Irritant Contact Dermatitis
Latex allergy is an all-encompassing term used to describe hypersensitivity reactions to products containing natural rubber latex from the Hevea brasiliensis tree and affects approximately 1% to 2% of the general population.1 Although latex gloves are the most widely known culprits, several other commonly used products can contain natural rubber latex, including adhesive tape, balloons, condoms, rubber bands, paint, tourniquets, electrode pads, and Foley catheters.2 The term latex allergy often is used as a general diagnosis, but there are in fact 3 distinct mechanisms by which individuals may develop an adverse reaction to latex-containing products: irritant contact dermatitis, allergic contact dermatitis (type IV hypersensitivity) and true latex allergy (type I hypersensitivity).
Irritant Contact Dermatitis
Irritant contact dermatitis, a nonimmunologic reaction, occurs due to mechanical factors (eg, friction) or contact with chemicals, which can have irritating and dehydrating effects. Individuals with irritant contact dermatitis do not have true latex allergy and will not necessarily develop a reaction to products containing natural rubber latex. Incorrectly attributing these irritant contact dermatitis reactions to latex allergy and simply advising patients to avoid all latex products (eg, use nitrile gloves rather than latex gloves) will not address the underlying problem. Rather, these patients must be informed that the dermatitis is a result of a disruption to the natural, protective skin barrier and not an allergic reaction.
Allergic Contact Dermatitis
Allergic contact dermatitis to rubber is caused by a type IV (delayed) hypersensitivity reaction and is the result of exposure to the accelerators present in rubber products in sensitive individuals. Individuals experiencing this type of reaction typically develop localized erythema, pruritus, and urticarial lesions 48 hours after exposure.3 Incorrectly labeling this problem as latex allergy and recommending nonlatex rubber substitutes (eg, hypoallergenic gloves) likely will not be effective, as these nonlatex replacement products contain the same accelerators as do latex gloves.
True Latex Allergy
The most severe form of latex allergy, often referred to as true latex allergy, is caused by a type I (immediate) hypersensitivity reaction mediated by immunoglobulin E (IgE) antibodies. Individuals experiencing this type of reaction have a systemic response to latex proteins that may result in fulminant anaphylaxis. Individuals with true latex allergy must absolutely avoid latex products, and substituting nonlatex products is the most effective approach.
Latex Reactions in Medical Practice
The varying propensity of certain populations to develop latex allergy has been well documented; for example, the prevalence of hypersensitivity in patients with spina bifida ranges from 20% to 65%, figures that are much higher than those reported in the general population.3 This hypersensitivity in patients with spina bifida most likely results from repeated exposure to latex products during corrective surgeries and diagnostic procedures early in life. Atopic individuals, such as those with allergic rhinitis, eczema, and asthma, have a 4-fold increased risk for developing latex allergy compared to nonatopic individuals.4 The risk of latex allergy among health care workers is increased due to increased exposure to rubber products. One study found that the risk of latex sensitization among health care workers exposed to products containing latex was 4.3%, while the risk in the general population was only 1.37%.1 Those at highest risk for sensitization include dental assistants, operating room personnel, hospital housekeeping staff, and paramedics or emergency medical technicians.3 However, sensitization documented on laboratory assessment does not reliably correlate with symptomatic allergy, as many patients with a positive IgE test do not show clinical symptoms. Schmid et al4 demonstrated that a 1.3% prevalence of clinically symptomatic latex allergy among health care workers may approximate the prevalence of latex allergy in the general population. In a study by Brown et al,5 although 12.5% of anesthesiologists were found to be sensitized to latex, only 2.4% had clinically symptomatic allergic reactions.
Testing for Latex Allergy
Several diagnostic tests are available to establish a diagnosis of type I sensitization or true latex allergy. Skin prick testing is an in vivo assay and is the gold standard for diagnosing IgE-mediated type I hypersensitivity to latex. The test involves pricking the skin of the forearm and applying a commercial extract of nonammoniated latex to monitor for development of a wheal within several minutes. The skin prick test should be performed in a health care setting equipped with oxygen, epinephrine, and latex-free resuscitation equipment in case of anaphylaxis following exposure. Although latex skin prick testing is the gold standard, it is rarely performed in the United States because there is no US Food and Drug Administration–approved natural rubber latex reagent.3 Consequently, physicians who wish to perform skin prick testing for latex allergy are forced to develop improvised reagents from the H brasiliensis tree itself or from highly allergenic latex gloves. Standardized latex allergens are commercially available in Europe.
The most noninvasive method of latex allergy testing is an in vitro assay for latex-specific IgE antibodies, which can be detected by either a radioallergosorbent test (RAST) or enzyme-linked immunosorbent assay (ELISA). The presence of antilatex IgE antibodies confirms sensitization but does not necessarily mean the patient will develop a symptomatic reaction following exposure. Due to the unavailability of a standardized reagent for the skin prick test in the United States, evaluation of latex-specific serum IgE levels may be the best alternative. While the skin prick test has the highest sensitivity, the sensitivity and specificity of latex-specific serum IgE testing are 50% to 90% and 80% to 87%, respectively.6
The wear test (also known as the use or glove provocation test), can be used to diagnose clinically symptomatic latex allergy when there is a discrepancy between the patient’s clinical history and results from skin prick or serum IgE antibody testing. To perform the wear test, place a natural rubber latex glove on one of the patient’s fingers for 15 minutes and monitor the area for development of urticaria. If there is no evidence of allergic reaction within 15 minutes, place the glove on the whole hand for an additional 15 minutes. The patient is said to be nonreactive if a latex glove can be placed on the entire hand for 15 minutes without evidence of reaction.3
Lastly, patch testing can differentiate between irritant contact and allergic contact (type IV hypersensitivity) dermatitis. Apply a small amount of each substance of interest onto a separate disc and place the discs in direct contact with the skin using hypoallergenic tape. With type IV latex hypersensitivity, the skin underneath the disc will become erythematous with developing papulovesicles, starting between 2 and 5 days after exposure. The Figure outlines the differentiation of true latex allergy from irritant and allergic contact dermatitis and identifies methods for making these diagnoses.
