User login
Low paravalvular leak shown in real-world registry of Sapien 3 recipients
PARIS – The initial report from a large, prospective registry documenting outcomes in recipients of the Edwards Sapien 3 transcatheter aortic valve in real-world clinical practice confirm that the excellent results seen earlier in the rarified, randomized clinical trial setting are routinely reproducible in everyday practice, Dr. Olaf Wendler said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
“The big thing with the Sapien 3 is the reduction in paravalvular leakage. Our rate of moderate or severe paravalvular leakage in the registry at 30 days is only 3.1%, with 73.6% of patients having no or only trace paravalvular leakage,” reported Dr. Wendler, professor of cardiothoracic surgery at King’s College Hospital, London.

The mean gradient improved from 43.8 mm Hg at baseline to 11.7 mm Hg at discharge, while the effective orifice area climbed from 0.72 to 1.67 cm2.
He presented 30-day outcomes from the SOURCE 3 registry for 1,947 recipients of the Edwards Sapien 3 valve during transcatheter aortic valve replacement at 80 European centers in 10 countries. Their average age was 81.6 years. Updates will continue during a planned 5 years of prospective follow-up.
“This will be a very rich dataset for subgroup analyses. We now have a number of procedural variables we can analyze over time. We will have good data to get to better outcomes in terms of how best to perform the procedure if you do it with a Sapien valve,” he explained.
Among the key findings: The lower profile of the Sapien 3 delivery system, compared with earlier iterations of the Sapien valve, enabled 87% of patients in the registry to undergo TAVR via transfemoral access. And, in these 1,695 patients, whose mean logistic EuroSCORE was 17.8, the 30-day rates of all-cause mortality and disabling stroke were just 1.9% and 0.5%, respectively.
“I think there are not many series that have shown better results than these,” Dr. Wendler commented.
The non–transfemoral-access group is a very different, higher surgical risk cohort with lots more comorbid conditions. Their mean logistic EuroSCORE was 21.8. Yet in this group, the 30-day all-cause mortality and disabling stroke rates were still only 4% and 0.8%.
The transfemoral access group had markedly lower rates of life-threatening bleeding (4%), new-onset atrial fibrillation (4.8%), extended ventilation (3.5%), stage 2-3 acute kidney injury (0.8%), plus a 2-day shorter mean hospital length of stay.
Sixty percent of transfemoral access patients had their TAVR done under conscious sedation. These 1,018 patients constitute the largest dataset ever treated using conscious sedation with one valve system. The conscious sedation group had significantly lower baseline rates of carotid artery disease, prior coronary artery bypass graft surgery, and heart failure than did patients who received general anesthesia. At 30 days post-TAVR, the conscious sedation group had significantly lower rates of extended ventilation and postdilatation, and they received less contrast volume than did the general anesthesia group. However, they had a significantly higher incidence of stroke: 1.7%, compared with 0.6% for the general anesthesia group.
“Why that is the case we don’t know at the moment. We need to look into this more in the future,” the surgeon said.
A particularly impressive finding was how well Sapien 3 valve recipients with a low left ventricular ejection fraction have done. For example, 30-day all-cause mortality in the 100 patients with a baseline LVEF below 30% was 3%, not significantly different from the 1.8% rate in the 1,198 subjects with an LVEF greater than 50% or the 2.6% in patients with an LVEF of 30%-50%.
“Three percent all-cause mortality with an ejection fraction below 30% is just remarkable from my point of view,” Dr. Wendler said.
The 778 who didn’t undergo balloon aortic valvuloplasty prior to Sapien 3 implantation did not fare any worse than did those who did in terms of 30-day all-cause mortality or all strokes, but they did have significantly higher rates of life-threatening bleeding and major vascular complications.
The SOURCE 3 registry is sponsored by Edwards Lifesciences. Dr. Wendler serves as a consultant to the company.
PARIS – The initial report from a large, prospective registry documenting outcomes in recipients of the Edwards Sapien 3 transcatheter aortic valve in real-world clinical practice confirm that the excellent results seen earlier in the rarified, randomized clinical trial setting are routinely reproducible in everyday practice, Dr. Olaf Wendler said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
“The big thing with the Sapien 3 is the reduction in paravalvular leakage. Our rate of moderate or severe paravalvular leakage in the registry at 30 days is only 3.1%, with 73.6% of patients having no or only trace paravalvular leakage,” reported Dr. Wendler, professor of cardiothoracic surgery at King’s College Hospital, London.
The mean gradient improved from 43.8 mm Hg at baseline to 11.7 mm Hg at discharge, while the effective orifice area climbed from 0.72 to 1.67 cm2.
He presented 30-day outcomes from the SOURCE 3 registry for 1,947 recipients of the Edwards Sapien 3 valve during transcatheter aortic valve replacement at 80 European centers in 10 countries. Their average age was 81.6 years. Updates will continue during a planned 5 years of prospective follow-up.
“This will be a very rich dataset for subgroup analyses. We now have a number of procedural variables we can analyze over time. We will have good data to get to better outcomes in terms of how best to perform the procedure if you do it with a Sapien valve,” he explained.
Among the key findings: The lower profile of the Sapien 3 delivery system, compared with earlier iterations of the Sapien valve, enabled 87% of patients in the registry to undergo TAVR via transfemoral access. And, in these 1,695 patients, whose mean logistic EuroSCORE was 17.8, the 30-day rates of all-cause mortality and disabling stroke were just 1.9% and 0.5%, respectively.
“I think there are not many series that have shown better results than these,” Dr. Wendler commented.
The non–transfemoral-access group is a very different, higher surgical risk cohort with lots more comorbid conditions. Their mean logistic EuroSCORE was 21.8. Yet in this group, the 30-day all-cause mortality and disabling stroke rates were still only 4% and 0.8%.
The transfemoral access group had markedly lower rates of life-threatening bleeding (4%), new-onset atrial fibrillation (4.8%), extended ventilation (3.5%), stage 2-3 acute kidney injury (0.8%), plus a 2-day shorter mean hospital length of stay.
Sixty percent of transfemoral access patients had their TAVR done under conscious sedation. These 1,018 patients constitute the largest dataset ever treated using conscious sedation with one valve system. The conscious sedation group had significantly lower baseline rates of carotid artery disease, prior coronary artery bypass graft surgery, and heart failure than did patients who received general anesthesia. At 30 days post-TAVR, the conscious sedation group had significantly lower rates of extended ventilation and postdilatation, and they received less contrast volume than did the general anesthesia group. However, they had a significantly higher incidence of stroke: 1.7%, compared with 0.6% for the general anesthesia group.
“Why that is the case we don’t know at the moment. We need to look into this more in the future,” the surgeon said.
A particularly impressive finding was how well Sapien 3 valve recipients with a low left ventricular ejection fraction have done. For example, 30-day all-cause mortality in the 100 patients with a baseline LVEF below 30% was 3%, not significantly different from the 1.8% rate in the 1,198 subjects with an LVEF greater than 50% or the 2.6% in patients with an LVEF of 30%-50%.
“Three percent all-cause mortality with an ejection fraction below 30% is just remarkable from my point of view,” Dr. Wendler said.
The 778 who didn’t undergo balloon aortic valvuloplasty prior to Sapien 3 implantation did not fare any worse than did those who did in terms of 30-day all-cause mortality or all strokes, but they did have significantly higher rates of life-threatening bleeding and major vascular complications.
The SOURCE 3 registry is sponsored by Edwards Lifesciences. Dr. Wendler serves as a consultant to the company.
PARIS – The initial report from a large, prospective registry documenting outcomes in recipients of the Edwards Sapien 3 transcatheter aortic valve in real-world clinical practice confirm that the excellent results seen earlier in the rarified, randomized clinical trial setting are routinely reproducible in everyday practice, Dr. Olaf Wendler said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
“The big thing with the Sapien 3 is the reduction in paravalvular leakage. Our rate of moderate or severe paravalvular leakage in the registry at 30 days is only 3.1%, with 73.6% of patients having no or only trace paravalvular leakage,” reported Dr. Wendler, professor of cardiothoracic surgery at King’s College Hospital, London.
The mean gradient improved from 43.8 mm Hg at baseline to 11.7 mm Hg at discharge, while the effective orifice area climbed from 0.72 to 1.67 cm2.
He presented 30-day outcomes from the SOURCE 3 registry for 1,947 recipients of the Edwards Sapien 3 valve during transcatheter aortic valve replacement at 80 European centers in 10 countries. Their average age was 81.6 years. Updates will continue during a planned 5 years of prospective follow-up.
“This will be a very rich dataset for subgroup analyses. We now have a number of procedural variables we can analyze over time. We will have good data to get to better outcomes in terms of how best to perform the procedure if you do it with a Sapien valve,” he explained.
Among the key findings: The lower profile of the Sapien 3 delivery system, compared with earlier iterations of the Sapien valve, enabled 87% of patients in the registry to undergo TAVR via transfemoral access. And, in these 1,695 patients, whose mean logistic EuroSCORE was 17.8, the 30-day rates of all-cause mortality and disabling stroke were just 1.9% and 0.5%, respectively.
“I think there are not many series that have shown better results than these,” Dr. Wendler commented.
The non–transfemoral-access group is a very different, higher surgical risk cohort with lots more comorbid conditions. Their mean logistic EuroSCORE was 21.8. Yet in this group, the 30-day all-cause mortality and disabling stroke rates were still only 4% and 0.8%.
The transfemoral access group had markedly lower rates of life-threatening bleeding (4%), new-onset atrial fibrillation (4.8%), extended ventilation (3.5%), stage 2-3 acute kidney injury (0.8%), plus a 2-day shorter mean hospital length of stay.
Sixty percent of transfemoral access patients had their TAVR done under conscious sedation. These 1,018 patients constitute the largest dataset ever treated using conscious sedation with one valve system. The conscious sedation group had significantly lower baseline rates of carotid artery disease, prior coronary artery bypass graft surgery, and heart failure than did patients who received general anesthesia. At 30 days post-TAVR, the conscious sedation group had significantly lower rates of extended ventilation and postdilatation, and they received less contrast volume than did the general anesthesia group. However, they had a significantly higher incidence of stroke: 1.7%, compared with 0.6% for the general anesthesia group.
“Why that is the case we don’t know at the moment. We need to look into this more in the future,” the surgeon said.
A particularly impressive finding was how well Sapien 3 valve recipients with a low left ventricular ejection fraction have done. For example, 30-day all-cause mortality in the 100 patients with a baseline LVEF below 30% was 3%, not significantly different from the 1.8% rate in the 1,198 subjects with an LVEF greater than 50% or the 2.6% in patients with an LVEF of 30%-50%.
“Three percent all-cause mortality with an ejection fraction below 30% is just remarkable from my point of view,” Dr. Wendler said.
The 778 who didn’t undergo balloon aortic valvuloplasty prior to Sapien 3 implantation did not fare any worse than did those who did in terms of 30-day all-cause mortality or all strokes, but they did have significantly higher rates of life-threatening bleeding and major vascular complications.
The SOURCE 3 registry is sponsored by Edwards Lifesciences. Dr. Wendler serves as a consultant to the company.
AT EUROPCR 2016
Key clinical point: Only 3.1% of patients had moderate or severe paravalvular leak at 30 days after TAVR with the Sapien 3.
Major finding: The 30-day, all-cause mortality and disabling stroke rates were 1.9% and 0.5% in Sapien 3 valve recipients whose transcatheter aortic valve replacement was done by the transfemoral route.
Data source: A prospective multicenter European registry which includes 1,947 patients who underwent transcatheter aortic valve replacement with the Edwards Sapien 3 valve in real-world commercial settings.
Disclosures: The SOURCE 3 registry is sponsored by Edwards Lifesciences. Dr. Wendler serves as a consultant to the company.

VIDEO: Depression worsens newly diagnosed juvenile idiopathic arthritis
LONDON – Depression is relatively common among teenagers newly diagnosed with juvenile idiopathic arthritis, and adolescents with both disorders appeared to have a less complete response to their treatment in a study of 102 patients.
Juvenile idiopathic arthritis (JIA) that first manifests when a patient is a teenager comes at a “vulnerable time” that can drive the development and worsening of depression, and depression can potentially exacerbate inflammation and also interfere with treatment compliance, Dr. John Ioannou said at the European Congress of Rheumatology,
Depression and JIA can produce a “vicious cycle in which depression exacerbates the disease and the disease exacerbates depression,” explained Dr. Ioannou, a rheumatologist at University College Hospital in London.
Although no study results have yet identified an effective intervention for depression identified in teenagers with newly diagnosed JIA, the immediate message from these new findings is that clinicians must assess the psychological health of adolescents with JIA both when they are first diagnosed as well as at subsequent visits, and if depression is found it requires some sort of intervention, Dr. Ioannou said in an interview.
He and his associates studied 102 patients from the United Kingdom, who were newly diagnosed with JIA and were 11-16 years old at baseline and enrolled in the Childhood Arthritis Prospective Study (CAPS), a nationwide cohort of patients with childhood-onset arthritis of various types. The average age of the group they studied was just under 13 years old, 57% were girls, 52% had persistent oligoarticular arthritis, 30% had polyarticular arthritis, and 18% had enthesitis-related arthritis. All patients underwent assessment at baseline for depression using the Mood and Feelings Questionnaire and 15 (15%) had a score that flagged them as having “probable” depression.
This depression prevalence is about three- to fourfold higher than for an otherwise healthy group of similarly aged adolescents, Dr. Ioannou said.
At baseline, the subgroup of teens with depression had a significantly higher number of inflamed joints, restricted joints, and also more overall pain and disability as measured on the Childhood Health Assessment Questionnaire.
The 102 teens with JIA underwent follow-up assessment 1-3 years later, after they had received ongoing treatment for their JIA. At follow-up, standard JIA treatment had largely resulted in resolution of joint inflammation and movement restriction among all patients, including those with depression at baseline. However the adolescents who had both JIA and depression at entry continued to have significantly more pain and disability at follow-up than did the nondepressed JIA patients, suggesting a link between depression and refractory pain and disability in JIA patients, the researchers reported.
“We need to ensure that psychological assessments and support are available to all young people diagnosed with JIA, and that this is fully integrated into routine care” for newly diagnosed JIA patients, Dr. Ioannou said. He had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
Prior study results showed that about a third of adult patients with rheumatoid arthritis are depressed. There seems to be a vulnerability to depression among patients diagnosed with inflammatory arthritis. It makes sense that chronic arthritis can cause depression as it is a painful, debilitating, and long-term disease that is often nonremitting.