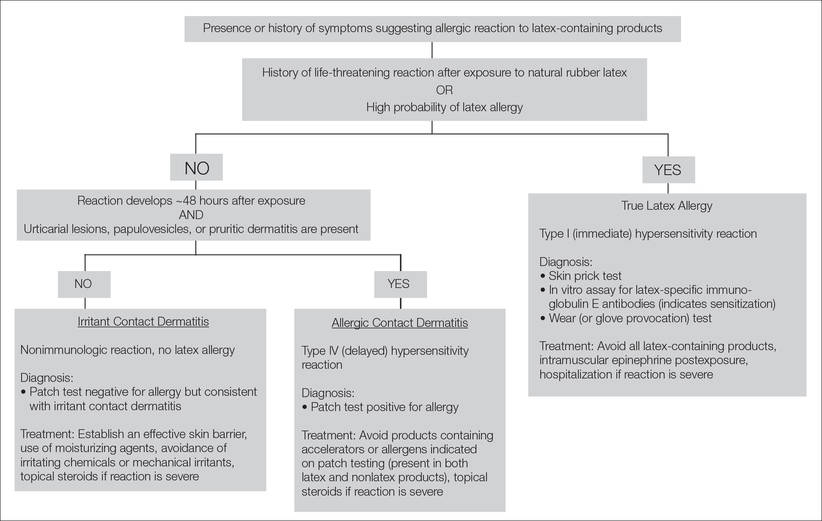
General Medical Protocol With Latex Reactions
To reduce the incidence of latex allergic reactions among health care workers and patients, Kumar2 recommends putting a protocol in place to document steps in preventing, diagnosing, and treating latex allergy. This protocol includes employee and patient education about the risks for developing latex allergy and the signs and symptoms of a reaction; available diagnostic testing; and alternative products (eg, hypoallergenic gloves) that are available to individuals with a known or suspected allergy. At-risk health care workers who have not been sensitized should be advised to avoid latex-containing products.3 Routine questioning and diagnostic testing may be necessary as part of every preoperative assessment, as there have been reported cases of anaphylaxis in patients with undocumented allergies.7 Anaphylaxis caused by latex allergy is the second leading cause of perioperative anaphylaxis, accounting for as many as 20% of cases.8 With the use of preventative measures and early identification of at-risk patients, the incidence of latex-related anaphylaxis is decreasing.8 Ascertaining valuable information about the patient’s medical history, such as known allergies to foods that have cross-reactivity to latex (eg, bananas, mango, kiwi, avocado), is one simple way of identifying a patient who should be tested for possible underlying latex allergy.8 Total avoidance of latex-containing products (eg, in the workplace) can further reduce the incidence of allergic reactions by decreasing primary sensitization and risk of exposure.
Conclusion
Patients claiming to be allergic to latex without documentation should be tested. The diagnostic testing available in the United States includes patch testing, wear (or glove provocation) testing, or assessment of IgE antibody titer. Accurate differentiation among irritant contact dermatitis, allergic contact dermatitis, and true latex allergy is paramount for properly educating patients and effectively treating these conditions. Additionally, distinguishing patients with true latex allergy from those who have been misdiagnosed can save resources and reduce health care costs.
- Bousquet J, Flahault A, Vandenplas O, et al. Natural rubber latex allergy among health care workers: a systematic review of the evidence. J Allergy Clin Immunol. 2006;118:447-454.
- Kumar RP. Latex allergy in clinical practice. Indian J Dermatol. 2012;57:66-70.
- Taylor JS, Erkek E. Latex allergy: diagnosis and management. Dermatol Ther. 2004;17:289-301.
- Schmid K, Christoph Broding H, Niklas D, et al. Latex sensitization in dental students using powder-free gloves low in latex protein: a cross-sectional study. Contact Dermatitis. 2002;47:103-108.
- Brown RH, Schauble JF, Hamilton RG. Prevalence of latex allergy among anesthesiologists: identification of sensitized but asymptomatic individuals. Anesthesiology. 1998;89:292-299.
- Pollart SM, Warniment C, Mori T. Latex allergy. Am Fam Physician. 2009;80:1413-1418.
- Duger C, Kol IO, Kaygusuz K, et al. A perioperative anaphylactic reaction caused by latex in a patient with no history of allergy. Anaesth Pain Intensive Care. 2012;16:71-73.
- Hepner DL, Castells MC. Anaphylaxis during the perioperative period. Anesth Analg. 2003;97:1381-1395.
Latex allergy is an all-encompassing term used to describe hypersensitivity reactions to products containing natural rubber latex from the Hevea brasiliensis tree and affects approximately 1% to 2% of the general population.1 Although latex gloves are the most widely known culprits, several other commonly used products can contain natural rubber latex, including adhesive tape, balloons, condoms, rubber bands, paint, tourniquets, electrode pads, and Foley catheters.2 The term latex allergy often is used as a general diagnosis, but there are in fact 3 distinct mechanisms by which individuals may develop an adverse reaction to latex-containing products: irritant contact dermatitis, allergic contact dermatitis (type IV hypersensitivity) and true latex allergy (type I hypersensitivity).
Irritant Contact Dermatitis
Irritant contact dermatitis, a nonimmunologic reaction, occurs due to mechanical factors (eg, friction) or contact with chemicals, which can have irritating and dehydrating effects. Individuals with irritant contact dermatitis do not have true latex allergy and will not necessarily develop a reaction to products containing natural rubber latex. Incorrectly attributing these irritant contact dermatitis reactions to latex allergy and simply advising patients to avoid all latex products (eg, use nitrile gloves rather than latex gloves) will not address the underlying problem. Rather, these patients must be informed that the dermatitis is a result of a disruption to the natural, protective skin barrier and not an allergic reaction.
Allergic Contact Dermatitis
Allergic contact dermatitis to rubber is caused by a type IV (delayed) hypersensitivity reaction and is the result of exposure to the accelerators present in rubber products in sensitive individuals. Individuals experiencing this type of reaction typically develop localized erythema, pruritus, and urticarial lesions 48 hours after exposure.3 Incorrectly labeling this problem as latex allergy and recommending nonlatex rubber substitutes (eg, hypoallergenic gloves) likely will not be effective, as these nonlatex replacement products contain the same accelerators as do latex gloves.
True Latex Allergy
The most severe form of latex allergy, often referred to as true latex allergy, is caused by a type I (immediate) hypersensitivity reaction mediated by immunoglobulin E (IgE) antibodies. Individuals experiencing this type of reaction have a systemic response to latex proteins that may result in fulminant anaphylaxis. Individuals with true latex allergy must absolutely avoid latex products, and substituting nonlatex products is the most effective approach.
Latex Reactions in Medical Practice
The varying propensity of certain populations to develop latex allergy has been well documented; for example, the prevalence of hypersensitivity in patients with spina bifida ranges from 20% to 65%, figures that are much higher than those reported in the general population.3 This hypersensitivity in patients with spina bifida most likely results from repeated exposure to latex products during corrective surgeries and diagnostic procedures early in life. Atopic individuals, such as those with allergic rhinitis, eczema, and asthma, have a 4-fold increased risk for developing latex allergy compared to nonatopic individuals.4 The risk of latex allergy among health care workers is increased due to increased exposure to rubber products. One study found that the risk of latex sensitization among health care workers exposed to products containing latex was 4.3%, while the risk in the general population was only 1.37%.1 Those at highest risk for sensitization include dental assistants, operating room personnel, hospital housekeeping staff, and paramedics or emergency medical technicians.3 However, sensitization documented on laboratory assessment does not reliably correlate with symptomatic allergy, as many patients with a positive IgE test do not show clinical symptoms. Schmid et al4 demonstrated that a 1.3% prevalence of clinically symptomatic latex allergy among health care workers may approximate the prevalence of latex allergy in the general population. In a study by Brown et al,5 although 12.5% of anesthesiologists were found to be sensitized to latex, only 2.4% had clinically symptomatic allergic reactions.
Testing for Latex Allergy
Several diagnostic tests are available to establish a diagnosis of type I sensitization or true latex allergy. Skin prick testing is an in vivo assay and is the gold standard for diagnosing IgE-mediated type I hypersensitivity to latex. The test involves pricking the skin of the forearm and applying a commercial extract of nonammoniated latex to monitor for development of a wheal within several minutes. The skin prick test should be performed in a health care setting equipped with oxygen, epinephrine, and latex-free resuscitation equipment in case of anaphylaxis following exposure. Although latex skin prick testing is the gold standard, it is rarely performed in the United States because there is no US Food and Drug Administration–approved natural rubber latex reagent.3 Consequently, physicians who wish to perform skin prick testing for latex allergy are forced to develop improvised reagents from the H brasiliensis tree itself or from highly allergenic latex gloves. Standardized latex allergens are commercially available in Europe.
The most noninvasive method of latex allergy testing is an in vitro assay for latex-specific IgE antibodies, which can be detected by either a radioallergosorbent test (RAST) or enzyme-linked immunosorbent assay (ELISA). The presence of antilatex IgE antibodies confirms sensitization but does not necessarily mean the patient will develop a symptomatic reaction following exposure. Due to the unavailability of a standardized reagent for the skin prick test in the United States, evaluation of latex-specific serum IgE levels may be the best alternative. While the skin prick test has the highest sensitivity, the sensitivity and specificity of latex-specific serum IgE testing are 50% to 90% and 80% to 87%, respectively.6
The wear test (also known as the use or glove provocation test), can be used to diagnose clinically symptomatic latex allergy when there is a discrepancy between the patient’s clinical history and results from skin prick or serum IgE antibody testing. To perform the wear test, place a natural rubber latex glove on one of the patient’s fingers for 15 minutes and monitor the area for development of urticaria. If there is no evidence of allergic reaction within 15 minutes, place the glove on the whole hand for an additional 15 minutes. The patient is said to be nonreactive if a latex glove can be placed on the entire hand for 15 minutes without evidence of reaction.3
Lastly, patch testing can differentiate between irritant contact and allergic contact (type IV hypersensitivity) dermatitis. Apply a small amount of each substance of interest onto a separate disc and place the discs in direct contact with the skin using hypoallergenic tape. With type IV latex hypersensitivity, the skin underneath the disc will become erythematous with developing papulovesicles, starting between 2 and 5 days after exposure. The Figure outlines the differentiation of true latex allergy from irritant and allergic contact dermatitis and identifies methods for making these diagnoses.
General Medical Protocol With Latex Reactions
To reduce the incidence of latex allergic reactions among health care workers and patients, Kumar2 recommends putting a protocol in place to document steps in preventing, diagnosing, and treating latex allergy. This protocol includes employee and patient education about the risks for developing latex allergy and the signs and symptoms of a reaction; available diagnostic testing; and alternative products (eg, hypoallergenic gloves) that are available to individuals with a known or suspected allergy. At-risk health care workers who have not been sensitized should be advised to avoid latex-containing products.3 Routine questioning and diagnostic testing may be necessary as part of every preoperative assessment, as there have been reported cases of anaphylaxis in patients with undocumented allergies.7 Anaphylaxis caused by latex allergy is the second leading cause of perioperative anaphylaxis, accounting for as many as 20% of cases.8 With the use of preventative measures and early identification of at-risk patients, the incidence of latex-related anaphylaxis is decreasing.8 Ascertaining valuable information about the patient’s medical history, such as known allergies to foods that have cross-reactivity to latex (eg, bananas, mango, kiwi, avocado), is one simple way of identifying a patient who should be tested for possible underlying latex allergy.8 Total avoidance of latex-containing products (eg, in the workplace) can further reduce the incidence of allergic reactions by decreasing primary sensitization and risk of exposure.
Conclusion
Patients claiming to be allergic to latex without documentation should be tested. The diagnostic testing available in the United States includes patch testing, wear (or glove provocation) testing, or assessment of IgE antibody titer. Accurate differentiation among irritant contact dermatitis, allergic contact dermatitis, and true latex allergy is paramount for properly educating patients and effectively treating these conditions. Additionally, distinguishing patients with true latex allergy from those who have been misdiagnosed can save resources and reduce health care costs.
Latex allergy is an all-encompassing term used to describe hypersensitivity reactions to products containing natural rubber latex from the Hevea brasiliensis tree and affects approximately 1% to 2% of the general population.1 Although latex gloves are the most widely known culprits, several other commonly used products can contain natural rubber latex, including adhesive tape, balloons, condoms, rubber bands, paint, tourniquets, electrode pads, and Foley catheters.2 The term latex allergy often is used as a general diagnosis, but there are in fact 3 distinct mechanisms by which individuals may develop an adverse reaction to latex-containing products: irritant contact dermatitis, allergic contact dermatitis (type IV hypersensitivity) and true latex allergy (type I hypersensitivity).
Irritant Contact Dermatitis
Irritant contact dermatitis, a nonimmunologic reaction, occurs due to mechanical factors (eg, friction) or contact with chemicals, which can have irritating and dehydrating effects. Individuals with irritant contact dermatitis do not have true latex allergy and will not necessarily develop a reaction to products containing natural rubber latex. Incorrectly attributing these irritant contact dermatitis reactions to latex allergy and simply advising patients to avoid all latex products (eg, use nitrile gloves rather than latex gloves) will not address the underlying problem. Rather, these patients must be informed that the dermatitis is a result of a disruption to the natural, protective skin barrier and not an allergic reaction.
Allergic Contact Dermatitis
Allergic contact dermatitis to rubber is caused by a type IV (delayed) hypersensitivity reaction and is the result of exposure to the accelerators present in rubber products in sensitive individuals. Individuals experiencing this type of reaction typically develop localized erythema, pruritus, and urticarial lesions 48 hours after exposure.3 Incorrectly labeling this problem as latex allergy and recommending nonlatex rubber substitutes (eg, hypoallergenic gloves) likely will not be effective, as these nonlatex replacement products contain the same accelerators as do latex gloves.
True Latex Allergy
The most severe form of latex allergy, often referred to as true latex allergy, is caused by a type I (immediate) hypersensitivity reaction mediated by immunoglobulin E (IgE) antibodies. Individuals experiencing this type of reaction have a systemic response to latex proteins that may result in fulminant anaphylaxis. Individuals with true latex allergy must absolutely avoid latex products, and substituting nonlatex products is the most effective approach.
Latex Reactions in Medical Practice
The varying propensity of certain populations to develop latex allergy has been well documented; for example, the prevalence of hypersensitivity in patients with spina bifida ranges from 20% to 65%, figures that are much higher than those reported in the general population.3 This hypersensitivity in patients with spina bifida most likely results from repeated exposure to latex products during corrective surgeries and diagnostic procedures early in life. Atopic individuals, such as those with allergic rhinitis, eczema, and asthma, have a 4-fold increased risk for developing latex allergy compared to nonatopic individuals.4 The risk of latex allergy among health care workers is increased due to increased exposure to rubber products. One study found that the risk of latex sensitization among health care workers exposed to products containing latex was 4.3%, while the risk in the general population was only 1.37%.1 Those at highest risk for sensitization include dental assistants, operating room personnel, hospital housekeeping staff, and paramedics or emergency medical technicians.3 However, sensitization documented on laboratory assessment does not reliably correlate with symptomatic allergy, as many patients with a positive IgE test do not show clinical symptoms. Schmid et al4 demonstrated that a 1.3% prevalence of clinically symptomatic latex allergy among health care workers may approximate the prevalence of latex allergy in the general population. In a study by Brown et al,5 although 12.5% of anesthesiologists were found to be sensitized to latex, only 2.4% had clinically symptomatic allergic reactions.