|
| Mitchel L. Zoler/Frontline Medical News Susan Barlett, Ph.D. |
Good evidence also suggests that people who are depressed are more vulnerable to develop rheumatoid arthritis or other autoimmune diseases.
The important new findings reported by Dr. Ioannou and his associates underscore the importance of providing psychological support to adolescents newly diagnosed with juvenile idiopathic arthritis. Among its many effects, depression is one of the few robust predictors of nonadherence to medical treatments by patients. Patients who are depressed are less likely to take their medications as prescribed. Depressed patients are also more likely to smoke because tobacco smoking can produce some depression relief. But smoking also contributes to the development and worsening of rheumatoid arthritis and likely other forms of inflammatory arthritis.
Susan Bartlett, Ph.D., is a psychologist and clinical epidemiologist at McGill University, Montreal, who specializes in chronic diseases including arthritis. She had no disclosures. She made these comments during a press conference.
Prior study results showed that about a third of adult patients with rheumatoid arthritis are depressed. There seems to be a vulnerability to depression among patients diagnosed with inflammatory arthritis. It makes sense that chronic arthritis can cause depression as it is a painful, debilitating, and long-term disease that is often nonremitting.
|
|
| Mitchel L. Zoler/Frontline Medical News Susan Barlett, Ph.D. |
Good evidence also suggests that people who are depressed are more vulnerable to develop rheumatoid arthritis or other autoimmune diseases.
The important new findings reported by Dr. Ioannou and his associates underscore the importance of providing psychological support to adolescents newly diagnosed with juvenile idiopathic arthritis. Among its many effects, depression is one of the few robust predictors of nonadherence to medical treatments by patients. Patients who are depressed are less likely to take their medications as prescribed. Depressed patients are also more likely to smoke because tobacco smoking can produce some depression relief. But smoking also contributes to the development and worsening of rheumatoid arthritis and likely other forms of inflammatory arthritis.
Susan Bartlett, Ph.D., is a psychologist and clinical epidemiologist at McGill University, Montreal, who specializes in chronic diseases including arthritis. She had no disclosures. She made these comments during a press conference.
Prior study results showed that about a third of adult patients with rheumatoid arthritis are depressed. There seems to be a vulnerability to depression among patients diagnosed with inflammatory arthritis. It makes sense that chronic arthritis can cause depression as it is a painful, debilitating, and long-term disease that is often nonremitting.
|
|
| Mitchel L. Zoler/Frontline Medical News Susan Barlett, Ph.D. |
Good evidence also suggests that people who are depressed are more vulnerable to develop rheumatoid arthritis or other autoimmune diseases.
The important new findings reported by Dr. Ioannou and his associates underscore the importance of providing psychological support to adolescents newly diagnosed with juvenile idiopathic arthritis. Among its many effects, depression is one of the few robust predictors of nonadherence to medical treatments by patients. Patients who are depressed are less likely to take their medications as prescribed. Depressed patients are also more likely to smoke because tobacco smoking can produce some depression relief. But smoking also contributes to the development and worsening of rheumatoid arthritis and likely other forms of inflammatory arthritis.
Susan Bartlett, Ph.D., is a psychologist and clinical epidemiologist at McGill University, Montreal, who specializes in chronic diseases including arthritis. She had no disclosures. She made these comments during a press conference.
LONDON – Depression is relatively common among teenagers newly diagnosed with juvenile idiopathic arthritis, and adolescents with both disorders appeared to have a less complete response to their treatment in a study of 102 patients.
Juvenile idiopathic arthritis (JIA) that first manifests when a patient is a teenager comes at a “vulnerable time” that can drive the development and worsening of depression, and depression can potentially exacerbate inflammation and also interfere with treatment compliance, Dr. John Ioannou said at the European Congress of Rheumatology,
Depression and JIA can produce a “vicious cycle in which depression exacerbates the disease and the disease exacerbates depression,” explained Dr. Ioannou, a rheumatologist at University College Hospital in London.
Although no study results have yet identified an effective intervention for depression identified in teenagers with newly diagnosed JIA, the immediate message from these new findings is that clinicians must assess the psychological health of adolescents with JIA both when they are first diagnosed as well as at subsequent visits, and if depression is found it requires some sort of intervention, Dr. Ioannou said in an interview.
He and his associates studied 102 patients from the United Kingdom, who were newly diagnosed with JIA and were 11-16 years old at baseline and enrolled in the Childhood Arthritis Prospective Study (CAPS), a nationwide cohort of patients with childhood-onset arthritis of various types. The average age of the group they studied was just under 13 years old, 57% were girls, 52% had persistent oligoarticular arthritis, 30% had polyarticular arthritis, and 18% had enthesitis-related arthritis. All patients underwent assessment at baseline for depression using the Mood and Feelings Questionnaire and 15 (15%) had a score that flagged them as having “probable” depression.
This depression prevalence is about three- to fourfold higher than for an otherwise healthy group of similarly aged adolescents, Dr. Ioannou said.
At baseline, the subgroup of teens with depression had a significantly higher number of inflamed joints, restricted joints, and also more overall pain and disability as measured on the Childhood Health Assessment Questionnaire.
The 102 teens with JIA underwent follow-up assessment 1-3 years later, after they had received ongoing treatment for their JIA. At follow-up, standard JIA treatment had largely resulted in resolution of joint inflammation and movement restriction among all patients, including those with depression at baseline. However the adolescents who had both JIA and depression at entry continued to have significantly more pain and disability at follow-up than did the nondepressed JIA patients, suggesting a link between depression and refractory pain and disability in JIA patients, the researchers reported.
“We need to ensure that psychological assessments and support are available to all young people diagnosed with JIA, and that this is fully integrated into routine care” for newly diagnosed JIA patients, Dr. Ioannou said. He had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
LONDON – Depression is relatively common among teenagers newly diagnosed with juvenile idiopathic arthritis, and adolescents with both disorders appeared to have a less complete response to their treatment in a study of 102 patients.
Juvenile idiopathic arthritis (JIA) that first manifests when a patient is a teenager comes at a “vulnerable time” that can drive the development and worsening of depression, and depression can potentially exacerbate inflammation and also interfere with treatment compliance, Dr. John Ioannou said at the European Congress of Rheumatology,
Depression and JIA can produce a “vicious cycle in which depression exacerbates the disease and the disease exacerbates depression,” explained Dr. Ioannou, a rheumatologist at University College Hospital in London.
Although no study results have yet identified an effective intervention for depression identified in teenagers with newly diagnosed JIA, the immediate message from these new findings is that clinicians must assess the psychological health of adolescents with JIA both when they are first diagnosed as well as at subsequent visits, and if depression is found it requires some sort of intervention, Dr. Ioannou said in an interview.
He and his associates studied 102 patients from the United Kingdom, who were newly diagnosed with JIA and were 11-16 years old at baseline and enrolled in the Childhood Arthritis Prospective Study (CAPS), a nationwide cohort of patients with childhood-onset arthritis of various types. The average age of the group they studied was just under 13 years old, 57% were girls, 52% had persistent oligoarticular arthritis, 30% had polyarticular arthritis, and 18% had enthesitis-related arthritis. All patients underwent assessment at baseline for depression using the Mood and Feelings Questionnaire and 15 (15%) had a score that flagged them as having “probable” depression.
This depression prevalence is about three- to fourfold higher than for an otherwise healthy group of similarly aged adolescents, Dr. Ioannou said.
At baseline, the subgroup of teens with depression had a significantly higher number of inflamed joints, restricted joints, and also more overall pain and disability as measured on the Childhood Health Assessment Questionnaire.
The 102 teens with JIA underwent follow-up assessment 1-3 years later, after they had received ongoing treatment for their JIA. At follow-up, standard JIA treatment had largely resulted in resolution of joint inflammation and movement restriction among all patients, including those with depression at baseline. However the adolescents who had both JIA and depression at entry continued to have significantly more pain and disability at follow-up than did the nondepressed JIA patients, suggesting a link between depression and refractory pain and disability in JIA patients, the researchers reported.
“We need to ensure that psychological assessments and support are available to all young people diagnosed with JIA, and that this is fully integrated into routine care” for newly diagnosed JIA patients, Dr. Ioannou said. He had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
AT THE EULAR 2016 CONGRESS
Key clinical point: Pain and disability often persist despite effective antirheumatic treatment in depressed adolescents with juvenile idiopathic arthritis.
Major finding: Disability and pain levels remained significantly elevated among JIA teens with depression, compared with JIA teens without baseline depression.
Data source: The 102 adolescents enrolled in the Childhood Arthritis Prospective Study with juvenile idiopathic arthritis, including 15 patients with depression at baseline.
Disclosures: Dr. Ioannou had no disclosures.

Artificial pancreas can improve inpatient glycemic control in type 2 diabetes
NEW ORLEANS – Having people with type 2 diabetes mellitus use an artificial pancreas during hospitalization has the potential to improve control of their glycemia when compared with conventional insulin therapy, based on the results of a small study of inpatients in the United Kingdom.
“This is the first study to show that automated subcutaneous closed-loop insulin delivery without meal-time insulin is feasible and safe in patients with insulin-treated type 2 diabetes in the general wards,” Dr. Hood Thabit of the University of Cambridge (England) reported at the ADA annual scientific sessions. “Closed-loop [delivery] increased time in target, with reduced glucose variability and reduced time spent hyperglycemic without actually increasing time spent hypoglycemic,” he said.
The study involved 40 general ward inpatients evenly assigned to the closed-loop system and conventional insulin therapy for 72 hours.