Testing for Latex Allergy
Several diagnostic tests are available to establish a diagnosis of type I sensitization or true latex allergy. Skin prick testing is an in vivo assay and is the gold standard for diagnosing IgE-mediated type I hypersensitivity to latex. The test involves pricking the skin of the forearm and applying a commercial extract of nonammoniated latex to monitor for development of a wheal within several minutes. The skin prick test should be performed in a health care setting equipped with oxygen, epinephrine, and latex-free resuscitation equipment in case of anaphylaxis following exposure. Although latex skin prick testing is the gold standard, it is rarely performed in the United States because there is no US Food and Drug Administration–approved natural rubber latex reagent.3 Consequently, physicians who wish to perform skin prick testing for latex allergy are forced to develop improvised reagents from the H brasiliensis tree itself or from highly allergenic latex gloves. Standardized latex allergens are commercially available in Europe.
The most noninvasive method of latex allergy testing is an in vitro assay for latex-specific IgE antibodies, which can be detected by either a radioallergosorbent test (RAST) or enzyme-linked immunosorbent assay (ELISA). The presence of antilatex IgE antibodies confirms sensitization but does not necessarily mean the patient will develop a symptomatic reaction following exposure. Due to the unavailability of a standardized reagent for the skin prick test in the United States, evaluation of latex-specific serum IgE levels may be the best alternative. While the skin prick test has the highest sensitivity, the sensitivity and specificity of latex-specific serum IgE testing are 50% to 90% and 80% to 87%, respectively.6
The wear test (also known as the use or glove provocation test), can be used to diagnose clinically symptomatic latex allergy when there is a discrepancy between the patient’s clinical history and results from skin prick or serum IgE antibody testing. To perform the wear test, place a natural rubber latex glove on one of the patient’s fingers for 15 minutes and monitor the area for development of urticaria. If there is no evidence of allergic reaction within 15 minutes, place the glove on the whole hand for an additional 15 minutes. The patient is said to be nonreactive if a latex glove can be placed on the entire hand for 15 minutes without evidence of reaction.3
Lastly, patch testing can differentiate between irritant contact and allergic contact (type IV hypersensitivity) dermatitis. Apply a small amount of each substance of interest onto a separate disc and place the discs in direct contact with the skin using hypoallergenic tape. With type IV latex hypersensitivity, the skin underneath the disc will become erythematous with developing papulovesicles, starting between 2 and 5 days after exposure. The Figure outlines the differentiation of true latex allergy from irritant and allergic contact dermatitis and identifies methods for making these diagnoses.
General Medical Protocol With Latex Reactions
To reduce the incidence of latex allergic reactions among health care workers and patients, Kumar2 recommends putting a protocol in place to document steps in preventing, diagnosing, and treating latex allergy. This protocol includes employee and patient education about the risks for developing latex allergy and the signs and symptoms of a reaction; available diagnostic testing; and alternative products (eg, hypoallergenic gloves) that are available to individuals with a known or suspected allergy. At-risk health care workers who have not been sensitized should be advised to avoid latex-containing products.3 Routine questioning and diagnostic testing may be necessary as part of every preoperative assessment, as there have been reported cases of anaphylaxis in patients with undocumented allergies.7 Anaphylaxis caused by latex allergy is the second leading cause of perioperative anaphylaxis, accounting for as many as 20% of cases.8 With the use of preventative measures and early identification of at-risk patients, the incidence of latex-related anaphylaxis is decreasing.8 Ascertaining valuable information about the patient’s medical history, such as known allergies to foods that have cross-reactivity to latex (eg, bananas, mango, kiwi, avocado), is one simple way of identifying a patient who should be tested for possible underlying latex allergy.8 Total avoidance of latex-containing products (eg, in the workplace) can further reduce the incidence of allergic reactions by decreasing primary sensitization and risk of exposure.
Conclusion
Patients claiming to be allergic to latex without documentation should be tested. The diagnostic testing available in the United States includes patch testing, wear (or glove provocation) testing, or assessment of IgE antibody titer. Accurate differentiation among irritant contact dermatitis, allergic contact dermatitis, and true latex allergy is paramount for properly educating patients and effectively treating these conditions. Additionally, distinguishing patients with true latex allergy from those who have been misdiagnosed can save resources and reduce health care costs.
- Bousquet J, Flahault A, Vandenplas O, et al. Natural rubber latex allergy among health care workers: a systematic review of the evidence. J Allergy Clin Immunol. 2006;118:447-454.
- Kumar RP. Latex allergy in clinical practice. Indian J Dermatol. 2012;57:66-70.
- Taylor JS, Erkek E. Latex allergy: diagnosis and management. Dermatol Ther. 2004;17:289-301.
- Schmid K, Christoph Broding H, Niklas D, et al. Latex sensitization in dental students using powder-free gloves low in latex protein: a cross-sectional study. Contact Dermatitis. 2002;47:103-108.
- Brown RH, Schauble JF, Hamilton RG. Prevalence of latex allergy among anesthesiologists: identification of sensitized but asymptomatic individuals. Anesthesiology. 1998;89:292-299.
- Pollart SM, Warniment C, Mori T. Latex allergy. Am Fam Physician. 2009;80:1413-1418.
- Duger C, Kol IO, Kaygusuz K, et al. A perioperative anaphylactic reaction caused by latex in a patient with no history of allergy. Anaesth Pain Intensive Care. 2012;16:71-73.
- Hepner DL, Castells MC. Anaphylaxis during the perioperative period. Anesth Analg. 2003;97:1381-1395.
- Bousquet J, Flahault A, Vandenplas O, et al. Natural rubber latex allergy among health care workers: a systematic review of the evidence. J Allergy Clin Immunol. 2006;118:447-454.
- Kumar RP. Latex allergy in clinical practice. Indian J Dermatol. 2012;57:66-70.
- Taylor JS, Erkek E. Latex allergy: diagnosis and management. Dermatol Ther. 2004;17:289-301.
- Schmid K, Christoph Broding H, Niklas D, et al. Latex sensitization in dental students using powder-free gloves low in latex protein: a cross-sectional study. Contact Dermatitis. 2002;47:103-108.
- Brown RH, Schauble JF, Hamilton RG. Prevalence of latex allergy among anesthesiologists: identification of sensitized but asymptomatic individuals. Anesthesiology. 1998;89:292-299.
- Pollart SM, Warniment C, Mori T. Latex allergy. Am Fam Physician. 2009;80:1413-1418.
- Duger C, Kol IO, Kaygusuz K, et al. A perioperative anaphylactic reaction caused by latex in a patient with no history of allergy. Anaesth Pain Intensive Care. 2012;16:71-73.
- Hepner DL, Castells MC. Anaphylaxis during the perioperative period. Anesth Analg. 2003;97:1381-1395.
Practice Points
- The term latex allergy often is used as a general diagnosis to describe 3 types of reactions to natural rubber latex, including irritant contact dermatitis, allergic contact dermatitis (type IV hypersensitivity reaction), and true latex allergy (type I hypersensitivity reaction).
- The latex skin prick test is considered the gold standard for diagnosis of true latex allergy, but this method is not available in the United States. In vitro assay for latex-specific immunoglobulin E antibodies is the best alternative.
The “Impossible” Diagnosis
I was taught—and still believe—that obtaining a thorough history can direct you to a good working diagnosis. About 20 years ago, while in the Navy, I had a patient who showed me that I should not be fooled by a history that does not fit the current presentation.
The patient was a 34-year-old sailor with right-side knee pain, occurring intermittently for a long time but worsening in recent months. The pain did not prevent him from running, performing in the Navy’s semi-annual fitness test, or participating in departmental physical fitness activities.
However, his pain worsened after he was assigned to a ship, which required him to ascend and descend the steep shipboard stairs or ladders. He also complained of some intermittent buckling or “giving out.” But he was quite clear when he stated that he had sustained no recent injury to explain his condition.
His history was notable for an injury he sustained six years earlier, while running. Although he could not remember the exact mechanism of injury, he recalled that his knee hurt and was swollen the next day. He was seen in medical, where he was given crutches, modified duty, and ibuprofen for a few days. After a relatively short time, his activity returned to normal.
I had seen a lot of knee pain on board ship, mostly of the patellar tendonitis or patellofemoral syndrome types, that could often be treated conservatively with temporary duty modification to avoid aggravating activity. More serious injuries—such as meniscal, collateral, or cruciate ligament tears—were associated with recent or acute injuries and a history including a suspicious mechanism of injury.
This patient’s complete knee exam was largely unremarkable, except his anterior drawer test seemed to have no distinct endpoint. When I compared the results with his asymptomatic left knee, I could not appreciate any difference.
So I relayed to him my thought process: If he had done something serious to his knee six years ago, it probably would have manifested sooner. As other clinicians did previously, I treated him conservatively with duty limitations and advised him that if he failed to improve soon, I would refer him to an orthopedist for a second opinion.
Well, he did not improve soon. Since he was still concerned, I provided the referral, without obtaining an MRI.
To perhaps everyone’s surprise—but most definitely mine—the patient was diagnosed with a complete ACL tear by the orthopedist (again, without MRI). He was scheduled for surgery at a later date.
What surprised me most was that someone could perform the way he was required to perform in the Navy for six years with a torn ACL. As a result of this case, I have not let a remote history of injury cloud my judgment since!
I was taught—and still believe—that obtaining a thorough history can direct you to a good working diagnosis. About 20 years ago, while in the Navy, I had a patient who showed me that I should not be fooled by a history that does not fit the current presentation.
The patient was a 34-year-old sailor with right-side knee pain, occurring intermittently for a long time but worsening in recent months. The pain did not prevent him from running, performing in the Navy’s semi-annual fitness test, or participating in departmental physical fitness activities.
However, his pain worsened after he was assigned to a ship, which required him to ascend and descend the steep shipboard stairs or ladders. He also complained of some intermittent buckling or “giving out.” But he was quite clear when he stated that he had sustained no recent injury to explain his condition.
His history was notable for an injury he sustained six years earlier, while running. Although he could not remember the exact mechanism of injury, he recalled that his knee hurt and was swollen the next day. He was seen in medical, where he was given crutches, modified duty, and ibuprofen for a few days. After a relatively short time, his activity returned to normal.
I had seen a lot of knee pain on board ship, mostly of the patellar tendonitis or patellofemoral syndrome types, that could often be treated conservatively with temporary duty modification to avoid aggravating activity. More serious injuries—such as meniscal, collateral, or cruciate ligament tears—were associated with recent or acute injuries and a history including a suspicious mechanism of injury.
This patient’s complete knee exam was largely unremarkable, except his anterior drawer test seemed to have no distinct endpoint. When I compared the results with his asymptomatic left knee, I could not appreciate any difference.
So I relayed to him my thought process: If he had done something serious to his knee six years ago, it probably would have manifested sooner. As other clinicians did previously, I treated him conservatively with duty limitations and advised him that if he failed to improve soon, I would refer him to an orthopedist for a second opinion.
Well, he did not improve soon. Since he was still concerned, I provided the referral, without obtaining an MRI.
To perhaps everyone’s surprise—but most definitely mine—the patient was diagnosed with a complete ACL tear by the orthopedist (again, without MRI). He was scheduled for surgery at a later date.
What surprised me most was that someone could perform the way he was required to perform in the Navy for six years with a torn ACL. As a result of this case, I have not let a remote history of injury cloud my judgment since!
I was taught—and still believe—that obtaining a thorough history can direct you to a good working diagnosis. About 20 years ago, while in the Navy, I had a patient who showed me that I should not be fooled by a history that does not fit the current presentation.
The patient was a 34-year-old sailor with right-side knee pain, occurring intermittently for a long time but worsening in recent months. The pain did not prevent him from running, performing in the Navy’s semi-annual fitness test, or participating in departmental physical fitness activities.
However, his pain worsened after he was assigned to a ship, which required him to ascend and descend the steep shipboard stairs or ladders. He also complained of some intermittent buckling or “giving out.” But he was quite clear when he stated that he had sustained no recent injury to explain his condition.
His history was notable for an injury he sustained six years earlier, while running. Although he could not remember the exact mechanism of injury, he recalled that his knee hurt and was swollen the next day. He was seen in medical, where he was given crutches, modified duty, and ibuprofen for a few days. After a relatively short time, his activity returned to normal.
I had seen a lot of knee pain on board ship, mostly of the patellar tendonitis or patellofemoral syndrome types, that could often be treated conservatively with temporary duty modification to avoid aggravating activity. More serious injuries—such as meniscal, collateral, or cruciate ligament tears—were associated with recent or acute injuries and a history including a suspicious mechanism of injury.
This patient’s complete knee exam was largely unremarkable, except his anterior drawer test seemed to have no distinct endpoint. When I compared the results with his asymptomatic left knee, I could not appreciate any difference.
So I relayed to him my thought process: If he had done something serious to his knee six years ago, it probably would have manifested sooner. As other clinicians did previously, I treated him conservatively with duty limitations and advised him that if he failed to improve soon, I would refer him to an orthopedist for a second opinion.
Well, he did not improve soon. Since he was still concerned, I provided the referral, without obtaining an MRI.
To perhaps everyone’s surprise—but most definitely mine—the patient was diagnosed with a complete ACL tear by the orthopedist (again, without MRI). He was scheduled for surgery at a later date.
What surprised me most was that someone could perform the way he was required to perform in the Navy for six years with a torn ACL. As a result of this case, I have not let a remote history of injury cloud my judgment since!
HU noninferior to transfusion for stroke prevention in SCD

Photo courtesy of ASH
ORLANDO, FL—Hydroxyurea (HU) is noninferior to chronic blood transfusions for reducing the risk of stroke in children with sickle cell disease (SCD), results of the TWiTCH trial suggest.
The trial showed that daily doses of HU lower the transcranial Doppler (TCD) blood velocity in children with SCD to a similar degree as blood transfusions, thereby decreasing the risk of stroke.
Because of these findings, the trial was terminated early, in November of last year.
Last week, results from TWiTCH were presented at the 2015 ASH Annual Meeting (abstract 3*) and published in The Lancet. The study was funded by the National Heart Lung and Blood Institute.
“Stroke . . . is one of the most severe and catastrophic clinical events that occurs in children with sickle cell, with serious motor and cognitive sequelae,” said study investigator and ASH presenter Russell E. Ware, MD, of Cincinnati Children’s Hospital Medical Center in Ohio.
“With the advent of TCD, we now have the ability to identify high-risk children and use chronic transfusion therapy to prevent primary stroke.”
Dr Ware noted that results of the STOP trial showed that chronic transfusion reduced the risk of stroke in high-risk children with SCD, but the transfusions could not be stopped. The STOP 2 trial confirmed this, showing that stopping transfusions led to an increase in TCD blood velocity and stroke risk.