Dr. Thabit said hyperglycemia in hospital patients is a common problem that’s poorly managed. “There’s an unmet need for an effective and safe glucose control, specifically in the underserved and understudied population of type 2 diabetes in the general wards,” he said. The use of the closed-loop system in inpatients with type 2 diabetes “remains untested until now,” Dr. Thabit added.
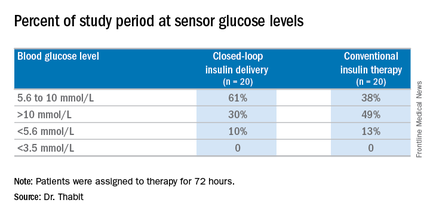
The 20 patients randomized to the closed-loop system spent an average of 61% of the whole study period within the sensor glucose target vs. 38% of those on conventional insulin therapy. The closed-loop patients also used comparable insulin daily on average: 62.6 U (±36.3 U) vs. 66.0 U (±39.6 U), Dr. Thabit said.
He noted that those on the closed-loop system did not have to announce meals to the control algorithm, or give any meal-time insulin – “we didn’t want to trouble our nurses with this, due to the increasing workload that health care professionals in the hospital currently face,” he said – and showed “significantly improved” nighttime control of glucose while “simultaneously reducing the risk of nocturnal hypoglycemia”. “The closed loop may potentially be an effective and safe tool to manage hospital inpatient hyperglycemia in this particularly underserved population of patients whilst easing the burden of health care professionals in hospital,” he said.
The cost is not insignificant. The pump and sensor devices together with related consumables can cost up to £6,000 (about $8,600), but he did note the artificial pancreas device itself is reusable. The cost of the automated closed-loop glucose control system can also potentially be offset by the reduced time of health care professionals spent managing inpatient hyperglycemia safely. The study investigators are in the process of planning a larger trial, Dr. Thabit said.
Dr. Thabit had no financial disclosures. Some coauthors disclosed relationships with Novo Nordisk; Medtronic MiniMed; Becton, Dickinson and Co.; Abbott Diabetes Care; Roche Pharmaceuticals; Cell Novo; Animas; Eli Lilly; B. Braun Melsungen; Sanofi-Aventis Deutschland; and Profil Institute for Clinical Research.
NEW ORLEANS – Having people with type 2 diabetes mellitus use an artificial pancreas during hospitalization has the potential to improve control of their glycemia when compared with conventional insulin therapy, based on the results of a small study of inpatients in the United Kingdom.
“This is the first study to show that automated subcutaneous closed-loop insulin delivery without meal-time insulin is feasible and safe in patients with insulin-treated type 2 diabetes in the general wards,” Dr. Hood Thabit of the University of Cambridge (England) reported at the ADA annual scientific sessions. “Closed-loop [delivery] increased time in target, with reduced glucose variability and reduced time spent hyperglycemic without actually increasing time spent hypoglycemic,” he said.
The study involved 40 general ward inpatients evenly assigned to the closed-loop system and conventional insulin therapy for 72 hours.
Dr. Thabit said hyperglycemia in hospital patients is a common problem that’s poorly managed. “There’s an unmet need for an effective and safe glucose control, specifically in the underserved and understudied population of type 2 diabetes in the general wards,” he said. The use of the closed-loop system in inpatients with type 2 diabetes “remains untested until now,” Dr. Thabit added.
The 20 patients randomized to the closed-loop system spent an average of 61% of the whole study period within the sensor glucose target vs. 38% of those on conventional insulin therapy. The closed-loop patients also used comparable insulin daily on average: 62.6 U (±36.3 U) vs. 66.0 U (±39.6 U), Dr. Thabit said.
He noted that those on the closed-loop system did not have to announce meals to the control algorithm, or give any meal-time insulin – “we didn’t want to trouble our nurses with this, due to the increasing workload that health care professionals in the hospital currently face,” he said – and showed “significantly improved” nighttime control of glucose while “simultaneously reducing the risk of nocturnal hypoglycemia”. “The closed loop may potentially be an effective and safe tool to manage hospital inpatient hyperglycemia in this particularly underserved population of patients whilst easing the burden of health care professionals in hospital,” he said.
The cost is not insignificant. The pump and sensor devices together with related consumables can cost up to £6,000 (about $8,600), but he did note the artificial pancreas device itself is reusable. The cost of the automated closed-loop glucose control system can also potentially be offset by the reduced time of health care professionals spent managing inpatient hyperglycemia safely. The study investigators are in the process of planning a larger trial, Dr. Thabit said.
Dr. Thabit had no financial disclosures. Some coauthors disclosed relationships with Novo Nordisk; Medtronic MiniMed; Becton, Dickinson and Co.; Abbott Diabetes Care; Roche Pharmaceuticals; Cell Novo; Animas; Eli Lilly; B. Braun Melsungen; Sanofi-Aventis Deutschland; and Profil Institute for Clinical Research.
NEW ORLEANS – Having people with type 2 diabetes mellitus use an artificial pancreas during hospitalization has the potential to improve control of their glycemia when compared with conventional insulin therapy, based on the results of a small study of inpatients in the United Kingdom.
“This is the first study to show that automated subcutaneous closed-loop insulin delivery without meal-time insulin is feasible and safe in patients with insulin-treated type 2 diabetes in the general wards,” Dr. Hood Thabit of the University of Cambridge (England) reported at the ADA annual scientific sessions. “Closed-loop [delivery] increased time in target, with reduced glucose variability and reduced time spent hyperglycemic without actually increasing time spent hypoglycemic,” he said.
The study involved 40 general ward inpatients evenly assigned to the closed-loop system and conventional insulin therapy for 72 hours.
Dr. Thabit said hyperglycemia in hospital patients is a common problem that’s poorly managed. “There’s an unmet need for an effective and safe glucose control, specifically in the underserved and understudied population of type 2 diabetes in the general wards,” he said. The use of the closed-loop system in inpatients with type 2 diabetes “remains untested until now,” Dr. Thabit added.
The 20 patients randomized to the closed-loop system spent an average of 61% of the whole study period within the sensor glucose target vs. 38% of those on conventional insulin therapy. The closed-loop patients also used comparable insulin daily on average: 62.6 U (±36.3 U) vs. 66.0 U (±39.6 U), Dr. Thabit said.
He noted that those on the closed-loop system did not have to announce meals to the control algorithm, or give any meal-time insulin – “we didn’t want to trouble our nurses with this, due to the increasing workload that health care professionals in the hospital currently face,” he said – and showed “significantly improved” nighttime control of glucose while “simultaneously reducing the risk of nocturnal hypoglycemia”. “The closed loop may potentially be an effective and safe tool to manage hospital inpatient hyperglycemia in this particularly underserved population of patients whilst easing the burden of health care professionals in hospital,” he said.
The cost is not insignificant. The pump and sensor devices together with related consumables can cost up to £6,000 (about $8,600), but he did note the artificial pancreas device itself is reusable. The cost of the automated closed-loop glucose control system can also potentially be offset by the reduced time of health care professionals spent managing inpatient hyperglycemia safely. The study investigators are in the process of planning a larger trial, Dr. Thabit said.
Dr. Thabit had no financial disclosures. Some coauthors disclosed relationships with Novo Nordisk; Medtronic MiniMed; Becton, Dickinson and Co.; Abbott Diabetes Care; Roche Pharmaceuticals; Cell Novo; Animas; Eli Lilly; B. Braun Melsungen; Sanofi-Aventis Deutschland; and Profil Institute for Clinical Research.
AT THE ADA ANNUAL SCIENTIFIC SESSIONS
Key clinical point:Hospitalized patients in the general ward with type 2 diabetes mellitus can maintain better glycemic control by using an artificial pancreas than by taking conventional therapy.
Major finding: Patients on the closed-loop system spent an average of 61% of the study period within the sensor glucose target vs. 38% for those on conventional insulin therapy.
Data source: A single-center study of 40 patients randomized to wear the artificial pancreas or take conventional therapy.
Disclosures: Dr. Thabit had no financial disclosures. Some coauthors disclosed relationships with Novo Nordisk; Medtronic MiniMed; Becton, Dickinson and Co.; Abbott Diabetes Care; Roche Pharmaceuticals; Cell Novo; Animas; Eli Lilly; B. Braun Melsungen; Sanofi-Aventis Deutschland; and Profil Institute for Clinical Research.

AAP, NASPAG issue joint guidance on menstruation management in teens with disabilities
For the first time, the American Academy of Pediatrics is offering guidance on managing menstruation and sexuality education in adolescents with disabilities.
Written jointly with the North American Society for Pediatric and Adolescent Gynecology, the clinical report offers guidance on options for menstrual management, sexual education and expression, and protection from sexual abuse. The report gives guidance regarding the care of adolescents with physical and/or intellectual abilities, but not for those with psychiatric illnesses (Pediatrics. 2016 April. doi: 10.1542/peds.2016-0295).
“Taking care of teens with disabilities and figuring out what to do with menstrual management has been all over the map,” Dr. Cora Collette Breuner, chair of the AAP’s committee on adolescence, said in an interview. “So, we tried to clarify it and help clinicians know what to do and when.”

A particular concern for the two groups was the threat of adverse events. “We wanted to cover what’s safe and what’s not in menstruation management, especially around bone health and thromboembolic events,” said Dr. Breuner, a professor of pediatrics and adolescent medicine at the University of Washington, Seattle.
Some of the information will not surprise clinicians, but there are some data that will perhaps come as news, said Dr. Breuner, including the recommendation that long-acting reversible contraception (LARC), such as the levonorgestrel intrauterine device or the progesterone implant rod should be considered first-line management therapies. “We point out that a number of studies show that these are safe.”
The report emphasizes offering anticipatory guidance before menses begins, noting that most teens with disabilities mature at the same rate as teens without disabilities. The report does not recommend premenarchal suppression in these patients, because doing so can interfere with normal bone growth. Such suppression also prevents patients and their families and caregivers from discovering that coping with the onset of menses is perhaps not as difficult as they might fear, the report states.
Although combined oral contraceptives are not contraindicated in teens with mobility issues, to guard against the threat of thromboembolic events in teens who use wheelchairs, the report recommends taking a thorough family history to rule out inherited thrombophilia. Otherwise, the recommendation is to prescribe the lowest-dose estrogen with a first- or second-generation progestin, as these are associated with lower rates of venous thrombotic events.
The guidance states that if cycles are creating difficulties in the patient’s life, “as determined by health care providers, patients, and families,” then menstrual management is appropriate. Even though it may take up to 3 years before a menstrual cycle becomes regular, the report cites irregularities caused by certain medications can be reason enough for menstrual management. Specific drugs noted include those affecting the dopaminergic system, valproic acid, and medications that elevate prolactin. Teens with obesity, seizure disorders, and polycystic ovary syndrome also can experience higher rates of irregularity.
The report also warns against the assumption that teens with disabilities are asexual or uninterested in sex. When appropriate, they should be offered the same confidential conversations about sexuality as are recommended for all teenagers by the AAP and the American College of Obstetricians and Gynecologists. “Teenagers with physical disabilities are just as likely to be sexually active as their peers and have a higher incidence of sexual abuse,” the report states. It is typically when a patient is cognitively impaired that consent to confidential services may require “discussion about legal guardianship or medical power of attorney status for families,” according to the report.
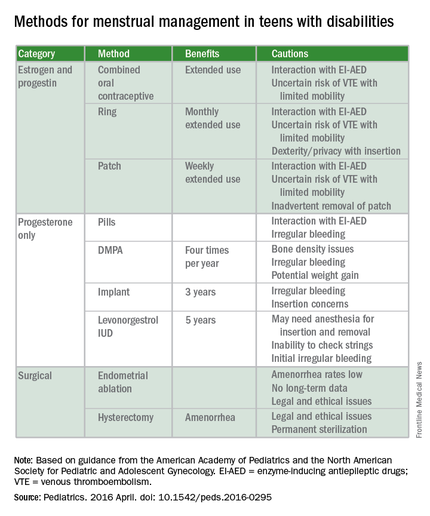
The report’s comprehensive review of four main menstrual management techniques – estrogen-containing, progestin-only, nonhormonal methods, and surgical requests and options – begins with the caveat that regardless of the method used, the threat of abuse or sexually transmitted infections remain. When a patient’s family or caregivers request suppression of menarche in a patient, stating fears of abuse or pregnancy, further investigation into the patient’s circumstances is warranted, the report states.
“It’s always worth reminding physicians that in this cohort, endometrial ablation can have legal implications, and it’s not recommended in this age group,” Dr. Breuner said.
On average, 1.5 hormonal methods are tried before achieving management goals, according to the report. Data cited in the study showed that at 42%, oral contraception is the preferred method of menstrual suppression, followed by the patch at 20%. Expectant management was third at 15%, followed by DMPA (depot medroxyprogesterone acetate) at 12%. The least utilized method was the levonorgestrel intrauterine device at 3%. No data were provided for the implantable contraceptive rod.
The clinical report is a companion document to another AAP clinical report, “Sexuality of Children and Adolescents with Developmental Disabilities” (Pediatrics. 2006. doi: 10.1542/peds.2006-1115).
AAP guidance on these matters in teens with psychiatric illnesses is expected to be issued within a few years, Dr. Breuner said.
There was no external funding and the authors have no relevant financial disclosures.
On Twitter @whitneymcknight
Although this guideline focuses on menstrual management and the guidance for you to help teens with disabilities through the pubertal transition, it’s very important to put this topic also into the context of sexuality. I think you have a great opportunity to do this because, often, you already have developed long-term relationships with these teenagers and their families, so the trust is already there. You should be the one to ensure all patients have appropriate sex education and help families with this.