Because transfusions must be continued indefinitely and are associated with morbidity, an alternative stroke prevention strategy is needed, Dr Ware said. He and his colleagues conducted the TWiTCH trial to determine if HU would fit the bill.
Study design
For this phase 3 study, the researchers compared 24 months of transfusions to HU in children with SCD and abnormal TCD velocities. Study enrollment began in September 2011 and ended in April 2013.
All eligible children had received at least 12 months of transfusions prior to enrollment. They were randomized 1:1 to continue receiving transfusions or to receive the maximum-tolerated dose (MTD) of HU.
In the transfusion arm, the goal was to keep hemoglobin S levels below 30%, and iron overload was managed with daily oral chelation.
In the HU arm, the drug was escalated to the MTD, and children continued receiving transfusions until the MTD was achieved. Iron overload was managed with monthly phlebotomy.
The study had a noninferiority design, and the primary endpoint was the 24-month TCD velocity (with a noninferiority margin of 15 cm/sec). TCD velocities were obtained every 12 weeks and reviewed centrally. Local researchers were masked to the results.
Results
In all, 121 children were randomized—61 to transfusions and 60 to HU. Patient characteristics—baseline TCD velocities, age, duration of transfusion, etc.—were well balanced between the treatment arms.
“The average age of the patients was 9 or 10 years old, with about 3 or 4 years of transfusions coming in to the study,” Dr Ware noted.
In the transfusion arm, the children maintained a hemoglobin level of about 9 g/dL and hemoglobin S levels of less than 30%. Most patients received chelation with deferasirox at 26 ±6 mg/kg/day.
In the HU arm, 57 of 60 patients reached the MTD, which was 27 ± 4 mg/kg/day, on average. The median transfusion overlap was 6 months, the average absolute neutrophil count was 3.5 ± 1.6 x 109/L, the average hemoglobin was about 9 g/dL, and fetal hemoglobin rose to about 25%. There were 756 phlebotomy procedures performed in 54 children.
“[In the HU arm,] very quickly after enrollment, the sickle hemoglobin rises, as the transfusions are weaned,” Dr Ware noted.
“Commensurately, the hemoglobin F rises as a protection. The neutrophil count and reticulocyte count drops, and those curves [counts in the HU and transfusion arms] diverge fairly quickly. The serum ferritin [curves] diverged as well.”
Early termination and noninferiority
Interim data analyses were scheduled to take place after one-third of the patients had exited the study and after two-thirds had exited. The first interim analysis demonstrated noninferiority, and the trial was closed early. An analysis was repeated after half of the patients had exited the study, and the trial was terminated.
At that point, 42 children had completed 24 months of treatment in the transfusion arm, 11 patients had truncated treatment, and 8 had early exits. Forty-one patients had completed 24 months of therapy in the HU arm, 13 had truncated treatment, and 6 had early exits.
The final TCD velocity (mean ± standard error) was 143 ± 1.6 cm/sec in the transfusion arm and 138 ± 1.6 cm/sec in the HU arm. The P value for noninferiority (in the intent-to-treat population) was 8.82 x 10-16. By post-hoc analysis, the P value for superiority was 0.023.
Secondary endpoints
There were 29 new neurological events during the trial—12 in the transfusion arm and 17 in the HU arm. There were no new strokes, but there were 6 new transient ischemic attacks—3 in each arm.
There were no new cerebral infarcts in either arm. But there was 1 new progressive vasculopathy in the transfusion arm. And 1 child in the transfusion arm was withdrawn from the study for increasing TCD (>240 cm/sec).
Iron overload improved more in the HU arm than the transfusion arm, with a greater average change in both serum ferritin (P<0.001) and liver iron concentration (P=0.001).
Serious adverse events were more common in the HU arm than the transfusion arm—23 events in 9 patients and 10 events in 6 patients, respectively. But none of these events were thought to be related to study treatment or procedures.
The most common serious adverse event in both groups was vaso-occlusive pain—11 events in 5 HU-treated patients and 3 events in 1 transfusion-treated patient.
Dr Ware noted that there were no secondary leukemias associated with HU in this trial, and there is “a cumulative body of evidence” spanning 20 years that suggests the drug is not carcinogenic in this patient population. ![]()
*Data in the abstract differ from data presented at the meeting.
Photo courtesy of ASH
ORLANDO, FL—Hydroxyurea (HU) is noninferior to chronic blood transfusions for reducing the risk of stroke in children with sickle cell disease (SCD), results of the TWiTCH trial suggest.
The trial showed that daily doses of HU lower the transcranial Doppler (TCD) blood velocity in children with SCD to a similar degree as blood transfusions, thereby decreasing the risk of stroke.
Because of these findings, the trial was terminated early, in November of last year.
Last week, results from TWiTCH were presented at the 2015 ASH Annual Meeting (abstract 3*) and published in The Lancet. The study was funded by the National Heart Lung and Blood Institute.
“Stroke . . . is one of the most severe and catastrophic clinical events that occurs in children with sickle cell, with serious motor and cognitive sequelae,” said study investigator and ASH presenter Russell E. Ware, MD, of Cincinnati Children’s Hospital Medical Center in Ohio.
“With the advent of TCD, we now have the ability to identify high-risk children and use chronic transfusion therapy to prevent primary stroke.”
Dr Ware noted that results of the STOP trial showed that chronic transfusion reduced the risk of stroke in high-risk children with SCD, but the transfusions could not be stopped. The STOP 2 trial confirmed this, showing that stopping transfusions led to an increase in TCD blood velocity and stroke risk.
Because transfusions must be continued indefinitely and are associated with morbidity, an alternative stroke prevention strategy is needed, Dr Ware said. He and his colleagues conducted the TWiTCH trial to determine if HU would fit the bill.
Study design
For this phase 3 study, the researchers compared 24 months of transfusions to HU in children with SCD and abnormal TCD velocities. Study enrollment began in September 2011 and ended in April 2013.
All eligible children had received at least 12 months of transfusions prior to enrollment. They were randomized 1:1 to continue receiving transfusions or to receive the maximum-tolerated dose (MTD) of HU.
In the transfusion arm, the goal was to keep hemoglobin S levels below 30%, and iron overload was managed with daily oral chelation.
In the HU arm, the drug was escalated to the MTD, and children continued receiving transfusions until the MTD was achieved. Iron overload was managed with monthly phlebotomy.
The study had a noninferiority design, and the primary endpoint was the 24-month TCD velocity (with a noninferiority margin of 15 cm/sec). TCD velocities were obtained every 12 weeks and reviewed centrally. Local researchers were masked to the results.
Results
In all, 121 children were randomized—61 to transfusions and 60 to HU. Patient characteristics—baseline TCD velocities, age, duration of transfusion, etc.—were well balanced between the treatment arms.
“The average age of the patients was 9 or 10 years old, with about 3 or 4 years of transfusions coming in to the study,” Dr Ware noted.
In the transfusion arm, the children maintained a hemoglobin level of about 9 g/dL and hemoglobin S levels of less than 30%. Most patients received chelation with deferasirox at 26 ±6 mg/kg/day.
In the HU arm, 57 of 60 patients reached the MTD, which was 27 ± 4 mg/kg/day, on average. The median transfusion overlap was 6 months, the average absolute neutrophil count was 3.5 ± 1.6 x 109/L, the average hemoglobin was about 9 g/dL, and fetal hemoglobin rose to about 25%. There were 756 phlebotomy procedures performed in 54 children.
“[In the HU arm,] very quickly after enrollment, the sickle hemoglobin rises, as the transfusions are weaned,” Dr Ware noted.
“Commensurately, the hemoglobin F rises as a protection. The neutrophil count and reticulocyte count drops, and those curves [counts in the HU and transfusion arms] diverge fairly quickly. The serum ferritin [curves] diverged as well.”
Early termination and noninferiority
Interim data analyses were scheduled to take place after one-third of the patients had exited the study and after two-thirds had exited. The first interim analysis demonstrated noninferiority, and the trial was closed early. An analysis was repeated after half of the patients had exited the study, and the trial was terminated.
At that point, 42 children had completed 24 months of treatment in the transfusion arm, 11 patients had truncated treatment, and 8 had early exits. Forty-one patients had completed 24 months of therapy in the HU arm, 13 had truncated treatment, and 6 had early exits.
The final TCD velocity (mean ± standard error) was 143 ± 1.6 cm/sec in the transfusion arm and 138 ± 1.6 cm/sec in the HU arm. The P value for noninferiority (in the intent-to-treat population) was 8.82 x 10-16. By post-hoc analysis, the P value for superiority was 0.023.
Secondary endpoints
There were 29 new neurological events during the trial—12 in the transfusion arm and 17 in the HU arm. There were no new strokes, but there were 6 new transient ischemic attacks—3 in each arm.
There were no new cerebral infarcts in either arm. But there was 1 new progressive vasculopathy in the transfusion arm. And 1 child in the transfusion arm was withdrawn from the study for increasing TCD (>240 cm/sec).
Iron overload improved more in the HU arm than the transfusion arm, with a greater average change in both serum ferritin (P<0.001) and liver iron concentration (P=0.001).
Serious adverse events were more common in the HU arm than the transfusion arm—23 events in 9 patients and 10 events in 6 patients, respectively. But none of these events were thought to be related to study treatment or procedures.
The most common serious adverse event in both groups was vaso-occlusive pain—11 events in 5 HU-treated patients and 3 events in 1 transfusion-treated patient.
Dr Ware noted that there were no secondary leukemias associated with HU in this trial, and there is “a cumulative body of evidence” spanning 20 years that suggests the drug is not carcinogenic in this patient population. ![]()
*Data in the abstract differ from data presented at the meeting.
Photo courtesy of ASH
ORLANDO, FL—Hydroxyurea (HU) is noninferior to chronic blood transfusions for reducing the risk of stroke in children with sickle cell disease (SCD), results of the TWiTCH trial suggest.
The trial showed that daily doses of HU lower the transcranial Doppler (TCD) blood velocity in children with SCD to a similar degree as blood transfusions, thereby decreasing the risk of stroke.
Because of these findings, the trial was terminated early, in November of last year.
Last week, results from TWiTCH were presented at the 2015 ASH Annual Meeting (abstract 3*) and published in The Lancet. The study was funded by the National Heart Lung and Blood Institute.
“Stroke . . . is one of the most severe and catastrophic clinical events that occurs in children with sickle cell, with serious motor and cognitive sequelae,” said study investigator and ASH presenter Russell E. Ware, MD, of Cincinnati Children’s Hospital Medical Center in Ohio.
“With the advent of TCD, we now have the ability to identify high-risk children and use chronic transfusion therapy to prevent primary stroke.”
Dr Ware noted that results of the STOP trial showed that chronic transfusion reduced the risk of stroke in high-risk children with SCD, but the transfusions could not be stopped. The STOP 2 trial confirmed this, showing that stopping transfusions led to an increase in TCD blood velocity and stroke risk.
Because transfusions must be continued indefinitely and are associated with morbidity, an alternative stroke prevention strategy is needed, Dr Ware said. He and his colleagues conducted the TWiTCH trial to determine if HU would fit the bill.
Study design
For this phase 3 study, the researchers compared 24 months of transfusions to HU in children with SCD and abnormal TCD velocities. Study enrollment began in September 2011 and ended in April 2013.
All eligible children had received at least 12 months of transfusions prior to enrollment. They were randomized 1:1 to continue receiving transfusions or to receive the maximum-tolerated dose (MTD) of HU.
In the transfusion arm, the goal was to keep hemoglobin S levels below 30%, and iron overload was managed with daily oral chelation.
In the HU arm, the drug was escalated to the MTD, and children continued receiving transfusions until the MTD was achieved. Iron overload was managed with monthly phlebotomy.
The study had a noninferiority design, and the primary endpoint was the 24-month TCD velocity (with a noninferiority margin of 15 cm/sec). TCD velocities were obtained every 12 weeks and reviewed centrally. Local researchers were masked to the results.
Results
In all, 121 children were randomized—61 to transfusions and 60 to HU. Patient characteristics—baseline TCD velocities, age, duration of transfusion, etc.—were well balanced between the treatment arms.
“The average age of the patients was 9 or 10 years old, with about 3 or 4 years of transfusions coming in to the study,” Dr Ware noted.
In the transfusion arm, the children maintained a hemoglobin level of about 9 g/dL and hemoglobin S levels of less than 30%. Most patients received chelation with deferasirox at 26 ±6 mg/kg/day.
In the HU arm, 57 of 60 patients reached the MTD, which was 27 ± 4 mg/kg/day, on average. The median transfusion overlap was 6 months, the average absolute neutrophil count was 3.5 ± 1.6 x 109/L, the average hemoglobin was about 9 g/dL, and fetal hemoglobin rose to about 25%. There were 756 phlebotomy procedures performed in 54 children.
“[In the HU arm,] very quickly after enrollment, the sickle hemoglobin rises, as the transfusions are weaned,” Dr Ware noted.
“Commensurately, the hemoglobin F rises as a protection. The neutrophil count and reticulocyte count drops, and those curves [counts in the HU and transfusion arms] diverge fairly quickly. The serum ferritin [curves] diverged as well.”
Early termination and noninferiority
Interim data analyses were scheduled to take place after one-third of the patients had exited the study and after two-thirds had exited. The first interim analysis demonstrated noninferiority, and the trial was closed early. An analysis was repeated after half of the patients had exited the study, and the trial was terminated.
At that point, 42 children had completed 24 months of treatment in the transfusion arm, 11 patients had truncated treatment, and 8 had early exits. Forty-one patients had completed 24 months of therapy in the HU arm, 13 had truncated treatment, and 6 had early exits.
The final TCD velocity (mean ± standard error) was 143 ± 1.6 cm/sec in the transfusion arm and 138 ± 1.6 cm/sec in the HU arm. The P value for noninferiority (in the intent-to-treat population) was 8.82 x 10-16. By post-hoc analysis, the P value for superiority was 0.023.
Secondary endpoints
There were 29 new neurological events during the trial—12 in the transfusion arm and 17 in the HU arm. There were no new strokes, but there were 6 new transient ischemic attacks—3 in each arm.
There were no new cerebral infarcts in either arm. But there was 1 new progressive vasculopathy in the transfusion arm. And 1 child in the transfusion arm was withdrawn from the study for increasing TCD (>240 cm/sec).
Iron overload improved more in the HU arm than the transfusion arm, with a greater average change in both serum ferritin (P<0.001) and liver iron concentration (P=0.001).
Serious adverse events were more common in the HU arm than the transfusion arm—23 events in 9 patients and 10 events in 6 patients, respectively. But none of these events were thought to be related to study treatment or procedures.