|
Dr. Elisabeth Quint |
For some of these teens who are cognitively impaired, the initial conversations about sex may focus more on safety and abuse prevention. For example, which parts of their body should not be touched by other people. You can help the families really be the educators. Parents can be the ones to teach their kids how to protect themselves by rehearsing the answers to questions like, “What do you do if someone touches you? Who do you tell? Where do you go? What if it happens at school?” As part of the safety aspects, you also can help families assess whether the patient will be able to have a consensual sexual relationship. It’s the teens who have mild cognitive impairment that I worry about most, because often they are friendly and open to people, and can be taken advantage of. You just want to make sure they have the right information at their appropriate level.
Adolescents with physical disabilities are going to be just as interested in sex as any other teens and should be helped with any potential issues that they may have around that issue. They will likely get sex education in schools, but are still often viewed as not interested in sex or sexually active, and they may not get the usual confidential teen questions or appropriate screenings. Menstrual management and sexuality education both are important aspects of reproductive health care for teens with disabilities.
Dr. Elisabeth Quint, lead author of the AAP clinical report “Menstrual Management for Adolescents,” is a clinical professor of obstetrics and gynecology at the University of Michigan, Ann Arbor. She is also a past president of the North American Society for Pediatric and Adolescent Gynecology.
Although this guideline focuses on menstrual management and the guidance for you to help teens with disabilities through the pubertal transition, it’s very important to put this topic also into the context of sexuality. I think you have a great opportunity to do this because, often, you already have developed long-term relationships with these teenagers and their families, so the trust is already there. You should be the one to ensure all patients have appropriate sex education and help families with this.
|
|
Dr. Elisabeth Quint |
For some of these teens who are cognitively impaired, the initial conversations about sex may focus more on safety and abuse prevention. For example, which parts of their body should not be touched by other people. You can help the families really be the educators. Parents can be the ones to teach their kids how to protect themselves by rehearsing the answers to questions like, “What do you do if someone touches you? Who do you tell? Where do you go? What if it happens at school?” As part of the safety aspects, you also can help families assess whether the patient will be able to have a consensual sexual relationship. It’s the teens who have mild cognitive impairment that I worry about most, because often they are friendly and open to people, and can be taken advantage of. You just want to make sure they have the right information at their appropriate level.
Adolescents with physical disabilities are going to be just as interested in sex as any other teens and should be helped with any potential issues that they may have around that issue. They will likely get sex education in schools, but are still often viewed as not interested in sex or sexually active, and they may not get the usual confidential teen questions or appropriate screenings. Menstrual management and sexuality education both are important aspects of reproductive health care for teens with disabilities.
Dr. Elisabeth Quint, lead author of the AAP clinical report “Menstrual Management for Adolescents,” is a clinical professor of obstetrics and gynecology at the University of Michigan, Ann Arbor. She is also a past president of the North American Society for Pediatric and Adolescent Gynecology.
Although this guideline focuses on menstrual management and the guidance for you to help teens with disabilities through the pubertal transition, it’s very important to put this topic also into the context of sexuality. I think you have a great opportunity to do this because, often, you already have developed long-term relationships with these teenagers and their families, so the trust is already there. You should be the one to ensure all patients have appropriate sex education and help families with this.
|
|
Dr. Elisabeth Quint |
For some of these teens who are cognitively impaired, the initial conversations about sex may focus more on safety and abuse prevention. For example, which parts of their body should not be touched by other people. You can help the families really be the educators. Parents can be the ones to teach their kids how to protect themselves by rehearsing the answers to questions like, “What do you do if someone touches you? Who do you tell? Where do you go? What if it happens at school?” As part of the safety aspects, you also can help families assess whether the patient will be able to have a consensual sexual relationship. It’s the teens who have mild cognitive impairment that I worry about most, because often they are friendly and open to people, and can be taken advantage of. You just want to make sure they have the right information at their appropriate level.
Adolescents with physical disabilities are going to be just as interested in sex as any other teens and should be helped with any potential issues that they may have around that issue. They will likely get sex education in schools, but are still often viewed as not interested in sex or sexually active, and they may not get the usual confidential teen questions or appropriate screenings. Menstrual management and sexuality education both are important aspects of reproductive health care for teens with disabilities.
Dr. Elisabeth Quint, lead author of the AAP clinical report “Menstrual Management for Adolescents,” is a clinical professor of obstetrics and gynecology at the University of Michigan, Ann Arbor. She is also a past president of the North American Society for Pediatric and Adolescent Gynecology.
For the first time, the American Academy of Pediatrics is offering guidance on managing menstruation and sexuality education in adolescents with disabilities.
Written jointly with the North American Society for Pediatric and Adolescent Gynecology, the clinical report offers guidance on options for menstrual management, sexual education and expression, and protection from sexual abuse. The report gives guidance regarding the care of adolescents with physical and/or intellectual abilities, but not for those with psychiatric illnesses (Pediatrics. 2016 April. doi: 10.1542/peds.2016-0295).
“Taking care of teens with disabilities and figuring out what to do with menstrual management has been all over the map,” Dr. Cora Collette Breuner, chair of the AAP’s committee on adolescence, said in an interview. “So, we tried to clarify it and help clinicians know what to do and when.”
A particular concern for the two groups was the threat of adverse events. “We wanted to cover what’s safe and what’s not in menstruation management, especially around bone health and thromboembolic events,” said Dr. Breuner, a professor of pediatrics and adolescent medicine at the University of Washington, Seattle.
Some of the information will not surprise clinicians, but there are some data that will perhaps come as news, said Dr. Breuner, including the recommendation that long-acting reversible contraception (LARC), such as the levonorgestrel intrauterine device or the progesterone implant rod should be considered first-line management therapies. “We point out that a number of studies show that these are safe.”
The report emphasizes offering anticipatory guidance before menses begins, noting that most teens with disabilities mature at the same rate as teens without disabilities. The report does not recommend premenarchal suppression in these patients, because doing so can interfere with normal bone growth. Such suppression also prevents patients and their families and caregivers from discovering that coping with the onset of menses is perhaps not as difficult as they might fear, the report states.
Although combined oral contraceptives are not contraindicated in teens with mobility issues, to guard against the threat of thromboembolic events in teens who use wheelchairs, the report recommends taking a thorough family history to rule out inherited thrombophilia. Otherwise, the recommendation is to prescribe the lowest-dose estrogen with a first- or second-generation progestin, as these are associated with lower rates of venous thrombotic events.
The guidance states that if cycles are creating difficulties in the patient’s life, “as determined by health care providers, patients, and families,” then menstrual management is appropriate. Even though it may take up to 3 years before a menstrual cycle becomes regular, the report cites irregularities caused by certain medications can be reason enough for menstrual management. Specific drugs noted include those affecting the dopaminergic system, valproic acid, and medications that elevate prolactin. Teens with obesity, seizure disorders, and polycystic ovary syndrome also can experience higher rates of irregularity.
The report also warns against the assumption that teens with disabilities are asexual or uninterested in sex. When appropriate, they should be offered the same confidential conversations about sexuality as are recommended for all teenagers by the AAP and the American College of Obstetricians and Gynecologists. “Teenagers with physical disabilities are just as likely to be sexually active as their peers and have a higher incidence of sexual abuse,” the report states. It is typically when a patient is cognitively impaired that consent to confidential services may require “discussion about legal guardianship or medical power of attorney status for families,” according to the report.
The report’s comprehensive review of four main menstrual management techniques – estrogen-containing, progestin-only, nonhormonal methods, and surgical requests and options – begins with the caveat that regardless of the method used, the threat of abuse or sexually transmitted infections remain. When a patient’s family or caregivers request suppression of menarche in a patient, stating fears of abuse or pregnancy, further investigation into the patient’s circumstances is warranted, the report states.
“It’s always worth reminding physicians that in this cohort, endometrial ablation can have legal implications, and it’s not recommended in this age group,” Dr. Breuner said.
On average, 1.5 hormonal methods are tried before achieving management goals, according to the report. Data cited in the study showed that at 42%, oral contraception is the preferred method of menstrual suppression, followed by the patch at 20%. Expectant management was third at 15%, followed by DMPA (depot medroxyprogesterone acetate) at 12%. The least utilized method was the levonorgestrel intrauterine device at 3%. No data were provided for the implantable contraceptive rod.
The clinical report is a companion document to another AAP clinical report, “Sexuality of Children and Adolescents with Developmental Disabilities” (Pediatrics. 2006. doi: 10.1542/peds.2006-1115).
AAP guidance on these matters in teens with psychiatric illnesses is expected to be issued within a few years, Dr. Breuner said.
There was no external funding and the authors have no relevant financial disclosures.
On Twitter @whitneymcknight
For the first time, the American Academy of Pediatrics is offering guidance on managing menstruation and sexuality education in adolescents with disabilities.
Written jointly with the North American Society for Pediatric and Adolescent Gynecology, the clinical report offers guidance on options for menstrual management, sexual education and expression, and protection from sexual abuse. The report gives guidance regarding the care of adolescents with physical and/or intellectual abilities, but not for those with psychiatric illnesses (Pediatrics. 2016 April. doi: 10.1542/peds.2016-0295).
“Taking care of teens with disabilities and figuring out what to do with menstrual management has been all over the map,” Dr. Cora Collette Breuner, chair of the AAP’s committee on adolescence, said in an interview. “So, we tried to clarify it and help clinicians know what to do and when.”
A particular concern for the two groups was the threat of adverse events. “We wanted to cover what’s safe and what’s not in menstruation management, especially around bone health and thromboembolic events,” said Dr. Breuner, a professor of pediatrics and adolescent medicine at the University of Washington, Seattle.
Some of the information will not surprise clinicians, but there are some data that will perhaps come as news, said Dr. Breuner, including the recommendation that long-acting reversible contraception (LARC), such as the levonorgestrel intrauterine device or the progesterone implant rod should be considered first-line management therapies. “We point out that a number of studies show that these are safe.”
The report emphasizes offering anticipatory guidance before menses begins, noting that most teens with disabilities mature at the same rate as teens without disabilities. The report does not recommend premenarchal suppression in these patients, because doing so can interfere with normal bone growth. Such suppression also prevents patients and their families and caregivers from discovering that coping with the onset of menses is perhaps not as difficult as they might fear, the report states.
Although combined oral contraceptives are not contraindicated in teens with mobility issues, to guard against the threat of thromboembolic events in teens who use wheelchairs, the report recommends taking a thorough family history to rule out inherited thrombophilia. Otherwise, the recommendation is to prescribe the lowest-dose estrogen with a first- or second-generation progestin, as these are associated with lower rates of venous thrombotic events.
The guidance states that if cycles are creating difficulties in the patient’s life, “as determined by health care providers, patients, and families,” then menstrual management is appropriate. Even though it may take up to 3 years before a menstrual cycle becomes regular, the report cites irregularities caused by certain medications can be reason enough for menstrual management. Specific drugs noted include those affecting the dopaminergic system, valproic acid, and medications that elevate prolactin. Teens with obesity, seizure disorders, and polycystic ovary syndrome also can experience higher rates of irregularity.
The report also warns against the assumption that teens with disabilities are asexual or uninterested in sex. When appropriate, they should be offered the same confidential conversations about sexuality as are recommended for all teenagers by the AAP and the American College of Obstetricians and Gynecologists. “Teenagers with physical disabilities are just as likely to be sexually active as their peers and have a higher incidence of sexual abuse,” the report states. It is typically when a patient is cognitively impaired that consent to confidential services may require “discussion about legal guardianship or medical power of attorney status for families,” according to the report.
The report’s comprehensive review of four main menstrual management techniques – estrogen-containing, progestin-only, nonhormonal methods, and surgical requests and options – begins with the caveat that regardless of the method used, the threat of abuse or sexually transmitted infections remain. When a patient’s family or caregivers request suppression of menarche in a patient, stating fears of abuse or pregnancy, further investigation into the patient’s circumstances is warranted, the report states.
“It’s always worth reminding physicians that in this cohort, endometrial ablation can have legal implications, and it’s not recommended in this age group,” Dr. Breuner said.
On average, 1.5 hormonal methods are tried before achieving management goals, according to the report. Data cited in the study showed that at 42%, oral contraception is the preferred method of menstrual suppression, followed by the patch at 20%. Expectant management was third at 15%, followed by DMPA (depot medroxyprogesterone acetate) at 12%. The least utilized method was the levonorgestrel intrauterine device at 3%. No data were provided for the implantable contraceptive rod.
The clinical report is a companion document to another AAP clinical report, “Sexuality of Children and Adolescents with Developmental Disabilities” (Pediatrics. 2006. doi: 10.1542/peds.2006-1115).
AAP guidance on these matters in teens with psychiatric illnesses is expected to be issued within a few years, Dr. Breuner said.
There was no external funding and the authors have no relevant financial disclosures.
On Twitter @whitneymcknight
FROM PEDIATRICS

Consider Fusobacterium in culture-negative pharyngitis
BOSTON – An underappreciated cause of bacterial pharyngitis had a similar clinical presentation to group A Streptococcus (GAS), although prevalence was low in the population of 300 pediatric patients in a single-site study.
The 10 patients (3.3%) who had positive cultures for Fusobacterium necrophorum were about as likely as those with GAS to have fever, sore throat, exudate, and absence of cough. GAS cultures were positive in 57 (19%) of the patients.

F. necrophorum is a common cause of serious bacterial pharyngitis, especially in adolescents and young adults. The gram-negative species, an obligate anaerobe, is a cause of Lemierre’s syndrome, and “has recently been identified to be an important pathogen of bacterial pharyngitis with higher prevalence than group A Streptococcus (GAS) in adolescents and young adults,” wrote Tam Van, Ph.D., and her colleagues in a poster presented at the annual meeting of the American Society for Microbiology.
To examine the prevalence and disease characteristics of F. necrophorum in the emergency department patient population at Children’s Hospital of Los Angeles, Dr Van, a medical microbiology fellow at the hospital, and her colleagues enrolled 300 patients with pharyngitis aged 1-20 years (mean, 7.8 years).
All patients’ throats were swabbed, and investigators conducted a rapid antigen detection test (RADT) for group A beta-hemolytic Streptococcus and cultured samples for Streptococcus on a blood agar plate, according to usual care; samples also were cultured anaerobically and tested via polymerase chain reaction (PCR) for F. necrophorum.
A total of 67 patients had positive culture or PCR results for both species. Fifteen of the RADT tests were positive, while 57 cultures returned positive for GAS growth. Nine of the 10 positive F. necrophorum PCR tests correlated with positive culture results for that species.
Luckily, said Dr. Van, penicillin is an effective treatment for F. necrophorum, although it’s a gram-negative bacterium, so if a patient is coinfected with F. necrophorum and GAS, or treated for GAS empirically, then standard of care treatment should be effective, she said. However, since the species is associated with serious complications such as Lemierre’s disease, close follow-up and a low threshold for aggressive treatment are warranted if F. necrophorum is suspected or identified.
The relatively low positive culture rate of 3.3% for F. necrophorum in the study population was a bit surprising, Dr. Van said in an interview but was perhaps accounted for by the relatively young age of the Children’s Hospital Los Angeles patients. “Previous reports looked at adolescents and young adults,” wrote Dr. Van and her colleagues, while two-thirds of the patients in their study were under the age of 10 years. “This may contribute to the difference in prevalence.”
“Although rare, recovery of F. necrophorum correlated with true signs and symptoms of bacterial pharyngitis,” wrote Dr. Van and her colleagues. Serious pharyngitis with a negative rapid test and culture for group A Streptococcus should prompt clinical suspicion for F. necrophorum, especially in older adolescents and young adults, said Dr. Tam.
Dr. Tam and her coauthors reported no outside sources of funding and reported no relevant financial disclosures.
On Twitter @karioakes
BOSTON – An underappreciated cause of bacterial pharyngitis had a similar clinical presentation to group A Streptococcus (GAS), although prevalence was low in the population of 300 pediatric patients in a single-site study.
The 10 patients (3.3%) who had positive cultures for Fusobacterium necrophorum were about as likely as those with GAS to have fever, sore throat, exudate, and absence of cough. GAS cultures were positive in 57 (19%) of the patients.
F. necrophorum is a common cause of serious bacterial pharyngitis, especially in adolescents and young adults. The gram-negative species, an obligate anaerobe, is a cause of Lemierre’s syndrome, and “has recently been identified to be an important pathogen of bacterial pharyngitis with higher prevalence than group A Streptococcus (GAS) in adolescents and young adults,” wrote Tam Van, Ph.D., and her colleagues in a poster presented at the annual meeting of the American Society for Microbiology.
To examine the prevalence and disease characteristics of F. necrophorum in the emergency department patient population at Children’s Hospital of Los Angeles, Dr Van, a medical microbiology fellow at the hospital, and her colleagues enrolled 300 patients with pharyngitis aged 1-20 years (mean, 7.8 years).
All patients’ throats were swabbed, and investigators conducted a rapid antigen detection test (RADT) for group A beta-hemolytic Streptococcus and cultured samples for Streptococcus on a blood agar plate, according to usual care; samples also were cultured anaerobically and tested via polymerase chain reaction (PCR) for F. necrophorum.
A total of 67 patients had positive culture or PCR results for both species. Fifteen of the RADT tests were positive, while 57 cultures returned positive for GAS growth. Nine of the 10 positive F. necrophorum PCR tests correlated with positive culture results for that species.
Luckily, said Dr. Van, penicillin is an effective treatment for F. necrophorum, although it’s a gram-negative bacterium, so if a patient is coinfected with F. necrophorum and GAS, or treated for GAS empirically, then standard of care treatment should be effective, she said. However, since the species is associated with serious complications such as Lemierre’s disease, close follow-up and a low threshold for aggressive treatment are warranted if F. necrophorum is suspected or identified.
The relatively low positive culture rate of 3.3% for F. necrophorum in the study population was a bit surprising, Dr. Van said in an interview but was perhaps accounted for by the relatively young age of the Children’s Hospital Los Angeles patients. “Previous reports looked at adolescents and young adults,” wrote Dr. Van and her colleagues, while two-thirds of the patients in their study were under the age of 10 years. “This may contribute to the difference in prevalence.”
“Although rare, recovery of F. necrophorum correlated with true signs and symptoms of bacterial pharyngitis,” wrote Dr. Van and her colleagues. Serious pharyngitis with a negative rapid test and culture for group A Streptococcus should prompt clinical suspicion for F. necrophorum, especially in older adolescents and young adults, said Dr. Tam.
Dr. Tam and her coauthors reported no outside sources of funding and reported no relevant financial disclosures.
On Twitter @karioakes
BOSTON – An underappreciated cause of bacterial pharyngitis had a similar clinical presentation to group A Streptococcus (GAS), although prevalence was low in the population of 300 pediatric patients in a single-site study.
The 10 patients (3.3%) who had positive cultures for Fusobacterium necrophorum were about as likely as those with GAS to have fever, sore throat, exudate, and absence of cough. GAS cultures were positive in 57 (19%) of the patients.
F. necrophorum is a common cause of serious bacterial pharyngitis, especially in adolescents and young adults. The gram-negative species, an obligate anaerobe, is a cause of Lemierre’s syndrome, and “has recently been identified to be an important pathogen of bacterial pharyngitis with higher prevalence than group A Streptococcus (GAS) in adolescents and young adults,” wrote Tam Van, Ph.D., and her colleagues in a poster presented at the annual meeting of the American Society for Microbiology.
To examine the prevalence and disease characteristics of F. necrophorum in the emergency department patient population at Children’s Hospital of Los Angeles, Dr Van, a medical microbiology fellow at the hospital, and her colleagues enrolled 300 patients with pharyngitis aged 1-20 years (mean, 7.8 years).
All patients’ throats were swabbed, and investigators conducted a rapid antigen detection test (RADT) for group A beta-hemolytic Streptococcus and cultured samples for Streptococcus on a blood agar plate, according to usual care; samples also were cultured anaerobically and tested via polymerase chain reaction (PCR) for F. necrophorum.
A total of 67 patients had positive culture or PCR results for both species. Fifteen of the RADT tests were positive, while 57 cultures returned positive for GAS growth. Nine of the 10 positive F. necrophorum PCR tests correlated with positive culture results for that species.
Luckily, said Dr. Van, penicillin is an effective treatment for F. necrophorum, although it’s a gram-negative bacterium, so if a patient is coinfected with F. necrophorum and GAS, or treated for GAS empirically, then standard of care treatment should be effective, she said. However, since the species is associated with serious complications such as Lemierre’s disease, close follow-up and a low threshold for aggressive treatment are warranted if F. necrophorum is suspected or identified.
The relatively low positive culture rate of 3.3% for F. necrophorum in the study population was a bit surprising, Dr. Van said in an interview but was perhaps accounted for by the relatively young age of the Children’s Hospital Los Angeles patients. “Previous reports looked at adolescents and young adults,” wrote Dr. Van and her colleagues, while two-thirds of the patients in their study were under the age of 10 years. “This may contribute to the difference in prevalence.”
“Although rare, recovery of F. necrophorum correlated with true signs and symptoms of bacterial pharyngitis,” wrote Dr. Van and her colleagues. Serious pharyngitis with a negative rapid test and culture for group A Streptococcus should prompt clinical suspicion for F. necrophorum, especially in older adolescents and young adults, said Dr. Tam.
Dr. Tam and her coauthors reported no outside sources of funding and reported no relevant financial disclosures.
On Twitter @karioakes
AT ASM MICROBE 2016
Key clinical point: Fusobacterium necrophorum has a similar presentation to group A Streptococcus (GAS) pharyngitis.
Major finding: Pediatric patients with F. necrophorum pharyngitis were about as likely as those with GAS to have fever, exudates, adenopathy, and no cough.
Data source: 300 pediatric emergency department patients with pharyngitis who received antigen testing, cultures, and PCR to identify both causative agents.
Disclosures: The study investigators reported no disclosures.