The most common serious adverse event in both groups was vaso-occlusive pain—11 events in 5 HU-treated patients and 3 events in 1 transfusion-treated patient.
Dr Ware noted that there were no secondary leukemias associated with HU in this trial, and there is “a cumulative body of evidence” spanning 20 years that suggests the drug is not carcinogenic in this patient population. ![]()
*Data in the abstract differ from data presented at the meeting.
Approach can help reduce VTE after surgery
Photo by Piotr Bodzek
New research suggests that individualized feedback is more effective than group instructions for helping general surgery residents prevent venous thromboembolism (VTE) in their patients.
The single-center study showed that regular, one-on-one feedback and written report cards helped ensure the use of correct VTE prophylaxis more effectively than the usual group lectures that newly minted surgeons receive as part of their training.
These results were published in Annals of Surgery.
The study, conducted between July 2013 and March 2014, involved 49 general surgery residents in their first through fifth year of training at Johns Hopkins Hospital in Baltimore, Maryland.
For the first 3 months, residents received no personalized feedback. For the following 3 months, they received an electronic score card via email detailing their individual performance, including how many times they prescribed the appropriate VTE prophylaxis, how many times they failed to do so, and how they fared compared with others.
For the next 3 months, all residents continued to receive monthly scores, but subpar performers—those who failed to prescribe appropriate treatment to every single patient they cared for—also received one-on-one coaching from a senior resident.
In the span of 6 months, this approach decreased—from 3 to 0—the number of preventable complications among surgery patients (complications occurring in patients who didn’t receive appropriate VTE prophylaxis).
In the 3-month period prior to deploying the personalized feedback strategy, 7 out of 865 surgical patients developed complications. Three of the 7 cases were subsequently identified as preventable. In comparison, there were no such preventable complications after residents received individualized feedback.
As a result of the feedback, the number of patients getting appropriate treatment increased from 89% to 96%.
The number of residents who performed at 100% (prescribing the correct treatment to every patient all the time) went up from 22 (45%) to 38 (78%). Most of the prescription failures—19 out of 28 such cases—were clustered in a group of 4 residents.
“Our results show that personalized, concrete feedback can be a form of forced introspection that improves self-awareness and decision-making on clotting prophylaxis,” said Elliott Haut, MD, PhD, of the Johns Hopkins University School of Medicine.
Beyond that, Dr Haut and his colleagues believe these results illustrate the notion that simple interventions can be harnessed to foster learning and improve performance among any frontline clinician.
“Speaking more broadly, why stop with residents? Why stop with anticlotting prophylaxis?” Dr Haut asked. “If our findings are borne out by larger studies, this approach could be harnessed to improve training and outcomes for anyone who touches a patient, from nurses to physicians to physical therapists.” ![]()
Photo by Piotr Bodzek
New research suggests that individualized feedback is more effective than group instructions for helping general surgery residents prevent venous thromboembolism (VTE) in their patients.
The single-center study showed that regular, one-on-one feedback and written report cards helped ensure the use of correct VTE prophylaxis more effectively than the usual group lectures that newly minted surgeons receive as part of their training.
These results were published in Annals of Surgery.
The study, conducted between July 2013 and March 2014, involved 49 general surgery residents in their first through fifth year of training at Johns Hopkins Hospital in Baltimore, Maryland.
For the first 3 months, residents received no personalized feedback. For the following 3 months, they received an electronic score card via email detailing their individual performance, including how many times they prescribed the appropriate VTE prophylaxis, how many times they failed to do so, and how they fared compared with others.
For the next 3 months, all residents continued to receive monthly scores, but subpar performers—those who failed to prescribe appropriate treatment to every single patient they cared for—also received one-on-one coaching from a senior resident.
In the span of 6 months, this approach decreased—from 3 to 0—the number of preventable complications among surgery patients (complications occurring in patients who didn’t receive appropriate VTE prophylaxis).
In the 3-month period prior to deploying the personalized feedback strategy, 7 out of 865 surgical patients developed complications. Three of the 7 cases were subsequently identified as preventable. In comparison, there were no such preventable complications after residents received individualized feedback.
As a result of the feedback, the number of patients getting appropriate treatment increased from 89% to 96%.
The number of residents who performed at 100% (prescribing the correct treatment to every patient all the time) went up from 22 (45%) to 38 (78%). Most of the prescription failures—19 out of 28 such cases—were clustered in a group of 4 residents.
“Our results show that personalized, concrete feedback can be a form of forced introspection that improves self-awareness and decision-making on clotting prophylaxis,” said Elliott Haut, MD, PhD, of the Johns Hopkins University School of Medicine.
Beyond that, Dr Haut and his colleagues believe these results illustrate the notion that simple interventions can be harnessed to foster learning and improve performance among any frontline clinician.
“Speaking more broadly, why stop with residents? Why stop with anticlotting prophylaxis?” Dr Haut asked. “If our findings are borne out by larger studies, this approach could be harnessed to improve training and outcomes for anyone who touches a patient, from nurses to physicians to physical therapists.” ![]()
Photo by Piotr Bodzek
New research suggests that individualized feedback is more effective than group instructions for helping general surgery residents prevent venous thromboembolism (VTE) in their patients.
The single-center study showed that regular, one-on-one feedback and written report cards helped ensure the use of correct VTE prophylaxis more effectively than the usual group lectures that newly minted surgeons receive as part of their training.
These results were published in Annals of Surgery.
The study, conducted between July 2013 and March 2014, involved 49 general surgery residents in their first through fifth year of training at Johns Hopkins Hospital in Baltimore, Maryland.
For the first 3 months, residents received no personalized feedback. For the following 3 months, they received an electronic score card via email detailing their individual performance, including how many times they prescribed the appropriate VTE prophylaxis, how many times they failed to do so, and how they fared compared with others.
For the next 3 months, all residents continued to receive monthly scores, but subpar performers—those who failed to prescribe appropriate treatment to every single patient they cared for—also received one-on-one coaching from a senior resident.
In the span of 6 months, this approach decreased—from 3 to 0—the number of preventable complications among surgery patients (complications occurring in patients who didn’t receive appropriate VTE prophylaxis).
In the 3-month period prior to deploying the personalized feedback strategy, 7 out of 865 surgical patients developed complications. Three of the 7 cases were subsequently identified as preventable. In comparison, there were no such preventable complications after residents received individualized feedback.
As a result of the feedback, the number of patients getting appropriate treatment increased from 89% to 96%.
The number of residents who performed at 100% (prescribing the correct treatment to every patient all the time) went up from 22 (45%) to 38 (78%). Most of the prescription failures—19 out of 28 such cases—were clustered in a group of 4 residents.
“Our results show that personalized, concrete feedback can be a form of forced introspection that improves self-awareness and decision-making on clotting prophylaxis,” said Elliott Haut, MD, PhD, of the Johns Hopkins University School of Medicine.
Beyond that, Dr Haut and his colleagues believe these results illustrate the notion that simple interventions can be harnessed to foster learning and improve performance among any frontline clinician.
“Speaking more broadly, why stop with residents? Why stop with anticlotting prophylaxis?” Dr Haut asked. “If our findings are borne out by larger studies, this approach could be harnessed to improve training and outcomes for anyone who touches a patient, from nurses to physicians to physical therapists.” ![]()