Debunking Psoriasis Myths: Can Diet Clear Psoriasis?
Myth: Psoriasis Can Be Treated By Eating or Avoiding Certain Foods
Patients who Google “diet and psoriasis” are flooded with search results of diets claiming to cure psoriasis. This misinformation is dangerous for patients, as there is no scientific evidence that any specific psoriasis diet can treat the condition. Patients may wish to improve their diet to prevent comorbidities such as cardiovascular disease and metabolic syndrome. Even though it may not be a cure, encouraging patients to eat healthy is never a bad thing.
In a 2014 analysis of psoriasis, obesity, body mass index (BMI), and diet literature, an increased risk for psoriasis development in the setting of obesity was discussed. There is evidence suggesting that a BMI greater than 30 kg/m2 may potentially play a role in the ability to achieve a full therapeutic effect of psoriasis therapy. “This could be for two possible reasons,” Debbaneh et al reported. “It may be a consequence of decreased drug distribution into the body due to increased body mass, or it may be a consequence of increased pro-inflammatory cytokine release as a result of increased adipocyte count.” However, this finding may be treatment specific. For example, higher body weight was an independent predictor of response to ustekinumab, providing the rationale for offering 2 weight-based dosing regimens of the drug. Overweight and obese patients also were less likely to experience clearance with adalimumab. However, studies have found no association between BMI and biologic treatment.
Weight loss through a low-calorie diet has been reported to achieve a greater reduction in psoriasis severity and a slower rebound of disease. “Interestingly, studies have shown that caloric restriction in obese subjects lowers the level of circulating inflammatory cytokines,” reported Debbaneh et al. “This may contribute to the observed beneficial effect in psoriatic disease.”
In patients whose disease has had a significant impact on quality of life, it is important that they are consulting resources online that will help them maintain a healthy lifestyle. The National Psoriasis Foundation provides useful information on diet and psoriasis, emphasizing that diet is not going to cure psoriatic disease but eating healthier can only help.
Expert Commentary
Many of my psoriasis patients ask me what should they avoid eating to prevent the psoriasis from worsening, or what did they eat to cause psoriasis to occur in the first place. I stress to patients that what they eat is not likely the cause of their psoriasis nor will avoiding certain foods prevent a flare. However, alcohol use may induce a psoriasis flare.
—Jashin J. Wu, MD (Los Angeles, California)
Debbaneh M, Millsop JW, Bhatia BK, et al. Diet and psoriasis: part I. impact of weight loss interventions. J Am Acad Dermatol. 2014;71:133-140.
National Psoriasis Foundation. Diet and psoriasis. https://www.psoriasis.org/about-psoriasis/treatments/alternative/diet-supplements. Accessed June 20, 2016.
Myth: Psoriasis Can Be Treated By Eating or Avoiding Certain Foods
Patients who Google “diet and psoriasis” are flooded with search results of diets claiming to cure psoriasis. This misinformation is dangerous for patients, as there is no scientific evidence that any specific psoriasis diet can treat the condition. Patients may wish to improve their diet to prevent comorbidities such as cardiovascular disease and metabolic syndrome. Even though it may not be a cure, encouraging patients to eat healthy is never a bad thing.
In a 2014 analysis of psoriasis, obesity, body mass index (BMI), and diet literature, an increased risk for psoriasis development in the setting of obesity was discussed. There is evidence suggesting that a BMI greater than 30 kg/m2 may potentially play a role in the ability to achieve a full therapeutic effect of psoriasis therapy. “This could be for two possible reasons,” Debbaneh et al reported. “It may be a consequence of decreased drug distribution into the body due to increased body mass, or it may be a consequence of increased pro-inflammatory cytokine release as a result of increased adipocyte count.” However, this finding may be treatment specific. For example, higher body weight was an independent predictor of response to ustekinumab, providing the rationale for offering 2 weight-based dosing regimens of the drug. Overweight and obese patients also were less likely to experience clearance with adalimumab. However, studies have found no association between BMI and biologic treatment.
Weight loss through a low-calorie diet has been reported to achieve a greater reduction in psoriasis severity and a slower rebound of disease. “Interestingly, studies have shown that caloric restriction in obese subjects lowers the level of circulating inflammatory cytokines,” reported Debbaneh et al. “This may contribute to the observed beneficial effect in psoriatic disease.”
In patients whose disease has had a significant impact on quality of life, it is important that they are consulting resources online that will help them maintain a healthy lifestyle. The National Psoriasis Foundation provides useful information on diet and psoriasis, emphasizing that diet is not going to cure psoriatic disease but eating healthier can only help.
Expert Commentary
Many of my psoriasis patients ask me what should they avoid eating to prevent the psoriasis from worsening, or what did they eat to cause psoriasis to occur in the first place. I stress to patients that what they eat is not likely the cause of their psoriasis nor will avoiding certain foods prevent a flare. However, alcohol use may induce a psoriasis flare.
—Jashin J. Wu, MD (Los Angeles, California)
Myth: Psoriasis Can Be Treated By Eating or Avoiding Certain Foods
Patients who Google “diet and psoriasis” are flooded with search results of diets claiming to cure psoriasis. This misinformation is dangerous for patients, as there is no scientific evidence that any specific psoriasis diet can treat the condition. Patients may wish to improve their diet to prevent comorbidities such as cardiovascular disease and metabolic syndrome. Even though it may not be a cure, encouraging patients to eat healthy is never a bad thing.
In a 2014 analysis of psoriasis, obesity, body mass index (BMI), and diet literature, an increased risk for psoriasis development in the setting of obesity was discussed. There is evidence suggesting that a BMI greater than 30 kg/m2 may potentially play a role in the ability to achieve a full therapeutic effect of psoriasis therapy. “This could be for two possible reasons,” Debbaneh et al reported. “It may be a consequence of decreased drug distribution into the body due to increased body mass, or it may be a consequence of increased pro-inflammatory cytokine release as a result of increased adipocyte count.” However, this finding may be treatment specific. For example, higher body weight was an independent predictor of response to ustekinumab, providing the rationale for offering 2 weight-based dosing regimens of the drug. Overweight and obese patients also were less likely to experience clearance with adalimumab. However, studies have found no association between BMI and biologic treatment.
Weight loss through a low-calorie diet has been reported to achieve a greater reduction in psoriasis severity and a slower rebound of disease. “Interestingly, studies have shown that caloric restriction in obese subjects lowers the level of circulating inflammatory cytokines,” reported Debbaneh et al. “This may contribute to the observed beneficial effect in psoriatic disease.”
In patients whose disease has had a significant impact on quality of life, it is important that they are consulting resources online that will help them maintain a healthy lifestyle. The National Psoriasis Foundation provides useful information on diet and psoriasis, emphasizing that diet is not going to cure psoriatic disease but eating healthier can only help.
Expert Commentary
Many of my psoriasis patients ask me what should they avoid eating to prevent the psoriasis from worsening, or what did they eat to cause psoriasis to occur in the first place. I stress to patients that what they eat is not likely the cause of their psoriasis nor will avoiding certain foods prevent a flare. However, alcohol use may induce a psoriasis flare.
—Jashin J. Wu, MD (Los Angeles, California)
Debbaneh M, Millsop JW, Bhatia BK, et al. Diet and psoriasis: part I. impact of weight loss interventions. J Am Acad Dermatol. 2014;71:133-140.
National Psoriasis Foundation. Diet and psoriasis. https://www.psoriasis.org/about-psoriasis/treatments/alternative/diet-supplements. Accessed June 20, 2016.
Debbaneh M, Millsop JW, Bhatia BK, et al. Diet and psoriasis: part I. impact of weight loss interventions. J Am Acad Dermatol. 2014;71:133-140.
National Psoriasis Foundation. Diet and psoriasis. https://www.psoriasis.org/about-psoriasis/treatments/alternative/diet-supplements. Accessed June 20, 2016.

Challenging ‘dogma’ of allografts in infectious endocarditis
When a patient undergoes aortic valve replacement for infective endocarditis, conventional thinking holds that cardiac surgeons should use homografts because they have greater resistance to infection, but a recent study of more than 300 cases at two academic medical centers concluded that homografts may not necessarily offer such a benefit.
The study, published in the June issue of the Journal of Thoracic and Cardiovascular Surgery (2016;151:1239-48), involved 304 consecutive adult patients on whom 30-40 different surgeons performed operations for active infective endocarditis (IE) in the aortic valve from 2002 to 2014.
“Our findings suggest that patient-specific factors, such as age and implant preference, as well as technical reconstructive considerations, should drive prosthetic choice, rather than surgical dogma,” said Joon Bum Kim, Ph.D., of Massachusetts General Hospital, Harvard Medical School, both in Boston, and Asan Medical Center in Seoul, Korea, and his colleagues.
The study found that cardiac surgeons favored homografts over conventional prostheses when the patient had prosthetic valve endocarditis (58.1% vs. 28.8%) and methicillin-resistant Staphylococcus aureus (25.6% vs. 12.1%), both significant differences.
“No significant benefit to the use of homografts was demonstrable with regard to resistance to reinfection in the setting of IE,” Dr. Kim and his colleagues said.
Because reinfection after valve replacement for IE is such a strong concern, the debate over which prosthesis is best has ensued for decades. The researchers pointed out that the evidence favoring autologous or allogeneic tissue over synthetic material in the infective field is weak, mostly built on single-armed observational studies without comparison to conventional prosthesis.
With that in mind, the researchers pooled data from two institutions to compare short- and long-term results for homograft vs. conventional prosthetic valves in patients with IE. In this study group, 86 (28.3%) had homografts, 139 (45.7%) had xenograft prostheses, and 79 (26%) mechanical prostheses. The homograft group had more than twice the rate of early death than did the conventional group – 19.8% vs. 9.2%, a significant difference (P = .019).
During follow-up, which ranged from 4.7 to 72.6 months, 60 patients (19.7%) of the total group died and 23 (7.7%) experienced reinfection, but rates did not vary between the homograft and conventional prosthesis groups, Dr. Kim and his colleagues reported.
Demographics were similar between the three groups with a few exceptions Those who received the mechanical prostheses were younger (mean age, 47.2 years vs. 55.6 and 59.8 for the homograft and xenograft groups, respectively), had lower rates of diabetes (5.1% vs. 10.5% and 12.2%) and had less-severe disease based on New York Heart Association functional class III or IV scores (34.2% vs. 54.7% and 53.2%). The types of IE pathogens also differed among the three groups; methicillin-resistant staphylococci was most common in the homograft group (25.6%), whereas the viridans group streptococci was the leading cause of IE in the mechanical (38% ) and xenograft groups (25.2% ).
The use of homografts involves a highly complex operation, typically requiring a complete aortic root replacement, which “may be the major drawback in recommending it to patients already at high risk of operative mortality,” the investigators wrote. The durability of homografts makes their use limited for younger patients, and such grafts are somewhat scarce and require cryopreservation. “Therefore, the notion that homografts are required may in practice present an obstacle to appropriate surgical management of patients who have IE,” Dr. Kim and his coauthors wrote. All patients but one in the homograft group received aortic arch replacement (98.8%) whereas 30 of the patients in the conventional group did so (13.8%).
The study findings are consistent with an earlier comparative study (Ann. Thorac. Surg. 2012;93:480-07), according to Dr. Kim and his colleagues. “These findings suggest that patient-specific factors, such as patient preferences and technical considerations, should be the principal drivers of choices of valve prostheses,” they said. “Furthermore, lack of access to homografts should not be considered an obstacle to surgical therapy for this serious condition.”
Coauthor Dr. Sundt disclosed that he is a consultant for Thrasos Therapeutics. Dr. Kim and the other coauthors had no financial disclosures.
The study by Dr. Kim and his colleagues joins a series of reports questioning conventional thinking on the use of homografts to prevent recurrent infective endocarditis (IE), but their propensity matching does not account for surgeon bias in selecting a prosthesis, Dr. James K. Kirklin of the University of Alabama at Birmingham said in his invited commentary (J Thorac. Cardiovasc Surg. 2016 May;151:1230-1).
For example, surgeon preference may account for the wide disparity in full root replacements, depending on the type of prosthesis, Dr. Kirklin said. “Some experienced homograft surgeons have preferred the intra-aortic cylinder technique or infracoronary implantation, which avoids the short-term and longer-term complexities of full root replacement and has demonstrated long-term structural durability equivalent to that of the full root replacement,” he said.
Also, experienced homograft surgeons may prefer the homograft for its resistance to infection and adaptability to severe root infection in individual patients, particularly in those with severe infection with an abscess. And he cautioned against the study’s implication that conventional prostheses are equivocal in the setting of IE.
“Of considerable importance, however, is the evidence-based conclusion that surgical referral of routine surgical aortic valve endocarditis to a center experienced with aortic homograft surgery is not necessary, and a justifiable expectation is that aortic valve endocarditis requiring operation can be safely and appropriately managed in centers with standard aortic valve surgery experience who do not have access to or experience with aortic valve homografts,” Dr. Kirklin concluded.
Dr. Kirklin had no financial relationships to disclose.

|
Dr. Christopher M. Feindel |
The series by Dr. Kim and his colleagues, one of the largest of acute infective endocarditis to date, provides further evidence that the type of prosthesis used in surgery for IE involving the aortic valve probably does not affect long-term outcomes or reinfection rates, Dr. Christopher M. Feindel of the University of Toronto said in his invited commentary (J Thorac Cardiovasc Surg. 2016 May;151:1249-50).
However, Dr. Feindel said, “numerous confounding factors” inherent in any observational study could raise questions about the conclusion.
“This article delivers an important message, although not all surgeons will agree with the statistical approach taken by Dr. Kim and his colleagues,” Dr. Feindel said. The propensity scoring method the study used lacked all baseline variables that affect treatment choice and outcomes, “a crucial assumption for effective use of the propensity score,” he said. However, given the multitude of variables in patients with acute and complex IE, he said most surgeons would be hard pressed to accept that’s even possible in the model the study used.
Dr. Feindel also said a close examination of the 115 patients who underwent root replacement would have been “very instructional,” and the lack of follow-up on valve-related complications in almost 25% of the patients is another limitation of the study.
Nonetheless, the conclusions of Dr. Kim and his colleagues are “reasonable,” Dr. Feindel said. “Clearly, this article contributes important additional information to the surgical management of IE that will help guide surgeons, especially when it comes to prosthesis of choice,” he concluded. “It is up to the reader to decide whether this report finally puts to rest the “dogma” that homografts should preferentially be used in the setting of IE.”
Dr. Feindel had no relationships to disclose.
The study by Dr. Kim and his colleagues joins a series of reports questioning conventional thinking on the use of homografts to prevent recurrent infective endocarditis (IE), but their propensity matching does not account for surgeon bias in selecting a prosthesis, Dr. James K. Kirklin of the University of Alabama at Birmingham said in his invited commentary (J Thorac. Cardiovasc Surg. 2016 May;151:1230-1).
For example, surgeon preference may account for the wide disparity in full root replacements, depending on the type of prosthesis, Dr. Kirklin said. “Some experienced homograft surgeons have preferred the intra-aortic cylinder technique or infracoronary implantation, which avoids the short-term and longer-term complexities of full root replacement and has demonstrated long-term structural durability equivalent to that of the full root replacement,” he said.
Also, experienced homograft surgeons may prefer the homograft for its resistance to infection and adaptability to severe root infection in individual patients, particularly in those with severe infection with an abscess. And he cautioned against the study’s implication that conventional prostheses are equivocal in the setting of IE.
“Of considerable importance, however, is the evidence-based conclusion that surgical referral of routine surgical aortic valve endocarditis to a center experienced with aortic homograft surgery is not necessary, and a justifiable expectation is that aortic valve endocarditis requiring operation can be safely and appropriately managed in centers with standard aortic valve surgery experience who do not have access to or experience with aortic valve homografts,” Dr. Kirklin concluded.
Dr. Kirklin had no financial relationships to disclose.
|
|
Dr. Christopher M. Feindel |
The series by Dr. Kim and his colleagues, one of the largest of acute infective endocarditis to date, provides further evidence that the type of prosthesis used in surgery for IE involving the aortic valve probably does not affect long-term outcomes or reinfection rates, Dr. Christopher M. Feindel of the University of Toronto said in his invited commentary (J Thorac Cardiovasc Surg. 2016 May;151:1249-50).
However, Dr. Feindel said, “numerous confounding factors” inherent in any observational study could raise questions about the conclusion.
“This article delivers an important message, although not all surgeons will agree with the statistical approach taken by Dr. Kim and his colleagues,” Dr. Feindel said. The propensity scoring method the study used lacked all baseline variables that affect treatment choice and outcomes, “a crucial assumption for effective use of the propensity score,” he said. However, given the multitude of variables in patients with acute and complex IE, he said most surgeons would be hard pressed to accept that’s even possible in the model the study used.
Dr. Feindel also said a close examination of the 115 patients who underwent root replacement would have been “very instructional,” and the lack of follow-up on valve-related complications in almost 25% of the patients is another limitation of the study.
Nonetheless, the conclusions of Dr. Kim and his colleagues are “reasonable,” Dr. Feindel said. “Clearly, this article contributes important additional information to the surgical management of IE that will help guide surgeons, especially when it comes to prosthesis of choice,” he concluded. “It is up to the reader to decide whether this report finally puts to rest the “dogma” that homografts should preferentially be used in the setting of IE.”
Dr. Feindel had no relationships to disclose.
The study by Dr. Kim and his colleagues joins a series of reports questioning conventional thinking on the use of homografts to prevent recurrent infective endocarditis (IE), but their propensity matching does not account for surgeon bias in selecting a prosthesis, Dr. James K. Kirklin of the University of Alabama at Birmingham said in his invited commentary (J Thorac. Cardiovasc Surg. 2016 May;151:1230-1).
For example, surgeon preference may account for the wide disparity in full root replacements, depending on the type of prosthesis, Dr. Kirklin said. “Some experienced homograft surgeons have preferred the intra-aortic cylinder technique or infracoronary implantation, which avoids the short-term and longer-term complexities of full root replacement and has demonstrated long-term structural durability equivalent to that of the full root replacement,” he said.
Also, experienced homograft surgeons may prefer the homograft for its resistance to infection and adaptability to severe root infection in individual patients, particularly in those with severe infection with an abscess. And he cautioned against the study’s implication that conventional prostheses are equivocal in the setting of IE.
“Of considerable importance, however, is the evidence-based conclusion that surgical referral of routine surgical aortic valve endocarditis to a center experienced with aortic homograft surgery is not necessary, and a justifiable expectation is that aortic valve endocarditis requiring operation can be safely and appropriately managed in centers with standard aortic valve surgery experience who do not have access to or experience with aortic valve homografts,” Dr. Kirklin concluded.
Dr. Kirklin had no financial relationships to disclose.
|
|
Dr. Christopher M. Feindel |
The series by Dr. Kim and his colleagues, one of the largest of acute infective endocarditis to date, provides further evidence that the type of prosthesis used in surgery for IE involving the aortic valve probably does not affect long-term outcomes or reinfection rates, Dr. Christopher M. Feindel of the University of Toronto said in his invited commentary (J Thorac Cardiovasc Surg. 2016 May;151:1249-50).
However, Dr. Feindel said, “numerous confounding factors” inherent in any observational study could raise questions about the conclusion.
“This article delivers an important message, although not all surgeons will agree with the statistical approach taken by Dr. Kim and his colleagues,” Dr. Feindel said. The propensity scoring method the study used lacked all baseline variables that affect treatment choice and outcomes, “a crucial assumption for effective use of the propensity score,” he said. However, given the multitude of variables in patients with acute and complex IE, he said most surgeons would be hard pressed to accept that’s even possible in the model the study used.
Dr. Feindel also said a close examination of the 115 patients who underwent root replacement would have been “very instructional,” and the lack of follow-up on valve-related complications in almost 25% of the patients is another limitation of the study.
Nonetheless, the conclusions of Dr. Kim and his colleagues are “reasonable,” Dr. Feindel said. “Clearly, this article contributes important additional information to the surgical management of IE that will help guide surgeons, especially when it comes to prosthesis of choice,” he concluded. “It is up to the reader to decide whether this report finally puts to rest the “dogma” that homografts should preferentially be used in the setting of IE.”
Dr. Feindel had no relationships to disclose.
When a patient undergoes aortic valve replacement for infective endocarditis, conventional thinking holds that cardiac surgeons should use homografts because they have greater resistance to infection, but a recent study of more than 300 cases at two academic medical centers concluded that homografts may not necessarily offer such a benefit.
The study, published in the June issue of the Journal of Thoracic and Cardiovascular Surgery (2016;151:1239-48), involved 304 consecutive adult patients on whom 30-40 different surgeons performed operations for active infective endocarditis (IE) in the aortic valve from 2002 to 2014.
“Our findings suggest that patient-specific factors, such as age and implant preference, as well as technical reconstructive considerations, should drive prosthetic choice, rather than surgical dogma,” said Joon Bum Kim, Ph.D., of Massachusetts General Hospital, Harvard Medical School, both in Boston, and Asan Medical Center in Seoul, Korea, and his colleagues.
The study found that cardiac surgeons favored homografts over conventional prostheses when the patient had prosthetic valve endocarditis (58.1% vs. 28.8%) and methicillin-resistant Staphylococcus aureus (25.6% vs. 12.1%), both significant differences.
“No significant benefit to the use of homografts was demonstrable with regard to resistance to reinfection in the setting of IE,” Dr. Kim and his colleagues said.
Because reinfection after valve replacement for IE is such a strong concern, the debate over which prosthesis is best has ensued for decades. The researchers pointed out that the evidence favoring autologous or allogeneic tissue over synthetic material in the infective field is weak, mostly built on single-armed observational studies without comparison to conventional prosthesis.
With that in mind, the researchers pooled data from two institutions to compare short- and long-term results for homograft vs. conventional prosthetic valves in patients with IE. In this study group, 86 (28.3%) had homografts, 139 (45.7%) had xenograft prostheses, and 79 (26%) mechanical prostheses. The homograft group had more than twice the rate of early death than did the conventional group – 19.8% vs. 9.2%, a significant difference (P = .019).
During follow-up, which ranged from 4.7 to 72.6 months, 60 patients (19.7%) of the total group died and 23 (7.7%) experienced reinfection, but rates did not vary between the homograft and conventional prosthesis groups, Dr. Kim and his colleagues reported.
Demographics were similar between the three groups with a few exceptions Those who received the mechanical prostheses were younger (mean age, 47.2 years vs. 55.6 and 59.8 for the homograft and xenograft groups, respectively), had lower rates of diabetes (5.1% vs. 10.5% and 12.2%) and had less-severe disease based on New York Heart Association functional class III or IV scores (34.2% vs. 54.7% and 53.2%). The types of IE pathogens also differed among the three groups; methicillin-resistant staphylococci was most common in the homograft group (25.6%), whereas the viridans group streptococci was the leading cause of IE in the mechanical (38% ) and xenograft groups (25.2% ).
The use of homografts involves a highly complex operation, typically requiring a complete aortic root replacement, which “may be the major drawback in recommending it to patients already at high risk of operative mortality,” the investigators wrote. The durability of homografts makes their use limited for younger patients, and such grafts are somewhat scarce and require cryopreservation. “Therefore, the notion that homografts are required may in practice present an obstacle to appropriate surgical management of patients who have IE,” Dr. Kim and his coauthors wrote. All patients but one in the homograft group received aortic arch replacement (98.8%) whereas 30 of the patients in the conventional group did so (13.8%).
The study findings are consistent with an earlier comparative study (Ann. Thorac. Surg. 2012;93:480-07), according to Dr. Kim and his colleagues. “These findings suggest that patient-specific factors, such as patient preferences and technical considerations, should be the principal drivers of choices of valve prostheses,” they said. “Furthermore, lack of access to homografts should not be considered an obstacle to surgical therapy for this serious condition.”
Coauthor Dr. Sundt disclosed that he is a consultant for Thrasos Therapeutics. Dr. Kim and the other coauthors had no financial disclosures.
When a patient undergoes aortic valve replacement for infective endocarditis, conventional thinking holds that cardiac surgeons should use homografts because they have greater resistance to infection, but a recent study of more than 300 cases at two academic medical centers concluded that homografts may not necessarily offer such a benefit.
The study, published in the June issue of the Journal of Thoracic and Cardiovascular Surgery (2016;151:1239-48), involved 304 consecutive adult patients on whom 30-40 different surgeons performed operations for active infective endocarditis (IE) in the aortic valve from 2002 to 2014.
“Our findings suggest that patient-specific factors, such as age and implant preference, as well as technical reconstructive considerations, should drive prosthetic choice, rather than surgical dogma,” said Joon Bum Kim, Ph.D., of Massachusetts General Hospital, Harvard Medical School, both in Boston, and Asan Medical Center in Seoul, Korea, and his colleagues.
The study found that cardiac surgeons favored homografts over conventional prostheses when the patient had prosthetic valve endocarditis (58.1% vs. 28.8%) and methicillin-resistant Staphylococcus aureus (25.6% vs. 12.1%), both significant differences.
“No significant benefit to the use of homografts was demonstrable with regard to resistance to reinfection in the setting of IE,” Dr. Kim and his colleagues said.
Because reinfection after valve replacement for IE is such a strong concern, the debate over which prosthesis is best has ensued for decades. The researchers pointed out that the evidence favoring autologous or allogeneic tissue over synthetic material in the infective field is weak, mostly built on single-armed observational studies without comparison to conventional prosthesis.
With that in mind, the researchers pooled data from two institutions to compare short- and long-term results for homograft vs. conventional prosthetic valves in patients with IE. In this study group, 86 (28.3%) had homografts, 139 (45.7%) had xenograft prostheses, and 79 (26%) mechanical prostheses. The homograft group had more than twice the rate of early death than did the conventional group – 19.8% vs. 9.2%, a significant difference (P = .019).
During follow-up, which ranged from 4.7 to 72.6 months, 60 patients (19.7%) of the total group died and 23 (7.7%) experienced reinfection, but rates did not vary between the homograft and conventional prosthesis groups, Dr. Kim and his colleagues reported.
Demographics were similar between the three groups with a few exceptions Those who received the mechanical prostheses were younger (mean age, 47.2 years vs. 55.6 and 59.8 for the homograft and xenograft groups, respectively), had lower rates of diabetes (5.1% vs. 10.5% and 12.2%) and had less-severe disease based on New York Heart Association functional class III or IV scores (34.2% vs. 54.7% and 53.2%). The types of IE pathogens also differed among the three groups; methicillin-resistant staphylococci was most common in the homograft group (25.6%), whereas the viridans group streptococci was the leading cause of IE in the mechanical (38% ) and xenograft groups (25.2% ).
The use of homografts involves a highly complex operation, typically requiring a complete aortic root replacement, which “may be the major drawback in recommending it to patients already at high risk of operative mortality,” the investigators wrote. The durability of homografts makes their use limited for younger patients, and such grafts are somewhat scarce and require cryopreservation. “Therefore, the notion that homografts are required may in practice present an obstacle to appropriate surgical management of patients who have IE,” Dr. Kim and his coauthors wrote. All patients but one in the homograft group received aortic arch replacement (98.8%) whereas 30 of the patients in the conventional group did so (13.8%).
The study findings are consistent with an earlier comparative study (Ann. Thorac. Surg. 2012;93:480-07), according to Dr. Kim and his colleagues. “These findings suggest that patient-specific factors, such as patient preferences and technical considerations, should be the principal drivers of choices of valve prostheses,” they said. “Furthermore, lack of access to homografts should not be considered an obstacle to surgical therapy for this serious condition.”
Coauthor Dr. Sundt disclosed that he is a consultant for Thrasos Therapeutics. Dr. Kim and the other coauthors had no financial disclosures.
FROM THE JOURNAL OF THORACIC AND CARDIOVASCULAR SURGERY
Key clinical point: Use of homografts showed no significant benefit, compared with conventional prosthetic valves when the patient has infective endocarditis involving the aortic valve.
Major finding: The homograft group had more than twice the rate of early death than the conventional group, 19.8% vs. 9.2%, but in longer-term follow-up, the survival rates did not differ between groups.
Data source: 304 consecutive adult patients from the perspective database of two tertiary academic centers who had surgery for active infective endocarditis involving the aortic valve from 2002 to 2014.
Disclosures: Coauthor Dr. Sundt, disclosed he is a consultant for Thrasos Therapeutics. Dr. Kim and the other coauthors had no financial disclosures.
Debunking Psoriasis Myths: Do Biologics Cause Cancer?
Myth: Biologics Cause Cancer
Biologics generally are safe and well-tolerated therapies; however, due to their immunosuppressive properties, the risk for lymphoma has been of potential concern, leading patients to believe that biologics cause cancer. The risk of some cancers, including some solid cancers, hematologic cancers, and skin cancers, appears to be increased in patients with psoriasis, possibly associated with chronic inflammation. Some psoriasis therapies may increase the risk for malignancy, including phototherapy with psoralen plus UVA, cyclosporine, and methotrexate.
Many studies have supported a favorable safety profile for biologics in terms of the risk for developing malignancy. In a 2015 analysis of 12,093 patients enrolled in PSOLAR (Psoriasis Longitudinal Assessment and Registry), none of the biologics were found to be associated with increased risk for malignancy.
In psoriasis patients with existing or prior malignancies, the benefits of biologic therapy to improve quality of life often outweigh the negligible risks for malignancy. However, coordinated care with oncology is recommended for psoriasis patients with a history of prior malignancy.
General recommendations from the American Academy of Dermatology indicate one should carefully consider the decision to use a tumor necrosis factor (TNF) antagonist in patients with a history of malignancy, particularly lymphoma. Short-term treatment with biologics (up to 4 years) appears to be safe with respect to lymphoma risk, especially with TNF-α inhibitors. The potential risk for melanoma, cutaneous T-cell lymphoma, and nonmelanoma skin cancer in patients treated with TNF inhibitors also has been raised.
Expert Commentary
OBSERVE-5 was a 5-year phase 4, prospective, multicenter surveillance registry of 2510 psoriasis patients with at least a baseline dose of etanercept (Kimball et al, 2015). There was no increased risk for cancer when compared to the Truven Health MarketScan database, which is a proxy for the general population.
ESPRIT is an ongoing, 10-year, international, prospective, observational registry of 6059 psoriasis patients with at least a baseline dose of adalimumab (Menter et al). There are no signals of increased risk for cancer.
We do not have enough numbers of psoriasis patients on secukinumab or ixekizumab yet, but their phase 3 trials also do not seem to indicate an increased risk for cancer.
—Jashin J. Wu, MD (Los Angeles, California)
American Academy of Dermatology. Psoriasis: TNF inhibitors general recommendations. https://www.aad.org/practice-tools/quality-care/clinical-guidelines/psoriasis/biologics/tnf-inhibitors-recommendations. Accessed June 14, 2016.
Dommasch E, Gelfand JM. Is there truly a risk of lymphoma from biologic therapies? Dermatol Ther. 2009;22:418-430.
Kimball AB, Rothman KJ, Kricorian G, et al. OBSERVE-5: Observational postmarketing safety surveillance registry of etanercept for the treatment of psoriasis final 5-year results. J Am Acad Dermatol. 2015;72:115-122.
Kimball AB, Schenfeld J, Accortt NA, et al. 5-year incidence rates of malignancies in psoriasis patients compared with the general US population. Poster presented at: Summer Meeting of the American Academy of Dermatology; August 6-10, 2014; Chicago, IL.
Menter A, Thaçi D, Papp KA, et al. Five-year analysis from the ESPRIT 10-year postmarketing surveillance registry of adalimumab treatment for moderate to severe psoriasis. J Am Acad Dermatol. 2015;73:410-419.e6.
Papp K, Gottlieb AB, Naldi L, et al. Safety surveillance for ustekinumab and other psoriasis treatments from the Psoriasis Longitudinal Assessment and Registry (PSOLAR). J Drugs Dermatol. 2015;14:706-714.
Patel S, Patel T, Kerdel FA. The risk of malignancy or progression of existing malignancy in patients with psoriasis treated with biologics: case report and review of the literature. Int J Dermatol. 2016;55:487-493.
Myth: Biologics Cause Cancer
Biologics generally are safe and well-tolerated therapies; however, due to their immunosuppressive properties, the risk for lymphoma has been of potential concern, leading patients to believe that biologics cause cancer. The risk of some cancers, including some solid cancers, hematologic cancers, and skin cancers, appears to be increased in patients with psoriasis, possibly associated with chronic inflammation. Some psoriasis therapies may increase the risk for malignancy, including phototherapy with psoralen plus UVA, cyclosporine, and methotrexate.
Many studies have supported a favorable safety profile for biologics in terms of the risk for developing malignancy. In a 2015 analysis of 12,093 patients enrolled in PSOLAR (Psoriasis Longitudinal Assessment and Registry), none of the biologics were found to be associated with increased risk for malignancy.
In psoriasis patients with existing or prior malignancies, the benefits of biologic therapy to improve quality of life often outweigh the negligible risks for malignancy. However, coordinated care with oncology is recommended for psoriasis patients with a history of prior malignancy.
General recommendations from the American Academy of Dermatology indicate one should carefully consider the decision to use a tumor necrosis factor (TNF) antagonist in patients with a history of malignancy, particularly lymphoma. Short-term treatment with biologics (up to 4 years) appears to be safe with respect to lymphoma risk, especially with TNF-α inhibitors. The potential risk for melanoma, cutaneous T-cell lymphoma, and nonmelanoma skin cancer in patients treated with TNF inhibitors also has been raised.
Expert Commentary
OBSERVE-5 was a 5-year phase 4, prospective, multicenter surveillance registry of 2510 psoriasis patients with at least a baseline dose of etanercept (Kimball et al, 2015). There was no increased risk for cancer when compared to the Truven Health MarketScan database, which is a proxy for the general population.
ESPRIT is an ongoing, 10-year, international, prospective, observational registry of 6059 psoriasis patients with at least a baseline dose of adalimumab (Menter et al). There are no signals of increased risk for cancer.
We do not have enough numbers of psoriasis patients on secukinumab or ixekizumab yet, but their phase 3 trials also do not seem to indicate an increased risk for cancer.
—Jashin J. Wu, MD (Los Angeles, California)
Myth: Biologics Cause Cancer
Biologics generally are safe and well-tolerated therapies; however, due to their immunosuppressive properties, the risk for lymphoma has been of potential concern, leading patients to believe that biologics cause cancer. The risk of some cancers, including some solid cancers, hematologic cancers, and skin cancers, appears to be increased in patients with psoriasis, possibly associated with chronic inflammation. Some psoriasis therapies may increase the risk for malignancy, including phototherapy with psoralen plus UVA, cyclosporine, and methotrexate.
Many studies have supported a favorable safety profile for biologics in terms of the risk for developing malignancy. In a 2015 analysis of 12,093 patients enrolled in PSOLAR (Psoriasis Longitudinal Assessment and Registry), none of the biologics were found to be associated with increased risk for malignancy.
In psoriasis patients with existing or prior malignancies, the benefits of biologic therapy to improve quality of life often outweigh the negligible risks for malignancy. However, coordinated care with oncology is recommended for psoriasis patients with a history of prior malignancy.
General recommendations from the American Academy of Dermatology indicate one should carefully consider the decision to use a tumor necrosis factor (TNF) antagonist in patients with a history of malignancy, particularly lymphoma. Short-term treatment with biologics (up to 4 years) appears to be safe with respect to lymphoma risk, especially with TNF-α inhibitors. The potential risk for melanoma, cutaneous T-cell lymphoma, and nonmelanoma skin cancer in patients treated with TNF inhibitors also has been raised.
Expert Commentary
OBSERVE-5 was a 5-year phase 4, prospective, multicenter surveillance registry of 2510 psoriasis patients with at least a baseline dose of etanercept (Kimball et al, 2015). There was no increased risk for cancer when compared to the Truven Health MarketScan database, which is a proxy for the general population.
ESPRIT is an ongoing, 10-year, international, prospective, observational registry of 6059 psoriasis patients with at least a baseline dose of adalimumab (Menter et al). There are no signals of increased risk for cancer.
We do not have enough numbers of psoriasis patients on secukinumab or ixekizumab yet, but their phase 3 trials also do not seem to indicate an increased risk for cancer.
—Jashin J. Wu, MD (Los Angeles, California)
American Academy of Dermatology. Psoriasis: TNF inhibitors general recommendations. https://www.aad.org/practice-tools/quality-care/clinical-guidelines/psoriasis/biologics/tnf-inhibitors-recommendations. Accessed June 14, 2016.
Dommasch E, Gelfand JM. Is there truly a risk of lymphoma from biologic therapies? Dermatol Ther. 2009;22:418-430.
Kimball AB, Rothman KJ, Kricorian G, et al. OBSERVE-5: Observational postmarketing safety surveillance registry of etanercept for the treatment of psoriasis final 5-year results. J Am Acad Dermatol. 2015;72:115-122.
Kimball AB, Schenfeld J, Accortt NA, et al. 5-year incidence rates of malignancies in psoriasis patients compared with the general US population. Poster presented at: Summer Meeting of the American Academy of Dermatology; August 6-10, 2014; Chicago, IL.
Menter A, Thaçi D, Papp KA, et al. Five-year analysis from the ESPRIT 10-year postmarketing surveillance registry of adalimumab treatment for moderate to severe psoriasis. J Am Acad Dermatol. 2015;73:410-419.e6.
Papp K, Gottlieb AB, Naldi L, et al. Safety surveillance for ustekinumab and other psoriasis treatments from the Psoriasis Longitudinal Assessment and Registry (PSOLAR). J Drugs Dermatol. 2015;14:706-714.
Patel S, Patel T, Kerdel FA. The risk of malignancy or progression of existing malignancy in patients with psoriasis treated with biologics: case report and review of the literature. Int J Dermatol. 2016;55:487-493.
American Academy of Dermatology. Psoriasis: TNF inhibitors general recommendations. https://www.aad.org/practice-tools/quality-care/clinical-guidelines/psoriasis/biologics/tnf-inhibitors-recommendations. Accessed June 14, 2016.
Dommasch E, Gelfand JM. Is there truly a risk of lymphoma from biologic therapies? Dermatol Ther. 2009;22:418-430.
Kimball AB, Rothman KJ, Kricorian G, et al. OBSERVE-5: Observational postmarketing safety surveillance registry of etanercept for the treatment of psoriasis final 5-year results. J Am Acad Dermatol. 2015;72:115-122.
Kimball AB, Schenfeld J, Accortt NA, et al. 5-year incidence rates of malignancies in psoriasis patients compared with the general US population. Poster presented at: Summer Meeting of the American Academy of Dermatology; August 6-10, 2014; Chicago, IL.
Menter A, Thaçi D, Papp KA, et al. Five-year analysis from the ESPRIT 10-year postmarketing surveillance registry of adalimumab treatment for moderate to severe psoriasis. J Am Acad Dermatol. 2015;73:410-419.e6.
Papp K, Gottlieb AB, Naldi L, et al. Safety surveillance for ustekinumab and other psoriasis treatments from the Psoriasis Longitudinal Assessment and Registry (PSOLAR). J Drugs Dermatol. 2015;14:706-714.
Patel S, Patel T, Kerdel FA. The risk of malignancy or progression of existing malignancy in patients with psoriasis treated with biologics: case report and review of the literature. Int J Dermatol. 2016;55:487-493.
FDA reports shortage of doxorubicin for injection, initiates importation
A critical shortage of doxorubicin hydrochloride 50 mg powder for injection has been reported to the Food and Drug Administration.
Doxorubicin is approved to treat acute lymphoblastic leukemia, acute myeloid leukemia, breast cancer, gastric cancer, ovarian cancer, neuroblastoma, and other cancer types.

To increase availability, the pharmaceutical company Hospira (a Pfizer company) is coordinating with the FDA to import the drug from Ahmedabad, India, where it is manufactured by Zydus Hospira Oncology Private Ltd. at an FDA-inspected facility that is in compliance with current good manufacturing practice requirements.
“It is important to note that there are substantive differences in the format and content of the labeling between the U.S.-approved doxorubicin hydrochloride for injection, USP and the Hospira Limited’s doxorubicin hydrochloride 50 mg powder for injection,” Hospira reported in a letter to health care providers.
To place an order or to get questions answered, contact Hospira directly by calling customer care at 1-877-946-7747 (Mondays-Fridays, 7 a.m.-6 p.m. Central time).
For clinical inquiries, contact Hospira Medical Communications at 1-800-615-0187 or email [email protected].
According to the letter, adverse events or quality problems associated with use of this product should be reported by calling Hospira Global Complaint Management by phone, 1-800-441-4100; by sending an email to [email protected]; or by submitting a report online to Medwatch.
On Twitter @jessnicolecraig
A critical shortage of doxorubicin hydrochloride 50 mg powder for injection has been reported to the Food and Drug Administration.
Doxorubicin is approved to treat acute lymphoblastic leukemia, acute myeloid leukemia, breast cancer, gastric cancer, ovarian cancer, neuroblastoma, and other cancer types.
To increase availability, the pharmaceutical company Hospira (a Pfizer company) is coordinating with the FDA to import the drug from Ahmedabad, India, where it is manufactured by Zydus Hospira Oncology Private Ltd. at an FDA-inspected facility that is in compliance with current good manufacturing practice requirements.
“It is important to note that there are substantive differences in the format and content of the labeling between the U.S.-approved doxorubicin hydrochloride for injection, USP and the Hospira Limited’s doxorubicin hydrochloride 50 mg powder for injection,” Hospira reported in a letter to health care providers.
To place an order or to get questions answered, contact Hospira directly by calling customer care at 1-877-946-7747 (Mondays-Fridays, 7 a.m.-6 p.m. Central time).
For clinical inquiries, contact Hospira Medical Communications at 1-800-615-0187 or email [email protected].
According to the letter, adverse events or quality problems associated with use of this product should be reported by calling Hospira Global Complaint Management by phone, 1-800-441-4100; by sending an email to [email protected]; or by submitting a report online to Medwatch.
On Twitter @jessnicolecraig
A critical shortage of doxorubicin hydrochloride 50 mg powder for injection has been reported to the Food and Drug Administration.
Doxorubicin is approved to treat acute lymphoblastic leukemia, acute myeloid leukemia, breast cancer, gastric cancer, ovarian cancer, neuroblastoma, and other cancer types.
To increase availability, the pharmaceutical company Hospira (a Pfizer company) is coordinating with the FDA to import the drug from Ahmedabad, India, where it is manufactured by Zydus Hospira Oncology Private Ltd. at an FDA-inspected facility that is in compliance with current good manufacturing practice requirements.
“It is important to note that there are substantive differences in the format and content of the labeling between the U.S.-approved doxorubicin hydrochloride for injection, USP and the Hospira Limited’s doxorubicin hydrochloride 50 mg powder for injection,” Hospira reported in a letter to health care providers.
To place an order or to get questions answered, contact Hospira directly by calling customer care at 1-877-946-7747 (Mondays-Fridays, 7 a.m.-6 p.m. Central time).
For clinical inquiries, contact Hospira Medical Communications at 1-800-615-0187 or email [email protected].
According to the letter, adverse events or quality problems associated with use of this product should be reported by calling Hospira Global Complaint Management by phone, 1-800-441-4100; by sending an email to [email protected]; or by submitting a report online to Medwatch.
On Twitter @jessnicolecraig

New HCV test approach could cut costs, streamline diagnosis
Substituting a less-expensive hepatitis C core antigen test into the standard two-step process for diagnosing active hepatitis C virus (HCV) infection could streamline and cut the cost of HCV detection.
The standard two-step approach – detection of HCV antibodies followed by nucleic acid testing (NAT) as a marker for HCV viremia – may be improved by replacing NAT with the HCV core antigen (HCVcAg) test, according to the results of a study published in Annals of Internal Medicine.
Dr. J. Morgan Freiman, of the Boston Medical Center, and her colleagues conducted a systematic literature review to identify 44 case-control, cross-sectional, cohort, or randomized trials that compared any of five HCVcAg screening tests with a NAT reference standard.
The investigators performed a meta-analysis to assess the sensitivity (proportion of samples with a positive NAT and HCVcAg) and specificity (proportion of samples with a negative NAT and HCVcAg) associated with the five HCVcAg tests, as well as how they correlated with NAT-derived HCV RNA levels greater than 3,000 IU/mL.
The two best-performing HCVcAg tests of the five assessed were the Abbott ARCHITECT HCV Ag and the Ortho HCV Ag ELISA, based on their sensitivity (93.4%and 93.2%, respectively), specificity (98.8% and 99.2%, respectively), and positive (80.6 and 116.5, respectively) and negative (0.06 and 0.06, respectively) likelihood ratios.
Although limited data were available, the results of three quantitative studies showed that Abbott ARCHITECT HCVcAg was well correlated with HCV RNA levels greater than 3,000 IU/mL.
“This systematic review concludes that a well-performing HCVcAg test can achieve similar diagnostic accuracy to NAT for identification of active HCV infection when the viral load exceeds 3,000 IU/mL,” Dr. Freiman and her colleagues noted.
HCVcAg testing should be researched further as a potentially viable and less-expensive alternative to NAT, the investigators said, with the goal of simplifying detection at the point of care and increasing the rate of patient diagnosis (Ann Intern Med. 2016 Jun 21; doi: 10.7326/M16-0065).
The National Institutes of Health funded the study. Dr. White disclosed grant support from the funding source. Dr. Ongarello and Dr. Denkinger reported relationships with the Foundation for Innovative New Diagnostics. No additional authors reported conflicts of interest.
Substituting a less-expensive hepatitis C core antigen test into the standard two-step process for diagnosing active hepatitis C virus (HCV) infection could streamline and cut the cost of HCV detection.
The standard two-step approach – detection of HCV antibodies followed by nucleic acid testing (NAT) as a marker for HCV viremia – may be improved by replacing NAT with the HCV core antigen (HCVcAg) test, according to the results of a study published in Annals of Internal Medicine.
Dr. J. Morgan Freiman, of the Boston Medical Center, and her colleagues conducted a systematic literature review to identify 44 case-control, cross-sectional, cohort, or randomized trials that compared any of five HCVcAg screening tests with a NAT reference standard.
The investigators performed a meta-analysis to assess the sensitivity (proportion of samples with a positive NAT and HCVcAg) and specificity (proportion of samples with a negative NAT and HCVcAg) associated with the five HCVcAg tests, as well as how they correlated with NAT-derived HCV RNA levels greater than 3,000 IU/mL.
The two best-performing HCVcAg tests of the five assessed were the Abbott ARCHITECT HCV Ag and the Ortho HCV Ag ELISA, based on their sensitivity (93.4%and 93.2%, respectively), specificity (98.8% and 99.2%, respectively), and positive (80.6 and 116.5, respectively) and negative (0.06 and 0.06, respectively) likelihood ratios.
Although limited data were available, the results of three quantitative studies showed that Abbott ARCHITECT HCVcAg was well correlated with HCV RNA levels greater than 3,000 IU/mL.
“This systematic review concludes that a well-performing HCVcAg test can achieve similar diagnostic accuracy to NAT for identification of active HCV infection when the viral load exceeds 3,000 IU/mL,” Dr. Freiman and her colleagues noted.
HCVcAg testing should be researched further as a potentially viable and less-expensive alternative to NAT, the investigators said, with the goal of simplifying detection at the point of care and increasing the rate of patient diagnosis (Ann Intern Med. 2016 Jun 21; doi: 10.7326/M16-0065).
The National Institutes of Health funded the study. Dr. White disclosed grant support from the funding source. Dr. Ongarello and Dr. Denkinger reported relationships with the Foundation for Innovative New Diagnostics. No additional authors reported conflicts of interest.
Substituting a less-expensive hepatitis C core antigen test into the standard two-step process for diagnosing active hepatitis C virus (HCV) infection could streamline and cut the cost of HCV detection.
The standard two-step approach – detection of HCV antibodies followed by nucleic acid testing (NAT) as a marker for HCV viremia – may be improved by replacing NAT with the HCV core antigen (HCVcAg) test, according to the results of a study published in Annals of Internal Medicine.
Dr. J. Morgan Freiman, of the Boston Medical Center, and her colleagues conducted a systematic literature review to identify 44 case-control, cross-sectional, cohort, or randomized trials that compared any of five HCVcAg screening tests with a NAT reference standard.
The investigators performed a meta-analysis to assess the sensitivity (proportion of samples with a positive NAT and HCVcAg) and specificity (proportion of samples with a negative NAT and HCVcAg) associated with the five HCVcAg tests, as well as how they correlated with NAT-derived HCV RNA levels greater than 3,000 IU/mL.
The two best-performing HCVcAg tests of the five assessed were the Abbott ARCHITECT HCV Ag and the Ortho HCV Ag ELISA, based on their sensitivity (93.4%and 93.2%, respectively), specificity (98.8% and 99.2%, respectively), and positive (80.6 and 116.5, respectively) and negative (0.06 and 0.06, respectively) likelihood ratios.
Although limited data were available, the results of three quantitative studies showed that Abbott ARCHITECT HCVcAg was well correlated with HCV RNA levels greater than 3,000 IU/mL.
“This systematic review concludes that a well-performing HCVcAg test can achieve similar diagnostic accuracy to NAT for identification of active HCV infection when the viral load exceeds 3,000 IU/mL,” Dr. Freiman and her colleagues noted.
HCVcAg testing should be researched further as a potentially viable and less-expensive alternative to NAT, the investigators said, with the goal of simplifying detection at the point of care and increasing the rate of patient diagnosis (Ann Intern Med. 2016 Jun 21; doi: 10.7326/M16-0065).
The National Institutes of Health funded the study. Dr. White disclosed grant support from the funding source. Dr. Ongarello and Dr. Denkinger reported relationships with the Foundation for Innovative New Diagnostics. No additional authors reported conflicts of interest.
FROM ANNALS OF INTERNAL MEDICINE
Key clinical point: The diagnostic rate of chronic hepatitis C virus may be increased by point of care hepatitis C virus core antigen assessment.
Major finding: For patients with chronic HCV infection and viral loads exceeding 3,000 IU/mL, an HCVcAg screen performed as well as nucleic acid testing.
Data sources: Case-control, cross-sectional, cohort, or randomized trials that compared any of five HCVcAg tests with a NAT reference standard.
Disclosures: The National Institutes of Health funded the study. Dr. White disclosed grant support from the funding source. Dr. Ongarello and Dr. Denkinger reported relationships with the Foundation for Innovative New Diagnostics. No additional authors reported conflicts of interest.